
Home » NDA (Page 3)
Category Archives: NDA
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Chelsea Therapeutics Announces FDA Advisory Committee to Review Northera(Droxidropa, 23651-95-8)

droxidropa
CHARLOTTE, N.C., Oct. 9, 2013 (GLOBE NEWSWIRE) — Chelsea Therapeutics International, Ltd. today announced that the U.S. Food and Drug Administration (FDA) has notified the Company that the New Drug Application (NDA) seeking approval to market Northera (droxidopa), an orally active synthetic precursor of norepinephrine, for the treatment of symptomatic neurogenic orthostatic hypotension (NOH) will be reviewed by the Cardiovascular and Renal Drug Advisory Committee (CRDAC). The meeting is tentatively scheduled for January 14, 2014 read more here————
http://www.drugs.com/nda/northera_131009.html
old article cut paste
FDA Deems Resubmission a Complete Response; PDUFA Date Set as
February 14, 2014
CHARLOTTE, N.C., Sept. 4, 2013 (GLOBE NEWSWIRE) — Chelsea Therapeutics International, Ltd. (Nasdaq:CHTP) today announced that the U.S. Food and Drug Administration (FDA) has acknowledged receipt of the New Drug Application (NDA) resubmission seeking approval to market NORTHERA(TM) (droxidopa), an orally active synthetic precursor of norepinephrine
read all at
http://www.pharmalive.com/chelsea-therapeutics-announces-fda-acceptance-of-northera-nda-resubmission
L-DOPS (L-threo-dihydroxyphenylserine; Droxidopa; SM-5688) is a psychoactive drug and synthetic amino acid precursor which acts as a prodrug to the neurotransmitters norepinephrine (noradrenaline) and epinephrine (adrenaline).[1] Unlike norepinephrine and epinephrine themselves, L-DOPS is capable of crossing the protective blood–brain barrier (BBB).[1]
Neurogenic orthostatic hypotension (NOH),[2] as well as NOH associated with multiple system atrophy (MSA), familial amyloid polyneuropathy (FAP), pure autonomic failure (PAF), and Parkinson’s disease (PD).
Intradialytic hypotension (IDH) or hemodialysis-induced hypotension.
Hypotension associated with fibromyalgia syndrome (FMS) and chronic fatigue syndrome (CFS).[3]
History
L-DOPS was developed by Sumitomo Pharmaceuticals under the trade name Droxidopa for the treatment of hypotension, including NOH,[2] and NOH associated with various disorders such as MSA, FAP, and PD, as well as IDH. The drug has been used in Japan and some surrounding Asian areas for these indications since 1989. Following a merge with Dainippon Pharmaceuticals in 2006, Dainippon Sumitomo Pharma licensed L-DOPS to Chelsea Therapeutics to develop and market it worldwide except in Japan, Korea, China, and Taiwan.
Clinical trials
Though L-DOPS has been used in Japan and Southeast Asia already for some time, it is also currently in clinical trials at the phase III point in the United States (U.S.), Canada, Australia, and throughout Europe. Provided L-DOPS successfully completes clinical trials, it could be approved for the treatment of NOH as early as 2011.[4] Additionally, phase II clinical trials for IDH are also underway. Chelsea Therapeutics obtained orphan drug status (ODS) for L-DOPS in the U.S. for NOH, and that of which associated with PD, PAF, and MSA, and is the pharmaceutical company developing it in that country.
- Goldstein, DS (2006). “L-Dihydroxyphenylserine (L-DOPS): a norepinephrine prodrug”. Cardiovasc Drug Rev 24 (3-4): 189–203. doi:10.1111/j.1527-3466.2006.00189.x. PMID 17214596.
- Mathias, Christopher J (2008). “L-dihydroxyphenylserine (Droxidopa) in the treatment of orthostatic hypotension”. Clin Auton Res 18 (Supplement 1): 25–29.doi:10.1007/s10286-007-1005-z.
- Crofford, LJ (2008). “Pain management in fibromyalgia”. Curr Opin Rheumatol 20 (3): 246–250. doi:10.1097/BOR.0b013e3282fb0268. PMID 18388513.
- Search of: “Droxidopa” – List Results – ClinicalTrials.gov
- Robertson, David (2008). “The pathophysiology and diagnosis of orthostatic hypotension”. Clin Auton Res 18 (Supplement 1): 2–7. doi:10.1007/s10286-007-1004-0.
FDA, EMA Accept Omeros Ophthalmology Product NDA
OMS302
US and European Regulators Accept for Review OMS302 Marketing Applications
— OMS302 Remains on Track for Planned 2014 Commercial Launch —
SEATTLE, Oct. 2, 2013 /PRNewswire/ — Omeros Corporation (NASDAQ: OMER) announced today that the New Drug Application (NDA) for its ophthalmology product, OMS302, has been confirmed for filing by the U.S. Food and Drug Administration (FDA), which means that the application, submitted in July of this year, is sufficiently complete to permit a substantive review. The company also announced that its Marketing Authorization Application (MAA) for OMS302, submitted last month, has been validated by the European Medicines Agency (EMA). Validation of the MAA confirms that the submission package is administratively complete and is ready for formal review by Europe’s Committee for Medicinal Products for Human Use (CHMP).
read all at
http://www.pharmalive.com/fda-ema-accept-omeros-opthamology-product-nda
FDA accepts new drug application for investigational compound Epanova for the treatment of severe hypertriglyceridaemia
LONDON, Sept. 18, 2013 – AstraZeneca today announced that the US Food and Drug Administration (FDA) has accepted for review a New Drug Application (NDA) for EpanovaTM, an investigational compound for the treatment for patients with severe hypertriglyceridaemia (triglyceride levels greater than or equal to 500mg/dL). The NDA submission for Epanova was filed by Omthera Pharmaceuticals, now a wholly-owned subsidiary of AstraZeneca, as a 505(b)(1) application in July 2013. The Prescription Drug User Fee Act (PDUFA) goal date for the FDA is 5 May 2014.http://www.pharmalive.com/fda-accepts-astrazeneca-nda-for-epanova
Sanofi to withdraw the lixisenatide New Drug Application (NDA) in the U.S., The company plans to resubmit the NDA in 2015, after completion of the ELIXA CV study.
lixisenatide
Sanofi Provides Update on Lixisenatide New Drug Application in U.S.
Paris, France – September 12, 2013 – Sanofi (EURONEXT: SAN and NYSE: SNY) announced today its decision to withdraw the lixisenatide New Drug Application (NDA) in the U.S., which included early interim results from the ongoing ELIXA cardiovascular (CV) outcomes study. The company plans to resubmit the NDA in 2015, after completion of the ELIXA CV study.
The decision to withdraw the lixisenatide application follows discussions with the U.S. Food and Drug Administration (FDA) regarding its proposed process for the review of interim data. Sanofi believes that potential public disclosure of early interim data, even with safeguards, could potentially compromise the integrity of the ongoing ELIXA study. Sanofi’s decision is not related to safety issues or deficiencies in the NDA………………………read all at
http://www.pharmalive.com/sanofi-pulls-diabetes-drug-nda
EU

Gilead Submits New Drug Application to U.S. FDA for Idelalisib for the Treatment of Indolent Non-Hodgkin’s Lymphoma
CAL 101, IDELALISIB
SEPT 2013
Gilead Submits New Drug Application to U.S. FDA for Idelalisib for the Treatment of Indolent Non-Hodgkin’s Lymphoma
FOSTER CITY, Calif.–(BUSINESS WIRE)–Sep. 11, 2013– Gilead Sciences, Inc. today announced that the company has submitted a New Drug Application (NDA) to the U.S. Food and Drug Administration (FDA) for approval of idelalisib, an investigational, targeted, oral inhibitor of PI3K delta, for the treatment of indolent non-Hodgkin’s lymphoma (iNHL). The data submitted in this NDA support the use of idelalisib for patients with iNHL that is refractory (non-responsive) to rituximab and to alkylating-agent-containing chemotherapy.
read all at
Idelalisib ….US FDA Accepts NDA for Gilead’s Idelalisib for the Treatment of Refractory Indolent Non-Hodgkin’s Lymphoma
JANUARY 14, 2014 8:35 AM / LEAVE A COMMENT

An antineoplastic agent and p110delta inhibitor
(S)-2-(1-(9H-purin-6-ylamino)propyl)-5-fluoro-3-phenylquinazolin-4(3H)-one
Icos (Originator)
- CAL-101
- GS-1101
- Idelalisib
- UNII-YG57I8T5M0
M.Wt: 415.43
Formula: C22H18FN7O
CAS No.: 870281-82-6
CAL-101 Solubility: DMSO ≥80mg/mL Water <1.2mg/mL Ethanol ≥33mg/mL
5-Fluoro-3-phenyl-2-[(1S)-1-(7H-purin-6-ylamino)propyl]-4(3H)-quinazolinone
idelalisib
Idelalisib (codenamed GS-1101 or CAL-101) is a drug under investigation for the treatment of chronic lymphocytic leukaemia. It is in Phase III clinical trials testing drug combinations with rituximab and/or bendamustine as of 2013. The substance acts as aphosphoinositide 3-kinase inhibitor; more specifically, it blocks P110δ, the delta isoform of the enzyme phosphoinositide 3-kinase.[1][2]
GDC-0032 is a potent, next-generation beta isoform-sparing PI3K inhibitor targeting PI3Kα/δ/γ with IC 50 of 0.29 nM/0.12 nM/0.97nM,> 10 fold over Selective PI3K [beta].
GS-1101 is a novel, orally available small molecule inhibitor of phosphatidylinositol 3-kinase delta (PI3Kdelta) develop by Gilead and is waiting for registration in U.S. for the treatment of patients with indolent non-Hodgkin’s lymphoma that is refractory (non-responsive) to rituximab and to alkylating-agent-containing chemotherapy and for the treatment of chronic lymphocytic leukemia. The compound is also in phase III clinical evaluation for the treatment of elderly patients with previously untreated small lymphocytic lymphoma (SLL) and acute myeloid leukemia. Clinical trials had been under way for the treatment of inflammation and allergic rhinitis; however, no recent development has been reported. Preclinical studies have shown that GS-1101 has desirable pharmaceutical properties. The compound was originally developed by Calistoga Pharmaceuticals, acquired by Gilead on April 1, 2011.
clinical trials, click link
http://clinicaltrials.gov/search/intervention=CAL-101%20OR%20GS-1101%20OR%20Idelalisib
FOSTER CITY, Calif.–(BUSINESS WIRE)–Jan. 13, 2014– Gilead Sciences, Inc. (Nasdaq: GILD) announced today that the U.S. Food and Drug Administration (FDA) has accepted for review the company’s New Drug Application (NDA) for idelalisib, a targeted, oral inhibitor of PI3K delta, for the treatment of refractory indolent non-Hodgkin’s lymphoma (iNHL). FDA has granted a standard review for the iNHL NDA and has set a target review date under the Prescription Drug User Fee Act (PDUFA) of September 11, 2014.
The NDA for iNHL, submitted on September 11, 2013, was supported by a single arm Phase 2 study (Study 101-09) evaluating idelalisib in patients with iNHL that is refractory (non-responsive) to rituximab and to alkylating-agent-containing chemotherapy. Following Gilead’s NDA submission for iNHL, FDA granted idelalisib a Breakthrough Therapy designation for relapsed chronic lymphocytic leukemia (CLL). The FDA grants Breakthrough Therapy designation to drug candidates that may offer major advances in treatment over existing options. Gilead submitted an NDA for idelalisib for the treatment of CLL on December 6, 2013.
About Idelalisib
Idelalisib is an investigational, highly selective oral inhibitor of phosphoinositide 3-kinase (PI3K) delta. PI3K delta signaling is critical for the activation, proliferation, survival and trafficking of B lymphocytes and is hyperactive in many B-cell malignancies. Idelalisib is being developed both as a single agent and in combination with approved and investigational therapies.
Gilead’s clinical development program for idelalisib in iNHL includes Study 101-09 in highly refractory patients and two Phase 3 studies of idelalisib in previously treated patients. The development program in CLL includes three Phase 3 studies of idelalisib in previously treated patients. Combination therapy with idelalisib and GS-9973, Gilead’s novel spleen tyrosine kinase (Syk) inhibitor, also is being evaluated in a Phase 2 trial of patients with relapsed or refractory CLL, iNHL and other lymphoid malignancies.
Additional information about clinical studies of idelalisib and Gilead’s other investigational cancer agents can be found at http://www.clinicaltrials.gov. Idelalisib and GS-9973 are investigational products and their safety and efficacy have not been established.
About Indolent Non-Hodgkin’s Lymphoma
Indolent non-Hodgkin’s lymphoma refers to a group of largely incurable slow-growing lymphomas that run a relapsing course after therapy and can lead ultimately to life-threatening complications such as serious infections and marrow failure. Most iNHL patients are diagnosed at an advanced stage of disease, and median survival from time of initial diagnosis for patients with the most common form of iNHL, follicular lymphoma, is 8 to 10 years. The outlook for refractory iNHL patients is significantly poorer.
About Gilead Sciences
Gilead Sciences is a biopharmaceutical company that discovers, develops and commercializes innovative therapeutics in areas of unmet medical need. The company’s mission is to advance the care of patients suffering from life-threatening diseases worldwide. Headquartered in Foster City, California, Gilead has operations in North and South America, Europe and Asia Pacific.
The delta form of PI3K is expressed primarily in blood-cell lineages, including cells that cause or mediate hematologic malignancies, inflammation, autoimmune diseases and allergies. By specifically inhibiting only PI3K delta, a therapeutic effect is exerted without inhibiting PI3K signalling that is critical to the normal function of healthy cells. Extensive studies have shown that inhibition of other PI3K forms can cause significant toxicities, particularly with respect to glucose metabolism, which is essential for normal cell activity.
In 2011, orphan drug designation was assigned to GS-1101 in the U.S. for the treatment of CLL. In 2013, several orphan drug designations were assigned to the compound in the E.U. and U.S.: for the treatment of follicular lymphoma, for the treatment of mucosa-associated lymphoid tissue lymphoma (MALT), for the treatment of nodal marginal zone lymphoma, for the treatment of splenic marginal zone lymphoma, and for the treatment of chronic lymphocytic leukemia/small lymphocytic lymphoma. Orphan drug designation was also assigned in the U.S. for the treatment of lymphoplasmacytic lymphoma with or without Walenstom’s macroglobulinemia and, in the E.U., for the treatment of Waldenstrom’s macroglobulinemia (lymphoplasmacytic lymphoma).
Later in 2013, some of these orphan drug designations were withdrawn in the E.U.; for the treatment of chronic lymphocytic leukemia / small lymphocytic lymphoma, for the treatment of extranodal marginal-zone lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma), for the treatment of of nodal marginal-zone lymphoma and for the treatment of splenic marginal-zone lymphoma. In 2013, the FDA granted a breakthrough therapy designation for the treatment of chronic lymphocytic leukemia.
- H. Spreitzer (13 May 2013). “Neue Wirkstoffe – Ibrutinib und Idelalisib”. Österreichische Apothekerzeitung (in German) (10/2013): 34.
- Wu, M.; Akinleye, A.; Zhu, X. (2013). “Novel agents for chronic lymphocytic leukemia”.Journal of Hematology & Oncology 6: 36. doi:10.1186/1756-8722-6-36.PMC 3659027. PMID 23680477.
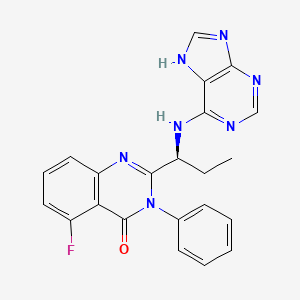
CAL-101 is an Oral Delta Isoform-Selective PI3 Kinase Inhibitor.
| US8207153 | 6-27-2012 | QUINAZOLINONES AS INHIBITORS OF HUMAN PHOSPHATIDYLINOSITOL 3-KINASE DELTA |
| US2012015964 | 1-20-2012 | QUINAZOLINONES AS INHIBITORS OF HUMAN PHOSPHATIDYLINOSITOL 3-KINASE DELTA |
| US2011306622 | 12-16-2011 | METHODS OF TREATING HEMATOLOGICAL DISORDERS WITH QUINAZOLINONE COMPOUNDS IN SELECTED SUBJECTS |
| US7932260 | 4-27-2011 | Quinazolinones as Inhibitors of Human Phosphatidylinositol 3-Kinase Delta |
| US2011044942 | 2-25-2011 | METHODS OF TREATMENT FOR SOLID TUMORS |
| US2010256167 | 10-8-2010 | QUINAZOLINONES AS INHIBITORS OF HUMAN PHOSPHATIDYLINOSITOL 3-KINASE DELTA |
| US2010202963 | 8-13-2010 | THERAPIES FOR HEMATOLOGIC MALIGNANCIES |
| WO2005113556A1 * | 12 May 2005 | 1 Dec 2005 | Icos Corp | Quinazolinones as inhibitors of human phosphatidylinositol 3-kinase delta |
| WO2005117889A1 * | 12 Nov 2004 | 15 Dec 2005 | Didier Bouscary | Methods for treating and/or preventing aberrant proliferation of hematopoietic |
| WO2005120511A1 * | 4 Jun 2005 | 22 Dec 2005 | Joel S Hayflick | Methods for treating mast cell disorders |
| WO2006089106A2 * | 16 Feb 2006 | 24 Aug 2006 | Icos Corp | Phosphoinositide 3-kinase inhibitors for inhibiting leukocyte accumulation |
| US20060106038 * | 25 May 2005 | 18 May 2006 | Icos Corporation | Methods for treating and/or preventing aberrant proliferation of hematopoietic cells |
The synthesis of a compound in accordance with formula I is first exemplified using steps A-E below, which provide a synthetic procedure for compound 107, the structure of which is shown below.

(107) is idelalisib
……………….
Synthesis of 2-fluoro-6-nitro-N-phenyl-benzamide (108)
Step A: A solution of 2-fluoro-6- nitrobenzoic acid (100 g, 0.54 mol) and dimethylformamide (5 mL) in dichloromethane (600 mL) was treated dropwise with oxalyl chloride (2 M in dichloromethane, 410 mL, 0.8 mol, 1.5 eq) over 30 min. After stirring 2 h at room temperature, the reaction was concentrated to an orange syrup with some solids present. The syrup was dissolved in dry dioxane (80 mL) and slowly added to a suspension of aniline (49 mL, 0.54 mol, 1 eq) and sodium bicarbonate (90 g, 1.08 mol, 2 eq) in a mixture of dioxane (250 mL) and water (250 mL) at 6 0C. The temperature reached 27°C at the end of the addition. After 30 min, the reaction mixture was treated with water (1.2 L). The precipitate was collected by vacuum filtration, washed with water (300 mL) , air dried in the funnel, and dried in vacuo at 50°C for 24 h to afford an off-white solid product (139 g, 99%). 1H NMR (300 MHz, DMSO-d6) δ 10.82 (s, IH), 8.12 (d, J = 7.7 Hz, IH), 7.91-7.77 (m, 2H), 7.64 (d, J = 7.7 Hz, 2H), 7.38 (t, J = 7.9 Hz, 2H), 7.15 > (t, J = 7.4 Hz, IH), ESI-MS m/z 261 (MH+). The reaction described above and compound 108 are shown below.

………………………..
Synthesis of(S) – [1- (2-fluoro-6-nitro-benzoyl) -phenyl-aminocarbonyl] – propyl-carbamic acid tert-butyl ester (109)
Step B: A suspension of compound 108 (0.5 mol) and dimethylformamide (5 mL) in thionyl chloride (256 mL, 2.5 mol, 5 eq) was stirred at 85°C for 5 hours. The reaction mixture was concentrated in vacuo to a brown syrup. The syrup was dissolved in dichloromethane (200 mL) and was slowly added to a solution of N-BOC-L-2-aminobutyric acid (112 g, 0.55 mol, 1.1 eq) and triethylamine (77 mL, 0.55 mol, 1.1 eq) in dichloromethane (600 mL) at 10 0C. After stirring at room temperature for 3 h, salts were removed by filtration, and the solution was washed with 100 mL of water, saturated sodium bicarbonate, water, 5% citric acid, and saturated sodium chloride. The organic phase was dried with magnesium sulfate and concentrated to a red syrup. The syrup was dissolved in dichloromethane (450 mL) and purified by flash chromatography on a silica gel plug (15 x 22 cm, 4 L dry silica) eluted with hexanes/ethyl acetate (10%, 8 L; 15%, 8 L; 20%, 8 L; 25%, 4 L) to yield the compound 109 as an off-white solid (147 g, 66%). 1H NMR (300 MHz, DMSO-d6) δ 8.13 (d, J = 8.0 Hz, IH), 7.84 (t, J = 8.6 Hz, IH), 7.78- 7.67 (m, IH), 7.65-7.49 (m, 3H), 7.40-7.28 ( m, 2H), 7.19 (d, J = 7.5 Hz, IH), 4.05 (broad s, IH), 1.75- 1.30 (m, 2H), 1.34 (s, 9H), 0.93 (broad s, 3H). ESI- MS m/z 446.3 (MH+) . The reaction described above and compound 109 are shown below.

Synthesis of(S) – [1- (5-fluoro-4-oxo-3-phenyl-3 , 4-dihydro-quinazolin-2- yl) -propyl] -carbamic acid tert-butyl ester (110)
Step C: A solution of compound 109 (125 mmol, 1 eq) in acetic acid (500 mL) was treated with zinc dust (48.4 g, 740 mmol, 6 eq) added in 3 portions, and the reaction mixture was allowed to cool to below 35°C between additions. After stirring for 2 h at ambient temperature, solids were filtered off by vacuum filtration and washed with acetic acid (50 mL) . The filtrate was concentrated in vacuo, dissolved in EtOAc (400 mL) , washed with water (300 mL) , and the water layer was extracted with EtOAc (300 mL) . The combined organic layers were washed with water (200 mL) , sat’d sodium bicarbonate (2 x 200 mL) , sat’d NaCl (100 mL) , dried with MgSO4, and concentrated to a syrup. The syrup was dissolved in toluene (200 mL) and purified by flash chromatography on a silica gel plug (13 x 15 cm, 2 L dry silica) eluted with hexanes/ethyl acetate (10%, 4 L; 15%, 4 L; 17.5%, 8 L; 25%, 4 L) to yield compound 110 as an off-white foamy solid (33.6 g, 69%). 1H NMR (300 MHz, DMSO-d6) δ 7.83 (td, J = 8.2, 5.7 Hz, IH), 7.64-7.48 (m, 5H), 7.39 (broad d, J = 7.6 Hz, IH), 7.30 (dd, J = 8.3 Hz, IH), 7.23 (d, J = 7.6 Hz, IH), 4.02-3.90 (m, IH), 1.76-1.66 (m, IH), 1.62-1.46 (m, IH), 1.33 (s, 9H), 0.63 (t, J= 7.3 Hz, 3H). ESI-MS m/z 398.3 (MH+). The reaction described above and compound 110 are shown below.

…………..
Syn of (S) -2- (1-amino-propyl) -5-fluoro-3-phenyl-3H-quinazolin-4- one (111)
Step D: A solution of compound 110 (85 mmol) in dichloromethane (60 mL) was treated with trifluoroacetic acid (60 mL) . The reaction mixture was stirred for 1 h, concentrated in vacuo, and partitioned between dichloromethane (150 mL) and 10% K2CO3 (sufficient amount to keep the pH greated than 10) . The aqueous layer was extracted with additional dichloromethane (100 raL) , and the combined organic layers were washed with water (50 mli) and brine (50 mL) . After drying with Mg SO4, the solution was concentrated to provide compound 111 as an off-white solid (22 g, 88%) . 1H NMR (300 MHz,
CDCl3) δ 7.73-7.65 (m, IH), 7.62-7.49 (m, 4H), 7.32- 7.22 (m, 2H), 7.13-7.06 (m, IH), 3.42 (dd, J= 7.5, 5.2 Hz, IH), 1.87-1.70 (m, IH), 1.58-1.43 (m, IH), 0.80 (t, J = 7.4 Hz, 3H) . ESI-MS m/z 298.2 (MH+) . The reaction described above and compound 111 are shown below.

………………
syn of (S) -5-fluoro-3-phenyl-2- [1- (9H-purin-6-ylamino) -propyl] – 3H-quinazolin-4-one (107)
Step E: A suspension of compound 111(65.6 mmol, 1 eq) , 6-bromopurine (14.6 g, 73.4 mmol, 1.1 eq) , and DIEA (24.3 mL, 140 mmol, 2 eq) in tert- butanol (40 mL) was stirred for 24 h at 800C. The reaction mixture was concentrated in vacuo and treated with water to yield a solid crude product that was collected by vacuum filtration, washed with water, and air dried. Half of the obtained solid crude product was dissolved in MeOH (600 mL) , concentrated onto silica gel (300 mL dry) , and purified by flash chromatography (7.5 x 36 cm, eluted with 10 L of 4% MeOH/CH2Cl2) to yield a solid product. The solid product was then dissolved in EtOH (250 mL) and concentrated in vacuo to compound 107 idelalisib as a light yellow solid (7.2 g, 50%).
1H NMR (300 MHz, 80 0C, DMSO-d5) δ 12.66 (broad s, IH), 8.11 (s, IH), 8.02 (broad s, IH), 7.81-7.73 (m, IH),7.60-7.42 (m, 6H), 7.25-7.15 (m, 2H), 4.97 (broad s, IH), 2.02-1.73 (m, 2H), 0.79 (t, J= 7.3 Hz, 3H).
ESI-MS m/z 416.2 (MH+).
C, H, N elemental analysis (C22Hi8N7OF-EtOH- 0.4 H2O).
Chiral purity 99.8:0.2 (S:R) using chiral HPLC (4.6 x 250 mm Chiralpak ODH column, 20 °C, 85:15 hexanes : EtOH, 1 rnL/min, sample loaded at a concentration of 1 mg/mL in EtOH) . The reaction described above and compound 107 idelalisib are shown below.

| WO2001030768A1 * | 26 Oct 2000 | 3 May 2001 | Gustave Bergnes | Methods and compositions utilizing quinazolinones |
| WO2001081346A2 * | 24 Apr 2001 | 1 Nov 2001 | Icos Corp | Inhibitors of human phosphatidyl-inositol 3-kinase delta |
| WO2003035075A1 * | 27 Aug 2002 | 1 May 2003 | Icos Corp | Inhibitors of human phosphatidyl-inositol 3-kinase delta |
| WO2005016348A1 * | 13 Aug 2004 | 24 Feb 2005 | Jason Douangpanya | Method of inhibiting immune responses stimulated by an endogenous factor |
| WO2005016349A1 * | 13 Aug 2004 | 24 Feb 2005 | Thomas G Diacovo | Methods of inhibiting leukocyte accumulation |
| WO2005067901A2 * | 7 Jan 2005 | 28 Jul 2005 | Carrie A Northcott | Methods for treating and preventing hypertension and hypertension-related disorders |
Merck Announces FDA Acceptance of New Drug Application for Investigational Fertility Treatment

corifollitropin alfa
WHITEHOUSE STATION, N.J.–(BUSINESS WIRE)–Merck (NYSE:MRK), known as MSD outside the United States and Canada, today announced that the New Drug Application (NDA) for its investigational fertility treatment, corifollitropin alfa, has been accepted for standard review by the U.S. Food and Drug Administration (FDA). Merck is seeking FDA approval of corifollitropin alfa for Controlled Ovarian Stimulation (COS) in women participating in assisted reproductive technology.
If approved, corifollitropin alfa would be the first sustained follicular stimulant for use in a fertility treatment regimen.
read all at
http://www.pharmalive.com/fda-accepts-mercks-fertility-treatment-nda

Corifollitropin alfa
Merck received approval on February 15, 2010 from the European Commission for ELONVA (corifollitropin alfa) a long lasting single injection fusion protein lacking LH activity. Only one injection is required for the first seven days, replacing the first seven daily injections of conventional FSH. Initial results demonstrates similar pregnancy rates as daily recombinant FSH injections.[7][8]
- ref 7 N. P. Koper, R. Boostanfar, P. Devroey, B. C. Fauser, P. C. IJzerman-Boon, B. M. J. L. Mannaerts. Global ClinicalDevelopment, Organon, Part of Schering-Plough Corporation, Oss, Netherlands; Huntington Reproductive Center, Tarzana, CA; Center of Reproductive Medicine, Dutch-speaking Free University, Brussels, Belgium; University Medical Center Utrecht, Utrecht, Netherlands; Biometrics, NV Organon, Part of Schering-Plough Corporation, Oss, Netherlands. “Corifollitropin alfa demonstrates similar pregnancy rates as compared to daily recombinant FSH treatment in a controlled ovarian stimulation regimen for IVF/ICSI.” Fertility and Sterility, 90:page S75.
- ref 8 ^ Devroey P, Boostanfar R, Koper NP, Mannaerts BM, Ijzerman-Boon PC, Fauser BC, 2009. “A double-blind, non-inferiority RCT comparing corifollitropin alfa and recombinant FSH during the first seven days of ovarian stimulation using a GnRH antagonist protocol.” Human Reproduction, 2009, August 14, [Epub ahead of print]. PMID 19684043.
In May2013, MSD launched ELONVA® (corifollitropin alfa injection) – a new treatment for fertility, – in Singapore. Approved for controlled ovarian stimulation in combination with a GnRH antagonist for the development of multiple follicles, Corifollitropin alfa injection is the first sustained follicle stimulant. A single subcutaneous injection of the recommended dose of corifollitropin alfa injection may replace the first seven injections of any
Findings showed that other failed repeated treatments may lead to depression, anxiety, sexua conventional daily recombinant follicle stimulating hormone (rFSH) preparation in a controlled ovarian stimulation treatment cycle. Simplified fertility treatment with Elonva not only helps to reduce the emotional and physical burden of fertility, it may also reduce dropout rates and potentially improve the overall chances of pregnancy.
l anxiety/difficulty, relationship problems with partner, family and friends, increased sense of self-blame and guilt, particularly for the partner experiencing fertility problem. ”By reducing the number of daily injections, the
availability of corifollitropin alfa injection is a positive step towards helping reduce the burden of fertility treatment for women experiencing difficulty conceiving. Simplifying fertility treatment with new modalities of treatment and new medication may encourage more infertile couple to embark
on treatment earlier when the wife’s age is younger and ovarian reserve better.” said Dr Loh Seong Feei, Medical Director of Thomson Fertility Centre
FDA Accepts Nuvo’s New Drug Application for Review
Nuvo Research Inc. announced that its U.S. licensee for PENNSAID@ (diclofenac sodium topical solution) 1.5% w/w and PENNSAID 2% (diclofenac sodium topical solution) 2% w/w,
Mallinckrodt has advised that the U.S. Food and Drug Administration has accepted for filing and review the New Drug Application (NDA) for PENNSAID 2% submitted by Mallinckrodt on August 7, 2013.
FDA Grants Priority Review To New Drug Application For MNK-795
FDA Grants Priority Review To New Drug Application For MNK-795 Submitted By Depomed Licensee Mallinckrodt
Controlled Substance Analgesic Combination Product Uses Depomed’s Proprietary Acuform® Technology
NEWARK, Calif., July 29, 2013 /PRNewswire/ — Depomed, Inc. (NASDAQ:DEPO) announced today that the U. S. Food and Drug Administration (FDA) has accepted for filing a New Drug Application (NDA) from Mallinckrodt (NYSE: MNK) for MNK-795. MNK-795 is a controlled-release oral formulation of oxycodone and acetaminophen that has been studied for the management of moderate to severe acute pain where the use of an opioid analgesic is appropriate. MNK-795 is formulated with Depomed’s Acuform® drug delivery technology.
http://www.pharmalive.com/fda-grants-priority-review-to-new-drug-application-for-mnk-795
The clinical benefit of high-dose toremifene for metastatic breast cancer

http://www.ncbi.nlm.nih.gov/pubmed/23863727
Source
Dept. of Surgery, Saga University Faculty of Medicine.
Abstract
Introduction: Toremifene(TOR)is a selective estrogen receptor modulator(SERM). A high dose of 120 mg TOR(HD-TOR) has been used for recurrent breast cancer in Japan, but there is still insufficient evidence regarding the efficacy of HD-TOR. Patients and methods: HD-TOR was administered for recurrent or metastatic breast cancer between January 2003 and May 2012. The primary end point of the study was the tumor response rate. Bone metastasis cases were excluded from the efficacy analysis, but were included in the safety population. Results: A total of 21 patients registered in the study and the 2 patients with bone metastasis only were excluded from the efficacy analysis. The median follow-up period was 8. 3 months. None of the patients in the study had a CR, 4 had a PR(21. 1%), 9 had SD(47. 4%), and 6 had PD(31. 6%). Eight of the 9 SD cases had a long-term SD. The ORR was 21. 1% and the CB rate was 63. 2%. The median TTP of CB cases was 18. 3 months. None of the patients discontinued treatment because of a grade 3 or grade 4 adverse effects. Conclusion: In summary, the current study showed that HD-TOR may lead to a CB for recurrent breast cancer in first- or second-line treatment rather than thirdline. In particular, HD-TOR may give a benefit in highly endocrine-sensitive cases.

toremifene
Toremifene citrate is an oral selective estrogen receptor modulator (SERM) which helps oppose the actions of estrogen in the body. Licensed in the United States under the brand name Fareston, toremifene citrate is FDA-approved for use in advanced (metastatic)breast cancer. It is also being evaluated for prevention of prostate cancer under the brand name Acapodene.[1]
In 2007 the pharmaceutical company GTx, Inc was conducting two different phase 3clinical trials; First, a pivotal Phase clinical trial for the treatment of serious side effects ofandrogen deprivation therapy (ADT) (especially vertebral/spine fractures and hot flashes, lipid profile, and gynecomastia) for advanced prostate cancer, and second, a pivotal Phase III clinical trial for the prevention of prostate cancer in high risk men with high gradeprostatic intraepithelial neoplasia, or PIN. Results of these trials are expected by first quarter of 2008[2]
An NDA for the first application (relief of prostate cancer ADT side effects) was submitted in Feb 2009,[3] and in Oct 2009 the FDA said they would need more clinical data, e.g. another phase III trial.[4]
- Price N, Sartor O, Hutson T, Mariani S. Role of 5a-reductase inhibitors and selective estrogen receptor modulators as potential chemopreventive agents for prostate cancer.Clin Prostate Cancer 2005;3:211-4. PMID 15882476
- “GTx’s Phase III Clinical Development of ACAPODENE on Course Following Planned Safety Review” (Press release). GTx Inc. 2007-07-12. Retrieved 2006-07-14.
- “GTx Announces Toremifene 80 mg NDA Accepted for Review by FDA” (Press release).
- “GTx and Ipsen End Prostate Cancer Collaboration due to Costs of FDA-Requested Phase III Study”. 2 Mar 2011.

J and J Submits Leukemia Drug, Ibrutinib for Approval

IBRUTINIB
1-[(3R)-3-[4-amino-3-(4-phenoxyphenyl)pyrazolo[3,4-d]pyrimidin-1-yl]piperidin-1-yl]prop-2-en-1-one
New Drug Application Submitted to U.S. FDA for Ibrutinib in the Treatment of Two B-Cell Malignancies
If approved, ibrutinib will address a high unmet need in relapsed/refractory chronic lymphocytic leukemia and relapsed/refractory mantle cell lymphoma
RARITAN, N.J., July 10, 2013
Janssen Research & Development, LLC announced the submission of a New Drug Application for ibrutinib to the U.S. Food and Drug Administration (FDA) for its use in the treatment of previously treated patients with chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL), and for its use in the treatment of previously treated patients with mantle cell lymphoma (MCL). The regulatory submission for ibrutinib is supported by data from two pivotal Phase 2 studies, one in relapsed/refractory CLL/SLL (PCYC-1102) and one in relapsed/refractory MCL (PCYC-1104), both of which were published in The New England Journal of Medicine online on June 19, 2013. Ibrutinib is a novel Bruton’s tyrosine kinase (BTK) inhibitor being jointly developed by Janssen and Pharmacyclics, Inc. for the treatment of B-cell malignancies.
If approved, ibrutinib would be the first in a class of oral BTK inhibitors and is one of the first medicines to file for FDA approval via the new Breakthrough Therapy Designation pathway. Ibrutinib will be co-commercialized in the U.S. by Janssen Biotech, Inc. and Pharmacyclics.
“The FDA submission is another important milestone for ibrutinib since we formed our strategic partnership with Pharmacyclics just 18 months ago,” said Peter F. Lebowitz, M.D., Ph.D., Global Oncology Head, Janssen. “Both companies recognize that there is great unmet need among these patient populations, and together in close collaboration with the FDA, as part of its Breakthrough Therapy Designation pathway, we have been able to accelerate the ibrutinib development program for the benefit of patients.”
About Chronic Lymphocytic Leukemia
Chronic Lymphocytic Leukemia (CLL) is a slow-growing blood cancer that starts in the white blood cells (lymphocytes), most commonly from B-cells. CLL is the second most common adult leukemia. Approximately 16,000 patients in the US are diagnosed each year with CLL. The prevalence of CLL is approximately 113,000 in the US. The disease is a chronic disease of the elderly with an average survival of about 5 years. Patients commonly receive multiple lines of treatment over the course of their disease.
In CLL the genetic mutation 17p deletion occurs when the short arm of chromosome 17 is missing. Del 17p is associated with abnormalities of a key tumor suppressor gene, TP53, which results in poor response to chemoimmunotherapy and worse treatment outcomes. It occurs in about 7% of treatment naive CLL patients and is estimated to be approximately 20% to 40% of relapsed or refractory patients harboring the mutation.
About Ibrutinib
Ibrutinib , previously publicly known as PCI-32765, is an experimental drug candidate for the treatment of various types of cancer. It was first synthesized at Celera Genomics as a selective inhibitor of Bruton’s tyrosine kinase (Btk).It was later discovered to have anti-lymphoma properties in vivo by scientists at Pharmacyclics, Inc.Ibrutinib is currently under development by Pharmacyclics, Inc and Johnson & Johnson‘sJanssen Pharmaceutical division for chronic lymphocytic leukemia, mantle cell lymphoma,diffuse large B-cell lymphoma, and multiple myeloma. It also has potential effects against autoimmune arthritis.
Janssen Biotech, Inc. and Pharmacyclics entered a collaboration and license agreement in December 2011 to co-develop and co-commercialize ibrutinib. Ibrutinib was designed to specifically target and selectively inhibit an enzyme called Bruton’s tyrosine kinase (BTK). BTK is a key mediator of at least three critical B-cell pro-survival mechanisms occurring in parallel – regulation of apoptosis, adhesion, and cell migration and homing. Through these multiple signals, BTK regulation helps to direct malignant B-cells to lymphoid tissues, thus allowing access to a micro environment necessary for survival.
The effectiveness of ibrutinib alone or in combination with other treatments is being studied in several B-cell malignancies, including chronic lymphocytic leukemia/small lymphocytic lymphoma, mantle cell lymphoma, diffuse large B-cell lymphoma, follicular lymphoma, Waldenstrom’s macroglobulinemia and multiple myeloma. To date five Phase III trials have been initiated with ibrutinib and a total of 26 trials are currently registered on www.clinicaltrials.gov.
About Pharmacyclics
Pharmacyclics® is a clinical-stage biopharmaceutical company focused on developing and commercializing innovative small-molecule drugs for the treatment of cancer and immune mediated diseases. Our mission and goal is to build a viable biopharmaceutical company that designs, develops and commercializes novel therapies intended to improve quality of life, increase duration of life and resolve serious unmet medical healthcare needs; and to identify promising product candidates based on scientific development and administrational expertise, develop our products in a rapid, cost-efficient manner and pursue commercialization and/or development partners when and where appropriate.
Presently, Pharmacyclics has three product candidates in clinical development and several preclinical molecules in lead optimization. The Company is committed to high standards of ethics, scientific rigor, and operational efficiency as it moves each of these programs to viable commercialization.
The Company is headquartered in Sunnyvale, California and is listed on NASDAQ under the symbol PCYC. To learn more about how Pharmacyclics advances science to improve human healthcare visit at http://www.pharmacyclics.com.
