
Home » FDA 2024
Category Archives: FDA 2024
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Levacetylleucine
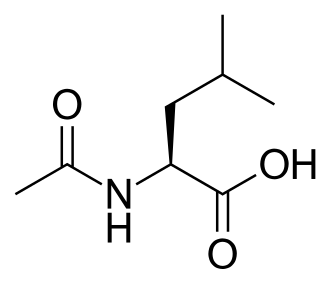
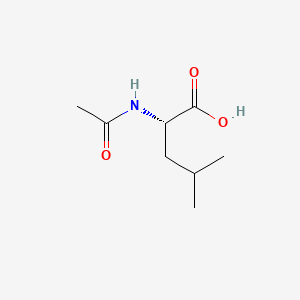

Levacetylleucine
WeightAverage: 173.212
Monoisotopic: 173.105193347
Chemical FormulaC8H15NO3
- N-Acetyl-L-leucine
- CAS 1188-21-2
- acetyl-L-leucine
- Ac-Leu-OH
- N-Acetylleucine
- NSC 206316
- UNII-E915HL7K2O
NSC-206316
(2S)-2-acetamido-4-methylpentanoic acid
FDA APPROVED 9/24/2024, To treat Niemann-Pick disease type C
Press Release
Drug Trials Snapshot
- Originator University of Munich; University of Oxford
- Developer IntraBio
- Class Acetamides; Amino acids; Esters; Neuroprotectants; Pentanoic acids; Small molecules; Vestibular disorder therapies
- Mechanism of Action Calcium channel modulators
- Orphan Drug StatusYes – Tay-Sachs disease; Niemann-Pick disease type C; Ataxia telangiectasia
Registered Niemann-Pick disease type C
- Phase IIIAtaxia telangiectasia
- Phase IISandhoff disease; Tay-Sachs disease
18 Mar 2025Phase-III clinical trials in Ataxia telangiectasia (In adolescents, In children, In the elderly, In adults) in Switzerland, Slovakia, Spain, Germany, USA, United Kingdom (PO) (NCT06673056)
- 04 Nov 2024IntraBio plans a phase III trial for Ataxia telangiectasia (In children, In adolescents, In adults, In elderly) in the US, Germany, Slovakia, Spain and Switzerland (PO, Suspension) in March 2025 (NCT06673056)
- 24 Sep 2024Registered for Niemann-Pick disease type C (In adolescents, In children, In adults) in USA (PO)
Levacetylleucine (N-acetyl-L-leucine), sold under the brand name Aqneursa, is a medication used for the treatment of neurological manifestations of Niemann-Pick disease type C.[1][2] Levacetylleucine is a modified version of the amino acid leucine.[1] It is the L-form of acetylleucine. It is taken by mouth.[1]
The most common side effects include abdominal pain, difficulty swallowing, upper respiratory tract infections, and vomiting.[1][2]
Levacetylleucine was approved for medical use in the United States in September 2024.[1][2][3] Levacetylleucine is the second medication approved by the US Food and Drug Administration (FDA) for the treatment of Niemann-Pick disease type C.[2] The FDA considers it to be a first-in-class medication.[4]
DATA
N-acetyl-D, L-leucine is the active ingredient of Tanganil ® which helps treat vertigo attacks.

N-Acetyl-D, L-leucine
Unlike the majority of chemical syntheses of active principles where it is desirable to separate the enanti omers and / or to retain the selective stereo information during the synthesis steps, the synthesis of N-acetyl-D, L-leucine is carried out from L-leucine and therefore involves a racemization step. This racemization takes place before the acetylation step, via a Schiff base formed in situ with salicylic aldehyde (Yamada et al., J. Org. Chem., 1983 48, 843- 846).

Two competitive reactions are then involved: the acetylation of leucine, the main reaction, where acetic anhydride reacts with the amine function of leucinate of sodium to give N-acetyleucinate and the hydrolysis of acetic anhydride to acetic acid, a side reaction described below.

This synthesis has a molar yield of 70%. The limiting steps are essentially the secondary reaction of hydrolysis of acetic anhydride and the step of isolation of the racemized leucine before the acetylation reaction. Indeed, on an industrial scale, the quantities of products brought into play for isolations prove to be very restrictive.
There is therefore a real need to develop a new process for the preparation of N-actéyl-D, L-leucine which is faster and more economical.
The inventors thus discovered that the racemization step could be carried out after the L-leucine acetylation step making it possible to avoid a step of isolating the intermediate product and that this process could be carried out in continuous flow. Du Vigneaud & Meyer (J. Biol Chem, 1932, 98, 295-308) had already shown that it was possible to racemize different acetylated amino acids by bringing them into the presence of acetic anhydride for several hours. However, no examples had been made with acetyl leucine. By attempting to reproduce this process with acetyl-leucine, the inventors have thus found that this racemization reaction did not give satisfactory results with acetyl-leucine because of a competitive hydrolysis reaction of acetic anhydride. used. The inventors have also surprisingly discovered that the racemization reaction of N-acetyl-L-leucine could be improved by producing it in a continuous flow. It seems indeed that the realization of this continuous flow process allows better control of the mixing of the reagents and therefore to better control the reaction. The inventors have also shown that the racemization of N-acetyl-L-Leucine in continuous flow was obtained in a very short time of the order of a few minutes.
Furthermore, there is also a need to develop a new method of acetylation of leucine for the preparation of N-actyle-leucine which is faster and more economical. The inventors have discovered that the acetylation reaction of leucine can be improved by making it in a continuous flow. The process according to the invention gives good yields, in a very short time and using fewer reagents compared to the method known hitherto.
Indeed, DeWitt et al. (J Am Chem Soc (1951) 73 (7) 3359-60) described the preparation of N-acetyl-L-Leucine by reacting L-Leucine with 3 molar equivalents of acetic anhydride and sodium hydroxide for 2 hours 20 minutes. . N-acetyl-L-leucine is then obtained in a yield of only 70-80%. In addition, the authors of this publication clearly indicated that a molar ratio between L-Leucine and acetic anhydride below 2 resulted in much lower yields.
SYNTHESIS
H. D. DeWitt and A. W. Ingersoll. The Preparation of Pure N-Acetyl-L-leucine and L-Leucine. Journal of the American Chemical Society 1951 73 (7), 3359-3360. DOI: 10.1021/ja01151a108
PATENT
https://patents.google.com/patent/WO2012038515A1/en
EXAMPLES
A. Acetylation of L-Leucine in Continuous Flow

A. L. Study of the molar ratio of acetic anhydride to leucine
The objective of this study is to define the necessary molar ratio of acetic anhydride so that the acetylation reaction with acetic anhydride is complete and is not disadvantageous by competition with the acetic anhydride hydrolysis reaction. In this study, the residence time in the reactor / exchanger (1 process plate) was set at 9 seconds, for a temperature of the reaction medium of between 25 and 30 ° C.
The ratio range studied is between 0.9 and 2.0 molar equivalents. The optimum is obtained for a ratio between 1.20 and 2.00, more particularly between 1.30 and 1.60. Below this ratio, the acetylation reaction is disadvantageous compared to the acetic hydrolysis reaction. Beyond this, the drop in pH (acid instead of base) also disadvantages the acetylation reaction.
EXAMPLES 1-10:
A solution of sodium L-leucinate, for passage in continuous flow reactor, is prepared in the following manner: 700 g of L-leucine are dissolved in a solution of 576 g of sodium hydroxide and 3.5 liters of Demineralized Water. This solution is the main fluid process. The reaction between this solution and the acetic anhydride is carried out in a continuous flow in a Boostec® reactor, made of silicon carbide. The reactor / exchanger is configured with an injection-type process plate comprised between two utility plates. The volume of the process plate is 10 mL. The temperature in the reactor is maintained by the circulation of a coolant heated by a thermostatic bath. The transformation of L-leucine to N-acetyl-L-leucine is monitored online by quantitative Raman spectroscopy. This method of analysis is calibrated beforehand with solutions of known concentration prepared with pure L-leucine and N-acetyl-L-leucine.
Example 1
The temperature of the thermostated bath is set at 25 ° C. The sodium leucinate solution and pure acetic anhydride are introduced into the reactor at respective flow rates set at 4.06 kg.h -1 and 0.42 kg h -1 . These flow rates correspond to a molar ratio of acetic anhydride to leucine of 0.91 equivalents. The total flow rate is therefore 4.48 kg.h -1 , which corresponds to a residence time (equivalent to the reaction time) of 8.7 s The yield of acetyl-L-leucinate determined by Raman spectroscopy online at the outlet of the reactor is 40% Example 2:
The temperature of the thermostated bath is set at 25 ° C. The sodium leucinate solution and pure acetic anhydride are introduced into the reactor at respective flow rates set at 3.95 kg · h -1 and 0.45 kg · h -1 . These flow rates correspond to a molar ratio of acetic anhydride to leucine of 1.01 equivalents. The total flow rate is therefore 4.40 kg.h -1 , which corresponds to a residence time of 8.9 S. The yield of acetyl-L-leucinate determined by in-line Raman spectroscopy at the outlet of the reactor is 52.degree. %.
Example 3
The temperature of the thermostated bath is set at 25 ° C. The sodium leucinate solution and pure acetic anhydride are introduced into the reactor at respective flow rates set at 3.89 kg · h -1 and 0.52 kg · h -1 . These flow rates correspond to a molar ratio of acetic anhydride to leucine of 1.18 equivalents. The total flow rate is therefore 4.41 kg.h -1 , which corresponds to a residence time of 8.9 S. The yield of acetyl-L-leucinate determined by in-line Raman spectroscopy at the outlet of the reactor is 57.degree. %. Example 4
The temperature of the thermostated bath is set at 25 ° C. The sodium leucinate solution and pure acetic anhydride are introduced into the reactor at respective flow rates set at 3.82 kg. h -1 and 0.57 kg h -1 . These flow rates correspond to a molar ratio of acetic anhydride to leucine of 1.32 equivalents. The total flow is therefore 4.39 kg. h “1 , which corresponds to a residence time of 8.9 S. The yield of acetyl-L-leucinate determined by in-line Raman spectroscopy at the outlet of the reactor is 83%.
Example 5
The temperature of the thermostated bath is set at 25 ° C. The sodium leucinate solution and pure acetic anhydride are introduced into the reactor at respective rates set at 3.64 kg. h -1 and 0.55 kg h -1 . These flow rates correspond to a molar ratio of acetic anhydride to leucine of 1.34 equivalents. The total flow is therefore 4, 19 kg. h “1 , which corresponds to a residence time of 9.4 s The yield of acetyl-L-leucinate determined by in-line Raman spectroscopy at the outlet of the reactor is 98%.
Example 6
The temperature of the thermostated bath is set at 25 ° C. The sodium leucinate solution and pure acetic anhydride are introduced into the reactor at respective rates set at 3.66 kg. h 1 and 0.62 kg h -1 . These flow rates correspond to a molar ratio of acetic anhydride to leucine of 1.50 equivalents. The total flow is therefore 4.28 kg. h “1 , which corresponds to a residence time of 9.2 s The yield of acetyl-L-leucinate determined by in-line Raman spectroscopy at the outlet of the reactor is 96%.
The temperature of the thermostated bath is set at 25 ° C. The sodium leucinate solution and pure acetic anhydride are introduced into the reactor at respective flow rates fixed at 3.67 kg. h -1 and 0.64 kg h -1 . These flow rates correspond to a molar ratio of acetic anhydride to leucine of 1.54 equivalents. The total flow is therefore 4.31 kg. h “1 , which corresponds to a residence time of 9.1 sec The yield of acetyl-L-leucinate determined by in-line Raman spectroscopy at the outlet of the reactor is 100%. Example 8
The temperature of the thermostated bath is set at 25 ° C. The sodium leucinate solution and pure acetic anhydride are introduced into the reactor at respective flow rates set at 3.63 kg. h -1 and 0.73 kg h -1 . These flow rates correspond to a molar ratio of acetic anhydride to leucine of 1.78 equivalents. The total flow is therefore 4.36 kg. h “1 , which corresponds to a residence time of 9.0 s The yield of acetyl-L-leucinate determined by in-line Raman spectroscopy at the outlet of the reactor is 90%.
PATENT
https://patents.google.com/patent/CN104592052A/en
Example 1:
100gL-leucine adds 1000ML2NNaOH rising temperature for dissolving, adds 1ML salicylic aldehyde, 95 degree of insulations of intensification 3 hours, recording optically-active is 0, be cooled to 5 degree and keep, dripping 80ML diacetyl oxide, dropwise maintenance 0.5 hour, be warmed up to 60 degree, add proper amount of active carbon decolouring, add 160ML HCl and adjust PH 2.5, be cooled to 4 degree, suction filtration, the 118g. of oven dry
Example 2:
100gL-leucine adds 1200ML 2NNaOH rising temperature for dissolving, adds 3ML salicylic aldehyde, 95 degree of insulations of intensification 3 hours, recording optically-active is 0, be cooled to 5 degree and keep, dripping 80ML diacetyl oxide, dropwise maintenance 0.5 hour, be warmed up to 60 degree, add proper amount of active carbon decolouring, add the 3.0. that 180ML HCl adjusts PH, be cooled to 4 degree, suction filtration, the 110g. of oven dry
Example 3:
100gL-leucine adds 1000ML 2NNaOH rising temperature for dissolving, adds 2ML salicylic aldehyde, 95 degree of insulations of intensification 3 hours, recording optically-active is 0, be cooled to 5 degree and keep, dripping 80ML diacetyl oxide, dropwise maintenance 0.5 hour, be warmed up to 60 degree, add proper amount of active carbon decolouring, add 180ML HCl and adjust PH 3.0, be cooled to 4 degree, suction filtration, the 120g. of oven dry
Medical uses
Levacetylleucine is indicated for the treatment of neurological manifestations of Niemann-Pick disease type C in people weighing at least 15 kilograms (33 lb).[1][2]
Adverse effects
The most common side effects include abdominal pain, difficulty swallowing, upper respiratory tract infections, and vomiting.[2]
Levacetylleucine may cause embryo-fetal harm if used during pregnancy.[1][2]
History
The safety and efficacy of levacetylleucine for the treatment of Niemann-Pick disease type C were evaluated in a randomized, double-blind, placebo-controlled, two-period, 24-week crossover study.[2] The duration was twelve weeks for each treatment period.[2] The study enrolled 60 participants.[2] To be eligible for the study participants had to be four years of age or older with a confirmed diagnosis of Niemann-Pick disease type C and at least mild disease-related neurological symptoms.[2] Participants could receive miglustat, an enzyme inhibitor, as background treatment in the study.[2]
The US Food and Drug Administration (FDA) granted the application for levacetylleucine priority review, fast track, orphan drug, and rare pediatric disease designations.[2] The FDA granted approval of Aqneursa to IntraBio Inc.[2]
Society and culture
Legal status
Levacetylleucine was approved for medical use in the United States in September 2024.[1][2][5]
Names
Levacetylleucine is the international nonproprietary name.[6]
Research
Levacetylleucine is being studied for the treatment of GM2 gangliosidoses (Tay-Sachs and Sandhoff diseases),[7] ataxia-telangiectasia,[8] Lewy body dementia,[9] amyotrophic lateral sclerosis, restless legs syndrome, multiple sclerosis, and migraine.[10]
References
- ^ Jump up to:a b c d e f g h i “Aqneursa- levacetylleucine granule, for suspension”. DailyMed. 24 September 2024. Retrieved 5 October 2024.
- ^ Jump up to:a b c d e f g h i j k l m n o “FDA Approves New Drug to Treat Niemann-Pick Disease, Type C”. U.S. Food and Drug Administration (Press release). 24 September 2024. Retrieved 25 September 2024.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “IntraBio Announces U.S. FDA Approval of Aqneursa for the Treatment of Niemann-Pick Disease Type C”. IntraBio (Press release). 25 September 2024. Retrieved 26 September 2024.
- ^ New Drug Therapy Approvals 2024 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2025. Archived from the original on 21 January 2025. Retrieved 21 January 2025.
- ^ “Novel Drug Approvals for 2024”. U.S. Food and Drug Administration (FDA). 1 October 2024. Retrieved 29 November 2024.
- ^ World Health Organization (2024). “International nonproprietary names for pharmaceutical substances (INN): proposed INN: list 131”. WHO Drug Information. 38 (2). hdl:10665/378367. ISBN 9789240098558.
- ^ Martakis K, Claassen J, Gascon-Bayari J, Goldschagg N, Hahn A, Hassan A, et al. (March 2023). “Efficacy and Safety of N-Acetyl-l-Leucine in Children and Adults With GM2 Gangliosidoses”. Neurology. 100 (10): e1072 – e1083. doi:10.1212/WNL.0000000000201660. PMC 9990862. PMID 36456200.
- ^ Fields T, Patterson M, Bremova-Ertl T, Belcher G, Billington I, Churchill GC, et al. (January 2021). “A master protocol to investigate a novel therapy acetyl-L-leucine for three ultra-rare neurodegenerative diseases: Niemann-Pick type C, the GM2 gangliosidoses, and ataxia telangiectasia”. Trials. 22 (1): 84. doi:10.1186/s13063-020-05009-3. PMC 7821839. PMID 33482890.
- ^ Passmore P (15 April 2014). A clinical trial to test amlodipine as a new treatment for vascular dementia. ISRCTN registry (Report). doi:10.1186/isrctn31208535.
- ^ Strupp M, Bayer O, Feil K, Straube A (February 2019). “Prophylactic treatment of migraine with and without aura with acetyl-DL-leucine: a case series”. Journal of Neurology. 266 (2): 525–529. doi:10.1007/s00415-018-9155-6. PMID 30547273. S2CID 56148131.
Further reading
- Churchill GC, Strupp M, Factor C, Bremova-Ertl T, Factor M, Patterson MC, et al. (August 2021). “Acetylation turns leucine into a drug by membrane transporter switching”. Scientific Reports. 11 (1): 15812. Bibcode:2021NatSR..1115812C. doi:10.1038/s41598-021-95255-5. PMC 8338929. PMID 34349180.
- Bremova-Ertl T, Ramaswami U, Brands M, Foltan T, Gautschi M, Gissen P, et al. (February 2024). “Trial of N-Acetyl-l-Leucine in Niemann-Pick Disease Type C”. The New England Journal of Medicine. 390 (5): 421–431. doi:10.1056/NEJMoa2310151. PMID 38294974.
- Tifft CJ (February 2024). “N-Acetyl-l-Leucine and Neurodegenerative Disease”. The New England Journal of Medicine. 390 (5): 467–470. doi:10.1056/NEJMe2313791. PMID 38294981.
External links
- Clinical trial number NCT05163288 for “A Pivotal Study of N-Acetyl-L-Leucine on Niemann-Pick Disease Type C” at ClinicalTrials.gov
- Bremova-Ertl T, Ramaswami U, Brands M, Foltan T, Gautschi M, Gissen P, Gowing F, Hahn A, Jones S, Kay R, Kolnikova M, Arash-Kaps L, Marquardt T, Mengel E, Park JH, Reichmannova S, Schneider SA, Sivananthan S, Walterfang M, Wibawa P, Strupp M, Martakis K: Trial of N-Acetyl-l-Leucine in Niemann-Pick Disease Type C. N Engl J Med. 2024 Feb 1;390(5):421-431. doi: 10.1056/NEJMoa2310151. [Article]
- Fields T, M Bremova T, Billington I, Churchill GC, Evans W, Fields C, Galione A, Kay R, Mathieson T, Martakis K, Patterson M, Platt F, Factor M, Strupp M: N-acetyl-L-leucine for Niemann-Pick type C: a multinational double-blind randomized placebo-controlled crossover study. Trials. 2023 May 29;24(1):361. doi: 10.1186/s13063-023-07399-6. [Article]
- FDA Approved Drug Products: Aqneursa (levacetylleucine) for oral suspension (September 2024) [Link]
- FDA News Release: FDA Approves New Drug to Treat Niemann-Pick Disease, Type C [Link]
| Clinical data | |
|---|---|
| Trade names | Aqneursa |
| Other names | IB1001 |
| AHFS/Drugs.com | Aqneursa |
| License data | US DailyMed: Levacetylleucine |
| Pregnancy category | Not recommended |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1188-21-2 |
| PubChem CID | 70912 |
| DrugBank | DB16956 |
| ChemSpider | 1918 |
| UNII | E915HL7K2O |
| KEGG | D12967 |
| ChEBI | CHEBI:17786 |
| ChEMBL | ChEMBL56021 |
| PDB ligand | LAY (PDBe, RCSB PDB) |
| CompTox Dashboard (EPA) | DTXSID6045870 |
| ECHA InfoCard | 100.013.370 |
| Chemical and physical data | |
| Formula | C8H15NO3 |
| Molar mass | 173.212 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
/////////Levacetylleucine, Aqneursa, Niemann-Pick disease type C, FDA 2024, APPROVALS 2024, N-Acetyl-L-leucine, 1188-21-2, acetyl-L-leucine, Ac-Leu-OH, N-Acetylleucine, NSC 206316, UNII-E915HL7K2O, ORPHAN DRUG, NSC-206316, NSC 206316
Trospium chloride



Trospium chloride
CAS
47608-32-2
10405-02-4
WeightAverage: 392.518
Monoisotopic: 392.22202025
Chemical FormulaC25H30NO3
T4Y8ORK057
- 73954-17-3
- 8-Benziloyloxy-6,10-ethano-5-azoniaspiro(4.5)decane chloride
- 3-[(2-hydroxy-2,2-diphenylacetyl)oxy]-8lambda5-azaspiro[bicyclo[3.2.1]octane-8,1′-pyrrolidin]-8-yliumchloride
- spiro[8-azoniabicyclo[3.2.1]octane-8,1′-azolidin-1-ium]-3-yl 2-hydroxy-2,2-diphenylacetate;chloride
- SMR002533165
- spiro[8-azoniabicyclo[3.2.1]octane-8,1′-azolidin-1-ium]-3-yl 2-hydroxy-2,2-diphenylacetate;chloride
FDA 2024, Cobenfy 9/26/2024, To treat schizophrenia
Press Release
Drug Trials Snapshot
Trospium chloride is a muscarinic antagonist used to treat overactive bladder.[3] It has side effects typical of this class of drugs, namely dry mouth, stomach upset, and constipation; these side effects cause problems with people taking their medicine as directed. However it doesn’t cause central nervous system side effects like some other muscarinic antagonists.[4]
Chemically it is a quaternary ammonium cation which causes it to stay in periphery rather than crossing the blood–brain barrier.[5] It works by causing the smooth muscle in the bladder to relax.[3]
It was patented in 1966 and approved for medical use in 1974.[6] It was first approved in the US in 2004, and an extended release version was brought to market in 2007. It became generic in the EU in 2009, and the first extended-release generic was approved in the US in 2012.
SYN

Tropium chloride is one of the azoniaspironortropine derivatives and is used for the treatment of urinary bladder dysfunction due to bladder dysfunction, night urination, overactive bladder, and urinary incontinence. Useful compounds. The chemical name of the tropium chloride is (1R, 3R, 5S) -3-[(hydroxydiphenylacetyl) oxy] spiro [8-azoniabiscyclo [3,2,1] octane-8,1 ‘ -Pyrrolidinium] chloride ((1R, 3R, 5S) -3-[(Hydroxydiphenylacetyl) oxy] spiro [8-azoniabicyclo [3,2,1] octane-8,1’-pyrrolidinium] chloride) It is represented by Formula (1).

As a method for preparing the thromium chloride, US Patent No. 3,480,626 (1969) is prepared in the form of a free base of nortropine benzilate represented by the formula (2) as an intermediate, as shown in Scheme 1 below Thereafter, it is reacted with 1,4-dichlorobutane of the formula (3) to synthesize a thromium chloride, which is then recrystallized in ethanol-ether to disclose a two-step process for obtaining the thromium chloride. However, the method does not use a base, has a long reaction time, a low yield (about 46%), and instead of intramolecular cyclization, positions 1 and 4 of butane represented by the formula (4) as side reactants. There is a disadvantage in that a large amount of the compound in the form of substituted 1,4-nortropin benzylate is produced.

PATENT
https://patents.google.com/patent/KR20090076081A/en

Example 1 Preparation of Tropium Chloride
In a 1 L reactor equipped with a stirrer, 100 g of nortropin benzylate hydrochloride, 59 ml of 1,4-dichlorobutane, 89 ml of 1,8-diazabicyclo and 5 ml of 1,8-diazabicyclo [5,4,0] undec-7-ene and 500 ml of acetonitrile The reaction was carried out at 60 ° C. for 2 hours. Thin-Layer Chromatography (TLC) confirmed the completion of the reaction, when the reaction was complete, cooled to 5 ℃, stirred for 1 hour at the same temperature, the resulting crystals were filtered, dried at 60 ℃, white 92.6 g (yield: 81%) of the target compound were obtained. The 1 H-NMR (D 2 O, 400 MHz) data of the obtained compound are as follows: δ 1.34 to 1.36 (2H, d), 1.80 to 1.87 (2H, m), 1.98 (4H, s), 2.44 to 2.48 (2H , d), 3.21-3.24 (2H, t), 3.43-3.46 (2H, t), 3.56 (2H, s), 5.12-5.13 (1H, t), 7.31-7.74 (10H, m).
PATENT
https://patents.google.com/patent/CN102718760B/en


Medical uses
Trospium chloride is used for the treatment of overactive bladder with symptoms of urge incontinence and frequent urination.[3][4][2]
It should not be used with people who retain urine, who have severe digestive conditions, myasthenia gravis, narrow-angle glaucoma, or tachyarrhythmia.[3]
It should be used with caution in people who have problems with their autonomous nervous system (dysautonomia) or who have gastroesophageal reflux disease, or in whom fast heart rates are undesirable, such as people with hyperthyroidism, coronary artery disease and congestive heart failure.[3]
There are no adequate and well-controlled studies of trospium chloride in pregnant women and there are signs of harm to the fetus in animal studies. The drug was excreted somewhat in the milk of nursing mothers.[3] The drug was studied in children.[3]
Side effects
Side effects are typical of gastrointestinal effects of anticholinergic drugs, and include dry mouth, indigestion, and constipation. These side effects lead to problems with adherence, especially for older people.[4] The only CNS side effect is headache, which was very rare. Tachycardia is a rare side effect.[3]
Pharmacology
Mechanism of action
| Target | Affinity (Ki, nM) | Species |
|---|---|---|
| M1 | 3.5 | Human |
| M2 | 1.1 | Human |
| M3 | 1.0 | Human |
| M4 | 1.4 | Human |
| M5 | 6.0 | Human |
| Notes: Values are Ki, unless otherwise specified. The smaller the value, the more strongly the drug binds to the site. | ||
Trospium chloride is a muscarinic antagonist. Trospium chloride blocks the effect of acetylcholine on muscarinic receptors organs that are responsive to the compounds, including the bladder.[3] Its parasympatholytic action relaxes the smooth muscle in the bladder.[4] Receptor assays showed that trospium chloride has negligible affinity for nicotinic receptors as compared to muscarinic receptors at concentrations obtained from therapeutic doses.[3] The drug has high and similar affinity for all five of the muscarinic acetylcholine receptor subtypes, including the M1, M2, M3, M4, and M5 receptors.[9][10][11]
Pharmacokinetics
After oral administration, less than 10% of the dose is absorbed. Mean absolute bioavailability of a 20 mg dose is 9.6% (range: 4.0 to 16.1%). Peak plasma concentrations (Cmax) occur between 5 and 6 hours post-dose. Mean Cmax increases greater than dose-proportionally; a 3-fold and 4-fold increase in Cmax was observed for dose increases from 20 mg to 40 mg and from 20 mg to 60 mg, respectively. AUC exhibits dose linearity for single doses up to 60 mg. Trospium chloride exhibits diurnal variability in exposure with a decrease in Cmax and AUC of up to 59% and 33%, respectively, for evening relative to morning doses.[12]
Administration with a high fat meal resulted in reduced absorption, with AUC and Cmax values 70 to 80% lower than those obtained when trospium chloride was administered while fasting. Therefore, it is recommended that trospium chloride should be taken at least one hour prior to meals or on an empty stomach.[12]
Protein binding ranged from 50 to 85% when concentration levels of trospium chloride (0.5 to 50 ng/mL) were incubated with human serum in vitro. The 3H-trospium chloride ratio of plasma to whole blood was 1.6:1. This ratio indicates that the majority of 3H-trospium chloride is distributed in plasma. The apparent volume of distribution for a 20 mg oral dose is 395 (± 140) liters.[12]
The metabolic pathway of trospium in humans has not been fully defined. Of the 10% of the dose absorbed, metabolites account for approximately 40% of the excreted dose following oral administration. The major metabolic pathway is hypothesized as ester hydrolysis with subsequent conjugation of benzylic acid to form azoniaspironortropanol with glucuronic acid. Cytochrome P450 is not expected to contribute significantly to the elimination of trospium. Data taken from in vitro human liver microsomes investigating the inhibitory effect of trospium on seven cytochrome P450 isoenzyme substrates (CYP1A2, 2A6, 2C9, 2C19, 2D6, 2E1, and 3A4) suggest a lack of inhibition at clinically relevant concentrations.[12]
The plasma half-life for trospium chloride following oral administration is approximately 20 hours. After oral administration of an immediate-release formulation of 14C-trospium chloride, the majority of the dose (85.2%) was recovered in feces and a smaller amount (5.8% of the dose) was recovered in urine; 60% of the radioactivity excreted in urine was unchanged trospium. The mean renal clearance for trospium (29 L/hour) is 4-fold higher than average glomerular filtration rate, indicating that active tubular secretion is a major route of elimination for trospium. There may be competition for elimination with other compounds that are also renally eliminated.[12]
Chemistry
Anticholinergic drugs used to treat overactive bladder were all amines as of 2003. Quaternary ammonium cations in general are more hydrophilic than other amines and don’t cross membranes well, so they tend to be poorly absorbed from the digestive system, and to not cross the blood–brain barrier. Oxybutynin, tolterodine, darifenacin, and solifenacin are tertiary amines while trospium chloride and propantheline are quaternary amines.[5]
History
The synthesis of trospium was described by scientists from Dr. Robert Pfleger Chemische Fabrik GmbH, Heinz Bertholdt, Robert Pfleger, and Wolfram Schulz, in US. Pat. No. 3,480,626 (the US equivalent to DE119442), and its activity was first published in the literature in 1967.[13][14]
The first regulatory approval was granted in Germany in August 1999 to Madaus AG for Regurin 20 mg Tablets.[15]: 13 Madaus is considered the originator for regulatory filings worldwide.[16] The German filing was recognized throughout Europe under the Mutual Recognition Procedure.[15]: 13
Madaus licensed the US rights to trospium chloride to Interneuron in 1999 and Interneuron ran clinical trials in the US to win FDA approval.[17][18] Interneuron changed its name to Indevus in 2002[19] Indevus entered into a partnership with Odyssey Pharmaceuticals, a subsidiary of Pliva, to market the drug in April 2004,[20] and won FDA approval for the drug, which it branded as Sanctura, in May 2004.[21][22] The approval earned Indevus a milestone payment of $120M from Pliva, which had already paid Indevus $30 million at signing; the market for overactive bladder therapies was estimated to be worth $1.1 billion in 2004.[23] In 2005 Pliva exited the relationship, selling its rights to Esprit Pharma,[24] and in September 2007 Allergan acquired Esprit, and negotiated a new agreement with Indevus under which Allergan would completely take over the US manufacturing, regulatory approvals, and marketing.[25] A month before, Indevus had received FDA approval for an extended release formulation that allowed once a day dosing, Sanctura XR.[26] Indevus had developed intellectual property around the extended release formulation which it licensed to Madaus for most of the world.[25]
In 2012 the FDA approved the first generic version of the extended release formulation, granting approval to the ANDA that Watson Pharmaceuticals had filed in 2009.[27] Annual sales in the US at that time were $67M.[28] European patents had expired in 2009.[29]
As of 2016, the drug is available worldwide under many brand names and formulations, including oral, extended release, suppositories, and injections.[1]
Society and culture
Marketing rights to the drug became subject to parallel import litigation in Europe in the case of Speciality European Pharma Ltd v Doncaster Pharmaceuticals Group Ltd / Madaus GmbH (Case No. A3/2014/0205) which was resolved in March 2015. Madaus had exclusively licensed the right to use the Regurin trademark to Speciality European Pharma Ltd. In 2009, when European patents expired on the drug, Doncaster Pharmaceuticals Group, a well known parallel importer, which had been selling the drug in the UK under another label, Ceris, which was used in France, began to put stickers on their packaging with the Regurin name. Speciality and Madaus sued and initially won based on the argument that 90% of prescriptions were already generic, but Doncaster appealed and won the appeal based on the argument that it could not charge a premium with a generic label. The case has broad implications for trade in the EU.[29][30]
Research
In 2007 Indevus partnered with Alkermes to develop and test an inhaled form of trospium chloride as a treatment for COPD; it was in Phase II trials at that time.[31]
Reference
- ^ Jump up to:a b “International brands of trospium”. Drugs.com. Retrieved 13 May 2016.
- ^ Jump up to:a b FDA “Trospium chloride label” (PDF). U.S. Food and Drug Administration. January 2011.
- ^ Jump up to:a b c d e f g h i j “Regurin XL 60mg”. UK eMC. 3 July 2015.
- ^ Jump up to:a b c d Biastre K, Burnakis T (February 2009). “Trospium chloride treatment of overactive bladder”. Ann Pharmacother. 43 (2): 283–95. doi:10.1345/aph.1L160. PMID 19193592. S2CID 20102756.
- ^ Jump up to:a b Pak RW, Petrou SP, Staskin DR (December 2003). “Trospium chloride : a quaternary amine with unique pharmacologic properties”. Curr Urol Rep. 4 (6): 436–40. doi:10.1007/s11934-003-0023-1. PMID 14622495. S2CID 4512769.
- ^ Fischer J, Ganellin CR (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 446. ISBN 9783527607495.
- ^ Liu T (2020). “BindingDB BDBM50540489 Flotros::IP-631::IP631::Regurin::Regurin xl::Sanctura::Sanctura xr::Spasmo-lyt::Trospium chloride::Uraplex”. Journal of Medicinal Chemistry. 63 (11): 5763–5782. doi:10.1021/acs.jmedchem.9b02100. PMC 8007111. PMID 32374602. Retrieved 28 October 2024.
- ^ Del Bello F, Bonifazi A, Giorgioni G, Piergentili A, Sabbieti MG, Agas D, et al. (June 2020). “Novel Potent Muscarinic Receptor Antagonists: Investigation on the Nature of Lipophilic Substituents in the 5- and/or 6-Positions of the 1,4-Dioxane Nucleus”. J Med Chem. 63 (11): 5763–5782. doi:10.1021/acs.jmedchem.9b02100. PMC 8007111. PMID 32374602.
- ^ Peretto I, Petrillo P, Imbimbo BP (November 2009). “Medicinal chemistry and therapeutic potential of muscarinic M3 antagonists”. Med Res Rev. 29 (6): 867–902. doi:10.1002/med.20158. PMID 19399831.
- ^ Pak RW, Petrou SP, Staskin DR (December 2003). “Trospium chloride: a quaternary amine with unique pharmacologic properties”. Curr Urol Rep. 4 (6): 436–440. doi:10.1007/s11934-003-0023-1. PMID 14622495.
- ^ Rosa GM, Bauckneht M, Scala C, Tafi E, Leone Roberti Maggiore U, Ferrero S, et al. (November 2013). “Cardiovascular effects of antimuscarinic agents in overactive bladder”. Expert Opin Drug Saf. 12 (6): 815–827. doi:10.1517/14740338.2013.813016. PMID 23800037.
- ^ Jump up to:a b c d e Doroshyenko O, Jetter A, Odenthal KP, Fuhr U (2005). “Clinical pharmacokinetics of trospium chloride”. Clin Pharmacokinet. 44 (7): 701–20. doi:10.2165/00003088-200544070-00003. PMID 15966754. S2CID 10968270.
- ^ US 6974820 which cites US 3480626 and Bertholdt H, Pfleger R, Schulz W (1967). “[On azoniaspire-compounds. 2. Preparation of esterified azoniaspire-compounds of nortropan-3-alpha- or 3-beta-ol (1)]”. Arzneimittelforschung. 17 (6): 719–26. PMID 5632538.
- ^ DE patent 1194422, Bertholdt H, Pfleger R, Schulz W, “[Verfahren zur Herstellung von Azoniaspironortropanderivaten] (A process for preparing azonia-spirono-tropane derivatives)”, issued 10 June 1965, assigned to Dr. Robert Pfleger Chemische Fabrik GmbH
- ^ Jump up to:a b “Trospium Chloride 20mg Film-Coated Tablets, Public Assessment Report” (PDF). Medicines and Healthcare products Regulatory Agency. 7 April 2011.
- ^ “Trospium chloride”. AdisInsight. Springer Nature Switzerland AG.
- ^ Miller J (23 September 2002). “Indevus to apply for new drug status for incontinence drug”. Boston Business Journal.
- ^ Herper M (25 September 2002). “A Biotech Phoenix Could Be Rising”. Forbes.
- ^ “Indevus Pharmaceuticals, Inc., Formerly Interneuron, to Begin Trading on Nasdaq”. Indevus Press Release. 2 April 2002.
- ^ “Indevus and PLIVA Sign Co-Promotion and Licensing Agreement for SANCTURA -Trospium Chloride”. Indevus Press Release. 7 April 2004. Archived from the original on 27 August 2021. Retrieved 14 May 2016.
- ^ “Sanctura (trospium chloride)”. CenterWatch. Archived from the original on 5 August 2019. Retrieved 13 May 2016.
- ^ “Indevus Announces FDA Approval Of Sanctura”. Indevus Press Release. 28 May 2004.
- ^ Osterweil N (28 May 2004). “FDA approves Indevus’ Sanctura”. for First Word Pharma.
- ^ “Novartis, P&G enter agreement for OAB drug”. Urology Times. 21 July 2005.
- ^ Jump up to:a b “Indevus Announces Allergan as New Partner for Sanctura Brand”. Indevus Press Release. 19 September 2007.
- ^ “Indevus’ Sanctura XR approved by US FDA”. The Pharma Letter. 13 August 2007.
- ^ “ANDA 091289 approval letter” (PDF). U.S. Food and Drug Administration. 12 October 2012.
- ^ “Watson’s Generic Sanctura XR Receives FDA Approval”. Watson Press Release. 12 October 2012.
- ^ Jump up to:a b “Court takes a permissive approach to parallel importers within the EU”. Lexology. 6 March 2015.
- ^ R.P.C. (2015) 132 (7): 521-540. doi: 10.1093/rpc/rcv039
- ^ “Alkermes, Indevus testing COPD drug”. UPI. 25 April 2007.
External links
Trospium chloride at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
| Clinical data | |
|---|---|
| Pronunciation | /ˈtroʊspiəm/ TROHS-pee-əm |
| Trade names | Regurin, Sanctura, others[1] |
| AHFS/Drugs.com | Monograph |
| Routes of administration | By mouth |
| Drug class | Antimuscarinic (peripherally selective) |
| ATC code | G04BD09 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[2]In general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Protein binding | 50–85% |
| Elimination half-life | 20 hours |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 10405-02-4 |
| PubChem CID | 107979 |
| DrugBank | DB00209 |
| ChemSpider | 10482307 |
| UNII | 1E6682427E |
| ChEBI | CHEBI:32270 |
| ChEMBL | ChEMBL1201344 |
| CompTox Dashboard (EPA) | DTXSID7023724 |
| ECHA InfoCard | 100.030.784 |
| Chemical and physical data | |
| Formula | C25H30ClNO3 |
| Molar mass | 427.97 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
- Trospium [Link]
- FDA drug approval: Trospium [Link]
- FDA Approved Drug Products: Cobenfy (xanomeline tartrate/trospium chloride) capsules for oral use (September 2024) [Link]
- DailyMed Label: TROSPIUM CHLORIDE oral capsule, extended release [Link]
///////Trospium chloride, Cobenfy, APPROVALS 2024, FDA 2024, SMR002533165
Flurpiridaz F 18



Flurpiridaz F 18
WeightAverage: 367.84
Monoisotopic: 367.1328329
Chemical FormulaC18H22ClFN2O3
- 863887-89-2
- Bms 747158-02
2-tert-butyl-4-chloro-5-[[4-(2-(18F)fluoranylethoxymethyl)phenyl]methoxy]pyridazin-3-one
- 2-tert-butyl-4-chloro-5-[[4-(2-(18F)fluoranylethoxymethyl)phenyl]methoxy]pyridazin-3-one
- 2-TERT-BUTYL-4-CHLORO-5-((4-((2-(18F)FLUOROETHOXY)METHYL)PHENYL)METHOXY)PYRIDAZIN-3(2H)-ONE
- 3(2H)-PYRIDAZINONE, 4-CHLORO-2-(1,1-DIMETHYLETHYL)-5-((4-((2-(FLUORO-18F)ETHOXY)METHYL)PHENYL)METHOXY)-
- UNII-TY3V24C029
FDA APPROVED 9/27/2024, Flyrcado, A radioactive diagnostic drug to evaluate for myocardial ischemia and infarction
Flurpiridaz (18F), sold under the brand name Flyrcado, is a cyclotron-produced radioactive diagnostic agent for use with positron emission tomography (PET) myocardial perfusion imaging under rest or stress (pharmacologic or exercise).[3] Flurpiridaz (18F) It is given by intravenous injection.[3]
The most common adverse reactions include dyspnea (shortness of breath), headache, angina pectoris (severe pain in the chest), chest pain, fatigue, ST segment changes, flushing, nausea, abdominal pain, dizziness, and arrhythmia (irregular heartbeat).[3]
Flurpiridaz (18F) was approved for medical use in the United States in September 2024.[3][4][5][6]
PATENT
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US9687571 | No | 2017-06-27 | 2032-11-01 |  |
| US9603951 | No | 2017-03-28 | 2031-05-02 | |
| US9161997 | No | 2015-10-20 | 2026-02-04 | |
| US8936777 | No | 2015-01-20 | 2031-06-30 | |
| US8226929 | No | 2012-07-24 | 2028-06-21 | |
| US7344702 | No | 2008-03-18 | 2026-05-26 | |
SYN
https://ejnmmipharmchem.springeropen.com/articles/10.1186/s41181-022-00182-z


Chemistry
Synthesis of precursor of [18F]Flurpiridaz (7) [2-(4-((1-tert-Butyl-5-chloro-6-oxo-1,6-dihydropyridazine-4-yloxy)methyl)benzyloxy)ethyl-4- methylbenzensulfonate] (6)
Precursor 6 was synthesized according to the literature procedures with few changes (Purohit et al. 2008; Nagel 2014) Briefly, to a mixture of mucochloric acid (1) (1.18 g, 6.98 mmol) and Na2CO3 (0.33 g, 3.11 mmol) in 15 ml of distilled water was added tert-butylhydrazine hydrochloride (0.86 g, 6.90 mmol) in ice-water bath and reaction mixture was stirred for about 4 h. White precipitate was washed by water and dried under reduced vacuum after filtration. Then, 13.2 ml of benzene and acetic acid (1.86 g, 30,95 mmol) were added and reaction was kept at 40 °C for 4 h. Organic phase was extracted with 10 ml of water and washed by 5 ml of 1.25 M NaOH(aq), 5 ml of 5 M HCl(aq) and 10 ml of water respectively. 0.83 g of DCP (2) was obtained as an orange solid. 1.0 g of DCP (2) (4,53 mmol) was dissolved in 15 ml of dry DMF, 1,4-phenylene dimethanol (3.2 g, 23.16 mmol) and Cs2CO3 (6.0 g, 18.41 mmol) were slowly added to the solution and reaction was stirred at 68 °C under nitrogen atmosphere for about 6 h and allowed to be cooled down to room temperature. Crude product was extracted with CHCl3/water several times and evaporated under vacuum. Residue was subjected to flash column chromatography (silica gel 40 g, EtOAc/Hexane 3:2) and 0.91 g of compound 3 was obtained as white solid. Then, 0.91 g of an alcoholic compound 3 was dissolved in 15 ml of freshly distilled dichloromethane and 0.14 ml of PBr3 was slowly added to the solution. The reaction was carried out at room temperature for about 2 h under nitrogen atmosphere. Crude product was extracted with 30 ml of water and dried under vacuum. White solid product 4 was successfully obtained in a quantitative yield without further purification for next step. KOtBu (0.28 g, 2.49 mmol) and 11.2 ml of ethylene glycol were stirred at room temperature under nitrogen atmosphere. Then, 0.95 g of bromide compound 4 dissolved in 8 ml of dry THF was added slowly into the reaction mixture and the reaction was stirred at 60 °C for overnight. After cooling to room temperature, THF was evaporated and residue was extracted with CHCl3/water several times. Organic phase was evaporated under vacuum and residue was submitted to flash column chromatograpy (silica gel 40 g, EtOAc/Hexane 2:1) and 0.86 g of compound 5 was obtained as colorless oil in quantitative yield. Finally, to a mixture of 0.85 g of compound 5 and tosyl chloride (690 mg, 3.62 mmol) in 6 ml of dry dichloromethane, 0.64 ml of DIPEA and 4-(dimethylamino) pyridine (445 mg, 3.64 mmol) were added and reaction was carried out at room temperature for 2.5 h under nitrogen atmosphere. Dichloromethane was evaporated and crude product was directly subjected to flash column chromatograpy (silica gel 45 g, EtOAc/Hexane 2:1). 0.9 g of pure tosylate 6 (precursor of [18F]Flurpiridaz) was obtained by recrystallisation in dichloromethane at + 4 °C. Tosylate 6 was further purified through semipreparative HPLC for an accurate spectroscopic characterization (Fig. 1). Anal. Calcd for C25H29ClN2O6S: C, 57.63; H, 5.61; Cl, 6.80; N, 5.38; S, 6.15. Found: C, 57.86; H, 5.84; Cl, 7.03; N, 5.66; S, 6.34.
1H NMR ((CDCl3, 400 MHz) δ (ppm)): 7,80 (d, J = 9.1 Hz, 2H); 7,73 (s, 1H); 7,39 (d, J = 9.1 Hz, 2H); 5,29 (s, 2H,); 4,49 (s, 2H,); 4,20–4,19 (m, 2H); 3,70–3,65 (m, 2H); 2,42 (s, 3H); 1,60 (s, 9H).
Synthesis of [18F]Flurpiridaz (7)
Preliminary studies & synthesis of [19F]Flurpiridaz (7) (Cold runs)
Materials KF, Ethanol, and Acetonitrile were obtained from Sigma Aldrich. Kryptofix K2.2.2./K2CO3 (22 mg Kryptofix K2.2.2., 7 mg K2CO3, 300 µl acetonitrile and 300 µl pure water), TBA-HCO3 (0.075 M) solution and QMA Cartridges were from ABX. Sep-Pak C18 Plus Light Cartridge was from Waters.
Methods
Firstly, consecutive cold syntheses of [19F]Flurpiridaz (7) were performed using stable isotope fluorine-19 and optimum reaction parameters were tried to be determined.
Eluent solution-I (Kryptofix K2.2.2./K2CO3)
50 mg of KF was dissolved in 2 mL of ultrapure water and directly passed through the preconditioned QMA cartridge. The QMA cartridge was rinsed with 5 mL of ultrapure water and dried with N2. [19F]F trapped on the QMA cartridge was eluted into the reaction vial with 600 µL of Kryptofix K2.2.2./K2CO3 solution. Solvents in the reaction vial were removed at 100 °C, [19F]F and Kryptofix K2.2.2./K2CO3 were dried gently. Then, 10 mg of precursor 6 dissolved in 2 mL of anhydrous acetonitrile was added to the reaction vial and the mixture was sealed and heated at 95 °C for 10 min. The reaction solution was diluted with 5 ml of ultrapure water and directly passed through a preconditioned C-18 cartridge. C-18 cartridge was rinsed with 5 mL of ultrapure water and dried with air. Finally, C-18 cartridge was eluted with 5 mL of ethanol and transferred into the final product vial. The final product was diluted with 5 mL of ultrapure water (n = 3) and analyzed by HPLC (described in HPLC analysis of precursor 6) to determine its composition.
Chromatogram analysis indicated that four different separate peaks were observed. The unreacted precursor 6 was detected around 11.55 min. The other two peaks were detected between 8 and 9 min. Another major peak around 5.5 min was detected. It was concluded that the chemical yield of product 7 was low due to the majority of side-product formations
Medical uses
Flurpiridaz (18F) is indicated for positron emission tomography myocardial perfusion imaging under rest or stress (pharmacologic or exercise) in adults with known or suspected coronary artery disease to evaluate for myocardial ischemia and infarction.[2][3]
History
Flurpiridaz F-18 is a fluorine 18-labeled agent developed by Lantheus Medical Imaging for the diagnosis of coronary artery disease.[7]
The efficacy and safety of flurpiridaz (18F) were evaluated in two prospective, multicenter, open-label clinical studies in adults with either suspected CAD (Study 1: NCT03354273) or known or suspected CAD (Study 2: NCT01347710).[3] Study 1 evaluated the sensitivity (ability to designate an imaged patient with disease as positive) and specificity (ability to designate an imaged patient without disease as negative) of flurpiridaz (18F) for the detection of significant CAD in subjects with suspected CAD who were scheduled for invasive coronary angiography (ICA).[3] Across three flurpiridaz (18F) imaging readers, estimates of sensitivity ranged from 74% to 89% and estimates of specificity ranged from 53% to 70% for CAD defined as at least 50% narrowing of an artery.[3]
Study 2 evaluated the sensitivity and specificity of flurpiridaz (18F) for the detection of significant CAD in subjects with known or suspected CAD who had ICA without intervention within 60 days prior to imaging or were scheduled for ICA.[3] Across three flurpiridaz (18F) imaging readers, estimates of sensitivity ranged from 63% to 77% and estimates of specificity ranged from 66% to 86% for CAD defined as at least 50% narrowing of an artery.[3]
Society and culture
Legal status
Flurpiridaz (18F) was approved for medical use in the United States in September 2024.[2][3]
Names
Flurpiridaz (18F) is the international nonproprietary name.[8]
References
- ^ “Flurpiridaz F 18”. AMA Finder. Retrieved 27 September 2024.
- ^ Jump up to:a b c d e “Flyrcado (flurpiridaz F 18) injection, for intravenous use” (PDF). U.S. Food and Drug Administration (FDA). Retrieved 27 September 2024.
- ^ Jump up to:a b c d e f g h i j k “FDA approves imaging drug for evaluation of myocardial ischemia”. U.S. Food and Drug Administration (FDA). 27 September 2024. Retrieved 27 September 2024. This article incorporates text from this source, which is in the public domain.
- ^ “Drug Approval Package: Flyrcado Injection”. U.S. Food and Drug Administration (FDA). 25 October 2024. Retrieved 21 January 2025.
- ^ “Novel Drug Approvals for 2024”. U.S. Food and Drug Administration. 1 October 2024. Retrieved 8 November 2024.
- ^ New Drug Therapy Approvals 2024 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2025. Archived from the original on 21 January 2025. Retrieved 21 January 2025.
- ^ “Flurpiridaz F-18”. Inxight Drugs. Retrieved 27 September 2024.
- ^ World Health Organization (2011). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 65”. WHO Drug Information. 25 (1). hdl:10665/74623.
Further reading
- Maddahi J, Agostini D, Bateman TM, Bax JJ, Beanlands RS, Berman DS, et al. (October 2023). “Flurpiridaz F-18 PET Myocardial Perfusion Imaging in Patients With Suspected Coronary Artery Disease”. Journal of the American College of Cardiology. 82 (16): 1598–1610. doi:10.1016/j.jacc.2023.08.016. PMID 37821170.
- Matsumoto N (2023). “Progress of 18F-flurpiridaz in Clinical Trials”. Annals of Nuclear Cardiology. 9 (1): 91–93. doi:10.17996/anc.23-00011. PMC 10696143. PMID 38058576.
External links
- Clinical trial number NCT03354273 for “An International Study to Evaluate Diagnostic Efficacy of Flurpiridaz (18F) Injection PET MPI in the Detection of Coronary Artery Disease (CAD)” at ClinicalTrials.gov
- Clinical trial number NCT01347710 for “A Phase 3 Multi-center Study to Assess PET Imaging of Flurpiridaz F 18 Injection in Patients With CAD” at ClinicalTrials.gov
- Maddahi J, Agostini D, Bateman TM, Bax JJ, Beanlands RSB, Berman DS, Dorbala S, Garcia EV, Feldman J, Heller GV, Knuuti JM, Martinez-Clark P, Pelletier-Galarneau M, Shepple B, Tamaki N, Tranquart F, Udelson JE: Flurpiridaz F-18 PET Myocardial Perfusion Imaging in Patients With Suspected Coronary Artery Disease. J Am Coll Cardiol. 2023 Oct 17;82(16):1598-1610. doi: 10.1016/j.jacc.2023.08.016. [Article]
- Berman DS, Maddahi J, Tamarappoo BK, Czernin J, Taillefer R, Udelson JE, Gibson CM, Devine M, Lazewatsky J, Bhat G, Washburn D: Phase II safety and clinical comparison with single-photon emission computed tomography myocardial perfusion imaging for detection of coronary artery disease: flurpiridaz F 18 positron emission tomography. J Am Coll Cardiol. 2013 Jan 29;61(4):469-477. doi: 10.1016/j.jacc.2012.11.022. Epub 2012 Dec 19. [Article]
- Maddahi J, Lazewatsky J, Udelson JE, Berman DS, Beanlands RSB, Heller GV, Bateman TM, Knuuti J, Orlandi C: Phase-III Clinical Trial of Fluorine-18 Flurpiridaz Positron Emission Tomography for Evaluation of Coronary Artery Disease. J Am Coll Cardiol. 2020 Jul 28;76(4):391-401. doi: 10.1016/j.jacc.2020.05.063. [Article]
- Patel KK, Singh A, Bateman TM: The Potential of F-18 Flurpiridaz PET/CT Myocardial Perfusion Imaging for Precision Imaging. Curr Cardiol Rep. 2022 Aug;24(8):987-994. doi: 10.1007/s11886-022-01713-5. Epub 2022 May 26. [Article]
- FDA Approved Drug Products: FLYRCADO (flurpiridaz F 18) injection, for intravenous use [Link]
/////////////Flurpiridaz F 18, Flyrcado, APPROVALS 2024, FDA 2024, Bms 747158-02, BMS 747158-02, BM-747158-02, BMS747158-02
| Clinical data | |
|---|---|
| Trade names | Flyrcado |
| Other names | NMB58, BMS-747158-02, flurpiridaz F-18, flurpiridaz F 18[1] (USAN US) |
| AHFS/Drugs.com | Flyrcado |
| License data | US DailyMed: Flurpiridaz |
| Routes of administration | Intravenous |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 863887-89-2 |
| PubChem CID | 11405965 |
| DrugBank | DB18773 |
| ChemSpider | 9580860 |
| UNII | TY3V24C029 |
| KEGG | D10009 |
| CompTox Dashboard (EPA) | DTXSID00235517 |
| Chemical and physical data | |
| Formula | C18H22Cl[18F]N2O3[2] |
| Molar mass | 367.8 [2] |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
Inavolisib



Inavolisib
WeightAverage: 407.378
Monoisotopic: 407.140510438
Chemical FormulaC18H19F2N5O4
- GDC-0077
- CAS 2060571-02-8
- GDC0077
- RG6114
- WHO 11204
- GDC 0077
- GDC-0077
- RG-6114
- RG6114
- RO-7113755
- RO7113755
FDA APPROVED, 10/10/2024, Itovebi, To treat locally advanced or metastatic breast cancer
Drug Trials Snapshot
(2S)-2-[[2-[(4S)-4-(difluoromethyl)-2-oxo-1,3-oxazolidin-3-yl]-5,6-dihydroimidazo[1,2-d][1,4]benzoxazepin-9-yl]amino]propanamide
- (2S)-2-((2-((4S)-4-(difluoromethyl)-2-oxo-3-oxazolidinyl)-5,6-dihydroimidazo(1,2-D)(1,4)benzoxazepin-9-yl)amino)propanamide
- propanamide, 2-((2-((4S)-4-(difluoromethyl)-2-oxo-3-oxazolidinyl)-5,6-dihydroimidazo(1,2-D)(1,4)benzoxazepin-9-yl)amino)-, (2S)-
Inavolisib, sold under the brand name Itovebi, is an anti-cancer medication used for the treatment of breast cancer.[2][3] It is an inhibitor and degrader of mutant phosphatidylinositol 3-kinase (PI3K) alpha.[4] The PI3K-mediated signalling pathway has shown to play an important role in the development of tumours as dysregulation is commonly associated with tumour growth and resistance to antineoplastic agents and radiotherapy.[5]
The most common adverse reactions include decreased neutrophils, decreased hemoglobin, increased fasting glucose, decreased platelets, decreased lymphocytes, stomatitis, diarrhea, decreased calcium, fatigue, decreased potassium, increased creatinine, increased ALT, nausea, decreased sodium, decreased magnesium, rash, decreased appetite, COVID-19 infection, and headache.[3]
Inavolisib was approved for medical use in the United States in October 2024.[3][6][7]
SYN
Hanan EJ, Braun MG, Heald RA, MacLeod C, Chan C, Clausen S, Edgar KA, Eigenbrot C, Elliott R, Endres N, Friedman LS, Gogol E, Gu XH, Thibodeau RH, Jackson PS, Kiefer JR, Knight JD, Nannini M, Narukulla R, Pace A, Pang J, Purkey HE, Salphati L, Sampath D, Schmidt S, Sideris S, Song K, Sujatha-Bhaskar S, Ultsch M, Wallweber H, Xin J, Yeap S, Young A, Zhong Y, Staben ST: Discovery of GDC-0077 (Inavolisib), a Highly Selective Inhibitor and Degrader of Mutant PI3Kalpha. J Med Chem. 2022 Dec 22;65(24):16589-16621. doi: 10.1021/acs.jmedchem.2c01422. Epub 2022 Dec 1.





PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US215633239&_cid=P11-M9XU5W-08686-1


Example 101 (S)-2-((2-((S)-4-(Difluoromethyl)-2-oxooxazolidin-3-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl)amino)propanamide 101
Step 1: 4-Bromo-2-hydroxybenzaldehyde
Step 2: 5-Bromo-2-(1H-imidazol-2-yl)phenol
Step 3: 9-Bromo-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepine
Step 4: 9-Bromo-2,3-diiodo-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepine
Step 5: 9-Bromo-2-iodo-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepine
Step 6: (R)-2,2-Dimethyl-[1,3]dioxolane-4-carbaldehyde
Step 7: (R)-4-Difluoromethyl-2,2-dimethyl-[1,3]dioxolane
Step 8: (R)-3-(tert-Butyldimethylsilanyloxy)-1,1-difluoropropan-2-ol
Step 9: ((S)-2-Azido-3,3-difluoropropoxy)-tert-butyldimethylsilane
Step 10: (S)-1-(tert-Butyldimethylsilanyloxymethyl)-2,2-difluoroethylamine
Step 12: (S)-3-(9-Bromo-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-2-yl)-4-(difluoromethyl)oxazolidin-2-one
Step 13: (S)-2-((2-((S)-4-(Difluoromethyl)-2-oxooxazolidin-3-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl)amino)propanamide
Medical uses
Inavolisib is indicated in combination with palbociclib and fulvestrant for the treatment of adults with endocrine-resistant, PIK3CA-mutated, hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative, locally advanced or metastatic breast cancer, as detected by an FDA-approved test, following recurrence on or after completing adjuvant endocrine therapy.[3]
Side effects
The most common adverse reactions include decreased neutrophils, decreased hemoglobin, increased fasting glucose, decreased platelets, decreased lymphocytes, stomatitis, diarrhea, decreased calcium, fatigue, decreased potassium, increased creatinine, increased ALT, nausea, decreased sodium, decreased magnesium, rash, decreased appetite, COVID-19 infection, and headache.[3]
History
Efficacy was evaluated in INAVO120 (NCT04191499), a randomized, double-blind, placebo-controlled, multicenter trial in 325 participants with endocrine-resistant, PIK3CA-mutated HR-positive, HER2-negative locally advanced or metastatic breast cancer whose disease progressed during or within twelve months of completing adjuvant endocrine therapy and who had not received prior systemic therapy for locally advanced or metastatic disease.[3] Primary endocrine resistance was defined as relapse while on the first two years of adjuvant endocrine therapy (ET) and secondary endocrine resistance was defined as relapse while on adjuvant ET after at least two years or relapse within twelve months of completing adjuvant ET.[3]
Structure, reactivity, and synthesis
Inavolisib is a synthetic, organic, small compound (the full structure can be seen here).[8] When binding to phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha (p110α), inavolisib’s carbonyl group can accept a hydrogen bond from the Tyr836 (conserved) in p110α. The difluoromethyl group can interact with the hydroxyl group presented on Ser774 (conserved) in p110α, which is 3.2Å nearer than of which on the equivalent residue Ser754 in p110δ. Additionally, the amide group can interact with Gln859 (non-conserved). This results in a very high selectivity regarding PI3Kα isoforms.[4][9]
Compared to similar PI3K inhibiting compounds, inavolisib has a higher thermodynamic aqueous solubility that proved advantageous in the formulation process and aiding greater consistency in predictions of absorption.[4]
Inavolisibcan be developed as a derivative of 1,3-oxazole[10] or by means of stereo-controlled N-arylation of alpha-amino acids.[11]
Metabolism and biotransformation
Inavolisib is orally administered, though there is little knowledge about its metabolism.[12]However, absorption, metabolism, and excretion data of taselisib, a molecule with a related chemical scaffold, suggest moderately slow absorption into the systemic circulation, metabolism to play a minor role in drug clearance, and biliary excretion to be the main route of excretion.[13]
Molecular mechanisms of action
Inavolisib is a selective PI3K-p110α (PIK3CA) inhibitor, which may offer antineoplastic functionality.[8] Therefore, it may serve as a new addition to combination therapy with conventional cancer treatment, such as chemotherapy. Combining inavolisib with palbociclib and fulvestrant might improve treatment of breast cancer.[14]
Next to its inhibitory enzymatic ability, it is suggested that inavolisib binds to – and activates degradation of – mutated forms of p110α. Members of the PI3K family regulate cellular processes such as cell growth and proliferation, survival, remodelling, and intracellular transport of organelles.[15] PI3K also plays an essential role for the immune system.
The class I isoform PI3K alpha (PI3Kα) is often times expressed in solid tumours through gene amplification or activated mutations.[4] Mutations in PI3Kα can often be found in cancer cells, especially HR+ breast cancer, which causes a disruption of the PI3K pathway. This leads to increased tumour growth and metastasis. One of the most common mutations can be found in PIK3CA, which plays a significant role in tumour cell proliferation.
In preclinical studies, inavolisib has shown to specifically initiate the degradation of this p110α oncogene with the help of proteasomes.[16] After binding to the mutant PI3Kα, inavolisib blocks phosphorylation of PIP2 to PIP3, thereby stopping downstream signalling.[17]
Consequently, biomarkers in the PI3K pathway are reduced, cell proliferation inhibited, and the rate of PIK3CA-mutant breast cancer apoptosis increased (in comparison to the wild type). The exact mechanism of action of inhibitors like inavolisib on mutated PI3Kα and the inhibitors’ influence on mutant structures are still unknown.[18]
Toxicity
Inavolisib is able to induce a cytotoxic response but this is directed towards tumour cells that contain the PI3K mutation, thereby inhibiting further tumour growth and leading to cell loss.[19]
Society and culture
Legal status
In October 2024, the US Food and Drug Administration (FDA) approved inavolisib for the treatment of PIK3CA-mutant breast cancer based on the results from the INAVO120 trial.[3][6][20][21] The drug application was granted priority review and breakthrough therapy designations by the FDA.[3]
Names
Inavolisib is the international nonproprietary name.[22][23]
Inavolisib is sold under the brand name Itovebi.[3]
Research
Due to inavolisib’s ability to inhibit the PI3K pathway through HER2-dependent degradation, it is undergoing clinical trials to potentially make use of it as an antineoplastic (anti-cancer) drug to treat breast cancer.[4][24][17]
References
- ^ “Register of Innovative Drugs”. Health Canada. 3 November 2006. Retrieved 17 April 2025.
- ^ Jump up to:a b “Itovebi- inavolisib tablet, film coated”. DailyMed. 11 October 2024. Retrieved 11 November 2024.
- ^ Jump up to:a b c d e f g h i j “FDA approves inavolisib with palbociclib and fulvestrant for endocrine-resistant, PIK3CA-mutated, HR-positive, HER2-negative, advanced breast cancer”. U.S. Food and Drug Administration (FDA). 10 October 2024. Retrieved 11 October 2024. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d e Hanan EJ, Braun MG, Heald RA, MacLeod C, Chan C, Clausen S, et al. (December 2022). “Discovery of GDC-0077 (Inavolisib), a Highly Selective Inhibitor and Degrader of Mutant PI3Kα”. Journal of Medicinal Chemistry. 65 (24). American Chemical Society (ACS): 16589–16621. doi:10.1021/acs.jmedchem.2c01422. PMID 36455032. S2CID 254149451.
- ^ “CID 124173720, Inavolisib”. PubChem. National Center for Biotechnology Information, U.S. National Library of Medicine. Retrieved 21 September 2023.
- ^ Jump up to:a b “Novel Drug Approvals for 2024”. U.S. Food and Drug Administration (FDA). 1 October 2024. Retrieved 29 November 2024.
- ^ New Drug Therapy Approvals 2024 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2025. Archived from the original on 21 January 2025. Retrieved 21 January 2025.
- ^ Jump up to:a b “inavolisib — Ligand page”. IUPHAR/BPS Guide to Pharmacology. Retrieved 21 September 2023.
- ^ Vanhaesebroeck B, Perry MW, Brown JR, André F, Okkenhaug K (October 2021). “PI3K inhibitors are finally coming of age”. Nature Reviews. Drug Discovery. 20 (10). Springer Science and Business Media LLC: 741–769. doi:10.1038/s41573-021-00209-1. PMC 9297732. PMID 34127844.
- ^ Chen J, Lv S, Liu J, Yu Y, Wang H, Zhang H (December 2021). “An Overview of Bioactive 1,3-Oxazole-Containing Alkaloids from Marine Organisms”. Pharmaceuticals. 14 (12). MDPI AG: 1274. doi:10.3390/ph14121274. PMC 8706051. PMID 34959674.
- ^ Han C, Kelly SM, Cravillion T, Savage SJ, Nguyen T, Gosselin F (2019). “Synthesis of PI3K inhibitor GDC-0077 via a stereocontrolled N-arylation of α-amino acids”. Tetrahedron. 75 (32). Elsevier BV: 4351–4357. doi:10.1016/j.tet.2019.04.057. ISSN 0040-4020. S2CID 150262658.
- ^ “Inavolisib: Uses, Interactions, Mechanism of Action”. DrugBank. 20 May 2019. DB15275. Retrieved 21 September 2023.
- ^ Ma S, Cho S, Sahasranaman S, Zhao W, Pang J, Ding X, et al. (April 2023). “Absorption, Metabolism, and Excretion of Taselisib (GDC-0032), a Potent β-Sparing PI3K Inhibitor in Rats, Dogs, and Humans”. Drug Metabolism and Disposition. 51 (4): 436–450. doi:10.1124/dmd.122.001096. PMID 36623882.
- ^ “A trial looking at a new drug called inavolisib for breast cancer that has spread (WO41554)”. Cancer Research UK. 22 June 2021. Retrieved 21 September 2023.
- ^ Koyasu S (April 2003). “The role of PI3K in immune cells”. Nature Immunology. 4 (4). Springer Science and Business Media LLC: 313–319. doi:10.1038/ni0403-313. PMID 12660731. S2CID 9951653.
- ^ Hong R, Edgar K, Song K, Steven S, Young A, Hamilton P, et al. (15 February 2018). “Abstract PD4-14: GDC-0077 is a selective PI3Kalpha inhibitor that demonstrates robust efficacy in PIK3CA mutant breast cancer models as a single agent and in combination with standard of care therapies”. Cancer Research. 78 (4_Supplement). American Association for Cancer Research (AACR): PD4–14–PD4–14. doi:10.1158/1538-7445.sabcs17-pd4-14. ISSN 0008-5472.
- ^ Jump up to:a b “Inavolisib (PI3K alpha inhibitor)”. Genentech. Retrieved 21 September 2023.
- ^ Menteş M, Karakuzulu BB, Uçar GB, Yandım C (August 2022). “Comparative molecular dynamics analyses on PIK3CA hotspot mutations with PI3Kα specific inhibitors and ATP”. Computational Biology and Chemistry. 99. Elsevier BV: 107726. doi:10.1016/j.compbiolchem.2022.107726. PMID 35842959. S2CID 250404770.
- ^ Song KW, Edgar KA, Hanan EJ, Hafner M, Oeh J, Merchant M, et al. (January 2022). “RTK-Dependent Inducible Degradation of Mutant PI3Kα Drives GDC-0077 (Inavolisib) Efficacy”. Cancer Discovery. 12 (1). American Association for Cancer Research (AACR): 204–219. doi:10.1158/2159-8290.cd-21-0072. PMC 9762331. PMID 34544753.
- ^ “FDA Approves Genentech’s Itovebi, a Targeted Treatment for Advanced Hormone Receptor-Positive, HER2-Negative Breast Cancer With a PIK3CA Mutation” (Press release). Genentech. 10 October 2024. Retrieved 11 October 2024 – via Business Wire.
- ^ “U.S. Food and Drug Administration Approves FoundationOne Liquid CDx as a Companion Diagnostic for Itovebi (inavolisib) to Identify Patients with Hormone Receptor-Positive, HER2-Negative Breast Cancer with a PIK3CA Mutation” (Press release). Foundation Medicine. 11 October 2024. Retrieved 11 October 2024 – via Business Wire.
- ^ World Health Organization (2020). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 84”. WHO Drug Information. 34 (3). hdl:10665/340680.
- ^ World Health Organization (2023). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 90”. WHO Drug Information. 37 (3). hdl:10665/373341.
- ^ Vanhaesebroeck B, Burke JE, Madsen RR (January 2022). “Precision Targeting of Mutant PI3Kα in Cancer by Selective Degradation”. Cancer Discovery. 12 (1). American Association for Cancer Research (AACR): 20–22. doi:10.1158/2159-8290.cd-21-1411. PMC 7612218. PMID 35022207.
External links
- Clinical trial number NCT04191499 for “A Study Evaluating the Efficacy and Safety of Inavolisib + Palbociclib + Fulvestrant vs Placebo + Palbociclib + Fulvestrant in Patients With PIK3CA-Mutant, Hormone Receptor-Positive, Her2-Negative, Locally Advanced or Metastatic Breast Cancer (INAVO120)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Itovebi |
| Other names | GDC-0077, RG6114, Ro7113755 |
| AHFS/Drugs.com | Itovebi |
| License data | US DailyMed: Inavolisib |
| Routes of administration | By mouth |
| Drug class | PI3K inhibitor |
| ATC code | None |
| Legal status | |
| Legal status | CA: ℞-only[1]US: ℞-only[2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2060571-02-8 |
| PubChem CID | 124173720 |
| IUPHAR/BPS | 9636 |
| DrugBank | DB15275 |
| ChemSpider | 59718498 |
| UNII | L4C1UY2NYH |
| KEGG | D11942 |
| ChEMBL | ChEMBL4650215 |
| PDB ligand | X3N (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C18H19F2N5O4 |
| Molar mass | 407.378 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
//////////Inavolisib, FDA 2024, APPROVALS 2024, GDC-0077, 2060571-02-8, GDC0077, RG6114, WHO 11204, GDC 0077, GDC-0077, RG-6114, RG6114, RO-7113755, RO7113755
Vanzacaftor



Vanzacaftor
- CAS 2374124-49-7
- COM1POP492
- VX-121
- 617.8 g/mol, C32H39N7O4S
FDA APPROVED vanzacaftor, tezacaftor, and deutivacaftor, 12/20/2024, Alyftrek , To treat cystic fibrosis
(14S)-8-[3-(2-dispiro[2.0.24.13]heptan-7-ylethoxy)pyrazol-1-yl]-12,12-dimethyl-2,2-dioxo-2λ6-thia-3,9,11,18,23-pentazatetracyclo[17.3.1.111,14.05,10]tetracosa-1(22),5(10),6,8,19(23),20-hexaen-4-one
Vanzacaftor (VX-121) is an orally active noval corrector of Cystic fibrosis transmembrane conductance regulator (CFTR). Vanzacaftor improves processing and trafficking of CFTR protein as well as increases chloride transport in triple combined with Tezacaftor (HY-15448) and Deutivacaftor. Vanzacaftor-Tezacaftor-Deutivacaftor is safe and well tolerated, improving lung function, respiratory symptoms, and CFTR function with cystic fibrosis, which is promising for research in the field of cystic fibrosis diseases.
| Cystic fibrosis (CF) is a recessive genetic disease that affects approximately 70,000 children and adults worldwide. Despite progress in the treatment of CF, there is no cure. |
PATENTS
https://patentscope.wipo.int/search/en/detail.jsf?docId=US356967369&_cid=P12-M9W6P5-06241-1
Example 104: Preparation of (14S)-8-[3-(2-{dispiro[2.0.2.1]heptan-7-yl}ethoxy)-1H-pyrazol-1-yl]-12,12-dimethyl(20-deuterio)-2λ6-thia-3,9,11,18,23-pentaazatetracyclo[17.3.1.111,14.05,10]tetracosa-1(23),5,7,9,19,21-hexaene-2,2,4-trione (Compound 300)

Step 1: (14S)-8-[3-(2-{Dispiro[2.0.2.1]heptan-7-yl}ethoxy)-1H-pyrazol-1-yl]-12,12-dimethyl-2,2,4-trioxo-2λ6-thia-3,9,11,18,23-pentaazatetracyclo[17.3.1.111,14.05,10]tetracosa-1(23),5,7,9,19,21-hexaen-20-yl 4-methylbenzene-1-sulfonate

To a stirred solution of (14S)-8-[3-(2-{dispiro[2.0.2.1]heptan-7-yl}ethoxy)-1H-pyrazol-1-yl]-20-hydroxy-12,12-dimethyl-2λ 6-thia-3,9,11,18,23-pentaazatetracyclo[17.3.1.111,14.05,10]tetracosa-1 (23),5,7,9,19,21-hexaene-2,2,4-trione (150 mg, 0.2367 mmol) in anhydrous dichloromethane (3.000 mL) was added 4-methylbenzenesulfonyl chloride (58 mg, 0.3042 mmol), triethylamine (80 μL, 0.5740 mmol) and catalytic amount of N,N-dimethylpyridin-4-amine (10 mg, 0.08185 mmol). The reaction mixture was stirred at room temperature overnight. The reaction mixture was quenched with saturated aqueous ammonium chloride solution and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated. The resultant brown residue was purified by silica gel column chromatography using a shallow gradient 100% hexanes to 100% ethyl acetate to afford (14S)-8-[3-(2-{dispiro[2.0.2.1]heptan-7-yl}ethoxy)-1H-pyrazol-1-yl]-12,12-dimethyl-2,2,4-trioxo-2λ 6-thia-3,9,11,18,23-pentaazatetracyclo[17.3.1.111,14.05,10]tetracosa-1(23),5,7,9,19,21-hexaen-20-yl 4-methylbenzene-1-sulfonate (120 mg, 51%) as a white solid. ESI-MS m/z calc. 787.28217, found 788.42 (M+1) +; Retention time: 1.39 min (LC Method J).
Step 2: (14S)-8-[3-(2-{Dispiro[2.0.2.1]heptan-7-yl}ethoxy)-1H-pyrazol-1-yl]-12,12-dimethyl(20-deuterio)-2λ6-thia-3,9,11,18,23-pentaazatetracyclo[17.3.1.111,14.05,10]tetracosa-1(23),5,7,9,19,21-hexaene-2,2,4-trione (Compound 300)

A solution of (14S)-8-[3-(2-{dispiro[2.0.2.1]heptan-7-yl}ethoxy)-1H-pyrazol-1-yl]-12,12-dimethyl-2,2,4-trioxo-2λ 6-thia-3,9,11,18,23-pentaazatetracyclo[17.3.1.111,14.05,10]tetracosa-1(23),5,7,9,19,21-hexaen-20-yl 4-methylbenzene-1-sulfonate (120 mg, 0.1523 mmol) in dry N,N-dimethylformamide (1 mL) was purged with nitrogen for 5 min using a balloon. Then, dichloronickel; triphenyl-phosphane (30 mg, 0.04586 mmol) and tricyclohexylphosphane (34 mg, 0.1212 mmol) were added. The resultant green solution was stirred for 5 min under nitrogen atmosphere and tetradeuterioboranuide (sodium salt) (87 mg, 2.079 mmol) was added in one portion. The resultant dark reddish brown mixture was stirred at room temperature for 1 h. Additional dichloronickel; triphenylphosphane (30 mg, 0.04586 mmol), tricyclohexylphosphane (34 mg, 0.1212 mmol) and tetradeuterioboranuide (sodium salt) (87 mg, 2.079 mmol) were added and the mixture was stirred at room temperature under nitrogen overnight. The reaction mixture was diluted with water and extracted with ethyl acetate. The organic layer was dried over magnesium sulfate, filtered and evaporated. The resultant residue was dissolved in dimethyl sulfoxide and filtered through a Whatman filter disc (puradisc 25 TF) and the filtrate was purified by reverse phase HPLC-MS using a dual gradient run from 50%-99% mobile phase B over 15.0 min (mobile phase A=water (5 mM hydrochloric acid), mobile phase B=acetonitrile) to afford (14S)-8-[3-(2-{dispiro[2.0.2.1]heptan-7-yl}ethoxy)-1H-pyrazol-1-yl]-12,12-dimethyl(20-deuterio)-2λ 6-thia-3,9,11,18,23-pentaazatetracyclo[17.3.1.111,14.05,10]tetracosa-1(23),5,7,9,19,21-hexaene-2,2,4-trione (Compound 300) (35 mg, 37%) as a white solid. 1H NMR (400 MHz, dimethyl sulfoxide-d 6) δ 12.52 (s, 1H), 8.20 (d, J=2.8 Hz, 1H), 7.81 (d, J=8.2 Hz, 1H), 7.56 (d, J=7.1 Hz, 1H), 7.04 (d, J=7.2 Hz, 1H), 6.98 (s, 1H), 6.90 (d, J=8.1 Hz, 1H), 6.08 (d, J=2.7 Hz, 1H), 4.25-4.17 (m, 2H), 3.92 (d, J=12.5 Hz, 1H), 3.17 (s, 1H), 2.94 (d, J=13.2 Hz, 1H), 2.72 (s, 1H), 2.20-2.06 (m, 1H), 1.81 (q, J=6.6 Hz, 4H), 1.60 (s, 3H), 1.56 (d, J=13.5 Hz, 2H), 1.51 (s, 3H), 1.46 (d, J=6.5 Hz, 1H), 1.36-1.26 (m, 1H), 1.23 (s, 1H), 0.87-0.76 (m, 4H), 0.70-0.59 (m, 2H), 0.50 (dd, J=8.0, 4.3 Hz, 2H). ESI-MS m/z calc. 618.2847, found 619.25 (M+1) +; Retention time: 1.28 min (LC Method J).
- US11866450, Compound 195
- US11866450, Compound 252
- US11866450, Compound 253
- US11866450, Compound 274
- US11866450, Compound 298
- US11866450, Compound 301
- [1]. Uluer AZ, et al. Safety and efficacy of vanzacaftor-tezacaftor-deutivacaftor in adults with cystic fibrosis: randomised, double-blind, controlled, phase 2 trials[J]. Lancet Respir Med. 2023 Jun;11(6):550-562. [Content Brief][2]. Kolski-Andreaco A, et al. Potentiation of BKCa channels by cystic fibrosis transmembrane conductance regulator (CFTR) correctors VX-445 and VX-121[J]. J Clin Invest. 2024 Jul 2:e176328. [Content Brief]
//////Vanzacaftor, Alyftrek , cystic fibrosis, COM1POP492, VX-121, FDA 2024, APPROVALS 2024
#Vanzacaftor, #Alyftrek , #cystic fibrosis, #COM1POP492, #VX-121, #FDA 2024, #APPROVALS 2024
Probenecid



Probenecid
- 57-66-9
- 4-(Dipropylsulfamoyl)benzoic acid
- Probenecid acid
- Benemid
4-(dipropylsulfamoyl)benzoic acid
C13H19NO4S, 285.359
HC 5006- NSC-18786
FDA APPROVED, 10/25/2024, sulopenem etzadroxil, probenecid, Orlynvah, To treat uncomplicated urinary tract infections (uUTI)
Drug Trial Snapshot
Probenecid, also sold under the brand name Probalan, is a medication that increases uric acid excretion in the urine. It is primarily used in treating gout and hyperuricemia.
Probenecid was developed as an alternative to caronamide[1] to competitively inhibit renal excretion of some drugs, thereby increasing their plasma concentration and prolonging their effects.
Experimental Properties
| Property | Value | Source |
|---|---|---|
| melting point (°C) | 195 °C | PhysProp |
| water solubility | 27.1 mg/L | Not Available |
| logP | 3.21 | HANSCH,C ET AL. (1995) |
| pKa | 3.4 | SANGSTER (1994) |
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US12109197 | No | 2024-10-08 | 2039-04-01 | |
| US11554112 | No | 2023-01-17 | 2039-04-01 | |
| US11478428 | No | 2022-10-25 | 2039-12-23 | |
| US7795243 | No | 2010-09-14 | 2029-06-03 | |

PATENT
https://patents.google.com/patent/CN103613521A/en
At present, the production technique of probenecid mainly contains two kinds:
(1) p-methyl benzenesulfonic acid-dipropyl amine method
Take p-methyl benzenesulfonic acid as raw material, through potassium bichromate or potassium permanganate oxidation, then react generation with chlorsulfonic acid generation sulfonating chlorinating to carboxyl benzene sulfonyl chloride, amidate action occurs then in organic solvent and obtain the finished product probenecid.Reaction process route is as follows:

This technique in a large number with an organic solvent, seriously polluted; Heavy metal recovery and treatment cost are high; Chlorsulfonic acid transportation, storage and use are dangerous large, and acid mist is obvious.Along with the increasing of environmental protection pressure, people increase severely day by day to the concern of environment, and this route is substantially in end-of-life state.
(2) to methyl benzenesulfonamide-Halopropane method
To methyl benzenesulfonamide, through potassium bichromate or potassium permanganate oxidation, be P―Carboxybenzenesulfonamide, under the effect of alkali, with Halopropane generation alkylated reaction, after acidifying, obtain probenecid.Reaction process route is as follows:

This process using sodium dichromate 99 or potassium permanganate oxidation are to methyl benzenesulfonamide, and yield is on the low side (lower than 50%).In addition, the waste water that contains chromium or manganese is difficult to dispose, and these have all seriously restricted further developing of this technique.
Reaction scheme of the present invention is as follows:

embodiment 1
(1) diazotization reaction
Get 68.6g para-amino benzoic acid (0.5mol), 250g water and 127.4ml hydrochloric acid (31%, 1.25mol) join in 2000ml there-necked flask, in ice-water bath, stir, be cooled to 0-5 ℃, drip sodium nitrite solution (34.5g Sodium Nitrite, 0.5mol, be dissolved in 190g water), control temperature at 10-20 ℃, it is 4 hours that time for adding is controlled, after dropping finishes, at this temperature, continue reaction 1 hour, obtain diazotization reaction liquid.
(2) sulfonating chlorinating reaction
In 5000ml there-necked flask, add 250g water, 765ml hydrochloric acid (31%, 7.5mol), in ice-water bath, stir, be cooled to-5 ℃, start to pass into liquid sulfur dioxide, control temperature at-3–1 ℃, when passing into 64g sulfurous gas (1mol), sulfurous gas absorbs complete, obtains sulfonating chlorinating reagent.
In sulfonating chlorinating reagent, add diazotization reaction liquid, adding the time control of diazotization reaction liquid is 5 hours, is warming up to gradually 5-10 ℃, continues reaction 8 hours at this temperature; Filtration obtains 121g to carboxyl benzene sulfonyl chloride.
(3) synthetic probenecid reaction
In 1000ml there-necked flask, add 350g water, 152g dipropyl amine (1.5mol), open and stir, when temperature is greater than 15 ℃, start to divide gradually 40 batches add step (2) gained to carboxyl benzene sulfonyl chloride, temperature control 40-50 ℃, adds and at this temperature, stirs 3 hours continuing after carboxyl benzene sulfonyl chloride.Drip hydrochloric acid (31%), regulate pH value to 2-3, continue to stir 1 hour.Filter, obtain 135g probenecid crude product, put in 500ml pure water, agitator treating 1 hour, heavy 122.8g after filtering, being dried, yield 86.2%(is in para-amino benzoic acid), purity 98.2%.
embodiment 2
(1) diazotization reaction
Get 68.6g para-amino benzoic acid (0.5mol), 250g water and 152.9ml hydrochloric acid (31%, 1.5mol) join in 2000ml there-necked flask, in ice-water bath, stir, be cooled to 0-5 ℃, drip sodium nitrite solution (36.0g Sodium Nitrite, 0.52mol, be dissolved in 190g water), control temperature at 0-10 ℃, it is 3 hours that time for adding is controlled, after dropping finishes, at this temperature, continue reaction 1 hour, obtain diazotization reaction liquid.
(2) sulfonating chlorinating reaction
In 5000ml there-necked flask, add 250g water, 887ml hydrochloric acid (31%, 8.7mol), in ice-water bath, stir, be cooled to-5 ℃, start to pass into liquid sulfur dioxide, control temperature at 0-5 ℃, when passing into 112g sulfurous gas (1.75mol), sulfurous gas absorbs complete, obtains sulfonating chlorinating reagent.
In sulfonating chlorinating reagent, add diazotization reaction liquid, adding the time control of diazotization reaction liquid is 4 hours, is warming up to gradually 5-15 ℃, continues reaction 5 hours at this temperature; Filtration obtains 150g to carboxyl benzene sulfonyl chloride.
(3) synthetic probenecid reaction
In 1000ml there-necked flask, add 350g water, 192g dipropyl amine (1.9mol), open and stir, when temperature is greater than 15 ℃, start to divide gradually 35 batches add step (2) gained to carboxyl benzene sulfonyl chloride, temperature control 40-50 ℃, adds and at this temperature, stirs 2 hours continuing after carboxyl benzene sulfonyl chloride.Drip hydrochloric acid (31%), regulate pH value to 2-3, continue to stir 1 hour.Filter, obtain 155.4g probenecid crude product, put in 500ml pure water, agitator treating 1 hour, heavy 129.5g after filtering, being dried, yield 90.9%(is in para-amino benzoic acid), purity 98.7%.
embodiment 3
(1) diazotization reaction
Get 68.6g para-amino benzoic acid (0.5mol), 250g water and 203.9ml hydrochloric acid (31%, 2mol) join in 2000ml there-necked flask, in ice-water bath, stir, be cooled to-10–5 ℃, drip sodium nitrite solution (38.0g Sodium Nitrite, 0.55mol, be dissolved in 190g water), control temperature at 0-10 ℃, it is 5 hours that time for adding is controlled, after dropping finishes, at this temperature, continue reaction 1 hour, obtain diazotization reaction liquid.
(2) sulfonating chlorinating reaction
In 5000ml there-necked flask, add 250g water, 968ml hydrochloric acid (31%, 9.5mol), in ice-water bath, stir, be cooled to-5 ℃, start to pass into liquid sulfur dioxide, control temperature at 5-10 ℃, when passing into 160g sulfurous gas (2.5mol), sulfurous gas absorbs complete, obtains sulfonating chlorinating reagent.
In sulfonating chlorinating reagent, add diazotization reaction liquid, adding the time control of diazotization reaction liquid is 3 hours, is warming up to gradually 10-15 ℃, continues reaction 20 hours at this temperature; Filtration obtains 146.7g to carboxyl benzene sulfonyl chloride, needn’t be dried, and directly enters next step reaction.
(3) synthetic probenecid reaction
In 1000ml there-necked flask, add 350g water, 202g dipropyl amine (2mol), open to stir, when temperature is greater than 30 ℃, start to divide gradually 30 batches add step (2) gained to carboxyl benzene sulfonyl chloride, temperature control 40-50 ℃, adds and at this temperature, stirs 4 hours continuing after carboxyl benzene sulfonyl chloride.Drip hydrochloric acid (31%), regulate pH value to 2-3, continue to stir 1 hour.Filtration obtains 151.7g probenecid crude product, puts in 500ml pure water, and agitator treating 1 hour, heavy 128.5g after filtering, being dried, yield 90.2%(is in para-amino benzoic acid), purity 98.8%.Medical uses
Probenecid is primarily used to treat gout and hyperuricemia.
Probenecid is sometimes used to increase the concentration of some antibiotics and to protect the kidneys when given with cidofovir. Specifically, a small amount of evidence supports the use of intravenous cefazolin once rather than three times a day when it is combined with probenecid.[2]
It has also found use as a masking agent,[3] potentially helping athletes using performance-enhancing substances to avoid detection by drug tests.
Adverse effects
Mild symptoms such as nausea, loss of appetite, dizziness, vomiting, headache, sore gums, or frequent urination are common with this medication. Life-threatening side effects such as thrombocytopenia, hemolytic anemia, leukemia and encephalopathy are extremely rare.[4] Theoretically probenecid can increase the risk of uric acid kidney stones.
Drug interactions
Some of the important clinical interactions of probenecid include those with captopril, indomethacin, ketoprofen, ketorolac, naproxen, cephalosporins, quinolones, penicillins, methotrexate, zidovudine, ganciclovir, lorazepam, and acyclovir. In all these interactions, the excretion of these drugs is reduced due to probenecid, which in turn can lead to increased concentrations of these.[5]
Pharmacology
Pharmacodynamics
In gout, probenecid competitively inhibits the reabsorption of uric acid through the organic anion transporter (OAT) at the proximal tubules. This leads to preferential reabsorption of probenecid back into plasma and excretion of uric acid in urine,[6] thus reducing blood uric acid levels and reducing its deposition in various tissues.
Probenecid also inhibits pannexin 1.[7] Pannexin 1 is involved in the activation of inflammasomes and subsequent release of interleukin-1β causing inflammation. Inhibition of pannexin 1 thus reduces inflammation, which is the core pathology of gout.[7]
Pharmacokinetics
In the kidneys, probenecid is filtered at the glomerulus, secreted in the proximal tubule and reabsorbed in the distal tubule. Probenicid lowers the concentration of certain drugs in urine drug screens by reducing renal excretion of these drugs.
Historically, probenecid has been used to increase the duration of action of drugs such as penicillin and other beta-lactam antibiotics. Penicillins are excreted in the urine at proximal and distal convoluted tubules through the same organic anion transporter (OAT) as seen in gout. Probenecid competes with penicillin for excretion at the OAT, which in turn increases the plasma concentration of penicillin.[8]
History
During World War II, probenecid was used to extend limited supplies of penicillin. This use exploited probenecid’s interference with drug elimination (via urinary excretion) in the kidneys and allowed lower doses of penicillin to be used.[9]
Probenecid was added to the International Olympic Committee‘s list of banned substances in January 1988, due to its use as a masking agent.[10]
References
- ^ Mason RM (June 1954). “Studies on the effect of probenecid (benemid) in gout”. Annals of the Rheumatic Diseases. 13 (2): 120–130. doi:10.1136/ard.13.2.120. PMC 1030399. PMID 13171805.
- ^ Cox VC, Zed PJ (March 2004). “Once-daily cefazolin and probenecid for skin and soft tissue infections”. The Annals of Pharmacotherapy. 38 (3): 458–463. doi:10.1345/aph.1d251. PMID 14970368. S2CID 11449580.
- ^ Morra V, Davit P, Capra P, Vincenti M, Di Stilo A, Botrè F (December 2006). “Fast gas chromatographic/mass spectrometric determination of diuretics and masking agents in human urine: Development and validation of a productive screening protocol for antidoping analysis”. Journal of Chromatography A. 1135 (2): 219–229. doi:10.1016/j.chroma.2006.09.034. hdl:2318/40201. PMID 17027009. S2CID 20282106.
- ^ Kydd AS, Seth R, Buchbinder R, Edwards CJ, Bombardier C (November 2014). “Uricosuric medications for chronic gout”. The Cochrane Database of Systematic Reviews (11): CD010457. doi:10.1002/14651858.CD010457.pub2. PMC 11262558. PMID 25392987.
- ^ Cunningham RF, Israili ZH, Dayton PG (March–April 1981). “Clinical pharmacokinetics of probenecid”. Clinical Pharmacokinetics. 6 (2): 135–151. doi:10.2165/00003088-198106020-00004. PMID 7011657. S2CID 24497865.
- ^ “Probenecid”. PubChem. U.S. National Library of Medicine. Retrieved 2022-06-12.
- ^ Jump up to:a b Silverman W, Locovei S, Dahl G (September 2008). “Probenecid, a gout remedy, inhibits pannexin 1 channels”. American Journal of Physiology. Cell Physiology. 295 (3): C761 – C767. doi:10.1152/ajpcell.00227.2008. PMC 2544448. PMID 18596212.
- ^ Ho RH (January 2010). “4.25 – Uptake Transporters”. In McQueen CA, Kim RB (eds.). Comprehensive Toxicology (Second ed.). Oxford: Elsevier. pp. 519–556. doi:10.1016/B978-0-08-046884-6.00425-5. ISBN 978-0-08-046884-6.
- ^ Butler D (November 2005). “Wartime tactic doubles power of scarce bird-flu drug”. Nature. 438 (7064): 6. Bibcode:2005Natur.438….6B. doi:10.1038/438006a. PMID 16267514.
- ^ Wilson W, Derse E, eds. (2001). Doping in Elite Sport: The Politics of Drugs in the Olympic Movement. Human Kinetics. p. 86. ISBN 0-7360-0329-0.
| Clinical data | |
|---|---|
| Trade names | Probalan |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a682395 |
| Routes of administration | By mouth |
| ATC code | M04AB01 (WHO) |
| Legal status | |
| Legal status | In general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Protein binding | 75-95% |
| Elimination half-life | 2-6 hours (dose: 0.5-1 g) |
| Excretion | kidney (77-88%) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 57-66-9 |
| PubChem CID | 4911 |
| IUPHAR/BPS | 4357 |
| DrugBank | DB01032 |
| ChemSpider | 4742 |
| UNII | PO572Z7917 |
| KEGG | D00475 |
| ChEMBL | ChEMBL897 |
| CompTox Dashboard (EPA) | DTXSID9021188 |
| ECHA InfoCard | 100.000.313 |
| Chemical and physical data | |
| Formula | C13H19NO4S |
| Molar mass | 285.36 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
/////////probenecid, APPROVALS 2024, FDA 2024, Orlynvah, HC 5006, NSC-18786
#probenecid, #APPROVALS 2024, #FDA 2024, #Orlynvah, #HC 5006, #NSC-18786
Sulopenem



Sulopenem
- 120788-07-0
- CP-70429
- 349.5 g/mol, C12H15NO5S3
- XX514BJ1XW
- PF-03709270
- PF03709270
(5R,6S)-6-[(1R)-1-hydroxyethyl]-7-oxo-3-[(1R,3S)-1-oxothiolan-3-yl]sulfanyl-4-thia-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid
- (5R,6S)-6-((1R)-1-HYDROXYETHYL)-7-OXO-3-(((1R,3S)-1-OXOTETRAHYDRO-1H-1.LAMBA.(SUP 4)-THIOPHEN-3-YL)SULFANYL)-4-THIA-1-AZABICYCLO(3.2.0)HEPT-2-ENE-2-CARBOXYLIC ACID
- (5R,6S)-6-((1R)-1-Hydroxyethyl)-7-oxo-3-(((3S)-tetrahydro-3-thienyl)thio)-4-thia-1-azabicyclo(3.2.0)hept-2-ene-2-carboxylic acid, (R)-S-oxide
- 4-THIA-1-AZABICYCLO(3.2.0)HEPT-2-ENE-2-CARBOXYLIC ACID, 6-((1R)-1-HYDROXYETHYL)-7-OXO-3-(((1R,3S)-TETRAHYDRO-1-OXIDO-3-THIENYL)THIO)-, (5R,6S)-
- 4-THIA-1-AZABICYCLO(3.2.0)HEPT-2-ENE-2-CARBOXYLIC ACID, 6-(1-HYDROXYETHYL)-7-OXO-3-((TETRAHYDRO-3-THIENYL)THIO)-, S-OXIDE, (5R-(3(1R*,3S*),5.ALPHA.,6.ALPHA.(R*)))-
FDA APPROVED sulopenem etzadroxil, probenecid, 10/25/2024, To treat uncomplicated urinary tract infections (uUTI)
Drug Trial Snapshot
Sulopenem (CP-70,429) is a thiopenem antibiotic derivative from the penem family, which unlike most related drugs is orally active. It was developed in Japan in the 1990s, and has been approved to treat uncomplicated urinary tract infections in combination with probenecid (Brand name: Orlynvah). It has reached Phase III clinical trials on several occasions and continues to be the subject of ongoing research into potential applications, especially in the treatment of multiple drug resistant urinary tract infections.[1][2][3][4][5]
In October 2024, the US Food and Drug Administration approved sulopenem etzadroxil with probenecid combination for the treatment of urinary tract infections caused by Escherichia coli, Klebsiella pneumoniae, or Proteus mirabilis in adult women with limited alternative oral antibiotic options. The combination was developed by Iterum Therapeutics under the trade name ORLYNVAH™.[6]


JP 1995278137; US 5013729; WO 8808845, J Org Chem 1992,57(16),4352-61
1) The reaction of L-aspartic acid (I) with NaNO2, NaBr and H2SO4 gives 2(S)-bromosuccinic acid (II), which is reduced with methyl sulfide borane complex in THF, yielding 2(S)-bromobutane-1,4-diol (III). The cyclization of (III) with Cs2CO3 in methylene chloride affords (R)-(2-hydroxyethyl)oxirane (IV), which is acylated with methanesulfonyl chloride to the corresponding mesylate (V). The cyclization of (V) with Na2S in acetonitrile/water gives 3(R)-hydroxythiolane (VI), which is acylated with p-toluenesulfonyl chloride, affording the corresponding tosylate (VII). The controlled oxidation of (VII) with potassium peroxymonosulfate (oxone) gives 3(R)-(p-toluenesulfonyloxy)thiolane-1(R)-oxide (VIII), which by reaction with potassium thioacetate in acetone is converted to 3(S)-(acetylthio)thiolane 1(R)-oxide (IX). The reaction of (IX) with NaOEt and CS2 in ethanol yields the trithiocarbonate (X), which is condensed with the chloroazetidinone (XI), yielding the trithiocarbonate ester (XII). The condensation of (XII) with 2-chloroallyloxalyl fluoride (XIII) by means of diisopropylethylamine in methylene chloride affords the substituted oxalamic ester (XIV), which is cyclized by means of triethyl phosphite in refluxing chloroform to the fully protected penem derivative (XV). The reaction of (XV) with tetrabutylammonium fluoride (TBAF) in THF eliminates the protecting tert-butyldimethylsilyl group, yielding the chloroallyl ester (XVI), which is treated with triphenylphosphine and sodium 2-ethylhexanoate in dichloromethane to obtain the corresponding sodium salt (XVII). Finally, this compound is treated with HCl in cool water.

US 4921972
2) The intermediate 3(R)-(p-toluenesulfonyloxy)thiolane (VII) can be obtained by two other synthetic pathways: a) The racemic 2-hydroxy-4-(methylsulfanyl)butyric acid ethyl ester (XVIII) is submitted to optical resolution with Pseudomonas fluorescens lipase in toluene/water, yielding the corresponding 2(R)-hydroxy ester (XIX), which is reduced with NaBH4 in THF/water to afford 4-(methylsulfanyl)butane-1,2(R)-diol (XX). The acylation of (XX) with p-toluenesulfonyl chloride and pyridine yields the ditosylate (XXI), which is cyclized in refluxing benzene to give 1(R)-methyl-3(R)-(p-toluenesulfonyloxy)thiolanium p-toluenesulfonate (XXII). Finally, this compound is treated with trifluoroacetic acid in pyridine to afford the thiolane (VII), already described. b) The reduction of 4-chloro-3(R)-hydroxybutyric acid methyl ester (XXIII) with lithium borohydride in THF gives 4-chlorobutane-1,3(R)-diol (XXIV), which is tosylated as before, yielding the bis(tosyloxy) derivative (XXV). Finally, this compound is cyclized with Na2S in hot acetonitrile/water to afford the thiolane (VII), already described.

https://pubsapp.acs.org/cen/coverstory/88/8836cover.html
References
- ^ Minamimura M, Taniyama Y, Inoue E, Mitsuhashi S (July 1993). “In vitro antibacterial activity and beta-lactamase stability of CP-70,429 a new penem antibiotic”. Antimicrobial Agents and Chemotherapy. 37 (7): 1547–1551. doi:10.1128/AAC.37.7.1547. PMC 188011. PMID 8363389.
- ^ Hamilton-Miller JM (November 2003). “Chemical and microbiologic aspects of penems, a distinct class of beta-lactams: focus on faropenem”. Pharmacotherapy. 23 (11): 1497–1507. doi:10.1592/phco.23.14.1497.31937. PMID 14620395. S2CID 43705118.
- ^ Ednie LM, Appelbaum PC (May 2009). “Antianaerobic activity of sulopenem compared to six other agents”. Antimicrobial Agents and Chemotherapy. 53 (5): 2163–2170. doi:10.1128/AAC.01557-08. PMC 2681565. PMID 19223615.
- ^ Bader MS, Loeb M, Leto D, Brooks AA (April 2020). “Treatment of urinary tract infections in the era of antimicrobial resistance and new antimicrobial agents”. Postgraduate Medicine. 132 (3): 234–250. doi:10.1080/00325481.2019.1680052. PMID 31608743. S2CID 204545734.
- ^ Veeraraghavan B, Bakthavatchalam YD, Sahni RD (December 2021). “Oral Antibiotics in Clinical Development for Community-Acquired Urinary Tract Infections”. Infectious Diseases and Therapy. 10 (4): 1815–1835. doi:10.1007/s40121-021-00509-4. PMC 8572892. PMID 34357517.
- ^ “Iterum Therapeutics Receives U.S. FDA Approval of ORLYNVAH™ (Oral Sulopenem) for the Treatment of Uncomplicated Urinary Tract Infections”. Iterum Therapeutics plc. 2024-10-25. Retrieved 2024-10-25.
| Clinical data | |
|---|---|
| ATC code | None |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 120788-07-0 |
| PubChem CID | 9950244 |
| DrugBank | DB15284 |
| ChemSpider | 8125855 |
| UNII | XX514BJ1XW |
| KEGG | D05969 |
| CompTox Dashboard (EPA) | DTXSID20869656 |
| Chemical and physical data | |
| Formula | C12H15NO5S3 |
| Molar mass | 349.43 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
FDA Approved Drug Products: Orlynvah (sulopenem etzadroxil and probenecid) tablets for oral use (October 2024) [Link]
FDA News Release: FDA approves new treatment for uncomplicated urinary tract infections in adult women who have limited or no alternative oral antibiotic treatment options [Link]
//////Sulopenem, Orlynvah, FDA 2024, APPROVALS 2024, CP-70,429, 120788-07-0, CP-70429, XX514BJ1XW, PF-03709270, PF03709270
#Sulopenem, #Orlynvah, #FDA 2024, #APPROVALS 2024, #CP-70,429, #120788-07-0, #CP-70429, #XX514BJ1XW, #PF-03709270, #PF03709270
Landiolol


Landiolol
- 133242-30-5
- ONO-1101
- Ono 1101
- WHO 7516
FDA APPROVED 11/22/2024, Rapiblyk, To treat supraventricular tachycardia
C25H39N3O8
509.6 g/mol
[(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]methyl 3-[4-[(2S)-2-hydroxy-3-[2-(morpholine-4-carbonylamino)ethylamino]propoxy]phenyl]propanoate
- [(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]methyl 3-[4-[(2S)-2-hydroxy-3-[2-(morpholine-4-carbonylamino)ethylamino]propoxy]phenyl]propanoate
- UNII-62NWQ924LH
- (S-(R*,R*))-(2,2-Dimethyl-1,3-dioxolan-4-yl)methyl 4-(2-hydroxy-3-((2-((4-morpholinylcarbonyl)amino)ethyl)amino)propoxy)benzenepropanoate
- Benzenepropanoic acid, 4-((2S)-2-hydroxy-3-((2-((4-morpholinylcarbonyl)amino)ethyl)amino)propoxy)-, ((4S)-2,2-dimethyl-1,3-dioxolan-4-yl)methyl ester
- (-)-((S)-2,2-Dimethyl-1,3-dioxolan-4-yl)methyl p-((S)-2-hydroxy-3-((2-(4-morpholinecarboxamido)ethyl)amino)propoxy)hydrocinnamate
- Benzenepropanoic acid, 4-(2-hydroxy-3-((2-((4-morpholinylcarbonyl)amino)ethyl)amino)propoxy)-, (2,2-dimethyl-1,3-dioxolan-4-yl)methyl ester, (S-(R*,R*))-

Landiolol hydrochloride- 144481-98-1
- Landiolol HCl
- ONO 1101 hydrochloride
- Onoact
Landiolol, sold under the brand name Onoact among others, is a medication used for the treatment of tachycardia, atrial fibrillation, and atrial flutter.[1][4] It is a beta-adrenergic blocker;[4] an ultra short-acting, β1-superselective intravenous adrenergic antagonist, which decreases the heart rate effectively with less negative effect on blood pressure or myocardial contractility.[6][7] In comparison to other beta blockers, landiolol has the shortest elimination half-life (3 to 4 minutes), ultra-rapid onset of effect (heart rate begins to decrease immediately after completion of administration), and predictable effectiveness with inactive metabolites (heart rate returns to baseline levels at 30 min after completion of landiolol hydrochloride administration).[8] The pure S-enantiomer structure of landiolol is believed to develop less hypotensive side effects in comparison to other β-blockers. This has a positive impact on the treatment of patients when reduction of heart rate without decrease in arterial blood pressure is desired.[9] It is used as landiolol hydrochloride.
Landiolol was approved for medical use in Japan in 2002,[10][11] in Canada in November 2023,[1] and in the United States in November 2024.[12][13][14]
Syn
- Landiolol 1 is a potent cardioselective beta-blocker with ultrarapid action, used as an arrhythmic agent in the form of the hydrochloride salt.
- [0004]The synthesis of Landiolol 1 is disclosed in US 5013734 , JP 3302647 , CN 100506814 , JP 2539734 and Chemical & Pharmaceutical Bulletin 1992, 40 (6) 1462-1469. The main synthetic route for the preparation of Landiolol is reported in the following scheme:

The synthesis of landiolol appeared in an earlier patent in 1990. Esterification of 3- (4-hydroxyphenyl)propionic acid (141) with 2,2-dimethyl- 1,3-dioxolan-4-ylmethyl chloride (142) in DMSO gave desired ester 143 in 57% yield. Treatment of phenol 143 with bromo epoxide 144 in the present of K2CO3 afforded ether 145 in 76% yield. Epoxide 145 was then reacted with free amine 146 via a neucleophilic ring opening process to provide landiolol (14).

Yield:144481-98-1 95.9%
Reaction Conditions:
with hydrogenchloride in ethyl acetate at 5 – 10; for 2 h;
Steps:
1.6 Preparation of Lantilolol Hydrochloride
Add Lantilolol (10g, 19.62mmol) and 100mL of ethyl acetate to the reaction flask. The temperature of the ice-water bath is lowered below 5 ° C, and a temperature of 10-18 ° C is added dropwise to a 15-18% HCl-ethyl acetate solution 4.63g A large amount of solid was gradually precipitated, dripped, stirred below 10 ° C for 2h, filtered, washed with ethyl acetate, and dried under vacuum at 50 ° C to obtain 10.28 g of a white solid with a yield of 95.9% and an HPLC purity of 99.85%.
References:
CN110483470,2019,A Location in patent:Paragraph 0031; 0045-0047
EP2687521,2014,A1
https://patents.google.com/patent/EP2687521B1/en

- [0025]Typically, to activate the salen catalyst, preferably (R,R)-N,N’-bis(3,5-di-tert-butylsalicylidene)-1,2-ciclohexanediamino cobalt 16 is reacted with 1.0 ÷ 3.0 equivalents of a carboxylic acid, preferably 4-nitrobenzoic acid 17, preferably 1.5 ÷ 2.5 equivalents. The reaction is carried out in a polar aprotic solvent, preferably dichloromethane, at a temperature of 10 ÷ 40°C, preferably at a temperature of 20 ÷ 30°C. 4 ÷ 15 Volumes of solvent are used, preferably 7 ÷ 12 volumes with respect to the amount of (R,R)-N,N’-bis(3,5-di-tert-butylsalicylidene)-1,2-ciclohexanediamino cobalt 16. After a dark brown color appears, the solvent is removed thereby obtaining the catalyst the in active form. This is then added with 10 ÷ 100 equivalents of starting product (S)-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl 3-(4-hydroxyphenyl)propanoate 3, preferably 20 ÷ 50 equivalents, then with a polar aprotic solvent, preferably methyl tert-butyl ether (MTBE). 1 ÷ 5 Volumes of solvent are used, preferably 2 ÷ 3 volumes with respect to the amount of (S)-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl 3-(4-hydroxyphenyl)propanoate 3. Afterwards, 2.0 ÷ 3.0 equivalents of a compound of formula 4, typically epichlorohydrin, are added, preferably 2.0 ÷ 2,5 equivalents. The reaction is carried out at a temperature of 10 ÷ 40°C, preferably at a temperature of 20 ÷ 30°C. The reaction is monitored by UPLC analysis using a C18 column and water / acetonitrile containing 1% formic acid as the eluent phase. After completion of the reaction, water and toluene are added and phases are separated. The organic phase is then distilled to recover (S)-epichlorohydrin and washed with dilute sodium hydroxide. The organic phase is then concentrated to small volume, added with a polar solvent, acetonitrile or methanol, preferably acetonitrile, concentrated again to small volume to remove toluene and finally added with 5 ÷ 30 volumes of a polar solvent, such as acetonitrile or methanol, preferably acetonitrile. The suspension is filtered thus recovering the catalyst and the resulting solution can be directly used in step b, or (S)-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl 3-(4-((2R)-3-chloro-2-hydroxypropoxy)phenyl)-propanoate 5 can be isolated as an oil that can be stored at room temperature for some days. In order to obtain (S)-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl 3-(4-((2R)-3-chloro-2-hydroxypropoxy)phenyl)propanoate 5 as an oil, the solution in the polar solvent is added with decolorizing filter aid, the obtained suspension is filtered and the resulting solution is evaporated to dryness. (S)-(2,2-Dimethyl-1,3-dioxolan-4-yl)methyl 3-(4-((2R)-3-chloro-2-hydroxy-propoxy)phenyl)propanoate 5 is obtained as an oil.
- [0026]Typically, (S)-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl 3-(4-((2R)-3-chloro-2-hydroxypropoxy)phenyl)propanoate 5 obtained in step a, either isolated as an oil or directly from the polar solvent solution, is reacted with an inorganic base, preferably potassium carbonate, in an amount of 1.0 ÷ 6.0 equivalents, 3.0 ÷ 4.0 equivalents, in the presence of a ionic inorganic catalyst, preferably potassium iodide, in catalytic amounts (0.05 ÷ 0.20 eq). Thereafter, 2-(morpholine-4-carboxamido)ethanamine as base or a salt thereof, such as the oxalate or the hydrochloride, preferably the oxalate, is added in an amount of 1.0 ÷ 4.0 equivalents, preferably 2.0 ÷ 3.0 equivalents. The reaction is carried out in a polar solvent, preferably acetonitrile, at a temperature 20 ÷ 85°C, preferably 60 ÷ 85°C. 5 ÷ 30 Volumes of solvent are used, preferably 10 ÷ 20 volumes with respect to the amount of (S)-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl 3-(4-((2R)-3-chloro-2-hydroxypropoxy)-phenyl)propanoate 5. The reaction is monitored by UPLC analysis using a C 18 column and water / acetonitrile containing 1% formic acid as the eluent. After completion of the reaction, ethyl acetate and water are added and the phases separated. The organic phase is then extracted with water at pH 2 ÷ 5, preferably 3 ÷ 4. The phases are separated and the aqueous phase is extracted again with ethyl acetate at pH 8 ÷ 13, preferably 9 ÷ 12. The solvent of the organic phase is then replaced with 2 ÷ 20 volumes of a polar solvent, such as isopropanol, and the resulting solution can be directly used in step c, or Landiolol 1 can be isolated. For this purpose, the solvent is removed or replaced with a polar solvent, for example diisopropyl ether, to promote solidification, then the solvent is stripped from the resulting suspension thereby obtaining Landiolol 1 as an oil which solidifies in time.
- [0027]Typically, Landiolol 1 obtained in step b directly from the polar solvent solution or by dissolution of the isolated product is directly salified to give Landiolol hydrochloride 2, preferably with hydrochloric acid. Salification is carried out in a polar solvent, preferably isopropanol, in amounts of 2 ÷ 20 volumes of solvent, preferably 5 ÷ 10 volumes with respect to the amount of Landiolol 1. After addition of the acid, the solvent is evaporated off and the product is crystallized by adding 1 ÷ 20 volumes of a polar solvent, preferably acetone. The suspension is filtered and the solid is dried at 25 ÷ 35°C under vacuum for 12 hours to obtain Landiolol hydrochloride 2. The enantiomeric excess of the final product is analyzed using a Chiralcel OD column and hexane / ethanol as the eluent phase containing diethylamine.
- [0028]The process of the invention is particularly advantageous in that is effected without isolating any intermediates. Intermediate 5 is obtained with high purity in very high yields under very mild reactions conditions. Furthermore, the starting material 4 in which X is chlorine (epichlorohydrin), is very inexpensive and easily commercially available. The catalysts used are commercially available at low costs and can be easily recovered by simple filtration. Surprisingly, the reaction to give Landiolol 1 starting from the novel intermediate 5 in which X is chlorine provides a markedly higher yield than those obtained with most processes mentioned in the background of the invention, which conversely start from intermediate 7. The resulting Landiolol 1 can be directly converted to Landiolol hydrochloride 2 in good overall yields, with no further purifications neither intermediate steps. The resulting Landiolol hydrochloride 2 has very high enantiomeric purity.
- [0029]Furthermore, the process of the invention allows to recover (S)-epichlorohydrin 12, which is a high added value product that can also be used in the synthesis of Landiolol 1 according to the following scheme, to prepare compound 3:
- [0030]The synthesis of intermediate 15 from 12 in very high yields is described in literature in a number of publications. Some publications which the disclose it are the following: Catalysis Communications, 8(12), 2087-2095; 2007; CN100506814 ; Journal of Molecular Catalysis A: Chemical, 236(1-2), 72-76; 2005; Chinese Journal of Chemistry, 23(9), 1275-1277; 2005; Synthetic Communications, 35(11), 1441-1445; 2005; Synthetic Communications, 31(22), 3411-3416; 2001; Chemistry Letters, (11), 2019-22; 1990; Khimiya Geterotsiklicheskikh Soedinenii, (1), 33-6; 1991. The synthesis of 3 in high yields starting from 15 and 19 is described in CN100506814 . A further publication disclosing it is US5013734 . Both publications have already been mentioned in the background of the invention for the synthesis of Landiolol 1.
- [0031]The invention is illustrated in detail by the following examples.
- [0032]
- [0033]A suspension of (R,R)-N,N’-bis(3,5-di-tert-butylsalicylidene)-1,2-ciclohexanediamino cobalt (16) (50 mg, 0.0828 mmol) in MTBE (1 ml) is added with acetic acid (10 mg, 0.166 mmol). The mixture is left under stirring for 1 h at 20-25°C until a dark color appears. Afterwards, (S)-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl 3-(4-hydroxyphenyl)propanoate (3), (500 mg, 1.78 mmol), then epichlorohydrin (compound of formula 4 in which X is chlorine) (340 mg, 3.56 mmol) are added thereto. The mixture is left under stirring at 20-25°C, monitoring by UPLC. After completion of the reaction, water (5 ml) and toluene (5 ml) are added, the phases are separated and the solvent and (S)-epichlorohydrin are removed from the organic phase under reduced pressure to obtain 600 mg (90.4%) of a dark oil.
- [0034]
- [0035]A suspension of (R,R)-N,N’-bis(3,5-di-tert-butylsalicylidene)-1,2-ciclohexanediamino cobalt (16) (1,9 g, 3.19 mmol) in dichloromethane (20 ml) is added with 4-nitrobenzoic acid 17 (1.1 g, 6.38 mmol). The mixture is left under stirring for 1 h at 20-25°C until a dark color appears. The solvent is replaced with MTBE (30 ml), subsequently (S)-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl 3-(4-hydroxyphenyl)propanoate (3), (18 g, 63,8 mmol) and then epichlorohydrin (compound of formula 4 in which X is chlorine) (13,4 g, 140 mmol) are added. The mixture is left under stirring at 20-25°C, monitoring by UPLC. After completion of the reaction, toluene (300 ml) and water (150 ml) are added and the phases are separated. The organic phase is evaporated to dryness thereby recovering the enriched (S)-epichlorohydrin. Toluene (300 ml) and 10% NaOH (100 ml) are added. The phases are separated, the resulting solution is concentrated to a volume of about 50 ml, added with 100 ml of acetonitrile, concentrated to a volume of 50 ml and finally added with 250 ml of acetonitrile. Decolorizing filter aid (2.5 g) is added, the mixture is left under stirring for 15′ and the suspension is filtered. The filtrate is evaporated to dryness to obtain 23.7 g (99,6%) of a red-brownish oil.
- [0036]A suspension of (R,R)-N,N’-bis(3,5-di-tert-butylsalicylidene)-1,2-ciclohexanediamino cobalt (16) (470 mg, 0.780 mmol) in dichloromethane (5 ml) is added with 4-nitrobenzoic acid 17 (270 mg, 1.56 mmol). The mixture is left under stirring for 45′ at 20-25°C until a dark color appears. The resulting solution is concentrated to a volume of about 2 ml, added with 5 ml of MTBE, concentrated to a volume of 2 ml and finally added with 6 ml of MTBE, subsequently with (S)-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl 3-(4-hydroxyphenyl)propanoate (3), (5 g, 17.7 mmol) and then with epichlorohydrin (compound of formula 4 in which X is chlorine) (3.7 g, 38.9 mmol). The mixture is left under stirring at 20-25°C, monitoring by UPLC. After completion of the reaction, toluene (25 ml) and water (25 ml) are added and the phases are separated. The organic phase is evaporated to dryness thereby recovering the enriched (S)-epichlorohydrin. Acetonitrile (25 ml) is added and the suspension is filtered thereby recovering the catalyst. The resulting solution is concentrated to a volume of about 5 ml, added with 15 ml of toluene, then concentrated to a volume of 5 ml and finally added with 20 ml of toluene and decolorizing filter aid (20.0 g). The mixture is left under stirring for 15′ and the suspension is filtered. The filtrate is evaporated to dryness to obtain 6.1 g (92.4%) of a yellow oil.
LC-MS (ESI+) [M+H]+ = 373
1H-NMR (CDCl3) (chemical shifts expressed in ppm with respect to TMS): 1,37 (3H, s, CH3); 1,43 (3H, s, CH3); 2,65 (2H, t, J = 7 Hz, CH2-Ar); 2,83 (1H, bs, OH); 2,91 (2H, t, J = 7 Hz, CH2-CO); 3,66 – 3,81 (3H, m, CH in 4 oxolane and CH2-Cl); 4.00 – 4,25 (6H, m, CH in 4 oxolane, CH2-OCO, CH2-OAr and CH in 5 oxolane); 4,25 (1H, m, CH-OH); 6,84 and 7,13 (4H, system AA’XX’, aromatics).
13C-NMR (CDCl3) (ppm): 25,3 (CH3); 26,6 (CH3); 29,9 (CH2); 35,8 (CH2); 45,9 (CH2-Cl); 64,6 (CH2); 66,2 (CH2); 68,5 (CH2); 69,7 (CH); 73,4 (CH); 109,7; 114,5 (CH); 129,3 (CH); 133,1; 156,7; 172,6 (COOR).
Elemental analysis: C, 58.3%; H, 6.9%; Cl, 9.3%; O, 25.5%. (% calculated: C, 58.0; H, 6.8; Cl, 9.5; O, 25.7).
FT-IR (UATR, cm-1): 3456, 2987, 2936, 1733, 1612, 1512, 1372, 1241, 1154, 1041,828,741,720. - [0037]
- [0038]A suspension of (S)-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl 3-(4-((2R)-3-chloro-2-hydroxypropoxy)phenyl)propanoate (5) prepared according Example 3 (0.50 g, 0.00134 mol) in isopropanol (10 ml) is added with 2-(morpholine-4-carboxamido)ethanamino hydrochloride (18) (1.4 g, 0.00670 mol), heated to 30-35°C and dropwise added with 30% NaOH, keeping pH at 10-11. The mixture is left under stirring at 35-40°C, monitoring by UPLC. After completion of the reaction, ethyl acetate (20 ml) and water (20 ml) are added and the phases are separated. The organic phase is added with water (20 ml) and adjusted to pH 3-4 with hydrochloric acid. The phases are separated and the resulting aqueous phase is then adjusted to pH 10-11 with sodium hydroxide and re-extracted with ethyl acetate (20 ml). The solvent is then evaporated off under reduced pressure to obtain 0.38 g (55.6%) of a pale yellow oil which solidifies in time to a pale yellow solid.
- [0039]
- [0040]A solution of (S)-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl 3-(4-((2R)-3-chloro-2-hydroxypropoxy)phenyl)propanoate (5) prepared according to Example 3 (0.30 g, 0.805 mmol) in acetonitrile (6.0 ml) is added with potassium carbonate 0.45 g (3.22 mmol), and KI 0.013 g (0.0805 mmol), then refluxed for 2 h and added with 2-(morpholine-4-carboxamido)ethanamino oxalate (6) (0.64 g, 2.42 mmol). The mixture is refluxed under stirring, monitoring by UPLC. After completion of the reaction, ethyl acetate (10 ml) and water (10 ml) are added and the phases are separated. The organic phase is added with water (10 ml) and adjusted to pH 4-5 with hydrochloric acid, the phases are separated and the resulting aqueous phase is then adjusted to pH 11-12 with sodium hydroxide and re-extracted with ethyl acetate (10 ml). Then the solvent is evaporated off under reduced pressure to obtain 0.29 g (70.7%) of a pale yellow oil which solidifies in time to a pale yellow solid.
LC-MS (ESI+) [M+H]+ = 510
1H-NMR (CDCl3) (chemical shifts expressed in ppm with respect to TMS) (assigned based on the hetero correlation HSQC spectrum): 1.36 (3H, s, CH3); 1.42 (3H, s, CH3); 2.63 (2H, t, J = 7 Hz, CH2-Ar); 2.75 – 2.93 (8H, m, CH2-CO, CH-CH 2 -NH, CH2-CH 2 -NH, NH and OH); 3.35 (6H, m, 2CH2-N morpholine and CH 2 -NH); 3.65 (4H, m, 2CH2-O morpholine), 3.68 (1H, m, CH in 4 oxolane); 3.94 (2H, bd, CH2-OAr); 4.00 – 4.20 (4H, m, CH in 4 oxolane, CH2-OCO and CH in 5 oxolane); 4.25 (1H, m, CH-OH); 5.21 (1H, bt, NH carbamate); 6.83 and 7.11 (4H, system AA’XX’, aromatics).
13C-NMR (CDCl3) (ppm) (multiplicity was assigned by DEPT-135): 25.3 (CH3); 26.6 (CH3); 29.9 (CH2); 35.8 (CH2); 40.2 (CH2); 43.8 (CH2-N morpholine); 49.2 (CH2); 51.5 (CH2); 64.6 (CH2); 66.2 (CH2); 66.4 (CH2-O morpholine); 68.3 (CH); 70.3 (CH2); 73.4 (CH); 109.7; 114.4 (CH); 129.2 (CH); 132.8; 157.0; 158.0; 172.5 (COOR).
FT-IR (UATR, cm-1): 3350. 2858, 1735, 1626, 1512, 1454, 1371, 1244, 1153, 1115, 1040. 829, 733. - [0041]
- [0042]A solution of Landiolol (1) prepared according to Example 5 (100 mg, 0.196 mmol) in isopropanol (6.0 ml) is added with 18% isopropanol hydrochloric acid (40 mg, 0.197 mmol). The solvent is then evaporated off under reduced pressure and the residue is crystallized from acetone (2 ml). The suspension is filtered and the crystal is dried at 25°C for 12 h to obtain 80 mg (74.7%) of a white solid.
- [0043]
- [0044]A suspension of (R,R)-N,N’-bis(3,5-di-tert-butylsalicylidene)-1,2-ciclohexanediamino cobalt (16) (47 mg, 0.0780 mmol) in dichloromethane (1 ml) is added with 4-nitrobenzoic acid 17 (27 mg, 0.156 mmol). The mixture is left under stirring for 45′ at 20-25°C until a dark color appears. The resulting solution is concentrated to a volume of about 0.5 ml, added with 0.5 ml of MTBE, concentrated to a volume of 0.5 ml and finally added with 0.5 ml of MTBE, then with (S)-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl 3-(4-hydroxyphenyl)propanoate (3), (0.5 g, 1,77 mmol) and then with epichlorohydrin (compound of formula 4 in which X is chlorine) (0.37 g, 3,89 mmol). The mixture is left under stirring at 20-25°C, monitoring by UPLC. After completion of the reaction, toluene (10 ml) and water (10 ml) are added and the phases are separated. The organic phase is evaporated recovering the enriched (S)-epichlorohydrin, then added again with toluene (10 ml) and washed with 10% NaOH (10 ml). The resulting solution is concentrated to a volume of about 2 ml, added with 5 ml of acetonitrile, concentrated to a volume of 2 ml and finally added with 10 ml of acetonitrile). The suspension is filtered thus recovering the catalyst and the solution is added with potassium carbonate 0.79 g (5,64 mmol), and KI 0.026 g (0.161 mmol), refluxed for 2 h, then added with 2-(morpholine-4-carboxamido)ethanamino oxalate (6) (1.07 g, 4.03 mmol). The mixture is refluxed under stirring, monitoring by UPLC. After completion of the reaction, ethyl acetate (20.0 ml) and water (20 ml) are added and the phases are separated. The organic phase is then adjusted to pH 4-5 with hydrochloric acid and extracted with water (20 ml). The phases are separated and the resulting aqueous phase is then adjusted to pH 11-12 with sodium hydroxide and re-extracted with ethyl acetate (20 ml). The resulting solution is concentrated to a volume of about 5 ml, added with 20 ml of isopropanol, concentrated to a volume of 5 ml and finally added with 30 ml of isopropanol, then 18% isopropanol hydrochloric acid (0.24 g, 1.18 mmol). The solvent is then evaporated off under reduced pressure and the residue is crystallized from acetone (10 ml). The suspension is filtered and the crystal is dried at 25°C for 12 h to obtain 0.48 g (49.7% total, enantiomeric purity: 99.8%) of a white solid.
m.p.: 126°C (from literature 123-127°C)
LC-MS (ESI+) [M+H]+ = 510
FT-IR (UATR, cm-1): 3265, 2941, 2789, 2419, 1723, 1615, 1538, 1515, 1435, 1371, 1260. 1242, 1196, 1118, 1047, 887, 838, 821, 771.
Medical uses
Landiolol is indicated as an antiarrhythmic agent to treat
- Supraventricular tachycardia and for the rapid control of ventricular rate in patients with atrial fibrillation or atrial flutter in perioperative, postoperative, or other circumstances where short-term control of the ventricular rate with a short acting agent is desirable.
- Non-compensatory sinus tachycardia where, in the physician’s judgment the rapid heart rate requires specific intervention.
Landiolol has been approved for the treatment of ventricular fibrillation or ventricular tachycardia in Japan.
In the United States, landiolol is indicated for the short-term reduction of ventricular rate in adults with supraventricular tachycardia including atrial fibrillation and atrial flutter.[4]
Landiolol can be used as first-line treatment for acute ventricular rate control in patients with atrial fibrillation (Level I recommendation- 2020 Guidelines of the European Society of Cardiology[15]).
Mode of action
The drug acts as an ultra-short-acting β1-selective blocking agent. It is rapidly hydrolyzed to an inactive form by both carboxylesterase in the liver and pseudocholinesterase in the plasma, resulting in an elimination half-life of about four minutes.[16] Landiolol is a highly selective beta-1-adrenoreceptor antagonist (the selectivity for beta-1-receptor blockade is 255 times higher than for beta-2-receptor blockade) that inhibits the positive chronotropic effects of the catecholamines adrenaline and noradrenaline on the heart, where beta-1-receptors are predominantly located. Landiolol, as other beta-blockers, is thought to reduce the sympathetic drive, resulting in reduction in heart rate, decrease in spontaneous firing of ectopic pacemakers, slowing the conduction and increase the refractory period of the AV node. Landiolol does not exhibit any membrane-stabilizing activity or intrinsic sympathomimetic activity in vitro. In preclinical and clinical studies, landiolol controlled tachycardia in an ultra-short acting manner with a fast onset and offset of action and further demonstrated anti-ischaemic and cardioprotective effects.[17] To date, landiolol has the shortest plasma half-time and the highest cardio-selectivity among β-blockers in clinical use. The selectivity of landiolol for β1-receptor blockade is 255 times higher than for β2-receptor blockade. In comparison, Metoprolol, has a much less cardioselectivity (landiolol is 100 times more cardioselective than metoprolol,[18] and 8 times more cardioselectove than esmolol[19]), and sixty times longer half-life (3–4 hours comparing to 3–4 minutes in case of landiolol). FDA points out that CYP2D6 poor metabolizers will have decreased cardioselectivity for metoprolol due to increased metoprolol blood levels, since the gene variation reduces the conversion of metoprolol to inactive metabolites leading to almost 5-fold higher plasma concentrations of metoprolol.[20]
Activation of β2 adrenergic receptors contributes to bronchial dilation and acceleration of alveolar fluid clearance in the pulmonary airway system. Consequently, a cardio-selective β1-blocker with limited effect on β2-receptor decreases the heart rate without the pulmonary adverse effects in patients with COPD or Asthma. Pharmacological stimulation of β2 receptors increases coronary blood flow in healthy humans and in patients with mildly atherosclerotic coronary arteries. Thus, not only does a cardio-selective β1-blocker reduce myocardial oxygen demand during exercise, but it also unveils β2-receptor-mediated coronary exercise hyperemia, while reducing the heart rate selectively. Interestingly, landiolol does not possess any sodium and calcium antagonistic properties, which makes it a more suitable cardio-selective β-blocker for patients with heart failure due to its lesser potency for negative inotropy, while offering higher potency for heart rate reduction. Contrary to landiolol, exposure to other β-blockers such as esmolol amplifies the re-expression of β-receptors which explains the drug tolerance effect seen during long-term esmolol infusion. Long term exposure of cells to betablockers which act as pharmacochaperones will raise the total surface level of β1-adrenergic receptors, resulting in exaggerating responses to endogenous agonists such as catecholamines, if the treatment is suddenly stopped. This phenomenon has been described as the betablocker withdrawal rebound. However, landiolol lacks appreciable pharmacochaperoning activity, as landiolol can hardly permeate cell membranes due to its large polar surface area.
Biotransformation
Landiolol is metabolised via hydrolysis of the ester moiety. In vitro and in vivo data suggest that landiolol is mainly metabolised in the plasma by pseudocholinesterases and carboxylesterases. Hydrolysis releases a ketal (the alcoholic component) that is further cleaved to yield glycerol and acetone, and the carboxylic acid component (metabolite M1), which subsequently undergoes beta-oxidation to form metabolite M2 (a substituted benzoic acid). The beta-1-adrenoreceptor blocking activity of landiolol metabolites M1 and M2 is 1/200 or less of the parent compound indicating a negligible effect on pharmacodynamics taking into account the maximum recommended landiolol dose and infusion duration.
Neither landiolol nor the metabolites M1 and M2 showed inhibitory effects on the metabolic activity of different cytochrome P450 molecular species (CYP1A2, 2C9, 2C19, 2D6 and 3A4) in vitro. The cytochrome P450 content was not affected in rats after repeated intravenous administration of landiolol. There are no data on a potential effect of landiolol or its metabolites on CYP P450 induction or time dependent inhibition available.
| IV β-Blocker | max. elimination half-life (min) | cardio-selectivity (β1/β2) | metabilization |
|---|---|---|---|
| Landiolol | 4 | 250 | pseudocholinesterases |
| Esmolol | 9 | 30 | ery-esterases |
| Metoprolol | 420 | 3 | cytochrom P2D6 (Leber) |
History
The beneficial effects of landiolol have been demonstrated in over sixty clinical trials (pubmed search -August 2018). Landiolol was generally well tolerated, with a relatively low risk of hypotension and bradycardia. Most clinical trials with landiolol have been conducted in peri-operative settings for the treatment or prophylaxis of supraventricular tachycardia or tachyarrhythmia before or after cardiac and non-cardiac surgeries. Randomized clinical trials have been published to compare landiolol with placebo<[21][22][23] diltiazem,[24] and amiodaron[25] in patients with or without heart failure. Case reports on the use of landiolol after myocardial infarction,[26] refractory electrical storm[27] have been published. The fast turnover of landiolol will diminish most adverse events due to self-limiting administration. Landiolol may be cardio-protective in septic rats by normalizing coronary microcirculation through blockage of sepsis-induced decrease in expression of VEGF signaling system but independent of inflammatory cytokines.
The efficacy and safety of landiolol in septic shock has been investigated in a multi-center prospective randomized controlled trial, and the results of the study have been published in the renown Journal Lancet Respiratory in 2020, demonstrating clinical impact of landiolol in sepsis patients through significant reduction of new-onset arrhythmia and keeping the patients within the target heart rate range.
Furthermore, landiolol demonstrated a positive clinical impact regarding ventilation-free days, ICU-free days and hospital-free days. Patients in the landiolol group had a survival rate of 88% by day 28, in contrast to a mortality rate of 20% in the control group by day 28. These are very important findings which may include landiolol in the standard of care for sepsis patients, since tachycardia and atrial fibrillation are key prognostic factors for sepsis. Additionally, tachycardia exceeding 100 beats per min (bpm) on admission to an intensive care unit (ICU) is a risk factor for worsening prognosis.[28]
A publication in the Journal of Cardiology illustrated in a prospective real-world setting, the safety and effectiveness of landiolol for the treatment of atrial fibrillation or atrial flutter in chronic heart failure (over one thousand patients at 209 medical institutions throughout Japan). In this survey, which is one of the largest studies ever performed in patients with chronic heart failure requiring intravenous rate control, report of serious hypotension was in less than 1% of patients, which highlights the cardio-selectivity of landiolol with limited effect on blood pressure. Noteworthy, over 70% of patients were in the NYHA class III or IV (35% NYHA IV), and close to 50% had a LVEF below 40%. The median time to first return to sinus rhythm after administration of landiolol was 14 hours, and the median highest infusion rate was 3 μg/kg/min.[29]
The excellent tolerance of landiolol at lower dosage (3–5 μg/kg/min) allows to initiate prophylactic use during surgery and post-operatively. Landiolol prophylaxis is associated with reduced incidence of postoperative atrial fibrillation without triggering adverse events related to a beta-blockade. Optimized infusion scheme with continuing landiolol infusion in the post-operative period seems to be associated with better response, while infusion limited to the intraoperative period may not be sufficient[30]
Society and culture
Legal status
Landiolol was approved for medical use in Japan in 2002,[10] in Canada in November 2023,[1] and in the United States in November 2024.[13]
Brand names
It is sold under various brand names including Rapibloc, Raploc, Runrapiq, Landibloc, Onoact, Corbeta, and Rapiblyk.
References
- ^ Jump up to:a b c d “Summary Basis of Decision for Sibboran”. Health Canada. 2 July 2024. Retrieved 12 October 2024.
- ^ “Details for: Sibboran”. Health Canada. 20 November 2023. Retrieved 3 March 2024.
- ^ “Regulatory Decision Summary for Sibboran”. Drug and Health Products Portal. 21 December 2022. Retrieved 2 April 2024.
- ^ Jump up to:a b c d https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/217202s000lbl.pdf
- ^ “List of nationally authorised medicinal products Active substance: landiolol” (PDF). Procedure no.: PSUSA/00010570/201802. Retrieved 12 October 2024.
- ^ Ikeshita K, Nishikawa K, Toriyama S, Yamashita T, Tani Y, Yamada T, et al. (2008). “Landiolol has a less potent negative inotropic effect than esmolol in isolated rabbit hearts”. Journal of Anesthesia. 22 (4): 361–6. doi:10.1007/s00540-008-0640-4. PMID 19011773. S2CID 5731527.
- ^ Wada Y, Aiba T, Tsujita Y, Itoh H, Wada M, Nakajima I, et al. (April 2016). “Practical applicability of landiolol, an ultra-short-acting β1-selective blocker, for rapid atrial and ventricular tachyarrhythmias with left ventricular dysfunction”. Journal of Arrhythmia. 32 (2): 82–8. doi:10.1016/j.joa.2015.09.002. PMC 4823575. PMID 27092187.
- ^ Atarashi H, Kuruma A, Yashima M, Saitoh H, Ino T, Endoh Y, et al. (August 2000). “Pharmacokinetics of landiolol hydrochloride, a new ultra-short-acting beta-blocker, in patients with cardiac arrhythmias”. Clinical Pharmacology and Therapeutics. 68 (2): 143–50. doi:10.1067/mcp.2000.108733. PMID 10976545. S2CID 46146913.
- ^ Iguchi S, Iwamura H, Nishizaki M, Hayashi A, Senokuchi K, Kobayashi K, et al. (June 1992). “Development of a highly cardioselective ultra short-acting beta-blocker, ONO-1101”. Chemical & Pharmaceutical Bulletin. 40 (6): 1462–9. doi:10.1248/cpb.40.1462. PMID 1356643.
- ^ Jump up to:a b “Ono Submits an Application of Onoact for Intravenous Infusion 50mg/150mg, a Short-Acting Selective β1 Blocker, in Japan for Additional Indication of Tachyarrhythmia in Pediatric Patients with Low Cardiac Function for a Partial Change in Approved Items of”. Ono Pharmaceutical. 28 October 2021. Retrieved 29 November 2024.
- ^ “A Short-Acting Selective β1 Blocker, Onoact for Intravenous Infusion 50mg/150mg Approved for Additional Indication of Tachyarrhythmia in Pediatric Patients with Low Cardiac Function in Japan”. Ono Pharmaceutical (Press release). 24 August 2022. Retrieved 28 November 2024.
- ^ “U.S. FDA Approves AOP Health’s Rapiblyk (landiolol) for Atrial Fibrillation and Atrial Flutter in the Critical Care Setting” (Press release). AOP Health. 27 November 2024. Retrieved 28 November 2024 – via Business Wire.
- ^ Jump up to:a b “Novel Drug Approvals for 2024”. U.S. Food and Drug Administration (FDA). 1 October 2024. Retrieved 29 November 2024.
- ^ New Drug Therapy Approvals 2024 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2025. Archived from the original on 21 January 2025. Retrieved 21 January 2025.
- ^ Hindricks G, Potpara T, Dagres N, Arbelo E, Bax JJ, Blomström-Lundqvist C, et al. (February 2021). “2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS): The Task Force for the diagnosis and management of atrial fibrillation of the European Society of Cardiology (ESC) Developed with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC”. European Heart Journal. 42 (5): 373–498. doi:10.1093/eurheartj/ehaa612. hdl:1887/3279676. PMID 32860505.
- ^ Circ J. 2016 Apr 25;80(5):1106-7
- ^ “Rapibloc Summary of Product Characteristics” (PDF). Archived from the original (PDF) on 16 June 2019. Retrieved 11 September 2018.
- ^ Baker JG (February 2005). “The selectivity of beta-adrenoceptor antagonists at the human beta1, beta2 and beta3 adrenoceptors”. British Journal of Pharmacology. 144 (3): 317–22. doi:10.1038/sj.bjp.0706048. PMC 1576008. PMID 15655528.
- ^ Okajima M, Takamura M, Taniguchi T (August 2015). “Landiolol, an ultra-short-acting β1-blocker, is useful for managing supraventricular tachyarrhythmias in sepsis”. World Journal of Critical Care Medicine. 4 (3): 251–7. doi:10.5492/wjccm.v4.i3.251. PMC 4524822. PMID 26261777.
- ^ Dean L (2017). “Metoprolol Therapy and CYP2D6 Genotype”. In Pratt VM, McLeod HL, Rubinstein WS, et al. (eds.). Medical Genetics Summaries. National Center for Biotechnology Information (NCBI). PMID 28520381. Bookshelf ID: NBK425389.
- ^ Ojima T, Nakamori M, Nakamura M, Katsuda M, Hayata K, Kato T, et al. (July 2017). “Randomized clinical trial of landiolol hydrochloride for the prevention of atrial fibrillation and postoperative complications after oesophagectomy for cancer”. The British Journal of Surgery. 104 (8): 1003–1009. doi:10.1002/bjs.10548. PMID 28444964. S2CID 1079409.
- ^ Xiao J, He P, Zou Q, Zhao Y, Xue Z, Deng X, et al. (March 2015). “Landiolol in the treatment of the intraoperative supraventricular tachycardia: a multicenter, randomized, double-blind, placebo-controlled study”. Journal of Clinical Anesthesia. 27 (2): 120–8. doi:10.1016/j.jclinane.2014.07.003. PMID 25434501.
- ^ Sezai A, Minami K, Nakai T, Hata M, Yoshitake I, Wakui S, et al. (June 2011). “Landiolol hydrochloride for prevention of atrial fibrillation after coronary artery bypass grafting: new evidence from the PASCAL trial”. The Journal of Thoracic and Cardiovascular Surgery. 141 (6): 1478–87. doi:10.1016/j.jtcvs.2010.10.045. PMID 21269646.
- ^ Sakamoto A, Kitakaze M, Takamoto S, Namiki A, Kasanuki H, Hosoda S (2012). “Landiolol, an ultra-short-acting β₁-blocker, more effectively terminates atrial fibrillation than diltiazem after open heart surgery: prospective, multicenter, randomized, open-label study (JL-KNIGHT study)”. Circulation Journal. 76 (5): 1097–101. doi:10.1253/circj.CJ-11-1332. PMID 22361918.
- ^ Shibata SC, Uchiyama A, Ohta N, Fujino Y (April 2016). “Efficacy and Safety of Landiolol Compared to Amiodarone for the Management of Postoperative Atrial Fibrillation in Intensive Care Patients”. Journal of Cardiothoracic and Vascular Anesthesia. 30 (2): 418–22. doi:10.1053/j.jvca.2015.09.007. PMID 26703973.
- ^ Kiyokuni M, Konishi M, Sakamaki K, Kawashima C, Narikawa M, Doi H, et al. (October 2016). “Beneficial effect of early infusion of landiolol, a very short-acting beta-1 adrenergic receptor blocker, on reperfusion status in acute myocardial infarction”. International Journal of Cardiology. 221: 321–6. doi:10.1016/j.ijcard.2016.07.076. PMID 27404699.
- ^ Kanamori K, Aoyagi T, Mikamo T, Tsutsui K, Kunishima T, Inaba H, et al. (2015). “Successful Treatment of Refractory Electrical Storm With Landiolol After More Than 100 Electrical Defibrillations”. International Heart Journal. 56 (5): 555–7. doi:10.1536/ihj.15-102. PMID 26346519.
- ^ Kakihana Y, Nishida O, Taniguchi T, Okajima M, Morimatsu H, Ogura H, et al. (2020). “Efficacy and safety of landiolol, an ultra-short-acting β1-selective antagonist, for treatment of sepsis-related tachyarrhythmia (J-Land 3S): A multicentre, open-label, randomised controlled trial”. The Lancet Respiratory Medicine. 8 (9): 863–872. doi:10.1016/S2213-2600(20)30037-0. PMID 32243865.
- ^ Yamashita T, Nakasu Y, Mizutani H, Sumitani K (2019). “A prospective observational survey on landiolol in atrial fibrillation/Atrial flutter patients with chronic heart failure – AF-CHF landiolol survey”. Journal of Cardiology. 74 (5): 418–425. doi:10.1016/j.jjcc.2019.05.012. PMID 31255463.
- ^ Balik M, Sander M, Trimmel H, Heinz G (2018). “Landiolol for managing post-operative atrial fibrillation”. European Heart Journal Supplements. 20 (Suppl A): A10 – A14. doi:10.1093/eurheartj/sux036. PMC 5909769. PMID 30188958.
Further reading
Shiga T (June 2022). “Benefits and safety of landiolol for rapid rate control in patients with atrial tachyarrhythmias and acute decompensated heart failure”. European Heart Journal Supplements. 24 (Suppl D): D11 – D21. doi:10.1093/eurheartjsupp/suac023. PMC 9190747. PMID 35706898.
- Rao SJ, Kanwal A, Kanwal A, Danilov A, Frishman WH (2024). “Landiolol: An Ultra-Short-Acting β-Blocker”. Cardiology in Review. 32 (5): 468–472. doi:10.1097/CRD.0000000000000555. PMID 37185629.
| showvteBeta blockers (C07) |
|---|
| Clinical data | |
|---|---|
| Trade names | Onoact, others |
| Other names | ONO-1101 |
| AHFS/Drugs.com | Rapiblyk |
| License data | US DailyMed: Landiolol |
| Routes of administration | Intravenous |
| Drug class | Antiarrhythmic |
| ATC code | C07AB14 (WHO) |
| Legal status | |
| Legal status | CA: ℞-only[1][2][3]US: ℞-only[4]Rx-only[5] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 133242-30-5 144481-98-1 |
| PubChem CID | 114905164457 |
| ChemSpider | 102855 |
| UNII | 62NWQ924LHG8HQ634Y17 |
| KEGG | D12410 D01847 |
| CompTox Dashboard (EPA) | DTXSID10158026 |
| Chemical and physical data | |
| Formula | C25H39N3O8 |
| Molar mass | 509.600 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
/////Landiolol, Rapiblyk, FDA 2024, APPROVALS 2024, supraventricular tachycardia, 133242-30-5, ONO-1101, Ono 1101, WHO 7516
Iomeprol



Iomeprol
- 78649-41-9
- Iomeprolum
- Iomeron
- Iomeprolo
WeightAverage: 777.089
Monoisotopic: 776.8541
Chemical FormulaC17H22I3N3O8
FDA APPROVED, 11/27/2024, Iomervu, For use as a radiographic contrast agent
1-N,3-N-bis(2,3-dihydroxypropyl)-5-[(2-hydroxyacetyl)-methylamino]-2,4,6-triiodobenzene-1,3-dicarboxamide
- N1,N3-Bis(2,3-dihydroxypropyl)-5-(2-hydroxy-N-methylacetamido)-2,4,6-triiodoisophthalamide
- 1-N,3-N-bis(2,3-dihydroxypropyl)-5-[(2-hydroxyacetyl)-methylamino]-2,4,6-triiodobenzene-1,3-dicarboxamide
- DTXCID2028987
- E7337
- N,N’-bis(2,3-dihydroxypropyl)-5-[glycoloyl(methyl)amino]-2,4,6-triiodoisophthalamide
Iomeprol, sold under the brand name Imeron among others, is a medication used as a radiocontrast agent in X-ray imaging.[1][2][3]
Iomeprol was approved for medical use in the United States in November 2024.[1][4][5]
Experimental Properties
| Property | Value | Source |
|---|---|---|
| melting point (°C) | 285-291 | https://www.chemos.de/import/data/msds/GB_en/78649-41-9-A0017152-GB-en.pdf |
| boiling point (°C) | 198 | https://datasheets.scbt.com/sds/eghs/en/sc-211652.pdf |
the first synthesis method is to take 5-amino-2, 4, 6-triiodoisophthalic acid as a starting material, methylate amino to chlorinate the starting material to prepare diacyl chloride, then use acetoxy acetyl chloride to carry out acylation reaction, then carry out amidation reaction with 3-amino-1, 2-propylene glycol, and finally use sodium hydroxide to hydrolyze the product to obtain iomeprol, as shown in the synthesis scheme 1
U.S. Patent No. 4,364,921 discloses three preparation processes of iopromide. One of them is shown in the following reaction scheme 1.
[Reaction scheme 1]

U.S. patent No. 4,364,921 are those basically under the same concept as shown in the following reaction schemes 3 and 4.
[Reaction scheme 3]

[Reaction scheme 4]

References:

M F C Co., Ltd.;Park Yung-ho;Hwang Seong-gwan;Park Jang-ha;Seo Rak-seok;Kim Gyeong-deok KR2020/77762, 2020, A Location in patent:Paragraph 0133-0136
Yield:78649-41-9 95%
Reaction Conditions:
with sodium hydroxide in N,N-dimethyl acetamide at 10 – 60; for 16 h;
Steps:
6 Synthesis of Iomeprole
Prepared N,N-bis(2,3-dihydroxypropyl)-5-(2-hydroxyacetamido)-2,4,6-triiodoisophthalamide 20 g (0.026 mol) in a reactor and 60 g of N,N-dimethylacetamide was added, followed by heating and stirring at 60 °C to dissolve.1.3 g (0.033 mol) of caustic soda was added to the reactor and stirred to dissolve. The reactor was cooled to 10 °C or less, and 4.5 g (0.032 mol) of iodomethane was added to the reactor, followed by stirring at 25 °C for 14 to 16 hours. After confirming the completion of the reaction, acetic acid was added to neutralize the reaction solution.The solvent was removed through concentration under reduced pressure at 60 to 65 °C and 60 g of ethanol was added to crystallize. The wet body was obtained by filtration and dried under reduced pressure in an oven at 60 °C for 12 hours to obtain iomeprol (yield 95%, purity 99.8%).
Iomeprol (CAS NO.: ), with its systematic name of , N,N’-bis(2,3-dihydroxypropyl)-5-((hydroxyacetyl)methylamino)-2,4,6-triiodo-, could be produced through many synthetic methods.
Following is one of the synthesis routes:
Firstly, 5-Amino-2,4,6-triiodo-1,3-benzenedicarboxylic acid (I) is treated with chloride to produce the dichloride (II). Secondly, compound (II) is treated with acetoxyacetyl chloride to afford compound (III), which is methylated with iodomethane. Lastly, condensation with 3-amino-1,2-propanediol of the N-methyl derivative (IV) thus obtained, followed by deacetylation with alkali metal hydroxide, yields iomeprol.

PATENT
https://patents.google.com/patent/WO2009134030A1/en
The process for preparing iopromide of formula (1) according to the present invention is shown in the following reaction scheme 5.
[Reaction scheme 5]

Step 1
5-amino-2,4,6-triiodoisophthalic acid dichloride of formula (2) is used as a starting material. The compound of formula (2) is reacted with methoxyacetyl chloride in dimethylacetamide solvent to produce 5-methoxyacetylamino-2,4,6-triiodoisophthalic acid dichloride of formula (3) which is then used without an additional purification procedure for the next step. The compound of formula (3) is reacted with 2,3-dihydroxypropylamine in dimethylacetamide solvent in the presence of triethylamine to form 5-methoxyacetylamino-2,4,6-triiodoisophthalic acid (2,3-dihydroxypropyl)amide chloride of formula (4), as shown in the following reaction scheme 6.
[Reaction scheme 6]

In the above reaction scheme, if 2,3-dihydroxypropylamine is used preferably in 0.6 to 1 equivalents, more preferably in 0.7 equivalents, the compound of formula (4) can be obtained in reasonable yield with minimizing the generation of the compound of formula (5) which is a bismer by-product.
In addition, since the unreacted compound of formula (3) existing in the filtrate obtained along with the compound of formula (4) can be recycled to the next batch without an additional recovery procedure, the loss of the yield of iopromide which occurs during the removal procedure of the bismer by-product generated in a large amount in conventional process can be prevented ultimately.
Step 2
The compound of formula (4) is reacted with acetic anhydride in acetic acid solvent in the presence of sulfuric acid as a catalyst to convert into the compound of formula (19). Sulfuric acid of preferably 0.01 to 0.2 moles, more preferably 0.05 to 0.1 moles per 1 molar reaction is added at a temperature of preferably from 0 to 30°C, more preferably from 5 to 25°C. Acetic anhydride of preferably 0.19 to 1 L, more preferably 0.35 to 0.7 L per 1 molar reaction is used.
5-methoxyacetylamino-2,4,6-triiodoisophthalic acid-N,N’-bis-(2,3-diacetoxypropyl) diamide of the compound of following formula (21), which is generated by a simultaneous conversion of the bismer by-product of formula (5) already produced in the step 1 with the conversion of the compound of formula (4) into the compound of formula (19), is easily removed by the simple crystallization procedure of the compound of formula (19). That is, the by-product of the compound of formula (21) can be removed even without an additional removal procedure. It consequently means that the bismer by-product of the compound of formula (5) which is hard to remove according to conventional process can be effectively removed in the present invention even without any additional purification procedures.
[Formula 5]

[Formula 21]

Step 3
The compound of formula (19) is reacted with 2,3-dihydroxy-N-methypropylamine in dimethylacetamide solvent in the presence of triethylamine as a base to convert into the compound of formula (20). By the hydrolysis of the compound of formula (20) in aqueous NaOH solution without further purification therefor, iopromide of formula (1) can be obtained.
The present invention will be explained more specifically by the following examples. However, the examples are not intended to limit the scope of the present invention thereto.
Example 1: Synthesis of 5-methoxyacetylamino-2,4,6-triiodoisophthalic acid (2,3-dihydroxypropyl)amide chloride (Formula 4)
5-amino-2,4,6-triiodoisophthalic acid dichloride (13.7 kg, 23 mol) was dissolved in dimethylacetamide (17.2 kg) and the mixture was cooled to 15 ℃ . Methoxyacetyl chloride (3.74 kg, 34.5 mol) was added dropwise thereto for 2 hours, and then the mixture was stirred for 15 hours. After confirming the disappearance of the starting material by HPLC analysis for reaction, methylene chloride (45.7 kg) and water (11.5 kg) were subsequently added to the reaction mixture upon being stirred, and then the stirring was stopped and the layers became separated. The obtained organic layer was washed with aqueous sodium bicarbonate solution and concentrated by distillation under reduced pressure. To a solution of the obtained concentrate dissolved in dimethylacetamide (43.1 kg), triethylamine (1.95 kg, 19.32 mol) was added and then a solution of 2,3-dihydroxypropylamine (1.47 kg, 16.13 mol) dissolved in dimethylacetamide (10.78 kg) was added dropwise thereto for 5 hours with maintaining 0 to 5 ℃ . After additional 3 hours with stirring, the reaction mixture was concentrated by distillation under reduced pressure and to the concentrate, methylene dichloride (213.33 kg) was added dropwise for 5 hours to form solid. The solid was filtered and the title compound was obtained as a white solid (10.98 kg, yield 66.1 %).
1 H NMR(dmso-d6, 500MHz) 10.2,10.06(2s, 1H); 8.79, 8.71, 8.63 (3t, 1H); 4.5~4.0(br, 2H); 4.04, 4.00(2s, 2H); 3.71~3.66(m, 1H); 3.48, 3.47(2s, 3H); 3.40~3.36(m, 2H); 3.36~3.27(m, 1H); 3.2~3.09 (m, 1H)
Example 2: Synthesis of 5-methoxyacetylamino-2,4,6-triiodoisophthalic acid (2,3-diacetoxypropyl)amide chloride (Formula 19)
5-methoxyacetylamino-2,4,6-triiodoisophthalic acid (2,3-dihydroxypropyl)amide chloride (9.97 kg, 13.8 mol) was dispersed in acetic acid, and then anhydrous acetic acid (7.45 kg) was added thereto and the mixture was cooled to 5 ℃ . Sulfuric acid (135 g) was slowly added thereto and the mixture was stirred for 1 hour. To the obtained clear solution, sodium acetate trihydrate (376 g) was added and dissolved at 0 to 5 ℃ . And then water (96.6 kg) was added for 3 hours with maintaining 0 to 10 ℃ to produce solid. The produced solid was filtered and the title compound was obtained as a white solid (10.04 kg, yield 90.2 %).
1 H NMR(dmso-d6, 500MHz) 10.12, 9.99(2s, 1H); 8.91, 8.80(2t, 1H); 5.10~5.06(m, 1H); 4.32~4.27(m, 1H); 4.20~4.16(m, 1H); 4.01, 4.00(2s, 2H); 3.52~3.37(m, 2H); 3.47, 3.46(2s, 3H); 2.02(s, 6H)
Example 3: Synthesis of 5-methoxyacetylamino-2,4,6-triiodoisophthalic acid [(2,3-dihydroxy-N-methylpropyl)-(2,3-dihydroxypropyl)]diamide (iopromide)
5-methoxyacetylamino-2,4,6-triiodoisophthalic acid (2,3-diacetoxypropyl)amide chloride (7.23 kg, 8.97 mol) was dissolved in dimethylacetamide (12.6 kg), and triethylamine (1.95 kg, 19.32 mol) was added thereto and a solution of 2,3-dihydroxy-N-methylpropylamine (943 g, 8.97 mol) dissolved in dimethylacetamide (4.2 kg) was added dropwise thereto at room temperature. After additional 2 hours with stirring, the solution was concentrated by distillation under reduced pressure. To an aqueous solution of the obtained 5-methoxyacetylamino-2,4,6-triiodoisophthalic acid [(2,3-dihydroxy-N-methylpropyl)-(2,3-diacetoxypropyl)]diamide dissolved in water, a solution of sodium hydroxide (897 g, 22.43 mol) dissolved in water was added and the reaction mixture was stirred for 10 hours with maintaining 20 to 25 ℃ . The reaction solution was passed through cation exchange resin column and anion exchange resin column to produce a colorless and transparent aqueous solution. The obtained aqueous solution was distilled under reduced pressure to remove water completely, crystallized in ethanol and filtered to obtain a white crystalline iopromide (6.032 kg, yield 85 %).
1 H NMR(dmso-d6, 500MHz) 10.07, 10.03, 9.97, 9.90(4s,1H); 8.66, 8.57, 8.52(3t, 1H); 4.76~4.74(m, 1H); 4.72, 4.67(2t, 1H); 4.59~4.58(m, 1H); 4.54~4.44(m, 1H); 4.00(s, 2H); 3.89~3.88(m, 1H); 3.69~3.68(m, 2H); 3.47(s, 3H); 3.44~3.38(m, 4H); 3.23~3.17(m, 3H), 2.85~2.83(4s, 3H)
Example 4: Synthesis of 5-methoxyacetylamino-2,4,6-triiodoisophthalic acid [(2,3-dihydroxy-N-methylpropyl)-(2,3-diacetoxypropyl)]diamide (Formula 20)
5-methoxyacetylamino-2,4,6-triiodoisophthalic acid (2,3-diacetoxypropyl)amide chloride (80.65 g, 0.1 mol) was dissolved in dimethylacetamide (140.5 g), and then triethylamine (11.13 g, 0.11 mol) was added thereto and a solution of 2,3-dihydroxy-N-methylpropylamine (10.5 g, 0.1 mol) dissolved in dimethylacetamide (46.9 g) was added dropwise thereto at room temperature. After additional 2 hours with stirring, the produced solid was filtered and then the filtrate was concentrated by distillation under reduced pressure. To the obtained concentrate, diethyl ether was added dropwise to form solid. The formed solid was filtered and the title compound was obtained as a white solid (85.8 g, yield 98 %).
1 H NMR(dmso-d6, 500MHz) 10.10, 10.06, 10.00, 9.92(4s,1H); 8.93, 8.83, 8.78(3m, 1H); 5.09(br, 1H); 4.78~4.74(m, 1H); 4.62~4.58(m, 1H); 4.34~4.26(m, 1H); 4.22~4.16(m, 1H); 4.00(s, 2H); 3.89(br, 1H); 3.72~3.65,(m, 1H); 3.49~3.40(br, 2H);3.47(s, 3H); 3.48~3.38(m, 2H); 3.22~3.12,(m, 1H); 3.07~3.04(m, 1H); 2.87~2.82(m, 2H); 2.03(s, 3H); 2.02(s, 3H)
PATENT
https://patents.google.com/patent/RU2563645C2/ru
Scheme 3

Example 1Preparation of a compound of formula 2 in which both substituents R and R’ are -NH-CH
2 -CH(OH)CH
2 OH groups using a starting solution heated to 60
° C.In a 2 L four-necked jacketed reactor equipped with a mechanical stirrer, reflux condenser and combination pH/temperature electrode, an aqueous solution of the sodium salt of 3,5-disubstituted phenol 1 corresponding to a phenol concentration of 22.8% (w/w) (1175 g solution; 0.816 mol; pH 9.6) was heated to 60 °C and then solid I
2 (250.6 g; 0.988 mol) was added in one portion. When the pH spontaneously dropped to 5, a 50% (w/w) aqueous solution of HIO
3 (173.6 g; 0.494 mol) was slowly added over 2 h. The reaction mixture was maintained at 60 °C for an additional 4 h, during which time the pH spontaneously remained in the range of 5-5.5. The red solution was cooled to 25°C and quenched by adding 18% (w/w) aqueous sodium bisulfite solution until the color was lost and the oxidation-reduction potential, measured with a suitable electrode, reached a stable negative value in the range from 0 to -20 mV.During quenching of the reaction mixture, the pH was maintained at 5 by adding minimal amounts of 30% (w/w) aqueous NaOH solution.HPLC analysis (the results of which are shown in Fig. 1) showed the degree of conversion of the starting compound to 3,5-disubstituted-2,4,6-triiodophenol 2b to be >98% (by area % on the HPLC chromatogram), and the resulting solution was used in the next stage of the synthesis without any further processing.Example 2Preparation of a compound of formula 2 in which both substituents R and R’ are -NH-CH
2 -CH(OH)CH
2 OH groups using a starting solution heated to 40
° CIn a 2 L jacketed four-neck reactor equipped with a mechanical stirrer, reflux condenser and combination pH/temperature electrode, solid I
2 (250.6 g, 0.988 mol) was added in one portion to an aqueous solution of the sodium salt of 3,5-disubstituted phenol 1 corresponding to a phenol concentration of 22.8% (w/w) (1175 g solution, 0.816 mol; pH 9.6) heated to 40 °C. When the pH spontaneously dropped to 5, a 50% (w/w) aqueous solution of HIO
3 (173.6 g, 0.494 mol) was slowly added over 3 h. The reaction mixture was then heated for 2 h at 40°C, 1 h at 50°C and 1 h at 60°C, during which time the pH spontaneously remained in the range of 5-5.5. The resulting red solution was cooled to 25°C, the pH was adjusted to 7 and maintained at this level by adding 30% (w/w) aqueous NaOH solution during quenching, which was carried out by adding 18% (w/w) aqueous sodium bisulfite solution until the color was lost and the oxidation-reduction potential, measured with a suitable electrode, reached stable negative values in the range of -20 to -50 mV.HPLC analysis showed the conversion of the starting compound to 3,5-disubstituted-2,4,6-triiodophenol 2b to be >98% (based on area % of the HPLC chromatogram), and the resulting solution was used in the next step of the synthesis without any further treatment.Example 3Preparation of a compound of formula 2 in which both substituents R and R’ are -NH-CH
2 -CH(OH)CH
2 OH groups using a stock solution heated to 30
° C and quenching the reaction mixture with bisulfite at pH5 in the final stepIn a 4 L four-necked jacketed reactor equipped with a mechanical stirrer, reflux condenser and combination pH/temperature electrode, an aqueous solution of the sodium salt of 3,5-disubstituted phenol 1 corresponding to a phenol concentration of 22.8% (w/w) (1175 g of solution; 0.816 mol; pH 9.6) was diluted with H
2 O (1054 g), heated to 30 °C and then solid I
2 (250.6 g; 0.988 mol) was added in one portion. When the pH spontaneously dropped to 5, a 50% (w/w) aqueous solution of HIO
3 (173.6 g; 0.494 mol) was slowly added over 4 h. The temperature of the reaction mixture was raised to 60°C and maintained at this temperature for a further 4 h, with the pH spontaneously remaining in the range of 5-5.5. The resulting red solution was cooled to 25°C and quenched by adding 18% (w/w) aqueous sodium bisulfite solution, maintaining the pH at 5 by adding 30% (w/w) aqueous NaOH solution until the color was lost and the oxidation-reduction potential, measured with a suitable electrode, reached stable negative values in the range of 0 to -20 mV.HPLC analysis showed the conversion of the starting compound to 3,5-disubstituted-2,4,6-triiodophenol 2b to be >98% (based on area % of the HPLC chromatogram), and the resulting solution was used in the next step of the synthesis without any further treatment.Example 4Preparation of a compound of formula 2 in which both substituents R and R’ are -NH-CH
2 -CH(OH)CH
2 OH groups using a starting solution heated to 30
° C and quenching the reaction mixture with bisulfite at pH7 in the final stepIn a 4 L four-necked jacketed reactor equipped with a mechanical stirrer, reflux condenser and combination pH/temperature electrode, an aqueous solution of the sodium salt of 3,5-disubstituted phenol 1 corresponding to a phenol concentration of 22.8% (w/w) (1175 g solution; 0.816 mol; pH 9.6) was diluted with H
2 O (1054 g), heated to 30 °C and then solid I
2 (250.6 g; 0.988 mol) was added in one portion. When the pH spontaneously dropped to 5, a 50% (w/w) aqueous solution of HIO
3 (173.6 g; 0.494 mol) was slowly added over 4 h. The temperature of the reaction mixture was raised to 60°C and maintained at this temperature for a further 4 h, during which time the pH spontaneously remained in the range of 5-5.5. The resulting red solution was cooled to 25°C, the pH was adjusted to 7 and maintained at this level by adding 30% (w/w) aqueous NaOH solution during quenching, which was carried out by adding 18% (w/w) aqueous sodium bisulfite solution until the color was lost and the oxidation-reduction potential, measured with a suitable electrode, reached stable negative values in the range of -20 to -50 mV.HPLC analysis showed the conversion of the starting compound to 3,5-disubstituted-2,4,6-triiodophenol 2b to be >98% (based on area % of the HPLC chromatogram), and the resulting solution was used in the next step of the synthesis without any further treatment.Example 5Preparation of a compound of formula 2, in which both substituents R and R’ are -NH-CH
2 -CH(OH)CH
2 OH groups, using a starting solution at room temperature (approximately 20
° C)In a 4 L four-necked jacketed reactor equipped with a mechanical stirrer, reflux condenser and combination pH/temperature electrode, an aqueous solution of 3,5-disubstituted phenol 1 sodium salt corresponding to a phenol concentration of 22.8% (w/w) (1175 g solution; 0.816 mol; pH 9.6) was first diluted with H
2 O (1054 g) maintaining the temperature at 20 °C, and then solid I
2 (250.6 g; 0.988 mol) was added in one portion. The resulting solution was then heated to 40 °C and when the pH spontaneously dropped to 5, a 50% (w/w) aqueous solution of HIO
3 (173.6 g; 0.494 mol) was slowly added over 4 h . The temperature of the reaction mixture was raised to 60°C over 2 h and maintained at this temperature for a further 3 h, during which time the pH spontaneously remained in the range of 5-5.5. The red solution was then cooled to 25°C, the pH was adjusted to 7 and maintained at this level by adding 30% (w/w) aqueous NaOH solution, and quenched with sodium bisulfite (18% (w/w) aqueous solution) until color loss and stable negative values (in the range of -20 to -50 mV) of the oxidation-reduction potential were reached, measured with suitable redox electrodes.HPLC analysis showed the conversion of the starting compound to 3,5-disubstituted-2,4,6-triiodophenol 2b to be >98% (based on area % of the HPLC chromatogram), and the resulting solution was used in the next step of the synthesis without any further treatment.Example 6Preparation of a compound of formula 2 in which the substituent R is a -NH-CH
2 -CH(OH)CH
2 OH group and R’ is -NH-CH(CH
2 OH)
2 using a starting solution heated to 60
° C.In a 1 L jacketed four-neck reactor equipped with a mechanical stirrer, reflux condenser, and combination pH/temperature electrode, N-(2,3-dihydroxypropyl)-N’-[2-hydroxy-1-(hydroxymethyl)ethyl]-5-hydroxy-1,3-benzenedicarboxamide (100.3 g, 0.305 mol) was dissolved in H
2 O (430 g) and converted to the corresponding sodium salt by adding 30% (w/w) NaOH (40.6 g, 0.305 mol) (pH 9.5). The solution was heated to 60 °C and solid I
2 (93.1 g, 0.367 mol) was added in one portion; When the pH spontaneously dropped to 5, 50% (w/w) aqueous HIO3 (64.5 g, 0.183 mol) was added slowly over 2 h
. The reaction temperature was maintained at 60 °C for a further 4 h, during which time the pH of the reaction mixture spontaneously remained in the range 5-5.5. The resulting red solution was cooled to 25 °C and quenched by adding 18% (w/w) aqueous sodium bisulfite solution, maintaining the pH at 5, by adding 30% (w/w) aqueous NaOH until colourlessness and a stable negative oxidation-reduction potential, measured with a suitable oxidation-reduction electrode, in the range 0 to -20 mV.HPLC analysis (Fig. 2) showed the degree of conversion of the starting compound to N-(2,3-dihydroxypropyl)-N’-[2-hydroxy-1-(hydroxymethyl)ethyl]-5-hydroxy-2,4,6-triiodo-1,3-benzenedicarboxamide >98% (by area % on the HPLC chromatogram), and the resulting solution was used in the next step of the synthesis without any additional processing.Example 7Preparation of a compound of formula 2 in which both substituents R and R’ are -NH-CH(CH
2 OH)
2 groups using a starting solution heated to 60
° C.In a 0.5 L jacketed four-neck reactor equipped with a mechanical stirrer, reflux condenser, and combination pH/temperature electrode, N,N’-bis[2-hydroxy-1-(hydroxymethyl)ethyl]-5-hydroxy-1,3-benzenedicarboxamide (50 g, 0.152 mol) was dissolved in H
2 O (215 g) and converted to the corresponding sodium salt by adding 30% (w/w) NaOH (20.3 g, 0.152 mol) (pH 9.5). The solution was heated to 60 °C and solid I
2 (46.4 g, 0.183 mol) was added in one portion; When the pH spontaneously dropped to 5, 50% (w/w) aqueous HIO3 (32.2 g, 0.091 mol) was added slowly over 2 h
. The reaction temperature was maintained at 60 °C for an additional 4 h, during which time the pH of the reaction mixture spontaneously remained in the range 5-5.5. The resulting red solution was cooled to 25 °C and quenched by adding 18% (w/w) aqueous sodium bisulfite solution, maintaining the pH at 5, with 30% (w/w) aqueous NaOH until colorlessness and a stable negative oxidation-reduction potential (in the range of -20 to -50 mV), as measured by a suitable oxidation-reduction electrode.HPLC analysis showed the conversion of the starting compound to N,N’-bis[2-hydroxy-1-(hydroxymethyl)ethyl]-5-hydroxy-2,4,6-triiodo-1,3-benzenedicarboxamide to be >98% (based on area % of the HPLC chromatogram), and the resulting solution was used in the next step of the synthesis without any further processing.Comparative example 1This test was carried out to evaluate the efficiency of the iodination reaction disclosed by Patil et al., in ARKIVOC, 2006, 104 and Tetrahedron Lett., 2005, 46, 7179.In a 50 mL three-necked round-bottomed flask equipped with a thermometer and a reflux condenser, solid 3,5-disubstituted phenol 1 (16.4 g, 50 mmol) was suspended in ethanol (30 mL). Then, to the resulting suspension, heated to 38-40 °C, solid I
2 (15.2 g, 60 mmol) was added in one portion and a solution of HIO
3 (5.3 g, 30 mmol) in H
2 O (3 mL) was added in the indicated order over 5 min. The resulting dark brown mixture was stirred at 38-40 °C for about 1 h and then the change in appearance of the reaction mixture was recorded, which turned into a clear dark brown solution. The reaction mixture was maintained at the above temperature for a total of 3.5 h, then cooled to room temperature, which caused crystallization of the pale yellow solid product. After 15 h at room temperature, the solid was isolated by filtration and dried to give the desired 3,5-disubstituted-2,4,6-triiodophenol (12.1 g, 17 mmol). Yield 34.3%.During the iodination reaction, the reaction mixture was analyzed by HPLC. In particular, the first analysis was performed 1.5 hours after the start of iodination (the reaction time was chosen based on the literature sources cited), and its results are shown in Fig. 3, and the second analysis was performed after another 2 hours (the total reaction time was 3.5 hours), and its results are shown in Fig. 4. The results show that even after 3.5 hours, the conversion is not complete and a significant amount (13% based on the area on the HPLC chromatogram) of the original substrate is still present. On the other hand, a longer reaction time leads to the formation of a significant amount of impurities, which are decay products, which are easily detected after 3.5 hours of reaction (Fig. 4). This is certainly a factor that adversely affects the reaction yields. However, the low reaction yields can also be attributed to the solubility of 3,5-disubstituted-2,4,6-triiodophenol 2b in an alcohol medium, as confirmed by the analysis of the mother liquor shown in Fig. 5, which interferes with the quantitative isolation of the iodination product.In this regard, the increase in both the reaction yield and the product purity due to the use of an aqueous medium and reaction conditions, expressed in the above results, is evident from a comparison of Figs. 3-5 with Figs. 1 and 2, which show chromatograms (HPLC) of crude reaction solutions (Examples 1 and 6, respectively) obtained using the method of the present invention.
PATENT
https://patents.google.com/patent/KR20200032280A/en
[Scheme 3]

Example One.
Iomeproletic Produce
5- (2-hydroxyacetamido) -N, N’-bis (2, 3-dihydroxypropyl) -2, 4, 6-triiodoisophthalamide (1b) 5g (1 equivalent) and 3.6 g (5 equivalents) of calcium chloride was added to 25 g of methanol together, and dissolved at reflux at room temperature or 70 ° C. for 60 minutes.
After cooling the temperature of the solution to 10 ℃ to 15 ℃ 0.3g (0.62 equivalents) of calcium hydroxide was added and stirred at the same temperature for 1 hour.
2.48 g (3 equivalents) of dimethyl sulfate was added to the reaction solution and stirred for 3 hours at the same temperature until the reaction was completed.
After completion of the reaction, 1 mL of HCl (35%) was added to acidify, 25 mg of 2-butanol was added, stirred at a temperature of 70 to 80 ° C. for 2 hours, cooled, filtered and washed with 2-butanol to obtain an iomeprole crude product.
The above prepared omeprolol was added to a mixture of 25 mL of methanol and 10 mL of water, heated to 50 ° C. to dissolve, put 20 mL of 2-butanol, refluxed at 90 ° C. for 3 hours, cooled to room temperature, and the resulting crystal was filtered.
After washing with 2-butanol and drying under reduced pressure at 90 ° C. for 12 hours, 4.17 g of Iomeprole (HPLC: 99.3%) was obtained.
Example 2.
Preparation of Iomeprole
5- (2-hydroxyacetamido) -N, N’-bis (2, 3-dihydroxypropyl) -2, 4, 6-triiodoisophthalamide (1b) 5g (1 equivalent) and 3.6 g (5 equivalents) of calcium chloride was added to 25 g of methanol together and refluxed at 70 ° C for 30 minutes to dissolve.
After cooling the solution to 0-5 ° C., 0.3 g (0.62 equivalents) of calcium hydroxide was added and stirred at the same temperature for 1 hour.
2.48 g (3 equivalents) of dimethyl sulfate was added to the reaction solution and stirred for 7 hours at the same temperature until the reaction was completed.
After completion of the reaction, 1 mL of HCl (35%) was added to acidify, 30 mL of 2-butanol was added, refluxed at a temperature of 70-80 ° C. for 2 hours, stirred, filtered and washed with 2-butanol to obtain an iomeprole crude product.
After adding the above prepared omeprolol to a mixture of 25 mL of methanol and 10 mL of water, the temperature was raised to 50 ° C. to dissolve, 20 mL of 2-butanol was added, refluxed at 90 ° C. for 3 hours, cooled to room temperature, and the resulting crystal was filtered. .
After washing with 2-butanol and drying under reduced pressure at 90 ° C. for 12 hours, 4.21 g of Iomeprole (HPLC: 99.1%) was obtained.
Comparative example 1 (Inorganic chloride Non-addition )
After adding 5- (2-hydroxyacetamido) -N, N’-bis (2, 3-dihydroxypropyl) -2, 4, 6-triiodoisophthalamide (1b) to 25 g of methanol It was refluxed for 30 minutes.
After the temperature of the turbid solution was cooled to 10 ° C to 15 ° C, 0.3 g (0.62 equivalent) of calcium hydroxide was added and stirred at the same temperature for 1 hour.
2.48 g (3 equivalents) of dimethyl sulfate was added to the reaction solution and stirred at the same temperature for 5 hours.
As a result of reactivity review by HPLC, synthesis of iomeprole progressed 5%, 5- (2-hydroxyacetamido) -N, N’-bis (2, 3-dihydroxypropyl) -2, 4, 6- Triiodoisophthalamide (1b) was found to be 90% or more remaining, so that the reactivity was very low.
Comparative example 2 (Inorganic base Non-addition )
5- (2-hydroxyacetamido) -N, N’-bis (2, 3-dihydroxypropyl) -2, 4, 6-triiodoisophthalamide (1b) and 3.6 g of calcium chloride are methanol After adding to 25 g, the mixture was refluxed for 30 minutes to dissolve.
After the temperature of the solution was cooled from 10 ° C to 15 ° C, 2.48 g (3 equivalents) of dimethyl sulfate was added to the reaction solution and stirred at the same temperature for 5 hours.
As a result of reactivity review by HPLC, synthesis of iomeprole proceeds 0.5%, 5- (2-hydroxyacetamido) -N, N’-bis (2, 3-dihydroxypropyl) -2, 4, 6- Triiodoisophthalamide (1b) was found to be very low reactivity with more than 99% remaining.
PATENT
https://patents.google.com/patent/CN102363600B/en
.Synthetic route is seen formula 1:

Embodiment
Embodiment 1
5-methylamino-2,4,6-triiodo m-phthaloyl chloride synthetic:
In the there-necked flask that agitator and reflux condensing tube are housed, 76g(0.133mol under the room temperature) 5-methylamino-2,4,6-triiodo m-phthalic acid is dissolved in 250mL(2.53mol) in the ethyl acetate, after waiting to stir, add 29mL(0.4mol) sulfur oxychloride, be warming up to 50 ℃ of acyl chloride reaction temperature then, stir and finished reaction in 6 hours.After treating that ethyl acetate and sulfur oxychloride boil off under the decompression, residue boils off solvent and washs through frozen water after adding the 100mL ethyl acetate, dry product 68.9g, yield is that 84.9%(is with 5-methylamino-2,4,6-triiodo m-phthalic acid meter), m.p.167 ~ 169 ℃.
Embodiment 2
5-methylamino-2,4,6-triiodo m-phthaloyl chloride synthetic:
The acyl chloride reaction temperature is 80 ℃, and in 3 hours reaction times, all the other are operated with embodiment 1.
Embodiment 3
5-[N-methyl-2-chloracetyl amido]-2,4,6-triiodo m-phthaloyl chloride synthetic:
In reaction flask, add 36.6g(0.06mol under the room temperature) 5-methylamino-2,4,6-triiodo m-phthaloyl chloride and 80mL N,N-dimethylacetamide, heating in water bath to 50 ℃, after the stirring and dissolving, be cooled to 10 ℃, begin to drip 10.2g(0.09mol) chloroacetyl chloride, dropwising the back heats up 50 ℃, stirred 3 hours, and be cooled to 10 ℃, be added dropwise to the 150mL frozen water.Filter, through the frozen water washing, dry product 38.7g, yield be 94%(with 5-methylamino-2,4,6-triiodo m-phthaloyl chloride meter), m.p.194 ~ 196 ℃.
Embodiment 4
5-[N-methyl-2-chloracetyl amido]-2,4,6-triiodo m-phthaloyl chloride synthetic:
The chlorine acylation temperature is 90 ℃, and in 1 hour reaction times, all the other are operated with embodiment 3.
Embodiment 5
5-[N-methyl-2-chloracetyl amido]-N, N ‘-two (2,3-dihydroxypropyl)-2,4,6-three iodo-1,3-benzenedicarboxamide synthetic:
In reaction flask, add 41.2g(0.06mol under the room temperature) 5-[N-methyl-2-chloracetyl amido]-2,4,6-triiodo m-phthaloyl chloride and 80mL N, the N-N,N-DIMETHYLACETAMIDE after the stirring and dissolving, is cooled to 10 ℃, add 16g(0.16mol) triethylamine and 14.6g(0.16mol) 3-amino-1, the 2-propylene glycol is heated to 20 ℃ then, stirs and finishes reaction in 15 hours, be chilled to after-filtration below 10 ℃, after evaporated under reduced pressure, residue is dissolved in the 160mL methyl alcohol with filtrate, adds 200mL water and stirs evenly, leave standstill, with the solid filtering of separating out, the washing, dry product 43.4g, yield is that 91%(is with 5-[N-methyl-2-chloracetyl amido]-2,4,6-triiodo m-phthaloyl chloride meter), m.p.204 ~ 207 ℃.
Embodiment 6
5-[N-methyl-2-chloracetyl amido]-N, N ‘-two (2,3-dihydroxypropyl)-2,4,6-three iodo-1,3-benzenedicarboxamide synthetic:
The amidate action temperature is 50 ℃, and in 8 hours reaction times, all the other are operated with embodiment 5.
Embodiment 7
5-[N-methyl-glycolamide base]-N, N ‘-two (2,3-dihydroxypropyl)-2,4,6-three iodo-1,3-benzenedicarboxamide synthetic:
In reaction flask, add 47.7g(0.06mol) 5-[N-methyl-2-chloracetyl amido]-N, N ‘-two (2,3-dihydroxypropyl)-2,4,6-three iodo-1,3-benzenedicarboxamide, 250mL water, stir and to add 26g(0.32mol down) sodium acetate, back flow reaction is after 24 hours, the pressure reducing and steaming solvent.The residue dissolve with methanol filters, and filtrate boils off methyl alcohol, and vacuum-drying gets solid, is dissolved in the 150mL water, adds gac 1.8g, and reflux 30min filters.Filtrate is successively respectively by 732 Zeo-karbs and 717 anionite-exchange resin, and pressure reducing and steaming solvent again is after the resistates vacuum-drying, add 180mL ethanol and carry out recrystallization, get white solid 40.1g, HPLC detects purity greater than 99.0%, and yield is that 86%(is with 5-[N-methyl-2-chloracetyl amido]-N, N ‘-two (2, the 3-dihydroxypropyl)-2,4,6-three iodo-1,3-benzenedicarboxamide meter), m.p.〉280 ℃. 1H-NMR?(DMSO-D 6)δ(ppm):2.91(s,4H),3.21~3.36(m,4H),?3.41~3.65(m,4H),?3.85(s,2H),?3.98(m,2H),?4.1~?4.5(m,5H),?8.2~?8.4(d,?2H); 13C-NMR(D 2O)δ(ppm)33.2,44.7,60.7,64.6,71.2,90.1,99.8,99.9,145.5,150.5,150.6,171.3,171.4,173.9。
PATENT
https://patents.google.com/patent/CN114213273A/en
synthetic route is as follows:

Example 1
The synthesis process of iomeprol with 5-amido-N, N’ -bis (2, 3-dihydroxypropyl) -2,4, 6-triiodo-1, 3-benzenedicarboxamide as initial material includes successive chloroacetylation, methylation, hydrolysis and hydroxylation to synthesize iomeprol product, and includes the following steps:

the specific process comprises 4 reaction steps:
s1, chloroacetylation (preparation of 5- (2-chloroacetamido) -2,4, 6-triiodo-N, N’ -bis (2, 3-bis (2-chloroacetoxy) propyl) -isophthalamide):
12g of 5-amino-N.N’ -bis (2, 3-dihydroxypropyl) -2,4, 6-triiodoisophthalamide was dissolved in 24g N, N-dimethylacetamide and stirred at room temperature. 12g of chloroacetyl chloride were added while controlling the temperature below 60 ℃. After the addition, the temperature is kept between 50 and 60 ℃, the stirring is carried out for 4 hours, and after the reaction is finished, the vacuum concentration is carried out below 65 ℃ until the reaction is dried. 36ml of ethyl acetate and 36ml of a 5% aqueous solution of sodium hydrogencarbonate were added to the concentrate to conduct extraction. The aqueous layer after separation was extracted twice with 10ml of ethyl acetate. The organic solutions thus extracted were mixed, 15ml of 5% saline was added thereto, and the mixture was washed, and after recovering the organic solution layer, magnesium sulfate was added to remove water, followed by filtration and distillation under reduced pressure to obtain 18.5g of an oily substance.
S2, methylation reaction (preparation of 5- (N-methyl-2-chloroacetamido) -2,4, 6-triiodo-N, N’ -bis (2, 3-bis (2-chloroacetoxy) propyl) -isophthalamide):
dissolving 18.5g of the oily substance in the previous step by using 37ml of acetone, cooling to 0 ℃, adding 3.8g of potassium carbonate, keeping the temperature and stirring, dropwise adding 5.8g of dimethyl sulfate while stirring, keeping the temperature and reacting for 8 hours, after the reaction is finished, carrying out vacuum concentration to dryness at 60 ℃, dissolving the concentrated solution in 50ml of ethyl acetate and 50ml of water, and adding 10ml of ethyl acetate into the separated water layer for secondary extraction. The organic solutions thus extracted were mixed, 15ml of 5% saline was added thereto, and the mixture was washed, and after recovering the organic solution layer, magnesium sulfate was added to remove water, followed by filtration and distillation under reduced pressure to obtain 19.3g of an oily substance.
S3, ester hydrolysis reaction (preparation of 5- (N-methyl-2-chloroacetamido) -2,4, 6-triiodo-N, N’ -bis (2, 3-dihydroxypropyl) -isophthalamide):
the above oily substance was dissolved in 20ml of methanol, 6.0g of water was added, 30.0g of 30% aqueous sodium hydroxide solution was added, and the reaction was carried out at 15 ℃ for 4 hours, and after the completion of the reaction, methanol was distilled off under reduced pressure to obtain an aqueous solution containing the objective compound.
S4 hydroxylation reaction (preparation of 5- (N-methyl-2-hydroxyacetamido) -2,4, 6-triiodo-N, N’ -bis (2, 3-dihydroxypropyl) -isophthalamide, i.e. iomeprol):
adding 6.0g of sodium acetate into the aqueous solution, heating to reflux for 24 hours, distilling under reduced pressure to remove the solvent, adding methanol into the residue to dissolve, filtering, evaporating the methanol from the filtrate, dissolving the residue in pure water, adding activated carbon, heating to reflux for 30 minutes, filtering, sequentially passing the filtrate through cation resin and anion resin, evaporating to remove the solvent, adding 70ml of ethanol to recrystallize, filtering, drying under reduced pressure at 50 ℃ to obtain 8.5g of white solid, wherein the HPLC purity is 99.2%, and the molar yield is 64%.
Example 2
S1, chloroacetylation (preparation of 5- (2-chloroacetamido) -2,4, 6-triiodo-N, N’ -bis (2, 3-bis (2-chloroacetoxy) propyl) -isophthalamide):
500g of 5-amino-N.N’ -bis (2, 3-dihydroxypropyl) -2,4, 6-triiodoisophthalamide was dissolved in 1000g N, N-dimethylacetamide and stirred at room temperature. 500g of chloroacetyl chloride was added while controlling the temperature below 60 ℃. After the addition, the temperature is kept between 50 and 60 ℃, the stirring is carried out for 4 hours, and after the reaction is finished, the vacuum concentration is carried out below 65 ℃ until the reaction is dried. 1500ml of ethyl acetate and 1500ml of 5% aqueous sodium bicarbonate solution were added to the concentrate to extract. The aqueous layer after separation was extracted twice with 480ml of ethyl acetate. The organic solutions thus extracted were mixed, and 650ml of 5% brine was added thereto, followed by washing, and after recovering the organic solution layer, magnesium sulfate was added to remove water, followed by filtration and distillation under reduced pressure to obtain 772.6g of an oil.
S2, methylation reaction (preparation of 5- (N-methyl-2-chloroacetamido) -2,4, 6-triiodo-N, N’ -bis (2, 3-bis (2-chloroacetoxy) propyl) -isophthalamide):
dissolving 772.6g of the oily matter in the previous step by 1545ml of acetone, cooling to 0 ℃, adding 158.5g of potassium carbonate, keeping the temperature and stirring, dropwise adding 241.5g of dimethyl sulfate while stirring, keeping the temperature and reacting for 8 hours, after the reaction is finished, vacuum concentrating at 60 ℃ to dryness, dissolving the concentrated solution in 1500ml of ethyl acetate and 1500ml of water, and adding 480ml of ethyl acetate into the separated water layer for secondary extraction. The organic solutions thus extracted were mixed, and then 650ml of 5% saline was added thereto, followed by washing, and after recovering the organic solution layer, magnesium sulfate was added thereto to remove water, followed by filtration and distillation under reduced pressure to obtain 805.7g of an oily substance.
S3, ester hydrolysis reaction (preparation of 5- (N-methyl-2-chloroacetamido) -2,4, 6-triiodo-N, N’ -bis (2, 3-dihydroxypropyl) -isophthalamide):
the above oil was dissolved in 800ml of methanol, 250g of water was added, 1250g of a 30% aqueous solution of sodium hydroxide was added, and the reaction was carried out at 15 ℃ for 4 hours, and after the completion of the reaction, methanol was distilled off under reduced pressure to obtain an aqueous solution containing the objective compound.
S4 hydroxylation reaction (preparation of 5- (N-methyl-2-hydroxyacetamido) -2,4, 6-triiodo-N, N’ -bis (2, 3-dihydroxypropyl) -isophthalamide, i.e. iomeprol):
adding 250g of sodium acetate into the aqueous solution, heating to reflux for 24 hours, distilling under reduced pressure to remove the solvent, adding methanol into the residue to dissolve, filtering, evaporating the filtrate to remove the methanol, dissolving the residue in pure water, adding activated carbon, heating to reflux for 60 minutes, filtering, sequentially passing the filtrate through cation resin and anion resin, evaporating to remove the solvent, adding 3000ml of ethanol to recrystallize, filtering, drying under reduced pressure at 50 ℃ to obtain 365.0g of white solid, wherein the HPLC purity is 99.1%, and the molar yield is 66%.
Compared with two synthesis methods in patent EP0026281A1, the synthesis method of iomeprol developed by the invention does not use thionyl chloride, reduces the difficulty of waste gas treatment, does not use acetoxy acetyl chloride, and ensures the process stability. Meanwhile, the used starting material (compound II) is a common intermediate of other contrast agents iohexol and ioversol, so that corresponding supporting construction is reduced.
Therefore, the synthetic route designed by the invention has the advantages of mild reaction conditions, stable quality, high yield, low cost, environmental protection and suitability for industrial production.
The embodiments are described in a progressive manner, each embodiment focuses on differences from other embodiments, and the same or similar parts among the embodiments are referred to each other. The device disclosed by the embodiment corresponds to the method disclosed by the embodiment, so that the description is simple, and the relevant points can be referred to the method part for description.
Side effects
It is classified as a water-soluble, nephrotrophic, low osmolar X-ray contrast medium.[2] Low osmolar non-ionic agents are better tolerated and less likely to cause side effects than the high osmolar ionic agents.[2]
Society and culture
Iomeprol is not metabolized in the human body but excreted in unchanged form.[medical citation needed] It is decomposed slowly and can therefore accumulate in the environment.[6]
Legal status
Iomeprol was approved for medical use in the United States in November 2024.[1][4]
Brand names
Iomeprol is sold under the brand name Iomervu.[1]
References
- ^ Jump up to:a b c d e https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/216017s000,216017s000lbl.pdf
- ^ Jump up to:a b c Rossiter D (2014). South African medicines formulary (11th ed.). Rondebosch, South Africa: Health and Medical Pub. Group .of the South African Medical Association. ISBN 978-1-875098-30-9. OCLC 869772940.
- ^ Haberfeld H, ed. (2020). Austria-Codex (in German). Vienna: Österreichischer Apothekerverlag. Iomeron 300 mg J/ml-Infusionsflasche.
- ^ Jump up to:a b “Novel Drug Approvals for 2024”. U.S. Food and Drug Administration (FDA). 1 October 2024. Retrieved 29 November 2024.
- ^ New Drug Therapy Approvals 2024 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2025. Archived from the original on 21 January 2025. Retrieved 21 January 2025.
- ^ Pfundstein P, Martin C, Schulz W, Seitz W, Ruth KM, Wille A, et al. (January 2015). “IC-ICP/MS-Analytik”. GIT Labor-Fachzeitschrift (in German): 29–31.
- Dooley M, Jarvis B: Iomeprol: a review of its use as a contrast medium. Drugs. 2000 May;59(5):1169-86. doi: 10.2165/00003495-200059050-00013. [Article]
- Katayama H, Spinazzi A, Fouillet X, Kirchin MA, Taroni P, Davies A: Iomeprol: current and future profile of a radiocontrast agent. Invest Radiol. 2001 Feb;36(2):87-96. doi: 10.1097/00004424-200102000-00004. [Article]
- Rosati G: Clinical pharmacology of iomeprol. Eur J Radiol. 1994 May;18 Suppl 1:S51-60. doi: 10.1016/0720-048x(94)90094-9. [Article]
- EMC Summary of Product Characteristics: Iomeron (iomeprol) solution for injection [Link]
- FDA Approved Drug Products: IOMERVU (iomeprol) injection, for intra-arterial or intravenous use [Link]
- Medsafe NZ: IOMERON® (iomeprol) safety data sheet [Link]
| showvteContrast media (V08) |
|---|
| Clinical data | |
|---|---|
| Trade names | Iomervu, others |
| License data | US DailyMed: Iomeprol |
| Routes of administration | Intravenous, intra-arterial |
| ATC code | V08AB10 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1]In general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Metabolism | none |
| Elimination half-life | 109±20 min |
| Excretion | Kidney |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 78649-41-9 |
| PubChem CID | 3731 |
| DrugBank | DB11705 |
| ChemSpider | 3600 |
| UNII | 17E17JBP8L |
| KEGG | D01719 |
| ChEBI | CHEBI:31710 |
| CompTox Dashboard (EPA) | DTXSID1049061 |
| Chemical and physical data | |
| Formula | C17H22I3N3O8 |
| Molar mass | 777.089 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
////////////Iomeprol, Iomervu, FDA 2024, APPROVALS 2024, 78649-41-9, Iomeprolum, Iomeron, Iomeprolo, UNII-17E17JBP8L, E-7337, E 7337, E-7337, E7337, XRAY CONTRAST AGENT
Crinecerfont



Crinecerfont
CAS 752253-39-7
SSR125543
SSR 125543
SSR-125543, WHO 10958, UNII-MFT24BX55I, 06-RORI,NBI-74788
- (S)-4-(2-chloro-4-methoxy-5-methylphenyl)-N-(2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl)-5-methyl-N-(prop-2-yn-1-yl)thiazol-2-amine
- 2-Thiazolamine, 4-(2-chloro-4-methoxy-5-methylphenyl)-N-((1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl)-5-methyl-N-2-propynyl-
- 2-Thiazolamine, 4-(2-chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]-5-methyl-N-2-propyn-1-yl
FDA APPROVED 12/13/2024, Crenessity, To treat classic congenital adrenal hyperplasia
Press Release
4-(2-chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]-5-methyl-N-prop-2-ynyl-1,3-thiazol-2-amine
WeightAverage: 483.04
Monoisotopic: 482.1594906
Chemical FormulaC27H28ClFN2OS

CAS No. : 321839-75-2
| Molecular Weight | 519.50 |
|---|---|
| Formula | C27H29Cl2FN2OS |
Crinecerfont, sold under the brand name Crenessity, is a medication used for the treatment of congenital adrenal hyperplasia.[1] It is a corticotropin-releasing factor type 1 receptor (CRF1R) antagonist developed to treat classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency (21OHD).[1] It is taken by mouth.[1]
The most common side effects of crinecerfont in adults include fatigue, dizziness, and arthralgia (joint pain).[2] For children, the most common side effects include headache, abdominal pain, and fatigue.[2]
Crinecerfont was approved for medical use in the United States in December 2024.[2][3] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication.[4]
A medication used to reduce the amount of steroid replacement required in patients with a genetic disease that causes, amongst other symptoms, a steroid deficiency.
- OriginatorSanofi
- DeveloperNeurocrine Biosciences; Sanofi
- ClassAmines; Antidepressants; Anxiolytics; Chlorobenzenes; Cyclopropanes; Fluorobenzenes; Halogenated hydrocarbons; Phenyl ethers; Small molecules; Thiazines; Thiazoles
- Mechanism of ActionCorticotropin releasing factor receptor 1 antagonists
- Orphan Drug StatusYes – Congenital adrenal hyperplasia
- MarketedCongenital adrenal hyperplasia
- DiscontinuedMajor depressive disorder; Post-traumatic stress disorders
20 Dec 2024Launched for Congenital adrenal hyperplasia (Adjunctive treatment, In adolescents, In children) in USA (PO)
- 20 Dec 2024Launched for Congenital adrenal hyperplasia (Adjunctive treatment, In adolescents, In children, In the elderly, In adults) in USA (PO)
- 20 Dec 2024Launched for Congenital adrenal hyperplasia (Adjunctive treatment, In the elderly, In adults) in USA (PO)



SYN

https://patents.google.com/patent/US12128033
Example processes and certain intermediates of the present invention are shown in Scheme I to Scheme VII below.
A representative Coupling-Step of 2-cyclopropylacetic acid (Compound
1A) with N,O-dimethylhydroxylamine or a salt thereof in the presence of a coupling-step reagent (e.g., 1,1′-carbonyldiimidazole), a coupling-step base (e.g., triethylamine), and a coupling-step solvent (e.g., dichloromethane) to prepare 2-cyclopropyl-N-methoxy-N-methylacetamide (Compound
2A) is provided below in Scheme I.

A representative Reacting-Step between 2-cyclopropyl-N-methoxy-N-methylacetamide (Compound
2A) with an organomagnesium reagent of 4-bromo-2-fluoro-1-methylbenzene in the presence of a reacting-step solvent (e.g., tetrahydrofuran (THF)) to prepare 2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethan-1-one (Compound
3A) is provided below in Scheme II.

A representative Condensing-Step of 2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethan-1-one (Compound
3A) with a Compound of Formula (Ic) or a salt thereof, in the presence of a condensing-step acid (e.g., p-toluenesulfonic acid) and a condensing-step solvent (e.g., toluene) to prepare a Compound of Formula (Ie) is provided below in Scheme III.

-
- wherein:
- R1c, R2c, and R3c are each independently selected from: H, C1-C6 alkoxy, C1-C6 alkyl, C1-C6 haloalkyl, and halogen.
A representative Reducing-Step of a Compound of Formula (Ie) in the presence of a reducing-catalyst (e.g., sponge nickel and Pd/Cu—C), hydrogen, and a reducing-step solvent (e.g., ethanol) to prepare a Compound of Formula (Ig) is provided below in Scheme IV.

-
- wherein:
- R1c, R2c, and R3c are each independently selected from: H, C1-C6 alkoxy, C1-C6 alkyl, C1-C6 haloalkyl, and halogen.
A representative Deprotecting-Step of a Compound of Formula (Ig), or a salt thereof, in the presence of a deprotecting-catalyst (e.g., Pd), hydrogen, and a deprotecting-step solvent (e.g., ethanol) to prepare (S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethan-1-amine (Compound
6A) or a salt thereof is provided below in Scheme V.

-
- wherein:
- R1c, R2c, and R3c are each independently selected from: H, C1-C6 alkoxy, C1-C6 alkyl, C1-C6 haloalkyl, and halogen.
A representative Cyclizing-Step of (S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethan-1-amine (Compound
6A) or a salt thereof, with 1-(2-chloro-4-methoxy-5-methylphenyl)-2-thiocyanatopropan-1-one (Compound
8A) or a tautomeric form thereof, in the presence of a cyclizing-step solvent (e.g., n-heptane) to prepare (S)-4-(2-chloro-4-methoxy-5-methylphenyl)-N-(2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl)-5-methylthiazol-2-amine (Compound
9A) or a salt thereof is provided below in Scheme VI.

A representative Alkylating-Step of (S)-4-(2-chloro-4-methoxy-5-methylphenyl)-N-(2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl)-5-methylthiazol-2-amine (Compound
9A) or a salt thereof, with a Compound of Formula (Ii), wherein LG is suitable leaving group (e.g., Br), in the presence of an alkylating-step solvent (e.g., methyl tert-butyl ether (MTBE), toluene, and mixtures thereof), a phase-transfer catalyst (e.g., tetra-n-butylammonium bromide (TBAB)), an alkylating-step base (e.g., potassium hydroxide), and water to prepare 4-(2-chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]-5-methyl-N-prop-2-ynyl-1,3-thiazol-2-amine (Compound 1) or a pharmaceutically acceptable salt thereof is provided below in Scheme VII.

One aspect of the present invention includes every combination of one or more process steps and intermediates related thereto used in the preparation of 4-(2-chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]-5-methyl-N-prop-2-ynyl-1,3-thiazol-2-amine (Compound 1), and/or pharmaceutically acceptable salts, and crystalline forms thereof, such as those processes exemplified by Schemes I, II, III, IV, V, VI, VII, and VII (supra) and Compounds contained therein.
8A was previously described in International Publication Number WO2010/125414 by Sanofi-Aventis.Example 1: Preparation of 4-(2-chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]-5-methyl-N-prop-2-ynyl-1,3-thiazol-2-amine (Compound 1), See FIG. 5 for a general synthetic schemeStep1A: Preparation of 2-Cyclopropyl-N-methoxy-N-methylacetamide (Compound 2A)

A suspension of 1,1′-carbonyldiimidazole (CDI, 152.6 kg, 1.01 eq.) in DCM (682 kg, 513 L, 7.3 w/w relative to 2-cyclopropylacetic acid) was treated with a solution of 2-cyclopropylacetic acid (Compound
1A, 93.6 kg, 1 eq.) in DCM (248 kg, 186 L, 2.65 w/w) over at least 1 h, keeping the temperature ≤25° C. and compensating for significant effervescence. The resulting mixture was stirred for 15 min at 22° C. and then N,O-dimethylhydroxylamine-HCl (93.6 kg, 1.03 eq.) was added in portions, keeping the temperature ≤30° C. Subsequently, triethylamine (46.4 kg, 0.49 eq.) was added to the stirring mixture at 20-25° C. The resulting mixture was stirred at 22° C. at least 1 h. The mixture was washed once with KHSO4 solution (0.24 M, 357.1 kg, 0.09 eq.), once with KHSO4 solution (0.40 M, 365.4 kg, 0.15 eq.), once with KHSO4 solution (0.80 M, 384.5 kg, 0.30 eq.), and once with NaHCO3 solution (0.60 M, 393.1 kg, 0.24 eq.). Residual DCM was removed by two put-and-takes of THF (166.6 kg, 1.78 w/w) and vacuum distillation (50-60° C., to minimum volume/until distillation stops) to provide Compound
2A. THF (333.2 kg. 3.56 w/w) was added and the yield was determined by correcting for the LOD and GC-FID purity of the sample (131.5 kg, 98.2% corrected). 1H-NMR (400 MHz, DMSO-d6) δ (ppm) −0.01-0.03 (m, 2H), 0.32-0.36 (m, 2H), 0.81-0.90 (br m, 1H), 2.18 (d, J=6.80 Hz, 2H), 2.97 (s, 3H), 3.53 (s, 3H). ESI-MS: 144.0 [M+H]+.Step 1B: Preparation of 2-Cyclopropyl-1-(3-fluoro-4-methylphenyl)ethan-1-one (Compound 3A)

Mg (turnings, 28.6 kg, 1.37 eq.) were suspended in THF (244.7 kg, 2.0 w/w) and DIBAL-H (1 M in n-heptane, 18.9 kg, 0.03 eq.) was added dropwise at 30° C. The resulting mixture was stirred at 30° C. for at least 10 min and then 4-bromo-2-fluoro-1-methylbenzene (neat, 21.1 kg, 0.13 eq.) was added over at least 30 min at 30-50° C. Subsequently, the mixture was treated with a solution of 4-bromo-2-fluoro-1-methylbenzene (191.6 kg, 1.18 eq.) in THF (414.5 kg, 3.37 w/w) at 30-50° C. over 3 h or less. The mixture was stirred at 30° C. for at least 1 h. The mixture was cooled to 12-18° C. and subsequently treated with 2-cyclopropyl-N-methoxy-N-methylacetamide (Compound
2A, 123.0 kg, 1 eq., 25.9% w/w solution in THF) over at least 1 h at 15-25° C. The resulting mixture was stirred at 20-25° C. for at least 1 h. The stirring mixture was then treated with aqueous HCl (3 M, 10.3% w/w, 668.9 kg, 2.24 eq.) at 10-25° C. and the resulting mixture was stirred at least 2 h until no Mg turnings were observed (check pH 3.0-3.5). The layers were separated, and the aqueous layer discarded. The organic layer was distilled at 55-65° C. and 400 mbar until distillation halts. Heptane (290.3 kg, 2.36 w/w) was added. The layers were separated, and the organic layer was washed once with NaHCO3 solution (0.63 M, 211.6 kg, 0.15 eq.) and once with NaCl solution (2.57 M, 213.0 kg, 0.55 eq.). The residual solvents were removed by vacuum distillation at 58-62° C. until distillation stops and then one put-and-take of toluene (275.5 kg, 2.24 w/w) at 107-117° C. until distillation stops. Toluene (275.5 kg, 2.24 w/w) was added and the yield was determined by correcting for the LOD and GC-FID purity of the sample (150.7 kg, 91.3% corrected). 1H NMR (400 MHz, DMSO-d6) β (ppm) 0.07-0.21 (m, 2H), 0.40-0.54 (m, 2H), 1.02 (ttt, J=8.16, 8.16, 6.68, 6.68, 4.86, 4.86 Hz, 1H), 2.30 (d, J=1.77 Hz, 3H), 2.91 (d, J=6.57 Hz, 2H), 7.44 (t, J=7.83 Hz, 1H), 7.57-7.78 (m, 2H). ESI-MS: 193.1 [M+H]+.Step 1C: Preparation of (S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)-N-(1-phenylethyl)ethan-1-imine (Compound 4A)

A mixture of 2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethan-1-one (Compound
3A, 150.7 kg, 1 eq., as a 27.6% w/w solution in toluene), (S)-(−)-1-phenylethylamine (112.9 kg, 1.19 eq.), and p-toluenesulfonic acid (7.4 kg, 0.05 eq.) was heated to reflux at 110-120° C. for 23-25 h in a reactor set up in a Dean-Stark configuration. The solvent was then removed at 125-135° C. under atmospheric pressure until distillation halts and a portion of toluene (275 kg, 2.24 w/w) was added to afford a suspension. The suspension was heated to reflux at 110-120° C. for 23-25 h. The mixture was cooled to 22° C. and washed twice with aqueous NH4Cl (10%, 301.2 kg, 0.72 eq.) and once with aqueous NaHCO3 (5%, 301.2 kg, 0.23 eq., check pH 8-9). The solvent was removed at 125-135° C. and atmospheric pressure to a target volume of 256 L, the mixture was filtered over CELITE®, and the cake was washed with toluene (25 kg). The resulting mixture containing Compound 4A was used directly in the next step without further isolation. The yield was determined by correcting for the LOD and GC-FID purity of the sample (208.4 kg, 90.0% corrected). EL-MS: 294.1 [M−H]*, 190.1 [M-C6H5CH(CH3)]+, 105.1 [C6H5CH(CH3)]+.Step 1D: Preparation of (S)-2-Cyclopropyl-1-(3-fluoro-4-methylphenyl)-N—((S)-1-phenylethyl)ethan-1-amine (Compound 5A) as the Hydrochloride Salt

Sponge nickel catalyst (144 kg, 0.70 w/w, shipped as a 50% w/w suspension in water) was added to a hydrogenation reactor, equipped with a dip tube capable of removing material from the top of the mass inside, minimizing the amount of water introduced. The supernatant was discarded, ethanol (329.3 kg, 1.58 w/w, anhydrous) was added, the suspension was stirred and then allowed to settle. This process was repeated four more times and the supernatant is checked; ≤1% H2O w/w (Karl Fisher (KF)). Compound 4A (208.4 kg, 1 eq., as a 62.6% solution in toluene) was added to the mixture in the hydrogenation reactor. Ethanol (389.4 kg, 1.86 w/w) was used to rinse the addition flask into the hydrogenation reactor. The hydrogenation reactor was pressurized/depressurized twice with nitrogen (2 bar), twice with hydrogen (5 bar), and then pressurized with hydrogen (9.8-10.2 bar). The resulting mixture was heated to 33-37° C. and stirred for 17-19 h. The system was depressurized/pressurized three times with nitrogen (1 bar). The suspension was filtered and washed three times with ethanol (total amount, 493.8 kg, 2.37 w/w). The filtrate was combined with HCl (concentrated, 83.4 kg, 1.07 eq.) and the resulting mixture stirred 25-35 min at 20-24° C. The mixture was concentrated by distillation at 78-80° C. and atmospheric pressure to remove water with a distillate target volume of 1167 L (5.6 L/kg based on imine Compound 4A) and the KF of the solution checked (≤1.5% H2O w/w). The mixture was stirred at 48-52° C. for 55-65 min, then 68-72° C. for 55-65 min, then cooled to 20-24° C. at a rate of 12° C./h and stirred for 25-35 min, then cooled to 0-4° C. at a rate of 10° C./h and stirred for 55-65 min. The suspension was filtered, the cake was washed twice with precooled ethanol (total amount, 329.2 kg, 1.58 w/w, 0° C.), and the collected solid was dried at 40° C. to afford Compound
5A as the HCl salt (156.5 kg, 66.4% uncorrected). 1H NMR (400 MHz, DMSO-d6) δ (ppm) −0.33–0.06 (m, 2H), 0.11-0.31 (m, 3H), 1.57 (d, J=6.57 Hz, 3H), 1.95 (br t, J=7.07 Hz, 2H), 2.26 (d, J=1.26 Hz, 3H), 3.68 (br d, J=7.83 Hz, 1H), 3.92 (br t, J=6.44 Hz, 1H), 6.98 (dd, J=7.71, 1.14 Hz, 1H), 7.28-7.36 (m, 2H), 7.37-7.50 (m, 5H). EST-MS: 298.2 m/z [M+H]+.Step 1E: Preparation of (S)-2-Cyclopropyl-1-(3-fluoro-4-methylphenyl)ethan-1-amine (Compound 6A) as the Hydrochloride Salt

5A (HCl salt, 156.5 kg, 1.00 eq.) and Pd/C (7.8 kg, 10% Pd basis) were added to an inerted hydrogenation reactor. The reactor was then pressurized/depressurized twice with nitrogen (2 bar) and then methanol (494.5 kg, 3.16 w/w) was added. The reactor was depressurized/pressurized three times with nitrogen (2 bar) then three times with hydrogen (5 bar), pressurized with hydrogen (9.8-10.2 bar), heated to 58-62° C. and stirred for 7-9 h. The reaction mixture was cooled to 20-24° C. The reactor was depressurized/pressurized three times with nitrogen (1 bar) and the suspension was filtered and washed three times with methanol (total amount, 492.9 kg, 3.15 w/w). The solution was concentrated at 63-67° C. and atmospheric pressure to a distillate target volume of 1408 L (9.0 L/kg Compound
6A), n-Heptane (1173.8 kg, 7.5 w/w) was added and the resulting mixture was heated to reflux at 65-80° C. and atmospheric pressure in Dean-Stark configuration to remove methanol. The suspension was cooled to 31-35° C. and filtered, the cake washed with n-heptane (147.1 kg, 0.94 w/w), and the solid dried at 40° C. to provide Compound
6A as the HCl salt (101.0 kg, 93.8% uncorrected, 99.6% ee). 1H NMR (400 MHz, DMSO-d6) δ (ppm) −0.12-0.14 (m, 2H), 0.26-0.42 (m, 2H), 0.44-0.55 (m, 1H), 1.70-1.83 (m, 2H), 2.23 (d, J=1.52 Hz, 3H), 4.24 (t, J=7.33 Hz, 1H), 7.22-7.29 (m, 1H), 7.29-7.36 (m, 1H), 7.40 (dd, J=10.99, 1.39 Hz, 1H). ESI-MS: 194.2 [M+H]+, 177.0 [M-NH2]+.Step 1F: Preparation of (S)-4-(2-chloro-4-methoxy-5-methylphenyl)-N-(2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl)-5-methylthiazol-2-amine (Compound 9A)

A mixture of n-heptane (146 kg), water (142 kg), Compound
6A (HCl salt, 57.4 kg), and aqueous sodium hydroxide (30% w/w, 41.0 kg) was stirred together. The layers were partitioned, and the aqueous layer removed. The organic layer was washed with water (170 kg) and the layers partitioned. The organic layer was set aside, n-Heptane (145 kg) and 1-(2-chloro-4-methoxy-5-methylphenyl)-2-thiocyanatopropan-1-one (Compound
8A, 66.1 kg, the preparation of Compound
8A has been previously described in International Publication Number WO2010/125414) were added to the reactor and heated to 85° C. The previously set aside organic layer containing the free base of Compound
6A was added at 84-85° C. to the reactor and rinsed with n-heptane (20 kg). The resulting mixture was stirred for 2 h at 83° C. Subsequently, the solvent was switched to methanol by four put-and-take additions/vacuum distillations of methanol (180 kg) at 55° C. with the target volume being 287 L remaining in the reactor. The suspension was cooled to 5° C. and water (570 kg) was added over 4 h at 5-10° C., with the first 60 kg added very slowly. The suspension was aged 2 h at 5° C. and then isolated by filtration, washed with a mixture of methanol/water (91/115 kg) and then a mixture of methanol/water (134/57 kg). The yellow solid was dried at 25° C. and 1 mbar for 17 h then 40° C. and 1 mbar for 22 h to afford Compound
9A (97.4 kg, 87.5% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm −0.01-0.14 (m, 2H), 0.29-0.42 (m, 2H), 0.61-0.73 (m, 1H), 1.47 (dt, J=13.83, 6.85 Hz, 1H), 1.76 (dt, J=13.89, 7.20 Hz, 1H), 2.00 (s, 3H), 2.11 (s, 3H), 2.19 (d, J=1.01 Hz, 3H), 3.82 (s, 3H), 4.54 (q, J=7.58 Hz, 1H), 7.00 (s, 1H), 7.06 (d, J=0.76 Hz, 1H), 7.08-7.14 (m, 2H), 7.18-7.23 (m, 1H), 7.89 (d, 1=8.08 Hz, 1H). ESI-MS: 445.3 m/z [M+H]+.Step 1G: Preparation of 4-(2-chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]-5-methyl-N-prop-2-ynyl-1,3-thiazol-2-amine (Compound 1)

A mixture of MTBE (279 kg), tetra-n-butylammonium bromide (10.5 kg), and Compound
9A (95.4 kg) were heated at 60° C. external temperature for 30 min and then cooled to 0° C. Aqueous potassium hydroxide (52.4% w/w, 364 kg) and propargyl bromide (39.4 kg as an 80% w/w solution in toluene, 1.19 eq.) were added at 0-5° C. The propargyl bromide additional funnel was washed with MTBE (25 kg) and the biphasic mixture was aged 14.5 h at 4-6° C. with vigorous stirring. Subsequently, water (191 kg) was added and the aqueous layer was discharged at 20° C. The organic layer was washed twice with water (382 kg) and once with aqueous acetic acid (5.26% w/w, 190 kg) at 20° C. The mixture is polish filtered, rinsed with ethanol (11 kg) and then the solvent switched to ethanol by 3 put-and-take additions/vacuum distillations of ethanol (300 kg) at 25-30° C. for the first cycle and then 35-40° C. with the target volume of each cycle being 250 L remaining in the reactor. Ethanol (164 kg) was added and the mixture heated at 60° C. external for 0.5 h before it was cooled to 25° C. in 1 h and seeded with authentic Form I (free base) of Compound 1 (0.340 kg) which can be prepared as described below in Example 2 and Example 3. The suspension was aged for 5 h, cooled to 0° C. in 2 h, aged 12 h, filtered, and washed twice with ethanol (24 kg each) pre-cooled to 0° C. The white solid was dried at 40° C. and 1 mbar for 19 h to yield 80.15 kg of Compound 1 (77.2% yield). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 0.14 (qt, J=8.59, 4.42 Hz, 2H), 0.29-0.48 (m, 2H), 0.61-0.82 (m, 1H), 1.89 (dt, J=14.08, 6.98 Hz, 1H), 2.07 (br d, J=7.83 Hz, 1H), 2.10 (s, 3H), 2.14 (s, 3H), 2.20 (d, J=1.01 Hz, 3H), 3.11 (t, J=2.27 Hz, 1H), 3.83 (s, 3H), 3.94-4.22 (m, 2H), 5.26 (t, J=7.58 Hz, 1H), 7.05 (s, 1H), 7.10-7.36 (m, 4H). ESI-MS: 483.2 m/z [M+H]+.Example 2: Preparation of 4-(2-chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]-5-methyl-N-prop-2-ynyl-1,3-thiazol-2-amine (Compound 1)
A mixture of MTBE (2 mL), tetra-n-butylammonium bromide (110 mg), and Compound
9A (1.003 g) at 0° C. was treated with aqueous potassium hydroxide (52.4% w/w, 1.80 mL, 2.73 g) and propargyl bromide (405 mg as an 80% w/w solution in toluene) maintaining the temperature at 0-5° C. The resulting biphasic mixture was aged 23 h at 4-6° C. Subsequently, water (2 mL) and MTBE (2 mL) were added and the aqueous layer was discharged. The organic layer was washed twice with water (4 mL) and once with aqueous acetic acid (5% w/w, 2 mL) at 20° C. Ethanol (4 mL) was added and then the solvent was switched to ethanol by 3 put-and-take additions/vacuum distillations of ethanol (6 mL) at 35-40° C. with the target volume of each cycle being 2 mL remaining in the vessel, except for the third cycle where the mixture was concentrated to dryness. Ethanol (4 mL) was added to the vessel and the mixture heated at 60° C. (external) for 0.5 h before it was cooled to 20° C. in 1 h and aged 18 h. The resulting suspension was cooled to 0° C., aged 6 h, filtered, and washed twice with ethanol (2 mL each) pre-cooled to 0° C. to afford a solid. The solid was dried at 40° C. under vacuum to afford Compound 1 (506 mg, 46% yield) as Form I. The 1H NMR and ESI-MS data matches that as described above in Example 1, Step 1G.Example 3: Preparation of 4-(2-chloro-4-methoxy-5-methylphenyl)-N-1(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]-5-methyl-N-prop-2-ynyl-1,3-thiazol-2-amine (Compound 1)
A mixture of MTBE (40 mL), tetra-n-butylammonium bromide (1.1 g), and Compound
9A (10.0 g) was heated to 45° C., aged for 10 min, then cooled to 0° C. The solution was treated with aqueous potassium hydroxide (52.4% w/w, 38.2 g) and propargyl bromide (3.36 g as an 80% w/w solution in toluene) maintaining the temperature at 0-5° C. The resulting biphasic mixture stirred vigorously for 16 h at 4-6° C. Subsequently, water (20 mL) was added and the aqueous layer was discharged. The organic layer was washed twice with water (40 mL) and once with aqueous acetic acid (5.2% w/w, 20 mL) at 20° C. The solvent was switched to ethanol by 4 put-and-take additions/vacuum distillations of ethanol (15 mL) at 35-40° C. with the target volume of each cycle being 15 mL remaining in the vessel. The solution was weighed to approximate the amount of ethanol remaining, and ethanol (26 mL) was added to the vessel to bring the total amount of ethanol to 40 mL. The solution was cooled to 4° C. and stirred for 45 min to afford a suspension. The suspension was heated to 38° C. in 15 min, aged 10 min, then cooled to 20° C. over 14 h. The suspension was cooled to 0° C., aged 1.5 h, filtered, and the solids washed twice with ethanol (7.5 mL each) pre-cooled to 0° C. The solid was dried at 40° C. under vacuum to afford Compound 1 (8.27 g, 76% yield) as Form I. The 1H NMR and ESL-MS data matches that as described above in Example 1, Step 1G.
The crystalline free base Compound
1, Form I was characterized by X-ray powder diffraction (XRPD) (FIG. 1
, Table 2) and DSC (FIG. 2
). The DSC indicated the crystalline Compound
1, Form I has an onset of melt (temperature) at about 83.7° C. (76.6 J/g). The Thermogravimetric Analysis (TGA) (FIG. 2
) of the crystalline free base exhibited substantially no weight loss (about 0.2%) from room temperature to ˜125° C. indicating Form I for the free base of Compound
1 is anhydrous.
Medical uses
Crinecerfont is indicated as adjunctive treatment to glucocorticoid replacement to control androgens in people four years of age and older with classic congenital adrenal hyperplasia.[1][2]
Adverse effects
The US Food and Drug Administration prescription label for crinecerfont has a warning for acute adrenal insufficiency or adrenal crisis.[2]
History
Crinecerfont’s approval is based on two randomized, double-blind, placebo-controlled trials in 182 adults and 103 children with classic congenital adrenal hyperplasia.[2] In the first trial, 122 adults received crinecerfont twice daily and 60 received placebo twice daily for 24 weeks.[2] After the first four weeks of the trial, the glucocorticoid dose was reduced to replacement levels, then adjusted based on levels of androstenedione, an androgen hormone.[2] The primary measure of efficacy was the change from baseline in the total glucocorticoid daily dose while maintaining androstenedione control at the end of the trial.[2] The group that received crinecerfont reduced their daily glucocorticoid dose by 27% while maintaining control of androstenedione levels, compared to a 10% daily glucocorticoid dose reduction in the group that received placebo.[2]
In the second trial, 69 children received crinecerfont twice daily and 34 received placebo twice daily for 28 weeks.[2] The primary measure of efficacy was the change from baseline in serum androstenedione at week four.[2] The group that received crinecerfont experienced a statistically significant reduction from baseline in serum androstenedione, compared to an average increase from baseline in the placebo group.[2] At the end of the trial, children assigned to crinecerfont were able to reduce their daily glucocorticoid dose by 18% while maintaining control of androstenedione levels compared to an almost 6% daily glucocorticoid dose increase in children assigned to placebo.[2]
The US Food and Drug Administration (FDA) granted the application for crinecerfont fast track, breakthrough therapy, orphan drug, and priority review designations.[2] The FDA granted the approval of Crenessity to Neurocrine Biosciences, Inc.[2]
Society and culture
Legal status
Crinecerfont was approved for medical use in the United States in December 2024.[1][2][5]
Names
Crinecerfont is the international nonproprietary name.[6]
Crinecerfont is sold under the brand name Crenessity.[1]
References
- ^ Jump up to:a b c d e f g “Crenessity- crinecerfont; capsule Crenessity- crinecerfont solution”. DailyMed. 1 December 2024. Retrieved 25 January 2025.
- ^ Jump up to:a b c d e f g h i j k l m n o p q “FDA Approves New Treatment for Congenital Adrenal Hyperplasia”. U.S. Food and Drug Administration (FDA) (Press release). 1 October 2024. Retrieved 16 December 2024. This article incorporates text from this source, which is in the public domain.
- ^ “Novel Drug Approvals for 2024”. U.S. Food and Drug Administration (FDA). 1 October 2024. Retrieved 20 December 2024.
- ^ New Drug Therapy Approvals 2024 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2025. Archived from the original on 21 January 2025. Retrieved 21 January 2025.
- ^ “Neurocrine Biosciences Announces FDA Approval of Crenessity (crinecerfont), a First-in-Class Treatment for Children and Adults With Classic Congenital Adrenal Hyperplasia” (Press release). Neurocrine Biosciences. 13 December 2024. Retrieved 16 December 2024 – via PR Newswire.
- ^ World Health Organization (2019). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 82”. WHO Drug Information. 33 (3). hdl:10665/330879.
Further reading
- Auchus, Richard; Chan, Jean; Farber, Robert; Fechner, Patricia; Giri, Nagdeep; Nokoff, Natalie; et al. (1 November 2022). “OR18-4 Crinecerfont (NBI-74788), a Novel CRF1 Receptor Antagonist, Lowers Adrenal Androgens and Precursors in Adolescents with Classic Congenital Adrenal Hyperplasia”. Journal of the Endocrine Society. 6 (Supplement_1): A618. doi:10.1210/jendso/bvac150.1281. PMC 9625506.
- Auchus, Richard J; Sarafoglou, Kyriakie; Fechner, Patricia Y; Vogiatzi, Maria; Giri, Nagdeep; Roberts, Eiry; et al. (8 May 2020). “OR25-03 The Effects of Crinecerfont (NBI-74788), a Novel CRF1 Receptor Antagonist, on Adrenal Androgens and Precursors in Patients with Classic Congenital Adrenal Hyperplasia: Results from A Multiple-Dose Phase 2 Study”. Journal of the Endocrine Society. 4 (Supplement_1): OR25-03. doi:10.1210/jendso/bvaa046.221. PMC 7209526.
- Auchus, Richard J; Sarafoglou, Kyriakie; Fechner, Patricia Y; Vogiatzi, Maria G; Imel, Erik A; Davis, Shanlee M; et al. (17 February 2022). “Crinecerfont Lowers Elevated Hormone Markers in Adults With 21-Hydroxylase Deficiency Congenital Adrenal Hyperplasia”. The Journal of Clinical Endocrinology & Metabolism. 107 (3): 801–812. doi:10.1210/clinem/dgab749. PMC 8851935. PMID 34653252.
- Newfield, Ron S; Sarafoglou, Kyriakie; Fechner, Patricia Y; Nokoff, Natalie J; Auchus, Richard J; Vogiatzi, Maria G; et al. (18 October 2023). “Crinecerfont, a CRF1 Receptor Antagonist, Lowers Adrenal Androgens in Adolescents With Congenital Adrenal Hyperplasia”. The Journal of Clinical Endocrinology & Metabolism. 108 (11): 2871–2878. doi:10.1210/clinem/dgad270. PMC 10583973. PMID 37216921.
External links
- “Crinecerfont (Code C174708)”. NCI Thesaurus.
- Clinical trial number NCT03525886 for “Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of NBI-74788 in Adults With Congenital Adrenal Hyperplasia” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Crenessity |
| Other names | SSR-125543, NBI-74788 |
| AHFS/Drugs.com | Crenessity |
| License data | US DailyMed: Crinecerfont |
| Routes of administration | By mouth |
| Drug class | Corticotropin-releasing factor type 1 receptor antagonist |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 752253-39-7 |
| PubChem CID | 5282340 |
| DrugBank | DB18518 |
| ChemSpider | 4445507 |
| UNII | MFT24BX55I |
| KEGG | D12366 |
| ChEBI | CHEBI:34969 |
| ChEMBL | ChEMBL291657 |
| CompTox Dashboard (EPA) | DTXSID10996687 |
| Chemical and physical data | |
| Formula | C27H28ClFN2OS |
| Molar mass | 483.04 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
- Prete A, Auchus RJ, Ross RJ: Clinical advances in the pharmacotherapy of congenital adrenal hyperplasia. Eur J Endocrinol. 2021 Nov 30;186(1):R1-R14. doi: 10.1530/EJE-21-0794. [Article]
- Yogi A, Kashimada K: Current and future perspectives on clinical management of classic 21-hydroxylase deficiency. Endocr J. 2023 Oct 30;70(10):945-957. doi: 10.1507/endocrj.EJ23-0075. Epub 2023 Jun 29. [Article]
- FDA Approved Drug Products: Crenessity (crinecerfont) capsules/solution for oral administration (December 2024) [Link]
- FDA News Release: FDA Approves New Treatment for Congenital Adrenal Hyperplasia [Link]
///////Crinecerfont, Crenessity, FDA 2024, APPROVALS 2024, 752253-39-7, SSR125543, SSR 125543, SSR-125543, WHO 10958, 06-RORI, NBI-74788, ORPHAN DRUG

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}