
Home » FDA 2017 (Page 5)
Category Archives: FDA 2017
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
FDA approves new eczema drug Dupixent (dupilumab)
The U.S. Food and Drug Administration today approved Dupixent (dupilumab) injection to treat adults with moderate-to-severe eczema (atopic dermatitis). Dupixent is intended for patients whose eczema is not controlled adequately by topical therapies, or those for whom topical therapies are not advisable. Dupixent can be used with or without topical corticosteroids.
“FDA’s approval of Dupixent demonstrates our commitment to approving new and innovative therapies for patients with skin disease,” said Julie Beitz, M.D., director of the Office of Drug Evaluation III in the FDA’s Center for Drug Evaluation and Research. “Eczema can cause significant skin irritation and discomfort for patients, so it is important to have a variety of treatment options available to patients, including those patients whose disease is not controlled by topical therapies.”
Atopic dermatitis, a chronic inflammatory skin disease, is often referred to as “eczema,” which is a general term for several types of inflammation of the skin. Atopic dermatitis is the most common of the many types of eczema; onset typically begins in childhood and can last through adulthood. The cause of atopic dermatitis is a combination of genetic, immune and environmental factors. In atopic dermatitis, the skin develops red, scaly and crusted bumps, which are extremely itchy. Scratching leads to swelling, cracking, “weeping” clear fluid, and finally, coarsening and thickening of the skin.
Dupixent is administered as an injection under the skin. Dupixent’s active ingredient is an antibody (dupilumab) that binds to a protein [interleukin-4 (IL-4) receptor alpha subunit (IL-4Ra)], that causes inflammation. By binding to this protein, Dupixent is able to inhibit the inflammatory response that plays a role in the development of atopic dermatitis.
The safety and efficacy of Dupixent were established in three placebo-controlled clinical trials with a total of 2,119 adult participants with moderate-to-severe atopic dermatitis not adequately controlled by topical medication(s). Overall, participants who received Dupixent achieved greater response, defined as clear or almost clear skin, and experienced a reduction in itch after 16 weeks of treatment.
Dupixent can cause side effects such as serious allergic reactions and eye problems, such as pink eye (conjunctivitis) and inflammation of the cornea (keratitis). If patients experience new or worsening eye symptoms such as redness, itching, pain or visual changes, they should consult a health care provider. The most common side effects include injection site reactions; cold sores in the mouth or on the lips; and eye and eyelid inflammation, including redness, swelling and itching.
The safety and efficacy of Dupixent have not been established in the treatment of asthma. Patients who also have asthma should not adjust or stop their asthma treatment without talking to their physicians.
The FDA granted the application for Dupixent Priority Review and Breakthrough Therapy designation.
The FDA granted the approval of Dupixent to Regeneron Pharmaceuticals, Inc.
FDA approves first treatment Bavencio (avelumab)for rare form of skin cancer


March 23, 2017
Release
The U.S. Food and Drug Administration today granted accelerated approval to Bavencio (avelumab) for the treatment of adults and pediatric patients 12 years and older with metastatic Merkel cell carcinoma (MCC), including those who have not received prior chemotherapy. This is the first FDA-approved treatment for metastatic MCC, a rare, aggressive form of skin cancer.
“While skin cancer is one of the most common cancers, patients with a rare form called Merkel cell cancer have not had an approved treatment option until now,” said Richard Pazdur, M.D., acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research and director of the FDA’s Oncology Center of Excellence. “The scientific community continues to make advances targeting the body’s immune system mechanisms for the treatment of various types of cancer. These advancements are leading to new therapies—even in rare forms of cancer where treatment options are limited or non-existent.”
According to the National Cancer Institute, approximately 1,600 people in the United States are diagnosed with MCC every year. While the majority of patients present with localized tumors that can be treated with surgical resection, approximately half of all patients will experience recurrence, and more than 30 percent will eventually develop metastatic disease. In patients with metastatic MCC, the cancer has spread beyond the skin into other parts of the body.
Bavencio targets the PD-1/PD-L1 pathway (proteins found on the body’s immune cells and some cancer cells). By blocking these interactions, Bavencio may help the body’s immune system attack cancer cells.
Bavencio received an Accelerated Approval, which enables the FDA to approve drugs for serious conditions to fill an unmet medical need using clinical trial data that is thought to predict a clinical benefit to patients. Further clinical trials are required to confirm Bavencio’s clinical benefit and the sponsor is currently conducting these studies.
Today’s approval of Bavencio was based on data from a single-arm trial of 88 patients with metastatic MCC who had been previously treated with at least one prior chemotherapy regimen. The trial measured the percentage of patients who experienced complete or partial shrinkage of their tumors (overall response rate) and, for patients with a response, the length of time the tumor was controlled (duration of response). Of the 88 patients who received Bavencio in the trial, 33 percent experienced complete or partial shrinkage of their tumors. The response lasted for more than six months in 86 percent of responding patients and more than 12 months in 45 percent of responding patients.
Common side effects of Bavencio include fatigue, musculoskeletal pain, diarrhea, nausea, infusion-related reactions, rash, decreased appetite and swelling of the limbs (peripheral edema). The most common serious risks of Bavencio are immune-mediated, where the body’s immune system attacks healthy cells or organs, such as the lungs (pneumonitis), liver (hepatitis), colon (colitis), hormone-producing glands (endocrinopathies) and kidneys (nephritis). In addition, there is a risk of serious infusion-related reactions. Patients who experience severe or life-threatening infusion-related reactions should stop using Bavencio. Women who are pregnant or breastfeeding should not take Bavencio because it may cause harm to a developing fetus or a newborn baby.
The FDA granted this application Priority Review and Breakthrough Therapydesignation. Bavencio also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted accelerated approval of Bavencio to EMD Serono Inc.

| Monoclonal antibody | |
|---|---|
| Type | ? |
| Source | Human |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| ChemSpider |
|
| UNII | |
| KEGG | |
Avelumab (MSB0010718C) is a fully human monoclonal PD-L1antibody of isotypeIgG1, currently in development by Merck KGaA, Darmstadt, Germany & Pfizer for use in immunotherapy, especially for treatment of Non-small-cell lung carcinoma (NSCLC) .[1]
Mechanism of action
Avelumab binds to the PD ligand 1 and therefore inhibits binding to its receptor programmed cell death 1 (PD-1). Formation of a PD-1/PD-L1 receptor/ligand complex leads to inhibition of CD8+ T cells, and therefore inhibition of an immune reaction. Immunotherapy aims at ceasing this immune blockage by blocking those receptor ligand pairs. In the case of avelumab, the formation of PD-1/PDL1 ligand pairs is blocked and CD8+ T cell immune response should be increased. PD-1 itself has also been a target for immunotherapy.[2] Therefore, avelumab belongs to the group of Immune checkpoint blockade cancer therapies.
Clinical trials
As of May 2015, according to Merck KGaA, Darmstadt, Germany & Pfizer, avelumab has been in Phase Iclinical trials for bladder cancer, gastric cancer, head and neck cancer, mesothelioma, NSCLC, ovarian cancer and renal cancer. For Merkel-cell carcinoma, Phase II has been reached and for NSCLC there is also a study already in Phase III.[1]
Merkel-cell carcinoma
On March 23, 2017, the U.S. Food and Drug Administration granted accelerated approval to avelumab (BAVENCIO, EMD Serono, Inc.) for the treatment of adults and pediatric patients 12 years and older with metastatic Merkel cell carcinoma (MCC).
Approval was based on data from an open-label, single-arm, multi-center clinical trial (JAVELIN Merkel 200 trial) demonstrating a clinically meaningful and durable overall response rate (ORR). All patients had histologically confirmed metastatic MCC with disease progression on or after chemotherapy administered for metastatic disease.
ORR was assessed by an independent review committee according to Response Evaluation Criteria in Solid Tumors (RECIST) 1.1. The ORR was 33% (95% confidence interval [CI]: 23.3, 43.8), with 11% complete and 22% partial response rates. Among the 29 responding patients, the response duration ranged from 2.8 to 23.3+ months with 86% of responses durable for 6 months or longer. Responses were observed in patients regardless of PD-L1 tumor expression or presence of Merkel cell polyomavirus.
Safety data were evaluated in 1738 patients who received avelumab, 10 mg/kg, every 2 weeks. The most common serious adverse reactions to avelumab are immune-mediated adverse reactions (pneumonitis, hepatitis, colitis, adrenal insufficiency, hypo- and hyperthyroidism, diabetes mellitus, and nephritis) and life-threatening infusion reactions. Among the 88 patients enrolled in the JAVELIN Merkel 200 trial, the most common adverse reactions were fatigue, musculoskeletal pain, diarrhea, nausea, infusion-related reaction, rash, decreased appetite, and peripheral edema. Serious adverse reactions that occurred in more than one patient in the trial were acute kidney injury, anemia, abdominal pain, ileus, asthenia, and cellulitis.
The recommended dose and schedule of avelumab is 10 mg/kg as an intravenous infusion over 60 minutes every 2 weeks. All patients should receive premedication with an antihistamine and acetaminophen prior to the first four infusions of avelumab.
Full prescribing information for avelumab is available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761049s000lbl.pdf
References
- ^ Jump up to:a b Merck-Pfizer Alliance. “Merck-Pfizer Alliance Avelumab Fact Sheet” (PDF). Retrieved 2 December 2015.
- Jump up^ Hamid, O; Robert, C; Daud, A; Hodi, F. S.; Hwu, W. J.; Kefford, R; Wolchok, J. D.; Hersey, P; Joseph, R. W.; Weber, J. S.; Dronca, R; Gangadhar, T. C.; Patnaik, A; Zarour, H; Joshua, A. M.; Gergich, K; Elassaiss-Schaap, J; Algazi, A; Mateus, C; Boasberg, P; Tumeh, P. C.; Chmielowski, B; Ebbinghaus, S. W.; Li, X. N.; Kang, S. P.; Ribas, A (2013). “Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma”. New England Journal of Medicine. 369 (2): 134–44. doi:10.1056/NEJMoa1305133. PMC 4126516
 . PMID 23724846.
. PMID 23724846.
//////////fda 2017, Bavencio, avelumab, EMD Serono Inc., Priority Review, Breakthrough Therapy designation. Orphan Drug designation, skin cancer
UPDATE ON EMA
| Bavencio : EPAR – Summary for the public | EN = English | 13/10/2017 |
Product details
| Name | Bavencio |
|---|---|
| Agency product number | EMEA/H/C/004338 |
| Active substance | avelumab |
| International non-proprietary name(INN) or common name | avelumab |
| Therapeutic area | Neuroendocrine Tumors |
| Anatomical therapeutic chemical (ATC) code | L01XC31 |
| Additional monitoring | This medicine is under additional monitoring. This means that it is being monitored even more intensively than other medicines. For more information, see medicines under additional monitoring. |
| Treatment of rare diseases | This medicine has an “orphan designation” which means that it is used to treat life-threatening or chronically debilitating conditions that affect no more than five in 10,000 people in the European Union, or are medicines which, for economic reasons, would be unlikely to be developed without incentives. |
| Conditional Approval | Sometimes, the CHMP recommends that a medicine be given ‘conditional approval’. This happens when the Committee has based its positive opinion on data which, while not yet comprehensive, indicate that the medicine’s benefits outweigh its risks.
The company is given obligations to fulfil, such as the performance of further studies. The approval is renewed on a yearly basis until all obligations have been fulfilled, and is then converted from a conditional approval into a normal approval. Conditional approvals can only be granted for medicines that satisfy an ‘unmet medical need’, meaning the medicine is intended to be used for a disease or condition for which no treatment is readily available, and it is therefore important that patients have early access to the medicine concerned. |
Publication details
| Marketing-authorisation holder | Merck Serono Europe Limited |
|---|---|
| Revision | 1 |
| Date of issue of marketing authorisation valid throughout the European Union | 18/09/2017 |
Contact address:
Merck Serono Europe Limited
56 Marsh Wall
London E14 9TP
United Kingdom
FDA approves drug Xadago (Safinamide, сафинамид , سافيناميد , 沙非胺 , ) to treat Parkinson’s disease
Safinamide
- Molecular Formula C17H19FN2O2
- Average mass 302.343 Da
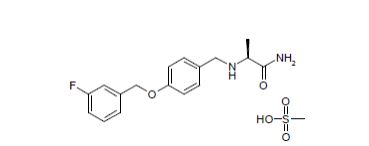
(+)-(S)-2-[[p-[(m-fluorobenzyl)oxy]benzyl]amino]propionamide monomethanesulfonate
Propanamide, 2-[[[4-[(3-fluorophenyl)methoxy]phenyl]methyl]amino]-, (2S)-, methanesulfonate
| Molecular Weight | 398.45 |
| Formula | C17H19FN2O2 ● CH4O3S |
CAS 202825-46-5 (Safinamide Mesylate)
Safinamide is a white to off-white, non-hygroscopic crystalline solid. It shows pH dependent solubility in aqueous buffers due to the secondary amine moiety, being soluble at acidic pH and practically insoluble at neutral pH.
It is freely soluble in de-ionized water, methanol and DMSO but practically insoluble in non-polar organic solvents.
Safinamide is chiral and possesses a single stereogenic centre.
Three crystalline forms are known. The anhydrous form selected for commercialisation is the most thermodynamically stable form, whilst the others are either not physiologically relevant or have very similar dissolution profiles. SOURCE EMA
Safinamide methanesulfonate was approved by European Medicine Agency (EMA) on Feb 22, 2015. It was developed by Newron and Zambon, then marketed as Xadago® by Zambon in EU.
FDA approved March 21, 2017,
- Chemistry Review(s) (PDF) for correct structure
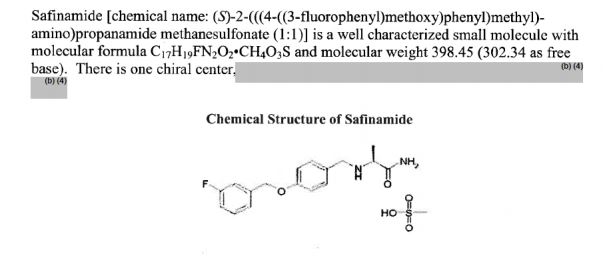
- Chemistry Review(s) (PDF) for correct structure
Safinamide is a unique molecule with a novel dual mechanism of action based on the enhancement of the dopaminergic function (through potent reversible inhibition of MAO-B and of dopamine uptake) and inhibition of the excessive release of glutamate. It is indicated for the treatment of Parkinson’s disease (PD).
Xadago® is available as film-coated tablet for oral use, containing Eq. 50 mg/100 mg of free Safinamide. The recommended dose is 50 mg or 100 mg once daily.
March 21, 2017, Release
The U.S. Food and Drug Administration today approved Xadago (safinamide) tablets as an add-on treatment for patients with Parkinson’s disease who are currently taking levodopa/carbidopa and experiencing “off” episodes. An “off” episode is a time when a patient’s medications are not working well, causing an increase in Parkinson’s symptoms, such as tremor and difficulty walking.
“Parkinson’s is a relentless disease without a cure,” said Eric Bastings, M.D., deputy director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research. “We are committed to helping make additional treatments for Parkinson’s disease available to patients.”
An estimated 50,000 Americans are diagnosed with Parkinson’s disease each year, according to the National Institutes of Health, and about one million Americans have the condition. The neurological disorder typically occurs in people over age 60, though it can occur earlier, when cells in the brain that produce a chemical called dopamine become impaired or die. Dopamine helps transmit signals between the areas of the brain that produce smooth, purposeful movement – such as eating, writing, and shaving. Early symptoms of the disease are subtle and occur gradually. In some people, Parkinson’s disease progresses more quickly than in others.
The efficacy of Xadago in treating Parkinson’s disease was shown in a clinical trial of 645 participants who were also taking levodopa and were experiencing “off” time. Those receiving Xadago experienced more beneficial “on” time, a time when Parkinson’s symptoms are reduced, without troublesome uncontrolled involuntary movement (dyskinesia), compared to those receiving a placebo. The increase in “on” time was accompanied by a reduction in “off” time and better scores on a measure of motor function assessed during “on” time than before treatment.
In another clinical trial of 549 participants, the participants adding Xadago to their levodopa treatment had more “on” time without troublesome uncontrolled involuntary movement compared to those taking a placebo, and also had better scores on a measure of motor function assessed during “on” time than before treatment.
Certain patients should not take Xadago. These include patients who have severe liver problems, or who take a medicine used to treat a cough or cold called dextromethorphan. It also should not be taken by patients who take another medicine called a monoamine oxidase inhibitor (MAOI) because it may cause a sudden severe increase in blood pressure, or by those who take an opioid drug, St. John’s wort, certain antidepressants (such as serotonin-norepinephrine reuptake inhibitors, tricyclics, tetracyclics, and triazolopyridines), or cyclobenzaprine, because it may cause a life-threatening reaction called serotonin syndrome.
The most common adverse reactions observed in patients taking Xadago were uncontrolled involuntary movement, falls, nausea, and trouble sleeping or falling asleep (insomnia).
Serious, but less common, risks include the following: exacerbated high blood pressure (hypertension); serotonin syndrome when used with MAOIs, antidepressants, or opioid drugs; falling asleep during activities of daily living; hallucinations and psychotic behavior; problems with impulse control/compulsive behaviors; withdrawal-emergent hyperpyrexia (fever) and confusion; and retinal pathology.
The FDA granted approval of Xadago to Newron Pharmaceuticals.
Safinamide (INN; brand name Xadago) is a drug indicated for the treatment of Parkinson’s disease with monoamine oxidase B inhibiting and other methods of action.[2] It was approved in Europe in February 2015,[3] and in the United States on March 21, 2017[4]. It has also been tested for the use in patients with restless legs syndrome (RLS), but no study results have been published.

Medical uses
Safinamide has been approved by the European Medicines Agency for the treatment of adult patients with idiopathic Parkinson’s disease as add-on therapy to a stable dose of levodopa (L-dopa) alone or in combination with other Parkinson drugs in patients with mid-to-late-stage fluctuating disease.[5]
Contraindications
Safinamide is contraindicated in patients with severe liver impairment, with albinism, retinitis pigmentosa, severe diabetic neuropathy, uveitis and other disorders of the retina. Combination with other monoamine oxidase (MAO) inhibitors and pethidine is also contraindicated.[6]
Adverse effects
Common adverse events in clinical trials (in more than 1% of patients) included nausea, dizziness, tiredness, sleeplessness, orthostatic hypotension (low blood pressure), and headache. There was no significant difference in the occurrence of these effects between safinamide and placebo treated patients.[6][7]
In experiments with rats (but not in those with monkeys), retinopathies have been observed.[1][8]
Overdose
Expected overdose effects are hypertension (high blood pressure), orthostatic hypotension, hallucinations, psychomotor agitation, nausea, vomiting, and dyskinesia. In studies, a singe patient was suspected to have overdosed for a month; symptoms were confusion, drowsiness and mydriasis (dilation of the pupils) and subsided completely after the drug was discontinued. No specific antidote is available.[6]
Interactions
As a MAO inhibitor, safinamide can theoretically cause hypertensive crises, serotonin syndrome and other severe side effects when combined with other MAO inhibitors or with drugs that are known to interact with MAO inhibitors, such as pethidine, dextromethorphan, selective serotonin reuptake inhibitors (SSRIs), serotonin–noradrenaline reuptake inhibitors (SNRIs), tricyclic and tetracyclic antidepressants. An interaction with tyramine, a substance found in various foods, could be expected by the same reasoning but has been excluded in studies.[6]
Another theoretical interaction is with drugs with affinity to the transporter protein ABCG2 (also known as BCRP), such as pitavastatin, pravastatin, ciprofloxacin, methotrexat, and diclofenac; a study with the latter has shown no clinical relevance.[9] A study testing possible interactions with amidase inhibitors is part of the post-authorisation development plan.[1] There are no relevant interactions related to cytochrome P450 (CYP) liver enzymes, although one inactivation pathway of safinamide seems to be mediated by CYP3A4.[6]
Pharmacology
Mechanisms of action
Like the older antiparkinson drugs selegiline and rasagiline, safinamide is a selective monoamine oxidase B inhibitor, reducing degradation of dopamine; in contrast to the other two, its action is reversible. Safinamide also inhibits glutamate release[7][10] and dopamine reuptake.[11] Additionally, it blocks sodium and calcium channels,[10][12] the relevance of which for its antiparkinson action is however unknown.[6]
Pharmacokinetics
Safinamide is absorbed quickly and nearly completely from the gut and reaches highest blood plasma concentrations after 1.8 to 2.8 hours. There is no relevant first-pass metabolism; total bioavailability is 95%. The substance is bound to plasma proteins to 88–90%.[6]
The metabolism is not well understood. The principal step is mediated by amidases which have not been identified, and produces safinamide acid (NW-1153). Other relevant metabolites are O-debenzylated safinamide (NW-1199),[9] the N-dealkylated amine which is then oxidized to a carboxylic acid (NW-1689), and the glucuronide of the latter.[6][13] In tests with liver microsomes, dealkylation seemed to be mediated by CYP3A4, but other CYP enzymes appear to be involved as well. Safinamide acid binds to the organic anion transporter 3 (OAT3), but this has probably no clinical relevance. Safinamide itself transiently binds to ABCG2. No other transporter affinities have been found in preliminary studies.[6]
Safinamide is eliminated, mainly (>90%) in form of its metabolites, via the kidney, with an elimination half-life of 20 to 30 hours. Only 1.5% are found in the stool.[6]

Metabolism pathways of safinamide.[9][13] Enzymes: CYP = cytochrome P450, MAO-A = monoamine oxidase A, ALDH = aldehyde dehydrogenases, UGT = UDP-glucuronosyltransferases. Gluc = acyl glucuronide.
History
The compound was originally discovered at Farmitalia-Carlo Erba, which was acquired by Pharmacia in 1993. In 1995, Pharmacia merged with Upjohn. Safinamide was first disclosed in 1998.[14] In the course of a major restructuring in the same year, all rights for safinamide were transferred to the newly formed company Newron Pharmaceuticals, which developed the drug until it was sold to Merck KGaA in 2006.[15]
In 2007, a Phase III clinical trial was started, scheduled to run until 2011.[16] In October 2011 Merck, now Merck-Serono, announced that they would give all rights to develop the compound back to Newron because they wanted to prioritise other projects and had corrected their estimates for safinamide’s market potential downwards.[17]
The US Food and Drug Administration (FDA) refused to file Newron’s application in 2014 on formal grounds.[18] Newron re-applied in December 2014.[19] In spring 2015, the European Medicines Agency (EMA) approved the drug. Safinamide is the first antiparkinson medication to be approved for ten years.[8]
Research
Potential additional uses might be restless legs syndrome (RLS) and epilepsy.[20] They were being tested in Phase II trials in 2008, but no results are available.

(+)-(S)-2-[[p-[(m-fluorobenzyl)oxy]benzyl]amino]propionamide monomethanesulfonate
Propanamide, 2-[[[4-[(3-fluorophenyl)methoxy]phenyl]methyl]amino]-, (2S)-, methanesulfonate
| Molecular Weight | 398.45 |
| Formula | C17H19FN2O2 ● CH4O3S |
CAS 202825-46-5 (Safinamide Mesylate)
Safinamide is a white to off-white, non-hygroscopic crystalline solid. It shows pH dependent solubility in aqueous buffers due to the secondary amine moiety, being soluble at acidic pH and practically insoluble at neutral pH.
It is freely soluble in de-ionized water, methanol and DMSO but practically insoluble in non-polar organic solvents.
Safinamide is chiral and possesses a single stereogenic centre.
Three crystalline forms are known. The anhydrous form selected for commercialisation is the most thermodynamically stable form, whilst the others are either not physiologically relevant or have very similar dissolution profiles.SOURCE EMA
Safinamide methanesulfonate was approved by European Medicine Agency (EMA) on Feb 22, 2015. It was developed by Newron and Zambon, then marketed as Xadago® by Zambon in EU.
FDA approved March 21, 2017
Safinamide is a unique molecule with a novel dual mechanism of action based on the enhancement of the dopaminergic function (through potent reversible inhibition of MAO-B and of dopamine uptake) and inhibition of the excessive release of glutamate. It is indicated for the treatment of Parkinson’s disease (PD).
Xadago® is available as film-coated tablet for oral use, containing Eq. 50 mg/100 mg of free Safinamide. The recommended dose is 50 mg or 100 mg once daily.
SYNTHESIS
 |
|
| Safinamide has been obtained by reductocondensation of 4-(3-fluorobenzyloxy)benzaldehyde (I) with L-alaninamide (II) by means of sodium cyanoborohydride in methanol.EP 0400495; EP 0426816; JP 1992500215; US 5236957; US 5391577; US 5502079; WO 9014334 |
CLIP
http://pubs.rsc.org/en/content/articlehtml/2016/sc/c6sc00197a

Scheme 2 Synthesis and isolation of [18F]safinamide, [18F]FMT, and [18F]mFBG.
PATENT
Safinamide (NW- 1015, FCE-26743A, PNU- 151774E) is a sodium channel blocker, a calcium channel modulator, a monoamino oxidase B (MAO-B) inhibitor, a glutamate release inhibitor and a dopamine metabolism modulator. Safinamide is useful in the treatment of CNS disorders, in particular of epilepsy, Parkinson’s disease, Alzheimer’s disease, depression, restless legs syndrome and migraine (WO 90/ 14334, WO 2004/089353, WO 2005/ 102300 and WO 2004/062655). Ralfinamide (NW- 1029, FCE-26742A, PNU-0154339E) is a sodium channel blocker useful in the treatment of pain conditions, including chronic pain and neuropathic pain, migraine, bipolar disorders, depressions, cardiovascular, inflammatory, urogenital, metabolic and gastrointestinal disorders (WO 99/35125, WO 03/020273, WO 2004/062655, WO 2005/018627, WO 2005/070405, WO 2005/ 102300).
In particular, safinamide is specifically described in WO 90/ 14334. Safinamide, its R-enantiomer, their racemic mixture and their salts with pharmaceutically acceptable acids and the use thereof for the preparation of pharmaceutical compositions active as anti-epileptic, anti-Parkinson, neuroprotective, antidepressant, antispastic and/or hypnotic agents are specifically claimed in WO 90/ 14334. Ralfinamide is specifically described in WO 90/ 14334. Ralfinamide, its R- enantiomer, their racemic mixture and their salts with pharmaceutically acceptable acids and their use thereof for the preparation of pharmaceutical compositions active as anti-epileptic, anti-Parkinson, neuroprotective, antidepressant, antispastic and/or hypnotic agent are comprised by the claims of WO 90/ 14334.
Moreover, the use as analgesics of safinamide, ralfinamide, the respective R-enantiomers, the respective racemic mixtures and their salts with pharmaceutically acceptable acids is claimed in WO 99/035125. WO 2006/027052 A2 specifically discloses and claims the use of the single R-enantiomer of ralfinamide i.e., (R)-2-[4-(2- fluorobenzyloxy)benzylamino]propanamide (I’b), and its salts with pharmaceutically acceptable acids as a selective sodium and calcium channel modulator for the selective treatment of pathological affections wherein sodium or calcium channel mechanism(s) play(s) a pathological role, including pain, migraine, inflammatory processes affecting all body systems, disorders affecting skin and related tissue, disorders of the respiratory system, disorders of the immune and endocrinological systems, gastrointestinal, and urogenital disorders, wherein the therapeutical activity of said compound is substantially free from any MAO inhibitory side effect or exhibits significantly reduced MAO inhibitory side effect.
It has now been discovered that the large scale preparations of safinamide and ralfinamide according to the methods described in the prior art, contain two undesired impurities, i.e., respectively, (S)-2-[3-(3- fluorobenzyl)-4-(3-fluorobenzyloxy)-benzylamino]propanamide (Ha) and (S)- 2-[3-(2-fluorobenzyl)-4-(2-fluorobenzyloxy)-benzylamino]propanamide (lib), and their salt, in particular the respective methanesulfonates (lie) and (Hd)
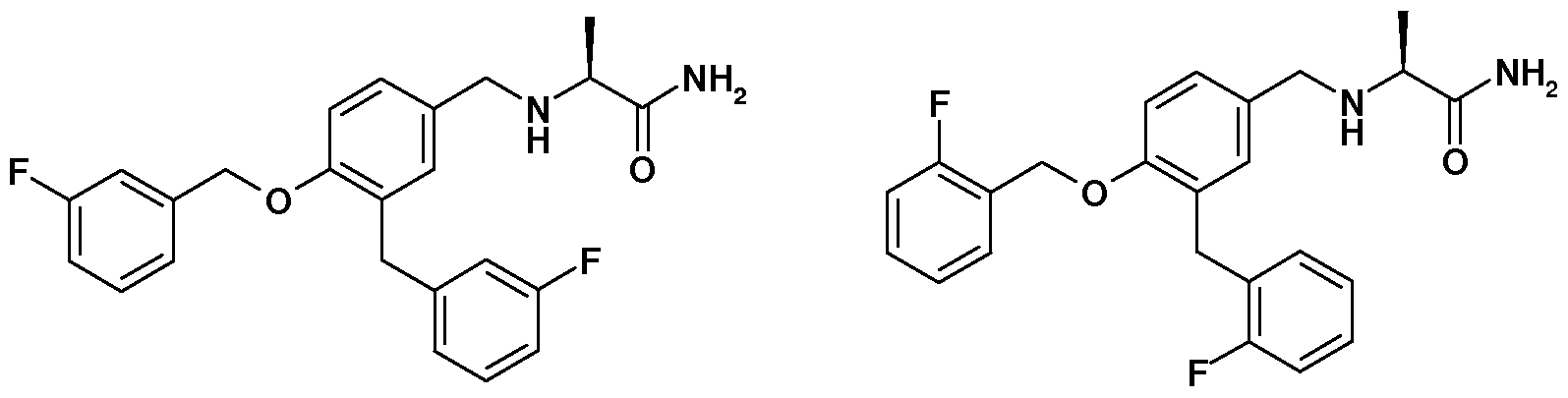
(Ha) (lib)
The same situation occurs with the preparation according the prior art methods for the R-enantiomers (I’a) and (I’b) of, respectively, safinamide and ralfinamide, the respective racemic mixtures (Ia, I’a) and (Ib, I’b), and the salts thereof with pharmaceutically acceptable acids, (I’c), (I’d) and the respective racemic mixtures (Ic, I’c) and (Id, I’d) in particular the methanesulfonates, which result to be contaminated by the respective R isomers (Il’a), (Il’b), (II’c), and (Il’d) of the above identified impurities (Ha), (lib), (lie) and (Hd) or the respective racemic mixtures (Ha, Il’a), (lib, Il’b), (Hc, II’c) and (Hd, Il’d).

PATENT
Parkinson’s disease (PD) is a progressive neurodegenerative disease characterized by bradykinesia, rigidity, resting tremor, and ataxia. These symptoms are caused by decreased dopamine release in the striatum. Clinically, PD is defined by presence of Lewy bodies, intracellular neuronal inclusions in the substantia nigra and at other sites in the brain. Estimated prevalence of this disease is 100 to 200 per 100,000 population including males and females across the entire age group. Current treatment for PD comprises dopaminergic medications that include levodopa, dopamine agonists (DAs), monoamine oxidase-B (MAO-B) inhibitors. Figure 1 provides few examples of pharmaceutically important benzyloxy-benzylamine derivatives. Many of these benzyl oxy-benzylamines with various amine functions were studied and has been patented as sodium channel blockers. Among them, safinamide ((5)-N2– {4-[3- fluorobenzyl)oxy] benzyl}- alaninamide methanesulfonate) is a noted example which is under phase III clinical trials for treatment of Parkinson’s disease. Its mechanism of action is manifold which comprise MAO-B and dopamine uptake inhibition. Further, safinamide is believed to block voltage-dependent sodium channels, modulates calcium channels and reduction of glutamate release in the central nervous system. WOl 998003472 discloses serinamide, glycinamide, alaninamide and phenylalaninamide derivatives of a compound (I). These compounds (I) are useful for the treatment of neurological diseases.
EP2474521 discloses high purity degree (S)-2-[4-(3-fluorobenzyloxy)- benzylamino]propanamide (safinamide) or (S)-2-[4-(2-fluorobenzyloxy)- benzylamino]propanamide (ralfinamide) or a salt thereof with a pharmaceutically acceptable acid with a content of the respective impurity (S)-2-[3-(3-fluorobenzyl)-4-(3- fluorobenzyloxy)-benzylamino]propanamide or (S)-2-[3-(2-fluorobenzyl)-4-(2- fluorobenzyloxy)-benzylamino]propanamide.
US2009149544 relates to novel alpha- aminoamide derivatives, their pharmaceutically acceptable salts, solvates, and hydrates thereof. The application also provides compositions comprising a compound and the use of such compositions in methods of treating diseases and conditions that are beneficially treated by administering an inhibitor of monoamine oxidase type B (MAO-B) and/or a sodium (Na.sup.+) channel blocker, and/or a calcium (Ca.sup.2+) channel modulator.
The strategy employed in the art to prepare benzyloxy-benzylamine derivatives including safinamide or its analogue ralfinamide is chiral pool approach starting from L-alaniriamide and reductively aminating with 4-(3-fluorobenzyloxy) benzaldehyde. Although this method is very simple and straightforward, it suffers from several serious drawbacks, such as need to use toxic reagents such as sodium cyanoborohydride and further formation of toxic by-products such as hydrogen cyanide and sodium cyanide and other toxic impurities in large-scale production Importantly, the possibility of generating a range of safinamide analogues by means of the chiral-pool approach is limited in terms of the structure and stereochemistry of the products because of inadequacies in the availability of D-alaninamide and its analogues
Hence, the developments of newer methods for the preparation of compounds of formula (I) comprising safinamide and related analogues are highly desirable
Example 2: Synthesis of (R)-l-(benzyIoxy)propan-2-ol [(R)-compound 3]
To a solution of (7? benzyl glycidyl ether [fR)-compound 2] (4 g, 24.4 mmol) in dry THF (10 mL) at 0 °C, a pre-cooled solution of lithium aluminium hydride (1.4 g, 36.6 mmol) in anhydrous THF (10 mL) was added slowly with stirring under nitrogen. After 60 min, the reaction mixture was quenched with 1 ml of water and 1 ml of 15 % NaOH solution and the content was stirred for 15 min. The inorganic precipitate was filtered, washed with ethyl acetate and the solvent evaporated under reduced pressure. The residue was purified by a short filtration column to afford (-fl)-compound 3 as a colorless oil (3.8 g, 95%); [a]22D = -14.5 (c 2, CHC13); IR (CHC13): vmax3418, 3087, 3063, 3030, 2963, 2924, 1952, 1873, 1600, 1495, 1454, 1363, 1244, 1099, 1028, 918, 808, 698 cm“1; Ή NMR (200 MHz, CDC13): δΗ 1.13 (d, J = 6.3 Hz, 3H), 2.5 (bs, 1H), 3.23-3.32 (dd, J = 9.8, 1.3 Hz, 1H), 3.43-3.49 (dd, J = 9.45, 3.2 Hz, 1H), 3.91-4.03 (m, 1H), 4.55 (s, 2H), 7.25-7.37 (m, 5H); I3C NMR (50 MHz, CDC13): 5C 137.8 (C), 128.3 (CH, 2 carbons), 127.7 (CH, 3 carbons), 75.7 (CH2), 73.2 (CH2), 66.4 (CH), 18.6 (CH3); MS: m/z 189 [M+Na]+.
Example 3: Synthesis of (S)-((2-azidopropoxy)methyl)benzene [(S)- compound 4]
To a stirred solution of secondary alcohol ( )-compound 3 (3 g, 18.1 mmol) in dry dichloromethane (25 mL), Et3N (3.1 mL, 21.7 mmol) at 0 °C was added, followed by drop wise addition of mesyl chloride (1.8 mL, 21.7 mmol). The reaction mixture was stirred at 0°C for 2 hours, subsequently at room temperature for 3 hours under a nitrogen atmosphere. After completion of the reaction (indicated by TLC), the reaction mixture was diluted with dichloromethane and washed with a saturated solution of sodium bicarbonate (30 mL) and water (2 x 10 mL). The organic layer was separated, dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure to give the O-mesyl compound (4.3 g; crude).
To a solution of the crude 0-mesyl compound (4 g, 16.37 mmol) in dry DMF (10 mL), sodium azide (1.6 g, 24.55 mmol) was added and the reaction mixture was heated at 60°C for 6 hours under nitrogen atmosphere. After completion of the reaction (indicated by TLC), water (10 mL) was added to the reaction mixture, then extracted with ethyl acetate (2 x 15 mL). The combined organic layers were washed with brine solution, dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure. Purification of the crude residue was done by column chromatography (silica gel, petroleum ether/EtOAc, 95:5) to yield (¾)-compound 4 as a colorless oil. (2.8 g; 89%); [a]22D = +6.1 (c 1.3, CHC13); IR (CHC13): vmax 3394, 3032, 2977, 2864, 2500, 2104, 1724, 1641 , 1496, 1454, 1363, 1269, 1 101 , 913, 698 αη ‘,Ή NMR (200 MHz, CDC13): δΗ 1.20 (d, J = 6.7 Hz, 3H), 3.39-3.54 (m, 2H), 3.61-3.77 (m, 1H), 4.57 (s, 2H), 7.25-7.39 (m, 5H); 13C NMR (50 MHz, CDC13): 5C 137.8 (C), 128.4 (CH, 2 carbons), 127.7 (CH), 127.5 (CH, 2 carbons), 73.7 (CH2), 73.2 (CH2), 56.9 (CH), 16.1 (CH3);MS: m/z 214 [M+Na]+.
Example 4: Synthesis of (S)-N-(l-hydroxypropan-2-yl)-2-nitrobenzenesulfonamide [(S)- compound 5]
To a solution of ^-compound 4 (2.5 g, 13.1 mmol) in methanol (15 mL), trifluoroacetic acid (2 mL) and palladium hydroxide on activated carbon (0.05 g, 10-20 wt %) were added and the reaction mixture was stirred under hydrogen (60 psi) for 8 hours. After completion of the reaction (indicated by TLC), the catalyst was filtered over a plug of celite and the solvent was evaporated under reduced pressure to half of its volume which was basified with 2.5 M methanolic NaOH. Evaporation of the remaining solvent under reduced pressure was done followed by filtration of the residue through a short bed of basic alumina (eluent; MeOH) to obtain the amino alcohol as a pale brown oil (0.94 g, crude) which was subjected to the next reaction without further purification.
To a solution of amino alcohol (0.9 g, 1 1.98 mmol) in dry dichloromethane (5 mL), 2-nitrobenzenesulfonylchloride (3.2 g, 14.37 mmol) in dichloromethane (8 mL) and triethylamine (2.6 mL, 17.97 mmol) at 0 °C were slowly added under nitrogen atmosphere. The solution was stirred for 2 hours. After completion of the reaction (indicated by TLC), water (10 mL) was added to the reaction mixture, then extracted with dichloromethane (2 x 15 mL). The combined organic layers were washed with brine solution, dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure. Purification of the crude residue was done by column chromatography (silica gel, petroleum ether/EtOAc, 60:40) to yield (S)- compound 5 as a pale yellow oil (2.33 g, 75% ); [a]22D = +80.2 (c 2.1, CHClj); IR (CHC13): vmax 3546, 3367, 3022, 2883, 2401, 1594, 1542, 1412, 1362, 1216, 1170, 1 125, 1059, 971, 854, 668 cm“1; ]H NMR (200 MHz, CDC13): δΗ 1.13 (d, J = 6.5 Hz, 3H), 2.16 (bs, 1H), 3.45-3.70 (m, 3H), 5.61 (d, J = 6.6 Hz, 1H), 7.73-7.80 (m, 2H), 7.86-7.91 (m, 1H), 8.13-8.22 (m, 1H); 13C NMR (50 MHz, CDC13): 5C 147.8 (C), 134.4 (C), 133.7 (CH), 133.0 (CH), 130.9 (CH), 125.5 (CH), 66.2 (CH2), 52.5 (CH), 17.8 (CH3); MS: m/z 283 [M+Na]+.
Example 5: Synthesis of l-fluoro-3-(iodomethyl)benzene ( compound 7)
To a stirred solution of triphenyl phosphine (4.15 g, 15.85 mmol), imidazole (1.1 g, 15.85 mmol) in dry dichloromethane (20 mL), iodine (4.8 g, 19.02 mmol) at 0°C was added and the solution was stirred for 5 min. To this, 3-fluoro benzyl alcohol (compound 6) (2 g, 15.85 mmol) dissolved in dichloromethane (5 mL) was added drop wise over 10 min and the stirring was continued for 1 hour with exclusion of light. After completion of the reaction (indicated by TLC), the reaction mixture was quenched by addition of an aqueous Na2S203 solution (15 mL), then extracted with dichloromethane (2 x 20 mL). The combined organic layers were washed with brine solution, dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure. Purification of the crude residue was done by column chromatography (silica gel, petroleum ether/EtOAc, 95:5) to yield compound 7 as a colorless oil (3.5 g, 95% ); (IR (CHC13): vmax 3460, 3060, 2965, 1695, 1613, 1593, 1482, 1446, 1259, 1 156, 1068, 944, 871, 782, 736, 686 cm“1 ; Ή NMR (200 MHz, CDC13): δΗ 4.42 (s, 2H), 6.89-6.99 (m, 1H), 7.05-7.17 (m, 2H), 7.21-7,29 (m, 1H); 13C NMR (50 MHz, CDC13): 6C 165.0 (C), 141.6 (C), 130.2 (CH), 124.4 (CH), 1 15.9 (CH), 1 14.7 (CH), 3.9 (C¾).
Example 6: Synthesis of (4-((3-flurobenzyl)oxy)phenyl)methanol (compound 8)
To a stirred solution of 4-(hydroxymethyl)phenol (1.57 g, 12.7 mmol) and K2C03 (8.8 g, 63.55 mmol) in dry acetonitrile (25 mL), compound 7 (3 g, 12.7 mmol) in acetonitrile was slowly added and the reaction mixture was heated at 70°C for 6 hours. After completion of the reaction (indicated by TLC), water (20 mL) was added to the reaction mixture, then extracted with ethylacetate (3 x 20 mL). The combined organic layers were washed with brine solution, dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure. Purification of the crude residue was done by column chromatography (silica gel, petroleum ether/EtOAc, 70:30) to yield compound 8 as a colorless solid (2.7 g, 91% ); mp 63-65 °C; IR (CHC13): vmax 3422, 3017, 1612, 1512, 1489, 1381, 1216, 1 174, 1020, 829, 668 cm“1; Ή NMR (200 MHz, CDC13): δΗ 4.61 (s, 2H), 5.06 (s, 2H), 6.91-6.98 (m, 2H), 7.00-7.06 (m, 1H), 7.12-7.20 (m, 2H), 7.25-7.37 (m, 3H); 13C NMR (50 MHz, CDC13): 5C 165.4 (C), 160.5 (C), 158.0 (C), 139.6 (C), 133.5 (CH), 130.2 (CH), 128.7 (CH, 2 carbons), 122.7 (CH), 1 14.8 (CH, 2 carbons), 1 13.9 (CH), 69.1 (CH2), 64.9 (CH2); MS: m/z 255 [M+Na]+.
Example 7: Synthesis of l-fluoro-3-((4-(iodomethyl)phenoxy)methyI)benzene (compound 9)
To a stirred solution of triphenyl phosphine (2.82 g, 10.8 mmol), imidazole (0.73 g, 10.76 mmol) in dry dichloromethane (20 mL), iodine (3.27 g, 12.9 mmol) at 0 °C was added and the solution was stirred for 5 min. To this, compound 8 (2.5 g, 10.8 mmol) dissolved in dichloromethane (5 mL) was added drop wise over 10 min and the stirring was continued for 1 hour with exclusion of light. After completion of the reaction (indicated by TLC), the reaction mixture was quenched by addition of an aqueous Na2S203 solution (15 mL), then extracted with dichloromethane (2 x 20 mL). The combined organic layers were washed with brine solution, dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure. Purification of the crude residue was done by column chromatography (silica gel, petroleum ether/EtOAc, 95:5) to yield compound 9 as a colorless oil (3.4 g, 93%); IR (CHC13): vmax 3503, 3033, 2925, 2089, 1607, 1509, 1488, 1381, 1301, 1250, 1 155, 1079, 944, 869, 776, 684 cm“1; 1H NMR (200 MHz, CDC13): δΗ 4.47 (s, 2H), 5.04 (s, 2H), 6.85-6.91 (m, 2H), 6.96-7.02 (m, 1H), 7.05-7.12 (m, 1H), 7.16-7.20 (m, 1H), 7.29-7.40 (m, 3H).
,3C NMR (50 MHz, CDC13): 6C 165.4 (C), 160.5 (C), 158.1 (C), 131.9 (C), 130.2 (CH), 130.1 (CH, 2 carbons), 122.7 (CH), 1 15.1 (CH, 2 carbons), 1 14.7 (CH), 1 13.9 (CH), 69.2 (CH2), 6.33 (CH2).
Example 8: Synthesis of (S)-N-(4-((3-flurobenzyl)oxy)benzyl)-N-(l-hydroxypropan-2-yl)-2-nitrobenzenesulfonamide [(S)-compound 10]
To a stirred solution of (^-compound 5 (1 g, 3.8 mmol) and K2C03 (2.65 g, 19.2 mmol) in dry acetonitrile (25 mL), compound 9 (1.84 g, 5.4 mmol) in acetonitrile was slowly added and the reaction mixture was heated at 70°C for 72 hours. After completion of the reaction (indicated by TLC), water (20 mL) was added to the reaction mixture, then extracted with ethylacetate (3 15 mL). The combined organic layers were washed with brine solution, dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure. Purification of the crude residue was done by column chromatography (silica gel, petroleum ether/EtOAc, 80:20) to yield (¾)-compound 10 as a colorless oil (1.46 g, 80% ); [a]22D = +5.4 (c 1.5, CHC13); IR (CHC13): vmax 3445, 3020, 2928, 2400, 1613, 1544, 1512, 1453, 1371, 1216, 1 162, 1029, 852, 668 cm“1; 1H NMR (200 MHz, CDC13): δΗ 1.07 (d, J = 6.9 Hz, 3H), 1.91 (t, J = 5.2 Hz, 1H), 3.41-3.53 (m, 2H), 4.05-4.22 (m, 1H), 4.37-4.57 (m, 2H), 5.02 (m, 2H), 6.87 (d, J = 8.53 Hz, 2H), 6.97-7.12 (m, 2H), 7.20 (d, J = 7.2 Hz, 2H), 7.32 (d, J = 8.7 Hz, 2H), 7.47-7.67 (m, 3H), 7.89 (d, J = 8.09 Hz, 1H); 13C NMR (50 MHz, CDC13): 6C 165.5 (C), 160.6 (C), 158.4 (C), 147.7 (C), 139.6 (C), 134.1 (C), 133.4 (CH), 131.6 (CH), 131.4 (CH), 130.3 (CH), 129.7 (CH, 2 carbons), 124.1 (CH), 122.8 (CH), 115.1 (CH), 114. 9 (CH, 2 carbons), 114.0 (CH), 69.2 (CH2), 64.3 (CH2), 56.2 (CH), 46.9 (CH2), 15.4 (CH3); MS: m/z 497 [M+Na]+.
Example 9: Synthesis of (S)-2-(N-(4-((3-fluorobenzyl)oxy)benzyl)-2-nitrophenylsulfonamido) propanoic acid [(S)-compound 11]
A mixture of (S compound 10 (1.25 g, 2.6 mmol), TEMPO (0.028 g, 0.18 mmol), acetonitrile (20 mL), and sodium phosphate buffer (16 mL, 0.67 M, pH 6.7) was heated to 35°C. Next, sodium chlorite (0.47 g dissolved in 2 mL water, 7.9 mmol) and diluted bleach (4-6%, 0.09 mL diluted in 1 mL water) were added simultaneously over 1 hour. The reaction mixture was stirred at 35°C until the reaction was complete (3 hours, TLC), then cooled to room temperature. Water (30 mL) was added and the pH adjusted to 8 with 2 M NaOH. The reaction was quenched by pouring it into ice cold Na2S03 solution maintained at <20°C. After stirring for 30 min at room temperature, ethyl acetate (20 mL) was added and the stirring was continued for an additional 15 min. The organic layer was separated and discarded. More ethyl acetate (20 mL) was added, and the aqueous layer was acidified with 1 M HC1 to pH 3-4. The organic layer was separated, washed with water (2 x 15 mL), brine and concentrated under reduced pressure to afford the carboxylic acid (S -compound 1 1 (1.1 g, 85%); [ ]22ο = -20.4 (c 1.1, CHC13); IR (CHC13): vmax 3398, 3095, 1718, 1612, 1591, 1543, 1512, 1489, 1457, 1371, 1303, 1251, 1163, 1059, 900, 852, 831 , 778, 684 cm“1; 1H NMR (200 MHz, CDC13): 8H 1.44 (d, J = 7.3 Hz, 3H), 4.23 (d, J = 15.6 Hz, 1H), 4.64 (d, J = 15.6 Hz, 1H), 4.82-4.90 (q, J = 7.4 Hz, 1H), 4.92 (s, 2H), 6.68 (d, J = 8.6 Hz, 2H), 6.89-7.01 (m, 2H), 7.07-7.13 (m, 3H), 7.18-7.33 (m, 2H), 7.43-7.55 (m, 3H), 8.81 (bs, 1H); 13C NMR (50 MHz, CDC13): 5C 176.5 (CO), 165. 0 (C), 158.0 (C), 147.4 (C), 139.4 (C), 134.1 (C), 133.2 (CH), 131.4 (CH), 130.3 (CH), 129.9 (CH, 2 carbons), 128.4 (C), 124.1
(CH), 122.6 (CH), 1 15.0 (CH), 114.6 (CH, 2 carbons), 1 14.3 (CH), 1 13.8 (CH) 69.1 (CH2), 56.1 (CH), 49.0 (CH2), 16.8 (CH3); MS: m/z 51 1 [M+Na .
Example 10: Synthesis of (S)-2-(N-(4-((3-fluorobenzyI)oxy)benzyl)-2-nitrophenylsulfonamido) propanamide [(S)- compound 12]
To a solution of carboxylic acid (¾)-compound 1 1 (1 g, 2.04 mmol) and triethyl amine (0.34 mL, 2.4 mmol) in dry THF (20 mL), ethyl chloroformate (0.21 mL, 2.2 mmol) at 0 °C was added under nitrogen atmosphere. After 1 hour, ammonium hydroxide (25% w/v aqueous solution, 1.4 mL, 10.2 mmol) was added and the resulting reaction mixture was stirred at room temperature for 16 hours. After completion of the reaction, potassium carbonate (0.29 g, 2.1 mmol) was added and the reaction mixture was filtered, and washed with ethylacetate. The solvent was removed under reduced pressure and the crude product was subjected to column chromatography (silica gel, petroleum ether/EtOAc, 50:50) to obtain sulfonamide (Sj-compound 12 as a colorless oil (0.9 g, 91%); [a]22D = -32.1 (c 1.2, CHC13); IR (CHC13): vmax 3472, 1961 , 161 1, 1592, 1542, 1511, 1449, 1371, 1304, 1243, 1 163, 1060, 1029, 895, 852, 684 cm“1; Ή NMR (200 MHz, CDC13): δΗ 1.43 (d, J = 7.1 Hz, 3H), 4.44 (d, J = 15.4 Hz, 1H), 4.59 (d, J = 15.5 Hz, 1H), 4.60-4.71 (q, J= 7.0 Hz, 1 H), 5.01 (s, 2H), 5.50 (bs, 1H), 6.31 (bs, 1H), 6.78 (d, J = 8.71 Hz, 2H), 6.98-7.1 1 (m, 2H), 7.15-7.22 (m, 3H), 7.31-7.45 (m, 2H), 7.59-7.64 (m, 3H);13C NMR (50 MHz, CDC13): 5C 172.3 (CO), 165.5 (C), 158.2 (C), 147.5 (C), 139.6 (C), 139.4 (C), 133.6 (CH), 131.7 (CH), 130.5 (CH, 2 carbons),130.3 (CH), 128.1 (C), 124.2 (CH), 122.7 (CH), 1 15.1 (CH), 1 14.7 (CH, 2 carbons),1 14.4 (CH), 1 13.9 (CH), 69.0 (CH2), 55.7 (CH), 48.3 (CH2), 14.9 (CH3); MS: m/z 510 [M+Na]+.
Example 11: Synthesis of (S)-2-((4-((3-fluorobenzyl)oxy) benzyl) amino) propanamide [(S)-compound of formula I]
To a solution of sulfonamide (S)- compound 12 (0.8 g, 1.64 mmol), potassium carbonate (0.56 g, 4.9 mmol) in dry DMF (10 mL), thiophenol (0.2 mL, 1.9 mmol) was added. The reaction mixture was vigorously stirred for 6 hours. After completion of the reaction (indicated by TLC), water (10 mL) was added to the reaction mixture, then extracted with ethylacetate (2 x 20 mL). The combined organic layers were washed with brine solution, dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure. Purification of the crude residue was done by column chromatography (silica gel, petroleum ether/EtOAc, 60:40) to yield (S) -compound of formula I as a colorless solid (0.43 g, 86% ); mp 207-09 °C; [a]22D = +3.89 (c 1.55, CHC13); IR (CHC13): vmax 3341, 2970, 2927, 2853, 1648, 1592, 1512, 1489, 1445, 1406, 1384, 1254, 1176, 1 137, 1030, 953, 928, 829, 680 cm“1; Ή NMR (200 MHz, CDC13): δΗ 1.34 (d, J = 6.9 Hz, 3H), 2.49 (bs, 2H), 3.19-3.30 (q, J = 6.8 Hz, 1H), 3.63-3.78 (dd, J = 19.4, 3.9 Hz, 2H), 5.05 (s, 2H), 5.85 (bs, 1H), 6.95 (d, J = 8.7 Hz, 2H), 7.00-7.06 (m, 1H), 7.13-7.24 (m, 4H), 7.29-7.40 (m, 1H). 13C NMR (50 MHz, CDC13): 8C 178.3 (CO), 165.4 (C), 157.7 (C), 139.6 (C), 132.1 (C), 130.2 (CH), 129.3 (CH, 2 carbons), 122.7 (CH), 1 14.9 (CH, 2 carbons), 1 14.6 (CH), 1 13.9 (CH), 69.2 (CH2), 57.5 (CH), 51.9 (CH2), 19.6 (CH3); MS: m/z 302 [M]+, 325 [M+Na]+.
Example 12: Synthesis of (S)-Safinamide mesylate
To a stirred solution of (^-compound of formula I (0.1 g, 0.33 mmol) in ethylacetate (3 mL) at 70°C, methanesulfonic acid (0.02 mL, 0.33 mmol) was added and the reaction mixture was stirred for 2 hours. Subsequently, the temperature was lowered to 35°C and the stirring was continued for additional 1 hour. The solvent was evaporated under reduced pressure and the residue was filtered through a short bed of basic alumina [eluent: EtOAc/MeOH; (95:5)] to obtain safinamide mesylate as a white solid (0.11 g, 90%); mp 209-10 °C [lit.7mp 210]; [a]22D = +9.6 (c 1.1, AcOH); {lit.7 [a] D = +12.9 (c 1.1, AcOH)} ee >98% [The ee of safinamide mesylate was determined by chiral HPLC analysis; Chiralcel OD-RH (150 x 4.6 mm) column; eluent:
Methanol/ Acetonitrile/Buffer-TEAP, pH 3 (20: 10:70); flow rate 0.5 mL/min (780 psi); detector: 224 nm] [f¾)-isomer tR = 1 1.55 min, (SJ-isomer tR = 12.94 min].

PAPERS
Synthesis2014, 46, 1751-1756.

N2-{4-[(3-Fluorobenzyl)oxy]benzyl}-L-alaninamide [(S)-14] BASE FORM
PhSH (0.2 mL, 1.9 mmol) was added to a solution of sulfonamide (S)-13 (0.8 g, 1.64 mmol) and K2CO3 (0.56 g, 4.9 mmol) in anhyd DMF (10 mL), and the mixture was vigorously stirred for 6 h. When the reaction was complete (TLC), H2O (10 mL) was added and the mixture was extracted with EtOAc (2 × 20 mL). The organic layers were combined, washed with brine (2 × 10), dried (Na2SO4), filtered, and concentrated under reduced pressure. The crude residue was purified by column chromatography [silica gel, PE–EtOAc(60:40)] to give a colorless solid; yield: 0.43 g (86%); mp 207–09 °C;
[α]D22 +3.89 (c 1.55, CHCl3).
IR (CHCl3): 3341, 2970, 2927, 2853, 1648, 1592, 1512, 1489, 1445,1406, 1384, 1254, 1176, 1137, 1030, 953, 928, 829, 680 cm–1.
1H NMR (200 MHz, CDCl3): δH = 1.34 (d, J = 6.9 Hz, 3 H), 2.49 (brs, 2 H), 3.19–3.30 (q, J = 6.8 Hz, 1 H), 3.71 (dd, J = 19.4, 3.9 Hz, 2H), 5.05 (s, 2 H), 5.85 (br s, 1 H), 6.95 (d, J = 8.7 Hz, 2 H), 7.00–7.06 (m, 1 H), 7.13–7.24 (m, 4 H), 7.29–7.40 (m, 1 H).
13C NMR (50 MHz, CDCl3): δC = 178.3 (CO), 165.4 (C), 157.7 (C),139.6 (C), 132.1 (C), 130.2 (CH), 129.3 (CH, 2 C), 122.7 (CH), 114.9 (CH, 2 C), 114.6 (CH), 113.9 (CH), 69.2 (CH2), 57.5 (CH),51.9 (CH2), 19.6 (CH3).
MS: m/z = 302 [M]+, 325 [M + Na]+.
(S)-Safinamide Mesylate (1)
MsOH (0.02 mL, 0.33 mmol) was added to a stirred solution of sulfonamide (S)-14 (0.1 g, 0.33 mmol) in EtOAc (3 mL) at 70 °C, and the mixture was stirred for 2 h. The temperature was then lowered to 35 °C, and the mixture was stirred for an additional 1 h. The solvent was evaporated under reduced pressure and the residue was filtered
through a short bed of basic alumina with elution by EtOAc–MeOH; (95:5) to give a white solid; yield: 0.11 g (90%);
mp 209–210 °C [Lit.7a 210 °C];
[α]D22 +9.6 (c 1.1, AcOH); {Lit.7 [α]D22+12.9 (c 1.1, AcOH)}.
Chiral HPLC: column: Chiralcel OD-RH (150 × 4.6 mm); eluent:MeOH–MeCN–buffer-TEAP (pH 3) (20:10:70); flow rate: 0.5mL/min (780 psi); detector: 224 nm [(R)-isomer: tR = 11.55 min;
(S)-isomer: tR = 12.94 min]; ee >98%.
7a) Pevarello, P.; Bonsignori, A.; Dostert, P.;
Heidempergher, F.; Pinciroli, V.; Colombo, M.; McArthur,
R. A.; Salvati, P.; Post, C.; Fariello, R. G.; Varasi, M. J. Med.
Chem. 1998, 41, 579.
PAPER

Chin. J. Pharmas.2012, 43, 161-163.

 …………….BASE
…………….BASE
 …………MESYLATE
…………MESYLATE
PAPER
J. Med. Chem. 2007, 50, 4909-4916.

(S)-2-[6-(3-Fluorobenzyloxy)-3,4-dihydro-1H-isoquinolin-2-yl]-propionamide (21). The title compound was obtained using the same procedure described for the synthesis of (R)-2-[6-(3-fluorobenzyloxy)-3,4-dihydro-1H-isoquinolin-2-yl]propionamide, starting from 6-(3-fluorobenzyloxy)-1,2,3,4-tetrahydroisoquinoline (0.24 g, 0.95 mmol) and (R)-2-amino-1-methyl-2-oxoethyl-2-nitrobenzenesulfonate (0.52 g, 1.9 mmol). After column chromatography
purification using 99:1 DCM/MeOH as eluent, 0.075 g (24% yield) of the title compound was obtained as a pure white solid. Mp 153- 154 °C. 1H NMR (CDCl3) ä 1.35 (d, 3H, J ) 7.0), 2.67-2.97 (m, 4H), 3.28 (q, 1H, J ) 7.0), 3.64 (d, 1H, J ) 14.2), 3.77 (d, 1H, J ) 14.2), 5.05 (s, 2H), 5.36 (br, 1H), 6.74 (d, 1H, J ) 2.5), 6.79 (dd, 1H, J ) 8.5, 2.5), 6.97 (d, 1H, J ) 8.5), 6.99-7.06 (m, 1H), 7.06-7.24 (m, 3H), 7.30-7.40 (m, 1H).
J. Med. Chem.1998, 41, 579-590.

References
- “Summary of the risk management plan (RMP) for Xadago (safinamide)” (PDF). European Medicines Agency. January 2015.
- Fariello, RG (2007). “Safinamide”. Neurotherapeutics. 4 (1): 110–116. doi:10.1016/j.nurt.2006.11.011. PMID 17199024.
- “EPAR Summary for the Public for Xadago” (PDF). European Medicines Agency. February 2015.
- “After an odyssey of setbacks, FDA finally green-lights Newron’s Parkinson’s drug Xadago”. endpts.com. Retrieved 2017-03-21.
- Lawrence, Janna (2015-01-19). “Safinamide recommended for approval as Parkinson’s disease therapy”. The Pharmaceutical Journal. Royal Pharmaceutical Society. Retrieved 2015-01-19.
- Haberfeld, H, ed. (2015). Austria-Codex (in German). Vienna: Österreichischer Apothekerverlag.
- H. Spreitzer (14 April 2014). “Neue Wirkstoffe – Safinamid”. Österreichische Apothekerzeitung (in German) (8/2014): 30.
- Klement, A (18 July 2016). “Xadago”. Österreichische Apothekerzeitung (in German) (15/2016): 10.
- “Summary of Product Characteristics for Xadago” (PDF). European Medicines Agency. 24 February 2015.
- ^ Jump up to:a b Caccia, C; Maj, R; Calabresi, M; Maestroni, S; Faravelli, L; Curatolo, L; Salvati, P; Fariello, RG (2006). “Safinamide: From molecular targets to a new anti-Parkinson drug”. Neurology. 67 (7 Suppl 2): S18–23. doi:10.1212/wnl.67.7_suppl_2.s18. PMID 17030736.
- Merck Serono: Vielversprechende Daten zur kognitiven Wirkung von Safinamid bei Parkinson im Frühstadium. (German) 8 June 2007.
- Pevarello, P; Bonsignori, A; Caccia, C; Amici, R; Salvati, P; Fariello, RG; McArthur, RA; Varasi, M (1999). “Sodium channel activity and sigma binding of 2-aminopropanamide anticonvulsants”. Bioorganic & Medicinal Chemistry Letters. 9 (17): 2521–2524. doi:10.1016/s0960-894x(99)00415-1.
- ^ Jump up to:a b Krösser, Sonja; Marquet, Anne; Gallemann, Dieter; Wolna, Peter; Fauchoux, Nicolas; Hermann, Robert; Johne, Andreas (2012). “Effects of ketoconazole treatment on the pharmacokinetics of safinamide and its plasma metabolites in healthy adult subjects”. Biopharmaceutics & Drug Disposition. 33 (9): 550. doi:10.1002/bdd.1822. PMID 23097240.
- Jump up^ Pevarello, P; Bonsignori, A; Dostert, P; Heidempergher, F; Pinciroli, V; Colombo, M; McArthur, RA; Varasi, M (1998). “Synthesis and Anticonvulsant Activity of a New Class of 2-[(Arylalkyl)amino]alkanamide Derivatives”. Journal of Medicinal Chemistry. 41 (4): 579–590. doi:10.1021/jm970599m. PMID 9484507.
- Jump up^ “Wichtigste Ergebnisse der Langzeitstudie mit Safinamid als Begleittherapie zu Levodopa bei Parkinson im fortgeschrittenen Stadium” [Major results from the long-term study of safinamide as add-on to levodopa for late-stage Parkinson] (in German). Merck KGaA. 4 November 2010.
- Jump up^ Study of Safinamide in Early Parkinson’s Disease as Add-on to Dopamine Agonist (MOTION)
- Jump up^ Merck Returns Rights for Safinamide to Newron, 21 October 2011.
- Jump up^ “Information about FDA Refusal to File” (PDF). Newron. 29 July 2014.
- “Information about FDA re-application” (PDF). Newron. 29 December 2014.
- Chazot, PL (2007). “Drug evaluation: Safinamide for the treatment of Parkinson’s disease, epilepsy and restless legs syndrome”. Current Opinion in Investigational Drugs. 8 (7): 570–579. PMID 17659477.
 |
|
| Clinical data | |
|---|---|
| Trade names | Xadago |
| AHFS/Drugs.com | UK Drug Information |
| Pregnancy category |
|
| Routes of administration |
Oral |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 95% |
| Protein binding | 88–90% |
| Metabolism | Amidases, glucuronidation |
| Biological half-life | 20–30 hrs |
| Excretion | 76% renal, 1.5% faeces |
| Identifiers | |
| Synonyms | EMD-1195686, PNU-15774E; (2S)-2-[[4-[(3-fluorophenyl)methoxy]phenyl] methylamino]propanamide |
| CAS Number | |
| PubChemCID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| ECHA InfoCard | 100.120.167 |
| Chemical and physical data | |
| Formula | C17H19FN2O2 |
| Molar mass | 302.34 g/mol |
| 3D model (Jmol) | |
//////////Xadago, safinamide, Newron Pharmaceuticals, FDA 2017, Parkinson’s disease, 133865-89-1 , сафинамид , سافيناميد, 沙非胺, EMD-1195686, ZP-034, FCE-28073(R-isomer), PNU-151774E, NW-1015, FCE-26743
C[C@H](NCC1=CC=C(OCC2=CC=CC(F)=C2)C=C1)C(N)=O
Novartis Kisqali® (ribociclib, LEE011) receives FDA approval as first-line treatment for HR+/HER2- metastatic breast cancer in combination with any aromatase inhibitor


![]()
- Approved based on a first-line Phase III trial that met its primary endpoint of progression-free survival (PFS) at interim analysis due to superior efficacy compared to letrozole alone[1]
- At this interim analysis, Kisqali plus letrozole reduced risk of disease progression or death by 44% over letrozole alone, and demonstrated tumor burden reduction with a 53% overall response rate[1]
- Kisqali plus letrozole showed treatment benefit across all patient subgroups regardless of disease burden or tumor location[1]
- At a subsequent analysis with additional follow-up and progression events, a median PFS of 25.3 months for Kisqali plus letrozole and 16.0 months for letrozole alone was observed[2]
Basel, March 13, 2017 – The US Food and Drug Administration (FDA) has approved Kisqali®(ribociclib, formerly known as LEE011) in combination with an aromatase inhibitor as initial endocrine-based therapy for treatment of postmenopausal women with hormone receptor positive, human epidermal growth factor receptor-2 negative (HR+/HER2-) advanced or metastatic breast cancer.
Kisqali is a CDK4/6 inhibitor approved based on a first-line Phase III trial that met its primary endpoint early, demonstrating statistically significant improvement in progression-free survival (PFS) compared to letrozole alone at the first pre-planned interim analysis[1]. Kisqali was reviewed and approved under the FDA Breakthrough Therapy designation and Priority Review programs.
“Kisqali is emblematic of the innovation that Novartis continues to bring forward for people with HR+/HER2- metastatic breast cancer,” said Bruno Strigini, CEO, Novartis Oncology. “We at Novartis are proud of the comprehensive clinical program for Kisqali that has led to today’s approval and the new hope this medicine represents for patients and their families.”
The FDA approval is based on the superior efficacy and demonstrated safety of Kisqali plus letrozole versus letrozole alone in the pivotal Phase III MONALEESA-2 trial. The trial, which enrolled 668 postmenopausal women with HR+/HER2- advanced or metastatic breast cancer who received no prior systemic therapy for their advanced breast cancer, showed that Kisqali plus an aromatase inhibitor, letrozole, reduced the risk of progression or death by 44 percent over letrozole alone (median PFS not reached (95% CI: 19.3 months-not reached) vs. 14.7 months (95% CI: 13.0-16.5 months); HR=0.556 (95% CI: 0.429-0.720); p<0.0001)[1].
More than half of patients taking Kisqali plus letrozole remained alive and progression free at the time of interim analysis, therefore median PFS could not be determined[1]. At a subsequent analysis with additional 11-month follow-up and progression events, a median PFS of 25.3 months for Kisqali plus letrozole and 16.0 months for letrozole alone was observed[2]. Overall survival data is not yet mature and will be available at a later date.
“In the MONALEESA-2 trial, ribociclib plus letrozole reduced the risk of disease progression or death by 44 percent over letrozole alone, and more than half of patients (53%) with measurable disease taking ribociclib plus letrozole experienced a tumor burden reduction of at least 30 percent. This is a significant result for women with this serious form of breast cancer,” said Gabriel N. Hortobagyi, MD, Professor of Medicine, Department of Breast Medical Oncology, The University of Texas MD Anderson Cancer Center and MONALEESA-2 Principal Investigator. “These results affirm that combination therapy with a CDK4/6 inhibitor like ribociclib and an aromatase inhibitor should be a new standard of care for initial treatment of HR+ advanced breast cancer.”
Kisqali is taken with or without food as a once-daily oral dose of 600 mg (three 200 mg tablets) for three weeks, followed by one week off treatment. Kisqali is taken in combination with four weeks of any aromatase inhibitor[1].
Breast cancer is the second most common cancer in American women[3]. The American Cancer Society estimates more than 250,000 women will be diagnosed with invasive breast cancer in 2017[3]. Up to one-third of patients with early-stage breast cancer will subsequently develop metastatic disease[4].
Novartis is committed to providing patients with access to medicines, as well as resources and support to address a range of needs. The Kisqali patient support program is available to help guide eligible patients through the various aspects of getting started on treatment, from providing educational information to helping them understand their insurance coverage and identify potential financial assistance options. For more information, patients and healthcare professionals can call 1-800-282-7630.
The full prescribing information for Kisqali can be found at https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/kisqali.pdf(link is external).
About Kisqali® (ribociclib)
Kisqali (ribociclib) is a selective cyclin-dependent kinase inhibitor, a class of drugs that help slow the progression of cancer by inhibiting two proteins called cyclin-dependent kinase 4 and 6 (CDK4/6). These proteins, when over-activated, can enable cancer cells to grow and divide too quickly. Targeting CDK4/6 with enhanced precision may play a role in ensuring that cancer cells do not continue to replicate uncontrollably.
Kisqali was developed by the Novartis Institutes for BioMedical Research (NIBR) under a research collaboration with Astex Pharmaceuticals.
About the MONALEESA Clinical Trial Program
Novartis is continuing to assess Kisqali through the robust MONALEESA clinical trial program, which includes two additional Phase III trials, MONALEESA-3 and MONALEESA-7, that are evaluating Kisqali in multiple endocrine therapy combinations across a broad range of patients, including premenopausal women. MONALEESA-3 is evaluating Kisqali in combination with fulvestrant compared to fulvestrant alone in postmenopausal women with HR+/HER2- advanced breast cancer who have received no or a maximum of one prior endocrine therapy. MONALEESA-7 is investigating Kisqali in combination with endocrine therapy and goserelin compared to endocrine therapy and goserelin alone in premenopausal women with HR+/HER2- advanced breast cancer who have not previously received endocrine therapy.
About Novartis in Advanced Breast Cancer
For more than 25 years, Novartis has been at the forefront of driving scientific advancements for breast cancer patients and improving clinical practice in collaboration with the global community. With one of the most diverse breast cancer pipelines and the largest number of breast cancer compounds in development, Novartis leads the industry in discovery of new therapies and combinations, especially in HR+ advanced breast cancer, the most common form of the disease.
Kisqali® (ribociclib) Important Safety Information
Kisqali® (ribociclib) can cause a heart problem known as QT prolongation. This condition can cause an abnormal heartbeat and may lead to death. Patients should tell their healthcare provider right away if they have a change in their heartbeat (a fast or irregular heartbeat), or if they feel dizzy or faint. Kisqali can cause serious liver problems. Patients should tell their healthcare provider right away if they get any of the following signs and symptoms of liver problems: yellowing of the skin or the whites of the eyes (jaundice), dark or brown (tea-colored) urine, feeling very tired, loss of appetite, pain on the upper right side of the stomach area (abdomen), and bleeding or bruising more easily than normal. Low white blood cell counts are very common when taking Kisqali and may result in infections that may be severe. Patients should tell their healthcare provider right away if they have signs and symptoms of low white blood cell counts or infections such as fever and chills. Before taking Kisqali, patients should tell their healthcare provider if they are pregnant, or plan to become pregnant as Kisqali can harm an unborn baby. Females who are able to become pregnant and who take Kisqali should use effective birth control during treatment and for at least 3 weeks after the last dose of Kisqali. Do not breastfeed during treatment with Kisqali and for at least 3 weeks after the last dose of Kisqali. Patients should tell their healthcare provider about all of the medicines they take, including prescription and over-the-counter medicines, vitamins, and herbal supplements since they may interact with Kisqali. Patients should avoid pomegranate or pomegranate juice, and grapefruit or grapefruit juice while taking Kisqali. The most common side effects (incidence >=20%) of Kisqali when used with letrozole include white blood cell count decreases, nausea, tiredness, diarrhea, hair thinning or hair loss, vomiting, constipation, headache, and back pain. The most common grade 3/4 side effects in the Kisqali + letrozole arm (incidence >2%) were low neutrophils, low leukocytes, abnormal liver function tests, low lymphocytes, and vomiting. Abnormalities were observed in hematology and clinical chemistry laboratory tests.
Please see the Full Prescribing Information for Kisqali, available at https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/kisqali.pdf(link is external).
About Novartis
Novartis provides innovative healthcare solutions that address the evolving needs of patients and societies. Headquartered in Basel, Switzerland, Novartis offers a diversified portfolio to best meet these needs: innovative medicines, cost-saving generic and biosimilar pharmaceuticals and eye care. Novartis has leading positions globally in each of these areas. In 2016, the Group achieved net sales of USD 48.5 billion, while R&D throughout the Group amounted to approximately USD 9.0 billion. Novartis Group companies employ approximately 118,000 full-time-equivalent associates. Novartis products are sold in approximately 155 countries around the world. For more information, please visit http://www.novartis.com.
Novartis is on Twitter. Sign up to follow @Novartis and @NovartisCancer at http://twitter.com/novartis(link is external) and http://twitter.com/novartiscancer (link is external)
For Novartis multimedia content, please visit www.novartis.com/news/media-library
For questions about the site or required registration, please contact media.relations@novartis.com
References
[1] Kisqali (ribociclib) Prescribing information. East Hanover, New Jersey, USA: Novartis Pharmaceuticals Corporation; March 2016.
[2] Novartis Data on File
[3] American Cancer Society. How Common Is Breast Cancer? Available at https://www.cancer.org/cancer/breast-cancer/about/how-common-is-breast-cancer.html(link is external). Accessed January 23, 2017.
[4] O’Shaughnessy J. Extending survival with chemotherapy in metastatic breast cancer. The Oncologist. 2005;10(Suppl 3):20-29.
рибоциклиб , ريبوسيكليب , 瑞波西利
Ribociclib « New Drug Approvals
////////////////Novartis, Kisqali®, ribociclib, LEE011, FDA 2017, HR+/HER2- metastatic breast cancer, рибоциклиб , ريبوسيكليب , 瑞波西利
FDA approves first treatment Noctiva (Desmopressin acetate) nasal spray for frequent urination at night due to overproduction of urine

Desmopressin acetate
March 3, 2017
The U.S. Food and Drug Administration today approved Noctiva (desmopressin acetate) nasal spray for adults who awaken at least two times per night to urinate due to a condition known as nocturnal polyuria (overproduction of urine during the night). Noctiva is the first FDA-approved treatment for this condition.
“Today’s approval provides adults who overproduce urine at night with the first FDA-approved therapeutic option to help reduce the number of times a night they wake up to urinate,” said Hylton V. Joffe, M.D., M.M.Sc., director of the Division of Bone, Reproductive, and Urologic Products in the FDA’s Center for Drug Evaluation and Research. “It is important to know that Noctiva is not approved for all causes of night-time urination, so patients should discuss their symptoms with their health care provider who can determine the underlying cause of the night-time urination and whether Noctiva is right for them.”
Nocturia (wakening at night to urinate) is a symptom that can be caused by a wide variety of conditions, such as congestive heart failure, poorly controlled diabetes mellitus, medications, or diseases of the bladder or prostate. Before considering Noctiva, health care providers should evaluate each patient for possible causes for the nocturia, and optimize the treatment of underlying conditions that may be contributing to the night-time urination. Because Noctiva is approved only for adults with nocturia caused by nocturnal polyuria, health care providers should confirm overproduction of urine at night with a 24-hour urine collection, if one has not been obtained previously. Health care providers should also be mindful of underlying conditions that can cause nocturia, but that make treatment with Noctiva unsafe, such as excessive drinking of fluids or symptomatic congestive heart failure.
Noctiva is taken daily, approximately 30 minutes before going to bed. It works by increasing the absorption of water through the kidneys, which leads to less urine production.
Noctiva’s efficacy was established in two 12-week, randomized, placebo-controlled trials in 1,045 patients 50 years of age and older with nocturia due to nocturnal polyuria. Although these trials showed a small reduction in the average number of night-time urinations with Noctiva compared to placebo, more patients treated with Noctiva were able to at least halve their number of night-time urinations, and patients treated with Noctiva had more nights with one or fewer night-time urinations.
Noctiva is being approved with a boxed warning and a Medication Guide because it can cause low sodium levels in the blood (hyponatremia). Severe hyponatremia can be life-threatening if it is not promptly diagnosed and treated, leading to seizures, coma, respiratory arrest or death. Health care providers should make sure the patient’s sodium level is normal before starting Noctiva, and should check sodium levels within one week and approximately one month after starting treatment and periodically thereafter. The lower Noctiva dose is recommended as the starting dose for those who may be at risk for hyponatremia, such as the elderly. Noctiva should not be used in patients at increased risk of severe hyponatremia, such as those with excessive fluid intake, those who have illnesses that can cause fluid or electrolyte imbalances, certain patients with kidney damage, and in those using certain medicines, known as loop diuretics or glucocorticoids.
Noctiva should also not be used in patients with symptomatic congestive heart failure or uncontrolled hypertension because fluid retention can worsen these underlying conditions. Use of Noctiva should be discontinued temporarily in patients with certain nasal conditions such as colds or allergies until those conditions have resolved.
Noctiva is also not recommended for the treatment of nocturia in pregnant women. Nocturia is usually related to normal changes in pregnancy that do not require treatment with Noctiva. Noctiva should not be used in children.
The most common side effects of Noctiva in clinical trials included nasal discomfort, cold symptoms (nasopharyngitis), nasal congestion, sneezing, high or increased blood pressure, back pain, nose bleeds, bronchitis and dizziness.
Although there are other FDA-approved medications that also contain desmopressin, none of those medications are approved to treat nocturia.
Noctiva is marketed by Milford, Pennsylvania-based Renaissance Lakewood, LLC for Serenity Pharmaceuticals, LLC.
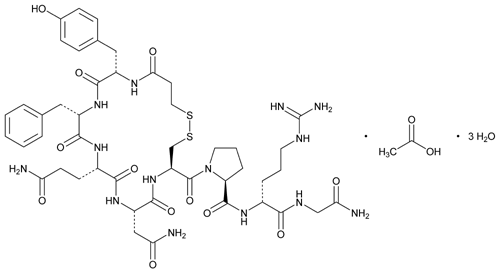
C48H68N14O14S2 C48H68N14O14S2·xH2O
(anhydrous) ![]()
![]()
![]()
![]()
![]() 1129.27
1129.27![]()
![]()
![]() [62288-83-9].
[62288-83-9].
1-(3-Mercaptopropionic acid)-8-D-arginine-vasopressin monoacetate (salt).

oxopentan-2-yl]-1-[4-(2-amino-2-oxoethyl)-7-(3-amino-3-oxopropyl)-10-benzyl-13-[(4-hydroxyphenyl)methyl]-3,6,9,12,15-pentaoxo-18,19-dithia-2,5,8,11,14-pentazacycloicosane-1-carbonyl]pyrrolidine-2-carboxamide;
Synonyms: 3-MERCAPTOPROPIONYL-TYR-PHE-GLN-ASN-CYS-PRO-D-ARG-GLY-NH2 ACETATE SALT;DDAVP ACETATE;[DEAMINO-CYS1,D-ARG8]-VASOPRESSIN ACETATE SALT;DESMOPRESSIN MONOACETATE;DESMORESSIN ACETATE;Mpr-Tyr-Phe-Gln-Asn-Cys-Pro-D-Arg-Gly-NH2(S-S:1-5);DESMOPRESSIN ACETATE;DESMOPRESSIN ACETATE SALT;
The Molecular Weight of Desmopressin Acetate(62288-83-9): 1129.27




Analytica Chimica Acta (2006), 572, (2), 197-204
Abstract
A monolithic column was prepared using l-phenylalanine as template and a covalent approach through the formation of Schiff base with o-phthalaldehyde (OPA). OPA, allylmercaptan, l-phenylalanine, and triethylamine were stirred at first, then methacrylic acid, 2-vinylpyridine, ethyleneglycol dimethacrylate, α,α-azobisisobutyronitrile, and 1-propanol were added to the reaction mixture. The resulting material was introduced into a capillary column. Following thermal polymerization, the template was then extracted with a mixture of HCl and methanol. The column was employed for the capillary electrochromatographic separation of oligopeptides. A capillary column of 75 (50) cm × 75 μm ID with a mobile phase of phosphate buffer (pH 7.0, 40 mM)/methanol (5%, v/v), an applied voltage of +15 kV, and detection at 214 nm, could baseline separate angiotensin I, angiotensin II, [Sar1, Thr8] angiotensin, oxytocin, vasopressin, tocinoic acid, β-casomorphin bovine, β-casomorphin human, and FMRF amide within 20 min. The separation behavior of the templated polymer was also compared with that of the non-templated polymer. As a result, it can be concluded that the electrochromatographic separation of this set of peptides was mediated by a combination of electrophoretic migration and chromatographic retention involving hydrophobic, hydrogen bonding, electrostatic as well as the Schiff base formation with OPA in the cavity of the templated polymer.
PATENT
CN 101372504
WO 2010119450
IN 2009CH00794
CN 103102395
CN 103467574
CN 105131079
CN 104761619
Desmopressin acetate is a structural analogue of natural arginine vasopressin, which is the result of two changes in the chemical structure of natural hormones. The structure is as follows:
M $ a-Tyr-Phe-Gln-Asn-C such as -Pro-D-Arg-GIy-N
Desmopressin acetate has a good hemostatic effect and does not produce side effects of pressurization. Mainly used to treat central diabetes insipidus, hemophilia and therapeutic control of bleeding and preoperative bleeding prevention. Good results and small side effects.
In the existing synthetic method of desmopressin acetate, liquid phase synthesis to produce more waste, the reaction time is long, each coupling an amino acid need to be purified, post-processing cumbersome, low yield, is not conducive to Industrial production.
Solid phase synthesis method, Chinese Patent CN 101372505, CN103992389 using Sieber Amide Resin or Rink Amide AM Resin one by one coupling to obtain linear peptide resin, and then solid-phase oxidation resin, cleavage and purification of desmopressin acetate. Chinese Patent CN103102395, CN102863513 Using Sieber Amide Resin or Rink AM Resin, linear peptide resin was obtained by coupling one by one, and liquid desulfurization was obtained after lysis to obtain desmopressin.
| Patent ID | Patent Title | Submitted Date | Granted Date |
|---|---|---|---|
| US8765152 | Pharmaceutical or neutraceutical formulation | 2010-02-25 | 2014-07-01 |
| Cited Patent | Filing date | Publication date | Applicant | Title | |
|---|---|---|---|---|---|
| US005726287 | Title not available | ||||
| US005990273 | Title not available | ||||
| US20060276626 | May 2, 2006 | Dec 7, 2006 | Avi Tovi | Methods for the production of peptide derivatives | |
| WO2004092202A1 | Apr 5, 2004 | Oct 28, 2004 | Novetide, Ltd. | Process for production of cyclic peptides |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN102863513A * | Sep 12, 2012 | Jan 9, 2013 | 无锡市凯利药业有限公司 | Preparation method of desmopressin acetate |
Oxymetazoline, оксиметазолин , أوكسيميتازولين , 羟甲唑啉
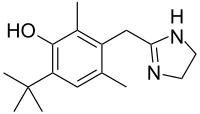
Oxymetazoline
1491-59-4 CAS NUMBER
- Molecular FormulaC16H24N2O
- Average mass260.375 Da
Oxymetazoline is a selective α1 adrenergic receptor agonist and α2 adrenergic receptorpartial agonist. It is a topical decongestant, used in the form of oxymetazoline hydrochloride. It was developed from xylometazoline at E. Merck Darmstadt by Fruhstorfer in 1961.[1]Oxymetazoline is generally available as a nasal spray.

Oxymetazoline HCl; CAS 2315-02-8
Oxymetazoline hydrochloride is a vasoconstrictor. Chemically it is 3-[(4,5-dihydro-1H-imidazol-2-yl)methyl]6-(1,1,-dimethylethyl)-2,4-dimethylphenolmono-hydrochloride. Its molecular weight is 296.8 for the hydrochloride salt and 260.4 for the free base. It is freely soluble in water and ethanol and has a partition coefficient of 0.1 in octanol/water. Its structural formula is:
 |
Medical uses
Oxymetazoline is available over-the-counter as a topical decongestant in the form of oxymetazoline hydrochloride in nasal sprays such as Afrin, Operil, Dristan, Dimetapp, oxyspray, Facimin, Nasivin, Nostrilla, Sudafed OM, Vicks Sinex, Zicam, SinuFrin, and Mucinex Full Force.[2]
Due to its vasoconstricting properties, oxymetazoline is also used to treat nose bleeds[3][4]and eye redness due to minor irritation (marketed as Visine L.R. in the form of eye drops).[5]
Company:
Allergan
Approval Status:
Approved January 2017
Specific Treatments:
facial erythema associated with rosacea
Therapeutic Areas
Dermatology
Find Related Trials for The Following Conditions
Rosacea
General Information
Rhofade (oxymetazoline hydrochloride) is an alpha1A adrenoceptor agonist. Oxymetazoline acts as a vasoconstrictor.
Rhofade is spccifically indicated for the topical treatment of persistent facial erythema associated with rosacea in adults.
Rhofade is supplied as a cream for topical administration. Apply a pea-sized amount of Rhofade cream, once daily in a thin layer to cover the entire face (forehead, nose, each cheek, and chin) avoiding the eyes and lips. Wash hands immediately after applying Rhofade cream.

Side effects and special considerations
Rebound congestion
It is recommended that oxymetazoline not be used for more than three days, as rebound congestion, or rhinitis medicamentosa, may occur.[6] Patients who continue to use oxymetazoline beyond this point may become dependent on the medication to relieve their chronic congestion.
Effects of benzalkonium chloride
Some studies have found that benzalkonium chloride, a common additive to oxymetazoline nasal sprays, may damage nasal epithelia and exacerbate rhinitis medicamentosa. However, the majority of studies find benzalkonium chloride to be a safe preservative.[7]
Use in pregnancy
The Food and Drug Administration places oxymetazoline in category C, indicating risk to the fetus cannot be ruled out. While it has been shown that a single dose does not significantly alter either maternal or fetal circulation,[8] this subject has not been studied extensively enough to draw reliable conclusions.
Overdose
If accidentally ingested, standard methods to remove unabsorbed drugs should be considered.[clarification needed] There is no specific antidote for oxymetazoline, although its pharmacological effects may be reversed by α adrenergic antagonists such as phentolamine. In the event of a possibly life-threatening overdose (such as a hypertensive crisis), benzodiazepines should be considered to decrease the likelihood of seizures and convulsions, as well as reduce anxiety and to lower blood pressure. In children, oxymetazoline may produce profound central nervous system depression due to stimulation of central α2receptors and imidazoline receptors, much like clonidine.
Pharmacology
Mechanism of action
Oxymetazoline is a sympathomimetic that selectively agonizes α1 and, partially, α2 adrenergic receptors.[9] Since vascular beds widely express α1 receptors, the action of oxymetazoline results in vasoconstriction. In addition, the local application of the drug also results in vasoconstriction due to its action on endothelial postsynaptic α2 receptors; systemic application of α2 agonists, in contrast, causes vasodilation because of centrally-mediated inhibition of sympathetic tone via presynaptic α2 receptors.[10] Vasoconstriction of vessels results in relief of nasal congestion in two ways: first, it increases the diameter of the airway lumen; second, it reduces fluid exudation from postcapillary venules.[11] It can reduce nasal airway resistance (NAR) up to 35.7% and nasal mucosal blood flow up to 50%.[12]
Pharmacokinetics
Imidazolines are sympathomimetic agents, with primary effects on α adrenergic receptors and little if any effect on β adrenergic receptors. Oxymetazoline is readily absorbed orally. Effects on α receptors from systemically absorbed oxymetazoline hydrochloride may persist for up to 7 hours after a single dose. The elimination half-life in humans is 5–8 hours. It is excreted unchanged both by the kidneys (30%) and in feces (10%).
History
The oxymetazoline brand Afrin was first sold as a prescription medication in 1966. After finding substantial early success as a prescription medication, it became available as an over-the-counter drug in 1975. Schering-Plough did not engage in heavy advertising until 1986.[13]From the late 1980s to mid 1990s, Afrin featured in many notable television advertisements. Some of these commercials showed men, women, and children using other brands of nasal sprays, and then standing upside down or hanging upside down from playground equipment to prevent their nasal spray from dripping out. This was juxtaposed with Afrin users having no problems.
Society and culture
Brand names
Brand names include Afrin, Dristan, Nasivin, Nezeril, Nostrilla, Logicin, Vicks Sinex, Visine L.R., Sudafed OM, Zicam, SinuFrin and Mucinex Sinus-Max.


References
- Jump up^ German Patent 1,117,588
- Jump up^ “Oxymetazoline: Drug Information Provided by Lexi-Comp: Merck Manual Professional”. Merck.com. Retrieved 2013-04-15.
- Jump up^ Katz, Robert I.; Hovagim, Alec R.; Finkelstein, Harvey S.; Grinberg, Yair; Boccio, Remigio V.; Poppers, Paul J. (1990). “A comparison of cocaine, lidocaine with epinephrine, and oxymetazoline for prevention of epistaxis on nasotracheal intubation”. Journal of Clinical Anesthesia. 2 (1): 16–20. doi:10.1016/0952-8180(90)90043-3. PMID 2310576.
- Jump up^ Krempl, G. A.; Noorily, A. D. (1995). “Use of oxymetazoline in the management of epistaxis”. The Annals of otology, rhinology, and laryngology. 104 (9 Pt 1): 704–6.PMID 7661519.
- Jump up^ “VISINE® Original Red Eye Drops | VISINE® products”. Visine.com. Retrieved2013-04-15.
- Jump up^ Ramey, J. T.; Bailen, E; Lockey, R. F. (2006). “Rhinitis medicamentosa”. Journal of investigational allergology & clinical immunology. 16 (3): 148–55. PMID 16784007.
- Jump up^ Marple, B; Roland, P; Benninger, M (2004). “Safety review of benzalkonium chloride used as a preservative in intranasal solutions: An overview of conflicting data and opinions”. Otolaryngology – Head and Neck Surgery. 130 (1): 131–41.doi:10.1016/j.otohns.2003.07.005. PMID 14726922.
- Jump up^ Rayburn, W. F.; Anderson, J. C.; Smith, C. V.; Appel, L. L.; Davis, S. A. (1990).“Uterine and fetal Doppler flow changes from a single dose of a long-acting intranasal decongestant”. Obstetrics and gynecology. 76 (2): 180–2. PMID 2196495.
- Jump up^ Westfall Thomas C, Westfall David P, “Chapter 6. Neurotransmission: The Autonomic and Somatic Motor Nervous Systems” (Chapter). Brunton LL, Lazo JS, Parker KL: Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11e: http://www.accessmedicine.com/content.aspx?aID=954433.
- Jump up^ Biaggioni Italo, Robertson David, “Chapter 9. Adrenoceptor Agonists & Sympathomimetic Drugs” (Chapter). Katzung BG: Basic & Clinical Pharmacology, 11e: http://www.accessmedicine.com/content.aspx?aID=4520412.
- Jump up^ Widdicombe, John (1997). “Microvascular anatomy of the nose”. Allergy. 52 (40 Suppl): 7–11. doi:10.1111/j.1398-9995.1997.tb04877.x. PMID 9353554.
- Jump up^ Bende, M.; Löth, S. (2007). “Vascular effects of topical oxymetazoline on human nasal mucosa”. The Journal of Laryngology & Otology. 100 (3): 285–8.doi:10.1017/S0022215100099151. PMID 3950497.
- Jump up^ Dougherty, Phillip H. (20 October 1986). “Advertising; Afrin Goes After Users Of Nasal Decongestants”. The New York Times. The New York Times Company. Retrieved 2015-03-30.
 |
|
| Clinical data | |
|---|---|
| Trade names | Afrin, Ocuclear, Drixine |
| AHFS/Drugs.com | Monograph |
| Pregnancy category |
|
| Dependence liability |
Moderate |
| Routes of administration |
Intranasal |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Metabolism | Kidney (30%), fecal (10%) |
| Biological half-life | 5–6 hours |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| ECHA InfoCard | 100.014.618 |
| Chemical and physical data | |
| Formula | C16H24N2O |
| Molar mass | 260.375 g·mol−1 |
| 3D model (Jmol) | |
| Melting point | 301.5 °C (574.7 °F) |
/////////Oxymetazoline, оксиметазолин , أوكسيميتازولين , 羟甲唑啉 , Rhofade, oxymetazoline hydrochloride, alpha1A adrenoceptor agonist, vasoconstrictor
CC1=CC(=C(C(=C1CC2=NCCN2)C)O)C(C)(C)C.Cl
FDA approves Odactra for house dust mite allergies

March 1, 2017
Release
The U.S. Food and Drug Administration today approved Odactra, the first allergen extract to be administered under the tongue (sublingually) to treat house dust mite (HDM)-induced nasal inflammation (allergic rhinitis), with or without eye inflammation (conjunctivitis), in people 18 through 65 years of age.
“House dust mite allergic disease can negatively impact a person’s quality of life,” said Peter Marks, M.D., Ph.D., director of the FDA’s Center for Biologics Evaluation and Research. “The approval of Odactra provides patients an alternative treatment to allergy shots to help address their symptoms.”
House dust mite allergies are a reaction to tiny bugs that are commonly found in house dust. Dust mites, close relatives of ticks and spiders, are too small to be seen without a microscope. They are found in bedding, upholstered furniture and carpeting. Individuals with house dust mite allergies may experience a cough, runny nose, nasal itching, nasal congestion, sneezing, and itchy and watery eyes.
Odactra exposes patients to house dust mite allergens, gradually training the immune system in order to reduce the frequency and severity of nasal and eye allergy symptoms. It is a once-daily tablet, taken year round, that rapidly dissolves after it is placed under the tongue. The first dose is taken under the supervision of a health care professional with experience in the diagnosis and treatment of allergic diseases. The patient is to be observed for at least 30 minutes for potential adverse reactions. Provided the first dose is well tolerated, patients can then take Odactra at home. It can take about eight to 14 weeks of daily dosing after initiation of Odactra for the patient to begin to experience a noticeable benefit.
The safety and efficacy of Odactra was evaluated in studies conducted in the United States, Canada and Europe, involving approximately 2,500 people. Some participants received Odactra, while others received a placebo pill. Participants reported their symptoms and the need to use symptom-relieving allergy medications. During treatment, participants taking Odactra experienced a 16 to 18 percent reduction in symptoms and the need for additional medications compared to those who received a placebo.
The most commonly reported adverse reactions were nausea, itching in the ears and mouth, and swelling of the lips and tongue. The prescribing information includes a boxed warning that severe allergic reactions, some of which can be life-threatening, can occur. As with other FDA-approved allergen extracts administered sublingually, patients receiving Odactra should be prescribed auto-injectable epinephrine. Odactra also has a Medication Guide for distribution to the patient.
Odactra is manufactured for Merck, Sharp & Dohme Corp., (a subsidiary of Merck and Co., Inc., Whitehouse Station, N.J.) by Catalent Pharma Solutions Limited, United Kingdom.
(sublingually) to treat house dust mite (HDM)-induced nasal inflammation (allergic rhinitis), with or without eye inflammation (conjunctivitis), in people 18 through 65 years of age
/////////////Odactra, Merck, Sharp & Dohme Corp, Catalent Pharma Solutions Limited, United Kingdom, FDA 2017, approves, house dust mite allergies
FDA approves Xermelo (telotristat ethyl) for carcinoid syndrome diarrhea

Telotristat ethyl
Molecular Formula, C27-H26-Cl-F3-N6-O3,
Molecular Weight, 574.9884,
RN: 1033805-22-9
UNII: 8G388563M
LX 1032
(2S)-2-Amino-3-[4-[2-amino-6-[[(1R)-1-[4-chloro-2-(3-methylpyrazol-1-yl)phenyl]-2,2,2-trifluoroethyl]oxy]pyrimidin-4-yl]phenyl]propionic acid ethyl ester
Ethyl-4-(2-amino-6-{(1R)-1-[4-chlor-2-(3-methyl-1H-pyrazol-1-yl)phenyl]-2,2,2-trifluorethoxy}-4-pyrimidinyl)-L-phenylalaninat

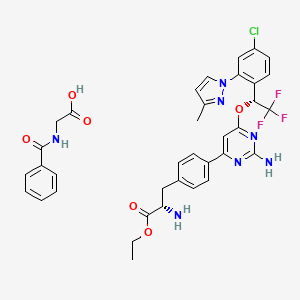
CAS: 1137608-69-5 (etiprate), LX 1606
Chemical Formula: C36H35ClF3N7O6
Molecular Weight: 754.16
- LX 1032 hippurate
- LX 1606



Carcinoid syndrome is a cluster of symptoms sometimes seen in people with carcinoid tumors. These tumors are rare, and often slow-growing. Most carcinoid tumors are found in the gastrointestinal tract. Carcinoid syndrome occurs in less than 10 percent of patients with carcinoid tumors, usually after the tumor has spread to the liver. The tumors in these patients release excess amounts of the hormone serotonin, resulting in diarrhea. Complications of uncontrolled diarrhea include weight loss, malnutrition, dehydration, and electrolyte imbalance.
“Today’s approval will provide patients whose carcinoid syndrome diarrhea is not adequately controlled with another treatment option,” said Julie Beitz, M.D., director of the Office of Drug Evaluation III in the FDA’s Center for Drug Evaluation and Research.
Xermelo, in a regimen with SSA therapy, is approved in tablet form to be taken orally three times daily with food. Xermelo inhibits the production of serotonin by carcinoid tumors and reduces the frequency of carcinoid syndrome diarrhea.
The safety and efficacy of Xermelo were established in a 12-week, double-blind, placebo-controlled trial in 90 adult participants with well-differentiated metastatic neuroendocrine tumors and carcinoid syndrome diarrhea. These patients were having between four to 12 daily bowel movements despite the use of SSA at a stable dose for at least three months. Participants remained on their SSA treatment, and were randomized to add placebo or treatment with Xermelo three times daily. Those receiving Xermelo added on to their SSA treatment experienced a greater reduction in average bowel movement frequency than those on SSA and placebo. Specifically, 33 percent of participants randomized to add Xermelo on to SSA experienced an average reduction of two bowel movements per day compared to 4 percent of patients randomized to add placebo on to SSA.
The most common side effects of Xermelo include nausea, headache, increased levels of the liver enzyme gamma-glutamyl transferase, depression, accumulation of fluid causing swelling (peripheral edema), flatulence, decreased appetite and fever. Xermelo may cause constipation, and the risk of developing constipation may be increased in patients whose bowel movement frequency is less than four bowel movements per day. Patients treated with a higher than recommended dosage of Xermelo developed severe constipation in clinical trials. One patient required hospitalization and two other patients developed complications of either intestinal perforation or intestinal obstruction. Patients should be monitored for severe constipation. If a patient experiences severe constipation or severe, persistent or worsening abdominal pain, they should discontinue Xermelo and contact their healthcare provider.
The FDA granted this application fast track designation and priority review. The drug also received orphan drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
Xermelo is manufactured by Woodlands, Texas-based Lexicon Pharmaceuticals, Inc.
SYNTHESIS…….WO 2011100285

5.67. Synthesis of (S)-2-Amino-3-[4-(2-amino-6-{R-l-[4-chloro-2-(3-methyl-pyrazol-l-yll- phenyll-2,2,2-trifluoro-ethoxy)-pyrimidin-4-yl)-phenyll-propionic acid ethyl ester

The title compound was prepared stepwise, as described below:
Step 1: Synthesis of l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanone. To a 500 ml 2 necked RB flask containing anhydrous methanol (300 ml) was added thionyl chloride (29.2 ml, 400 mmol) dropwise at 0-5°C (ice water bath) over 10 minutes. The ice water bath was removed, and 2-bromo-4-chloro-benzoic acid (25 g, 106 mmol) was added. The mixture was heated to mild reflux for 12h. Progress of the reaction was monitored by TLC and LCMS. After completion of the reaction, the reaction mixture was concentrated. Crude product was dissolved in dichloromethane (DCM, 250 ml), washed with water (50 ml), sat. aq. NaHC03 (50 ml), brine (50 ml), dried over sodium sulfate, and concentrated to give the 2- bromo-4-chloro-benzoic acid methyl ester (26 g, 99 %), which was directly used in the following step.
2-Bromo-4-chloro-benzoic acid methyl ester (12.4 g, 50 mmol) in toluene (200 ml) was cooled to -70°C, and trifluoromethyl trimethyl silane (13 ml, 70 mmol) was added.
Tetrabutylamonium fluoride (1M, 2.5 ml) was added dropwise, and the mixture was allowed to warm to room temperature over 4h, after which it was stirred for 10 hours at room temperature. The reaction mixture was concentrated to give the crude [l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-l-methoxy-ethoxy]-trimethyl-silane. The crude intermediate was dissolved in methanol (100 ml) and 6N HCI (100 ml) was added. The mixture was kept at 45-50°C for 12h. Methanol was removed, and the crude was extracted with dichloromethane (200 ml). The combined DCM layer was washed with water (50 ml), NaHC03 (50 ml), brine (50 ml), and dried over sodium sulfate. Removal of solvent gave a crude product, which was purified by ISCO column chromatography, using 1-2% ethyl acetate in hexane as solvent, to afford l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanone (10 g, 70%). !H-NMR (300 MHz, CDC ): δ (ppm) 7.50 (d,lH), 7.65(d,lH), 7.80(s,lH).
Step 2: Synthesis of R-l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanol. To catechol borane (1M in THF 280 ml, 280 mmol) in a 2L 3-necked RB flask was added S-2-methyl-CBS oxazaborolidine (7.76 g, 28 mmol) under nitrogen, and the resulting mixture was stirred at room temperature for 20 min. The reaction mixture was cooled to -78°C (dry ice/acetone bath), and 1-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanone (40 g, 139 mmol) in THF (400 ml) was added dropwise over 2 hours. The reaction mixture was allowed to warm to -36°C, and was stirred at that temperature for 24 hours, and further stirred at -32 °C for another 24h. 3N NaOH (250 ml) was added, and the cooling bath was replaced by ice-water bath. Then 30 % hydrogen peroxide in water (250 ml) was added dropwise over 30 minutes. The ice water bath was removed, and the mixture was stirred at room temperature for 4 hours. The organic layer was separated, concentrated and re-dissolved in ether (200 ml). The aqueous layer was extracted with ether (2 x 200 ml). The combined organic layers were washed with IN aq. NaOH (4 x 100 ml), brine, and dried over sodium sulfate. Removal of solvent gave crude product which was purified by column chromatography using 2 to 5% ethyl acetate in hexane as solvent to give desired alcohol 36.2 g (90 %, e.e. >95%). The alcohol (36.2 g) was crystallized from hexane (80 ml) to obtain R-l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanol 28.2 g (70 %; 99-100 % e.e.). !H-NMR (400 MHz, CDCIs) δ (ppm) 5.48 (m, 1H), 7.40 (d, 1H), 7.61 (d, 2H).
Step 3: Synthesis of R-l-[4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyll-2.2.2-trifluoro-ethanol. R-l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanol (15.65 g, 54.06 mmol), 3-methylpyrazole (5.33 g, 65 mmol), Cul (2.06 g, 10.8 mmol), 2CO3 (15.7 g, 113.5 mmol), (lR,2R)-N,N’-dimethyl-cyclohexane-l,2-diamine (1.54 g, 10.8 mmol) and toluene (80 ml) were combined in a 250 ml pressure tube and heated to 130°C (oil bath temperature) for 12 hours. The reaction mixture was diluted with ethyl acetate and washed with H2O (4 x 100 ml), brine, and dried over sodium sulfate. Removal of solvent gave a crude product, which was purified by ISCO column chromatography using 5-10 % ethyl acetate in hexane as solvent to get R-l-[4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyl]-2,2,2-trifluoro-ethanol (13.5 g; 86 %). i-H-NMR (400 MHz, CDC ): δ (ppm) 2.30(s, 3H), 4.90(m, 1H), 6.20(s, 1H), 6.84(d, 1H), 7.20(s, 1H), 7.30(d, 1H), 7.50(d, 1H).
Step 4: Synthesis of (S)-2-Amino-3- 4-(2-amino-6-fR-l-[4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyll^^^-trifluoro-ethoxyl-pyrimidin^-yll-phenvD-propionic acid ethyl ester. R-l-[4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyl]-2,2,2-trifluoro-ethanol (17.78 g, 61.17 mmol), (S)-3-[4-(2-amino-6-chloro-pyrimidine-4-yl)-phenyl]-2-tert-butoxycarbonylamino-propionic acid (20.03 g, 51 mmol), 1,4-dioxane (250 ml), and CS2CO3 (79.5 g, 244 mmol) were combined in a 3-necked 500 ml RB flask and heated to 100°C (oil bath temperature) for 12-24 hours. The progress of reaction was monitored by LCMS. After the completion of the reaction, the mixture was cooled to 60°C, and water (250 ml) and THF (400 ml) were added. The organic layer was separated and washed with brine (150 ml). The solvent was removed to give crude BOC protected product, which was taken in THF (400 ml), 3N HCI (200 ml). The mixture was heated at 35-40 °C for 12 hours. THF was removed in vacuo. The remaining aqueous layer was extracted with isopropyl acetate (2x 100 ml) and concentrated separately to recover the unreacted alcohol (3.5 g). Traces of remaining organic solvent were removed from the aqueous fraction under vacuum.
To a 1L beaker equipped with a temperature controller and pH meter, was added H3PO4 (40 ml, 85 % in water) and water (300 ml) then 50 % NaOH in water to adjust pH to 6.15. The temperature was raised to 58 °C and the above acidic aqueous solution was added dropwise into the buffer with simultaneous addition of 50 % NaOH solution in water so that the pH was maintained between 6.1 to 6.3. Upon completion of addition, precipitated solid was filtered and washed with hot water (50-60°C) (2 x 200 ml) and dried to give crude (S)-2-amino-3-[4-(2-amino-6-[R-l-[4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyl]-2,2,2-trifluoro-ethoxy}-pyrimidin-4-yl)-phenyl}^ propionic acid (26.8 g; 95 %). LCMS and HPLC analysis indicated the compound purity was about 96-97 %.
To anhydrous ethanol (400 ml) was added SOC (22 ml, 306 mmol) dropwise at 0-5°C.
Crude acid (26.8 ) from the above reaction was added. The ice water bath was removed, and the reaction mixture was heated at 40-45°C for 6-12 hours. After the reaction was completed, ethanol was removed in vacuo. To the residue was added ice water (300 ml), and extracted with isopropyl acetate (2 x 100 ml). The aqueous solution was neutralized with saturated Na2C03 to adjust the pH to 6.5. The solution was extracted with ethyl acetate (2 x 300 ml). The combined ethyl acetate layer was washed with brine and concentrated to give 24 g of crude ester (HPLC purity of 96-97 %). The crude ester was then purified by ISCO column chromatography using 5 % ethanol in DCM as solvent to give (S)-2-amino-3-[4-(2-amino-6-{R-l-[4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyl]-2,2,2-trifluoro-ethoxy}-pyrimidin-4-yl)-phenyl}-propionic acid ethyl ester (20.5g; 70 %; HPLC purity of 98 %). LCMS M+l = 575. !H-NMR (400 MHz, CDsOD): δ (ppm) 1.10 (t, 3H), 2.25 (s, 3H), 2.85 (m, 2H), 3.65 (m, IH), 4.00 (q, 2H), 6.35 (s, IH), 6.60 (s, IH), 6.90 (m, IH), 7.18 (d, 2H), 7.45 (m, 2H), 7.70 (d, IH), 7.85 (m, 3H).
SYNTHESIS OF INTERMEDIATE
WO 2009048864

https://google.com/patents/WO2009048864A1?cl=en
6.15. Preparation of 6SV3-(4-(2-Amino-6-chloropyrimidin-4-yl)phenyl)-2- (fert-butoxycarbonylamino)propanoic Acid Using the Lithium Salt of (S)-2-(te^-butoxycarbonylamino)-3-(4-(4,4,5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)phenyl)propanoic Acid

During preparation of compound 7, the isolation of the free acid can be optionally omitted. Thus, an aqueous solution of the lithium salt of compound 7 in 100 ml water, prepared from 5.0 g of Boc-Tyr-OMe (4, 17 mmol), was mixed 2-amino-4,6- dichloropyrimidine (3.3 g, 1.2 eq), potassium bicarbonate (5.0 g, 3 eq), bis(triphenylphosphine)palladium(II) dichloride (60 mg, 0.5 mol%), and 100 ml ethanol. The resulting mixture was heated at 700C for 5 hours. Additional 2-amino-4,6- dichloropyrimidine (1.1 g, 0.4 eq) was added and heating was continued at 7O0C for an additional 2 hours. HPLC analysis showed about 94% conversion. Upon cooling and filtration, the filtrate was analyzed by HPLC against a standard solution of compound 8. The assay indicated 3.9 g compound 8 was contained in the solution (59% yield from compound 4).
6.16. Alternative Procedure for Preparation of (S)-3-(4-f2-Amino-6- chloropyrimidin-4-yl)phenyl)-2-(fe^-butoxycarbonylamino)propanoic Acid Using Potassium Carbonate as Base

The boronic acid compound 11 (Ryscor Science, Inc., North Carolina, 1.0 g, 4.8 mmol) and potassium carbonate (1.32 g, 2 eq) were mixed in aqueous ethanol (15 ml ethanol and 8 ml water). Di-ter£-butyldicarbonate (1.25 g, 1.2 eq) was added in one portion. After 30 minutes agitation at room temperature, HPLC analysis showed complete consumption of the starting compound 11. The 2-amino-4,6- dichloropyrimidine (1.18 g, 1.5 eq) and the catalyst bis(triphenylphosphine)palladium(II) dichloride (34 mg, 1 mol%) were added and the resulting mixture was heated at 65-700C for 3 hours. HPLC analysis showed complete consumption of compound 12. After concentration and filtration, HPLC analysis of the resulting aqueous solution against a standard solution of compound 8 showed 1.26 g compound 8 (67% yield).
6.17. Alternative procedure for preparation of (5)-3-(4-(2-Amino-6-

The boronic acid compound 11 (10 g, 48 mmol) and potassium bicarbonate (14.4 g, 3 eq) were mixed in aqueous ethanol (250 ml ethanol and 50 ml water). Oi-tert- butyldicarbonate (12.5 g, 1.2 eq) was added in one portion. HPLC analysis indicated that the reaction was not complete after overnight stirring at room temperature. Potassium carbonate (6.6 g, 1.0 eq) and additional di-te/t-butyldicarbonate (3.1 g, 0.3 eq) were added. After 2.5 hours agitation at room temperature, HPLC analysis showed complete consumption of the starting compound 11. The 2-amino-4,6-dichloropyrimidine (11.8 g, 1.5 eq) and the catalyst bis(triphenylphosphine)-palladium(II) dichloride (0.34 g, 1 mol%” were added and the resulting mixture was heated at 75-8O0C for 2 hours. HPLC analysis showed complete consumption of compound 12. The mixture was concentrated under reduced pressure and filtered. The filtrate was washed with ethyl acetate (200 ml) and diluted with 3 : 1 THF/MTBE (120 ml). This mixture was acidified to pH about 2.4 by 6 N hydrochloric acid. The organic layer was washed with brine and concentrated under reduced pressure. The residue was precipitated in isopropanol, filtered, and dried at 500C under vacuum to give compound 8 as an off-white solid (9.0 g, 48% yield). Purity: 92.9% by HPLC analysis. Concentration of the mother liquor yielded and additional 2.2 g off-white powder (12% yield). Purity: 93.6% by HPLC analysis
PATENT
https://www.google.com/patents/WO2013059146A1?cl=en
This invention is directed to solid pharmaceutical dosage forms in which an active pharmaceutical ingredient (API) is (S)-ethyl 2-amino-3-(4-(2-amino-6-((R)-l-(4-chloro-2-(3- methyl-lH-pyrazol-l-yl)phenyl)-2,2,2-trifluoroethoxy)pyrimidin-4-yl)phenyl)propanoate
(telotristat):

or a pharmaceutically acceptable salt thereof. The compound, its salts and crystalline forms can be obtained by methods known in the art. See, e.g., U.S. patent no. 7,709,493.
PATENT
http://www.google.co.in/patents/WO2008073933A2?cl=en
6.19. Synthesis of (S)-2-Amino-3-r4-q-amino-6-{R-l-r4-chloro-2-(3-methyl- Pyrazol-l-yl)-phenyll-2,2,2-trifluoro-ethoxy}-pyrimidin-4-yl)-phenyll- propionic acid ethyl ester

The title compound was prepared stepwise, as described below: Step 1 : Synthesis of l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanone. To a 500 ml 2 necked RB flask containing anhydrous methanol (300 ml) was added thionyl chloride (29.2 ml, 400 mmol) dropwise at 0-50C (ice water bath) over 10 min. The ice water bath was removed, and 2-bromo-4-chloro-benzoic acid (25 g, 106 mmol) was added. The mixture was heated to mild reflux for 12h. Progress of the reaction was monitored by TLC and LCMS. After completion of the reaction, the reaction mixture was concentrated. Crude product was dissolved in dichloromethane (DCM, 250 ml), washed with water (50 ml), sat. aq. NaHCO3 (50 ml), brine (50 ml), dried over sodium sulfate, and concentrated to give the 2- bromo-4-chloro-benzoic acid methyl ester (26 g, 99 %), which was directly used in the following step.
2-Bromo-4-chloro-benzoic acid methyl ester (12.4 g, 50 mmol) in toluene (200 ml) was cooled to -700C, and trifluoromethyl trimethyl silane (13 ml, 70 mmol) was added. Tetrabutylamonium fluoride (IM, 2.5 ml) was added dropwise, and the mixture was allowed to warm to room temperature over 4h, after which it was stirred for 1Oh at room temperature. The reaction mixture was concentrated to give the crude [l-(2-bromo-4-chloro-phenyl)-2,2,2- trifluoro-l-methoxy-ethoxy]-trimethyl-silane. The crude intermediate was dissolved in methanol (100 ml) and 6N HCl (100 ml) was added. The mixture was kept at 45-500C for 12h. Methanol was removed, and the crude was extracted with dichloromethane (200 ml). The combined DCM layer was washed with water (50 ml), NaHCO3 (50 ml), brine (50 ml), and dried over sodium sulfate. Removal of solvent gave a crude product, which was purified by ISCO column chromatography, using 1-2% ethyl acetate in hexane as solvent, to afford 1- (2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanone (10 g, 70%). 1H-NMR (300 MHz, CDCl3): δ (ppm) 7.50 (d,lH), 7.65(d,lH), 7.80(s,lH).
Step 2: Synthesis of R-l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanol. To catechol borane (IM in THF 280 ml, 280 mmol) in a 2L 3-necked RB flask was added S-2- methyl-CBS oxazaborolidine (7.76 g, 28 mmol) under nitrogen, and the resulting mixture was stirred at room temperature for 20 min. The reaction mixture was cooled to -78°C (dry ice/acetone bath), and l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanone (40 g, 139 mmol) in THF (400 ml) was added dropwise over 2h. The reaction mixture was allowed to warm to -36°C, and was stirred at that temperature for 24 h, and further stirred at -32°C for another 24h. 3N NaOH (250 ml) was added, and the cooling bath was replaced by ice-water bath. Then 30 % hydrogen peroxide in water (250 ml) was added dropwise over 30 minutes. The ice water bath was removed, and the mixture was stirred at room temperature for 4h. The organic layer was separated, concentrated and re-dissolved in ether (200 ml). The aqueous layer was extracted with ether (2 x 200 ml). The combined organic layers were washed with IN aq. NaOH (4 x 100 ml), brine, and dried over sodium sulfate. Removal of solvent gave crude product which was purified by column chromatography using 2 to 5% ethyl acetate in hexane as solvent to give desired alcohol 36.2 g (90 %, e.e. >95%). The alcohol (36.2 g) was crystallized from hexane (80 ml) to obtain R-l-(2-bromo-4-chloro- phenyl)-2,2,2-trifiuoro-ethanol 28.2 g (70 %; 99-100 % e.e.). 1H-NMR (400 MHz, CDCl3) δ (ppm) 5.48 (m, IH), 7.40 (d, IH), 7.61 (d, 2H). Step 3: Synthesis of R-l-r4-chloro-2-(3-methyl-pyrazol-l-vπ-phenyl1-2.2.2-trifluoro- ethanol. R-l-(2-bromo-4-chloro-phenyl)-2,2,2-trifluoro-ethanol (15.65g, 54.06 mmol), 3- methylpyrazole (5.33 g, 65 mmol), CuI (2.06 g, 10.8 mmol), K2CO3 (15.7 g, 113.5 mmol), (lR,2R)-N,N’-dimethyl-cyclohexane-l,2-diamine (1.54 g, 10.8 mmol) and toluene (80 ml) were combined in a 250 ml pressure tube and heated to 1300C (oil bath temperature) for 12 h. The reaction mixture was diluted with ethyl acetate and washed with H2O (4 x 100 ml), brine, and dried over sodium sulfate. Removal of solvent gave a crude product, which was purified by ISCO column chromatography using 5-10 % ethyl acetate in hexane as solvent to get R-I- [4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyl]-2,2,2-trifluoro-ethanol (13.5 g; 86 %). 1H-NMR (400 MHz, CDCl3): δ (ppm) 2.30(s, 3H), 4.90(m, IH), 6.20(s, IH), 6.84(d, IH), 7.20(s, IH), 7.30(d, IH), 7.50(d, IH).
Step 4: Synthesis of (S)-2-Amino-3- r4-(2-amino-6- (R-I- r4-chloro-2-(3-methyl- pyrazol- 1 -ylVphenyl~|-2,2.,2-trifluoro-ethoxy| -pyrimidin-4-yl)-phenyU -propionic acid ethyl ester. R-l-[4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyl]-2,2,2-trifluoro-ethanol (17.78 g, 61.17 mmol), (S)-3-[4-(2-amino-6-chloro-pyrimidine-4-yl)-phenyl]-2-tert- butoxycarbonylamino-propionic acid (20.03 g, 51 mmol), 1,4-dioxane (250 ml), and Cs2CO3 (79.5 g, 244 mmol) were combined in a 3-necked 500 ml RB flask and heated to 1000C (oil bath temperature) for 12-24 h. The progress of reaction was monitored by LCMS. After the completion of the reaction, the mixture was cooled to 600C, and water (250 ml) and THF (400 ml) were added. The organic layer was separated and washed with brine (150 ml). The solvent was removed to give crude BOC protected product, which was taken in THF (400 ml), 3N HCl (200 ml). The mixture was heated at 35-400C for 12h. THF was removed in vacuo. The remaining aqueous layer was extracted with isopropyl acetate (2x 100 ml) and concentrated separately to recover the unreacted alcohol (3.5 g). Traces of remaining organic solvent were removed from the aqueous fraction under vacuum.
To a IL beaker equipped with a temperature controller and pH meter, was added H3PO4 (40 ml, 85 % in water) and water (300 ml) then 50 % NaOH in water to adjust pH to 6.15. The temperature was raised to 58°C and the above acidic aqueous solution was added dropwise into the buffer with simultaneous addition of 50 % NaOH solution in water so that the pH was maintained between 6.1 to 6.3. Upon completion of addition, precipitated solid was filtered and washed with hot water (50-600C) (2 x 200 ml) and dried to give crude (S)-2- amino-3-[4-(2-amino-6-{R-l-[4-chloro-2-(3-methyl-pyrazol-l-yl)-phenyl]-2,2,2-trifluoro- ethoxy}-pyrimidin-4-yl)-phenyl} -propionic acid (26.8 g; 95 %). LCMS and HPLC analysis indicated the compound purity was about 96-97 %. To anhydrous ethanol (400 ml) was added SOCl2 (22 ml, 306 mmol) dropwise at 0-
5°C. Crude acid (26.8 g ) from the above reaction was added. The ice water bath was removed, and the reaction mixture was heated at 40-450C for 6-12h. After the reaction was completed, ethanol was removed in vacuo. To the residue was added ice water (300 ml), and extracted with isopropyl acetate (2 x 100 ml). The aqueous solution was neutralized with saturated Na2CO3 to adjust the pH to 6.5. The solution was extracted with ethyl acetate (2 x 300 ml). The combined ethyl acetate layer was washed with brine and concentrated to give 24 g of crude ester (HPLC purity of 96-97 %). The crude ester was then purified by ISCO column chromatography using 5 % ethanol in DCM as solvent to give (S)-2-amino-3-[4-(2- amino-6- (R- 1 -[4-chloro-2-(3-methyl-pyrazol- 1 -yl)-phenyl]-2,2,2-trifluoro-ethoxy} – pyrimidin-4-yl)-phenyl} -propionic acid ethyl ester (20.5g; 70 %; HPLC purity of 98 %). LCMS M+l = 575. 1H-NMR (400 MHz, CD3OD): δ (ppm) 1.10 (t, 3H), 2.25 (s, 3H), 2.85 (m, 2H), 3.65 (m, IH), 4.00 (q, 2H), 6.35 (s, IH), 6.60 (s, IH), 6.90 (m, IH), 7.18 (d, 2H), 7.45 (m, 2H), 7.70 (d, IH), 7.85 (m, 3H).
PATENT
WO 2011056916
https://www.google.com/patents/WO2011056916A1?cl=en
PATENT
WO 2010065333
CLIP,……..PL CHECK ERROR



REFERENCES
Kulke, M.H.; Hoersch, D.; Caplin, M.E.; et al.
Telotristat ethyl, a tryptophan hydroxylase inhibitor for the treatment of carcinoid syndrome
J Clin Oncol 2017, 35(1): 14
| WO2010056992A1 * | Nov 13, 2009 | May 20, 2010 | The Trustees Of Columbia University In The City Of New York | Methods of preventing and treating low bone mass diseases |
| US7709493 | May 20, 2009 | May 4, 2010 | Lexicon Pharmaceuticals, Inc. | 4-phenyl-6-(2,2,2-trifluoro-1-phenylethoxy)pyrimidine-based compounds and methods of their use |
| US20090088447 * | Sep 25, 2008 | Apr 2, 2009 | Bednarz Mark S | Solid forms of (s)-ethyl 2-amino-3-(4-(2-amino-6-((r)-1-(4-chloro-2-(3-methyl-1h-pyrazol-1-yl)phenyl)-2,2,2-trifluoroethoxy)-pyrimidin-4-yl)phenyl)propanoate and methods of their use |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US9199994 | Sep 5, 2014 | Dec 1, 2015 | Karos Pharmaceuticals, Inc. | Spirocyclic compounds as tryptophan hydroxylase inhibitors |
| US9512122 | Sep 1, 2015 | Dec 6, 2016 | Karos Pharmaceuticals, Inc. | Spirocyclic compounds as tryptophan hydroxylase inhibitors |
///////////telotristat ethyl, fast track designation,priority review,orphan drug designation, Xermelo , Woodlands, Texas-based, Lexicon Pharmaceuticals, Inc, fda 2017, LX 1606, LX 1032
O=C(OCC)[C@@H](N)Cc1ccc(cc1)c2cc(nc(N)n2)O[C@H](c3ccc(Cl)cc3n4ccc(C)n4)C(F)(F)F
O=C(OCC)[C@@H](N)CC1=CC=C(C2=NC(N)=NC(O[C@H](C3=CC=C(Cl)C=C3N4N=C(C)C=C4)C(F)(F)F)=C2)C=C1.O=C(O)CNC(C5=CC=CC=C5)=O
Deflazacort
Deflazacort
- CAS 14484-47-0
- Molecular Formula C25H31NO6
- Average mass 441.517 Da
- 5’βH-Pregna-1,4-dieno[17,16-d]oxazole-3,20-dione, 11β,21-dihydroxy-2′-methyl-, 21-acetate (8CI)
- (11β,16β)-21-(Acetyloxy)-11-hydroxy-2′-methyl-5’H-pregna-1,4-dieno[17,16-d]oxazole-3,20-dione
- 2H-Naphth[2′,1′:4,5]indeno[1,2-d]oxazole, 5’H-pregna-1,4-dieno[17,16-d]oxazole-3,20-dione deriv.
- Azacort
- Azacortinol
- Calcort
- DL 458IT
- Deflan
| Optical Rotatory Power | +62.3 ° | Conc: 0.5 g/100mL; Solv: chloroform (67-66-3); Wavlength: 589.3 nm |
…………..REF, “Drugs – Synonyms and Properties” data were obtained from Ashgate Publishing Co. (US)Hoechst Marion Roussel (now Aventis Pharma) has developed and launched Deflazacort (Dezacor; Flantadin; Lantadin; Calcort) a systemic corticosteroid developed for the treatment of a variety of inflammatory conditions .
In March 1990, the drug was approved in Spain, and by January 2013, the drug had been launched by FAES Farma . By the end of 1999, the product had been launched in Germany, Italy, Belgium, Switzerland and South Korea
Deflazacort is a corticosteroid first launched in 1985 by Guidotti in Europe for the oral treatment of allergic asthma, rheumatoid arthritis, arthritis, and skin allergy.
In 2017, an oral formulation developed at Marathon Pharmaceuticals was approved by the FDA for the treatment of Duchenne’s muscular dystrophy in patients 5 years of age and older.
Deflazacort (trade name Emflaza or Calcort among others) is a glucocorticoid used as an anti-inflammatory and immunosuppressant.
In 2013, orphan drug designation in the U.S. was assigned to the compound for the treatment of Duchenne’s muscular dystrophy. In 2015, additional orphan drug designation in the U.S. was assigned for the treatment of pediatric juvenile idiopathic arthritis (JIA) excluding systemic JIA.
Also in 2015, deflazacort was granted fast track and rare pediatric disease designations in the U.S. for the treatment of Duchenne’s muscular dystrophy.
| Deflazacort is a glucocorticoid used as an anti-inflammatory and immunosuppressant. It was approved in February, 2017 by the FDA for use in treatment of Duchenne muscular dystrophy (trade name Emflaza). |
- Aventis Pharma (Originator), Lepetit (Originator), Guidotti (Licensee), Shire Laboratories (Licensee)
February 9, 2017 FDA approved
The U.S. Food and Drug Administration today approved Emflaza (deflazacort) tablets and oral suspension to treat patients age 5 years and older with Duchenne muscular dystrophy (DMD), a rare genetic disorder that causes progressive muscle deterioration and weakness. Emflaza is a corticosteroid that works by decreasing inflammation and reducing the activity of the immune system.
Corticosteroids are commonly used to treat DMD across the world. This is the first FDA approval of any corticosteroid to treat DMD and the first approval of deflazacort for any use in the United States.
“This is the first treatment approved for a wide range of patients with Duchenne muscular dystrophy,” said Billy Dunn, M.D., director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research. “We hope that this treatment option will benefit many patients with DMD.”
DMD is the most common type of muscular dystrophy. DMD is caused by an absence of dystrophin, a protein that helps keep muscle cells intact. The first symptoms are usually seen between 3 and 5 years of age and worsen over time. The disease often occurs in people without a known family history of the condition and primarily affects boys, but in rare cases it can affect girls. DMD occurs in about one of every 3,600 male infants worldwide.
People with DMD progressively lose the ability to perform activities independently and often require use of a wheelchair by their early teens. As the disease progresses, life-threatening heart and respiratory conditions can occur. Patients typically succumb to the disease in their 20s or 30s; however, disease severity and life expectancy vary.
The effectiveness of deflazacort was shown in a clinical study of 196 male patients who were 5 to 15 years old at the beginning of the trial with documented mutation of the dystrophin gene and onset of weakness before age 5. At week 12, patients taking deflazacort had improvements in a clinical assessment of muscle strength across a number of muscles compared to those taking a placebo. An overall stability in average muscle strength was maintained through the end of study at week 52 in the deflazacort-treated patients. In another trial with 29 male patients that lasted 104 weeks, deflazacort demonstrated a numerical advantage over placebo on an assessment of average muscle strength. In addition, although not statistically controlled for multiple comparisons, patients on deflazacort appeared to lose the ability to walk later than those treated with placebo.
The side effects caused by Emflaza are similar to those experienced with other corticosteroids. The most common side effects include facial puffiness (Cushingoid appearance), weight gain, increased appetite, upper respiratory tract infection, cough, extraordinary daytime urinary frequency (pollakiuria), unwanted hair growth (hirsutism) and excessive fat around the stomach (central obesity).
Other side effects that are less common include problems with endocrine function, increased susceptibility to infection, elevation in blood pressure, risk of gastrointestinal perforation, serious skin rashes, behavioral and mood changes, decrease in the density of the bones and vision problems such as cataracts. Patients receiving immunosuppressive doses of corticosteroids should not be given live or live attenuated vaccines.
The FDA granted this application fast track designation and priority review. The drug also received orphan drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The sponsor is receiving a rare pediatric disease priority review voucher under a program intended to encourage development of new drugs and biologics for the prevention and treatment of rare pediatric diseases. A voucher can be redeemed by a sponsor at a later date to receive priority review of a subsequent marketing application for a different product. This is the ninth rare pediatric disease priority review voucher issued by the FDA since the program began.
Emflaza is marketed by Marathon Pharmaceuticals of Northbrook, Illinois.
Medical uses
The manufacturer lists the following uses for deflazacort:[1]
- Acute interstitial nephritis
- Anaphylaxis
- Asthma
- Autoimmune haemolytic anaemia
- Bullous pemphigoid
- Mixed connective tissue disease (other than systemic sclerosis)
- Crohn’s disease
- Dermatomyositis
- Idiopathic thrombocytopenic purpura
- Juvenile chronic arthritis
- Severe hypersensitivity reactions
- Immunosuppression in transplantation
- Acute and lymphatic leukaemia
- Malignant lymphoma
- Multiple myeloma
- Rheumatoid arthritis
- Polymyalgia rheumatica
- Nephrotic syndrome
- Pemphigus
- Polyarteritis nodosa
- Pyoderma gangrenosum
- Sarcoidosis
- Systemic lupus erythematosus
- Ulcerative colitis
In the United States, deflazacort is only FDA-approved for the treatment of Duchenne muscular dystrophy in people over the age of 5.



Adverse effects
Deflazacort carries the risks common to all corticosteroids, including immune suppression, decreased bone density, and endocrine insufficiency. In clinical trials, the most common side effects (>10% above placebo) were Cushing’s-like appearance, weight gain, and increased appetite.[2]
Pharmacology
Mechanism of action
Deflazacort is an inactive prodrug which is metabolized rapidly to the active drug 21-desacetyldeflazacort.[3]
Relative potency
Deflazacort’s potency is around 70–90% that of prednisone.[4] A 2017 review found its activity of 7.5 mg of deflazacort is approximately equivalent to 25 mg cortisone, 20 mg hydrocortisone, 5 mg of prednisolone or prednisone, 4 mg of methylprednisolone or triamcinolone, or 0.75 mg of betamethasone or dexamethasone. The review noted that the drug has a high therapeutic index, being used at initial oral doses ranging from 6 to 90 mg, and probably requires a 50% higher dose to induce the same demineralizing effect as prednisolone. Thus it has “a smaller impact on calcium metabolism than any other synthetic corticosteroid, and therefore shows a lower risk of growth rate retardation in children and of osteoporosis” in the elderly, and comparatively small effects on carbohydrate metabolism, sodium retention, and hypokalemia.[5]
History
In January 2015, the FDA granted fast track status to Marathon Pharmaceuticals to pursue approval of deflazacort as a potential treatment for Duchenne muscular dystrophy, a rare, “progressive and fatal disease” that affects boys.[6] Although deflazacort was approved by the FDA for use in treatment of Duchenne muscular dystrophy on February 9, 2017,[7][8] Marathon CEO announced on February 13, 2017 that the launch of deflazacort (Emflaza) would be delayed amidst controversy over the steep price Marathon was asking for the drug – $89,000-a-year. In Canada the same drug can be purchased for around $1 per tablet.[9] Marathon has said that Emflaza is estimated to cost $89,000/year which is “roughly 70 times” more than it would cost overseas.[10] Deflazacort is sold in the United Kingdom under the trade name Calcort;[4] in Brazil as Cortax, Decortil, and Deflanil; in India as Moaid, Zenflav, Defolet, DFZ, Decotaz, and DefZot; in Bangladesh as Xalcort; in Panama as Zamen; Spain as Zamene; and in Honduras as Flezacor.[11]
SYNTHESIS

Worlddrugtracker drew this
1 Protection of the keto groups in pregna-1,4-diene derivative with NH2NHCOOMe using HCOOH, yields the corresponding methyl ester.
2 Cleavage of epoxide with NH3 in DMAc/DMF gives amino-alcohol,
3 which on esterification with acetic anhydride in the presence of AcOH furnishes acetate.
4 Cyclization of amine using NaOH, Na2CO3 or K2CO3 produces oxazoline derivative ,
5 which is finally deprotected with HCl to afford Deflazacort
SYNTHESIS FROM CHEMDRUG

The cyclization of 17alpha-azido-3beta,16alpha-acetoxy-5alpha-pregnane-11,20-dione (I) by hydrogenation with H2 over Pt in methanol, followed by a treatment with 10% HCl gives 3beta-hydroxy-5alpha-pregnane-11,20-dione-[17alpha,16alpha-d]-2′-methyloxazoline (II), which is converted into the semicarbazone (III) by treatment with semicarbazide hydrochloride (A) and pyridine in refluxing methanol. The reduction of one ketonic group of (III) with NaBH4 in refluxing ethanol yields the dihydroxy-semicarbazone (IV), which is hydrolyzed with 10% HCl in refluxing methanol to afford the ketodiol (V). The oxidation of (V) with cyclohexanone and aluminum isopropoxide in refluxing toluene gives 11beta-hydroxy-5alpha-pregnane-3,20-dione-[17alpha,16alpha-d]-2′-methyloxazoline (VI). The dehydrogenation of (VI) by treatment with Br2 in dioxane-acetic acid, followed by treatment with Li2CO3 in DMF at 140 C yields the corresponding 1,4-diene derivative (VII). Finally, the reaction of (VII) with I2 by means of azobisisobutyronitrile in CH2Cl2 affords the corresponding 21-iodo compound, which is then acetylated with triethylammonium acetate in refluxing acetone.

The monoacetylation of (V) with acetic anhydride and pyridine at 100 C gives the 3-acetoxy-11-hydroxy compound (IX), which is dehydrated by treatment with methanesulfonyl chloride and then with sodium acetate yielding 3beta-acetoxy-5alpha-pregn-9(11)-ene-20-one-[17alpha,16alpha-d]-2′-methyloxazoline (X). The hydrolysis of (X) with KOH in refluxing methanol affords the corresponding hydroxy compound (XI), which is acetoxylated by treatment with I2 and AZBN as before giving the iodo derivative (XII), and then with triethylammonium acetate also as before, yielding 3beta-hydroxy-21-acetoxy-5alpha-pregn-9(11)-ene-20-one-[17alpha,16alpha-d]-2′-methyloxazoline (XIII). The oxidation of (XIII) with CrO3 in acetone yields the 3,20-diketone (XIV), which by treatment with Br2 and Li2CO3 as before is dehydrogenated affording the 1,4,9(11)-pregnatriene (XV). Finally, the reaction of (XV) with N-bromoacetamide in THF yields 9alpha-bromo-11beta-hydroxy-21-acetoxy-5alpha-pregna-1,4-dieno-3,20-dione-[17alpha,16alpha-d]-2′-methyloxazoline (XVI), which is then debrominated by reaction with chromous acetate and butanethiol in DMSO.
PAPER
Journal of Medicinal Chemistry (1967), 10(5), 799-802
Steroids Possessing Nitrogen Atoms. III. Synthesis of New Highly Active Corticoids. [17α,16α,-d]Oxazolino Steroids
PATENT
CN 105622713
PATENT CN 106008660
MACHINE TRANSLATED FROM CHINESE may seem funny



Description of the drawings
[0007] Figure 1 is a map of the traditional method of the combination process;
Figure 2 is a two-step method of the present invention.
detailed description
[0008] In order to more easily illustrate the gist and spirit of the present invention, the following examples illustrate:
Example 1
A: Preparation of hydroxylamine
In a 100 ml three-necked flask, 20 g of 16 (17) a-epoxy prednisolone, 30 ml of DMF, 300 ml of chloroform was added and incubated at 30-35 ° C with 8 g of ammonia gas at 1-2 atmospheres Reaction 16 ~ 20 hours, TLC detection reaction end point, after the reaction, the vacuum exhaust ammonia gas, add 3x100ml saturated brine washing 3 times, plus 10ml pure water washing times, then, under reduced pressure to chloroform to dry, add 200ml Ethyl acetate, Ig activated carbon, stirring reflux 60-90 minutes, cooling to 50-55 degrees, hot filter, l-2ml ethyl acetate washing carbon, combined filtrate and lotion, and then below 500C concentrated under pressure 95 % Of ethyl acetate, the system cooled to -5-0 ° C, stirring crystallization 2 ~ 3 hours, filter, 0.5-lml ethyl acetate washing, lotion and filtrate combined sets of approved; filter cake below 70 ° C Drying, get hydroxylamine 18.2g, HPLC content of 99.2%, weight loss of 91%.
[0009] B: Preparation of terracavir
Add 10 g of hydroxylamine, 150 ml of glacial acetic acid and 150 ml of acetic anhydride in a 100 ml three-necked flask. Add 5 g of concentrated sulfuric acid under stirring at room temperature. The reaction was carried out at 30-35 ° C for 12-16 hours. TLC confirmed the end of the reaction. Add 500ml of pure water, and adjust the pH of 7.5.5 with liquid alkali, cool to 10 ~ 15 ° C, stirring crystallization 2-3 hours, filtration, washing to neutral, combined filtrate and lotion, pretreated into Waste water treatment tank, filter cake below 70 V drying, Texaco can be special crude 112.5g, HPLC content of 98.2%, the yield of 112.5% ο the above terracotta crude dissolved in 800ml of alcohol, add 5g activated carbon, Decolorization 1-1.5 hours, hot filter, 10ml alcohol detergent cake, lotion and filtrate combined, atmospheric pressure recovery of about 90% of the alcohol, and then cooled to -5-0 ° C, frozen crystal 2-3 hours, Filtration, filter cake with 4-5ml alcohol washing, 70 ° C below drying, digoxin special product 89.2g, melting point 255.5-256.0 degrees, HPLC content of 99.7%, yield 89.2%. The mother liquor is recycled with solvent and crude.
[0010] Example II
A: Preparation of hydroxylamine
In a 100 ml three-necked flask, 20 g of 16 (17) a-epoxy prednisolone, 120 ml of toluene was added and incubated at 30-35 ° C with 8 g of ammonia and 16 to 20 at atmospheric pressure The reaction was carried out in the presence of 3 x 50 ml of saturated brine and 50 ml of pure water was added. Then, the toluene was dried under reduced pressure to dryness, and 200 ml of ethyl acetate, Ig activated carbon was added, and the mixture was stirred. Reflux 60-90 minutes, cool to 50-55 ° C, hot filter, l2ml ethyl acetate wash carbon, combined filtrate and lotion, and then below 500C under reduced pressure 95% ethyl acetate, the system cooling To 5-0C, stirring crystallization 2 ~ 3 hours, filter, 0.5-lml ethyl acetate washing, lotion and filtrate combined sets of the next batch; filter cake 70 ° C below drying, hydroxylamine 18.0g, HPLC content 99.1%, 90% by weight.
[0011] B: Preparation of terracavir
Add 10 g of hydroxylamine, 500 ml of chloroform and 150 ml of acetic anhydride in a 100 ml three-necked flask, add 5 g of p-toluenesulfonic acid under stirring at room temperature, and incubate at 30-35 ° C for 12-16 hours. TLC confirms the reaction end, After the addition of 500ml of pure water, and with the liquid alkali pH 7.55, down to 10 ~ 15 ° C, stirring 0.5_1 hours, separate the water layer, washed to neutral, combined with water and lotion, pretreated into Waste water treatment tank, organic layer under reduced pressure concentrated chloroform to near dry, adding 200ml hexane, reflux 0.5-1 hours, slowly cooling to -5 ~ O0C, stirring crystallization 2-3 hours, filter, filter cake with 4-5ml Alcohol washing, the filtrate and lotion combined apply to the next batch, the filter cake below 70 ° C drying, Texaco can crude 110.5g, HPLC content of 98.4%, the yield of 110.5%. The above-mentioned diltiazem crude product dissolved in 800ml alcohol, add 5g activated carbon, temperature reflux bleaching 1-1.5 hours, hot filter, 10ml alcohol washing cake, lotion and filtrate combined, atmospheric pressure recovery of about 90% of the alcohol And then cooled to -500C, frozen crystallization for 2-3 hours, filtration, filter cake with 4-5ml alcohol washing, 70 ° C the following drying, digester can special products 88.6g, melting point 255.0-256.0 degrees, HPLC content of 99.5%, the yield of 88.6%. The mother liquor is recycled with solvent and crude.
[0012] Example 3
A: Preparation of hydroxylamine
Add 20 g of 16 (17) a-epoxy prednisolone to 120 ml of ethanol in a 100 ml three-necked flask and incubate at 30-35 ° C with stirring to give Sg ammonia at 16 to 20 hours , TLC test reaction end point, after the reaction, vacuum exhaust ammonia gas, concentrated ethanol to the near dry, cooling, adding 300ml chloroform, stirring dissolved residue, and then add 3x100ml saturated brine washing, plus 10ml pure water washing, washing And then concentrated to reduce the chloroform to dry, add 200ml of ethyl acetate, Ig activated carbon, stirring reflux 60-90 minutes, cooling to 50-55 ° C, hot filter, l2ml ethyl acetate washing carbon, combined filtrate and lotion And then concentrated below 50 ° C to 95% ethyl acetate under reduced pressure. The system was cooled to -5-0 0C, stirred for 2 to 3 hours, filtered, 0.5-l of ethyl acetate, washed and filtrate The filter cake was dried at 70 ° C, 18.6 g of hydroxylamine, 99.5% of HPLC, and 93% by weight.
[0013] B: Preparation of terracavir
In a 100ml three-necked flask, add 10g of hydroxylamine, 500ml toluene, 150ml acetic anhydride, stirring at room temperature by adding 5g concentrated sulfuric acid, insulation at 30-35 degrees stirring reaction 12-16 hours, TLC confirmed the end of the reaction, after the reaction, Add 500ml of pure water, and liquid pH adjustment pH 7.5, cooling to 1 ~ 15 ° C, stirring 0.5-1 hours, the water layer, washed to neutral, combined with water and lotion, pretreated into the wastewater The cells were dried and the organic layer was concentrated to dryness under reduced pressure. 200 ml of hexane was added and refluxed
0.5-1 hours, slowly cool to -5 ~ O0C, stirring crystallization 2-3 hours, filtration, filter cake with 4-5ml hexane, the filtrate and lotion combined apply to the next batch, filter cake below 70 ° C Drying, digoxin crude 112.5g, HPLC content of 97.4%, the yield of 112,5% ο will be the above terracotta crude dissolved in 800ml of alcohol, add 5g activated carbon, heating reflux bleaching 1-1.5 hours, while Hot filter, 10ml alcohol detergent cake, lotion and filtrate combined, atmospheric pressure recovery of about 90% of the alcohol, and then cooled to -500C, frozen crystallization for 2-3 hours, filter, filter cake with 4-5ml alcohol Washing, 70 ° C below the dry, Diges can special products 86.2g, melting point 255.5-256.0 degrees, HPLC content of 99.8%, the yield of 86.2%. The mother liquor is recycled with solvent and crude.
PATENT
https://www.google.com/patents/CN101418032A?cl=en


Example 1
21- bromo -ll (3- hydroxy – pregna–l, 4- diene -3, 20-dione [170, 16o-d] -2′- methyl-oxazoline (4) Preparation:
A dry fitted with a thermometer, a reflux condenser, magnetically stirred flask was added 250mL three compound (2) (19.17 g; Fw: 383.48; 50 mmol), N- bromosuccinimide (9.79 g; Fw: 178.00; 55 mmol), 150 ml of ether; then ammonium acetate (0.39 g; Fw: 77.08; 0.005 mmol) added to the system. System continues to stir at 20 ° C 0.5 h, the reaction is complete. After completion of the reaction was filtered to remove the white precipitate cake was washed with 50 mL of dichloromethane, and the combined organic Xiangde pale yellow clear liquid, the solvent was evaporated under reduced pressure to give a pale yellow solid 21.27 g, yield: 92%, HPLC content of greater than 95%.
Example 2
21- bromo -lip- hydroxy – pregna–l, 4- diene -3, 20-dione [17 “16o-d] -2′- methyl-oxazoline (4) Preparation:
A dry fitted with a thermometer, a reflux condenser, magnetically stirred flask were added sequentially 250mL three compound (2) (19.17 g; Fw: 383.48; 50 mmol), N- bromosuccinimide (9.79 g; Fw : 178.00; 55 mmol), 150 ml of toluene; then ammonium acetate (0.39 g; Fw: 77.08; 0.005 mmol) added to the system. System continues to stir at 110 ° C 5 h, the reaction is complete. After completion of the reaction was cooled to room temperature, the white precipitate was removed by filtration cake was washed with 50 mL of dichloromethane, and the combined organic Xiangde pale yellow clear liquid, concentrated under reduced pressure to remove the solvent to give a pale yellow solid 19.65 g, yield: 85%, HPLC content greater than 95%.
Example 3
21 Jie bromo -11 – hydroxy – pregna-1,4-diene -3, 20-dione [17a, 16o-d] -2′- methyl-oxazoline (4) Preparation:
A dry fitted with a thermometer, a reflux condenser, magnetically stirred flask were added sequentially 250mL three compound (2) (19.17 g; Fw: 383.48; 50 mmol), 1,3- dibromo-5,5-dimethyl- Hein (35.74 g; Fw: 285.94; 125 mmol), 150 ml of ether; then ammonium acetate (0.39 g; Fw: 77.08; 0.005 mmol) added to the system. System Stirring was continued at reflux for 3 h, the reaction was completed. After completion of the reaction a white precipitate was removed by filtration and the cake was washed with 50 mL of diethyl ether, and the combined organic Xiangde pale yellow clear liquid, concentrated under reduced pressure to remove the solvent to give a pale yellow solid 16.18 g, yield: 70%, HPLC content greater than 92%.
Example 4
21- bromo -11 Jie – hydroxy – pregna-1,4-diene -3, 20- dione [17c, 16o-d] -2′- methyl-oxazoline (4) Preparation:
A dry fitted with a thermometer, a reflux condenser, magnetically stirred flask were added sequentially 250mL three compound (2) (19.17 g; Fw: 383.48; 50 mmol), 1,3- dibromo-5,5-dimethyl- Hein (35.74 g; Fw: 285.94; 125 mmol), 150 ml dichloromethane; followed by ammonium acetate (0.039 g; Fw: 77.08; 0.0005 mmol) added to the system. System Stirring was continued at reflux for 24 h, the reaction was completed. After completion of the reaction a white precipitate was removed by filtration and the cake was washed with 50 mL of diethyl ether, and the combined organic Xiangde pale yellow clear liquid, concentrated under reduced pressure to remove the solvent to give a pale yellow solid 16.41 g, yield: 71%, HPLC content of greater than 92. / 0.
Example 5
Deflazacort Preparation:
In a nitrogen-filled dry fitted with a thermometer, magnetic stirring and a reflux condenser 100 mL three-necked flask was charged with Compound (4) (11.56 g; Fw: 462.38; 25 mmol), followed by addition of sodium acetate (8.20g; Fw: 82.03; lOOmmol), 50 mL methanol was added to the system.
Then tetrabutylammonium bromide (O. 81g; Fw: 322.38; 2.5 mmol). Warmed to 50 ° C with stirring
48 h. Until after the completion of the reaction was cooled to room temperature. After completion of the reaction, temperature of the system was cooled to room temperature, the system was supplemented with chloroform 50mL, filtered, and the filter cake was washed with small amount of chloroform and then to confirm that no product was dissolved, and the combined organic phases, the organic phase washed with 10% aqueous sodium carbonate paint 3 times, saturated sodium chloride once. The organic phase was dried over anhydrous sodium sulfate, the inorganic salt was removed to give a pale yellow liquid, was concentrated to dryness, purified ethyl acetate to give the product 9.93g, yield 90%, HPLC content> 990/0.
Example 6
Deflazacort Preparation –
In a nitrogen-filled dry fitted with a thermometer, magnetic stirring and a reflux condenser 100 mL three-necked flask was charged with Compound (4) (11.56 g; Fw: 462.38; 25 mmol), followed by addition of anhydrous potassium acetate (3.68g; Fw: 98.14; 37.5 mmol), 50 mL acetone was added to the system. Followed by tetrabutylammonium iodide (0.10g; Fw: 369.37; 0.25 mmol). Heated to reflux with stirring 2h. Until after the completion of the reaction was cooled to room temperature. After completion of the reaction, temperature of the system was cooled to room temperature, the system was supplemented with chloroform 50mL, filtered, and the filter cake was washed with small amount of chloroform and then to confirm that no product was dissolved, and the combined organic phases, the organic phase was washed 3 times with 10% aqueous sodium carbonate , washed once with saturated sodium chloride. The organic phase was dried over anhydrous sodium sulfate, the inorganic salt was removed to give a pale yellow liquid, was concentrated to dryness, ethyl acetate was purified to give the product 10.93 g, yield 99%, HPLC content> 99%.
Example 7
Deflazacort Preparation:
In a nitrogen-filled dry fitted with a thermometer, magnetic stirring and a reflux condenser 100 mL three-necked flask was charged with Compound (4) (11.56 g; Fw: 462.38; 25 mmol), followed by addition of anhydrous potassium acetate (3.68g; Fw: 98.14; 37.5 mmol), 50 mL acetonitrile was added to the system. Followed by tetrabutylammonium iodide (0.10g; Fw: 369.37; 0.25 mmol). Heated to reflux with stirring 2h. Until after the completion of the reaction was cooled to room temperature. After completion of the reaction, temperature of the system was cooled to room temperature, the system was supplemented with chloroform 50mL, filtered, and the filter cake was washed with small amount of chloroform and then to confirm that no product was dissolved, and the combined organic phases, the organic phase was washed 3 times with 10% aqueous sodium carbonate , washed once with saturated sodium chloride. The organic phase was dried over anhydrous sodium sulfate, the inorganic salt was removed to give a pale yellow liquid, was concentrated to dryness, ethyl acetate was purified to give the product 10.93 g, yield 99%, HPLC content> 99%.
Example 8
Deflazacort Preparation:
In a nitrogen-filled dry fitted with a thermometer, magnetic stirring and a reflux condenser 100 mL three-necked flask was charged with Compound (4) (11.56 g; Fw: 462.38; 25 mmol), followed by addition of anhydrous potassium acetate (2.45g; Fw: 98.14; 25 mmol), the N, N- dimethylformamide, 50 mL added to the system. Followed by tetrabutylammonium iodide (O.IO g; Fw: 369.37; 0.25 mmol). Warmed to 120. C stirring 2h. Until after the completion of the reaction was cooled to room temperature. After completion of the reaction, temperature of the system was cooled to room temperature, the system was supplemented with chloroform 50mL, filtered, and the filter cake was washed with small amount of chloroform and then to confirm that no product was dissolved, and the combined organic phases, the organic phase was washed 3 times with 10% aqueous sodium carbonate , washed once with saturated sodium chloride. The organic phase was dried over anhydrous sodium sulfate, the inorganic salt was removed to give a pale yellow liquid, was concentrated to dryness, ethyl acetate was purified to give the product 10.93 g, yield 99%, HPLC content> 99o / q.
PATENT
https://www.google.com/patents/WO1997021722A1?cl=zh
compound (llβ,16β)-21-(acetyloxy)-11- hydroxy-2 ‘ -methyl-5 ‘H-pregna-1, -dieno[17 , 16-d Joxazole- 3,20-dione, also known, and hereinafter referred to, with the INN (International Nonproprietary Name) deflazacort. Deflazacort is represented by the following formula I

Deflazacort is employed in therapy aince some years as a calcium-sparing corticoid agent. This compound belongs to the more general class of pregneno-oxazolines, for which anti-inflammatory, glucocorticoid and hormone-like pharmacological activities are reported. Examples of compounds of the above class, comprising deflazacort, are disclosed in US 3413286, where deflazacort is referred to as llβ-21-dihydroxy-2 ‘ -methyl-5 ‘ βH-pregna-1,4-dieno.17 , 16- d]oxazole-3,20-dione 21-acetate.
According to the process disclosed by US 3413286, deflazacort is obtained from 5-pregnane-3β-ol-ll , 20- dione-2 ‘-methyloxazoline by 2 , -dibromination with Br2– dioxane, heating the product in the presence of LiBr- iC03 for obtaining the 1,4-diene, and converting this latter into the 21-iodo and then into the desired 21- acetyloxy compound. By hydrolysis of deflazacort, the llβ-21-dihydroxy-2 ‘ -methyl-5 ‘βH-pregna-1, -dieno[ 17 , 16- d-]oxazoline-3, 20-dione of formula II is obtained:

The compound of formula II is preferably obtained according to a fermentation process disclosed in
EP-B-322630; in said patent, the compound of formula II is referred to as llβ-21-dihydroxy-2 ‘-methyl-5 ‘ βH- pregna-1,4-dieno[17,16-d-]oxazoline-3,20-dione.
The present invention provides a new advantageous single-step process for obtaining deflazacort, by acetylation of the compound of formula II.
CLIP

tructure of deflazacort and its forced degradation product (A), chromatogram plot of standard deflazacort (B), contour plot of deflazacort (C). Deflazacort was found to be a stable drug under stress condition such as thermal, neutral and oxidative condition. However, the forceddegradation study on deflazacort showed that the drug degraded under alkaline, acid and photolytic conditions.

Mass fragmentation pathway for degradant product of deflazacort.
PATENT

Example 1: Protective reaction To the reaction flask was added 20 g of 1,4-diene-11? -hydroxy-16,17-epoxy_3,20-dione pregnone (Formula I) 20% of the aqueous solution of glacial acetic acid 300g, stirring 5 minutes, temperature 10 ° C ~ 15 ° C, adding ethyl carbazate 14g, temperature control 30 ° C reaction 6 hours; TLC detection reaction is complete, cooling to 0 ° C ~ 5 ° C for 2 hours, until dry, washed to neutral; 60 ° C vacuum dry to dry creatures 20. 5g; on P, oxazoline ring reaction The above protective products into the reaction bottle, add 41ml Of the DMAC dissolved, temperature 25 ~ 30 ° C, access to ammonia, to keep the reaction bottle micro-positive pressure, the reaction of 32 hours, atmospheric pressure exhaust ammonia and then decompression pumping ammonia for 30 minutes; 5 ° C, temperature 5 ~ 0 ° C by adding 5ml glacial acetic acid, then add 21ml acetic anhydride, heated to 35 ° C reaction 4 hours, the sample to confirm the reaction completely; slowly add 5% sodium hydroxide solution 610ml and heated to 60 ~ 70 ° C reaction 2 hours; point plate to confirm the end of the reaction, cooling to 50 ° C, half an hour by adding refined concentrated hydrochloric acid 40ml, insulation 50 ~ 55 ° C reaction 10 hours; to the end of the reaction temperature to room temperature, chloroform Extraction, drying and filtration, concentration of at least a small amount of solvent, ethyl acetate entrained twice, leaving a small amount of solvent, frozen crystallization filter high purity [17a, 16a-d] terfu Kete intermediate. Example 2: Protective reaction 20 g of 1,4-diene-l1-la-hydroxy-16,17-epoxy_3,20_dione progestin (Formula I) was added to the reaction flask and 15% Formic acid solution 300g, stirring for 5 minutes, temperature 10 ~ 15 ° C, adding methyl carbazate 12g, temperature control 30 ° C reaction 5 hours to test the end of the reaction, cooling to O ~ 5 ° C stirring 2 hours crystallization, Suction to dry, washed to neutral; 60 ° C vacuum drying to dry protection of 20g; on P, oxazoline ring reaction The protection of the reaction into the reaction flask, add 30ml of DMF dissolved, temperature control 25 ~ 30 ° C, access to ammonia, keep the reaction bottle in the micro-positive pressure, reaction 30 hours, atmospheric pressure exhaust ammonia and then decompression pumping ammonia for 30 minutes, ice water cooled to 5 ° C, temperature 5 ~ 10 ° C add 5ml of glacial acetic acid, then add 20ml acetic anhydride, heated to 30 ° C reaction for 5 hours to confirm the reaction is complete; slowly add 20% sodium carbonate aqueous solution 500ml and heated to 60 ~ 70 ° C reaction 4 hours, the point plate to confirm the reaction The temperature of 55 ~ 60 ° C for 10 hours; to be the end of the reaction temperature to room temperature, chloroform extraction, drying and filtration, concentration of a small amount of solvent, acetic acid isopropyl The ester was entrained twice, leaving a small amount of solvent, frozen and crystallized to obtain high purity [17a, 16a-d] oxazoline residues. [0024] Example 3: Protective reaction 20 g of I, 4-diene-16,17-epoxy-3,11,20-triketone pregnone (Formula I) was added to the reaction flask and 20% Formic acid solution 300g, stirring for 5 minutes, temperature 10 ~ 15 ° C, adding hydrazine carbamate 15g, temperature control 30 ° C reaction 5 hours to test the end of the reaction, cooling to O ~ 5 ° C stirring 2 hours crystallization, To the dry, washed to neutral; 60 ° C vacuum drying to dry protection of 22g; on P, oxazoline ring reaction of the protection of the reaction into the bottle, add 30ml of DMAC dissolved temperature control 35 ~ 40 ° C, access to ammonia, keep the reaction bottle in the micro-positive pressure, reaction 40 hours, atmospheric pressure exhaust ammonia and then decompression pumping ammonia for 30 minutes, ice water cooling to 5 ° C, temperature 5 ~ 10 ° C add 5ml of glacial acetic acid, then add 20ml acetic anhydride, heated to 40 ° C reaction 5 hours to confirm the reaction is complete; slowly add 20% potassium carbonate aqueous solution 500ml and heated to 60 ~ 70 ° C reaction 7 hours, the point plate to confirm the reaction The temperature of the reaction to the end of the temperature to room temperature, chloroform extraction, drying filter, concentrated to a small amount of solvent, acetic acid isopropyl The ester was entrained twice, leaving a small amount of solvent, frozen and crystallized to obtain high purity [17a, 16a-d] oxazoline residues.
PATENT

xample 1
[0028] A 30 g 16, 17 α- epoxy – pregn -20- substituting methyl hydrazine -3-acetyl-1,4-diene, 11- dione (a) and 150 mL of chloroform and 15 mLDMF mixed, pressure reactor, stirring ammonia gas to the reactor pressure to 0.15 MPa (during ventilation control the reaction temperature at 10-15 ° C), 30 ° C heat reaction, TLC track the progress of the reaction. Completion of the reaction, the material was transferred to a glass reaction flask, the temperature of the material to be reduced to below 10 ° C, add acetic acid adjusted to pH 5 to 6, the solvent was removed under reduced pressure; reaction flask was added 30 mL of acetic acid, 30 g of acetic anhydride, The reaction temperature was controlled at 30 ° C, the reaction 6 hours, the reaction mixture was poured into cold 500 mL10% sodium hydroxide solution, stirred for 1 hour, filtration to give product 30.6 g, 102% mass yield, product by HPLC , a purity of 95.2%.
[0029] Example 2
[0030] A 30 g 16, 17 α- epoxy – pregn -20- substituting methyl hydrazine -3-acetyl-1,4-diene, 11- dione (a) and 150 mL of chloroform and 30 mL of pyridine were mixed, added pressure reactor, stirring ammonia gas to the reactor pressure to 0. 15 MPa (during ventilation control the reaction temperature at 10~15 ° C), 15 ° C heat reaction, TLC track the progress of the reaction. Completion of the reaction, the material was transferred to a glass reaction flask, the temperature of the material to be reduced to below 10 ° C, add acetic acid adjusted to pH 5 to 6, the solvent was removed under reduced pressure; reaction flask was added 30 mL of acetic acid, 30 g of acetic anhydride, The reaction temperature was controlled at 30 ° C, the reaction 6 hours, the reaction mixture was poured into cold 500 mL10% sodium hydroxide solution, stirred for 1 hour, filtration to give product 28.6 g, yield 95% by mass, product by HPLC , a purity of 94.8%.
[0031] Example 3
[0032] A 30 g 16, 17 α- epoxy – pregn -20- substituting methyl hydrazine -3-acetyl-1,4-diene, 11- dione (a) and 150 mL of chloroform and 30 mLDMF mixed, pressure reactor, stirring ammonia gas to the reactor pressure to 0.15 MPa (during ventilation control the reaction temperature at 10~15 ° C), 40 ° C heat reaction, TLC track the progress of the reaction.Completion of the reaction, the material was transferred to a glass reaction flask, the temperature of the material to be reduced to below 10 ° C, add acetic acid adjusted to pH 5 to 6, the solvent was removed under reduced pressure; reaction flask was added 30 mL of acetic acid, 30 g of acetic anhydride, The reaction temperature was controlled at 30 ° C, the reaction for 6 hours. The reaction mixture was poured into cold 500 mL10% sodium hydroxide solution, stirred for 1 hour, filtration to give the product 31.2 g, yield 104% quality products by HPLC , a purity of 95.4%.
[0033] Example 4
[0034] A 30 g 16, 17 α- epoxy – pregn -20- substituting methyl hydrazine -3-acetyl-1,4-diene, 11- dione (a) and 150 mL of chloroform and 30 mLDMF mixed, pressure reactor, stirring ammonia gas to the reactor pressure to 0.5 MPa (during ventilation control the reaction temperature at 10~15 ° C), 40 ° C heat reaction, TLC track the progress of the reaction. Completion of the reaction, the material was transferred to a glass reaction flask, the temperature of the material to be reduced to below 10 ° C, add acetic acid adjusted to pH 5 to 6, the solvent was removed under reduced pressure; reaction flask was added 30 mL of acetic acid, 30 g of acetic anhydride, The reaction temperature was controlled at 30 ° C, the reaction 6 hours, the reaction mixture was poured into cold 500 mL10% sodium hydroxide solution, stirred for 1 hour, filtration to give the product 31. I g, 102% mass yield, product by by HPLC, the purity was 95.2%.
[0035] Example 5
[0036] A 30 g 16, 17 α- epoxy – pregn -20- substituting methyl hydrazine -3-acetyl-1,4-diene, 11- dione (a) and 150 mL of chloroform and 30 mLDMF mixed, pressure reactor, stirring ammonia gas to the reactor pressure to 0.15 MPa (during ventilation control the reaction temperature at 10~15 ° C), 40 ° C heat reaction, TLC track the progress of the reaction. Completion of the reaction, the material was transferred to a glass reaction flask, the temperature of the material to be reduced to below 10 ° C, add acetic acid adjusted to pH 5 to 6, the solvent was removed under reduced pressure; reaction flask was added 60 mL of acetic acid, 15 g of acetic anhydride, The reaction temperature was controlled at 30 ° C, the reaction 6 hours, the reaction mixture was poured into cold 500 mL10% sodium hydroxide solution, stirred for 1 hour, filtration to give the product 29. 5 g, yield 98% by mass, the product of by HPLC, purity of 95%.
[0037] Example 6
[0038] A 30 g 16, 17 α- epoxy – pregn -20- substituting methyl hydrazine -3-acetyl-1,4-diene, 11- dione (a) and 150 mL of chloroform and 30 mLDMF mixed, pressure reactor, stirring ammonia gas to the reactor pressure to 0.15 MPa (during ventilation control the reaction temperature at 10~15 ° C), 40 ° C heat reaction, TLC track the progress of the reaction. The reaction was complete, the material was transferred to a glass reaction flask until the material temperature drops below 10 ° C, plus acetic acid to adjust the pH to 5 to 6, the solvent was removed under reduced pressure; the reaction flask was added 30 mL of acetic acid, 30 g of maleic dianhydride, the reaction temperature was controlled at 30 ° C, the reaction 6 hours, the reaction mixture was poured into cold 500 mL10% sodium hydroxide solution, stirred for 1 hour, filtration to give the product 30 g, 100% mass yield, product by HPLC purity of 95.2%.
[0039] Example 7
[0040] A 30 g 16, 17 α- epoxy – pregn -20- substituting methyl hydrazine -3-acetyl-1,4-diene, 11- dione (a) and 150 mL of chloroform and 30 mLDMF mixed, pressure reactor, stirring ammonia gas to the reactor pressure to 0.15 MPa (during ventilation control the reaction temperature at 10~15 ° C), 40 ° C heat reaction, TLC track the progress of the reaction. Completion of the reaction, the material was transferred to a glass reaction flask, the temperature of the material to be reduced to below 10 ° C, add acetic acid adjusted to pH 5 to 6, the solvent was removed under reduced pressure; reaction flask was added 30 mL of acetic acid, 30 g of propionic anhydride, The reaction temperature was controlled at 30 ° C, the reaction for 6 hours. The reaction mixture was poured into cold 500 mL10% sodium hydroxide solution, stirred for 1 hour, filtration to give the product 27.6 g, 92% yield of quality products by HPLC , a purity of 93.5%.
[0041] Example 8
[0042] A 30 g 16, 17 α- epoxy – pregn -20- substituting methyl hydrazine -3-acetyl-1,4-diene, 11- dione (a) and 150 mL of chloroform and 30 mLDMF mixed, pressure reactor, stirring ammonia gas to the reactor pressure to 0.15 MPa (during ventilation control the reaction temperature at 10~15 ° C), 40 ° C heat reaction, TLC track the progress of the reaction. Completion of the reaction, the material was transferred to a glass reaction flask, the temperature of the material to be reduced to below 10 ° C, add acetic acid adjusted to pH 5 to 6, the solvent was removed under reduced pressure; reaction flask was added 30 mL of acetic acid, 30 g of acetic anhydride, The reaction temperature is controlled at 50 ° C, the reaction for 6 hours. The reaction mixture was poured into cold 500 mL10% sodium hydroxide solution, stirred for 1 hour, filtration to give the product 29.8 g, 99% yield of quality products by HPLC , a purity of 94.8%.
References
- Jump up^ “Refla: deflazacort” (PDF).
- Jump up^http://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208684s000,208685s000lbl.pdf
- Jump up^ Möllmann, H; Hochhaus, G; Rohatagi, S; Barth, J; Derendorf, H (1995). “Pharmacokinetic/pharmacodynamic evaluation of deflazacort in comparison to methylprednisolone and prednisolone”. Pharmaceutical Research. 12 (7): 1096–100. PMID 7494809.
- ^ Jump up to:a b “Calcort”. electronic Medicines Compendium. June 11, 2008. Retrieved on October 28, 2008.
- Jump up^ Luca Parente (2017). “Deflazacort: therapeutic index, relative potency and equivalent doses versus other corticosteroids”. BMC Pharmacol Toxicol. doi:10.1186/s40360-016-0111-8.
- Jump up^ Ellen Jean Hirst (January 19, 2015), Duchenne muscular dystrophy drug could get OK for U.S. sales in 2016, The Chicago Tribune, retrieved February 13, 2017,
has been shown to prolong lives … a progressive and fatal disease that has no drug treatment available in the US
- Jump up^ “FDA approves drug to treat Duchenne muscular dystrophy”. http://www.fda.gov. 2017-02-09. Retrieved 2017-02-10.
- Jump up^ “Marathon Pharmaceuticals to Charge $89,000 for Muscular Dystrophy Drug”. http://www.wsj.com. 2017-02-10. Retrieved 2017-02-10.
- Jump up^ Clifton Sy Mukherjee (February 10, 2017). “Brainstorm Health Daily”. Retrieved February 13, 2017.
- Jump up^ Joseph Walker and Susan Pulliam (February 13, 2017), Marathon Pharmaceuticals to Charge $89,000 for Muscular Dystrophy Drug After 70-Fold Increase, The Wall Street Journal, retrieved February 13, 2017,
FDA-approved deflazacort treats rare type of disease affecting boys
- Jump up^ “Substâncias: DEFLAZACORT” (in Portuguese). Centralx. 2008. Retrieved on October 28, 2008.
 |
|
| Clinical data | |
|---|---|
| Trade names | Emflaza, Calcort, others |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration |
By mouth |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Protein binding | 40% |
| Metabolism | By plasma esterases, to active metabolite |
| Biological half-life | 1.1–1.9 hours (metabolite) |
| Excretion | Renal (70%) and fecal (30%) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| ECHA InfoCard | 100.034.969 |
| Chemical and physical data | |
| Formula | C25H31NO6 |
| Molar mass | 441.517 g/mol |
| 3D model (Jmol) | |
| CN102746358A * | Apr 22, 2011 | Oct 24, 2012 | 天津金耀集团有限公司 | Novel technology for synthesis of pregnane 21-bit bromide |
| CN102746358B * | Apr 22, 2011 | Feb 10, 2016 | 天津金耀集团有限公司 | 一种合成孕甾21位溴化物的工艺 |
| CN102936274A * | Nov 12, 2012 | Feb 20, 2013 | 浙江仙居君业药业有限公司 | Preparation method for [17alpha, 16alpha-d] methyl oxazoline |
| CN102936274B * | Nov 12, 2012 | Apr 1, 2015 | 江西君业生物制药有限公司 | Preparation method for [17alpha, 16alpha-d] methyl oxazoline |
///////FDA 2017, Emflaza, Calcort, Deflazacort, orphan drug designation, FAST TRACK
|
[H][C@@]12C[C@@]3([H])[C@]4([H])CCC5=CC(=O)C=C[C@]5(C)[C@@]4([H])[C@@]([H])(O)C[C@]3(C)[C@@]1(N=C(C)O2)C(=O)COC(C)=O
|
FDA approves new psoriasis drug Siliq (brodalumab)
FDA approves new psoriasis drug
The U.S. Food and Drug Administration today approved Siliq (brodalumab) to treat adults with moderate-to-severe plaque psoriasis. Siliq is administered as an injection.
For Immediate Release
February 15, 2017
Release
The U.S. Food and Drug Administration today approved Siliq (brodalumab) to treat adults with moderate-to-severe plaque psoriasis. Siliq is administered as an injection.
Siliq is intended for patients who are candidates for systemic therapy (treatment using substances that travel through the bloodstream, after being taken by mouth or injected) or phototherapy (ultraviolet light treatment) and have failed to respond, or have stopped responding to other systemic therapies.
“Moderate-to-severe plaque psoriasis can cause significant skin irritation and discomfort for patients, and today’s approval provides patients with another treatment option for their psoriasis,” said Julie Beitz, M.D., director of the Office of Drug Evaluation III in the FDA’s Center for Drug Evaluation and Research. “Patients and their health care providers should discuss the benefits and risks of Siliq before considering treatment.”
Psoriasis is a skin condition that causes patches of skin redness and flaking. Psoriasis is an autoimmune disorder that occurs more commonly in patients with a family history of the disease, and most often begins in people between the ages of 15 and 35. The most common form of psoriasis is plaque psoriasis, in which patients develop thick, red skin with flaky, silver-white scales.
Siliq’s active ingredient (brodalumab) binds to a protein that causes inflammation, inhibiting the inflammatory response that plays a role in the development of plaque psoriasis.
Siliq’s safety and efficacy were established in three randomized, placebo-controlled clinical trials with a total of 4,373 adult participants with moderate-to-severe plaque psoriasis who were candidates for systemic therapy or phototherapy. More patients treated with Siliq compared to placebo had skin that was clear or almost clear, as assessed by scoring of the extent, nature and severity of psoriatic changes of the skin.
Suicidal ideation and behavior, including completed suicides, have occurred in patients treated with Siliq during clinical trials. Siliq users with a history of suicidality or depression had an increased incidence of suicidal ideation and behavior compared to users without this history. A causal association between treatment with Siliq and increased risk of suicidal ideation and behavior has not been established.
Because of the observed risk of suicidal ideation and behavior, the labeling for Siliq includes a Boxed Warning and the drug is only available through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the Siliq REMS Program. Notable requirements of the Siliq REMS Program include the following:
- Prescribers must be certified with the program and counsel patients about this risk. Patients with new or worsening symptoms of depression or suicidality should be referred to a mental health professional, as appropriate.
- Patients must sign a Patient-Prescriber Agreement Form and be made aware of the need to seek medical attention should they experience new or worsening suicidal thoughts or behavior, feelings of depression, anxiety or other mood changes.
- Pharmacies must be certified with the program and must only dispense to patients who are authorized to receive Siliq.
Siliq is also approved with a Medication Guide to inform patients of the risk of suicidal ideation and behavior, and that because Siliq is a medication that affects the immune system, patients may have a greater risk of getting an infection, or an allergic or autoimmune condition. Patients with Crohn’s disease should not use Siliq. Health care providers should also evaluate patients for tuberculosis (TB) infection prior to initiating treatment with Siliq. Health care providers should not administer Siliq to patients with active TB infection, and should avoid immunizations with live vaccines in patients being treated with Siliq.
The most common adverse reactions reported with the use of Siliq include joint pain (arthralgia), headache, fatigue, diarrhea, throat pain (oropharyngeal pain), nausea, muscle pain (myalgia), injection site reactions, influenza, low white blood cell count (neutropenia) and fungal (tinea) infections.
Siliq is marketed by Bridgewater, New Jersey-based Valeant Pharmaceuticals.

{kind=link}