
Home » EU 2023
Category Archives: EU 2023
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
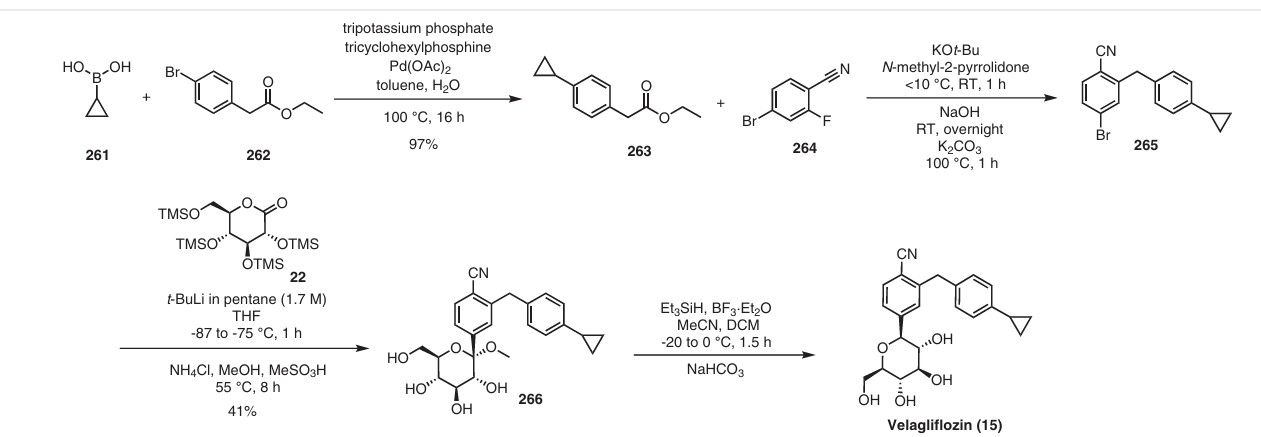
Velagliflozin
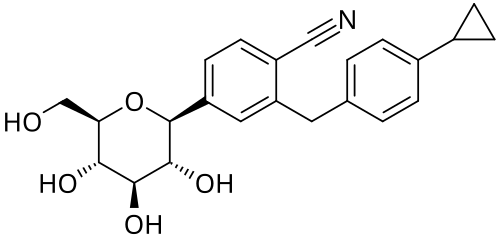
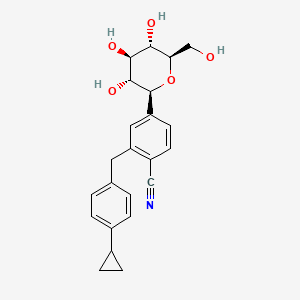
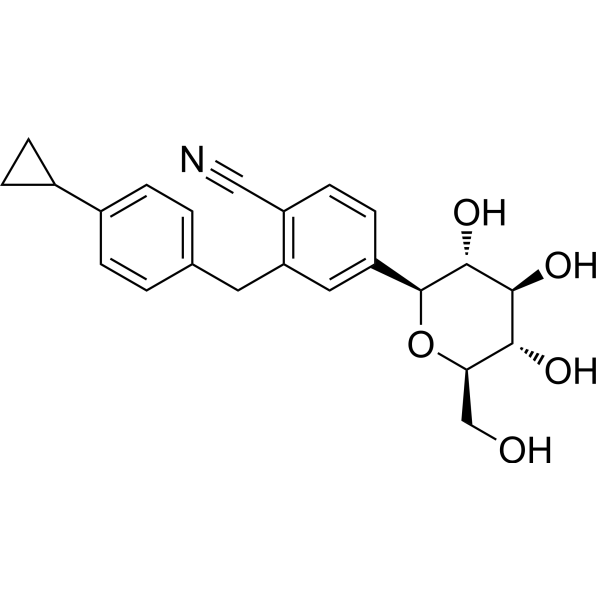
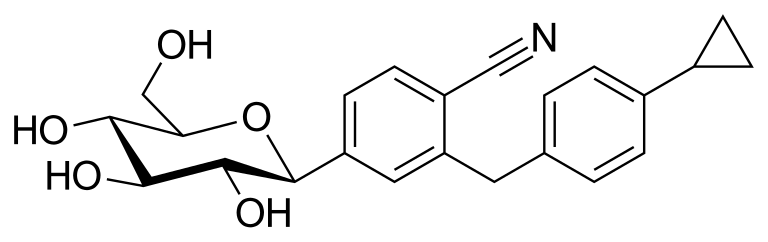
Velagliflozin
VETERINARY DRUG
- Cas 946525-65-1
- FV2YU8SL0P
- 2-((4-cyclopropylphenyl)methyl)-4-((2S,3R,4R,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl)benzonitrile
- 2-((4-Cyclopropylphenyl)methyl)-4-beta-D-glucopyranosylbenzonitrile
- 395.4 g/mol, C23H25NO5
2-[(4-cyclopropylphenyl)methyl]-4-[(2S,3R,4R,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]benzonitrile
- 2-((4-CYCLOPROPYLPHENYL)METHYL)-4-.BETA.-D-GLUCOPYRANOSYLBENZONITRILE
- BENZONITRILE, 2-((4-CYCLOPROPYLPHENYL)METHYL)-4-.BETA.-D-GLUCOPYRANOSYL-
Velagliflozin L-proline H2O
Velagliflozin, sold under the brand name Senvelgo, is an antidiabetic medication used for the treatment of cats.[2][4][5] Velagliflozin is a sodium-glucose cotransporter 2 (SGLT2) inhibitor.[6] It is taken by mouth.[2]
Velagliflozin is the active ingredient of the first oral liquid medication approved by the Food and Drug Administration for the treatment of diabetes in cats. This compound belongs to the known class of sodium-glucose cotransporter 2 inhibitors approved to treat diabetes in human.
- Application: NADA 141-568Drug: Senvelgo®Active Ingredient(s): VelagliflozinCompany: Boehringer lngelheim Animal Health USA Inc.Patent(s): 7776830 (Exp: 05/01/2027); 8557782 (Exp: 05/01/2027); 9145434 (Exp: 09/07/2033); 10617666 (Exp: 06/06/2035); 11896574 (Exp: 12/17/2034); 10220017 (Exp: 09/29/2036); 10709683 (Exp: 08/24/2036); 11225500 (Exp: 12/17/2038)
- [Indication for Use] To improve glycemic control in otherwise healthy cats with diabetes mellitus not previously treated with insulin.Application: NADA 141-568Active Ingredient(s): VelagliflozinCompany: Boehringer lngelheim Animal Health USA Inc.Freedom of Information: FOIA Summary 14320Approval Date: August 10, 2023
APPROVALS 2023, GDA 2023, EU 2023, EMA 2023, SENVELGO
Velagliflozin (brand name Senvelgo) is a veterinary medication approved for treating diabetes in cats, not humans.
Approved countries and years for velagliflozin:
- United States (US): Approved by the FDA in August 2023.
- European Union (EU): Received marketing authorization in November 2023.
- Switzerland: Approved in 2023.
- Great Britain: Approved in 2023.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US310904480&_cid=P11-METCZG-99171-1
SYN
US7776830
https://patentscope.wipo.int/search/en/detail.jsf?docId=US41880220&_cid=P11-METD0X-00376-1
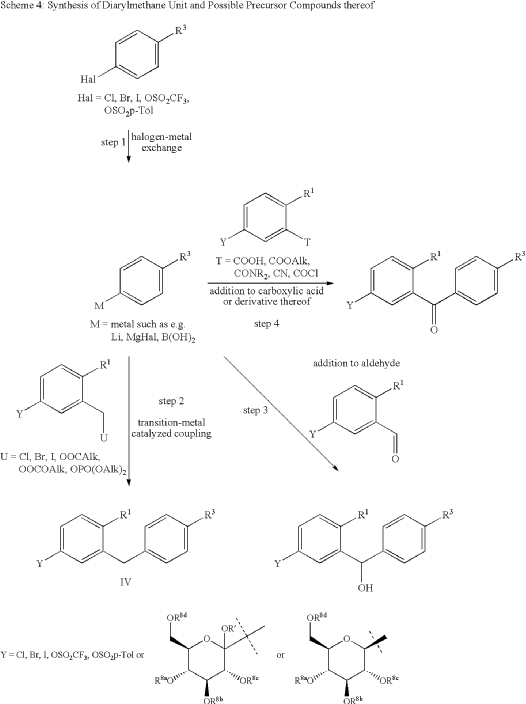
| The following compound is obtained analogously to Example XXIV: |
(1) 1-Cyano-2-(4-cyclopropyl-benzyl)-4-(β-D-glucopyranos-1-yl)-benzene
EXAMPLE 17
2-(4-Cyclopropyl-benzyl)-4-(β-D-glucopyranos-1-yl)-benzonitrile
| The compound is obtained according to example 6 using 4-cyclopropyl-phenylboronic acid as the coupling partner. |
SYN
WO2007128749
https://patents.google.com/patent/WO2007128749A1/en
The following compound is obtained analogously to Example XXIV:
(1 ) 1 -Cvano-2-(4-cvclopropyl-benzyl)-4-(3-D-glucopyranos-1 -vD-benzene

Mass spectrum (ESI“): m/z = 413 [M+H] + Advantageously, the reduction of the anomeric carbon center of the appropriate intermediate obtained during the synthesis of this compound is conducted with the oxygen functionalities on the pyranose ring protected. Preferred protective groups are benzyl, p-methoxybenzyl, trimethylsilyl, triethylsilyl, terfbutyldimethylsilyl, triisopropylsilyl and allyl.
Example XXV

1-Cyano-2-(4-cyclopropyl-benzyl)-4-(tetra-O-acetyl-β-D-glucopyranos-1-yl)-benzene To a flask charged with a stir bar, 4-(2,3,4,6-tetra-O-acetyl-D-glucopyranos-1-yl)-2-(4- trifluoromethylsulfonyloxy-benzyl)-benzonitrile (4.4 g), degassed toluene (12 ml.) and degassed water (8 ml.) and kept under argon atmosphere is added cyclopropylboronic acid (0.20 g), potassium phosphate (5.0 g), tricyclohexylphosphine (0.19 g) and at last palladium(ll)acetate (76 mg). The mixture is stirred at 1 10 °C for 6 h meanwhile cyclopropylboronic acid is added after each hour (5x 0.20 g). After cooling to room temperature, the mixture is diluted with aqueous sodium hydrogen carbonate solution and extracted with ethyl acetate. The combined extracts are dried (sodium sulphate) and the solvent is removed under reduced pressure. The residue is chromatographed on silica gel (cyclohexane/ethyl acetate 20:1 -> 1 :1 ). Yield: 3.2 g (87% of theory ) Mass spectrum (ESI+): m/z = 581 [M+NH4] +
Example XXVI

4-(1 -Hvdroxy-cvclopropyD-phenylboronic acid A 3.0 M solution of ethylmagnesium bromide in diethylether (7.6 ml.) is added to a stirred solution of titanium(IV) isopropoxide (2.2 ml.) in diethylether (70 ml.) chilled to -78 °C. The resultant solution is stirred at -78 °C for 1.5 h, before 4-(4,4,5,5-tetramethyl-[1 ,3,2]dioxa borolan-2-yl)-benzoic acid methyl ester (2.0 g) is added. The reaction mixture is warmed to ambient temperature and stirred for an additional 12 h. Then, 1 M aqueous hydrochloric acid is added and the resulting mixture is extracted with ethyl acetate. The combined organic extracts are dried (sodium sulphate) and the solvent is evaporated. The residue is dissolved in acetone (60 ml.) and 0.1 M aqueous NH4OAc solution (50 ml.) followed by NaIO4 (2.3 g) is added. The resulting reaction mixture is stirred at room temperature for 18 h. After removal of the acetone, the residue is extracted with ethyl acetate. The combined extracts are dried (sodium sulphate) and the solvent is evaporated. The residue is purified by chromatography on silicagel (cyclohexane/ethyl acetate). Yield: 0.45 g (33% of theory) Mass spectrum (ESI“): m/z = 223 [M+HCOO]“ Preparation of the end compounds:
Example 17: 2-(4-Cyclopropyl-benzyl)-4-(β-D-glucopyranos-1-yl)-benzonitrile

Mass spectrum (ESI+): m/z = 413 [M+NH4]+
The compound is obtained according to example 6 using 4-cyclopropyl-phenylboronic acid as the coupling partner.
Yield: 83% of theory
Alternatively this compound is obtained as described in Example XXIV(I ).
The compound of example 17 is also obtained by employing the following procedure:
A solution of 2-(4-cyclopropyl-benzyl)-4-(2,3,4,6-tetra-O-acetyl-D-glucopyranos-1 -yl)- benzonitrile (0.80 g) in methanol (5 ml.) and THF (5 ml.) is treated with aqueous potassium hydroxide solution (4 mol/l, 5 ml_). The reaction solution is stirred at ambient temperature for 1 h and then neutralized with 1 M hydrochloric acid. The organic solvents are evaporated and the residue is diluted with brine and extracted with ethyl acetate. The organic extracts are dried (sodium sulphate) and the solvent is removed. The residue is chromatographed on silica gel (dichloromethane/methanol 1 :0 -> 9:1 ). Yield: 0.54 g (96% of theory)
SYN
Synthesis 2024, 56, 906–943
In 2007, Boehringer-Ingelheim Vetmedica GmbH pioneered the development of velagliflozin (15), subsequently submitting a patent application in the United States with the identification number US7776830B2.72a More recently, through clinical investigations, this compound has demonstrated its efficacy as an SGLT2 inhibitor, proving adept at curtailing glucose reabsorption, encouraging glucosuria,
and leading to reductions in both blood glucose and insulin levels.
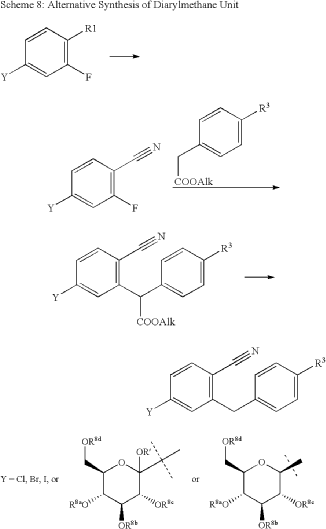
The initial synthesis of velagliflozin (15) was also disclosed in the above patent,72a and in patent
WO2007128749A1.72b The synthesis, depicted in Scheme46, comprises of nine-steps starting with the readily available raw material 2-bromo-5-iodobenzoic acid (250), which undergoes reduction using LiBH4 to form the corresponding alcohol 251. Subsequently, chlorination is carried out using thionyl chloride, resulting in the formation of chloride 252. O-Alkylation of phenol with compound 252 is
then conducted in a basic medium, yielding intermediate 253.The C-glycosylation of 253 with 2,3,4,6-tetrakis-O(trimethylsilyl)-D-glucopyranone 22 in the presence of turbo Grignard reagent (isopropylmagnesium chloride and LiCl) and methanesulfonic acid in methanol gives compound
254 with an impressive 93% yield. The hydroxy group of in termediate 254 is protected using acetic anhydride, and themethoxy group is subsequently removed via Lewis acid (BF3·Et2O, Et3SiH) treatment, providing compound 255 in a yield of 60%. A metal-catalyzed cyano group installation is then performed on intermediate 255, leading to the formation of compound 256 in 84% yield. The subsequent steps involve benzylic bromination followed by coupling with cyclopropylphenyl boronic acid 260, resulting in the formation of intermediate 258. Finally, deacetylation of intermediate 258 using aqueous KOH produces the desired product
The overall yield obtained for velagliflozin (15) is calculated to be 11.3%, with this synthetic route providing a systematic and efficient approach. The highlight of the route is high-yielding chemical transformations. However, the drawback is the use of two palladium-mediated couplings
that increase the possibility of leaching of the toxic metal in scale-up batches. Additionally, the synthetic route requires a large number of chemical transformations and not best suited for commercial production.
The same authors reported an alternative method (Scheme 47) for the synthesis of velagliflozin (15) in the product patent.72 The aglycone intermediate 265 is accessed in two steps starting from ethyl 2-(4-bromophenyl)acetate (262). O-Glycosylation takes place with the aglycone
4-bromo-2-(4-cyclopropylbenzyl)benzonitrile (265) using 2,3,4,6-tetrakis-O-(trimethylsilyl)-D-glucopyranone 22 in the presence of tert-butyllithium in pentane (1.7 M), resulting in the formation of compound 266. Reduction of compound 266 using boron trifluoride–diethyl etherate yields
the final API velagliflozin (15). This truncated synthetic route is well suited for scale-up due to the significantly low er number of transformations compared to the previous route. Unfortunately, the specific yields were not clearly in dicated for this process. This method presents an alternative approach to the synthesis of velagliflozin (15), providing a potential pathway for its preparation in 5 steps with
an overall yield of 40%.
(72) (a) Eckhardt, M.; Himmelsbach, F.; Eickelmann, P.; Sauer, A.;
Thomas, L. US7776830B2, 2010. (b) Eckhardt, M.; Himmelsbach,
F.; Eickelmann, P.; Sauer, A.; Thomas, L. WO2007128749A1,
2007.



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
Medical uses
Velagliflozin is indicated to improve glycemic control in otherwise healthy cats with diabetes not previously treated with insulin.[2][4][6]
References
- “Notice: Multiple additions to the Prescription Drug List (PDL) [2024-10-18]”. Health Canada. 18 October 2024. Retrieved 25 October 2024.
- “Senvelgo- velagliflozin solution”. DailyMed. 8 November 2023. Retrieved 13 December 2023.
- “Senvelgo Product information”. Union Register of veterinary medicinal products. 22 November 2023. Retrieved 29 August 2024.
- “NADA 141-568 Senvelgo (velagliflozin oral solution) Cats”.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - Cook AK, Behrend E (January 2025). “SGLT2 inhibitor use in the management of feline diabetes mellitus”. Journal of Veterinary Pharmacology and Therapeutics. 48 Suppl 1 (Suppl 1): 19–30. doi:10.1111/jvp.13466. PMC 11736986. PMID 38954371.
- “Dear Veterinarian Letter regarding important safety conditions associated with the use of Senvelgo (velagliflozin oral solution) for improving glycemic control in certain cats with diabetes mellitus”. U.S. Food and Drug Administration. 4 December 2023. Retrieved 13 December 2023. This article incorporates text from this source, which is in the public domain.
| Clinical data | |
|---|---|
| Trade names | Senvelgo |
| License data | US DailyMed: Velagliflozin |
| Routes of administration | By mouth |
| ATCvet code | QA10BK90 (WHO) |
| Legal status | |
| Legal status | CA: ℞-only[1]US: ℞-only[2]EU: Rx-only[3] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 946525-65-1 |
| PubChem CID | 24862817 |
| ChemSpider | 58827717 |
| UNII | FV2YU8SL0PEQE2P2T77I |
| Chemical and physical data | |
| Formula | C23H25NO5 |
| Molar mass | 395.455 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
- SGLT2 inhibitors: a novel therapy for cognitive impairment via multifaceted effects on the nervous systemPublication Name: Translational NeurodegenerationPublication Date: 2024-08-09PMCID: PMC11312905PMID: 39123214DOI: 10.1186/s40035-024-00431-y
- Demographic, morphologic, hormonal and metabolic factors associated with the rate of improvement from equine hyperinsulinaemia-associated laminitisPublication Name: BMC Veterinary ResearchPublication Date: 2022-01-18PMCID: PMC8764787PMID: 35042535DOI: 10.1186/s12917-022-03149-z
- The efficacy and safety of velagliflozin over 16 weeks as a treatment for insulin dysregulation in poniesPublication Name: BMC Veterinary ResearchPublication Date: 2019-02-26PMCID: PMC6390376PMID: 30808423DOI: 10.1186/s12917-019-1811-2
- The sodium-glucose co-transporter 2 inhibitor velagliflozin reduces hyperinsulinemia and prevents laminitis in insulin-dysregulated poniesPublication Name: PLOS ONEPublication Date: 2018-09-13PMCID: PMC6136744PMID: 30212530DOI: 10.1371/journal.pone.0203655
- Effects of the sodium‐glucose cotransporter 2 (<scp>SGLT</scp>2) inhibitor velagliflozin, a new drug with therapeutic potential to treat diabetes in catsPublication Name: Journal of Veterinary Pharmacology and TherapeuticsPublication Date: 2017-11-15PMID: 29139146DOI: 10.1111/jvp.12467
/////////Velagliflozin, APPROVALS 2023, GDA 2023, EU 2023, EMA 2023, SENVELGO, DIABETES, SENVELGO,
Vamorolone
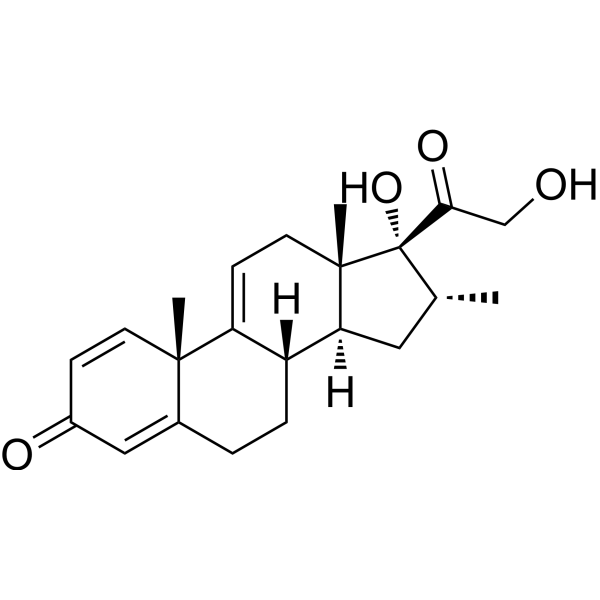
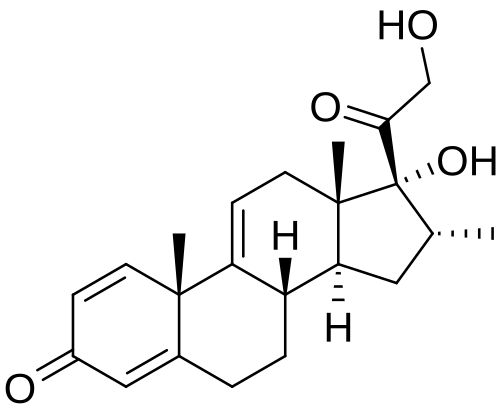
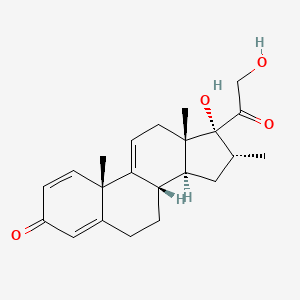

Vamorolone
CAS 13209-41-1
| Molecular Weight | 356.46 |
|---|---|
| Formula | C22H28O4 |
- Agamree
- 17,21-Dihydroxy-16alpha-methylpregna-1,4,9(11)-triene-3,20-dione
- VBP-15 free alcohol
- (8S,10S,13S,14S,16R,17R)-17-hydroxy-17-(2-hydroxyacetyl)-10,13,16-trimethyl-7,8,12,14,15,16-hexahydro-6H-cyclopenta[a]phenanthren-3-one
- (16alpha)-17,21-dihydroxy-16-methylpregna-1,4,9(11)-triene-3,20-dione
- (8S,10S,13S,14S,16R,17R)-17-hydroxy-17-(2-hydroxyacetyl)-10,13,16-trimethyl-7,8,12,14,15,16-hexahydro-6H-cyclopenta(a)phenanthren-3-one
- DTXCID601356317
- (1R,2R,3aS,3bS,9aS,11aS)-1-hydroxy-1-(2-hydroxyacetyl)-2,9a,11a-trimethyl-1H,2H,3H,3aH,3bH,4H,5H,7H,9aH,11H,11aH-cyclopenta(a)phenanthren-7-one
Vamorolone (VBP15) is a first-in-class, orally active dissociative steroidal anti-inflammatory agent and membrane-stabilizer. Vamorolone improves muscular dystrophy without side effects. Vamorolone shows potent NF-κB inhibition and substantially reduces hormonal effects.
Vamorolone, sold under the brand name Agamree, is a synthetic corticosteroid, which is used for the treatment of Duchenne muscular dystrophy.[4][5][6][7][8] It is taken by mouth.[1] It is a dual atypical glucocorticoid and antimineralocorticoid.[9]
The most common adverse reactions include cushingoid features, psychiatric disorders, vomiting, increased weight, and vitamin D deficiency.[10]
Vamorolone was approved for medical use in the United States in October 2023,[11][10] and in the European Union in December 2023.[2][3]
Vamorolone is a novel and fully synthetic glucocorticoid developed by Santhera Pharmaceuticals. It is used to manage inflammation and immune dysregulation in patients with Duchenne muscular dystrophy (DMD), a neuromuscular disorder characterized by the insidious regression of neuromuscular function and the most common form of muscular dystrophy in the United States. Corticosteroid therapy is the current standard of care for DMD despite relatively high rates of adverse effects. Vamorolone is positioned as having a more tolerable adverse effect profile than other corticosteroids owing to its unique receptor binding profile, thus providing an additional treatment option in patients for whom corticosteroid adverse effects are intolerable or otherwise unacceptable. Vamorolone was approved by the FDA in October 2023 for the management of DMD in patients ≥2 years of age. In December 2023, it was approved in the EU for the treatment of patients ≥4 years of age.
PATENT
https://patents.google.com/patent/WO2023016817A1/en
Vamorolone is currently produced from the commercially available 3TR (Tetraene acetate) – see Scheme 2. In step a, TMS imidazole, MeMgCI and THF are added to 3TR, with subsequent addition of CuAc2, H2O, DMPU, MeMgCI and THF in step b. Under treatment with peracetic acid in Toluene from compound 2 the intermediate 3 is formed in step c. After treatment with NaHSCO3 and TFA (step d), EtOAc and heptane (step e) and acetonitrile trituration (step f) HBr in CH2CI2 is added (step g) and MeOH (step h) is used for crystallization to form Acetyl- Vamorolone 4. Acetyl-Vamorolone is deacetylated with K2CO3 in MeOH, followed by HCI to obtain Vamorolone (step i). The synthesis is disclosed in Bioorganic & Medicinal Chemistry, Volume 21 , Issue 8, 15 April 2013, Pages 2241-2249.
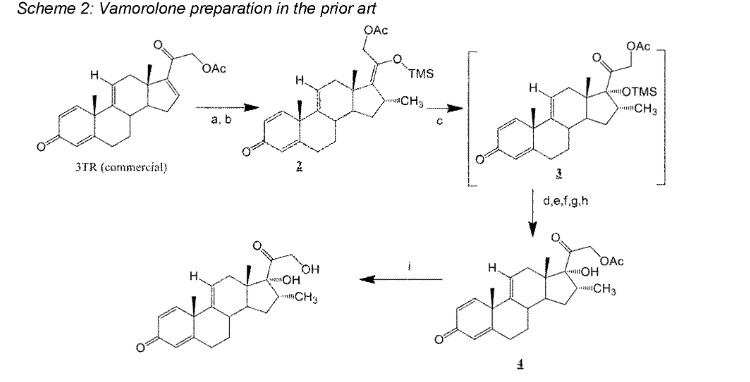
Example 2: Synthesis of the present invention
Scheme C: Route of Synthesis of Vamorolone from 8-DM

Vamorolone was synthesized in three synthetic steps from commercially available 8-DM.
The synthetic route started with the acetylation of 8-DM using acetic anhydride and catalytic DMAP in THF, followed by crystallization of 8-DM Acetate after aqueous quench. Then, a deoxygenation reaction converted 8-DM Acetate directly into Vamorolone Acetate. This deoxygenation proceeded via initial formation of an iodohydrin with excess aq. HI, followed by simultaneous I2 and H2O-elimination to give Vamorolone Acetate. During the reaction, partial de-acetylation occurred (20-25%) and therefore re-acetylation with acetic anhydride was necessary. After completed re-acetylation, Vamorolone Acetate was directly crystallized by addition of H2O. Finally, the acetate group is cleaved under basic conditions to give crude Vamorolone, which was recrystallized from iPrOH to obtain the pure product.
2.1 Acetylation

A 10 L glass dj ( double jacketed reactor )-reactor was charged with 8-DM (490 g, 1.32 mol, 1.0 eq.) and DMAP (16.1 g, 0.132 mmol, 0.10 eq.). THF (1.25 L, 2.5 vol.) was added at IT = 20-25 °C. Then, AC2O (201 g, 187 mL, 1.97 mol, 1.5 eq.) was added dropwise over 20-40 min, keeping IT below 30 °C during the addition. After complete addition, the reaction mixture was stirred at IT = 20-25 °C for 30 min. IPC control by LC/MS indicated >99% conversion of 8-DM to 8-DM Acetate.
The reaction mixture was quenched by dropwise addition of H2O (4.9 L, 10 vol.) over 30-45 min, keeping IT below 25 °C. The resulting aqueous suspension was aged at IT = 20-25 °C for 1 h. The product was filtered off, washed with H2O (3 x 0.5 L), and dried on a rotary evaporator (900-10 mbar, 65 °C bath temperature) to provide 8-DM Acetate (539 g, 1.30 mol, 99% yield, >99% a/a, 98% w/w) as a white solid (cryst 1#1).
Analytical Data:
LC/MS column: Zorbax RRHD SB-Aq, 2.1x50mm, 1.8pm
Program: G_005%B_TFA_0,800ml_2,00min
Eluent A: Water/TFA 100:0.04, Eluent B: Acetonitrile
IPC preparation for LC/MS
10 microliter in 1 mL H2O:MeCN 1 :1
Conversion was determined with respect to consumption of 8-DM relative to formation of 8- DM Acetate.
Detected mass: [M+1]= 373.19 for 8-DM and [M+1] = 415,19 8-DM Acetate. 2.2 Deoxygenation with HI

A 10 L glass dj-reactor was charged with 8-DM Acetate (500 g, 1.21 mol, 1.0 eq.). Toluene (2.5 L, 5 vol.) was added. The suspension was cooled to IT = 0-5 °C and then a solution of 57% aqueous HI (1 .08 kg, 637 mL, 4.83 mol, 4.0 eq.) in AcOH (1.25 L, 2.5 vol.) was added via peristaltic pump over 45-60 min, keeping IT below 5 °C during the addition. The resulting dark purple to brown solution was stirred at IT = 3-5 °C for 24 h. IPC control by LC/MS indicated >98% conversion of 8-DM Acetate/intermediate iodohydrin to Vamorolone Acetate/Vamorolone.
The reaction mixture was quenched by dropwise addition of 25% aq. Na2SO3 solution (2.0 L, 4 vol.) over 10-20 min, keeping IT below 15 °C. After complete addition, EtOAc (1.0 L, 2 vol.) was added and the biphasic mixture was warmed to IT = 15-20 °C. Stirring was stopped and the phases were separated (Organic Phase 1 and aqueous Phase 1 ; goal pH of the aqueous Phase 1 : 2; aqueous Phase 1 disposed). 25% aq. Na2SO3 solution (1.25 L, 2.5 vol.) was added to Organic Phase 1 and the biphasic mixture was stirred at IT = 15-20 °C for 5 min, stirring was stopped and phases separated (Organic Phase 1 and aqueous Phase 2; goal pH aqueous Phase 2: 4-5; aqueous Phase 2 disposed). 25% aq. Na2SO3 solution (1.25 L, 2.5 vol.) was added to Organic Phase 1 and the biphasic mixture was stirred at IT = 15-20 °C for 5 min, stirring was stopped and phases separated (Organic Phase 1 and aqueous Phase 3; goal pH aqueous Phase 3: 5-6; aqueous Phase 3 disposed). H2O (0.5 L, 1.0 vol.) was added to Organic Phase 1 and the biphasic mixture was stirred at IT = 15-20 °C for 5 min, stirring was stopped and phases separated (Organic Phase 1 and aqueous Phase 4; goal pH aqueous Phase 4: 5-6; aqueous Phase 4 disposed).
A slight vacuum was applied to the double-jacketed reactor (100-150 mbar), containing Organic Phase 1 , and toluene was distilled off at 70 °C jacket temperature (ET) from the reaction mixture with continuous addition of MeCN, and the distillation continued until target residual toluene value has been reached (goal: less than 5% toluene according to 1 H-NMR of reaction mixture. Final volume in reactor after distillation: ca. 3.5 L (7.5 vol.).
Once toluene was removed, the vacuum was broken with N2 and resulting fine suspension cooled to IT = 20-25 °C. At this point, the amount of Vamorolone was assessed by IPC (typical ratio: Vamorolone Acetate to Vamorolone: 75:25; x = 25% a/a). DMAP (3.7 g, 0.0302 mol, 0.025 eq.) was added, followed by slow addition of AC2O (61.6 g, 57 mL, 0.603 mol, 0.5 eq.) over 5-10 min at IT = 20-25 °C. After complete addition of AC2O, the reaction mixture was stirred for 30 min at IT = 20-25 °C. IPC control by LC/MS indicated ≤ 2% a/a Vamorolone (ratio: Vamorolone Acetate to Vamorolone: 98.5:1.5).
The reaction mixture was quenched by slow addition of H2O (4.9 L, 10 vol.) over 15-30 min, keeping IT below 25 °C. The resulting aqueous suspension was cooled to IT = 0-5 °C and aged at this temperature for 2 h. The product was filtered off, washed with H2O/MeCN 4:1 (2 x 0.5 L), and dried on a rotary evaporator (900-10 mbar, 65 °C bath temperature) to provide Vamorolone Acetate (301 g, 0.76 mol, 63% yield, 98% a/a, 98% w/w) as an off-white solid (cryst 1#1).
Over the course of the reaction, partial de-acetylation of Vamorolone Acetate to Vamorolone was observed (between 20-25% a/a). Therefore, after aq. workup and solvent switch to MeCN, the ratio of Vamorolone Acetate to Vamorolone was assessed by LC/MS (in % a/a), and the following amounts of DMAP and Ac2O were added: x = amount of Vamorolone in % a/a (e.g. x = 20% a/a)
DMAP eq. = (0.1 -x)/100 (e.g. 0.02 eq.)
Ac2O eq. = (2.0-x)/100 (e.g. 0.40 eq.)
Analytical Data
LC/MS column: Zorbax RRHD SB-Aq, 2.1x50mm, 1.8pm
Program: G_005%B_TFA_0,800ml_2,00min
Eluent A: Water/TFA 100:0.04, Eluent B: Acetonitrile
IPC preparation for LC/MS
10 microliter in 1 mL H2O:MeCN 1 :1
Conversion was determined with respect to consumption of the sum of (8-DM Acetate + intermediate iodohydrin) relative to the sum of (Vamorolone Acetate + Vamorolone). Detected mass: [M+1] = 415,19 for 8-DM Acetate, [M+1]= 357,28 for Vamorolone, 399,20 for
Vamorolone Acetate and 543,12 for intermediate lodohydrin
2.3 De-Acetylation

A 10 L glass dj-reactor was charged with Vamorolone Acetate (280 g, 0.703 mol, 1.0 eq.). MeOH (1.54 L, 5.5 vol.) was added. The suspension was cooled to IT = 0-5 °C and then a solution of K2CO3 (107 g, 0.773 mol, 1.1 eq.) in H2O (0.7 L, 2.5 vol.) was added dropwise via peristaltic pump over 20-40 min, keeping IT below 10 °C during the addition. After complete addition, the reaction mixture was warmed IT = 20-25 °C and stirred for 5 h. IPC control by LC/MS indicated 99.3% conversion of Vamorolone Acetate to Vamorolone.
The reaction mixture was cooled to IT = 15-17 °C and quenched by dropwise addition of 1 M aq. HCI (950 mL, 0.95 mol, 1.35 eq.) over 20-40 min, keeping IT below 20 °C during the addition (goal pH: 5-6). The resulting aqueous suspension was aged at IT = 15-20 °C for 12 h. The product was filtered off, washed with H2O/MeOH 2:1 (3 x 0.3 L), and dried on a rotary evaporator (900-10 mbar, 65 °C bath temperature) to provide Vamorolone (241.5 g, 0.68 mol, 96% yield, >99% a/a, 98% w/w) as a slightly yellow solid (crude 1#1).
Analytical Data
LC/MS column: Zorbax RRHD SB-Aq, 2.1x50mm, 1.8pm
Program: G_005%B_TFA_0,800ml_2,00min
Eluent A: Water/TFA 100:0.04, Eluent B: Acetonitrile
IPC preparation for LC/MS
10 microliter in 1 mL H2O:MeCN 1 :1
Conversion was determined with respect to consumption of Vamorolone Acetate relative to formation of Vamorolone. 2.4 Recrystallization

A 10 L glass dj-reactor was charged with Vamorolone (230 g, 0.645 mol, 1.0 eq.). iPrOH (5 L, 22 vol.) was added. The suspension was heated to reflux (jacket temperature ET = 97 °C) and stirred until complete dissolution of Vamorolone occurred (10-15 min on this scale).
After complete dissolution, the clear yellow solution was slowly cooled to IT = 0-5 °C over the course of 12 h and then aged at IT = 0-5 °C for 1 h. The recrystallized product was filtered off, washed with cold iPrOH (2 x 250 mL), and dried on a rotary evaporator (900-10 mbar, 65 °C bath temperature) to provide Vamorolone (201 g, 87% recovery, >99% a/a, 99% w/w) as an off white glimmery solid (cryst 1#1).
Iso-propanol (iPrOH) was found to the best solvent for recrystallization with excellent purity upgrading properties (by rejection of impurities), although a high dilution is necessary to completely dissolve the crude Vamorolone at reflux temperature. Higher concentrations for the recrystallization satisfactory results are obtainable using mixtures of isopropanol and water. Maximum solubility of Vamorolone was determined to be at reflux of a 80:20 (isopropanol : water) mixture.
PATENT
https://patents.google.com/patent/US20200281942A1/en
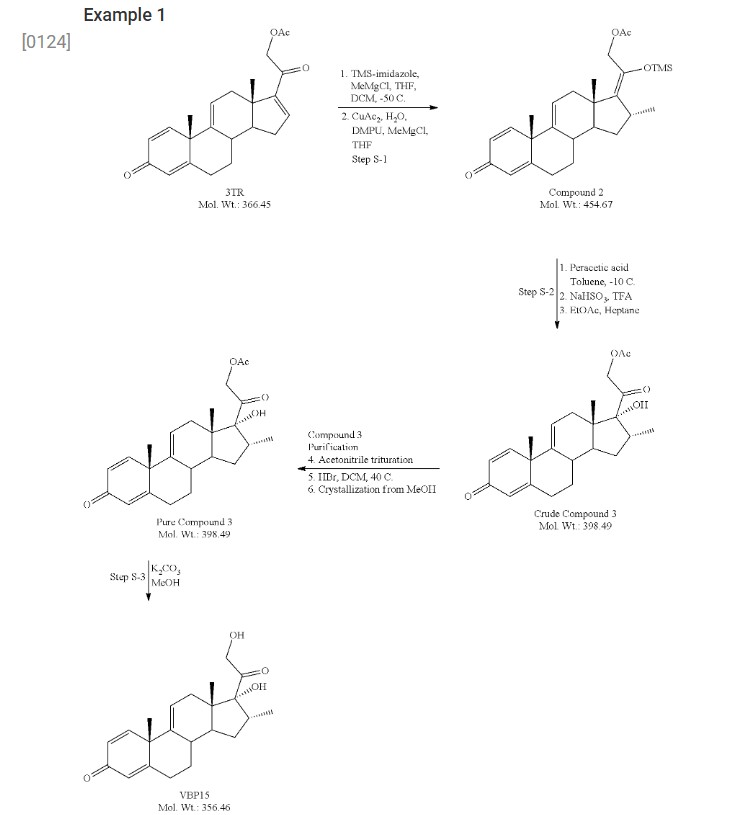
- [0124]
- [0125]3-TR (100 g, 273 mmol), dichloromethane (DCM, 500 mL) and tetrahydrofuran (THF, 400 mL) were charged to a reaction flask under nitrogen. To this was charged trimethylsilyl imidazole (TMS-imidazole, 65.3 g, 466 mmol, 1.7 eq). The resulting mixture was stirred at room temperature for 3 hours.
- [0126]In a separate flask, copper acetate monohydrate (5.4 g, 27 mmol), tetrahydrofuran (400 ml) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (DMPU, 53.3 g, 416 mmol) were combined and stirred at room temperature for approximately 3 hours. The blue mixture was subsequently cooled to −50° C., and to this was added methyl magnesium chloride solution (27 ml, 3.0 M in THF, 82 mmol) dropwise. After 30 minutes, the mixture had formed a deep blue, sticky “ball.”
- [0127]The 3-TR/TMS-imidazole mixture was cooled to −50° C. and to this was charged the copper acetate/DMPU solution above via canula. The residual sticky mass from the copper acetate/DMPU mixture was dissolved using DCM (50 mL) and also transferred.
- [0128]Methyl magnesium chloride (123.2 mL, 3.0 M solution in THF, 368 mmol) was added dropwise over 45 minutes to the combined reaction mixtures, which were then allowed to stir for 2 hours at −50° C. Subsequent HPLC analysis showed complete consumption of starting material. The mixture was allowed to warm to room temperature overnight, with stirring.
- [0129]Toluene (800 mL) was added to the mixture, followed by 5% acetic acid solution (600 mL). The aqueous layer was removed and discarded. The acetic acid wash was repeated. The organic layer was washed with brine (400 mL), 5% sodium bicarbonate solution (400 mL×2), followed by a brine wash (400 mL). The organic solution was dried over sodium sulfate, then concentrated to dryness under reduced pressure. The product was recovered as a viscous, light golden oil. Mass recovery was 146 grams (119% of theoretical).
- [0130]Compound 2 (92 g, 202 mmol) and toluene (1000 mL, 10.9 vol) were charged to a reaction flask under nitrogen and the solution was cooled to −10° C. A 32 wt % solution of peracetic acid in acetic acid (60 mL, 283 mmol, 1.4 eq) was added dropwise over about 30 min maintaining the temperature at −10° C. The reaction was held for approximately 20 h (HPLC showed 75% Cmpd 3, Cmpd 2 1.5%, 6% diastereomer; 5% epoxide). Starting at −10° C., a 20% aqueous solution of sodium bisulfite (920 mL, 10 vol) was added carefully via addition funnel, keeping the temperature below 10° C. Trifluoroacetic acid (16 mL, 202 mmol, 1 eq) was added and the mixture was held for 3 h at 0-5° C. to complete desilylation (endpoint by HPLC). The lower aqueous layer was drained, and the organic layer was washed with a saturated solution of sodium bicarbonate (3×250 mL), followed by water (1×250 mL), and brine (1×150 mL). The organic layer was then dried over Na2SO4, filtered and concentrated to a pasty solid (89 g). The residue was taken up in 1.5 vol of EtOAc and transferred to neat heptane (19 vol) to precipitate crude Cmpd 3 as an off-white solid (50 g, 62.5% yield; HPLC 79% Cmpd 3, 5.6% epoxide, 1.7% diastereomer). The crude Cmpd 3 (48.5 g) was triturated in hot acetonitrile (2 vol) at 60° C. for 4 h, and then gradually cooled to ambient temperature overnight. The mixture was filtered using the recycled filtrate to rinse and wash the wet cake. After drying, the recovery was 64.3% (31.2 g; HPLC 93.5% Cmpd 3, 3.3% epoxide). To remove the epoxide impurity, the 31 Cmpd 3 was dissolved in DCM (250 mL, 8 vol) and a solution of 48% HBr in water was added (7.5 mL). The mixture was heated at 40° C. for 1 h (HPLC<0.3% epoxide). The mixture was cooled and transferred to a separatory funnel. The lower aqueous layer (brown) was removed and the upper organic layer was washed with water (200 mL), saturated NaHCO3 (150 mL), and brine (100 mL). The organic layer was dried over Na2SO4, filtered, and concentrated to a tan foam (32 g, ˜100% recovery). Methanol (64 mL, 2 vol) was added to the 32 g foam forming a slurry. To this was added a 1:1 solution of MeOH:water (60 mL, 2 vol) dropwise. The slurry cooled to slightly below ambient temperature and filtered using recycled filtrate to rinse and wash the wet cake. The solids were dried to constant weight, affording 26.1 g Cmpd 3 (81% recovery; HPLC 97.8%). The overall yield for Step 2 was 32.5%.
- [0131]Compound 3 (26 g, 65 mmol) and MeOH (156 mL, 6 vol) were mixed in a reaction flask and cooled to 0-5° C. A solution of K2CO3 (9.9 g, 72 mmol, 1.1 eq) in water (65 mL) was added dropwise, and the mixture was allowed to gradually warm to ambient temperature overnight. Analysis by HPLC showed 2.5% SM and another 5 mol % K2CO3 was added and the mixture stirred for another day (HPLC endpoint 1.1% Cmpd 3). The mixture was neutralized to pH 7 with 1.5 M HCl (53 mL) and ˜25% of the MeOH (30 g) was removed under vacuum to maximize recovery. After stirring for 2 days, the product was isolated by filtration using the recycled filtrate to aid transferring the wet cake to the funnel. The wet cake was dried under vacuum, affording 19.3 g VBP15 (83% yield) as an off-white powder. Analysis of the solids by HPLC showed 98.8% purity with 0.6% Cmpd 3 as the only major impurity.
- [0132]Power X-Ray Diffraction (pXRD)
- [0133]The solid samples were examined using X-ray diffractometer (Bruker D8 advance). The system is equipped with highly-parallel x-ray beams (Gobel Mirror) and LynxEye detector. The samples were scanned from 3 to 40°2θ, at a step size 0.02°2θ and a time per step of 19.70 seconds. The tube voltage and current were 45 kV and 40 mA, respectively. The sample was transferred from sample container onto zero background XRD-holder and gently ground.
Syn
EuropeanJournalofMedicinalChemistry265(2024)116124
Vamorolone (Agamree)
On October 26, 2023, Vamorolone, developed jointly by Santhera Pharmaceuticals and ReveraGen BioPharma, has received FDA approval to treat DMD in patients aged 2 years and older [1]. DMD is a prevalent neuromuscular disorder in childhood, ranking among the most common.
This condition is caused by mutations in the gene responsible for producing the dystrophin protein, which plays a crucial role in maintaining muscle integrity. Moreover, DMD is an X-linked genetic disorder [69]. Vamorolone is a novel steroidal anti-inflammatory and membrane-stabilizing agent that can be taken orally. The distinction between it and traditional corticosteroid drugs lies in its capacity to
specifically activate particular signaling pathways of corticosteroids. In individuals diagnosed with DMD, the primary mechanism through which corticosteroid drugs exhibit their effectiveness is by exerting
anti-inflammatory effects. However, the secondary activities of corticosteroids can lead to adverse effects that impact the overall well-being of patients. Vamorolone has the ability to decrease the occurrence of
adverse effects while still preserving the therapeutic effectiveness of corticosteroids in individuals with DMD [70].
Preparation of Vamorolone is depicted in Scheme 19, which began with commercially available steroid 3 TR VAMO-001 [71]. Copper catalyzed addition of VAMO-001 with trimethylsilyl chloride (TMSCl)
gave silyl enol ether VAMO-002. VAMO-002 was oxidized by peracetic acid in acetic acid to yield intermediate VAMO-003, which was deprotected and hydrolyzed to obtain Vamorolone.
[69] D. Duan, N. Goemans, S. Takeda, E. Mercuri, A. Aartsma-Rus, Duchenne muscular
dystrophy, Nat. Rev. Dis. Prim. 7 (2021) 13.
[70] M. Guglieri, P.R. Clemens, S.J. Perlman, E.C. Smith, I. Horrocks, R.S. Finkel, J.
K. Mah, N. Deconinck, N. Goemans, J. Haberlova, V. Straub, L.J. Mengle-Gaw, B.
D. Schwartz, A.D. Harper, P.B. Shieh, L. De Waele, D. Castro, M.L. Yang, M.
M. Ryan, C.M. McDonald, M. Tulinius, R. Webster, H.J. McMillan, N.L. Kuntz, V.
K. Rao, G. Baranello, S. Spinty, A.M. Childs, A.M. Sbrocchi, K.A. Selby,
M. Monduy, Y. Nevo, J.J. Vilchez-Padilla, A. Nascimento-Osorio, E.H. Niks, I.J.
M. de Groot, M. Katsalouli, M.K. James, J. van den Anker, J.M. Damsker,
A. Ahmet, L.M. Ward, M. Jaros, P. Shale, U.J. Dang, E.P. Hoffman, Efficacy and
safety of vamorolone vs placebo and prednisone among boys with duchenne
muscular dystrophy: a randomized clinical trial, JAMA Neurol. 79 (2022)
1005–1014.
[71] E.K.M. Reeves, E.P. Hoffman, K. Nagaraju, J.M. Damsker, J.M. McCall, VBP15:
preclinical characterization of a novel anti-inflammatory delta 9,11 steroid,
Bioorg. Med. Chem. 21 (2013) 2241–2249.

Syn
J. Med. Chem. 2025, 68, 2147−2182
Vamorolone (Agamree). Developed by Santhera and ReveraGen BioPharma, the corticosteroid vamorolone (9) was approved for the treatment of Duchenne muscular dystrophy in October 2023.
70 Traditional corticosteroid treatment has been hampered by safety concerns including decreased bone mineral density and increased muscle atrophy. 71−73 Vamorolone is structurally distinct from other corticosteroids such as prednisone (Figure 3). 74 Removalofthe11βcarbonylmaintains binding to the glucocorticoid receptor but results in mineralocorticoid receptor antagonism; prednisone is a
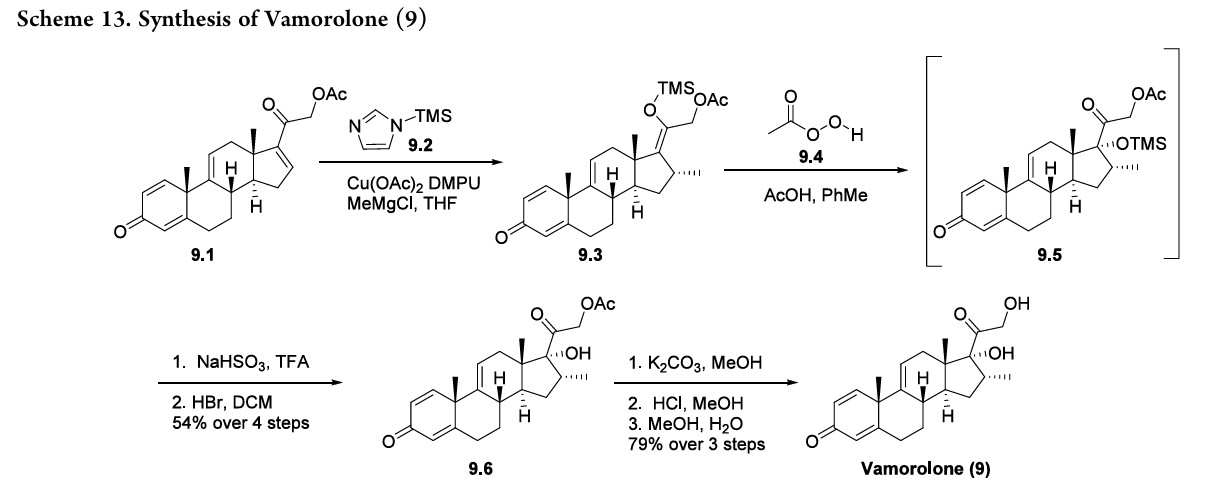
mineralocorticoid receptor agonist. 75,76 This also results in decreased glucocorticoid receptor-drive transactivation, ultimately improving the safety profile of vamorolone as compared to other corticosteroid therapies. 74 The synthesis of vamorolone (9) as disclosed by ReveraGen BioPharma is summarized in Scheme 13. 77 Readily available steroid 9.1 was subjected to copper-catalyzed Michael addition.
Thein situ generated enolate was trapped using TMS-imidazole 9.2, providing the silyl enol ether 9.3. Treatment of crude 9.3 with peracetic acid 9.4 resulted in oxidized intermediate 9.5. Quenching of the peracetic acid and silyl deprotection afforded the protected steroid 9.6 in 54% yield from 9.1. Finally, K2CO3 mediated acetate deprotection of 9.6, neutralization and methanol/water crystallization provided vamorolone (9) in 79% yield over three steps.
(70) Keam, S. J. Vamorolone: first approval. Drugs 2024, 84, 111−
117.
(71) Hoffman, E. P.; Nader, G. A. Balancing muscle hypertrophy and
atrophy. Nat. Med. 2004, 10, 584−585.
(72) Hoffman, E. P.; Reeves, E.; Damsker, J.; Nagaraju, K.; McCall, J.
M.; Connor, E. M.; Bushby, K. Novel approaches to corticosteroid
treatment in Duchennemusculardystrophy.Phys.Med.Rehabil. Clin. N.
Am. 2012, 23, 821−828.
(73) Singh, A.; Schaeffer, E. K.; Reilly, C. W. Vertebral fractures in
Duchenne muscular dystrophy patients managed with Deflazacort. J.
Pediatr. Orthop. 2018, 38, 320−324.
(74) Liu, X.; Wang, Y.; Gutierrez, J. S.; Damsker, J. M.; Nagaraju, K.;
Hoffman, E. P.; Ortlund, E. A. Disruption of a key ligand-H-bond
network drives dissociative properties in vamorolone for Duchenne
muscular dystrophy treatment. Proc. Natl. Acad. Sci. U. S. A. 2020, 117,
24285−24293.
(75) Heier, C. R.; Yu, Q.; Fiorillo, A. A.; Tully, C. B.; Tucker, A.;
Mazala, D. A.; Uaesoontrachoon, K.; Srinivassane, S.; Damsker, J. M.;
Hoffman, E.P.; et al. Vamorolone targets dual nuclear receptors to treat
inflammation and dystrophic cardiomyopathy. Life Sci. Alliance 2019, 2,
No. e201800186.
(76)Boger, D.L.Thedifferenceasingleatomcanmake:synthesisand
design at the chemistry−biology interface. J. Org. Chem. 2017, 82,
11961−11980.
(77) Reeves, E. K. M.; Hoffman, E. P.; Nagaraju, K.; Damsker, J. M.;
McCall, J. M. VBP15: Preclinical characterization of a novel anti
inflammatory delta 9,11 steroid. Bioorg. Med. Chem. 2013, 21, 2241−
2249.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
PATENTS
Drugs 2024, 84, 111−117.
Nat. Med. 2004, 10, 584−585.
Clin. N.Am. 2012, 23, 821−828.
Pediatr. Orthop. 2018, 38, 320−324.
Proc. Natl. Acad. Sci. U. S. A. 2020, 117,24285−24293.
Life Sci. Alliance 2019, 2,No. e201800186.
J. Org. Chem. 2017, 82,11961−11980.
Bioorg. Med. Chem. 2013, 21, 2241−2249.
Neurol. 79 (2022)1005–1014
Bioorg. Med. Chem. 21 (2013) 2241–2249
Nat. Rev. Dis. Prim. 7 (2021) 13
Chemistry
Vamorolone is a synthetic corticosteroid and is also known by the chemical name 17α,21-dihydroxy-16α-methylpregna-1,4,9(11)-triene-3,20-dione or as 16α-methyl-9,11-dehydroprednisolone. It is a derivative of cortisol (hydrocortisone) and prednisolone (1,2-dehydrocortisol).
Anti-inflammatory drugs of the corticosteroid class show a carbonyl (=O) or hydroxyl (-OH) group on the C11 carbon of the steroid backbone. In contrast, vamorolone contains a Δ9,11 double bond between the C9 and C11 carbons. This change in structure has been shown to remove a molecular contact site with the glucocorticoid receptor, and leads to dissociative properties.[12]
History
In phase I clinical trials of adult volunteers, vamorolone was shown to be safe and well tolerated, with blood biomarker data suggesting possible loss of safety concerns of the corticosteroid class.[13]
In phase IIa dose-ranging clinical trial of 48 children with Duchenne muscular dystrophy (2 weeks on drug, 2 weeks off drug), vamorolone was shown to be safe and well tolerated, and showed blood biomarker data consistent with a myofiber membrane stabilization and anti-inflammatory effects, and possible loss of safety concerns.[14] These children continued on to a 24-week open-label extension study at the same doses, and this showed dose-dependent improvement of motor outcomes, with 2.0 and 6.0 mg/kg/day suggesting benefit.[15] These same children continued on a long-term extension study with dose escalations, and this suggested continued clinical improvement through 18-months treatment.[16]
Population pharmacokinetics (PK) of vamorolone was shown to fit to a 1-compartment model with zero-order absorption, with both adult men and young boys showing dose-linearity of PK parameters for the doses examined, and no accumulation of the drug during daily dosing. Apparent clearance averaged 2.0 L/h/kg in men and 1.7 L/h/kg in boys. Overall, vamorolone exhibited well-behaved linear PK, with similar profiles in healthy men and boys with DMD, moderate variability in PK parameters, and absorption and disposition profiles similar to those of classical glucocorticoids.[17] Exposure/response analyses have suggested that the motor outcome of time to stand from supine velocity showed the highest sensitivity to vamorolone, with the lowest AUC value providing 50% of maximum effect (E50 = 186 ng·h/mL), followed by time to climb 4 stairs (E50 = 478 ng·h/mL), time to run/walk 10 m (E50 = 1220 ng·h/mL), and 6-minute walk test (E50 = 1770 ng·h/mL). Week 2 changes of proinflammatory PD biomarkers showed exposure-dependent decreases. The E50 was 260 ng·h/mL for insulin-like growth factor-binding protein 2, 1200 ng·h/mL for matrix metalloproteinase 12, 1260 ng·h/mL for lymphotoxin α1/β2, 1340 ng·h/mL for CD23, 1420 ng·h/mL for interleukin-22-binding protein, and 1600 ng·h/mL for macrophage-derived chemokine/C-C motif chemokine 22.[18]
A trial titled “Efficacy and Safety of Vamorolone Over 48 Weeks in Boys With Duchenne Muscular Dystrophy” published in March 2024 found vamorolone (Agamree) at a dose of 6 mg/kg/d showed maintenance of improvement for all motor outcomes to week 48. There was also significant improvement in linear growth after crossover in the prednisone to vamorolone 6 mg/kg/d group, and quick reversal of prednisone-induced decline in bone turnover biomarkers in each crossover group.[19]
The US Food and Drug Administration (FDA) approved vamorolone based on evidence from a single clinical trial of 121 boys with DMD who were 4 to <7 years of age. The trial (Study 1) was conducted at 33 sites in 11 countries in Australia, Belgium, Canada, the Czech Republic, Spain, the United Kingdom, Greece, Israel, Netherlands, Sweden, and the United States.[10] In addition to Study 1, safety was also evaluated in a separate, open-label study of children with DMD aged 2 to <4 years (N=16) and children with DMD aged 7 to <18 years (N=16).[10]
Society and culture
Legal status
Santhera Pharmaceuticals signed an agreement with Catalyst Pharmaceuticals for the North American commercialization of vamorolone in July 2023.[20]
In October 2023, the FDA approved vamorolone (Agamree; Catalyst Pharmaceuticals) for the treatment of Duchenne muscular dystrophy.[11][21][22]
In October 2023, the Committee for Medicinal Products for Human Use adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Agamree, intended for the treatment of Duchenne muscular dystrophy.[2] The applicant for this medicinal product is Santhera Pharmaceuticals (Deutschland) GmbH.[2] Vamorolone was approved for medical use in the European Union in December 2023.[2][3]
Brand names
Vamorolone is the international nonproprietary name.[23]
Vamorolone is sold under the brand name Agamree.[1][2][3] Agamree (vamorolone) is a dissociative steroid that selectively binds to the glucocorticoid receptor to exert anti-inflammatory and immunosuppressive effects. Vamorolone also inhibits mineralocorticoid receptor activation by aldosterone.[24]
References
- “Agamree- vamorolone kit”. DailyMed. 26 October 2023. Retrieved 20 November 2023.
- “Agamree EPAR”. European Medicines Agency. 12 October 2023. Retrieved 27 December 2023. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- “Agamree Product information”. Union Register of medicinal products. 15 December 2023. Retrieved 26 December 2023.
- “Vamorolone – ReveraGen Biopharma”. AdisInsight. Springer Nature Switzerland AG. Archived from the original on 7 October 2017. Retrieved 2 July 2017.
- Reeves EK, Hoffman EP, Nagaraju K, Damsker JM, McCall JM (April 2013). “VBP15: preclinical characterization of a novel anti-inflammatory delta 9,11 steroid”. Bioorganic & Medicinal Chemistry. 21 (8): 2241–2249. doi:10.1016/j.bmc.2013.02.009. PMC 4088988. PMID 23498916.
- Heier CR, Damsker JM, Yu Q, Dillingham BC, Huynh T, Van der Meulen JH, et al. (October 2013). “VBP15, a novel anti-inflammatory and membrane-stabilizer, improves muscular dystrophy without side effects”. EMBO Molecular Medicine. 5 (10): 1569–1585. doi:10.1002/emmm.201302621. PMC 3799580. PMID 24014378.
- Dadgar S, Wang Z, Johnston H, Kesari A, Nagaraju K, Chen YW, et al. (October 2014). “Asynchronous remodeling is a driver of failed regeneration in Duchenne muscular dystrophy”. The Journal of Cell Biology. 207 (1): 139–158. doi:10.1083/jcb.201402079. PMC 4195829. PMID 25313409.
- Damsker JM, Conklin LS, Sadri S, Dillingham BC, Panchapakesan K, Heier CR, et al. (September 2016). “VBP15, a novel dissociative steroid compound, reduces NFκB-induced expression of inflammatory cytokines in vitro and symptoms of murine trinitrobenzene sulfonic acid-induced colitis”. Inflammation Research. 65 (9): 737–743. doi:10.1007/s00011-016-0956-8. PMID 27261270. S2CID 18698831.
- Heier CR, Yu Q, Fiorillo AA, Tully CB, Tucker A, Mazala DA, et al. (February 2019). “Vamorolone targets dual nuclear receptors to treat inflammation and dystrophic cardiomyopathy”. Life Sci Alliance. 2 (1): e201800186. doi:10.26508/lsa.201800186. PMC 6371196. PMID 30745312.
- “Drug Trials Snapshots: Agamree”. U.S. Food and Drug Administration (FDA). 16 February 2024. Archived from the original on 18 February 2024. Retrieved 14 March 2024. This article incorporates text from this source, which is in the public domain.
- “Drug Approval Package: Agamree”. U.S. Food and Drug Administration (FDA). 7 November 2023. Archived from the original on 13 November 2023. Retrieved 13 November 2023.
- Liu X, Wang Y, Gutierrez JS, Damsker JM, Nagaraju K, Hoffman EP, et al. (September 2020). “Disruption of a key ligand-H-bond network drives dissociative properties in vamorolone for Duchenne muscular dystrophy treatment”. Proceedings of the National Academy of Sciences of the United States of America. 117 (39): 24285–24293. Bibcode:2020PNAS..11724285L. doi:10.1073/pnas.2006890117. PMC 7533876. PMID 32917814.
- Hoffman EP, Riddle V, Siegler MA, Dickerson D, Backonja M, Kramer WG, et al. (June 2018). “Phase 1 trial of vamorolone, a first-in-class steroid, shows improvements in side effects via biomarkers bridged to clinical outcomes”. Steroids. 134: 43–52. doi:10.1016/j.steroids.2018.02.010. PMC 6136660. PMID 29524454.
- Conklin LS, Damsker JM, Hoffman EP, Jusko WJ, Mavroudis PD, Schwartz BD, et al. (October 2018). “Phase IIa trial in Duchenne muscular dystrophy shows vamorolone is a first-in-class dissociative steroidal anti-inflammatory drug”. Pharmacological Research. 136: 140–150. doi:10.1016/j.phrs.2018.09.007. PMC 6218284. PMID 30219580.
- Hoffman EP, Schwartz BD, Mengle-Gaw LJ, Smith EC, Castro D, Mah JK, et al. (September 2019). “Vamorolone trial in Duchenne muscular dystrophy shows dose-related improvement of muscle function”. Neurology. 93 (13): e1312 – e1323. doi:10.1212/WNL.0000000000008168. PMC 7011869. PMID 31451516.
- Smith EC, Conklin LS, Hoffman EP, Clemens PR, Mah JK, Finkel RS, et al. (September 2020). “Efficacy and safety of vamorolone in Duchenne muscular dystrophy: An 18-month interim analysis of a non-randomized open-label extension study”. PLOS Medicine. 17 (9): e1003222. doi:10.1371/journal.pmed.1003222. PMC 7505441. PMID 32956407.
- Mavroudis PD, van den Anker J, Conklin LS, Damsker JM, Hoffman EP, Nagaraju K, et al. (July 2019). “Population Pharmacokinetics of Vamorolone (VBP15) in Healthy Men and Boys With Duchenne Muscular Dystrophy”. Journal of Clinical Pharmacology. 59 (7): 979–988. doi:10.1002/jcph.1388. PMC 6548694. PMID 30742306.
- Li X, Conklin LS, van den Anker J, Hoffman EP, Clemens PR, Jusko WJ (October 2020). “Exposure-Response Analysis of Vamorolone (VBP15) in Boys With Duchenne Muscular Dystrophy”. Journal of Clinical Pharmacology. 60 (10): 1385–1396. doi:10.1002/jcph.1632. PMC 7494537. PMID 32434278.
- “Efficacy and Safety of Vamorolone Over 48 Weeks in Boys With Duchenne Muscular Dystrophy: A Randomized Controlled Trial”. PMID 38335499.
{{cite web}}: Missing or empty|url=(help) - Deswal P. “Santhera and Catalyst to market DMD drug vamorolone in North America”. Pharmaceutical Technology.
- “FDA Approves Vamorolone for Treatment of Duchenne Muscular Dystrophy in Patients Aged 2 Years and Older”. Pharmacy Times. 26 October 2023. Archived from the original on 27 October 2023. Retrieved 27 October 2023.
- “Santhera Receives U.S. FDA Approval of Agamree (vamorolone) for the Treatment of Duchenne Muscular Dystrophy” (Press release). Santhera Pharmaceuticals Holding AG. 27 October 2023. Archived from the original on 31 October 2023. Retrieved 13 November 2023 – via GlobeNewswire.
- World Health Organization (2017). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 77”. WHO Drug Information. 31 (1). hdl:10665/330984.
- “Agamree for the Treatment of Duchenne Muscular Dystrophy, US”. Clinicaltrials Arena. Retrieved 11 February 2025.
External links
Clinical trial number NCT03439670 for “A Study to Assess the Efficacy and Safety of Vamorolone in Boys With Duchenne Muscular Dystrophy (DMD)” at ClinicalTrials.gov
- [1]. Heier CR, et al. VBP15, a novel anti-inflammatory and membrane-stabilizer, improves muscular dystrophy without side effects. EMBO Mol Med. 2013 Oct;5(10):1569-85. [Content Brief][2]. Dillingham BC, et al. VBP15, a novel anti-inflammatory, is effective at reducing the severity of murine experimental autoimmune encephalomyelitis. Cell Mol Neurobiol. 2015 Apr;35(3):377-387. [Content Brief][3]. Heier CR, et al. Vamorolone targets dual nuclear receptors to treat inflammation and dystrophic cardiomyopathy. Life Sci Alliance. 2019 Feb 11;2(1). pii: e201800186. [Content Brief]
| Clinical data | |
|---|---|
| Trade names | Agamree |
| Other names | VBP; VBP-15; 17α,21-Dihydroxy-16α-methylpregna-1,4,9(11)-triene-3,20-dione |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a624005 |
| License data | US DailyMed: Vamorolone |
| Routes of administration | By mouth |
| ATC code | H02AB18 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1]EU: Rx-only[2][3] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 13209-41-1 |
| PubChem CID | 3035000 |
| DrugBank | DB15114 |
| ChemSpider | 2299335 |
| UNII | 8XP29XMB43 |
| KEGG | D11000 |
| ChEBI | CHEBI:228304 |
| ChEMBL | ChEMBL2348780 |
| CompTox Dashboard (EPA) | DTXSID60927527 |
| ECHA InfoCard | 100.032.874 |
| Chemical and physical data | |
| Formula | C22H28O4 |
| Molar mass | 356.462 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
////////////Vamorolone, VBP 15, APPROVALS 2023, EMA 2023, FDA 2023, 8XP29XMB43, AGAMREE, EU 2023
Zilucoplan
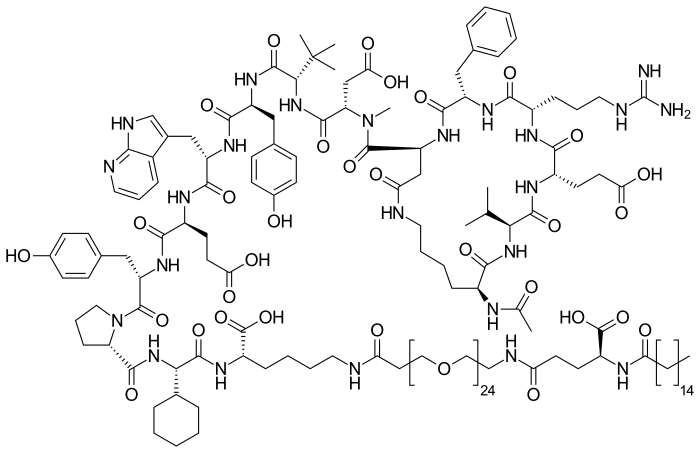
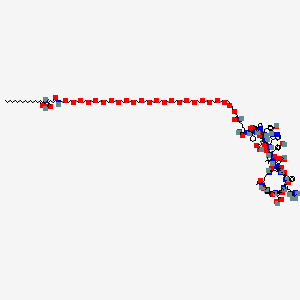
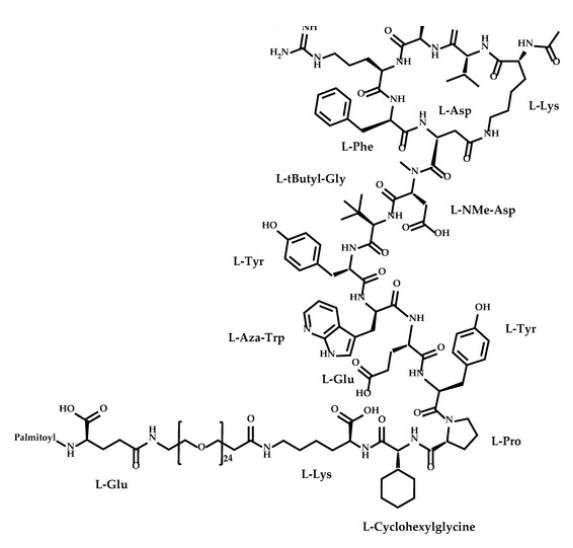
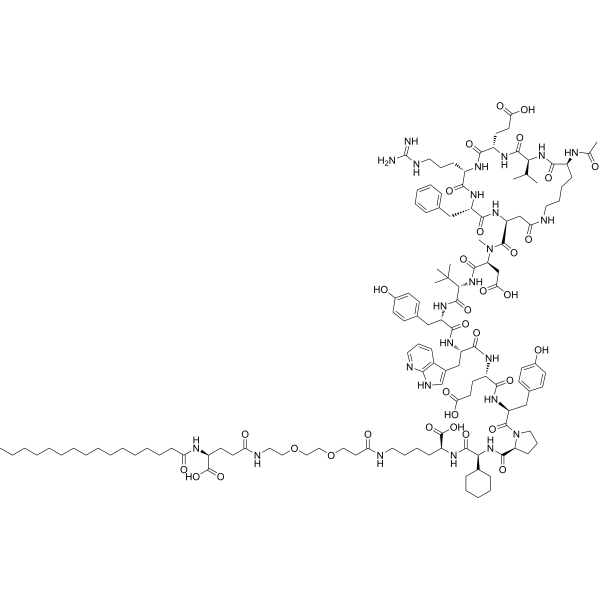
Zilucoplan
CAS 1841136-73-9
YG391PK0CC, RA101495, WHO 10602
3562 g/mol, C172H278N24O55
Zilucoplan lso designated as RA101495, is the active principle of Zilbrysq®, commercialized by UCB Pharma S.A. It is a 3.5 kDa synthetic macrocyclic peptide composed of 15 amino acid residues, including four unnatural amino acids [27]. The amino acid residues composition is: L-Lys, L-Val, L-Glu, L-Arg, L-Phe, L-Asp, L-L-NMe-Asp, L-tButyl-Gly, L-Tyr, L-7-aza-Trp, L-Glu, L-Tyr, L-Pro, L-Cyclohexyl-Gly, and L-Lys.
N2-ACETYL-L-LYSYL-L-VALYL-L-.ALPHA.-GLUTAMYL-L-ARGINYL-L-PHENYLALANYL-L-.ALPHA.-ASPARTYL-N-METHYL-L-.ALPHA.-ASPARTYL-3-METHYL-L-VALYL-L-TYROSYL-3-(1H-PYRROLO(2,3-B)PYRIDIN-3-YL)-L-ALANYL-L-.ALPHA.-GLUTAMYL-L-TYROSYL-L-PROLYL-(2S)-2-CYCLOHEXYLGLYCYL-N6-(3
POLY(OXY-1,2-ETHANEDIYL), ALPHA-(2-(((4S)-4-CARBOXY-1-OXO-4-(1-OXOHEXADECYL)BUTYL)AMINO)ETHYL)-OMEGA-HYDROXY-, 15-ETHER WITH N-ACETYL-L-LYSYL-L-VALYL-L-ALPHA-GLUTAMYL-L-ARGINYL-L-PHENYLALANYL-L-ALPHA-ASPARTYL-N-METHYL-L-ALPHA-ASPARTYL-3-METHYL-
(2S)-2-[[(2S)-2-[[(2S)-1-[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S,5S,8S,11S,14S,22S)-22-acetamido-11-benzyl-8-(3-carbamimidamidopropyl)-5-(2-carboxyethyl)-3,6,9,12,16,23-hexaoxo-2-propan-2-yl-1,4,7,10,13,17-hexazacyclotricosane-14-carbonyl]-methylamino]-3-carboxypropanoyl]amino]-3,3-dimethylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-3-(1H-pyrrolo[2,3-b]pyridin-3-yl)propanoyl]amino]-4-carboxybutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]pyrrolidine-2-carbonyl]amino]-2-cyclohexylacetyl]amino]-6-[3-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[[(4S)-4-carboxy-4-(hexadecanoylamino)butanoyl]amino]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]propanoylamino]hexanoic acid
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Zilucoplan Sodium | Not Available | Not Available | FUSMWKLQHKXKHI-WHKBRXDJSA-J |
FDA 10/17/2023, Zilbrysq, To treat generalized myasthenia gravis in adults who are anti-acetylcholine receptor (AChR) antibody positive
Drug Trials Snapshot
Zilucoplan, sold under the brand name Zilbrysq, is a medication used for the treatment of generalized myasthenia gravis.[6][9][10] It is a complement inhibitor that is injected subcutaneously (under the skin).[6]
Zilucoplan is a cyclic peptide that binds to the protein complement component 5 (C5) and inhibits its cleavage into C5a and C5b.[11]
Zilucoplan was approved for medical use in the United States in October 2023,[6][12] in the European Union in December 2023,[7] and in Australia in July 2024.[1]
Zilucoplan is a 15 amino-acid, synthetic macrocyclic peptide with formula C172H278N24O55. Its sodium salt is used for the treatment of generalised myasthenia gravis (a disease that leads to muscle weakness and tiredness) in adults whose immune system produces antibodies against acetylcholine receptors. It has a role as a complement component 5 inhibitor and an immunosuppressive agent. It is a macrocycle, a homodetic cyclic peptide and a polyether. It is a conjugate acid of a zilucoplan(4-).
PATENT
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US11752190 | No | 2023-09-12 | 2035-06-12 |  |
| US11014965 | No | 2021-05-25 | 2035-06-12 | |
| US10435438 | No | 2019-10-08 | 2035-06-12 | |
| US10208089 | No | 2019-02-19 | 2035-06-12 | |
| US10106579 | No | 2018-10-23 | 2035-06-12 | |
| US10835574 | No | 2020-11-17 | 2035-06-12 | |
| US11535650 | No | 2022-12-27 | 2035-06-12 | |
| US10562934 | No | 2020-02-18 | 2035-06-12 | |
| US11965040 | No | 2024-04-23 | 2035-06-12 | |
PAPER
https://www.mdpi.com/2813-2998/3/2/18
References
- ^ Jump up to:a b c “Zilbrysq (zilucoplan)”. Therapeutic Goods Administration (TGA). 24 September 2024. Retrieved 12 October 2024.
- ^ “Therapeutic Goods (Poisons Standard—June 2024) Instrument 2024”. Federal Register of Legislation. 30 May 2024. Retrieved 10 June 2024.
- ^ “Zilbrysq (UCB Australia Pty Ltd T/A UCB Pharma Division of UCB Australia)”. Therapeutic Goods Administration (TGA). 13 September 2024. Retrieved 15 September 2024.
- ^ “Notice: Multiple additions to the Prescription Drug List (PDL) [2024-08-13]”. Health Canada. 13 August 2024. Retrieved 15 August 2024.
- ^ “Regulatory Decision Summary for Zilbrysq”. Drug and Health Products Portal. 11 July 2024. Retrieved 27 December 2024.
- ^ Jump up to:a b c d e “Zilbrysq- zilucoplan injection, solution”. DailyMed. 19 July 2024. Retrieved 15 September 2024.
- ^ Jump up to:a b c d “Zilbrysq EPAR”. European Medicines Agency. 1 December 2023. Retrieved 11 December 2023.
- ^ “Zilbrysq Product information”. Union Register of medicinal products. 4 December 2023. Archived from the original on 11 December 2023. Retrieved 11 December 2023.
- ^ Howard JF, Kaminski HJ, Nowak RJ, Wolfe GI, Benatar MG, Ricardo A, et al. (April 2018). “RA101495, a subcutaneously administered peptide inhibitor of complement component 5 (C5) for the treatment of generalized myasthenia gravis (gMG): Phase 1 results and phase 2 design (S31. 006)”. Neurology. 90 (15 Supplement). doi:10.1212/WNL.90.15_supplement.S31.006. S2CID 56969245. Archived from the original on 22 February 2022. Retrieved 24 June 2021.
- ^ Howard JF, Vissing J, Gilhus NE, Leite MI, Utsugisawa K, Duda PW, et al. (May 2021). “Zilucoplan: An Investigational Complement C5 Inhibitor for the Treatment of Acetylcholine Receptor Autoantibody-Positive Generalized Myasthenia Gravis”. Expert Opinion on Investigational Drugs. 30 (5): 483–493. doi:10.1080/13543784.2021.1897567. hdl:11250/2770699. PMID 33792453. S2CID 232482753.
- ^ Ricardo A, Arata M, DeMarco S, Dhamnaskar K, Hammer R, Fridkis-Hareli M, et al. (2015). “Preclinical Evaluation of RA101495, a Potent Cyclic Peptide Inhibitor of C5 for the Treatment of Paroxysmal Nocturnal Hemoglobinuria”. Blood. 126 (23): 939. doi:10.1182/blood.V126.23.939.939.
- ^ “Novel Drug Approvals for 2023”. U.S. Food and Drug Administration (FDA). 22 December 2023. Archived from the original on 8 January 2023. Retrieved 27 December 2023.
- ^ Jump up to:a b “Zilbrysq: Pending EC decision”. European Medicines Agency. 15 September 2023. Archived from the original on 26 September 2023. Retrieved 24 September 2023. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Zilucoplan Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). Archived from the original on 17 October 2023. Retrieved 19 October 2023.
- ^ “EU/3/22/2650: Orphan designation for the treatment of myasthenia gravis”. European Medicines Agency. 15 September 2023. Archived from the original on 29 January 2023. Retrieved 24 September 2023.
- ^ World Health Organization (2018). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 80”. WHO Drug Information. 32 (3). hdl:10665/330907.
External links
- Clinical trial number NCT04115293 for “Safety, Tolerability, and Efficacy of Zilucoplan in Subjects With Generalized Myasthenia Gravis (RAISE)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Zilbrysq |
| Other names | RA101495 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a624002 |
| License data | US DailyMed: Zilucoplan |
| Pregnancy category | AU: D[1] |
| Routes of administration | Subcutaneous |
| Drug class | Complement inhibitor |
| ATC code | L04AJ06 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)[2][3][1]CA: ℞-only[4][5]US: ℞-only[6]EU: Rx-only[7][8] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1841136-73-9 |
| PubChem CID | 133083018 |
| DrugBank | DB15636 |
| ChemSpider | 71115966 |
| UNII | YG391PK0CC |
| KEGG | D12357 |
| ChEBI | CHEBI:229659 |
| Chemical and physical data | |
| Formula | C172H278N24O55 |
| Molar mass | 3562.229 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
/////////zilucoplan, Zilbrysq, FDA 2023, APPROVALS 2023, EU 2023, EMA 2023, RA101495, RA 101495, WHO 10602
Tofersen

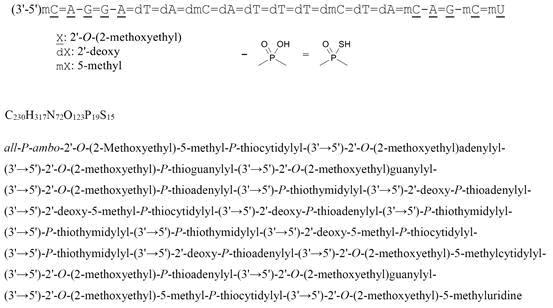
all-P-ambo-2′-O-(2-Methoxyethyl)-5-methyl-P-thiocytidylyl-(3’→5′)-2′-O-(2-methoxyethyl)adenylyl-(3’→5′)-2′-O-(2-methoxyethyl)-P-thioguanylyl-(3’→5′)-2′-O-(2-methoxyethyl)guanylyl-(3’→5′)-2′-O-(2-methoxyethyl)-P-thioadenylyl-(3’→5′)-P-thiothymidylyl-(3’→5′)-2′-deoxy-P-thioadenylyl-(3’→5′)-2′-deoxy-5-methyl-P-thiocytidylyl-(3’→5′)-2′-deoxy-P-thioadenylyl-(3’→5′)-P-thiothymidylyl-(3’→5′)-P-thiothymidylyl-(3’→5′)-P-thiothymidylyl-(3’→5′)-2′-deoxy-5-methyl-P-thiocytidylyl-(3’→5′)-P-thiothymidylyl-(3’→5′)-2′-deoxy-P-thioadenylyl-(3’→5′)-2′-O-(2-methoxyethyl)-5-methylcytidylyl-(3’→5′)-2′-O-(2-methoxyethyl)-P-thioadenylyl-(3’→5′)-2′-O-(2-methoxyethyl)guanylyl-(3’→5′)-2′-O-(2-methoxyethyl)-5-methyl-P-thiocytidylyl-(3’→5′)-2′-O-(2-methoxyethyl)-5-methyluridine
C230H317N72O123P19S15 : 7127.86
[2088232-70-4]

Tofersen
CAS 2088232-70-4
FDA APPROVED 4/25/2023, Qalsody
- BIIB 067
- BIIB067
- Formula
C230H317N72O123P19S15
Molar mass
7127.85 g·mol−1
- Antisense Oligonucleotide Inhibitor Of The Expression Of Superoxide Dismutase 1 Gene
- DNA, D((2′-O-(2-METHOXYETHYL))M5RC-SP-(2′-O-(2-METHOXYETHYL))RA-(2′-O-(2-METHOXYETHYL))RG-SP-(2′-O-(2-METHOXYETHYL))RG-(2′-O-(2-METHOXYETHYL))RA-SP-T-SP-A-SP-M5C-SP-A-SP-T-SP-T-SP-T-SP-M5C-SP-T-SP-A-SP-(2′-O-(2-METHOXYETHYL))M5RC-(2′-O-(2-METHOXYETHYL))R
- IONIS SOD1Rx
To treat amyotrophic lateral sclerosis in adults who have a SOD1 gene mutation
Drug Trials Snapshot
A nucleic acid-based drug indicated for the treatment of a specific type of amyotrophic lateral sclerosis.
Tofersen, sold under the brand name Qalsody, is a medication used for the treatment of amyotrophic lateral sclerosis (ALS).[3] Tofersen is an antisense oligonucleotide that targets the production of superoxide dismutase 1, an enzyme whose mutant form is commonly associated with amyotrophic lateral sclerosis. It is administered as an intrathecal injection.[3]
The most common side effects include fatigue, arthralgia (joint pain), increased cerebrospinal (brain and spinal cord) fluid white blood cells, and myalgia (muscle pain).[3]
Tofersen was approved for medical use in the United States in April 2023,[3][6] and in the European Union in May 2024.[4] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication.[7]
| Clinical data | |
|---|---|
| Trade names | Qalsody |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a623024 |
| License data | US DailyMed: Tofersen |
| Routes of administration | Intrathecal |
| ATC code | N07XX22 (WHO) |
| Legal status | |
| Legal status | CA: ℞-only[1]US: ℞-only[2][3]EU: Rx-only[4][5] |
| Identifiers | |
| CAS Number | 2088232-70-4 |
| DrugBank | DB14782 |
| UNII | 2NU6F9601K |
| KEGG | D11811 |
| Chemical and physical data | |
| Formula | C230H317N72O123P19S15 |
| Molar mass | 7127.85 g·mol−1 |
References
- ^ “Register of Innovative Drugs”. Health Canada. 3 November 2006. Retrieved 17 April 2025.
- ^ “Qalsody- tofersen injection”. DailyMed. 25 April 2023. Archived from the original on 8 May 2023. Retrieved 10 June 2023.
- ^ Jump up to:a b c d e f g h i j k l “FDA approves treatment of amyotrophic lateral sclerosis associated with a mutation in the SOD1 gene” (Press release). U.S. Food and Drug Administration (FDA). 25 April 2023. Archived from the original on 25 April 2023. Retrieved 25 April 2023. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d “Qalsody EPAR”. European Medicines Agency (EMA). 22 February 2024. Retrieved 24 February 2024. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b “Qalsody PI”. Union Register of medicinal products. 3 June 2024. Retrieved 7 September 2024.
- ^ “FDA Grants Accelerated Approval for Qalsody (tofersen) for SOD1-ALS, a Major Scientific Advancement as the First Treatment to Target a Genetic Cause of ALS” (Press release). Biogen. 25 April 2023. Archived from the original on 25 April 2023. Retrieved 25 April 2023 – via GlobeNewswire.
- ^ Jump up to:a b New Drug Therapy Approvals 2023 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2024. Archived from the original on 10 January 2024. Retrieved 9 January 2024.
- ^ Liu A (1 May 2019). “Biogen’s antisense ALS drug shows promise in early clinical trial”. FierceBiotech. Archived from the original on 2 February 2023. Retrieved 25 April 2023.
- ^ Langreth R (22 March 2023). “Biogen’s ALS Drug Gets Partial Backing From FDA Panel”. Bloomberg News. Retrieved 25 April 2023.
- ^ “FDA approves drug which helps to slow progression of rare form of MND”. http://www.sheffield.ac.uk. 28 April 2023. Retrieved 16 May 2024.
- ^ Berdyński M, Miszta P, Safranow K, Andersen PM, Morita M, Filipek S, et al. (January 2022). “SOD1 mutations associated with amyotrophic lateral sclerosis analysis of variant severity”. Scientific Reports. 12 (1): 103. Bibcode:2022NatSR..12..103B. doi:10.1038/s41598-021-03891-8. PMC 8742055. PMID 34996976.
- ^ Constantino A (25 April 2023). “FDA grants accelerated approval for Biogen ALS drug that treats rare form of the disease”. CNBC. Archived from the original on 25 April 2023. Retrieved 25 April 2023.
- ^ Constantino A (22 March 2023). “FDA advisors vote against effectiveness of Biogen’s ALS drug for rare and aggressive form of the disease”. CNBC. Archived from the original on 10 April 2023. Retrieved 25 April 2023.
- ^ Robins R (25 April 2023). “F.D.A. Approves Drug for Rare Form of A.L.S.” The New York Times. Archived from the original on 25 April 2023. Retrieved 25 April 2023.
- ^ “New treatment for rare motor neuron disease recommended for approval”. European Medicines Agency (EMA) (Press release). 23 February 2024. Retrieved 24 February 2024.
////////////tofersen, Qalsody, FDA 2023, APPROVALS 2023, EU 2024, EMA 2024, BIIB 067, BIIB067, IONIS SOD1Rx

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}