

| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2017095472 | NOVEL N-ACYL-(3-SUBSTITUTED)-(8-SUBSTITUTED)-5, 6-DIHYDRO-[1, 2, 4]TRIAZOLO[4, 3-a]PYRAZINES AS SELECTIVE NK-3 RECEPTOR ANTAGONISTS, PHARMACEUTICAL COMPOSITION, METHODS FOR USE IN NK-3 RECEPTOR-MEDIATED DISORDERS |
2016-12-07
|
|
| US2016318941 | SUBSTITUTED [1, 2, 4]TRIAZOLO[4, 3-a]PYRAZINES AS SELECTIVE NK-3 RECEPTOR ANTAGONISTS |
2016-07-08
|
|
| US2017298070 | NOVEL CHIRAL SYNTHESIS OF N-ACYL-(3-SUBSTITUTED)-(8-SUBSTITUTED)-5, 6-DIHYDRO-[1, 2, 4]TRIAZOLO[4, 3-A]PYRAZINES |
2015-09-25
|
|
| US9422299 | NOVEL N-ACYL-(3-SUBSTITUTED)-(8-SUBSTITUTED)-5, 6-DIHYDRO-[1, 2, 4]TRIAZOLO[4, 3-a]PYRAZINES AS SELECTIVE NK-3 RECEPTOR ANTAGONISTS, PHARMACEUTICAL COMPOSITION, METHODS FOR USE IN NK-3 RECEPTOR-MEDIATED DISORDERS |
2015-04-23
|
2015-08-20
|
| US2018111943 | NOVEL N-ACYL-(3-SUBSTITUTED)-(8-SUBSTITUTED)-5, 6-DIHYDRO-[1, 2, 4]TRIAZOLO[4, 3-a]PYRAZINES AS SELECTIVE NK-3 RECEPTOR ANTAGONISTS, PHARMACEUTICAL COMPOSITION, METHODS FOR USE IN NK-3 RECEPTOR-MEDIATED DISORDERS |
2017-10-27
|
Home » APPROVALS 2023 (Page 4)
Category Archives: APPROVALS 2023
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Quizartinib dihydrochloride, キザルチニブ塩酸塩
Quizartinib dihydrochloride
キザルチニブ塩酸塩
| Formula |
C29H32N6O4S. 2HCl
|
|---|---|
| CAS |
1132827-21-4
|
| Mol weight |
633.5891
|
JAPAN, PMDA APPROVED,. 2019/6/18, Vanflyta, To use as part of a treatment regimen for newly diagnosed acute myeloid leukemia that meets certain criteria
Drug Trials Snapshot
fda 2023, 7/20/2023
Quizartinib (AC220) is a small molecule receptor tyrosine kinase inhibitor, originated Ambit Biosciences, and acquired by Daiichi Sankyo, that is currently under development for the treatment of acute myeloid leukaemia. Its molecular target is FLT3, also known as CD135 which is a proto-oncogene.[1]
Flt3 mutations are among the most common mutations in acute myeloid leukaemia due to internal tandem duplication of Flt3. The presence of this mutation is a marker of adverse outcome.
Mechanism
Specifically, Quizartinib selectively inhibits class III receptor tyrosine kinases, including FMS-related tyrosine kinase 3 (FLT3/STK1), colony-stimulating factor 1 receptor (CSF1R/FMS), stem cell factor receptor (SCFR/KIT), and platelet derived growth factor receptors (PDGFRs).
Mutations cause constitutive action of Flt3 resulting in inhibition of ligand-independent leukemic cell proliferation and apoptosis.
Clinical trials
It reported good results in 2012 from a phase II clinical trial for refractory AML – particularly in patients who went on to have a stem cell transplant.[2]
As of 2017 it has completed 5 clinical trials and another 7 are active.[3]
SYN

References
- ^ Chao, Qi; Sprankle, Kelly G.; Grotzfeld, Robert M.; Lai, Andiliy G.; Carter, Todd A.; Velasco, Anne Marie; Gunawardane, Ruwanthi N.; Cramer, Merryl D.; Gardner, Michael F.; James, Joyce; Zarrinkar, Patrick P.; Patel, Hitesh K.; Bhagwat, Shripad S. (2009). “Identification of N-(5-tert-Butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazol-2-yl]phenyl}urea Dihydrochloride (AC220), a Uniquely Potent, Selective, and Efficacious FMS-Like Tyrosine Kinase-3 (FLT3) Inhibitor”. Journal of Medicinal Chemistry. 52 (23): 7808–7816. doi:10.1021/jm9007533.
- ^ Drug Tames Refractory AML. ASH Dec 2012
- ^ Quizartinib studies
 |
|
| Names | |
|---|---|
| IUPAC name
1-(5-(tert-Butyl)isoxazol-3-yl)-3-(4-(7-(2-morpholinoethoxy)benzo[d]imidazo[2,1-b]thiazol-2-yl)phenyl)urea
|
|
| Other names
AC220
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChEBI | |
| ChEMBL | |
| ChemSpider | |
| KEGG | |
| UNII | |
|
CompTox Dashboard (EPA)
|
|
| Properties | |
| C29H32N6O4S | |
| Molar mass | 560.67 g·mol−1 |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
/////////////Quizartinib dihydrochloride, キザルチニブ塩酸塩, JAPAN 2019
OLD POST

QUIZARTINIB
1-(5-(tert-Butyl)isoxazol-3-yl)-3-(4-(7-(2-morpholinoethoxy)benzo[d]imidazo[2,1-b]thiazol-2-yl)phenyl)urea
N-(5-tert-butyl-isoxazol-3-yl)-N’-{ 4- [7-(2-morpholin-4-yl-ethoxy)imidazo [2, 1 -b] [ 1 ,3 ]benzothiazol-2-yl]phenyl } urea
FOR acute myeloidLeukemia,
950769-58-1 CAS
| CAS | 950769-58-1 (free base) 1132827-21-4 (2HCl) |
| Formula | C29H32N6O4S |
| MW | 560.7 |
| Synonim | AC220, AC-010220 ASP-2689 |
PATENTS
U.S. Provisional Patent App. No. 60/743,543, filed March 17, 2006, U.S. Patent App. No. 11/724,992, filed March 16, 2007, and U.S. Patent App. Publication No. 2007/0232604, published October 4, 2007,

BioNews TexasAmbit initiates QUANTUM-R Phase 3 clinical trial of quizartinib in FLT3-ITD …News-Medical.net… the treatment of both newly diagnosed and relapsed FLT3-ITD positive and negative AML patients.
Both the U.S. Food and Drug Administration (FDA) and European Commission have granted orphan drug designation to quizartinib for the treatment of AML.AML, High Risk MDS Therapy
see

Quizartinib
Ambit Biosciences
13 MAY 2013
Ambit Biosciences (NASDAQ:AMBI) is a biotech company that focuses on treatments that inhibit kinases, which are drivers for diseases such as cancer. Three drugs are in development, with the lead one being quizartinib — a Phase 2B trial treatment for acute myeloid leukemia. However, AMBI’s collaboration agreement with Astellas Pharma is set to expire in September, and if it is not replaced, it could mean a delay in Phase 3 trials for quizartinib. Keep in mind that AMBI generated $23.8 million in collaboration revenues last year.
Quizartinib (AC220) is a small molecule receptor tyrosine kinase inhibitor that is currently under development by Ambit Biosciencesfor the treatment of acute myeloid leukaemia. Its molecular target is FLT3, also known as CD135 which is a proto-oncogene.[1]

AC-220 is an angiogenesis inhibitor that antagonizes several proteins involved in vascularization. It was engineered by Ambit Biosciences using KinomeScan technology to potently target FLT3, KIT, CSF1R/FMS, RET and PDGFR kinases. Ambit is developing oral AC-220 in phase III clinical studies for the treatment of relapsed/refractory acute myeloid leukemia (AML) patients with the FMS-like tyrosine kinase-3 (FLT3)-ITD mutation. Early clinical trials are also ongoing for the treatment of advanced solid tumors, for the treatment of refractory or relapsed myelodysplasia, in combination with induction and consolidation chemotherapy for previously-untreated de novo acute myeloid leukemia, and as a maintenance therapy of AML following hematopoietic stem cell transplantation (HSCT). In 2009, orphan drug designation was received both in the U.S. and in the EU for the treatment of AML. In 2009, Ambit Biosciences and Astellas Pharma have entered into a worldwide agreement to jointly develop and commercialize the drug candidate for the treatment of cancer and non-oncology indications. This agreement was terminated in 2013.
Flt3 mutations are among the most common mutations in acute myeloid leukaemia due to internal tandem duplication of Flt3. The presence of this mutation is a marker of adverse outcome.
| Quizartinib is a small molecule with potential anticancer activity. Quizartinib is a selective inhibitor of class III receptor tyrosine kinases, including FMS-related tyrosine kinase 3 (FLT3/STK1), stem cell factor receptor (SCFR / KIT), colony-stimulating factor 1 receptor (CSF1R/FMS) and platelet-derived growth factor receptors (PDGFRs .) Able to inhibition of ligand-independent cell proliferation and apoptosis. Mutations in FLT3 are the most frequent genetic alterations in acute myeloid leukemia (AML) and occur in approximately 30% of cases of AML. | |
| Quizartinib представляет собой малую молекулу с потенциальной противораковой активностью. Quizartinib является селективным ингибитором класса III рецепторов тирозин киназ, в том числе FMS-связанных тирозинкиназы 3 (FLT3/STK1), фактор стволовых клеток рецепторов (SCFR / KIT), колониестимулирующий фактор 1 рецепторов (CSF1R/FMS) и тромбоцитарный рецепторов фактора роста (PDGFRs). Способен к торможению лиганд-независимой клеточной пролиферации и апоптоза. Мутации в FLT3 являются наиболее частыми генетическими изменениями в остром миелобластном лейкозе (ОМЛ) и встречаются примерно в 30% случаев ОМЛ. |
Mechanism
Specifically, Quizartinib selectively inhibits class III receptor tyrosine kinases, including FMS-related tyrosine kinase 3 (FLT3/STK1), colony-stimulating factor 1 receptor (CSF1R/FMS), stem cell factor receptor (SCFR/KIT), and platelet derived growth factor receptors (PDGFRs).
Mutations cause constitutive action of Flt3 leading to resulting in inhibition of ligand-independent leukemic cell proliferation and apoptosis.

Clinical trials
It had good results in a phase II clinical trial for refractory AML – particularly in patients who went on to have a stem cell transplant.[2]

………………………..
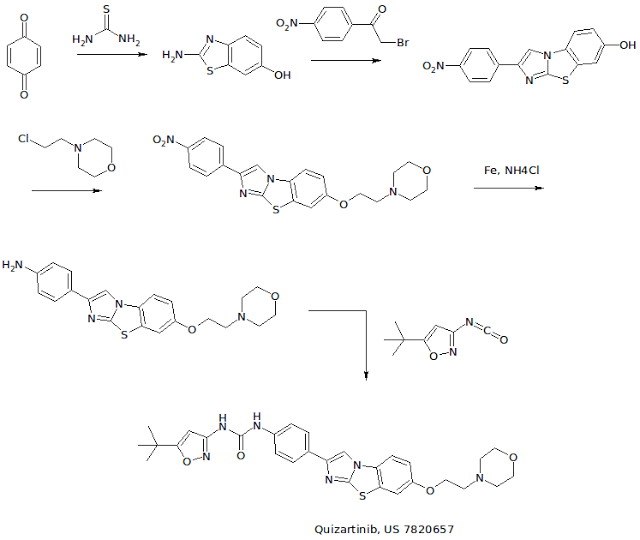
WO 2007109120 COMPD B1
EXAMPLE 3: PREPARATION OF N-(5-TERT-BUTYL-ISOXAZOL-3-YL)-N’-{4-[7-(2- MORPHOLIN-4-YL-ETHOXY)IMIDAZO[2,1 -B3[1 ,3]BENZOTHIAZOL-2-YL]PHENYL}UREA [Compound B1]
[00426] A. The intermediate 2-amino-1,3-benzothiazol-6-ol was prepared according to a slightly modified literature procedure by Lau and Gompf. J. Org. Chem. 1970, 35, 4103-4108. To a stirred solution of thiourea (7.6 g, 0.10 mol) in a mixture of 200 ml_ ethanol and 9 ml_ concentrated hydrochloric acid was added a solution of 1 ,4-benzoquinone (21.6 g, 0.20 mol) in 400 mL of hot ethanol. The reaction was stirred for 24 hours at room temperature and then concentrated to dryness. The residue was triturated with hot acetonitrile and the resulting solid was filtered and dried.
[00427] The free base was obtained by dissolving the hydrochloride salt in water, neutralizing with sodium acetate, and collecting the solid by filtration. The product (2-amino-1 ,3-benzothiazol-6-ol) was obtained as a dark solid that was pure by LCMS (M+H = 167) and NMR. Yield: 13.0 g (78 %). NMR (DMSOd6) £7.6 (m, 2H ), 6.6 (d, 1H).
[00428] B. To prepare the intermediate 2-(4-nitrophenyl)imidazo[2,1- b][1 ,3]benzothiazoI-7-ol, 2-amino-1 ,3-benzothiazol-6-ol, (20.0 g, 0.12 mol) and 2-bromo-4′-nitroacetophenone (29.3 g, 0.12 mol) were dissolved in 600 mL ethanol and heated to reflux overnight. The solution was then cooled to 00C in an ice-water bath and the product was collected by vacuum filtration. After drying under vacuum with P2O5 , the intermediate (2-(4- nitrophenyl)imidazo[2,1-_D][1,3]benzothiazol-7-ol) was isolated as a yellow solid. Yield: 17.0 g (46 %) NMR (DMSO-CT6) δ 10 (s, 1 H), 8.9 (s, 1H), 8.3 (d, 2H), 8.1 (d, 2H), 7.8 (d, 1 H), 7.4 (s, 1 H), 6.9 (d, 1 H). [00429] C. To make the 7-(2-morpholin-4-yl-ethoxy)-2-(4-nttro- phenyl)imidazo[2,1-£>][1 ,3]benzothiazo!e intermediate: 2-(4- nitrophenyl)imidazo[2,1-jb][1 ,3]benzothiazol-7-ol, (3.00 g, 9.6 mmol) was suspended in 100 mL dry DMF. To this mixture was added potassium carbonate (4.15 g, 30 mmol, 3 eq), chloroethyl morpholine hydrochloride (4.65 g, 25 mmol, 2.5 eq) and optionally tetrabutyl ammonium iodide (7.39 g, 2 mmol). The suspension was then heated to 900C for 5 hours or until complete by LCMS. The mixture was cooled to room temperature, poured into 800 mL water, and allowed to stand for 1 hour. The resulting precipitate was collected by vacuum filtration and dried under vacuum. The intermediate, (7-(2- morpholin-4-yl-ethoxy)-2-(4-nitro-phenyl)imidazo[2,1-jb][1 ,3]benzothiazole) was carried on without further purification. Yield: 3.87 g (95 %) NMR (DMSO-Cf6) δ 8.97 (s, 1 H), 8.30 (d, 2H), 8.0 (d, 2H), 7.9 (d, 1 H), 7.7 (s, 1 H), 7.2 (d, 1 H), 4.1 (t, 2H), 5.6 (m, 4H), 2.7 (t, 2H).
[00430] D. To make the intermediate 7-(2-morpholin-4-yl-ethoxy)-2-(4- amino-phenyl)!midazo[2, 1 -b][1 ,3]benzothiazole: To a suspension of 7-(2- morpholin-4-yl-ethoxy)-2-(4-nitro-phenyl)imidazo[2,1-ib][1 ,3]benzothiazole (3.87g, 9.1 mmol) in 100 ml_ isopropyl alcohol/water (3:1 ) was added ammonium chloride (2.00 g, 36.4 mmol) and iron powder (5.04 g, 90.1 mmol). The suspension was heated to reflux overnight with vigorous stirring, completion of the reaction was confirmed by LCMS. The mixture was filtered through Celite, and the filtercake was washed with hot isopropyl alcohol (150 ml_). The filtrate was concentrated to approximately 1/3 of the original , volume, poured into saturated sodium bicarbonate, and extracted 3 times with dichloromethane. The combined organic phases were dried over MgSO4 and concentrated to give the product as an orange solid containing a small amount (4-6 %) of starting material. (Yield: 2.75 g 54 %). 80% ethanol/water may be used in the place of isopropyl alcohol /water — in which case the reaction is virtually complete after 3.5 hours and oniy traces of starting material are observed in the product obtained. NMR (DMSO-d6) δ 8.4 (s, 1 H), 7.8 (d, 1 H), 7.65 (d, 1 H), 7.5 (d, 2H), 7.1 (d, 1 H), 6.6 (d, 2H), 4.1 (t, 2H)1.3.6 (m, 4H), 2.7 (t, 2H).
[00431] E. A suspension of 7-(2-morpholin-4-yl-ethoxy)-2-(4-amino- phenyl)imidazo[2,1-b][1 ,3]benzothiazole (4.06 g, 10.3 mmol) and 5-tert- butylisoxazole-3-isocyanate (1.994 g, 12 mmol) in toluene was heated at 120 0C overnight. The reaction was quenched by pouring into a mixture of methylene chloride and water containing a little methanol and neutralized with saturated aqueous NaHCO3 solution. The aqueous phase was extracted twice with methylene chloride, the combined organic extracts were dried over MgSO4 and filtered. The filtrate was concentrated to about 20 ml volume and ethyl ether was added resulting in the formation of a solid. The precipitate was collected by filtration, washed with ethyl ether, and dried under vacuum to give the free base. Yield: 2.342 g (41 %) NMR (DMSO-Cf6) £9.6 (br, 1H), 8.9 (br, 1H), 8.61 (s, 1H), 7.86 (d, 1 H), 7.76 (d, 2H), 7.69 (d, 1 H), 7.51 (d, 2H), 7.18 (dd, 1H), 6.52 (s, 1H), 4.16 (t, 2H), 3.59 (t, 4H), 3.36 (overlapping, 4H), 2.72 (t, 2H), 1.30 (s, 9H). NMR (CDCI3) £9.3 (br, 1H), 7.84 (m, 4H), 7.59 (d, 2H), 7.49 (d, 1 H), 7.22 (d, 1 H), 7.03 (dd, 1 H)1 5.88 (s, 1 H), 4.16 (t, 2H), 3.76 (t, 4H), 2.84 (t, 2H), 2.61 (t, 4H), 1.37 (s, 9H).
[00432] F. For the preparation of the hydrochloride salt, N-(5-tert-butyl- isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2, 1 – b][1 ,3]benzothiazol-2-yl]phenyI}urea hydrochloride, the free base was dissolved in a mixture of 20 ml methylene chloride and 1 ml methanol. A solution of 1.0 M HCI in ethyl ether (1.1 eq.) was added dropwise, followed by addition of ethyl ether. The precipitate was collected by filtration or centrifugation and washed with ethyl ether to give the hydrochloride salt. Yield: 2.44 g (98 %) NMR (DMSO-d6) £11-0 (br, 1 H), 9.68 (s, 1H), 9.26 (s, 1H), 8.66 (s, 1 H), 7.93 (d, 1H), 7.78 (m, 3H), 7.53 (d, 2H), 7.26 (dd, 1H), 6.53 (S, 1 H), 4.50 (t, 2H), 3.97 (m, 2H), 3.81 (t, 2H), 3.6 (overlapping, 4H)13.23 (m, 2H)1 1.30 (s, 9H).
[00433] G. Alternatively, Compound B1 may be made by taking the intermediate from Example 4B and reacting it with chloroethyl morpholine hydrochloride under conditions described in Step C. [00434] H . Λ/-(5-tert-butyl-isoxazol-3-yl)-Λ/’-{4-[5-(2-morpholin-4-yl- ethoxy)imidazo[2,1-6][1 ,3]benzothiazol-2-yl]phenyl}urea hydrochloride, a compound having the general formula (I) where R1 is substituted on the 5 position of the tricyclic ring, was prepared in the manner described in Steps A- F but using the cyciization product 2-amino-benzothiazol-4-ol with 2-bromo-4′- nitroacetophenone in Step A. 1H NMR (DMSO-d6) δ 11.6 (br, 1 H)1 9.78 (br, 1H), 9.56 (br, 1 H), 8.64 (s, 1H)1 7.94 (d, 2H), 7.70 (s, 1H)1 7.56 (d, 2H), 7.45 (t, 1 H), 7.33 (d, 1H), 6.54 (s, 1 H), 4.79 (t, 2H), 3.87 (m, 6H), 3.60 (m, 2H), 3.34 (m, 2H)1 1.30 (s, 9H); LC-MS: ESI 561 (M+H)+. [Compound B11] [00435] I. N-(5-tert-butyl-isoxazol-3-yl)-N’-{4-[6-(2-morpholin-4-yl- ethoxy)imidazo[2,1-b][1 ,3]benzothiazol-2-yl]phenyl}urea hydrochloride [Compound B12] was also prepared by first preparing the benzothiazole starting material, 5 methoxy-benzothiazol-2yl~amine: [00436] To prepare the 5-methoxy-benzothiazol-2-ylamine starting material: To a suspension of (3-methoxy-phenyl)-thiourea (1.822g, 10 mmol) in CH2CI2 (20 ml_) at 0 0C was added dropwise a solution of bromine (1.76 g, 11 mmol) in 10 ml of trichloromethane over a period of thirty minutes. The reaction was stirred for 3 hours at room temperature then heated to 3 hours to reflux for one hour. The precipitate was filtered and washed with dichloromethane. The solid was suspended in saturated NaHCOsand extracted with CH2CI2. The extract was dried over MgSO4 and concentrated to give a white solid (1.716 g, 95%).
………………….
WO 2011056939
N-(5-ieri-butyl- isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l-&][l,3]benzothiazol-2- yl]phenyl}urea (I), or a pharmaceutically acceptable salt, solvate, hydrate, or polymorph thereof. N-(5-ieri-Butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- / ][!, 3]benzo

N- (5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- &][l,3]benzo-thiazol-2-yl]phenyl}urea (I), or a pharmaceutically acceptable salt, solvate, hydrate, or polymorph thereof, comprising any one, two, three, four, five, six, seven of the steps of:
(A) converting 2-amino-6-alkoxybenzothiazole (II), wherein R1 is a suitable phenolic hydroxyl protecting ;

(II) (III)
(B) reacting 2-amino-6-hydroxybenzothiazole (III) with compound (IV), wherein X is a leaving group, to yield 2-(4-nitrophenyl)imidazo[2,l-b]benzothiazol-7-ol (V);

(C) reacting 2-(4-nitrophenyl)imidazo[2,l-b]benzothiazol-7-ol (V) with compound (VI), wherein X2 is a leaving group, to yield 7-(2-morpholin-4-yl-ethoxy)-2-(4- nitrophenyl)imidazo[ -b]benzothiazole (VII);


(D) reducing 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo[2, 1- bjbenzothiazole (VII) to yield 7-(2-morpholin-4-yl-ethoxy)-2-(4- am

(E) converting 3-amino-5-£er£-butyl isoxazole (IX) to a 5-?er?-butylisoxazol-3- ylcarbamate derivative (X), wherein R2 is optionally substituted aryl, heteroaryl, alkyl, or cycloalkyl;

(IX) (X)
(F) reacting 7-(2-morpholin-4-yl-ethoxy)-2-(4-aminophenyl)imidazo[2,l- bjbenzothiazole (VIII) with a 5-£er£-butylisoxazol-3-ylcarbamate derivative (X) to yield N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- &][l,3]benzo-

(G) converting N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2,l-&][l,3]benzo-thiazol-2-yl]phenyl}urea to an acid addition salt of N- (5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- b] [ 1 ,3]benzo-thiazol-2-yl]phenyl } urea.
[00128] In certain embodiments, provided herein are processes for the preparation of N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- &][l,3]benzo-thiazol-2-yl]phenyl}urea, or a pharmaceutically acceptable salt, solvate, hydrate, or polymorph thereof, as depicted in Scheme 1, wherein R1, R2, X1, and X2 are defined herein elsewhere. In specific embodiments, provided herein are processes for the preparation of N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2,l-&][l,3]benzo-thiazol-2-yl]phenyl}urea (I), or a pharmaceutically acceptable salt, solvate, hydrate, or polymorph thereof, comprising any one, two, three, four, five, six, seven, of the Steps A, B, C, D, E, F, and G, as depicted in Scheme 1.
Scheme 1 :

A. Preparation of 2-amino-6-hydroxybenzothiazole
1. Example A-l[00252] To a 1-L 3-necked round bottom flask fitted with a condenser, heating mantle, and mechanical stirrer was charged aqueous hydrobromic acid (48%, 632 mL, 5.6 mol, 10 equiv). 2-Amino-6-methoxybenzothiazole (100 g, 0.55 mol, 1 equiv) was added to the above flask over 15 minutes. The reaction temperature was raised slowly to reflux (105-110 °C). A clear dark brown colored solution was observed at about 80 °C. The reflux was continued at 105-110 °C for about 4 hr. The progress of the reaction was monitored by HPLC. When 2-amino-6-methoxybenzothiazole was less than 2%, the reaction was substantially complete.
[00253] The reaction mass was cooled to 0-5 °C and at this point precipitation of a solid was observed. The mixture was maintained at 0-5 °C for 0.5 hr and filtered, and the cake was pressed to remove HBr. The wet cake was transferred to a 2-L round bottom flask fitted with a mechanical stirrer. Saturated aqueous sodium bicarbonate solution (-1500 mL) was added slowly at ambient temperature, whereupon considerable frothing was observed. The pH of the solution was found to be about 6.5 to 7. The mixture was stirred for 0.5 hr at ambient temperature and filtered. The filter cake was washed with water (500 mL), dried on the filter and then under vacuum at 30-35 °C for 10-12 hr to give the product 2-amino-6-hydroxybenzothiazole (82 g, 89% yield, HPLC purity = 99%). JH NMR (DMSO-if6, 500 MHz): δ 7.12 (d, 1H), 7.06 (S, 2H, NH2), 7.01 (d, 1H), 6.64 (dd, 1H); MS (m/z) = 167.1 [M+ + 1].
[00254] Table: Summary of Plant Batches

[00255] HPLC chromatographic conditions were as follows: The column used was XTerra RP8, 250 X 4.6 mm, 5μ or equivalent. Mobile Phase A was buffer, prepared by mixing 3.48 g of dipotassium hydrogen phosphate in 1.0 L of water, and adjusting the pH to 9.0 with phosphoric acid. Mobile Phase B was methanol. The flow rate was 1.0 mL/minute. Detection was set at UV 270 nm. The injection volume was 20 μΐ^, and the sample was diluted with a diluent (Mobile Phase A : Mobile Phase B = 70:30). Test solution was prepared by weighing accurately about 25 mg of sample and transferring it into a 100 mL volumetric flask, dissolving with 20-30 mL of diluent, making up the volume to the mark with diluent, and mixing. The HPLC was performed by separately injecting equal volumes of blank and test solution, and recording the chromatogram for all injections. The purity was calculated by area normalization method.
[00256] Table: HPLC Method

2. Example A-2
[00257] 2-Amino-6-methoxybenzothiazole was reacted with hot aqueous HBr at a temperature of >70 °C for about 3 hours and then the clear solution was cooled to ambient temperature overnight. The precipitated solids were collected, dissolved in hot water and the pH was adjusted to between 4.5-5.5. The resultant solids were collected, dried and re-crystallized from isopropanol. Second crop material was collected. The solids were vacuum dried to give 2-amino-6-hydroxybenzothiazole.
[00258] The reaction progress was monitored by thin layer chromatography (TLC). The product was isolated as a white solid, with 99.4% purity (HPLC area %). JH NMR (300 MHz, DMSO-if6) was collected, which conformed to structure.
3. Example A-3
[00259] A 22-L 3-neck round bottom flask was equipped with a mechanical agitator, thermocouple probe, a reflux condenser, and a heating mantle. The flask was charged with hydrobromic acid (14 L, 123.16 mol, 13.10 equiv). Heating was initiated and 2- amino-6-methoxybenzothiazole was added (1.7 kg, 9.4 mol, 1.00 equiv) over 10 minutes with stirring. The heating of the reaction mixture was continued to reflux, and maintained (>107 °C) for approximately 5 hours. The reaction mixture turned into a clear solution between 75 °C and 85 °C. The reaction progress was monitored by TLC until no starting material was observed (A -0.5 mL reaction mixture aliquot was diluted with -0.5 mL water as a clear solution, neutralized with sodium acetate to pH -5 and extracted with 1 mL dichloromethane. The organic layer was spotted: 5%
methanol/dichloromethane; Rf (product) = 0.35; Rf (starting material) = 0.40).
[00260] The reaction mixture was cooled to – 20 °C (overnight). White solids precipitated. The solids were filtered on a polypropylene filter and pressed to remove as much hydrobromic acid from the solids as possible to facilitate the subsequent pH adjustment step. The slightly wet crude product was dissolved in hot (50 °C) water (5 L). The clear solution was filtered to remove any insoluble material present, and the solids were washed with 50 °C water. The filtrate was cooled to 10 °C. The cooled filtrate was neutralized with sodium acetate (- 1.0 kg) to pH 4.5 to 5.5 with vigorous stirring. A thick white solid precipitated. The solids were collected by filtration, and washed with cool (-10 °C) water (2 x 1.0 L) and air dried.
[00261] The wet crude product was slurried in hot (50 °C) isopropanol (3 L) briefly and allowed to stand in a cool room (-5 °C) overnight. The solids were collected by filtration and washed with methyl ferf-butylether (2 x 500 mL). The solids were dried in a vacuum oven overnight (<30 mm Hg) at 30 °C (first crop). Yield: 1068 g (68%), white solid. HPLC: 99.4% (area). JH NMR (300 MHz, DMSO- ) conformed to structure.
[00262] The organic filtrate was collected in a total volume of 1.0 L, cooled to 10 °C. The off-white solids were precipitated and collected by filtration. The solids were dried in a vacuum oven overnight (<30 mm Hg) at 30 °C (second crop). Yield: 497 g (32%), off-white solid. HPLC: 99.8% (area).
[00263] The overall yield combining the first crop and the second crop was 1565 g, (99%).
B. Preparation of 2-(4-nitrophenyl)imidazo[2,l-b]benzothiazol-7-ol

1. Example B-l[00264] A 3-L 3-neck round bottom flask fitted with a condenser, a heating mantle, and a mechanical stirrer was charged with H-butanol (1.5 L), followed by 2-amino-6- hydroxybenzothiazole (75 g, 0.45 mol, 1.0 equiv), 2-bromo-4′-nitroacetophenone (121 g, 0.50 mol, 1.1 equiv), and sodium bicarbonate (41.6 g, 0.50 mol, 1.0 equiv). The reaction temperature was gradually raised to reflux and maintained at reflux (110-115 °C) for 2-3 hr. During the temperature increase, the reaction mass turned into a clear solution and then immediately changed into an orange colored suspension at 65-75 °C. The progress of the reaction was monitored by HPLC analysis every 1 hr (reaction mass sample was submitted to QC). When the level of 2-amino-6-hydroxybenzothiazole was less than 2%, the reaction was substantially complete.
[00265] The reaction mass was slowly cooled to 50-60 °C and then further cooled to 0-5 °C and stirred for 15 min. The precipitated solids were collected by filtration, and dried on the filter. The wet cake was transferred in to a 1-L round bottom flask, and water (600 mL) was added. The suspension was stirred for 0.5 hr and filtered, and the solid was dried on the filter. The wet cake was again taken in to a 1-L round bottom flask and stirred with acetone (200 mL). The slurry was filtered and washed with acetone (2 X 100 mL), and the solid was dried on the filter, unloaded and further dried in a vacuum oven at ambient temperature to give the product 2-(4-nitrophenyl)imidazo[2,l- b]benzothiazol-7-ol (V) (120 g, 85.7% yield, HPLC purity = 98.7%). JH NMR (DMSO- d6, 500 MHz): δ 9.96 (s, 1H, OH), 8.93 (s, 1H), 8.27 (d, 2H), 8.06 (d, 2H), 7.78 (d, 1H), 7.38 (d, 1H), 6.97 (dd, 1H); MS (m/z) = 312 [M+ + 1].
[00266] Table: Summary of Plant Batches

* Input of 2-amino-6-hydroxybenzothiazole (III)
[00267] HPLC chromatographic conditions were as follows: The column used was XTerra RP8, 250 X 4.6 mm, 5μ or equivalent. Mobile Phase A was buffer, prepared by mixing 3.48 g of dipotassium hydrogen phosphate in 1.0 L of water, and adjusting the H to 9.0 with phosphoric acid. Mobile Phase B was methanol. The flow rate was 1.0 mL/minute. Detection was set at UV 235 nm. The injection volume was 10 μΐ^. The blank was prepared by transferring 200 μΐ. of DMSO and 200 μΐ. of 2M NaOH into a 10 mL volumetric flask, making up the volume to the mark with methanol, and mixing. The test solution was prepared by weighing accurately about 10 mg of sample and transferring it into a 50 mL volumetric flask, dissolving with 1 mL of DMSO and 1 mL of 2M NaOH, sonicating to dissolve, making up the volume to the mark with methanol, and mixing. The HPLC was performed by separately injecting equal volumes of blank and test solution, and recording the chromatogram for all injections. The purity was calculated by area normalization method.
[00268] Table: HPLC Method

2. Example B-2
[00269] A 50-L 3-neck round bottom flask was equipped with a mechanical agitator, a thermocouple probe, a reflux condenser, and a heating mantle. The flask was charged with 2-amino-6-hydroxybenzothiazole (1068 g, 6.43 mol, 1.0 equiv) and ethanol (200 proof, 32.0 L), and the suspension was stirred for 10 minutes. 2-Bromo-4- nitroacetophenone (1667 g, 6.49 mol, 1.01 equiv) was added in one portion. The reaction mixture was heated to reflux (78 °C). The reflux was maintained for approximately 25 hours, resulting in a yellow suspension. The reaction progress was monitored by TLC (20% methanol/ethyl acetate; Rf (product) = 0.85; Rf (starting material) = 0.30). TLC indicated -50% 2-amino-6-hydroxybenzothiazole after 24 hours of reflux. Tetrabutylammonium iodide (10 g) was added and reflux was maintained for an additional 12 hours. TLC indicated -50% starting material still present. Coupling was seen to occur at both the thiazole and the amine.
[00270] The reaction mixture was cooled to 0-5 °C. The solids were collected by filtration, and the yellow solid was washed with ethanol (200 proof, 2 X 1.0 L) and diethyl ether (2 X 1.5 L). The solids were dried in a vacuum oven (<10 mm Hg) at 40 °C. Yield: 930 g (46%), yellow solid. HPLC: 99.5% (area). JH NMR (300 MHz, DMSO-i¾) conformed to structure.
3. Example B-3
[00271] The reaction of Step B was carried out on multiple runs, varying solvents, temperature, and base. The results were summarized in the table below. The product (V) was isolated as yellow or green solids, with 1H NMR consistent with the structure and a purity of greater than about 98% by HPLC analysis.
[00272] Table: Reaction Condition Screening

TBAI = Tetrabutylammonium Iodide
C. Preparation of 7-(2-morpholin-4-yl-ethoxy)-2-(4- nitrophenyl)imidazo[2, 1 -bjbenzothiazole

1. Example C-l
[00273] To a 2000-L glass-lined (GL) reactor was charged DMF (298 kg), and the agitator was started. Under a nitrogen blanket, the reactor was charged with 2-(4- nitrophenyl)imidazo[2,l-&]benzothiazol-7-ol (36.8 kg, 118.2 mol, 1.0 equiv), 4-(2- chloroethyl)morpholine hydrochloride (57.2-66.0 kg, 307.3-354.6 mol, 2.6-3.0 equiv), tetrabutylammonium iodide (8.7 kg, 23.6 mol, 0.2 equiv) and potassium carbonate (49.0 kg, 354.6 mol, 3.0 equiv). The resulting yellow suspension was heated and stirred at 90 + 5 °C for at least 15 minutes, then the temperature was increased to 110 + 5 °C. The mixture was stirred for at least 1 hour and then sampled. The reaction was deemed complete if 2-(4-nitrophenyl) imidazo[2,l-&]benzothiazol-7-ol was <1% by HPLC. If the reaction was not complete, the heating was continued and the reaction sampled. If the reaction was incomplete after 6 hours, additional 4-(2-chloroethyl)morpholine hydrochloride may be charged. In general, additional charges of 4-(2- chloroethyl)morpholine hydrochloride had not been necessary at the given scale.
[00274] The reactor was cooled to 20 + 5 °C and charged with water (247 kg) and acetone (492 kg). The mixture was agitated at 20 + 5 °C for at least 1 hour. The product (VII) was isolated by filtration or centrifuge, and washed with water and acetone, and then dried in a vacuum oven at 45 °C to constant weight to give a yellow solid (46.2 kg, 92% yield, HPLC purity = 97.4% by area). JH NMR (300 MHz, DMSO- ) conformed to structure.
2. Example C-2
[00275] 2-(4-Nitrophenyl)imidazo[2, l-b]benzothiazol-7-ol, 4-(2-chloroethyl)- morpholine hydrochloride, potassium carbonate, and tetrabutylammonium iodide were added to N,N-dimethylformamide forming a yellow suspension that was heated at a temperature of >50 °C for over 3 hours. The reaction was cooled and the solids were collected, slurried into water, filtered, slurried into acetone, filtered and washed with acetone to give yellow solids that were dried under vacuum to give 7-(2-morpholin-4-yl- ethoxy)-2-(4-nitrophenyl)imidazo[2,l-b]benzothiazole.
[00276] The reaction progress was monitored by thin layer chromatography (TLC). The product was isolated as a yellow solid, with 99% purity (HPLC area %), and a water content of 0.20%. Infrared (IR) spectrum was collected, which conformed to structure.
3. Example C-3
[00277] A 50-L 3-neck round bottom flask was equipped with a mechanical agitator, a thermocouple probe, a drying tube, a reflux condenser, and a heating mantle. The flask was charged with 2-(4-nitrophenyl)imidazo [2,l-&]benzothiazol-7-ol (1.770 kg, 5.69 mol, 1.0 equiv), N,N-dimethylformamide (18.0 L), 4-(2-chloroethyl)morpholine hydrochloride (2.751 kg, 14.78 mol, 2.6 equiv), potassium carbonate (2.360 kg, 17.10 mol, 3.0 equiv), and tetrabutylammonium iodide (0.130 kg, 0.36 mol, 0.06 equiv) with stirring. The resulting yellow suspension was heated to about 90 °C to 95 °C, maintaining the temperature for approximately 5 hours. The reaction was monitored by TLC until no starting material was observed (20% methanol / ethyl acetate; Rf (product) = 0.15; Rf (starting material) = 0.85).
[00278] The reaction mixture was cooled to -10 °C, and the yellow solids were collected by filtration on a polypropylene filter pad. The solids were slurried in water (2 X 5 L) and filtered. The crude wet product was slurried in acetone (5 L), filtered, and the solids were rinsed with acetone (2 X 1.5 L). The solids were dried in a vacuum oven (<10 mm Hg) at 45 °C. Yield: 2.300 kg (95%), yellow solid. TLC: R/ = 0.15 (20% methanol / EtOAc). HPLC: 95.7% (area). JH NMR (300 MHz, DMSO-i¾) conformed to the structure.
[00279] Table: Yields from multiple batch runs

4. Example C-4
[00280] To a reactor were added 2-(4-nitrophenyl)imidazo [2,l-&]benzothiazol-7-ol (1.0 kg), 4-(2-chloroethyl)morpholine hydrochloride (1.6 kg), tetrabutylammonium iodide (0.24 kg), and potassium carbonate (1.3 kg, anhydrous, extra fine, hydroscopic). N,N-Dimethylformamide (DMF) (8.6 L) was added into the reactor. The DMF used had water content of no more than 0.05% w/w. The mixture was stirred for between 15 and 30 minutes to render a homogeneous suspension, which was heated to between 85 °C and 95 °C and stirred at between 85 °C and 95 °C for 15 to 30 minutes. The mixture was then heated to between 105 °C and 120 °C and stirred at between 105 °C and 120 °C (e.g. , 115 °C) until the reaction was complete (as determined by HPLC to monitor the consumption of starting material). In some embodiments, if necessary (e.g. , if after 6 hours the reaction was not complete as indicated by HPLC analysis), additional 4-(2- chloroethyl)morpholine hydrochloride (0.03 kg) may be added and the reaction mixture stirred at between 105 °C and 120 °C (e.g. , 115 °C) until reaction completion.
[00281] The reaction mixture was cooled to between 20 °C and 30 °C (e.g. , over a period of 3 hours). To another reactor was added deionized water (7.6 L) and acetone (15 L). The mixture of water and acetone was then added into the reaction mixture while maintaining the temperature at between 20 °C and 30 °C. The mixture was then stirred for 1 to 2 hours at a temperature of between 20 °C and 30 °C. The mixture was filtered, and the solid was washed with deionized water (e.g. , about 45x deionized water) until pH of washes was below 8. The solid was then washed with acetone (9.7 L). The solid was dried under vacuum at a temperature of less than 50 °C until the water content by Karl-Fischer was less than 0.30% w/w and TGA curve showed a mass loss of less than 0.30% w/w at about 229 °C (sampling approximately every 6 hours). The desired product was obtained in about 89% yield having about 99% purity by HPLC.
5. Example C-5
[00282] To a reactor were added 2-(4-nitrophenyl)imidazo [2, l-&]benzothiazol-7-ol (1.0 kg), 4-(2-chloroethyl)morpholine hydrochloride (1.6 kg), and potassium carbonate (1.3 kg, anhydrous, extra fine, hydroscopic). N,N-Dimethylformamide (DMF) (8.6 L) was added into the reactor. The DMF used had water content of no more than 0.05% w/w. The mixture was stirred for between 15 and 30 minutes to render a homogeneous suspension, which was heated to between 95 °C and 120 °C (e.g. , between 100 °C and 105 °C) and stirred at between 95 °C and 120 °C (e.g. , 105 °C) until the reaction was complete (as determined by HPLC to monitor the consumption of starting material). In some embodiments, if necessary (e.g. , if after 6 hours the reaction was not complete as indicated by HPLC analysis), additional 4-(2-chloroethyl)morpholine hydrochloride (0.03 kg) and potassium carbonate (0.024 kg) may be added and the reaction mixture stirred at between 100 °C and 120 °C (e.g. , 105 °C) until reaction completion.
[00283] The reaction mixture was cooled to between 60 °C and 70 °C over a period of at least 60 minutes. Industrial water (6 L) was added to the reactor. The reaction mixture was cooled to between 20 °C and 30 °C. Acetone (6 L) was added to the reactor. The mixture was stirred for 1 to 2 hours at a temperature of between 20 °C and 30 °C. The mixture was filtered, and the solid was washed with industrial water (e.g. , about 45 x industrial water) until pH of washes was below 8. The solid was then washed with acetone (9.7 L). The solid was dried under vacuum at a temperature of less than 50 °C, until the water content by Karl-Fischer was less than 0.30% w/w and TGA curve showed a mass loss of less than 0.30% w/w at about 229 °C (sampling approximately every 6 hours).
6. Example C-6
[00284] To a reactor is added 2-(4-nitrophenyl)imidazo [2, l-&]benzothiazol-7-ol (1.0 kg), 4-(2-chloroethyl)morpholine hydrochloride (1.6 kg), and potassium carbonate (1.3 kg, anhydrous, extra fine, hydroscopic). N,N-Dimethylformamide (DMF) (8.6 L) is added into the reactor. The DMF has a water content of no more than 0.05% w/w. The mixture is stirred for between 15 and 30 minutes to render a homogeneous suspension, which is heated to between 95 °C and 120 °C (e.g. , between 100 °C and 105 °C) and stirred at between 95 °C and 120 °C (e.g. , 105 °C) until the reaction is complete (as determined by HPLC to monitor the consumption of starting material). In some embodiments, if necessary (e.g. , if after 6 hours the reaction is not complete as indicated by HPLC analysis), additional 4-(2-chloroethyl)morpholine hydrochloride (0.03 kg) and potassium carbonate (0.024 kg) may be added and the reaction mixture stirred at between 100 °C and 120 °C (e.g. , 105 °C) until reaction completion.
[00285] The reaction mixture is cooled to between 20 °C and 30 °C (e.g. , over a period of 3 hours). To another reactor is added deionized water (7.6 L) and acetone (15 L). The mixture of water and acetone is then added into the reaction mixture while maintaining the temperature at between 20 °C and 30 °C. The mixture is then stirred for 1 to 2 hours at a temperature of between 20 °C and 30 °C. The mixture is filtered, and the solid is washed with deionized water (e.g. , about 45x deionized water) until pH of washes is below 8. The solid is then washed with acetone (9.7 L). The solid is dried under vacuum at a temperature of less than 50 °C until the water content by Karl-Fischer is less than 0.30% w/w and TGA curve shows a mass loss of less than 0.30% w/w at about 229 °C (sampling approximately every 6 hours). D. Preparation of 7-(2-morpholin-4-yl-ethoxy)-2-(4- aminophenyl)imidazo [2, 1 -bjbenzothiazole

[00286] To a 200-L high pressure (HP) reactor was charged a slurry of 7-(2- morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo [2,l-&]benzothiazole (VII) (7.50 kg, 17.7 mol, 1.0 equiv) in methanol (30 kg). The container was rinsed with additional methanol (10 kg) and the rinse was charged to the reactor. The reactor was then charged with THF (67 kg) and methanol (19 kg). The contents were agitated and the reactor was flushed with nitrogen by alternating nitrogen and vacuum. Vacuum was applied to the reactor and Raney Ni catalyst (1.65 kg, 0.18 wt. equiv) was charged through a sample line. Water (1 kg) was charged through the sample line to rinse the line. The reactor was flushed with nitrogen by alternating nitrogen and vacuum. The reactor was then vented and heated to 50 °C. The reactor was closed and pressurized with hydrogen gas to 15 psi keeping the internal temperature below 55 °C. The reactor was vented and re- pressurized a total of 5 times, then pressurized to 150 psi with hydrogen gas. The contents were agitated at 50 °C for at least 4 hours. At this point a hydrogen uptake test was applied: The reactor was re-pressurized to 150 psi and checked after 1 hour. If a pressure drop of more than 5 psi was observed, the process was repeated. Once the pressure drop remained < 5 psi, the reactor was vented and sampled. The reaction was deemed complete when 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo [2,1- 6]benzothiazole (VII) was < 0.5% by HPLC.
[00287] The reactor was flushed with nitrogen as shown above. The 200-L HP reactor was connected to the 2000-L GL reactor passing through a bag filter and polish filter. The bag filter and polish filter were heated with steam. The 200-L HP reactor was pressurized (3 psi nitrogen) and its contents were filtered into the 2000-L reactor. The filtrates were hot. The 200-L reactor was vented and charged with THF (67 kg) and methanol (59 kg), the reactor agitated, and filtered into the 2000-L GL reactor.
[00288] A total of 6 reductions (46.2 kg processed) were carried out and the combined batches were concentrated by vacuum distillation (without exceeding an internal temperature of 40 °C) to a volume of -180 L. The reactor was cooled to 20 °C and charged with heptane (250 kg) and again vacuum distilled to a volume of -180 L. The reactor was charged with heptane (314 kg) and agitated at 20 °C for at least 1 hour, and then the product was isolated by centrifugation or collection on a Nutsche filter, washing with heptanes (2-5 kg per portion for centrifugation, followed by a 10-20 kg heptanes rinse of the reactor; or 94 kg for Nutsche filtration, rinsing the reactor first). The cake was blown dry, transferred to a vacuum oven and dried to constant weight maintaining a temperature < 50 °C to give the desired product (VIII) (34.45 kg, 80% yield, HPLC purity = 97.9%).
2. Example D-2
[00289] 7-(2-Morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo[2,l-b]benzothiazole was dissolved into methanol and THF and placed in a hydrogenator. Raney nickel was added and the vessel was pressurized with hydrogen and stirred for >24 hours. The reaction mixture was concentrated to a thick paste and diluted with methyl ferf-butyl ether. The resulting solids were filtered and washed with methyl ferf-butyl ether and dried under vacuum to give 7-(2-morpholin-4-yl-ethoxy)-2-(4-aminophenyl) imidazo [2, 1 -bjbenzothiazole.
[00290] The reaction progress was monitored by thin layer chromatography (TLC). The product was isolated as a yellow solid, with 99% purity (HPLC area %). IR was collected, which conformed to structure.
3. Example D-3
[00291] Into a 5-gallon autoclave, 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitrophenyl) imidazo[2,l-&]benzothiazole (580 g, 1.37 mol, 1.0 equiv), THF (7.5 L), methanol (7.5 L, AR) and -55 g of decanted Raney nickel catalyst were added. The reaction vessel was purged with nitrogen (3 X 50 psi) and hydrogen (2 X 50 psi), with stirring briefly under pressure and then venting. The autoclave was pressurized with hydrogen (150 psi). The mixture was stirred and the hydrogen pressure was maintained at 150 psi for over 24 hours with repressurization as necessary. The reaction progress was monitored by TLC (10% methanol / chloroform with 1 drop ammonium hydroxide; Rf (product) 0.20; Rf (SM) 0.80). The reaction was substantially complete when the TLC indicated no starting material present, typically after 24 hours of stirring at 150 psi. The hydrogenation was continued at 150 psi for a minimum of 4 hours or until completion if starting material was still present after the initial 4 hours.
[00292] The reaction mixture was filtered through a Buchner funnel equipped with a glass fiber filter topped with a paper filter. Unreacted starting material was removed together with the catalyst. The filtrate was concentrated to a total volume of 0.5 L, and the residue was triturated with methyl ferf-butyl ether (0.5 L). The resultant solids were collected by filtration, and washed with methyl ferf-butyl ether (0.3 L) (first crop).
[00293] The filtrate was concentrated to dryness and the residue was diluted with methyl ferf-butyl ether (2 L). The resultant solids were collected by filtration, washing with methyl ferf-butyl ether (0.5 L) (second crop).
[00294] The solids were dried in a vacuum oven (<10 mm Hg) at 25 °C. Yield: 510 g (95%), beige solid. TLC: R/ 0.2 (10% methanol / chloroform with one drop of ammonium hydroxide). HPLC: 99.0% (area). JH NMR (300 MHz, DMSO-i¾) conformed to the structure.
[00295] Table: Yields from multiple batch runs

4. Example D-4
[00296] The reaction of Step D was carried out in multiple runs under various conditions, such as, e.g. , varying catalyst loading, concentration of reactant, reaction temperature, and/or workup procedures. The results are summarized in the table below.

Description Run # l Run # 2 Run # 3 Run # 4 Run # 5Rxn Temp (°C) RT RT RT RT RT
Rxn Time (Hr) 24 hr 24 hr 24 hr 24 hr 24 hr
Filtered the Filtered the solution
Filtered the Filtered the Filtered the
solution through through celite. The solution through solution through solution through
celite, washed celite filter cake celite, celite, celite,
with THF, refluxed in THF concentrated, concentrated, concentrated,
concentrated, washed with hot solvent exchanged solvent exchanged solvent exchanged
Work Up solvent exchanged THF, concentrated, with heptane, with heptane, with heptane,
with heptane, solvent exchanged stirred the solids stirred the solids stirred the solids
stirred the solids with heptane, stirred and filtered and filtered and filtered
and filtered the solids and washed with washed with washed with
washed with filtered washed with heptane heptane heptane
heptane heptane
Produce (VIII) 1.9 g 3.88 g 1.11 g 2.6 g 4.4 g
Yield 88% 83.4% 56 94.6%
HPLC purity 95.6% 77.5% 91% 93.8%

5. Example D-5
[00297] To a pressure reactor under nitrogen atmosphere was added a slurry of Raney Nickel in water (0.22 kg) (e.g. about 0.14 kg dry catalyst in water) and the charging line was rinsed with deionized water (0.13 L). To another reactor (Reactor B) were added methanol (5.05 L) and 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo [2, 1- &]benzothiazole (1.0 kg), and the mixture was stirred for between 15 and 30 minutes to render a homogenous suspension. The suspension was transferred to the pressure reactor. Reactor B was washed with methanol (4.88 L) and the wash was transferred to the pressure reactor. Tetrahydrofuran (10.1 L) was added to the pressure reactor.
Hydrogen was charged to the pressure reactor to a pressure of between 2.0 bar and 3.0 bar. The reactor was heated to a temperature of between 45 °C and 55 °C. Hydrogen was then charged to the pressure reactor to a pressure of between 6.0 bar and 7.0 bar. The mixture was stirred at a temperature of between 45 °C and 55 °C (e.g. , 50 °C), while maintaining the hydrogen pressure between 6.0 bar and 7.0 bar until reaction completion (as determined by HPLC to monitor the consumption of starting material).
[00298] The mixture was filtered while maintaining the temperature at between 35 °C and 50 °C. The pressure reactor and the filter were washed with a mixture of THF (10.1 L) and methanol (9.93 L) preheated to a temperature of between 45 °C and 55 °C (e.g. , 50 °C). The combined filtrate was concentrated to a volume of between 2.4 L and 2.8 L under vacuum at a temperature of no more than 40 °C, and a precipitate was formed. Methanol (7.5 L) was added. The slurry was cooled to a temperature of between 5 °C and -5 °C (e.g. , over 3 hours) and stirred for at least 1 hour (e.g. , for 3 hours) while maintaining the temperature at between 5 °C and -5 °C. The suspension was filtered. The solid was washed with methanol (2 X 1.2 L). The solid was then dried under vacuum at a temperature of less than 50 °C until the methanol and THF contents were each less than 3000 ppm as analyzed by GC (e.g. , less than 1500 ppm). The desired product was obtained in about 88.5% yield having about 99% purity by HPLC.
E. Preparation of phenyl 5-£er£-butylisoxazol-3-ylcarbamate

[00299] The jacket temperature of a 200-L glass-lined (GL) reactor was set to 20 °C. To the reactor was charged 5-ieri-butylisoxazole-3-amine (15.0 kg, 107.0 mol, 1.0 equiv), then K2C03 (19.5 kg, 141.2 mol, 1.3 equiv) and anhydrous THF (62 kg).
Agitation was started and then phenyl chloroformate (17.6 kg, 112.4 mol, 1.05 equiv) was charged. The charging line was rinsed with additional anhydrous THF (5 kg). The reaction was agitated at 20 + 5 °C for at least 3 hours then sampled. The reaction was deemed complete if 5-£er£-butylisoxazole-3-amine was < 2% by HPLC. If the reaction was not complete after 6 hours, additional K2CO3 and phenyl chloroformate may be added to drive the reaction to completion.
[00300] Once complete, the reaction was filtered (Nutsche). The filter was rinsed with THF (80 kg). The filtrate was vacuum distilled without exceeding an internal temperature of 40 °C until -50 L remained. Water (188 kg) and ethanol (45 L) were charged, and the mixture was agitated for at least 3 hours with a jacket temperature of 20 °C. The resulting solid was isolated by centrifugation or collection on a Nutsche filter, rinsed with water (2-5 kg for each centrifugation portion; 30 kg for Nutsche filtration) and blow-dried. The solid was then dried to constant weight in a vacuum oven (45 °C) to give the desired product (19.4 kg, 92% yield, HPLC purity = 97.4%). On an 800 g scale, 1559 g of the desired product (98% yield) was obtained with a 99.9% HPLC purity. JH NMR (DMSO-i¾) δ 11.17 (s, 1H); 7.4 (m, 2H); 7.2 (m, 3H); 1.2 (s, 9H). LCMS (M+H)+ 261.
F. Preparation of N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2, 1 -b] [ 1 ,3 ]benzothiazol-2-yl]phenyl } urea

1. Example F-l
[00301] The jacket of a 2000-L GL reactor was set to 20 °C and the reactor was charged with 7-(2-morpholin-4-yl-ethoxy)-2-(4-aminophenyl)imidazo[2,l- &]benzothiazole (26.7 kg, 67.8 mol, 1.0 equiv), 3-amino-5-?-butylisoxazole phenyl carbamate (19.4 kg, 74.5 mol, 1.1 equiv), DMAP (0.5 kg, 4.4 mol, 0.06 equiv), and DCM (anhydrous, 260 kg). Agitation was started, triethylamine (1.0 kg, 10.2 mol, 0.15 equiv) was charged followed by additional DCM (5 kg) through the charging line. The reaction was heated to reflux (-40 °C) and agitated for at least 20 hours with complete dissolution observed followed by product crystallizing from solution after -30 minutes. The reaction was sampled and deemed complete when HPLC analysis showed a ratio of compound (VIII) to compound (I) < 1%.
[00302] The reactor was cooled to 0 °C and stirred for at least 2 hours. The content of the reactor were isolated by centrifuge. Each portion was rinsed with 2-3 kg of cold (0 °C) DCM and spun dry for at least 5 minutes with a 10 psi nitrogen purge. For the final portion, the reactor was rinsed with 10 kg of cold (0 °C) DCM and transferred to the centrifuge where it was spun dry for at least 5 minutes with a 10 psi nitrogen purge. The combined filter cakes were transferred to a vacuum tray dryer and dried to constant weight at 50 °C and at least >20 inches of Hg to give the desired product (I) (35.05 kg, 92% yield, HPLC purity = 98.8%). Phenol was the major impurity detected (0.99%); and three other impurities (<0.10%) were detected. JH NMR (300 MHz, DMSO- ) conformed to structure.
2. Example F-2
[00303] A variety of solvents were used in the reaction of Step F to optimize for better yields and purity profiles. The contents of the symmetrical urea impurity (XI) were compared and summarized in the table below:

http://www.google.com/patents/WO2011056939A1?cl=en SE THIS FOR DELETED CLIPS


4. Example F-4
[00305] To Reactor A under a nitrogen atmosphere was added 7-(2-morpholin-4-yl- ethoxy)-2-(4-aminophenyl)imidazo[2,l-&]benzothiazole (1 kg) and DMAP (0.02 kg). To Reactor B under a nitrogen atmosphere was added 3-amino-5-?-butylisoxazole phenyl carbamate (0.73 kg) and DCM (5.6 L). The DCM used had a water content of less than 0.05 % w/w. The mixture in Reactor B was stirred until dissolution. The solution was transferred into Reactor A (the solution can be filtered into Reactor A to remove any insoluble impurities in the carbamate starting material), and the mixture was stirred in Reactor A. Reactor B was washed with DCM (0.8 L) and the wash was transferred into Reactor A. Reactor A was washed with DCM (0.9 L). To Reactor A was added triethylamine (0.1 L) and the charging vessel and lines were rinsed with DCM (0.1 L) into Reactor A. The mixture was then heated to reflux and stirred at reflux until reaction completion (as determined by HPLC to monitor the consumption of starting material).
[00306] The reaction mixture was cooled (e.g. , over 2 to 3 hours) to a temperature of between -5 °C and 5 °C (e.g. , 0 °C). The mixture was then stirred for 2 to 3 hours at a temperature of between -5 °C and 5 °C (e.g. , 0 °C). The suspension was filtered. The solid was washed with cool DCM (2 X 1.5 L) (pre-cooled to a temperature of between -5 °C and 5 °C). The solid was dried under vacuum at a temperature of less than 45 °C until the DCM content was less than 1000 ppm (e.g., below 600 ppm) as analyzed by GC. The desired product was obtained having about 99% purity by HPLC.
G. Preparation of N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2, l-b] [1 ,3]benzothiazol-2-yl]phenyl }urea dihydrochloride

1. Example G-l
[00307] The jacket of a 2000-L GL reactor was set to 20 °C and the reactor was charged with N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo [2, 1-&][1, 3]benzothiazol-2-yl]phenyl}urea (35.0 kg, 62.4 mol, 1.0 equiv) followed by methanol (553 kg). Agitation was started and the reaction mixture was heated to reflux (-65 °C). Concentrated aqueous HC1 (15.4 kg, 156.0 mol, 2.5 equiv) was charged rapidly (<5 minutes) and the charge line was rinsed into the reactor with methanol (12 kg). Addition of less than 2.0 equivalents of HC1 normally resulted in the formation of an insoluble solid. The reaction mixture was heated at reflux for at least 1 hour. Upon HC1 addition, the slurry dissolved and almost immediately the salt started to crystallize, leaving insufficient time for a polish filtration.
[00308] The reactor was cooled to 20 °C and the product was isolated by filtration (Nutsche) rinsing the reactor and then the cake with methanol (58 kg). The solid was then dried in a vacuum oven (50 °C) to constant weight to give the desired
dihydrochloride salt (35 kg, 89% yield, HPLC purity = 99.94%). JH NMR (300 MHz, DMSO-i¾) conformed to structure.
2. Example G-2
[00309] Concentrated HC1 was added to a suspension of N-(5-ieri-butyl-isoxazol-3- yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l-&][l,3]benzothiazol-2- yl]phenyl}urea in warm methanol forming a solution that slowly began to precipitate. The reaction mixture was refluxed for over 2 hours and then stirred overnight at ambient temperature. The dihydrochloride salt was collected and dried under vacuum.
3. Example G-3
[00310] A 50-L 3-neck round bottom flask was equipped with a mechanical agitator, a thermocouple probe, a nitrogen inlet, a drying tube, a reflux condenser, an addition funnel, and a heating mantle. The flask was charged with N-(5-ieri-butyl-isoxazol-3-yl)- N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l-&][l,3]benzothiazol-2-yl]phenyl}urea (775 g, 1.38 mol, 1.0 equiv) and MeOH (40 L, AR). The resulting off-white suspension was heated to reflux (68 °C). A clear solution did not form. HC1 (37% aqueous) (228 mL, 3.46 mol, 2.5 equiv) was added over 5 minutes at 68 °C. The reaction mixture turned into a clear solution and then a new precipitate formed within approximately 3 minutes. Heating was continued at reflux for approximately 5 hours. The reaction mixture was allowed to cool to ambient temperature overnight. The off-white solids were collected by filtration on a polypropylene filter, washing with MeOH (2 X 1 L, AR). [00311] Two lots of material prepared in this manner were combined (740 g and 820 g). The combined solids were slurried in methanol (30 L) over 30 minutes at reflux and allowed to cool to the room temperature. The solids were collected by filtration on a polypropylene filter, rinsing with methanol (2 X 1.5 L). The solids were dried in a vacuum oven (<10 mm Hg) at 40 °C. Yield: 1598 g (84%), off-white solid. HPLC: 98.2% (area). MS: 561.2 (M+l)+. JH NMR (300 MHz, DMSO-i¾) conformed to the structure. Elemental Analysis (EA): Theory, 54.97 %C; 5.41 %H; 13.26 %N; 5.06 %S; 11.19 %C1; Actual, 54.45 %C; 5.46 %H; 13.09 %N; 4.99 %S; 10.91 %C1.
4. Example G-4
[00312] Into a 50-L 3-neck round bottom flask equipped with a mechanical stirrer, a heating mantle, a condenser and a nitrogen inlet, were charged N-(5-ieri-butyl-isoxazol- 3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l-&][l,3]benzothiazol-2- yl]phenyl}urea (1052.4 g, 1.877 mol, 1.0 equiv) and methanol (21 L). The reactor was heated and stirred. At an internal temperature of > 50 °C, cone. HC1 (398.63 mL, 4.693 mol, 2.5 equiv) was charged over 5 minutes through an addition funnel. During the addition, the mixture changed from a pale yellow suspension to a white suspension. The internal temperature was 55 °C at the conclusion of the addition. The mixture was heated to reflux for 1 hour, then heating was discontinued and the mixture was allowed to cool to room temperature. The mixture was filtered in two portions, and each filter cake was washed with methanol (2 X 1 L), transferred to trays and dried in a vacuum oven (45 °C) to constant weight. The dried trays were combined to produce 1141.9 g of the salt (96% yield, 99.1 % HPLC purity, 10.9% chloride by titration).
H. Analytical Data
1. N-(5-ieri-butyl-isoxazol-3-yl)-N’-{ 4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2, l-&] [l ,3]benzothiazol-2-yl]phenyl}urea
dihydrochloride
[00314] A batch of about 30 grams of N-(5-ieri-butyl-isoxazol-3-yl)-N’- {4-[7-(2- morpholin-4-yl-ethoxy)imidazo[2, l-&] [l ,3]benzothiazol-2-yl]phenyl}urea
dihydrochloride was prepared using the methods described herein. This lot was
prepared in accordance with the requirements for production of clinical Active
Pharmaceutical Ingredients (APIs) under GMP conditions. The analytical data for this batch was obtained, and representative data were provided herein. [00315] Summary of analytical data for the dihydrochloride salt.


………………………
EXAMPLE 1. SYNTHESIS OF N-(5-TERT-BUTYL-ISOXAZOL-3-YU- N>-{4-f7-(2-MORPHOLIN-4- YL-ETHOXY)IMID AZO[2,1- BlH,31BENZOTHIAZOL-2-YL|PHENYLiUREA (“COMPOUND Bl”)
[00357] A. The intermediate 2-amino-l,3-benzothiazol-6-ol was prepared according to a slightly modified literature procedure by Lau and Gompf: J. Org. Chem. 1970, 35, 4103- 4108. To a stirred solution of thiourea (7.6 g, 0.10 mol) in a mixture of 200 mL ethanol and 9 mL concentrated hydrochloric acid was added a solution of 1 ,4-benzoquinone (21.6 g, 0.20 mol) in 400 mL of hot ethanol. The reaction was stirred for 24 hours at room temperature and then concentrated to dryness. The residue was triturated with hot acetonitrile and the resulting solid was filtered and dried.
[00358] The free base was obtained by dissolving the hydrochloride salt in water, neutralizing with sodium acetate, and collecting the solid by filtration. The product (2- amino-l,3-benzothiazol-6-ol) was obtained as a dark solid that was pure by LCMS (M+H = 167) and NMR. Yield: 13.0 g (78 %). NMR (DMSO-^) Sl.6 (m, 2H), 6.6 (d, IH). [00359] B. To prepare the 2-(4-nitrophenyl)imidazo[2,l-b][l,3]benzothiazol-7-ol intermediate, 2-amino-l,3-benzothiazol-6-ol (20.0 g, 0.12 mol) and 2-bromo-4′- nitroacetophenone (29.3 g, 0.12 mol) were dissolved in 600 mL ethanol and heated to reflux overnight. The solution was then cooled to O0C in an ice-water bath and the product was collected by vacuum filtration. After drying under vacuum with P2O5 , the intermediate (2- (4-nitrophenyl)imidazo[2,l-£][l,3]benzothiazol-7-ol) was isolated as a yellow solid. Yield: 17.0 g (46 %) NMR (DMSO-(I6) δ 10 (s, IH), 8.9 (s, IH), 8.3 (d, 2H), 8.1 (d, 2H), 7.8 (d, IH), 7.4 (s, IH), 6.9 (d, IH).
[00360] C. To make the 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitro-phenyl)imidazo[2,l-
6][l,3]benzothiazole intermediate: 2-(4-nitrophenyl)imidazo[2,l-6][l,3]benzothiazol-7-ol,
NYI-4144519vl 84 (3.00 g, 9.6 mmol) was suspended in 100 mL dry DMF. To this mixture was added potassium carbonate (4.15 g, 30 mmol, 3 eq), chloroethyl morpholine hydrochloride (4.65 g, 25 mmol, 2.5 eq) and optionally tetrabutyl ammonium iodide (7.39 g, 2 mmol). The suspension was then heated to 900C for 5 hours or until complete by LCMS. The mixture was cooled to room temperature, poured into 800 mL water, and allowed to stand for 1 hour. The resulting precipitate was collected by vacuum filtration and dried under vacuum. The intermediate, (7-(2-morpholin-4-yl-ethoxy)-2-(4-nitro-phenyl)imidazo[2, 1 – b][\, 3]benzothiazole) was carried on without further purification. Yield: 3.87 g (95 %) NMR (DMSO-d6) δ 8.97 (s, IH), 8.30 (d, 2H), 8.0 (d, 2H), 7.9 (d, IH), 7.7 (s, IH), 7.2 (d, IH), 4.1 (t, 2H), 5.6 (m, 4H), 2.7 (t, 2H).
[00361] D. To make the intermediate 7-(2-morpholin-4-yl-ethoxy)-2-(4-amino- phenyl)imidazo[2,l-b][l,3]benzothiazole: To a suspension of 7-(2-morpholin-4-yl-ethoxy)- 2-(4-nitro-phenyl)imidazo[2,l -b][\ , 3]benzothiazole (3.87g, 9.1 mmol) in 100 mL isopropyl alcohol/water (3:1) was added ammonium chloride (2.00 g, 36.4 mmol) and iron powder (5.04 g, 90.1 mmol). The suspension was heated to reflux overnight with vigorous stirring, completion of the reaction was confirmed by LCMS. The mixture was filtered through Celite, and the filtercake was washed with hot isopropyl alcohol (150 mL). The filtrate was concentrated to approximately 1/3 of the original volume, poured into saturated sodium bicarbonate, and extracted 3 times with dichloromethane. The combined organic phases were dried over MgSO4 and concentrated to give the product as an orange solid containing a small amount (4-6 %) of starting material. (Yield: 2.75 g 54 %). 80% ethanol/water may be used in the place of isopropyl alcohol /water – in which case the reaction is virtually complete after 3.5 hours and only traces of starting material are observed in the product obtained. NMR (DMSO-Λfc) δ 8.4 (s, IH), 7.8 (d, IH), 7.65 (d, IH), 7.5 (d, 2H), 7.1 (d, IH), 6.6 (d, 2H), 4.1 (t, 2H), 3.6 (m, 4H), 2.7 (t, 2H).
[00362] E. A suspension of 7-(2-morpholin-4-yl-ethoxy)-2-(4-amino- phenyl)imidazo[2,l-b][l,3]benzothiazole (4.06 g, 10.3 mmol) and 5-tert-butylisoxazole-3- isocyanate (1.994 g, 12 mmol) in toluene was heated at 120 0C overnight. The reaction was quenched by pouring into a mixture of methylene chloride and water containing a little methanol and neutralized with saturated aqueous NaHCO3 solution. The aqueous phase was extracted twice with methylene chloride, the combined organic extracts were dried over
NYI-4144519vl 85 MgSO4 and filtered. The filtrate was concentrated to about 20 ml volume and ethyl ether was added resulting in the formation of a solid. The precipitate was collected by filtration, washed with ethyl ether, and dried under vacuum to give the free base of Compound B 1. Yield: 2.342 g (41 %) NMR (DMSO-J6) £9.6 (br, IH), 8.9 (br, IH), 8.61 (s, IH), 7.86 (d, IH), 7.76 (d, 2H), 7.69 (d, IH), 7.51 (d, 2H), 7.18 (dd, IH), 6.52 (s, IH), 4.16 (t, 2H), 3.59 (t, 4H), 3.36 (overlapping, 4H), 2.72 (t, 2H), 1.30 (s, 9H). NMR (CDCl3) £9.3 (br, IH), 7.84 (m, 4H), 7.59 (d, 2H), 7.49 (d, IH), 7.22 (d, IH), 7.03 (dd, IH), 5.88 (s, IH), 4.16 (t, 2H), 3.76 (t, 4H), 2.84 (t, 2H), 2.61 (t, 4H), 1.37 (s, 9H).
6.2 EXAMPLE 2. ALTERNATIVE SYNTHESIS QF N-(5-TERT-BUTYL- ISOXAZQL-3- YL)-N -{4-[7-q-MORPHOLIN-4- YL- ETHOXYUMID AZOf2,l-BUl,31BENZOTHIAZOL-2- YLIPHENYLIUREA (“COMPOUND Bl”)
[00363] A. To a suspension of the intermediate 2-(4-Nitrophenyl)imidazo[2,l- b][l,3]benzothiazol-7-ol from Example IB (2.24 g, 7.2 mmol) in ethanol (40 mL) was added SnCl2 1H2O (7.9Og, 35 mmol) and heated to reflux. Concentrated HCl was added to the reaction mixture and the precipitate formed gradually. The reaction mixture was heated to reflux for 20 hours and then allowed to cool to room temperature. The solution was poured into ice and neutralized with 10% NaOH and adjusted to approximately pH 6. The organic phase was extracted three times with ethyl acetate (80 mL x 3). Extracts were dried over MgSθ4 and concentrated to give a yellow solid. (1.621 g, 80%). The solid was recrystallized from methanol to give a pure product (1.355 g, 67%).
[00364] B. To a suspension of the intermediate from Step 2A (0.563 g, 2 mmol) in toluene (30 mL) was added 5-tert-butylisoxazole-3-isocyanate (0.332g, 2 mmol) and heated to reflux overnight. LC-MS analysis showed presence of the intermediate but no trace of 5- tert-butylisoxazole-3-isocyanate and an additional 0.166 g of the isocyanate was added. The reaction was again heated to reflux overnight. Completion of reaction was verified by LC- MS. The solvent was removed and the resulting mixture was dissolved in methanol which was removed to give the second intermediate as a solid.
[00365] The mixture was dissolved in CH2Cl2 (150 mL) and washed with saturated
NaHCO3. The organic layer was dried over MgSO4, concentrated, and purified by silica gel chromatography three times, first using a methanol/CH2Cl2 gradient, the second time using a
NYI-4144519vl 86 hexane/ethyl acetate gradient followed by a methanol/ethyl acetate gradient, and a third time using a methanol/CH2Cl2 gradient.
[00366] C. To a suspension of the intermediate from Step 2B (0.1 10 g, 0.25 mmol) in
THF (5mL) was added Ph3P (0.079g, 0.3 mmol), diisopropylazodicarboxylate (0.06 Ig, 0.3 mmol) and 4-morpholinoethanol (0.039 g, 0.3 mmol). The reaction mixture was stirred at room temperature overnight. Completion of the reaction was verified by LC-MS. The solvent was removed and the final product was purified using silica gel chromatography, with methanol in CH2Cl2 (0.030g, 21%).
6.3 EXAMPLE 3. BULK SYNTHESIS OF N-(5-TERT-BUTYL- ISOXAZOL-3-YL)-N’-f4-[7-(2-MORPHOLIN-4-YL- ETHOXY^IMID AZO[2α-BUlJlBENZOTHIAZOL-2- YLlPHENYLiUREA (“COMPOUND Bl”)
[00367] A multi-step reaction scheme that was used to prepare bulk quantities of
Compound Bl is depicted in FIG. 66a and FIG. 66b, and is described further below. [00368] Step 1 : Preparation of 2- Amino-6-hydroxybenzothiazole (Intermediate 1). 2-
Amino-6-methoxybenzothiazole is reacted with hot aqueous HBr for about 3 hrs and then the clear solution is cooled to ambient temperature overnight. The precipitated solids are collected, dissolved in hot water and the pH is adjusted to between 4.5-5.5. The resultant solids are collected, dried and recrystallized from Isopropanol. Second crop material is collected. The solids are vacuum dried to give Intermediate 1.
[00369] Step 2: Preparation of 2-(4-Nitrophenyl) imidazo [2J-b]benzothiazol-7-ol
(Intermediate 2). 2-Amino-6-hydroxybenzothiazole, 2-Bromo-4-nitroacetophenone and absolute Ethanol are added together and heated to reflux for approximately 24 hours. Tetrabutylammonium iodide is added and the reaction is refluxed an additional 12 hours. The resulting yellow suspension is cooled and the solids collected and washed with Ethanol and Diethyl ether. The solids are dried under vacuum to give Intermediate 2. [00370] Step 3: Preparation of 7-(2-Morpholin-4-yl-ethoxy)-2-(4-nitrophenyl) imidazo
[2,1-b] benzothiazole (Intermediate 3). Intermediate 2, 4-(2-Chloroethyl)morpholine hydrochloride, Potassium carbonate and Tetrabutylammonium iodide are added to N,N- Dimethylformamide forming a yellow suspension that is heated for over 3 hours. The reaction is cooled and the solids are collected, slurried into water, filtered, slurried into
NYl-4 l4451′)v l 87 acetone, filtered and washed with Acetone to give yellow solids that are dried under vacuum to give Intermediate 3.
[O0371] Step 4: Preparation of 7-(2-Moφholin-4-yl-ethoxy)-2-(4-aminophenyl) imidazo [2,1 -b] benzothiazole (Intermediate 4). Intermediate 3 is dissolved into Methanol and THF and placed in a Hydrogenator. Raney Nickel is added and the vessel is pressurized with Hydrogen and stirred for >24 hrs. The reaction mixture is concentrated to a thick paste and diluted with Methyl tert-butyl ether. The resulting solids are filtered and washed with Methyl tert-butyl ether and dried under vacuum to give Intermediate 4. [O0372] Step 5: Preparation of {[5-(tert-Butyl) isoxazol-3-vnatnino}-N-{4-r7-(2- morpholin-4-yl-ethoxy)(4-hvdroimidazolo[2J-blbenzothiazol-2-yl)]phenyl|carboxamide (Compound Bl). 3 -Amino- 5 -tert-butyl isoxazole in Methylene chloride is added to a vessel containing toluene which is cooled to approx 0 0C. Triphosgene is then added and the reaction mixture is cooled to below -15 0C. Triethylamine is added, followed by Intermediate 4. The mixture is heated to distill off the Methylene chloride and then heated to over 60 0C for over 12 hours and cooled to 50-60 °C. The resulting solids are filtered, washed with Heptane, slurried with 4% sodium hydroxide solution, and filtered. The solids are then washed with Methyl tert-butyl ether followed by Acetone and dried under vacuum to give Compound Bl.
6.4 EXAMPLE 4. EXAMPLES OF PREPARATION OF COMPOUND Bl HCL SALT
[00373] Example A: For the preparation of a hydrochloride salt of Compound Bl5 N-
(5-tert-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- b][l,3]benzothiazol-2-yl]phenyl}urea hydrochloride, the free base was dissolved in a mixture of 20 ml methylene chloride and 1 ml methanol. A solution of 1.0 M HCl in ethyl ether (1.1 eq.) was added dropwise, followed by addition of ethyl ether. The precipitate was collected by filtration or centrirugation and washed with ethyl ether to give a hydrochloride salt of Compound Bl. Yield: 2.44 g (98 %) NMR (DMSO-^) S X 1.0 (br, IH), 9.68 (s, IH), 9.26 (s, IH), 8.66 (s, IH), 7.93 (d, IH), 7.78 (m, 3H), 7.53 (d, 2H), 7.26 (dd, IH), 6.53 (s, IH), 4.50 (t, 2H), 3.97 (m, 2H), 3.81 (t, 2H), 3.6 (overlapping, 4H), 3.23 (m, 2H), 1.30 (s, 9H). [00374] Example B: Concentrated HCl is added to a suspension of Compound Bl in warm methanol forming a solution that slowly begins to precipitate. The reaction mixture is
NYI-4144519vl 88 refluxed for over 2 hrs and then stirred overnight at ambient temperature. The HCl salt is collected and dried under vacuum.
[00375] Example C: Materials: {[5-(tert-Butyl) isoxazol-3-yl]amino}-N-{4-[7-(2- morpholin-4-yl-ethoxy)(4-hydroimidazolo[2,l-6]benzothiazol-2-yl)] phenyl }carboxamide (775 g, 1.38 mol, 1.0 eq); HCl 37% aqueous (288 mL, 3.46 mol, 2.5 eq); Methanol (MeOH, AR) (40L). Procedure: (Step 1) Equipped a 5OL 3-neck round bottom flask with a mechanical agitator, thermocouple probe, Nitrogen inlet, drying tube, reflux condenser, addition funnel and in a heating mantle. (Step 2) Charged the flask with {[5-(tert-Butyl) isoxazol-3-yl] amino}-N-{4-[7-(2-morpholin-4-yl-ethoxy)(4-hydroimidazolo[2,l- b]benzothiazol-2-yl)] phenyl jcarboxamide (775g) and MeOH, AR (40L). Heat the resulting off-white suspension to reflux (680C). A clear solution did not form. (Step 3) Added HCl (37% aqueous) (228 mL) over 5 minutes at 68°C. The reaction mixture turned into a clear solution and then a new precipitate formed within approximately 3 minutes. Continued heating at reflux for approximately 5 hours. Allowed the reaction mixture to cool to ambient temperature overnight. (Step 4) Collected the off-white solids by filtration onto a polypropylene filter, washing the solids with MeOH, AR (2 x 1 L). (Step 5) Combined two lots of material prepared in this manner (74Og and 82Og). Slurried the combined solids in Methanol (30 L) over 30 minutes at reflux and cool to the room temperature. (Step 6) Collected the solids by filtration onto a polypropylene filter, rinsing with Methanol (2 x 1.5L). (Step 7) Dried the solids in a vacuum oven (<10mniHg) at 400C. Yield: 1598 g (84%), off-white solid; HPLC: 98.2% (area); MS: 561.2 (M+l); IH NMR: conforms (300 MHz, DMSO-d6); Elemental Analysis (EA): Theory = 54.97 %C; 5.41 %H; 13.26 %N; 5.06 %S; 11.19 %C1; Actual = 54.45 %C; 5.46 %H; 13.09 %N; 4.99 %S; 10.91 %C1.
NYl-4I44519v! 89 [00376] Examples of Compound Bl HCl salt synthesis

[00377] Example D: In a 50-L 3-neck round bottom flask equipped with a mechanical stirrer, heating mantle, condenser and nitrogen inlet was charged Compound Bl (1052.4 g, 1.877 mol, 1.00 equiv.) and methanol (21 L). The reactor was heated and stirred. At an internal temperature > 50 0C, cone. HCl (398.63 mL, 4.693 mol, 2.5 equiv.) was charged over 5 minutes through an addition funnel. With the addition, the reaction changed from a pale yellow suspension to a white suspension. The internal temperature was 55 0C at the conclusion of the addition. The reaction was heated to reflux for 1 hour, then heating discontinued and the reaction allowed to cool to room temperature. The reaction was filtered in two portions, each filter cake washed with methanol (2 x 1 L), transferred to trays and dried in a vacuum oven (45 0C) to constant weight. The dried trays were combined to produce 1141.9 g, 96% yield, 99.1 % HPLC purity, 10.9% chloride by titration.
Solid Forms Comprising the HCl Salt of Compound Bl 6.6.2.1 Preparation of Solid Forms

6.6.2.2 Cold Precipitation Experiments

NYl-4144519vl 102 6.6.2.3 Slurry Experiments
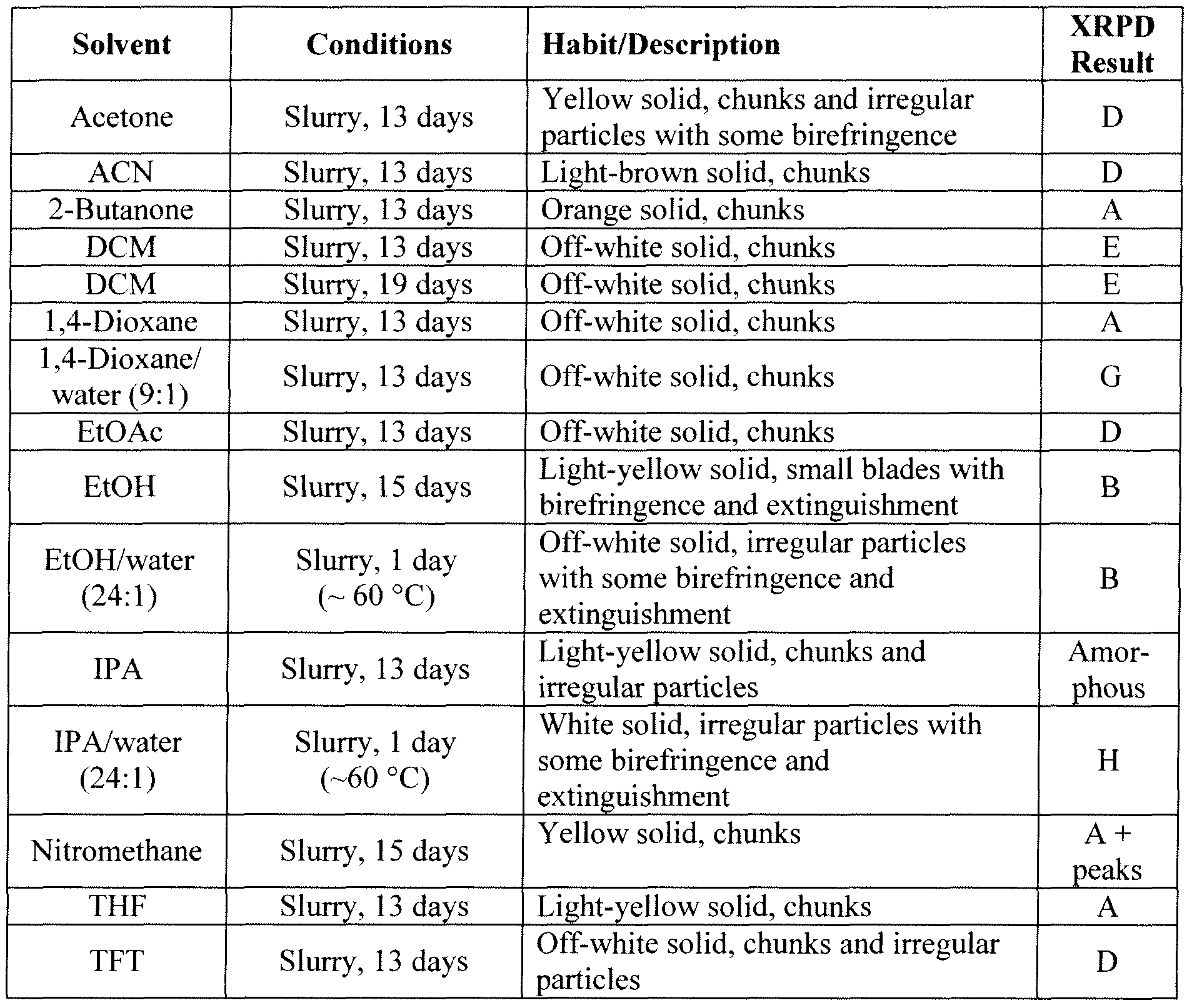
NYI-41445 l9vl 103 6.6.2.4 Additional Preparation of Solid Forms Comprising the HCI Salt of Compound Bl

NYl-4144519v l 104

NYM 144519vl 105

N Y l -4 1 4 4 5 1 9 v l 1 0 6

NYI-4I44519vi 107

N V I 4 1 4 4 5 1 9 1 0 8

“Abbreviations in Table: CC = crash cool, CP = crash precipitation, EtOAc = ethyl acetate, FE = fast evaporation, VD = vapor diffusion, IPA = isopropanol, MEK = methyl ethyl ketone (2-butanone), RE = rotary evaporation, RT = room (ambient) temperature, SC = slow cool, SE = slow evaporation, THF = tetrahydrofuran, TFE = 2,2,2=trifluoroethanol.
6.6.2.5 Scale-up Experiments of Involving Crystal Forms Comprising the HCl Salt of Compound Bl

NYI-4144519v l 109

Abbreviations in Table: CC = crash cool, CP = crash precipitation, EtOAc = ethyl acetate, FE = fast evaporation, IPA = isopropanol, MEK = methyl ethyl ketone (2-butanone), RE = rotary evaporation, RT = room (ambient) temperature, SC = slow cool, SE = slow evaporation, THF = tetrahydrofuran, TFE = 2,2,2=trifluoroethanol.
……………………
Identification of N-(5-tert-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazol-2-yl]phenyl}urea dihydrochloride (AC220), a uniquely potent, selective, and efficacious FMS-like tyrosine kinase-3 (FLT3) inhibitor
J Med Chem 2009, 52(23): 7808
http://pubs.acs.org/doi/full/10.1021/jm9007533

N-(5-tert-Butyl-isoxazol-3-yl)-N′-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazol-2-yl]phenyl}urea Dihydrochloride (7): General Procedure D
A suspension of 2-(4-aminophenyl)-7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazole (19c) (4.06 g, 10.3 mmol) and 5-tert-butyl-isoxazole-3-isocyanate (5) (1.994 g, 12 mmol) in toluene (60 mL) was heated at 120 °C overnight. The reaction was quenched with a mixture of dichloromethane and water containing a little methanol, and the mixture was neutralized with saturated aqueous NaHCO3. The aqueous phase was extracted twice with dichloromethane, and the combined organic extracts were dried over MgSO4 and filtered. The filtrate was concentrated to a volume of about 20 mL and ethyl ether was added, resulting in the formation of a solid. The precipitate was collected by filtration, washed with ethyl ether, and dried under vacuum to give the free base of 7 (2.342 g, 41%).
1H NMR (DMSO-d6) δ 9.6 (br, 1H), 8.9 (br, 1H), 8.61 (s, 1H), 7.86 (d, J = 8.9 Hz, 1H), 7.76 (d, J = 8.0 Hz, 2H), 7.69 (d, J = 1.3 Hz, 1H), 7.51 (d, J = 8.0 Hz, 2H), 7.18 (dd, J = 1.3 and 8.9 Hz, 1H), 6.52 (s, 1H), 4.16 (t, J = 5.7 Hz, 2H), 3.59 (t, J = 4.2 Hz, 4H), 3.36 (overlapping, 4H), 2.72 (t, J = 5.7 Hz, 2H), 1.30 (s, 9H).
General Procedure E for Preparation of Hydrochloride Salt
The free base was dissolved in a mixture of dichloromethane (20 mL) and methanol (1 mL). A solution of 1.0 M HCl in ethyl ether (1.1 equiv for all compounds except 7, for which 2.5 equiv were used) was added dropwise, followed by addition of ethyl ether. The precipitate was collected by filtration to give
N-(5-tert-butyl-isoxazol-3-yl)-N′-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazol-2-yl]phenyl}urea dihydrochloride (7) (2.441 g, 98%).
1H NMR (DMSO-d6) δ 11.0 (br, 1H), 9.68 (s, 1H), 9.26 (s, 1H), 8.66 (s, 1H), 7.93 (d, J = 8.9 Hz, 1H), 7.78 (m, 3H), 7.53 (d, J = 8.7 Hz, 2H), 7.26 (dd, J = 2.4 and 8.9 Hz, 1H), 6.53 (s, 1H), 4.50 (t, J = 4.1 Hz, 2H), 3.97 (m, 2H), 3.81 (t, J = 12.1 Hz, 2H), 3.6 (overlapping, 4H), 3.23 (m, 2H), 1.30 (s, 9H). LC-MS (ESI) m/z 561 (M + H)+.
Anal. (C29H32N6O4S·2HCl) C, H, N. C: calcd 54.97; found 54.54. H: calcd 5.22; found 5.87. N: calcd 13.26; found 13.16.
References
- Chao, Qi; Sprankle, Kelly G.; Grotzfeld, Robert M.; Lai, Andiliy G.; Carter, Todd A.; Velasco, Anne Marie; Gunawardane, Ruwanthi N.; Cramer, Merryl D.; Gardner, Michael F.; James, Joyce; Zarrinkar, Patrick P.; Patel, Hitesh K.; Bhagwat, Shripad S. (2009). “Identification of N-(5-tert-Butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazol-2-yl]phenyl}urea Dihydrochloride (AC220), a Uniquely Potent, Selective, and Efficacious FMS-Like Tyrosine Kinase-3 (FLT3) Inhibitor”. Journal of Medicinal Chemistry 52 (23): 7808–7816.
- Drug Tames Refractory AML. ASH Dec 2012
- NMR……….http://file.selleckchem.com/downloads/nmr/S152601-AC-220-HNMR-Selleck.pdf
- HPLC………http://file.selleckchem.com/downloads/hplc/S152601-AC-220-HPLC-Selleck.pdf


Fezolinetant, фезолинетант , فيزولينيتانت , 非唑奈坦 ,
Fezolinetant ESN-364
- Molecular FormulaC16H15FN6OS
- Average mass358.393 Da
-
Methanone, [(8R)-5,6-dihydro-8-methyl-3-(3-methyl-1,2,4-thiadiazol-5-yl)-1,2,4-triazolo[4,3-a]pyrazin-7(8H)-yl](4-fluorophenyl)-UNII:83VNE45KXXфезолинетант [Russian] [INN]فيزولينيتانت [Arabic] [INN]非唑奈坦 [Chinese] [INN]
(4-Fluorophenyl)[(8R)-8-methyl-3-(3-methyl-1,2,4-thiadiazol-5-yl)-5,6-dihydro[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl]methanone
10205
1629229-37-3 [RN]
83VNE45KXX
FDA APPROVED 5/12/2023, Veozah
- Originator Euroscreen
- Developer Ogeda
- Class Pyrazines; Small molecules; Triazoles
- Mechanism of Action Gonadal steroid hormone modulators; Neurokinin 3 receptor antagonists
- Phase II Hot flashes; Polycystic ovary syndrome; Uterine leiomyoma
- Preclinical Weight gain
- DiscontinuedBenign prostatic hyperplasia; Endometriosis
- 14 Sep 2018 Ogeda completes a phase II trial in Hot flashes (In the elderly, In adults) in USA (PO) (NCT03192176)
- 23 May 2018 Astellas Pharma completes a phase I trial in Polycystic ovary syndrome (In volunteers) in Japan (PO) (NCT03436849)
- 22 Feb 2018 Phase-I clinical trials in Polycystic ovary syndrome (In volunteers) in Japan (PO) (NCT03436849)
Fezolinetant (INN; former developmental code name ESN-364) is a small-molecule, orally active, selective neurokinin-3 (NK3) receptorantagonist which is under development by Ogeda (formerly Euroscreen) for the treatment of sex hormone-related disorders.[1][2] As of May 2017, it has completed phase I and phase IIa clinical trials for hot flashes in postmenopausal women.[1] Phase IIa trials in polycystic ovary syndrome patients are ongoing.[1] In April 2017, it was announced that Ogeda would be acquired by Astellas Pharma.[3]
Ogeda (formerly Euroscreen ) is developing fezolinetant, an NK3 antagonist, for treating endometriosis, benign prostate hyperplasia, polycystic ovary syndrome, uterine fibroids and hot flashes. In November 2018, drug was listed under phase II development for PCOS, uterine fibroids and hot flashes in company’s pipeline. In October 2018, the company was proceeding to phase III study preparation, and regulatory filings were expected in 2021 or later .
Fezolinetant shows high affinity for and potent inhibition of the NK3 receptor in vitro (Ki = 25 nM, IC50 = 20 nM).[2] Loss-of-function mutations in TACR and TACR3, the genes respectively encoding neurokinin B and its receptor, the NK3 receptor, have been found in patients with idiopathic hypogonadotropic hypogonadism.[2] In accordance, NK3 receptor antagonists like fezolinetant have been found to dose-dependently suppress luteinizing hormone (LH) secretion, though not that of follicle-stimulating hormone (FSH), and consequently to dose-dependently decrease estradiol and progesterone levels in women and testosterone levels in men.[4] As such, they are similar to GnRH modulators, and present as a potential clinical alternative to them for use in the same kinds of indications.[5]However, the inhibition of sex hormone production by NK3 receptor inactivation tends to be less complete and “non-castrating” relative to that of GnRH modulators, and so they may have a reduced incidence of menopausal-like side effects such as loss of bone mineral density.[4][5]
Unlike GnRH modulators, but similarly to estrogens, NK3 receptor antagonists including fezolinetant and MLE-4901 (also known as AZD-4901, formerly AZD-2624) have been found to alleviate hot flashes in menopausal women.[6][7] This would seem to be independent of their actions on the hypothalamic–pituitary–gonadal axis and hence on sex hormone production.[6][7] NK3 receptor antagonists are anticipated as a useful clinical alternative to estrogens for management of hot flashes, but with potentially reduced risks and side effects.[6][7]
PATENT
WO2011121137
hold protection in most of the EU states until 2031 and expire in the US in 2031.
PATENT
US 20170095472

PATENT
WO2016146712
PATENT
WO-2019012033
Novel deuterated analogs of fezolinetant , processes for their preparation and compositions comprising them are claimed. Also claims are their use for treating pain, convulsion, obesity, inflammatory disease including irritable bowel syndrome, emesis, asthma, cough, urinary incontinence, reproduction disorders, testicular cancer and breast cancer. Further claims are processes for the preparation of fezolinetant. claiming use of NK3R antagonist eg fezolinetant, for treating pathological excess body fat or prevention of obesity.
Fezolinetant was developed as selective antagonist of NK-3 receptor and is useful as therapeutic compound, particularly in the treatment and/or prevention of sex-hormone dependent diseases. Fezolinetant corresponds to (R)-(4-fluorophenyl)-(8-methyl-3-(3-memyl-l,2,4-miacMazol-5-yl)-5,6-dmy(ko-[l,2,4]trizolo[4,3-a]pyrazin-7(8H)-yl)methanone and is described in WO2014/154895.
Drug-drug interactions are the most common type of drug interactions. They can decrease how well the medications works, may cause serious unexpected side effects, or even increase the blood level and possible toxicity of a certain drug.
Drug interaction may occur by pharmacokinetic interaction, during which one drug affects another drug’s absorption, distribution, metabolism, or excretion. Regarding metabolism, it should be noted that drugs are usually eliminated from the body as either the unchanged drug or as a metabolite. Enzymes in the liver, usually the cytochrome P450s (CYPs) enzymes, are often responsible for metabolizing drugs. Therefore, determining the CYP profile of a drug is of high relevancy to determine if it will affect the activity of CYPs and thus if it may lead to drug-drug interactions.The five most relevant CYPs for drug-drug interaction are CYP3A4, 2C9, 2C19, 1A2 and 2D6, among which isoforms 3A4, 2C9 and 2C19 are the major ones. The less a drug inhibits these CYPs, the less drug-drug interactions would be expected.
Therefore, it is important to provide drugs that present the safest CYP profile in order to minimize as much as possible the potential risks of drug-drug interactions.Even if fezolinetant possesses a good CYP profile, providing analogs of fezolinetant with a further improved CYP profile would be valuable for patients.
In a completely unexpected way, the Applicant evidenced that deuteration of fezolinetant provides a further improved CYP profile, especially on isoforms CYP 2C9 and 2C19. This was evidenced for the deuterated form (R)-(4-fluorophenyl)-(8-methyl-3-(3-(memyl-d.?)-l,2,4-miacttazol-5-y ^yl)methanone, hereafter referred to as “deuterated fezolinetant”.
Importantly, deuterated fezolinetant retains the biological activity of fezolinetant as well as its lipophilic efficiency.
Deuterated fezolinetant also presents the advantage to enable improvement of the in vivo half -life of the drug. For example, half -life is increased by a factor 2 in castrated monkeys, compared to fezolinetant.
Synthetic scheme
Deuterated fezolinetant may be synthesized using the methodology described following schemes (Part A and Part B):
Part A: Preparation of deuterated key intermediate (ii)
Part B: Synthesis of deuterated fezolinetant using intermediate (ii)
Synthesis of deuterated fezolinetant was performed through key intermediate (ii). Part A corresponds to the synthesis of intermediate (ii). Part B leads to deuterated fezolinetant (d3-fezolinetant), using intermediate (ii), using procedures adapted from WO2014/154895.
Experimental details
Part A – Step 1): Formation of d3-acetamide (b)
To i¾-acetic acid (a) (10 g, 1 equiv.) in DCM (100 mL) CDI (25.3 g, 1 equiv.) was added and the resultant mixture stirred at RT for 30 min, thereupon ammonia gas was bubbled through the reaction mixture for 40 min at 0-5 °C. Thereafter the bubbling was stopped, the mixture was filtered and the filtrate was evaporated under reduced pressure to give 30.95 g crude product that was purified using flash chromatography on silica to furnish 6.65 g (yield: 73 %) deuterated acetamide (b) was obtained (GC (column RTX-1301 30 m x 0.32 mm x 0.5 μπι) Rt 7.4 min, 98 %).
Part A – Step 2): Ring closure leading to compound (c)
<¾-Acetamide (b) (3.3 g, 1 equiv.) and chlorocarbonylsulfenyl chloride (CCSC) (8.4 g, 1.2 equiv.) were combined in 1,2-dichloroethane (63 mL), and refluxed for 4.5 h. CCSC can be prepared as per the procedure described in Adeppa et al. (Synth. Commun., 2012, Vol. 42, pp. 714-721). The volatiles were then removed to obtain 6.60 g (102 % yield) oxathiazolone (c) product as a yellow oil. The product was analyzed by GC (Rt= 7.8 min, 97 ). 13C NMR (CDC13): 16.0, 158.7, 174.4 ppm.
Part A – Step 3): formation of compound (d)
To oxathiazolone (c) (6.6 g, 1 equiv) in rn-xylene (231 mL) methyl cyanoformate (14.70 g, 3.2 equiv.) was added. The mixture was stirred at 130 °C for 19 h and thereafter the volatiles removed under reduced pressure at 50 °C to obtain 4.53 g brown oil (yield: 51 %). The product (d) was analyzed by GC (Rt = 11.8 min, 81 %) and mass spectrometry (M+H = 162).
Part A – Step 4): formation of intermediate (ii)
The ester (d) obtained above (3.65 g, lequiv.) was dissolved in ethanol (45 mL). The undissolved material was filtered off then hydrazine hydrate (2.3 mL, 1.15 equiv. 55w/w in H20) was added to the stirred solution. Thick suspension formed in minutes, the suspension was stirred for 45 min, filtered and washed with EtOH (3 mL) to furnish intermediate (ii) a pale yellow solid (2.43 g, 55 % yield). Mass spectrometry (M+H = 162, M+Na = 184); ¾ NMR (cfe-DMSO): 4.79 ppm (br s, 2H), 10.55 ppm (br s, 1H); 13C NMR (fife-DMSO): 17.4 ppm, 155.6 ppm, 173.4 ppm, 183.0 ppm.
Part B – Step a): formation of compound (iii)
Intermediate (i) was prepared as described in WO2014/154895.
Intermediate (ii) (490 mg, 3.04 mmol) and compound (i) (1.0 g (87 mol 1.3 content), 2.97 mmol) were taken up in MeOH and the reaction mixture was stirred at a temperature ranging from 55°C to 70°C for a period of time ranging from 6 hours to 8 hours. The reaction was deemed complete by TLC. The reaction mixture was evaporated and the crude product was purified by flash chromatography on silica in DCM : MeOH eluent to afford 1.13 g (97 % yield) of compound (iii) as a yellow oil. JH NMR (CDC13): δ (ppm) 7.26 (d, 1H), 6.48-6.49 (2H), 4.50 (m, 1H), 4.30 (m, 1H), 4.09 (m, 1H), 3.94 (d, 1H), 3.80 (s, 6H), 3.61 (d, 1H), 3.22 (m, 1H), 2.75 (m, 1H), 1.72 (d, 3H); Mass spectrometry (M+H = 390, 2M+Na = 801). Chiral LC (column: Chiralpak IC, 250 x 4.6 mm – eluent: MTBE MeOH DEA 98/2/0.1) 99.84 .
Part B – Step b): deprotection leading to compound (iv)
Intermediate (iii) prepared above (1.05 g, 2.7 mmol) was dissolved in DCM and washed with aq. NaOH. The organic phase was dried, then TFA (1.56 mL, 2.3 g, 7.5 equiv.) was added at RT. The resulting solution was stirred at RT for 2 h. The reaction was monitored by TLC. After completion of the reaction water was added to the reaction mixture, and the precipitate filtered and washed with water. The phases were separated, the pH of the aq. phase was adjusted to pH 13 by addition of 20 % aq. NaOH. NaCl was then added to the aqueous solution that was then extracted with DCM. The organic phase was evaporated under reduced pressure to give 504 mg of compound (iv) (78 % yield). ¾ NMR (cfe-DMSO): δ (ppm) 4.42 (m, 1H), 4.10 (m, 2H), 3.0 (m, 1H), 2.82 (m, 1H), 1.46 (d, 3H). 13C NMR (rf6-DMSO): δ (ppm) 174.8, 173.4, 156.2, 145.0, 48.1, 45.7, 40.7, 19.1. Mass spectrometry (M+H = 240, 2M+Na = 501).
Part B – Step c): acylation and recrystallization to form deuterated fezolinetant
Intermediate (iv) (450 mg, 1.88 mmol) was dissolved in DCM, then sat. aq. NaHC03 was added and the mixture was stirred for 30 min. To this mixture 4-fluorobenzoyl chloride (v) (220 1 equiv.) was added dropwise at RT. The reaction was stirred for a period of time ranging from about 20 min to overnight at RT and reaction progress monitored by TLC. After completion the phases were separated, the organic phase was washed with water, dried over MgS04, filtered and evaporated under reduced pressure to give 745 mg crude <i3-fezolinetant (110 % yield). The crude product was purified by flash chromatography using MeOH : DCM together with a second batch, then
crystallized (EtOH H20) before final analysis. ¾ NMR (d6-DMSO): δ (ppm) 7.60 (m, 2H), 7.33 (m, 2H), 5.73 (m, 1H), 4.68 (dd, 1H), 4.31 (m, 1H), 4.06 (m, 1H), 3.65 (m, 1H), 1.61 (d, 3H). 13C NMR (d6-DMSO): δ (ppm) 174.4, 173.5, 168.7, 163.7, 161.8, 154.1, 144.9, 131.6, 129.5, 115.5, 44.7, 18.7. Isotopic purity based on an intense molecular ion observed at m/z = 362.2 Da is estimated as approximately 100 % isotopic purity. Chiral purity (LC) (column: Chiralpak IC, 250 x 4.6 mm – eluent: n-hexane/EtOH DEA 80/20/0.1) >99.9 %. A single crystal X-ray structure of the deuterated fezolinetant final product was obtained (Figure 1) that confirmed the structure of the compound as well as the stereochemistry.
References
- ^ Jump up to:a b c http://adisinsight.springer.com/drugs/800039455
- ^ Jump up to:a b c Hoveyda, Hamid R.; Fraser, Graeme L.; Dutheuil, Guillaume; El Bousmaqui, Mohamed; Korac, Julien; Lenoir, François; Lapin, Alexey; Noël, Sophie (2015). “Optimization of Novel Antagonists to the Neurokinin‑3 Receptor for the Treatment of Sex-Hormone Disorders (Part II)”. ACS Medicinal Chemistry Letters (6): 736-740. doi:10.1021/acsmedchemlett.5b00117.
- ^ http://www.prnewswire.com/news-releases/astellas-to-acquire-ogeda-sa-300433141.html
- ^ Jump up to:a b Fraser GL, Ramael S, Hoveyda HR, Gheyle L, Combalbert J (2016). “The NK3 Receptor Antagonist ESN364 Suppresses Sex Hormones in Men and Women”. J. Clin. Endocrinol. Metab. 101 (2): 417–26. doi:10.1210/jc.2015-3621. PMID 26653113.
- ^ Jump up to:a b Fraser GL, Hoveyda HR, Clarke IJ, Ramaswamy S, Plant TM, Rose C, Millar RP (2015). “The NK3 Receptor Antagonist ESN364 Interrupts Pulsatile LH Secretion and Moderates Levels of Ovarian Hormones Throughout the Menstrual Cycle”. Endocrinology. 156 (11): 4214–25. doi:10.1210/en.2015-1409. PMID 26305889.
- ^ Jump up to:a b c http://www.medscape.com/viewarticle/878262
- ^ Jump up to:a b c https://www.clinicalleader.com/doc/ogeda-announces-positive-fezolinetant-treatment-menopausal-flashes-0001
External links
 |
|
| Clinical data | |
|---|---|
| Synonyms | ESN-364 |
| Routes of administration |
By mouth |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C16H15FN6OS |
| Molar mass | 358.40 g·mol−1 |
| 3D model (JSmol) | |
////////////////Fezolinetant, ESN-364, фезолинетант , فيزولينيتانت , 非唑奈坦 , Phase II, Hot flashes, Polycystic ovary syndrome, Uterine leiomyoma, Euroscreen, Ogeda, FDA 2023, APPROVALS 2023, Veozah
Smiles
C[C@H]1N(CCn2c1nnc2c3nc(C)ns3)C(=O)c4ccc(F)cc4
“ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This is a compilation for educational purposes only. P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent

READ
ANTHONY MELVIN CRASTO

DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE


Join me on twitter
Join me on google plus  Googleplus
Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
CALL +919323115463 INDIA
//////////////
Sparsentan, PS433540, RE-021

Sparsentan (PS433540, RE-021)
- C32H40N4O5S
- Average mass592.749
FDA APPROVED 2023/2/17, Filspari
4′-((2-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl)-N-(4,5-dimethylisoxazol-3-yl)-2′-(ethoxymethyl)-[1,1′-biphenyl]-2-sulfonamide
4′-[(2-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methvn-N-(3,4- dimethyl-5-isoxazolyl)-2′-ethoxymethyl [ 1 , l’-biphenyll -2-sulfonamide
Sparsentan
PS433540; RE-021, formerly known as DARA
CAS :254740-64-2
4-[(2-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-N-(4,5- dimethylisoxazol-3-yl)-2-(ethoxymethyl)biphenyl-2-sulfonamide
Mechanism of Action:acting as both an Endothelin Receptor Antagonist (ERA) and Angiotensin Receptor Blocker (ARB).
Indication: Focal Segmental Glomerulosclerosis (FSGS).Focal Segmental Glomerulosclerosis (FSGS) is a rare and severe nephropathy which affects approximately 50,000 patients in the United States. Most cases of FSGS are pediatric.
Development Stage: Phase II
Developer:Retrophin, Inc
- OriginatorBristol-Myers Squibb
- DeveloperRetrophin
- ClassAntihypertensives; Isoxazoles; Small molecules; Spiro compounds; Sulfonamides
- Mechanism of ActionAngiotensin type 1 receptor antagonists; Endothelin A receptor antagonists
- Orphan Drug Status Yes – Focal segmental glomerulosclerosis
-
- 09 Jan 2015 Sparsentan receives Orphan Drug status for Focal segmental glomerulosclerosis in USA
- 31 Dec 2013 Phase-II/III clinical trials in Focal segmental glomerulosclerosis in USA (PO)
- 07 May 2012I nvestigation in Focal segmental glomerulosclerosis in USA (PO)
Sparsentan is an investigational therapeutic agent which acts as both a selective endothelin receptor antagonist and an angiotensin receptor blocker. Retrophin is conducting the Phase 2 DUET trial of Sparsentan for the treatment of FSGS, a rare and severe nephropathy that is a leading cause of end-stage renal disease. There are currently no therapies approved for the treatment of FSGS in the United States. Ligand licensed worldwide rights of Sparsentan (RE-021) to Retrophin in 2012 .The Food and Drug Administration (FDA) has granted orphan drug designation for Retrophins sparsentan for the treatment of focal segmental glomerulosclerosis (FSGS) in January 2015.
In 2006, the drug candidate was licensed to Pharmacopeia by Bristol-Myers Squibb for worldwide development and commercialization. In 2012, a license was obtained by Retrophin from Ligand. In 2015, Orphan Drug Designation was assigned by the FDA for the treatment of focal segmental glomerulosclerosis.
Sparsentan, also known as RE-021, BMS346567, PS433540 and DARA-a, is a Dual angiotensin II and endothelin A receptor antagonist. Retrophin intends to develop RE-021 for orphan indications of severe kidney diseases including Focal Segmental Glomerulosclerosis (FSGS) as well as conduct proof-of-concept studies in resistant hypertension and diabetic nephropathy. RE-021, with its unique dual blockade of angiotensin and endothelin receptors, is expected to provide meaningful clinical benefits in mitigating proteinuria in indications where there are no approved therapies
Sparsentan, sold under the brand name Filspari, is a medication used for the treatment of primary immunoglobulin A nephropathy.[1] Sparsentan is an endothelin and angiotensin II receptor antagonist.[1][4] It is taken by mouth.[1]
The most common side effects include swelling of the extremities, low blood pressure, dizziness, high blood potassium, anemia, injury to the kidney, and increased liver enzymes in the blood.[5]
It was approved for medical use in the United States in February 2023.[5][6][7] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication.[8]
PATENT
WO 2000001389
https://www.google.co.in/patents/WO2000001389A1?cl=en


Example 41
4′- [(2-Butyl-4-oxo- 1.3-diazaspiro [4.4! non- l-en-3-yl)methyll -N-(3.4- dimethyl-5-isoxazolyl)-2′-hydroxymethyl[l, l’-biphenyl! -2-sulfonamide

A. 4′-[(2-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methyll-N-(3.4- dimethyl-5-isoxazolyl)-N-[(2-trimethylsilylethoxy)methyl]-2′- hydroxym ethyl [1, l’-biphenyl] -2-sulfonamide P14 (243 mg, 0.41 mmol) was used to alkylate 2-butyl-4-oxo-l,3- diazaspiro[4.4]non-l-ene hydrochloride according to General Method 4. 41A (100 mg, 35% yield) was isolated as a slightly yellow oil after silica gel chromatography using 1:1 hexanes/ethyl acetate as eluant. B. 4′- [(2-Butyl-4-oxo- 1 ,3-diazaspiro [4.41 non- l-en-3-yl)methvn -N-0.4- dimethyl-5-isoxazolyl)-2′-hydroxymethyl[l,l’-biphenyn-2- sulfonamide
Deprotection of 41A (100 mg, 0.14 mmol) according to General Method 8 (ethanol) gave the title compound as white solid in 46% yield following silica gel chromatography (96:4 methanol/chloroform eluant):
MS m/e 565 (ESI+ mode); HPLC retention time 3.21 min (Method A);
HPLC purity >98%.
Example 42
4′-[(2-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methvn-N-(3,4- dimethyl-5-isoxazolyl)-2′-ethoxymethyl [ 1 , l’-biphenyll -2-sulfonamide

A. 4′- [(2-Butyl-4-oxo- 1 ,3-diazaspiro [4.41 non- l-en-3-yl)methyll -N-(3 ,4- dimethyl-5-isoxazolyl)-N-[(2-methoxyethoxy)methyll-2′- hvdroxym ethyl [1 , l’-biphenyl] -2-sulfonamide
Triethylsilane (6 ml) and TFA (6 ml) were added to a solution of 5F (960 mg, 1.5 mmol) in 15 ml dichloromethane at RT. The mixture was stirred at RT for 2 h and was then concentrated. The residue was taken up in ethyl acetate and was washed successively with aqueous sodium bicarbonate, water, and brine. The organic layer was dried over sodium sulfate and concentrated. The residue was chromatographed on silica gel using 100:2 dichloromethane/methanol to afford 42A (740 mg, 77%) as a colorless gum. Rf=0.13, silica gel, 100:5 dichloromethane/methanol. B. 4′- [(2-Butyl-4-oxo- 1.3-diazaspiro [4.41 non- l-en-3-yl)methyll -N-(3.4- dimethyl-5-isoxazolyl)-N-r(2-methoxyethoxy)methyll-2′- ethoxymethyl[l.l’-biphenyll-2-sulfonamide A mixture of 42A (100 mg, 0.15 mmol), iodoethane (960 mg, 6.1 mmol) and silver (I) oxide (180 mg, 0.77 mmol) in 0.7 ml DMF was heated at 40 ° C for 16 h.. Additional iodoethane (190 mg, 1.2 mmol) and silver (I) oxide (71 mg, 0.31 mmol) were added and the reaction mixture was heated at 40 ° C for an additional 4 h. The mixture was diluted with 1:4 hexanes/ethylacetate and was then washed with water and brine. The organic layer was dried over sodium sulfate and was then concentrated. The residue was chromatographed on silica gel using 200:3 dichloromethane/methanol as eluant to afford 42B (51mg, 49%) as a colorless gum. Rf=0.35, silica gel, 100:5 dichloromethane/methanol.
C. 4,-[(2-Butyl-4-oxo-1.3-diazaspirof4.41non-l-en-3-yl)methyll-N-(3.4- dimethyl-5-isoxazolyl )-2′-ethoxym ethyl [ 1. l’-biphenyll -2-sulfonamide
42B (51 mg) was deprotected according to General Method 7 to afford the title compound in 80% yield following preparative reverse-phase HPLC purification: white solid; m.p. 74-80 ° C (amorphous); IH NMR (CDCL, )δ0.87(tr, J=7Hz, 3H), 0.99(tr, J=7Hz, 3H), 1.32(m, 2H), 1.59(m, 2H), 1.75-2.02(m, 11H), 2.16(s, 3H), 2.35(m, 2H), 3.38 (m, 2H), 4.23(m, 2H), 4.73(s, 2H), 7.11-7.85 (m, 7H); MS m/e 593 (ESI+ mode); HPLC retention time 18.22 min. (Method E); HPLC purity >97%.
PATENT
WO 2001044239
http://www.google.co.in/patents/WO2001044239A2?cl=en
……………………
Dual angiotensin II and endothelin A receptor antagonists: Synthesis of 2′-substituted N-3-isoxazolyl biphenylsulfonamides with improved potency and pharmacokinetics
J Med Chem 2005, 48(1): 171
J. Med. Chem., 2002, 45 (18), pp 3829–3835
DOI: 10.1021/jm020138n
 BMS 248360 A DIFFERENT COMPD
BMS 248360 A DIFFERENT COMPDThe ETA receptor antagonist (2) (N-(3,4-dimethyl-5-isoxazolyl)-4‘-(2-oxazolyl)-[1,1‘-biphenyl]-2-sulfonamide, BMS-193884) shares the same biphenyl core as a large number of AT1 receptor antagonists, including irbesartan (3). Thus, it was hypothesized that merging the structural elements of 2 with those of the biphenyl AT1 antagonists (e.g., irbesartan) would yield a compound with dual activity for both receptors. This strategy led to the design, synthesis, and discovery of (15) (4‘-[(2-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-N-(3,4-dimethyl-5-isoxazolyl)-2‘-[(3,3-dimethyl-2-oxo-1-pyrrolidinyl)methyl]-[1,1‘-biphenyl]-2-sulfonamide, BMS-248360) as a potent and orally active dual antagonist of both AT1 and ETAreceptors. Compound 15 represents a new approach to treating hypertension.

Scheme 2 a DIFFERENT COMPD
a (a) DIBAL, toluene; (b) NaBH4, MeOH; (c) (Ph)3P, CBr4, THF (51% from 9); (d) compound 7, NaH, DMF; (e) 1 N HCl; (f) compound 4, (Ph3P)4Pd, aqueous Na2CO3, EtOH/toluene; (g) 6 N aqueous HCl/EtOH (60% from 10); (h) 13, sodium triacetoxy borohydride, AcOH, (i) diisopropylcarbodiimide, CH2Cl2 (31% from 12).
PATENT
WO 2010135350
http://www.google.com/patents/WO2010135350A2?cl=en
Compound 1 :

Scheme IV

Scheme V

Formula IV 1
Scheme VII

Formula Vl

A solution of 2-(2,4-dimethylphenyl)benzenesulfonic acid (Compound 12) (0.5 g, 1.9 mmol) in 50 mL of anhydrous acetonitrile was prepared and transferred to a round-bottom flask. After flushing with nitrogen gas, N-bromosuccinimide (0.75 g, 4.2 mmol) was added followed by 50 mg (0.2 mmol) of benzoyl peroxide. The solution was heated at reflux for 3 hours. The solvent was removed in-vacuo and the resulting syrup purified by silica gel chromatography (1 :1 hexanes/EtOAc) to yield Compound 13 as a white solid. 1H NMR (500 MHz, CD3CN) 8.12 (d, J = 7.5 Hz, IH), 7.92 (t, J = 7.5 Hz, IH), 7.78 (d, J= 7.5 Hz, IH), 7.74-7.71 (m, 2H), 7.68-7.65 (m, 2H), 5.12 (s, 2H), 4.70 (s, 2H). Example 4 2-(4-Bromomethyl-2-ethoxymethylphenyl)benzenesulfonic acid (Compound 14)

A solution of 20 mg (0.058 mmol) of (l-bromomethylbenzo[3,4- d])benzo[l,2-f]-2-oxa-l,l-dioxo-l-thiocycloheptane (Compound 13) in ethanol was stirred at elevated temperature until the starting material was consumed to give crude product (compound 14) that was used directly in the next step without isolation or purification.
Example 5
2-(4-((2-Butyl-4-oxo-l,3-diazaspiro[4.4]non-l-en-3-yl)methyl>2- ethoxymethylphenyl)benzenesulfonic acid (Compound 15)

To the above ethanol solution of crude 2-(4-bromomethyl-2- ethoxymethylphenyl)benzenesulfonic acid (Compound 14) described in Example 4 was added approximately 25 mL of anhydrous DMF. The ethanol was removed from the system under reduced pressure. Approximately 15 mg (0.065 mmol) of 2-butyl-l,3- diazaspiro[4.4]non-l-en-4-one (compound 7 in Scheme IV) was added followed by 300 μL of a IM solution of lithium bis-trimethylsilylamide in THF. The solution was allowed to stir at room temperature for 3 hours. The solvents were removed under reduced pressure and the remaining residue purified by preparative RP-HPLC employing a Cl 8 column and gradient elution (H2O:MeCN) affording the title compound as a white solid; [M+H]+ calcd for C27H34N2O5S 499.21, found, 499.31 ; 1H NMR (500 MHz, CD3CN) 8.04 (t, J= 5.5 Hz, IH), 7.44-7.10 (m, 2H), 7.28 (s, IH), 7.22 (d, J= 8.0 Hz, 2H), 7.08- 7.04 (m, 2H), 4.74 (br s, 2H), 4.32 (d, J= 13.0 Hz IH), 4.13 (d, J= 13.0 Hz IH), 3.40- 3.31 (m, 2H), 2.66 (t, J= 8 Hz, 2H), 2.18-2.13 (m, 5H), 1.96-1.90 (m, 2H obscured by solvent), 1.48 (m, 2H), 1.27 (s, J= 7 Hz, 2H), 1.16 (t, J= 7 Hz, 3H), 0.78 (t, J= 7.5 Hz, 3H).
Example 6
2-(4-((2-Butyl-4-oxo-l,3-diazaspiro[4.4]non-l-en-3-yl)methyl>2- ethoxymethylphenyl)benzenesulfonyl chloride (Compound 16)

To a solution of DMF (155 μL, 2 mmol, 2 equiv.) in dichloromethane (5 mL) at 0 0C was added dropwise oxalyl chloride (175 μL, 2 mmol, 2 equiv.) followed by a dichloromethane (5 mL) solution of 2-(4-((2-butyl-4-oxo-l,3-diazaspiro[4.4]non-l- en-3-yl)methyl)-2-ethoxymethylphenyl)benzenesulfonic acid (Compound 15) (0.50 g, 1.0 mmol). The resulting mixture was stirred at 0 0C for ~2 hours, diluted with additional dichloromethane (25 mL), washed with saturated sodium bicarbonate solution (10 mL), water (10 mL), and brine (10 mL), dried over sodium sulfate, and then concentrated to give crude sulfonyl chloride (compound 16) that was used without purification.
Example 7
N-(3,4-Dimethyl-5-isoxazolyl)-2-(4-(2-butyl-4-oxo-l,3-diazospiro[4.4]non-l-en- 3yl)methyl-2-ethoxymethylphenyl)phenylsulfonamide (Compound 1)

[0062] To a solution of 5-amino-3,4-dimethylisoxazole (60 mg, 0.54 mmol) in THF at -60 °C was added dropwise potassium tert-butoxide (1 mL of 1 M solution) followed by a solution of crude 2-(4-((2-butyl-4-oxo-l,3-diazaspiro[4.4]non-l-en-3- yl)methyl)-2-ethoxymethylphenyl)benzenesulfonyl chloride (Compound 16) (0.28 g, 0.54 mmol) in THF (4 mL). The resulting mixture was stirred at about -60 °C for 1 hour, allowed to warm to room temperature overnight, and then quenched with IN HCl solution to about pH 4. Standard workup of extraction with ethyl acetate, washing with water, drying, and concentration provided the final compounds as a white solid. 1H NMR (400 MHz, CDCl3) 8.03 (dd, J = 8.0 and 1.2, IH), 7.60 (td, J = 7.5 and 1.5, IH), 7.50 (td, J = 7.7 and 1.5, IH), 7.36 (s, IH), 7.28 (d, J= 2.1, 1 H), 7.25 (dd, J = 7.5 and 1.2, IH), 7.09 (dd, J= 7.9 and 1.6, IH), 6.61 (bs, IH), 4.77 (AB quartet, J= 15.5 and 8.1, 2H), 4.18 (AB quartet, J= 12.0 and 35, 2H), 3.45-3.32 (m, 2H), 2.39 (t, J= 7.5, 2H), 2.26 (s, 3H), 2.02- 1.84 (m, 8H), 1.82 (s, 3H), 1.63 (quint, J = 7.5, 2H), 1.37 (sextet, J = 7.3, 2H), 1.07 (t, J = 7.0, 3H), and 0.90 (t J= 7.3, 3H).
Example 8 l-Bromo-2-ethoxymethyl-4-hydroxymethylbenzene (Compound 17)

To a solution of ethyl 4-bromo-3-ethoxymethylbenzoate (9.4 g, 33 mmol) in toluene (56 mL) at about -10 0C was added 51 g of a 20% diisobutylaluminum hydride solution in toluene (ca. 70 mmol). The reaction was stirred at the same temperature for about 30 minutes until the reduction was completed, and then quenched with icy 5% NaOH solution to keep the temperature below about 10 °C. Organic phase of the resulting mixture was separated and the aqueous phase was extracted with toluene. The combined organic phase was concentrated in vacuo to a final volume of ~60 mL toluene solution of l-bromo-2-ethoxymethyl-4-hydroxymethylbenzene (Compound 17) that was used in next step without purification.
Example 9 l-Bromo-2-ethoxymethyl-4-methanesulfonyloxymethylbenzene (Compound 18)

To a solution of 1 -bromo-2-ethoxymethyl-4-hydroxymethylbenzene (Compound 17) (8.4 g, 33 mmol) in toluene (60 mL) prepared in Example 8 at about -10 °C was added methanesulfonyl chloride (7.9 g, 68 mmol). The reaction was stirred at the same temperature for about 30 minutes until the reduction was completed, and then quenched with icy water to keep the temperature at about 0 °C. The organic layer was separated and washed again with icy water to provide a crude product solution of 1 – bromo-2-ethoxymethyl-4-methanesulfonyloxymethylbenzene (Compound 18) that was used without purification.
Example 10
1 -Bromo-4-((2-butyl-4-oxo- 1 ,3 -diazaspiro [4.4]non- 1 -en-3 -yl)methy l)-2- ethoxymethylbenzene bisoxalic acid salt (Compound 19)

To the crude solution of 1 -bromo-2-ethoxymethyl-4- methanesulfonyloxymethylbenzene (Compound 18) (1 1 g, 33 mmol) in toluene (80 mL) prepared in Example 9 was added a 75% solution of methyltributylammonium chloride in water (0.47 mL). The resulting mixture was added to a solution of 2-butyl-4-oxo-l,3- diazaspiro[4.4]non-l-ene (compound 7 in Scheme VI) (7.5 g, 32 mmol) in dichloromethane (33 mL) pretreated with a 10 M NaOH solution (23 mL). The reaction mixture was stirred at room temperature for 2 hours until compound 18 was not longer detectable by HPLC analysis and then was quenched with water (40 mL). After stirring about 10 minutes, the organic layer was separated and aqueous layer was extracted with toluene. The combined organic phase was washed with water and concentrated to a small volume. Filtration through a silica gel pad using ethyl acetate as solvent followed by concentration yielded 1 -bromo-4-((2-buty 1-4-oxo- 1 ,3 -diazaspiro [4.4]non- 1 -en-3 – yl)methyl)-2-ethoxymethylbenzene as a crude oil product.
The crude oil was dissolved in ethyl acetate (22 mL) and warmed to around 50 °C. Anhydrous oxalic acid (4.6 g) was added to the warm solution at once and the resulting mixture was stirred until a solution was obtained. The mixture was cooled gradually and the bisoxalic acid salt (compound 19) was crystallized. Filtration and drying provided pure product (compound 19) in 50-60% yield from ethyl 4-bromo-3- ethoxymethylbenzoate in 3 steps. 1H NMR (400 MHz, CDCl3) 12.32 (bs, 4H), 7.58 (d, J = 7.8, IH), 7.36 (s, IH), 7.12 (d, J= 7.8, IH), 4.90 (s, 2H), 4.56 (s, 2H), 3.68 (q, J= 7.5, 2H), 2.87-2.77 (m, 2H), 2.40-1.95 (m, 8H), 1.62-1.53 (m, 2H), 1.38-1.28 (m, 4H), and 1.82 (t, J= 7.5, 3H).
Example 11
N-(3,4-Dimethyl-5-isoxazolyl)-2-(4-(2-butyl-4-oxo-l,3-diazospiro[4.4]non-l-en- 3yl)methyl-2-ethoxymethylphenyl)phenylsulfonamide (Compound 1)

To a suspension of l-bromo-4-((2-butyl-4-oxo-l,3-diazaspiro[4.4]non- l-en-3-yl)methyl)-2-ethoxymethylbenzene bisoxalic acid salt (Compound 19) (5.0 g, 8.3 mmol) in toluene (20 niL) under nitrogen was added water (30 mL) and pH was adjusted to 8-9 by addition of a 2 M NaOH solution at room temperature. The organic phase was separated and mixed with 2-(N-(3,4-dimethyl-5-isoxazolyl)-N- methoxymethylamino)sulfonylphenylboronic acid pinacol ester (Scheme VII, Formula IX, where R8is methoxymethyl and M = boronic acid pinacol ester) (3.6 g, 8.5 mmol), bis(dibenzylideneacetone)palladium(0) (Pd(dba)2) (0.12 g), and a standard phosphine ligand. After a 2 M sodium carbonate solution was added, the reaction mixture was warmed to 70 0C and stirred until the reaction was complete by HPLC analysis. The reaction was cooled to room temperature and quenched with water, and then separated in phases. The organic phase was treated with activated carbon, filtered through a pad of silica gel, and was concentrated to afford a crude mixture.
The crude reaction mixture was dissolved in ethanol (40 mL) after palladium catalyst was removed and was treated with 6 M HCl solution (ca. 40 mL). The mixture was warmed to 75-80 °C and stirred for about 2 hours until the reaction was completed by HPLC analysis. After the mixture was cooled to room temperature, the pH of the mixture was adjusted to 8 by addition of 10 M NaOH solution. The mixture was stirred for 2 more hours and the pH was adjusted to 6 by adding 2 M HCl and the crystal seeds. Filtration of the crystalline solid followed by drying provided N-(3,4-dimethyl-5- isoxazolyl)-2-(4-(2-butyl-4-oxo-l,3-diazospiro[4.4]non-l-en-3yl)methyl-2- ethoxymethylphenyl)phenylsulfonamide (Compound 1) as a white solid.1H NMR (400 MHz, CDCIa) 8.03 (dd, J= 8.0 and 1.2, IH), 7.60 (td, J = 7.5 and 1.5, IH), 7.50 (td, J = 7.7 and 1.5, IH), 7.36 (s, IH), 7.28 (d, J= 2.1, 1 H), 7.25 (dd, J = 7.5 and 1.2, IH), 7.09 (dd, J= 7.9 and 1.6, IH), 6.61 (bs, IH), 4.77 (AB quartet, J= 15.5 and 8.1, 2H), 4.18 (AB quartet, J= 12.0 and 35, 2H), 3.45-3.32 (m, 2H), 2.39 (t, J= 7.5, 2H), 2.26 (s, 3H), 2.02- 1.84 (m, 8H), 1.82 (s, 3H), 1.63 (quint, J= 7.5, 2H), 1.37 (sextet, J= 7.3, 2H), 1.07 (t, J = 7.0, 3H), and 0.90 (t J= 7.3, 3H).
| US20040002493 * | Aug 20, 2001 | Jan 1, 2004 | Kousuke Tani | Benzoic acid derivatives and pharmaceutical agents comprising the same as active ingredient |
| US20070054806 * | Sep 6, 2006 | Mar 8, 2007 | Bayer Cropscience Gmbh | Novel sulfonamide-comprising solid formulations |
| US20070054807 * | Sep 8, 2006 | Mar 8, 2007 | Bayer Cropscience Gmbh | Storage-stable formulations of sulfonamides |
.
 |
|
| Clinical data | |
|---|---|
| Trade names | Filspari |
| Other names | RE-021, PS433540 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a623018 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
By mouth |
| ATC code | |
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| UNII | |
| KEGG | |
| ChEBI | |
| ECHA InfoCard | 100.275.317 |
| Chemical and physical data | |
| 3D model (JSmol) | |
|
show
|
|
|
show
|
|
References
- ^ Jump up to:a b c d e f “Filspari- sparsentan tablet, film coated”. DailyMed. 17 February 2023. Retrieved 6 March 2023.
- ^ Jump up to:a b c d “Filspari EPAR”. European Medicines Agency (EMA). 22 February 2024. Retrieved 24 February 2024. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b “Filspari Product information”. Union Register of medicinal products. 23 April 2024. Retrieved 7 September 2024.
- ^ Chiu AW, Bredenkamp N (September 2023). “Sparsentan: A First-in-Class Dual Endothelin and Angiotensin II Receptor Antagonist”. The Annals of Pharmacotherapy. 58 (6): 645–656. doi:10.1177/10600280231198925. PMID 37706310. S2CID 261743204.
- ^ Jump up to:a b c d e f g h i j k l m n o p q “Drug Trials Snapshots: Filspari”. U.S. Food and Drug Administration (FDA). 17 February 2023. Retrieved 7 September 2024.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Travere Therapeutics Announces FDA Accelerated Approval of Filspari (sparsentan), the First and Only Non-immunosuppressive Therapy for the Reduction of Proteinuria in IgA Nephropathy” (Press release). Travere Therapeutics. 17 February 2023. Retrieved 17 February 2023 – via GlobeNewswire.
- ^ Syed YY (April 2023). “Sparsentan: First Approval”. Drugs. 83 (6): 563–568. doi:10.1007/s40265-023-01864-x. PMC 10232600. PMID 37022667.
- ^ New Drug Therapy Approvals 2023 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2024. Archived from the original on 10 January 2024. Retrieved 9 January 2024.
- ^ “PHARMACOPEIA LAUNCHES STUDY OF DARA COMPOUND | FDAnews”. http://www.fdanews.com.
- ^ “Ligand Licenses DARA Program to Retrophin”. investor.ligand.com. 21 February 2012.
- ^ https://www.fiercebiotech.com/biotech/retrophin-sheds-shkreli-connection-new-name-travere-therapeutics.
{{cite news}}: Missing or empty|title=(help) - ^ “Ongoing Non-malignant Hematological, Neurological, and Other Disorder Indications Accelerated Approvals”. U.S. Food and Drug Administration (FDA). 21 August 2024. Retrieved 7 September 2024.
- ^ “Travere Therapeutics Announces Full FDA Approval of Filspari (sparsentan), the Only Non-Immunosuppressive Treatment that Significantly Slows Kidney Function Decline in IgA Nephropathy” (Press release). Travere Therapeutics. 5 September 2024. Retrieved 7 September 2024 – via GlobeNewswire.
- ^ “Despite trial scare, Travere’s Filspari gains full FDA nod in kidney disease showdown with Novartis”. fiercepharma.com.
External links
- Clinical trial number NCT03762850 for “A Study of the Effect and Safety of Sparsentan in the Treatment of Patients With IgA Nephropathy (PROTECT)” at ClinicalTrials.gov
SYN
https://doi.org/10.1021/acs.jmedchem.4c02079
J. Med. Chem. 2025, 68, 2147−2182
Sparsentan (Filspari). Sparsentan (27), marketed by Travere Therapeutics, is an oral, dual endothelin angiotensin receptor antagonist that received accelerated USFDA approval in February 2023 for reducing proteinuria in adults with primary immunoglobulin A (IgA) nephropathy who are at risk of rapid
disease progression.205206,207 Also known as Berger’s disease, IgAnephropathy is an immune-complex mediated disease characterized by deposits of IgA in the kidneys, resulting in inflammation and damage which can eventually lead to kidney failure. Typical treatment of IgA nephropathy has focused
on supportive care to slow kidney decline, for example, lowering blood pressure, reducing proteinuria, and minimizing lifestyle risk factors; immunosuppressive therapy has also been utilized, though it is controversial and carries risks.208 Sparsentan is the first nonimmunosuppressive treatment for IgA nephropathy and has received first-in-class and orphan drug designations. Accelerated approval was based on reduction of proteinuria (which is a risk factor for disease progression) during interim
analysis in phase III clinical trials. 209 endothelin type A (ETASparsentan blocks ) and angiotensin II type 1 receptors(AT1), interrupting the signaling pathway that contributes to disease progression. 210
The structure of the drug combines 211,212 elements that target both of these receptor types.
213 Thesynthesis of sparsentan (27), as shown in Scheme 50 and Scheme 51, was disclosed by Retrophin Pharmaceuticals (now Travere Therapeutics). Its telescoped sequences and isolation of intermediates as salts suggest that this route may be suitable for large-scale manufacturing.
The synthesis of the spirocyclic imidazolinone intermediate 27.7 is shown in Scheme 50.
Displacement of the benzylic bromide in 27.1 with sodium ethoxide produced ether 27.2. Reduction of the ester with sodium borohydride and zinc chloride yielded alcohol 27.3 which was then converted to mesylate 27.4. Reaction with spirocyclic imidazolinone 27.5 under phase transfer conditions
yielded 27.6 whichwasisolatedasthebisoxalatesalt (27.7).The sequence from 27.1 to 27.7 is telescoped, and no yields were given in the patent.
The construction of the biphenyl framework is shown in Scheme 51. Treatment of aryl bromide 27.8 with n-BuLi and triisopropyl borate followed by reaction with pinacol yielded boronic ester 27.9. Intermediates 27.7 and 27.9 were coupled via a Suzuki reaction to form the biphenyl which was isolated as
the camphorsulfonate salt (27.10). The synthesis was finished with deprotection of the methoxymethyl group under acidic conditions followed by recrystallization from isopropanol and heptane to yield sparsentan (27).
(206) Donadio, J. V.; Grande, J. P. IgA nephropathy. N. Engl. J. Med.2002, 347, 738−748.
(207) Fabiano, R. C. G.; Pinheiro, S. V. B.; Simões e Silva, A. C.Immunoglobulin A nephropathy: a pathophysiology view. Inflammation Res. 2016, 65, 757−770.
(208) Floege, J.; Rauen, T.; Tang, S. C. W. Current treatment of IgAnephropathy. Springer Semin. Immunopathol. 2021, 43, 717−728.
(209) Rovin, B.H.; Barratt, J.; Heerspink, H. J. L.; Alpers, C. E.; Bieler,S.; Chae, D.-W.; Diva, U. A.; Floege, J.; Gesualdo, L.; Inrig, J. K.; et al.Efficacy and safety of sparsentan versus irbesartan in patients with IgA
nephropathy (PROTECT): 2-year results from a randomised, active controlled, phase 3 trial. Lancet 2023, 402, 2077−2090.
(210) Komers, R.; Plotkin, H. Dual inhibition of renin-angiotensin aldosterone system and endothelin-1 in treatment of chronic kidney disease. Am. J. Physiol.: Regul., Integr. Comp. Physiol. 2016, 310, R877−
R884.
(211) Murugesan, N.; Tellew, J. E.; Gu, Z.; Kunst, B. L.; Fadnis, L.;Cornelius, L. A.; Baska, R. A. F.; Yang, Y.; Beyer, S. M.; Monshizadegan, H.; et al. Discovery of N-isoxazolyl biphenylsulfonamides as potent dual
angiotensin II and endothelin A receptor antagonists. J. Med. Chem.2002, 45, 3829−3835.
(212) Murugesan, N.; Gu, Z.; Fadnis, L.; Tellew, J. E.; Baska, R. A. F.; Yang, Y.; Beyer, S. M.; Monshizadegan, H.; Dickinson, K. E.; Valentine,M.T.; et al. Dual angiotensin II and endothelin A receptor antagonists:
synthesis of 2′-substituted N-3-isoxazolyl biphenylsulfonamides withimproved potencyandpharmacokinetics. J. Med. Chem. 2005, 48, 171−179.
(213) Komers, R.; Shih, A. Biphenyl sulfonamide compounds for the treatment of kidney diseases or disorders. WO 2018071784, 2018.
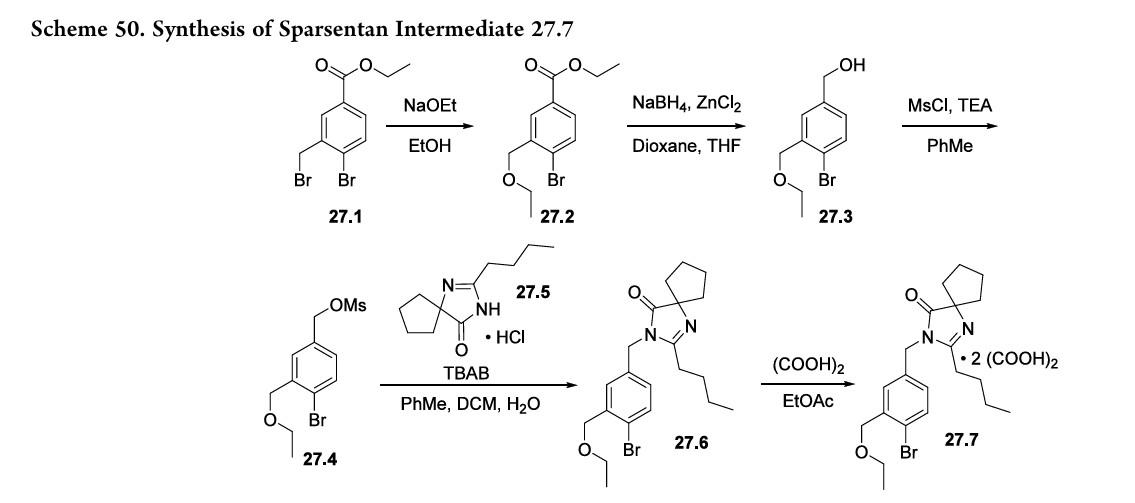

//////////////Sparsentan, PS433540, RE-021, Bristol-Myers Squibb, ORPHAN DRUG, Retrophin, FDA 2023, APPROVALS 2023
O=S(C1=CC=CC=C1C2=CC=C(CN3C(CCCC)=NC4(CCCC4)C3=O)C=C2COCC)(NC5=NOC(C)=C5C)=O,
BEXAGLIFLOZIN


Bexagliflozin
THR1442; THR-1442, EGT 0001442; EGT1442
CAS :1118567-05-7
(2S,3R,4R,5S,6R)-2-[4-chloro-3-({4-[2- (cyclopropyloxy) ethoxy] phenyl} methyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H- pyran-3,4,5-triol
D-Glucitol, 1,5-anhydro-1-C-(4-chloro-3-((4-(2-(cyclopropyloxy)ethoxy)phenyl)methyl)phenyl)-, (1S)-
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
1-[4-Chloro-3-[4-[2-(cyclopropyloxy)ethoxy]benzyl]phenyl]-1-deoxy-beta-D-glucopyranose
1,5-Anhydro-1(S)-[4-chloro-3-[4-[2-(cyclopropyloxy)ethoxy]benzyl]phenyl]-D-glucitol
(1S)-1,5-anhydro-1-C-[4-chloro-3-({4-[2- (cyclopropyloxy)ethoxy]phenyl}methyl)phenyl]-D-glucitol
Chemical Formula: C24H29ClO7
Exact Mass: 464.16018
Mechanism of Action:SGLT2 inhibitor, Sodium-glucose transporter 2 inhibitors
Indication:Type 2 diabetes
FDA APPROVED
| 1/20/2023 |
To improve glycemic control in adults with type 2 diabetes mellitus as an adjunct to diet and exercise
Drug Trials Snapshot
Phase II
Developer:Theracos, Inc.
| Conditions | Phases | Recruitment | Interventions | Sponsor/Collaborators |
|---|---|---|---|---|
| Diabetes Mellitus Type 2 | Phase 2 | Completed | Drug: EGT0001442|Drug: Placebo capsules to match EGT0001442 | Theracos |
| Diabetes Mellitus | Phase 2 | Completed | Drug: EGT0001442|Drug: Placebo | Theracos |
| Type 2 Diabetes Mellitus | Phase 3 | Not yet recruiting | Drug: Bexagliflozin|Drug: Placebo | Theracos |
| Diabetes Mellitus, Type 2 | Phase 2|Phase 3 | Recruiting | Drug: Bexagliflozin tablets | Theracos |

 DIPROLINE COMPLEX
DIPROLINE COMPLEX
Bexagliflozin diproline
RN: 1118567-48-8, C24-H29-Cl-O7.2C5-H9-N-O2
Molecular Weight, 695.2013
L-Proline, compd. with (1S)-1,5-anhydro-1-C-(4-chloro-3-((4-(2-(cyclopropyloxy)ethoxy)phenyl)methyl)phenyl)-D-glucitol (2:1)
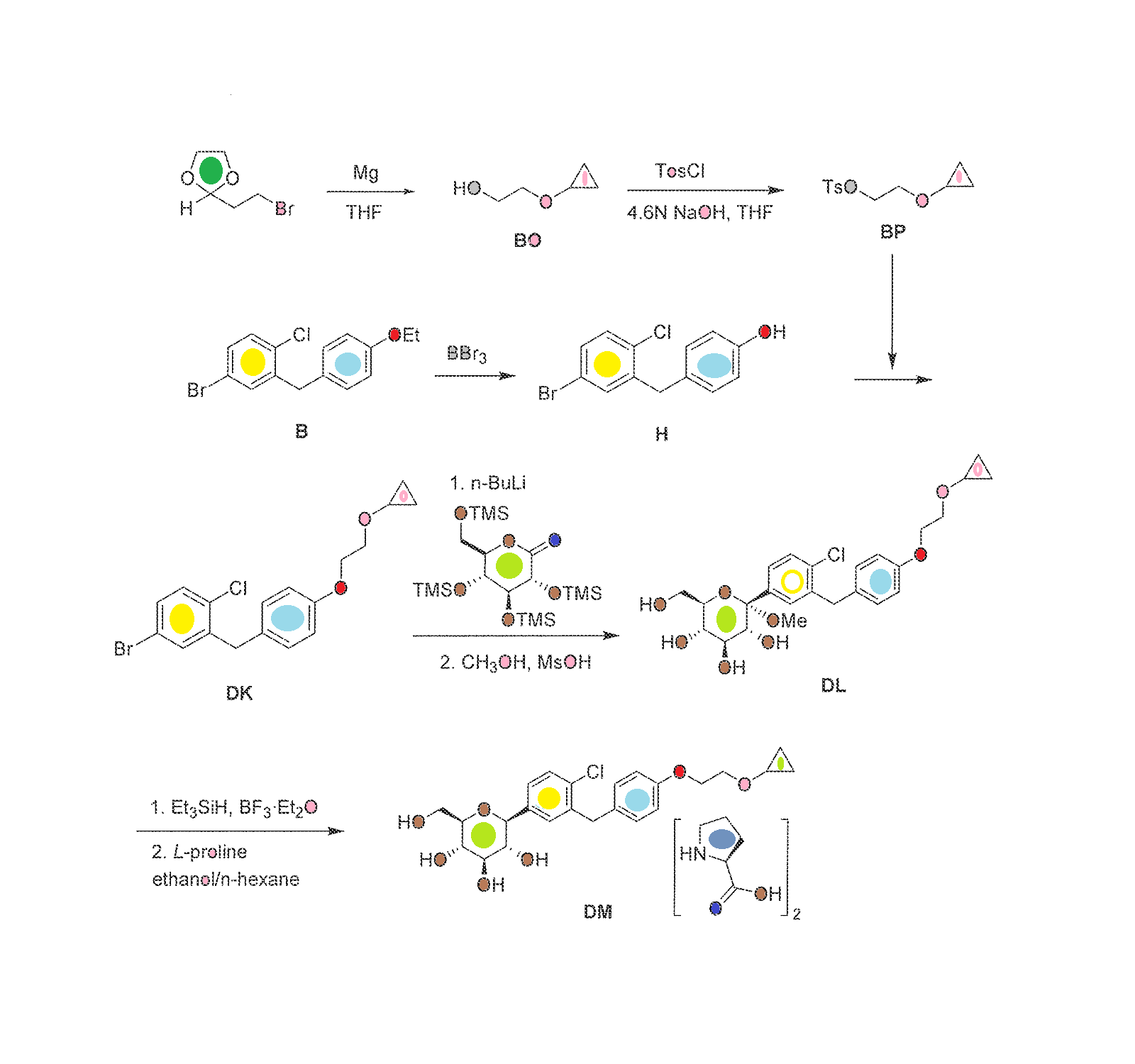

Bexagliflozin [(2S,3R,4R,5S,6R)-2-[4-chloro-3-({4-[2-(cyclopropyloxy) ethoxy] phenyl} methyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol] is an orally administered drug for the treatment of Type 2 Diabetes Mellitus (T2DM) and is classified as a Sodium Glucose co-Transporter 2 (SGLT2) Inhibitor. It is in Phase 2b study to evaluate the effect of bexagliflozin tablets in subjects with type 2 diabetes mellitus.
Bexagliflozin, also known as EGT1442, is a potent and selective SGLT2 inhibitor, attenuates blood glucose and HbA(1c) levels in db/db mice and prolongs the survival of stroke-prone rats. The IC(50) values for EGT1442 against human SGLT1 and SGLT2 are 5.6μM and 2nM, respectively. In normal rats and dogs a saturable urinary glucose excretion was produced with an ED(50) of 0.38 and 0.09mg/kg, respectively. EGT1442 showed favorable properties both in vitro and in vivo and could be beneficial to the management of type 2 diabetic patients.
One promising target for therapeutic intervention in diabetes and related disorders is the glucose transport system of the kidneys. Cellular glucose transport is conducted by either facilitative (“passive”) glucose transporters (GLUTs) or sodium-dependent (“active”) glucose cotransporters (SGLTs). SGLTl is found predominantly in the intestinal brush border, while SGLT2 is localized in the renal proximal tubule and is reportedly responsible for the majority of glucose reuptake by the kidneys.
Recent studies suggest that inhibition of renal SGLT may be a useful approach to treating hyperglycemia by increasing the amount of glucose excreted in the urine (Arakawa K, et al., Br J Pharmacol 132:578-86, 2001; Oku A, et al., Diabetes 48:1794-1800, 1999).

The potential of this therapeutic approach is further supported by recent findings that mutations in the SGL T2 gene occur in cases of familial renal glucosuria, an apparently benign syndrome characterized by urinary glucose excretion in the presence of normal serum glucose levels and the absence of general renal dysfunction or other disease (Santer R, et al., J Am Soc Nephrol 14:2873-82, 2003). Therefore, compounds which inhibit SGLT, particularly SGL T2, are promising candidates for use as antidiabetic drugs.
Compounds previously described as useful for inhibiting SGLT include C-glycoside derivatives (such as those described in US6414126, US20040138439, US20050209166, US20050233988, WO2005085237, US7094763, US20060009400, US20060019948, US20060035841, US20060122126, US20060234953, WO2006108842, US20070049537 and WO2007136116), O-glycoside derivatives (such as those described in US6683056, US20050187168, US20060166899, US20060234954, US20060247179 and US20070185197), spiroketal-glycoside derivatives (described in WO2006080421), cyclohexane derivatives (such as those described in WO2006011469), and thio- glucopyranoside derivatives (such as those described in US20050209309 and WO2006073197).

PATENT
WO 2009026537……………PRODUCT PATENT
http://www.google.co.in/patents/WO2009026537A1?cl=en


Example 19
[0289] The synthesis of compound BQ within the invention is given below.
[0290] Preparation of 2-cyclopropoxyethanol (Intermediate BO)
To a suspension of Mg powder (0.87 g, 36.1 mmol) and iodine (catalytic) in THF (4 mL) was added slowly BrCH2CH2Br (4.6 g, 24.5 mmol) in THF (8 mL). The exothermic reaction was cooled in an ice-bath. After complete addition OfBrCH2CH2Br, a solution of 2- (2-bromoethyl)-l,3-dioxolane (1 g, 5.6 mmol) was added dropwise. The reaction mixture was then kept at reflux for 24 h, quenched by addition of aqueous NH4Cl, and extracted with DCM. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated to give crude intermediate BO (400 mg) as yellow oil. [0292] Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (Intermediate BP)
Ts0^°V
To a solution of 2-cyclopropoxyethanol (400 mg, 3.92 mmol) in DCM (10 niL) were added TsCl (821 mg, 4.31 mmol) and Et3N (0.6 mL, 4.31 mmol). The reaction was stirred at room temperature overnight. Then, IN HCl was added, and the reaction was extracted with DCM. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated to give a yellow oil. The oil was purified by preparative TLC to obtain intermediate BP (50 mg) as a yellow oil.
Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2- cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Compound BQ)

To a solution of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-hydroxybenzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (intermediate Dl) (30 mg, 0.08 mmol) in anhydrous DMF (1 mL) were added 2-cyclopropoxyethyl 4-methylbenzenesulfonate (intermediate BP) (20 mg, 0.08 mmol) and Cs2CO3 (52 mg, 0.16 mmol). The mixture was stirred at room temperature for 12 h. Then the reaction mixture was poured into water, extracted with EA, washed with brine, dried with anhydrous Na2SO4 and concentrated to an oil. The oil was purified by preparative HPLC to obtain compound BQ (11 mg) as a colorless oil. 1H NMR (CD3OD): δ 7.30 (m, 3H), 7.11 (d, J= 8.8 Hz, 2H), 6.82 (d, J= 8.8 Hz, 2H), 4.13 (m, 5H), 3.85 (m, 3H), 3.81 (m, IH), 3.40 (m, 4H), 3.30 (m, IH), 0.52 (m, 4H); MS ESI (m/z) 465 (M+H)+, calc. 464.

Example 33
The synthesis of complex DM within the invention is outlined in FIG. 30, with the details given below.
Preparation of 2-cyclopropoxyethanol (Intermediate BO)
To a suspension of Mg powder (86.7 g, 3.6 mol) and I2 (catalytic) in anhydrous THF (0.7 L) was added slowly 1,2-dibromoethane (460 g, 2.4 mol) in anhydrous THF (2 L) at a rate that maintained the reaction temperature between 40-55° C. A solution of 2-(2-bromoethyl)-1,3-dioxolane (100 g, 0.56 mol) in anhydrous THF (750 mL) was added dropwise, and the reaction mixture was kept at 40-55° C. for 16 h. The reaction was quenched by addition of an aqueous solution of ammonium chloride. The mixture was extracted with methylene chloride. The organic layer was dried over sodium sulfate, and concentrated to give intermediate BO (27 g) as yellow oil, which was used in the next step without further purification.
Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (Intermediate BP)
To a stirred solution of sodium hydroxide (32 g, 0.8 mol) in water (180 mL) and THF (180 mL) was added crude 2-cyclopropoxyethanol from the previous step (27 g, 0.26 mol) at −5 to 0° C. A solution of p-toluenesulfonyl chloride (52 g, 0.27 mol) in THF (360 mL) was added dropwise, and the reaction mixture was kept at −5 to 0° C. for 16 h. The reaction mixture was then incubated at room temperature for 30 min, the organic layer was separated and the aqueous layer was extracted with ethyl acetate (2×1.0 L). The combined organic layers were washed with brine, dried over Na2SO4 and concentrated to get the crude intermediate BP as a yellow oil (53.3 g), which was used for the preparation of intermediate DK below without further purification.
Preparation of 4-(5-bromo-2-chlorobenzyl)phenol (Intermediate H)

To a stirred solution of 4-bromo-1-chloro-2-(4-ethoxybenzyl)benzene (intermediate B) (747 g, 2.31 mol) in dichloromethane was added slowly boron tribromide (1.15 kg, 4.62 mol) at −78° C. The reaction mixture was allowed to warm to room temperature. When the reaction was complete as measured by TLC, the reaction was quenched with water. The mixture was extracted with dichloromethane. The organic layer was washed with an aqueous solution of saturated sodium bicarbonate, then with water, and then with brine, and dried over Na2SO4. The residue was concentrated and then recrystallized in petroleum ether to obtain intermediate H as a white solid (460 g, yield 68%). 1H NMR (CDCl3, 400 MHz): δ 7.23˜7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.79 (d, J=8.8 Hz, 2H), 5.01 (s, 1H), 4.00 (s, 2H).
Preparation of 4-bromo-1-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (Intermediate DK)

A mixture of 4-(5-bromo-2-chlorobenzyl)phenol (56.7 g, 210 mmol) and Cs2CO3 (135 g, 420 mmol) in DMF (350 mL) was stirred at room temperature for 30 min, and then 2-cyclopropoxyethyl 4-methylbenzenesulfonate (crude intermediate BP from the second preceeding step above) (53.3 g, 210 mmol) was added. The reaction mixture was stirred at room temperature overnight, and then diluted with water (3 L) and extracted with EtOAc. The organic layer was washed with water, then with brine, and dried over Na2SO4. The residue was concentrated and then purified by flash column chromatography on silica gel (eluent PE:EA=10:1) to give intermediate DK as a liquid (51 g, yield 64%). 1H NMR (CDCl3, 400 MHz): δ 7.22˜7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.88 (d, J=8.8 Hz, 2H), 4.10 (t, J=4.8 Hz, 2H), 3.86 (t, J=4.8 Hz, 2H), 3.38-3.32 (m, 1H), 0.62-0.66 (m, 2H), 0.49-0.52 (m, 2H).
Preparation of (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (Intermediate DL)

To a stirred solution of 4-bromo-1-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (213 g) in anhydrous THF/toluene (1:2 v/v, 1.7 L) under argon was added n-BuLi (2.5 M in hexane, 245.9 mL) dropwise at −60±5° C. The mixture was stirred for 30 min, and then transferred to a stirred solution of (3R,4S,5R,6R)-3,4,5-tris(trimethylsilyloxy)-6-((trimethylsilyloxy)methyl)tetrahydro-2H-pyran-2-one (310.5 g) in toluene (1.6 L) at −60±5° C. The reaction mixture was continuously stirred at −60±5° C. for 1 before quenching with an aqueous solution of saturated ammonium chloride (1.5 L). The mixture was allowed to warm to room temperature and stirred for 1 h. The organic layer was separated and the water layer was extracted with ethyl acetate (3×500 mL). The combined organic layers were washed with brine (1 L), dried over Na2SO4, and concentrated. The residue was dissolved in methanol (450 mL), and methanesulfonic acid (9.2 mL) was added at 0° C. The solution was allowed to warm to room temperature and stirred for 2.0 h. The reaction was quenched with an aqueous solution of sodium bicarbonate (50 g) in water (500 mL) and then additional water (900 mL) was added. The mixture was extracted with ethyl acetate (3×1.0 L). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated. The crude product was used in the next step without further purification.
Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex (Complex DM)

To a stirred solution of crude (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol from the previous step in CH2Cl2/CH3CN (1:1, 1.3 L) at −5° C. was added triethylsilane (28.2 mL, 563 mmol), followed by BF3.Et2O (52.3 mL, 418.9 mmol). The reaction was stirred for 16 h while the temperature was allowed to warm gradually to room temperature. The reaction was quenched by addition of an aqueous solution of saturated sodium bicarbonate to pH 8.0. The organic volatiles were removed under vacuum. The residue was partitioned between ethyl acetate (2.25 L) and water (2.25 L). The organic layer was separated, washed with brine, dried over Na2SO4 and concentrated to give the crude product (230 g, purity 82.3%). To the crude product was added L-proline (113.7 g) in EtOH/H2O (15:1 v/v, 2.09 L), and the mixture was stirred at 80° C. for 1 h until it became a clear solution. Hexane (3.0 L) was added dropwise over 50 min, while the temperature was maintained at about 60° C. The reaction mixture was stirred overnight at room temperature. The solid was filtered and washed with EtOH/H2O (15:1 v/v, 2×300 mL), hexane (2×900 mL), and dried at 45° C. under vacuum for 10 h to give pure complex DM as a white solid (209 g; HPLC purity 99.2% (UV)). 1H NMR (CD3OD, 400 MHz): δ 7.25˜7.34 (m, 3H), 7.11 (d, J=8.8 Hz, 2H), 6.84 (d, J=8.8 Hz, 2H), 4.03-4.11 (m, 5H), 3.96-4.00 (m, 2H), 3.83-3.90 (m, 3H), 3.68-3.72 (m, 1H), 3.36-3.46 (m, 6H), 3.21-3.30 (m, 3H), 2.26-2.34 (m, 2H), 2.08-2.17 (m, 2H), 1.94-2.02 (m, 4H), 0.56-0.57 (m, 2H), 0.52-0.53 (m, 2H).
Crystalline complex DM was analyzed by X-ray powder diffraction using CuKα1 radiation. The diffraction pattern is shown inFIG. 31 and summarized in Table 1 (only peaks up to 30° in 2θ are listed). The melting point of complex DM was determined by differential scanning calorimetry (DSC) as 151±1° C. (evaluated as onset-temperature; heating from 50° C. to 200° C. at 10° C./min). The DSC spectrum is shown in FIG. 32.
Preparation of (3R,4R,5S,6R)-2-(4-chloro-3-(4-hydroxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Intermediate D)
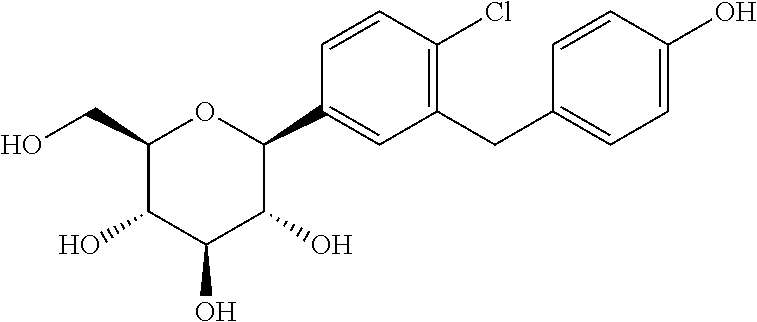
To a stirred solution of (3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Intermediate C) (2 g, 5.9 mmol) in dichloromethane was added BBr3 (14.6 mL, 1 M) dropwise at −78° C. After the addition was complete, the mixture was allowed to warm to 0° C. and held at this temperature for 2 h. When LC-MS showed that no starting material remained, the mixture was cooled to −78° C. again, and quenched with water. When the temperature was stable, saturated NaHCO3 solution was added. The mixture was evaporated under reduced pressure, and the residue was extracted with EtOAc. The organic layer was washed with NaHCO3 and brine, dried over Na2SO4, evaporated and purified to obtain intermediate D (0.7 g).
In addition, for use in the synthesis of certain compounds of the invention, the 2S isomer (intermediate D1) and the 2R isomer (intermediate D2) of intermediate D were separated by preparative LC-MS. Intermediate D1: 1H NMR (CD3OD): δ 7.30 (m, 3H), 6.97 (d, 2H, J=6.8 Hz), 6.68 (d, 2H, J=6.8 Hz), 4.56 (s, 1H), 4.16 (s, 1H), 3.91˜4.02 (m, 5H), 3.79 (m, 1H), 3.64 (m, 1H). Intermediate D2: 1H NMR (CD3OD): δ 7.29˜7.33 (m, 3H), 7.00 (d, 2H, J=6.8 Hz), 6.70 (d, 2H, J=6.8 Hz), 4.58 (d, 1H, J=4.0 Hz), 3.96˜4.02 (m, 4H), 3.93˜3.95 (m, 1H), 3.81˜3.85 (m, 1H), 3.64˜3.69 (m, 1H).
PATENT
http://www.google.com/patents/US20130267694

Example 14 Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol crystals
This example describes preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol by crystallization of ((2S,3R,4R,5S,6R)-2-(4-chloro-3-(442-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol bis(L-proline) complex in methanol/water solvent mixture.

(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (1.3 kg) was added to a propylene drum (25 L) and methanol (3.6 kg) and water (1.3 kg) and the mixture was stirred until the solids dissolved. The solution was filtered through filter membrane (Millipore, 0.45 μm) into a clean glass reactor (50 L). The mixture was refluxed for 30 min and water (7.2 kg) was added over 1.0 h while maintaining the temperature between 50 and 65° C. The mixture was slowly cooled to ˜42° C. over 2 h. A suspension of seed crystal (26 g) in cold (−5° C.) mixture of methanol/water (78 mL, 2.8/6.5 (w/w)) and the slow cooling was continued to −5° C. over 12 h. The suspension was stirred for another 5 h and was filtered. The solid was slurried with cold water and filtered (0 to 5° C., 3×2.6 kg). The filter cake was dried under reduced pressure for 24 h until the loss on drying was no more than 0.5% to give a white solid (825 g, 92% yield, 99.3% pure by \HPLC-0001).
Example 15 Preparation of 4-(2-Chloro-5-Iodobenzyl)Phenol
This example describes preparation of 4-(2-chloro-5-iodobenzyl)phenol using gaseous hydrobromic acid.

Preparation of (2-chloro-5-iodophenyl)methan-1-ol

A 250 mL of 4-necked flask equipped with thermometer and mechanical stirring was charged with NaBH4 (4.16 g, 0.11 mol) and THF (60 mL) under argon. After cooling to 0˜5° C. with stirring, a solution of iodine in THF (12.7 g I2 in 25 mL THF) was added slowly dropwise over 30 min and the reaction temperature was maintained below 10° C. After the addition was completed, a solution of 2-chloro-5-iodobenzoic acid (15.0 g, 50 mmol) in THF (20 mL) was added dropwise over 30 min and kept the reaction temperature below 10° C. After stirring for another 3 h at 20˜25° C., the reaction mixture was heated to reflux for additional 16 h and monitored by TLC (PE/EA=1:1, Rf=0.2). The mixture was cooled to 20˜25° C. and poured into ice water (100 mL), extracted with ethyl acetate (2×100 mL), washed with water (2×100 mL), brine (100 mL), concentrated and the residue was purified by flash chromatography (PE:EA=20:1 as eluant, 200 mL) to give an off-white solid. Yield: 10.0 g (70%) MS ESI (m/z): 269 [M+1]+.
Preparation of 4-(2-Chloro-5-Iodobenzyl)Phenol

A 100 mL of 4-necked flask equipped with thermometer and mechanical stirrer was charged with (2-chloro-5-iodophenyl)methanol (268.5 mg, 1 mmol), anhydrous ZnCl2 (136.3 mg, 1 mmol), dichloromethane (5.0 mL) and n-hexane (29 mL) under argon. After stirring for 10 min at 20 to 25° C., HBr (gas) was bubbled into the mixture for 10 min and a solution of phenol (197.6 mg, 2.1 mmol) in dry dichloromethane (3.0 mL) was added dropwise over 30 min. After bubbling HBr for additional 2 h, the mixture was refluxed for 3 days. The conversion was about 65%. The mixture was quenched with ice water (50 mL), extracted with ethyl acetate (2×30 mL), washed with water (2×30 mL), brine (30 mL), concentrated and the residue was purified by flash chromatography (PE:EA=25:1 as eluant, 200 mL) to give an off-white solid. Yield: 180 mg (52%). 1H NMR (CDCl3, 400 MHz): δ 7.44 (d, J=8.4 Hz, 2H), 7.03˜7.09 (m, 3H), 6.77 (d, J=8.4 Hz, 2H), 4.76 (s, 1H), 3.95 (s, 2H), 3.82 (s, 2H). MS ESI (m/z): 345 [M+1]+. 13C NMR (CDCl3, 100 MHz): δ 154.1, 141.4, 139.5, 136.6, 134.2, 131.2, 130.9, 130.1, 115.5, 91.67, 38.07.
Example 16 Preparation of 2-(4-(2-Cyclopropoxyethoxy)Benzyl)-1-Chloro-4-Iodobenzene
This example describes the preparation of 2-(4-(2-cyclopropoxyethoxy)benzyl)-1-chloro-4-iodobenzene via coupling of the 4-(2-chloro-5-iodobenzyl)phenol with 2-cyclopropoxyethyl 4-methylbenzenesulfonate.

Under nitrogen a 500 L glass-lined reactor was charged with acetone (123 kg) with stirring (120 RPM), 4-(2-chloro-5-iodobenzyl)phenol (19.37 kg, 0.056 kmol), 2-cyclopropoxyethyl 4-methylbenzenesulfonate (15.85 kg, 0.062 kmol), cesium carbonate (18.31 kg, 0.0562 kmol) powder, potassium carbonate (23.3 kg, 0.169 kmol) powder and TBAI (4.15 kg, 0.011 kmol). After stirring for 4045 h at 40° C., TLC (PE:EA=4:1, Rf=0.3) showed that starting material was consumed. The mixture was cooled to 20˜25° C.
The reaction mixture was filtered over diatomite (28 kg) and the filter cake was washed with acetone (2×31 kg). The combined filtrates were transferred to a 500 L glass-lined reactor and concentrated. The residue was dissolved in ethyl acetate (175 kg, washed with water (2×97 kg) and concentrated until the volume was about 100 L and was transferred to a 200 L glass-lined reactor and continued to concentrate to get about 22.5 kg of crude material.
The crude material was dissolved in methanol/n-hexane (10:1, 110 kg) under refluxing for 30 min with stirring (100 RPM) until it was a clear solution. The mixture was cooled to 5 to 10° C. and some crystal seeds (20 g) were added. The suspension was stirred for another 5 h at 5 to 10° C. The mixture was filtered at 0 to 5° C. and the filter cake was washed with pre-cooled methanol/n-hexane (10:1, 5° C., 2×11 kg). The filter cake was dried under at 15 to 20° C. for 15 h to give off-white to white solid. Yield: 18.1 kg, 75%. Melting Point: 31° C. (DSC onset). 1H NMR (CDCl3, 400 MHz): δ 7.45˜7.50 (m, 2H), 7.09˜7.12 (m, 3H), 6.88 (d, J=8.8 Hz, 2H), 4.11 (t, J=5.2 Hz, 2H), 3.99 (s, 2H), 3.88 (t, J=5.2 Hz, 2H), 3.40˜3.44 (m, 1H), 0.63˜0.67 (m, 2H), 0.49˜0.54 (m, 1H). MS ESI (m/z): 429 [M+1]+. 13C NMR (CDCl3, 100 MHz): δ 157.5, 141.5, 139.5, 136.6, 134.2, 131.2, 130.8, 129.9, 114.9, 91.66, 69.00, 67.13, 53.72, 38.08, 5.63.
Example 9 Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex
This example describes preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex by co-crystallization of ((2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol with L-proline in ethanol/water/n-heptane solvent mixture.
The crude (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (2.5 kg) was added to a glass reactor containing ethanol (95%, 16 kg) and L-proline (1.24 kg) and the mixture was refluxed for 1 h. While keeping the temperature above 60° C., n-heptane (8.5 kg) was added over 40 min. The mixture was slowly cooled to 25 to 20° C. and stirred at this temperature for 10 h. The mixture was filtered and the solids were washed with cold (−5° C.) ethanol (95%, 2×2.5 L) and n-heptane (2×5 L) and the solids were dried under reduced pressure at 55 to 65° C. for 20 h to give a white solid (3.03 kg, 81% yield, 99.4% pure by HPLC-0001).
Example 7 Preparation of ((2S,3R,4R,5S,6R)-2-(4-Chloro-3-(4-(2-Cyclopropoxyethoxy)Benzyl)Phenyl)-6-(Hydroxymethyl)Tetrahydro-2H-Pyran-3,4,5-triol
This example describes preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol by removal of the anomeric OH or OMe.

(2S,3R,4S,5S,6R)-2-(4-Chloro-3-(4-(2-Cyclopropoxyethoxy)Benzyl)Phenyl)-6-(Hydroxymethyl)-2-Methoxytetrahydro-2H-Pyran-3,4,5-Triol Solution
A 30 L glass reactor equipped with a thermometer was charged with crude (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (1.15 kg), DCM (2.3 kg) and acetonitrile (1.4 kg), and the mixture was magnetically stirred until all the solids dissolved under nitrogen sparging. The solution was cooled to ˜−15° C.
Triethylsilane Solution:
BF3.Et2O (1.2 kg) was added to a cold (−20 to −15° C.) solution of triethysilane (1.08 kg) dichloromethane (2.3 kg) and acetonitrile (1.4 kg) with nitrogen sparging.
The cold (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol solution was added to the cold triethylsilane solution at such a rate to maintain the temperature between −20 and −15° C. (˜2 to 3 h).
The reaction mixture was stirred for another 2 to 3 h and then quenched by addition of an aqueous solution of sodium bicarbonate (7.4% w/w, 7.8 kg) and the reaction mixture was stirred for about 15 min. The solvents were removed under reduced pressure (2 h, temperature below 40° C.). The residue was partitioned between ethyl acetate (6.9 kg) and water (3.9 kg). The layers were separated and the aqueous layer was extracted with ethyl acetate (2×3.5 kg). The combined organic layers were washed with brine (2×3.8 kg) and the solvents were removed under reduced pressure. Anhydrous ethanol (2.3 kg) was added and concentrated to give the crude product of the title compound (1 kg, 90% yield, 90% HPLC-0001) as yellow solid.
PATENT
WO 2011153953
https://www.google.com/patents/WO2011153953A1?cl=en
Example 1. Preparation of (2S.iR. R.5S.6R)-2-(4-chloro-3-(4-(2-cvclopropoxyethoxy) benzyl)phenyl)-6-(hvdroxymethyl)tetrahvdro-2H-pyran-3,4,5-triol, bis(X-proline) complex


Example 1A
Preparation of 2-cyclopropoxyethanol (1)

To a suspension of Mg powder (86.7 g, 3.6 mol) and iodine (cat) in anhydrous THF (0.7 L) was added slowly 1,2-dibromoethane (460 g, 2.4 mol) in anhydrous THF (2 L) slowly at a rate as to keep the internal temperature between 40-55 °C. After the addition, a solution of 2-(2-bromoethyl)-l,3-dioxolane (lOOg, 0.56 mol) in anhydrous THF (750 mL) was added dropwise. The reaction mixture was kept at 40-55 °C for 16h and was quenched by addition of aqueous solution of ammonium chloride. The mixture was extracted with methylene chloride. The organic layer was dried over sodium sulfate, and concentrated to give the title product (27 g) as yellow oil, which was directly used without further purification.
Example IB
Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (2)

To a stirred solution of sodium hydroxide (32 g, 0.8 mol) in water (180 mL) and THF (180 mL) was added Example 1A (27 g, 0.26 mol) at -5 to 0 °C. Afterwards, a solution of ji?-toluenesulfonyl chloride (52 g, 0.27 mol) in THF (360 mL) was added dropwise. The reaction mixture was kept at -5 to 0 °C for 16 h. The reaction mixture was then kept at room temperature for 30 min. The organic layer was separated and the aqueous layer was extracted with ethyl acetate (2×1.0 L). The combined organic layers were washed with brine, dried over Na2S04 and concentrated to get the crude product as yellow oil (53.3 g). It was used directly without further purification.
Example 1C
Preparation of 4-(5-bromo-2-chlorobenzyl)phenol (3)

To a stirred solution of 4-bromo-l-chloro-2-(4-ethoxybenzyl)benzene (747 g, 2.31 mol) in dichloromethane was added boron tribromide (1.15 kg, 4.62 mol) slowly at -78 °C. The reaction mixture was allowed to rise to room temperature. When the reaction was complete as measure by TLC, the reaction was quenched with water. The mixture was extracted with dichloromethane. The organic layer was washed with aqueous solution of saturated sodium bicarbonate, water, brine, dried over Na2S04, and concentrated. The residue was recrystallized in petroleum ether to give the title compound as a white solid (460 g, yield 68%). 1H NMR (CDC13, 400MHz): δ 7.23-7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.79 (d, J=8.8 Hz, 2H), 5.01 (s, 1H), 4.00 (s, 2H).
Example ID
Preparation of 4-bro -l-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (4)

A mixture of Example 1C (56.7 g, 210 mmol) and Cs2C03 (135 g, 420 mmol) in DMF (350 mL) was stirred at room temperature for 0.5 h. Example IB (53.3 g, 210 mmol) was added. The reaction mixture was stirred at room temperature overnight. It was diluted with water (3 L) and extracted with EtOAc. The organic layer was washed with water, brine, dried over Na2S04, and concentrated. The residue was purified by flash column
chromatography on silica gel eluting with petroleum ether:ethyl acetate (10:1) to give the title compound as liquid (51 g, yield 64%). 1H NMR (CDC13, 400MHz): δ 7.22-7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.88 (d, J=8.8 Hz, 2H), 4.10 (t, J=4.8 Hz, 2H), 3.86 (t, J=4.8 Hz, 2H), 3.38-3.32 (m, 1H), 0.62-0.66 (m, 2H), 0.49-0.52(m, 2H).
Example IE
Preparation of (25,5R, S,55,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6-(hydroxymethyl)-2-metlioxytetraliydro-2H-pyran-3,4,5-triol (5)

To a stirred solution of Example ID (213 g) in anhydrous THF/toluene (1 :2 (v/v), 1.7 L) under argon was added n-BuLi (2.5 M hexane, 245.9 mL) drop wise at -60 ± 5 °C. The mixture was stirred for 30 min. before transferred to a stirred solution of 2,3,4,6-tetra-O- trimethylsilyl-P-Z -glucolactone (310.5 g) in toluene (1.6 L) at -60 ± 5 °C. The reaction mixture was continuously stirred at -60 ± 5 °C for 1 h before quenching with aqueous solution of saturated ammonium chloride (1.5 L). Then mixture was allowed to warm to room temperature and stirred for 1 h. The organic layer was separated and the water layer was extracted with ethyl acetate (3×500 niL). The combined organic layers were washed with brine (1 L), dried over Na2S04, and concentrated. The residue was dissolved in methanol (450 mL) and methanesulfonic acid (9.2 mL) was added at 0 °C. The solution was allowed to warm to room temperature and stirred for 20 h. It was quenched with aqueous solution of sodium bicarbonate (50 g) in water (500 mL) and additional water (900 mL) was added. The mixture was extracted with ethyl acetate (3×1.0 L). The combined organic layers were washed with brine, dried over Na2S04, concentrated and used directly in the next step without further purification.
Example IF
Preparation of (25,5R, R,55,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(Z-proline) complex (7)

To stirred solution of Example IE in CH2C12/CH3CN (650 mL:650 mL) at -5 °C was added triethylsilane (28.2 mL, 563 mmol), and followed by BF3-Et20 (52.3 mL, 418.9 mmol). The reaction was stirred for 16 h while the temperature was allowed to warm to room temperature gradually. The reaction was quenched with aqueous solution of saturated sodium bicarbonate to pH 8.0. The organic volatiles were removed under vacuum. The residue was partitioned between ethyl acetate (2.25 L) and water (2.25 L). The organic layer was separated, washed with brine, dried over Na2S04 and concentrated to give the crude product 6 (230 g, purity 82.3%). This product and L-proline (113.7 g) in EtOH/H20 (15:1 v/v, 2.09 L) was stirred at 80 °C for 1 h when it became a clear solution. Hexane (3.0 L) was added dropwise into the above hot solution over 50 min, with the temperature being kept at about 60 °C. The reaction mixture was stirred overnight at room temperature. The solid was filtered and washed with EtOH/ H20 (15:1 (v/v), 2×300 mL), hexane (2×900 mL), and dried at 45 °C under vacuum for 10 h to give the pure title compound 7 as a white solid (209 g).
Purity (HPLC) 99.2% (UV). 1H NMR (CD3OD, 400 MHz): δ 7.25—7.34 (m, 3H), 7.11 (d, J = 8.8 Hz, 2H), 6.84 (d, J= 8.8 Hz, 2H), 4.03-4.11 (m, 5H), 3.96-4.00 (m, 2H), 3.83-3.90 (m, 3H), 3.68-3.72 (m, 1H), 3.36-3.46 (m, 6H), 3.21-3.30 (m, 3H), 2.26-2.34 (m, 2H), 2.08-2.17 (m, 2H), 1.94-2.02 (m, 4H), 0.56-0.57 (m, 2H), 0.52-0.53(m, 2H).
Example 2. Direct Preparation of Crystalline Compound 8 from Complex 7
This example illustrates the preparation of a crystalline form of (2S, 3R, 4R, 5S, 6R)-2- (4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H- pyran-3,4,5-triol.

To a 5.0 L 4-necked flask equipped with a mechanical stirrer was added the starting co-crystal (150.0 g) and methanol (300 mL). The mixture was stirred at room temperature with mechanical stirring (anchor agitator, 2-blades 9 cm) until a cloudy solution/suspension formed, to which distilled water (1500 mL) was added dropwise at a rate of -12.5 mL/min. As the mixture warmed from the exotherm of adding water to methanol, the mixture became clear after adding about 1/5 to 1/3 of the water. After the addition was completed the reaction was stirred continuously at 80 rpm for another 5 h. The reaction mixture was filtered over medium-speed filter paper and the filter cake was washed with distilled water (450 mL and then 300 mL) and dried under vacuum using an oil pump (~6 mm Hg) at 45 °C for 48 hours to give the target product as a white crystalline solid (94.2 g, 93.9% yield, purity (HPLC): 99.3%).
Example 5. Indirect Preparation of Crystalline Compound 8 from Complex 7

[0113] To a 200 L glass lined reactor equipped with a double-tier paddle agitator and a glass condenser was added sequentially complex 7 (7.33 kg), ethyl acetate (67.5 kg) and pure water (74.0 kg). The mixture was heated to reflux and stirred at reflux for 30 min. The reaction mixture was cooled to approximately 50 °C and the organic layer was separated and the aqueous layer was extracted with ethyl acetate (34.0 kg). The combined organic layers were washed with pure water (3×74.0 kg) (IPC test showed that the IPC criteria for L-proline residue was met after three water washes). The mixture was concentrated at 40 °C under vacuum (-15 mmHg) for 3 h until the liquid level dropped below the lower-tier agitator paddle. The mixture (18 kg) was discharged and transferred to a 20L rotary evaporator. The mixture was concentrated under vacuum (40 °C, ~5 mmHg) to a minimum volume. The remaining trace amount of ethyl acetate was removed azeotropically at 40 °C under vacuum with methanol (10 kg). The residue was dried under vacuum of an oil pump (~6 mmHg) at 40 °C for 10 h to give 8 as a white amorphous solid (4.67 kg, purity (HPLC): 99.2%) which was used in the next step without further purification.
The recrystallization was accomplished by the following steps. To a 100 L glass line reactor equipped with a double-tier paddle agitator and a glass condenser was added the above amorphous 8 (4.67 kg) and methanol (18.0 kg). The mixture was refluxed at 70 °C for 30 min until a clear solution formed, to which pure water (45.0 kg) was added over 2 hours. After the addition was completed (the reaction temperature was 41 °C), the reaction mixture was cooled to room temperature and stirred at room temperature for 15 hours. The reaction mixture was filtered and the wet cake was washed with pure water (2×15 kg) and dried under vacuum at 55-60 °C for 12 hours to give the target product as an off-white crystalline solid (3.93 kg, yield: 84% in two steps; purity (HPLC): 99.7%).
Example 6. Direct Preparation of Crystalline Compound 8 from Amorphous 8

A 5 L 4-neck flask was charged with 8 (amorphous), 116 g, and methanol (580 mL). The reaction mixture was heated to 60 C with mechanical stirring and the solution became clear. Water (2320 mL) was added dropwise to the reaction solution at 40 mL/min at 50 °C. The reaction mixture was stirred overnight at room temperature. The reaction mixture was filtered and the filter cake was washed with water (2×200 mL), dried under vacuum at 55 °C for 12 hours, to afford white crystalline 8. Yield is 112.8 g (97.2%).
References:
1. Clinical Trial, A Dose Range Finding Study to Evaluate the Effect of Bexagliflozin Tablets in Subjects With Type 2 Diabetes Mellitus. NCT02390050 (retrieved on 26-03-2015).
| WO2008144346A2 * | May 15, 2008 | Nov 27, 2008 | Squibb Bristol Myers Co | Crystal structures of sglt2 inhibitors and processes for their preparation | |||||||||||||||
| WO2009026537A1 * | Aug 22, 2008 | Feb 26, 2009 | Theracos Inc | Benzylbenzene derivatives and methods of use | |||||||||||||||
| CN1407990A * | Oct 2, 2000 | Apr 2, 2003 | 布里斯托尔-迈尔斯斯奎布公司 | C-aryl glucoside sgltz inhibitors
|
| WO2010022313A2 * | Aug 21, 2009 | Feb 25, 2010 | Theracos, Inc. | Processes for the preparation of sglt2 inhibitors |
////////BEXAGLIFLOZIN, APPROVALS 2023, FDA 2023
c1cc(ccc1Cc2cc(ccc2Cl)[C@H]3[C@@H]([C@H]([C@@H]([C@H](O3)CO)O)O)O)OCCOC4CC4
SYN
https://doi.org/10.1021/acs.jmedchem.4c02079J.Med.Chem.2025,68,2147−2182
Bexagliflozin (Brenzavvy). Bexagliflozin (3) was discoveredanddevelopedbyTheracosBioforthetreatmentof
type2diabetesmellitus.28Bexagliflozinisasodium-dependent glucose cotransporter 2 (SGLT2) inhibitor. Inhibition of SGLT2 reduces blood sugar without stimulating insulin release.29 Bexagliflozin shows >2000-fold selectivity forSGLT2 over SGLT1 and demonstrated improvement inglycemiccontrolwithaoncedaily,20mgdose.28Since 2011, there have been 11 therapeutics targeting
SGLT2.30Thesedrugsexhibit commonstructural features(abiarylmethaneandglycoside)andlikelyfacesimilarsynthetic challenges.31 The medicinal chemistry efforts to identifybexagliflozinweredisclosedintheprimaryliterature.32Apatent fromTheracos, Inc. in2013describedasyntheticapproachto bexagliflozinonmultikilogramscale.33Slightvariations inthe
reactionconditions,yieldandisolationstrategyofintermediates wereincludedinthepatent.Theimplementationoftelescoping intheprocessislikelyduetopoorcrystallinityofintermediates,
whichmaybeacommonchallengetootherSGLT2inhibitors.31
Anotherpatent disclosedbyPiramal Enterprises suggesteda
similarbondformationstrategybut includedanacetylationof bexagliflozinprior tothefinal isolation inorder toprovidea crystallinesolid.34
Bexagliflozinwas assembled by cryogenicmetal halogen exchangeof aryl iodide3.1with turboGrignard(i-PrMgCl·LiCl)andsubsequentadditiontoprotectedgluconolactone3.2
whichwaspreparedbytreatmentofD-(+)-glucono-1,4-lactonewithTMSClandNMMinTHFin94%yield(Scheme4).WhentheGrignardadditionwascomplete,thereactionwasquenchedand a solution of the product inEtOAcwas treatedwith
activated carbon, filtered, concentrated, and diluted with methanol.ThissolutionwastreatedwithconcentratedHCl to remove thesilyl protectinggroupsandprovidecrudemethyl ketal3.3inyields rangingfrom79to95%.Themethyl ketal
functionalitywasreducedusingtriethylsilaneandBF3·Et2Oin DCMandMeCNatcryogenictemperaturestoprovidecrude bexagliflozin (3) as a solid after concentrating the reaction mixture. Alternatively, a larger-scale demonstration of this processinthepatenttelescopedasolutionofcrudebexagliflozin toformabis-L-prolinecomplexinethanol,water,andheptane,
whichwasisolatedasacrystallinesolidin81%yield.Thiswas convertedto the free formin82%yieldbycrystallization in methanolandwater.Arecrystallizationofbexagliflozin(3)was
reported in 92% yield. Details on stereoselectivity of this
approachwerenotdisclosed.
Amilligram-togram-scaleconstructionofthearyliodide3.1 wasalsodisclosedintheTheracospatent from2013(Scheme 5).33First,carboxylicacid3.5wasreducedtoprimaryalcohol
3.6using sodiumborohydride and iodine. Next, the diaryl methanecorewas assembledbyFriedel−Crafts alkylationof phenol with3.6 after activationwithHBr andZnCl2. This reactionwasdemonstratedonmilligramscaleandachieved65% conversion, with 52% isolated yield after chromatographic purification.Analternativeapproachtoabromovariantofaryl iodide3.7waspresentedina2009patentfromTheracos,where Friedel−Craftsacylationprovidedtheanalogousbenzophenone intermediatewhichwas thensubsequentlyreduced.35Finally,alkylationofthephenolwasconductedusingthetosylatedether
3.8toprovidearyl iodide3.1in75%yieldonkilogramscale.A syntheticapproachtothetosylatedetherwasprovidedinthe earlyTheracospatent,35wherecyclopropylether formationin 3.10wasgeneratedviaGrignardformationandrearrangement of 2-(2-bromoethyl)-1,3-dioxolane 3.9 (Scheme 6). The primary alcohol 3.10was protectedas the tosylate3.8and employedinthealkylationstepwithoutpurification.Noyields wereprovided.

(28) Hoy, S. M. Bexagliflozin: first approval. Drugs 2023, 83, 447−
453.
(29) Hsia, D. S.; Grove, O.; Cefalu, W. T. An update on sodium
glucose co-transporter-2 inhibitors for the treatment of diabetes
mellitus. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 73−79.
(30) Guo, Y.-Y.; Zhang, J.-Y.; Sun, J.-F.; Gao, H. A comprehensive
review of small-molecule drugs for the treatment of type 2 diabetes
mellitus: Synthetic approaches and clinical applications. Eur. J. Med.
Chem. 2024, 267, No. 116185.
(31) Aguillón, A. R.; Mascarello, A.; Segretti, N. D.; de Azevedo, H. F.
Z.; Guimaraes, C. R. W.; Miranda, L. S. M.; de Souza, R. O. M. A.
Synthetic strategies toward SGLT2 inhibitors. Org. Process Res. Dev.
2018, 22, 467−488.
(32) Xu, B.; Feng, Y.; Cheng, H.; Song, Y.; Lv, B.; Wu, Y.; Wang, C.;
Li, S.; Xu, M.; Du, J.; et al. C-aryl glucosides substituted at the 4′
position as potent and selective renal sodium-dependent glucose co
transporter 2 (SGLT2) inhibitors for the treatment of type 2 diabetes.
Bioorg. Med. Chem. Lett. 2011, 21, 4465−4470.
(33) Xu, B.; Lv, B.; Xu, G.; Seed, B.; Roberge, J. Y. Process for the
preparation of benzyl-benzene C-glycosides via coupling reaction as
potential SGLT2 inhibitors. US 20130267694, 2013.
(34) Gharpure, M.; Sharma, S. K.; Vishwasrao, S.; Vichare, P.; Varal,
D. Aprocess for the preparation of SGLT2 inhibitor and intermediates
thereof. WO 2018207113, 2018.
(35) Song, Y.; Chen, Y.; Cheng, H.; Li, S.; Wu, Y.; Feng, Y.; Lv, B.; Xu,
B.; Seed, B.; Hadd, M. J.; et al. Preparation of benzylbenzene glycoside
derivatives as antidiabetic agents. WO 2009026537, 2009.
.
European Journal of Medicinal Chemistry
Volume 265, 5 February 2024, 116124
https://doi.org/10.1016/j.ejmech.2024.116124
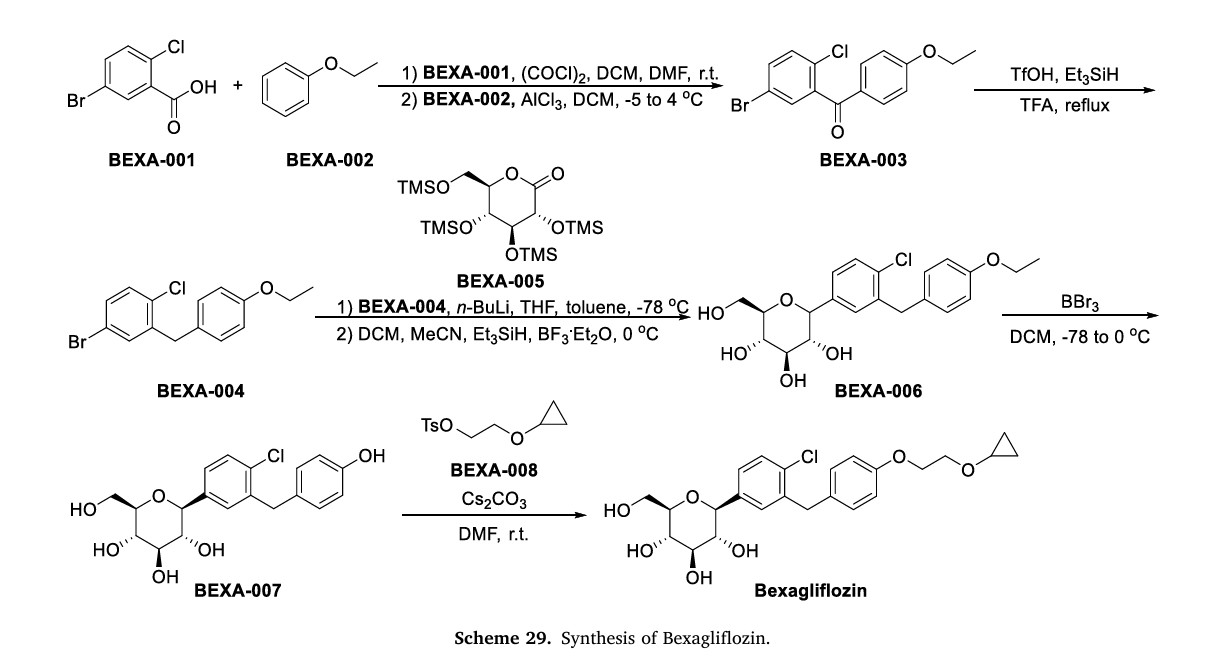
Bexagliflozin (Brenzavvy)
On January 20, 2023, the FDA granted approval to Bexagliflozin, a medication developed by Theracos Inc, for the treatment of type 2 diabetes mellitus (T2DM) [104–106]. The SGLT2 inhibitor Bexagliflozin
can increase energy expenditure, reduce fluid retention, and increase urinary glucose excretion by inhibiting SGLT2 in renal tubular epithelial cells [106]. SGLT2 inhibitors have significant advantages compared to other drugs: (1) they can lower both pre-meal and post-meal blood sugar levels (not all drugs can lower both); (2) they have a lower risk of hypoglycemia as they do not stimulate insulin secretion; (3) they have adiuretic effect due to their primary action on the renal tubules, which
lowers systolic blood pressure; (4) research has shown that SGLT2 in hibitors have therapeutic effects on diabetic kidney disease [107,108].
The process of synthesizing Bexagliflozin started by conducting theFriedel-Crafts acylation of ethoxybenzene (BEXA-002) with 5-bromo-2-chlorobenzoic acid (BEXA-001) (Scheme 29) [109]. This reaction produced ketone BEXA-003. Subsequently, the carbonyl reduction of BEXA-003 was carried out using trifluoromethanesulfonic acid (TfOH),triethylsilane, and TFA. This step yielded BEXA-004. Next, n-butyllithium (n-BuLi) and pyrone BEXA-005 were combined with BEXA-004 at78◦C. This reaction produced an intermediate, which was thenreacted with triethylsilane and BF◦3⋅Et2O at 0C. The final product obtained from this reaction was BEXA-006, which contained a sugar ring.
BEXA-006 underwent dealkylation upon treatment with boron tribromide, resulting in the formation of BEXA-007, which was a phenol.
Subsequently, BEXA-007 was alkylated using 2-cyclopropoxyethyl4-methylbenzenesulfonate (BEXA-008) to yield Bexagliflozin.
[104] S.M. Hoy, Bexagliflozin: first approval, Drugs 83 (2023) 447–453.
[105] W. Zhang, A. Welihinda, J. Mechanic, H. Ding, L. Zhu, Y. Lu, Z. Deng, Z. Sheng,
B. Lv, Y. Chen, J.Y. Roberge, B. Seed, Y.X. Wang, EGT1442, a potent and selectiveSGLT2 inhibitor, attenuates blood glucose and HbA(1c) levels in db/db mice and
prolongs the survival of stroke-prone rats, Pharmacol. Res. 63 (2011) 284–293.
[106] O. Azzam, R. Carnagarin, L.M. Lugo-Gavidia, J. Nolde, V.B. Matthews, M.
P. Schlaich, Bexagliflozin for type 2 diabetes: an overview of the data, Expet Opin.
Pharmacother. 22 (2021) 2095–2103.
[107] B.F. Palmer, D.J. Clegg, Kidney-protective effects of SGLT2 inhibitors, Clin. J. Am.
Soc. Nephrol. 18 (2023) 279–289.
[108] M. Singh, A. Kumar, Risks associated with SGLT2 inhibitors: an overview, Curr.
Drug Saf. 13 (2018) 84–91.
[109] Y. Song, Y. Chen, H. Cheng, S. Li, Y. Wu, Y. Feng, B. Lv, B. Xu, B. Seed, M.J. Hadd,
J. Du, C. Wang, J.Y. Roberge, Preparation of Benzylbenzene Glycoside Derivatives
as Antidiabetic Agents, 2009. WO2009026537A1.
.



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
Momelotinib


Momelotinib
414.47, C23H22N6O2,
1056634-68-4
FDA 2023, Ojjaara,
| To treat intermediate or high-risk myelofibrosis in adults with anemia Drug Trials Snapshot |
N-(Cyanomethyl)-4-[2-(4-morpholin-4-ylanilino)pyrimidin-4-yl]benzamide
N-(Cyanomethyl)-4-[2-[4-(4-morpholinyl)phenylamino]pyrimidin-4-yl]benzamide
Jak2 tyrosine kinase inhibitor; Jak1 tyrosine kinase inhibitor
Inflammatory disease; Myelofibrosis; Myeloproliferative disorder; Pancreatic ductal adenocarcinoma; Polycythemia vera
CYT 387; CYT-387; momelotinib)
GS-0387
CYT387 sulfate saltCAS No: 1056636-06-6
 CYT387 Mesylate CAS No: 1056636-07-7
CYT387 Mesylate CAS No: 1056636-07-7
DI HCL SALT 1380317-28-1
Momelotinib, sold under the brand name Ojjaara among others, is an anticancer medication used for the treatment of myelofibrosis.[5] It is a Janus kinase inhibitor and it is taken by mouth.[5]
The most common adverse reactions include dizziness, fatigue, bacterial infection, hemorrhage, thrombocytopenia, diarrhea, and nausea.[8]
Momelotinib was approved for medical use in the United States in September 2023,[5][8][9] and in the European Union in January 2024.[6][10]
CYT387 is an ATP-competitive small molecule JAK1 / JAK2 inhibitor with IC50 of 11 and 18 nM for JAK1 and JAK2, respectively. CYT387 is useful for treatment of myeloproliferative disorders and anti-cancer.
CYT-387 is a potent, orally administered JAK1/JAK2/ Tyk2 inhibitor in phase III clinical studiest at Gilead for the treatment of post-polycythemia vera, for the treatment of primary myelofibrosis and for the treatment of post-essential thrombocythemia. Phase II studies are also ongoing, in combination with gemcitabine and nab-paclitaxel, in adults with untreated metastatic pancreatic ductal adenocarcinoma.
The compound possesses an excellent selectivity and safety profile. In 2010 and 2011, orphan drug designation was assigned by the FDA and the EMA, respectively, for the treatment of myelofibrosis. In 2011, orphan drug designation was assigned by the EMA for the treatment of post-essential thrombocythemia myelofibrosis and for the treatment of post-polycythemia vera myelofibrosis.
PAT
http://www.google.com.ar/patents/US8486941?cl=ja
N-(cyanomethyl)-4-(2-(4-morpholinophenylamino)pyrimidin-4-yl)benzamide
| 3 |
 |
414.18 | 1H NMR (300 MHz, d6-DMSO): δ 9.47 (1 H, s), 9.32 (1 H, t, J = 5.5 Hz), 8.54 (1 H, d, J = 5.0 Hz), 8.27 (2 H, d, J = 8.7 Hz), 8.02 (2 H, d, J = 8.2 Hz), 7.67 (2 H, d, J = 9.1 Hz), 7.41 (1 H, d, J = 5.5 Hz), 6.93 (2 H, d, J = 9.1 Hz), 4.36 (2 H, d, J = 5.5 Hz), 3.75 (4 H, m), 3.05 (4 H, m). | m/z 415.3 [M + H]+ | N-(cyanomethyl)-4-(2-(4- morpholinophenylamino)pyrimidin- 4-yl)benzamide |
Example 1Synthesis of Compound 3
A mixture of 4-ethoxycarbonylphenyl boronic acid (23.11 g, 119 mmol), 2,4-dichloropyrimidine (16.90 g, 113 mmol), toluene (230 mL) and aqueous sodium carbonate (2 M, 56 mL) was stirred vigorously and nitrogen was bubbled through the suspension for 15 minutes. Tetrakis(triphenylphosphine)palladium[0] (2.61 g, 2.26 mmol) was added. Nitrogen was bubbled through for another 10 min., the mixture was heated to 100° C., then at 75° C. overnight. The mixture was cooled, diluted with ethyl acetate (200 mL), water (100 mL) was added and the layers were separated. The aqueous layer was extracted with ethyl acetate (100 ml) and the two organic extracts were combined. The organics were washed with brine, filtered through sodium sulfate, concentrated, and the resultant solid was triturated with methanol (100 mL) and filtered. The solids were washed with methanol (2×30 mL) and air dried. This material was dissolved in acetonitrile (150 mL) and dichloromethane (200 mL), stirred with MP.TMT Pd-scavenging resin (Agronaut part number 800471) (7.5 g) over 2 days. The solution was filtered, the solids were washed with dichloromethane (2×100 mL), and the filtrate concentrated to give ethyl 4-(2-chloropyrimidin-4-yl)benzoate as an off-white solid (17.73 g, 60%)—additional washing with dichloromethane yielded a further 1.38 g and 0.5 g of product. 1H NMR (300 MHz, d6-DMSO) δ 8.89 (1H, d, J=5.0 Hz); 8.32 (2H, d, J=8.7 Hz); 8.22 (1H, d, J=5.5 Hz); 8.12 (2H, d, J=8.7 Hz); 4.35 (2H, q, J=7.1 Hz); 1.34 (3H, t, J=7.1 Hz); LC-ESI-MS (method B): rt 7.3 min.; m/z 263.0/265.0 [M+H]+.
A mixture of ethyl 4-(2-chloropyrimidin-4-yl)benzoate (26.15 g, 99.7 mmol) and 4-morpholinoaniline (23.10 g, 129.6 mmol) was suspended in 1,4-dioxane (250 mL). p-Toluenesulfonic acid monohydrate (17.07 g, 89.73 mmol) was added. The mixture was heated at reflux for 40 h., cooled to ambient temperature, concentrated then the residue was partitioned between ethyl acetate and 1:1 saturated sodium bicarbonate/water (1 L total). The organic phase was washed with water (2×100 mL) and concentrated. The aqueous phase was extracted with dichloromethane (3×200 mL). The material which precipitated during this workup was collected by filtration and set aside. The liquid organics were combined, concentrated, triturated with methanol (200 mL) and filtered to yield additional yellow solid. The solids were combined, suspended in methanol (500 mL), allowed to stand overnight then sonicated and filtered. The solids were washed with methanol (2×50 mL) to give, after drying, ethyl 4-(2-(4-morphonlinophenylamino)pyrimidin-4-yl)benzoate (35.39 g, 88%). 1H NMR (300 MHz, d6-DMSO) δ 9.49 (1H, s); 8.54 (1H, d, J=5.0 Hz); 8.27 (2H, d, J=8.7 Hz); 8.10 (2H, d, J=8.7 Hz), 7.66 (2H, d, J=9.1 Hz); 7.38 (1H, d, J=5.0 Hz); 6.93 (2H, d, J=8.7 Hz); 4.35 (2H, q, J=6.9 Hz), 3.73 (4H, m); 3.04 (4H, m); 1.34 (3H, t, J=6.9 Hz); LC-ESI-MS (method B): rt 7.5 min.; m/z 404.1 [M+H].
A solution of ethyl 4-(2-(4-morpholinophenylamino)pyrimidin-4-yl)benzoate (35.39 g, 87.6 mmol) in 3:1 methanol/tetrahydrofuran (350 mL) was treated with lithium hydroxide (4.41 g, 183.9 mmol) in water (90 mL). The mixture was heated at reflux for 2 h., cooled, concentrated and acidified with hydrochloric acid (2M, 92.5 mL, 185 mmol). The dark precipitate was filtered, washed with water, and dried under vacuum. The solid was ground to a powder with a mortar and pestle, triturated with methanol (500 mL) then filtered again to yield 4-(2-(4-morpholinophenylamino)pyrimidin-4-yl)benzoic acid as a muddy solid. This material was washed with ether, air dried overnight, and ground to a fine powder with mortar and pestle. On the basis of mass recovery (34.49 g) the yield was assumed to be quantitative. 1H NMR (300 MHz, d6-DMSO) δ 9.47 (1H, s); 8.53 (1H, d, J=5.2 Hz); 8.24 (2H, d, J=8.5 Hz); 8.08 (2H, d, J=8.8 Hz), 7.66 (2H, d, J=9.1 Hz); 7.37 (1H, d, J=5.2 Hz); 6.93 (2H, d, J=9.1 Hz); 3.73 (4H, m); 3.04 (4H, m). LC-ESI-MS (method C): rt 7.3 min.; m/z 377.1 [M+H]+.
To a suspension of 4-(2-(4-morpholinophenylamino)pyrimidin-4-yl)benzoic acid (theoretically 32.59 g, 86.6 mmol) in DMF (400 mL) was added triethylamine (72.4 mL, 519.6 mmol, 6 eq.) The mixture was sonicated to ensure dissolution. Aminoacetonitrile hydrochloride (16.02 g, 173.2 mmol) was added followed by N-hydroxybenzotriazole (anhydrous, 14.04 g, 103.8 mmol) and 1-ethyl-3-(dimethylaminopropyl)carbodiimide hydrochloride (19.92 g, 103.8 mmol). The suspension was stirred vigorously overnight. The solvent was evaporated under reduced pressure, the residue was diluted with 5% sodium bicarbonate (400 mL) and water (300 mL), giving a yellow solid, which was broken up and filtered. The solids were washed several times with 100 mL portions of water, triturated with hot methanol/dichloromethane (500 mL, 1:1), concentrated to a volume of approximately 300 mL), cooled and filtered. The solids were washed with cold methanol (3×100 mL), ether (200 mL) and hexane (200 mL) prior to drying to afford
Compound 3 (31.69 g, 88%). M.p. 238-243° C.
Microanalysis: Found C, 66.52; H, 5.41; N, 20.21. C23H26N6O10S2 requires C, 66.65; H, 5.35; N, 20.28%.
13C NMR (75.5 MHz, d6-DMSO) δ 166.04, 162.34, 160.26, 159.14, 146.14, 139.87, 134.44, 132.73, 127.80, 126.84, 120.29, 117.49, 115.50, 107.51, 66.06, 49.16, 27.68.
1H NMR GIVEN ABOVE
Example 6Salt Formation from Compound 3
Compound 3 (10.0 g) was suspended in methanol (1 L). Concentrated sulfuric acid (10.52 g, 90% w/w) was added dropwise to the stirring solution. A clear brown solution resulted and a solid lump formed. The solution was filtered quickly then allowed to continue stirring for 3 h (a second precipitate appeared within minutes). After this time the pale yellow precipitate was collected by filtration, washed with methanol (10 mL) then dried under vacuum overnight to afford 4-(4-(4-(4-(cyanomethylcarbamoyl)phenyl)pyrimidin-1-ium-2-ylamino)phenyl)morpholin-4-ium hydrogensulfate, as a pale yellow solid (10.20 g, 69%). m.p. 205° C. Microanalysis: Found C, 45.18; H, 4.36; N, 13.84; S, 10.24. C23H26N6O10S2 requires C, 45.24; H, 4.29; N, 13.76; S 10.50%. 1H NMR (300 MHz, d6-DMSO) δ 9.85 (br. s, 1H), 9.34 (t, J=5.4 Hz, 1H), 8.59 (d, J=5.2 Hz, 1H), 8.27 (d, J=8.5 Hz, 2H), 8.03 (d, J=8.5 Hz, 2H), 7.83 (d, J=8.4 Hz, 2H), 7.50 (d, J=5.2 Hz, 1H), 7.34 (br. s, 2H), 4.36 (d, J=5.4 Hz, 2H), 3.89 (br. s, 4H), 3.37 (br. s, 4H); 13C NMR (75.5 MHz, d6-DMSO) δ 166.07, 163.36, 159.20, 158.48, 140.19, 139.34, 136.45, 134.89, 128.00, 127.22, 121.13, 119.89, 117.59, 109.05, 64.02, 54.04, 27.82. LC-ESI-MS (method D): rt 10.0 min.; m/z 415.1 [M+H]+.
Compound 3 (0.25 g) was suspended in methanol (25 ml). Methane sulfonic acid (0.255 g) was added dropwise to the stirring solution and a clear brown solution resulted. The solution was allowed to stir for 3 h, after which the volume was reduced to 9 ml. The resultant precipitate was collected and dried under vacuum for 8 h to afford 4-(4-(4-(4-(cyanomethylcarbamoyl)phenyl)pyrimidin-1-ium-2-ylamino)phenyl)morpholin-4-ium methanesulfonate as a pale yellow solid (0.22 g). m.p. 208° C. 1H NMR (300 MHz, d6-DMSO) δ 9.83 (br. s, 1H), 9.35 (t, J=5.3 Hz, 1H), 8.59 (d, J=5.1 Hz, 1H), 8.28 (d, J=8.5 Hz, 2H), 8.04 (d, J=8.5 Hz, 2H), 7.83 (d, J=9.0 Hz, 2H), 7.50 (d, J=5.5 Hz, 1H), 7.31 (d, J=9.0 Hz, 2H), 4.36 (d, J=5.5 Hz, 2H), 3.88 (m, 4H), 3.35 (br. s, 4H), 2.36 (s, 6H); LC-ESI-MS (method D): rt 10.2 min.; m/z 415.3 [M+H]+.
Compound 3 (0.50 g) was suspended in methanol (45 ml). A freshly prepared solution of hydrochloric acid in methanol (2.6 ml, HCl conc. 40 mg/ml) was added dropwise to the stirring solution and a clear brown solution resulted. The solution was allowed to stir for 2 h, then the resultant precipitate was collected, washed with methanol (5 ml) and dried under vacuum for 8 h to afford 4-(4-(4-(4-(cyanomethylcarbamoyl)phenyl)pyrimidin-1-ium-2-ylamino)phenyl)morpholin-4-ium chloride a pale yellow solid (0.30 g). m.p. 210° C. 1H NMR (300 MHz, d6-DMSO) 1H NMR (300 MHz, DMSO) δ 9.92 (br. s, 1H), 9.42 (t, J=5.3, 1H), 8.62 (d, J=4.8, 1H), 8.29 (d, J=8.1, 2H), 8.06 (d, J=8.1, 2H), 7.89 (d, J=9.0, 2H), 7.53 (br. s, 3H), 4.36 (d, J=5.4, 2H), 3.82 (br. s, 4H), 3.43 (br. s, 4H)
LC-ESI-MS (method D): rt 10.3 min.; m/z 415.3 [M+H]+.
PAT
WO 2014114274
References on CYT387
. [1] A Pardanani et al CYT387, a Selective JAK1 / JAK2 inhibitor: in vitroassessment of kinase selectivity and preclinical s using Cell lines and Primary cells from polycythemia vera Patients Leukemia (2009) 23, 1441-1445
Abstract
Somatic mutations in Janus kinase 2 (JAK2), including JAK2V617F, result in dysregulated JAK-signal transducer and activator transcription (STAT) signaling, which is implicated in myeloproliferative neoplasm (MPN) pathogenesis. CYT387 is an ATP-competitive small molecule that potently inhibits JAK1 / JAK2 kinases ( IC (50) = 11 and 18 nM, respectively), with significantly less activity against other kinases, including JAK3 (IC (50) = 155 nM). CYT387 inhibits growth of Ba / F3-JAK2V617F and human erythroleukemia (HEL) cells ( IC (50) approximately 1500 nM) or Ba / F3-MPLW515L cells (IC (50) = 200 nM), but has considerably less activity against BCR-ABL harboring K562 cells (IC = 58 000 nM). Cell lines harboring mutated JAK2 alleles (CHRF-288-11 or Ba / F3-TEL-JAK2) were inhibited more potently than the corresponding pair harboring mutated JAK3 alleles (CMK or Ba / F3-TEL-JAK3), and STAT-5 phosphorylation was inhibited in HEL cells with an IC (50) = 400 nM. …
[2]. Tyner Jeffrey W. et al CYT387, a novel JAK2 inhibitor, induces Hematologic Responses and normalizes inflammatory cytokines in murine myeloproliferative neoplasms Blood June 24, 2010vol. no 115. 255232-5240
Abstract
Activating alleles of Janus kinase 2 (JAK2) SUCH as JAK2 (V617F) are Central to the pathogenesis of myeloproliferative neoplasms (MPN), suggesting Small molecule inhibitors targeting JAK2 That May be therapeutically Useful. IDENTIFIED We have an aminopyrimidine derivative ( CYT387), which inhibits JAK1, JAK2, and tyrosine kinase 2 (TYK2) at low nanomolar concentrations, with few additional targets. Between 0.5 and 1.5muM CYT387 caused growth suppression and apoptosis in JAK2-dependent hematopoietic cell lines, while nonhematopoietic cell lines were unaffected. In a murine MPN model, CYT387 normalized white cell counts, hematocrit, spleen size, and restored physiologic levels of inflammatory cytokines. Despite the hematologic responses and reduction of the JAK2 (V617F) allele burden, JAK2 (V617F) cells persisted and MPN recurred upon cessation of treatment, suggesting JAK2 inhibitors That May be Unable to Eliminate JAK2 (V617F) cells, Consistent with Preliminary results from Clinical Trials of JAK2 inhibitors in myelofibrosis. …
[3]. Sparidans RW, Durmus S, Xu N, Schinkel AH, Schellens JH, Beijnen JH.Liquid chromatography-tandem mass spectrometric assay for the JAK2 inhibitor CYT387 in plasma.J Chromatogr B Analyt Technol Biomed Life Sci 2012 May 1; 895-896:. 174-7 Epub 2012 Mar 23..
abstract
A quantitative bioanalytical Liquid Chromatography-Tandem Mass spectrometric (LC-MS / MS) assay for the JAK2 inhibitor CYT387 WAS Developed and validated. Plasma samples Were Treated using pre-Protein precipitation with acetonitrile containing cediranib as Internal Standard. The extract WAS Directly Injected into the chromatographic system after dilution with water. This system consisted of a sub-2 μm particle, trifunctional bonded octadecyl silica column with a gradient using 0.005% (v / v) of formic acid in a mixture of water and methanol. The eluate was transferred into the electrospray interface with positive ionization and the analyte was detected in the selected reaction monitoring mode of a triple quadrupole mass spectrometer. The assay was validated in a 0.25-1000 ng / ml calibration range. Within day precisions were 3.0-13.5%, BETWEEN Day Precisions 5.7% and 14.5%. Accuracies Were BETWEEN 96% and 113% for the Whole Calibration range. The Drug WAS stable under All Relevant Analytical Conditions. Finally, the assay successfully WAS Used to ASSESS Drug Levels in mice.
[4] . Monaghan KA, Khong T, Burns CJ, Spencer A.The novel JAK inhibitor CYT387 suppresses Multiple Signalling pathways, and induces apoptosis in Prevents Proliferation phenotypically Diverse myeloma cells.Leukemia 2011 Dec; 25 (12):. 1891-9.
Abstract
Janus kinases (JAKs) are involved in various signalling pathways exploited by malignant cells. In multiple myeloma (MM), the interleukin-6 / JAK / signal transducers and activators of transcription (IL-6 / JAK / STAT) pathway has been the focus of research for a number of years and IL-6 has an established role in MM drug resistance. JAKs therefore make a rational drug target for anti-MM therapy. CYT387 is a novel, orally bioavailable JAK1 / 2 inhibitor, which has recently been described. This preclinical evaluation of CYT387 for treatment of MM demonstrated that CYT387 was able to prevent IL-6-induced phosphorylation of STAT3 and greatly decrease IL-6- and insulin-like growth factor-1-induced phosphorylation of AKT and extracellular signal-regulated kinase in human myeloma cell lines (HMCL). CYT387 inhibited MM proliferation in a time- and dose-dependent manner in 6/8 HMCL, and this was not abrogated by the addition of exogenous IL-6 (3/3 HMCL). Cell cycling was inhibited with a G (2) / M accumulation of cells, and apoptosis was induced by CYT387 in all HMCL tested (3/3). CYT387 synergised in killing HMCL when used in combination with the conventional anti-MM therapies melphalan and bortezomib. Importantly, WAS Also apoptosis induced in Primary Patient MM cells (N = 6) with CYT387 as a single agent, and synergy WAS Seen Again when Combined with Conventional therapies.
[5]. Tyner JW, Bumm TG, Deininger J, Wood L, Aichberger KJ, Loriaux MM, Druker BJ, Burns CJ, Fantino E, Deininger MW.CYT387, a novel JAK2 inhibitor, induces hematologic responses and normalizes inflammatory cytokines in murine myeloproliferative neoplasms.Blood 2010 Jun 24; 115 (25):. 5232- 40. Epub 2010 Apr 12.
Abstract
Activating alleles of Janus kinase 2 (JAK2) SUCH as JAK2 (V617F) are Central to the pathogenesis of myeloproliferative neoplasms (MPN), suggesting Small molecule inhibitors targeting JAK2 That May be therapeutically Useful. We have IDENTIFIED an aminopyrimidine derivative (CYT387), which inhibits JAK1, JAK2, and tyrosine kinase 2 (TYK2) at low nanomolar concentrations, with few additional targets. Between 0.5 and 1.5muM CYT387 caused growth suppression and apoptosis in JAK2-dependent hematopoietic cell lines, while nonhematopoietic cell lines were unaffected. In a murine MPN model, CYT387 normalized white cell counts, hematocrit, spleen size, and restored physiologic levels of inflammatory cytokines. Despite the hematologic responses and reduction of the JAK2 (V617F) allele burden, JAK2 (V617F) cells persisted and MPN recurred upon cessation of treatment, suggesting that JAK2 inhibitors may be unable to eliminate JAK2 (V617F) cells, consistent with preliminary results from clinical trials of JAK2 inhibitors in myelofibrosis. While the clinical benefit of JAK2 inhibitors may be substantial, not the least due to reduction of inflammatory cytokines and symptomatic improvement, our data add to increasing evidence that kinase inhibitor monotherapy of malignant disease is not curative, suggesting a need for drug combinations to optimally target the malignant cells.
JAKs are kinases which phosphorylate a group of proteins called Signal Transduction and Activators of Transcription or STATs. When phosphorylated, STATs dimerize, translocate to the nucleus and activate expression of genes which lead to, amongst other things, cellular proliferation.
The central role played by the JAK family of protein tyrosine kinases in the cytokine dependent regulation of both proliferation and end function of several important cell types indicates that agents capable of inhibiting the JAK kinases are useful in the prevention and chemotherapeutic treatment of disease states dependent on these enzymes. Potent and specific inhibitors of each of the currently known four JAK family members will provide a means of inhibiting the action of the cytokines that drive immunological and inflammatory diseases.
Myeloproliferative disorders (MPD) include, among others, polycythemia vera (PV), primary myelofibrosis, thrombocythemia, essential thrombocythemia (ET), idiopathic myelofibrosis (IMF), chronic myelogenous leukemia (CML), systemic mastocystosis (SM), chronic neutrophilic leukemia (CNL), myelodisplastic syndrome (MDS) and systemic mast cell disease (SMCD). JAK2 is a member of the JAK family of kinases in which a specific mutation (JAK2V617F) has been found in 99% of polycythemia vera (PV) patients and 50% of essential thrombocytopenia (ET) and idiopathic myelofibrosis (MF). This mutation is thought to activate JAK2, giving weight to the proposition that a JAK2 inhibitor will be useful in treating these types of diseases.
Asthma is a complex disorder characterized by local and systemic allergic inflammation and reversible airway obstruction. Asthma symptoms, especially shortness of breath, are a consequence to airway obstruction, and death is almost invariably due to asphyxiation. Airway Hyper Responsiveness (AHR), and mucus hyper secretion by goblet cells are two of the principle causes of airway obstruction in asthma patients. Intriguingly recent work in animal experimental models of asthma has underscored the importance of IL-13 as a key player in the pathology of asthma. Using a specific IL-13 blocker, it has been demonstrated that IL-13 acts independently of IL-4 and may be capable of inducing the entire allergic asthma phenotype, without the induction of IgE (i.e. in a non-atopic fashion). This and other models have pointed to an important second tier mechanism for elicitating the pathophysiology of asthma, that is not dependent on the production of IgE by resident B-cells or the presence of eonisophils. A direct induction of AHR by IL-13, represents an important process that is likely to be an excellent target for intervention by new therapies. A contemplated effect of a JAK2 inhibitor to the lungs would result in the suppression of the local release of IL-13 mediated IgE production, and therefore reduction in histaminine release by mast cells and eosinophils. This and other consequences of the absence of IL-13 indicate that many of the effects of asthma may be alleviated through administration of a JAK2 inhibitor to the lungs.
Chronic Obstructive Pulmonary Disease (COPD) is a term which refers to a large group of lung diseases which can interfere with normal breathing. Current clinical guidelines define COPD as a disease state characterized by airflow limitation which is not fully reversible. The airflow limitation is usually both progressive and associated with an abnormal inflammatory response of the lungs to noxious particles and gases, particularly cigarette smoke and pollution. Several studies have pointed to an association between increased production of IL-13 and COPD, lending support to the proposition that the potential alleviation of asthma symptoms by use of a JAK2 inhibitor, may also be achieved in COPD. COPD patients have a variety of symptoms including cough, shortness of breath, and excessive production of sputum. COPD includes several clinical respiratory syndromes including chronic bronchitis and emphysema.
Chronic bronchitis is a long standing inflammation of the bronchi which causes increased production of mucus and other changes. The patient’s symptoms are cough and expectoration of sputum. Chronic bronchitis can lead to more frequent and severe respiratory infections, narrowing and plugging of the bronchi, difficult breathing and disability.
Emphysema is a chronic lung disease which affects the alveoli and/or the ends of the smallest bronchi. The lung loses its elasticity and therefore these areas of the lungs become enlarged. These enlarged areas trap stale air and do not effectively exchange it with fresh air. This results in difficult breathing and may result in insufficient oxygen being delivered to the blood. The predominant symptom in patients with emphysema is shortness of breath.
Additionally, there is evidence of STAT activation in malignant tumors, among them lung, breast, colon, ovarian, prostate and liver cancer, as well as Hodgkins lymphoma, multiple myeloma and hepatocellular carcinoma. Chromosomal translocations involving JAK2 fusions to Tel, Bcr and PCM1 have been described in a number of hematopoietic malignancies including chronic myelogenous leukemia (CML), acute myelogenous leukemia (AML), chronic eosinophilic leukemia (CEL), myelodisplastic syndrome (MDS), myeloproliferative disease (MPD) and acute lymphocytic leukemia (ALL). This suggests treatment of hyperproliferative disorders such as cancers including multiple myeloma; prostate, breast and lung cancer; Hodgkin’s Lymphoma; CML; AML; CEL; MDS; ALL; B-cell Chronic Lymphocytic Leukemia; metastatic melanoma; glioma; and hepatoma, by JAK inhibitors is indicated.
Potent inhibitors of JAK2, in addition to the above, will also be useful in vascular disease such as hypertension, hypertrophy, cardiac ischemia, heart failure (including systolic heart failure and diastolic heart failure), migraine and related cerebrovascular disorders, stroke, Raynaud’s phenomenon, POEMS syndrome, Prinzmetal’s angina, vasculitides, such as Takayasu’s arteritis and Wegener’s granulomatosis, peripheral arterial disease, heart disease and pulmonary arterial hypertension.
Pulmonary arterial hypertension (PAH) is a pulmonary vascular disease affecting the pulmonary arterioles resulting in an elevation in pulmonary artery pressure and pulmonary vascular resistance but with normal or only mildly elevated left-sided filling pressures. PAH is caused by a constellation of diseases that affect the pulmonary vasculature. PAH can be caused by or associated with collagen vascular disorders such as systemic sclerosis (scleroderma), uncorrected congenital heart disease, liver disease, portal hypertension, HIV infection, Hepatitis C, certain toxins, splenectomy, hereditary hemorrhagic teleangiectasia, and primary genetic abnormalities. In particular, a mutation in the bone morphogenetic protein type 2 receptor (a TGF-b receptor) has been identified as a cause of familial primary pulmonary hypertension (PPH). It is estimated that 6% of cases of PPH are familial, and that the rest are “sporadic.” The incidence of PPH is estimated to be approximately 1 case per 1 million population. Secondary causes of PAH have a much higher incidence. The pathologic signature of PAH is the plexiform lesion of the lung which consists of obliterative endothelial cell proliferation and vascular smooth muscle cell hypertrophy in small precapillary pulmonary arterioles. PAH is a progressive disease associated with a high mortality. Patients with PAH may develop right ventricular (RV) failure. The extent of RV failure predicts outcome. The JAK/STAT pathway has recently been implicated in the pathophysiology of PAH. JAKs are kinases which phosphorylate a group of proteins called Signal Transduction and Activators of Transcription or STATs. When phosphorylated, STATs dimerize, translocate to the nucleus and activate expression of genes which lead to proliferation of endothelial cells and smooth muscle cells, and cause hypertrophy of cardiac myocytes. There are three different isoforms of JAK: JAK1, JAK2, and JAK3. Another protein with high homology to JAKs is designated Tyk2. An emerging body of data has shown that the phosphorylation of STAT3, a substrate for JAK2, is increased in animal models of PAH. In the rat monocrotaline model, there was increased phosphorylation of the promitogenic transcription factor STAT3. In this same study pulmonary arterial endothelial cells (PAECs) treated with monocrotaline developed hyperactivation of STAT3. A promitogenic agent or protein is an agent or protein that induces or contributes to the induction of cellular proliferation. Therefore, one effect of JAK2 inhibition would be to decrease proliferation of endothelial cells or other cells, such as smooth muscle cells. A contemplated effect of a JAK2 inhibitor would be to decrease the proliferation of endothelial cells or other cells which obstruct the pulmonary arteriolar lumen. By decreasing the obstructive proliferation of cells, a JAK2 inhibitor could be an effective treatment of PAH.
Additionally the use of JAK kinase inhibitors for the treatment of viral diseases and metabolic diseases is indicated.
Although the other members of the JAK family are expressed by essentially all tissues, JAK3 expression appears to be limited to hematopoetic cells. This is consistent with its essential role in signalling through the receptors for IL-2, IL4, IL-7, IL-9 and IL-15 by non-covalent association of JAK3 with the gamma chain common to these multichain receptors. Males with X-linked severe combined immunodeficiency (XSCID) have defects in the common cytokine receptor gamma chain (gamma c) gene that encodes a shared, essential component of the receptors of interleukin-2 (IL-2), IL-4, IL-7, IL-9, and IL-15. An XSCID syndrome in which patients with either mutated or severely reduced levels of JAK3 protein has been identified, suggesting that immunosuppression should result from blocking signalling through the JAK3 pathway. Gene Knock out studies in mice have suggested that JAK3 not only plays a critical role in B and T lymphocyte maturation, but that JAK3 is constitutively required to maintain T cell function. Taken together with the biochemical evidence for the involvement of JAK3 in signalling events downstream of the IL-2 and IL-4 receptor, these human and mouse mutation studies suggest that modulation of immune activity through the inhibition of JAK3 could prove useful in the treatment of T-cell and B-cell proliferative disorders such as transplant rejection and autoimmune diseases. Conversely undesired inhibition of JAK3 could have a devastating effect on the immune status of an individual treated with drug.
Although the inhibition of various types of protein kinases, targeting a range of disease states, is clearly beneficial, it has been to date demonstrated that the identification of a compound which is selective for a protein kinase of interest, and has good “drug like” properties such as high oral bioavailability, is a challenging goal. In addition, it is well established that the predictability of inhibition, or selectivity, in the development of kinase inhibitors is quite low, regardless of the level sequence similarity between the enzymes being targeted.
The challenges in developing therapeutically appropriate JAK2 inhibitors for use in treatment kinase associated diseases such as immunological and inflammatory diseases including organ transplants; hyperproliferative diseases including cancer and myeloproliferative diseases; viral diseases; metabolic diseases; and vascular diseases include designing a compound with appropriate specificity which also has good drug-likeliness.
There is therefore a continuing need to design and/or identify compounds which specifically inhibit the JAK family of kinases, and particularly compounds which may preferentially inhibit one of the JAK kinases relative to the other JAK kinases, particularly JAK2. There is a need for such compounds for the treatment of a range of diseases.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- “Omjjara (GlaxoSmithKline Australia Pty Ltd)”. Therapeutic Goods Administration (TGA). 14 January 2025. Retrieved 20 January 2025.
- https://www.tga.gov.au/resources/artg/442230 [bare URL]
- “Notice: Multiple additions to the Prescription Drug List (PDL) [2024-12-20]”. Health Canada. 20 December 2024. Retrieved 21 December 2024.
- “Ojjaara product information”. Health Canada. 8 November 2024. Retrieved 27 December 2024.
- “Ojjaara- momelotinib tablet”. DailyMed. U.S. National Library of Medicine. 15 September 2023. Archived from the original on 30 November 2023. Retrieved 20 September 2023.
- “Omjjara EPAR”. European Medicines Agency. 5 August 2011. Retrieved 18 March 2024.
- “Omjjara Product information”. Union Register of medicinal products. 26 January 2024. Retrieved 18 March 2024.
- “FDA Roundup: September 19, 2023”. U.S. Food and Drug Administration (FDA) (Press release). 19 September 2023. Archived from the original on 21 September 2023. Retrieved 20 September 2023. This article incorporates text from this source, which is in the public domain.
- “Novel Drug Approvals for 2023”. U.S. Food and Drug Administration (FDA). 15 September 2023. Archived from the original on 21 January 2023. Retrieved 20 September 2023. This article incorporates text from this source, which is in the public domain.
- “GSK’s Omjjara Authorized in EU for Treating Myelofibrosis With Anemia”. MarketWatch. Retrieved 30 January 2024.
- Pardanani A, Lasho T, Smith G, Burns CJ, Fantino E, Tefferi A (August 2009). “CYT387, a selective JAK1/JAK2 inhibitor: in vitro assessment of kinase selectivity and preclinical studies using cell lines and primary cells from polycythemia vera patients”. Leukemia. 23 (8): 1441–1445. doi:10.1038/leu.2009.50. PMID 19295546. S2CID 26947444.
- “Omjjara: Pending EC decision”. European Medicines Agency (EMA). 10 November 2023. Archived from the original on 29 November 2023. Retrieved 5 December 2023.
External links
- Clinical trial number NCT04173494 for “A Study of Momelotinib Versus Danazol in Symptomatic and Anemic Myelofibrosis Patients (MOMENTUM)” at ClinicalTrials.gov
- Clinical trial number NCT01969838 for “Momelotinib Versus Ruxolitinib in Subjects With Myelofibrosis (Simplify 1)” at ClinicalTrials.gov
 |
|
| Names | |
|---|---|
| Preferred IUPAC name
N-(Cyanomethyl)-4-{2-[4-(morpholin-4-yl)anilino]pyrimidin-4-yl}benzamide
|
|
Other names
|
|
| Identifiers | |
|
|
|
3D model (JSmol)
|
|
| ChEBI | |
| ChEMBL | |
| ChemSpider | |
| DrugBank |
|
| KEGG | |
|
PubChem CID
|
|
| UNII |
|
|
CompTox Dashboard (EPA)
|
|
| Properties | |
| C23H22N6O2 | |
| Molar mass | 414.469 g·mol−1 |
| Pharmacology | |
| L01EJ04 (WHO) | |
| By mouth | |
| Legal status | |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
| Clinical data | |
|---|---|
| Other names | Momelotinib hydrochloride hydrate (JAN JP), Momelotinib dihydrochloride (USAN US) |
| License data |
|
| Identifiers | |
| PDB ligand | |
| CompTox Dashboard (EPA) | |
//////////Momelotinib, APPROVALS 2023, FDA 2023, Ojjaara, high-risk myelofibrosis, anemia, APPROVALS 2024, EU 2024, EMA 2024
REF
European Journal of Medicinal Chemistry 265 (2024) 116124
Scheme 13 illustrates the synthesis of Momelotinib Dihydrochloride [48]. The Pd(PPh3) 4-catalyzed Suzuki coupling reaction between 2,4-dichloropyrimidine (MOME-001) and boronic acid MOME-002
resulted in the formation of MOME-003. Subsequently, MOME-003 underwent a substitution reaction with aniline MOME-004 in the presence of p-toluenesulfonic acid (TsOH), yielding MOME-005.
MOME-005 was hydrolyzed by lithium hydroxide, leading to the formation of carboxylic acid MOME-006. MOME-006 underwent amidation with 2-aminoacetonitrile hydrochloride (MOME-007) to produce
Momelotinib.
[48] G.D. Smith, R. Fida, M.M. Kowalski, N-(cyanomethyl)-4-[2-[[4-(4-morpholinyl)
phenyl]amino]-4-pyrimidinyl]-benzamide [CYT387] or a Related Compound,
2012. WO2012071612A1.
.
