
Home » APPROVALS 2023 (Page 2)
Category Archives: APPROVALS 2023
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Durlobactam
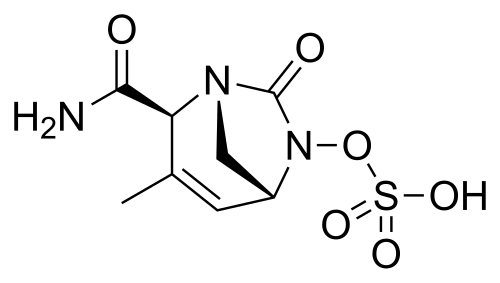
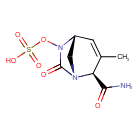
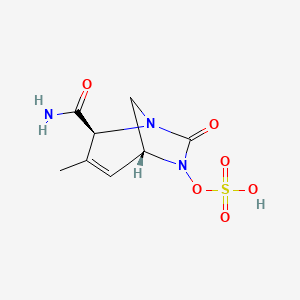
Durlobactam
CAS 1467829-71-5
WeightAverage: 277.25
Monoisotopic: 277.03685626
Chemical FormulaC8H11N3O6S
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Durlobactam sodium | F78MDZ9CW9 | 1467157-21-6 | WHHNOICWPZIYKI-IBTYICNHSA-M |
FDA 5/23/2023, Xacduro, To treat hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia caused by susceptible isolates of Acinetobacter baumannii-calcoaceticus complex
Press Release
Drug Trials Snapshots
(2S,5R)-2-CARBAMOYL-3-METHYL-7-OXO-1,6-DIAZABICYCLO(3.2.1)OCT-3-EN-6-YL SULFATE
SULFURIC ACID, MONO((2S,5R)-2-(AMINOCARBONYL)-3-METHYL-7-OXO-1,6-DIAZABICYCLO(3.2.1)OCT-3-EN-6-YL) ESTER
ETX 2514, ETX-2514, ETX2514, WHO 10824
Durlobactam is a non-beta-lactam, beta-lactamase inhibitor used to treat hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia.
Durlobactam is a beta-lactamase inhibitor used in combination with sulbactam to treat susceptible strains of bacteria in the genus Acinetobacter[1] It is an analog of avibactam.
The combination therapy sulbactam/durlobactam was approved for medical use in the United States in May 2023.[1]
PATENT
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US9309245 | No | 2016-04-12 | 2033-04-02 |  |
| US9623014 | No | 2017-04-18 | 2033-04-02 | |
| US9968593 | No | 2018-05-15 | 2035-11-17 | |
| US10376499 | No | 2019-08-13 | 2035-11-17 | |
SYN
https://doi.org/10.1021/acs.jmedchem.4c02079
J. Med. Chem. 2025, 68, 2147−2182
Durlobactam (1) is a copackaged antibiotic combination being developed by Entasis Therapeutics for the treatment of infections caused by Acinetobacter baumannii-calcoaceticus. 13,14
Entasis Therapeutics obtained the worldwide development rights for durlobactam (1) from AstraZeneca in 2015.13 The drug combination was approved by the USFDA in 2023 for use in patients 18 years of age and older as an intravenous infusion.13 Acinetobacter baumannii is a critical bacterial pathogen that has
become highly resistant to various β-lactam antibiotics for Gram-negative infections, including penicillin.15,16 The inventors targeted β-lactam resistance via coadministration of a β-lactamase inhibitor to restore the activity of β-lactam antibiotics. Sulbactam is a β-lactam antibiotic that inhibits penicillin binding proteins (PBP 1 and 3) essential for cell wall synthesis. Durlobactam is a β-lactamase inhibitor that protects sulbactam from degradation by Ambler class A, C, and D serine β-lactamases produced byAcinetobacter baumannii-calcoaceticus. Durlobactam binds covalently with these β-lactamases by
carbamoylating the active site serines, thus safeguarding sulbactam from enzymatic degradation.17,18 The covalent bond between durlobactam and the active site serine isreversible due to the ability of sulfated amine of durlobactam to recyclize back into urea. This allows durlobactam to exchange from one
enzyme molecule to another via a mechanism known as acylation exchange
(13) Keam, S. J. Sulbactam/Durlobactam: first approval. Drugs 2023, 83, 1245−1252.
(14) El-Ghali, A.; Kunz Coyne, A. J.;Caniff, K.; Bleick,C.; Rybak, M. J.Sulbactam-durlobactam: a novel β-lactam-β-lactamase inhibitor combination targeting carbapenem-resistant Acinetobacter baumannii
infections. Pharmacotherapy 2023, 43, 502−513.
(15) O’Donnell, J.; Tanudra, A.; Chen, A.; Miller, A. A.; McLeod, S.M.; Tommasi, R. In vitro pharmacokinetics/pharmacodynamics of the β-lactamase inhibitor, durlobactam, in combination with sulbactam against Acinetobacter baumannii-calcoaceticus complex. Antimicrob.Agents Chemother. 2024, 68, e00312-23.
(16) Arya, R.; Goldner, B. S.; Shorr, A. F. Novel agents in development for multidrug-resistant Gram-negative infections: potential new options facing multiple challenges. Curr. Opin. Infect. Dis. 2022, 35, 589−594.
(17) Shapiro, A. B.; Moussa, S. H.; McLeod, S. M.; Durand-Réville, T.; Miller, A. A. Durlobactam, a new diazabicyclooctane β-lactamase inhibitor for the treatment of Acinetobacter infections in combination
with Sulbactam. Front. Microbiol. 2021, 12, No. 709974.
(18) Iyer, R.; Moussa, S. H.; Durand-Reville, T. F.; Tommasi, R.; Miller, A. Acinetobacter baumannii OmpA is a selective antibiotic permeant porin. ACS Infect. Dis. 2018, 4, 373−381.
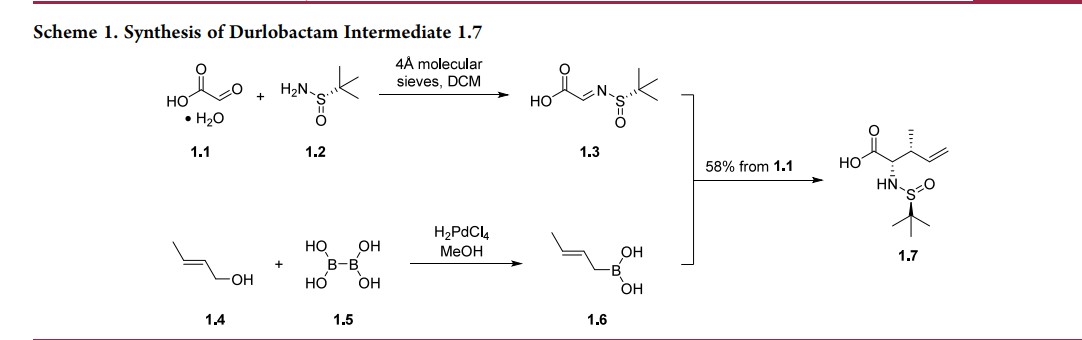
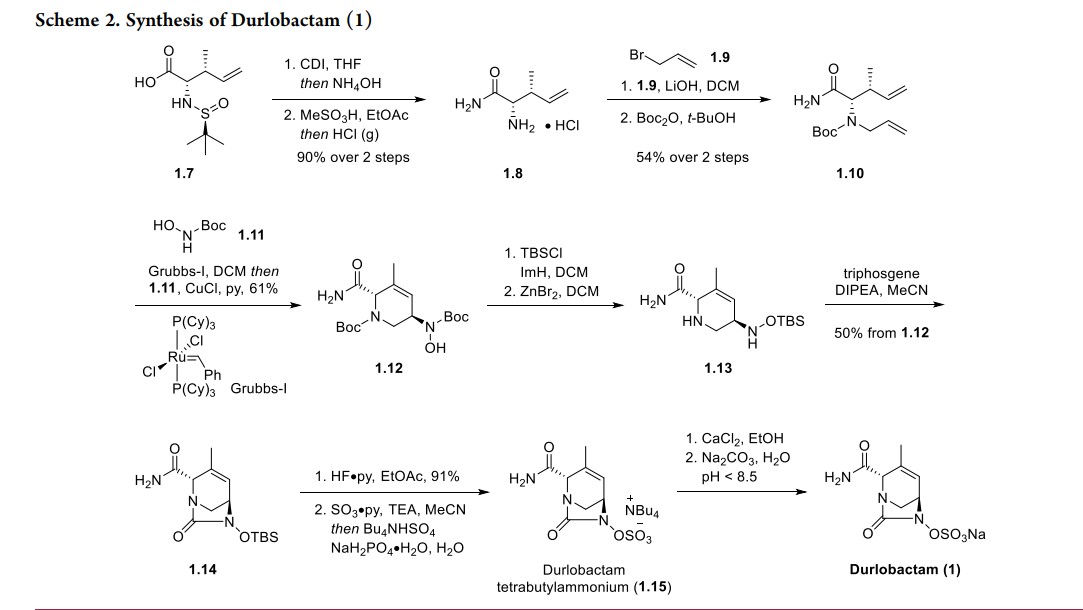
The route below was chosen as it was demonstrated on a multikilogram scale (Scheme 1), although some reagents (e.g.,triphosgene) are not typical for large-scale manufacturing.20,22 The synthesis commenced with the condensation of glyoxylic acid monohydrate (1.1) with (S)-tert-butylsulfinamide (1.2) to generate a solution of 2-(tert-butylsulfinylimino)acetic acid 1.3. In parallel, commercially available trans-crotyl alcohol (1.4) was treated with diboronic acid (1.5) in the presence of a palladium catalyst to produce a solution of crotylboronic acid 1.6. These two solutions were mixed to afford chiral α-amino acid 1.7 in
58% overall yield. Diastereo- and enantioselectivity were not reported for the transformation.
Conversion of 1.7 into durlobactam sodium (1) is described in Scheme 2. First, the carboxylic acid 1.7 was converted to an amide and the sulfinamide was removed to afford amino amide 1.8 as an HCl salt. The primary amine in 1.8 was subsequently alkylated with allyl bromide (1.9) and the resulting allyl amine
was protected with Boc anhydride to provide olefin metathesis precursor 1.10. Bisolefin 1.10 was then subjected to Grubbs first-generation catalyst (Grubbs-I) to generate a tetrahydropyridine precursor, which participated in a one-pot nitroso-ene reaction with N-Boc hydroxylamine (1.11) to produce allyl
hydroxylamine 1.12 in 61% overall yield. This key transformation efficiently installed the amine stereocenter required for formation of the bridged urea. Next, the hydroxyl moiety in 1.12 was protected as the TBS ether and the two Boc groups were removed with ZnBr2 to unveil bis-amine 1.13. The intramolecular urea formation was accomplished by the treatment with triphosgene to generate diazabicyclooctene 1.14 in 50% yield over 3 steps. The TBS ether was then removed, and the hydroxyl urea intermediate was treated with sulfur trioxide-pyridine complex and tetrabutylammonium
hydrogen sulfate to afford durlobactam tetrabutylammonium salt 1.15. Finally, tetrabutylammonium durlobactam 1.15 was converted to a calcium salt and subsequently to the targeted sodium salt providing 1. The authors mentioned that the salt formations were required to improve the purity of the final API
(>99%), however, the yields of these steps were not reported.22
(19) McGuire, H.; Bist, S.; Bifulco, N.; Zhao, L.; Wu, Y.; Huynh, H.; Xiong, H.; Comita-Prevoir, J.; Dussault, D.; Geng, B.; et al. Preparation of oxodiazabicyclooctenyl hydrogen sulfate derivatives for use as betalactamase inhibitors. WO 2013150296, 2013.
(20) Basarab, G. S.; Moss, B.; Comita-Prevoir, J.; Durand-Reville, T. F.; Gauthier, L.; O’Donnell, J.; Romero, J.; Tommasi, R.; Verheijen, J.C.; Wu, F.; et al. Preparation of substituted 2-(1,6-diazabicyclo[3.2.1]-
oct-3-en-6-yloxy)acetates as beta-lactamase inhibitors. WO2018053215, 2018.
(21) Durand-Reville, T. F.;Comita-Prevoir, J.; Zhang, J.; Wu, X.; MayDracka, T. L.; Romero, J. A. C.; Wu, F.; Chen, A.; Shapiro, A. B.; Carter, N. M.; et al. Discovery of an orally available diazabicyclooctane
inhibitor (ETX0282) of class A, C, and D serine β-lactamases. J. Med.Chem. 2020, 63, 12511−12525.
(22) Durand-Reville, T. F.; Wu, F.; Liao, X.; Wang, X.; Zhang, S.Preparation of Durlobactam crystalline forms. WO 2023206580, 2023
syn
https://www.mdpi.com/1424-8247/15/3/384
Synthesis of Durlobactam
Chemically, durlobactam is [(2S,5R)-2-carbamoyl-3-methyl-7-oxo-1,6-diazabicyclo [3.2.1] oct-3-en-6-yl] hydrogen sulfate which can be prepared from the key intermediate hydroxyurea 6-hydroxy-3-methyl-7-oxo-1,6-diaza-bicyclo [3.2.1] oct-3-ene-2-carboxylic acid amide I, which is the structural isomer of III prepared to synthetize ETX-1317 [101]. Then, according to Scheme 15, compound 1 obtained in the synthesis of III (Scheme 14) was reacted with penta-1,3-diene in place of isoprene, and, by an aza-Diels−Alder reaction, compound 2 was obtained.
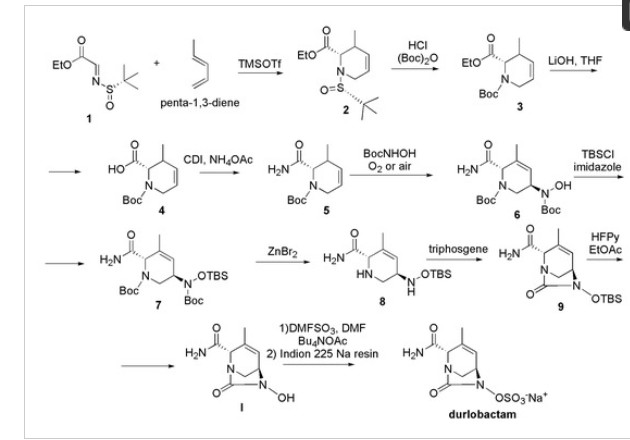
Scheme 15. Synthesis of durlobactam (C8H11N3O6S, MW = 277.36, IUPAC name, [(2S,5R)-2-carbamoyl-3-methyl-7-oxo-1,6-diazabicyclo [3.2.1] oct-3-en-6-yl] hydrogen sulphate.
Compound 2 underwent deprotection of the tert-butyl sulfinyl group to afford 3, subsequently Boc protected, to give compound 4. The saponification of the ester followed by amide coupling using ammonium acetate afforded compound 5. The reaction of alkene 5 with N-Boc-hydroxylamine in the presence of oxygen or air gave the desired compound 6 in a single step. Compound 6 was then protected with TBS group, using TBSCl to afford 7, which was Boc deprotected using zinc bromide obtaining compound 8. Cyclization of the diamine 8 with tri-phosgene provided the corresponding cyclic urea 9, which was TBS deprotected with HFPy to give the key intermediate I. This compound was then immediately sulfated with the DMF:SO3 complex to obtain the sulfate, which was isolated as its tetrabutylammonium salt 10 by reacting with tetrabutylammonium acetate. The tetrabutylammonium salt was converted to durlobactam in the form of sodium salt by passing 10 through a column filled with Indion 225 sodium resin.
REF
https://sioc-journal.cn/Jwk_yjhx/EN/abstract/abstract350784.shtml
References
- ^ Jump up to:a b “FDA Approves New Treatment for Pneumonia Caused by Certain Difficult-to-Treat Bacteria”. U.S. Food and Drug Administration (Press release). 24 May 2023. Retrieved 24 May 2023.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain.
Further reading
- Shapiro AB, Moussa SH, McLeod SM, Durand-Réville T, Miller AA (2021). “Durlobactam, a New Diazabicyclooctane β-Lactamase Inhibitor for the Treatment of Acinetobacter Infections in Combination With Sulbactam”. Frontiers in Microbiology. 12: 709974. doi:10.3389/fmicb.2021.709974. PMC 8328114. PMID 34349751.
- Papp-Wallace KM, McLeod SM, Miller AA (May 2023). “Durlobactam, a Broad-Spectrum Serine β-lactamase Inhibitor, Restores Sulbactam Activity Against Acinetobacter Species”. Clinical Infectious Diseases. 76 (Supplement_2): S194 – S201. doi:10.1093/cid/ciad095. PMC 10150275. PMID 37125470.
| Clinical data | |
|---|---|
| Other names | ETX2514 |
| Routes of administration | Intravenous |
| Drug class | Antibacterial, beta-lactamase inhibitor |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only co-packaged with sulbactam |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1467829-71-5 |
| PubChem CID | 89851852 |
| DrugBank | DB16704DBSALT003190 |
| ChemSpider | 5761778471060725 |
| UNII | PSA33KO9WAF78MDZ9CW9 |
| KEGG | D11591D11592 |
| ChEMBL | ChEMBL4298137ChEMBL4297378 |
| Chemical and physical data | |
| Formula | C8H11N3O6S |
| Molar mass | 277.25 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
/////////Durlobactam, Xacduro, FDA 2023, APPROVED 2023, ETX 2514, ETX-2514, ETX2514, WHO 10824
Syn
European Journal of Medicinal Chemistry 291 (2025) 117643
Durlobactam, developed by Entasis Therapeutics, is a novel β-lactamase inhibitor designed to combat multidrug-resistant (MDR) Acinetobacter baumannii infections [83]. It is co-formulated with sulbactam, a
β-lactam antibiotic, and marketed under the brand name XACDURO. In 2024, the NMPA approved XACDURO for the treatment of hospital-acquired bacterial pneumonia (HABP) and ventilator-associated bacterial pneumonia (VABP) caused by susceptible isolates of Acinetobacter baumannii-calcoaceticus complex in adults [84]. Durlobactam inhibits a broad spectrum of β-lactamases, including class A, C, and D enzymes, which are commonly produced by A. baumannii. By protecting sulbactam from enzymatic degradation, it restores sulbactam’s antibacterial activity against these resistant pathogens. The
clinical efficacy of sulbactam-durlobactam was demonstrated in the PhaseIII ATTACK trial (NCT03894046), a randomized, active-controlled study comparing sulbactam-durlobactam to colistin in patients with infections caused by carbapenem-resistant A. baumannii [85]. In this trial, the primary efficacy endpoint was achieved. It demonstrated non-inferiority in terms of 28-day all-cause mortality. The mortality rate in the sulbactam – durlobactam group was 19.0 %, while that in the colistin group reached 32.3 %. Moreover, the incidence of nephrotoxicity was remarkably lower in the sulbactam-durlobactam
group. From the perspective of toxicity, sulbactam-durlobactam was typically well-tolerated by the subjects. The most common adverse reactions included liver function test abnormalities, diarrhea, and hypokalemia. Notably, the incidence of nephrotoxicity was lower compared to colistin, highlighting a more favorable safety profile. The approval of XACDURO provides a targeted therapeutic option for managing severe infections caused by MDR A. baumannii, addressing a critical need in the
treatment of these challenging pathogens [86–88].
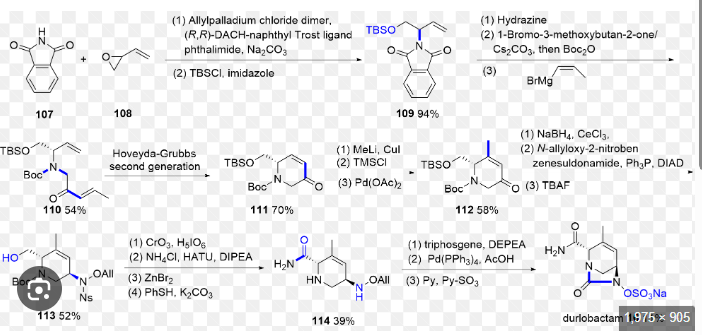
The synthetic route of Durlobactam, shown in Scheme 20, commences with a Grignard substitution between Durl-001 and Durl-002, affording Durl-003 [89]. This intermediate undergoes Diels-Alder
cyclization to form Durl-004, followed by reduction to Durl-005. Mitsunobu reaction of Durl-005 generates Durl-006, which is subjected to sequential deprotections yielding Durl-007 and subsequently Durl-008. Amidation of Durl-008 produces Durl-009, followed by TBAF-mediated deprotection to afford Durl-010. Oxidation of Durl-010Ngives carboxylic acid Durl-011, which undergoes amidation to form
Durl-012. Palladium-catalyzed coupling of Durl-012 produces Durl-013, with final ion exchange affording Durlobactam.
83-89
[83] A.B. Shapiro, S.H. Moussa, S.M. McLeod, T. Durand-R´ eville, A.A. Miller,
Durlobactam, a new diazabicyclooctane β-Lactamase inhibitor for the treatment of
acinetobacter infections in combination with sulbactam, Front. Microbiol. 12
(2021) 709974.
[84] G. Granata, F. Taglietti, F. Schiavone, N. Petrosillo, Durlobactam in the treatment
of multidrug-resistant Acinetobacter baumannii infections: a systematic review, J. Clin. Med. 11 (2022) 3258.
[85] K.M. Papp-Wallace, S.M. McLeod, A.A. Miller, Durlobactam, a broad-spectrum
serine β-lactamase inhibitor, restores sulbactam activity against acinetobacter
species, Clin. Infect. Dis. 76 (2023) S194–s201.
[86] Sulbactam and Durlobactam, Drugs and Lactation Database (Lactmed®), National
Institute of Child Health and Human Development, Bethesda (MD), 2006.
[87] S.J. Keam, Sulbactam/durlobactam: first approval, Drugs 83 (2023) 1245–1252.
[88] Y. Fu, T.E. Asempa, J.L. Kuti, Unraveling sulbactam-durlobactam: insights into its
role in combating infections caused by Acinetobacter baumannii, Expert Rev. Anti
Infect. Ther. 23 (2024) 1–12.
[89] H. McGuire, S. Bist, N. Bifulco, L. Zhao, Y. Wu, H. Huynh, H. Xiong, J. Comita-
Prevoir, D. Dussault, B. Geng, B. Chen, T. Durand-Reville, S. Guler, Preparation of
Oxodiazabicyclooctenyl Hydrogen Sulfate Derivatives for Use as beta-lactamase
Inhibitors, 2013 US9623014B2.
Sulbactam



Sulbactam
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Sulbactam benzathine | 49MU89FVBV | 83031-43-0 | YSEPFTSCLHUBNH-HFKSPEPWSA-N |
| Sulbactam sodium | DKQ4T82YE6 | 69388-84-7 | NKZMPZCWBSWAOX-IBTYICNHSA-M |
WeightAverage: 233.242
Monoisotopic: 233.035793157
Chemical FormulaC8H11NO5S
(2S,5R)-3,3-dimethyl-4,4,7-trioxo-4λ6-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid
Betamaze, Penicillanic Acid Sulfone, Sulbactamum, CP 45899, CP-45899, CP45899
FDA 2023, Xacduro, 5/23/2023, To treat hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia caused by susceptible isolates of Acinetobacter baumannii-calcoaceticus complex
Press Release
Drug Trials Snapshots
Sulbactam is a β-lactamase inhibitor. This drug is given in combination with β-lactam antibiotics to inhibit β-lactamase, an enzyme produced by bacteria that destroys the antibiotics.[1]
It was patented in 1977 and approved for medical use in 1986.[2]
Sulbactam is a beta (β)-lactamase inhibitor and a derivative of the basic penicillin nucleus. When given in combination with β-lactam antibiotics, sulbactam produces a synergistic effect as it blocks the enzyme responsible for drug resistance by hydrolyzing β-lactams.
PATENT
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US9309245 | No | 2016-04-12 | 2033-04-02 | |
| US9623014 | No | 2017-04-18 | 2033-04-02 | |
| US9968593 | No | 2018-05-15 | 2035-11-17 | |
| US10376499 | No | 2019-08-13 | 2035-11-17 | |
doi:10.1016/S0040-4039(00)89275-8

SYN
European Journal of Medicinal Chemistry
Volume 265, 5 February 2024, 116124
https://doi.org/10.1016/j.ejmech.2024.116124
On May 23, 2023, the FDA granted approval to Xacduro for the treatment of Baumannii-sensitive strains causing hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia in
patients aged 18 years or older [4]. Xacduro consists of Sulbactam and Durlobactam. Sulbactam, a medication with a similar structure to Penicillin, has the ability to eliminate Acinetobacter baumannii. On the other hand, Durlobactam shields Sulbactam from being broken down by enzymes that may be produced by Acinetobacter baumannii [5].
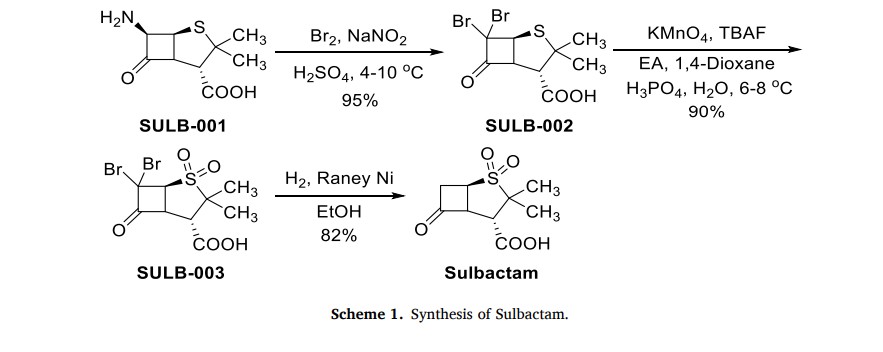
The production process of Sulbactam started with 6-aminopenicilanic acid (6-APA) (SULB-001) as the starting material (Scheme 1) [6]. It underwent bromination reaction with sodium nitrite and bromine
in the presence of sulfuric acid. Then, SULB-002 was oxidized by potassium permanganate to obtain sulfone SULB-003. Finally, the sulfonewas catalytically hydrogenated and dehalogenated in the presence of Raney nickel to get Sulbactam
[5] A. El-Ghali, A.J. Kunz Coyne, K. Caniff, C. Bleick, M.J. Rybak, Sulbactamdurlobactam: a novel β-lactam-β-lactamase inhibitor combination targeting
carbapenem-resistant Acinetobacter baumannii infections, Pharmacotherapy 43
(2023) 502–513.
[6] Z.M. Song, W. Liu, J. Yang, Y. Sun, Improvement on the synthetic process of
sulbactam, Chin. J
PATENT
https://patents.google.com/patent/CN101967155A/en
Embodiment 1
In the four-hole boiling flask of 2000ML, add 600ML methylene dichloride and 180ML2.5N sulfuric acid, stirring is cooled to below 0 ℃, add 28ML bromine and 25g Sodium Nitrite, 0 ± 0.2 ℃, gradation adds 40g 6-APA, and controlled temperature is lower than 5 ℃, stirring reaction 1h, be cooled to then below 0 ℃, 20% aqueous solution of sodium bisulfite of dropping below 0 ℃ leaves standstill phase-splitting to the color fade of bromine, water 100ML dichloromethane extraction 3 times, merge organic phase, with 100ML saturated sodium-chloride water solution washing 2 times, obtain 6, the 6-dibromo penicillanic acid;
To go up 6, the 6-dibromo penicillanic acid changes in the 2000ML beaker mutually. and add 250ML distilled water and stir, be cooled to below 5 ℃, drip 4NNaHCO 3The aqueous solution leaves standstill phase-splitting to pH=7, organic phase extracts 3 times with the 80ML deionized water, merge water, water changes in the 2000ML four-hole boiling flask, stirring is cooled to 0 ℃, and beginning dropping oxidizing agent (44g KMn04+10.8ML H3P04+700MLH20 stirring and dissolving) dripped in 30 minutes, controlled temperature is lower than 10 ℃ in the dropping process, keep 0~5 ℃ then, stirring reaction 1h adds the 500ML ethyl acetate, drip 6N sulfuric acid to pH=1.25, be cooled to 0 ℃, slowly add the color fade of 27.5% hydrogen peroxide (about 45g) to KMn04, during continue to keep pH=1.25 with 6N sulfuric acid, controlled temperature is lower than 10 ℃, reaction 10mi n filters, and adds sodium-chlor in the filtrate to no longer dissolving, leave standstill the branch phase of anhydrating, water 250ML ethyl acetate extraction 4 times merge organic phase, and wash 2 times with the 100ML saturated sodium-chloride water solution, organic phase contains 6, the acid of 6-dibromo sulbactam;
To go up 6,6-dibromo sulbactam acid organic phase changes in the 2000ML four-hole boiling flask, adds 350ML water, is cooled to below 5 ℃, uses 4N NaHCO 3The aqueous solution is transferred pH to 5.0, and add 25ML methyl alcohol, add the 26g zinc powder in batches, and drip 6N sulfuric acid maintenance pH:4.5~5.5, after adding zinc powder, keep stirring reaction 4h below 5 ℃, keep pH=4.5~5.5 with 6N sulfuric acid simultaneously, filter, with 25ML ethyl acetate and 25ML water washing, merging filtrate is transferred pH to 2.0 with 6N sulfuric acid, add sodium-chlor to water insoluble till, leave standstill the branch phase of anhydrating, water merges organic phase with 150ML ethyl acetate extraction 4 times, washs to redness with the 50ML-100ML5% potassium permanganate solution earlier at organic layer and does not take off, again with 150ML saturated sodium-chloride water solution washing 2 times, layering, organic layer add the 2g activated carbon decolorizing, the 15g anhydrous magnesium sulfate drying, suction filtration, be evaporated to feed liquid and be creamy white, cool to 0 ℃ after centrifuging, after the oven dry product Sulbactam (sulbactam acid) 32g, the product yield is 74%, the product colour pure white was placed 30 days the color no change under the room temperature.
Embodiment 2
In the reactor of 2000L, add 600L methylene dichloride and 180L2.5N sulfuric acid, stirring is cooled to below 0 ℃, add 28L bromine and 25Kg Sodium Nitrite, 0 ± 0.2 ℃, gradation adds 40Kg 6-APA, and controlled temperature is lower than 5 ℃, stirring reaction 1h, be cooled to then below 0 ℃, 20% aqueous solution of sodium bisulfite of dropping below 0 ℃ leaves standstill phase-splitting to the color fade of bromine, water 100L dichloromethane extraction 3 times, merge organic phase, with 100L saturated sodium-chloride water solution washing 2 times, obtain 6, the 6-dibromo penicillanic acid;
Will on obtain 6, the 6-dibromo penicillanic acid changes in the 2000L reactor mutually. add the 250L tap water and stir, be cooled to below 5 ℃, drip 4NNaHCO 3The aqueous solution leaves standstill phase-splitting to pH=7, organic phase extracts 3 times with the 80L deionized water, merge water, water changes in the 2000L reactor, stirring is cooled to 0 ℃, and beginning dropping oxidizing agent (44Kg KMn04+10.8L H3P04+700LH20 stirring and dissolving) dripped in 30 minutes, controlled temperature is lower than 10 ℃ in the dropping process, keep 0~5 ℃ then, stirring reaction 1h adds the 500L ethyl acetate, drip 6N sulfuric acid to pH=1.25, be cooled to 0 ℃, slowly add 28% hydrogen peroxide (about 44Kg) color fade to KMn04, during continue to keep pH=1.25 with 6N sulfuric acid, controlled temperature is lower than 10 ℃, reaction 10mi n filters, and adds sodium-chlor in the filtrate to no longer dissolving, leave standstill the branch phase of anhydrating, water 250L ethyl acetate extraction 4 times merge organic phase, and wash 2 times with the 100L saturated sodium-chloride water solution, organic phase contains 6, the acid of 6-dibromo sulbactam;
To go up organic phase and contain 6, the acid of 6-dibromo sulbactam changes in the 2000L reactor, adds 350L water, is cooled to below 5 ℃, uses 4N NaHCO 3The aqueous solution is transferred pH to 5.0, and add 25L methyl alcohol, add the 26Kg zinc powder in batches, and drip 6N sulfuric acid maintenance pH:4.5~5.5, after adding zinc powder, keep stirring reaction 4h below 5 ℃, keep pH=4.5~5.5 with 6N sulfuric acid simultaneously, filter, with 25L ethyl acetate and 25L water washing, merging filtrate is transferred pH to 2.0 with 6N sulfuric acid, add sodium-chlor to water insoluble till, leave standstill the branch phase of anhydrating, water merges organic phase with 150L ethyl acetate extraction 4 times, washs to redness with the 30-50L10% potassium permanganate solution earlier at organic layer and does not take off, again with 150L saturated sodium-chloride water solution washing 2 times, layering, organic layer add the 2Kg activated carbon decolorizing, the 15Kg anhydrous magnesium sulfate drying, suction filtration, be evaporated to feed liquid and be creamy white, cool to 0 ℃ after centrifuging, after the oven dry product Sulbactam (sulbactam acid) 31.5Kg, the product yield is 72.8%, the product colour pure white was placed 30 days the color no change under the room temperature.
Embodiment 3
In the four-hole boiling flask of 1000ML, add 300ML methylene dichloride and 90ML2.5N Hydrogen bromide, stirring is cooled to below 0 ℃, add 14ML bromine and 12.5g Sodium Nitrite, 0 ± 0.2 ℃, gradation adds 20g 6-APA, and controlled temperature is lower than 5 ℃, stirring reaction 1h, be cooled to then below 0 ℃, 20% aqueous solution of sodium bisulfite of dropping below 0 ℃ leaves standstill phase-splitting to the color fade of bromine, water 50ML dichloromethane extraction 3 times, merge organic phase, with 50ML saturated sodium-chloride water solution washing 2 times, obtain 6, the 6-dibromo penicillanic acid;
To go up 6, the 6-dibromo penicillanic acid changes in the 1000ML beaker mutually. and add 125ML distilled water and stir, be cooled to below 5 ℃, drip 4NNaHCO 3The aqueous solution leaves standstill phase-splitting to pH=7, organic phase extracts 3 times with the 40ML deionized water, merge water, water changes in the 1000ML four-hole boiling flask, stirring is cooled to 0 ℃, and beginning dropping oxidizing agent (22g KMn04+5.4ML H3P04+300MLH20 stirring and dissolving) dripped in 30 minutes, controlled temperature is lower than 10 ℃ in the dropping process, keep 0~5 ℃ then, stirring reaction 1h adds the 250ML ethyl acetate, drip 6N sulfuric acid to pH=1.25, be cooled to 0 ℃, slowly add 25% hydrogen peroxide (about 29g) color fade to KMn04, during continue to keep pH=1.25 with 6N sulfuric acid, controlled temperature is lower than 10 ℃, reaction 10mi n filters, and adds sodium-chlor in the filtrate to no longer dissolving, leave standstill the branch phase of anhydrating, water 125ML ethyl acetate extraction 4 times merge organic phase, and wash 2 times with the 50ML saturated sodium-chloride water solution, organic phase contains 6, the acid of 6-dibromo sulbactam.
To go up organic phase and contain 6, the acid of 6-dibromo sulbactam changes in the 1000ML four-hole boiling flask, adds 175ML water, is cooled to below 5 ℃, uses 4N NaHCO 3The aqueous solution is transferred pH to 5.0, and add 12.5ML methyl alcohol, add the 13g zinc powder in batches, and drip 6N sulfuric acid maintenance pH:4.5~5.5, after adding zinc powder, keep stirring reaction 4h below 5 ℃, keep pH=4.5~5.5 with 6N sulfuric acid simultaneously, filter, with 12.5ML ethyl acetate and 12.5ML water washing, merging filtrate is transferred pH to 2.0 with 6N sulfuric acid, add sodium-chlor to water insoluble till, leave standstill the branch phase of anhydrating, water merges organic phase with 75ML ethyl acetate extraction 4 times, washs to redness with the 15ML-35ML7% potassium permanganate solution earlier at organic layer and does not take off, again with 75ML saturated sodium-chloride water solution washing 2 times, layering, organic layer add the 1g activated carbon decolorizing, the 7.5g anhydrous magnesium sulfate drying, suction filtration, be evaporated to feed liquid and be creamy white, cool to 0 ℃ after centrifuging, after the oven dry product Sulbactam (sulbactam acid) 15.9g, the product yield is 73.5%, the product colour pure white was placed 30 days the color no change under the room temperature.
PATENT
https://patents.google.com/patent/US4420426A/en
Medical uses
The combination ampicillin/sulbactam (Unasyn) is available in the United States.[3]
The combination cefoperazone/sulbactam (Sulperazon) is available in many countries but not in the United States.[4]
The co-packaged combination sulbactam/durlobactam was approved for medical use in the United States in May 2023.[5]
Mechanism
Sulbactam is primarily used as a suicide inhibitor of β-lactamase, shielding more potent beta-lactams such as ampicillin.[6] Sulbactam itself contains a beta-lactam ring, and has weak antibacterial activity by inhibiting penicillin binding proteins (PBP) 1 and 3, but not 2.[7]
References
- ^ Totir MA, Helfand MS, Carey MP, Sheri A, Buynak JD, Bonomo RA, Carey PR (August 2007). “Sulbactam forms only minimal amounts of irreversible acrylate-enzyme with SHV-1 beta-lactamase”. Biochemistry. 46 (31): 8980–8987. doi:10.1021/bi7006146. PMC 2596720. PMID 17630699.
- ^ Fischer J, Ganellin CR (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 492. ISBN 9783527607495.
- ^ “Unasyn- ampicillin sodium and sulbactam sodium injection, powder, for solution”. DailyMed. U.S. National Library of Medicine. 29 March 2023. Retrieved 25 May 2023.
- ^ “Sulperazon”. drugs.com.
- ^ “FDA Approves New Treatment for Pneumonia Caused by Certain Difficult-to-Treat Bacteria”. U.S. Food and Drug Administration (Press release). 24 May 2023. Retrieved 24 May 2023.
- ^ Crass RL, Pai MP (February 2019). “Pharmacokinetics and Pharmacodynamics of β-Lactamase Inhibitors”. Pharmacotherapy. 39 (2): 182–195. doi:10.1002/phar.2210. PMID 30589457. S2CID 58567725.
- ^ Penwell WF, Shapiro AB, Giacobbe RA, Gu RF, Gao N, Thresher J, et al. (March 2015). “Molecular mechanisms of sulbactam antibacterial activity and resistance determinants in Acinetobacter baumannii”. Antimicrobial Agents and Chemotherapy. 59 (3): 1680–1689. doi:10.1128/AAC.04808-14. PMC 4325763. PMID 25561334.
Further reading
Singh GS (January 2004). “Beta-lactams in the new millennium. Part-II: cephems, oxacephems, penams and sulbactam”. Mini Reviews in Medicinal Chemistry. 4 (1): 93–109. doi:10.2174/1389557043487547. PMID 14754446.
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| MedlinePlus | a693021 |
| Routes of administration | Intravenous, intramuscular |
| ATC code | J01CG01 (WHO) |
| Legal status | |
| Legal status | UK: POM (Prescription only) |
| Pharmacokinetic data | |
| Protein binding | 29% |
| Elimination half-life | 0.65–1.20 hrs |
| Excretion | Mainly kidneys (41–66% within 8 hrs) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 68373-14-8 |
| PubChem CID | 130313 |
| ChemSpider | 115306 |
| UNII | S4TF6I2330 |
| KEGG | D08533 |
| ChEBI | CHEBI:9321 |
| ChEMBL | ChEMBL403 |
| CompTox Dashboard (EPA) | DTXSID1023605 |
| ECHA InfoCard | 100.063.506 |
| Chemical and physical data | |
| Formula | C8H11NO5S |
| Molar mass | 233.24 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Melting point | 148 to 151 °C (298 to 304 °F) |
| showSMILES | |
| showInChI | |
//////////Sulbactam, Xacduro, FDA 2023, APPROVALS 2023, Betamaze, Penicillanic Acid Sulfone, Sulbactamum, CP 45899, CP-45899, CP45899
Perfluorhexyloctane



WeightAverage: 432.269
Monoisotopic: 432.112266666Chemical FormulaC14H17F13
Perfluorhexyloctane
- 133331-77-8
- MIEBO
- Tetradecane, 1,1,1,2,2,3,3,4,4,5,5,6,6-tridecafluoro-
- 7VYX4ELWQM
- NOV03, NOV 03
- 1-(perfluorohexyl)octane
- F6H8
- NOV03
- Perfluorohexyloctane
1,1,1,2,2,3,3,4,4,5,5,6,6-tridecafluorotetradecane
FDA APPROVED 8/16/2023, Sohonos, To reduce the volume of new heterotopic ossification in adults and pediatric patients (aged 8 years and older for females and 10 years and older for males) with fibrodysplasia ossificans progressiva
Drug Trials Snapshot
Perfluorohexyloctane is a fluoroalkane that is tetradecane in which all of the hydrogen atoms at positions 1, 2, 3, 4, 5, and 6 have been replaced by fluorine atoms. It is an ophthalmic solution used to treat the signs and symptoms of dry eye disease. It has a role as an ophthalmology drug and a nonionic surfactant. It is a fluorohydrocarbon and a fluoroalkane. It derives from a hydride of a tetradecane.
Perfluorohexyloctane (branded as Evotears, Miebo,[a] and Novatears, among others) is a medication used for the treatment of dry eye disease.[4] It is a semifluorinated alkane.[4]
Perfluorohexyloctane has been available in multiple markets since 2015 under the brand names Evotears and Novatears,[5] and was additionally approved for medical use in the United States in May 2023 under the brand name Miebo.[4][6] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication.[7]
PATENT
Show 102550100 entries
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US11357738 | No | 2022-06-14 | 2036-09-29 | |
| US10058615 | No | 2018-08-28 | 2033-09-12 | |
| US10369117 | No | 2019-08-06 | 2033-09-12 | |
| US10449164 | No | 2019-10-22 | 2033-09-12 | |
| US10507132 | No | 2019-12-17 | 2037-06-21 | |
| US10576154 | No | 2020-03-03 | 2033-09-12 | |
SYN
https://www.scientificupdate.com/process-chemistry-articles/not-a-dry-eye-in-the-house



Medical uses
Perfluorohexyloctane is indicated for the treatment of the signs and symptoms of dry eye disease.[4][8][9]
Availability
Perfluorohexyloctane is sold as an over-the-counter medication under the brand names Evotears and Novatears in multiple countries,[10] costing around NZ$34.00, A$30, and €30 for a one-month supply.
In the US, perfluorohexyloctane is sold under the brand name Miebo; a prescription is required.
Notes and references
- ^ “Notice: Multiple additions to the Prescription Drug List (PDL) [2024-10-18]”. Health Canada. 18 October 2024. Retrieved 25 October 2024.
- ^ “Miebo product information”. Health Canada. 4 September 2024. Retrieved 27 December 2024.
- ^ “Regulatory Decision Summary for Miebo”. Drug and Health Products Portal. 4 September 2024. Retrieved 27 December 2024.
- ^ Jump up to:a b c d e “Miebo- perfluorohexyloctane solution”. DailyMed. 18 May 2023. Retrieved 8 June 2023.
- ^ “URSAPHARM GmbH and Novaliq GmbH Announce European Partnership Agreement” (Press release). Retrieved 15 February 2024.
- ^ “Bausch + Lomb and Novaliq Announce FDA Approval of Miebo (Perfluorohexyloctane Ophthalmic Solution) for the Treatment of the Signs and Symptoms of Dry Eye Disease” (Press release). Bausch + Lomb Corporation. 18 May 2023. Retrieved 8 June 2023 – via Business Wire.
- ^ New Drug Therapy Approvals 2023 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2024. Archived from the original on 10 January 2024. Retrieved 9 January 2024.
- ^ Ballesteros-Sánchez A, De-Hita-Cantalejo C, Sánchez-González MC, Jansone-Langine Z, de Sotomayor MA, Culig J, et al. (October 2023). “Perfluorohexyloctane in dry eye disease: A systematic review of its efficacy and safety as a novel therapeutic agent”. The Ocular Surface. 30: 254–262. doi:10.1016/j.jtos.2023.10.001. hdl:11441/151762. PMID 37813152. S2CID 263802332.
- ^ Sheppard JD, Evans DG, Protzko EE (November 2023). “A review of the first anti-evaporative prescription treatment for dry eye disease: perfluorohexyloctane ophthalmic solution”. The American Journal of Managed Care. 29 (14 Suppl): S251 – S259. doi:10.37765/ajmc.2023.89464. PMID 37930231. S2CID 265032840.
- ^ “In Australia, NovaTears Eye Drops Are Available on the Pharmaceutical Benefits Scheme (PBS) from Now On” (Press release). Retrieved 15 February 2024.
Further reading
- Azhar A, Taimuri MA, Oduoye MO, Sumbal A, Sheikh A, Iqbal A, et al. (September 2024). “MEIBO (perfluorohexyloctane): a novel approach to treating dry eye disease”. Annals of Medicine and Surgery (2012). 86 (9): 5292–5298. doi:10.1097/MS9.0000000000002322. PMC 11374244. PMID 39239035.
| Clinical data | |
|---|---|
| Trade names | Evotears Miebo (/ˈmaɪboʊ/ MY-bow) Novatears |
| Other names | NOV03; 1-(perfluorohexyl)octane |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a623054 |
| License data | US DailyMed: Perfluorohexyloctane |
| Routes of administration | Eye drops |
| ATC code | None |
| Legal status | |
| Legal status | CA: ℞-only[1][2][3]US: ℞-only[4] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 133331-77-8 |
| PubChem CID | 10477896 |
| DrugBank | DB17823 |
| ChemSpider | 8653305 |
| UNII | 7VYX4ELWQM |
| KEGG | D12604 |
| ChEBI | CHEBI:229658 |
| CompTox Dashboard (EPA) | DTXSID20440585 |
| Chemical and physical data | |
| Formula | C14H17F13 |
| Molar mass | 432.269 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
////////Perfluorhexyloctane, Sohonos, APPROVALS 2023, FDA 2023, NOV03, NOV 03, MIEBO, 1-perfluorohexyl)octane, F6H8, NOV03, Perfluorohexyloctane
Tofersen

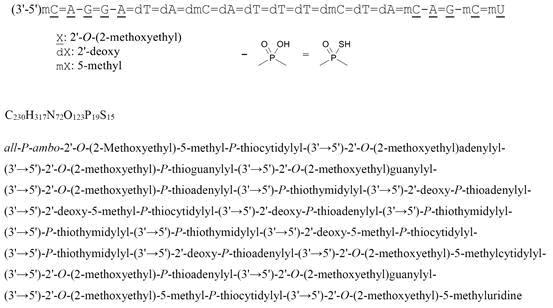
all-P-ambo-2′-O-(2-Methoxyethyl)-5-methyl-P-thiocytidylyl-(3’→5′)-2′-O-(2-methoxyethyl)adenylyl-(3’→5′)-2′-O-(2-methoxyethyl)-P-thioguanylyl-(3’→5′)-2′-O-(2-methoxyethyl)guanylyl-(3’→5′)-2′-O-(2-methoxyethyl)-P-thioadenylyl-(3’→5′)-P-thiothymidylyl-(3’→5′)-2′-deoxy-P-thioadenylyl-(3’→5′)-2′-deoxy-5-methyl-P-thiocytidylyl-(3’→5′)-2′-deoxy-P-thioadenylyl-(3’→5′)-P-thiothymidylyl-(3’→5′)-P-thiothymidylyl-(3’→5′)-P-thiothymidylyl-(3’→5′)-2′-deoxy-5-methyl-P-thiocytidylyl-(3’→5′)-P-thiothymidylyl-(3’→5′)-2′-deoxy-P-thioadenylyl-(3’→5′)-2′-O-(2-methoxyethyl)-5-methylcytidylyl-(3’→5′)-2′-O-(2-methoxyethyl)-P-thioadenylyl-(3’→5′)-2′-O-(2-methoxyethyl)guanylyl-(3’→5′)-2′-O-(2-methoxyethyl)-5-methyl-P-thiocytidylyl-(3’→5′)-2′-O-(2-methoxyethyl)-5-methyluridine
C230H317N72O123P19S15 : 7127.86
[2088232-70-4]

Tofersen
CAS 2088232-70-4
FDA APPROVED 4/25/2023, Qalsody
- BIIB 067
- BIIB067
- Formula
C230H317N72O123P19S15
Molar mass
7127.85 g·mol−1
- Antisense Oligonucleotide Inhibitor Of The Expression Of Superoxide Dismutase 1 Gene
- DNA, D((2′-O-(2-METHOXYETHYL))M5RC-SP-(2′-O-(2-METHOXYETHYL))RA-(2′-O-(2-METHOXYETHYL))RG-SP-(2′-O-(2-METHOXYETHYL))RG-(2′-O-(2-METHOXYETHYL))RA-SP-T-SP-A-SP-M5C-SP-A-SP-T-SP-T-SP-T-SP-M5C-SP-T-SP-A-SP-(2′-O-(2-METHOXYETHYL))M5RC-(2′-O-(2-METHOXYETHYL))R
- IONIS SOD1Rx
To treat amyotrophic lateral sclerosis in adults who have a SOD1 gene mutation
Drug Trials Snapshot
A nucleic acid-based drug indicated for the treatment of a specific type of amyotrophic lateral sclerosis.
Tofersen, sold under the brand name Qalsody, is a medication used for the treatment of amyotrophic lateral sclerosis (ALS).[3] Tofersen is an antisense oligonucleotide that targets the production of superoxide dismutase 1, an enzyme whose mutant form is commonly associated with amyotrophic lateral sclerosis. It is administered as an intrathecal injection.[3]
The most common side effects include fatigue, arthralgia (joint pain), increased cerebrospinal (brain and spinal cord) fluid white blood cells, and myalgia (muscle pain).[3]
Tofersen was approved for medical use in the United States in April 2023,[3][6] and in the European Union in May 2024.[4] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication.[7]
| Clinical data | |
|---|---|
| Trade names | Qalsody |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a623024 |
| License data | US DailyMed: Tofersen |
| Routes of administration | Intrathecal |
| ATC code | N07XX22 (WHO) |
| Legal status | |
| Legal status | CA: ℞-only[1]US: ℞-only[2][3]EU: Rx-only[4][5] |
| Identifiers | |
| CAS Number | 2088232-70-4 |
| DrugBank | DB14782 |
| UNII | 2NU6F9601K |
| KEGG | D11811 |
| Chemical and physical data | |
| Formula | C230H317N72O123P19S15 |
| Molar mass | 7127.85 g·mol−1 |
References
- ^ “Register of Innovative Drugs”. Health Canada. 3 November 2006. Retrieved 17 April 2025.
- ^ “Qalsody- tofersen injection”. DailyMed. 25 April 2023. Archived from the original on 8 May 2023. Retrieved 10 June 2023.
- ^ Jump up to:a b c d e f g h i j k l “FDA approves treatment of amyotrophic lateral sclerosis associated with a mutation in the SOD1 gene” (Press release). U.S. Food and Drug Administration (FDA). 25 April 2023. Archived from the original on 25 April 2023. Retrieved 25 April 2023. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d “Qalsody EPAR”. European Medicines Agency (EMA). 22 February 2024. Retrieved 24 February 2024. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b “Qalsody PI”. Union Register of medicinal products. 3 June 2024. Retrieved 7 September 2024.
- ^ “FDA Grants Accelerated Approval for Qalsody (tofersen) for SOD1-ALS, a Major Scientific Advancement as the First Treatment to Target a Genetic Cause of ALS” (Press release). Biogen. 25 April 2023. Archived from the original on 25 April 2023. Retrieved 25 April 2023 – via GlobeNewswire.
- ^ Jump up to:a b New Drug Therapy Approvals 2023 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2024. Archived from the original on 10 January 2024. Retrieved 9 January 2024.
- ^ Liu A (1 May 2019). “Biogen’s antisense ALS drug shows promise in early clinical trial”. FierceBiotech. Archived from the original on 2 February 2023. Retrieved 25 April 2023.
- ^ Langreth R (22 March 2023). “Biogen’s ALS Drug Gets Partial Backing From FDA Panel”. Bloomberg News. Retrieved 25 April 2023.
- ^ “FDA approves drug which helps to slow progression of rare form of MND”. http://www.sheffield.ac.uk. 28 April 2023. Retrieved 16 May 2024.
- ^ Berdyński M, Miszta P, Safranow K, Andersen PM, Morita M, Filipek S, et al. (January 2022). “SOD1 mutations associated with amyotrophic lateral sclerosis analysis of variant severity”. Scientific Reports. 12 (1): 103. Bibcode:2022NatSR..12..103B. doi:10.1038/s41598-021-03891-8. PMC 8742055. PMID 34996976.
- ^ Constantino A (25 April 2023). “FDA grants accelerated approval for Biogen ALS drug that treats rare form of the disease”. CNBC. Archived from the original on 25 April 2023. Retrieved 25 April 2023.
- ^ Constantino A (22 March 2023). “FDA advisors vote against effectiveness of Biogen’s ALS drug for rare and aggressive form of the disease”. CNBC. Archived from the original on 10 April 2023. Retrieved 25 April 2023.
- ^ Robins R (25 April 2023). “F.D.A. Approves Drug for Rare Form of A.L.S.” The New York Times. Archived from the original on 25 April 2023. Retrieved 25 April 2023.
- ^ “New treatment for rare motor neuron disease recommended for approval”. European Medicines Agency (EMA) (Press release). 23 February 2024. Retrieved 24 February 2024.
////////////tofersen, Qalsody, FDA 2023, APPROVALS 2023, EU 2024, EMA 2024, BIIB 067, BIIB067, IONIS SOD1Rx
Leniolisib



Leniolisib
CAS 1354690-24-6
WeightAverage: 450.466
Monoisotopic: 450.199108558
Chemical FormulaC21H25F3N6O2
CDZ-173-NX- CDZ173
- CDZ173-NX
1-[(3S)-3-({6-[6-methoxy-5-(trifluoromethyl)pyridin-3-yl]-5H,6H,7H,8H-pyrido[4,3-d]pyrimidin-4-yl}amino)pyrrolidin-1-yl]propan-1-one
FDA APPROVED Joenja, 3/24/2023, To treat activated phosphoinositide 3-kinase delta syndrome
Drug Trials Snapshot
Leniolisib (INN[3][4]), sold under the brand name Joenja, is a medication used for the treatment of activated phosphoinositide 3-kinase delta syndrome (APDS).[2][5] It is a kinase inhibitor[2][6] that is taken by mouth.[2]
The most common side effects include headache, sinusitis, and atopic dermatitis.[5]
Leniolisib was approved for medical use in the United States in March 2023.[5][7][8] It is the first approved medication for the treatment of activated PI3K delta syndrome.[5] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication.[9]
PATENT
https://patents.google.com/patent/US8653092B2/en
PATENT
https://patentscope.wipo.int/search/en/WO2012004299
Example 67 was prepared according the general procedure described in scheme 4
Example 67: 1 -{(S)-3-[6-(6-Methoxy-5-trifluoromethyl-pyridin-3-yl)-5,6,7,8-tetrahydro-pyrido[4,3-d]pyrimidin-4-ylamino]-pyrrolidin-1-yl}-propan-1-one
To a solution of (S)-3-[6-(6-methoxy-5-trifluoromethyl-pyridin-3-yl)-5,6,7,8-tetrahydro-pyrido[4,3-d]pyrimidin-4-ylamino]-pyrrolidine-1 -carboxylic acid tert-butyl ester (intermediate 24) (13.4 g, 27.1 mmol) in CH2CI2 (100 mL), was added TFA (41 .8 mL) and the mixture stirred at rt for 1 h. Concentrated in vacuo and partitioned between 2M NaOH(aq) (300 mL) and CH2CI2 (200 mL). The organic phase was separated and the aqueous phase extracted with CH2CI2 (2 x 200 mL). The organic phases were combined, dried (MgS04) and
evaporated in vacuo to give a brown foam. The foam was dissolved in CH2CI2 (50 mL) and was added simultaneously portionwise with sat.NaHC03(aq) (50 mL) to a vigourously stirring solution of propionyl chloride (2.63 g, 28.5 mmol) in CH2CI2 (50 mL) at rt. The resulting biphasic mixture was stirred at rt for 1 h. Further propionyl chloride (0.566g, 6.12 mmol) was added and continued stirring vigorously for 20 min. The organic layer was separated and the aqueous layer extracted with CH2CI2 (100 mL). The organic layers were combined, dried (MgS04) and concentrated in vacuo to give a brown gum. The gum was stirred in EtOAc (100 mL) and the resulting solid filtered (9.4 g). The mother liquors were concentrated in vacuo and purified by column chromatography through a Biotage® amino silica gel eluting with EtOAc / MeOH, 100/0 to 90/10 to give a yellow foam which was then stirred in EtOAc (20 mL) and the resulting solid filtered (870 mg). Both batches of solids were combined and stirred in refluxing EtOAc (50 mL) for 1 h. Filtered to give 1-{(S)-3-[6-(6-methoxy-5-trifluoromethyl-pyridin-3-yl)-5,6,7,8-tetrahydro-pyrido[4,3-d]pyrimidin-4-ylamino]-pyrrolidin-1 -yl}-propan-1 -one as a colourless solid (9.42 g, 76% yield). 1 H NMR (400 MHz, DMSO-d6, 298K) δ ppm 0.95-1.05 (m, 3H) 1 .87-2.32 (m, 4H) 2.77-2.86 (m, 2H) 3.25-3.88 (m, 6H) 3.93 (s, 3H) 3.98 (s, 2H) 4.55-4.80 (m, 1 H) 6.70-6.80 (m, 1 H, N-H) 7.86-7.92 (m, 1 H) 8.27-8.33 (m, 1 H) 8.33-8.37 (m, 1 H) LCMS: [M+H]+=451.0, Rt (6)= 1.49 min.
Alternative synthesis for example 67
A solution of (S)-3-[6-(6-methoxy-5-trifluoromethyl-pyridin-3-yl)-5,6,7,8-tetrahydro-pyrido[4,3-d]pyrimidin-4-ylamino]-pyrrolidine-1-carboxylic acid tert-butyl ester (intermediate 24) (29.04 g, 58.73 mmol) in 2-Me-THF (100 mL) was dropwise added into aqueous HCI solution (150 mL, 31 %) over 15 min. The reaction mixture was partitioned between water (300 mL) and isopropyl acetate (100 mL) and the upper organic phase was discarded. The aqueous phase was partitioned between 25% NaOH (aq) (200 g) and 2-Me-THF (200 mL), and the organic phase was collected and dried. Triethylamine (16.32 mL, 1 17.48 mmol) was added into the organic phase followed by dropwise addition of propionyl chloride (6.0 g, 64.6 mmol) at 0 °C. The resulting mixture was stirred at 0 °C for 1 h. The reaction mixture was washed with water (1 10 mL) and the resulting organic phase was concentrated in vacuo to give a brown gum.
The residue was recrystallized with isopropanol and methyl tert-butyl ether to give 1 -{(S)-3- [6-(6-methoxy-5-trifluoromethyl-pyridin-3-yl)-5,6,7,8-tetrahydro-pyrido[4,3-d]pyrimidin-4- ylamino]-pyrrolidin-1-yl}-propan-1 -one as a colourless solid (17.2 g, 65% yield).
Crystallization of Example 67 by heating in acetonitrile/water
2.0 g of Example 67 (4.440 mol) were dissolved in 10 mL of acetonitrile and 0.5 mL of water at 75°C. The solution was allowed to cool down to rt within 30 min resulting in a suspension. The mixture was stirred for 16 h at rt. The crystals were collected by filtration. The filter cake was washed 2 times with 1 mL of acetonitrile and afterwards dried for 16 h at 24°C and ca. 10 mbar vacuum. Elementary analysis of the material showed a waterless form.
PAPER
https://pubs.acs.org/doi/10.1021/acsmedchemlett.7b00293
ACS Medicinal Chemistry Letters
Cite this: ACS Med. Chem. Lett. 2017, 8, 9, 975–980
https://doi.org/10.1021/acsmedchemlett.7b00293
The predominant expression of phosphoinositide 3-kinase δ (PI3Kδ) in leukocytes and its critical role in B and T cell functions led to the hypothesis that selective inhibitors of this isoform would have potential as therapeutics for the treatment of allergic and inflammatory disease. Targeting specifically PI3Kδ should avoid potential side effects associated with the ubiquitously expressed PI3Kα and β isoforms. We disclose how morphing the heterocyclic core of previously discovered 4,6-diaryl quinazolines to a significantly less lipophilic 5,6,7,8-tetrahydropyrido[4,3-d]pyrimidine, followed by replacement of one of the phenyl groups with a pyrrolidine-3-amine, led to a compound series with an optimal on-target profile and good ADME properties. A final lipophilicity adjustment led to the discovery of CDZ173 (leniolisib), a potent PI3Kδ selective inhibitor with suitable properties and efficacy for clinical development as an anti-inflammatory therapeutic. In vitro, CDZ173 inhibits a large spectrum of immune cell functions, as demonstrated in B and T cells, neutrophils, monocytes, basophils, plasmocytoid dendritic cells, and mast cells. In vivo, CDZ173 inhibits B cell activation in rats and monkeys in a concentration- and time-dependent manner. After prophylactic or therapeutic dosing, CDZ173 potently inhibited antigen-specific antibody production and reduced disease symptoms in a rat collagen-induced arthritis model. Structurally, CDZ173 differs significantly from the first generation of PI3Kδ and PI3Kγδ-selective clinical compounds. Therefore, CDZ173 could differentiate by a more favorable safety profile. CDZ173 is currently in clinical studies in patients suffering from primary Sjögren’s syndrome and in APDS/PASLI, a disease caused by gain-of-function mutations of PI3Kδ.
Synthesis and full characterization of (S)-1-(3-((6-(6-methoxy-5-(trifluoromethyl)pyridin-3-yl)-
5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-yl)amino)pyrrolidin-1-yl)propan-1-one (3h, CDZ173,
leniolisib)
TFA (41.8 mL) was added to a solution of (S)-3-[6-(6-methoxy-5-trifluoromethyl-pyridin-3-yl)-5,6,7,8-
tetrahydro-pyrido[4,3-d]pyrimidin-4-ylamino]-pyrrolidine-1-carboxylic acid tert-butyl ester (13.4 g,
27.1 mmol) in CH2Cl2 (100 mL), and the mixture was stirred at RT for 1 h. After that time, the mixture
was concentrated under reduced pressure, and the residue was partitioned between NaOH (aqu., 2M,
300 mL) and CH2Cl2 (200 mL). The organic phase was separated, and the aqueous phase was extracted
with CH2Cl2 (2 x 200 mL). The combined organic phases were dried (MgSO4) and concentrated under
reduced pressure. The resulting brown foam was dissolved in CH2Cl2 (50 mL) and added simultaneously with a NaHCO3 solution (aqu., saturated) (50 mL) to a vigorously stirring solution of propionyl chloride (2.63 g, 28.5 mmol) in CH2Cl2 (50 mL) at RT. The resulting biphasic mixture was stirred at RT for
1h. Additional propionyl chloride (0.566 g, 6.12 mmol) was added, and vigorous stirring was continued
for 20 min. The organic layer was separated and the aqueous layer extracted with CH2Cl2 (100 mL). The
combined organic layers were dried (MgSO4) and concentrated under reduced pressure. The resulting
brown gum was stirred in EtOAc (100 mL) and the resulting solid was filtered (9.4 g). The mother liquors were concentrated under reduced pressure and purified by column chromatography through a Biotage®
amino silica gel eluting with EtOAc / MeOH, 100/0 to 90/10. After concentration under reduced
pressure, the resulting yellow foam was stirred in EtOAc (20 mL) and the resulting solid was filtered
(870 mg). Both batches of solids were combined and stirred in refluxing EtOAc (50 mL) for 1h. The
resulting solid was filtered to give the title compound as a colorless solid (9.42 g, 76%). 1H NMR (400
MHz, DMSO-d6, 298K, ca. 1:1 mixture of rotamers) δ ppm 8.35 (m, 1H) 8.30 (m, 1H) 7.89 (m, 1H)
6.80-6.70 (m, 1H, N-H) 4.80-4.55 (m, 1H) 3.93 (s, 3H) 3.98 (s, 2H) 3.88-3.25 (m, 6H) 2.86-2.75 (m,
2H) 2.32-1.87 (m, 4H) 1.05-0.95 (m, 3H); 13C-NMR (150 MHz, DMSO-d6, 298K, ca. 1:1 mixture of
rotamers, data given for cis-isomer): δ ppm 171.3, 158.6, 158.1, 155.5, 153.6, 141.3, 138.0, 125.7,
123.3, 111.1, 109.7, 53.7, 50.8, 49.4, 45.8, 45.8, 44.3, 31.2, 29.7, 26.6, 8.97; LCMS method 1: Rt 1.49
min, calcd for C21H26F3N6O2 [M+H]+
451.2, found 451.0, HRMS (ESI+) calcd for C21H26F3N6O2
[M+H]+ 451.20693, found 451.20642
REF

REF
https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2024.1337436/full

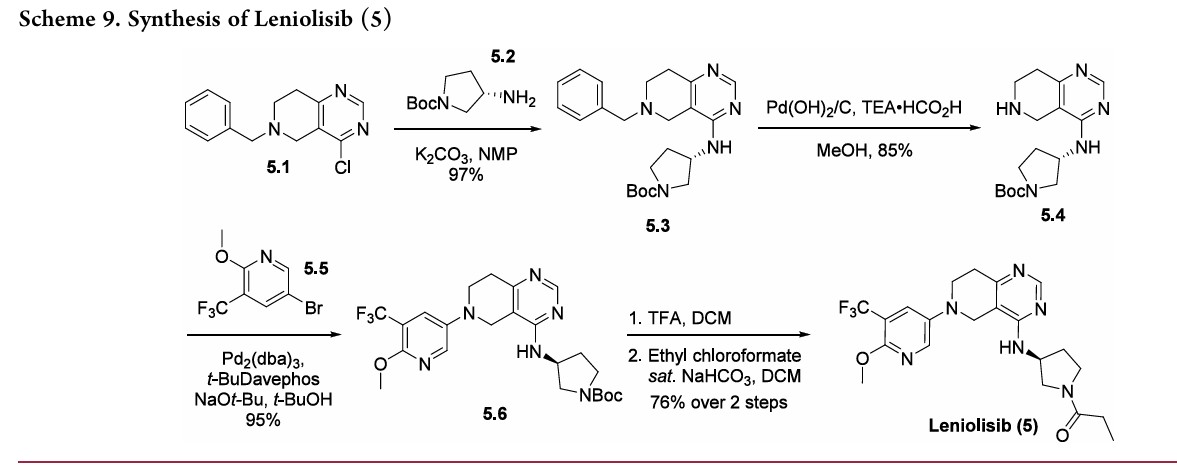
-Benzyl-4-chloro-5,6,7,8-tetrahydro-pyrido[4,3-d]pyrimidine (compound 1) is coupled with (S)-tert-butyl 3-aminopyrrolidine-1-carboxylate (compound 2) in the presence of triethylamine at 120 °C for 42 h to give compound 3 a 93% yield. The benzyl group is deprotected with 20% palladium hydroxide on carbon and ammonium formate in methanol at 65 °C for 2 h to give compound 4 a 66% yield. Compound 4 is coupled with 5-bromo-2-methoxy-3-(trifluoromethyl)pyridine (compound 5) in the presence of sodium –tert-butoxide, tris(dibenzylideneacetone)dipalladium(0), 2-di-t-butylphosphino-2′-(N,N-dimethylamino)biphenyl in tert-butanol at 100 °C for 5 h to give compound 6 a 74% yield. Deprotection of the Boc group in DCM/TFA, followed by coupling with propionyl chloride in the presence of sodium bicarbonate in DCM at room temperature for 1 h gives the final compound 7 (leniolisib) a 76% yield.
REF
https://www.sciencedirect.com/science/article/abs/pii/S0223523424000047

REF
J. Med. Chem. 2025, 68, 2147−2182
Leniolisib (Joenja). Leniolisib (5), is a twice-daily, orally available selective phosphoinositide 3-kinase-delta
(PI3Kδ) inhibitor developed by Novartis and in-licensed by Pharming Group NV for the treatment of activated phosphoinositide 3-kinase-delta syndrome (APDS). APDS is a primary immunodeficiency caused by mutations in PI3Kδ catalytic (PIK3CD) or regulatory (PIK3R1) subunits. The loss or gain of function of these subunits results in hyperactivity of the PI3Kδ pathway which can result in infections, lymphoprolif
eration, autoimmunity, increased risk of malignant lymphoma and early mortality. 44−46
Current treatment strategies include immunosuppressives such as corticosteroids, antiviral, and antibiotic therapies, stem cell transplantation, and immunoglobulin replacement therapy. However, none of these therapeutic strategies treats the underlying hyperactivity of the PI3Kδpathway. Thus, the approval of leniolisib by the USFDA in March 2023 provided a significant breakthrough therapy for patients 12 years and older.47 48
A concise synthetic route to leniolisib has been disclosed by Novartis,beginning with commercially available tetrahydropyridopyrimidine 5.1 (Scheme 9). An SNAr reaction with amine 5.2 furnished intermediate 5.3 in good yield. Transfer hydrogenation with Pd(OH)2 on carbon to remove the benzyl
group gave free amine 5.4, setting up the system for a Buchwald−Hartwig amination with bromide 5.5 to produce 5.6 in good yield. Protecting group removal and subsequent acylation with ethyl chloroformate provided leniolisib (5) in 76% yield over two steps.
(44) Hoegenauer, K.; Soldermann, N.; Stauffer, F.; Furet, P.;
Graveleau, N.; Smith, A. B.; Hebach, C.; Hollingworth, G. J.; Lewis,
I.; Gutmann, S.; et al. Discovery and pharmacological characterization
of novel quinazoline-based PI3K delta-selective inhibitors. ACS Med.
Chem. Lett. 2016, 7, 762−767.
(45) Hoegenauer, K.; Soldermann, N.; Zécri, F.; Strang, R. S.;
Graveleau, N.; Wolf, R. M.; Cooke, N. G.; Smith, A. B.; Hollingworth,
G. J.; Blanz, J.; et al. Discovery of CDZ173 (Leniolisib), representing a
structurally novel class of PI3K delta-selective inhibitors. ACS Med.
Chem. Lett. 2017, 8, 975−980.
(46) Duggan, S.; Al-Salama, Z. T. Leniolisib: first approval. Drugs
2023, 83, 943−948.
(47) Pharming announces US FDA approval of Joenja® (leniolisib) as
the first and only treatment indicated for APDS. Pharming, March 24, 2023 https://www.pharming.com/news/pharming-announces-us-fda
approval-joenja-leniolisib-first-and-only-treatment-indicated-apds (ac
cessed February 2024).
(48) Fernandes Gomes dos Santos, P. A.; Hogenauer, K.;
Hollingworth, G.; Soldermann, N.; Stowasser, F.; Tufilli, N.; Zecri, F.
Solid forms and salts of tetrahydro-pyrido-pyrimidine derivatives. WO
2013001445 A1, 2013
Ref
https://www.sciencedirect.com/science/article/abs/pii/S0223523424000047
Leniolisib developed by Novartis Pharma AG, was approved on March 24, 2023, making it the first treatment drug for APDS [3]. APDS is an immunodeficiency disorder that primarily occurs due to mutations in the gene responsible for encoding phosphotidylinsitol-3-kinase δ(PI3Kδ). These mutations enhance the function of PI3Kδ, resulting in impaired immune response and heightened vulnerability to infections.
Leniolisib is capable of inhibiting the hyperactive PI3Kδ enzyme by obstructing the active binding site within the p110δ subunit [54]. Inisolated enzyme assays conducted without cells, the selectivity of PI3K-δ
was found to be higher compared to PI3Kα(28-fold), PI3K-β (43-fold),and PI3K-γ (257-fold), as well as other enzymes in the kinome [54].Leniolisib demonstrated the ability to decrease phosphoinositide-3-kinase/protein kinase B (pAKT) pathway activity and suppress the growth and activation of B and T cell subsets in cell-based experiments. Leniolisib effectively blocks the signaling pathways responsible for the excessive production of phosphatidylinositol 3,4,5-trisphosphate (PIP3), overactivation of the downstream
mammalian target of rapamycin (mTOR)/protein kinase B (AKT) pathway, and the imbalanced functioning of B and T cells [55].
One representative approach of Leniolisib is depicted in Scheme 15 [55]. Pyrrolidine LENI-003 was obtained by nucleophilic substitution of aminopyrrolidine LENI-001 and the 4-Cl of LENI-002 under alkaline conditions, and LENI-004 was obtained by debenzylation of LENI-003 under palladium hydroxide/carbon. LENI-004 and 5-bromo-2-methox y-3-(trifluoromethyl)pyridine (LENI-005) were coupled to obtain LENI-006. LENI-006 was deprotected by TFA and further condensed with acyl chloride to obtain Leniolisib.
54] V.K. Rao, S. Webster, V. Dalm, A. ˇ Sediv´ a, P.M. van Hagen, S. Holland, S.
D. Rosenzweig, A.D. Christ, B. Sloth, M. Cabanski, A.D. Joshi, S. de Buck,
J. Doucet, D. Guerini, C. Kalis, I. Pylvaenaeinen, N. Soldermann, A. Kashyap,
G. Uzel, M.J. Lenardo, D.D. Patel, C.L. Lucas, C. Burkhart, Effective “activated
PI3Kδ syndrome”-targeted therapy with the PI3Kδ inhibitor leniolisib, Blood 130
(2017) 2307–2316.
[55] K. Hoegenauer, N. Soldermann, F. Z´ ecri, R.S. Strang, N. Graveleau, R.M. Wolf, N.
G. Cooke, A.B. Smith, G.J. Hollingworth, J. Blanz, S. Gutmann, G. Rummel,
A. Littlewood-Evans, C. Burkhart, Discovery of CDZ173 (Leniolisib), representing
a structurally novel class of PI3K delta-selective inhibitors, ACS Med. Chem. Lett.
8 (2017) 975–980.[55] K. Hoegenauer, N. Soldermann, F. Z´ ecri, R.S. Strang, N. Graveleau, R.M. Wolf, N.
G. Cooke, A.B. Smith, G.J. Hollingworth, J. Blanz, S. Gutmann, G. Rummel,
A. Littlewood-Evans, C. Burkhart, Discovery of CDZ173 (Leniolisib), representing
a structurally novel class of PI3K delta-selective inhibitors, ACS Med. Chem. Lett.
8 (2017) 975–980.

,
References
- ^ “Joenja (Ballia Holdings Pty Ltd)”. Therapeutic Goods Administration (TGA). 16 April 2025. Retrieved 3 May 2025.
- ^ Jump up to:a b c d e f “Joenja- leniolisib tablet, film coated”. DailyMed. 29 March 2023. Retrieved 20 June 2023.
- ^ World Health Organization (2016). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 76”. WHO Drug Information. 30 (3). hdl:10665/331020.
- ^ World Health Organization (2017). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 77”. WHO Drug Information. 31 (1). hdl:10665/330984.
- ^ Jump up to:a b c d e f g h i j “FDA approves first treatment for activated phosphoinositide 3-kinase delta syndrome”. U.S. Food and Drug Administration (FDA) (Press release). 24 March 2023. Retrieved 24 March 2023. This article incorporates text from this source, which is in the public domain.
- ^ Duggan S, Al-Salama ZT (July 2023). “Leniolisib: First Approval”. Drugs. 83 (10): 943–948. doi:10.1007/s40265-023-01895-4. PMID 37256490. S2CID 258989663.
- ^ Jump up to:a b “US FDA approves Pharming’s immune disorder drug”. Reuters. Archived from the original on 24 March 2023. Retrieved 24 March 2023.
- ^ “Pharming announces US FDA approval of Joenja (leniolisib) as the first and only treatment indicated for APDS” (PDF). Pharming Group N.V. (Press release). 24 March 2023. Retrieved 25 March 2023.
- ^ New Drug Therapy Approvals 2023 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2024. Archived from the original on 10 January 2024. Retrieved 9 January 2024.
![]() This article incorporates text from this source, which is in the public domain: Bing Chat output modified to create the initial revision of this article. 25 March 2023. – via Microsoft
This article incorporates text from this source, which is in the public domain: Bing Chat output modified to create the initial revision of this article. 25 March 2023. – via Microsoft
External links
Clinical trial number NCT02435173 for “Study of Efficacy of CDZ173 in Patients With APDS/PASLI” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Joenja |
| Other names | CDZ173 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a623016 |
| License data | US DailyMed: Leniolisib |
| Routes of administration | By mouth |
| Drug class | Antineoplastic |
| ATC code | L03AX22 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)[1]US: ℞-only[2] |
| Identifiers | |
| CAS Number | 1354690-24-6as salt: 1354691-97-6 |
| DrugBank | DB16217 |
| ChemSpider | 52083264 |
| UNII | L22772Z9CP |
| KEGG | D11158as salt: D11159 |
| ChEMBL | ChEMBL3643413 |
| PDB ligand | 9NQ (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C21H25F3N6O2 |
| Molar mass | 450.466 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
//////////leniolisib, Joenja, FDA 2023, APPROVALS 2023, CDZ-173-NX, CDZ173, CDZ173-NX



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
Rezafungin



Rezafungin
CAS 1396640-59-7
WeightAverage: 1226.411
Monoisotopic: 1225.602719729
Chemical FormulaC63H85N8O17
FDA APPROVED 3/22/2023, Rezzayo, To treat candidemia and invasive candidiasis
Drug Trials Snapshot
2-[[(3S,6S,9S,11R,15S,18S,20R,21R,24S,25S,26S)-6-[(1S,2S)-1,2-dihydroxy-2-(4-hydroxyphenyl)ethyl]-11,20,25-trihydroxy-3,15-bis[(1R)-1-hydroxyethyl]-26-methyl-2,5,8,14,17,23-hexaoxo-18-[[4-[4-(4-pentoxyphenyl)phenyl]benzoyl]amino]-1,4,7,13,16,22-hexazatricyclo[22.3.0.09,13]heptacosan-21-yl]oxy]ethyl-trimethylazanium
- Rezafungin ion
- Rezafungin cation
- CD-101
- SP-3025
- G013B5478J
Rezafungin, sold under the brand name Rezzayo (by Melinta Therapeutics), is a medication used for the treatment of invasive candidiasis.[2] It is an echinocandin antifungal[1][4] that acts as a fungal β-glucan synthase inhibitor.[5]
Rezafungin was approved for medical use in the United States in March 2023,[1][6][5] and in the European Union in December 2023.[2][3]

CAS No. : 1631754-41-0
Rezafungin acetate (Synonyms: Biafungin acetate; CD101 acetate; SP-3025 acetate)
Rezafungin acetate (Biafungin acetate) is a next-generation, broad-spectrum, and long-lasting echinocandin. Rezafungin acetate shows potent antifungal activity against Candida spp., Aspergillus spp., and Pneumocystis spp..
SYN
https://doi.org/10.1021/acs.jmedchem.4c02079
J. Med. Chem. 2025, 68, 2147−2182
Rezafungin (Rezzayo). Rezafungin (2) is a secondgeneration echinocandin that was discovered by Seachaid
Pharmaceuticals and developed by Cidera Therapeutics. The once weekly intravenously administered drug is used to treat candidemia and invasive candidiasis and to prevent invasive fungal diseases in blood and bone marrow transplant patients.23
Rezafungin was designed to improve the pharmacokinetic properties of the USFDA-approved first-generation echinocandins anidulafungin, caspofungin, and micafungin, enabling less frequent dosing. Mechanistically, echinocandins exert their antifungal activity by inhibiting β-(1→3)-glucan synthase, a
transmembrane protein complex essential for the synthesis of an important polysaccharide component of the fungal cell wall.
This noncompetitive inhibition destabilizes the cell wall, leading to osmotic imbalance and fungal cell death.24 Rezafungin was approved by the USFDA in March 2023 for use in patients 18 years and older.25
An elegant semisynthesis of rezafungin from anidulafungin (2.1) was reported by Cidera Therapeutics that circumvented chemical instability including potential racemization of the
parent compound (Scheme 3).26,27 The semisynthetic sequence26 begins with boronate formation between the 1,2-diol of 2.1 and 3,4-dimethoxyphenylborane (2.2) utilizing azeotropic distillation, maintaining a constant volume of THF. Addition of a solution of choline chloride, TFA, and TFAA in
MeCN to the slurry of boronate ester 2.3 gave the choline ether. Selective ether formation at the hemiaminal hydroxyl group occurred due to its increased reactivity compared to the other free hydroxyls in the compound.27 The specific boronate ester used in this sequence was found to be beneficial at minimizing the amount of a diastereomer impurity (at the hemiaminal) formed in the choline conjugation, though the authors of the patent shared that this was unexpected given the remote boronic
acid from the hemiaminal that participated in conjugation. A 95:5 α:β selectivity of the conjugation was achieved under acidic conditions, and preferential crystallization of the α-isomer while
maintaining solution equilibrium enabled control of the βisomer to less than 2.0%. Work up of the reaction using ammonium acetate and ammonium hydroxide provided crude
rezafungin. Ion exchange chromatography was used to remove3,4-dimethyoxyphenyl boronic acid, eluting with ammonium acetate to afford rezafungin (2). Using this synthetic sequence, a purity of 98.49% was reported with only minor amounts of racemization observed (0.77% undesired diastereomer and
0.51% unwanted epimer at the benzylic center).
(23) Syed, Y. Y. Rezafungin: first approval. Drugs 2023, 83, 833−840.
(24) Denning, D. W. Echinocandins: a new class of antifungal. J.
Antimicrob. Chemother. 2002, 49, 889−891.
(25) Cidara Therapeutics and Melinta Therapeutics announce FDA
approval of RezzayoTM (Rezafungin for injection) for the treatment of
candidemia and invasive candidiasis. Cidera Therapeutics, March 22,
- https://www.cidara.com/news/cidara-therapeutics-andmelinta-therapeutics-announce-fda-approval-of-rezzayo-rezafunginfor-injection-for-the-treatment-of-candidemia-and-invasivecandidiasis/ (accessed February 2024).
(26) Cidara Therapeutics. Synthesis of echinocandin antifungal agent.
WO 2019241626 A1, 2019.
(27) Jamison, J. A.; LaGrandeur, L. M.; Rodriguez, M. J.; Turner, W.
W.; Zeckner, D. J. The synthesis and antifungal activity of nitrogen
containing hemiaminal ethers of LY303366. J. Antibiot. (Tokyo) 1998,
51, 239−42

.
SYN
Hughes, D., et al. (2022). Synthesis of echinocandin antifungal agent. (U.S. Patent No. 11,524,980 B2). U.S. Patent and Trademark Office. https://patentimages.storage.googleapis.com/34/d5/c2/1a8cdcfb3fe3db/US11524980.pdf
https://patentscope.wipo.int/search/en/detail.jsf?docId=US327113930&_cid=P11-MAORN7-73998-1
Example 9. Synthesis of Compound 1 from the 3,4-dimethoxyphenylboronate Ester of Anidulafungin—Coupling in the Presence of TFAA
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US10702573 | No | 2020-07-07 | 2033-03-14 | |
| US9526835 | No | 2016-12-27 | 2033-03-14 | |
| US8722619 | No | 2014-05-13 | 2032-03-02 | |
| US11197909 | No | 2021-12-14 | 2038-07-14 | |
| US11654196 | No | 2023-05-23 | 2032-03-02 | |
| US11712459 | No | 2023-08-01 | 2037-03-15 | |
| US11819533 | No | 2023-11-21 | 2038-07-11 | |



Medical uses
In the United States, rezafungin is indicated in adults who have limited or no alternative options for the treatment of candidemia and invasive candidiasis.[1]
In the European Union, rezafungin is indicated for the treatment of invasive candidiasis in adults.[2]
Rezafungin, while remaining a hydrophilic compound, exhibits a volume of distribution more than twice that of caspofungin.[7] This pharmacokinetic property has supported its investigation for the treatment of deep-seated Candida infections, including osteomyelitis.[8][9]
Legal status
Rezafungin was approved for medical use in the United States in March 2023,[1][10][11] The FDA granted the application for rezafungin orphan drug, fast track, and priority review designations.[12]
In October 2023, the Committee for Medicinal Products for Human Use of the European Medicines Agency adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Rezzayo, intended for the treatment of invasive candidiasis in adults.[2] The applicant for this medicinal product is Mundipharma GmbH.[2] Rezafungin was approved for medical use in the European Union in December 2023.[3]
Rezafungin is a member of the family of echinocandins that inhibits 1,3-beta-D-glucan synthase. It is developed by Cidara Therapeutics and approved for the treatment of candidaemia and invasive candidiasis in patients aged >= 18 years who have limited or no alternative treatment options. It is an echinocandin, a quaternary ammonium ion, an antibiotic antifungal drug, an azamacrocycle, a homodetic cyclic peptide and an aromatic ether.
Brand names
Rezafungin is the international nonproprietary name.[13]
Rezafungin is sold under the brand name Rezzayo.[2]
References
- ^ Jump up to:a b c d e “Rezzayo- rezafungin injection, powder, lyophilized, for solution”. DailyMed. 8 June 2023. Retrieved 26 December 2023.
- ^ Jump up to:a b c d e f g “Rezzayo EPAR”. European Medicines Agency (EMA). 12 October 2023. Retrieved 27 December 2023. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b c “Rezzayo Product information”. Union Register of medicinal products. 22 December 2023. Retrieved 26 December 2023.
- ^ Zhao Y, Perlin DS (September 2020). “Review of the Novel Echinocandin Antifungal Rezafungin: Animal Studies and Clinical Data”. Journal of Fungi. 6 (4): 192. doi:10.3390/jof6040192. PMC 7712954. PMID 32998224.
- ^ Jump up to:a b Syed YY (June 2023). “Rezafungin: First Approval”. Drugs. 83 (9): 833–840. doi:10.1007/s40265-023-01891-8. PMID 37212966. S2CID 258831091.
- ^ “Rezzayo approved by FDA amid rapid Candida auris spread”. thepharmaletter.com. 23 March 2023.
- ^ Albanell-Fernández M (January 2025). “Echinocandins Pharmacokinetics: A Comprehensive Review of Micafungin, Caspofungin, Anidulafungin, and Rezafungin Population Pharmacokinetic Models and Dose Optimization in Special Populations”. Clinical Pharmacokinetics. 64 (1): 27–52. doi:10.1007/s40262-024-01461-5. PMC 11762474. PMID 39707078.
- ^ Grasselli Kmet N, Luzzati R, Monticelli J, Babich S, Conti J, Bella SD (March 2025). “Salvage therapy of complicated Candida albicans spondylodiscitis with Rezafungin”. European Journal of Clinical Microbiology & Infectious Diseases. doi:10.1007/s10096-025-05117-5. PMID 40163284.
- ^ Viceconte G, Buonomo AR, Esposito N, Cattaneo L, Somma T, Scirocco MM, et al. (April 2024). “Salvage Therapy with Rezafungin for Candida parapsilosis Spondylodiscitis: A Case Report from Expanded Access Program”. Microorganisms. 12 (5): 903. doi:10.3390/microorganisms12050903. PMC 11123963. PMID 38792732.
- ^ “Novel Drug Approvals for 2023”. U.S. Food and Drug Administration (FDA). 22 December 2023. Retrieved 27 December 2023.
- ^ “Drug Approval Package: Rezzayo”. U.S. Food and Drug Administration (FDA). 18 April 2023. Retrieved 27 December 2023.
- ^ New Drug Therapy Approvals 2023 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2024. Archived from the original on 10 January 2024. Retrieved 9 January 2024.
- ^ World Health Organization (2018). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 79”. WHO Drug Information. 32 (1). hdl:10665/330941.
External links
- “Rezafungin Injection”. U.S. Food and Drug Administration. 18 April 2023.
| Clinical data | |
|---|---|
| Trade names | Rezzayo |
| Other names | Biafungin; CD101 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a623021 |
| License data | US DailyMed: Rezafungin |
| Routes of administration | Intravenous |
| Drug class | Antifungal |
| ATC code | J02AX08 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1]EU: Rx-only[2][3] |
| Pharmacokinetic data | |
| Excretion | Feces |
| Identifiers | |
| CAS Number | 1396640-59-7 |
| PubChem CID | 78318119 |
| DrugBank | DB16310 |
| UNII | G013B5478J |
| KEGG | D11197 |
| ChEBI | CHEBI:229680 |
| Chemical and physical data | |
| Formula | C63H85N8O17+ |
| Molar mass | 1226.412 g·mol−1 |
- Lamoth F: Novel Therapeutic Approaches to Invasive Candidiasis: Considerations for the Clinician. Infect Drug Resist. 2023 Feb 22;16:1087-1097. doi: 10.2147/IDR.S375625. eCollection 2023. [Article]
- Miesel L, Lin KY, Ong V: Rezafungin treatment in mouse models of invasive candidiasis and aspergillosis: Insights on the PK/PD pharmacometrics of rezafungin efficacy. Pharmacol Res Perspect. 2019 Nov 20;7(6):e00546. doi: 10.1002/prp2.546. eCollection 2019 Dec. [Article]
- Thompson GR 3rd, Soriano A, Cornely OA, Kullberg BJ, Kollef M, Vazquez J, Honore PM, Bassetti M, Pullman J, Chayakulkeeree M, Poromanski I, Dignani C, Das AF, Sandison T, Pappas PG: Rezafungin versus caspofungin for treatment of candidaemia and invasive candidiasis (ReSTORE): a multicentre, double-blind, double-dummy, randomised phase 3 trial. Lancet. 2023 Jan 7;401(10370):49-59. doi: 10.1016/S0140-6736(22)02324-8. Epub 2022 Nov 25. [Article]
- Ong V, Wills S, Watson D, Sandison T, Flanagan S: Metabolism, Excretion, and Mass Balance of [(14)C]-Rezafungin in Animals and Humans. Antimicrob Agents Chemother. 2022 Jan 18;66(1):e0139021. doi: 10.1128/AAC.01390-21. Epub 2021 Oct 18. [Article]
- FDA Approved Drug Products: REZZAYO (rezafungin) injection for intravenous use (March 2023) [Link]
- Globe News Wire: Cidara Therapeutics and Melinta Therapeutics Announce FDA Approval of REZZAYO (rezafungin for injection) for the Treatment of Candidemia and Invasive Candidiasis [Link]
- EMA Summary of Product Characteristics: REZZAYO (rezafungin) solution for infusion [Link]
//////////Rezafungin, Rezzayo, APROVALS 2023, FDA 2023, Rezafungin ion, Rezafungin cation, CD 101, SP 3025, G013B5478J
Omaveloxolone



Omaveloxolone
CAS
1474034-05-3
N-[(4aS,6aR,6bS,8aR,12aS,14aR,14bS)-11-cyano-2,2,6a,6b,9,9,12a-heptamethyl-10,14-dioxo-1,2,3,4,4a,5,6,6a,6b,7,8,8a,9,10,12a,14,14a,14b-octadecahydropicen-4a-yl]-2,2-difluoropropanamide
N-[(4aS,6aR,6bS,8aR,12aS,14aR,14bS)-11-cyano-2,2,6a,6b,9,9,12a-heptamethyl-10,14-dioxo-1,3,4,5,6,7,8,8a,14a,14b-decahydropicen-4a-yl]-2,2-difluoropropanamide
FDA 2023, 2/28/2023, To treat Friedrich’s ataxia
Drug Trials Snapshot
WeightAverage: 554.723
Monoisotopic: 554.331999611
Chemical FormulaC33H44F2N2O3
- RTA 408
- RTA-408
- OriginatorDartmouth College; University of Texas M. D. Anderson Cancer Center
- DeveloperBiogen
- ClassAnalgesics; Anti-inflammatories; Antineoplastics; Eye disorder therapies; Neuroprotectants; Small molecules; Triterpenes
- Mechanism of ActionNF-E2-related factor 2 stimulants
- Orphan Drug StatusYes – Friedreich’s ataxia; Malignant melanoma
- MarketedFriedreich’s ataxia
- Phase IIMitochondrial disorders; Ocular inflammation; Ocular pain
- Phase I/IIMalignant melanoma
- PreclinicalBrain disorders
- DiscontinuedDuchenne muscular dystrophy; Non-small cell lung cancer; Radiation-induced skin damage
- 08 Apr 2025Biogen completes a phase I pharmacokinetics trial (In volunteers) in USA (PO) (NCT06612879)
- 17 Mar 2025Registered for Friedreich’s ataxia (In adolescents, In adults) in Canada (PO)
- 18 Oct 2024Biogen initiates enrolment in a phase I pharmacokinetics trial (In volunteers) in USA (PO) (NCT06612879)
Omaveloxolone, sold under the brand name Skyclarys, is a medication used for the treatment of Friedreich’s ataxia.[2][5] It is taken by mouth.[2]
The most common side effects include an increase in alanine transaminase and an increase of aspartate aminotransferase, which can be signs of liver damage, headache, nausea, abdominal pain, fatigue, diarrhea and musculoskeletal pain.[5]
Omaveloxolone was approved for medical use in the United States in February 2023,[2][5][6][7][8] and in the European Union in February 2024.[3] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication.[9]
SYNTHESIS

PATENT
Sheikh, AY et al. (2018). Bardoxolonmethyl-2,2-difluoropropionamide derivatives, polymorphe forms and procedures for use thereof. DK/EP 2989114 T3. Danish Patent and Trademark Office. Available at https://patentimages.storage.googleapis.com/51/87/43/97d0fb3e69ee73/DK2989114T3.pdf
https://patentscope.wipo.int/search/en/detail.jsf?docId=EP159939262&_cid=P21-MAKI10-93498-1

[0164] Reagents and conditions: (a) (PhO) 2PON 3 (DPPA), triethylamine, toluene, 0 °C for 5 minutes, then ambient temperature overnight, ∼94%; (b) benzene, 80 °C for 2 hours; (c) HCl, CH 3CN, ambient temperature for 1 hour; (d) CH 3CF 2CO 2H, dicyclohexylcarbodiimide, 4-(dimethylamino)pyridine, CH 2Cl 2, ambient temperature overnight, 73% from RTA 401 (4 steps).
[0165]Compound 1: RTA 401 (20.0 g, 40.6 mmol), triethylamine (17.0 mL, 122.0 mmol), and toluene (400 mL) were added into a reactor and cooled to 0 °C with stirring. Diphenyl phosphoryl azide (DPPA) (13.2 mL, 61.0 mmol) was added with stirring at 0 °C over 5 minutes, and the mixture was continually stirred at room temperature overnight (HPLC-MS check shows no RTA 401 left). The reaction mixture was directly loaded on a silica gel column and purified by column chromatography (silica gel, 0% to 5% ethyl acetate in CH 2Cl 2) to give compound 1 (19.7 g, ∼94%, partially converted into compound 2) as a white foam.
[0166]Compound 2: Compound 1 (19.7 g, ∼38.1 mmol) and benzene (250 mL) were added into a reactor and heated to 80 °C with stirring for 2 hours (HPLC-MS check shows no compound 1 left). The reaction mixture was concentrated at reduced pressure to afford crude compound 2 as a solid residue, which was used for the next step without purification.
[0167]Compound 3: Crude compound 2 (≤38.1 mmol) and CH 3CN (200 mL) were added into a reactor and cooled to 0 °C with stirring. HCl (12 N, 90 mL) was added at 0 °C over 1 minute, and the mixture was continually stirred at room temperature for 1 hour (HPLC-MS check shows no compound 2 left). The reaction mixture was cooled to 0 °C and 10% NaOH (∼500 mL) was added with stirring. Then, saturated NaHCO 3 (1 L) was added with stirring. The aqueous phase was extracted by ethyl acetate (2×500 mL). The combined organic phase was washed by H 2O (200 mL), saturated NaCl (200 mL), dried over Na 2SO 4, and concentrated to afford crude compound 3 (16.62 g) as a light yellow foam, which was used for the next step without purification.
[0168]RTA 408: Crude amine 3 (16.62 g, 35.9 mmol), CH 3CF 2CO 2H (4.7388 g, 43.1 mmol), and CH 2Cl 2 (360 mL) were added into a reactor with stirring at room temperature. Then, dicyclohexylcarbodiimide (DCC) (11.129 g, 53.9 mmol) and 4-(dimethylamino)pyridine (DMAP) (1.65 g, 13.64 mmol) were added and the mixture was continually stirred at room temperature overnight (HPLC-MS check shows no compound 3 left). The reaction mixture was filtered to remove solid by-products, and the filtrate was directly loaded on a silica gel column and purified by column chromatography (silica gel, 0% to 20% ethyl acetate in hexanes) twice to give compound RTA 408 (16.347 g, 73% from RTA 401 over 4 steps) as a white foam: 1H NMR (400 MHz, CD 3Cl) δ ppm 8.04 (s, 1H), 6.00 (s, 1H), 5.94 (s, br, 1H), 3.01 (d, 1H, J = 4.8 Hz), 2.75-2.82 (m, 1H), 1.92-2.18 (m, 4H), 1.69-1.85 (m, 7H), 1.53-1.64 (m, 1H), 1.60 (s, 3H), 1.50 (s, 3H), 1.42 (s, 3H), 1.11-1.38 (m, 3H), 1.27 (s, 3H), 1.18 (s, 3H), 1.06 (s, 3H), 1.04 (s, 3H), 0.92 (s, 3H); m/z 555 (M+1).
SYNTHESIS
J. Med. Chem. 2025, 68, 2147−2182
Omaveloxolone (Skyclarys). Omaveloxolone (6) was approved in February 2023 for the treatment of Friedreich’s Ataxia (FRDA), a genetic, neurodegenerative disease. Patients with FRDA have lowered activity of the frataxin gene (FXN), attributed to an expansion of a guanine-adenine-adenine (GAA)
triplet. The resulting decrease in frataxin limits the production of iron−sulfur clusters, leading to accumulation of iron in the mitochondria and oxidative stress which in turn leads to cell damageanddeath.49
Omaveloxoloneactivates the nuclear factor erythroid 2-related factor 2 (Nrf2), an important pathway in
oxidative stress. It acts by preventing ubiquitination and subsequent degradation of Nrf2, keeping levels high enough to counteract the oxidative stress associated with FRDA. 50
Omaveloxolone was developed by Reata Pharmaceuticals (which was acquired by Biogen in September 2023) and was granted orphan drug, fast track, priority review, and rare pediatric disease designations. 51Omaveloxolone (6) is a semisynthetic triterpenoid based on the oleanolic acid scaffold.52
advanced intermediate 6.1,The synthesis started from the53also known as CDDO orbardoxolone, which has individually been investigated fortherapeutic benefits from Nrf2 activation (Scheme 10).
Treatment of acid 6.1 with DPPA produced the azide, and subsequent heating in benzene generated isocyanate 6.2 via aCurtius rearrangement. Hydrolysis with aqueous acid generated amine 6.3, and an amidation with 2,2-difluoropropanoic acid produced omaveloxolone (6). A yield of 73% over the sequence was reported, and intermediates were used crude with no purification between steps.
(49) Ghanekar, S. D.; Miller, W. W.; Meyer, C. J.; Fenelon, K. J.;
Lacdao, A.; Zesiewicz, T. A. Orphan drugs in development for the
treatment of Friedreich’s ataxia: focus on omaveloxolone. Degener.
Neurol. Neuromuscular Dis. 2019, 9, 103−107.
(50) Abeti, R.; Baccaro, A.; Esteras, N.; Giunti, P. Novel Nrf2-inducer
prevents mitochondrial defects and oxidative stress in Friedreich’s
ataxia models. Front. Cell. Neurosci. 2018, 12, 188.
(51) Lee,A.Omaveloxolone:first approval. Drugs 2023, 83, 725−729.
(52) Anderson, E.; Decker, A.; Liu, X. Synthesis, pharmaceutical use,
and characterization of crystalline forms of 2,2-difluoropropionamide
derivatives of bardoxolone methyl. WO 2013163344, 2013.
(53) Honda, T.; Rounds, B. V.; Gribble, G. W.; Suh, N.; Wang, Y.;
Sporn, M. B. Design and synthesis of 2-cyano-3,12-dioxoolean-1,9
dien-28-oic acid, a novel and highly active inhibitor of nitric oxide
production in mouse macrophages. Bioorg. Med. Chem. Lett. 1998, 8,
2711−2714.

SYN
European Journal of Medicinal Chemistry 265 (2024) 116124
Omaveloxolone (Skyclarys)
Omaveloxolone was granted FDA approval on February 28, 2023, to treat Friedrich’s ataxia in individuals aged 16 and older [2]. Omaveloxolone possesses antioxidant and anti-inflammatory properties, making it a semi-synthetic triterpenoid compound. It has the ability to function as a stimulator of nuclear factor-erythroid 2 related factor 2(Nrf2), a transcription factor that reduces oxidative stress. In individuals
suffering from FA, a genetic disorder characterized by mitochondrial dysfunction, the Nrf2 pathway is compromised, leading to a decrease in Nrf2 activity. Hence, Omaveloxolone, an Nrf2 activator, can be
employed as a therapeutic option for the management of these in dividuals [23].The process route of Omaveloxolone is described below in Scheme 724]. The substitution reaction of carboxylic acid OMAV-001 with diphenylphosphoryl azide (DPPA) gave the acyl azide OMAV-002,which underwent Curtius-rearrangement under heating conditions to produce isocyanate OMAV-003. The amine OMAV-004 was obtained under acidic conditions. OMAV-004 was condensed with 2,2-difluoro propionic acid to obtain the final product Omaveloxolone.
[23] B.L. Probst, I. Trevino, L. McCauley, R. Bumeister, I. Dulubova, W.C. Wigley, D.
A. Ferguson, RTA 408, A novel synthetic triterpenoid with broad anticancer and
anti-inflammatory activity, PLoS One 10 (2015) e0122942.
[24] E. Anderson, A. Decker, X. Liu Synthesis, Pharmaceutical Use, and
Characterization of Crystalline Forms of 2,2-difluoropropionamide Derivatives of
Bardoxolone Methyl, 2013. WO2013163344.

.
Medical uses
Omaveloxolone is indicated for the treatment of Friedreich’s ataxia.[2][5]
Friedreich’s ataxia causes progressive damage to the spinal cord, peripheral nerves, and the brain, resulting in uncoordinated muscle movement, poor balance, difficulty walking, changes in speech and swallowing, and a shortened lifespan.[5] The condition can also cause heart disease.[5] This disease tends to develop in children and teenagers and gradually worsens over time.[5]
Although rare, Friedreich’s ataxia is the most common form of hereditary ataxia in the United States, affecting about one in every 50,000 people.[5]
Mechanism of action
The mechanism of action of omaveloxolone and its related compounds has been demonstrated to be through a combination of activation of the antioxidative transcription factor Nrf2 and inhibition of the pro-inflammatory transcription factor NF-κB.[10]
Nrf2 transcriptionally regulates multiple genes that play both direct and indirect roles in producing antioxidative potential and the production of cellular energy (i.e., adenosine triphosphate or ATP) within the mitochondria. Consequently, unlike exogenously administered antioxidants (e.g., vitamin E or Coenzyme Q10), which provide a specific and finite antioxidative potential, omaveloxolone, through Nrf2, broadly activates intracellular and mitochondrial antioxidative pathways, in addition to pathways that may directly increase mitochondrial biogenesis (such as PGC1α) and bioenergetics.[11]
History
Omaveloxolone is a second generation member of the synthetic oleanane triterpenoid compounds and in clinical development by Reata Pharmaceuticals. Preclinical studies have demonstrated that omaveloxolone possesses antioxidative and anti-inflammatory activities[10][12] and the ability to improve mitochondrial bioenergetics.[11] Omaveloxolone is under clinical investigation for a variety of indications, including Friedreich’s ataxia, mitochondrial myopathies, immunooncology, and prevention of corneal endothelial cell loss following cataract surgery.
The efficacy and safety of omaveloxolone was evaluated in a 48-week randomized, placebo-controlled, and double-blind study [Study 1 (NCT02255435)] and an open-label extension.[5] Study 1 enrolled 103 individuals with Friedreich’s ataxia who received placebo (52 individuals) or omaveloxolone 150 mg (51 individuals) for 48 weeks.[5] Of the research participants, 53% were male, 97% were white, and the mean age was 24 years at study entry.[5] Nine (18%) patients were younger than age 18.[5] The primary objective was to evaluate the change in the modified Friedreich’s Ataxia Rating Scale (mFARS) score compared to placebo at week 48.[5] The mFARS is a clinical assessment that measures disease progression, namely swallowing and speech (bulbar), upper limb coordination, lower limb coordination, and upright stability.[5] Individuals receiving omaveloxolone performed better on the mFARS than people receiving placebo.[5]
The US Food and Drug Administration (FDA) granted the application for omaveloxolone orphan drug, fast track, priority review, and rare pediatric disease designations.[5][9]
Society and culture
Legal status
Omaveloxolone was approved for medical use in the United States in February 2023.[2][5]
In December 2023, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Skyclarys, intended for the treatment of Friedreich’s ataxia.[3] The applicant for this medicinal product is Reata Ireland Limited.[3] Omaveloxolone was approved for medical use in the European Union in February 2024.[3][4]
References
- ^ “Register of Innovative Drugs”. Health Canada. 3 November 2006. Retrieved 17 April 2025.
- ^ Jump up to:a b c d e f “Skyclarys- omaveloxolone capsule”. DailyMed. 12 May 2023. Archived from the original on 1 July 2023. Retrieved 16 December 2023.
- ^ Jump up to:a b c d e “Skyclarys EPAR”. European Medicines Agency (EMA). 14 December 2023. Archived from the original on 15 December 2023. Retrieved 16 December 2023. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b “Skyclarys product information”. Union Register of medicinal products. 12 February 2024. Retrieved 19 February 2024.
- ^ Jump up to:a b c d e f g h i j k l m n o p q “FDA approves first treatment for Friedreich’s ataxia”. U.S. Food and Drug Administration (FDA). 28 February 2023. Archived from the original on 1 March 2023. Retrieved 28 February 2023. This article incorporates text from this source, which is in the public domain.
- ^ “Reata Pharmaceuticals Announces FDA Approval of Skyclarys (Omavaloxolone), the First and Only Drug Indicated for Patients with Friedreich’s Ataxia”. Reata Pharmaceuticals Inc. (Press release). 28 February 2023. Archived from the original on 1 March 2023. Retrieved 28 February 2023.
- ^ Lee A (June 2023). “Omaveloxolone: First Approval”. Drugs. 83 (8): 725–729. doi:10.1007/s40265-023-01874-9. PMID 37155124. S2CID 258567442. Archived from the original on 9 December 2023. Retrieved 16 December 2023.
- ^ Subramony SH, Lynch DL (May 2023). “A Milestone in the Treatment of Ataxias: Approval of Omaveloxolone for Friedreich Ataxia”. Cerebellum. 23 (2): 775–777. doi:10.1007/s12311-023-01568-8. PMID 37219716. S2CID 258843532.
- ^ Jump up to:a b New Drug Therapy Approvals 2023 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2024. Archived from the original on 10 January 2024. Retrieved 9 January 2024.
- ^ Jump up to:a b Reisman SA, Lee CY, Meyer CJ, Proksch JW, Ward KW (July 2014). “Topical application of the synthetic triterpenoid RTA 408 activates Nrf2 and induces cytoprotective genes in rat skin”. Archives of Dermatological Research. 306 (5): 447–454. doi:10.1007/s00403-013-1433-7. PMID 24362512. S2CID 25733020.
- ^ Jump up to:a b Neymotin A, Calingasan NY, Wille E, Naseri N, Petri S, Damiano M, et al. (July 2011). “Neuroprotective effect of Nrf2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis”. Free Radical Biology & Medicine. 51 (1): 88–96. doi:10.1016/j.freeradbiomed.2011.03.027. PMC 3109235. PMID 21457778.
- ^ Reisman SA, Lee CY, Meyer CJ, Proksch JW, Sonis ST, Ward KW (May 2014). “Topical application of the synthetic triterpenoid RTA 408 protects mice from radiation-induced dermatitis”. Radiation Research. 181 (5): 512–520. Bibcode:2014RadR..181..512R. doi:10.1667/RR13578.1. PMID 24720753. S2CID 23906747.
External links
Clinical trial number NCT02255435 for “RTA 408 Capsules in Patients With Friedreich’s Ataxia – MOXIe” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Skyclarys |
| Other names | RTA 408 |
| AHFS/Drugs.com | Monograph |
| License data | US DailyMed: Omaveloxolone |
| Routes of administration | By mouth |
| ATC code | N07XX25 (WHO) |
| Legal status | |
| Legal status | CA: ℞-only[1]US: ℞-only[2]EU: Rx-only[3][4] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1474034-05-3 |
| PubChem CID | 71811910 |
| IUPHAR/BPS | 7573 |
| DrugBank | DB12513 |
| ChemSpider | 34980948 |
| UNII | G69Z98951Q |
| KEGG | D10964 |
| ChEBI | CHEBI:229661 |
| CompTox Dashboard (EPA) | DTXSID101138251 |
| Chemical and physical data | |
| Formula | C33H44F2N2O3 |
| Molar mass | 554.723 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
- Zesiewicz TA, Hancock J, Ghanekar SD, Kuo SH, Dohse CA, Vega J: Emerging therapies in Friedreich’s Ataxia. Expert Rev Neurother. 2020 Dec;20(12):1215-1228. doi: 10.1080/14737175.2020.1821654. Epub 2020 Sep 21. [Article]
- Jiang Z, Qi G, Lu W, Wang H, Li D, Chen W, Ding L, Yang X, Yuan H, Zeng Q: Omaveloxolone inhibits IL-1beta-induced chondrocyte apoptosis through the Nrf2/ARE and NF-kappaB signalling pathways in vitro and attenuates osteoarthritis in vivo. Front Pharmacol. 2022 Sep 27;13:952950. doi: 10.3389/fphar.2022.952950. eCollection 2022. [Article]
- Shekh-Ahmad T, Eckel R, Dayalan Naidu S, Higgins M, Yamamoto M, Dinkova-Kostova AT, Kovac S, Abramov AY, Walker MC: KEAP1 inhibition is neuroprotective and suppresses the development of epilepsy. Brain. 2018 May 1;141(5):1390-1403. doi: 10.1093/brain/awy071. [Article]
- Probst BL, Trevino I, McCauley L, Bumeister R, Dulubova I, Wigley WC, Ferguson DA: RTA 408, A Novel Synthetic Triterpenoid with Broad Anticancer and Anti-Inflammatory Activity. PLoS One. 2015 Apr 21;10(4):e0122942. doi: 10.1371/journal.pone.0122942. eCollection 2015. [Article]
- Lynch DR, Farmer J, Hauser L, Blair IA, Wang QQ, Mesaros C, Snyder N, Boesch S, Chin M, Delatycki MB, Giunti P, Goldsberry A, Hoyle C, McBride MG, Nachbauer W, O’Grady M, Perlman S, Subramony SH, Wilmot GR, Zesiewicz T, Meyer C: Safety, pharmacodynamics, and potential benefit of omaveloxolone in Friedreich ataxia. Ann Clin Transl Neurol. 2018 Nov 10;6(1):15-26. doi: 10.1002/acn3.660. eCollection 2019 Jan. [Article]
- Zighan M, Arkadir D, Douiev L, Keller G, Miller C, Saada A: Variable effects of omaveloxolone (RTA408) on primary fibroblasts with mitochondrial defects. Front Mol Biosci. 2022 Aug 12;9:890653. doi: 10.3389/fmolb.2022.890653. eCollection 2022. [Article]
- FDA Approved Drug Products: SKYCLARYS (omaveloxolone) capsules for oral use (February 2023) [Link]
- EMA Approved Drug Products: Skyclarys (omaveloxolone) Oral Capsules [Link]
- Health Canada Approved Drug Products: SKYCLARYS (Omaveloxolone) Capsules For Oral Use [Link]
///////////Omaveloxolone, Skyclarys, Friedrich’s ataxia, FDA 2023, APPROVALS 2023, RTA 408, RTA-408, omaveloxolona, RTA 408, 63415, PP415, orphan drug, fast track, priority review, rare pediatric disease
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Elacestrant


Elacestrant
(6R)-6-[2-[ethyl-[[4-[2-(ethylamino)ethyl]phenyl]methyl]amino]-4-methoxyphenyl]-5,6,7,8-tetrahydronaphthalen-2-ol
(6R)-6-{2-[ethyl({4-[2-(ethylamino)ethyl]phenyl}methyl)amino]-4-methoxyphenyl}-5,6,7,8-tetrahydronaphthalen-2-ol
FDA 1/27/2023, Orserdu
WeightAverage: 458.646
Monoisotopic: 458.293328472
Chemical FormulaC30H38N2O2
To treat estrogen receptor-positive, human epidermal growth factor receptor 2-negative, ESR1-mutated, advanced or metastatic breast cancer with disease progression following at least one line of endocrine therapy
Drug Trials Snapshot
- CAS 722533-56-4
- RAD-1901
- ER-306323
- FM6A2627A8
- WHO 10247
Elacestrant, sold under the brand name Orserdu, is a selective estrogen receptor degrader (SERD) used in the treatment of breast cancer.[1][4] It is taken by mouth.[1][4]
Elacestrant is an antiestrogen that acts as an antagonist of estrogen receptors, which are the biological targets of endogenous estrogens like estradiol.[1] The most common side effects of elacestrant include body pain, nausea and vomiting, increased serum lipids, elevated liver enzymes, fatigue, decreased hemoglobin, raised creatinine, decreased appetite, diarrhea, headache, constipation, abdominal pain, and hot flashes.[2]
Elacestrant was approved for medical use in the United States in January 2023,[1][2][5][6] and in the European Union in September 2023.[3][7]
PATENTS
Cruskie MP, et al. (2019). Polymorphic forms of RAD1901-2HCl (U.S. Patent No. 10,385,008 B2). U.S. Patent and Trademark Office. https://patentimages.storage.googleapis.com/42/82/b6/e9fcbbbd08054e/US10385008.pdf
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US10071066 | No | 2018-09-11 | 2034-10-10 | |
| US10385008 | No | 2019-08-20 | 2038-01-05 | |
| US10420734 | No | 2019-09-24 | 2034-10-10 | |
| US10745343 | No | 2020-08-18 | 2038-01-05 | |
| US11779552 | No | 2023-10-10 | 2034-10-10 | |
| US11819480 | No | 2023-11-21 | 2036-11-29 | |
| US7612114 | No | 2009-11-03 | 2026-08-18 | |
| US8399520 | No | 2013-03-19 | 2023-12-25 | |

PATENT
https://patents.google.com/patent/US10385008B2/en
Medical uses
Elacestrant is indicated for the treatment of postmenopausal women or adult men with estrogen receptor (ER)-positive, human epidermal growth factor receptor 2 (HER2)-negative, ESR1–mutated, advanced or metastatic breast cancer with disease progression following at least one other line of endocrine therapy.[2][4]
Pharmacology
Pharmacodynamics
Elacestrant is an antiestrogen that acts as an antagonist of estrogen receptors, specifically targeting the estrogen receptor alpha (ERα), which is the biological target of endogenous estrogens like estradiol.[1] Additionally, elacestrant is a selective estrogen receptor degrader (SERD), meaning it induces the degradation of ERα.[1][8]
Pharmacokinetics
Elacestrant has an oral bioavailability of approximately 10%.[1] Its plasma protein binding exceeds 99% and remains independent of concentration.[1] Elacestrant is metabolized in the liver, primarily by the cytochrome P450 enzyme CYP3A4 and to a lesser extent by CYP2A6 and CYP2C9.[1] The elimination half-life of elacestrant is 30 to 50 hours.[1] It is excreted primarily in feces (82%) and to a lesser extent in urine (7.5%).[1]
History
The efficacy of elacestrant was evaluated in the EMERALD trial, which was a randomized, open-label, active-controlled, multicenter study involving 478 postmenopausal women and men with ER-positive, HER2-negative advanced or metastatic breast cancer. Among them, 228 participants had ESR1 mutations. Eligible participants had experienced disease progression on one or two prior lines of endocrine therapy, including one line with a CDK4/6 inhibitor, and could have received up to one prior line of chemotherapy in the advanced or metastatic setting.[2]
Participants were randomly assigned in a 1:1 ratio to receive either elacestrant 345 mg orally once daily or investigator’s choice of endocrine therapy. The choices for the control arm included fulvestrant, or an aromatase inhibitor. Randomization was stratified based on whether the ESR1 mutation was detected or not, prior treatment with fulvestrant, and presence of visceral metastasis.[2]
The FDA granted the application for elacestrant priority review and fast track designations.[2]
Research
It is a nonsteroidal combined selective estrogen receptor modulator (SERM) and selective estrogen receptor degrader (SERD) (described as a “SERM/SERD hybrid (SSH)”) that was discovered by Eisai and is under development by Radius Health and Takeda for the treatment estrogen receptor (ER)-positive advanced breast cancer.[9] Elacestrant has dose-dependent, tissue-selective estrogenic and antiestrogenic activities, with biphasic weak partial agonist activity at the ER at low doses and antagonist activity at higher doses.[10] It shows agonistic activity on bone and antagonistic activity on breast and uterine tissues.[11] Unlike the SERD fulvestrant, elacestrant is able to readily cross the blood-brain-barrier into the central nervous system, where it can target breast cancer metastases in the brain,[10][11] and is orally bioavailable and does not require intramuscular injection.[10][11]
References
- ^ Jump up to:a b c d e f g h i j k l m n o p q “Orserdu- elacestrant tablet, film coated”. DailyMed. 8 February 2023. Archived from the original on 11 February 2023. Retrieved 11 February 2023.
- ^ Jump up to:a b c d e f g “FDA approves elacestrant for ER-positive, HER2-negative, ESR1-mutated advanced or metastatic breast cancer”. U.S. Food and Drug Administration (FDA). 27 January 2023. Archived from the original on 2 February 2023. Retrieved 1 February 2023. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b “Orserdu Product information”. Union Register of medicinal products. 18 September 2023. Retrieved 1 October 2023.
- ^ Jump up to:a b c d “Orserdu EPAR”. European Medicines Agency (EMA). 9 October 2023. Retrieved 9 October 2023.
- ^ https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2023/217639Orig1s000ltr.pdf Archived 2023-02-02 at the Wayback Machine This article incorporates text from this source, which is in the public domain.
- ^ “Stemline Therapeutics Inc., a wholly owned subsidiary of Menarini Group, Receives Approval from U.S. FDA for Orserdu (elacestrant) as the First and Only Treatment Specifically Indicated for Patients with ESR1 Mutations in ER+, HER2- Advanced or Metastatic Breast Cancer”. Radius (Press release). 31 January 2023. Archived from the original on 2 February 2023. Retrieved 1 February 2023.
- ^ “EC approves Menarini Group’s Orserdu for advanced or metastatic breast cancer”. PMLive. 21 September 2023. Retrieved 22 September 2023.
- ^ Lloyd MR, Wander SA, Hamilton E, Razavi P, Bardia A (2022). “Next-generation selective estrogen receptor degraders and other novel endocrine therapies for management of metastatic hormone receptor-positive breast cancer: current and emerging role”. Therapeutic Advances in Medical Oncology. 14: 17588359221113694. doi:10.1177/17588359221113694. PMC 9340905. PMID 35923930.
- ^ Clinical trial number NCT03778931 for “Phase 3 Trial of Elacestrant vs. Standard of Care for the Treatment of Patients With ER+/HER2- Advanced Breast Cancer” at ClinicalTrials.gov
- ^ Jump up to:a b c Wardell SE, Nelson ER, Chao CA, Alley HM, McDonnell DP (October 2015). “Evaluation of the pharmacological activities of RAD1901, a selective estrogen receptor degrader”. Endocrine-Related Cancer. 22 (5): 713–724. doi:10.1530/ERC-15-0287. PMC 4545300. PMID 26162914.
- ^ Jump up to:a b c Garner F, Shomali M, Paquin D, Lyttle CR, Hattersley G (October 2015). “RAD1901: a novel, orally bioavailable selective estrogen receptor degrader that demonstrates antitumor activity in breast cancer xenograft models”. Anti-Cancer Drugs. 26 (9): 948–956. doi:10.1097/CAD.0000000000000271. PMC 4560273. PMID 26164151.
| Clinical data | |
|---|---|
| Pronunciation | /ˌɛləˈsɛstrənt/ EL-ə-SES-trənt |
| Trade names | Orserdu |
| Other names | RAD-1901; ER-306323 |
| License data | US DailyMed: Elacestrant |
| Routes of administration | By mouth |
| ATC code | L02BA04 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1][2]EU: Rx-only[3][4] |
| Pharmacokinetic data | |
| Bioavailability | ~10%[1] |
| Protein binding | >99%[1] |
| Metabolism | Liver (major: CYP3A4, minor: CYP2A6, CYP2C9)[1] |
| Elimination half-life | 30–50 hours[1] |
| Excretion | Feces (82%), urine (7.5%)[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 722533-56-4 |
| PubChem CID | 23642301 |
| DrugBank | DB06374 |
| ChemSpider | 57583807 |
| UNII | FM6A2627A8 |
| KEGG | D11671 |
| ChEMBL | ChEMBL4297509 |
| PDB ligand | I0V (PDBe, RCSB PDB) |
| CompTox Dashboard (EPA) | DTXSID901045846 |
| ECHA InfoCard | 100.312.890 |
| Chemical and physical data | |
| Formula | C30H38N2O2 |
| Molar mass | 458.646 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
/////////Elacestrant, Orserdu, FDA 2023, APPROVALS 2023, FM6A2627A8, WHO 10247, ER 306323, RAD 1901, RAD1901
Pirtobrutinib



Pirtobrutinib
- CAS 2101700-15-4
- JAYPIRCA
- RXC-005
- LY3527727
- LOXO-305
- WHO 11681
- WeightAverage: 479.436
- Monoisotopic: 479.158052208
- Chemical FormulaC22H21F4N5O3
5-amino-3-[4-[[(5-fluoro-2-methoxybenzoyl)amino]methyl]phenyl]-1-[(2S)-1,1,1-trifluoropropan-2-yl]pyrazole-4-carboxamide
FDA 2023, 1/27/2023, Jaypirca
To treat relapsed or refractory mantle cell lymphoma in adults who have had at least two lines of systemic therapy, including a BTK inhibitor
Drug Trials Snapshot
Pirtobrutinib, sold under the brand name Jaypirca, is an anticancer medication that is used to treat mantle cell lymphoma.[1][2][4] It inhibits B cell lymphocyte proliferation and survival by binding and inhibiting Bruton’s tyrosine kinase (BTK).[5] It is taken by mouth.[1]
The most common adverse reactions include fatigue, musculoskeletal pain, diarrhea, edema, dyspnea, pneumonia, and bruising.[4][6] The most common adverse reactions when used to treat chronic lymphocytic leukemia or small lymphocytic leukemia include fatigue, bruising, cough, musculoskeletal pain, COVID-19, diarrhea, pneumonia, abdominal pain, dyspnea, hemorrhage, edema, nausea, pyrexia, and headache.[7]
Pirtobrutinib was approved for medical use in the United States in January 2023,[4][8][9][10] and in the European Union in November 2023.[2]
PATENTS
Guisot, N. (2017). Compounds useful as kinase inhibitors (WO 2017/103611 A1). World Intellectual Property Organization. https://patentimages.storage.googleapis.com/d7/16/21/9300e49071a21a/WO2017103611A1.pdf
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017103611&_cid=P10-MAG7OA-80884-1


[00381] Example 120: 5-amino-3-[4-[[(5-fluoro-2-methoxy-benzoyl)amino]methyl]phenyl]-1- (2,2,2-trifluoro-1 -methyl-ethyl)pyrazole-4-carboxamide
N-[(2,2,2-Trifluoro-1-methyl-ethylidene)aminolbenzamide
General procedure S, benzhydrazide (49.9 mmol) and 1,1,1- trifluoroacetone (74.9 mmol) gave, after washing, the titled compound as a white solid. UPLC-MS (ES + , Short acidic): 1.45 min, m/z 230.9 [M+H] +
-Amino-3-[4-[[(5-fluoro-2-methoxy-benzoyl)amino]methyl]phenyl]-1-(2,2,2-trifluoro-1-methyl-ethyl)pyrazole-4-carboxamide
General procedure M, N-[[4-[5-amino-4-cyano-1-(2,2,2-trifluoro-1-methyl-ethyl)pyrazol-3-yl]phenyl]methyl]-5-fluoro-2-methoxy-benzamide (0.83 mmol) gave, after purification, the titled compound (0.42 mmol) as a white solid. UPLC-MS (ES + , Short acidic): 1.55 min, m/z 480.1 [M+H] + . UPLC-MS (ES + , Long acidic): 3.57 min, m/z 480.1 [M+H] + . 1 H NMR (400 MHz, DMSO-d 6 , δ): 8.84 (t, J = 6.1 Hz, 1H), 7.52 (dd, J = 9.2, 3.3 Hz, 1H), 7.48-7.41 (m, 4H), 7.37-7.32 (m, 1H), 7.19 (dd, J = 9.1, 4.3 Hz, 1H), 6.67 (s, 2H), 5.35-5.24 (m, 1H), 4.56 (d, J = 6.0 Hz, 2H), 3.90 (s, 3H), 1.62 (d, J = 6.9 Hz, 3H).
MORE



Medical uses
In the United States, pirtobrutinib is indicated to treat relapsed or refractory mantle cell lymphoma after at least two lines of systemic therapy, including a Bruton’s tyrosine kinase (BTK) inhibitor.[1][11] In December 2023, the US Food and Drug Administration (FDA) expanded the indication for pirtobrutinib to include the treatment of adults with chronic lymphocytic leukemia or small lymphocytic leukemia.[7][12]
In the European Union, pirtobrutinib is indicated for the treatment of mantle cell lymphoma.[2]
Mechanism of action
B cells are white cells of the lymphocyte subtype that produce antibodies, but when some of them grow uncontrollably they can be a cause of cancer. A key enzyme in B cell stimulation and survival is BTK, and pirtobrutinib inhibits BTK in a way that is different from the prototypical BTK inhibitor ibrutinib by binding in a different way that avoids a genetic change (mutation at active site cysteine residue C481 in BTK) that can make some tumors less responsive to ibrutinib.[5]
History
Pirtobrutinib is manufactured by Eli Lilly and Company and was approved by the US Food and Drug Administration in January 2023, for the treatment of mantle cell lymphoma that has become refractory to other BTK inhibitors.[13]
Efficacy was evaluated in BRUIN (NCT03740529), an open-label, multicenter, single-arm trial of pirtobrutinib monotherapy that included 120 participants with mantle cell lymphoma previously treated with a Bruton’s tyrosine kinase (BTK) inhibitor.[4] Participants had a median of three prior lines of therapy, with 93% having two or more prior lines.[4] The most common prior Bruton’s tyrosine kinase inhibitors received were ibrutinib (67%), acalabrutinib (30%), and zanubrutinib (8%); 83% had discontinued their last Bruton’s tyrosine kinase inhibitor due to refractory or progressive disease.[4] The trial was conducted at 49 sites in 10 countries in the United States, Europe, Australia, and Asia.[6] The same trial was used to assess safety and efficacy.[6]
Efficacy was evaluated in BRUIN (NCT03740529], an open-label, international, single-arm, multicohort trial that included 108 participants with chronic lymphocytic leukemia or small lymphocytic lymphoma previously treated with at least two prior lines of therapy, including a Bruton’s tyrosine kinase (BTK) inhibitor and a B-cell lymphoma-2 (BCL-2) inhibitor.[7] Participants received a median of five prior lines of therapy (range: 2 to 11).[7] Seventy-seven percent of participants discontinued the last BTK inhibitor for refractory or progressive disease.[7] Pirtobrutinib was administered orally at 200 mg once daily and was continued until disease progression or unacceptable toxicity.[7]
Society and culture
Legal status
In April 2023, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a conditional marketing authorization for the medicinal product Jaypirca, intended for the treatment of relapsed or refractory mantle cell lymphoma (MCL).[14] The applicant for this medicinal product is Eli Lilly Nederland B.V.[14] Pirtobrutinib was approved for medical use in the European Union in November 2023.[2]
References
- ^ Jump up to:a b c d “Jaypirca- pirtobrutinib tablet, coated”. DailyMed. 27 January 2023. Archived from the original on 11 February 2023. Retrieved 11 February 2023.
- ^ Jump up to:a b c d e “Jaypirca EPAR”. European Medicines Agency (EMA). 20 November 2023. Archived from the original on 22 November 2023. Retrieved 22 November 2023.
- ^ “Jaypirca Product information”. Union Register of medicinal products. 31 October 2023. Archived from the original on 22 November 2023. Retrieved 22 November 2023.
- ^ Jump up to:a b c d e f “FDA grants accelerated approval to pirtobrutinib for relapsed or refractory mantle cell lymphoma”. FDA. 27 January 2023. Archived from the original on 28 January 2023. Retrieved 28 January 2023. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b Aslan B, Kismali G, Iles LR, Manyam GC, Ayres ML, Chen LS, et al. (May 2022). “Pirtobrutinib inhibits wild-type and mutant Bruton’s tyrosine kinase-mediated signaling in chronic lymphocytic leukemia”. Blood Cancer Journal. 12 (5): 80. doi:10.1038/s41408-022-00675-9. PMC 9123190. PMID 35595730.
- ^ Jump up to:a b c “Drug Trials Snapshots: Jaypirca”. U.S. Food and Drug Administration (FDA). 27 January 2023. Retrieved 13 May 2024.
- ^ Jump up to:a b c d e f “FDA grants accelerated approval to pirtobrutinib for chronic lymphocytic leukemia and small lymphocytic lymphoma”. U.S. Food and Drug Administration (FDA). 1 December 2023. Archived from the original on 3 December 2023. Retrieved 3 December 2023. This article incorporates text from this source, which is in the public domain.
- ^ “U.S. FDA Approves Jaypirca (pirtobrutinib), the First and Only Non-Covalent (Reversible) BTK Inhibitor, for Adult Patients with Relapsed or Refractory Mantle Cell Lymphoma After at Least Two Lines of Systemic Therapy, Including a BTK Inhibitor” (Press release). Eli Lilly. 27 January 2023. Archived from the original on 30 January 2023. Retrieved 31 January 2023 – via PR Newswire.
- ^ Keam SJ (April 2023). “Pirtobrutinib: First Approval”. Drugs. 83 (6): 547–553. doi:10.1007/s40265-023-01860-1. PMID 37004673. S2CID 257912433. Archived from the original on 19 November 2023. Retrieved 19 November 2023.
- ^ Telaraja D, Kasamon YL, Collazo JS, Leong R, Wang K, Li P, et al. (August 2023). “FDA Approval Summary: Pirtobrutinib for Relapsed or Refractory Mantle Cell Lymphoma”. Clinical Cancer Research. 30 (1): OF1 – OF6. doi:10.1158/1078-0432.CCR-23-1272. PMC 10841293. PMID 37624619. S2CID 265965744.
- ^ De SK (October 2023). “Pirtobrutinib: First Non-covalent Tyrosine Kinase Inhibitor for Treating Relapsed or Refractory Mantle Cell Lymphoma in Adults”. Current Medicinal Chemistry. 31. doi:10.2174/0109298673251030231004052822. PMID 37818564. S2CID 263828536.
- ^ “Jaypirca (pirtobrutinib) Now Approved by U.S. FDA for the Treatment of Adult Patients with Chronic Lymphocytic Leukemia or Small Lymphocytic Lymphoma Who Have Received at Least Two Lines of Therapy, Including a BTK Inhibitor and a BCL-2 Inhibitor” (Press release). Eli Lilly. 1 December 2023. Archived from the original on 3 December 2023. Retrieved 3 December 2023 – via PR Newswire.
- ^ “FDA approves Eli Lilly’s drug for rare blood cancer”. Reuters. 27 January 2023. Archived from the original on 28 January 2023.
- ^ Jump up to:a b “Jaypirca: Pending EC decision”. European Medicines Agency. 26 April 2023. Archived from the original on 26 April 2023. Retrieved 27 April 2023. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
Further reading
- Mato AR, Woyach JA, Brown JR, Ghia P, Patel K, Eyre TA, et al. (July 2023). “Pirtobrutinib after a Covalent BTK Inhibitor in Chronic Lymphocytic Leukemia”. The New England Journal of Medicine. 389 (1): 33–44. doi:10.1056/NEJMoa2300696. hdl:11585/960014. PMID 37407001. S2CID 259358462.
External links
- “Pirtobrutinib”. National Cancer Institute. 16 February 2023.
- “Pirtobrutinib”. NCI Drug Dictionary.
| Clinical data | |
|---|---|
| Trade names | Jaypirca |
| Other names | LOXO-305 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a623012 |
| License data | US DailyMed: Pirtobrutinib |
| Routes of administration | By mouth |
| Drug class | Protein kinase inhibitor |
| ATC code | L01EL05 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1]EU: Rx-only[2][3] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2101700-15-4 |
| PubChem CID | 129269915 |
| DrugBank | DB17472 |
| ChemSpider | 114875989 |
| UNII | JNA39I7ZVB |
| KEGG | D12050 |
| ChEBI | CHEBI:229212 |
| ChEMBL | ChEMBL4650485 |
| PDB ligand | Y7W (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C22H21F4N5O3 |
| Molar mass | 479.436 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
- Jensen JL, Mato AR, Pena C, Roeker LE, Coombs CC: The potential of pirtobrutinib in multiple B-cell malignancies. Ther Adv Hematol. 2022 Jun 16;13:20406207221101697. doi: 10.1177/20406207221101697. eCollection 2022. [Article]
- Aslan B, Kismali G, Iles LR, Manyam GC, Ayres ML, Chen LS, Gagea M, Bertilaccio MTS, Wierda WG, Gandhi V: Pirtobrutinib inhibits wild-type and mutant Bruton’s tyrosine kinase-mediated signaling in chronic lymphocytic leukemia. Blood Cancer J. 2022 May 20;12(5):80. doi: 10.1038/s41408-022-00675-9. [Article]
- Alu A, Lei H, Han X, Wei Y, Wei X: BTK inhibitors in the treatment of hematological malignancies and inflammatory diseases: mechanisms and clinical studies. J Hematol Oncol. 2022 Oct 1;15(1):138. doi: 10.1186/s13045-022-01353-w. [Article]
- Mato AR, Shah NN, Jurczak W, Cheah CY, Pagel JM, Woyach JA, Fakhri B, Eyre TA, Lamanna N, Patel MR, Alencar A, Lech-Maranda E, Wierda WG, Coombs CC, Gerson JN, Ghia P, Le Gouill S, Lewis DJ, Sundaram S, Cohen JB, Flinn IW, Tam CS, Barve MA, Kuss B, Taylor J, Abdel-Wahab O, Schuster SJ, Palomba ML, Lewis KL, Roeker LE, Davids MS, Tan XN, Fenske TS, Wallin J, Tsai DE, Ku NC, Zhu E, Chen J, Yin M, Nair B, Ebata K, Marella N, Brown JR, Wang M: Pirtobrutinib in relapsed or refractory B-cell malignancies (BRUIN): a phase 1/2 study. Lancet. 2021 Mar 6;397(10277):892-901. doi: 10.1016/S0140-6736(21)00224-5. [Article]
- Wang E, Mi X, Thompson MC, Montoya S, Notti RQ, Afaghani J, Durham BH, Penson A, Witkowski MT, Lu SX, Bourcier J, Hogg SJ, Erickson C, Cui D, Cho H, Singer M, Totiger TM, Chaudhry S, Geyer M, Alencar A, Linley AJ, Palomba ML, Coombs CC, Park JH, Zelenetz A, Roeker L, Rosendahl M, Tsai DE, Ebata K, Brandhuber B, Hyman DM, Aifantis I, Mato A, Taylor J, Abdel-Wahab O: Mechanisms of Resistance to Noncovalent Bruton’s Tyrosine Kinase Inhibitors. N Engl J Med. 2022 Feb 24;386(8):735-743. doi: 10.1056/NEJMoa2114110. [Article]
- FDA Approved Drug Products: JAYPIRCA (pirtobrutinib) tablets for oral use [Link]
- BioSpace: U.S. FDA Approves Jaypirca (pirtobrutinib), the First and Only Non-Covalent (Reversible) BTK Inhibitor, for Adult Patients with Relapsed or Refractory Mantle Cell Lymphoma After at Least Two Lines of Systemic Therapy, Including a BTK Inhibitor [Link]
//////////////Jaypirca, FDA 2023, APPROVALS 2023, Pirtobrutinib, RXC-005, LY3527727, LOXO-305, LOXO 305, WHO 11681
Capivasertib
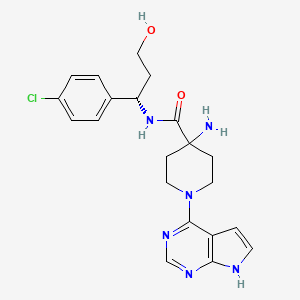
Capivasertib
C21H25ClN6O2
428.915
- 1143532-39-1
AZD 5363
4-amino-N-[(1S)-1-(4-chlorophenyl)-3-hydroxypropyl]-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide
(S)-4-AMINO-N-(1-(4-CHLOROPHENYL)-3-HYDROXYPROPYL)-1-(7H-PYRROLO[2,3-D]PYRIMIDIN-4-YL)PIPERIDINE-4-CARBOXAMIDE
FDA APPROVED 11/16/2023, To treat breast cancer that meets certain disease criteria, Truqap
Capivasertib, sold under the brand name Truqap, is an anti-cancer medication used for the treatment of breast cancer.[1][2]
The most common adverse reactions include diarrhea, cutaneous adverse reactions, increased random glucose, decreased lymphocytes, decreased hemoglobin, increased fasting glucose, nausea, fatigue, decreased leukocytes, increased triglycerides, decreased neutrophils, increased creatinine, vomiting, and stomatitis.[3]
In November 2023, capivasertib was approved in the United States for people with hormone receptor-positive, human epidermal growth factor receptor 2-negative breast cancer when used in combination with fulvestrant.[3][4][5]
Capivasertib is a novel pyrrolopyrimidine derivative, and an orally available inhibitor of the serine/threonine protein kinase AKT (protein kinase B) with potential antineoplastic activity. Capivasertib binds to and inhibits all AKT isoforms. Inhibition of AKT prevents the phosphorylation of AKT substrates that mediate cellular processes, such as cell division, apoptosis, and glucose and fatty acid metabolism. A wide range of solid and hematological malignancies show dysregulated PI3K/AKT/mTOR signaling due to mutations in multiple signaling components. By targeting AKT, the key node in the PIK3/AKT signaling network, this agent may be used as monotherapy or combination therapy for a variety of human cancers.
Medical uses
Capivasertib, used in combination with fulvestrant (Faslodex), is indicated for adults with hormone receptor-positive, human epidermal growth factor receptor 2-negative locally advanced or metastatic breast cancer with one or more PIK3CA/AKT1/PTEN-alterations, as detected by an FDA-approved test, following progression on at least one endocrine-based regimen in the metastatic setting or recurrence on or within twelve months of completing adjuvant therapy.[1][3]
History
Efficacy was evaluated in CAPItello-291 (NCT04305496), a randomized, double-blind, placebo-controlled, multicenter trial in 708 participants with locally advanced or metastatic HR-positive, HER2-negative breast cancer, of which 289 participants had tumors with PIK3CA/AKT1/PTEN-alterations.[3] All participants were required to have progression on aromatase inhibitor-based treatment.[3] Participants could have received up to two prior lines of endocrine therapy and up to one line of chemotherapy for locally advanced or metastatic disease.[3]
PATENT
EXAMPLE 9: (S)-4-AMINO-N-(1-(4-CHLOROPHENYL)-3-HYDROXYPROPYL)-1-(7H-PYRROLO[2,3-D]PYRIMIDIN-4-YL)PIPERIDINE-4-CARBOXAMIDE (E9)
EXAMPLE 9 ALTERNATIVE ROUTE 1: (S)-4-AMINO-N-(1-(4-CHLOROPHENYL)-3-HYDROXYPROPYL)-1-(7H-PYRROLO[2,3-D]PYRIMIDIN-4-YL)PIPERIDINE-4-CARBOXAMIDE
EXAMPLE 9 ALTERNATIVE ROUTE 2: (S)-4-AMINO-N-(1-(4-CHLOROPHENYL)-3-HYDROXYPROPYL)-1-(7H-PYRROLO[2,3-D]PYRIMIDIN-4-YL)PIPERIDINE-4-CARBOXAMIDE
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
//////////
| Clinical data | |
|---|---|
| Trade names | Truqap |
| Other names | AZD-5363 |
| AHFS/Drugs.com | Truqap |
| License data | US DailyMed: Capivasertib |
| Routes of administration | By mouth |
| Drug class | Threonine kinase inhibitor |
| ATC code | L01EX27 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1143532-39-1 |
| PubChem CID | 25227436 |
| DrugBank | DB12218 |
| ChemSpider | 28189073 |
| UNII | WFR23M21IE |
| KEGG | D11371 |
| ChEMBL | ChEMBL2325741 |
| PDB ligand | 0XZ (PDBe, RCSB PDB) |
| CompTox Dashboard (EPA) | DTXSID40150710 |
| ECHA InfoCard | 100.208.066 |
| Chemical and physical data | |
| Formula | C21H25ClN6O2 |
| Molar mass | 428.92 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
References
- ^ Jump up to:a b c “Truqap- capivasertib tablet, film coated”. DailyMed. 16 November 2023. Archived from the original on 20 November 2023. Retrieved 20 November 2023.
- ^ Turner NC, Oliveira M, Howell SJ, Dalenc F, Cortes J, Gomez Moreno HL, et al. (June 2023). “Capivasertib in Hormone Receptor–Positive Advanced Breast Cancer”. New England Journal of Medicine. 388 (22): 2058–2070. doi:10.1056/NEJMoa2214131. PMID 37256976. S2CID 259002400.
- ^ Jump up to:a b c d e f “FDA approves capivasertib with fulvestrant for breast cancer”. U.S. Food and Drug Administration. 16 November 2023. Archived from the original on 17 November 2023. Retrieved 17 November 2023.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Oncology (Cancer) / Hematologic Malignancies Approval Notifications”. U.S. Food and Drug Administration. 16 November 2023. Archived from the original on 17 November 2023. Retrieved 17 November 2023.
- ^ “Truqap (capivasertib) plus Faslodex approved in the US for patients with advanced HR-positive breast cancer”. AstraZeneca (Press release). 17 November 2023. Archived from the original on 17 November 2023. Retrieved 17 November 2023.
External links
- Clinical trial number NCT04305496 for “Capivasertib+Fulvestrant vs Placebo+Fulvestrant as Treatment for Locally Advanced (Inoperable) or Metastatic HR+/HER2- Breast Cancer (CAPItello-291)” at ClinicalTrials.gov
///////Capivasertib, Truqap, FDA 2023, APPROVALS 2023, AZD 5363
NC1(CCN(CC1)C1=C2C=CNC2=NC=N1)C(=O)N[C@@H](CCO)C1=CC=C(Cl)C=C1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}