
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 9)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Sendegobresib
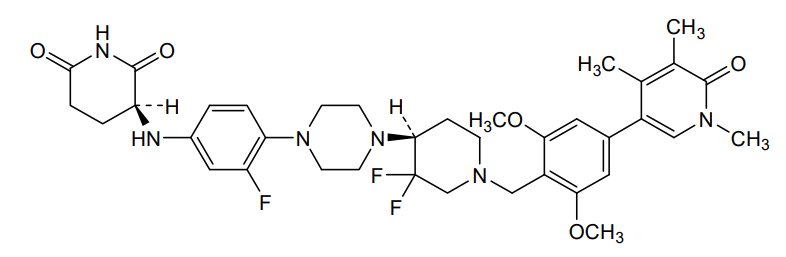
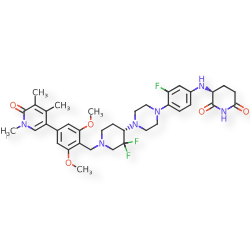
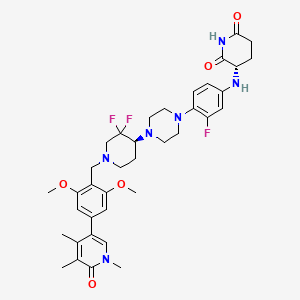
Sendegobresib
CAS 2704617-96-7
MFC37H45F3N6O5, 710.79
2,6-PIPERIDINEDIONE, 3-((4-(4-((4S)-1-((4-(1,6-DIHYDRO-1,4,5-TRIMETHYL-6-OXO-3-PYRIDINYL)-2,6-DIMETHOXYPHENYL)METHYL)-3,3-DIFLUORO-4-PIPERIDINYL)-1-PIPERAZINYL)-3-FLUOROPHENYL)AMINO)-, (3S)-
(3S)-3-[4-[4-[(4S)-1-[[2,6-dimethoxy-4-(1,4,5-trimethyl-6-oxo-3-pyridinyl)phenyl]methyl]-3,3-difluoropiperidin-4-yl]piperazin-1-yl]-3-fluoroanilino]piperidine-2,6-dione

bromodomain-containing protein 9 (BRD9) degradation inducer, antineoplastic, AW8PEP3VZ3, CFT 8634, ORPHAN DRUG
Sendegobresib is an orally bioavailable heterobifunctional protein degrader of bromodomain-containing protein 9 (BRD9; sarcoma antigen NY-SAR-29; rhabdomyosarcoma antigen MU-RMS-40.8), with potential antineoplastic activity. Sendegobresib is comprised of an E3 ligase-binding moiety and a BRD9-binding moiety. Upon oral administration, sendegobresib targets and binds to BRD9 with its BRD9-binding moiety. Upon BRD9 binding, the E3 ligase-binding moiety binds to cereblon (CRBN), a component of the CRL4-CRBN E3 ubiquitin ligase complex, which directs proteins for destruction, resulting in the proteasome-mediated degradation of BRD9. This leads to an inhibition of the growth of tumor cells that rely on BRD9 for survival. BRD9, a component of one form of the Brg/Brahma-Associated Factor (BAF) complex, is needed for the survival of certain cancer cells due to mutations.
A Study to Assess the Safety and Tolerability of CFT8634 in Locally Advanced or Metastatic SMARCB1-Perturbed Cancers, Including Synovial Sarcoma and SMARCB1-Null Tumors
CTID: NCT05355753
Phase: Phase 1
Status: Terminated
Date: 2024-12-17
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US355912448&_cid=P11-MINYJY-62955-1
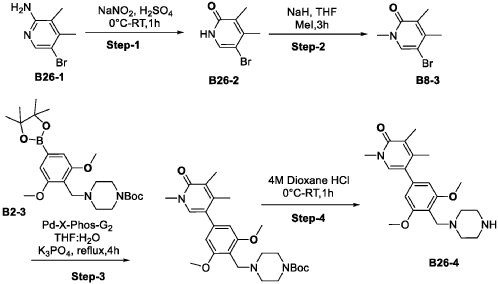
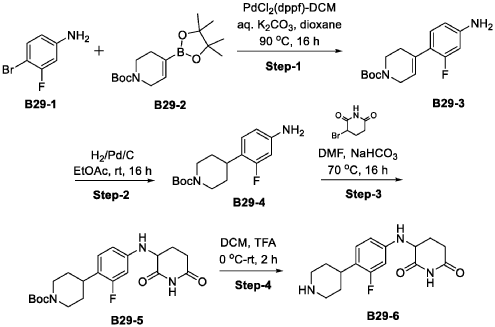
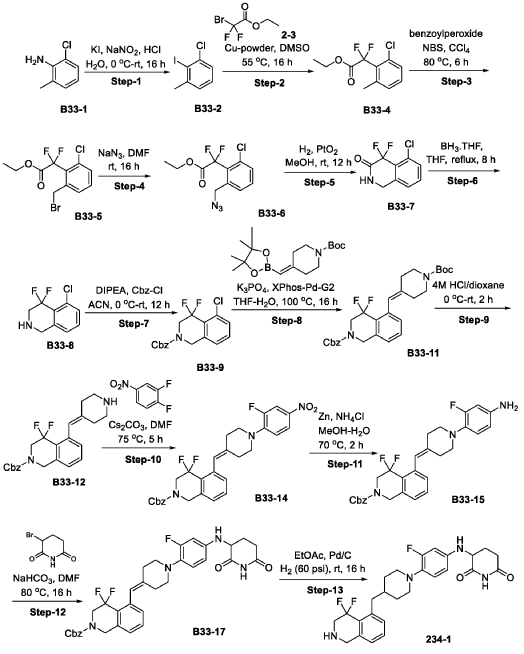
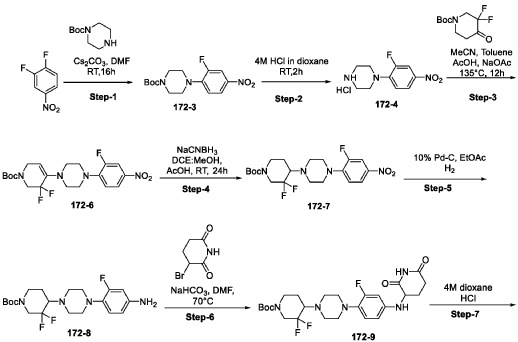
Synthesis of Compound 172
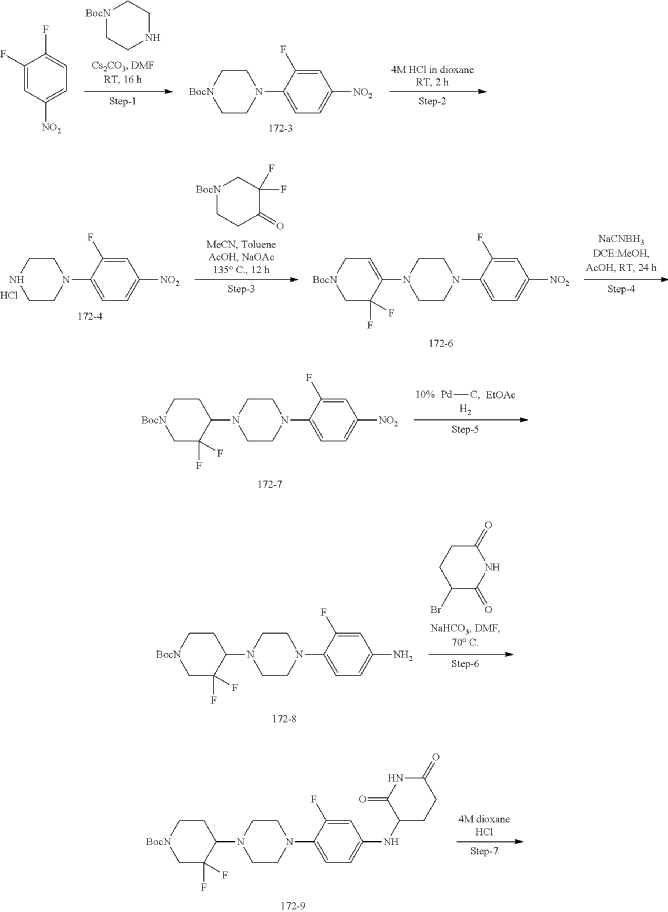
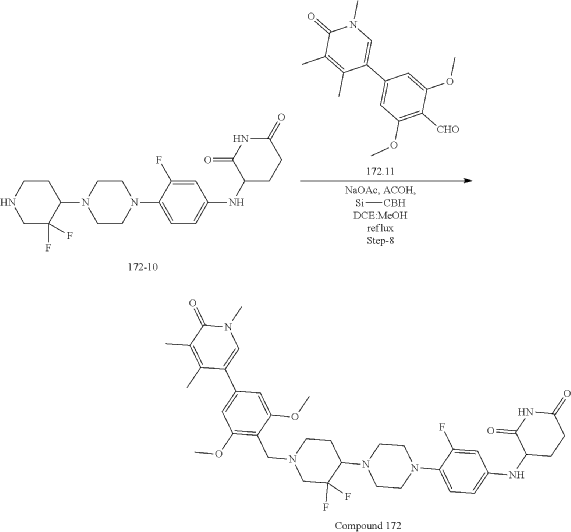
| Step-1: To a stirred solution of compound tert-butyl piperazine-1-carboxylate (85.40 g, 536.82 mmol) in DMF (500 mL) was added cesium carbonate (262.4 g, 805.4 mmol) and stirred for 15 min before adding 1,2-difluoro-4-nitro-benzene (100 g, 536.82 mmol). The reaction mixture stirred at RT for 16 h while monitoring by TLC. After completion, the reaction mass was quenched with ice flakes and the precipitated solid was filtered, dried under vacuum to afford tert-butyl 4-(2-fluoro-4-nitro-phenyl) piperazine-1-carboxylate 172-3 (152 g, 88.85% yield, 97.94% purity) as a yellow solid. |
PAT
PAT
PAT
- Enhanced hyt-induced protein degradation using lipid nanoparticle deliveryPublication Number: US-2023414723-A1Priority Date: 2020-10-26
- Compounds for targeted degradation of brd9Publication Number: WO-2021178920-A1Priority Date: 2020-03-05
- Compounds for targeted degradation of brd9Publication Number: US-2022098194-A1Priority Date: 2020-03-05
- Compounds for targeted degradation of brd9Publication Number: US-2023060334-A1Priority Date: 2020-03-05
- Compounds for targeted degradation of BRD9Publication Number: US-11691972-B2Priority Date: 2020-03-05Grant Date: 2023-07-04
- Selected compounds for targeted degradation of brd9Publication Number: US-2024245677-A1Priority Date: 2021-09-09
- Exosome-based cancer assaysPublication Number: US-11938164-B2Priority Date: 2021-04-07Grant Date: 2024-03-26
- Exosome-based cancer assaysPublication Number: US-2022331390-A1Priority Date: 2021-04-07
- Exosome-based cancer assaysPublication Number: WO-2022216765-A1Priority Date: 2021-04-07
- Enhanced hyt-induced protein degradation using lipid nanoparticle deliveryPublication Number: WO-2022093809-A1Priority Date: 2020-10-26
- Directed degron molecules and applications thereofPublication Number: WO-2023081400-A1Priority Date: 2021-11-04
- Directed degron molecules and applications thereofPublication Number: WO-2023081400-A9Priority Date: 2021-11-04
- Directed degron molecules and applications thereofPublication Number: EP-4426687-A1Priority Date: 2021-11-04
- Selected compounds for targeted degradation of brd9Publication Number: WO-2023039208-A1Priority Date: 2021-09-09
- Selected compounds for targeted degradation of brd9Publication Number: EP-4398904-A1Priority Date: 2021-09-09



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
/////////Sendegobresib, antineoplastic, AW8PEP3VZ3, CFT 8634, ORPHAN DRUG
Segigratinib, Ratangratinib
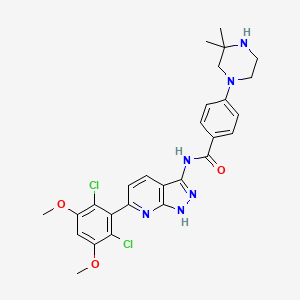
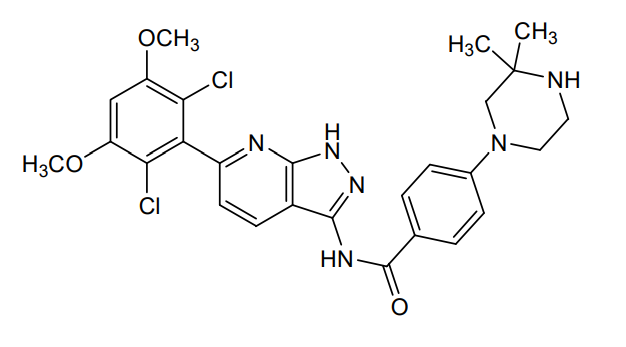
Segigratinib, Ratangratinib
CAS 1882873-93-9
MF C27H28Cl2N6O3 MW 555.5 g/mol
N-[6-(2,6-dichloro-3,5-dimethoxyphenyl)-1H-pyrazolo[3,4-b]pyridin-3-yl]-4-(3,3-dimethylpiperazin-1-yl)benzamide
N-[6-(2,6-dichloro-3,5-dimethoxyphenyl)-1H-pyrazolo[5,4-b]pyridin-3-yl]-4-(3,3-dimethylpiperazin-1-yl)benzamide
fibroblast growth factor receptor tyrosine kinase inhibitor, antineoplastic, 3D 185, Ratangratinib, 3D-185, G0Z5E4YTB4, HH 185
Ratangratinib is an orally bioavailable inhibitor of the fibroblast growth factor receptor (FGFR) types 1, 2, and 3 (FGFR1/2/3) and colony stimulating factor 1 receptor (CSF1R; CSF-1R; CD115; M-CSFR), with potential immunomodulatory and antineoplastic activities. Upon administration, ratangratinib binds to and inhibits FGFR1/2/3, which may result in the inhibition of FGFR1/2/3-mediated signal transduction pathways. This inhibits proliferation in FGFR1/2/3-overexpressing tumor cells. 3D185 also targets and binds to CSF1R, thereby blocking CSF1R activation and CSF1R-mediated signaling. This inhibits the activities of tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs), and prevents immune suppression in the tumor microenvironment (TME). This enhances antitumor T-cell immune responses and inhibits the proliferation of tumor cells. FGFR, a family of receptor tyrosine kinases (RTKs) upregulated in many tumor cell types, plays a key role in cellular proliferation, migration and survival. CSF1R, also known as macrophage colony-stimulating factor receptor (M-CSFR) and CD115 (cluster of differentiation 115), is a cell-surface receptor that plays major roles in tumor cell proliferation and metastasis.
Efficacy and Safety of 3D185 Monotherapy in Subjects With Previously Treated Locally Advanced or Metastatic Cholangiocarcinoma
CTID: NCT05039892
Phase: Phase 2
Status: Not yet recruiting
Date: 2025-05-20
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016026445&_cid=P20-MIMK7T-68502-1
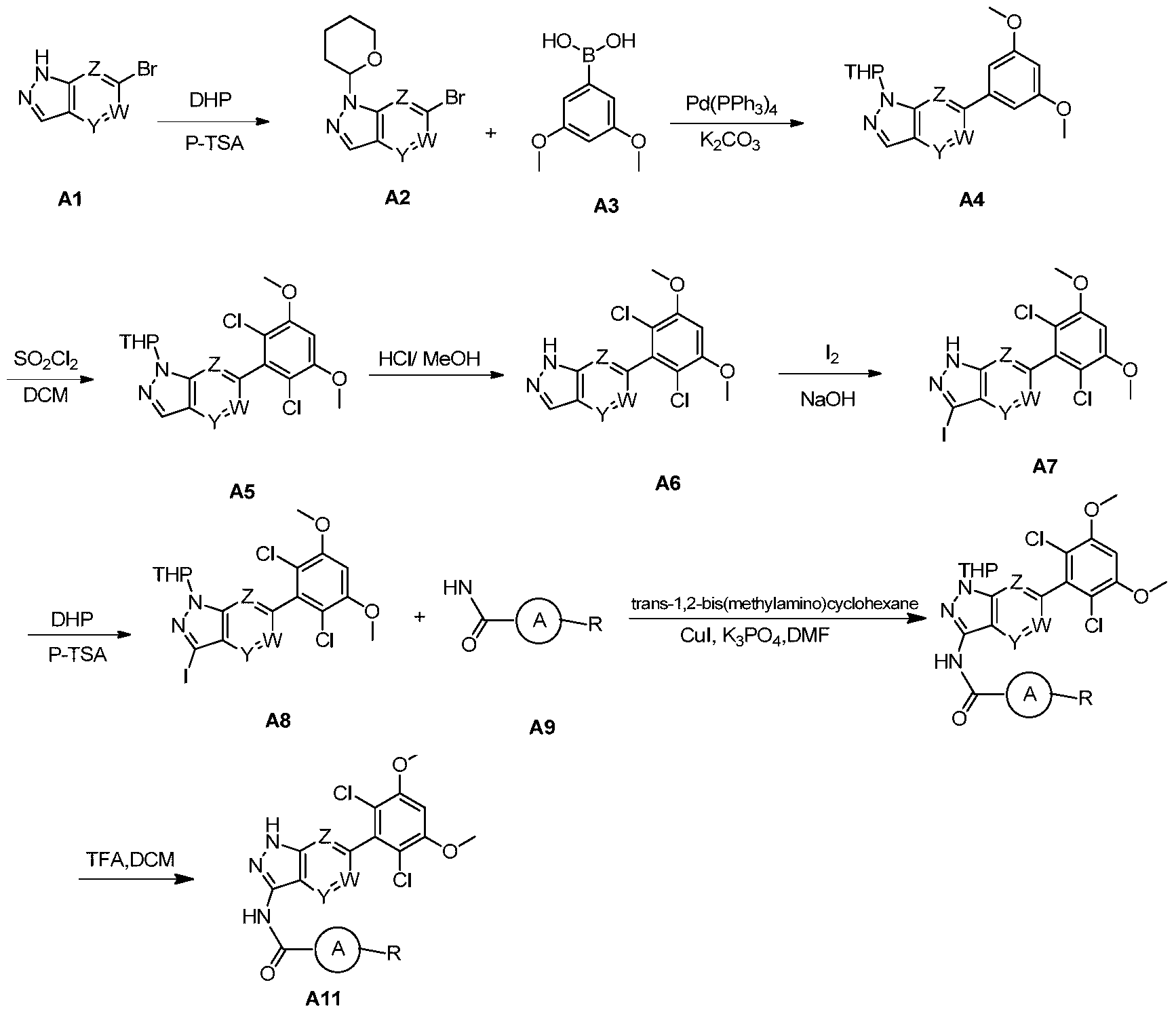
N-(6-(2,6-dichloro-3,5-dimethoxyphenyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)-6-(3,3-dimethylpiperazin-1-yl)nicotinamide
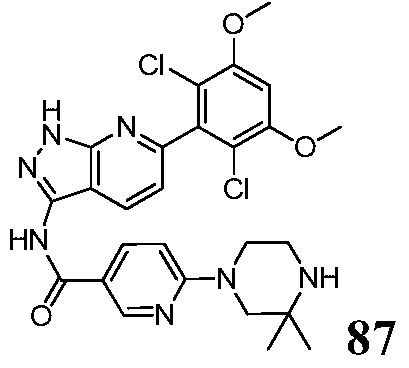
1H NMR(DMSO-d6,400MHz)δppm 13.39(s,1H),10.86(s,1H),8.81(d,1H,J=2.0Hz),8.40(d,1H,J=8.0Hz),8.16(dd,1H,J 1=2.4Hz,J 2=2.4Hz),7.08(t,2H,J=8.4Hz),6.90(d,1H,J=9.2Hz),3.99(s,6H),3.60(t,2H,J=4.0Hz),3.43(s,2H),2.82(t,2H,J=4.4Hz),1.04(s,6H).LCMS:556.2[M+H] +,RT=1.21min。
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US204149576&_cid=P20-MIMK4B-66027-1
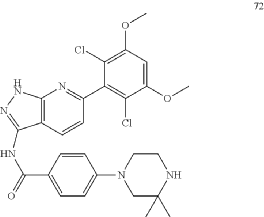
1H NMR (d-MeOD, 400 MHz) δ ppm 8.52 (d, J=8.0 Hz, 1H), 8.03 (d, J=8.0 Hz, 2H), 7.15-7.13 (m, 3H), 6.94 (s, 1H), 3.99 (s, 6H), 3.58-3.57 (m, 2H), 3.44-3.40 (m, 4H), 10.50 (s, 6H).
PAT
- Indazole compounds as fgfr kinase inhibitor, preparation and use thereofPublication Number: EP-3184521-A1Priority Date: 2014-08-19
- Indazole compounds as FGFR kinase inhibitor, preparation and use thereofPublication Number: US-10562900-B2Priority Date: 2014-08-19Grant Date: 2020-02-18
- Indazole compounds as fgfr kinase inhibitor, preparation and use thereofPublication Number: US-2017275291-A1Priority Date: 2014-08-19
- Indazole compounds as fgfr kinase inhibitor, preparation and use thereofPublication Number: WO-2016026445-A1Priority Date: 2014-08-19
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
////////segigratinib, antineoplastic, 3D 185, Ratangratinib, 3D-185, G0Z5E4YTB4, HH 185
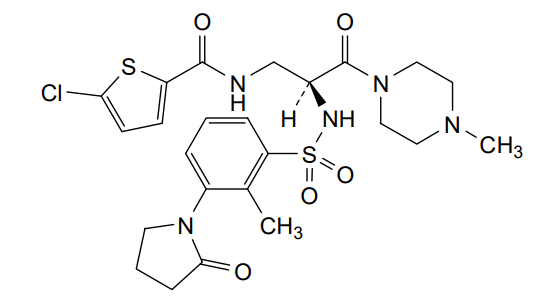
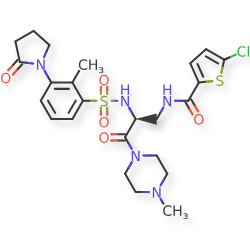
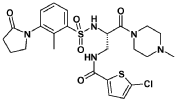
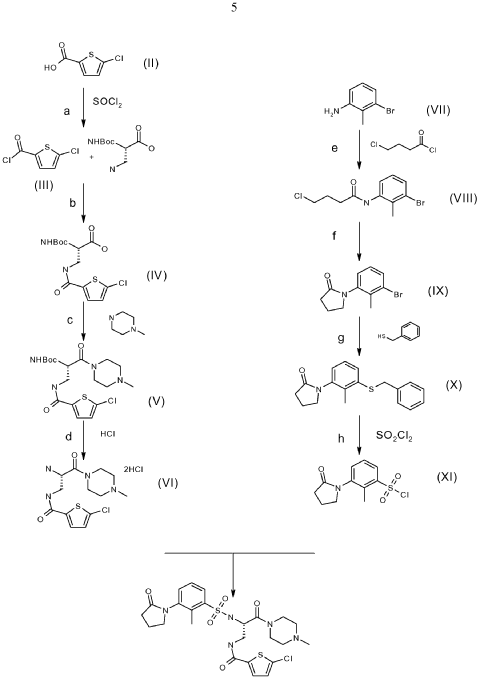
Segatroxaban
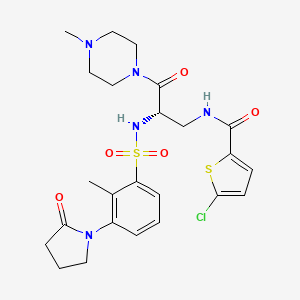
Segatroxaban
CAS 1184300-63-7
MF C24H30ClN5O5S2 MW568.11
5-chloro-N-{(2S)-2-[2-methyl-3-(2-oxopyrrolidin-1-yl)benzene1-sulfonamido]-3-(4-methylpiperazin-1-yl)-3-oxopropyl}thiophene-2-carboxamide
2-THIOPHENECARBOXAMIDE, 5-CHLORO-N-((2S)-2-(((2-METHYL-3-(2-OXO-1-PYRROLIDINYL)PHENYL)SULFONYL)AMINO)-3-(4-METHYL-1-PIPERAZINYL)-3-OXOPROPYL)-5-CHLORO-N-((2S)-2-(((2-METHYL-3-(2-OXO-1-PYRROLIDINYL)PHENYL)SULFONYL)AMINO)-3-(4-METHYL-1-PIPERAZINYL)-3-OXOPROPYL)-2-THIOPHENECARBOXAMIDE5-CHLOROTHIOPHENE-2-CARBOXYLIC ACID N-((S)-2-(((2-METHYL-3-(2-OXOPYRROLIDIN-1-YL)PHENYL)SULFONYL)AMINO)-3-(4-METHYLPIPERAZIN-1-YL)-3-OXOPROPYL)AMIDE5-CHLORO-N-((2S)-2-(2-METHYL-3-(2-OXOPYRROLIDIN-1-YL)BENZENE-1-SULFONAMIDO)-3-(4-METHYLPIPERAZIN-1-YL)-3-OXOPROPYL)THIOPHENE-2-CARBOXAMIDE
blood coagulation factors Xa and IIa (thrombin) inhibitor, 53FM6EUY9U, SAR107375, SAR 107375
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2009103440&_cid=P10-MILGON-12468-1
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014174102&_cid=P10-MILGJE-09654-1
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023031083&_cid=P10-MILGQR-13553-1
PAT
Chlorothiophene-amides as inhibitors of coagulation factors xa and thrombin
Publication Number: WO-2009103440-A1
Priority Date: 2008-02-21
- Tartrate salt of 5-chloro-thiophene-2-carboxylic acid [(S)-2-[2-methyl-3-(2-oxo-pyrrolidin-1-yl)-benzenesulfonylamino]-3-(4-methyl-piperazin-1-yl)-3-oxo-propyl]amidePublication Number: US-9637479-B2Priority Date: 2013-04-26Grant Date: 2017-05-02
- Tartrate salt of 5-chloro-thiophene-2-carboxylic acid [(s)-2-[methyl-3-(2-oxo-pyrrolidin-1-yl)-benzenesulfonylamino]-3-(4-methyl piperazin-1 -yl)-3-oxo-propryl]amidePublication Number: WO-2014174102-A1Priority Date: 2013-04-26
- Chlorothiophene-amides as inhibitors of coagulation factors xa and thrombinPublication Number: EP-2254881-A1Priority Date: 2008-02-21
- Chlorothiophene-amides as inhibitors of coagulation factors xa and thrombinPublication Number: EP-2254881-B1Priority Date: 2008-02-21Grant Date: 2012-09-12
- Chlorothiophene-amides as inhibitors of coagulation factors xa and thrombinPublication Number: US-2011112075-A1Priority Date: 2008-02-21
- Substituted S-alaninate derivativesPublication Number: US-11912692-B2Priority Date: 2021-09-03Grant Date: 2024-02-27
- Substituted s-alaninate derivativesPublication Number: WO-2023031083-A1Priority Date: 2021-09-03
- A method for detecting isomers in SAR107375 by high performance liquid chromatographyPublication Number: CN-112666269-BPriority Date: 2019-10-16Grant Date: 2022-12-30
- Tartrate salt of 5-chloro-thiophene-2-carboxylic acid [(s)-2-[methyl-3-(2-oxo-pyrrolidin-1-yl)-benzenesulfonylamino]-3-(4-methyl-piperazin-1 -yl)-3-oxo-propyl]amidePublication Number: AU-2014259378-B2Priority Date: 2013-04-26Grant Date: 2018-08-30
- Tartrate salt of 5-chloro-thiophene-2-carboxylic acid [(s)-2-[methyl-3-(2-oxo-pyrrolidin-1-yl)-benzenesulfonylamino]-3-(4-methyl-piperazin-1-yl)-3-oxo-propyl]amidePublication Number: US-2016102082-A1Priority Date: 2013-04-26
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////////segatroxaban, 53FM6EUY9U, SAR107375, SAR 107375
Rupitasertib
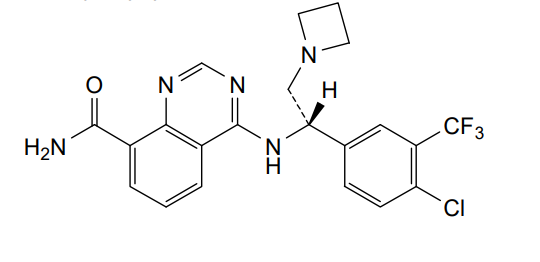
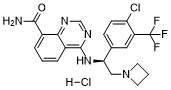
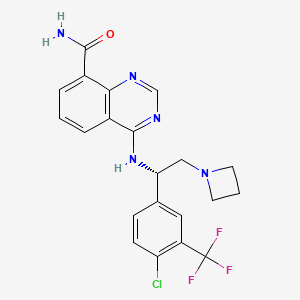
Rupitasertib
CAS 1379545-95-5
MF C21H19ClF3N5O 449.9 g/mol
4-({(1S)-2-(azetidin-1-yl)-1-[4-chloro-3-(trifluoromethyl)phenyl]ethyl}amino)quinazoline-8-carboxamide
4-[[(1S)-2-(azetidin-1-yl)-1-[4-chloro-3-(trifluoromethyl)phenyl]ethyl]amino]quinazoline-8-carboxamide
serine/ threonine kinase inhibitor, antineoplastic, EMD SERONO, Gastric cancer; HER2 positive breast cancer; Solid tumours, M2698 HCl, M2698 hydrochloride, MSC2363318A, MSC 2363318A, MSC-2363318A, M2698, M-269, M 2698. Rupitasertib HCl, 0DXG50I4WD
- OriginatorEMD Serono
- DeveloperEMD Serono; Evexta Bio
- ClassAntineoplastics; Small molecules
- Mechanism of Action70 kDa ribosomal protein S6 kinase inhibitors; Proto-oncogene protein c-akt inhibitors
- PreclinicalGlioblastoma; HER2 negative breast cancer
- No development reportedGastric cancer; HER2 positive breast cancer; Solid tumours
- 28 Oct 2025No recent reports of development identified for preclinical development in Gastric-cancer in France (PO)
- 28 Jun 2025No recent reports of development identified for phase-I development in HER2-positive-breast-cancer(Combination therapy, Late-stage disease, Metastatic disease) in USA (PO)
- 28 Jun 2025No recent reports of development identified for phase-I development in Solid-tumours(Combination therapy, Late-stage disease) in USA (PO)
- First-in-Human Dose Escalation Trial in Subjects With Advanced Malignancies
- CTID: NCT01971515
- Phase: Phase 1
- Status: Completed
- Date: 2018-09-19
Rupitasertib is an orally available inhibitor of the serine/threonine protein kinases ribosomal protein S6 Kinase (p70S6K) and Akt (protein kinase B), with potential antineoplastic activity. Upon administration, rupitasertib binds to and inhibits the activity of p70S6K and Akt. This prevents the activation of the PI3K/Akt/p70S6K signaling pathway and inhibits tumor cell proliferation in cancer cells that have an overactivated PI3K/Akt/p70S6K signaling pathway. Constitutive activation and dysregulated signaling of the PI3K/Akt/p70S6K pathway are frequently associated with tumorigenesis of many tumor types; targeting multiple kinases in this pathway is more efficacious than targeting a single kinase.
An optimized S6K inhibitor to overcome limitations of PAM pathway inhibitors
In just over 20 years, protein kinase inhibitors have changed the face of oncology and opened the new eras of targeted therapies and precision medicine. However, with few exceptions, no patient can be cured by one of these drugs alone. Today, scientists seek to develop novel kinase inhibitors[1] with improved efficacy and the potential to overcome resistances. The dual S6K AKT1/3 inhibitor rupitasertib (formerly DIACC3010, acquired from Merck KGaA, Darmstadt, Germany) has both of these characteristics and reaches brain metastases. After successfully completing a Phase I trial in patients with advanced/refractory solid tumors, including breast cancer, the drug candidate will be evaluated in a Phase 2/3 trial in ER+ HER2 breast cancer, which is expected to start in 2024.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2012069146&_cid=P10-MIJPKI-12294-1
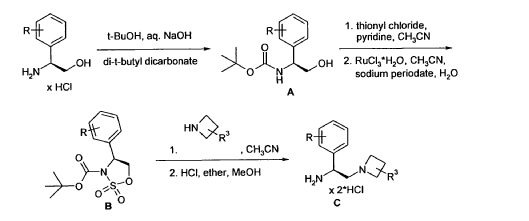
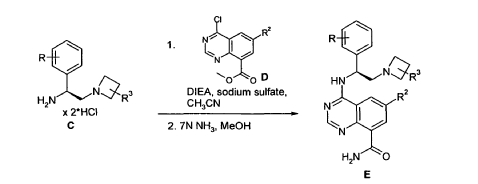
Example 4 was prepared following the general synthesis of A-E starting with (S)-2- amino-2-(3,4-di-fluoro-phenyl)-ethanol.LCMS [384.20 (M+1)]. 1H NMR (DMSO-d6, ppm) 1.92 (2H), 2.75 (1H), 2.93 (1H), 3.15 (4H), 5.43 (1H), 7.34 (2H), 7.53 (1H), 7.68 (1H), 7.81 (1H), 8.58 (4H), 10.30 (1H).
4-[(S)-2-Azetidin-1-yl-1-(4-chloro-3-trifluoromethylphenyl)-ethylamino]-guinazoline-8- carboxylic acid amide (5)
IC50 P70S6K [nM]: 0.9
pS6 MDA-MB-468 [nM]: 11
Akt1 IC50 [nM]: 1.4
Aurora B IC50 [nM]: 100
PAT
- Quinazoline carboxamide azetidinesPublication Number: SG-190318-A1Priority Date: 2010-11-24
- Quinazoline carboxamide azetidinesPublication Number: US-2013252942-A1Priority Date: 2010-11-24
- Quinazoline carboxamide azetidinesPublication Number: US-8946247-B2Priority Date: 2010-11-24Grant Date: 2015-02-03
- SMAC Mimetic for Treating Myelodysplastic SyndromesPublication Number: US-2015158908-A1Priority Date: 2009-07-02
- Methods of treating a ras protein-related disease or disorderPublication Number: US-2025049810-A1
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
- p70S6K/Akt dual inhibitor DIACC3010 is efficacious in preclinical models of gastric cancer alone and in combination with trastuzumabPublication Name: Scientific ReportsPublication Date: 2023-09-25PMCID: PMC10520030PMID: 37749105DOI: 10.1038/s41598-023-40612-9
- TTD: Therapeutic Target Database describing target druggability informationPublication Name: Nucleic Acids ResearchPublication Date: 2023-09-15PMCID: PMC10767903PMID: 37713619DOI: 10.1093/nar/gkad751
- Identification of Clinical Candidate M2698, a Dual p70S6K and Akt Inhibitor, for Treatment of PAM Pathway-Altered CancersPublication Name: Journal of Medicinal ChemistryPublication Date: 2021-10-01PMID: 34596404DOI: 10.1021/acs.jmedchem.1c01087
- Phase 1 study of M2698, a p70S6K/AKT dual inhibitor, in patients with advanced cancerPublication Name: Journal of Hematology & OncologyPublication Date: 2021-08-18PMCID: PMC8371902PMID: 34407844DOI: 10.1186/s13045-021-01132-z
- M2698 is a potent dual-inhibitor of p70S6K and Akt that affects tumor growth in mouse models of cancer and crosses the blood-brain barrierPublication Name: American journal of cancer researchPublication Date: 2016PMCID: PMC4859885PMID: 27186432
////////////Rupitasertib, antineoplastic, EMD SERONO, Gastric cancer; HER2 positive breast cancer; Solid tumours, M2698 HCl, M2698 hydrochloride, MSC2363318A, MSC 2363318A, MSC-2363318A, M2698, M-269, M 2698. Rupitasertib HCl, 0DXG50I4WD
Rogocekib
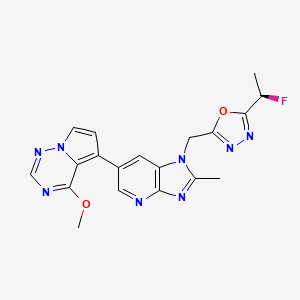
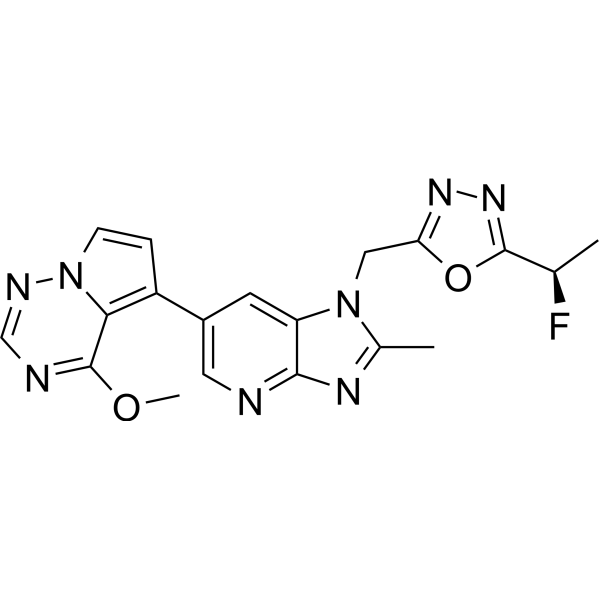
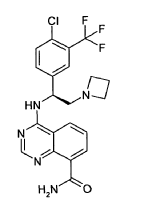
Rogocekib
CAS 2144751-78-8
MF C19H17FN8O2 MW 408.39
1-({5-[(1R)-1-fluoroethyl]-1,3,4-oxadiazol-2-yl}methyl)-6-(4-methoxypyrrolo[2,1-f][1,2,4]triazin-5-yl)-2-methyl1H-imidazo[4,5-b]pyridine
2-[(1R)-1-fluoroethyl]-5-[[6-(4-methoxypyrrolo[2,1-f][1,2,4]triazin-5-yl)-2-methylimidazo[4,5-b]pyridin-1-yl]methyl]-1,3,4-oxadiazole
dual specificity protein kinase CLK (CDC2-like kinase)inhibitor, antineoplastic, CTX 712, XE88VQP94E
Rogocekib is an orally effective CLK 2 inhibitor, with an IC50 of 1.4 nM, showing anti-tumor activity.
Rogocekib is an orally bioavailable inhibitor of CLK family kinases, with potential antineoplastic activity. Upon oral administration, rogocekib binds to and inhibits the activity of CLK family kinases, thereby inhibiting the phosphorylation of serine/arginine-rich (SR) domain-containing splicing factors (SFs). This modulates RNA splicing, prevents the expression of certain tumor-associated genes, and inhibits tumor cell proliferation. In many cancer cells, core spliceosome proteins, including SF3B1, U2 small nuclear ribonucleoprotein auxiliary factor 1 (U2AF1), serine/arginine-rich splicing factor 2 (SRSF2) and U2 small nuclear ribonucleoprotein auxiliary factor subunit-related protein 2 (ZRSR2), are mutated and aberrantly activated leading to a dysregulation of mRNA splicing. CLK family kinases, an evolutionarily conserved group of kinases, phosphorylates various SR proteins including SR domain-containing SFs.
SYN
https://pubs.acs.org/doi/10.1021/acsmedchemlett.5c00412
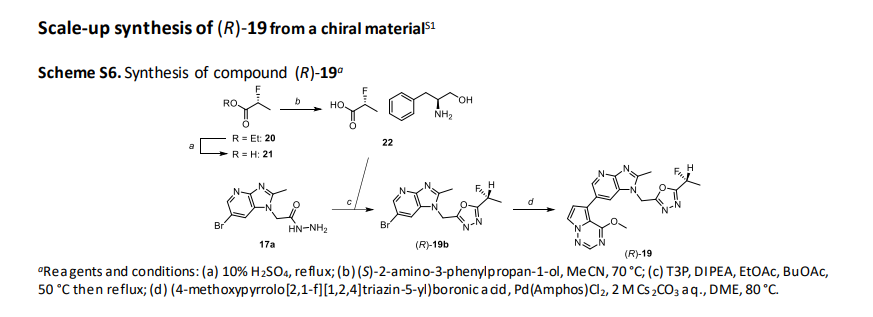
(R)-2-fluoropropanoic acid (21)
(R)-Ethyl 2-fluoropropanoate (20) (95 g, 791 mmol) was suspended in 10% sulfuric acid (950 mL), and heated and
refluxed for 3 h. After cooled, sodium chloride was added to saturate the aqueous layer, and the aqueous layer
was extracted with TBME (900 mL x4). The obtained organic layer was dried over MgSO4, and concentrated under
reduced pressure to give the title compound (124 g, 791 mmol calcd as quant., containing TBME).
1H NMR (300 MHz, DMSO-d6) δ 1.35-1.56 (3H, m), 4.91-5.21 (1H, m), 13.19 (1H, brs).
(S)-2-amino-3-phenylpropane-1-ol (R)-2-fluoropropanoate (22)
To a solution of (S)-2-amino-3-phenylpropan-1-ol (119 g, 787 mmol) in EtOH (360 mL) and MeCN (1090 mL) was
added dropwise a solution of 21 (791 mmol, theoretically calcd as quant.) in MeCN (1090 mL) at 65° C to 70° C.
The mixture was stirred at 60° C for 1 h, and further stirred at room temperature for 1 h. Precipitated crystals were
collected by filtration, and washed with MeCN (500 mL) to obtain white crystals (170 g, 699 mmol, 89%).
The obtained crystals(140 g, 575 mmol) were dissolved in EtOH (700 mL) at 60° C, and to the solution was added
MeCN (4200 mL) at 58° C to 65° C. The mixture was stirred at 60° C for 1 h. The mixture was cooled to room
temperature, and then stirred overnight at room temperature. The obtained solid was collected by filtration, and
washed with MeCN to obtain give the title compound (109 g, 448 mmol, 78%) as a white crystal.
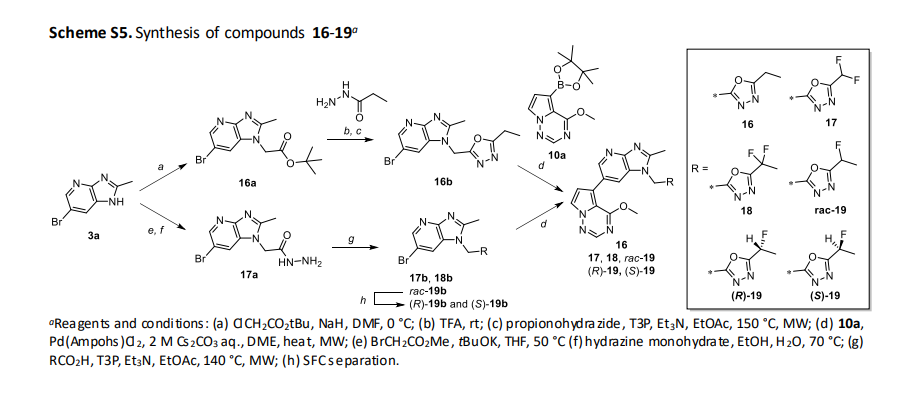
(R)-2-((6-bromo-2-methyl-1H-imidazo[4,5-b]pyridin-1-yl)methyl)-5-(1-fluoroethyl)-1,3,4-oxadiazole ((R)-19b)
22 (109 g, 448 mmol) was dissolved in 1M HCl aq. (1500 mL) and brine (1500 mL) and extracted with TBME (1000
mL x4). The organic layer was dried over MgSO4 and concentrated in vacuo to give free salt of 22 (i.e., 21) as a
colorless oil. 50 wt% T3P in EtOAc (419 mL, 704 mmol) was added to a suspension of the above material, 17a (100
g, 351.97 mmol), and DIPEA (246 mL, 1408 mmol) in BuOAc (3000 mL) at room temperature. After being stirred at
50 °C for 30 min, 50 wt% T3P in EtOAc (210 mL, 351.97 mmol) was added to the mixture and then the mixture was
heated and refluxed for 3 h. After cooling, to the mixture was added sat NaHCO3 aq. (3000 mL), then the insoluble
material was removed by filtration. The filtrate was extracted with EtOAc (1500 mL x2). The organic layer was
separated, washed with water and brine, then passed through NH silica gel eluted with EtOAc. The residue was
concentrated in vacuo and the resulting precipitate was washed with IPE (3000 mL) to give the title compound
(57.8 g, 170 mmol, 48.3%) as an off-white solid.
1H NMR (300 MHz, DMSO-d6) δ 1.62-1.79 (3H, m), 2.62 (3H, s), 5.83-6.14 (3H, m), 8.38 (1H, d, J = 1.9 Hz), 8.45 (1H,
d, J = 1.9 Hz). MS m/z 340.0, 341.9 [M+H]+
.
1-((5-((1R)-1-fluoroethyl)-1,3,4-oxadiazol-2-yl)methyl)-6-(4-methoxypyrrolo[2,1-f][1,2,4]triazin-5-yl)-2-methyl1H-imidazo[4,5-b]pyridine ((R)-19, CTX-712)
A mixture of (4-methoxypyrrolo[2,1-f][1,2,4]triazin-5-yl)boronic acid (79 g, 409.39 mmol), (R)-19b (100 g, 294
mmol), Pd(Amphos)Cl2 (2.00 g, 2.97 mmol), 2 M Cs2CO3 aq. (295 mL, 590 mmol) and DME (2000 mL) was stirred at
80 °C for 1 h. After cooled to 50 °C, the mixture was diluted with THF (1000 mL). The mixture was poured into
NaHCO3 aq. (1600 mL) and extracted with EtOAc (1000 mL x3). The organic layer was separated, washed with 5%
ammonia aq. (1600 mLx2) and brine (1600 mL), dried over MgSO4 and concentrated in vacuo to give a yellow solid.
To the solution of obtained solid in THF (8000 mL) and water (200 mL) was added NH silica gel (2400 g) and stirred
for 3.5 h at room temperature. The insoluble material was removed by filtration and washed with THF (15 L). The
filtrate was concentrated in vacuo to give a yellow solid. The solid was washed with TBME to give the title
compound (98 g, 240 mmol, 82 %) as a pale yellow solid. A mixture of the above material (115 g, 270 mmol) and
activated carbon (Ecosorb, 33 g) in EtOH/water = 9/1 (2200 mL) and water (1100 mL) was stirred at 55 °C for 1 h.
The insoluble material was removed by filtration, and washed EtOH (550 mL). The resultant solution was diluted
with water (1600 mL) at 55 °C and stirred at room temperature overnight. After cooled to 5 °C, the mixture was
stirred for 3 h. The solid was collected by filtration and washed with EtOH/water = 1/1 (1000 mL) to give a
colorless crystal (88 g, 207 mmol, 77% as a water adduct).
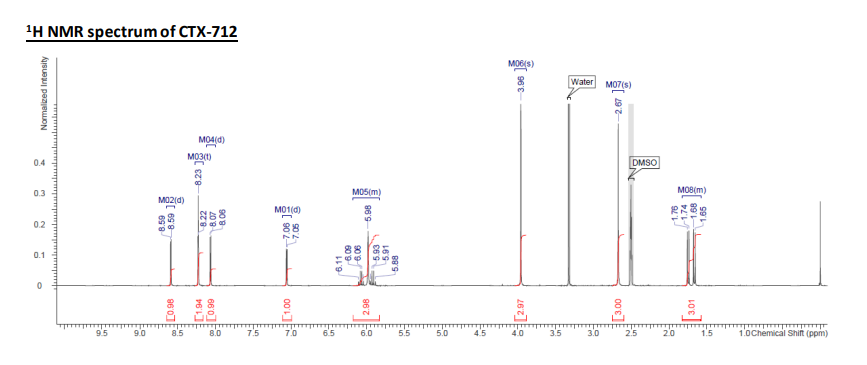
1H NMR (300 MHz, DMSO-d6) δ 1.58-1.82 (3H, m), 2.67 (3H, s), 3.96 (3H, s), 5.83-6.18 (3H, m), 7.06 (1H, d, J = 2.7
Hz), 8.06 (1H, d, J = 2.7 Hz), 8.23 (2H, t, J = 1.0 Hz), 8.59 (1H, d, J = 2.0 Hz). MS m/z 409.1 [M+H]+
.
PAT
Patent document 1:
WO 2010/016526
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2010016526&_cid=P10-MIIA44-38372-1
WO 2011/096535
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023190967&_cid=P10-MII9ZT-35263-1
SYN
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=JP275206879&_cid=P10-MII9SJ-29591-1
PAT
- Condensed Heterocyclic CompoundsPublication Number: KR-102431405-B1Priority Date: 2016-04-28Grant Date: 2022-08-10
- COMPOUND, MEDICINE, AND, USE OF THE COMPOUND OR SALT THEREOFPublication Number: BR-112018072039-B1Priority Date: 2016-04-28
- Fused Heterocyclic CompoundsPublication Number: CN-109415384-BPriority Date: 2016-04-28Grant Date: 2022-01-11
- Condensed heterocyclic compoundPublication Number: EP-3450436-B1Priority Date: 2016-04-28Grant Date: 2022-07-27
- Fused heterocyclic compoundPublication Number: US-11390634-B2Priority Date: 2016-04-28Grant Date: 2022-07-19
- condensed heterocyclic compoundPublication Number: ES-2927529-T3Priority Date: 2016-04-28Grant Date: 2022-11-08
- CONDENSED HETEROCYCLIC COMPOUNDPublication Number: HR-P20221277-T1Priority Date: 2016-04-28
- Fused heterocyclic compoundPublication Number: US-10577382-B2Priority Date: 2016-04-28Grant Date: 2020-03-03
- Fused heterocyclic compoundPublication Number: US-2019106437-A1Priority Date: 2016-04-28
- Fused heterocyclic compoundPublication Number: US-2020140462-A1Priority Date: 2016-04-28
- Fused heterocyclic compoundPublication Number: US-10981934-B2Priority Date: 2016-04-28Grant Date: 2021-04-20
- Fused heterocyclic compoundPublication Number: US-2021115067-A1Priority Date: 2016-04-28
- Medicament for treatment and/or prevention of cancerPublication Number: WO-2024048541-A1Priority Date: 2022-08-30
- Biomarker for treatment of solid cancer by imidazo[4,5-b]pyridine derivativePublication Number: WO-2023190967-A1Priority Date: 2022-03-31
- Fused heterocyclic compoundPublication Number: CA-3021185-A1Priority Date: 2016-04-28
- Condensed heterocyclic compoundPublication Number: EP-3450436-A1Priority Date: 2016-04-28
- Fused heterocyclic compoundsPublication Number: JP-WO2017188374-A1Priority Date: 2016-04-28
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
- [1]. Akinori Yoda, et al. CTX-712, a Novel Clk Inhibitor Targeting Myeloid Neoplasms with SRSF2 Mutation. Blood. (2021) 205–206[2]. Zhen Qin, et al. Development of Cdc2-like Kinase 2 Inhibitors: Achievements and Future Directions. J Med Chem. 2021 Sep 23;64(18):13191-13211. [Content Brief]
///////rogocekib, CTX 712, XE88VQP94E
Riselcaftor
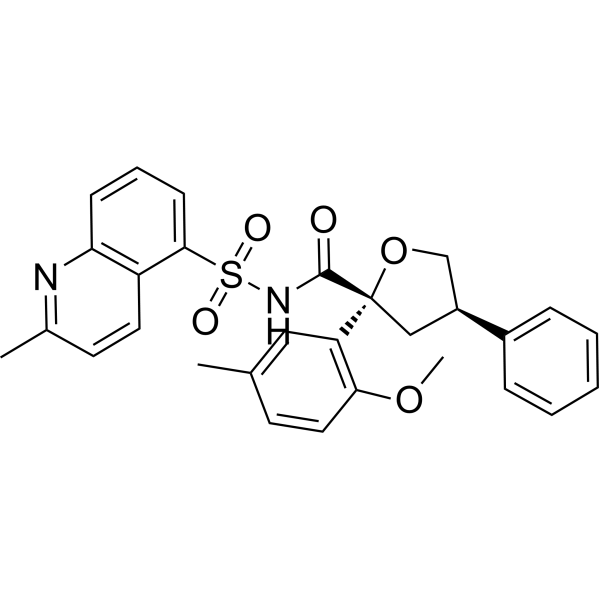
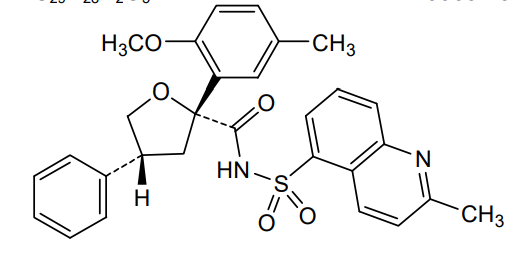
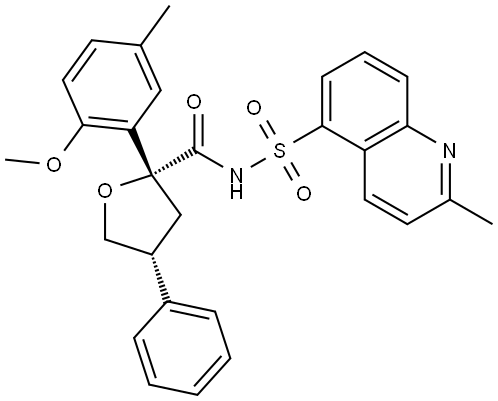
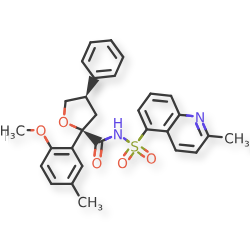
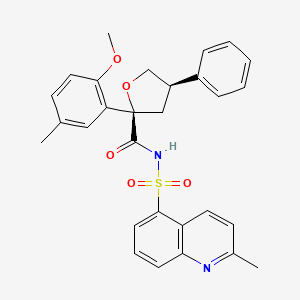
Riselcaftor
CAS 2799652-36-9
MF C29H28N2O5S MW 516.61
(2R,4R)-2-(2-methoxy-5-methylphenyl)-N-(2-methylquinoline-5-sulfonyl)-4-phenyloxolane-2-
carboxamide
(2R,4R)-2-(2-methoxy-5-methylphenyl)-N-(2-methylquinolin-5-yl)sulfonyl-4-phenyloxolane-2-carboxamide
cystic fibrosis transmembrane regulator (CFTR)protein modulator, 726GWJ6KQQ
Riselcaftor (Example 33) is a CFTR modulator, with an EC50 of 20.1 nM in human bronchial epithelial cells. Riselcaftor can be used for research of cystic fibrosis.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US367940046&_cid=P11-MIH63N-23616-1
Example 33
(2R,4R)-2-(2-methoxy-5-methylphenyl)-N-(2-methylquinoline-5-sulfonyl)-4-phenyloxolane-2-carboxamide
PAT
- Modulators of the Cystic Fibrosis Transmembrane Conductance Regulator Protein and Methods of UsePublication Number: US-2022211692-A1Priority Date: 2021-01-06
- Modulators of the cystic fibrosis transmembrane conductance regulator protein and methods of usePublication Number: WO-2022150174-A1Priority Date: 2021-01-06
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////Riselcaftor, 726GWJ6KQQ
Pudafensine
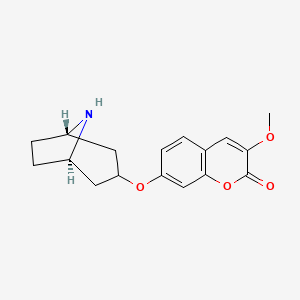
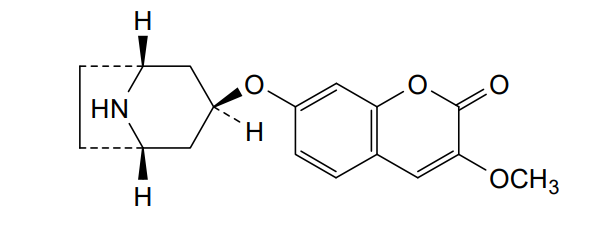
Pudafensine
CAS 1320346-14-2
MFC17H19NO4 MW 301.34 g/mol
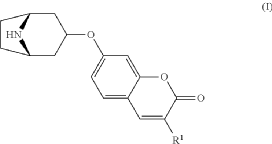
7-{[(1R,3s,5S)-8-azabicyclo[3.2.1]octan-3-yl]oxy}-3-methoxy2H-1-benzopyran-2-one
monoamine reuptake inhibitor, erectile dysfunction, neuropathic pain, NS18313, NS 18313, L9NG7US8GE, IP2015, IP 2015
Pudafensine is a monoamine reuptake inhibitor being developed as a potential treatment for erectile dysfunction (ED) and neuropathic pain. As a drug candidate, it works by preferentially inhibiting the reuptake of dopamine and serotonin. It is designed to be a first-line treatment for patients with organic ED who are not adequately served by existing therapies like PDE5 inhibitors.
How it works
- Pudafensine is a monoamine reuptake inhibitor that increases the levels of dopamine and serotonin in the brain by preventing their reabsorption into neurons.
- It has been shown in animal models and human trials to improve erectile function and reduce pain, including neuropathic pain.
Potential uses
- Erectile Dysfunction (ED): Pudafensine is being investigated for its potential to help men with organic ED who do not respond well to or cannot tolerate current treatments. Phase IIb clinical trial results are expected in late 2023.
- Neuropathic Pain: A clinical trial on pain involving pudafensine indicated it reduced allodynia and was well-tolerated with a favorable safety profile compared to pregabalin.
Development status
- Initiator Pharma is developing pudafensine as an oral tablet.
- Phase IIb studies for erectile dysfunction and Phase II studies for neuropathic pain have been completed, with positive results.
- The company is exploring its use in treating patients who are inadequately treated with existing medications.
Erectile dysfunction (ED)
Pudafensine, Initiator’s most advanced drug program has successfully demonstrated efficacy in a Clinicial Phase 2a Proof-of-Concept study and in a Phase 2b study to treat patients who suffer from organic erectile dysfunction (ED) that do not respond or cannot tolerate the currently marketed drugs in the PDE5i class (e.g. Viagra®, Cialis®, Levitra®).
Pudafensine strengthens the natural erection response by having a dual-action, both a central effect initiating erection and a peripheral effect potentiating erection through smooth muscle relaxation. Pudafensine is aimed for treatment of organic erectile dysfunction in patients who have erectile dysfunction (ED) due to abnormalities of the penile arteries and/or veins. Most common risk factors for organic ED are diabetes, overweight, lack of exercise, high cholesterol, high blood pressure, and cigarette smoking. Since Initiator Pharma was founded and pudafensine acquired, all preclinical development of the drug candidate to enable an application for clinical trials (CTA) has been carried out by the company’s auspices. Pudafensine is developed as a tablet that is taken orally on-demand. It is the company’s goal to be able to create a new “First-Line” treatment (recommended treatment) for the large group of men who have organic erectile dysfunction, who are sub-optimally treated with PDE5i products or for whom PDE5i treatment is contraindicated.
In Q4 2023 positive results from the Phase IIb clinical trial with pudafensine (IP2015) was announced. The Phase 2b trial is a randomized, double-blind, placebo-controlled, parallel-dosing group trial studying the efficacy and safety of high and low doses of pudafensine (IP2015) and placebo in otherwise healthy patients suffering from moderate to severe ED. The study comprises 130 patients divided into 3 parallel arms receiving a higher and a lower dose of pudafensine and placebo, respectively, with treatment duration of 4 weeks with frequent assessments of erectile dysfunction, safety and pharmacokinetics. The study has been conducted at the MAC clinical sites in the UK.
The study demonstrated statistically significant efficacy on the primary endpoint (related to improvements in intercourse settings) compared to placebo [p=0.034] and baseline [p=0.046]. Furthermore, the results were consistent throughout the study. Several other clinical endpoints related to improved intercourse activities (obtained from the International Index of Erectile Function Questionnaire, IIEF-15) demonstrated significant effects compared to the baseline. The frequency and type of adverse effects were mild to moderate and comparable to those observed in the placebo group. There was no reporting of critical safety observations.
Neuropathic pain
Pudafensine have shown effects in a human model of pain ie. in a clinical Phase I study in healthy subjects dosed with the drug pudafensine and challenged with a pain-inducing ingredient (capsaicin).
The Phase I study was a randomized, double blind, placebo controlled study in 24 healthy male subjects, investigating the effects on pain measures (hyperalgesia, allodynia, and subjects pain rating) of single doses of pudafensine, pregabalin as an active control, and placebo. The pain was induced by intradermal capsaicin. Pudafensine demonstrated a statistically significant effect on allodynia (p=0.049) and showed a dose-dependent effect on the measured pain parameters. Pregabalin (p=0.083) and IP2015 (p=0.051) tended to reduce hyperalgesia, although the effects on hyperalgesia were not statistically significant compared to placebo-treated subjects.
Syn
US20130040985
https://patentscope.wipo.int/search/en/detail.jsf?docId=US76705962&_cid=P22-MIFE0H-55553-1
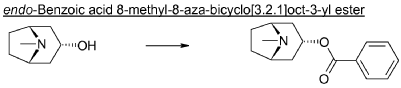
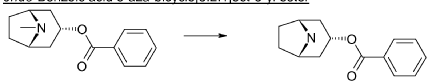
endo-Benzoic acid 8-methyl-8-aza-bicyclo[3.2.1]oct-3-yl ester
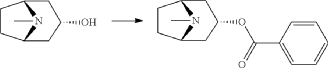
Benzoylchloride (84.3 g, 600 mmol) was added during 30 min at <30° C. to a mixture of tropine (70.6 g, 500 mmol), potassium tert-butoxide (67.3 g, 600 mmol) and THF (500 ml). The mixture was stirred at room temperature for 2 h. Water (1 L) was added followed by extraction with diethylether (2×500 ml). The organic phase was washed twice with water (2×200 ml) followed by a solution of saturated aqueous sodium chloride (200 ml). The ether phase was dried and hydrochloric acid in ethanol (170 ml, 3 M) was added. The precipitated hydrochloride was filtered and washed with diethylether. The free base was obtained by adding an excess of aqueous ammonia followed by extraction with a mixture of ethylacetate and diethylether. Yield 66.8 g (54%).
endo-Benzoic acid 8-aza-bicyclo[3.2.1]oct-3-yl ester
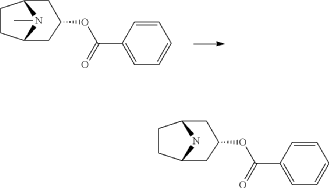
2,2,2-Trichloroethylchloroformate (75.0 ml, 544 mmol) was added dropwise to a mixture of endo-benzoic acid 8-methyl-8-aza-bicyclo[3.2.1]oct-3-yl ester (66.8 g, 272 mmol) and dry toluene (500 ml). The mixture was allowed to stir for 1 h at room temperature, followed by 15 h at 100° C. Water (250 ml) was added followed by stirring 1 h. The phases were separated and the organic phase was washed twice with water (2×200 ml). The mixture of the intermediate 3-benzoyloxy-8-aza-bicyclo[3.2.1]octane-8-carboxylic acid trichloromethyl ester, was dried and evaporated. Acetic acid (350 ml) was added followed by addition of zinc (53.4 g, 817 mmol) over 3 h time period. Water (100 ml) was added, cooled by adding ice and made alkaline by adding concentrated aqueous ammonia (ca: 400 ml) and the mixture was extracted with dichloromethane (2×300 ml). Yield 44.5 g (61%).
endo-3-Benzoyloxy-8-aza-bicyclo[3.2.1]octane-8-carboxylic acid tert-butyl ester
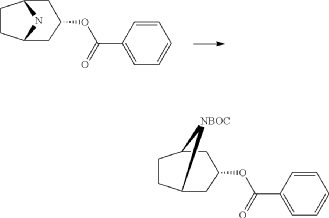
Di-tert-butyl-dicarbonate (39.9 g, 183 mmol) solved in THF (100 ml) was added to a stirred mixture of endo-benzoic acid 8-aza-bicyclo[3.2.1]oct-3-yl ester (44.5 g, 166.4 mmol), triethylamine (67.4 g, 666 mmol) and THF (250 ml) during 0.5 h at room temperature, followed by stirring for 1 h. Water (1 L) was added and the mixture was extracted with diethylether (2×300 ml). The collected ether phase was washed twice with water (2×200 ml), dried and evaporated. Yield 60.1 g (100%).
endo-3-Hydroxy-8-aza-bicyclo[3.2.1]octane-8-carboxylic acid tert-butyl ester
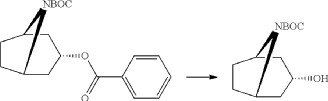
A mixture of endo-3-benzoyloxy-8-aza-bicyclo[3.2.1]octane-8-carboxylic acid tert-butyl ester (55.0 g, 166 mmol), potassium hydroxide (11.2 g 199 mmol) and ethanol (99%, 400 ml) was stirred for 3 days at room temperature. Potassium benzoate was separated by filtration and the filtrate was evaporated. Diethylether (200 ml) was added and remaining potassium benzoate was separated by filtration and the filtrate was evaporated. The product was triturated with petroleum. Yield 30.0 g (80%). Mp 139.5-140.8° C.
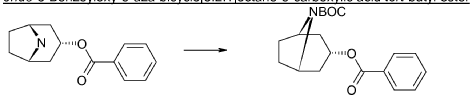
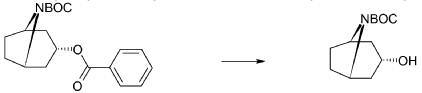
xample 1
Exo-tert-butyl-3-(3-methoxy-2-oxo-chromen-7-yl)oxy-8-azabicyclo[3.2.1]octane-8-carboxylate (Intermediate)
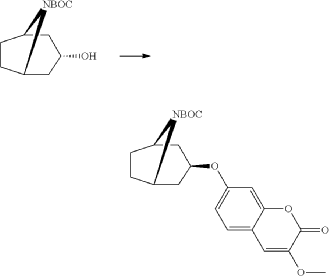
Triphenylphosphine (1.15 g, 4.37 mmol) was solved in toluene (20 ml) and cooled to <20° C. Diethylazodicarboxylate (40% in toluene) (2.0 ml, 4.37 mmol) was added to the mixture below 20° C., followed by stirring for 10 minutes. endo-3-Hydroxy-8-aza-bicyclo[3.2.1]octane-8-carboxylic acid tert-butyl ester (0.828 g, 3.64 mmol) was added and after 10 minutes 7-hydroxy-3-methoxy-chromen-2-one (0.70 g, 3.64 mmol) (prepared according to J. Med. Chem. 1999, 42, p2662-2672) was added to the mixture. The temperature raised to 25° C. due to an exothermic reaction. The mixture precipitates. The mixture was allowed to stir for 15 h at room temperature. Water (20 ml) and sodium hydroxide (0.5 ml, 4 M) was added followed by stirring. The mixture was cooled on an ice-bath, filtered and washed with water and diethylether. Yield 0.92 g (63%).
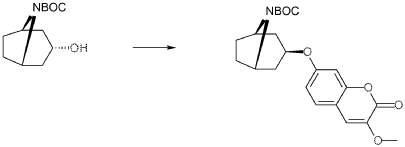
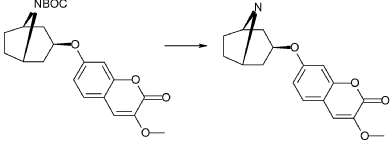
Exo-7-[(-8-azabicyclo[3.2.1]octan-3-yl)oxy]-3-methoxy-chromen-2-one hydrochloride (Compound 1.1)
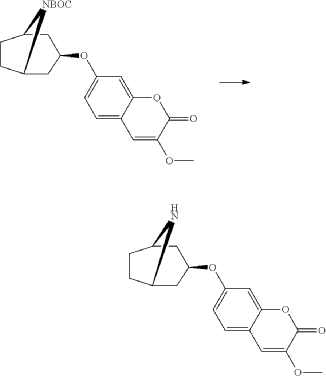
Exo-tert-butyl-3-(3-methoxy-2-oxo-chromen-7-yl)oxy-8-azabicyclo[3.2.1]octane-8-carboxylate (0.92 g, 2.29 mmol) and hydrogen chloride (15 ml, 1 M) in acetic acid was mixed as a solution and stirred at room-temperature and precipitated after a few minutes. The product was filtered and washed with diethylether. Yield 0.48 g (62%). LC-ESI-HRMS of [M+H]+ shows 302.13856 Da. Calc. 302.138689 Da, dev. −0.4 ppm.
Syn
WO2011092061
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2011092061&_cid=P22-MIFE80-61015-1
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2024008808&_cid=P22-MIFDSB-50229-1

SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2024089247&_cid=P22-MIFDSB-50229-1
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2024146892&_cid=P22-MIFDSB-50229-1
PAT
- Compound for treatment of erectile dysfunctionPublication Number: WO-2024146892-A1Priority Date: 2023-01-03
- Compound for treatment of painPublication Number: WO-2024089247-A1Priority Date: 2022-10-28
- Compound for treatment of female sexual dysfunctionPublication Number: WO-2024008808-A1Priority Date: 2022-07-08
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////////Pudafensine, monoamine reuptake inhibitor, erectile dysfunction, neuropathic pain, NS18313, NS 18313, L9NG7US8GE, IP2015, IP 2015
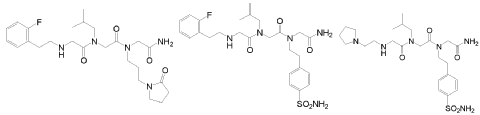
Privosegtor
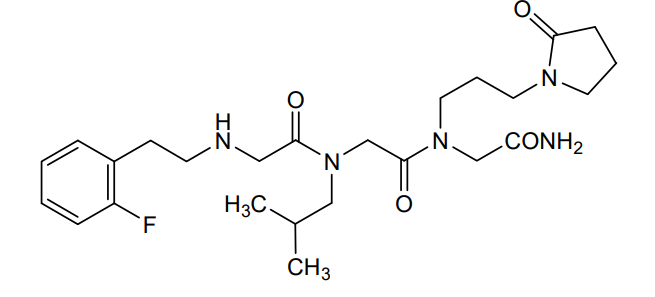
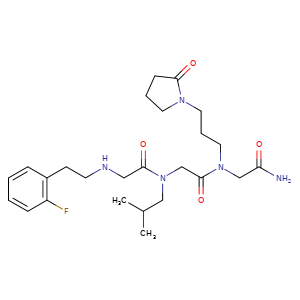
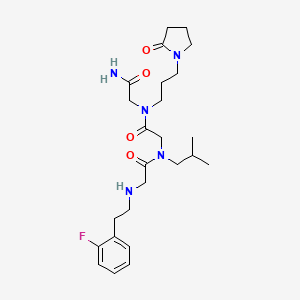
Privosegtor
CAS 1361200-34-1
MF C25H38FN5O4, MW 491.6 g/mol
GLYCINAMIDE, N-(2-(2-FLUOROPHENYL)ETHYL)GLYCYL-N-(2-METHYLPROPYL)GLYCYL-N2-(3-(2-OXO-1-PYRROLIDINYL)PROPYL)-
N-(2-(2-FLUOROPHENYL)ETHYL)GLYCYL-N-(2-METHYLPROPYL)GLYCYL-N2-(3-(2-OXO-1-PYRROLIDINYL)PROPYL)GLYCINAMIDE
N-(2-(2-FLUOROPHENYL)ETHYL)GLYCYL-N-(2-METHYLPROPYL)GLYCYL-N2-(3-(2-OXOPYRROLIDIN-1-YL)PROPYL)GLYCINAMIDE
N-[2-(2-fluorophenyl)ethyl]glycyl-N-(2-methylpropyl)glycyl-N2[3-(2-oxopyrrolidin-1-yl)propyl]glycinamide
serum/ glucocorticoid-regulated kinase 2 (Sgk2) activator, Phase 2, Optic neuritis, orphan drug, BN-201, BN 201, G-79, G 79, KCN37L7EIH
- OriginatorBionure
- DeveloperBionure; Oculis Pharma
- ClassAnti-inflammatories; Antiglaucomas; Eye disorder therapies; Neuroprotectants; Peptides; Small molecules
- Mechanism of ActionBrain derived neurotrophic factor agonists; Insulin-like growth factor I stimulants; Neuron modulators; Serum-glucocorticoid regulated kinase stimulants
- Orphan Drug StatusYes – Optic neuritis
- Phase IIOptic neuritis
- PreclinicalMultiple sclerosis; Neurotrophic keratopathy
- No development reportedGlaucoma; Neuromyelitis optica
- 06 Oct 2025Oculis Holding plans the PIONEER-2 trial in Optic neuritis in first half of 2026
- 06 Oct 2025Oculis Holding plans the PIONEER-3 trial in Optic nerve disorders in mid-2026
- 06 Oct 2025Oculis Holding completes End-of-phase II meeting with US FDA and receives positive feedback for registrational PIONEER program in Optic neuritis and Optic nerve disorders
OCS-05 in Patients With Optic Neuritis
CTID: NCT04762017
Phase: Phase 2
Status: Completed
Date: 2025-09-22
N-[2-[(2-amino-2-oxoethyl)-[3-(2-oxopyrrolidin-1-yl)propyl]amino]-2-oxoethyl]-2-[2-(2-fluorophenyl)ethylamino]-N-(2-methylpropyl)acetamide (BN201) is a small peptide molecule, a first-in-class neuroprotective compound. BN201 promotes the survival of cultured neural cells when subjected to oxidative stress or when deprived of trophic factors. BN201 promotes neuronal differentiation, the differentiation of precursor cells to mature oligodendrocytes in vitro, and the myelination of new axons. BN201 modulates several kinases participating in the insulin growth factor 1 pathway including serum-glucocorticoid kinase and midkine, inducing the phosphorylation of NDRG1 and the translocation of the transcription factor Foxo3 to the cytoplasm. In vivo, BN201 prevents axonal and neuronal loss, and it promotes remyelination in models of multiple sclerosis, chemically induced demyelination, and glaucoma. Bionure, a spin-off from Hospital Clínic de Barcelona that is based in California, is developing BN201 for multiple sclerosis, acute optic neuritis (AON) and glaucoma. BN201 was granted with orphan designation status for optic neuritis by the FDA. Optic neuritis is often an early sign of multiple sclerosis. The efficacy, safety, and capacity of the drug to cross the blood-brain barrier have been demonstrated in animal models, but the drug has not yet entered clinical testing.
PAT
Agonists of neurotrophin receptors and their use as medicaments
Publication Number: WO-2012028959-A1
Priority Date: 2010-08-31
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2012028959&_cid=P10-MIDYQ0-58943-1
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021084013&_cid=P10-MIDYSN-60542-1
In another embodiment, optionally in combination with one or more features of the various embodiments described above or below throughout all the description, the compound of formula (I) is selected from the group consisting of G79 ([N-(2-(2′-fluorophenyl)ethyl)- glycyl]-[N-(2-methylpropyl)-glycyl]-N-[3-(2′-oxopyrrolidinyl)-propyl]glycinamide, BN201 , Chemical Formula: C25H38FN5O4; MW 491.5987), G-80 ([N-(2-(2′-fluorophenyl)ethyl)- glycyl]-[N-(2-methyl-propyl)glycyl]-N-[2-(4′-sulfamoyl-phenyl)ethyl]glycinamide, BN 119, Chemical Formula: C26H36FN5O5S; MW 549.658) and G81 ([N-(2-(1 -pyrrolidinyl)ethyl)- glycyl]-[N-(2-methyl-propyl)glycyl]-N-[2-(4′-sulfamoyl-phenyl)ethyl]glycinamide, BN 120, Chemical Formula: C24H4oN6OS; MW 524.6766):
G79 (BN201) G80 (BN119) G81 (BN120)
Compounds of formula (I) can be prepared as disclosed in WO2012028959.
PAT
- Agonists of Neurotrophin Receptors and Their Use as MedicamentsPublication Number: US-2012052094-A1Priority Date: 2010-08-31
- Agonists of Neurotrophin Receptors and Their Use as MedicamentsPublication Number: US-2015005239-A1Priority Date: 2010-08-31
- Agonists of neurotrophin receptors and their use as medicamentsPublication Number: US-2017121367-A1Priority Date: 2010-08-31
- Agonists of neurotrophin receptors and their use as medicamentsPublication Number: US-8791076-B2Priority Date: 2010-08-31Grant Date: 2014-07-29
- Agonists of neurotrophin receptors and their use as medicamentsPublication Number: US-9453047-B2Priority Date: 2010-08-31Grant Date: 2016-09-27
- Combination Therapy Methods, Compositions and KitsPublication Number: KR-20220109378-APriority Date: 2019-07-03
- Combination therapy methods, compositions and kitsPublication Number: US-2022378866-A1Priority Date: 2019-07-03
- Agonists of neurotrophin receptors and their use as medicamentsPublication Number: EP-2611775-A1Priority Date: 2010-08-31
- Agonists of neurotrophin receptors and their use as medicamentsPublication Number: EP-2611775-B1Priority Date: 2010-08-31Grant Date: 2016-03-16
- Agonists of neurotrophin receptors and their use as medicamentsPublication Number: US-10106577-B2Priority Date: 2010-08-31Grant Date: 2018-10-23
- Combination therapy methods, compositions and kitsPublication Number: WO-2021001464-A1Priority Date: 2019-07-03
- Combination therapy methods, compositions and kitsPublication Number: AU-2020298782-A1Priority Date: 2019-07-03
- Combination therapy methods, compositions and kitsPublication Number: CN-114206329-APriority Date: 2019-07-03
- Combination therapy methods, compositions and kitsPublication Number: EP-3993784-A1Priority Date: 2019-07-03
- Combination therapy methods, compositions and kitsPublication Number: JP-2022539999-APriority Date: 2019-07-03
- Boron-nitrogen compound, organic electroluminescence composition, and organic electroluminescence device containing samePublication Number: WO-2022121951-A1Priority Date: 2020-12-10
- New treatment regimen for the treatment of neurological diseases or conditionsPublication Number: WO-2021084013-A1Priority Date: 2019-10-30
- Novel Therapeutic Approaches for the Treatment of Neurological Diseases or ConditionsPublication Number: CN-115052595-APriority Date: 2019-10-30
- New treatment regimen for the treatment of neurological diseases or conditionsPublication Number: EP-4051263-A1Priority Date: 2019-10-30
- New treatment regiment for the treatment of neurological diseases or conditionsPublication Number: US-2022387385-A1Priority Date: 2019-10-30
- A plant zinc-increasing compound inoculant and its preparation method and applicationPublication Number: CN-117286034-APriority Date: 2023-09-11
- A plant zinc-enhancing composite bacterial agent and its preparation method and applicationPublication Number: CN-117286034-BPriority Date: 2023-09-11Grant Date: 2024-11-15
- Compound, pharmaceutical composition comprising the same, and process for synthesizing the samePublication Number: TW-202432095-APriority Date: 2022-12-22
- Synthesis of small molecule agonists of neuroptrophinPublication Number: WO-2024133860-A1Priority Date: 2022-12-22
- Boron-nitrogen compound, organic electroluminescent composition and organic electroluminescent device containing samePublication Number: WO-2022121920-A1Priority Date: 2020-12-10
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
- Development and validation of PAMPA-BBB QSAR model to predict brain penetration potential of novel drug candidatesPublication Name: Frontiers in PharmacologyPublication Date: 2023-12-01PMCID: PMC10722238PMID: 38108064DOI: 10.3389/fphar.2023.1291246
- A Phase 1 randomized study on the safety and pharmacokinetics of OCS-05, a neuroprotective disease modifying treatment for Acute Optic Neuritis and Multiple SclerosisPublication Name: Scientific ReportsPublication Date: 2023-03-29PMCID: PMC10060579PMID: 36991169DOI: 10.1038/s41598-023-32278-0
- Retrospective assessment of rat liver microsomal stability at NCATS: data and QSAR modelsPublication Name: Scientific ReportsPublication Date: 2020-11-26PMCID: PMC7693334PMID: 33244000DOI: 10.1038/s41598-020-77327-0
- A High-Throughput Screen of a Library of Therapeutics Identifies Cytotoxic Substrates of P-glycoproteinPublication Name: Molecular PharmacologyPublication Date: 2019-11PMCID: PMC6790066PMID: 31515284DOI: 10.1124/mol.119.115964
- Predictive models of aqueous solubility of organic compounds built on A large dataset of high integrityPublication Name: Bioorganic & Medicinal ChemistryPublication Date: 2019-07-15PMCID: PMC8274818PMID: 31176566DOI: 10.1016/j.bmc.2019.05.037
/////////Privosegtor, Phase 2, Optic neuritis, orphan drug, BN-201, BN 201, G-79, G 79, KCN37L7EIH
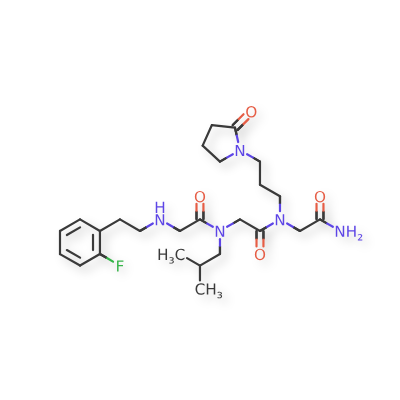
Pregabalin naproxencarbil
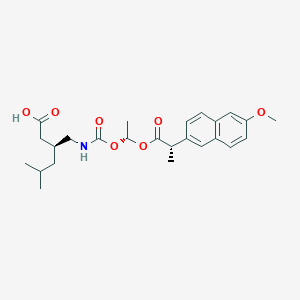
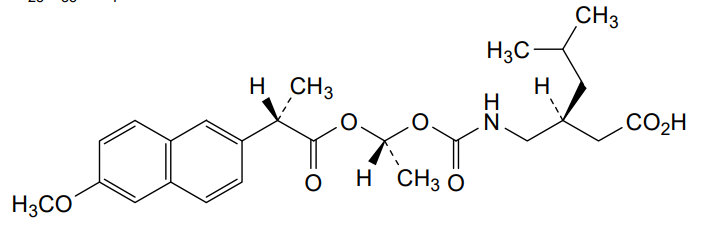
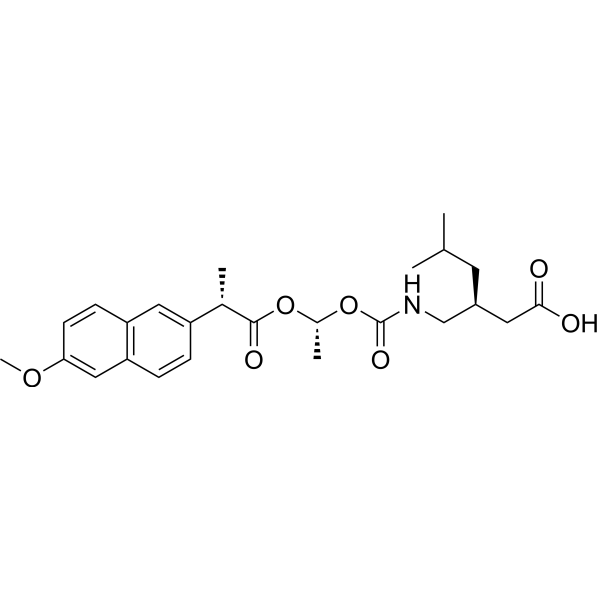
Pregabalin naproxencarbil
CAS 1221072-91-8
MF C25H33NO7 MW459.5 g/mol
(3S)-3-[({[(1R)-1-{[(2S)-2-(6-methoxynaphthalen-2-yl)propanoyl]oxy}ethoxy]carbonyl}amino)methyl]-5-
methylhexanoic acid
(3S)-3-[[[(1R)-1-[(2S)-2-(6-methoxynaphthalen-2-yl)propanoyl]oxyethoxy]carbonylamino]methyl]-5-methylhexanoic acid
gabamimetic, analgesic, ZVG8DDT3FJ
- OriginatorXgene Pharmaceutical
- ClassAminobutyric acids; Analgesics; Antiepileptic drugs; Antipyretics; Antirheumatics; Anxiolytics; Drug conjugates; Gabapentinoids; Naphthaleneacetic acids; Neuroprotectants; Nonsteroidal anti-inflammatories; Small molecules
- Mechanism of ActionCACNA2D1 protein modulators; Cyclooxygenase inhibitors
- Phase II/IIIPostoperative pain
- Phase IIAcute pain; Cancer pain; Pain
- Phase I/IIBack pain; Neuropathic pain
- No development reportedDiabetic neuropathies
- 15 Jul 2025XG005 licensed to NeuroGen in China, Hong Kong, and Macau
- 31 Dec 2024Efficacy and adverse events data from phase-II/III trial in Postoperative pain released by Xgene Pharmaceutical
- 25 Oct 2024Xgene Pharmaceutical completes the phase II/III trial in Postoperative pain in USA (PO)
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019219000&_cid=P22-MID0L3-49648-1
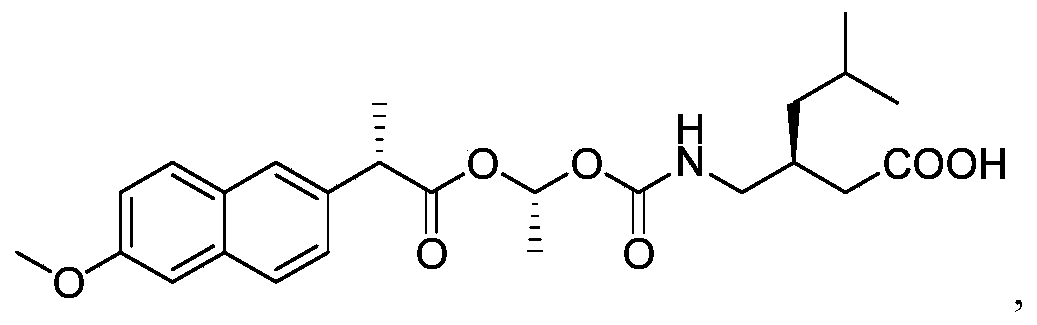
Example 1: Exemplary synthesis of the compound of Formula I and crystallization thereof
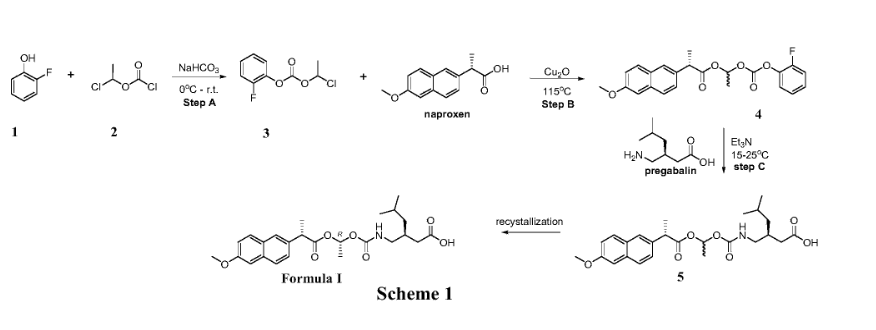
Step A: Synthesis of 1-chloroethyl 2-fluorophenyl carbonate (3)
[0306]
A suitable reaction vessel was charged with water and sodium bicarbonate followed by the starting material 2-fluoro-phenol (1) . The mixture was cooled to a temperature of about 0~5℃ and 1-chloroethyl chloroformate (2) was added slowly while maintaining the temperature at 0~5℃. The temperature was raised to about 15 ± 5℃. When the reaction was judged complete by the disappearance of 2-fluoro-phenol (criteria: ≤ 2.0%, by HPLC) the reaction was worked up. n-Heptane was added and the organic phase was separated, washed with water and brine. The solution was concentrated, then toluene was added and the solution was concentrated again. The toluene addition and the concentration cycle were repeated once more.
[0307]
Step B: Synthesis of (S) – ( (R, S) -1- ( (2-fluorophenoxy) carbonyloxy) ethyl 2- (6-methoxynaphthalen-2-yl) propanoate (4)
[0308]
To a solution of naproxen in toluene in a suitable reaction vessel, 1-chloroethyl 2-fluorophenyl carbonate (3) and cuprous oxide was added. The temperature of the mixture was raised to about 115 ±5℃. When the reaction was judged complete by the disappearance of 1-chloroethyl 2-fluorophenyl carbonate (criteria: ≤ 2.5%, by HPLC) the reaction was worked up. Methyl tert-butyl ether was added at about 50 ± 5℃. The resulting mixture was filtered and the filtrate was collected at about 25 ± 5℃. Purified water was added into the filtrate and then the mixture was cooled to about 0 ± 5℃. The mixture was alkalified with ammonium hydroxide to a pH of about 9~11 and the organic phase was separated and washed with ammonium hydroxide and brine. The solution was concentrated, then acetonitrile was added and the solution was concentrated again. The acetonitrile addition and the concentration cycle were repeated some more times until the residual toluene was not more than 10% (by GC method) .
[0309]
Step C: Synthesis of (S) -3- ( ( ( (R) -1- ( (S) -2- (6-methoxynaphthalen-2-yl) propanoyloxy) ethoxy) carbonyl-amino) methyl) -5-methylhexanoic acid (the compound of Formula I)
[0310]
To a solution of the mixture of (S) – ( (R, S) -1- ( (2-fluorophenoxy) carbonyloxy) ethyl 2- (6-methoxynaphthalen-2-yl) propanoate (4) in acetonitrile and methyl tert-butyl ether in a suitable reaction vessel, purified water and pregabalin was charged. Triethylamine was added slowly while maintaining the temperature at about 15 ± 5℃. The temperature was raised to about 25 ± 3℃ for reaction. When the reaction was judged complete by the disappearance of (S) – ( (R, S) -1- ( (2-fluorophenoxy) carbonyloxy) ethyl 2- (6-methoxynaphthalen-2-yl) propanoate (criteria: ≤ 0.5%, by HPLC) the reaction was worked up. The resulting mixture was acidified with KHSO 4to pH 3~5 and extracted with methyl tert-butyl ether. The combined organic layers were washed with purified water and brine. The organic phase was then concentrated. Isopropanol was added and the solution was concentrated again. The isopropanol addition and the concentration cycle was repeated once or more times until the total residual acetonitrile and methyl tert-butyl ether was not more than 5% (by GC method) . n-Heptane was added into the mixture at about 40~45 ℃ and stirred and then the temperature was gradually lowered at set appropriate intervals to crystallize (S) -3- ( ( ( (R) -1- ( (S) -2- (6-methoxynaphthalen-2-yl) propanoyloxy) ethoxy) carbonyl-amino) methyl) -5-methylhexanoic acid (5) from the system. When the precipitation was complete, the heterogeneous mixture was centrifuged and the solid was collected.
[0311]
The crude product of (S) -3- ( ( ( (R) -1- ( (S) -2- (6-methoxynaphthalen-2-yl) propanoyloxy) ethoxy) carbonyl-amino) methyl) -5-methylhexanoic acid (5) was added to a solution of isopropanol and water in a suitable vessel. The mixture was stirred while raising the temperature to 45 ± 3 ℃ until all the solid was dissolved, then the temperature was lowered gradually at set appropriate intervals to recrystallize (S) -3- ( ( ( (R) -1- ( (S) -2- (6-methoxynaphthalen-2-yl) propanoyloxy) ethoxy) carbonyl-amino) methyl) -5-methylhexanoic acid from the system. When the precipitation had stopped, the heterogeneous mixture was centrifuged and the expected pure (S) -3- ( ( ( (R) -1- ( (S) -2- (6-methoxynaphthalen-2-yl) propanoyloxy) ethoxy) carbonyl-amino) methyl) -5-methylhexanoic acid (Formula I) was collected.
Example 2: Alternative synthetic route of (S) -3- ( ( ( (R) -1- ( (S) -2- (6-methoxynaphthalen-2-yl) propanoyloxy) ethoxy) carbonyl-amino) methyl) -5-methylhexanoic acid
PAT
- Crystalline form of 1-(acyloxy)-alkyl carbamate drug complex of naproxen and pregabalinPublication Number: JP-7441181-B2Priority Date: 2018-05-14Grant Date: 2024-02-29
- Crystalline forms of 1-(acyloxy)-alkyl carbamate drug conjugates of naproxen and pregabalinPublication Number: TW-I837128-BPriority Date: 2018-05-14Grant Date: 2024-04-01
- Method for preparing 1-(acyloxy)-alkyl carbamate drug complex of naproxen and pregabalin and intermediate thereofPublication Number: KR-102750784-B1Priority Date: 2018-05-14Grant Date: 2025-01-06
- GABA conjugates and methods of use thereofPublication Number: US-9186341-B2Priority Date: 2008-10-08Grant Date: 2015-11-17
- Crystalline forms of 1-(acyloxy)-alkyl carbamate drug conjugates of naproxen and pregabalinPublication Number: US-2021221768-A1Priority Date: 2018-05-14
- Crystalline morphology of the 1- (acyloxy) -alkylcarbamate drug complex of naproxen and pregabalinPublication Number: JP-2021530434-APriority Date: 2018-05-14
- Crystalline forms of 1-(acyloxy)-alkyl carbamate drug conjugates of naproxen and pregabalinPublication Number: AU-2019271799-B2Priority Date: 2018-05-14Grant Date: 2023-10-12
- Crystalline forms of 1-(acyloxy)-alkyl carbamate drug conjugates of naproxen and pregabalinPublication Number: EP-4227293-A2Priority Date: 2018-05-14
- Crystalline forms of 1-(acyloxy)-alkyl carbamate drug conjugates of naproxen and pregabalinPublication Number: EP-4227293-A3Priority Date: 2018-05-14
- Crystalline forms of 1-(acyloxy)-alkyl carbamate drug conjugates of naproxen and pregabalinPublication Number: AU-2019271799-A1Priority Date: 2018-05-14
- crystalline forms of 1- (acyloxy) -alkyl carbamate drug conjugates of naproxen and pregabalinPublication Number: BR-112020022885-A2Priority Date: 2018-05-14
- Crystalline forms of 1-(acyloxy)-alkyl carbamate drug conjugates of naproxen and pregabalinPublication Number: CN-112424158-APriority Date: 2018-05-14
- Crystalline forms of 1-(acyloxy)-alkyl carbamate drug conjugates of naproxen and pregabalinPublication Number: EP-3793974-A1Priority Date: 2018-05-14
- Crystalline form of 1-(acyloxy)-alkyl carbamate drug complex of naproxen and pregabalinPublication Number: KR-20210013081-APriority Date: 2018-05-14
- Naproxen and pregabalin 1-(acyloxy)-alkyl carbamate drug conjugate purification processPublication Number: TW-202436285-APriority Date: 2022-11-24
- Naproxen and pregabalin 1- (acyloxy) -alkyl carbamate drug conjugate purification processPublication Number: WO-2024109817-A1Priority Date: 2022-11-24
- Crystalline forms of 1-(acyloxy)-alkyl carbamate drug conjugates of naproxen and pregabalinPublication Number: WO-2019219000-A1Priority Date: 2018-05-14
- Process for making 1-(acyloxy)-alkyl-carabmate drug conjugates of naproxen and pregabalinPublication Number: CA-3099775-A1Priority Date: 2018-05-14
- Naproxen (NAPROXEN) and pregabalin (PREGABALIN) 1-(acetyl)-alkylcarbamate drug conjugate crystalline formPublication Number: TW-202016065-APriority Date: 2018-05-14
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
.//////////Pregabalin naproxencarbil, gabamimetic, analgesic, ZVG8DDT3FJ
Sevabertinib
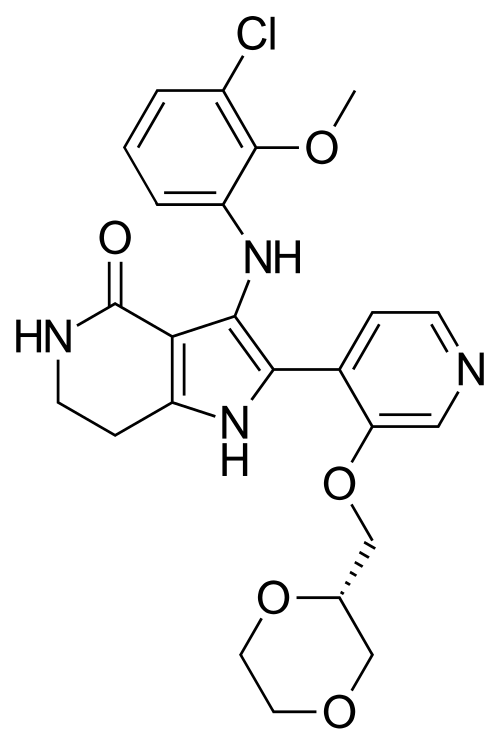
Sevabertinib
CAS 2521285-05-0
MF C24H25ClN4O5, 484.9 g/mol
3-(3-chloro-2-methoxyanilino)-2-[3-[[(2S)-1,4-dioxan-2-yl]methoxy]-4-pyridinyl]-1,5,6,7-tetrahydropyrrolo[3,2-c]pyridin-4-one
11/19/2025, FDA 2025, APPROVALS 2025, Hyrnuo, 2A7VPM5RWH, BAY-2927088, BAY 2927088
To treat locally advanced or metastatic non-squamous non-small cell lung cancer with tumors that have activating HER2 tyrosine kinase domain activating mutations in patients who received a systemic therapy
Sevabertinib, sold under the brand name Hyrnuo, is an anti-cancer medication used for the treatment of non-small cell lung cancer.[1] Sevabertinib is a kinase inhibitor.[1] It is taken by mouth.[1]
Sevabertinib was approved for medical use in the United States in November 2025.[2]
SYN
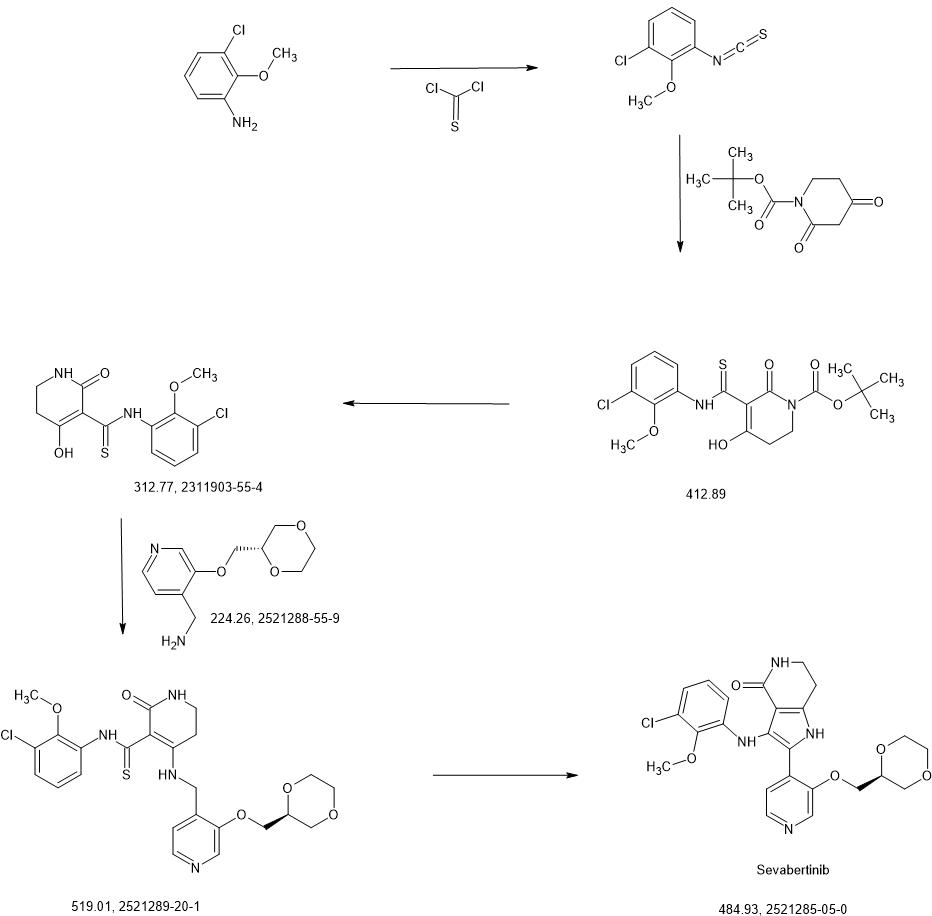
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020216781&_cid=P22-MICIVF-33261-1
Intermediate 3-1
1-chloro-3-isothiocyanato-2-methoxybenzene
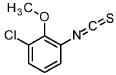
3-chloro-2-methoxyaniline (CAS 511 14-68-2, 8.4 ml, 63 mmol) was solved in DCM (100 ml) and sat. sodium bicarbonate solution (100 ml) was added. To the ice cooled mixture was slowly added thiophosgene (5.4 ml, 70 mmol). The reaction was stirred at 0°C for 2 h. At RT the DCM layer was separated and washed with sat. sodium bicarbonate solution, filtered through a hydrophobic filter and concentrated under reduced pressure to give the title compound (12.97 g, 100 % yield) which was used directly in the next step.
1H-NMR (400MHz, DMSO-de): d [ppm]= 7.51 (dd, 1 H), 7.35 (dd, 1 H), 7.20 (t, 1 H), 3.85 -3.91 (m, 3H).
Intermediate 4-1
tert- butyl 5-[(3-chloro-2-methoxyphenyl)carbamothioyl]-4-hydroxy-6-oxo-3,6-dihydropyridine-1(2/-/)-carboxylate
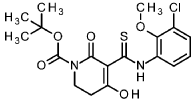
To an ice-cooled solution of 1-chloro-3-isothiocyanato-2-methoxybenzene (intermediate 3-1 , 4.00 g, 20.0 mmol) and tert- butyl 2,4-dioxopiperidine-1-carboxylate (CAS 845267-78-9, 4.27 g, 20.0 mmol) in acetonitrile (92 ml) was added dropwise DBU (4.5 ml, 30 mmol). The reaction was stirred at RT overnight. To the reaction mixture was added ice-water (200 ml_) and cone. HCI (2 ml_). The mixture was stirred for 20 min. and extracted with DCM. The organic phase was filtered over a water-repellent filter, conentrated under reduced pressure and purified by flash chromatography (silica, hexane / EtOAc gradient 0-50 %) to give 6.54 g of the title compound (71 % yield).
1H-NMR (400MHz, DMSO-de): d [ppm]= 13.36 (br s, 1 H), 7.73 (d, 1 H), 7.47 (dd, 1 H), 7.22 (t, 1 H), 3.76 – 3.82 (m, 5H), 2.88 (t, 2H), 1.48 (s, 9H).
LC-MS (method 1): Rt = 1.49 min; MS (ESIpos): m/z = 413.1 [M+H]+
Intermediate 5-1
A/-(3-chloro-2-methoxyphenyl)-4-hydroxy-2-oxo-1 ,2,5,6-tetrahydropyridine-3-carbothioamide
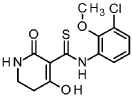
To a solution of tert- butyl 5-[(3-chloro-2-methoxyphenyl)carbamothioyl]-4-hydroxy-6-oxo-3,6-dihydropyridine-1 (2/-/)-carboxylate (intermediate 4-1 , 6.54 g, 15.8 mmol) in dichloromethane (94 ml) was added TFA (12 ml, 160 mmol) and the mixture was stirred 1.5 h at RT. The reaction mixture was concentrated under reduced pressure and the residue was solved in EtOAc and washed with sat. sodium bicarbonate solution and brine. The organic layer was filtered through a hydrophobic filter and the filtrate was dried to dryness. The residue was purified by flash chromatography (silica, hexane / EtOAc gradient 20-100 %) to give 4.06 g of the title compound (78 % yield).
1H-NMR (400 MHz, DMSO-de): d [ppm]= 16.45 (d, 1 H), 14.69 (s, 1 H), 14.33 (s, 1 H), 9.37 (br s, 1 H), 8.18 (br s, 1 H), 7.76 – 7.87 (m, 1 H), 7.37 – 7.45 (m, 1 H), 7.15 – 7.23 (m, 1 H), 3.73 – 3.76 (m, 3H), 3.43 (td, 1 H), 3.27 – 3.32 (m, 1 H), 2.79 (t, 1 H), 2.59 – 2.69 (m, 1 H).
LC-MS (method 1): Rt = 1.19 min; MS (ESIpos): m/z = 313 [M+H]+
ntermediate 6-2
A/-(3-chloro-2-methoxyphenyl)-4-{[(3-{[(2S)-1 ,4-dioxan-2-yl]methoxy}pyridin-4-yl)methyl]amino}-2-oxo-1 ,2,5,6-tetrahydropyridine-3-carbothioamide
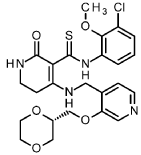
A mixture of A/-(3-chloro-2-methoxyphenyl)-4-hydroxy-2-oxo-1 ,2,5,6-tetrahydropyridine-3-carbothioa ide (intermediate 5-1 , 866 mg, 2.77 mmol) and 1-(3-{[(2S)-1 ,4-dioxan-2-yl]methoxy}pyridin-4-yl)methanamine (intermediate 2-8, 776 mg, 80% purity, 2.77 mmol) in ACN (22 ml) was treated with A/,0-bis(trimethylsilyl)acetamide (2.05 ml, 8.6 mmol, CAS 10416-59-8) and stirred at 80°C for 4 h. The reaction mixture was concentrated under reduced pressure and purified by flash chromatography (silica, DCM / EtOH gradient 0-20%) to give 1.23 g (95% purity, 81 % yield) of the title compound.
1H-NMR (400MHz, DMSO-d6): d [ppm]= 2.78 (t, 2H), 3.16 (td, 2H), 3.40 – 3.54 (m, 3H), 3.59 – 3.69 (m, 2H), 3.71 (s, 3H), 3.73 – 3.79 (m, 1 H), 3.83 – 3.95 (m, 2H), 4.16 (t, 2H), 4.67 (d, 2H), 7.11 (t, 1 H), 7.27 – 7.33 (m, 2H), 7.73 (br s, 1 H), 7.81 (dd, 1 H), 8.24 (d, 1 H), 8.39 (s, 1 H), 13.69 (s, 1 H), 14.79 (s, 1 H).
LC-MS (method 2): Rt = 1.09 min; MS (ESIpos): m/z = 519 [M+H]+
Example 2
3-(3-chloro-2-methoxyanilino)-2-(3-{[(2S)-1 ,4-dioxan-2-yl]methoxy}pyridin-4-yl)-1 ,5,6,7-tetrahydro-4H-pyrrolo[3,2-c]pyridin-4-one (Stereoisomer 1)

The title compound from example 1 (140 mg) was separated into enantiomers by preparative chiral HPLC to give title compound (enantiomer 1 , 27 mg at Rt = 14.0 – 17.0 min) and enantiomer 2 (25 mg at Rt = 20.0 – 24.8 min, see example 3).
Preparative chiral HPLC method:
Instrument: Labomatic HD5000, Labocord-5000; Gilson GX-241 , Labcol Vario 4000; column: Cellulose SB 5m, 250×30 mm; eluent A: hexane + 0.1 vol. % diethylamine (99 %); eluent B: 2-propanol; isocratic: 50 % A + 50 % B; flow 50 ml/min; UV: 254 nm.
Analytical chiral HPLC method:
Instrument: Agilent HPLC 1260; column: Cellulose SB 3m, 100×4.6 mm; eluent A: hexane + 0.1 vol. % diethylamine (99 %); eluent B: 2-propanol; isocratic: 50 % A + 50 % B, flow 1.4 ml/min; temperature: 25°C; UV: 254 nm
Analytical chiral HPLC: Rt = 4.49 min.
Optical rotation:[a]D = 1.7° +/- 0.98° (c = 3.6 mg/2 ml, methanol)
Enantioselective synthesis confirmed the title compound as 3-(3-chloro-2-methoxyanilino)-2-(3-{[(2S)-1 ,4-dioxan-2-yl]methoxy}pyridin-4-yl)-1 ,5,6,7-tetrahydro-4/-/-pyrrolo[3,2-c]pyridin-4-one. 872 mg (95% purity, 72% yield) of the title compound were prepared in analogy to example 1 using A/-(3-chloro-2-methoxyphenyl)-4-{[(3-{[(2S)-1 ,4-dioxan-2-yl]methoxy}pyridin-4-yl)methyl]amino}-2-oxo-1 ,2,5,6-tetrahydropyridine-3-carbothioamide (intermediate 6-2, 1.23 g, 2.36 mmol) as starting material, followed by purification with preparative HPLC (method 10, gradient: 0.00-0.50 min 15% B, 0.50-6.00 min 15-55% B).
1H-NMR (400MHz, DMSO-d6): d [ppm]= 2.86 (t, 2H), 3.38 – 3.47 (m, 3H), 3.53 (td, 1 H), 3.69
– 3.78 (m, 2H), 3.83 (dd, 1 H), 3.88 (s, 3H), 3.90 (m, 1 H), 3.98 – 4.08 (m, 1 H), 4.12 – 4.18 (m, 1 H), 4.28 (dd, 1 H), 6.12 – 6.17 (quin, 1 H), 6.66 – 6.71 (m, 2H), 7.16 (s, 1 H), 7.28 (d, 1 H),
7.52 (s, 1 H), 8.04 (d, 1 H), 8.39 (s, 1 H), 11.07 (s, 1 H).
Analytical chiral HPLC: Rt = 4.46 min.
Optical rotation:[a]D = -12.5° +/- 0.52° (c = 5.6 mg/ l, chloroform)
PAT
- 4H-pyrrolo[3,2-c]pyridin-4-one compoundPublication Number: CN-114127064-BPriority Date: 2019-04-24Grant Date: 2023-12-26
- 4H-Pyrrolo[3,2-c]pyridin-4-one compoundsPublication Number: CN-117946100-APriority Date: 2019-04-24
- 4H-pyrrolo [3,2-c ] pyridin-4-one compoundsPublication Number: CN-117986251-APriority Date: 2019-04-24
- 4h-pyrrolo[3,2-c]pyridin-4-one compoundsPublication Number: TW-I849114-BPriority Date: 2019-04-24Grant Date: 2024-07-21
- 4H-pyrrolo[3,2-c]pyridin-4-one compoundPublication Number: KR-20220004103-APriority Date: 2019-04-24
- 4H-PYRROLO[3,2-C]PYRIDIN-4-ONE COMPOUNDSPublication Number: PE-20220254-A1Priority Date: 2019-04-24
- 4H-Pyrrolo [3,2-C] Pyridine-4-one CompoundPublication Number: JP-2022532850-APriority Date: 2019-04-24
- 4h-pyrrolo[3,2-c]pyridin-4-one compoundsPublication Number: US-2022298157-A1Priority Date: 2019-04-24
- 4H-PYRROL[3,2-C]PYRIDIN-4-ONE, ITS USES, PHARMACEUTICAL COMPOSITION, AND KIT OF PARTSPublication Number: BR-112021019998-B1Priority Date: 2019-04-24
- 4h-pyrrolo[3,2-c]pyridin-4-one compoundsPublication Number: WO-2020216781-A1Priority Date: 2019-04-24
- 4h-pyrrolo[3,2-c]pyridin-4-one compoundsPublication Number: TW-202106683-APriority Date: 2019-04-24
- 4H-pyrrolo(3,2-c)pyridin-4-one compoundsPublication Number: AU-2020262221-A1Priority Date: 2019-04-24
- 4H-pyrrolo [3,2-c ] pyridin-4-one compoundsPublication Number: CN-114127064-APriority Date: 2019-04-24
- 4h-pyrrolo[3,2-c]pyridin-4-one compoundsPublication Number: EP-3959211-A1Priority Date: 2019-04-24
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Medical uses
Sevabertinib is indicated for the treatment of adults with locally advanced or metastatic non-squamous non-small cell lung cancer whose tumors have HER2 (ERBB2) tyrosine kinase domain activating mutations.[1][2]
Adverse effects
The US prescribing information includes warnings and precautions for diarrhea, hepatotoxicity, interstitial lung disease/pneumonitis, ocular toxicity, pancreatic enzyme elevation, and embryo-fetal toxicity.[2]
History
Efficacy was evaluated in people with unresectable or metastatic, non-squamous non-small cell lung cancer with HER2 (ERBB2) tyrosine kinase domain activating mutations who had received prior systemic therapy and received sevabertinib in SOHO-01 (NCT05099172), an open-label, single-arm, multi-center, multi-cohort clinical trial.[2] HER2 (ERBB2) activating mutations were determined in tumor tissue or plasma by local laboratories prior to enrollment.[2]
The US Food and Drug Administration granted the application for sevabertinib priority review, breakthrough therapy, and orphan drug designations.[2]
Society and culture
Legal status
Sevabertinib was approved for medical use in the United States in November 2025.[3][4]
Names
Sevabertinib is the international nonproprietary name.[5]
Sevabertinib is sold under the brand name Hyrnuo.[1][3]
References
- “HYRNUO (sevabertinib) tablets, for oral use” (PDF). Bayer HealthCare Pharmaceuticals Inc. U.S. Food and Drug Administration.
- “FDA grants accelerated approval to sevabertinib for non-squamous non-small cell lung cancer”. U.S. Food and Drug Administration (FDA). 19 November 2025. Retrieved 21 November 2025.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - “U.S. FDA Approves Hyrnuo (sevabertinib) for Previously Treated Patients with HER2-Mutated Locally Advanced or Metastatic Non-Squamous Non-Small Cell Lung Cancer” (Press release). Bayer. 20 November 2025. Retrieved 21 November 2025 – via Business Wire.
- “U.S. FDA grants accelerated approval to Bayer’s Hyrnuo (sevabertinib) for patients with previously treated advanced HER2-mutant non-small cell lung cancer”. Bayer (Press release). 20 November 2025. Retrieved 21 November 2025.
- World Health Organization (2025). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 93”. WHO Drug Information. 39 (1). hdl:10665/381075.
Further reading
- Le X, Kim TM, Loong HH, Prelaj A, Goh BC, Li L, et al. (November 2025). “Sevabertinib in Advanced HER2-Mutant Non-Small-Cell Lung Cancer”. The New England Journal of Medicine. 393 (18): 1819–1832. doi:10.1056/NEJMoa2511065. PMID 41104928.
- Siegel F, Siegel S, Kotýnková K, Karsli Uzunbas G, Korr D, Tomono H, et al. (October 2025). “Sevabertinib, a Reversible HER2 Inhibitor with Activity in Lung Cancer”. Cancer Discovery: OF1 – OF14. doi:10.1158/2159-8290.CD-25-0605. PMID 41090369.
External links
- “Sevabertinib”. NCI Drug Dictionary.
- “Sevabertinib ( Code – C185187 )”. EVS Explore.
- Clinical trial number NCT05099172 for “First in Human Study of BAY2927088 in Participants Who Have Advanced Non-small Cell Lung Cancer (NSCLC) With Mutations in the Genes of Epidermal Growth Factor Receptor (EGFR) and/or Human Epidermal Growth Factor Receptor 2 (HER2)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Hyrnuo |
| Other names | BAY2927088, sevabertinib hydrate (JAN JP) |
| License data | US DailyMed: Sevabertinib |
| Routes of administration | By mouth |
| Drug class | Antineoplastic |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| CAS Number | 2521285-05-0 |
| PubChem CID | 155234713 |
| DrugBank | DB21667 |
| ChemSpider | 129786615 |
| UNII | 2A7VPM5RWH |
| KEGG | D13098 |
| Chemical and physical data | |
| Formula | C24H25ClN4O5 |
| Molar mass | 484.94 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
/////sevabertinib, FDA 2025, APPROVALS 2025, Hyrnuo, 2A7VPM5RWH, BAY-2927088, BAY 2927088

{kind=link}