
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 7)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Zerencotrep



Zerencotrep
CAS 1628287-16-0
MF C23H20ClF3N4O5, MW 524.88
7-[(4-chlorophenyl)methyl]-1-(3-hydroxypropyl)-3-methyl-8-[3-(trifluoromethoxy)phenoxy]purine-2,6-dione
7-[(4-chlorophenyl)methyl]-1-(3-hydroxypropyl)-3-
methyl-8-[3-(trifluoromethoxy)phenoxy]-3,7-dihydro1H-purine-2,6-dione
transient receptor potential channel 4 and 5 (TRPC4, TRPC5) inhibitor, Pico 145, HC 608, HMIMSYLCWQ
Pico145 (HC-608) is a remarkable inhibitor of TRPC1/4/5 channels, inhibits (-)-englerin A-activated TRPC4/TRPC5 channels, with IC50s of 0.349 and 1.3 nM in cells, and shows no effect on TRPC3, TRPC6, TRPV1, TRPV4, TRPA1, TRPM2, TRPM8.
Zerencotrep is a small molecule drug. The usage of the INN stem ‘-cotrep’ in the name indicates that Zerencotrep is a transient receptor potential canonical channel 5 (TRPC5) antagonist. Zerencotrep has a monoisotopic molecular weight of 524.11 Da.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014143799&_cid=P22-MJF454-30876-1


The following examples 7a through 7k were prepared using the method of example 6, step 1.
Example 7a 7-(4-chlorobenzyl)-l -(3-hydroxypropyl)-3-methyl-8-(3-(trifluoromethoxy)phenoxy)-l -purine-2,6(3H,7H)-dione

The title compound was prepared using the method of example 6, step 1 and purified
preparative HPLC to give 7-(4-chlorobenzyl)-l -(3-hydroxypropyl)-3-methyl-8-(3-
(trifluoromethoxy)phenoxy)-lH-purine-2,6(3H,7H)-dione (10 mg, 17.3% yield) as white solid. lH-NMR (CD3OD) δ 7.57-7.53(t, IH), 7.46-7.44(d, 2H), 7.37-7.33(m, 4H), 7.26-7.24(d, IH), 5.49(s, 2H), 4.13-4.09(t, IH), 3.64-3.60(t, 2H), 3.42(s, 3H), 1.89-1.86(m, 2H). LCMS retention time 3.059 min; LCMS MH+ 525.
PAT
- Side chain unsaturated 1α-hydroxyvitanim D homologsPublication Number: US-5250523-APriority Date: 1988-04-29Grant Date: 1993-10-05
- Antiviral methods utilizing ribofuranosylthiazolo[4,5-d]pyrimdine derivativesPublication Number: US-4880784-APriority Date: 1987-12-21Grant Date: 1989-11-14
- NEW NUCLEOSIDES AND NUCLEOTIDES AND PROCEDURES FOR THE PREPARATION.Publication Number: NO-893343-LPriority Date: 1987-12-21
- New amidino derivativesPublication Number: JP-H01131145-APriority Date: 1987-09-21
- Oligonucleotides, a process for preparing the same and their application as mediators of the action of interferonPublication Number: US-4476301-APriority Date: 1982-04-29Grant Date: 1984-10-09



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
- Pico145 inhibits TRPC4-mediated mICAT and postprandial small intestinal motilityPublication Name: Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapiePublication Date: 2023-12PMID: 37857250DOI: 10.1016/j.biopha.2023.115672
- Human TRPC5 structures reveal interaction of a xanthine-based TRPC1/4/5 inhibitor with a conserved lipid binding sitePublication Name: Communications BiologyPublication Date: 2020-11-23PMCID: PMC7683545PMID: 33230284DOI: 10.1038/s42003-020-01437-8
- Discovery of a Potent and Selective TRPC5 Inhibitor, Efficacious in a Focal Segmental Glomerulosclerosis ModelPublication Name: ACS Medicinal Chemistry LettersPublication Date: 2019-10-22PMCID: PMC6862342PMID: 31749913DOI: 10.1021/acsmedchemlett.9b00430
- Potent, selective, and subunit‐dependent activation of TRPC5 channels by a xanthine derivativePublication Name: British Journal of PharmacologyPublication Date: 2019-09-06PMCID: PMC6811774PMID: 31277085DOI: 10.1111/bph.14791
- Review of Transient Receptor Potential Canonical (TRPC5) Channel Modulators and DiseasesPublication Name: Journal of Medicinal ChemistryPublication Date: 2019-04-03PMID: 30943030DOI: 10.1021/acs.jmedchem.8b01954
///////////zerencotrep, Pico 145, HC 608, HMIMSYLCWQ
Zemprocitinib


Zemprocitinib
CAS 2417414-44-7
MF C16H19N5O2S MW 345.4 g/mol
N-[3-(3,5,8,10-tetrazatricyclo[7.3.0.02,6]dodeca-1,4,6,8,11-pentaen-3-yl)-1-bicyclo[1.1.1]pentanyl]propane-1-sulfonamide
N-[3-(imidazo[4,5-d]pyrrolo[2,3-b]pyridin-1(6H)-yl)bicyclo[1.1.1]pentan-1-yl]propane-1-sulfonamide
Janus kinase inhibitor, anti-inflammatory, LNK 01001, LG6MM3RP86
Zemprocitinib (also known as LNK01001) is a selective Janus kinase (JAK) 1 inhibitor, a type of small molecule drug being developed for inflammatory and autoimmune conditions like rheumatoid arthritis, atopic dermatitis, and ankylosing spondylitis. It works by blocking the JAK1 enzyme, reducing the inflammatory signals that cause these diseases, and has shown promising results in clinical trials, with development reaching Phase 3.
Key Aspects:
- Drug Class: JAK1 Inhibitor.
- Mechanism: Blocks Janus Kinase 1, a key enzyme in inflammatory pathways.
- Developer: Initially Lynk Pharmaceuticals.
- Potential Uses: Rheumatoid Arthritis, Atopic Dermatitis, Ankylosing Spondylitis, Psoriasis, Alopecia Areata.
- Development Stage: Reached Phase 3 clinical trials for several indications.
- Chemical Info: CAS: 2417414-44-7; Formula: C16H19N5O2S.
In Summary:
Zemprocitinib is an investigational drug targeting inflammation by inhibiting JAK1, with potential to treat various autoimmune disorders, showing strong efficacy in early clinical trials for conditions like rheumatoid arthritis.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US347660217&_cid=P21-MJDP3D-82397-1
Example 1



Step 1. 4-Chloro-1-tosyl-1H-pyrrolo[2,3-b]pyridine (1b)
Step 2. 4-Chloro-5-nitro-1-tosyl-1H-pyrrolo[2,3-b]pyridine (1c)
Step 3. Tert-butyl 3-((5-nitro-1-tosyl-1H-pyrrolo[2,3-b]pyridin-4-yl)amino)bicyclo[1.1.1]pentane-1-carboxylate (Id)
Step 4. Tert-butyl 3-((5-amino-1-tosyl-1H-pyrrolo[2,3-b]pyridin-4-yl)amino)bicyclo[1.1.1]pentane-1-carboxylate (le)
Step 5. Tert-butyl 3-(6-tosylimidazo[4,5-d]pyrrolo[2,3-b]pyridin-1(6H)-yl)bicyclo[1.1.1]pentane-1-carboxylate (1f)
Step 6. 3-(6-Tosylimidazo[4,5-d]pyrrolo[2,3-b]pyridin-1(6LF)-yl)bicyclo[1.1.1]pentane-1-carboxylic acid (1g)
Step 7. Tert-butyl (3-(6-tosylimidazo[4,5-d]pyrrolo[2,3-b]pyridin-1(6LF)-yl)bicyclo[1.1.1]pentan-1-yl)carbamate (1h)
Step 8. Tert-butyl (3-(imidazo[4,5-d]pyrrolo[2,3-b]pyridin-1(6H)-yl)bicyclo[1.1.1]pentan-1-yl)carbamate (1i)
Step 9. 3-(Imidazo[4,5-d]pyrrolo[2,3-b]pyridin-1(6H)-yl)bicyclo[1.1.1]pentan-1-amine 2,2,2-trifluoroacetate (1j)
Step 10. N-(3-(Imidazo[4,5-d]pyrrolo[2,3-b]pyridin-1(6H)-yl)bicyclo[1.1.1]pentan-1-yl)propane-1-sulfonamide (1)
PAT
- Tricyclic janus kinase 1 inhibitors, and compositions and methods thereofPublication Number: ES-2993867-T3Priority Date: 2018-11-01Grant Date: 2025-01-10
- Tricyclic janus kinase 1 inhibitors, and compositions and methods thereofPublication Number: JP-2024147699-APriority Date: 2018-11-01
- Tricyclic janus kinase 1 inhibitors, and compositions and methods thereofPublication Number: EP-3856742-B1Priority Date: 2018-11-01Grant Date: 2024-10-02
- Tricyclic janus kinase 1 inhibitors, and compositions and methods thereofPublication Number: US-2022009927-A1Priority Date: 2018-11-01
- Tricyclic janus kinase 1 inhibitors, and compositions and methods thereofPublication Number: US-2023357247-A1Priority Date: 2018-11-01
- Tricyclic janus kinase 1 inhibitors, and compositions and methods thereofPublication Number: US-2023339950-A1Priority Date: 2018-11-01
- Tricyclic Janus kinase 1 inhibitors, and compositions and methods thereofPublication Number: AU-2019372677-B2Priority Date: 2018-11-01Grant Date: 2024-05-30
- Tricyclic janus kinase 1 inhibitors, and compositions and methods thereofPublication Number: TW-202432555-APriority Date: 2018-11-01
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////Zemprocitinib, Janus kinase inhibitor, anti-inflammatory, LNK 01001, LG6MM3RP86
Zemirciclib



Zemirciclib
CAS 2057509-72-3
MF C22H28ClN5O2, 429.9 g/mol
(1S,3R)-3-acetamido-N-[5-chloro-4-(5,5-dimethyl-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl)pyridin-2-
yl]cyclohexane-1-carboxamide
(1S,3R)-3-acetamido-N-[5-chloro-4-(5,5-dimethyl-4,6-dihydropyrrolo[1,2-b]pyrazol-3-yl)pyridin-2-yl]cyclohexane-1-carboxamide
(1S,3R)-3-acetamido-N-(5-chloro-4-(5,5-dimethyl-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl)pyridin-2-yl)cyclohexanecarboxamide
cyclin-dependent kinase inhibitor, antineoplastic, AZD 4573, UNII-E5XSP3X68B
Zemirciclib is a selective, short-acting inhibitor of the serine/threonine cyclin-dependent kinase 9 (CDK9), the catalytic subunit of the RNA polymerase II (RNA Pol II) elongation factor positive transcription elongation factor b (PTEF-b; PTEFb), with potential antineoplastic activity. Upon intravenous administration, zemirciclib binds to and blocks the phosphorylation and kinase activity of CDK9, thereby preventing PTEFb-mediated activation of RNA Pol II, leading to the inhibition of gene transcription of various anti-apoptotic proteins. This induces cell cycle arrest and apoptosis, and leads to a reduction in tumor cell proliferation. CDK9 regulates elongation of transcription through phosphorylation of RNA polymerase II at serine 2 (p-Ser2-RNAPII). It is upregulated in various tumor cell types and plays a key role in the regulation of Pol II-mediated transcription of anti-apoptotic proteins. Tumor cells are dependent on anti-apoptotic proteins for their survival.
AZD-4573 is a small molecule drug with a maximum clinical trial phase of II and has 1 investigational indication.
- AZD4573 in Novel Combinations With Anti-cancer Agents in Patients With Advanced Blood CancerCTID: NCT04630756Phase: Phase 1/Phase 2Status: CompletedDate: 2025-04-09
- AZD4573 as Monotherapy or in Combinations With Anti-cancer Agents in Patients With r/r PTCL or r/r cHLCTID: NCT05140382Phase: Phase 2Status: CompletedDate: 2024-08-28
- Study to Assess Safety, Tolerability, Pharmacokinetics and Antitumor Activity of AZD4573 in Relapsed/Refractory Haematological MalignanciesCTID: NCT03263637Phase: Phase 1Status: CompletedDate: 2021-10-22
SYN
- Large-Scale Synthesis of AZD4573Publication Name: SynfactsPublication Date: 2022-05-17DOI: 10.1055/s-0041-1738312
- From Structure Modification to Drug Launch: A Systematic Review of the Ongoing Development of Cyclin-Dependent Kinase Inhibitors for Multiple Cancer TherapyPublication Name: Journal of Medicinal ChemistryPublication Date: 2022-04-29PMID: 35485642DOI: 10.1021/acs.jmedchem.1c02064
- Lessons Learned from Past Cyclin-Dependent Kinase Drug Discovery EffortsPublication Name: Journal of Medicinal ChemistryPublication Date: 2022-03-02PMID: 35235745DOI: 10.1021/acs.jmedchem.1c02190
- Discovery of AZD4573, a Potent and Selective Inhibitor of CDK9 That Enables Short Duration of Target Engagement for the Treatment of Hematological MalignanciesPublication Name: Journal of Medicinal ChemistryPublication Date: 2020-12-11PMID: 33306391DOI: 10.1021/acs.jmedchem.0c01754
- A comprehensive insight on the recent development of Cyclic Dependent Kinase inhibitors as anticancer agentsPublication Name: European Journal of Medicinal ChemistryPublication Date: 2020-10-01PMID: 32707525DOI: 10.1016/j.ejmech.2020.112571
- Recent Developments in the Biology and Medicinal Chemistry of CDK9 Inhibitors: An UpdatePublication Name: Journal of Medicinal ChemistryPublication Date: 2020-08-31PMID: 32866383DOI: 10.1021/acs.jmedchem.0c00744
- AZD4573 Is a Highly Selective CDK9 Inhibitor That Suppresses MCL-1 and Induces Apoptosis in Hematologic Cancer CellsPublication Name: Clinical cancer research : an official journal of the American Association for Cancer ResearchPublication Date: 2020-02-14PMID: 31699827DOI: 10.1158/1078-0432.ccr-19-1853
- A New CDK9 Inhibitor on the Block to Treat Hematologic MalignanciesPublication Name: Clinical cancer research : an official journal of the American Association for Cancer ResearchPublication Date: 2020-02-14PMID: 31843752DOI: 10.1158/1078-0432.ccr-19-3670
- Cyclin dependent kinase (CDK) inhibitors as anticancer drugs: Recent advances (2015–2019)Publication Name: Bioorganic & Medicinal Chemistry LettersPublication Date: 2019-10-15PMID: 31477350DOI: 10.1016/j.bmcl.2019.126637
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017001354&_cid=P22-MJC84G-87476-1


Example 14: (1S,3R)-3-acetamido-N-(5-chloro-4-(5,5-dimethyl-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl)pyridin-2-yl)cyclohexanecarboxamide


PAT
- COMPOUNDS DERIVED FROM POLYCYCLIC AMIDE AS CDK9 INHIBITORS, COMPOSITION AND THEIR USESPublication Number: BR-122019013677-B1Priority Date: 2015-06-29
- Polycyclic amide derivatives as CDK9 inhibitorsPublication Number: KR-102663113-B1Priority Date: 2015-06-29Grant Date: 2024-05-02
- Methods of treating a ras protein-related disease or disorderPublication Number: US-2025049810-A1
- Chemical compoundsPublication Number: TW-I723028-BPriority Date: 2015-06-29Grant Date: 2021-04-01
- Chemical compoundsPublication Number: US-2021171541-A1Priority Date: 2015-06-29
- POLYCYCLIC AMIDA DERIVATIVES AS CDK9 INHIBITORSPublication Number: HR-P20211970-T1Priority Date: 2015-06-29
- Pyridine and pyrimidine derivativesPublication Number: US-11352369-B2Priority Date: 2015-06-29Grant Date: 2022-06-07
- Chemical compoundsPublication Number: US-2022340592-A1Priority Date: 2015-06-29
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
/////////Zemirciclib, cyclin-dependent kinase inhibitor, antineoplastic, AZD 4573, UNII-E5XSP3X68B
Zelenirstat



Zelenirstat
CAS 1215011-08-7
MF C24H30Cl2N6O2S, 537.5 g/mol
2,6-dichloro-N-[1,5-dimethyl-3-(2-methylpropyl)-1Hpyrazol-4-yl]-4-[2-(piperazin-1-yl)pyridin-4-yl]benzene-1-sulfonamide
N-myristoyltransferase inhibitor, antineoplastic, PCLX 001, DDD86481, CCI 002, DDD 86481
Zelenirstat (PCLX-001) is an investigational, oral small-molecule drug that inhibits N-myristoyltransferases (NMTs), enzymes crucial for adding fatty acids to proteins, a process vital for cell signaling and membrane attachment. Developed by Pacylex Pharmaceuticals, it’s being tested for various cancers, showing promise in hematologic cancers like AML and lymphomas, as well as solid tumors, by disrupting cancer cell survival and growth, with early trials indicating good safety and potential efficacy.
How it works:
- Targets NMT enzymes: Zelenirstat blocks NMT1 and NMT2, preventing myristoylation (adding a fatty acid) to proteins.
- Disrupts cancer cell processes: This inhibition interferes with essential cell signaling and stability, especially in cancer cells where NMT expression is altered, leading to cell death (apoptosis).
- Affects mitochondrial function: It also disrupts mitochondrial complex I and oxidative phosphorylation, vital for leukemia stem cell survival, notes Pacylex Pharmaceuticals.
Development & Status:
- Orphan Drug Status: Granted for Acute Myeloid Leukemia (AML).
- Clinical Trials: A Phase 1 trial demonstrated good safety and early signs of activity in patients with advanced solid tumors and lymphomas, leading to further development.
- New Drug Class: It represents a novel approach to cancer treatment, distinct from many existing therapies.
Potential Applications:
- Acute Myeloid Leukemia (AML)
- B-cell Lymphomas (like Diffuse Large B-Cell Lymphoma)
- Colorectal Carcinoma
- Other cancers, including breast, lung, bladder, and pancreatic cancers, show sensitivity in preclinical models.
Zelenirstat, also known as PCLX-001, is an investigational new drug that is being evaluated for the treatment of cancer and as an antiviral agent. It is a small molecule inhibitor targets both N-myristoyltransferase 1 (NMT1) and N-myristoyltransferase 2 (NMT2) proteins, which are responsible for myristoylation. Its dual mechanism of action disrupts both cell signaling and energy production in cancer cells.
Zelenirstat is a strong pan-N myristoyl transferase inhibitor, which prevents addition of myristic acid into penultimate glycine of protein with myristoylation signal, and initially has been introduced as anti-tumor drug.[1][2][3] It has completed phase I clinical trial and is going through escalation phase.[4] Its prototype DDD85646 as well as other NMT inhibitors such as IMP-1088 have strong antiviral activities against viruses that required myristoylated proteins to complete their life cycle, including hemorrhagic viruses, such as lassa and argentinian virus, and pox viruses, such as vaccinia and monkeypox.[5][6]
Zelenirstat is an orally bioavailable inhibitor of the enzyme N-myristoyl transferase (NMT), with potential antineoplastic activity. Upon oral administration, zelenirstat targets and binds to NMT, especially NMT type 2 (NMT2). This prevents NMT-mediated signaling and myristoylation. This inhibits proliferation of certain cancer cells in which NMT expression is lost. Zelenirstat also inhibits B-cell receptor (BCR) signaling and reduces the levels of Src-family tyrosine kinases (SFKs). NMTs mediate myristoylation, a key process by which the fatty acid myristate is added to proteins and allows proteins to interact with cell membranes and become part of the cell signaling system. NMT expression is lost in numerous cancers, such as blood cancer cells, thereby making these cells more sensitive to zelenirstat compared to normal cells. The loss of NMT expression may promote tumorigenesis.
Mechanism of action
Zelenirstat acts by inhibiting NMT I and II enzymes, which are required to complete the myristoylation of proteins. Without myristoylation, these proteins are targeted for proteasomal degradation.[7]
PCLX-001 is a first-in-kind N-Myristoyltransferase (NMT) inhibitor being developed by [Pacylex Pharmaceuticals](https://pacylex.com). Current studies have shown that PCLX-001 works differently than other known cancer drugs and has high activity and positive results in breast, lung, bladder and pancreas cancers.
- Study of PCLX-001 in R/R Advanced Solid Malignancies and B-cell LymphomaCTID: NCT04836195Phase: Phase 1Status: CompletedDate: 2025-04-17
- Study of Oral PCLX-001 in R/R Acute Myeloid LeukemiaCTID: NCT06613217Phase: Phase 1Status: RecruitingDate: 2025-03-10
REF
- Novel, First-in-Human, Oral PCLX-001 Treatment in a Patient with Relapsed Diffuse Large B-Cell LymphomaPublication Name: Current oncology (Toronto, Ont.)Publication Date: 2022-03-13PMCID: PMC8947478PMID: 35323358DOI: 10.3390/curroncol29030158
- N-myristoyltransferase proteins in breast cancer: prognostic relevance and validation as a new drug targetPublication Name: Breast Cancer Research and TreatmentPublication Date: 2021-01-04PMCID: PMC7940342PMID: 33398478DOI: 10.1007/s10549-020-06037-y
- Targeting N-myristoylation for therapy of B-cell lymphomasPublication Name: Nature CommunicationsPublication Date: 2020-10-22PMCID: PMC7582192PMID: 33093447DOI: 10.1038/s41467-020-18998-1
- Emerging New Targets for the Treatment of Resistant Fungal InfectionsPublication Name: Journal of Medicinal ChemistryPublication Date: 2018-01-02PMID: 29294275DOI: 10.1021/acs.jmedchem.7b01413
- Interrogating the Roles of Post-Translational Modifications of Non-Histone ProteinsPublication Name: Journal of Medicinal ChemistryPublication Date: 2017-05-15PMID: 28505447DOI: 10.1021/acs.jmedchem.6b01817
SYN
DDD 86481
https://patentscope.wipo.int/search/en/detail.jsf?docId=US73438944&_cid=P12-MJAUPA-00022-1
INTERMEDIATE 23A
4-Bromo-2,6-dichloro-N-(3-isobutyl-1,5-dimethyl-1H-pyrazol-4-yl)-benzenesulfonamide
EXAMPLE DDD86481
2,6-Dichloro-N-(3-isobutyl-1,5-dimethyl-1H-pyrazol-4-yl)-4-(2-piperazin-1-yl-pyridin-4-yl)-benzenesulfonamide
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2010026365&_cid=P12-MJAUOO-99381-1
PAT
N-myristoyl transferase inhibitors
Publication Number: WO-2010026365-A1
Priority Date: 2008-09-02
- N-myristoyl transferase inhibitorsPublication Number: ES-2546865-T3Priority Date: 2008-09-02Grant Date: 2015-09-29
- N-Myristoyl Transferase InhibitorsPublication Number: US-2011312921-A1Priority Date: 2008-09-02
- N-myristoyl transferase inhibitorsPublication Number: US-2016060224-A1Priority Date: 2008-09-02
- N-myristoyl transferase inhibitorsPublication Number: US-9156811-B2Priority Date: 2008-09-02Grant Date: 2015-10-13
- N-myristoyl transferase inhibitorsPublication Number: US-9828346-B2Priority Date: 2008-09-02Grant Date: 2017-11-28
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Gamma JM, Liu Q, Beauchamp E, Iyer A, Yap MC, Zak Z, et al. (January 2025). “Zelenirstat Inhibits N-Myristoyltransferases to Disrupt Src Family Kinase Signaling and Oxidative Phosphorylation, Killing Acute Myeloid Leukemia Cells”. Molecular Cancer Therapeutics. 24 (1): 69–80. doi:10.1158/1535-7163.MCT-24-0307. PMC 11694064. PMID 39382188.
- Sangha R, Jamal R, Spratlin J, Kuruvilla J, Sehn LH, Beauchamp E, et al. (August 2024). “A first-in-human phase I trial of daily oral zelenirstat, a N-myristoyltransferase inhibitor, in patients with advanced solid tumors and relapsed/refractory B-cell lymphomas”. Investigational New Drugs. 42 (4): 386–393. doi:10.1007/s10637-024-01448-w. PMC 11327210. PMID 38837078.
- Sangha RS, Jamal R, Spratlin J, Kuruvilla J, Sehn LH, Weickert M, et al. (June 2024). “Final results of a first-in-human phase I dose escalation trial of daily oral zelenirstat, a n-myristoyltransferase inhibitor, in patients with advanced solid tumors and relapsed/refractory B-cell lymphomas”. Journal of Clinical Oncology. 42 (16_suppl): 3082. doi:10.1200/JCO.2024.42.16_suppl.3082. ISSN 0732-183X.
- Spratlin JL, Sangha RS, Jamal R, Beauchamp E, Berthiaume LG, Mackey JR (20 January 2024). “A first-in-human, open-label, phase I trial of daily oral zelenirstat, an NMT inhibitor, in patients with relapsed/refractory advanced cancer including gastrointestinal cancers”. Journal of Clinical Oncology. 42 (3_suppl): 129–129. doi:10.1200/jco.2024.42.3_suppl.129. Retrieved 19 January 2025.
- Witwit H, Betancourt CA, Cubitt B, Khafaji R, Kowalski H, Jackson N, et al. (August 2024). “Cellular N-Myristoyl Transferases Are Required for Mammarenavirus Multiplication”. Viruses. 16 (9): 1362. doi:10.3390/v16091362. PMC 11436053. PMID 39339839.
- Witwit H, Cubitt B, Khafaji R, Castro EM, Goicoechea M, Lorenzo MM, et al. (January 2025). “Repurposing Drugs for Synergistic Combination Therapies to Counteract Monkeypox Virus Tecovirimat Resistance”. Viruses. 17 (1): 92. doi:10.3390/v17010092. ISSN 1999-4915. PMC 11769280.
- Witwit H, Betancourt CA, Cubitt B, Khafaji R, Kowalski H, Jackson N, et al. (August 2024). “Cellular N-Myristoyl Transferases Are Required for Mammarenavirus Multiplication”. Viruses. 16 (9): 1362. doi:10.3390/v16091362. PMC 11436053. PMID 39339839.
| Clinical data | |
|---|---|
| Other names | PCLX-001 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1215011-08-7 |
| PubChem CID | 58561243 |
| DrugBank | DB15567 |
| ChemSpider | 35034199 |
| UNII | 5HY8BYC3Q6 |
| ChEMBL | ChEMBL3357685 |
| Chemical and physical data | |
| Formula | C24H30Cl2N6O2S |
| Molar mass | 537.50 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
////////zelenirstat, N-myristoyltransferase inhibitor, antineoplastic, PCLX 001, DDD86481, CCI 002, DDD 86481
Zavolosotine




Zavolosotine
CAS 2604416-66-0
MF C20H18F5N5O MW439.38
4-[(3S)-3-aminopyrrolidin-1-yl]-6-cyano-5-(3,5-difluorophenyl)-N-[(2S)-1,1,1-trifluoropropan-2-yl]pyridine-3-carboxamide
4-[(3S)-3-aminopyrrolidin-1-yl]-6-cyano-5-(3,5-difluorophenyl)-N-[(2S)-1,1,1-trifluoropropan-2-yl]pyridine-3-carboxamide
4-[(3S)-3-aminopyrrolidin-1-yl]-6-cyano-5-(3,5-difluorophenyl)- N-[(2S)-1,1,1-trifluoropropan-2-yl]pyridine3-carboxamide
somatostatin receptor agonist, 275EAX4XXX
Zavolosotine (Compound 1) is an orally active agonist for somatostatin receptor type 5 (SST5) with EC50 <1 nM. Zavolosotine inhibits insulin and glucagon secretion, increases levels of glucagon in blood in rat model.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022177988&_cid=P20-MJ9E0I-92373-1

SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US318018214&_cid=P20-MJ9DV5-88499-1

Example 4. 4-[(3S)-3-aminopyrrolidin-1-yl]-6-cyano-5-(3,5-difluorophenyl)-N-[(2S)-1,1,1-trifluoropropan-2-yl]pyridine-3-carboxamide (Compound 1-71)

| Step 4-1, preparation of tert-butyl (S)-(1-(2-chloro-5-formylpyridin-4-yl)pyrrolidin-3-yl)carbamate: to a DMF (70 mL) solution was added 4,6-dichloronicotinaldehyde (6.8 g, 1.0 Eq, 39 mmol), tert-butyl (S)-pyrrolidin-3-ylcarbamate (7.6 g, 1.1 Eq, 41 mmol) and TEA (16 mL, 3.1 Eq, 120 mmol). The resulting mixture was stirred at 50° C. for 4 hours. The reaction crude was quenched with water (100 mL) and extracted with ethyl acetate (3×40 mL). The organic layers were combined, washed with brine, dried and concentrated under vacuum. The remaining residue was purified by silica gel chromatography eluting with ethyl acetate/petroleum ether (1/3) to afford tert-butyl (S)-(1-(2-chloro-5-formylpyridin-4-yl)pyrrolidin-3-yl)carbamate (5.3 g, 42%) as a yellow solid. MS (M+H) +=326.2. |
PAT
- Nonpeptide somatostatin type 5 receptor agonists and uses thereofPublication Number: US-2022144802-A1Priority Date: 2019-08-14
- Non-peptide somatostatin type 5 receptor agonists and uses thereofPublication Number: JP-2022544055-APriority Date: 2019-08-14
- Nonpeptide somatostatin type 5 receptor agonists and uses thereofPublication Number: US-11479540-B2Priority Date: 2019-08-14Grant Date: 2022-10-25
- Nonpeptide somatostatin type 5 receptor agonists and uses thereofPublication Number: TW-I841768-BPriority Date: 2019-08-14Grant Date: 2024-05-11
- Non-peptide somatostatin type 5 receptor agonists and uses thereofPublication Number: JP-7611893-B2Priority Date: 2019-08-14Grant Date: 2025-01-10
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////zavolosotine, somatostatin receptor agonist, 275EAX4XXX
Zamzetoclax



Zamzetoclax
CAS 2388470-64-0
MF C38H46ClN5O6S MW736.32
N-[(3′R,4S,6′R,7′S,8′E,11′S)-7-chloro-7′-methoxy-11′-methyl-13′,15′-dioxospiro[2,3-dihydro-1H-naphthalene-4,22′-20-oxa-13λ6-thia-1,14-diazatetracyclo[14.7.2.03,6.019,24]pentacosa-8,13,16(25),17,19(24)-pentaene]-13′-yl]-3-methoxy-1-methylpyrazole-4-carboxamide
- 1H-Pyrazole-4-carboxamide, N-[(1’S,10S,12S,14E,16S,16aR,18aR)-6′-chloro-3′,4′,8,11,12,13,16,16a,17,18,18a,19-dodecahydro-16-methoxy-12-methyl-10-oxido-8-oxospiro[5,7-etheno-1H-10lambda4-cyclobut[i][1,4]oxazepino[3,4-f][1,2,7]thiadiazacyclohexadecine-2(3H),1′(2’H)-naphthalen]-10-yl]-3-methoxy-1-methyl-
- N-[(1’S,10S,12S,14E,16S,16aR,18aR)-6′-Chloro-3′,4′,8,11,12,13,16,16a,17,18,18a,19-dodecahydro-16-methoxy-12-methyl-10-oxido-8-oxospiro[5,7-etheno-1H-10lambda4-cyclobut[i][1,4]oxazepino[3,4-f][1,2,7]thiadiazacyclohexadecine-2(3H),1′(2’H)-naphthalen]-10-yl]-3-methoxy-1-methyl-1H-pyrazole-4-carboxamide

B-cell lymphoma 2 (Bcl-2) inhibitor, antineoplastic, RRS8GZU2UN
Zamzetoclax (compound 1) is a potential Mcl-1 inhibitor.
Publication Name: Journal of Medicinal Chemistry
Publication Date: 2023-04-28
PMID: 37114951
DOI: 10.1021/acs.jmedchem.2c01953
SYN
WO 2019/222112
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019222112&_cid=P12-MJ7Z15-98773-1
Example 154

[0447] Example 154 was synthesized in the same manner as Example 18 using 3-methoxy-1-methyl-1H-pyrazole-4-carboxylic acid and Example 109. Example 109 (620 mg, 1.04 mmol) was dissolved in dichloromethane (12 mL). 3-Methoxy-1-methyl-1H-pyrazole-4-carboxylic acid (324 mg, 2.08 mmol, 2 equiv.) and N-(3-dimethylaminopropyl)-N¢-ethylcarbodiimide hydrochloride (400 mg, 2.08 mmol, 2 equiv.) were added. The reaction mixture was stirred for 5 minutes at room temperature before DMAP (253 mg, 2.08 mmol, 2 equiv.) was added in a single portion. The reaction mixture was stirred overnight at room temperature and the progress of the reaction was monitored by LCMS. Upon completion, the reaction mixture was concentrated under reduced pressure, and the residue was purified by Gilson reverse phase prep HPLC (60-100% ACN/H2O with 0.1% TFA) to give Example 154.1H NMR (400 MHz, methanol-d4) d 8.07 (s, 1H), 7.76 (d, J = 8.6 Hz, 1H), 7.34 (d, J = 8.2 Hz, 1H), 7.22– 7.10 (m, 3H), 6.92 (d, J = 8.2 Hz, 1H), 6.20– 6.05 (m, 1H), 5.63 (dd, J = 15.5, 8.0 Hz, 1H), 4.10 (d, J = 12.0 Hz, 1H), 4.06 (s, 4H), 3.91– 3.83 (m, 1H), 3.82 (s, 3H), 3.79 (s, 1H), 3.72 (d, J = 14.4 Hz, 1H), 3.38 (d, J = 14.5 Hz, 1H), 3.30 (s, 3H), 3.09 (dd, J = 15.1, 10.0 Hz, 1H), 2.89– 2.72 (m, 2H), 2.51 (d, J = 26.7 Hz, 2H), 2.24 (dd, J = 10.9, 6.0 Hz, 2H), 2.12 (d, J = 13.7 Hz, 1H), 2.02– 1.70 (m, 4H), 1.54– 1.40 (m, 1H), 1.14 (d, J = 6.1 Hz, 3H). LCMS-ESI+ (m/z): calcd for C38H46ClN5O6S: 735.28; found: 735.94.
SYN
WO 2019/222112 discloses novel 3′,4,4′,5-tetrahydro-2H,2′H-spiro[benzo[b][1,4]oxazepine-3,1′-naphthalene] derivatives that are active against MCL-1. For example, Compound 1 (below) has been shown to be an effective MCL-1 inhibitor

SYN
SYN
PAT
- Mcl-1 inhibitorsPublication Number: US-2019352271-A1Priority Date: 2018-05-14
- Mcl-1 inhibitorsPublication Number: US-2020331870-A1Priority Date: 2018-05-14
- MCL-1 inhibitorsPublication Number: US-10988451-B2Priority Date: 2018-05-14Grant Date: 2021-04-27
- MCL-1 inhibitorsPublication Number: US-11643400-B2Priority Date: 2018-05-14Grant Date: 2023-05-09
- Mcl-1 inhibitorsPublication Number: US-2023312490-A1Priority Date: 2018-05-14
- Processes and intermediates for preparing mcl1 inhibitorsPublication Number: US-2023013713-A1Priority Date: 2019-11-26
- Processes and intermediates for preparing MCL1 inhibitorsPublication Number: US-11760736-B2Priority Date: 2019-11-26Grant Date: 2023-09-19
- Mcl1 inhibitorsPublication Number: US-2021171543-A1Priority Date: 2019-11-12
- Mcl1 inhibitorsPublication Number: US-2023348494-A1Priority Date: 2019-11-12
- MCL-1 inhibitorsPublication Number: US-10703733-B2Priority Date: 2018-05-14Grant Date: 2020-07-07
- Salts and polymorphs of certain mcl-1 inhibitorsPublication Number: US-2023357274-A1Priority Date: 2022-05-04
- Combination mcl-1 inhibitors with anti-body drug conjugatesPublication Number: US-2022409736-A1Priority Date: 2021-06-11
- Processes and intermediates for preparing mcl1 inhibitorsPublication Number: US-2022177409-A1Priority Date: 2020-11-19
- Processes and intermediates for preparing mcl1 inhibitorsPublication Number: US-2021179570-A1Priority Date: 2019-11-26
- Processes and intermediates for preparing MCL1 inhibitorsPublication Number: US-11325891-B2Priority Date: 2019-11-26Grant Date: 2022-05-10
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////zamzetoclax, B-cell lymphoma 2 (Bcl-2) inhibitor, antineoplastic, RRS8GZU2UN
Vilzemetkib


Vilzemetkib
CAS 1363402-44-1
MF C36H36F2N4O5 MW 642.7 g/mol
1-N‘-[4-[7-[[1-(cyclopentylamino)cyclopropyl]methoxy]-6-methoxyquinolin-4-yl]oxy-3-fluorophenyl]-1-N-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide
- 1,1-Cyclopropanedicarboxamide, N-[4-[[7-[[1-(cyclopentylamino)cyclopropyl]methoxy]-6-methoxy-4-quinolinyl]oxy]-3-fluorophenyl]-N’-(4-fluorophenyl)-
- 1-N’-[4-[7-[[1-(cyclopentylamino)cyclopropyl]methoxy]-6-methoxyquinolin-4-yl]oxy-3-fluorophenyl]-1-N-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide
- 1363402-44-1
- N-[4-[[7-[[1-(Cyclopentylamino)cyclopropyl]methoxy]-6-methoxy-4-quinolinyl]oxy]-3-fluorophenyl]-N’-(4-fluorophenyl)-1,1-cyclopropanedicarboxamide

hepatocyte growth factor receptor inhibitor, antineoplastic, AL 2846, FJ4Y6XP24Y
Vilzemetkib (also known as AL2846) is an investigational, orally active small-molecule drug that acts as a potent inhibitor of the c-Met receptor tyrosine kinase, a protein often overexpressed in cancers, aiming to block tumor growth, survival, and spread by disrupting key cellular signals. It’s being studied in clinical trials, often in combination with other agents like TQB2450 (a PD-L1 inhibitor), for advanced cancers such as esophageal and liver cancer, showing promise in immunotherapy-resistant patients.
How it Works:
- Targets c-Met: Vilzemetkib binds to the c-Met protein, preventing its phosphorylation (activation).
- Blocks Signaling: This action disrupts downstream pathways crucial for cancer cell proliferation, survival, invasion, metastasis, and new blood vessel formation (angiogenesis).
Development & Use:
- Developer: Developed by Advenchen Laboratories.
- Status: Investigational drug, currently in clinical trials.
- Research Focus: Studied for cancers like esophageal squamous cell carcinoma (ESCC) and hepatocellular carcinoma (HCC).
Key Information:
- Chemical Name: 1,1-Cyclopropanedicarboxamide, N-[4-[[7-[[1-(cyclopentylamino)cyclopropyl]methoxy]-6-methoxy-4-quinolinyl]oxy]-3-fluorophenyl]-N′-(4-fluorophenyl)-.
- Purpose: Potential anti-cancer (antineoplastic) activity.
- OriginatorAdvenchen Laboratories
- DeveloperAdvenchen Laboratories; Chia Tai Tianqing Pharmaceutical Group
- ClassAntineoplastics; Small molecules
- Mechanism of ActionReceptor protein-tyrosine kinase antagonists
- Phase IIINon-small cell lung cancer; Thyroid cancer
- Phase IILung cancer; Ovarian cancer
- Phase I/IIColorectal cancer; Neurofibromatosis 1; Pancreatic cancer
- No development reportedSolid tumours
- 28 Oct 2025No recent reports of development identified for phase-I development in Solid-tumours(Combination therapy, In the elderly, Late-stage disease, Second-line therapy or greater, In adults) in China (PO, Capsule)
- 10 Oct 2025700363489: CTP push: KDM and HE updated
- 26 Aug 2025Chemical structure information added.
Vilzemetkib is an orally bioavailable small molecule inhibitor of the oncoprotein c-Met (hepatocyte growth factor receptor; HGFR), with potential antineoplastic activity. Upon oral administration vilzemetkib targets and binds to the c-Met protein, prevents c-Met phosphorylation and disrupts c-Met-dependent signal transduction pathways. This may induce cell death in tumor cells overexpressing c-Met protein or expressing constitutively activated c-Met protein. c-Met protein is overexpressed or mutated in many tumor cell types and plays key roles in tumor cell proliferation, survival, invasion, metastasis, and tumor angiogenesis.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US73570351&_cid=P20-MJ6JF6-22611-1



EXAMPLE 6
N-(4-(7-((1-(cyclopentylamino)cyclopropyl)methoxy)-6-methoxyquinolin-4-yloxy)-3-fluoro-phenyl)-N-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide
| The title compound was prepared by similar manner to Example 3, by using cyclopentanone instead of tetrahydro-4H-pyran-4-one. Mass: (M+1), 643 |
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022268158&_cid=P20-MJ6JJ7-25153-1
WO2012034055 discloses N-(4-((7-((1-(cyclopentylamino)cyclopropyl)methoxy)-6-methoxyquinolone-4-yl)oxy)-3-fluorophenyl)-N-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide (hereinafter referred to as compound (I)) as a c-Met kinase inhibitor and its use in inhibiting tyrosine kinase activity. Compound (I) is a novel class of compounds with excellent pharmacological properties, capable of inhibiting the activity of various protein tyrosine kinases, such as c-Met, VEGFr, EGFr, c-kit, PDGF, FGF, SRC, Ron, Tie2, etc. This disclosure relates to the treatment of neurofibromatosis type I with compound (I).

SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2012034055&_cid=P20-MJ6JLU-26634-1
Example 6
N-(4-(7-((1-(cyclopentylamino)cyclopropyl)methoxy)-6-methoxyquinolin-4-yloxy)-3-fluoro-phenyl)-N-(4-fluorophenyl)cyclopropane- 1,1-dicarboxamide
The title compound was prepared by similar manner to Example 3, by using cyclopentanone instead of tetrahydro-4H-pyran-4-one. Mass: (M + 1), 643
PAT
- Crystal of compound as c-met kinase inhibitor and preparation method therefor and use thereofPublication Number: EP-4279138-A2Priority Date: 2018-03-02
- Compounds As c-Met Kinase InhibitorsPublication Number: US-2012123126-A1Priority Date: 2010-09-12
- Compounds as c-Met kinase inhibitorsPublication Number: US-8664244-B2Priority Date: 2010-09-12Grant Date: 2014-03-04
- Compounds as c-met kinase inhibitorsPublication Number: WO-2012034055-A2Priority Date: 2010-09-12
- Use of compound as c-met kinase inhibitor for treatment of neurofibromatosis type iPublication Number: WO-2022268158-A1Priority Date: 2021-06-23
- Combined pharmaceutical composition of c-met kinase inhibitor and anti-pd-l1 antibodyPublication Number: US-2023263795-A1Priority Date: 2020-06-02
- Crystal of compound as c-met kinase inhibitor and preparation method therefor and use thereofPublication Number: EP-3766870-A1Priority Date: 2018-03-02
- Crystalline of compound as c-met kinase inhibitor and preparation method therefor and use thereofPublication Number: US-2021047272-A1Priority Date: 2018-03-02
- Crystalline of compound as c-Met kinase inhibitor and preparation method therefor and use thereofPublication Number: US-11279676-B2Priority Date: 2018-03-02Grant Date: 2022-03-22
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
////////Vilzemetkib, hepatocyte growth factor receptor inhibitor, antineoplastic, AL 2846, FJ4Y6XP24Y
Vicadrostat
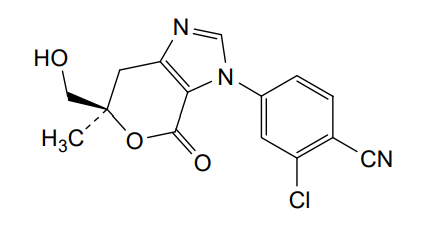
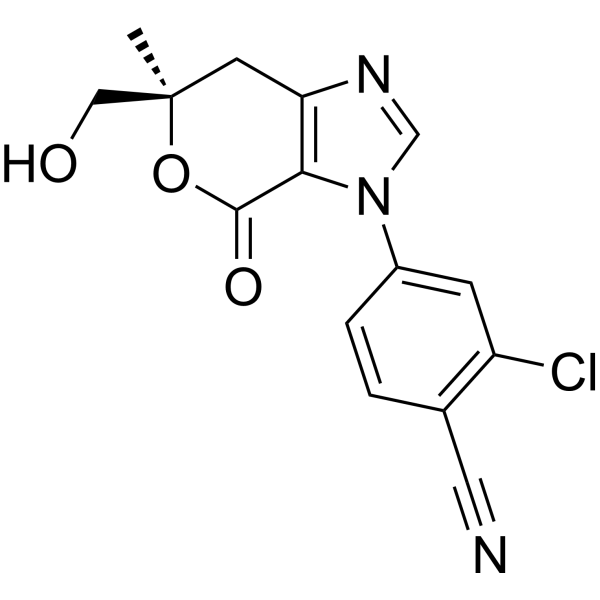
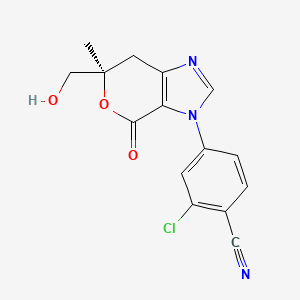
Vicadrostat
CAS 1868065-21-7
MF C15H12ClN3O3 MW 317.73
- 2-CHLORO-4-((6R)-6-(HYDROXYMETHYL)-6-METHYL-4-OXO-6,7-DIHYDROPYRANO(3,4-D)IMIDAZOL-3(4H)-YL)BENZONITRILE
- 2-Chloro-4-[(6R)-6,7-dihydro-6-(hydroxymethyl)-6-methyl-4-oxopyrano[3,4-d]imidazol-3(4H)-yl]benzonitrile
- 2-chloro-4-[(6R)-6-(hydroxymethyl)-6-methyl-4-oxo-7H-pyrano[3,4-d]imidazol-3-yl]benzonitrile
2-chloro-4-[(6R)-6-(hydroxymethyl)-6-methyl-4-oxo-6,7-dihydropyrano[3,4-d]imidazol-3(4H)-yl]benzonitrile
aldosterone synthase inhibitor, BI 690517, AF4VW4GA3H
Vicadrostat is an aldosterone synthase inhibitor (IC50=48 nM). Vicadrostat can be used for research in kidney diseases and cardiovascular diseases
Vicadrostat (BI 690517) is an investigational drug by Boehringer Ingelheim that selectively blocks aldosterone synthase, reducing excess aldosterone linked to kidney, heart, and metabolic diseases like chronic kidney disease (CKD) and heart failure. Currently in Phase III trials (EASi-KIDNEY and EASi-HF), it’s being tested alone and with empagliflozin (an SGLT2 inhibitor) to reduce proteinuria and improve heart/kidney health, showing promise in reducing albuminuria.
What it is
- Type: A highly selective Aldosterone Synthase Inhibitor (ASI).
- Mechanism: Blocks the enzyme that makes aldosterone, a hormone that causes fluid retention and damage in heart/kidney conditions.
What it’s for
- Conditions: Investigated for Chronic Kidney Disease (CKD) and Heart Failure with Preserved Ejection Fraction (HFpEF).
- Goal: To reduce high aldosterone levels, organ damage, and slow disease progression, particularly in interconnected cardiovascular and renal conditions.
How it’s being studied
- Combination Therapy: Key trials combine vicadrostat with empagliflozin (Jardiance).
- Promising Results: A Phase II trial showed significant reduction in urine protein (albuminuria) when combined with empagliflozin.
- Clinical Trials: Undergoing large Phase III trials (EASi-KIDNEY and EASi-HF) to confirm its efficacy and safety.
Key benefit
- Offers a potential new treatment by targeting aldosterone, addressing multiple interconnected organ systems (heart, kidney, metabolism) simultaneously.
- OriginatorBoehringer Ingelheim
- Class2 ring heterocyclic compounds; Alcohols; Benzonitrile; Chlorinated hydrocarbons; Imidazoles; Pyrones; Small molecules; Urologics
- Mechanism of ActionCytochrome P-450 CYP11B2 inhibitors
- Phase IIICardiovascular disorders; Heart failure; Hypertension; Renal failure; Type 2 diabetes mellitus
- No development reportedDiabetic nephropathies
- 28 Oct 2025No recent reports of development identified for phase-I development in Renal-failure(In volunteers) in Netherlands (IV)
- 28 Oct 2025No recent reports of development identified for phase-I development in Renal-failure(In volunteers) in Netherlands (PO)
- 08 Sep 2025Boehringer Ingelheim initiates a phase I trial (In volunteers, Combination therapy) in Germany (NCT07133399)
- A Study to Test Whether Vicadrostat in Combination With Empagliflozin Helps People With Chronic Kidney DiseaseCTID: NCT06926660Phase: Phase 2Status: RecruitingDate: 2025-11-28
- A Study to Test Whether Vicadrostat (BI 690517) in Combination With Empagliflozin Helps People With Heart Failure and a Weak Pumping Function of the Left Side of the HeartCTID: NCT06935370Phase: Phase 3Status: RecruitingDate: 2025-11-26
- A Study to Test Whether Vicadrostat in Combination With Empagliflozin Helps People With Heart FailureCTID: NCT06424288Phase: Phase 3Status: RecruitingDate: 2025-11-26
- A Study to Test Vicadrostat (BI 690517) Taken Together With Empagliflozin in People With Type 2 Diabetes, High Blood Pressure, and Cardiovascular DiseaseCTID: NCT07064473Phase: Phase 3Status: RecruitingDate: 2025-11-26
- A Study in Healthy Men to Compare the Amount of Vicadrostat and Empagliflozin in the Blood When Taken Separately and TogetherCTID: NCT07035457Phase: Phase 1Status: CompletedDate: 2025-08-20
SYN
compound 29 A [WO2016014736A1]
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016014736&_cid=P12-MJ3WOZ-69028-1
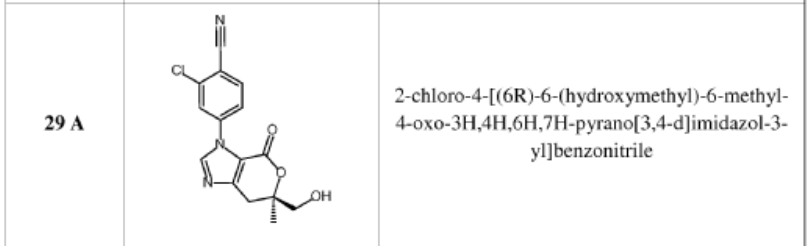
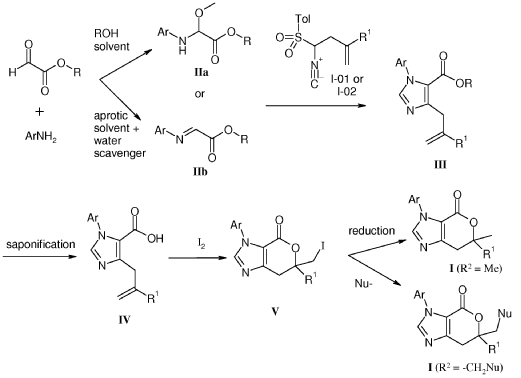
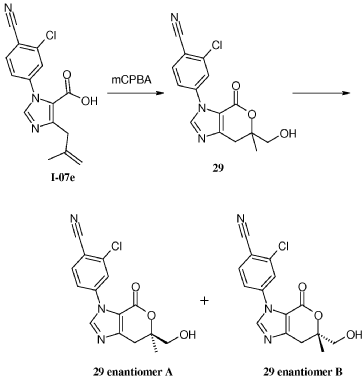
Example 8: Synthesis of 2-chloro-4-[(6R)-6-(hydroxymethyl)-6-methyl-4-oxo-3H,4H,6H,7H-pyrano[3,4-d]imidazol-3-yl]benzonitrile (29 enantiomer A) and 2-chloro-4-[(6S)-6-(hydroxymethyl)-6-methyl-4-oxo-3H,4H,6H,7H-pyrano[3,4-d]imidazol-3-yl]benzonitrile (29 enantiomer B)
29 enan
A mixture of 0.50 g (1.7 mmol) of I-07e and 0.56 g (2.5 mmol) of 77% m-CPBA (m-chloroperoxybenzoic acid) in 10 mL of CH2CI2 is stirred fori 6 h. EtOAc (200 mL) and 20 mL of 10% Na2S03 are added. The mixture is washed twice with 50 mL of NaHC03 and the washes are extracted with 50 mL of CH2C12. The organic extracts are combined, dried with MgS04, filtered and concentrated to give 507 mg of racemic 29 as a pale yellow solid. Chiral
chromatography of 507 mg (LUX 5u Cellulose 4, 28% EtOH:C02, 80 g/min, 120 bar, 40 °C) delivers 238 mg of 29 enantiomer A and 230 mg of 29 enantiomer B. The absolute
stereochemistry for compounds 29 A and 29 B were determined by high resolution single crystal X-ray crystallography structure determination and careful examination of the Flack parameter on the refined structures (H.D. Flack and G. Bernardinelli, 2008, Chirality, 20, 681-690).
The following compounds are prepared from the appropriate olefin I-07c and n in the same manner as 29 enantiomers A & B.
3- (3,4-dichlorophenyl)-6-(hydroxymethyl)-6-methyl-3H,4H,6H,7H-pyrano[3,4- d]imidazol-4-one (30 enantiomers A & B) from I-07c.(RegisPack, 25% (EtOH + 1% iPrNH2):C02, 80 mL/min, 100 bar, 25 °C)
4- [6-(hydroxymethyl)-6-methyl-4-oxo-3H,4H,6H,7H-pyrano[3,4-d]imidazol-3-yl]-3- methylbenzonitrile (31 enantiomers A & B) from I-07n. (LUX 5u Cellulose 4, 25% EtOH:C02, 90 g/min, 120 bar, 40 °C)
SYN
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2025174790&_cid=P12-MJ3WOZ-69028-1
PAT
- Aldosterone synthase inhibitorsPublication Number: US-2016024105-A1Priority Date: 2014-07-24
- Aldosterone synthase inhibitorsPublication Number: US-9334285-B2Priority Date: 2014-07-24Grant Date: 2016-05-10
- Aldosterone synthase inhibitorsPublication Number: WO-2016014736-A1Priority Date: 2014-07-24
- Aldosterone synthase inhibitorsPublication Number: KR-102378648-B1Priority Date: 2014-07-24Grant Date: 2022-03-28
- Aldosterone synthase inhibitors for treating chronic kidney diseasePublication Number: US-2025049763-A1
- Aldosterone synthase inhibitors for treating chronic kidney diseasePublication Number: US-2023181538-A1Priority Date: 2021-12-14
- Aldosterone synthase inhibitorsPublication Number: EP-3172212-A1Priority Date: 2014-07-24
- Aldosterone synthase inhibitorsPublication Number: EP-3172212-B1Priority Date: 2014-07-24Grant Date: 2018-06-13
- Aldosterone synthase inhibitorPublication Number: JP-2017521466-APriority Date: 2014-07-24
- Aldosterone synthase inhibitorPublication Number: JP-6250862-B2Priority Date: 2014-07-24Grant Date: 2017-12-20
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
[1]. Jennifer Burke, et al. Aldosterone synthase inhibitors.WO2016014736.2018-09-07
- Targeting aldosterone to improve cardiorenal outcomes: from nonsteroidal mineralocorticoid receptor antagonists to aldosterone synthase inhibitorsPublication Name: Current opinion in nephrology and hypertensionPublication Date: 2025-02-27PMID: 40012539DOI: 10.1097/mnh.0000000000001067
- The potential for improving cardio-renal outcomes in chronic kidney disease with the aldosterone synthase inhibitor vicadrostat (BI 690517): a rationale for the EASi-KIDNEY trialPublication Name: Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association – European Renal AssociationPublication Date: 2024-11-12PMCID: PMC12209857PMID: 39533115DOI: 10.1093/ndt/gfae263
- Emerging Therapies for Treatment-Resistant Hypertension: A Review of Lorundrostat and Related Selective Aldosterone Synthase InhibitorsPublication Name: Cardiology in ReviewPublication Date: 2024-02-15PMID: 38358268DOI: 10.1097/crd.0000000000000665
- Author response for ‘Aldosterone synthase inhibitor (BI 690517) therapy for people with diabetes and albuminuric chronic kidney disease: A multicentre, randomized, double‐blind, placebo‐controlled, Phase I trial’Publication Name: Diabetes, Obesity and MetabolismPublication Date: 2024-01-16PMID: 38497241DOI: 10.1111/dom.15518
//////////vicadrostat, aldosterone synthase inhibitor, BI 690517, AF4VW4GA3H
Varegacestat

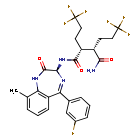
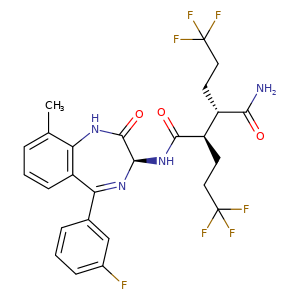
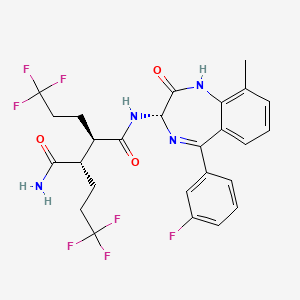
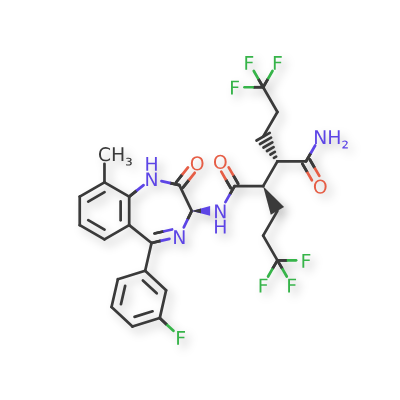
Varegacestat
CAS 1584647-27-7
MF C26H25F7N4O3 MW574.5
(2R,3S)-N1-[(3S)-5-(3-fluorophenyl)-9-methyl-2-oxo-2,3-dihydro-1H1,4-benzodiazepin-3-yl]-2,3-bis(3,3,3-trifluoropropyl)butanediamide
(2R,3S)-N1-((3S)-5-(3-FLUOROPHENYL)-2,3-DIHYDRO-9-METHYL-2-OXO-1H-1,4-BENZODIAZEPIN-3-YL)-2,3-BIS(3,3,3-TRIFLUOROPHENYL)BUTANEDIAMIDE
(2R,3S)-N1-((3S)-5-(3-FLUOROPHENYL)-9-METHYL-2-OXO-2,3-DIHYDRO-1H-1,4-BENZODIAZEPIN-3-YL)-2,3-BIS(3,3,3-TRIFLUOROPHENYL)BUTANEDIAMIDE
gamma-secretase inhibitor, antineoplastic, AL102, BMS 986115, LSK1L593UU, AL 102
BMS-986115 has been used in trials studying the treatment of Various Advanced Cancer.
Varegacestat is an orally bioavailable, gamma secretase (GS) and pan-Notch inhibitor, with potential antineoplastic activity. Upon administration, varegacestat binds to GS and blocks the proteolytic cleavage and release of the Notch intracellular domain (NICD), which would normally follow ligand binding to the extracellular domain of the Notch receptor. This prevents both the subsequent translocation of NICD to the nucleus to form a transcription factor complex and the expression of Notch-regulated genes. This results in the induction of apoptosis and the inhibition of growth of tumor cells that overexpress Notch. Overexpression of the Notch signaling pathway plays an important role in tumor cell proliferation and survival. The integral membrane protein GS is a multi-subunit protease complex that cleaves single-pass transmembrane proteins, such as Notch receptors, at residues within their transmembrane domains and leads to their activation
AL 102 (previously known as BMS 986115), was developed as an orally active a gamma-secretase and pan-Notch inhibitor. The drug participated in phase I clinical trials in solid tumor patients. The drug was safe and well-tolerated and stabilized disease for more than six months in 14% of patients, however, Bristol-Myers Squibb terminated the study because of the changes in the business objectives. Ayala, an Israeli biotech company, licensed rights for the development of AL 102 from Bristol-Myers Squibb. In December 2018, Ayala in collaborating with Novartis decided to investigate AL102 for treatment of multiple myeloma. Ayala studied AL102, an inhibitor of the Notch pathway, in blood cancers. It is known that the pathway regulates cell-fate determination during development and maintains adult tissue balance. Cumulative evidence indicates that Notch is overactive in multiple myeloma and participates in its onset and progression.
SYN
PATENTS
PATENT
https://www.google.com/patents/US20140087992
Example 1(2R,3S)—N-((3S)-5-(3-Fluorophenyl)-9-methyl-2-oxo-2,3-dihydro-1H-1,4-benzodiazepin-3-yl)-2,3-bis(3,3,3-trifluoropropyl)succinamide
Intermediate 1A: (2S,3R)-tert-Butyl 6,6,6-trifluoro-3-(((S)-5-(3-fluorophenyl)-9-methyl-2-oxo-2,3-dihydro-1H-benzo[e][1,4]diazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoate

In a 100 mL round-bottomed flask, a solution of Intermediate B-1 (1683 mg, 5.94 mmol), Et3N (1.656 mL, 11.88 mmol), and Intermediate S-1 in DMF (20 mL) was treated with o-benzotriazol-1-yl-N,N,N′,N′-tetramethyluronium tetrafluoroborate (3815 mg, 11.88 mmol) and stirred at room temperature for 1 hour. The reaction mixture was diluted with water and saturated aqueous NaHCO3. An off white precipitate formed and was filtered and washed with water. The resulting solid was dried on the filter under a stream of nitrogen to give Intermediate 1A (3.7 g, 99% yield). MS (ES): m/z=632.4[M+H+]; HPLC: RT=3.635 min Purity=98%. (H2O/MeOH with TFA, CHROMOLITH® ODS S5 4.6×50 mm, gradient=4 min, wavelength=220 nm). 1H NMR (400 MHz, methanol-d4) δ 7.53 (t, J=4.5 Hz, 1H), 7.46-7.30 (m, 3H), 7.28-7.23 (m, 1H), 7.23-7.18 (m, 2H), 5.37 (s, 1H), 2.88 (td, J=10.4, 3.4Hz, 1H), 2.60 (td, J=10.2, 4.1 Hz, 1H), 2.54-2.40 (m, 1H), 2.47 (s, 3H), 2.33-2.12 (m, 3H), 1.98-1.69 (m, 4H), 1.51 (s, 9H).
Intermediate 1B: (2S,3R)-6,6,6-Trifluoro-3-(((S)-5-(3-fluorophenyl)-9-methyl-2-oxo-2,3-dihydro-1H-benzo[e][1,4]diazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoic acid

In a 250 mL round-bottomed flask, a solution of Intermediate 1A (3.7 g, 5.86 mmol) in DCM (25 mL) was treated with TFA (25 mL) and the resulting pale orange solution was stirred at room temperature for 1.5 hours. The reaction mixture was then concentrated to give Intermediate 1B. HPLC: RT=3.12 min (H2O/MeOH with TFA, CHROMOLITH® ODS S5 4.6×50 mm, gradient=4 min, wavelength=220 nm). MS (ES): m/z=576.3 (M+H)+. 1H NMR (400 MHz, methanol-d4) δ 7.54 (t, J=4.5 Hz, 1H), 7.49-7.29 (m, 3H), 7.28-7.15 (m, 3H), 5.38 (br. s., 1H), 2.89 (td, J=10.3, 3.7 Hz, 1H), 2.67 (td, J=9.9, 4.2Hz, 1H), 2.56-2.38 (m, 1H), 2.48 (s, 3H), 2.34-2.13 (m, 3H), 2.00-1.71 (m, 4H).
Example 1
In a 250 mL round-bottomed flask, a solution of Intermediate 1B (4.04 g, 5.86 mmol) in THF (50 mL) was treated with ammonia (2M in iPrOH) (26.4 mL, 52.7 mmol), followed by HOBT (1.795 g, 11.72 mmol) and EDC (2.246 g, 11.72 mmol). The resulting white suspension was stirred at room temperature overnight. The reaction mixture was diluted with water and saturated aqueous NaHCO3. The resulting solid was filtered, rinsed with water and then dried on the filter under a stream of nitrogen. The crude product was suspended in 20 mL of iPrOH and stirred at room temperature for 20 min and then filtered and washed with iPrOH and dried under vacuum to give 2.83 g of solid. The solid was dissolved in refluxing EtOH (100 mL) and slowly treated with 200 mg activated charcoal added in small portions. The hot mixture was filtered through CELITE® and rinsed with hot EtOH. The filtrate was reduced to half volume, allowed to cool and the white precipitate formed was filtered and rinsed with EtOH to give 2.57 g of white solid. A second recrystallization from EtOH (70 mL) afforded Example 1 (2.39 g, 70% yield) as a white solid. HPLC: RT=10.859 min (H2O/CH3CN with TFA, Sunfire C18 3.5 μm, 3.0×150 mm, gradient=15 min, wavelength=220 and 254 nm); MS (ES): m/z=575.3 [M+H+]; 1H NMR (400 MHz, methanol-d4) δ 7.57-7.50 (m, 1H), 7.47-7.30 (m, 3H), 7.29-7.15 (m, 3H), 5.38 (s, 1H), 2.85-2.75 (m, 1H), 2.59 (td, J=10.5, 4.0 Hz, 1H), 2.53-2.41 (m, 4H), 2.31-2.10 (m, 3H), 1.96-1.70 (m, 4H).
PATENT
https://www.google.com/patents/WO2014047372A1?cl=en


Scheme 3


XII XI
Scheme 4

Intermediate S-l : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid

Intermediate S-IA: 3,3,3-Trifluoro ropyl trifluoromethanesulfonate

[00180] To a cold (-25 °C) stirred solution of 2,6-lutidine (18.38 mL, 158 mmol) in DCM (120 mL) was added Tf20 (24.88 mL, 147 mmol) over 3 min, and the mixture was stirred for 5 min. To the reaction mixture was added 3,3,3-trifluoropropan-l-ol (12 g, 105 mmol) over an interval of 3 min. After 2 hr, the reaction mixture was warmed to room temperature and stirred for 1 hr. The reaction mixture was concentrated to half its volume, then purified by loading directly on a silica gel column (330g ISCO) and the product was eluted with DCM to afford Intermediate S-IA (13.74 g, 53%) as a colorless oil. 1H NMR (400 MHz, CDC13) δ ppm 4.71 (2 H, t, J= 6.15 Hz), 2.49-2.86 (2 H, m).
Intermediate S-1B: (4S)-4-Benzyl-3-(5,5,5-trifluoropentanoyl)-l,3-oxazolidin-2-one

[00181] To a stirring solution of 5,5,5-trifluoropentanoic acid (14.76 g, 95 mmol) and DMF (0.146 rriL) in DCM (50 mL) was slowly added oxalyl chloride (8.27 mL, 95 mmol). After 2h, the mixture was concentrated to dryness. A separate flask was changed with (S)-4-benzyloxazolidin-2-one (16.75 g, 95 mmol) in THF (100 mL) and then cooled to -78 °C. To the solution was slowly added n-BuLi (2.5M, 37.8 mL, 95 mmol) over 10 min, stirred for 10 min, and then a solution of the above acid chloride in THF (50 mL) was slowly added over 5 min. The mixture was stirred for 30 min, and then warmed to room temperature. The reaction was quenched with sat aq NH4C1. Next, 10% aq LiCl was then added to the mixture, and the mixture was extracted with Et20. The organic layer was washed with sat aq NaHC03 then with brine, dried (MgSC^), filtered and concentrated to dryness. The residue was purified by Si02 chromatography (ISCO, 330 g column, eluting with a gradient from 100% hexane to 100% EtOAc) to afford the product Intermediate S-IB; (25.25 g, 85%): 1H NMR (400 MHz, CDC13) δ ppm 7.32-7.39 (2 H, m), 7.30 (1 H, d, J= 7.05 Hz), 7.18-7.25 (2 H, m), 4.64-4.74 (1 H, m), 4.17-4.27 (2 H, m), 3.31 (1 H, dd, J= 13.35, 3.27 Hz), 3.00-3.11 (2 H, m), 2.79 (1 H, dd, J= 13.35, 9.57 Hz), 2.16-2.28 (2 H, m), 1.93-2.04 (2 H, m).
Intermediate S-IC: tert- utyl (3R)-3-(((4S)-4-benzyl-2-oxo-l,3-oxazolidin-3- yl)carbonyl)-6,6,6-trifluoroh xanoate

[00182] To a cold (-78 °C), stirred solution of Intermediate S-IB (3.03 g, 9.61 mmol) in THF (20 mL) was added NaHMDS (1.0M in THF) (10.6 mL, 10.60 mmol) under a nitrogen atmosphere. After 2 hours, tert-butyl 2-bromoacetate (5.62 g, 28.8 mmol) was added neat via syringe at -78 °C and stirring was maintained at the same temperature. After 6 hours, the reaction mixture was warmed to room temperature. The reaction mixture was partitioned between saturated NH4C1 and EtOAc. The organic phase was separated, and the aqueous phase was extracted with EtOAc (3x). The combined organics were washed with brine, dried (Na2s04), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (Teledyne ISCO
CombiFlash Rf, 5% to 100% solvent A/B = hexanes/EtOAc, REDISEP® Si02 120g). Concentration of the appropriate fractions provided Intermediate S-1C (2.79 g, 67.6%) as a colorless viscous oil: 1H NMR (400 MHz, CDC13) δ ppm 7.34 (2 H, d, J= 7.30 Hz), 7.24-7.32 (3 H, m), 4.62-4.75 (1 H, m, J= 10.17, 6.89, 3.43, 3.43 Hz), 4.15-4.25 (3 H, m), 3.35 (1 H, dd, J= 13.60, 3.27 Hz), 2.84 (1 H, dd, J= 16.62, 9.57 Hz), 2.75 (1 H, dd, J = 13.35, 10.07 Hz), 2.47 (1 H, dd, J= 16.62, 4.78 Hz), 2.11-2.23 (2 H, m), 1.90-2.02 (1 H, m), 1.72-1.84 (1 H, m), 1.44 (9 H, s).
Intermediate S-ID: (2R)-2-( -tert-Butoxy-2-oxoethyl)-5,5,5-trifluoropentanoic acid

[00183] To a cool (0 °C), stirred solution of Intermediate S-1C (2.17 g, 5.05 mmol) in THF (50 mL) and water (15 mL) was added a solution of LiOH (0.242 g, 10.11 mmol) and H202 (2.065 mL, 20.21 mmol) in H20 (2 mL). After 10 min, the reaction mixture was removed from the ice bath, stirred for lh, and then cooled to 0 °C. Saturated aqueous NaHCC”3 (25 mL) and saturated aqueous Na2s03 (25 mL) were added to the reaction mixture, and the mixture was stirred for 10 min, and then partially concentrated. The resulting mixture was extracted with DCM (2x), cooled with ice and made acidic with cone. HC1 to pH 3. The mixture was saturated with solid NaCl, extracted with EtOAc (3x), and then dried over MgS04, filtered and concentrated to a colorless oil to afford Intermediate S-ID, 1.2514g, 92%): 1H NMR (400 MHz, CDCI3) δ ppm 2.83-2.95 (1 H, m), 2.62-2.74 (1 H, m), 2.45 (1 H, dd, J= 16.62, 5.79 Hz), 2.15-2.27 (2 H, m), 1.88-2.00 (1 H, m), 1.75-1.88 (1 H, m), 1.45 (9 H, s). Intermediate S-l : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and Intermediate S-1E: (2R,3R)-3-(tert-butoxycarbonyl)- 6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid
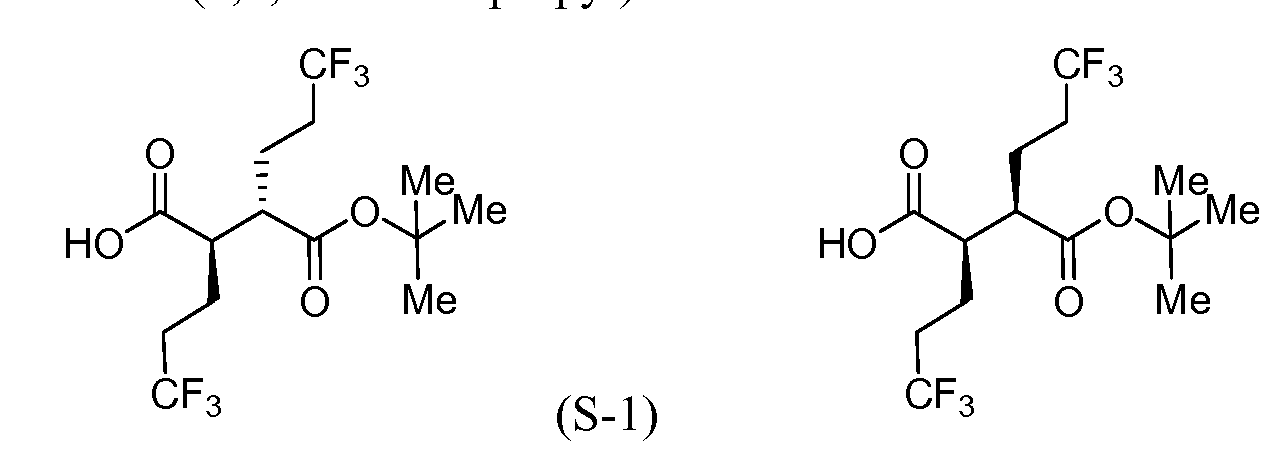
(S-1E)
[00184] To a cold (-78 °C) stirred solution of Intermediate S-1D (5 g, 18.50 mmol) in THF (60 mL) was slowly added LDA (22.2 mL, 44.4 mmol, 2.0M) over 7 min. After stirring for 2 hr, Intermediate S- 1 A (6.38 g, 25.9 mmol) was added to the reaction mixture over 3 min. After 60 min, the reaction mixture was warmed to -25 °C
(ice/MeOH/dry ice) and stirred for an additional 60 min at which time sat aq NH4C1 was added. The separated aqueous phase was acidified with IN HC1 to pH 3, and then extracted with Et20. The combined organic layers were washed with brine (2x), dried over MgS04, filtered and concentrated to provide a 1 :4 (II :I1E) mixture (as determined by 1H NMR) of Intermediate S-l and Intermediate S-1E (6.00 g, 89%) as a pale yellow solid. 1H NMR (500 MHz, CDC13) δ ppm 2.81 (1 H, ddd, J = 10.17, 6.32, 3.85 Hz), 2.63- 2.76 (1 H, m), 2.02-2.33 (4 H, m), 1.86-1.99 (2 H, m), 1.68-1.85 (2 H, m), 1.47 (9 H, s).
[00185] To a cold (-78 °C), stirred solution of a mixture of Intermediate S-l and Intermediate S-1E (5.97 g, 16.30 mmol) in THF (91 mL) was added LDA (19 mL, 38.0 mmol, 2.0M in THF/hexane/ethyl benzene) dropwise via syringe over 10 min (internal temperature never exceeded -65 °C, J-KEM® probe in reaction solution). The mixture was stirred for 15 min, and then warmed to room temperature (24 °C water bath), stirred for 15 min, and then cooled to -78 °C for 15 min. To the reaction mixture was added Et2AlCl (41 mL, 41.0 mmol, 1M in hexane) via syringe (internal temperature never exceeded -55 °C), and the mixture was stirred for 10 min, and then warmed to room temperature (24 °C bath) for 15 min and then back to -78 °C for 15 min. Meanwhile, a 1000 mL round bottom flask was charged with MeOH (145 mL) and precooled to -78 °C. With vigorous stirring the reaction mixture was transferred via cannula over 5 min to the MeOH. The flask was removed from the bath, ice was added followed by the slow addition of IN HC1 (147 mL, 147 mmol). Gas evolution was observed as the HC1 was added. The reaction mixture was allowed to warm to room temperature during which the gas evolution subsided. The reaction mixture was diluted with EtOAc (750 mL), saturated with NaCl, and the organic phase was separated, washed with a solution of potassium fluoride (8.52 g, 147 mmol) and IN HC1 (41 mL, 41.0 mmol) in water (291 mL), brine (100 mL), and then dried (Na2s04), filtered and concentrated under vacuum. 1H NMR showed the product was a 9: 1 mixture of Intermediate S-l and Intermediate S- 1E. The enriched mixture of Intermediate S-l and Intermediate S-1E (6.12 g, >99% yield) was obtained as a dark amber solid: 1H NMR (400 MHz, CDC13) δ ppm 2.64-2.76 (2 H, m), 2.04-2.35 (4 H, m), 1.88-2.00 (2 H, m), 1.71-1.83 (2 H, m), 1.48 (9 H, s).
Alternate procedure to make Intermediate S-l :
Intermediate S-IF: (2R,3 -1 -Benzyl 4-tert-butyl 2,3-bis(3,3,3-trifluoropropyl)succinate

[00186] To a stirred solution of a 9: 1 enriched mixture of Intermediate S-l and Intermediate S-1E (5.98 g, 16.33 mmol) in DMF (63 mL) were added potassium carbonate (4.06 g, 29.4 mmol) and benzyl bromide (2.9 mL, 24.38 mmol), the mixture was then stirred overnight at room temperature. The reaction mixture was diluted with EtOAc (1000 mL), washed with 10% LiCl (3×200 mL), brine (200 mL), dried (Na2S04), filtered, concentrated, and then dried under vacuum. The residue was purified by Si02 chromatography using a toluene:hexane gradient. Diastereomerically purified
Intermediate S-IF (4.81g, 65%) was obtained as a colorless solid: 1H NMR (400 MHz, chloroform-d) δ 7.32-7.43 (m, 5H), 5.19 (d, J= 12.10 Hz, 1H), 5.15 (d, J= 12.10 Hz, 1H), 2.71 (dt, J= 3.52, 9.20 Hz, 1H), 2.61 (dt, J= 3.63, 9.63 Hz, 1H), 1.96-2.21 (m, 4H), 1.69-1.96 (m, 3H), 1.56-1.67 (m, 1H), 1.45 (s, 9H).
Intermediate S-l : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid

[00187] To a solution of Intermediate S-1F (4.81 g, 10.54 mmol) in MeOH (100 mL) was added 10% palladium on carbon (wet, Degussa type, 568.0 mg, 0.534 mmol) in a H2– pressure flask. The vessel was purged with N2 (4x), then purged with H2 (2x), and finally, pressurized to 50 psi and shaken overnight. The reaction vessel was
depressurized and purged with nitrogen. The mixture was filtered through CELITE®, washed with MeOH and then concentrated and dried under vacuum. Intermediate S-1 (3.81 g, 99% yield)) was obtained as a colorless solid: 1H NMR (400 MHz, chloroform-d) δ 2.62-2.79 (m, 2H), 2.02-2.40 (m, 4H), 1.87-2.00 (m, 2H), 1.67-1.84 (m, 2H), 1.48 (s, 9H).
Alternate procedure to make Intermediate S-1 :
Intermediate S-1 : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid

[00188] Intermediate S-1 as a mixture with Intermediate S-IE was prepared in a similar procedure as above from Intermediate S-1D to afford a 1 :2.2 mixture of
Intermediate S-1 and Intermediate S-IE (8.60 g, 23.48 mmol), which was enriched using LDA (2.0 M solution in THF, ethyl benzene and heptane, 28.2 mL, 56.4 mmol) and diethyl aluminum chloride (1.0 M solution in hexane, 59 mL, 59.0 mmol) in THF (91 mL). After workup as described above, the resulting residue was found to be a 13.2: 1 (by 1H NMR) mixture of Intermediate S-1 and Intermediate S-IE, which was treated as follows: The crude material was dissolved in MTBE (43 mL). Hexanes (26 mL) were slowly charged to the reaction mixture while maintaining a temperature below 30 °C. The reaction mixture was stirred for 10 min. Next, tert-butylamine (2.7 mL, 1.1 eq) was charged slowly over a period of 20 minutes while maintaining a temperature below 30 °C. This addition was observed to be exothermic. The reaction mixture was stirred for 2 hrs below 30 °C and then filtered. The solid material was washed with 5:3 MTBE: hexane (80 mL), and the filtrate was concentrated and set aside. The filtered solid was dissolved in dichloromethane (300 mL), washed with IN HC1 (lOOmL), and the organic layer was washed with brine (100 mL x 2), and then concentrated under reduced pressure below 45 °C to afford Intermediate S-l (5.46 g, 64%).
A second alternate procedure for preparing Intermediate S-l :
Intermediate S-1G: tert- utyl 5,5,5-trifluoropentanoate

[00189] To a stirred solution of 5,5,5-trifluoropentanoic acid (5 g, 32.0 mmol) in THF (30 mL) and hexane (30 mL) at 0 °C, was added tert-butyl 2,2,2-trichloroacetimidate (11.46 mL, 64.1 mmol). The mixture was stirred for 15 min at 0 °C. Boron trifluoride etherate (0.406 mL, 3.20 mmol) was added and the reaction mixture was allowed to warm to room temperature overnight. To the clear reaction mixture was added solid NaHC03 (5 g) and stirred for 30 min. The mixture was filtered through MgSC^ and washed with hexanes (200 mL). The solution was allowed to rest for 45 min, and the resulting solid material was removed by filtering on the same MgSC^ filter again, washed with hexanes (100 mL) and concentrated under reduced pressure without heat. The volume was reduced to about 30 mL, filtered through a clean fritted funnel, washed with hexane (5 mL), and then concentrated under reduced pressure without heat. The resulting neat oil was filtered through a 0.45μιη nylon membrane filter disk to provide Intermediate S-1G (6.6 g, 31.4 mmol 98% yield) as a colorless oil: 1H NMR (400 MHz, CDC13) δ ppm 1.38 (s, 9 H) 1.74-1.83 (m, 2 H) 2.00-2.13 (m, 2 H) 2.24 (t, J= 7.28 Hz, 2 H). Intermediate S-1H: (4S)-4-(Propan-2-yl)-3-(5,5,5-trifluoropentanoyl)-l,3-oxazolidin-2- one

[00190] To a stirred solution of 5,5,5-trifluoropentanoic acid (5.04 g, 32.3 mmol) in DCM (50 mL) and DMF (3 drops) was added oxalyl chloride (3.4 mL, 38.8 mmol) dropwise over 5 min. The solution was stirred until all bubbling subsided. The reaction mixture was concentrated under reduced pressure to give pale yellow oil. To a separate flask charged with a solution of (4S)-4-(propan-2-yl)-l,3-oxazolidin-2-one (4.18 g, 32.4 mmol) in THF (100 mL) at -78 °C was added n-BuLi (2.5M in hexane) (13.0 mL, 32.5 mmol) dropwise via syringe over 5 min. After stirring for 10 min, the above acid chloride, dissolved in THF (20 mL), was added via cannula over 15 min. The reaction mixture was warmed to 0 °C, and was allowed to warm to room temperature as the bath warmed and stirred overnight. To the reaction mixture was added saturated NH4C1, and the mixture was extracted with EtOAc (2x). The combined organics were washed with brine, dried (Na2s04), filtered and concentrated under reduced pressure. The crude material was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 5% to 60% solvent A/B = hexanes/EtOAc, REDISEP® Si02 120g). Concentration of the appropriate fractions provided Intermediate S-1H (7.39 g, 86%) as a colorless oil: 1H NMR (400 MHz, CDC13) δ ppm 4.44 (1 H, dt, J= 8.31, 3.53 Hz), 4.30 (1 H, t, J= 8.69 Hz), 4.23 (1 H, dd, J= 9.06, 3.02 Hz), 2.98-3.08 (2 H, m), 2.32-2.44 (1 H, m, J= 13.91, 7.02, 7.02, 4.03 Hz), 2.13-2.25 (2 H, m), 1.88-2.00 (2 H, m), 0.93 (3 H, d, J= 7.05 Hz), 0.88 (3 H, d, J= 6.80 Hz).
Intermediate S-1I: (2S,3R)-tert-Butyl 6,6,6-trifluoro-3-((S)-4-isopropyl-2- oxooxazolidine-3-carbonyl)-2-(3,3,3-trifluoropropyl)hexanoate, and Intermediate S-U: (2R,3R)-tert-Butyl 6,6,6-trifluoro-3-((S)-4-isopropyl-2-oxooxazolidine-3-carbonyl)-2- (3 ,3 ,3 -trifluoropropyl)hexanoate

[00191] To a cold (-78 °C), stirred solution of diisopropylamine (5.3 mL, 37.2 mmol) in THF (59 mL) under a nitrogen atmosphere was added n-BuLi (2.5M in hexane) (14.7 mL, 36.8 mmol). The mixture was then warmed to 0 °C to give a 0.5M solution of LDA. A separate vessel was charged with Intermediate S-1H (2.45 g, 9.17 mmol). The material was azeotroped twice with benzene (the RotoVap air inlet was fitted with a nitrogen inlet to completely exclude humidity), and then toluene (15.3 mL) was added. This solution was added to a flask containing dry lithium chloride (1.96 g, 46.2 mmol). To the resultant mixture, cooled to -78 °C, was added the LDA solution (21.0 mL, 10.5 mmol) and the mixture was stirred at -78 °C for 10 min, then warmed to 0 °C for 10 min., and then cooled to -78 °C. To a separate reaction vessel containing Intermediate S-1G (3.41 g, 16.07 mmol), also azeotroped twice with benzene, was added toluene (15.3 mL), cooled to -78 °C and LDA (37.0 mL, 18.5 mmol) was added. The resulting solution was stirred at -78 °C for 25 min. At this time the enolate derived from the ester was transferred via cannula into the solution of the oxazolidinone enolate and stirred at -78 °C for an additional 5 min, at which time the septum was removed and solid powdered bis(2- ethylhexanoyloxy)copper (9.02 g, 25.8 mmol) was rapidly added to the reaction vessel and the septum was replaced. The vessel was immediately removed from the cold bath and immersed into a warm water bath (40 °C) with rapid swirling and with a concomitant color change from the initial turquoise to brown. The reaction mixture was stirred for 20 min, was then poured into 5% aqueous NH4OH (360 mL) and extracted with EtOAc (2x). The combined organics were washed with brine, dried (Na2s04), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 0% to 60% solvent A/B = hexanes/EtOAc, REDISEP® Si02 120g). Concentration of the appropriate fractions provided a mixture of Intermediate S- II and Intermediate S-1J (2.87 g, 66%) as a pale yellow viscous oil. 1H NMR showed the product was a 1.6: 1 mixture of diastereomers S-1LS-1J as determined by the integration of the multiplets at 2.74 and 2.84 ppm: 1H NMR (400 MHz, CDC13) δ ppm 4.43-4.54 (2 H, m), 4.23-4.35 (5 H, m), 4.01 (1 H, ddd, J= 9.54, 6.27, 3.51 Hz), 2.84 (1 H, ddd, J = 9.41, 7.28, 3.64 Hz), 2.74 (1 H, ddd, J= 10.29, 6.27, 4.02 Hz), 2.37-2.48 (2 H, m, J = 10.38, 6.98, 6.98, 3.51, 3.51 Hz), 2.20-2.37 (3 H, m), 1.92-2.20 (8 H, m), 1.64-1.91 (5 H, m), 1.47 (18 H, s), 0.88-0.98 (12 H, m). Intermediate S-1 : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and Intermediate S-IE: (2R,3R)-3-(tert-Butoxycarbonyl)- 6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid

(S-IE)
[00192] To a cool (0 °C), stirred solution of Intermediate S-1I and Intermediate S-1 J (4.54 g, 9.51 mmol) in THF (140 mL) and water (42 mL) were sequentially added hydrogen peroxide (30% in water) (10.3 g, 91 mmol) and LiOH (685.3 mg, 28.6 mmol). The mixture was stirred for 1 hr. At this time the reaction vessel was removed from the cold bath and then stirred for 1.5 hr. To the reaction mixture were added saturated NaHC03 (45 mL) and saturated Na2s03 (15 mL), and then the mixture was partially concentrated under reduced pressure. The resulting crude solution was extracted with DCM (3x). The aqueous phase was acidified to pH~l-2 with IN HC1, extracted with DCM (3x) and then EtOAc (lx). The combined organics were washed with brine, dried (Na2s04), filtered and concentrated under reduced pressure to provide a mixture of Intermediates S-1 and S-IE (3.00 g, 86%) as a colorless oil: 1H NMR (400 MHz, CDC13) δ ppm 2.76-2.84 (1 H, m, diastereomer 2), 2.64-2.76 (3 H, m), 2.04-2.35 (8 H, m), 1.88- 2.00 (4 H, m), 1.71-1.83 (4 H, m), 1.48 (9 H, s, diastereomer 1), 1.46 (9 H, s,
diastereomer 2); 1H NMR showed a 1.7: 1 mixture of S-1E:S-1F by integration of the peaks for the t-butyl groups. Intermediate S-1 : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and Intermediate S-IF: (2R,3R)-3-(fert-Butoxycarbonyl)- 6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid

[00193] To a cold (-78 °C) stirred solution of diisopropylamine (1.7 mL, 11.93 mmol) in THF (19 mL) under a nitrogen atmosphere was added n-BuLi (2.5M in hexanes) (4.8 mL, 12.00 mmol). The mixture was stirred for 5 min and then warmed to 0 °C. In a separate vessel, to a cold (-78 °C) stirred solution of the mixture of Intermediates S-1 and S-1E (1.99 g, 5.43 mmol) in THF (18 mL) was added the LDA solution prepared above via cannula slowly over 25 min. The mixture was stirred for 15 min, then warmed to room temperature (placed in a 24 °C water bath) for 15 min, and then again cooled to -78 °C for 15 min. To the reaction mixture was added Et2AlCl (1M in hexane) (11.4 mL, 11.40 mmol) via syringe. The mixture was stirred for 10 min, warmed to room
temperature for 15 min and then cooled back to -78 °C for 15 min. Methanol (25 mL) was rapidly added, swirled vigorously while warming to room temperature, and then concentrated to ~l/4 the original volume. The mixture was dissolved in EtOAc and washed with IN HC1 (50 mL) and ice (75 g). The aqueous phase was separated and extracted with EtOAc (2x). The combined organics were washed with a mixture of KF (2.85g in 75 mL water) and IN HC1 (13 mL) [resulting solution pH 3-4], then with brine, dried (Na2s04), filtered and concentrated under reduced pressure to give a 9: 1 (S-LS-1E) enriched diastereomeric mixture (as determined by 1H NMR) of Intermediate S-1 and Intermediate S-1E (2.13 g, >99%) as a pale yellow viscous oil: 1H NMR (400 MHz, CDC13) δ ppm 2.64-2.76 (2 H, m), 2.04-2.35 (4 H, m), 1.88-2.00 (2 H, m), 1.71-1.83 (2 H, m), 1.48 (9 H, s).
Intermediate S-2: (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3- fluoropropyl)hexanoic acid

Intermediate S-2: (2R,3S)-3-(tert-Butoxycarbonyl)-7,7,7-trifluoro-2-(3,3,3- trifluoropropyl)heptanoic acid, and Intermediate S-2A: (2R,3R)-3-(tert-Butoxycarbonyl)- 7,7,7-trifluoro-2-(3,3,3-trifluoropropyl)heptanoic acid

(S-2A)
[00194] To a cold (-78 °C), stirred solution of Intermediate S-1D (1.72 g, 6.36 mmol) in THF (30 mL) was slowly added LDA (7.32 mL, 14.6 mmol) over 7 min. After stirring for 1 h, 4,4,4-trifluorobutyltrifluoromethanesulfonate (2.11 g, 8.11 mmol) was added to the reaction mixture over 2 min. After 15 min, the reaction mixture was warmed to -25 °C (ice/MeOH/dry ice) for lh, and then cooled to -78 °C. After 80 min, the reaction was quenched with a saturated aqueous NH4C1 solution (10 mL). The reaction mixture was further diluted with brine and the solution was adjusted to pH 3 with IN HC1. The aqueous layer was extracted with ether. The combined organics were washed with brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure to provide a mixture of Intermediates S-2 and S-2A (2.29 g, 95%) as a colorless oil. 1H NMR (400MHz, chloroform-d) δ 2.83-2.75 (m, 1H), 2.64 (ddd, J = 9.9, 6.7, 3.6 Hz, 1H), 2.32-2.03 (m, 5H), 1.98-1.70 (m, 3H), 1.69-1.52 (m, 3H), 1.50-1.42 (m, 9H). 1H NMR showed a 1 :4.5 mixture (S-2:S-2A) of diastereomers by integration of the peaks for the t- Bu groups.
Intermediate S-2: (2R,3S)-3-(fert-Butoxycarbonyl)-7,7,7-trifluoro-2-(3,3,3- trifluoropropyl)heptanoic acid, and Intermediate S-2A: (2R,3R)-3-(tert-Butoxycarbonyl)- 7,7,7-trifluoro-2-(3,3,3-trifluoropropyl)heptanoic acid

[00195] A mixture of Intermediate S-2 and Intermediate S-2A (2.29 g, 6.02 mmol) was dissolved in THF (38 mL) to give a colorless solution which was cooled to -78 °C. Then, LDA (7.23 mL, 14.5 mmol) (2.0M in heptane/THF/ethylbenzene) was slowly added to the reaction mixture over 3 min. After stirring for 15 min, the reaction mixture was placed in a room temperature water bath. After 15 min the reaction mixture was placed back in a -78 °C bath and then diethylaluminum chloride (14.5 mL, 14.5 mmol) (1M in hexane) was added slowly over 5 min. The reaction mixture was stirred at -78 °C. After 15 min, the reaction mixture was placed in a room temperature water bath for 10 min, and then cooled back to -78 °C. After 15 min, the reaction was quenched with MeOH (30.0 mL, 741 mmol), removed from the -78 °C bath and concentrated. To the reaction mixture was added ice and HC1 (60.8 mL, 60.8 mmol) and the resulting mixture was extracted with EtOAc (2x 200 mL). The organic layer was washed with potassium fluoride (3.50g, 60.3 mmol) in 55 mL H20 and 17.0 mL of IN HC1. The organics were dried over anhydrous magnesium sulfate and concentrated under reduced pressure to provide an enriched mixture of Intermediate S-2 and Intermediate S-2A (2.25g, 98% yield) as a light yellow oil. 1H NMR (400MHz, chloroform-d) δ 2.83-2.75 (m, 1H), 2.64 (ddd, J= 9.9, 6.7, 3.6 Hz, 1H), 2.32-2.03 (m, 5H), 1.98-1.70 (m, 3H), 1.69-1.52 (m, 3H), 1.50-1.42 (m, 9H). 1H NMR showed a 9: 1 ratio in favor of the desired diastereomer Intermediate S-2.
Intermediate S-2B: (2R,3S)-1 -Benzyl 4-tert-butyl 2,3-bis(4,4,4-trifluorobutyl)succinate

[00196] To a stirred 9: 1 mixture of Intermediate S-2 and Intermediate S-2A (2.24 g, 5.89 mmoL) and potassium carbonate (1.60 g, 11.58 mmoL) in DMF (30 mL) was added benzyl bromide (1.20 mL, 10.1 mmoL)). The reaction mixture was stirred at room temperature for 19 h. The reaction mixture was diluted with ethyl acetate (400 mL) and washed with 10% LiCl solution (3 x 100 mL), brine (50 mL), and then dried over anhydrous magnesium sulfate, filtered and concentrated to dryness under vacuum. The residue was purified by flash chromatography (Teledyne ISCO CombiFlash 0%> to 100% solvent A/B = hexane/EtOAc, REDISEP® Si02 220 g, detecting at 254 nm, and monitoring at 220 nm). Concentration of the appropriate fractions provided Intermediate S-2B (1.59 g, 57.5%). HPLC: RT = 3.863 min (CHROMOLITH® SpeedROD column 4.6 x 50 mm, 10-90% aqueous methanol over 4 minutes containing 0.1% TFA, 4 mL/min, monitoring at 220 nm), 1H NMR (400MHz, chloroform-d) δ 7.40-7.34 (m, 5H), 5.17 (d, J= 1.8 Hz, 2H), 2.73-2.64 (m, 1H), 2.55 (td, J= 10.0, 3.9 Hz, 1H), 2.16-1.82 (m, 5H), 1.79-1.57 (m, 3H), 1.53-1.49 (m, 1H), 1.45 (s, 9H), 1.37-1.24 (m, 1H).
Intermediate S-2: (2R,3S)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(4,4,4- trifluorobutyl)hexanoic acid

[00197] To a stirred solution of Intermediate S-2B (1.59 g, 3.37 mmoL) in MeOH (10 mL) and EtOAc (10 mL) under nitrogen was added 10%> Pd/C (510 mg). The atmosphere was replaced with hydrogen and the reaction mixture was stirred at room temperature for 2.5 h. The palladium catalyst was filtered off through a 4 μΜ polycarbonate film and rinsed with MeOH. The filtrate was concentrated under reduced pressure to give intermediate S-2 (1.28 g, 99%). 1H NMR (400MHz, chloroform-d) δ 2.76-2.67 (m, 1H), 2.65-2.56 (m, 1H), 2.33-2.21 (m, 1H), 2.17-2.08 (m, 3H), 1.93 (dtd, J= 14.5, 9.9, 5.2 Hz, 1H), 1.84-1.74 (m, 2H), 1.70-1.52 (m, 3H), 1.48 (s, 9H).
Intermediate A- 1 : (2-Amino-3 -methylphenyl)(3 -fluorophenyl)methanone

Intermediate A-1 A: 2-Amino- -methoxy-N,3-dimethylbenzamide

[00198] In a 1 L round-bottomed flask was added 2-amino-3-methylbenzoic acid (11.2 g, 74.1 mmol) and Ν,Ο-dimethylhydroxylamine hydrochloride (14.45 g, 148 mmol) in DCM (500 mL) to give a pale brown suspension. The reaction mixture was treated with Et3N (35 mL), HOBT (11.35 g, 74.1 mmol) and EDC (14.20 g, 74.1 mmol) and then stirred at room temperature for 24 hours. The mixture was then washed with 10% LiCl, and then acidified with IN HCl. The organic layer was washed successively with 10%> LiCl and aq NaHC03. The organic layer was decolorized with charcoal, filtered, and the filtrate was dried over MgSC^. The mixture was filtered and concentrated to give 13.22 g (92% yield) of Intermediate A-1A. MS(ES): m/z = 195.1 [M+H+]; HPLC: RT = 1.118 min. (H20/MeOH with TFA, CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm); 1H NMR (500MHz, chloroform-d) δ 7.22 (dd, J= 7.8, 0.8 Hz, 1H), 7.12-7.06 (m, 1H), 6.63 (t, J= 7.5 Hz, 1H), 4.63 (br. s., 2H), 3.61 (s, 3H), 3.34 (s, 3H), 2.17 (s, 3H).
Intermediate A- 1 : (2-Amino-3 -methylphenyl)(3 -fluorophenyl)methanone

[00199] In a 500 mL round-bottomed flask, a solution of l-fluoro-3-iodobenzene (13.61 mL, 116 mmol) in THF (120 mL) was cooled in a -78 °C bath. A solution of n- BuLi, (2.5M in hexane, 46.3 mL, 116 mmol) was added dropwise over 10 minutes. The solution was stirred at -78 °C for 30 minutes and then treated with a solution of
Intermediate A-1 A (6.43 g, 33.1 mmol) in THF (30 mL). After 1.5 hours, the reaction mixture was added to a mixture of ice and IN HCl (149 mL, 149 mmol) and the reaction flask was rinsed with THF (5 ml) and combined with the aqueous mixture. The resulting mixture was diluted with 10% aq LiCl and the pH was adjusted to 4 with IN NaOH. The mixture was then extracted with Et20, washed with brine, dried over MgS04, filtered and concentrated. The resulting residue was purified by silica gel chromatography (220g ISCO) eluting with a gradient from 10% EtOAc/hexane to 30% EtOAc/hexane to afford Intermediate A-l (7.11 g, 94% yield) as an oil. MS(ES): m/z = 230.1 [M+H+]; HPLC: RT = 2.820 min Purity = 99%. (H20/MeOH with TFA, CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm).
Intermediate B-1 : (S)-3-Amino-5-(3-fluorophenyl)-9-methyl-lH-benzo[e][l,4]diazepin- 2(3H)-one

Intermediate B-1 A: (S)-Benzyl (5-(3-fluorophenyl)-9-methyl-2-oxo-2,3-dihydro benzo[e] [ 1 ,4]diazepin-3-yl)carbamate

(B-1A)
[00225] In a 1 L round-bottomed flask, a solution of 2-(lH-benzo[d][l,2,3]triazol-l- yl)-2-((phenoxycarbonyl)amino)acetic acid (J. Org. Chem., 55:2206-2214 (1990)) (19.37 g, 62.0 mmol) in THF (135 mL) was cooled in an ice/water bath and treated with oxalyl chloride (5.43 mL, 62.0 mmol) and 4 drops of DMF. The reaction mixture was stirred for 4 hours. Next, a solution of Intermediate A- 1 (7.11 g, 31.0 mmol) in THF (35 mL) was added and the resulting solution was removed from the ice/water bath and stirred at room temperature for 1.5 hours. The mixture was then treated with a solution of ammonia, (7M in MeOH) (19.94 mL, 140 mmol). After 15 mins, another portion of ammonia, (7M in MeOH) (19.94 mL, 140 mmol) was added and the resulting mixture was sealed under N2 and stirred overnight at room temperature. The reaction mixture was then concentrated to ~l/2 volume and then diluted with AcOH (63 mL) and stir at room temperature for 4 hours. The reaction mixture was then concentrated, and the residue was diluted with 500 mL water to give a precipitate. Hexane and Et20 were added and the mixture was stirred at room temperature for 1 hour to form an orange solid. Et20 was removed under a stream of nitrogen and the aqueous layer was decanted. The residue was triturated with 40 mL of iPrOH and stirred at room temperature to give a white precipitate. The solid was filtered and washed with iPrOH, then dried on a filter under a stream of nitrogen to give racemic Intermediate B-1A (5.4 g, 41.7%yield).
[00226] Racemic Intermediate B-1A (5.9 g, 14.3 mmol) was resolved using the Chiral SFC conditions described below. The desired stereoisomer was collected as the second peak in the elution order: Instrument: Berger SFC MGIII, Column: CHIRALPAK® IC 25 x 3 cm, 5 cm; column temp: 45 °C; Mobile Phase: C02/MeOH (45/55); Flow rate: 160 mL/min; Detection at 220 nm.
[00227] After evaporation of the solvent, Intermediate B-1A (2.73 g, 46% yield) was obtained as a white solid. HPLC: RT = 3.075 min. (H20/MeOH with TFA,
CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm).
Chiral HPLC RT: 8.661 min (AD, 60% (EtOH/MeOH)/heptane) > 99%ee. MS(ES): m/z = 418.3 [M+H+];1H NMR (500MHz, DMSO-d6) δ 10.21 (s, 1H), 8.38 (d, J= 8.3 Hz, 1H), 7.57-7.47 (m, 2H), 7.41-7.29 (m, 8H), 7.25-7.17 (m, 2H), 5.10-5.04 (m, 3H), 2.42 (s, 3H).
Intermediate B-l : (S)-3-Amino-5-(3-fluorophenyl)-9-methyl-lH-benzo[e][l,4]diazepin- 2(3H)-one.
[00228] In a 100 mL round-bottomed flask, a solution of Intermediate B-1A (2.73 g, 6.54 mmol) in acetic acid (12 mL) was treated with HBr, 33% in HOAc (10.76 mL, 65.4 mmol) and the mixture was stirred at room temperature for 1 hour. The solution was diluted with Et20 to give a yellow precipitate. The yellow solid was filtered and rinsed with Et20 under nitrogen. The solid was transferred to 100 mL round bottom flask and water was added (white precipitate formed). The slurry was slowly made basic with saturated NaHC03. The resulting tacky precipitate was extracted with EtOAc. The organic layer was washed with water, dried over MgS04, and then filtered and
concentrated to dryness to give Intermediate B-l (1.68 g, 91% yield) as a white foam solid. MS(ES): m/z = 284.2 [M+H+]; HPLC: RT = 1.72 min (H20/MeOH with TFA, CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm). 1H NMR (400MHz, DMSO-d6) δ 10.01 (br. s., 1H), 7.56-7.44 (m, 2H), 7.41-7.26 (m, 3H), 7.22-7.11 (m, 2H), 4.24 (s, 1H), 2.55 (br. s., 2H), 2.41 (s, 3H). [00229] The compounds listed below in Table 6 (Intermediates B-2 to B-3) were prepared according to the general synthetic procedure described for Intermediate B-l , using the starting materials Intermediate A- 10 and Intermediate A-4, respectively.
Example 1
(2R,3S)-N-((3S)-5-(3-Fluorophenyl)-9-methyl-2-oxo-2,3-dihydro-lH-l,4-benzodiazepin- 3-yl)-2, -bis(3,3,3-trifluoropropyl)succinamide

Intermediate 1A: (2S,3R)-tert-Butyl 6,6,6-trifluoro-3-(((S)-5-(3-fluorophenyl)-9-methyl- 2-0X0-2, 3-dihydro-lH-benzo[e][l,4]diazepin-3-yl)carbamoyl)-2-(3,3 ,3- trifluoropropyl)hexanoat

[00240] In a 100 mL round-bottomed flask, a solution of Intermediate B-l (1683 mg, 5.94 mmol), Et3N (1.656 mL, 11.88 mmol), and Intermediate S-l in DMF (20 mL) was treated with o-benzotriazol-l-yl-A .A .N’.N’-tetramethyluronium tetrafluoroborate (3815 mg, 11.88 mmol) and stirred at room temperature for 1 hour. The reaction mixture was diluted with water and saturated aqueous NaHC03. An off white precipitate formed and was filtered and washed with water. The resulting solid was dried on the filter under a stream of nitrogen to give Intermediate 1A (3.7 g, 99% yield). MS(ES): m/z =
632.4[M+H+]; HPLC: RT = 3.635 min Purity = 98%. (H20/MeOH with TFA,
CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm). 1H NMR (400MHz, methanol-d4) δ 7.53 (t, J = 4.5 Hz, 1H), 7.46-7.30 (m, 3H), 7.28-7.23 (m, 1H), 7.23-7.18 (m, 2H), 5.37 (s, 1H), 2.88 (td, J = 10.4, 3.4 Hz, 1H), 2.60 (td, J =
10.2, 4.1 Hz, 1H), 2.54-2.40 (m, 1H), 2.47 (s, 3 H), 2.33-2.12 (m, 3H), 1.98-1.69 (m, 4H), 1.51 (s, 9H). Intermediate IB: (2S,3R)-6,6,6-Trifluoro-3-(((S)-5-(3-fluorophenyl)-9-methyl-2-oxo-
2,3-dihydro-lH-benzo[e][l,4]diazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoic acid

[00241] In a 250 mL round-bottomed flask, a solution of Intermediate 1A (3.7 g, 5.86 mmol) in DCM (25 mL) was treated with TFA (25 mL) and the resulting pale orange solution was stirred at room temperature for 1.5 hours. The reaction mixture was then concentrated to give Intermediate IB. HPLC: RT = 3.12 min (H20/MeOH with TFA, CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm).
MS(ES): m/z = 576.3 (M+H)+. 1H NMR (400MHz, methanol-d4) δ 7.54 (t, J= 4.5 Hz, 1H), 7.49-7.29 (m, 3H), 7.28-7.15 (m, 3H), 5.38 (br. s., 1H), 2.89 (td, J= 10.3, 3.7 Hz, 1H), 2.67 (td, J= 9.9, 4.2 Hz, 1H), 2.56-2.38 (m, 1H), 2.48 (s, 3 H), 2.34-2.13 (m, 3H), 2.00-1.71 (m, 4H).
Example 1 :
[00242] In a 250 mL round-bottomed flask, a solution of Intermediate IB (4.04 g, 5.86 mmol) in THF (50 mL) was treated with ammonia (2M in iPrOH) (26.4 mL, 52.7 mmol), followed by HOBT (1.795 g, 11.72 mmol) and EDC (2.246 g, 11.72 mmol). The resulting white suspension was stirred at room temperature overnight. The reaction mixture was diluted with water and saturated aqueous NaHC03. The resulting solid was filtered, rinsed with water and then dried on the filter under a stream of nitrogen. The crude product was suspended in 20 mL of iPrOH and stirred at room temperature for 20 min and then filtered and washed with iPrOH and dried under vacuum to give 2.83 g of solid. The solid was dissolved in re fluxing EtOH(100 mL) and slowly treated with 200 mg activated charcoal added in small portions. The hot mixture was filtered through CELITE® and rinsed with hot EtOH. The filtrate was reduced to half volume, allowed to cool and the white precipitate formed was filtered and rinsed with EtOH to give 2.57 g of white solid. A second recrystallization from EtOH (70 mL) afforded Example 1 (2.39 g, 70% yield) as a white solid. HPLC: RT = 10.859 min (H20/CH3CN with TFA, Sunfire C18 3.5μπι, 3.0x150mm, gradient = 15 min, wavelength = 220 and 254 nm); MS(ES): m/z = 575.3 [M+H+]; 1H NMR (400MHz, methanol-d4) δ 7.57-7.50 (m, 1H), 7.47-7.30 (m, 3H), 7.29-7.15 (m, 3H), 5.38 (s, 1H), 2.85-2.75 (m, 1H), 2.59 (td, J= 10.5, 4.0 Hz, 1H), 2.53-2.41 (m, 4H), 2.31-2.10 (m, 3H), 1.96-1.70 (m, 4H).
SEE
WO2012129353A1 *Mar 22, 2012Sep 27, 2012Bristol-Myers Squibb CompanyBis(fluoroalkyl)-1,4-benzodiazepinone compounds
PAPER RELATED
Structure–activity relationships in a series of (2-oxo-1,4-benzodiazepin-3-yl)-succinamides identified highly potent inhibitors of γ-secretase mediated signaling of Notch1/2/3/4 receptors. On the basis of its robust in vivo efficacy at tolerated doses in Notch driven leukemia and solid tumor xenograft models, 12 (BMS-906024) was selected as a candidate for clinical evaluation.
Discovery of Clinical Candidate BMS-906024: A Potent Pan-Notch Inhibitor for the Treatment of Leukemia and Solid Tumors
Ashvinikumar V. Gavai*†, Claude Quesnelle†, Derek Norris†, Wen-Ching Han†, Patrice Gill†, Weifang Shan†, Aaron Balog†, Ke Chen§, Andrew Tebben†, Richard Rampulla†, Dauh-Rurng Wu†, Yingru Zhang†, Arvind Mathur†,Ronald White†, Anne Rose†, Haiqing Wang†, Zheng Yang†, Asoka Ranasinghe†, Celia D’Arienzo†, Victor Guarino†, Lan Xiao†, Ching Su†, Gerry Everlof†, Vinod Arora‡, Ding Ren Shen†, Mary Ellen Cvijic†, Krista Menard†, Mei-Li Wen†, Jere Meredith‡, George Trainor†, Louis J. Lombardo†, Richard Olson‡, Phil S. Baran§,John T. Hunt†, Gregory D. Vite†, Bruce S. Fischer†, Richard A. Westhouse†, and Francis Y. Lee†
†Bristol-Myers Squibb Research and Development, Princeton, New Jersey 08543, United States
‡Bristol-Myers Squibb Research and Development, 5 Research Parkway, Wallingford, Connecticut 06492, United States
§ Department of Chemistry, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037,United StatesACS Med. Chem. Lett.
, 2015, 6 (5), pp 523–527
DOI: 10.1021/acsmedchemlett.5b00001, http://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.5b00001
*Phone: 609-252-5091. E-mail: ashvinikumar.gavai@bms.com.
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
PAPER RELATED

An enantioselective synthesis of (S)-7-amino-5H,7H-dibenzo[b,d]azepin-6-one (S–1) is described. The key step in the sequence involved crystallization-induced dynamic resolution (CIDR) of compound 7 using Boc-d-phenylalanine as a chiral resolving agent and 3,5-dichlorosalicylaldehyde as a racemization catalyst to afford S–1 in 81% overall yield with 98.5% enantiomeric excess.
Crystallization-Induced Dynamic Resolution toward the Synthesis of (S)-7-Amino-5H,7H-dibenzo[b,d]-azepin-6-one: An Important Scaffold for γ-Secretase Inhibitors
Sukhen Karmakar†, Vijay Byri†, Ashvinikumar V. Gavai‡, Richard Rampulla‡, Arvind Mathur‡, and Anuradha Gupta*†
† Department of Discovery Synthesis, Biocon Bristol-Myers Squibb Research Centre, Biocon Park, Bommasandra IV Phase, Jigani Link Road, Bengaluru 560099, India
‡Bristol-Myers Squibb Company, P.O Box 4000, Princeton, New Jersey 08543-4000, United StatesOrg. Process Res. Dev.
, Article ASAP
DOI: 10.1021/acs.oprd.6b00207, http://pubs.acs.org/doi/suppl/10.1021/acs.oprd.6b00207
*E-mail: anuradha.gupta@syngeneintl.com.
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2000007995A1 * | Aug 7, 1999 | Feb 17, 2000 | Du Pont Pharmaceuticals Company | SUCCINOYLAMINO LACTAMS AS INHIBITORS OF Aβ PROTEIN PRODUCTION |
| WO2000038618A2 * | Dec 23, 1999 | Jul 6, 2000 | Du Pont Pharmaceuticals Company | SUCCINOYLAMINO BENZODIAZEPINES AS INHIBITORS OF Aβ PROTEIN PRODUCTION |
| WO2001060826A2 * | Feb 16, 2001 | Aug 23, 2001 | Bristol-Myers Squibb Pharma Company | SUCCINOYLAMINO CARBOCYCLES AND HETEROCYCLES AS INHIBITORS OF Aβ PROTEIN PRODUCTION |
| US6737038 * | May 17, 2000 | May 18, 2004 | Bristol-Myers Squibb Company | Use of small molecule radioligands to discover inhibitors of amyloid-beta peptide production and for diagnostic imaging |
| US7053084 | Feb 17, 2000 | May 30, 2006 | Bristol-Myers Squibb Company | Succinoylamino benzodiazepines as inhibitors of Aβ protein production |
| US7456172 | Jan 13, 2006 | Nov 25, 2008 | Bristol-Myers Squibb Pharma Company | Succinoylamino benzodiazepines as inhibitors of Aβ protein production |
| US20030134841 * | Nov 1, 2002 | Jul 17, 2003 | Olson Richard E. | Succinoylamino lactams as inhibitors of A-beta protein production |
| US20120245151 * | Mar 22, 2012 | Sep 27, 2012 | Bristol-Myers Squibb Company | Bisfluoroalkyl-1,4-benzodiazepinone compounds |
//////////varegacestat, BMS-986115, BMS 986115, 3,5-dichlorosalicylaldehyde, Alzheimer’s disease, Boc-D-phenylalanine, CIDR;dibenzoazepenone, DKR; Notch inhibitors, Notch inhibitor, SAR, T-acute lymphoblastic leukemia, triple-negative breast cancer, γ-secretase inhibitor, PHASE 1, BMS, Bristol-Myers Squibb, Ashvinikumar Gavai, 1584647-27-7, LSK1L593UU, AL 102
Tigozertinib
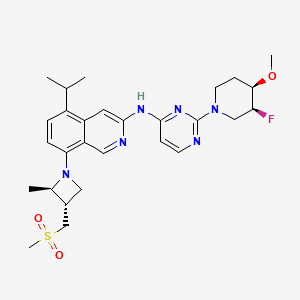
Tigozertinib
CAS 2660250-10-0
MF C28H37FN6O3S MW 556.7
3-Isoquinolinamine, N-[2-[(3S,4R)-3-fluoro-4-methoxy-1-piperidinyl]-4-pyrimidinyl]-5-(1-methylethyl)-8-[(2R,3S)-2-methyl-3- [(methylsulfonyl)methyl]-1-azetidinyl]-
N-{2-[(3S,4R)-3-fluoro-4-methoxypiperidin-1-yl]pyrimidin-4-yl}-8-{(2R,3S)-3-[(methanesulfonyl)methyl]-2-methylazetidin-1-yl}-5-(propan-2-yl)isoquinolin-3-amine
N-[2-[(3S,4R)-3-fluoro-4-methoxypiperidin-1-yl]pyrimidin-4-yl]-8-[(2R,3S)-2-methyl-3-(methylsulfonylmethyl)azetidin-1-yl]-5-propan-2-ylisoquinolin-3-amine
N-(2-((3S,4R)-3-fluoro-4-methoxypiperidin-l-yl)pyrimidin-4-yl)-5-isopropyl-8-(3-(methylsulfonylmethyl)azetidin-l-yl)isoquinolin-3-amine
epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor, antineoplastic, PA4PTH5HL9, BLU 945
Tigozertinib (BLU-945) is currently under investigation in clinical trial NCT04862780 (Phase 1/2 Study Targeting EGFR Resistance Mechanisms in NSCLC) for the treatment of NSCLC.
Tigozertinib is a fourth-generation, orally bioavailable, mutant-selective, epidermal growth factor receptor (EGFR) inhibitor, with potential antineoplastic activity. Upon oral administration, tigozertinib targets, binds to and inhibits the activity of EGFR with C797S triple mutations including ex19del/T790M/C797S and L858R/T790M/C797S, thereby preventing EGFR-mediated signaling. This may both induce cell death and inhibit tumor growth in EGFR-overexpressing tumor cells. EGFR, a receptor tyrosine kinase mutated in many tumor cell types, plays a key role in tumor cell proliferation and tumor vascularization. BLU-945 inhibits mutated forms of EGFR with C797S mutation, which prevents covalent bond formation with third-generation EGFR inhibitors leading to drug resistance. BLU-945 may have enhanced anti-tumor effects in tumors with C797S-mediated resistance when compared to other EGFR tyrosine kinase inhibitors.Tigozertinib is a fourth-generation, orally bioavailable, mutant-selective, epidermal growth factor receptor (EGFR) inhibitor, with potential antineoplastic activity. Upon oral administration, tigozertinib targets, binds to and inhibits the activity of EGFR with C797S triple mutations including ex19del/T790M/C797S and L858R/T790M/C797S, thereby preventing EGFR-mediated signaling. This may both induce cell death and inhibit tumor growth in EGFR-overexpressing tumor cells. EGFR, a receptor tyrosine kinase mutated in many tumor cell types, plays a key role in tumor cell proliferation and tumor vascularization. BLU-945 inhibits mutated forms of EGFR with C797S mutation, which prevents covalent bond formation with third-generation EGFR inhibitors leading to drug resistance. BLU-945 may have enhanced anti-tumor effects in tumors with C797S-mediated resistance when compared to other EGFR tyrosine kinase inhibitors.
- First-in-Human, Phase 1b/2a Trial of a Multipeptide Therapeutic Vaccine in Patients With Progressive GlioblastomaCTID: NCT04116658Phase: Phase 1/Phase 2Status: CompletedDate: 2025-11-28
- (SYMPHONY) Phase 1/2 Study Targeting EGFR Resistance Mechanisms in NSCLCCTID: NCT04862780Phase: Phase 1Status: TerminatedDate: 2025-02-10
- A Novel Therapeutic Vaccine (EO2401) in Metastatic Adrenocortical Carcinoma, or Malignant Pheochromocytoma/ParagangliomaCTID: NCT04187404Phase: Phase 1/Phase 2Status: TerminatedDate: 2024-11-12
REF
- Synthesis of BLU-945, an EGFR Mutant Inhibitor for Treatment-Resistant Non-Small-Cell Lung CancerPublication Name: SynfactsPublication Date: 2022-09-20DOI: 10.1055/s-0041-1738705
- Discovery of BLU-945, a Reversible, Potent, and Wild-Type-Sparing Next-Generation EGFR Mutant Inhibitor for Treatment-Resistant Non-Small-Cell Lung CancerPublication Name: Journal of Medicinal ChemistryPublication Date: 2022-07-15PMCID: PMC9340769PMID: 35838760DOI: 10.1021/acs.jmedchem.2c00704
- Effects of antithymocyte serum on lymph node cells participating in the graft-vs-host reactionPublication Name: Cellular ImmunologyPublication Date: 1976-06-01PMID: 7359DOI: 10.1016/0008-8749(76)90132-5
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021133809&_cid=P11-MJ29N8-15768-1
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021133809&_cid=P11-MJ292P-02302-1
Example 5, Compound 117: Synthesis of (3S,4R)-3-fluoro-l-(4-(5-isopropyl-8-((2R,3S)-2-methyl-3-(methylsulfonylmethyl)azetidin-l-yl)isoquinolin-3-ylamino)pyrimidin-2-yl)-4-methylpiperidin-4-ol
To a solution of 3-chloro-8-[(2R,3S)-3-(methanesulfonylmethyl)-2-methylazetidin-l-yl]-5-(propan-2-yl)isoquinoline(28 g,76.3mmol, from step 1 of Example 3), (3R,4S)-l-(4-aminopyrimidin-2-yl)-3-fluoro-4-methylpiperidin-4-ol(17.2g,76.3mmol, peak 1 from Example B12), CS2CO3 (49.8 g, 152 mmol),C-phos (4.27 g, 9.15mmol, 2-dicyclohexylphosphino-2’,6’-bis(N,N-dimethylamino)biphenyl) and Pd2(dba)3 (3.94 g, 3.81 mmol) in dioxane (400 mL) was heated to 100 °C for 16 h under N2 atmosphere. The mixture reaction was filtered and the filtrate was concentration under vacuum. The residue was applied onto a silica gel column with EA/PE (2: 1) to give product 28.8 g (67%) as a light-yellow solid.
OTHERS

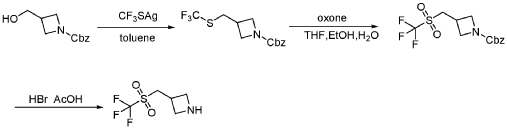
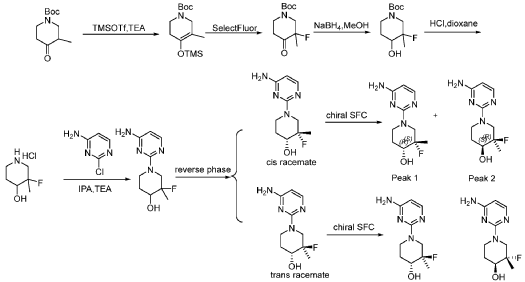
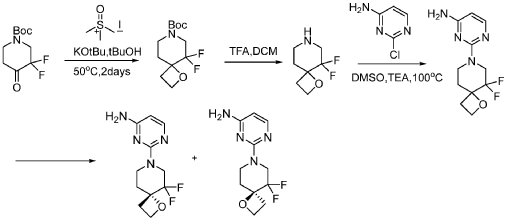
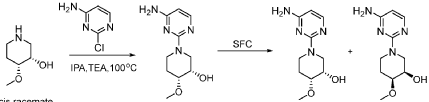
PAT
- Crystalline forms of cftr modulatorsPublication Number: US-2025051326-A1
- Small molecule inhibitors of dyrk/clk and uses thereofPublication Number: US-2025051325-A1
- RNAi agents for inhibiting expression of HIF-2 alpha (EPAS1), compositions thereof, and methods of usePublication Number: US-12221610-B2Grant Date: 2025-02-11
- Silicomagnesium-aluminate-hydrate gel and method of preparing the samePublication Number: US-3476692-APriority Date: 1966-02-04Grant Date: 1969-11-04
- InsecticidesPublication Number: DE-1542900-A1Priority Date: 1963-06-22
- Manufacture of conditioning compound for ground meat productsPublication Number: US-2634212-APriority Date: 1951-05-04Grant Date: 1953-04-07
- Kras modulators and uses thereofPublication Number: US-2025051365-A1
- Inhibitors of the myst family of lysine acetyl transferasesPublication Number: US-2025051343-A1
- Novel acid compoundsPublication Number: US-3945994-APriority Date: 1973-02-08Grant Date: 1976-03-23
- Production method of new phosphate ester compoundPublication Number: JP-S5930718-B2Priority Date: 1971-08-17
- Connection of a semiconductor device with a power sourcePublication Number: DE-2015923-A1Priority Date: 1969-04-23
- Ultrasonic doppler instrumentPublication Number: US-3732532-APriority Date: 1967-08-03Grant Date: 1973-05-08
- Electromechanical transducerPublication Number: US-3396285-APriority Date: 1966-08-10Grant Date: 1968-08-06
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////Tigozertinib, antineoplastic, PA4PTH5HL9, BLU 945

{kind=link}
{kind=link}