
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 55)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
VX 148
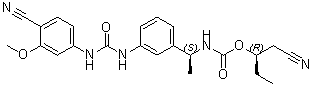
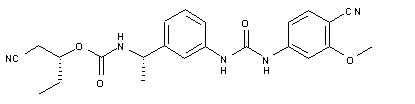
VX 148
297730-05-3
Name: VX-148
CAS#: 297730-05-3
Chemical Formula: C23H25N5O4
Exact Mass: 435.19065
Molecular Weight: 435.48
Elemental Analysis: C, 63.44; H, 5.79; N, 16.08; O, 14.70
| Molecular Weight | 435.48 |
| Formula | C23H25N5O4 |
| CAS No. | 297730-05-3 (VX 148); |
| Chemical Name | Carbamic acid, N-[(1S)-1-[3-[[[(4-cyano-3-methoxyphenyl)amino]carbonyl]amino]phenyl]ethyl]-, (1R)-1-(cyanomethyl)propyl ester |
- OriginatorVertex Pharmaceuticals
- ClassAntipsoriatics
- Mechanism of ActionInosine monophosphate dehydrogenase inhibitors
- DiscontinuedPsoriasis; Transplant rejection; Viral infections
- 13 Nov 2003Interim data from a media release have been added to the adverse events and Skin Disorders therapeutic trials sections
- 23 May 2003Vertex Pharmaceuticals has completed enrolment in a phase IIa trial for Psoriasis in Iceland
- 24 Dec 2002Phase-II clinical trials in Psoriasis in Iceland (unspecified route)
VX-148 is a second-generation, orally administered inhibitor of inosine monophosphate dehydrogenase (IMPDH). The IMPDH enzyme plays a key role in regulating immune response and proliferation of specific cell types, including lymphocytes. VX-148 is a developed for the treatment of autoimmune diseases.
Investigated for use/treatment in autoimmune diseases, psoriasis and psoriatic disorders, and viral infection.
VX-148 is a novel, uncompetitive IMPDH inhibitor with a K(i) value of 6 nM against IMPDH type II enzyme. VX-148 is slightly more potent than mycophenolic acid and VX-497 in inhibiting the proliferation of mitogen-stimulated primary human lymphocytes (IC(50) value of ~80 nM). The inhibitory activity of VX-148 is alleviated in the presence of exogenous guanosine. VX-148 does not inhibit proliferation of nonlymphoid cell types such as fibroblasts, indicating selectivity for inhibition of IMPDH activity. VX-148 is orally bioavailable in rats and mice; oral administration of VX-148 inhibits primary antibody response in mice in a dose-dependent manner with an ED(50) value of 38 mg/kg b.i.d. VX-148 significantly prolongs skin graft survival at 100 mg/kg b.i.d. in mice.
SYN
WO 0056331
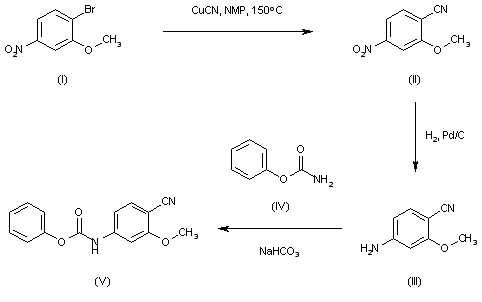
The intermediate carbamate (V) has been obtained as follows. The reaction of 4-bromo-3-methoxynitrobenzene (I) with CuCN in NMP at 150 C gives 2-methoxy-4-nitrobenzonitrile (II), which is reduced with H2 over Pd/C in ethyl acetate to yield 4-amino-2-methoxybenzonitrile (III). Finally, this compound is condensed with phenyl carbamate (IV) by means of NaHCO3 in ethyl acetate to afford the desired carbamate intermediate (V).
SYN
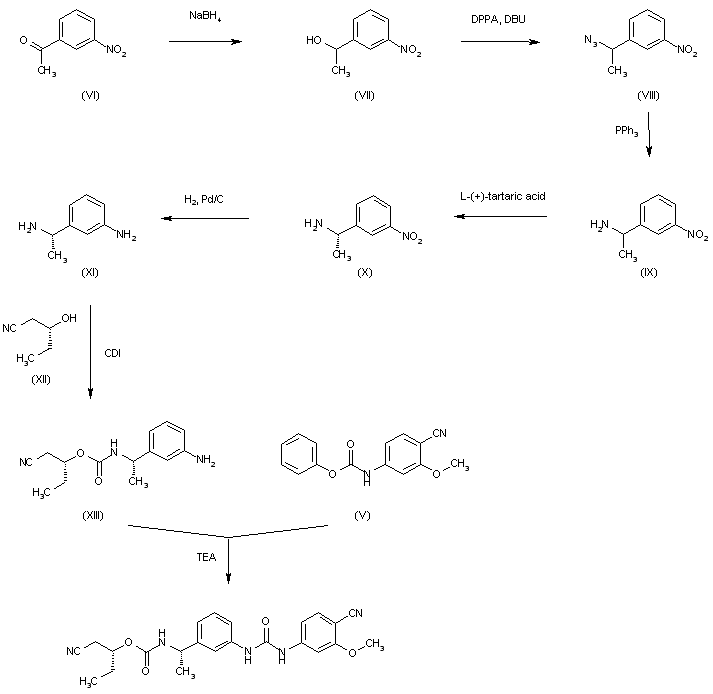
The reduction of 3-nitroacetophenone (VI) by means of NaBH4 in ethanol gives 1-(3-nitrophenyl)ethanol (VII), which is treated with DPPA and DBU in hot toluene to yield the azido derivative (VIII). The reduction of (VIII) with PPh3 in THF/water affords 1-(3-nitrophenyl)ethylamine (IX) as a racemic mixture that is submitted to optical resolution with L-(+)-tartaric acid to provide the desired (S)-isomer (X). The reduction of the nitro group of (X) by means of H2 over Pd/C in methanol gives 1(S)-(3-aminophenyl)ethylamine (XI), which is condensed with 2(R)-hydroxypentanenitrile (XII) and CDI to yield the carbamate (XIII). Finally, this compound is condensed with intermediate carbamate (V) by means of TEA in hot ethyl acetate to afford the target urea.
- Jain J, Almquist SJ, Heiser AD, Shlyakhter D, Leon E, Memmott C, Moody CS, Nimmesgern E, Decker C: Characterization of pharmacological efficacy of VX-148, a new, potent immunosuppressive inosine 5′-monophosphate dehydrogenase inhibitor. J Pharmacol Exp Ther. 2002 Sep;302(3):1272-7. [Article]
////////////VX 148, phase 2
O=C(O[C@H](CC)CC#N)N[C@H](C1=CC=CC(NC(NC2=CC=C(C#N)C(OC)=C2)=O)=C1)C

NEW DRUG APPROVALS
one time
$10.00
VX- ? (3-[2-(4-fluorophenyl)-lH-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2- oxo-pyrrolidin-3-yl ] propanamide)
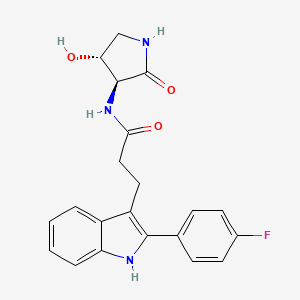
VX- ?
CAS 2446817-72-5
HYDRATE 2446818-26-2
Acetic acid, 1-methylethyl ester 2446818-27-3
C21 H20 F N3 O3, 381.4
1H-Indole-3-propanamide, 2-(4-fluorophenyl)-N-[(3S,4R)-4-hydroxy-2-oxo-3-pyrrolidinyl]-
3-[2-(4-fluorophenyl)-lH-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2- oxo-pyrrolidin-3-yl ] propanamide
use in treating focal segmental glomerulosclerosis (FSGS) and/or non-diabetic kidney disease (NDKD).

NEW DRUG APPROVALS
one time
$10.00
PATENT
SOLID FORMS OF APOL1 INHIBITOR AND METHODS OF USING SAME
Compound I is disclosed as Compound 87 in U.S. Provisional Application No.62/780,667 filed on December 17, 2018, U.S. Application No. 16/717,099 filed onDecember 17, 2019, and PCT International Application No. PCT/US2019/066746 filed on December 17, 2019, the entire contents of each of which are incorporated herein by reference.
Compound I, which can be employed in the treatment of diseases mediated by APOLl, such as FSGS and NDKD
Example 1. Synthesis of Compound
Preparation of Compound I and Forms Thereof
Compound I Compound I /‘– PrOAc solvate Form A
n-pentanol/
n-heptane
Compound I
Form B
Step 1. Synthesis of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid (C101)
[00156] To a mixture of C104 (100.0 g, 1.0 equiv) and phenyl hydrazine hydrochloride (72.2 g, 1.05 eqiv) was charged AcOH (800 mL, 8 vol). The mixture was agitated and heated to 85 °C for 16 hours. The batch was cooled to 22 °C. A vacuum was applied and the batch distill at <70 °C to ~3 total volumes. The batch was cooled to 19- 25 °C. The reactor was charged with iPrOAc (800 mL, 8 vol) and then charged with water (800 mL, 8 vol). The internal temperature was adjusted to 20 – 25 °C and the biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and the phases allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. 1 N HC1 (500 mL, 5 vol) was charged to the reactor. The internal temperature was adjusted to 20 – 25 °C, and the
biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and phases were allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. The reactor was charged with 1 N HC1 (500 mL, 5 vol). The internal temperature was adjusted to 20 – 25 °C, and the biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and phases were allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. Water (500 mL, 5 vol) was charged to the reactor. The internal temperature was adjusted to 20 – 25 °C, and the biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and phases were allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. Water (500 mL, 5 vol) was charged to the reactor. The internal temperature was adjusted to 20 – 25 °C, and the biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and phases were allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. The organic phase was distilled under vacuum at <75 °C to 3 total volumes. The reactor was charged with toluene (1000 mL, 10 vol). The organic phase was distilled under vacuum at <75 °C to 5 total volumes. The reactor was charged with toluene (1000 mL, 10 vol). The organic phase was distilled under vacuum at <75 °C to 5 total volumes. The resulting slurry was heated to an internal temperature of 85 °C until complete dissolution of solids was achieved. The mixture was allowed to stir for 0.5 h at 85 °C and then cooled to an internal temperature of 19 – 25 °C over 5 h. The mixture was allowed to stir at 25 °C for no less than 2 h. The slurry was filtered. The filter cake was washed with toluene (1 x 2 vol (200 mL) and 1 x 1.5 vol (150 mL)). The solids were dried under vacuum with nitrogen bleed at 60 °C to afford product C101 (95.03 g, 70%).
Step 2. Synthesis of Compound I
[00157] A mixture of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid C101 (50 g, 1.0 equiv), S2 hydrochloride (28.3 g, 1.05 equiv), and CDMT (34.1 g, 1.1 equiv) was charged with 2-MeTHF (200 mL, 4 vol) and DMF (50 mL, 1 vol) and the mixture was agitated. The internal temperature adjusted to <13 °C. The reactor was charged with NMM (64.5 g, 3.5 equiv) over 1 h, while maintaining internal temperature <20 °C. The internal temperature was adjusted to 25 °C and the batch was stirred at that temperature for 14 h. The batch was cooled to 10 °C and charged with water (250 mL, 5 vol) while keeping the internal temperature <20 °C. The batch was then warmed to 20 – 25 °C. Stirring was stopped, and the phases allowed to separate for 10 min. The lower aqueous phase was removed. The aqueous layer was back extracted with 2-MeTHF (2 x 200 mL, 2 x 4 vol) at
20 – 25 °C. The combined organic phases were washed with 1 N HC1 (500 mL, 10 vol) at 20 – 25 °C by mixing for 10 min and settling for 10 min. The lower aqueous phase was removed. The organic phases were washed with 0.25 N HC1 (2 x 250 mL, 2 x 5 vol) at 20 – 25 °C by mixing for 10 min and settling for 10 min for each wash. Lower aqueous phases were removed after each wash. The organic phase was washed with water (250 mL, 5 vol) at 20 – 25 °C by mixing for 10 min and settling for 10 min. The reactor was charged with 20 wt % Nuchar RGC® and stirred for 4 h. The reaction mixture was filtered through a pad of celite®. The reactor and celite® pad were rinsed with 2-MeTHF. The combined organics were distilled under vacuum at <50 °C to 5 total volumes. The reactor was charged with iPrOAc (500 mL, 10 vol). The organic phase was distilled under vacuum at <50 °C to 5 total volumes. The mixture was charged with additional iPrOAc (400 mL, 8 vol) and distillation under vacuum was repeated. The mixture was charged with additional iPrOAc (250 mL, 5 vol), heated to an internal temperature of 75 °C and stirred for 5 h. The slurry was cooled to 25 °C, over 5 h and stirred for no less than 12 h. The slurry was filtered and the filter cake washed with iPrOAc (2 x 50 mL, 2 x 1 vol). The solids were dried under vacuum with nitrogen bleed at 55 – 60 °C to afford Compound I as an iPrOAc solvate (60.38 g including 9.9% w/w iPrOAc, 80.8% yield).
Recrystallization to Form A of Compound I
[00158] Compound I as an iPrOAc solvate (17.16 g after correction for iPrOAc content, 1.0 equiv) was charged to a reactor. A mixture of IP A (77 mL, 4.5 vol) and water (137 mL, 8 vol) were charged to the reactor. The slurry was heated to an internal temperature of 75 °C. The batch was cooled to an internal temperature of 25 °C over 10 h and then stirred at 25 °C for at least 12 h. The slurry was filtered. The filter cake was washed with 36/64 IP A/water (2 x 52 mL, 2 x 3 vol). The solids were dried under vacuum with nitrogen bleed at 60 °C to afford Compound I as a neat, crystalline form (Form A, 15.35 g, 89%).
[00159] The X-ray powder diffractogram of Compound I Form A (FIG. 50) was acquired at room temperature using a PANalytical Empyrean diffractometer equipped with PIXcel ID detector. The peaks are listed in Table A below.
Table A. XRPD of Form A of Compound I
|
I
PATENT
- WO2020131807
Alternative Preparation I of Compound 87 (Indole preparation route C)
Step 1. Synthesis of 2-(4-fluorophenyl)-lH-indole (C98)
[00401] To a stirred suspension of indole (5 g, 42.7 mmol) and (4- fluorophenyl)boronic acid (8.96 g, 64.0 mmol) in AcOH (200 mL) was
added Pd(OAc)2.Trimer (1.44 g, 6.4 mmol) and the mixture stirred at room temperature for 16 h under 02-balloon pressure. Then the reaction mixture was filtered through a Celite® pad, washed with EtOAc (500 mL). The filtrates were washed with water, sat. NaHC03 solution, brine solution, then dried over Na2S04 and concentrated under reduced pressure. Purification by silica gel chromatography (Gradient: 0-10 % EtOAc in heptane) yielded the product afforded 2-(4-fluorophenyl)-lH-indole (5.5 g, 61 %). ‘H NMR (300 MHz, DMSO-de) 5 11.51 (s, 1H), 7.9 (t, J = 5.4 Hz, 2H), 7.52 (d, J = 7.8 Hz, 1H), 7.39 (d, J = 8.1 Hz, 1H), 7.30 (t, J = 8.7 Hz, 2H), 7.09 (t, J = 12 Hz, 1H), 6.99 (t, J = 7.5 Hz, 1H), 6.86 (s, 1H). LCMS m/z 212.4 [M+H]+.
Step 2. Synthesis of methyl (E)-3-[2-(4-fluorophenyl)-lH-indol-3-yl]prop-2-enoate (C99)
[00402] 2-(4-fluorophenyl)-lH-indole (1.0 g, 4.76 mmol) and methyl 3,3-dimethoxypropanoate (0.81 mL, 5.7 mmol) were suspended in dichloromethane (15 mL). Trifluoroacetic acid (2.00 mL, 26 mmol) was added rapidly via syringe, resulting in a clear brown solution. The reaction mixture was heated to 40 °C for three hours. The reaction was diluted with dichloromethane (15 mL) to give an amber solution which was washed with saturated aqueous NaHCCh (25 mL) to yield a bright yellow/light amber biphasic mixture. The phases were separated and the organic layer was washed with saturated NaHCCh (30 mL), then dried (MgSCh) and filtered. The mixture was concentrated under a nitrogen stream overnight. The crude product was obtained as a yellow powder. The product was dissolved in minimum 2-MeTHF and pentane added until the suspension became lightly cloudy. The suspension was allowed to stand overnight, and the precipitate was filtered off. The filter cake was washed with heptane (2 x 15 mL), and dried in vacuo at 40 °C to afford the product as a yellow powder. Methyl (E)-3-[2-(4-fluorophenyl)-lH-indol-3-yl]prop-2-enoate (1.30 g, 86 %). ¾ NMR (300 MHz, Chloroform -if) d 8.41 (s, 1H), 8.01 – 7.95 (m, 1H), 7.92 (d, J = 16.0 Hz,
1H), 7.58 – 7.50 (m, 2H), 7.46 – 7.41 (m, 1H), 7.33 – 7.27 (m, 2H), 7.22 (t, J = 8.6 Hz, 2H), 6.59 (d, J = 16.0 Hz, 1H), 3.79 (s, 3H). LCMS m/z 295.97 [M+H]+.
Step 3. Synthesis of methyl 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoate (CIOO)
[00403] To a solution of methyl (E)-3-[2-(4-fluorophenyl)-lH-indol-3-yl]prop-2-enoate (7 g, 0.02 mol) in EtOAc (350 mL) was added Palladium on carbon (4 g, 10 %w/w, 0.004 mol) and stirred at room temperature for 2 h under an atmosphere of H2 (bladder pressure). The reaction mixture was filtered through a pad of Celite® and washed with EtOAc (400 mL). The filtrates was concentrated to afford methyl 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoate (7.1 g, 100 %). 1H MR (300 MHz, DMSO-<fc) 5 11.2 (s, 1H), 7.65 (q, J = 5.4 Hz, 2H), 7.54 (d, J = 8.1 Hz, 1H), 7.36 (t, J = 9.0 Hz, 3H), 7.10 (t, J = 8.1 Hz, 1H), 7.02 (t, J = 7.8 Hz, 1H), 3.53 (s, 3H), 3.10 (t, J = 15.9 Hz, 2H), 2.63 (t, J = 15.9 Hz, 2H). LCMS m/z 298.21 [M+H]+. The product was used directly in the subsequent step without further purification.
Step 4. Synthesis of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid (C101)
[00404] To stirred solution of methyl 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoate (14.4 g, 0.05mol) in THF (300 mL), MeOH (300 mL) and H2O (250 mL) was cooled to -10°C. LiOH.H20 (10.1 g, 0.24 mol) was slowly added in a portion-wise manner. The reaction mixture was allowed to stir at room temperature for 16 h. The mixture was
evaporated and ice cold water (200 mL) was added, pH was adjusted to pH- 2 with 1M HC1 (400 mL, Cold solution). The mixture was stirred for 10 minutes, filtered and dried to afford 3-[2-(4-fhiorophenyl)-lH-indol-3-yl]propanoic acid (12.9 g, 94 %). ‘H NMR (400 MHz, DMSCMJ) 5 12.11 (s, 1H), 11.18 (s, 1H), 7.65 (q, J = 5.2 Hz, 2H), 7.56 (d, J = 7.6 Hz, 1H), 7.36 (t, J = 8.8 Hz, 3H), 7.10 (t, J = 8 Hz, 1H), 7.01 (t, J = 8 Hz, 1H), 3.06 (t, J = 16.4 Hz, 2H), 2.55 (t, J = 16 Hz, 2H). LCMS m/z 284.21 [M+H]+.
Step 5. Synthesis of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2- oxo-pyrrolidin-3-yl ] propanamide (87)
[00405] A mixture of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid C101 (40 g, 120.0 mmol) and (3S,4R)-3-amino-4-hydroxy-pyrrolidin-2-one (Hydrochloride salt) S2 (23.8 g, 156.0 mmol) in DMF (270 mL) was stirred at room temperature for 5 minutes. CDMT (27.2 g, 154.9 mmol) and NMM (53 mL, 482.1 mmol) were added and the mixture was stirred at room temperature for 2 h. The mixture was poured into water (140 mL) and then stirred for 1 h at room temperature, then filtered and washing the solids with water (50 mL). The solids were dissolved in 1 : 1 IP A/water (-400 mL, until all solids dissolved) with heating (reflux) and stirring. The mixture was allowed to cool slowly to room temperature overnight. The mixture was cooled to 0 oC and stirred to break up crystals for filtration. The crystals were then filtered off, rinsed with cold 1 : 1 IP A/water to afford a tan solid (45 g). The solid was dissolved in IPA (200 mL) and heated to 80 °C to dissolve the solid. Activated charcoal (10 g) was added and the mixture was heated with stirring for 30 minutes. The mixture was filtered through Celite ® and solvent removed under reduced pressure. A mixture of 40:60 IP A/water (350 mL) was added to the solid and the mixture was heated until all solids dissolved. The mixture was cooled to room temperature over 5 h. Solids precipitated within the mixture. The mixture was then cooled to 0 °C and stirred for 1 h. The solids were filtered off and air dried on funnel for 1 h, then in a vacuum at 55 °C overnight to afford the product. 3-[2-(4-fluorophenyl)-lH-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2-oxo-pyrrolidin-3-yl]propanamide (36.6 g, 79 %). ¾ NMR (300 MHz, Methanol-i¾) d 7.63 (ddt, J= 8.6, 5.1, 2.7 Hz, 3H), 7.35 (dt, J= 8.1, 1.0 Hz, 1H), 7.25 – 7.16 (m, 2H), 7.11 (ddd, J= 8.1, 7.0, 1.3 Hz, 1H), 7.03 (ddd, J = 8.0, 7.0, 1.2 Hz, 1H), 4.34 (td, J= 7.6, 6.8 Hz, 1H), 4.22 (d, J= 7.7 Hz, 1H), 3.55 (dd, J= 9.9, 7.5 Hz, 1H), 3.26 – 3.18 (m, 2H), 3.10 (dd, J= 9.9, 6.8 Hz, 1H), 2.69 – 2.59 (m, 2H). LCMS m/z 382.05 [M+H]+. The
product contained 0.23 % IPA by weight by NMR (1439 ppm IPA by residual solvent analysis). Purity is 99.5 % by (qNMR).
Alternative Preparation II of Compound 87 ( Indole Preparation route D)
Step 1. Synthesis of 5-(4-fluorophenyl)-5-oxo-pentanoic acid (Cl 04)
[00406] To a stirred suspension of AlCb(13.9 g, 0.10 mol) in dichloromethane (50 mL) was added a solution of tetrahydropyran-2,6-dione (5.93 g, 0.05
mol) in dichloromethane (100 mL) at 0 °C over a period of 15 minutes and stirred for 30 min. Then to the reaction mixture was added fluorobenzene (5 g, 0.05 mol) at 0 °C over a period of 15 min, gradually allowed to room temperature and stirred for 16 h. Then the reaction mixture was added to ice water (50 mL) under stirring. The resulting solid was filtered to afford a light yellow solid. The solid was diluted with 3 % NaOH solution (50 mL) and dichloromethane (50 mL). The aqueous layer was separated and acidified with IN HC1 at 0 °C. The mixture was then extracted with EtOAc (100 mL), dried over Na2SC>4, and concentrated under reduced pressure. The solid was then washed with pentane and dried to afford 5-(4-fluorophenyl)-5-oxo-pentanoic acid as an off white solid. (6 g, 53 %). ¾ NMR (300 MHz, DMSO-^) d 12.07 (s, 1H), 8.06 (d, J = 6 Hz, 1H), 8.02 (d, J = 5.4 Hz, 1H), 7.36 (t, J = 8.7 Hz, 2H), 3.06 (t, J = 12 Hz,
2H), 2.31 (t, J = 7.2 Hz, 2H), 1.86-1.78 (m, 2H). LCMS m/z 211.18 [M+H]+.
Step 2. Synthesis of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid (Cl 01) [00407] Phenylhydrazine (Hydrochloride salt) (375.7 g, 2.6 mol) was combined with the 5-(4-fluorophenyl)-5-oxo-pentanoic acid (507.7 g, 2.4 mol) in a 12 L three-necked round-bottomed flask equipped with an overhead stirrer, temperature probe, and reflux condenser. AcOH (5 L) was added. The stirring was initiated and ZnCk (605 g, 4.44 mol) was added. The white suspension rapidly thickened after a few minutes (due to formation of the hydrazine intermediate). Approx. 500 mL of extra AcOH was added to aid stirring. The reaction was then heated to 100 °C for three hours. The reaction was cooled to room temperature and poured into water (approx. 6 L). The mixture was extracted with EtOAc (approx 8 L). The extract was washed with water, dried
(MgS04), filtered, and evaporated in vacuo to afford a golden yellow solid. The solid was triturated with approx. 4 L of 10 % EtOAc/DCM and filtered. The filter cake was washed with 50 % dichloromethane/heptane (approx 1 L). The filter cake was dissolved in 40 % EtOAc/dichloromethane (approx. 2L) and filtered over a plug of silica gel. The plug was eluted with 40 % EtOAc/ dichloromethane until the product had been eluted (checked by TLC (25 % EtOAc/ dichloromethane)). The filtrate was evaporated in vacuo to afford 382.6 g of an off-white solid (Crop 1). All filtrates were combined and evaporated in vacuo. The remaining solid was dissolved in 10 %
EtOAc/dichloromethane (approx. 1 L) and chromatographed on a 3 kg silica gel cartridge on the ISCO Torrent (isocratic gradient of 10 % EtOAc/dichloromethane). Product fractions were combined and evaporated in vacuo to afford a yellow solid that was slurried with dichloromethane, cooled under a stream of nitrogen, and filtered. The filter cake was washed with 50 % dichloromethane/heptane and dried in vacuo to afford 244.2 g of product (Crop 2). Altogether, both crops afforded 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid (626.8 g, 93 %). ¾ NMR (300 MHz, DMSO-i/e) d 12.15 (s, 1H), 11.20 (s, 1H), 7.74 – 7.62 (m, 2H), 7.57 (d, J = 7.8 Hz, 1H), 7.47 – 7.28 (m, 3H), 7.11 (ddd, J = 8.1, 7.0, 1.2 Hz, 1H), 7.02 (ddd, J = 7.9, 7.0, 1.1 Hz, 1H), 3.17 – 2.85 (m, 2H), 2.61 – 2.52 (m, 2H) ppm. 19F NMR (282 MHz, DMSO-i/e) d -114.53 ppm. LCMS m/z 284.15 [M+H]+.
Step 3. Synthesis of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2- oxo-pyrrolidin-3-yl ] propanamide (87)
[00408] A 3-L three neck RBF under nitrogen was equipped with a 150 mL addition funnel and thermocouple, then loaded with 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid (77.2 g, 228.6 mmol), (3S,4R)-3-amino-4-hydroxy-pyrrolidin-2-one
(Hydrochloride salt) (36.6 g, 239.9 mmol) and CDMT (44.2 g, 251.7 mmol). DMF (320 mL) was added and the orange slurry was cooled to -5 °C (acetone/brine/dry ice). NMM (88 mL, 800.4 mmol) was added via a funnel over 75 minutes to keep the internal temp <0 °C. The slurry was stirred at between -10 and 0 °C for 1 hour, then allowed to warm to ambient temperature progressively over 2 hours. Additional reagents were added (10 % of the initial quantities), and the mixture was stirred overnight at ambient temperature. Water (850 mL) was added over 60 minutes, maintaining the internal temperature at <25 °C (ice bath). This slow water addition allows for complete dissolution of any visible salt before precipitation of the product. The resulting thick slurry was stirred at ambient temperature overnight. The solid was recovered by filtration and washed with water (3 x 500 mL). The solid was dried under a stream of air at ambient temperature, then purified by crystallization.
Crystallization of 3- [2-( 4-fluorophenyl)-lH-indol-3-yl ]-N-[ ( 3S, 4R)-4-hydroxy-2-oxo- pyrrolidin-3-yl ] propanamide (87)
[00409] Under nitrogen atmosphere, a 2-L, 3 -neck flask equipped with addition funnel and thermocouple was charged with a light brown suspension of the crude 3-[2-(4-fluorophenyl)-lH-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2-oxo-pyrrolidin-3-yljpropanamide (89.5 g) in IPA (225 mL, 2.5 vol). The slurry was heated to 50 °C and water (675 mL, 7.5 vol) was added until near-complete dissolution of solid was observed. The temperature was adjusted to 70 °C-to achieve full dissolution, yielding a clear amber solution. After 30 minutes, the heat source was removed and the mixture was cooled to ambient temperature over the weekend, stirring gently while maintaining the nitrogen atmosphere. The solid was recovered by filtration, washed with IPA:H20 = 1 :2 (2 x 300 mL, 2 x 3.3 vol) dried under a stream of air overnight to afford the product. 3-[2-(4-fluorophenyl)-lH-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2-oxo-pyrrolidin-3-yl]propanamide (84.8 g, 92 %). ¾ NMR (300 MHz, DMSO-^) d 11.19 (s, 1H), 8.23 (d, J= 7.5 Hz, 1H), 7.77 (s, 1H), 7.72 – 7.63 (m, 2H), 7.60 (d, J= 7.8 Hz, 1H), 7.41 -7.31 (m, 3H), 7.12 (ddd, J= 8.1, 7.0, 1.2 Hz, 1H), 7.03 (ddd, J= 8.0, 7.0, 1.1 Hz, 1H), 5.49 (d, J= 5.0 Hz, 1H), 4.20 – 4.06 (m, 2H), 3.38 (s, 1H), 3.11 – 3.00 (m, 2H), 2.92 (dd, J= 9.4, 6.6 Hz, 1H). LCMS m/z 382.15 [M+H]+.
Crystallization of 3- [2-( 4-fluorophenyl)-lH-indol-3-yl J-N-[ ( 3S, 4R)-4-hydroxy-2-oxo- pyrrolidin-3-yl ] propanamide (87)
[00410] A 2-L, 3-neck flask equipped with addition funnel and thermocouple was charged with a light brown suspension of the crude 3-[2-(4-fluorophenyl)-lH-indol-3- yl]-N-[(3S,4R)-4-hydroxy-2-oxo-pyrrolidin-3-yl]propanamide in IPA (225 mL, 1 vol). The slurry was heated to 50 °C and water (675 mL, 3 vol) was added until near- complete dissolution of solid observed (mL). Temperature was increased to 70 °C under nitrogen (full dissolution, yielding a clear amber solution). After 30 minutes, the heat was removed and the mixture cooled to ambient temperature over the weekend, stirring gently under nitrogen atmosphere. The solid was recovered by filtration and washed with IPAiLLO = 1 :2 (2 x 300 mL).The solid was dried under a stream of air overnight to afford the product. 3-[2-(4-fluorophenyl)-lH-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2-oxo- pyrrolidin-3-yl]propanamide (84.8 g, 92 %). ¾ NMR (300 MHz, DMSO-i/e) d 11.19 (s, 1H), 8.23 (d, J= 7.5 Hz, 1H), 7.77 (s, 1H), 7.72 – 7.63 (m, 2H), 7.60 (d, J= 7.8 Hz,
1H), 7.41 – 7.31 (m, 3H), 7.12 (ddd, J= 8.1, 7.0, 1.2 Hz, 1H), 7.03 (ddd, 7= 8.0, 7.0,
1.1 Hz, 1H), 5.49 (d, J= 5.0 Hz, 1H), 4.20 – 4.06 (m, 2H), 3.38 (s, 1H), 3.11 – 3.00 (m, 2H), 2.92 (dd, J= 9.4, 6.6 Hz, 1H). LCMS m/z 382.15 [M+H]+.
Large Scale Preparation of Compound 87
/- PrOAc solvate Form A
Step 1. Synthesis of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid (C101)
[00411] To a mixture of C104 (100.0 g, 1.0 equiv) and phenyl hydrazine hydrochloride (72.2 g, 1.05 eqiv) was charged AcOH (800 mL, 8 vol). The mixture was agitated and heated to 85 °C for 16 hours. The batch was cooled to 22 °C. A vacuum was applied and the batch distill at <70°C to ~3 total volumes. The batch was cooled to 19- 25 °C. The reactor was charged with iPrOAc (800 mL, 8 vol) and then charged with water (800 mL, 8 vol). The internal temperature was adjusted to 20 – 25 °C and the biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and the phases allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. 1 N HC1 (500 mL, 5 vol) was charged to the reactor. The internal temperature was adjusted to 20 – 25 °C, and the biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and phases were allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. The reactor was charged with 1 N HC1 (500 mL, 5 vol). The internal temperature was adjusted to 20 – 25 °C, and the biphasic mixture was stirred for no less than 0.5 h.
Stirring was stopped and phases were allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. Water (500 mL, 5 vol) was charged to the reactor.
The internal temperature was adjusted to 20 – 25 °C, and the biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and phases were allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. Water (500 mL, 5 vol) was charged to the reactor. The internal temperature was adjusted to 20 – 25 °C, and the biphasic mixture was stirred for no less than 0.5 h. Stirring was stopped and phases were allowed to separate for no less than 0.5 h. The lower aqueous layer was removed. The organic phase was distilled under vacuum at <75 °C to 3 total volumes. The reactor was charged with toluene (1000 mL, 10 vol). The organic phase was distilled under vacuum at <75 °C to 5 total volumes. The reactor was charged with toluene (1000 mL, 10 vol). The organic phase was distilled under vacuum at <75 °C to 5 total volumes. The resulting slurry was heated to an internal temperature of 85 °C until complete dissolution of solids was achieved. The mixture was allowed to stir for 0.5 h at 85 °C and then cooled to an internal temperature of 19 – 25 °C over 5 h. The mixture was allowed to stir at 25 °C for no less than 2 h. The slurry was filtered. The filter cake was washed with toluene (1 x 2 vol (200 mL) and 1 x 1.5 vol (150 mL)). The solids were dried under vacuum with nitrogen bleed at 60 °C to afford product C101 (95.03 g, 70%).
Purification of Compound 87 by Recrystallization to Form A
[00412] Compound 87 as an iPrOAc solvate (17.16 g after correction for iPrOAc content, 1.0 equiv) was charged to a reactor. A mixture of IP A (77 mL, 4.5 vol) and water (137 mL, 8 vol) were charged to the reactor. The slurry was heated to an internal temperature of 75 °C. The batch was cooled to an internal temperature of 25 °C over 10 h and then stirred at 25 °C for at least 12 h. The slurry was filtered. The filter cake was washed with 36/64 IP A/water (2 x 52 mL, 2 x 3 vol). The solids were dried under vacuum with nitrogen bleed at 60 °C to afford Compound 87 as a neat, crystalline form (Form A, 15.35 g, 89%).
Synthetic Procedure
[00413] A mixture of 3-[2-(4-fluorophenyl)-lH-indol-3-yl]propanoic acid C101 (50 g, 1.0 equiv), S2 hydrochloride (28.3 g, 1.05 equiv), and CDMT (34.1 g, 1.1 equiv) was charged with 2-MeTHF (200 mL, 4 vol) and DMF (50 mL, 1 vol) and the mixture was agitated. The internal temperature adjusted to <13 °C. The reactor was charged with NMM (64.5 g, 3.5 equiv) over 1 h, while maintaining internal temperature <20 °C. The internal temperature was adjusted to 25 °C and the batch was stirred at that temperature for 14 h. The batch was cooled to 10 °C and charged with water (250 mL, 5 vol) while keeping the internal temperature <20 °C. The batch was then warmed to 20 – 25 °C. Stirring was stopped, and the phases allowed to separate for 10 min. The lower aqueous phase was removed. The aqueous layer was back extracted with 2-MeTHF (2 x 200 mL, 2 x 4 vol) at 20 – 25 °C. The combined organic phases were washed with 1 N HC1 (500 mL, 10 vol) at 20 – 25 °C by mixing for 10 min and settling for 10 min. The lower aqueous phase was removed. The organic phases were washed with 0.25 N HC1 (2 x 250 mL, 2 x 5 vol) at 20 – 25 °C by mixing for 10 min and settling for 10 min for each wash. Lower aqueous phases were removed after each wash. The organic phase was washed with water (250 mL, 5 vol) at 20 – 25 °C by mixing for 10 min and settling for 10 min. The reactor was charged with 20 wt % Nuchar RGC® and stirred for 4 h. The reaction mixture was filtered through a pad of celite®. The reactor and celite® pad were rinsed with 2-MeTHF. The combined organics were distilled under vacuum at <50 °C to 5 total volumes. The reactor was charged with iPrOAc (500 mL, 10 vol). The organic phase was distilled under vacuum at <50 °C to 5 total volumes. The mixture was charged with additional iPrOAc (400 mL, 8 vol) and distillation under vacuum was repeated. The mixture was charged with additional iPrOAc (250 mL, 5 vol), heated to an internal
temperature of 75 °C and stirred for 5 h. The slurry was cooled to 25 °C, over 5 h and stirred for no less than 12 h. The slurry was filtered and the filter cake washed with iPrOAc (2 x 50 mL, 2 x 1 vol). The solids were dried under vacuum with nitrogen bleed at 55 – 60 °C to afford Compound 87 as an iPrOAc solvate (60.38 g including 9.9% w/w iPrOAc, 80.8% yield).
Form A of Compound 87
[00414] Compound 87 hydrate form was converted to the dehydrated, neat crystalline form (Form A) after drying.
Hydrate Form A of Compound 87
[00415] A mixture of IP A (4.5 vol) and water (8 vol) was added to compound 87
(iPrOAc solvate containing ~2.5 – 11 wt% iPrOAc, 1.0 equiv). The slurry was heated to an internal temperature of 75 °C and filtered hot. The filtrate was cooled to 25 °C for at least 12 h. The slurry was filtered. The filter cake was washed with 36/64 IP A/water (2 x 3 vol). The solids were dried under vacuum with nitrogen bleed at 55 – 60 °C. The product was isolated as Hydrate form.
IPAC Solvate of Compound 87:
[00416] The large scale synthesis described above provided an iPrOAc solvate containing ~2.5 – 11 wt% iPrOAc after drying.
Amorphous Form of Compound 87
[00417] ~lg of compound 87 was dissolved in 22mL of acetone. The solution was evaporated using a Genevac. The resulted solid was dried at 60C under vacuum overnight. The dried solid was amorphous form.
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| WO-2020131807-A1 | Inhibitors of apol1 and methods of using same | 2018-12-17 | |
| US-2020377479-A1 | Inhibitors of apol1 and methods of using same | 2018-12-17 |
///////////
O=C(N[C@@H]1C(=O)NC[C@H]1O)CCc1c2ccccc2[NH]c1c1ccc(F)cc1
SIMILAR

predicted
VX 147
cas 2446816-88-0 predicted
O=C(N[C@@H]1C(=O)NC[C@H]1O)CCc1c2cc(F)cc(F)c2[NH]c1c1ccc(F)cc1
- OriginatorVertex Pharmaceuticals
- ClassSmall molecules; Urologics
- Mechanism of ActionApolipoprotein L1 inhibitors
- Orphan Drug StatusNo
- New Molecular EntityYes
Highest Development Phases
- Phase IIFocal segmental glomerulosclerosis
- Phase IKidney disorders
Most Recent Events
- 14 Apr 2020Phase-II clinical trials in Focal segmental glomerulosclerosis in USA (PO) (EudraCT2020-000185-42) (NCT04340362)
- 31 Dec 2019Vertex Pharmaceuticals completes phase I clinical trial in Focal segmental glomerulosclerosis and Kidney disorders (In volunteers) in USA (PO)
- 05 Aug 2019Vertex Pharmaceuticals plans a phase II proof-of-concept trial for focal segmental glomerulosclerosis in 2020
| NCT Number ICMJE | NCT04340362 |
|---|---|
| Other Study ID Numbers ICMJE | VX19-147-101 2020-000185-42 ( EudraCT Number ) |
PROPOFOL
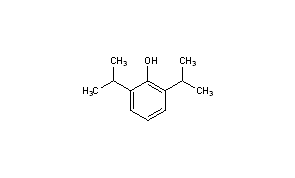
PropofolCAS Registry Number: 2078-54-8
CAS Name: 2,6-Bis(1-methylethyl)phenolAdditional Names: 2,6-diisopropylphenol; disoprofol
Manufacturers’ Codes: ICI-35868
Trademarks: Ansiven (Abbott); Diprivan (AstraZeneca); Disoprivan (AstraZeneca); Rapinovet (Schering-Plough Vet.)Molecular Formula: C12H18OMolecular Weight: 178.27Percent Composition: C 80.85%, H 10.18%, O 8.97%
Literature References: Prepn: A. J. Kolka et al.,J. Org. Chem.21, 712 (1956); 22, 642 (1957); G. G. Ecke, A. J. Kolka, US2831898 (1958 to Ethyl Corp.); T. J. Kealy, D. D. Coffman, J. Org. Chem.26, 987 (1961); B. E. Firth, T. J. Rosen, US4447657 (1984 to Universal Oil Products). Chromatographic study: J. K. Carlton, W. C. Bradbury, J. Am. Chem. Soc.78, 1069 (1956). Animal studies: J. B. Glen, Br. J. Anaesth.52, 731 (1980).Pharmacokinetics: H. K. Adam et al.,ibid. 743; idem,ibid.55, 97 (1983). Determn in blood: eidem,J. Chromatogr.223, 232 (1981). Comparative studies vs other injectable anesthetics: B. Kay, D. K. Stephenson, Anaesthesia35, 1182 (1980); D. V. Rutter et al.,ibid. 1188. Use in i.v. anesthesia: E. Major et al.,ibid.37, 541 (1982). Cardiovascular effects: D. Al-Khudhairi et al.,ibid. 1007. Pharmacology of emulsion formulation: J. B. Glen, S. C. Hunter, Br. J. Anaesth.56, 617 (1984). Series of articles on pharmacology and clinical experience: Postgrad. Med. J.61, Suppl. 3, 1-169 (1985).
Properties: bp30 136°. bp17 126°. mp 19°. nD20 1.5134. nD25 1.5111. d20 0.955.Melting point: mp 19°Boiling point: bp30 136°; bp17 126°Index of refraction:nD20 1.5134; nD25 1.5111Density: d20 0.955Therap-Cat: Anesthetic (intravenous).Therap-Cat-Vet: Intravenous anesthetic (dogs and cats).Keywords: Anesthetic (Intravenous).SYN

Prepn: A. J. Kolka et al., J. Org. Chem. 21, 712 (1956); 22, 642 (1957); G. G. Ecke, A. J. Kolka, US 2831898 (1958 to Ethyl Corp.); T. J. Kealy, D. D. Coffman, J. Org. Chem. 26, 987 (1961); B. E. Firth, T. J. Rosen, US 4447657 (1984 to Universal Oil Products).SYN

SYNhttps://pubs.acs.org/doi/pdf/10.1021/op400300t

A commercially viable manufacturing process for propofol (1) is described. The process avoids acid–base neutralization events during isolation of intermediate, 2,6-di-isopropylbenzoic acid (3) and crude propofol, and thus simplifies the synthesis on industrial scale to a considerable extent. Syntheses of five impurities/related substances (USP and EP) are also described.


SYN

SYN

Propofol is used during surgeries for sedation and an injectable grade with purity > 99.90% is desired by the medical community. An embodiment of the present invention provides an economically feasible, industrial process for the manufacture of high purity injectable grade Propofol. An embodiment of the present invention relates to a process and novel strategy for purification of 2,6-diisopropylphenol (Propofol) and similar products.
[0003] Propofol is a sterile injectable drug that appears in the USP, EP and IP Monographs. Drug product is manufactured by using high purity drug substance 2,6-di-isopropylphenol commonly known as Propofol.
[0004] Propofol is used to put patients to sleep and keep them asleep during general anesthesia for surgery or other medical procedures. It is used in adults as well as children 2 months and older. Propofol is frequently used as a sedative, and has a rapid onset of action and a short recovery period. Propofol slows the activity of brain and nervous system. Propofol is also used to sedate a patient who is under critical care and needs a mechanical ventilator (breathing machine). Propofol is a hypnotic alkylphenol derivative. When formulated for intravenous induction of sedation and hypnosis during anaesthesia, Propofol facilitates inhibitory neurotransmission mediated by gamma- Aminobutyric acid (GABA). Propofol is associated with minimal respiratory depression and has a short half-life with a duration of action of 2 to 10 minutes.
[0005] Propofol is commonly used parenteral anesthetic agent in the United States, extensively used for minor and outpatient surgical procedures because of its rapid onset and reversal of action, and in intensive care units (ICUs) for maintaining coma. Propofol has been associated with rare instances of idiosyncratic acute liver injury; in addition, prolonged high dose Propofol therapy can cause the “Propofol infusion syndrome” which is marked by brady arrhythmias, metabolic acidosis, rhabdomyolysis, hyperlipidemia and an enlarged or fatty liver.
[0006] Friedel-Craft’s alkylation of phenol using propylene gas in the presence of Lewis acid (LA) catalysts is a commonly used method for the synthesis of Propofol and is well documented in a number of publications and patents [Ecke, G. G., Kolka, A. J. US 2,831,898 A, 1958. Firth, B. E., Rosen, T. J. US 4,447,657, 1984. Akio, T., Yoshiaki, I., Hidekichi, H., Kiyoji, K., Takashi, K., Masanobu, M. EP 0169359A1, 1986. Ecke, G. G., Kolka, A. J. US 3,271,314, 1966. Napolitano, J. P. US 3,367,981 A, 1968. Goddard L. E. US 3,766,276, 1973. Firth, B. E. US 4,275,248, 1981, etc.]
[0007] A number of patents and published literature describe the manufacture of Propofol. US. Pat. No. 4,275,248; W0200034218; EP169359; US. Pat. No. 3,367,981; US. Pat. No.
3,271,314; US. Pat. No. 3,766,276; US. Pat. No. 2,831,898; US.Pat.No.2,207,753; GB1318100; U.S. Pat. No. 4,391,998; US. Pat. No. 4,774, 368; US. Pat. No. 5,589,598; US. Pat. No. 6,362,234; etc. EP 0511947, discloses purification of Propofol that is obtained by alkylation of phenol and purified by crystallization at -10 to -20°C (melting point of Propofol is 18°C). This patent also describes purification using non-polar solvents such as Petroleum ether or Hexane, where solvent residue is removed by distillation or evaporation and finally Propofol is obtained using fractional distillation under high vacuum.
[0008] Continuous separation of a mixture of Propofol with phenolic impurities and methanol is described in an U.S. Pat. No. 5,264,085. U.S. Pat. No. 5,705,039 describes the purification of impure 2,6-diisopropylphenol first using continuous distillation and then distilling pure Propofol under high vacuum.
[0009] Patent CN103360219A describes purification wherein 2,6-diisopropyl phenol is reacted with benzoyl chloride to generate ‘benzoic acid-2, 6-diisopropyl benzene ester’, which is then purified to yield Propofol. The patent discloses that an adsorbent is added at the rectifying stage, so that impurities with similar chemical structures and boiling points are effectively removed; the content of a single impurity in the product is not higher than 0.01%; the total impurity is not higher than 0.05%.
[0010] CN105601477A describes purification of Propofol wherein crude Propofol is purified with three-stage distillation method; the crude Propofol enters feeding tank protected by nitrogen and is charged into first-stage film distillation system through pump; then the product is fed to second-stage molecular distillation system and low boiling point impurities are removed; finally, the processed product is charged into third-stage molecular distiller through a pump, high-boiling-point impurities are separated, and the colourless or yellowish high-purity Propofol is obtained.
[0011] In another prior art disclosure, after completion of the reaction, the final product is isolated and purified by high-vacuum distillation. Alkylation of phenol using propylene gas at high pressure and high temperature is reported. Several impurities like 2,4-diisopropyl and 2,4,6-triisopropyl phenol are the major side products along with the corresponding Isopropyl ether. All these impurities need to be controlled at a limit of NMT 0.05% or less in the final API for it to be pharmaceutically acceptable. In another prior art disclosure, isopropanol was used as the propylating agent instead of direct propylene gas. In this method propylene is generated in situ using IPA and strong acid like sulfuric acid and catalysts like Aluminoslicate [See Baltalksne, A. E.; Zitsmanis, A. H. SU 443019, 1974. Jain, K. P., Edaki, D. U., Minhas H. S., Minhas G. S. WO/2011/ 161687 Al, 2011. Wu, M. US 4,391,998, 1983]
[0012] Another method is to use of protected phenol, where 4-chloro or 4-carboxylic acid substituted phenol is used as starting material along with Isopropanol in sulfuric acid, followed by removal of the 4-substituent to give Propofol [Baltalksne, A. E.; Zitsmanis, A. H. SU 443019, 1974. Jain, K. P., Edaki, D. U., Minhas H. S., Minhas G. S. WO/2011/ 161687 Al, 2011. Wu, M. US 4,391,998, 1983. Tsutsumi, S.; Yoshizawa, T.; Koyama, K. Nippon Kagaku Zasshi 1956, 77, 737-738. Paiocchi, M. US 5,589,598, 1996. Nieminen, K., Essen, P. US 5,175,376, 1992. Keller, S., Schlegel, J. WO/2012/152665 Al, 2012.] The final purification is carried out by high- vacuum distillation to get highly pure Propofol. Since the para position is blocked, related impurities such as 2,4-isopropyl and 2,4,6-triisopropyl derivatives are avoided. In this approach, intermediate is purified before converting to crude Propofol using either de-chlorination by hydrogenation or de-carboxylation before vacuum distillation for final purification.
[0013] It is reported in the literature that 4-hydroxybenzoic acid is used as starting material for alkylation with isopropyl alcohol in sulfuric acid. In that method 2,6-diisopropyl-4-hydroxy benzoic acid gets formed, which is extracted in toluene either in presence of an acid or the impurities are extracted in toluene under alkaline condition. The decarboxylation is carried out using solvents like monoethylene glycol or ethoxyethanol at high temperature. At the end of decarboxylation, crude Propofol is isolated by extracting into toluene. The advantage is Propofol does not form sodium salt under the conditions, but all other acidic impurities form sodium salt and thus do not get extracted in toluene. The toluene containing Crude Propofol is distilled to recover toluene and then vacuum distilled to obtain pure Propofol. [Chen, T; Chen, X.; Bois-Choussy, M.; Zhu, J. J. Am. Chem. Soc. 2006, 128, 87-89. Lau, S.; Keay, B. Can. J. Chem. 2001, 79, 1541-1545]
[0014] In summary, strategies disclosed in prior art for the production of 2,6-diisopropylphenol (Propofol) predominantly involve synthesis starting from phenol or by using protected 4-position of phenol like, 4-hydroxybenzoic acid, 4-chlorophenol (references: Baltalksne, A. E.; Zitsmanis, A. H. SU 443019, 1974. Jain, K. P., Edaki, D. U., Minhas H. S., Minhas G. S. WO/2011/ 161687 Al, 2011. Wu, M. US 4,391,998, 1983. Tsutsumi, S.; Yoshizawa, T.; Koyama, K. Nippon Kagaku Zasshi 1956, 77, 737-738. Paiocchi, M. US 5,589,598, 1996. Nieminen, K., Essen, P. US 5,175,376, 1992. Keller, S., Schlegel, J. WO/2012/152665 Al, 2012). Processes described in the literature generally propose purification of crude 2,6-diisopropylphenol by ‘high vacuum distillation or molecular distillation’.
[0015] The phenols are susceptible to oxidation, formation of polymeric and color impurities. There are processes where repeated vacuum distillation has been carried out to obtain desired purity of product. Sometimes, to reduce the oxidized and colored impurities, reduction of impurities by catalytic hydrogenation is also used.
[0016] Propofol that does not meet Pharmaceutical grade may be manufactured by several processes generally known to persons of skill in the art and described in prior art, but purification of Propofol to consistently achieve high purity required for the injectable drug substance using an economical and industrial process remains a challenge.
Example 1:
[0033] Commercially available concentrated sulfuric acid (30 Kg) was diluted with water (2.26 Kg) at low temperature (0-15°C). Methyl 4-hydroxybenzoate (5 Kg 32.79 mol.) was added to this diluted sulfuric acid at 5 to 10 °C with stirring. After complete addition, isopropyl alcohol (5.9 Kg 98.16 mol.) was gradually added to the reaction content, controlling the temperature at 0-15 °C. The reaction mixture was then heated at 60-70°C and continued to complete di-isopropylation and ester hydrolysis to yield methyl-4-hydroxybenzoate. The conversion was checked on TLC or by HPLC for the complete conversion of methyl-4 hydroxybenzoate to 3, 5 -Diisopropyl 4-hydroxybenzoic acid.
[0034] The reaction contents were cooled at room temperature and carefully charged into a stirred, precooled mixture of water (50 L) and Toluene (40 L) at (0 to 5°C). The mixture was stirred and maintained below 15°C for about 30 to 60 minutes.
[0035] The content was then heated at 25 to 30°C, stirred for 30 min., allowed to settle into two layers. The water layer was extracted again with toluene and discarded. The toluene layers, containing the product 3, 5-Diisopropyl 4-hydroxybenzoic acid, were combined and extracted with about 25 L of 10 % NaOH. The aqueous layer containing the sodium salt of 3, 5 -Di-isopropyl 4-hydroxybenzoic acid was acidified with concentrated HC1 (about 9 Kg) to precipitate 3, 5-Diisopropyl 4-hydroxybenzoic acid, filtered, and washed with water (about 50 L) to yield 3, 5 -diisopropyl 4-hydroxybenzoic acid (about 45-60 %)
[0036] To the mixture of 3, 5-diisopropyl 4-hydroxybenzoic acid (3 Kg, 13.5 mol.) in ethylene glycol (5.0 Kg, 80.55 mol.) was added sodium hydroxide (1.25 Kg, 31.25 mol.) for decarboxylation. The reaction mixture was heated at 145 ± 5°C till completion of
decarboxylation by monitoring using TLC or HPLC (or solubility in bicarbonate of precipitated product). After complete decarboxylation, the reaction mixture was cooled at 40 to 45 °C, under nitrogen environment and diluted with water (about 15 L) and allowed to settle. The oily product layer was separated and washed with water (6L) to isolate crude Propofol (i.e., 2,6-diisopropyl phenol) and stored under nitrogen. The isolated volatile Crude Propofol (along with carry over ppm ethylene glycol and NaOH) was then subjected to steam distillation purification process as described below.
[0037] The Crude Propofol is purified by using one of the steam distillation processes as described below.
[0038] The Crude Propofol layer is added to purified water in a reactor (preferably glass lined reactor), and slowly heated to boiling to co-distil Pure Propofol along with water under normal atmospheric pressure and the high volatile initial fraction is isolated first. The biphasic layers of main distillate, are separated and the liquid layer of Propofol is treated with dehydrating agent to absorb dissolved moisture in Pure Propofol under nitrogen or argon. The transparent Pure Propofol liquid layer is then filtered through ultrafme Micron filter (0.45 and 0.2 micron) under nitrogen (or argon) pressure and isolated in controlled environment to obtain pharmaceutical injectable grade Pure Propofol of more than 99.90% purity.
[0039] The Crude Propofol liquid layer is charged into a reactor with steam distillation arrangement, like steam purging dip tube, column, heat exchanger and receivers. Pure steam is purged in the reactor at controlled pressure to co-distil Pure Propofol with water. The layers are allowed settle and water layer is kept aside for recirculation. The transparent Pure Propofol transparent liquid layer is then filtered through ultrafme Micron filter (0.45 and 0.2 micron) under nitrogen (or argon) pressure and isolated in controlled environment to obtain pharmaceutical injectable grade Pure Propofol of more than 99.90% purity.
[0040] The Crude Propofol layer is added to purified water in a reactor (preferably glass lined or Hastelloy reactor) and slowly heated at boiling to co-distil Pure Propofol along with water under mild vacuum. The biphasic layers are separated and the liquid layer of Propofol is treated with dehydrating agent to absorb dissolved moisture in Pure Propofol under nitrogen (or argon). The transparent Pure Propofol liquid layer is then filtered through ultrafme Micron filter (0.45 and 0.2 micron) under nitrogen (or argon) pressure and isolated in controlled environment to obtain pharmaceutical injectable grade Pure Propofol of more than 99.90% purity.
[0041] The Crude Propofol layer is added to reactor containing purified water and 0.1 to 1% antioxidant and 0.1 to 0.5% sodium hydroxide and slowly heated to boiling to co-distil Pure Propofol along with water. The biphasic layers are separated and the liquid layer of Propofol is treated or passed through column packed with dehydrating agent to absorb dissolved moisture in Pure Propofol. The transparent Pure Propofol liquid layer is then filtered through ultrafme Micron filter (0.45 and 0.2 micron) under nitrogen (or argon) pressure and isolated in controlled environment to obtain pharmaceutical injectable grade Pure Propofol of more than 99.90% purity.
[0042] The crude Propofol liquid layer is treated with preferably neutral or basic activated carbon (about 2-5%) and filtered under nitrogen. The filtered liquid is collected, under nitrogen, in distillation reactor containing purified water is slowly heated to boiling to co-distil Pure Propofol along with water under normal pressure or mild vacuum. The co-distilled biphasic layers are separated and the liquid layer of Propofol, is treated under nitrogen, with or passed through column packed with dehydrating agent to absorb dissolved moisture trapped in Pure Propofol. The transparent Pure Propofol liquid layer is then filtered through ultrafme Micron filter (0.45 and 0.2 micron) under nitrogen (or argon) pressure and isolated in controlled environment to obtain pharmaceutical injectable grade Pure Propofol of more than 99.90% purity.
Example No. 2:
[0043] Friedel-Crafts reaction was performed as described in Example 1. Decarboxylation was performed by using KOH instead of NaOH by following the same procedure as described in Example 1.
Example No. 3:
[0044] Decarboxylation was performed as per operations described in Example 1. After complete decarboxylation, the reaction mixture was cooled at 40 to 45°C, under nitrogen environment and diluted with water (about 15 L) The biphasic mixture subjected to steam distillation by any of the purification methods described in Example 1.
Example No. 4:
[0045] Friedel-Crafts reaction was performed as described in Example 1. The reaction contents were cooled at room temperature and carefully charged at 0 to 5°C into a sodium hydroxide solution to basic pH at stirred. The aqueous alkaline solution was extracted twice with toluene or hexane. The aqueous layer was acidified with HC1 to precipitate 3, 5-diisopropyl-4-hydroxybenzoic acid. The wet product was washed with water, dried and decarboxylated using sodium hydroxide in ethylene glycol as solvent at 145±5°C. The reaction contents were cooled to room temperature, diluted with water and acidified and then Crude Propofol was extracted twice in toluene. The toluene layer was washed with water, bicarbonate and with water then distilled to obtain crude oily layer of Propofol (>99% pure). This Crude Propofol was then purified by using purification steam distillation by any of the purification methods described in Example 1.
Example 5:
[0046] Continuous steam distillation of crude Propofol by purging pure steam. Continuous steam distillation of Crude Propofol was carried out using a feed pump for feeding liquid Crude Propofol (prepared by one of the processes described in this application or other literature) to the steam distillation system connected to a pure steam generator. Steam at 1-10 kg pressure was purged in the steam distillation system at controlled rate and the co-distilled Pure Propofol with water was cooled using heat exchanger and continuous separator. The residue was discharged from bottom valve at defined time intervals. The water layer was recycled to steam generator and Pure Propofol was dehydrated, filtered and collected in controlled environment as described in Example 1.
Propofol, marketed as Diprivan, among other names, is a short-acting medication that results in a decreased level of consciousness and a lack of memory for events.[4] Its uses include the starting and maintenance of general anesthesia, sedation for mechanically ventilated adults, and procedural sedation.[4] It is also used for status epilepticus if other medications have not worked.[4] It is given by injection into a vein, and the maximum effect takes about two minutes to occur and typically lasts five to ten minutes.[4] Propofol is also used for medical assistance in dying in Canada.[5]
Common side effects of propofol include an irregular heart rate, low blood pressure, a burning sensation at the site of injection and the cessation of breathing.[4] Other serious side effects may include seizures, infections due to improper use, addiction, and propofol infusion syndrome with long-term use.[4] The medication appears to be safe for use during pregnancy but has not been well studied for use in this case.[4] It is not recommended for use during a cesarean section.[4] It is not a pain medication, so opioids such as morphine may also be used,[6] however whether or not they are always needed is not clear.[7] Propofol is believed to work at least partly via a receptor for GABA.[4]
Propofol was discovered in 1977 and approved for use in the United States in 1989.[4][8] It is on the World Health Organization’s List of Essential Medicines[9] and is available as a generic medication.[4] It has been referred to as milk of amnesia (a play on “milk of magnesia“), because of the milk-like appearance of the intravenous preparation, and because of its tendency to suppress memory recall.[10][11] Propofol is also used in veterinary medicine for anesthesia.[12][13]
Medical uses
Anesthesia
To induce general anesthesia, propofol is the drug used almost always, having largely replaced sodium thiopental.[14][6] It can also be administered as part of an anesthesia maintenance technique called total intravenous anesthesia, using either manually programmed infusion pumps or computer-controlled infusion pumps in a process called target controlled infusion (TCI). Propofol is also used to sedate individuals who are receiving mechanical ventilation but not undergoing surgery, such as patients in the intensive care unit.[15][16] In critically ill patients, propofol is superior to lorazepam both in effectiveness and overall cost.[17] Propofol is relatively inexpensive compared to medications of similar use due to shorter ICU stay length.[17] One of the reasons propofol is thought to be more effective (although it has a longer half-life than lorazepam) is because studies have found that benzodiazepines like midazolam and lorazepam tend to accumulate in critically ill patients, prolonging sedation.[17] Propofol has also been suggested as a sleep aid in critically ill adults in the ICU, however, the effectiveness of this medicine at replicating the mental and physical aspects of sleep for people in the ICU are not clear.[16]
Propofol is often used instead of sodium thiopental for starting anesthesia because recovery from propofol is more rapid and “clear”.[citation needed]
Propofol can be run through a peripheral IV or central line. Propofol is frequently paired with fentanyl (for pain relief) in intubated and sedated people.[18] Both are compatible in IV form.[18]
Procedural sedation
Propofol is also used for procedural sedation. Its use in these settings results in a faster recovery compared to midazolam.[19] It can also be combined with opioids or benzodiazepines.[20][21][22] Because of its rapid induction and recovery time, propofol is also widely used for sedation of infants and children undergoing MRI.[23] It is also often used in combination with ketamine with minimal side effects.[24]
COVID-19
In March 2021, the U.S. Food and Drug Administration (FDA) issued an emergency use authorization (EUA) for Propofol‐Lipuro 1% to maintain sedation via continuous infusion in people greater than age sixteen with suspected or confirmed COVID‑19 who require mechanical ventilation in an intensive care unit ICU setting.[25][26][27][28] In the circumstances of this public health emergency, it would not be feasible to require healthcare providers to seek to limit Fresenius Propoven 2% Emulsion or Propofol-Lipuro 1% only to be used for patients with suspected or confirmed COVID‑19; therefore, this authorization does not limit use to such patients.[28]
Other uses
Executions
The US state of Missouri added propofol to its execution protocol in April 2012. However, Governor Jay Nixon halted the first execution by the administration of a lethal dose of propofol in October 2013 following threats from the European Union to limit the drug’s export if it were used for that purpose.[29][30] The United Kingdom had already banned the export of medicines or veterinary medicines containing propofol to the United States.[31]
Recreational use
Recreational use of the drug via self-administration has been reported[32][33] but is relatively rare due to its potency and the level of monitoring required for safe use.[citation needed] Critically, a steep dose-response curve makes recreational use of propofol very dangerous, and deaths from self-administration continue to be reported.[34][35] The short-term effects sought via recreational use include mild euphoria, hallucinations, and disinhibition.[36][37]
Recreational use of the drug has been described among medical staff, such as anesthetists who have access to the drug.[38][39] It is reportedly more common among anesthetists on rotations with short rest periods, as usage generally produces a well-rested feeling.[40] Long-term use has been reported to result in addiction.[38][41]
Attention to the risks of off-label use of propofol increased in August 2009 due to the Los Angeles County coroner’s conclusion that music icon Michael Jackson died from a mixture of propofol and the benzodiazepine drugs lorazepam, midazolam, and diazepam on June 25, 2009.[42][43][44][45] According to a July 22, 2009 search warrant affidavit unsealed by the district court of Harris County, Texas, Jackson’s physician, Conrad Murray, administered 25 milligrams of propofol diluted with lidocaine shortly before Jackson’s death.[43][44][46] Even so, as of 2016, propofol was not on a US Drug Enforcement Administration schedule.[40][47]
Side effects
One of propofol’s most common side effects is pain on injection, especially in smaller veins. This pain arises from activation of the pain receptor, TRPA1,[48] found on sensory nerves and can be mitigated by pretreatment with lidocaine.[49] Less pain is experienced when infused at a slower rate in a large vein (antecubital fossa). Patients show considerable variability in their response to propofol, at times showing profound sedation with small doses.
Additional side effects include low blood pressure related to vasodilation, transient apnea following induction doses, and cerebrovascular effects. Propofol has more pronounced hemodynamic effects relative to many intravenous anesthetic agents.[50] Reports of blood pressure drops of 30% or more are thought to be at least partially due to inhibition of sympathetic nerve activity.[51] This effect is related to the dose and rate of propofol administration. It may also be potentiated by opioid analgesics.[52] Propofol can also cause decreased systemic vascular resistance, myocardial blood flow, and oxygen consumption, possibly through direct vasodilation.[53] There are also reports that it may cause green discolouration of the urine.[54]
Although propofol is heavily used in the adult ICU setting, the side effects associated with propofol seem to be of greater concern in children. In the 1990s, multiple reported deaths of children in ICUs associated with propofol sedation prompted the FDA to issue a warning.[55]
As a respiratory depressant, propofol frequently produces apnea. The persistence of apnea can depend on factors such as premedication, dose administered, and rate of administration, and may sometimes persist for longer than 60 seconds.[56] Possibly as the result of depression of the central inspiratory drive, propofol may produce significant decreases in respiratory rate, minute volume, tidal volume, mean inspiratory flow rate, and functional residual capacity.[50]
Diminishing cerebral blood flow, cerebral metabolic oxygen consumption, and intracranial pressure are also characteristics of propofol administration.[57] In addition, propofol may decrease intraocular pressure by as much as 50% in patients with normal intraocular pressure.[58]
A more serious but rare side effect is dystonia.[59] Mild myoclonic movements are common, as with other intravenous hypnotic agents. Propofol appears to be safe for use in porphyria, and has not been known to trigger malignant hyperpyrexia.[citation needed]
Propofol is also reported to induce priapism in some individuals,[60][61] and has been observed to suppress REM sleep stage and to worsen the poor sleep quality in some patients.[62]
As with any other general anesthetic agent, propofol should be administered only where appropriately trained staff and facilities for monitoring are available, as well as proper airway management, a supply of supplemental oxygen, artificial ventilation, and cardiovascular resuscitation.[63]
Because of its lipid base, some hospital facilities require the IV tubing (of continuous propofol infusions) to be changed after 12 hours. This is a preventive measure against microbial growth and infection.[64]
Propofol infusion syndrome
Main article: Propofol infusion syndrome
A rare, but serious, side effect is propofol infusion syndrome. This potentially lethal metabolic derangement has been reported in critically ill patients after a prolonged infusion of high-dose propofol, sometimes in combination with catecholamines and/or corticosteroids.[65]
Interactions
The respiratory effects of propofol are increased if given with other respiratory depressants, including benzodiazepines.[66]
Pharmacology
Pharmacodynamics
Propofol has been proposed to have several mechanisms of action,[67][68][69] both through potentiation of GABAA receptor activity and therefore acting as a GABAA receptor positive allosteric modulator, thereby slowing the channel-closing time. At high doses, propofol may be able to activate GABAA receptors in the absence of GABA, behaving as a GABAA receptor agonist as well.[70][71][72] Propofol analogs have been shown to also act as sodium channel blockers.[73][74] Some research has also suggested that the endocannabinoid system may contribute significantly to propofol’s anesthetic action and to its unique properties.[75] EEG research upon those undergoing general anesthesia with propofol finds that it causes a prominent reduction in the brain’s information integration capacity.[76]
Pharmacokinetics

A 20 ml ampoule of 1% propofol emulsion, as sold in Australia by Sandoz
Propofol is highly protein-bound in vivo and is metabolised by conjugation in the liver.[77] The half-life of elimination of propofol has been estimated to be between 2 and 24 hours. However, its duration of clinical effect is much shorter, because propofol is rapidly distributed into peripheral tissues. When used for IV sedation, a single dose of propofol typically wears off within minutes. Propofol is versatile; the drug can be given for short or prolonged sedation, as well as for general anesthesia. Its use is not associated with nausea as is often seen with opioid medications. These characteristics of rapid onset and recovery along with its amnestic effects[78] have led to its widespread use for sedation and anesthesia.
History
John B. Glen, a veterinarian and researcher at Imperial Chemical Industries (ICI) spent 13 years developing propofol, an effort which led to the awarding to him of the prestigious 2018 Lasker Award for clinical research. Propofol was originally developed as ICI 35868. It was chosen for development after extensive evaluation and structure–activity relationship studies of the anesthetic potencies and pharmacokinetic profiles of a series of ortho-alkylated phenols.[79]
First identified as a drug candidate in 1973, clinical trials followed in 1977, using a form solubilised in cremophor EL.[80] However, due to anaphylactic reactions to cremophor, this formulation was withdrawn from the market and subsequently reformulated as an emulsion of a soya oil/propofol mixture in water. The emulsified formulation was relaunched in 1986 by ICI (now AstraZeneca) under the brand name Diprivan. The currently available preparation is 1% propofol, 10% soybean oil, and 1.2% purified egg phospholipid as an emulsifier, with 2.25% glycerol as a tonicity-adjusting agent, and sodium hydroxide to adjust the pH. Diprivan contains EDTA, a common chelation agent, that also acts alone (bacteriostatically against some bacteria) and synergistically with some other antimicrobial agents. Newer generic formulations contain sodium metabisulfite or benzyl alcohol as antimicrobial agents. Propofol emulsion is a highly opaque white fluid due to the scattering of light from the tiny (about 150-nm) oil droplets it contains: Tyndall Effect.
Developments
A water-soluble prodrug form, fospropofol, has been developed and tested with positive results. Fospropofol is rapidly broken down by the enzyme alkaline phosphatase to form propofol. Marketed as Lusedra, this formulation may not produce the pain at injection site that often occurs with the conventional form of the drug. The U.S. Food and Drug Administration (FDA) approved the product in 2008.[81] However fospropofol is a Schedule IV controlled substance with the DEA ACSCN of 2138 in the United States unlike propofol.[82]
By incorporation of an azobenzene unit, a photoswitchable version of propofol (AP2) was developed in 2012, that allows for optical control of GABAA receptors with light.[83] In 2013, a propofol binding site on mammalian GABAA receptors has been identified by photolabeling using a diazirine derivative.[84] Additionally, it was shown that the hyaluronan polymer present in the synovia can be protected from free-radical depolymerization by propofol.[85]

NEW DRUG APPROVALS
ONE TIME
$10.00
References
- ^ “Propofol”. Drugs.com. Retrieved 2 January 2019.
- ^ Ruffle JK (November 2014). “Molecular neurobiology of addiction: what’s all the (Δ)FosB about?”. Am J Drug Alcohol Abuse. 40 (6): 428–437. doi:10.3109/00952990.2014.933840. PMID 25083822. S2CID 19157711.
Propofol is a general anesthetic, however its abuse for recreational purpose has been documented (120). Using control drugs implicated in both ΔFosB induction and addiction (ethanol and nicotine), similar ΔFosB expression was apparent when propofol was given to rats. Moreover, this cascade was shown to act via the dopamine D1 receptor in the NAc, suggesting that propofol has abuse potential (119)
- ^ “Diprivan- propofol injection, emulsion”. DailyMed. Retrieved 17 April 2021.
- ^ Jump up to:a b c d e f g h i j k l m n “Propofol”. The American Society of Health-System Pharmacists. Archived from the original on 9 October 2016. Retrieved 21 January 2017.
- ^ Divisions of Family Practice Medical Assistance in Dying (MAiD): Protocols and Procedures Handbook.
- ^ Jump up to:a b Miner, JR; Burton, JH (August 2007). “Clinical practice advisory: Emergency department procedural sedation with propofol”. Annals of Emergency Medicine. 50 (2): 182–7. doi:10.1016/j.annemergmed.2006.12.017. PMID 17321006.
- ^ Wakai, A; Blackburn, C; McCabe, A; Reece, E; O’Connor, G; Glasheen, J; Staunton, P; Cronin, J; Sampson, C; McCoy, SC; O’Sullivan, R; Cummins, F (29 July 2015). “The use of propofol for procedural sedation in emergency departments”. The Cochrane Database of Systematic Reviews. 7 (7): CD007399. doi:10.1002/14651858.CD007399.pub2. PMC 6517206. PMID 26222247.
- ^ Miller’s Anesthesia (8 ed.). Elsevier Health Sciences. 2014. p. 920. ISBN 9780323280112.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ Euliano TY, JS (2004). “A brief pharmacology related to anesthesia”. Essential anesthesia: from science to practice. Cambridge, UK: Cambridge University Press. p. 173. ISBN 978-0-521-53600-4. Retrieved 2 June 2009.
- ^ MD, David M. Novick (2017). A Gastroenterologist’s Guide to Gut Health: Everything You Need to Know About Colonoscopy, Digestive Diseases, and Healthy Eating. Rowman & Littlefield. p. 15. ISBN 9781442271999.
- ^ “Anesthesia Medications”. Veterinary Dentistry for the Small Animal Technician. Hoboken: Wiley. 2013. ISBN 9781118694800.
- ^ “PropoFlo (propofol) for Animal Use”. Drugs.com. Retrieved 13 February 2019.
- ^ “Discovery and development of propofol, a widely used anesthetic”. The Lasker Foundation. Retrieved 8 September2020.
Propofol is used today to initiate anesthesia in nearly 100% of general anesthesia cases worldwide.
- ^ Barr, Juliana (1995). “Propofol”. International Anesthesiology Clinics. 33 (1): 131–154. doi:10.1097/00004311-199500000-00008. ISSN 0020-5907.
- ^ Jump up to:a b Lewis, Sharon R.; Schofield-Robinson, Oliver J.; Alderson, Phil; Smith, Andrew F. (8 January 2018). “Propofol for the promotion of sleep in adults in the intensive care unit”. The Cochrane Database of Systematic Reviews. 1: CD012454. doi:10.1002/14651858.CD012454.pub2. ISSN 1469-493X. PMC 6353271. PMID 29308828.
- ^ Jump up to:a b c Cox, CE; Reed, SD; Govert, JA; Rodgers, JE; Campbell-Bright, S; Kress, JP; Carson, SS (March 2008). “Economic evaluation of propofol and lorazepam for critically ill patients undergoing mechanical ventilation”. Crit Care Med. 36 (3): 706–14. doi:10.1097/CCM.0B013E3181544248. PMC 2763279. PMID 18176312.
- ^ Jump up to:a b Isert, Peter R.; Lee, Doris; Naidoo, Daya; Carasso, Melanie L.; Kennedy, Ross A. (June 1996). “Compatibility of propofol, fentanyl, and vecuronium mixtures designed for potential use in anesthesia and patient transport”. Journal of Clinical Anesthesia. 8 (4): 329–336. doi:10.1016/0952-8180(96)00043-8. PMID 8695138.
- ^ McQuaid, KR.; Laine, L. (May 2008). “A systematic review and meta-analysis of randomized, controlled trials of moderate sedation for routine endoscopic procedures”. Gastrointest Endosc. 67 (6): 910–23. doi:10.1016/j.gie.2007.12.046. PMID 18440381.
- ^ Canadian National Forumulary 2010
- ^ Appleton & Lange Nursing Drug Guide, 1999
- ^ Numorphan® (oxymorphone) package insert (English), Endo 2009
- ^ Machata, AM; Willschke, H; Kabon, B; Kettner, SC; Marhofer, P (August 2008). “Propofol-based sedation regimen for infants and children undergoing ambulatory magnetic resonance imaging”. British Journal of Anaesthesia. 101 (2): 239–43. doi:10.1093/bja/aen153. PMID 18534971.
- ^ Yan, JW; McLeod, SL; Iansavitchene, A (20 August 2015). “Ketamine-Propofol Versus Propofol Alone for Procedural Sedation in the Emergency Department: A Systematic Review and Meta-analysis”. Academic Emergency Medicine. 22 (9): 1003–13. doi:10.1111/acem.12737. PMID 26292077.
- ^ https://www.bbraunusa.com/content/dam/b-braun/us/website/company/covid-files/210319_Propofol_EUA_Submission_to_FDA_hcp.pdf
- ^ https://www.fda.gov/media/146680/download
- ^ https://www.fda.gov/media/146681/download
- ^ Jump up to:a b “Emergency Use Authorization”. U.S. Food and Drug Administration (FDA). Retrieved 17 April 2021.
- ^ Death Row Improvises, Lacking Lethal Mix Archived 8 July 2017 at the Wayback Machine, By RICK LYMAN, New York Times, 18 August 2013
- ^ After EU threats, Missouri halts execution by Propofol injectionArchived 12 October 2013 at the Wayback Machine Al Jazeera America 12 October 2013
- ^ Article 4A of Export Control Order 2008 – provisions supplementing “the torture Regulation”
- ^ Riezzo I, Centini F, Neri M, Rossi G, Spanoudaki E, Turillazzi E, Fineschi V (2009). “Brugada-like EKG pattern and myocardial effects in a chronic propofol abuser”. Clin Toxicol. 47 (4): 358–63. doi:10.1080/15563650902887842. PMID 19514884. S2CID 22531823.
- ^ Belluck, Pam (6 August 2009). “With High-Profile Death, Focus on High-Risk Drug”. New York Times. Archived from the original on 11 November 2011. Retrieved 7 August 2009.
- ^ Iwersen-Bergmann S, Rösner P, Kühnau HC, Junge M, Schmoldt A (2001). “Death after excessive propofol abuse”. International Journal of Legal Medicine. 114 (4–5): 248–51. CiteSeerX 10.1.1.528.7395. doi:10.1007/s004149900129. PMID 11355404. S2CID 25963187.
- ^ Kranioti EF, Mavroforou A, Mylonakis P, Michalodimitrakis M (22 March 2007). “Lethal self-administration of propofol (Diprivan): A case report and review of the literature”. Forensic Science International. 167 (1): 56–8. doi:10.1016/j.forsciint.2005.12.027. PMID 16431058.
- ^ In Sweetman SC (Ed.). Martindale: The Complete Drug Reference 2005. 34th Edn London pp. 1305–7
- ^ Baudoin Z. General anesthetics and anesthetic gases. In Dukes MNG and Aronson JK (Eds.). Meyler’s Side Effects of Drugs 2000. 14th Edn Amsterdam pp. 330
- ^ Jump up to:a b Roussin A, Montastruc JL, Lapeyre-Mestre M (21 October 2007). “Pharmacological and clinical evidences on the potential for abuse and dependence of propofol: a review of the literature”. Fundamental and Clinical Pharmacology. 21 (5): 459–66. doi:10.1111/j.1472-8206.2007.00497.x. PMID 17868199. S2CID 22477291.
- ^ C.F. Ward, 2008, Propofol: Dancing with a “White Rabbit”Archived 8 September 2017 at the Wayback Machine, CSA Bulletin, pp. 61–63, accessed 24 November 2014.
- ^ Jump up to:a b Charatan F (2009). “Concerns mount over recreational use of propofol among US healthcare professionals”. BMJ. 339: b3673. doi:10.1136/bmj.b3673. PMID 19737827. S2CID 9877560.
- ^ Bonnet U, Harkener J, Scherbaum N (June 2008). “A case report of propofol dependence in a physician”. J Psychoactive Drugs. 40(2): 215–7. doi:10.1080/02791072.2008.10400634. PMID 18720673. S2CID 15779389.
- ^ Moore, Solomon (28 August 2009). “Jackson’s Death Ruled a Homicide”. The New York Times. Archived from the original on 14 November 2013.
- ^ Jump up to:a b Surdin, Ashley (25 August 2009). “Coroner Attributes Michael Jackson’s Death to Propofol”. The Washington Post. Archivedfrom the original on 9 November 2012. Retrieved 22 May 2010.
- ^ Jump up to:a b Itzkoff, Dave (24 August 2009). “Coroner’s Findings in Jackson Death Revealed”. The New York Times. Archived from the original on 11 June 2010. Retrieved 22 May 2010.
- ^ “Jackson’s Death: How Dangerous Is Propofol?”. Time. 25 August 2009. Archived from the original on 25 July 2010. Retrieved 22 May 2010.
- ^ “Michael Jackson search warrant”. Scribd. Archived from the original on 5 March 2016. Retrieved 12 August 2015.
- ^ DEA may limit drug eyed in Jackson case. Associated Press.15 July 2009.
- ^ Matta, J. A.; Cornett, P. M.; Miyares, R. L.; Abe, K.; Sahibzada, N.; Ahern, G. P. (2008). “General anesthetics activate a nociceptive ion channel to enhance pain and inflammation”. Proceedings of the National Academy of Sciences. 105 (25): 8784–8789. doi:10.1073/pnas.0711038105. PMC 2438393. PMID 18574153.
- ^ “Propofol Drug Information, Professional”. m drugs.com. Archived from the original on 23 January 2007. Retrieved 2 January 2007.
- ^ Jump up to:a b Sebel, PS; Lowden, JD (1989). “Propofol: a new intravenous anesthetic”. Anesthesiology. 71 (2): 260–77. doi:10.1097/00000542-198908000-00015. PMID 2667401. S2CID 34331379.
- ^ Robinson, B; Ebert, T; O’Brien, T; et al. (1997). “Mechanisms whereby propofol mediates peripheral vasodilation in humans (1997)”. Anesthesiology. 86 (1): 64–72. doi:10.1097/00000542-199701000-00010. PMID 9009941. S2CID 31288656.
- ^ “New awakening in anaesthesia—at a price”. Lancet. 329 (8548): 1469–70. 1987. doi:10.1016/s0140-6736(87)92214-8. S2CID 28545161.
- ^ Larijani, G; Gratz, I; Afshar, M; et al. (1989). “Clinical pharmacology of propofol: an intravenous anesthetic agent [published erratum appears in DICP 1990 Jan; 24: 102]”. DICP. 23(10): 743–9. doi:10.1177/106002808902301001. PMID 2683416. S2CID 43010280.
- ^ Jung SL, Hyun SJ, Byeong JP (2013). “Green discoloration of urine after propofol infusion”. Korean Journal of Anesthesiology. 65 (2): 177–9. doi:10.4097/kjae.2013.65.2.177. PMC 3766788. PMID 24024005.
- ^ Parke, T. J.; Stevens, J. E.; Rice, A. S.; Greenaway, C. L.; Bray, R. J.; Smith, P. J.; Waldmann, C. S.; Verghese, C. (12 September 1992). “Metabolic acidosis and fatal myocardial failure after propofol infusion in children: five case reports”. BMJ. 305 (6854): 613–616. doi:10.1136/bmj.305.6854.613. ISSN 0959-8138. PMC 1883365. PMID 1393073.
- ^ Langley, M; Heel, R (1988). “Propofol. A review of its pharmacodynamic and pharmacokinetic properties and use as an intravenous anaesthetic”. Drugs. 35 (4): 334–72. doi:10.2165/00003495-198835040-00002. PMID 3292208.
- ^ Bailey, J; Mora, C; Shafer, S (1996). “Pharmacokinetics of propofol in adult patients undergoing coronary revascularization”. Anesthesiology. 84 (6): 1288–97. doi:10.1097/00000542-199606000-00003. PMID 8669668. S2CID 26019589.
- ^ Reilly, C; Nimmo, W (1987). “New intravenous anaesthetics and neuromuscular blocking drugs. A review of their properties and clinical use”. Drugs. 34 (1): 115–9. doi:10.2165/00003495-198734010-00004. PMID 3308413. S2CID 46973781.
- ^ Schramm, BM; Orser, BA (2002). “Dystonic reaction to propofol attenuated by benztropine (Cogentin)”. Anesth Analg. 94 (5): 1237–40. doi:10.1097/00000539-200205000-00034. PMID 11973196.
- ^ Vesta, Kimi; Shaunta’ Martina; Ellen Kozlowski (25 April 2009). “Propofol-Induced Priapism, a Case Confirmed with Rechallenge”. The Annals of Pharmacotherapy. 40 (5): 980–982. doi:10.1345/aph.1G555. PMID 16638914. S2CID 36563320.
- ^ Fuentes, Ennio; Silvia Garcia; Manuel Garrido; Cristina Lorenzo; Jose Iglesias; Juan Sola (July 2009). “Successful treatment of propofol-induced priapism with distal glans to corporal cavernosal shunt”. Urology. 74 (1): 113–115. doi:10.1016/j.urology.2008.12.066. PMID 19371930.
- ^ Eumorfia Kondili; Christina Alexopoulou; Nectaria Xirouchaki; Dimitris Georgopoulos (2012). “Effects of propofol on sleep quality in mechanically ventilated critically ill patients: a physiological study”. Intensive Care Medicine. 38 (10): 1640–1646. doi:10.1007/s00134-012-2623-z. PMID 22752356. S2CID 21206446.
- ^ “AstraZeneca – United States Home Page” (PDF). .astrazeneca-us.com. Archived from the original (PDF) on 4 October 2011. Retrieved 8 June 2013.
- ^ Kim, MD, FACEP, Tae Eung; Shankel, MD, Tamara; Reibling, PhD, MA, Ellen T.; Paik, MSN, RN, Jacqueline; Wright, PhD, RN, Dolores; Buckman, PhD, RN, Michelle; Wild, MS, RN, Kathi; Ngo, MS, Ehren; Hayatshahi, PharmD, Alireza (1 January 2017). “Healthcare students interprofessional critical event/disaster response course”. American Journal of Disaster Medicine. 12 (1): 11–26. doi:10.5055/ajdm.2017.0254. ISSN 1932-149X. PMID 28822211.
- ^ Vasile B, Rasulo F, Candiani A, Latronico N (2003). “The pathophysiology of propofol infusion syndrome: a simple name for a complex syndrome”. Intensive Care Medicine. 29 (9): 1417–25. doi:10.1007/s00134-003-1905-x. PMID 12904852. S2CID 23932736.
- ^ Doheny, Kathleen; Louise Chang; Hector Vila Jr (24 August 2009). “Propofol Linked to Michael Jackson’s Death”. WebMD. Archived from the original on 28 August 2009. Retrieved 26 August 2009.
- ^ Trapani G, Altomare C, Liso G, Sanna E, Biggio G (February 2000). “Propofol in anesthesia. Mechanism of action, structure-activity relationships, and drug delivery”. Curr. Med. Chem. 7 (2): 249–71. doi:10.2174/0929867003375335. PMID 10637364.
- ^ Kotani, Y; Shimazawa, M; Yoshimura, S; Iwama, T; Hara, H (Summer 2008). “The experimental and clinical pharmacology of propofol, an anesthetic agent with neuroprotective properties”. CNS Neuroscience and Therapeutics. 14 (2): 95–106. doi:10.1111/j.1527-3458.2008.00043.x. PMC 6494023. PMID 18482023.
- ^ Vanlersberghe, C; Camu, F (2008). Propofol. Handbook of Experimental Pharmacology. 182. pp. 227–52. doi:10.1007/978-3-540-74806-9_11. ISBN 978-3-540-72813-9. PMID 18175094.
- ^ Trapani, G; Latrofa, A; Franco, M; Altomare, C; Sanna, E; Usala, M; Biggio, G; Liso, G (1998). “Propofol analogues. Synthesis, relationships between structure and affinity at GABAA receptor in rat brain, and differential electrophysiological profile at recombinant human GABAA receptors”. Journal of Medicinal Chemistry. 41 (11): 1846–54. doi:10.1021/jm970681h. PMID 9599235.
- ^ Krasowski MD, Jenkins A, Flood P, Kung AY, Hopfinger AJ, Harrison NL (April 2001). “General anesthetic potencies of a series of propofol analogs correlate with potency for potentiation of gamma-aminobutyric acid (GABA) current at the GABA(A) receptor but not with lipid solubility”. J. Pharmacol. Exp. Ther. 297 (1): 338–51. PMID 11259561.
- ^ Krasowski, MD; Hong, X; Hopfinger, AJ; Harrison, NL (2002). “4D-QSAR analysis of a set of propofol analogues: mapping binding sites for an anesthetic phenol on the GABA(A) receptor”. Journal of Medicinal Chemistry. 45 (15): 3210–21. doi:10.1021/jm010461a. PMC 2864546. PMID 12109905.
- ^ Haeseler G, Leuwer M (March 2003). “High-affinity block of voltage-operated rat IIA neuronal sodium channels by 2,6 di-tert-butylphenol, a propofol analogue”. Eur J Anaesthesiol. 20 (3): 220–4. doi:10.1017/s0265021503000371. PMID 12650493. S2CID 25072723.
- ^ Haeseler, G; Karst, M; Foadi, N; Gudehus, S; Roeder, A; Hecker, H; Dengler, R; Leuwer, M (September 2008). “High-affinity blockade of voltage-operated skeletal muscle and neuronal sodium channels by halogenated propofol analogues”. British Journal of Pharmacology. 155 (2): 265–75. doi:10.1038/bjp.2008.255. PMC 2538694. PMID 18574460.
- ^ Fowler CJ (February 2004). “Possible involvement of the endocannabinoid system in the actions of three clinically used drugs”. Trends Pharmacol. Sci. 25 (2): 59–61. doi:10.1016/j.tips.2003.12.001. PMID 15106622.
- ^ Lee, U; Mashour, GA; Kim, S; Noh, GJ; Choi, BM (2009). “Propofol induction reduces the capacity for neural information integration: implications for the mechanism of consciousness and general anesthesia”. Conscious. Cogn. 18 (1): 56–64. doi:10.1016/j.concog.2008.10.005. PMID 19054696. S2CID 14699319.
- ^ Favetta P, Degoute CS, Perdrix JP, Dufresne C, Boulieu R, Guitton J (2002). “Propofol metabolites in man following propofol induction and maintenance”. British Journal of Anaesthesia. 88(5): 653–8. doi:10.1093/bja/88.5.653. PMID 12067002.
- ^ Veselis RA, Reinsel RA, Feshchenko VA, Wroński M (October 1997). “The comparative amnestic effects of midazolam, propofol, thiopental, and fentanyl at equisedative concentrations”. Anesthesiology. 87 (4): 749–64. doi:10.1097/00000542-199710000-00007. PMID 9357875. S2CID 30185553.
- ^ James, R; Glen, JB (December 1980). “Synthesis, biological evaluation, and preliminary structure-activity considerations of a series of alkylphenols as intravenous anesthetic agents”. Journal of Medicinal Chemistry. 23 (12): 1350–1357. doi:10.1021/jm00186a013. ISSN 0022-2623. PMID 7452689.
- ^ Foundation, Lasker. “Discovery and development of propofol, a widely used anesthetic”. The Lasker Foundation. Retrieved 25 July 2020.
- ^ “Drugs@FDA: FDA Approved Drug Products”. U.S. Food and Drug Administration (FDA). Archived from the original on 13 August 2014. Retrieved 8 June 2013.
- ^ “Archived copy” (PDF). Archived (PDF) from the original on 17 April 2014. Retrieved 15 June 2014. pp. 3. accessed 23. January 2016
- ^ Stein M, et al. (September 2012). “Azo-Propofols: Photochromic Potentiators of GABAA Receptors”. Angewandte Chemie International Edition. 51 (42): 15000–4. doi:10.1002/anie.201205475. PMC 3606271. PMID 22968919.
- ^ Yip G, Z.-W Chen, Edge C J, Smith E H, Dickinson R, Hohenester, E, Townsend R R, Fuchs K, Sieghart W, Evers A S, Franks N P (September 2013). “A propofol binding site on mammalian GABAAreceptors identified by photolabeling”. Nature Chemical Biology. 9 (11): 715–720. doi:10.1038/nchembio.1340. PMC 3951778. PMID 24056400.
- ^ Kvam C, Granese D, Flaibani A, Pollesello P, Paoletti S (1993). “Hyaluronan can be protected from free-radical depolymerization by 2, 6-diisopropylphenol, a novel radical scavenger”. Biochem. Biophys. Res. Commun. 193 (3): 927–33. doi:10.1006/bbrc.1993.1714. PMID 8391811.
External links
| Wikimedia Commons has media related to Propofol. |
- “Propofol”. Drug Information Portal. U.S. National Library of Medicine.
- GB patent 1472793, John B Glen & Roger James, “Pharmaceutical Compositions”, published 1977-05-04, assigned to Imperial Chemical Industries Ltd
| Clinical data | |
|---|---|
| Trade names | Diprivan, others[1] |
| AHFS/Drugs.com | Monograph |
| License data | US DailyMed: Propofol |
| Pregnancy category | AU: C |
| Dependence liability | Physical: very low (seizures) Psychological: no data |
| Addiction liability | Moderate[2] |
| Routes of administration | Intravenous |
| ATC code | N01AX10 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)CA: ℞-onlyUK: POM (Prescription only)US: ℞-only [3]In general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Bioavailability | NA |
| Protein binding | 95–99% |
| Metabolism | Liver glucuronidation |
| Onset of action | 15–30 seconds[4] |
| Elimination half-life | 1.5–31 hours[4] |
| Duration of action | ~5–10 minutes[4] |
| Excretion | Liver |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2078-54-8 |
| PubChem CID | 4943 |
| IUPHAR/BPS | 5464 |
| DrugBank | DB00818 |
| ChemSpider | 4774 |
| UNII | YI7VU623SF |
| KEGG | D00549 |
| ChEBI | CHEBI:44915 |
| ChEMBL | ChEMBL526 |
| CompTox Dashboard (EPA) | DTXSID6023523 |
| ECHA InfoCard | 100.016.551 |
| Chemical and physical data | |
| Formula | C12H18O |
| Molar mass | 178.275 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
/////////////PROPOFOL
IOHEXOL
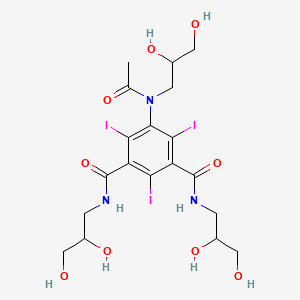
IOHEXOLCAS Registry Number: 66108-95-0N1,N3-bis(2,3-dihydroxypropyl)-5-[N-(2,3-dihydroxypropyl)acetamido]-2,4,6-triiodobenzene-1,3-dicarboxamide
CAS Name: 5-[Acetyl(2,3-dihydroxypropyl)amino]-N,N¢-bis(2,3-dihydroxypropyl)-2,4,6-triiodo-1,3-benzenedicarboxamideAdditional Names:N,N¢-bis(2,3-dihydroxypropyl)-5-[N-(2,3-dihydroxypropyl)acetamido]-2,4,6-triiodoisophthalamide
Manufacturers’ Codes: Win-39424; Compd 545Trademarks: Omnipaque (GE Healthcare)
Molecular Formula: C19H26I3N3O9Molecular Weight: 821.14Percent Composition: C 27.79%, H 3.19%, I 46.36%, N 5.12%, O 17.54%
Literature References: Nonionic radio-contrast medium. Prepn: V. Nordal, H. Holtermann, DE2726196; eidem,US4250113 (1977, 1981 both to Nyegaard). HPLC-UV determn in plasma: R. S. Soman et al., J. Chromatogr. B816, 339 (2005).
Pharmacology and toxicology: Acta Radiol.Suppl. 362, 1-134 (1980). Acute toxicity: S. Salvesen, ibid. 73. Fibrillatory potential in dogs: G. L. Wolf et al.,Invest. Radiol.16, 320 (1981).Comparative clinical studies in coronary angiography: G. B. J. Mancini et al.,Am. J. Cardiol.51, 1218 (1983); I. D. Sullivan et al.,Br. Heart J.51, 643 (1984); M. A. Bettmann et al.,Radiology153, 583 (1984). Review: T. Almén, Acta Radiol.Suppl. 366, 9-19 (1983).
Properties: Crystals from butanol, mp 174-180°. Sol in water. Stable in aqueous solutions. Viscosity (cP): 6.2 at 37°; 12.6 at 20° (c = 200 mg Iodine/ml). LD50 in male, female rats, mice (g Iodine/kg): 15.0, 12.3, 24.3, 25.1 i.v. (Salvesen).
Melting point: mp 174-180°
Toxicity data: LD50 in male, female rats, mice (g Iodine/kg): 15.0, 12.3, 24.3, 25.1 i.v. (Salvesen)Therap-Cat: Diagnostic aid (radiopaque medium).Keywords: Diagnostic Aid (Radiopaque Medium).
Synthesis ReferenceXiu C. Wang, Steve A. Chamberlin, Ashok V. Bhatia, Gregg E. Robinson, John Hufnagel, “Process for the preparation of iohexol.” U.S. Patent US5705692, issued December, 1985.
Iohexol, sold under the trade name Omnipaque among others, is a contrast agent used for X-ray imaging.[1] This includes when visualizing arteries, veins, ventricles of the brain, the urinary system, and joints, as well as during computed tomography (CT scan).[1] It is given by mouth, injection into a vein, or into a body cavity.[2]
Iohexol is a contrast agent for intrathecal administration used in myelography and contrast enhancement for computerized tomography.
Side effects include vomiting, skin flushing, headache, itchiness, kidney problems, and low blood pressure.[1] Less commonly allergic reactions or seizures may occur.[1] Allergies to povidone-iodine or shellfish do not affect the risk of side effects more than other allergies.[3] Use in the later part of pregnancy may cause hypothyroidism in the baby.[4] Iohexol is an iodinated non-ionic radiocontrast agent.[1] It is in the low osmolar family.[5]
Iohexol was approved for medical use in 1985.[6] It is on the World Health Organization’s List of Essential Medicines.[7][2]
Chemistry
The osmolality of iohexol ranges from 322 mOsm/kg—approximately 1.1 times that of blood plasma—to 844 mOsm/kg, almost three times that of blood.[8] Despite this difference, iohexol is still considered a low-osmolality contrast agent; the osmolality of older agents, such as diatrizoate, may be more than twice as high.[9]
Society and culture
Names
It is sold under the brand names Omnipaque[10] and Hexopaque. It is also sold as a density gradient medium under the names Accudenz, Histodenz and Nycodenz.[11][12]
Formulations
It is available in various concentrations, from 140[citation needed] to 350[13] milligrams of iodine per milliliter.
PATENT
https://patents.google.com/patent/WO2005003080A1/en#:~:text=Primary%20production%20of%20iohexol%20involves,and%20a%20thorough%20purification%20stage.&text=The%20solvent%20is%20then%20evaporated,and%20recrystallised%20twice%20from%20butanol.The present invention relates to a process for the manufacture of iohexol, 5-[N- (2,3- dihydroxypropyl) -acetamido]-N,N’-bis(2,3 -dihydroxypropyl)-2,4,6- triiodoisophtalamide.Iohexol is the non-proprietory name of the chemical drug substance of a non-ionic iodinated X-ray contrast agent marketed under the trade name OMNIPAQUE®. OMNIPAQUE® is one of the most used agents in diagnostic X-ray procedures.The manufacture of such non-ionic contrast agents involves the production of the chemical drug substance (referred to as primary production) followed by formulation into the drug product (referred to as secondary production). Primary production of iohexol involves a multistep chemical synthesis and a thorough purification stage. For a commercial drug product it is important for the primary production to be efficient and economical and to provide a drug substance fulfilling the specifications.The final step in the synthesis of iohexol is a N-alkylation step in which 5-(acetamido)-N,N’-bis(2,3-dihydroxypropyl)-2,4,6 triiodoisophtalamide (hereinafter 5- Acetamide) is reacted in the liquid phase with an alkylating agent to introduce the 2,3-dihydroxypropyl group at the nitrogen of the 5-acetamido group. Following this reaction, iohexol is isolated from the reaction mixture and purified by crystallisation and treatment with ion exchange resins.The manufacture of iohexol is disclosed for example in US-4,250,113 which is hereby incorporated by reference. In the last step of the multistep chemical synthesis crude iohexol is obtained from the reaction between 5-Acetamide and 1-chloro-2,3- propandiol at ambient temperature in propylene glycoi and in the presence of sodium methoxide. The solvent is then evaporated and crude iohexol is obtained. The crude product is evaporated to dryness and recrystallised twice from butanol.Several suggestions to improve the N-alkylation and the purification steps have been published. WO-A-98/08804 discloses the use of 2-methoxy-ethanol and optionally isopropanol both in the alkylation step of 5-Acetamide and in the purification of crude iohexol. WO-A-02/083623 discloses the purification of crude iohexol using 1- methoxy-2-propanol as the solvent optionally in a mixture with other solvents.The N-alkylation step where 5-Acetamide in solution is reacted with an alkylation agent such as e.g. 1-chloro-2,3-propandiol to introduce the 2,3-dihydroxypropyl group at the nitrogen of the 5-acetamido group is illustrated in Scheme 1 :

5-Acetamide Iohexol5-acatamido-N,N’-bis(2,3-dihydroxypropyl)- 5-[N-(2,3-dihydroxypropyl)acetamido]- 2,4,6-triiodoisophtalamide N,N’-bis(2,3-dihydroxypropyl)- 2,4,6-triiodoisophtalamideScheme 1.The N-alkylation step is challenging because O-alkylated by-products can also be formed when the alkylation occurs at the oxygen atoms of the hydroxy groups. It is therefore a desire to limit the formation of these O-alkylated by-products and thereby to limit their presence in the final purified iohexol. The upper limit for values for O- alkylated by-products in the end product is fixed by the European Pharmacopea to 0.6% (HPLC by area).The O-alkylated by-products are removed to the degree desired or necessary by recrystallisation steps. Further unidentified by-products also referred to as impurities are also formed during the alkylation reaction and must be reduced to a tolerable level. In addition the solvents used should be easily available, be environmentally friendly and be of low toxicity.There is therefore a need to identify a solvent that can be used in the N-alkylation reaction and that fulfil the desiderata mentioned above. It is further desired to improve the overall process including the N-alkylation step and the purification step in the manufacture of iohexol. If the crude product obtained by the N-alkylation step is to be re-crystallised from a solvent that is different from the solvent used in the N- alkylation step, then the reaction solvent must first be removed e.g. by evaporation to dryness. It is known from crystallisation theory and experience that even small quantities of residual solvents from previous steps may cause a crystallisation process to get out of control due to changes in its supersaturation conditions, and thorough removal of the reaction solvent is an important step. Solvent removal is an energy consuming operation which also risks degradation of the product due to exposure to elevated temperature.Example 1 : Synthesis of iohexol in 1-methoxy-2-propanol/methanol1-methoxy-2-propanol (44 ml), methanol (19 ml) and sodium hydroxide (4.87 g) was added to a jacketed glass reactor and stirred for about 15 minutes at 25°C. 5-Acetamide (70 g) was added to the reactor, and the mixture stirred overnight at 45°C, before it was allowed to cool to 25°C. 1-chloro-2,3-propanediol (12.43 g) was added to the solution. After 1.5 hours, more 1-chloro-2,3-propanediol (0.83 g) was added, and the reaction was allowed to proceed for 24 hours. HPLC analysis (water/acetonitrile) of the reaction mixture gave the following results:Iohexol 98.1 %5-Acetamide 1.17 % O-alkylated substances 0.58 %Other impurities 0.1 %Example 2: Synthesis of iohexol in 1 -methoxy-2-propanol/water1-methoxy-2-propanol (63 ml), water (7 ml) and sodium hydroxide (4.50 g) was added to a jacketed glass reactor and stirred for about 15 minutes at 25°C. 5-Acetamide (70 g) was added to the reactor, and the mixture stirred overnight at 45°C, before it was allowed to cool to 35°C. 1-chloro-2,3-propanediol (11.39 g) was added to the solution. After 3 hours, more 1-chloro-2,3-propanediol (0.83 g) was added, and the reaction was allowed to proceed for 24 hours. HPLC analysis (water/acetonitrile) of the reaction mixture gave the following results:Iohexol 98.3 % 5-Acetamide 0.68 %O-alkylated substances 0.81 %Other impurities 0.3 % Example 3: Alkylation and crystallisation in solutions containing 1-methoxy-2- propanol1-methoxy-2-propanol (63 L), methanol (27 L) and sodium hydroxide (6.96 kg) was added to a 500 L reactor and stirred until all solids were dissolved and the temperature was below 30°C. 5-Acetamide (100 kg) was added to the reactor, and the mixture stirred overnight at 45°C before it was allowed to cool to 25°C. 1-chloro- 2,3-propanediol (16.76 kg) was added to the clear solution. After 1.5 hours, more 1- chloro-2,3-propanediol (1.18 kg) was added, and the reaction was allowed to proceed for 30 hours. HPLC analysis (water/acetonitrile) of the reaction mixture gave the following results:Iohexol 97.9 % 5-Acetamide 0.9 %O-alkylated substances 0.83 %Other impurities 0.4 %The reaction was stopped by addition of hydrochloric acid (650 ml), and the reaction mixture diluted with a mixture of 1-methoxy-2-propanol (53 L) and methanol (13 L). The mixture was filtered, and the salts on the filter washed with methanol (3×10 L). The combined filtrate and wash was diluted with water (22 L) and treated with cationic ion exchange resin (AMB 200C, 80 L) and anionic ion exchange resin (IRA 67, 80 L) to a salt content of 0.006 w/w %. The solution was filtered, and the ion exchange resins washed in several stages with a mixture of water (160 L) and methanol (85 L). The combined filtrate and wash was concentrated under reduced pressure to a volume of 155 L. One half of this was taken further to crystallisation as described below.Water was removed from the solution by azeotropic distillation. The volume was held at a constant level by replacing the distillate by 1-methoxy-2-propanol (80 L). At water content of 0.16 Ukg iohexol, further 1-methoxy-2-propanol (159 L) was added, and the solution seeded with iohexol crystals (0.26 kg). After stirring at reflux overnight, the volume of the solution was reduced by 42 L by distillation under reduced pressure (300-600 mbar). The temperature was set to 90°C, which was held for 3 hours before cooling to 60°C over 3 hours. The crystallisation mixture was stirred overnight at 60°C, filtered and washed with isopropanol (90 L, 6 portions). The yield was 48.4 kg (as dry powder), corresponding to 88-weight % corrected for seeding material and samples. HPLC analysis (water/acetonitrile) of the crystals gave the following results:Iohexol 99.3 %5-Acetamide 0.15 %O-alkylated substances 0.45 %Other impurities 0.11 %
PAPERhttps://www.quickcompany.in/patents/a-new-process-for-the-synthesis-of-high-pure-iohexol-and-its-intermediatesPATENThttps://patents.google.com/patent/WO2005003080A1/enThe present invention relates to a process for the manufacture of iohexol, 5-[N- (2,3- dihydroxypropyl) -acetamido]-N,N’-bis(2,3 -dihydroxypropyl)-2,4,6- triiodoisophtalamide.Iohexol is the non-proprietory name of the chemical drug substance of a non-ionic iodinated X-ray contrast agent marketed under the trade name OMNIPAQUE®. OMNIPAQUE® is one of the most used agents in diagnostic X-ray procedures.The manufacture of such non-ionic contrast agents involves the production of the chemical drug substance (referred to as primary production) followed by formulation into the drug product (referred to as secondary production). Primary production of iohexol involves a multistep chemical synthesis and a thorough purification stage. For a commercial drug product it is important for the primary production to be efficient and economical and to provide a drug substance fulfilling the specifications.The final step in the synthesis of iohexol is a N-alkylation step in which 5-(acetamido)-N,N’-bis(2,3-dihydroxypropyl)-2,4,6 triiodoisophtalamide (hereinafter 5- Acetamide) is reacted in the liquid phase with an alkylating agent to introduce the 2,3-dihydroxypropyl group at the nitrogen of the 5-acetamido group. Following this reaction, iohexol is isolated from the reaction mixture and purified by crystallisation and treatment with ion exchange resins.The manufacture of iohexol is disclosed for example in US-4,250,113 which is hereby incorporated by reference. In the last step of the multistep chemical synthesis crude iohexol is obtained from the reaction between 5-Acetamide and 1-chloro-2,3- propandiol at ambient temperature in propylene glycoi and in the presence of sodium methoxide. The solvent is then evaporated and crude iohexol is obtained. The crude product is evaporated to dryness and recrystallised twice from butanol.Several suggestions to improve the N-alkylation and the purification steps have been published. WO-A-98/08804 discloses the use of 2-methoxy-ethanol and optionally isopropanol both in the alkylation step of 5-Acetamide and in the purification of crude iohexol. WO-A-02/083623 discloses the purification of crude iohexol using 1- methoxy-2-propanol as the solvent optionally in a mixture with other solvents.The N-alkylation step where 5-Acetamide in solution is reacted with an alkylation agent such as e.g. 1-chloro-2,3-propandiol to introduce the 2,3-dihydroxypropyl group at the nitrogen of the 5-acetamido group is illustrated in Scheme 1 :
5-Acetamide Iohexol5-acatamido-N,N’-bis(2,3-dihydroxypropyl)- 5-[N-(2,3-dihydroxypropyl)acetamido]- 2,4,6-triiodoisophtalamide N,N’-bis(2,3-dihydroxypropyl)- 2,4,6-triiodoisophtalamideScheme 1.The N-alkylation step is challenging because O-alkylated by-products can also be formed when the alkylation occurs at the oxygen atoms of the hydroxy groups. It is therefore a desire to limit the formation of these O-alkylated by-products and thereby to limit their presence in the final purified iohexol. The upper limit for values for O- alkylated by-products in the end product is fixed by the European Pharmacopea to 0.6% (HPLC by area).The O-alkylated by-products are removed to the degree desired or necessary by recrystallisation steps. Further unidentified by-products also referred to as impurities are also formed during the alkylation reaction and must be reduced to a tolerable level. In addition the solvents used should be easily available, be environmentally friendly and be of low toxicity.There is therefore a need to identify a solvent that can be used in the N-alkylation reaction and that fulfil the desiderata mentioned above. It is further desired to improve the overall process including the N-alkylation step and the purification step in the manufacture of iohexol. If the crude product obtained by the N-alkylation step is to be re-crystallised from a solvent that is different from the solvent used in the N- alkylation step, then the reaction solvent must first be removed e.g. by evaporation to dryness. It is known from crystallisation theory and experience that even small quantities of residual solvents from previous steps may cause a crystallisation process to get out of control due to changes in its supersaturation conditions, and thorough removal of the reaction solvent is an important step. Solvent removal is an energy consuming operation which also risks degradation of the product due to exposure to elevated temperature.Example 1 : Synthesis of iohexol in 1-methoxy-2-propanol/methanol1-methoxy-2-propanol (44 ml), methanol (19 ml) and sodium hydroxide (4.87 g) was added to a jacketed glass reactor and stirred for about 15 minutes at 25°C. 5-Acetamide (70 g) was added to the reactor, and the mixture stirred overnight at 45°C, before it was allowed to cool to 25°C. 1-chloro-2,3-propanediol (12.43 g) was added to the solution. After 1.5 hours, more 1-chloro-2,3-propanediol (0.83 g) was added, and the reaction was allowed to proceed for 24 hours. HPLC analysis (water/acetonitrile) of the reaction mixture gave the following results:Iohexol 98.1 %5-Acetamide 1.17 % O-alkylated substances 0.58 %Other impurities 0.1 %Example 2: Synthesis of iohexol in 1 -methoxy-2-propanol/water1-methoxy-2-propanol (63 ml), water (7 ml) and sodium hydroxide (4.50 g) was added to a jacketed glass reactor and stirred for about 15 minutes at 25°C. 5-Acetamide (70 g) was added to the reactor, and the mixture stirred overnight at 45°C, before it was allowed to cool to 35°C. 1-chloro-2,3-propanediol (11.39 g) was added to the solution. After 3 hours, more 1-chloro-2,3-propanediol (0.83 g) was added, and the reaction was allowed to proceed for 24 hours. HPLC analysis (water/acetonitrile) of the reaction mixture gave the following results:Iohexol 98.3 % 5-Acetamide 0.68 %O-alkylated substances 0.81 %Other impurities 0.3 % Example 3: Alkylation and crystallisation in solutions containing 1-methoxy-2- propanol1-methoxy-2-propanol (63 L), methanol (27 L) and sodium hydroxide (6.96 kg) was added to a 500 L reactor and stirred until all solids were dissolved and the temperature was below 30°C. 5-Acetamide (100 kg) was added to the reactor, and the mixture stirred overnight at 45°C before it was allowed to cool to 25°C. 1-chloro- 2,3-propanediol (16.76 kg) was added to the clear solution. After 1.5 hours, more 1- chloro-2,3-propanediol (1.18 kg) was added, and the reaction was allowed to proceed for 30 hours. HPLC analysis (water/acetonitrile) of the reaction mixture gave the following results:Iohexol 97.9 % 5-Acetamide 0.9 %O-alkylated substances 0.83 %Other impurities 0.4 %The reaction was stopped by addition of hydrochloric acid (650 ml), and the reaction mixture diluted with a mixture of 1-methoxy-2-propanol (53 L) and methanol (13 L). The mixture was filtered, and the salts on the filter washed with methanol (3×10 L). The combined filtrate and wash was diluted with water (22 L) and treated with cationic ion exchange resin (AMB 200C, 80 L) and anionic ion exchange resin (IRA 67, 80 L) to a salt content of 0.006 w/w %. The solution was filtered, and the ion exchange resins washed in several stages with a mixture of water (160 L) and methanol (85 L). The combined filtrate and wash was concentrated under reduced pressure to a volume of 155 L. One half of this was taken further to crystallisation as described below.Water was removed from the solution by azeotropic distillation. The volume was held at a constant level by replacing the distillate by 1-methoxy-2-propanol (80 L). At water content of 0.16 Ukg iohexol, further 1-methoxy-2-propanol (159 L) was added, and the solution seeded with iohexol crystals (0.26 kg). After stirring at reflux overnight, the volume of the solution was reduced by 42 L by distillation under reduced pressure (300-600 mbar). The temperature was set to 90°C, which was held for 3 hours before cooling to 60°C over 3 hours. The crystallisation mixture was stirred overnight at 60°C, filtered and washed with isopropanol (90 L, 6 portions). The yield was 48.4 kg (as dry powder), corresponding to 88-weight % corrected for seeding material and samples. HPLC analysis (water/acetonitrile) of the crystals gave the following results:Iohexol 99.3 %5-Acetamide 0.15 %O-alkylated substances 0.45 %Other impurities 0.11 %
PatentCN109134289https://patents.google.com/patent/CN109134289A/en

N-Acylation of 5-amino-N,N’-bis(2,3-dihydroxypropyl)-2,4,6-triiodoisophthalamide (1) with acetic anhydride (2) in the presence of p-TsOH gives 5-(acetylamino)-N,N’-bis(2,3-dihydroxypropyl)-2,4,6-triiodoisophthalamide (3) , which upon condensation with glycidol using NaOMe in 2-methoxyethanol at 90 °C or epichlorohydrin by means of NaHCO3 in propylene glycol at 85 °C or 3-chloropropane-1,2-diol (5) using aqueous NaOH furnishes the iohexol .(7) synthesis of IodixanolModus ponens (I) compound (200g, 0.28mol) be added 1L there-necked flask in, thereto be added acetic anhydride (207g, 2.03mol), acetic acid (103.3mL), p-methyl benzenesulfonic acid monohydrate (1g, 5.42mmol), finishes reaction solution being heated to 60 DEG C Start to react, keep the temperature 30 minutes after reacting liquid temperature reaches 120-125 DEG C, cooling is concentrated into after can just stirring thereto It is added 50%v/v (600mL), is slowly added dropwise thereto into 50%w/v sodium hydrate aqueous solution, by adding in reaction process The mode of 50%w/v sodium hydrate aqueous solution keeps the pH of reaction solution between 11~12, and reaction temperature is maintained at 40-45 DEG C, Reaction is finished, and concentrated hydrochloric acid is added into reaction solution and adjusts pH3-4, and stirring filters after 3.0 hours, and filter cake is washed with water to neutrality, dries It is dry, obtain white solid 187g, yield 88.2%, HPLC98.14%.Go step obtained solid (150g, 0.2mol) be added there-necked flask in, thereto be added sodium hydroxide (14.4g, 0.36mol), purified water (300mL), epoxychloropropane (27.9g, 0.30mol) finish 30-35 DEG C of reaction 72.0 hours, instead It should finish, adjust pH3-4, Iodixanol HPLC purity 72.5%, Iohexol HPLC11.3% with concentrated hydrochloric acid.(4) synthesis of IohexolModus ponens (I) compound (200g, 0.28mol) be added 1L there-necked flask in, thereto be added acetic anhydride (432g, 4.23mol) flows back 3.0 hours, be then concentrated under reduced pressure into p-methyl benzenesulfonic acid monohydrate (1g, 5.42mmol), agitating and heating It can just stir, be added portionwise into reaction solution methanol (25g), methanol is added after 1.0 hours in stirring thereto again (140g) is finished and is stirred to react 1.0 hours, and being concentrated under reduced pressure into can just stir, and purified water (20g) then is added thereto, 60 DEG C are finished to be stirred overnight.Reaction solution is cooled to 30 DEG C hereinafter, extracting reaction solution 200mL, stirring is lower will with 50%w/v sodium hydrate aqueous solution Reaction solution pH is adjusted to 12, the addition 1- chloro- 2 into reaction solution, 3-propanediol (20g, 0.18mol), passes through benefit in reaction process The mode of 50%w/v sodium hydrate aqueous solution is added to keep the pH of reaction solution between 11~12, after reaction 12.0 hours thereto Add 1- chloro- 2,3-propanediol (3g, 29.29mmol) finishes that the reaction was continued 48.0 hours, and reaction solution samples HPLC detection, iodine Mykol purity is 89.9%.(5) synthesis of IoversolModus ponens (I) compound (200g, 0.28mol) is added in 1L there-necked flask, and N-Methyl pyrrolidone is added thereto Chloracetyl chloride (200mL) is added in (200mL) thereto under stirring, finish 50-53 DEG C and react 3.0 hours, and reaction is finished, and is cooled to 20 DEG C, reaction solution is slowly added in methanol (2000mL).It finishing, flows back 9.0 hours, reaction is finished, and is cooled to 25 DEG C, it filters, Filter cake is washed with methanol, and drying obtains white solid 177g, yield 79.8%, HPLC purity 98.3%.It takes previous step obtained solid (150g, 0.19mol) to be added in 1L there-necked flask, purified water 300mL is added thereto, Acetic acid sodium trihydrate (183g, 1.34mol) finishes back flow reaction, by adding 50%w/v sodium hydroxide water in reaction process The mode of solution keeps the pH of reaction solution between 5-6, and reaction is finished, and concentrated hydrochloric acid is added into reaction solution, adjusts pH3-4, stirring It being filtered after 3.0 hours, filter cake is with purifying water washing to neutrality, and drying obtains white solid 127g, yield 86.7%, HPLC98.4%.It takes step obtained solid (100g, 0.13mol), is added in 1L there-necked flask, purified water 300mL, chlorine are added thereto Change sodium (46.5g, 0.796mol), finish, be warming up to 50 DEG C, 10N sodium hydrate aqueous solution (39.3mL) and 2- are added thereto Chlorethanol (63.5g, 0.79mol) finishes 48-52 DEG C of heat preservation and reacts 5.0 hours, and reaction is finished, and concentrated hydrochloric acid is added thereto and adjusts PH6.5, reaction solution HPLC detection, Iohexol purity 89.7%.(6) synthesis of IopentolModus ponens (I) compound (200g, 0.28mol) be added 1L there-necked flask in, thereto be added acetic anhydride (432g, 4.23mol) flows back 3.0 hours, be then concentrated under reduced pressure into p-methyl benzenesulfonic acid monohydrate (1g, 5.42mmol), agitating and heating It can just stir, be added portionwise into reaction solution methanol (25g), methanol (140g) is added thereto again after stirring 1.0 hours, It finishes and is stirred to react 1.0 hours, being concentrated under reduced pressure into can just stir, and purified water (20g) then is added thereto, finishes 60 DEG C It is stirred overnight.Reaction solution is cooled to 30 DEG C hereinafter, extracting reaction solution 200mL, stirring is lower will with 50%w/v sodium hydrate aqueous solution Reaction solution pH is adjusted to 12, and the chloro- 3- methoxy-2-propanol (22.5g, 0.18mol) of 1-, reaction process are added into reaction solution In keep the pH of reaction solution between 11~12 by way of adding 50%w/v sodium hydrate aqueous solution, react 12.0 hours Add 1- chloro- 2 thereto afterwards, 3-propanediol (3.4g, 29.29mmol) finishes that the reaction was continued 48.0 hours, reaction solution sampling HPLC detection, Iopentol purity are 91.3%.(7) synthesis of IodixanolModus ponens (I) compound (200g, 0.28mol) be added 1L there-necked flask in, thereto be added acetic anhydride (207g, 2.03mol), acetic acid (103.3mL), p-methyl benzenesulfonic acid monohydrate (1g, 5.42mmol), finishes reaction solution being heated to 60 DEG C Start to react, keep the temperature 30 minutes after reacting liquid temperature reaches 120-125 DEG C, cooling is concentrated into after can just stirring thereto It is added 50%v/v (600mL), is slowly added dropwise thereto into 50%w/v sodium hydrate aqueous solution, by adding in reaction process The mode of 50%w/v sodium hydrate aqueous solution keeps the pH of reaction solution between 11~12, and reaction temperature is maintained at 40-45 DEG C, Reaction is finished, and concentrated hydrochloric acid is added into reaction solution and adjusts pH3-4, and stirring filters after 3.0 hours, and filter cake is washed with water to neutrality, dries It is dry, obtain white solid 187g, yield 88.2%, HPLC98.14%.Go step obtained solid (150g, 0.2mol) be added there-necked flask in, thereto be added sodium hydroxide (14.4g, 0.36mol), purified water (300mL), epoxychloropropane (27.9g, 0.30mol) finish 30-35 DEG C of reaction 72.0 hours, instead It should finish, adjust pH3-4, Iodixanol HPLC purity 72.5%, Iohexol HPLC11.3% with concentrated hydrochloric acid.To sum up, method of the invention is easy to operate, and (III) three obtained formula (I), formula (II) or formula intermediate can be made For the raw material for synthesizing diodone, not by-product truly;Importantly, general sieve of synthesis iodine that can be convenient Amine does not have the generation of two acylated by-products, and compared with original grinds the production technology of medicine, process route is entirely different, high income, cost It is low, a kind of very effective, completely new approach is provided for industrialized production Iopromide, is had a extensive future.
Patent
Publication numberPriority datePublication dateAssigneeTitleWO1998008804A1 *1996-08-291998-03-05Nycomed Imaging AsProcess for iohexol manufactureUS5847212A *1997-04-211998-12-08Abbott LaboratoriesProcess for the preparation of iohexolWO1999026916A1 *1997-11-261999-06-03Nycomed Imaging AsN-alkylation of 5-amino-2,4,6-triiodo-isophthalamidesFamily To Family CitationsITMI20010773A1 *2001-04-112002-10-11Chemi SpaProcess for the production of high purity iohexole
Non-Patent
TitleHAAVALDSEN J ET AL: “X-RAY CONTRAST AGENTS. I. SYNTHESIS OF SOME DERIVATIVES OF 5-AMINO-2, 4, 6-TRIIODOISOPHTHLAMIDE”, ACTA PHARMACEUTICA SUECICA, XX, XX, vol. 20, no. 3, 1983, pages 219 – 232, XP002052827, ISSN: 0001-6675 *
Publication numberPriority datePublication dateAssigneeTitleWO2007013816A1 *2005-07-292007-02-01Ge Healthcare AsContinuous crystallisation process of iodinated phenyl derivativesWO2007060380A1 *2005-11-242007-05-31Hovione Inter LtdProcess for the manufacture of iohexolJP2009502910A *2005-07-292009-01-29ジーイー・ヘルスケア・アクスイェ・セルスカプMethod for continuous crystallization of iodinated phenyl derivativesCN101195587B *2006-12-192010-07-21浙江尖峰海洲制药有限公司Production method for lodixanol hydrolysateUS8766002B22009-11-262014-07-01Imax Diagnostic Imaging Holding LimitedPreparation and purification of iodixanolNO342021B1 *2005-07-292018-03-12Ge Healthcare AsContinuous crystallization processFamily To Family CitationsWO2011041275A1 *2009-09-302011-04-07Mallinckrodt Inc.Alkylation of triiodo-substituted arylamides in an aqueous mixed solvent systemES2680019T3 *2010-12-212018-09-03Ge Healthcare AsDesalination of a composition comprising a contrast agentUS20140065076A1 *2012-08-302014-03-06Otsuka Pharmaceutical Co. Ltd.Container with concentrated substance and method of using the same* Cited by examiner, † Cited by third party, ‡ Family to family citation
Similar Documents
PublicationPublication DateTitleEP1641743B12008-11-12Process for iohexol manufactureKR101188596B12012-10-05Preparation of iodixanolEP1960349B12015-11-18Purification of iodixanolEP1966110B12013-04-24Purification process of iodixanolJP5536087B22014-07-02Method for producing iodinated contrast agentUS5948940A1999-09-07Process for iohexol manufactureUS7541494B22009-06-02Process for the manufacture of iohexolEP2277855B12011-11-09Crystallization of iodixanol using millingCA2707173C2011-08-02Crystallization of iodixanol in isopropanol and methanolRU2173315C22001-09-10Method of preparing ionexolCA2710577C2012-09-18Crystallization of iodixanol using milling

NEW DRUG APPROVALS
one time
$10.00
References
- ^ Jump up to:a b c d e World Health Organization (2009). Stuart MC, Kouimtzi M, Hill SR (eds.). WHO Model Formulary 2008. World Health Organization. pp. 317–8. hdl:10665/44053. ISBN 9789241547659.
- ^ Jump up to:a b Hamilton, Richart (2015). Tarascon Pocket Pharmacopoeia 2015 Deluxe Lab-Coat Edition. Jones & Bartlett Learning. p. 171. ISBN 9781284057560.
- ^ ACR Manual on Contrast Media v10.3. 2017 (PDF). American College of Radiology. 2017. p. 6. ISBN 9781559030120. Archived (PDF) from the original on 1 January 2018. Retrieved 1 January 2018.
- ^ Briggs, Gerald G.; Freeman, Roger K.; Yaffe, Sumner J. (2011). Drugs in Pregnancy and Lactation: A Reference Guide to Fetal and Neonatal Risk. Lippincott Williams & Wilkins. p. 761. ISBN 9781608317080. Archived from the original on 1 January 2017.
- ^ Sutton, David; Young, Jeremy W. R. (2012). A Short Textbook of Clinical Imaging. Springer Science & Business Media. p. 235. ISBN 9781447117551. Archived from the original on 1 January 2017.
- ^ Broe, Marc E. de; Porter, George A.; Bennett, William M.; Verpooten, G. A. (2013). Clinical Nephrotoxins: Renal Injury from Drugs and Chemicals. Springer Science & Business Media. p. 325. ISBN 9789401590884. Archived from the original on 1 January 2017.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ GE Healthcare (May 2006). “Omnipaque (Iohexol) injection. Product label”. DailyMed. U.S. National Library of Medicine. Retrieved 28 March 2007.
- ^ Amersham Health (April 2006). “Hypaque (Diatrizoate Meglumine and Diatrizoate Sodium) injection, solution. Product label”. DailyMed. U.S. National Library of Medicine. Archived from the original on 23 May 2011. Retrieved 29 March 2007.
- ^ “Omnipaque” (PDF). Ireland: Health Products Regulatory Authority. January 2018. Retrieved 31 July 2020.
- ^ “HistoDenz (D2158)” Archived 2015-11-20 at the Wayback Machine, product information sheet, Sigma-Aldrich. Accessed on line 19 November 2015.
- ^ “Nycodenz®: A universal density gradient medium” Archived 2015-02-26 at the Wayback Machine, Axis-Shield Density Gradient Media. Accessed 19 November 2015.
- ^ Haberfeld H, ed. (2020). Austria-Codex (in German). Vienna: Österreichischer Apothekerverlag. Omnipaque 350 mg J/ml Infusionsflasche.
External links
- “Iohexol”. Drug Information Portal. U.S. National Library of Medicine.
- “Iohexol Injection, Oral, Rectal Advanced Patient Information”. Drugs.com. 13 January 2019. Retrieved 3 February 2020.
| Clinical data | |
|---|---|
| Trade names | Omnipaque, Hexopaque, Oraltag, others |
| Other names | 5-[N-(2,3-Dihydroxypropyl)acetamido]-2,4,6-triiodo-N,N’-bis(2,3-dihydroxypropyl)isophthalamide |
| AHFS/Drugs.com | Micromedex Detailed Consumer Information |
| License data | US DailyMed: Iohexol |
| Routes of administration | intrathecal, intravascular, by mouth, intracavital, rectal |
| ATC code | V08AB02 (WHO) |
| Legal status | |
| Legal status | US: ℞-onlyIn general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Protein binding | Low |
| Metabolism | Nil |
| Elimination half-life | Variable |
| Excretion | Kidney, unchanged |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 66108-95-0 |
| PubChem CID | 3730 |
| DrugBank | DB01362 |
| ChemSpider | 3599 |
| UNII | 4419T9MX03 |
| KEGG | D01817 |
| ChEBI | CHEBI:31709 |
| ChEMBL | ChEMBL1200455 |
| CompTox Dashboard (EPA) | DTXSID6023157 |
| ECHA InfoCard | 100.060.130 |
| Chemical and physical data | |
| Formula | C19H26I3N3O9 |
| Molar mass | 821.142 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Melting point | 174 to 180 °C (345 to 356 °F) |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
////////////IOHEXOL, Win-39424, Compd 545, Omnipaque, Oraltag, GE Healthcare, X RAY CONTRAST AGENTS, WIN 39424
CC(=O)N(CC(O)CO)C1=C(I)C(C(=O)NCC(O)CO)=C(I)C(C(=O)NCC(O)CO)=C1I
Avalglucosidase alfa
QQGASRPGPR DAQAHPGRPR AVPTQCDVPP NSRFDCAPDK AITQEQCEAR GCCYIPAKQG
LQGAQMGQPW CFFPPSYPSY KLENLSSSEM GYTATLTRTT PTFFPKDILT LRLDVMMETE
NRLHFTIKDP ANRRYEVPLE TPRVHSRAPS PLYSVEFSEE PFGVIVHRQL DGRVLLNTTV
APLFFADQFL QLSTSLPSQY ITGLAEHLSP LMLSTSWTRI TLWNRDLAPT PGANLYGSHP
FYLALEDGGS AHGVFLLNSN AMDVVLQPSP ALSWRSTGGI LDVYIFLGPE PKSVVQQYLD
VVGYPFMPPY WGLGFHLCRW GYSSTAITRQ VVENMTRAHF PLDVQWNDLD YMDSRRDFTF
NKDGFRDFPA MVQELHQGGR RYMMIVDPAI SSSGPAGSYR PYDEGLRRGV FITNETGQPL
IGKVWPGSTA FPDFTNPTAL AWWEDMVAEF HDQVPFDGMW IDMNEPSNFI RGSEDGCPNN
ELENPPYVPG VVGGTLQAAT ICASSHQFLS THYNLHNLYG LTEAIASHRA LVKARGTRPF
VISRSTFAGH GRYAGHWTGD VWSSWEQLAS SVPEILQFNL LGVPLVGADV CGFLGNTSEE
LCVRWTQLGA FYPFMRNHNS LLSLPQEPYS FSEPAQQAMR KALTLRYALL PHLYTLFHQA
HVAGETVARP LFLEFPKDSS TWTVDHQLLW GEALLITPVL QAGKAEVTGY FPLGTWYDLQ
TVPIEALGSL PPPPAAPREP AIHSEGQWVT LPAPLDTINV HLRAGYIIPL QGPGLTTTES
RQQPMALAVA LTKGGEARGE LFWDDGESLE VLERGAYTQV IFLARNNTIV NELVRVTSEG
AGLQLQKVTV LGVATAPQQV LSNGVPVSNF TYSPDTKVLD ICVSLLMGEQ FLVSWC
(Disulfide bridge:26-53, 36-52, 47-71, 477-502, 591-602, 882-896)
Avalglucosidase alfa
アバルグルコシダーゼアルファ (遺伝子組換え)
Avalglucosidase alfa (USAN/INN);
Avalglucosidase alfa (genetical recombination) (JAN);
Avalglucosidase alfa-ngpt
To treat late-onset Pompe disease
| Formula | C4490H6818N1197O1299S32 |
|---|---|
| CAS | 1802558-87-7 |
| Mol weight | 99375.4984 |
FDA APPROVED Nexviazyme, 2021/8/6, Enzyme replacement therapy product
Treatment of Pompe disease
Biologic License Application (BLA): 761194
Company: GENZYME CORP
https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-pompe-diseaseFor Immediate Release:August 06, 2021
Today, the U.S. Food and Drug Administration approved Nexviazyme (avalglucosidase alfa-ngpt) for intravenous infusion to treat patients 1 year of age and older with late-onset Pompe disease.
Patients with Pompe disease have an enzyme deficiency that leads to the accumulation of a complex sugar, called glycogen, in skeletal and heart muscles, which cause muscle weakness and premature death from respiratory or heart failure. Normally, glycogen—the stored form of glucose—breaks down to release glucose into the bloodstream to be used as fuel for the cells.
“Pompe disease is a rare genetic disease that causes premature death and has a debilitating effect on people’s lives,” said Janet Maynard, M.D., deputy director of the Office of Rare Diseases, Pediatrics, Urologic and Reproductive Medicine in the FDA’s Center for Drug Evaluation and Research. “Today’s approval brings patients with Pompe disease another enzyme replacement therapy option for this rare disease. The FDA will continue to work with stakeholders to advance the development of additional new, effective and safe therapies for rare diseases, including Pompe disease.”
Nexviazyme, an enzyme replacement therapy, is an intravenous medication that helps reduce glycogen accumulation. The effectiveness of Nexviazyme for the treatment of Pompe disease was demonstrated in a study of 100 patients who were randomized to take Nexviazyme or another FDA-approved enzyme replacement therapy for Pompe disease. Treatment with Nexviazyme improved lung function similar to the improvement seen with the other therapy.
The most common side effects included headache, fatigue, diarrhea, nausea, joint pain (arthralgia), dizziness, muscle pain (myalgia), itching (pruritus), vomiting, difficulty breathing (dyspnea), skin redness (erythema), feeling of “pins and needles” (paresthesia) and skin welts (urticaria). Serious reactions included hypersensitivity reactions like anaphylaxis and infusion-associated reactions, including respiratory distress, chills and raised body temperature (pyrexia). Patients susceptible to fluid volume overload or with compromised cardiac or respiratory function may be at risk for serious acute cardiorespiratory failure.
The FDA granted this application Fast Track, Priority Review and Breakthrough Therapy designations. Nexviazyme also received an orphan drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases. The FDA granted the approval of Nexviazyme to Genzyme Corporation.
###
NEW DRUG APPROVALS
one time
$10.00
FDA grants priority review for avalglucosidase alfa, a potential new therapy for Pompe disease
- The FDA decision date for avalglucosidase alfa, an investigational enzyme replacement therapy, is set for May 18, 2021
- Regulatory submission based on positive data from two trials in patients with late-onset and infantile-onset Pompe disease, respectively
- Avalglucosidase alfa received FDA Breakthrough Therapy and Fast Track designations for the treatment of people with Pompe Disease
- Pompe disease, a rare degenerative muscle disorder, affects approximately 3,500 people in the U.S.
- Milestone reinforces 20+year commitment to Pompe disease community
PARIS – November 18, 2020 – The U.S. Food and Drug Administration (FDA) has accepted for priority review the Biologics License Application (BLA) for avalglucosidase alfa for long-term enzyme replacement therapy for the treatment of patients with Pompe disease (acid α-glucosidase deficiency). The target action date for the FDA decision is May 18, 2021.
Avalglucosidase alfa is an investigational enzyme replacement therapy designed to improve the delivery of acid alpha-glucosidase (GAA) enzyme to muscle cells, and if approved, would offer a potential new standard of care for patients with Pompe disease.
In October, the European Medicines Agency accepted for review the Marketing Authorization Application for avalglucosidase alfa for long-term enzyme replacement therapy for the treatment of patients with Pompe disease. The Medicines and Healthcare Products Regulatory Agency in the UK has granted Promising Innovative Medicine designation for avalglucosidase alfa.
“The hallmarks of Pompe disease are the relentless and debilitating deterioration of the muscles, which causes decreased respiratory function and mobility,” said Karin Knobe, Head of Development for Rare Diseases and Rare Blood Disorders at Sanofi. “Avalglucosidase alfa is specifically designed to deliver more GAA enzyme into the lysosomes of the muscle cells. We have been greatly encouraged by positive clinical trial results in patients with late-onset and infantile-onset Pompe disease.”
Pompe disease is a rare, degenerative muscle disorder that can impact an individual’s ability to move and breathe. It affects an estimated 3,500 people in the U.S. and can manifest at any age from infancy to late adulthood.i
The BLA is based on positive data from two trials:
- Pivotal Phase 3, double-blind, global comparator-controlled trial (COMET), which evaluated the safety and efficacy of avalglucosidase alfa compared to alglucosidase alfa (standard of care) in patients with late-onset Pompe disease. Results from this trial were presented during a Sanofi-hosted virtual scientific session in June 2020 and in October 2020 at World Muscle Society and the American Association of Neuromuscular and Electrodiagnostic Medicine.
- The Phase 2 (mini-COMET) trial evaluated the safety and exploratory efficacy of avalglucosidase alfa in patients with infantile-onset Pompe disease previously treated with alglucosidase alfa. Results from this trial were presented at the WORLDSymposium, in February 2020.
Delivery of GAA to Clear Glycogen
Pompe disease is caused by a genetic deficiency or dysfunction of the lysosomal enzyme GAA, which results in build-up of complex sugars (glycogen) in muscle cells throughout the body. The accumulation of glycogen leads to irreversible damage to the muscles, including respiratory muscles and the diaphragm muscle supporting lung function, and other skeletal muscles that affect mobility.
To reduce the glycogen accumulation caused by Pompe disease, the GAA enzyme must be delivered into the lysosomes within muscle cells. Research led by Sanofi has focused on ways to enhance the delivery of GAA into the lysosomes of muscle cells by targeting the mannose-6-phosphate (M6P) receptor that plays a key role in the transport of GAA.
Avalglucosidase alfa is designed with approximately 15-fold increase in M6P content, compared to standard of care alglucosidase alfa, and aims to help improve cellular enzyme uptake and enhance glycogen clearance in target tissues.ii The clinical relevance of this difference has not been confirmed.
Avalglucosidase alfa is currently under clinical investigation and its safety and efficacy have not been evaluated by any regulatory authority worldwide.
| About Sanofi Sanofi is dedicated to supporting people through their health challenges. We are a global biopharmaceutical company focused on human health. We prevent illness with vaccines, provide innovative treatments to fight pain and ease suffering. We stand by the few who suffer from rare diseases and the millions with long-term chronic conditions. With more than 100,000 people in 100 countries, Sanofi is transforming scientific innovation into healthcare solutions around the globe. Sanofi, Empowering Life |
/////////Avalglucosidase alfa, FDA 2021, Nexviazyme, APPROVALS 2021, PEPTIDE, Enzyme replacement therapy , Pompe disease, アバルグルコシダーゼアルファ (遺伝子組換え), Fast Track, Priority Review, Breakthrough Therapy, orphan drug designation, genzyme, sanofi
ONO-2910

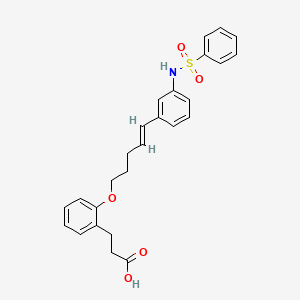
ONO-2910
CAS 2410177-35-2
3- [2-[(E) -5- [3- (benzenesulfonamide) phenyl] penta-4-enoxy] phenyl] propanoic acid
3- [2-[(E) -5- [3- (benzenesulfonamido) phenyl] penta-4-enoxy] phenyl] propanoic acidC26 H27 N O5 S465.56Benzenepropanoic acid, 2-[[(4E)-5-[3-[(phenylsulfonyl)amino]phenyl]-4-penten-1-yl]oxy]-
ONO Pharmaceuticals is developing ONO-2910 , the lead from a program of novel transient receptor potential cation channel 4/5 inhibitors, for treating peripheral neuropathy. In April 2021, a phase II trial in patients with diabetic polyneuropathy was initiated.
PATENT
CN112513011-BENZENE DERIVATIVE
| Example 84: 3-[2-[(E)-5-[3-(Benzenesulfonamido)phenyl]pent-4-enyloxy]phenyl]propionic acid |
| [Chemical formula 52] |
| |
| To a solution of the compound (146 mg) produced in Example 83 in THF (0.5 mL) and methanol (0.1 mL), 1M aqueous lithium hydroxide solution (0.5 mL) was added, and the mixture was stirred at 50°C for 8 hours. 1M hydrochloric acid was added to make it acidic, and it was extracted with ethyl acetate. After drying the organic layer over sodium sulfate, it was concentrated under reduced pressure to obtain the title compound (105 mg) having the following physical properties. |
| HPLC retention time (min): 1.10 |
| 1 H-NMR(CD 3 OD): δ 1.95-2.03, 2.41-2.46, 2.57-2.61,2.92-2.95, 4.03-4.06, 6.24, 6.36, 6.86, 6.90-6.95, 7.06-7.08, 7.11-7.19, 7.45-7.49, 7.55, 7.75 -7.78. |

NEW DRUG APPROVALS
ONE TIME
$10.00
PATENT
WO-2021153690
Novel crystalline forms of 3-[2-[(E)-5-[3-(benzenesulfonamide) phenyl] penta-4-enoxy] phenyl] propanoic acid act as neuroprotective, useful for treating neurological disorders eg chronic inflammatory demyelinating polyneuritis, Guillain-Barre syndrome and allergic angiitis.Example 1:
Sulfuric acid (0.26 mL) is added to a solution of isopropyl 3- (2-hydroxyphenyl) propanoate 3,4-dihydrocoumarin (50.0 g) in isopropyl alcohol (500 mL), and the reaction mixture is mixed at room temperature for 2 hours. Stirred. The reaction mixture was concentrated under reduced pressure, and the obtained residue was diluted with ethyl acetate. The mixture was washed with saturated aqueous sodium hydrogen carbonate solution, water and saturated brine, dried over sodium sulfate, and concentrated under reduced pressure to give the title compound (73.2 g) having the following physical properties.
1 1 H-NMR (CDCl 3 ): δ 1.20, 2.66-2.70, 2.87-2.91, 4.95-5.08, 6.86-6.91, 7.06-7.15, 7.35.
Example 2: Isopropyl 3- (2- (pent-4-in-1-yloxy) phenyl) propanoate In a solution of the compound (3.00 g) prepared in Example 1 in N, N-dimethylacetamide (25 mL) at room temperature. Cesium carbonate (9.39 g) was added at the same temperature, and the mixture was stirred at the same temperature for 15 minutes. 5-Chloro-1-pentyne (CAS Registry Number: 14267-92-6) (1.63 g) was added to the reaction solution at room temperature, and the mixture was stirred at 60 ° C. for 3 hours. Water was added to the reaction solution, and the mixture was extracted with diethyl ether. The organic layer was dried over sodium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane: ethyl acetate = 1: 0 → 5: 1) to give the title compound (2.40 g) having the following physical property values.
HPLC retention time (minutes): 1.13.Example 3: Isopropyl (E) -3- (2-((5- (4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) penta-4-en-1-yl) Il) Oxy) Phenyl) Propanoate In
a heptane (2 mL) solution of the compound (1.00 g) prepared in Example 2, 4,4,5,5-tetramethyl-1,3,2-dioxaborolane (1. 17 g) and 4-dimethylaminobenzoic acid (60.2 mg) were added, and the mixture was stirred at 100 ° C. for 4 hours. The reaction solution was cooled to room temperature and then concentrated. The obtained residue was purified by silica gel column chromatography (hexane: ethyl acetate = 20: 1 → 4: 1) to give the title compound (503 mg) having the following physical characteristics.
HPLC retention time (minutes): 1.38.Example 3 (1):
Pyridine (0.95 mL), N, N-dimethyl in a solution of N- (3-bromophenyl) benzenesulfonamide 3-bromoaniline (1.02 g) in dichloromethane (20 mL) at 0 ° C. Aminopyridine (hereinafter abbreviated as DMAP) (72.4 mg) and benzenesulfonyl chloride (1.10 g) were added, and the mixture was stirred at room temperature for 2 hours. After concentrating the reaction solution, the obtained residue is purified by silica gel column chromatography (hexane: ethyl acetate = 9: 1 → 2: 1) to give the title compound (1.96 g) having the following physical properties. rice field.
HPLC retention time (minutes): 0.98.
Example 4: Isopropyl (E) -3-(2-((5- (3- (phenylsulfonamide) phenyl) penta-4-en-1-yl) oxy) phenyl) propanoate The
compound prepared in Example 3. In a solution of (180 mg) in THF (3 mL), the compound (168 mg) prepared in Example 3 (1), chloro (2-dicyclohexylphosphino-2′, 4′, 6′-triisopropyl-1,1′- Biphenyl) [2- (2′-amino-1,1′-biphenyl)] palladium (II) (0.035 g) and a 2M tripotassium phosphate aqueous solution (0.67 mL) were added, and the mixture was stirred at 60 ° C. for 1 hour. .. The reaction solution was cooled to room temperature, water was added, and the mixture was extracted with ethyl acetate. The organic layer was dried over sodium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane: ethyl acetate = 7: 1 → 2: 1) to give the title compound (113 mg) having the following physical characteristics.
HPLC retention time (minutes): 1.24
Example 5: 3- [2-[(E) -5- [3- (benzenesulfonamide) phenyl] penta-4-enoxy] phenyl] propanoic acid
[Chemical 2]
A 1 M aqueous lithium hydroxide solution (0.5 mL) was added to a solution of the compound (146 mg) prepared in Example 4 in THF (0.5 mL) and methanol (0.1 mL), and the mixture was stirred at 50 ° C. for 8 hours. It was acidified by adding 1M hydrochloric acid and extracted with ethyl acetate. The organic layer was dried over sodium sulfate and concentrated under reduced pressure to give the title compound (105 mg) having the following physical characteristics.
Form: Amorphous
HPLC retention time (minutes): 1.101
1 H-NMR (CD 3 OD): δ 1.95-2.03, 2.41-2.46, 2.57-2.61, 2.92-2.95, 4.03-4.06, 6.24, 6.36, 6.86, 6.90-6.95, 7.06-7.08, 7.11-7.19, 7.45-7.49, 7.55, 7.75-7.78.
PATENT
WO2020027150
https://patents.google.com/patent/WO2020027150A1/en
Example 83: Isopropyl (E) -3- (2-((5- (3- (phenylsulfonamido) phenyl) penta-4-en-1-yl) oxy) phenyl) propanoate The compound prepared in Example 82 Compound (168 mg) prepared in Example 9 and chloro (2-dicyclohexylphosphino-2 ′, 4 ′, 6′-triisopropyl-1,1′-biphenyl) [180 mg) in THF (3 mL) solution were added. 2- (2′-Amino-1,1′-biphenyl)] palladium (II) (0.035 g) and a 2M aqueous solution of tripotassium phosphate (0.67 mL) were added, and the mixture was stirred at 60 ° C. for 1 hour. After cooling the reaction solution to room temperature, water was added, and the mixture was extracted with ethyl acetate. The organic layer was dried over sodium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane: ethyl acetate = 7: 1 → 2: 1) to give the title compound (113 mg) having the following physical data.
HPLC retention time (min): 1.24.Example 84: 3- [2-[(E) -5- [3- (benzenesulfonamido) phenyl] penta-4-enoxy] phenyl] propanoic acid
To a solution of the compound prepared in Example 83 (146 mg) in THF (0.5 mL) and methanol (0.1 mL) was added a 1 M aqueous lithium hydroxide solution (0.5 mL), and the mixture was stirred at 50 ° C. for 8 hours. The mixture was acidified with 1M hydrochloric acid and extracted with ethyl acetate. The organic layer was dried over sodium sulfate and concentrated under reduced pressure to give the title compound (105 mg) having the following physical data.
HPLC retention time (min): 1.10
1 H-NMR (CD 3 OD): δ 1.95-2.03, 2.41-2.46, 2.57-2.61, 2.92-2.95, 4.03-4.06, 6.24, 6.36, 6.86, 6.90-6.95, 7.06-7.08, 7.11-7.19, 7.45 -7.49, 7.55, 7.75-7.78.

///////////ONO-2910, ONO 2910, PHASE 2,
O=S(=O)(Nc1cc(\C=C\CCCOc2ccccc2CCC(=O)O)ccc1)c1ccccc1
Bemiparin

Bemiparin
- AVE 5026
- Adomiparin
- Ardeparin
- Arteven
- Bemiparin
- CY 216
- CY 222
- Centaxarin
- Certoparin
- Clevarin
- Clivarin
- Clivarine
- Dalteparin
- Deligoparin
- F 202
- FR 860
- Fluxum
- Fragmin A
- Fragmin B
- Fraxiparin
- Gammaparin
- H 5284
- H 9399
- Hapacarin
- Heparin subcutan
- Heparin sulfate
- Heparinic acid
- Heparins
- KB 101
- Leparan
- LipoHep Forte
- Livaracine
- M 118
- M 118REH
- M 402
- M 402 (heparin)
- Mono-embolex
- Multiparin
- Nadroparin
- Nadroparine
- Necuparanib
- Novoheparin
- OP 386
- OP 622
- Octaparin
- Pabyrn
- Parnaparin
- Parvoparin
- Reviparin
- Sandoparin
- Semuloparin
- Subeparin
- Sublingula
- Tafoxiparin
- Tinzaparin
- Triofiban
- Vetren
- Vitrum AB
- α-Heparin
cas 91449-79-5
Bemiparin (trade names Ivor and Zibor, among others) is an antithrombotic and belongs to the group of low molecular weight heparins (LMWH).[1]
Bemiparin is an ultra-low molecular weight heparin (ultra-LMWH) used to prevent thromboembolism following surgery and extracorporeal clotting during dialysis.
Rovi and Archimedes (a wholly owned subsidiary of ProStrakan), have developed and launched bemiparin, a Factor Xa inhibitor for the injectable treatment and prevention of thrombosis.
low or very low molecular weight heparins (eg bemiparin sodium) with a high anti-factor Xa activity for the treatment of deep vein thrombosis.
Bemiparin is an antithrombotic and belongs to the group of drugs known as the low molecular weight heparins (LMWH). Like semuloparin, bemiparin is classified as an ultra-LMH because of its low mean molecular mass of 3600 daltons, which is a unique property of this class 1. These heparins have lower anti-thrombin activity than the traditional low molecular weight heparins and act mainly on factor-Xa, reducing the risk of bleeding due to selectivity for this specific clotting factor. Interestingly, current research is underway for the potential benefit of bemiparin in the treatment of tumors and diabetic foot ulcers 12,1.
Laboratorios Farmaceuticos Rovi has developed and launched Enoxaparina Rovi, a biosimilar version of enoxaparin sodium, an injectable low-molecular-weight fraction of heparin, for the prophylaxis of venous thromboembolism.
PATENT
WO2018015463
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018015463
claiming a method for analyzing glycosaminoglycans, heparins and their derivatives in a compound comprising a monosaccharide residues present in heparin (eg bemiparin sodium) chains by identification and relative quantification of its characteristic signals by1H NMR one-dimensional nuclear magnetic resonance and/or 1H-13C HSQC two-dimensional nuclear magnetic resonance, using dimethylmalonic acid as internal reference
PATENT
CN-110092848
https://patents.google.com/patent/CN110092848A/enEmbodiment 1Experimental raw used and instrument are as follows in embodiment 1:Refined heparin sodium (ZH160712 quality of lot meets CP2015), benzethonium chloride, purified water, 40% (W/V) trimethoxy Base methanolic ammonium hydroxide, methylene chloride, methanol, 10% (W/V) sodium acetate methanol solution, 30% hydrogen peroxide, medicinal second Alcohol, sodium chloride, glass reaction pot (5000ml) three-necked flask 500ml, digital display heat-collecting magnetic stirring device, beaker, freeze dryer (on Hai Dongfulong) etc..A kind of preparation method of Bemiparin sodium of the present invention, the following steps are included:1. at salt1.1 weigh, dissolution, react1.1.1 the refined heparin sodium for weighing 10g is poured into tank, and the purified water of 100ml is added into reactor tank, is stirred to molten Solution is complete.1.1.2 25g benzethonium chloride is added in beaker, 125ml purified water stirring and dissolving is added.1.1.3 benzethonium chloride solution is added slowly with stirring in the heparin sodium aqua in reactor tank, time for adding 4.5h controls 35 DEG C of feed liquid temperature, continues stirring 2 hours, stops stirring and stands 2 hours, then as far as possible by supernatant liquid Removing.1.2 washings, centrifugation, drying:1.2.1 300ml purified water is added into residue precipitating suspended matter to wash in three times, then starts to wash for the first time, 20 DEG C of feed liquid temperature of control is stirred 1 hour, is stopped stirring and is stood 2 hours, repeats the above operation twice.1.2.2 supernatant liquid is removed, filters and be washed with water under stirring, record slurry amount, collect sediment.1.2.3 final gained sediment is uniformly divided in stainless steel disc, is transferred in heated-air circulation oven, adjust temperature 40 DEG C of degree, dry 6h crushes solid with Universalpulverizer after then 60 DEG C of dry range estimations are not glued to solid, smashed solid Body continues to be transferred in heated-air circulation oven, until loss on drying≤2.0%.Rewinding obtains heparin-benzyl rope ammonium salt about 32g, does Dry weightless 1.5%.2. degradation2.1 weighingBy above-mentioned 30g heparin-benzyl rope ammonium salt in 500ml three-necked flask, the methylene chloride of 150ml is added into reactor tank It is added in three-necked flask.2.2 dissolutions: three-necked flask is put into digital display heat-collecting magnetic stirring device, is heated to 33 DEG C and is stirred to having dissolved Entirely.2.3 degradations: being added 40% (W/V) trimethoxy methanolic ammonium hydroxide of 20.4ml in Xiang Shangshu solution, puts down Respectively 4 additions, it is for 24 hours that interval time is added every time.It after the 4th is added, then reacts for 24 hours, amounts to reaction 96h, during reaction Maintain 34 DEG C of temperature.2.4 terminate reaction: above-mentioned reaction solution being cooled to 20 DEG C, 180ml10% (W/V) sodium acetate methanol is added thereto Solution stirs 30min, filters to obtain its precipitating.2.5 washings: washing above-mentioned sediment with 300ml methanol solution, dry bemiparin crude product about 9g.3. purification3.1 will be above-mentioned dry that 9g bemiparin crude product pours into tank, and the purified water of 90ml, stirring are added into reactor tank It is complete to dissolution.3.2 adjust material liquid pH 9.5 with 20% sodium hydroxide solution.0.54ml hydrogen peroxide is added to be stirred to react at 20 DEG C 7.5 hours, through 0.22 μm of micro porous filtration.3.3 1.8g sodium chloride is added into feed liquid, then uses 4mol/L hydrochloric acid flavouring liquid pH to 6.5, is added into feed liquid 450ml medicinal alcohol stops stirring after stirring 30 minutes, places 4 hours.3.4 take supernatant away, and 90ml purified water is added, and stirring adjusts PH6.5 to dissolving completely, through 0.22 μm of micro porous filtration, Sabot freeze-drying.After 3.5 freeze-drying 36h, collection material weighing 7g.Three, the primary quality measure statistics of gained bemiparin
| Serial number | Project | Control standard | Testing result |
| 1 | Weight average molecular weight | 3000~4200 | 3650 |
| 2 | Molecular weight is greater than 6000 constituent content | < 15% | 12.9% |
| 3 | Constituent content of the molecular weight less than 2000 | < 35% | 36.7% |
| 4 | Molecular weight is between 2000~6000 constituent contents | 50%~75% | 50.4% |
| 5 | Anti-Xa activity | 80~120IU/mg | 116IU/mg |
| 6 | Anti- IIa activity | 5~20IU/mg | 14.6IU/mg |
| 7 | The anti-anti- IIa of Xa/ | ≥7 | 7.95 |
Embodiment 2Experimental raw used and instrument are as follows in embodiment 1:Refined heparin sodium (ZH180912 quality of lot meets CP2015), benzethonium chloride, purified water, 40% (W/V) trimethoxy Base methanolic ammonium hydroxide, methylene chloride, methanol, 10% (W/V) sodium acetate methanol solution, 30% hydrogen peroxide, medicinal second Alcohol, sodium chloride, glass reaction pot (10000ml, 30000L), three-necked flask 500ml, digital display heat-collecting magnetic stirring device, beaker, Freeze dryer (Shanghai Dong Fulong) etc..A kind of preparation method of Bemiparin sodium of the present invention, the following steps are included: 1. one-tenth salt1.1 weigh, dissolution, react1.1.1 the refined heparin sodium for weighing 500g is poured into tank, the purified water of 5000ml is added into reactor tank, stirring is extremely Dissolution is complete.1.1.2 1250g benzethonium chloride is added in beaker, 6300ml purified water stirring and dissolving is added.1.1.3 benzethonium chloride solution is added slowly with stirring in the heparin sodium aqua in reactor tank, time for adding 5h controls 35 DEG C of feed liquid temperature, continues stirring 2 hours, stops stirring and stands 2 hours, then as far as possible by supernatant liquid It removes.1.2 washings, centrifugation, drying:1.2.1 5000ml purified water is added into residue precipitating suspended matter to wash in three times, then starts to wash for the first time, 30 DEG C of feed liquid temperature of control is stirred 1 hour, is stopped stirring and is stood 2 hours, repeats the above operation twice.1.2.2 supernatant liquid is removed, filters and be washed with water under stirring, record slurry amount, collect sediment.1.2.3 final gained sediment is uniformly divided in stainless steel disc, is transferred in heated-air circulation oven, adjust temperature 45 DEG C of degree, dry 6h crushes solid with Universalpulverizer after then 70 DEG C of dry range estimations are not glued to solid, smashed solid Body continues to be transferred in heated-air circulation oven, until loss on drying≤2.0%.Rewinding obtains heparin-benzyl rope ammonium salt about 1505g, Loss on drying 1.0%.2. degradation2.1 weighingBy above-mentioned 1500g heparin-benzyl rope ammonium salt in 30L glass reaction kettle, the methylene chloride of 7500ml is added thereto.2.2 dissolutions: leading to hot water for its interlayer, is heated to 33~36 DEG C and stirs complete to dissolving.2.3 degradations: being added 40% (W/V) trimethoxy methanolic ammonium hydroxide of 1020ml in Xiang Shangshu solution, puts down Respectively 4 additions, it is for 24 hours that interval time is added every time.It after the 4th is added, then reacts for 24 hours, amounts to reaction 96h, during reaction Maintain 35 DEG C of temperature.2.4 terminate reaction: above-mentioned reaction solution being cooled to 20 DEG C, 9000ml10% (W/V) sodium acetate first is added thereto Alcoholic solution stirs 30min, filters to obtain its precipitating.2.5 washings: washing above-mentioned sediment with 15000ml methanol solution, dry bemiparin crude product about 400g.3. purification3.1 will be above-mentioned dry that 400g bemiparin crude product pours into tank, and the purified water of 4000ml is added into reactor tank, Stirring is complete to dissolving.3.2 adjust material liquid pH 9.5 with 20% sodium hydroxide solution.24ml hydrogen peroxide is added, and at 30 DEG C to be stirred to react 7 small When, through 0.22 μm of micro porous filtration.3.3 8g sodium chloride is added into feed liquid, then uses 4mol/L hydrochloric acid flavouring liquid pH to 6.5, is added into feed liquid 20000ml medicinal alcohol stops stirring after stirring 30 minutes, places 4 hours.3.4 take supernatant away, and 4000ml purified water is added, and stirring adjusts PH6.5, through 0.22 μm of micropore mistake to dissolving completely Filter, sabot freeze-drying.After 3.5 freeze-drying 36h, collection material weighing 350g.Three, the primary quality measure statistics of gained bemiparin
NEW DRUG APPROVALS
one time
$10.00
PATENT
WO-2021152192
https://patentscope.wipo.int/search/en/detail.jsf;jsessionid=9D96E01E1CE8B8107A83A95B4B344DD3.wapp2nC?docId=WO2021152192&tab=PCTDESCRIPTION
Use of a composition comprising low or very low molecular weight heparins (eg bemiparin sodium) with a high anti-factor Xa activity for the treatment of deep vein thrombosis.
Heparin belongs to the glycosaminoglycan family and is a polysaccharide of animal origin, which is extracted from the intestine or lungs of mammals (cow, lamb, pig) and is used in human therapies for the prevention and treatment of thromboembolic diseases . It is well known that the use of heparin is accompanied by very annoying bleeding effects and its daily administration, three subcutaneous or intravenous injections, constitutes a very considerable inconvenience.
During the course of the last few years, different chemical methods have been used to depolymerize heparin, such as:
– treatment with sodium nitrite in an acid medium,
– alkaline treatment of asters,
– use of free radicals generated in the presence of hydrogen peroxide,
– treatment of a quaternary ammonium salt of heparin in a non-aqueous medium with a strong base according to a beta elimination mechanism.
These methods make it possible to obtain, with variable yields, mixtures of heparin fragments in which the average molecular weight and anticoagulant activity vary according to the procedure and operating conditions. Low molecular weight heparins (LMWH) described in the state of the art or commercialized are obtained according to different depolymerization procedures. Their average molecular weights (Mw) are in the range of 3,600 and 7,500 Daltons.
It is now recognized that the antithrombotic activity of LMWH is mainly due to its ability to activate antithrombin III, a plasma protein and potent inhibitor of activated factor X and thrombin. In this way, it is possible to measure the antithrombotic activity of heparin by means of specific tests to determine the inhibition of these factors.
Research carried out by different authors shows that heparin fragments or oligosaccharides, with short chains of average molecular weight <4,800 Daltons, have a selective action on activated factor X and not on thrombin, in determinations using methods of the Pharmacopoeia. .
It has been found that if very low molecular weight fragments are required that have strong anti-factor Xa activity, it is preferable to use a selective depolymerization technique in non-aqueous medium, as described in US patent 9,981,955, which respects the antithrombin III binding site.
The document EP 1070503 A1 describes the controlled depolymerization of heparin using a process in a non-aqueous medium that makes it possible to obtain a family of LMWH that are obtained enriched in low molecular weight oligosaccharides that have a high anti-factor Xa activity and a low anti-factor lia activity, and which can be represented by the general formula:
in which:
n can vary between 1 and 12,
Ri = H or S0 3 Na,
R 2 = SOsNao COCH 3 ,
Said very low molecular weight heparin is obtained by selective depolymerization of heparin in a non-aqueous medium according to a beta elimination procedure.
Medical uses
Bemiparin is used for the prevention of thromboembolism after surgery, and to prevent blood clotting in the extracorporeal circuit in haemodialysis.[2]
Contraindications
The medication is contraindicated in patients with a history of heparin-induced thrombocytopenia with or without disseminated intravascular coagulation; acute bleeding or risk of bleeding; injury or surgery of the central nervous system, eyes or ears; severe liver or pancreas impairment; and acute or subacute bacterial endocarditis.[2]
Interactions
No interaction studies have been conducted. Drugs that are expected to increase the risk of bleeding in combination with bemiparin include other anticoagulants, aspirin and other NSAIDs, antiplatelet drugs, and corticosteroids.[2]
Chemistry
Like semuloparin, bemiparin is classified as an ultra-LMWH because of its low molecular mass of 3600 g/mol on average.[3] (Enoxaparin has 4500 g/mol.) These heparins have lower anti-thrombin activity than classical LMWHs and act mainly on factor Xa, reducing the risk of bleeding.[4]
References
- ^ Chapman TM, Goa KL (2003). “Bemiparin: a review of its use in the prevention of venous thromboembolism and treatment of deep vein thrombosis”. Drugs. 63 (21): 2357–77. doi:10.2165/00003495-200363210-00009. PMID 14524738.
- ^ Jump up to:a b c Austria-Codex (in German). Vienna: Österreichischer Apothekerverlag. 2018. Ivor 2500 IE Anti-Xa/0,2 ml Injektionslösung in Fertigspritzen.
- ^ Planès A (September 2003). “Review of bemiparin sodium–a new second-generation low molecular weight heparin and its applications in venous thromboembolism”. Expert Opinion on Pharmacotherapy. 4 (9): 1551–61. doi:10.1517/14656566.4.9.1551. PMID 12943485. S2CID 13566575.
- ^ Jeske WP, Hoppensteadt D, Gray A, Walenga JM, Cunanan J, Myers L, Fareed J, Bayol A, Rigal H, Viskov C (October 2011). “A common standard is inappropriate for determining the potency of ultra low molecular weight heparins such as semuloparin and bemiparin”. Thrombosis Research. 128 (4): 361–7. doi:10.1016/j.thromres.2011.03.001. PMID 21458847.
External links
- bemiparin at the US National Library of Medicine Medical Subject Headings (MeSH)
| Clinical data | |
|---|---|
| Trade names | Badyket, Ivor, Hibor, Zibor, others |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration | Subcutaneous injection (except for haemodialysis) |
| ATC code | B01AB12 (WHO) |
| Pharmacokinetic data | |
| Bioavailability | 96% (estimated) |
| Elimination half-life | 5–6 hours |
| Identifiers | |
| CAS Number | 91449-79-5 |
| DrugBank | DB09258 |
| ChemSpider | none |
| Chemical and physical data | |
| Molar mass | 3600 g/mol (average) |
| (what is this?) (verify) |
- Chapman TM, Goa KL: Bemiparin: a review of its use in the prevention of venous thromboembolism and treatment of deep vein thrombosis. Drugs. 2003;63(21):2357-77. [Article]
- Planes A: Review of bemiparin sodium–a new second-generation low molecular weight heparin and its applications in venous thromboembolism. Expert Opin Pharmacother. 2003 Sep;4(9):1551-61. [Article]
- Jeske WP, Hoppensteadt D, Gray A, Walenga JM, Cunanan J, Myers L, Fareed J, Bayol A, Rigal H, Viskov C: A common standard is inappropriate for determining the potency of ultra low molecular weight heparins such as semuloparin and bemiparin. Thromb Res. 2011 Oct;128(4):361-7. doi: 10.1016/j.thromres.2011.03.001. Epub 2011 Apr 2. [Article]
- Sanchez-Ferrer CF: Bemiparin: pharmacological profile. Drugs. 2010 Dec 14;70 Suppl 2:19-23. doi: 10.2165/1158581-S0-000000000-00000. [Article]
- Hoffman M, Monroe DM: Coagulation 2006: a modern view of hemostasis. Hematol Oncol Clin North Am. 2007 Feb;21(1):1-11. doi: 10.1016/j.hoc.2006.11.004. [Article]
- Antonijoan RM, Rico S, Martinez-Gonzalez J, Borrell M, Valcarcel D, Fontcuberta J, Barbanoj MJ: Comparative pharmacodynamic time-course of bemiparin and enoxaparin in healthy volunteers. Int J Clin Pharmacol Ther. 2009 Dec;47(12):726-32. [Article]
- Irish Medicines Board: Bemiparin [Link]
- Hibor-Bemiparin Sodium [Link]
- Zibor 2,500 IU Solution for Injection [Link]
- Injectable drugs guide [Link]
- Thrombosis Advisors- Factor Xa inhibitor [Link]
- Anti-tumor effects of bemiparin in HepG2 and MIA PaCa-2 cells [Link]
- Bemiparin, an effective and safe low molecular weight heparin: a review [Link]
- Bemiparin sodium [Link]
Patent
Publication numberPriority datePublication dateAssigneeTitleUS4981955A *1988-06-281991-01-01Lopez Lorenzo LDepolymerization method of heparinEP0293539B1 *1987-01-051994-06-08Laboratorios Farmaceuticos Rovi, S.A.Process for the depolymerization of heparin for obtaining heparin with a low molecular weight and having an antithrombotic activityCN1379781A *1999-10-222002-11-13阿文蒂斯药物股份有限公司Novel oligosaccharides, preparation method and pharmaceutical composition containing sameCN102399306A *2010-09-092012-04-04上海喜恩医药科技发展有限公司Preparation method of heparin-derived polysaccharide mixtureCN105693886A *2016-04-192016-06-22常州市蓝勖化工有限公司Preparation method of heparin sodiumCN106467577A *2015-08-212017-03-01苏州融析生物科技有限公司A kind of pulmonis Bovis seu Bubali Enoxaparin Sodium and preparation method and applicationCN106977627A *2017-05-162017-07-25苏州二叶制药有限公司A kind of Enoxaparin production method of sodiumCN109575156A *2018-11-052019-04-05上海宝维医药技术有限公司A kind of purification process of low molecular weight heparinFamily To Family Citations
////////////Bemiparin sodium, Bemiparin
TRK 700
![1-[4-(Dimethylamino)piperidin-1-yl]-3-(1-methylimidazol-2-yl)propan-1-one.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=71738795&t=l)
TRK-700
CAS 1463432-16-7C14 H24 N4 O264.371-Propanone, 1-[4-(dimethylamino)-1-piperidinyl]-3-(1-methyl-1H-imidazol-2-yl)-
1-[4-(dimethylamino)piperidin-1-yl]-3-(1-methylimidazol-2-yl)propan-1-one
- 1-[4-(Dimethylamino)-1-piperidinyl]-3-(1-methyl-1H-imidazol-2-yl)-1-propanone
- OriginatorToray Industries
- ClassAnalgesics
- Mechanism of ActionUndefined mechanism
- Phase IIPostherpetic neuralgia
- PreclinicalPeripheral nervous system diseases
- 12 Sep 2018Pharmacodynamics data from a preclinical trial in Peripheral neuropathy presented at the 17th World Congress on Pain (WCP-2018)
- 01 Jul 2017Toray Industries completes a phase II trial for Postherpetic neuralgia (In adults, In the elderly) in Japan (PO) (NCT02701374)
- 21 May 2017Toray Industries completes a phase I drug-drug interaction trial in Healthy volunteers in Japan (PO) (NCT03043248)
developed by Toray for treating neuropathic pain and investigating for fibromyalgia. In August 2021, this drug was reported to be in phase 1 clinical development.
PATENT
WO 2016136944
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016136944

(Reference Example 22) Synthesis of (E) -methyl 3- (1-methyl-1H-imidazol-2-yl) acrylate:
[Chemical 56]
1-methyl-1H-imidazol-2-carbaldehyde (10.0 g, Methyl (triphenylphosphoranylidene) acetate (33.4 g, 99.9 mmol) was added to a solution of 90.8 mmol) in dichloromethane (240 mL) at room temperature, and the mixture was stirred for 16 hours and then concentrated under reduced pressure. The residue was washed with a mixed solvent of hexane / dichloromethane = 19/1, and the washing liquid was concentrated. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give (E) -methyl 3- (1-methyl-1H-imidazol-2-yl) acrylate as a white solid (11.9 g, 71. 6 mmol, 79%).
1 H-NMR (400 MHz, CDCl 3 ) δ: 3.76 (3H, s), 3.81 (3H, s), 6.82 (1H, d, J = 15.6 Hz), 6.98 (1H, brs), 7.16 (1H, brs), 7.53 (1H, d, J = 15.6Hz).
ESI-MS: m / z = 167 (M + H) + .
(Reference Example 27) Synthesis of 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan-1-one:
[Chemical 61]
(E) )-Methyl 3- (1-methyl-1H-imidazol-2-yl) acrylate (0.180 g, 1.08 mmol) in ethanol (4.0 mL) solution of palladium-carbon (10% wet, 15 mg) at room temperature In a hydrogen atmosphere, the mixture was stirred for 4 hours. The reaction mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure. Methanol (1.0 mL) was added to the obtained residue at room temperature to dissolve it, and the mixture was cooled to 0 ° C. An aqueous sodium hydroxide solution (1.0 N, 1.19 mL, 1.19 mmol) was added to the reaction solution at 0 ° C., the mixture was stirred at room temperature for 2 hours, and then concentrated under reduced pressure. Chloroform (10.0 mL) was added to the obtained residue at room temperature to dissolve it. Add diisopropylethylamine (0.568 mL, 3.25 mmol), HBTU (0.616 g, 1.63 mmol) and 4- (dimethylamino) piperidine (0.125 g, 0.975 mmol) to the reaction solution at room temperature, and add the reaction solution. The mixture was stirred at the same temperature for 16 hours. A saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with chloroform. The organic layer was washed with a 10% aqueous sodium chloride solution, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by flash column chromatography (NH silica gel, chloroform / methanol) and 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propane. -1-one (0.179 g, 0.68 mmol, 63%) was obtained as a colorless oil.
1 1 H-NMR (400 MHz, CDCl 3) δ: 1.29-1.43 (2H, m), 1.80-1.88 (2H, m), 2.27 (6H, s), 2.29-2.38 (1H, m), 2.54-2.63 (1H, m), 2.88-3.04 ( 5H, m), 3.62 (3H, s), 3.98-4.05 (1H, m), 4.57-4.65 (1H, m), 6.79 (1H, d, J = 1.2 Hz), 6.91 (1H, d, J = 1.2 Hz).
ESI-MS: m / z = 265 (M + H) + .
(Comparative Example 1) Synthesis of 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan-1-one hydrochloride:
[Chemical 66]
1- (4- (Dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan-1-one (1.50 g, 5.67 mmol) diethyl ether (60) A dioxane solution of hydrogen chloride (4.0 M, 3.69 mL, 14.8 mmol) was added to the (0.0 mL) solution at 0 ° C. The reaction mixture was stirred at the same temperature for 1 hour and then at room temperature for 30 minutes. The precipitated white solid was collected by filtration, washed with diethyl ether (100 mL), dried at room temperature for 36 hours, and then 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-). Imidazole-2-yl) propan-1-one hydrochloride (1.41 g, 4.18 mmol, 74%) (hereinafter, the compound of Comparative Example 1) was obtained as a white solid.
1 1 H-NMR (400 MHz, D 2 O) δ: 1.53-1.80 (2H, m), 2.12-2.23 (2H, m), 2.68-2.80 (1H, m), 2.88 (6H, s), 3.01- 3.08 (2H, m), 3.15-3.26 (3H, m), 3.47-3.58 (1H, m), 3.84 (3H, s), 4.08-4.16 (1H, m), 4.50-4.59 (1H, m), 7.29-7.33 (2H, m).
ESI-MS; 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) as propan-1-one : m / z = 265 (M + H) + .
(Comparative Example 2) Synthesis of 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan-1-one sulfate monohydrate:
[Chemical 67]
1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan-1-one (6.72 g, 25.4 mmol) Concentrated sulfuric acid (2.49 g, 25.4 mmol), water (1.83 g, 102 mmol) and 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl) in a DMSO (100 mL) solution. Seed crystals (50 mg, 0.13 mmol) of -1H-imidazol-2-yl) propan-1-one sulfate monohydrate were added at 80 ° C. The reaction was stirred at the same temperature for 2.5 hours, at 50 ° C. for 2.5 hours and at room temperature for 15 hours. The precipitated white solid was collected by filtration, washed successively with DMSO (20 mL) and methyl ethyl ketone (40 mL), dried at room temperature, and then 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl). -1H-imidazol-2-yl) propan-1-one sulfate monohydrate (8.42 g, 22.1 mmol, 87%) (hereinafter, the compound of Comparative Example 2) was obtained as white crystals.
1 1 H-NMR (400 MHz, DMSO-d 6)) δ: 1.36 (1H, m), 1.58 (1H, m), 1.95 (2H, br), 2.44-2.57 (1H, m), 2.65 (6H, s), 2.74-2.88 (4H, m), 3.00 (1H, t, J = 12.0 Hz), 3.22 (1H, m), 3.61 (3H, s), 4.02 (1H, d, J = 14.0 Hz), 4.47 (1H, d, J = 12.8 Hz), 6.87 (1H, d, J = 1.2 Hz), 7.11 (1H, d, J = 1.2 Hz).
ESI-MS; 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-) As 1H-imidazol-2-yl) propan-1-one: m / z = 265 (M + H) + .
NEW DRUG APPROVALS
ONE TIME
$10.00
PATENT
WO-2021153744
PATENT
WO-2021153743
Novel crystalline polymorphic form of 1-(4-(dimethylamino) piperidin-1-yl)-3-(1-methyl-1H-imidazol-2-yl)propan-1-one, useful as an analgesic in treating neuropathic pain and/or fibromyalgia.Pain is an experience with unpleasant sensations and emotions that occurs when or may cause tissue damage. Pain is mainly classified into nociceptive pain, neuropathic pain or psychogenic pain according to its cause. In addition, fibromyalgia is known as pain of unknown cause.
Neuropathic pain is pathological pain caused by dysfunction of the peripheral or central nervous system itself, and is caused by direct damage or compression of nervous tissue even though nociceptors are not stimulated. It refers to the pain that occurs. As a therapeutic agent for neuropathic pain, an anticonvulsant, an antidepressant, anxiolytic, or an antiepileptic drug such as gabapentin or pregabalin is used.
Fibromyalgia is a disease in which systemic pain is the main symptom and neuropsychiatric symptoms and autonomic nervous system symptoms are secondary symptoms. Pregabalin approved in the United States and Japan, duloxetine and milnacipran approved in the United States are mainly used as therapeutic agents for fibromyalgia, and non-approved agents for fibromyalgia are not approved. It has also been used for steroidal anti-inflammatory agents, opioid compounds, antidepressants, anticonvulsants and antiepileptic drugs. However, the therapeutic effects of non-steroidal anti-inflammatory drugs and opioid compounds are generally considered to be low (Non-Patent Document 1).
On the other hand, Patent Document 1 discloses that certain substituted piperidins have cardiotonic activity, and Patent Document 2 discloses that an imidazole derivative exhibits an FXa inhibitory effect. Patent Document 3 suggests that the substituted piperidins may have a medicinal effect on overweight or obesity, and Patent Documents 4 to 6 and Non-Patent Document 2 indicate that the imidazole derivative has an analgesic effect. It is disclosed.
In addition, the quality of pharmaceutical products needs to be maintained over a long period of time such as distribution and storage, and the compound as an active ingredient is required to have high chemical and physical stability. Therefore, as the active ingredient of a pharmaceutical product, a crystal that can be expected to have higher stability than an amorphous substance is generally adopted. Further, if crystals are obtained, a purification effect due to recrystallization during production can be expected. Further, it is preferable to have low hygroscopicity from the viewpoint of maintaining stability and handling during manufacturing, storage, formulation and analysis of the drug substance. In addition, since a drug needs to be dissolved in the digestive tract in order to exhibit its medicinal effect, it is preferable that the drug has excellent solubility, which is a physical property contrary to stability.
In order to obtain crystals of a compound that is an active ingredient of a pharmaceutical product, it is necessary to study various conditions for precipitating crystals from the solution. It is common to carry out crystallization under the condition of being dissolved in.
Patent documents
Patent Document 1: French Patent Invention No. 2567885
Patent Document 2: Japanese Patent Application Laid-Open No. 2006-0083664
Patent Document 3: International Publication No. 2003/031432
Patent Document 4: International Publication No. 2013/147160
Patent Document 5: International Publication No. 2015/046403
Patent Document 6: International Publication No. 2016/136944
Non-patent literature
Non-Patent Document 1: Okifuji et al., Pain and Therapy, 2013, Volume 2, p. 87-104
Non-Patent Document 2: Takahashi et al., Toxicological Pathology, 2019, Vol. 47. p. 494-503
Compound (I) was synthesized by the method described in the following reference example. For the compounds used in the synthesis of the reference example compounds for which the synthesis method is not described, commercially available compounds were used.
(Reference Example 4) Synthesis of amorphous compound (I):
[Chemical formula 2] 2 of
crude ethyl 3- (1-methyl-1H-imidazol-2-yl) propanol (5.00 g, 27.4 mmol) Aqueous sodium hydroxide solution (1.0N, 30.2 mL, 30.2 mmol) was added to a solution of -propanol (55 mL) at 0 ° C., and the mixture was stirred at room temperature for 12 hours. 2-Propanol (220 mL) was added to the reaction solution at room temperature, and crude 4- (dimethylamino) piperidine (3.17 g, 24.7 mmol) and DMT-MM (8.35 g, 30.2 mmol) were added at room temperature to react. The liquid was stirred at the same temperature for 3 hours. A 10% aqueous sodium chloride solution and a 1.0N aqueous sodium hydroxide solution were added to the reaction mixture, and the mixture was extracted with chloroform. The organic layer was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to give compound (I) (6.98 g) as an amorphous substance.
1 1 H-NMR (400 MHz, CDCl 3 ) δ: 1.29-1.43 (2H, m), 1.80-1.88 (2H, m), 2.27 (6H, s), 2.29-2.38 (1H, m), 2.54-2.63 (1H, m), 2.88-3.04 (5H, m), 3.62 (3H, s), 3.98-4.05 (1H, m), 4.57-4.65 (1H, m), 6.79 (1H, d, J = 1.2 Hz) ), 6.91 (1H, d, J = 1.2 Hz).
ESI-MS: m / z = 265 (M + H) + .
(Reference Example 5) Synthesis of crude 4- (dimethylamino) piperidine:
[Chemical
formula 3] 1-benzyloxycarbonyl-4- (dimethylamino) piperidine (20.1 g, 77.0 mmol) in methanol (154.0 mL) Palladium-carbon (10% wet, 2.01 g) was added thereto, and the mixture was stirred at room temperature for 19 hours under a hydrogen atmosphere. The reaction mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure to give a crude product of 4- (dimethylamino) piperidine (9.86 g).
(Reference Example 6) Synthesis of crude ethyl 3- (1-methyl-1H-imidazol-2-yl) propanoate:
[Chemical
formula 4] Sodium hydride (55%, 4.36 g, 100 mmol) aqueous solution and tetrahydrofuran (150 mL) To the mixture was added triethylphosphonoacetate (19.1 mL, 95.0 mmol) at 0 ° C. After stirring the reaction solution for 20 minutes, a solution of 1-methyl-1H-imidazol-2-carbaldehyde (10.0 g, 91.0 mmol) in tetrahydrofuran (150 mL) was added at 0 ° C., and then ethanol (30 mL) was added in the same manner. The mixture was added at temperature and stirred at room temperature for 2 hours. A 10% aqueous sodium chloride solution was added to the reaction mixture, and the mixture was extracted with dichloromethane. The organic layer was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, chloroform / methanol). After adding methanol (310 mL) to the residue, palladium-carbon (10% wet, 1.40 g) was added, and the mixture was stirred at room temperature for 3 hours under a hydrogen atmosphere. The reaction mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure to obtain a crude product (14.2 g) of ethyl 3- (1-methyl-1H-imidazol-2-yl) propanoate.
(Reference Example 7) Synthesis of 1-benzyloxycarbonyl-4- (dimethylamino) piperidine:
[Chemical
formula 5] dichloromethane (55.7 mL) of 1-benzyloxycarbonyl-4-oxopiperidine (13.0 g, 55.7 mmol) ) Solution of dimethylamine in tetrahydrofuran (2.0 M, 34.8 mL, 69.7 mmol), acetic acid (0.32 mL, 5.6 mmol) and sodium triacetoxyborohydride (4.8 g, 22.6 mmol). Added at ° C. After stirring the reaction solution at the same temperature for 30 minutes, sodium triacetoxyborohydride (4.8 g, 22.6 mmol) was added at 0 ° C. The reaction mixture was stirred at the same temperature for 30 minutes, sodium triacetoxyborohydride (8.1 g, 38.2 mmol) was added at 0 ° C., and the mixture was stirred at room temperature for 12 hours. The reaction solution was cooled to 0 ° C. A saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with chloroform. The organic layer was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, n-hexane / ethyl acetate) and then again by flash chromatography (silica gel, chloroform / methanol) to obtain 1-benzyloxycarbonyl-4- (dimethylamino) piperidine (dimethylamino) piperidine. 13.6 g, 51.8 mmol, 93%) was obtained as a colorless oil.
1 1 H-NMR (400 MHz, CDCl 3) δ: 1.34-1.46 (2H, m), 1.78-1.86 (2H, m), 2.28 (6H, s), 2.29-2.34 (1H, m), 2.75-2.85 (2H, m), 4.14-4.28 ( 2H, m), 5.12 (2H, s), 7.29-7.36 (5H, m).
ESI-MS: m / z = 263 (M + H) + .
(Reference Example 8) Synthesis of 1-benzyloxycarbonyl-4-oxopiperidine:
[Chemical
formula 6] Hydrochloride (130 mL) and water (130 mL) of 4-piperidinone hydrochloride monohydrate (10.0 g, 65.1 mmol) Sodium carbonate (13.8 g, 130.2 mmol) and benzyl chloroformate (8.79 mL, 61.8 mmol) were added to the mixed solution with and at 0 ° C., and the mixture was stirred at room temperature for 3 hours. The reaction mixture was extracted with ethyl acetate. The organic layer was washed with 10% aqueous sodium chloride solution, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, n-hexane / ethyl acetate) to give 1-benzyloxycarbonyl-4-oxopiperidine (13.1 g, 56.2 mmol, 86%) as a colorless oil.
1 1 H-NMR (400 MHz, CDCl 3 ) δ: 2.42-2.50 (4H, m), 3.78-3.82 (4H, m), 5.18 (2H, s), 7.32-7.38 (5H, m).
(Example 1) Production of A-type crystal of
compound (I): Amorphous compound (6.98 g) of compound (I) prepared in Reference Example 4 is purified and concentrated with chloroform / methanol by silica gel column chromatography. After that, the wall surface of the flask was rubbed with a spartel and mechanical stimulation was applied to obtain A-type crystals of compound (I) as a powder. For the obtained crystals, measurement of powder X-ray diffraction using a powder X-ray diffractometer (Rigaku Co., Ltd .; 2200 / RINT ultima + PC) and TG-DTA using a TG-DTA device (Rigaku Co., Ltd .; TG8120) Was done. The results of these measurements are shown in FIGS. 1 and 2.
Diffraction angle 2θ: 5.9, 16.5, 17.7, 20.8, 26.7 °
Endothermic peak: 55 ° C
PATENT
WO2013147160
Example 1 Synthesis of 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan-1-one:
[Chemical 27]
(E) )-Methyl 3- (1-methyl-1H-imidazol-2-yl) acrylate (0.180 g, 1.08 mmol) in ethanol (4.0 mL) solution of palladium-carbon (10% wet, 15 mg) at room temperature In a hydrogen atmosphere, the mixture was stirred for 4 hours. The reaction mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure. Methanol (1.0 mL) was added to the obtained residue at room temperature to dissolve it, and the mixture was cooled to 0 ° C. An aqueous sodium hydroxide solution (1.0 N, 1.19 mL, 1.19 mmol) was added to the reaction solution at 0 ° C., the mixture was stirred at room temperature for 2 hours, and then concentrated under reduced pressure. Chloroform (10.0 mL) was added to the obtained residue at room temperature to dissolve it. Add diisopropylethylamine (0.568 mL, 3.25 mmol), HBTU (0.616 g, 1.63 mmol) and 4- (dimethylamino) piperidine (0.125 g, 0.975 mmol) to the reaction solution at room temperature, and add the reaction solution. The mixture was stirred at the same temperature for 16 hours. A saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with chloroform. The organic layer was washed with a 10% aqueous sodium chloride solution, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography (NH silica gel, chloroform / methanol) and 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan- 1-one (0.179 g, 0.68 mmol, 63%) (hereinafter, the compound of Example 1) was obtained as a colorless oil.
1 1 H-NMR (400 MHz, CDCl 3) δ: 1.29-1.43 (2H, m), 1.80-1.88 (2H, m), 2.27 (6H, s), 2.29-2.38 (1H, m), 2.54-2.63 (1H, m), 2.88-3.04 ( 5H, m), 3.62 (3H, s), 3.98-4.05 (1H, m), 4.57-4.65 (1H, m), 6.79 (1H, d, J = 1.2 Hz), 6.91 (1H, d, J = 1.2 Hz).
ESI-MS: m / z = 265 (M + H) + .
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| WO-2016136944-A1 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | |
| JP-WO2013147160-A1 | Cyclic amine derivatives and their pharmaceutical use | 2012-03-29 | |
| TW-201350119-A | Cyclic amine derivatives and their medical uses | 2012-03-29 | |
| WO-2013147160-A1 | Cyclic amine derivative and use thereof for medical purposes | 2012-03-29 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| RU-2667062-C1 | Dynamic cyclic amine and pharmaceutical application thereof | 2015-02-27 | 2018-09-14 |
| TW-201639826-A | Cyclic amine derivatives and their medical uses | 2015-02-27 | |
| TW-I682927-B | Cyclic amine derivatives and their medical uses | 2015-02-27 | 2020-01-21 |
| US-10173999-B2 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | 2019-01-08 |
| US-2018065950-A1 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| EP-3263565-A1 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | |
| EP-3263565-B1 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | 2019-06-26 |
| ES-2744785-T3 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | 2020-02-26 |
| JP-6569671-B2 | Cyclic amine derivatives and their pharmaceutical use | 2015-02-27 | 2019-09-04 |
| JP-WO2016136944-A1 | Cyclic amine derivatives and their pharmaceutical use | 2015-02-27 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| WO-2019189781-A1 | Agent for inhibiting rise in intraneuronal calcium concentration | 2018-03-30 | |
| AU-2016224420-A1 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | |
| AU-2016224420-B2 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | 2019-08-22 |
| CA-2977614-A1 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | |
| CN-107250128-B | Cyclic amine derivatives and its medical usage | 2015-02-27 | 2019-07-26 |
//////////TRK-700, phase 1, neuropathic pain, fibromyalgia, toray
O=C(CCc1nccn1C)N1CCC(CC1)N(C)C
Rutoside, Rutin


Rutoside
RUTIN
- Molecular FormulaC27H30O16
- Average mass610.518
- рутозид [Russian] [INN]ルチン [Japanese]روتوسيد [Arabic] [INN]芦丁 [Chinese] [INN]
CAS 153-18-4
- C.I. 75730
- NSC-9220
2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-3-[(2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-[[(2R,3R,4R,5R,6S)-3,4,5-trihydroxy-6-methyloxan-2-yl]oxymethyl]oxan-2-yl]oxychromen-4-one


Rutin trihydrate | CAS 250249-75-3 RutinCAS Registry Number: 153-18-4
CAS Name: 3-[[6-O-(6-Deoxy-a-L-mannopyranosyl)-b-D-glucopyranosyl]oxy]-2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-4H-1-benzopyran-4-one
Additional Names: rutoside; quercetin-3-rutinoside; 3,3¢,4¢,5,7-pentahydroxyflavone-3-rutinoside; melin; phytomelin; eldrin; ilixathin; sophorin; globularicitrin; paliuroside; osyritrin; osyritin; myrticolorin; violaquercitrin
Trademarks: Birutan (Merck KGaA)
Molecular Formula: C27H30O16Molecular Weight: 610.52Percent Composition: C 53.12%, H 4.95%, O 41.93%
Literature References: Identity with ilixanthin: Schindler, Herb, Arch. Pharm.288, 372 (1955). Found in many plants, especially the buckwheat plant (Fagopyrum esculentum Moench., Polygonaceae) which contains about 3% (dry basis): Couch et al.,Science103, 197 (1946). From tobacco (Nicotiana tabacum L., Solanaceae) Couch, Krewson, U.S. Dept. Agr., Eastern Regional Res. Lab.AIC-52 (1944).
In forsythia [Forsythia suspensa (Thunb.) Vahl. var. fortunei (Lindl.) Rehd., Oleaceae], in hydrangea (Hydrangea paniculata Sieb., Saxifragaceae), in pansies (Viola sp., Violaceae).
General extraction procedure: BeilsteinXXXI, 376. From leaves of Eucalyptus macroryncha F. v. Muell., Myrtaceae: Attree, Perkin, J. Chem. Soc.1927, 234.
Industrial production from Eucalyptus spp.: Humphreys, Econ. Bot.18, 195 (1964).
Structure: Zemplén, Gerecs, Ber.68B, 1318 (1935).
Synthesis: Shakhova et al.,Zh. Obshch. Khim.32, 390 (1962), C.A.58, 1426e (1963). Rutin is hydrolyzed by rhamnodiastase from the seed of Rhamnus utilis Decne, Rhamnaceae (Chinese buckthorn); emulsin is not effective: Bridel, Charaux, Compt. Rend.181, 925 (1925). Toxicity data: Harrison et al.,J. Am. Pharm. Assoc.39, 557 (1950). Book: J. Q. Griffith, Jr., Rutin and Related Flavonoids (Mack, Easton, Pa., 1955).
Comprehensive description: T. I. Khalifa et al.,Anal. Profiles Drug Subs.12, 623-681 (1983).UV






Properties: Pale yellow needles from water, gradual darkening on exposure to light. The crystals contain 3 H2O and become anhydr after 12 hrs at 110° and 10 mm Hg. Anhydr rutin browns at 125°, becomes plastic at 195-197°, and dec 214-215° (with effervescence). [a]D23 +13.82° (ethanol); [a]D23 -39.43° (pyridine). Anhydr rutin is hygroscopic. One gram dissolves in about 8 liters water, about 200 ml boiling water, 7 ml boiling methanol. Sol in pyridine, formamide and alkaline solns; slightly sol in alcohol, acetone, ethyl acetate. Practically insol in chloroform, carbon bisulfide, ether, benzene, petr solvents. Dil solns give green color with ferric chloride. Rutin is colored brown by tobacco enzyme under experimental conditions: Neuberg, Kobel, Naturwissenschaften23, 800 (1935). LD50 i.v. in mice: 950 mg/kg (propylene glycol soln) (Harrison).
Optical Rotation: [a]D23 +13.82° (ethanol); [a]D23 -39.43° (pyridine)
Toxicity data: LD50 i.v. in mice: 950 mg/kg (propylene glycol soln) (Harrison)Therap-Cat: Capillary protectant.Keywords: Vasoprotectant.
C13




MASS

Rutin, also called rutoside, quercetin-3-O-rutinoside and sophorin, is the glycoside combining the flavonol quercetin and the disaccharide rutinose (α-L-rhamnopyranosyl-(1→6)-β-D-glucopyranose). It is a citrus flavonoid found in a wide variety of plants including citrus.
Rutin, also called rutoside, is the glycoside flavonoid found in a certain fruits and vegetables. Most rutine-rich foods are capers, olives, buckwheat (whole grain flour), asparagus, raspberry.In a clinical trial, rutin was found to aid control of intraocular pressure in patients with primary open angle glaucoma. As a component of dietary supplement Phlogenzym, rutin is used for treatment of osteoarthritis. Rutin is also used for treatment of post-surgical swelling of the arm after breast cancer surgery. Traditionally, rutin is used to prevent mucositis due to cancer treatment, to treat blood vessel disease such as varicose veins, bleeding, hemorrhoids.
Occurrences
Rutin is one of the phenolic compounds found in the invasive plant species Carpobrotus edulis and contributes to the antibacterial[3] properties of the plant.
Its name comes from the name of Ruta graveolens, a plant that also contains rutin.
Various citrus fruit peels contain 32 to 49 mg/g of flavonoids expressed as rutin equivalents.[4]
Citrus leaves contain rutin at concentrations of 11 and 7 g/kg in orange and lime trees respectively.[5]
Metabolism
The enzyme quercitrinase can be found in Aspergillus flavus.[6] It is an enzyme in the rutin catabolic pathway.[7]
In food
Rutin is a citrus flavonoid glycoside found in many plants including buckwheat,[8] the leaves and petioles of Rheum species, and asparagus. Tartary buckwheat seeds have been found to contain more rutin (about 0.8–1.7% dry weight) than common buckwheat seeds (0.01% dry weight).[8] Rutin is one of the primary flavonols found in ‘clingstone’ peaches.[9] It is also found in green tea infusions.[10]
Approximate rutin content per 100g of selected foods, in milligrams per 100 milliliters:[11]
| Numeric | Alphabetic |
|---|---|
| 332 | Capers, spice |
| 45 | Olive [Black], raw |
| 36 | Buckwheat, whole grain flour |
| 23 | Asparagus, raw |
| 19 | Black raspberry, raw |
| 11 | Red raspberry, raw |
| 9 | Buckwheat, groats, thermally treated |
| 6 | Buckwheat, refined flour |
| 6 | Greencurrant |
| 6 | Plum, fresh |
| 5 | Blackcurrant, raw |
| 4 | Blackberry, raw |
| 3 | Tomato (Cherry), whole, raw |
| 2 | Prune |
| 2 | Fenugreek |
| 2 | Marjoram, dried |
| 2 | Tea (Black), infusion |
| 1 | Grape, raisin |
| 1 | Zucchini, raw |
| 1 | Apricot, raw |
| 1 | Tea (Green), infusion |
| 0 | Apple |
| 0 | Redcurrant |
| 0 | Grape (green) |
| 0 | Tomato, whole, raw |
Research
Rutin (rutoside or rutinoside)[12] and other dietary flavonols are under preliminary clinical research for their potential biological effects, such as in reducing post-thrombotic syndrome, venous insufficiency, or endothelial dysfunction, but there was no high-quality evidence for their safe and effective uses as of 2018.[12][13][14][needs update] As a flavonol among similar flavonoids, rutin has low bioavailability due to poor absorption, high metabolism, and rapid excretion that collectively make its potential for use as a therapeutic agent limited.[12]
Biosynthesis
The biosynthesis pathway of rutin in mulberry (Morus alba L.) leaves begins with phenylalanine, which produces cinnamic acid under the action of phenylalanine ammonia lyase (PAL). Cinnamic acid is catalyzed by cinnamic acid-4-hydroxylase (C4H) and 4-coumarate-CoA ligase (4CL) to form p–coumaroyl-CoA. Subsequently, chalcone synthase (CHS) catalyzes the condensation of p-coumaroyl-CoA and three molecules of malonyl-CoA to produce naringenin chalcone, which is eventually converted into naringenin flavanone with the participation of chalcone isomerase (CHI). With the action of flavanone 3-hydroxylas (F3H), dihydrokaempferol (DHK) is generated. DHK can be further hydroxylated by flavonoid 3´-hydroxylase (F3’H) to produce dihydroquercetin (DHQ), which is then catalyzed by flavonol synthase (FLS) to form quercetin. After quercetin is catalyzed by UDP-glucose flavonoid 3-O-glucosyltransferase (UFGT) to form isoquercitrin, finally, the formation of rutin from isoquercitrin is catalyzed by flavonoid 3-O-glucoside L-rhamnosyltransferase.[15]

SYN
https://www.sciencedirect.com/science/article/abs/pii/S100184171300017X
The compound 2 was synthesized for the first time by highly selective esterification reaction and fully characterized. The by-products of the reaction were complex, which brought out many considerable difficulties in separation and purification of the target product. Our work was the first in using the improved pyrogallol autoxidation method to test the antioxidant activities of these two flavonoids compounds in vitro and discovered that the compound 2 was much more effective as a free radical scavenger than the compound 1.

SYN
Synthesis of Rutin
The synthesis of rutin can be achieved according to the following three schemes. These schemes differ in the synthesis of ouercetin (the aglycone moiety of rutin).
Scheme 1: Kostanecki — et al. 1904 (33 ). Based upon the Claisen reaction between 2-hydroxy4, 6-dimethoxyacetophenone [l] and 3, 4-dimethoxybenzaldehyde [2] to give the intermediate [3] which upon treatment with HC1, cyclization occurs to give 5, 7, 3 , which upon treatment with F2SO4 enolisation occurs to give 5, 7, 1’3 ,’4 -tetramethoxyflavonol [6]. tion with HI affords quercetin [7].
Scheme 2: Robinson et al. 1926 (34 ). , ‘4 -tetramethoxyflavonone [4]. Oximination affords [5] Demethyla- — Condensation ofw-methoxypholoroacetophenone [I] with veratric acid anhydride [2] in the presence of the potassium salt of veratric acid to give the diarylester [3]. On hydrolysis with alcoholic KOH affords 5, 7-dihydroxy-3, /3 , ‘4 -trimethoxyf lavone [ 41 , which on demethylation with HI gives quercetin [5].
Scheme 3: Shakhova et al. 1962 (35), complete synthesis of rutin. W-methoxyphloroacetophenone [2] was condensed with 0-benzylvanillinic acid, anhydride [ 13 in triethylamine to give 5 , 7-dihydroxy-4 -benzyloxy-3, /3 -dimethoxyf lavone [3]. On treatment with AcOH-HC1 mixture gave 5, 7, ‘4 -trihydroxy-3,’3 -dimethoxyflavone [4]. Demethylation of the latter with HI yielded (about 802) quercetin [5]. Ouercetin potassium salt [6] was produced upon treating [5] with AcOK in ethanol. Levoglucosan [7] was acetylated with Ac20 in the presence of AcONa to give 2, 3, 4-triacetyllevoglucosan [8] which with TIC14 gave 1-chloro-2, 3, 4-triacetyl Dglucose [9]. L-rhamnose tetraacetate [lo] treated with TiBr4 in CHC13 gave 1-bromo-2, 3, I-triacetyl-L-rhamnose [ll]. [lo] + [11] heated with Hg (OAC)~ in C6H6 gave (53x) CC – acetochloro-f3-l-L-rhamnosido-6-D-glucose [12]. [12] was treated with AgOAc and acetylated with Ac20 to prodilce (68.703 B-heptaacet yl-f3-1-L-rhamnos ido-6-D-glucose [13]. This with 33% HBr in AcOH gave (61%) d – acetobromo-~-l-L-rhamnosido-6-D-glucose [14]. [14] and quercetin potassium salt [6] were dissolved in NH40H which was evaporated and treated with methanol andpurified over a chromatographic column packed with polycaprolactum resin to give rutin [151.









References
- ^ Merck Index, 12th Edition, 8456
- ^ Krewson CF, Naghski J (Nov 1952). “Some physical properties of rutin”. Journal of the American Pharmaceutical Association. 41 (11): 582–7. doi:10.1002/jps.3030411106. PMID 12999623.
- ^ van der Watt E, Pretorius JC (2001). “Purification and identification of active antibacterial components in Carpobrotusedulis L.”. Journal of Ethnopharmacology. 76 (1): 87–91. doi:10.1016/S0378-8741(01)00197-0. PMID 11378287.
- ^ [1] p. 280 Table 1
- ^ [2] p.8 fig. 7
- ^ quercitrinase on www.brenda-enzymes.org
- ^ Tranchimand S, Brouant P, Iacazio G (Nov 2010). “The rutin catabolic pathway with special emphasis on quercetinase”. Biodegradation. 21 (6): 833–59. doi:10.1007/s10532-010-9359-7. PMID 20419500. S2CID 30101803.
- ^ Jump up to:a b Kreft S, Knapp M, Kreft I (Nov 1999). “Extraction of rutin from buckwheat (Fagopyrum esculentumMoench) seeds and determination by capillary electrophoresis”. Journal of Agricultural and Food Chemistry. 47 (11): 4649–52. doi:10.1021/jf990186p. PMID 10552865.
- ^ Chang S, Tan C, Frankel EN, Barrett DM (Feb 2000). “Low-density lipoprotein antioxidant activity of phenolic compounds and polyphenol oxidase activity in selected clingstone peach cultivars”. Journal of Agricultural and Food Chemistry. 48 (2): 147–51. doi:10.1021/jf9904564. PMID 10691607.
- ^ Malagutti AR, Zuin V, Cavalheiro ÉT, Henrique Mazo L (2006). “Determination of Rutin in Green Tea Infusions Using Square-Wave Voltammetry with a Rigid Carbon-Polyurethane Composite Electrode”. Electroanalysis. 18 (10): 1028–1034. doi:10.1002/elan.200603496.
- ^ “foods in which the polyphenol Quercetin 3-O-rutinoside is found”. Phenol-Explorer v 3.6. June 2015.
- ^ Jump up to:a b c “Flavonoids”. Micronutrient Information Center, Linus Pauling Institute, Oregon State University, Corvallis, Oregon. November 2015. Retrieved 25 February 2018.
- ^ Morling, J. R; Yeoh, S. E; Kolbach, D. N (November 2018). “Rutosides for treatment of post-thrombotic syndrome”. Cochrane Database of Systematic Reviews. 11 (11): CD005625. doi:10.1002/14651858.CD005625.pub4. PMC 6517027. PMID 30406640.
- ^ Martinez-Zapata, M. J; Vernooij, R. W; Uriona Tuma, S. M; Stein, A. T; Moreno, R. M; Vargas, E; Capellà, D; Bonfill Cosp, X (2016). “Phlebotonics for venous insufficiency”. Cochrane Database of Systematic Reviews. 4: CD003229. doi:10.1002/14651858.CD003229.pub3. PMC 7173720. PMID 27048768.
- ^ Yu X, Liu J, Wan J, Zhao L, Liu Y, Wei Y, Ouyang Z. Cloning, prokaryotic expression, and enzyme activity of a UDP-glucose flavonoid 3-o-glycosyltransferase from mulberry (Morus alba L.) leaves. Phcog Mag 2020;16:441-7
| Names | |
|---|---|
| IUPAC name3′,4′,5,7-Tetrahydroxy-3-[α-L-rhamnopyranosyl-(1→6)-β-D-glucopyranosyloxy]flavone | |
| Preferred IUPAC name(42S,43R,44S,45S,46R,72R,73R,74R,75R,76S)-13,14,25,27,43,44,45,73,74,75-Decahydroxy-76-methyl-24H-3,6-dioxa-2(2,3)-[1]benzopyrana-4(2,6),7(2)-bis(oxana)-1(1)-benzenaheptaphane-24-one | |
| Other namesRutoside (INN) Phytomelin Sophorin Birutan Eldrin Birutan Forte Rutin trihydrate Globularicitrin Violaquercitrin Quercetin rutinoside | |
| Identifiers | |
| CAS Number | 153-18-4 |
| 3D model (JSmol) | Interactive image |
| ChemSpider | 4444362 |
| DrugBank | DB01698 |
| ECHA InfoCard | 100.005.287 |
| KEGG | C05625 |
| PubChem CID | 5280805 |
| RTECS number | VM2975000 |
| UNII | 5G06TVY3R7 |
| CompTox Dashboard (EPA) | DTXSID3022326 |
| showInChI | |
| showSMILES | |
| Properties | |
| Chemical formula | C27H30O16 |
| Molar mass | 610.521 g·mol−1 |
| Appearance | Solid |
| Melting point | 242 °C (468 °F; 515 K) |
| Solubility in water | 12.5 mg/100 mL[1] 13 mg/100mL[2] |
| Pharmacology | |
| ATC code | C05CA01 (WHO) |
| Hazards | |
| NFPA 704 (fire diamond) |  200 200 |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references |
/////////Rutoside, RUTIN, рутозид , ルチン , روتوسيد , 芦丁 , C.I. 75730, NSC 9220,
CC1C(C(C(C(O1)OCC2C(C(C(C(O2)OC3=C(OC4=CC(=CC(=C4C3=O)O)O)C5=CC(=C(C=C5)O)O)O)O)O)O)O)O

NEW DRUG APPROVALS
ONE TIME
$10.00
Anifrolumab
(Heavy chain)
EVQLVQSGAE VKKPGESLKI SCKGSGYIFT NYWIAWVRQM PGKGLESMGI IYPGDSDIRY
SPSFQGQVTI SADKSITTAY LQWSSLKASD TAMYYCARHD IEGFDYWGRG TLVTVSSAST
KGPSVFPLAP SSKSTSGGTA ALGCLVKDYF PEPVTVSWNS GALTSGVHTF PAVLQSSGLY
SLSSVVTVPS SSLGTQTYIC NVNHKPSNTK VDKRVEPKSC DKTHTCPPCP APEFEGGPSV
FLFPPKPKDT LMISRTPEVT CVVVDVSHED PEVKFNWYVD GVEVHNAKTK PREEQYNSTY
RVVSVLTVLH QDWLNGKEYK CKVSNKALPA SIEKTISKAK GQPREPQVYT LPPSREEMTK
NQVSLTCLVK GFYPSDIAVE WESNGQPENN YKTTPPVLDS DGSFFLYSKL TVDKSRWQQG
NVFSCSVMHE ALHNHYTQKS LSLSPGK
(Lihgt chain)
EIVLTQSPGT LSLSPGERAT LSCRASQSVS SSFFAWYQQK PGQAPRLLIY GASSRATGIP
DRLSGSGSGT DFTLTITRLE PEDFAVYYCQ QYDSSAITFG QGTRLEIKRT VAAPSVFIFP
PSDEQLKSGT ASVVCLLNNF YPREAKVQWK VDNALQSGNS QESVTEQDSK DSTYSLSSTL
TLSKADYEKH KVYACEVTHQ GLSSPVTKSF NRGEC
(Disulfide bridge: H22-96, H144-H200, H220-L215, H226-H’226, H229-H’229, H261-H321, H367-H425, H’22-H’96, H’144-H’200, H’220-L’215, H’261-H’321, H’367-H’425, L23-L89, L135-L195, L’23-L’89, L’135-L’195)
Anifrolumab
| アニフロルマブ (遺伝子組換え) |
FDA APPROVED 2021/7/30, Saphnelo
- MEDI 546
| Formula | C6444H9964N1712O2018S44 |
|---|---|
| Cas | 1326232-46-5 |
| Mol weight | 145117.1846 |
| Immunomodulator, Anti-IFN-type 1 receptor antibody | |
| Disease | Systemic lupus erythematosus |
|---|
Monoclonal antibody
Treatment of systemic lupus erythematosus (SLE)
- OriginatorMedarex
- DeveloperAstraZeneca; Medarex; MedImmune
- ClassAntirheumatics; Monoclonal antibodies; Skin disorder therapies
- Mechanism of ActionInterferon alpha beta receptor antagonists
- RegisteredSystemic lupus erythematosus
- Phase IILupus nephritis
- DiscontinuedRheumatoid arthritis; Scleroderma
- 02 Jul 2021Phase-III clinical trials in Systemic lupus erythematosus in USA (SC) (NCT04877691)
- 25 Jun 2021AstraZeneca plans a phase III trial in Systemic lupus erythematosus (Adjunctive treatment) in the China, Hong Kong, South Korea, Philipines, Taiwan and Thailand (IV, Infusion), in July 2021 (NCT04931563)
- 02 Jun 2021Pharmacokinetic, efficacy and adverse events data from a phase II TULIP-LN1 trial in Lupus nephritis presented at the 22nd Annual Congress of the European League Against Rheumatism (EULAR-2021)
Anifrolumab, sold under the brand name Saphnelo, is a monoclonal antibody used for the treatment of systemic lupus erythematosus (SLE).[1][2] It binds to the type I interferon receptor, blocking the activity of type I interferons such as interferon-α and interferon-β.[medical citation needed]
Anifrolumab was approved for medical use in the United States in August 2021.[1][3][4][5]
Anifrolumab is a monoclonal antibody that inhibits type 1 interferon receptors, indicated in the treatment of moderate to severe systemic lupus erythematosus.
Anifrolumab, or MEDI-546, is a type 1 interferon receptor (IFNAR) inhibiting IgG1κ monoclonal antibody indicated in the treatment of adults with moderate to severe systemic lupus erythematosus.7,11 The standard therapy for systemic lupus erythematosus consists of antimalarials like hydroxychloroquine, glucocorticoids like dexamethasone, and disease modifying antirheumatic drugs like methotrexate.8,11
Three monoclonal antibodies (anifrolumab, rontalizumab, and sifalimumab) that target the type 1 interferon pathway entered clinical trials as potential treatments for systemic lupus erythematosus, but so far only anifrolumab has been approved.3
The design of early clinical trials of anti-interferon treatments such as anifrolumab, rontalizumab, and sifalimumab have come under criticism.3 The design of the clinical trials use different definitions of autoantibody positivity, making comparison between trials difficult; all trials involve large portions of patients also using corticosteroids, which may alter patient responses in the experimental and placebo groups; and patient populations were largely homogenous, which may have increased the odds of success of the trial.3
Anifrolumab has also been investigated for the treatment of Scleroderma.1
Anifrolumab was granted FDA approval on 30 July 2021.11
Adverse effects
The most common adverse effect was shingles, which occurred in 5% of patients in the low-dose group, to 10% in the high-dose group, and to 2% in the placebo group. Overall adverse effect rates were comparable in all groups.[6]
History
The drug was developed by MedImmune, a unit of AstraZeneca, which chose to move anifrolumab instead of sifalimumab into phase III trials for lupus in 2015.[7][8][9]
Clinical trial results
Anifrolumab failed to meet its endpoint of significant reduction in disease as assessed by the SLE Responder Index 4 instrument in the TULIP 1 phase III trial.[10] This multi-center, double-blind, placebo-controlled study followed adults with moderate to severe SLE over the course of one year. Preliminary results were announced on 31 August 2018.
Names
Anifrolumab is the international nonproprietary name (INN).[11]
References
- ^ Jump up to:a b chttps://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761123s000lbl.pdf
- ^ Statement On A Nonproprietary Name Adopted By The USAN Council – Anifrolumab, American Medical Association.
- ^https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2021/761123Orig1s000ltr.pdf
- ^ https://www.astrazeneca.com/media-centre/press-releases/2021/saphnelo-approved-in-the-us-for-sle.html
- ^ “Saphnelo (anifrolumab) Approved in the US for Moderate to Severe Systemic Lupus Erythematosus” (Press release). AstraZeneca. 2 August 2021. Retrieved 2 August 2021 – via Business Wire.
- ^ Spreitzer H (29 August 2016). “Neue Wirkstoffe – Anifrolumab”. Österreichische Apothekerzeitung (in German) (18/2016).
- ^ “Press release: New Hope for Lupus Patients”. MedImmune. 11 August 2015. Archived from the original on 31 July 2017.
- ^ “Anifrolumab”. NHS Specialist Pharmacy Service. Retrieved 31 July 2017.
- ^ “Anifrolumab”. AdisInsight. Retrieved 31 July 2017.
- ^ “Update on TULIP 1 Phase III trial for anifrolumab in systemic lupus erythematosus”. http://www.astrazeneca.com. Retrieved 2019-02-05.
- ^ World Health Organization (2014). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 71”. WHO Drug Information. 28 (1). hdl:10665/331151.
Further reading
- Anderson E, Furie R (April 2020). “Anifrolumab in systemic lupus erythematosus: current knowledge and future considerations”. Immunotherapy. 12 (5): 275–86. doi:10.2217/imt-2020-0017. PMID 32237942.
External links
- “Anifrolumab”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT01438489 for “A Study of the Efficacy and Safety of MEDI-546 in Systemic Lupus Erythematosus” at ClinicalTrials.gov
- Clinical trial number NCT02446912 for “Efficacy and Safety of Two Doses of Anifrolumab Compared to Placebo in Adult Subjects With Active Systemic Lupus Erythematosus” at ClinicalTrials.gov
- Clinical trial number NCT02446899 for “Efficacy and Safety of Anifrolumab Compared to Placebo in Adult Subjects With Active Systemic Lupus Erythematosus” at ClinicalTrials.gov
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | Interferon α/β receptor |
| Clinical data | |
| Trade names | Saphnelo |
| Other names | MEDI-546, anifrolumab-fnia |
| License data | US DailyMed: Anifrolumab |
| Routes of administration | Intravenous |
| Drug class | type I interferon receptor antagonist (IFN) |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| CAS Number | 1326232-46-5 |
| DrugBank | DB11976 |
| ChemSpider | none |
| UNII | 38RL9AE51Q |
| KEGG | D11082 |
| Chemical and physical data | |
| Formula | C6444H9964N1712O2018S44 |
| Molar mass | 145119.20 g·mol−1 |
- Goldberg A, Geppert T, Schiopu E, Frech T, Hsu V, Simms RW, Peng SL, Yao Y, Elgeioushi N, Chang L, Wang B, Yoo S: Dose-escalation of human anti-interferon-alpha receptor monoclonal antibody MEDI-546 in subjects with systemic sclerosis: a phase 1, multicenter, open label study. Arthritis Res Ther. 2014 Feb 24;16(1):R57. doi: 10.1186/ar4492. [Article]
- Peng L, Oganesyan V, Wu H, Dall’Acqua WF, Damschroder MM: Molecular basis for antagonistic activity of anifrolumab, an anti-interferon-alpha receptor 1 antibody. MAbs. 2015;7(2):428-39. doi: 10.1080/19420862.2015.1007810. [Article]
- Massarotti EM, Allore HG, Costenbader K: Editorial: Interferon-Targeted Therapy for Systemic Lupus Erythematosus: Are the Trials on Target? Arthritis Rheumatol. 2017 Feb;69(2):245-248. doi: 10.1002/art.39985. [Article]
- Furie R, Khamashta M, Merrill JT, Werth VP, Kalunian K, Brohawn P, Illei GG, Drappa J, Wang L, Yoo S: Anifrolumab, an Anti-Interferon-alpha Receptor Monoclonal Antibody, in Moderate-to-Severe Systemic Lupus Erythematosus. Arthritis Rheumatol. 2017 Feb;69(2):376-386. doi: 10.1002/art.39962. [Article]
- Tummala R, Rouse T, Berglind A, Santiago L: Safety, tolerability and pharmacokinetics of subcutaneous and intravenous anifrolumab in healthy volunteers. Lupus Sci Med. 2018 Mar 23;5(1):e000252. doi: 10.1136/lupus-2017-000252. eCollection 2018. [Article]
- Riggs JM, Hanna RN, Rajan B, Zerrouki K, Karnell JL, Sagar D, Vainshtein I, Farmer E, Rosenthal K, Morehouse C, de Los Reyes M, Schifferli K, Liang M, Sanjuan MA, Sims GP, Kolbeck R: Characterisation of anifrolumab, a fully human anti-interferon receptor antagonist antibody for the treatment of systemic lupus erythematosus. Lupus Sci Med. 2018 Apr 5;5(1):e000261. doi: 10.1136/lupus-2018-000261. eCollection 2018. [Article]
- Bui A, Sanghavi D: Anifrolumab . [Article]
- Trindade VC, Carneiro-Sampaio M, Bonfa E, Silva CA: An Update on the Management of Childhood-Onset Systemic Lupus Erythematosus. Paediatr Drugs. 2021 Jul;23(4):331-347. doi: 10.1007/s40272-021-00457-z. Epub 2021 Jul 10. [Article]
- Ryman JT, Meibohm B: Pharmacokinetics of Monoclonal Antibodies. CPT Pharmacometrics Syst Pharmacol. 2017 Sep;6(9):576-588. doi: 10.1002/psp4.12224. Epub 2017 Jul 29. [Article]
- Koh JWH, Ng CH, Tay SH: Biologics targeting type I interferons in SLE: A meta-analysis and systematic review of randomised controlled trials. Lupus. 2020 Dec;29(14):1845-1853. doi: 10.1177/0961203320959702. Epub 2020 Sep 22. [Article]
- FDA Approved Drug Products: Saphnelo (Anifrolumab-fnia) Intravenous Injection [Link]
 //////////Anifrolumab, Saphnelo, FDA 2021, APPROVALS 2021, peptide, Monoclonal antibody, アニフロルマブ (遺伝子組換え) , MEDI 546, AstraZeneca, Medarex, MedImmune
//////////Anifrolumab, Saphnelo, FDA 2021, APPROVALS 2021, peptide, Monoclonal antibody, アニフロルマブ (遺伝子組換え) , MEDI 546, AstraZeneca, Medarex, MedImmune
NEW DRUG APPROVALS
one time
$10.00

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}