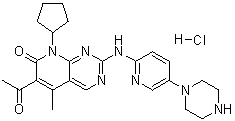
CHEMICAL NAMES
1. Pyrido[2,3-d]pyrimidin-7(8H)-one, 6-acetyl-8-cyclopentyl-5-methyl-2-[[5-(1-
piperazinyl)-2-pyridinyl]amino]-
2. 6-acetyl-8-cyclopentyl-5-methyl-2-{[5-(piperazin-1-yl)pyridin-2-
yl]amino}pyrido[2,3-d]pyrimidin-7(8H)-one
MOLECULAR FORMULA C24H29N7O2
MOLECULAR WEIGHT 447.5
TRADEMARK None as yet
SPONSOR Pfizer Inc.
CODE DESIGNATION PD-0332991
CAS#: 571190-30-2 (PD0332991); 827022-32-2 (PD0332991 HCl salt)
http://www.ama-assn.org/resources/doc/usan/palbociclib.pdf FOR STRUCTURE AND DETAILS
Palbociclib, also known as PD0332991, is an orally available pyridopyrimidine-derived cyclin-dependent kinase (CDK) inhibitor with potential antineoplastic activity. PD-0332991 selectively inhibits cyclin-dependent kinases (particularly Cdk4/cyclin D1 kinase), which may inhibit retinoblastoma (Rb) protein phosphorylation; inhibition of Rb phosphorylation prevents Rb-positive tumor cells from entering the S phase of the cell cycle (arrest in the G1 phase), resulting in suppression of DNA replication and decreased tumor cell proliferation. PD 0332991 is a highly specific inhibitor of cyclin-dependent kinase 4 (Cdk4) (IC50 = 0.011 μmol/L) and Cdk6 (IC50 = 0.016 μmol/L), having no activity against a panel of 36 additional protein kinases. Check for active clinical trials or closed clinical trials using this agent. (NCI Thesaurus)
Date: April 10, 2013
Pfizer Inc. said that its experimental pill for advanced, often deadly breast cancer has been designated as a breakthrough therapy by the Food and Drug Administration.
The breakthrough designation, created under legislation enacted last summer to fund and improve operations of the FDA, is meant to speed up development and review of experimental treatments that are seen as big advances over existing therapies for serious diseases. Pfizer is working with the agency to determine exactly what research results it will need to apply for approval of the drug.
Palbociclib is being evaluated as an initial treatment for the biggest subgroup of postmenopausal women whose breast cancer is locally advanced or has spread elsewhere in the body. About 60% of women with such advanced breast cancer have tumors classified as ER+, or estrogen-receptor positive, but HER2-, or lacking an excess of the growth-promoting protein HER2.
Estrogen-receptor positive tumors have proteins inside and on the surface of their cells to which the estrogen hormone can attach and then fuel growth of cells. These tumors tend to grow slowly and can be fought with drugs that block estrogen’s effects.
Meanwhile, about 80% of breast cancer tumor cells are HER2 negative. That means that unlike HER2 positive tumors, they don’t produce too much of the HER2 protein, which makes tumors grow and spread more aggressively than in other breast cancer types.
New York-based Pfizer is currently running a late-stage study of palbociclib at multiple centers, comparing its effects when used in combination with letrozole with the effects of letrozole alone.
Letrozole, sold under the brand name Femara for about the past 15 years, is a pill that works by inhibiting aromatase. That’s an enzyme in the adrenal glands that makes estrogen.
According to Pfizer, palbociclib targets enzymes called cyclin dependent kinases 4 and 6. By inhibiting those enzymes, the drug has been shown in laboratory studies to block cell growth and suppress copying of the DNA of the cancer cells.
Pfizer, which has made research on cancer medicines a priority in recent years, also is testing palbociclib as a treatment for other cancers.
| Highlight of recent study using PD-0332991 |
Phase I study of PD-0332991: Forty-one patients were enrolled. DLTs were observed in five patients (12%) overall; at the 75, 125, and 150 mg once daily dose levels. The MTD and recommended phase II dose of PD 0332991 was 125 mg once daily. Neutropenia was the only dose-limiting effect. After cycle 1, grade 3 neutropenia, anemia, and leukopenia occurred in five (12%), three (7%), and one (2%) patient(s), respectively. The most common non-hematologic adverse events included fatigue, nausea, and diarrhea. Thirty-seven patients were evaluable for tumor response; 10 (27%) had stable disease for ≥4 cycles of whom six derived prolonged benefit (≥10 cycles). PD 0332991 was slowly absorbed (median T(max), 5.5 hours), and slowly eliminated (mean half-life was 25.9 hours) with a large volume of distribution (mean, 2,793 L). The area under the concentration-time curve increased linearly with dose. Using an E(max) model, neutropenia was shown to be proportional to exposure. CONCLUSIONS:
PD 0332991 warrants phase II testing at 125 mg once daily, at which dose neutropenia was the sole significant toxicity. (Source: Clin Cancer Res; 18(2); 568-76.)
Phase I study of PD-0332991 in 3-week cycles (Schedule 2/1): Six patients had DLTs (18%; four receiving 200 mg QD; two receiving 225 mg QD); the MTD was 200 mg QD. Treatment-related, non-haematological adverse events occurred in 29 patients (88%) during cycle 1 and 27 patients (82%) thereafter. Adverse events were generally mild-moderate. Of 31 evaluable patients, one with testicular cancer achieved a partial response; nine had stable disease (≥10 cycles in three cases). PD 0332991 was slowly absorbed (mean T(max) 4.2 h) and eliminated (mean half-life 26.7 h). Volume of distribution was large (mean 3241 l) with dose-proportional exposure. Using a maximum effective concentration model, neutropenia was proportional to exposure. CONCLUSION: PD 0332991 was generally well tolerated, with DLTs related mainly to myelosuppression. The MTD, 200 mg QD, is recommended for phase II study. (source: Br J Cancer. 2011 Jun 7;104(12):1862-8)
1: Flaherty KT, Lorusso PM, Demichele A, Abramson VG, Courtney R, Randolph SS, Shaik MN, Wilner KD, O’Dwyer PJ, Schwartz GK. Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer. Clin Cancer Res. 2012 Jan 15;18(2):568-76. doi: 10.1158/1078-0432.CCR-11-0509. Epub 2011 Nov 16. PubMed PMID: 22090362.
2: Smith D, Tella M, Rahavendran SV, Shen Z. Quantitative analysis of PD 0332991 in mouse plasma using automated micro-sample processing and microbore liquid chromatography coupled with tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2011 Oct 1;879(27):2860-5. doi: 10.1016/j.jchromb.2011.08.009. Epub 2011 Aug 16. PubMed PMID: 21889427.
3: Katsumi Y, Iehara T, Miyachi M, Yagyu S, Tsubai-Shimizu S, Kikuchi K, Tamura S, Kuwahara Y, Tsuchiya K, Kuroda H, Sugimoto T, Houghton PJ, Hosoi H. Sensitivity of malignant rhabdoid tumor cell lines to PD 0332991 is inversely correlated with p16 expression. Biochem Biophys Res Commun. 2011 Sep 16;413(1):62-8. doi: 10.1016/j.bbrc.2011.08.047. Epub 2011 Aug 17. PubMed PMID: 21871868; PubMed Central PMCID: PMC3214763.
4: Schwartz GK, LoRusso PM, Dickson MA, Randolph SS, Shaik MN, Wilner KD, Courtney R, O’Dwyer PJ. Phase I study of PD 0332991, a cyclin-dependent kinase inhibitor, administered in 3-week cycles (Schedule 2/1). Br J Cancer. 2011 Jun 7;104(12):1862-8. doi: 10.1038/bjc.2011.177. Epub 2011 May 24. PubMed PMID: 21610706; PubMed Central PMCID: PMC3111206.
5: Nguyen L, Zhong WZ, Painter CL, Zhang C, Rahavendran SV, Shen Z. Quantitative analysis of PD 0332991 in xenograft mouse tumor tissue by a 96-well supported liquid extraction format and liquid chromatography/mass spectrometry. J Pharm Biomed Anal. 2010 Nov 2;53(3):228-34. doi: 10.1016/j.jpba.2010.02.031. Epub 2010 Feb 26. PubMed PMID: 20236782.
6: Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, Ginther C, Atefi M, Chen I, Fowst C, Los G, Slamon DJ. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11(5):R77. doi: 10.1186/bcr2419. PubMed PMID: 19874578; PubMed Central PMCID: PMC2790859.
7: Menu E, Garcia J, Huang X, Di Liberto M, Toogood PL, Chen I, Vanderkerken K, Chen-Kiang S. A novel therapeutic combination using PD 0332991 and bortezomib: study in the 5T33MM myeloma model. Cancer Res. 2008 Jul 15;68(14):5519-23. doi: 10.1158/0008-5472.CAN-07-6404. PubMed PMID: 18632601.
8: Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, Albassam M, Zheng X, Leopold WR, Pryer NK, Toogood PL. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004 Nov;3(11):1427-38. PubMed PMID: 15542782.

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....