

Crystallization-Induced Dynamic Resolution toward the Synthesis of (S)-7-Amino-5H,7H-dibenzo[b,d]-azepin-6-one: An Important Scaffold for γ-Secretase Inhibitors
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 143)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Register Today for the ACS Symposium in India on Recent Advances in Drug Development, 11-12 November 2016 in Hyderabad, India

![]()
Inaugural ACS Industry Symposium, 11-12 November 2016 in Hyderabad, India
Recent Advances in Drug Development
Register Today for the ACS Symposium in India on Recent Advances in Drug Development
To view this email as a web page, go here.
Register now for the inaugural ACS Industry Symposium, 11-12 November 2016 in Hyderabad, India. Be sure to secure your seat today as rates will increase on 27 October!
http://acssymposium.org.in/
The theme of the Symposium is Recent Advances in Drug Development. The event will feature lectures by the world’s leading researchers and experts in the pharma industry, including:
- Dr. Peter Senter of Seattle Genetics
- Dr. Jagath Reddy Junutula of Cellerant Therapeutics, Inc.
- Dr. Ming-Wei Wang of the Shanghai Institute of Materia Medica, Chinese Academy of Sciences
This is an exclusive event being organized in partnership with Dr. Reddy’s Laboratories for pharma professionals throughout India. Space is limited so register today!
Please visit our website to learn more about the speakers and the program.
Register today to ensure your access to the ACS Industry Symposium. We look forward to seeing you in Hyderabad in November.
CAS
2540 Olentangy River Rd Columbus, OH 43202 US
![]()
CAS
2540 Olentangy River Rd Columbus, OH 43202 US
![]()



Inaugural ACS Industry Symposium, 11-12 November 2016 in Hyderabad, India
Recent Advances in Drug Development
/////// ACS Symposium, Recent Advances in Drug Development, 11-12 November 2016, Hyderabad, India, dr reddys, cas
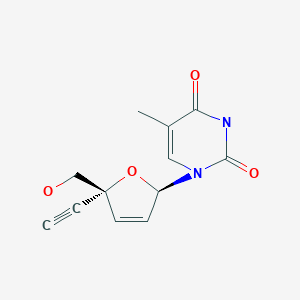
BMS 986001, Censavudine, Festinavir
BMS 986001
Censavudine, Festinavir
Has anti-HIV activity. IN PHASE 2
CAS: 634907-30-5, UNII: 6IE83O6NGA, OBP 601, 4′-Ethynyl D4T, 4′-Ed4T, TDK-4-114
Molecular Formula, C12-H12-N2-O4, Molecular Weight, 248.2368
2′,3′-Didehydro-3′-deoxy-4′-ethynylthymidine,
1-((2R,5R)-5-Ethynyl-5-(hydroxymethyl)-2H-furan-2-yl)-5-methyl-pyrimidine-2,4-dione,
2′,3′-Didehydro-3′-deoxy-4′-ethynylthymidine
INNOVATOR= YALE UNIVERSITY
![]()
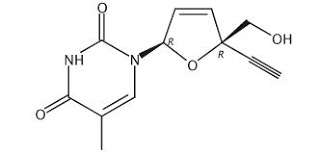
Festinavir is a nucleoside reverse transcriptase inhibitor
(NRTI) which is being developed for the treatment of HIV infection. The drug has shown considerable efficacy in early development, and with perhaps less toxicity than some other NRTIs, such as the drug stavudine (marketed under the trade name ZERIT®).
Festinavir has the chemical form and the structural formula:

Festinavir was developed by Yale University in conjunction with two Japanese research scientists, and is protected by U.S. Patent No. 7,589,078, the contents of which are incorporated herein by reference. The ‘078 patent sets forth the synthesis of the primary compound, and other structural analogs. In addition, Oncolys BioPharma, Inc. of Japan has now published US 2010/0280235 for the production of 4′ ethynyl D4T. As starting raw material, the Oncolys method utilizes a substituted furan compound, furfuryl alcohol. In another publication by Nissan Chemical Industries of Japan, and set forth in WO 201 1/099443, there is disclosed a method for producing a beta-dihydrofuran deriving compound or a beta-tetrahydrofuran deriving compound. In this process, a diol compound is used as the starting material. Nissan has also published WO 2011/09442
directed to a process for the preparation of a β-glycoside compound. Two further publications, each to Hamari Chemicals of Japan, WO 2009/1 19785 and
WO 2009/125841, set forth methods for producing and purifying ethynyl thymide compounds. Pharmaset, Inc. of the U.S. has also published US 2009/0318380,
WO 2009/005674 and WO 2007/038507 for the production of 4’ -nucleoside analogs for treating HIV infection. Reference is also made to the BMS application entitled
“Sulfilimine and Sulphoxide Methods for Producing Festinavir” filed as a PCT application, PCT/US2013/042150 on May 22, 2013 (now WO2013/177243).
PAPER
Haraguchi, Kazuhiro; Bioorganic & Medicinal Chemistry Letters 2003, V 13(21), PG 3775-3777
http://dx.doi.org/10.1016/j.bmcl.2003.07.009
http://www.sciencedirect.com/science/article/pii/S0960894X0300831X
Compounds having methyl, vinyl, and ethynyl groups at the 4′-position of stavudine (d4T: 2′,3′-didehydro-3′-deoxythymidine) were synthesized. The compounds were assayed for their ability to inhibit the replication of HIV in cell culture. The 4′-ethynyl analogue (15) was found to be more potent and less toxic than the parent compound stavudine.
Graphic


- Physical data for 15 are as follows: solid (mp 207–209 °C);
- UV (MeOH) λmax 264 nm (ε 10800), λmin 235 nm (ε 4800);
- 1H NMR (CDCl3) δ 1.83 (3H, s, Me), 2.63 (1H, s, C≡CH), 3.47 (1H, br, OH), 3.88 (1H, d,Jgem=12.5 Hz, H-5′a), 3.96 (1H, d, Jgem=12.5 Hz, H-5′b), 5.91 (1H, dd, J1′,2′=1.1 Hz and J2′,3′=5.9 Hz, H-2′), 6.30 (1H, dd, J1′,3′=2.0 Hz and J2′,3′=5.9 Hz, H-3′), 7.16–7.17 (1H, m, H-1′), 7.44 (1H, d, J6,Me=1.1 Hz, H-6), 9.06 (1H, br, NH);
- FAB-MS m/z 249 (M++H). Anal. calcd for C12H12N2O4·1/6H2O: C, 57.37; H, 4.95; N, 11.15. Found: C, 57.36; H, 4.69; N, 10.98.
- PAPER
- Scalable Synthesis of the Potent HIV Inhibitor BMS-986001 by Non-Enzymatic Dynamic Kinetic Asymmetric Transformation (DYKAT)
Angewandte Chemie, International Edition (2015), 54, (24), 7185-7188. - http://onlinelibrary.wiley.com/doi/10.1002/anie.201502290/abstract
- http://onlinelibrary.wiley.com/store/10.1002/anie.201502290/asset/supinfo/anie_201502290_sm_miscellaneous_information.pdf?v=1&s=9c516d28bb61a8b090de88c2a75f5f50f060aaa9
-
Scalable Synthesis of the Potent HIV Inhibitor BMS-986001 by Non-Enzymatic Dynamic Kinetic Asymmetric Transformation (DYKAT)†
Described herein is the synthesis of BMS-986001 by employing two novel organocatalytic transformations: 1) a highly selective pyranose to furanose ring tautomerization to access an advanced intermediate, and 2) an unprecedented small-molecule-mediated dynamic kinetic resolution to access a variety of enantiopure pyranones, one of which served as a versatile building block for the multigram, stereoselective, and chromatography-free synthesis of BMS-986001. The synthesis required five chemical transformations and resulted in a 44 % overall yield.

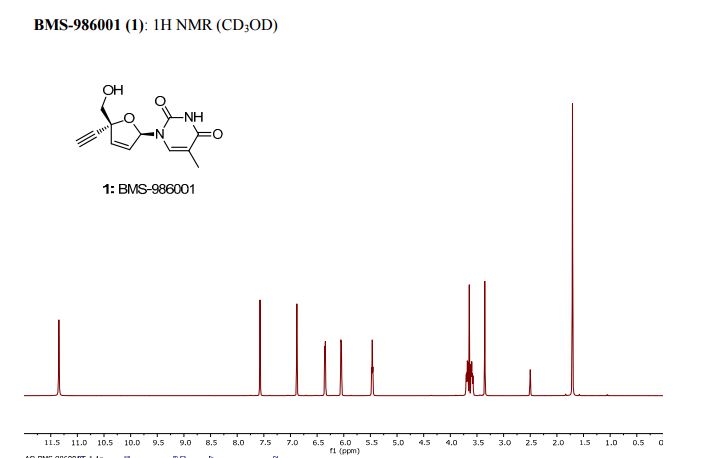
white crystalline solid. 1: Rf = 0.8 (silica, MeOH:CH2Cl2,1:4);
M.P. = 196-207°C;
1 H NMR (d6-DMSO, 500 MHz): δ = 11.34 (s, 1 H), 6.88 (s, 1 H), 6.35 (d, J = 6.0 Hz, 6.05 (d, J = 6.0 Hz, 1 H), 5.45 (t, J = 5.5 Hz, 1 H), 3.69 (dd, J = 12.0, 1.5 Hz, 1 H), 3.64 (s, 1 H), 3.59 (dd, J = 12.0, 1.5 Hz, 1 H) 1.70 (s, 3 H) ppm;
13C NMR (d6-DMSO, 125 MHz): δ = 163.85, 150.82, 136.81, 135.54, 127.13, 109.04, 88.94, 86.60, 81.45, 77.39, 65.76, 12.23 ppm;
HRMS calcd for C12H12N2O4H+ [M + H+] 249.09 found 249.08.

PATENT
WO 2014172264
https://www.google.ch/patents/WO2014172264A1?cl=en
invention:

Step#l: Acetal Formation

Compound 1
85% yield
The starting material is 5-methylurdine, which is commercially available. The first step of the process is an acetal formation. 5-methyluridine is utilized and is treated with H2SO4 and acetaldehyde. Other acids available to the scientist, such as perchloric acid, will also work for this transformation. The solvent utilized for this step is acetonitrile (ACN), and other solvents may also be utilized as well. Once the starting material is consumed, a slurry is obtained and the product can be simply filtered off and dried to provide Compound 1 as a solid.

Acetal formation
Preparation of l-((3aR,4R,6R,6aR)-6-(hydroxymethyl)-2-methyltetrahydrofuro [3,4-d] [1,3] dioxol-4-yl)-5-methylpyrimidine-2,4(lH,3H)-dione
The following were added to a flask: 5-methyluridine (10 g, 38.70 mmol), acetonitrile (20 mL) and 70% perchloric acid (4.01 mL, 47.63 mmol). A solution of acetaldehyde (3.26 mL, 58.10 mmol) in acetonitrile (20 mL) was added dropwise over 1 h. The resulting solution was allowed to stir at 20 °C for 18 h. The resulting slurry was filtered and dried (50 °C, 25 mmHg) to afford Acetal (9.30 g, 84% yield) as white solid
XH NMR (400MHz, DMSO-d6) δ = 11.39 (s, 1H), 7.72 – 7.63 (m, 1H), 5.82 (d, J=3.0 Hz, 1H), 5.21 – 5.07 (m, 2H), 4.84 (dd, J=6.6, 2.5 Hz, 1H), 4.68 (dd, J=6.6, 3.0 Hz, 1H), 4.12 – 4.05 (m, 1H), 3.65 – 3.51 (m, 2H), 3.36 (s, 2H), 1.77 (s, 3H), 1.37 (d, J=5.1 Hz, 3H) 13C NMR (101MHz, DMSO-d6) δ = 163.77, 150.32, 137.64, 109.39, 104.50, 90.79, 86.16, 83.83, 81.37, 61.25, 19.76, 12.06
Step #2: Acetate protection

Compound 2
85% yield
The next step of the sequence is installation of a 4-biphenylacetate. Without being bound by any particular theory, this protecting step may be chosen for two reasons:
1) To provide a solid intermediate that can be easily isolated, and
2) Act as a directing group in the next step (set forth later on).
This reaction consists of reacting Compound 1 with 4-biphenyl acid chloride and pyridine in acetonitrile. In this reaction, pyridine is preferred as it allows the reaction to occur only at the -OH moiety of the molecule. It should also be noted that other polar solvents could be used, but acetonitrile allowed the desired product Compound 2 to be isolated as s solid.

Ac lation
Preparation of ((3aR,4R,6R,6aR)-2-methyl-6-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)tetrahydrofuro[3,4-d] [l,3]dioxol-4-yl)methyl [1,1′-biphenyl]-4-carboxylate.
Acetal (9.30 g, 32 mmol) was dissolved into acetonitrile (100 mL). Pyridine (1.3 eq) was added followed by the addition of 4-biphenylcarbonyl chloride (1.05 eq). The solution was heated to 50 °C and held for 2 h. The slurry was cooled to 20 °C and held for 2 h. The slurry was filtered and washed with acetonitrile (100 mL). The solids were dried (50 °C, 25 mmHg) to Compound 2 (85% yield).
XH NMR (400MHz, CHLOROFORM-d) δ = 8.10 (d, J=8.1 Hz, 2H), 7.62 (d, J=7.6 Hz, 2H), 7.67 (d, J=8.1 Hz, 2H), 7.55 – 7.36 (m, 3H), 7.09 (s, 1H), 5.71 (s, 1H), 5.26 (q, J=4.7 Hz, 1H), 5.03 (dd, J=6.6, 2.0 Hz, 1H), 4.91 (dd, J=6.7, 3.2 Hz, 1H), 4.73 – 4.63 (m, 1H), 4.61 – 4.50 (m, 2H), 2.02 (s, 3H), 1.85 – 1.76 (m, 3H), 1.52 (d, J=4.8 Hz, 3H)
1JC MR (101MHz, CHLOROFORM-d) δ = 164.02, 161.94, 148.20, 144.18, 137.85, 135.89, 128.20, 127.05, 126.36, 126.30, 125.35, 125.26, 1 14.49, 109.20, 103.88, 92.51, 83.36, 83.29, 79.87, 75.45, 75.13, 74.81, 62.54, 17.92, 10.32, -0.01

With the acetal and 4-biphenylacetate groups in place, the next reaction is a regioselective acetal opening utilizing TMSOTf (Trimethylsilyl trifluoromethane sulfonate, or other available Lewis acids)/Et3N to afford the corresponding silyl ether, which is cleaved in situ, to afford the 2-vinyloxy compound as Compound 3. Compound 3 may be prepared in a step-wise fashion (shown below), but in order to reduce the number of steps, it is possible to take Compound 3 and selectively form the desired 2-vinyl oxy regioisomer Compound 3. Those skilled in the art may recognize that the 4-biphenylacetate can be important to obtain high selectivity for this transformation.


![]()
Although a variety of Lewis acids may be utilized, TMSOTf is generally found to be more effective. Et3 is also a preferred reactant, as other amine bases are generally less effective. The ratio of TMSOTf to Ets is preferably within the range of about 1 : 1.3; if the reaction medium became acidic, Compound 3 would revert back to Compound 2. In terms of solvents, DCM (Dichloromethane) may be particularly effective, but toluene, CF3-PI1, sulfolane, and DCE (Dichloroethene) are also effective. The reaction can be worked up using aqueous acid, preferably K2HP04, or methanolic NH4F to quench the reaction, as well as remove the TMS-ether in situ.

TMSOTf-opening
Preparation of ((2R,3R,4R,5R)-3-hydroxy-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)-4-(vinyloxy)tetrahydrofuran-2-yl)methyl [1,1′-biphenyl]-4-carboxylate
Compound 2 (20 g, 43.06 mmol) was dissolved into DCM (160 mL). Triethylamine (78 mL, 560 mmol) was added followed by the addition of TMSOTf (80.30 mL, 431 mmol). This solution was heated to 45 °C and held there until complete by HPLC analysis (6 h). Once complete, this solution was added to ammonium acetate (66.40 g, 861 mmol) in water (200 mL). After stirring for 20 min, the layers were separated. The organics were concentrated and the resulting residue was dissolved into EtOAc (200 mL). The organics were washed with the following solution (potassium phosphate monobasic (118 g, 861 mmol) in water (400 mL). The organics were then dried ( a2S04), filtered and concentrated. The resulting residue was purified by column chromatography [Silica gel; 20% to 90% EtOAc in Hexanes] to afford Compound 3 (15.8 g, 79% yield) as a solid.
XH NMR (400MHz, CHLOROFORM-d) 6 = 9.18 (br. s., IH), 8.18 – 8.06 (m, 2H), 7.73 -7.56 (m, 4H), 7.55 – 7.38 (m, 3H), 7.24 (d, J=1.3 Hz, IH), 6.59 (dd, J=14.0, 6.4 Hz, IH), 5.81 (d, J=2.0 Hz, IH), 4.84 (dd, J=12.6, 2.5 Hz, IH), 4.63 (dd, J=12.5, 4.2 Hz, IH), 4.59 – 4.44 (m, 3H), 4.40 – 4.26 (m, 2H), 1.70 (d, J=1.0 Hz, 3H)
13C MR (101MHz, CHLOROFORM-d) δ = 166.13, 163.65, 150.00, 149.67, 146.39, 139.66, 135.67, 130.16, 129.01, 128.40, 128.06, 127.32, 127.28, 111.43, 91.93, 89.44, 81.60, 80.19, 69.32, 63.06, 12.32
Step #4: Iodiiiation

Compound 4
Compound 3 75% yie|d
Next, Compound 3 is transformed into the iodide compound which is Compound 4. This can be accomplished by treating Compound 3 with (2.0 eq), PPI13 (2.0 eq.) and imidazole (4.0 eq). Other methods to install the iodide may also be utilized, such as mesylation/Nal, etc., but these may be less preferred. In addition, other halogen-bearing compounds such as Br2 and CI2 may be considered by the skilled scientist. Premixing imidazole, , and PPh3, followed by addition of Compound 3 in THF and heating at 60 °C allows smooth conversion to Compound 4. It is highly preferred to add all reagents prior to the addition of Compound 3; if not, the vinyloxy group will be cleaved. Other solvents, such as 2-MeTHF and PhMe may be utilized, but THF often provides the best yield.

Iodiiiation
Preparation of ((2R,3S,4S,5R)-3-iodo-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)-4-(vinyloxy)tetrahydrofuran-2-yl)methyl [l,l’-biphenyl]-4-carboxylate
The following were added to a flask: imidazole (8.79 g, 129 mmol),
triphenylphosphine (16.94 g, 65 mmol), iodine 16.39 g, 65 mmol) and THF (525 mL). A solution of Compound 3 (15 g, 32 mmol) in THF (375 mL) was added. The solution was heated to 60 °C and was held at 60 °C for 4 h. Once complete by HPLC analysis (4 h), the solution was concentrated and the residue was purified by column chromatography [Silica gel; 10% to 60% EtOAc in Hexanes] to afford Compound 4 (17.0 g, 92% yield) as a solid.
XH NMR (400MHz, CHLOROFORM-d) δ = 9.25 (br. s., IH), 8.16 (d, J=8.3 Hz, 2H), 7.75 – 7.61 (m, 5H), 7.54 – 7.40 (m, 3H), 7.32 – 7.24 (m, 2H), 7.23 – 7.16 (m, 2H), 6.56 -6.45 (m, IH), 6.06 (d, J=1.5 Hz, IH), 4.89 (s, IH), 4.66 (dd, J=12.0, 6.9 Hz, IH), 4.56 (dd, J=12.0, 3.9 Hz, IH), 4.46 (d, J=4.0 Hz, IH), 4.39 – 4.26 (m, 2H), 4.13 (dt, J=7.1, 3.8 Hz, 1H), 2.06 – 1.97 (m, 3H)
1JC MR (101MHz, CHLOROFORM-d) δ = 165.96, 163.94, 150.27, 149.29, 146.28, 139.81, 137.88, 135.84, 130.37, 129.06, 129.01, 128.34, 128.25, 127.94, 127.31, 127.22, 125.32, 1 11.07, 91.37, 90.32, 89.18, 78.43, 69.15, 25.81, 21.49, 12.71
Step #5: Iodide Elimination

Compound 4
The next step of the sequence is to install the allyic moiety. Heating a solution of Compound 4 in toluene in the presence of DABCO (l,4-Diazabicyclo[2.2.2]octane) allows for elimination of the iodide. Other solvents, such as THF and DCE may be utilized, but toluene often provides the best conversion and yield. Other amine bases may be used in this transformation, but generally DABCO is preferred.

Elimination
Preparation of ((4R,5R)-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-l (2H)-yl)-4-(vinyloxy)-4,5-dihydrofuran-2-yl)methyl [l,l’-biphenyl]-4-carboxylate
Compound 4 (17 g, 30 mmol) was dissolved into toluene (255 niL), and DABCO (10 g, 89 mmol) was added. The solution was heated to 90 °C and held there for 2 h. Once complete, the organics were washed with sat. aq. a2S203 (200 mL). The organics were then dried ( a2S04), filtered, and concentrated. The resulting residue was purified by column chromatography [Silica gel; 5% to 60% EtOAc in Hexanes] to yield
Compound 5 (10.9, 85% yield) as a foam.
XH NMR (400MHz, CHLOROFORM-d) δ = 8.93 (br. s., IH), 8.18 – 8.11 (m, 2H), 7.75 -7.61 (m, 5H), 7.55 – 7.39 (m, 4H), 6.95 (d, J=1.0 Hz, IH), 6.54 (d, J=2.0 Hz, IH), 6.46 (dd, J=14.3, 6.7 Hz, IH), 5.53 (d, J=2.5 Hz, IH), 5.09 (d, J=2.8 Hz, IH), 5.04 (d, J=6.6 Hz, 2H), 4.29 (dd, J=14.3, 2.4 Hz, IH), 4.23 (dd, J=6.7, 2.4 Hz, IH), 1.88 (d, J=1.0 Hz, 3H)
1JC MR (101MHz, CHLOROFORM-d) δ = 165.73, 159.58, 149.10, 146.49, 139.70, 134.51, 132.17, 132.07, 131.94, 131.92, 130.30, 129.01, 128.56, 128.44, 128.40, 127.73, 127.30, 127.28, 112.50, 99.16, 90.57, 90.23, 84.81, 58.68, 12.44
Step #6: Claisen Rearrangement

An important reaction in the sequence is the Claisen rearrangement. This reaction is utilized to install the quaternary stereocenter and the olefin geometry in the ring. Heating Compound 5 in benzonitrile at 190 °C for 2-3 hours allows for smooth conversion to Compound 6, and after chromatography, a 90% yield can be achieved.
Toluene (110 °C, 8 h) also works to provide the desired Compound 6 as a solid by simply cooling the reaction to 20 °C (no chromatography). Other solvents with boiling points over about 100°C may also be utilized.

Claisen Rearrangement
Preparation of ((2S,5R)-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)-2-(2-oxoethyl)-2,5-dihydrofuran-2-yl)methyl [l,l’-biphenyl]-4-carboxylate
Compound 5 (1 mmol) was dissolved into benzonitrile (10 mL). The solution was heated to 190 °C for 3 h. After cooling to 20 °C, the solution was purified by column chromatography [silica gel, 50:50 Hexanes:EtOAc] to afford Compound 6 (1 mmol).
Alternatively, Compound 5 (1 mmol) was dissolved into toluene (10 mL). The solution was heated to 110 °C and held for 12 h. Upon cooling to 20 °C, a slurry formed. The solids were filtered, washed (PhMe) and dried (50 °C, 25 mmHg) to afford
Compound 6 (1 mmol) as a white solid.
XH NMR (400MHz, CHLOROFORM-d) δ = 9.84 (t, J=1.8 Hz, 1H), 8.53 (br. s., 1H), 8.13 – 8.03 (m, J=8.3 Hz, 2H), 7.73 – 7.67 (m, 2H), 7.67 – 7.60 (m, 2H), 7.56 – 7.38 (m, 3H), 7.14 (d, J=1.3 Hz, 1H), 7.04 (t, J=1.5 Hz, 1H), 6.57 (dd, J=6.1, 2.0 Hz, 1H), 6.02 (dd, J=5.9, 1.1 Hz, 1H), 4.68 – 4.52 (m, 2H), 3.06 – 2.89 (m, 2H), 1.59 (d, J=1.0 Hz, 3H)
13C MR (101MHz, CHLOROFORM-d) δ = 198.33, 165.83, 163.35, 150.65, 146.56, 139.63, 136.24, 135.02, 130.21, 129.04, 128.44, 127.86, 127.49, 127.41, 127.28, 111.59, 90.03, 89.61, 67.33, 50.06, 12.06
ne Formation via elimination of Enol Nonaflate

The alkyne formation is performed by first treating Compound 6 with TMSCl (Trimethylsilyl chloride)/Et3N. NfF (Nonafluoro- 1 -butanesulfonyl fluoride) and P-base () are then added at -20 °C. After warming to 20 °C, the desired alkyne Compound 7 can be isolated in about 80 % yield. Initially, TMSCl is presumed to react at the NH moiety. NfF/P-base then reacts with the aldehyde to form the enol Nonaflate. Upon warming to 20 °C in the presence of P-base, the enol Nonaflate eliminates smoothly to the alkyne Compound 7. Without the TMSCl/Et3N, the yields are only -25%.

Alkyne formation
Preparation of ((2R,5R)-2-ethynyl-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)-2,5-dihydrofuran-2-yl)methyl [l,l’-biphenyl]-4-carboxylate
Compound 6 (1 g, 2.24 mmol) was dissolved into DMF (Dimethylformamide) (5 mL). (Other polar solvents could also have been used.) Triethylamine (406 uL, 2.91 mmol) was added and the solution was cooled to 0 °C. TMSCl (314 uL, 2.46 mmol) was added and the solution was allowed to stir at 0 °C for 30 min. The solution was then cooled to -20 °C, and NfF (484 uL, 2.69 mmol) was added and the solution was allowed to stir at -20 °C for 5 min. Phosphazane P l-base (1.54 mL, 4.93 mmol) was added
dropwise over 20 min. The solution was then allowed to warm to 20 °C and held for 20 h. The solution was then poured into water (50 mL) and extracted with DCM (100 mL). The organics were concentrated and the resulting residue was purified by column chromatography [Silica gel; 10% to 60% EtOAc in Hexanes] to afford Compound 7 (816 mg, 85% yield) as a solid.
XH NMR (400MHz, DMSO-d6) δ = 11.46 (s, 1H), 8.08 – 7.97 (m, J=8.6 Hz, 2H), 7.92 -7.80 (m, 2H), 7.73 (d, J=7.1 Hz, 2H), 7.59 – 7.39 (m, 3H), 7.06 (d, J=1.0 Hz, 1H), 6.89 (d, J=1.5 Hz, 1H), 6.61 (dd, J=5.6, 2.0 Hz, 1H), 6.23 (dd, J=5.6, 1.0 Hz, 1H), 4.66 (d, J=12.1 Hz, lH), 4.57 (d, J=11.6 Hz, 1H), 3.87 (s, 1H), 1.37 (s, 3H)
13C MR (101MHz, DMSO-d6) δ = 164.89, 163.57, 150.61, 145.13, 138.73, 135.30, 134.40, 129.94, 129.12, 128.49, 127.84, 127.78, 127.18, 126.98, 110.01, 89.37, 83.69, 80.01, 78.23, 66.89, 11.46

90% yield
The final step of the sequence is to remove the aromatic ester protecting group. This consists of hydrolysis by NaOH in aq. THF solution. The API is extracted into THF and then crystallized from THF/PhMe.

Deprotection
Preparation of l-((2R,5R)-5-ethynyl-5-(hydroxymethyl)-2,5-dihydrofuran-2-yl)-5-methylpyrimidine-2,4(lH,3H)-dione (Ed4T)
Compound 7 (10 g, 23.40 mmol) was dissolved into THF (100 mL). 3N NaOH (10 mL) was added. The solution was allowed to stir at 20 °C for 12 h. The layers were split and the organics were kept. The organics were concentrated to reach a KF <1 wt%. Toluene (100 mL) was added, and solids crashed out of solution. The solids were filtered and washed with Toluene (100 mL). The solids were then dried (50 °C, 25 mmHg) to afford Festinavir (5.21 g, 90% yield) as a white solid.
XH NMR (400MHz, DMSO-d6) δ = 1 1.36 (s, 1H), 7.58 (s, 1H), 6.89 (s, 1H), 6.36 (d, J=6.1 Hz, 1H), 6.05 (d, J=6.1 Hz, 1H), 5.48 (t, J=5.6 Hz, 1H), 3.78 – 3.49 (m, 3H), 3.46 3.31 (m, 1H), 1.71 (s, 3H)
1JC MR (101MHz, DMSO-d6) δ = 163.80, 150.76, 136.75, 135.47, 127.06, 108.98, 88.87, 86.52, 81.37, 77.33, 65.68, 12.17.
PAPER
Tetrahedron (2009), 65(36), 7630-7636.
Volume 65, Issue 36, 5 September 2009, Pages 7630–7636

Synthesis of (±)-4′-ethynyl-5′,5′-difluoro-2′,3′-dehydro-3′-deoxy- carbocyclic thymidine: a difluoromethylidene analogue of promising anti-HIV agent Ed4T
PAPER
Nucleophilic Substitution at the 4‘-Position of Nucleosides: New Access to a Promising Anti-HIV Agent 2‘,3‘-Didehydro-3‘-deoxy-4‘-ethynylthymidine
School of Pharmaceutical Sciences, Showa University, 1-5-8 Hatanodai, Shinagawa-ku, Tokyo 142-8555, Japan
J. Org. Chem., 2006, 71 (12), pp 4433–4438
DOI: 10.1021/jo060194m
Journal of Organic Chemistry (2006), 71(12), 4433-4438.
http://pubs.acs.org/doi/abs/10.1021/jo060194m

For the synthesis of 2‘,3‘-didehydro-3‘-deoxy-4‘-ethynylthymidine (8: 4‘-Ed4T), a recently reported promising anti-HIV agent, a new approach was developed. Since treatment of 1-(2,5-dideoxy-β-l–glycero-pent-4-enofuranosyl)thymine with Pb(OBz)4 allowed the introduction of the 4‘-benzoyloxy leaving group, nucleophilic substitution at the 4‘-position became feasible for the first time. Thus, reaction between the 4‘-benzoyloxy derivative (14) and Me3SiC⋮CAl(Et)Cl as a nucleophile led to the isolation of the desired 4‘-“down”-ethynyl derivative (18) stereoselectively in 62% yield. As an application of this approach, other 4‘-substituted nucleosides, such as the 4‘-allyl (24a) and 4‘-cyano (26a) derivatives, were synthesized using organosilicon reagents. In these instances, pretreatment of 14 with MeAlCl2 was necessary.

PATENTS
US75890782009-09-15Anti-viral nucleoside analogs and methods for treating viral infections, especially HIV infections
| Patent ID | Date | Patent Title |
|---|---|---|
| US2016060252 | 2016-03-03 | 5-METHYLURIDINE METHOD FOR PRODUCING FESTINAVIR |
| US2015140610 | 2015-05-21 | SULFILIMINE AND SULPHOXIDE METHODS FOR PRODUCING FESTINAVIR |
| US2015104511 | 2015-04-16 | Pharmaceutical Antiretroviral Combinations Comprising Lamivudine, Festinavir and Nevirapine |
| US8927237 | 2015-01-06 | Method for producing acyloxypyranone compound, method for producing alkyne compound, and method for producing dihydrofuran compound |
| US2012322995 | 2012-12-20 | beta-DIHYDROFURAN DERIVING COMPOUND, METHOD FOR PRODUCING beta-DIHYDROFURAN DERIVING COMPOUND OR beta-TETRAHYDROFURAN DERIVING COMPOUND, beta-GLYCOSIDE COMPOUND, METHOD FOR PRODUCING beta GLYCOSIDE COMPOUND, AND METHOD FOR PRODUCING 4′-ETHYNYL D4T AND ANALOGUE COMPOUNDS THEREOF |
| US2012252751 | 2012-10-04 | ANTI-VIRAL NUCLEOSIDE ANALOGS AND METHODS FOR TREATING VIRAL INFECTIONS, ESPECIALLY HIV INFECTIONS |
| US8193165 | 2012-06-05 | Anti-viral nucleoside analogs and methods for treating viral infections, especially HIV infections |
| US2011312880 | 2011-12-22 | POTENT CHIMERIC NRTI-NNRTI BIFUNCTIONAL INHIBITORS OF HIV-1 REVERSE TRANSCRIPTASE |
| US2011054164 | 2011-03-03 | PRODUCTION PROCESS OF ETHYNYLTHYMIDINE COMPOUNDS FROM 5-METHYLURIDINE AS A STARTING MATERIAL |
| US2010280235 | 2010-11-04 | METHOD FOR PRODUCING 4’ETHYNYL d4T |
/////////BMS 986001, 634907-30-5, UNII: 6IE83O6NGA, OBP 601, 4′-Ethynyl D4T, 4′-Ed4T, TDK-4-114, PHASE 2
Cc1cn(c(=O)[nH]c1=O)[C@H]2C=C[C@](O2)(CO)C#C
QbD Presentations
Organizational Initiatives Towards Developing Greener Processes for Generic Active Pharmaceutical Ingredients
– Dr. Vilas H. Dahanukar, Chief Scientist-Process R&D, Integerated Product Development Organization, Dr. Reddy’s Laboratories Ltd., India


presented at
4th Industrial Green Chemistry World Convention & Ecosystem (IGCW-2015) on 4th – 5th December 2015
A PRESENTATION
 The presentation will load below
The presentation will load below
Please scroll with mouse to see
A PRESENTATION
Innovative Techniques, To Synthesize Breakthrough Molecules, See DOE On pae 4 onwards
The presentation will load below
IF YOU HAVE TROUBLE VIEWING SEE…….http://www.allfordrugs.com/qbd-presentations/
Quality by Design Questions to Consider How can we maximize the benefits to the industry and other stakeholders? How can we ensure that this will speed.http://player.slideplayer.com/13/3802357/
http://player.slideplayer.com/13/3802357/
Quality by Design (QbD) N. Vidyashankar 1Quality by Design (QbD), FICCI, 19th Jan 2012.http://player.slideplayer.com/16/5119457/
IF YOU HAVE TROUBLE VIEWING SEE…….http://www.allfordrugs.com/qbd-presentations/
Quality by Design (QbD) N. Vidyashankar 1Quality by Design (QbD), FICCI, 19th Jan 2012.http://player.slideplayer.com/16/5119457/
First to file (FTF) regulatory challenge to QbD adoption 1.http://player.slideplayer.com/26/8584360/
IF YOU HAVE TROUBLE VIEWING SEE…….http://www.allfordrugs.com/qbd-presentations/
First to file (FTF) regulatory challenge to QbD adoption 1.http://player.slideplayer.com/26/8584360/
WHAT YOU NEED TO KNOW…..
Quality by Design in Drug Product Development
Introduction to drug product development – setting the scene
- Drug product development at a glance – from first in man to marketing authorization
- Pharmaceutical QbD: Quo vadis?
- Application of QbD principles to drug product development
Expectations from regulatory agencies
- Regulatory initiatives and approaches for supporting emerging technologies
- Concepts of Real Time Release Testing (Draft Annex 17 EU GMP Guideline)
- Harmonization of regulatory requirements (QbD parallel-assessment FDA-EMA, ICH Q8 -> Draft Q12?)
- Regulatory expectations: Lessons learned from applications so far
Knowledge Management
- Knowledge Management (KM) System – Definition and Reason
- Knowledge Management Cycle
- Explicit and Tacit Knowledge – The Knowledge Spiral
- Correlation between KM and other Processes
- Enabling Knowledge Management
- Knowledge Review – integral part of the Management Review (ICH Q10)
Quality Risk Assessment and Control Strategy
- Objectives of Quality Risk Assessment (QRA) as part of development
- Overview to risk assessment tools
- Introduction of Process Risk Map
- Introduction of risk based control strategy development
QbD Toolbox: Case studies DoE, PAT, and Basic Statistics
- Value-added use of QbD tools – generic approaches and tailored solutions
- Case studies and examples for different unit operations and variable problems
Reports and Documentation
- Development Reports
- Transfer protocols and reports
- Control Strategy and link to the submission dossier
Wrap-up & Final Discussion
The concepts and tools used over the two days will be summarized and future implications and opportunities of applying QbD principles to process development will be discussed. Delegates will be given time to ask questions on how they can apply what they have learned to their own drug product development and manufacturing.
Workshop Process Risk Map & link to Control Strategy
Based on a risk assessment tool tailored to cover development needs, delegates will work on case studies of process development for a solid oral dosage form.
From QTPP and CQA to relationship analysis of process parameters and material attributes
Process mapping for integrated documentation of the development work
Process Risk Map as a tool for development-focussed risk assessment
Quality by Design in API Manufacturing
General framework and key elements of QbD for APIs – background and potential strategies
- What is it all about?
- What are the benefits?
- When and how should you use it?
- Practical examples with typical points of discussion
How to identify and control Critical Quality Attributes (CQAs) in API synthesis – a risk-based approach to developing a control strategy
- Severity assessment of quality attributes
- Impact levels for critical process parameters (CPPs) and critical material attributes (CMAs)
- Considerations for the API Starting material
- Design of an effective risk-based control strategy
- Examples
How to provide information on the development of the API manufacturing process – dossier requirements
- What should be done at which stage?
- Which information is relevant for the dossier?
- What are the key-points to be considered for APIs (NCE/Biotech) and their formulations
- Typical questions from Authorities
Process Evaluation and Design Space
- Changing Validation Approach
- Validation Life Cycle
- Design Space Concept
Application of PAT in the API industry
- PAT at development stages of a QbD-based development
- PAT as part of the Control Strategy in a GMP environment
- Practical examples of PAT implementations at a commercial scale in a GMP environmen
t
Control strategies – Case studies and examples
- HA definitions
- Why and When is a control strategy needed
- Different types/elements of a control strategy
- Practical examples
///////////
ENZYMES AS GREEN CATALYSTS FOR PHARMACUETICAL INDUSTRY

ENZYMES AS GREEN CATALYSTS FOR PHARMACUETICAL INDUSTRY
‘Green’ Catalysts for ‘greener’ reactions
– Dr. Dinesh Nair, Regional Business Manager at Novozymes South Asia Pvt. Ltd

/////////Novozymes, ENZYMES, GREEN CATALYSTS, PHARMACEUTICAL INDUSTRY, ‘Green’ Catalysts, ‘greener’ reactions
BMS-986115

BMS-986115
CAS 1584647-27-7
(2R,3S)-N-((3S)-5-(3-Fluorophenyl)-9-methyl-2-oxo-2,3-dihydro-lH-l,4-benzodiazepin- 3-yl)-2, -bis(3,3,3-trifluoropropyl)succinamide
MW: 574.4945, C26-H25-F7-N4-O3, UNII: LSK1L593UU
10-Nitrooleate, CTK3B7458, CTK3C3167, 9-Octadecenoic acid, 10-nitro-, 875685-46-4, AG-L-63109, 9-Octadecenoic acid, 10-nitro-, (9E)-, 88127-53-1
FOR advanced solid tumors
- Originator Bristol-Myers Squibb
- Class Antineoplastics
- Mechanism of Action Amyloid precursor protein secretase inhibitors; Notch signalling pathway inhibitors
- Phase I Solid tumours
Most Recent Events
- 30 Aug 2016Bristol-Myers Squibb terminates a phase I trial for Solid tumours (late-stage disease, second-line therapy or greater) in USA, Australia and Canada (NCT01986218)
- 25 Jan 2016Bristol-Myers Squibb completes enrolment in its phase I trial for Solid tumours in USA, Australia and Canada (NCT01986218)
- 31 Dec 2013Phase-I clinical trials in Solid tumours (late-stage disease) in Canada & Australia (Oral)
DETAILS WILL BE UPDATED SOON………….
BMS-986115 is an orally bioavailable, gamma secretase (GS) and pan-Notch inhibitor, with potential antineoplastic activity. Upon administration, GS/pan-Notch inhibitor BMS 986115 binds to GS and blocks the proteolytic cleavage and release of the Notch intracellular domain (NICD), which would normally follow ligand binding to the extracellular domain of the Notch receptor. This prevents both the subsequent translocation of NICD to the nucleus to form a transcription factor complex and the expression of Notch-regulated genes. This results in the induction of apoptosis and the inhibition of growth of tumor cells that overexpress Notch. Overexpression of the Notch signaling pathway plays an important role in tumor cell proliferation and survival
| Ashvinikumar V. Gavai, George V. Delucca,Daniel O’MALLEY, Patrice Gill, Claude A. Quesnelle, Brian E. Fink, Yufen Zhao,Francis Y. Lee, | |
| Applicant | Bristol-Myers Squibb Company |

Ashvinikumar Gavai

Claude Quesnelle
Senior Research Investigator/Chemist at Bristol-Myers Squibb

RICHARD LEE


Patrice Gill
Research scientist at BMS

Dan O’Malley (Rice University)
Currently: Bristol-Myers Squibb
PICTURES WILL BE UPDATED………….
Useful for the treatment of conditions related to the Notch pathway, such as cancer and other proliferative diseases.
Notch signaling has been implicated in a variety of cellular processes, such as cell fate specification, differentiation, proliferation, apoptosis, and angiogenesis. (Bray, Nature Reviews Molecular Cell Biology, 7:678-689 (2006); Fortini, Developmental Cell 16:633-647 (2009)). The Notch proteins are single-pass heterodimeric transmembrane molecules. The Notch family includes 4 receptors, NOTCH 1-4, which become activated upon binding to ligands from the DSL family (Delta-like 1, 3, 4 and Jagged 1 and 2).
The activation and maturation of NOTCH requires a series of processing steps, including a proteolytic cleavage step mediated by gamma secretase, a multiprotein complex containing Presenilin 1 or Presenilin 2, nicastrin, APH1, and PEN2. Once NOTCH is cleaved, NOTCH intracellular domain (NICD) is released from the membrane. The released NICD translocates to the nucleus, where it functions as a transcriptional activator in concert with CSL family members (RBPSUH, “suppressor of hairless”, and LAG1). NOTCH target genes include HES family members, such as HES- 1. HES- 1 functions as transcriptional repressors of genes such as HERP 1 (also known as HEY2), HERP2 (also known as HEY1), and HATH1 (also known as ATOH1).
The aberrant activation of the Notch pathway contributes to tumorigenesis. Activation of Notch signaling has been implicated in the pathogenesis of various solid tumors including ovarian, pancreatic, as well as breast cancer and hematologic tumors such as leukemias, lymphomas, and multiple myeloma. The role of Notch inhibition and its utility in the treatment of various solid and hematological tumors are described in Miele, L. et al, Current Cancer Drug Targets, 6:313-323 (2006); Bolos, V. et al, Endocrine Reviews, 28:339-363 (2007); Shih, I.-M. et al, Cancer Research, 67: 1879- 1882 (2007); Yamaguchi, N. et al., Cancer Research, 68: 1881-1888 (2008); Miele, L., Expert Review Anti-cancer Therapy, 8: 1 197-1201 (2008); Purow, B., Current Pharmaceutical Biotechnology, 10: 154-160 (2009); Nefedova, Y. et al, Drug Resistance Updates, 1 1 :210-218 (2008); Dufraine, J. et al, Oncogene, 27:5132-5137 (2008); and Jun, H.T. et al, Drug Development Research, 69:319-328 (2008).
There remains a need for compounds that are useful as Notch inhibitors and that have sufficient metabolic stability to provide efficacious levels of drug exposure. Further, there remains a need for compounds useful as Notch inhibitors that can be orally or intravenously administered to a patient.
U.S. Patent No. 7,053,084 Bl discloses succinoylamino benzodiazepine compounds useful for treating neurological disorders such as Alzheimer’s Disease. The reference discloses that these succinoylamino benzodiazepine compounds inhibit gamma secretase activity and the processing of amyloid precursor protein linked to the formation of neurological deposits of amyloid protein. The reference does not disclose the use of these compounds in the treatment of proliferative diseases such as cancer.
Applicants have found potent compounds that have activity as Notch inhibitors and have sufficient metabolic stability to provide efficacious levels of drug exposure upon intravenous or oral administration. These compounds are provided to be useful as pharmaceuticals with desirable stability, bioavailability, therapeutic index, and toxicity values that are important to their drugability.
![]()

PATENTS
PATENT
https://www.google.com/patents/US20140087992
Example 1(2R,3S)—N-((3S)-5-(3-Fluorophenyl)-9-methyl-2-oxo-2,3-dihydro-1H-1,4-benzodiazepin-3-yl)-2,3-bis(3,3,3-trifluoropropyl)succinamide
Intermediate 1A: (2S,3R)-tert-Butyl 6,6,6-trifluoro-3-(((S)-5-(3-fluorophenyl)-9-methyl-2-oxo-2,3-dihydro-1H-benzo[e][1,4]diazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoate

In a 100 mL round-bottomed flask, a solution of Intermediate B-1 (1683 mg, 5.94 mmol), Et3N (1.656 mL, 11.88 mmol), and Intermediate S-1 in DMF (20 mL) was treated with o-benzotriazol-1-yl-N,N,N′,N′-tetramethyluronium tetrafluoroborate (3815 mg, 11.88 mmol) and stirred at room temperature for 1 hour. The reaction mixture was diluted with water and saturated aqueous NaHCO3. An off white precipitate formed and was filtered and washed with water. The resulting solid was dried on the filter under a stream of nitrogen to give Intermediate 1A (3.7 g, 99% yield). MS (ES): m/z=632.4[M+H+]; HPLC: RT=3.635 min Purity=98%. (H2O/MeOH with TFA, CHROMOLITH® ODS S5 4.6×50 mm, gradient=4 min, wavelength=220 nm). 1H NMR (400 MHz, methanol-d4) δ 7.53 (t, J=4.5 Hz, 1H), 7.46-7.30 (m, 3H), 7.28-7.23 (m, 1H), 7.23-7.18 (m, 2H), 5.37 (s, 1H), 2.88 (td, J=10.4, 3.4Hz, 1H), 2.60 (td, J=10.2, 4.1 Hz, 1H), 2.54-2.40 (m, 1H), 2.47 (s, 3H), 2.33-2.12 (m, 3H), 1.98-1.69 (m, 4H), 1.51 (s, 9H).
Intermediate 1B: (2S,3R)-6,6,6-Trifluoro-3-(((S)-5-(3-fluorophenyl)-9-methyl-2-oxo-2,3-dihydro-1H-benzo[e][1,4]diazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoic acid

In a 250 mL round-bottomed flask, a solution of Intermediate 1A (3.7 g, 5.86 mmol) in DCM (25 mL) was treated with TFA (25 mL) and the resulting pale orange solution was stirred at room temperature for 1.5 hours. The reaction mixture was then concentrated to give Intermediate 1B. HPLC: RT=3.12 min (H2O/MeOH with TFA, CHROMOLITH® ODS S5 4.6×50 mm, gradient=4 min, wavelength=220 nm). MS (ES): m/z=576.3 (M+H)+. 1H NMR (400 MHz, methanol-d4) δ 7.54 (t, J=4.5 Hz, 1H), 7.49-7.29 (m, 3H), 7.28-7.15 (m, 3H), 5.38 (br. s., 1H), 2.89 (td, J=10.3, 3.7 Hz, 1H), 2.67 (td, J=9.9, 4.2Hz, 1H), 2.56-2.38 (m, 1H), 2.48 (s, 3H), 2.34-2.13 (m, 3H), 2.00-1.71 (m, 4H).
Example 1
In a 250 mL round-bottomed flask, a solution of Intermediate 1B (4.04 g, 5.86 mmol) in THF (50 mL) was treated with ammonia (2M in iPrOH) (26.4 mL, 52.7 mmol), followed by HOBT (1.795 g, 11.72 mmol) and EDC (2.246 g, 11.72 mmol). The resulting white suspension was stirred at room temperature overnight. The reaction mixture was diluted with water and saturated aqueous NaHCO3. The resulting solid was filtered, rinsed with water and then dried on the filter under a stream of nitrogen. The crude product was suspended in 20 mL of iPrOH and stirred at room temperature for 20 min and then filtered and washed with iPrOH and dried under vacuum to give 2.83 g of solid. The solid was dissolved in refluxing EtOH (100 mL) and slowly treated with 200 mg activated charcoal added in small portions. The hot mixture was filtered through CELITE® and rinsed with hot EtOH. The filtrate was reduced to half volume, allowed to cool and the white precipitate formed was filtered and rinsed with EtOH to give 2.57 g of white solid. A second recrystallization from EtOH (70 mL) afforded Example 1 (2.39 g, 70% yield) as a white solid. HPLC: RT=10.859 min (H2O/CH3CN with TFA, Sunfire C18 3.5 μm, 3.0×150 mm, gradient=15 min, wavelength=220 and 254 nm); MS (ES): m/z=575.3 [M+H+]; 1H NMR (400 MHz, methanol-d4) δ 7.57-7.50 (m, 1H), 7.47-7.30 (m, 3H), 7.29-7.15 (m, 3H), 5.38 (s, 1H), 2.85-2.75 (m, 1H), 2.59 (td, J=10.5, 4.0 Hz, 1H), 2.53-2.41 (m, 4H), 2.31-2.10 (m, 3H), 1.96-1.70 (m, 4H).
PATENT
https://www.google.com/patents/WO2014047372A1?cl=en


Scheme 3


XII XI
Scheme 4

Intermediate S-l : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid

Intermediate S-IA: 3,3,3-Trifluoro ropyl trifluoromethanesulfonate

[00180] To a cold (-25 °C) stirred solution of 2,6-lutidine (18.38 mL, 158 mmol) in DCM (120 mL) was added Tf20 (24.88 mL, 147 mmol) over 3 min, and the mixture was stirred for 5 min. To the reaction mixture was added 3,3,3-trifluoropropan-l-ol (12 g, 105 mmol) over an interval of 3 min. After 2 hr, the reaction mixture was warmed to room temperature and stirred for 1 hr. The reaction mixture was concentrated to half its volume, then purified by loading directly on a silica gel column (330g ISCO) and the product was eluted with DCM to afford Intermediate S-IA (13.74 g, 53%) as a colorless oil. 1H NMR (400 MHz, CDC13) δ ppm 4.71 (2 H, t, J= 6.15 Hz), 2.49-2.86 (2 H, m).
Intermediate S-1B: (4S)-4-Benzyl-3-(5,5,5-trifluoropentanoyl)-l,3-oxazolidin-2-one

[00181] To a stirring solution of 5,5,5-trifluoropentanoic acid (14.76 g, 95 mmol) and DMF (0.146 rriL) in DCM (50 mL) was slowly added oxalyl chloride (8.27 mL, 95 mmol). After 2h, the mixture was concentrated to dryness. A separate flask was changed with (S)-4-benzyloxazolidin-2-one (16.75 g, 95 mmol) in THF (100 mL) and then cooled to -78 °C. To the solution was slowly added n-BuLi (2.5M, 37.8 mL, 95 mmol) over 10 min, stirred for 10 min, and then a solution of the above acid chloride in THF (50 mL) was slowly added over 5 min. The mixture was stirred for 30 min, and then warmed to room temperature. The reaction was quenched with sat aq NH4C1. Next, 10% aq LiCl was then added to the mixture, and the mixture was extracted with Et20. The organic layer was washed with sat aq NaHC03 then with brine, dried (MgSC^), filtered and concentrated to dryness. The residue was purified by Si02 chromatography (ISCO, 330 g column, eluting with a gradient from 100% hexane to 100% EtOAc) to afford the product Intermediate S-IB; (25.25 g, 85%): 1H NMR (400 MHz, CDC13) δ ppm 7.32-7.39 (2 H, m), 7.30 (1 H, d, J= 7.05 Hz), 7.18-7.25 (2 H, m), 4.64-4.74 (1 H, m), 4.17-4.27 (2 H, m), 3.31 (1 H, dd, J= 13.35, 3.27 Hz), 3.00-3.11 (2 H, m), 2.79 (1 H, dd, J= 13.35, 9.57 Hz), 2.16-2.28 (2 H, m), 1.93-2.04 (2 H, m).
Intermediate S-IC: tert- utyl (3R)-3-(((4S)-4-benzyl-2-oxo-l,3-oxazolidin-3- yl)carbonyl)-6,6,6-trifluoroh xanoate

[00182] To a cold (-78 °C), stirred solution of Intermediate S-IB (3.03 g, 9.61 mmol) in THF (20 mL) was added NaHMDS (1.0M in THF) (10.6 mL, 10.60 mmol) under a nitrogen atmosphere. After 2 hours, tert-butyl 2-bromoacetate (5.62 g, 28.8 mmol) was added neat via syringe at -78 °C and stirring was maintained at the same temperature. After 6 hours, the reaction mixture was warmed to room temperature. The reaction mixture was partitioned between saturated NH4C1 and EtOAc. The organic phase was separated, and the aqueous phase was extracted with EtOAc (3x). The combined organics were washed with brine, dried (Na2s04), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (Teledyne ISCO
CombiFlash Rf, 5% to 100% solvent A/B = hexanes/EtOAc, REDISEP® Si02 120g). Concentration of the appropriate fractions provided Intermediate S-1C (2.79 g, 67.6%) as a colorless viscous oil: 1H NMR (400 MHz, CDC13) δ ppm 7.34 (2 H, d, J= 7.30 Hz), 7.24-7.32 (3 H, m), 4.62-4.75 (1 H, m, J= 10.17, 6.89, 3.43, 3.43 Hz), 4.15-4.25 (3 H, m), 3.35 (1 H, dd, J= 13.60, 3.27 Hz), 2.84 (1 H, dd, J= 16.62, 9.57 Hz), 2.75 (1 H, dd, J = 13.35, 10.07 Hz), 2.47 (1 H, dd, J= 16.62, 4.78 Hz), 2.11-2.23 (2 H, m), 1.90-2.02 (1 H, m), 1.72-1.84 (1 H, m), 1.44 (9 H, s).
Intermediate S-ID: (2R)-2-( -tert-Butoxy-2-oxoethyl)-5,5,5-trifluoropentanoic acid

[00183] To a cool (0 °C), stirred solution of Intermediate S-1C (2.17 g, 5.05 mmol) in THF (50 mL) and water (15 mL) was added a solution of LiOH (0.242 g, 10.11 mmol) and H202 (2.065 mL, 20.21 mmol) in H20 (2 mL). After 10 min, the reaction mixture was removed from the ice bath, stirred for lh, and then cooled to 0 °C. Saturated aqueous NaHCC”3 (25 mL) and saturated aqueous Na2s03 (25 mL) were added to the reaction mixture, and the mixture was stirred for 10 min, and then partially concentrated. The resulting mixture was extracted with DCM (2x), cooled with ice and made acidic with cone. HC1 to pH 3. The mixture was saturated with solid NaCl, extracted with EtOAc (3x), and then dried over MgS04, filtered and concentrated to a colorless oil to afford Intermediate S-ID, 1.2514g, 92%): 1H NMR (400 MHz, CDCI3) δ ppm 2.83-2.95 (1 H, m), 2.62-2.74 (1 H, m), 2.45 (1 H, dd, J= 16.62, 5.79 Hz), 2.15-2.27 (2 H, m), 1.88-2.00 (1 H, m), 1.75-1.88 (1 H, m), 1.45 (9 H, s). Intermediate S-l : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and Intermediate S-1E: (2R,3R)-3-(tert-butoxycarbonyl)- 6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid
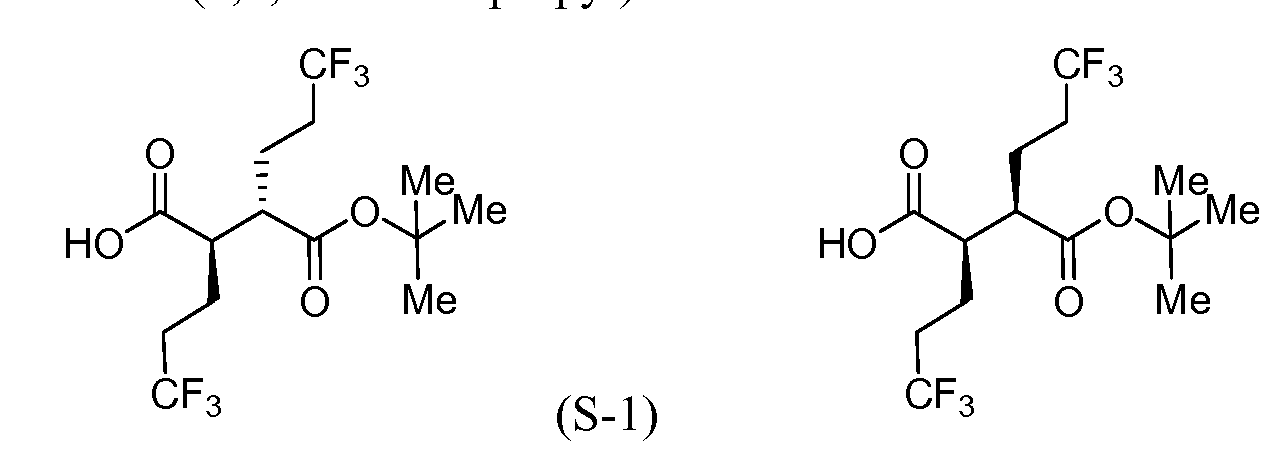
(S-1E)
[00184] To a cold (-78 °C) stirred solution of Intermediate S-1D (5 g, 18.50 mmol) in THF (60 mL) was slowly added LDA (22.2 mL, 44.4 mmol, 2.0M) over 7 min. After stirring for 2 hr, Intermediate S- 1 A (6.38 g, 25.9 mmol) was added to the reaction mixture over 3 min. After 60 min, the reaction mixture was warmed to -25 °C
(ice/MeOH/dry ice) and stirred for an additional 60 min at which time sat aq NH4C1 was added. The separated aqueous phase was acidified with IN HC1 to pH 3, and then extracted with Et20. The combined organic layers were washed with brine (2x), dried over MgS04, filtered and concentrated to provide a 1 :4 (II :I1E) mixture (as determined by 1H NMR) of Intermediate S-l and Intermediate S-1E (6.00 g, 89%) as a pale yellow solid. 1H NMR (500 MHz, CDC13) δ ppm 2.81 (1 H, ddd, J = 10.17, 6.32, 3.85 Hz), 2.63- 2.76 (1 H, m), 2.02-2.33 (4 H, m), 1.86-1.99 (2 H, m), 1.68-1.85 (2 H, m), 1.47 (9 H, s).
[00185] To a cold (-78 °C), stirred solution of a mixture of Intermediate S-l and Intermediate S-1E (5.97 g, 16.30 mmol) in THF (91 mL) was added LDA (19 mL, 38.0 mmol, 2.0M in THF/hexane/ethyl benzene) dropwise via syringe over 10 min (internal temperature never exceeded -65 °C, J-KEM® probe in reaction solution). The mixture was stirred for 15 min, and then warmed to room temperature (24 °C water bath), stirred for 15 min, and then cooled to -78 °C for 15 min. To the reaction mixture was added Et2AlCl (41 mL, 41.0 mmol, 1M in hexane) via syringe (internal temperature never exceeded -55 °C), and the mixture was stirred for 10 min, and then warmed to room temperature (24 °C bath) for 15 min and then back to -78 °C for 15 min. Meanwhile, a 1000 mL round bottom flask was charged with MeOH (145 mL) and precooled to -78 °C. With vigorous stirring the reaction mixture was transferred via cannula over 5 min to the MeOH. The flask was removed from the bath, ice was added followed by the slow addition of IN HC1 (147 mL, 147 mmol). Gas evolution was observed as the HC1 was added. The reaction mixture was allowed to warm to room temperature during which the gas evolution subsided. The reaction mixture was diluted with EtOAc (750 mL), saturated with NaCl, and the organic phase was separated, washed with a solution of potassium fluoride (8.52 g, 147 mmol) and IN HC1 (41 mL, 41.0 mmol) in water (291 mL), brine (100 mL), and then dried (Na2s04), filtered and concentrated under vacuum. 1H NMR showed the product was a 9: 1 mixture of Intermediate S-l and Intermediate S- 1E. The enriched mixture of Intermediate S-l and Intermediate S-1E (6.12 g, >99% yield) was obtained as a dark amber solid: 1H NMR (400 MHz, CDC13) δ ppm 2.64-2.76 (2 H, m), 2.04-2.35 (4 H, m), 1.88-2.00 (2 H, m), 1.71-1.83 (2 H, m), 1.48 (9 H, s).
Alternate procedure to make Intermediate S-l :
Intermediate S-IF: (2R,3 -1 -Benzyl 4-tert-butyl 2,3-bis(3,3,3-trifluoropropyl)succinate

[00186] To a stirred solution of a 9: 1 enriched mixture of Intermediate S-l and Intermediate S-1E (5.98 g, 16.33 mmol) in DMF (63 mL) were added potassium carbonate (4.06 g, 29.4 mmol) and benzyl bromide (2.9 mL, 24.38 mmol), the mixture was then stirred overnight at room temperature. The reaction mixture was diluted with EtOAc (1000 mL), washed with 10% LiCl (3×200 mL), brine (200 mL), dried (Na2S04), filtered, concentrated, and then dried under vacuum. The residue was purified by Si02 chromatography using a toluene:hexane gradient. Diastereomerically purified
Intermediate S-IF (4.81g, 65%) was obtained as a colorless solid: 1H NMR (400 MHz, chloroform-d) δ 7.32-7.43 (m, 5H), 5.19 (d, J= 12.10 Hz, 1H), 5.15 (d, J= 12.10 Hz, 1H), 2.71 (dt, J= 3.52, 9.20 Hz, 1H), 2.61 (dt, J= 3.63, 9.63 Hz, 1H), 1.96-2.21 (m, 4H), 1.69-1.96 (m, 3H), 1.56-1.67 (m, 1H), 1.45 (s, 9H).
Intermediate S-l : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid

[00187] To a solution of Intermediate S-1F (4.81 g, 10.54 mmol) in MeOH (100 mL) was added 10% palladium on carbon (wet, Degussa type, 568.0 mg, 0.534 mmol) in a H2– pressure flask. The vessel was purged with N2 (4x), then purged with H2 (2x), and finally, pressurized to 50 psi and shaken overnight. The reaction vessel was
depressurized and purged with nitrogen. The mixture was filtered through CELITE®, washed with MeOH and then concentrated and dried under vacuum. Intermediate S-1 (3.81 g, 99% yield)) was obtained as a colorless solid: 1H NMR (400 MHz, chloroform-d) δ 2.62-2.79 (m, 2H), 2.02-2.40 (m, 4H), 1.87-2.00 (m, 2H), 1.67-1.84 (m, 2H), 1.48 (s, 9H).
Alternate procedure to make Intermediate S-1 :
Intermediate S-1 : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid

[00188] Intermediate S-1 as a mixture with Intermediate S-IE was prepared in a similar procedure as above from Intermediate S-1D to afford a 1 :2.2 mixture of
Intermediate S-1 and Intermediate S-IE (8.60 g, 23.48 mmol), which was enriched using LDA (2.0 M solution in THF, ethyl benzene and heptane, 28.2 mL, 56.4 mmol) and diethyl aluminum chloride (1.0 M solution in hexane, 59 mL, 59.0 mmol) in THF (91 mL). After workup as described above, the resulting residue was found to be a 13.2: 1 (by 1H NMR) mixture of Intermediate S-1 and Intermediate S-IE, which was treated as follows: The crude material was dissolved in MTBE (43 mL). Hexanes (26 mL) were slowly charged to the reaction mixture while maintaining a temperature below 30 °C. The reaction mixture was stirred for 10 min. Next, tert-butylamine (2.7 mL, 1.1 eq) was charged slowly over a period of 20 minutes while maintaining a temperature below 30 °C. This addition was observed to be exothermic. The reaction mixture was stirred for 2 hrs below 30 °C and then filtered. The solid material was washed with 5:3 MTBE: hexane (80 mL), and the filtrate was concentrated and set aside. The filtered solid was dissolved in dichloromethane (300 mL), washed with IN HC1 (lOOmL), and the organic layer was washed with brine (100 mL x 2), and then concentrated under reduced pressure below 45 °C to afford Intermediate S-l (5.46 g, 64%).
A second alternate procedure for preparing Intermediate S-l :
Intermediate S-1G: tert- utyl 5,5,5-trifluoropentanoate

[00189] To a stirred solution of 5,5,5-trifluoropentanoic acid (5 g, 32.0 mmol) in THF (30 mL) and hexane (30 mL) at 0 °C, was added tert-butyl 2,2,2-trichloroacetimidate (11.46 mL, 64.1 mmol). The mixture was stirred for 15 min at 0 °C. Boron trifluoride etherate (0.406 mL, 3.20 mmol) was added and the reaction mixture was allowed to warm to room temperature overnight. To the clear reaction mixture was added solid NaHC03 (5 g) and stirred for 30 min. The mixture was filtered through MgSC^ and washed with hexanes (200 mL). The solution was allowed to rest for 45 min, and the resulting solid material was removed by filtering on the same MgSC^ filter again, washed with hexanes (100 mL) and concentrated under reduced pressure without heat. The volume was reduced to about 30 mL, filtered through a clean fritted funnel, washed with hexane (5 mL), and then concentrated under reduced pressure without heat. The resulting neat oil was filtered through a 0.45μιη nylon membrane filter disk to provide Intermediate S-1G (6.6 g, 31.4 mmol 98% yield) as a colorless oil: 1H NMR (400 MHz, CDC13) δ ppm 1.38 (s, 9 H) 1.74-1.83 (m, 2 H) 2.00-2.13 (m, 2 H) 2.24 (t, J= 7.28 Hz, 2 H). Intermediate S-1H: (4S)-4-(Propan-2-yl)-3-(5,5,5-trifluoropentanoyl)-l,3-oxazolidin-2- one

[00190] To a stirred solution of 5,5,5-trifluoropentanoic acid (5.04 g, 32.3 mmol) in DCM (50 mL) and DMF (3 drops) was added oxalyl chloride (3.4 mL, 38.8 mmol) dropwise over 5 min. The solution was stirred until all bubbling subsided. The reaction mixture was concentrated under reduced pressure to give pale yellow oil. To a separate flask charged with a solution of (4S)-4-(propan-2-yl)-l,3-oxazolidin-2-one (4.18 g, 32.4 mmol) in THF (100 mL) at -78 °C was added n-BuLi (2.5M in hexane) (13.0 mL, 32.5 mmol) dropwise via syringe over 5 min. After stirring for 10 min, the above acid chloride, dissolved in THF (20 mL), was added via cannula over 15 min. The reaction mixture was warmed to 0 °C, and was allowed to warm to room temperature as the bath warmed and stirred overnight. To the reaction mixture was added saturated NH4C1, and the mixture was extracted with EtOAc (2x). The combined organics were washed with brine, dried (Na2s04), filtered and concentrated under reduced pressure. The crude material was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 5% to 60% solvent A/B = hexanes/EtOAc, REDISEP® Si02 120g). Concentration of the appropriate fractions provided Intermediate S-1H (7.39 g, 86%) as a colorless oil: 1H NMR (400 MHz, CDC13) δ ppm 4.44 (1 H, dt, J= 8.31, 3.53 Hz), 4.30 (1 H, t, J= 8.69 Hz), 4.23 (1 H, dd, J= 9.06, 3.02 Hz), 2.98-3.08 (2 H, m), 2.32-2.44 (1 H, m, J= 13.91, 7.02, 7.02, 4.03 Hz), 2.13-2.25 (2 H, m), 1.88-2.00 (2 H, m), 0.93 (3 H, d, J= 7.05 Hz), 0.88 (3 H, d, J= 6.80 Hz).
Intermediate S-1I: (2S,3R)-tert-Butyl 6,6,6-trifluoro-3-((S)-4-isopropyl-2- oxooxazolidine-3-carbonyl)-2-(3,3,3-trifluoropropyl)hexanoate, and Intermediate S-U: (2R,3R)-tert-Butyl 6,6,6-trifluoro-3-((S)-4-isopropyl-2-oxooxazolidine-3-carbonyl)-2- (3 ,3 ,3 -trifluoropropyl)hexanoate

[00191] To a cold (-78 °C), stirred solution of diisopropylamine (5.3 mL, 37.2 mmol) in THF (59 mL) under a nitrogen atmosphere was added n-BuLi (2.5M in hexane) (14.7 mL, 36.8 mmol). The mixture was then warmed to 0 °C to give a 0.5M solution of LDA. A separate vessel was charged with Intermediate S-1H (2.45 g, 9.17 mmol). The material was azeotroped twice with benzene (the RotoVap air inlet was fitted with a nitrogen inlet to completely exclude humidity), and then toluene (15.3 mL) was added. This solution was added to a flask containing dry lithium chloride (1.96 g, 46.2 mmol). To the resultant mixture, cooled to -78 °C, was added the LDA solution (21.0 mL, 10.5 mmol) and the mixture was stirred at -78 °C for 10 min, then warmed to 0 °C for 10 min., and then cooled to -78 °C. To a separate reaction vessel containing Intermediate S-1G (3.41 g, 16.07 mmol), also azeotroped twice with benzene, was added toluene (15.3 mL), cooled to -78 °C and LDA (37.0 mL, 18.5 mmol) was added. The resulting solution was stirred at -78 °C for 25 min. At this time the enolate derived from the ester was transferred via cannula into the solution of the oxazolidinone enolate and stirred at -78 °C for an additional 5 min, at which time the septum was removed and solid powdered bis(2- ethylhexanoyloxy)copper (9.02 g, 25.8 mmol) was rapidly added to the reaction vessel and the septum was replaced. The vessel was immediately removed from the cold bath and immersed into a warm water bath (40 °C) with rapid swirling and with a concomitant color change from the initial turquoise to brown. The reaction mixture was stirred for 20 min, was then poured into 5% aqueous NH4OH (360 mL) and extracted with EtOAc (2x). The combined organics were washed with brine, dried (Na2s04), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 0% to 60% solvent A/B = hexanes/EtOAc, REDISEP® Si02 120g). Concentration of the appropriate fractions provided a mixture of Intermediate S- II and Intermediate S-1J (2.87 g, 66%) as a pale yellow viscous oil. 1H NMR showed the product was a 1.6: 1 mixture of diastereomers S-1LS-1J as determined by the integration of the multiplets at 2.74 and 2.84 ppm: 1H NMR (400 MHz, CDC13) δ ppm 4.43-4.54 (2 H, m), 4.23-4.35 (5 H, m), 4.01 (1 H, ddd, J= 9.54, 6.27, 3.51 Hz), 2.84 (1 H, ddd, J = 9.41, 7.28, 3.64 Hz), 2.74 (1 H, ddd, J= 10.29, 6.27, 4.02 Hz), 2.37-2.48 (2 H, m, J = 10.38, 6.98, 6.98, 3.51, 3.51 Hz), 2.20-2.37 (3 H, m), 1.92-2.20 (8 H, m), 1.64-1.91 (5 H, m), 1.47 (18 H, s), 0.88-0.98 (12 H, m). Intermediate S-1 : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and Intermediate S-IE: (2R,3R)-3-(tert-Butoxycarbonyl)- 6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid

(S-IE)
[00192] To a cool (0 °C), stirred solution of Intermediate S-1I and Intermediate S-1 J (4.54 g, 9.51 mmol) in THF (140 mL) and water (42 mL) were sequentially added hydrogen peroxide (30% in water) (10.3 g, 91 mmol) and LiOH (685.3 mg, 28.6 mmol). The mixture was stirred for 1 hr. At this time the reaction vessel was removed from the cold bath and then stirred for 1.5 hr. To the reaction mixture were added saturated NaHC03 (45 mL) and saturated Na2s03 (15 mL), and then the mixture was partially concentrated under reduced pressure. The resulting crude solution was extracted with DCM (3x). The aqueous phase was acidified to pH~l-2 with IN HC1, extracted with DCM (3x) and then EtOAc (lx). The combined organics were washed with brine, dried (Na2s04), filtered and concentrated under reduced pressure to provide a mixture of Intermediates S-1 and S-IE (3.00 g, 86%) as a colorless oil: 1H NMR (400 MHz, CDC13) δ ppm 2.76-2.84 (1 H, m, diastereomer 2), 2.64-2.76 (3 H, m), 2.04-2.35 (8 H, m), 1.88- 2.00 (4 H, m), 1.71-1.83 (4 H, m), 1.48 (9 H, s, diastereomer 1), 1.46 (9 H, s,
diastereomer 2); 1H NMR showed a 1.7: 1 mixture of S-1E:S-1F by integration of the peaks for the t-butyl groups. Intermediate S-1 : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and Intermediate S-IF: (2R,3R)-3-(fert-Butoxycarbonyl)- 6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid

[00193] To a cold (-78 °C) stirred solution of diisopropylamine (1.7 mL, 11.93 mmol) in THF (19 mL) under a nitrogen atmosphere was added n-BuLi (2.5M in hexanes) (4.8 mL, 12.00 mmol). The mixture was stirred for 5 min and then warmed to 0 °C. In a separate vessel, to a cold (-78 °C) stirred solution of the mixture of Intermediates S-1 and S-1E (1.99 g, 5.43 mmol) in THF (18 mL) was added the LDA solution prepared above via cannula slowly over 25 min. The mixture was stirred for 15 min, then warmed to room temperature (placed in a 24 °C water bath) for 15 min, and then again cooled to -78 °C for 15 min. To the reaction mixture was added Et2AlCl (1M in hexane) (11.4 mL, 11.40 mmol) via syringe. The mixture was stirred for 10 min, warmed to room
temperature for 15 min and then cooled back to -78 °C for 15 min. Methanol (25 mL) was rapidly added, swirled vigorously while warming to room temperature, and then concentrated to ~l/4 the original volume. The mixture was dissolved in EtOAc and washed with IN HC1 (50 mL) and ice (75 g). The aqueous phase was separated and extracted with EtOAc (2x). The combined organics were washed with a mixture of KF (2.85g in 75 mL water) and IN HC1 (13 mL) [resulting solution pH 3-4], then with brine, dried (Na2s04), filtered and concentrated under reduced pressure to give a 9: 1 (S-LS-1E) enriched diastereomeric mixture (as determined by 1H NMR) of Intermediate S-1 and Intermediate S-1E (2.13 g, >99%) as a pale yellow viscous oil: 1H NMR (400 MHz, CDC13) δ ppm 2.64-2.76 (2 H, m), 2.04-2.35 (4 H, m), 1.88-2.00 (2 H, m), 1.71-1.83 (2 H, m), 1.48 (9 H, s).
Intermediate S-2: (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3- fluoropropyl)hexanoic acid

Intermediate S-2: (2R,3S)-3-(tert-Butoxycarbonyl)-7,7,7-trifluoro-2-(3,3,3- trifluoropropyl)heptanoic acid, and Intermediate S-2A: (2R,3R)-3-(tert-Butoxycarbonyl)- 7,7,7-trifluoro-2-(3,3,3-trifluoropropyl)heptanoic acid

(S-2A)
[00194] To a cold (-78 °C), stirred solution of Intermediate S-1D (1.72 g, 6.36 mmol) in THF (30 mL) was slowly added LDA (7.32 mL, 14.6 mmol) over 7 min. After stirring for 1 h, 4,4,4-trifluorobutyltrifluoromethanesulfonate (2.11 g, 8.11 mmol) was added to the reaction mixture over 2 min. After 15 min, the reaction mixture was warmed to -25 °C (ice/MeOH/dry ice) for lh, and then cooled to -78 °C. After 80 min, the reaction was quenched with a saturated aqueous NH4C1 solution (10 mL). The reaction mixture was further diluted with brine and the solution was adjusted to pH 3 with IN HC1. The aqueous layer was extracted with ether. The combined organics were washed with brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure to provide a mixture of Intermediates S-2 and S-2A (2.29 g, 95%) as a colorless oil. 1H NMR (400MHz, chloroform-d) δ 2.83-2.75 (m, 1H), 2.64 (ddd, J = 9.9, 6.7, 3.6 Hz, 1H), 2.32-2.03 (m, 5H), 1.98-1.70 (m, 3H), 1.69-1.52 (m, 3H), 1.50-1.42 (m, 9H). 1H NMR showed a 1 :4.5 mixture (S-2:S-2A) of diastereomers by integration of the peaks for the t- Bu groups.
Intermediate S-2: (2R,3S)-3-(fert-Butoxycarbonyl)-7,7,7-trifluoro-2-(3,3,3- trifluoropropyl)heptanoic acid, and Intermediate S-2A: (2R,3R)-3-(tert-Butoxycarbonyl)- 7,7,7-trifluoro-2-(3,3,3-trifluoropropyl)heptanoic acid

[00195] A mixture of Intermediate S-2 and Intermediate S-2A (2.29 g, 6.02 mmol) was dissolved in THF (38 mL) to give a colorless solution which was cooled to -78 °C. Then, LDA (7.23 mL, 14.5 mmol) (2.0M in heptane/THF/ethylbenzene) was slowly added to the reaction mixture over 3 min. After stirring for 15 min, the reaction mixture was placed in a room temperature water bath. After 15 min the reaction mixture was placed back in a -78 °C bath and then diethylaluminum chloride (14.5 mL, 14.5 mmol) (1M in hexane) was added slowly over 5 min. The reaction mixture was stirred at -78 °C. After 15 min, the reaction mixture was placed in a room temperature water bath for 10 min, and then cooled back to -78 °C. After 15 min, the reaction was quenched with MeOH (30.0 mL, 741 mmol), removed from the -78 °C bath and concentrated. To the reaction mixture was added ice and HC1 (60.8 mL, 60.8 mmol) and the resulting mixture was extracted with EtOAc (2x 200 mL). The organic layer was washed with potassium fluoride (3.50g, 60.3 mmol) in 55 mL H20 and 17.0 mL of IN HC1. The organics were dried over anhydrous magnesium sulfate and concentrated under reduced pressure to provide an enriched mixture of Intermediate S-2 and Intermediate S-2A (2.25g, 98% yield) as a light yellow oil. 1H NMR (400MHz, chloroform-d) δ 2.83-2.75 (m, 1H), 2.64 (ddd, J= 9.9, 6.7, 3.6 Hz, 1H), 2.32-2.03 (m, 5H), 1.98-1.70 (m, 3H), 1.69-1.52 (m, 3H), 1.50-1.42 (m, 9H). 1H NMR showed a 9: 1 ratio in favor of the desired diastereomer Intermediate S-2.
Intermediate S-2B: (2R,3S)-1 -Benzyl 4-tert-butyl 2,3-bis(4,4,4-trifluorobutyl)succinate

[00196] To a stirred 9: 1 mixture of Intermediate S-2 and Intermediate S-2A (2.24 g, 5.89 mmoL) and potassium carbonate (1.60 g, 11.58 mmoL) in DMF (30 mL) was added benzyl bromide (1.20 mL, 10.1 mmoL)). The reaction mixture was stirred at room temperature for 19 h. The reaction mixture was diluted with ethyl acetate (400 mL) and washed with 10% LiCl solution (3 x 100 mL), brine (50 mL), and then dried over anhydrous magnesium sulfate, filtered and concentrated to dryness under vacuum. The residue was purified by flash chromatography (Teledyne ISCO CombiFlash 0%> to 100% solvent A/B = hexane/EtOAc, REDISEP® Si02 220 g, detecting at 254 nm, and monitoring at 220 nm). Concentration of the appropriate fractions provided Intermediate S-2B (1.59 g, 57.5%). HPLC: RT = 3.863 min (CHROMOLITH® SpeedROD column 4.6 x 50 mm, 10-90% aqueous methanol over 4 minutes containing 0.1% TFA, 4 mL/min, monitoring at 220 nm), 1H NMR (400MHz, chloroform-d) δ 7.40-7.34 (m, 5H), 5.17 (d, J= 1.8 Hz, 2H), 2.73-2.64 (m, 1H), 2.55 (td, J= 10.0, 3.9 Hz, 1H), 2.16-1.82 (m, 5H), 1.79-1.57 (m, 3H), 1.53-1.49 (m, 1H), 1.45 (s, 9H), 1.37-1.24 (m, 1H).
Intermediate S-2: (2R,3S)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(4,4,4- trifluorobutyl)hexanoic acid

[00197] To a stirred solution of Intermediate S-2B (1.59 g, 3.37 mmoL) in MeOH (10 mL) and EtOAc (10 mL) under nitrogen was added 10%> Pd/C (510 mg). The atmosphere was replaced with hydrogen and the reaction mixture was stirred at room temperature for 2.5 h. The palladium catalyst was filtered off through a 4 μΜ polycarbonate film and rinsed with MeOH. The filtrate was concentrated under reduced pressure to give intermediate S-2 (1.28 g, 99%). 1H NMR (400MHz, chloroform-d) δ 2.76-2.67 (m, 1H), 2.65-2.56 (m, 1H), 2.33-2.21 (m, 1H), 2.17-2.08 (m, 3H), 1.93 (dtd, J= 14.5, 9.9, 5.2 Hz, 1H), 1.84-1.74 (m, 2H), 1.70-1.52 (m, 3H), 1.48 (s, 9H).
Intermediate A- 1 : (2-Amino-3 -methylphenyl)(3 -fluorophenyl)methanone

Intermediate A-1 A: 2-Amino- -methoxy-N,3-dimethylbenzamide

[00198] In a 1 L round-bottomed flask was added 2-amino-3-methylbenzoic acid (11.2 g, 74.1 mmol) and Ν,Ο-dimethylhydroxylamine hydrochloride (14.45 g, 148 mmol) in DCM (500 mL) to give a pale brown suspension. The reaction mixture was treated with Et3N (35 mL), HOBT (11.35 g, 74.1 mmol) and EDC (14.20 g, 74.1 mmol) and then stirred at room temperature for 24 hours. The mixture was then washed with 10% LiCl, and then acidified with IN HCl. The organic layer was washed successively with 10%> LiCl and aq NaHC03. The organic layer was decolorized with charcoal, filtered, and the filtrate was dried over MgSC^. The mixture was filtered and concentrated to give 13.22 g (92% yield) of Intermediate A-1A. MS(ES): m/z = 195.1 [M+H+]; HPLC: RT = 1.118 min. (H20/MeOH with TFA, CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm); 1H NMR (500MHz, chloroform-d) δ 7.22 (dd, J= 7.8, 0.8 Hz, 1H), 7.12-7.06 (m, 1H), 6.63 (t, J= 7.5 Hz, 1H), 4.63 (br. s., 2H), 3.61 (s, 3H), 3.34 (s, 3H), 2.17 (s, 3H).
Intermediate A- 1 : (2-Amino-3 -methylphenyl)(3 -fluorophenyl)methanone

[00199] In a 500 mL round-bottomed flask, a solution of l-fluoro-3-iodobenzene (13.61 mL, 116 mmol) in THF (120 mL) was cooled in a -78 °C bath. A solution of n- BuLi, (2.5M in hexane, 46.3 mL, 116 mmol) was added dropwise over 10 minutes. The solution was stirred at -78 °C for 30 minutes and then treated with a solution of
Intermediate A-1 A (6.43 g, 33.1 mmol) in THF (30 mL). After 1.5 hours, the reaction mixture was added to a mixture of ice and IN HCl (149 mL, 149 mmol) and the reaction flask was rinsed with THF (5 ml) and combined with the aqueous mixture. The resulting mixture was diluted with 10% aq LiCl and the pH was adjusted to 4 with IN NaOH. The mixture was then extracted with Et20, washed with brine, dried over MgS04, filtered and concentrated. The resulting residue was purified by silica gel chromatography (220g ISCO) eluting with a gradient from 10% EtOAc/hexane to 30% EtOAc/hexane to afford Intermediate A-l (7.11 g, 94% yield) as an oil. MS(ES): m/z = 230.1 [M+H+]; HPLC: RT = 2.820 min Purity = 99%. (H20/MeOH with TFA, CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm).
Intermediate B-1 : (S)-3-Amino-5-(3-fluorophenyl)-9-methyl-lH-benzo[e][l,4]diazepin- 2(3H)-one

Intermediate B-1 A: (S)-Benzyl (5-(3-fluorophenyl)-9-methyl-2-oxo-2,3-dihydro benzo[e] [ 1 ,4]diazepin-3-yl)carbamate

(B-1A)
[00225] In a 1 L round-bottomed flask, a solution of 2-(lH-benzo[d][l,2,3]triazol-l- yl)-2-((phenoxycarbonyl)amino)acetic acid (J. Org. Chem., 55:2206-2214 (1990)) (19.37 g, 62.0 mmol) in THF (135 mL) was cooled in an ice/water bath and treated with oxalyl chloride (5.43 mL, 62.0 mmol) and 4 drops of DMF. The reaction mixture was stirred for 4 hours. Next, a solution of Intermediate A- 1 (7.11 g, 31.0 mmol) in THF (35 mL) was added and the resulting solution was removed from the ice/water bath and stirred at room temperature for 1.5 hours. The mixture was then treated with a solution of ammonia, (7M in MeOH) (19.94 mL, 140 mmol). After 15 mins, another portion of ammonia, (7M in MeOH) (19.94 mL, 140 mmol) was added and the resulting mixture was sealed under N2 and stirred overnight at room temperature. The reaction mixture was then concentrated to ~l/2 volume and then diluted with AcOH (63 mL) and stir at room temperature for 4 hours. The reaction mixture was then concentrated, and the residue was diluted with 500 mL water to give a precipitate. Hexane and Et20 were added and the mixture was stirred at room temperature for 1 hour to form an orange solid. Et20 was removed under a stream of nitrogen and the aqueous layer was decanted. The residue was triturated with 40 mL of iPrOH and stirred at room temperature to give a white precipitate. The solid was filtered and washed with iPrOH, then dried on a filter under a stream of nitrogen to give racemic Intermediate B-1A (5.4 g, 41.7%yield).
[00226] Racemic Intermediate B-1A (5.9 g, 14.3 mmol) was resolved using the Chiral SFC conditions described below. The desired stereoisomer was collected as the second peak in the elution order: Instrument: Berger SFC MGIII, Column: CHIRALPAK® IC 25 x 3 cm, 5 cm; column temp: 45 °C; Mobile Phase: C02/MeOH (45/55); Flow rate: 160 mL/min; Detection at 220 nm.
[00227] After evaporation of the solvent, Intermediate B-1A (2.73 g, 46% yield) was obtained as a white solid. HPLC: RT = 3.075 min. (H20/MeOH with TFA,
CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm).
Chiral HPLC RT: 8.661 min (AD, 60% (EtOH/MeOH)/heptane) > 99%ee. MS(ES): m/z = 418.3 [M+H+];1H NMR (500MHz, DMSO-d6) δ 10.21 (s, 1H), 8.38 (d, J= 8.3 Hz, 1H), 7.57-7.47 (m, 2H), 7.41-7.29 (m, 8H), 7.25-7.17 (m, 2H), 5.10-5.04 (m, 3H), 2.42 (s, 3H).
Intermediate B-l : (S)-3-Amino-5-(3-fluorophenyl)-9-methyl-lH-benzo[e][l,4]diazepin- 2(3H)-one.
[00228] In a 100 mL round-bottomed flask, a solution of Intermediate B-1A (2.73 g, 6.54 mmol) in acetic acid (12 mL) was treated with HBr, 33% in HOAc (10.76 mL, 65.4 mmol) and the mixture was stirred at room temperature for 1 hour. The solution was diluted with Et20 to give a yellow precipitate. The yellow solid was filtered and rinsed with Et20 under nitrogen. The solid was transferred to 100 mL round bottom flask and water was added (white precipitate formed). The slurry was slowly made basic with saturated NaHC03. The resulting tacky precipitate was extracted with EtOAc. The organic layer was washed with water, dried over MgS04, and then filtered and
concentrated to dryness to give Intermediate B-l (1.68 g, 91% yield) as a white foam solid. MS(ES): m/z = 284.2 [M+H+]; HPLC: RT = 1.72 min (H20/MeOH with TFA, CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm). 1H NMR (400MHz, DMSO-d6) δ 10.01 (br. s., 1H), 7.56-7.44 (m, 2H), 7.41-7.26 (m, 3H), 7.22-7.11 (m, 2H), 4.24 (s, 1H), 2.55 (br. s., 2H), 2.41 (s, 3H). [00229] The compounds listed below in Table 6 (Intermediates B-2 to B-3) were prepared according to the general synthetic procedure described for Intermediate B-l , using the starting materials Intermediate A- 10 and Intermediate A-4, respectively.
Example 1
(2R,3S)-N-((3S)-5-(3-Fluorophenyl)-9-methyl-2-oxo-2,3-dihydro-lH-l,4-benzodiazepin- 3-yl)-2, -bis(3,3,3-trifluoropropyl)succinamide

Intermediate 1A: (2S,3R)-tert-Butyl 6,6,6-trifluoro-3-(((S)-5-(3-fluorophenyl)-9-methyl- 2-0X0-2, 3-dihydro-lH-benzo[e][l,4]diazepin-3-yl)carbamoyl)-2-(3,3 ,3- trifluoropropyl)hexanoat

[00240] In a 100 mL round-bottomed flask, a solution of Intermediate B-l (1683 mg, 5.94 mmol), Et3N (1.656 mL, 11.88 mmol), and Intermediate S-l in DMF (20 mL) was treated with o-benzotriazol-l-yl-A .A .N’.N’-tetramethyluronium tetrafluoroborate (3815 mg, 11.88 mmol) and stirred at room temperature for 1 hour. The reaction mixture was diluted with water and saturated aqueous NaHC03. An off white precipitate formed and was filtered and washed with water. The resulting solid was dried on the filter under a stream of nitrogen to give Intermediate 1A (3.7 g, 99% yield). MS(ES): m/z =
632.4[M+H+]; HPLC: RT = 3.635 min Purity = 98%. (H20/MeOH with TFA,
CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm). 1H NMR (400MHz, methanol-d4) δ 7.53 (t, J = 4.5 Hz, 1H), 7.46-7.30 (m, 3H), 7.28-7.23 (m, 1H), 7.23-7.18 (m, 2H), 5.37 (s, 1H), 2.88 (td, J = 10.4, 3.4 Hz, 1H), 2.60 (td, J =
10.2, 4.1 Hz, 1H), 2.54-2.40 (m, 1H), 2.47 (s, 3 H), 2.33-2.12 (m, 3H), 1.98-1.69 (m, 4H), 1.51 (s, 9H). Intermediate IB: (2S,3R)-6,6,6-Trifluoro-3-(((S)-5-(3-fluorophenyl)-9-methyl-2-oxo-
2,3-dihydro-lH-benzo[e][l,4]diazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoic acid

[00241] In a 250 mL round-bottomed flask, a solution of Intermediate 1A (3.7 g, 5.86 mmol) in DCM (25 mL) was treated with TFA (25 mL) and the resulting pale orange solution was stirred at room temperature for 1.5 hours. The reaction mixture was then concentrated to give Intermediate IB. HPLC: RT = 3.12 min (H20/MeOH with TFA, CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm).
MS(ES): m/z = 576.3 (M+H)+. 1H NMR (400MHz, methanol-d4) δ 7.54 (t, J= 4.5 Hz, 1H), 7.49-7.29 (m, 3H), 7.28-7.15 (m, 3H), 5.38 (br. s., 1H), 2.89 (td, J= 10.3, 3.7 Hz, 1H), 2.67 (td, J= 9.9, 4.2 Hz, 1H), 2.56-2.38 (m, 1H), 2.48 (s, 3 H), 2.34-2.13 (m, 3H), 2.00-1.71 (m, 4H).
Example 1 :
[00242] In a 250 mL round-bottomed flask, a solution of Intermediate IB (4.04 g, 5.86 mmol) in THF (50 mL) was treated with ammonia (2M in iPrOH) (26.4 mL, 52.7 mmol), followed by HOBT (1.795 g, 11.72 mmol) and EDC (2.246 g, 11.72 mmol). The resulting white suspension was stirred at room temperature overnight. The reaction mixture was diluted with water and saturated aqueous NaHC03. The resulting solid was filtered, rinsed with water and then dried on the filter under a stream of nitrogen. The crude product was suspended in 20 mL of iPrOH and stirred at room temperature for 20 min and then filtered and washed with iPrOH and dried under vacuum to give 2.83 g of solid. The solid was dissolved in re fluxing EtOH(100 mL) and slowly treated with 200 mg activated charcoal added in small portions. The hot mixture was filtered through CELITE® and rinsed with hot EtOH. The filtrate was reduced to half volume, allowed to cool and the white precipitate formed was filtered and rinsed with EtOH to give 2.57 g of white solid. A second recrystallization from EtOH (70 mL) afforded Example 1 (2.39 g, 70% yield) as a white solid. HPLC: RT = 10.859 min (H20/CH3CN with TFA, Sunfire C18 3.5μπι, 3.0x150mm, gradient = 15 min, wavelength = 220 and 254 nm); MS(ES): m/z = 575.3 [M+H+]; 1H NMR (400MHz, methanol-d4) δ 7.57-7.50 (m, 1H), 7.47-7.30 (m, 3H), 7.29-7.15 (m, 3H), 5.38 (s, 1H), 2.85-2.75 (m, 1H), 2.59 (td, J= 10.5, 4.0 Hz, 1H), 2.53-2.41 (m, 4H), 2.31-2.10 (m, 3H), 1.96-1.70 (m, 4H).
SEE
WO2012129353A1 *Mar 22, 2012Sep 27, 2012Bristol-Myers Squibb CompanyBis(fluoroalkyl)-1,4-benzodiazepinone compounds
PAPER RELATED
Structure–activity relationships in a series of (2-oxo-1,4-benzodiazepin-3-yl)-succinamides identified highly potent inhibitors of γ-secretase mediated signaling of Notch1/2/3/4 receptors. On the basis of its robust in vivo efficacy at tolerated doses in Notch driven leukemia and solid tumor xenograft models, 12 (BMS-906024) was selected as a candidate for clinical evaluation.
Discovery of Clinical Candidate BMS-906024: A Potent Pan-Notch Inhibitor for the Treatment of Leukemia and Solid Tumors
†Bristol-Myers Squibb Research and Development, Princeton, New Jersey 08543, United States
‡Bristol-Myers Squibb Research and Development, 5 Research Parkway, Wallingford, Connecticut 06492, United States
§ Department of Chemistry, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037,United States
ACS Med. Chem. Lett., 2015, 6 (5), pp 523–527
DOI: 10.1021/acsmedchemlett.5b00001, http://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.5b00001
*Phone: 609-252-5091. E-mail: ashvinikumar.gavai@bms.com.

Patent
http://www.google.co.in/patents/WO2012129353A1?cl=en
PATENT RELATED






PATENTS RELATED
Clip RELATED
For some disease targets, an indirect approach may be best. Or so Ashvinikumar V. Gavai and his colleagues atBristol-Myers Squibbfound in their quest toward a potential cancer drug. Gavai unveiled BMS-906024, which is an experimental—and slightly roundabout—treatment for a number of cancers, including breast, lung, and colon cancers, and leukemia.
Cancers have a tendency to relapse or to become resistant to treatments that once worked. Research at BMS and elsewhere had suggested that a family of proteins called Notch is implicated in that resistance and in cancer progression more generally. Gavai, director of oncology chemistry at BMS in Princeton, N.J., and his team set out to block Notch family signaling.
Notch family members lack enzymatic activity, so blocking them directly is difficult. Instead, BMS developed inhibitors of an enzyme that is essential for activating Notch signaling—γ-secretase.

Company: Bristol-Myers Squibb
Target: pan-Notch
Disease: breast, lung, colon cancer; leukemia
Interfering with Notch, even in this indirect way, can have detrimental effects on the gastrointestinal tract. Only two of the four Notch family members are linked to that side effect, Gavai says. But he and his team think their drug will be most effective if it acts on all four family members roughly equally—a so-called pan-Notch inhibitor. By selecting a molecule that’s well tolerated in animals and carefully scheduling doses of the drug in humans, it could be possible to minimize side effects, he says.
The BMS team relied on Notch signaling assays in leukemia and breast cancer cell lines to find leads. They soon learned that for their molecules to work, three chiral centers had to be in the S,R,Sconfiguration. After that, they strove to make the molecules last in the bloodstream. They removed an isobutyl group and tweaked some other parts of their candidate’s succinamide side chain. It was tough to retain both a long half-life and activity against Notch, Gavai told C&EN. “You’d optimize one and lose the other.”
His team threaded the needle with BMS-906024. Their studies with mice suggest that a dose of 4–6 mg once a week could be effective in people. That’s lower than doses being tested for other Notch-targeted agents, according to the website clinicaltrials.gov. The mouse studies also back the idea that Notch is involved in cancer drug resistance and suggest that Notch could be a target for taking on cancer stem cells, which are notoriously resistant to chemotherapy.
BMS-906024 is in Phase I clinical trials, both alone and in combination with other agents. Patients with colon, lung, breast, and other cancers are receiving intravenous doses of the compound to determine its safety and optimum dose ranges.

(From left, front row) Gavai, Weifeng Shan, (second row) Aaron Balog, Patrice Gill, Gregory Vite, (third row) Francis Lee, Claude Quesnelle, (rear row) Wen-Ching Han, Richard Westhouse.
Credit: Catherine Stroud Photography
http://cen.acs.org/articles/91/i16/BMS-906024-Notch-Signaling-Inhibitor.html

PAPER RELATED

An enantioselective synthesis of (S)-7-amino-5H,7H-dibenzo[b,d]azepin-6-one (S–1) is described. The key step in the sequence involved crystallization-induced dynamic resolution (CIDR) of compound 7 using Boc-d-phenylalanine as a chiral resolving agent and 3,5-dichlorosalicylaldehyde as a racemization catalyst to afford S–1 in 81% overall yield with 98.5% enantiomeric excess.
† Department of Discovery Synthesis, Biocon Bristol-Myers Squibb Research Centre, Biocon Park, Bommasandra IV Phase, Jigani Link Road, Bengaluru 560099, India
‡Bristol-Myers Squibb Company, P.O Box 4000, Princeton, New Jersey 08543-4000, United States
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.6b00207, http://pubs.acs.org/doi/suppl/10.1021/acs.oprd.6b00207
*E-mail: anuradha.gupta@syngeneintl.com.
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2000007995A1 * | Aug 7, 1999 | Feb 17, 2000 | Du Pont Pharmaceuticals Company | SUCCINOYLAMINO LACTAMS AS INHIBITORS OF Aβ PROTEIN PRODUCTION |
| WO2000038618A2 * | Dec 23, 1999 | Jul 6, 2000 | Du Pont Pharmaceuticals Company | SUCCINOYLAMINO BENZODIAZEPINES AS INHIBITORS OF Aβ PROTEIN PRODUCTION |
| WO2001060826A2 * | Feb 16, 2001 | Aug 23, 2001 | Bristol-Myers Squibb Pharma Company | SUCCINOYLAMINO CARBOCYCLES AND HETEROCYCLES AS INHIBITORS OF Aβ PROTEIN PRODUCTION |
| US6737038 * | May 17, 2000 | May 18, 2004 | Bristol-Myers Squibb Company | Use of small molecule radioligands to discover inhibitors of amyloid-beta peptide production and for diagnostic imaging |
| US7053084 | Feb 17, 2000 | May 30, 2006 | Bristol-Myers Squibb Company | Succinoylamino benzodiazepines as inhibitors of Aβ protein production |
| US7456172 | Jan 13, 2006 | Nov 25, 2008 | Bristol-Myers Squibb Pharma Company | Succinoylamino benzodiazepines as inhibitors of Aβ protein production |
| US20030134841 * | Nov 1, 2002 | Jul 17, 2003 | Olson Richard E. | Succinoylamino lactams as inhibitors of A-beta protein production |
| US20120245151 * | Mar 22, 2012 | Sep 27, 2012 | Bristol-Myers Squibb Company | Bisfluoroalkyl-1,4-benzodiazepinone compounds |
//////////BMS-986115, BMS 986115, 3,5-dichlorosalicylaldehyde, Alzheimer’s disease, Boc-D-phenylalanine, CIDR;dibenzoazepenone, DKR; Notch inhibitors, Notch inhibitor, SAR, T-acute lymphoblastic leukemia, triple-negative breast cancer, γ-secretase inhibitor, PHASE 1, BMS, Bristol-Myers Squibb, Ashvinikumar Gavai, 1584647-27-7, UNII: LSK1L593UU
Cc1cccc2c1NC(=O)[C@H](N=C2c3cccc(c3)F)NC(=O)[C@H](CCC(F)(F)F)[C@H](CCC(F)(F)F)C(=O)N
BMS 906024

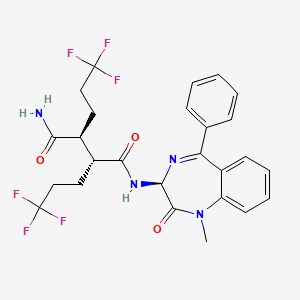

BMS 906024
cas 1401066-79-2
- MF C26H26F6N4O3
- MW 556.500
(2R,3S)-N-[(3S)-1-Methyl-2-oxo-5-phenyl-2,3-dihydro-1H-1,4-benzodiazepin-3-yl]-2,3-bis(3,3,3-trifluoropropyl)succinamide
Butanediamide, N1-((3S)-2,3-dihydro-1-methyl-2-oxo-5-phenyl-1H-1,4-benzodiazepin-3-yl)-2,3-bis(3,3,3-trifluorophenyl)-, (2R,3S)-
(2R,35)-N-((35)-l-Methyl-2-oxo-5-phenyl-2,3-dihydro-lH-l,4-benzodiazepin-3-yl)-3- (2,2,2-trifluoroethyl)-2-(3,3,3-trifluoropropyl)succinamide
| Claude Quesnelle, Soong-Hoon Kim, Francis Lee, Ashvinikumar Gavai | |
| Applicant | Bristol-Myers Squibb Company |

Claude Quesnelle
Senior Research Investigator/Chemist at Bristol-Myers Squibb
RICHARD LEE
BMS-906024 is a novel, potent Notch receptor inhibitor . Cancers have a tendency to relapse or to become resistant to treatments that once worked. A family of proteins called Notch is implicated in that resistance and in cancer progression more generally. BMS-906024 is in Phase I clinical trials, both alone and in combination with other agents. Patients with colon, lung, breast, and other cancers are receiving intravenous doses of the compound to determine its safety and optimum dose ranges.
New Phase I drug structure by Bristol-Myers Squibb disclosed at the spring 2013 American Chemical Society meeting in New Orleans to treat breast, lung, and colon cancers and leukemia.[1] The drug works as an pan-Notch inhibitor. The structure is one of a set patented in 2012,[2] and it currently being studied in clinical trials.[3][4]
useful for the treatment of conditions related to the Notch pathway, such as cancer and other proliferative diseases.
Notch signaling has been implicated in a variety of cellular processes, such as cell fate specification, differentiation, proliferation, apoptosis, and angiogenesis. (Bray, Nature Reviews Molecular Cell Biology, 7:678-689 (2006); Fortini, Developmental Cell 16:633-647 (2009)). The Notch proteins are single-pass heterodimeric transmembrane molecules. The Notch family includes 4 receptors, NOTCH 1-4, which become activated upon binding to ligands from the DSL family (Delta-like 1, 3, 4 and Jagged 1 and 2).
The activation and maturation of NOTCH requires a series of processing steps, including a proteolytic cleavage step mediated by gamma secretase, a multiprotein complex containing Presenilin 1 or Presenilin 2, nicastrin, APH1, and PEN2. Once NOTCH is cleaved, NOTCH intracellular domain (NICD) is released from the membrane. The released NICD translocates to the nucleus, where it functions as a transcriptional activator in concert with CSL family members (RBPSUH, “suppressor of hairless”, and LAG1). NOTCH target genes include HES family members, such as HES- 1. HES- 1 functions as transcriptional repressors of genes such as HERP 1 (also known as HEY2), HERP2 (also known as HEY1), and HATH1 (also known as ATOH1).
The aberrant activation of the Notch pathway contributes to tumorigenesis. Activation of Notch signaling has been implicated in the pathogenesis of various solid tumors including ovarian, pancreatic, as well as breast cancer and hematologic tumors such as leukemias, lymphomas, and multiple myeloma. The role of Notch inhibition and its utility in the treatment of various solid and hematological tumors are described in Miele, L. et al, Current Cancer Drug Targets, 6:313-323 (2006); Bolos, V. et al, Endocrine Reviews, 28:339-363 (2007); Shih, I.-M. et al, Cancer Research, 67: 1879- 1882 (2007); Yamaguchi, N. et al., Cancer Research, 68: 1881-1888 (2008); Miele, L., Expert Review Anti-cancer Therapy, 8: 1 197-1201 (2008); Purow, B., Current Pharmaceutical Biotechnology, 10: 154-160 (2009); Nefedova, Y. et al, Drug Resistance Updates, 1 1 :210-218 (2008); Dufraine, J. et al, Oncogene, 27:5132-5137 (2008); and Jun, H.T. et al, Drug Development Research, 69:319-328 (2008).
There remains a need for compounds that are useful as Notch inhibitors and that have sufficient metabolic stability to provide efficacious levels of drug exposure. Further, there remains a need for compounds useful as Notch inhibitors that can be orally or intravenously administered to a patient.
U.S. Patent No. 7,053,084 Bl discloses succinoylamino benzodiazepine compounds useful for treating neurological disorders such as Alzheimer’s Disease. The reference discloses that these succinoylamino benzodiazepine compounds inhibit gamma secretase activity and the processing of amyloid precursor protein linked to the formation of neurological deposits of amyloid protein. The reference does not disclose the use of these compounds in the treatment of proliferative diseases such as cancer.
Applicants have found potent compounds that have activity as Notch inhibitors and have sufficient metabolic stability to provide efficacious levels of drug exposure upon intravenous or oral administration. These compounds are provided to be useful as pharmaceuticals with desirable stability, bioavailability, therapeutic index, and toxicity values that are important to their drugability.
![]()
PAPER

Structure–activity relationships in a series of (2-oxo-1,4-benzodiazepin-3-yl)-succinamides identified highly potent inhibitors of γ-secretase mediated signaling of Notch1/2/3/4 receptors. On the basis of its robust in vivo efficacy at tolerated doses in Notch driven leukemia and solid tumor xenograft models, 12 (BMS-906024) was selected as a candidate for clinical evaluation.
Discovery of Clinical Candidate BMS-906024: A Potent Pan-Notch Inhibitor for the Treatment of Leukemia and Solid Tumors
†Bristol-Myers Squibb Research and Development, Princeton, New Jersey 08543, United States
‡Bristol-Myers Squibb Research and Development, 5 Research Parkway, Wallingford, Connecticut 06492, United States
§ Department of Chemistry, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037,United States
ACS Med. Chem. Lett., 2015, 6 (5), pp 523–527
DOI: 10.1021/acsmedchemlett.5b00001, http://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.5b00001
*Phone: 609-252-5091. E-mail: ashvinikumar.gavai@bms.com.
(2R,3S)-N-((3S)-1-methyl-2-oxo-5-phenyl-2,3-dihydro-1H-1,4- benzodiazepin-3-yl)-2,3-bis(3,3,3-trifluoropropyl)succinamide
colorless solid: HPLC: RT = 9.60 min (HPLC Method D). Chiral LC/Analytical SFC conditions: Column: LuxCellulose-2 (0.46 x 25cm), Mobile phase: 10% methanol in CO2, Flow rate: 3 mL/min, wavelength: 220 nm; Temp.: 35C. RT = 9.21 min, Purity = 99.95%.
MS (ES): m/z = 557 [M+H]+ ;
1H NMR (400 MHz, DMSO-d6) 9.54 (1H, d, J = 7.28 Hz), 7.71 – 7.80 (1H, m), 7.68 (2H, d, J = 8.78 Hz), 7.50 – 7.62 (3H, m), 7.45 (2H, t, J = 7.28 Hz), 7.29 – 7.40 (2H, m), 7.15 (1H, s), 5.30 (1H, d, J = 7.28 Hz), 3.39 (3H, s), 2.74 – 2.86 (1H, m), 2.02 -2.32 (3H, m), 1.45 – 1.79 (4H, m);
[]D = -107.0° (5.73 mg/mL, DMSO).
Elemental analysis: Theoretical: C: 54.11%; H: 4.70%; N: 10.06%; Actual: C: 54.06%; H: 4.90%; N: 10.08%.
Karl Fisher Moisture: 0.48.
HPLC Method D: Sunfire C18 3.5um, 3.0x150mm column, solvent A: 5% acetonitrile – 95% water – 0.05% TFA, solvent B: 95% acetonitrile – 5% water – 0.05% TFA, flow=0.5 mL/min, gradient from 10%B to 100%B over 15min, 254 nm detector.
Patent
http://www.google.co.in/patents/WO2012129353A1?cl=en
Example 1
(2R,35)-N-((35′)-l-Methyl-2-oxo-5-phenyl-2,3-dihydro-lH-l,4-benzodiazepin-3-yl)-2,3- b -trifluoropropy l)succinamide
Preparation 1A: tert-Butyl 5, -trifluoropentanoate

[00219] To a stirred solution of 5,5,5-trifluoropentanoic acid (5 g, 32.0 mmol) in THF (30 mL) and hexane (30 mL) at 0 °C, was added tert-butyl 2,2,2-trichloroacetimidate (11.46 mL, 64.1 mmol). The mixture was stirred for 15 min at 0 °C. Boron trifluoride etherate (0.406 mL, 3.20 mmol) was added and the reaction mixture was allowed to warm to room temperature overnight. To the clear reaction mixture was added solid aHC03 (5 g) and stirred for 30 min. The mixture was filtered through MgS04 and washed with hexanes (200 mL). The solution was allowed to rest for 45 min, and the resulting solid material was removed by filtering on the same MgS04 filter again, washed with hexanes (100 mL) and concentrated under reduced pressure without heat. The volume was reduced to about 30 mL, filtered through a clean fritted funnel, washed with hexane (5 mL), and then concentrated under reduced pressure without heat. The resulting neat oil was filtered through a 0.45μηι nylon membrane filter disk to provide tert-butyl 5,5,5- trifluoropentanoate (6.6 g, 31.4 mmol 98% yield) as a colorless oil: XH NMR (400 MHz, CDC13) δ ppm 1.38 (s, 9 H) 1.74-1.83 (m, 2 H) 2.00-2.13 (m, 2 H) 2.24 (t, J=7.28 Hz, 2 H).
Preparation IB: (45)-4-(Propan-2- l)-3-(5,5,5-trifluoropentanoyl)-l,3-oxazolidin-2-one

[00220] To a stirred solution of 5,5,5-trifluoropentanoic acid (5.04 g, 32.3 mmol) in DCM (50 mL) and DMF (3 drops) was added oxalyl chloride (3.4 mL, 38.8 mmol) dropwise over 5 min and the solution was stirred until all bubbling subsided. The reaction mixture was concentrated under reduced pressure to give pale yellow oil. To a separate flask charged with a solution of (45)-4-(propan-2-yl)-l,3-oxazolidin-2-one (4.18 g, 32.4 mmol) in THF (100 mL) at -78 °C was added n-BuLi (2.5M in hexane) (13.0 mL, 32.5 mmol) dropwise via syringe over 5 min. After stirring for 10 min, the above acid chloride dissolved in THF (20 mL) was added via cannula over 15 min. The reaction mixture was warmed to 0 °C, and was allowed to warm to room temperature as the bath warmed and stirred overnight. To the reaction mixture was added saturated NH4CI, and then extracted with EtOAc (2x). The combined organics were washed with brine, dried (Na2S04), filtered and concentrated under reduced pressure. The crude material was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 5% to 60% solvent A/B=hexanes/EtOAc, REDISEP® S1O2 120g). Concentration of appropriate fractions provided Preparation IB (7.39 g, 86%) as a colorless oil: XH NMR (400 MHz, CDC13) δ ppm 4.44 (1 H, dt, J=8.31, 3.53 Hz), 4.30 (1 H, t, J=8.69 Hz), 4.23 (1 H, dd, J=9.06, 3.02 Hz), 2.98-3.08 (2 H, m), 2.32-2.44 (1 H, m, J=13.91, 7.02, 7.02, 4.03 Hz), 2.13-2.25 (2 H, m), 1.88-2.00 (2 H, m), 0.93 (3 H, d, J=7.05 Hz), 0.88 (3 H, d, J=6.80 Hz). Preparation 1C: (25′,3R)-tert-Butyl 6,6,6-trifluoro-3-((5)-4-isopropyl-2-oxooxazolidine- 3 -carbonyl)-2-(3 ,3,3 -trifluoropropyl)hexanoate, and
Preparation ID: (2R,3R)-tert-Butyl 6,6,6-trifluoro-3-((5)-4-isopropyl-2-oxooxazolidine- 3 -carbonyl)- -(3 ,3 ,3 -trifluoropropyl)hexanoate

(1 C) (1 D)
[00221] To a cold (-78 °C), stirred solution of diisopropylamine (5.3 mL, 37.2 mmol) in THF (59 mL) under nitrogen atmosphere was added n-BuLi (2.5M in hexane) (14.7 mL, 36.8 mmol), then warmed to 0 °C to give a 0.5M solution of LDA. A separate vessel was charged with Preparation IB (2.45 g, 9.17 mmol), the material was azeotroped twice with benzene (the RotoVap air inlet was fitted with nitrogen inlet to completely exclude humidity) then toluene (15.3 mL) was added. This solution was added to a flask containing dry lithium chloride (1.96 g, 46.2 mmol). To the resultant mixture, cooled to -78 °C, was added LDA solution (21.0 mL, 10.5 mmol) and stirred at -78 °C for 10 min, warmed to 0 °C for 10 min then recooled to -78 °C. To a separate reaction vessel containing Preparation 1A (3.41 g, 16.07 mmol), also azeotroped twice with benzene, was added toluene (15.3 mL), cooled to -78 °C and LDA (37.0 mL, 18.5 mmol) was added, the resulting solution was stirred at -78° for 25 min. At this time the enolate derived from the ester was transferred via cannula into the solution of the oxazolidinone enolate, stirred at -78 °C for an additional 5 min at which time the septum was removed and solid powdered bis(2-ethylhexanoyloxy)copper (9.02 g, 25.8 mmol) was rapidly added to the reaction vessel and the septum replaced. The vessel was immediately removed from the cold bath and immersed into a warm water bath (40 °C) with rapid swirling with a concomitant color change from the initial turquoise to brown. The reaction mixture was stirred for 20 min, was poured into 5% aqueous NH4OH (360 mL) and extracted with EtOAc (2x). The combined organics were washed with brine, dried (Na2S04), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 0% to 60% solvent A/B=hexanes/EtOAc, REDISEP® S1O2 120g). Concentration of appropriate fractions provided Preparation 1C (2.87 g, 66%) as pale yellow viscous oil. XH NMR showed the product was a 1.6: 1 mixture of diastereoisomers 1C: 1D as determined by the integration of the multiplets at 2.74 & 2.84 ppm: XH NMR (400 MHz, CDC13) δ ppm 4.43-4.54 (2 H, m), 4.23-4.35 (5 H, m), 4.01 (1 H, ddd, J=9.54, 6.27, 3.51 Hz), 2.84 (1 H, ddd, J=9.41, 7.28, 3.64 Hz), 2.74 (1 H, ddd, J=10.29, 6.27, 4.02 Hz), 2.37-2.48 (2 H, m, J=10.38, 6.98, 6.98, 3.51, 3.51 Hz), 2.20-2.37 (3 H, m), 1.92-2.20 (8 H, m), 1.64-1.91 (5 H, m), 1.47 (18 H, s), 0.88-0.98 (12 H, m). Preparation IE: (2R,35)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and
Preparation IF: (2R,3R)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid

(1 E) (1 F)
[00222] To a cool (0 °C), stirred solution of Preparation 1C and ID (4.54 g, 9.51 mmol) in THF (140 mL) and water (42 mL) was sequentially added hydrogen peroxide (30% in water) (10.3 g, 91 mmol) and LiOH (685.3 mg, 28.6 mmol) and the mixture was stirred for 1 hr. At this time the reaction vessel was removed from the cold bath and then stirred for 1.5 hr. The reaction was judged complete by HPLC. To the reaction mixture was added saturated NaHC03 (45 mL) and saturated a2S03(15 mL), and then partially concentrated under reduced pressure. The resulting crude solution was extracted with DCM (3x). The aqueous phase was acidified to pH~l-2 with IN HC1, extracted with DCM (3x) and EtOAc (lx). The combined organics were washed with brine, dried (Na2S04), filtered and concentrated under reduced pressure to provide a mixture of Preparation IE and IF (3.00 g, 86%) as colorless oil: XH NMR (400 MHz, CDC13) δ ppm 2.76-2.84 (1 H, m, diastereoisomer 2), 2.64-2.76 (3 H, m), 2.04-2.35 (8 H, m), 1.88-2.00 (4 H, m), 1.71-1.83 (4 H, m), 1.48 (9 H, s, diastereoisomer 1), 1.46 (9 H, s, diastereoisomer 2); XH NMR showed a 1.7: 1 mixture of 1E: 1F by integration of the peaks for the ?-butyl groups.
Preparation IE: (2R,35)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and
Preparation IF: (2R,3R)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid

(1 E) (1 F)
[00223] To a cold (-78 °C), stirred solution of diisopropylamine (1.7 mL, 11.93 mmol) in THF (19 mL) under nitrogen atmosphere was added n-BuLi (2.5M in hexanes) (4.8 mL, 12.00 mmol). The mixture was stirred for 5 min and then warmed to 0 °C. In a separate vessel, to a cold (-78 °C) stirred solution of the mixture of Preparation IE and IF (1.99 g, 5.43 mmol) in THF (18 mL) was added the LDA solution prepared above via cannula slowly over 25 min. The mixture was stirred for 15 min, then warmed to room temperature (placed in a 24 °C water bath) for 15 min, and then again cooled to -78 °C for 15 min. To the reaction mixture was added Et2AlCl (1M in hexane) (11.4 mL, 1 1.40 mmol) via syringe, stirred for 10 min, warmed to room temperature for 15 min and then cooled back to -78 °C for 15 min. Methanol (25 mL) was rapidly added, swirled vigorously while warming to room temperature, then concentrated to ~l/4 original volume. The mixture was dissolved in EtOAc and washed with IN HCl (50 mL) and ice (75 g). The aqueous phase was separated, extracted with EtOAc (2x). The combined organics were washed with a mixture of KF (2.85g in 75 mL water) and IN HCl (13 mL) [resulting solution pH 3-4], then with brine, dried (Na2S04), filtered and concentrated under reduced pressure to give a 9: 1 (IE: IF) enriched diastereoisomeric mixture (as determined by XH NMR) of Preparation IE and Preparation IF (2.13 g, >99%) as a pale yellow viscous oil: XH NMR (400 MHz, CDC13) δ ppm 2.64-2.76 (2 H, m), 2.04-2.35 (4 H, m), 1.88-2.00 (2 H, m), 1.71-1.83 (2 H, m), 1.48 (9 H, s). Preparation 1 G: (35)-3 -Amino- 1 -methyl-5-phenyl- 1 ,3 -dihydro-2H- 1 ,4-benzodiazepin-2- one, and
Preparation 1H: (3R)-3 -Amino- 1 -methyl-5-phenyl- 1 ,3-dihydro-2H- 1 ,4-benzodiazepin-2- one

(1G) (1 H)
[00224] Racemic 3-amino-l-methyl-5-phenyl-l,3-dihydro-2H-l,4-benzodiazepin-2- one (Rittle, K.E. et al, Tetrahedron Letters, 28(5):521-522 (1987)) was prepared according to the literature procedure. The enantiomers were separated under chiral-SFC conditions using the following method: CHIRALPAK® AS-H 5×25; Mobile phase: 30% MeOH+ 0.1% DEA in C02; Flow rate: 280 mL/min; Pressure: 100 bar; Temperature: 35 °C.
[00225] Obtained the S-enantiomer (Preparation 1G): HPLC: RT=1.75 min (30% MeOH + 0.1% DEA in C02 on CHIRALPAK® AS-H 4.6×250 mm, 3 mL/min, 35 °C, 100 bar, 230 nm, ΙΟμΙ injection); ¾ NMR (400 MHz, CDC13) δ ppm 7.58-7.63 (2 H, m), 7.55 (1 H, ddd, J=8.50, 7.1 1, 1.76 Hz), 7.40-7.47 (1 H, m), 7.34-7.40 (3 H, m), 7.31 (1 H, dd, J=7.81, 1.51 Hz), 7.14-7.22 (1 H, m), 4.46 (1 H, s), 3.44 (3 H, s), 3.42 (2 H, s); [a]D= -155° (c=1.9, MeOH) (Lit. Rittle, K.E. et al, Tetrahedron Letters, 28(5):521-522 (1987): [a]D=-236°).
[00226] Also obtained the R-enantiomer (Preparation 1H): HPLC: RT=1.71 min; [a]D=+165° (c=2.1, MeOH) (Lit [a]D= +227°).
Alternate procedure to make Preparation 1 G:
Preparation 1G»CSA salt: (35)-3-Amino-l-methyl-5-phenyl-l,3-dihydro-2H-l,4- benzodiazepin-2-one, (15)-(+)-10-camphorsulfonic acid salt

[00227] Preparation lG’CSA was prepared from racemic 3-amino-l-methyl-5-phenyl- l,3-dihydro-2H-l,4-benzodiazepin-2-one (9.98g, 37.6 mmol) (prepared according to the literature as shown above) according to the literature procedure (Reider, P.J. et al, J. Org. Chem., 52:955-957 (1987)). Preparation lG’CSA (16.91g, 99%) was obtained as a colorless solid: Optical Rotation: [a]D = -26.99° (c=l, H20) (Lit. [a]D = -27.8° (c=l,
H20))
Preparation II: tert-Butyl (25,,3R)-6,6,6-trifluoro-3-(((35)-l-methyl-2-oxo-5-phenyl-2,3- dihydro- 1 H- 1 ,4-benzodiazepin-3 -yl)carbamoyl)-2-(3 ,3 ,3 -trifluoropropyl)hexanoate, and Preparation 1J: tert-Butyl (2R,3R)-6,6,6-trifluoro-3-(((35)-l-methyl-2-oxo-5-phenyl-2,3- dihydro- 1 H- 1 ,4-benzodiazepin-3 -yl)carbamoyl)-2-(3 ,3 ,3-trifluoropropyl)hexanoate

(11) (U)
[00228] To a stirred solution of Preparation 1G (1.45 g, 5.47 mmol) and a 9: 1 mixture of Preparation IE and IF (1.989 g, 5.43 mmol) in DMF (19 mL) was added O- benzotriazol-l-yl-N,N,N’,N’-tetra-methyluronium tetrafluoroborate (1.79 g, 5.57 mmol) and triethylamine (3.0 mL, 21.52 mmol) and stirred overnight. The reaction was judged complete by LCMS. The reaction mixture was poured into water (125 mL) and the precipitated solid was collected by filtration, washed with water and air dried to provide an 8: 1 mixture of Preparation II and Preparation 1J (2.95 g, 89%) as a cream solid: MS (ES): m/z= 614 [M+H]+;XH NMR (400 MHz, CDC13) δ ppm 7.55-7.65 (3 H, m), 7.44- 7.52 (2 H, m), 7.35-7.45 (4 H, m), 5.52 (1 H, d, J=8.03 Hz), 3.48 (3 H, s), 2.63 (2 H, ddd, J=9.35, 3.95, 3.76 Hz), 2.14-2.25 (4 H, m), 1.90-2.03 (3 H, m), 1.69-1.82 (1 H, m), 1.51 (9 H, s).
Preparation IK: (25,,3R)-6,6,6-Trifluoro-3-(((35)-l-methyl-2-oxo-5-phenyl-2,3-dihydro- lH-l,4-benzodiazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoic acid, and
Preparation 1L: (2R,3R)-6,6,6-Trifluoro-3-(((35)-l-methyl-2-oxo-5-phenyl-2,3-dihydro- 1 H- 1 ,4-

(1 K) (1 L)
[00229] To a cool (0 °C), stirred solution of the above mixture of Preparation II and Preparation 1 J (2.95 g, 4.81 mmol) in DCM (20 mL) was added TFA (20 mL, 260 mmol). The reaction mixture was stirred for lhr, then allowed to warm to room temperature and stirred for 2.5 hr. The reaction was judged complete by LCMS. The reaction mixture was diluted with toluene (50 mL) and concentrated under reduced pressure. The residue mixture was redissolved in toluene (50 mL) and concentrated under reduced pressure then dried under high vacuum. The crude product was dissolved in DCM, S1O2 (15g) was added, concentrated, then was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 0% to 45% solvent A/B=DCM/EtOAc, REDISEP® S1O2 80g). Concentration of appropriate fractions provided a mixture of Preparation IK and Preparation 1L (2.00 g, 75%) as a cream solid: HPLC: RT=2.770 min
(CHROMOLITH® SpeedROD 4.6 x 50 mm (4 min grad) eluting with 10-90% aqueous MeOH over 4 minutes containing 0.1% TFA, 4 mL/min, monitoring at 254 nm); MS (ES): m/z= 558 [M+H]+; XH NMR (400 MHz, CDC13) δ ppm 8.32 (1 H, d, J=8.03 Hz), 7.65-7.71 (1 H, m), 7.50-7.60 (3 H, m), 7.41-7.49 (2 H, m), 7.39 (1 H, dd, J=7.91, 1.63 Hz), 7.23-7.35 (2 H, m), 5.59 (1 H, d, J=8.03 Hz), 3.51 (3 H, s), 2.81 (1 H, ddd, J=10.54, 6.90, 3.64 Hz), 2.67-2.76 (1 H, m), 2.22-2.33 (3 H, m), 1.99-2.12 (3 H, m), 1.85-1.94 (1 H, m), 1.79 (1 H, ddd, J=13.87, 7.84, 3.64 Hz). Example 1 :
[00230] To a stirred solution of an 8: 1 mixture of Preparation IK and Preparation 1L (3.46 g, 6.21 mmol) in DMF (25 mL) under nitrogen atmosphere was added ammonium chloride (3.32 g, 62.1 mmol), EDC (3.55 g, 18.52 mmol), HOBT (2.85 g, 18.61 mmol), and triethyl amine (16 mL, 1 15 mmol) and stirred overnight. The reaction was judged complete by LCMS. The reaction mixture was poured into water (200 mL) with vigorous swirling and then allowed to sit. The solid was collected by filtration, washed with water, allowed to dry to afford 3.6 g colorless solid. The solid was purified by preparative SFC chromatography (Lux-Cellulose-2 (3x25cm), 8% methanol in CO2, 140ml/min @220nm and 35 °C; Sample: 3.6g in 50cc methanol, conc.=70mg/ml, Stack injection:
0.5cc/9.2min). Fractions containing product were concentrated, dried overnight under vacuum. Obtained Example 1 (2.74 g, 79%) as a colorless solid (Crystal Form -1): HPLC: RT=9.601 min (H20/CH3CN with TFA, Sunfire CI 8 3.5um, 4.6x150mm, 4.6x150mm, gradient = 15 min, wavelength = 220 and 254 nm). MS (ES): m/z= 557 [M+H]+; XH NMR (400 MHz, DMSO-d6) δ ppm 9.54 (1 H, d, J=7.28 Hz), 7.71-7.80 (1 H, m), 7.68 (2 H, d, J=8.78 Hz), 7.50-7.62 (3 H, m), 7.45 (2 H, t, J=7.28 Hz), 7.29-7.40 (2 H, m), 7.15 (1 H, br. s.), 5.30 (1 H, d, J=7.28 Hz), 3.39 (3 H, s), 2.74-2.86 (1 H, m), 2.02-2.32 (3 H, m), 1.45-1.79 (4 H, m); [a]D = -107.0° (5.73 mg/mL, DMSO).
[00231] Crystal Form A-2 was prepared by adding approximately 1 mg of Example 1 to approximately 0.7 mL of acetone/acetonitrile/water solution (2:2: 1). A mixture of colorless needles and thin blades crystals were obtained after one day of slow evaporation of the solution at room temperature. The thin blade crystals were separated to provide crystal Form A-2.
[00232] Crystal Form EA-3 was prepared by adding approximately 1 mg of Example 1 to approximately 0.7 mL of ethyl acetate/heptane solution (1 : 1). Colorless blade crystals were obtained after three days of slow evaporation of the solution at room temperature.
[00233] Crystal Form THF-2 was obtained by adding approximately 5 mg of Example 1 to approximately 0.7 mL of THF/water solution (4: 1). Colorless blade-like crystals were obtained after one day of solvent evaporation at room temperature.
Alternate Procedure to Make Example 1 : Preparation 1M: 3,3,3-Trifluoropropyl trifluoromethanesulfonate

[00234] To a cold (-25 °C), stirred solution of 2,6-lutidine (18.38 mL, 158 mmol) in CH2CI2 (120 mL) was added Tf20 (24.88 mL, 147 mmol) over 3 min, and stirred for 5 min. To the reaction mixture was added 3,3,3-trifluoropropan-l-ol (12 g, 105 mmol) over an interval of 3 min. After 2 hr, the reaction mixture was warmed to room temperature and stirred for 1 hr. The reaction mixture was concentrated to half volume, then purified by loading directly on silica gel column (330g ISCO) and eluted with CH2C12. Obtained Preparation 1M (13.74 g, 53%) as a colorless oil. XH NMR (400 MHz, CDCI3) δ ppm 4.71 (2 H, t, J=6.15 Hz), 2.49-2.86 (2 H, m).
Preparation IN: (45)-4-Benzyl- -(5,5,5-trifluoropentanoyl)-l,3-oxazolidin-2-one

[00235] Preparation IN was prepared from 5,5,5-trifluoropentanoic acid (3.35 g, 21.46 mmol) and (45)-4-benzyl-l,3-oxazolidin-2-one (3.80 g, 21.46 mmol) by the general methods shown for Preparation IB. Preparation IN (5.67 g, 84%) was obtained as a colorless viscous oil: XH NMR (400 MHz, CDC13) δ ppm 7.32-7.39 (2 H, m), 7.30 (1 H, d, J=7.05 Hz), 7.18-7.25 (2 H, m), 4.64-4.74 (1 H, m), 4.17-4.27 (2 H, m), 3.31 (1 H, dd, J=13.35, 3.27 Hz), 3.00-3.1 1 (2 H, m), 2.79 (1 H, dd, J=13.35, 9.57 Hz), 2.16-2.28 (2 H, m), 1.93-2.04 (2 H, m).
Preparation 10: tert-Butyl (3R)-3-(((45)-4-benzyl-2-oxo-l,3-oxazolidin-3-yl)carbonyl)- 6,6,6-trifluorohexanoate

[00236] To a cold (-78 °C), stirred solution of Preparation IN (3.03 g, 9.61 mmol) in THF (20 mL) was added NaHMDS (1.0M in THF) (10.6 mL, 10.60 mmol) under nitrogen atmosphere. After 2 hours, tert-butyl 2-bromoacetate (5.62 g, 28.8 mmol) was added neat via syringe at -78 °C and stirring was maintained at the same temperature. After 6 hours, the reaction mixture was warmed to room temperature. The reaction mixture was partitioned between saturated NH4C1 and EtOAc. The organic phase was separated, and the aqueous was extracted with EtOAc (3x). The combined organics were washed with brine, dried (Na2S04), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 5% to 100% solvent A/B=hexanes/EtO Ac, REDISEP® Si02 120g). Concentration of appropriate fractions provided Preparation 10 (2.79 g, 67.6%) as a colorless viscous oil: XH NMR (400 MHz, CDC13) δ ppm 7.34 (2 H, d, J=7.30 Hz), 7.24-7.32 (3 H, m), 4.62- 4.75 (1 H, m, J=10.17, 6.89, 3.43, 3.43 Hz), 4.15-4.25 (3 H, m), 3.35 (1 H, dd, J=13.60, 3.27 Hz), 2.84 (1 H, dd, J=16.62, 9.57 Hz), 2.75 (1 H, dd, J=13.35, 10.07 Hz), 2.47 (1 H, dd, J=16.62, 4.78 Hz), 2.1 1-2.23 (2 H, m), 1.90-2.02 (1 H, m), 1.72-1.84 (1 H, m), 1.44 (9 H, s). -2-(2-tert-Butoxy-2-oxoethyl)-5,5,5-trifluoropentanoic acid

[00237] Preparation IP was prepared from Preparation 10 (2.79 g, 6.50 mmol) by the general methods shown for Preparation IE. Preparation IP (1.45 g, 83%) was obtained as a colorless oil: XH NMR (400 MHz, CDC13) δ ppm 2.83-2.95 (1 H, m), 2.62-2.74 (1 H, m), 2.45 (1 H, dd, J=16.62, 5.79 Hz), 2.15-2.27 (2 H, m), 1.88-2.00 (1 H, m), 1.75-1.88 (1 H, m), 1.45 (9 H, s). Preparation IE: (2R,35′)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and
Preparation IF: (2R,3R)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid

(1 E) (1 F)
[00238] To a cold (-78 °C), stirred solution of Preparation IP (5.44 g, 20.13 mmol) in THF (60 mL) was slowly added LDA (24.60 mL, 44.3 mmol) over 7 min. After stirring for 2 hr, Preparation 1M (6.44 g, 26.2 mmol) was added to the reaction mixture over 3 min. After 45 min, the reaction mixture was warmed to -25 °C bath (ice/MeOH/dry ice) for 1 hr, and then warmed to 0 °C. After 45 min, Preparation 1M (lg) was added and the reaction mixture was stirred for 20 min. The reaction was quenched with water and IN NaOH and was extracted with (¾(¾. The organic layer was again extracted with IN NaOH (2x) and the aqueous layers were combined. The aqueous layer was cooled in ice/water bath and then acidified with concentrated HCl to pH 2. Next, the aqueous layer was extracted with EtOAc. The combined organics were washed with brine, dried over anhydrous sodium sulphate, and concentrated under reduced pressure. The residue was dried under high vacuum to provide a 1 :5 (IE: IF) mixture (as determined by XH NMR) of Preparation IE and Preparation IF (5.925 g, 80%) as a pale yellow solid. XH NMR (500 MHz, CDC13) 8 ppm 2.81 (1 H, ddd, J=10.17, 6.32, 3.85 Hz), 2.63-2.76 (1 H, m), 2.02- 2.33 (4 H, m), 1.86-1.99 (2 H, m), 1.68-1.85 (2 H, m), 1.47 (9 H, s).
Preparation IE: (2R,35)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and
Preparation IF: (2R,3R)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid

(1 E) (1 F)
[00239] A mixture of Preparation IE and Preparation IF (64 mg, 1.758 mmol) was taken in THF (6 mL) to give a colorless solution which was cooled to -78 °C. Then, LDA (2.149 mL, 3.87 mmol) (1.8M in heptane/THF/ethylbenzene) was slowly added to the reaction mixture over 10 min. After stirring for 15 min the reaction mixture was placed in a room temperature water bath. After 15 min the reaction mixture was placed back in -78 °C bath and then diethylaluminum chloride (3.87 mL, 3.87 mmol) (1M in hexane) was added slowly over 5 min. The reaction mixture was stirred at -78 °C. After 15 min the reaction mixture was placed in a room temperature water bath for 10 min and then cooled back to -78 °C bath. After 15 min the reaction was quenched with MeOH (8 mL, 198 mmol), removed from the -78 °C bath and concentrated. To the reaction mixture was added ice and HC1 (16 mL, 16.00 mmol), followed by extraction with EtOAc (2x). The organic layer was washed with potassium fluoride (920 mg, 15.84 mmol) (in 25 mL FLO) and HC1 (4.5 mL, 4.50 mmol). The organics were dried over anhydrous magnesium sulphate and concentrated under reduced pressure to provide a 9: 1 (IE: IF) enriched mixture of Preparation IE and Preparation IF (540 mg, 1.583 mmol, 90% yield) as light yellow/orange solid. ¾ NMR (400 MHz, CDC13) δ ppm 2.64-2.76 (2 H, m), 2.04-2.35 (4 H, m), 1.88-2.00 (2 H, m), 1.71-1.83 (2 H, m), 1.48 (9 H, s). It was converted to Example 1 by the sequence of reactions as outlined above.
Alternate procedure to make Preparation IE:
Preparation 1Q: (2R,35)- -Benzyl 4-tert-butyl 2,3-bis(3,3,3-trifluoropropyl)succinate

(1Q) [00240] A clean and dry 5 L four neck round bottom flask equipped with mechanical stirring, thermometer socket and nitrogen bubbler at room temperature was charged with Ν,Ν-dimethyl formamide (2.07 L), a 1.2: 1 mixture of Preparation IE and Preparation IF (207 g, 0.5651 moles), potassium carbonate (1 17.1 g, 0.8476 moles) followed by benzyl bromide (116 g, 0.6781 moles) over 15-20 min. The reaction mixture was stirred for 2-3 hr. After completion of the reaction, the reaction mixture was concentrated to dryness at 50-55 °C under vacuum. Ethyl acetate (3.1 L, 30 Vol.) was charged into the concentrated reaction mass and then washed with water (2.07 L), brine (0.6 L) then dried over anhydrous sodium sulfate (207 g), filtered and concentrated to dryness at 40-45 °C under vacuum. The residue was dissolved in dichloromethane (1.035 L, 5 vol.) and then absorbed onto silica gel (60-120) (607 g, 3.0 w/w), then was purified with column chromatography using petroleum ether and ethyl acetate as solvents. After pooling several batches, Preparation 1Q (235 g) was obtained. HPLC purity: 99.77%, Preparation IE: (2R,35)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid

[00241] A clean and dry 2 L autoclave was charged with methanol (540 mL) and was purged with nitrogen for 5-10 minutes. To the autoclave was added 10% palladium on carbon (12 g, 20%), purged with nitrogen once again for 5-10 min then was charged with Preparation 1Q (60g, 0.1315 moles), the autoclave was flushed with methanol (60mL) and stirred for 4-6 hr at 20-25 °C under 5Kg hydrogen pressure. After completion of the reaction, the reaction mass was filtered through CELITE®, washed with methanol (180 mL), dried with anhydrous sodium sulfate (60 g), filtered and concentrated to dryness at 45-50 °C under vacuum. Obtained Preparation IE (45.8 g, 95%) as a colorless solid: HPLC purity: 98.9%.
Alternate procedure to make Preparation IE: Preparation IE: (2R,35)-3-(te^Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid

[00242] Preparation IE was prepared in a procedure identical as above from a mixture of Preparations IE and IF (200g, 0.5460 moles) using LDA (1.8 M solution in THF, ethyl benzene and heptane) (698mL, 2.3equiv.) and diethyl aluminum chloride (1.0 M solution in hexane) (1256mL, 2.3equiv) in THF (2.0L). After workup as explained above, the resulting residue was treated as follows: The crude material was added to a 2L four neck round bottom flask, followed by the addition of MTBE (1.0L) charged below 30 °C. The resulting mixture was stirred for 5-10 minutes to obtain a clear solution.
Hexanes (600mL) was charged to the reaction mixture at a temperature below 30 °C. The reaction mixture was stirred for 10 min. Next, tert-butylamine (43.8g, l. leq) was charged slowly over a period of 15 minutes below 30 °C. This addition was observed to be exothermic. The reaction mixture was stirred for 2 hrs below 30 °C and filtered. The solid material was washed with 5:3 MTBE: hexane (200mL), the filtrate was
concentrated and transferred to an amber color bottle. The filtered solid was dissolved in dichloromethane (2.0L), washed with IN HC1 (2.0), the organic layer was washed with brine (1.0L x 2), then was concentrated under reduced pressure below 45 °C. This material was found to be 91.12% pure. The material was repurified by the above t- butylamine crystallization purification procedure. Obtained Preparation IE (78 g, 39%): HPLC purity: 99.54%.
Alternate procedure to make Example 1 :
Preparation II: tert-Butyl (25,,3R)-6,6,6-trifluoro-3-(((35)-l-methyl-2-oxo-5-phenyl-2,3- dihydro- 1 H- 1 ,4-benzodiazepin-3 -yl)carbamoyl)-2-(3 ,3 ,3 -trifluoropropyl)hexanoate

[00243] A clean and dry 2 L four neck round bottom flask equipped with mechanical stirring, thermometer socket and nitrogen bubbler was charged with N,N- dimethylformamide (457 mL), Preparation IE (45.7g, 0.1248moles) and Preparation lG’CSA (62.08g, 0.1248moles) under nitrogen atmosphere at 20-25 °C. The reaction mixture was stirred for 15-20 minutes to make clear solution at 20-25 °C. To the reaction mixture was added TBTU (48.16g, 0.1498 moles) at 20-25 °C followed by triethylamine (50.51g, 0.4992 moles) over 15-20 minutes at 20-25 °C. The reaction mixture was stirred for 60-120 minutes at 20-25 °C under nitrogen atmosphere. After completion of the reaction, the reaction was quenched into water (1.37L, 30 Vol.) at 20-25 °C under stirring. The reaction mixture was stirred for 30 minutes at 20-25 °C. The reaction mixture was filtered and washed with water (228 mL). The resulting solid material was dissolved in ethyl acetate (457 mL), washed with water (2×137 mL), brine (137 mL), and then dried with anhydrous sodium sulfate (45.7g). Activated charcoal (9.14 g, 20%) was charged into the reaction mixture and stirred for 30 minutes. The mixture was filtered through CELITE® bed and 1 micron filter cloth, washed charcoal bed with ethyl acetate (137 mL), concentrated to 1.0 Vol. stage and then petroleum ether (457 mL, 10 Vol.) was charged and stirred for 30 minutes at 20-25 °C. The solid was collected by filtration, washed with petroleum ether (137 mL) and then dried under vacuum at 40-45 °C for 8 hr until loss on drying was less than 3.0%. Obtained Preparation II (65.2 g, 85%): HPLC purity: 98.26%.
Preparation IK: (25,,3R)-6,6,6-Trifluoro-3-(((35)-l-methyl-2-oxo-5-phenyl-2,3-dihydro- 1 H- 1 ,4-benzodiazepin-3 -yl)carbamoyl)-2-(3 ,3 ,3 -trifluoropropyl)hexanoic acid

[00244] A clean and dry 3 L four neck round bottom flask equipped with mechanical stirring, thermometer socket and nitrogen bubbler was charged with dichloromethane (980 mL) under nitrogen atmosphere followed by Preparation II (140 g, 0.2282 moles) at 20-25 °C. The reaction mixture was cooled to 0-5 °C and trifluoroacetic acid (980 mL) was charged slowly for 30-40 minutes. The resulting mixture was stirred for 2 hr at 0-5 °C under nitrogen atmosphere. The reaction temperature was raised to 20 to 25 °C, and the reaction mixture was stirred for 1-2 hr at 20 to 25 °C. After completion of the reaction, the reaction mixture was concentrated to dryness at 50 to 55 °C under vacuum. Toluene (3×700 mL,) was charged into the concentrated reaction mass, and then distilled off at 50 to 55 °C under vacuum. After complete concentration from toluene, ethyl acetate (280 mL) was charged into the reaction mass at 20 to 25 °C, stirred for 60 minutes, then the solid was collected by filtration, washed with ethyl acetate (140 mL), dried under vacuum at 50 to 55 °C for 12 hr until loss on drying was less than 2.0%. Obtained Preparation IK (106 g, 84%): HPLC purity: 98.43%.
Example 1 :
[00245] A reaction vessel was charged with Preparation IK (30 g, 53.81 mmol), HOBt (8.7g, 64.38 mmol), and THF (150 mL) at room temperature. To the homogeneous solution was added EDCI (12.4g, 64.68 mmol), stirred for 15 min, then cooled to 8 °C. To the reaction mixture was added ammonia (2M in IP A) (81 mL, 162 mmol) over 5 min so as to maintain a temperature below 10 °C. The resulting heavy slurry was stirred for 10 min, warmed to room temperature over 30 min, then stirred for 4 hr. At the completion of the reaction, water (230 mL) was slowly added over 15 min to maintain a temperature below 20 °C, and then stirred for 2 hr. The solid was collected by filtration, washed with water (3X60 mL), then dried under vacuum 48 hr at 55 °C. The above crude product was charged into a 1 L 3 -necked round flask. IP A (200 mL) was added, then heated to 80 °C resulting in a homogeneous solution. Water (170 mL) was slowly added (15 min) to maintain an internal temperature >75 °C. The resulting slurry was stirred and cooled to room temperature for 2 hr. The solid was collected by filtration, washed with water (2 X 50 mL), then dried under vacuum (55 °C for 24 h, and 30 °C for 48 h).
Obtained Example 1 (23.4 g, 78% yield): HPLC purity: 99.43%.
Example 2 NOT SAME
WITHOUT METHYL GROUP
(2R,35)-N-((35)-2-Oxo-5-phenyl-2,3-dihydro-lH-l,4-benzodiazepin-3-yl)-2,3-bis(3,3,3- trifluoropropyl)succinamide

Preparation 2A: (35)-3-Amino-5-phenyl-l,3-dihydro-2H-l,4-benzodiazepin-2-one, and Preparation 2B: -3-Amino-5-phenyl-l,3-dihydro-2H-l,4-benzodiazepin-2-one

(2A) (2B)
[00246] Racemic 3-amino-5-phenyl-l,3-dihydro-2H-l,4-benzodiazepin-2-one (J. Med. Chem., 49:231 1-2319 (2006), compound# 5) was prepared according to the literature procedure. The enantiomers were separated on Berger SFC MGIII Column: Lux 25X3 cm, 5cm; Mobile phase: 30% MeOH+ 0.1% DEA in C02; Flow rate: 150 mL/min;
Temperature: 40 °C; Detector wavelength: 250 nM. Obtained the S-enantiomer
Preparation 2A as a white solid: XH NMR (400 MHz, DMSO-d6) δ ppm 10.67 (1 H, br. s.), 7.58 (1 H, td, J=7.65, 1.76 Hz), 7.37-7.53 (5 H, m), 7.23-7.30 (2 H, m), 7.14-7.22 (1 H, m), 4.23 (1 H, s), 2.60 (2 H, br. s.); HPLC: RT=3.0625 min (30% MeOH + 0.1% DEA in C02 on OD-H Column, 3 mL/min, 35 °C, 96 bar, 230 nm, ΙΟμΙ inj); [a]D = -208.3° (5.05 mg/niL, MeOH). Also obtained the R-enantiomer Preparation 2B as an off white solid: HPLC: RT=3.970 min; [a]D = 182.1° (2.01 mg/mL, MeOH).
Preparation 2C: tert-Butyl (25,,3R)-6,6,6-trifluoro-3-(((35)-2-oxo-5-phenyl-2,3-dihydro- 1 H- 1 ,4-benzodiazepin-3 -yl)carbamoyl)-2-(3 ,3 ,3 -trifluoropropyl)hexanoate, and
Preparation 2D: tert-Butyl (2R,3R)-6,6,6-trifluoro-3-(((35)-2-oxo-5-phenyl-2,3-dihydro- 1 H- -benzodiazepin-3 -yl)carbamoyl)-2-(3 ,3 ,3 -trifluoropropyl)hexanoate

(2C) (2D)
[00247] Preparation 2C was prepared from Preparation 2A (564 mg, 2.244 mmol) and a mixture of Preparation IE and Preparation IF (822 mg, 2.244 mmol) according to the general procedure shown for Preparation II. Obtained Preparation 2C and Preparation 2D (1.31 g, 97%): HPLC: RT=3.443 min (CHROMOLITH® ODS 4.6 x 50 mm (4 min grad) eluting with 10-90% aqueous MeOH over 4 minutes containing 0.% TFA, 4 mL/min, monitoring at 220 nm); MS (ES): m/z= 600.3 [M+H]+.
Preparation 2E: (25′,3R)-6,6,6-Trifluoro-3-(((35)-2-oxo-5-phenyl-2,3-dihydro-lH-l,4- benzodiazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoic acid, and
Preparation 2F: (2R,3R)-6,6,6-Trifluoro-3-(((35)-2-oxo-5-phenyl-2,3-dihydro-lH-l,4- benzodiazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoic acid

(2E) (2F) [00248] A mixture of Preparation 2E and Preparation 2F was prepared from a mixture of Preparation 2C and Preparation 2D (1.3 lg, 2.185 mmol) by the general methods shown for Preparation IK. Obtained a mixture of Preparation 2E and Preparation 2F (1.18 g, 99%): HPLC: RT=2.885 min (CHROMOLITH® ODS 4.6 x 50 mm (4 min grad) eluting with 10-90% aqueous MeOH over 4 minutes containing 0.% TFA, 4 mL/min, monitoring at 220 nm). MS (ES): m/z= 544.2 [M+H]+.
Example 2:
[00249] Example 2 was prepared from a mixture of Preparation 2E and Preparation 2F (354 mg, 0.651 mmol) by the general methods shown for Example 1. After separation of the diastereoisomers, Example 2 was obtained (188 mg, 52%) as a white solid: HPLC: RT=9.063 min (H20/CH3CN with TFA, Sunfire C18 3.5um, 4.6x150mm, 4.6x150mm, gradient = 15 min, wavelength = 220 and 254 nm); MS (ES): m/z= 543 [M+H]+; XH NMR (400 MHz, DMSO-d6) δ ppm 10.87 (1 H, br. s.), 9.50-9.55 (1 H, m), 7.62-7.69 (2 H, m), 7.40-7.57 (5 H, m), 7.29-7.36 (2 H, m), 7.22-7.28 (1 H, m), 7.16 (1 H, br. s.), 5.25 (1 H, d), 3.30-3.32 (1 H, m), 2.75-2.86 (1 H, m), 2.44-2.48 (1 H, m), 2.06-2.34 (3 H, m), 1.51- 1.77 (4 H, m); [a]D = -114.4° (8.04 mg/mL, DMSO).
[00250] Crystal Form M2- 1 was prepared by adding approximately 1 mg of Example 2 to approximately 0.7 mL of MeOH/fluorobenzene solution (3 : 1). Colorless plate-like crystals were obtained after 2 days of solvent evaporation at room temperature.
PATENT

Example 1
(2R,3S)—N-((3S)-1-Methyl-2-oxo-5-phenyl-2,3-dihydro-1H-1,4-benzodiazepin-3-yl)-2,3-bis(3,3,3-trifluoropropyl)succinamide

Preparation 1A: tert-Butyl 5,5,5-trifluoropentanoate

Preparation 1B: (4S)-4-(Propan-2-yl)-3-(5,5,5-trifluoropentanoyl)-1,3-oxazolidin-2-one

Preparation 1C: (2S,3R)-tert-Butyl 6,6,6-trifluoro-3-((S)-4-isopropyl-2-oxooxazolidine-3-carbonyl)-2-(3,3,3-trifluoropropyl)hexanoate, and
Preparation 1D: (2R,3R)-tert-Butyl 6,6,6-trifluoro-3-((S)-4-isopropyl-2-oxooxazolidine-3-carbonyl)-2-(3,3,3-trifluoropropyl)hexanoate

Preparation 1E: (2R,3S)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid, and
Preparation 1F: (2R,3R)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid

Preparation 1E: (2R,3S)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid, and
Preparation 1F: (2R,3R)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid

Preparation 1G: (3S)-3-Amino-1-methyl-5-phenyl-1,3-dihydro-2H-1,4-benzodiazepin-2-one, and
Preparation 1H: (3R)-3-Amino-1-methyl-5-phenyl-1,3-dihydro-2H-1,4-benzodiazepin-2-one

Alternate Procedure to Make Preparation 1G
Preparation 1G•CSA salt: (3S)-3-Amino-1-methyl-5-phenyl-1,3-dihydro-2H-1,4-benzodiazepin-2-one, (1 S)-(+)-10-camphorsulfonic acid salt

Preparation 1I: tert-Butyl (2S,3R)-6,6,6-trifluoro-3-(((3S)-1-methyl-2-oxo-5-phenyl-2,3-dihydro-1H-1,4-benzodiazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoate, and
Preparation 1J: tert-Butyl (2R,3R)-6,6,6-trifluoro-3-(((3S)-1-methyl-2-oxo-5-phenyl-2,3-dihydro-1H-1,4-benzodiazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoate

Preparation 1K: (2S,3R)-6,6,6-Trifluoro-3-(((3S)-1-methyl-2-oxo-5-phenyl-2,3-dihydro-1H-1,4-benzodiazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoic acid, and
Preparation 1L: (2R,3R)-6,6,6-Trifluoro-3-(((3S)-1-methyl-2-oxo-5-phenyl-2,3-dihydro-1H-1,4-benzodiazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoic acid

Example 1
Alternate Procedure to Make Example 1
Preparation 1M: 3,3,3-Trifluoropropyl trifluoromethanesulfonate

Preparation 1N: (4S)-4-Benzyl-3-(5,5,5-trifluoropentanoyl)-1,3-oxazolidin-2-one

Preparation 1O: tert-Butyl (3R)-3-(((4S)-4-benzyl-2-oxo-1,3-oxazolidin-3-yl)carbonyl)-6,6,6-trifluorohexanoate

Preparation 1P: (2R)-2-(2-tert-Butoxy-2-oxoethyl)-5,5,5-trifluoropentanoic acid

Preparation 1E: (2R,3S)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid, and
Preparation 1F: (2R,3R)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid

Preparation 1E: (2R,3S)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid, and
Preparation 1F: (2R,3R)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid

Alternate Procedure to Make Preparation 1E
Preparation 1Q: (2R,3S)-1-Benzyl 4-tert-butyl 2,3-bis(3,3,3-trifluoropropyl)succinate

Preparation 1E: (2R,3S)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid

Alternate Procedure to Make Preparation 1E
Preparation 1E: (2R,3S)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid

Alternate Procedure to Make Example 1
Preparation 1I: tert-Butyl (2S,3R)-6,6,6-trifluoro-3-(((3S)-1-methyl-2-oxo-5-phenyl-2,3-dihydro-1H-1,4-benzodiazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoate

Preparation 1K: (2S,3R)-6,6,6-Trifluoro-3-(((3S)-1-methyl-2-oxo-5-phenyl-2,3-dihydro-1H-1,4-benzodiazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoic acid

Example 1
PATENTS
Clip
For some disease targets, an indirect approach may be best. Or so Ashvinikumar V. Gavai and his colleagues atBristol-Myers Squibbfound in their quest toward a potential cancer drug. Gavai unveiled BMS-906024, which is an experimental—and slightly roundabout—treatment for a number of cancers, including breast, lung, and colon cancers, and leukemia.
Cancers have a tendency to relapse or to become resistant to treatments that once worked. Research at BMS and elsewhere had suggested that a family of proteins called Notch is implicated in that resistance and in cancer progression more generally. Gavai, director of oncology chemistry at BMS in Princeton, N.J., and his team set out to block Notch family signaling.
Notch family members lack enzymatic activity, so blocking them directly is difficult. Instead, BMS developed inhibitors of an enzyme that is essential for activating Notch signaling—γ-secretase.
Company: Bristol-Myers Squibb
Target: pan-Notch
Disease: breast, lung, colon cancer; leukemia
Interfering with Notch, even in this indirect way, can have detrimental effects on the gastrointestinal tract. Only two of the four Notch family members are linked to that side effect, Gavai says. But he and his team think their drug will be most effective if it acts on all four family members roughly equally—a so-called pan-Notch inhibitor. By selecting a molecule that’s well tolerated in animals and carefully scheduling doses of the drug in humans, it could be possible to minimize side effects, he says.
The BMS team relied on Notch signaling assays in leukemia and breast cancer cell lines to find leads. They soon learned that for their molecules to work, three chiral centers had to be in the S,R,Sconfiguration. After that, they strove to make the molecules last in the bloodstream. They removed an isobutyl group and tweaked some other parts of their candidate’s succinamide side chain. It was tough to retain both a long half-life and activity against Notch, Gavai told C&EN. “You’d optimize one and lose the other.”
His team threaded the needle with BMS-906024. Their studies with mice suggest that a dose of 4–6 mg once a week could be effective in people. That’s lower than doses being tested for other Notch-targeted agents, according to the website clinicaltrials.gov. The mouse studies also back the idea that Notch is involved in cancer drug resistance and suggest that Notch could be a target for taking on cancer stem cells, which are notoriously resistant to chemotherapy.
BMS-906024 is in Phase I clinical trials, both alone and in combination with other agents. Patients with colon, lung, breast, and other cancers are receiving intravenous doses of the compound to determine its safety and optimum dose ranges.
(From left, front row) Gavai, Weifeng Shan, (second row) Aaron Balog, Patrice Gill, Gregory Vite, (third row) Francis Lee, Claude Quesnelle, (rear row) Wen-Ching Han, Richard Westhouse.
Credit: Catherine Stroud Photography
http://cen.acs.org/articles/91/i16/BMS-906024-Notch-Signaling-Inhibitor.html
clip

BMS-906024
Company: Bristol-Myers Squibb
Meant to treat: cancers including breast, lung, colon, and leukemia
Mode of action: pan-Notch inhibitor
Medicinal chemistry tidbit: The BMS team used an oxidative enolate heterocoupling en route to the candidate– a procedure from Phil Baran’s lab at Scripps Research Institute. JACS 130, 11546
Status in the pipeline: Phase I
Relevant documents: WO 2012/129353
PAPER
An enantioselective synthesis of (S)-7-amino-5H,7H-dibenzo[b,d]azepin-6-one (S–1) is described. The key step in the sequence involved crystallization-induced dynamic resolution (CIDR) of compound 7 using Boc-d-phenylalanine as a chiral resolving agent and 3,5-dichlorosalicylaldehyde as a racemization catalyst to afford S–1 in 81% overall yield with 98.5% enantiomeric excess.
† Department of Discovery Synthesis, Biocon Bristol-Myers Squibb Research Centre, Biocon Park, Bommasandra IV Phase, Jigani Link Road, Bengaluru 560099, India
‡Bristol-Myers Squibb Company, P.O Box 4000, Princeton, New Jersey 08543-4000, United States
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.6b00207, http://pubs.acs.org/doi/suppl/10.1021/acs.oprd.6b00207
*E-mail: anuradha.gupta@syngeneintl.com.
References
- Jump up^ C. Drahl, Liveblogging First-Time Disclosures of Drug Structures from #ACSNOLA, 2013, http://cenblog.org/the-haystack/2013/04/liveblogging-first-time-disclosures-of-drug-structures-from-acsnola/
- Jump up^ http://patentscope.wipo.int/search/en/detail.jsf?docId=WO2012129353
- Jump up^ http://clinicaltrials.gov/show/NCT01653470
- Jump up^ http://clinicaltrials.gov/show/NCT01292655
1. Quesnelle, Claude; Kim, Soong-Hoon; Lee, Francis; Gavai, Ashvinikumar. Bis(fluoroalkyl)-1,4-benzodiazepinone compounds as Notch receptor inhibitors and their preparation and use in the treatment of cancer. PCT Int. Appl. (2012), WO 2012129353 A1 20120927.
| Patent ID | Date | Patent Title |
|---|---|---|
| US2016060232 | 2016-03-03 | BIS(FLUOROALKYL)-1, 4-BENZODIAZEPINONE COMPOUNDS |
| US2016022723 | 2016-01-28 | COMBINATION THERAPY FOR THE TREATMENT OF PROLIFERATIVE DISEASES |
| US2016008316 | 2016-01-14 | USE OF DIANHYDROGALACTITOL AND ANALOGS OR DERIVATIVES THEREOF IN COMBINATION WITH PLATINUM-CONTAINING ANTINEOPLASTIC AGENTS TO TREAT NON-SMALL-CELL CARCINOMA OF THE LUNG AND BRAIN METASTASES |
| US2016009785 | 2016-01-14 | NOVEL FUSION MOLECULES AND USES THEREOF |
| US2015284342 | 2015-10-08 | BIS(FLUOROALKYL)-1, 4-BENZODIAZEPINONE COMPOUNDS |
| US2015232491 | 2015-08-20 | PRODRUGS OF 1, 4-BENZODIAZEPINONE COMPOUNDS |
| US8968741 | 2015-03-03 | Anti-CD22 antibodies and immunoconjugates and methods of use |
| US2014357605 | 2014-12-04 | BIS(FLUOROALKYL)-1, 4-BENZODIAZEPINONE COMPOUNDS |
| US8822454 | 2014-09-02 | Bisfluoroalkyl-1, 4-benzodiazepinone compounds |
| US8629136 | 2014-01-14 | Bisfluoroalkyl-1, 4-benzodiazepinone compounds |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(2R,3S)-N-[(3S)-1-Methyl-2-oxo-5-phenyl-2,3-dihydro-1H-1,4-benzodiazepin-3-yl]-2,3-bis(3,3,3-trifluoropropyl)succinamide
|
|
| Identifiers | |
| PubChem | CID 66550890 |
| ChemSpider | 28536138 |
| Chemical data | |
| Formula | C26H26F6N4O3 |
| Molar mass | 556.500 g/mol |
///////////////3,5-dichlorosalicylaldehyde, Alzheimer’s disease, Boc-D-phenylalanine, CIDR;dibenzoazepenone, DKR; Notch inhibitors, Notch inhibitor, SAR, T-acute lymphoblastic leukemia, triple-negative breast cancer, γ-secretase inhibitor, PHASE 1, BMS, Bristol-Myers Squibb, 1401066-79-2, Ashvinikumar Gavai
CN1c2ccccc2C(=N[C@@H](C1=O)NC(=O)[C@H](CCC(F)(F)F)[C@H](CCC(F)(F)F)C(=O)N)c3ccccc3

Patent US8377886 – Use of gamma secretase inhibitors and notch …
Figure US08377886-20130219-C00003. gamma secretase inhibitor

RO4929097 | γ-secretase inhibitor – Cellagen Technology
RO4929097 | γ-secretase inhibitor

YO-01027 (Dibenzazepine) | gamma-secretase inhibitor – Cellagen …
YO-01027 (Dibenzazepine) | gamma-secretase inhibitor

Semagacestat (LY450139) | Gamma-secretase inhibitor | Read Reviews …
Semagacestat (LY450139) Chemical Structure
MCC 950
MCC 950
256373-96-3 (sodium salt); 210826-40-7 (free form).
MCC950; CP-456773; CAS 210826-40-7; DSSTox_CID_27301; DSSTox_RID_82252; DSSTox_GSID_47301;
CP-456,773; CRID3
1-(1,2,3,5,6,7-hexahydro-s-indacen-4-yl)-3-[4-(2-hydroxypropan-2-yl)furan-2-yl]sulfonylurea
| C20H24N2O5S | |
| Molecular Weight: | 404.47996 g/mol |
|---|
CP-456773, also known as MCC950 and CRID3, is a potent and selective cytokine release inhibitor and NLRP3 inflammasome inhibitor for the treatment of inflammatory diseases. CP-456773 inhibits interleukin 1β (IL-1β) secretion and caspase 1 processing. MCC950 blocked canonical and noncanonical NLRP3 activation at nanomolar concentrations. MCC950 specifically inhibited activation of NLRP3 but not the AIM2, NLRC4 or NLRP1 inflammasomes. MCC950 reduced interleukin-1β (IL-1β) production in vivo and attenuated the severity of experimental autoimmune encephalomyelitis (EAE), a disease model of multiple sclerosis. MCC950 is a potential therapeutic for NLRP3-associated syndromes, including autoinflammatory and autoimmune diseases, and a tool for further study of the NLRP3 inflammasome in human health and disease.

| Formula | C20H23N2NaO5S |
|---|---|
| MW | 426.5 |
| CAS | 256373-96-3 |
sodium ((1,2,3,5,6,7-hexahydro-s-indacen-4-yl)carbamoyl)((4-(2-hydroxypropan-2-yl)furan-2-yl)sulfonyl)amide

PAPER
Identification, Synthesis, and Biological Evaluation of the Major Human Metabolite of NLRP3 Inflammasome Inhibitor MCC950
† Institute for Molecular Bioscience, The University of Queensland, Brisbane, Queensland 4072, Australia
‡ School of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, Queensland 4072, Australia
ACS Med. Chem. Lett., Article ASAP
DOI: 10.1021/acsmedchemlett.6b00198
*E-mail: uqarob15@uq.edu.au. Fax: +61-7-3346-2090. Phone: +61-7-3346-2204., *E-mail: m.cooper@uq.edu.au. Fax: +61-7-3346-2090. Phone: +61-7-3346-2044.
Abstract

MCC950 is an orally bioavailable small molecule inhibitor of the NOD-like receptor pyrin domain-containing protein 3 (NLRP3) inflammasome that exhibits remarkable activity in multiple models of inflammatory disease. Incubation of MCC950 with human liver microsomes, and subsequent analysis by HPLC–MS/MS, revealed a major metabolite, where hydroxylation of MCC950 had occurred on the 1,2,3,5,6,7-hexahydro-s-indacene moiety. Three possible regioisomers were synthesized, and coelution using HPLC–MS/MS confirmed the structure of the metabolite. Further synthesis of individual enantiomers and coelution studies using a chiral column in HPLC–MS/MS showed the metabolite was R-(+)- N-((1-hydroxy-1,2,3,5,6,7-hexahydro-s-indacen-4-yl)carbamoyl)-4-(2-hydroxypropan-2-yl)furan-2-sulfonamide (2a). Incubation of MCC950 with a panel of cytochrome P450 enzymes showed P450s 2A6, 2C9, 2C18, 2C19, 2J2, and 3A4 catalyze the formation of the major metabolite 2a, with a lower level of activity shown by P450s 1A2 and 2B6. All of the synthesized compounds were tested for inhibition of NLRP3-induced production of the pro-inflammatory cytokine IL-1β from human monocyte derived macrophages. The identified metabolite 2a was 170-fold less potent than MCC950, while one regioisomer had nanomolar inhibitory activity. These findings also give first insight into the SAR of the hexahydroindacene moiety.
PATENT
WO 2001019390
http://www.google.co.in/patents/WO2001019390A1?cl=en
Synthesis of precursors will be update soon……………

Novel synthesis of 1-(1,2,3,5,6,7-hexahydro-s-indacen-4-yl)-3-[4-(1-hydroxy-1-methylethyl)furan-2-sulfonyl]urea, an antiinflammatory agentPAPER
Synthetic Communications (2003), 33, (12), 2029-2043.
http://dx.doi.org/10.1081/SCC-120021029
Abstract
A novel synthesis of the anti-inflammatory agent 1-(1,2,3,5,6,7- hexahydro-s-indacen-4-yl)-3-[4-(1-hydroxy-1-methyl-ethyl)-furan-2-sulfonyl] urea 1 is described. Sulfonamide 5 was prepared starting from ethyl 3-furoate 2. Key steps were a one-pot sulfonylation with chlorosulfonic acid in methylene chloride followed by pyridinium salt formation and reaction with phosphorus pentachloride to provide ethyl 2-(chlorosulfonyl)-4-furoate 7. This sulfonyl chloride was treated with ammonium bicarbonate to form sulfonamide 8, followed by treatment with excess methyl magnesium chloride to provide 4-(1-hydroxy-1-methyl-ethyl)-furan-2-sulfonamide 5. 4-Isocyanato-1,2,3,5,6,7-hexahydro-s-indacene 16 was prepared from indan in five steps. The formation of the desired sulfonyl urea was carried out both with the isolated isocyanate 16 and via an in situ method.
1-(1,2,3,5,6,7-Hexahydro-s-indacen-4-yl)-3-[4-(1-hydroxy-1-methylethyl)-furan-2-sulfonylurea 1 has been in development for treatment of inflammation. [1] The synthetic route to furan sulfonamide 5 used by its discoverer Mark Dombroski in Medicinal Chemistry is shown in Sch. 1. The starting material was ethyl 3-furoate 2. This was treated with excess methyl magnesium chloride to provide the 3-furanyl-tertiary alcohol 3. Furan alcohol 3 was deprotonated with methyl lithium followed by s-butyl lithium at low temperature and reacted with liquid sulfur dioxide to generate sulfinic acid 4. Without isolation, sulfinic acid 4 was oxidized to sulfonamide 5 with hydroxylamine O-sulfinic acid via a procedure described by workers at Merck.[2] We were interested in finding a synthesis of furan sulfonamide 5 and its conversion to sulfonylurea 1 that would be suitable for scale up. In this article, we describe the discovery of a better bulk process to sulfonamide 5 from the same starting material and a procedure to form the desired sulfonylurea without isolating the isocyanate of 4-amino-1,2,3,5,6,7-hexahydro-s-indacene.




1-(1,2,3,5,6,7-Hexahydro-s-indacen-4-yl)-3-[4-(1-hydroxy- 1-methyl-ethyl)-furan-2-sulfonyl Urea (1) ………….. anhydrous sodium salt weighed 4.9 g. mp 239 C. 1 H NMR (D2O, 400 MHz) 7.35 (s, 1), 6.81 (s, 1), 6.65 (s, 1), 2.53 (m, 4), 2.41 (m, 4), 1.73 (m, 4), 1.31 (s, 6). 13C NMR (D2O, 100 MHz) 159.87, 151.82, 143.89, 140.49, 138.77, 134.87, 129.58, 118.38, 112.02, 68.24, 32.67, 30.10, 29.53, 25.34. Anal. calcd. for C20H23N2O5SNa: C, 56.33; H, 5.44; N, 6.57; S, 7.52. Found: C, 56.19; H, 5.40; N, 6.34; S, 7.42. N
CLIPS


Dr Rebecca Coll, PostDoctoral Researcher, Inflammasome Lab, UQ Fellow
Rebecca completed her PhD research under Prof. Luke O’Neill in Trinity College Dublin at one of the leading laboratories in the innate immunity field. For her work on the regulation of TLR signalling she received the International Endotoxin and Innate Immunity Society Young Investigator Award in 2012. However, her main research focus has been inflammasomes and their therapeutic targeting by small molecule drugs. Her recent first author publication on MCC950 in Nature Medicine has been widely acclaimed (the subject of seven commentaries in leading journals and attention from 24 international news outlets) and is already a highly cited paper. She joined the Schroder group in May 2014 with the goal of defining the molecular target of MCC950 as part of a broader collaboration between the Schroder, Cooper and O’Neill labs.
Email: r.coll@imb.uq.edu.au
Office Telephone: +61 7 3346 2351
Lab Telephone: +61 7 3346 2071
A collaboration between scientists from Dublin’s Trinity College (Ireland) and the University of Queensland (Australia) identified a compound able to inhibit an inflammatory process common to many diseases, including Alzheimer’s disease. The study entitled “A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases” was published on line in the journal Nature Medicine.
Pathogenesis of several diseases, including Alzheimer’s, have a strong inflammatory component. Inflammatory processes can be triggered by molecules of the NOD-like receptor (NLR) family such as NLRP3. Once activated, this molecule leads to a cascade of events known as the NLRP3 inflammasome that ultimately causes the production of inflammatory factors. Aberrant activation of NLRP3 is responsible for increased inflammatory responses in complex diseases such as multiple sclerosis, Muckle-Wells syndrome, type 2 diabetes, Alzheimer’s disease and atherosclerosis.
Targeting this molecule can overcome the side effects of other anti-inflammatory drugs commonly used: “Drugs like aspirin or steroids can work in several diseases, but can have side effects or be ineffective. What we have found is a potentially transformative medicine, which targets what appears to be the common disease-causing process in a myriad of inflammatory diseases,” said Luke A J O’Neill, one of the team leaders.
Previous studies identified NLRP3 inhibitors, though neither very potent nor specific. This research team now identified a specific inhibitor of NLRP3 inflammasome, the molecule MCC950. They observed that it inhibits NLRP3 in mouse models of multiple sclerosis with consequent attenuation of disease progression. MCC950 also blocks production of inflammatory factors in blood samples from patients with a severe inflammatory disorder, Muckle-Wells syndrome. These results demonstrated the pharmaceutical potential of this specific NLRP3 inhibitor.
“MCC950 is blocking what was suspected to be a key process in inflammation. There is huge interest in NLRP3 both among medical researchers and pharmaceutical companies and we feel our work makes a significant contribution to the efforts to find new medicines to limit it,” said Rebecca Coll, the paper’s first author.
The researchers were able to demonstrate the potential of MCC950 in multiple sclerosis, an inflammatory disease of the central nervous system (CNS). However, the target for MCC950 is strongly implicated in other diseases of the CNS such as Alzheimer’s and Parkinson’s diseases indicating that it has the potential to treat all of these conditions. The fact that MCC950 can be orally administered further enhances the potential of this molecule as a therapeutic drug.
“MCC950 is able to be given orally and will be cheaper to produce than current protein-based treatments, which are given daily, weekly, or monthly by injection. Importantly, it will also have a shorter duration in the body, allowing clinicians to stop the anti-inflammatory action of the drug if the patient ever needed to switch their immune response back to 100% in order to clear an infection.” said Matt Cooper, chemist and also co-senior author in this study.
REFERENCES
1: Shao BZ, Xu ZQ, Han BZ, Su DF, Liu C. NLRP3 inflammasome and its inhibitors: a review. Front Pharmacol. 2015 Nov 5;6:262. doi: 10.3389/fphar.2015.00262. eCollection 2015. Review. PubMed PMID: 26594174; PubMed Central PMCID: PMC4633676.
2: Baker PJ, Boucher D, Bierschenk D, Tebartz C, Whitney PG, D’Silva DB, Tanzer MC, Monteleone M, Robertson AA, Cooper MA, Alvarez-Diaz S, Herold MJ, Bedoui S, Schroder K, Masters SL. NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. Eur J Immunol. 2015 Oct;45(10):2918-26. doi: 10.1002/eji.201545655. Epub 2015 Aug 24. PubMed PMID: 26173988.
3: Krishnan SM, Dowling JK, Ling YH, Diep H, Chan CT, Ferens D, Kett MM, Pinar A, Samuel CS, Vinh A, Arumugam TV, Hewitson TD, Kemp-Harper BK, Robertson AA, Cooper MA, Latz E, Mansell A, Sobey CG, Drummond GR. Inflammasome activity is essential for one kidney/deoxycorticosterone acetate/salt-induced hypertension in mice. Br J Pharmacol. 2016 Feb;173(4):752-65. doi: 10.1111/bph.13230. Epub 2015 Jul 31. PubMed PMID: 26103560; PubMed Central PMCID: PMC4742291.
4: Groß CJ, Groß O. The Nlrp3 inflammasome admits defeat. Trends Immunol. 2015 Jun;36(6):323-4. doi: 10.1016/j.it.2015.05.001. Epub 2015 May 16. PubMed PMID: 25991463.
5: Coll RC, Robertson AA, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, Vetter I, Dungan LS, Monks BG, Stutz A, Croker DE, Butler MS, Haneklaus M, Sutton CE, Núñez G, Latz E, Kastner DL, Mills KH, Masters SL, Schroder K, Cooper MA, O’Neill LA. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015 Mar;21(3):248-55. doi: 10.1038/nm.3806. Epub 2015 Feb 16. PubMed PMID: 25686105; PubMed Central PMCID: PMC4392179.
| Patent ID | Date | Patent Title |
|---|---|---|
| US2016008420 | 2016-01-14 | Treatment Of HIV-1 Infection And AIDS |
| US2015343011 | 2015-12-03 | Treatment Of HIV-1 Infection And AIDS |
| US2005064519 | 2005-03-24 | Methods of using GST-Omega-2 |
| US2003143230 | 2003-07-31 | Combination of an IL-1/18 inhibitor with a TNF inhibitor for the treatment of inflammation |
| EP0964849 | 2003-06-04 | SULFONYL UREA DERIVATIVES AND THEIR USE IN THE CONTROL OF INTERLEUKIN-1 ACTIVITY |
| US6433009 | 2002-08-13 | Sulfonyl urea derivatives and their use in the control of interleukin-1 activity |
| US6166064 | 2000-12-26 | Sulfonyl urea derivatives and their use in the control of interleukin-1 activity |
| US6022984 | 2000-02-08 | Efficient synthesis of furan sulfonamide compounds useful in the synthesis of new IL-1 inhibitors |
| EP0976742 | 2000-02-02 | A synthesis of furan sulfonamide compounds useful in the synthesis of IL-1 inhibitors |
| WO9832733 | 1998-07-30 | SULFONYL UREA DERIVATIVES AND THEIR USE IN THE CONTROL OF INTERLEUKIN-1 ACTIVITY |
/////////cytochrome P450, inflammasome, MCC950, metabolite, microsome, NLRP3, MCC950, CP-456,773, CRID3, 256373-96-3, 210826-40-7 ,
CC(C)(C1=COC(=C1)S(=O)(=O)NC(=O)NC2=C3CCCC3=CC4=C2CCC4)O
BMT-145027
BMT-145027
CAS ?
MF C23H14ClF3N4
MW: 438.0859
3-(4-chloro-3-(trifluoromethyl)phenyl)-4-cyclopropyl-6-phenyl-1H-pyrazolo[3,4-b]pyridine-5-carbonitrile
3-(4-chloro-3- (trifluoromethyl)phenyl)-4-cyclopropyl-6-phenyl-1H-pyrazolo[3,4-b]pyridine-5-carbonitrile
1H NMR (600 MHz, DMSO-d6) δ = 14.46 (br. s., 1H), 8.24 (s, 1H), 8.14 (d, J=8.1 Hz, 1H), 7.88 (d, J=8.3 Hz, 1H), 7.84 (dd, J=6.1, 2.7 Hz, 2H), 7.61 – 7.55 (m, 3H), 2.50 – 2.45 (m, 1H), 0.74 – 0.68 (m, 2H), 0.65 – 0.59 (m, 2H).
13C NMR (126 MHz, DMSO-d6) δ 160.5, 155.0, 153.0, 144.1, 138.3, 135.4, 133.9, 132.0, 131.2, 130.3, 129.7, 128.9, 128.9, 128.8, 127.0 (q, J=30.5 Hz), 118.1, 112.4, 103.9, 14.6, 9.4.
LCMS (method A) tR, 2.01 min, MS Anal. Calcd. for [M+H]+ C23H15ClF3N4: 439.09; found: 439.15.
LC/MS HPLC methods: method A: Column: Phenomenex Luna 30 x 2.0 mm 3um; Mobile Phase A: 10:90 acetonitrile:water with 0.1% TFA; Mobile Phase B: 90:10 acetonitrile:water with 0.1% TFA; Temperature: 40 °C; Gradient: 0-100% B over 2 min; Flow: 1 mL/min.
DETAILS WILL BE UPDATED…………



Senior Research Investigator II at Bristol-Myers Squibb
Highly effective leader seeking to apply innovative thinking and critical analysis to strategy and scientific challenges. Diverse educational background, including recent MBA studies, provides foundation for excellent communication, collaboration, and team building across organizational functions. Experience includes 13 years of cutting-edge scientific research in a global work environment, specializing in the fields of organic chemistry and drug discovery.
Experience

Senior Research Investigator II
Bristol-Myers Squibb
July 2014 – Present (2 years 4 months)Wallingford, CT
Oncology Discovery Chemistry, Program: Bromodomain and Extra-Terminal Inhibitor Program, undisclosed target
BMT-145027 is a potent mGluR5 PAM with no inherent mGluR5 agonist activity. BMT-145027 is a non-MPEP site PAM to demonstrate in vivo efficacy. BMT-145027 has mGluR5 PAM EC50 = 47 nM, with fold shit = 3.5, and is effective in mouse NOR. The metabotropic glutamate receptor 5 (mGluR5) is an attractive target for the treatment of schizophrenia due to its role in regulating glutamatergic signaling in association with the N-methyl-D-aspartate receptor (NMDAR).

The metabotropic glutamate receptor 5 (mGluR5) is an attractive target for the treatment of schizophrenia due to its role in regulating glutamatergic signaling in association with the N-methyl-d-aspartate receptor (NMDAR). We describe the synthesis of 1H-pyrazolo[3,4-b]pyridines and their utility as mGluR5 positive allosteric modulators (PAMs) without inherent agonist activity. A facile and convergent synthetic route provided access to a structurally diverse set of analogues that contain neither the aryl-acetylene-aryl nor aryl-methyleneoxy-aryl elements, the predominant structural motifs described in the literature. Binding studies suggest that members of our new chemotype do not engage the receptor at the MPEP and CPPHA mGluR5 allosteric sites. SAR studies culminated in the first non-MPEP site PAM, 1H-pyrazolo[3,4-b]pyridine 31 (BMT-145027), to improve cognition in a preclinical rodent model of learning and memory.
Development of 1H-Pyrazolo[3,4-b]pyridines as Metabotropic Glutamate Receptor 5 Positive Allosteric Modulators
Matthew D. Hill*, Haiquan Fang, Jeffrey M. Brown, Thaddeus Molski, Amy Easton, Xiaojun Han, Regina Miller, Melissa Hill-Drzewi, Lizbeth Gallagher, Michele Matchett, Michael Gulianello, Anand Balakrishnan, Robert L. Bertekap, Kenneth S. Santone, Valerie J. Whiterock, Xiaoliang Zhuo, Joanne J. Bronson, John E. Macor, and Andrew P. Degnan
Research and Development, Bristol-Myers Squibb, 5 Research Parkway, Wallingford, Connecticut 06492-7660, United States
ACS Med. Chem. Lett., Article ASAP
DOI: 10.1021/acsmedchemlett.6b00292, http://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.6b00292
*Tel: 1-203-677-7102. Fax: 1-203-677-7884. E-mail: matthew.hill@bms.com.

SIMILAR STR
1929593-12-3
C23 H15 F3 N4, 404.39
1H-Pyrazolo[3,4-b]pyridine-5-carbonitrile, 4-cyclopropyl-6-phenyl-3-[4-(trifluoromethyl)phenyl]-
A Multicomponent Approach to Highly Substituted 1H-Pyrazolo[3,4-b]pyridines
Synthesis (2016), 48, (14), 2201-2204.
Synthesis (2016), 48, (14), 2201-2204.
A Multicomponent Approach to Highly Substituted 1H-Pyrazolo[3,4-b]pyridines
- Bristol-Myers Squibb Research and Development, 5 Research Parkway, Wallingford, CT 06492-7660, USA Email:matthew.hill@bms.com
DOI: 10.1055/s-0035-1562230



Compound 12 (500 mg, 65% yield: 1H NMR (500 MHz, DMSO-d6 δ 14.41 (br. s., 1H, 7.86 – 7.80 (m, 3H, 7.76 (dt, J=7.1, 1.6 Hz, 1H, 7.61 – 7.51 (m, 5H, 2.48 – 2.45 (m, 1H, 0.73 – 0.66 (m, 2H, 0.62 – 0.57 (m, 2H. 13C NMR (400 MHz, DMSO-d6 δ 159.98, 154.67, 152.15, 144.68, 137.75, 135.74, 132.69, 129.87, 129.71, 129.14, 128.99, 128.34, 128.24, 128.21, 117.61, 111.88, 103.05, 14.01, 8.75. IR (film: 3228 (s, 3052 (w, 2228 (m, 1581 (s, 1555 (s, 1503 (w, 1447 (m, 1284 (m cm–1. HRMS (ESI: m/z [M+H]+ calcd for C22H16N4Cl: 371.1058; found: 371.1053.
Compound 13 (103 mg, 28% yield: 1H NMR (500 MHz, DMSO-d6 δ 14.50 (br. s., 1H, 8.03 (d, J=7.9 Hz, 2H, 7.92 – 7.80 (m, 4H, 7.63 – 7.55 (m, 3H, 2.51 (br. s., 1H, 0.65 (d, J=7.6 Hz, 2H, 0.56 (d, J=4.3 Hz, 2H. MS (ESI: m/z = 405.15 [M+H]+.
///////////BMT-145027, glutamat, mGluR5, novel object recognition, positive allosteric modulator, schizophrenia
c1(c(c(c2c(n1)nnc2c3ccc(c(c3)C(F)(F)F)Cl)C4CC4)C#N)c5ccccc5
ClC(C=C1)=C(C(F)(F)F)C=C1C2=NNC3=C2C(C4CC4)=C(C#N)C(C5=CC=CC=C5)=N3
GDUFA: FDA’s new Guidance on Self-Identification of Generic Drug Manufacturers

GDUFA: FDA’s new Guidance on Self-Identification of Generic Drug Manufacturers
FDA’s new Guidance requesting generic drug manufacturers who want to export to the USA to self-identify has recently been published in a finalised form. Read more here about what types of generic drug manufacturers are affected and which company data are required by the FDA.
The GDUFA (Generic Drug User Fee Amendments) is a legislative package which came into force in 2012 and entitles the US-American FDA to collect fees from generic drug manufacturers, who strive for a marketing authorisation for the American market. An annual fee has to be paid after the successful registration.
The core of the document is the obligation to “Self-Identify” for those companies that have to submit essential site-related information to the FDA. The details of this self-identification are set in a Guidance for Industry entitled “Self-Identification of Generic Drug Facilities, Sites, and Organizations” published on 22 September 2016 by the FDA in the finalised form.
The Guidance describes the following elements:
1. Which types of generic facilities, sites, and organizations are required to self-identify?
2. What information is requested?
3. What technical standards are to be used for electronically submitting the requested information?
4. What is the penalty for failing to self-identify?
Hereinafter, you will find a short summary of these four topics:
1. Companies that manufacture finished generic medicinal products for human use or the APIs for them, or both are required to self-identify as well as companies that package the finished generic drug into the primary container and label it. Besides, sites that – pursuant to a contract with the applicant (generic drug manufacturer) – repack/redistribute the finished drug from a primary container into a different primary container are also required to submit a self-identification as well as sites that perform bioequivalence/bioavailability studies. Last but not least, the obligation to self-identify also concerns sites that are listed in the application dossier as contract laboratories for the sampling and performing of analytical testing.
2. Essential data are: the D-U-N-S number (a unique nine-digit sequence specific for each site / each distinct physical location of an entity), the “Facility Establishment Identifier, FEI” (an identifier used by the FDA for the planning and tracking of inspections) and general information with regard to the facility (company owner, type of business operation, contact data, information about the manufacture of non generic drugs).
3. The HLS standard (Health Level Seven Structured Product Labeling) requested for generic applications (ANDAs) has to be also used for the submission of self-identification information. A detailed description of this standard can be found in the Guidance “Providing Regulatory Submissions in Electronic Format – Drug Establishment Registration and Drug Listing“.
4. Companies that fail to self-identify do not have to expect an explicit penalty. However, such a failure leads to two drawbacks: first, the likelihood of a site inspection by the FDA prior to approval is higher. The second drawback which is much more serious is that all the APIs or finished drugs from a manufacturer who hasn’t self-identified are deemed misbranded. For the FDA, such products are not allowed for importation in the USA.
To the satisfaction of the FDA, the regulations set in the GDUFA and the provisions laid down in the new Guidance represent a major contribution to an enhanced transparency in particular of complex supply chains.
//////////GDUFA, FDA, new Guidance, Self-Identification, Generic Drug Manufacturers
Glecaprevir (ABT-493)

Glecaprevir (ABT-493), A-1282576.0
(3aR,7S,10S,12R,21E,24aR)-7-tert-butyl-N-((1R,2R)-2-(difluoromethyl)-1-((1-methylcyclopropane-1-sulfonyl)carbamoyl)cyclopropyl)-20,20-difluoro-5,8-dioxo-2,3,3a,5,6,7,8,11,12,20,23,24a-dodecahydro-1H,10H-9,12-methanocyclopenta(18,19)(1,10,17,3,6)trioxadiazacyclononadecino(11,12-b)quinoxaline-10-carboxamide
Cyclopropanecarboxamide, N-((((1R,2R)-2-((4,4-difluoro-4-(3-hydroxy-2-quinoxalinyl)-2-buten-1-yl)oxy)cyclopentyl)oxy)carbonyl)-3-methyl-L-valyl-(4R)-4-hydroxy-L-prolyl-1-amino-2-(difluoromethyl)-N-((1-methylcyclopropyl)sulfonyl)-, cyclic (1->2)-ether, (1R,2R)-
CAS RN: 1365970-03-1
UNII: K6BUU8J72P
Molecular Formula, C38-H46-F4-N6-O9-S
Molecular Weight, 838.8724
Classification Code, Treatment of Chronic Hepatitis C Infection
(1R,14E,18R,22R,26S,29S)-N-[(1R,2R)-2-(Difluormethyl)-1-{[(1-methylcyclopropyl)sulfonyl]carbamoyl}cyclopropyl]-13,13-difluor-26-(2-methyl-2-propanyl)-24,27-dioxo-2,17,23-trioxa-4,11,25,28-tetraazapent ;acyclo[26.2.1.03,12.05,10.018,22]hentriaconta-3,5(10),6,8,11,14-hexaen-29-carboxamid
(1R,14E,18R,22R,26S,29S)-N-[(1R,2R)-2-(Difluoromethyl)-1-{[(1-methylcyclopropyl)sulfonyl]carbamoyl}cyclopropyl]-13,13-difluoro-26-(2-methyl-2-propanyl)-24,27-dioxo-2,17,23-trioxa-4,11,25,28-tetraazape ;ntacyclo[26.2.1.03,12.05,10.018,22]hentriaconta-3,5(10),6,8,11,14-hexaene-29-carboxamide
| Class | Antivirals (small molecules) |
|---|---|
| Mechanism Of Action | Hepatitis C virus NS3 protein inhibitors |
| Who Atc Codes | J05A-E (Protease inhibitors) |
| Ephmra Codes | J5B1 (Viral hepatitis products) |
| Indication | Hepatitis C, Renal Impairment, Hepatic impairment |
- Originator AbbVie; Enanta Pharmaceuticals
- Developer AbbVie
- Class Antivirals; Aza compounds; Cyclic ethers; Cyclopentanes; Cyclopropanes; Quinoxalines; Small molecules
- Mechanism of Action Hepatitis C virus NS3 protein inhibitors
- Phase II Hepatitis C
Most Recent Events
- 18 Apr 2016 Pooled efficacy and adverse event data from the phase II SURVEYOR-I and SURVEYOR-2 trials for Hepatitis C presented at The International Liver Congress™ 2016 (ILC-2016)
- 15 Apr 2016 Updated efficacy data from a phase II MAGELLAN 1 study were reported by Enanta Pharmaceuticals
- 15 Apr 2016 Updated safety and efficacy data from a phase II MAGELLAN 1 study were presented at the International Liver Congress™ (ILC-2016)
- OCT 2016, US FDA grants breakthrough therapy designation to AbbVie’s G/P to treat HCV
AbbVie’s investigational, pan-genotypic regimen of glecaprevir (ABT-493) / pibrentasvir (ABT-530) (G/P) has received breakthrough therapy designation from the US Food and Drug Administration (FDA) to treat chronic hepatitis C virus (HCV).

HCV is the principal cause of non-A, non-B hepatitis and is an increasingly severe public health problem both in the developed and developing world. It is estimated that the virus infects over 200 million people worldwide, surpassing the number of individuals infected with the human immunodeficiency virus (HIV) by nearly five fold. HCV infected patients, due to the high percentage of individuals inflicted with chronic infections, are at an elevated risk of developing cirrhosis of the liver, subsequent hepatocellular carcinoma and terminal liver disease. HCV is the most prevalent cause of hepatocellular cancer and cause of patients requiring liver transplantations in the western world.
There are considerable barriers to the development of anti-HCV therapeutics, which include, but are not limited to, the persistence of the virus, the genetic diversity of the virus during replication in the host, the high incident rate of the virus developing drug-resistant mutants, and the lack of reproducible infectious culture systems and small-animal models for HCV replication and pathogenesis. In a majority of cases, given the mild course of the infection and the complex biology of the liver, careful consideration must be given to antiviral drugs, which are likely to have significant side effects.
Only two approved therapies for HCV infection are currently available. The original treatment regimen generally involves a 3-12 month course of intravenous interferon-α (IFN-α), while a new approved second-generation treatment involves co-treatment with IFN-α and the general antiviral nucleoside mimics like ribavirin. Both of these treatments suffer from interferon related side effects as well as low efficacy against HCV infections. There exists a need for the development of effective antiviral agents for treatment of HCV infection due to the poor tolerability and disappointing efficacy of existing therapies.
In a patient population where the majority of individuals are chronically infected and asymptomatic and the prognoses are unknown, an effective drug would desirably possess significantly fewer side effects than the currently available treatments. The hepatitis C non-structural protein-3 (NS3) is a proteolytic enzyme required for processing of the viral polyprotein and consequently viral replication. Despite the huge number of viral variants associated with HCV infection, the active site of the NS3 protease remains highly conserved thus making its inhibition an attractive mode of intervention. Recent success in the treatment of HIV with protease inhibitors supports the concept that the inhibition of NS3 is a key target in the battle against HCV.
HCV is a flaviridae type RNA virus. The HCV genome is enveloped and contains a single strand RNA molecule composed of circa 9600 base pairs. It encodes a polypeptide comprised of approximately 3010 amino acids.
The HCV polyprotein is processed by viral and host peptidase into 10 discreet peptides which serve a variety of functions. There are three structural proteins, C, E1 and E2. The P7 protein is of unknown function and is comprised of a highly variable sequence. There are six non-structural proteins. NS2 is a zinc-dependent metalloproteinase that functions in conjunction with a portion of the NS3 protein. NS3 incorporates two catalytic functions (separate from its association with NS2): a serine protease at the N-terminal end, which requires NS4A as a cofactor, and an ATP-ase-dependent helicase function at the carboxyl terminus. NS4A is a tightly associated but non-covalent cofactor of the serine protease.
The NS3/4A protease is responsible for cleaving four sites on the viral polyprotein. The NS3-NS4A cleavage is autocatalytic, occurring in cis. The remaining three hydrolyses, NS4A-NS4B, NS4B-NS5A and NS5A-NS5B all occur in trans. NS3 is a serine protease which is structurally classified as a chymotrypsin-like protease. While the NS serine protease possesses proteolytic activity by itself, the HCV protease enzyme is not an efficient enzyme in terms of catalyzing polyprotein cleavage. It has been shown that a central hydrophobic region of the NS4A protein is required for this enhancement. The complex formation of the NS3 protein with NS4A seems necessary to the processing events, enhancing the proteolytic efficacy at all of the sites.
A general strategy for the development of antiviral agents is to inactivate virally encoded enzymes, including NS3, that are essential for the replication of the virus. Current efforts directed toward the discovery of NS3 protease inhibitors were reviewed by S. Tan, A. Pause, Y. Shi, N. Sonenberg, Hepatitis C Therapeutics: Current Status and Emerging Strategies, Nature Rev. Drug Discov. 1, 867-881 (2002).
PATENT
Yat Sun Or, Jun Ma, Guoqiang Wang, Jiang Long, Bin Wang
Example 6Compound of Formula VIII, Wherein

Step 6a

The acid 1-6a (21 mg, 0.0356 mmol) was dissolved in DCM (1.5 ml), and to this solution was added sulfonamide 1-7e (13.0 mg, 0.0463 mmol), HATU (17.6 mg, 0.0462 mmol) and DIPEA (12.4 ul, 0.0712 mmol). The mixture was stirred for 3 h, and then diluted with DCM. The organic layer was washed with 1 N HCl, water, brine, dried and concentrated in vacuo. The residue was purified by HPLC to afford the title compound. MS-ESI m/z 839.41 (M+H)+.
REFERENCES
http://aac.asm.org/content/60/3/1546.full
////////////glecaprevir, ABT-493, US FDA, breakthrough therapy designation, AbbVie’s G/P, treat HCV , PHASE 2, A-1282576.0, 1365970-03-1, US 20120070416
CC1(CC1)S(=O)(=O)NC(=O)[C@]2(C[C@H]2C(F)F)NC(=O)[C@@H]3C[C@@H]4CN3C(=O)[C@@H](NC(=O)O[C@@H]5CCC[C@H]5OC/C=C/C(c6c(nc7ccccc7n6)O4)(F)F)C(C)(C)C

US FDA grants breakthrough therapy designation to AbbVie’s G/P to treat HCV

4 October 2016
AbbVie’s investigational, pan-genotypic regimen of glecaprevir (ABT-493) / pibrentasvir (ABT-530) (G/P) has received breakthrough therapy designation from the US Food and Drug Administration (FDA) to treat chronic hepatitis C virus (HCV).
The HCV is a bloodborne virus commonly transmitted through injecting drug use due to the sharing of injection equipment, reuse or inadequate sterilisation of medical equipment, and the transfusion of unscreened blood and blood products.
The designation facilitates the use of AbbVie’s G/P to treat chronic HCV patients who failed previous therapy with direct-acting antivirals (DAAs) in genotype 1 (GT1), including therapy with an NS5A inhibitor and / or protease inhibitor.
AbbVie research and development executive vice-president Michael Severino said: “AbbVie is committed to advancing HCV care and addressing areas of continued unmet need for people living with chronic HCV.
“AbbVie is committed to advancing HCV care and addressing areas of continued unmet need for people living with chronic HCV.”
“The FDA’s breakthrough therapy designation is an important step in our effort to bring our pan-genotypic regimen to market, which we are also investigating as an eight-week path to virologic cure for the majority of patients.”
AbbVie said that G/P is currently in Phase III trials evaluating the safety and efficacy of the regimen across all major HCV genotypes (genotypes 1-6).
Figures released by the World Health Organisation revealed that an estimated 700,000 people die each year from hepatitis C-related liver diseases.
There is currently no vaccine for hepatitis C, although research in this area is underway at present.

