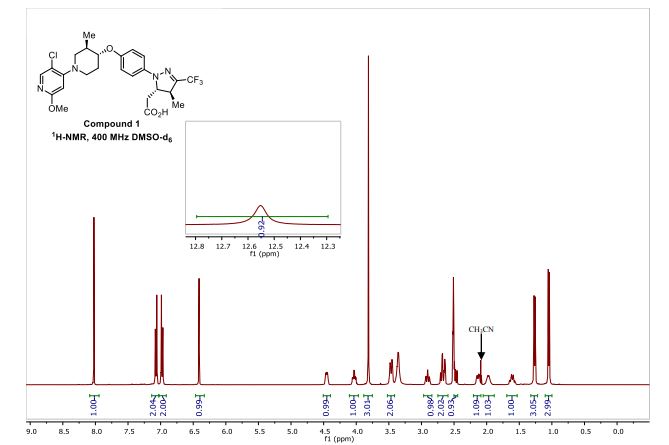
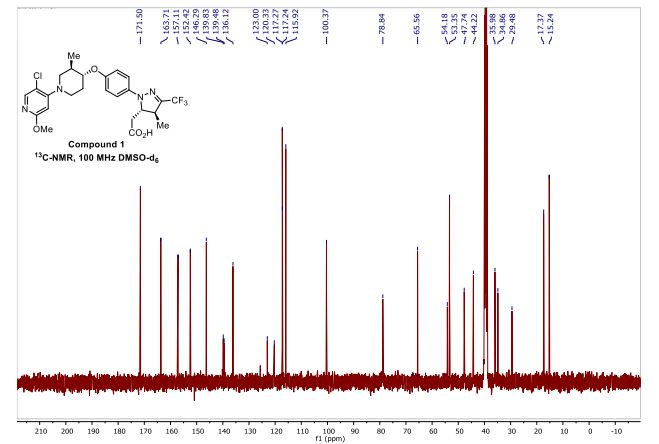
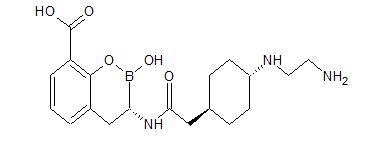
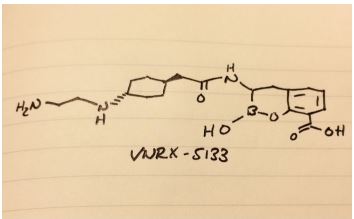
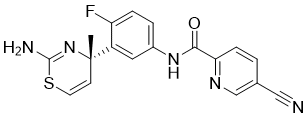
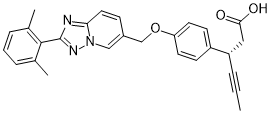
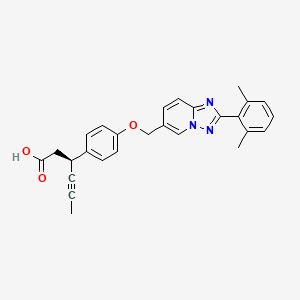
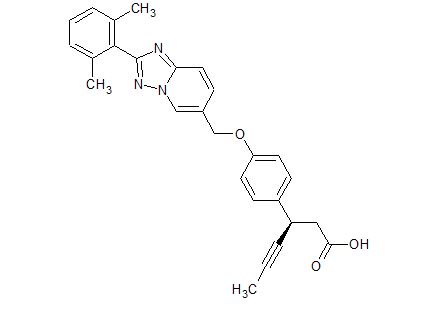
Reporter’s notes: Completing another set of back-to-back presentations on the same target, Watterson revealed another BTK inhibitor also in Phase II clinical trials. Chemists made BMS-986195 in seven steps, and the molecule showed high levels of BTK inactivation in mice. The team aimed to develop an effective compound that required low doses and that had low metabolic degradation.
Patent
WO 2016065226
Inventor Saleem AhmadJoseph A. TinoJohn E. MacorAndrew J. TebbenHua GongQingjie LiuDouglas G. BattKhehyong NguScott Hunter WattersonWeiwei GuoBertrand Myra Beaudoin
Original Assignee Bristol-Myers Squibb Company
https://patents.google.com/patent/WO2016065226A1/en
PATENT
WO 2018045157
https://patentscope.wipo.int/search/en/detail.jsf;jsessionid=E81EF2BDB127473D100AAA55455FC42B.wapp1nA?docId=WO2018045157&recNum=1&maxRec=&office=&prevFilter=&sortOption=&queryString=&tab=PCTDescription








otein kinases, the largest family of human enzymes, encompass well over 500 proteins. Btk is a member of the Tec family of tyrosine kinases, and is a regulator of early B-cell development, as well as mature B-cell activation, signaling, and survival.
B-cell signaling through the B-cell receptor (BCR) leads to a wide range of biological outputs, which in turn depend on the developmental stage of the B-cell. The magnitude and duration of BCR signals must be precisely regulated. Aberrant BCR-mediated signaling can cause dysregulated B-cell activation and/or the formation of pathogenic auto-antibodies leading to multiple autoimmune and/or inflammatory diseases. Mutation of Btk in humans results in X-linked agammaglobulinaemia (XLA). This disease is associated with the impaired maturation of B-cells, diminished immunoglobulin production, compromised T-cell-independent immune responses and marked attenuation of the sustained calcium signal upon BCR stimulation.
Evidence for the role of Btk in allergic disorders and/or autoimmune disease and/or inflammatory disease has been established in Btk-deficient mouse models. For example, in standard murine preclinical models of systemic lupus erythematosus (SLE), Btk deficiency has been shown to result in a marked amelioration of disease progression. Moreover, Btk deficient mice are also resistant to developing collagen-induced arthritis and are less susceptible to Staphylococcus-induced arthritis.
A large body of evidence supports the role of B-cells and the humoral immune system in the pathogenesis of autoimmune and/or inflammatory diseases. Protein-based therapeutics (such as Rituxan) developed to deplete B-cells, represent an important approach to the treatment of a number of autoimmune and/or inflammatory diseases.
Because of Btk’s role in B-cell activation, inhibitors of Btk can be useful as inhibitors of B-cell mediated pathogenic activity (such as autoantibody production).
Btk is also expressed in mast cells and monocytes and has been shown to be important for the function of these cells. For example, Btk deficiency in mice is associated with impaired IgE -mediated mast cell activation (marked diminution of T F-alpha and other inflammatory cytokine release), and Btk deficiency in humans is associated with greatly reduced TNF-alpha production by activated monocytes.
Thus, inhibition of Btk activity can be useful for the treatment of allergic disorders and/or autoimmune and/or inflammatory diseases including, but not limited to: SLE, rheumatoid arthritis, multiple vasculitides, idiopathic thrombocytopenic purpura (ITP), myasthenia gravis, allergic rhinitis, multiple sclerosis (MS), transplant rejection, type I diabetes, membranous nephritis, inflammatory bowel disease, autoimmune hemolytic anemia, autoimmune thyroiditis, cold and warm agglutinin diseases, Evan’s syndrome, hemolytic uremic syndrome/thrombotic thrombocytopenic purpura (HUS/TTP), sarcoidosis, Sjogren’s syndrome, peripheral neuropathies (e.g., Guillain-Barre syndrome), pemphigus vulgaris, and asthma.
In addition, Btk has been reported to play a role in controlling B-cell survival in certain B-cell cancers. For example, Btk has been shown to be important for the survival of BCR-Abl-positive B-cell acute lymphoblastic leukemia cells. Thus inhibition of Btk activity can be useful for the treatment of B-cell lymphoma and leukemia.
In view of the numerous conditions that are contemplated to benefit by treatment involving modulation of protein kinases, it is immediately apparent that new compounds capable of modulating protein kinases such as Btk and methods of using these compounds should provide substantial therapeutic benefits to a wide variety of patients.
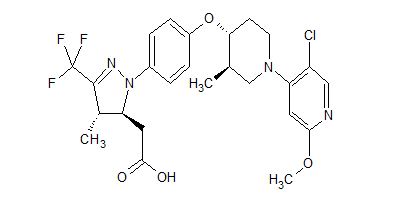
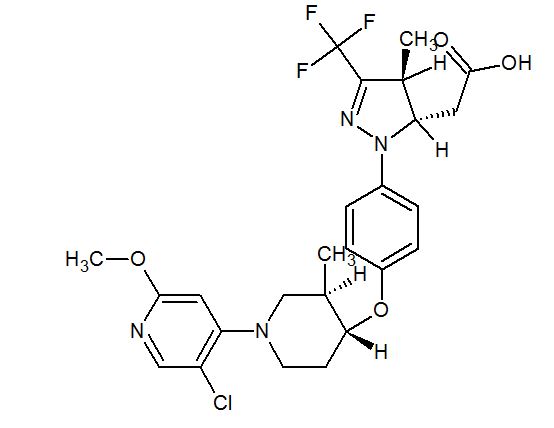
WO 2016/065226 discloses indole carboxamide compounds useful as Btk inhibitors, including (S)-4-(3-(but-2-ynamido)piperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide (Example 223), which has the structure:

Also disclosed is multistep synthesis process for preparing (S)-4-(3-(but-2-ynamido) piperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide.
There are difficulties associated with the adaptation of the multistep synthesis disclosed in WO 2016/065226 to larger scale synthesis, such as production in a pilot plant or a manufacturing plant for commercial production. Further, there is a continuing need to find a process that has few synthesis steps, provides higher yields, and/or generates less waste.
Applicants have discovered a new synthesis process for the preparation of (S)-4-(3-(but-2-ynamido)piperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide that has fewer synthesis steps and/or provides higher yields than the process disclosed in WO 2016/065226. Furthermore, this process contains no metal-catalyzed steps, no genotoxic intermediates, and is adaptable to large scale manufacturing.
EXAMPLE 1
(S)-4-(3-(but-2-ynamido)piperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide

Step 1 : Preparation of Methyl (S)-2-amino-4-(3-((tert-butoxycarbonyl)amino)piperidin-l-yl)-5-fluorobenz
To a 250 mL ChemGlass reactor were charged methyl 2-amino-4,5-difluoro-benzoate (11.21 g, 59.90 mmol), tert-butyl N-[(3S)-3-piperidyl]carbamate (10 g, 49.930 mmol), potassium phosphate, dibasic (10.44 g, 59.94 mmol), and dimethyl sulfoxide (100 mL, 1400 mmol). The resulting thin slurry was heated to 95 to 100 °C and agitated at this temperature for 25 hours. The mixture was cooled to 50 °C. Methanol (100 mL) was added and followed by slow addition of water (50 mL). The mixture was aged at 50 °C for 30 minutes to result in a thick white slurry. Additional water (150 mL) was slowly charged to the above mixture and agitated at 50 °C for 1 hour. The slurry was cooled to 20 °C in 1 hour and aged at this temperature for 4 hours. The slurry was filtrated. The wet cake washed with 25% MeOH in water (30 mL), water (100 mL) and dried under vacuum at 60 °C for 24 h. Methyl (S)-2-amino-4-(3-((tert-butoxycarbonyl)amino) piperidin-l-yl)-5-fluorobenzoate was obtained as a white solid (7 g, yield: 72.5%). ¾ MR (400MHz, METHANOLS) δ 7.34 (d, J=14.6 Hz, 1H), 6.27 (d, J=7.3 Hz, 1H), 3.83-3.71 (s, 3H), 3.68-3.57 (m., 1H), 3.50 -3.40 (m 1H), 3.39 -3.31 (m, 1H), 3.31-3.26 (m, 1H), 2.86-2.70 (m, 1H), 2.64 (t, J=10.0 Hz, 1H), 1.97-1.84 (m, 1H), 1.84-1.74 (m, 1H), 1.73-1.61 (m, 1H), 1.44 (s, 9H), 1.38 (m, 1H). LC-MS [M+H] 368.
Step 2: Preparation of Methyl (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxylate
To a reactor were charged methyl (S)-2-amino-4-(3-((tert-butoxycarbonyl)amino) piperidin-l-yl)-5-fluorobenzoate (5.0 g), DPPOH (diphenyl phosphate, 6.81 g, 2 eq) and 3-hydroxybutanone (1.2 eq, 1.44 g), followed by addition of isopropyl acetate (100 mL, 20 mL/g). The mixture was allowed to warm up to 70 to 75 °C, resulting in a yellow solution. The solution was stirred at 70 to 75 °C for 30 h to complete the cyclization.
Water (2 mL) was added and the mixture was aged at 70 °C over 24 h to remove the Boc group. The mixture was cooled to room temperature. Next, aqueous 20% K3PO4 solution (50 mL) was added and the mixture was stirred for 15 min. The organic layer was separated and washed with water (50 mL). The organic layer was then concentrated under vacuum (200 Torr) to -50 mL. The resulting slurry was stirred at 50 °C for 2 h and then heptane (100 mL) was added over 1 h. The mixture was cooled to room
temperature, stirred for 20 h, and then filtered. The cake was washed with heptane (50 mL). Methyl (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxylate, DPPOH salt was obtained as a light yellow solid. The wet-cake was added to a reactor. Isopropyl acetate (100 mL) was added, followed by addition of aqueous K3PO4 solution (4 g in water 50 mL). The mixture was stirred at room temperature for -half-hour, resulting in a two phase clear solution (pH >10 for aqueous). The organic layer was separated and washed with water (50 mL), and then concentrated under vacuum to a volume of 15 mL. The resulting slurry was stirred at room temperature for 4 h, then heptane (75 mL) was added over 1 h. The mixture was aged at room temperature for 24 h, then concentrated to a volume to -50 mL. The slurry was filtered. The cake was washed with heptane 20 mL and dried under vacuum at 50 °C for 24 h. Methyl (S)-4-(3- aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxylate was obtained as a light yellow solid (2.76 g, yield: 69%). ¾ NMR (400MHz, DMSO-d6) δ 10.64 (s, 1H), 7.33 (d, J=13.7 Hz, 1H), 3.89 (s, 3H), 3.14 (br. m., 1H), 3.07-2.90 (m, 2H), 2.84 (br. m., 1H), 2.70 (br. m., 1H), 2.35 (s, 3H), 2.33 (s, 3H), 1.87 (br. m., 1H), 1.67 (br. m., 3H). LC-MS: M+H= 320.
Alternative Preparation
Step 2: Preparation of ethyl (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxylate trifluoroacetic acid salt
To a reactor were charged ethyl (S)-2-amino-4-(3-((tert-butoxycarbonyl)amino) piperidin-l-yl)-5-fluorobenzoate (1.0 g, limiting reagent), DPPOH (diphenyl phosphate, 1.97 g, 3.0 eq) and 3-hydroxybutanone (1.4 eq, 0.32 g), followed by addition of toluene (20 mL, 20 mL/g). The mixture was allowed to warm up to 80-90 °C, resulting in a yellow solution. The solution was stirred at 80-90 °C for 10 h to complete the
cyclization. Water (0.4 mL, 0.4 ml/g) was added and the mixture was aged at 80-90 °C for 8 hours. The mixture was cooled to room temperature. Next, aqueous 20% K3PO4 solution (15 mL, 15 mL/g) was added and the mixture was stirred for 0.5 hour. The organic layer was separated and the aqueous layer was washed with toluene (7.5 mL, 7.5 mL/g). To combined organic layers water (10 mL, 10 mL/g) was added and the mixture was stirred for 0.5 hour. The organic layer was separated. To the organic layer water (10 mL, 10 mL/g) was added and the mixture was stirred for 0.5 hour. The organic layer was separated. The organic layer was concentrated under vacuum (100 Torr) to 8 mL (8 ml/g). Following concentration the reaction mixture was cooled to 20-25 °C and MTBE (20 mL, 20 mL/g) was added. Trifluoroacetic acid (1.2 eq., 0.36 g) was slowly added to make the salt maintaining temperature at 20-25 °C. The resulting slurry was aged for 4 hours and then filtered. The filtered solids are washed with MTBE (8 mL, 8 mL/g) and the cake
was dried under vacuum at 50 °C. (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxylate trifluoroacetic acid salt was obtained as a white to tan crystalline material (85% yield, 1.0 g). ¾ NMR (400 MHz, DMSO-d6) δ 10.74 (s, 1H), 8.16-7.88 (m, 2H), 7.37 (d, 7=13.6 Hz, 1H), 4.38 (q, 7=7.1 Hz, 2H), 3.18-3.01 (m, 3H), 2.96 (br s, 1H), 2.35 (s, 6H), 2.30 (s, 1H), 2.12 (br d, 7=9.3 Hz, 1H), 1.78 (br s, 2H), 1.45-1.31 (m, 4H), 1.10 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ 165.1, 165.1, 158.4, 158.1, 135.4, 134.7, 134.6, 132.2, 128.8, 128.2, 126.9, 126.8, 118.7, 115.7, 110.6, 110.3,108.7, 108.6, 106.6, 106.5, 83.5, 79.8, 60.5, 54.9, 51.7, 48.7, 47.2, 28.4, 26.8, 23.6, 14.2, 11.1, 10.2
Step 3A: Preparation of (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide
A 40 mL vial was charged with methyl (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxylate (1.5 g, 4.70 mmol), followed by the addition of N,N-dimethylformamide (12.0 mL, 8.0 mL/g). The vial was purged with N2. Formamide (1.49 mL, 37.6 mmol) was added followed by sodium methoxide solution in methanol (35 wt%, 1.29 mL, 3.76 mmol). The resulting solution was heated at 50 °C over 8 hours. The reaction mixture was cooled down to room temperature and the reaction was quenched with water (12.0 mL, 8.0 mL/g). 2-methyltetrahydrofuran (30 mL, 20 mL/g) was added to the mixture. The mixture was shaken vigorously. The layers were separated and the aqueous layer was extracted with 2-methyltetrahydrofuran (15 mL, 10 mL/g) two more times. Organic extracts were then washed with brine and water (15 mL each, 10 mL/g). The organic layer was evaporated. Solids were dried in vacuo at 60 °C to afford (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide as a yellow solid (1.04 g, 69% yield). ¾ NMR (500MHz, DMSO-d6) δ 10.60 (br. s.,
1H), 7.91 (br. s., 1H), 7.40 (d, 7=14.0 Hz, 1H), 7.32 (br. s., 1H), 3.10 (br. s., 1H), 2.98 (br. s., 2H), 2.82 (br. s., 1H), 2.68 (br. s., 1H), 2.34 (br. s., 3H), 2.30 (br. s., 3H), 1.88 (br. s., 1H), 1.67 (br. s., 2H), 1.45 (br. s., 2H), 1.05 (br. s., 1H). LCMS [M+H] 305.24.
Step 3B: Alternative Preparation of (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide
A 100 mL Hastelloy high pressure EasyMax reactor was charged with methyl (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxylate (1.5 g, 4.70 mmol), followed by addition of 7 N ammonia solution in methanol (45.0 mL, 30.0 mL/g) followed by addition of l,3,4,6,7,8-hexahydro-2H-pyrimido[l,2-a]pyrimidine (1.33 g, 9.39 mmol). The reactor was sealed and purged with N2 three times. The reactor was then heated to 80 °C for 24 hrs. The reaction mixture was cooled to room temperature and the vessel contents were purged with N2 three times. Volatiles were concentrated to ~6 mL (4 mL/g) and water (24 mL, 16 mL/g) was added. The yellow precipitate was collected and filtered. The precipitate was washed with methanol/water mixture (20:80 v/v, 6 mL, 4 mL/g), and then water (18 mL, 12 mL/g). The solids were dried in vacuo at 60 °C to afford (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide as a yellow crystalline material (0.93 g, 62% yield). ¾ MR (500MHz, DMSO-de) δ 10.60 (br. s., 1H), 7.91 (br. s., 1H), 7.40 (d, J=14.0 Hz, 1H), 7.32 (br. s., 1H), 3.10 (br. s., 1H), 2.98 (br. s., 2H), 2.82 (br. s., 1H), 2.68 (br. s., 1H), 2.34 (br. s., 3H), 2.30 (br. s., 3H), 1.88 (br. s., 1H), 1.67 (br. s., 2H), 1.45 (br. s., 2H), 1.05 (br. s., 1H). LCMS [M+H] 305.24.
Alternative Preparation:
Step 3C: Preparation of (,S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide 2-butynoic acid salt
Ethyl (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxylate trifluoroacetic acid salt (1.0 g, limiting reagent) and formamide (5 mL, 5 mL/g) were added to a nitrogen inerted reactor. The temperature was maintained at 20-25 °C. To the reactor was added a solution of 20 wt% potassium t-butoxide in THF. The reaction mixture was allowed to sit for 6 hours. To reaction mixture was added Me-THF (15 mL, 15 mL/g) and 12.5 wt % aqueous NaCl (5 mL, 5 mL/g). The reaction mixture was stirred for 0.5 hour. The organic layer was separated, 5 wt% aqueous NaCl (1 mL, 1 mL/g) and 0.25 N aqueous NaOH (4 mL, 4 mL/g) were added, and then stirred for 0.5 hour. The organic layer was separated and 5 wt% aqueous NaCl (5 mL, 5 mL/g) was added, the mixture was stirred for 0.5 hour, and organic phase was separated. The rich organic phase was dried distillation at a pressure of 100 mtorr with Me-THF to obtain KF in 1.5-4wt% range at 5 mL Me-THF volume. The volume was adjusted to 15 mL Me-THF by adding Me-THF (10 mL, 10 mL/g) and EtOH (4 mL, 4 mL/g). Next, 2-butynoic acid (1.0 eq., 0.19 g) was added and the mixture was agitated for 10 hrs. The resulting slurry was filtered. The cake was washed with Me-THF (10 mL, 10 mL/g) and dried under vacuum at 75 °C to afford (,S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide 2-butynoic acid salt (0.7 g, 80% yield) as white crystalline powder. ¾ NMR (400 MHz, DMSO-d6) δ 10.68 (s, 1H), 7.98 (br s, 1H), 7.50-7.32 (m, 2H), 3.32 (br d, J=8.6 Hz, 2H), 3.21 (br t, J=10.5 Hz, 1H), 3.13-2.89 (m, 3H), 2.32 (d, J=5.1 Hz, 5H), 2.11 (br d, J=10.9 Hz, 1H), 1.81-1.67 (m, 4H), 1.55-1.28 (m, 1H).
Step 4A: Preparation of (S)-4-(3-(but-2-ynamido)piperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide
To Reactor-1 was charged N,N-dimethylformamide (DMF, 12.77 kg, 13.5 L). Reactor-1 was purged with N2 to inert. (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide (3.0 kg, 1.0 equiv) was charged followed by 2-butynoic acid (0.854 kg, 1.04 equiv). Reactor-1 was rinsed with DMF (1.42 kg, 1.5 L). The mixture was sparged with N2 for 20 min. Triethylamine (2.99 kg, 3.0 equiv) was charged followed by a DMF rinse (1.42 kg, 1.5 L). TBTU (O-(Benzotriazol-l-yl)-N,N,N’,N’-tetramethyluronium tetrafluorob orate, 3.256 kg, 1.04 equiv) was charged followed by a DMF rinse (1.42 kg, 1.5 L). The reaction mixture was agitated for 1.5 h at 20 °C. MeTHF (46.44 kg, 60 L) was charged to the batch. The reaction was quenched with LiCl (20 wt%, 26.76 kg, 24 L) at 20 °C. The bottom aqueous layer was discharged as waste. The organic layer was washed with 2N HCl solution (24.48 kg, 24 L), 10 wt% sodium bicarbonate solution (25.44 kg, 24 L) and deionized water (24.0 kg, 24 L). THF (26.61 kg, 30 L) was charged into Reactor-1. The rich organic stream in MeTHF/TFIF was polish filtered. The stream was distilled down to 15 L at 75-100 Torn Constant volume distillation was carried out at 15 L with THF feed (39.92 kg, 45 L). The stream was heated to 60 °C for 1 hr and cooled to 50 °C. MTBE (33.30 kg, 45 L) was charged slowly over 2 h. The slurry was aged at 50 °C for 4 h and cooled to 20 °C over 2 h, and aged at 20 °C for >2 h. The 1st drop slurry was filtered and was rinsed with MTBE (8.88 kg, 12 L) twice. Wet cake was dried under vacuum 60 to 70 °C at 25 mbar overnight (>15 h). Reactor-1 was thoroughly cleaned with IPA. The dry cake was charged into Reactor-1 followed by the charge of IPA (47.10 kg, 60 L). The batch was heated to 60 °C to achieve full dissolution and cooled to 40 °C. Rich organic (24 L) was transferred to Reactor-2 for crystallization. The stream was distilled at 24 L constant volume and 100 mbar using remaining rich organic from reactor-1 as distillation feed. Following distillation completion, the batch was heated to 60 °C, aged at 60 °C for 2 h, cooled to 20 °C over 2 h, and aged at 20 °C over 2 h. The slurry was filtered. IPA (1.18 kg) was used to rinse the reactor and washed the cake. The wet cake was dried under vacuum at 70 °C and 25 mbar for >15 h. The dry cake (2.196 kg, 63.2% yield) was discharged as an off-white crystalline solid. ¾ NMR (400MHz, DMSO-d6): δ 10.62 (s, 1H), 8.48 (d, J= 7.1 Hz, 1H), 7.91 (s, 1H), 7.39 (d, J=7.4 Hz, 1H), 7.33 (s, 1H), 3.88 (m, 1H), 3.11 (t, J= 8.0 Hz, 1H), 3.0 (m, 1H), 2.96 (m, 1H), 2.78 (t, J= 10.0 Hz, 1H), 2.35 (s, 3H), 2.30 (s, 3H), 1.92 (s, 3H), 1.86 (m, 1H), 1.31 (m, 1H), 1.70 (m, 2H); 13C NMR (400 MHz, DMSO-d6): δ 168.2, 153.2, 151.9, 134.4, 133.2, 132.1, 126.5, 112.3, 108.4, 106.0, 82.3, 75.7, 56.9, 51.9, 46.3, 29.7, 24.4, 11.1, 10.2, 3.0; LC-MS: M+H= 371.2.
Step 4B: Alternative preparation of (S)-4-(3-(but-2-ynamido)piperidin-l-yl)-5-fluoro-2,3-dimeth -lH-indole-7-carboxamide
To Reactor-1 was charged N,N-dimethylformamide (DMF 4.5 mL, 4.5 mL/g). Reactor-1 was purged with N2 to inert. (,S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide 2-butynoic acid salt (1.0 g, limiting reagent) was charged followed by 2-butynoic acid (0.065g, 0.3 equiv.). The mixture was inerted with N2 for 20 min. N-methylmorpholine (0.78 g, 3.0 equiv) was charged. Next,
diphenylphosphinic chloride (0.79 g, 1.3 equiv) was charged over 0.5 h while maintaining the reaction temperature at 20-25 °C. The reaction mixture was agitated for 1.5 hour at 20 °C. Me-THF (14 mL, 14 mL/g) was charged to the reaction mixture. The reaction was quenched with the addition of aqueous NaCl (12.5 wt%, 6 mL, 6 mL/g) at 20 °C. The bottom aqueous layer was discharged as waste. Aqueous NaCl (12.5 wt%, 6 mL, 6 mL/g) at 20 °C was added to the organic layer, stirred for 0.5 hour and the bottom aqueous layer was discharged to waste. Deionized water (6 mL, 6 mL/g) was charged to the organic layer, stirred for 0.5 hour and the bottom aqueous layer was discharged to waste. THF (8 mL, 8 mL/g) was charged into Reactor-1 and the mixture was
concentrated under vacuum to remove Me-THF and water, and reconstituted in 4 L/kg of THF. The mixture was heated to 60 °C and stirred for 1 hour; the temperature was reduced to 50 °C and MTBE (12 mL, 12 mL/g) was added. The mixture was aged for 4 hours while maintaining the temperature of 50 °C and then cooled to room temperature. The solids were filtered and washed with MTBE (6.5 mL, 6.5 mL/g). The solids of crude were dried at 70 °C under vacuum for 12 hours.
Crude (S)-4-(3-(but-2-ynamido)piperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide was charged to Reactor-2, followed by THF (12 mL, 12 mL/g). The mixture was stirred for 0.5 hour. The solution was polish filtered. The solution was concentrated under vaccuum to remove THF and reconstituted in EtOH (7 mL, 7 mL/g). (S)-4-(3-(but-2-ynamido)piperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide seeds (0.01 g, 0.01 g/g) were added, the mixture was heated to 60 °C and aged for 2 hours, n-heptane (21 mL, 21 mL/g) was added slowly over 4 hours. The mixture was aged for additional 2 hours at 60 °C, followed by cooldown to room temperature. The slurry was filtered, washed with n-heptane (6 mL, 6 mL/g), and dried under vacuum at 70 °C for 12 hours. The dry cake (0.68 g, 71% yield) was discharged as an off-white crystalline solid. ¾ NMR (400MHz, DMSO-d6): δ 10.62 (s, 1H), 8.48 (d, J= 7.1 Hz, 1H), 7.91 (s, 1H), 7.39 (d, J=7.4 Hz, 1H), 7.33 (s, 1H), 3.88 (m, 1H), 3.11 (t, J= 8.0 Hz, 1H), 3.0 (m, 1H), 2.96 (m, 1H), 2.78 (t, J= 10.0 Hz, 1H), 2.35 (s, 3H), 2.30 (s, 3H), 1.92 (s, 3H), 1.86 (m, 1H), 1.31 (m, 1H), 1.70 (m, 2H); 13C MR (400 MHz, DMSO-d6): δ 168.2, 153.2, 151.9, 134.4, 133.2, 132.1, 126.5, 112.3, 108.4, 106.0, 82.3, 75.7, 56.9, 51.9, 46.3, 29.7, 24.4, 11.1, 10.2, 3.0; LC-MS: M+H= 371.2.
Applicants have discovered a new synthesis process for the preparation of (S)-4- (3-(but-2-ynamido)piperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide which offers significant advantages.
The new synthesis process utilizes fewer synthesis steps (4 vs 8) than the process disclosed in WO 2016/065226.
Additionally, the process of the present invention provided (S)-4-(3-(but-2-ynamido)piperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide at an overall
yield of 22% (step 1 : 73.%, step 2: 69%, step 3 : 69%, step 4: 63%). In comparison, (S)-4-(3-(but-2-ynamido)piperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide was prepared according to the process of WO 2016/065226, which provided (S)-4-(3-(but-2-ynamido)piperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide at an overall yield of 2.9% yield (step 1 : 91%, step 2: 71%, step 3 : 35%, step 4: 88%, step 5: 80%, step 6: 29%, step 7: 99%, step 8: 63%).
Furthermore, the process of the present invention does not include any transition metal-catalyzed steps, no genotoxic intermediates, and is adaptable to large scale manufacturing. In comparison, the process disclosed in WO 2016/065226 employed lead (Pb) in process step (8) and included a potentially genotoxic hydrazine intermediate in process step 8.
The process of the present invention has an estimated manufacturing cycle time of approximately 6 months versus a estimated manufacturing cycle time of approximately 12 months for the process disclosed in WO 2016/065226.
REFERENCE

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....












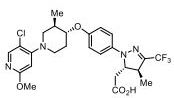
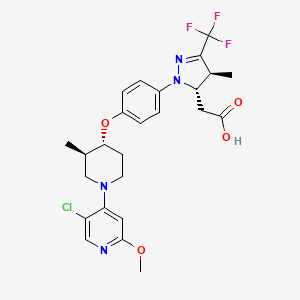



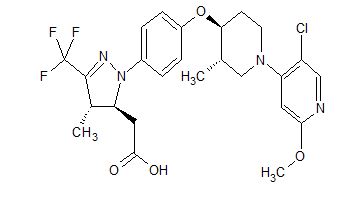






 Rick Ewing, Head, External Partnerships, Discovery Chemistry and Molecular Technologies at Bristol-Myers Squibb
Rick Ewing, Head, External Partnerships, Discovery Chemistry and Molecular Technologies at Bristol-Myers Squibb