
Home » 2016 (Page 46)
Yearly Archives: 2016
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
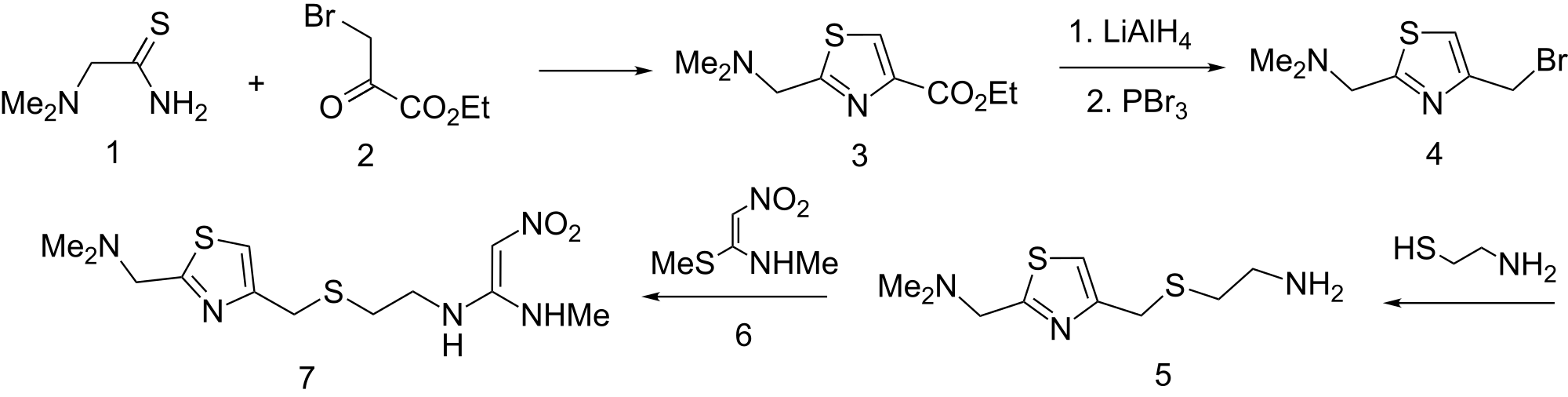
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Zydus Cadila, New Patent,US 20160039759, PERAMPANEL

PERAMPANEL

Zydus Cadila, New Patent,US 20160039759, PERAMPANEL
(US20160039759) PROCESS FOR THE PREPARATION OF PERAMPANEL
CADILA HEALTHCARE LIMITED
Sanjay Jagdish DESAI
Jayprakash Ajitsingh Parihar
Kuldeep Natwarlal Jain
Sachin Ashokrao Patil

Perampanel, a non-competitive AMPA receptor antagonist, is the active ingredient of FYCOMPA® tablets (U.S) which is approved as an adjunctive therapy for the treatment of partial on-set seizures with or without secondarily generalized seizures in patients with aged 12 years and older. Chemically, Perampanel is 5′-(2-cyanophenyl)-1′-phenyl-2,3′-bipyridinyl-6′(1′H)-one, with an empirical formula C23H15N30 and molecular weight 349.384 g/mol which is represented by Formula (I).

U.S. Pat. No. 6,949,571 B2 discloses perampanel and its various processes for preparation thereof.
U.S. Pat. No. 7,759,367 B2 discloses the pharmaceutical composition of perampanel and an immunoregulatory agent and their uses.
U.S. Pat. No. 8,304,548 B2 discloses the reaction of 5′-bromo-1′-phenyl-[2,3′-bipyridin]-6′(1′H)-one with 2-(1,3,2-dioxaborinan-yl)benzonitrile in the presence of palladium compound, a copper compound, a phosphorus compound and a base to form perampanel of Formula (I). Also discloses the crystalline hydrate, anhydrous crystal Form I, anhydrous crystal Form III, & anhydrous crystal Form V of perampanel of Formula (I).
U.S. Pat. No. 7,803,818 B2 discloses an amorphous form of perampanel. U.S. Pat. No. 7,718,807 B2 discloses salts of perampanel. International (PCT) publication No. WO 2013/102897 A1 discloses anhydrous crystalline Form III, V & VII of perampanel.
U.S. PG-Pub. No. 2013/109862 A1 discloses the method for preparing 2-alkoxy-5-(pyridin-2-yl)pyridine, which is an intermediate for preparing perampanel key starting material 5-(2′-pyridyl)-2-pyridone.
U.S. Pat. No. 7,524,967 B2 discloses the preparation of 5-(2′-pyridyl)-2-pyridone, an intermediate in the preparation perampanel.
International (PCT) publication No. WO 2014/023576 A1 discloses the preparation of cyanophenyl boronic acid, an intermediate in the preparation perampanel.
The prior-art processes suffer with problems of poor yield and requirement of chromatographic purification or series of crystallizations which further reduces the overall yield of the final product, which is overcome by the process of the present invention.









 Pankaj Patel, chairman, Zydus Cadila
Pankaj Patel, chairman, Zydus Cadila
EXAMPLES
The present invention is further illustrated by the following examples which is provided merely to be exemplary of the invention and do not limit the scope of the invention. Certain modification and equivalents will be apparent to those skilled in the art and are intended to be included within the scope of the present invention.
Example-A: Preparation of 5-(2-pyridyl)-1,2-dihydropyridin-2-one In a 500 mL round bottom flask, equipped with a mechanical stirrer, thermometer and an addition funnel, a solution of 188.80 g 5-bromo-2-methoxypyridine in 190 mL tetrahydrofuran and 12.92 g pyridine-2-yl boronic acid were added and refluxed. The reaction mixture was cooled to 25-30° C. and aqueous solution of hydrochloric acid was added and stirred for 1 hour. The reaction mixture was neutralized with aqueous sodium hydroxide and extracted with tetrahydrofuran.
The organic layer was washed with saline water, dried over anhydrous magnesium sulfate, and then evaporated to obtain the titled compound.
Example-1
Preparation of 3-bromo-5-(2-pyridyl)-1,2-dihydropyridin-2-one
In a 2 L round bottom flask, equipped with a mechanical stirrer, thermometer and an addition funnel, 201.5 g 5-(2-pyridyl)-1,2-dihydropyridin-2-one, 208.3 g N-bromosuccinimide and 1300 mL N,N-dimethylforamide were stirred at 25-30° C. for 2-3 hours. After completion of the reaction, the reaction mixture was poured into water and stirred for 30 min. The precipitate was filtered, washed with N,N-dimethylforamide and dried at 50° C. to obtain 230 g title compound.
Example-2
Preparation of 3-bromo-5-2-pyridyl)-1-phenyl-1,2-dihydropyridine-2-one
In a 500 mL round bottom flask, equipped with a mechanical stirrer, thermometer and an addition funnel, a solution of 18.75 g 3-bromo-5-(2-pyridyl)-1,2-dihydropyridin-2-one in 300 mL methylene dichloride, 18.36 g 1-phenyl boronic acid, 3.47 g palladium triphenylphosphine and 10 mL triethyl amine were added and the reaction mixture was stirred for 1 hour at 25-35° C. The reaction mixture was filtered and the filtrate was evaporated to dryness. The residue was crystallised from ethyl acetate to obtain the title compound.
Example-3
Preparation of Perampanel
In a 1 L round bottom flask, equipped with a mechanical stirrer, thermometer and an addition funnel, a suspension of 188 g 3-bromo-5-(2-pyridyl)-1-phenyl-1,2-dihydropyridine-2-one, 161.2 g 2-(1,3,2-dioxaborinan-2-yl)benzonitrile, 3.0 g tetrakis(triphenylphosphine)-palladium(0), 10 mL triethylamine (10 mL) in 300 mL methylene dichloride were stirred at 25-30° C. for 12 hours. To the reaction mixture was added 5 mL conc. aqueous ammonia, 10 mL water and 40 mL ethyl acetate. The separated organic layer was washed with water and saturated saline solution and dried over magnesium sulfate. The solvent was removed under vacuum. Ethyl acetate was added to the residue and heated obtain clear solution. n-hexane was added to this solution and cooled to 25-30° C. The obtained solid was filtered and washed with ethyl acetate and dried to obtain perampanel.
Example-4
Preparation of 3-Bromo-5-(2-pyridyl)-1,2-dihydropyridin-2-one
In a 2 L round bottom flask, equipped with a mechanical stirrer, thermometer and an addition funnel, 100 g 5-(2-pyridyl)-1,2-dihydropyridin-2-one, 108.5 g N-bromosuccinimide and 500 mL N,N-dimethylforamide were stirred at 30-35° C. for 3 hours. 100 mL water was added to the reaction mixture at 5-15° C. and stirred at 30-35° C. for 1 hour. The solid obtained was filtered, washed with water and dried to obtain 129 g 3-bromo-5-(2-pyridyl)-1,2-dihydropyridin-2-one.
Example-5
Preparation of 3-bromo-5-(2-pyridyl)-1-phenyl-1,2-dihydropyridine-2-one
In a 2 L round bottom flask, equipped with a mechanical stirrer, thermometer and an addition funnel, 100 g 3-bromo-5-(2-pyridyl)-1,2-dihydropyridin-2-one, 72.8 g phenylboronic acid and 500 mL N,N-dimethylformamide were added at 30-35° C. and stirred. 11.9 g copper acetate and 15.7 g pyridine were added and air was purged into the reaction mixture and stirred for 16 hours at 30-35° C. After the completion of the reaction, the reaction mixture was poured into 1200 mL aqueous ammonia at 10-15° C. and stirred for 2 hours at 30-35° C. The obtained solid was filtered, washed with water and dried to obtain 120 g 3-bromo-5-(2-pyridyl)-1-phenyl-1,2-dihydropyridine-2-one.
Example-6
Purification of 3-bromo-5-(2-pyridyl)-1-phenyl-1,2-dihydropyridine-2-one
In a 1 L round bottom flask, equipped with a mechanical stirrer, thermometer and an addition funnel, 100 g 3-bromo-5-(2-pyridyl)-1-phenyl-1,2-dihydropyridine-2-one and 500 mL isopropyl alcohol were stirred at 60-65° C. for 30 min. The reaction mixture was cooled to 20-25° C. and stirred for 30 min. The reaction mixture was filtered, washed with isopropanol and dried to obtain 96 g pure 3-bromo-5-(2-pyridyl)-1-phenyl-1,2-dihydropyridine-2-one.
Example-7
Preparation of Perampanel
In a 1 L round bottom flask, equipped with a mechanical stirrer, thermometer and an addition funnel, 100 g 3-bromo-5-(2-pyridyl)-1-phenyl-1,2-dihydropyridine-2-one and 125 g 2-(1,3,2-dioxaborinan-2-yl)benzonitrile and 1500 mL N,N-dimethylformamide were added under inert atmosphere. 44 g potassium carbonate and 4.2 g palladium tetrakis were added and stirred at 115-125° C. for 3 hours. The solvent was removed under vacuum. Ethyl acetate was added to the residue and the organic layer was distilled off to obtain perampanel (78 g).

////////Zydus Cadila, New Patent,US 20160039759, PERAMPANEL
Benfotiamine, бенфотиамин , بينفوتيامين , 苯磷硫胺 , ベンフォチアミン

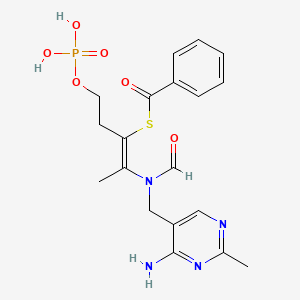
Benfotiamine
S-[(Z)-2-[(4-amino-2-methylpyrimidin-5-yl)methyl-formylamino]-5-phosphonooxypent-2-en-3-yl] benzenecarbothioate
Benphothiamine; Betivina; Biotamin; Neurostop; Nitanevril; cas 22457-89-2
C19H23N4O6PS MF
466.447882 g/mol MW
- Benzenecarbothioic acid, S-[2-[[(4-amino-2-methyl-5-pyrimidinyl)methyl]formylamino]-1-[2-(phosphonooxy)ethyl]-1-propenyl] ester (9CI)
- Benzoic acid, thio-, S-ester with N-[(4-amino-2-methyl-5-pyrimidinyl)methyl]-N-(4-hydroxy-2-mercapto-1-methyl-1-butenyl)formamide dihydrogen phosphate (ester) (8CI)
- Formamide, N-[(4-amino-2-methyl-5-pyrimidinyl)methyl]-N-(4-hydroxy-2-mercapto-1-methyl-1-butenyl)-, S-benzoate O-(dihydrogen phosphate) (8CI)
- 8088CB
- BTMP
- Benfothiamine
- Benfotiamine
- Benzoylthiamine O-monophosphate
- Benzoylthiamine monophosphate
- Berdi
- Betivina
- Bietamine
- Biotamin
- Milgamma
- N-[(4-Amino-2-methyl-5-pyrimidinyl)methyl]-N-(4-hydroxy-2-mercapto-1-methyl-1-butenyl)formamide S-benzoate O-phosphate
- Neurostop
- Nitanevril
- S-Benzoylthiamine O-monophosphate
- S-Benzoylthiamine monophosphate
- Tabiomyl
- Vitanevril
- EINECS:245-013-4
- LD50:2200 mg/kg (M, i.v.); 15 g/kg (M, p.o.)
- ATC:A11DA03
Benfotiamine (rINN, or S-benzoylthiamine O-monophosphate) is a synthetic S-acyl derivative of thiamine (vitamin B1).
It has been licensed for use in Germany since 1993 under the trade name Milgamma. (Combinations with pyridoxine or cyanocobalamin are also sold under this name.) It is prescribed there for treating sciatica and other painful nerve conditions.[1]
It is marketed as a medicine and/or dietary supplement, depending on the respective Regulatory Authority.[citation needed]


Uses
Benfotiamine is primarily marketed as an antioxidant dietary supplement. In a clinical study with six patients, benfotiamine lowered AGE by 40%.[2]
Benfotiamine may be useful for the treatment of diabetic retinopathy, neuropathy, and nephropathy however “Most of the effects attributed to benfotiamine are extrapolated from in vitro and animal studies. Unfortunately apparent evidences from human studies are scarce and especially endpoint studies are missing. Therefore additional clinical studies are mandatory to explore the therapeutic potential of benfotiamine in both diabetic and non-diabetic pathological conditions”.[3] It is thought that treatment with benfotiamine leads to increased intracellular thiamine diphosphate levels,[3] a cofactor of transketolase. This enzyme directs advanced glycation and lipoxidation end products (AGE’s, ALE’s) substrates to the pentose phosphate pathway, thus reducing tissue AGEs.[4][5][6][7][8]


Pharmacology
After absorption, benfotiamine can be dephosphorylated by cells bearing an ecto-alkaline phosphatase to the lipid-soluble S-benzoylthiamine.[9] Benfotiamine should not be confused with allithiamine, a naturally occurring thiamine disulfide derivative with a distinct pharmacological profile.[10]
PATENT
The Benfotiamine, disclosed in US pat. no. 19623064000 US english names: S-benzoylthiamine O-monophosphate common name: Benfotiamine, chemical name: S − 2-[ [ (2-methyl-4-amino-5-pyrimidinyl) methyl ]-propionylamino ]-5-phosphonato-2-pentene-3-thiol benzoate, formula C 19 H 23 N 406 PS molecular weight 466.45 the following structural formula:

Chemical composition of the same species, in various physico-chemical conditions, crystallization into two or more different structure of the crystalline phenomenon, also referred to as polymorphs or homogeneous an image drug polymorph is a common phenomenon of drug discovery, drug quality is an important factor. Various polymorphs have different physical properties such as appearance, melting point, hardness, dissolution rate, chemical stability, mechanical stability, etc. differences, these differences in the physical properties of the sometimes affect the stability of the drug, bioavailability, even the drug availability. Thus, in drug development, it should be fully considered drug poly-type problems, the type of study and control in drug development of significant research content.
The benfotiamine, vitamin B 1 lipid-soluble derivatives, improved water-soluble vitamins B1 low bioavailability of disadvantages, increased blood and tissues. Thiamine concentration, thereby enhancing efficacy. The primary application to the following aspects (1) for thiamine deficiency disease prevention and treatment; (2) vitamin B 1 demand increases, from the food uptake is not sufficient make-up, fatigue, hyperthyroidism, gestation, lactation, vigorous manual labor, etc.); (3) for the treatment of non-l 酒性 lopinavir, grams of brain disease; (4) for the treatment of foot disease; (5) for the disease, the speculative and thiamine deficiency and metabolic disorders associated with treatment, such as: neuropathic pain; muscle pain, joint pain ; Peripheral-inflammatory, peripheral nerve
The paralysis; myocardial metabolism disorders, constipation, gastrointestinal motility dysfunction. The benfotiamine as vitamin B 1 supplemental agents have been in the united states, japan, europe, etc worldwide market. Recent studies have shown that, benfotiamine in diabetic peripheral neuropathy and retinopathy of significant therapeutic effect. In addition, our studies, benfotiamine may also be applied to the prevention and treatment of alzheimer’s disease, and aging.
Alzheimer’s disease (Altheimer’s disease, AD) is a cognitive, behavioral disorders is the primary clinical manifestations progressive neurodegenerative diseases, an age-related disorders, with age, their prevalence is a significant rise. 我国 the number of people in excess of 600 million AD patients, it is contemplated that in 2050 worldwide by the year AD patient may exceed 3000 million people as the medical scientific development, severe affect human health, mortality is a leading significant diseases such as cancer, stroke, cardiovascular disease, exhibit a decrease in mortality year by year, and AD mortality the rendering large increase in . In addition, alzheimer’s disease course long, the disabling rate is high, thus, alzheimer’s disease will be the 21 st century threaten both human diseases the most serious. It is estimated that worldwide by the year AD 2010 for medical costs up to 6040 of millions of dollars, the same global of the gross national product of 1%
China and the USA, the world there have been the following two classes of drugs approved for AD treatment: cholinesterase inhibitors and N-methyl D-aspartate (NMDA) receptor antagonist are both improved AD patient symptoms, slow disease progression does not prevent or reverse the progression of a disease. The benfotiamine by inhibiting the sugar synthase kinase -3 (Glycogen synthase kinase -3, GSK -3) activity, decrease in brain beta-amyloid protein (beta-amyloid, alpha beta) the deposition and tau protein phosphorylation, reduce alzheimer’s disease, pathological damage.
Presently available, benfotiamine primarily in the form of tablets and powders is administered in the form of, all formulations are not related to the benfotiamine feedstock form has not yet been the benfotiamine crystalline be systematically studied, the present US pat. no. first for benfotiamine of systematic study of various forms, illustrating different form benfotiamine characteristics and their feasibility. As a pharmaceutical agent
Thiamine derivatives such as thiamine monophosphate dihydrate and S-benzoyl thiamine monophosphate compounds and salts thereof are useful as therapeutics and nutrients. Further, some of the thiamine derivatives are known to be biochemically important compounds.
There are processes known where thiamine derivatives have been prepared by reacting thiamine with polyphosphoric acid derived from phosphoric acid; heating of the two gives a mixture of thiamine phosphate comprising thiamine 0- monophosphate, thiamine O-diphosphate, thiamine O-triphosphate and thiamine O-polyphosphate. Each polyphosphate may be isolated from the reaction mixture. However, the above prior process is commercially unpractical, because yields of the desired phosphates isolated are extremely poor.
In another process, orthophosphoric acid is used as phosphorylating agent to convert thiamine hydrochloride into thiamine monophosphate. The process demands heating acids like orthophosphoric acid which is considered to be highly unsafe up to 270° C. Moreover, this prior art process does not produce the desired product immediately after work up. The process requires 7 days to get all higher phosphates derivatives eq. Thiamine diphosphate (cocarboxylase) and thiamine triphosphate to convert into thiamine monophosphate. The prepared thiamine monophosphate dihydrate is converted into S-benzoyl thiamine monophosphate by reaction with benzoyl chloride or dibenzyl sulfide or Sodium benzoyl thiosulfate. Due to such drastic conditions the intermediate purity and in turn the product purity is extremely poor requiring series of purification steps.
In yet another process, compounds like P205 which are unsafe from operation point of view since it requires changing into orthophosphoric acid at considerably high temperature. This process gives the mixture of mono, di, tri and tetra phosphate derivatives of thiamine. The process provides that even after hydrolysis, the reaction mass contains only 61 -62% desired thiamine monophosphate dihydrate along with 31 -32 % cocarboxylase and 2-3 % thiamine triphosphate. Further, it requires their separation by means of techniques like Ion Exchange which is again troublesome and not advantageous from process point of view.
Thus, the processes known hitherto for the production of such thiamine derivatives such as thiamine monophosphate dihydrate and S-benzoyl thiamine monophosphate compounds and salts thereof have cost and/or lower yield with poor quality disadvantages. The prior art processes also suffer in that the workup of reaction mass is tedious which eventually increases the manufacturing cost.

PATENT
Preparation of Thiamine monophosphate
To the solution of 2500 g of Polyphosphoric acid and 25 g sodium pyrophosphate was added 700 g thiamine hydrochloride chloride and the mixture was heated slowly at 120 Deg C. After the ceasing of HCI gas evolution, which generally takes 3-4 hours. It was maintained for 2 hours and was further cooled to 50 Deg C. HPLC analysis shown the following analysis 7% of cocarboxylase ( thiamine diphosphate)
88.0% of thiamine monophosphate
1 .0% of thiamine triphosphate
1 .5% of thiamine chloride After cooling hot deminerahzed water was added and the reaction mass heated to 90 Deg C. The reaction mass was maintained at this temperature for 5-6 hours.
HPLC analysis of reaction mass shown the following analysis. 1 % of cocarboxylase
95% of thiamine monophosphate
1 .0% of thiamine chloride The reaction mass is allowed to cool up to 25 Deg C. To this was added 4000 ml Tri-n-butyl amine and 5000 ml chloroform. The two layer formed was separated and product was recovered from aqueous layer by adding 5000 ml methanol by filtration. Dry weight of thiamine monophosphate dihydrate was 862 g (almost 100 % yield ) of purity 99 % by HPLC. Melting point 198-200 Deg C.
EXAMPLE 2
Preparation of S-benzoyl thiamine monophosphate 100 g thiamine monophosphate dihydrate was added to 300 ml water and cooled up to 0 to 5 Deg C. 10 % caustic solution is run in to this solution to make pH 8 – 10. 75 g of benzoyl chloride was added drop wise to the mixture with stirring within 4 hours and maintained the reaction mixture alkaline by occasional addition of 25% aqueous sodium hydroxide. After the completion of reaction, the mass is concentrated to dryness and the product was isolated by adding acetone. The precipitated solid was filtered and dried. Dry weight of product was 75 g.
EXAMPLE 3 Preparation of Lithium salt of S-benzoyl thiamine monophosphate
To a mixture of 12 g of S-benzoyl thiamine O-monophosphate dihydrate with 35 ml of water is added with stirring and ice cooling a 10% solution of lithium hydroxide to adjust pH to about 8.0. The resulting solution is filtered, added with acetone and allowed to stand at cold place to precipitate crystals of Lithium salt of S-benzoyl thiamine O-monophosphate. The crystals are filtered and dissolved in a small amount of water. Acetone is then added to the solution to give a recrystallization of purified product, which is dried in vacuum oven. Yield -9 g and MP – decomposes at about 190°C. EXAMPLE 4
Preparation of Barium salt of S-benzoyl thiamine monophosphate
To a mixture of 15 g of S-benzoyl thiamine O-monophosphate dihydrate with 35 ml of water is added with stirring and ice cooling a 10% solution of Barium hydroxide to adjust pH to about 8.0. The resulting solution is filtered, added with acetone and allowed to stand at cold place to precipitate crystals of Barium salt of S-benzoyl thiamine O-monophosphate. The crystals are filtered and dissolved in a small amount of water. Acetone is then added to the solution to give a recrystallization of purified product, which is dried in vacuum oven. Yield 9 g and melt with decomposition at 180 Deg C. EXAMPLE 5
Preparation of Magnesium salt of S-benzoyl thiamine monophosphate
To a mixture of 15 g of S-benzoyl thiamine O-monophosphate dihydrate with 35 ml of water is added with stirring and ice cooling a 10% solution of Magnesium hydroxide to adjust pH to about 8.0. The resulting solution is filtered, added with acetone and allowed to stand at cold place to precipitate crystals of Magnesium salt of S-benzoyl thiamine O-monophosphate. The crystals are filtered and dissolved in a small amount of water. Acetone is then added to the solution to give a recrystallization of purified product, which is dried in vacuum oven. Yield 9 g and melt with decomposition at 205 Deg C.
WO 2014059702
PATENT
IN 2014MU03690
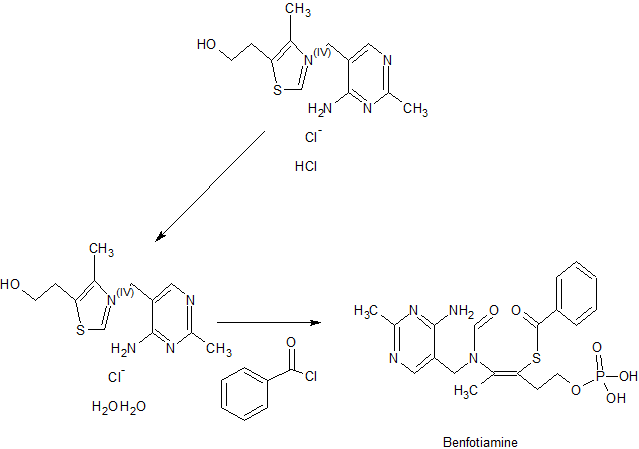
PATENT
CN 104418889
CN 103772432
PATENT

http://www.google.com/patents/CN103772432A?cl=en
Example 1:
Was added to the reaction kettle 4000kg polyphosphoric acid, heated to 100 ~ 120 ° C, the vitamin BI 1000kg batches added to the reaction dad, add after kept at this temperature range 8 hours, was added water quenching 3000kg off after the reaction, the temperature was raised to 80-90 ° C hydrolysis of 10 hours; cooled to room temperature, was added to the kettle 5000kg trioctylamine mixture of methyl tert-butyl ether = WPA / 1/1; aqueous phase 5000kg methanol to precipitate a solid, centrifuged to obtain a monoester 1200kg vitamin BI phosphoric acid crude; the 1200kg Vitamin `prime BI phosphate monoester crude in 6000kg water mixed beating, down to O ~ 5 ° C, dropping liquid in this temperature range adjusting the PH value of the base system to 12.0 ~ 14.0; PH after adjustment to ensure that the reactor temperature 10 ~ 25 ° C within 1200kg of benzoyl chloride was added dropwise, after the addition is complete heat the reaction to completion; filtered and the filtrate adjust PH from 3.5 to 4.0 precipitated solid was isolated and dried to give a white solid 1200kg, namely benfotiamine. Yield: 77.38%, Purity: 98.70% ο
Example 2:
Was added to the reaction kettle 5000kg polyphosphoric acid, heated to 80 ~ 100 ° C, the vitamin BI 1000kg batches added to the reaction dad, add after kept at this temperature range 6 hours, was added water quenching 5000kg off after the reaction was heated to reflux for 5 hours hydrolysis; cooled to room temperature, the autoclave was added to the mixture was extracted twice 4000kg trioctylamine / methyl tert-butyl ether = 1/1; aqueous phase 6000kg ethanol precipitation The solid obtained by centrifugation vitamin BI phosphate monoester 1200kg crude; after 1200kg vitamin BI crude phosphate monoester product mixing beating in 6000kg water, down to O ~ 5 ° C, solution of caustic soda adjust PH value system in this temperature range to 10.0 ~ 12.0; PH adjusting finished, to ensure the reactor temperature 10 ~ 25 ° C within 1200kg of benzoyl chloride was added dropwise, after the addition is complete heat the reaction to completion; filtered, the solid was filtered, the filtrate was adjusted to 3.5 ~ PH value 4.0 precipitated solid was isolated and dried to give a white solid 1250kg, namely benfotiamine. Yield: 80.61%, Purity: 98.50% ο
Example 3:
After the reactor was added 3000kg polyphosphoric acid, heated to 90 ~ 110 ° C, the vitamin BI 1000kg batches added to the reaction dad, add after the insulation in this temperature range for 5 hours, 5000kg of water quenching off after the reaction, the temperature was raised to 90-100 ° C hydrolysis 5 hours; cooled to room temperature, was added to the kettle 5000kg trioctylamine methyl tert-butyl ether mixture was extracted twice = / 1/1; aqueous phase Join 7000kg acetone precipitate a solid, mono- 1230kg centrifuged to obtain crude vitamin BI phosphoric acid; vitamin BI after 1200kg crude phosphate monoester product mixing beating in 6000kg water, down to O ~ 5 ° C, solution of caustic soda adjusted within this temperature range System PH value to 11.0 ~ 13.0; PH after adjustment to ensure that the temperature of the reactor was added dropwise within 10 ~ 25 ° C within 1200kg benzoyl chloride, and after the addition is complete heat to the completion of the reaction; filtered, the filtrate was adjusted to 3.5 PH value to 4.0 precipitated solid was isolated and dried to give a white solid 1240kg, namely benfotiamine. Yield: 79.96%, Purity: 98.50% ο
Example 4
Was added to the reaction kettle 4000kg polyphosphoric acid, heated to 100 ~ 120 ° C, the vitamin BI 1000kg batches added to the reaction dad, add after kept at this temperature range for 4 hours, water quenching 8000kg off after the reaction, the temperature was raised to 90 – 110 ° C hydrolysis seven hours; cooled to room temperature, was added to the kettle 4000kg trioctylamine / methyl tert-butyl ether mixture was extracted phosphoric = 1/1; aqueous phase 6000kg methanol precipitated solid was centrifuged to give 1200kg vitamin BI phosphate monoester crude; the 1200kg vitamin BI phosphate monoester crude 6000kg water were mixed after beaten, cooled to O ~ 5 ° C, caustic soda was added dropwise at this temperature adjustment range of the system PH value to 9.0 ~ 11.0; PH adjustment finished, the reactor temperature to ensure solution of 10 ~ 25 ° C within 1200kg benzoyl chloride, and after the addition is complete heat to the completion of the reaction; filtered, the filtrate was adjusted to PH value
3.5 to 4.0 precipitated solid was isolated and dried to give a white solid 1260kg, namely benfotiamine. Yield: 81.24%, Purity: 98.70% ο
Example 5
Was added to the reaction kettle 5000kg polyphosphoric acid, heated to 110 ~ 130 ° C, the vitamin BI 1000kg batches added to the reaction dad, add after kept at this temperature range for 3 hours, water quenching 10000kg off after the reaction, the temperature was raised to 110 – 120 ° C under reflux for 3 hours hydrolysis; cooled to room temperature, the mixture was extracted phosphoric acid was added to the kettle 3000kg trioctylamine / methyl tert-butyl ether = 1/1; aqueous phase `6000kg ethanol was added to precipitate a solid, obtained by centrifugation 1200kg vitamin BI phosphate monoester crude; after 1200kg vitamin BI phosphate monoester crude mixing beating in 6000kg water, down to O ~ 5 ° C, solution of caustic soda in this temperature range adjusting the PH value of the system to the 8.0 ~ 10.0; PH adjusting finished, 1200kg of benzoyl chloride was added dropwise to ensure the kettle temperature within 10 ~ 25 ° C, after the addition is complete heat the reaction to completion; filtered, the filtrate was adjusted to PH value 3.5 to 4.0 precipitated solid was isolated and dried to give a white solid 1230kg, namely benfotiamine. Yield: 79.31%, purity: 98.60% ο
PATENT

http://www.google.com/patents/CN102911208A?cl=en
Example I: Phosphorus oxychloride 15. 33g (O. Imol) was added to the water 10. 8mL, placed in an ice bath with stirring O. 5 hours was added portionwise thiamine 26. 53g (O. lmol), warmed to 50 ° C followed by stirring for 2 hours, cooled to room temperature to obtain a solution of phosphorus thiamine, thiamine HPLC phosphorus content of 91.36%, adjusted with 15% NaOH solution to pH 8_9 the solution was added 28. Ilg (O. 2mol) benzoyl chloride, the 0_5 ° C under stirring, monitoring the reaction solution and pH changes, the pH value is stable, does not change when the reaction liquid PH, stirring was continued for I hour the reaction, the solution was adjusted to pH 3. 5-4. 0, suction filtration to give 33. 58g benfotiamine white solid. Yield 71.9%.
MP: 164-165 ° C; H1 NMR (400MHz, CDCl3): 2.18 (s, 3H), 2.56 (s, 3H), 2 58 (t, / = 6 7,2H.), 4.. 33 (t, / = 6.7,2H), 4. 83 (s, 2H), 7. 44 (m, 2H), 7. 57 (dd, / = 7. 3, J = I. 5, 1H), 7. 60 (m, 2H), 7. 70 (s, 1H), 8. 67 (s, 1H).
Example 2: Phosphorus oxychloride 15. 33g (O. lmol) was added to a 7. 2mL of water, placed in an ice bath with stirring O. 5 hours was added portionwise thiamine 21. 23g (O. OSmol), warmed to 60 ° C followed by stirring for 2 hours, cooled to room temperature to obtain a solution of phosphorus thiamine, thiamine HPLC phosphorus content of 92.37%, adjusted with 15% NaOH solution to pH 8_9 the solution was added 28. Ilg (O. 2mol) benzoyl chloride, stirred at 0-5 ° C, and monitoring the pH of the reaction solution changes, stable pH, the reaction solution PH does not change when the stirring was continued for I hour the reaction, the solution pH adjusted to 3. 5-4. 0, suction filtration to give 27. 69g benfotiamine white solid. Yield 74.2%.
MP: 164-165 ° C; H1 NMR (400MHz, CDCl3):.. 2.18 (s, 3H), 2 56 (s, 3H), 2 58 (t, / = 6 7,2H.), 4. 33 (t, / = 6.7,2H), 4. 83 (s, 2H), 7. 44 (m, 2H), 7. 57 (dd, / = 7. 3, / = 1. 5, 1H ), 7. 60 (m, 2H), 7. 70 (s, 1H), 8. 67 (s, 1H).
Example 3: Phosphorus oxychloride 15. 33g (O. lmol) was added to a 3. 6mL of water, placed in an ice bath with stirring O. 5 hours was added portionwise thiamine 15. 92g (O. 06mol), warmed to 70 ° C followed by stirring for 2 hours, cooled to room temperature to obtain a solution of phosphorus thiamine, thiamine HPLC phosphorus content of 93.23%, adjusted with 15% NaOH solution to pH 8_9 the solution was added 28. Ilg (O. 2mol) benzoyl chloride, stirred at 0-5 ° C, and monitoring the pH of the reaction solution changes, stable pH, the reaction solution PH does not change when the stirring was continued for I hour the reaction, the solution pH adjusted to 3. 5-4. 0, filtration, benfotiamine was a white solid 23. 71g. Yield 84.7%.
MP: 164-165 ° C; H1 NMR (400MHz, CDCl3): 2.18 (s, 3H), 2.56 (s, 3H), 2 58 (t, / = 6 7,2H.), 4.. 33 (t, / = 6.7,2H), 4. 83 (s, 2H), 7. 44 (m, 2H), 7. 57 (dd, / = 7. 3, / = 1. 5, 1H), 7. 60 (m, 2H), 7. 70 (s, 1H), 8. 67 (s, 1H).
Example 4: Phosphorus oxychloride 15. 33g (O. lmol) was added to a 7. 2mL of water, placed in an ice bath with stirring O. 5 hours was added portionwise thiamine 10. 62g (O. 04mol), warmed to 80 ° C followed by stirring for 2 hours, cooled to room temperature to obtain a solution of phosphorus thiamine, thiamine HPLC phosphorus content of 95.26%, adjusted with 15% NaOH solution to pH 8_9 the solution was added 28. Ilg (O. 2mol) benzoyl chloride, stirred at 0-5 ° C, and monitoring the pH of the reaction solution changes, stable pH, the reaction solution PH does not change when the stirring was continued for I hour the reaction, the solution pH adjusted to 3. 5-4. 0, filtration, benfotiamine was a white solid 15. 22g. Yield 85.2%.
MP: 164-165 ° C; H1 NMR (400MHz, CDCl3): 2.18 (s, 3H), 2.56 (s, 3H), 2 58 (t, / = 6 7,2H.), 4.. 33 (t, / = 6.7,2H), 4. 83 (s, 2H), 7. 44 (m, 2H), 7. 57 (dd, / = 7. 3, / = 1. 5, 1H), 7. 60 (m, 2H), 7. 70 (s, 1H), 8. 67 (s, 1H).

PATENT
http://www.google.com/patents/CN103724374A?cl=en

Synthesis I) thiamine monophosphate hydrochloride
In the reaction flask was added phosphate, thiamine hydrochloride, phosphorous pentoxide was added and stirred to dissolve, controlling the reaction temperature to complete the reaction thiamine hydrochloride, was added and stirring was continued after dropwise addition of concentrated hydrochloric acid hydrolysis of purified water was added dropwise acetone crystallization dropwise at raising grain, filtration, washed with acetone crystal, vacuum drying intermediates thiamine monophosphate hydrochloride;

2) Synthesis of crude benfotiamine
In the reaction flask thiamine monophosphate hydrochloride, dissolved in purified water, sodium hydroxide was added dropwise to adjust the pH to alkaline and steady, benzoyl chloride, sodium hydroxide was added dropwise while controlling alkaline pH, to control the temperature of the reaction pH remained stable, the end of the reaction, concentrated hydrochloric acid was added and extracted twice with ethyl acetate, the aqueous phase of sodium hydroxide was added dropwise until the pH is acidic, crystal seeding planting, filtration, purified water and acetone crystal, vacuum drying crude benfotiamine;

See also
References
- 1 “BBC news story: Back pain drug ‘may aid diabetics'”. BBC News. 18 February 2003.
- 2
- J Lin, A Alt, J Liersch, RG Bretzel, M Brownlee (May 2000). “Benfotiamine Inhibits Intracellular Formation of Advanced Glycation End Products in vivo” (PDF). Diabetes. 49 (Suppl1) (A143): 583.
- 3
- Balakumar P, Rohilla A, Krishan P, Solairaj P, Thangathirupathi A (2010). “The multifaceted therapeutic potential of benfotiamine”. Pharmacol Res 61 (6): 482–8. doi:10.1016/j.phrs.2010.02.008. PMID 20188835.
- 4
- Since AGEs are the actual agents productive of diabetic complications, in theory, if diabetic patients could block the action of AGEs completely by benfotiamine, strict blood sugar control, with its disruption of lifestyle and risks to health and life by severe hypoglycemic episodes, could be avoided, with revolutionary implications for the treatment of diabetes. Hammes, HP; Du, X; Edelstein, D; Taguchi, T; Matsumura, T; Ju, Q; Lin, J; Bierhaus, A; Nawroth, P; Hannak, D; Neumaier, M; Bergfeld, R; Giardino, I; Brownlee, M (2003). “Benfotiamine blocks three major pathways of hyperglycemic damage and prevents experimental diabetic retinopathy”. Nat Med 9 (3): 294–299. doi:10.1038/nm834.
- 5
- Stirban A, Negrean M, Stratmann B; et al. (2007). “Adiponectin decreases postprandially following a heat-processed meal in individuals with type 2 diabetes: an effect prevented by benfotiamine and cooking method”. Diabetes Care 30 (10): 2514–6. doi:10.2337/dc07-0302. PMID 17630265.
- 6
- Stracke H, Hammes HP, Werkmann D; et al. (2001). “Efficacy of benfotiamine versus thiamine on function and glycation products of peripheral nerves in diabetic rats”. Exp. Clin. Endocrinol. Diabetes 109 (6): 330–6. doi:10.1055/s-2001-17399. PMID 11571671.
- 7
- Stirban A, Negrean M, Stratmann B; et al. (2006). “Benfotiamine prevents macro- and microvascular endothelial dysfunction and oxidative stress following a meal rich in advanced glycation end products in individuals with type 2 diabetes”. Diabetes Care 29 (9): 2064–71. doi:10.2337/dc06-0531. PMID 16936154.
- 8
- Babaei-Jadidi R, Karachalias N, Ahmed N, Battah S, Thornalley PJ (2003). “Prevention of incipient diabetic nephropathy by high-dose thiamine and benfotiamine”. Diabetes 52 (8): 2110–20. doi:10.2337/diabetes.52.8.2110. PMID 12882930.
- 9
- Yamazaki, M (1968). “Studies on the absorption of S-benzoylthiamine O-monophosphate : (I) Metabolism in tissue homogenates”. Vitamins 38 (1): 12–20.
- 10
Volvert, M.L.; Seyen, S.; Piette, M.; Evrard, B.; Gangolf, M.; Plumier, J.C.; Bettendorff, L. (2008). “Benfotiamine, a synthetic S-acyl thiamine derivative, has different mechanisms of action and a different pharmacological profile than lipid-soluble thiamine disulfide derivatives”. BMC Pharmacology 8 (1): 10. doi:10.1186/1471-2210-8-10. PMC 2435522. PMID 18549472.
External links
| CN101654464A * | Jul 28, 2009 | Feb 24, 2010 | 湖北华中药业有限公司;湖北制药有限公司 | Method for synthesizing vitamin B1 phosphatic monoester |
| CN102766163A * | Jun 29, 2012 | Nov 7, 2012 | 暨明医药科技(苏州)有限公司 | Synthesis method of phosphate monoester of vitamin B1 |
| CN102911208A * | Sep 25, 2012 | Feb 6, 2013 | 同济大学 | Method for synthesizing benfotiamine |
| CA682778A * | Mar 24, 1964 | Sankyo Kabushiki Kaisha | S-benzoylthiamine o-monophosphate and a process for preparing the same | |
| US3507854 * | Apr 7, 1965 | Apr 21, 1970 | Sankyo Co | Process for preparing thiamine derivatives |
| CN103772432A * | Jan 3, 2014 | May 7, 2014 | 湖北瑞锶科技有限公司 | Production method of benfotiamine |
| CN103772432B * | Jan 3, 2014 | Jan 20, 2016 | 湖北瑞锶科技有限公司 | 一种苯磷硫胺的生产方法 |
| Patent | Submitted | Granted |
|---|---|---|
| Topical compositions comprising benfotiamine and pyridoxamine [US7666442] | 2006-03-02 | 2010-02-23 |
| METHODS OF USING BENFOTIAMINE AND PYRIDOXAMINE COMPOSITIONS [US2010151061] | 2010-06-17 | |
| Topical delivery of trace metals for skin care [US7569558] | 2006-08-17 | 2009-08-04 |
| METHODS OF IDENTIFYING CRITICALLY ILL PATIENTS AT INCREASED RISK OF DEVELOPMENT OF ORGAN FAILURE AND COMPOUNDS FOR THE TREATMENT HEREOF [US2014322207] | 2014-07-11 | 2014-10-30 |
| Protein Carrier-Linked Prodrugs [US2014323402] | 2012-08-10 | 2014-10-30 |
| ANTINEURITIC PHARMACEUTICAL COMBINATION AND COMPOSITIONS [US2014323428] | 2012-12-14 | 2014-10-30 |
| METHODS FOR IMPROVING MEDICAL THERAPIES [US2014335074] | 2012-12-13 | 2014-11-13 |
| LONG LASTING BREATH MINT [US2014335139] | 2014-05-13 | 2014-11-13 |
| High-Loading Water-Soluable Carrier-Linked Prodrugs [US2014296257] | 2012-08-10 | 2014-10-02 |
| PYRAZOLE-AMIDE COMPOUNDS AND PHARMACEUTICAL USE THEREOF [US2014296315] | 2014-03-14 | 2014-10-02 |
| Patent | Submitted | Granted |
|---|---|---|
| HYDRATE AND CRYSTAL OF FLUORENE COMPOUNDS [US2014296316] | 2014-03-14 | 2014-10-02 |
| Cysteine Peptide-Containing Health Drink [US2014302171] | 2012-10-11 | 2014-10-09 |
| Delaying the Progression of Diabetes [US2014303079] | 2012-05-08 | 2014-10-09 |
| FIBRONECTIN BASED SCAFFOLD DOMAIN PROTEINS THAT BIND TO MYOSTATIN [US2014309163] | 2014-05-12 | 2014-10-16 |
| METHODS AND COMPOSITIONS FOR CORRECTION OF ORGAN DYSFUNCTION [US2014274957] | 2014-03-13 | 2014-09-18 |
| COMPOUNDS FOR IMPROVED VIRAL TRANSDUCTION [US2014234278] | 2012-09-28 | 2014-08-21 |
| TOPICAL DERMAL DELIVERY COMPOSITIONS USING SELF ASSEMBLING NANOPARTICLES WITH CETYLATED COMPONENTS [US2014234428] | 2013-02-15 | 2014-08-21 |
| Polymeric Hyperbranched Carrier-Linked Prodrugs [US2014243254] | 2012-08-10 | 2014-08-28 |
| ENCAPSULATED OILS [US2014023688] | 2013-07-12 | 2014-01-23 |
| COMPOSITIONS, KITS AND METHODS FOR NUTRITION SUPPLEMENTATION [US2014023751] | 2013-09-27 | 2014-01-23 |
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
S-[2-{[(4-Amino-2-methylpyrimidin-5-yl)methyl] (formyl)amino}-5-(phosphonooxy)pent-2-en-3-yl] benzenecarbothioate
|
|
| Clinical data | |
| Trade names | Milgamma |
| AHFS/Drugs.com | International Drug Names |
| Legal status | |
| Routes of administration |
Oral |
| Identifiers | |
| CAS Number | 22457-89-2 |
| ATC code | A11DA03 |
| PubChem | CID 3032771 |
| ChemSpider | 2297665 |
| UNII | Y92OUS2H9B |
| ChEBI | CHEBI:41039 |
| ChEMBL | CHEMBL1491875 |
| Synonyms | S-Benzoylthiamine O-monophosphate |
| Chemical data | |
| Formula | C19H23N4O6PS |
| Molar mass | 466.448 g/mol |
- Hamanaka, Wataru; JP 37011040 B 1962
- (3) Koltunova, V. I.; Zhurnal Obshchei Khimii 1969, V39(1), P102-9
- (4) GB 896089 1962 CAPLUS
- (5) “Drugs – Synonyms and Properties” data were obtained from Ashgate Publishing Co. (US)
- (6) Sunagawa, Genshun; JP 37013483 B 1962
///////benfotiamine, бенфотиамин , بينفوتيامين , 苯磷硫胺 , ベンフォチアミン
O=P(O)(O)OCCC(/SC(=O)c1ccccc1)=C(/N(C=O)Cc2cnc(nc2N)C)C
MELOXICAM

Meloxicam ;
351.40, C14H13N3O4S2, MP 255 °C
(8E)-8-[hydroxy-[(5-methyl-1,3-thiazol-2-yl)amino]methylidene]-9-methyl-10,10-dioxo-10$l^{6}-thia-9-azabicyclo[4.4.0]deca-1,3,5-trien-7-one;
4-Hydroxy-2-methyl-N-(5-methyl-2-thiazolyl)-2H-1,2-benzothiazine-3-carboxamide 1,1-dioxide;
CAS 133687-22-6; Mobec;Mobic (TN);
2H-1,2-Benzothiazine-3-carboxamide, 4-hydroxy-2-methyl-N-(5-methylthiazolyl)-, 1,1-dioxide;
The IUPAC name of Meloxicam is (3E)-3-[hydroxy-[(5-methyl-1,3-thiazol-2-yl)amino]methylidene]-2-methyl-1,1-dioxo-1λ6,2-benzothiazin-4-one. With the CAS registry number 71125-38-7, it is also named as 2H-1,2-Benzothiazine-3-carboxamide, 4-hydroxy-2-methyl-N-(5-methylthiazolyl)-, 1,1-dioxide.
Uses of Meloxicam: this chemical is a nonsteroidal anti-inflammatory drug with analgesic and fever reducer effects. And it inhibits cyclooxygenase that can be used as an anti-inflammatory. Additionally, it can be used for the treatment of rheumatoid arthritis and osteoarthritis.
In Europe, where the product has been available since the early 1990s, it is also prescribed and licensed for other anti-inflammatory benefits including relief from both acute and chronic pain in dogs and cats. For many years, both injectable and oral (liquid and tablet) formulations of meloxicam have been licensed for use in dogs, and injectable ones for use in cats. In June 2007, a new oral version of Metacam was licensed in Europe for the long-term relief of pain in cats. As of June 2008, Meloxicam is registered for long term use in cats in Australia, New Zealand, and throughout Europe. ‘Metacam oral suspension 1.5 is not approved or recommended (according to the manufacture insert) for use in cats in the U.S.

1H NMR DMSOD6

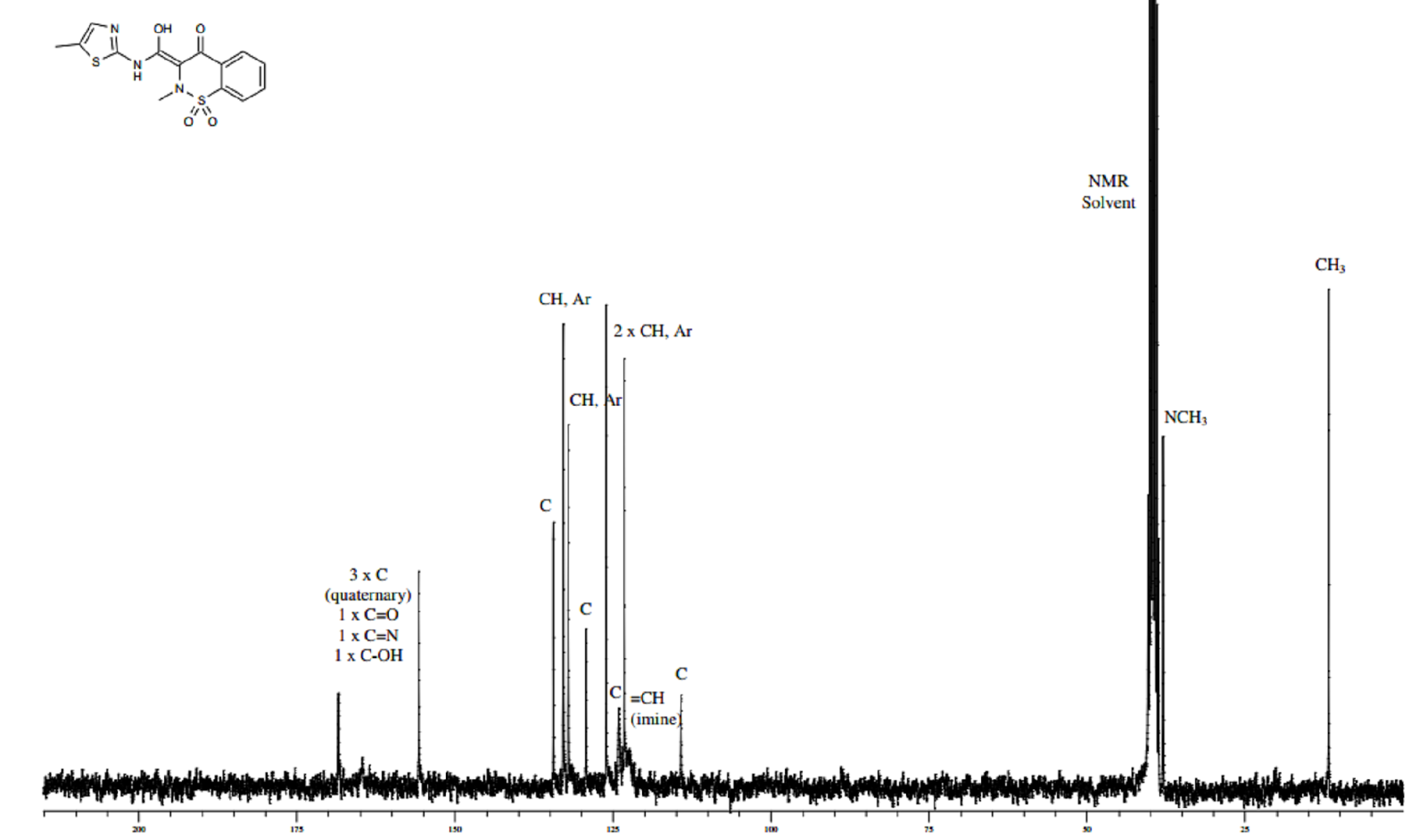
13C NMR DMSOD6
Meloxicam is a nonsteroidal anti-inflammatory drug (NSAID) with analgesic and fever reducer effects. It is a derivative of oxicam, closely related to piroxicam, and falls in the enolic acid group of NSAIDs.[2] It was developed by Boehringer-Ingelheim. Meloxicam starts to relieve pain about 30–60 minutes after administration.[3]
Mechanism of action
Meloxicam blocks cyclooxygenase (COX), the enzyme responsible for converting arachidonic acid into prostaglandin H2—the first step in the synthesis of prostaglandins, which are mediators of inflammation. Meloxicam has been shown, especially at its low therapeutic doses, selectively to inhibit COX-2 over COX-1.[1]
Meloxicam concentrations in synovial fluid range from 40% to 50% of those in plasma. The free fraction in synovial fluid is 2.5 times higher than in plasma, due to the lower albumin content in synovial fluid as compared to plasma. The significance of this penetration is unknown,[2] but it may account for the fact that it performs exceptionally well in treatment of arthritis in animal models.[4]
Side effects
Meloxicam use can result in gastrointestinal toxicity and bleeding, headaches, rash, and very dark or black stool (a sign of intestinal bleeding). Like other NSAIDs, its use is associated with an increased risk of cardiovascular events such as heart attack and stroke.[5]It has fewer gastrointestinal side effects than diclofenac,[6] piroxicam,[7] naproxen,[8] and perhaps all other NSAIDs which are not COX-2 selective.[6] Although meloxicam does inhibit thromboxane A, it does not appear to do so at levels that would interfere withplatelet function.
A pooled analysis of randomized, controlled studies of meloxicam therapy of up to 60 days duration found that meloxicam was associated with a statistically significantly lower number of thromboembolic complications than the NSAID diclofenac (0.2% versus 0.8% respectively) but a similar incidence of thromboembolic events to naproxen and piroxicam.[9]
Potential serious cardiovascular side effects
Persons with hypertension, high cholesterol, or diabetes are at risk for cardiovascular side effects. Persons with family history of heart disease, heart attack or stroke must tell their treating physician as the potential for serious cardiovascular side effects is significant.[10][11]
Veterinary use
Meloxicam is also used in the veterinary field, most commonly in dogs and cats, but also sees off-label use in other animals such as cattle and exotics.[12][13] The U.S. Food and Drug Administration sent a Notice of Violation to the manufacturer for its promotional materials which included promotion of the drug for off-label use.[14] In the U.S. the drug is indicated for management of pain and inflammation associated with osteoarthritis in dogs only. In Europe, where the product has been available since the early 1990s,[citation needed] it is also prescribed and licensed for other anti-inflammatory benefits including relief from both acute and chronic pain in dogs. Side effects in animals are similar to those found in humans; the principal side effect is gastrointestinal irritation (vomiting, diarrhea and ulceration). Rarer but important side effects include liver and kidney toxicity.
Since 2003, the oral (liquid) formulations of meloxicam have been licensed in the U.S for use in dogs only,[15] with the January 2005 product insert specifically warning in bold-face type: “Do not use in cats.”[16] An injectable formulation for use in dogs was approved by the FDA in November 2003,[17] with a formulation for cats, for surgical use only, approved in October 2004.[18]
In the U.S., per the manufacturer’s clinical instructions as of July 2010, injectable meloxicam is indicated in operative use with felines as a single, one-time dose only, with specific and repeated warnings not to administer a second dose.[19] In June 2007, a new oral version of meloxicam was licensed in Europe for the long-term relief of pain in cats. As of June 2008, meloxicam is registered for long term use in cats in Australia, New Zealand, and throughout Europe. A peer-reviewed journal article cites feline overdose of NSAIDs, including meloxicam, as being a cause of severe kidney damage in cats.[20]
The pharmacokinetics of meloxicam have been investigated in koalas (Phascolarctos cinereus).[21]
Meloxicam has been investigated as an alternative to Diclofenac by the RSPB to prevent deaths of vultures.
Preparation of Meloxicam: this chemical can be prepared by Methyl 4-hydroxy-2-methyl-(2H)-1,2-benzothiazine-3-carboxylate-1,1-dioxide and 2-Amino-5-methylthiazole. The yield is 74 %.



References
- Noble, S; Balfour, JA (March 1996). “Meloxicam.”. Drugs 51 (3): 424–30; discussion 431–32. doi:10.2165/00003495-199651030-00007. PMID 8882380.
- “Meloxicam official FDA information, side effects, and uses”. Drugs.com. March 2010. Retrieved 17 March 2010.
- Auvinet, B; Ziller, R; Appelboom, T; Velicitat, P (November–December 1995). “Comparison of the onset and intensity of action of intramuscular meloxicam and oral meloxicam in patients with acute sciatica.”. Clinical Therapeutics 17 (6): 1078–98.doi:10.1016/0149-2918(95)80086-7. PMID 8750399.
- Engelhardt, G; Homma, D; Schlegel, K; Utzmann, R; Schnitzler, C (Oct 1995). “Anti-inflammatory, analgesic, antipyretic and related properties of meloxicam, a new non-steroidal anti-inflammatory agent with favourable gastrointestinal tolerance”. Inflammation Research 44 (10): 423–433. doi:10.1007/BF01757699. PMID 8564518.
- Stamm O, Latscha U, Janecek P, et al. (January 1976). “Development of a special electrode for continuous subcutaneous pH measurement in the infant scalp”. Am. J. Obstet. Gynecol. 124 (2): 193–5. PMID 2012.
- Hawkey, C; Kahan, A; Steinbrü, K; Alegre, C; Baumelou, E; Bégaud, B; Dequeker, J; Isomäki, H; et al. (Sep 1998). “Gastrointestinal tolerability of meloxicam compared to diclofenac in osteoarthritis patients”. Rheumatology 37 (9): 937–945(9).doi:10.1093/rheumatology/37.9.937.
- Dequeker, J; Hawkey, C; Kahan, A; Steinbruck, K; Alegre, C; Baumelou, E; Begaud, B; Isomaki, H; et al. (1998). “Improvement in gastrointestinal tolerability of the selective cyclooxygenase (COX)-2 inhibitor, meloxicam, compared with piroxicam: results of the Safety and Efficacy Large-scale Evaluation of COX- inhibiting Therapies (SELECT) trial in osteoarthritis”. The British Journal of Rheumatology 37 (9): 946–51.doi:10.1093/rheumatology/37.9.946. PMID 9783758.
- Wojtulewski, JA; Schattenkirchner, M; Barceló, P; Le Loët, X; Bevis, PJR; Bluhmki, E; Distel, M. “A Six-Month Double-Blind Trial to Compare the Efficacy and Safety of Meloxicam 7.5 mg Daily and Naproxen 750 mg Daily in Patients with Rheumatoid Arthritis”.Rheumatology. 35, Supplement 1: 22–8. doi:10.1093/rheumatology/35.suppl_1.22.
- Singh, G; Lanes, S; Steinbrü, G; Triadafilopoulos (2004). “Gastrointestinal tolerability of meloxicam compared to diclofenac in osteoarthritis patients”. Am J Med 117 (9): 100–6.doi:10.1016/j.amjmed.2004.03.012. PMID 15234645.
- “Medline Plus”. Nlm.nih.gov. Retrieved 15 November 2014.
- “Drugs.com”. Drugs.com. Retrieved 15 November 2014.
- Off-label use discussed in: Arnold Plotnick MS, DVM, ACVIM, ABVP, Pain Management using Metacam, and Stein, Robert, Perioperative Pain Management Part IV, Looking Beyond Butorphanol, Sep 2006, Veterinary Anesthesia & Analgesia Support Group.
- For off-label use example in rabbits, see Krempels, Dana, Hind Limb Paresis and Paralysis in Rabbits, University of Miami Biology Department.
- US FDA Notice of Violation for off-label use promotion, April 2005.
- “NADA 141-213: New Animal Drug Application Approval (for Metacam (meloxicam) 0.5 mg/mL and 1.5 mg/mL Oral Suspension)” (PDF). US Food and Drug Administration. April 15, 2003. Retrieved 24 July 2010.
- Metacam Client Information Sheet, product description: “Non-steroidal anti-inflammatory drug for oral use in dogs only”, and in the “What Is Metacam” section in bold-face type: “Do not use in cats.”, January 2005.
- “Metacam 5 mg/mL Solution for Injection” (PDF). Fda.gov. Retrieved 15 November2014.
- “Metacam 5 mg/mL Solution for Injection, Supplemental Approval” (PDF). Fda.gov. October 28, 2004. Retrieved 15 November 2014.
- See the manufacturer’s FAQ on its website, and its clinical dosing instructions for cats.
- Merola, Valentina, DVM, DABT, and Dunayer Eric, MS, VMD, DABT, The 10 most common toxicoses in cats, Toxicology Brief, Veterinary Medicine, pp. 340–342, June, 2006.
- Kimble, B.; Black, L. A.; Li, K. M.; Valtchev, P.; Gilchrist, S.; Gillett, A.; Higgins, D. P.; Krockenberger, M. B.; Govendir, M. (2013). “Pharmacokinetics of meloxicam in koalas ( ) after intravenous, subcutaneous and oral administration”. Journal of Veterinary Pharmacology and Therapeutics 36 (5): 486–493. doi:10.1111/jvp.12038.PMID 23406022.
External links
- Manufacturer’s Official Product Website for the Veterinary formulations
- Manufacturer’s United States Division website for the Veterinary formulations
- FDA Metacam
- Meloxicam Side Effects
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
4-hydroxy-2-methyl-N-(5-methyl-2-thiazolyl)-2H-1,2-benzothiazine-3-carboxamide-1,1-dioxide.
|
|
| Clinical data | |
| Trade names | Mobic |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a601242 |
| Pregnancy category |
|
| Legal status | |
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | 89%[1] |
| Protein binding | 99.4%[1] |
| Metabolism | Hepatic (CYP2C9 and 3A4-mediated)[1] |
| Biological half-life | 20 hours[1] |
| Excretion | Urine and faeces equally[1] |
| Identifiers | |
| CAS Number | 71125-38-7 |
| ATC code | M01AC06 |
| PubChem | CID 5281106 |
| IUPHAR/BPS | 7220 |
| DrugBank | DB00814 |
| ChemSpider | 10442740 |
| UNII | VG2QF83CGL |
| KEGG | D00969 |
| ChEBI | CHEBI:6741 |
| ChEMBL | CHEMBL599 |
| PDB ligand ID | MXM (PDBe, RCSB PDB) |
| Chemical data | |
| Formula | C14H13N3O4S2 |
| Molar mass | 351.403 g/mol |
/////
Cc1cnc(s1)NC(=O)C\3=C(/O)c2ccccc2S(=O)(=O)N/3C
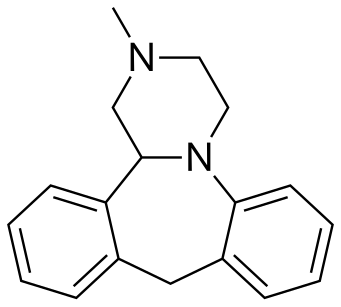
MIANSERIN

MIANSERIN
Mianserin (brand names: Depnon (IN), Lantanon (ZA), Lerivon (AR, BE, CZ, PL, RU, SK), Lumin (AU), Norval (UK), Tolvon (AU, HK†, IE†,NZ, SG†), Tolmin (DK); where † indicates discontinued products) is a psychoactive drug of the tetracyclic antidepressant (TeCA) therapeutic family. It is classified as a noradrenergic and specific serotonergic antidepressant (NaSSA) and has antidepressant,anxiolytic (anti-anxiety), hypnotic (sedating), antiemetic (nausea and vomiting-attenuating), orexigenic (appetite-stimulating), andantihistamine effects.
It is not approved for use in the US, but its analogue, mirtazapine, is. Mianserin was the first antidepressant to reach the UK market that was less dangerous than the tricyclic antidepressants in overdose.[3]

Medical uses
When used for the treatment of depression, its efficacy appears comparable to that of amitriptyline, citalopram, clomipramine,dothiepin, doxepin, fluoxetine, flupenthixol, fluvoxamine, imipramine, moclobemide, nortriptyline, paroxetine, and trazodone.[1][4]Mianserin received TGA approval in May 1996.[5]
Similarly to its analogue, mirtazapine, mianserin has been tried as an augmentation strategy in treatment-resistant depression with some success.[6] Mianserin has been tried, similarly to mirtazapine, as an adjunct in schizophrenia and has been found to reduce negative and cognitive symptoms.[7][8][9]
Mianserin has demonstrated efficacy as a monotherapy for the treatment of Parkinson’s disease psychosis in an open-label clinical trial.[10]
Interactions
CYP2D6 inhibitors such as the selective serotonin reuptake inhibitors (SSRIs), quinidine, ritonavir, etc. would likely raise plasma levels of mianserin and hence could lead to mianserin toxicity. Conversely, CYP2D6 inducers would likely lead to reduced mianserin plasma concentrations and hence potentially diminish the therapeutic effects of mianserin.[1]
Withdrawal
Abrupt or rapid discontinuation of mianserin may provoke a withdrawal, the effects of which may include depression, anxiety, panic attacks,[14] decreased appetite or anorexia,insomnia, diarrhea, nausea and vomiting, and flu-like symptoms, such as allergies or pruritus, among others.
Pharmacology
Mianserin is an antagonist/inverse agonist of the H1, 5-HT1D, 5-HT2A, 5-HT2B, 5-HT2C, 5-HT3, 5-HT6, 5-HT7, α1-adrenergic, and α2-adrenergic receptors, and also inhibits thereuptake of norepinephrine.[16][17] As a high affinity H1 receptor inverse agonist, mianserin has strong antihistamine effects (sedation, weight gain, etc.). Contrarily, it has negligible affinity for the mACh receptors, and thus lacks anticholinergic properties. It was recently found to be a weak (Ki = 1.7 μM, EC50 = 0.53 μM) κ-opioid receptor partial agonist.[18]
In addition, mianserin also appears to be a potent antagonist of the neuronal octopamine receptor.[19] What implications this may have on mood are currently unknown, however octopamine has been implicated in the regulation of sleep, appetite and insulin production and therefore may theoretically contribute to the overall side effect profile of mianserin.[20][21]
Blockade of the H1 and α1-adrenergic receptors has sedative effects,[2] and also antagonism of the 5-HT2A and α1-adrenergic receptors inhibits activation of intracellularphospholipase C (PLC), which seems to be a common target for several different classes of antidepressants.[22] By antagonizing the somatodendritic and presynaptic α2-adrenergic receptors which function predominantly as inhibitory autoreceptors and heteroreceptors, mianserin disinhibits the release of norepinephrine, dopamine, serotonin, andacetylcholine in various areas of the brain and body.
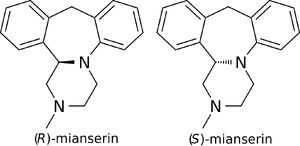
Enantioselectivity

(S)-mianserin
(S)-(+)-Mianserin is approximately 200–300 times more active than its enantiomer (R)-(−)-mianserin.


http://www.beilstein-journals.org/bjoc/single/articleFullText.htm?publicId=1860-5397-11-164
(14bS)-(+)-1,2,3,4,10,14b-Hexahydro-2-methyldibenzo[c,f]pyrazino[1,2-a]azepine (1)
(S)-(+)-1 in the form of solidifying oil; during purification step a small degree of product decomposition was observed; []D 23 = +469.2 (c 1, CHCl3); []D 23 = +436.5 (c 1, EtOH) {[9] []D 23 = +450 (c 0.26, EtOH)}; []D 23 = +428.0 (c 0.5, MeOH) {[5] []D 25 = +469.0 (c 1, MeOH)}.
Enantiomeric purity was determined by HPLC analysis (Chiracel OD-H, hexane:2- propanol = 80:20, 1ml/min, S isomer 5.6min).
IR (CCl4): 3064, 3022, 2939, 2794, 1492, 1446, 1251, 1132 cm–1 ;
1H NMR (500 MHz, CDCl3): δ 2.37-2.42 (m, 4 H), 2.46 (t, J = 10.5 Hz, 1 H), 2.92 (dt, J1 = 2.0 Hz, J2 = 11.0 Hz, 1 H), 3.02 (dd, J1 = 1.5 Hz, J2 = 11.0 Hz, 1 H), 3.25-3.28 (m, 1 H), 3.30 (d, J = 13.0 Hz, 1 H, methylene bridge), 3.42 (td, J1 = 3.0 Hz, J2 = 11.0 Hz, 1 H), 4.14 (dd, J1 = 2.0 Hz, J2 = 10.0 Hz, 1 H,methine), 4.81 (d, J = 13.0 Hz, 1 H, methylene bridge), 6.87 (td, J1 = 1.0 Hz, J2 = 7.5 Hz, 1 H, Ar), 7.00-7.02 (m, 2 H, Ar), 7.05-7.13 (m, 4 H, Ar), 7.16 (td, J1 = 1.5 Hz, J2 = 7.5 Hz, 1 H, Ar);
13C NMR (125 MHz, CDCl3): δ 38.8, 45.6, 51.0, 55.4, 64.6, 66.2, 119.0, 122.3, 126.5, 126.6, 127.0, 127.3, 128.1, 129.5, 137.1, 139.3, 139.8, 148.4.
HRMS (ESI): m/z calcd for C18H21N2 [M+H]+ : 265.1705; found: 265.1712.
(±)-1,2,3,4,10,14b-Hexahydro-2-methyldibenzo[c,f]pyrazino[1,2-a]azepine (1)
The racemate was prepared in the same manner as pure enantiomer; mp = 109.5- 110.5 °C ([10] mp = 111–113 °C). The HPLC analysis (Chiracel OD-H, hexane/2- propanol = 80:20, 1mL/min, R isomer 5.0 min and S isomer 5.6 min)
SYN 1

The title compound has been synthesized by several procedures. Acylation of 2-benzylaniline (I) by chloroacetyl chloride (II) gave chloroacetamide (III). Subsequent cyclization of amide (III) under Vilsmeier conditions furnished the dibenzoazepine (IV). Nucleophilic substitution of the chlorine atom of (IV) by methylamine led to amine (V). The imine function of (V) was reduced with either LiAlH4 or NaBH4 to the diamine (VI), which was further converted into the fused diketopiperazine (VII) upon heating with diethyl oxalate. The amide groups of (VII) were then reduced by means of borane in THF, yielding the target tetracyclic diamine, which was finally isolated as the corresponding hydrochloride salt……US 3534041
SYN 2

In a further procedure, styrene oxide (XV) was condensed with 2-(benzylamino)ethanol (XXVIII) to give amino diol (XXIX). After chlorination of (XXIX) using SOCl2 and DMAP, dichloro derivative (XXX) was condensed with 2-aminobenzyl alcohol (X) yielding piperazine (XXXI). Cyclization of (XXXI) in hot sulfuric acid afforded the tetracyclic compound (XXXII). The N-benzyl group of (XXXII) was then removed by treatment with butyl chloroformate producing carbamate (XXXIII), which was further hydrolyzed and decarboxylated to (XXXIV) under basic conditions. Finally, methylation of the secondary amine (XXXIV) was performed by reductive alkylation with formaldehyde either in the presence of formic acid under Leuckart-Wallach conditions or by catalytic hydrogenation
DE 4305659; EP 0612745
SYN 3

In a different method, reaction of styrene oxide (XV) with methylamine provided amino alcohol (XVI), which was further condensed with ethylene oxide (XVII) to afford amino diol (XVIII). Alternatively, diol (XVIII) was prepared by a more direct procedure by condensation of epoxide (XV) with 2-(methylamino)ethanol (XIX). Chlorination of (XVIII) employing SOCl2 yielded the dichloro derivative (XX), which was subsequently condensed with 2-aminobenzyl alcohol (X) leading to piperazine (XXI). Cyclization of (XXI) to the title compound was accomplished by treatment with hot polyphosphoric acid. Optionally, alcohol (XXI) was converted to chloride (XXII), which was then cyclized in the presence of AlCl3. In a related method, alcohol (XXI) was esterified with AcOH, and the resultant acetate ester (XXIII) was then cyclized in the presence of polyphosphoric acid……US 4217452

The key intermediate (XXI) was also prepared by several related procedures. Chlorination of aminoalcohol (XVI) gave chloro amine (XXIV), which was condensed with 2-aminobenzyl alcohol (X) to afford diamine (XXV). Then, alkylation of diamine (XXV) with dibromoethane (XIII) in hot pyridine gave rise to the target piperazine (XXI). Alternatively, diamine (XXV) was condensed with ethyl chloroacetate or with diethyl oxalate to produce the mono- or dioxopiperazines (XXVII) and (XXVI), respectively, which were then reduced to (XXI) by means of LiAlH4. Cyclization of alcohol (XXI) to the title compound was achieved by treatment with concentrated sulfuric acid
SYN5

FR 2647114
Treatment of alpha-chlorophenylacetyl chloride (VIII) with methylamine provided the corresponding chloro amide (IX), which was subsequently condensed with 2-aminobenzyl alcohol (X) to afford amino alcohol (XI). Cyclization of (XI) in the presence of AlCl3 led to the dibenzoazepine (XII). This was converted to the tetracyclic compound (XIV) by reaction with dibromoethane (XIII) in the presence of Na2CO3. Reduction of the amide carbonyl group of (XIV) by means of LiAlH4 furnished the title compound. In a related strategy, amide (XII) was initially reduced to diamine (VI) by using LiAlH4. Subsequent condensation of (VI) with dibromoethane (XIII) led to the target tetracyclic derivative
OTHER……….


References
- Truven Health Analytics, Inc. DRUGDEX® System (Internet) [cited 2013 Sep 29]. Greenwood Village, CO: Thomsen Healthcare; 2013.
- Merck Sharp & Dohme (Australia) Pty Limited. “Tolvon Product Information”(PDF). GuildLink Pty Ltd.
- Walker, R; Whittlesea, C, ed. (2007) [1994]. Clinical Pharmacy and Therapeutics (4th ed.). Edinburgh: Churchill Livingstone Elsevier. ISBN 978-0-7020-4293-5.
- Wakeling A (April 1983). “Efficacy and side effects of mianserin, a tetracyclic antidepressant”. Postgrad Med J 59 (690): 229–31. doi:10.1136/pgmj.59.690.229.PMC 2417496. PMID 6346303.
- AlphaPharm. “Lumin Mianserin hydrochloride product information” (PDF). GuildLink Pty Ltd.
- Ferreri M, Lavergne F, Berlin I, Payan C, Puech AJ (January 2001). “Benefits from mianserin augmentation of fluoxetine in patients with major depression non-responders to fluoxetine alone”. Acta Psychiatr Scand 103 (1): 66–72. doi:10.1111/j.1600-0447.2001.00148.x. PMID 11202131.
- Poyurovsky, M; Koren, D; Gonopolsky, I; Schneidman, M; Fuchs, C; Weizman, A; Weizman, R (March 2003). “Effect of the 5-HT2 antagonist mianserin on cognitive dysfunction in chronic schizophrenia patients: an add-on, double-blind placebo-controlled study”. European Neuropsychopharmacology 13 (2): 123–128. doi:10.1016/S0924-977X(02)00155-4. PMID 12650957.
- Shiloh, R; Zemishlany, Z; Aizenberg, D; Valevski, A; Bodinger, L; Munitz, H; Weizman, A (March 2002). “Mianserin or placebo as adjuncts to typical antipsychotics in resistant schizophrenia”. International Clinical Psychopharmacology 17 (2): 59–64.doi:10.1097/00004850-200203000-00003. PMID 11890187.
- Mizuki, Y; Kajimura, N; Imai, T; Suetsugi, M; Kai, S; Kaneyuki, H; Yamada, M (April 1990). “Effects of mianserin on negative symptoms in schizophrenia”. International Clinical Psychopharmacology 5 (2): 83–95. doi:10.1097/00004850-199004000-00002.PMID 1696292.
- Ikeguchi, K; Kuroda, A (1995). “Mianserin treatment of patients with psychosis induced by antiparkinsonian drugs”. European Archives of Psychiatry and Clinical Neuroscience 244(6): 320–324. doi:10.1007/BF02190411. PMID 7772616.
- “Australian Medicines Handbook”. Australian Medicines Handbook Pty Ltd. 2013.
- British National Formulary (BNF) (65th ed.). Pharmaceutical Press. p. 1120.ISBN 978-0857110848.
- Mianserin Hydrochloride. Martindale: The Complete Drug Reference (The Royal Pharmaceutical Society of Great Britain). 5 December 2011. Retrieved 3 November 2013.
- Kuniyoshi M, Arikawa K, Miura C, Inanaga K (June 1989). “Panic anxiety after abrupt discontinuation of mianserin”. Jpn. J. Psychiatry Neurol. 43 (2): 155–9. doi:10.1111/j.1440-1819.1989.tb02564.x. PMID 2796025.
- Taylor D, Paton C, Kapur S, Taylor D. The Maudsley prescribing guidelines in psychiatry. 11th ed. Chichester, West Sussex: John Wiley & Sons; 2012.
- Leonard B, Richelson H (2000). “Synaptic Effects of Antidepressants: Relationship to Their Therapeutic and Adverse Effects”. In Buckley JL, Waddington PF. Schizophrenia and Mood Disorders: The New Drug Therapies in Clinical Practice. Oxford: Butterworth-Heinemann. pp. 67–84. ISBN 978-0-7506-4096-1.
- Müller G (8 May 2006). “Target Family-directed Masterkeys in Chemogenomics”. In Kubinyi H, Müller G, Mannhold R, Folkers G. Chemogenomics in Drug Discovery: A Medicinal Chemistry Perspective. John Wiley & Sons. p. 25. ISBN 978-3-527-60402-9. Retrieved 13 May 2012.
- Olianas MC, Dedoni S, Onali P (November 2012). “The atypical antidepressant mianserin exhibits agonist activity at κ-opioid receptors”. Br. J. Pharmacol. 167 (6): 1329–41.doi:10.1111/j.1476-5381.2012.02078.x. PMID 22708686.
- Roeder T (November 1990). “High-affinity antagonists of the locust neuronal octopamine receptor”. Eur. J. Pharmacol. 191 (2): 221–4. doi:10.1016/0014-2999(90)94151-M.PMID 2086239.
- Crocker A, Sehgal A (September 2008). “Octopamine regulates sleep in drosophila through protein kinase A-dependent mechanisms”. J. Neurosci. 28 (38): 9377–85.doi:10.1523/JNEUROSCI.3072-08a.2008. PMC 2742176. PMID 18799671.
- Bour S, Visentin V, Prévot D, Carpéné C (September 2003). “Moderate weight-lowering effect of octopamine treatment in obese Zucker rats”. J. Physiol. Biochem. 59 (3): 175–82.doi:10.1007/BF03179913. PMID 15000448.
- Dwivedi Y, Agrawal AK, Rizavi HS, Pandey GN (December 2002). “Antidepressants reduce phosphoinositide-specific phospholipase C (PI-PLC) activity and the mRNA and protein expression of selective PLC beta 1 isozyme in rat brain”. Neuropharmacology 43(8): 1269–79. doi:10.1016/S0028-3908(02)00253-8. PMID 12527476.
- Roth, BL; Driscol, J (12 January 2011). “PDSP Ki Database”. Psychoactive Drug Screening Program (PDSP). University of North Carolina at Chapel Hill and the United States National Institute of Mental Health. Retrieved 13 October 2013.
Further reading
- Peet M, Behagel H (1978). “Mianserin: a decade of scientific development”. Br. J. Clin. Pharmacol. 5 Suppl 1: 5S–9S. PMC 1429213. PMID 623702.
External links
![]()

 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(±)-2-methyl-1,2,3,4,10,14b-hexahydrodibenzo[c,f]pyrazino[1,2-a]azepine
|
|
| Clinical data | |
| Trade names | Bolvidon (discontinued), Tolvon |
| AHFS/Drugs.com | International Drug Names |
| Pregnancy category |
|
| Legal status | |
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | 20–30%[1] |
| Protein binding | 95%[1] |
| Metabolism | Hepatic (mediated byCYP2D6; most metabolism occurs via aromatic hydroxylation, N-oxidation and N-demethylation)[1] |
| Biological half-life | 21–61 hours[2] |
| Excretion | Renal (4–7%) Faecal (14–28%)[1] |
| Identifiers | |
| CAS Number | 24219-97-4 |
| ATC code | N06AX03 |
| PubChem | CID 4184 |
| IUPHAR/BPS | 135 |
| DrugBank | DB06148 |
| ChemSpider | 4040 |
| UNII | 250PJI13LM |
| KEGG | D08216 |
| ChEBI | CHEBI:51137 |
| ChEMBL | CHEMBL6437 |
| Chemical data | |
| Formula | C18H20N2 |
| Molar mass | 264.365 |
///////////MIANSERIN
c42c(N3C(c1ccccc1C2)CN(C)CC3)cccc4
NIZATIDINE
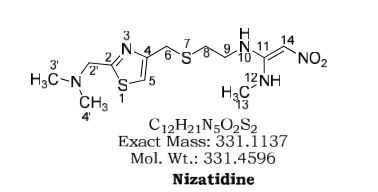
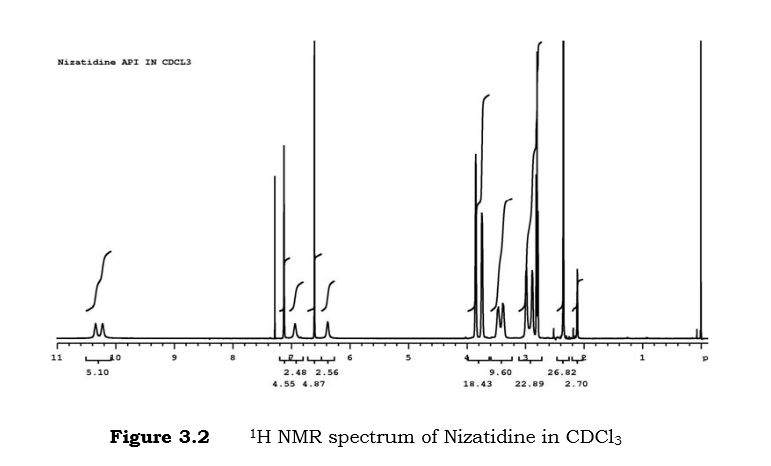

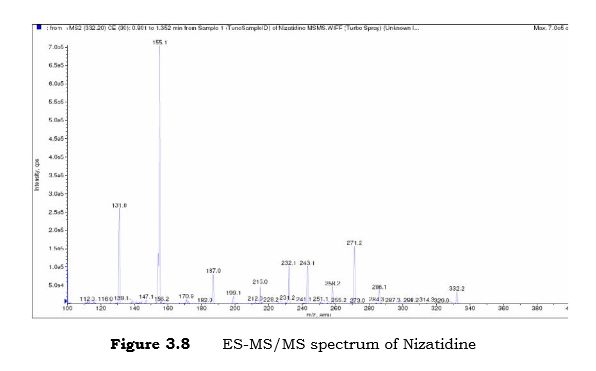

Nizatidine is a histamine H2 receptor antagonist that inhibits stomach acid production, and is commonly used in the treatment of peptic ulcer disease and gastroesophageal reflux disease. It was developed by Eli Lilly and is marketed under the brand names Tazac and Axid.
Clinical use
Nizatidine is used to treat duodenal ulcers, gastric ulcers, and gastroesophageal reflux disease (GERD/GORD), and to prevent stress ulcers.[1]
Adverse effects
Side effects are uncommon, usually minor, and include diarrhea, constipation, fatigue, drowsiness, headache, and muscle aches.[1]
History and development
Nizatidine was developed by Eli Lilly, and was first marketed in 1987. It is considered to be equipotent with ranitidine and differs by the substitution of a thiazole ring in place of the furan ring in ranitidine. In September 2000, Eli Lilly announced they would sell the sales and marketing rights for Axid to Reliant Pharmaceuticals.[2] Subsequently, Reliant developed the oral solution of Axid, marketing this in 2004, after gaining approval from the U.S. Food and Drug Administration (FDA).[3] However, a year later, they sold rights of the Axid Oral Solution (including the issued patent[4] protecting the product) to Braintree Laboratories.[5]
Nizatidine proved to be the last new histamine H2 receptor antagonist introduced prior to the advent of proton pump inhibitors.’
Nizatidine, the systematic chemical name of which is N-[2-[[[2-[ imemylammo)memyl]-4-tl iazolyl]memyl]mio]e yl]–N’- methyl-2-nitro-l,l-ethenecliamine, which has the formula (I).This compound is a histamine H2-receptor antagonist which is useful as anti- ulcer agents capable of inmbiting gastric acid secretion in mammals.

United States Patent No. 4,375,547; 4587344, 4777260; 4,904,792 and 5334725 discloses Nizatidine and other related products. The synthesis of nizatidine disclosed in US patent No. 4,904,792 involves a multi-step process. The first step of the process comprises reacting dimethylaminotmoacetamide hydrochloride with ethyl bromopyruvate to obtain 2-(dinιethylaminon ethyl)-4-thiazolecarboxylate. Reduction of this 4- tbiazolecarboxylate derivative with lithium triethylborohydride gives 2-
(<-Umethylaminoπιethyl)-4-tI-ύazolenιethanol, which is then converted into 4- (2-ammoetϊhyl)ti omethyl-2-d by reacting with
2-aminoethanethiol hydrochloride (cysteamine hydrochloride). This 2- ό-imetihylan-ιinoπιethylthiazol derivative is then converted into Nizatidine by reacting .with N-met-hyl-l-methyltHo-2-mt-coet-hyleneamine in the presence of an acid United States Patent No. 4,382,090 describes a method to prepare 4-
(2-aminoethyl)tMome1_hyl-2-din ethylaminon etihyltl iazol by fusing 4- cmoronιe yl-2-d- nethylaminonιet-hylthiazole with cysteamine hydrochloride at above 100 °C.
United States Patent No. 4,468,517 described a method to prepare 4- cldoronιethyl-2-<-ιimethylaminon et-hylt-lιiazole. The method described in this patent involved reaction of dimet-hylaminotmoacetamide hydrochloride with 1,3-dichloroacetone in haloalkane (1,2-dichloroethane) as a solvent to obta 4-cHoromethyl^-hydroxy-2-dimet^ This 2-thiazoline derivative is then dehydrated with a dehydrating agent like PC13, PBr3, SOCl2, POCl3 etc., to get 4-chloromethyl-2- din etihyl-in monietihylthiazole.
European Patent Application EP 0,515,121 and EP 0,960,880 describe the process for the preparation of 2-(dim.et-hylarninomethyl)-4- thiazolemethanol. The process consists of reacting (-Umethylaminothioacetamide hydrochloride with 1,3-dichloroacetone in toluene to get 4-chlorometiιyl-4-hyαioxy-2-d-methylaminomethyl-2- thiazoline, which is then reacted with alkali metal base in an inert solvent such as toluene to get 2-(dimethylam-m.omethyl)-4-thiazolemethanol.
The methods described in United States Patent No. 4,468,517 for the synthesis of 4-chloromethyl-4-hy( oxy-2-dimethyl-ui-momethyl-2- thiazoline, requires complete evaporation of the solvent 1,2-dichloroethane to get the crude product; it is then washed with ethyl acetate to obtain a pure product. Evaporation of the solvent to complete dryness is an inconvenient and inappropriate operation in large-scale manufacturing. Such evapprations in large-scale operations would produce the solids as lumps; further washing such lumps with solvents would be ineffective due to improper -mixing of -the solid -with solvent. The method described in EP 0,515,121 and EP 0,960,880 for the synthesis of 4-cHorometlιyl-4-hyc oxy*-2-α_im requires isolation of the product from the reaction mixture by precipitation of the product from the mother liquor by the addition of petroleum ether. The crude product obtained by the precipitation is then subjected to an additional purification step by crystallisation from toluene.
A number of procedures are described for the preparation of dimethylammotmoacetamide. Examples are Japanese Patent No. JP 62,273,948, JP 62,273,949, JP 02,264,755 and Org. Prep. Proced. Int., 1992, 24, P.66-7. All the procedures described in the literature- or the preparation of dirnethylaminotitioacetamide from dimethylam oacetomtrile involve the use of hydrogen sulfide under pressure in the presence of promoters or catalysts. The disadvantage with the use of hydrogen sulfide is the difficulty it poses in handling commercial quantities, as it is a very toxic gas. The object of the present invention is to provide an improved manufactxiring process for 4-chloromethyl-4-hydr xy-2- di–netihylam omethyl-2-tibiazoline..
 .
.
SYN2
The cyclization of dimethylaminothioacetamide (I) with ethyl bromopyruvate (II) in refluxing ethanol gives ethyl 2-(dimethylaminomethyl)-4-thiazolecarboxylate (III), which is reduced with lithium triethyl borohydride in THF yielding 2-(dimethylaminomethyl)-4-thiazolemethanol (IV). The condensation of (IV) with 2-aminoethanethiol (V) by means of 48% HBr affords 2-(dimethylaminomethyl)-4-(2-aminoethylthiomethyl)thiazole (VI), which is finally condensed with 1-(methylthio)-2-nitro-N-methylethyleneamine (VII) in water.

PATENT
http://www.google.com/patents/WO2004069817A1?cl=en
Example No: 1 Preparation of dirnethylaniinothioacetaniide hydrochloride Into water (3000 ml), phosphorus pentasulfi.de (1302 g; 2.93 mol) and dimethylam oacetonitrile (1000 g; 11.88 mol) are added one after another at 10°C. The mixture is then slowly warmed to 70°C and maintained for 3 hrs to complete the reaction. The reaction mixture is then cooled to 20°C and sodiu hydroxide (53% w/w, 2200 g, 29.15 mol) is added into it below 20°C. The reaction mixture is then warmed to 50°C and extracted with toluene (2 x 2000 l). Isopropanolic hydrochloric acid (12% w/w; 3700 ml) is added into the extract at 25 to 30°C to adjust the pH to 2 and the mass stirred for 1 h to precipitate the product. The slurry is filtered, washed with isopropyl alcohol (1000 ml) and dried to get (1360 g) dimethyl ammotMoacetamide hydrochloride. Yield = 74.0%, HPLC purity = 97.6% Example No: 2
Preparation of 4-chloromethyl-4-hydr oxy-2-dimethylaminomethyl-2- thiazoline
Dimethylam othioacetamide hydrochloride (1000 g; 6.472 mol) is suspended in diisopropyletiier (4000 ml). Added into this suspension is sodium bicarbonate (1200 g; 14.28 mol) and sodium sulphate (1000 g). The slurry is heated to 55-60° C and stirred for 1 hr. Into this suspension is added 1,3 dichloroacetone (1000 g; 7.87 mol) dissolved in diisopropylether (1000 ml). The reaction is continued at 50-55° C for 2 h. The progress of the reaction is monitored by a qualitative HPLC analysis. Upon completion of the reaction, the reaction mixture is* filtered hot at 50-55° C to remove insoluble inorganic salts. The mother liquor is cooled slowly to 0-5° C to crystallize out the product. The product is then filtered and washed with precooled diisopropylether (250 ml). The product is dried at 50° C under reduced pressure to obtain 1120 g. Yield = 83%; HPLC purity = 98.2%. The following example illustrates the process to convert this pure 4- cHoromethyl-4-hyσ-roxy-2-ά-imet^^ Nizatidine. Example No 3: Preparation of N- [2- [ [ [2- [(Dimethylaι-nino)methyl] -4- thiazolyl] methyl] thio] ethyl] -N’-methyl-2-nitro-l,l-ethenediamine. A. Preparation of 4-chloromethyl-2-ααmethylam onιethylthiazole Hydrochloride.
Thionyl chloride (430 ml; 5.9 mol) is added into chloroform (1000 ml) and cooled to 20° C. Into this solution is added 4-chloromethyl-4- hyά^oxy-2-dinιethylam ome yl-2-thiazoline (1000 g; 4.79 mol), dissolved in chloroform (4000 ml). The reaction mixture is further gradually heated to 60-65° C and maintained at this temperature till qualitative HPLC analysis shows the completion of the reaction. The reaction mixture is then cooled slowly to 30° C to get the product crystallized out. The product is filtered, washed and dried under reduced pressure to obtain 900 g of pure product. Yield = 83.3 %. B. Preparation of 4-(2-am oethyl)thiomethyl-2- ά-imethylam omethylthiazole.
2-A-minoethanetl iol hydrochloride (cysteamine hydrochloride, 520 g; 4.5 mol) is suspended in water (500 ml). This suspension is cooled to 5° C and sodium hydroxide solution (45 % w/w, 870 ml; 14.7 mol) is added into it at 5-10° C. Into this suspension, hydroxylamine sulphate (100 g; 0.6 mol) is added and stirred. A solution of 4-chloromethyl-2- di-n ethyl- inomethylthiazole hydrochloride (1000 g; 4.43 mol) dissolved in water (1250 ml) is prepared separately. This solution is added into the said suspension below 10° C and the reaction continued at 10° C for another 1 h. The completion of the reaction is determined by qualitative HPLC. The reaction mixture is then diluted with water (2000 ml), heated to 40-45° C and extracted with toluene (2 x 2000 ml). The toluene extract is treated with activated carbon at 40-45° C for 30 min. Activated carbon is removed by filtration through hyflo bed and evaporated toluene from the filtrate under reduced pressure at 60° C to obtain 910 g of the product. Yield = 88 %. C. Preparation of N-(2-(((2-(Dimethylamino)methyl)-4- tltiazolyl)m.ethyl)tltio)elhyl)-N’-methyl-2-nitro-l ,1 -etheneά-iamine (Nizatidine).
N-methyl-l-methyltHo-2-mtroethyleneamine (NMSM, 610 g; 4.12 mol) is mixed with water (1500 ml), and the mixture is cool to 20-25° C. 4- (2-Am-hoethyl)d omethyl-2-<^ (1000 g; 4.32 mol) dissolved in water (1500 ml) is added into this suspension at 20-25° C. The reaction mixture is warmed to 30-35° C and continued the reaction for 8 h. The progress of the reaction is monitored by qualitative HPLC analysis. The reaction mixture is extracted with toluene (2 x 1000 ml), and the aqueous layer is treated with activated carbon (50 g) at 55-60° C for 30 min. Activated carbon is removed by filtration through hyflo bed and the aqueous filtrate is extracted with chloroform (4 x 1000 ml)rThe cHorόform extract is concentrated under reduced pressure at less than 50° C; ethyl acetate (3000 ml) is added into the concentrate and reconcentrated. Acetone (300 ml), ethyl acetate (300 ml) is added into the concentrate and cooled to 0-5° C to crystallize the product. The product is filtered, washed with precooled ethyl acetate (250 ml), and dried to obtain pure Nizatidine 1160 g. Yield = 81.0%; HPLC purity -= 99.3%.
References
- [1] Archived May 26, 2008 at the Wayback Machine
- [2] Archived December 26, 2013 at the Wayback Machine
- “United States Patent: 6930119”. Patft.uspto.gov. Retrieved 2015-10-11.
- [3] Archived August 14, 2007 at the Wayback Machine
External links
| US4468517 * | May 12, 1983 | Aug 28, 1984 | Eli Lilly And Company | Synthesis of thiazoles |
| US5457206 * | Jul 1, 1994 | Oct 10, 1995 | Eli Lilly And Company | Process for preparing intermediates to nizatidine and related compounds |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2015002150A1 | Jun 30, 2014 | Jan 8, 2015 | Shin Nippon Biomedical Laboratories, Ltd. | Novel compound, organic cation transporter 3 detection agent, and organic cation transporter 3 activity inhibitor |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(E)-1-N‘-[2-[[2-[(dimethylamino)methyl]-1,3-thiazol-4-yl]methylsulfanyl]ethyl]-1-N-methyl-2-nitroethene-1,1-diamine
|
|
| Clinical data | |
| Trade names | Axid |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a694030 |
| Licence data | US FDA:link |
| Pregnancy category |
|
| Legal status | |
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | >70% |
| Protein binding | 35% |
| Metabolism | Hepatic |
| Biological half-life | 1–2 hours |
| Excretion | Renal |
| Identifiers | |
| CAS Number | 76963-41-2 |
| ATC code | A02BA04 |
| PubChem | CID 3033637 |
| IUPHAR/BPS | 7248 |
| DrugBank | DB00585 |
| ChemSpider | 2298266 |
| UNII | P41PML4GHR |
| KEGG | D00440 |
| ChEBI | CHEBI:7601 |
| ChEMBL | CHEMBL653 |
| Chemical data | |
| Formula | C12H21N5O2S2 |
| Molar mass | 331.46 g/mol |
VASICINE, (peganine)
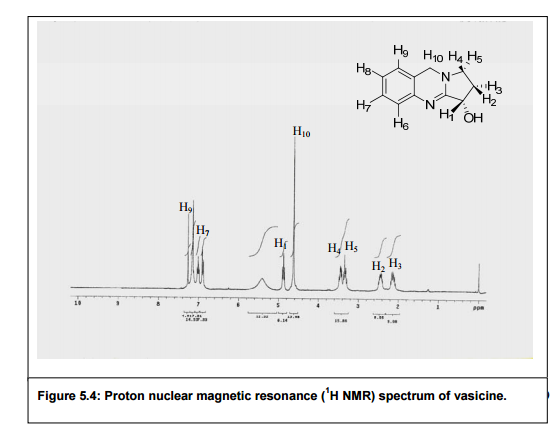 .
.
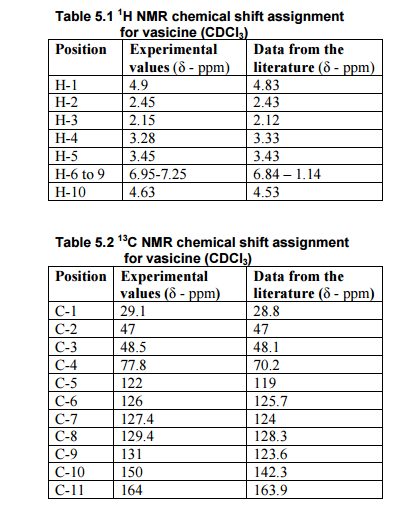
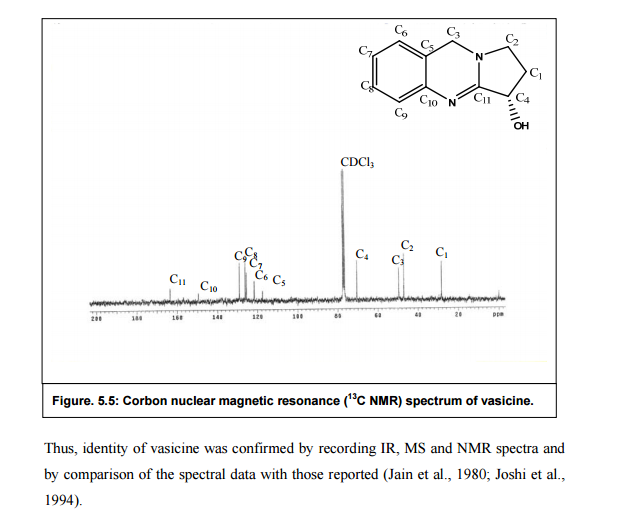


Vasicine (peganine) is a quinazoline alkaloid. It is the active compound of Justicia adhatoda, after which the chemical is named.
Vasicine has been compared to theophylline both in vitro and in vivo.[1] It has also been studied in combination with the related alkaloid vasicinone. Both the alkaloids in combination (1:1) showed pronounced bronchodilatory activity in vivo and in vitro.[2] Both alkaloids are also respiratory stimulants.[2] Vasicine has a cardiac–depressant effect, while vasicinone is a weak cardiac stimulant; the effect can be normalized by combining the alkaloids.[2][3] Vasicine is reported to have a uterine stimulant effect.[3]
Vasicine
Synonym Peganine
Biological Sources It is obtained from the leaves of Adhatoda vasica (L.) Nees (Acanthaceae) (Malabar Nut, Adotodai, Paveltia); and the seeds of Peganum harmala L. (Rutaceae) (Harmel, Syrian Rue, African Rue).
Chemical Structure
1, 2, 3, 9-Tetrahydropyrrolo [2, 1-b] quinazoline-3-ol; (C11H12N2O).
Isolation It is isolated from the leaves of Adhatoda vasica* and also from the seeds of Peganum harmala** by adopting the standard methods of isolation described earlier in this chapter.
Characteristic Features
dl-Form: 1. It is obtained as needles from ethanol having mp 210°C.
- It sublimes on being subjected to high vacuum.
- It is soluble in acetone, alcohol, chloroform; and slightly soluble in water, ether and
benzene.
l-Form: 1. It is obtained as needles from ethanol with mp 212°C.
- Its specific rotation [α ]D14-2540(C = 2.4 in CHCl3); [α ]D14–14 62° (C = 2.4 in ethanol).
Note: In dilute HCl it is obtained as its dextrorotatory form.
Identification Tests
- Hydrochloride dihydrate derivative is obtained as needles having mp 208°C (dry).
- Hydroiodide dihydrate derivative is formed as needles with mp 195°C (dry).
- Methiodide derivative is obtained as needles from methanol having mp 187°C.
- Acetyl vasicine derivative (C11H11N2O COCH3) is formed as crystals having mp 123°C and bp0.01 230-240°C.
Uses
- It is mostly used as an expectorant and bronchodilator.
- It also shows oxytocic properties very similar to those exhibited by oxytocin and methyl ergometrine.
- Vasicine also shows abortifacient action which is due to the release of prostaglandins.
Biosynthesis of Vasicine Various studies in Peganum harmala have evidently revealed vasicine (peganine) to be derived from the anthranilic acid, while the remaining portion of the structure comprising of a pyrrolidine ring provided by ornithine. The probable mechanism of vasicine skeleton may be explained by virtue of the nucleophilic attack from the N-atom present in anthranilate upon the pyrrolidinium cation, ultimately followed by amide formation. However, interestingly this pathway is not being adopted in Justicia adhatoda.

Vasaka
http://www.himalayawellness.com/products/pharmaceuticals/vasaka.htm
Effective respiratory care
Vasaka (Malabar Nut Tree/Adhatoda zeylanica) is well known in Ayurveda for its beneficial effects in respiratory ailments, particularly as an expectorant in bronchitis. The leaves, flowers, fruits and roots are used extensively for treating cold, cough, whooping-cough, chronic bronchitis and asthma.
Vasaka grows throughout India, up to an altitude of 1,300 meters.
Active constituents:
Vasaka contains the pyrroquinazoline alkaloids, including vasicine, vasicol and vasinone along with other minor constituents. Vasicine and vasinone are the major bioactive constituents of Vasaka which have bronchodilatory and antitussive properties.
The alkaloids present in the plant show significant protection against allergen-induced bronchial obstruction.
Herb Functions:
Respiratory care: Vasaka exhibits anti-inflammatory, antitussive and bronchodilatory action which eases congestion and coughing by helping loosen and thin mucus in airways. Vasaka relieves dyspnea by dilating the airways and improves overall lung functions. The herb is an excellent supportive therapy for symptomatic relief in tuberculosis and pulmonary infections.
Indications
- Productive cough
- Bronchitis
- Bronchial asthma
Contraindications:
None
Recommended dose:
One capsule, twice a day or as directed by your physician
Composition:
Each capsule contains 250mg extract of Vasaka
Note: Since Himalaya’s Pure Herbs are in capsule form, some children below 14 years may find it difficult to swallow them. For this reason, Pure Herbs are recommended for children ages 14 and above.
The information on this page is not intended to be a substitute for professional medical advice. Do not use this information to diagnose or treat your problem without consulting your doctor.

http://kumarncsirihbt.weebly.com/publications.html

Adhatoda Vasica (Justicia Adhatoda) – Malabar Nut, Vasa, Vasaka …
Presentation “Herbal drugs for health Herbal drugs for health …
References
- Nepali, Kunal; Sharma, Sahil; Ojha, Ritu; Dhar, Kanaya Lal (2012). “Vasicine and structurally related quinazolines”. Medicinal Chemistry Research 22 (1): 1–15. doi:10.1007/s00044-012-0002-5. ISSN 1054-2523.
- Avula, B.; et al. (2008). “Quantitative determination of vasicine and vasicinone in Adhatoda vasica by high performance capillary electrophoresis” (PDF). Die Pharmazie – An International Journal of Pharmaceutical Sciences 63 (1): 20–22. doi:10.1691/ph.2008.7175.
- ^ Jump up to:a b Rajani, M; Soni, S; Anandjiwala, Sheetal; Patel, G (2008). “Validation of different methods of preparation of Adhatoda vasica leaf juice by quantification of total alkaloids and vasicine”. Indian Journal of Pharmaceutical Sciences 70 (1): 36. doi:10.4103/0250-474X.40329.ISSN 0250-474X.
.png) |
|
| Names | |
|---|---|
| IUPAC name
1,2,3,9-Tetrahydropyrrolo[2,1-b]quinazolin-3-ol
|
|
| Other names
Peganine
|
|
| Identifiers | |
| 6159-56-4 | |
| Jmol interactive 3D | Image |
| PubChem | 72610 |
| Properties | |
| C11H12N2O | |
| Molar mass | 188.23 g·mol−1 |
| Melting point | 210 °C (410 °F; 483 K) |
| Solubility in acetone, alcohol, chloroform | Soluble |
//////
ENJOY SOME ANIMATIONS







ILAPRAZOLE, IY-81149

Ilaprazole
cas 172152-36-2;
Iy 81149; IY-81149; 2-{[(4-methoxy-3-methylpyridin-2-yl)methyl]sulfinyl}-6-(1h-pyrrol-1-yl)-1h-benzimidazole; Aldenon;
MW 366.437, MF C19 H18 N4 O2 S
2-[(4-methoxy-3-methylpyridin-2-yl)methylsulfinyl]-6-pyrrol-1-yl-1H-benzimidazole
Ilaprazole is a proton pump inhibitor (PPI) used in the treatment of dyspepsia, peptic ulcer disease, gastroesophageal reflux disease and duodenal ulcer.
Ilaprazole is chemically known as 2-[[(4-methoxy-3-methyl-2-pyridinyl)methyl]sulfinyl]-6-(lH-pyrrol-l-yl)-lH-benzimidazole
Il-Yang Il Yang Pharmaceutical Company, Ltd.………. Innovator
launched in 2008 by Livzon in China
![]()

Ilaprazole (trade name Noltec) is a proton pump inhibitor (PPI) used in the treatment of dyspepsia, peptic ulcer disease (PUD),gastroesophageal reflux disease (GORD/GERD) and duodenal ulcer. It is available in strengths of 5, 10, and 20 mg.
Clinical studies show that ilaprazole is at least as potent a PPI as omeprazole when taken in equivalent doses. Studies also showed that ilaprazole significantly prevented the development of reflux oesophagitis.
Ilaprazole is developed by Il-Yang Pharmaceutical (Korea), and is still under clinical trials with US FDA. It has launched in Korea and China for the treatment of gastric ulcer, duodenal ulcer, gastroesophageal reflux disease and erosive esophagitis.[1]
Ilaprazole is a substituted benzimidazole proton pump inhibitor first launched in 2008 by Livzon in China for the oral treatment of peptic ulcers. The compound was also being evaluated in early clinical trials at Il-Yang for the treatment of gastroesophageal reflux disease (GERD), but no recent development has been reported. In 2009, development of the compound was discontinued by Takeda Pharmaceuticals North America for the treatment of esophagitis due to a phase II study which did not meet its predefined endpoint.
The drug has been shown to significantly inhibit acute gastric erosion induced by indomethacin, ethanol or stress, acute mepirizole induced duodenal ulcers, and to accelerate the healing of acetic acid induced chronic ulcers through a H+/K+-ATPase inhibition mechanism.
In September 2005, TAP (a joint venture established between Abbott and Takeda which was dissolved in 2008) and Il-Yang signed a license agreement, granting the latter development and distribution rights to the drug candidate worldwide outside of Korea and China.
Il-Yang Pharm. Co., Ltd., Korea has developed a Novel PPI, i.e. racemic 5-(1H-pyrrol-1-yl)- 2[[(3-methyl-4-methoxy-2-Pyridyl)-methyl]sulfinyl]-benzimidaziole[1,2], which shows superior anti-ulcer effects as compared to Omeprazole in the treatment of GORD(gastro-oesophageal reflux diseases), gastric ulcer and duodenal ulcer (KR 179,401 and US 5,703,097). Gastric and duodenal ulcers are a gastrointestinal disease caused by various factors such as mental stress, dietary habit, intake of irritable food, and the like. The direct cause of peptic ulcers is damage to the gastric membrane due to excessive secretion of gastric acid.
Since their introduction in the late 1980s, proton pump inhibitors have improved the treatment of various acid-related gastrointestinal (GI) disorders, including gastroesophageal reflux disease (GERD), peptic ulcer disease, Zollinger-Ellison Syndrome (ZES), ulcers, and nonsteroidal anti-inflammatory drug (NSAID)-induced gastropathy. GERD encompasses three disease categories: non-erosive reflux disease (NERD), erosive esophagitis, and Barrett’s esophagus. ZES is caused by a gastrin-secreting tumor of the pancreas that stimulates the acid-secreting cells of the stomach to maximal activity. Proton pump inhibitors have also be used to treat ulcers such as duodenal, gastric, and NSAID-associated gastric/duodenal ulcers.
As antisecretory drugs, proton pump inhibitors are currently the recommended first line therapy, being viewed as more effective than other treatments. In general, proton pump inhibitors offer superior gastric acid suppression over histamine H2-receptor blockers. The use of proton pump inhibitors by patients who suffer from gastric acid-related disorders is generally believed to have led to an increase in their quality of life, productivity, and overall well being.
Proton pump inhibitors are also used to treat extra-esophageal manifestations of GERD (asthma, hoarseness, chronic cough, non-cardiac chest pain), and with antibiotics for Helicobacter pylori eradication. The goals of GERD management are threefold: prompt and sustained symptom control, healing of the injured esophageal mucosa and prevention of GERD-related complications (including stricture Formation, Barrett’s esophagus, and/or adenocarcinoma). Pharmacological therapy with proton pump inhibitors Forms the basis of both acute and long-term management of GERD. Proton pump inhibitors provide effective relief of symptoms and healing of the esophagitis, as well as sustaining long-term remission.
Although therapeutic efficacy is the primary concern for a therapeutic agent, the solid-state form, as well as the salt form, and the properties unique to the particular form of a drug candidate are often equally important to its development. Each solid state form (crystalline or amorphous) of a drug candidate can have different physical and chemical properties, for example, solubility, stability, or the ability to be reproduced. These properties can impact the ultimate pharmaceutical dosage form, the optimization of manufacturing processes, and absorption in the body. Moreover, finding the most adequate form for further drug development can reduce the term and the cost of that development.
Ilaprazole, 2[[(4-methoxy-3-methyl-2-pyridinyl)-methyl]sulfinyl]-5-(1H-pyrrol-1-yl) 1H-Benzimidazole, is a substituted benzimidazole that acts as a proton pump inhibitor. Ilaprazole selectively and irreversibly inhibits gastric acid secretion through inhibition of the hydrogen-potassium adenosine triphosphatase (H+K+-ATPase) (proton pump) mechanism. Inhibition of the proton pump occurs by formation of disulfide covalent bonds with accessible cysteines on the enzyme. Ilaprazole has a prolonged duration of action that persists after their elimination from plasma. See, for example, U.S. Pat. Nos. 5,703,097 and 6,280,773, which are incorporated herein by reference.
Ilaprazole has the empirical formula C19H18N4O2S having a molecular weight of 366.44 daltons. Ilaprazole is a chiral molecule and has the following structural Formula (I):

Ilaprazole, like all proton pump inhibitors, possesses the unique feature of a chiral sulfur atom, S*. This can be depicted as follows with the lone pair of electrons on the chiral sulfur atom occupying one position in each stereoisomer, as shown below:

The absolute structure and absolute confirmation of (−)-S-ilaprazole was made through single crystal structure determination and is shown below. See Example 7 of co-pending U.S. application Ser. No. 11/966,808 of Brackett et al. entitled, “Solid State Forms of Enantiopure Ilaprazole” filed Dec. 28, 2007, herein incorporated by reference in its entirety.

Thus, its complimentary enantiomer is (+)-R-ilaprazole, as shown below.


SYN 1
EP 0696281; JP 1997503000; US 5703097; WO 9523140

The condensation of 2-(chloromethyl)-4-methoxy-3-methylpyridine (I) with 5-(1-pyrrolidinyl)benzimidazole-2-thiol (II) by means of NaOH in hot methanol gives 2-(4-methoxy-3-methyl-2-pyridylmethylsulfanyl)-5-(1-pyrrolidinyl)benzimidazole (III), which is finally oxidized with m-chloroperbenzoic acid (MCPBA) in chloroform.
SYN 2
J Med Chem 1992,35(6),1049
http://pubs.acs.org/doi/pdf/10.1021/jm00084a010

3-Methoxy-2-methylpyridine (VII), prepared by methylation of 2-methyl-3-pyridinol (VI), was converted to the N-oxide (VIII) employing peracetic acid. Nitration of the pyridine N-oxide (VIII) with concentrated nitric acid gave the 4-nitro derivative (IX). Subsequent displacement of the nitro group of (IX) by sodium methoxide led to the dimethoxypyridine N-oxide (X). Rearrangement of the N-oxide group of (X) in hot acetic anhydride produced the acetoxymethyl pyridine (XI). After basic hydrolysis of the acetate ester (XI), the resultant hydroxymethyl pyridine (XII) was chlorinated by SOCl2, yielding chloride (XIII). Condensation between mercapto benzimidazole (V) and the chloromethyl pyridine (XIII) in ethanolic NaOH led to the sulfide adduct (XIV). This was finally oxidized to the desired sulfoxide by using meta-chloroperbenzoic acid in CH2Cl2. The oxidation of sulfide (XIV) has also been performed employing sodium perborate, sodium percarbonate in the presence of ammonium molybdate, or tert-butyl hydroperoxide in the presence of vanadyl acetylacetonate.

The synthesis of IY-81149 can be obtained according to Scheme 22875502a. The oxidation of 2,3-lutidine (I) with hydrogen peroxide in acetic acid affords 2,3-dimethylpyridine-N-oxide (II), which is treated with sulfuric acid and nitric acid to give the corresponding nitro compound (III). The treatment of (III) with NaOH in methanol gives 2,3-dimethyl-4-methoxypyridine-N-oxide (IV), which is reacted with acetic acid and acetic anhydride and oxidized in refluxing NaOH, yielding 3-methyl-4-methoxypyridine-2-methanol (V). The chlorination of (V) with thionylchloride in CH2Cl2 affords 3-methyl-4-methoxy-2-chloromethylpyridine (VI). The reaction of 2-mercapto-5-nitrobenzimidazole (VII) with iron and concentrated HCl in refluxing ethanol and water gives monoamine (VIII), which by condensation with 2,5-dimethoxytetrahydrofuran (IX) in acetic acid yields 2-mercapto-5-(1-pyrrolyl)benzimidazole (X). The condensation of (VI) with (X) by means of NaOH in methanol gives 2-[(4-methoxy-3-methyl-2-pyridinyl)methylsulfanyl]-5-(1H-pyrrol-1-yl)-1H-benzimidazole (XI), which is finally treated with m-chloroperoxybenzoic acid (m-CPBA) in chloroform.
PATENT
http://www.google.com/patents/WO2013114232A1?cl=en
Ilaprazole is a proton pump inhibitor (PPI) used in the treatment of dyspepsia, peptic ulcer disease, gastroesophageal reflux disease and duodenal ulcer.
Ilaprazole is chemically known as 2-[[(4-methoxy-3-methyl-2-pyridinyl)methyl]sulfinyl]-6-(lH-pyrrol-l-yl)-lH-benzimidazole havin following structure

There are very few patent documents related to crystallization of ilaprazole.
The example 2 of Indian Patent No. 183088 describes crystallization of ilaprazole from mixture of ethyl acetate and ether.
Indian patent application No. 3607/DELNP/2009 discloses various crystalline forms: A, B, E, F, I of ilaprazole and process for their preparation. The crystalline form B of ilaprazole is obtained by crystallization from acetone/triethylamine in a refrigerator for 11 days. The form B is characterized by peaks at 12.6 and 18.1 degree 2Θ in X-ray powder differactogram.
Another Indian patent application No. 3634/DELNP/2009 discloses various solvates of ilaprazole, these are crystalline form C (1,4-dioxane hemisolvate), D (tetrahydrofuran hemisolvate), G (methanol solvate), K (hydrate) of ilaprzole and process for their preparation.
International Patent Publication No. WO 2011/071314 discloses process for the preparation of Form A and Form B. The process for the preparation of Form A involves conversion of ilaprazole to its inorganic salt followed by neutralization with suitable acid in a solvent. The process for preparation of Form B requires use of multiple solvents for the crystallization.
The earlier processes crystallization of ilaprazole has following disadvantages:
i) process is laborious due to concentration of solvent carried out several times;
ii) difficult to obtain the pharmaceutically acceptable purity; and
iii) time consuming.
The physical or chemical properties of a drug can vary depending on the crystalline form of the drug, and such physical and chemical properties can greatly influence a suitable dosage form of the drug, the optimization of a process for preparing the drug, and the in vivo absorption of the drug. The discovery of the most appropriate crystalline form of a drug in a procedure for developing the drug enables the development time and cost to be reduced.
Patent
http://www.google.co.in/patents/US8592599
FIG. 22 is the proton NMR spectrum of racemic ilaprazole, Form F.
FIG. 23 is the solid state 13C CP/MAS ssNMR spectrum of racemic ilaprazole, Form F.
FIG. 24 is the IR spectrum of racemic ilaprazole, Form F.
FIG. 25 is the RAMAN spectrum of racemic ilaprazole, Form F.




References
PatentSubmittedGranted
Solid dosage form comprising proton pump inhibitor and suspension made thereof [US2006134210]2006-06-22
Optimally stabilized microgranule comprising 5-pyrrolyl-2-pyridylmethylsulfinylbenzimidazole derivative [US6280773]2001-08-28
Method and system for dosing a pharmaceutical sample in a packaging machine [US7536843]2007-07-262009-05-26
Parenteral Formulation Comprising Proton Pump Inhibitor Sterilized in its Final Container by Ionizing Radiation [US2009111856]2009-04-30
SOLID STATE FORMS OF ENANTIOPURE ILAPRAZOLE [US2008200515]2008-08-21
Injection [US2009036406]2009-02-05
Pharmaceutical compositions of ilaparazole [US2008050444]2008-02-28
Substituted sulfoxide compounds, methods for preparing the same and use thereof [US2006217423]2006-09-28
CRYSTALLINE FORMS OF SOLVATED ILAPRAZOLE [US7989632]2008-08-212011-08-02
SOLID STATE FORMS OF RACEMIC ILAPRAZOLE [US7999110]2008-08-212011-08-16
| Patent | Submitted | Granted |
|---|---|---|
| SOLID DOSAGE FORM COMPRISING PROTON PUMP INHIBITOR AND SUSPENSION MADE THEREOF [US2013273168] | 2013-06-05 | 2013-10-17 |
| METHOD AND APPARATUS FOR PRODUCING OXIDIZED COMPOUND [US2014100370] | 2013-10-31 | 2014-04-10 |
| New Combination Dosage Form [US2007122470] | 2007-05-31 | |
| Agents for the treatment of lower abdominal disorders [US2006235053] | 2004-05-04 | 2006-10-19 |
| Use of proton pump inhibitors for the treatment of noncardiac chest pain [US2005154026] | 2003-03-11 | 2005-07-14 |
| Use of proton pump inhibitors for the treatment of airway disorders [US2005131026] | 2003-03-11 | 2005-06-16 |
| Solid Dosage Form Comprising Proton Pump Inhibitor and Suspension Made Thereof [US2008020053] | 2005-12-20 | 2008-01-24 |
| Synthesis of prazole compounds [US8895271] | 2010-12-08 | 2014-11-25 |
| ORALLY-DISINTEGRATING SOLID PREPARATION [US2015037423] | 2014-10-21 | 2015-02-05 |
| Patent | Submitted | Granted |
|---|---|---|
| SUBSTITUTED SULFOXIDE COMPOUNDS, METHODS FOR PREPARING THE SAME AND USE THEREOF [US8017784] | 2008-09-25 | 2011-09-13 |
| SUBSTITUTED BENZIMIDAZOLES [US2008255200] | 2008-10-16 | |
| Method and Apparatus for Producing Oxidized Compound [US2008262235] | 2008-10-23 | |
| ORALLY DISINTEGRATING SOLID PREPARATION [US2010316709] | 2010-12-16 | |
| Prodrugs of proton pump inhibitors including the 1h-imidazo[4,5-b] pyridine moiety [US2010317689] | 2010-12-16 | |
| SOLID STATE FORMS OF RACEMIC ILAPRAZOLE [US2011046184] | 2011-02-24 | |
| PROCESS FOR PREPARING INTERMEDIATE COMPOUND FOR SYNTHESIZING AN ANTIULCERANT [US2011071302] | 2011-03-24 | |
| SOLID STATE FORMS OF RACEMIC ILAPRAZOLE [US2011082174] | 2011-04-07 | |
| Oral Pharmaceutical Dosage Form Comprising as Active Ingredients a Proton Pump Inhibitor together with Acetyl Salicyclic Acid [US2010178334] | 2010-07-15 | |
| Prodrugs of proton pump inhibitors including the (1h-pyrrol-1-yl)-1h-benzimidazole moiety [US2010113524] | 2010-05-06 |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
2-[(RS)-[(4-methoxy-3-methylpyridin-2-yl)methyl]sulfinyl]-5-(1H-pyrrol-1-yl)-1H-benzimidazole
|
|
| Clinical data | |
| Trade names | Noltec |
| Routes of administration |
Oral |
| Identifiers | |
| CAS Number | 172152-36-2 |
| ATC code | None |
| PubChem | CID 214351 |
| ChemSpider | 185839 |
| UNII | 776Q6XX45J |
| ChEMBL | CHEMBL2106370 |
| Chemical data | |
| Formula | C19H18N4O2S |
| Molar mass | 366.436820 g/mol |
/////////IY-81149, ILAPRAZOLE
CC1=C(C=CN=C1CS(=O)C2=NC3=C(N2)C=C(C=C3)N4C=CC=C4)OC
Revised Ph. Eur. Chapter on Raman Spectroscopy introducing PAT

The revised General Ph. Eur. Chapter on Raman Spectroscopy (2.2.48) comes into operation on April 1, 2016. The Chapter now includes hand-held devices and adaption to PAT purposes. Find out more about the revised Ph. Eur. Raman Spectroscopy chapter.
The revision of the General Ph. Eur. Chapter on Raman Spectroscopy (2.2.48) comes into operation on April 1, 2016.
The revised Chapter has been published in Ph. Eur. Supplement 8.7. It has been completely rewritten to include now available handheld devices and adaption to Process Analytical Technology (PAT). Raman Spectroscopy has received more and more attention in pharmaceutical industry. Hand-held instruments are suitable for rapid identification purposes for example in the incoming goods control of raw and packaging materials.
Hand-held instruments require different tolerances for wavenumber scale verification than benchtop models. Therefore, an inter-laboratory study was organized and the results have been published in a corresponding paper titled “Rationale for the update of the European Pharmacopoeia general chapter 2.2.48. Raman Spectroscopy” in Pharmeuropa Bio & Scientific Notes 2015.
In addition, the chapter has been revised to focus more on the potential use of Raman Spectroscopy in a PAT environment. Raman Spectrometry is increasingly used for PAT and chemical imaging applications. Therefore, a chapter on Chemical Imaging is also under elaboration. The draft chapter on Chemical Imaging will be published for public consultation in Pharmeuropa 28.2 (April 2016).
For more information please visit the Newsroom on the EDQM website
///////////////Raman Spectroscopy, PAT
Palbociclib, Sun Pharmaceutical Industries Ltd, New patent, WO 2016016769

Palbociclib
WO2016016769, A PROCESS FOR THE PREPARATION OF PALBOCICLIB
SUN PHARMACEUTICAL INDUSTRIES LIMITED [IN/IN]; Sun House, Plot No. 201 B/1 Western Express Highway Goregaon (E) Mumbai, Maharashtra 400 063 (IN)
TYAGI, Vipin; (IN).
MOHAMMAD, Kallimulla; (IN).
RAI, Bishwa Prakash; (IN).
PRASAD, Mohan; (IN)

The present invention relates to a process for the preparation of palbociclib utilizing a silyl-protected crotonic acid derivative to produce a silyl-protected 5-(1-methyl-3 carboxy-prop-1-en-1-yl)-2-chloro-piperazine followed by intramolecular cyclization of the compound the piperazine intermediate to produce 2-chloro-8-cyclopentyl-5-methyl-8H-pyrido[2,3-d]pyrimidin-7-one which is then converted to palbociclib.
Sun Pharma managing director Dilip Shanghvi.
Palbociclib chemically is 6-acetyl-8-cyclopentyl-5-methyl-2-[[5-(l-piperazinyl)-2-pyridinyl]amino]pyrido 2,3-d]pyrimidin-7(8H)-one, represented by the Formula I.

Formula I
U.S. Patent No. 6,936,612 discloses palbociclib and a process for the preparation of its hydrochloride salt.
U.S. Patent No. 7,781,583 discloses a process for the preparation of palbociclib, wherein 2-chloro-8-cyclopentyl-5-methyl-8H-pyrido[2,3-d]pyrimidin-7-one of Formula II

Formula II
prepared by reacting 5-bromo-2-chloro-N-cyclopentylpyrimidin-4-amine of Formula III

Formula III
with crotonic acid.
U.S. Patent No. 7,863,278 discloses polymorphs of various salts of palbociclib and processes for their preparation.
A first aspect of the present invention provides a process for the preparation of a compound of Formula IV,

Formula IV
wherein R is trimethylsilyl, dimethylsilyl, or fert-butyldimethylsilyl
comprising reacting a crotonic acid derivative of Formula V

Formula V
wherein R is trimethylsilyl, dimethylsilyl, or fert-butyldimethylsilyl
with a compound of Formula III

Formula III
in the presence of a palladium catalyst, a base, and optionally a ligand to give a compound of Formula IV.
A second aspect of the present invention provides a process for the preparation of palbociclib of Formula I,

Formula I
a) reacting a crotonic acid derivative of Formula V,

Formula V
wherein R is trimethylsilyl, dimethylsilyl, or fert-butyldimethylsilyl with a compound of Formula III

Formula III
in the presence of a palladium catalyst, a base, and optionally a ligand to give a compound of Formula IV

Formula IV
wherein R is trimethylsilyl, dimethylsilyl, or fert-butyldimethylsilyl; and b) converting the compound of Formula IV to palbociclib of Formula I.
A third aspect of the present invention provides a process for the preparation of a compound of Formula II

Formula II
a) reacting a crotonic acid derivative of Formula V,

Formula V
wherein R is trimethylsilyl, dimethylsilyl, or fert-butyldimethylsilyl with a compound of Formula III

Formula III
in the presence of a palladium catalyst, a base, and optionally a ligand to give a compound of Formula IV,

Formula IV
wherein R is trimethylsilyl, dimethylsilyl, or fert-butyldimethylsilyl; and b) intramolecular cyclization of the compound of Formula IV to give a
compound of Formula II.
A fourth aspect of the present invention provides a process for the preparation of palbociclib of Formula I

Formula I
comprising:
a) reacting a crotonic acid derivative of Formula V,

Formula V
wherein R is trimethylsilyl, dimethylsilyl, or fert-butyldimethylsilyl with a compound of Formula III

Formula III
in the presence of a palladium catalyst, a base, and optionally a ligand to give a compound of Formula IV

Formula IV
wherein R is trimethylsilyl, dimethylsilyl, or fert-butyldimethylsilyl;
intramolecular cyclization of the compound of Formula IV to give a compound of Formula II; and

Formula II
converting the compound of Formula II to palbociclib of Formula I.
EXAMPLES
Preparation of 2-chloro-8 -cyclopentyl-5 -methyl-8H-pyrido Γ2.3 – lpyrimidin-7-one (Formula II)
Step a: Preparation of trimethylsilyl (2£)-but-2-enoate (Formula V, when R is trimethylsilyl)
Crotonic acid (18.68 g) was taken in dichloromethane (80 mL) at room
temperature to obtain a solution. Hexamethyldisilazane (HMDS) (21 g) followed by imidazole (0.4 g) was added to the solution at room temperature under stirring. The reaction mixture was refluxed for 2 hours. Dichloromethane was recovered completely under vacuum at 45°C. Dichloromethane (200 mL) was again added to the reaction mixture, and then recovered completely under vacuum at 45°C. The colorless liquid obtained was taken as such for next step.
Step b: Preparation of 2-chloro-8-cyclopentyl-5-methyl-8H-pyrido[2,3-i/|pyrimidin-7-one (Formula II)
Method A
Trimethylsilyl (2£)-but-2-enoate (obtained from step a) and diisopropylethylamine (52 mL) were added to a solution of 5-bromo-2-chloro-N-cyclopentylpyrimidin-4-amine (20 g, Formula III) in tetrahydrofuran (100 mL) at room temperature under a nitrogen atmosphere. The reaction system was degassed under vacuum and then flushed with nitrogen; this evacuation procedure was repeated three times. Trans-dichlorobis(acetonitrile) palladium (II) (0.970 g) followed by the addition of tri-o-tolylphosphine (0.770 g) was added to the reaction mixture under a nitrogen atmosphere.
The reaction system was again degassed under vacuum and then flushed with nitrogen; this evacuation procedure was repeated three times. The reaction mixture was heated at 75°C to 80°C overnight. The progress of the reaction was monitored by thin layer chromatography (TLC) (60% ethyl acetate/toluene). Trans-dichlorobis(acetonitrile) palladium (II) (0.725 g) was again added followed by the addition of tri-o-tolylphosphine (0.725 g) to the reaction mixture at 75°C to 80°C. The reaction mixture was heated at 75°C to 80°C for 4 hours. After completion of the reaction, acetic anhydride (17 mL) was added, and then the mixture was stirred at 75°C to 80°C for 3 hours. The reaction mixture was cooled to room temperature. Dichloromethane (100 mL) and IN hydrochloric acid (100 mL) were added and then the mixture was stirred for 10 minutes. The layers were separated and the aqueous layer was re-extracted with dichloromethane (40 mL) and separated. The combined organic layers were washed with a 5% sodium bicarbonate solution (200 mL) at room temperature. The organic layer was separated and activated carbon (2 g) was added to the mixture. The mixture was stirred for 20 minutes at room temperature. The mixture was filtered through a Hyflo® bed and then washed with dichloromethane (40 mL). The organic layer was evaporated under vacuum to obtain a residue. Isopropyl alcohol (80 mL) was added to the residue and the solvent was evaporated under reduced pressure until 40 mL of isopropyl alcohol remained. Isopropyl alcohol (40 mL) was again added to the mixture, and then the solvent was evaporated under reduced pressure until 20 mL of isopropyl alcohol remained. The mixture was stirred for 3 hours at room temperature. The product was filtered, thenwashed with isopropyl alcohol (20 mL), and then dried under vacuum at 45°C to obtain the title compound.
Yield: 0.535% w/w
Chromatographic purity: 99.51%
Method B
Trimethylsilyl (2£)-but-2-enoate (obtained from step a) and diisopropylethylamine (26.5 mL) were added to a solution of 5-bromo-2-chloro-N-cyclopentylpyrimidin-4-amine (Formula III, 10 g) in tetrahydrofuran (50 mL) at room temperature under a nitrogen atmosphere. The reaction system was degassed under vacuum and then flushed with nitrogen; this evacuation procedure was repeated three times. Trans-dichlorobis(acetonitrile) palladium (II) (1.39 g) followed by the addition of tri-o- tolylphosphine (1.1 g) was added to the reaction mixture under a nitrogen atmosphere. The reaction system was degassed under vacuum and then flushed with nitrogen; this evacuation procedure was repeated three times. The reaction mixture was heated at 75 °C to 80°C overnight. After completion of the reaction, acetic anhydride (20 mL) was added, and then the mixture was stirred at 75°C to 80°C for 3 hours. The reaction mixture was cooled to room temperature. Dichloromethane (50 mL) and IN hydrochloric acid (50 mL) were added, and then the mixture was stirred for 10 minutes. The layers were separated and the aqueous layer was re-extracted with dichloromethane (20 mL) and separated. The combined organic layers were washed with a 5% sodium bicarbonate solution (200 mL) at room temperature. The organic layer was separated and activated carbon (1 g) was added to the mixture. The mixture was stirred for 20 minutes at room temperature. The mixture was filtered through a Hyflo® bed and then washed with dichloromethane (20 mL). The organic layer was evaporated under vacuum to obtain a residue. Isopropyl alcohol (40 mL) was added to the residue and then the solvent was evaporated under reduced pressure until 20 mL of isopropyl alcohol remained. Isopropyl alcohol (20 mL) was again added to the mixture and then the solvent was evaporated under reduced pressure until 20 mL of isopropyl alcohol remained. The mixture was stirred for 3 hours at room temperature. The product was filtered and washed with isopropyl alcohol (10 mL), and then dried under vacuum at 45°C to obtain the title compound.
Yield: 0.46% w/w
Chromatographic purity: 98.1%
/////Palbociclib, Sun Pharmaceutical Industries Ltd, New patent, WO 2016016769
WO2016016279, NEW PATENT, DOLUTEGRAVIR SODIUM, LEK PHARMACEUTICALS D.D. , SANDOZ AG

WO2016016279, NOVEL HYDRATES OF DOLUTEGRAVIR SODIUM
LEK PHARMACEUTICALS D.D. [SI/SI]; Verovskova 57 1526 Ljubljana (SI).
SANDOZ AG [CH/CH]; Lichtstrasse 35 CH-4056 Basel (CH)
HOTTER, Andreas; (AT).
THALER, Andrea; (AT).
LEBAR, Andrija; (SI).
JANKOVIC, Biljana; (SI).
NAVERSNIK, Klemen; (SI).
KLANCAR, Uros; (SI).
ABRAMOVIC, Zrinka; (SI)
The present invention relates to novel hydrates of sodium dolutegravir and their methods of preparation. In addition, the invention relates to a novel crystalline form of sodium dolutegravir, which is a useful intermediate for the preparation of one of the new hydrates. The invention also relates to the use of the new hydrates for the production of pharmaceutical compositions.
Finally, the invention relates to pharmaceutical compositions comprising an effective amount of the novel hydrates, oral dosage forms comprising these pharmaceutical compositions, a process for preparing said oral dosage forms, and the use of such pharmaceutical compositions or dosage forms in the treatment of retroviral infections such as HIV infections -1.
Dolutegravir, chemically designated (4f?, 12aS)-/V-(2,4-difluorobenzyl)-7-hydroxy-4-methyl-6,8-dioxo-3,4,6,8, 12, 12a-hexahydro-2H-pyrido[1 ‘,2’:4,5]pyrazino[2, 1- ?][1 ,3]oxazine-9-carboxamide, is a human immunodeficiency virus type 1 (HIV-1 ) integrase strand transfer inhibitor (INSTI) indicated in combination with other a nti retroviral agents for the treatment of HIV-1 infection. The marketed finished dosage form (TIVICAY™) contains dolutegravir as its sodium salt, chemically denominated sodium (4f?,12aS)-9-((2,4-difluorobenzyl)carbamoyl)-4-methyl-6,8-dioxo-3,4,6,8,12, 12a-hexahydro-2H-pyrido[1 ‘,2’:4,5]pyrazino[2, 1- ?][1 ,3]oxazin-7-olate, which is represented by the following general chemical formula (I):

(I)
WO 2010/068253 A1 discloses a monohydrate and an anhydrous form of dolutegravir sodium as well as a crystalline form of the free compound. Processes for the preparation of said forms are also provided in the application.
WO 2013/038407 A1 discloses amorphous dolutegravir sodium and processes for preparing the same.
Hydrates of pharmaceutical drug substances are of particular interest as they provide new opportunities for preparing novel pharmaceutical compositions with improved quality, activity and/or compliance. This is due to the fact that hydrates have different physicochemical properties compared to their anhydrous counterparts such as melting point, density, habitus, chemical and physical stability, hygroscopicity, dissolution rate, solubility, bioavailability etc., which influence the formulation process and also impact the final drug product.
If an anhydrous form is selected, phase changes during the formulation process induced by hydrate formation must be avoided. This can be particularly difficult if for example wet granulation is used with a substance that is able to form hydrates like dolutegravir sodium.
Hence, a stable hydrate of dolutegravir sodium would allow to easily formulate dolutegravir sodium in a controlled manner and subsequently also facilitate storage and packaging.
However, the so far known dolutegravir sodium monohydrate disclosed in WO 2010/068253 A1 shows excessive water uptake when exposed to moisture and on the other hand already dehydrates below 30% relative humidity.
Therefore, there is a need for hydrates of dolutegravir sodium with improved physicochemical properties, e.g. for hydrates which are stable over a broad humidity range, in particular for hydrates absorbing only low amounts of water at elevated humidity and on the other hand preserving their crystal structure also at dry conditions. In addition, there is a need for pharmaceutical compositions comprising these hydrates, and thus also for hydrates that allow for improved formulation of dolutegravir sodium in pharmaceutical compositions.
SUMMARY OF THE INVENTION
The present invention relates to novel hydrates of dolutegravir sodium and to processes for their preparation. Specifically, the present invention provides crystalline forms of dolutegravir sodium of formula (I) according to respective claims 1 , 5 and 6, with preferred embodiments being set forth in sub-claim 2. The present invention also provides processes for their preparation according to respective claims 3, 7 and 8, with preferred process embodiments being set forth in sub-claim 4. The present invention further provides the uses according to claims 9 and 16, and a pharmaceutical composition according to claim 10, and preferred embodiments thereof according to sub-claims 1 1 and 12. The present invention also provides a process for the preparation of the pharmaceutical composition according to claim
13, and preferred embodiments thereof according to sub-claim 14. The pharmaceutical composition for therapeutic use is set forth in claim 15.
The novel hydrates are physically and chemically stable over a broad humidity range, show only low water uptakes when exposed to moisture and are even stable at dry conditions. Therefore, the novel hydrates are especially suitable for the preparation of pharmaceutical compositions, e.g. in terms of time and costs.
In particular, it has been found that crystal Form HxA exhibits improved properties which allow for improved formulation of Form HxA in pharmaceutical compositions.
In addition, the present invention relates to a novel crystalline form of dolutegravir sodium, which, for the first time, allows the preparation of one of the novel hydrates and is therefore a valuable intermediate.
///////////
WO2016016279, NEW PATENT, DOLUTEGRAVIR SODIUM, LEK PHARMACEUTICALS D.D. , SANDOZ AG

{kind=link}