
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
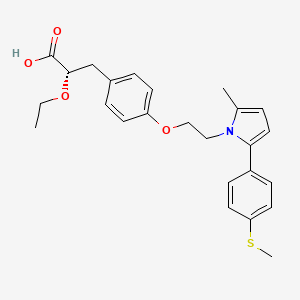
Saroglitazar, Lipaglyn by Zydus Cadila

(2S)-2-Ethoxy-3-[4-(2-{2-methyl-5-[4-(methylsulfanyl)phenyl]-1H-pyrrol-1-yl}ethoxy)phenyl]propanoic acid
(αS)-α-Ethoxy-4-[2-[2-methyl-5-[4-(methylthio)phenyl]-1H-pyrrol-1-yl]ethoxy]benzenepropanoic Acid
alpha-ethoxy-4-(2-(2-methyl-5-(4-methylthio)phenyl))-1H-pyrrol-1-yl)ethoxy))benzenepropanoic acid
alpha-ethoxy-4-(2-(2-methyl-5-(4-methylthio)phenyl))-1H-pyrrol-1-yl)ethoxy))benzenepropanoic acid magnesium salt
(2S)-2-ethoxy-3-[4-[2-[2-methyl-5-(4-methylsulfanylphenyl)pyrrol-1-yl]ethoxy]phenyl]propanoic acid
Benzenepropanoic acid, α-ethoxy-4-[2-[2-methyl-5-[4-(methylthio)phenyl]-1H-pyrrol-1-yl]ethoxy]-, (αS)-
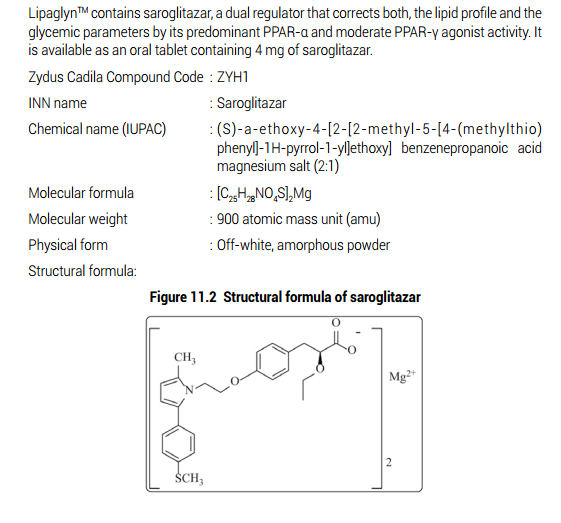
ZYH1 compound
Cas no 495399-09-2
Saroglitazar, Lipaglyn
| Molecular Weight | 439.56706 g/mol |
|---|---|
| Molecular Formula | C25H29NO4S |
Cadila Healthcare Ltd innovator
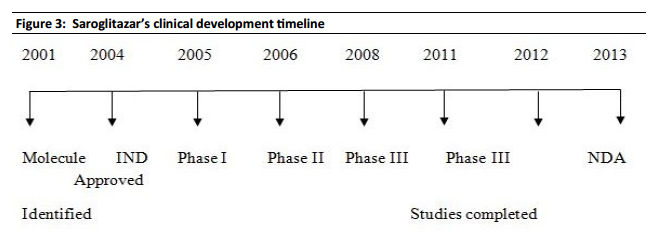
Zydus-Cadila has developed and launched saroglitazar (ZYH-1; Lipaglyn; structure shown), a lipid metabolism modulator, a potent PPAR-alpha agonist with relatively weak PPAR-gamma activity, an insulin sensitizer (glucose-lowering agent), for the once-daily oral treatment of metabolic disorders, including diabetic dyslipidemia and hypertriglyceridemia . The company is also developing saroglitazar for the potential treatment of lipodystrophy, nonalcoholic steatohepatitis (NASH) and type II diabetes.
In June 2013, the Drug Controller General of India (DCGI) approved the drug for launch in India ; in September 2013, the drug was launched. In May 2014, a phase III trial for lipodystrophy was initiated . In January 2015, a phase III study for NASH was initiated .
In February 2015, phase III development was ongoing in type II diabetes . In November 2015, a phase II trial was planned in the US . In June 2016, the US FDA approved the company’s plan to initiate a phase II trial of saroglitazar in patients with NASH .
In June 2012, the company was seeking to outlicense the drug for regional/global partnerships.
By June 2012, an NDA filing had been made for dyslipidemia . In June 2013, the DCGI approved the drug for launch in India . By September 2013, the drug was launched for dyslipidemia and hypertriglyceridemia .
Saroglitazar was approved by the Drug Controller General of India (DCGI) on June 5, 2013. It was developed and marketed as Lipaglyn® by Zydus cadila.
Saroglitazar is novel first in class drug which acts as a dual PPAR agonist at the subtypes α (alpha) and γ (gamma) of the peroxisome proliferator-activated receptor (PPAR). Agonist action at PPARα lowers high blood triglycerides, and agonist action on PPARγ improves insulin resistance and consequently lowers blood sugar. It is indicated for for the treatment of diabetic dyslipidemia and hypertriglyceridemia with type 2 diabetes mellitus not controlled by statin therapy.
Lipaglyn® is available as tablet for oral use, containing 4 mg of free Saroglitazar. The recommended dose is 4 mg orally once daily.
Zydus-Cadila has developed and launched saroglitazar for treating diabetic dyslipidemia and hypertriglyceridemia.
In September 2013, saroglitazar was launched in India for treating dyslipidemia and hypertriglyceridemia.
As of March 2015, Zydus-Cadila is developing saroglitazar for treating nonalcoholic steatohepatitis and type II diabetes (both in phase III clinical trials).


Saroglitazar (INN, trade name Lipaglyn) is a drug for the treatment of type 2 diabetes mellitus and dyslipidemia. It is approved for use in India by the Drug Controller General of India.[1] Saroglitazar is indicated for the treatment of diabetic dyslipidemia andhypertriglyceridemia with type 2 diabetes mellitus not controlled by statin therapy. In clinical studies, saroglitazar has demonstrated reduction of triglycerides (TG), LDL cholesterol, VLDL cholesterol, non-HDL cholesterol and an increase in HDL cholesterol a characteristic hallmark of atherogenic diabetic dyslipidemia (ADD). It has also shown favorable Anti-diabetic medication property by reducing the fasting plasma glucose and HBA1c in diabetes patients. The recommended dose of saroglitazar is one tablet of 4 mg once a day.
In February 2013, Saroglitazar became the first glitazar that has been approved by any FDA for clinical use. Saroglitazar is marketed under the trade name Lipaglyn and developed by Zydus Cadila. Saroglitazar (2 and 4 mg q.d.) is currently approved in India by Drug Controller General of India (DCGI ) for the management of diabetic dyslipidemia and hypertriglyceridemia in T2DM not controlled by statin therapy. Lipaglyn provides the option of a once-daily oral therapy for the patients suffering from diabetic dyslipidemia.
Saroglitazar has another first attached to it. It is the first indigenously developed NCE by any Indian company; in this case Zydus Cadila.
Lipaglyn is indicated 4 mg (or 2 mg where such a need arise) oral dose once daily.
Saroglitazar Synthesis
http://ayurajan.blogspot.in/2016/01/saroglitazar.html
WO2003009841A1:

Identification:

| 1H NMR (Estimated) for Saroglitazar |
Experimental: 1H NMR: 1.14 (3H, t, J = 6.9Hz); 2.37 (3H, s); 2.48 (3H, s); 2.92-3.06 (2H, m); 3.32-3.42 (1H, m); 3.57-3.64 (1H, m); 3.9 (2H, t, J=6.36 Hz); 4.0 (1H, dd); 4.28(2H, t, J = 6.2 Hz); 5.9 (1H, d, J = 3.3 Hz); 6.08 (1H, d, J = 3.38 Hz); 6.6 (2H, d, J = 8.5Hz); 7.1(2H, d, J = 8.5Hz); 7.26 (2H, d, J = 8.4Hz); 7.3 (2H, d, J = 8.34Hz)



Details see below
Mechanism of action
Saroglitazar is novel first in class drug which acts as a dual PPAR agonist at the subtypes α (alpha) and γ (gamma) of theperoxisome proliferator-activated receptor (PPAR). Agonist action at PPARα lowers high blood triglycerides, and agonist action onPPARγ improves insulin resistance and consequently lowers blood sugar.[2]
Efficacy
Being a dual PPAR agonist, Saroglitazar (Lipaglyn) helps in controlling blood glucose and Lipid parameters especially high triglycerides and high non HDL-Cholesterol.[3] Lipaglyn effectively reduces triglycerides and non HDL-C and controlles high blood sugar, a typical situation in Insulin Resistance condition.[4][5]
Safety
Saroglitazar has not demonstrated any of the adverse effects like weight gain and edema that are usually identified with similar molecules like the glitazone class of drugs.[6] Because it is an insulin sensitizer, Saroglitazar (Lipaglyn) has less potential for hypoglycemia. No major serious adverse events have been reported; however, long-term cardiovascular safety has not been established.[7]
| Saroglitazar, is a drug for the treatment of diabetic dyslipidemia and hypertriglyceridemia with Type 2 diabetes mellitus not controlled by statin therapy. Its trade name is Lipaglyn. It is also a 1,2-Diarylpyrroles derivative, which can be used in the preparation of Nonsteroidal anti-inflammatory drugs (NSAIDs). |
| References: Khanna, I. K., et al.: J. Med. Chem., 40, 1619 (1997) |
_MoA.png)
PAPER
A new enantioselective synthesis of (S)-2-ethoxy-3-(4-hydroxyphenyl)propanoic acid esters (EEHP and IEHP), useful pharmaceutical intermediates of PPAR agonists
Tetrahedron Lett 2014, 55(21): 3223
http://www.sciencedirect.com/science/article/pii/S0040403914006200

PATENT
WO 2003009841
http://www.google.co.in/patents/WO2003009841A1?cl=en
PATENT
US 20030236254
http://www.google.com/patents/US20030236254
PATENT
US 20140099333
http://www.google.com/patents/US20140099333
PATENT
http://patentscope.wipo.int/search/en/WO2014174524
 (I)
(I)
The compound as claimed in claim 1 wherein R is -SMe and M+ is Mg+2.
The compound of claim 1 is Saroglitazar.
wherein ‘R’ is selected from hydroxy, hydroxyalkyl, acyl, alkoxy, alkylthio, thioalkyl, aryloxy, arylthio and M+ represents suitable metal cations such as Na+, K+, Ca+2, Mg+2 and the like. r .
PATENT
3-Aryl-2-hydroxy propanoic acid derivatives serve as a key intermediate for the synthesis of many pharmaceutically important compounds especially, peroxime proliferator activated receptor (PPAR) agonist.
Optically active 3-aryl-2-alkoxy propanoic acid and its esters, particularly, ethyl (2S)-2-ethoxy-3-(4-hydroxyphenyl)propanoate (EEHP) and isopropyl (2S)-2-ethoxy-3-(4-hydroxyphenyl)propanoate (IEHP) are versatile chiral pharmacophores present in many pharmaceutically important compounds, especially in peroxisome proliferator activated receptor (PPAR) agonists that have beneficial effects in treating Type 2 diabetes.
Several PPAR agonists, in particular PPAR α/γ dual agonists, commonly termed as glitazars (Ragaglitazar, Tesaglitazar, Navaglitazar etc.), as shown in the figure below were developed by many pharmaceutical companies that have a potential application in the treatment of Type 2 diabetes and dyslipidemia.
However, many of these drugs were discontinued due to their undesirable side effects, but some of them still have great potential [For example, Saraglitazar (LipaglynTM) developed by Zydus Cadila got approval in India for the treatment of diabetic dyslipidemia or hypertriglyceridemia]. Several PPAR α/γ agonists possessing chiral (S)-l moieties are shown below.

Tesaglitazar Naveglitazar
In addition, these derivatives find an application in photosensitive materials, sweetening agents, treatment of certain eating disorders etc. Therefore, these compounds have attracted a great deal of attention of synthetic chemists and different methods of preparation of the compound of formula (S)-l have been extensively studied.
Generally, the reported protocols for the synthesis involve chiral pool approaches starting from L-tyrosine and its derivatives (Refer WO 02/24625, US 6559335B2, WO 2003/027084), asymmetric synthesis (Org. Lett. 2005, 7, 1947, US 2007/0149804) and resolution processes using chiral amines or enzymes (WO 2000/026200, WO 2001/11073, Org. Process Res. Dev. 2003, 7, 82, Org. Process Res. Dev. 2004, 8, 838, Tetrahedron Asymmetry 2009, 20, 2594).
Some of these methods have disadvantages such as expensive chiral starting materials and catalysts, low enantioselectivity and overall yields, problems associated with the O-alkylation step which often leads to the loss of optical purity, and many others.
The processes described in WO20026200 (Rao et. al.) uses benzyl bromide for benzylation, which is highly lachrymatory. Again, in the processes described, the debenzylation of the final intermediate was done by using Pd/C under pressure, which escalates the process economics.
WO2003024915 describes a process for the preparation 3-aryl-2-hydroxy propanoic acid derivatives from 3-(4-hydroxyphenyl)-2-oxopropanoic acid.
WO 2003008362 describes 3-Aryl-2-hydroxy propanoic acid derivatives of formula I and the preparation thereof.

wherein Rland R2 may be same or different and represent hydrogen or (CI- C6) alkyl.
The process is depicted in Scheme 1 below.
Scheme 1

In another process variant as in Scheme 2, WO’362 discloses a process for the preparation of novel 3-aryl-2 -hydroxy propanol and their derivatives of the formula (I)

wherein OR and OR together form a substituted or unsubstituted 5 membered cyclic structure containing carbon and oxygen atoms, which comprises: i) reducing the compound of formula (III) where R represents hydrogen or alkyl group, R3 represents benzyl to a compound of formula (IV) where R3 represents benzyl, ii) cyclizing the compound of formula (IV) to a compound of formula (V) where ORl and OR2 together form a substituted or unsubstituted 5 membered cyclic structure containing carbon and oxygen atoms and R3 represents benzyl and iii) debenzylating the compound of formula (V) in the presence of metal catalysts to yield pure compound of formula (I).
Scheme 2

Both the processes described in WO’362 result in poor overall yield and further fail to describe the preparation of compound of formula V using different alkylating agents. This document exemplifies the compound of formula V with similar ether groups as it fails to teach selective alkylation of formula IV.
WO2005019152 discloses an improved process for the preparation of compound of the general formula (la) and (lb).

Wherein, Rl represent H or (C1-C6) alkyl group such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl, t-butyl and the like. R2 represents (Ci-Ce) alkyl group such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl, t- butyl and the like. R3 represents H, protecting groups such as benzyl, substituted benzyl, (C1-C3) alkyl and like.
The compound of general formula (la) is prepared according to the following schemes 3 and 4.
Scheme 3

Both the processes start with selective O-alkylation or O-aralkylation of L-Tyrosine of formula (2a) using a base, a chelating agent, an alkyl or aralkyl halide in the presence of solvents to obtain the compound of formula (3a), which is diazotized to obtain formula (4a) which upon dialkylation using an excess of alkylating agent and excess base, in presence of suitable solvent to obtain optically pure compound of formula (la). Alternatively, compound of formula (4a) may be selectively esterified to obtain compound of formula (5a), which is subsequently O-alkylated to obtain compound of formula (la) (Scheme 2).
However, the above processes have many disadvantages such as multistep synthesis including protection & deprotection and low overall yield. Further, low temperature diazotization on industrial scale is not viable. Moreover, the starting material is very expensive and hence escalates the process.
In the light of the foregoing, development of a new, alternate enantio-selective synthetic route to these important chiral intermediates, which are simple and can preserve the optical purity at the C-2 carbon of 3-Aryl-2-hydroxy propanoic acid derivatives, is highly desirable. There is a need for an efficient process for synthesis of 3-Aryl-2-hydroxy propanoic acid derivatives of formula (S)-l in high enantiopurity and good overall yield from commercially available starting material.

OR

Synthesis of saroglitazar
1. 2-Bromo-1-[4-(methylthio)phenyl]ethanone is condensed with methyl acetoacetate in the presence of NaOMe and Na2SO4 in toluene, to give alpha-keto methyl ester ,
2. This alpha-keto methyl ester ,is hydrolyzed and decarboxylated by means of NaOH in MeOH/toluene at 50 °C giving diketone .
3. Diketone is subjected to Paal-Knorr reaction with ethanolamine in the presence of pivallic acid in toluene at 110 °C to yield pyrrole primary alcohol derivative .
4. Sulfonylation of this pyrrole primary alcohol with MsCl in the presence of Et3N,
5. O-alkylation of mesylate with ethyl 2(S)-ethoxy-3-(4-hydroxyphenyl)propionate in the presence of K2CO3, optionally in the presence of 18-crown-6 in toluene/THF at 80 °C provides ether.
6. Finally, hydrolysis of ethyl ester using NaOH in H2O affords the target saroglitazar.
PATENT
saroglitazar magnesium alongwith its intermediates may be prepared by the reaction scheme- 1, scheme-2 and scheme-3 as shown below, which is also the scope of the present invention.

Scheme-1

EXAMPLES
Example-l:
Preparation of methanesulfonic acid 2-r2-methyl-5-(4-methylsulfanyl-phenyl)-pyrrol-l-yl]-ethyl ester (Al)

In a 5 Liter three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, sodium methoxide (165 g) and toluene (1000.0 ml) were added under nitrogen environment and cooled to 8°C to 12°C. Methyl acetoacetate (331.55 g) was added dropwise and stirred for 1 hour. 2-bromo-l-(4-methyl sulfonyl phenyl) ethanone (500.0 g) compound (El) in toluene (1500.0 ml) and sodium sulfate
(75.0 g) mixture was stirred for 10 min and filtered at 25° to 35°C. The filtrate as obtained was added dropwise into the previous reaction mixture and stirred at 30°C to 35°C for 30 min. The organic layer was collected and washed with 10% sodium bicarbonate solution. The separated organic layer was collected and washed with water. 2-[2-(4-Methyl sulfanyl-phenyl)-2-oxo-ethyl]-3-oxo-butynic acid methyl ester as obtained in toluene layer is diluted with methanol (2500 ml) and sodium hydroxide solution (89.75 g) in water (2500 ml) was added and heated to 50° to 55°C for 1 hour. The layers were separated and the toluene layer was collected and heated to 45° to 55°C and charcoalized. The reaction mixture was filtered and pivalic acid (57.3 g) and ethanol amine (143.9 g) were added and heated to 105° to 1 15°C for removing water azeotropically. The toluene layer was separated and triethyl amine (271.85 g) was added at 25° to 35°C and the reaction mixture was cooled to 10° to 20°C. Methane sulphonyl chloride (282.5 g) was added dropwise, and stirred for 2 hours and heated to 35° to 45°C. The reaction mixture was filtered and washed with toluene. Toluene was distilled out completely under the vacuum to obtain the residue. The residue was dissolved in toluene (1500 mL) and used for further process.
ExampIe-2:
Preparation of methanesulfonic acid 2-f2-methyl-5-(4-methylsulfanyl-pheny0-pyrrol- 1-viyethyl ester (Al)

In a 250 mL three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, 4-(methylthio)benzaldehyde (10 g), methyl vinyl ketone (3.63 g), triethylamine (9.95 g) and 3-methyl-5-(2-hydroxyethyl)-4-methyI thiazolium iodide (stetter
catalyst) (2.8 g) were heated to 70°C to 80°C and maintained overnight. The reaction mixture was cooled to room temperature and ethanol (100 mL) was added. The reaction mixture was stirred for 30 min and filtered. The product was washed with ethanol and dried to obtain 1 ,4-diketo compound (CI).
1 ,4-diketo compound (CI) obtained above and toluene (50 mL) were heated to 45° to 55°C and charcoalized. The reaction mixture was filtered and pivalic acid (5.7 g) and ethanol amine (14.4 g) were added and heated to 105° to 1 15°C and cooled to 25°C. Triethyl amine (27.2 g) was added at 25° to 35°C and the reaction mixture was cooled to 10° to 20°C. Methane sulphonyl chloride (28.3 g) was added dropwise, and stirred for 2 hours and heated to 35° to 45°C. The reaction mixture was filtered and washed with toluene. Toluene was distilled out completely under the vacuum, methanol (2500 ml) was added and heated to 55° to 65 °C and charcoalized for 30 min. The reaction mixture was filtered and washed with methanol. The reaction mixture was cooled to 25° to 35°C and stirred for 30 min. Reaction mass was further cooled to -5° to 5°C and filtered. The wet-cake was washed with methanol and dried to obtain compound (Al). The compound (Al) was characterized as crystalline solid by x-ray powder diffraction (FIG.2).
Example-3:
Purification of methanesulfonic acid 2-r2-methyl-5-(4-methylsulfanyl-phenyl)-pyrrol-l-yl]-ethyl ester (Al)
In a 250 mL three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, 70 g methanesulfonic acid 2-[2-methyl-5-(4-methylsulfanyl-phenyl)-pyrrol-l -yl]-ethyl ester (Al) and 420 mL ethyl acetate were added at 25°C. The reaction mixture was stirred for 30 min to obtain clear solution. 3.5 g charcoal was added and stirred for 30 min. The reaction mixture was filtered and washed with ethyl acetate. The filtrate was concentrated and 315 mL methanol was added. The reaction mixture was stirred for 2 hours at 25°C and cooled to 0°C. The product precipitated was filtered and washed with methanol to obtain crystalline
compound (Al). The compound (Al) was characterized as crystalline solid by x-ray powder diffraction (FIG.3).
Example-4:
Preparation of saroglitazar magnesium (T)

In a 5 Liter three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, 2-ethoxy-3-(4-hydroxy-phenyl)-propionic acid ethyl ester (A) (100.0 g) and toluene (1300.0 ml) were charged and reaction mixture was heated to 45° to 55°C. Potassium carbonate (58.0 g) was added and stirred for 30 min. Toluene solution of methanesulfonic acid 2-[2-methyl-5-(4-methylsulfanyl-phenyl)-pyrrol- 1 -yl]-ethyl ester (Al) (150.24 g) obtained in example- 1, 18-Crown-6 (5.0 g) and THF (200.0 ml) were added and heated to 75°C to 85°C for 36 hour, The reaction mixture was cooled to 25° to 35°C and water (1000.0 ml) was added and stirred for 15 min. The separated aqueous layer was treated with toluene (200.0 ml) and stirred for 15 min. The organic, layers were combined and washed with caustic solution (600.0 ml). The separated organic layer was washed with water (600.0 ml) and characoalized with HP-120 (5.0 g) charcoal and stirred for 30 min and filtered. The filtrate was added sodium hydroxide 20.14 g solution in water (200.0 ml) and the reaction mixture was stirred for 3 hours. The reaction mixture was diluted with water (1800.0 ml) and stirred for 15 min. The separated aqueous layer was washed with n-butyl acetate. The separated aqueous layer was added magnesium acetate tetrahydrate solution (90.0 g) in water (100.0 ml) and stirred for 1 hour. The aqueous layer was extracted with methylene dichloride (2000 ml). The separated organic layer was washed with sodium chloride solution and charcoalized. The charcoalized solution was filtered and filtrate was distilled to remove toluene completely. The residue was diluted with toluene (1000 ml) and stirred for 30 min. The organic solution was added into n-heptane (1500 mL) and stirred for 3 hours. The product was filtered and washed with n-heptane and dried in vacuum tray dryer at 25°C to 30°C for 3 hours. The product was sieved through 0.5 mm sieve and milled through jet-milled. The product was further dried in vacuum tray drier at 40°C to 50°C for 6 hours followed by drying at 55°C to 65°C for 40 hours to obtain amorphous saroglitazar magnesium (I). The compound is characterized by x-ray power diffraction (FIG.l).
The reaction of methanesulfonic acid 2-[2-methyl-5-(4-methylsulfanyl-phenyl)-pyrrol-l-yl]-ethyl ester (Al) and 2-ethoxy-3-(4-hydroxy-phenyl)-propionic acid ethyl ester (A) may also be performed in similar manner as above in absence of phase transfer catalyst 18-Crown-6.
ExampIe-5:
Preparation of saroglitazar (S)-(-)-phenyl ethylamine salt:

In a 250 mL three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, residue-A obtained in example- 1 and ethanol (400 mL) were stirred for 15 min. Sodium hydroxide 20.14 g solution in water (200.0 ml) was added and the reaction mixture was stirred for 3 hours. The reaction mixture was diluted with water (1800.0 ml) and stirred for 15 min. The separated aqueous layer was washed with isopropyl acetate (400 mL). The separated aqueous layer was diluted with isopropyl acetate (500 mL) and acidified with cone. HCI at adjust the pH 2-3. The separated aqueous layer was washed with isopropyl acetate. The combined organic layer was treated with (S)-(-)-phenyl ethylamine (55.94 g) and stirred for 2 hours at 25°C and 30 min at 45°C. The reaction mixture was cooled to 0°C and stirred for 2 hours, filtered and washed with isopropyl acetate. The wet-cake was dried to obtain saroglitazar phenyl ethylamine salt.
ExampIe-6:
Preparation of saroglitazar magnesium from saroglitazar (SH-)-phenyl ethylamine salt:
In a 250 mL three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, saroglitazar phenyl ethylamine wet-cake obtained in example-7 and isopropyl acetate (800 mL) were added at 25°C. The reaction mixture was diluted with water (400.0 ml) and acidified with cone. HCI at adjust the pH 2-3. The separated aqueous layer was washed with isopropyl acetate. The combined organic layer was treated with sodium hydroxide solution (20.14 g) in water (200 mL) and stirred for 30 min. The separated aqueous layer was treated with magnesium acetate tetrahydrate (2.29 g) in water (5 mL) solution and stirred for 60 min. The reaction mixture was extracted with methylene dichloride (800 mL). The methylene dichloride was complete removed by distillation under vacuum below 40°C to obtain the residue. The residue was diluted with methylene dichloride (50 ml) and stirred for 30 min. The organic solution was added into n-heptane (1500 mL) and stirred for 3 hours. The product was filtered and washed with n-heptane and dried in vacuum tray dryer at 25°C to 30°C for 3 hours. The product was sieved through 0.5 mm sieve and milled through jet-milled. The product was further dried in vacuum tray drier at 40°C to 50°C for 6 hours followed by drying at 55°C to 65°C for 40 hours to obtain substantially amorphous saroglitazar magnesium (I). The compound is characterized by x-ray power diffraction (FIG.l).

PATENT
WO 2015029066
Dwivedi, Shri Prakash Dhar; Singh, Ramesh Chandra; Patel, Vikas; Desai, Amar Rajendra
Cadila Healthcare Ltd
Polymorphic form of pyrrole derivative and intermediate thereof
Pyrrole derivative of present invention is chemically 2-ethoxy-3-(4-(2-(2-methyl-5-(4-(methylthio)phenyl)-lH-pyrrol-l-yl)ethoxy)pKenyl)propanoate, which may be optically active or racemic and its pharmaceutically acceptable salts, hydrates, solvates, polymorphs or intermediates thereof. The INN name for pyrrole derivative is Saroglitazar® which is magnesium salt of pyrrole compound of Formula (I), having below chemical structure.

The present invention relates to Saroglitazar free acid of Formula (IA) or its pharmaceutically acceptable salts, pharmaceutically acceptable solvates, pharmaceutically acceptable esters, stereoisomers, tautomers, analogs and derivs. thereof. The present invention also provides an amorphous form of saroglitazar free acid and processes of prepn. thereof. The present invention also provides pharmaceutical compn. comprising an amorphous form saroglitazar magnesium.
Amorphous forms of saroglitazar free acid and its salt form are claimed. Also claims the process for the synthesis the same compound. Useful for treating obesity, hyperlipidemia and hypercholesteremia. Picks up from WO2015011730, claiming the stable composition comprising saroglitazar magnesium or its derivatives. Zydus-Cadila has developed and launched saroglitazar for treating diabetic dyslipidemia and hypertriglyceridemia.
In September 2013, saroglitazar was launched for treating dyslipidemia and hypertriglyceridemia.
As of March 2015, Zydus-Cadila is developing saroglitazar for treating nonalcoholic steatohepatitis and type II diabetes (both in phase III clainical trials).
Pyrrole derivative of present invention is chemically 2-ethoxy-3-(4-(2-(2-methyl- 5-(4-(methylthio)phenyl)-lH-pyrrol-l-yl)ethoxy)ph’enyl)propanoate, which may be optically active or racemic and its pharmaceutically acceptable salts, hydrates, solvates, polymorphs or intermediates thereof. The INN name for pyrrole derivative is Saroglitazar® which is magnesium salt of pyrrole compound o f saroglitazar,
the process comprising: 5WO 2015/029066 PCT/IN2014/000551 (a) dissolving saroglitazar magnesium of Formula (I) in one or more organic solvents to obtain a solution, (b) adding the solution in one or more o f anti-solvent at temperature from about -80°C to about 150°C to obtain saroglitazar magnesium o f Formula (I); and (c) obtaining the amorphous saroglitazar magnesium by removal of anti-solvent.
Example-1: Preparation of saroglitazar magnesium (Ί) In a 5 Liter three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, 2-ethoxy-3-(4-hydroxy-phenyl)- propionic acid ethyl ester (A) (100.0 g) and cyclohexane (1300.0 ml) were charged and reaction mixture was heated to 45° to 55°C. Potassium carbonate (58.0 g) was added and stirred for 30 min. methanesulfonic acid 2-[2-methyl-5-(4-methyIsulfanyl-phenyl)-pyrroll-yl]-ethyl ester (A l) (150.24 g) and THF (200.0 ml) were added and heated to 75°C to 85°C for 36 hour. The reaction mixture was cooled to 25° to 35°C and water (1000.0 ml) was added and stirred for 15 min. The separated aqueous layer was treated with cyclohexane (200.0 ml) and stirred for 15 min. The organic layers were combined and washed with caustic solution (600.0 ml). The separated organic layer was washed with water (600.0 ml) and characoalized with (5.0 g) charcoal and stirred for 30 min and filtered. The filtrate was distilled to remove cyclohexane and the residue was collected (residue-A). The residue-A as obtained was treated with ethanol (400.0 ml) and stirred for 15 min. Sodium hydroxide 20.14 g solution in water (200.0 ml) was added and the reaction mixture was stirred for 3 hours. The reaction mixture was diluted with water (1800.0 ml) and stirred for 15 min. The separated aqueous layer was washed with n-butyl acetate. The separated aqueous layer was added magnesium acetate tetrahydrate solution (90.0 g) in water (100.0 ml) and stirred for I hour. The aqueous layer was extracted with methylene dichloride (200 ml). The separated organic layer was washed with sodium chloride solution and charcoalized. The charcoalized solution was filtered and filtrate was distilled to remove methylene dichloride completely. The residue was diluted with methylene dichloride (1000 ml) and stirred for 30 min. The organic solution was added into n-heptane (1500 mL) and stirred for 3 hours. The product was filtered and washed with n-heptane and dried in vacuum tray dryer at 25°C to 30°C for 3 hours. The product was sieved through 0.5 mm sieve and milled through jet-milled. The product was further dried in vacuum tray drier at 40°C to 50°C for 6 hours followed by drying at 55°C to 65°C for 40 hours to obtain substantially amorphous saroglitazar magnesium (I). The compound is characterized by x-ray power diffraction (FIG.I).
PATENT
| WO/2015/011730 |
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015011730
The present invention relates to the stable pharmaceutical composition of a suitable hypolipidemic agent. Preferably, the present invention discloses novel formulations of the compound of formula (I), or pharmaceutically acceptable salts of compounds of formula (I). More particularly the present invention relates to the stable pharmaceutical composition of compounds of formula (I) comprising compounds of formula (I) or its pharmaceutically acceptable salts, wherein the pH of the formulation is maintained above 7. formula (I)
The compounds of formula (I) are new synthetic compounds having hypolipidemic activity. The compounds of formula (I) are used primarily for triglyceride lowering, with concomitant beneficial effect on glucose lowering and cholesterol lowering.
The structural formula of compounds of formula (I) is shown below.

wherein ‘R’ is selected from hydroxy, hydroxyalkyl, acyl, alkoxy, alkylthio, thioalkyl, aryloxy, arylthio and M+ represents suitable metal cations such as Na+, K+, Ca+2, Mg+2 and the like. Preferably, R is selected from alkylthio or thioalkyl groups; most preferably R represents -SCH3.The Mg+2 salt is preferred. The compounds of formula (I) are generally insoluble in water, but freely soluble in dimethyl sulfoxide, dichloromethane & slightly soluble in methanol and IPA.
1H NMR PREDICT


13c NMR PREDICT


“ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This article is a compilation for educational purposes only.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
/////////////////
O=C(O)[C@@H](OCC)Cc3ccc(OCCn1c(ccc1C)c2ccc(SC)cc2)cc3
REFERENCES
- “Zydus Group launches new diabetic drug”. The Times of India. Jun 6, 2013.
- “Lipaglyn (Saroglitazar) for Treating Hypertriglycerdemia in Type II Diabetes, India”. Drug Development and Technology.
- “The nuances of atherogenic dyslipidemia in diabetes: focus on triglycerides and current management strategies.”. Indian Heart Journal.
- “Observational Study of Effects of Saroglitazar on Glycaemic and Lipid Parameters on Indian Patients with Type 2 Diabetes”. SCIENTIFIC REPORTS.
- “From ‘Make in India’ to ‘Made in India’: the saroglitazar story.”. Indian Heart Journal.
- “Observational study to evaluate the safety and efficacy of saroglitazar in Indian diabetic dyslipidemia patients.”. Indian Heart Journal.
- Munigoti, SrinivasaP; Harinarayan, CV (2014). “Role of Glitazars in atherogenic dyslipidemia and diabetes: Two birds with one stone?”. Indian Journal of Endocrinology and Metabolism 18 (3): 283. doi:10.4103/2230-8210.131134. PMC 4056123.PMID 24944919.
Indian Pat. Appl. (2015), IN 2013MU02905
WO 2015033357
WO 2015150565
WO 2015001573
IN 2013MU02828
WO 2015029066
IN 2013MU01910
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2003009841A1 * | Jul 25, 2002 | Feb 6, 2003 | Cadila Healthcare Ltd | Novel pyrroles having hypolipidemic hypocholesteremic activities, process for their preparation and pharmaceutical compositions containing them and their use in medicine |
| WO2012104869A1 | Jan 30, 2012 | Aug 9, 2012 | Cadila Healthcare Limited | Treatment for lipodystrophy |
| INMU19102013A | Title not available | |||
| US6987123 | Aug 10, 2001 | Jan 17, 2006 | Cadila Healthcare Limited | Heterocyclic compounds, their preparation, pharmaceutical compositions containing them and their use in medicine |
| US7041837 | Jul 19, 2002 | May 9, 2006 | Cadilla Healthcare Limited | Heterocyclic compounds having hypolipidemic, hypocholesteremic activities process for their preparation and pharmaceutical compositions containing them and their use in medicine |
| US7323491 | Mar 1, 2004 | Jan 29, 2008 | Cadila Healthcare Limited | Heterocyclic compounds, their preparation, pharmaceutical compositions containing them and their use in medicine |
| US8110598 | Feb 7, 2012 | Cadila Healthcare Limited | Heterocyclic compounds, their preparation, pharmaceutical compositions containing them and their use in medicine | |
| US8212057 | Jul 25, 2002 | Jul 3, 2012 | Cadila Healthcare Limited | Pyrroles having hypolipidemic hypocholesteremic activities, process for their preparation and pharmaceutical compositions containing them and their use in medicine |
| US20110275669 | Nov 10, 2011 | Cadilla Healthcare Limited | Novel pyrroles having hypolipidemic hypocholesteremic activities, process for their preparation and pharmaceutical compositions containing them and their use in medicine |
 |
| Zydus Cadila chairman and MD Pankaj R. Patel (centre) and deputy managing director Sharvil P. Patel (left) in Mumbai on Wednesday. (PTI)JUNE 5, 2013 |
Cadila banks on diabetes drug
Calcutta Telegraph
It generally takes around 10-15 years for a drug to be developed from the time of its discovery In the case of Lipaglyn, the molecule was identified in 2001, and Phase III clinical trials was completed around four years ago. While Zydus has not yet …http://www.telegraphindia.com/1130606/jsp/business/story_16976915.jsp
Mumbai, June 5: Cadila Healthcare will launch a homegrown drug against diabetes by the third quarter of this year.
The Drug Controller General of India has approved its drug — Lipaglyn — to treat “diabetic dyslipidemia”.
Diabetic dyslipidemia is a condition where a person is diabetic and has elevated levels of total cholesterol. Over 80 per cent of diabetic patients are dyslipidemic.
http://www.telegraphindia.com/1130606/jsp/business/story_16976915.jsp
Zydus Cadila said it is looking for partnership to market its new chemical entity (NCE) Lipaglyn, to be used for treating a type of diabetes in developed and developing markets. “Lipaglyn is the first glitazar to be approved in the world and the first NCE discovered and developed indigenously by an Indian pharma company.
The new drug is expected to be launched in Q3 of this fiscal in the country,” Zydus Cadila Chairman and Manging Director Pankaj Patel told reporters.
The company has spent USD 250 million in developing Lipaglyn and aims to spend another USD 150-200 million to launch the drug in overseas markets in next 3-5 years period, Patel said, adding that the company is looking for marketing partnerships.
“We expect this to be a blockbuster drug, which means over USD 1 billion sales a year, when the drug is sold globally, he said. The market for this drug is estimated at Rs 100 crore in the local market over the next three years and having market potential size of over USD 30 billion in the world market, he said.
Zydus Cadila took about eight years to develop the molecule and conducted clinical trials on more than 1,000 patients in India, Patel said, adding that the company is yet to finalise the price, but believes that it will be reasonably priced in the local market.
The company said that the Indian drug regulator Drug Controller General of India (DCGI) has approved Lipaglyn to be used for treating ‘diabetic dyslipidemia’.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(2S)-2-Ethoxy-3-[4-(2-{2-methyl-5-[4-(methylsulfanyl)phenyl]-1H-pyrrol-1-yl}ethoxy)phenyl]propanoic acid
|
|
| Clinical data | |
| Trade names | Lipaglyn |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral |
| Identifiers | |
| CAS Number | 495399-09-2 |
| ATC code | None |
| PubChem | CID 60151560 |
| ChemSpider | 32079086 |
| Chemical data | |
| Formula | C25H29NO4S |
| Molar mass | 439.56 g/mol |
by WORLD DRUG TRACKER
DR ANTHONY
do not miss out on updates
see my update at https://newdrugapprovals.org/2015/03/09/saroglitazar-magnesium-new-patent-wo-2015029066-cadila-healthcare-ltd/ 9 may 2015
Lipaglyn (Saroglitazar) won a lot of support at the 75th Anniversary Conference of the American Diabetes Association. Lipaglyn is currently under Phase III clinical development for treatment of Non Alcoholic SteatoHepatitis (NASH), a serious liver disease and an unmet healthcare need, globally. There is currently no drug approved for treating NASH. Lipaglyn is already approved in India for the treatment of diabetic dyslipidemia


Speaking on the development, Mr. Pankaj R. Patel, Chairman and Managing Director, Zydus Cadila said, “These new robust scientific data on the safety and efficacy of Lipaglyn
(Saroglitazar) being presented at the 75th Annual Scientific Sessions of the American Diabetes Association (ADA) reflect our continued commitment to millions of patients living with Diabetes, Dyslipidemia, Non-alcoholic fatty liver disease (NAFLD) and Non-alcoholic steatohepatitis (NASH).”
Zydus Cadila, a leading global healthcare provider, today announced that new scientific and clinical data on Saroglitazar will be presented at the 75th Annual Scientific Sessions of the American Diabetes Association (ADA) in Boston, Massachusetts, USA from 5thto 9th June, 2015. Several analyses of real-world patient data of Saroglitazar will also be presented. The abstracts are available on theADA website.
Lipaglyn – The world’s first drug for treating Diabetic Dyslipidemia combines lipid and glucose lowering effects in one single molecule.

Pankaj Patel, chairman and MD, Cadila Healthcare Ltd

Zydus is an innovation-led global healthcare provider that discovers, manufactures and markets a broad range of healthcare therapies. The group employs over 19,000 people worldwide including over 1200 scientists engaged in research and is dedicated to creating healthier communities globally.
With a strong research pipeline of NCEs, biologics and vaccines, the group became India’s first pharmaceutical company to launch its own indigenously researched therapy Lipaglyn which is also the world’s first approved therapy for diabetic dyslipidaemia. Exemptia, the world’s first biosimilar of Adalimumab is also a product of Zydus innovation. Zydus also collaborates with partners to support and make therapies affordable and accessible to communities across the world.
As a leading healthcare provider, it aims to become a global research-based pharmaceutical company by 2020.




Pankaj R. Patel (left), Chairman & Managing Director, Zybus Cadila,

Ganesh Nayak, Chief Operating Officer and Executive Director, Zydus Cadila


Zydus Cadila has announced a breakthrough in the anti-diabetic drug Lipaglyn. Lipaglyn – The world’s first drug for treating Diabetic Dyslipidemia combines lipid and glucose lowering effects in one single molecule.
The Zydus Group announced a breakthrough in its research efforts with Lipaglyn (Saroglilazar), a novel drug targeted at bridging an unmet healthcare need for treating Diabetic Dyslipidemia or Hypertriglyeeridemia in Type II diabetes, not controlled by statins alone. The drug has been approved for launch in India by the Drug Controller General of India (DCGI). With a novel action that offers lipid and glucose lowering effects in one molecule, Lipaglyn is the first Glitazar to be approved anywhere in the world.
“Lipaglyn provides patients suffering from diabetic dyslipidemia the option of a once-daily oral therapy that has a beneficial effect on both lipid parameters as well as glycemic control,” said Pankaj R. Fatel, Chairman and Managing Director, Zydus Cadila. “It has always been our dream to take a molecule right from the concept stage up to its launch. Today, we have realized this dream. It is an important breakthrough and I would like to dedicate this to all the Indian research scientists in the Held of drug discovery,” Patel added,
Diabetic Dyslipidemia is a condition where a person is diabetic and has elevated levels of the total cholesterol, the “bad” low-density lipoprotein (LDL) cholesterol and the triglycerides and a decrease in the “good” high-density lipoprotein (HDL) cholesterol concentration in the blood. Optimal LDL cholesterol levels ibr adults with diabetes are less than 100 mg/dh, optimal HDL cholesterol levels are equal to or greater than 40 mg/dL, and desirable triglycerides levels are less than 150 mg/dLT LipaglynrM, a non-thiazoKdinedione, is the first therapy to be approved for this condition,
World over, it is estimated that 30% of all deaths occur due lo cardiovascular diseases (CVD). In India, one out of every five persons is at serious risk of developing CVD, Research has shown that diabetes is one of the major risk factors of CVD. India has a population of nearly 65 million diabetics and 77 million prc-diabctics, 85 – 97% of the diabetes patients suffer from dyslipidemia or lipid abnormalities. Hence, addressing the problem of diabetes and dyslipidemia is crucial in tackling the health risk posed by CVD.
Discovered by the Zydus Research Centre, the dedicated NCE research arm of the Zydus group, LipaglynrM is a best-in-class innovation, designed to have a unique cellular mechanism of action following an extensive structure-activity relationship study initiated in the year 2000, Lipaglyn1M has a predominant affinity to PPAR alpha isoform and moderate affinity to PPAR gamma isoform of PPAR nuclear receptor subfamily. The molecule has shown beneficial effects on lipids and glyeemic control without side effects. This molecule underwent extensive pre-clinical characterisation and the I.ND was submitted in the year 2004,
As a part of the clinical development programme, extensive Phase-I, Phase-II and Phase-Ill clinical trials were conducted to evaluate the phamacokinetics, pharmacodynamics, efficacy and safety of Lipaglyn. The new drug application for Lipaglyn1 was based on a comprehensive clinical development programme spanning eight years.
Results from the first Phase III programme with Pioglitazone as a comparator drug in diabetes patients showed that the 4 mg dose of Lipaglyn led to a reduction of triglycerides and LDL (bad) cholesterol, and an increase in HDL (good) cholesterol and also showed a reduction in Fasting Plasma Glucose and glycosylated haemoglobin (HbAlc) thereby confirming its beneficial effects of both lipid and glyeemic control in diabetic patients,
In the second Phase III study, Lipaglyn was studied in diabetic dyslipidemic patients insufficiently controlled with statin therapy. The results from this study confirmed that Lipaglyn had a pronounced beneficial effect on both the lipid and glyeemic parameters in these subjects.
In both the studies, Lipaglyn was well tolerated and had a better safety profile than the comparators. Importantly Lipaglyn1 M has a non-renal route of elimination, and did not show adverse events like edema, weight gain, myopathies or derangement of liver and/or kidney functions, thus making it sale and efficacious. LipaglynIM is recommended for once daily administration as 4 mg tablets.
Zydus will offer a dedicated LipaglynIM support programme to patients and earegivers, The programme shall provide important support and information regarding access, adherence, education and thereby help patients to start and appropriately manage their disease and therapy over time.
About Lipaglyn
Lipaglyn[TM] (Saroglitazar) was launched in September 2013 in India, for treating Hypertriglyceridemia and Diabetic Dyslipidemia in Patients with Type 2 Diabetes not controlled by statins. Since then, more than 80,000 patients are availing this drug with a prescriber base over 3500 diabetologists, cardiologists and physicians. Lipaglyn[TM] helps in a reduction of triglycerides and LDL (bad) cholesterol, and an increase in HDL (good) cholesterol and has also shown a reduction in Fasting Plasma Glucose and glycosylated haemoglobin (HbA1c), thereby confirming its beneficial effects on both lipid and glycemic control in diabetic patients. Lipaglyn[TM] is a prescription medicine, and can be taken only under the advice and guidance of a registered medical practitioner.
About Zydus
Zydus Cadila is an innovative, global pharmaceutical company that discovers, manufactures and markets a broad range of healthcare therapies, including small molecule drugs, biologic therapeutics and vaccines. The group employs over 16,500 people worldwide including over 1200 scientists engaged in R & D and is dedicated to creating healthier communities globally. As a leading healthcare provider, it aims to become a global research based pharmaceutical company by 2020.
References
Zydus to present new scientific data on Lipaglyn in the US
New Delhi, Jun 8 (UNI) Healthcare services provider, Zydus Cadila today said the new scientific and clinical data on Lipaglyn (Saroglitazar) will be presented at the 75th annual scientific sessions of the American Diabetes Association (ADA) in Boston, Massachusetts, US from 5th to 9th June,2015.
Read more at http://www.uniindia.com/news/business-economy/zydus-to-present-new-scientific-data-on-lipaglyn-in-the-us/84440.html
READ …..https://newdrugapprovals.org/2013/06/07/cadila-banks-on-diabetes-druglipaglynsaroglitazar/
http://lipaglyn.com/downloads/Lipaglyn_Product_Monograph.pdf
http://www.ijpcs.net/sites/default/files/IJPCS_3_1_02_0.pdf
http://onlinelibrary.wiley.com/doi/10.1002/prp2.136/pdf
////////////
CCO[C@@H](Cc1ccc(cc1)OCCn2c(ccc2c3ccc(cc3)SC)C)C(=O)O
CCOC(CC1=CC=C(C=C1)OCCN2C(=CC=C2C3=CC=C(C=C3)SC)C)C(=O)O
Zydus Cadila’s, Lipaglyn (Saroglitazar) won a lot of support at the 75th Anniversary Conference of the American Diabetes Association
Lipaglyn (Saroglitazar) won a lot of support at the 75th Anniversary Conference of the American Diabetes Association. Lipaglyn is currently under Phase III clinical development for treatment of Non Alcoholic SteatoHepatitis (NASH), a serious liver disease and an unmet healthcare need, globally. There is currently no drug approved for treating NASH. Lipaglyn is already approved in India for the treatment of diabetic dyslipidemia
Speaking on the development, Mr. Pankaj R. Patel, Chairman and Managing Director, Zydus Cadila said, “These new robust scientific data on the safety and efficacy of Lipaglyn
(Saroglitazar) being presented at the 75th Annual Scientific Sessions of the American Diabetes Association (ADA) reflect our continued commitment to millions of patients living with Diabetes, Dyslipidemia, Non-alcoholic fatty liver disease (NAFLD) and Non-alcoholic steatohepatitis (NASH).”
Zydus Cadila, a leading global healthcare provider, today announced that new scientific and clinical data on Saroglitazar will be presented at the 75th Annual Scientific Sessions of the American Diabetes Association (ADA) in Boston, Massachusetts, USA from 5thto 9th June, 2015. Several analyses of real-world patient data of Saroglitazar will also be presented. The abstracts are available on theADA website.
Lipaglyn – The world’s first drug for treating Diabetic Dyslipidemia combines lipid and glucose lowering effects in one single molecule.
Pankaj Patel, chairman and MD, Cadila Healthcare Ltd
Zydus is an innovation-led global healthcare provider that discovers, manufactures and markets a broad range of healthcare therapies. The group employs over 19,000 people worldwide including over 1200 scientists engaged in research and is dedicated to creating healthier communities globally.
With a strong research pipeline of NCEs, biologics and vaccines, the group became India’s first pharmaceutical company to launch its own indigenously researched therapy Lipaglyn which is also the world’s first approved therapy for diabetic dyslipidaemia. Exemptia, the world’s first biosimilar of Adalimumab is also a product of Zydus innovation. Zydus also collaborates with partners to support and make therapies affordable and accessible to communities across the world.
As a leading healthcare provider, it aims to become a global research-based pharmaceutical company by 2020.
Pankaj R. Patel (left), Chairman & Managing Director, Zybus Cadila,
Ganesh Nayak, Chief Operating Officer and Executive Director, Zydus Cadila
Zydus Cadila has announced a breakthrough in the anti-diabetic drug Lipaglyn. Lipaglyn – The world’s first drug for treating Diabetic Dyslipidemia combines lipid and glucose lowering effects in one single molecule.
The Zydus Group announced a breakthrough in its research efforts with Lipaglyn (Saroglilazar), a novel drug targeted at bridging an unmet healthcare need for treating Diabetic Dyslipidemia or Hypertriglyeeridemia in Type II diabetes, not controlled by statins alone. The drug has been approved for launch in India by the Drug Controller General of India (DCGI). With a novel action that offers lipid and glucose lowering effects in one molecule, Lipaglyn is the first Glitazar to be approved anywhere in the world.
“Lipaglyn provides patients suffering from diabetic dyslipidemia the option of a once-daily oral therapy that has a beneficial effect on both lipid parameters as well as glycemic control,” said Pankaj R. Fatel, Chairman and Managing Director, Zydus Cadila. “It has always been our dream to take a molecule right from the concept stage up to its launch. Today, we have realized this dream. It is an important breakthrough and I would like to dedicate this to all the Indian research scientists in the Held of drug discovery,” Patel added,
Diabetic Dyslipidemia is a condition where a person is diabetic and has elevated levels of the total cholesterol, the “bad” low-density lipoprotein (LDL) cholesterol and the triglycerides and a decrease in the “good” high-density lipoprotein (HDL) cholesterol concentration in the blood. Optimal LDL cholesterol levels ibr adults with diabetes are less than 100 mg/dh, optimal HDL cholesterol levels are equal to or greater than 40 mg/dL, and desirable triglycerides levels are less than 150 mg/dLT LipaglynrM, a non-thiazoKdinedione, is the first therapy to be approved for this condition,
World over, it is estimated that 30% of all deaths occur due lo cardiovascular diseases (CVD). In India, one out of every five persons is at serious risk of developing CVD, Research has shown that diabetes is one of the major risk factors of CVD. India has a population of nearly 65 million diabetics and 77 million prc-diabctics, 85 – 97% of the diabetes patients suffer from dyslipidemia or lipid abnormalities. Hence, addressing the problem of diabetes and dyslipidemia is crucial in tackling the health risk posed by CVD.
Discovered by the Zydus Research Centre, the dedicated NCE research arm of the Zydus group, LipaglynrM is a best-in-class innovation, designed to have a unique cellular mechanism of action following an extensive structure-activity relationship study initiated in the year 2000, Lipaglyn1M has a predominant affinity to PPAR alpha isoform and moderate affinity to PPAR gamma isoform of PPAR nuclear receptor subfamily. The molecule has shown beneficial effects on lipids and glyeemic control without side effects. This molecule underwent extensive pre-clinical characterisation and the I.ND was submitted in the year 2004,
As a part of the clinical development programme, extensive Phase-I, Phase-II and Phase-Ill clinical trials were conducted to evaluate the phamacokinetics, pharmacodynamics, efficacy and safety of Lipaglyn. The new drug application for Lipaglyn1 was based on a comprehensive clinical development programme spanning eight years.
Results from the first Phase III programme with Pioglitazone as a comparator drug in diabetes patients showed that the 4 mg dose of Lipaglyn led to a reduction of triglycerides and LDL (bad) cholesterol, and an increase in HDL (good) cholesterol and also showed a reduction in Fasting Plasma Glucose and glycosylated haemoglobin (HbAlc) thereby confirming its beneficial effects of both lipid and glyeemic control in diabetic patients,
In the second Phase III study, Lipaglyn was studied in diabetic dyslipidemic patients insufficiently controlled with statin therapy. The results from this study confirmed that Lipaglyn had a pronounced beneficial effect on both the lipid and glyeemic parameters in these subjects.
In both the studies, Lipaglyn was well tolerated and had a better safety profile than the comparators. Importantly Lipaglyn1 M has a non-renal route of elimination, and did not show adverse events like edema, weight gain, myopathies or derangement of liver and/or kidney functions, thus making it sale and efficacious. LipaglynIM is recommended for once daily administration as 4 mg tablets.
Zydus will offer a dedicated LipaglynIM support programme to patients and earegivers, The programme shall provide important support and information regarding access, adherence, education and thereby help patients to start and appropriately manage their disease and therapy over time.
About Lipaglyn
Lipaglyn[TM] (Saroglitazar) was launched in September 2013 in India, for treating Hypertriglyceridemia and Diabetic Dyslipidemia in Patients with Type 2 Diabetes not controlled by statins. Since then, more than 80,000 patients are availing this drug with a prescriber base over 3500 diabetologists, cardiologists and physicians. Lipaglyn[TM] helps in a reduction of triglycerides and LDL (bad) cholesterol, and an increase in HDL (good) cholesterol and has also shown a reduction in Fasting Plasma Glucose and glycosylated haemoglobin (HbA1c), thereby confirming its beneficial effects on both lipid and glycemic control in diabetic patients. Lipaglyn[TM] is a prescription medicine, and can be taken only under the advice and guidance of a registered medical practitioner.
About Zydus
Zydus Cadila is an innovative, global pharmaceutical company that discovers, manufactures and markets a broad range of healthcare therapies, including small molecule drugs, biologic therapeutics and vaccines. The group employs over 16,500 people worldwide including over 1200 scientists engaged in R & D and is dedicated to creating healthier communities globally. As a leading healthcare provider, it aims to become a global research based pharmaceutical company by 2020.
References
Zydus to present new scientific data on Lipaglyn in the US
New Delhi, Jun 8 (UNI) Healthcare services provider, Zydus Cadila today said the new scientific and clinical data on Lipaglyn (Saroglitazar) will be presented at the 75th annual scientific sessions of the American Diabetes Association (ADA) in Boston, Massachusetts, US from 5th to 9th June,2015.
Read more at http://www.uniindia.com/news/business-economy/zydus-to-present-new-scientific-data-on-lipaglyn-in-the-us/84440.html
READ …..https://newdrugapprovals.org/2013/06/07/cadila-banks-on-diabetes-druglipaglynsaroglitazar/
http://lipaglyn.com/downloads/Lipaglyn_Product_Monograph.pdf
http://www.ijpcs.net/sites/default/files/IJPCS_3_1_02_0.pdf
http://onlinelibrary.wiley.com/doi/10.1002/prp2.136/pdf
//////
Should Equipment Status Identification Labels be retained with the Batch Record?

Should Equipment Status Identification Labels be retained with the Batch Record?
Keeping equipment status identification labels with the batch record provides additional confirmation during the review process. But is it required?
Keeping equipment status identification labels with the batch record or other files is often done to provide additional confirmation during review of the record. It supports verification that certain equipment was cleaned before usage for manufacturing. But is it required?
The U.S. Food and Drug Administration FDA has answered this question in an Q&A Document. Assuming each major piece of equipment has a unique “Cleaning and Use Log” that is adequately retained, these “quick reference” equipment labels can be discarded according the agency. FDA sees “no value in the retention of such labels in addition to the required equipment log or batch record documentation. The labels serve a valuable, temporary purpose of positively identifying the current status of equipment and the material under process. Any status label should be correct, legible, readily visible, and associated with the correct piece of equipment. The information on the temporary status label should correspond with the information recorded in the equipment cleaning and use log, or the previous batch record for non-dedicated equipment.”
However, as said before, it might be useful keeping these labels in a batch record. Many companies are doing so; not because it is a requirement but it is a helpful and reliable practice.
/////////
Selurampanel, BGG 492
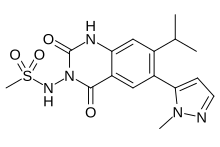
Selurampanel, BGG492,
cas 912574-69-7
Chemical Formula: C16H19N5O4S
Exact Mass: 377.1158
UNII-7WG1MR7DAR;
N-(7-isopropyl-6-(1-methyl-1H-pyrazol-5-yl)-2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl)methanesulfonamide
N-[7-Isopropyl-6-(1-methyl-1H-pyrazol-5-yl)-2,4-dioxo-1,2,3,4-tetrahydroquinazolin-3-yl]methanesulfonamide
PHASE 2 , FOR EPILEPSY, TITINUS
NOVARTIS INNOVATOR
Selurampanel (INN, code name BGG492) is a drug closely related to the quinoxalinedione series which acts as a competitive antagonist of the AMPA and kainate receptors and, as of 2015, is being investigated in clinical trials by Novartis for the treatment ofepilepsy.[1][2][3] It has also been studied in the acute treatment of migraine, and was found to produce some pain relief, but with a relatively high rate of side effects.[4]
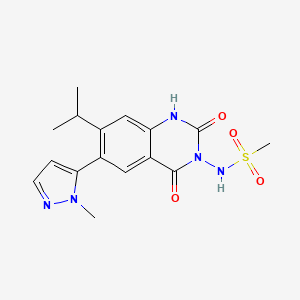
PATENT
Example 44: N-[7-IsopropyI-6-(l-methyl-lH-pyrazol-4-yl)-2,4-dioxo-l,4-dihydro-2H-quinazoIin-3-yl]-methanesulfonamide
2-Amino-4-isopropyl-5-(2-methyl-2H-pyrazol-3-yl)-benzoic acid methyl ester

The 2-amino-5-iodo-4-isopropyl-benzoic acid methyl ester required for the coupling reaction described below was prepared according to the procedures described in WO 2004/033435 Al.
The l-methyl-5-tributylstannanyl-lH-pyrazole required for the coupling reaction was prepared according to the procedure described above.
2-Amino-5-iodo-4-isopropyl-benzoic acid methyl ester (300 mg, 0.94 mmol) and l-methyl-5-tributylstannanyl-lH-pyrazole (523 mg, 1.5 equiv) were weighed in air and added in a flame-dried flask. [Bistriphenylphosphine]dichloropalladium (67.3 mg, 0.1 equiv) was added and the flask was closed by a septum. Dioxane (1 mL) was added and the mixture was stirred for 18 h (TLC control) at 100 0C. The mixture was dissolved with EtOAc, filtered and evaporated to dryness. The crude product was purified by flash chromatography (hexanes to EtOAc / hexanes (4:6)) to yield 2-amino-4-isopropyl-5-(2-methyl-2H- pyrazol-3-yl)-benzoic acid methyl ester (169 mg, 66%) as a yellow solid. (ESI-MS: m/z 21 A [M+H]+, rt 5.20 min).
2-(4-Chloro-phenoxycarbonylamino)-4-isopropyl-5-(2-methyl-2H-pyrazol-3-yl)-benzoic acid methyl ester

4-Chlorophenyl-chloroformate (88 μL, 1.1 equiv) was added to a solution of 2-amino-4-isopropyl-5-(2~ methyl-2H-pyrazol-3-yl)-benzoic acid methyl ester (156 mg, 0.57 mmol) in dioxane (1.5 mL). The mixture was stirred for 2 h (TLC control) at 80 0C. The mixture was evaporated to dryness. The obtained yellow solid was used in the next step without further purification, (rt 6.77 min)
N-[7-Isopropyl-6-(2-methyl-2H-pyrazol-3 -yl)-2,4-dioxo- 1 ,4-dihydro-2H-quinazolin-3 -yl] -methanesulfonamide

CH3SO2NHNH2 (79.5 mg, 1.1 equiv) and J-Pr2NEt (225 μL, 2 equiv) were added to a solution of 2-(4-chloro-phenoxycarbonylamino)-4-isopropyl-5-(2-methyl-2H-pyrazol-3-yl)-benzoic acid methyl ester (281 mg, 0.65 mmol) in dioxane (8 mL). The mixture was stirred for 16 h (TLC control) at 80 0C. The mixture was evaporated to dryness. The crude product was purified by flash chromatography (MeOH / DCM (1:9)) to provide N-[7-isopropyl-6-(2-methyl-2H-pyrazol-3 ~yl)-2,4-dioxo- 1 ,4-dihydro-2H-quinazolin-3 -yl]-methanesulfonamide as a white solid (120 mg, 48%) (ESI-MS: m/z 378 [M+H]+, rt 4.20 min).
| Patent | Submitted | Granted |
|---|---|---|
| Substituted 1H-quinazoline-2,4-diones useful as AMPA receptor ligands [US7655666] | 2008-06-26 | 2010-02-02 |
| N-(2,4-dioxo-6-(tetrahydrofuran-2-yl)-7-(trifluoromethyl)-1,4-dihydro-2H-quinazolin-3-yl)methanesulfonamide [US8012988] | 2010-06-10 | 2011-09-06 |
| 2,4-DIOXO-1,4-DIHYDRO-2H-QUINAZOLIN-3-YL-SULFONAMIDE DERIVATIVES [US2013053381] | 2011-05-18 | 2013-02-28 |
| Use of 1H-quinazoline-2,4-diones [US2013090346] | 2012-09-05 | 2013-04-11 |
| Use of 1H-quinazoline-2,4-diones [US2013096145] | 2011-06-24 | 2013-04-18 |
| Use of 1H-quinazoline-2,4-diones [US2014163050] | 2014-02-12 | 2014-06-12 |
| FOMULATION COMPRISING 1 H-QUINAZOLINE-2, 4-DIONE AMPA RECEPTOR ANTAGONISTS, IN THE FORM OF IMMEDIATE RELEASE TABLETS AND PREPARATION THEREOF [US2012263791] | 2010-12-21 | 2012-10-18 |
| Use of 1H-Quinazoline-2,4-Diones [US2014018376] | 2010-10-20 | 2014-01-16 |
| 1-H-QUINAZOLINE-2, 4-DIONES FOR USE IN THE TREATMENT OF NEURONAL CEROID LIPOFUSCINOSIS [US2012122903] | 2010-07-23 | 2012-05-17 |
References
- Faught, Edward (2014). “BGG492 (selurampanel), an AMPA/kainate receptor antagonist drug for epilepsy”. Expert Opinion on Investigational Drugs 23 (1): 107–113.doi:10.1517/13543784.2014.848854. ISSN 1354-3784.
- Belcastro, Vincenzo; Verrotti, Alberto (2015). “Novel Molecular Targets for Drug-Treatment of Epilepsy”: 183–199.doi:10.1007/978-3-319-12283-0_10.
- Hanada, Takahisa (2014). “The AMPA receptor as a therapeutic target in epilepsy: preclinical and clinical evidence”. Journal of Receptor, Ligand and Channel Research: 39.doi:10.2147/JRLCR.S51475. ISSN 1178-699X.
- Gomez-Mancilla B, Brand R, Jürgens TP, et al. (February 2014). “Randomized, multicenter trial to assess the efficacy, safety and tolerability of a single dose of a novel AMPA receptor antagonist BGG492 for the treatment of acute migraine attacks”. Cephalalgia 34 (2): 103–13.doi:10.1177/0333102413499648. PMID 23963355.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N-[7-Isopropyl-6-(2-methylpyrazol-3-yl)-2,4-dioxo-1H-quinazolin-3-yl]methanesulfonamide
|
|
| Identifiers | |
| CAS Number | 912574-69-7 |
| ATC code | None |
| PubChem | CID 45381907 |
| ChemSpider | 32698379 |
| Chemical data | |
| Formula | C16H19N5O4S |
| Molar mass | 377.418 g/mol |
see……..http://apisynthesisint.blogspot.in/2016/02/selurampanel-bgg-492.html
////Selurampanel, BGG492, 912574-69-7
CC(C)c1cc2c(cc1c3ccnn3C)c(=O)n(c(=O)[nH]2)NS(=O)(=O)C
CS(=O)(NN1C(NC2=C(C=C(C3=CC=NN3C)C(C(C)C)=C2)C1=O)=O)=O
ONL 1204 a small molecule peptide

OR

ONL 1204
CAS 1349038-53-4
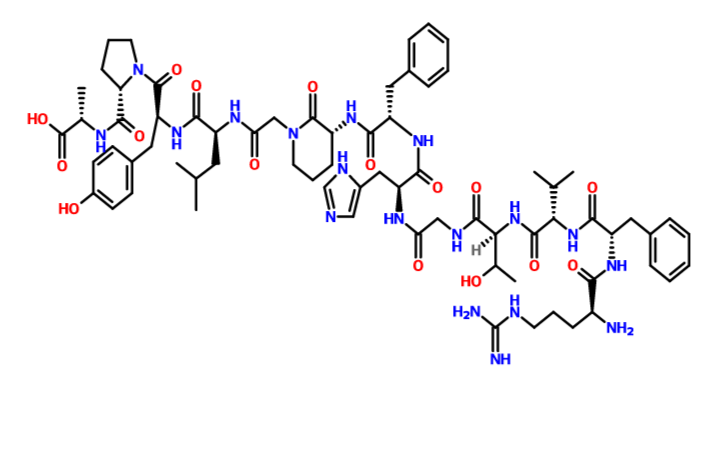
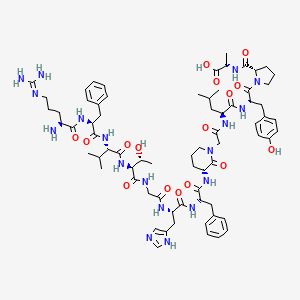
(2S)-2-[[(2S)-1-[(2S)-2-[[(2S)-2-[[2-[(3R)-3-[[(2S)-2-[[(2S)-2-[[2-[[(2S,3R)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-amino-5-(diaminomethylideneamino)pentanoyl]amino]-3-phenylpropanoyl]amino]-3-methylbutanoyl]amino]-3-hydroxybutanoyl]amino]acetyl]amino]-3-(1H-imidazol-5-yl)propanoyl]amino]-3-phenylpropanoyl]amino]-2-oxopiperidin-1-yl]acetyl]amino]-4-methylpentanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]pyrrolidine-2-carbonyl]amino]propanoic acid
His-His- Ile-Tyr-Leu-Gly-Ala-Val-Asn-Tyr-Ile-Tyr-NH2
ONL Therapeutics Inc.
Fas receptor (CD95)
Peptide, Retinal detachment, OPTHALMIC DRUGS
C71 H100 N18 O16, 1461.66
L-Histidyl-L-histidyl-L-isoleucyl-L-tyrosyl-L-leucylglycyl-L-alanyl-L-valyl-L-asparaginyl-L-tyrosyl-L-isoleucyl-L-tyrosinamide
RFVTGHFXGL YPA
ORPHAN DRUG DESIGNATION DATA
His-His- Ile-Tyr-Leu-Gly-Ala-Val-Asn-Tyr-Ile-Tyr-NH2
01/13/2016
Treatment of retinal detachment
ONL Therapeutics, Inc
1600 Huron Parkway
Second Floor
Ann Arbor, Michigan 48109…….http://www.accessdata.fda.gov/scripts/opdlisting/oopd/OOPD_Results_2.cfm?Index_Number=501215
![]()
ONL1204, ONL’s lead therapeutic candidate, is a first-in-class small molecule peptide designed to protect key retinal cells, including photoreceptors, against the apoptosis (programmed cell death) that occurs in a range of retinal diseases and conditions. It is this death of these retinal cells that is the root cause of vision loss and the leading cause of blindness.
Researchers have shown that ONL1204 effectively inhibits the Fas pathway; one of the body’s primary mechanisms for inducing programmed cell death (apoptosis). Specifically, the compound’s activity inhibits the Fas receptor, blocks the activation of the Fas pathway, and prevents the apoptosis cascade which results in the death of key retinal cells, including photoreceptor.
While initial development efforts for ONL1204 are focused on retinal detachment, preclinicalin vivo data, along with a growing body of literature, support potential application in age-related macular degeneration (AMD) and other chronic retinal diseases. Combined, the estimated market for the initial indications that ONL plans to target is >$12 billion globally.
ONL Therapeutics, Inc., a biopharmaceutical company developing novel therapies for preserving sight in a range of retinal diseases, today announced that the United States Food and Drug Administration (FDA) has granted orphan drug designation to ONL1204 for the treatment of retinal detachment. ONL1204 is a novel, first-in-class small molecule peptide designed to protect key retinal cells, including photoreceptors, from cell death that occurs in a range of retinal diseases and conditions. Death of these retinal cells is the root cause of vision loss and the leading cause of blindness. ONL expects to advance ONL1204 into clinical trials for retinal detachment patients in 2016.
Retinal detachment occurs when the retina is separated from the underlying layer of cells called the retinal pigment epithelium (RPE). The RPE provides nutritional support to the highly-active photoreceptors in the retina. When there is a detachment, the photoreceptors no longer receive these nutrients and undergo cell death processes that dramatically impact a patient’s vision. Retinal detachments occur in approximately 50,000 people each year in the United States and affect people of all ages, although risk increases as people reach fifty years of age.
Patients experiencing a retinal detachment are normally treated by surgical reattachment of the retina to reconnect the photoreceptors with the RPE and prevent additional loss of vision. However, these procedures do not address the photoreceptor death and vision loss, which can be significant, that occurs prior to surgery. ONL1204 will be delivered to patients upon diagnosis and is intended to block photoreceptor cells from dying until surgery can be completed.
“When retinal detachments involve the center of vision called the macula, more than a third of patients have final best corrected vision of 20/60 or worse after successful surgery,” said David Zacks, M.D., Ph.D., co-founder and chief science officer of ONL Therapeutics. “Those are truly poor outcomes from successful surgeries. We are very pleased the FDA has recognized this need and that ONL is the only company to have received an orphan designation for this disease. It reinforces our belief that ONL1204 can play a key role in preventing vision loss in these patients by protecting their photoreceptors.”
The FDA’s Orphan Drug Designation program provides certain incentives for companies developing therapeutics to treat rare diseases or conditions that affect less than 200,000 individuals in the US. A drug candidate and its developer must meet several key criteria in order to qualify for, and obtain, orphan drug status. Once a drug has received orphan drug designation, the developer qualifies for a range of benefits, including federal grants, tax credits, reduction in certain regulatory fees, and the potential for seven years of market exclusivity for the drug following FDA marketing approval.
About ONL Therapeutics
ONL Therapeutics (ONL) is a biopharmaceutical company committed to protecting and improving the vision of patients with retinal disease. By advancing a novel breakthrough technology designed to protect key retinal cells from Fas-mediated cell death, ONL is pioneering an entirely new approach to preserving sight. The death of key retinal cells is the root cause of vision loss and leading cause of blindness, and is implicated in a wide range of retinal diseases, including retinal detachment and both the wet and dry forms of age related macular degeneration (AMD).
read
FDA grants orphan status for ONL Therapeutics’ ONL1204 to treat retinal detachment
The US Food and Drug Administration (FDA) has granted orphan drug designation for ONL Therapeutics’ first-in-class small molecule peptide, ONL1204, for the treatment of retinal detachment.
/////
Use smiles
N[C@@H](CCCNC(=N)N)C(=O)N[C@@H](Cc1ccccc1)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)O)C(=O)NCC(=O)N[C@@H](Cc2cncn2)C(=O)N[C@@H](Cc3ccccc3)C(=O)N[C@@H]6CCCN(CC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](Cc4ccc(O)cc4)C(=O)N5CCC[C@H]5C(=O)N[C@@H](C)C(=O)O)C6=O
OR
CC(C)CC(C(=O)NC(CC1=CC=C(C=C1)O)C(=O)N2CCCC2C(=O)NC(C)C(=O)O)NC(=O)CN3CCCC(C3=O)NC(=O)C(CC4=CC=CC=C4)NC(=O)C(CC5=CN=CN5)NC(=O)CNC(=O)C(C(C)O)NC(=O)C(C(C)C)NC(=O)C(CC6=CC=CC=C6)NC(=O)C(CCCN=C(N)N)N
OR
C[C@@H](CC)[C@H](NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@H](CC(=O)N)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)CNC(=O)[C@H](CC(C)C)NC(=O)[C@H](Cc2ccc(O)cc2)NC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@H](Cc3cncn3)N)Cc4cncn4)[C@@H](C)CC)C(C)C)C(=O)N[C@@H](Cc5ccc(O)cc5)C(N)=O
Biocon’s Rosuvastatin Calcium tablets get EU approval to treat hyperlipidemia

Biocon’s Rosuvastatin Calcium tablets get EU approval to treat hyperlipidemia
Indian biopharmaceutical company Biocon has received approval from the European Commission for its Rosuvastatin Calcium tablets to treat hyperlipidemia or mixed dyslipidemia.

Indian biopharmaceutical company Biocon has received approval from the European Commission for its Rosuvastatin Calcium tablets to treat hyperlipidemia or mixed dyslipidemia.
Hyperlipidemia is a common genetic disorder that increases lipids and/or lipoproteins levels in the blood.
The first generic formulation approval will allow Biocon to sell Rosuvastatin Calcium 5mg, 10mg, 20mg and 40mg tablets in more than 15 European countries, starting in fiscal 2017.
The company plans to collaborate with regional partners to market the drug; a generic equivalent of Crestor tablets.
Biocon chairperson and managing director Kiran Mazumdar-Shaw said: “This is indeed a proud moment for Biocon’s Small Molecules business.
 Biocon chairperson and managing director Kiran Mazumdar-Shaw
Biocon chairperson and managing director Kiran Mazumdar-Shaw
“This approval paves the way for Biocon to launch Rosuvastatin Calcium tablets in several European countries.”
The approval will allow the company to address the $1.2bn opportunity in the EU. It will also make it easier for the company to market its products in emerging markets, where regulatory clearances are primarily based on approvals given by regulators in the US / EU.
Biocon was the first generic company to receive a certificate of suitability (CEP) for Rosuvastatin Calcium API from the European Directorate for the Quality of Medicines (EDQM).
CEP certification indicates that an API is suitable for use in medicinal products in the EU.
Biocon CEO and joint managing director Dr Arun Chandavarkar said: “The European approval for Biocon’s generic version of Rosuvastatin Calcium underscores Biocon’s unique strengths in the chronic therapies space and our compliance with global standards that enable us to achieve the highest quality standards for all our products.
“It augurs well for this nascent business, which will be one of our growth drivers in the coming years.”
The company plans to boost its generic formulations business with a target of 20-25 filings over the next few years.
Additionally, Biocon is developing a new facility in Bengaluru, in the Indian state of Karnataka, where it will produce oral solid dosage formulations.
 Biocon CEO and joint managing director Dr Arun Chandavarkar
Biocon CEO and joint managing director Dr Arun Chandavarkar
/////////
Vismodegib

Vismodegib
2-Chloro-N-(4-chloro-3-pyridin-2-ylphenyl)-4-methylsulfonylbenzamide
Vismodegib; 879085-55-9; GDC-0449; 2-chloro-N-(4-chloro-3-(pyridin-2-yl)phenyl)-4-(methylsulfonyl)benzamide; Erivedge; HhAntag691; CUR-691
GDC-449
Hh-Antag691
HhAntag
R-3616
RG-3616
421.29706 g/mol
LAUNCHED 2012
Vismodegib is a Hedgehog Pathway Inhibitor. The mechanism of action of vismodegib is as a Smoothened Receptor Antagonist.
Hedgehog Antagonist GDC-0449 is an orally bioavailable small molecule with potential antineoplastic activity. Hedgehog antagonist GDC-0449 targets the Hedgehog signaling pathway, blocking the activities of the Hedgehog-ligand cell surface receptors PTCH and/or SMO and suppressing Hedgehog signaling. The Hedgehog signaling pathway plays an important role in tissue growth and repair; aberrant constitutive activation of Hedgehog pathway signaling and uncontrolled cellular proliferation may be associated with mutations in the Hedgehog-ligand cell surface receptors PTCH and SMO.
NMR from net

Vismodegib is an active pharmaceutical ingredient produced by Genentech (Roche) and sold under the trade name Erivedge® (which contains crystalline Vismodegib as the active ingre-dient). Erivedge® is an oral Hedgehog signaling pathway inhibitor approved for the treatment of basal-cell carcinoma (BCC).
Developed and launched by Roche and its subsidiary Genentech, under license from Curis. Family members of the product Patent of vismodegib (WO2006028958),
![]()
Vismodegib was first disclosed in WO Patent Publication No. 06/028959. Vismodegib, chem-ically 2-Chloro-N-(4-chloro-3-pyridin-2-ylphenyl)-4-methylsulfonylbenzamide, is represented by the following structure:

Vismodegib (trade name Erivedge) is a drug for the treatment of basal-cell carcinoma (BCC). The approval of vismodegib on January 30, 2012, represents the first Hedgehog signaling pathway targeting agent to gain U.S. Food and Drug Administration (FDA) approval.[1] The drug is also undergoing clinical trials for metastatic colorectal cancer, small-cell lung cancer, advanced stomach cancer, pancreatic cancer, medulloblastoma and chondrosarcoma as of June 2011.[2] The drug was developed by thebiotechnology/pharmaceutical company Genentech, which is headquartered at South San Francisco, California, USA.
Indication
Vismodegib is indicated for patients with basal cell carcinoma (BCC) which has metastasized to other parts of the body, relapsed after surgery, or cannot be treated with surgery or radiation.[3] [4]
Mechanism of action
The substance acts as a cyclopamine-competitive antagonist of the smoothened receptor (SMO) which is part of the hedgehog signaling pathway.[2] SMO inhibition causes the transcription factors GLI1 and GLI2 to remain inactive, which prevents the expression of tumor mediating genes within the hedgehog pathway.[5] This pathway is pathogenetically relevant in more than 90% of basal-cell carcinomas.[6]
PAPER
Bioorg Med Chem Lett 2009, 19(19): 5576
http://www.sciencedirect.com/science/article/pii/S0960894X10012709

Figure 1.
Schematic for the discovery of 2 (GDC-0449) from 1, and the inspiration for further analogs 3 and 4
CN 103910671
http://www.google.com/patents/CN103910671A?cl=en
In embryonic development, Hedgehog signaling in cell differentiation, tissue development and organogenesis play an important role. In the adult body, Hedgehog signaling pathway is mainly in slumber, but when abnormal tissue growth and self-healing, Hedgehog pathway may be activated. With the in-depth study of the tumor, the presence of numerous evidence of abnormal tumor occurrence and the close relationship between Hedgehog signaling pathway, such as sporadic basal cell carcinoma, medulloblastoma, small cell lung cancer and gastrointestinal cancer and other diseases, therefore Hedgehog signaling pathway targeted anti-cancer therapy inhibitors become hot.
Vismodegib chemical name 2_ chlorine -N_ (4_ chlorine _3_ (_2_ pyridyl) phenyl) _4_ (methylsulfonyl) benzamide, is by Roche’s Genentech (Genentech) Hedgehog pathway inhibitors developed, and can be inhibited by binding seven transmembrane protein Smoothened (Smo), thereby preventing signal transduction. Vismodegib capsule in January 2012 I was approved and listed by the US Food and Drug Administration, under the trade name Erivedge, for the treatment of adults with the most common type of skin cancer – basal cell carcinoma. This medicine is not intended for surgery or radiotherapy of cancer and basal cell skin cancer locally advanced patients have been transferred. This was the first drug approved for the treatment of basal cell carcinoma.

W02006028958 Vismodegib disclose the following synthesis route:
Route One Negishi coupling reactions

wherein, X1 is chloro, bromo or iodo; X2 is bromo, iodo or tosylate. The route to the 2-halo-pyridine as starting material an organic zinc compound, and then prepared by Negishi coupling reaction to give 2- (2-chloro-5-nitrophenyl) pyridine. 2- (2-chloro-5-nitrophenyl) pyridine in turn through a reduction reaction with acylation reaction, to give the final product Vismodegib. The key coupling step of the route using an organic zinc reagent required to react under strict anhydrous, anaerobic conditions.
The second route Suzuki coupling reaction [0010]

wherein, X2 is bromo, iodo or tosylate. The route from 3-halo-4-chloro-nitrobenzene as raw material, and 2-chloro-5-nitrophenyl boronic acid pinacol ester, and then reacted with a 2-halo-pyridine was prepared to give 2- (2-chloro 5-nitrophenyl) pyridine. 2- (2-chloro-5-nitrophenyl) pyridine then after reduction and acylation reaction, to give the final product Vismodegib. The key coupling step of the route using the Suzuki coupling reaction, organic boron reagent price to use expensive, high production costs.
The route three Suzuki coupling reaction

wherein, X2 is bromo, iodo or tosylate. Similar to the second route, the route is still critical coupling step using a Suzuki coupling reaction, the same need to use expensive organic boron reagents, higher production costs.
route four Stille coupling reaction

The route to 2-p-toluenesulfonyl pyridine as starting material, is reacted with an organotin reagent, prepared to give pyridin-2-yl trimethyltin, then by Stille coupling reaction, was prepared to give 2- (2-chloro – 5- nitrophenyl) pyridine, followed by reduction reaction, acylation prepared to give Vismodegib. The key step of the route using the Stille coupling reaction, this step need to use expensive and toxic organotin reagents, and the need to carry out the reaction under strict anhydrous, anaerobic conditions.
A process for preparing 2-chloro -N- (4- chloro-3- (pyridin-2-yl) phenyl) -4- (methylsulfonyl) benzamide, comprising: a compound of formula III was prepared as a compound of Formula II;

Then, the compound of formula II with a compound of formula I, to give 2-chloro -N- (4- chloro-3- (pyridin-2-yl) phenyl) -4- (methylsulfonyl) benzamide;

Wherein, R1 is halogen or hydroxy, preferably chlorine, or a hydroxyl group.
2. A process for preparing 2-chloro -N- (4- chloro-3- (pyridin-2-yl) phenyl) -4- (methylsulfonyl) benzamide, comprising:

Wherein, X is halogen, preferably bromo or iodo condition is halo or hydroxy, preferably chlorine, or a hydroxyl group.
3. A process for preparing 2-chloro -N- (4- chloro-3- (pyridin-2-yl) phenyl) -4- (methylsulfonyl) benzamide, comprising:

Wherein, X is halogen, preferably bromo or iodo condition is halo or hydroxy, preferably chlorine, or a hydroxyl group.
Example 1: N–oxo-2- (2-chloro-5-nitrophenyl) pyridine

[0108] To a 100mL three-necked flask were added 30mmoll- oxopyrido, 10mmol2- bromo-1-chloro-4-nitrobenzene, 12mmol potassium carbonate, 0.05mmol tri-butyl acetate button and 0.15mmol phosphorus tetrafluoroborate salt, 40ml of toluene, IS gas exchange three times, under argon at reflux for 2 days, then the reaction mixture was poured into 100mL of ethyl acetate, filtered, and the filtrate was washed with saturated brine, dried and the solvent was distilled off under reduced pressure, column chromatography (mobile phase V / V: methanol / dichloromethane = 1/50), fractions were collected and the solvent was distilled off under reduced pressure to give a pale yellow solid, yield 60%.
1HMffi (500Hz, DMS0_d6): 8.35 (m, 3H), 7.90 (d, 1Η), 7.62 (q, 1Η), 7.55 (m, 1Η), 7.48 (m, 1Η);
MS: 251.1,253.1 ([Μ + Η] +).
2 Example: Ν–oxo-2- (2-chloro-5-nitrophenyl) pyridine

To a 100mL three-necked flask 30mmoll- oxopyrido, 10mmol2- bromo-1-chloro-4-nitrobenzene, 12mmol of potassium carbonate, 0.05mmol iodide and 0.1Ommoll, 10- Fei Luo Jie morpholine, 40ml of xylene, an argon gas exchange three times, under argon at reflux for 2 days, cooled to room temperature and then the reaction system was poured into 100mL methylene chloride, filtered and the filtrate washed with saturated brine, dried, filtered, The filtrate solvent was distilled off under reduced pressure, column chromatography (mobile phase V / V: methanol / dichloromethane = 1/50) to give a pale yellow solid, yield 42%. .
3 Example: 2- (2-chloro-5-nitrophenyl) pyridine

After 3.0mmol N- oxo added to 100mL of Lord vial _2_ (2_ chloro _5_ nitrophenyl) pyrazole 唳, 15mmol phosphorus trichloride and 30ml of chloroform was heated at reflux for 12h, the reaction It was poured into 100mL of water and extracted with ethyl acetate (50ml X 2), and the combined organic phase was dried and the solvent was distilled off under reduced pressure, column chromatography (mobile phase V / V: petroleum ether / ethyl acetate = 20/1) , fractions were collected, the solvent was distilled off under reduced pressure to give a white solid, yield 95%.
1Hnmr (SooHzJDCI3): 8.78 (d, 1H), 8.51 (d, 1H), 8.20 (m, 1H), 7.85 (m, 1H), 7.72 (d, 1H), 7.65 (d, 1H), 7.40 (m, 1H);
MS: 235.1,237.1 ([M + H] +).
4 Example 2: Preparation 4_ chlorine _3_ (topiramate 唳 _2_ yl) aniline

To a vial was added 100mL of Lord 20mmol2- (2- chloro-5-nitrophenyl) pyridine 唳, 50ml of acetic acid, heated to 80 ° C and stirred, and then slowly added IOOmmol iron, reaction 0.5h The reaction solution was poured into 200ml water and extracted with dichloromethane (150ml X 3), the combined organic phases, the organic phase was washed with saturated sodium carbonate solution (50ml X 3), the organic phase was dried, evaporated under reduced pressure to give the crude product, n-propyl alcohol weight crystallized to give a pale yellow solid, yield 75%.
1HMflUSOOHz, DMS0_d6): 8.63 (m, 1H), 7.84 (m, 1H), 7.56 (d, 1H), 7.37 (m, 1H),
7.13 (d, 1H), 6.76 (d, 1H), 6.61 (q, 1H), 5.32 (s, 2H);
MS: 205.1,207.1 ([M + H] +).
5 Example: 4-chloro-3- (pyridin 唳-2-yl) aniline

to 100mL of God-shaped flask 20mmol2_ (2_ chlorine _5_ nitrophenyl) pyridine Jie set, 50ml of methanol, Ig activated carbon, 2mmol FeOOH and 60mmol85% of hydrazine hydrate, heated to reflux and stirred for 6 ~ 8h, after the completion of the reaction, was filtered, spin-dry the solvent, dissolved in 150ml of dichloromethane, the organic phase was washed with saturated sodium bicarbonate solution (20ml X3), the organic phase was dried, evaporated under reduced pressure to give the crude product was recrystallized from n-propanol to give a pale yellow solid, yield 96%.
6 Example 2: Preparation 4_-chloro-3- (2-yl) aniline

20mmol N- oxo added to 100mL eggplant-shaped flask _2_ (2_ chloro _5_ nitrophenyl) pyridine, 50ml of acetic acid, heated to 80 ° C and stirred, and then iron powder was slowly added IOOmmol After 0.5h the reaction the reaction solution was poured into 200ml water and extracted with dichloromethane (150ml X3), the combined organic phases were washed with saturated sodium carbonate solution (50ml X3), the organic phase was dried, evaporated under reduced pressure to give the crude product, n-propanol recrystallized to give a white solid, yield 70%.
Preparation 7.Α ~ chlorine -3_ (topiramate 唳 2-yl) aniline [0130] Example

20mmol N- oxo added to 100mL eggplant type flask _2_ (2_ chloro _5_ nitrophenyl) pyridine, 50ml of methanol, Ig active carbon, 2mmol FeOOH 60mmol85% hydrazine hydrate and heated to reflux and stirred for 6 ~ 8h, after the completion of the reaction, was filtered, spin-dry the solvent, dissolved in 150ml of dichloromethane, washed with saturated aqueous sodium bicarbonate solution, the organic phase (20mlX3), the organic phase was dried, evaporated under reduced pressure to give the crude product, n-propyl alcohol weight crystallized to give a white solid, yield 82%.
Vismodegib Preparation: 8 Example

In the Lord 50ml vial, the 1.50mmol2- chloro-4-methanesulfonyl-chloride in 15ml of dry tetrahydrofuran, cooled to ice bath O ~ 10 ° C, a solution of 4-chloro-3 – (pyridin-2-yl) aniline in anhydrous tetrahydrofuran (1.47mmol / 10ml), triethylamine was added dropwise and then finished 2.5mmol of dropwise addition, the reaction at room temperature 4h, the reaction was completed, the reaction system was poured into 50ml water and stirred, precipitated solid was filtered, washed with water, and dried to give a white solid product, yield 88%.
1HNMR (500Hz, DMS0_d6): 10.90 (s, 1H), 8.70 (d, 1H), 8.12 (d, 1H), 8.01 (t, 2H), 7.92 (m, 2H), 7.74 (q, 1H ), 7.69 (d, 1H), 7.58 (d, 1H), 7.44 (m, 1H), 3.34 (s, 3H).
MS: 421.1,423.1 ([M + H] +).
Vismodegib Preparation: 9 Example

In 50ml vial of God, will 1.50mmol2_ chlorine _4_ methylsulfonyl benzoic acid, 1.47mmol4_ chlorine _3_ (batch 唳 2-yl) aniline and triethylamine were dissolved in 25ml 2.5mmol anhydrous tetrahydrofuran in an ice bath to cool to O ~ 10 ° C, was added in portions N, N ‘- dicyclohexyl carbodiimide (DCC) 1.50mmol, After the addition, the reaction at room temperature 6h, after the reaction, white solid was removed by filtration, the filtrate was poured into 50ml water and stirred, precipitated solid was filtered, washed with water, and dried to give a white solid product, yield 84%.
Vismodegib Preparation: 10 [0141] Example

In 50ml eggplant-shaped flask, 1.50mmol2- chloro-4-methanesulfonyl-benzoic acid was dissolved in 15ml of dichloromethane, cooled to ice bath O ~ 5 ° C, thionyl chloride was added dropwise 3.0mmol After stirring at room temperature 30min, removed by rotary evaporation dichloromethane and excess thionyl chloride, 15ml of anhydrous tetrahydrofuran was added, the ice bath was cooled to O ~ 10 ° C, solution of 4-chloro-3- (pyridin-2- yl) aniline in anhydrous THF (1.47mmol / 10ml), triethylamine was added dropwise and then finished 2.5mmol of dropwise addition, the reaction at room temperature 4h, the reaction was completed, the reaction was poured into 50ml water system and stirring, the precipitated solid was filtered, washed with water, and dried to give a white solid product, yield 88%.
PATENT
CN 103910672
http://www.google.com/patents/CN103910672A?cl=en
Vismodegib PreparatioN

In 50ml eggplant-shaped flask, 1.50mmol2- chloro-4-methanesulfonyl-benzoic acid was dissolved in 15ml of dichloromethane, cooled to ice bath O ~ 5 ° C, thionyl chloride was added dropwise 3.0mmol After stirring at room temperature 30min, removed by rotary evaporation dichloromethane and excess thionyl chloride, 15ml of anhydrous tetrahydrofuran was added, the ice bath was cooled to O ~ 10 ° C, solution of 4-chloro-3- (pyridin-2- yl) aniline in anhydrous THF (1.47mmol / 10ml), triethylamine was added dropwise and then finished 2.5mmol of dropwise addition, the reaction at room temperature 4h, the reaction was completed, the reaction was poured into 50ml water system and stirring, the precipitated solid was filtered, washed with water, and dried to give a white solid product, yield 88%.
PATENT
WO2006028958
https://www.google.co.in/patents/WO2006028958A2?cl=en
Example 1 General Procedure
Compounds of examples 2-51 were prepared according to the following general procedures.
A: Suzuki Coupling Procedure

2 M aq. Potassium carbonate (5.0 eq) and 4:1 toluene :ethanol mixture (2.5 mL) were added to a microwave vial charged with the appropriate boronate ester (2.6 eq), aryl halide (0.35 mmol, 1.0 eq), and Pd(PPh3)4 (0.04 eq). The vial was sealed and heated with stirring in the microwave to 160 0C for ten minutes. The solution was poured onto 2 M aq. Sodium hydroxide (20 mL), extracted with ethyl acetate (2 x 20 mL), dried (MgSO4), and concentrated. Purification of the crude product by chromatography on silica gel (conditions given below) afforded the desired product.
B: Negishi Coupling Procedure

X = I or Br R = H, 3-Me, 4-Me5 5-Me, 6-Me
Aryl zinc bromide (0.5 M in THF, 2.5 eq) was added to an oven-dried microwave vial charged with the appropriate aryl halide (1.0 eq) and Pd(PPh3)4 (0.04 eq). The vial was sealed and heated with stirring in the microwave to 140 0C for 10 minutes. The crude reaction mixture was concentrated and purified by chromatography on silica gel (conditions given below) to afford the desired product.
C: Iron Reduction of Aryl Nitro Group

R = I or pyridin-2-yl
The appropriate nitro aryl (1 mmol, 1 eq) in AcOH/EtOH (1:1, 0.42 M) was added slowly to a solution of Iron powder (6.0 eq) in AcOH/EtOH (1:2, 2 M) at 60 °C. The solution was stirred at 70 0C for 30-60 minutes. The reaction mixture was cooled to 23 0C, filtered through celite, washed with ethyl acetate, and concentrated. The oily residue was dissolved in ethyl acetate (30 mL), washed with saturated aq. NaHCO3 (2 x 15 rnL) and water (2 x 10 niL), dried (MgSO4), and concentrated. The oily residue was used with out further purification.
D: Amide Bond Formation

R = I or pyridin-2-yI
Acid chloride (1.05-1.1 eq) was added to a solution of aniline (1.0 eq) and TEA (1.1-1.5 eq) in methylene chloride at the indicated temperature. The solution was stirred for 0.5-3 hours, poured onto saturated aq. NaHCO3, extracted twice with methylene chloride, dried (MgSO4), and concentrated. Purification of the crude product by chromatography on silica gel (conditions given below) afforded the desired product.
E: EDC Amide Bond Formation

R = I or pyridin-2-yl
Carboxylic acid (1.1 eq) was added to a solution of aniline (1.0 eq) and EDC (1.4 eq) in methylene chloride (0.7 M in aniline). The solution was stirred at 23 0C for 2 hours, poured onto a 1 :1 mixture of saturated aq. NH4Cl and water, extracted twice with methylene chloride, dried (MgSO4), and concentrated. Purification of the crude product by chromatography on silica gel (conditions given below) afforded the desired product. F: addition of amines to 2-chloropyridine

NHRR’ = ethanolamine, analine, benzylamine, 2-methylpropylamine, N-methylpiperazine, morpholine, 2-morpholinoethylamine
Primary or secondary amine (5 eq) in either BuOH or a mixture of BuOH/ethylene gylcol was heated to 170 to 220 0C for 20 min in a sealed tube. The BuOH was removed under reduced pressure. In cases where ethylene glycol was used, the reaction was diluted with water, and the product was extracted into ethyl acetate, dried (MgSO^, and concentrated. The crude residue was purified by reverse phase HPLC to afford the desired product.
G: Amide bond coupling with HATU
HATU, DIPEA, DMF NaOH or NaHCO3

ethyl acetate extraction

Aniline (1.0 eq) was added to a mixture of carboxylic acid (1.1 eq), HATU (1.1 eq) and DIPEA (2 eq) in DMF (0.1 – 0.2 M). After stirring overnight, the reaction mixture was diluted with 0.1 N sodium hydroxide or saturated NaHCθ3, extracted into ethyl acetate and the combined organic layers were washed with brine. The organic layer was dried (MgSO4), concentrated and the crude mixture was purified by reverse phase HPLC. H: Preparation of sulfonamide benzoic acids

Chlororsulfonylbenzoic acid (1.0 eq) was added to a solution of amine (1.1 eq) in 10-20% DEPEA/methanol (1 M) at 4 0C. After 1 h, the reaction mixture was concentrated, and the crude residue was purified by reverse phase HPLC.
I : Stannylation of 2-pyridyl triflates

A solution of tetrakis-triphenylphosphinepalladium (0.04 eq.) in toluene (1 mL) was added to degassed solution of aryltriflate (1 eq), bis-trialkyltin (1.05 eq), and lithium chloride (3 eq) in dioxane. Heated to reflux for 2 hours, cooled to 23 0C, diluted with ethyl acetate, washed with 10% NH4θH(aq) and brine, dried (MgSO4) and concentrated. The crude material was used without further purification.
J: Stannylation of substituted pyridines

ιMmβco3 n-Butyl lithium (6 eq, 2.5 M in hexanes) was added dropwise to a solution of dimethylaminoethanol (3 eq) in hexane at 0 0C. The solution was stirred at 0 0C for thirty minutes before dropwise addition of the substituted pyridine (1 eq). The solution was stirred at 0 0C for an additional hour, then cooled to -78 0C. A solution of trialkyltin in hexane was added dropwise. The solution was stirred at -78 0C for thirty minutes, warmed to 0 0C, quenched with water, extracted twice with ether, dried (MgSO4), and concentrated. K: Stille Coupling

Palladium catalyst (0.02 eq) was added to a degassed solution of aryliodide (1 eq), arylstannane (2 eq), and triphenylphosphine (0.16 eq) in NMP. Heated in the microwave to 130 0C for 15 minutes. The reaction mixture was diluted with ethylacetate, washed with 10% NH4θH(aq) and brine, dried (MgSC>4), concentrated and purified by silica gel chromatography.
L: Synthesis of alky lethers

A solution of hydroxypyridine (1 eq), alkyliodide (excess), and cesium carbonate in NMP was heated in the microwave to 1000C for ten minutes. The reaction mixture was diluted with ethylacetate, washed with 10% NH4θH(aq) and brine, dried (MgSC^), concentrated and purified by silica gel chromatography.
M: Methyl Ester Saponification

The methyl ester (leq) was hydrolyzed with LiOH (2eq) in 50/50 THF/water mix. Upon completion of the reaction the THF was evaporated under reduced pressure and the solution is acidified with HCl to pH 2. The resultant solid was filtered and dried to give the pure acid.
N: Bromination in the presence of a free acid functionality
The paramethylbenzoic acid (leq) was combined with Benzoyl Peroxide (O.leq) and N- Bromosuccinimde (0.9eq) in a solution of 5%AcOH in Benzene and heated in the microwave at 120°C for 5-15minutes. The product was separated from the starting material and di-bromo product via ISCO flash chromatography with an ethyl acetate (with 1% AcOH) and hexanes solvent system.
O: Sodium Methanesulfinate displacement of Bromine

To the bromine starting material (leq) was added sodium methanesulfinate (2eq) in DMF and heated to 120°C in the microwave for 5 minutes. Alternatively, the reaction was heated to 60°C in an oil bath for several hours until completed. Reaction mixture was concentrated under reduced pressure and extracted in ethyl acetate and water. The organic layer was dried over Magnesium Sulfate, filtered and concentrated in vacuo to yield generic methylsulfone.
P: Amine displacement of Bromine

To the bromo starting material (leq) was added appropriate amine (3eq) in either DMSO or BuOH and stirred at room temperature until complete. For less nucleophilic amines or anilines, the reactions were forced to completion using microwave conditions ranging from 150°-170°C for 15 minutes. Crude reactions were concentrated to dryness and either extracted with ethyl acetate and saturated bicarbonate if the reaction resulted in an intermediate or purified via HPLC if the reaction resulted in a final product.
Q: Thiol displacement of halogen

The paramethylbromo benzoate (leq) was treated with Potassium (or Cesium) Carbonate (1.5eq) and appropriate thiol derivative (l,leq) in DMF (or CH3CN) and stirred overnight at room temperature. The DMF was evaporated in vacuo and the reaction was extracted with ethyl acetate and water. The organic layer was dried over Magnesium Sulfate , filtered and concentrated to yield the thiol or derivatized thiol compound.
R: Oxone Oxidation
oxone 2:1 MeOHTH2O


Derivatized thiol (leq) was dissolved in MeOH while Oxone (2eq) was seperately dissolved in half the amount of water. Once all the oxone was dissolved, the solution was added to the thiol in MeOH solution at once and stirred until complete. The MeOH was evaporated in vacuo and the remaining water was extracted twice with Ethyl Acetate. The organic layer was dried over Magnesium Sulfate and concentrated to yield the sulfone.
S: Thio lysis of epoxides at alumina surfaces

A mixture of epoxides (1.0 eq), thiophenol (1.5 eq) and neutral aluminum oxide (~70 eq) in diethyl ether was stirred for 3 h at room temperature while being monitored by TLC. The reaction mixture was filtered through Celite, washed with ethyl acetate and concentrated. Purified by silica gel chromatography (0-40% ethyl acetate/hexane) to yield β -hydroxysulfide product.
T: Conversion of nitrile group to carboxylic acid

R
A solution of benzonitrile (1.0 eq) and sodium hydroxide (2.0 eq) in H2O was heated to 120 ° C for 2h. The reaction mixture was cooled to room temperature and acidified with HCl to pH 2. The resulting solid was filtered to afford the pure acid product.
U. Alkylation of phenols

The phenol was dissolved in DMF (1.0 ml). Cesium carbonate (1.0 eq.) and an alkyl bromide or alkyl iodide (1.0 to 2.0 eq.) were added, and the reaction was stirred at room temperature for 18 hrs or 5O0C for 1 to 24 hours. The reaction was quenched in water, and extracted with ethyl acetate twice. The organic extracts were washed with water once, brine once, dried with MgSC>4, and evaporated to a crude oil which was purified on reverse phase HPLC.
V. Amide bond formation with an acid chloride and an aniline

The aniline was dissolved in THF (1.5 ml) and dichloromethane (1.5 ml). MP-Carbonate (1.5 eq.) and an acid chloride (1.1 eq.) were added, and the solution was stirred at room temperature for 18 hours. The reaction was diluted with methanol and dichloromethane, and filtered to remove the MP-Carbonate. The mother liquors were evaporated to a solid and purified by reverse phase HPLC.
W. Amidine formation from an imidate

A solution of freshly formed imidate in methanol was treated with a primary or secondary amine (1.5 eq.) at room temperature for 18 hours. The methanol was removed on a rotary evaporator and the residue purified by reverse phase HPLC.
Example 37 2-chloro-N-(4-chloro-3-(pyridin-2-yl)phenyl)-4-(methylsulfonyl)benzamide

Procedure G was used to couple 4-chloro-3-(pyridin-2-yl)aniline (50 mg) and 2-chloro-4- methylsulfonylbenzoic acid to produce 2-chloro-N-(4-chloro-3-(pyridin-2-yl)phenyl)-4- (methylsulfonyl)benzamide. MS (Ql) 421.0 (M)+. The product was then dissolved in 1 Ν HCI solution followed by freebasing with 0.5 Ν NaOH solution (pH to 11). The resulting precipitate was filtered and vacuum-dry.
Procedure D may also be used to couple 4-chloro-3-(pyridin-2-yl)aniline and 2-chloro-4- (methylsulfonyl)benzoyl chloride to produce 2-chloro-N-(4-chloro-3-(pyridin-2-yl)phenyl)-4-
(methylsulfonyl)benzamide which is collected by suction filtration and the HCl salt is washed with
Et2O (or alternatively with MTBE). This material is freebased using EtOAc/aq NaHCO3 and the organics are dried and concentrated to the solid freebase. This material is then crystallized from acetone :EtOAc (80:20, approx lOmL/g) which is then finally recrystallized from hot slurry of iPrOAc. 2-chloro-N-(4-chloro-3-(pyridin-2-yl)phenyl)-4-(methylsulfonyl)benzamide HCl salt may also be dissolved in distilled water followed by freebasing with 0.5 N NaOH solution (pH to 11) and filtering and vacuum drying the precipitate.
Patent
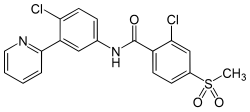
WO 2016020324, BASF AG, vismodegib , new patent
WO2016020324, MULTI-COMPONENT CRYSTALS OF VISMODEGIB AND SELECTED CO-CRYSTAL FORMERS OR SOLVENTS
BASF SE [DE/DE]; 67056 Ludwigshafen (DE)
VIERTELHAUS, Martin; (DE).
CHIODO, Tiziana; (DE).
SALVADOR, Beate; (DE).
VOSSEN, Marcus; (DE).
HAFNER, Andreas; (CH).
HINTERMANN, Tobias; (CH).
WEISHAAR, Walter; (DE).
HELLMANN, Rolf; (DE)
![]()
The present invention primarily relates to multi-component crystals comprising a compound of formula 1 and a second compound selected from the group consisting of co-crystal formers and sol-vents. The invention is further related to pharmaceutical compositions comprising such multi-component crystals. Furthermore, the invention relates to processes for preparing said multi-component crystals. The invention also relates to several aspects of using said multi-component crystals or pharmaceutical compositions to treat a disease.
Developed and launched by Roche and its subsidiary Genentech, under license from Curis. Family members of the product Patent of vismodegib (WO2006028958),
Vismodegib was first disclosed in WO Patent Publication No. 06/028959. Vismodegib, chem-ically 2-Chloro-N-(4-chloro-3-pyridin-2-ylphenyl)-4-methylsulfonylbenzamide, is represented by the following structure:
formula 1
Vismodegib is an active pharmaceutical ingredient produced by Genentech (Roche) and sold under the trade name Erivedge® (which contains crystalline Vismodegib as the active ingre-dient). Erivedge® is an oral Hedgehog signaling pathway inhibitor approved for the treatment of basal-cell carcinoma (BCC).
The present invention primarily relates to multi-component crystals comprising a compound of formula 1 (cf. above) and a second compound selected from the group consisting of co-crystal formers and solvents.
The invention is further related to pharmaceutical compositions comprising said multi-component crystals. Furthermore, the invention also relates to processes for preparing said multi-component crystals. The invention also relates to several aspects of using said multi-component crystals or pharmaceutical compositions to treat a disease. Further details as well as further aspects of the present invention will be described herein below.
Vismodegib is a BCS class II compound with a high permeability but a low solubility where enhanced solubility or dissolution rates can lead to a significant advantage in respect to bio-availability.
Vismodegib is known to exist as crystalline free base. Salts of Vismodegib are men-tioned in US 7,888,364 B2 but not specified. In particular, the HCI salt is mentioned as intermediate but not characterized. Co-crystals or solvates are not reported at all.
The solubility of Vismodegib is reported to be 0.1 μg/mL at pH 7 and 0.99 mg/mL at pH 1 for Erivedge®. The absolute bio-availability after single dose is reported to be 31.8 % and the ex-posure is not linear at single doses higher than 270 mg. Erivedge® capsules do not have a food label. The estimated elimination half-life (t1/2) after continuous once-daily dosing is 4 days and 12 days after a single dose treatment (Highlights of Prescribing Information: ERIVEDGE® (vismodegib) capsule for oral use; Revised: 01/2012).
The discovery and preparation of new co-crystals or solvates offer an opportunity to improve the performance profile of a pharmaceutical product. It widens the reservoir of techniques/materials that a formulation scientist can use for designing a new dosage form of an active pharmaceutical ingredient (API) with improved characteristics. One of the most important characteristics of an API such as Vismodegib is the bio-availability which is often determined by the aqueous solubility.
A compound like Vismodegib may give rise to a variety of crystalline forms having dis-tinct crystal structures and physical characteristics like melting point, X-ray diffraction pattern, infrared spectrum, Raman spectrum and solid state NMR spectrum. One crystalline form may give rise to thermal behavior different from that of another crystalline form. Thermal behavior can be measured in the laboratory by such techniques as capillary melting point, thermogravimetry (TG), and differential scanning calorimetry (DSC) as well as content of sol-vent in the crystalline form, which have been used to distinguish polymorphic forms.
Multi-component crystals comprising Vismodegib and selected co-crystal formers or solvents may improve the dissolution kinetic profile and allow to control the hygrosco-picity of Vismodegib.
Therefore, there is a need for multi-component crystals comprising Vismodegib that avoid the above disadvantages. In particular, it is an object of the present invention to provide multi-component crystals of Vismodegib with optimized manufacture, formula-tion, stability and/or biological efficacy
.
Example 1 :
314 mg Vismodegib and 86 mg maleic acid are suspended in toluene saturated with maleic acid for 2 d, filtered and dried.
TG data shows a mass loss of about 2.3 wt % between 100 and 1 18 °C which is attributed to rest solvent. DSC data shows a single endothermal peak with an onset of about 1 15 °C (99 J/g).
H-NMR spectroscopy indicates a molar ratio of Vismodegib to maleic acid of about 1 :1 .3. However single crystal X-ray data confirms a ratio of 1 :2 (Table 1 ).


update……………
Vismodegib Synthesis
WO2009126863A2: also see Ref. 1. It all started from here.

Identification:

| 1H NMR (Estimated) for Vismodegib |
Experimental: 1H NMR (400MHz, CDCl3) δ (ppm): 9.58 (bs, 1H), 8.43 (d, J = 4.7Hz, 1H), 8.03 (dd, J = 2.6, 8.7Hz, 1H), 7.90 (d, J = 1.6Hz, 1H), 7.67-7.78 (m, 4H), 7.60 (d, J = 8.0Hz, 1H), 7. 51 (d, J = 8.8Hz, 1H), 7.23-7.24 (m, 1H), 3.01 (s, 3H).
UPDATES…….
Manufacturing Development and Genotoxic Impurity Control Strategy of the Hedgehog Pathway Inhibitor Vismodegib

The development work toward the robust and efficient manufacturing process to vismodegib, the active pharmaceutical ingredient (API) in Erivedge, is described. The optimization of the four-stage manufacturing process was designed to produce the API with the required critical quality attributes: (1) the selective catalytic hydrogenation reduction of the nitro compound 3 to the corresponding aniline 4 while minimizing the formation of potential genotoxic (mutagenic) impurities; (2) the control of the polymorphic phase and multipoint specification for particle size distribution.
Vismodegib


1H

13C



////////////////
References
- “Vismodegib, First Hedgehog Inhibitor, Approved for BCC Patients”.
- “Molecule of the Month”. June 2011.
- “FDA approves Erivedge (vismodegib) capsule, the first medicine for adults with advanced basal cell carcinoma”.
- Lacroix, Marc (2014). Targeted Therapies in Cancer. Hauppauge , NY: Nova Sciences Publishers. ISBN 978-1-63321-687-7.
- “Vismodegib (GDC-0449) Smoothened Inhibitor – BioOncology”.
- H. Spreitzer (4 July 2011). “Neue Wirkstoffe – Vismodegib”. Österreichische Apothekerzeitung (in German) (14/2011): 10.
- FDA Professional Drug Information
External links
- Erivedge® (vismodegib), a prescription oral medica on approved for advanced basal cell carcinoma treatment
- Efficacy and Safety of Vismodegib
- Food and Drug Administration (FDA) approved vismodegib
PatentSubmittedGranted
Pyridyl inhibitors of hedgehog signalling [US7888364]2006-03-232011-02-15
PYRIDYL INHIBITORS OF HEDGEHOG SIGNALLING [US2009281089]2009-11-12
ANTI-HEDGEHOG ANTIBODIES [US8030454]2010-01-072011-10-04
PYRIDYL INHIBITORS OF HEDGEHOG SIGNALLING [US2011092461]2011-04-21
PYRIDYL INHIBITORS OF HEDGEHOG SIGNALLING [US2012094980]2011-10-142012-04-19
COMBINATION THERAPY WITH NANOPARTICLE COMPOSITIONS OF TAXANE AND HEDGEHOG INHIBITORS [US2013045240]2010-08-252013-02-21
COMBINATION THERAPY WITH NANOPARTICLE COMPOSITIONS OF TAXANE AND HEDGEHOG INHIBITORS [US2014072630]2013-02-282014-03-13
Acyl guanidine derivatives modulating the hedgehog protein signaling pathway [US8889678]2010-07-192014-11-18
COMBINATION THERAPY [US2012184529]2012-01-032012-07-19
METHOD OF INHIBITING DYRK1B [US2014371251]2014-06-182014-12-18
USE OF SUBSTITUTED HEXITOLS INCLUDING DIANHYDROGALACTITOL AND ANALOGS TO TREAT NEOPLASTIC DISEASE AND CANCER STEM AND CANCER STEM CELLS INCLUDING GLIOBLASTOMA MULTIFORME AND MEDULLOBLASTOMA [US2014377336]2013-01-222014-12-25
SHH Regulation and Methods Thereof [US2012082623]2011-09-302012-04-05
NOVEL 2-PIPERIDIN-1-YL-ACETAMIDE COMPOUNDS FOR USE AS TANKYRASE INHIBITORS [US2015025070]2012-07-132015-01-22
Compositions and Methods for Modulating Neuron Degeneration and Neuron Guidance [US2011065645]2010-09-102011-03-17
SMOOTHENED ANTAGONISM FOR THE TREATMENT OF HEDGEHOG PATHWAY-RELATED DISORDERS [US2014200217]2014-01-242014-07-17
| CN101072755A * | Sep 2, 2005 | Nov 14, 2007 | 遗传技术研究公司 | Pyridyl inhibitors of hedgehog signalling |
| CN102731373A * | Jul 19, 2012 | Oct 17, 2012 | 南京药石药物研发有限公司 | Preparation method of intermediate of antitumor drug GDC-0449 (vismodegib) |
| US20080132698 * | Nov 30, 2006 | Jun 5, 2008 | University Of Ottawa | Use of N-oxide compounds in coupling reactions |
| US20090076266 * | Sep 10, 2008 | Mar 19, 2009 | The University Of Houston System | Copper-catalyzed c-h bond arylation |
NON-PATENT CITATIONS
| Reference | ||
|---|---|---|
| 1 | * | GEORGETTE M. CASTANEDO,等: “Second generation 2-pyridyl biphenyl amide inhibitors of the hedgehog pathway“, 《BIOORGANIC & MEDICINAL CHEMISTRY LETTERS》, vol. 20, 15 September 2010 (2010-09-15), pages 6748 – 6753 |
| 2 | * | 曹萌,等: “Vismodegib 的合成“, 《第十一届全国青年药学工作者最新科研成果交流会论文集》, 21 June 2012 (2012-06-21) |
| 3 | * | 耿一丁: “Vismodegib“, 《中国药物化学杂志》, vol. 22, no. 3, 20 June 2012 (2012-06-20) |
| 4 | * | 邢其毅,等: “《基础有机化学》”, 31 December 2005, article “201310019450.0“, pages: 896-897 |
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
2-Chloro-N-(4-chloro-3-pyridin-2-ylphenyl)-4-methylsulfonylbenzamide
|
|
| Clinical data | |
| Trade names | Erivedge |
| AHFS/Drugs.com | monograph |
| Licence data | EMA:Link, US FDA:link |
| Pregnancy category |
|
| Legal status | |
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | 31.8% |
| Protein binding | >99% |
| Metabolism | <2% metabolised byCYP2C9, CYP3A4, CYP3A5 |
| Biological half-life | 4 days (continuous use), 12 days (single dose) |
| Excretion | Faeces (82%), urine (4.4%) |
| Identifiers | |
| CAS Number | 879085-55-9 |
| ATC code | L01XX43 |
| PubChem | CID 24776445 |
| IUPHAR/BPS | 6975 |
| DrugBank | DB08828 |
| ChemSpider | 23337846 |
| UNII | 25X868M3DS |
| ChEBI | CHEBI:66903 |
| ChEMBL | CHEMBL473417 |
| Synonyms | GDC-0449, RG-3616 |
| Chemical data | |
| Formula | C19H14Cl2N2O3S |
| Molar mass | 421.30 g/mol |
SEE…http://apisynthesisint.blogspot.in/2016/02/vismodegib.html
/////
CS(=O)(=O)C1=CC(=C(C=C1)C(=O)NC2=CC(=C(C=C2)Cl)C3=CC=CC=N3)Cl
CS(=O)(=O)C1=CC(=C(C=C1)C(=O)NC2=CC(=C(C=C2)Cl)C3=CC=CC=N3)Cl
Drug Discovery, Hit to Lead
//////
WO 2016020324, BASF AG, Vismodegib , New patent
WO 2016020324, BASF AG, vismodegib , new patent
WO2016020324, MULTI-COMPONENT CRYSTALS OF VISMODEGIB AND SELECTED CO-CRYSTAL FORMERS OR SOLVENTS
BASF SE [DE/DE]; 67056 Ludwigshafen (DE)
VIERTELHAUS, Martin; (DE).
CHIODO, Tiziana; (DE).
SALVADOR, Beate; (DE).
VOSSEN, Marcus; (DE).
HAFNER, Andreas; (CH).
HINTERMANN, Tobias; (CH).
WEISHAAR, Walter; (DE).
HELLMANN, Rolf; (DE)
![]()
The present invention primarily relates to multi-component crystals comprising a compound of formula 1 and a second compound selected from the group consisting of co-crystal formers and sol-vents. The invention is further related to pharmaceutical compositions comprising such multi-component crystals. Furthermore, the invention relates to processes for preparing said multi-component crystals. The invention also relates to several aspects of using said multi-component crystals or pharmaceutical compositions to treat a disease.
Developed and launched by Roche and its subsidiary Genentech, under license from Curis. Family members of the product Patent of vismodegib (WO2006028958),
Vismodegib was first disclosed in WO Patent Publication No. 06/028959. Vismodegib, chem-ically 2-Chloro-N-(4-chloro-3-pyridin-2-ylphenyl)-4-methylsulfonylbenzamide, is represented by the following structure:
formula 1
Vismodegib is an active pharmaceutical ingredient produced by Genentech (Roche) and sold under the trade name Erivedge® (which contains crystalline Vismodegib as the active ingre-dient). Erivedge® is an oral Hedgehog signaling pathway inhibitor approved for the treatment of basal-cell carcinoma (BCC).
The present invention primarily relates to multi-component crystals comprising a compound of formula 1 (cf. above) and a second compound selected from the group consisting of co-crystal formers and solvents.
The invention is further related to pharmaceutical compositions comprising said multi-component crystals. Furthermore, the invention also relates to processes for preparing said multi-component crystals. The invention also relates to several aspects of using said multi-component crystals or pharmaceutical compositions to treat a disease. Further details as well as further aspects of the present invention will be described herein below.
Vismodegib is a BCS class II compound with a high permeability but a low solubility where enhanced solubility or dissolution rates can lead to a significant advantage in respect to bio-availability.
Vismodegib is known to exist as crystalline free base. Salts of Vismodegib are men-tioned in US 7,888,364 B2 but not specified. In particular, the HCI salt is mentioned as intermediate but not characterized. Co-crystals or solvates are not reported at all.
The solubility of Vismodegib is reported to be 0.1 μg/mL at pH 7 and 0.99 mg/mL at pH 1 for Erivedge®. The absolute bio-availability after single dose is reported to be 31.8 % and the ex-posure is not linear at single doses higher than 270 mg. Erivedge® capsules do not have a food label. The estimated elimination half-life (t1/2) after continuous once-daily dosing is 4 days and 12 days after a single dose treatment (Highlights of Prescribing Information: ERIVEDGE® (vismodegib) capsule for oral use; Revised: 01/2012).
The discovery and preparation of new co-crystals or solvates offer an opportunity to improve the performance profile of a pharmaceutical product. It widens the reservoir of techniques/materials that a formulation scientist can use for designing a new dosage form of an active pharmaceutical ingredient (API) with improved characteristics. One of the most important characteristics of an API such as Vismodegib is the bio-availability which is often determined by the aqueous solubility.
A compound like Vismodegib may give rise to a variety of crystalline forms having dis-tinct crystal structures and physical characteristics like melting point, X-ray diffraction pattern, infrared spectrum, Raman spectrum and solid state NMR spectrum. One crystalline form may give rise to thermal behavior different from that of another crystalline form. Thermal behavior can be measured in the laboratory by such techniques as capillary melting point, thermogravimetry (TG), and differential scanning calorimetry (DSC) as well as content of sol-vent in the crystalline form, which have been used to distinguish polymorphic forms.
Multi-component crystals comprising Vismodegib and selected co-crystal formers or solvents may improve the dissolution kinetic profile and allow to control the hygrosco-picity of Vismodegib.
Therefore, there is a need for multi-component crystals comprising Vismodegib that avoid the above disadvantages. In particular, it is an object of the present invention to provide multi-component crystals of Vismodegib with optimized manufacture, formula-tion, stability and/or biological efficacy
.
Example 1 :
314 mg Vismodegib and 86 mg maleic acid are suspended in toluene saturated with maleic acid for 2 d, filtered and dried.
TG data shows a mass loss of about 2.3 wt % between 100 and 1 18 °C which is attributed to rest solvent. DSC data shows a single endothermal peak with an onset of about 1 15 °C (99 J/g).
H-NMR spectroscopy indicates a molar ratio of Vismodegib to maleic acid of about 1 :1 .3. However single crystal X-ray data confirms a ratio of 1 :2 (Table 1 ).

Pfizer’s Fosdagrocorat, PF-04171327 for Rheumatoid Arthritis
Fosdagrocorat, PF-04171327,
CAS 1044535-58-1
(2R,4aS,10aR)-4a-Benzyl-7-((2-methylpyridin-3-yl)carbamoyl)-2-(trifluoromethyl)-1,2,3,4,4a,9,10,10a-octahydrophenanthren-2-yl dihydrogen phosphate
2-Phenanthrenecarboxamide, 4b,5,6,7,8,8a,9,10-octahydro-N-(2-methyl-3-pyridinyl)-4b-(phenylmethyl)-7-(phosphonooxy)-7-(trifluoromethyl)-, (4bS,7R,8aR)-
(2R,4aS,10aR)-4a-benzyl-7-((2-methylpyridin-3-yl)carbamoyl)-2-(trifluoromethyl)-1,2,3,4,4a,9,10,10a-octahydrophenanthren-2-yl dihydrogen phosphate
MF C29H30F3N2O5P
Exact Mass: 574.1844
- PF 04171327
- PF-04171327
- UNII-HPI19004QS
- Selective Glucocorticoid Receptor Modulator
phase 2 .Rheumatoid Arthritis
Glucocorticoid receptor modulators
Pfizer
- 03 Sep 2015Phase II development of fosdagrocorat is ongoing
- 01 Jun 2014Pfizer completes a phase II trial in Rheumatoid arthritis in US, Bulgaria, Colombia, the Czech Republic, Germany, Hungary, India, South Korea, Malaysia, Mexico, Poland, Romania, Russia, Serbia, Slovakia, South Africa, Spain and the Ukraine (NCT01393639)
- 30 Sep 2011Phase-II clinical trials in Rheumatoid arthritis in Bulgaria, Colombia, Germany, India, Malaysia, Mexico, Poland, Romania and South Africa (PO)
Fosdagrocorat, also known as PF-04171327, a dissociated agonist of the glucocorticoid receptor (DAGR), a selective high-affinity partial agonist of the GR with potent anti-inflammatory activity at exposures that provide less undesirable effects on bone and glucose metabolism compared with prednisone (pred).
Glucocorticoid receptor modulators are glucocorticoid receptor ligands that are used to treat a variety of conditions because of their powerful anti-inflammatory, antiproliferative and immunomodulatory activity. J. Miner, et al., Expert Opin. Investig. Drugs (2005) 14(12):1527-1545.
Examples of glucocorticoid receptor modulators include dexamethasone, prednisone, prednisolone, RU-486, and as described in WO 2000/66522 and WO 2004/005229.
Treatment with glucocorticoid receptor modulators is often associated with side effects, such as bone loss and osteoporosis.
Identifying a glucocorticoid receptor modulator that is efficacious, potent, and has mitigated side-effects fulfills a medical need.
SYNTHESIS COMING…………
PATENT
WO 2008093227/US 20100286214
https://www.google.com/patents/WO2008093227A1?cl=en
SCHEME A

The 1 (/?)-Benzyl-5-bromo-9(S)-hydro-10(R)-hydroxy-10(R)-methyl-tricyclo[7.3.1.02‘7]trideca-2,4,6-trien-13-one of Formula A-8 was prepared using the protocol described in Scheme A, which is generally disclosed in WO 00/66522. Ph depicts Phenyl. Bn depicts Benzyl. Compound A-1 can be purchased (for example, VOUS and Riverside; CAS No. 4133-35-1 ). Compound A-2 can be prepared as described in Org. Syn. 1971 , 51 , 109-112.
SCHEME B


The (4βS,7R,8αR)-4β-benzyl-7-hydroxy-Λ/-(2-methylpyridin-3-yl)-7-(trifluoromethyl)-4b,5,6,7,8α,9,10-octahydrophenanthrene-2-carboxamide was prepared as described in Scheme B.
SCHEME C
 The (2R,4αS, 10αR)-4α-benzyl-7-((2-methylpyridin-3-yl)carbamoyl)-2-(trifluoromethyl)-1 ,2,3,4,4α,9,10,10α-octahydrophenanthren-2-yl dihydrogen phosphate of C-3 was prepared as described in Scheme C. Bn depicts benzyl.
The (2R,4αS, 10αR)-4α-benzyl-7-((2-methylpyridin-3-yl)carbamoyl)-2-(trifluoromethyl)-1 ,2,3,4,4α,9,10,10α-octahydrophenanthren-2-yl dihydrogen phosphate of C-3 was prepared as described in Scheme C. Bn depicts benzyl.
SCHEME D


The (2R,4αS,10αR)-4α-benzyl-7-((2-methylpyridin-3-yl)carbamoyl)-2-(trifluoromethyl)-1 ,2,3,4,4α,9,10,10α-octahydrophenanthren-2-yl dihydrogen phosphate of C-3 was prepared as described in Scheme D. Bn depicts benzyl. Ph depicts phenyl.
SCHEME E


The (2R,4αS, 10αR)-4α-benzyl-7-((2-methylpyridin-3-yl)carbamoy[)-2-(trifluoromethyl)-1 ,2,3,4,4α,9,10,10α-octahydrophenanthren-2-yl dihydrogen phosphate of C-3 was prepared as described in Scheme E. Bn depicts benzyl. Ph depicts phenyl.
Starting Material A-8 is 1(R)~Benzyl-5-bromo-9(S)-hydro-10(R)-hydroxy-10(R)-methyl-tricyclo[7.3.1.02‘7]trideca-2,4,6-trien-13-one as depicted by the following formula:

Preparation 1 : (S)-4a-benzyl-7-bromo-2-ethoxy-3,4,4a,9-tetrahydrophenanthrene

Starting Material A-8 (450 g; 1.17 moles) was dissolved in ethanol (4.5 L) at ambient temperature. 21% sodium ethoxide in ethanol (44 mL; 0.12 moles) was added and the mixture was heated to reflux for three hours. Once the Starting Material A-8 was consumed, the reaction mixture was chilled to -250C. Acetyl chloride (250 mL; 3.51 moles) was slowly added to the mixture while the temperature was maintained near -25°C. After the addition was complete, the mixture was warmed to O0C and held there until the intermediate enone was consumed. The mixture was slurry at this point. 21 % sodium ethoxide in ethanol (1.31 L; 3.51 moles) was added to the mixture while the temperature was maintained between -5°C and 50C. If the mixture was not basic, more sodium ethoxide was added. The temperature of the mixture was increased to 25°C and then diluted with water (5.9 L). The mixture was filtered and the solid was washed with water (3 X). The title compound (440 g; 85 area %) was obtained as a beige solid. 1H NMR (DMSO) δ ppm: 1.27 (t, 3H), 1.65 (dt, 1 H), 2.06 (d, 1 H), 2.21 (dd, 1 H)1 2.49 (m, 1 H), 2.65 (m, 2H), 2.89 (m, 2H), 3.85 (q, 2H), 5.45 (m, 2H), 6.44 (d, 2H), 6.98 (t, 2H), 7.06 (m, 2H), 7.25 (d, 1 H), 7.33 (dd, 1 H).
Preparation 2: (S)-4a-benzyl-7-bromo-2,2-(1,2-ethylenedioxy)-1,2,3,4,4a,9-hexahydrophenanthrene

The (S)-4α-benzyl-7-bromo-2-ethoxy-3,4,4α,9-tetrahydrophenanthrene (1270 g; 3.2 moles; 85 area %, which may be prepared as described in Preparation 1 ) was dissolved in toluene (6.45 L). The ethylene glycol (898 mL; 16.1 moles) and p-toluenesulfonic acid (6.1 g; 0.03 moles) were added and the reaction heated to reflux. Solvent (1 L) was distilled from the mixture and replaced with fresh toluene (1 L). This distillation process was repeated twice more. More p-toluenesulfonic acid (6.1 g) was added each time fresh toluene was added. During the reaction, two intermediates (detected by LC) were formed as the substrate was converted into product. The end point of the reaction was an equilibrium point between the two intermediates and the product. Once the endpoint was reached, the mixture was cooled to ambient temperature. The mixture was washed with 0.5 M NaOH (2 L). The phases separated quickly and both were dark with a small rag layer. The mixture was washed with water (2 L). The phases
separated very slowly. The mixture was dried by azeotropic distillation. Methanol (4 L) was added to the mixture and solvent (4 L) was distilled from the mixture. The methanol addition and solvent distillation were repeated twice more. Methanol was added to the mixture and precipitation occurred a few minutes later. More methanol (4 L) was added to the mixture and then brought to reflux. After 30 minutes, the mixture was cooled to 00C. The mixture was filtered and the solid was washed with chilled methanol (2 X 2L). The solid was dried in a vacuum oven at 65°C. The title compound (882 g; 98 area %) was obtained as a beige solid. 1H NMR (DMSO) δ ppm: 1.71 (m, 2H), 2.06 (m, 2H), 2.31 (dd, 1 H), 2.39 (m, 1 H), 2.68 (d, 1 H), 2.77 (m, 1 H), 2.86 (dd, 1 H), 3.36 (d, 1 H), 3.86 (m, 4H), 5.45 (m, 1 H), 6.50 (m, 2H), 7.00 (m, 4H), 7.37 (dd, 1 H), 7.44 (d, 1 H).
Preparation 3: (S)-methyl 4β-benzyl-7,7-(1,2-ethylenedioxy)-4β,5,6,7,8,10-hexahydrophenanthrene-2-carboxylate

The (S)-4α-benzyl-7-bromo-2,2-(1 ,2-ethylenedioxy)-1 ,2,3,4,4α,9-hexahydrophenanthrene (719 g; 1.75 moles, which may be prepared as described in Preparation 2) was dissolved in tetrahydrofuran (7.19 L) and chilled to -7O0C. The 1.6 M n-butyl lithium in hexane (2270 mL; 2.27 moles) was added at a rate such that the temperature was maintained below -6O0C. The mixture held an additional 15 minutes after the addition. Carbon dioxide (108 g; 2.45 moles) was added while the temperature was maintained below -60°C. The mixture held an additional 15 minutes after the addition. The mixture was warmed to ambient temperature. Solvent (7 L) was distilled from the mixture at atmospheric pressure. DMF (7 L) was added to the mixture. The mixture was cooled to ambient temperature. Methyl iodide (152 mL; 2.45 moles) was added and the mixture was held until the reaction was completed (~1 hour). The mixture was heated to 7O0C and solvent was distilled by gradually reducing the pressure to 70 mmHg. Once distillation had ceased, the mixture was cooled to room
temperature. Water (6.5 L) was slowly added to the mixture to precipitate the product. The mixture was filtered and the solid washed with water (3 X). The solid was dried on the filter. The crude product (736 g; 74 area %) was obtained as a beige solid. The product was purified by chromatography. 463 g of product was recovered from the chromatography. This material was separated from n-heptane (6130 mL). 394 g of the title compound was recovered. Another 70 g of title compound was recovered from the mother liquor by chromatography. 1H NMR (DMSO) δ ppm: 1.74 (m, 2H), 2.10 (m, 2H)1 2.33 (dd, 1 H), 2.45 (m, 1 H), 2.72 (d, 1 H), 2.79 (m, 1 H), 2.94 (dd, 1 H), 3.40 (d, 1 H), 3.87 (m, 7H), 5.49 (m, 1 H), 6.47 (m, 2H), 6.93 (m, 2H), 7.01 (m, 1 H), 7.42 (d, 1 H), 7.64 (d, 1 H), 7.79 (dd, 1 H).
Preparation 4: (4βS,8α/?)-methyl 4β-benzyl-7,7-(1,2-ethylenedioxy)-4β,5,6,7,8,8α,9,10-octahydrophenanthrene-2-carboxylate

The (S)-methyl 4β-benzyl-7,7-(1 ,2-ethylenedioxy)-4β,5,6,7,8,10-hexahydrophenanthrene-2-carboxylate (201 g; 0.515 moles, which may be prepared as described in Preparation 3) and 50 ml of ethylene glycol was dissolved in toluene (2.0 L) in an autoclave. To this was added 10 grams of a 5% Pd/C (dry catalyst). The autoclave was then sealed and purged with nitrogen (three cycles) followed by hydrogen (three cycles). The reaction was run for 18 hours with a pressure of 80 psig and temperature of 50 0C. HPLC analysis for completion and selectivity (typical selectivity’s are: 95 to 5, Trans to Cis). The suspension was filtered through Celite® to remove the catalyst and the toluene solution is concentrated at 50 0C, under vacuum, to
approximately 200 ml. While still at 50 0C, 1 L of 1-butanol was added and the solution heated to 60 0C, until clear. Upon cooling, the resulting solid title compound was isolated by vacuum filtration (196 grams; 97%; Trans to Cis 95.75 to 4.24). 1H NMR (300 MHz, CDCI3) δ ppm: 7.79 (bs, 1 H1 Ar-H), 7.47 (d, J= 9 Hz, 1 H, Ar-H), 7.13-7.05 (cm, 3H, Ar-H), 6.56-6.53 (cm, 2H, Ar-H), 6.43 (d, J= 9 Hz, 1 H, Ar-H), 4.04-3.93 (cm, 4H, 2-CH2), 3.89 (s, 3H, CH3),3.08-3.03 (cm, 3H, CH2, CH-H), 2.63 (d, J= 15 Hz, CH-H), 2.22-1.72 (cm, 8H, 4-CH2), 1.57 (cm, 1 H, CH-H).; 13CNMR (CDCI3, δ): 167.7, 149.2, 137.7, 136.4, 131.1 , 130.5, 127.8, 127.7, 127.4, 126.3, 125.5, 108.9, 64.6, 64.5, 52.1 , 40.5, 39.8, 38.3, 35.8, 31.6, 30.3, 27.9, 24.6.
Preparation 5: (4βS,8α/?)-methyl 4β-benzyl-7-oxo-4β,5,6,7,8,8α,9,10-octahydrophenanthrene-2-carboxylate

ThΘ (4βS,8αR)-mΘthyl 4β-benzyl-7,7-(1 ,2-ethylenΘdioxy)-4β,5,6,7,8,8α,9,10-octahydrophenanthrene-2-carboxylate (150 g, 382 mmol, which may be prepared as described in Preparation 4) was dissolved in dichloromethane (630 ml). Water (270 ml) was added with stirring followed by trifluoroacetic acid (73 ml. 1150 mmol) via drop funnel over 30 minutes, maintaining the internal temperature below 3O0C. After the addition was complete, the reaction was heated at 4O0C for 2 hours. In process check indicated incomplete reaction with around 9% (area percent) starting material. The layers were separated and fresh water (270 ml) and trifluoroacetic acid (31 ml) was added. The reaction mixture was heated at 4O0C for 1 hour. This process was continued until the starting material was consumed. The organic phase was washed with 5% aqueous sodium bicarbonate (300 ml), water (300 ml) and dried over MgSO4 and concentrated to dryness to give 126.4 g of the title compound (representing a 95% yield). 1H NMR (DMSO) δ ppm: 7.70 (s, 1 H), 7.37 (d, J=8.4 Hz, 1 H), 7.11 (m, 3H), 6.6 (d, J= 5.70 Hz, 2H), 6.45 (d, J=8.4 Hz, 1H), 3.80 (s, 3H), 3.80 (m, 2H), 3.04-1.48 (m, 11 H).
Preparation 6: (4βS,7f?,8α/?)-methyl 4β-benzyl-7-hydroxy-7-(trifluoromethyl)-4β,5J6,7,8,8α,9,10-octahydrophenanthrene-2-carboxylate

The (4βS,8αf?)-methyl 4β-benzyl-7-oxo-4β,5,6,7,8I8α,9,10-octahydrophenanthrene-2-carboxylate (118g, 0.339 mole, which may be prepared as described in Preparation 5) dissolved in dichloromethane was chilled to -5O0C. The solution became turbid. 1.0 M Tetrabutylammonium fluoride a solution in THF (3.4 ml, 0.003 mol) was added with no appreciable temperature change. Trifluorotrimethylsilane (79 ml, 0.51 mol) was added over 20 minutes with a color change to bright orange to light red in color. The reaction mixture was held at -50 0C for about 2 hours and then allowed to warm to 0 0C.
Tetrabutylammonium fluoride (340 ml, 0.34 moles) was added very slowly at 0 0C, to the reaction mixture over 45 minutes. An exotherm was observed with gas evolution. The reaction mixture was stirred 10 minutes and HPLC analysis indicated complete desilylialation. Water (1 L) was added to the reaction mixture and with vigorous stirring and allowed to warm to room temperature. The organic layer was washed with water (1 L). The organic layer was concentrated and chromatographed to produce 72 g, 51 % of the title compound, with an additional 32 g of impure product. 1H NMR (DMSO) δ ppm: 7.70 (s, 1 H), 7.37 (d, J=8.1 Hz, 1 H)1 7.09 (m, 3H), 6.5 (dd, J=1.2, 6.6 Hz, 2H), 6.38 (d, J=8.4 Hz, 1 H), 3.80 (s, 3H), 3.80 (m, 2H), 3.09-1.21 (m, 13H).
Preparation 7: (4βS,7/?,8α/?)-methyl 4β-benzyl-7-(bis(benzyloxy)phosphoryloxy)-7-(trifluoromethyl)-4β,5,6,7,8,8α,9,10-octahydrophenanthrene-2-carboxylate

The (4βS,7R,8αf?)-methyl 4β-benzyl-7-hydroxy-7-(trifluoromethyl)-4β)5,6,7)8,8α,9,10-octahydrophenanthrene-2-carboxylate (5.0 g; 11.9 mmol, which may be prepared as in Preparation 6) and 5-methyltetrazole (3.6 g; 43.0 mmol) were mixed together in dichloromethane (50 mL) at ambient temperature. Dibenzylphosphoramidite (8.3 mL; 25.1 mmol) was added and the mixture was stirred until the reaction was completed (1 hour). The mixture was chilled to 00C and 30% hydrogen peroxide (10 mL) was added. The reaction was stirred until the oxidation was completed (30 minutes). The aqueous phase was separated from the organic phase. The organic phase was washed with 10% sodium meta-bisulfite (50 ml_). The organic phase was dried with anhydrous magnesium sulfate and concentrated. The crude product was purified by silica gel chromatography with 15% ethyl acetate in hexanes. The purified title compound (8.41 g; 94% yield) was obtained as a colorless oil that contained 6% ethyl acetate by weight. 1H NMR (DMSO): δ 1.31 (t, 1 H), 1.63-1.92 (m, 3H), 2.05-2.35 (m, 3H), 2.63 (d, 1 H), 2.75-3.16 (m, 4H), 3.80 (s, 3H), 5.13 (m, 4H), 6.43 (d, 1 H), 6.49 (m, 2H), 7.04-7.17 (m, 3H), 7.33-7.42 (m, 12H), 7.71 (d, 1 H).
Preparation 8: dibenzyl (2f?,4αS,10αR)-4α-benzyl-7-((2-methylpyridin-3-o yl)carbamoyl)-2-(trifluoromethyl)-1 ,2,3,4,4α,9,10,10α-octahydrophenanthren-2-yI phosphate

The (4βS,7R,8αf?)-methyl 4β-benzyl-7-(bis(benzyloxy)phosphoryloxy)-7- (trifluoromethyl)-4β,5,6,7,8,8α,9,10-octahydrophenanthrene-2-carboxylate (7.9 g; 11.6 5 mmol, which may be prepared as in Preparation 7) and 3-amino-2-picoline (1.3 g; 12.2 mmol) were mixed together in tetrahydrofuran (80 ml_) and chilled to 0°C. The 1 M solution of lithium bis(trimethylsilyl)amide in tetrahydrofuran (24 ml_; 24.4 mmol) was added while maintaining the temperature below 100C. The mixture was stirred for 30 minutes. Water (50 mL) was added to the reaction mixture. The mixture was extracted with ethyl acetate. The organic extract was washed with water. The organic phase was dried with anhydrous magnesium sulfate and concentrated. The crude product was purified by silica gel chromatography with 70% ethyl acetate in hexanes. The purified title compound (6.79 g; 68% yield) was obtained as a yellow gum that contained 6% ethyl acetate by weight. 1H NMR (DMSO): δ 1.33 (t, 1 H), 1.66-1.93 (m, 3H), 2.08-2.34 (m, 3H), 2.41 (s, 3H), 2.68 (d, 1 H), 2.76-3.19 (m, 4H), 5.14 (m, 4H), 6.47 (d, 1 H), 6.56 (m, 2H), 7.07-7.19 (m, 3H), 7.20-7.53 (m, 12H), 7.71 (d, 1 H), 7.76 (s, 1 H), 8.32 (d, 1 H), 9.93 (s, 1 H).
Example 1 : (4βS,7/?,8αR)-4β-benzyl-7-hydroxy-W-(2-methylpyridin-3-yl)-7-(trifluoromethyl)-4β,5,6,7,8,8α,9,10-octahydrophenanthrene-2-carboxamide

The (4βS,7ft,8αR)-methyl 4β-benzyl-7-hydroxy-7-(trifluoromethyl)-4β,5,6,7,8,8α,9,10-octahydrophenanthrene-2-carboxylate (10 g; 23.9 mmol, which may be prepared as described in Preparation 6), and 3-amino-2-picoline (2.71 g; 25.1 mmol) were dissolved in toluene (200 ml_). The 1 M lithium bis(trimethylsilyl)amide in tetrahydrofuran (74.1 mL; 74.1 mmol) was added at a rate such that the temperature was maintained below 350C. There was a mild exotherm and a solid precipitated during the addition. The mixture was held an additional 30 minutes after the addition. Water (250 mL) was added to the mixture. There was a mild exotherm and the solid dissolved. Ethyl acetate (50 mL) was added to the mixture to ensure the product did not precipitate. Stirring was stopped to allow the phases to separate. The aqueous phase was removed. The organic phase was washed with water (250 mL). Solvent (230 mL) was distilled at atmospheric pressure from the organic phase. The mixture was cooled to ambient temperature. The mixture was filtered and the solid was washed with toluene (2 times) followed by heptane (2 times). The solid was dried in a vacuum oven at 700C. The title compound of the present example (10 g) was obtained as a beige solid. 1H NMR (DMSO) δ ppm: 1.32 (m, 1 H), 1.82 (m, 4H), 2.10 (m, 4H), 2.41 (s, 3H), 2.68 (d, 1 H), 3.08 (m, 3H), 6.00 (s, 1H), 6.43 (d, 1 H), 6.59 (m, 2H), 7.12 (m, 3H), 7.25 (dd, 1H), 7.44 (dd, 1H), 7.71 (dd, 1 H), 7.75 (d, 1 H), 8.31 (dd, 1 H), 9.91 (s, 1 H).
Example 2: (2f?,4αS,10αR)-4α-benzyl-7-((2-methylpyridin-3-yl)carbamoyl)-2-(trifluoromethyl)-i ,2,3,4,4α,9,10,1 Oα-octahydrophenanthren-2-yl dihydrogen phosphate

The dibenzyl (2R,4αS, 10αR)-4α-bθnzyl-7-((2-methylpyridin-3-yl)carbamoyl)-2-(trifluoromethyl)-1 ,2,3,4,4a,9,10,10a-octahydrophenanthren-2-yl phosphate (6 g; 7.9 mmol, which may be prepared as described in Preparation 8) was dissolved in methanol (120 ml_). 5% palladium on carbon (63% water) (1.3 g; 0.4 mmol) was added to the mixture. The mixture was treated with hydrogen (50 psi) at room temperature. The reaction stalled with 12% of the monobenzylic intermediate remaining. The mixture was filtered through a pad of Celite®. Fresh catalyst (1.3 g) was added to the solution and resubmitted to the hydrogenation conditions. Once the reaction was completed, the mixture was filtered through a pad of Celite®. The solution was concentrated to about 60 ml_ by distillation and not by using a rotary evaporator. During the distillation a white solid precipitated. The mixture was cooled to ambient temperature. The mixture was filtered and the solid washed with methanol. The solid was dried in a vacuum oven at 700C. The compound of the present example (3.36 g; 75% yield) was obtained as a white solid and had an LC purity of 98 area %. 1H NMR (DMSO): δ 1.33 (t, 1 H)1 1.69-1.98 (m, 3H), 2.07-2.29 (m, 3H)1 2.42 (s, 3H), 2.61-2.80 (m, 2H)1 2.93-3.19 (m, 3H)1 3.30 (d, 1 H), 6.50 (d, 1 H), 6.64 (m, 2H), 7.08-7.20 (m, 3H), 7.29 (dd, 1 H), 7.48 (dd, 1 H), 7.75 (dd, 2H), 8.33 (dd, 1 H), 9.96 (s, 1 H).
PATENT
WO 2008093236
http://www.google.co.in/patents/WO2008093236A1?cl=en
Example 1 : (4βS,7/?,8α/?)-4β-benzyl-7-hydroxy-N-(2-methylpyridin-3-yl)-7- (trifluoromethyl)-4β,5,6,7,8,8α,9,10-octahydrophenanthrene-2-carboxamide

The (4βS,7R,8α/?)-methyl 4β-benzyl-7-hydroxy-7-(trifluoromethyl)-4β,5,6J7,8,δα,9, 10- octahydrophenanthrene-2-carboxylate (10 g; 23.9 mmol, which may be prepared as described in Preparation 6), and 3-amino-2-picoline (2.71 g; 25.1 mmol) were dissolved in toluene (200 ml_). The 1 M lithium bis(trimethylsilyl)amide in tetrahydrofuran (74.1 ml_; 74.1 mmol) was added at a rate such that the temperature was maintained below 350C. There was a mild exotherm and a solid precipitated during the addition. The mixture was held an additional 30 minutes after the addition. Water (250 ml_) was added to the mixture. There was a mild exotherm and the solid dissolved. Ethyl acetate (50 ml_) was added to the mixture to ensure the product did not precipitate. Stirring was stopped to allow the phases to separate. The aqueous phase was removed. The organic phase was washed with water (250 ml_). Solvent (230 ml_) was distilled at atmospheric pressure from the organic phase. The mixture was cooled to ambient temperature. The mixture was filtered and the solid was washed with toluene (2 times) followed by heptane (2 times). The solid was dried in a vacuum oven at 700C. The title compound of the present example (10 g) was obtained as a beige solid. 1H NMR (DMSO) δ ppm: 1.32 (m, 1H), 1.82 (m, 4H), 2.10 (m, 4H), 2.41 (s, 3H), 2.68 (d, 1 H), 3.08 (m, 3H), 6.00 (s, 1 H), 6.43 (d, 1 H), 6.59 (m, 2H), 7.12 (m, 3H), 7.25 (dd, 1 H), 7.44 (dd, 1 H), 7.71 (dd, 1 H), 7.75 (d, 1 H), 8.31 (dd, 1 H), 9.91 (s, 1 H).
Example 2: (2f?,4αS,10α/?)-4α-benzyl-7-((2-methylpyridin-3-yl)carbamoyl)-2- (trifluoromethyl)-1,2,3,4,4α,9,10,10α-octahydrophenanthren-2-yl dihydrogen phosphate

The dibenzyl (2R,4αS,10αR)-4α-benzyl-7-((2-methylpyridin-3-yl)carbamoyl)-2- (trifluoromethyl)-1 ,2,3,4,4a,9,10,10a-octahydrophenanthren-2-yl phosphate (6 g; 7.9 mmol, which may be prepared as described in Preparation 8) was dissolved in methanol (120 ml_). 5% palladium on carbon (63% water) (1.3 g; 0.4 mmol) was added to the mixture. The mixture was treated with hydrogen (50 psi) at room temperature. The reaction stalled with 12% of the monobenzylic intermediate remaining. The mixture was filtered through a pad of Celite®. Fresh catalyst (1.3 g) was added to the solution and resubmitted to the hydrogenation conditions. Once the reaction was completed, the mixture was filtered through a pad of Celite®. The solution was concentrated to about 60 ml_ by distillation and not by using a rotary evaporator. During the distillation a white solid precipitated. The mixture was cooled to ambient temperature. The mixture was filtered and the solid washed with methanol. The solid was dried in a vacuum oven at 7O0C. The compound of the present example (3.36 g; 75% yield) was obtained as a white solid and had an LC purity of 98 area %. 1H NMR (DMSO): δ 1 .33 (t, 1 H), 1 .69- 1.98 (m, 3H), 2.07-2.29 (m, 3H), 2.42 (s, 3H), 2.61 -2.80 (m, 2H), 2.93-3.19 (m, 3H), 3.30 (d, 1 H), 6.50 (d, 1 H), 6.64 (m, 2H), 7.08-7.20 (m, 3H), 7.29 (dd, 1 H), 7.48 (dd, 1 H), 7.75 (dd, 2H), 8.33 (dd, 1 H), 9.96 (s, 1 H).
REFERENCES
https://www.pfizer.com/sites/default/files/product-pipeline/July%2028%202015%20Pipeline%20Update.pdf
https://clinicaltrials.gov/ct2/show/NCT00938587
////////
Cc1c(cccn1)NC(=O)c2ccc3c(c2)CC[C@H]4[C@]3(CC[C@@](C4)(C(F)(F)F)OP(=O)(O)O)Cc5ccccc5
O=P(O)(O[C@@]1(C(F)(F)F)C[C@@]2([H])CCC3=C(C=CC(C(NC4=CC=CN=C4C)=O)=C3)[C@]2(CC5=CC=CC=C5)CC1)O
