
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Phase 3-AMG 145 for hyperlipidaemia and mixed dyslipidaemia
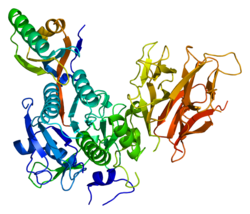
AMG 145
Amgen Limited.
AMG 145 is a fully-human monoclonal antibody which targets proprotein convertase subtilisin/kexin type 9 (PCSK9).
It is intended for use in the reduction of elevated total cholesterol (TC), low-density lipoprotein cholesterol (LDL-C), apolipoprotein B (apo-B), non-high density lipoprotein cholesterol (non-HDL-C) and lipoprotein A (Lp(a)) in patients with primary hyperlipidaemia and mixed dyslipidaemia.
It is also intended for use as an adjunct to other lipid lowering therapies in patients with primary Ho-FH.
Dyslipidaemias include a wide range of lipid abnormalities and disturbances in lipid metabolism that lead to changes in plasma lipoprotein function and/or levels. Along with other cardiovascular risk factors, this may lead to the development of atherosclerosis. TC and LDL-C levels constitute the primary targets of therapy as evidence showing that reducing TC and LDL-C can prevent cardiovascular disease (CVD) is strong and compelling(1). However, other dyslipidaemias also predispose to premature CVD. The atherogenic lipid triad consisting of increased very low density lipoprotein (VLDL) remnants manifested as mildly elevated triglycerides (TG), increased small dense low-density lipoprotein (LDL) particles, and reduced high-density lipoprotein-cholesterol (HDL-C) levels is a common pattern found in premature CVD.
Hypercholesterolaemia is defined as the presence of high concentrations of cholesterol in the blood(2). Blood cholesterol has a log-linear relationship to the risk of CVD and is a key modifiable risk factor. In high-income countries, blood cholesterol levels >3.8mmol/L(b) are estimated to be responsible for more than 50% of CVD associated events(3). Primary hypercholesterolaemia is associated with an underlying genetic cause. This may be a specific genetic defect, as in familial hypercholesterolaemia (FH), or the interaction of multiple genes with dietary and other risk factors (non-familial hypercholesterolaemia).
FH is often transmitted as a codominant trait, with two principle forms described: homozygous-FH (Ho-FH) and heterozygous-FH (He-FH) in which either both or one of the pair of LDL-C receptor genes is defective or mutated with reduced activity. FH results in markedly elevated LDL-C levels, with other forms of cholesterol remaining normal. He-FH is often clinically silent and may be diagnosed at any age following a complete lipid analysis. Untreated, He-FH typically leads to symptomatic CVD by the fourth or fifth decade of life(2,4). The more severe homozygous form may be manifest from an early age, and is characterised by extravascular cholesterol deposits, cutaneous or tendon xanthomas, LDL-C levels >3.3 g/L(b) and arteriopathy.
Paragazole Excels as Breast Cancer Treatment
PFIZER’S Palbociclib Granted Breakthrough Label by the Food and Drug Administration
CHEMICAL NAMES
1. Pyrido[2,3-d]pyrimidin-7(8H)-one, 6-acetyl-8-cyclopentyl-5-methyl-2-[[5-(1-
piperazinyl)-2-pyridinyl]amino]-
2. 6-acetyl-8-cyclopentyl-5-methyl-2-{[5-(piperazin-1-yl)pyridin-2-
yl]amino}pyrido[2,3-d]pyrimidin-7(8H)-one
MOLECULAR FORMULA C24H29N7O2
MOLECULAR WEIGHT 447.5
TRADEMARK None as yet
SPONSOR Pfizer Inc.
CODE DESIGNATION PD-0332991
CAS#: 571190-30-2 (PD0332991); 827022-32-2 (PD0332991 HCl salt)
http://www.ama-assn.org/resources/doc/usan/palbociclib.pdf FOR STRUCTURE AND DETAILS
Palbociclib, also known as PD0332991, is an orally available pyridopyrimidine-derived cyclin-dependent kinase (CDK) inhibitor with potential antineoplastic activity. PD-0332991 selectively inhibits cyclin-dependent kinases (particularly Cdk4/cyclin D1 kinase), which may inhibit retinoblastoma (Rb) protein phosphorylation; inhibition of Rb phosphorylation prevents Rb-positive tumor cells from entering the S phase of the cell cycle (arrest in the G1 phase), resulting in suppression of DNA replication and decreased tumor cell proliferation. PD 0332991 is a highly specific inhibitor of cyclin-dependent kinase 4 (Cdk4) (IC50 = 0.011 μmol/L) and Cdk6 (IC50 = 0.016 μmol/L), having no activity against a panel of 36 additional protein kinases. Check for active clinical trials or closed clinical trials using this agent. (NCI Thesaurus)
Date: April 10, 2013
Pfizer Inc. said that its experimental pill for advanced, often deadly breast cancer has been designated as a breakthrough therapy by the Food and Drug Administration.
The breakthrough designation, created under legislation enacted last summer to fund and improve operations of the FDA, is meant to speed up development and review of experimental treatments that are seen as big advances over existing therapies for serious diseases. Pfizer is working with the agency to determine exactly what research results it will need to apply for approval of the drug.
Palbociclib is being evaluated as an initial treatment for the biggest subgroup of postmenopausal women whose breast cancer is locally advanced or has spread elsewhere in the body. About 60% of women with such advanced breast cancer have tumors classified as ER+, or estrogen-receptor positive, but HER2-, or lacking an excess of the growth-promoting protein HER2.
Estrogen-receptor positive tumors have proteins inside and on the surface of their cells to which the estrogen hormone can attach and then fuel growth of cells. These tumors tend to grow slowly and can be fought with drugs that block estrogen’s effects.
Meanwhile, about 80% of breast cancer tumor cells are HER2 negative. That means that unlike HER2 positive tumors, they don’t produce too much of the HER2 protein, which makes tumors grow and spread more aggressively than in other breast cancer types.
New York-based Pfizer is currently running a late-stage study of palbociclib at multiple centers, comparing its effects when used in combination with letrozole with the effects of letrozole alone.
Letrozole, sold under the brand name Femara for about the past 15 years, is a pill that works by inhibiting aromatase. That’s an enzyme in the adrenal glands that makes estrogen.
According to Pfizer, palbociclib targets enzymes called cyclin dependent kinases 4 and 6. By inhibiting those enzymes, the drug has been shown in laboratory studies to block cell growth and suppress copying of the DNA of the cancer cells.
Pfizer, which has made research on cancer medicines a priority in recent years, also is testing palbociclib as a treatment for other cancers.
| Highlight of recent study using PD-0332991 |
Phase I study of PD-0332991: Forty-one patients were enrolled. DLTs were observed in five patients (12%) overall; at the 75, 125, and 150 mg once daily dose levels. The MTD and recommended phase II dose of PD 0332991 was 125 mg once daily. Neutropenia was the only dose-limiting effect. After cycle 1, grade 3 neutropenia, anemia, and leukopenia occurred in five (12%), three (7%), and one (2%) patient(s), respectively. The most common non-hematologic adverse events included fatigue, nausea, and diarrhea. Thirty-seven patients were evaluable for tumor response; 10 (27%) had stable disease for ≥4 cycles of whom six derived prolonged benefit (≥10 cycles). PD 0332991 was slowly absorbed (median T(max), 5.5 hours), and slowly eliminated (mean half-life was 25.9 hours) with a large volume of distribution (mean, 2,793 L). The area under the concentration-time curve increased linearly with dose. Using an E(max) model, neutropenia was shown to be proportional to exposure. CONCLUSIONS:
PD 0332991 warrants phase II testing at 125 mg once daily, at which dose neutropenia was the sole significant toxicity. (Source: Clin Cancer Res; 18(2); 568-76.)
Phase I study of PD-0332991 in 3-week cycles (Schedule 2/1): Six patients had DLTs (18%; four receiving 200 mg QD; two receiving 225 mg QD); the MTD was 200 mg QD. Treatment-related, non-haematological adverse events occurred in 29 patients (88%) during cycle 1 and 27 patients (82%) thereafter. Adverse events were generally mild-moderate. Of 31 evaluable patients, one with testicular cancer achieved a partial response; nine had stable disease (≥10 cycles in three cases). PD 0332991 was slowly absorbed (mean T(max) 4.2 h) and eliminated (mean half-life 26.7 h). Volume of distribution was large (mean 3241 l) with dose-proportional exposure. Using a maximum effective concentration model, neutropenia was proportional to exposure. CONCLUSION: PD 0332991 was generally well tolerated, with DLTs related mainly to myelosuppression. The MTD, 200 mg QD, is recommended for phase II study. (source: Br J Cancer. 2011 Jun 7;104(12):1862-8)
| References |
1: Flaherty KT, Lorusso PM, Demichele A, Abramson VG, Courtney R, Randolph SS, Shaik MN, Wilner KD, O’Dwyer PJ, Schwartz GK. Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer. Clin Cancer Res. 2012 Jan 15;18(2):568-76. doi: 10.1158/1078-0432.CCR-11-0509. Epub 2011 Nov 16. PubMed PMID: 22090362.
2: Smith D, Tella M, Rahavendran SV, Shen Z. Quantitative analysis of PD 0332991 in mouse plasma using automated micro-sample processing and microbore liquid chromatography coupled with tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2011 Oct 1;879(27):2860-5. doi: 10.1016/j.jchromb.2011.08.009. Epub 2011 Aug 16. PubMed PMID: 21889427.
3: Katsumi Y, Iehara T, Miyachi M, Yagyu S, Tsubai-Shimizu S, Kikuchi K, Tamura S, Kuwahara Y, Tsuchiya K, Kuroda H, Sugimoto T, Houghton PJ, Hosoi H. Sensitivity of malignant rhabdoid tumor cell lines to PD 0332991 is inversely correlated with p16 expression. Biochem Biophys Res Commun. 2011 Sep 16;413(1):62-8. doi: 10.1016/j.bbrc.2011.08.047. Epub 2011 Aug 17. PubMed PMID: 21871868; PubMed Central PMCID: PMC3214763.
4: Schwartz GK, LoRusso PM, Dickson MA, Randolph SS, Shaik MN, Wilner KD, Courtney R, O’Dwyer PJ. Phase I study of PD 0332991, a cyclin-dependent kinase inhibitor, administered in 3-week cycles (Schedule 2/1). Br J Cancer. 2011 Jun 7;104(12):1862-8. doi: 10.1038/bjc.2011.177. Epub 2011 May 24. PubMed PMID: 21610706; PubMed Central PMCID: PMC3111206.
5: Nguyen L, Zhong WZ, Painter CL, Zhang C, Rahavendran SV, Shen Z. Quantitative analysis of PD 0332991 in xenograft mouse tumor tissue by a 96-well supported liquid extraction format and liquid chromatography/mass spectrometry. J Pharm Biomed Anal. 2010 Nov 2;53(3):228-34. doi: 10.1016/j.jpba.2010.02.031. Epub 2010 Feb 26. PubMed PMID: 20236782.
6: Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, Ginther C, Atefi M, Chen I, Fowst C, Los G, Slamon DJ. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11(5):R77. doi: 10.1186/bcr2419. PubMed PMID: 19874578; PubMed Central PMCID: PMC2790859.
7: Menu E, Garcia J, Huang X, Di Liberto M, Toogood PL, Chen I, Vanderkerken K, Chen-Kiang S. A novel therapeutic combination using PD 0332991 and bortezomib: study in the 5T33MM myeloma model. Cancer Res. 2008 Jul 15;68(14):5519-23. doi: 10.1158/0008-5472.CAN-07-6404. PubMed PMID: 18632601.
8: Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, Albassam M, Zheng X, Leopold WR, Pryer NK, Toogood PL. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004 Nov;3(11):1427-38. PubMed PMID: 15542782.
AYURVEDA–Manjishta, Rubia cordifolia

| Rubia cordifolia | |
|---|---|
 |
|
| Rubia cordifolia | |
| Scientific classification | |
| Kingdom: | Plantae |
| (unranked): | Angiosperms |
| (unranked): | Eudicots |
| (unranked): | Asterids |
| Order: | Gentianales |
| Family: | Rubiaceae |
| Tribe: | Rubieae |
| Genus: | Rubia |
| Species: | R. cordifolia |
| Binomial name | |
| Rubia cordifolia L. |
|
Rubia cordifolia, often known as Common Madder or Indian Madder, is a species of flowering plant in the coffee family, Rubiaceae. It has been cultivated for a red pigment derived from roots.
Common names of this plant include Manjistha in Sanskrit, Marathi, Kannada and Bengali, Majith in Hindi and Gujarati, Tamaralli in Telugu, Manditti in Tamil.
Description
It can grow to 1.5 m in height. The evergreen leaves are 5–10 cm long and 2–3 cm broad, produced in whorls of 4-7 starlike around the central stem. It climbs with tiny hooks at the leaves and stems. The flowers are small (3–5 mm across), with five pale yellow petals, in dense racemes, and appear from June to August, followed by small (4–6 mm diameter) red to black berries. The roots can be over 1 m long, up to 12 mm thick. It prefers loamy soils with a constant level of moisture. Madders are used as food plants for the larvae of some Lepidoptera species including Hummingbird hawk moth.
Uses
Rubia cordifolia was an economically important source of a red pigment in many regions of Asia, Europe and Africa. It was extensively cultivated from antiquity until the mid nineteenth century. The plant’s roots contain an organic compound called Alizarin, that gives its red colour to a textile dye known as Rose madder. It was also used as a colourant, especially for paint, that is referred to as Madder lake. The substance was also derived other species; Rubia tinctorum, also widely cultivated, and the Asiatic species Rubia argyi (H. Léveillé & Vaniot) H. Hara ex Lauener (synonym = Rubia akane Nakai,[1] based on the Japanese Aka (アカ or あか) = red, and ne (ネ or ね) = root). The invention of a synthesized duplicate, an anthracene compound called alizarin, greatly reduced demand for the natural derivative.[2]
The roots of Rubia cordifolia are also the source of a medicine used in Ayurveda, this is commonly known in Ayurvedic Sanskrit as Manjistha (or Manjista or Manjishta) and the commercial product in Hindi as Manjith.[3]
It is known as btsod (Tibetan: བཙོད་) in Traditional Tibetan Medicine where it is used to treat blood disorders; spread heat (Tibetan: འགྲམས་ཚད་), excess heat in the lungs, kidneys, and intestines; reduce swelling; and is a component of the three reds (Tibetan: དམར་གསུམ་), a subcompound included in many Tibetan preparations in order to remove excess heat in the blood.[4]
In Traditional Chinese Medicine it is known as qiàn cǎo gēn (茜草根).
The following properties were described in various cellular and animal models:
- Iwatsuki, K., T. Yamazaki, D. E. Boufford and H. Ohba. 1993. Flora of Japan IIIa: 232.
- “Material Name: madder”. material record. Museum of Fine Arts, Boston. November 2007. Retrieved 2009-01-01.
- R. Daman, S. Bhandari, B. Singh and Brij Lal; S. Pathania (2006). “Comparative Studies of Rubia cordifolia L. and its Commercial Samples”. Ethnobotanical Leaflets (11): 179–188.
- Gyatso, Thinley; Hakim, Chris (2010). Essentials of Tibetan traditional medicine. Berkeley, Calif.: North Atlantic Books. p. 167. ISBN 978-1-55643-867-7.
- Joshan Rani S., Nagarauk R., Anuradha P. “Antibacterial properties of extracts of Indian medicinal plants: Syzygium alternifolium, phyllanthus niruri and rubia cordifolia” Biomedical and Pharmacology Journal 2010 3:1 (123-128)
- Divakar K., Pawar A.T., Chandrasekhar S.B., Dighe S.B., Divakar G.”Protective effect of the hydro-alcoholic extract of Rubia cordifolia roots against ethylene glycol induced urolithiasis in rats” Food and Chemical Toxicology 2010 48:4 (1013-1018)
- Caldecott, Todd (2006). Ayurveda: The Divine Science of Life. Elsevier/Mosby. ISBN 0-7234-3410-7. Contains a detailed monograph on Rubia cordifolia (Manjishta) as well as a discussion of health benefits and usage in clinical practice. Available online at http://www.toddcaldecott.com/index.php/herbs/learning-herbs/306-manjishta
Manjishta, considered to be the best all around herbal blood purifier according to Ayurvedic Text, Manjishta Rasayana cools and helps detoxify the blood. Manjishta relieves pain caused by inflammation such as a tooth ache, helps stop bleeding, and helps remove obstructions in the blood stream, liver, and kidneys. It can assist all inflammatory conditions of the blood and female reproductive system. It can help dissolve abnormal growths in the tissues. Manjishta can improve blood flow and promote healing of broken bones and tissue damaged by injury or infection. It may help with sexually transmitted diseases.
Ayurveda is a holistic healing science which comprises of two words, Ayu and Veda. Ayu means life and Veda means knowledge or science. So the literal meaning of the word Ayurveda is the science of life. Ayurveda is a science dealing not only with treatment of some diseases but is a complete way of life. Ayurveda is the ancient Indian medical science, the origin of which can be traced back to the Vedas, which are the oldest available classics of the world. Vedas are the ancient books of knowledge, or science, from India.
They contain practical and scientific information on various subjects beneficial to the humanity like health, philosophy, engineering, astrology etc. Ayurveda combines physical, psychological and spiritual therapies in an approach to health, that has addressed itself to the fundamental principles of good health and longevity. It has developed a tradition of medicine and a system of treatment based on the inherent ability of the human body to rejuvenate, to heal and to restore its natural balance.
Ayurveda is based on a system of Tridosha or Three Humours which classifies all individual constitutions of people, diseases, herbs and other non-herbal remedies and therapies according to whether they are Vata (air or nerve oriented), Kapha (water or mucoid type) or Pitta (fire type) [5] .
Herbs that have pungent, sour and salty flavors stimulate fire; herbs that are astringent (drying) and bitter stimulate vata-air, or the nerve centered humour; herbs that are sweet, salty and sour stimulate or increase Kapha-water, or the mucoid humour.
In contrast, herbs that are sweet, sour and salty flavored ameliorate Vata-air, which means that they have a particular affinity for the nervous system. Herbs that are astringent, sweet and bitter ameliorate Pitta-fire, meaning that they are soothing and anti-inflammatory.
Finally herbs that are pungent, bitter and astringent ameliorate Kapha-water, which means they tend to increase digestive fire, expel and dry excessive fluid build up in the system, including clearing excessive fat from the body, and the accumulation of cholesterol and other fatty deposits in the veins and arteries of the body.
This Indian system of medicine has laid down principles and methods of treatment for various diseases including chronic illnesses where there is no definite curative treatment, and symptomatic relief is the only existing treatment option.
Origin
Ayurvedic medicine originated in the early civilizations of India some 3,000-5,000 years ago. It is mentioned in the Vedas, the ancient religious and philosophical texts that are the oldest surviving literature in the world, which makes Ayurvedic medicine the oldest surviving healing system.
According to the texts, Ayurveda was conceived by enlightened wise men as a system of living harmoniously and maintaining the body so that mental and spiritual awareness could be possible. Medical historians believe that Ayurvedic ideas were transported from ancient India to China and were instrumental in the development of Chinese medicine.
Ayurveda is a science based on ancient Indian philosophy. The Vedas encompass the whole knowledge of the Universe. There are four Vedas, namely, Rigveda, Yajurveda, Samaveda and Atharvaveda. Amongst these, the Atharvaveda mainly deals with different facets of health.
The main body of Ayurveda is found in the fourth Veda – the Artharvaveda. Ayurveda is an offspring of the Atharvaveda and is also considered as the fifth Veda. Ayurveda is recognized as an upa or supplementary Veda in its own right. It contains the description of various diseases and their aetiology, and recommends the correct diet and behaviour regimen to counter those diseases.
Mythology has it that Brahma, the creator, imparted the knowledge of Ayurveda to Prajapati Daksha who, in turn, passed it on to the Ashwinikumara twins who were the physicians to the gods. The Ashwinikumaras then offered this knowledge to Lord Indra. Lord Indra instructed Dhanwantari to spread this invaluable science of longevity on the earth. Sushruta, a renowned surgeon and student of Dhanwantari, wrote his famous compendium on surgery – the Sushruta Samhita. The credit for the famous treatise on general medicine, the Charaka Samhita, goes to Charaka who probably lived sometime between the second century B.C. and the second century A.D.. Sushruta Samhita and Charaka Samhita are the two ancient treatises on which Ayurveda is based.
Ayurvedic philosophy provides a link between the living and non-living matters of the universe and indicates the origin of human and plant life from the five basic elements which are earth, water, fire, air and ether.
Principles of Ayurveda
Ayurveda is bestowed upon us by our ancestors, who were eminent and wiser and having insight into our being. Basically Ayurveda is Health promotive – preventive – curative and nutritive – all self contained.
The two principle objectives of Ayurveda are :
(a.) “Swasthyas swasthya rakshanam” – To prolong life and promote perfect health ( add years to life and life to years )
(b.) “Aturasya vikar prashamanamcha” – To completely eradicate the disease and dysfunction of the body.
Ayurveda takes the individual as whole and seeks to re-establish harmony between all the constituents in the body. Perfect balance of the tripod – Mind, Body and Spirit means perfect health.
To understand Ayurvedic treatment, it is necessary to have an idea how the Ayurvedic system views the body. The basic life force in the body is prana, which is also found in the elements and is similar to the Chinese notion of chi.
In Ayurveda, there are five basic elements that contain prana: earth, water, fire, air, and ether. These elements interact and are further organized in the human body as three main categories or basic physiological principles in the body that govern all bodily functions known as the doshas. The three doshas are vata, pitta, and kapha. Each person has a unique blend of the three doshas, known as the person’s prakriti, which is why Ayurvedic treatment is always individualized. In Ayurveda, disease is viewed as a state of imbalance in one or more of a person’s doshas, and an Ayurvedic physician strives to adjust and balance them, using a variety of techniques.
SPOTLIGHT-Linaclotide, Linzess, Ironwood Pharmaceuticals,

Drug: Linzess
Generic molecule: linaclotide
Company: Ironwood Pharmaceuticals
Approval date: Aug. 30,2012
851199-59-2 CAS NO
L-Cysteinyl-L-cysteinyl-L-glutamyl-L-tyrosyl-L-cysteinyl-L-cysteinyl-L-asparaginyl-L-prolyl-L-alanyl-L-cysteinyl-L-threonylglycyl-L-cysteinyl-L-tyrosine cyclo(1-6),(2-10),(5-13)-tris(disulfide)
Linaclotide is a peptide consisting of 14 amino acids. The sequence is
H–Cys1–Cys2–Glu3–Tyr4–Cys5–Cys6–Asn7–Pro8–Ala9–Cys10–Thr11–Gly12–Cys13–Tyr14–OH
There are three disulfide bonds: Between Cys1 and Cys6, between Cys2 and Cys10, and between Cys5 and Cys13.[8]
Linaclotide (marketed under the trade name Linzess) is an experimentalpeptide agonist of guanylate cyclase 2C that is undergoing clinical trials for use in treating abdominal pain in patients with irritable bowel syndrome (IBS) accompanied by constipation. The drug also looks promising in the treatment of gastroparesis, chronic intestinal pseudo-obstruction (CIPO), andinertia coli as well.[1] The drug was developed by Ironwood Pharmaceuticals, based in Cambridge, Massachusetts.
Linaclotide was approved by the FDA on August 30, 2012 for the treatment of chronic idiopathic constipation and to treat irritable bowel syndrome with constipation (IBS-C) in adults.[2] It became available in US pharmacies on December 17, 2012. [3] That same month, it was forecast by market research firm Decision Resources to achieve blockbuster status by 2021.[4]
The National Institutes of Health (NIH) estimates that as many as 20% of Americans may experience signs of irritable bowel syndrome, with approximately one-third of those affected experiencing constipation often accompanied by abdominal pain, affecting as many as 10 million Americans.Laxatives can assist with constipation but don’t treat pain, while use ofopiates to treat pain can aggravate constipation. While low-cost laxatives and pain killers would likely be tried first, linaclotide targets both associated conditions in a once-daily pill and could be used if standard treatments are unsuccessful in treating symptoms, though it would likely cost as much as several dollars per day.[5]
The approval of partner Ironwood’s linaclotide in late August is one of many reasons Forest Labs has been oft-cited as a takeover target in biopharma. Forest markets the drug, which is OK’d for chronic idiopathic constipation and to treat irritable bowel syndrome with constipation. Morgan Stanley has estimated potential peak sales at $2 billion.

- Tadataka Yamada, ed. (2011). Textbook of Gastroenterology. John Wiley & Sons. ISBN 9781444359411.
- “FDA approves Linzess to treat certain cases of irritable bowel syndrome and constipation”. 30 Aug 2012.
- “Ironwood and Forest Announce U.S. Availability of LINZESS”. 17 Dec 2012.
- “Constella/Linzess Will Achieve Blockbuster Sales of $1.2 Billion in 2021 in the Irritable Bowel Syndrome Drug Market”. 19 Dec 2012.
- Pollack, Andrew. “Drug for Irritable Bowel Achieves Goals in Trial”, The New York Times, September 13, 2010. Accessed September 14, 2010.
- Jeffrey M Johnston , Caroline B Kurtz , Douglas A Drossman , Anthony J Lembo , Brenda I Jeglinski , James E MacDougall , Stephen M Antonelli & Mark G Currie . “Pilot Study on the Effect of Linaclotide in Patients With Chronic Constipation”, The American Journal of Gastroenterology 104, 125–132 (1 January 2009) | doi:10.1038/ajg.2008.59. Accessed September 15, 2010.
- Staff. “Daily International Pharma Alert”, FDANews, September 17, 2007, Vol. 4 No. 182. Accessed September 15, 2010.
- Albericio, F; Giraud, M; Gongora, M; Paradis, M; Tulla-Puche, J; Werbitzky, O. Solid-Phase Synthesis of the Cys-rich Peptide Linaclotide.
Bausch + Lomb Receives FDA Approval for Prolensa, Bromfenac

bromfenac
april 2013
Bausch + Lomb, the global eye health company, today announced that the U.S. Food and Drug Administration (FDA) has approved the company’s New Drug Application (NDA) for Prolensa (bromfenac ophthalmic solution) 0.07 percent prescription eye drop, an innovative once-daily nonsteroidal anti-inflammatory drug (NSAID) for the treatment of postoperative inflammation and reduction of ocular pain in patients who have undergone cataract surgery. Prolensa will be available in 1.6ml and 3ml bottle sizes.
Prolensa provides powerful and rapid resolution of inflammation and pain by leveraging the unique potency of the bromfenac molecule in a formulation designed to facilitate ocular penetration. The advanced formulation allows for a lower concentration of bromfenac in a once daily dosing regimen. Prolensa is a solution that does not require shaking to deliver a consistent dose in each drop.
“The data show that once-daily dosing with Prolensa provides powerful and rapid control of inflammation and pain following cataract surgery, confirming the potency of this NSAID and the benefits of the new formulation,” said Steven M. Silverstein, M.D., FACS, founder of Silverstein Eye Centers in Kansas City, MO. “Prolensa reduces the amount of medication placed on the healing eye while maintaining a high degree of efficacy and ocular comfort.”
The efficacy of Prolensa was evaluated in two randomized, double-masked, vehicle-controlled studies of patients undergoing cataract surgery. Each randomized patient received Prolensa or vehicle starting with one drop into the surgical eye on the day prior to and the day of surgery, and for 14 days following surgery. The primary efficacy endpoint was complete clearing of ocular inflammation (assessed by the summed ocular inflammation score, SOIS, which includes cells and flare) by day 15. The secondary efficacy endpoint was the number of subjects that were pain free on day one after surgery.
About Bausch + Lomb
Bausch + Lomb is a leading global eye health company that is solely focused on protecting, enhancing, and restoring people’s eyesight. Our core businesses include ophthalmic pharmaceuticals, contact lenses and lens care products, and ophthalmic surgical devices and instruments. We globally develop, manufacture and market one of the most comprehensive product portfolios in our industry, which are available in more than 100 countries. Founded in 1853, our company is headquartered in Rochester, NY, and employs more than 11,000 people worldwide.
Prolensa™ is a trademark of Bausch & Lomb Incorporated or its affiliates.
Bromfenac is a non-steroidal anti-inflammatory drug (NSAID) marketed in the US as an ophthalmic solution (current brand name Bromday, prior formulation brand name Xibrom, which has since been discontinued.) by ISTA Pharmaceuticals for short-term, local use. Bromday® is the once-daily formulation of bromfenac, while Xibrom®, which has since been discontinued, was the twice-daily formulation. Bromfenac is indicated for the treatment of ocular inflammation and pain after cataract surgery, though it may be prescribed in an off-label manner by the physician.
For ophthalmic use, bromfenac has been prescribed more than 20,000,000 times across the world. As an eye drop, it has been available since 2000, starting in Japan where it was sold as Bronuck®. It was first FDA approved for use in the United States in 2005 and it was marketed as Xibrom®, twice-daily. October 2010 was the FDA approval of the new once-daily formulation of bromfenac called Bromday®. The bromfenac molecule will be marketed in Europe and other worldwide markets with agreements from Bausch & Lomb, Croma Pharma, and other companies.
Bromfenac was formerly marketed in the United States by Wyeth-Ayerst in an oral formulation called Duract® for short-term relief of pain (less than 10 days at a time). It was brought to market in July, 1997, and was withdrawn June 22, 1998 following numerous reports of hepatotoxicity in patients who had taken the medication for longer than the recommended 10-day period. The dose was one 25 mg capsule every 6 to 8 hours, or two capsules if taken with a high-fat meal, up to a maximum of 150 mg per day.

GSK licenses out Tykerb in China, signs Singapore pact with A*Star

LAPATINIB
Lapatinib (INN), used in the form of lapatinib ditosylate, (USAN) (Tykerb/Tyverb, GSK) is an orally active drug for breast cancer and other solid tumours.It is a dual tyrosine kinase inhibitor which interrupts the HER2/neu and epidermal growth factor receptor (EGFR) pathways. It is used in combination therapy for HER2-positive breast cancer. It is used for the treatment of patients with advanced or metastatic breast cancer whose tumors overexpress HER2 (ErbB2)
Expanding its Asia focus, GlaxoSmithKline has signed up Hong Kong-based Eddingpharm to sell its breast cancer drug Tykerb in China, having just entered into an alliance with Singapore’s A*Star to develop new medicines for emerging markets.
First up, Eddingpharm is to acquire exclusive rights in mainland China to import, market and promote Tykerb (lapatinib), which has recently been given the green light by the country’s state Food and Drug Administration. Specifically the drug is approved for use in combination with Roche’s Xeloda (capecitabine) for the treatment of patients with metastatic breast cancer whose tumours overexpress HER2 and who have received prior therapy with an anthracycline, a taxane and another Roche drug, Herceptin (trastuzumab).
Xin Ni, chief executive at Eddingpharm, said the deal, the financials for which have not been disclosed, is an important milestone, not least because “this is the first time a Chinese pharmaceutical company will participate in the launch of a proprietary global oncology drug”. He added that the partnership with GSK “helps us fully leverage our rich market experience and mature marketing platforms to open a fast track for Tykerb’s China launch”.
Tykerb has been approved in the aforementioned indication in more than 100 countries and Eddingpharm noted that in China there are 170,000 new cases of breast cancer diagnosed each year.
The deal came a day after GSK linked up with the Institute of Chemical and Engineering Sciences (ICES), owned by the Agency for Science, Technology and Research (A*Star) in Singapore.
A five-year strategic agreement has been signed to develop new evidence-based formulations (EBFs) specifically for emerging markets. The latter are medicines which are reformulated to provide additional patient benefit.
Keith Carpenter, executive director at ICES, which has been working with GSK since 2003, said that this latest deal “provides an opportunity for us to further our research and deepen our capabilities in formulation science with skilled scientists and technical expertise”. He added that the venture “will enable us to develop future scientists and laboratory analysts with the right skills to grow this industry in Singapore”.
Duncan McKay, head of emerging markets and Asia Pacific R&D at GSK, said that within the firm’s portfolio of off-patent products, “EBFs are an important part of our growth strategy”. He went on to say that “our hope is that together with ICES, we will create a sustainable, scalable model to meet both specific market conditions and patient requirements”.
Tapping into Singapore scientific talent
Commenting on the agreement, Kevin Lai, director of biomedical sciences at the Singapore Economic Development Board, said the deal is a strong endorsement of the country’s “scientific talent and capabilities”. He added that it also “further reinforces Singapore’s offering as the base for companies to generate insights and develop new solutions and market access strategies for the fast-growing emerging markets”.
GSK is a major player in Singapore, which is home to the drug giant’s headquarters for the emerging markets and Asia Pacific. It also has an R&D facility in Biopolis, two global manufacturing and supply sites (Jurong and Quality Road), a manufacturing facility for its Stiefel unit and a state-of-the-art vaccines plant in Tuas.
A*Star oversees 14 biomedical sciences and physical sciences and engineering research institutes, plus six consortia and centres and says it supports Singapore’s “key economic clusters by providing intellectual, human and industrial capital to its partners in industry”.
Links
AYURVEDA- BITTER MELON (Momordica charantia)

BITTER MELON (Momordica charantia): This edible gourd should be every physician’s “go-to” plant for the 16 million or more Americans with high-normal glucose readings or ‘boderline diabetic/metabolic syndrome patients.
Preliminary evidence suggests bitter melon’s hypoglycemic action can be explained through several independent mechanisms: for one, it has been shown to increase peripheral glucose oxidation as well as glucose tolerance and insulin signaling in induced insulin resistance models (Sridhar MG, et al: Br J Nutr. 2008;99(4):806-12. Basch E, et al. Am J Health Syst Pharm. 2003;60:356-9). It also decreases hepatic gluconeogenesis, while increasing glycogen synthesis.
Bitter Melon increases insulin output from the pancreas, and it also provides a unique compound called polypeptide-P, which is an insulin mimetic with a similar structure to bovine insulin (Krawinkel MB, Keding GB. Nutr Rev. 2006;64(7 Pt 1):331-7).
 Bitter Melon slices.
Bitter Melon slices.Compounds produced by this intriguing gourd have been shown to reduce triglyceride levels in a dose-dependent manner in animal trials (Jayasooriya AP, et al. J Ethnopharmacol. 2000;72:331-6). Though we don’t yet have human data corroborating this effect, the animal studies suggest that bitter melon may have a role in reducing cardiovascular risk, particularly in people with diabetes or metabolic syndrome.
Bitter melon products are typically standardized to their constituents, momordicosides and charantin, and usually dispensed in 500-600 mg doses, twice daily, following meals. As it does have an insulin mimetic action, it may be necessary to adjust the dose of concurrently prescribed hypoglycemic drugs.
DRUG SPOTLIGHT-Ambrisentan

Ambrisentan
(2S)-2-[(4,6-dimethylpyrimidin-2-yl)oxy]-3-methoxy- 3,3-diphenylpropanoic acid
177036-94-1 cas no
Ambrisentan (U.S. trade name Letairis; E.U. trade name Volibris; India trade namepulmonext by MSN labs ) is a drug indicated for use in the treatment of pulmonary hypertension.
It functions as an endothelin receptor antagonist, and is selective for the type A endothelin receptor (ETA).[1] Once daily oral ambrisentan 2.5 to 10 mg/day significantly improved exercise capacity (6-minute walk distance) compared with placebo in two double-blind, multicenter trials (ARIES-1 & ARIES-2).[2]
Ambrisentan was approved for sale by the U.S. Food and Drug Administration (FDA) on June 15, 2007 for the once-daily treatment of pulmonary arterial hypertension.[3][4][5] It was later approved by the European Medicines Agency for use in the EU on April 2008.[6]Ambrisentan had previously been designated an orphan drug by both the FDA and the European Commission, in August 2004 and May 2005 respectively.[7]
Ambrisentan is indicated for the treatment of pulmonary arterial hypertension (WHO Group 1) in patients with WHO class II or III symptoms to improve exercise capacity and delay clinical worsening.
The LETAIRIS Education and Access Program (LEAP) is a program to help physicians and patients learn about the risks of LETAIRIS, including the serious risks of liver injury and birth defects.
LEAP works by:
- Providing information to prescribers on the risks of LETAIRIS
- Providing comprehensive education to patients and assistance with obtaining LETAIRIS
- Requiring enrollment of both prescriber and patient in LEAP
- Controlling dispensing through a specialized distribution network (specialty pharmacies)
- Letairis website run by Gilead Sciences
- Prescribing information
- Information on the LETAIRIS Education and Access Program (LEAP)
- Vatter H, Seifert V (2006). “Ambrisentan, a non-peptide endothelin receptor antagonist”. Cardiovasc Drug Rev 24 (1): 63–76.doi:10.1111/j.1527-3466.2006.00063.x. PMID 16939634.
- Frampton JE. Ambrisentan. American Journal of Cardiovascular Drugs August 1, 2011; 11 (4): 215-226.Link text
- Pollack, Andrew (2007-06-16). “Gilead’s Drug Is Approved to Treat a Rare Disease”. New York Times. Archived from the original on 20 June 2007. Retrieved 2007-05-25.
- “U.S. Food and Drug Administration Approves Gilead’s Letairis Treatment of Pulmonary Arterial Hypertension” (Press release).Gilead Sciences. 2007-06-15. Retrieved 2007-06-16.
- “FDA Approves New Orphan Drug for Treatment of Pulmonary Arterial Hypertension” (Press release). Food and Drug Administration. 2007-06-15. Archived from the original on 23 June 2007. Retrieved 2007-06-22.
- “GlaxoSmithKline’s Volibris (ambrisentan) receives authorisation from the European Commission for the treatment of Functional Class II and III Pulmonary Arterial Hypertension” (Press release). GlaxoSmithKline. 2008-04-25. Archived from the original on 30 April 2008. Retrieved 2008-04-29.
- Waknine, Yael (2005-05-09). “International Approvals: Ambrisentan, Oral-lyn, Risperdal”. Medscape. Retrieved 2007-06-16.


Patent EP2547663A1


READ MORE ON SNTAN SERIES……http://medcheminternational.blogspot.in/p/sentan-series.html
DRUG SPOTLIGHT–Treprostinil

Treprostinil (marketed under the trade names Remodulin for infusion and Tyvaso for inhalation) is a synthetic analog of prostacyclin (PGI2).
Treprostinil sodium, Uniprost, LRX-15, U-62840, UT-15, BW-15AU, 15AU81, Remodulin
289480-64-4, 81846-19-7 (free acid

During the 1960s a U.K. research team, headed by Professor John Vane began to explore the role of prostaglandins in anaphylaxis and respiratory diseases. Working with a team from the Royal College of Surgeons, Vane discovered that aspirin and other oral anti-inflammatory drugs worked by inhibiting the synthesis of prostaglandins. This finding opened the door to a broader understanding of the role of prostaglandins in the body.

Vane and a team from the Wellcome Foundation had identified a lipid mediator they called “PG-X,” which inhibited platelet aggregation. PG-X, which later would become known as prostacyclin, was 30 times more potent than any other known anti-aggregatory agent.
By 1976, Vane and fellow researcher Salvador Moncada published the first paper on prostacyclin, in the scientific journal Nature. The collaboration produced a synthetic molecule which was given the name epoprostenol. But like native prostacyclin, the structure of the epoprostenol molecule proved to be unstable in solution, prone to rapid degradation. This presented a challenge for both in vitro experiments and clinical applications. To overcome this challenge, the research team that discovered prostacyclin was determined to continue the research in an attempt to build upon the success they had seen with the prototype molecule. The research team synthesized nearly 1,000 analogs.
Treprostinil has demonstrated a unique effect on PPAR gamma, a transcription factor important in vascular pathogenesis as a mediator of proliferation, inflammation and apoptosis. Through a complementary, yet cyclic AMP-independent pathway, treprostinil activates PPARs, another mechanism that contributes to the anti-growth benefits of the prostacyclin class.
Treprostinil is indicated for the treatment of pulmonary arterial hypertension in patients with NYHA Class II-IV symptoms to diminish symptoms associated with exercise.[1] It may be administered as a continuous subcutaneous infusion or continuous intravenous infusion; however, because of the risks associated with chronic indwelling central venous catheters, including serious blood stream infections, continuous intravenous infusion should be reserved for patients who are intolerant of the subcutaneous route, or in whom these risks are considered warranted.
In patients with pulmonary arterial hypertension requiring transition from epoprostenol sodium (Flolan), treprostinil is indicated to diminish the rate of clinical deterioration. The risks and benefits of each drug should be carefully considered prior to transition.
The pharmacokinetics of continuous subcutaneous treprostinil are linear over the dose range of 1.25 to 125 ng/kg/min (corresponding to plasma concentrations of about 15 pg/mL to 18,250 pg/m) and can be described by a two-compartment model. Dose proportionality at infusion rates greater than 125 ng/kg/min has not been studied.
The major effects of treprostinil are vasodilation of arteries in the pulmonary (lung) and body. Treprostinil also inhibits platelet aggregation.
Treprostinil may be administered as a continuous subcutaneous infusion or continuous intravenous infusion via a small infusion pumpthat the patient must wear at all times. Treprostinil can be given subcutaneously by continuous infusion using an infusion set connected to an infusion pump, but also may be given intravenously via a central venous catheter if the patient is unable to tolerate subcutaneous administration because of severe site pain or reaction.
1. Remodulin Full Prescribing Information US Patent No. 5,153,222
2. UT_OpinVEvid_FEB09v.1
3. Cost-minimization analysis of treprostinil vs. epoprostenol as an alternate to oral therapy nonresponders for the treatment of pulmonary arterial hypertension L. Narine, L. K. Hague, J. H. Walker, C. Vicente, R. Schilz, O. Desjardins, T. R. Einarson and M. Iskedjian

…..
(+)-Treprostinil (also known as UT-15) is the active ingredient in Remodulin®, a commercial drug approved by FDA for the treatment of pulmonary arterial hypertension (PAH). It was first described in U.S. Pat. No. 4,306,075. Treprostinil is a stable analog of prostacyclin (PGI2) belonging to a class of compounds known as benzindene prostacyclins, which are useful pharmaceutical compounds possessing activities such as platelet aggregation inhibition, gastric secretion reduction, lesion inhibition, and bronchodilation.

U.S. Pat. No. 5,153,222 describes use of treprostinil for treatment of pulmonary hypertension. Treprostinil is approved for the intravenous as well as subcutaneous route, the latter avoiding potential septic events associated with continuous intravenous catheters. U.S. Pat. Nos. 6,521,212 and 6,756,033 describe administration of treprostinil by inhalation for treatment of pulmonary hypertension, peripheral vascular disease and other diseases and conditions. U.S. Pat. No. 6,803,386 discloses administration of treprostinil for treating cancer such lung, liver, brain, pancreatic, kidney, prostate, breast, colon and head-neck cancer. U.S. patent application publication No. 2005/0165111 discloses treprostinil treatment of ischemic lesions. U.S. Pat. No. 7,199,157 discloses that treprostinil treatment improves kidney functions. U.S. Pat. No. 7,879,909 discloses treprostinil treatment of neuropathic foot ulcers. U.S. publication No. 2008/0280986 discloses treprostinil treatment of pulmonary fibrosis, interstitial lung disease with treprostinil and asthma. U.S. Pat. No. 6,054,486 discloses treatment of peripheral vascular disease with treprostinil. U.S. patent application publication No. 2009/0036465 discloses combination therapies comprising treprostinil. U.S. publication No. 2008/0200449 discloses delivery of treprostinil using a metered dose inhaler. U.S. Pat. Nos. 7,417,070, 7,384,978 and 7,544,713 as well as U.S. publications Nos. 2007/0078095, 2005/0282901, and 2008/0249167 describe oral formulations of treprostinil and other prostacyclin analogs as well as their use for treatment of a variety of conditions. U.S. provisional application No. 61/354,949 filed Jun. 15, 2010 discloses the use of orally administered treprostinil for treatment of Raynaud’s phenomenon, systemic sclerosis and digital ischemic lesions.
Treprostinil and other prostacyclin derivatives have been prepared as described in Moriarty, et al in J. Org. Chem. 2004, 69, 1890-1902, Drug of the Future, 2001, 26(4), 364-374, U.S. Pat. Nos. 4,306,075, 6,441,245, 6,528,688, 6,700,025, 6,765,117, 6,809,223 and US Publication No. 2009/0163738. The entire teaching of these documents are incorporated herein by reference in their entirety. The methods described in these patent documents, however, do not describe a feasible production method for producing stereochemically pure treprostinil because, for example, the methods require the use of expensive reagents and tedious chromatographic purification techniques. Therefore, there is a need in the art for an economical, efficient and simplified method for preparing treprostinil and its synthetic intermediates.

NMR
The 1HNMR and HPLC of the samples were compared with reference UT-15 and were identical; 1H NMR (CDCl3, 300 MHz) δ 0.90 (t, 3H, 6 Hz), 1.05-1.78 (m, 13H), 2.85-2.85-2.98 (m, 1H), 2.03 2.12 (m, 1H), 2.21-2.32 (m, 1H), 2.45-2.53 (m, 1H), 2.61-2.81 (m, 3H), 3.52 (br s, 1H), 3.58-3.69 (m, 1H), 4.62 (s, 2H), 6.69 (d, 1H, J=8 Hz), 6.78 (d, 1H, J=8 Hz), 7.04 (dd, 1H, J=8 Hz).
J. Org. Chem. 2004, 69, 1890-1902
mp 126−127 °C;
[α]25D +52.6 (c 0.453, MeOH), [α]25D + 34.0° (c 0.457, EtOH).
IR 3385, 2928, 2856, 1739, 1713, 1585, and 779 cm–1;
1H NMR (CDCl3, 300 MHz) δ 0.87 (t, 3 H, J = 6 Hz), 1.21−1.86 (m, 13H), 2.02−2.44 (m, 4H), 3.42−3.76 (m, 3H), 3.81 (s, 2H), 3.82−3.94 (m, 1H), 4.63−4.68 (m, 1H), 4.88−4.92 (m, 1H), 4.94−4.98 (m, 1H), 4.99−5.02 (m, 1H), 5.60 (s, 1H), 5.92−6.06 (m, 1H), 6.85 (d, 1H, J = 6 Hz), 7.20−7.27 (m, 1H), 7.31−7.37 (m, 1H);
13C NMR (MeOH, 75 MHz) δ 13.1, 22.4, 25.1, 25.3, 28.3, 31.8, 32.7, 33.2, 34.7, 36.9, 40.7, 41.0, 51.3, 65.2, 71.6, 76.3, 109.5, 121.1, 125.8, 127.4, 140.8, 155.2, 171.5; UV, λmax MeOH, 217 nm;
HPLC, Hypersil ODS column (4.6 × 250 mm2), 5 μm; flow rate 2.0 mL/min; mobile phase A, water (60%):acetonitrile (40%):trifluoroacetic acid (0.1%), and mobile phase B, water (22%):acetonitrile (78%):trifluoroacetic acid (0.1%); retention time, 15 min (purity 99.7%). Anal. Calcd for C23H34O5: C, 70.74; H, 8.78. Found: C, 70.41; H, 8.83.
