
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Ayurveda- Turmeric, Antiarthritic properties

Turmeric (Curcuma longa) is a rhizomatous herbaceous perennial plant of the ginger family, Zingiberaceae. It is native to tropical South Asia and needs temperatures between 20°C and 30°C (68°F and 86°F) and a considerable amount of annual rainfall to thrive. Plants are gathered annually for their rhizomes, and propagated from some of those rhizomes in the following season. In Vietnam, turmeric is called “nghệ”, “củ nghệ”.
When not used fresh, the rhizomes are boiled for several hours and then dried in hot ovens, after which they are ground into a deep orange-yellow powder commonly used as a spice in curries and other South Asian and Middle Eastern cuisine, for dyeing, and to impart color to mustard condiments. Its active ingredient is curcumin and it has a distinctly earthy, slightly bitter, slightly hot peppery flavor and a mustardy smell



3. Prevented breast cancer from spreading to the lungs in mice.
4. May prevent melanoma and cause existing melanoma cells to commit suicide.
5. Reduces the risk of childhood leukemia.
6. Is a natural liver detoxifier.
7. May prevent and slow the progression of Alzheimer’s disease by removing amyloyd plaque buildup in the brain.
8. May prevent metastases from occurring in many different forms of cancer.
9. It is a potent natural anti-inflammatory that works as well as many anti-inflammatory drugs but without the side effects.
10. Has shown promise in slowing the progression of multiple sclerosis in mice.
11. Is a natural painkiller and cox-2 inhibitor.
12. May aid in fat metabolism and help in weight management.
13. Has long been used in Chinese medicine as a treatment for depression.
14. Because of its anti-inflammatory properties, it is a natural treatment for arthritis and rheumatoid arthritis.
15. Boosts the effects of chemo drug paclitaxel and reduces its side effects.
16. Promising studies are underway on the effects of turmeric on pancreatic cancer.
17. Studies are ongoing in the positive effects of turmeric on multiple myeloma.
18. Has been shown to stop the growth of new blood vessels in tumors.
19. Speeds up wound healing and assists in remodeling of damaged skin.
20. May help in the treatment of psoriasis and other inflammatory skin conditions.

AYURVEDA-SHILAJIT, ANTIAGEING PROPERTIES
A composition comprising Shilajit or an extract thereof in a vitamin and mineral preparation. Shilajit is a compact mass of vegetable organic matter, composed of a gummy matrix interspersed with vegetable fibres and minerals. Substances which have been identified in Shilajit include moisture, gums, albuminoids, calcium, potassium, nitrogen, silica, resin, vegetable matter, magnesium, sulphur, iron, chloride, phosphorous, iodine, glycosides, tannic acid, benzoic acid and a number of vitamins and enzymes.
Shilajit is a natural exudate ejected from rocks during hot weather in the lower Himalayas, Vindhya and other mountain tracts and Nepal, or it may be a tar formed in the earth from the decomposition of vegetable substances. (See the Indian Materia Medica, pages 23 to 32 for a detailed discussion of the composition and properties of Shilajit). It is a compact mass of vegetable organic matter, composed of a gummy matrix interspersed with vegetable fibres and minerals. Shilajit also contains benzoic acid, a compound which, along with its derivatives, has been used as a component of nutritional vitamin and mineral preparations.

Ancient Sanskrit holy texts, over 3,000 years old, make reference to a mysterious substance called shilajit, which they describe as the “destroyer of weakness.” The texts list its powerful health and spiritual benefits and the positive changes that shilajit brought in the lives of those who used it. The sacred substance was prescribed for thousands of years for many different health problems and became a powerful tool in Ayurvedic medicine. There is some indication that shilajit may have been the priceless soma of the Eastern alchemists.
The rediscovery of the power of shilajit is said to have been made by Himalayan villagers observing large white monkeys migrate to the mountains in the warm summer months. The monkeys were seen to be chewing a semi-soft substance that flowed from between layers of rock. The villagers attributed the monkey’s great strength, longevity and wisdom to the strange substance. They began to consume it themselves and reported a broad spectrum of improvements in health. It seemed to give them more energy, relieve digestive problems, Increase sex drive, improve memory and cognition, improve diabetes, reduce allergies, improve the quality and quantity of life and it seemed to cure all diseases.
The ancient Vedic text Rig Veda states that soma “has mountains and stones for its body” and “dwells within the mountainous rock where it grows.” Mountainous rocks are the “abode of soma,” and it is “plucked from between the rocks by mountain dwellers and brought to the priests-alchemists who prepared the soma by washing and grinding and cooking.” Soma was considered the elixir of immortality, the secret substance used by alchemists to perfect both body and mind.
Shilajit must be harvested from sacred cliff sides high in the Himalayan Mountains of Nepal. Millions of years ago, before the Himalayas were formed, a lush garden flourished in a vast fertile valley. The vegetation in that primeval garden became trapped and preserved as the movement of the continents caused that valley to become the tallest mountain range in the world. Today, millions of years later the monsoon rains and extreme freeze and thaw conditions work together to crack large rock formations, exposing the precious shilajit. Because of its ancient nature, the vegetation was never exposed to any type of fertilizer, pesticide, herbicide, or pollution. The native Nepali people collect and carry this gift of nature down the mountain, where it is alchemically processed into a potent, high-quality extract.
This ancient wisdom was passed from generation to generation among the Indian and Nepali alchemists and holy men, but it escaped the notice of the Western medical establishment until the last days of the twentieth century, when explorer John Anderson heard of the amazing benefits of this substance and refused to give up the search until he found its source. He journeyed throughout India and Nepal until he learned of the perilous harvesting the raw shilajit from the cliffs. He also documented the reams of Sanskrit studies showing the rare plant’s benefits. He spoke firsthand with more than fifty Indian and Nepalese researchers that have been studying the wonderful effects of shilajit and perfecting the processes for delivering the purest, most concentrated shilajit ever know to man.
Over sixty years of clinical research have shown that shilajit has positive effects on humans. It increases longevity, improves memory and cognitive ability, reduces allergies and respiratory problems, reduces stress, and relieves digestive troubles. It is anti-inflammatory, antioxidant, and eliminates free radicals. The research proves that shilajit increases immunity, strength, and endurance, and lives up to its ancient reputation as the “destroyer of weakness.”
Technically, shilajit is an exudate that is pressed out from layers of rock in the most sacred and highest mountains in Nepal and other areas. It is composed of humus and organic plant material that has been compressed by layers of rock. Humus is formed when soil microorganisms decompose animal and plant material into elements usable by plants. Plants are the source of all our food and humus is the source of plant food. Unlike other soil humus, shilajit humus consists of 60-80% organic mass.
DRUG SPOTLIGHT – HALOPERIDOL

HALOPERIDOL
CAS No:- [52-86-8]
IUPAC Name:- 4-[4-(4-Chlorophenyl)-4-hydroxy-1-piperidinyl]-1-(4-fluorophenyl)-1-butanone
MW: 375.86, C21H23ClFNO2
Drug information:- Haloperidol is an anti-psychotic drug. This butyrophenone compound is used for the treatment of schizophrenia and other psychotic disorders in adults and childrens.
Haloperidol was discovered by Paul Janssen. It was developed in 1958 at the Belgian company Janssen Pharmaceutica and submitted to the first of clinical trials in Belgium later that year.
Haloperidol was approved by the U.S. Food and Drug Administration (FDA) on April 12, 1967; it was later marketed in the U.S. and other countries under the brand name Haldol by McNeil Laboratories.
Haloperidol is a dopamine inverse agonist of the typical antipsychotic class of medications. It is a butyrophenone derivative and has pharmacological effects similar to the phenothiazines.
Haloperidol is an older antipsychotic used in the treatment of schizophrenia and acutepsychotic states and delirium. A long-acting decanoate ester is used as an injection given every four weeks to people with schizophrenia or related illnesses who have poor adherence to medication regimens and suffer frequent relapses of illness, or to overcome the drawbacks inherent to its orally administered counterpart that burst dosage increases risk or intensity of side effects. In some countries, such as the United States of America, injections of antipsychotics such as haloperidol can be ordered by a court at the request of a psychiatrist.
Haloperidol is sold under the tradenames Aloperidin, Bioperidolo, Brotopon, Dozic,Duraperidol (Germany), Einalon S, Eukystol, Haldol (common tradename in the US and UK), Halosten, Keselan, Linton, Peluces, Serenace, Serenase, andSigaperidol


Conditions:-
i. Fluorobenzene, aluminum chloride, carbon disulfide, room temperature, 2 h,
ii. 4-(p-chlorophenyl)piperadine-4-ol, potassium iodide, toluene, 100 – 110 ºC
Arena Pharmaceuticals Initiates Phase 1 Clinical Trial of APD334 for Autoimmune Diseases

April 5, 2013
Arena Pharmaceuticals, Inc. announced today the initiation of dosing in a Phase 1 clinical trial of APD334, a novel oral drug candidate that targets the sphingosine 1-phosphate subtype 1 (S1P1) receptor for the potential treatment of autoimmune diseases.
This randomized, double-blind and placebo-controlled Phase 1 trial will evaluate the safety, tolerability and pharmacokinetics of single-ascending doses of APD334 in up to 64 healthy adult volunteers.
“We are pleased to expand our clinical-stage pipeline by initiating a Phase 1 trial of APD334, and look forward to advancing this novel compound through our validated development platform,” said William R. Shanahan, M.D., Arena’s Senior Vice President and Chief Medical Officer. “APD334’s selectivity for the S1P1 receptor has the potential to improve upon the adverse event profile of currently available treatments for a spectrum of autoimmune diseases.”
About Autoimmune Diseases
Autoimmune diseases are characterized by an inappropriate immune response against substances and tissues that are normally present in the body. In an autoimmune reaction, a person’s antibodies and immune cells target healthy tissues, triggering an inflammatory response. Reducing the immune and/or inflammatory response is an important goal in the treatment of autoimmune disease.
About APD334
APD334 is an orally available drug candidate discovered by Arena that targets the S1P1 receptor for the potential treatment of a number of conditions related to autoimmune diseases, including multiple sclerosis, psoriasis and rheumatoid arthritis. S1P1 receptors have been demonstrated to be involved in the modulation of several biological responses, including lymphocyte trafficking from lymph nodes to the peripheral blood. By isolating lymphocytes in lymph nodes, fewer immune cells are available in the circulating blood to effect tissue damage. Arena has optimized APD334 as a potent and selective small molecule S1P1 receptor agonist that reduces the severity of disease in preclinical autoimmune disease models.
About Arena Pharmaceuticals
Arena is a biopharmaceutical company focused on discovering, developing and commercializing novel drugs that target G protein-coupled receptors, or GPCRs, to address unmet medical needs. BELVIQ® (lorcaserin HCl), Arena’s internally discovered drug, was approved by the US Food and Drug Administration in June 2012 and is under review for regulatory approval in additional territories. Arena’s US operations are located in San Diego, California, and its operations outside of the United States, including its commercial manufacturing facility, are located in Zofingen, Switzerland. For more information, visit Arena’s website at www.arenapharm.com.
Arena Pharmaceuticals® and Arena® are registered service marks of Arena Pharmaceuticals, Inc. BELVIQ® is a registered trademark of Arena Pharmaceuticals GmbH
Drug spotlight, Celecoxib from G. D. Searle Company


CELECOXIB
4-[5-(4- methylphenyl)-3-(trifluoromethyl)-lH-pyrazol-l-yl]benzenesulfonamide
169590-42-5
mp…157-159 deg C
Celecoxib is a sulfonamide non-steroidal anti-inflammatory drug (NSAID) and selective COX-2 inhibitor used in the treatment of osteoarthritis, rheumatoid arthritis, acute pain, painful menstruation and menstrual symptoms, and to reduce numbers of colon and rectum polyps in patients with familial adenomatous polyposis. It is marketed by Pfizer. It is known under the brand name Celebrex or Celebra for arthritis and Onsenal for polyps. Celecoxib is available by prescription in capsule form.
Celecoxib was discovered developed by G. D. Searle & Company and was approved by the FDA on December 31, 1998. It was co-promoted by Monsanto Company (parent company of Searle) and Pfizer under the brand name Celebrex. Monsanto merged with Pharmacia, from which the Medical Research Division was acquired by Pfizer, giving Pfizer ownership of Celebrex. The drug was at the core of a major patent dispute that was resolved in Searle’s favor (later Pfizer) in 2004. In University of Rochester v. G.D. Searle & Co., 358 F.3d 916 (Fed. Cir. 2004), the University of Rochester claimed that United States Pat. No. 6,048,850 (which claimed a method of inhibiting COX-2 in humans using a compound, without actually disclosing what that compound might be) covered drugs such as celecoxib. The court ruled in favor of Searle, holding in essence that the University had claimed a method requiring, yet provided no written description of, a compound that could inhibit COX-2 and therefore the patent was invalid.

After the withdrawal of rofecoxib (Vioxx) from the market in September 2004, Celebrex enjoyed a robust increase in sales. However, the results of the APC trial in December of that year raised concerns that Celebrex might carry risks similar to those of Vioxx, and Pfizer announced a moratorium on direct-to-consumer advertising of Celebrex soon afterwards. After a significant drop, sales of Celebrex have recovered, and reached $2 billion in 2006.[6] Pfizer resumed advertising Celebrex in magazines in 2006, and resumed television advertising in April 2007 with an unorthodox, 2 1⁄2-minute advertisement which extensively discussed the adverse effects of Celebrex in comparison with other anti-inflammatory drugs. The ad drew criticism from the consumer advocacy group Public Citizen, which called the ad’s comparisons misleading. Pfizer has responded to Public Citizen’s concerns with assurances that they are truthfully advertising the risk and benefits of Celebrex as set forth by the FDA.
In late 2007, Pfizer released another U.S. television ad for Celebrex, which also discussed celecoxib’s adverse effects in comparison with those of other anti-inflammatory drugs.
Daniel L. Simmons of Brigham Young University, who discovered the COX-2 enzyme, is suing Pfizer to be credited with discovery of the technique in 1989 that eventually led to the drug, and for $1 billion USD. The company has made about $30 billion from the drug as of 2006. A settlement was finally reached in April 2012.
Celecoxib is licensed for use in osteoarthritis, rheumatoid arthritis, acute pain, painful menstruation and menstrual symptoms, ankylosing spondylitis and to reduce the number of colon and rectal polyps in patients with familial adenomatous polyposis. It was originally intended to relieve pain while minimizing the gastrointestinal adverse effects usually seen with conventional NSAIDs. In practice, its primary indication is in patients who need regular and long term pain relief; there is probably no advantage to using celecoxib for short term or acute pain relief over conventional NSAIDs, except in the situation where non-selective NSAIDs or aspirin cause cutaneous reactions (urticaria or “hives”). In addition, the pain relief offered by celecoxib is similar to that offered by paracetamol (acetaminophen).
Synthesis
https://www.google.com/patents/WO2010095024A2?cl=en
US 5,466,823, also discloses a process for the preparation of Celecoxib, which comprises reacting 4-methylacetophenone (II) with 1-ethyltrifluoroacetate (III) in the presence of methyl t-butyl ether and sodium methoxide, followed by recrystallisation from isooctane to produce l-(4-methylphenyl)-4,4,4-trifluorobutane-l ,3-dione (IV), which is further condensed with 4-hydrazinophenylsulfonamide hydrochloride (V) in the presence of ethanol to produce crude Celecoxib, which is recrystallised from ethyl acetate and isooctane to give Celecoxib (I),
The process is as shown in Scheme -I below:

HI rv

The synthesis of celecoxib was first described in 1997 by a team of researchers at Searle Research and Development. Celecoxib is synthesized by a Claisen condensation reaction of an acetophenone with N-(trifluoroacetyl)imidazole catalyzed by the strong base, sodium bis(trimethylsilyl)amide to produce a 1,3-dicarbonyl adduct. Condensation of the diketone with (4-sulfamoylaphenyl)hydrazine produces the 1,5-diarylpyrazole drug moiety.
Scheme-I The above process involves isolation of the intermediate l-(4-methylphenyl)-4,4,4- tiϊfluorobutane-l ,3-dione (IV) by crystallization, before condensing with 4- sulphonamido-phenylhydrazine, which adds to the cost and complexity of the synthesis.
Further, the above process proceeds with less selectivity to Celecoxib, which is having about 4 wt. % of regioisomer (VI) by-product under commercial conditions.
US 6, 150,534 discloses a process for the preparation of Celecoxib, which comprises, condensing l-(4-methylphenyl)-4,4,4-trifluorobutane-l ,3-dione (IV) with 4- sulphonamido-phenylhydrazine in presence of an amide solvent at controlled temperature to produce amide solvate of Celecoxib, which is further desolvated by recrystallization from isopropanol and water.
The above process also involves isolation of the intermediate l-(4-methylphenyl)- 4,4,4-trifluorobutane-l ,3-dione (IV) by crystallization, before condensing with 4- sulphonamido-phenylhydrazine,
US 5,892,053 discloses a process for the preparation of Celecoxib by condensing 4- methylacetophenone (II) with 1-ethyltrifluoro acetate (III) to produce l-(4- methylphenyl)-4,4,4-trifluoiObutane-l ,3-dione (IV), which is further reacted with 4- hydrazinophenylsulfonamide (V) in presence of aqueous mixture of alcohol and acid to produce Celecoxib.
US 6,579,988 discloses a preparation of Celecoxib via novel intermediate compound of formula VII. Formula VII

US 2007/0004924 Al discloses a process for the preparation of Celecoxib by condensing l-(4-methylphenyl)-4,4,4-trifluorobutane-1.3-dione (IV) with 4- hydrazinophenylsulfonamide (V) in presence of a solvent system containing an organic solvent, the salt of the 4-sulphonamidophenylhydrazine having a solubility in the organic solvent at least 0.05 M. ‘
US 2008/0234491 Al discloses the condensation of l-(4-methylphenyl)-4,4,4- trifluorobutane-l,3-dione (IV) with 4-hydrazinophenylsulfonamide (V) or its acid addition salts in the presence of a solvent medium comprising an alkyl ester, water or mixtures thereof to produce Celecoxib. Further, crystallization of crude Celecoxib is carried out in toluene alone.
l-(4-Methylphenyl)-4,4,4-trifluorobutane-l,3-dione (IV) is condensed with 4- hydrazinophenylsulfonamide (V) or its acid addition salt in a solvent selected from water, inert organic solvent to produce 4-[5-(4-methylphenyl)-3-(trifluoiOmethyl)-lH- pyrazol-l-yl]benzenesulfonamide (Celecoxib) of Formula I. The acid addition salts of compound of the formula IV includes, but are not limited to, hydrochloride, hydrobromide, sulfate, nitrate, oxalate, mesylate, methane sulfonate, and tartrate, preferably, hydrochloride salt. The suitable inert organic solvents for the above reaction include but are not limited to ketone solvents, such as acetone, methyl ethyl ketone, methyl isobutyl ketone, n-butanone, and tertiary-butyl ketone; nitrile solvents, such as acetonitrile. and propionitrile; halogenated solvents, such as dichloromethane, ethylene dichloride, and chloroform; esters, such as ethyl acetate, n-propylacetate, isopropyl acetate, and tertiary-butyl acetate; aprotic polar solvents, such as N,N- dimethylformamide, dimethylsulfoxide, and N,N-dimethylacetamide; ethers, such as diisopropyl ether, tetrahydrofuran and 1,4-dioxane; hydrocarbon solvents, such as cyclohexane, toluene and xylene; and mixtures thereof. The preferred solvent is water. The reaction may be performed at a temperature ranging from about 25°C to about reflux temperature of the solvent or mixture of solvents used for the reaction. The above reaction is conducted in presence of an acid selected from aqueous hydrochloric acid, aqueous sulfuric acid, p-toluene sulfonic acid, trifluoroacetic acid, and acetic acid to maintain the pH of the reaction mixture is below 7. More preferably, aqueous HCl is added. Crude Celecoxib (I) produced may be isolated by precipitation of compound from the reaction mixture, which may be performed by cooling the reaction mixture, followed by addition of an organic solvent selected from alcohols such as methanol, ethanol, isopropanol or aromatic hydrocarbons such as toluene, xylene, ethyl benzene and mixtures thereof solvents. The preferred solvent is mixture of methanol and toluene.
It has been observed that preparation of Celecoxib (I) using above reaction conditions results in regioisomer of compound (VI) to less than 2.5% by HPLC analysis.
EXAMPLE 1
Stage-1:
Preparation of l-(4-methylphenyl)-4,4,4-trifluorobutane-l,3-dione (IV)
4-Methylacetophenone (50 g, 0.373 mol) was dissolved in toluene (250 ml) and 30% methanolic sodium methoxide solution (80.6 g, 0.447 mol), followed by 1- ethyltrifluoro acetate (63.58 g, 0.447 mol) were added at 25-3O0C. Temperature of the reaction mass was raised to 55-600C and stirred ~ 4 hr to complete the reaction. The reaction mass was cooled to 20-250C and washed with 10% aqueous hydrochloric acid (200 ml). The layers were separated and concentrated the organic layer at 50-550C under reduced pressure to produce 80 g of l-(4-methylphenyl)-4,4,4-trifluoiObutane- 1,3-dione (IV) as an oily mass.
Stage-2:
Preparation of 4-[5-(4-methylphenyl)-3-(trifluorornethyl)-lh-pyrazol-l- yljbenzenesulfonamide (Celecoxib) (I) l-(4-Methylphenyl)-4,4,4-trifluorobutane-l,3-dione (IV) (80 g, 0.348 mol), 4- hydrazinophenylsulfonamide (V) (77.74 g, 0.348 mol) and concentrated hydrochloric acid (18.6 g) were added to DM water (500 ml) and heated to 98-1000C. The mass was stirred for 4 hr to complete the reaction. The reaction mass was cooled to 70-75 C and a mixture of toluene (600 ml) and methanol (10 ml) was added to the reaction mass. After 1 hr stirring at 70-750C, the reaction mass was cooled to 20-250C, the product was filtered and λvashed with toluene (100 ml) followed by DM water (200 ml). The product obtained was dried at 55-600C under reduced pressure to produce 1 15 g of Celecoxib crude. Chromatographic purity: 99%(by PTPLC, by area normalization)
……………………..

The synthesis of celecoxib was first described in 1997 by researchers at Searle Research and Development. It is synthesized by a Claisen condensation reaction of an acetophenone with N-(trifluoroacetyl)imidazole catalyzed by the strong base, sodium bis(trimethylsilyl)amide to produce a 1,3-dicarbonyl adduct. Condensation of the diketone with (4-sulfamoylaphenyl)hydrazine produces the 1,5-diarylpyrazole drug moiety.
Penning TD, Talley JJ, Bertenshaw SR, Carter JS, Collins PW, Docter S, Graneto MJ, Lee LF, Malecha JW, Miyashiro JM, Rogers RS, Rogier DJ, Yu SS, Burton EG, Cogburn JN, Gregory SA, Koboldt CM, Perkins WE, Seibert K, Veenhuizen AW, Zhang YY, Isakson PC (1997). “Synthesis and Biological Evaluation of the 1.5 Diarylpyrazole Class of Cyclooxygenase-2 Inhibitors: Identification of 4-[5-(4-Methylphenyl)-3-(trifluoromethyl)-1H-pyrazole-1-yl]benzenesulfonamide (SC-58634, Celecoxib)”. Journal of Medicinal Chemistry40 (9): 1347–1365. doi:10.1021/jm960803q. PMID 9135032.

……………………………….


- The condensation of 4-methylacetophenone (I) with ethyl trifluoroacetate (II) by means of NaOMe in refluxing methanol gives 4,4,4-trifluoro-1-(4-methylphenyl)butane-1,3-dione, which is cyclized with 4-hydrazinophenylsulfonamide (III) in refluxing ethanol.
…………………
http://www.google.com/patents/US7759497
In U.S. Pat. Nos. 5,892,053 and 5,910,597, Zhi et al. describe a scalable two step process for the preparation of pyrazoles from the condensation of diketones and hydrazines. In the first step, a diketone is formed by the treatment of a ketone with base and ester in a suitable solvent. In the second step, the diketone is solubilized in an aqueous alcohol and condensed with a hydrazine to form the pyrazole product. This two step process has been used on a commercial scale for the preparation of celecoxib (4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazole-1-yl]benzenesulfonamide) sold under the trademark CELEBREX® by Pharmacia Corporation as shown in the following reaction:

While this synthetic approach proceeds with high selectivity to celecoxib, about 2-5 wt. % of regioisomer and hydroxyregioisomer by-products are formed under commercial conditions.

The regioisomer and hydroxyregioisomer by-products must be separated from celecoxib in a purification step to enable the celecoxib to meet purity requirements. The separation is typically done through a crystallization step in which celecoxib preferentially crystallizes while the regioisomer and hydroxyregioisomer by-products predominantly remain in solution. The celecoxib crystals are then removed from the resultant slurry and separated from impurities by solid-liquid separation techniques known to those skilled in the art, such as centrifugation or filtration.
Under commercial conditions used to date, of the two by-products, regioisomer is selectively formed over hydroxyregioisomer. This is problematic, however, since the regioisomer is generally more difficult to separate through crystallization from celecoxib than is the hydroxyregioisomer, and regioisomer concentrations of greater than about 1% typically require two crystallizations to achieve desired celecoxib purity. The second crystallization adds time to the manufacturing process and thus negatively impacts product throughput. Additionally, a second crystallization reduces yield as some celecoxib remains uncrystallized and is not recovered from the liquid phase.

Example 7Preparation of Celecoxib with Hydrazine Reactant Containing Water
To a 250 mL reactor which had been purged with nitrogen and which had been fitted with a mechanical stirrer and a chilled condenser was charged while stirring, isopropyl alcohol (50.75 g), ethyltrifluoroacetate (37.95 g), sodium methoxide (25% in methanol, 53.28 g) and 4′-methylacetophenone (27.43 g). The reaction mixture was heated to 50-55° C. and held for at least 2 hours. To a separate 1 L reactor which had been purged with nitrogen and fitted with a mechanical stirrer and a chilled condenser, was charged 4-SAPH•HCl (45.96 g), isopropyl alcohol (101.2 g), water (74 g) and trifluoroacetic acid (23.43 g). The 4-SAPH•HCl was heated to 50° C. with agitation. At the completion of the 2 hour reaction period, the contents of the first reactor was transferred to the second reactor containing the 4-SAPH•HCl over a period of at least five minutes and the reaction mixture was then brought to 55° C. and maintained at that temperature for at least 30 minutes. The pH of the reaction mixture was then adjusted to be within the range of 3 to 9 followed by the addition of water (95 g). The contents were then heated to 65° C. and the pH was again adjusted to be within the range of 3 to 9. Water (90 g) was then added to the mixture over a time period of about 20 minutes while maintaining the temperature at about 65° C. The reaction mixture was then cooled to about 20° C. over a period of 12 to 14 hours to produce celecoxib (62-65 g) with less than 0.05% regio-isomer and undetectable regioisomer.
Example 8Preparation of Celecoxib with Anhydrous Hydrazine Reactant
To a 250 mL reactor which had been purged with nitrogen and which had been fitted with a mechanical stirrer and a chilled condenser was charged while stirring, isopropyl alcohol (50.75 g), ethyltrifluoroacetate (37.95 g), sodium methoxide (25% in methanol, 53.28 g) and 4′-methylacetophenone (27.43 g). The reaction mixture was heated to 50-55° C. and held for at least 2 hours. To a separate 1 L reactor which had been purged with nitrogen and fitted with a mechanical stirrer and a chilled condenser, was charged 4-SAPH•HCl (45.96 g), isopropyl alcohol (101.2 g) and trifluoroacetic acid (23.43 g). The 4-SAPH•HCl was heated to 50° C. with agitation. At the completion of the 2 hour reaction period, the contents of the first reactor was transferred to the second reactor containing the 4-SAPH•HCl over a period of at least five minutes and the reaction mixture was then brought to 55° C. and maintained at that temperature for at least 30 minutes. The pH of the reaction mixture was then adjusted to be within the range of 3 to 9 followed by the addition of water (95 g). The contents were then heated to 65° C. and the pH was again adjusted to be within the range of 3 to 9. Water (90 g) was then added to the mixture over a time period of about 20 minutes while maintaining the temperature at about 65° C. The reaction mixture was then cooled to about 20° C. over a period of 12 to 14 hours to produce celecoxib (62-65 g) with less than 0.05% regio-isomer. Analysis of the reaction mixture prior to initiation of crystallization indicated that the regio-isomer content was less than 0.5 mole percent of the reaction products.
Example 9Preparation of Celecoxib by Addition of Diketone Salt to 4-SAPH-HCl
To a 250 mL reactor, fitted with a mechanical stirrer and maintained under a nitrogen atmosphere, was added isopropyl alcohol (54.8 g, 0.912 moles), ethyl trifluoroacetate (38.0 g, 0.267 moles) and 25% sodium methoxide in methanol (53.3 g, 0.246 moles). To the agitated reactor was added 4-methylacetophenone (27.6 g, 0.206 moles). The reaction mixture was heated to 50° C. and maintained for 2 hours. To a second (1 liter) reactor was added 4-sulphamidophenyl hydrazine hydrochloride (46.0 g, 0.206 moles), isopropyl alcohol (101.3 g, 1.685 moles) and trifluoroacetic acid (11.7 g, 0.103 moles) with stirring. The reaction mixture was heated to approximately 45° C. Upon completion of the 2-hour reaction period in the 250 mL reactor, the contents was added to the second reactor over approximately 10 minutes. The reaction mixture maintained at 55° C. for 30 minutes. The pH was adjusted with 50% aqueous sodium hydroxide to a pH of 5-6. The reaction mixture was heated to 65° C. and water was added (95 g, 5.3 moles). The pH was again adjusted with 50% aqueous sodium hydroxide to a value of 5-6. Water (90 g, 5.0 moles) was added over 20 minutes while maintaining the temperature at 65° C. The reaction mixture was then cooled over 9 hours to 20° C. The reaction mixture was filtered, washed twice with 50% aqueous isopropyl alcohol and dried in a vacuum over for 16 hours to yield celecoxib (65.6 g) whose HPLC retention time was identical to that of authentic celecoxib. Regio-isomer was not detected by HPLC.
Example 10Preparation of Celecoxib by the Addition of 4-SAPH-HCl to Diketone
To a 1 L reactor fitted with a mechanical stirrer and maintained under a nitrogen atmosphere, was added isopropyl alcohol (54.7 g, 0.912 moles), ethyl trifluoroacetate (37.7 g, 0.267 moles), and 25% sodium methoxide in methanol (53.3 g, 0.247 moles). To the agitated reactor was added 4-methylacetophenone (27.32 g, 0.205 moles). The reaction mixture was heated to 50° C. and maintained for 2 hours. Trifluoroacetic acid (36.69, 0.321 moles) was added to the reaction mixture over a period of five minutes. 4-SAPH-HCl (46.0 g, 0.205 moles) was added through a power addition funnel over a period of 10 minutes. The reaction mixture was brought to 55° C. and maintained for one hour. Isopropyl alcohol (81.5 g, 1.36 moles) was added followed by the addition of 50% sodium hydroxide (18.5 g, 0.231 moles) to achieve a pH of 7. Water (87.8 g, 4.88 moles) was added and the reaction mixture heated to 65° C. Water (90.0 g 5.00 moles) was added over ten minutes. The reaction mixture was cooled to 20° C. over nine hours. The slurry was filtered and washed twice with 50% (weight) aqueous isopropyl alcohol (100 g). The solid was dried in a vacuum oven for 16 hours to yield celecoxib (67.2 g) whose HPLC retention time was identical to that of authentic material. Regio-isomer was not detected by HPLC.
4-[5-(4-Methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide (Celecoxib)
E-mail: prataprp@ drreddys.com. Fax: 914044346285. Telephone: 9989997176


HPLC Conditions:
 29.2 min30.9 min
29.2 min30.9 minhttps://newdrugapprovals.org/2013/04/07/drug-spotlight-celecoxib-from-g-d-searle-company/
Celecoxib extraction
Celecoxib was extracted from Celebra™ 100
mg and 200 mg capsules. Ten capsules were
crushed to fine powder, using agate mortar and
pestle, transferred to a 50 ml volumetric flask
and diluted to volume with methanol. The solution
was shaked for 5 min and filtered. The
residue was recrystallized from acetonitrile after
evaporation of the solvent in a water bath at 50
°C, under a stream of nitrogen.
Thin layer Chromatography (TLC) In this procedure it was used silica gel 60 F254 plates (20 x 20 cm) with a thickness of 0.25 mm. All plates used were commercially prepared by MERCK (lot # 040422153). The mobile phase used to develop the system consisted of chloroform-ethyl acetate-ether (10:5:1, v/v/v). Just in order to verify the selectivity of the proposed system, another COX-2 inhibitor (rofecoxib) with similar properties was used. 20 µl of celecoxib reference substance and extracted from tablets and rofecoxib solutions (50 µg.ml–1) were spotted on the TLC plates, and transferred to a developing tank containing the mobile phase. The plate was then examined under UV light (254 and 365 nm).
Thin-layer chromatography One of the most effective screening methods is the thin-layer chromatography (TLC), which is the simplest of all the widely used chromatographic methods to perform. In the determination of this method, different chromatography systems were tested and analyzed according to the classification proposed by Moffat 13, which divide the drugs in three categories: acid, basic and neutrals based on their polarity and acid characteristics. This procedure is important in
order to increase the information obtained only by changing the mobile phase, and thus resulting in a significant change in selectivity. These chromatographic systems consisted of mixtures of the following solvents: chloroformacetonitrile, ethyl acetate, acetone and methanol, in different concentrations. Nevertheless, all the mobile phases tested were not adequate for the proper identification of the drug. With the objective to develop more reliable chromatographic method, a modification in the system tested was proposed, in order to improve the chromatographic resolution. The mobile phase used consisted of chloroform-ethyl acetate-ether (10:5:1 v/v/v). The system was chosen due to its sensitivity, simplicity and efficacy
The preference to use commercially prepared silica gel plates was due to its durability and homogeneity of the absorbent layer. A sample solution of the working standard and the drug sample (50 (µg ml–1) were spotted onto a silica gel plate with a micropipette and the chromatogram was developed by placing the plate in a tank containing the mobile phase. Following development the individual solute spots were identified under UV lamp (254 and 365 nm). The spots in the drug sample and the working standard presented similar Rf values. The Rf value obtained for drug sample and the working standard were 0.45 and for rofecoxib 0.32.
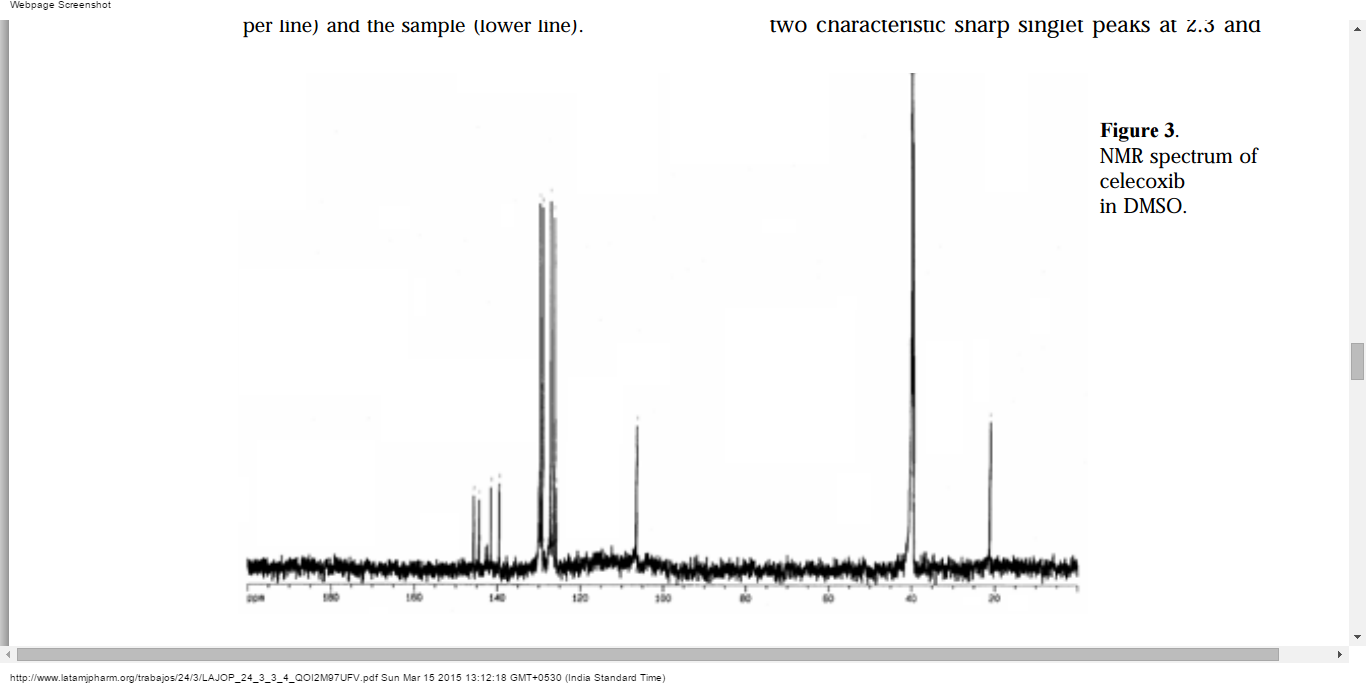
NMR
The spectrum shown in Figure 3 possesses two characteristic sharp singlet peaks at 2.3 and3.3 ppm that belong to the methyl and sulfonamide protons of celecoxib. The spectrum also reveals peaks from 7.3 to 7.9, which is due to the protons of aromatic groups. The characteristics peaks from the working standard agree well with those observed in the samples, considering the fact that a constant shift is observed in all peaks 11, 12.
UV

IR
1150 – 1350 S = O stretching (sulfonamide group)
1550 – 1600 N – H stretching
3300 – 3500 NH2 stretching


1H NMR PREDICT

13 C NMR

(4) (a) Matsuo, M.; Tsuji, K.; Konishi, N.; Nakamura, K. EP patent 0,418,845, A1, 1990. (b) Matsuo, M.; Tsuji, K.; Konishi, N.; Ogino, T. EP patent 0,554,829, A1, 1993. (c) Nishiwaki, T. Bull. Chem. Soc. Jpn. 1969, 42, 3024. (d) Soliman, R.; Feid-allah, H. J. Pharm. Sci. 1980, 70, 602. (e) Wright, J. B.; Dulin, W. E.; Markillie, J. H. J. Med. Chem. 1963, 7, 102. (f) Habeeb, A. G.; Rao, P. N. P.; Knaus, E. E. J. Med. Chem. 2001, 44, 3039. (g) Szabo, G.; Fischer, J.; Kis-Varga, A.; Gyires, K. J. Med. Chem. 2008, 51, 142. (h) Oh, L. M. Tetrehedron Lett. 2006, 47, 7943.
(5) Talley, J. J.; Penning, T. D.; Collins, P. W.; Rogier, D. J.; Malecha, J. W.; Miyashiro, J. M.; Bertenshaw, S. R.; Khanna, I. K.; Graneto, M. J.; Rogers, R. S.; Carter, J. S. US patent 5,466,823, 1995.
(6) Penning, T. D.; Talley, J. J.; Bertenshaw, S. R.; Carter, J. S.; Collins, P. W.; Docter, S.; Graneto, M. J.; Lee, L. F.; Malecha, J. W.; Miyashiro, J. M.; Rogers, R. S.; Rogier, D. J.; Yu, S. S.; Anderson, G. D.; Burton, E. G.; Cogburn, J. N.; Gregory, S. A.; Koboldt, C. M.; Perkins, W. E.; Seibert, K.; Veenhuizen, A. W.; Zhang, Y. Y.; Isakson, P. C. J. Med. Chem. 1997, 40, 1347.
7 Talley, J. J.; Penning, T. D.; Collins, P. W.; Rogier, D. J.; Malecha, J. W.; Miyashiro, J. M.; Bertenshaw, S. R.; Khanna, I. K.; Graneto, M. J.; Rogers, R. S.; Carter, J. S.; Docter, S. H.; Yu, S. S. US patent 6,586,603, B1, 2003.
(8) (a) O′ Shea, P. ; Tillyer, R. D. ; Wang, X. ; Clas, S. D. ; Dalton, C. US patent 6,150,534, 2000. (b) O′ Shea, P. ; Tillyer, R. D. ; Wang, X. ; Clas, S. D. ; Dalton, C. US patent 6,232,472, 2001.
(9) (a) Zhi, B. ; Newaz, M. US patent 5,,892,053, 1999. (b) Zhi, B. ; Newaz, M. US patent 5,910,597, 1999.
10(a) Letendre, L. J.; McGhee, W. D.; Snoddy, C.; Klemm, G.; Graud, H. T. US patent 7,141,678, 2006. (b) Letendre, L. J.; McGhee, W. D.; Snoddy, C.; Klemm, G.; Graud, H. T. US patent 2007/0,004, 924 A1, 2007.
(11) ICH harmonized tripartite guideline, Impurities in New Drug Substances Q3A (R2), current step 4 version dated 25 October2006.
(12) Ahlstrom, M. M.; Ridderstrom, M.; Zamora, I.; Luthman, K. J. Med. Chem. 2007, 50, 4444. (13) Soliman, R. J. Med. Chem. 1979, 22, 321.
SYNTHESIS
 +
+
![]() CELECOXIB
CELECOXIB
EUCLISES PHARMACEUTICALS, INC.; MARTINEZ, Eduardo, J.; TALLEY, John, J.; JEROME, Kevin, D.; BOEHM, Terri, L. Patent: WO2014/12074 A2, 2014 ; Location in patent: Paragraph 00209; 00210
2
+
![]()
 +
+
Reddy, Anumula Raghupathi; Sampath, Alla; Goverdhan, Gilla; Yakambaram, Bojja; Mukkanti, Kagga; Reddy, Padi Pratap Organic Process Research and Development, 2009 , vol. 13, # 1 p. 98 – 101
3
Prabhakaran, Jaya; Underwood, Mark D.; Parsey, Ramin V.; Arango, Victoria; Majo, Vattoly J.; Simpson, Norman R.; Van Heertum, Ronald; Mann, J. John; Kumar, J.S. Dileep Bioorganic and Medicinal Chemistry, 2007 , vol. 15, # 4 p. 1802 –
4
Li, Feng; Nie, Jing; Sun, Long; Zheng, Yan; Ma, Jun-An Angewandte Chemie – International Edition, 2013 , vol. 52, # 24 p. 6255 – 6258
5
Synlett, , vol. 1997, # 4 p. 375 – 377
6
Tetrahedron Letters, , vol. 52, # 45 p. 6000 – 6002
7
Bioorganic and Medicinal Chemistry Letters, , vol. 21, # 22 p. 6636 – 6640
8

![]() celecoxib
celecoxib
Angewandte Chemie – International Edition, , vol. 52, # 24 p. 6255 – 6258
9
Tetrahedron Letters, , vol. 52, # 45 p. 6000 – 6002

![]()
celecoxib
10Angewandte Chemie – International Edition, , vol. 52, # 24 p. 6255 – 6258

![]()
11
Bioorganic and Medicinal Chemistry, , vol. 15, # 4 p. 1802 – 1807

![]()
..celecoxib
………
http://www.google.com/patents/WO2014012074A2?cl=en
4,4,4-Trifluoro-l-(p-tolyl)butane-l,3-dione (C01)

25% sodium methoxide in methanol (51.3 ml, 223.5 mmol) and ethyl
trifluoroacetate (24.4 ml, 204.9 mmol) were dissolved in 110 mL methyl tert-butyl ether under N2, at room temperature. 4′-methyl acetophenone (25.0 ml, 186.3 mmol) was added and stirred at room temperature overnight. The reaction was washed with 3M HC1 and dried over magnesium sulfate. The solution was then evaporated and the resulting oil dried under vacuum overnight. The resulting light orange crystalline solid was washed with cold isooctane and dried under vacuum to yield an off white crystalline solid (37.3 g, 87% yield). LC tr=3.49 minutes (C- 18 column, 5 to 95% acetonitrile/water over 6 minutes at 1.7 mL/min with detection 254 nm, at 23 °C).
4-(5-(p-Tolyl)-3-(trifluoromethyl)-lH-pyrazol-l-yl)benzenesulfonamide (C02)

] C01 (23.55 g, 102.3 mmol) was refluxed with 4-sulphonamidophenyl
hydrazine HCl (23.95 g, 127.9 mmol) in 700 mL ethanol overnight. The reaction was evaporated, dissolved in 700 mL ethyl acetate, washed with water and brine, dried over magnesium sulfate and evaporated to -100 mL ethyl acetate. The product was crystalized by the addition of ~ 400 mL isooctane. After 15 minutes, the white crystalline solid was broken up, washed with isooctane and dried under vacuum (35.15 g, 90% yield). 1H NMR (400 MHz, CDC13) δ 7.94-7.91 (m, 2H), 7.51-7.49 (m, 2H), 7.21-7.20 (m, 2H), 7.15-7.13 (m, 2H), 6.77 (s, 1H), 2.41 (s, 3H). LC tr=4.27 minutes (C-18 column, 5 to 95% acetonitrile/water over 6 minutes at 1.7 mL/min with detection 254 nm, at 23° C). ES(neg)MS m/z 380 (M-H calcd for C17H14F3N302S requires 380).


http://www.slideshare.net/kennytirorx/celebrex-ms-report
………………………………….
http://www.google.co.in/patents/WO2011055233A2?cl=en
The preparation and use as COX-2 inhibitors of benzenesulfonamide derivatives such as celecoxib is described in US Patent No. 5,466,823. Processes for the preparation of celecoxib are also described in U.S. Patent Nos. 5,760,068 and 5,910,597.
As per the process exemplified in the U.S. Patent No. 5,760,068 (hereinafter referred to as the Ό68 patent), celecoxib is prepared by the reaction of l-(4-methylphenyl)- 4,4,4-trifluorobutane-l,3-dione with 4-sulphonamidophenyl hydrazine hydrochloride in absolute ethanol at reflux temperature under argon for 24 hours. The resulting mass is cooled to room temperature, followed by filtering and concentrating the reaction mixture to afford an orange solid, which is then recrystallized from a solvent system containing methylene chloride/hexane to produce the product as a pale yellow solid (melting point: 157° – 159°C).
The recrystallization process for preparing celecoxib described in the ‘608 patent suffers from disadvantages since the recrystallization process requires large volumes of solvents (more than 20 volumes each of methylene chloride and hexane solvents per gram of celecoxib), which is not commercially and environmentally, advisable for scale up operations. Moreover, the use of methylene chloride is hazardous to the environment and human health. The use of n-hexane is not advisable because it causes an ignition and fire risk due to its electrostatic charge accumulation property.
PCT Publication No. WO 01/42222 (hereinafter referred to as the ‘222 application) discloses three polymorphic forms (Form I, Form II and Form III) of celecoxib, pharmaceutical compositions, and methods of use thereof. The crystalline forms are characterized by powder X-ray diffraction (P-XRD), differential scanning calorimetry (DSC) and Infrared (IR) spectroscopy. The ‘222 application further teaches that the crystalline Form III of celecoxib is more thermodynamically stable than Form I and Form II. The ‘222 application also teaches that crystalline Form III of celecoxib is produced by crystallization of celecoxib from a solvent comprising isopropanol and water, for example, as described in U.S. Patent No. 5,910,597.
According to the ‘222 application, the polymorphic Form I is characterized by an X-ray powder diffraction pattern having peaks expressed as 2-theta at about 5.5, 5.7, 7.2 and 16.6 degrees; a melting point of about 162.5°C to about 163°C; a differential scanning calorimetry (DSC) endotherm maximum at about 163.3°C; and an Infra Red (IR) spectrum with peaks at about 3256 and 3356 cm-1. The polymorphic Form II is characterized by an X- ray powder diffraction pattern having peaks expressed as 2-theta at about 10.3, 13.8 and 17.7 degrees, a melting point of about 161°C to about 162°C; a differential scanning calorimetry (DSC) endotherm maximum at about 162°C. The polymorphic Form III is characterized by a melting point of about 160.8°C.
However, it has been observed by the present inventors that the celecoxib obtained after crystallization from isopropanol and water is fluffier resulting in low bulk density and poor flow properties. Moreover, it has also been observed that the particles of the crystalline form III of celecoxib obtained by the aforementioned crystallization processes are static or cohesive thereby increasing the difficulties of formulation scientists.
PCT Publication No. WO 01/42221 discloses an amorphous form of celecoxib, and processes for preparing amorphous celecoxib using crystallization inhibitors. Amorphous celecoxib exhibits an apparent glass transition at 111.4°C (onset).
EP Patent No. 1167355 (hereinafter referred to as the ‘355 patent) discloses a crystalline form, designated as Form I, of celecoxib, processes for the preparation, and pharmaceutical compositions thereof. The crystalline form is characterized by powder X-ray diffraction (P-XRD) and scanning electron microscopy (SEM). According to the ‘355 patent, the crystalline Form I is characterized by an X-ray powder diffraction pattern having peaks expressed as 2-theta at about 14.8, 16.05, 17.875, 19.615, 21.455, 22.080, 22.385, 23.425, 25.33 and 29.355 degrees. The crystalline Form I is further characterized by an X-ray powder diffraction pattern having additional peaks expressed as 2-theta at about 10.67, 10.97, 12.985, 13.855, 18.340, 18.685, 20.425, 20.67, 23.185, 24.51, 24.93, 25.73, 26.915, 27.63, 28.185, 29.955, 30.375, 31.405, 34.915, 35.585, 37.895, 44.070 and 45.250 degrees. The ‘355 patent teaches that the crystalline Form I has improved properties over prior art crystal form which is used for formulating celecoxib as disclosed in International Application No. WO 95/15316. The ‘355 patent teaches that the prior art crystal form (designated as Form II) has several disadvantages, caused by its crystal structure, since it has low bulk density and a crystal morphology that tends to form long cohesive needles.
According to the ‘355 patent, the celecoxib crystalline Form I is prepared by dissolving celecoxib in a solvent system comprising at least one amide solvent selected from the group consisting of N,N-dimethylformamide, Ν,Ν-dimethylacetamide, and mixtures thereof; and isolating the crystals of Form I by adding a non-solvent, especially water, to the solution.
The process for the preparation of the celecoxib crystalline Form I described in the ‘355 patent also suffers from drawbacks since the use of amide solvents in the purification/crystallization of celecoxib leads to the formation of solvates (for example, the formation of solvates of celecoxib with amide solvents such as dimethylacetamide and dimethylformamide can be found in the preparative examples of the WO 01/42222). It is well known that the removal of these residual amide solvents from the celecoxib crystalline form is very difficult and requires high temperatures.
Formation of solvates of celecoxib with amide solvents such as dimethylacetamide and dimethylformamide is also described in PCT Publication No. WO2005/014546. Moreover, celecoxib obtained by the crystallization process using amide solvents described in the ‘355 patent does not have satisfactory purity for pharmaceutical use.
The solvated forms of celexicob are not acceptable from regulatory point of view since they include substantial amounts of organic solvents, and thus are not acceptable for clinical use. It is well known that impurities and residual solvents in celecoxib or any active pharmaceutical ingredient (API) are undesirable and might be harmful. Purity standards are set by regulatory authorities with the intention of ensuring that an API is as free of impurities and residual solvents as possible, and, thus, are as safe as possible for clinical use. Furthermore, it is required to control the levels of these impurities in the final drug compound obtained by the manufacturing process and to ensure that the impurities and/or residual solvents are present in the lowest possible levels.
celecoxib characterized by a powder X-ray diffraction (P-XRD) pattern having peaks (designated as d-values) at about 16.0, 15.3, 12.3, 10.6 ± 0.2 A. According to the ‘340 application, the crystalline Form N of celecoxib is prepared by suspending celecoxib Form III in a hydrocarbon solvent selected from the group consisting of n-tetradecane, and n-decane, heating the suspension at high temperatures (about 165°C) while stirring, stirring the resulting emulsion at the same high temperature, followed by cooling to 145°C. The resulting mass is then reheated to about 165°C, followed by cooling to about 110°C, filtering the separated crystals, and drying at 100°C under the vacuo for 12 hours to produce celecoxib Form N.
The crystallization process for preparing celecoxib described in the ‘340 application also suffers from disadvantages since the processes involve tedious and cumbersome procedures such as the use of high boiling point solvents, large amounts of solvents (about 20 volumes of high boiling point solvents per gram of celecoxib), high temperatures (about 165°C), high drying temperatures (about 100°C), and prolonged periods of drying at a high temperature, resulting in formation of unwanted impurities, thereby making the process industrially unfeasible.
PCT Publication No. WO 05/089511 discloses a hydrate of celecoxib sodium salt characterized by a powder X-ray diffraction (P-XRD) pattern having peaks at 3.05, 8.91 and 10.77 degrees 2-theta.
PCT Publication No. WO 2006079923A1 discloses a crystalline Form IV of celecoxib characterized by a powder X-ray diffraction (P-XRD) pattern having peaks at about 4.46, 13.13, 18.29, 20.21, 21.83 and 26.24 degrees 2-theta. [0020] Based on the aforementioned drawbacks, the prior art crystallization processes may be unsuitable for the preparation of celecoxib, especially in crystalline Form III, in commercial scale operations.
| Patent | Submitted | Granted |
|---|---|---|
| 5-oxo-pyrrolidine-2-carboxylic acid hydroxamide derivatives [EP1004578] | 2000-05-31 | 2004-02-25 |
| ANALGESIC COMPOSITIONS COMPRISING ANTI-EPILEPTIC COMPOUNDS AND METHODS OF USING SAME [EP1011658] | 2000-06-28 | 2005-12-07 |
| SYNERGISTIC ANALGESIC COMBINATION OF OPIOID ANALGESIC AND CYCLOOXYGENASE-2 INHIBITOR [EP1014886] | 2000-07-05 | 2004-11-24 |
| BIARYLALKANOIC ACIDS AS CELL ADHESION INHIBITORS [EP1017382] | 2000-07-12 | 2006-03-01 |
| BIOADHESIVE COMPOSITIONS AND METHODS FOR TOPICAL ADMINISTRATION OF ACTIVE AGENTS [EP1021204] | 2000-07-26 | 2005-12-28 |
| Implantable medical device with enhanced biocompatibility and biostability [EP1023879] | 2000-08-02 | 2005-04-06 |
| NOVEL COMBINATION [EP1027052] | 2000-08-16 | |
| CYCLIC AMINO ACID DERIVATIVES AS CELL ADHESION INHIBITORS [EP1033983] | 2000-09-13 | |
| SUBSTITUTED BETA-ALANINE DERIVATIVES AS CELL ADHESION INHIBITORS [EP1034164] | 2000-09-13 | 2004-05-19 |
| Delayed-release oral formulation of dihydroxy open acid simvastatin and salts and esters thereof [EP1036563] | 2000-09-20 |
| Patent | Submitted | Granted |
|---|---|---|
| Crystalline hydrated dihydroxy open-acid simvastatin calcium salt [EP1036783] | 2000-09-20 | 2003-05-21 |
| CYCLOOXYGENASE-2 INHIBITION CYCLOOXYGENASE-2 INHIBITION [EP1039914] | 2000-10-04 | |
| Dioxocyclopentyl hydroxamic acids [EP1041072] | 2000-10-04 | 2003-07-16 |
| PHARMACEUTICAL ORAL DOSAGE FORM COMPRISING A COMBINATION OF AN OPIOID AGONIST AND NALTREXONE [EP1041987] | 2000-10-11 | 2006-04-19 |
| Method and compositions for the treatment and prevention of pain and inflammation [US2005101563] | 2005-05-12 | |
| Compositions of a cyclooxygenase-2 selective inhibitor and a neurotrophic factor-modulating agent for the treatment of central nervous system mediated disorders [US2005148589] | 2005-07-07 | |
| Compositions and methods of treatment involving peroxisome proliferator-activated receptor-gamma agonists and cyclooxygenase-2 selective inhibitors [US2003220374] | 2003-11-27 | |
| Methods for treating carbonic anhydrase mediated disorders [US2003220376] | 2003-11-27 | |
| Method of using a Cox-2 inhibitor and a 5-HT1A receptor modulator as a combination therapy [US2004147581] | 2004-07-29 | |
| Tyrosine kinase inhibitors [US2002137755] | 2002-09-26 |
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO1995015316A1 | 14 Nov 1994 | 8 Jun 1995 | Stephen R Bertenshaw | Substituted pyrazolyl benzenesulfonamides for the treatment of inflammation |
| WO2001042221A1 | 6 Dec 2000 | 14 Jun 2001 | Michael J Hageman | Solid-state form of celecoxib having enhanced bioavailability |
| WO2001042222A1 | 1 Dec 2000 | 14 Jun 2001 | Leonard J Ferro | Polymorphic crystalline forms of celecoxib |
| WO2002000627A1 * | 26 Jun 2001 | 3 Jan 2002 | Mehmet Bahar | A crystalline form of celecoxib |
| WO2005014546A1 | 8 Aug 2003 | 17 Feb 2005 | Hetero Drugs Ltd | Novel crystalline forms of celecoxib |
| WO2005089511A2 | 17 Mar 2005 | 29 Sep 2005 | Transform Pharmaceuticals Inc | Novel pharmaceutical forms, and methods of making and using the same |
| WO2006051340A1 | 21 Jul 2005 | 18 May 2006 | Pliva Istrazivanje I Razvoj D | Novel form of celecoxib |
| WO2006079923A2 | 19 Jan 2006 | 3 Aug 2006 | Pharmacia & Upjohn Co Llc | Form iv crystalline celecoxib |
| EP1167355A1 | 15 Mar 2001 | 2 Jan 2002 | Fako Ilaclari A.S. | A crystalline form of celecoxib |
| US5466823 | 30 Nov 1993 | 14 Nov 1995 | G.D. Searle & Co. | Antiinflammatory agents; FDA Orange book listed patent for Celebrex |
| US5760068 | 14 Nov 1994 | 2 Jun 1998 | G.D. Searle & Co. | Substituted pyrazolyl benzenesulfonamides for the treatment of inflammation |
| US5910597 | 14 Oct 1998 | 8 Jun 1999 | G.D. Searle & Co. | Process for preparing 3-haloalkyl-1H-pyrazoles |
………..

Dr Raghupathi Reddy Anumula
Managing Director- Chemikos Laboratories Private Limited
-
Managing Director
Chemikos Laboratories Private Limited
August 2014 – Present (Hyderabad Area, India
Director- API development
Dr. Reddy’s Laboratories
July 2012 – July 2014 (2 years 1 month)Hyderabad Area, India
Managing the three teams consisting of 50 members and guiding them to deliver the projects in time without any major surprises.

Associate Director- API development
Dr. Reddy’s Laboratories
October 2006 – June 2012 (5 years 9 months)Hyderabad Area, India
New product initiation, development ,Regulatory filing and launch support for API’s.
New product initiation, development ,Regulatory filing and launch support for API’s.
1994 – 2006
Shire begins Phase II ‘Vascugel’ trials
![]()
April 05,2012
Shire has kicked off two mid-stage trials assessing Vascugel – which is being developed by Shire under the name SRM003 – for enhancing blood vessel repair in patients with end-stage renal disease (ESRD) receiving haemodialysis.
Before patients can undergo haemodialysis, an arteriovenous (AV) access site must be created where blood can be removed, filtered and returned to the body.
In most cases, AV access is achieved through either an AV fistula (AVF), where the vein is connected directly to the artery, or an AV graft, where the vein and artery are connected via a synthetic tube.
Shire notes that there are around 100,000 AV fistulas and 60,000 AV grafts occurring annually in the US. But complications – such as infection, blood clots, and narrowing of the vessel – are common and frequently lead to access failure.
In fact, an estimated 60% of AV grafts fail after one year, requiring an additional procedure to restore flow or to create another access site, Shire said.
AYURVEDA SPOTLIGHT- BRAHMI
Bacopa monnieri
| Bacopa monnieri | |
|---|---|
 |
|
| Scientific classification | |
| Kingdom: | Plantae |
| (unranked): | Angiosperms |
| (unranked): | Eudicots |
| (unranked): | Asterids |
| Order: | Lamiales |
| Family: | Plantaginaceae |
| Genus: | Bacopa |
| Species: | B. monnieri |
Bacopa monnieri (waterhyssop, brahmi, thyme-leafed gratiola, water hyssop) is a perennial, creeping herb whose habitat includes wetlands and muddy shores. Brahmi is also the name given to Centella asiatica, particularly in North India, and Kerala where it is also identified in Malayalam as muttil (മുത്തിള്) or kodakan. This identification of brāhmī as C. asiatica has been in use for long in northern India, as Hēmādri’s Commentary on Aṣṭāṅgahṛdayaṃ (Āyuṛvēdarasāyanaṃ) treats maṇḍūkapaṛṇī (C. asiatica) as a synonym of brahmi, although that may be a case of mistaken identification that was introduced during the 16th century.
Brahmi been used by Indian Ayurvedic medical practitioners for almost 3000 years. The earliest chronicled mention of Brahmi is in several ancient Ayurvedic treatises including the Caraka Samhita (6th century A.D.), in which it is recommended in formulations for the management of a range of mental conditions including anxiety, poor cognition and lack of concentration, and the Bravprakash Var-Prakarana (16th century A.D.).
Description

Bacopa monnieri in Hyderabad, India.
The leaves of this plant are succulent and relatively thick. Leaves are oblanceolate and are arranged oppositely on the stem. The flowers are small and white, with four or five petals. Its ability to grow in water makes it a popular aquarium plant. It can even grow in slightly brackish conditions. Propagation is often achieved through cuttings.
Ecology
It commonly grows in marshy areas throughout India, Nepal, Sri Lanka, China, Taiwan, and Vietnam, and is also found in Florida, Hawaii and other southern states of the USA where it can be grown in damp conditions by the pond or bog garden.
Traditional uses
It has been used in traditional Ayurvedic treatment for epilepsy and asthma. It is also used in Ayurveda for ulcers, tumors, ascities, enlarged spleen, indigestion, inflammations, leprosy, anemia, and biliousness. This plant can be grown even as hydroponics using almost simple water.
Chemical constituents
Bacopa monnieri has many chemical constituents including alkaloids (brahmine and herpestine), saponins (d-mannitol and hersaponin, acid A, and monnierin), flavonoids (luteolin and apigenin). It also contains significant amounts of betulic acid, stigmasterol, beta-sitosterol, and bacopasaponins (bacosides A, bacosides B, bacopaside II, bacopaside I, bacopaside X, bacopasaponin C, bacopaside N2). The minor components include bacopasaponin F, bacopasaponin E, bacopaside N1, bacopaside III, bacopaside IV, and bacopaside V).
Pharmacology of chemical constituents
In rats, bacosides A enhance antioxidant defenses, increasing superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX) activity.Laboratory studies on rats indicate that extracts of the plant improve memory capacity. Some studies in mice suggest that ingestion of Bacopa for a 12 week period can significantly improve cognitive ability by accelerating the rate of learning and enhanced memory.The sulfhydryl and polyphenol components of Bacopa monnieri extract have also been shown to impact the oxidative stress cascade by scavenging reactive oxygen species, inhibiting lipoxygenase activity and reducing divalent metals.This mechanism of action may explain the effect of Bacopa monniera extract in reducing beta amyloid deposits in a mouse model of Alzheimer’s disease. B. monnieri has a demonstrated ability to reverse diazepam-induced amnesia in the Morris water maze test. The mechanism of this action is unknown. In some trials, bacopacide extract did not restore or enhance memory formation, but improved retention. In others, including a randomized clinical trial of 98 healthy older people (over 55 years), Bacopa significantly improved memory acquisition and retention. A 2012 systematic review found some evidence to suggest that Bacopa improves memory free recall, but there was a lack of evidence for enhancement of other cognitive abilities.
Brahmi may regulate antibody production by augmenting both Th1 and Th2 cytokine production.It may also cause a lower heart rate, and increase secretions in the stomach, intestines, and urinary tract. The increase in secretions may irritate ulcers and urinary tract obstructions.
No safety studies have been performed on brahmi’s use in humans. When a preparation of the plant was evaluated for safety and tolerability it showed no adverse effects but there were some reports of mild gastrointestinal symptoms.
However, participants in a 2001 double-blind study published in Psycho pharmacology experienced side effects including nausea, weakness and dry mouth while taking brahmi, notes the University of Michigan Health System. Brahmi could potentially cause elevated thyroid-hormone levels and decreased sperm counts. Therefore, taking brahmi should be avoided if you have a thyroid condition or are taking thyroid replacement therapies and other medications that affect thyroid function.
Aqueous extract of Bacopa monnieri (Brahmi) has been reported to reversibly suppress spermatogenesis and fertility in male mice with at a dose of 250 mg/kg body weight/day for 28 and 56 days(equivalent to 1.54 g/day for a 76kg male, when properly controlling for animal to human conversions ) Parameters of motility, viability, morphology, and number of spermatozoa in cauda epididymidis returned to baseline 56 days after treatment cessation.
The plant is known by many names in many international languages, including:
- ബ്രഹ്മി in Malayalam
- நீர்ப்பிரமி (Niirpirami)/ Valaarai in Tamil
- ผักมิ (Phak mi), พรมมิ (Phrommi) in Thai
- Lunuwila in Sinhalese (Sri Lanka)
- ae’ ae’ in Hawaiian (Hawaii)
- Rau Đắng in Vietnamese
- פְּשֵטָה שרועה (“psheta sru’a”) in Hebrew\
- conclusions
- Brahmi is a plant that has been used in traditional Indian medicine (Ayurveda). Be careful not to confuse brahmi (Bacopa monnieri) with gotu kola and other natural medicines that are also sometimes called brahmi.Brahmi is used for Alzheimer’s disease, improving memory, anxiety, attention deficit-hyperactivity disorder (ADHD), allergic conditions, irritable bowel syndrome, and as a general tonic to fight stress.People also take brahmi to treat backache, hoarseness, mental illness, epilepsy, joint pain, and sexual performance problems in both men and women. It is also sometimes used as a “water pill.”
-
Brahmi Benefits
Brahmi is considered a nootropic agent, which is the term given to a drug that improves mental functions such as cognition, memory, intelligence, motivation, attention, and concentration. Brahmi has been used in ayurvedic medicine and in traditional treatments for a number of disorders, particularly those involving anxiety, intellect, and poor memory.
Recent major scientific reviews of the plant suggest that it has prominent action on the central nervous system, where it improves understanding, memory, intellect, and speech, and corrects aberrations of emotions, mood, and personality in an individual. Based on the results of human clinical trials, the nootropic effects of Brahmi are thought to manifest after chronic dosing (i.e. 12 weeks) rather than acute (i.e. single day). Studies in humans have shown that chronic administration of Brahmi results in improvements in working memory, visual information processing, learning rate and anxiety.
In India, Brahmi is currently recognized as being effective in the treatment of mental illness and epilepsy. In certain parts of India, Brahmi is believed to be an aphrodisiac; in Sri Lanka, under the name of Loonooweella, Brahmi is prescribed for fevers; in the Philippines, it is used as a diuretic.
-
How does it work?
Brahmi might increase certain brain chemicals that are involved in thinking, learning, and memory. Some research suggests that it might also protect brain cells from chemicals involved in Alzheimer’s disease.
PHASE 3-Afatinib for locally advanced, recurrent or metastatic head and neck squamous cell carcinoma

Afatinib
Boehringer Ingelheim Pharmaceuticals.
Afatinib (Tomtovok; BIBW2992) Boehringer Ingelheim Pharmaceuticals., is a selective, potent, irreversible inhibitor of the ErbB family. The compound irreversibly binds to its targets and has potential for broader anti-tumour activity against receptors with acquired mutations which are resistant to first-generation inhibitors. Afatinib is intended for treatment of locally advanced, recurrent or metastatic HNSCC. Afatinib is administered orally at a starting dose of 40mg once daily. The dose may be increased to 50mg and/or reduced to 30mg, or 20mg depending on response and adverse effects
afatinib may provide an additional treatment option for this patient group who currently have limited effective therapeutic options. It has the potential to increase length of survival in this patient group.
Afatinib, Tomtovok, previously Tovok) is a candidate drug against non-small cell lung carcinoma (NSCLC), developed by Boehringer Ingelheim.As of July 2012, it is undergoing Phase III clinical trials for this indication and breast cancer, as well as Phase II trials for prostate and head and neck cancer, and a Phase I glioma trial. Afatinib is not a first-line treatment; it is only used after other therapies have failed.
In October 2010 a Phase III trial in NSCLC patients called Lux-Lung 5 began with this drug.Fall 2010 interim results suggested the drug extended progression-free survivalthreefold compared to placebo, but did not extend overall survival. In May 2012, the Phase IIb/III trial Lux-Lung 1 came to the same conclusion.
Phase II results for breast cancer that over-expresses the protein human epidermal growth factor receptor 2 (Her2-positive breast cancer) were described as promising by the authors, with 19 of 41 patients achieving benefit from afatinib. Double-blind Phase III trials are under way to confirm or refute this finding. Her2-negative breast cancers showed limited or no response to the drug.

Afatinib covalently binds to cysteine number 797 of the epidermal growth factor receptor (EGFR) via a Michael addition (IC50 = 0.5 nM).
Phase III trial of lupus drug Benlysta (belimumab) in patients with ANCA (Anti-neutrophil Cytoplasmic Antibodies) positive vasculitis

3 mar 2013
GSK, the company said it has kicked off a Phase III trial of its lupus drug Benlysta (belimumab) in patients with ANCA (Anti-neutrophil Cytoplasmic Antibodies) positive vasculitis, a condition characterised by inflammation of the blood vessels.
The multicentre, multi-national, randomised, double-blind study will assess the drug’s efficacy and safety in combination with azathioprine for the maintenance of remission in patients with a particular type of vascultitic disease called ANCA associated Vasculitis (Granulomatosis with Polyangiitis (Wegener’s) or microscopic polyangiitis).

Belimumab (trade name Benlysta, previously known as LymphoStat-B) is a human monoclonal antibody that inhibits B-cell activating factor (BAFF), also known as B-lymphocyte stimulator (BLyS) B cells are responsible for part of the normal immune response, and also for the over-aggressive immune response in autoimmune diseases like systemic lupus erythematosus (SLE).
Belimumab is approved in the United States, Canada and Europe for treatment of SLE. However, the major phase III trials excluded the more severe cases of SLE with kidney and brain damage, so its effectiveness has not been demonstrated in those cases. A Phase III study for SLE patients with kidney disease is now recruiting.
U.S. F.D.A. reviewers were concerned that belimumab is only “marginally” effective, and that there were more deaths in the treatment group.
Phase II trials of belimumab for rheumatoid arthritis were unsuccessful. Phase II trials for Sjögren’s Syndrome were more successful.
Belimumab was developed by Human Genome Sciences (HGS) and Cambridge Antibody Technology. GlaxoSmithKline acquired HGS, took belimumab through Phase III clinical trials, and markets belimumab.

Phase 3-Volasertib for acute myeloid leukaemia in patients ineligible for intensive induction therapy

Volasertib (BI 6727) for Acute myeloid leukaemia. is a cell cycle kinase inhibitor of polo-like kinases 1, 2 and 3. Volasertib inhibits cancer growth by disrupting cell division and inducing cell death. Volasertib is administered as a 350mg one hour intravenous (IV) infusion on days 1 and 15 of a 28 day cycle in combination with low-dose cytarabine (LDAC), administered via subcutaneous injection (SC) at 20mg twice daily on days 1-10 of a 28 day cycle.
Acute myeloid leukaemia (AML): previously untreated; patients considered ineligible for intensive remission induction therapy – first line; in combination with low-dose cytarabine
volasertib (also known as BI 6727) is a small molecule inhibitor of the PLK1 (polo-like kinase 1) protein being developed by Boehringer Ingelheim for use as an anti-cancer agent. Volasertib is the second in a novel class of drugs called dihydropteridinone derivatives.
