
Home » Posts tagged 'RAVUCONAZOLE'
Tag Archives: RAVUCONAZOLE
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
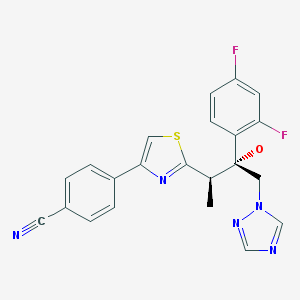
Fosravuconazole in phase 1 for the treatment of fungal infections.

Fosravuconazole
Phosphoric acid 2(R)-[4-(4-cyanophenyl)thiazol-2-yl]-1(R)-(2,4-difluorophenyl)-1-(1H-1,2,4-triazol-1-ylmethyl)propyoxymethyl monoester
(2R,3R)-3-r4-(4-cyanophenyl)thiazol-2-yll-2-(2,4-difluorophenyl)- 1 -(1 H- 1 ,2,4- triazol-l-yl)-2-[(dihydrogen phosphonoxy)methoxylbutane
BEF-1224
BMS-379224
E-1224
Phosphoric acid 2(R)-[4-(4-cyanophenyl)thiazol-2-yl]-1(R)-(2,4-difluorophenyl)-1-(1H-1,2,4-triazol-1-ylmethyl)propyoxymethyl monoester bis(L-lysine) salt is used as drug
The azole antifungal agent E-1224 is a prodrug of ravuconazole. In 2009, originator Eisai licensed E-1224 to Drugs for Neglected Diseases Initiative for the treatment of American trypanosomiasis (Chagas disease) in Latin America and the Caribbean. DNDi was conducting phase II clinical trials with the prodrug for this indication, however, development of the compound has been discontinued due to lack of sustained efficacy. Ravuconazole was originally licensed by Eisai to Bristol-Myers Squibb (BMS). BMS developed the drug’s prodrug, referred to by BMS as BMS-379224. For strategic reasons, BMS did not pursue development of the compound. In 2010, E-1224 was licensed exclusively to Brain Factory for development, commercialization and sublicense in Japan for the treatment of fungal infections.
About Ravuconazole and Ravuconazole Prodrug
The compound on the left is ravuconazole; the compound on the right is the dihydrogen phosphonoxy methoxy derived ravuconazole prodrug which has improved solubility and bioavailability.

……………………………………………………………
WO 2001052852
http://www.google.com/patents/WO2001052852A1?cl=en
Triazole antifungal compounds are well known in the prior art. Of the several classes known, one particularly potent class contains a tertiary hydroxyl group. For example, U. S. Patent 5,648,372 discloses that (2R,3R)-3-[4-(4- cyanophenyl)thiazol-2-yl]-2-(2,4-difluorophenyl)- 1 -( 1 H- 1 ,2,4-triazol- 1 -yl)- butan-2-ol has anti-fungal activity.

The utility of this class of compounds is limited by their low water solubility. For example, the solubility of the above triazole compound in water at pH 6.8 is 0.0006 mg/mL. This greatly impedes developing suitable parenteral dosage forms.
One method of addressing this problem was disclosed in European Patent Application 829478, where the water solubility of an azole antifungal agent was increased by attaching a linked amino-acid to the azole portion of the molecule

Alternatively, WO 97/28169 discloses that a phosphate moiety can be attached directly to the tertiary hydroxyl portion of the anti-fungal compound, e.g. the compound having the formula

U.S. Patent 5,707,977 and WO 95/19983 disclose water soluble prodrugs having the general formula

wherein X is OP(O)(OH)2 or an easily hydrolyzable ester OC(O)RNR l’rR>2.
WO 95/17407 discloses water-soluble azole prodrugs of the general formula

wherein X is P(O)(OH)2, C(O)-(CHR’)n-OP(O)(OH)2 or C(O)-(CHR’)π
-(OCHR,CHR1)mOR2.
WO 96/38443 discloses water-soluble azole prodrugs of the general formula

U.S. Patent 5,883,097 discloses water-soluble amino acid azole prodrugs such as the glycine ester

The introduction of the phosphonooxymethyl moiety into hydroxyl containing drugs has been disclosed as a method to prepare water-soluble prodrugs of hydroxyl containing drugs.
European Patent Application 604910 discloses phosphonooxymethyl taxane derivatives of the general formula

wherein at least one of R1 ‘, R2″, R3′, R6′ or R7′ is OCH2OP(O)(OH)2.
European Patent Application 639577 discloses phosphonooxymethyl taxane derivatives of the formula T-[OCH2(OCH2)mOP(O)(OH)2]n wherein T is a taxane moiety bearing on the C13 carbon atom a substituted 3-amino-2- hydroxypropanoyloxy group; n is 1, 2 or 3; m is 0 or an integer from 1 to 6 inclusive, and pharmaceutically acceptable salts thereof. WO 99/38873 discloses O-phosphonooxymethyl ether prodrugs of a diaryl 1,3,4-oxadiazolone potassium channel opener.
Golik, J. et al, Bioorganic & Medicinal Chemistry Letters, 1996, 6:1837- 1842 discloses novel water soluble prodrugs of paclitaxel such as


EXAMPLE 1
(2R,3R)-3-r4-(4-cyanophenyl)thiazol-2-yll-2-(2,4-difluorophenyl)- 1 -(1 H- 1 ,2,4- triazol-l-yl)-2-[(dihydrogen phosphonoxy)methoxylbutane, sodium salt

(2R,3R)-3-r4-(4-cyanophenyl)thiazol-2-yll-2-(2,4-difluorophenyl)-l-(lH- 1 ,2,4-triazol- 1 -yl)-2-[(di-tert-butyl phosphonoxy)methoxy1butane

To a solution of (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-2-(2,4- difluorophenyl)-l-(lH-l,2,4-triazol-l-yl)butan-2-ol, II, (8.74 g, 20 mmol) in THF (40 mL) under a nitrogen atmosphere was added sodium hydride (0.80 g, 60% in oil, 20 mmol) at rt. The resulting mixture was stirred at rt for 0.25 h and then di- tert-butyl chloromethyl phosphate, III (10.3 g, 40 mmol) was added. The reaction mixture was heated at 50 °C for 16 h. The reaction mixture was then allowed to cool to rt and was concentrated under reduced pressure. The residue was dissolved in Et2O and was washed with H2O and brine. The organic layer was dried over MgSO4 and was concentrated under reduced pressure to obtain 17.0 g of crude subtitled compound. IV, as a gum. A small portion of this crude compound was purified by reverse phase chromatography on C- 18. The column was eluted with 30% CH3CN/H2O, 38% CH3CN/H2O, 45% CH3CN/H2O and then 50% CH3CN/Η2O. The product containing fractions were concentrated under reduced pressure in order to remove CH3CN. The resulting aqueous layer was then extracted with Et2O. The Et O layers were washed with brine, dried and concentrated under reduced pressure to afford purified subtitled compound, IV, as a white solid. 1H NMR (300 MHz, CDC13): δ 8.35 (s, 1H), 7.98 (d, 2H, J=9), 7.76 (s, 1H), 7.71 (d, 2H, J=9), 7.63 (s, 1H), 7.36-7.27 (m, 1H), 6.86-6.78 (m, 2H), 5.53 (dd, 1H, J=28,6), 5.53 (dd, 1H, J=9,6), 5.17 (d, 1H, J=15), 5.03 (d, 1H, J=15), 4.01 (q, 1H, J=7), 1.47 (s, 9H), 1.45 (s, 9H), 1.37 (d, 3H, J=7). MS [ESI+ (M+H)+] 660.2 obs. B. (2R,3R)-3-r4-(4-cyanoρhenyl)thiazol-2-yll-2-(2,4-difluorophenyl)-l-(lH- 1 ,2,4-triazol-l-yl)-2-[(dihydrogen phosphonoxy)methoxy]butane, sodium saltdeprotection


The crude (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-2-(2,4- difluoropheny 1)- 1 -( 1 H- 1 ,2 ,4-triazol- 1 -y l)-2- [(di-tert-buty 1 phosphonoxy)methoxy]butane, IV, (17 g) was dissolved in CH C1 (100 mL). To this solution was added TFA (50 mL) and the reaction mixture was stirred at rt for 0.25 h. The reaction mixture was then concentrated under reduced pressure. To the residue was added H2O (200 mL), Et2O (100 mL) and EtOAc (100 mL). The pH of the aqueous layer was adjusted to 7.6 by addition of solid Na2CO3 and then the organic and aqueous layers were separated. The aqueous layer was then subjected to reverse phase chromatography on 400 g of C-18 eluted with H2O to 5% CH3CN/Η2O. The product containing fractions were concentrated under reduced pressure, frozen and lyophilized to afford 1.5 g of the subtitled compound, I, as a white solid. (1.5 g, 12% over two steps). Η NMR (500 MHz, D2O) δ 8.91 (s, IH), 7.92 (s, IH), 7.81 (d, 2H, J=8), 7.80 (s, IH), 7.77 (d, 2H, J=8), 7.21 (dd, IH, J=15,9), 6.99 (ddd, IH, J=9,9,2), 6.91 (ddd, IH, J=9,9,2), 5.35 (dd, IH, J=6,6), 5.29 (d, IH, J=15), 5.21 (dd, IH, J=6,6), 5.19 (d, IH, J=15), 3.86 (q, IH, J=7), and 1.35 (d, 3H, J=7); MS [(ESI“ (M-HV 546.1]; Anal. Calcd for C23Hi8F2N5θ5SιPι Na2/3.5 H2O: C, 42.21 : H, 3.85: N, 10.70: Na, 7.03. Found: C, 42.32: H, 3.83: N, 10.60: Na, 7.04.
Di-tert-butyl chloromethyl phosphate, III:
Di-tert-butyl chloromethyl phosphate, III, may be made by any of the following methods.
Method 1
Silver di-t-butyl phosphate (6.34 g, 20 mmol), which was prepared by mixing di- t-butyl phosphate (obtained from di-t-butyl phosphite by the method of Zwierzak and Kluba, Tetrahedron, 1971 , 27, 3163) with one equivalent of silver carbonate in 50% aqueous acetonitrile and by lyophilizing to dryness, was placed together with chloroiodomethane (35 g, 200 mmol) in benzene and stirred at room temperature for 18 hrs. The reaction mixture was filtered and the filtrate concentrated under reduced pressure. The residue was chromatographed on silica and eluted with 2:1 hexanes-ethyl acetate. Appropriate fractions were concentrated to dryness to obtain the subtitled compound III (3.7 g, 71% yield): H NMR (CDCI3) δ 5.63 (d, 2H, J=17), 1.51 (s, 18H); MS (MH+ = 259).
Method 2
Tetrabutylammonium di-t-butyl phosphate was prepared by dissolving di-t-butyl phosphate [ 20g, 94 mmol (obtained from di-t-butyl phosphite by the method of Zwierzak and Kluba, Tetrahedron, 1971, 27, 3163)] in methanolic tetrabutylammonium hydroxide (47 mL of 1M solution, 47 mmol). The reaction mixture had a temperature of 23 °C and pH of 4.33. The pH of the reaction mixture was adjusted to 6.5-7.0 by addition of methanolic tetrabutylammonium hydroxide (48 mL of 1M solution, 48 mmol) over 0.2 h. The reaction mixture was stirred for 0.5 h at approximately 26 °C and then was concentrated under reduced pressure at a bath temperature below 40 °C. The crude residue was azeotroped three times by adding toluene (3×100 mL) and then the mixture was concentrated under reduced pressure. The crude residue was then triturated in cold hexanes (0°C) for 1 h and then the solid was collected by filtration, washed with a minimum amount of cold hexanes and dried to give a first crop of tetrabutylammonium di-t-butyl phosphate as a white solid. (24. Og). The mother liquor was concentrated under reduced pressure and then triturated in cold hexanes (20 mL) for 1 h. The solid was collected by filtration, washed with a minimum amount of cold hexanes and dried to give a second crop of tetrabutylammonium di-t-butyl phosphate as a white solid. [(8.5g), 32.5g total (77%)]. A solution of tetrabutylammonium di-t-butyl phosphate (218 g, 480 mmol) in benzene (200 mL) was added dropwise to stirred chloroiodomethane (800g, 4535 mmol) over 1.5 h at rt. The reaction mixture was stirred an additional 1.5 h at rt and then was concentrated under reduced pressure. The oily residue was dissolved in Et2O and filtered to remove white solids which had precipitated. The organic layer was washed with saturated NaHCO3 and H O/brine (1/1). The organic layer was then dried over magnesium sulfate, filtered and concentrated under reduced pressure to yield a red brown oil (320 g). The red brown oil was subjected to chromatography on silica gel (800g) eluted with 20% EtOAc/Hexanes, 25% EtOAc/Hexanes then 30% EtOAc/Hexanes. The product containing fractions were concentrated under reduced pressure to yield a golden oil. The oil was diluted with CH2C12 (30 mL) , concentrated under reduced pressure and then dried under vacuum to yield the subtitled compound III (61.3g, 49% yield). 1H NMR (Benzene-d6) δ 5.20 (2H, d, J=15), 1.22 (18H, s).
Method 3
Iodochloromethane (974 g, 402 mL, 5.53 mol) at 25°C was treated with tetrabutylammonium di-t-butylphosphate (250 g, 0.553 mol). The phosphate was added portion wise over 10 minutes. The heterogeneous mixture became a clear pink solution after approximately 15 minutes. The mixture was stirred for three hours, and the iodochloromethane was then removed by rotary evaporation with a bath temperature of <30°C. The residue was taken up in 1 L t-butyl methyl ether and stirred for 15 minutes to precipitate tetrabutylammonium iodide by-product. Tetrabutylammonium iodide was removed by vacuum filtration through a sintered glass funnel. The filtrate was concentrated by rotary evaporation to an oil which contained a 5:1 mixture of III and undesired dimer impurity

III”
The mixture can be purified by a silica gel chromatography to obtain III as pure compound in ~60% yield as an oil.
EXAMPLE 2
(2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-2-(2,4-difluorophenyl)-l-(lH-l,2,4- triazol- 1 -yl)-2- (dihydrogen phosphonoxy)methoxy]butane
A. An oven dried, 1L round-bottom flask equipped with a mechanical stirrer, nitrogen inlet adapter, pressure-equalizing addition funnel fitted with a rubber septum and temperature probe was charged with sodium hydride (2.89 g, 0.069 mol, 60%) and THF (50 mL). To this stirred suspension, (2R,3R)-3-[4-(4- cyanophenyl)thiazol-2-yl]-2-(2,4-difluorophenyl)- 1 -( 1 H- 1 ,2,4-triazol- 1 -yl)butan- 2-ol, II, (10 g, 0.023 mol) in 30 mL of THF was added dropwise over 20 minutes at room temperature. After stirring for 45 minutes, a solution of iodine (2.99 g, 0.0115 mol) in THF (30 mL)) was added dropwise over 10 minutes followed by dropwise addition of compound di tert butylchloromethyl phosphate, III (13.29 g, 0.035 mol, -68% purity) over 15 minutes. The reaction mixture was stirred for 4 hours at about 41 °C to complete the reaction. The completion of the reaction was judged by in-process HPLC. The reaction mixture was poured into ice cold water (100 mL). The aqueous phase was separated and extracted with ethyl acetate (3 x 50 mL) and the combined organic extract was washed with 10% sodium thiosulfite (50 mL), water (50 mL), brine (50 mL), dried over magnesium sulfate and concentrated under reduced pressure to give pale yellow oil (22.8 g, In-process HPLC: ~ 97% pure). The crude product was used “as is” in step B.
B. To a round-bottom flask equipped with magnetic stirrer, cooling bath, pH probe and N2 inlet-outlet was charged the product of Step A above (7.5 g) in CH2C12 (23 mL) and cooled to 0 °C. To this stirred solution, trifluoroacetic acid (8.8 mL) was added slowly and stirred for 3 h to complete the reaction. The completion of the reaction was judged by in-process HPLC. The reaction mixture was poured into a cold solution of 2N NaOH (64 mL). The reaction mixture was extracted with t-butyl acetate (2 x 65 mL) to remove all the organic impurities. The aqueous layer containing the title product as bis sodium salt was treated with activated charcoal (10 g) and filtered through a bed of Celite. The clear filtrate was acidified with IN HC1 to pH 2.5. The free acid, the title product, was extracted into ethyl acetate (2 x 50 mL). The combined organic layer was washed with water, dried over MgSO4) filtered, and the filtrate concentrated under reduced pressure to afford 3.39 g of crude title product.
EXAMPLE 3
Bis lysine salt of (2R,3R)-3-r4-(4-cyanophenyl)thiazol-2-yl]-2-(2,4- difluorophenyl)- 1 -( 1 H- 1 ,2,4-triazol- 1 -yl)-2-[(dihydrogen phosphonoxy)methoxy]butane
The above obtained title product from Example 2 was dissolved in methanol (75 mL) and to this L-lysine (1.8 g) was added and heated at 60 °C for 4.5 h. The hot reaction mixture was filtered through a bed of Celite. The filtrate was concentrated to about 5 mL, mixed with ethanol (100 mL) and heated to 65 °C to crystallize the bis lysine salt. The salt was collected on a Buchner funnel and dried under vacuum to afford 3.71 g of the title compound as an off white crystalline solid.
About Eisai Co., Ltd.
Eisai Co., Ltd. is a research-based human health care (hhc) company that discovers, develops, and markets products throughout the world. Eisai focuses its efforts in three therapeutic areas: integrative neuroscience, including neurology and psychiatric medicines; integrative oncology, which encompasses oncotherapy and supportive-care treatments; and vascular and immunological reactions. Eisai contributes to the well-being of people around the world through a global network of research facilities, manufacturing sites and marketing subsidiaries. For more information about Eisai Co., Ltd., please visit http://www.eisai.co.jp/index-e.html.
ref
BMS-379224, a water-soluble prodrug of ravuconazole
42nd Intersci Conf Antimicrob Agents Chemother (ICAAC) (September 27-30, San Diego) 2002, Abst F-817
| WO2000030655A1 * | Nov 17, 1999 | Jun 2, 2000 | Squibb Bristol Myers Co | Water soluble prodrugs of azole compounds |
| WO2006118351A1 | May 1, 2006 | Nov 9, 2006 | Eisai Co Ltd | Mono-lysine salts of azole compounds |
| WO2012060448A1 | Nov 4, 2011 | May 10, 2012 | Eisai R&D Management Co., Ltd. | Combined pharmaceutical composition as antifungal agent |
| CN101341160B | Dec 20, 2006 | Jan 25, 2012 | 卫材R&D管理有限公司 | Process for production of water-soluble azole prodrug |
| EP1345915A1 * | Oct 18, 2001 | Sep 24, 2003 | Bristol-Myers Squibb Company | Improved process for water soluble azole compounds |
| EP2291084A1 * | May 20, 2009 | Mar 9, 2011 | Neurogesx, Inc. | Carbonate prodrugs and methods of using the same |
| US7230023 | Aug 20, 2003 | Jun 12, 2007 | Sankyo Company, Limited | Triazole compound containing a phosphonate group |
| US8735376 | May 20, 2009 | May 27, 2014 | Acorda Therapeutics, Inc. | Carbonate prodrugs and methods of using the same |
some animations
![]()



RAVUCONAZOLE

- BMS 207147
- ER 30346
- Ravuconazole
- UNII-95YH599JWV
4-[2-[(1R,2R)-2-(2,4-Difluorophenyl)-2-hydroxy-1-methyl-3-(1H-1,2,4-triazol-1-yl)propyl]-4-thiazolyl]benzonitrile



 |
|
| Systematic (IUPAC) name | |
|---|---|
| 4-[2-[(2R,3R)-3-(2,4-Difluorophenyl)-3-hydroxy-4-(1,2,4-triazol-1-yl)butan-2-yl]-1,3-thiazol-4-yl]benzonitrile | |
| Clinical data | |
| Legal status |
PHASE 2 AS ON SEPT 2014
|
| Identifiers | |
| CAS number | 182760-06-1 |
| ATC code | None |
| PubChem | CID 467825 |
| NIAID ChemDB | 057176 |
| Chemical data | |
| Formula | C22H17F2N5OS |
| Mol. mass | 437.465086 g/mol |

DRUG PROCESS…do not miss this

http://www.drugprocess.com/pdf/Isavuconazole_DPLA_ProcessSummary.pdf =++++++++++++++++++++++
…………………………………………………
Thiazole antifungals. III. Stereocontrolled synthesis of an optically active triazolymethyloxirane precursor to antifungal oxazolidine derivatives
Chem Pharm Bull 1991, 39(9): 2241
https://www.jstage.jst.go.jp/article/cpb1958/39/9/39_9_2241/_pdf
……………………………………………………
Optically active antifungal azoles. I. Synthesis and antifungal activity of (2R,3R)-2-(2,4-difluorophenyl)-3-mercapto-1-(1H-1,2,4-triazol-1-yl)-2-butanol and its stereoisomers
Chem Pharm Bull 1993, 41(6): 1035
https://www.jstage.jst.go.jp/article/cpb1958/41/6/41_6_1035/_pdf
………………………………………………………….
A novel route for chiral synthesis of the triazole antifungal ER-30346
Chem Pharm Bull 1998, 46(7): 1125
https://www.jstage.jst.go.jp/article/cpb1958/46/7/46_7_1125/_pdf
……………………………………………………….

ER-30346 is synthesized by thiazole ring formation of (2R, 3R) -3- (2,4-difluorophenyl) -3-hydroxy-2-methyl-4- (1H-1,2,4-triazol-1-yl ) thiobutanamide (I) and 4-bromoacetylbenzonitrile (II) by means of reflux in methanol. The thioamide (I) is obtained with excellent yield from a chiral nitrile (III) by heating with diethyl dithiophosphate in aqueous medium.




The nitrile (III), a chiral key intermediate of this synthesis, can be obtained by two different synthetic routes as follows: Route-b: The starting material of this route is methyl (S) -3-hydroxy-2-methylpropionate (VIII ), which contains one additional carbon between the hydroxyl group and the 2-position carbon of (R) -lactate, the starting material of route-a. The hydroxyl group of (VIII) is protected by triphenylmethyl group. Then, 2,4 -difluorophenyl moiety is introduced to give the ketone (X). Direct conversion of the ketone (X) to the oxirane (XIV) by dimethylsulfoxonium methylide, the same condition for compound (IV) in route-a, does not proceed. The oxirane (XIV) having desired stereochemistry is obtained via oxidation reaction. The ketone (X) is converted to the exomethylene (XI) by Wittig reaction. The stereoselective oxidation of (XI) is achieved by means of osmium tetroxide in the presence of 4-methylmorpholine N-oxide to give the diol (XII) in 58% yield after separation of its epimer by column chromatography. After methanesulfonylation of the primary alcohol of (XII), a triazole moiety is introduced and the triphenylmethyl group is deprotected. Then, the primary hydroxyl group of (XVI) is oxidized under Swern oxidation condition to give the aldehyde (XVII), which is converted to the chiral nitrile intermediate (III) by means of heating with hydroxylamine-O-sulfonic acid.

The synthesis of (2S, 3S) -3- (2,4-difluorophenyl) -3-hydroxy-2-methyl-4- (1,2,4-triazol-1-yl) butyronitrile (XV), a key intermediate the synthesis of ER-30346 has been described: The tritylation of 3-hydroxy-2 (S) -methylpropionic acid methyl ester (I) with trityl chloride in hot pyridine gives the trityl ether (II), which is hydrolyzed with LiOH in H2O / THF / methanol yielding the free acid (III). The esterification of (III) with 2-mercaptopyridine (IV) by means of dicyclohexylcarbodiimide (DCC) in dichloromethane gives the thioester (V), which is treated with 2,4-difluorophenylmagnesium bromide (VI) in THF yielding the propiophenone (VII), which by treatment with methyltriphenylphosphonium bromide / NaH in THF is converted into the methylene derivative (VIII). The oxidation of (VIII) with OsO4 and N-methylmorpholine oxide in acetone affords, after column chromatography, the chiral diol (IX), which is monomesylated with mesyl chloride / triethylamine in dichlormethane giving the monoester (X). The reaction of (X) with 1,2,4-triazol (XI) and NaH in DMF yields (2R, 3S) -2- (2,4-difluorophenyl) -3-methyl-1- (1,2,4-triazol-1-yl) -4- (triphenylmethoxy) -2-butanol (XII), which is detritylated with p-toluenesulfonic acid in methanol affording the diol (XIII). The oxidation of (XIII) with oxalyl chloride / DMSO in dichloromethane gives the aldehyde (XIV), which is finally treated with hydroxylamine-O-sulfonic acid in water yielding the desired bytyronitrile intermediate (XV) already referenced.

http://www.google.com/patents/WO2011042827A1?cl=en
Example 1
(2R,3R)-3-i4-(4-cvanophenyl)thiazol-2-yl1-1 -(1 H-1 ,2,4-triazol-1 -yl)-2-(2,4-difluorophenyl)- butan-2-ol
To a solution of racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1 -(1 H-1 ,2,4-triazol-1 -yl)-2-(2,4- difluorophenyl)-butan-2-ol (43.7 g) in acetone (800 ml) a solution of (1 R)-10- camphorsulfonic acid (23 g) in methanol (300 ml) was added and the mixture was heated under reflux until a clear solution was obtained. The solution was slowly cooled to rt, seeded with crystals of the title enantiomeric salt and let overnight. The solid was collected by filtration, washed with acetone and dried to provide (2R,3R)-3-[4-(4- cyanophenyl)thiazol-2-yl]-1 -(1 H-1 ,2,4-triazol-1 -yl)-2-(2,4-difluorophenyl)-butan-2-ol (1 R)- 10-camphorsulfonate as white solid. This crude salt was then taken up in methylenechloride (100 ml) and water (ca. 100 ml) and the mixture was basified with aqueous sodium hydroxide solution. The organic layer was separated and the aqueous phase washed twice with methylenechloride (50 ml) and combined. The organic phases were then washed twice with water (2×50 ml), dried with sodium sulfate, filtrated and the solvent removed under reduced pressure. The crude product was then mixed with isopropanol (ca. 150 ml), heated for 10 min, cooled to 0° C and stirred for ca. 2 hrs. The product was collected, washed with isopropanol and dried under reduced pressure to provide the enantiomerically pure title compound (17.5 g, 41 % yield, 99.1 % ee); m.p. 164-166° C; [a]=-30° (c=1 , methanol, 25° C); NMR (CDCI3): 1 .23(3H, d, J=8 Hz), 4.09(1 H, q, J=8Hz), 4.26(1 H, d, J=14Hz), 4.92(1 H, d, J=14Hz), 5.75(1 H, s), 6.75- 6.85(2H, m), 7.45-7.54(2H, m), 7.62(1 H, s), 7.69(1 H, s), 7.75(1 H, d, J=8Hz), 7.86(1 H, s), 8.03(1 H,d,J=8Hz). The analytical data were identical with published (US5648372 and Chem. Pharm. Bull. 1998, 46, 623-630).
…………………………
http://www.google.com/patents/WO1999045008A1?cl=en
Example 1
a) Preparation of (2R)-2′,5′-Difluoro-2-(3,4,5,6-tetrahydro-
2H-pyran-2-yloxy)-propiophenone A mixture of magnesium ( 7.25 g, 0.298 mol ) and iodine ( catalytic amount ) and l-bromo-2,5-difluorobenzene ( 20.0 g, 0.178 mol ) in THF ( 250ml ) was vigously stirred. The color of iodine was disappeared and the inner temperature rose up to 65°C. To this mixture was added additional l-bromo-2,5-difluorobenzene ( 30.0 g, 0.267 mol ) dropwise to maintain the inner temperature from 50 to 55°C over 45min. The resulting mixture was stirred at 55°C for 30min. then at r.t. for lhr. The – 21 –
mixture was cooled down to -5°C. To this mixture was added a solution of.4-[(2R)-2-(3,4,5,6-Tetrahydro-2H-pyran-2-yloxy)propionyl] morpholine ( 52.5 g, 0.216 mol ) in THF ( 150ml ) dropwise over 40min. And the resulting mixture was stirred at r.t. for 4hrs. The reaction mixture was cooled down to 5°C and saturated NH4C1 aq. ( 100ml ) was added carefully. The whole was diluted with H20 ( 600ml ) and extracted with EtOAc ( 400ml + 200ml x 2 ). The combined organic layer was dried over Na2S04 and concentrated in vacuo. The residue was chromatographed on silica gel ( n-hexane : EtOAc = 10 :1 ~ 5 : 1 ) to give (2R)-2′,5′- Difluoro-2-(3,4,5,6-tetrahydro-2H-pyran-2-yloxy)-propiophenone (47.3 g,
81 % ) as pale yellow syrup.
Physical form : colorless oil; FAB-MS: m/z 271(M+H)+; Η-NMR(CDCl;j): 1.42~1.90(9H,m),3.32~3.40(lHxl/2,m),3.69~3.77(lHxl/2,m),3.86~3.94 (lHxl/2,m),4.66(lHxl/2,t,J=3.6Hz),4.75(lHxl/2,t,J=3.6Hz),4.87(lHxl/2, q,J=6.6Hz),5.11(lHxl/2,q,J=6.9Hz),7.08~7.25(2H,m),7.49~7.55(lH,m).
b) Preparation of 2-(2,5-Difluorophenyl)-2-[(lR)-l-(3,4,5,6,- tetr ahy dro-2H-pyran-2-yloxy ) ethyl] oxir ane To a stirred mixture of NaH ( 60% in oil, 9.1g, 0.228mol ) in DMSO
(300ml ) was added portionwise trimethylsulfoxonium iodide ( 53.9g, 0.245 mol ) at the inner teperature with the range from 15°C to 18°C. over 20min. The ice bath was removed and the mixtuer was stirred at r.t. for 3hrs. The mixture was cooled down to 10°C. To this mixture was added a solution of (2R)-2′,5′-Difluoro-2-(3,4,5,6-tetrahydro-2H-pyran-2- yloxy)-propiophenone ( 47.3 g , 0.175 mol ) in DMSO (150ml ) dropwise over 20min. The resulting mixture was stirred at r.t. for 4hrs. The reaction mixture was poured into ice-water ( 800ml ). The whole was extracted with EtOAc ( 400ml + 200ml x 2 ). The combined organic layer was washed with brine, dried over Na2S04 and concentrated in vacuo.
The residue was chromatograkkphed on silicagel ( n-hexane : EtOAc = – 22 –
8 : 1 ~ 5 : 1 ) to give 2-(2,5-Difluorophenyl)-2-[(lR)-l-(3,4,5,6,- tetrahydro-2H-pyran-2-yloxy)ethyl]oxirane (48.3 g, 97 % ). Physical form : pale yellow syrup, EI-MS: m/z 284 (M)+ ; 1H-NMR(CDC13): 1.15(3Hxl/2,dd,J=6.6,1.3Hz), 1.24(3Hxl/2,dd, J=6.6,1.3Hz), 1.52-1.87 (6H,m),2.83~2,90(lH,m),3.07
(lHxl/2,d,J=5.3Hz),3.36(lHxl/2,d,J=5.6Hz), 3.48~3.56(lH,m),3.82~3.92 (lH,m),4.00~4.16(lH,m),4.73~4.92(lH,m), 6.96~7.02(lH,m),7.09~7.15 (lH,m).
c) Preparation of (3R)-2-(2,5-difluorophenyl)-3-(3,4,5,6- tetrahydro-2H-pyran-2-yloxy)-l-(lH-l,2,4-triazol-l-yl)-2-butanol
To a stirred suspension of NaH ( 60 % in oil, 21.0 g, 0.525 mol ) in DMF (300ml ) was added portionwise 1,2,4-triazole ( 43.3 g, 0.627 mol ) at the inner temperature from 2°C to 11°C over 30min. The resulting mixture was stirred at r.t. for l.δhrs. To this mixture was added a solution of 2-(2,5-Difluorophenyl)-2-[(lR)-l-(3,4,5,6-tetrahydro-2H- pyran-2-yloxy)ethyl]oxirane ( 48.3 g, 0.170 mol ) in DMF ( 50 ml ). The mixture was stirred at 60°C for lhr. and then at 65°C for 14hrs. The reaction mixture was cooled down to 10°C and then poured into ice- water (800 mL ). The resulting mixture was extracted with EtOAc
(400ml + 200ml x 2 ). The combined organic layer was dried over Na2S04 and concentrated in vacuo. The residue was chromatographed on silicagel ( n-hexane : EtOAc = 4 : 1 ~ 1 : 5 ) to give (3R)-2-(2,5- difluorophenyl)-3-(3,4,5,6-tetrahydro-2H-pyran-2-yloxy)-l-(lH-l,2,4- triazol-l-yl)-2-butanol ( 43.9 g, 73 % ) and recovered starting material
(13.2 g, 27 % ).
Physical form : colorless syrup ; FAB-MS: m/z 354 (M+H)+ ; Η- NMR(CDCl3): 1.00(3Hxl/2,d,J=6.6Hz),1.13(3Hxl/2,d,J=6.6Hz), 1.42~1.88(6H,m),3.38~3.60 (lH,m),3.80~4.00(lH,m),4.32~5.02(5H,m),6.83~6.99 (2H,m),7.14-7.21
(lH,m),7.73(lHxl/2,s),7.74(lHxl/2,s),7.92(lHxl/2,s),7.95(lHxl/2,s). – 23 –
d) Preparation of (2R,3R)-2-(2,5-difluorophenyl)-l-(lH-l,2,4- triazol-l-yl)-2,3-butanediol
A mixture of (3R)-2-(2,5-difluorophenyl)-3-(3,4,5,6-tetrahydro-2H- pyran-2-yloxy)-l-(lH-l,2,4-triazol-l-yl)-2-butanol ( 43.9 g, 0.124 mol ) and PPTS ( 15.6 g, 62.1 mmol ) in EtOH ( 400ml ) was stirred at 55°C for 5hrs. The mixture was was evaporated to remove solvent down to 100ml. The residue was poured into ice-aqueous NaHC03 ( 500ml ). The whole was extracted with EtOAc ( 400ml + 200ml x 2 ). The combined organic layer was dried over Na2S04 and concentrated in vacuo. The residue was chromatographed on silicagel (CH2C12 : MeOH = 20 : 1) to give (2R,3R)-2-(2,5-difluorophenyl)-l-(lH-l,2,4-triazol-l-yl)-2,3- butanediol (18.0 g, 54 % ). Physical form : colorless syrup ; FAB-MS: m/z 270 (M+H)’ ; ‘H- NMR(CDC13): 0.99(3H,d,J=6.6Hz),2.61(lH,d,J=10.6Hz), 4.31-4.36
(lH,m),4.79,4.88
(2H,ABq,J=14.5Hz),4.84(lH,s),6.84~6.99(2H,m),7.13~7.19(lH,m),7.84(l H,s),7.85(lH,s).
e) Preparation of (2R,3S)-2-(2,5-Difluorophenyl)-3-methyl-2-
[ ( 1H- 1 ,2,4-triazol-l -yl) -methyl] -oxir ane
To a cold ( 0°C ) and stirred solution of (2R,3R)-2-(2,5-difluorophenyl)- l-(lH-l,2,4-triazol-l-yl)-2,3-butanediol ( 35.0 g, 0.130 mol ) and triethylamine ( 54.8 ml, 0.393 mol ) in CH2C12 ( 500ml ) was added a mesylchloride ( 12.1 ml, 0.156 mol ) dropwise over 5min. The resulting mixture was stirred at r.t. for l.δhrs. The reaction mixture was poured into ice-water ( 300ml ). The resulting mixture was shaken well and the organic layer was separated. The aqueous layer was further extracted with CH2C12 ( 150ml x 2 ). All the organic layers were combined, dried over Na2SO4 and concentrated in vacuo to give mesylate ( 46.7 g ) as crude syrup. The obtained mesylate was dissolved in MeOH ( 500ml ) – 24 –
and the solution was cooled down to 0°C. To this solution was added 28% NaOMe methanol solution (29.0 ml ). The mixture was stirred at 0°C for 50min. The reaction mixture was evaporated to reduce the volume of the solvent down to 150 ml. The residue was poured into ice- water ( 300ml ). The resulting mixture was extracted with ethylacetate (300ml + 200ml x 2 ). The combined organic layer was dried over Na.,S0 and concentrated in vacuo. The residue was cromatographed on silicagel (hexane : EtOAc = 1 : 3 ) to give (2R,3S)-2-(2,5-Difluorophenyl)- 3-methyl-2-[(lH-l,2,4-triazol-l-yl)-methyl]-oxirane (30.3 g, 93 %).
Physical form : white solid ; FAB-MS : m z 252 (M+H)+ ; ]H- NMR(CDC13): 1.64(3H,d,J=5.6Hz),3.19(lH,q,J=5.6Hz),4.42,4.97 (2H,ABq,J=14.8Hz), 6.75~6.81(lH,m),6.89~7.01(2H,m),7.83(lH,s),7.98 UH,s).
f) Preparation of (2S,3R)-3-(2,5-Difluoro-phenyl)-3-hydroxy-
2-methyl-4-[l,2,4]triazol-l-yl-butyronitrile
A mixture of (2R,3S)-2-(2,5-Difluorophenyl)-3-methyl-2-[(lH-l,2,4- triazol-l-yl)-methyl]-oxirane ( 30.3 g, 0.121 mol ), trimethylsilylcyanide ( 65.0 ml ) and MgO ( 24.5 g ) in o-xylene ( 400 ml ) was stirred at 130°C for lOhrs. To this mixture was added additional trimethylsilylcyanide (20.0 ml ) and MgO ( 8.5 g ) and the resulting mixture was stirred at 130°C further for 6hrs. The reaction mixture was cooled down to r.t. The precipitate was filtered off and washed with CH2C12. The filtrate was concentrated in vacuo to give crude brown syrup.
This crude syrup was dissolved in THF ( 600ml ) and the solution was cooled down to 0°C. To this mixture was added 1.0 M tetra n- butylammoniumfluoride THF solution ( 133ml, 0.133 mol ) dropwise over 5min. The mixture was stirred at r.t. for 50min. The solvent was removed under reduced pressure down to 150ml. The residue was poured into ice-water ( 400ml ). The resulting mixture was extracted – 25 –
with EtOAc ( 300ml + 200ml x 2 ). The combined organic layer was dried over Na2SO4 and concentrated in vacuo. The residue was chromatographed on silicagel ( n-hexane : EtOAc = 1 : 3 ) to give (2S,3R)-3-(2,5-Difluoro-phenyl)-3-hydroxy-2-methyl-4-[l,2;4]triazol-l-yl- butyronitrile ( 30.5 g, 91 % ).
Physical form : colorless syrup ; FAB-MS : m/z 279 (M+H)+ ; Η- NMR(CDCl3): 1.19(3H,d,J=7.3Hz),3.33(lH,q,J=7.3Hz),4.82,5.00 (2H,ABq,J=13.9Hz), 5.56(lH,brs),6.89~7.04(2H,m),7.12~7.19(lH,m),7.85(lH,s),7.86(lH,s).
g) Preparation of (2R,3R)-3-(2,5-Difluoro-phenyl)-3-hydroxy-
2-methyl-4- [ 1 ,2,4] triazol-1 -ylthiob tyramide
A mixture of (2S,3R)-3-(2,5-Difluoro-phenyl)-3-hydroxy-2-methyl-4- [l,2,4]triazol-l-yl-butyronitrile ( 30–5 S> O.llOmol ), diethyldithio- phospate ( 235 ml ) and H2O ( 110 ml ) was stirre at 80°C for 2hrs. The reaction mixture was cooled down to r.t. n-Hexane ( 400ml ) and water (200 ml ) was added. The whole was shaken well and the aqueous layer was separated. The remaining organic layer was further extracted with H20 ( 100ml x 3 ). All the aqueous layer was combined. Cooled down to
0°C and neutralized and basified ( PH8 ) with NaHC03. This basic(PH8) aqueous layer was extracted with EtOAc ( 300ml + 100ml x 3 ). The combined organic layer was dried over Na2S04 and concentrated in vacuo to give dark brown syrup. By addition of CH2C12 ( 100ml ) to this crude syrup, precipitate was formed. The precipitate was filtered and washed with CH2C12-hexane ( 5 : 1 mixture ) to give (2R,3R)-3-(2,5-Difluoro-phenyl)-3-hydroxy-2-methyl-4-[l,2,4]triazol-l- ylthiobutyramide ( 19.2 g, 56 % ) as white powder. On the oter hand, the filtrate was concentrated in vacuo and the residue was chromatographed on silica gel ( Wako-gel C-300, CH2C12 : MeOH = 20 :
1 ) to give additional (2R,3R)-3-(2,5-Difluoro-phenyl)-3-hydroxy-2- – 26 –
methyl-4-[l,2,4]triazol-l-ylthiobutyramide ( 7.46 g, 22 % ) as pale brown amorphous powder.
Physical form : White solid ; FAB-MS : m/z 313 (M+H)+ ; ‘H-NMR (CDC13): 1.12(3H,d,J=7.3Hz),3.74(lH,q,J=7.3Hz), 4.55,5.12 (2H,ABq,J=14.5Hz), 5.84(lH,s),6.85~7.02(2H,m),7.15-7.22(lH,m),7.80
(1H,S),7.89(1H,S), 7.89(lH,brs),8.43(lH,brs).
h) Preparation of 4-{2-[(lR,2R)-2-(2,5-Difluoro-phenyl)-2- hydroxy-l-methyl-3-[l,2,4]triazol-l-yl-propyl]-thiazol-4-yl}- benzonitrile
A mixture of (2R,3R)-3-(2,5-Difluoro-phenyl)-3-hydroxy-2-methyl-4- [l,2,4]triazol-l-ylthiobutyramide ( 26.7 g, 85.4 mmol ) and a-bromo-4′- cyano-acetophenone ( 24.0 g, 0.107 mol ) in EtOH ( 500ml ) was refluxed for lhr. The reaction mixture was cooled down to r.t. And the solvent was removed under reduced pressure down to 150ml. The residue was poured into in to cold ( 0°C ) saturated NaHC03 aq. ( 400ml ). The resulting mixture was extracted with EtOAc ( 300ml + 150 ml x 2 ). The combined organic layer was washed with brine (200ml ), dried over Na2S04 and concentrated in vacuo. The residue was chromatographed on silica gel ( Wako-gel C-300, Hexane : EtOAc = 1 : 2 ) to give 4-{2-
[(lR,2R)-2-(2,5-Difluoro-phenyl)-2-hydroxy-l-methyl-3-[l,2,4]triazol-l- yl-propyl]-thiazol-4-yl}-benzonitrile ( 32.0 g, 86 % ).
Physical form : colorless heavy syrup ; ESI-MS : m/z 437 (M)+ ; ‘H-
NMR(CDCl3): 1.25(3H,d,J=7.3Hz),4.12(lH,q,J=7.3Hz),4.26,4.96 (2H,Abq,J=14.5Hz), 5.75(lH,s),6.89~7.07(2H,m),7.23~7.29(lH,m),7.65
(lH,s),7.71(lH,s),7.75,8.02 (4H,Abq,J=8.6Hz),7.85(lH,s). – 27 –
i) Preparation of 4-{4-[(tert-Butoxycarbonyl-methyl-amino)- acetoxy]-3,5-dimethyl-benzyl}-l-[(2R,3R)-3-[4-(4-cyano-phenyl)- thiazol-2-yl]-2-(2,5-difluoro-phenyl)-2-hydroxy-butyl]-lH- [l,2,4]triazol-4-ium bromide A mixture of 22.7mg of 4-{2-[(lR,2R)-2-(2,5-Difluoro-phenyl)-2-hydroxy- l-methyl-3-[l,2,4]triazol-l-yl-propyl]-thiazol-4-yl}-benzonitrile and 25.0mg of 4-tert-butoxycarbonyl-methyl-aminoacetoxy-3,5-dimethyl- benzyl bromide in CH3CN(1.5mL) was refluxed over 15hrs. The solvent was evaporated in vacuo and the residue was chromatographed on silica gel (Wakogel C-200, solvent:CH2Cl MeOH=10/l) to give 4-{4-[(tert-
Butoxycarbonyl-methyl-amino)-acetoxy]-3,5-dimethyl-benzyl}-l- [(2R,3R)-3-[4-(4-cyano-phenyl)-thiazol-2-yl]-2-(2,5-difluoro-phenyl)-2- hydroxy-butyl]-lH-[l,2,4]triazol-4-ium bromide (36.0mg, 84% as colorless heavy syrup) ; FAB-MS : m/z 743 (M-Br)’ ; Η-NMR(CDC1S): 1.23(3H,d,J=7.3Hz),
1.47(9H,s),2.14(6H,s),3.03(3H,s),4.15(lH,q,J=7.3Hz),4.25(2H,s), 4.98,5.16(2H,ABq,J=13.9Hz),5.39~5.54(2H,m),6.27(lH,s),6.89-7.07(4H, m),7.24~7.27(lH,m),7.58(lH,s),7.73,8.06(4H,ABq,J=8.58),8.07(lH,s),ll. 26 (lH,s).
j) Preparation of l-{(2R,3R)-3-[4-(4-cyano-phenyl)-thiazol-2-yl]- 2-(2,5-difluoro-phenyl)-2-hydroxy-butyl}-4-(3,5-dimethyl-4- methylaminoacetoxy-benzyl)-lH-[l,2,4]triazol-4-ium bromide To a solution of 36mg of 4-{4-[(tert-Butoxycarbonyl-methyl-amino)- acetoxy]-3,5-dimethyl-benzyl}-l-[(2R,3R)-3-[4-(4-cyano-phenyl)-thiazol-
2-yl]-2-(2,5-difluoro-phenyl)-2-hydroxy-butyl]-lH-[l,2,4]triazol-4-ium bromide in ethylacetate(2ml) was added dropwise 4N HC1 ethylacetate solution(lmL) and the mixture was stirred at r.t. for 4hrs.The precipitate was “filtered and washed with diethylether to give 1- {(2R,3R)-3-[4-(4-cyano-phenyl)-thiazol-2-yl]-2-(2,5-difluoro-phenyl)-2- hydroxy-butyl}-4-(3,5-dimethyl-4-methylaminoacetoxy-benzyl)-lH- – 28 –
[l,2,4]triazol-4-ium bromide (24.5mg, 74% as HC1 salt and as white solid) ;
FAB-MS : m/z 643 (M-Br)+ ; Η-NMR(DMSO-d): 1.19(3H,d,J=7.3Hz), 2.11(6H,s),2.64(3H,s),4.15(lH,q,J=7.3Hz),4.41(2H,s),4.74,5.04(2H,ABq,J =14.5Hz),5.40(2H,s),6.76(lH,brs),7.10(2H,s),7.20~7.38(2H,m), 7.94,8.21
(4H,ABq,J=8.25),8.45(lH,s),9.07(lH,s),9.50(lH,brs),10.17(lH,s).
………………………
http://www.google.co.in/patents/US5648372
OR
http://www.google.co.in/patents/EP1394142A1
COMPD 21

- Example 88:Preparation of a compound of the structural formula:
-

-
2-(2,4-Difluorophenyl)-3-thioamide-1-(1H-1,2,4-triazol-1-yl)-2-butanol (the raw material 2) (156 mg) was dissolved in EtOH (2 ml), and 2-bromo-4′-cyanoacetophenone (the raw material 3) (224 mg) was added to the solution, followed by heating and refluxing for 1 hour. The liquid reaction mixture was neutralized with a saturated aqueous solution of NaHCO3 and subjected to extraction with AcOEt. After the extract was washed with H2O and then a saturated aqueous solution of NaCl and dried over MgSO4, AcOEt was distilled out. The resultant residue was purified by chromatography on silica gel (SiO2: 20 g, eluted with CH2Cl2 and then with 1% solution of MeOH in CH2Cl2), and then crystallized from IPE, thereby obtaining the intended compound (109 mg). Physical properties of this compound are described below.
- mp:
- 196-197°C.
- NMR:
- δ solvent (CDCl3)
1.23(3H,d,J=8.0Hz), 4.09(1H,q,J=8.0Hz), 4.26(1H,d,J=14.3Hz), 4.92(1H,d,J=14.3Hz), 5.74(1H,s), 6.78-6.85(2H,m), 7.48-7.54(1H,m), 7.64(1H,s), 7.69(1H,s), 7.75(1H,d,J=8.1Hz), 7.85(1H,s), 8.03(1H,d,J=8.1Hz). - MS:
- MH+ = 438.

References
- National Cancer Institute. Ravuconazole in Preventing Fungal Infections in Patients Undergoing Allogeneic Stem Cell Transplantation. In: ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). 2000- [cited 2010 Feb 18]. Available from:http://clinicaltrials.gov/ct2/show/NCT00064311?term=ravuconazole&spons_ex=Y&rank=1 NLM Identifier: NCT00064311.
- The Aspergillus Website, Pasqualotto AC, Denning DW. Ravuconazole. Date accessed: 2010 Feb 18.
- Pasqualotto AC, Thiele KO, Goldani LZ (2010). “Novel triazole antifungal drugs: focus on isavuconazole, ravuconazole and albaconazole”. Curr Opin Investig Drugs 11 (2): 165–74. PMID 20112166.
- Pfaller, M. A.; Messer, S. A.; Hollis, R. J.; Jones, R. N.; Sentry Participants, Group (2002). “Antifungal Activities of Posaconazole, Ravuconazole, and Voriconazole Compared to Those of Itraconazole and Amphotericin B against 239 Clinical Isolates of Aspergillus spp. and Other Filamentous Fungi: Report from SENTRY Antimicrobial Surveillance Program, 2000”. Antimicrobial Agents and Chemotherapy46 (4): 1032. doi:10.1128/AAC.46.4.1032-1037.2002. PMC 127116. PMID 11897586.

Literature References:
Ergosterol biosynthesis inhibitor. Prepn (stereochemistry unspecified): T. Naito et al, EP 667346; eidem,US 5648372 (1995, 1997 both to Eisai); of optically acitve form: A. Tsuruoka et al., Chem. Pharm. Bull. 46, 623 (1998). Chiral synthesis: Y. Kaku et al., ibid. 1125.
In vitro comparative antifungal spectrum: J. C. Fung-Tomc et al., Antimicrob. Agents Chemother. 42, 313 1998. Antifungal activity in candidosis: K. V. Clemons, D. A. Stevens, ibid. 45, 3433 (2001); in aspergillosis: W. R. Kirkpatrick et al., J. Antimicrob. Chemother. 49, 353 (2002).
Clinical evaluation in onychomycosis: A. K. Gupta et al., J. Eur. Acad. Dermatol. Venereol. 19, 437 (2005).
Review of development and therapeutic potential: S. Arikan, J. H. Rex, Curr. Opin. Invest. Drugs 3, 555-561 (2002).
Extras you may need

http://www.google.com/patents/WO2011042827A1?cl=en
Scheme 1 :

The manufacturing process for Isavuconazole is similar: Since Isavuconazole differentiates from Ravuconazole by only another fluorine substitution on the aromatic ring (2,5- instead of 2,4-difluorophenyl), the identical synthesis has been used (US 6300353 from October 9, 2001 and Bioorg. & Med. Chem. Lett. 13, 191 (2003)). Consequently, also this manufacturing process, based on (R)-lactic acid, faces the same problems: to many steps, extremely low overall yield and in addition to US patent 6300353 claims even already known step as novel (claim 36).
Recent attempts to improve this concept as reported in WO 2007/062542 (Dec.1 , 2005), using less expensive, natural configured (S)-lactic acid, also failed: As already reported in US 6133485 and in US 2003/0236419, the second chiral center was formed from an optically active allyl alcohol prepared in a few steps from (S)-lactic acid. This allyl alcohol was subjected to Sharpless diastereoselective epoxidation providing first an opposite configured, epimeric epoxy alcohol which had to be then epimerized in an additional inversion step yielding finally the desired epoxy alcohol as the known precursor for Isavuconazole (US 6300353). It is obvious that this process using less expensive (S)- lactic acid makes the entire process with an inversion step even more complex than the original approach.
Elegant and more efficient process has been claimed in US 2004/0176432 from June 26, 2001 ) in which both chiral centers have been formed simultaneously, diastereo- and enantio-selectively pure in one single reaction step using chiral (R)-2-butynol as a chiral precursor in the presence of Pd(ll)-catalyst and diethyl zinc (Scheme 2).
Scheme 2:

Since water soluble, (R)-2-butynol is expensive, recently identical process has been published, in which instead of (R)-2-butynol less water soluble and therefore, less expensive (R)-4-phenyl-3-butyn-2-ol was used (Synthetic Commun. 39, 161 1 (2009)). Nevertheless, as incorrectly stated there, this process does not provide better diastereoselectivity than the original process using (R)-2-butynol: On the contrary disadvantage of this process is a very bad atom economy because huge phenyl group of (R)-4-phenyl-3-butyn-2-ol has to be “disposed” in oxidation step by the conversion of triple bond into carboxylic acid function.
……………………………
http://www.google.com/patents/WO2014023623A1?cl=en
The invention relates to a process for the manufacture of a
diastereomerically and enantiomerically enriched ester intermediate for isavuconazole or ravuconazole.
Isavuconazole and ravuconazole are triazole antifungal compounds. Processes for the manufacture of isavuconazole and ravuconazole were disclosed in patents WO99/45008, WO2007/062542 and WO03/002498 to Basilea. In WO2011/042827 a process for the manufacture of enantiomerically pure antifungal azoles such as ravuconazole and isavuconazole is disclosed, wherein a classical resolution of a racemic mixture is performed by the addition of an enantiopure chiral acid, then collection of the desired diastereomer followed by conversion of the salt into the enantiomerically pure form of the desired compound by treatment with a base or an ion-exchange resin. The disadvantages of using such classical resolution are that the chiral auxiliary needs to be applied in near stoichiometric amounts, and that additional process steps are required for recovery of these relatively high amounts of chiral reagent as well as for converting the salt into the free enantiopure product.
http://www.google.com/patents/US8076494
Reaction Scheme 1:

