

An efficient process for the preparation of 1-(2-methoxyphenoxy)-2,3-epoxypropane, a key intermediate for the synthesis of ranolazine is described.
Home » Posts tagged 'Ranolazine'
Tag Archives: Ranolazine
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Ranolazine Intermediate, An Efficient Synthesis of 1-(2-Methoxyphenoxy)-2,3-epoxypropane: Key Intermediate of β-Adrenoblockers

http://pubs.acs.org/doi/suppl/10.1021/op300056k
Preparation of 1-(2-Methoxyphenoxy)-2,3-epoxypropane 4.
To a stirring solution of 2-methoxy phenol 2 (10 kg, 80.55 mol) and water (40 L) at about 30 °C was added sodium hydroxide (1.61 kg, 40.25 mol) and water (10 L). After stirring for 30−45 min, epichlorohydrin 3 (22.35 kg, 241.62 mol) was added and stirred for 10−12 h at 25−35 °C. Layers were separated, and water (40 L) was added to the organic layer (bottom layer) containing product. Sodium hydroxide solution (3.22 kg, 80.5 mol) and water (10 L) were added at 27 °C and stirred for 5−6 h at 27 °C.
The bottom product layer was separated and washed with sodium hydroxide solution (3.0 kg 75 mol) and water (30 L). Excess epichlorohydrin (3) was recovered by distillation of the product layer at below 90 °C under vacuum (650−700 mmHg) to give 13.65 kg (94%) of title compound with 98.3% purity by HPLC, 0.2% of 2- methoxy phenol 2, 0.1% of epichlorohydrin 3, 0.1% of chlorohydrin 11, 0.3% of dimer 12 and 0.3% of dihydroxy 13.
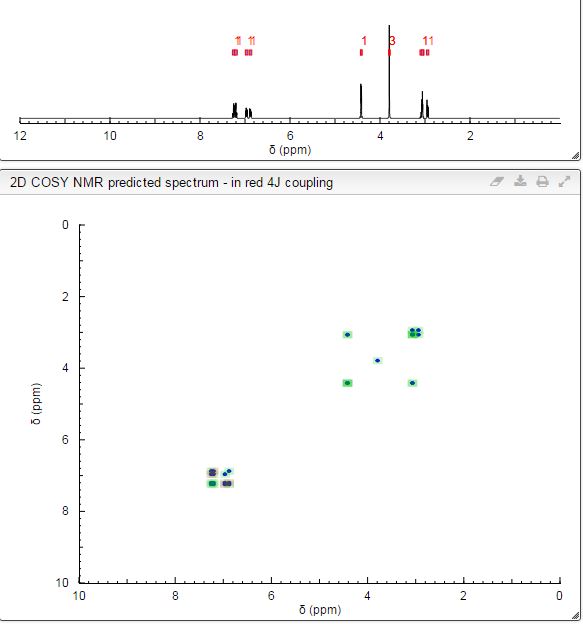
1 H NMR (400 MHz, CDCl3, δ) 6.8−7.0 (m, 4H), 4.3 (dd, J = 5.6 Hz, 5.4 Hz, 1H), 3.8 (dd, J = 5.6 Hz, 5.3 Hz, 1H), 3.7 (s, 3H), 3.2−3.4 (m, 1H), 2.8 (dd, J = 5.6 Hz, 5.4 Hz, 1H), 2.7 (dd, J = 5.6 Hz, 5.3 Hz, 1H);
IR (KBr, cm−1 ) 2935 (C−H, aliphatic), 1594 and 1509 (CC, aromatic), 1258 and 1231 (C−O−C, aralkyl ether), 1125 and 1025 (C−O−C, epoxide);
MS (m/z) 181 (M+ + H).
Compound Details
| Properties | |
| MWt | 180.2 |
| MF | C10H12O3 |
CAS 2210-74-4
| Glycidyl 2-methoxyphenyl ether | |
| Guaiacol glycidyl ether |
1H NMR PREDICT


13C NMR PREDICT



COSY PREDICT

CREDIT……….http://www.molbase.com/en/synthesis_2210-74-4-moldata-95563.html

DR REDDYS LABORATORIES
An Efficient Synthesis of 1-(2-Methoxyphenoxy)-2,3-epoxypropane: Key Intermediate of β-Adrenoblockers
† Innovation Plaza, IPD, R&D, Dr. Reddy’s Laboratories Ltd., Survey Nos. 42, 45,46, and 54, Bachupally, Qutubullapur – 500073, Andhra Pradesh, India
‡ Institute of Science and Technology, Center for Environmental Science, JNT University, Kukatpally, Hyderabad – 500 072, Andhra Pradesh, India
Org. Process Res. Dev., 2012, 16 (10), pp 1660–1664
DOI: 10.1021/op300056k
Publication Date (Web): September 14, 2012
Copyright © 2012 American Chemical Society
*Telephone: +91 4044346000. Fax: +91 4044346285. E-mail: rakeshwarb@drreddys.com.
////////////////1-(2-Methoxyphenoxy)-2,3-epoxypropane, β-Adrenoblockers, ranolazine
COc2ccccc2OCC1CO1
OTHER COMPD


Ranolazine, 雷诺嗪

Ranolazine
雷诺嗪
- MF C24H33N3O4
- MW 427.536
Approvals FDA 2006, EMA 2008 for chronic angina
Sponsor/Developer: Gilead
Mechanism of action: Late sodium current inhibitor
Indication (Phase): Type 2 diabetes (Phase III)
A Phase 3 Study of Ranolazine in Subjects With Type 2 Diabetes Who Are Not Well Controlled on Metformin Alone (currently recruiting participants as of August 2012, ClinicalTrials.gov Identifier: NCT01555164, see the link here)
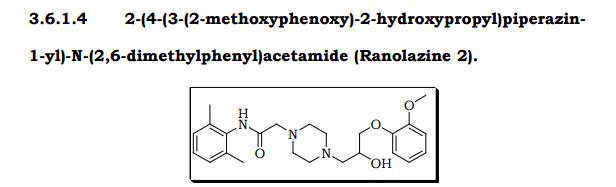
Chemical Name of Ranolazine: (RS)-N-(2,6-dimethylphenyl)-2-[4-[2-hydroxy-3-(2-methoxyphenoxy)-propyl]piperazin-1-yl]acetamide
N-(2,6-dimethylphenyl)-2-[4-[2-hydroxy-3-(2-methoxyphenoxy)propyl]piperazin-1-yl]acetamide
1-Piperazineacetamide, N-(2,6-dimethylphenyl)-4-[2-hydroxy-3-(2-methoxyphenoxy)propyl]-
CAS 95635-55-5 [RN]
QA-2943
Ranexa®
Ranexa, Ranolazine
Ranexa;CVT 303;RS 43285-003
| Solubility (25°C) * | In vitro | DMSO | 86 mg/mL (201.15 mM) |
|---|---|---|---|
| Ethanol | 20 mg/mL (46.77 mM) | ||
| Water | <1 mg/mL (<1 mM) | ||
| In vivo |
Clinical Trial Information( data from http://clinicaltrials.gov, updated on 2016-07-30)
| NCT Number | Recruitment | Conditions | Sponsor /Collaborators |
Start Date | Phases |
|---|---|---|---|---|---|
| NCT02829034 | Recruiting | Pulmonary Hypertension | University of Pennsylvania|Brigham and Womens Hospital|Un …more | July 2016 | — |
| NCT02817932 | Recruiting | Healthy Male Individuals | A.Menarini Asia-Pacific Holdings Pte Ltd | March 2016 | Phase 1 |
| NCT02687269 | Not yet recruiting | Myocardial Stunning | Policlinico Universitario Agostino Gemelli | March 2016 | Phase 4 |
| NCT02653833 | Recruiting | Muscular Dystrophy | Cedars-Sinai Medical Center | December 2015 | Phase 0 |
| NCT02611596 | Not yet recruiting | Silent Myocardial Ischemia|Type 2 Diabetes | Walter Reed National Military Medical Center | November 2015 | — |
view more CLICK
CLIP
Active Substance
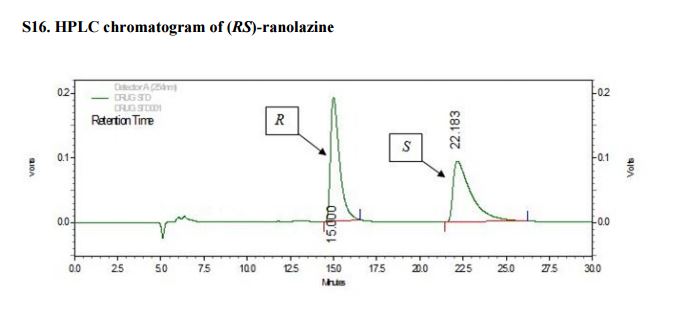
The chemical name of ranolazine is (±)-N-(2,6-dimethylphenyl)-4-[2-hydroxy-3-(2- methoxyphenoxy)propyl] piperazineacetamide. Ranolazine is a white to off-white solid, very slightly soluble in water. It is freely soluble in aqueous buffered solutions at pH levels below 4.4 and soluble in several organic solvents e.g. dichloromethane and methanol. The chemical structure is well characterised by means of elemental analysis, UV, IR, 1 H-NMR, 13C-NMR chemical ionization, electron impact mass spectra and x-ray diffraction. Ranolazine exhibits a chiral center and is obtained as a racemic mixture that consists of a 1:1 ratio of (R) and (S) enantiomers. This is confirmed by demonstrating that ranolazine does not exhibit any optical rotation of plane polarized light in polarimeter measurements. Both enantiomers exhibit pharmacological activity. Regarding polymorphism, crystallisation studies were conducted using different solvents, crystallization conditions and vapor diffusion experiments. In these studies three crystalline forms named as Form I, Form II, Form III and one amorphous form were identified. Form I is the only one that was thermodynamically stable, Form II and Form III are kinetically unstable. The synthetic process used for the synthesis of ranolazine has been shown to produce only Form I. Extreme conditions that are not relevant to the synthetic process are required to convert ranolazine to other solid-state forms (amorphous and two other crystalline forms, Form II and Form III)
Manufacture
Ranolazine is manufactured using a three step synthetic process followed by purification, drying and milling. The starting materials are 2,6-dimethylaniline (2,6-DMA), chloroacetyl chloride (CAC), piperazine dihydrochloride and guaiacol glicydil ether (GGE). The synthetic process has been adequately described the critical process parameters have been identified and are controlled with appropriate in-process controls. Data from four validation batches have been provided that demonstrate that the manufacturing process is capable to consistently produce batches of active substance that comply with the predefined specifications. A detailed discussion about potential impurities and their origin has been provided in line with ICH Guideline Q3A(R). Three specified impurities arising from the route of synthesis and one arising from the staring materials have been identified. There are also eight unspecified potential impurities.
Ranolazine, its enantiomers, and three metabolites (RS-88390, RS-89961, and RS-88772) were shown to have moderate affinity for α1A-and α1B-adrenergic receptors. Ranolazine, its S-enantiomer, and the same three metabolites had a similar affinity for β1-adrenergic receptors, with the R-enantiomer having no significant binding activity. The affinity of ranolazine for β2-adrenergic receptors was slightly lower, with the S-enantiomer and metabolites RS-88390 and RS-88772 having a similar affinity as the racemate. The metabolite RS-89961 had a higher affinity for β2-adrenergic receptors, whereas the R-enantiomer had no significant binding activity.
………CLICK FOR REFERNCE
also

Ranolazine HCl
N-(2,6-二甲基苯基)-4-[2-羟基-3-(2-甲氧苯氧基)丙基]-1-哌嗪乙酰胺盐酸盐
| CAS | 95635-56-6 |
|---|---|
| Molecular Formula | C24H35Cl2N3O4 |
| MW | 500.46 |
Ranolazine, developed by CV Therapeutics whom Gilead Sciences bought in 2009, is also sold under the trade name Ranexa for the treatment of chronic angina (chest pain).
Ranolazine, a partial fatty acid oxidation inhibitor available that is also a late sodium channel inhibitor as an oral extended-release tablet, has been developed and launched by Gilead Palo Alto (formerly CV Therapeutics; CVT), a wholly owned subsidiary of Gilead Sciences, under license from Roche Bioscience (formerly Syntex)
Ranolazine, sold under the trade name Ranexa by Gilead Sciences, is a drug to treat angina that was first approved in 2006.
Angina also known as Angina pectoris is indication for heart disease caused by lack of blood circulation to the heart. The most widespread reason for the angina is Atherosclerosis. In coronary heart disease patients, arteries become narrow and stiff when compared with the healthy heart arteries. These narrow and stiff arteries cause difficulties to reach oxygen rich blood for heart. About 17 million Americans are suffering with coronary heart diseases and about 9 millions are suffering with chronic angina. Ranolazine is the one of the medicament used to manage chronic angina, developed by Roche Bioscience (formerly Syntex) and marketed by CV Therapeutics. USFDA was approved Ranolazine 2 under brand name of Ranexa® in January 27, 2006. Subsequently European medical agency (EMEA) approved in July 09, 2008. Latter on it was approved in few other developing countries. Ranexa ® is available in market in the form of 500 mg and 1000 mg film coated tablet and the maximum daily dosage should be less than 2.0g. Over dosage of Ranexa ® lead to dizziness, nausea, and vomiting. Worldwide sales of Ranexa® by December 2011 is about 400 millions USD (~2000 crores) with the consumption of 1, 00, 678 kg. Major contribution is from USA i.e. about 300 millions USD. ……..CLICK
(5) (a) Kluge, A. F.; Clark, R. D.; Strosberg, A. M.; Pascal, J. G.; Whiting, R. United states patent, US 4,567,264, 1986. (b) Kluge, A. F.; Clark, R. D.; Strosberg, A. M.; Pascal, J. C.; Whiting, R. L. European patent, EP 0,126,449, 1987. (c) Kluge, A. F.; Clark, R. D.; Strosberg, A. M.; Pascal, J. C.; Whiting, R. Canadian patent, CA 1256874, 1987.

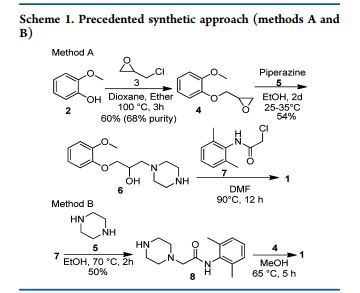
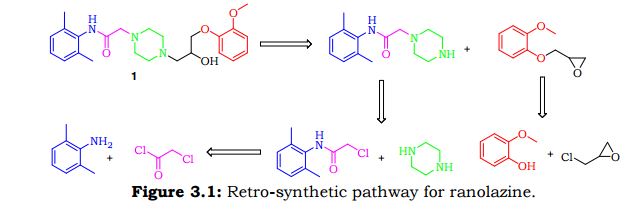
Amongst the various synthetic routes described for the preparation of Ranolazine, some of the key approaches are discussed here under. Kluge.F.A et al 5 have reported two synthetic approaches for preparation of Ranolazine 2 using commercially available 2-Methoxy phenol 25 and 2, 6-dimethyl aniline 20 as key starting materials. The first synthetic route commenced with the synthesis of methyl oxirane derivative 27. Key intermediate methyl oxirane derivative 27 was synthesized from 25 and epichlorohydrin 26 in presence of NaOH employing Williamson reaction conditions. Thus obtained 27 treated with piperazine 23 in ethanol to obtain hydroxyl piperazine derivative 33. Thereafter, reaction of hydroxyl piperazine derivative 33 with phenyl acetamide derivative 22 in dimethylformamide afforded dihydrochloride salt of ranolazine 2, which was treated with ammonia to furnish ranolazine 2(Scheme 3.1).

Second synthetic path way for the preparation of ranolazine involves the condensation of piperazinyl acetamide intermediate 24 and methyl oxirane 27 in mixture of methanol and toluene (Scheme 3.2).

Mingfieng.S et al reported7 similar approach for the synthesis of Ranolazine 2 utilizing hydroxy propyl halide intermediate 94 instead of methyl oxirane compound 27. The requisite hydroxy propyl halide intermediate 94 prepared by reacting 2-methoxy phenol 25 with 1, 3- dichloropropan-2-ol 93 in presence of NaOH and mixture of ethanol & water as shown in Scheme 3.3.
(7) Lisheng, W.; Xiaoyu, F.; Hong-yuan, Z. Journal of Guangxi University (Natural Science Edition), 2003, 28, 301-303.
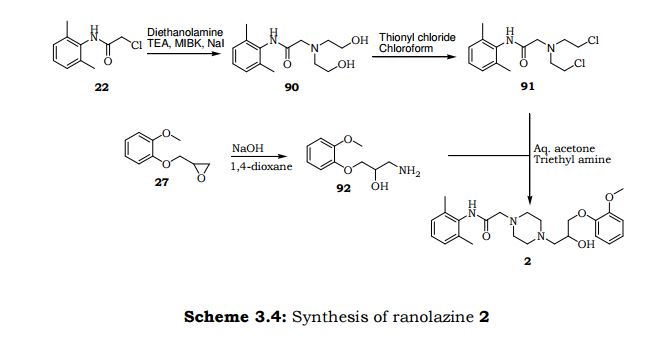
Eva.C.A et al.6 discovered an alternative synthetic path way for preparation of Ranolazine. As depicted in Scheme 3.3 reaction of phenyl acetamide derivative 22 with diethanolamine in presence of triethylamine and subsequent chlorination using thionyl chloride furnishes dichloro compound 91. Condensation of dichloro compound 91 with amino isopropanol derivative 92 provided Ranolazine 2. Amino isopropanol derivative 92 is achieved by reaction of methyl oxirane compound 27 with ammonia.
(6) Agai-Csongor, E.; Gizur, T.; Haranyl, K.; Trischler, F.; DemeterSzabo, A.; Csehi, A.; Vajda, E.; Szab-Koml si, G. European patent, EP 483932 A1, 1992.
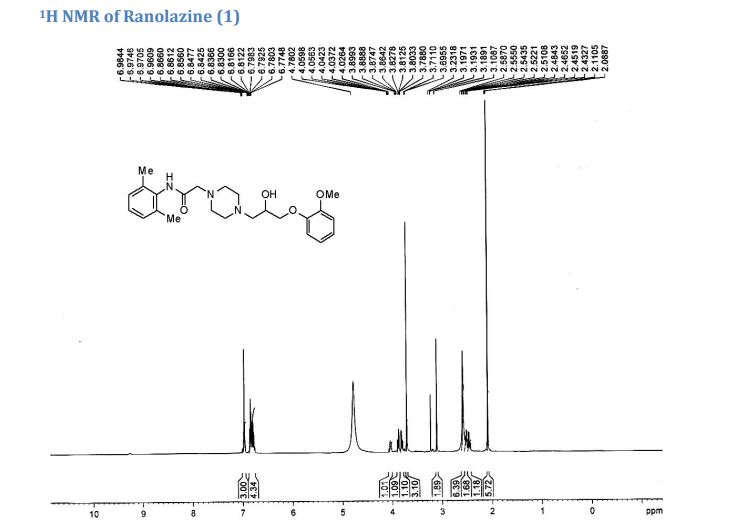
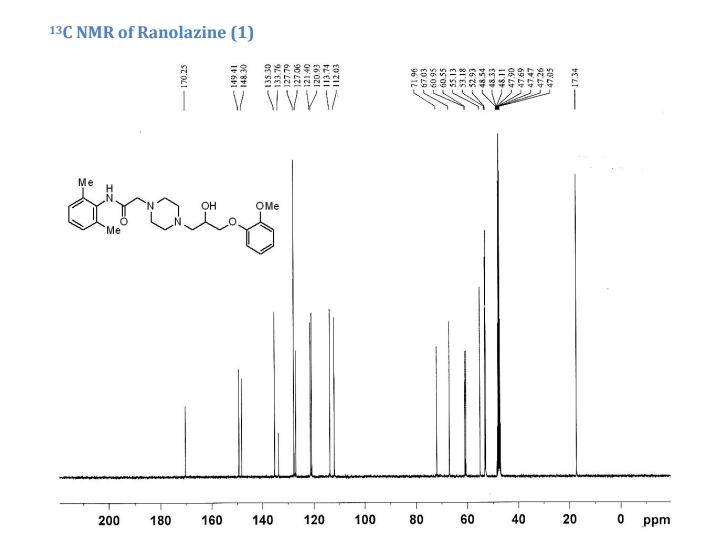
2 with 99.9% purity.
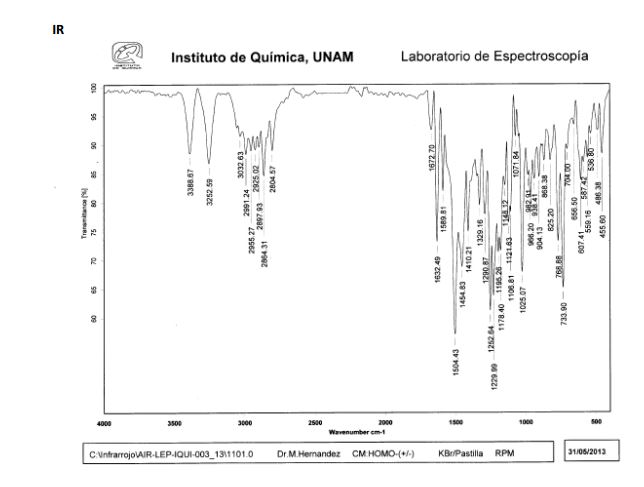
IR (KBr, cm–1): 3331 (Amine, NH), 3002 (Aromatic, =CH), 2955, 2936 and 2834 (Ali, CH), 1686 (Amide, C=O), 1592 and 1495 (Aromatic, C═C), 1254 and 1022 (Ether, C-O-C) & 1125 (C-N).
1H NMR (500 MHz, DMSO–d6): δH 9.1 (s, 1H, N-H), 6.8-7.1 (m, 6H, ArH), 4.8 (s, 1H, OH), 3.9 (s, 1H, CH), 3.8-3.9 (dd, 2H, J=6.5 Hz, 10.7 Hz, CH2), 3.8 (s, 3H, CH3), 3.1 (s, 2H, CH2), 2.4-2.6 (m, 10H, CH2) 2.1 (s, 6H, CH3).
13C NMR (500 MHz, DMSO–d6): 18.23, 39.16, 39.83, 39.50, 39.76, 39.87, 53.18, 53.31, 55.50, 61.13, 61.44, 66.63, 71.96, 112.37, 113.64, 120.74, 120.03, 126.32, 127.62, 134.97, 135.06, 148.36, 149.17, 167.97.
M/S (m/z): 428.4(M+ + H).
CHN analysis: Anal. Calcd for C24H33N3O4 (427.54): C 67.42, H 7.78, N 9.83.; Found: C 67.62 H 7.47, N 9.68.
Title: Ranolazine
CAS Registry Number: 95635-55-5
CAS Name: N-(2,6-Dimethylphenyl)-4-[2-hydroxy-3-(2-methoxyphenoxy)propyl]-1-piperazineacetamide
Additional Names: (±)-4-[2-hydroxy-3-(o-methoxyphenoxy)propyl]-1-piperazineaceto-2¢,6¢-xylidide; (±)-1-[3-(2-methoxyphenoxy)-2-hydroxypropyl]-4-[N-(2,6-dimethylphenyl)carbamoylmethyl]piperazine
Trademarks: Ranexa (CV Therapeutics)
Molecular Formula: C24H33N3O4
Molecular Weight: 427.54
Percent Composition: C 67.42%, H 7.78%, N 9.83%, O 14.97%
Literature References: Anti-ischemic agent which modulates myocardial metabolism. Prepn: A. F. Kluge et al., EP 126449;eidem, US 4567264 (1984, 1986 both to Syntex). HPLC resolution of enantiomers: E. Delée et al., Chromatographia 24, 357 (1987). Clinical trial in angina: B. R. Chaitman et al., J. Am. Coll. Cardiol. 43, 1375 (2004). Review of pharmacology and clinical development: J. G. McCormack et al., Gen. Pharmacol. 30, 639-645 (1998); R. S. Schofield, J. A. Hill, Expert Opin. Invest. Drugs11, 117-123 (2002).
Derivative Type: Dihydrochloride
CAS Registry Number: 95635-56-6
Manufacturers’ Codes: RS-43285
Molecular Formula: C24H33N3O4.2HCl
Molecular Weight: 500.46
Percent Composition: C 57.60%, H 7.05%, N 8.40%, O 12.79%, Cl 14.17%
Properties: White crystalline powder from methanol/ether, mp 164-166°. Readily sol in water.
Melting point: mp 164-166°
Therap-Cat: Antianginal.


Medical uses
Ranolazine is used to treat chronic angina.[1] It may be used concomitantly with β blockers, nitrates, calcium channel blockers,antiplatelet therapy, lipid-lowering therapy, ACE inhibitors, and angiotensin receptor blockers.[2]

Contraindications
Some contraindications for ranolazine are related to its metabolism and are described under Drug Interactions. Additionally, in clinical trials ranolazine slightly increased QT interval in some patients[3] and the FDA label contains a warning for doctors to beware of this effect in their patients.[2] The drug’s effect on the QT interval is increased in the setting of liver dysfunction; thus it is contraindicated in persons with mild to severe liver disease.[4]

Side effects
The most common side effects are dizziness (11.8%) and constipation (10.9%).[1] Other side effects include headache and nausea.[3]
Biological Activity
| Description | Ranolazine is a calcium uptake inhibitor via the sodium/calcium channel, used to treat chronic angina. | |||||
|---|---|---|---|---|---|---|
| Targets | Calcium channel [1] | |||||
| In vitro | Ranolazine is found to bind more tightly to the inactivated state than the resting state of the sodium channel underlying I(NaL), with apparent dissociation constants K(dr)=7.47 mM and K(di)=1.71 mM, respectively. Ranolazine at 5 mM and 10 mM reversibly shortens the duration of TCs and abolishes the after contraction.[1] Ranolazine inhibits the late component of INa and attenuates prolongation of action potential duration when late INa is increased, both in the absence and presence of IK-blocking drugs. Ranolazine (10 mM) reduces by 89% the 13.6-fold increase in variability of APD caused by 10 nM ATX-II. [2] | |||||
| In vivo | Ranolazine significantly and reversibly shortens the action potential duration (APD) of myocytes stimulated at either 0.5 or 0.25 Hz in a concentration-dependent manner in left ventricular myocytes of dogs. [1] Ranolazine (10 mM) significantly increases glucose oxidation 1.5-fold to 3-fold under conditions in which the contribution of glucose to overall ATP production is low (low Ca, high FA, with insulin), high (high Ca, low Fa, with pacing), or intermediate in working heart of rats. Ranolazine (10 mM) similarly increases glucose oxidation in normoxic Langendorff hearts (high Ca, low FA; 15 mL/min) of rats. Ranolazine significantly improves functional outcome in reperfused ischemic working hearts, which is associated with significant increases in glucose oxidation. [3] | |||||
| Features | ||||||
Conversion of different model animals based on BSA (Value based on data from FDA Draft Guidelines)
| Species | Mouse | Rat | Rabbit | Guinea pig | Hamster | Dog |
| Weight (kg) | 0.02 | 0.15 | 1.8 | 0.4 | 0.08 | 10 |
| Body Surface Area (m2) | 0.007 | 0.025 | 0.15 | 0.05 | 0.02 | 0.5 |
| Km factor | 3 | 6 | 12 | 8 | 5 | 20 |
| Animal A (mg/kg) = Animal B (mg/kg) multiplied by | Animal B Km |
| Animal A Km |
For example, to modify the dose of resveratrol used for a mouse (22.4 mg/kg) to a dose based on the BSA for a rat, multiply 22.4 mg/kg by the Km factor for a mouse and then divide by the Km factor for a rat. This calculation results in a rat equivalent dose for resveratrol of 11.2 mg/kg.
| Rat dose (mg/kg) = mouse dose (22.4 mg/kg) × | mouse Km(3) | = 11.2 mg/kg |
| rat Km(6) |
References
[1] Undrovinas AI, et al. J Cardiovasc Electrophysiol,?006, 17 Suppl 1, S169-S177.
[2] Song Y, et al. J Cardiovasc Pharmacol,?004, 44(2), 192-199.3]
3 Baptista T, et al. Circulation,?996, 93(1), 135-142.
Drug interactions
Ranolazine is metabolized mainly by the CYP3A enzyme. It also inhibits another metabolizing enzyme, cytochrome CYP2D6.[2] For this reason, the doses of ranolazine and drugs that interact with those enzymes need to be adjusted when they are used by the same patient.
Ranolazine should not be used with drugs like ketoconazole, clarithromycin, and nelfinavir that strongly inhibit CYP3A nor with drugs that activate CYP3A like rifampin and phenobarbital.[2]
For drugs that are moderate CYP3A inhibitors like diltiazem, verapamil, erythromycin, the dose of ranolazine should be reduced.[2]
Drugs that are metabolized by CYP2D6 like tricyclic antidepressants may need to be given at reduced doses when administered with ranolazine.[2]
Mechanism of action
Ranolazine inhibits persistent or late inward sodium current (INa) in heart muscle[5] in a variety of voltage-gated sodium channels.[6] Inhibiting that current leads to reductions in elevated intracellular calcium levels. This in turn leads to reduced tension in the heart wall, leading to reduced oxygen requirements for the muscle.[3] The QT prolongation effect of ranolazine on the surface electrocardiogram is the result of inhibition of IKr, which prolongs the ventricular action potential.[2]
Legal status
Ranolazine was approved by the FDA in January 2006, for the treatment of patients with chronic angina as a second-line treatment in addition to other drugs.[3] In 2007 the label was updated to make ranolazine a first-line treatment, alone or with other drugs.[3] In April 2008 ranolazine was approved by the European EMEA for use in angina.[7]
History
In 1996, CV Therapeutics licensed the North American and European rights to ranolazine from Syntex, a subsidiary of Roche, which had discovered the drug and had developed it through Phase II trials in angina.[8] In 2006, CV Therapeutics acquired the remaining worldwide rights to ranolazine from Roche.[9] In 2008 CV Therapeutics exclusively licensed rights for ranolazine in Europe and some other countries to Menarini.[10] In 2009, Gilead acquired CV Therapeutics.[11] In 2013 Gilead expanded the partnership with Menarini to include additional countries, including those in Asia.[12]
Ranolazine (CAS NO.: 95635-55-5), with its systematic name of 1-Piperazineacetamide, N-(2,6-dimethylphenyl)-4-(2-hydroxy-3-(2-methoxyphenoxy)propyl)-, could be produced through many synthetic methods.
Following is one of the synthesis routes:
The acylation of 2,6-dimethylaniline (II) with chloroacetyl chloride in the presence of triethylamine in dichloromethane affords N-(2,6-dimethylphenyl) chloroacetamide (III), which is condensed with piperidine (IV) in refluxing ethanol to yield N-(2,6-dimethylphenyl)-2-piperazinoacetamide IV). At last, this compound is condensed with 3-(2-methoxyphenoxy)-12-epoxypropane (VI) in refluxing methanol toluene.

CLIP
Paper
*Corresponding authors
aDepartment of Medicinal Chemistry, National Institute of Pharmaceutical Education and Research (NIPER), Sector 67, S. A. S. Nagar 160 062, Punjab, India
E-mail: akchakraborti@niper.ac.in,akchakraborti@rediffmail.com
E-mail: akchakraborti@niper.ac.in,akchakraborti@rediffmail.com
Green Chem., 2013,15, 756-767
DOI: 10.1039/C3GC36997H
A novel strategy of ‘all water chemistry’ is reported for a concise total synthesis of the novel class anti-anginal drug ranolazine in its racemic (RS) and enantiopure [(R) and (S)] forms. The reactions at the crucial stages of the synthesis are promoted by water and led to the development of new water-assisted chemistries for (i) catalyst/base-free N-acylation of amine with acyl anhydride, (ii) base-free N-acylation of amine with acyl chloride, (iii) catalyst/base-free one-pot tandem N-alkylation and N-Boc deprotection, and (iv) base-free selective mono-alkylation of diamine (e.g., piperazine). The distinct advantages in performing the reactions in water have been demonstrated by performing the respective reactions in organic solvents that led to inferior results and the beneficial effect of water is attributed to the synergistic electrophile and nucleophile dual activation role of water. The new ‘all water’ strategy offers two green processes for the total synthesis of ranolazine in two and three steps with 77 and 69% overall yields, respectively, and which are devoid of the formation of the impurities that are generally associated with the preparation of ranolazine following the reported processes.

Damodara Naidu Kommi
Prof. Asit K. Chakraborti








PATENT
https://www.google.com/patents/US20130090475
Ranolazine, chemically known as (±)-N-(2,6-dimethylplenyl)-4-[2-hydroxy-3-(2-methoxyphenoxy)propyl]-1-piperazineacetamide, is represented by the formula as given below.

Ranolazine, a novel agent used to treat angina pectoris type coronary heart disease, was developed by American CV Therapeutica Company (now known as Gilead Sciences Company). Ranolazine has firstly been appeared on the market in US in 2006 and could be used to treat myocardial infarction, congestive heart disease, angina and arhythmia etc. The mechanism of action of ranolazine is to inhibit partial fatty acid oxidation, which changes fatty acid oxidation to glucose oxidation in heart, and thereby reduces the cardiac oxygen consumption. Ranolazine is the only antianginal agent without changing heart rate or blood pressure.
The processses for the preparation of ranolazine, which could be roughly divided into two types as shown in FIG. 1 and FIG. 2, were disclosed in International Application Publication No. WO 2010/025370, WO 2010/023687, WO 2009/153651, WO 2008/139492, WO 2008/047388, WO 2006/008753, Chinese patent No. CN101560196, CN101544617, CN1915982, the United States patent No. US2008312247, the publication China Pharmacist, 2007, 10(12), 1176-1177, Chinese Journal of Medicinal Chemistry, 2003, 13(5), 283-285, and Chinese Journal of Pharmaceuticals, 2004, 35(11): 641-642.
The process described in FIG. 1 (method 1) involves reacting [(2,6-dimethylphenyl)-carbamylmethyl]-peperazine with 1-(2-methoxyphenoxy)-2,3-epoxypropane to obtain ranolazine, in which comprises the steps of:
a) condensing 2,6-xylidine with chloroacetyl chloride in the presence of base to get amide, which is further reacted with piperazine by a substitution reaction of N-monoalkylation to get N-(2,6-dimethylphenyl)-1-piperazineacetamide, and
b) condensing guaiacol with epoxy chloropropane to get 1-(2-methoxyphenoxy)-2,3-epoxypropane.
As the condensation is carried out in the alkaline environment, the epoxy ring becomes easy to open loop, and thus the products comprise mixtures of open-looped and looped form, thereby requiring rigorous separation conditions and being difficult to achieve the desired purity in the following reaction.
The process described in FIG. 2 (method 2) involves reacting 2-chloro-N-(2,6-dimethylphenyl)-acetamide with 1-(2-methoxyphenoxy)-3-(N-piperazine)-2-hydroxypropane to get ranolazine, in which comprises the steps of:
a) condensing 2,6-xylidine with chloroacetyl chloride in the presence of base to get 2-chloro-N-(2,6-dimethylphenyl)-acetamide, and
b) condensing guaiacol with epoxy chloropropane to get 1-(2-methoxyphenoxy)-2,3-epoxypropane, which is further reacted with piperazine to get 1-(2-methoxyphenoxy)-3-(N-piperazine)-2-hydroxypropane.
As the condensation is carried out in the alkaline environment, the epoxy ring becomes easy to open loop, and thus the products comprise mixtures of open-looped and looped form, thereby requiring rigorous separation conditions and being difficult to achieve the desired purity in the following reaction. The monosubstitution reaction of N-alkylation reacted with peperazine is further difficult to be controlled to produce the desired products.
Compared with method 2, method 1 could be easier to be industrialized as the quality of intermediates obtained by method 1 could be easier to be controlled and also the method 1 could be easier to be operated. But in the repeated experiments, it was found that it still had a lot of difficulties in realizing the industrialization by method 1 although it could be easier to be operated as there are mixtures including open-looped and looped products rather than single product produced when guaiacol (o-methoxyphenol) was reacted with epoxy chloropropane, so the operation of distillatory separation would still need very high temperature (above 250° C.) and very low vacuum degree (5 mm Hg) with the disadvantages of high energy consumption, high facilities investment and tedious operation. And in the following condensation reaction, there are a lot of products were produced during the reaction so as to make the quality of the products hard to be controlled.


Example 1Preparation of N-(2,6-dimethylphenyl)-1-piperazinylacetamide1.1: Preparation of 2-chloro-N-(2,6-dimethylphenyl)-acetamid

30.5 g (0.252 mol) of 2,6-xylidine, 100 ml of ethyl acetate, 26.5 g (0.25 mol) of sodium carbonate were successively added into a 250 ml of 3-neck flask and placed in an ice-water bath. 36.5 g (0.323 mol) of chloroacetyl chloride was dissolved in 50 ml of ethyl acetate and then the mixture was dropwise added into the 3-neck flask till completion. The ice-water bath was removed and the reaction was carried out for 3 h at the room temperature. The reaction product was slowly added 100 ml of water in an ice-water bath, stirred for 10 min and filtered. The filter cake as white needle solid was washed and dried under vacuum to get 46.3 g of 2-chloro-N-(2,6-dimethylphenyl)-acetamide having a yield of 93%
1.2: Preparation of N-(2,6-dimethylphenyl)-1-piperazinylacetamide

58.3 g (0.3 mol) of piperazine hexahydrate was dissolved in 230 ml of ethanol and 50.0 g (0.25 mol) of 2-chloro-N-(2,6-dimethylphenyl)-acetamide was subsquently added. The mixture was heated under reflux for 3 h till completion. The reaction product was cooled to room temperature and filtered. The filter was concentrated under reduced pressure and 80 ml of water was added. The mixture was extracted with dichloromethane and the organic layer was concentrated under vacuum at 60° C. to get 39.4 g of N-(2,6-dimethylphenyl)-1-piperazinylacetamide having a yield of 63%. 1HNMR (CDCl3): 2.23˜2.27,s, 6H, 2.67,s, 4H, 2.96˜2.98,t, 4H, 3.19˜3.21,s, 2H, 7.08˜7.26,m, 3H, 8.69,s, 1H.
Example 2Preparation of Ring-Opening Halide2.1: Preparation of 1-chloro-3-(2-methoxyphenoxy)-2-propylalcohol

26 g (0.65 mol) of sodium hydroxide, 150 ml of water, 150 ml of ethanol, 62 g (0.5 g) of guaiacol were successively added into a reaction flask and 103 g (0.8 mol) of 1,3-dichloro-2-propylalcohol was slowly dropwise added till completion. The mixture was heated up to 45° C. for 24 h. The reaction product was extracted three times with 150 ml of dichloromethane each and the organic layer was combined, dried with anhydrous magnesium chloride and distilled under reduced pressure. The fraction at 160° C. and a pressure of 2 kp was collected to get 73.6 g of faint yellow liquid having a yield of 68%. 1HNMR (CDCl3): 3.44˜3.46,d, 1H, 3.69-3.78,dd, 2H, 3.85,s, 3H, 4.11˜4.12,d, 2H; 4.18˜4.22 μm, 1H, 6.89˜7.00,m, 4H. The result confirmed that the yellow liquid was 1-chloro-3-(2-methoxyphenoxy)-2-propylalcohol.
2.2: Preparation of 1-bromo-3-(2-methoxyphenoxy)-2-propylalcohol

26 g (0.65 mol) of sodium hydroxide, 150 ml of water, 150 ml of ethanol, 62 g (0.5 g) of guaiacol were successively added into a reaction flask and 174.4 g (0.8 mol) of 1,3-dibromo-2-propylalcohol was slowly dropwise added till completion. The mixture was heated up to 45° C. for 10 h. The reaction product was extracted three times with 150 ml of dichloromethane each and the organic layer was combined, dried with anhydrous magnesium chloride and distilled under reduced pressure. The fraction at 160° C. and a pressure of 2 kp was collected to get 103 g of faint yellow liquid of 1-bromo-3-(2-methoxyphenoxy)-2-propylalcohol having a yield of 79%.
Example 3Preparation of Ranolazine3.1: 1-chloro-3-(2-methoxyphenoxy)-2-propylalcohol as a raw material

2.5 g (0.01 mol) of 1-chloro-3-(2-methoxyphenoxy)-2-propylalcohol, 3.1 g (0.012 mol) of N-(2,6-dimethylphenyl)-1-piperazinylacetamide, 4.1 g (0.03 mol) of potassium carbonate, 25 ml of methanol and 50 ml of toluene were successively added into a reaction flask and heated under reflux for 4.5 h till completion.
The fraction whose main ingredient was methanol was collected by atmospheric distillation at boiling point of 62-68° C. and then filtrated. The filtrate was washed with 3N HCl to get 50 ml of liquid having a pH of 1-2 and further treated with 50 ml of saturated sodium carbonate solution to adjust pH to 9-10. The product was extracted three times with 20 ml of dichloromethane each and the lower organic phase was combined. After the dichloromethane was removed by distillation under reduced pressure and rotary evaporation, the yellow viscous liquid was obtained and then further dissolved in about 10 ml of methonal. The tetrahydrofuran was then dropwise added under reflux till turbidity. The product was slowly crystallized with cooling and filtrated to get 3.42 g of white solid having a yield of 80.1% by vacuum drying at 40° C
1HNMR (CDCl3): 2.22,s, 6H, 2.60˜2.62,t, 4H, 2.75,s, 6H, 3.21,s, 2H, 3.45,s, 3H; 3.85,s, 3H, 4.02˜4.04,t, 2H, 4.16,s, 1H, 6.88˜6.90,t, 2H, 6.91˜6.96,m, 2H, 7.08˜7.1,m, 3H, 8.65,s, 1H. The result confirmed that the compound obtained is ranolazine. Purity by HPLC (area normalization method): 99.1%.
PATENT
https://www.google.com/patents/CN102875490A?cl=zh
Ranolazine piperazine derivatives, chemical name: (±) -1- [3- (2_ methoxyphenoxy) -2_-hydroxypropyl] -4- [N- (2, 6- dimethylphenyl) carbamoylmethyl] piperazine. Ranolazine is a novel antianginal drugs, which can provide metabolic myocardial protection at the cellular level by improving myocardial energy, while heart rate, blood pressure and hemodynamic impact, has a good prospect. [0004] Currently, the literature synthetic routes ranolazine can be grouped into three: a route: literature (Wolff HeartFailure Reviews, 2002,7 (2): 187- 203.) Using 2_ [N, N- two – ( 2-chloroethyl) amino] -2,6-dimethyl-acetanilide and 3- (2-methoxyphenoxy) -2-propanol of the -I-, amino cyclization to synthesize the desired product. The advantage of this method is to avoid the use of large amounts of piperazine, but the drawback is six steps required to complete the reaction step is long, the total yield is low, is not applicable to industrial production. Route II: literature (US, 4567264; LI Shu-chun Chinese Journal of Medicinal Chemistry, 2003, 13 (5): 283-285) piperazine used directly as the raw material, the advantage of a four-step reaction process is shorter, but due to the direct use of piperazine N- (2,6- dimethylphenyl) -2-chloro – acetamide (2) the reaction, in order to avoid generating disubstituted compound and increased the yield dropping proportion piperazine, piperazine need to consume a large amount. Route III: Document (Qin Mingli, Xinyang Normal University, 2007,20 (2): 226-229) synthetic routes and route only difference is that two different priorities on the piperazine ring substituted on. After two routes have two places noteworthy: how to avoid the generation of disubstituted compounds and the compound (4), (it) is purified.

Synthesis of Compound (3)
In the synthesis of the compound (3), since piperazine simultaneous introduction of two groups, by changing the reaction conditions, to seek optimal reaction molar ratio, in order to optimize the synthesis process, to improve the yield. Since the formation of crystalline anhydrous piperazine water easily precipitated in the solvent methylene chloride, anhydrous conditions so the need to control and make the feed ratio of I: 2 Avoid disubstituted product formation. Methanol can also be used as solvent to avoid precipitation of piperazine, and generates less disubstituted, but did not significantly increase yield (61.5%), it is still producing less toxic with methylene chloride as the solvent, control anhydrous conditions. Removed by filtration and the compound (3) excess piperazine, after the solvent is evaporated, dissolved in water, filtered off disubstituted extracted with methylene chloride, in high purity in the latter studies, may be mono-substituted piperazine as the raw material, and then and then removing the protecting group, thereby avoiding the generation of double substitution also improves the yield.
Synthesis [0008] Compound (5)
When the use of trifluoroacetic acid deprotection, since the compound (4) itself has two salt-forming groups, so the need to increase the TFA feeding, paper, compound (4): trifluoroacetic acid = 1: 6 feeding, the reaction was stirred at room temperature for two hours after the end, and then try to solvent evaporated to dryness, a small amount of ethyl acetate was added and then repeatedly evaporated with divisible trifluoroacetic acid. Finally ethanol: petroleum ether = 1: 1.4 was recrystallized to give compound (5).
Synthesis [0009] Compound (I),
Document (Mcaroon, J Med Chem, 1981,24 (11): 1320- 1328) with methanol – toluene system, literature (US, 4567264) with DMF system. Considering the safety, environmental protection, price, cost, industrial production and other factors, we use isopropyl alcohol as a solvent. In this step, less side reaction byproducts concentrated in raw materials, in strict accordance with the reaction so after molar ratio, TLC detection, should be enough to make up the raw materials, to minimize raw material residues, reducing the difficulty of recrystallization.
[0010]
Specific implementation methods
Synthesis below with embodiments of the present invention will be further described in Example a N- (2,6- dimethylphenyl) -2-chloroacetamide (2)
In 3000ml three-neck flask, into 2,6-dimethylaniline (45. 53 g, 0. 375 mol), toluene (750 ml), sodium carbonate (39. 75g, 0. 375 mol), water (750 ml ), with vigorous stirring slowly added dropwise chloroacetyl chloride (50. 90 g, 0. 45mol), temperature 20~35 ° C (ice water bath). During the reaction, TLC detection reaction process. After completion of the reaction, ice-water bath cooling and crystallization, filtration, washed with toluene, recrystallized from 50% ethanol to give the compound (2), white needles (64. 53g, yield of 86. 9%, mp: 148 ~149 ° C).
Synthesis Example Two N-BOC’s [0011] implementation
In three 250ml flask inputs piperazine (3. 07g, 0. 0356mol), dichloromethane 50ml, piperazine with vigorous stirring to dissolve. Was slowly added dropwise while piperazine (2. 99g, 0. 0347mol dissolved in 50ml of methylene chloride), a BOC anhydride (7. 30g, was dissolved in 50ml of methylene chloride), temperature (Γ 5 ° C. After the addition was complete, the reaction was stirred overnight .TLC detection process. after completion of the reaction, a white solid was filtered off. the filtrate was concentrated, dissolved in water IOOml, a white solid was filtered off. the filtrate with dichloromethane (50ml X3 times). the organic layer was dried over anhydrous sodium sulfate , the drying agent was removed by filtration and the filtrate evaporated to give the compound (3), white needle crystals 4. 07g, yield 65. 3%, 1H-NMR (CDCL3):.. 3. 75 (s, 4H), 2 86 ~2. 91 (m, 4H,), I. 99 Cs, 1H), I. 45 (s, 9H).
[0012] Example (2,6-dimethylphenyl) Synthesis of (N-B0C piperazinyl) acetamide (4) of the three N- -1-.
[0013] In 150ml three-necked flask was added N-BOC piperazine (3) (5. 40g, O. 0289mol), the compound (2) (5. 71g, 0.0289mo, potassium carbonate (4. OOgO. 0202mol) in dry ethanol 10ml, was heated 4h, TLC detection progress of the reaction. after completion of the reaction, water was added 10ml, extracted with ethyl acetate (30mlX2). The organic layer was dried over anhydrous sodium sulfate, filtered off and the filtrate was concentrated and dried U. homogeneous, with ethyl .: petroleum ether = 1: 32 recrystallized compound (4) (white solid, 8 Olg, yield 79. 6%, mp: 119~120 ° C; 1H-NMR3 (s, 7. 09, 3H, Ar-H), 3. 50 (q, 4H, J = 4. 8), 3. 22 (s, 2H), 2.64 (q, 4H, J = 4.8), 2. 23 (s, 6H, 2 X CH3), 1.611 (s, 9H, 3X CH3);.. 13CNMR (167.95,154.43,134.78,133.35,128.14,127.08,79.83,61.65,53.40,43.37,26 24,18 47).
[0014] Fourth Embodiment N- (2,6-dimethylphenyl) -1-piperazine acetamide put in 50ml round bottom flask N- (2,6-dimethylphenyl) -1 – (N-BOC piperazine) -acetamide (4) (. 4 30g, O. 121mol), trifluoroacetic acid (8. 24g, 0 0722mol.), ethyl acetate 6ml, was stirred at room temperature under reflux for 2h, TLC detection reaction process . After completion of the reaction, the solvent evaporated to dryness to give a white solid. With ethanol: petroleum ether = 1: 14 recrystallized compound
(5), a white powder (2. 82g, yield 92. 5%, mp:. 130~131 .., 1H-NMR3 9. 573 (s, IN-H), 9 043 (s, 2XN- H), 7 · 187~7. 087 (t, 3X Ar-H), 3. 66 (s, 4H), 3. 27 (s, 2H), 3. 07 (s, 4Η) ^ _
2. 142 (s, 6Η, 2 X CH3).
Four cases of ranolazine dihydrochloride (I) Synthesis of [0015] implementation
In three 150ml round bottom flask was added the compound (3) (5. OOg, O. 02mol), isopropyl alcohol (35. Oml), was slowly added dropwise at the reflux temperature of the compound (5) (4. 14g, 0. 023mol ), continued under reflux conditions I. 5h, TLC detection progress of the reaction, the reaction was complete, cooled and added to the reactor 9. Oml 12mol / L of concentrated hydrochloric acid solution was adjusted to pH 2 and concentrated to near dryness to give bright yellow brown liquid, repeatedly adding ethanol, rotary evaporation to a white solid. Absolute ethanol and recrystallized to give compound (the I), as a white solid (6. 80g, yield 78. 7%, mp: 217 ~219 ° C (Dec) j1H-NMR (DMS0-d6): 10. 17 ( s, 1H, -CONH-), 7.21 ~6.87 (m, 7H, Ar-H), 4. 42 (m, 1H, -OCH2CHCH2-), 4. 23 (s, 2H, -CH2N), 4. 00 ~3.92 (m, 2H, -OCH2CHCH2), 3. 77 (s, 3H, -OCH3), 2. 67~2. 50 (m, 8H, 2 X -NCH2CH2N-), 2. 33 ~I. 91 ( m, 2H, -OCH2CHCH2), 2. 17 (s, 6H, 2 X CH3); MS (m / e): 427. 54).
CLIP

An in silico modelling based biocatalytic approach for the synthesis of drugs and drug intermediates in enantiopure forms is a rationalized methodology over the organo-chemical routes. In this study, enzyme-ligand based docking was carried out using (RS)-ranolazine, as the model drug for the screening of a suitable biocatalyst for the kinetic resolution of the racemic drug. The differential interaction of the two enantiomers with the lipase was analyzed on the basis of docking score and H-bond interaction with the amino acid residues, which helped to define the trans-esterification mechanism. Ranolazine [N-(2,6-dimethylphenyl)-2-[4-(2-hydroxy)-3-(2-methoxyphenoxy)propylpiperazin-1-yl]acetamide], an anti-anginal drug, significantly reduces the frequency of anginal attack and has also been used for the treatment of ventricular arrhythmias, and bradycardia. Various lipases were examined via computational as well as wet lab screening and Candida antartica lipase in the form of CLEA was the most efficient one for the (S)-selective kinetic resolution of (RS)-ranolazine, with highest conversion and enantiomeric excess. This is the first report of the chemo-enzymatic synthesis of (S)-ranolazine where the whole drug molecule was used for lipase catalysis. The present study showed that the combination of in silico studies and a classical wet lab approach could change the paradigm of biocatalysis.



In silico approach towards lipase mediated chemoenzymatic synthesis of (S)-ranolazine, as an anti-anginal drug
*
Corresponding authors
a
Department of Pharmaceutical Technology (Biotechnology), National Institute of Pharmaceutical Education and Research, Sec-67, S. A. S. Nagar-160062, India
E-mail: ucbanerjee@niper.ac.in
E-mail: ucbanerjee@niper.ac.in
RSC Adv., 2016,6, 49150-49157
DOI: 10.1039/C6RA06879K


CLIP

![Image result for Synthesis of Ranolazine Derivatives Containing the (1S,4S)-2,5-Diazabicyclo[2.2.1]Heptane Moiety and Their Evaluation as Vasodilating Agents](https://www.researchgate.net/profile/Patricia_Demare2/publication/259824588/viewer/AS:183135694237696@1420674360238/background/2.png)


OTHER NMR…….http://onlinelibrary.wiley.com/store/10.1111/cbdd.12285/asset/supinfo/cbdd12285-sup-0001-SupplementaryData.pdf?v=1&s=1c11a72432d0627b201f1bd37dab7ef913b0ff1f
OF Epimer (S,S,S)-5, Epimer (S,S,R)-5
PATENT
Ranolazine is marketed under the brand name Ranexa® and is indicated for the treatment of chronic angina. Ranexa may be used with beta-blockers, nitrates, calcium channel blockers, anti-platelet therapy, lipid-lowering therapy, ACE inhibitors, and angiotensin receptor blockers. Ranolazine is a racemic mixture, chemically described as 1-piperazineacetamide, N-(2, 6-dimethylphenyl)-4-[2-hydroxy-3-(2-methoxyphenoxy) propyl]-, (±)- indicated by compound of formula (1).

(1)
U.S. Patent No. 4,567,264 teaches two methods for the preparation process of Ranolazine. Method 1 disclosed reaction of 2-methoxyphenol compound of formula (2) with epichlorohydrin in presence of water, dioxane and NaOH to obtain l-(2-methoxyphenoxy)-2, 3-epoxypropane compound of formula (3) which is condensed with piperazine in presence of ethanol to obtain 2-(2-methoxyphenoxy)-l-(piperazin-l-yl) ethanol compound of formula (4). Reacting 2, 6-Dimethylaniline compound of formula (5) with chloroacetyl chloride in presence of TEA and MDC to obtain 2-chloro-N-(2,6-dimethylphenyl) acetamide compound of formula (6). Compound of formula (4) was condensed with compound of formula (6) in presence of dimethylformamide to obtain Ranolazine compound of formula (1). The method (1) is depicted below as scheme (I).

Scheme (I) (1)
US ‘264 taught another method for preparation of Ranolazine by condensing compound of formula (6) with piperazine in presence of ethanol to obtain N-(2, 6-dimethylphenyl)-2-(piperazin-l-yl) acetamide compound of formula (7). Compound of formula (3) was condensed with compound of formula (7) in presence of mixture of methanol and toluene at reflux temperature. The obtained Ranolazine is purified by column chromatography on silica gel. Excess of hydrochloric acid in methanol was added to get dihydrochloride salt of Ranolazine which was converted into its free base by suspending it in ether and stirred with excess of dilute aqueous potassium carbonate to get Ranolazine free base. The scheme is depicted below by Scheme (II).



Scheme (II) (!)
EP0483932A1 disclosed condensation of condensation of N, N-bis (2-chloro ethyl)-amino]-2,6-dimethyl acetanilide compound of formula (9) with l-[3-(2-methoxyphenoxy)-2-hydroxy]propylamine compound of formula (8) to obtain Ranolazine base. The base was purified by column chromatography; hydrochloride salt was formed by treating with methanolic HCI. The detailed impurity profile study was not reported for Ranolazine. The synthetic scheme is depicted below in scheme (III).

Chinese patent application No.102875490 disclosed condensation of compound of formula (6) with N-Boc-piperazine to obtain compound of formula (10) in the presence of K2CO3 in EtOH, removal of Boc group by means of TFA in EtOAc gives compound of formula (7) which is then converted into Ranolazine. The synthetic scheme is depicted below in scheme (IV).


Scheme (IV)
Organic Process Research & Development 2012, 16, 748-754 disclosed condensation of compound of formula (6) with piperazine in methanol to produce compound of formula (7), in which unwanted solid bis alkylated compound of formula (11) was filtered. The resulting filtrate pH adjusted to 5.0-5.5 with 44% phosphoric acid solution to recover piperazine monophosphate monohydrate salt. The compound of formula (7) was extracted with MDC.
PCT application No. 2008/047388 disclosed a process for the preparation Ranolazine, by reacting 2, 6-dimethyl aniline with Chloroacetyl chloride in the presence of base in water. The resulting amide intermediate is reacted with piperazine, and the resulting piperazine derivative is further condensed with l-(2-methoxyphenoxy)-2,3-epoxypropane in an inert solvent to produce crude Ranolazine, which is further purified by crystallizing from organic solvents selected from alcohols or aromatic hydrocarbons. Ranolazine obtained in the disclosed art does not have satisfactory purity for pharmaceutical use. Unacceptable amounts of impurities are generally formed along with Ranolazine. In addition, the processes involve the additional step of column chromatographic purifications, which are generally undesirable for large-scale operations.
As described above the cited literature processes suffer from many drawbacks like use of excess amount of piperazine during the reaction, which is difficult to handle in large scale; generation of large amount of effluent due to excessive use of piperazine, that is difficult to recover and recycle; Ranolazine obtained as an oil is difficult to handle in large scale production and laborious chromatographic
techniques are used for purification of Ranolazine.
It is observed that pharmaceutically acceptable salts of Ranolazine when prepared from impure Ranolazine do not meet the pharmaceutical acceptable quality. There is therefore, an unfulfilled need to provide industrially feasible process for the preparation of Ranolazine free base and its acid addition salt with high purity. The present invention provides Ranolazine of high purity by using phosphate salt of piperazine to prepare Ranolazine. In this process, excess of unreacted piperazine is easy to recover and recycle in the next reactions. Thus it is easy to avoid the generation of large amount of effluent due to reuse of piperazine, which are generally desirable for large-scale operations thereby making the process commercially feasible.
All the available literature uses unprotected piperazine and protected piperazine leading to formation of dimer impurities which are difficult to remove from the product and also resulting in poor overall yield of the product. The maximum daily dosage of Ranolazine is 2 g; therefore, known and unknown impurities must be controlled below 0.05% in the final drug substance.
From the above known fact our main target is:
1. To study the detailed impurity profile to and to control the formation of all the impurities below the desired limit (NMT 0.05%).
2. To obtained the Cost effective process by utilizing the maximum consumption of piperazine in the form of piperazine monophosphate salt there by reducing formation of unwanted impurities and also reusing recovered piperazine.
All the available literature uses unprotected piperazine and protected piperazine leading to formation of dimer impurities which are difficult to remove from the product and also resulting in poor overall yield of the product.


EXAMPLES
The following examples are presented for illustration only, and are not intended to limit the scope of the invention or appended claims.
Example 1 :
Preparation of [(2, 6-Dimethylphenyl)-amino carbonyl methyl) chloride (6)
To 0.74 kg of potassium carbonate and 2.51ml of water, was added. 500 gm of 2,6-Dimethyl aniline in 1.25 L of Acetone at 0-5 °C. 650 gm of Chloroacetyl chloride was added to the reaction mixture below 5 °C and stirred for 3 hrs. 2500 ml of water was added, stirred for 1 hr, filtered the product, washed with water and dried at 75 °C to get [(2,6- Dimethylphenyl)-amino carbonyl methyl] chloride (6). Yield: 95%; purity >98%
Example 2:
Preparation of l-(2-Methoxy phenoxy)-2, 3-epoxy propane (3)
Added 2.5 L of water to R.B Flask, 80 gms of NaOH was added and stirred to dissolve. Added 500 gms of Guaiacol, 1.12 Kg of Epichlorohydrine and stirred at 25-350C for 5-6 h. The organic layer was separated. To the Epichlorohydrine layer charged 160 gms NaOH dissolved in 2.5 L of water and stirred at 25-30°C for 3-4 h. The organic layer was separated and washed with 150 gms NaOH dissolved in 1.5 L of water. Excess Epichlorohydrine was recovered by distillation of the product layer at 90°C under vacuum (600-700 mmHg) to give 650-680 gms of oil. To the crude oil was added 3.0 L of Isopropanol and cooled to 0°C and filtered the product to get l-(2- Methoxy phenoxy)-2,3-epoxy propane (3).
Yield: 80%; purity >98%.
Example 3:
Preparation of piperazine monophosphate monohydrate
Added 1000 ml of water to R.B Flask 109 gms piperazine was added and stirred to dissolve. pH was adjusted to 5.0-5.5 with O-phosphoric acid. After stirring for 1-2-h at room temperature. Filtered the reaction mass and solid was isolated as piperazine monophosphate monohydrate.
Example 4:
Preparation of compound of formula (7)
Added 1000 ml of water to R.B Flask. 109 gms piperazine was added and stirred to dissolve. pH was adjusted to 5.0-5.5 with O-phosphoric acid. After stirring for 1-2- h at room temperature. Filtered the reaction mass and solid was isolated as piperazine monophosphate monohydrate and charged further to R.B Flask containing 1000 ml water. 100 gms of [(2,6-Dimethylphenyl)-amino carbonyl methyl)chloride (6) was added and heated the reaction mixture at reflux temperature for 7-8 h. Cooled the reaction mixture at 25-30°C and adjusted the pH to 5.5-6.0 with dilute sodium hydroxide solution filtered. Filtrate was washed with 100 ml x 2 methylene chloride and further basified with dilute sodium hydroxide solution and extracted with 500 ml x 3 methylene chloride to obtained compound of formula (7).
Example 5:
Preparation of Ranolazine
Added 1000 ml of water to R.B Flask 109 gms piperazine was added and stirred to dissolve. pH was adjusted to 5.0-5.5 with O-phosphoric acid, 100 gms of [(2,6-Dimethylphenyl)-amino carbonyl methyl)chloride (6) was added and heated the reaction mixture at reflux temperature for 7-8 h. Cooled the reaction mixture at 25-30°C and adjusted pH to 5.5-6.0 with dilute sodium hydroxide solution and filtered. Filtrate was washed with 100 ml x 2 methylene chloride and further basified with dilute sodium hydroxide solution and extracted with 500 ml x 3 methylene chloride. Combined organic layer was washed with saturated brine solution and 80 gm of l-(2-Methoxy phenoxy)-2, 3-epoxy propane (3) was added. Distilled out Methylene chloride under reduced pressure, added 500 ml methanol and refluxed for 5-6 h. Cooled the reaction mass to room temperature and added 500 ml water and cooled to 0°C. Filtered the product to get crude Ranolazine. Yield: 80%; purity >99%.
Example 6:
Preparation of Ranolazine from recovered piperazine monophosphate monohydrate
Added 1000 ml of water to R.B Flask 109 gms piperazine was added and stirred to dissolve. Added recovered piperazine monophosphate monohydrate and pH was adjusted to 5.0-5.5 with O-phosphoric acid, 100 gms of [(2,6-Dimethylphenyl)-amino carbonyl methyl)chloride (6) was added and heated the reaction mixture at reflux temperature for 7-8 h. Cooled the reaction mixture at 25-30°C and adjusted pH 5.5-6.0 with dilute sodium hydroxide solution and filtered. Filtrate was washed with 100 ml x 2 methylene chloride and further basified with dilute sodium
hydroxide solution and extracted with 500 ml x 3 methylene chloride. Combined organic layer was washed with saturated brine solution and 80 gm of l-(2-Methoxy phenoxy)-2,3-epoxy propane (3) was added. Distilled out Methylene chloride under reduced pressure, added 500 ml methanol and refluxed for 5-6 h. Cooled the reaction mass to room temperature and added 500 ml water and cooled to 0°C. Filtered the product to get crude Ranolazine. Yield: 80%; purity >99%.
Example 7:
Preparation of Ranolazine.
Added 1000 ml of water to R.B Flask 109 gms piperazine was added and stirred to dissolve. pH was adjusted to 5.0-5.5 with O-phosphoric acid. 100 gms of [(2,6-Dimethylphenyl)-amino carbonyl methyl)chloride (6) was added and heated the reaction mixture at reflux temperature for 7-8 h. Cooled the reaction mixture at 25-30°C, adjusted pH to 5.5-6.0 with dilute sodium hydroxide solution and filtered. Filtrate was washed with 100 ml x 2 methylene chloride and further basified with dilute sodium hydroxide solution and extracted with 500 ml x 3 methylene chloride. Combined organic layer was washed with saturated brine solution and 80 gm of l-(2-Methoxy phenoxy)-2,3-epoxy propane (3) was added. Distilled out Methylene chloride under reduced pressure, added 500 ml isopropyl alcohol, refluxed for 5-6 h. cooled the reaction mass to 0°C. Filtered the product to get crude Ranolazine. Yield: 80%; purity >98%.
Example 8:
Preparation of Ranolazine
Added 1000 ml of water to R.B Flask 109 gms piperazine was added and stirred to dissolve. pH was adjusted to 5.0-5.5 with O-phosphoric acid. After stirring for 1-2- h at room temperature. Filtered the reaction mass and solid was isolated as piperazine monophosphate monohydrate and charged further to R.B Flask containing 1000 ml water. 100 gms of [(2,6-Dimethylphenyl)-amino carbonyl methyl)chloride (6) was added and heated the reaction mixture at reflux temperature for 7-8 h. Cooled the reaction mixture at 25-30°C and adjusted the pH to 5.5-6.0 with dilute sodium hydroxide solution filtered. Filtrate was washed
with 100 ml x 2 methylene chloride and further basified with dilute sodium hydroxide solution and extracted with 500 ml x 3 methylene chloride. Combined organic layer was washed with saturated brine solution and 80 gm of l-(2-Methoxy phenoxy)-2,3-epoxy propane (3) was added. Distilled out Methylene chloride under reduced pressure, added 500 ml methanol and refluxed for 5-6 h. Cooled the reaction mass to room temperature, added 500 ml water, cooled to 0°C and filtered the product to get crude Ranolazine. Yield: 80%; purity >99%.
Example 9:
Purification of Ranolazine
Added 300 ml of methanol to R.B Flask, 100 gms of crude ranolazine piperazine and heated to dissolve. Added Activated charcoal and filtered the hot solution through hyflo and washed the hyflo with 100 ml methanol. Reaction mixture was cooled to room temperature. 200 ml water was added and was cooled further to 0-5°C. Filtered to afford pure Ranolazine. Yield: 90%; purity >99.9%.
PATENT
WO2006008753,
https://www.google.com/patents/WO2006008753A1?cl=en
US Patent 4567264 describes the preparation of Ranolazine base from basic stages by condensing [(2,6-dimethyphenyl) amino; carbonyl methyl] – chloride (II) with l-[3-(2-metlioxyphenoxy)-2- hydroxypropyl]piperazine.(III) The base was purified by column chromatography and isolated as oil. The hydrochloride salt was prepared in methanol using hydrochloric acid and the salt was isolated by addition of ether.

Ranolazine Base
EP 0483932 describes the preparation of Ranolazine base by condensation of α-[ N3N -bis (2-cWoroetiiyl)-amino]-2,6-dimetliylacetanilide hydrochloride (IV) with l-[3-(2-methoxy phenoxy)-2-hydroxy]-propylamine (V). The base was purified using column chromatography and hydrochloride was formed by treating with metholic hydrochloric acid and crystallized by addition of diethyl ether as co solvent to obtain a product with melting point 229- 230 0C.

Ranolazine base
It is a long standing need to avoid the formation of oil and obtain the product directly as solid there by eliminating laborious and expensive column chromatographic methods and achieving the higher yields of Ranolazine diliydrochloride. More over the prior art does not teach, any features such as polymorphic forms of the drug which may have varying pharmacological effects ‘
Example-1:
Preparation of l-[3-(2-Metkoxyphenoxy)-2-hydroxypropyl ] piperazine
100 gms l-(2-methoxyphenoxy)-2,3-epoxypropane was added in a 60 min at 0-5 0C to 192 gms of anhydrous piperazine dissolved in 500 ml methanol. Reaction mixture is stirred further for 2 Hrs at 0-5 0C. It is quenched in 400 ml DMW & filtered. The product is obtained by extraction with MDC from the saturated aqueous layer with sodium chloride. 65 gms of acetic acid and 400 ml water is added in the MDC layer. Aqueous layers was separated and basified with 100 ml liquor ammonia. The product was extracted with 500 ml methylene dichloride and isolated by evaporation of solvent. The residue was used as such in the next reaction.
Yield =80 gms. HPLC purity = 96-$k %.
ExampIe-2 r-
Preparation of crude (+)-l-[3~(2-Methoxyphenoxy)-2-hydroxypropyl]-4- [N-(2,6-dimethylphenyl)carbamoylmethyl] piperazine dihydrochloride.
A mixture of 90 gms l-[3-(2-Memoxyphenoxy)-2-hydroxypropyl ] piperazine, 85 gms [(2,6-dimethylphenyl) aminocarbonyl methyl)chloride, 120 gms anhydrous potassium carbonate and 3.6 gms sodium iodide in 260 ml dimethyl formamide is stirred at room temperature (30-35 0C) for 18 Hrs. The reaction mixture is quenched in 1600 ml water and extracted thrice with 300 ml methylene dichloride each time . Combined methylene dichloride layer is treated with a mixture of 1100 ml aqueous hydrochloric acid ( 35 %) & 900 ml water. Acidic aqueous layer is basified with ammonia, extracted with methylene dichloride and solvent is evaporated to get Ranolazine base. ; Yield = 140 gms ,
The above Ranolazine base is taken in 2160 j ml j acetone and 100 hydrochloric acid gas dissolved in isopropyl alcohol is added at room temperature till pH is acidic. The precipitated dihydrochloride compound is Filtered, is washed with acetone to give the Ranolazine dihydrochloride Yield = 144 gm.
Example-3 :-
Preparation of Crystalline (+)-l-[3-(2-Methoxyphenoxy)-2- hydroxypropyl]-4-[N-(2,6-diniethylphenyl)carbamoylmethyl] piperazine dihydrochloride.
100 gms of Crude (+)-l-[3-(2-Methoxyphenoxy)-2-hydroxypropyl]-4-[N- (2,6-dimemylplienyl)caitamoyhnetliyl] piperazine dihydrochloride is dissolved to get a clear solution in 500 ml methanol., The solution is cooled to room temperature and further cooled to 100C. The product is filtered, washed with 2 X 50 ml methanol and dried at 75 degree C for 10 Hrs. get crystalline Form -A of Ranolazine diliydrochloride] ;: characterized .by XRD & DSC as shown in Figure |I and II.
Example-4: –
Preparation of Amorphouse (+)-l-[3-(2-Methoxyphenoxy)-2- hydroxypropyl]-4-[N-(2,6-dimethylphenyl)carbamoylmethyl] piperazine dihydrochloride
100 gms Ranolazine diliydrochloride is added in 500 ml water and heated to get a clear solution. Water is distilled off under reduced pressure, the residue is cooled to room temperature to obtain, amorphous form characterized by a XRD pattern (Figure III ) and DSC (Figure IV) exhibiting a broad endotherm around 80 and exotherm bet 220-224 and followed by endotherm 150-156 0C.
Example-5: –
Preparation of Amorphouse ,(+)-l-[3-(J2-Methoxyphenoxy)-2- hydroxypropyl]-4-[N-(2,6-dimethylphenyl)carbampylitnethyl] piperazine dihydrochloride
100 gms Ranolazine dihydrochloride is added ;i| in 2000 ml ethanol containing 10 % water and heated to get a clear: solution. Solvent is distilled off under reduced pressure, the residue is cooled to room temperature to obtain amorphous form characterized by a XRD pattern (Figure m ) and DSC (Figure IV) exhibiting a broad endotherm around 80 and exotherm bet 220-224 and followed by endotherm 150-156 0C.
Example -6:~
Preparation of Ranolazine base from its di hydrochloride salt
20 gms Ranolazine dihydrohloride at room temperature is added to a mixture containing 150 ml water and 50 ml acetone and 20 ml liquor ammonia. It is stirred for two hrs. The precipitated base, was . filtered and dried under vacuum at 70 0C to get crystalline form of Ranolazine base characterized by XRD & DSC as shown in Figure V & VI. Yield = 12 gms.
CLIP
Improved Process for Ranolazine: An Antianginal Agent†
‡ Research and Development, Integrated Product Development, Innovation Plaza, Dr. Reddy’s Laboratories Ltd., Survey Nos. 42, 45, 46 and 54, Bachupally, Qutubullapur, Ranga Reddy-500 072, Andhra Pradesh, India
§ Research and Development, Macleods Pharmaceuticals Limited, G-2, Mahakali Caves Road, Shanthi Nagar, Andheri (E), Mumbai-400 093, Maharashtra, India
∥ Department of Chemistry, University College of Science, Osmania University, Hyderabad-500 007, Andhra Pradesh, India
Org. Process Res. Dev., 2012, 16 (5), pp 748–754
DOI: 10.1021/op300026r
Publication Date (Web): April 12, 2012,*E-mail: vummenthalapv@yahoo.co.in. Fax: +91-40-44346285. Telephone: +91-9849210408.
An improved process has been developed for the active pharmaceutical ingredient, ranolazine with 99.9% purity and 47% overall yield (including three chemical reactions and one recrystallization). Formation and control of all the possible impurities is described. All the solvents used in the process were recovered and reused. The unreacted piperazine is recovered as piperazine monophosphate monohydrate salt.

References
- Banon D et al. The usefulness of ranolazine for the treatment of refractory chronic stable angina pectoris as determined from a systematic review of randomized controlled trials. Am J Cardiol. 2014 Mar 15;113(6):1075-82. PMID 24462341
- “Ranexa (ranolazine) Extended-Release Tablets, for Oral Use. Full Prescribing Information”. Gilead Sciences, Inc. Foster City, CA 94404. Retrieved8 September 2016.
- ^ Jump up to:a b c d e Kloner RA, et al. Efficacy and safety of ranolazine in patients with chronic stable angina. Postgrad Med. 2013 Nov;125(6):43-52. PMID 24200760
- Jump up^ “FDA Approves New Treatment for Chest Pain”. FDA News. 2006-01-31. Retrieved2011-03-02.
- D Noble and P J Noble. Late sodium current in the pathophysiology of cardiovascular disease: consequences of sodium–calcium overload Heart. Jul 2006; 92(Suppl 4): iv1–iv5.PMID 16775091 PMCID 1861316
- Jump up^ Sokolov, S; Peters, CH; Rajamani, S; Ruben, PC (2013). “Proton-dependent inhibition of the cardiac sodium channel Nav1.5 by ranolazine” (PDF). Frontiers in Pharmacology. 4: 78. doi:10.3389/fphar.2013.00078. PMC 3689222
 . PMID 23801963. Retrieved8 September 2016.
. PMID 23801963. Retrieved8 September 2016. - Jump up^ EMEA Ranolazine page at the EMEA
- Jump up^ CV Therapeutics press release. April 1, 1996 CV Therapeutics Licenses Late-Stage Anti-Anginal Drug from Syntex (U.S.A.), an Affiliate of Roche Holding Ltd.
- Jump up^ CV Therapeutics, 22 June 2006 CV Therapeutics Acquires Rights to Ranolazine in Asia
- Thepharmaletter.com 22 September 2008 Italy’s Menarini to pay up to $385 million for rights to CV Thera’s Ranexa
- Jump up^ Reuters, via the New York Times. 12 March 2009. Gilead, a White Knight, to Buy CV Therapeutics
- Menarini press release. 18 June 2013 Memarii Group announces agreement with Gilead Sciences to commercialize Ranexa® (ranolazine) in 50 new countries
- http://shodhganga.inflibnet.ac.in/bitstream/10603/19311/11/11_chapter%203.pdf
External links
| CN1404471A * | Feb 22, 2001 | Mar 19, 2003 | Cv Therapeutics | Substituted piperazine compound |
| Reference | ||
|---|---|---|
| 1 | * | “Green Chemistry” 20,130,131 Damodara N. Kommi ET Al. ” All Water Chemistry ” for A Concise Total Synthesis of Novel, class at The Anti-anginal Drug (the RS), (R & lt), and (S) -ranolazine 756-767 1-9 Vol. 15, |
| 2 | * | “Tetrahedron Letters” 20080304 Sadula Sunitha et al. An efficient and chemoselective Br nsted acidic ionic liquid-catalyzed N-Boc protection of amines 2527-2532 1-9 Vol. 49, |
| 3 | * | N. KOMMI the ET AL .: DAMODARA ” ” All Water Chemistry “for A Concise Total Synthesis of Novel, class at The Anti-anginal Drug (the RS), (R & lt), and (S) -ranolazine “, “GREEN CHEMISTRY”, Vol. 15, 31 January 2013 (2013-01-31) , pages 756 – 767 |
| 4 | * | Sunitha the ET AL .: SADULA ” An Efficient and chemoselective Brønsted acidic Ionic Liquid-Catalyzed N of Boc-Protection of Amines “, “TETRAHEDRON LETTERS”, Vol 49, 4 March 2008 (2008-03-04), Pages 2527 -. 2532 |
| 5 | * | Qin Mingli et al: ” Study on the Synthesis of ranolazine ..”, “Xinyang Normal University: Natural Science”, vol 20, no 2, 30 April 2007 (2007-04-30), pages 226 – 229 |
RANEXA (ranolazine) Extended-release Tablets
Ranolazine is a racemic mixture, chemically described as 1-piperazineacetamide, N-(2,6-dimethylphenyl)-4-[2-hydroxy-3-(2-methoxyphenoxy)propyl]-, (±)-. It has an empirical formula of C24H33N3O4, a molecular weight of 427.54 g/mole, and the following structural formula:
 |
Ranolazine is a white to off-white solid. Ranolazine is soluble in dichloromethane and methanol; sparingly soluble in tetrahydrofuran, ethanol, acetonitrile, and acetone; slightly soluble in ethyl acetate, isopropanol, toluene, and ethyl ether; and very slightly soluble in water.
RANEXA tablets contain 500 mg or 1000 mg of ranolazine and the following inactive ingredients: carnauba wax, hypromellose, magnesium stearate, methacrylic acid copolymer (Type C), microcrystalline cellulose, polyethylene glycol, sodium hydroxide, and titanium dioxide. Additional inactive ingredients for the 500 mg tablet include polyvinyl alcohol, talc, Iron Oxide Yellow, and Iron Oxide Red; additional inactive ingredients for the 1000 mg tablet include lactose monohydrate, triacetin, and Iron Oxide Yellow.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(RS)-N-(2,6-Dimethylphenyl)-2-[4-[2-hydroxy-3-(2-methoxyphenoxy)-propyl]piperazin-1-yl]acetamide
|
|
| Clinical data | |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a606015 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
By mouth (tablets) |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 35 to 50% |
| Protein binding | ~62% |
| Metabolism | Extensive in liver (CYP3A,CYP2D6) and intestine |
| Biological half-life | 7 hours |
| Excretion | Renal (75%) and fecal (25%) |
| Identifiers | |
| CAS Number | 142387-99-3 |
| ATC code | C01EB18 (WHO) |
| PubChem | CID 56959 |
| IUPHAR/BPS | 7291 |
| DrugBank | DB00243 |
| ChemSpider | 51354 |
| UNII | A6IEZ5M406 |
| ChEBI | CHEBI:87681 |
| ChEMBL | CHEMBL1404 |
| Chemical data | |
| Formula | C24H33N3O4 |
| Molar mass | 427.537 g/mol |
| Chirality | Racemic mixture |
////////////////////Ranolazine, 盐酸雷诺嗪 ,雷诺嗪 , Antianginal
CLIP
Ranolazine (Ranexa™)
Ranolazine, developed by CV therapeutics after licensing it from Roche (Syntex), is a late stage sodium channel
blocker approved in March 2006 for the treatment of chronic angina. The compounds anti-angina and anti-ischemic affects do not depend on reductions in heart rate or blood pressure.
Because of the potential for QT prolongation, the drug is indicated for treating patients that do not get adequate response with other anti-anginal drugs [6,27].
Two syntheses, one from the inventors at Roche [28] and other from a group in Hungary [29], of Ranolazine have been described in the patent literature.
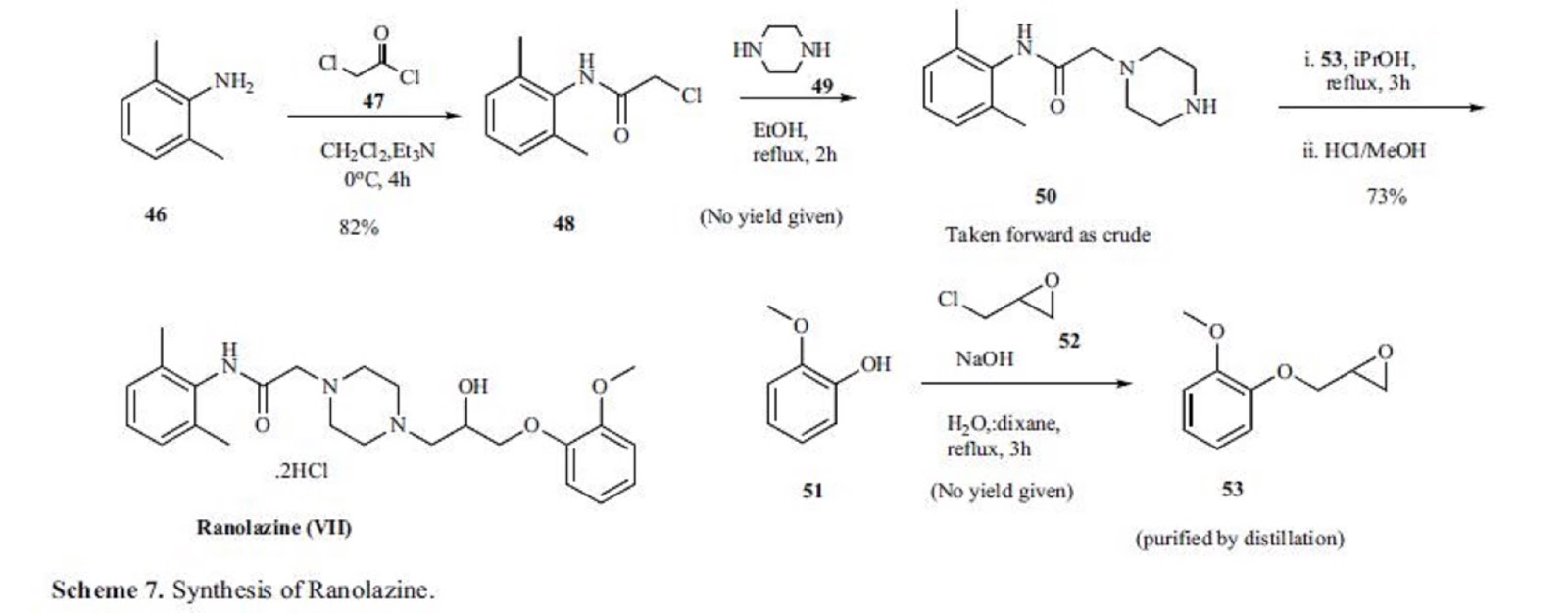
The original synthesis is highlighted in Scheme 7. Reaction of 2,6-dimethylaniline 46 with chloroacetyl chloride (47) in the presence of triethylamine for 4h at 0ºC gave amide 48 in 82% yield. This chloro amide 48 was reacted with piperazine in refluxing ethanol for 2 h to give piperazinyl amide 50.
Reaction of amide 50 with epoxide intermediate 53, prepared by reacting 2-methoxy phenol 51 with epichlorohydrin, in refluxing isopropanol for 3 h followed by treatment with HCl/methanol gave ranolazine dihydrochloride (VII) in 73% yield.
[6] Graul, A. I.; Prous, J. R. Drug News Perspect, 2007, 20, 17.
[27] Jones, R. IDrugs, 1999, 2, 1353.
[28] Kluge, A. F.; Clark, R. D.; Strosberg, A. M.; Pascal, J. C.; Whiting,R. L. EP-0126449 A1, 1984.
[29] Agai-Csongor, E.; Gizur, T.; Hasanyl, K.; Trischler, F.; Demeter-Sabo, A.; Csehi, A.; Vajda, E.; Szab-Komi si, G. EP-0483932 A1,1991.
