



Home » Posts tagged 'Priority review' (Page 5)
Tag Archives: Priority review
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
FDA approves new drug Doptelet (avatrombopag) for patients with chronic liver disease who have low blood platelets and are undergoing a medical procedure
Avatrombopag
https://newdrugapprovals.org/2015/08/24/avatrombopag/
The U.S. Food and Drug Administration today approved Doptelet (avatrombopag) tablets to treat low blood platelet count (thrombocytopenia) in adults with chronic liver disease who are scheduled to undergo a medical or dental procedure. This is the first drug approved by the FDA for this use.Continue reading.
May 21, 2018
Release
The U.S. Food and Drug Administration today approved Doptelet (avatrombopag) tablets to treat low blood platelet count (thrombocytopenia) in adults with chronic liver disease who are scheduled to undergo a medical or dental procedure. This is the first drug approved by the FDA for this use.
“Patients with chronic liver disease who have low platelet counts and require a procedure are at increased risk of bleeding,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Doptelet was demonstrated to safely increase the platelet count. This drug may decrease or eliminate the need for platelet transfusions, which are associated with risk of infection and other adverse reactions.”
Platelets (thrombocytes) are colorless cells produced in the bone marrow that help form blood clots in the vascular system and prevent bleeding. Thrombocytopenia is a condition in which there is a lower-than-normal number of circulating platelets in the blood. When patients have moderately to severely reduced platelet counts, serious or life-threatening bleeding can occur, especially during invasive procedures. Patients with significant thrombocytopenia typically receive platelet transfusions immediately prior to a procedure to increase the platelet count.
The safety and efficacy of Doptelet was studied in two trials (ADAPT-1 and ADAPT-2) involving 435 patients with chronic liver disease and severe thrombocytopenia who were scheduled to undergo a procedure that would typically require platelet transfusion. The trials investigated two dose levels of Doptelet administered orally over five days as compared to placebo (no treatment). The trial results showed that for both dose levels of Doptelet, a higher proportion of patients had increased platelet counts and did not require platelet transfusion or any rescue therapy on the day of the procedure and up to seven days following the procedure as compared to those treated with placebo.
The most common side effects reported by clinical trial participants who received Doptelet were fever, stomach (abdominal) pain, nausea, headache, fatigue and swelling in the hands or feet (edema). People with chronic liver disease and people with certain blood clotting conditions may have an increased risk of developing blood clots when taking Doptelet.
This product was granted Priority Review, under which the FDA’s goal is to take action on an application within six months where the agency determines that the drug, if approved, would significantly improve the safety or effectiveness of treating, diagnosing or preventing a serious condition.
The FDA granted this approval to AkaRx Inc.
//////////////Doptelet, avatrombopag, fda 2018, akarx, priority review,
FDA approves new uses for two drugs Tafinlar (dabrafenib) and Mekinist (trametinib) administered together for the treatment of BRAF-positive anaplastic thyroid cancer


FDA approves new uses for two drugs Tafinlar (dabrafenib) and Mekinist (trametinib) administered together for the treatment of BRAF-positive anaplastic thyroid cancer
The U.S. Food and Drug Administration approved Tafinlar (dabrafenib) and Mekinist (trametinib), administered together, for the treatment of anaplastic thyroid cancer (ATC) that cannot be removed by surgery or has spread to other parts of the body (metastatic), and has a type of abnormal gene, BRAF V600E (BRAF V600E mutation-positive). Continue reading.
May 4, 2018
Release
The U.S. Food and Drug Administration approved Tafinlar (dabrafenib) and Mekinist (trametinib), administered together, for the treatment of anaplastic thyroid cancer (ATC) that cannot be removed by surgery or has spread to other parts of the body (metastatic), and has a type of abnormal gene, BRAF V600E (BRAF V600E mutation-positive).
“This is the first FDA-approved treatment for patients with this aggressive form of thyroid cancer, and the third cancer with this specific gene mutation that this drug combination has been approved to treat,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “This approval demonstrates that targeting the same molecular pathway in diverse diseases is an effective way to expedite the development of treatments that may help more patients.”
Thyroid cancer is a disease in which cancer cells form in the tissues of the thyroid gland. Anaplastic thyroid cancer is a rare, aggressive type of thyroid cancer. The National Institutes of Health estimates there will be 53,990 new cases of thyroid cancer and an estimated 2,060 deaths from the disease in the United States in 2018. Anaplastic thyroid cancer accounts for about 1 to 2 percent of all thyroid cancers.
Both Tafinlar and Mekinist are also approved for use, alone or in combination, to treat BRAF V600 mutation-positive metastatic melanoma. Additionally, Tafinlar and Mekinist are approved for use, in combination, to treat BRAF V600E mutation-positive, metastatic non-small cell lung cancer.
The efficacy of Tafinlar and Mekinist in treating ATC was shown in an open-label clinical trial of patients with rare cancers with the BRAF V600E mutation. Data from trials in BRAF V600E mutation-positive, metastatic melanoma or lung cancer and results in other BRAF V600E mutation-positive rare cancers provided confidence in the results seen in patients with ATC. The trial measured the percent of patients with a complete or partial reduction in tumor size (overall response rate). Of 23 evaluable patients, 57 percent experienced a partial response and 4 percent experienced a complete response; in nine (64 percent) of the 14 patients with responses, there were no significant tumor growths for six months or longer.
The side effects of Tafinlar and Mekinist in patients with ATC are consistent with those seen in other cancers when the two drugs are used together. Common side effects include fever (pyrexia), rash, chills, headache, joint pain (arthralgia), cough, fatigue, nausea, vomiting, diarrhea, myalgia (muscle pain), dry skin, decreased appetite, edema, hemorrhage, high blood pressure (hypertension) and difficulty breathing (dyspnea).
Severe side effects of Tafinlar include the development of new cancers, growth of tumors in patients with BRAF wild-type tumors, serious bleeding problems, heart problems, severe eye problems, fever that may be severe, serious skin reactions, high blood sugar or worsening diabetes, and serious anemia.
Severe side effects of Mekinist include the development of new cancers; serious bleeding problems; inflammation of intestines and perforation of the intestines; blood clots in the arms, legs or lungs; heart problems; severe eye problems; lung or breathing problems; fever that may be severe; serious skin reactions; and high blood sugar or worsening diabetes.
Both Tafinlar and Mekinist can cause harm to a developing fetus; women should be advised of the potential risk to the fetus and to use effective contraception.
The FDA granted Priority Review and Breakthrough Therapy designation for this indication. Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases, was also granted for this indication.
The FDA granted this approval to Novartis Pharmaceuticals Corporation.
///////////////Tafinlar, dabrafenib, Mekinist, trametinib, fda 2018, Priority Review, Breakthrough Therapy designation, Orphan Drug designation, Novartis Pharmaceuticals Corporation,
Baloxavir marboxil, バロキサビルマルボキシル , балоксавир марбоксил , بالوكسافير ماربوكسيل , 玛巴洛沙韦 ,


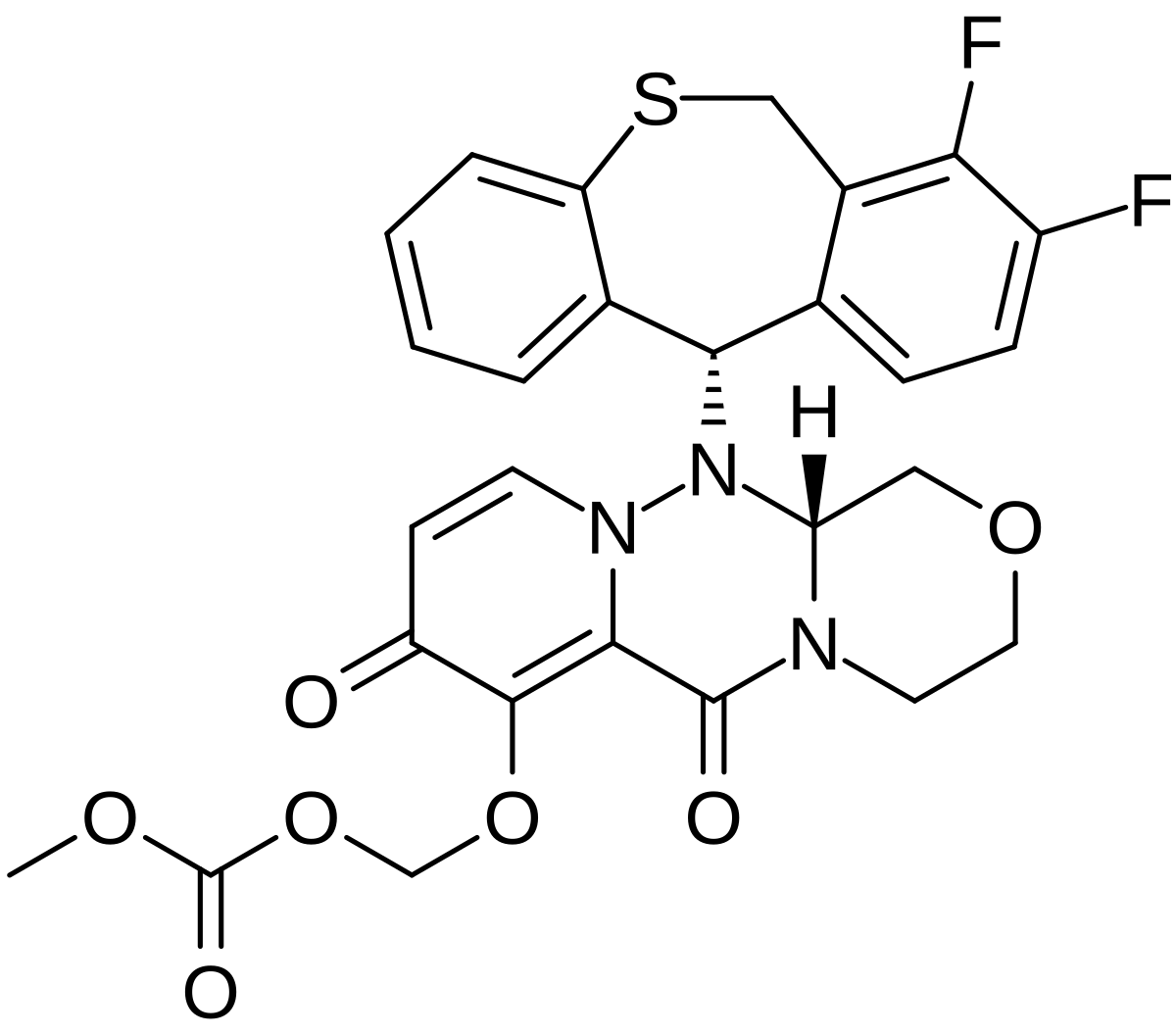
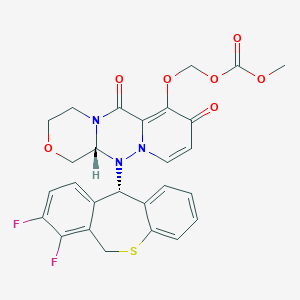
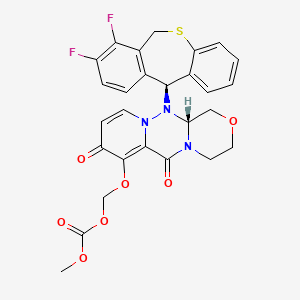
Baloxavir marboxil
バロキサビルマルボキシル
балоксавир марбоксил [Russian] [INN]
بالوكسافير ماربوكسيل [Arabic] [INN]
玛巴洛沙韦 [Chinese] [INN]
Carbonic acid, [[(12aR)-12-[(11S)-7,8-difluoro-6,11-dihydrodibenzo[b,e]thiepin-11-yl]-3,4,6,8,12,12a-hexahydro-6,8-dioxo-1H-[1,4]oxazino[3,4-c]pyrido[2,1-f][1,2,4]triazin-7-yl]oxy]methyl methyl ester
({(12aR)-12-[(11S)-7,8-Difluoro-6,11-dihydrodibenzo[b,e]thiepin-11-yl]-6,8-dioxo-3,4,6,8,12,12a-hexahydro-1H-[1,4]oxazino[3,4-c]pyrido[2,1-f][1,2,4]triazin-7-yl}oxy)methyl methyl carbonate
- (((12aR)-12-((11S)-7,8-Difluoro-6,11-dihydrodibenzo(b,E)thiepin-11-yl)-6,8-dioxo-3,4,6,8,12,12ahexahydro-1H-(1,4)oxazino(3,4-C)pyrido(2,1-F)(1,2,4)triazin-7-yl)oxy)methyl methyl carbonate
- Carbonic acid, (((12aR)-12-((11S)-7,8-difluoro-6,11-dihydrodibenzo(b,E)thiepin-11-yl)-3,4,6,8,12,12a-hexahydro-6,8-dioxo-1H-(1,4)oxazino(3,4-C)pyrido(2,1-F)(1,2,4)triazin-7-yl)oxy)methyl methyl ester
Antiviral
In Japan the product is indicated for treatment influenza types A and B in adults and children
RG-6152
- Originator Shionogi
- Developer Roche; Shionogi
- Class Antivirals; Dibenzothiepins; Esters; Pyridines; Small molecules; Triazines
- Mechanism of Action Endonuclease inhibitors
Highest Development Phases
- Marketed Influenza A virus infections; Influenza B virus infections
- Phase III Influenza virus infections
- Preclinical Influenza A virus H5N1 subtype
|
Xofluza (TN)
Antiviral
|
|
| Formula |
C27H23F2N3O7S
|
|---|---|
| Cas |
1985606-14-1
|
| Mol weight |
571.5492
|
| 2018/2/23 | PMDA | JAPAN | APPROVED | Baloxavir marboxil | Xofluza | Shionogi |

| バロキサビル マルボキシル Baloxavir Marboxil  C27H23F2N3O7S : 571.55 [1985606-14-1] |
![]()

https://chem.nlm.nih.gov/chemidplus/sid/1985606141
Baloxavir marboxil (trade name Xofluza, compound code S-033188/S-033447) is a medication being developed by Shionogi Co., a Japanese pharmaceutical company, for treatment of influenza A and influenza B. The drug was in late-stage trials in Japan and the United States as of early 2018, with collaboration from Roche AG.[1].
It was approved for sale in Japan on February 23, 2018.[2]
Baloxavir marboxil is a medication developed by Shionogi Co., a Japanese pharmaceutical company, for treatment of influenza A and influenza B. The drug was approved for use in Japan in February 2018 and is in late phase trials in the United States as of early 2018. Roche, which makes Tamiflu, has acquired the license to sell Xofluza internationally, but it may not be until 2019 that it could be available in the United States [7]. Interestingly, a study has determined that administering Baloxavir marboxil with neuraminidase inhibitors leads to a synergistic effect in influenza treatment

It is an influenza therapeutic agent (cap-dependent endonuclease inhibitor), characterized by only taking one dose. Unlike neuraminidase inhibitors such as oseltamivir (Tamiflu) and zanamivir (Relenza) that inhibit the action of neuraminidase, which liberates viruses from the infected cells surface, baloxavir marboxil may prevent replication by inhibiting the cap-dependent endonuclease activity of the viral polymerase.[3]
In October 2015, the Japanese Ministry of Health, Labour and Welfare granted Sakigake status to Shionogi’s baloxavir marboxil for A type or B -type influenza virus infection . In October 2015, the drug was designated for Priority Review by the Ministry of Health, Labour and Welfare, presumably for the treatment of A type or B -type influenza virus infection .
This drug is a CAP endonuclease inhibitor [1]. The influenza endonuclease is an essential subdomain of the viral RNA polymerase enzyme. CAP endonuclease processes host pre-mRNAs to serve as primers for viral mRNA and therefore has been a common target for studies of anti-influenza drugs.
Viral gene transcription is primed by short-capped oligonucleotides that are cleaved from host cell pre mRNA by endonuclease activity. Translation of viral mRNAs by the host ribosome requires that they are capped at the 5′ end, and this is achieved in cells infected with influenza virus by a “cap-snatching” mechanism, whereby the endonuclease cleaves 5′ caps from host mRNA which then act as primers for transcription.The N-terminal domain of PA subunit (PAN) has been confirmed to accommodate the endonuclease activity residues, which is highly preserved among subtypes of influenza A virus and is able to fold functionally [4]. Translation of viral mRNAs by the host ribosome requires that they are capped at the 5′ end, and this is achieved in cells infected with influenza virus by a “cap-snatching” mechanism, whereby the endonuclease cleaves 5′ caps from host mRNA which then act as primers for transcription. The endonuclease domain binds the N-terminal half of PA (PAN) and contains a two-metal (Mn2+) active site that selectively cleaves the pre-mRNA substrate at the 3′ end of a guanine [3].
The administration of a CAP endonuclease inhibitor, such as Baloxavir marboxil, prevents the above process from occurring, exhibiting its action at the beginning of the pathway before CAP endonuclease may exert its action
It achieves this by inhibiting the process known as cap snatching[4], which is a mechanism exploited by viruses to hijack the host mRNA transcription system to allow synthesis of viral RNAs.
Shionogi, in collaboration with licensee Roche (worldwide except Japan and Taiwan), have developed and launched baloxavir marboxil
In March 2018, Shionogi launched baloxavir marboxil for the treatment of influenza types A and B in Japan . In September 2017, Shionogi was planning to file an NDA in the US; in February 2018, the submission remained in preparation
By September 2016, baloxavir marboxil had been awarded Qualified Infectious Disease Product (QIDP) designation in the US
In March 2017, a multicenter, randomized, double-blind, parallel-group, phase III study (NCT02954354; 1601T0831; CAPSTONE-1) was initiated in the US, Canada and Japan to compare a single dose of baloxavir marboxil versus placebo or oseltamivir bid for 5 days in influenza patients aged from 12 to 64 years of age (n = 1494). The primary endpoint was the time to alleviation of symptoms (TTAS).
PATENTS
JP 5971830
Kawai, Makoto; Tomita, Kenji; Akiyama, Toshiyuki; Okano, Azusa; Miyagawa, Masayoshi
PATENTS
WO 2017104691
Shishido, Takao; Noshi, Takeshi; Yamamoto, Atsuko; Kitano, Mitsutaka
In Japanese Patent Application No. 2015-090909 (Patent No. 5971830, issued on Aug. 17, 2016, Registered Publication), a compound having a CEN inhibitory action and represented by the formula:
[Chemical Formula 2]
is described. Anti-influenza agents of six mechanisms are enumerated as drugs that can be used together with the above compounds. However, no specific combinations are described, nor is it disclosed nor suggested about the combined effect.
Synthesis Example 2
[formula 39]
Compound III-1 (1.00g, 2.07mmol) to a suspension of DMA (5 ml) of chloromethyl methyl carbonate (0.483 g, 3.10 mmol) and potassium carbonate (0 .572 g, 4.14 mmol) and potassium iodide (0.343 g, 2.07 mmol) were added, the temperature was raised to 50 ° C. and the mixture was stirred for 6 hours. Further, DMA (1 ml) was added to the reaction solution, and the mixture was stirred for 6 hours. The reaction solution was cooled to room temperature, DMA (6 ml) was added, and the mixture was stirred at 50 ° C. for 5 minutes and then filtered. 1 mol / L hydrochloric acid water (10 ml) and water (4 ml) were added dropwise to the obtained filtrate under ice cooling, and the mixture was stirred for 1 hour. The precipitated solid was collected by filtration and dried under reduced pressure at 60 ° C. for 3 hours to obtain compound II-4 (1.10 g, 1.93 mmol, yield 93%).
1 H-NMR (DMSO-D 6) δ: 2.91-2.98 (1 H, m), 3.24-3.31 (1 H, m), 3.44 (1 H, t, J = 10.4 Hz) J = 10.8, 2.9 Hz), 4.06 (1 H, d, J = 14.3 Hz), 4.40 (1 H, dd, J = 11.5, 2.8 Hz), 3.73 (3 H, s), 4.00 , 5.67 (1 H, d, J = 6.5 Hz), 5.72 (1 H, d, J = 11.8 Hz), 4.45 (1H, dd, J = 9.9, 2.9 Hz), 5.42 J = 8.0, 1.1 Hz), 7.14 – 7.18 (1 H, m ), 7.23 (1 H, d, J = 7.8 Hz), 7.37 – 7.44 (2 H, m)
PATENTS
JP 6212678
PATENTS
JP 6249434
JP 5971830
SYNTHESIS OF KEY INTERMEDIATE

SYNTHESIS OF KEY INTERMEDIATE

SYNTHESIS OF FINAL PRODUCT

Japan’s New Drug: One Pill May Stop The Flu in Just One Day
Isao Teshirogi, president and chief executive officer of Shionogi & Co., speaks during an interview in Tokyo, Japan. Photographer: Kiyoshi Ota/Bloomberg
One day, you may be able to stop flu viruses in your body in just one day with just one pill. Based on an announcement yesterday, that day may be someday very soon in May in Japan.
On Friday, Japanese pharmaceutical company Shionogi announced that the flu medication that they have developed, Xofluza, otherwise known as baloxavir marboxil (which sounds a bit like a Klingon General), has been approved to be manufactured and sold in Japan. Beginning in October 2015, the medication underwent priority review by Japan’s Ministry of Health, Labor, and Welfare. Shionogi filed for approval in the autumn of 2017. Compared to Tamiflu, which requires two doses each day for five days, apparently only a single dose of Xofluza will be needed to treat the flu. Even though Xofluza has received approval, people will have to wait until the Japanese national insurance sets a price for the medication, which according to Preetika Rana writing for the Wall Street Journal, may not occur until May.
Xofluza works via a different mechanism from neuroaminidase inhibitors like Tamiflu (oseltamivir) and Relenza (zanamivir). Flu viruses are like squatters in your home that then use the furniture and equipment in your home to reproduce. Yes, I know, that makes for a lovely picture. A flu infection begins when flu viruses reach your lungs. Each flu virus will enter a cell in your lungs and then use your cell’s genetic material and protein production machinery to make many, many copies of itself. In order to do this, the flu virus uses “cap-snatching”, which has nothing to do with bottle caps or Snapchat. The virus employs an endonuclease enzyme to clip off and steal the caps or ends of your messenger RNA and then re-purposes these caps to reproduce its own genetic material. After the virus has made multiple copies of itself, the resulting viruses implement another enzyme called a neuroaminidase to separate themselves from parts of the host cell and subsequently spread throughout the rest of your body to cause havoc. While Tamiflu, Relenza, and other neuroaminidase inhibitors try to prevent the neuroaminidase enzyme from working, Xofluza acts at an earlier step, stopping the “cap-snatching” by blocking the endonuclease enzyme.

In a clinical trial, Xofluza stopped an infected person from shedding flu virus sooner than Tamiflu. (Photo Illustration by Ute Grabowsky/Photothek via Getty Images)
By acting at an earlier step before the virus has managed to replicate, Xofluza could stop a flu virus infection sooner than neuroaminidase inhibitors. The results from Shionogi’s Phase III CAPSTONE-1 clinical trial compared Xofluza (then called Cap-dependent Endonuclease Inhibitor S-033188, which doesn’t quite roll off the tongue) with oseltamivir and placebo, with results being published in Open Forum Infectious Diseases. The study found that baloxavir marboxil (or Xofluza) stopped an infected person from shedding flu virus earlier (median 24 hours) than oseltamivir (median 72 hours). Those taking baloxavir marboxil also had lower measured amounts of viruses than those taking oseltamivir throughout the first 3 days of the infection. Baloxavir marboxil also seemed to shorten the duration of flu symptoms (median 53.7 hours compared to a median of 80.2 hours for those taking placebo). Since symptoms are largely your body’s reaction to the flu virus, you can begin shedding virus before you develop symptoms, and symptoms can persist even when you are no longer shedding the virus.
The key with any of these flu medications is early treatment, especially within the first 24 to 48 hours of infection, which may be before you notice any symptoms. Once the virus has replicated and is all over your body, your options are limited. The vaccine still remains the best way to prevent an infection.
In the words of Alphaville, this new drug could be big in Japan. While Xofluza won’t be available in time to help with the current flu season, this year’s particularly harsh flu season has highlighted the need for better ways to treat the flu. But will the United States see Xofluza anytime soon? Similar to Pokemon, Xofluza may need a year or two to reach the U.S. market. But one day, one pill and one day may be a reality in the U.S.
http://www.shionogi.co.jp/en/company/news/2018/pmrltj0000003nx1-att/e180223.pdf
XOFLUZA TM (Baloxavir Marboxil) Tablets 10mg/20mg Approved for the Treatment of Influenza Types A and B in Japan Osaka, Japan, February 23, 2018 – Shionogi & Co., Ltd. (Head Office: Osaka; President & CEO: Isao Teshirogi, Ph.D.; hereafter “Shionogi”) announced that XOFLUZATM (generic name: baloxavir marboxil) tablets 10mg/20mg was approved today by the Ministry of Health, Labour and Welfare for the treatment of Influenza Types A and B. As the cap-dependent endonuclease inhibitor XOFLUZATM suppresses the replication of influenza viruses by a mechanism different from existing anti-flu drugs, XOFLUZATM was designated for Sakigake procedure with priority review by the Ministry of Health, Labour, and Welfare of Japan in October 2015. Shionogi filed for approval to manufacture and sell XOFLUZATM in October 25, 2017. As the treatment with XOFLUZATM requires only a single oral dose regardless of age, it is very convenient, and is expected to improve adherence. XOFLUZATM is expected to be a new treatment option that can improve the quality of life in influenza patients. Shionogi will launch the product immediately after the National Health Insurance (NHI) price listing. Shionogi’s research and development targets infectious disease as one of its priority areas, and Shionogi have positioned “protecting people from the threat of infectious diseases” as one of its social mission targets. Shionogi strives constantly to bring forth innovative drugs for the treatment of infectious diseases, to protect the health of patients we serve.
References
- Jump up^ Rana, Preetika (10 February 2018). “Experimental Drug Promises to Kill the Flu Virus in a Day”. Wall Street Journal.
- Jump up^ “XOFLUZA (Baloxavir Marboxil) Tablets 10mg/20mg Approved For The Treatment Of Influenza Types A And B In Japan”. 23 February 2018 – via http://www.publicnow.com.
- Jump up^ Dias, Alexandre; Bouvier, Denis; Crépin, Thibaut; McCarthy, Andrew A.; Hart, Darren J.; Baudin, Florence; Cusack, Stephen; Ruigrok, Rob W. H. (2009). “The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit”. Nature. 458(7240): 914–918. doi:10.1038/nature07745. ISSN 0028-0836.
- Jump up^ “Cap snatching”.
 |
|
| Identifiers | |
|---|---|
| CAS Number | |
| PubChem CID | |
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C27H23F2N3O7S |
| Molar mass | 571.55 g·mol−1 |
| 3D model (JSmol) | |

Shionogi & Company, Limited(塩野義製薬株式会社 Shionogi Seiyaku Kabushiki Kaisha) is a Japanesepharmaceutical company best known for developing Crestor. Medical supply and brand name also uses Shionogi (“シオノギ”).
Shionogi has business roots that date back to 1878, and was incorporated in 1919. Among the medicines produced are for hyperlipidaemia, antibiotics, and cancer medicines.
In Japan it is particularly known as a producer of antimicrobial and antibiotics. Because of antibiotic resistance and slow growth of the antibiotic market, it has teamed up with US based Schering-Plough to become a sole marketing agent for its products in Japan.
Shionogi had supported the initial formation of Ranbaxy Pharmaceuticals, a generic manufacturer based in India. In 2012 the company became a partial owner of ViiV Healthcare, a pharmaceutical company specialising in the development of therapies for HIV.[3]
The company is listed on the Tokyo Stock Exchange and Osaka Securities Exchange and is constituent of the Nikkei 225 stock index.[4]
Medicines
- Claritin, An anti-histamine marketed in alliance with Schering-Plough.
- Crestor, cholesterol drug
- Nitrazepam, a short-term treatment for insomnia.
- Differin, a topical retinoid for acne.
- Moxifloxacin, antibacterial antiseptic that treats a number of infections
- Cymbalta, an SNRI class anti-depressant, marketed in alliance with Eli Lilly
- Osphena, an estrogen receptor agonist
Media
- Shionogi has a close relationship with Fuji Television Network, Inc., because Shionogi is the sponsor of “Music Fair” (as of 2018, aired on 17 TV stations including TV Oita System Co.) started in 1964.
- Shionogi was a main sponsor of Team Lotus during the age 1991/1994.[5]
References
- “Shionogi Company Profile”. Retrieved March 18, 2014.
- “Shionogi Annual Report 2013” (PDF). Retrieved March 18, 2014.
- “Shionogi and ViiV Healthcare announce new agreement to commercialise and develop integrase inhibitor portfolio”. viivhealthcare.com. Retrieved 18 March 2014.
- “Components:Nikkei Stock Average”. Nikkei Inc. Retrieved March 11,2014.
- Perry, Alan. “Sponsor Company Profiles”. Retrieved 25 April 2012.
External links
- Official Website (in English)
/////////Baloxavir marboxil, バロキサビルマルボキシル, JAPAN 2018, Xofluza, S-033188, S-033447, RG-6152, Qualified Infectious Disease Product, Priority Review, SAKIGAKE, балоксавир марбоксил , بالوكسافير ماربوكسيل , 玛巴洛沙韦 , Shionogi, roche
COC(=O)OCOC1=C2C(=O)N3CCOCC3N(N2C=CC1=O)C4C5=C(CSC6=CC=CC=C46)C(=C(C=C5)F)F
FDA approves new HIV treatment Trogarzo (ibalizumab-uiyk) for patients who have limited treatment options

![]()
Today, the U.S. Food and Drug Administration approved Trogarzo (ibalizumab-uiyk), a new type of antiretroviral medication for adult patients living with HIV who have tried multiple HIV medications in the past (heavily treatment-experienced) and whose HIV infections cannot be successfully treated with other currently available therapies (multidrug resistant HIV, or MDR HIV).Trogarzo is administered intravenously once every 14 days by a trained medical professional and used in combination with other antiretroviral medications. Continue reading.
March 6, 2018
Release
Today, the U.S. Food and Drug Administration approved Trogarzo (ibalizumab-uiyk), a new type of antiretroviral medication for adult patients living with HIV who have tried multiple HIV medications in the past (heavily treatment-experienced) and whose HIV infections cannot be successfully treated with other currently available therapies (multidrug resistant HIV, or MDR HIV).Trogarzo is administered intravenously once every 14 days by a trained medical professional and used in combination with other antiretroviral medications.
“While most patients living with HIV can be successfully treated using a combination of two or more antiretroviral drugs, a small percentage of patients who have taken many HIV drugs in the past have multidrug resistant HIV, limiting their treatment options and putting them at a high risk of HIV-related complications and progression to death,” said Jeff Murray, M.D., deputy director of the Division of Antiviral Products in the FDA’s Center for Drug Evaluation and Research. “Trogarzo is the first drug in a new class of antiretroviral medications that can provide significant benefit to patients who have run out of HIV treatment options. New treatment options may be able to improve their outcomes.”
The safety and efficacy of Trogarzo were evaluated in a clinical trial of 40 heavily treatment-experienced patients with MDR HIV-1 who continued to have high levels of virus (HIV-RNA) in their blood despite being on antiretroviral drugs. Many of the participants had previously been treated with 10 or more antiretroviral drugs. The majority of participants experienced a significant decrease in their HIV-RNA levels one week after Trogarzo was added to their failing antiretroviral regimens. After 24 weeks of Trogarzo plus other antiretroviral drugs, 43 percent of the trial’s participants achieved HIV RNA suppression.
The clinical trial focused on the small patient population with limited treatment options and demonstrated the benefit of Trogarzo in achieving reduction of HIV RNA. The seriousness of the disease, the need to individualize other drugs in the treatment regimen, and safety data from other trials were considered in evaluating the Trogarzo development program.
A total of 292 patients with HIV-1 infection have been exposed to Trogarzo IV infusion. The most common adverse reactions to Trogarzo were diarrhea, dizziness, nausea and rash. Severe side effects included rash and changes in the immune system (immune reconstitution syndrome).
The FDA granted this application Fast Track, Priority Review and Breakthrough Therapy designations. Trogarzo also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted approval of Trogarzo to TaiMed Biologics USA Corp.
Theratechnologies Announces FDA Approval of Breakthrough Therapy, Trogarzo™ (ibalizumab-uiyk) Injection, the First HIV-1 Inhibitor and Long-Acting Monoclonal Antibody for Multidrug Resistant HIV-1
- First HIV treatment approved with a new mechanism of action in more than 10 years
- Infused every two weeks, only antiretroviral treatment (ART) that does not require daily dosing
- Trogarzo™ has no drug-drug interactions and no cross-resistance with other ARTs
MONTREAL, March 6, 2018 /PRNewswire/ – Theratechnologies Inc. (Theratechnologies) (TSX: TH) and its partner TaiMed Biologics, Inc. (TaiMed) today announced that the U.S. Food and Drug Administration (FDA) has granted approval of Trogarzo™ (ibalizumab-uiyk) Injection. In combination with other ARTs, Trogarzo™ is indicated for the treatment of human immunodeficiency virus type 1 (HIV-1) infection in heavily treatment-experienced adults with multidrug resistant HIV-1 infection failing their current antiretroviral regimen.1
")
Trogarzo™ represents a critical new treatment advance as the first HIV therapy with a new mechanism of action approved in 10 years and proven effectiveness in difficult-to-treat patients with limited options. Unlike all other classes of ARTs, Trogarzo™ is a CD4-directed post-attachment HIV-1 inhibitor that binds to CD4+ receptors on host cells and blocks the HIV virus from infecting the cells.1
“Today’s approval of Trogarzo™ by the FDA is great news for people infected with difficult-to-treat multidrug resistant HIV. We look forward to bringing this much-needed therapy to patients in the U.S within six weeks,” said Luc Tanguay, President and Chief Executive Officer, Theratechnologies Inc. “We are grateful to the patients, investigators, as well as the FDA who supported the clinical development of Trogarzo™, and are helping address this critical unmet medical need.”
Trogarzo™ previously received Breakthrough Therapy and Orphan Drug designations as well as Priority Review status from the FDA, underscoring the significance of the treatment for this patient population.
“I witnessed some of the earliest cases of HIV and AIDS, at a time when the diagnosis was terrifying to patients because in many cases it was a death sentence,” said David Ho, M.D., chief scientific advisor of TaiMed and scientific director and CEO of the Aaron Diamond AIDS Research Center. “Since then, treatment advances and the discovery that combinations of ARTs was the best way to bring viral load below the level of detection have allowed most people to manage HIV like a chronic condition and live long, healthy lives. However, this is not the reality for people whose HIV is resistant to multiple drugs and whose viral load is not controlled, which is why TaiMed dedicated the past decade to advancing ibalizumab in the clinic. For these patients, it represents the next breakthrough.”
Up to 25,000 Americans with HIV are currently multidrug resistant, of which 12,000 are in urgent need of a new treatment option because their current treatment regimen is failing them and their viral load has risen to detectable levels, jeopardizing their health and making HIV transmittable.2-13 The best way to prevent the transmission of multidrug resistant HIV is to control the virus in those living with it. According to new guidance from the Centers for Disease Control and Prevention (CDC), the HIV virus cannot be transmitted if it is being fully suppressed.13
“I’ve struggled with multidrug resistant HIV for almost 30 years and it was completely debilitating to feel like I had run out of options – I made no long-term plans,” said Nelson Vergel, founder of the Program for Wellness Restoration (PoWeR) and Trogarzo™ patient. “Since starting treatment with Trogarzo™ six years ago and getting my viral load to an undetectable level, I have been my happiest, most productive self. Trogarzo™ is a new source of hope and peace of mind for people whose treatments have failed them, and I feel incredibly lucky to have been able to participate in the clinical trial program.”
TaiMed and Theratechnologies partnered on the development of Trogarzo™ so patients who can benefit from the treatment have access to it. For patients who need assistance accessing Trogarzo™ or who face challenges affording medicines, Theratechnologies has a team of patient care coordinators available to help. Patients can get assistance and expert support by contacting THERA patient support™ at 1-833-23-THERA (84372).
“In Phase 3 ibalizumab trials, we saw marked improvements in patients’ health who not only were heavily treatment-experienced and had limited remaining treatment options, but in cases they also had extremely high viral loads and significantly impaired immune systems,” said Edwin DeJesus, M.D., Medical Director for the Orlando Immunology Center. “As an investigator for ibalizumab clinical trials over nearly 10 years, it was remarkable and inspiring to see the dramatic effect ibalizumab had on such vulnerable patients. As a clinician, I am excited that we will now have another option with a different mechanism of action for our heavily pretreated patients who are struggling to keep their viral load below detection because their HIV is resistant to multiple drugs.”
Clinical Trial Findings
Clinical studies show that Trogarzo™, in combination with other ARTs, significantly reduces viral load and increases CD4+ (T-cell) count among patients with multidrug resistant HIV-1.
The Phase 3 trial showed:1
- Trogarzo™ significantly reduced viral load within seven days after the first dose of functional monotherapy and maintained the treatment response when combined with an optimized background regimen that included at least one other active ART for up to 24 weeks of treatment, while being safe and well tolerated.
- More than 80% of patients achieved the study’s primary endpoint – at least a 0.5 log10 (or 70%) viral load reduction from baseline seven days after receiving a 2,000 mg loading dose of Trogarzo™ and no adjustment to the failing background regimen.
- The average viral load reduction after 24 weeks was 1.6 log10 with 43% of patients achieving undetectable viral loads.
Patients experienced a clinically-significant mean increase in CD4+ T-cells of 44 cells/mm3, and increases varied based on T-cell count at baseline. Rebuilding the immune system by increasing T-cell count is particularly important as people with multidrug resistant HIV-1 often have the most advanced form of HIV.1
The most common drug-related adverse reactions (incidence ≥ 5%) were diarrhea (8%), dizziness (8%), nausea (5%) and rash (5%). No drug-drug interactions were reported with other ARTs or medications, and no cross-resistance with other ARTs were observed.1
About Trogarzo™ (ibalizumab-uiyk) Injection
Trogarzo™ is a humanized monoclonal antibody for the treatment of multidrug resistant HIV-1 infection. Trogarzo™ binds primarily to the second extracellular domain of the CD4+ T receptor, away from major histocompatibility complex II molecule binding sites. It prevents HIV from infecting CD4+ immune cells while preserving normal immunological function.
IMPORTANT SAFETY INFORMATION
Trogarzo™ is a prescription HIV medicine that is used with other antiretroviral medicines to treat human immunodeficiency virus-1 (HIV-1) infections in adults.
Trogarzo™ blocks HIV from infecting certain cells of the immune system. This prevents HIV from multiplying and can reduce the amount of HIV in the body.
Before you receive Trogarzo™, tell your healthcare provider if you:
- are pregnant or plan to become pregnant. It is not known if Trogarzo™ may harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known if Trogarzo™ passes into breast milk.
Tell your healthcare provider about all the medicines you take, including all prescription and over-the-counter medicines, vitamins, and herbal supplements.
Trogarzo™ can cause serious side effects, including:
Changes in your immune system (Immune Reconstitution Inflammatory Syndrome) can happen when you start taking HIV-1 medicines. Your immune system might get stronger and begin to fight infections that have been hidden in your body for a long time. Tell your health care provider right away if you start having new symptoms after starting your HIV-1 medicine.
The most common side effects of Trogarzo™ include:
- Diarrhea
- Dizziness
- Nausea
- Rash
Tell your healthcare provider if you have any side effect that bothers you or that does not go away. These are not all the possible side effects of Trogarzo™. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. You may also report side effects to at 1-833-23THERA (1-833-238-4372).
About Theratechnologies
Theratechnologies (TSX: TH) is a specialty pharmaceutical company addressing unmet medical needs to promote healthy living and an improved quality of life among HIV patients. Further information about Theratechnologies is available on the Company’s website at www.theratech.com and on SEDAR at www.sedar.com.
/////Trogarzo, ibalizumab-uiyk, fda 2018, Fast Track, Priority Review, Breakthrough Therapy designations, Orphan Drug designation
FDA approves new treatment Erleada (apalutamide) for a certain type of prostate cancer using novel clinical trial endpoint
The U.S. Food and Drug Administration today approved Erleada (apalutamide) for the treatment of patients with prostate cancer that has not spread (non-metastatic), but that continues to grow despite treatment with hormone therapy (castration-resistant). This is the first FDA-approved treatment for non-metastatic, castration-resistant prostate cancer. Continue reading.
February 14, 2018
Release
The U.S. Food and Drug Administration today approved Erleada (apalutamide) for the treatment of patients with prostate cancer that has not spread (non-metastatic), but that continues to grow despite treatment with hormone therapy (castration-resistant). This is the first FDA-approved treatment for non-metastatic, castration-resistant prostate cancer.
“The FDA evaluates a variety of methods that measure a drug’s effect, called endpoints, in the approval of oncology drugs. This approval is the first to use the endpoint of metastasis-free survival, measuring the length of time that tumors did not spread to other parts of the body or that death occurred after starting treatment,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “In the trial supporting approval, Erleada had a robust effect on this endpoint. This demonstrates the agency’s commitment to using novel endpoints to expedite important therapies to the American public.”
According to the National Cancer Institute (NCI) at the National Institutes of Health, prostate cancer is the second most common form of cancer in men in the U.S.. The NCI estimates approximately 161,360 men were diagnosed with prostate cancer in 2017, and 26,730 were expected to die of the disease. Approximately 10 to 20 percent of prostate cancer cases are castration-resistant, and up to 16 percent of these patients show no evidence that the cancer has spread at the time of the castration-resistant diagnosis.
Erleada works by blocking the effect of androgens, a type of hormone, on the tumor. These androgens, such as testosterone, can promote tumor growth.
The safety and efficacy of Erleada was based on a randomized clinical trial of 1,207 patients with non-metastatic, castration-resistant prostate cancer. Patients in the trial either received Erleada or a placebo. All patients were also treated with hormone therapy, either with gonadotropin-releasing hormone (GnRH) analog therapy or with surgery to lower the amount of testosterone in their body (surgical castration). The median metastasis-free survival for patients taking Erleada was 40.5 months compared to 16.2 months for patients taking a placebo.
Common side effects of Erleada include fatigue, high blood pressure (hypertension), rash, diarrhea, nausea, weight loss, joint pain (arthralgia), falls, hot flush, decreased appetite, fractures and swelling in the limbs (peripheral edema).
Severe side effects of Erleada include falls, fractures and seizures.
This application was granted Priority Review, under which the FDA’s goal is to take action on an application within 6 months where the agency determines that the drug, if approved, would significantly improve the safety or effectiveness of treating, diagnosing or preventing a serious condition.
The sponsor for Erleada is the first participant in the FDA’s recently-announced Clinical Data Summary Pilot Program, an effort to provide stakeholders with more usable information on the clinical evidence supporting drug product approvals and more transparency into the FDA’s decision-making process. Soon after approval, certain information from the clinical summary report will post with the Erleada entry on Drugs@FDA and on the new pilot program landing page.
The FDA granted the approval of Erleada to Janssen Pharmaceutical Companies.
//////////////fda 2018, Erleada, apalutamide, Priority Review, Janssen
FDA approves new treatment for certain digestive tract cancers Lutathera (lutetium Lu 177 dotatate)

lutetium Lu 177 dotatate
FDA approves new treatment for certain digestive tract cancers
The U.S. Food and Drug Administration today approved Lutathera (lutetium Lu 177 dotatate) for the treatment of a type of cancer that affects the pancreas or gastrointestinal tract called gastroenteropancreatic neuroendocrine tumors (GEP-NETs). This is the first time a radioactive drug, or radiopharmaceutical, has been approved for the treatment of GEP-NETs. Lutathera is indicated for adult patients with somatostatin receptor-positive GEP-NETs. Continue reading.\
January 26, 2018
Release
The U.S. Food and Drug Administration today approved Lutathera (lutetium Lu 177 dotatate) for the treatment of a type of cancer that affects the pancreas or gastrointestinal tract called gastroenteropancreatic neuroendocrine tumors (GEP-NETs). This is the first time a radioactive drug, or radiopharmaceutical, has been approved for the treatment of GEP-NETs. Lutathera is indicated for adult patients with somatostatin receptor-positive GEP-NETs.
“GEP-NETs are a rare group of cancers with limited treatment options after initial therapy fails to keep the cancer from growing,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “This approval provides another treatment choice for patients with these rare cancers. It also demonstrates how the FDA may consider data from therapies that are used in an expanded access program to support approval for a new treatment.”
GEP-NETs can be present in the pancreas and in different parts of the gastrointestinal tract such as the stomach, intestines, colon and rectum. It is estimated that approximately one out of 27,000 people are diagnosed with GEP-NETs per year.
Lutathera is a radioactive drug that works by binding to a part of a cell called a somatostatin receptor, which may be present on certain tumors. After binding to the receptor, the drug enters the cell allowing radiation to cause damage to the tumor cells.
The approval of Lutathera was supported by two studies. The first was a randomized clinical trial in 229 patients with a certain type of advanced somatostatin receptor-positive GEP-NET. Patients in the trial either received Lutathera in combination with the drug octreotide or octreotide alone. The study measured the length of time the tumors did not grow after treatment (progression-free survival). Progression-free survival was longer for patients taking Lutathera with octreotide compared to patients who received octreotide alone. This means the risk of tumor growth or patient death was lower for patients who received Lutathera with octreotide compared to that of patients who received only octreotide.
The second study was based on data from 1,214 patients with somatostatin receptor-positive tumors, including GEP-NETS, who received Lutathera at a single site in the Netherlands. Complete or partial tumor shrinkage was reported in 16 percent of a subset of 360 patients with GEP-NETs who were evaluated for response by the FDA. Patients initially enrolled in the study received Lutathera as part of an expanded access program. Expanded access is a way for patients with serious or immediately life-threatening diseases or conditions who lack therapeutic alternatives to gain access to investigational drugs for treatment use.
Common side effects of Lutathera include low levels of white blood cells (lymphopenia), high levels of enzymes in certain organs (increased GGT, AST and/or ALT), vomiting, nausea, high levels of blood sugar (hyperglycemia) and low levels of potassium in the blood (hypokalemia).
Serious side effects of Lutathera include low levels of blood cells (myelosuppression), development of certain blood or bone marrow cancers (secondary myelodysplastic syndrome and leukemia), kidney damage (renal toxicity), liver damage (hepatotoxicity), abnormal levels of hormones in the body (neuroendocrine hormonal crises) and infertility. Lutathera can cause harm to a developing fetus; women should be advised of the potential risk to the fetus and to use effective contraception. Patients taking Lutathera are exposed to radiation. Exposure of other patients, medical personnel, and household members should be limited in accordance with radiation safety practices.
Lutathera was granted Priority Review, under which the FDA’s goal is to take action on an application within six months where the agency determines that the drug, if approved, would significantly improve the safety or effectiveness of treating, diagnosing or preventing a serious condition. Lutathera also received Orphan Drugdesignation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted the approval of Lutathera to Advanced Accelerator Applications.
MORE FROM PUBLIC DOMAIN……………..
WATCH THIS SPACE FOR SYNTHESIS COMING
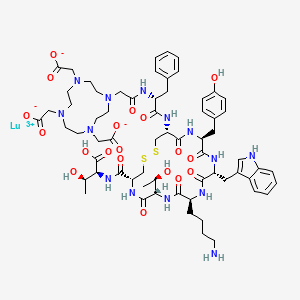
Dotatate lutenium Lu-177; 437608-50-9; DTXSID20195927
2-[4-[2-[[(2R)-1-[[(4R,7S,10S,13R,16S,19R)-10-(4-aminobutyl)-4-[[(1S,2R)-1-carboxy-2-hydroxypropyl]carbamoyl]-7-[(1R)-1-hydroxyethyl]-16-[(4-hydroxyphenyl)methyl]-13-(1H-indol-3-ylmethyl)-6,9,12,15,18-pentaoxo-1,2-dithia-5,8,11,14,17-pentazacycloicos-19-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-2-oxoethyl]-7,10-bis(carboxylatomethyl)-1,4,7,10-tetrazacyclododec-1-yl]acetate;lutetium(3+)

Lutetium-177
 Lutetium-177 has been quite a late addition as an isotope of significance to the nuclear medicine yet it is making big strides especially as a therapeutic radiopharmaceutical for neuroendocrine tumours in the form of 177Lu-DOTA-TATE on regular basis as described by Das & Pillai (2013). Lutetium-177 a lanthanide is an f block element that has a half-life of 6.7 days and decays mainly by beta emission to Hf-177, is accompanied by two gamma ray emissions. These radionuclide properties are very similar to those of I-131 which has long served as a therapeutic radionuclide, it was therefore not surprising that Lu-177 also emerged as a highly valuable radionuclide for similar applications,
There are several other upcoming applications especially for bone pain palliatiion. As a result of its convenient production logistics Lu-177 as discussed by Pillai et al (2003) is fast emerging a radionuclide of choice in radionuclide therapy (RNT).
Lu-177 can be prepared in a nuclear reactor by one of the two reactions given below :
176Lu(n,gamma)177Lu or
176Yb(n,gamma)177Yb –beta–> 177Lu
The former reaction has a high thermal neutron capture cross section and is presently the method adopted at our reactors in spite of the formation of long lived Lu-177m whose yield is very much low and is considered insignificant to cause any great concern.
Lutetium-177 Impact
 Recently there has been a rush of several research reviews and articles where Lu-177 holds the centre stage, for example, Banerjee et al (2015) have reviewed the chemistry and applications of Lu-177; Dash et al (2015) reviewed its production and available options; Knapp & Pillai (2015) highlighted its usefulness in cancer treatment and chronic diseases and Pillai and Knapp (2015) have discussed the evolving role of Lu-177 in nuclear medicine with this ready availability of Lu-177. Peptide receptor radionuclide therapy is one of the upcoming field of investigation where Lu-177 holds much promise among few other radionuclides. Indeed Lutetium-177 has covered a good distance especially for Therapeutic and as a palliative radiopharmaceutical.
Chemistry
Das et al (2014) have described the preparation of Lu-177 EDTMP kit.
Parus et al (2015) have discussed chemistry of bifunctional chelating agents for binding Lu-177.
Gupta et al (2014) have compiled methods of labelleing antibdoies with radioiodine and radiometals.
Applications
Limouris (2012) has reviewed applications in neuroendocrine tumors with focus on Liver metastasis. Das and Banerjee (2015) described the potential theranostic applications with Lu-177.
Anderson et al (1960) were among the first to use Lutetium (as chloride and citrate) in a clinical trial which were not so successful and did not encourage much promise. Keeling et al (1988) published their results with in vitro uptake of Lutetium hydroxylapatite particles. Lu-EDTMP as bone palliating agent by Ando et al (1998) soon followed, However the greatest impact was seen with the advent of a somatostatin analogue Lu-DOTATATE for targetting neuroendocrine tumors reported by Kwekkeboom et al (2001) and reviewed recently by Bodei et al (2013).
PRRNT – IAEA (2013) has brought out a human health series booklet on the subject with emphasis on neuroendocrine tumors.
177Lu Labelled Peptides in NET Kam et al (2012).
177Lu- DOTATATE – PRRNT – Bakker et al (2006)
177Lu-EDTMP – Bone Pain Palliation – Bahrami-Samani et al (2012)
177Lu-EDTMP – Pharmacokinetics, dosimetry and Therapeutic efficacy – Chakraborty S et al (2015)
177Lu-Hydroxylapatite – Radiosynovectomy – Kamalleshwaran et al. (2014) Shinto et al. (2015)
117Lu- Radioimmunotherapy – Kameshwaran et al (2015)
177Lu – Pretargeted Radioimmunotherapy (PRIT) Frost et al (2015).
More specific applications and additional information about the highly valuable therapeutic isotope would soon be added.
References and Notes
Anderson J, Farmer FT, Haggith JW, Hill M. (1960). The treatment of myelomatosis with Lutetium. Br J Radiol. 33:374-378.
Ando A, Ando L, Tonami N, Kinuya S, Kazuma K, Kataiwa A, Nakagawa M, Fujita N. (1998). 177Lu-EDTMP: a potential therapeutic bone agent. Nucl Med Commun. 19: 587-591.
Bahrami-Samani A, Anvari A, Jalilian AR, Shirvani-Arani S, Yousefnia H, Aghamiri MR, Ghannadi-Maragheh M. (2012). Production, Quality Control and Pharmacokinetic Studies of 177Lu-EDTMP for Human Bone Pain Palliation Therapy Trials. Iran J Pharm Res. 11:137-44.
Bakker WH, Breeman WAP, Kwekkeboom DJ, De Jong LC, Krenning EP. ((2006) Practical aspects of peptide receptor radionuclide therapy with [177Lu][DOTA0, Tyr3]octreotate. Q J Nucl Med Mol Imaging 50: 265-271.
Banerjee S, Pillai MR, Knapp FF (2015). Lutetium-177 Therapeutic Radiopharmaceuticals: Linking Chemistry, Radiochemistry, and Practical Applications. Chem Rev. 115: 2934-2974.
Bodei L, Mueller-Brand J, Baum RP, Pavel ME, Hörsch D, O’Dorisio MS, O’Dorisio TM, Howe JR, Cremonesi M, Kwekkeboom DJ, Zaknun JJ. (2013).The joint IAEA, EANM, and SNMMI practical guidance on peptide receptor radionuclide therapy (PRRNT) in neuroendocrine tumours. Eur J Nucl Med Mol Imaging. 2013 40:800-16.
Chakraborty S, Balogh L, Das T, Polyák A, Andócs G, Máthé D, Király R, Thuróczy J, Chaudhari PR, Jánoki GA, Jánoki G, Banerjee S, Pillai MR (2015). Evaluation of 177Lu-EDTMP in dogs with spontaneous tumor involving bone: Pharmacokinetics, dosimetry and therapeutic efficacy. Curr Radiopharm (ahead of Pub)
Das T, Banerjee S. (2015). Theranostic Applications of Lutetium-177 in Radionuclide Therapy. Curr Radiopharm. (ahead of print).
Das T , Sarma HD, Shinto A, Kamaleshwaran KK, Banerjee S. (2014). Formulation, Preclinical Evaluation, and Preliminary Clinical Investigation of an In-House Freeze-Dried EDTMP Kit Suitable for the Preparation of Lu-177-EDTMP. Cancer Biotherap Radiopharm. 29: (ahead of publication).
Das T, Pillai M.R.A. (2013).Options to meet the future global demand of radionuclides for radionuclide therapy. Nucl Med Biol. 40: 23-32.
Dash A, Pillai MR, Knapp FF Jr. (2015). Production of 177Lu for targeted radionuclide therapy : Available options. Nucl Med Mol Imaging. 49: 85-107.
Frost SH, Frayo SL, Miller BW, Orozco JJ, Booth GC, Hylarides MD, Lin Y, Green DJ, Gopal AK, Pagel JM, Bäck TA, Fisher DR, Press OW. (2015) Comparative efficacy of 177Lu and 90Y for anti-CD20 pretargeted radioimmunotherapy in murine lymphoma xenograft models. PLoS One. 2015 Mar 18;10(3):e0120561. Gupta S, Batra S, Jain M (2014) Antibody labeling with radioiodine and radiometals. Methods Mol Biol. 2014;1141:147-57.
IAEA (2013). Peptide receptor radionuclide therapy (PRRNT) for neuroendocrine tumors. IAEA Human Health Series No. 20., IAEA, Vienna.
Kam BLR, Teunissen JJM, Krenning EP, de Herder WW, Khan S, van Vliet EI, Kwekkeboom DJ. (2012). Lutetium-labelled peptides for therapy of neuroendocrine tumours. Eur J Nucl Med Mol Imaging 39 (Suppl 1):S103–S112.
Kamaleshwaran KK, Rajamani V, Thirumalaisamy SG, Chakraborty S, Kalarikal R, Mohanan V, Shinto AS.(2014).
Radiosynovectomy of the elbow joint synovitis in rheumatoid arthritis treated with Lutetium – 177 labeled hydroxylapatite (Lu-177HA) particulates; first case report and image of Lu -177 HA in the elbow joint. Indian J Nucl Med. 29:270-2.
Kameshwaran M, Pandey U, Dhakan C, Pathak K, Gota V, Vimalnath KV, Dash A, Samuel G. (2015) .Synthesis and Preclinical Evaluation of (177)Lu-CHX-A”-DTPA-Rituximab as a Radioimmunotherapeutic Agent for Non-Hodgkin’s Lymphoma. Cancer Biother Radiopharm. 2015 Aug;30(6):240-6Kwekkeboom DJ, Bakker WH, Kooij PP, Konijnenberg MW, Srinivasan A, Erion JL, Schmidt MA, Bugaj JL, de Jong M, Krenning EP.. (2001). [177Lu-DOTAOTyr3]octreotate: comparison with [111In-DTPAo]octreotide in patients.Eur J Nucl Med. 28: 1319-1325.
(Russ) Knapp FF, Pillai MR.(2015). Lutetium-177 Labeled Therapeutics: Emerging Importance for Cancer Treatment and Therapy of Chronic Disease. Curr Radiopharm. (ahead of Pub)
Parus JL, Pawlak D, Mikolajczak R, Duatti A. (2015) Chemistry and bifunctional chelating agents for binding 177Lu Curr Radiopharm (Ahead of Pub)
Limouris G. (2012) Neuroendocrine tumors: a focus on liver metastatic lesions. Front Oncol. 2:20 (Ahead of Pub) PMC article
Pillai MR, (Russ) Knapp FF. (2015). Evolving Important Role of Lutetium-177 for Therapeutic Nuclear Medicine Curr Radiopharm (ahead of print).
Pillai MR, Chakraborty S, Das T, Venkatesh M, Ramamoorthy N. (2003). Production logistics of 177Lu for radionuclide therapy. Appl Radiat Isot. 59: 109-118.
Shinto AS, Kamaleshwaran KK, Vyshakh K, Thirumalaisamy SG, Karthik S, Nagaprabhu VN, Vimalnath KV, Das T, Chakraborty S, Banerjee S. (2015) Radiosynovectomy of Painful Synovitis of Knee Joints Due to Rheumatoid Arthritis by Intra‑Articular Administration of 177Lu‑Labeled Hydroxyapatite Particulates: First Human Study and Initial Indian Experience. World J Nucl Med. 14: (ahead of print).
Videos
|
 |
|
| Names | |
|---|---|
| Other names
DOTA-(Tyr3)-octreotate
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChemSpider | |
|
PubChem CID
|
|
| Properties | |
| C65H90N14O19S2 | |
| Molar mass | 1,435.63 g·mol−1 |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
DOTA-TATE, DOTATATE or DOTA-octreotate is a substance which, when bound to various radionuclides, has been tested for the treatment and diagnosis of certain types of cancer, mainly neuroendocrine tumours.
Chemistry and mechanism of action
DOTA-TATE is an amide of the acid DOTA (top left in the image), which acts as a chelator for a radionuclide, and (Tyr3)-octreotate, a derivative of octreotide. The latter binds to somatostatin receptors, which are found on the cell surfaces of a number of neuroendocrine tumours, and thus directs the radioactivity into the tumour.
Usage examples
Gallium (68Ga) DOTA-TATE (GaTate[1]) is used for tumour diagnosis in positron emission tomography (PET).[2] DOTA-TATE PET/CT has a much higher sensitivitycompared to In-111 octreotide imaging.[1]
Lutetium (177Lu) DOTA-TATE[3] has been tested for the treatment of tumors such as carcinoid and endocrine pancreatic tumor. It is also known as Lutathera.[4]
Patients are typically treated with an intravenous infusion of 7.5 GBq of lutetium-177 octreotate. After about four to six hours, the exposure rate of the patient has fallen to less than 25 microsieverts per hour at one metre and the patients can be discharged from hospital.
A course of therapy consists of four infusions at three monthly intervals.[5]
Availability
Lu177 octreotate therapy is currently available under research protocols in five different medical centers in North America: Los Angeles (CA), Quebec City, (Qc), Birmingham, AL, Edmonton, (Ab), London, (On) as Houston (Tx) on clinical trial.[6] Medical centers in Europe also offer this treatment. For instance: Cerrahpasa Hospital in Turkey, Uppsala Centre of Excellence in Neuroendocrine Tumors in Sweden and Erasmus University in the Netherlands.[7] In Israel, treatment is available at Hadassah Ein Kerem Medical Center. In Australia, treatment is available at St George Hospital and Royal North Shore Hospital, Sydney;[8] the Royal Brisbane and Women’s Hospital in Brisbane [9], the Peter MacCallum Cancer Centre [1] and at the Department of Nuclear Medicine at Fremantle Hospital in Western Australia.[10] In Aarhus universitet hospital in Denmark. In the coming years such therapy will also become commercially available in Latvia, Riga – “Clinic of nuclear medicine”.
See also
- DOTATOC or edotreotide, a similar compound
References
- ^ Jump up to:a b c Hofman, M. S.; Kong, G.; Neels, O. C.; Eu, P.; Hong, E.; Hicks, R. J. (2012). “High management impact of Ga-68 DOTATATE (GaTate) PET/CT for imaging neuroendocrine and other somatostatin expressing tumours”. Journal of Medical Imaging and Radiation Oncology. 56 (1): 40–47. doi:10.1111/j.1754-9485.2011.02327.x. PMID 22339744.
- Jump up^ Breeman, W. A. P.; De Blois, E.; Sze Chan, H.; Konijnenberg, M.; Kwekkeboom, D. J.; Krenning, E. P. (2011). “68Ga-labeled DOTA-Peptides and 68Ga-labeled Radiopharmaceuticals for Positron Emission Tomography: Current Status of Research, Clinical Applications, and Future Perspectives”. Seminars in Nuclear Medicine. 41 (4): 314–321. doi:10.1053/j.semnuclmed.2011.02.001. PMID 21624565.
- Jump up^ Bodei, L.; Cremonesi, M.; Grana, C. M.; Fazio, N.; Iodice, S.; Baio, S. M.; Bartolomei, M.; Lombardo, D.; Ferrari, M. E.; Sansovini, M.; Chinol, M.; Paganelli, G. (2011). “Peptide receptor radionuclide therapy with 177Lu-DOTATATE: The IEO phase I-II study”. European Journal of Nuclear Medicine and Molecular Imaging. 38(12): 2125–2135. doi:10.1007/s00259-011-1902-1. PMID 21892623.
- Jump up^ Radiolabeled Peptide Offers PFS Benefit in Midgut NET
- Jump up^ Claringbold, P. G.; Brayshaw, P. A.; Price, R. A.; Turner, J. H. (2010). “Phase II study of radiopeptide 177Lu-octreotate and capecitabine therapy of progressive disseminated neuroendocrine tumours”. European Journal of Nuclear Medicine and Molecular Imaging. 38 (2): 302–311. doi:10.1007/s00259-010-1631-x. PMID 21052661.
- Jump up^ Clinical trial number NCT01237457 for “177Lutetium-DOTA-Octreotate Therapy in Somatostatin Receptor-Expressing Neuroendocrine Neoplasms” at ClinicalTrials.gov
- Jump up^ “PRRT Behandelcentrum Rotterdam”. PRRT Behandelcentrum Rotterdam. Erasmus Universiteit.
- Jump up^ http://www.swslhd.nsw.gov.au/liverpool/pet/PET.html
- Jump up^ https://agitg.org.au/control-nets-study-set-to-commence
- Jump up^ Turner, J. H. (2012). “Outpatient therapeutic nuclear oncology”. Annals of Nuclear Medicine. 26 (4): 289–97. doi:10.1007/s12149-011-0566-z. PMID 22222779.
- Freedman, N; Klein, M; Gross, D; Glasberg, S; Meirovitz, A; Maimon, O; Krausz, Y; Bar-Shalom, R (2014). “Lu177-DOTATATE therapy for NET: Does tumor dose predict response?”. J Nucl Med. 55 (Supplement 1).
//////////////Lutathera, lutetium Lu 177 dotatate, fda 2018, PRIORITY REVIEW, ORPHAN DRUG
CC(C1C(=O)NC(CSSCC(C(=O)NC(C(=O)NC(C(=O)NC(C(=O)N1)CCCCN)CC2=CNC3=CC=CC=C32)CC4=CC=C(C=C4)O)NC(=O)C(CC5=CC=CC=C5)NC(=O)CN6CCN(CCN(CCN(CC6)CC(=O)[O-])CC(=O)[O-])CC(=O)[O-])C(=O)NC(C(C)O)C(=O)O)O.[Lu+3]
FDA approves first drug for Eosinophilic Granulomatosis with Polyangiitis, a rare disease formerly known as the Churg-Strauss Syndrome
The U.S. Food and Drug Administration today expanded the approved use of Nucala (mepolizumab) to treat adult patients with eosinophilic granulomatosis with polyangiitis (EGPA), a rare autoimmune disease that causes vasculitis, an inflammation in the wall of blood vessels of the body. This new indication provides the first FDA-approved therapy specifically to treat EGPA. Continue reading.
December 12, 2017
Release
The U.S. Food and Drug Administration today expanded the approved use of Nucala (mepolizumab) to treat adult patients with eosinophilic granulomatosis with polyangiitis (EGPA), a rare autoimmune disease that causes vasculitis, an inflammation in the wall of blood vessels of the body. This new indication provides the first FDA-approved therapy specifically to treat EGPA.
According to the National Institutes of Health, EGPA (formerly known as Churg-Strauss syndrome) is a condition characterized by asthma, high levels of eosinophils (a type of white blood cell that helps fight infection), and inflammation of small- to medium-sized blood vessels. The inflamed vessels can affect various organ systems including the lungs, gastrointestinal tract, skin, heart and nervous system. It is estimated that approximately 0.11 to 2.66 new cases per 1 million people are diagnosed each year, with an overall prevalence of 10.7 to 14 per 1,000,000 adults.
“Prior to today’s action, patients with this challenging, rare disease did not have an FDA-approved treatment option,” said Badrul Chowdhury, M.D., Ph.D., director of the Division of Pulmonary, Allergy, and Rheumatology Products in the FDA’s Center for Drug Evaluation and Research. “The expanded indication of Nucala meets a critical, unmet need for EGPA patients. It’s notable that patients taking Nucala in clinical trials reported a significant improvement in their symptoms.”
The FDA granted this application Priority Review and Orphan Drug designations. Orphan Drug designation provides incentives to assist and encourage the development of drugs for rare diseases.
Nucala was previously approved in 2015 to treat patients age 12 years and older with a specific subgroup of asthma (severe asthma with an eosinophilic phenotype) despite receiving their current asthma medicines. Nucala is an interleukin-5 antagonist monoclonal antibody (IgG1 kappa) produced by recombinant DNA technology in Chinese hamster ovary cells.
Nucala is administered once every four weeks by subcutaneous injection by a health care professional into the upper arm, thigh, or abdomen.
The safety and efficacy of Nucala was based on data from a 52-week treatment clinical trial that compared Nucala to placebo. Patients received 300 milligrams (mg) of Nucala or placebo administered subcutaneously once every four weeks while continuing their stable daily oral corticosteroids (OCS) therapy. Starting at week four, OCS was tapered during the treatment period. The primary efficacy assessment in the trial measured Nucala’s treatment impact on disease remission (i.e., becoming symptom free) while on an OCS dose less than or equal to 4 mg of prednisone. Patients receiving 300 mg of Nucala achieved a significantly greater accrued time in remission compared with placebo. A significantly higher proportion of patients receiving 300 mg of Nucala achieved remission at both week 36 and week 48 compared with placebo. In addition, significantly more patients who received 300 mg of Nucala achieved remission within the first 24 weeks and remained in remission for the remainder of the 52-week study treatment period compared with patients who received the placebo.
The most common adverse reactions associated with Nucala in clinical trials included headache, injection site reaction, back pain, and fatigue.
Nucala should not be administered to patients with a history of hypersensitivity to mepolizumab or one of its ingredients. It should not be used to treat acute bronchospasm or status asthmaticus. Hypersensitivity reactions, including anaphylaxis, angioedema, bronchospasm, hypotension, urticaria, rash, have occurred. Patients should discontinue treatment in the event of a hypersensitivity reaction. Patients should not discontinue systemic or inhaled corticosteroids abruptly upon beginning treatment with Nucala. Instead, patients should decrease corticosteroids gradually, if appropriate.
Health care providers should treat patients with pre-existing helminth infections before treating with Nucala because it is unknown if Nucala would affect patients’ responses against parasitic infections. In addition, herpes zoster infections have occurred in patients receiving Nucala. Health care providers should consider vaccination if medically appropriate.
The FDA granted approval of Nucala to GlaxoSmithKline.
//////////////Nucala, mepolizumab, fda 2017, gsk, Eosinophilic Granulomatosis, Polyangiitis, Churg-Strauss Syndrome, Priority Review, Orphan Drug
FDA approves new treatment for certain advanced or metastatic breast cancers
FDA approves new treatment for certain advanced or metastatic breast cancers
|
|||
https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm578071.htm
(abemaciclib)
September 28, 2017
Release
The U.S. Food and Drug Administration today approved Verzenio (abemaciclib) to treat adult patients who have hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced or metastatic breast cancer that has progressed after taking therapy that alters a patient’s hormones (endocrine therapy). Verzenio is approved to be given in combination with an endocrine therapy, called fulvestrant, after the cancer had grown on endocrine therapy. It is also approved to be given on its own, if patients were previously treated with endocrine therapy and chemotherapy after the cancer had spread (metastasized).
“Verzenio provides a new targeted treatment option for certain patients with breast cancer who are not responding to treatment, and unlike other drugs in the class, it can be given as a stand-alone treatment to patients who were previously treated with endocrine therapy and chemotherapy,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research.
Verzenio works by blocking certain molecules (known as cyclin-dependent kinases 4 and 6), involved in promoting the growth of cancer cells. There are two other drugs in this class that are approved for certain patients with breast cancer, palbociclib approved in February 2015 and ribociclib approved in March 2017.
Breast cancer is the most common form of cancer in the United States. The National Cancer Institute at the National Institutes of Health estimates approximately 252,710 women will be diagnosed with breast cancer this year, and 40,610 will die of the disease. Approximately 72 percent of patients with breast cancer have tumors that are HR-positive and HER2-negative.
The safety and efficacy of Verzenio in combination with fulvestrant were studied in a randomized trial of 669 patients with HR-positive, HER2-negative breast cancer that had progressed after treatment with endocrine therapy and who had not received chemotherapy once the cancer had metastasized. The study measured the length of time tumors did not grow after treatment (progression-free survival). The median progression-free survival for patients taking Verzenio with fulvestrant was 16.4 months compared to 9.3 months for patients taking a placebo with fulvestrant.
The safety and efficacy of Verzenio as a stand-alone treatment were studied in a single-arm trial of 132 patients with HR-positive, HER2-negative breast cancer that had progressed after treatment with endocrine therapy and chemotherapy after the cancer metastasized. The study measured the percent of patients whose tumors completely or partially shrank after treatment (objective response rate). In the study, 19.7 percent of patients taking Verzenio experienced complete or partial shrinkage of their tumors for a median 8.6 months.
Common side effects of Verzenio include diarrhea, low levels of certain white blood cells (neutropenia and leukopenia), nausea, abdominal pain, infections, fatigue, low levels of red blood cells (anemia), decreased appetite, vomiting and headache.
Serious side effects of Verzenio include diarrhea, neutropenia, elevated liver blood tests and blood clots (deep venous thrombosis/pulmonary embolism). Women who are pregnant should not take Verzenio because it may cause harm to a developing fetus.
The FDA granted this application Priority Review and Breakthrough Therapydesignations.
The FDA granted the approval of Verzenio to Eli Lilly and Company.
//////////Verzenio, abemaciclib, fda 2017, metastatic breast cancers, Eli Lilly , Priority Review, Breakthrough Therapy designations, antibodies
FDA approves Mavyret (glecaprevir and pibrentasvir) for Hepatitis C

Glecaprevir

Pibrentasvir
08/03/2017 03:06 PM EDT
The U.S. Food and Drug Administration today approved Mavyret (glecaprevir and pibrentasvir) to treat adults with chronic hepatitis C virus (HCV) genotypes 1-6 without cirrhosis (liver disease) or with mild cirrhosis, including patients with moderate to severe kidney disease and those who are on dialysis. Mavyret is also approved for adult patients with HCV genotype 1 infection who have been previously treated with a regimen either containing an NS5A inhibitor or an NS3/4A protease inhibitor but not both.
The U.S. Food and Drug Administration today approved Mavyret (glecaprevir and pibrentasvir) to treat adults with chronic hepatitis C virus (HCV) genotypes 1-6 without cirrhosis (liver disease) or with mild cirrhosis, including patients with moderate to severe kidney disease and those who are on dialysis. Mavyret is also approved for adult patients with HCV genotype 1 infection who have been previously treated with a regimen either containing an NS5A inhibitor or an NS3/4A protease inhibitor but not both.
Mavyret is the first treatment of eight weeks duration approved for all HCV genotypes 1-6 in adult patients without cirrhosis who have not been previously treated. Standard treatment length was previously 12 weeks or more.
“This approval provides a shorter treatment duration for many patients, and also a treatment option for certain patients with genotype 1 infection, the most common HCV genotype in the United States, who were not successfully treated with other direct-acting antiviral treatments in the past,” said Edward Cox, M.D., director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research.
Hepatitis C is a viral disease that causes inflammation of the liver that can lead to diminished liver function or liver failure. According to the Centers for Disease Control and Prevention, an estimated 2.7 to 3.9 million people in the United States have chronic HCV. Some patients who suffer from chronic HCV infection over many years may have jaundice (yellowish eyes or skin) and complications, such as bleeding, fluid accumulation in the abdomen, infections, liver cancer and death.
There are at least six distinct HCV genotypes, or strains, which are genetically distinct groups of the virus. Knowing the strain of the virus can help inform treatment recommendations. Approximately 75 percent of Americans with HCV have genotype 1; 20-25 percent have genotypes 2 or 3; and a small number of patients are infected with genotypes 4, 5 or 6.
The safety and efficacy of Mavyret were evaluated during clinical trials enrolling approximately 2,300 adults with genotype 1, 2, 3, 4, 5 or 6 HCV infection without cirrhosis or with mild cirrhosis. Results of the trials demonstrated that 92-100 percent of patients who received Mavyret for eight, 12 or 16 weeks duration had no virus detected in the blood 12 weeks after finishing treatment, suggesting that patients’ infection had been cured.
Treatment duration with Mavyret differs depending on treatment history, viral genotype, and cirrhosis status.
The most common adverse reactions in patients taking Mavyret were headache, fatigue and nausea.
Mavyret is not recommended in patients with moderate cirrhosis and contraindicated in patients with severe cirrhosis. It is also contraindicated in patients taking the drugs atazanavir and rifampin.
Hepatitis B virus (HBV) reactivation has been reported in HCV/HBV coinfected adult patients who were undergoing or had completed treatment with HCV direct-acting antivirals, and who were not receiving HBV antiviral therapy. HBV reactivation in patients treated with direct-acting antiviral medicines can result in serious liver problems or death in some patients. Health care professionals should screen all patients for evidence of current or prior HBV infection before starting treatment with Mavyret.
The FDA granted this application Priority Review and Breakthrough Therapydesignations.
The FDA granted approval of Mavyret to AbbVie Inc.
////////// glecaprevir, pibrentasvir, fda 2017, Hepatitis C, AbbVie Inc, Priority Review, Breakthrough Therapy designations,
 |
|
| Clinical data | |
|---|---|
| Trade names | Maviret (combination with pibrentasvir) |
| Routes of administration |
By mouth |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| Synonyms | ABT-493 |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C38H46F4N6O9S |
| Molar mass | 838.87 g·mol−1 |
Glecaprevir (INN,[1] codenamed ABT-493) is a hepatitis C virus (HCV) nonstructural (NS) protein 3/4A protease inhibitor that was identified jointly by AbbVie and Enanta Pharmaceuticals. It is being developed as a treatment of chronic hepatitis C infection in co-formulation with an HCV NS5A inhibitor pibrentasvir. Together they demonstrated potent antiviral activity against major HCV genotypes and high barriers to resistance in vitro.[2]
On December 19, 2016, AbbVie submitted New Drug Application to U.S. Food and Drug Administration for glecaprevir/pibrentasvir (trade name Maviret) regimen for the treatment of all major genotypes (1–6) of chronic hepatitis C.[3]
References
- Jump up^ “International Nonproprietary Names for Pharmaceutical Substances (INN). Recommended International Nonproprietary Names: List 76” (PDF). World Health Organization. p. 503. Retrieved 25 February 2017.
- Jump up^ Lawitz, EJ; O’Riordan, WD; Asatryan, A; Freilich, BL; Box, TD; Overcash, JS; Lovell, S; Ng, TI; Liu, W; Campbell, A; Lin, CW; Yao, B; Kort, J (28 December 2015). “Potent Antiviral Activities of the Direct-Acting Antivirals ABT-493 and ABT-530 with Three-Day Monotherapy for Hepatitis C Virus Genotype 1 Infection”. Antimicrobial Agents and Chemotherapy. 60 (3): 1546–55. PMC 4775945
 . PMID 26711747. doi:10.1128/AAC.02264-15.
. PMID 26711747. doi:10.1128/AAC.02264-15. - Jump up^ “AbbVie Submits New Drug Application to U.S. FDA for its Investigational Regimen of Glecaprevir/Pibrentasvir (G/P) for the Treatment of All Major Genotypes of Chronic Hepatitis C”. AbbVie Inc. North Chicago, Illinois, U.S.A. December 19, 2016. Retrieved 25 February 2017.
 |
|
| Identifiers | |
|---|---|
| Synonyms | ABT-530 |
| CAS Number | |
| Chemical and physical data | |
| Formula | C57H65F5N10O8 |
| Molar mass | 1,113.20 g·mol−1 |
Pibrentasvir is an antiviral agent.[1] In the United States, it is approved for use with glecaprevir as the combination drug glecaprevir/pibrentasvir (Mavyret) for the treatment of hepatitis C.[2]
References
- Jump up^ Ng, Teresa I.; Krishnan, Preethi; Pilot-Matias, Tami; Kati, Warren; Schnell, Gretja; Beyer, Jill; Reisch, Thomas; Lu, Liangjun; Dekhtyar, Tatyana; Irvin, Michelle; Tripathi, Rakesh; Maring, Clarence; Randolph, John T.; Wagner, Rolf; Collins, Christine (2017). “In Vitro Antiviral Activity and Resistance Profile of the Next-Generation Hepatitis C Virus NS5A Inhibitor Pibrentasvir”. Antimicrobial Agents and Chemotherapy. 61 (5): e02558–16. PMID 28193664. doi:10.1128/AAC.02558-16.
- Jump up^ Linda A. Johnson (August 3, 2017). “FDA OKs new drug to treat all forms of hepatitis C”. Fox Business.
FDA approves Vosevi for Hepatitis C
07/18/2017
The U.S. Food and Drug Administration today approved Vosevi to treat adults with chronic hepatitis C virus (HCV) genotypes 1-6 without cirrhosis (liver disease) or with mild cirrhosis.
The U.S. Food and Drug Administration today approved Vosevi to treat adults with chronic hepatitis C virus (HCV) genotypes 1-6 without cirrhosis (liver disease) or with mild cirrhosis. Vosevi is a fixed-dose, combination tablet containing two previously approved drugs – sofosbuvir and velpatasvir – and a new drug, voxilaprevir. Vosevi is the first treatment approved for patients who have been previously treated with the direct-acting antiviral drug sofosbuvir or other drugs for HCV that inhibit a protein called NS5A.
“Direct-acting antiviral drugs prevent the virus from multiplying and often cure HCV. Vosevi provides a treatment option for some patients who were not successfully treated with other HCV drugs in the past,” said Edward Cox, M.D., director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research.
Hepatitis C is a viral disease that causes inflammation of the liver that can lead to diminished liver function or liver failure. According to the Centers for Disease Control and Prevention, an estimated 2.7 to 3.9 million people in the United States have chronic HCV. Some patients who suffer from chronic HCV infection over many years may have jaundice (yellowish eyes or skin) and develop complications, such as bleeding, fluid accumulation in the abdomen, infections, liver cancer and death.
There are at least six distinct HCV genotypes, or strains, which are genetically distinct groups of the virus. Knowing the strain of the virus can help inform treatment recommendations. Approximately 75 percent of Americans with HCV have genotype 1; 20-25 percent have genotypes 2 or 3; and a small number of patients are infected with genotypes 4, 5 or 6.
The safety and efficacy of Vosevi was evaluated in two Phase 3 clinical trials that enrolled approximately 750 adults without cirrhosis or with mild cirrhosis.
The first trial compared 12 weeks of Vosevi treatment with placebo in adults with genotype 1 who had previously failed treatment with an NS5A inhibitor drug. Patients with genotypes 2, 3, 4, 5 or 6 all received Vosevi.
The second trial compared 12 weeks of Vosevi with the previously approved drugs sofosbuvir and velpatasvir in adults with genotypes 1, 2 or 3 who had previously failed treatment with sofosbuvir but not an NS5A inhibitor drug.
Results of both trials demonstrated that 96-97 percent of patients who received Vosevi had no virus detected in the blood 12 weeks after finishing treatment, suggesting that patients’ infection had been cured.
Treatment recommendations for Vosevi are different depending on viral genotype and prior treatment history.
The most common adverse reactions in patients taking Vosevi were headache, fatigue, diarrhea and nausea.
Vosevi is contraindicated in patients taking the drug rifampin.
Hepatitis B virus (HBV) reactivation has been reported in HCV/HBV coinfected adult patients who were undergoing or had completed treatment with HCV direct-acting antivirals, and who were not receiving HBV antiviral therapy. HBV reactivation in patients treated with direct-acting antiviral medicines can result in serious liver problems or death in some patients. Health care professionals should screen all patients for evidence of current or prior HBV infection before starting treatment with Vosevi.
The FDA granted this application Priority Review and Breakthrough Therapydesignations.
The FDA granted approval of Vosevi to Gilead Sciences Inc
//////////////Vosevi, Gilead Sciences Inc, Priority Review, Breakthrough Therapy designations, fda 2017, sofosbuvir, velpatasvir , voxilaprevir, Hepatitis B
