
Home » Posts tagged 'MERCK' (Page 2)
Tag Archives: MERCK
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
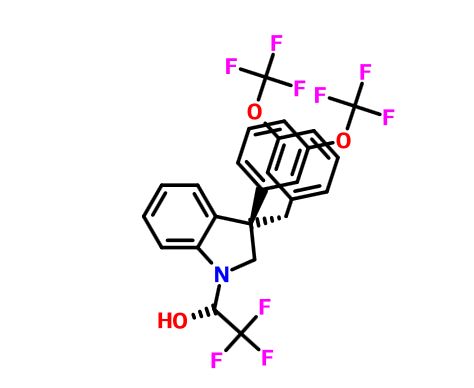
Merck’s Novel Indoline Cholesterol Ester Transfer Protein Inhibitors (CETP)
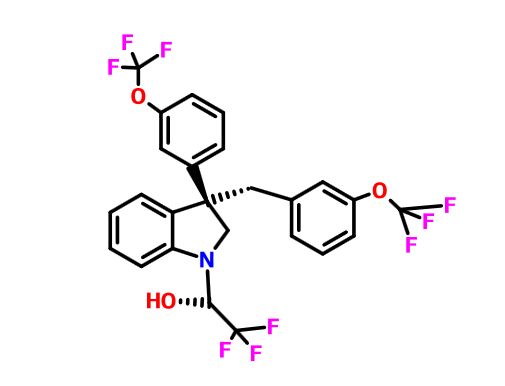
Indoline 7 as in ACS MEDCHEM LETTERS, DOI: 10.1021/acsmedchemlett.5b00404
and
eg 10 as in WO2015054088
(2R)- 1,1,1 -trifluoro-3-(3-(3-(trifluoromethoxy)benzyl)-3-(3- (trifluoromethoxy)-phenyl)indolin-l-yl)propan-2-ol.
1H-Indole-1-ethanol, 2,3-dihydro-3-[3-(trifluoromethoxy)phenyl]-3-[[3-(trifluoromethoxy)phenyl]methyl]-α-(trifluoromethyl)-, (αR)-
cas 1699732-96-1 R ISOMER
Merck Sharp & Dohme Corp. INNOVATOR

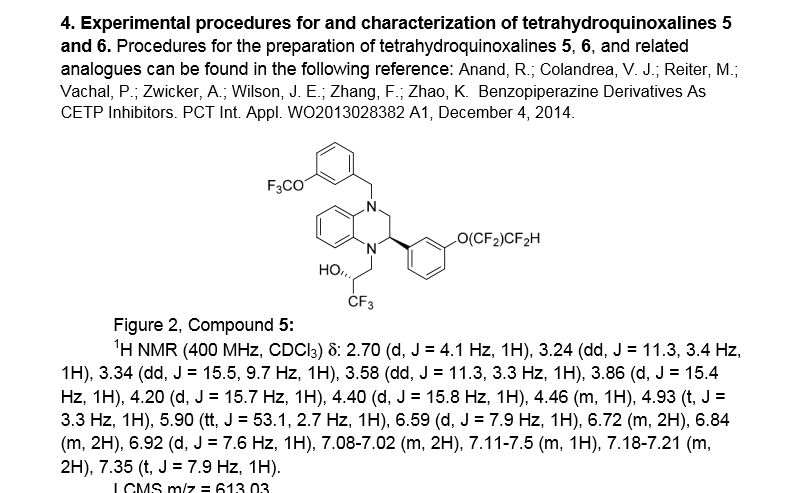
Using the collective body of known (CETP) inhibitors as inspiration for design, a structurally novel series of tetrahydroquinoxaline CETP inhibitors were discovered. An exemplar from this series, compound 5, displayed potent in vitro CETP inhibition and was efficacious in a transgenic cynomologus-CETP mouse HDL PD (pharmacodynamic) assay. However, an undesirable metabolic profile and chemical instability hampered further development of the series. A three-dimensional structure of tetrahydroquinoxaline inhibitor 6 was proposed from 1H NMR structural studies, and this model was then used in silico for the design of a new class of compounds based upon an indoline scaffold. This work resulted in the discovery of compound 7, which displayed potent in vitro CETP inhibition, a favorable PK–PD profile relative to tetrahydroquinoxaline 5, and dose-dependent efficacy in the transgenic cynomologus-CETP mouse HDL PD assay.
chemical compounds that inhibit cholesterol ester transfer protein (CETP) and are expected to have utility in raising HDL-C, lowering LDL-C, and in the treatment and prevention of atherosclerosis.
see………….http://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.5b00404
http://pubs.acs.org/doi/suppl/10.1021/acsmedchemlett.5b00404/suppl_file/ml5b00404_si_001.pdf
Discovery of Novel Indoline Cholesterol Ester Transfer Protein Inhibitors (CETP) through a Structure-Guided Approach
Atherosclerosis and its clinical consequences, including coronary heart disease
(CHD), stroke and peripheral vascular disease, represent a truly enormous burden to the health care systems of the industrialized world. In the United States alone, approximately 13 million patients have been diagnosed with CHD, and greater than one half million deaths are attributed to CHD each year. Further, this toll is expected to grow over the next quarter century as an epidemic in obesity and diabetes continues to grow.
It has long been recognized that in mammals, variations in circulating lipoprotein profiles correlate with the risk of atherosclerosis and CHD. The clinical success of HMG-CoA reductase inhibitors, especially the statins, in reducing coronary events is based on the reduction of circulating low density lipoprotein cholesterol (LDL-C), levels of which correlate directly with an increased risk for atherosclerosis. More recently, epidemiologic studies have
demonstrated an inverse relationship between high density lipoprotein cholesterol (HDL-C) levels and atherosclerosis, leading to the conclusion that low serum HDL-C levels are associated with an increased risk for CHD.
Metabolic control of lipoprotein levels is a complex and dynamic process involving many factors. One important metabolic control in man is the cholesteryl ester transfer protein (CETP), a plasma glycoprotein that catalyzes the movement of cholesteryl esters from HDL to the apoB containing lipoproteins, especially VLDL (see Hesler, C.B., et. al. (1987) Purification and characterization of human plasma cholesteryl ester transfer protein. J. Biol. Chem. 262(5), 2275-2282)). Under physiological conditions, the net reaction is a heteroexchange in which CETP carries triglyceride to HDL from the apoB lipoprotein and transports cholesterol ester from HDL to the apoB lipoprotein.
In humans, CETP plays a role in reverse cholesterol transport, the process whereby cholesterol is returned to the liver from peripheral tissues. Intriguingly, many animals do not possess CETP, including animals that have high HDL levels and are known to be resistant to coronary heart disease, such as rodents (see Guyard-Dangremont, V., et. al, (1998)
Phospholipid and cholesteryl ester transfer activities in plasma from 14 vertebrate species. Relation to atherogenesis susceptibility, Comp. Biochem. Physiol. B Biochem. Mol. Biol. 120(3), 517-525). Numerous epidemiologic studies correlating the effects of natural variation in CETP activity with respect to coronary heart disease risk have been performed, including studies on a small number of known human null mutations (see Hirano, K.-L, Yamashita, S. and Matsuzawa, Y. (2000) Pros and cons of inhibiting cholesteryl ester transfer protein, Curr. Opin. Lipidol. 11(6), 589-596). These studies have clearly demonstrated an inverse correlation between plasma HDL-C concentration and CETP activity (see Inazu, A., et. al. (2000) Cholesteryl ester transfer protein and atherosclerosis, Curr. Opin. Lipidol. 11(4), 389-396), leading to the hypothesis that pharmacologic inhibition of CETP lipid transfer activity may be beneficial to humans by increasing levels of HDL-C while lowering LDL-C.
Despite the significant therapeutic advance that statins such as simvastatin and atorvastatin represent, statins only achieve a risk reduction of approximately one-third in the treatment and prevention of atherosclerosis and ensuing atherosclerotic disease events.
Currently, few pharmacologic therapies are available that favorably raise circulating levels of HDL-C. Certain statins and some fibrates offer modest HDL-C gains. Niacin provides an effective therapy for raising HDL-C but suffers from patient compliance issues, due in part to side effects such as flushing. Drugs that inhibit CETP (CETP inhibitors) have been under development with the expectation that they will effectively raise HDL cholesterol levels and also reduce the incidence of atherosclerosis in patients. Torcetrapib was the first drug that was tested in a long-term outcomes clinical trial. The clinical trial of torcetrapib was terminated early due to a higher incidence of mortality in patients to whom torcetrapib and atorvastatin were administered concomitantly compared with patients who were treated with atorvastatin alone. The cause of the increased mortality is not completely understood, but it is not believed to be associated with the CETP inhibiting effects of the drug.
Two other drug candidates, dalcetrapib and anacetrapib, are currently being tested in Phase III clinical trials, including large scale outcomes trials. Data from the recently completed DEFINE Phase III trial of anacetrapib are promising. Patients who were being treated with anacetrapib along with baseline statin therapy showed an increase of HDL-C of 138% and a decrease of LDL-C of 40%> compared with patients who were treated with just a statin. See: N. Engl. J. Med. 2010: 363: 2406-15. The data in the DEFINE trial were sufficient to indicate that an increase in mortality for patients treated with anacetrapib is unlikely. Additional drug candidates are still being sought that may have properties that are advantageous compared with the CETP inhibitors that have so far been studied or are currently being studied. Such properties may include, for example, higher potency, reduced off-target activity, better pharmacodynamics, higher bioavailability, or a reduced food effect compared with many of the highly lipophilic compounds that have so far been studied. “Food effect” refers to the variability in exposure to the active drug that occurs depending on when the patient had last eaten, whether or not the drug is administered with food, and the fat content of the food.
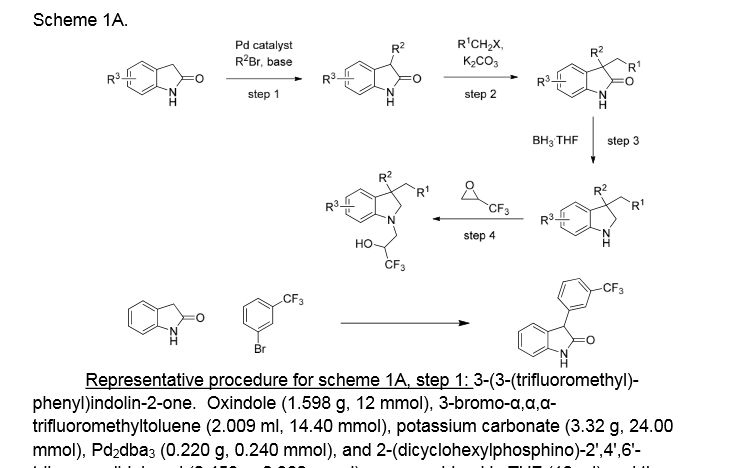
Example 18 as in patent

(R)- 1,1, 1 -trifluoro-3-((R)-4-(3-trifluoromethoxy)benzyl)-2-(3-(l, 1 ,2,2,-tetrafluoroethoxy)phenyl)-3,4- dihydroquinoxalin- 1 (2H)-yl)propan-2-ol
SPA: 15 nM
Example 18 was prepared from 2-bromo-l-(3-(l , 1 ,2,2,-tetrafluoroethoxy)phenyl)ethanone in three steps, using the reactions detailed in Schemes A6, A2 and Al . Spectral data are as follows: 1H NMR (400 MHz, CDC13) £2.70 (bd, J=4.1 Hz, IH), 3.24 (dd, J=l 1.3, 3.4 Hz, IH), 3.34 (dd, J=15.5, 9.7 Hz, IH), 3.58 (dd, J=l 1.3, 3.3 Hz, IH), 3.86 (d, J=15.4 Hz, IH), 4.20 (d, J=15.7 Hz, IH), 4.40 (d, J=15.8 Hz, IH), 4.46 (m, IH), 4.927 (t, J=3.3 Hz, IH), 5.90 (tt, J=53.1 , 2.7 Hz, IH), 6.59 (d, J= 7.9 Hz, IH), 6.72 (m, 2H), 6.84 (m, 2H), 6.92 (d, J=7.6 Hz, IH), 7.20 (m, 2H), 7.35 (t, J=7.9 Hz, IH), MS m/z = 613.03.
Scheme A12

Methyl 3 – { 1 – [(R)-3 ,3 ,3 -trifluoro-2-hy droxypropyl] -4- [3 -(trifluoromethoxy) benzyl]-l,2,3,4-tetrahydroquinoxalin-2-yl}benzoate (700 mg, 1.262 mmol) is made as described in Example 16 but with one stereochemical center unresolved. The compound was dissolved in MeOH (12.6mL), lithium hydroxide monohydrate (530 mg, 12.62 mmol) was added, and the reaction mixture was heated to 60°C for 4 hours. The crude mixture was dissolved in saturated ammonium chloride solution and extracted into EtOAc, the organic phase was dried with anhydrous magnesium sulfate, filtered, concentrated, and purified on a silica gel column with a 0-100% Hex/EtOAc gradient. The major peak was concentrated to afford 3-{l-[(R)-3,3,3-trifluoro-2-hydroxypropyl]-4-[3-(trifluoromethoxy)benzyl]-l,2,3,4-tetra-hydroquinoxalin-2-yl} benzoic acid. MS m/z = 541.09.

Patent
WO2015054088
http://google.com/patents/WO2015054088A1?cl=en
Scheme Al

Scheme A2

Scheme A3

R = Ar, NR2l C02R, CN, S02Me
es
es
SEE EXAMPLE ………SIMILAR BUT NOT SAME

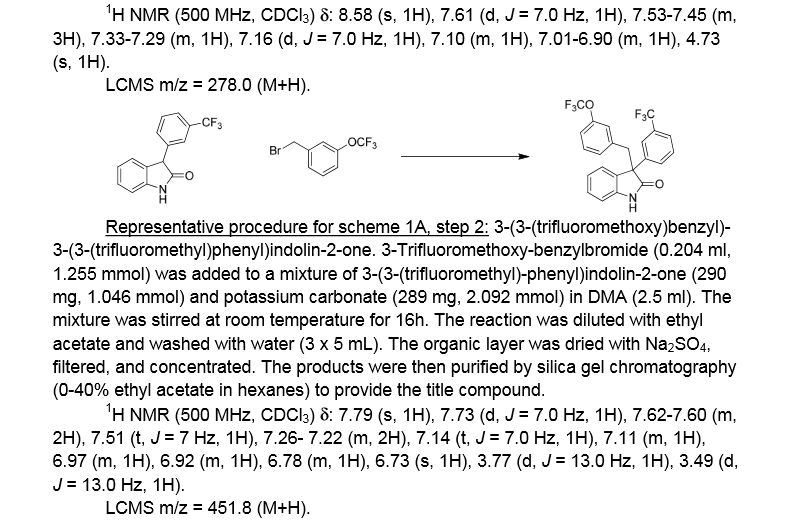
Example 1. (2R)- 1,1,1 -trifluoro-3-(3-(3-(trifluoromethoxy)benzyl)-3-(3- (trifluoromethyl)-phenyl)indolin-l-yl)propan-2-ol. This material was prepared according to Scheme Al, as described below.

3-(3-(trifluoromethyl)phenyl)indolin-2-one. Oxindole (1.598 g, 12 mmol), 3-bromo-a,a,a-trifluoromethyltoluene (2.009 ml, 14.40 mmol), potassium carbonate (3.32 g, 24.00 mmol), Pd2dba3 (0.220 g, 0.240 mmol), and 2-(dicyclohexylphosphino)-2′,4′,6′-triisopropylbiphenyl (0.458 g, 0.960 mmol) were combined in THF (12 ml) and the mixture was degassed with nitrogen. The solution was then heated to 80 °C for 18h. The mixture was cooled to room temperature, filtered through silica eluting with ethyl acetate, and concentrated. The material was then purified by silica gel chromatography (Biotage lOOg SNAP cartridge, 0-50% ethyl acetate in hexanes) to provide 3-(3-(trifluoromethyl)phenyl)indolin-2-one as a white solid.
1H NMR (500 MHz) δ 8.58 (s, 1H), 7.61 (d, J=7 Hz, 1H), 7.53-7.45 (m, 3H), 7.33-7.29 (m, 1H), 7.16 (d, J=7 Hz, 1H), 7.10 (m, 1H), 7.01-6.90 (m, 1H), 4.73 (s, 1H).

3 -(3 -(trifluoromethoxy)benzyl)-3 -(3 -(trifluoromethyl)phenyl)indolin-2-one . 3 -Trifluoromethoxy-benzylbromide (0.204 ml, 1.255 mmol) was added to a mixture of 3-(3-(trifluoromethyl)-phenyl)indolin-2-one (290 mg, 1.046 mmol) and potassium carbonate (289 mg, 2.092 mmol) (sodium carbonate may be used in place of potassium carbonate) in DMA (2.5 ml). The mixture was stirred at r.t. for 16h. The reaction was diluted with ethyl acetate and washed with water (3×5 mL). The organic layer was dried with Na2S04, filtered, and concentrated. The products were then purified by silica gel chromatography (Biotage 50g SNAP cartridge; 0-40%> ethyl acetate in hexanes) to provide 3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)-phenyl)indolin-2-one .
1H NMR (500 MHz) δ 7.79 (s, 1H), 7.73 (d, J=7 Hz, 1H), 7.62-7.60 (m, 2H), 7.51 (t, J=7 Hz, 1H), 7.26- 7.22 (m, 2H), 7.14 (t, J=7.0 Hz, 1H), 7.11 (m, 1H), 6.97 (m, 1H), 6.92 (m, 1H), 6.78 (m, 1H), 6.73 (s, 1H), 3.77 (d, J=13 Hz, 1H), 3.49 (d, J=13 Hz, 1H).
LCMS m/z = 451.8 (M+H)

3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)phenyl)indoline. Borane tetrahydrofuran complex (1.673 ml, 1.673 mmol) was added to a solution of 3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)phenyl)indolin-2-one (302 mg, 0.669 mmol) in THF (1.5 ml). The mixture was heated to 70 °C for 20h. The reaction was cooled to room temperature and quenched with saturated NH4C1 solution, and this mixture was stirred vigorously for 20 minutes. The product was extracted with ethyl acetate. The extracts were dried over Na2S04, filtered, and concentrated. The product was purified by silica gel chromatography (Biotage 25g SNAP cartridge, 0-50% ethyl acetate in hexanes) to provide 3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)phenyl)indoline. This material may also be used without purification in the final step of the sequence, epoxide opening.
1H NMR (500 MHz) δ 7.66 (s, IH), 7.59 (d, J=7 Hz, IH), 7.53 (d, J=7 Hz, IH), 7.45 (t, J=8 Hz, IH), 7.18-7.13 (m, 2H), 7.04 (d, J=8 Hz, IH), 6.98 (d, J=7 Hz, IH), 6.81 (t, J=7.5 Hz, IH), 6.71 (m, 2H), 6.60 (s, IH), 3.83 (m, IH), 3.75-3.73 (m, 2H), 3.46 (d, J=13 Hz, IH), 3.41 (d, J=13 Hz, IH).
= 437.9 (M+H)

(2R)- 1,1,1 -trifluoro-3-(3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)-phenyl)indolin-l-yl)propan-2-ol. (S)-2-(trifluoromethyl)oxirane (81 μΐ, 0.933 mmol) was added to a solution of 3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)phenyl)indoline (136 mg, 0.311 mmol) in l,l,l,3,3,3-hexafluoro-2-propanol (412 μΐ, 3.91 mmol). The reaction was stirred at room temperature overnight. The solvent was removed and the product was purified by silica gel chromatography (Biotage 25 g SNAP cartridge; 0-25% ethyl acetate in hexanes) to provide (2R)- 1 ,1,1 -trifluoro-3 -(3 -(3 -(trifluoromethoxy)benzyl)-3 -(3 -(trifluoromethyl)phenyl)indolin- 1 -yl)propan-2-ol.
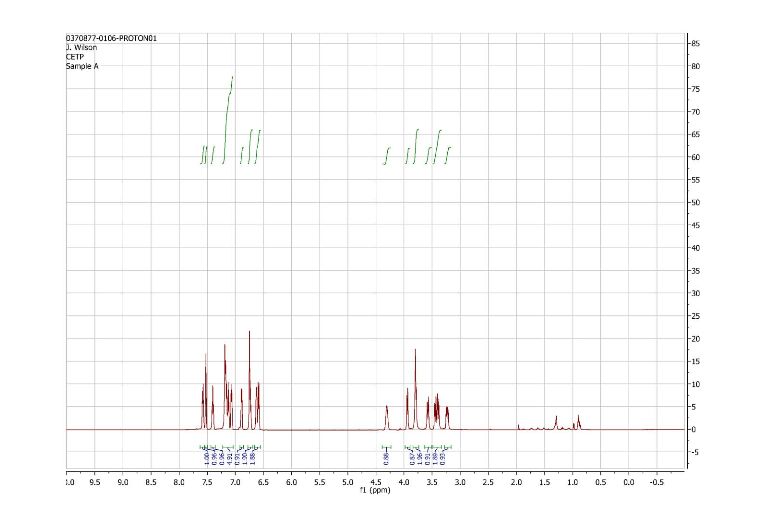
1H NMR (500 MHz) (mixture of diastereomers) δ 7.72 (s, 0.5 H), 7.69 (s, 0.5 H), 7.65 (d, J=6.5 Hz, 0.5 H), 7.61 (d, J=7.5 Hz, 0.5 H), 7.56 (s, 1H), 7.50 (m, 1H), 7.25-7.17 (m, 2H), 7.07 (broad s, 2H), 6.91-6.89 (m, 1H), 6.79-6.75 (m, 1H), 6.53 (m, 2H), 4.00 (broad s, 1H), 3.83 (d, J= 9 Hz, 0.5H), 3.77 (d, J=9 Hz, 0.5H), 3.59-3.55 (m, 1H), 3.45-3.43 (m, 1H), 3.39-3.29 (m, 2H), 3.21-3.15 (m, 1H), 2.32 (m, 0.5H), 2.15 (m, 0.5H).
LCMS m/z = 549.8 (M+H)
Examples 1-25, in the table below, were prepared according to Scheme Al in a
SEE EG 10…….(2R)- 1,1,1 -trifluoro-3-(3-(3-(trifluoromethoxy)benzyl)-3-(3- (trifluoromethoxy)-phenyl)indolin-l-yl)propan-2-ol.

ABOUT AUTHOR
Jonathan Wilson
Associate Principal Scientist at Merck

https://www.linkedin.com/in/jonathan-wilson-23206523
Experience
Associate Principal Scientist
Merck
October 2013 – Present (2 years 4 months)
Senior scientist
Merck
May 2009 – October 2013 (4 years 6 months)

Postdoctoral researcher
Princeton University
October 2007 – May 2009 (1 year 8 months)
Associate Medicinal Chemist
Merck
2000 – 2002 (2 years)
Education


///////CETP inhibition, cholesterol ester transfer protein, HDL, indoline, tetrahydroquinoxaline, merck, discovery
c21ccccc1N(C[C@@]2(c3cccc(c3)OC(F)(F)F)Cc4cc(ccc4)OC(F)(F)F)C(C(F)(F)F)O
FC(F)(F)Oc1cccc(c1)C3(CN(C[C@@H](O)C(F)(F)F)c2ccccc23)Cc4cccc(OC(F)(F)F)c4
see…………http://worlddrugtracker.blogspot.in/2016/01/mercks-novel-indoline-cholesterol-ester.html
Vibegron ビベグロン
, click to zoom.")
Vibegron, MK-4618, KRP 114V
update FDA APPROVED 12/23/2020, GEMTESA, To treat overactive bladder
Target-based Actions Beta 3 adrenoceptor agonist
Indications Overactive bladder; Urinary incontinence
UPDATE 2018/9/21 pmda Beova JAPAN 2018Kyorin Pharmaceutical, under license from Merck, is developing vibegron (phase II, September 2014) for the treating of overactive bladder. In July 2014, Merck has granted to Kyorin an exclusive license to develop, manufacture and commercialize vibegron in Japan.
MK-4618 is being developed in phase II clinical trials at Merck & Co. for the treatment of overactive bladder. The company had been developing the compound for the treatment of endocrine disorders and hypertension; however, recent progress reports are not available at present.
In 2014, Merck licensed the product to Kyorin for development and commercialization in Japan.
The function of the lower urinary tract is to store and periodically release urine. This requires the orchestration of storage and micturition reflexes which involve a variety of afferent and efferent neural pathways, leading to modulation of central and peripheral neuroeffector mechanisms, and resultant coordinated regulation of sympathetic and parasympathetic components of the autonomic nervous system as well as somatic motor pathways. These proximally regulate the contractile state of bladder (detrusor) and urethral smooth muscle, and urethral sphincter striated muscle.
β Adrenergic receptors (βAR) are present in detrusor smooth muscle of various species, including human, rat, guinea pig, rabbit, ferret, dog, cat, pig and non-human primate. However, pharmacological studies indicate there are marked species differences in the receptor subtypes mediating relaxation of the isolated detrusor; β1AR predominate in cats and guinea pig, β2AR predominate in rabbit, and β3AR contribute or predominate in dog, rat, ferret, pig, cynomolgus and human detrusor. Expression of βAR subtypes in the human and rat detrusor has been examined by a variety of techniques, and the presence of β3AR was confirmed using in situ hybridization and/or reverse transcription-polymerase chain reaction (RT-PCR). Real time quantitative PCR analyses of β1AR, β2AR and β3AR mRNAs in bladder tissue from patients undergoing radical cystectomy revealed a preponderance of β3AR mRNA (97%, cf 1.5% for β1AR mRNA and 1.4% for β2AR mRNA). Moreover, β3AR mRNA expression was equivalent in control and obstructed human bladders. These data suggest that bladder outlet obstruction does not result in downregulation of β3AR, or in alteration of β3AR-mediated detrusor relaxation. β3AR responsiveness also has been compared in bladder strips obtained during cystectomy or enterocystoplasty from patients judged to have normal bladder function, and from patients with detrusor hyporeflexia or hyperreflexia. No differences in the extent or potency of β3AR agonist mediated relaxation were observed, consistent with the concept that the β3AR activation is an effective way of relaxing the detrusor in normal and pathogenic states.
Functional evidence in support of an important role for the β3AR in urine storage emanates from studies in vivo. Following intravenous administration to rats, the rodent selective β3AR agonist CL316243 reduces bladder pressure and in cystomeric studies increases bladder capacity leading to prolongation of micturition interval without increasing residual urine volume.
Overactive bladder is characterized by the symptoms of urinary urgency, with or without urgency urinary incontinence, usually associated with frequency and nocturia. The prevalence of OAB in the United States and Europe has been estimated at 16 to 17% in both women and men over the age of 18 years. Overactive bladder is most often classified as idiopathic, but can also be secondary to neurological condition, bladder outlet obstruction, and other causes. From a pathophysiologic perspective, the overactive bladder symptom complex, especially when associated with urge incontinence, is suggestive of detrusor overactivity. Urgency with or without incontinence has been shown to negatively impact both social and medical well-being, and represents a significant burden in terms of annual direct and indirect healthcare expenditures. Importantly, current medical therapy for urgency (with or without incontinence) is suboptimal, as many patients either do not demonstrate an adequate response to current treatments, and/or are unable to tolerate current treatments (for example, dry mouth associated with anticholinergic therapy). Therefore, there is need for new, well-tolerated therapies that effectively treat urinary frequency, urgency and incontinence, either as monotherapy or in combination with available therapies. Agents that relax bladder smooth muscle, such as β3AR agonists, are expected to be effective for treating such urinary disorders.
PATENT
http://www.google.com/patents/WO2013062881A1?cl=en

EXAMPLE 3

To a three neck flask equipped with a N2 inlet, a thermo couple probe was charged pyrrolidine i-11 (10.0 g), sodium salt i-12 (7.87 g), followed by IPA (40 mL) and water (24 mL). 5 N HC1 (14.9 mL) was then slowly added over a period of 20 min to adjust pH = 3.3- 3.5, maintaining the batch temperature below 35 °C. Solid EDC hydrochloride (7.47 g) was charged in portions over 30 min. The reaction mixture was aged at RT for additional 0.5 – 1 h, aqueous ammonia (14%) was added dropwise to pH ~8.6. The batch was seeded and aged for additional 1 h to form a slurry bed. The rest aqueous ammonia (14%, 53.2 ml total) was added dropwise over 6 h. The resulting thick slurry was aged 2-3 h before filtration. The wet-cake was displacement washed with 30% IPA (30 mL), followed by 15% IPA (2 x 20mL) and water (2 X 20mL). The cake was suction dried under N2 overnight to afford 14.3 g of compound of Formula (I)-
1H NMR (DMSO) δ 10.40 (s, NH), 7.92 (d, J = 6.8, 1H), 7.50 (m, 2H), 7.32 (m, 2H), 7.29 (m, 2H), 7.21 (m, 1H), 7.16 (m, 2H), 6.24 (d, J = 6.8, 1H), 5.13 (dd, J = 9.6, 3.1, 1H), 5.08 (br s, OH), 4.22 (d, J = 7.2, 1H), 3.19 (p, J = 7.0, 1H), 3.16-3.01 (m, 3H), 2.65 (m, 1H), 2.59-2.49 (m, 2H), 2.45 (br s, NH), 2.16 (ddt, J = 13.0, 9.6, 3.1, 1H), 1.58 (m, 1H), 1.39 (m, 1H), 1.31-1.24 (m, 2H).
13C NMR (DMSO) δ 167.52, 165.85, 159.83, 154.56, 144.19, 136.48, 135.66, 129.16, 127.71, 126.78, 126.62, 119.07, 112.00, 76.71, 64.34, 61.05, 59.60, 42.22, 31.26, 30.12, 27.09, 23.82.
HPLC method – For monitoring conversion
Column: XBridge C18 cm 15 cm x 4.6 mm, 3.5 μιη particle size;
Column Temp. : 35 °C; Flow rate: 1.5 mL/min; Detection: 220 nm;
Mobile phase: A. 5 mM Na2B407.10 H20 B: Acetonitrile
Gradient:

HPLC method – For level of amide epimer detection
Column: Chiralpak AD-H 5 μηι, 250 mm x 4.6 mm.
Column Temp: 35 °C; Flow rate: 1.0 mL/min; Detection: 250 nm;
Mobile phase: Isocratic 30% Ethanol in hexanes + 0.1% isobutylamine
PATENT
WO 2009124167
http://www.google.com/patents/WO2009124167A1?cl=en
EXAMPLE 103
(6y)-N-r4-({(‘25′. 5R)-5-r(‘R)-hvdroxy(‘phenvnmethyl1pyrrolidin-2-yl}methvnphenyl1-4-oxo- 4,6J,8-tetrahydropyiτolori,2-α1pyrimidine-6-carboxamide

ter?-butyl(2R. 55f)-2-rCR)-hvdroxy(‘phenvnmethyl1-5-r4-(‘{r(‘65f)-4-oxo-4.6.7.8-
tetrahydropyrrolof 1.2-alpyrimidin-6- yl]carbonyl} amino)benzyl]pyrrolidine- 1 – carboxylate

To a solution of i-13a (21.4 g, 55.9 mmol) in N,N-dimethylformamide (100 ml) at O0C was added [(65)-4-oxo-4,6,7,8-tetrahydropyrrolo[l,2-α]pyrimidine-6-carboxylic acid (11.1 g, 61.5 mmol), followed by 1 -hydroxybenzotriazole (i-44, 7.55 g, 55.9 mmol), N-(3- dimethylaminopropyl)-Nl-ethylcarbodiimide hydrochloride (16.1 g, 84.0 mmol) and N,N- diisopropylethylamine (29.2 ml, 168 mmol). The reaction mixture was stirred from O0C to ambient temperature for 2 h. Water (600 ml) was added and it was extracted with dichloromethane (600 ml x 2). The combined organic layers were dried over Na2SO4. After removal of the volatiles, the residue was purified by using a Biotage Horizon® system (0-5% then 5% methanol with 10% ammonia/dichloromethane mixture) to afford the title compound which contained 8% of the minor diastereomer. It was further purified by supercritical fluid chromatography (chiral AS column, 40% methanol) to afford the title compound as a pale yellow solid (22.0 g, 72%). 1H NMR (CDCl3): δ 9.61 (s, IH), 7.93 (d, J = 6.6 Hz, IH), 7.49 (d, J = 8.4 Hz, 2H), 7.35-7.28 (m, 5H), 7.13 (d, J = 8.5 Hz, 2H), 6.40 (d, J = 6.7 Hz, IH), 5.36 (d, J = 8.6 Hz, IH), 4.38 (m, IH), 4.12-4.04 (m, 2H), 3.46 (m,lH), 3.15-3.06 (m, 2H), 2.91 (dd, J = 13.1, 9.0 Hz, IH), 2.55 (m, IH), 2.38 (m, IH), 1.71-1.49 (m, 13H). LC-MS 567.4 (M+23).
(6S)-N-\4-( U2S. 5R)-5-r(R)-hvdroxy(phenyl)methyl1pyrrolidin-2-
yl}methyl)phenyl1-4-oxo-4,6J,8-tetrahvdropyrrolori,2-α1pyrimidine-6- carboxamide

To a solution of the intermediate from Step A (2.50 g, 4.59 mmol) in dichloromethane (40 ml) was added trifluoroacetic acid (15 ml). The reaction mixture was stirred at ambient temperature for 1.5 h. After removal of the volatiles, saturated NaHCCh was added to make the PH value to 8-9. The mixture was then extracted with dichloromethane. The combined organic layers were dried over Na2SO4. After concentration, crystallization from methanol/acetonitrile afforded the title compound as a white solid (1.23g, 60%). 1H NMR (DMSO-Cl6): δ 10.40 (s, IH), 7.91 (d, J = 6.7 Hz, IH), 7.49 (d, J = 8.3 Hz, 2H), 7.32-7.26 (m, 4H), 7.21 (m, IH), 7.15 (d, J = 8.4 Hz, 2H), 6.23 (d, J = 6.7 Hz, IH), 5.11 (dd, J = 9.6, 2.9 Hz, IH), 5.10 (br, IH), 4.21 (d, J = 7.1 Hz, IH), 3.20-3.00 (m, 4H), 2.66-2.51 (m, 3H), 2.16 (m, IH), 1.57 (m, IH), 1.38 (m, IH), 1.29-1.23 (m, 2H). LC-MS 445.3 (M+l).
Using the Biological Assays described above, the human β3 functional activity of Example 103 was determined to be between 11 to 100 nM.
PATENT
CHECK STRUCTURE…………….CAUTION
http://www.google.com/patents/US8247415


CAUTION…………….
Example 103(6S)-N-[4-({(2S,5R)-5-[(R)-hydroxy(phenyl)methyl]pyrrolidin-2-yl}methyl)phenyl]-4-oxo-4,6,7,8-tetrahydropyrrolo[1,2-α]pyrimidine-6-carboxamide

Step A: tert-butyl(2R,5S)-2-[(R)-hydroxy(phenyl)methyl]-5-[4-({[(6S)-4-oxo-4,6,7,8-tetrahydropyrrolo[1,2-α]pyrimidin-6-yl]carbonyl}amino)benzyl]pyrrolidine-1-carboxylate

To a solution of i-13a (21.4 g, 55.9 mmol) in N,N-dimethylformamide (100 ml) at 0° C. was added [(6S)-4-oxo-4,6,7,8-tetrahydropyrrolo[1,2-α]pyrimidine-6-carboxylic acid (11.1 g, 61.5 mmol), followed by 1-hydroxybenzotriazole (i-44, 7.55 g, 55.9 mmol), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (16.1 g, 84.0 mmol) and N,N-diisopropylethylamine (29.2 ml, 168 mmol). The reaction mixture was stirred from 0° C. to ambient temperature for 2 h. Water (600 ml) was added and it was extracted with dichloromethane (600 ml×2). The combined organic layers were dried over Na2SO4. After removal of the volatiles, the residue was purified by using a Biotage Horizon® system (0-5% then 5% methanol with 10% ammonia/dichloromethane mixture) to afford the title compound which contained 8% of the minor diastereomer. It was further purified by supercritical fluid chromatography (chiral AS column, 40% methanol) to afford the title compound as a pale yellow solid (22.0 g, 72%). 1H NMR (CDCl3): δ 9.61 (s, 1H), 7.93 (d, J=6.6 Hz, 1H), 7.49 (d, J=8.4 Hz, 2H), 7.35-7.28 (m, 5H), 7.13 (d, J=8.5 Hz, 2H), 6.40 (d, J=6.7 Hz, 1H), 5.36 (d, J=8.6 Hz, 1H), 4.38 (m, 1H), 4.12-4.04 (m, 2H), 3.46 (m, 1H), 3.15-3.06 (m, 2H), 2.91 (dd, J=13.1, 9.0 Hz, 1H), 2.55 (m, 1H), 2.38 (m, 1H), 1.71-1.49 (m, 13H). LC-MS 567.4 (M+23).
Step B: (6S)-N-[4-({(2S,5R)-5-[(R)-hydroxy(phenyl)methyl]pyrrolidin-2-yl}methyl)phenyl]-4-oxo-4,6,7,8-tetrahydropyrrolo[1,2-α]pyrimidine-6-carboxamide

To a solution of the intermediate from Step A (2.50 g, 4.59 mmol) in dichloromethane (40 ml) was added trifluoroacetic acid (15 ml). The reaction mixture was stirred at ambient temperature for 1.5 h. After removal of the volatiles, saturated NaHCO3 was added to make the PH value to 8-9. The mixture was then extracted with dichloromethane. The combined organic layers were dried over Na2SO4. After concentration, crystallization from methanol/acetonitrile afforded the title compound as a white solid (1.23 g, 60%). 1H NMR (DMSO-d6): δ 10.40 (s, 1H), 7.91 (d, J=6.7 Hz, 1H), 7.49 (d, J=8.3 Hz, 2H), 7.32-7.26 (m, 4H), 7.21 (m, 1H), 7.15 (d, J=8.4 Hz, 2H), 6.23 (d, J=6.7 Hz, 1H), 5.11 (dd, J=9.6, 2.9 Hz, 1H), 5.10 (br, 1H), 4.21 (d, J=7.1 Hz, 1H), 3.20-3.00 (m, 4H), 2.66-2.51 (m, 3H), 2.16 (m, 1H), 1.57 (m, 1H), 1.38 (m, 1H), 1.29-1.23 (m, 2H). LC-MS 445.3 (M+1).
Using the Biological Assays described above, the human β3 functional activity of Example 103 was determined to be between 11 to 100 nM.
PATENT
WO2014150639
Step 6. Preparation of Compound 1-7 from Compound 1-6 and Compound A-2

To a three neck flask equipped with a N2 inlet, a thermo couple probe was charged pyrrolidine hemihydrate 1-6 (10.3 g), sodium salt A-2 (7.87 g), followed by IPA (40 mL) and water (24 mL). 5 N HC1 (14.9 mL) was then slowly added over a period of 20 minutes to adjust pH = 3.3-3.5, maintaining the batch temperature below 35°C. Solid EDC hydrochloride (7.47 g) was charged in portions over 30 minutes. The reaction mixture was aged at RT for additional 0.5 – 1 hour, aqueous ammonia (14%) was added dropwise to pH -8.6. The batch was seeded and aged for additional 1 hour to form a slurry bed. The rest aqueous ammonia (14%, 53.2 ml total) was added dropwise over 6 hours. The resulting thick slurry was aged 2-3 hours before filtration. The wet-cake was displacement washed with 30% IPA (30 mL), followed by 15% IPA (2 x 20mL) and water (2 X 20mL). The cake was suction dried under N2 overnight to afford 14.3 g of compound 1-7.
1H NMR (DMSO) δ 10.40 (s, NH), 7.92 (d, J = 6.8, 1H), 7.50 (m, 2H), 7.32 (m, 2H), 7.29 (m, 2H), 7.21 (m, 1H), 7.16 (m, 2H), 6.24 (d, J = 6.8, 1H), 5.13 (dd, J = 9.6, 3.1, 1H), 5.08 (br s, OH), 4.22 (d, J = 7.2, 1H), 3.19 (p, J = 7.0, 1H), 3.16-3.01 (m, 3H), 2.65 (m, 1H), 2.59-2.49 (m, 2H), 2.45 (br s, NH), 2.16 (ddt, J = 13.0, 9.6, 3.1, 1H), 1.58 (m, 1H), 1.39 (m, 1H), 1.31-1.24 (m, 2H).
13C NMR (DMSO) δ 167.52, 165.85, 159.83, 154.56, 144.19, 136.48, 135.66, 129.16, 127.71, 126.78, 126.62, 119.07, 112.00, 76.71, 64.34, 61.05, 59.60, 42.22, 31.26, 30.12, 27.09, 23.82.
The crystalline freebase anhydrous form I of Compound 1-7 can be characterized by XRPD by

PATENT
WO-2014150633
Merck Sharp & Dohme Corp
Process for preparing stable immobilized ketoreductase comprises bonding of recombinant ketoreductase to the resin in a solvent. Useful for synthesis of vibegron intermediates. For a concurrent filling see WO2014150639, claiming the method for immobilization of ketoreductase. Picks up from WO2013062881, claiming the non enzymatic synthesis of vibegron and intermediates.
PAPER
Discovery of Vibegron: A Potent and Selective β3 Adrenergic Receptor Agonist for the Treatment of Overactive Bladder
http://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.5b01372
http://pubs.acs.org/doi/suppl/10.1021/acs.jmedchem.5b01372/suppl_file/jm5b01372_si_001.pdf

The discovery of vibegron, a potent and selective human β3-AR agonist for the treatment of overactive bladder (OAB), is described. An early-generation clinical β3-AR agonist MK-0634 (3) exhibited efficacy in humans for the treatment of OAB, but development was discontinued due to unacceptable structure-based toxicity in preclinical species. Optimization of a series of second-generation pyrrolidine-derived β3-AR agonists included reducing the risk for phospholipidosis, the risk of formation of disproportionate human metabolites, and the risk of formation of high levels of circulating metabolites in preclinical species. These efforts resulted in the discovery of vibegron, which possesses improved druglike properties and an overall superior preclinical profile compared to MK-0634. Structure–activity relationships leading to the discovery of vibegron and a summary of its preclinical profile are described.



| Reference | ||
|---|---|---|
| 1 | H.P. Kaiser, et al., “Catalytic Hydrogenation of Pyrroles at Atmospheric Pressure“, J. Org. Chem., vol. 49, No. 22, p. 4203-4209 (1984). | |
ClinicalTrials.gov Web Site 2011, April 28
| WO2011043942A1 * | Sep 27, 2010 | Apr 14, 2011 | Merck Sharp & Dohme Corp. | Combination therapy using a beta 3 adrenergic receptor agonist and an antimuscarinic agent |
| US20090253705 * | Apr 2, 2009 | Oct 8, 2009 | Richard Berger | Hydroxymethyl pyrrolidines as beta 3 adrenergic receptor agonists |
| US20110028481 * | Apr 2, 2009 | Feb 3, 2011 | Richard Berger | Hydroxymethyl pyrrolidines as beta 3 adrenergic receptor agonists |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8642661 | Aug 2, 2011 | Feb 4, 2014 | Altherx, Inc. | Pharmaceutical combinations of beta-3 adrenergic receptor agonists and muscarinic receptor antagonists |
| US8653260 | Jun 20, 2012 | Feb 18, 2014 | Merck Sharp & Dohme Corp. | Hydroxymethyl pyrrolidines as beta 3 adrenergic receptor agonists |
| US20120202819 * | Sep 27, 2010 | Aug 9, 2012 | Merck Sharp & Dohme Corporation | Combination therapy using a beta 3 adrenergic receptor agonists and an antimuscarinic agent |
| US20020028835 | Jul 12, 2001 | Mar 7, 2002 | Baihua Hu | Cyclic amine phenyl beta-3 adrenergic receptor agonists |
| US20070185136 | Feb 2, 2007 | Aug 9, 2007 | Sanofi-Aventis | Sulphonamide derivatives, their preparation and their therapeutic application |
| US20110028481 | Apr 2, 2009 | Feb 3, 2011 | Richard Berger | Hydroxymethyl pyrrolidines as beta 3 adrenergic receptor agonists |
| WO2003072572A1 | Feb 17, 2003 | Sep 4, 2003 | Jennifer Anne Lafontaine | Beta3-adrenergic receptor agonists |
|
8-22-2012
|
Hydroxymethyl pyrrolidines as [beta]3 adrenergic receptor agonists
|
////////////C1CC(NC1CC2=CC=C(C=C2)NC(=O)C3CCC4=NC=CC(=O)N34)C(C5=CC=CC=C5)O
Sun Pharma, Merck & Co Inc ink pact for Tildrakizumab

Sep 17, 2014,
Under terms of the agreement, Sun Pharma will acquire worldwide rights to tildrakizumab for use in all human indications from Merck in exchange for an upfront payment of USD 80 million.
Pharma major Sun Pharmaceutical Industries today entered into a licensing agreement with Merck & Co Inc for investigational therapeutic antibody candidate, tildrakizumab to be used for treatment of plaque psoriasis. Under terms of the agreement, Sun Pharma will acquire worldwide rights to tildrakizumab for use in all human indications from Merck in exchange for an upfront payment of USD 80 million, the companies said in a joint statement. Tildrakizumab is being evaluated in Phase III registration trials for the treatment of chronic plaque psoriasis, a skin ailment. “Merck will continue all clinical development and regulatory activities, which will be funded by Sun Pharma. Upon product approval, Sun Pharma will be responsible for regulatory activities, including subsequent submissions, pharmacovigilance, post approval studies, manufacturing and commercialisation of the approved product,” it added.


Sun Pharma managing director Dilip Shanghvi.


| Monoclonal antibody | |
|---|---|
| Source | Humanized (from mouse) |
| Target | IL23 |
| Clinical data | |
| Legal status |
?
|
| Identifiers | |
| CAS number | 1326244-10-3 |
| ATC code | None |
| Chemical data | |
| Formula | C6426H9918N1698O2000S46 |
| Mol. mass | 144.4 kDa |
Tildrakizumab is a monoclonal antibody designed for the treatment of immunologically mediated inflammatory disorders.[1]
Tildrakizumab was designed to block interleukin-23, a cytokine that plays an important role in managing the immune system andautoimmune disease. Originally developed by Schering-Plough, this drug is now part of Merck‘s clinical program, following that company’s acquisition of Schering-Plough.
As of March 2014, the drug was in phase III clinical trials for plaque psoriasis. The two trials will enroll a total of nearly 2000 patients, and preliminary results are expected in June, 2015. [2][3]
References
Telmapitant……Tachykinin NK1 Antagonists
, click to zoom.")
Telmapitant
TELMAPITANT; Telmapitant (USAN); Telmapitant [USAN]; 552292-58-7; HJ5FE4153B; D10391
(5R,8S)-8-[[(1R)-1-[3,5-bis(trifluoromethyl)phenyl]ethoxy]methyl]-8-phenyl-1,3,9-triazaspiro[4.5]decane-2,4-dione
1,3,7-Triazaspiro[4.5]decane-2,4-dione, 8-[[(1R)-1-[3,5-bis(trifluoromethyl)phenyl]ethoxy]methyl]-8-phenyl-, (5R,8S)-
(5R,8S)-8-(((1R)-1-(3,5-Bis(Trifluoromethyl)phenyl)ethoxy)methyl)-8-phenyl-1,3,7- triazaspiro(4.5)decane-2,4-dione
1,3,7-Triazaspiro(4.5)decane-2,4-dione,
8-(((1R)-1-(3,5-bis(trifluoromethyl)phenyl)ethoxy)methyl)-8-phenyl-, (5R,8S)-
Molecular Formula: C24H23F6N3O3
Molecular Weight: 515.448139
cas 552292-58-7
Merck & Co. (innovator)
Treatment of Nausea and Vomiting,
SYNTHESIS
……………………………………….
US7902366
http://www.google.com/patents/US7902366
Example 43a Example 43b

Step 1:

To a suspension of lactol Compound 3 (60 g, 93.0 mmol, 1 equiv.) and Wittig Reagent (93.5 g, 200.0 mmol, 2.15 equiv.) in toluene (800 ml) stirred at −78° C. under N2, a solution of KHMDS (0.5M in toluene, 558 ml, 280.0 mmol, 3 equiv.) was added dropwise at −78° C. The cooling bath was removed and the yellow mixture was warmed to RT to form a red solution. The mixture was allowed to stir at 23° C. for further 1 h before being quenched with saturated NH4Cl solution. EtOAc was added and layers were separated. The separated aqueous layer was extracted with EtOAc (2×500 ml). The combined organic layers were dried (MgSO4) and filtered. Removal of solvents in vacuum followed by Biotage column chromatography [5% EtOAc-hexane to 10% EtOAc-hexane] gave alkene Compound 42 as white solid (40.5 g, 68%), Electrospray MS [M+1]+ 638.1. Continuous elution gave an impure cyclized product Compound 43.
Step 2:

A suspension of alkene Compound 42 (40.5 g, 64 mmol, 1 equiv.) and PtO2 (1.44 g, 6.4 mmol, 0.1 equiv.) in EtOH (400 ml) were stirred under a H2 balloon at 23° C. for 24 h. Another batch of PtO2 (1.44 g, 6.4 mmol, 0.1 equiv) was added and the mixture was stirred for another 24 h at 23° C. The catalyst was filtered via a pad of Celite. This solution of alkane Compound 44 was used in the next step without further purification.
Step 3:

p-TsOH.H2O (2.42 g, 13.0 mmol) was added to the ethanolic solution of alkane Compound 44 from above and the solution was heated to reflux for 4 h. The solution was cooled to RT and neutralized with Et3N. Solvents were removed in vacuum and EtOAc was added. Saturated NaHCO3 solution was added and layers were separated. The separated aqueous layer was extracted with EtOAc (300 ml×2). The combined organic layers were dried (MgSO4) and filtered. Removal of solvents in vacuum followed by Biotage column chromatography [10% ether-hexane] gave enamide Compound 45 (first batch) as yellow oil. Some intermediate and starting material were recovered as yellow oil by continuous elution with [50% EtOAc-hexane]. The yellow oil was dissolved in toluene and 10 mol % p-TsOH was added. The mixture was heated to reflux for 2 h and cooled to RT. Work up was as above and the combined enamide Compound 45 (25 g, 70%), Electrospray MS [M+1]+ 564.1, was obtained as yellow oil.
Step 4:

BH3.Me2S (13.6 ml, 133 mmo, 3.02 equiv) was added to a solution of enamide Compound 45 (25 g, 44.0 mmol,1 equiv.) in THF at 23° C. under N2. The mixture was stirred at 23° C. for 18 h and then cooled over an ice-water bath. A solution of NaOH (500 ml, 2N) was added slowly followed by a solution of H202 (500 ml, 30% aqueous). The mixture was allowed to stir from 0° C. to 23° C. for 18 h. Layers were separated and the separated aqueous layer was extracted with Et2O (500 ml×2). The combined organic layers were dried (MgSO4) and filtered. Removal of solvents in vacuum followed by Biotage column chromatography [hexane-EtOAc, 3:1 (v/v)] gave alcohol Compound 46 as colorless oil (19 g, 74%), Electrospray MS [M+1]+ 582.1.
Step 5:

Oxalyl chloride (5.7 ml, 65.3 mmol, 2 equiv.) was added to a solution of DMSO (9.3 ml, 131.0 mmol, 4 equiv.) in CH2Cl2 (300 ml) at −78° C. under N2. The mixture was stirred at −78° C. for 15 min before a solution of alcohol Compound 46 (19 g, 32.7 mmol. 1 equiv.) in CH2Cl2 (50 ml) was added. The mixture was stirred at −78° C. for a further 1 h and Et3N (32 ml, 228.9 mmol, 7 equiv.) was added. The cooling bath was removed and the mixture was warmed to RT before it was quenched with saturated NaHCO3 solution. Layers were separated and the aqueous was extracted with CH2Cl2 (300 ml×2). The combined organic layers were dried (MgSO4) and filtered. Removal of solvents in vacuum followed by Biotage column chromatography [hexane-ether, 4:1 (v/v)] gave ketone Compound 47 as colorless oil (15 g, 80%), Electrospray MS [M+1]+ 580.1.
Step 6:

EtOH (150 ml) was added to Cbz-ketone Compound 47 (15 g, 25.88 mmol, 1 equiv.), followed by NH4(CO3)2 (9.95 g, 103.5 mmol, 4 equiv.) and a solution of KCN (3.4 g, 51.77 mmol, 2 equiv.). The resulting mixture was heated at 58° C. under N2 for 72 h. TLC (1:1 EtOAc:hexane) revealed complete consumption of the starting material. The reaction mixture was cooled to RT and poured into sat. aq. NaHCO3 (200 ml) and extracted with EtOAc (3×200 ml). The combined organic layers were dried over MgSO4 and concentrated in vacuo to afford crude Cbz-hydantoin Compound 48 (16.5 g, 98%), Electrospray MS [M+1]+650.1. The crude material was used in the next reaction without further purification.
Step 7:
The crude Cbz-hydantoin Compound 48 (16.5 g, 25.4 mmol, 1 equiv.) was dissolved in MeOH (220 ml) and 20% Pd(OH)2—C (3.6 g) was added. The reaction mixture was shaken in a parr shaker under H2 atmosphere at 40 psi for 18 h. TLC (1:1 EtOAc:hexane) revealed complete consumption of the starting material. The reaction mixture was filtered through a pad of celite and the celite was washed with MeOH. The resulting solution was concentrated in vacuo. The crude product was purified by column chromatography on a Biotage (3:2, EtOAc:hex). Two major spots were collected. The less-polar spot corresponds to the isomer Example 43a (3 g, overall 20% over two steps), Electrospray MS [M+1]+ 516.1. The more polar spot corresponds to the isomer Example 43b (4.5 g, overall 30% over two steps), Electrospray MS [M+1]+ 516.1.
………………………………..
http://www.google.com/patents/WO2003051840A1?cl=en
Example 43a Example 43b

Step 1 :
Compound 3

To a suspension of lactol Compound 3 (60g, 93.0mmol, lequiv.) and Wittig Reagent (93. δg, 200.0mmol, 2.1 δequiv.) in toluene (800ml) stirred at -78°C under δ N2, a solution of KHMDS (O.δM in toluene, δδδml, 280.0mmol, 3equiv.) was added dropwise at -78°C. The cooling bath was removed and the yellow mixture was warmed to RT to form a red solution. The mixture was allowed to stir at 23°C for further 1 h before being quenched with saturated NH CI solution. EtOAc was added and layers were separated. The separated aqueous layer was extracted with EtOAc 0 (2 x δOOml). The combined organic layers were dried (MgSO ) and filtered.
Removal of solvents in vacuum followed by Biotage column chromatography [δ% EtOAc-hexane to 10% EtOAc-hexane] gave alkene Compound 42 as white solid (40. δg, 68%), Electrospray MS [M+1]+ 638.1. Continuous elution gave an impure cyclized product Compound 43. δ Step 2:
Compound 42

A suspension of alkene Compound 42 (40. δg, 64mmol, lequiv.) and PtO2 (1.44g, 6.4mmol, 0.1 equiv.) in EtOH (400ml) were stirred under a H2 balloon at 23°C for 24 h. Another batch of PtO2 (1.44g, 6.4mmol, 0.1 equiv) was added and the 0 mixture was stirred for another 24 h at 23°C. The catalyst was filtered via a pad of Celite. This solution of alkane Compound 44 was used in the next step without further purification. Step 3:
Compound 44

p-TsOH.H2O (2.42g, 13.0mmol) was added to the ethanolic solution of alkane
Compound 44 from above and the solution was heated to reflux for 4 h. The solution was cooled to RT and neutralized with Et3N. Solvents were removed in vacuum and EtOAc was added. Saturated NaHCO3 solution was added and layers
5 were separated. The separated aqueous layer was extracted with EtOAc (300ml x
2). The combined organic layers were dried (MgSO4) and filtered. Removal of solvents in vacuum followed by Biotage column chromatography [10% ether- hexane] gave enamide Compound 45 (first batch) as yellow oil. Some intermediate and starting material were recovered as yellow oil by continuous elution with 0 [50%EtOAc-hexane]. The yellow oil was dissolved in toluene and 10mol% p-TsOH was added. The mixture was heated to reflux for 2 h and cooled to RT. Work up was as above and the combined enamide Compound 45 (2δg, 70%), Electrospray
MS [M+1]+ 664.1 , was obtained as yellow oil.
Step 4:

BH3.Me2S (13.6ml, 133mmo, 3.02 equiv) was added to a solution of enamide Compound 45T25g, 44.0mmol, lequiv.) in THF at 23°C under N2. The mixture was stirred at 23°C for 18 h and then cooled over an ice-water bath. A solution of NaOH (600ml, 2N) was added slowly followed by a solution of H O2 (600ml, 30% 0 aqueous). The mixture was allowed to stir from 0°C to 23°C for 18 h. Layers were separated and the separated aqueous layer was extracted with Et.20 (600ml x 2). The combined organic layers were dried (MgSO4) and filtered. Removal of solvents in vacuum followed by Biotage column chromatography [hexane-EtOAc, 3:1 (v/v)] gave alcohol Compound 46 as colorless oil (19g, 74%), Electrospray MS [M+1]+ δ 582.1. Step 5:
Compound 46

Oxalyl chloride (δ.7ml, 6δ.3mmol, 2equiv.) was added to a solution of DMSO (9.3ml, 131.0mmol, 4equiv.) in CH2CI2 (300ml) at -78°C under N2. The mixture was 0 stirred at -78°C for 1 δ min before a solution of alcohol Compound 46 (19g, 32.7mmol. lequiv.) in CH2CI2 (50ml) was added. The mixture was stirred at -78°C for a further 1 h and Et3N (32ml, 228.9mmol, 7equiv.) was added. The cooling bath was removed and the mixture was warmed to RT before it was quenched with saturated NaHCO3 solution. Layers were separated and the aqueous was extracted with CH2CI2 (300ml x 2). The combined organic layers were dried (MgSO4) and filtered. Removal of solvents in vacuum followed by Biotage column chromatography [hexane-ether, 4:1 (v/v)] gave ketone Compound 47 as colorless oil (1δg, 80%), Electrospray MS [M+1]+ 680.1.

EtOH (150ml) was added to Cbz-ketone Compound 47 (15g, 2δ.88mmol, lequiv.), followed by NH (CO )2 (9.9δg, 103.5mmol, 4equiv.) and a solution of KCN (3.4g, 61.77mmoI, 2equiv.). The resulting mixture was heated at 68°C under N2 for 72 h. TLC (1 :1 EtOAc:hexane) revealed complete consumption of the starting
1δ material. The reaction mixture was cooled to RT and poured into sat. aq. NaHCO3 (200 ml) and extracted with EtOAc (3 x 200ml). The combined organic layers were dried over MgSO4 and concentrated in vacuo to afford crude Cbz-hydantoin Compound 48 (16.δg, 98%), Electrospray MS [M+1]+ 650.1. The crude material was used in the next reaction without further purification.
20 Step 7:
The crude Cbz-hydantoin Compound 48 (16.5g, 2δ.4mmol, lequiv.) was dissolved in MeOH (220ml) and 20% Pd(OH)2-C (3.6g) was added. The reaction mixture was shaken in a parr shaker under H2 atmosphere at 40 psi for 18 h. TLC (1 :1 EtOAc:hexane) revealed complete consumption of the starting material. The
26 reaction mixture was filtered through a pad of celite and the celite was washed with MeOH. The resulting solution was concentrated in vacuo. The crude product was purified by column chromatography on a Biotage (3:2, EtOAc:hex). Two major spots were collected. The less-polar spot corresponds to the isomer Example 43a (3 g, overall 20% over two steps), Electrospray MS [M+1]+ 616.1. The more polar spot
30 corresponds to the isomer Example 43b (4.6 g, overall 30% over two steps), Electrospray MS [M+1]+ 616.1.
|
4-29-2011
|
NK1 ANTAGONISTS
|
|
|
3-9-2011
|
NK1 antagonists
|
| English translation of Knabe, J., et al., “Racemates and Enantiomers of . . . ,” Pharmazie 52(12):912-919 (1997). | ||
| 2 | English translation of Schult, Karl E., et al., “Hydantoin-Derivate as Potential . . . ,” Eur. J. Med. Chem.-Chimica Therapeutics 13(1):25-31 (1978). | |
| 3 | English translation of Schult, Karl E., et al., “Hydantoin-Derivate as Potential . . . ,” Eur. J. Med. Chem.—Chimica Therapeutics 13(1):25-31 (1978). | |
| 4 | Knabe, J., et al., “Racemates and Enantiomers of Basic Substituted 5-Phenylhydantoins . . . ,” Pharmazie 52(12): 912-919 (1997). | |
| 5 | Oh, Chang-Hyun et al., “Synthesis of New Hydantoin-3-Acetic Acid Derivatives . . . ,” Bull. Korean Chem. Soc. 9(4):231-235 (1988). | |
| 6 | Shulte, Karl E., et al., “Hydantoin-Derivate als . . . ,” Eur. J. Med. Chem.-Chimica Therapeutica 13(1):25-31 (1978). | |
| 7 | Shulte, Karl E., et al., “Hydantoin-Derivate als . . . ,” Eur. J. Med. Chem.—Chimica Therapeutica 13(1):25-31 (1978). | |
| 8 | Wu, X. et al., “Generation of Cyclopenta [c] piperidines and Pyrrolo [3,4-c]piperidines- . . . ,” Tetrahedron 56(34): 6279-6290 (2000). | |
| 9 | * | Xiujuan Wu et al 2000. , Stereoselective transformation of 2H-1,4-Oxazin-2-ones into 2,(2),5,5-tri- and tetrasubstituted Analogues. . . |
| US6436928 * | Dec 14, 2000 | Aug 20, 2002 | Schering Corporation | Selective neurokinin antagonists |
| US6635639 * | Feb 13, 2002 | Oct 21, 2003 | Nps Allelix Corp. | Use of N-alkylamino-heterocylic compounds for the treatment of migraine |
| US7041682 * | Jul 2, 2003 | May 9, 2006 | Schering Corporation | Antiemetics, antidepressants, anxiolytic agents, antitussive agents |
| US7122677 * | Nov 12, 2002 | Oct 17, 2006 | Scherig Corporation | NK1 antagonists |
| US20060094720 * | Dec 15, 2005 | May 4, 2006 | Neng-Yang Shih | NK1 antagonists |
| US20060223804 * | Jun 30, 2005 | Oct 5, 2006 | Schering Corporation | NK1 antagonists |
| EP0790248A1 | Jan 20, 1997 | Aug 20, 1997 | Pfizer Limited | 3-Aza-piperidone- (tetrahydropyrimidin-2-one) and 3-oxa-piperidone (1,3 oxazin-2-one) derivatives, their preparation and their use as tachykinin/neurokinin antagonists |
key
Telmapitant, Merck, Tachykinin NK1 Antagonists
ORGANIC SPECTROSCOPY
Merck’s New Drug Application for an Investigational Intravenous (IV) Formulation of NOXAFIL® (posaconazole) Receives FDA Priority Review

Posaconazole, SCH 56592, Noxafil (Schering-Plough)
Posaconazole is a triazole antifungal drug that is used to treat invasive infections by Candida species and Aspergillus species in severely immunocompromised patients.
For prophylaxis of invasive Aspergillus and Candida infections in patients, 13 years of age and older, who are at high risk of developing these infections due to being severely immunocompromised as a result of procedures such as hematopoietic stem cell transplant (HSCT) recipients with graft-versus-host disease (GVHD), or due to hematologic malignancies with prolonged neutropenia from chemotherapy. Also for the treatment of oropharyngeal candidiasis, including oropharyngeal candidiasis refractory to itraconazole and/or fluconazole. Posaconazole is used as an alternative treatment for invasive aspergillosis, Fusarium infections, and zygomycosis in patients who are intolerant of, or whose disease is refractory to, other antifungals
Posaconazole is designated chemically as 4-[4-[4-[4-[[ (3R,5R)-5- (2,4-difluorophenyl)tetrahydro-5-(1H-1,2,4-triazol-1 -ylmethyl)-3-furanyl]methoxy]phenyl]-1 -piperazinyl]phenyl]-2-[ (1S,2S)-1 -ethyl-2- hydroxypropyl]-2,4-dihydro-3H-1,2,4-triazol-3-one with an empirical formula of C37H42F2N8O4 and a molecular weight of 700.8.
Posaconazole is used, for example, to prevent and/or treat invasive fungal infections caused by Candida species, Mucor species, Aspergillus species,Fusarium species, or Coccidioides species in immunocompromised patients and/or in patients where the disease is refractory to other antifungal agents such as amphothericin B, fluconazole, or itraconazole, and/or in patients who do not tolerate these antifungal agents.
CAS No. 171228-49-2
Posaconazole compounds have been described inU.S. Pat. Appl. No. 2003/0055067 for “Antifungal Composition with Enhanced Bioavailability,” U.S. Pat. Appl. No. 2004/0058974 for “Treating Fungal Infections,” and European Patent Publication1372394 (A1 ) for “Liquid Suspensions of Posaconazole (SCH 56592) with Enhanced Bioavailability for Treating Fungal Infections.”
| Synonyms: | Pcz;Pos;Noxafil;Sch 56592;Aids058495;Aids-058495;Posconazole;Posaconazole;Posaconazole for research;HYDROXYPROPYL]-2,4-DIHYDRO-3H-1,2,4-TRIAZOL-3-ONE |
| Molecular Formula: | C37H42F2N8O4 |
| Formula Weight: | 700.78 |
Merck’s New Drug Application for an Investigational Intravenous (IV) Formulation of NOXAFIL® (posaconazole) Receives FDA Priority Review
Marketing Authorization Application also Filed with the European Medicines Agency
WHITEHOUSE STATION, N.J., Nov. 18, 2013–(BUSINESS WIRE)–Merck (NYSE:MRK), known as MSD outside the United States and Canada, today announced that its New Drug Application for an investigational intravenous (IV) solution formulation of the company’s antifungal agent, NOXAFIL® (posaconazole), has been accepted for priority review by the U.S. Food and Drug Administration (FDA).http://www.pharmalive.com/mercks-noxafil-nda-gets-fda-priority-review
Posaconazole (CAS Registry Number 171228-49-2; CAS Name: 2,5-anhydro-1 ,3,4-trideoxy-2- C-(2,4-difluorophenyl)-4-[[4-[4-[4-[1-[(1S,2S)-1-ethyl-2-hydroxypropyl]-1 ,5-dihydro-5-oxo-4H- 1 ,2,4-triazol-4-yl]phenyl]-1-piperazinyl]phenoxy]methyl]-1-(1 H-1 ,2,4-triazol-1-yl)-D-threo-pentitol) which is represented by the following general formula (I)

(I)
is known as an antifungal agent. It is available as an oral suspension (40 mg/ml) under the trademark NOXAFIL® from Schering Corporation, Kenilworth, NJ. WO95/17407 and WO 96/38443 disclose the compound having the general formula (I) and its use in treating fungal infections. Various pharmaceutical compositions comprising posaconazole and being adapted for oral, topical or parenteral use are described e.g. in WO 02/80678, U.S. Patent No. 5,972,381 , U.S. Patent No. 5,834,472, U.S. Patent No. 4,957,730 and WO 2005/117831. As was mentioned above, WO 95/17407 and WO 96/38443 disclose the compound having the general formula (I). However, during prosecution of the subsequently filed European patent application no. 98951994.7, now European patent EP 1 021 439 B1 , the applicant declared that the methods disclosed in these publications only lead to the compound of formula (I) as an amorphous solid.
Polymorphism is a phenomenon relating to the occurrence of different crystal forms for one molecule. There may be several different crystalline forms for the same molecule with distinct crystal structures and distinct and varying physical properties like melting point, XRPD pattern, IR-spectrum and solubility profile. These polymorphs are thus distinct solid forms which share the molecular formula of the compound from which the crystals are made up, however, they may have distinct advantageous physical properties which can have a direct effect on the ability to process and/or manufacture the drug product, like flowability, as well as physical properties such as solubility, stability and dissolution properties which can have a direct effect on drug product stability, solubility, dissolution, and bioavailability.
Three polymorphic forms of posaconazole designated as forms I, Il and III are described and characterized in WO 99/18097 (US-B-6,713,481 , US-B-6,958,337). Crystalline forms Il and III were found to be unstable under the conditions investigated, so that crystalline form I was considered to be useful in the development of a pharmaceutical product.
A. K. Saksena et al., WO 9517407; eidem, US 5661151 (1995, 1997 both to Schering);
eidem, Tetrahedron Lett. 37, 5657 (1996).
SCH-56592, a novel orally active broad spectrum antifungal agent35th Intersci Conf Antimicrob Agents Chemother (Sept 17-20, San Francisco) 1995,Abst F61
seeSaksena, A.K.; Girijavallabhan, V.M.; Lovey, R.G.; Pike, R.E.; Wang, H.; Liu, Y.-T.; Ganguly, A.K.; Bennett, F. (Schering Corp.) EP 0736030; JP 1997500658; US 5661151; US 5703079; WO 9517407
Process for the preparation of triazolonesWO 9633178
Mono N-arylation of piperazine(III): Metal-catalyzed N-arylation and its application to the novel preparations of the antifungal posaconazole and its advanced intermediateTetrahedron Lett 2002,43(18),3359
Comparative antifungal spectrum: A. Cacciapuoti et al., Antimicrob. Agents Chemother. 44, 2017 (2000).
Pharmacokinetics, safety and tolerability: R. Courtney et al., ibid. 47, 2788 (2003).
HPLC determn in serum: H. Kim et al., J. Chromatogr. B 738, 93 (2000).
Review of development: A. K. Saksena et al. inAnti-Infectives: Recent Advances in Chemistry and Structure Activity Relationships (Royal Soc. Chem., Cambridge, 1997) pp 180-199; and clinical efficacy in fungal infections: R. Herbrecht, Int. J. Clin. Pract. 58, 612-624 (2004).
synthesis 1

……………..

Synthesis of intermediate (XX): The reaction of 2-chloro-2′,4′-difluoroacetophenone (I) with sodium acetate and NaI in DMF gives 2-acetoxy-2′,4′-difluoroacetophenone (II), which by methylenation with methyltriphenylphosphonium bromide and sodium bis(trimethylsilyl)amide in THF yields 2-(2,4-difluorophenyl)-2-propen-1-ol acetate ester (III). The hydrolysis of (III) with KOH in dioxane/water affords the corresponding alcohol (IV), which is regioselectively epoxidized with titanium tetraisopropoxide and L-(+)-diethyl tartrate in dichloromethane to (S)-(-)-2-(2,4-difluorophenyl)oxirane-2-methanol (V). The reaction of (V) with 1,2,4-triazole (VI) in DMF affords (R)-2-(2,4-difluorophenyl)-3-(1,2,4-triazol-1-yl)propane-1,2-diol (VII), which is selectively mesylated with methanesulfonyl chloride and triethylamine to the monomesylate (VIII). The cyclization of (VIII) with NaH in DMF gives the oxirane (IX), which is condensed with diethyl malonate (X) by means of NaH in DMSO to yield a mixture of (5R-cis)- and (5R-trans)-5-(2,4-difluorophenyl)-2-oxo-5-(1,2,4-triazol-1-ylmethyl) tetrahydrofuran-3-carboxylic acid ethyl ester (XI). The reduction of (XI) with NaBH4 and LiCl in ethanol affords (R)-4-(2,4-difluorophenyl)-2-(hydroxymethyl)-5-(1,2,4-triazol-1-yl) pentane-1,4-diol (XII), which is selectively tosylated with tosyl chloride and triethylamine in THF to the bistosylate (XIII). The cyclization of (XIII) by means of NaH in refluxing toluene gives (5R-cis)-5-(2,4-difluorophenyl)-5-(1,2,4-triazol-1-ylmethyl) tetrahydrofuran-3-methanol tosylate ester (XIV). The reaction of (XIV) with 1-(4-hydroxyphenyl)-4-(4-nitrophenyl)piperazine (XV) to obtain compound (XVI), and the following reaction sequence (XVI) to (XVII) to (XVIII) to (XIX) to (5R-cis)-4-[4-[4-[4-[5-(2,4-difluorophenyl)-5-(1,2,4-triazol-1-ylmethyl)tetrahydrofuran-3-ylmethoxy]phenyl]piperazin-1-yl]phenyl-3,4-dihydro-2H-1,2,4-triazol-3-one (XX) has been performed according to J Med Chem 1984, 27: 894-900.
………………….pat approved expiry
| United States | 5661151 | 1999-07-19 | 2019-07-19 |
| Canada | 2305803 | 2009-12-22 | 2018-10-05 |
| Canada | 2179396 | 2001-04-17 | 2014-12-20 |
| United States | 5703079 | 1994-08-26 | 2014-08-26 |
MORE INFO
| US Patent No | Patent expiry | |
|---|---|---|
| 5661151 | Jul 19, 2019 | |
| 5703079 | Aug 26, 2014 | |
| 6958337 | Oct 5, 2018 | |
| 8263600 | Apr 1, 2022 |
- Cornely OA, Maertens J, Winston DJ, Perfect J, Ullmann AJ, Walsh TJ, Helfgott D, Holowiecki J, Stockelberg D, Goh YT, Petrini M, Hardalo C, Suresh R, Angulo-Gonzalez D: Posaconazole vs. fluconazole or itraconazole prophylaxis in patients with neutropenia. N Engl J Med. 2007 Jan 25;356(4):348-59. Pubmed
- Ullmann AJ, Lipton JH, Vesole DH, Chandrasekar P, Langston A, Tarantolo SR, Greinix H, Morais de Azevedo W, Reddy V, Boparai N, Pedicone L, Patino H, Durrant S: Posaconazole or fluconazole for prophylaxis in severe graft-versus-host disease. N Engl J Med. 2007 Jan 25;356(4):335-47. Pubmed
- Bhattacharya M, Rajeshwari K, Dhingra B: Posaconazole. J Postgrad Med. 2010 Apr-Jun;56(2):163-7. Pubmed
- Frampton JE, Scott LJ: Posaconazole : a review of its use in the prophylaxis of invasive fungal infections. Drugs. 2008;68(7):993-1016.Pubmed
- Schiller DS, Fung HB: Posaconazole: an extended-spectrum triazole antifungal agent. Clin Ther. 2007 Sep;29(9):1862-86. Pubmed
- Kwon DS, Mylonakis E: Posaconazole: a new broad-spectrum antifungal agent. Expert Opin Pharmacother. 2007 Jun;8(8):1167-78.Pubmed
- Groll AH, Walsh TJ: Posaconazole: clinical pharmacology and potential for management of fungal infections. Expert Rev Anti Infect Ther. 2005 Aug;3(4):467-87. Pubmed
- Rachwalski EJ, Wieczorkiewicz JT, Scheetz MH: Posaconazole: an oral triazole with an extended spectrum of activity. Ann Pharmacother. 2008 Oct;42(10):1429-38. Epub 2008 Aug 19. Pubmed
- Li Y, Theuretzbacher U, Clancy CJ, Nguyen MH, Derendorf H: Pharmacokinetic/pharmacodynamic profile of posaconazole. Clin Pharmacokinet. 2010 Jun;49(6):379-96. doi: 10.2165/11319340-000000000-00000. Pubmed
MK 5172 a next Generation HCV NS3/4a Protease Inhibitor

1206524-85-7
Chemical Formula: C29H38N4O7
Exact Mass: 554.27405
Molecular Weight: 554.63462
Elemental Analysis: C, 62.80; H, 6.91; N, 10.10; O, 20.19
IUPAC/Chemical name:
(1aR,5S,8S,10R,22aR)-5-(1,1-Dimethylethyl)-1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-14-methoxy-3,6-dioxo-8H-7,10-methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxaline-8-carboxylic acid.
Development of a Practical, Asymmetric Synthesis of the Hepatitis C Virus Protease Inhibitor MK-5172.Org. Lett. 2013;
15: 4174-4177

MK-5172 is a hepatitis C virus protease inhibitor. Key steps in the synthesis depicted are (1) the regioselective SNAr reaction of dichloroquinoxaline A with prolinol derivative B and (2) construction of the 18-membered macrocycle using a macrolactamization (F → G).
Comment
The medicinal chemistry route to MK-5172 is based on a ring-closing metathesis strategy (S. Harper et al.ACS Med. Chem. Lett. 2012, 3, 332). The best regioselectivity (20:1) and minimization of double substitution in the SNAr reaction of A with B was achieved using 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as the base in polar solvents such as DMSO, NMP, or DMAc.
Merck Announces FDA Acceptance of New Drug Application for Investigational Fertility Treatment

corifollitropin alfa
WHITEHOUSE STATION, N.J.–(BUSINESS WIRE)–Merck (NYSE:MRK), known as MSD outside the United States and Canada, today announced that the New Drug Application (NDA) for its investigational fertility treatment, corifollitropin alfa, has been accepted for standard review by the U.S. Food and Drug Administration (FDA). Merck is seeking FDA approval of corifollitropin alfa for Controlled Ovarian Stimulation (COS) in women participating in assisted reproductive technology.
If approved, corifollitropin alfa would be the first sustained follicular stimulant for use in a fertility treatment regimen.
read all at
http://www.pharmalive.com/fda-accepts-mercks-fertility-treatment-nda

Corifollitropin alfa
Merck received approval on February 15, 2010 from the European Commission for ELONVA (corifollitropin alfa) a long lasting single injection fusion protein lacking LH activity. Only one injection is required for the first seven days, replacing the first seven daily injections of conventional FSH. Initial results demonstrates similar pregnancy rates as daily recombinant FSH injections.[7][8]
- ref 7 N. P. Koper, R. Boostanfar, P. Devroey, B. C. Fauser, P. C. IJzerman-Boon, B. M. J. L. Mannaerts. Global ClinicalDevelopment, Organon, Part of Schering-Plough Corporation, Oss, Netherlands; Huntington Reproductive Center, Tarzana, CA; Center of Reproductive Medicine, Dutch-speaking Free University, Brussels, Belgium; University Medical Center Utrecht, Utrecht, Netherlands; Biometrics, NV Organon, Part of Schering-Plough Corporation, Oss, Netherlands. “Corifollitropin alfa demonstrates similar pregnancy rates as compared to daily recombinant FSH treatment in a controlled ovarian stimulation regimen for IVF/ICSI.” Fertility and Sterility, 90:page S75.
- ref 8 ^ Devroey P, Boostanfar R, Koper NP, Mannaerts BM, Ijzerman-Boon PC, Fauser BC, 2009. “A double-blind, non-inferiority RCT comparing corifollitropin alfa and recombinant FSH during the first seven days of ovarian stimulation using a GnRH antagonist protocol.” Human Reproduction, 2009, August 14, [Epub ahead of print]. PMID 19684043.
In May2013, MSD launched ELONVA® (corifollitropin alfa injection) – a new treatment for fertility, – in Singapore. Approved for controlled ovarian stimulation in combination with a GnRH antagonist for the development of multiple follicles, Corifollitropin alfa injection is the first sustained follicle stimulant. A single subcutaneous injection of the recommended dose of corifollitropin alfa injection may replace the first seven injections of any
Findings showed that other failed repeated treatments may lead to depression, anxiety, sexua conventional daily recombinant follicle stimulating hormone (rFSH) preparation in a controlled ovarian stimulation treatment cycle. Simplified fertility treatment with Elonva not only helps to reduce the emotional and physical burden of fertility, it may also reduce dropout rates and potentially improve the overall chances of pregnancy.
l anxiety/difficulty, relationship problems with partner, family and friends, increased sense of self-blame and guilt, particularly for the partner experiencing fertility problem. ”By reducing the number of daily injections, the
availability of corifollitropin alfa injection is a positive step towards helping reduce the burden of fertility treatment for women experiencing difficulty conceiving. Simplifying fertility treatment with new modalities of treatment and new medication may encourage more infertile couple to embark
on treatment earlier when the wife’s age is younger and ovarian reserve better.” said Dr Loh Seong Feei, Medical Director of Thomson Fertility Centre
Merck and Lupin collaborate to co-market Merck’s Pneumovax 23 Pneumococcal polysacharide vaccine for Indian market
%200.5%20mL407.GIF)
Pneumococcal polysaccharide vaccine (PPSV) — the latest version is known asPneumovax 23 (PPV-23) — is the first pneumococcal vaccine, the first vaccine derived from a capsular polysaccharide, and an important landmark in medical history. The polysaccharide antigens were used to induce type-specific antibodies that enhanced opsonization, phagocytosis, and killing of pneumococci by phagocytic cells. The pneumococcal polysaccharide vaccine is widely used in high-risk adults. As a result, there have been important reductions in the incidence, morbidity, and mortality from pneumococcal pneumoniae and invasive pneumococcal disease.
First used in 1945, the tetravalent vaccine was not widely distributed, since its deployment coincided with the discovery of penicillin. In the 1970s, Robert Austrian championed the manufacture and distribution of a 14-valent PPSV. This evolved in 1983 to a 23-valent formulation (PPSV23). A significant breakthrough impacting the burden of pneumococcal disease was the licensing of a protein conjugate heptavalent vaccine (PCV7) beginning in February 2000.
Merck: Good Results in Alzheimer’s Trial

Processing of the amyloid precursor protein
Merck Presents Findings from Phase 1b Study of Investigational BACE Inhibitor, MK-8931, in Patients with Alzheimer’s Disease

Merck, known as MSD outside the United States and Canada, today announced the presentation of results from a Phase Ib study showing a dose-dependent decrease in β amyloid levels in cerebral spinal fluid (CSF) following administration of MK-8931, Merck’s investigational oral β-site amyloid precursor protein cleaving enzyme (BACE1 or β secretase) inhibitor, in patients with mild to moderate Alzheimer’s disease (AD). In the study, β amyloid levels were analyzed as a measure of BACE activity. The data were presented during an oral session at the Alzheimer’s Association International Conference (AAIC) in Boston, July 13-18 (Abstract O1-06-05).
http://www.pharmalive.com/merck-good-results-in-alzheimers-trial
| Beta-site APP-cleaving enzyme 1 | |||
|---|---|---|---|
 PDB rendering based on 1fkn |
Beta-secretase 1 (BACE1) also known as beta-site APP cleaving enzyme 1(beta-site amyloid precursor protein cleaving enzyme 1), memapsin-2(membrane-associated aspartic protease 2), and aspartyl protease 2 (ASP2) is an enzyme that in humans is encoded by the BACE1 gene.
β-Secretase is an aspartic-acid protease important in the formation of myelin sheaths in peripheral nerve cells. The transmembrane protein contains two active site aspartate residues in its extracellular protein domain and may function as a dimer.
