
Home » Posts tagged 'LNP 023'
Tag Archives: LNP 023
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Iptacopan


Iptacopan
1644670-37-0
422.525, C25H30N2O4
- 4-((2S,4S)-4-ethoxy-1-((5-methoxy-7-methyl-1H-indol-4-yl)methyl)piperidin-2-yl) benzoic acid
- BENZOIC ACID, 4-((2S,4S)-4-ETHOXY-1-((5-METHOXY-7-METHYL-1H-INDOL-4-YL)METHYL)-2-PIPERIDINYL)-
- Iptacopan
- LNP 023
- LNP-023
- LNP023
- NVP-LNP023
- NVP-LNP023-NX
Fda approved, To treat paroxysmal nocturnal hemoglobinuria, 12/5/2023, Fabhalta ‘CHINA 2024
Iptacopan is a small-molecule factor B inhibitor previously investigated as a potential treatment for the rare blood disease paroxysmal nocturnal hemoglobinuria (PNH) by inhibiting the complement factor B.1 Factor B is a positive regulator of the alternative complement pathway, where it activates C3 convertase and subsequently C5 convertase.2 This is of particular importance to PNH, where one of the disease hallmarks is the mutation of the PIGA gene. Due to this mutation, all progeny erythrocytes will lack the glycosyl phosphatidylinositol–anchored proteins that normally anchor 2 membrane proteins, CD55 and CD59, that protect blood cells against the alternative complement pathway.3 Additionally, iptacopan has the benefit of targeting factor B, which only affect the alternative complement pathway, leaving the classic and lectin pathway untouched for the body to still mount adequate immune responses against pathogens.2
On December 6th, 2023, Iptacopan under the brand name Fabhalta was approved by the FDA for the treatment of adults with PNH. This approval was based on favorable results obtained from the phase III APPL-PNH and APPOINT-PNH studies, where 82.3% and 77.5% of patients experienced a sustained hemoglobin improvement without transfusions respectively.5
Iptacopan , sold under the brand name Fabhalta, is a medication used for the treatment of paroxysmal nocturnal hemoglobinuria.[1] It is a complement factor B inhibitor that was developed by Novartis.[1] It is taken by mouth.[1]
Iptacopan was approved by the US Food and Drug Administration (FDA) for the treatment of adults with paroxysmal nocturnal hemoglobinuria in December 2023.[2][3]
Medical uses
Iptacopan is indicated for the treatment of adults with paroxysmal nocturnal hemoglobinuria.[1][4]
Side effects
The FDA label for iptacopan contains a black box warning for the risk of serious and life-threatening infections caused by encapsulated bacteria, including Streptococcus pneumoniae, Neisseria meningitidis, and Haemophilus influenzae type B.[1]
Research
In a clinical study with twelve participants, iptacopan as a single drug led to the normalization of hemolytic markers in most patients, and no serious adverse events occurred during the 12-week study.[5][6]
Iptacopan is also investigated as a drug in other complement-mediated diseases, like age-related macular degeneration and some types of glomerulopathies.[7]
PATENT
https://patents.google.com/patent/US9682968B2/en
Example-26Example-26a4-((2S,4S)-(4-ethoxy-1-((5-methoxy-7-methyl-1H-indol-4-yl)methyl)piperidin-2-yl))benzoic acid ((+) as TFA Salt)

A mixture of methyl 4-((2S,4S)-4-ethoxy-1-((5-methoxy-7-methyl-1H-indol-4-yl)methyl)piperidin-2-yl)benzoate, Intermediate 6-2b peak-1 (tr=1.9 min), (84 mg, 0.192 mmol) and LiOH in H2O (1 mL, 1 mmol) in THF (1 mL)/MeOH (2 mL) was stirred at room temperature for 16 h, and then concentrated. The resulting residue was purified by RP-HPLC (HC-A) to afford the title compound. Absolute stereochemistry was determined by comparison with enantiopure synthesis in Example-26c. 1H NMR (TFA salt, 400 MHz, D2O) δ 8.12 (d, J=8.19 Hz, 2H), 7.66 (br. d, J=8.20 Hz, 2H), 7.35 (d, J=3.06 Hz, 1H), 6.67 (s, 1H), 6.25 (d, J=3.06 Hz, 1H), 4.65 (dd, J=4.28, 11.49 Hz, 1H), 4.04 (d, J=13.00 Hz, 1H), 3.87-3.98 (m, 2H), 3.53-3.69 (m, 5H), 3.38-3.50 (m, 1H), 3.20-3.35 (m, 1H), 2.40 (s, 3H), 2.17-2.33 (m, 2H), 2.08 (br. d, J=15.70 Hz, 1H), 1.82-1.99 (m, 1H), 1.28 (t, J=7.03 Hz, 3H); HRMS calcd. for C26H31N2O3 (M+H)+ 423.2284, found 423.2263.
PATENT
Example 1
PAPER
https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.9b01870
The alternative pathway (AP) of the complement system is a key contributor to the pathogenesis of several human diseases including age-related macular degeneration, paroxysmal nocturnal hemoglobinuria (PNH), atypical hemolytic uremic syndrome (aHUS), and various glomerular diseases. The serine protease factor B (FB) is a key node in the AP and is integral to the formation of C3 and C5 convertase. Despite the prominent role of FB in the AP, selective orally bioavailable inhibitors, beyond our own efforts, have not been reported previously. Herein we describe in more detail our efforts to identify FB inhibitors by high-throughput screening (HTS) and leveraging insights from several X-ray cocrystal structures during optimization efforts. This work culminated in the discovery of LNP023 (41), which is currently being evaluated clinically in several diverse AP mediated indications.



a Reagents and conditions: (a) i PrMgCl·LiCl, Cbz-Cl, THF; (b) Zn, AcOH; (c) LiBH4, THF; (d) TBDPS-Cl, imidazole, DMF; (e) separation of diastereomers by flash chromatography; (f) TBAF, THF; (g) NaH, EtI, DMF; (h) Ba(OH)2, i PrOH, H2O; (i) K2CO3, MeI, DMF; (j) H2, Pd/C, MeOH; (k) (±)-50, DIPEA, DMA; (l) K2CO3, MeOH; then TMS-diazomethane, toluene, MeOH; (m) chiral SFC; (n) LiOH, H2O, MeOH, THF; (o) (2S,4S)-50, NaBH(OAc)3, DCE.
4-((2S,4S)-(4-Ethoxy-1-((5-methoxy-7-methyl-1H-indol-4- yl)methyl)piperidin-2-yl))benzoic Acid (41, LNP023). Step 1: tert-Butyl 4-(((2S,4S)-4-Ethoxy-2-(4-(methoxycarbonyl)phenyl)- piperidin-1-yl)methyl)-5-methoxy-7-methyl-1H-indole-1-carboxylate (58). To a solution of tert-butyl 4-formyl-5-methoxy-7-methyl1H-indole-1-carboxylate (57) (1.5 g, 5.18 mmol) and methyl 4- ((2S,4S)-4-ethoxypiperidin-2-yl)benzoate ((2S,4S)-50) (1.185 g, 4.50 mmol) in DCE (20 mL) was added NaBH(OAc)3 (3 g, 14.1 mmol), and this was stirred at rt for 21.5h. Additional tert-butyl 4-formyl-5- methoxy-7-methyl-1H-indole-1-carboxylate (57) (500 mg, 1.90 mmol) was added, and this was stirred for 20 h. The reaction was diluted with EtOAc, washed successively with 5% aqueous NaHCO3, H2O, and brine, dried over Na2SO4, filtered, and concentrated to provide the title compound (2.415 g, quant) which was used without further purification. MS (ESI+) m/z 537.4 (M + H). The absolutestereochemistry was ultimately determined via cocrystallization of 41 with the catalytic domain of FB. Step 2: 4-((2S,4S)-(4-Ethoxy-1-((5-methoxy-7-methyl-1H-indol-4- yl)methyl)piperidin-2-yl))benzoic Acid (41, LNP023). To a solution of tert-butyl 4-(((2S,4S)-4-ethoxy-2-(4-(methoxycarbonyl)phenyl)- piperidin-1-yl)methyl)-5-methoxy-7-methyl-1H-indole-1-carboxylate (58) (2.415 g, 4.50 mmol) in THF (10 mL) and MeOH (20 mL) was added 1 M LiOH in H2O (15 mL, 15 mmol), and this was stirred at 70 °C for 8 h. The reaction was cooled to rt, diluted with H2O, half saturated aqueous KHSO4 and citric acid, saturated with sodium chloride, then extracted with 9:1 DCM/TFE, dried with Na2SO4, filtered, and concentrated. RP-HPLC-B purification provided the title compound (730 mg, 38% for 2 steps). 1 H NMR (400 MHz, D2O) δ 7.96 (d, J = 8.0 Hz, 2H), 7.58 (d, J = 8.1 Hz, 2H), 7.30 (d, J = 3.2 Hz, 1H), 6.66 (s, 1H), 6.20 (s, 1H), 4.62−4.47 (m, 1H), 4.06 (d, J = 13.2 Hz, 1H), 3.97−3.76 (m, 2H), 3.66−3.48 (m, 5H), 3.43−3.29 (m, 1H), 3.26−3.15 (m, 1H), 2.35 (s, 3H), 2.31−2.11 (m, 2H), 2.00 (d, J = 15.4 Hz, 1H), 1.93−1.74 (m, 1H), 1.25−1.07 (m, 3H). HRMS calcd for C25H31N2O4 (M + H)+ 423.2284, found 423.2263. 4-((2S,4S)-(4-Ethoxy-1-((5-methoxy-7-methyl-1H-indol-4- yl)methyl)piperidin-2-yl))benzoic Acid Hydrochloride (41· HCl). To a solution of 41 (620 mg, 1.47 mmol) in H2O (10 mL) and acetonitrile (3 mL) was added 5 M aqueous HCl (0.5 mL, 2.5 mmol). The mixture was then lyophilized, and the resulting solid was suspended in i PrOH and heated to 70 °C. The mixture turned into a solution after 1.5 h and was then cooled to rt with stirring. After about 5 h, the mixture turned into a suspension and the solid was collected by filtration and dried under high vacuum at 50 °C to provide the title compound as the hydrochloride salt (450 mg, 65%). 1 H NMR (400 MHz, methanol-d4) δ 10.73 (s, 1H), 8.23 (d, J = 8.2 Hz, 2H), 7.74 (d, J = 8.3 Hz, 2H), 7.36−7.31 (m, 1H), 6.77 (s, 1H), 6.42−6.31 (m, 1H), 4.40−4.19 (m, 2H), 3.87−3.80 (m, 1H), 3.76 (s, 3H), 3.68− 3.50 (m, 4H), 3.45−3.38 (m, 1H), 2.51 (s, 3H), 2.30−2.18 (m, 2H), 2.13−1.89 (m, 2H), 1.31 (t, J = 7.0 Hz, 3H). MS (ESI+) m/z 423.3 (M + H).
SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Iptacopan (Fabhalta®), a first-in-class oral therapeutic agent discovered by Novartis, specifically targets the complement Factor B protein within the alternative complement system. NMPA granted
marketing authorization in 2024, indicated for complement inhibitor-naïve adult patients diagnosed with paroxysmal nocturnal hemoglobinuria (PNH) [75]. By competitively binding to the catalytic domain of
Factor B, the drug effectively blocks C3 convertase assembly, thereby suppressing downstream cleavage of C3 into its active fragments. This dual inhibitory action addresses both intravascular erythrocyte
destruction and extravascular hemolytic processes characteristic of PNHpathogenesis [76]. Clinical validation emerged from the multinational APPOINT-PNH study (ClinicalTrials.gov identifier NCT04820530), where treatment-naïve participants exhibited sustained hemoglobin
stabilization (≥12 g/dL) in 79.6 % of cases, achieving transfusion in dependence over 24 weeks. Secondary endpoints revealed significant improvements in fatigue scores and health-related quality metrics [77]. Safety monitoring identified encapsulated bacterial infection as critical risks, necessitating mandatory vaccination ≥2 weeks pre-treatment. Common treatment-emergent adverse events comprised transient gastrointestinal disturbances (nausea 18.3 %, diarrhea 14.7 %) and mild
cephalgia (22.1 %), with resolution typically occurring within 4 weeks [78].
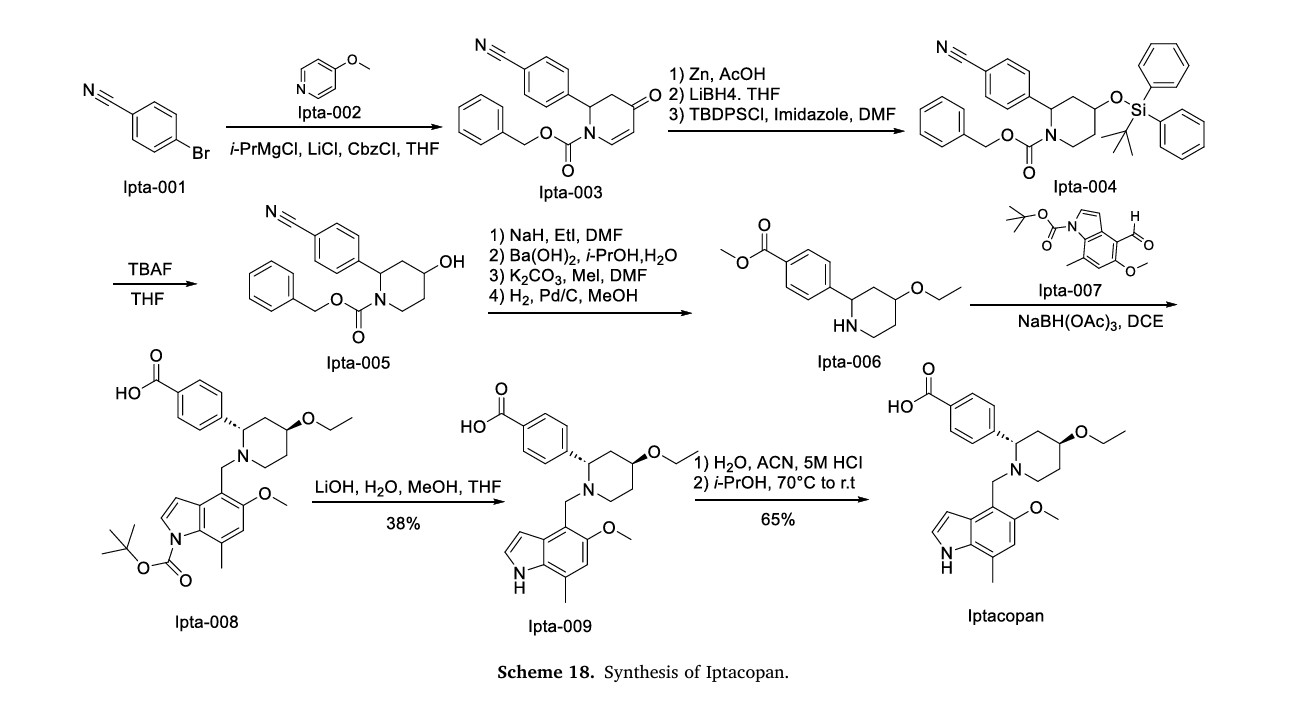
The synthetic pathway of Iptacopan, delineated in Scheme 18, initiates with nucleophilic substitution between Ipta-001 and Ipta-002, followed by Grignard coupling yielding Ipta-003 [79]. This intermedi
ate undergoes NaBH4-mediated reduction and TMSCl-induced silanization to afford Ipta-004. Acid-catalyzed TMS deprotection (HCl/MeOH) delivers Ipta-005, which progresses through sequential alkylation (methyl iodide/K2CO3 catalytic hydrogenation (H)/Pd–C), transesterification (EtONa), and to construct Ipta-006. Condensation with Ipta-007 and subsequent reduction forms Ipta-008. Strategic TFA-mediated Boc cleavage in DCM followed by HCl-induced salt formation in dioxane ultimately furnishes Iptacopan hydrochloride.
75-79
[75] Iptacopan, Drugs and Lactation Database (Lactmed®), National Institute of Child
Health and Human Development, Bethesda (MD), 2006.
[76] J.H. Jang, L. Wong, B.S. Ko, S.S. Yoon, K. Li, I. Baltcheva, P.K. Nidamarthy,
R. Chawla, G. Junge, E.S. Yap, Iptacopan monotherapy in patients with paroxysmal
nocturnal hemoglobinuria: a 2-cohort open-label proof-of-concept study, Blood
Adv 6 (2022) 4450–4460.
[77] A.M. Risitano, C. de Castro, B. Han, A.G. Kulasekararaj, J.P. Maciejewski,
P. Scheinberg, Y. Ueda, S. Vallow, G. Bermann, M. Dahlke, R. Kumar, R. Peffault de
Latour, Patient-reported improvements in patients with PNH treated with
iptacopan from two phase 3 studies, Blood Adv 9 (2025) 1816–1826.
[78] C.M. de Castro, B.J. Patel, Iptacopan for the treatment of paroxysmal nocturnal
hemoglobinuria, Expert Opin Pharmacother 25 (2024) 2331–2339.
[79] N. Mainolfi, T. Ehara, R.G. Karki, K. Anderson, A. Mac Sweeney, S.M. Liao, U.
A. Argikar, K. Jendza, C. Zhang, J. Powers, D.W. Klosowski, M. Crowley,
T. Kawanami, J. Ding, M. April, C. Forster, M. Serrano-Wu, M. Capparelli,
R. Ramqaj, C. Solovay, F. Cumin, T.M. Smith, L. Ferrara, W. Lee, D. Long,
M. Prentiss, A. De Erkenez, L. Yang, F. Liu, H. Sellner, F. Sirockin, E. Valeur,
P. Erbel, D. Ostermeier, P. Ramage, B. Gerhartz, A. Schubart, S. Flohr, N. Gradoux,
R. Feifel, B. Vogg, C. Wiesmann, J. Maibaum, J. Eder, R. Sedrani, R.A. Harrison,
M. Mogi, B.D. Jaffee, C.M. Adams, Discovery of 4-((2S,4S)-4-Ethoxy-1-((5-
methoxy-7-methyl-1H-indol-4-yl)methyl)piperidin-2-yl)benzoic acid (LNP023), a
factor B inhibitor specifically designed to be applicable to treating a diverse array
of complement mediated diseases, J. Med. Chem. 63 (2020) 5697–5722.
.
//////////////


AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
///////////
| Clinical data | |
|---|---|
| Trade names | Fabhalta |
| Other names | LNP023 |
| AHFS/Drugs.com | Fabhalta |
| License data | US DailyMed: Iptacopan |
| Routes of administration | By mouth |
| Drug class | Complement factor B inhibitor |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| CAS Number | 1644670-37-0 |
| PubChem CID | 90467622 |
| DrugBank | DB16200 |
| ChemSpider | 75533872 |
| UNII | 8E05T07Z6W |
| KEGG | D12251D12252 |
| ChEMBL | ChEMBL4594448 |
| PDB ligand | JGQ (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C25H30N2O4 |
| Molar mass | 422.525 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
References
- ^ Jump up to:a b c d e f “Fabhalta- iptacopan capsule”. DailyMed. 5 December 2023. Archived from the original on 10 December 2023. Retrieved 10 December 2023.
- ^ “Novartis receives FDA approval for Fabhalta (iptacopan), offering superior hemoglobin improvement in the absence of transfusions as the first oral monotherapy for adults with PNH”. Novartis (Press release). Archived from the original on 12 December 2023. Retrieved 6 December 2023.
- ^ “Novel Drug Approvals for 2023”. U.S. Food and Drug Administration (FDA). 6 December 2023. Archived from the original on 21 January 2023. Retrieved 10 December 2023.
- ^ https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2023/218276Orig1s000ltr.pdf Archived 10 December 2023 at the Wayback Machine
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jang JH, Wong L, Ko BS, Yoon SS, Li K, Baltcheva I, et al. (August 2022). “Iptacopan monotherapy in patients with paroxysmal nocturnal hemoglobinuria: a 2-cohort open-label proof-of-concept study”. Blood Advances. 6 (15): 4450–4460. doi:10.1182/bloodadvances.2022006960. PMC 9636331. PMID 35561315.
- ^ “Novartis Phase III APPOINT-PNH trial shows investigational oral monotherapy iptacopan improves hemoglobin to near-normal levels, leading to transfusion independence in all treatment-naïve PNH patients”. Novartis (Press release). Archived from the original on 12 December 2023. Retrieved 6 September 2023.
- ^ Schubart A, Anderson K, Mainolfi N, Sellner H, Ehara T, Adams CM, et al. (April 2019). “Small-molecule factor B inhibitor for the treatment of complement-mediated diseases”. Proceedings of the National Academy of Sciences of the United States of America. 116 (16): 7926–7931. Bibcode:2019PNAS..116.7926S. doi:10.1073/pnas.1820892116. PMC 6475383. PMID 30926668.
External links
- Clinical trial number NCT04558918 for “Study of Efficacy and Safety of Twice Daily Oral LNP023 in Adult PNH Patients With Residual Anemia Despite Anti-C5 Antibody Treatment (APPLY-PNH)” at ClinicalTrials.gov
- Clinical trial number NCT04820530 for “Study of Efficacy and Safety of Twice Daily Oral Iptacopan (LNP023) in Adult PNH Patients Who Are Naive to Complement Inhibitor Therapy (APPOINT-PNH)” at ClinicalTrials.gov
///////Iptacopan, fda 2023, approvals, 2023, paroxysmal nocturnal hemoglobinuria, 12/5/2023, Fabhalta , LNP 023, LNP-023, LNP023, NVP-LNP023, NVP-LNP023-NX

NEW DRUG APPROVALS
ONE TIME
$10.00
LNP 023

![4-[(2S,4S)-4-Ethoxy-1-[(5-methoxy-7-methyl-1H-indol-4-yl)methyl]piperidin-2-yl]benzoic acid.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=90467622&t=l)
LNP 023
CAS 1644670-37-0
ROTATION +
4-((2S,4S)-4-ethoxy-1-((5-methoxy-7-methyl-1H-indol-4-yl)methyl)piperidin-2-yl)benzoic acid
| M.Wt | 422.525 | |
| Formula | C25H30N2O4 | |
4-[(2S,4S)-4-ethoxy-1-[(5-methoxy-7-methyl-1H-indol-4-yl)methyl]piperidin-2-yl]benzoic acid
LNP023
RENRQMCACQEWFC-UGKGYDQZSA-N
PATENT US9682968, Example-26a
BDBM160475
4-((2S,4S)-(4-ethoxy-1-((5-methoxy-7-methyl-1H-indol-4-yl)methyl)piperidin-2-yl))benzoic acid
4-[(2~{S},4~{S})-4-ethoxy-1-[(5-methoxy-7-methyl-1~{H}-indol-4-yl)methyl]piperidin-2-yl]benzoic acid
LNP023 (LNP-023) is a highly potent, reversible, selective inhibitor of factor B (IC50=10 nM), the proteolytically active component of the C3 and C5 convertases.
LNP023 (LNP-023) is a highly potent, reversible, selective inhibitor of factor B (IC50=10 nM), the proteolytically active component of the C3 and C5 convertases; shows direct, reversible, and high-affinity binding to human FB with Kd of 7.9 nM in SPR assays, demonstrates potent inhibition of AP-induced MAC formation in 50% human serum with IC50 of 0.13 uM; shows no inhibition of factor D (FD), as well as classical or lectin complement pathway activation (up to 100 uM), and no significant effects (up to 10 μM) in a broad assay panel of receptors, ion channels, kinases, and proteases; blocks zymosan-induced MAC formation membrane attack complex (MAC) with IC50 of 0.15 uM, prevents KRN-induced arthritis in mice and is effective upon prophylactic and therapeutic dosing in an experimental model of membranous nephropathy in rats afer oral adminstration; also prevents complement activation in sera from C3 glomerulopathy patients and the hemolysis of human PNH erythrocytes.
Other Indication
Phase 2 Clinical

PATENT
WO 2015009616
https://patents.google.com/patent/WO2015009616A1/en
PATENT
https://patents.google.com/patent/US9682968B2/en
Example-26Example-26a4-((2S,4S)-(4-ethoxy-1-((5-methoxy-7-methyl-1H-indol-4-yl)methyl)piperidin-2-yl))benzoic acid ((+) as TFA Salt)
A mixture of methyl 4-((2S,4S)-4-ethoxy-1-((5-methoxy-7-methyl-1H-indol-4-yl)methyl)piperidin-2-yl)benzoate, Intermediate 6-2b peak-1 (tr=1.9 min), (84 mg, 0.192 mmol) and LiOH in H2O (1 mL, 1 mmol) in THF (1 mL)/MeOH (2 mL) was stirred at room temperature for 16 h, and then concentrated. The resulting residue was purified by RP-HPLC (HC-A) to afford the title compound. Absolute stereochemistry was determined by comparison with enantiopure synthesis in Example-26c. 1H NMR (TFA salt, 400 MHz, D2O) δ 8.12 (d, J=8.19 Hz, 2H), 7.66 (br. d, J=8.20 Hz, 2H), 7.35 (d, J=3.06 Hz, 1H), 6.67 (s, 1H), 6.25 (d, J=3.06 Hz, 1H), 4.65 (dd, J=4.28, 11.49 Hz, 1H), 4.04 (d, J=13.00 Hz, 1H), 3.87-3.98 (m, 2H), 3.53-3.69 (m, 5H), 3.38-3.50 (m, 1H), 3.20-3.35 (m, 1H), 2.40 (s, 3H), 2.17-2.33 (m, 2H), 2.08 (br. d, J=15.70 Hz, 1H), 1.82-1.99 (m, 1H), 1.28 (t, J=7.03 Hz, 3H); HRMS calcd. for C26H31N2O3 (M+H)+ 423.2284, found 423.2263.
PATENT
WO 2020016749
The present invention relates to a process for the preparation of phenylpiperidinyl indole derivatives. More particularly, the present invention relates to a process for the preparation of the compound of formula (I)
also referred to as 4-((2S,4S)-(4-ethoxy-1 -((5-methoxy-7-methyl-1 /-/-indol-4-yl)methyl)piperidin-2-yl))benzoic acid, or a pharmaceutically acceptable salt thereof, which is capable of inhibiting the activation of the alternative pathway of the complement system. The complement system plays a major role in the innate and adaptive immunity system and comprises a group of proteins that are normally present in an inactive state. These proteins are organized in three activation pathways: the classical, the lectin, and the alternative pathways (Holers, In Clinical Immunology: Principles and practice, ed. R.R. Rich, Mosby Press; 1996, 363-391 ). Molecules from microorganisms, antibodies or cellular components can activate these pathways resulting in the formation of protease complexes known as the C3-convertase and the C5-convertase. The classical pathway is a calcium / magnesium-dependent cascade, which is normally activated by the formation of antigen-antibody complexes. It can also be activated in an antibody-independent manner by the binding of C-reactive protein complexed to
ligand and by many pathogens including gram-negative bacteria. The alternative pathway is a magnesium-dependent cascade, which is activated by deposition and activation of C3 on certain susceptible surfaces (e.g. cell wall polysaccharides of yeast and bacteria, and certain biopolymer materials). The alternative pathway (AP) utilizes C3 fragments (C3b) to opsonize the pathogens hence targeting them for phagocytosis without the need for antibodies. Hyperactivity of the complement system, and in particular in its AP, plays a role in a large number of complement-driven diseases, such as C3 glomerulopathy (C3G), paroxysmal nocturnal hemoglobinuria (PNH) and IgA nephropathy (IgAN). Phenylpiperidinyl indole derivatives, such as compound of formula (I), or a pharmaceutically acceptable salt thereof, play a role in the inhibition of complement factor B, a known critical enzyme for activation of the alternative complement pathway (Lesavre et al J. Exp. Med. 1978, 148, 1498-1510; Volanakis et al New Eng. J. Med. 1985, 312, 395-401 ), which may also be a suitable target for the inhibition of the amplification of the complement pathways. The phenylpiperidinyl indole derivatives, such as compound of formula (I), or a pharmaceutically acceptable salt thereof, and a method for preparing such derivatives, are described in WO2015/009616. In particular, compound of formula (I) is described in example 26, of WO2015/009616. One of the drawbacks of the synthesis was the use of hazardous chemicals (such as sodium hydride, or dimethylacetamide, which represent safety concerns on a larger scale) and the poor enantio- and diastereo-selectivity of the steps, leading to unwanted stereoisomers.
Thus, there is a need to provide an alternative reaction route in a process for producing compound of formula (I), or a pharmaceutically acceptable salt thereof, generating less by products, and easier to handle on a large scale.
Scheme 1 , vide infra.
Compound of fformula (II)
ormu a ( )
formula (1)
Scheme 1
1. Asymmetric synthesis of compound of formula (II): .
One aspect of the present invention relates to an asymmetric process for preparing a compound of formula (II), or salt thereof, as outlined in Scheme 2 below, wherein the stereocenters in position 2 and in position 4 on the piperidine are obtained in high enantio- and diastereo-selectivity.
formula (ii)
Scheme 2
Example 1 : Synthesis of Benzyl-2-r4-(methoxycarbonyl)phenyl1-4-oxopiperidine-1 -carboxylate according to the following sequence:
Y
R = Methyl R = Methyl R =: Methyl
Step 1 : Synthesis of Benzyl-2-[4-(methoxycarbonyl)phenyl]-4-oxo-3, 4-dihydro pyridine-1(2W)-carboxylate (C3, wherein Pi = Cbz and R = methyl)
iPrMgCI (2N THF, 109.96 g, 54.98 ml_, 2.0 eq) was charged in a reactor. A solution of bis[2 -(N,N-dimethylaminoethyl)] ether (2.5 eq, 22.03 g, 137.46 mmol) in THF (24 ml.) was added at 15 – 25 °C. The mixture was stirred for 1 hour. A solution of C1 (20.17 g, 76.98 mmol, 1 .4 eq) in THF (102 ml.) was added slowly at 15 – 25 °C. The mixture was heated to 25 – 30 °C, stirred for more than 1 hour, and checked by HPLC. The mixture was cooled to -30 °C. A solution of C2 (methyl 4-iodobenzoate, 6.0 g, 54.98 mmol, 1 .0 eq) in THF (20 ml.) was added, followed by a solution of benzyl chloroformate (1 .15 eq, 10.79 g, 63.23 mmol) in THF (36 ml_). The mixture was stirred for 2 hours and quenched with AcOH (6.60 g, 109.96 mmol, 2 eq). Isopropyl acetate (60 ml.) was added. Hydrogen chloride (15%, 90 g) was added to adjust the pH = 1 – 2. The organic layer was separated and washed with brine (15%, 100 g), and concentrated. Isopropyl acetate (160 ml.) was added and concentrated to remove the THF. The crude product was recrystallized in Isopropyl
acetate (1 14 ml.) and n-heptane (120 ml_). The product was dried at 60 °C to provide C3 as light yellow solid (16.0 g, 79.65 % yield). 1 H-NMR (400 MHz, DMSO-d6) d (ppm) = 8.1 1 (dd, J=8.39, 1.01 Hz, 1 H), 7.91 (d, J=8.39 Hz, 2H), 7.33 – 7.37 (m, 6H), 5.82 (d, J= 7.20 Hz, 1 H), 5.20 – 5.35 (m, 3H) , 3.83 (s, 3H), 3.41 (br. s, 1 H), 3.31 (dd, J=16.64, 7.52 Hz, 1 H), 2.66 (br. d, J=16.55 Hz, 1 H).
Step 2: Synthesis of Benzyl-2-[4-(methoxycarbonyl)phenyl]-4-oxopiperidine-1 -carboxylate (C4, wherein Pi = Cbz and R = methyl)
A solution of C3 (25 g, 68.42 mmol, 1 .0 eq) in AcOH (200 ml.) was heated to 50 – 60 °C to form a clear solution. The solution was then cooled to 35 °C. Zn powder (13.42 g, 205.26 mmol, 3.0 eq) was added portionwise while keeping the inner temperature at 35 – 40 °C. After addition, the mixture was stirred for more than 8 hours and checked by HPLC. THF (250 ml.) was added. The mixture was cooled to 25 °C, filtered, and the filter cake was washed with THF (125 volume). The filtrate was concentrated to dryness. Isopropanol (375 ml.) was added. The solution was cooled to 0 – 5 °C. EDTA-4Na.2H20 (40 g) in water (200 ml.) was added. The mixture was neutralized to pH = 9 – 10 with 30% sodium hydroxide solution and stirred for 2 hours. The organic layer was collected, washed with brine (15%, 250 g) and concentrated to about 50 ml_. MTBE (100 ml.) was added and concentrated to about 50 ml_. MTBE (80 ml.) was added followed by n-heptane (20 ml.) dropwise. Then the mixture was cooled to 0 °C gradually. The mixture was filtered and the filter cake was dried to afford C4 as a light yellow solid (20.1 1 g, 80.0 % yield). 1 H NMR (400 MHz, CDCIs) d (ppm)= 7.99 (d, J=8.31 Hz, 2H), 7.27 – 7.39 (m, 7H), 5.83 (br. s, 1 H), 5.14 – 5.28 (m, 2H), 4.20 – 4.42 (m, 1 H), 3.92 (s, 3H), 3.12 – 3.33 (m, 1 H), 2.84 – 3.04 (m, 2H), 2.46 – 2.65 (m, 1 H), 2.23 – 2.45 (m, 1 H).
Example 2: Synthesis of Benzyl -4-hvdroxy-2-(4-(methoxycarbonyl)phenyl)piperidine-1-
carboxylate (C5. wherein Pi = Cbz and R = methyl)
P1 = Cbz P i = Cbz
R = Methyl R = Methyl
A 0.1 M pH = 7.0 PBS was prepared with disodium phosphate dodecahydrate (22.2 g), sodium dihydrogen phosphate dihydrate (6.2 g) and purified water (999 g). To a reactor equipped with a pH meter 0.1 M pH = 7.0 PBS (499 g), D-glucose (40.2 g, 233.14 mmol, 2.0 eq), NADP (EnzymeWorks, 0.72 g), GDH (EnzymeWorks, 0.41 g) and KRED-EW124 (EnzymeWorks, 2.05 g)
were added, followed by addition of emulsion of C4 (41 g, 1 1 1 .60 mmol, 1 .0 eq) in DMSO (102.5 ml_). The mixture was heated to JT < 45 °C, IT 41 ± 3 °C and stirred at IT 41 ± 3 °C for > 16 h while controlling pH 6.9-7.2 by adding 1 M sodium hydroxide solution. A mixture of NADP (0.29 g), GDH (0.16 g) and KRED-EW124 (0.82 g, #Enzyme Works Inc. China) in 0.1 M pH = 7.0 PBS (1 1 g) were charged and stirred at IT 41 ± 3 °C for > 20 hours. The reaction was monitored by HPLC.
The reaction was filtered to afford white wet cake. To a 1 .0 L Radleys reactor equipped with anchor agitator crude C5 wet cake (80 g) and acetonitrile (500 ml.) were charged. The mixture was stirred to form a light yellow suspension (700 RPM). The suspension was heated to IT = 70 ± 5 °C and stirred for 4 hours, and then cooled to IT = 25 ± 5 °C. The suspension was filtered and the cake was washed with acetonitrile (75 ml_). To a clean 500 ml. Radleys reactor equipped with anchor agitator the resulting mother liquor was charged. The mother liquid was concentrated to about 95 g, solvent exchanged with three portions of toluene (105 g) to 95 g residue. Toluene (170 g) was charged and the reaction was checked by GC (acetonitrile / (toluene + acetonitrile) < 1 .2%). The suspension was heated to IT = 80 ± 5 °C, held for 1 hour, cooled to IT = 45 ± 3 °C and adjusted the agitation speed to low mode. Sequential operations of seeding and aging for 2 hours, charging n-heptane (10.2 g) in 0.5 hours and aging for 1 hour, charging n-heptane (34 g) over 1 .5 hours and aging for 0.5 hours were carried out. The mixture was cooled to IT = 10 ± 3 °C over 7 hours and maintained at 10 ± 3 °C for 2 hours. The mixture was filtered and the cake was washed with cold mixed solvents of toluene (50 ml.) and n-heptane (10 ml.) to afford a light yellow solution of C5 (330 g, trans/cis = 90/10, assay 6.8%, yield 52%). The mother liquor was telescoped to the next step. 1 H-NMR (400 MHz, CDCI3, mixture of two isomers, data for the minor isomer is shown in brackets): d (ppm) = 7.99 (d, J=8.44 Hz, 2H) [7.92 (d, J=8.44 Hz, 0.04H)], 7.23 – 7.39 (m, 7H) [7.10 – 7.18 (m, 0.21 H)], 5.69 (br. s, 1 H) [5.40-5.42 (m, 0.1 1 H)], 5.19 (s, 2H) [5.14 (s, 0.23H)], 4.26 (br. d, J=13.33 Hz, 1 H) [4.18-4.20(m, 0.13H)], 3.91 (s, 3H) [3.90 (s, 0.4H)], 3.67 – 3.79 (m, 1 H) [3.38-3.45 (m, 0.1 1 H)], 2.83 (td, J=13.51 , 2.81 Hz, 1 H), 2.64 (br. d, J=13.33 Hz, 1 H) [2.41 -2.47 (m, 0.12H)], 1 .81-1 .91 (m, 2H) [2.17-2.22 (m, 0.12H)], 1 .72 – 1 .77 (m, 1 H), 1 .45 – 1 .56 (m, 1 H). HRMS: Calcd for C21 H24NO5 (M+H): 370.1654m, found 370.1662.
Example 3: Synthesis of Methyl 4-r(2S,4S)-4-ethoxypiperidin-2-yl1benzoate (Compound of formula according to the following sequence:
R = Methyl R = Methyl R = Methyl
Step 1 : Synthesis of Benzyl (4S)-4-((tert-butyldimethylsilyl)oxy)-2-(4-(methoxycarbonyl) phenyl)piperidine-1 -carboxylate (C8, wherein Pi = Cbz, P2 = TBS and R = methyl).
To a 500 ml. Radleys Reactor charged with C5 in a toluene/heptane solution (1 .0 eq, 145.67 g from previous step, assay 6.07%, 23.94 mmol). The solution was concentrated to about 25 g. Then dichloromethane (1 17.1 g) was charged and the solution was cooled to 23 ± 4 °C. To the clear solution, imidazole (3.42 g, 50.26 mmol, 2.1 eq) and TBS-CI (6.13 g, 40.69 mmol, 1 .7 eq) were introduced. The yellow suspension was stirred at 23 ± 4 °C for 10 hours. The reaction was monitored by HPLC. Then 10% Na2CC>3 (70.7 g) was charged and the mixture was stirred for 1 hours. The organic phase was washed with 5% brine (53 g) and concentrated to about 30 g. Then the solvent was exchange with toluene (45 g) to about 25 g. The residue was diluted with dichloromethane (66 g) and the mixture was filtered through a pad of 200-300 mesh silica gel (1 .66 g). The silica gel was eluted with another portion of dichloromethane (17.5 g). The eluent was concentrated and the residue was subjected to solvent exchange with acetonitrile (71 .1 g + 98.2 g) to 90 g (yield 100%). C8 in acetonitrile solution was used in the next step. 1 H-NMR (400 MHz, CDCI3, mixture of two isomers, data for the minor isomer is shown in brackets): d (ppm) = 8.01 (d, J=8.44 Hz, 2H) [7.94 (d, J=8.44 Hz, 0.17H)], 7.26 – 7.34 (m, 7H) [7.09 – 7.18 (m, 0.13H)], 5.65 (br. d, J=2.04 Hz, 1 H) [5.41 (br. d, J=2.04 Hz, 0.08H)], 5.19 (s, 2H) [5.13 (s, 0.16H)], 4.22 (br. d, J=13.69 Hz, 1 H) [4.10-4.14(m, 0.19H)], 3.92 (s, 3H) [3.90 (s, 0.3H)], 3.62 – 3.69 (m, 1 H) [3.43-3.50 (m, 0.08H)], 2.81 (td, J=13.54, 2.87 Hz, 1 H), 2.49 (br. d, J=13.57 Hz, 1 H) [2.31 -2.35 (m, 0.1 OH)], 1.84-1 .92 (m, 1 H) [2.08-2.14 (m, 0.07H)], 1 .74 – 1 .75 (m, 1 H), 1 .48 – 1 .59 (m, 1 H), 0.86 (s, 9H) [0.56 (s, 0.65H)], 0.03 (s, 3H) [0.09 (s, 0.27H)].
Step 2: Synthesis of Benzyl (4S)-4-ethoxy-2-(4-(methoxycarbonyl)phenyl)piperidine-1 -carboxylate (C9, wherein Pi = Cbz, R = methyl)
To a 250 ml. Radleys Reactor equipped with impeller agitator C8 in acetonitrile solution (135.5 g, assay 12.53%, 35.10 mmol) was charged and rinsed with acetonitrile (with 8.5 g). Et3SiH (12.25 g, 105.31 mmol, 3.0 eq) was charged. The reactor was cooled to IT = 4 ± 5 °C. TESOTf (1 .392 g,
5.265 mmol, 0.15 eq) was charged. A solution of 2,4,6-trimethyM ,3,5-trioxane (4.64 g, 35.10 mmol, 1 .0 eq) in acetonitrile (7.9 g) was added to the mixture in 60 min at IT = 4 ± 5 °C. After addition, the mixture was stirred for 15 min and followed by HPLC. To the reaction mixture was charged 5% aqueous Na2CC>3 (21 .22 g) and water (30 g). Followed by n-heptane (20.4 g) and the mixture was stirred at 25 ± 5 °C for 30 min. Phase cut and the bottom acetonitrile phase was collected. The acetonitrile phase was concentrated to about 65 g. MTBE (100.6 g) and 5% aqueous Na2CC>3 (43.44 g) were charged to the residual acetonitrile solution. The mixture was stirred for 30 min. The upper MTBE phase was collected and filtered via Charcoal film. The charcoal film was washed with MTBE (7.4 g). The mother liquor was concentrated to about 35 g. To the residue methanol (79.2 g) was charged and the solution was concentrated to 70 g. The solution was telescoped to the next step. 1 H NMR (400 MHz, CDCI3, mixture of two isomers, data for the minor isomer is shown in brackets) d (ppm) = 8.01 (d, J= 8.31 Hz, 2H) [7.96 (d, J= 8.31 Hz, 0.21 H)], 7.29 – 7.32 (m, 7H) [7.07 – 7.22 (m, 0.40H)], 5.68 (br. s, 1 H) [5.32 – 5.34 (m, 0.10H)], 5.19 (s, 2H) [5.1 1 (s, 0.19H)], 4.27 (br. d, J=13.08 Hz, 1 H) [4.05 – 4.14 (m, 0.15H)], 3.91 (s, 3H) [3.89 (s, 0.15H)], 3.41 – 3.54 (m, 2H) [3.14 – 3.25 (m, 0.21 )], 3.30 – 3.40 (m, 1 H) [3.86 – 3.75 (m, 0.13H)], 2.84 (td, J=13.51 , 2.81 Hz, 1 H), 2.66 (br. d, J=13.20 Hz, 1 H), 1 .62 – 1 .95 (m, 2H), 1 .40 – 1 .53 (m, 1 H), 1 .18 (t, J= 6.97 Hz, 3H).
Step3: Synthesis of Methyl 4-((4S)-4-ethoxypiperidin-2-yl)benzoate (removal of the protecting group Pi = Cbz – R = methyl)
To a 500 ml. autoclave charged with 10% Pd/C (50% wet, 3.83 g), C9 solution in methanol (assay 19.97%, 192 g, 96.46 mmol) and methanol (28 g). The reactor was purged with vacuum/H2, three times. The mixture was hydrogenated at 3 bar and at a temperature of 25 ± 4 °C for 4 hours. The mixture was filtered and the Pd/C cake was washed with methanol (20 g). The mother liquor was concentrated to 48 g, solvent swapped twice with 142 g isopropyl acetate to 106 g, cooled to 8 ± 5 °C, and 3% hydrogen chloride solution (90.2 g) was added. After phase separation, the aqueous phase was collected and washed with isopropyl acetate (86.4 g). To the aqueous phase MTBE (72 g) and 10% Na2C03 (99.2 g) were added. After phase separation, the aqueous phase was extracted with MTBE (72 g). The combined MTBE phase was washed with water (40 g). The MTBE solution was introduced into the next step. 1 H NMR (400 MHz, CDCI3, mixture of two isomers, data for the minor isomer is shown in brackets) d (ppm) = 7.96 (m, J= 8.31 Hz, 2H), 7.40 – 7.46 (m, 2H), 4.06 (dd, J=1 1 .62, 2.45 Hz, 1 H), 3.88 (s, 3H), 3.70 – 3.79 (m, 1 H) [3.64 – 3.69 (m, 0.12H)], 3.48 -3.56 (m, 2H) [3.38 – 3.45(m, 0.1 1 H)], 3.1 1 – 3.18 (m, 1 H) [3.21 – 3.26 (m, 0.1 1 H)], 2.88 – 2.97 (m, 1 H) [2.73 – 2.80 (m, 0.12H )], 1 .94 – 2.00 (m, 1 H) [ 2.14 – 2.19 (m, 0.10H)], 1.84 – 1 .89 (m, 1 H) [2.02 – 2.07 (m, 0.12H)], 1 .75 (S, 1 H), 1 .65 – 1 .70 (m, 1 H) [1 .45 – 1 .49 (m, 0.10H)], 1 .59 – 1 .64 (m, 1 H) [1 .36 – 1 .42 (m, 0.1 1 H)], 1 .22 – 1 .25 (t, 3H) [1 .17 – 1 .20 (t, J= 6.97, 0.24H)].
Step 4: Synthesis of Methyl 4-[(2S,4S)-4-ethoxypiperidin-2-yl]benzoate (Compound of formula (II) – R = methyl).
To a 500 ml. one neck flask was added the crude solution of step 3 (above) in MTBE (telescoped from last step, 1 10 g, assay 10.52%, light yellow solution, 43.95 mmol). The solution was concentrated to 18.4 g and the solvent was exchanged (JT = 60 °C) with 55 g of n-heptane twice to get 35 g yellow solution. The solution was transferred to 100 ml. Easy Max equipped with impeller agitator. The solution was heated to 50 °C with 300 RPM , aged for 30 min, cooled to 41 ± 2 °C and seed was added. The agitation was adjusted to low speed. The mixture was aged at 41 ± 2 °C for 2 hours, cooled to 35 ± 2 °C in 8 – 10 hours and then aged at 35 ± 2 °C for 1 – 2 hours n-heptane (7.9 g) was added dropwise. The agitation was adjusted to medium speed. The mixture was cooled to IT = 25 ± 2 °C in 1 hour and aged at 25 ± 2 °C for 10 – 20 minutes. The mixture was filtered. The filtrate was re-charged to the reactor for rinsing the solid on the reactor wall. The mixture was filtered and the filter cake was washed with pre-cooled (-5 °C) n-heptane (7.9g). The cake was dried at 40 °C for > 10 hours to afford 6.4 g of white solid (50% yield). 1H NMR (400 MHz, CDCIs) d (ppm) = 7.99 (m, J=8.31 Hz, 2H), 7.45 (m, J=8.19 Hz, 2H), 4.09 (dd, J=1 1 .62, 2.20 Hz, 1 H), 3.90 (s, 3H), 3.75 (t, J=2.81 Hz, 1 H), 3.53 (q, J= 6.97 Hz, 2H), 3.17 (td, J=12.13, 2.63 Hz, 1 H), 2.91 – 2.99 (m, 1 H), 1.99 (dd, J=13.57, 2.69 Hz, 1 H), 1 .88 (dt, J=13.79, 2.58 Hz, 1 H), 1 .69 – 1 .79 (m, 1 H), 1 .57 – 1 .68 (m, 2H), 1 .25 (t, J= 7.03 Hz, 3H).
Example 4: Enantioselective synthesis of compound according to the following
sequence:
Step 1 : Synthesis of Benzyl 4-oxo-3,4-dihydropyridine-1 (2H)-carboxylate (C6, wherein Pi = Cbz and R = methyl)
To a 2.0 L reactor, 4-methoxypyridine (C1 , 45.0 g, 412.39 mmol, 1 .0 eq) and methanol (900 ml.) were added. The mixture was cooled to -75 °C with dry ice/acetone bath. A solution of benzyl
chloroformate (73.86 g, 432.99 mmol, 1 .05 eq) in THF (90 ml.) was charged dropwise while keeping IT < -70 °C. The reaction was stirred for 1 hour to afford a white suspension at -70 °C. Sodium borohydride (16.38 g, 432.99 mmol, 1 .05 eq) was added in portions while keeping IT < -70 °C. The reaction was stirred at -70 °C for 2 hours. Water (200 g) was added and the cooling bath was removed. A solution of 36% hydrogen chloride (16.72 g, 164.95 mmol, 0.4 eq) in water (50 ml.) was added in 10 min at 0 – 5 °C and stirred for 1 hour. Then 20% Na2CC>3 (85.5 g) was added to adjust pH = 7 while maintained IT < 5 °C. Organic solvents were removed under vacuum. The resulting residue was extracted with dichloromethane (450 ml_). The dichloromethane phase was washed with 3wt% hydrogen chloride (151 ml.) and 3 wt% Na2C03 (151 ml_). After solvent exchange with MTBE, about 4 volume (180 ml) of the MTBE mixture was obtained. The mixture was heated to 50 °C to afford a solution and then cooled to 45 °C. Crystal seed of C6 was charged and the mixture was aged at 40 – 45 °C for 7 hours. The mixture was cooled to 10 – 15 °C in 3 hours. The white suspension was filtered and the wet cake was rinsed with cold MTBE (45 ml_). The cake was dried under vacuum at 40 – 50 °C for 2 hours to afford C6 as a white powder (91.56 g, 60% yield). 1H NMR (400 MHz, CDCI3): d (ppm) = 7.85 (br. s, 1 H), 7.37 – 7.43 (m, 5H), 5.43 (br. s, 1 H), 5.26 (s, 2H), 4.05 (t, J=7.34 Hz, 2H), 2.54 – 2.58 (m, 2H).
Step 2: Synthesis of Benzyl (S)-2-(4-(methoxycarbonyl)phenyl)-4-oxopiperidine-1 -carboxylate ((S)-C4, wherein Pi = Cbz and R = methyl)
Method 1 : A 500 ml Radleys reactor was purged 3 times with vacuum/N2. C6 (8 g, 34.60 mmol, 1.0 eq), C7 (9.34 g, 51.89 mmol, 1 .5 eq), tert- Amyl alcohol (160 ml.) and deionized water (16 ml.) were added. The mixture was stirred for > 40 minutes to give a clear colorless solution. The solution was purged 4 times with vacuum / N2 and bubbled with N2 via a syringe needle for 1 hour. To the colorless solution was charged the mixed solid of (S)-XylBINAP (0.381 g, 0.519 mmol, 0.015 eq) and Rh(Acac)(C2H4)2 (0.134 g, 0.519 mmol, 0.015 eq). The mixture was continued to bubble with N2 for 15 minutes and purged 4 times with vacuum / N2. The suspension was stirred for another 2 hours to dissolve (S)-XylBINAP. The reaction mixture was stirred at 55 ± 4 °C for 15 hours. The reaction was followed by HPLC. The mixture was cooled and treated with 7.7% sodium hypochlorite (1 g, 1 .04 mmol, 0.03 eq) for 1 .5 hours at 40 ± 4 °C. tert- Amyl alcohol was distilled off. The residue was extracted with isopropyl acetate (64 ml.) and ethyl acetate (8 ml.) and filtered. The organic phase was washed with 5% NaHC03 (50 g) then with 15% brine (40 g) at 50 ± 5 °C. Some solvents were removed and ethyl acetate (21 .6 g) was added. The solution was treated with Smopex-234 (1 .2 g) at IT =55 ± 5 °C for 2 hours then filtered via 200 – 300 mesh silica gel (1 .6 g). After solvent exchange with n-heptane, MTBE (44.4 g) was added. The mixture was cooled to IT = 42 ± 3 °C. (S)-C4 seed (10 mg) was added. The mixture was aged for 2 hours and cooled to IT =
31 ± 3 °C in 3 hours n-heptane (23.2 g) was then charged in 1 – 2 hours. The mixture was aged for 2 hours and cooled to IT = 20 ± 3 °C in 2 hours. The mixture was filtered and the cake was washed with a mixed solvent of MTBE (4.4 g) and n-heptane (4.1 g). Dried the wet cake at 60 °C for > 5 hours to afford (S)-C4 (7.63 g, 60% yield) as yellow powder. 1H NMR (400 MHz, CDCI3): d (ppm) = 7.99 (d, J=8.44 Hz, 2 H), 7.28 – 7.37 (m, 7H), 5.82 (br. s, 1 H), 5.14 – 5.28 (m, 2H), 4.30 (br. s, 1 H), 3.91 (s, 3H), 3.22 (br. d, J=8.31 Hz, 1 H), 2.84 – 3.03 (m, 2H), 2.46 – 2.64 (m, 1 H), 2.38 (br. d, J=16.26 Hz, 1 H).
Method 2: To a 500 ml Radleys reactor purged 3 times with vacuum/N2, C6 (8 g, 34.60 mmol, 1 .0 eq), C7 (9.34 g, 51 .89 mmol, 1 .5 eq), fe/f-Amyl alcohol (160 ml.) and deionized water (16 ml.) were added. The mixture was stirred for roughly 40 minutes to give a clear colorless solution. The solution was purged 4 times with vacuum / N2 and bubbled with N2 via a syringe needle for 1 hour. To the colorless solution, was charged the mixed solid of (R, R)-Ph-BPE-Rh(Acac) (0.005 eq., 0.122 g, 0.173 mmol). The mixture was continued to bubble with N2 for 15 minutes and purged with vacuum / N2. The reaction mixture was stirred at 55 ± 4 °C for 15 hours. The reaction was followed by HPLC. Tert- amyl alcohol was distilled off. The residue was extracted with isopropyl acetate (64 ml.) and ethyl acetate (8 ml_), and then filtered. The organic phase was washed with 5% NaHC03 (50 g), then with 15% brine (40 g) at 50 ± 5 °C. Some solvents were removed and ethyl acetate (21 .6 g) was added. The solution was treated with Smopex-234 (1 .2 g) at IT = 55 ± 5 °C for 2 hours then filtered via 200 – 300 mesh silica gel (1 .6 g). After solvent exchange with n-heptane, MTBE (44.4 g) was added. The mixture was cooled to IT = 42 ± 3 °C. (S)-C4 seed (10 mg) was added. The mixture was aged for 2 hours and cooled to IT = 31 ± 3 °C in 3 hours n-heptane (23.2 g) was then charged in 1 – 2 hours. The mixture was aged for 2 hours and cooled to IT = 20 ± 3 °C in 2 hours. The mixture was filtered and the cake was washed with a mixed solvent of MTBE (4.4 g) and n-heptane (4.1 g). The wet cake was dried at 60 °C for roughly 5 hours to afford (S)-C4 (10.17 g, 80% yield) as yellow powder. 1 H NMR (400 MHz, CDCI3) d (ppm) = 7.99 (d, J=8.44 Hz, 2 H), 7.28 – 7.37 (m, 7H), 5.82 (br. s, 1 H), 5.14 – 5.28 (m, 2H), 4.30 (br. s, 1 H), 3.91 (s, 3H), 3.22 (br. d, J=8.31 Hz, 1 H), 2.84 – 3.03 (m, 2H), 2.46 – 2.64 (m, 1 H), 2.38 (br. d, J=16.26 Hz, 1 H).
Method 3: To a 500 ml Radleys reactor purged 3 times with vacuum/N2. C6 (8 g, 34.60 mmol, 1 .0 eq), C7 (9.34 g, 51 .89 mmol, 1 .5 eq), tert- amyl alcohol (160 ml.) and deionized water (16 ml.) were added. The mixture was stirred for roughly 40 minutes to give a clear colorless solution. The solution was purged 4 times with vacuum / N2, and bubbled with N2 via a syringe needle for 1 hour. To the colorless solution was charged the mixed solid of (S)-XylBINAP-Rh(Acac) (0.01 eq., 0.324
g, 0.346 mmol). The mixture was continued to bubble with N2 for 15 minutes and purged with vacuum / N2. The reaction mixture was stirred at 55 ± 4 °C for 15 hours. The reaction was followed by HPLC. Tert- amyl alcohol was distilled off. The residue was extracted with isopropyl acetate (64 mL) and ethyl acetate (8 mL), and then filtered. The organic phase was washed with 5% NaHC03 (50 g), then with 15% brine (40 g) at 50 ± 5 °C. Some solvents were removed and ethyl acetate (21 .6 g) was added. The solution was treated with Smopex-234 (1 .2 g) at IT =55 ± 5 °C for 2 hours then filtered via 200 – 300 mesh silica gel (1 .6 g). After solvent exchange with n-heptane, MTBE (44.4 g) was added. The mixture was cooled to IT = 42 ± 3 °C. (S)-C4 seed (10 mg) was added. The mixture was aged for 2 hours and cooled to IT = 31 ± 3 °C in 3 hours n-heptane (23.2 g) was then charged in 1 – 2 hours. The mixture was aged for 2 hours and cooled to IT = 20 ± 3 °C in 2 hours. The mixture was filtered, and the cake was washed with a mixed solvent of MTBE (4.4 g) and n-heptane (4.1 g). The wet cake was dried at 60 °C for roughly 5 hours to afford (S)-C4 (10.30 g, 81 % yield) as yellow powder. 1H NMR (400 MHz, CDCI3) d (ppm) = 7.99 (d, J=8.44 Hz, 2 H), 7.28 – 7.37 (m, 7H), 5.82 (br. s, 1 H), 5.14 – 5.28 (m, 2H), 4.30 (br. s, 1 H), 3.91 (s, 3H), 3.22 (br. d, J=8.31 Hz, 1 H), 2.84 – 3.03 (m, 2H), 2.46 – 2.64 (m, 1 H), 2.38 (br. d, J=16.26 Hz, 1 H).
Example 5: Synthesis of Benzyl -4-hvdroxy-2-(4-(methoxycarbonyl)phenyl)piperidine-
1-carboxylate f(S)-C5, wherein Pi = Cbz and R = methyl)
R = Methyl R = Methyl
Preparation of 0.1 M PBS, pH 7.0, with 0.1 % TPGS buffer solution: To a 500 ml. Radleys reactor equipped with impeller agitator was charged Na2HP04.12H20 (8.63 g), NaH2P04.2H20 (2.41 g), Tap Water (388.6 g) and TPGS-750-M.001 (0.388 g). The mixture was stirred for > 3 hours at IT = 60 ± 5 °C and then cooled to IT = 51 ± 3 °C. 80 g of the buffer solution was taken from the reactor to a flask and cooled to < 35 °C. Check pH value of the buffer solution (7.0 ± 0.5). To the above Radleys reactor (S)-C4 (20.0 g, 54.4 mmol, 1 .0 eq), Isopropanol (16.36 g, 272.2 mmol, 5.0 eq) and 0.1 % TPGS buffer solution (60 g) were added. To a 25 mL flask was charged KRED-P3-G09 (0.4 g, #Codexis), NADP+ (0.1 g) and 0.1 % TPGS buffer solution (60 g) from the above flask. All the solid was dissolved. The solution of enzyme was charged to the 500 mL Reactor at IT =50 ± 5 °C. Rinsed the 25 mL flask with 0.1 % TPGS buffer (10 g) and transferred the solution to the 500 mL reactor at IT =50 ± 5 °C. The mixture was stirred with agitation speed > 500 RPM at 51 ± 3 °C for >
8 hours. The reaction was followed by HPLC. To the reactor 2-MeTHF (200 mL) was added and the mixture was stirred for > 60 minutes at 50 ± 5 °C. The mixture was held for > 50 minutes without agitation and the bottom aqueous phase was separated. The organic phase was washed twice with another 200 g of water at 50 ± 5 °C. The organic phase was concentrated to about 70 g. After solvent exchange with twice 158 g acetonitrile to give about 80 g solution, which was cooled to < 30 °C then filtered via MCC. MCC cake was washed with isopropyl acetate (40 mL/35.5 g) to afford (S)-C5 in a light color solution (1 14.3 g, assay 16.95% 96.34% yield). The acetonitrile / isopropyl acetate solution was telescoped to the next step directly. 1 H NMR (400 MHz, CDCI3): d (ppm) = 7.98 (d, J=8.44 Hz, 2H), 7.23 – 7.38 (m, 7H), 5.61 – 5.72 (m, 1 H), 5.18 (s, 2H), 4.23 (br. d, J=13.33 Hz, 1 H), 3.90 (s, 3H), 3.62 – 3.75 (m, 1 H), 2.81 (td, J=13.51 , 2.81 Hz, 1 H), 2.62 (br. d, J=13.33 Hz, 1 H), 2.45 (br. s, 1 H), 1 .79 – 1 .91 (m, 2H), 1 .41 – 1 .56 (m, 1 H).
Example 6: Asymmetric synthesis of Methyl 4-r(2S.4S)-4-ethoxypiperidin-2-yl1benzoate
(Compound of formula . or a salt thereof. – R= methyl) according to the following
sequence:
(S)-C5 (S)-C9 Compound of (Pi = Cbz) (Pi = Cbz, P2 = TBS) (Pi = Cbz) formula (II) R = Methvl R = Methyl R = Methyl R = Methyl
Step 1 : Synthesis of Benzyl (2S,4S)-4-{[tert-butyl(dimethyl)silyl]oxy}-2-[4-(methoxy carbonyl) phenyl]piperidine-1 -carboxylate ((S)-(C8), wherein Pi = Cbz, P2 = TBS, and R = methyl).
To a 500 ml Radleys Reactor was charged with (S)-C5 solution (in acetonitrile / isopropyl acetate, 271 .8 g, assay 14.72%, contained 40.0 g of (S)-C5, 108.31 mmol, 1 .0 eq) from the previous step. After solvent exchange with isopropyl acetate (159.8 g / 180 ml_), 100 g clear solution was obtained. Isopropyl acetate (176 g /198 ml_), imidazole (26.54 g, 389.90 mmol, 3.6 eq) and TBS-CI (27.75 g, 184.12 mmol, 1 .7 eq) were added. The yellow suspension was stirred at 55 ± 4 °C for 7 hours. The reaction was followed by HPLC. The reaction mixture was cooled to 23 ± 4 °C and filtered through MCC (2 g). The cake was washed with isopropyl acetate (88.8 g / 100 ml_). 6% NaHC03 (240 g) was added and the mixture was stirred for 20 minutes. The organic phase was washed with 5% brine (2×240 g) and concentrated to about 105 g. After solvent exchange with toluene (120 g / 135.4 ml_), 105 g solution was obtained. Dichloromethane (298 g / 224.5 ml.) was added and the solution was filtered via 200-300 mesh silica gel (4.4 g). The silica gel was eluted with another portion of dichloromethane (44 g / 33 ml_). The mother liquor was concentrated and the solvent was exchanged with acetonitrile (2×280 ml_, 442.4 g in total) to 100 g. The residue was diluted with acetonitrile (105 g / 132.9 ml.) to afford a light yellow solution (205 g, assay 25.55%, 100% yield), which was used for the next step directly. 1 H NMR (400 MHz, CDCI3) d (ppm) = 8.01 (d, J=8.44 Hz, 2 H), 7.23 – 7.37 (m, 7 H), 5.60 – 5.70 (m, 1 H), 5.18 (s, 2H), 4.22 (br. d, J=13.45 Hz, 1 H), 3.90 (s, 3H), 3.62 – 3.71 (m, 1 H), 2.82 (td, J=13.51 , 2.81 Hz, 1 H), 2.49 (br. d, J=13.45 Hz, 1 H), 1.83 – 1 .96 (m, 1 H), 1 .75 – 1 .80 (m, 1 H), 1 .47 – 1.60 (m, 1 H), 0.86 (s, 9H), 0.03 (s, 3H), 0.00 (s, 3H).
Step 2: Synthesis of Benzyl (2S, 4S)-4-ethoxy-2-[4-(methoxycarbonyl)phenyl]piperidine-1 -carboxylate ((S)-C9, wherein Pi = Cbz amd R = methyl)
To a 500 ml. Radleys Reactor equipped with impeller agitator (S)-C8 in an acetonitrile solution (170.8 g, assay 29.28%, 103.38 mmol, 1 .0 eq) and fresh acetonitrile (220 g) were charged, followed by Et3SiH (36.06 g, 310.13 mmol, 3.0 eq). The mixture was cooled to IT =4 ± 5 °C and TESOTf (5.47 g, 20.68 mmol, 0.2 eq) was charged. To the mixture was charged a solution of 2,4,6-trimethyl-1 ,3,5-trioxane (13.66 g, 103.38 mmol, 1 .0 eq) in acetonitrile (23 g) over 60 minutes at IT =4 ± 5 °C. Upon addition, the mixture was stirred for 15 minutes. The reaction was followed by HPLC. To the reaction mixture was charged 5% aqueous sodium hydroxide (16.54 g, 20.68 mmol, 0.2 eq) and 20 g water, followed by n-heptane (60 g). The mixture was stirred for 30 minutes at 20 ± 5 °C. The bottom acetonitrile phase was collected. To the acetonitrile phase was charged with MTBE (1 1 1 g) and 10% brine (300 g). The mixture was stirred for 30 minutes. The upper MTBE phase was washed with 10% brine (2×300 g), concentrated to 90 g. MTBE (185 g) and water (150 g) were charged. After phase separation at 38 ± 4 °C and solvent exchange of the organic layer with isopropyl acetate (2×266.4 g), 205 g solution was obtained, which was filtered through Charcoal film slowly. The charcoal film was washed with isopropyl acetate (22.2 g) to afford as a light yellow solution (223 g, 100% yield). The solution was telescoped to the next step directly. 1 H NMR (400 MHz, CDCI3) d (ppm) = 8.01 (d, J=8.44 Hz, 2H), 7.25 – 7.38 (m, 7H), 5.68 (br. s, 1 H), 5.19 (s, 2H), 4.27 (br. d, J=13.33 Hz, 1 H), 3.92 (s, 3H), 3.42 – 3.54 (m, 2H), 3.34 (ddd, J=10.88, 6.91 , 4.22 Hz, 1 H), 2.84 (td, J=13.51 , 2.81 Hz, 1 H), 2.66 (br. d, J=13.20 Hz, 1 H), 1 .96 (br. d, J=10.51 Hz, 1 H), 1 .75 – 1 .90 (m, 1 H), 1 .33 – 1 .53 (m, 1 H), 1 .18 (t, J= 6.97 Hz, 3H).
Step 3: Synthesis of Methyl 4-((2S,4S)-4-ethoxypiperidin-2-yl)benzoate (compound of Formula (II), or a salt thereof – R= methyl)
To a 500 ml. autoclave which was purged with vacuum / N2 (S)-C9 in an isopropyl acetate solution (278.4 g, assay 17.96%, 50 g of (S)-C9, 125.80 mmol) and 10% Pd/C (5.0 g, 50% wet) were
charged. The reactor was purged with vacuum / H2 and stirred for > 7 hours at 25 ± 5 °C. The reaction was followed by HPLC analysis. Filtered the reaction mixture via MCC (7.7 g) which was pre-washed with isopropyl acetate . Rinsed the reactor and MCC with isopropyl acetate (39 g). The mother liquor was combined to afford compound of formula (II) as a light yellow solution (315 g, assay 10.0%, 95.1 % yield). 1 H NMR (400 MHz, CDCI3) d (ppm) = 7.99 (m, J=8.31 Hz, 2H), 7.45 (m, J=8.19 Hz, 2H), 4.09 (dd, J=1 1 .62, 2.20 Hz, 1 H), 3.90 (s, 3H), 3.75 (t, J=2.81 Hz, 1 H), 3.53 (q, J= 6.97 Hz, 2H), 3.17 (td, J=12.13, 2.63 Hz, 1 H), 2.91 – 2.99 (m, 1 H), 1 .99 (dd, J=13.57, 2.69 Hz, 1 H), 1 .88 (dt, J=13.79, 2.58 Hz, 1 H), 1 .69 – 1 .79 (m, 1 H), 1 .57 – 1 .68 (m, 2H), 1 .25 (t, J= 7.03 Hz, 3H).
Step 4: Synthesis of the maleic salt of compound of formula (II) (R = methyl)
To a 500 ml. Radleys Reactor equipped with impeller agitator a solution of methyl 4-((2S,4S)-4-ethoxypiperidin-2-yl)benzoate (381 g, assay 10.03%, 145.12 mmol, 1 .0 eq) from the previous step was charged. The solution was concentrated to 281 g and fresh isopropyl acetate (28.6 g) was added. Then a solution of maleic acid (8.45 g, 72.56 mmol, 0.5 eq) in acetone (30.5 ml.) was added at 51 ± 3 °C in 30 minutes. After stirring for 15 minutes, a seed of the maleic salt of compound of formula (II) was added and the mixture was aged for 2 hours. A solution of maleic acid (8.45 g, 72.56 mmol, 0.5 eq) in acetone (30.5 ml.) was charged at 51 ± 3 °C in 60 minutes and the mixture was aged for 2 hours. The mixture was cooled to IT = 10 ± 3 °C in 6 hours and stirred for > 120 minutes. The mixture was filtered and the filter cake was washed with pre-cooled isopropyl acetate (44.4 g). The cake was dried under high vacuum at 55 °C for 5 – 12 hours to afford maleic salt of compound of formula (II) as white solid (49.8 g, Yield 90.4%). 1 H NMR (400 MHz, CDCIs) d (ppm) 9.35 – 9.78 (m, 2H), 8.02 (m, J=8.31 Hz, 2H), 7.58 (m, J=8.31 Hz, 2H), 6.17 (s, 2H), 4.56 (br. d, J=1 1.13 Hz, 1 H), 3.90 (s, 3H), 3.86 (s, 1 H), 3.48 – 3.57 (m, 2H), 3.38 – 3.44 (m, 2H), 2.42 (br. t, J=13.57 Hz, 1 H), 1 .98 – 2.20 (m, 3H), 1 .24 (t, J= 6.97 Hz, 3H).
The maleic salt of compound of formula (II) may be characterized by a x-ray powder diffraction pattern (XRPD) comprising four or more 2Q values (CuKa l=1 .5418 A) selected from the group consisting of 5.893, 6.209, 1 1 .704, 13.014, 16.403, 17.295, 17.592, 18.629, 18.942, 21 .044, 21 .733, 21 .737, 22.380, 23.528, 24.195, 26.013, 26.825, 29.017, 29.515, 32.250, 35.069, 35.590, and 37.932, measured at a temperature of about 22 °C and an x-ray wavelength, l, of 1 .5418 A.
Example 7: Synthesis of fert-butyl 4-formyl-5-methoxy-7-methyl-1 H-indole-1 -carboxylate
(Compound of formula (III), or a salt thereof) according to the following seguence:
Step 1 : Synthesis of 7-methyl-1 H-indol-5-ol (C11 )
To a 250 ml. flask equipped with a thermometer 3.4% Na2HP04 (100 g, pH = 8.91 ) was charged, followed by addition of Fremy’s salt (4.84 g, 2.4 eq). The mixture was stirred at 20 ± 5 °C until a clear solution was formed. A solution of 7-methylindoline in acetone (9.1 g, 1 1 %) was added in one portion. The mixture was stirred at 20 ± 5 °C for 1 .5 hours. Then sodium sulfite (0.38 g) was added. The mixture was extracted with ethyl acetate (100 ml. x 2) The combined organic extracts were dried over anhydrous sodium sulfate, filtered and concentrated. To the residue 20ml_ acetonitrile was added. The solution was used directly in the next step.
Step 2: Synthesis of fert-butyl 5-hydroxy-7-methyl-1 H-indole-1 -carboxylate (C12, wherein P3 = Boc)
The above as prepared solution was cooled to 0 ± 5 °C. DMAP (0.34 g, 0.4 eq) was charged followed by addition of (Boc)20 (4.9 g, 3.0 eq). The mixture was warmed to 20 ± 5 °C, stirred at 20 ± 5 °C for 30 minutes and concentrated. To the residue was added methanol (40 ml_). The mixture was cooled to 0 ± 5 °C. Potassium carbonate (5.1 g, 5.0 eq) was added. The mixture was stirred at 0 ± 5 °C for 4 hours, warmed to 20 ± 5 °C and stirred for additional 2 hours. The mixture was cooled to 0 ± 5 °C. Acetic acid (2 g) was added. pH was 7-8. The mixture was filtered and the filter cake was washed with methanol (10 mL x 2). The filtrate was concentrated and ethyl acetate (30 ml.) was added. The mixture was washed with water (20 ml.) and 5% brine (20 ml_). The organic layer was concentrated to afford a dark oil, which was slurried with (3:2) n-heptane: Ethyl acetate (5 g) to afford a yellow solid. The solid was collected by filtration and dried to give C12 as yellow solid. 27.4% isolate yield from C10. 1 H-NMR (400 MHz, DMSO-d6): d (ppm) = 9.13 (s, 1 H), 7.52 (d, J= 3.67 Hz, 1 H), 6.74 (d, J= 2.2 Hz, 1 H), 6.56 (m, 1 H), 6.50 (d, J= 3.67 Hz, 1 H), 2.45 (s, 3 H), 1.57 (s, 9 H). LCMS (m/z): positive mode 248.1 [M]+, LCMS (m/z): negative mode 246.1 [M-1 ]-.
Step 3: Synthesis of fert-butyl 4-formyl-5-hydroxy-7-methyl-1 H-indole-1 -carboxylate (C13, wherein P3 = Boc)
To a solution of fe/f-butyl 5-hydroxy-7-methyl-1 H-indole-1 -carboxylate (C12) (53.8% assay, 1 .0 g, 2.2 mmol) in THF (20 ml.) was added dropwise the solution of CH3MgBr in THF (1 N, 2.2 ml_, 2.2 mmol). The resulting mixture was stirred at 20 – 25 °C for 10 minutes. (CHO)n (0.2 g, 6.53 mmol)
was added to the mixture. The reaction mixture was heated to 65 – 70 °C and stirred for 1 hours. The reaction mixture was cooled to 20 – 25 °C. Saturated NH4CI (20 ml.) and MTBE (20 ml.) were added. The mixture was separated and the aqueous layer was extracted with MTBE (20 ml_). The organic layers were combined and concentrated to give compound C13 as yellow solid (0.7 g, 79% assay, 92% yield). 1H-NMR (400 MHz, DMSO-d6) d (ppm) = 10.74 (s, 1 H), 10.54 (s, 1 H), 7.82 (d, J= 4.0 Hz, 1 H), 7.34 (d, J= 4.0 Hz, 1 H), 6.81 (s, 1 H), 2.59 (s, 3H), 1 .65 (s, 9H). LCMS (m/z): positive mode 290.1 [M]+.
Step 4: Synthesis of fert-Butyl 4-formyl-5-methoxy-7-methyl-1 H-indole-1 -carboxylate (Compound of formula (III)).
To a solution of compound C13 (50 mg, 0.182 mmol) in dry DMF (3 ml.) was added K2CO3 (50.2 mg, 0.363 mmol). The mixture was stirred for 10 minutes and then dimethyl sulfate (25.2 mg, 0.20 mmol) was added. The reaction mixture was stirred for 1 hours and poured into ice-water (12 ml_). The mixture was filtered and the filter cake was washed with water. The cake was dried under vacuum to give tert- Butyl 4-formyl-5-methoxy-7-methyl-1 H-indole-1 -carboxylate (Compound of formula (III)) as pale solid (48 mg, 91 % yield). 1 H-NMR (400 MHz, DMSO-d6) d (ppm) = 10.51 (s, 1 H), 7.80 (d, J= 4.0 Hz, 1 H), 7.31 (d, J= 4.0 Hz, 1 H), 6.81 (s, 1 H), 3.95 (s, 3H), 2.61 (s, 3H), 1 .59 (s, 9H). LCMS (m/z): negative mode 274.1 [M-1 ]-.
Example 8: Synthesis of fert-butyl 4-formyl-5-methoxy-7-methyl-1 H-indole-1 -carboxylate
(Compound of formula (III), or a salt thereof) according to the following sequence:
f available (P3 = Boc)
formula (ill)
Step 1 : Synthesis of 5-(benzyloxy)-1 ,3-dimethyl-2 -nitrobenzene
To a solution of commercially available 3,5-dimethyl-4-nitrophenol (100.0 g, 590.4 mmol) in DMF (500 ml_), CS2CO3 (230.8 g, 708.5 mmol) was added and the resulting mixture was stirred for 10 minutes. Then, (bromomethyl)benzene (104.1 g, 590.4 mmol) was added dropwise to the mixture within 30 minutes. The reaction mixture was stirred at 20-25 °C for 1 hour, and then poured into ice-water (1800 ml_). The solid separated out was collected by filtration and washed with water (500 ml_). The cake was dissolved in ethyl acetate (500 ml.) and the solution was washed with a saturated solution of NaCI (50 ml_), was separated, and the solution was concentrated to give 5-(benzyloxy)-l ,3-dimethyl-2-nitrobenzene 2 (147 g, 97.8% yield) as brown solid. HPLC purity
99.7%. 1H-NMR (400 MHz, DMSO-d6) d (ppm) = 7.42 (m, 5 H), 6.94 (s, 2H), 5.16 (s, 2 H), 2.25 (s, 6 H); LCMS (m/z): negative mode 256.2 [M-1 ]-
Step 2: Synthesis of fert-butyl 5-hydroxy-7-methyl-1 H-indole-1 -carboxylate (C12, wherein P3 = Boc)
To a solution of 5-(benzyloxy)-1 ,3-dimethyl-2-nitrobenzene (60.0 g, 233.2 mmol, from Step 1) in DMF (300 ml.) were added DMF-DMA (87.8 g, 699.6 mmol) and pyrrolidine (50.3 g, 699.6 mmol). The solution was heated to 85-90 °C and stirred for 19 hours under nitrogen, then the mixture was cooled to 20-25 °C. The volatile components (DMF-DMA, pyrrolidine and DMF) were removed at 65-70 °C on a rotary evaporator. The crude mixture was dissolved in ethyl acetate (300 ml_), and Raney Nickel (6.0 g) was added. The reaction mixture was subjected to catalytic hydrogenation under atmospheric pressure, overnight. Then, the reaction mixture was put under nitrogen. The mixture was filtrated and the filtrate was concentrated to provide 5-(benzyloxy)-7-methyM H-indole as a black oil. 5-(benzyloxy)-7-methyl-1 H-indole was used without further purification into the next step.
5-(benzyloxy)-7-methyl-1 H-indole was dissolved in acetonitrile (300 ml_), (Boc)20 (53.6 g, 233.2 mmol) and DMAP (5.7 g, 46.6 mmol) were added. The reaction mixture was stirred at 20-25 °C for 1 hour. Acetonitrile was removed on a rotary evaporator, and the residual mixture was dissolved in ethyl acetate (300 ml_). The solution was washed with a saturated aqueous solution of NaHC03 and then concentrated to give a crude oil which was purified by column chromatography (Si02, 500 g) using a mixture of heptane / MTBE (1 :10) to provide the intermediate tert-butyl 5-(benzyloxy)-7-methyl-1 H-indole-1 -carboxylate as a brown oil (42.1 g, 49.2% yield). HPLC purity 93.5%. 1 H-NMR (400 MHz, DMSO-d6) d (ppm) = 7.59 (d, J= 3.67 Hz, 1 H), 7.40 (m, 5 H), 7.04 (d, J= 2.45 Hz, 1 H), 6.81 (d, J= 2.2 Hz, 1 H), 6.57 (d, J= 3.67 Hz, 1 H), 5.1 1 (s, 2 H), 2.51 (s, 3 H), 1.58 (s, 9 H). LCMS (m/z): negative mode 336.2 [M-1 ]- To a solution of intermediate tert-butyl 5-(benzyloxy)-7-methyl-1 H-indole-1-carboxylate (36.7 g, 100 mmol) in ethanol (250 mL), under nitrogen, 10% Pd/C (10.6 g, 10 mmol) and ammonium formate (6.8 g, 105 mmol) were added. The solution was heated to 45-50 °C and stirred for 5 hours under nitrogen. Then the mixture was cooled to room temperature, filtered, and the filtrate was concentrated to give a residue oil. The residual oil was dissolved in ethyl acetate (250 mL), the solution was washed with a saturated aqueous solution of NaCI (100 mL), the phases were separated. The organic layers were collected and concentrated. The obtained crude mixtures was slurried with a (1 :15) mixture of MTBE / Heptane (160 mL) for 2 hours. The precipitate was filtered and washed with heptane (50 mL). The cake was dried under vacuum to give tert-butyl 5-hydroxy-7-methyl-1 H-indole-1 -carboxylate (C12) as a tawny solid (21 .8 g, 87.2% yield). HPLC purity 97.7%. 1 H-NMR (400 MHz, DMS0-d6) d (ppm) = 9.13 (s, 1 H), 7.52 (d, J= 3.67 Hz, 1 H), 6.74 (d, J= 2.2 Hz, 1 H), 6.56 (m, 1 H), 6.50 (d, J= 3.67 Hz, 1 H), 2.45 (s, 3 H), 1 .57 (s, 9 H). LCMS (m/z): negative mode 246.2 [M-1 ]-
Step 3: Synthesis of fert-butyl 4-formyl-5-hydroxy-7-methyl-1 H-indole-1 -carboxylate (C13, wherein P3 = Boc)
To a mixture of MgCI2 (1 1 .6 g, 1 19.7 mmol) and (CHO)n (5.0 g, 159.6 mmol), in THF (150 ml), under nitrogen, triethylamine (17.8 ml_, 127.7 mmol) was added dropwise and the resulting mixture was stirred at 20-25 °C for 10 minutes. Then, tert-butyl 5-hydroxy-7-methyl-1 H-indole-1 -carboxylate (C12) (10.0 g, 39.9 mmol) was added to the mixture. The reaction mixture was heated to 65-70 °C and stirred for 3 hours. The reaction mixture was cooled to 20-25 °C, followed by addition of 2N HCI (70 ml) and isopropyl acetate (150 ml). The mixture was separated and the organic layer was washed with a 5% NaCI solution. Then, the solution was concentrated to give a crude solid. The solid was slurried with ethanol (100 ml.) for 1 hour. The solid precipitate was filtrated, and washed with ethanol (20 ml_). The cake was dried under vacuum to give tert-butyl 4-formyl-5-hydroxy-7-methyl-1 H-indole-1 -carboxylate (C13) as a tawny solid (7.2 g, 63.9% yield). HPLC purity 96.5%. The filtrate solution was concentrated to 20 mL, then stirred for 1 hour. The solid was filtrated, and washed with ethanol (5 mL). The cake was dried by vacuum to give an additional amount of tert-butyl 4-formyl-5-hydroxy-7-methyl-1 H-indole-1 -carboxylate (C13) as a tawny solid (1 .1 g, 95.3% assay, 9.5% yield.). HPLC purity 90.5%. 1 H-NMR (400 MHz, DMSO-d6) d (ppm) = 10.69 (s, 1 H), 10.47 (s, 1 H), 7.75 (d, J= 3.35 Hz, 1 H), 7.27 (d, J= 3.55 Hz, 1 H), 6.74 (s, 1 H), 2.51 (s, 3 H), 1 .59 (s, 9 H); LCMS (m/z): negative mode 274.2 [M-1 ]-.
Step 4: Synthesis of fert-Butyl 4-formyl-5-methoxy-7-methyl-1 H-indole-1 -carboxylate (Compound of formula (III)).
To a suspension of tert-butyl 4-formyl-5-hydroxy-7-methyl-1 H-indole-1 -carboxylate (C13) (6.0 g, 21 .3 mmol) in MeCN (60 mL), 50% K2C03 solution (20 mL) and dimethyl sulfate (2.26 mL, 23.4 mmol) were added. The resulting mixture was stirred at 35-40 °C for 3 hours. The reaction mixture was cooled to 20-25 °C and isopropyl acetate (30 mL) was added. The mixture was then extracted; the water layer was extracted with isopropyl acetate (15 mL), the organic layers were combined and concentrated to give a crude residual. The crude residual was dissolved in isopropyl acetate (60 mL), the solution was washed with a statured NH4CI solution, and then concentrated to give a crude product (6.6 g). The crude was slurried with ethyl acetate / Heptane (100 mL, 1/50) for 3 hours. The solid was filtrated, washed with heptane (20 mL). The cake was dried under vacuum to give tert-butyl 4-formyl-5-methoxy-7-methyl-1 H-indole-1 -carboxylate (Compound of formula (III))
as a pink solid (5.5 g, 87.8% yield). HPLC purity 99.3%. 1 H-NMR (400 MHz, DMSO-d6) d (ppm) = 10.52 (s, 1 H), 7.79 (d, J= 3.67 Hz, 1 H), 7.31 (d, J= 3.67 Hz, 1 H), 7.02 (s, 1 H) , 3.95 (s, 3 H), 2.61 (s, 3 H), 1 .60 (s, 9 H); LCMS (m/z): positive mode 290 [M]+.
Example 9: Synthesis of Compound of formula , or salt thereof (R = methyl).
Method 1 (Pa = Boc and R = methyl): To a vessel were added lr(CO)2acac (1 mg, 0.1 mol%), compound of formula (II) (maleic salt, 3 mmol, 1 .137g), compound of formula (III) (3 mmol, 0.867g) in 9 ml. of degassed ethanol. The autoclave was purged 3 times with nitrogen and 3 times with H2 under stirring (250 RPM). The reactions were run for 24 hours at 75 °C under 20 bar of H2 at 700 RPM. An aliquot of the reaction was diluted in methanol and was analyzed by HPLC. Compound of formula (C15) was obtained after 24 hours in 88% conversion.
Method 2 (Pa = Boc and R = methyl): To a vessel were added lrCI3, xH20 (0.05 mol%, 0.9 mg, anhydrous), compound of formula (II) (maleic salt, 6 mmol, 2.274 g ), compound of formula (III) (6 mmol, 1 .735g) in 12 ml. of degassed ethanol. The autoclave was purged 3 times with nitrogen and 3 times with carbon monoxide (CO) (250 RPM). The autoclave was pressurized with 1 bar of CO and 19 bar of H2 and run for 24 hours at 75 °C under 20 bar of H2 / CO at 700 RPM. An aliquot of the reaction was diluted in methanol and was analyzed by HPLC. Compound of formula (C15) was obtained after 24 hours in 62% conversion.
1H NMR (400 MHz, DMSO-d6) d ppm 8.13 (d, J=8.16 Hz, 2H), 7.77 (br. d, J=7.84 Hz, 2H), 7.62 -7.68 (m, 1 H), 6.85 (s, 1 H), 6.80 (d, J= 3.76 Hz, 1 H), 4.01 (s, 3H), 3.92 (s, 3H), 3.73 (br. s, 1 H), 3.55 – 3.67 (m, 4H), 3.39 – 3.42 (m, 1 H), 2.60 – 2.70 (m, 5H), 1 .99 – 2.02(br. d, 1 H), 1 .82 – 1.90 (m, 2H), 1.74 (s, 9H), 1 .64 – 1 70(m, 1 H), 1 .35 (t, J= 6.97 Hz, 3H).
1. Schubart A, et al. Proc Natl Acad Sci U S A. 2019 Mar 29. pii: 201820892.
Proceedings of the National Academy of Sciences of the United States of America (2019), 116(16), 7926-7931.
//////LNP 023, BDBM160475, ZINC223246892, HY-127105, CS-0093107, LNP023
O=C(O)c1ccc(cc1)[C@@H]4C[C@H](CCN4Cc2c(OC)cc(C)c3nccc23)OCC

{kind=link}
{kind=link}