
Home » Posts tagged 'liver fibrosis'
Tag Archives: liver fibrosis
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
TROPIFEXOR
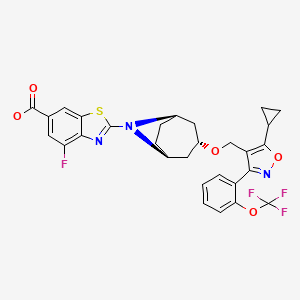

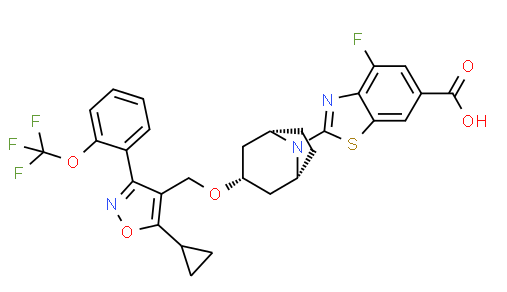
TROPIFEXOR
トロピフェクサー;
PHASE 2, NASH, PBC, liver fibrosis, bile acid diarrhea, non-alcoholic fatty liver disease
| Formula | C29H25F4N3O5S |
|---|---|
| CAS | 1383816-29-2 |
| Mol weight | 603.5845 |
TROPIFEXORLJN 452;LJN-452;LJN452;CS-2712;CPD1549;Tropifexor;Tropifexor (LJN452);LJN452;LJN452,Tropifexor;2-[(1R,3r,5S)-3-({5-cyclopropyl-3-[2-(trifluoromethoxy)phenyl]-1,2-oxazol-4-yl}methoxy)-8-azabicyclo[3.2.1]octan-8-yl]-4-fluoro-1,3-benzothiazole-6-carboxylic acidтропифексор [Russian] [INN]
تروبيفيكسور [Arabic] [INN]
曲匹法索 [Chinese] [INN]2-[(3-endo)-3-({5-Cyclopropyl-3-[2-(trifluormethoxy)phenyl]-1,2-oxazol-4-yl}methoxy)-8-azabicyclo[3.2.1]oct-8-yl]-4-fluor-1,3-benzothiazol-6-carbonsäure [German] [ACD/IUPAC Name]
2-[(3-endo)-3-({5-Cyclopropyl-3-[2-(trifluoromethoxy)phenyl]-1,2-oxazol-4-yl}methoxy)-8-azabicyclo[3.2.1]oct-8-yl]-4-fluoro-1,3-benzothiazole-6-carboxylic acid [ACD/IUPAC Name]
6-Benzothiazolecarboxylic acid, 2-[(3-endo)-3-[[5-cyclopropyl-3-[2-(trifluoromethoxy)phenyl]-4-isoxazolyl]methoxy]-8-azabicyclo[3.2.1]oct-8-yl]-4-fluoro- [ACD/Index Name]
Acide 2-[(3-endo)-3-({5-cyclopropyl-3-[2-(trifluorométhoxy)phényl]-1,2-oxazol-4-yl}méthoxy)-8-azabicyclo[3.2.1]oct-8-yl]-4-fluoro-1,3-benzothiazole-6-carboxylique [French] [ACD/IUPAC Name]
NMZ08KM76Z
Tropifexor fast facts
| CAS Reg. No. | 1383816-29-2 |
| Molar mass | 603.58 g/mol |
| Empirical formula | C29H25F4N3O5S |
| Appearance | White crystals |
| Melting point | 221 ºC |
| Water solubility | 6 mg/L |
| Efficacy | Anti-inflammatory, Farnesoid X receptor (FXR) agonist |
|---|---|
| Comment | Treatment of non-alcoholic steatohepatitis |
Novartis is developing tropifexor, a non-bile acid farnesoid X receptor agonist, and its analog LJP-305, for treating NASH, PBC, liver fibrosis, bile acid diarrhea and non-alcoholic fatty liver disease. In June 2021, this drug was reported to be in phase 2 clinical development.
Nonalcoholic steatohepatitis (NASH) is a liver disease that is becoming more prevalent as worldwide obesity and type 2 diabetes increase. It is characterized by accumulation of fat in the liver, inflammation, hepatocyte ballooning, and fibrosis.
Another liver disease, primary biliary cholangitis (PBC), is a cholestatic condition in which bile flow from the liver to the intestine is reduced or interrupted. It is thought to be autoimmune.
PBC is associated with decreased expression of the farnesoid X receptor (FXR), a ligand-activated nuclear receptor that is highly expressed in the liver and other organs. FXR is a key regulator of bile acid production, conjugation, and transport. FXR activation also suppresses lipogenesis; thus, it has been proposed as a treatment for NASH.
Recently, David C. Tully and colleagues at the Genomics Institute of the Novartis Research Foundation (San Diego) and the Novartis Institutes for Biomedical Research (Emeryville, CA) discovered tropifexor, a highly potent FXR agonist. They began by replacing an indole group in an existing partial FXR agonist with a 2-substituted benzothiazole-6-carboxylic acid, a change that resulted in a dramatic increase in potency. Further changes, including optimization of the benzothiazole substituent, resulted in more potent, orally bioavailable tropifexor.
Tropifexor is an investigational drug which acts as an agonist of the farnesoid X receptor (FXR). It was discovered by researchers from Novartis and Genomics Institute of the Novartis Research Foundation. Its synthesis and pharmacological properties were published in 2017.[1] It was developed for the treatment of cholestatic liver diseases and nonalcoholic steatohepatitis (NASH). In combination with cenicriviroc, a CCR2 and CCR5 receptor inhibitor, it is undergoing a phase II clinical trial for NASH and liver fibrosis.[2]
Rats treated orally with tropifexor (0.03 to 1 mg/kg) showed an upregulation of the FXR target genes, BSEP and SHP, and a down-regulation of CYP8B1. Its EC50 for FXR is between 0.2 and 0.26 nM depending on the biochemical assay.
The patent which covers tropifexor and related compounds was published in 2010.[3]
PATENT
WO-2021104022
Novel, stable crystalline polymorphic form II of tropifexor , useful for treating non-alcoholic steatohepatitis (NASH), fatty liver and primary biliary cholangitis (PBC).Tropifexor was originally developed by Novartis and then licensed to Pfizer for cooperative development. It is a non-steroidal FXR (farnesoid receptor) agonist, currently in clinical phase II of indications for NASH (non-alcoholic steatohepatitis), fatty liver and primary biliary cholangitis.
The structure of Tropifexor is shown in the following formula (1):
Drug polymorphism is a common phenomenon in drug development and an important factor affecting drug quality. Different crystal forms of the same drug may have significant differences in physical and chemical properties such as appearance, fluidity, solubility, storage stability, bioavailability, etc., and there may be great differences, which will affect the storage transfer, application, stability, and efficacy of the drug In order to obtain an effective crystal form that is conducive to production or pharmaceutical preparations, it is necessary to conduct a comprehensive investigation of the crystallization behavior of the drug to obtain a crystal form that meets the production requirements.
At present, there is no literature that discloses the crystal form of Tropifexor, and there is no related literature report.
The present invention obtains a new crystal form of the compound through a large number of experimental studies on the Tropifexor compound. The new crystal form has the advantages of high solubility, good stability, low moisture absorption, simple preparation process and easy operation, etc., and has excellent properties in industrial production. Superiority.Example 1 Preparation method of Tropifexor crystal form II[0049]After mixing 60.3 mg of Tropifexor and p-aminobenzoic acid (13.7 mg), they were added to ethanol (3.0 ml), stirred at 27° C. to obtain a clear solution, and then allowed to stand at room temperature for about 2 days to precipitate a solid product. It was filtered with suction and placed in a drying box at 50°C and vacuum dried to constant weight to obtain 51.3 mg of solid powder. The obtained crystal was detected by XPRD and confirmed to be Tropifexor crystal form II; its X-ray powder diffraction pattern was basically consistent with Fig. 1, its DSC pattern was basically the same as Fig. 2, and its TGA pattern was basically the same as Fig. 3.[0050]Example 2 Preparation method of Tropifexor crystal form II[0051]After mixing 60.3 mg of Tropifexor and p-hydroxybenzoic acid (13.8 mg), they were added to ethanol (3.0 ml), stirred at 27° C. to obtain a clear solution, and then allowed to stand at room temperature for about 2 days to precipitate a solid product. It was filtered with suction and placed in a drying box at 50°C and vacuum dried to constant weight to obtain 48.5 mg of solid powder. The obtained crystal was detected by XPRD and confirmed to be Tropifexor crystal form II; its X-ray powder diffraction pattern was basically consistent with Fig. 1, its DSC pattern was basically the same as Fig. 2, and its TGA pattern was basically the same as Fig. 3.[0052]Example 3 Preparation method of Tropifexor crystal form II[0053]After mixing 60.3 mg of Tropifexor and salicylic acid (13.8 mg), they were added to ethanol (3.0 ml), stirred at 27°C to obtain a clear solution, and then allowed to stand at room temperature for about 2 days to precipitate a solid product. Filter with suction and place in a drying box at 50°C and vacuum dry to constant weight to obtain 50.0 mg of solid powder. The obtained crystal was detected by XPRD and confirmed to be Tropifexor crystal form II; its X-ray powder diffraction pattern was basically consistent with Fig. 1, its DSC pattern was basically the same as Fig. 2, and its TGA pattern was basically the same as Fig. 3.[0054]Example 4 Preparation method of Tropifexor crystal form II[0055]After mixing 60.3 mg of Tropifexor and 2,4-dihydroxybenzoic acid (15.4 mg), they were added to ethanol (3.0 ml), stirred at 27°C to obtain a clear solution, and then allowed to stand at room temperature for about 2 days to precipitate a solid product. It was filtered with suction and placed in a drying box at 50°C and vacuum dried to constant weight to obtain 49.5 mg of solid powder. The obtained crystal was detected by XPRD and confirmed to be Tropifexor crystal form II; its X-ray powder diffraction pattern was basically consistent with Fig. 1, its DSC pattern was basically the same as Fig. 2, and its TGA pattern was basically the same as Fig. 3.
PATENT
WO2021104021 ,
claiming crystalline polymorphic form I of tropifexor,Example 1 Preparation method of Tropifexor crystal form I
50.0 mg of Tropifexor was added to ethanol (1.0 ml), heated to 60° C. and stirred to obtain a clear solution, and then water (3 ml) was added dropwise to the Tropifexor solution. Stir and precipitate solid product. It was filtered with suction and placed in a drying box at 50°C and vacuum dried to constant weight to obtain 38.5 mg of solid powder. The obtained crystal was detected by XPRD and confirmed to be Tropifexor crystal form I; its X-ray powder diffraction pattern was basically consistent with Figure 1, its DSC pattern was basically consistent with Figure 2, and its TGA pattern was basically consistent with Figure 3
PATENT
product pat, WO2012087519 , https://patents.google.com/patent/WO2012087519A1/en
has protection in the EU until November 2031, and expire in US in February 2032 with US154 extension.
PATENT
WO 2016097933
Example 1
2-r(1 R,3r,5S)-3-(f5-cvclopropyl-3-r2-(trifluoromethoxy)phenyll-1 ,2-oxazol-4-yl)methoxy)-8- azabicvcloi3.2.1 loctan-8-yll-4-fluoro-1 ,3-benzothiazole-6-carboxylic acid (1 -1 B) and
-r(1 R,3r,5S)-3-(f5-cvclopropyl-3-r2-(trifluoromethyl)phenyll-1 ,2-oxazol-4-yl)methoxy)-8-
R1a = OCF3 (1 -1A, 1 -1 B)
R a = CF3 (1-2A, 1-2B)
Methyl 2-[(1 R,3r,5S)-3-(i5-cvclopropyl-3-r2-(trifluoromethoxy)phenyll-1 ,2-oxazol-4- yl}methoxy)-8-azabicvcloi3.2.1 loctan-8-yll-4-fluoro-1 ,3-benzothiazole-6-carboxylate (1 -1 A). Into a 25-mL round-bottom flask equipped with a stir bar was added sequentially 4-(((1 R,3r,5S)- 8-azabicyclo[3.2.1 ]octan-3-yloxy)methyl)-5-cyclopropyl-3-(2-(trifluoromethoxy)phenyl)isoxazole (1 .29 mmol), N,N-dimethylacetamide (3.6 mL), cesium carbonate (3.31 mmol), and methyl 2- bromo-4-fluorobenzo[d]thiazole-6-carboxylate (3.87 mmol). After stirring the resulting slurry at room temperature for 10 minutes, the mixture was then warmed to 60 °C and stirred for 1 h. The reaction slurry was allowed to cool to room temperature, and was diluted with 200 mL of ethyl acetate and washed with water (3 χ 30 mL). The organic extracts were concentrated under vacuum and directly purified using normal phase silica gel chromatography (40 g silica column) with a 15 min gradient of 10 % to 60 % ethyl acetate/hexanes. Desired fractions were concentrated in vacuo, and the resulting residue crystallized upon standing to give methyl 2- [(1 R,3r,5S)-3-({5-cyclopropyl-3-[2-(trifluoromethoxy)phenyl]-1 ,2-oxazol-4-yl}methoxy)-8- azabicyclo[3.2.1 ]octan-8-yl]-4-fluoro-1 ,3-benzothiazole-6-carboxylate (1-1 A) as a white crystalline solid. MS (m/z) : 618.2 (M+1 ).
2-r(1 R,3r,5S)-3-(i5-cvclopropyl-3-r2-(trifluoromethoxy)phenyll-1 ,2-oxazol-4-yl}methoxy)- 8-azabicvcloi3.2.1 loctan-8-yll-4-fluoro-1 ,3-benzothiazole-6-carboxylic acid (1 -1 B). To a 25-mL round-bottom flask equipped with a stir bar was added the ester (0.89 mmol), THF (4 mL),
MeOH (2 mL), and 3 N aqueous KOH solution (1 mL, 3 mmol). The resulting homogenous solution was stirred for 1 hour at 70 °C, cooled to room temperature, and then quenched with AcOH (roughly 0.2 mL of glacial acetic, 3 mmol) until pH=6 was achieved (Whatman class pH strip paper). At this time the reaction was diluted with ethyl acetate (40 mL) and washed with water (3 5 mL). The ethyl acetate fraction was concentrated under vacuum to give to an oily residue. To the resulting oil was then added MeOH (6 mL). The oil quickly dissolved, then immediately began to crystallize. Upon standing for 2.5 hrs, the mother liquor was withdrawn and crystals washed (3 x 2 mL of ice cold MeOH). The crystals were dried via vacuum (10 mm Hg pressure at 45 °C overnight) and then recrystallized from acetonitrile, filtered, and dried under vacuum to give 2-[(1 R,3r,5S)-3-({5-cyclopropyl-3-[2-(trifluoromethoxy)phenyl]-1 ,2-oxazol-4-yl}methoxy)-8-azabicyclo[3.2.1 ]octan-8-yl]-4-fluoro-1 ,3-benzothiazole-6-carboxylic acid (1 -1 B). 2-[(1 R,3r,5S)-3-({5-cyclopropyl-3-[2-(trifluoromethyl)phenyl]-1 ,2-oxazol-4-yl}methoxy)-8-azabicyclo[3.2.1 ]octan-8-yl]-4-fluoro-1 ,3-benzothiazole-6-carboxylic acid (1 -2B).
Examples 1 -2A and the corresponding acid 1 -2B can be prepared following the same procedures, from the reaction of intermediate 4-((8-azabicyclo[3.2.1 ]octan-3-yloxy)methyl)-5-cyclopropyl-3-(2-(trifluoromethyl)phenyl)isoxazole.
PAPER
European journal of medicinal chemistry (2021), 209, 112910
https://www.sciencedirect.com/science/article/abs/pii/S0223523420308825

Abstract
Farnesoid X receptor (FXR) agonists are emerging as potential therapeutics for the treatment of various metabolic diseases, as they display multiple effects on bile acid, lipid, and glucose homeostasis. Although the steroidal obeticholic acid, a full FXR agonist, was recently approved, several side effects probably due to insufficient pharmacological selectivity impede its further clinical application. Activating FXR in a partial manner is therefore crucial in the development of novel FXR modulators. Our efforts focusing on isoxazole-type FXR agonists, common nonsteroidal agonists for FXR, led to the discovery a series of novel FXR agonists bearing aryl urea moieties through structural simplification of LJN452 (phase 2). Encouragingly, compound 11k was discovered as a potent FXR agonist which exhibited similar FXR agonism potency but lower maximum efficacy compared to full agonists GW4064 and LJN452 in cell-based FXR transactivation assay. Extensive in vitro evaluation further confirmed partial efficacy of 11k in cellular FXR-dependent gene modulation, and revealed its lipid-reducing activity. More importantly, orally administration of 11k in mice exhibited desirable pharmacokinetic characters resulting in promising in vivo FXR agonistic activity.
References
- ^ Tully DC, Rucker PV, Chianelli D, Williams J, Vidal A, Alper PB, et al. (December 2017). “Discovery of Tropifexor (LJN452), a Highly Potent Non-bile Acid FXR Agonist for the Treatment of Cholestatic Liver Diseases and Nonalcoholic Steatohepatitis (NASH)”. Journal of Medicinal Chemistry. 60 (24): 9960–9973. doi:10.1021/acs.jmedchem.7b00907. PMID 29148806.
- ^ Clinical trial number NCT03517540 for “Safety, Tolerability, and Efficacy of a Combination Treatment of Tropifexor (LJN452) and Cenicriviroc (CVC) in Adult Patients With Nonalcoholic Steatohepatitis (NASH) and Liver Fibrosis. (TANDEM)” at ClinicalTrials.gov
- ^ WO Application Filing 2012087519, Alper PB, Chianelli D, Mutnick D, Vincent P, Tully DC, “Compositions and methods for modulating fxr”, published 2012-06-28, assigned to Genomics Institute of the Novartis Research Foundation. Retrieved 17 May 2019.
| Clinical data | |
|---|---|
| ATC code | None |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1383816-29-2 |
| PubChem CID | 121418176 |
| UNII | NMZ08KM76Z |
| KEGG | D11548 |
| Chemical and physical data | |
| Formula | C29H25F4N3O5S |
| Molar mass | 603.59 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| show |
///////////TROPIFEXOR, トロピフェクサー, NOVARTIS, PHASE 2, тропифексор , تروبيفيكسور , 曲匹法索 , LJN 452, LJN-452, LJN452, CS-2712, CPD1549, Tropifexor, Tropifexor (LJN452), LJN452, LJN452, PHASE 2, NASH, PBC, liver fibrosis, bile acid diarrhea, non-alcoholic fatty liver disease
1ccc(c(c1)c2c(c(on2)C3CC3)CO[C@H]4C[C@H]5CC[C@@H](C4)N5c6nc7c(cc(cc7s6)C(=O)O)F)OC(F)(F)F

NEW DRUG APPROVALS
ONE TIME
$10.00
MALOTILATE, Malotilat

Malotilate, Malotilat
(Kantec; Hepation; NKK 105)
Diisopropyl 1,3-dithiol-2-ylidenemalonate
Nihon Nohyaku Co., Ltd. innovator
Malotilate (INN) is a drug used in the treatment of liver disease. It has been shown to facilitate liver regeneration in rats.[1]
DA-3857
NKK-105
| Systematic (IUPAC) name | |
|---|---|
| diisopropyl 1,3-dithiol-2-ylidenemalonate | |
| Clinical data | |
| AHFS/Drugs.com | International Drug Names |
| Legal status |
|
| Routes | Oral |
| Identifiers | |
| CAS number | 59937-28-9 |
| ATC code | None |
| PubChem | CID 4006 |
| UNII | RV59PND975 |
| Chemical data | |
| Formula | C12H16O4S2 |
| Mol. mass | 288.38 g/mol |
Brief background information
| Salt | ATC | Formula | MM | CAS |
|---|---|---|---|---|
| – | A02AD02 | C 12 H 16 O 4 S 2 | 288.39 g / mol | 59937-28-9 |
Application
-
hepatoprotector
-
in the treatment of liver diseases
Classes of substances
-
1,2-dithiolane and 1,2-dithiols
-
Esters
-
Anilides and other derivatives of malonic acid
-
-
-
It is known that there are a large number of patients who suffer from liver damages caused by various factors such as alcohol, malnutrition, viruses, chemicals, toxicants, etc. The liver diseases may generally be classified by their types into acute hepatitis, chronic hepatitis, liver cirrhosis, and fulminant hepatitis. It is said to be very difficult to treat these liver diseases. Namely, currently available methods for the treatment such as treatments with pharmaceuticals e.g. liver protective agents such as various vitamins, saccharides, amino acids, glutathione, glycyrrhizin, liver hydrolyzates or adrenocortical hormones; cholagogues; immunomodulaters; or antiviral substances against viral hepatitis, are all nothing more than symptomatic treatments, and they are not adequately effective for the treatment of the existing liver damages.
-
It has recently been reported that 1,3-dithiol derivatives represented by Malotilate as identified below, are effective for the treatment of liver damages (see Japanese Examined Patent Publications No. 18,576/1981, No. 18,577/1981 and No. 18,578/1981).

-
Other 1,3-dithiol derivatives similar to Malotilate with respect to structure and pharmaceutical properties are described in US-A-4,118,506, EP-A-99 329 and US-A-4,022,907.
-
As a result of extensive researches, the present inventors have found that certain novel 1,3-dithiol derivatives represented by the after-mentioned formula I, exhibit excellent activities for the treatment of a wide spectrum of liver damages, which are comparable or superior to the above-mentioned conventional 1,3-dithiol derivatives. The present invention has been accomplished on the basis of this discovery.
-
Namely, the present invention provides a 1,3-dithiol-2-ylidene derivative of the formula:



Synthesis pathway
| Synthesis a) |
|---|
    |
………………
US 4327223
http://www.google.co.in/patents/US4327223
EXAMPLE 1
Diisopropoxycarbonylketene disodium mercaptide crystals (8 g, 0.02 mol) was dissolved in 50 ml of dimethylsulfoxide, and 1,1,1-trichloroethane (2.7 g, 0.02 mol) and subsequently a 30% sodium hydroxide aqueous solution (2.7 g, 0.02 mol NaOH) were added thereto. Thus, reaction was carried out at 60° C. for 1 hour. The resulting mixture was poured into ice-water, and then extracted with benzene. Drying of the extract over anhydrous magnesium sulfate, distillation to remove benzene, and recrystallization from n-hexane gave 2.6 g of the object matter diisopropyl 1,3-dithiol-2-ylidene malonate; m.p. 60.5° C., yield 45%.
EXAMPLE 2
Diisopropyl malonate (18.8 g, 0.1 mol) and carbon disulfide (7.6 g, 0.1 mol) were dissolved in 200 ml of dimethylsulfoxide. Dropping thereto a 45% potassium hydroxide aqueous solution (31 g, 0.25 mol KOH) at 13°-17° C., gave a yellowish red solution containing diisopropoxycarbonylketene dipotassium mercaptide. At 20° C., 1,1,1-trichloroethane (26.6 g, 0.2 mol) was added, and 5 minutes after a 45% potassium hydroxide aqueous solution (18.6 g, 0.15 mol KOH) was dropped thereinto. The temperature was raised to 70° C. to carry out reaction for 30 minutes. The resulting mixture was poured into ice-water and then extracted with benzene. Drying of the extract over anhydrous magnesium sulfate, distillation to remove benzene, and recrystallization from n-hexane gave 23.6 g of the object matter diisopropyl 1,3-dithiol-2-ylidene malonate; m.p. 60.5° C., yield 82.1%.
EXAMPLE 3
Diisopropyl malonate (18.8 g, 0.1 mol) and carbon disulfide (7.6 g, 0.1 mol) were dissolved in 200 ml of dimethylsulfoxide. A 45% potassium hydroxide aqueous solution (49.6 g, 0.4 mol KOH) was dropped thereto at 15° C., then 1,1,1-trichloroethane (13.3 g, 0.1 mol) was added at 20° C., and reaction was carried out at 70° C. for 30 minutes. The resulting mixture was poured into ice-water and then extracted with benzene. Drying of the extract over anhydrous magnesium sulfate, distillation to remove benzene, and recrystallization from n-hexane gave 18.1 g of the object matter diisopropyl 1,3-diethiol-2-ylidene malonate; m.p. 60.5° C., yield 62.8%.
………………………………….
US 4035387
http://www.google.co.in/patents/US4035387
Example 1Synthesis of diisopropyl 1,3-dithiol-2-ylidene malonate (the compound 3)
1.1 Grams (0.03 mole) of 69% purity sodium hydride was suspended in 30 ml. of dry tetrahydrofuran. Into the resulting suspension, 5.6g (0.03 mole) of diisopropyl malonate was gradually dropped with ice-cooling. After completion of the generation of hydrogen gas, 8.2g (0.03 mole) of 2-methylthio-1,3-dithiolium iodide was added. The resulting mixture was heated under reflux for 1 hour, and then the reaction product was poured into a large amount of ice water to deposit crystals. The crystals were recovered by filtration, dried and then recrystallized from n-hexane to obtain 6.7g of white crystals, m.p. 59°-60° C., yield 77.5% .
The 2-methylthio-1,3-dithiolium iodide used as starting material was synthesized in the following manner;
44.4 Grams (0.2 mole) of 1,3-dithiol-2-thion-4,5-dicarboxylic acid was dissolved in 240 ml of nitromethane, and the resulting solution was heated to 80° C. Into this solution, 100 ml of iodomethyl was gradually dropped, and the resulting mixture was refluxed for 6 hours. After completion of the reaction, the formed crystals were recovered by filtration, washed with 100 ml of ether and then air-dried to obtain 48.4g of the desired compound, m.p. 114°-116° C. (decomp.), yield 87.0%.
………………………………………..
Fujinami, T.; et al. The preparation of cyclic dithia and thiaza compounds by the reaction of potassium carbonate with heterocumulenes and alkylene dibromides or carbonate catalyzed by organostannyl compounds
Bull Chem Soc Jpn 1982, 55(4): 1174
https://www.jstage.jst.go.jp/article/bcsj1926/55/4/55_4_1174/_pdf
…………………………………….
Trade Names
| Country | Trade name | Manufacturer |
|---|---|---|
| Japan | Kantek | Daiichi |
| Ukraine | No | No |
Formulations
-
200 mg tablets
Links
-
DOS 2,545,569 (Nihon Nohyaku; appl. 10.10.1975; J-prior. 18.10.1974, 22.10.1974).
-
US 4,035,387 (Nihon Nohyaku; 12.7.1977; J-prior. 18.10.1974, 22.10.1974).
1H NMR PREDICTIONS
WATCH OUT
13C NMR PREDICTIONS
References
- Bührer M, Le Cotonnec JY, Wermeille M, Bircher J (1986). “Treatment of liver disease with malotilate. A pharmacokinetic and pharmacodynamic phase II study in cirrhosis”. Eur. J. Clin. Pharmacol. 30 (4): 407–16. doi:10.1007/BF00607952.PMID 3743616.
- Siegers CP, Pauli V, Korb G, Younes M (August 1986). “Hepatoprotection by malotilate against carbon tetrachloride-alcohol-induced liver fibrosis”. Agents Actions 18 (5–6): 600–3. doi:10.1007/BF01964970. PMID 3766314.
- Younes M, Siegers CP (May 1985). “Effect of malotilate on paracetamol-induced hepatotoxicity”. Toxicol. Lett. 25 (2): 143–6.doi:10.1016/0378-4274(85)90074-8. PMID 4002245.
- Mayer, R.; et al.Synthesis of 1,3-dithiol-2-thiones (‘ Isotrithione’)
Angew Chem Int Ed 1964, 76(3): 143 - O’Connor, B.R.; Jones, F.N.Reactions of ethylene di- and trithiocarbonates with acetylenes. Anomalous reaction with bromocyanoacetylene to give a thioacyl bromide
J Org Chem 1970, 35(6): 2002 - Fujinami, T.; et al. The preparation of cyclic dithia and thiaza compounds by the reaction of potassium carbonate with heterocumulenes and alkylene dibromides or carbonate catalyzed by organostannyl compounds
Bull Chem Soc Jpn 1982, 55(4): 1174
Biological Activity of Malotilate
Malotilate is a Liver Protein Metabolism Improved Compound, Which Selectively INHIBIT the 5-lipoxygenase. IC50 Value : Target : 5-lipoxygenase in vitro : In an in vitro assay using RAT Invasion lung endothelial (RLE) cells, Invasion of tumor cells Which HAD BEEN treated with MT (10 ng / ml, 24 h) was not affected; however, when RLE cells had been treated with MT, invasion was significantly inhibited in three cell lines (SAS, Ca9-22 and HSC-4) and a tendency to inhibition WAS Also Observed in other Cell lines [1]. in Vivo : The Improvement Rates for choline esterase Were Significantly Greater Activity in the malotilate group than in the Control group Levels Significantly Increased Serum albumin in the malotilate group BUT not in the Control group. [2]. In the rats treated with MT for 19 days after iv inoculation of c-SST-2 cells, lung metastasis was also significantly suppressed [3]. Malotilate prevented increases in serum markers of type III and IV collagen synthesis as well as accumulation of the collagens, laminin and fibronectin in the Liver [4]. Toxicity : Malotilate cytotoxicity to PBMCs, Assessed by trypan blue dye Exclusion and lactate dehydrogenase (LDH) Release into the Culture Media, WAS found to be markedly Increased by the Addition of the NADPH generating system, indicating that metabolites play a significant role in toxicity [5].
References on Malotilate
[1] Shibata T, et al Inhibitory Effects of malotilate on in vitro Cell Invasion of lung endothelial monolayer by human oral squamous carcinoma cells Tumour Cell Biol 2000 Sep-Oct; 21 (5):….. 299-308 [2 …] Takase S, et al Effects of treatment on malotilate Alcoholic Liver disease Alcohol 1989 May-Jun; 6 (3):. 219-22. [3] Nagayasu H, et al Inhibitory Effects of malotilate on Invasion and.. Metastasis of RAT mammary carcinoma cells by modifying the Functions of Vascular endothelial cells Br J Cancer 1998 May; 77 (9):.. 1371-7. [4] Ryhanen L, et al The Effect of malotilate on type III and type.. . IV collagen, laminin and fibronectin Liver Metabolism in dimethylnitrosamine-induced fibrosis in the RAT J Hepatol 1996 Feb; 24 (2):. 238-45. [5] Nomura F, et al Detection of malotilate Toxicity in vitro with Peripheral.. . blood mononuclear cells as targets A preliminary report J Hepatol 1990 Jul; 11 (1):.. 65-9.

{kind=link}