


| Patent ID | Date | Patent Title |
|---|---|---|
| US2015290207 | 2015-10-15 | HETEROCYCLIC COMPOUNDS AND USES THEREOF |
| US2015225410 | 2015-08-13 | HETEROCYCLIC COMPOUNDS AND USES THEREOF |
| US2015111874 | 2015-04-23 | HETEROCYCLIC COMPOUNDS AND USES THEREOF |
Home » Posts tagged 'Immuno-oncology'
Tag Archives: Immuno-oncology
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
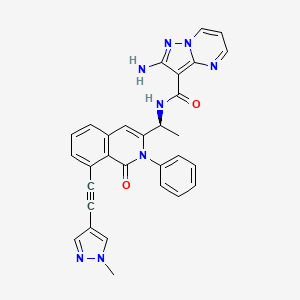
IPI-549

IPI-549
CAS 1693758-51-8
MF : C30H24N8O2
Molecular Weight: 528.576
(S)-2-amino-N-(1-(8-((1-methyl-1H-pyrazol-4-yl)ethynyl)-1-oxo-2-phenyl-1,2-dihydroisoquinolin-3-yl)ethyl)pyrazolo[1,5-a]pyrimidine-3-carboxamide
2-amino-N-[(1S)-1-[8-[2-(1-methylpyrazol-4-yl)ethynyl]-1-oxo-2-phenylisoquinolin-3-yl]ethyl]pyrazolo[1,5-a]pyrimidine-3-carboxamide
| Latest Stage of Development | Phase I |
| Standard Indication | Solid tumors |
| Indication Details | Treat solid tumors |
- Originator Intellikine
- Developer Infinity Pharmaceuticals
- ClassAntineoplastics; Small molecules
- Mechanism of ActionPhosphatidylinositol 3 kinase delta inhibitors; Phosphatidylinositol 3 kinase gamma inhibitors
- Phase I Solid tumours

Most Recent Events
- 18 Apr 2016 Pharmacodynamics data from a preclinical study in Solid tumours presented at the 107th Annual Meeting of the American Association for Cancer Research (AACR-2016)
- 01 Dec 2015 Phase-I clinical trials in Solid tumours (Monotherapy, Combination therapy, Late-stage disease, Second-line therapy or greater) in USA (PO)
IPI-549 is a potent and selective phosphoinositide-3-kinase (PI3Kγ) Inhibitor as an Immuno-Oncology Clinical Candidate (Kd = 0.29 nM). Bioactivity data of IPI-549: biochemcial IC50 (nM) for PI3K isoform: 3200 (α); 3500 (β); 16 (γ); and >8400 (δ) respectively. Cellar IC50 (nM) of IPI549 for PI3K isoform: 250 (α); 240 (β); 1.6 (γ); and 180 (δ) respectively. IPI-549 shows >100-fold selectivity over other lipid and protein kinases. IPI-549 demonstrates favorable pharmacokinetic properties and robust inhibition of PI3K-γ mediated neutrophil migration in vivo and is currently in Phase 1 clinical evaluation in subjects with advanced solid tumors.
Patent
WO 2015051244
https://www.google.co.in/patents/WO2015051244A1?cl=en
Scheme 1

Scheme 2

Example 1

[00657] Compound 4 was prepared in 3 steps from compound A according to the following procedures:
Compound A was prepared according to Method A. It was coupled to 2-((tert-butoxycarbonyl)amino)pyrazolo[l,5-a]pyrimidine-3-carboxylic acid according to the following procedure: Compound A (27.4 mmol, 1.0 equiv), HOBt hydrate (1.2 equiv), 2-((tert-butoxycarbonyl)amino)pyrazolo[l,5-a]pyrimidine-3-carboxylic acid (1.05 equiv) and
EDC (1.25 equiv) were added to a 200 mL round bottomed flask with a stir bar. N,N-Dimethylformamide (50 mL) was added and the suspension was stirred at RT for 2 min. Hunig’s base (4.0 equiv) was added and after which the suspension became homogeneous and was stirred for 22h resulting in the formation of a solid cake in the reaction flask. The solid mixture was added to water (600 mL) and stirred for 3h. The resulting cream colored solid was filtered and washed with water (2 x 100 mL) and dried. The solid was then dissolved in methylene chloride (40 mL) after which trifluoroacetic acid (10 equiv, 20 mL) was added and the reaction was stirred for 30 min at RT after which there is no more starting material by LC/MS analysis. The solution was then concentrated and coevaporated with a mixture of methylene choride/ethanol (1 : 1 v/v) and then dried under high vacuum overnight. The resulting solid was triturated with 60 mL of ethanol for lh and then collected via vacuum filtration. The beige solid was then neutralized with sodium carbonate solution (100 mL) and then transferred to a separatory funnel with methylene chloride (350 mL). The water layer was extracted with an additional 100 mL of methylene chloride. The combined organic layers were dried over sodium sulfate, filtered and concentrated under vacuum to provide a pale yellow solid that was purified using flash silica gel chromatography (Combiflash, 24g column, gradient of 0-5% methanol/methylene chloride) to provide amide B. ESI-MS m/z: 459.4 [M+H]+.
[00658] Amide B was placed in a sealed tube (0.67 mmol, 1.0 equiv) followed by dichlorobis(acetonitrile)palladium (15 mol%), X-Phos (45 mol%), and cesium carbonate (3.0 equiv) Propionitrile (5 mL) was added and the mixture was bubbled with Ar for 1 min. 4-Ethynyl-l -methyl- lH-pyrazole (1.24 equiv) was added and the resulting orange mixture was sealed and stirred in an oil bath at 85 oC for 1.5h. The resulting brownish-black mixture was allowed to cool at which point there was no more SM by LC/MS analysis. The mixture was then filtered through a short plug of cotton using acetonitrile and methylene chloride. The combined filtrates were concentrated onto silica gel and purified using flash silica gel chromatography (Combiflash, 4g column, gradient of 0-5% methylene chloride/methanol). The resulting material was further purified by reverse phase HPLC (15-90%o acetonitrile with 0.1%o formic acid/water with 0.1%o formic water) to provide desired compound 4. ESI-MS m/z: 529.5 [M+H]+.
PAPER
WO 2015143012
https://www.google.com/patents/WO2015143012A1?cl=en
PAPER
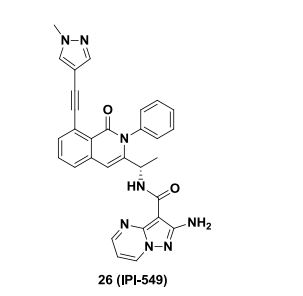
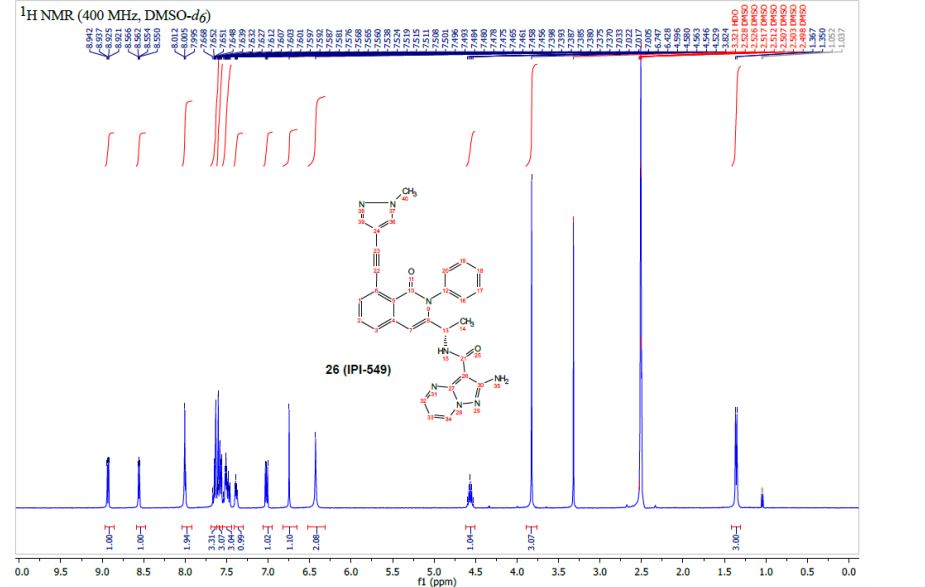
IPI-549 NMR 1H
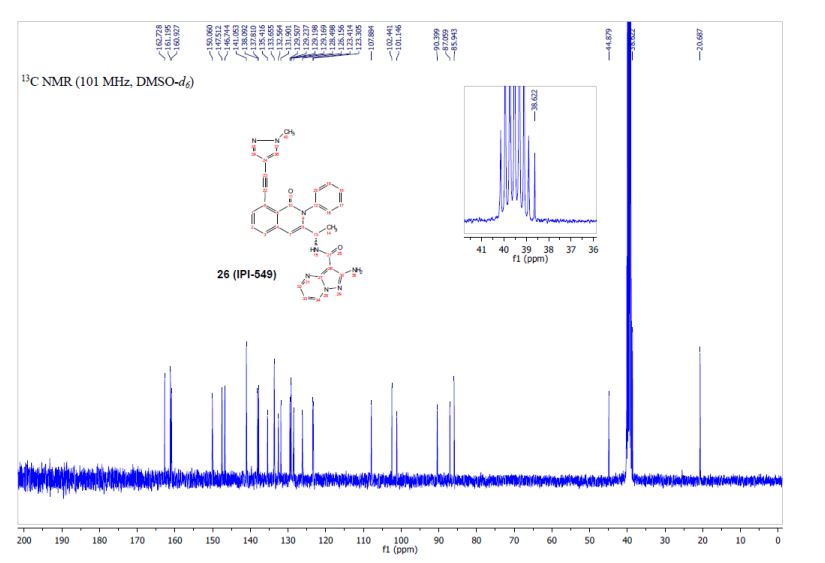
IPI-549 13C NMR
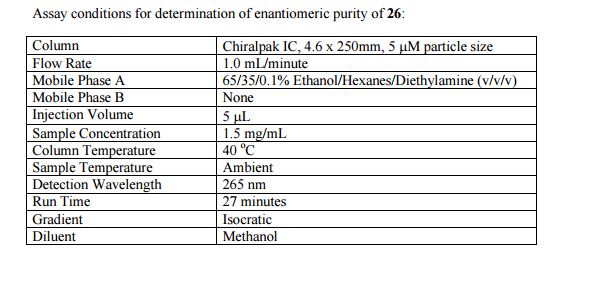
IPI-549 ASSAY


Compound 1 is coupled to 4-ethynyl-1-methyl-1H-pyrazole using the general procedure outlined above to provide compound 26 IPI-549, in 70% yield with >98% enantiomeric purity.
IPI-549
1H NMR (400 MHz, DMSO-d6) δ 8.92 (dd, J = 6.8, 1.7 Hz, 1H), 8.55 (dd, J = 4.5, 1.7 Hz, 1H), 8.00 (d, J=6.8 Hz, 1H), 8.00 (s, 1H), 7.69 – 7.54 (m, 5H), 7.53 – 7.43 (m, 3H), 7.41 – 7.35 (m, 1H), 7.01 (dd, J = 6.7, 4.5 Hz, 1H), 6.74 (s, 1H), 6.42 (s, 2H), 4.56 (quin, J = 6.8 Hz, 1H).), 3.82 (s, 3H), 1.35 (d, J = 6.8 Hz, 3H).
13C NMR (101 MHz, DMSO-d6) δ 162.73, 161.19, 160.93, 150.06, 147.51, 146.74, 141.05, 138.09, 137.81, 135.42, 133.66, 132.56, 131.90, 129.51, 129.24, 129.20, 129.17, 128.50, 126.16, 123.41, 123.31, 107.88, 102.44, 101.15, 90.40, 87.06, 85.94, 44.88, 38.62, 20.69.
ESI-HRMS: calcd for 529.2095 C30H25N8O2 (M+H)+ , found 529.2148.
[]D 22: +447.8o (c 1.007, DMSO)
COMPD1
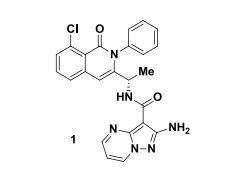
compound 1 in 95% yield.
1H NMR (400 MHz, CDCl3) 8.41 (dd, J = 6.8, 1.7 Hz, 1H), 8.37 (dd, J = 4.4, 1.7 Hz, 1H), 7.90 (d, J = 7.0 Hz, 1H), 7.50-7.34 (m, 5H), 7.34-7.27 (m, 2H), 6.76 (dd, J = 7.1, 4.9 Hz, 1H), 6.57 (s, 1H), 5.54 (broad s, 2H), 4.79 (quin, J = 6.9 Hz, 1H), 1.36 (d, J = 6.5 Hz, 3H);
ESI-HRMS: calcd for C24H20ClN6O2 459.1331 (M+H)+ , found 459.1386. HPLC Purity: 96% AUC.

Optimization of isoquinolinone PI3K inhibitors led to the discovery of a potent inhibitor of PI3K-γ (26 or IPI-549) with >100-fold selectivity over other lipid and protein kinases. IPI-549 demonstrates favorable pharmacokinetic properties and robust inhibition of PI3K-γ mediated neutrophil migration in vivo and is currently in Phase 1 clinical evaluation in subjects with advanced solid tumors.
Discovery of a Selective Phosphoinositide-3-Kinase (PI3K)-γ Inhibitor (IPI-549) as an Immuno-Oncology Clinical Candidate
Infinity Pharmaceuticals, Inc., 784 Memorial Drive, Cambridge, Massachusetts 02139, United States
ACS Med. Chem. Lett., 2016, 7 (9), pp 862–867
DOI: 10.1021/acsmedchemlett.6b00238
*E-mail: catherine.evans@infi.com., *E-mail: alfredo.castro@infi.com.
http://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.6b00238
CLIP
Infinity Expands Pipeline with Addition of IPI-549, an Immuno-Oncology Development Candidate for the Treatment of Solid Tumors
– IPI-549, a Selective PI3K-Gamma Inhibitor, Targets the Immune-Suppressive Tumor Microenvironment –
– Preclinical Data for IPI-549 Presented at CRI-CIMT-EATI-AACR – The Inaugural International Cancer Immunotherapy Conference –
CAMBRIDGE, Mass.–(BUSINESS WIRE)–Infinity Pharmaceuticals, Inc. (NASDAQ: INFI) today announced the expansion of its pipeline with the addition of IPI-549, an orally administered immuno-oncology development candidate that selectively inhibits phosphoinositide-3-kinase gamma (PI3K-gamma), for the treatment of solid tumors. Preclinical data demonstrating the potential of IPI-549 to disrupt the immune-suppressive tumor microenvironment and enable a heightened anti-tumor immune response are being presented today at CRI-CIMT-EATI-AACR – The Inaugural International Cancer Immunotherapy Conference: Translating Science into Survival Meeting in New York City. IPI-549 was discovered at Infinity and is expected to enter Phase 1 clinical development in early 2016.
“Infinity is committed to developing first-in-class and best-in-class medicines, and the expansion of our pipeline with the addition of IPI-549 represents an important step toward fulfilling our vision of building a sustainable biopharmaceutical company that brings meaningful medicines to patients”
“Infinity is committed to developing first-in-class and best-in-class medicines, and the expansion of our pipeline with the addition of IPI-549 represents an important step toward fulfilling our vision of building a sustainable biopharmaceutical company that brings meaningful medicines to patients,” stated Vito Palombella, Ph.D., Infinity’s chief scientific officer. “Infinity’s ability to internally develop a selective PI3K-gamma inhibitor provides us with a unique opportunity to explore the impact that PI3K-gamma inhibition has on disrupting the tumor microenvironment. We look forward to initiating the first clinical study of IPI-549 in patients with solid tumors.”
“I have had the pleasure of collaborating with Infinity’s discovery team and am excited to have worked with IPI-549 in my laboratory,” Jedd Wolchok, M.D., Ph.D., chief of Melanoma and Immunotherapeutics Service, Lloyd J. Old/Ludwig Chair in Clinical Investigation Department of Medicine and Ludwig Center, at Memorial Sloan Kettering Cancer Center and the principal investigator for the planned Phase 1 clinical study of IPI-549. “IPI-549 is a novel, small molecule immuno-oncology agent, and I am looking forward to leading the Phase 1 study for this program.”
IPI-549 inhibits immune suppressive macrophages within the tumor microenvironment, whereas other immunotherapies such as checkpoint modulators more directly target immune effector cell function. As such, IPI-549 may have the potential to treat a broad range of solid tumors and represents a potentially complementary approach to restoring anti-tumor immunity in combination with other immunotherapies such as checkpoint inhibitors.
Preclinical Data for IPI-549 Presented at CRI-CIMT-EATI-AACR – The Inaugural International Cancer Immunotherapy Conference
Today at the AACR meeting in New York City Infinity researchers are presenting preclinical data for IPI-549 in a poster entitled, “The potent and selective phosphoinositide-3-kinase-gamma inhibitor, IPI-549, inhibits tumor growth in murine syngeneic solid tumor models through alterations in the immune suppressive microenvironment.”
In vitro data showed that IPI-549 blocks both the migration of murine myeloid cells and the differentiation of myeloid cells to the M2 phenotype, which is a type of myeloid cell known to promote cancer growth and suppress anti-tumor immune responses. In vivo data in murine solid tumor models demonstrated that IPI-549 treatment also decreased tumor-associated myeloid cells found in the immune suppressive microenvironment. Additionally, IPI-549 treatment increased the number of intratumoral CD8+T-cells, which are known to play a role in inhibiting tumor growth.
IPI-549 has demonstrated dose-dependent, single-agent, anti-tumor activity in multiple solid tumor models, including murine models of lung, colon and breast cancer. Additionally, mice treated with IPI-549 in combination with checkpoint inhibitors showed greater tumor growth inhibition than either treatment as a monotherapy. Preclinical in vivo data also demonstrated that T-cells are required for the anti-tumor activity of IPI-549, which is a hallmark of immunotherapy.
Further details about the IPI-549 development program will be provided at Infinity’s R&D Day on Tuesday, October 6, 2015. R&D Day will be held in New York City from 7:30 a.m. to 12:00 p.m. ET. The event will be webcast beginning at 8:00 a.m. ET and can be accessed in the Investors/Media section of Infinity’s website, www.infi.com. A replay of the event will also be available.
Infinity is also developing duvelisib, an investigational, oral, dual inhibitor of PI3K-delta and PI3K-gamma. The PI3K pathway is also known to play a critical role in regulating the growth and survival of certain types of blood cancers. The investigational agent is being evaluated in registration-focused studies, including DYNAMOTM, a Phase 2 study in patients with refractory indolent non-Hodgkin lymphoma, DYNAMO+R, a Phase 3 study in patients with previously treated follicular lymphoma, and DUOTM, a Phase 3 study in patients with relapsed/refractory chronic lymphocytic leukemia. Duvelisib is an investigational compound and its safety and efficacy have not been evaluated by the U.S. Food and Drug Administration or any other health authority.
About Infinity Pharmaceuticals, Inc.
Infinity is an innovative biopharmaceutical company dedicated to discovering, developing and delivering best-in-class medicines to people with difficult-to-treat diseases. Infinity combines proven scientific expertise with a passion for developing novel small molecule drugs that target emerging disease pathways. For more information on Infinity, please refer to the company’s website at www.infi.com.
Clip
IPI-549-01-A phase 1/1b first in human study of IPI-549, a PI3K-γ inhibitor, as monotherapy and in combination with pembrolizumab in subjects with advanced solid tumors.
Subcategory:
Category:
Developmental Therapeutics—Immunotherapy
Meeting:
Session Type and Session Title:
Poster Session, Developmental Therapeutics—Immunotherapy
Abstract Number: TPS3111
Poster Board Number:
Board #425a
Citation:
J Clin Oncol 34, 2016 (suppl; abstr TPS3111)
Abstract:
Background: IPI-549 is a potential first-in-class potent and selective PI3K-γ inhibitor being developed as a novel orally administered immuno-oncology therapeutic in multiple cancer indications. Preclinical studies demonstrate a role for PI3K-γ in polarization of immune suppressive myeloid cells in the tumor microenvironment. Inhibition of PI3K-γ by IPI-549 enhanced antitumor immune responses and inhibited tumor growth in syngeneic solid tumor preclinical models. In addition, IPI-549 in combination with immune checkpoint inhibitors showed increased tumor growth inhibition compared to each single agent in multiple pre-clinical models. These data served as the scientific foundation for initiating a clinical trial testing IPI-549 as a potential immuno-oncology therapy. This first-in-human clinical study will evaluate the safety and tolerability, and determine the recommended Phase 2 dose (RP2D) of IPI-549 when administered as a monotherapy and in combination with pembrolizumab (NCT02637531) in solid tumors. Methods: This multi-part Phase 1/1b open-label trial will initiate with monotherapy dose escalation cohorts consisting of an accelerated dose escalation phase followed by a standard phase with a 3+3 design. Evaluation of the PK, PD, and safety data in these cohorts will lead to the determination of the maximum tolerated dose (MTD) and RP2D of IPI-549 monotherapy. Subsequently, combination dose escalation cohorts will be initiated in which the combination of IPI-549 and pembrolizumab will be evaluated. Expansion cohorts evaluating the safety, PK, PD, and preliminary clinical activity of IPI-549 as a monotherapy and in combination with pembrolizumab will occur following the dose escalation phase. All subjects in the trial will have advanced and/or metastatic carcinoma or melanoma, and will have failed to respond to standard therapies. Combination expansion cohorts will recruit subjects with non-small cell lung cancer or melanoma who must have received an anti-PD-1 or anti-PD-L1 antibody as their most recent treatment. This trial is currently enrolling patients in the US. Clinical trial information: NCT02637531
REFERENCES
Discovery of a Selective Phosphoinositide-3-Kinase (PI3K)-γ Inhibitor (IPI-549) as an Immuno-Oncology Clinical Candidate
Catherine A. Evans, Tao Liu, André Lescarbeau, Somarajan J. Nair, Louis Grenier, Johan A. Pradeilles, Quentin Glenadel, Thomas Tibbitts, Ann M. Rowley, Jonathan P. DiNitto, Erin E. Brophy, Erin L. O’Hearn, Janid A. Ali, David G. Winkler, Stanley I. Goldstein, Patrick O’Hearn, Christian M. Martin, Jennifer G. Hoyt, John R. Soglia, Culver Cheung, Melissa M. Pink, Jennifer L. Proctor, Vito J. Palombella, Martin R. Tremblay, and Alfredo C. Castro
Publication Date (Web): July 22, 2016 (Letter)
DOI: 10.1021/acsmedchemlett.6b00238
///////immuno-oncology, IPI-549, isoform selectivity, neutrophil migration, phosphoinositide-3-kinase, PI3K-gamma inhibitor, IPI 549, IPI549. PRECLINICAL
O=C1N(C2=CC=CC=C2)C([C@@H](NC(C3=C(N=CC=C4)N4N=C3N)=O)C)=CC5=CC=CC(C#CC6=CN(C)N=C6)=C51
AUNP-12 from Aurigene Discovery Technologies Limited
AUNP-12
AUR-012; Aurigene-012; NP-12, Aurigene; PD-1 inhibitor peptide (cancer), Aurigene; PD-1 inhibitor peptide (cancer), Aurigene/ Pierre Fabre; W-014A
| Latest Stage of Development | Preclinical |
| Standard Indication | Cancer (unspecified) |
| Indication Details | Treat cancer |
| Regulatory Designation | |
| Partner | Laboratoires Pierre Fabre S.A. |
Aurigene Discovery Technologies Limited

-
Programmed Cell Death 1 or PD-1 (also referred to as PDCD1) is a 50 to 55 kD type I membrane glycoprotein (Shinohara T et al, Genomics, 1994, Vol. 23, No. 3, pp. 704-706). PD-1 is a receptor of the CD28 superfamily that negatively regulates T cell antigen receptor signalling by interacting with the specific ligands and is suggested to play a role in the maintenance of self tolerance.
-
PD-1 peptide relates to almost every aspect of immune responses including autoimmunity, tumour immunity, infectious immunity, transplantation immunity, allergy and immunological privilege.
-
The PD-1 protein’s structure comprise of—
-
- an extracellular IgV domain followed by
- a transmembrane region and
- an intracellular tail
-
-
The intracellular tail contains two phosphorylation sites located in an immunoreceptor tyrosine-based inhibitory motif and an immunoreceptor tyrosine-based switch motif, which suggests that PD-1 negatively regulates TCR signals. Also, PD-1 is expressed on the surface of activated T cells, B cells, and macrophages, (Y. Agata et al., Int Immunol 8, 765, May 1996) suggesting that compared to CTLA-4 ((Cytotoxic T-Lymphocyte Antigen 4, also known as CD152 (Cluster of differentiation 152) is a protein that also plays an important regulatory role in the immune system), PD-1 more broadly negatively regulates immune responses.
-
PD-1 has two ligands, PD-L1 (Programmed Death Ligand for PDCD1L1 or B7-H1) (Freeman G J et al, Journal of Experimental Medicine, 2000, Vol. 19, No. 7, pp. 1027-1034) and PD-L2 (Programmed Death Ligand 2 or PDCD1L2 or B7-DC) (Latchman Y et al, Nature Immunology, 2001, Vol. 2, No. 3, pp. 261-267), which are members of the B7 family. PD-L1 is known to be expressed not only in immune cells, but also in certain kinds of tumour cell lines (such as monocytic leukaemia-derived cell lines, mast cell tumour-derived cell lines, hematoma-derived cell lines, neuroblastoma-derived cell lines, and various mammary tumour-derived cell lines) and in cancer cells derived from diverse human cancer tissues (Latchman Y et al, Nature Immunology, 2001, Vol. 2, No. 3, pp. 261-267) and on almost all murine tumour cell lines, including PA1 myeloma, P815 mastocytoma, and B16 melanoma upon treatment with IFN-γ (Y. Iwai et al., Proc Natl Acad Sci USA 99, 12293, Sep. 17, 2002 and C. Blank et al., Cancer Res 64, 1140, February, 2004). Similarly PD-L2 expression is more restricted and is expressed mainly by dendritic cells and a few tumour cell lines. PD-L2 expression has been verified in Hodgkin’s lymphoma cell lines and others. There is a hypothesis that some of the cancer or tumour cells take advantage from interaction between PD-1 and PD-L1 or PD-L2, for suppressing or intercepting T-cell immune responses to their own (Iwai Y et al, Proceedings of the National Academy of Science of the United States of America, 2002, Vol. 99, No. 19, pp. 12293-12297).
-
Tumour cells and virus (including HCV and HIV) infected cells are known to express the ligand for PD-1 (to create Immunosuppression) in order to escape immune surveillance by host T cells. It has been reported that the PD-1 gene is one of genes responsible for autoimmune diseases like systemic lupus erythematosis (Prokunina et al, Nature Genetics, 2002, Vol. 32, No. 4, 666-669). It has also been indicated that PD-1 serves as a regulatory factor for the onset of autoimmune diseases, particularly for peripheral self-tolerance, on the ground that PD-1-deficient mice develop lupus autoimmune diseases, such as glomerulonephritis and arthritis (Nishimura H et al, International Immunology, 1998, Vol. 10, No. 10, pp. 1563-1572; Nishimura H et al, Immunity, 1999, Vol. 11, No. 2, pp. 141-151), and dilated cardiomyopathy-like disease (Nishimura H et al, Science, 2001, Vol. 291, No. 5502, pp. 319-332).
-
Hence, in one approach, blocking the interaction of PD-1 with its ligand (PD-L1, PD-L2 or both) may provide an effective way for specific tumour and viral immunotherapy.
-
Wood et al in U.S. Pat. No. 6,808,710 discloses method for down modulating an immune response comprising contacting an immune cell expressing PD-1 with an antibody that binds to PD-1, in multivalent form, such that a negative signal is transduced via PD-1 to thereby down modulate the immune response. Such an antibody may be a cross-linked antibody to PD-1 or an immobilized antibody to PD-1.
-
Freeman et al in U.S. Pat. No. 6,936,704 and its divisional patent U.S. Pat. No. 7,038,013 discloses isolated nucleic acids molecules, designated B7-4 nucleic acid molecules, which encode novel B7-4 polypeptides, isolated B7-4 proteins, fusion proteins, antigenic peptides and anti-B7-4 antibodies, which co-stimulates T cell proliferation in vitro when the polypeptide is present on a first surface and an antigen or a polyclonal activator that transmits an activating signal via the T-cell receptor is present on a second, different surface.
-
There are some reports regarding substances inhibiting immunosuppressive activity of PD-1, or interaction between PD-1 and PD-L1 or PD-L2, as well as the uses thereof. A PD-1 inhibitory antibody or the concept of a PD-1 inhibitory peptide is reported in WO 01/14557, WO 2004/004771, and WO 2004/056875. On the other hand, a PD-L1 inhibitory antibody or a PD-L1 inhibitory peptide is reported in WO 02/079499, WO 03/042402, WO 2002/086083, and WO 2001/039722. A PD-L2 inhibitory antibody or a PD-L2 inhibitory peptide is reported in WO 03/042402 and WO 02/00730.
-
WO2007005874 describes isolated human monoclonal antibodies that specifically bind to PD-L1 with high affinity. The disclosure provides methods for treating various diseases including cancer using anti-PD-L1 antibodies.
-
US2009/0305950 describes multimers, particularly tetramers of an extracellular domain of PD-1 or PD-L1. The application describes therapeutic peptides.
-
Further, the specification mentions that peptides can be used therapeutically to treat disease, e.g., by altering co-stimulation in a patient. An isolated B7-4 or PD-1 protein, or a portion or fragment thereof (or a nucleic acid molecule encoding such a polypeptide), can be used as an immunogen to generate antibodies that bind B7-4 or PD-1 using standard techniques for polyclonal and monoclonal antibody preparation. A full-length B7-4 or PD-1 protein can be used, or alternatively, the invention provides antigenic peptide fragments of B7-4 or PD-1 for use as immunogens. The antigenic peptide of B7-4 or PD-1 comprises at least 8 amino acid residues and encompasses an epitope of B7-4 or PD-1 such that an antibody raised against the peptide forms a specific immune complex with B7-4 or PD-1. Preferably, the antigenic peptide comprises at least 10 amino acid residues, more preferably at least 15 amino acid residues, even more preferably at least amino acid residues, and most preferably at least 30 amino acid residues.
-
Freeman et al in U.S. Pat. No. 7,432,059 appears to disclose and claim methods of identifying compounds that up modulate T cell activation in the presence of a PD-1-mediated signal. Diagnostic and treatment methods utilizing compositions of the invention are also provided in the patent.
-
Further, Freeman et al in U.S. Pat. No. 7,709,214 appears to cover methods for up regulating an immune response with agents that inhibit the interactions between PD-L2 and PD-1.
-
Despite existence of many disclosures as discussed above, however, a significant unmet medical need still exists due to the lack of effective peptides or modified peptides as therapeutic agents as alternatives in the therapeutic area. It is known that synthetic peptides offer certain advantages over antibodies such as ease of production with newer technologies, better purity and lack of contamination by cellular materials, low immunogenicity, improved potency and specificity. Peptides may be more stable and offer better storage properties than antibodies. Moreover, often peptides possess better tissue penetration in comparison with antibodies, which could result in better efficacy. Peptides can also offer definite advantages over small molecule therapeutics counterparts such as lesser degree of toxicity and lower probability of drug-drug interaction.
-
The present invention therefore may provide the solution for this unmet medical need by offering novel synthetic peptide and its derivatives which are based on the PD1 ectodomain.


Aurigene team: (from left) Brahma Reddy V, Thomas Antony, Murali Ramachandra, Venkateshwar Rao G, Wesley Roy Balasubramanian, Kishore Narayanan, Samiulla DS, Aravind AB, and Shekar Chelur
Patent
http://www.google.com/patents/US20110318373
| 8. | SNTSESFK(SNTSESF)FRVTQLAPKAQIKE-NH2 (SEQ ID NO: 49) |
|

Example 2 Synthesis of

Synthesis of Linear Fragment—Fmoc-FRVTQLAPKAQIKE
-
Desiccated CLEAR-Amide resin ((100-200 mesh) 0.4 mmol/g, 0.5 g) was distributed in 2 polyethylene vessels equipped with a polypropylene filter. The linear peptide synthesis on solid phase were carried out automatically, using Symphony parallel synthesizer (PTI) using the synthesis programs mentioned in the table below. Swelling, C-terminal amino acid [Fmoc-Glu(OtBu)-OH] attachment and capping of the peptidyl resin was carried out as per the protocol in Table I. Subsequent amino acid coupling was carried out as mentioned in Table II. The amino acids used in the synthesis were Fmoc Phe-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Val-OH, Fmoc-Thr(OtBu)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Lys(Boc)-OH, Fmoc-Leu-OH, Fmoc-Ala-OH, Fmoc-Pro-OH, Fmoc-Ile-OH. After the completion of Fmoc-Phe-OH coupling the resin was taken out form peptide synthesiser and manual coupling was carried out as follows
-
Fmoc-Phe-OH peptidyl resin from automated synthesiser was pooled in to a glass vessel with frit. The Fmoc group of the peptidyl resin was deprotected by treating it twice with 20% (v/v) piperidine/DMF solution for 5 and 15 min (10 m L). The resin was washed with DMF (6×15 m L), DCM (6×15 m L) and DMF (6×15 m L). Kaiser test on peptide resin aliquot upon completion of Fmoc-deprotection was positive. Fmoc-Lys (Fmoc)-OH (0.48 g; 4 equiv. 0.8 m mol) in dry DMF was added to the deprotected resin and coupling was initiated with DIC (0.15 m L; 5 equiv, 1 m mol) and HOBT (0.08 g; 5 equiv, 0.6 m mol) in DMF. The concentration of each reactant in the reaction mixture was approximately 0.4 M. The mixture was rotated on a rotor at room temperature for 3 h. Resin was filtered and washed with DMF (6×15 mL), DCM (6×15 mL) and DMF (6×15 mL). Kaiser test on peptide resin aliquot upon completion of coupling was negative. The Fmoc group on the peptidyl resin is deprotected by treating it twice with 20% (v/v) piperidine/DMF solution for 5 and 15 min (15 mL). The resin was washed with DMF (6×15 mL), DCM (6×15 mL) and DMF (6×15 mL). Kaiser test on peptide resin aliquot upon completion of Fmoc-deprotection was positive. After the deprotection of Fmoc group on Fmoc-Lys(Fmoc)-attached peptidyl resin the peptide chain growth was carried out from both the free amino terminus suing 8 equivalent excess of amino acid (1.6 m mol, 8 equivalent excess of HOBt (0.22 g, 1.6 m mol) and 10 equivalent excess of DIC (0.32 m L, 2 m mol) relative to resin loading. The coupling was carried out at room temperature for 3 h. The amino acids coupled to the peptidyl resin were; Fmoc-Phe-OH (0.62 g; 8 equiv, 1.6 m mol), Fmoc-Ser (OtBu)-OH (0.62 g; 8 equiv, 1.6 m mol), Fmoc-Glu (OtBu)-OH (0.68 g; 8 equiv, 1.6 m mol), Fmoc-Ser (OtBu)-OH (0.62 g; 8 equiv, 1.6 m mol), Fmoc-Thr (OtBu)-OH (0.64 g; 8 equiv, 1.6 m mol), Fmoc-Asn (Trt)-OH (0.95 g; 8 equiv, 1.6 m mol) and N-terminus amino acids as Boc-Ser (OtBu)-OH (0.41 g; 8 equiv, 1.6 m mol) The peptidyl resin was cleaved as mentioned in procedure for cleavage using cleavage cocktail A to yield (565 mg), 70% yield. The crude material was purified by preparative HPLC on Zorbax Eclipse XDB-C18 column (9.4 mm×250 mm, 5 μm) with buffer A: 0.1% TFA/Water, buffer B: Acetonitrile. The peptide was eluted by gradient elution 0-5 min=5-10% buffer B, 10-20 min=29% buffer B with a flow rate of 7 mL/min. HPLC: (method 1): RT-12 min (96%); LCMS Calculated Mass: 3261.62, Observed Mass: 1631.6 [M/2+H]+; 1088 [M/3+H]+); 816.2[M/4+H]+;
STRUCTURE , READER DISCRETION IS NEEDED
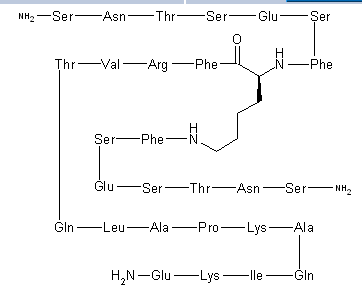
N2,N6-Bis(L-seryl-L-asparaginyl-L-threonyl-L-seryl-L-alpha-glutamyl-L-seryl-L-phenylalanyl)-L-lysyl-L-phenylalanyl-L-arginyl-L-valyl-L-threonyl-L-glutaminyl-L-leucyl-L-alanyl-L-prolyl-L-lysyl-L-alanyl-L-glutaminyl-L-isoleucyl-L-lysyl-L-alpha-glutamine
C142 H226 N40 O48, 3261.553
CAS 1353563-85-5,
L-α-Glutamine, N2,N6– bis(L-seryl-L-asparaginyl-L-threonyl-L-seryl-L-α-glutamyl-L- seryl-L-phenylalanyl)-L-lysyl-L-phenylalanyl-L-arginyl-L- valyl-L-threonyl-L-glutaminyl-L-leucyl-L-alanyl-L-prolyl-L- lysyl-L-alanyl-L-glutaminyl-L-isoleucyl-L-lysyl-
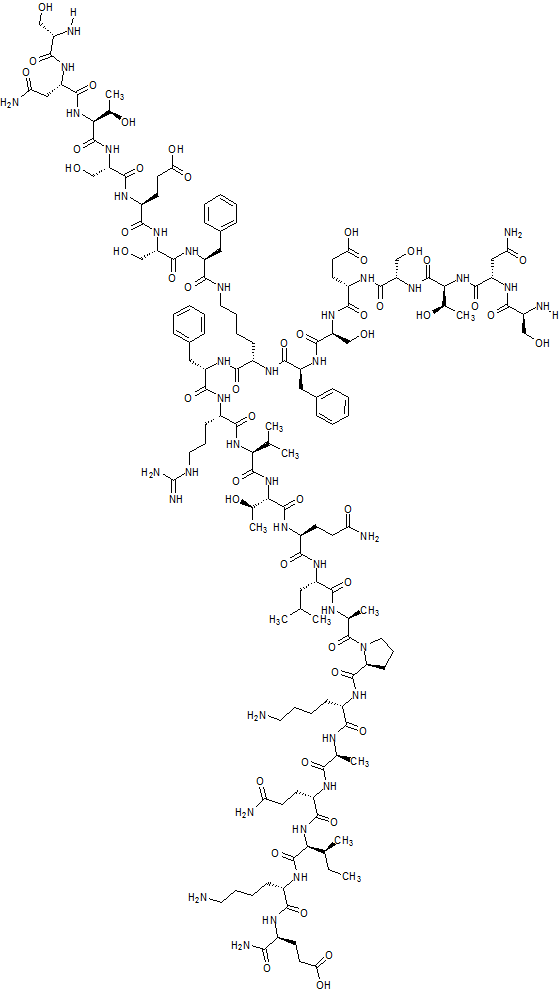
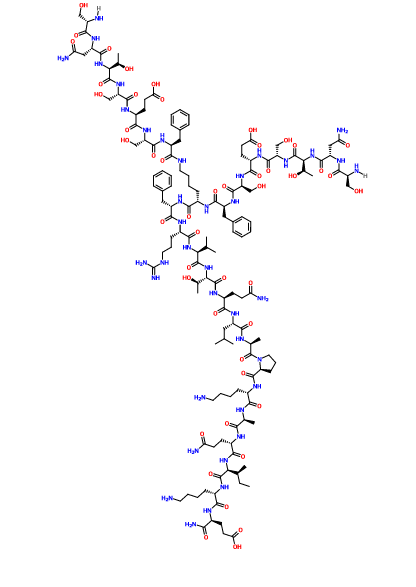
SEE ALSO
CAS 1353564-61-0,
L-α-Glutamine, N2,N6– bis(D-seryl-L-asparaginyl-L-threonyl-L-seryl-L-α-glutamyl-L- seryl-L-phenylalanyl)-L-lysyl-L-phenylalanyl-L-arginyl-L- valyl-L-threonyl-L-glutaminyl-L-leucyl-L-alanyl-L-prolyl-L- lysyl-L-alanyl-L-glutaminyl-L-isoleucyl-L-lysyl-
CAS 1353563-91-3
D-α-Glutamine, N2,N6– bis(D-seryl-D-asparaginyl-D-threonyl-D-seryl-D-α-glutamyl-D- seryl-D-phenylalanyl)-D-lysyl-D-phenylalanyl-D-arginyl-D- valyl-D-threonyl-D-glutaminyl-D-leucyl-D-alanyl-D-prolyl-D- lysyl-D-alanyl-D-glutaminyl-D-isoleucyl-D-lysyl-
US 2015087581
Compound 8 (SEQ ID NO: 49) SNTSESFK(SNTSESF)FRVTQLAPKAQIKE-NH2
Example 2Synthesis of Sequence Shown in SEQ ID NO: 49
Synthesis of Linear Fragment—Fmoc-FRVTQLAPKAQIKE
Desiccated CLEAR-Amide resin ((100-200 mesh) 0.4 mmol/g, 0.5 g) was distributed in 2 polyethylene vessels equipped with a polypropylene filter. The linear peptide synthesis on solid phase were carried out automatically, using Symphony parallel synthesizer (PTI) using the synthesis programs mentioned in the table below. Swelling, C-terminal amino acid [Fmoc-Glu(OtBu)-OH] attachment and capping of the peptidyl resin was carried out as per the protocol in Table I. Subsequent amino acid coupling was carried out as mentioned in Table II. The amino acids used in the synthesis were Fmoc Phe-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Val-OH, Fmoc-Thr(OtBu)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Lys(Boc)-OH, Fmoc-Leu-OH, Fmoc-Ala-OH, Fmoc-Pro-OH, Fmoc-Ile-OH. After the completion of Fmoc-Phe-OH coupling the resin was taken out form peptide synthesiser and manual coupling was carried out as follows.
Fmoc-Phe-OH peptidyl resin from automated synthesiser was pooled in to a glass vessel with frit. The Fmoc group of the peptidyl resin was deprotected by treating it twice with 20% (v/v) piperidine/DMF solution for 5 and 15 min (10 mL). The resin was washed with DMF (6×15 mL), DCM (6×15 mL) and DMF (6×15 mL). Kaiser test on peptide resin aliquot upon completion of Fmoc-deprotection was positive.
Fmoc-Lys (Fmoc)-OH (0.48 g; 4 equiv. 0.8 mmol) in dry DMF was added to the deprotected resin and coupling was initiated with DIC (0.15 mL; 5 equiv, 1 mmol) and HOBT (0.08 g; 5 equiv, 0.6 mmol) in DMF. The concentration of each reactant in the reaction mixture was approximately 0.4 M. The mixture was rotated on a rotor at room temperature for 3 h. Resin was filtered and washed with DMF (6×15 mL), DCM (6×15 mL) and DMF (6×15 mL). Kaiser test on peptide resin aliquot upon completion of coupling was negative. The Fmoc group on the peptidyl resin is deprotected by treating it twice with 20% (v/v) piperidine/DMF solution for 5 and 15 min (15 mL). The resin was washed with DMF (6×15 mL), DCM (6×15 mL) and DMF (6×15 mL). Kaiser test on peptide resin aliquot upon completion of Fmoc-deprotection was positive.
After the deprotection of Fmoc group on Fmoc-Lys(Fmoc)-attached peptidyl resin the peptide chain growth was carried out from both the free amino terminus suing 8 equivalent excess of amino acid (1.6 mmol, 8 equivalent excess of HOBt (0.22 g, 1.6 mmol) and 10 equivalent excess of DIC (0.32 mL, 2 mmol) relative to resin loading. The coupling was carried out at room temperature for 3 h. The amino acids coupled to the peptidyl resin were; Fmoc-Phe-OH (0.62 g; 8 equiv, 1.6 mmol), Fmoc-Ser (OtBu)-OH (0.62 g; 8 equiv, 1.6 mmol), Fmoc-Glu (OtBu)-OH (0.68 g; 8 equiv, 1.6 mmol), Fmoc-Ser (OtBu)-OH (0.62 g; 8 equiv, 1.6 mmol), Fmoc-Thr (OtBu)-OH (0.64 g; 8 equiv, 1.6 mmol), Fmoc-Asn (Trt)-OH (0.95 g; 8 equiv, 1.6 m mol) and N-terminus amino acids as Boc-Ser (OtBu)-OH (0.41 g; 8 equiv, 1.6 mmol) The peptidyl resin was cleaved as mentioned in procedure for cleavage using cleavage cocktail A to yield (565 mg), 70% yield. The crude material was purified by preparative HPLC on Zorbax Eclipse XDB-C18 column (9.4 mm×250 mm, 5 μm) with buffer A: 0.1% TFA/Water, buffer B:Acetonitrile. The peptide was eluted by gradient elution 0-5 min=5-10% buffer B, 10-20 min=29% buffer B with a flow rate of 7 mL/min. HPLC: (method 1): RT—12 min (96%); LCMS Calculated Mass: 3261.62, Observed Mass: 1631.6 [M/2+H]+; 1088 [M/3+H]+;); 816.2[M/4+H]+.
SMILES
O=C(N[C@@H](CCCCNC(=O)[C@H](Cc1ccccc1)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(=O)O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CO)N)[C@@H](C)O)C(=O)N[C@@H](Cc2ccccc2)C(=O)N[C@@H](CCCNC(=N)N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C)C(=O)N3CCC[C@H]3C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(=O)O)C(N)=O)[C@H](Cc4ccccc4)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(=O)O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CO)N)[C@@H](C)O
NEXT………..
CAS 1353564-65-4
C142 H226 N40 O48
L-α-Glutamine, L-seryl-L-asparaginyl-L-threonyl-L-seryl-L-α-glutamyl-L-seryl-L-phenylalanyl-N6– (L-seryl-D-asparaginyl-L-threonyl-L-seryl-L-α-glutamyl-L- seryl-L-phenylalanyl)-L-lysyl-L-phenylalanyl-L-arginyl-L- valyl-L-threonyl-L-glutaminyl-L-leucyl-L-alanyl-L-prolyl-L- lysyl-L-alanyl-L-glutaminyl-L-isoleucyl-L-lysyl-
- Molecular Weight, 3261.55
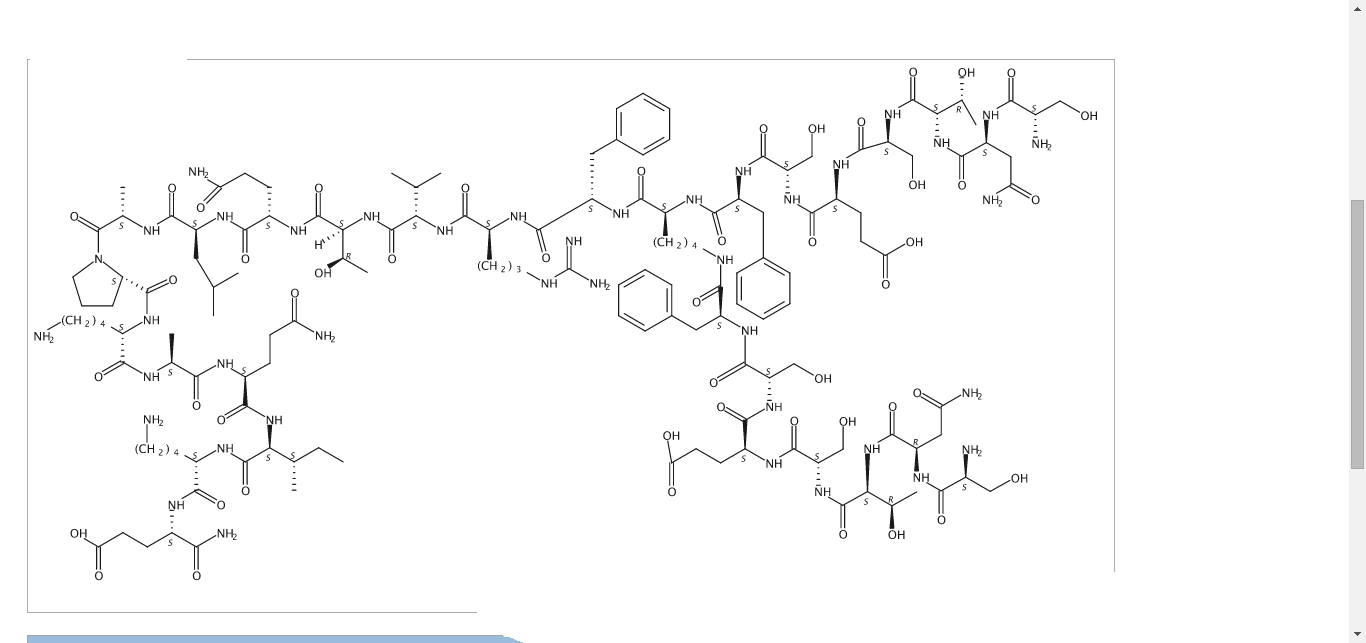
NEXT……….
CAS 1353564-31-4, C142 H226 N40 O48
L-α-Glutamine, L-seryl-L-asparaginyl-L-threonyl-L-seryl-L-α-glutamyl-L-seryl-L-phenylalanyl-N6– (D-seryl-D-asparaginyl-D-threonyl-D-seryl-D-α-glutamyl-D- seryl-D-phenylalanyl)-L-lysyl-L-phenylalanyl-L-arginyl-L- valyl-L-threonyl-L-glutaminyl-L-leucyl-L-alanyl-L-prolyl-L- lysyl-L-alanyl-L-glutaminyl-L-isoleucyl-L-lysyl-
USE ALL YOUR DISCRETION……………

Clips
Aurigene and Pierre Fabre Pharmaceuticals Announce a Licensing Agreement for a New Cancer Therapeutic in Immuno-oncology: AUNP12, an Immune Checkpoint Modulator Targeting the PD-1 Pathway
Pierre Fabre are thus reinforcing their oncology portfolio which already enjoys a combination of chemotherapies, monoclonal antibodies and immuno-conjugates assets at various development phases
Feb 13, 2014, 03:14 ET from Aurigene and Pierre Fabre Pharmaceuticals
CASTRES, France and BANGALORE, India, February 13, 2014 /PRNewswire/ —
Pierre Fabre, the third largest French pharmaceutical company, and Aurigene, a leading biotech company based in India, today announced that the two companies have entered into a collaborative license, development and commercialization agreement granting Pierre Fabre global Worldwide rights (excluding India) to a new immune checkpoint modulator, AUNP-12.
AUNP-12 offers a breakthrough mechanism of action in the PD-1 pathway compared to other molecules currently in development in the highly promising immune therapy cancer space. AUNP-12 is the only peptide therapeutic in this pathway and could offer more effective and safer combination opportunities with emerging and established treatment regimens. AUNP-12 will be in development for numerous cancer indications.
Under the terms of this agreement, Aurigene will receive an upfront payment from Pierre Fabre. Aurigene will also receive additional milestone payments based upon the continued development, regulatory progresses and commercialization of AUNP-12.
“We are pleased that Pierre Fabre see the PD-1 program as a strategic asset in their portfolio. Overall, the deal structure, in line with the financial terms that have been seen in this space, demonstrate the importance that Pierre Fabre attach to the program,” said CSN Murthy, CEO, Aurigene.
“The plans that Pierre Fabre have detailed for the development of this differentiated asset highlight the long-term opportunities for this novel cancer therapeutic,” added Murali Ramachandra, Sr VP, Research, Aurigene.
“This agreement, in the field of oncology, is fully consistent with our vision to build Pierre Fabre’s future in prescription drugs, from a combination of cutting-edge internal R&D capabilities and license partnerships with innovative biotech companies like Aurigene,” stated Bertrand Parmentier, CEO, Pierre Fabre.
“With this deal, Pierre-Fabre Pharmaceuticals are reinforcing their portfolio of oncology assets and capitalizing on their proven capabilities in developing biological compounds such as monoclonal antibodies and immuno-conjugates. We have been impressed by the science at Aurigene and encouraged by the differentiated profile reported for AUNP-12,” added Frédéric Duchesne, President, Pierre Fabre Pharmaceuticals.
About immuno-oncology
Immuno-oncology is an emerging field in cancer therapy, where the body’s own immune system is harnessed to fight against cancer. This approach of targeting cancer through immune response has had a breakthrough when robust and sustained responses were obtained only upon blocking the immune checkpoint targets (such as PD-1 and CTLA4). Recent successes in clinical trials performed with such therapies suggest that immunotherapy should be considered alongside surgery, chemotherapy, radiotherapy and targeted therapy as the fifth cornerstone of cancer treatment.
PD-1 (Programmed cell Death 1) is a receptor that negatively regulates T-cell activation by interacting with specific ligands PD-L1 and PD-L2. Tumor cells express these ligands and thereby escape from the action of T-cells.
About AUNP-12
AUNP-12 is a branched 29-amino acid peptide sequence engineered from the PD-L1/ L2 binding domain of PD-1 It blocks the PD-1/PD-L1, PD-1/PD-L2 and PD-L1/CD80 pathways. AUNP-12 is highly effective in antagonizing PD-1 signaling, with desirable in vivo exposure upon subcutaneous dosing. It inhibits tumor growth and metastasis in preclinical models of cancer and is well tolerated with no overt toxicity at any of the tested doses.
About Aurigene
Aurigene is a biotech focused on development of innovative small molecule and peptide therapeutics for Oncology and Inflammation; key focus areas for Aurigene are Immuno-oncology, Epigenetics and the Th17 pathway. Aurigene’s PD-1 program is the first of several peptide-based immune checkpoint programs that are at different stages of Discovery.
Aurigene has partnered with several big pharma and mid-pharma companies in the US and Europe, and has delivered multiple clinical compounds through these partnerships. With over 500 scientists, Aurigene has collaborated with 6 of the top 10 pharma companies.
Aurigene’s pre-clinical pipeline includes (1) Selective and pan-BET Bromodomain inhibitors (2) RoR gamma reverse agonists (3) EZH2 inhibitors (4) NAMPT inhibitors and (5) Several immune check point peptide inhibitor programs.
For more information: http://aurigene.com/
About Pierre Fabre:
Pierre Fabre is a privately-owned health care company created in 1961 by Mr Pierre Fabre. It is the second largest French independent pharmaceutical group with 2013 sales amounting to about €2 billion (yet to be audited) across 140 countries. The company is structured around two divisions: Pharmaceuticals (Prescription drugs, OTC, Oral care) and Dermo-cosmetics. Prescription drugs are organized around four main franchises: oncology, dermatology, women’s health and neuropsychiatry. Pierre Fabre employs some 10 000 people worldwide, including 1 300 in R&D. The company allocates about 20% of its pharmaceuticals sales to R&D and relies on more than 25 years of experience in the discovery, development and global commercialization of innovative drugs in oncology. Pierre Fabre has a long commitment to oncology and immunology with major R&D centers in France: the Pierre Fabre immunology Centre (CIPF) in Saint Julien en Genevois and the Pierre Fabre Research Institute (IRPF) located on the Toulouse-Oncopole campus which has been officially recognized as a National Center of Excellence for cancer research since 2012.
REFERENCES
http://slideplayer.com/slide/5760496/
P. Sasikumar, R. Shrimali, S. Adurthi, R. Ramachandra, L. Satyam, A. Dhudashiya, D. Samiulla, K. B. Sunilkumar and M. Ramachandra, “A novel peptide therapeutic targeting PD1 immune checkpoint with equipotent antagonism of both ligands and a potential for better management of immune-related adverse events,” Journal for ImmunoTherapy of Cancer, vol. 1, no. Suppl 1, O24, 2013.
P. G. N. Sasikumar, M. Ramachandra, S. K. Vadlamani, K. R. Vemula, L. K. Satyam, K. Subbarao, K. R. Shrimali and S. Kandepudu (Aurigene Discovery Technologies Ltd, Bangalore, India), “Immunosuppression modulating compounds”, US Patent application US 2011/0318373, 29 Dec 2011.
P. G. Sasikumar, L. K. Satyam, R. K. Shrimali, K. Subbarao, R. Ramachandra, S. Vadlamani, A. Reddy, A. Kumar, A. Srinivas, S. Reddy, S. Gopinath, D. S. Samiulla and M. Ramachandra, “Demonstration of anti-tumor efficacy in multiple preclinical cancer models using a novel peptide inhibitor (Aurigene-012) of the PD1 signaling pathway,” Cancer Research, vol. 72, no. 8 Suppl. 1, Abstract 2850, 2012.
P. G. N. Sasikumar, M. Ramachandra, S. K. Vadlamani, K. R. Shrimali and K. Subbarao, “Therapeutic compounds for immunomodulation” (Aurigene Discovery Technologies Ltd, Bangalore, India), PCT Patent Application WO 2012/168944, 13 Dec 2012.
P. G. N. Sasikumar and M. Ramachandra, “Immunomodulating cyclic compounds from the BC loop of human PD1” (Aurigene Discovery Technologies Ltd, Bangalore, India), PCT Patent Application WO/2013/144704, 3 Oct 2013.
P. G. N. Sasikumar, M. Ramachandra and S. S. S. Naremaddepalli, “Peptidomimetic compounds as immunomodulators” (Aurigene Discovery Technologies Ltd, Bangalore, India), US Patent Application US 2013/0237580, 12 Sep 2013.
A. H. Sharpe, M. J. Butte and S. Oyama (Harvard College), “Modulators of immunoinhibitory receptor PD-1, and methods of use thereof”, PCT Patent Application WO/2011/082400, 7 Jul 2011.
M. Cordingley, “Battle of PD-1 blockade is on”, February 7, 2014 : http://discoveryview.ca/battle-of-pd-1-blockade-is-on/ [Accessed 25 February 2014].
Mr. CSN Murthy
Chief Executive Officer, Aurigene Discovery Technologies Ltd.

Mr. CSN Murthy began his career with ICICI Ventures, India’s first Venture Capital fund. He was subsequently a management consultant to the Pharma and Chemical sectors. Later, he worked in the Business Development and General Management functions in Pharmaceutical companies, including as the Chief Operating Officer of Gland Pharma Ltd. CSN holds a Bachelors degree in Chemical Engineering from the Indian Institute of Technology (IIT), Madras and an MBA from the Indian Institute of Management (IIM), Bangalore.
Dr.Thomas Antony
Associate Research Director, Aurigene Discovery Technologies Ltd.

Dr.Thomas Antony did his Ph.D in Biophysical Chemistry from University of Delhi and had his postdoctoral training at Jawaharlal Nehru University- Delhi, The University of Medicine and Dentistry of New Jersey- USA, and Max Planck Institute for Biophysical Chemistry- Germany. He is the recipient of many research fellowships, including Max Planck Fellowship and Humboldt Research Fellowship. He has more than 20 years of research experience. Dr.Thomas has published 24 research papers and he is the co-author of three international patents. His core area of expertise is in assay development and screening. At Aurigene, Dr.Thomas leads the Biochemistry and Structural Biology Divisions. He was the coordinator of Aurigene-University of Malaya collaboration programs.
Dr. Kavitha Nellore
Associate Research Director, Aurigene Discovery Technologies Ltd.

Dr. Kavitha Nellore obtained her PhD in Bioengineering from Pennsylvania State University, USA. During this time, she was a fellow of the Huck’s Institute of Life Sciences specializing in Biomolecular Transport Dynamics. She has been at Aurigene for more than a decade, and is currently leading a group of cell biologists at both Bangalore and Kuala Lumpur. At Aurigene, she leads multiple drug discovery programs in the therapeutic areas of inflammation, oncology and immuno-oncology. She plays a key role in target selection as well as validation efforts to add to Aurigene’s pipeline. Kavitha also played a key role in coordinating the Aurigene-University of Malaya collaboration.
To print: Click here or Select File and then Print from your browser's menu “Strategic partnerships will boost drug discovery activities in India”Wednesday, May 16, 2007 08:00 IST
What are the factors led to the scores of partnerships in research and drug discovery space? Biology demands expertise in areas such as DMPK, cell and assay biology and vivo expertise, which is not as common as chemistry expertise in India. There is also a paradigm shift in assignments for CROs, as they are gearing up to gain higher value from existing projects, including Intellectual Property (IP) generation. Probably, this is where Aurigene has a head start over other CROs. Although we are in research services, our success is in IP generated work. We work with world-class companies like Novo Nordisk, Rheoscience, Debiopharm, Forest Labs and Merck Serono on integrated discovery projects. What do you think is the uniqueness of Aurigene that sets it apart from other companies? It is understood that a major chunk of the business for CROs is from global customers. What is the reason for this? The whole concept of drug discovery outsourcing business has caught on in the last 18-month. Could you give us an overview of the market? In India, CROs operate differently. The innovation driven companies are taking on ‘collaborative drug discovery’ projects, leveraging the mutual strengths each has to offer the other (cost and expertise respectively). Therefore, the complexion of business is different from that of the Western countries. We would see large outsourcing companies positioning themselves as offshore partners for pharma-biotech majors. They could also go in for a BOT (Build Operate Transfer) model. Under this model, companies will set-up the facility and manage it to make it a ‘centre of excellence’, focusing on certain disease segments. Another option is that some companies can take up their own programmes to develop compounds and enter into licensing partnerships. This is similar to the US and Europe biotech style of working. Both of these practices are likely to happen in India. How much do you acknowledge science and strategy in Aurigene’s growth? What according to you is the future of the drug discovery sector? |
 The Bangalore-based Aurigene Discovery Technologies Limited, an independent subsidiary of Dr Reddy’s, is now competing in a mature contract research organization space in the country. Established in 2003, Aurigene Discovery Technologies is a partnership focused collaborative discovery organisation. CSN Murthy is chief executive officer of the company. In an interaction with Nandita Vijay, Murthy cut a clear picture of the contract research scene. Excerpts:
The Bangalore-based Aurigene Discovery Technologies Limited, an independent subsidiary of Dr Reddy’s, is now competing in a mature contract research organization space in the country. Established in 2003, Aurigene Discovery Technologies is a partnership focused collaborative discovery organisation. CSN Murthy is chief executive officer of the company. In an interaction with Nandita Vijay, Murthy cut a clear picture of the contract research scene. Excerpts:
/////////AUNP-12, Aurigene, Pierre Fabre Pharmaceuticals, Licensing Agreement, New Cancer Therapeutic, Immuno-oncology, AUNP 12, Immune Checkpoint Modulator Targeting the PD-1 Pathway, PEPTIDES
FEW MORE COMPDS FROM PATENT
C142 H225 N39 O49
L-Glutamic acid, N2,N6-bis(L-seryl-L-asparaginyl-L-threonyl-L-seryl-L-α-glutamyl-L-seryl-L-phenylalanyl)-L-lysyl-L-phenylalanyl-L-arginyl-L-valyl-L-threonyl-L-glutaminyl-L-leucyl-L-alanyl-L-prolyl-L-lysyl-L-alanyl-L-glutaminyl-L-isoleucyl-L-lysyl-
3262.54, Sequence Length: 29, 22, 7
multichain; modified (modifications unspecified)
SNTSESFK FRVTQ LAPKAQIKE, 1353564-66-5
SNTSESF
C142 H225 N39 O49
L-Glutamic acid, N2,N6-bis(L-seryl-L-asparaginyl-L-threonyl-L-seryl-L-α-glutamyl-L-seryl-L-phenylalanyl)-L-lysyl-L-phenylalanyl-L-arginyl-L-valyl-L-threonyl-L-glutaminyl-L-leucyl-L-alanyl-L-prolyl-L-lysyl-L-alanyl-L-glutaminyl-L-isoleucyl-L-lysyl-
3262.54

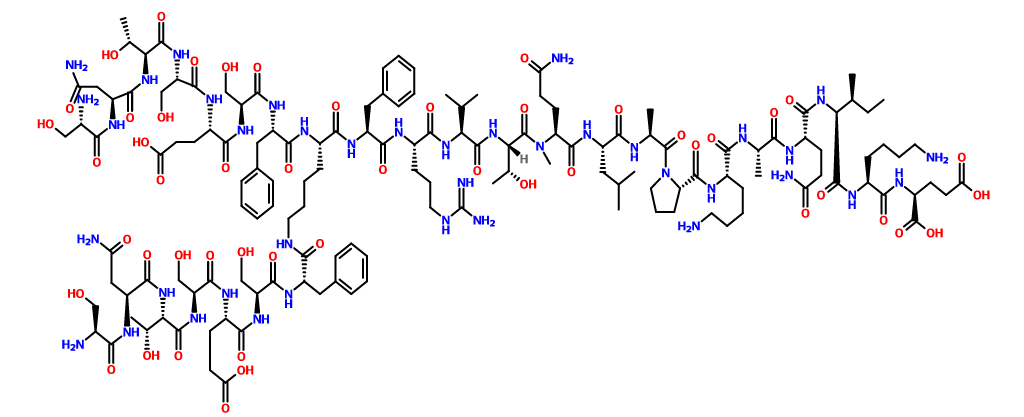
NEXT……………………
SNTSESFK FRVTQ LAPKAQI KE
SNTSESF
CAS 1353564-64-3
C142 H226 N40 O48
L-α-Glutamine, L-seryl-D-asparaginyl-L-threonyl-L-seryl-L-α-glutamyl-L-seryl-L-phenylalanyl-N6-(L-seryl-L-asparaginyl-L-threonyl-L-seryl-L-α-glutamyl-L-seryl-L-phenylalanyl)-L-lysyl-L-phenylalanyl-L-arginyl-L-valyl-L-threonyl-L-glutaminyl-L-leucyl-L-alanyl-L-prolyl-L-lysyl-L-alanyl-L-glutaminyl-L-isoleucyl-L-lysyl-
MW 3261.55, Sequence Length: 29, 22, 7
multichain; modified
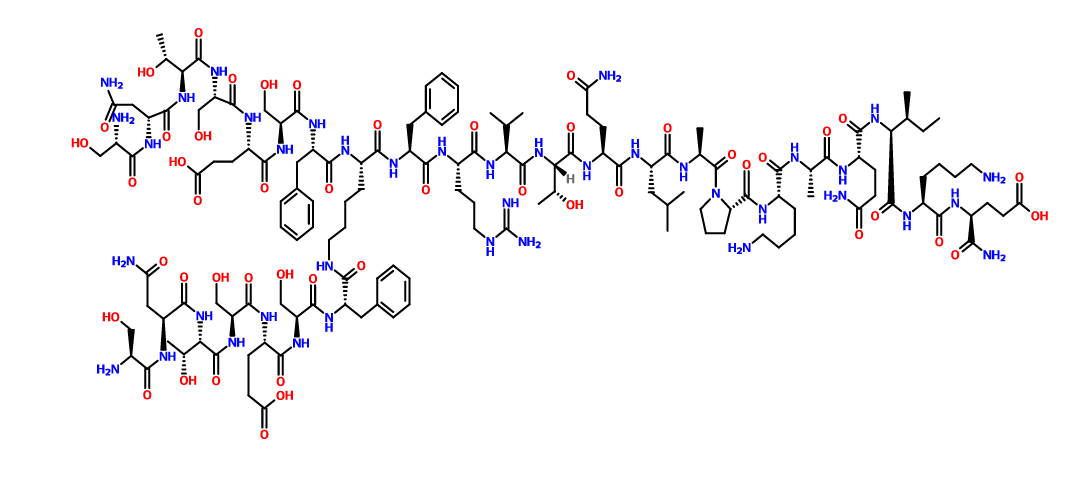

smiles
O=C(N[C@@H](CCCCNC(=O)[C@H](Cc1ccccc1)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(=O)O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(N)=O)NC(=O)[C@@H](N)CO)[C@@H](C)O)C(=O)N[C@@H](Cc2ccccc2)C(=O)N[C@@H](CCCNC(=N)N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C)C(=O)N3CCC[C@H]3C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(=O)O)C(N)=O)[C@H](Cc4ccccc4)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(=O)O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@@H](CC(N)=O)NC(=O)[C@@H](N)CO)[C@@H](C)O
NEXT……………..
CAS 1353564-60-9
C142 H226 N40 O48
L-α-Glutamine, D-seryl-L-asparaginyl-L-threonyl-L-seryl-L-α-glutamyl-L-seryl-L-phenylalanyl-N6-(L-seryl-L-asparaginyl-L-threonyl-L-seryl-L-α-glutamyl-L-seryl-L-phenylalanyl)-L-lysyl-L-phenylalanyl-L-arginyl-L-valyl-L-threonyl-L-glutaminyl-L-leucyl-L-alanyl-L-prolyl-L-lysyl-L-alanyl-L-glutaminyl-L-isoleucyl-L-lysyl-
3261.55
Sequence Length: 29, 22, 7multichain; modified
SNTSESFKFR VTQLAPKAQI KE
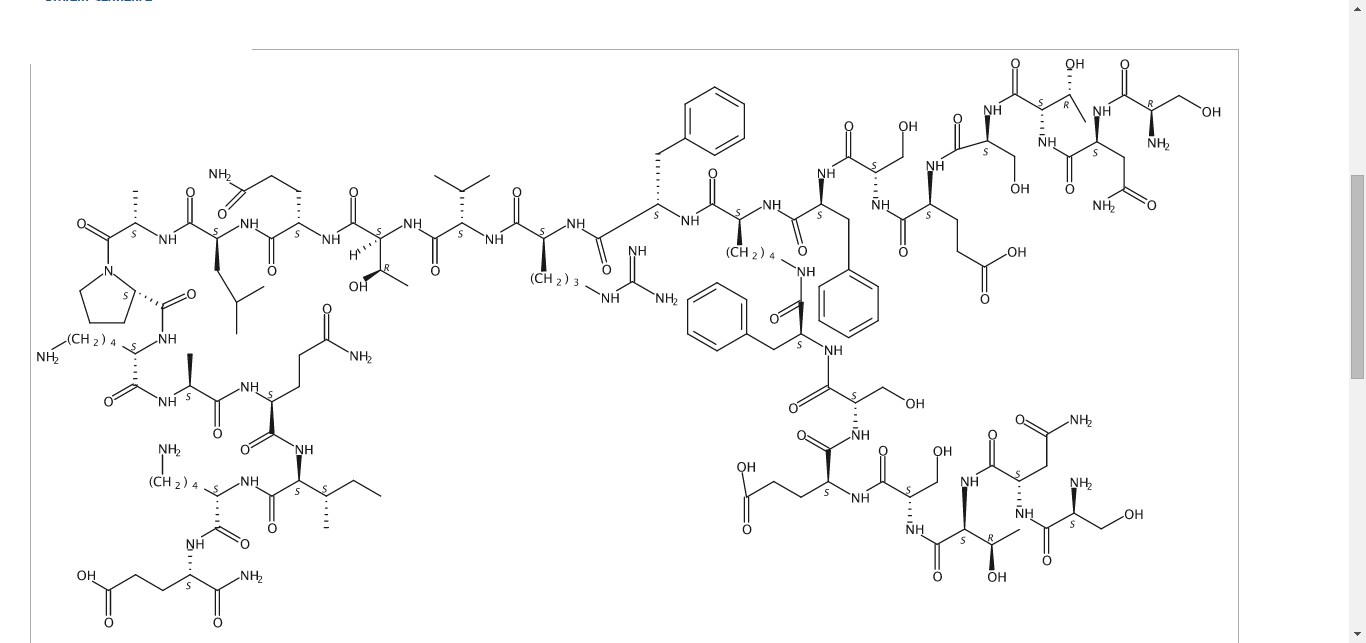
NRXT…………………….
. CAS 1353564-61-0
C142 H226 N40 O48
L-α-Glutamine, N2,N6-bis(D-seryl-L-asparaginyl-L-threonyl-L-seryl-L-α-glutamyl-L-seryl-L-phenylalanyl)-L-lysyl-L-phenylalanyl-L-arginyl-L-valyl-L-threonyl-L-glutaminyl-L-leucyl-L-alanyl-L-prolyl-L-lysyl-L-alanyl-L-glutaminyl-L-isoleucyl-L-lysyl-
3261.55
Sequence Length: 29, 22, 7multichain; modified
SNTSESFK FRVTQ LAPKAQI KE
SNTSESF

/////////////
