
Home » Posts tagged 'hepatocellular carcinoma'
Tag Archives: hepatocellular carcinoma
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
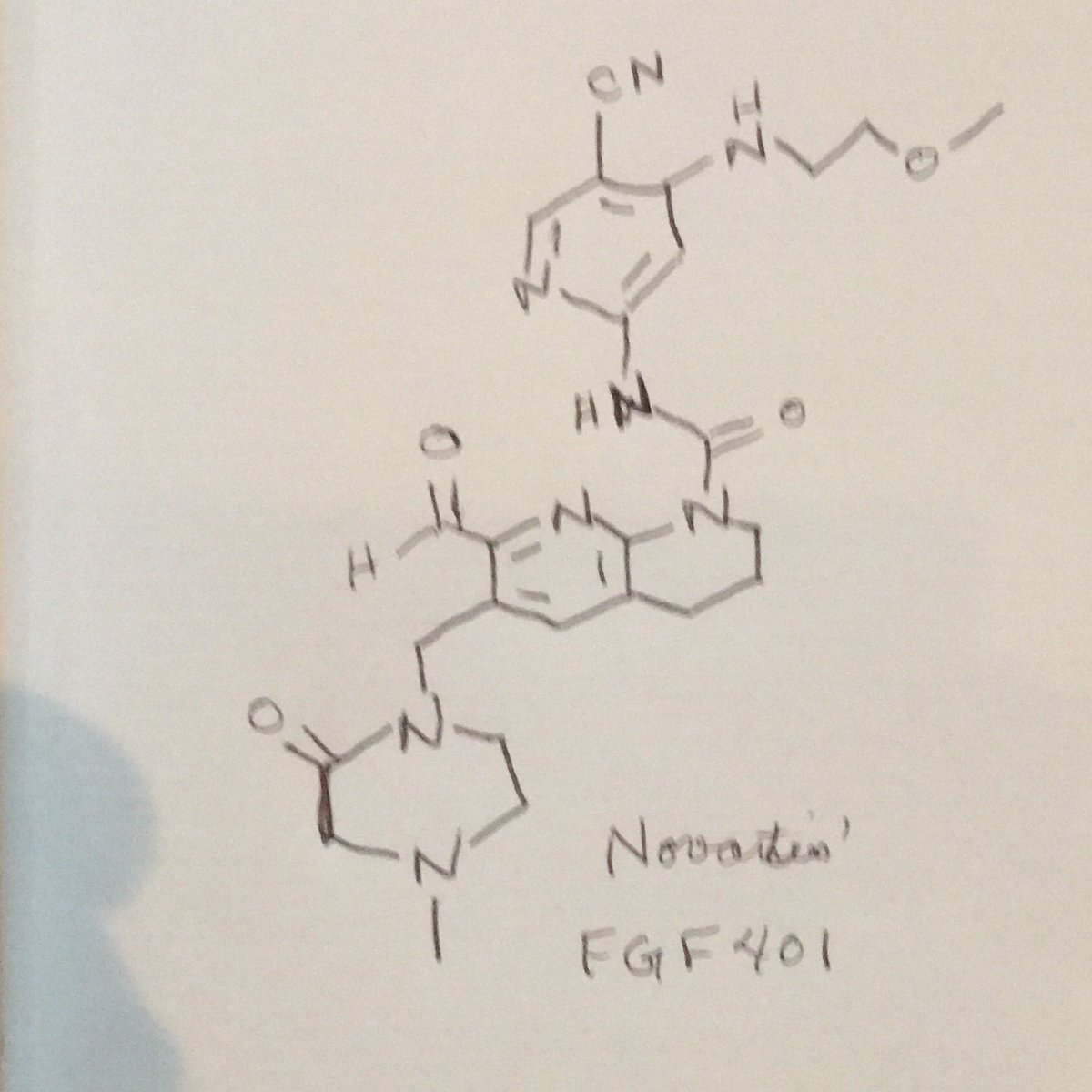
FGF 401


FGF 401
NVP-FGF-401
CAS 1708971-55-4
N-[5-Cyano-4-[(2-methoxyethyl)amino]-2-pyridinyl]-7-formyl-3,4-dihydro-6-[(4-methyl-2-oxo-1-piperazinyl)methyl]-1,8-naphthyridine-1(2H)-carboxamide
/V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide
Phase I/II Hepatocellular carcinoma; Solid tumours
- Originator Novartis
- Developer Novartis Oncology
- Class Antineoplastics
- Mechanism of Action Type 4 fibroblast growth factor receptor antagonists
- 26 Jan 2016 Phase-I/II clinical trials in Solid tumours and Hepatocellular carcinoma in USA, Hong Kong, Japan, Taiwan, France, Germany and Spain (PO)
- 26 Dec 2014 Phase-I/II clinical trials in Hepatocellular carcinoma in Singapore (PO)
- 26 Dec 2014 Phase-I/II clinical trials in Solid tumours in Singapore (PO)
Activation of FGFRs (fibroblast growth factor receptors) has an essential role in regulating cell survival, proliferation, migration and differentiation.1 Dysregulation of the FGFR signaling pathway has been associated with human cancer.1 FGFRs represent an important target for cancer therapeutics because a growing body of evidence indicates that they can act in an oncogenic fashion to promote multiple steps of cancer progression, including induction of mitogenic and survival signals
FGF-401 is a FGFR4 inhibitor in phase I/II clinical studies at Novartis for the treatment of positive FGFR4 and KLB expresion solid tumors and hepatocellular carcinoma
Normal growth, as well as tissue repair and remodeling, require specific and delicate control of activating growth factors and their receptors. Fibroblast Growth Factors (FGFs) constitute a family of over twenty structurally related polypeptides that are developmental^ regulated and expressed in a wide variety of tissues. FGFs stimulate proliferation, cell migration and differentiation and play a major role in skeletal and limb development, wound healing, tissue repair, hematopoiesis, angiogenesis, and tumorigenesis (reviewed in Ornitz, Novartis Found Symp 232: 63-76; discussion 76-80, 272-82 (2001)).
The biological action of FGFs is mediated by specific cell surface receptors belonging to the Receptor Protein Tyrosine Kinase (RPTK) family of protein kinases. These proteins consist of an extracellular ligand binding domain, a single transmembrane domain and an intracellular tyrosine kinase domain which undergoes phosphorylation upon binding of FGF. Four FGFRs have been identified to date: FGFR1 (also called Fig, fms-like gene, fit- 2, bFGFR, N-bFGFR or Cek1 ), FGFR2 (also called Bek-Bacterial Expressed Kinase-, KGFR, Ksam, Ksaml and Cek3), FGFR3 (also called Cek2) and FGFR4. All mature FGFRs share a common structure consisting of an amino terminal signal peptide, three extracellular immunoglobulin-like domains (Ig domain I, Ig domain II, Ig domain III), with an acidic region between Ig domains (the “acidic box” domain), a transmembrane domain, and intracellular kinase domains (Ullrich and Schlessinger, Cell 61 : 203,1990 ; Johnson and Williams (1992) Adv. Cancer Res. 60: 1 -41). The distinct FGFR isoforms have different binding affinities for the different FGF ligands.
Alterations in FGFRs have been associated with a number of human cancers including myeloma, breast, stomach, colon, bladder, pancreatic and hepatocellular carcinomas. Recently, it was reported that FGFR4 may play an important role in liver cancer in particular (PLoS One, 2012, volume 7, 36713). Other studies have also implicated FGFR4 or its ligand FGF19 in other cancer types including breast, glioblastoma, prostate, rhabdomyosarcoma, gastric, ovarian, lung, colon (Int. J. Cancer 1993; 54:378-382; Oncogene 2010; 29:1543-1552; Cancer Res 2010; 70:802-812; Cancer Res 201 1 ; 71 :4550-4561 ; Clin Cancer Res 2004; 10:6169-6178; Cancer Res 2013;
73:2551 -2562; Clin Cancer Res 2012; 18:3780-3790; J. Clin. Invest. 2009; 1 19:3395-3407; Ann Surg Oncol 2010; 17:3354-61 ; Cancer 201 1 ; 1 17:5304-13; Clin Cancer Res 2013; 19:809-820; PNAS 2013; 1 10:12426-12431 ; Oncogene 2008; 27:85-97).
Therapies involving FGFR4 blocking antibodies have been described for instance in
WO2009/009173, WO2007/136893, WO2012/138975, WO2010/026291 , WO2008/052798 and WO2010/004204. WO2014/144737 and WO2014/01 1900 also describe low molecular weight FGFR4 inhibitors.
in spite of numerous treatment options for patients with cancer, there remains a need for effective and safe therapeutic agents and a need for new combination therapies that can be administered for the effective long-term treatment of cancer.
Liver cancer or hepatic cancer is classified as primary liver cancer (i.e. cancer that forms in the tissues of the liver) and secondary liver cancer (i.e. cancer that spreads to the liver from another part of the body). According to the National Cancer Institute at the National Institutes of Health, the number of estimated new cases and deaths from liver and intrahepatic bile duct cancer in the United States in 2014 was 33,190 and 23,000, respectively. Importantly, the percent surviving five years or more after being diagnosed with liver and intrahepatic bile duct cancer is only about 16%.
It has now been found that a combination of /V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide in free form or in pharmaceutically acceptable salt form and at least one further active ingredient, as defined herein, shows synergistic combination activity in an in vitro cell proliferation assay as shown in the experimental section and may therefore be effective for the delay of progression or treatment of a proliferative disease, such as cancer, in particular liver cancer.
| Inventors | Nicole Buschmann, Robin Alec Fairhurst, Pascal Furet, Thomas Knöpfel, Catherine Leblanc, Robert Mah, Pierre NIMSGERN, Sebastien RIPOCHE, Lv LIAO, Jing XIONG, Xianglin ZHAO, Bo Han, Can Wang, |
| Applicant | Novartis Ag |

Nicole Buschmann

Drawn by worlddrugtracker, helping millions………………..
PATENT
WO 2015059668
https://www.google.com/patents/WO2015059668A1?cl=en
PATENT
A/-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1-yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide in citric acid salt form has the following structure:

Example 1 – A/-(5-cvano-4 (2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1-yl)methyl)-3,4-dihvdro-1 ,8-naphthyridine-1 (2H)-carboxamide in citric acid salt form (1 :1).

Step 1 : 2-(dimethoxymethyl)-1 ,8-naphthyridine.
The procedure described in J. Org. Chem., 2004, 69 (6), pp 1959-1966 was used. Into a 20 L 4-necked round-bottom flask was placed 2-aminopyridine-3-carbaldehyde (1000 g, 8.19 mol), 1 , 1-dimethoxypropan-2-one (1257 g, 10.64 mol), ethanol (10 L), and water (2 L). This was followed by the addition of a solution of sodium hydroxide (409.8 g, 10.24 mol) in water (1000 mL) drop wise with stirring at 0-15 °C. The solution was stirred for 3 h at 0-20 °C and then concentrated under vacuum. The resulting solution was extracted with 3×1200 mL of ethyl acetate and the organic layers were combined. The mixture was dried over sodium sulfate and concentrated under vacuum. The residue was washed with 3×300 mL of hexane and the solid was collected by filtration. This resulted in the title compound as a yellow solid. 1 H-NMR (400 MHz, DMSO-cf6) δ 9.1 1 (dd, 1 H), 8.53 (d, 1 H), 8.50 (dd, 1 H), 7.73 (d, 1 H), 7.67 (dd, 1 H), 5.44 (s, 1 H), 3.41 (s, 6H).
Step 2: 7-(dimethoxymethyl)-1 ,2,3,4-tetrahydro-1 ,8-naphthyridine.
The procedure described in J. Org. Chem. , 2004, 69 (6), pp 1959-1966 was used. Into a 5-L pressure tank reactor (5 atm) was placed 2-(dimethoxymethyl)-1 ,8-naphthyridine (200 g, 979 mmol), ethanol (3 L), Pt02 (12 g). The reactor was evacuated and flushed three times with nitrogen, followed by flushing with hydrogen. The mixture was stirred overnight at 23 °C under an
atmosphere of hydrogen. This reaction was repeated four times. The solids were filtered out and the resulting mixture was concentrated under vacuum to give the title compound as a yellow solid. 1 H-NMR (400 MHz, DMSO-d6) δ 7.14 (d, 1 H), 6.51 (d, 1 H), 6.47 – 6.41 (m, 1 H), 4.98 (s, 1 H), 3.28 -3.19 (m, 2H), 3.23 (s, 6H), 2.64 (t, 2H), 1 .73 – 1.79 (m, 2H).
Step 3: 6-bromo-7-(dimethoxymethyl)-1 ,2,3,4-tetrahydro-1 ,8-naphthyridine.
Into a 3 L 4-necked round-bottom flask was placed 7-(dimethoxymethyl)-1 ,2,3, 4-tetrahydro-1 ,8-naphthyridine (1 14.6 g, 550.3mmol) in acetonitrile (2 L). This was followed by the addition of NBS (103 g, 578 mol) in portions with stirring at 25 °C. The resulting solution was stirred for 30 min at 25 °C. The resulting mixture was concentrated under vacuum and the residue was diluted with 1000 mL of diethylether. The mixture was washed with 3×100 mL of ice/water. The aqueous phase was extracted with 2×100 mL of diethylether and the organic layers were combined. The resulting mixture was washed with 1×100 mL of brine, dried over sodium sulfate and concentrated under vacuum to give the title compound as a light yellow solid. LC-MS: (ES, m/z): 286.03 [M+H]+. 1 H-NMR: (300MHz, CDCI3) δ 1 .86 – 1 .94 (2H, m), 2.70 – 2.74 (2H, m), 3.9 – 3.43 (2H, m), 3.47 (6H, s), 5.23 (1 H, s), 5.58 (1 H, s), 7.29 (1 H, s).
Step 4: 2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1 ,8-naphthyridine-3-carbaldehyde.
To a solution of 6-bromo-7-(dimethoxymethyl)-1 ,2,3, 4-tetrahydro-1 ,8-naphthyridine (15.0 g, 52.2 mmol) in THF (400 mL) at -78 °C under argon, was added MeLi (1 .6 M in Et20, 32.6 mL, 52.2 mmol), the solution was stirred for 5 min, then n-BuLi (1 .6 M in hexane, 35.9 mL, 57.5 mmol) was added slowly and the solution was stirred for 20 min. THF (100 mL) was added to the reaction at -78 °C. Subsequently, n-BuLi (1 .6 M in hexane, 49.0 mL, 78 mmol) was added and the reaction mixture was stirred for 20 min, then again n-BuLi (1 .6 M in hexane, 6.53 mL, 10.45 mmol) was added and the mixture was stirred for 10 min at – 78 °C. DMF (2.10 mL, 27.2 mmol) was added and the reaction mixture was stirred at -78 °C for 45 min, then it was allowed to warm to room temperature, poured into sat. aq. NH4CI and extracted twice with DCM. The combined organic phases were dried over Na2S04, filtered and evaporated to give the title compound as an orange oil. (UPLC-MS 3) tR 0.63 min; ESI-MS 237.2 [M+H]+.
Step 5: ethyl 2-((2-((tert-butoxycarbonyl)amino)ethyl)(methyl)amino)acetate.
Ethyl bromoacetate (1.27 mL, 1 1 .48 mmol) was added to a mixture of tert-butyl (2-(methylamino)ethyl)carbamate (2.0 g, 1 1 .48 mmol), triethylamine (4.81 mL) and THF (24 mL) at 0 °C. After stirring 24 h at room temperature the reaction mixture was partitioned between saturated aqueous NaHC03 and DCM, extracted 2x with DCM, the organic layers dried over Na2S04 and
evaporated to give the title compound as a clear pale-yellow oil. 1H NMR (400 MHz, CDCI3) δ 5.20 (s, br, 1 H), 4.18 (q, 2H), 3.24 (s, 2H), 3.22 – 3.16 (m, 2H), 2.65 – 2.61 (m, 2H), 2.38 (s, 3H), 1 .42 (s, 9H), 1 .24 (t, 3H).
Step 6: ethyl 2-((2-aminoethyl)(methyl)amino)acetate dihydrochloride.
Concentrated hydrochloric acid (10 mL) was added to a solution of ethyl 2-((2-((tert-butoxycarbonyl)amino)ethyl)(methyl)amino)acetate (3.05 g, 1 1 .13 mmol) in THF (20 mL) and EtOH (100 mL) at room temperature. After stirring 1 h at room temperature the reaction mixture was evaporated, ethanol (20 mL) added, evaporated, further ethanol (50 mL) added and then stirred at 60 °C for 70 min. The cooled reaction mixture was then evaporated to give the title compound as a pale-yellow glass. 1 H NMR (400 MHz, DMSO-d6) δ 8.58 (s, br, 3H), 4.19 (q, 2H), 4.26 – 4.15 (m, 2H), 3.44 (s, br, 2H), 3.21 (s, br, 2H), 2.88 (s, 3H), 1 .21 (t, 3H).
Step 7: 1 -((2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1 ,8-naphthyridin-3-yl)methyl)-4-methylpiperazin-2-one.
Sodium triacetoxyborohydride (3.10 g, 14.61 mmol) was added to a mixture of 2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1 ,8-naphthyridine-3-carbaldehyde (obtained in step 4, 2.30 g, 9.74 mmol), ethyl 2-((2-aminoethyl)(methyl)amino)acetate dihydrochloride (obtained in step 6, 2.6 g, 14.61 mmol) and triethylamine (6.75 mL, 48.7 mmol) in 1 ,2-dichloroethane (20 mL) at room temperature. The reaction mixture was stirred for 21 h at room temperature and additional sodium triacetoxyborohydride (2.6 g, 9.74 mmol) was added. After a further 4 h stirring at room temperature, again additional sodium triacetoxyborohydride (1 .3 g, 4.87 mmol) was added and the reaction maintained at 4 °C for 2.5 days. The reaction mixture was then warmed to room temperature, saturated aqueous NaHC03 solution added, the mixture extracted with DCM (3x), the combined organic layers dried over Na2S04 and evaporated. The residue was applied to a 120 g RediSep® silica column as a DCM solution and purified by normal phase chromatography, eluting with a gradient from DCM to 10% MeOH in DCM. Product containing fractions were combined and evaporated to give the title compound as an orange foam. 1 H NMR (400 MHz, CDCI3) δ 7.08 (s, 1 H), 5.30 (s, br, 1 H), 5.20 (s, 1 H), 4.69 (s, 2H), 3.44 – 3.34 (m, 2H), 3.40 (s, 6H), 3.22 – 3.15 (m, 2H), 3.24 (s, 2H), 2.71 – 2.64 (m, 2H), 2.58 – 2.50 (m, 2H), 2.31 (s, 3H), 1 .98 – 1.82 (m, 2H). (UPLC-MS 6) tR 0.33; ESI-MS 335.3 [M+H]+.
Step 8: 4-fluoro-5-iodopyridin-2-amine.
A suspension of 4-fluoropyridin-2-amine (336 g, 2.5 mol) and NIS (745 g, 2.75 mol) in MeCN (9 L) was treated with TFA (1 14 g, 1 mol). The reaction mixture was then stirred at room temperature for 8 h. The reaction mixture was diluted with EtOAc (10 L), washed with sat. aq. Na2S203 (2 x 5 L), brine (4 x 5 L). The combined organic layers were dried over Na2S04, filtered and concentrated to get the crude product. The crude product was purified by recrystallization from EtOAc/pentane (1/10) to afford the title compound as a white solid. 1H NMR (400 MHz, DMSO-cf6) δ 8.14 (d, 1 H), 6.45 (s, 2H), 6.33 (d, 1 H).
Step 9: 6-amino-4-fluoronicotinonitrile.
4-fluoro-5-iodopyridin-2-amine (obtained in step 8, 240 g, 1 mol), zinc cyanide (125 g, 1.05 mol), zinc (13 g, 0.2 mol), Pd2(dba)3 (25 g, 25 mmol) and dppf (55 g, 0.1 mol) in DMA (800 mL) were degassed and charged into the round bottom flask under nitrogen. The mixture was stirred at 100 °C for 3 h. The reaction mixture was diluted with 5% NaHC03 (2 L), extracted with EtOAc (4 x 600 mL). The combined organic layers were washed with 5% NaOH (1 L), dried over Na2S04, concentrated to 700 mL. The resulting organic phase was eluted through silica gel column with EtOAc (1.7 L). The combined organic filtrate was washed with 2 M HCI (3 x 800 mL). The pH of the aqueous phase was adjusted to 10 with saturated NaHC03. The aqueous phase was extracted whit DCM (3 x 500 mL). The combined DCM was dried over Na2S04 and concentrated. The residue was further purified by column chromatography (eluted with pentane: EtOAc 10: 1 to 3:2) followed by recrystallization from pentane/EtOAc 3/1 to give the title compound as white solid. 1 H NMR (400 MHz, DMSO-d6) δ 8.40 (d, 1 H), 7.40 (s, 2H), 6.34 (d, 1 H).
Step 10: tert-butyl (4-chloro-5-cyanopyridin-2-yl)carbamate.
A mixture of 2,4-dichloro-5-cyanopyridine (1 Og, 57.8 mmol), fe/f-butyl carbamate (8.2 g, 70.5 mmol), Pd(OAc)2 (0.26 g, 1 .1 mmol), Xantphos (1 .34 g, 2.3mmol) and K2C03 (12 g, 87 mmol) in THF (150 mL) was degassed 3x with nitrogen. The mixture was then heated at 70 °C for 4-5 h and monitored by chromatography until complete conversion. Following completion of the reaction, additional THF (100 mL) was added and heated the mixture at 70 °C for additional 1 h and then cooled to room temperature. The suspension was then filtered through a pad of celite to remove the solid. The filtrate was then concentrated and azotropically distilled with ethyl acetete before filtering to give the title compound. 1 H NMR (DMSO-d6, 400 MHz): δ 10.82 (s, 1 H), 8.79 (s, 1 H), 8.09 (s, 1 H), 1 .49 (s, 9H).
Step 1 1 : fe/f-butyl N-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)carbamate.
A mixture of tert-butyl (4-chloro-5-cyanopyridin-2-yl)carbamate (obtained in step 10, 9.8 g, 38.6 mmol), 2-methoxyethylamine (5.8 g, 77.3 mmol) and DIPEA (6 g, 46.4 mmol) in DMSO (80 mL) was heated at 65-70 °C for 24 h and monitored by chromatography until complete conversion. The
solution was then cooled to room temperature and a white solid precipitated gradually. Water (20 mL) was then added slowly within 1 h. The suspension was stirred for a further 1 h, filtered and dried to give the title compound as a white solid. 1 H NMR (DMSO-d6, 400 MHz): δ 9.87 (s, 1 H), 8.18 (s, 1 H), 7.20 (s, 1 H), 6.86 (s, 9H), 3.51 (t, 2H), 3.36 (t, 2H), 3.28 (s, 3H), 1.47 (s, 9H).
Step 12: 6-amino-4-((2-methoxyethyl)amino)nicotinonitrile.
A solution of 6-amino-4-fluoronicotinonitrile (obtained in step 9, 1 .10 g, 8.02 mmol) in DMA (20 mL) was treated with 2-methoxyethylamine (2.07 mL, 24.1 mmol) and DIPEA (4.20 mL, 24.1 mmol), heated to 50 °C and stirred for 15 h. The reaction mixture was cooled to room temperature and concentrated. The crude material was purified by normal phase chromatography (24 g silica gel cartridge, heptanes/EtOAc 100:0 to 0:100). The product containing fractions were concentrated and dried under vacuum to give the title compound as an off-white solid.
An alternative synthesis of 6-amino-4-((2-methoxyethyl)amino)nicotinonitrile is outlined below:
To tert-butyl N-{5-cyano-4-[(2-methoxyethyl)amino]pyridin-2-yl}carbamate (obtained in step 1 1 , 7g) was added 30-36% aqueous HCI (40 mL), the mixture stirred at room temperature for 30 minutes and monitored by chromatography until complete conversion. The solution was then basified with 20-30% NaOH solution to pH=9-10 and filtered to give a white solid. The solid was added to ethyl acetate (15 mL) and heated to 50-55 °C to form a clear solution. The solution was then cooled to 3-6 °C, stirred for 2-3 h and filtered. The wet cake was then dried to give the title compound as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 7.92 (s, 1 H), 6.39 (s, 2H), 6.15 (t, 1 H), 5.61 (s, 1 H), 3.46 (t, 2H), 3.27 (s, 3H), 3.24 (q, 2H). (UPLC-MS 3) tR 0.62; ESI-MS 193.1 [M+H]+.
Step 13: N-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-(dimethoxymethyl)-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide.
A solution of 6-amino-4-((2-methoxyethyl)amino)nicotinonitrile (obtained in step 12, 481 mg, 2.50 mmol) in anhydrous DMF (1.5 mL) was added drop wise over 10 minutes to a mixture of di(1 H-1 ,2,4-triazol-1 -yl)methanone (410 mg, 2.50 mmol) and DMF (1 .5 mL) cooled at 0 °C. After stirring for 45 minutes at 0 °C the reaction mixture was allowed to warm to room temperature and after a further 90 minutes at room temperature a solution of 1 -((2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1 ,8-naphthyridin-3-yl)methyl)-4-methylpiperazin-2-one (obtained in step 7, 418 mg, 1.00 mmol) in DMF (2 mL) was added. The reaction mixture was stirred for 17.5 h at room temperature, quenched by the addition of MeOH and evaporated. The residue was applied to a 80 g RediSep® silica column as a DCM solution and purified by normal phase chromatography, eluting with a gradient from DCM to 2% MeOH in DCM. Product containing fractions were combined and evaporated to give the title compound as an orange foam. 1H NMR (400 MHz, DMSO-d6) δ 13.50 (s, 1 H), 8.27 (s,
1 H), 7.52 (s, 1 H), 7.39 (s, 1 H), 6.93 (t, 1 H), 5.45 (s, 1 H), 4.65 (s, 2H), 3.94 – 3.89 (m, 2H), 3.54 -3.50 (m, 2H), 3.40 – 3.35 (m, 2H), 3.38 (s, 6H), 3.29 (s, 3H), 3.20 – 3.16 (m, 2H), 3.05 (s, 2H), 2.86 – 2.80 (m, 2H), 2.61 – 2.55 (m, 2H), 2.22 (s, 3H), 1 .94 – 1 .88 (m, 2H). (UPLC-MS 6) tR 0.72; ESI-MS 553.3 [M+H]+.
Step 14: /V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-form
yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide
Concentrated hydrochloric acid (0.40 mL) was added to a solution of A/-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-(dimethoxymethyl)-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (obtained in step 13, 470 mg, 0.808 mmol) in THF (3 mL) and water (1 mL) at room temperature. After stirring for 3 h at room temperature saturated aqueous NaHC03 was added, the mixture extracted with DCM (3x), the organic layers dried over Na2S04 and evaporated. The residue was sonicated with EtOAc (6 mL) and pentane (6 mL) and then filtered. The white solid obtained was then dissolved in DCM (6 mL), EtOAc added (3 mL), the solution warmed, sealed and allowed to stand at room temperature for 2 h. Filtration and drying gave A/-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide as a white solid.
1 H NMR (400 MHz, DMSO-d6) δ 13.43 (s, 1 H), 10.06 (s, 1 H), 8.24 (s, 1 H), 7.49 (s, 1 H), 7.47 (s, 1 H), 6.96 (t, br, 1 H), 4.86 (s, 2H), 3.96 – 3.90 (m, 2H), 3.52 – 3.46 (m, 2H), 3.39 – 3.33 (m, 2H), 3.30 – 3.21 (m, 2H), 3.37 (s, 3H), 3.02 (s, 2H), 2.93 – 2.86 (m, 2H), 2.61 – 2.56 (m, 2H), 2.21 (s, 3H), 1 .95 – 1.85 (m, 2H). (UPLC-MS 6) tR0.70, ESI-MS 507.2, [M+H]+.
Step 15: A/-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide in citric acid form (1 :1 ).
A/-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (obtained in step 14, 4g, 7.896 mmol) was stirred in propionic acid (29.3 g, 29.60mL) at 70 °C until dissolution was complete (20 minutes). The solution was cooled to 55 °C and a solution of citric acid in acetone (23% w/w) was added to it. Separately, a seed suspension was prepared by adding acetone (0.2 g, 0.252mL) to A/-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide in citric acid form (0.0185 g, 0.026 mmol). The seed suspension was added to the solution at 50 °C and the resulting suspension was left to stir at 50 °C for 40 minutes. A further solution of citric acid in acetone (26.6g, 2.51 % w/w, 33.63 mL) was added to the reaction over 380 minutes. The resulting suspension was stirred for a further 120 minutes and cooled to 20 °C with stirring over 4 hours. The suspension was stirred for another 12 hours
before filtering the suspension under vacuum and washing the resulting solid with a propionic acid: acetone solution (1 : 1 , 7g, 7.96ml_) at room temperature. The solid was further washed with acetone (7g, 8.85ml_) at room temperature. The resulting solid was dried in an oven at 40 °C and 5mbar to give the title compound as a light orange solid (5.2g, 7.443 mmol). (mw 698.70), mp (DSC) 168.8 °C (onset).
XRPD analysis showed the same pattern as with particles obtained by a process described in PCT/I B2014/065585 (reference example 1 ) – see Figure 5.
Example 1a
Steps 1 to 14 were carried out as described in example 1 .
Step 15a: A/-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide in citric acid form (1 : 1 )
A/-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (obtained in step 14, 5g, 9.930 mmol) was stirred in propionic acid (33.5 g, 33.84ml_) at 60 °C. Once A/-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide had dissolved, anhydrous citric acid powder (0.19g, 0.9889 mmol) was added. The resulting suspension was heated to 70 °C and sonicated for 5 minutes to ensure full dissolution. The resulting solution was cooled to 50 °C and a solution of citric acid in ethyl acetate (3.7 g, 1 .3% citric acid in ethyl acetate) was added over 20 minutes. Seeds of N-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide in citric acid form (0.02 g) were added to the solution and the suspension was aged for 15 minutes. Another aliquot of citric acid in ethyl acetate (128g, 1 .3% citric acid in ethyl acetate) was added to the suspension over 1 1 .85hours. The suspension was left to stir for over 4 hours. The suspension was then filtered under vacuum (500mbar) and the resulting solid was washed firstly with a propionic acid: ethyl acetate solution (1 : 1 , 7g, 7.44ml_) at room temperature and then with ethyl acetate (12g, 13.38ml_) at room temperature. The resulting solid was dried in an oven at 40 °C and 5mbar to give the title compound as a light orange solid (6.3 g, 9.074 mmol).
XRPD analysis showed the same pattern as with particles obtained by a process described in PCT/I B2014/065585 (reference example 1 ) – see Figure 5.
Reference example 1 (described in PCT/IB2014/065585) – V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihvdro-1 ,8-naphthyridine-1 (2H)-carboxamide in citric acid form (1 :1 )
Steps 1 to 14 were carried out as described in example 1.
Reference Step 15 – /V-(5-cvano-4-((2-methoxyethyl‘)amino‘)pyridin-2-yl‘)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl‘)methyl‘)-3,4-dihvdro-1 ,8-naphthyridine-1 (2H‘)-carboxamide in citric acid form (1 :1 )
A solution of citric acid (96.9 mg) in acetone (5 mL) was prepared at room temperature (0.1 M). A portion of the 0.1 M citric acid in acetone solution (2 mL) was then added to a suspension of Λ/-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (100 mg) in acetone (4 mL) and the mixture sonicated for 1 minute then heated at 55 °C with stirring for 2 h before slowly cooling to room temperature. The white solid was then collected by filtration, washing 2x with acetone (2 mL), and dried for 18 h at 40 °C under vacuum to give the title salt.
Alternatively, N-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (6.5 g, 12.83 mmol) was placed in a 500ml 4-flask reactor. 49 mL of glacial acetic acid was added and the resulting suspension was stirred at 23 °C until a clear mixture was obtained. In a separate flask, anhydrous 2-hydroxypropane-1 ,2,3-tricarboxylic acid (2.59 g, 13.47 mmol, 1 .05 equiv.) was dissolved in 49 mL of glacial acetic acid at 50 °C until a clear solution was obtained. This solution was then added at 23°C to the N-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide solution previously prepared. This mixture was stirred for 30 min at 23 °C and then added dropwise over 1 h to 192 mL of ethyl acetate warmed to 75 °C. The temperature remained constant over the addition. At the end of the addition, the temperature of the mixture was cooled slowly to 23 °C and let 16h at this temperature under gentle stirring. The suspension was cooled to 5-10 °C and filtered. The cake was washed with 15 mL of ethyl acetate and 15 mL of acetone. The wet cake (ca 8.5g) was transferred in a 500 mL flask containing 192 mL of dry acetone. The resulting suspension was refluxed for 24h. The suspension was filtered and the cake was washed with 2 times 15 mL of dry acetone then dried at 50 °C under vacuum for several hours to give the title salt.
PATENT
The synthesis of /V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (abbreviated herein as CPi and also named as Example 83) and salts thereof is disclosed in PCT/IB2014/065585, the content of which are incorporated by reference, as described herein below:
Example 83: /V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide.
Concentrated hydrochloric acid (0.40 ml) was added to a solution of /V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-(dimethoxymethyl)-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (intermediate 80, 470 mg, 0.808 mmol) in THF (3 ml) and water (1 ml) at room temperature. After stirring for 3 h at room temperature saturated aqueous NaHC03 was added, the mixture extracted with DCM (3x), the organic layers dried over Na2S04 and evaporated. The residue was sonicated with EtOAc (6 ml) and pentane (6 ml) and then filtered. The white solid obtained was then dissolved in DCM (6 ml), EtOAc added (3 ml), the solution warmed, sealed and allowed to stand at room temperature for 2 h. Filtration and drying gave the title compound as a white solid.
1H NMR (400 MHz, DMSO-c/6) δ 13.43 (s, 1 H), 10.06 (s, 1 H), 8.24 (s, 1 H), 7.49 (s, 1 H), 7.47 (s, 1 H), 6.96 (t, br, 1 H), 4.86 (s, 2H), 3.96 – 3.90 (m, 2H), 3.52 – 3.46 (m, 2H), 3.39 – 3.33 (m, 2H), 3.30 – 3.21 (m, 2H), 3.37 (s, 3H), 3.02 (s, 2H), 2.93 – 2.86 (m, 2H), 2.61
– 2.56 (m, 2H), 2.21 (s, 3H), 1 .95 – 1 .85 (m, 2H).
(UPLC-MS 6) tR 0.70, ESI-MS 507.2, [M+H]+.
The following salts were prepared from the above free form form of /V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide by precipitation with the appropriate counterions.
Malate with 1 :1 stoichiometry (mw 640.66), mp (DSC) 181 .1 °C (onset): Acetone (2 ml) was added to a mixture of malic acid (26.4 mg, 0.197 mmol) and /V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (100 mg, 0.197 mmol) and the mixture heated on a mini-block with heating-cooling cycles from 55 to 5 °C for 7 repeat cycles (heating rate: 1 .5 °C/min, cooling rate: 0.25 °C/min). The white solid was collected by centrifugation and dried for 18 h at 40 °C to give the title salt.
Tartrate with 1 :0.5 stoichiometry (mw 581 .72), mp (DSC) 176.7 °C (onset). A solution of tartaric acid (75.7 mg) in methanol (5 ml) was prepared at room temperature (0.1 M). A portion of the 0.1 M tartaric acid in acetone solution (2 ml) was then added to a suspension of /V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (100 mg) in methanol (4 ml) and the mixture sonicated for 1 minute then heated at 55 °C with stirring for 2 h. The white solid was then collected by filtration, washing 2x with methanol (2 ml), and dried for 18 h at 40 °C under vacuum to give the title salt.
Tartrate with 1 :1 stoichiometry (mw 656.66), mp (DSC) 169.9 °C (onset): A solution of tartaric acid (75.7 mg) in acetone (5 ml) was prepared at room temperature (0.1 M). A portion of the 0.1 M tartaric acid in acetone solution (2 ml) was then added to a suspension of /V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (100 mg) in methanol (4 ml) and the mixture sonicated for 1 minute then heated at 55 °C with stirring for 2 h. The white solid was then collected by filtration, washing 2x with acetone (2 ml), and dried for 18 h at 40 °C under vacuum to give the title salt.
Citrate with 1 :0.5 stoichiometry (mw 602.73), mp (DSC) 168.4 °C (onset): A solution of citric acid (96.9 mg) in methanol (5 ml) was prepared at room temperature (0.1 M). A portion of the 0.1 M citric acid in methanol solution (2 ml) was then added to a suspension of /V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (100 mg) in methanol (4 ml) and the mixture sonicated for 1 minute then heated at 55 °C with
stirring for 2 h. The white solid was then collected by filtration, washing 2x with acetone (2 ml), and dried for 18 h at 40 °C under vacuum to give the title salt.
Citrate with 1 :1 stoichiometry (mw 698.70), mp (DSC) 168.8 °C (onset): A solution of citric acid (96.9 mg) in acetone (5 ml) was prepared at room temperature (0.1 M). A portion of the 0.1 M citric acid in acetone solution (2 ml) was then added to a suspension of /V-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (100 mg) in acetone (4 ml) and the mixture sonicated for 1 minute then heated at 55 °C with stirring for 2 h before slowly cooling to room temperature. The white solid was then collected by filtration, washing 2x with acetone (2 ml), and dried for 18 h at 40 °C under vacuum to give the title salt.
Alternatively, N-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide (6.5 g, 12.83 mmol) was placed in a 500ml 4-flask reactor. 49 ml of glacial acetic acid was added and the resulting suspension was stirred at 23 °C until a clear mixture was obtained. In a separate flask, anhydrous 2-hydroxypropane-1 ,2,3-tricarboxylic acid (2.59 g, 13.47 mmol, 1 .05 equiv.) was dissolved in 49 ml of glacial acetic acid at 50 °C until a clear solution was obtained. This solution was then added at 23°C to the N-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7-formyl-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide solution previously prepared. This mixture was stirred for 30 min at 23 °C and then added dropwise over 1 h to 192 ml of ethyl acetate warmed to 75 °C. The temperature remained constant over the addition. At the end of the addition, the temperature of the mixture was cooled slowly to 23 °C and let 16h at this temperature under gentle stirring. The suspension was cooled to 5-10 °C and filtered. The cake was washed with 15 ml of ethyl acetate and 15 ml of acetone. The wet cake (ca 8.5g) was transferred in a 500 ml flask containing 192 ml of dry acetone. The resulting suspension was refluxed for 24h. The suspension was filtered and the cake was washed with 2 times 15 ml of dry acetone then dried at 50 °C under vacuum for several hours to give the title salt.
Intermediate 80: N-(5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)-7- (dimethoxymethyl)-6-((4-methyl-2-oxopiperazin-1 -yl)methyl)-3,4-dihydro-1 ,8-naphthyridine-1 (2H)-carboxamide.
A solution of 6-amino-4-((2-methoxyethyl)amino)nicotinonitrile (intermediate 75, 481 mg, 2.50 mmol) in anhydrous DMF (1 .5 ml) was added drop wise over 10 minutes to a mixture of di(1 H-1 ,2,4-triazol-1 -yl)methanone (410 mg, 2.50 mmol) and DMF (1 .5 ml) cooled at 0 °C. After stirring for 45 minutes at 0 °C the reaction mixture was allowed to warm to room temperature and after a further 90 minutes at room temperature a solution of 1 -((2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1 ,8-naphthyridin-3-yl)methyl)-4-methylpiperazin-2-one (intermediate 81 , 418 mg, 1 .00 mmol) in DMF (2 ml) was added. The reaction mixture was stirred for 17.5 h at room temperature, quenched by the addition of MeOH and evaporated. The residue was applied to a 80 g RediSep® silica column as a DCM solution and purified by normal phase chromatography, eluting with a gradient from DCM to 2% MeOH in DCM. Product containing fractions were combined and evaporated to give the title compound as an orange foam. 1H NMR (400 MHz, DMSO-c/6) δ 13.50 (s, 1 H), 8.27 (s, 1 H), 7.52 (s, 1 H), 7.39 (s, 1 H), 6.93 (t, 1 H), 5.45 (s, 1 H), 4.65 (s, 2H), 3.94 – 3.89 (m, 2H), 3.54 – 3.50 (m, 2H), 3.40 – 3.35 (m, 2H), 3.38 (s, 6H), 3.29 (s, 3H), 3.20 – 3.16 (m, 2H), 3.05 (s, 2H), 2.86 – 2.80 (m, 2H), 2.61 – 2.55 (m, 2H), 2.22 (s, 3H), 1 .94 – 1 .88 (m, 2H). (UPLC-MS 6) tR 0.72; ESI-MS 553.3 [M+H]+.
Intermediate 81 : 1 -((2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1 ,8-naphthyridin-3-yl)methyl)-4-methylpiperazin-2-one.
Sodium triacetoxyborohydride (3.10 g, 14.61 mmol) was added to a mixture of 2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1 ,8-naphthyridine-3-carbaldehyde (intermediate 41 , 2.30 g, 9.74 mmol), ethyl 2-((2-aminoethyl)(methyl)amino)acetate dihydrochloride (intermediate 82, 2.6 g, 14.61 mmol) and triethylamine (6.75 ml, 48.7 mmol) in 1 ,2-dichloroethane (20 ml) at room temperature. The reaction mixture was stirred for 21 h at room temperature and additional sodium triacetoxyborohydride (2.6 g, 9.74 mmol) was added. After a further 4 h stirring at room temperature, again additional sodium triacetoxyborohydride (1 .3 g, 4.87 mmol) was added and the reaction maintained at 4 °C for 2.5 days. The reaction mixture was then warmed to room temperature, saturated aqueous NaHC03 solution added, the mixture extracted with DCM (3x), the combined organic layers dried over Na2S04 and evaporated. The residue was applied to a 120 g RediSep® silica column as a DCM solution and purified by normal phase chromatography, eluting with a gradient from DCM to 10% MeOH in DCM. Product containing fractions were combined and evaporated to give the title compound as an orange foam. 1H NMR (400 MHz, CDCI3) δ 7.08 (s, 1 H), 5.30 (s, br, 1 H), 5.20 (s, 1 H), 4.69 (s, 2H), 3.44 – 3.34 (m, 2H), 3.40 (s, 6H), 3.22 – 3.15 (m, 2H), 3.24 (s, 2H), 2.71 -2.64 (m, 2H), 2.58 – 2.50 (m, 2H), 2.31 (s, 3H), 1 .98 – 1 .82 (m, 2H). (UPLC-MS 6) tR 0.33; ESI-MS 335.3 [M+H]+.
Intermediate 82: ethyl 2-((2-aminoethyl)(methyl)amino)acetate dihydrochloride.
Concentrated hydrochloric acid (10 ml) was added to a solution of ethyl 2-((2-((tert-butoxycarbonyl)amino)ethyl)(methyl)amino)acetate (intermediate 83, 3.05 g, 1 1 .13 mmol) in THF (20 ml) and EtOH (100 ml) at room temperature. After stirring 1 h at room temperature the reaction mixture was evaporated, ethanol (20 ml) added, evaporated, further ethanol (50 ml) added and then stirred at 60 °C for 70 min. The cooled reaction
mixture was then evaporated to give the title compound as a pale-yellow glass. 1H NMR (400 MHz, DMSO-c/6) δ 8.58 (s, br, 3H), 4.19 (q, 2H), 4.26 – 4.15 (m, 2H), 3.44 (s, br, 2H), 3.21 (s, br, 2H), 2.88 (s, 3H), 1 .21 (t, 3H).
Intermediate 83: ethyl 2-((2-((tert-butoxycarbonyl)amino)ethyl)(methyl)amino)acetate.
Ethyl bromoacetate (1 .27 ml, 1 1 .48 mmol) was added to a mixture of tert-butyl (2-(methylamino)ethyl)carbamate (2.0 g, 1 1 .48 mmol), triethylamine (4.81 ml) and THF (24 ml) at 0 °C. After stirring 24 h at room temperature the reaction mixture was partitioned between saturated aqueous NaHC03 and DCM, extracted 2x with DCM, the organic layers dried over Na2S04 and evaporated to give the title compound as a clear pale-yellow oil. 1 H NMR (400 MHz, CDCI3) δ 5.20 (s, br, 1 H), 4.18 (q, 2H), 3.24 (s, 2H), 3.22 -3.16 (m, 2H), 2.65 – 2.61 (m, 2H), 2.38 (s, 3H), 1 .42 (s, 9H), 1 .24 (t, 3H).
Intermediate 41 : 2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1 ,8-naphthyridine-3-carbaldehyde.
To a solution of 6-bromo-7-(dimethoxymethyl)-1 ,2,3,4-tetrahydro-1 ,8-naphthyridine
(intermediate 12, 15.0 g, 52.2 mmol) in THF (400 ml) at -78 °C under argon, was added MeLi (1 .6 M in Et20, 32.6 ml, 52.2 mmol), the solution was stirred for 5 min, then n-BuLi (1 .6 M in hexane, 35.9 ml, 57.5 mmol) was added slowly and the solution was stirred for 20 min. THF (100 ml) was added to the reaction at – 78 °C. Subsequently, n-BuLi (1 .6 M in hexane, 49.0 ml, 78 mmol) was added and the reaction mixture was stirred for 20 min, then again n-BuLi (1 .6 M in hexane, 6.53 ml, 10.45 mmol) was added and the mixture was stirred for 10 min at – 78 °C. DMF (2.10 ml, 27.2 mmol) was added and the reaction mixture was stirred at -78 °C for 45 min, then it was allowed to warm to room
temperature, poured into sat. aq. NH4CI and extracted twice with DCM. The combined organic phases were dried over Na2S04, filtered and evaporated to give the title compound as an orange oil. (UPLC-MS 3) tR 0.63 min; ESI-MS 237.2 [M+H]+.
Intermediate 12: 6-bromo-7-(dimethoxymethyl)-1 ,2,3,4-tetrahydro-1 ,8-naphthyridine.
Into a 3 I 4-necked round-bottom flask was placed 7-(dimethoxymethyl)-1 ,2,3,4-tetrahydro-1 ,8-naphthyridine (intermediate 4, 1 14.6 g, 550.3mmol) in acetonitrile (2 I). This was followed by the addition of NBS (103 g, 578 mol) in portions with stirring at 25 °C. The resulting solution was stirred for 30 min at 25 °C. The resulting mixture was concentrated under vacuum and the residue was diluted with 1000 ml of diethylether. The mixture was washed with 3×100 ml of ice/water. The aqueous phase was extracted with 2×100 ml of diethylether and the organic layers were combined. The resulting mixture was washed with 1 x100 ml of brine, dried over sodium sulfate and concentrated under vacuum to give the title compound as a light yellow solid. LC-MS: (ES, m/z):
286.03 [M+H]+. 1H-NMR: (300MHz, CDCI3) δ 1 .86 – 1 .94 (2H, m), 2.70 – 2.74 (2H, m), 3.9 – 3.43 (2H, m), 3.47 (6H, s), 5.23 (1 H, s), 5.58 (1 H, s), 7.29 (1 H, s).
Intermediate 4: 7-(dimethoxymethyl)-1 ,2,3,4-tetrahydro-1 ,8-naphthyridine.
The procedure described in J. Org. Chem. , 2004, 69 (6), pp 1959-1966 was used. Into a 5-I pressure tank reactor (5 atm) was placed 2-(dimethoxymethyl)-1 ,8-naphthyridine (intermediate 5, 200 g, 979 mmol), ethanol (3 I), Pt02 (12 g). The reactor was evacuated and flushed three times with nitrogen, followed by flushing with hydrogen. The mixture was stirred overnight at 23 °C under an atmosphere of hydrogen. This reaction was repeated four times. The solids were filtered out and the resulting mixture was concentrated under vacuum to give the title compound as a yellow solid.
Intermediate 5: 2-(dimethoxymethyl)-1 ,8-naphthyridine.
The procedure described in J. Org. Chem. , 2004, 69 (6), pp 1959-1966 was used. Into a 20 I 4-necked round-bottom flask was placed 2-aminopyridine-3-carbaldehyde (1000 g, 8.19 mol), 1 ,1 -dimethoxypropan-2-one (1257 g, 10.64 mol), ethanol (10 I), and water (2 I). This was followed by the addition of a solution of sodium hydroxide (409.8 g, 10.24 mol) in water (1000 ml) drop wise with stirring at 0-15 °C. The solution was stirred for 3 h at 0-20 °C and then concentrated under vacuum. The resulting solution was extracted with 3×1200 ml of ethyl acetate and the organic layers were combined. The mixture was dried over sodium sulfate and concentrated under vacuum. The residue was washed with 3×300 ml of hexane and the solid was collected by filtration. This resulted in the title compound as a yellow solid. 1H-NMR (400 MHz, DMSO-c/6) δ 9.1 1 (dd, 1 H), 8.53 (d, 1 H), 8.50 (dd, 1 H), 7.73 (d, 1 H), 7.67 (dd, 1 H), 5.44 (s, 1 H), 3.41 (s, 6H).
Intermediate 75: 6-amino-4-((2-methoxyethyl)amino)nicotinonitrile.
A solution of 6-amino-4-fluoronicotinonitrile (intermediate 21 , 1 .10 g, 8.02 mmol) in DMA (20 ml) was treated with 2-methoxyethylamine (2.07 ml, 24.1 mmol) and DIPEA (4.20 ml_, 24.1 mmol), heated to 50 °C and stirred for 15 h. The reaction mixture was cooled to room temperature and concentrated. The crude material was purified by normal phase chromatography (24 g silica gel cartridge, heptanes/EtOAc 100:0 to 0:100). The product containing fractions were concentrated and dried under vacuum to give the title compound as an off-white solid.
An alternative synthesis of 6-amino-4-((2-methoxyethyl)amino)nicotinonitrile is outlined below:
To fe/ -butyl N-{5-cyano-4-[(2-methoxyethyl)amino]pyridin-2-yl}carbamate (intermediate 287, 7g) was added 30-36% aqueous HCI (40 ml), the mixture stirred at room temperature for 30 minutes and monitored by chromatography until complete conversion. The solution was then basified with 20-30% NaOH solution to pH=9-10 and filtered to give a white solid. The solid was added to ethyl acetate (15 ml) and heated to 50-55 °C to form a clear solution. The solution was then cooled to 3-6 °C, stirred for 2-3 h and filtered. The wet cake was then dried to give the title compound as a white solid. 1H NMR (400 MHz, DMSO-c/6) δ 7.92 (s, 1 H), 6.39 (s, 2H), 6.15 (t, 1 H), 5.61 (s, 1 H), 3.46 (t, 2H), 3.27 (s, 3H), 3.24 (q, 2H). (UPLC-MS 3) tR 0.62; ESI-MS 193.1 [M+H]+.
1H-NMR (400 MHz, DMSO-c/6) δ 7.14 (d, 1 H), 6.51 (d, 1 H), 6.47 – 6.41 (m, 1 H), 4.98 (s, 1 H), 3.28 – 3.19 (m, 2H), 3.23 (s, 6H), 2.64 (t, 2H), 1 .73 – 1 .79 (m, 2H).
Intermediate 21 : 6-amino-4-fluoronicotinonitrile.
4-fluoro-5-iodopyridin-2-amine (intermediate 22, 240 g, 1 mol), zinc cyanide (125 g, 1 .05 mol), zinc (13 g, 0.2 mol), Pd2(dba)3 (25 g, 25 mmol) and dppf (55 g, 0.1 mol) in DMA (800 ml) were degassed and charged into the round bottom flask under nitrogen. The mixture was stirred at 100 °C for 3 h. The reaction mixture was diluted with 5% NaHC03 (2 I), extracted with EtOAc (4 x 600 ml). The combined organic layers were washed with 5% NaOH (1 I), dried over Na2S04, concentrated to 700 ml. The resulting organic phase was eluted through silica gel column with EtOAc (1 .7 I). The combined organic filtrate was washed with 2 M HCI (3 x 800 ml). The pH of the aqueous phase was adjusted to 10 with saturated NaHC03. The aqueous phase was extracted whit DCM (3 x 500 ml). The combined DCM was dried over Na2S04 and concentrated. The residue was further purified by column chromatography (eluted with pentane: EtOAc 10:1 to 3:2) followed by recrystallization from pentane/EtOAc 3/1 to give the title compound as white solid. 1H NMR (400 MHz, DMSO-c/6) δ 8.40 (d, 1 H), 7.40 (s, 2H), 6.34 (d, 1 H).
Intermediate 22: 4-fluoro-5-iodopyridin-2-amine.
A suspension of 4-fluoropyridin-2-amine (336 g, 2.5 mol) and NIS (745 g, 2.75 mol) in MeCN (9 I) was treated with TFA (1 14 g, 1 mol). The reaction mixture was then stirred at room temperature for 8 h. The reaction mixture was diluted with EtOAc (10 I), washed with sat. aq. Na2S203 (2 x 5 I), brine (4 x 5 I). The combined organic layers were dried over Na2S04, filtered and concentrated to get the crude product. The crude product was purified by recrystallization from EtOAc/pentane (1/10) to afford the title compound as a white solid. 1H NMR (400 MHz, DMSO-c/6) δ 8.14 (d, 1 H), 6.45 (s, 2H), 6.33 (d, 1 H).
Intermediate 287: fe/ -butyl (5-cyano-4-((2-methoxyethyl)amino)pyridin-2-yl)carbamate.
A mixture of tert-butyl (4-chloro-5-cyanopyridin-2-yl)carbamate (intermediate 288, 9.8 g, 38.6 mmol), 2-methoxyethylamine (5.8 g, 77.3 mmol) and DIPEA (6 g, 46.4 mmol) in DMSO (80 ml) was heated at 65-70 °C for 24 h and monitored by chromatography until complete conversion. The solution was then cooled to room temperature and a white solid precipitated gradually. Water (20 ml) was then added slowly within 1 h. The suspension was stirred for a further 1 h, filtered and dried to give the title compound as a white solid. 1H NMR (DMSO-d6, 400 MHz): δ 9.87 (s, 1 H), 8.18 (s, 1 H), 7.20 (s, 1 H), 6.86 (s, 9H), 3.51 (t, 2H), 3.36 (t, 2H), 3.28 (s, 3H), 1 .47 (s, 9H).
Intermediate 288: tert-butyl (4-chloro-5-cyanopyridin-2-yl)carbamate.
A mixture of 2,4-dichloro-5-cyanopyridine (10g, 57.8 mmol), fe/ -butyl carbamate (8.2 g, 70.5 mmol), Pd(OAc)2 (0.26 g, 1 .1 mmol), Xantphos (1 .34 g, 2.3mmol) and K2C03 (12 g, 87 mmol) in THF (150 ml) was degassed 3x with nitrogen. The mixture was then heated at 70 °C for 4-5 h and monitored by chromatography until complete conversion. Following completion of the reaction, additional THF (100 ml) was added and heated the mixture at 70 °C for additional 1 h and then cooled to room temperature. The suspension was then filtered through a pad of celite to remove the solid. The filtrate was then concentrated and azotropically distilled with ethyl acetete before filtering to give the title compound. 1H NMR (DMSO-d6, 400 MHz): δ 10.82 (s, 1 H), 8.79 (s, 1 H), 8.09 (s, 1 H), 1 .49 (s, 9H).
/////////////FGF 401, 1708971-55-4, PHASE 1, Hepatocellular carcinoma, Solid tumours, Novartis, Novartis Oncology, Antineoplastics, Type 4 fibroblast growth factor receptor antagonists, NVP-FGF-401, Nicole Buschmann, Robin Alec Fairhurst, Pascal Furet, Thomas Knöpfel, Catherine Leblanc, Robert Mah, Pierre NIMSGERN, Sebastien RIPOCHE, Lv LIAO, Jing XIONG, Xianglin ZHAO, Bo Han, Can Wang,
Now in #MEDI 1st time disclosures Robin Fairhurst of @Novartis will also talk about an FGFR inhibitor. They are popular! #ACSSanFran
CN4CC(=O)N(Cc1cc(C=O)nc2N(CCCc12)C(=O)Nc3cc(NCCOC)c(C#N)cn3)CC4
Bayer HealthCare has obtained approval from the Japanese Ministry of Health, Labour and Welfare (MHLW) for its Nexavar (sorafenib) for treatment of patients with unresectable differentiated thyroid carcinoma.

Sorafenib
(4-(4-(3-(4-chloro-3-(trifluoromethyl)phenyl)ureido)phenoxy)-N-methylpicolinamide)
BAY 43-9006

Bayer HealthCare has obtained approval from the Japanese Ministry of Health, Labour and Welfare (MHLW) for its Nexavar (sorafenib) for treatment of patients with unresectable differentiated thyroid carcinoma.
Bayer HealthCare has obtained approval from the Japanese Ministry of Health, Labour and Welfare (MHLW) for its Nexavar (sorafenib) for treatment of patients with unresectable differentiated thyroid carcinoma.
Nexavar’s approval in Japan is supported by data from the multicentre, placebo-controlled Phase III DECISION (‘stuDy of sorafEnib in loCally advanced or metastatIc patientS with radioactive Iodine refractory thyrOid caNcer’) study.
The international Phase III DECISION study, which randomised a total of 417 patients, met its primary endpoint of extended progression-free survival. Safety and tolerability profile of sorafenib was generally consistent with the known profile of sorafenib.
The most common treatment-emergent adverse events in the sorafenib arm were hand-foot skin reaction, diarrhea, alopecia, weight loss, fatigue, hypertension and rash.
Nexavar was awarded orphan drug status by the MHLW for thyroid carcinoma in September 2013.
Sorafenib (co-developed and co-marketed by Bayer and Onyx Pharmaceuticals as Nexavar),[1] is a drug approved for the treatment of primary kidney cancer (advanced renal cell carcinoma), advanced primary liver cancer (hepatocellular carcinoma), and radioactive iodine resistant advanced thyroid carcinoma.
Medical uses
At the current time sorafenib is indicated as a treatment for advanced renal cell carcinoma (RCC), unresectable hepatocellular carcinomas (HCC) and thyroid cancer.[2][3][4][5]
Kidney cancer
An article in The New England Journal of Medicine, published January 2007, showed compared with placebo, treatment with sorafenib prolongs progression-free survival in patients with advanced clear cell renal cell carcinoma in whom previous therapy has failed. The median progression-free survival was 5.5 months in the sorafenib group and 2.8 months in the placebo group (hazard ratio for disease progression in the sorafenib group, 0.44; 95% confidence interval [CI], 0.35 to 0.55; P<0.01).[6] A few reports described patients with stage IV renal cell carcinomas that were successfully treated with a multimodal approach including neurosurgical, radiation, and sorafenib.[7] This is one of two TGA-labelled indications for sorafenib, although it is not listed on the Pharmaceutical Benefits Scheme for this indication.[5][8]
Liver cancer
At ASCO 2007, results from the SHARP trial[9] were presented, which showed efficacy of sorafenib in hepatocellular carcinoma. The primary endpoint was median overall survival, which showed a 44% improvement in patients who received sorafenib compared to placebo (hazard ratio 0.69; 95% CI, 0.55 to 0.87; p=0.0001). Both median survival and time to progression showed 3-month improvements. There was no difference in quality of life measures, possibly attributable to toxicity of sorafenib or symptoms related to underlying progression of liver disease. Of note, this trial only included patients with Child-Pugh Class A (i.e. mildest) cirrhosis. The results of the study appear in the July 24, 2008, edition of The New England Journal of Medicine. Because of this trial Sorafenib obtained FDA approval for the treatment of advanced hepatocellular carcinoma in November 2007.[10]
In a randomized, double-blind, phase II trial combining sorafenib with doxorubicin, the median time to progression was not significantly delayed compared with doxorubicin alone in patients with advanced hepatocellular carcinoma. Median durations of overall survival and progression-free survival were significantly longer in patients receiving sorafenib plus doxorubicin than in those receiving doxorubicin alone.[10] A prospective single-centre phase II study which included the patients with unresectable hepatocellular carcinoma (HCC)concluding that the combination of sorafenib and DEB-TACE in patients with unresectable HCC is well tolerated and safe, with most toxicities related to sorafenib.[11] This is the only indication for which sorafenib is listed on the PBS and hence the only Government-subsidised indication for sorafenib in Australia.[8] Along with renal cell carcinoma, hepatocellular carcinoma is one of the TGA-labelled indications for sorafenib.[5]
Thyroid cancer
A phase 3 clinical trial has started recruiting (November 2009) to use sorafenib for non-responsive thyroid cancer.[12] The results were presented at the ASCO 13th Annual Meeting and are the base for FDA approval. The Sorafenib in locally advanced or metastatic patients with radioactive iodine-refractory differentiated thyroid cancer: The Phase 3 DECISION trial showed significant improvement in progression-free survival but not in overall survival. However, as is known, the side effects were very frequent, specially hand and foot skin reaction.[13]
Adverse effects
Adverse effects by frequency
Note: Potentially serious side effects are in bold.
Very common (>10% frequency)
- Lymphopenia
- Hypophosphataemia[Note 1]
- Haemorrhage[Note 2]
- Hypertension[Note 3]
- Diarrhea
- Rash
- Alopecia (hair loss; occurs in roughly 30% of patients receiving sorafenib)
- Hand-foot syndrome
- Pruritus (itchiness)
- Erythema
- Increased amylase
- Increased lipase
- Fatigue
- Pain[Note 4]
- Nausea
- Vomiting[Note 5][14]
Common (1-10% frequency)
- Leucopoenia[Note 6]
- Neutropoenia[Note 7]
- Anaemia[Note 8]
- Thrombocytopenia[Note 9]
- Anorexia (weight loss)
- Hypocalcaemia[Note 10]
- Hypokalaemia[Note 11]
- Depression
- Peripheral sensory neuropathy
- Tinnitus[Note 12]
- Congestive heart failure
- Myocardial infarction[Note 13]
- Myocardial ischaemia[Note 14]
- Hoarseness
- Constipation
- Stomatitis[Note 15]
- Dyspepsia[Note 16]
- Dysphagia[Note 17]
- Dry skin
- Exfoliative dermatitis
- Acne
- Skin desquamation
- Arthralgia[Note 18]
- Myalgia[Note 19]
- Renal failure[Note 20]
- Proteinuria[Note 21]
- Erectile dysfunction
- Asthenia (weakness)
- Fever
- Influenza-like illness
- Transient increase in transaminase
Uncommon (0.1-1% frequency)
- Folliculitis
- Infection
- Hypersensitivity reactions[Note 22]
- Hypothyroidism[Note 23]
- Hyperthyroidism[Note 24]
- Hyponatraemia[Note 25]
- Dehydration
- Reversible posterior leukoencephalopathy
- Hypertensive crisis
- Rhinorrhoea[Note 26]
- Interstitial lung disease-like events[Note 27]
- Gastro-oesophageal reflux disease (GORD)
- Pancreatitis[Note 28]
- Gastritis[Note 29]
- Gastrointestinal perforations[Note 30]
- Increase in bilirubin leading, potentially, to jaundice[Note 31]
- Cholecystitis[Note 32]
- Cholangitis[Note 33]
- Eczema
- Erythema multiforme[Note 34]
- Keratoacanthoma[Note 35]
- Squamous cell carcinoma
- Gynaecomastia (swelling of the breast tissue in men)
- Transient increase in blood alkaline phosphatase
- INR abnormal
- Prothrombin level abnormal
- bulbous skin reaction[15]
Rare (0.01-0.1% frequency)
Mechanism of action
Sorafenib is a small molecular inhibitor of several tyrosine protein kinases (VEGFR and PDGFR) and Raf kinases (more avidly C-Raf than B-Raf).[16][17] Sorafenib also inhibits some intracellular serine/threonine kinases (e.g. C-Raf, wild-type B-Raf and mutant B-Raf).[10] Sorafenib treatment induces autophagy,[18] which may suppress tumor growth. However, autophagy can also cause drug resistance.[19]
History
Renal cancer
Sorafenib was approved by the U.S. Food and Drug Administration (FDA) in December 2005,[20] and received European Commission marketing authorization in July 2006,[21] both for use in the treatment of advanced renal cancer.
Liver cancer
The European Commission granted marketing authorization to the drug for the treatment of patients with hepatocellular carcinoma(HCC), the most common form of liver cancer, in October 2007,[22] and FDA approval for this indication followed in November 2007.[23]
In November 2009, the UK’s National Institute of Clinical Excellence declined to approve the drug for use within the NHS in England, Wales and Northern Ireland, stating that its effectiveness (increasing survival in primary liver cancer by 6 months) did not justify its high price, at up to £3000 per patient per month.[24] In Scotland the drug had already been refused authorization by the Scottish Medicines Consortium for use within NHS Scotland, for the same reason.[24]
In March 2012, the Indian Patent Office granted a domestic company, Natco Pharma, a license to manufacture generic Sorafenib, bringing its price down by 97%. Bayer sells a month’s supply, 120 tablets, of Nexavar for![]() 280000 (US$4,700). Natco Pharma will sell 120 tablets for
280000 (US$4,700). Natco Pharma will sell 120 tablets for ![]() 8800 (US$150), while still paying a 6% royalty to Bayer.[25][26] Under Indian Patents Act, 2005 and the World Trade Organisation TRIPS Agreement, the government can issue a compulsory license when a drug is not available at an affordable price.[27]
8800 (US$150), while still paying a 6% royalty to Bayer.[25][26] Under Indian Patents Act, 2005 and the World Trade Organisation TRIPS Agreement, the government can issue a compulsory license when a drug is not available at an affordable price.[27]
Thyroid Cancer
As of November 22, 2013, sorafenib has been approved by the FDA for the treatment of locally recurrent or metastatic, progressive differentiated thyroid carcinoma (DTC) refractory to radioactive iodine treatment.[28]
Research
Lung
In some kinds of lung cancer (with squamous-cell histology) sorafenib administered in addition to paclitaxel and carboplatin may be detrimental to patients.[29]
Brain (Recurrent Glioblastoma)
There is a phase I/II study at the Mayo Clinic[30] of sorafenib and CCI-779 (temsirolimus) for recurrent glioblastoma.
Desmoid Tumor (Aggressive Fibromatosis)
A study performed in 2011 showed that Sorafenib is active against Aggressive fibromatosis. This study is being used as justification for using Sorafenib as an initial course of treatment in some patients with Aggressive fibromatosis.[31]
Nexavar Controversy
In January 2014, Bayer’s CEO stated that Nexavar was developed for “western patients who [could] afford it”. At the prevailing prices, a kidney cancer patient would pay $96,000 (£58,000) for a year’s course of the Bayer-made drug. However, the cost of the Indian version of the generic drug would be around $2,800 (£1,700).[32]
Notes
- Low blood phosphate levels
- Bleeding; including serious bleeds such as intracranial and intrapulmonary bleeds
- High blood pressure
- Including abdominal pain, headache, tumour pain, etc.
- Considered a low (~10-30%) risk chemotherapeutic agent for causing emesis)
- Low level of white blood cells in the blood
- Low level of neutrophils in the blood
- Low level of red blood cells in the blood
- Low level of plasma cells in the blood
- Low blood calcium
- Low blood potassium
- Hearing ringing in the ears
- Heart attack
- Lack of blood supply for the heart muscle
- Mouth swelling, also dry mouth and glossodynia
- Indigestion
- Not being able to swallow
- Sore joints
- Muscle aches
- Kidney failure
- Excreting protein [usually plasma proteins] in the urine. Not dangerous in itself but it is indicative kidney damage
- Including skin reactions and urticaria (hives)
- Underactive thyroid
- Overactive thyroid
- Low blood sodium
- Runny nose
- Pneumonitis, radiation pneumonitis, acute respiratory distress, etc.
- Swelling of the pancreas
- Swelling of the stomach
- Formation of a hole in the gastrointestinal tract, leading to potentially fatal bleeds
- Yellowing of the skin and eyes due to a failure of the liver to adequately cope with the amount of bilirubin produced by the day-to-day actions of the body
- Swelling of the gallbladder
- Swelling of the bile duct
- A potentially fatal skin reaction
- A fairly benign form of skin cancer
- A potentially fatal abnormality in the electrical activity of the heart
- Swelling of the skin and mucous membranes
- A potentially fatal allergic reaction
- Swelling of the liver
- A potentially fatal skin reaction
- A potentially fatal skin reaction
- The rapid breakdown of muscle tissue leading to the build-up of myoglobin in the blood and resulting in damage to the kidneys
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
| 4-[4-[[4-chloro-3-(trifluoromethyl)phenyl]carbamoylamino] phenoxy]-N-methyl-pyridine-2-carboxamide |
|
| Clinical data | |
| Trade names | Nexavar |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a607051 |
| Licence data | EMA:Link, US FDA:link |
| Pregnancy cat. | D (AU) D (US) |
| Legal status | Prescription Only (S4) (AU) ℞-only (CA) POM (UK) ℞-only (US) |
| Routes | Oral |
| Pharmacokinetic data | |
| Bioavailability | 38–49% |
| Protein binding | 99.5% |
| Metabolism | Hepatic oxidation and glucuronidation (CYP3A4 & UGT1A9-mediated) |
| Half-life | 25–48 hours |
| Excretion | Faeces (77%) and urine (19%) |
| Identifiers | |
| CAS number | 284461-73-0 |
| ATC code | L01XE05 |
| PubChem | CID 216239 |
| DrugBank | DB00398 |
| ChemSpider | 187440 |
| UNII | 9ZOQ3TZI87 |
| KEGG | D08524 |
| ChEBI | CHEBI:50924 |
| ChEMBL | CHEMBL1336 |
| Synonyms | Nexavar Sorafenib tosylate |
| PDB ligand ID | BAX (PDBe, RCSB PDB) |
| Chemical data | |
| Formula | C21H16ClF3N4O3 |
| Mol. mass | 464.825 g/mol |

4-(4-{3-[4-chloro-3-(trifluoromethyl)phenyl]ureido}phenoxy)-Λ/2-methylpyridine-2- carboxamide is commonly known as sorafenib (I). Sorafenib is prepared as its tosylate salt. Sorafenib blocks the enzyme RAF kinase, a critical component of the RAF/MEK/ERK signaling pathway that controls cell division and proliferation; in addition, sorafenib inhibits the VEGFR-2/PDGFR-beta signaling cascade, thereby blocking tumor angiogenesis.
Sorafenib, marketed as Nexavar by Bayer, is a drug approved for the treatment of advanced renal cell carcinoma (primary kidney cancer). It has also received “Fast Track” designation by the FDA for the treatment of advanced hepatocellular carcinoma (primary liver cancer). It is a small molecular inhibitor of Raf kinase, PDGF (platelet-derived growth factor), VEGF receptor 2 & 3 kinases and c Kit the receptor for Stem cell factor.

Sorafenib and pharmaceutically acceptable salts thereof is disclosed in WO0042012. Sorafenib is also disclosed in WO0041698. Both these patents disclose processes for the preparation of sorafenib.
WO0042012 and WO0041698 describe the process as given in scheme I which comprises reacting picolinic acid (II) with thionyl chloride in dimethyl formamide (DMF) to form acid chloride salt (III). This salt is then reacted with methylamine dissolved in tetrahydrofuran (THF) to give carboxamide (IV). This carboxamide when further reacted with 4- aminophenol in anhydrous DMF and potassium tert-butoxide 4-(2-(N-methylcarbamoyl)-4- pyridyloxy)aniline (V) is formed. Subsequent reaction of this aniline with 4-chloro-3- (trifluoromethyl) phenyl isocyanate (Vl) in methylene chloride yields sorafenib (I). The reaction is represented by Scheme I as given below.
Scheme I

Picolini

Sorafenib (I)
WO2006034796 also discloses a process for the preparation of sorafenib and its tosylate salt. The process comprises reacting 2-picolinic acid (II) with thionyl chloride in a solvent inert toward thionyl chloride without using dimethyl formamide to form acid chloride salt (III). This acid salt on further reaction with aqueous solution methylamine or gaseous methylamine gives compound (IV). Compound (IV) is then reacted with 4-aminophenol with addition of a carbonate salt in the presence of a base to yield compound (V).
Compound (V) can also be obtained by reacting compound (IV) with 4-aminophenol in the presence of water with addition of a phase transfer catalyst. Compound (V) when reacted with 4-chloro-3-(trifluoromethyl) phenyl isocyanate (Vl) in a non-chlorinated organic solvent, inert towards isocyanate gives sorafenib (I). Sorafenib by admixing with p- toluenesulfonic acid in a polar solvent gives sorafenib tosylate (VII). The reaction is represented by Scheme Il as given below.
Scheme Il
P

A key step in the synthesis of sorafenib is the formation of the urea bond. The processes disclosed in the prior art involve reactions of an isocyanate with an amine. These isocyanate compounds though commercially available are very expensive. Further synthesis of isocyanate is very difficult which requires careful and skillful handling of reagents.
Isocyanate is prepared by reaction of an amine with phosgene or a phosgene equivalent, such as bis(trichloromethyl) carbonate (triphosgene) or trichloromethyl chloroformate (diphosgene). Isocyanate can also be prepared by using a hazardous reagent such as an azide. Also, the process for preparation of an isocyanate requires harsh reaction conditions such as strong acid, higher temperature etc. Further, this isocyanate is reacted with an amine to give urea.
Reactions of isocyanates suffer from one or more disadvantages. For example phosgene or phosgene equivalents are hazardous and dangerous to use and handle on a large scale. These reagents are also not environment friendly. Isocyanates themselves are thermally unstable compounds and undergo decomposition on storage and they are incompatible with a number of organic compounds. Thus, the use of isocyanate is not well suited for industrial scale application.
Sorafenib and its pharmaceutically acceptable salts and solvates are reported for the first time in WO0041698 (corresponding US 03139605) by Bayer. In the literature only one route is disclosed for the preparation of sorafenib. According to this route (Scheme-I), picolinic acid of formula III is reacted with thionyl chloride to give the 4-chloro derivative which on treatment

VII
Scheme-I with methanol gave the methyl ester of formula V. Compound of formula V is reacted with methylamine to get the corresponding amide of formula VL Compound of formula VI is reacted with 4-aminophenol to get the ether derivative of formula VII. Compound of formula VII is reacted with 4-chloro-3-trifluoromethylphenylisocyante to get sorafenib base of formula I. Overall yield of sorafenib in this process is 10% from commercially available 2-picolinic acid of formula II. Main drawback in this process is chromatographic purification of the intermediates involved in the process and low yield at every step.
Donald Bankston’s (Org. Proc. Res. Dev., 2002, 6, 777-781) development of an improved synthesis of the above basic route afforded sorafenib in an overall yield of 63% without involving any chromatographic purification. Process improvements like reduction of time in thionyl chloride reaction; avoid the isolation of intermediates of formulae IV and V5 reduction of base quantity in p-aminophenol reaction, etc lead to the simplification of process and improvement in yield of final compound of formula I.
Above mentioned improvements could not reduce the number of steps in making sorafenib of formula-I. In the first step all the raw materials are charged and heated to target temperature (72°C). Such a process on commercial scale will lead to sudden evolution of gas emissions such as sulfur dioxide and hydrogen chloride. Also, in the aminophenol reaction two bases (potassium carbonate and potassium t-butoxide) were used in large excess to accomplish the required transformation.
A scalable process for the preparation of sorafenib is disclosed in WO2006034796. In this process also above mentioned chemistry is used in making sorafenib of formula I. In the first step, catalytic quantity. of DMF used in the prior art process is replaced with reagents like hydrogen bromide, thionyl bromide and sodium bromide. Yield of required product remained same without any advantages from newly introduced corrosive reagents. Process improvements like change of solvents, reagents, etc were applied in subsequent steps making the process scalable. Overall yield of sorafenib is increased to 74% from the prior art 63% yield. Purity of sorafenib is only 95% and was obtained as light brown colored solid.
Main drawbacks in this process are production of low quality sorafenib and requirement of corrosive and difficult to handle reagents such as thionyl bromide and hydrogen bromide. Also, there is no major improvement in the yield of sorafenib.
Sorafenib tosylate ( Brand name: Nexavar ®, BAY 43-9006 other name, Chinese name: Nexavar, sorafenib, Leisha Wa) was Approved by U.S. FDA for the treatment of advanced kidney cancer in 2005 and liver cancer in 2007 .
Sorafenib, co-Developed and co-marketed by Germany-based Bayer AG and South San Francisco-based Onyx Pharmaceuticals , is an Oral Multi-kinase inhibitor for VEGFR1, VEGFR2, VEGFR3, PDGFRbeta, Kit, RET and Raf-1.
In March 2012 Indian drugmaker Natco Pharma received the first compulsory license ever from Indian Patent Office to make a generic Version of Bayer’s Nexavar despite the FACT that Nexavar is still on Patent. This Decision was based on the Bayer Drug being too expensive to most patients. The Nexavar price is expected to drop from $ 5,500 per person each month to $ 175, a 97 percent decline. The drug generated $ 934 million in global sales in 2010, according to India’s Patent Office.

Sorafenib tosylate
Chemical Name: 4-Methyl-3-((4 – (3-pyridinyl)-2-pyrimidinyl) amino)-N-(5 – (4-methyl-1H-imidazol-1-yl) -3 – (trifluoromethyl) phenyl) benzamide monomethanesulfonate, Sorafenib tosylate
CAS Number 475207-59-1 (Sorafenib tosylate ) , 284461-73-0 (Sorafenib)
References for the Preparation of Sorafenib References
1) Bernd Riedl, Jacques Dumas, Uday Khire, Timothy B. Lowinger, William J. Scott, Roger A. Smith, Jill E. Wood, Mary-Katherine Monahan, Reina Natero, Joel Renick, Robert N. Sibley; Omega-carboxyaryl Substituted diphenyl Ureas as RAF kinase inhibitors ; U.S. Patent numberUS7235576
2) Rossetto, Pierluigi; Macdonald, Peter, Lindsay; Canavesi, Augusto; Process for preparation of sorafenib and Intermediates thereof , PCT Int. Appl., WO2009111061
3) Lögers, Michael; gehring, Reinhold; Kuhn, Oliver; Matthäus, Mike; Mohrs, Klaus; müller-gliemann, Matthias; Stiehl, jürgen; berwe, Mathias; Lenz, Jana; Heilmann, Werner; Process for the preparation of 4 – {4 – [( {[4-chloro-3-(TRIFLUOROMETHYL) phenyl] amino} carbonyl) amino] phenoxy}-N-methylpyridine-2-carboxamide , PCT Int. Appl., WO2006034796
4) Shikai Xiang, Liu Qingwei, Xieyou Rong, sorafenib preparation methods, invention patent application Publication No. CN102311384 , Application No. CN201010212039
5) Zhao multiply there, Chenlin Jie, Xu Xu, MASS MEDIA Ji Yafei; sorafenib tosylate synthesis ,Chinese Journal of Pharmaceuticals , 2007 (9): 614 -616
Preparation of Sorafenib Tosylate (Nexavar) Nexavar, sorafenib Preparation of methyl sulfonate

Sorafenib (Sorafenib) chemical name 4 – {4 – [({[4 – chloro -3 – (trifluoromethyl) phenyl] amino} carbonyl) amino] phenoxy}-N-methyl-pyridine -2 – formamide by Bayer (Bayer) research and development, in 2005 the U.S. Food and Drug Administration (FDA) approval. Trade name Nexavar (Nexavar). This product is an oral multi-kinase inhibitor, for the treatment of liver cancer and kidney cancer.
Indian Patent Office in March this year for Bayer’s Nexavar in liver and kidney cancer drugs (Nexavar) has released a landmark “compulsory licensing” (compulsory license). Indian Patent Office that due to the high price Nexavar in India, the vast majority of patients can not afford the drug locally, thus requiring local Indian pharmaceutical company Natco cheap Nexavar sales. Nexavar in 2017 before patent expiry, Natco pay only Bayer’s pharmaceutical sales to 6% royalties. The move to make Nexavar patent drug prices, the supply price from $ 5,500 per month dropped to $ 175, the price reduction of 97%. Compulsory licensing in India for other life-saving drugs and patent medicines overpriced open a road, the Indian Patent Office through this decision made it clear that the patent monopoly does not guarantee that the price is too high. Nexavar is a fight against advanced renal cell carcinoma, liver cancer cure. In China, a box of 60 capsules of Nexavar price of more than 25,000 yuan. In accordance with the recommended dose, which barely enough to eat half of patients with advanced cancer. In September this year India a patent court rejected Bayer Group in India cheap drugmaker emergency appeal. Indian government to refuse patent medicine overpriced undo “compulsory licensing rules,” allowing the production of generic drugs Nexavar.
Sorafenat by Natco – Sorafenib – Nexavar – India natco Nexavar

Chemical Synthesis of Sorafenib Tosylate (Nexavar)
Sorafenib tosylate (brand name :Nexavar®, other name BAY 43-9006, was approved by US FDA for the treatment of kidney cancer in 2005 and advanced liver cancer in 2007.

US Patent US7235576, WO2006034796, WO2009111061 and Faming Zhuanli Shenqing(CN102311384) disclosed processes for preparation of sorafenib base and its salt sorafenib tosylate.
References
1)Bernd Riedl, Jacques Dumas, Uday Khire, Timothy B. Lowinger, William J. Scott, Roger A. Smith, Jill E. Wood, Mary-Katherine Monahan, Reina Natero, Joel Renick, Robert N. Sibley; Omega-carboxyaryl substituted diphenyl ureas as raf kinase inhibitors; US patent numberUS7235576
2)Rossetto, pierluigi; Macdonald, peter, lindsay; Canavesi, augusto; Process for preparation of sorafenib and intermediates thereof, PCT Int. Appl., WO2009111061
3)Lögers, michael; gehring, reinhold; kuhn, oliver; matthäus, mike; mohrs, klaus; müller-gliemann, matthias; stiehl, jürgen; berwe, mathias; lenz, jana; heilmann, werner; Process for the preparation of 4-{4-[({[4-chloro-3-(trifluoromethyl)phenyl]amino}carbonyl)amino]phenoxy}-n-methylpyridine-2-carboxamide, PCT Int. Appl., WO2006034796CN102311384, CN201010212039
Full Experimental Details for the preparation of Sorafenib Tosylate (Nexavar)
Synthesis of 4-(2-(N-methylcarbamoyl)-4-pyridyloxy)aniline.
A solution of 4-aminophenol (9.60 g, 88.0 mmol) in anh. DMF (150 mL) was treated with potassium tert-butoxide (10.29 g, 91.7 mmol), and the reddish-brown mixture was stirred at room temp. for 2 h. The contents were treated with 4-chloro- N -methyl-2-pyridinecarboxamide (15.0 g, 87.9mmol) and K2CO3 (6.50 g, 47.0 mmol) and then heated at 80°C. for 8 h. The mixture was cooled to room temp. and separated between EtOAc (500 mL) and a saturated NaCl solution (500 mL). The aqueous phase was back-extracted with EtOAc (300 mL). The combined organic layers were washed with a saturated NaCl solution (4×1000 mL), dried (Na2SO4) and concentrated under reduced pressure. The resulting solids were dried under reduced pressure at 35°C. for 3 h to afford 4-(2-(N-methylcarbamoyl)-4-pyridyloxy)aniline as a light-brown solid 17.9 g, 84%):. 1H-NMR (DMSO-d6) δ 2.77 (d, J = 4.8 Hz, 3H), 5.17 (br s, 2H), 6.64, 6.86 (AA’BB’ quartet, J = 8.4 Hz, 4H), 7.06 (dd, J = 5.5, 2.5 Hz, 1H), 7.33 (d, J = 2.5 Hz, 1H), 8.44 (d, J = 5.5 Hz; 1H), 8.73 (br d, 1H); HPLC ES-MS m/z 244 ((M+H)+).
Synthesis of 4-{4-[({[4-Chloro-3-(trifluoromethyl)phenyl]amino}carbonyl)amino]phenoxy}-N-methylpyridine-2-carboxamide (sorafenib)
4-(4-Aminophenoxy)-N-methyl-2-pyridinecarboxamide (52.3 kg, 215 mol) is suspended in ethyl acetate (146 kg) and the suspension is heated to approx. 40° C. 4-Chloro-3-trifluoromethylphenyl isocyanate (50 kg, 226 mol), dissolved in ethyl acetate (58 kg), is then added to such a degree that the temperature is kept below 60° C. After cooling to 20° C. within 1 h, the mixture is stirred for a further 30 min and the product is filtered off. After washing with ethyl acetate (30 kg), the product is dried under reduced pressure (50° C., 80 mbar). 93 kg (93% of theory) of the title compound are obtained as colorless to slightly brownish crystals. m.p. 206-208° C. 1H-NMR (DMSO-d6, 500 MHz): δ =2.79 (d, J=4.4 Hz, 3H, NCH3); 7.16 (dd, J=2.5, 5.6 Hz, 1H, 5-H); 7.18 (d, J=8.8 Hz, 2H, 3′-H, 5′-H); 7.38 (d, J=2.4 Hz, 1H, 3-H); 7.60-7.68 (m, 4H, 2′-H, 6′-H, 5″-H, 6″-H); 8.13 (d, J=1.9 Hz, 1H, 2″-H); 8.51 (d, J=5.6 Hz, 1H, 6-H); 8.81 (d, J=4.5 Hz, 1H, NHCH3); 9.05 (br. s, 1H, NHCO); 9.25 (br. s, 1H, NHCO) MS (ESI, CH3CN/H2O): m/e=465 [M+H]+.
Synthesis of Sorafenib Tosylate (Nexavar)
4-(4-{3-[4-chloro-3-(trifluoromethyl)phenyl]ureido}phenoxy)-N2-methylpyridine-2-carboxamide (sorafenib) (50g, 0.1076 mol) is suspended in ethyl acetate (500 g) and water (10g). The mixture is heated to 69°C within 0.5 h, and a filtered solution of p-toluenesulfonic acid monohydrate (3.26 g, 0.017 mol) in a mixture of water (0.65 g) and ethyl acetate (7.2 g) is added. After filtration a filtered solution of p-toluenesulfonic acid monohydrate (22g, 0.11 mol) in a mixture of ethyl acetate (48 g) and water (4.34 g) is added. The mixture is cooled to 23°C within 2 h. The product is filtered off, washed twice with ethyl acetate (92.5 g each time) and dried under reduced pressure. The sorafenib tosylate (65.5 g, 96% of theory) is obtained as colorless to slightly brownish crystals.
…………………..
http://www.google.com/patents/EP2195286A2?cl=en
Example 22: Synthesis of Sorafenib
Phenyl 4-chloro-3-(trifluoromethyl)phenylcarbamate (100 g, 0.3174 mol) and 4-(4- aminophenoxy)-N-methylpicolinamide (77.14 g, 0.3174 mol) were dissolved in N1N- dimethyl formamide (300 ml) to obtain a clear reaction mass. The reaction mass was agitated at 40-450C for 2-3 hours, cooled to room temperature and diluted with ethyl acetate (1000 ml). The organic layer was washed with water (250 ml) followed by 1N HCI (250ml) and finally with brine (250 ml). The organic layer was separated, dried over sodium sulfate and degassed to obtain solid. This solid was stripped with ethyl acetate and finally slurried in ethyl acetate (1000 ml) at room temperature. It was then filtered and vacuum dried to give (118 g) of 4-(4-(3-(4-chloro-3- (trifluoromethyl)phenyl)ureido)phenoxy)-N-methylpicolinamide (sorafenib base).
Example 23: Synthesis of 1-(4-chloro-3-(trifluoromethyl)phenyl)urea (Compound 4)
Sodium cyanate (1.7 g, 0.02mol) was dissolved in water (17ml) at room temperature to obtain a clear solution. This solution was then charged drop wise to the clear solution of 3- trifluoromethyl-4-chloroaniline (5 g, 0.025 mol) in acetic acid (25 ml) at 40°C-45°C within 1- 2 hours. The reaction mass was agitated for whole day and cooled gradually to room temperature. The obtained solid was filtered washed with water and vacuum dried at 500C to afford the desired product (5.8 g) i.e. 1-(4-chloro-3-(trifluoromethyl)phenyl)urea.
Example 24: Synthesis of Sorafenib
1-(4-chloro-3-(trifluoromethyl) phenyl)urea (15 g, 0.0628 mol), 1 ,8- diazabicyclo[5.4.0]undec-7-ene (11.75 ml, 0.078 mol) and 4-(4-aminophenoxy)-N- methylpicolinamide (15.27 g, 0.0628 mol) were mixed with dimethyl sulfoxide (45 ml) and the reaction mass was then heated to 110-1200C for 12-18 hours. The reaction mass was cooled to room temperature and quenched in water (250 ml). The quenched mass was extracted repeatedly with ethyl acetate and the combined ethyl acetate layer was then back washed with water. It was dried over sodium sulfate and evaporated under vacuum to obtain solid. The obtained solid was slurried in acetonitrile (150 ml) at ambient temperature and filtered to give 4-(4-(3-(4-chloro-3-(trifluoromethyl) phenyl) ureido) phenoxy)-N-methylpicolinamide (sorafenib base) (17.5 g).
………………………..
http://www.google.com/patents/WO2009054004A2?cl=en


EXAMPLES
Example 1
Preparation of l-(4-chloro-3-(trifluoromethyl)phenyI)-3-(4-hydroxyphenyl)urea Into a 250 ml, four-necked RB flask was charged 1O g of 4-aminophenol and 100 ml of toluene. A solution of 4-chloro-3-(trifluoromethyl)phenyl isocyante (20.4 g) in toluene (50 ml) was added to the reaction mass at 25-300C. The reaction mass was stirred at room temperature for 16 h. The reaction mass was filtered and washed the. solid with 50 ml of toluene. The wet material was dried in the oven at 50-60°C to get 29.8 g of title compound as white solid. M.P. is 218-222°C. IR (KBr): 3306, 1673, 1625, 1590, 1560, 1517, 1482, 1435, 1404, 1328, 1261, 1182, 1160, 1146, 1125, 1095, 1032, 884, 849, 832, 812, 766, 746, 724, 683, 539 and 434 cm“1.
Example 2 Preparation of sorafenib tosylate
Into a 100 ml, three-necked RB flask was charged 2.0 g of l-(4-chloro-3- (trifluoromethyl)-phenyl)-3-(4-hydroxyphenyl)urea and 10 ml of DMF. Potassium tert- butoxide (2.3 g) was added to the reaction mass and stirred for 45 min at RT. 4-Chlro-N- methylpicolinamide (1.14 g) and potassium carbonate (0.42 g) were added to the reaction mass and heated to 80°C. The reaction mass was maintained at 80-85°C for 8 h and cooled to 30°C. The reaction mass was poured into water and extracted with ethyl acetate. Ethyl acetate layer was washed with water, brine and dried over sodium sulphate. Solvent was distilled of under reduced pressure.
The crude compound (4.7 g) was dissolved in 10 ml of IPA and added 1.9 g of p- toluenesulfonic acid. The reaction mass was stirred at RT for 15 h and filtered. The wet solid was washed with 10 ml of IPA and dried at 50-60°C to get 3.4 g of title compound as off-white crystalline solid.
…………………..
A Scaleable Synthesis of BAY 43-9006: A Potent Raf Kinase Inhibitor for the Treatment of Cancer
http://pubs.acs.org/doi/abs/10.1021/op020205n

Urea 3 (BAY 43–9006), a potent Raf kinase inhibitor, was prepared in four steps with an overall yield of 63%. Significant process research enabled isolation of each intermediate and target without chromatographic purification, and overall yield increases >50% were observed compared to those from previous methods. This report focuses on improved synthetic strategies for production of scaled quantities of 3 for preclinical, toxicological studies. These improvements may be useful to assemble other urea targets as potential therapeutic agents to combat cancer.
References
- “FDA Approves Nexavar for Patients with Inoperable Liver Cancer” (Press release). FDA. November 19, 2007. Retrieved November 10, 2012.
- “Nexavar (sorafenib) dosing, indications, interactions, adverse effects, and more”. Medscape Reference. WebMD. Retrieved 26 December 2013.
- “NEXAVAR (sorafenib) tablet, film coated [Bayer HealthCare Pharmaceuticals Inc.]”. DailyMed. Bayer HealthCare Pharmaceuticals Inc. November 2013. Retrieved 26 December 2013.
- “Nexavar 200mg film-coated tablets – Summary of Product Characteristics (SPC) – (eMC)”. electronic Medicines Compendium. Bayer plc. 27 March 2013. Retrieved 26 December 2013.
- “PRODUCT INFORMATION NEXAVAR® (sorafenib tosylate)” (PDF). TGA eBusiness Services. Bayer Australia Ltd. 12 December 2012. Retrieved 26 December 2013.
- Escudier, B; Eisen, T; Stadler, WM; Szczylik, C; Oudard, S; Siebels, M; Negrier, S; Chevreau, C; Solska, E; Desai, AA; Rolland, F; Demkow, T; Hutson, TE; Gore, M; Freeman, S; Schwartz, B; Shan, M; Simantov, R; Bukowski, RM (January 2007). “Sorafenib in advanced clear-cell renal-cell carcinoma”. New England Journal of Medicine 356 (2): 125–34. doi:10.1056/NEJMoa060655. PMID 17215530.
- Walid, MS; Johnston, KW (October 2009). “Successful treatment of a brain-metastasized renal cell carcinoma”. German Medical Science 7: Doc28. doi:10.3205/000087. PMC 2775194. PMID 19911072.
- “Pharmaceutical Benefits Scheme (PBS) -SORAFENIB”. Pharmaceutical Benefits Scheme. Australian Government Department of Health. Retrieved 27 December 2013.
- Llovet, et al. (2008). “Sorafenib in Advanced Hepatocellular Carcinoma” (PDF). New England Journal of Medicine 359 (4): 378–90.
- Keating GM, Santoro A (2009). “Sorafenib: a review of its use in advanced hepatocellular carcinoma”. Drugs 69 (2): 223–40. doi:10.2165/00003495-200969020-00006. PMID 19228077.
- Pawlik TM, Reyes DK, Cosgrove D, Kamel IR, Bhagat N, Geschwind JF (October 2011). “Phase II trial of sorafenib combined with concurrent transarterial chemoembolization with drug-eluting beads for hepatocellular carcinoma”. J. Clin. Oncol. 29 (30): 3960–7. doi:10.1200/JCO.2011.37.1021. PMID 21911714.
- “Phase 3 Trial of Nexavar in Patients With Non-Responsive Thyroid Cancer”[dead link]
- [1]
- “Chemotherapy-Induced Nausea and Vomiting Treatment & Management”. Medscape Reference. WebMD. 3 July 2012. Retrieved 26 December 2013.
- Hagopian, Benjamin (August 2010). “Unusually Severe Bullous Skin Reaction to Sorafenib: A Case Report”. Journal of Medical Cases 1 (1): 1–3. doi:10.4021/jmc112e. Retrieved 11 February 2014.
- Smalley KS, Xiao M, Villanueva J, Nguyen TK, Flaherty KT, Letrero R, Van Belle P, Elder DE, Wang Y, Nathanson KL, Herlyn M (January 2009). “CRAF inhibition induces apoptosis in melanoma cells with non-V600E BRAF mutations”. Oncogene 28 (1): 85–94. doi:10.1038/onc.2008.362. PMC 2898184. PMID 18794803.
- Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M (October 2008). “Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling”. Mol. Cancer Ther. 7 (10): 3129–40. doi:10.1158/1535-7163.MCT-08-0013. PMID 18852116.
- Zhang Y (Jan 2014). “Screening of kinase inhibitors targeting BRAF for regulating autophagy based on kinase pathways.”. J Mol Med Rep 9 (1): 83–90. PMID 24213221.
- Gauthier A (Feb 2013). “Role of sorafenib in the treatment of advanced hepatocellular carcinoma: An update..”. Hepatol Res 43 (2): 147–154. doi:10.1111/j.1872-034x.2012.01113.x. PMID 23145926.
- FDA Approval letter for use of sorafenib in advanced renal cancer
- European Commission – Enterprise and industry. Nexavar. Retrieved April 24, 2007.
- “Nexavar® (Sorafenib) Approved for Hepatocellular Carcinoma in Europe” (Press release). Bayer HealthCare Pharmaceuticals and Onyx Pharmaceuticals. October 30, 2007. Retrieved November 10, 2012.
- FDA Approval letter for use of sorafenib in inoperable hepatocellular carcinoma
- “Liver drug ‘too expensive‘“. BBC News. November 19, 2009. Retrieved November 10, 2012.
- http://www.ipindia.nic.in/ipoNew/compulsory_License_12032012.pdf
- “Seven days: 9–15 March 2012”. Nature 483 (7389): 250–1. 2012. doi:10.1038/483250a.
- “India Patents (Amendment) Act, 2005”. WIPO. Retrieved 16 January 2013.
- [2]
- “Addition of Sorafenib May Be Detrimental in Some Lung Cancer Patients”
- ClinicalTrials.gov NCT00329719 Sorafenib and Temsirolimus in Treating Patients With Recurrent Glioblastoma
- “Activity of sorafenib against desmoid tumor/deep fibromatosis”
- “‘We didn’t make this medicine for Indians… we made it for western patients who can afford it‘“. Daily Mail Reporter. 24 Jan 2014.
External links
- Nexavar.com – Manufacturer’s website
- Prescribing Information – includes data from the key studies justifying the use of sorafenib for the treatment of kidney cancer (particularly clear cell renal cell carcinoma, which is associated with the von Hippel-Lindau gene)
- Patient Information from FDA
- Sorafenib in Treating Patients With Soft Tissue Sarcomas
- Sorafenib Sunitinib differences – diagram
- ClinicalTrials.gov NCT00217399 – Sorafenib and Anastrozole in Treating Postmenopausal Women With Metastatic Breast Cancer
- Cipla launches Nexavar generic at 1/10 of Bayer’s price
| Reference | ||
|---|---|---|
| 1 | * | D. BANKSTON ET AL.: “A Scalable Synthesis of BAY 43-9006: A Potent Raf Kinase Inhibitor for the Treatment of Cancer” ORGANIC PROCESS RESEARCH & DEVELOPMENT, vol. 6, no. 6, 2002, pages 777-781, XP002523918 cited in the application |
| 2 | * | PAN W ET AL: “Pyrimido-oxazepine as a versatile template for the development of inhibitors of specific kinases” BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, PERGAMON, ELSEVIER SCIENCE, GB, vol. 15, no. 24, 15 December 2005 (2005-12-15), pages 5474-5477, XP025314229 ISSN: 0960-894X [retrieved on 2005-12-15] |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2011036647A1 | Sep 24, 2010 | Mar 31, 2011 | Ranbaxy Laboratories Limited | Process for the preparation of sorafenib tosylate |
| WO2011036648A1 | Sep 24, 2010 | Mar 31, 2011 | Ranbaxy Laboratories Limited | Polymorphs of sorafenib acid addition salts |
| WO2011058522A1 | Nov 12, 2010 | May 19, 2011 | Ranbaxy Laboratories Limited | Sorafenib ethylsulfonate salt, process for preparation and use |
| WO2011092663A2 | Jan 28, 2011 | Aug 4, 2011 | Ranbaxy Laboratories Limited | 4-(4-{3-[4-chloro-3-(trifluoromethyl)phenyl]ureido}phenoxy)-n2-methylpyridine-2-carboxamide dimethyl sulphoxide solvate |
| WO2011113367A1 * | Mar 17, 2011 | Sep 22, 2011 | Suzhou Zelgen Biopharmaceutical Co., Ltd. | Method and process for preparation and production of deuterated ω-diphenylurea |
| US8552197 | Nov 12, 2010 | Oct 8, 2013 | Ranbaxy Laboratories Limited | Sorafenib ethylsulfonate salt, process for preparation and use |
| US8604208 | Sep 24, 2010 | Dec 10, 2013 | Ranbaxy Laboratories Limited | Polymorphs of sorafenib acid addition salts |
| US8609854 | Sep 24, 2010 | Dec 17, 2013 | Ranbaxy Laboratories Limited | Process for the preparation of sorafenib tosylate |
| US8618305 | Jan 28, 2011 | Dec 31, 2013 | Ranbaxy Laboratories Limited | Sorafenib dimethyl sulphoxide solvate |
| US8669369 | Mar 17, 2011 | Mar 11, 2014 | Suzhou Zelgen Biopharmaceutical Co., Ltd. | Method and process for preparation and production of deuterated Ω-diphenylurea |
