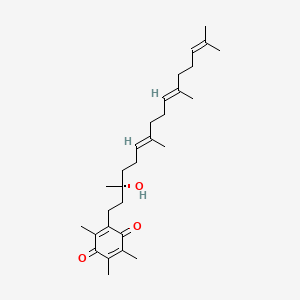
Graphical abstract

WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ]
Home » Posts tagged 'FAST TRACK' (Page 4)
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
PHASE 2, Non-alcoholic steatohepatitis, GILEAD
| Molecular Formula: | C28H31N3O8S |
|---|---|
| Molecular Weight: | 569.62604 g/mol |

| Company | Nimbus Therapeutics LLC |
| Description | Small molecule allosteric inhibitor of acetyl-coenzyme A carboxylase alpha (ACACA; ACC1) and acetyl-coenzyme A carboxylase beta (ACACB; ACC2) |
| Molecular Target | Acetyl-Coenzyme A carboxylase alpha (ACACA) (ACC1) ; Acetyl-Coenzyme A carboxylase beta (ACACB) (ACC2) |
| Mechanism of Action | Acetyl-coenzyme A carboxylase alpha (ACACA) (ACC1) inhibitor; Acetyl-coenzyme A carboxylase beta (ACACB) (ACC2) inhibitor |
| Therapeutic Modality | Small molecule |
In April 2016, Gilead Sciences and Nimbus Therapeutics, LLC announced that the companies have signed a definitive agreement under which Gilead will acquire Nimbus Apollo, Inc., a wholly-owned subsidiary of Nimbus Therapeutics, and its Acetyl-CoA Carboxylase (ACC) inhibitor program. Nimbus Therapeutics will receive an upfront payment of $400 million, with the potential to receive an additional $800 million in development-related milestones over time.
The Nimbus Apollo program includes the lead candidate NDI-010976, an ACC inhibitor, and other preclinical ACC inhibitors for the treatment of non-alcoholic steatohepatitis (NASH), and for the potential treatment of hepatocellular carcinoma (HCC) and other diseases.
In May 2016, Nimbus Therapeutics announced the recent closing of Gileads acquisition of Nimbus Apollo. The acquisitions completion triggered a $400 million upfront payment to Nimbus from Gilead.
In January 2016, fast track designation was assigned in the U.S. for this indication. In May 2016, Gilead Sciences acquired Nimbus Apollo from Nimbus Therapeutics, including its acetyl-CoA carboxylase (ACC) inhibitor program.
Gilead Sciences following the acquisition of Nimbus Apollo , is developing firsocostat , the lead from a program of acetyl-CoA carboxylase (ACC)-targeting compounds, for treating fatty liver disease including non-alcoholic steatohepatitis.
Nimbus compounds targeting liver disease in rat models
Data were presented by Geraldine Harriman, from Nimbus Therapeutics, from rat models using acetyl-CoA carboxylase (ACC) inhibitors NDI-010976 (ND-630) and N-654, which improved metabolic syndrome endpoints, decreased liver steatosis, decreased expression of inflammatory markers and improved fibrosis. The hepatotropic ACC inhibitor NDI-010976 had IC50 values of 2 and 7 nM for ACC1 and 2, respectively, EC50 values in HepG2 serum free and 10% serum of 9 and 66 nM, respectively, and 2-fold C2C12 fatty acid oxidation (FAOxn) stimulation at 200 nM. Rat FASyn (synthase), malonyl-CoA (liver) and malonyl-COA (muscle) respective ED50 values were 0.14 mg/kg po, 0.8 and 3 mg/kg. The rat respiratory quotient (RQ) MED was 3 mg/kg po. ADME data showed low multispecies intrinsic clearance (human, mouse, rat, dog, monkey). NDI-010976 was eliminated predominantly as the parent drug. Additionally, P450 inhibition was > 50 microM. In liver and muscle, NDI-010976 modulated key metabolic parameters including a dose-dependent reduction in the formation of the enzymatic product of acetyl coA carboxyloase malonyl coA; the ED50 value was lower in muscle. The drug also decreased FASyn dose dependently and increased fatty acid oxidation in the liver (EC50 = 0.14 mg/kg). In 28-day HS DIO rats, NDI-010976 favorably modulated key plasma and liver lipids, including decreasing liver free fatty acid, plasma triglycerides and plasma cholesterol; this effect was also seen in 37-day ZDF rats

http://www.google.com/patents/WO2013071169A1?cl=en
Example 76: Synthesis of 2-[l-[2-(2-methoxyphenyl)-2-(oxan-4-yloxy)ethyl]-5- methyl-6-(l,3-oxazol-2-yl)-2,4-dioxo-lH,2H,3H,4H-thieno[2,3-d]pyrimidin-3-yl]-2- methylpropanoic acid (1-181).

Synthesis of compound 76.1. Into a 250-mL 3 -necked round-bottom flask, purged and maintained with an inert atmosphere of nitrogen, was placed oxan-4-ol (86 g, 842.05 mmol, 2.01 equiv) and FeCl3 (10 g). This was followed by the addition of 57.2 (63 g, 419.51 mmol, 1.00 equiv) dropwise with stirring at 0 °C. The resulting solution was stirred for 3 h at room temperature. The resulting solution was diluted with 500 mL of H20. The resulting solution was extracted with 3×1000 mL of ethyl acetate and the organic layers combined. The resulting solution was extracted with 3×300 mL of sodium chloride (sat.) and the organic layers combined and dried over anhydrous sodium sulfate. The residue was applied onto a silica gel column with ethyl acetate/petroleum ether (1 : 10). This resulted in 22 g (21%) of 76.1 as a white solid.
Synthesis of compound 76.2. The enantiomers of 76.1 (22g) were resolved by chiral preparative HPLC under the following conditions (Gilson Gx 281): Column: Venusil Chiral OD-
H, 21.1 *25 cm, 5 μιη; mobile phase: hexanes (0.2% TEA) and ethanol (0.2% TEA) (hold at 10% ethanol (0.2%TEA) for 13 min); detector: UV 220/254 nm. 11.4 g (52%) of 76.2 were obtained as a white solid.
Synthesis of compound 76.3. Into a 500-mL 3-necked round-bottom flask, purged and maintained with an inert atmosphere of nitrogen, was placed 70.1 (12 g, 20.49 mmol, 1.00 equiv), tetrahydrofuran (200 mL), 76.2 (6.2 g, 24.57 mmol, 1.20 equiv) and DIAD (6.5 g, 32.18 mmol, 1.57 equiv). This was followed by the addition of a solution of triphenylphosphane (8.4 g, 32.03 mmol, 1.56 equiv) in tetrahydrofuran (100 mL) dropwise with stirring at 0 °C in 60 min. The resulting solution was stirred overnight at room temperature. The resulting mixture was concentrated under vacuum. The residue was applied onto a silica gel column with ethyl acetate/petroleum ether (1 :5). This resulted in 17 g (crude) of 76.3 as a white solid.
Synthesis of compound 76.4. Into a 500-mL 3-necked round-bottom flask, purged and maintained with an inert atmosphere of nitrogen, was placed 76.3 (17 g, crude), toluene (300 mL), Pd(PPh3)4 (1.7 g, 1.47 mmol, 0.07 equiv) and 2-(tributylstannyl)-l,3-oxazole (8.6 g, 24.02 mmol, 1.16 equiv). The resulting solution was stirred overnight at 110 °C. The resulting mixture was concentrated under vacuum. The residue was applied onto a silica gel column with ethyl acetate/petroleum ether (1 : 10). Purification afforded 6 g of 76.4 as a white solid.
Synthesis of compound 1-181. Into a 250-mL 3-necked round-bottom flask, was placed 76.4 (6 g, 7.43 mmol, 1.00 equiv), tetrahydrofuran (100 mL), TBAF (2.3 g, 8.80 mmol,
I .18 equiv). The resulting solution was stirred for 1 h at room temperature. The resulting mixture was concentrated under vacuum. The residue was applied onto a silica gel column with dichloromethane/methanol (50: 1). This resulted in 3.4 g (80%) of Compound 1-181 as a white solid.
Purification: MS (ES): m/z 570 (M+H)+, 592 (M+Na)+.
1H NMR (300 MHz, DMSO- d6): δ 1.22-1.36 (m, 2H), 1.62 (m, 8H), 2.75 (s, 3H), 3.20-3.39 (m, 3H), 3.48-3.58 (m, 2H), 3.80 (s, 3H), 3.85-4.20 (m, 2H), 5.30 (m, 1H), 7.03 (m, 2H), 7.33-7.50 (m, 3H), 8.2 (s, 1H).

Conference: 66th Annual Meeting of the American Association for the Study of Liver Diseases Conference Start Date: 13-Nov-2015
…candidates for minimizing IR injury in liver transplantation.Nimbus compounds targeting liver disease in rat modelsData were presented by Geraldine Harriman, from Nimbus Therapeutics, from rat models using acetyl-CoA carboxylase (ACC) inhibitors NDI-010976 (ND–630) and N-654, which improved metabolic syndrome endpoints, decreased liver steatosis, decreased expression of inflammatory markers and improved fibrosis. The hepatotropic ACC inhibitor NDI-010976 had IC50 values of 2 and 7 nM for ACC1 and 2, respectively…
REFERENCES
| WO-2014182943 |
| WO-2014182945 |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015203510 | 2015-07-23 | ACC INHIBITORS AND USES THEREOF |
| US2013123231 | 2013-05-16 | ACC INHIBITORS AND USES THEREOF |
WO2017151816 ,
CN 107629069
CN 107629069
CN 107151251
WO 2013071169
WO 2016112305
PATENT
WO-2018161022
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018161022&tab=PCTDESCRIPTION&maxRec=1000
Solid forms, including a salts (such as choline, diethylamine, NN-dibenzylethylenediamine, ethanolamine) or co-crystal, of firsocostat and compositions comprising them are claimed, which exhibits Acetyl-CoA carboxylase inhibitory activity and useful for treating ACC mediated diseases such as metabolic disorders, neurological disorders, and infectious diseases. Also claimed are process for preparing firsocostat and intermediates useful for preparing them are claimed.
The present disclosure provides forms of Compound I or a compound of formula (I) having the formula:
Compound I may be referred to by formula (I):
(I)
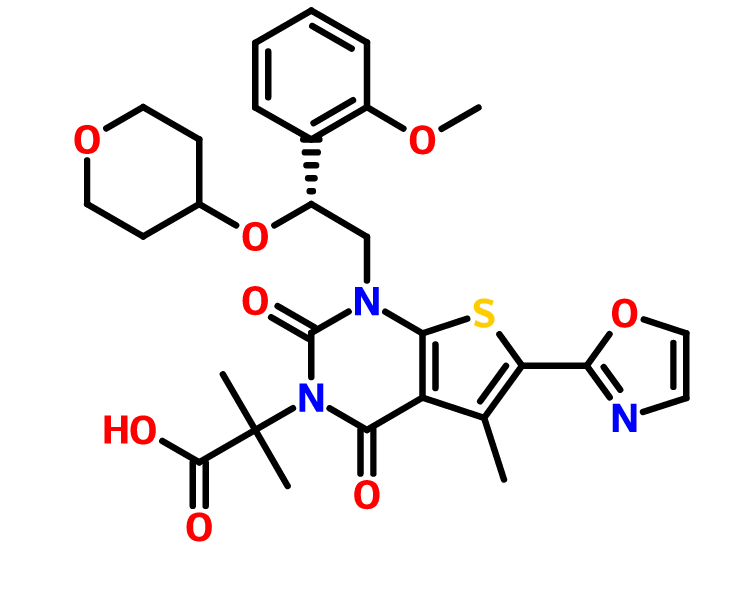
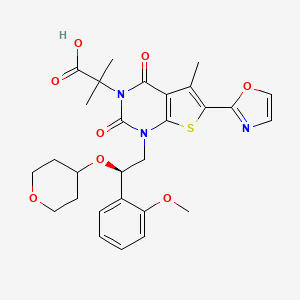
or its chemical name of (R)-2-(l-(2-(2-methoxyphenyl)-2-((tetrahydro-2H-pyran-4-yl)oxy)ethyl)-5-methyl-6-(oxazol-2-yl)-2,4-dioxo-l,2-dihydrothieno[2,3-d]pyrimidin-3(4H)-yl)-2-methylpropanoic acid. U.S. Patent No. 8,969,557 discloses that Compound I exhibits ACC inhibitory activity. In the present disclosure, compounds may be presented in the form of chemical structures or names.
Scheme 1 represents an exemplary synthesis of a compound of formula (F) and may be carried out according to the embodiments described herein.
Scheme 1
(E) (F)
Scheme 2
(E-1 ) (I)
Scheme 3
Step (g)
Scheme 4
scheme 5
Example 1 : Synthesis of Compound B-2
B-2
[0401] Compound A-2 was combined with Compound G-1 (about 1 equivalents (“equiv”)) with K2CO3 (about 2.3 equiv) in dimethylacetamide. The mixture was stirred at room temperature. The resulting mixture was then diluted with ethyl acetate and washed with water and brine. The organic layer was separated and concentrated to dryness, and the resulting product was purified by column chromatography (eluent: 0 to about 28% ethyl acetate:
heptanes). The resulting product was Compound B-2. ¾ NMR (300 MHz, CDCh): δ 7.92 (d, J
= 8.4 Hz, 1H), 7.57 (m, 1H), 7.06 (m, 2H), 5.20 (s, 2H), 4.00 (s, 3H), 2.42 (s, 3H), 1.77 (s, 6H), 1.44 (s, 9H).
Example 2: Synthesis of a compound of formula (C)
(B) (C)
[0402] Compound of formula (B) or Compound B (which may be prepared as described in Example 1) and a (S,S)-Ruthenium catalyst, such as a Ruthenium catalyst as described herein, or a suitable antipode of the Ruthenium catalyst, are combined in the presence of potassium tert-butoxide (“KO^-Bu”) and isopropanol and refluxed to yield a compound of formula (C) or Compound C. Compound C is isolated and purified by methods described herein.
Example 3: Synthesis of Compound D-1
C-1 D-1
[0403] To Compound C-1 in dichloromethane is added 4-bromotetrahydro-2H-pyran. Upon addition of an organic base, the reaction mixture is stirred ovemight to yield a compound of formula D-1 or Compound D-1. Compound D-1 is isolated and purified by the methods described herein.
Example 4: S
D-1 E-2
[0404] Oxazole in THF is cooled to between about -80 °C and about -60 °C. Then, ft-butyllithium in hexanes is added while maintaining the temperature of the reaction below about -60 °C. The mixture is stirred at this temperature for 90 minutes. Zinc (II) chloride is added, maintaining the temperature of the mixture below about -60 °C, and the mixture is stirred at that temperature for about one hour before warming to about 10-20 °C. Compound D-1 is added to the reactor followed by tetrakis(triphenylphosphine)palladium(0) (“Pd(PPh3)4”), and the temperature is adjusted to between about 55-65 °C. The mixture is stirred at that temperature for about 12 hours to yield Compound E-2. Compound E-2 is isolated and purified by the methods described herein.
Example 5: Synthesis of Compound I
[0405] A sulfuric acid solution was prepared by addition of concentrated sulfuric acid (47 g,
4.7 w/w Compound E-2) to water (12 g, 1.2 v/w Compound E-2) followed by a water (15 g, 1.5 v/w Compound E-2) rinse forward. 2-Propanol (37 g, 4.7 v/w Compound E-2) was slowly charged to a reactor containing sulfuric acid solution at about 9 °C while maintaining the reaction contents at no more than about 40 °C, and the solution was cooled to about 5 °C .
Compound E-2 (10 g, 1.0 equiv) was charged to the solution, followed by a 2-propanol rinse forward (2 g, 0.25 v/w E-2). The contents were cooled to about 7 °C and stirred for a minimum of about 21 hours. The contents were slowly added into water, and the slurry was agitated for about 30 minutes. The slurry was filtered, and the filter cake was washed and dried under vacuum for about 4 hours. The crude wet cake was charged back to the reactor, followed by additions of ethyl acetate (40 g, 4.4 v/w Compound E-2) and water (100 g, 10 v/w Compound E-2). The slurry was adjusted to pH at about 8-9 with an about 20 wt% sodium hydroxide solution at about 22 °C, and then agitated for about 30 minutes at about 22 °C. The solution was allowed to settle. The top organic layer was collected and the bottom aqueous layer was washed with ethyl acetate (40 g, 4.4 v/w Compound E-2) at about 22 °C for about 30 minutes. The solution was allowed to settle, and the top organic layer was removed. 2-Methyltetrahydrofuran (86 g, 10 v/w Compound E-2) was then added, was adjusted to pH at about 4-5 with an about 4 N HCl solution at about 22 °C. The solution was agitated for about 30 minutes at about 22 °C and then allowed to settle. The bottom aqueous layer was extracted with 2-methyltetrahydrofuran (52 g, 6 v/w Compound E-2) at about 22 °C for about 30 minutes. After the solution was allowed to settle, the bottom aqueous layer was removed. The organic layers were combined and distilled under vacuum (jacket at about < 45 °C) to about 4V pot volume. Ethanol (55.4 g, 7 v/w
Compound E-2) was added and the reaction as distilled (repeated twice). Ethanol was again added (23.7 g,3 v/w Compound E-2), followed by water (30 g, 3 v/w Compound E-2). The reaction was heated to about 75 °C and then cooled over about 4 hours to about 50 °C, then to about 0 °C over about 5 hours. The reaction was then aged and filtered, and the solid was washed with a precooled mixture of ethanol (9.5 g, 1.2 v/w Compound E-2) and water (6 g, 0.6 v/w Compound E-2). The resulting product was washed to afford Compound of formula (I). ¾ NMR (400 MHz, CDCh): δ 7.70 (s, 1H), 7.57 (dd, J= 1.6 Hz, J= 7.6 Hz, 1H), 7.29 (td, J= 1.6 Hz, J = 8.0 Hz, 1H), 7.23 (d, J= 0.4 Hz, 1H), 7.02 (t, J= 7.6 Hz, 1H), 6.86 (d, J= 8.4 Hz, 1H), 5.39 (dd, J= 5.6 Hz, J= 8.0 Hz, 1H), 4.17-4.14 (m, 1H), 4.04 (br, 1H), 3.86 (s, 3H), 3.78-3.67 (m, 2H), 3.46-3.40 (m, 1H), 3.37-3.32 (m, 2H), 2.85 (s, 3H), 1.87 (s, 3H), 1.83 (s, 3H), 1.75-1.72 (m, 2H), 1.59-1.51 (m, 1H), 1.48-1.39 (m, 1H).
Example 6: Synthesis of Compound J-l
Step (a): Formation of Compound P-l
[0406] 2-Methoxyphenylmagnesium bromide (1 M in THF, 1.0 equiv.) was added to a solution of diethyl oxalate (1.1 equiv.) in THF (250 mL) at about -20 °C over approximately 20 min. After aging for about 45 min at about -20 °C, the resulting slurry was quenched with saturated NH4CI (250 mL) and was diluted with water (200 mL). This mixture was extracted with EtO Ac (400 mL), and the organic phase was washed with brine (200 mL). The organic phase was concentrated and the solvent was exchanged to THF. The resulting THF solution was used in the next step as is. ¾ NMR (400 MHz, CDCh): δ 7.90 (m, 1H), 7.61 (m, 1H), 7.10 (t, J = 7.6 Hz, 1H), 7.01 (d, J= 8.4 Hz 1H), 4.41 (q, J= 7.1 Hz, 2H), 3.88 (s, 3H), 1.41 (t, J= 7.1 Hz, 3H).
Alternate Preparation Compound P-l:
[0407] Anisole (1.0 equiv.) in THF (15 mL) was cooled to about -20 °C, and 2.5 M n-BuLi/hexane (1.1 equiv.) was added. The mixture was allowed to warm to about 0 °C and aged for about 2 hours, then warmed to room temperature overnight. The solution was then added to a solution of diethyl oxalate (4.0 equiv.) in THF (10 mL) at about -20 °C. The mixture was allowed to warm to about room temperature and aged for approximately 2 hours, then cooled to about 0 °C and quenched via addition of saturated NH4CI (30 mL). This mixture was extracted with EtOAc, and the organic phase was washed with brine and dried over MgSCk
Concentration afforded Compound P-1.
Alternate Preparation Compound P-1:
[0408] 2-Bromoanisole (1.0 equiv.) in THF (63 mL) was cooled to about -65 °C and 2.5M ft-BuLi/hexanes (1.0 equiv) was added. After aging for approximately 1 h, diethyl oxalate (4.0 equiv.) was charged, and the reaction mixture was allowed to warm to about room temperature. After approximately 1 h at about room temperature, the reaction mixture was cooled to about 0 °C, quenched by addition of saturated NH4CI (50 mL), and diluted with EtOAc. The aqueous phase was separated and was extracted with EtOAc. The combined organic phases were washed with brine and dried over MgS04. Concentration under high vacuum afforded a product that was passed through a plug of silica gel to afford Compound P-1.
Step (b): Hydrolysis of Compound P-1 and salt conversion to Compound O-l:
P-1 0-1
[0409] The resulting solution of ketoester, compound P-1, in THF (about 1.0 equiv.) was cooled over an ice bath and 2N NaOH (1.36 equiv.) was added. The reaction was agitated at about 0 °C and after reaction completion, the reaction was then acidified by addition of 6N HC1 (57 mL) to about pH<l and extracted with EtOAc (500 mL). The organic phase was washed with brine (200 mL). The organic phase was concentrated and then solvent exchanged to EtOAc. The resulting solution was cooled to about 0 °C and solid KOlBu (1.0 equiv.). The slurry was agitated for approximately 4 h and the solids were filtered, rinsed with EtOAc, and dried overnight at about 60 °C under vacuum to afford Compound O-l . ¾ NMR (400 MHz, DMSO-d6): 5 7.61 (d, J= 7.6 Hz, 1H), 7.49 – 7.41 (m, 1H), 7.04 (d, J= 8.4 Hz 1H), 6.96 (t, J = 7.4 Hz, 1H), 3.73 (s, 3H).
Step (c): Reduction of Compound O-l to Compound N-1:
0-1 N-1
[0410] To triethylamine (3.6 equiv.) precooled to about 0 °C, was added formic acid (9.0 equiv.) over about 30 min while maintaining a temperature less than about 30 °C. Solid RuCl (i?,i?)-Ts-DENEB catalyst (0.07 mol%) followed by ketoacid potassium salt (1.0 equiv.) were then charged to the mixture of triethylarnine/forrnic acid. The resulting slurry was warmed to about 50 °C and was stirred under nitrogen until the reaction was complete. The reaction was cooled over an ice bath and quenched by the addition of water (76 mL) followed by 10N NaOH (128 mL) to pH>13. Water (30 mL) and iPrAc (130 mL) were added and the organic layer was separated, and the aqueous phase was extracted with iPrAc (2 χ 130 mL). The aqueous phase was cooled and was acidified with concentrated HC1. This was extracted with iPrAc several times and the combined organic extract was concentrated and solvent exchanged to toluene, filtered hot, and then cooled to about 30 °C over approximately 2 h, aged for approximately 1 h, then filtered to afford solids that were then slurry-rinsed with toluene (50 mL) at room temperature and filtered. The wet cake was dried to afford Compound N-1. ¾ NMR (400 MHz, CDCh): δ 7.44 (d, J = 7.6 Hz, 1H), 7.40 – 7.36 (m, 1H), 7.06 (t, J = 7.6 Hz 1H), 6.98 (d, J = 8.4 Hz, 1H), 5.41 (s, 1H), 3.94 (s, 3H).
Step (d): Spiroketalization to afford Compound L-1:
N-1 L-1
[0411] Compound N-1 (1.0 equiv.), tetrahydropyran-4-one (compound M, 1.1 equiv.), and MTBE (30 mL) were sequentially charged and cooled to about 0 °C. Boron trifluoride THF complex (1.4 equiv.) was added over about 10 mins. After reaction completion, the reaction was slowly quenched with a pre-mixed solution of sodium bicarbonate (3.66 g) and water (40 mL). The solution was warmed to about 20 °C and diluted with toluene (40 mL) and stirred until dissolved. Agitation was stopped and the aqueous layer removed. The organic layer was washed with water (20 mL) and removed. The organic layer was collected and reactor rinsed forward with toluene (4 mL) to yield Compound L-1. ¾ NMR (400 MHz, CDCh): δ 7.42 – 7.38 (m, 1H), 7.32 (dd, J = 7.5, 1.5 Hz, 1H), 7.03 (t, J = 7.5 Hz, 1H), 6.98 (d, J = 8.3 Hz, 1H), 5.52 (s, 1H), 3.97 – 3.79 (m, 7H), 2.18 – 1.97 (m, 4H).
Step (e): Reduction of Compound L-1 to Compound K-l :
L-1 K-1
[0412] A stock solution of spiroketal, compound L-1, in MeTHF/MTBE (1.0 equiv.) was charged to a reactor. The solution was then distilled to about 4 volumes. MeTHF (187 mL) was charged, and distilled down to about 5 volumes. The solution was cooled to about 20 °C. DCM (90 mL) was charged and the solution was cooled to about 10 °C and tert-butyl magnesium chloride (2 M in diethyl ether) (5.0 equiv.) was added over approximately 45 mins. Following addition, the contents were cooled to about 7 °C and aged overnight at about 10 °C, then to about 0 °C. A premixed solution of HC1 (45 mL) and water (126 mL) was then slowly added. The aqueous bottom layer was drained and the aqueous layer extracted with MeTHF (93 mL). The combined organic layers were washed with water (37 mL) and the remaining organic layer was distilled down to about 4 volumes. Isopropyl acetate (181 mL) was charged and the solution reduced to about 5 volumes. The reaction was cooled to about 72 °C and heptanes (58 mL) was charged and the solution was held for about 1 hour before cooling to about 0 °C over approximately 5 hours. The slurry was agitated at about 0 °C for >12 h and then filtered, rinsed with an isopropyl acetate (9 mL) and heptanes (18 mL) mixture, followed by water (54 mL). The solids were dried to yield compound K-l. ¾ NMR (400 MHz, CDCh): δ 8.49 (br. s, 1 H), 7.42 – 7.29 (m, 2H), 6.98 (t, J= 7.4 Hz, 1H), 6.92 (d, 8.3 Hz, 1H), 5.43 (s, 1H), 3.96 (dt, J = 11.5, 4.3 Hz, 1H), 3.89 (dt, J = 11.5, 4.3 Hz, 1H), 3.85 (s, 3H), 3.67 – 3.58 (m, 1H), 3.47 – 3.30 (m, 2H), 2.03 – 1.93 (m, 1H), 1.84 – 1.75 (m, 1H), 1.75 – 1.56 (m, 2H).
Step (f): Reduction of Com ound K-l to Compound J-1:
J-1
K-1
[0413] A solution of acid, compound K-l (1.0 equiv.), in THF (90 mL) was cooled to about 0 °C and NaBH4 (1.2 equiv.) was added followed by BF3 THF complex (1.5 equiv.). The solution was warmed to about 20 °C and agitated until the reaction was deemed complete. Upon completion, MeOH (24 mL) was added to the reaction mixture after adjusting the temperature to about 5 °C, and was stirred until the gas evolution ceased. EtOAc (102 mL) was charged followed by saturated NLUClaq solution (87 mL). The agitation was stopped and the aqueous layer was removed. The organic layer was distilled down to about 3 volumes under vacuum, and then heptane (46 mL) was charged. The resulting mixture was cooled to about 0 °C and agitated at this temperature for approximately 4 h before being filtered and rinsed with heptane (3 mL). The resulting solids were dried to yield compound J-1. ¾ NMR (400 MHz, CDCh): δ 7.42 (d, J = 7.2 Hz, 1H), 7.27 (m, 1H), 6.98 (m, 1H), 6.87 (d, J = 8.4 Hz, 1H), 5.06 (dd, J = 8.4, 2.8 Hz, 1H), 3.93 (m, 2H), 3.82 (s, 3H), 3.67 (m, 1H), 3.55 – 3.46 (m, 2H), 3.41 – 3.32 (m, 2H), 2.27 (d, J = 8.0 Hz, 1H), 2.01 (m, 1H), 1.80 – 1.70 (m, 1H), 1.65 (m, 2H).
Step (g): Alternate Direct Reduction of Compound L-1 to Compound J-1:
L-1 J-1
[0414] To a solution of ketal, compound L-1 (1 equiv.), in diglyme (0.7 mL) was added NaBH4 (3.6 equiv.) followed by BF3 THF complex (4.5 equiv.). Reaction mixture was agitated for about 18 hours and was quenched by dropwise addition of MeOH (1 mL) followed by saturated Ν¾(¾ solution (1 mL). EtOAc (2 mL) was added, shaken well and the aqueous layer was removed. Organic solvent was removed under reduced pressure to obtain the crude compound J-1.
Example 7: Alternate Synthesis to Compound N-1
Step (a): Addition of hydrogen cyanide to ortho-anisaldehyde, compound U-1, to form compound T-1
[0415] To an Eppendorf tube was added ort/ro-anisaldehyde, compound U-1 (1.0 equiv), followed by 0.4 M sodium acetate buffer pH 5 (0.25 mL) and fert-butyl methyl ether (0.75 mL). The mixture was shaken using a thermomixer at about 30 °C and about 1200 rpm to ensure
complete dissolution of the aldehyde. Once this was complete acetone cyanohydrin (1.15 equiv) is added to the reaction mixture followed by hydroxynitrilase enzyme (2 mg). The Eppendorf tube was shaken in a thermomixer at about 30 °C and about 1200 rpm overnight. The Eppendorf tube was then heated to about 60 °C at about 1400 rpm for about 15 mins in order to denature the enzyme before being cooled to about 30 °C. The Eppendorf tube was then centrifuged at about 13,400 rpm for about 15 mins in order to pellet the denatured enzyme from the organic layer. The organic layer was removed and concentrated to dryness to give crude compound T-l . ¾ NMR (400 MHz, CDCh): δ 7.45 – 7.39 (m, 2H), 7.04 – 6.96 (m, 2H), 5.63 (s 1H), 3.94 (s, 3H), 3.75 (br, 1H).
Step (b): Hydrolysis of c
T-1 N-1
[0416] Before starting the reaction the following stock solutions were prepared: A solution of the crude cyanohydrin (compound T-l) in DMSO (about 100 mg/mL); a solution of 50 mM potassium phosphate (pH 7) containing 2 mM dithiothreitol (DTT); and 1 mM ethylenediamine tetraacetic acid (EDTA). To an Eppendorf tube was added nitrilase enzyme (4 mg) followed by 1.1 mL of the reaction buffer solution and 0.05 mL of the solution containing the crude cyanohydrin (about 10 mg). The Eppendorf tube was shaken in a thermomixer at about 30 °C and about 1200 rpm overnight. The Eppendorf tube was then heated to about 60 °C at about 1400 rpm for about 15 mins in order to denature the enzyme before being cooled to about 30 °C once more. The Eppendorf tube was centrifuged at about 13,400 rpm for about 15 mins in order to pellet the denatured enzyme and then separate it from the supernatant. The supernatant was either sampled directly for reverse phase UPLC or extracted with DCM for normal phase HPLC. In the case of DCM extraction, after separating the layers the organic layer was concentrated to dryness before the appropriate diluent was added for normal phase HPLC. UPLC analysis showed a peak with retention time identical to a reference standard of compound N-1.
Example 8: Alternate S nthesis to Compound N-1
P-1 V-1 N-1
Step (a): Reduction of Compound P-1 to form 2 ‘-methoxy-ethyl mandelate, Compound V-1:
P-1 V-1
[0417] The following stock solutions were made prior to the start of the reaction: a solution of starting material in DMSO (about 100 mg/ mL), NADP+ or NAD+ in 0.1M phosphate buffer (as appropriate) (2 mg/mL), glucose dehydrogenase in 0.1 M phosphate buffer (4 mg/mL), and glucose in 0.1 M phosphate buffer (20 mg/mL). To an Eppendorf tube is charged the ketoreductase enzyme (2 mg) followed by 0.25 mL of buffer solution containing NAD(P)+, 0.25 mL of buffer solution containing glucose dehydrogenase (GDH) and 0.5 mL of buffer solution containing glucose. Finally, 0.05 mL of the stock solution containing the starting material, compound P-1 in DMSO is added. The Eppendorf tube was then shaken in a thermomixer at about 30 °C and about 1200 rpm overnight. The Eppendorf tube was then heated to about 60 °C at about 1400 rpm for about 15 mins in order to denature the enzymes before being cooled to about 30 °C. The Eppendorf tube was then centrifuged at about 13,400 rpm for about 15 mins in order to pellet the denatured enzyme and the supernatant removed. This was either sampled directly for reverse phase UPLC or extracted with DCM for normal phase HPLC. In the case of DCM extraction after separating the layers the organic layer was concentrated to dryness before the appropriate diluent was added for normal phase HPLC. UPLC analysis showed a peak with retention time identical to a reference standard of the product material.
Step (b) Hydrolysis of 2 ‘-methoxy-ethyl mandelate, compound V-1, to provide compound N-1:
V-1 N-1
[0418] A solution of 2′ -methoxy-ethyl mandelate (1.0 equiv.) in EtOH (30 mL) was cooled to about 0 °C and 1.25 M NaOH (30 mL) was slowly added. Upon reaction completion, the reaction was adjusted to about pH 1 with 1M HC1 (40 mL). The mixture was extracted three times with ethyl acetate (30 mL) and the combined organics were washed with a brine solution (25 mL). The combined organic layers were dried over sodium sulfate, filtered, and the solvent removed under vacuum to provide the product. NMR data reported as above.
CLIP
https://cen.acs.org/articles/94/i39/silent-liver-disease-epidemic.html

/////// ND 630, NDI 010976, ND-630, NDI-010976, NIMBUS, GILEAD, 1434635-54-7, PHASE 2
FIRSOCOSTAT, ND 630, GS-0976, NDI-010976, FAST TRACK, CS-6509
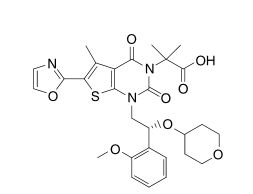
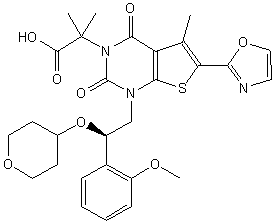
COc1ccccc1[C@H](CN2C(=O)N(C(=O)c3c(C)c(sc23)c4occn4)C(C)(C)C(=O)O)OC5CCOCC5
O=C(O)C(C)(C)N4C(=O)c1c(C)c(sc1N(C[C@H](OC2CCOCC2)c3ccccc3OC)C4=O)c5ncco5
Avoralstat, BCX4161,
CAS 918407-35-9
UNII: UX17773O15
513.5513, C28-H27-N5-O5
Hereditary angioedema (HAE)
Kallikrein inhibitor
BioCryst Pharmaceuticals
![]()
BioCryst is also investigating second-generation plasma kallikrein inhibitors to avoralstat, for treating HAE (in February 2016, this program was listed as being in preclinical development).
Prevent acute attacks in patients with hereditary angioedema (HAE); Treat hereditary angioedema (HAE)
U.S. – Fast Track (Treat hereditary angioedema (HAE));
U.S. – Orphan Drug (Prevent acute attacks in patients with hereditary angioedema (HAE))
26 Feb 2016Clinical trials in Hereditary angioedema (Prevention) in USA (PO, Hard-gelatin capsule) before February 2016
24 Feb 2016Discontinued – Phase-III for Hereditary angioedema (Prevention) in France (PO, Soft-gelatin capsule)
24 Feb 2016Discontinued – Phase-III for Hereditary angioedema (Prevention) in Germany (PO, Soft-gelatin capsule)

| Conditions | Interventions | Phases | Recruitment | Sponsor/Collaborators |
|---|---|---|---|---|
| Hereditary Angioedema|HAE | Drug: BCX4161|Drug: Placebo | Phase 2|Phase 3 | Recruiting | BioCryst Pharmaceuticals |
| Hereditary Angioedema | Drug: BCX4161|Drug: Placebo | Phase 2 | Completed | BioCryst Pharmaceuticals |
| Hereditary Angioedema | Drug: BCX4161 | Phase 1 | Completed | BioCryst Pharmaceuticals |
| Hereditary Angioedema | Drug: BCX4161 | Phase 1 | Completed | BioCryst Pharmaceuticals |
Avoralstat, also known as BCX-4161, is a potent and orally active Kallikrein inhibitor and Bradykinin inhibitor. Avoralstat may be potentially useful for treatment for Hereditary angioedema. Avoralstat inhibits plasma kallikrein and suppresses bradykinin production. Bradykinin is the mediator of acute swelling attacks in HAE patients.

Selective inhibitor of plasma kallikrein that subsequently suppresses bradykinin production
Hereditary angioedema (HAE) is a serious and potentially life-threatening rare genetic illness, caused by mutations in the C1-esterase inhibitor (C1 INH) gene, located on chromosome 11q. HAE is inherited as an autosomal dominant condition, although one quarter of diagnosed cases arise from a new mutation. HAE has been classed as an orphan disease in Europe, with an estimated prevalence of 1 in 50,000. Individuals with HAE experience recurrent acute attacks of painful subcutaneous or submucosal edema of the face, larynx, gastrointestinal tract, limbs or genitalia which, if untreated, may last up to 5 days. Attacks vary in frequency, severity and location and can be life-threatening. Laryngeal attacks, with the potential for asphyxiation, pose the greatest risk. Abdominal attacks are especially painful, and often result in exploratory procedures or unnecessary surgery. Facial and peripheral attacks are disfiguring and debilitating.
HAE has a number of subtypes. HAE type I is defined by C1 INH gene mutations which produce low levels of C1 -inhibitor, whereas HAE type II is defined by mutations which produce normal levels of ineffective C1 protein. HAE type III has separate pathogenesis, being caused by mutations in the F12 gene which codes for the serine protease known as Factor XII. Diagnostic criteria for distinguishing the subtypes of HAE, and distinguishing HAE from other angioedemas, can be found in Ann Allergy Asthma Immunol 2008; 100(Suppl 2): S30-S40 and J Allergy Clin Immunol 2004; 114: 629-37, incorporated herin by reference.
Current treatments for HAE fall into two main types. Older non-specific treatments including androgens and antifibrinolytics are associated with significant side effects, particularly in females. Newer treatments are based on an understanding of the molecular pathology of the disease, namely that C1 INH is the most important inhibitor of kallikrein in human plasma and that C1 INH deficiency leads to unopposed activation of the kallikrein-bradykinin cascade, with bradykinin the most important mediator of the locally increased vascular permeability that is the hallmark of an attack.
Approved therapies include purified plasma-derived C1 INH (Cinryze®, Berinert), the recombinant peptide kallikrein inhibitor ecallantide (Kalbitor®), and the bradykinin receptor B2 inhibitor iticabant (Firazyr®). All of the currently available targeted therapies are administered by intravenous or subcutaneous injection. There is currently no specific targeted oral chronic therapy for HAE.
There are many delivery routes for active pharmaceutical ingredients (APIs). Generally, the oral route of administration is favored. Oral administration provides a number of advantages, such as, but not limited to, patient convenience, flexibility of timing of administration, location of administration and non-invasiveness. Oral administration also provides more prolonged drug exposure compared with intermittent intravenous infusion, which may be important for drugs with schedule-dependent efficacy. For example, a drug with a short half-life can achieve a greater exposure time by either continuous infusion or by continuous oral dosing. The use of oral therapy further has the potential to reduce the cost of healthcare resources for inpatient and ambulatory patient care services.
In the pharmaceutical arts, it is known that a number of APIs cannot be administered effectively by the oral route. The main reasons why these compounds cannot be administered by the oral route are: a) rapid enzymatic and metabolic degradation; b) chemical and/or biological instability; c) low solubility in aqueous medium; and/or d) limited permeability in the gastrointestinal tract. For such compounds, non-oral routes of delivery, such as parenteral administration, mainly via intramuscular or subcutaneous injections, may be developed. However, non-oral administration poses a disadvantage for the patient as well as healthcare providers, and for this reason, it is important to develop alternative routes of administration for such compounds, such as oral routes of administration.
While the oral route of administration is the most convenient for the patient and the most economical, designing formulations for administration by the oral route involves many complications. Several methods are available to predict the ease by which an API may be formulated into a formulation suitable for administration by the oral route. Such methods include, but are not limited to, and Lipinski rule (also referred to as the Rule of Five) and the Biopharmaceutical Drug Disposition Classification System (BDDCS).
The BDDCS divides APIs into four classifications, depending on their solubility and permeability. Class I APIs have high solubility and high permeability; Class II APIs have low solubility and high permeability; Class III APIs have high solubility and low permeability; and Class IV APIs have low solubility and low permeability. APIs in higher classes in the BDDCS face greater challenges in formulating into an effective, pharmaceutically acceptable product than those in lower classes. Of the four classes, APIs falling into Class IV are the most difficult to formulate into a formulation for administration by the oral route that is capable of delivering an effective amount of the API as problems of both solubility and permeability must be addressed (note the BDDCS does not inherently address chemical stability). The role of BDDCS in drug development is described generally in L.Z. Benet J Pharm Sci. 2013, 102(1), 34-42.
Lipinski’s rule (described in Lipinski et al. Adv. Drug Deliv. Rev. 46 (1-3): 3-26) states, in general, that in order to develop a successful formulation for administration by the oral route, an API can have no more than one violation of the following criteria:
i) not more than 5 hydrogen bond donors (nitrogen or oxygen atoms with one or more hydrogen atoms)
ii) not more than 10 hydrogen bond acceptors (nitrogen or oxygen atoms) iii) a molecular mass less than 500 daltons
iv) an octanol-water partition coefficient log P not greater than 5.
J. Zhang et al. Medicinal Chemistry, 2006, 2, 545-553, describes a number of small molecule amidine compounds which have activity as inhibitors of kallikrein. The molecules described in this document fall into Class IV of the BDDCS as described above. The compounds are poorly soluble in aqueous and physiological fluids, and are poorly permeable as demonstrated by oral dosing in rats and in vitro experiments with Caco-2 cells.
Furthermore, 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid, one of the compounds described in Zhang et al., is a Class IV API and violates criteria iii) and iv) as set forth in the Lipinski Rule.
Furthermore, the compounds described in Zhang et al., including 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid, exhibit poor stability with respect to oxidation in air, to light
(photodegradation) and in aqueous and physiological fluids, as well as to elevated temperatures.
Therefore, the compounds described by Zhang et al. including, but not limited to, 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid, not only exhibit poor solubility and permeability characteristics, but also poor stability characteristics. As a result, such compounds are predicted to be especially difficult to formulate into an effective, orally deliverable
pharmaceutical composition that is capable of delivering an effective amount of the compound to a subject.
Polymorphism, the occurrence of different crystal forms, is a property of some molecules. A single molecule may give rise to a variety of polymorphs having distinct crystal structures and physical properties, such as, but not limited to, melting point, thermal behaviors (e.g. measured by thermogravimetric analysis (TGA), or differential scanning calorimetry (DSC), x-ray diffraction pattern, infrared absorption fingerprint, and solid state NMR spectrum. One or more of these techniques may be used to distinguish different polymorphic forms of a compound.
Discovering new polymorphic forms and solvates of a pharmaceutical product can provide alternate forms of the compound that display a number of desirable and advantageous properties, such as, but not limited to, ease of handling, ease of processing, ease of formulation, storage stability, and/or ease of purification. Further, new polymorphic forms and solvates of a pharmaceutically useful compound or salts thereof may further provide for improved pharmaceutical products, by providing compounds that are more soluble in a set of pharmaceutical excipients. Still further, the provision of new polymorphic forms and solvates of a pharmaceutically useful compound or salts thereof enlarges the repertoire of compounds that a formulation scientist has available for formulation optimization, for example by providing a pharmaceutical product with different properties, such as, but not limited to, improved processing characteristics, improved handling characteristics, improved solubility profiles, improved dissolution profile and/or improved shelf-life. Therefore, there is a need for additional polymorphs of pharmaceutically useful compounds, such as, but not limited to, 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6- (cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid and the compounds disclosed herein.
In one aspect, the present invention provides an oral formulation that is capable of delivering an effective amount of the amidine compounds described by Zhang et al. to a subject. In particular, the present invention provides an oral formulation that is capable of delivering an effective amount of 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid to a subject. In one specific aspect, the 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid is present in a particular crystal form designated Form A. In light of the art suggesting the difficulties in formulating such an oral formulation, this result was unexpected.
As described herein, the amidine compounds described in Zhang et al., including, but not limited to, 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6- (cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid (specifically including particular crystal Form A), may now be conveniently used in oral administration and further used in oral administration for the treatment of a number of diseases and conditions in a subject, such as, but not limited to, HAE as described herein.
![]()
May 16 is HAE awareness day
See BioCryst’s video regarding HAE to learn more
Avoralstat is being developed as an oral prophylactic treatment for patients suffering from Hereditary Angioedema (HAE). Avoralstat inhibits plasma kallikrein and suppresses bradykinin production. Bradykinin is the mediator of acute swelling attacks in HAE patients.
In May 2014 BioCryst, announced that the OPuS-1 (OralProphylaxiS-1) Phase 2a proof of concept clinical trial met its primary efficacy endpoint, several secondary endpoints and all other objectives established for the trial. OpuS-1 enrolled 24 HAE patients with a history of HAE attack frequency of at least 1 per week. Treatment with avoralstat demonstrated a statistically significant mean attack rate reduction of 0.45 attacks per week versus placebo, p<0.001. The mean attack rate per week was 0.82 on BCX4161 treatment, compared to 1.27 on placebo.
In December 2014, BioCryst initiated enrollment in OPuS-2 (Oral ProphylaxiS-2). OPuS-2 is a blinded, randomized, 12-week, three-arm, parallel cohort design trial evaluating the efficacy and safety of two different dose regimens of avoralstat administered three-times daily, 300 mg and 500 mg, compared with placebo. The primary efficacy endpoint for the trial will be the mean angioedema attack rate, which will be reported for each avoralstat dose group compared to placebo. The trial is being conducted in the U.S., Canada and Europe. On October 8, 2015, announced that it has completed enrollment of approximately 100 HAE patients with a history of moderately frequent to very frequent attacks in OPuS-2. BioCryst expects to report the OPuS-2 trial results in early 2016.
PATENT
WO200234711
http://www.google.com/patents/WO2002034711A1?cl=en
PATENT
PATENT
Examples
Example 1 – Synthesis of 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl- phenyll-6-(cvclopropylmethyl-carbarnoyl)-pyridine-2-carboxylic acid
The synthesis of the above compound and intermediates is described below. In this section, the following abbreviations are used:

The synthesis of starting material, (4-(benzyloxy)-2-formyl-5-methoxyphenyl)boronic acid (1f) is described in Scheme 1.
f 0HCY ° ΒΓΥΥ°

Preparation of 6-bromobenzofdl[1,3ldioxole-5-carbaldehvde (1b)

1a 1b
To a mixture of piperonal (1a) (498 g, 3.32 mol) in glacial acetic acid (1000 mL) was added a solution of bromine (200 mL, 3.89 mol) in glacial acetic acid (500 mL) over a period of 30 min and stirred at room temperature for 24h. The reaction mixture was poured into water (2000 mL) and the solid that separated was collected by filtration. The solid was dissolved in boiling ethanol (4000 mL) and cooled to room temperature. The solid obtained on cooling was collected by filtration to furnish 6-bromobenzo[d][1 ,3]dioxole-5-carbaldehyde (lb) (365 g, 48 %) as a white solid, MP 126 °C; HNMR (300 MHz, DMSO-d6): δ 10.06 (s, 1 H), 7.42 (s,1 H), 7.29 (s, 1 H), 6.20 (d, J=12.3, 2H); IR (KBr) 3434, 2866, 1673,1489, 1413, 259, 1112, 1031 , 925 cm“1; Analysis calculated for CeH5BrO3.O 25H C, 41.15; H, 2.37; Found: C, 41.07; H, 2.11.
Preparation of 2-bromo-5-hvdroxy-4-methoxybenzaldehyde (1c)

1c
A solution of potassium tert-butoxide (397 g, 3.36 mol) in DMSO (1.5 L) was heated at 50 °C for 30 min. Methanol (1.5 L) was added to it and continued heating at 50 °C for additional 30 min. To the hot reaction mixture was added 6-bromo-benzo[d][1,3]dioxole-5-carbaldehyde (1 b) (350g, 1.53 mol) and continued heating at 50 °C for 30 min. The reaction mixture was cooled to room temperature and quenched with water (2.3 L) and sodium hydroxide (61.2 g, 1.53 mol). The reaction mixture was washed with ether (2 x 1.5 L), acidified to pH 2 using cone. HCI and extracted with ethyl acetate ( 1 L). The ethyl acetate layers were combined and concentrated under vacuum to dryness. The residue obtained was treated with water (1.5 L) and ethyl acetate (1 L). The solid obtained was collected by filtration to furnish 2-bromo-5-hydroxy-4-methoxybenzaldehyde (1c) (97 g, 27.5% as a first crop). The layers from the filtrate were separated and aqueous layer was extracted with ethyl acetate (200 ml_). The ethyl acetate layers were combined dried over MgS04 and concentrated under vacuum to dryness to furnish 2-bromo-5-hydroxy-4-methoxybenzaldehyde (1c) (192 g, 54.4%, second crop) as an orange solid, MP 108 °C; ‘HNMR (300MHz, DMSO-cfe): S 10.00 (s, 1 H), 9.92 (s,1 H), 7.27 (s, 1 H), 7.26 (s, 1 H), 3.93 (s, 3H); IR (KBr) 3477, 2967, 2917,
2837, 2767, 2740, 1657, 1595, 1428, 1270, 1210, 1164, 1022 cm“‘; Analysis calculated for C8H7Br03.H20: C, 38.58; H, 3.64: Found: C, 38.60; H, 3.60.
Preparation of 5-(benzyloxy)-2-bromo-4-methoxybenzaldehvde ( d)

To a solution 2-bromo-5-hydroxy-4-methoxybenzaldehyde (1c) (120 g, 520 mmol) in DMF (1000 mL) was added potassium carbonate (79 g, 572 mmol) and benzyl bromide (68 mL, 572 mmol). The reaction mixture was stirred at room temperature overnight and quenched with water (3000 mL). The solid obtained was collected by filtration, washed with ether and dried under vacuum to furnish 5-(benzyloxy)-2-bromo-4-methoxybenzaldehyde (1d) (113.19 g, 67.9%) as a white solid, MP 144 °C;1HNMR (300 MHz, DMSO-c/6): δ 10.06 (s, 1H), 7.47-7.34 (m, 7H), 5.17 (s, 2H), 3.92 (s, 3H); IR (KBr) 2898, 2851 , 1673, 1592, 1502, 1437, 1402, 1264, 1210, 1158, 1017, 754 cm“1; Analysis calculated for C 5H13Br03: C, 56.10; H, 4.08; Found: C, 55.44; H, 4.08.
Preparation of 1-(benzyloxy)-4-bromo-5-(diethoxymethyl)-2-methoxybenzene (1e)
15 046578
146

1d 1e
To a solution of 5-(benzyloxy)-2-bromo-4-methoxybenzaldehyde (1d) (100 g, 311 mmol) in
ethanol (1500 mL) was added triethyl orthoformate (103 mL, 622 mmol), ammonium nitrate
(7.5 g, 93.3 mmol) and stirred at room temperature overnight. The reaction mixture was
treated with ether (1200 mL) and stirred for 15 min before filtration. The filtrate was
concentrated under vacuum to dryness to give 1-(benzyloxy)-4-bromo-5-(diethoxymethyl)-2-methoxybenzene (1e) (134 g) as a brown syrup; The product was used in the next step
without further purification; 1H N R (300 MHz, DMSO-cf6) δ 7.45 – 7.37 (m, 4H), 7.36 – 7.33
(m, 1 H), 7.17 – 7.14 (m, 1 H), 7.10 (s, 1 H), 5.10 (s, 2H), 3.80 (s, 3H), 3.58 – 3.33 (m, 5H),
1.13 – 1.07 (m, 6H); IR (KBr) 2974, 2879, 1601 , 1503, 1377, 1260, 1163, 1060 cm“1;
Analysis calculated for C19H23Br04: C, 57.73; H, 5.86; Found: C, 57.21 ; H, 5.94.
acid (1fi

To a solution of 1-(benzyloxy)-4-bromo-5-(diethoxymethyl)-2-methoxybenzene (1e) (120 g,
300 mmol) in dry ether (1000 mL) at -78 °C was added n-butyllithium (1.6 M solution in
hexanes, 244 mL, 390 mmol) over a period of 30 min and further stirred at -78 °C for 30 min.
A solution of tri-n-butylborate (110 mL, 405 mmol) in dry ether (300 mL) was added to this
solution at -78 °C over a period of 30 min. The reaction mixture was further stirred for 2 h at -78 °C and warmed to 0 °C. The reaction mixture was quenched with 3N HCI (300 mL) at 0
°C and heated at reflux for 1 h. After cooling to room temperature, the solid obtained was
collected by filtration washed with water (250 mL) dried in vaccum to afford (4-(benzyloxy)-2-formyl-5-methoxyphenyl)boronic acid (1f) (30.85 gm, 37.6% as a white solid. The organic
layer from above filtrate was extracted with 1.5 N NaOH (3 x 200 mL). The combined basic
extracts were acidified with cone. HCI (pH about 4). The solid obtained was collected by
filtration, washed with water and dried under vacuum to furnish a second crop of (4-(benzyloxy)-2-formyl-5-methoxyphenyl)boronic acid (1f) (22.3 g, 26%) as a light orange solid
MP 158 °C; 1H NMR (300 MHz, DMSO-cfe) δ 10.08 (s, 1 H), 7.52 (s, 1 H), 7.48 – 7.33 (m, 5H),
7.24 (s, 1H), 5.18 (s, 2H), 3.89 (s, 3H); 1H NMR (300 MHz, DMSO-d6/D20) δ 10.06 (s, 1H),
7.52 (s, 1H), 7.49 – 7.32 (m, 5H), 7.23 (s, 1 H), 5.18 (s, 2H), 3.89 (s, 3H); MS (ES+) 309.1 (M+Na); IR (KBr) 3335, 2937, 1647, 1545, 1388, 1348, 1268, 1146, 1095 cm-1; Analysis calculated for C15H15BO5.0.25H2O: C, 62.00; H, 5.38; Found: C, 61.77; H, 5.19.
Synthesis of methyl-6-(cvclopropylmethylcarbamoyl¾-3-ftrifluoromethylsulfonyloxyVpicolinate
The synthesis of the intermediate methyl 6-(cyclopropylmethylcarbamoyl)-3-(trifluoromethyl sulfonyloxy)picolinate (2h) is described in Scheme 2.

Preparation of 2-bromo-3-hvdroxy-6-methylpyridine (2b)

H3C N Br
2a 2b
To a solution of 3-hydroxy-6-methylpyridine (2a) (3000 g, 27.5 mol) in pyridine (24 L) cooled to 15 °C was added a solution of bromine (4.83 kg, 1.55 L, 30.2 mol) in pyridine (3 L) over a period of 50 min maintaining the internal temperature between 20 to 25 DC. After stirring for 19 h at room temperature the solvent was removed under vacuum and the residue was triturated with water. The solid separated was collected by filtration, washed with water and dried under vacuum to give 2-bromo-3-hydroxy-6-methylpyridine (2b) (3502 g, 67.7 %) as a light brown solid which was used as such without further purification; 1H NMR (300 MHz, DMSO-d6) δ 10.43 (s, 1H), 7.18 (d, J = 8.0 Hz, 1 H), 7.08 (d, J
MS (ES+) 188.35, 186.36 (M+1).
(2c)

2b 2c
A mixture of 2-bromo-3-hydroxy-6-methylpyridine (2b) (3000 g, 15.96 mol), anhydrous potassium carbonate (3308 g, 23.94 mol), and iodomethane (2.491 kg, 1.09 L, 17.556 mol) in 30 L of acetone was heated at 40 °C overnight. The reaction mixture was cooled to room temperature and filtered through Celite. Evaporation of the solvent followed by silica gel chromatography (Hexane: ethyl acetate = 7:3) afforded the desired compound, 2-bromo-3-methoxy-6-methylpyridine (2c) which was used as such for the next step; 1H NMR (300 MHz, DMSO-cfe) δ 7.42 (dd, J = 8.3, 1.5 Hz, 1H), 7.29 – 7.19 (m, 1H), 3.84 (d, J = 1.6 Hz, 3H), 2.37 (d, J = 1.7 Hz, 3H).

2c
2d
To a solution of 2-bromo-3-methoxy-6-methylpyridine (2c) (310 g, 1.53 mol) in 6000 mL of water at 60 °C was added KMnO, (725 g, 4.59 mol) in small portions over a 90 min period with vigorous mechanical stirring. A dark purple solution resulted. This solution was kept at 90 °C for a further 3 h and filtered through Celite while still hot to give a colourless filtrate.
After cooling, the aqueous solution was acidified to pH 1-2 by adding 6 N HCI. The white solid obtained was collected by filtration to give on drying 6-bromo-5-methoxy-2-pyridinecarboxylic acid (2d) (302g, 85%) of product, which was used as such in the next reaction without further purification. An analytical sample was obtained by recrystallization from methanol to give 6-bromo-5-methoxy-2-pyridinecarboxylic acid; 1H NMR (300 MHz, DMSO-tfe) δ 7.40 – 7.28 (m, 1H), 7.17 (d, J = 8.3 Hz, 1 H), 3.83 (d, J = 1.7 Hz, 3H).
Preparation of 6-bromo-N-(cvclopropylmethyl)-5-methoxypicolinamide (2e)

To a solution of 6-bromo-5-methoxy-2-pyridinecarboxylic acid (2d) (12 g, 52 mol) in pyridine (70 mL) was added EDCI (11.5 g, 59 mmol) and cyclopropylmethylamine (3.6 g, 52 mmol). The reaction mixture was stirred at room temperature overnight and then concentrated under vacuum. The reaction mixture was diluted with water (100 mL) and ethyl acetate (100 mL). The organic layer was separated and the water layer was extracted with ethyl acetate (2 x 100 mL). The organic layers were combined and washed with water (2 x 50 mL), brine (500 mL), dried over magnesium sulphate, filtered and concentrated under vacuum to furnish 10.43g of crude product. The crude product was converted into a slurry (silica gel 20 g) and purified by flash column chromatography (silica gel 230 g, eluting with 0-100% ethyl acetate in hexane) to yield compound 6-bromo-N-(cyclopropylmethyl)-5-methoxypicolinamide (2e) (8.02 g, 54%) as off white solid, mp 67-70 °C; 1HNMR (300 MHz, DMSO-d6) δ 8.51 (t, J = 5.8, 1 H), 8.02 (d, J = 8.4, 1 H), 7.65 (d, J = 8.5, 1 H), 3.96 (s, 3H), 3.14 (t, J = 6.5, 2H), 1.11 -0.99 (m, 1 H), 0.47 – 0.36 (m, 2H), 0.27 – 0.20 (m, 2H); MS (ES+) 307.0, 309.0 (100%
M+Na)
Preparation of methyl 6-(cvclopropylmethylcarbamoyl)-3-methoxypicolinate (2f)

To a solution of 6-bromo-N-(cyclopropylmethyl)-5-methoxypicolinamide (2e) (7.5 g, 27.6 mol) in methanol (300 mL) in a 2-L stainless steel bomb was added Pd(OAc)2(750 mg), 1 ,1-bis(diphenylphosphino)-ferrocene (750 mg), and triethylamine (3.9 mL, 27.6 mmol). The reaction mixture was vacuum flushed and charged with CO gas to 150 psi. The reaction mixture was and heated with stirring at 150°C overnight and cooled to room temperature. The catalyst was filtered through a pad of celite, and concentrated to dryness to furnish crude product. The crude was purified by flash column chromatography (silica gel 150 g,
eluting with, 0%, 5%, 10%, 20%, 30%, 50% ethyl acetate/hexanes (250 mL each) as eluents to give methyl 6-(cyclopropylmethyl-carbamoyl)-3-methoxypicolinate (2f) (6.29 g, 86.1 %) as a salmon coloured solid, MP 107 °C; 1HNMR (300 MHz, DMSO-cfe) δ 8.28 (t, J = 6.0, 1H), 7.91 (d, J = 8.8, 1H), 7.55 (d, J = 8.8, 1 H), 3.68 (s, 3H), 3.64 (s, 3H), 2.90 (t, J = 6.5, 2H), 0.89 – 0.68 (m, 1 H), 0.26 – 0.09 (m, 2H), 0.08 – 0.00 (m, 2H); MS (ES+) 287.1 (M+Na); IR (KBr) 3316, 2921 , 1730, 1659, 1534, 1472, 1432, 1315, 1272, 1228, 1189, 1099, 1003, 929, 846, 680 cm“1; Analysis calculated for C13H16 204: C, 59.08; H, 6.10; N, 10.60; Found: C, 58.70; H, 5.97; N, 10.23.
Preparation of 6-(cvclopropylmethylcarbamoyl 3-hvdroxypicolinic acid (2q)

2f 2g
Aluminium chloride method:
To a solution of methyl 6-(cyclopropylmethylcarbamoyl)-3-methoxypicolinate (2f) (0.16 mmol) in dichloromethane (840 mL) was added AICI3 (193 g, 1.5 mol). The reaction mixture was heated at reflux for 12 h under nitrogen. After slowly adding ~2L of 1 N HCI, the organic layer was separated. The aqueous layer was re-extracted several times with ethyl acetate/DME. The combined organic layer was washed with brine, dried (MgSO.4), and evaporated in vacuo to furnish crude 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid. To a solution of 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid was added a solution of acetyl chloride (1 10 mL) in methanol (1.1 L). The reaction mixture was stirred for 12 h at room temperature and then concentrated to dryness in vacuo. After co-evaporating once with methanol, the compound was purified by flash-column chromatography (silica gel, 500 g, eluted with chloroform and 3% methanol in chloroform) to furnish 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid (2g).
Boron tribromide method:
To a stirring solution of methyl 6-(cyclopropylmethylcarbamoyl)-3-ethoxypicolinate (2f) (58.0 g, 208 mmol) was added BBr3 (79 mL, 834 mmol) in CH2CI2 (1.3 L) at 0-5 °C. The reaction mixture was allowed to warm to room temperature and stirred for 18h. The reaction mixture was evaporated to dryness and anhydrous methanol (1 L) was added to the light yellowish solid residue. Insoluble solid was collected by filtration (36 g). Mother liquor was evaporated and co-evaporated with MeOH (2 x 200 mL). The insoluble solid (36 g) was treated with MeOH (500 mL) and acetyl chloride (50 mL) and stirred at room temperature for 18 h (at this point reaction mixture was clear). The mixture was evaporated to dryness and diluted with water and extracted with EtOAc. White solid that separated out from EtOAc layer was collected by filtration, washed with water (2 x 20 mL), dried in vacuo at 50 °C to afford 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid (2g) (5.36 g, 10 %) as a white solid, MP 92-95 °C. 1HNMR (DMSO-cfe) δ 11.04 (s, 1 H, exchangeable with D20), 8.37 (t, J = 6.0, 1 H, exchangeable with D20), 8.12 (d, J = 8.7 Hz, 1 H), 7.57 (d, J = 8.7 Hz, 1 H), 3.90 (m, 3 H), 3.15 (m, 2 H), 1.04 ( m, 1 H), 0.41 (m, 2 H), 0.24 (m, 2 H). IR (KBr): 3346, 3205, 1684 cm“1; MS (ES+): 251.1 (M+1); Analysis calculated for C12H14N2O4.0.1 H2O: C, 57.18; H, 5.67; N, 11.14; Found: C, 57.11 ; H, 5.61; N, 11.09.
Preparation of methyl-6-(cvclopropylmethylcarbamoyl)-3-(trifluoromethylsulfonyloxy) picolinate (2h

To a solution of 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid (2g) (28 mmol) in DMF (200 mL) were added triethylamine (12 mL, 84 mmol) and N-phenyl-bis(trifluoromethanesulfonimide) (12 g, 34 mmol). The reaction mixture was stirred for 1.5 h at room temperature and then poured into ice. After diluting with water and extracting with ethyl acetate, the aqueous phase was re-extracted, and then the combined organic layer was washed with water and concentrated under vacuum to give methyl-6-(cyclopropylmethylcarbamoyl)-3-(trifluoromethylsulfonyloxy)picolinate (2h), which was used in the next step without purification.
1H NMR (300 MHz, CDCI3) δ 8.50 (d, J = 8.6, 1 H), 8.07 (s, 1 H), 7.88 (d, J = 8.6, 1 H), 4.09 (d, J = 12.6, 3H), 3.48 – 3.24 (m, 2H), 1.18 – 1.01 (m, 1 H), 0.69 – 0.44 (m, 2H), 0.42 – 0.20 (m, 2H). MS (ES*): 405.17, 100%, M+Na.
Synthesis of 3-f2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyll-6-(cvclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid:
The synthesis of 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid (3i) is described as shown in Scheme 3.

3-f4-Benzyloxy-2-formyl-5-methoxy-phenylV6-(cvcloDroDvlmethvl-carbarnovn-pyridine-2-carboxylic acid methyl ester (3a)
5 046578
153

3a
To a solution of methyl-6-(cyclopropylmethylcarbamoyl)-3-(trifluoromethylsulfonyloxy)
picolinate (2h) (24.3g, 63 mmol) in DME (225 mL) were added water (25 mL), (4- (benzyloxy)-2-formyl-5-methoxyphenyl)boronic acid (1f) (27.3 g, 95 mmol), NaHC03(15.9 g,
5 189 mmol), and bis(triphenylphosphine)palladium(ll) chloride (0.885 g). The reaction
mixture was stirred at 70°C overnight under nitrogen. After extracting with ethyl acetate, the organic layer was washed with water and brine and dried (MgSO^), and then concentrated
under vacuum. The compound was purified by flash-column chromatography (silica gel, 300 g, eluting with 10%, 20%, 30% and 40% ethyl acetate in hexane) to furnish 3-(4-benzyloxy- 10 2-formyl-5-methoxy-phenyl)-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid
methyl ester (3a) (25 g, 83%) as off white solid, MP 48-50°C: 1H NMR (300 MHz, DMSO-cfe) δ 9.61(s, 1 H), 8.40 (d, J= 7.9 Hz, 1H), 8.14 (t, J= 5.0 Hz, 1H), 7.87 (d, J= 8.1 Hz, 1 H), 7.58
(s, 1H), 7.54-7.30 (m, 5H), 6.71 (s, 1 H), 5.24 (s, 2H), 3.93 (s, 3H), 3.70 (s, 3H), 3.45-3.34 (m,
2H), 1.19-1.05 (m, 1 H), 0.64-0.54 (m, 2H), 0.37-0.30 (m, 2H); IR ( Br) 1735, 1678, 1594,
15 1513, 1437, 1283, 1217, 1141, 1092 cm“1; MS (ES+) 497.29 (M+Na); Analysis calculated for
C27H2eN206: C, 68.34; H, 5.52; N, 5.90; Found; C, 68.16; H, 5.62; N, 5.80.
2-(6-(Cvclopropylmethylcarbamoyl)-2-(methoxycarbonyl)pyridin-3-vn-4-methoxy-5- vinylbenzoic acid (3b)

To a solution of 3-(4-benzyloxy-2-formyl-5-methoxy-phenyl)-6-(cyclopropylmethyl- carbamoyl)-pyridine-2-carboxylic acid methyl ester (3a) (24g, 50.6 mmol) in acetonitrile (50
mL), 2-methyl-2-propanol (350 mL), and water (125 mL) were added sodium dihydrogen
phosphate (12.5 g) and 2-methyl-2-butene (55 mL, 519 mmol). The reaction mixture was cooled in an ice bath and then sodium chlorite (28 g) was added. After stirring for 1 h, the reaction mixture was extracted with ethyl acetate and washed with water. The aqueous layer was re-extracted and then the combined organic layers were dried (MgS04). The solvent was evaporated in vacuo to furnish 5-(benzyloxy)-2-(6- ((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxybenzoic acid (3b) (29 g) which was used for the next step. MS (ES+): 513.24, (M+Na(; (ES ): 489.26, M-1.
Methyl 3-(4-(benzyloxy)-5-methoxy-2-(((2-methoxyethoxy)methoxytoarbonyltohenyl)-6-(cvclopropylmethylcarbamovnpicolinate (3c)

To a mixture of 5-(benzyloxy)-2-(6-(cyclopropylmethylcarbamoyl)-2-(methoxy-carbonyl)pyridin-3-yl)-4-methoxybenzoic acid (3b) (31 g, 63.2 mmol), and triethylamine (17.7 mL, 126.4 mmol) in dichloromethane (300 mL), was added MEM-chloride (9.03 mL, 79 mmol), and stirred at room temperature overnight. The reaction mixture was diluted with water and extracted with ethyl acetate. The organic layer was washed with water and dried over MgS04, filtered and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, 40 g) to furnish methyl 3-(4-(benzyloxy)-5-methoxy-2-(((2-methoxyethoxy)methoxy)carbonyl)phenyl)-6-(cyclopropylmethylcarbamoyl)picolinate (3c) (32.8 g, 89%) as a thick gum; H NMR (300 MHz, CDCI3) δ 8.35 (d, J = 8.0 Hz, 1 H), 8.15 (t, J = 5.7 Hz, 1 H), 7.78 (d, J = 8.0 Hz, 1H), 7.71 (s, 1H), 7.49 (d, J = 6.8 Hz, 2H), 7.36 (ddd, J = 7.5, 14.8, 22.4 Hz, 3H), 6.66 (s, 1 H), 5.37-5.13 (m, 4H), 3.90 (s, 3H), 3.69 (s, 3H), 3.60-3.49 (m, 2H), 3.49 (s, 2H), 3.39 (dd, J = 4.4, 8.4 Hz, 2H), 3.34 (s, 3H), 1.19-1.00 (m, 1H), 0.57 (q, J = 5.8 Hz, 2H), 0.38-0.25 (m, 2H). MS (ES+): 601.24 (M+Na); (ES“): 577.27 (M-1);1H NMR (300 MHz, DMSO-cfe) δ 8.69 (t, 7 = 6.1 Hz, 1H), 8.20 (d, J = 8.0 Hz, 1H), 7.97 (d, J = 8.0 Hz, 1 H), 7.63 (s, 1H), 7.41 (m, 5H), 6.92 (s, 1 H), 5.20 (m, 4H), 3.83 (s, 3H), 3.57 (s, 3H), 3.44 (m, 2H), 3:33 (m, 2H), 3.21 (m, 5H), 1.14 (m, 1H), 0.44 (m, 2H), 0.27 (m, 2H). IR (KBr):
1732, 1671 cm“1. MS (ES+): 601.1(M+Na); Analysis calculated for C31H 2Oe: C, 64.35; H, 5.92; N, 4.84; Found: C, 64.27; H, 6.04; N, 4.79.
Methyl 6-(cvclopropylmethylcarbamoyl)-3-(4-hvdroxy-5-methoxy-2-(((2-methoxyethoxy¾methoxy)carbonyl)phenyl)picolinate (3d)

3c 3d
To a solution of methyl 3-(4-(benzyloxy)-5-methoxy-2-(((2-methoxyethoxy)methoxy)-carbonyl)phenyl)-6-(cyclopropylmethylcarbamoyl)picolinate (3c) (32.8 g, 56.68 mmol) in ethanol (650 mL) was added 10% Pd/C (4 g) and hydrogenated at 45 psi for 5 h. The catalyst was removed by filtration through Celite and the filtrate was concentrated under vacuum to yield methyl 6-(cyclopropylmethylcarbamoyl)-3-(4-hydroxy-5-methoxy-2-(((2-methoxyethoxy)methoxy)carbonyl)phenyl)picolinate (3d) (31.87 g, 86%), which was pure enough to be used as such for the next step. An analytical sample of methyl 6-(cyclopropylmethylcarbamoyl)-3-(4-hydroxy-5-methoxy-2-(((2-methoxyethoxy) methoxy)carbonyl)phenyl)picolinate (3d) was obtained by purification of 350 mg of above crude using flash column chromatography (silica gel, eluting with ethyl acetate in hexane) to afford methyl 6-(cyclopropylmethyl-carbamoyl)-3-(4-hydroxy-5-methoxy-2-(((2-methoxyethoxy)methoxy)carbonyl)-phenyl)picolinate (3d) as a clear gum; 1HNMR (300 MHz, DMSO-d6) δ 9.74 (s, 1 H), 8.68 (t, J = 6.1 Hz, 1H), 8.18 (d, J = 8.0 Hz, 1 H), 7.95 (d, J = 8.0 Hz, 1H), 7.47 (s, 1H), 6.83 (s, 1H), 5.19 (s, 2H), 3.77 (m, 3H), 3.58 (s, 3H), 3.44 (m, 2H), 3.34 (m, 2H), 3.21 (m, 5H), 1.04 (m, 1 H), 0.44 (m, 2H), 0.27 (m, 2H); IR (KBr): 1731 , 1664 cm‘1. MS (ES*): 489.0 (M+1); Analysis calculated for C^e^O,,: C, 59.01; H, 5.78; N, 5.73; Found: C, 58.92; H, 6.15; N, 5.29.
6-(Cvclopropylmethylcarbamovn-3-(5-methoxy-2-(((2-methoxyethoxy^methoxy)-carbonyl)-4- (trifluoromethylsulfonyloxy)phenyl)picolinate (3e)

To a solution of methyl 6-(cyclopropylmethylcarbamoyl)-3-(4-hydroxy-5-methoxy-2-(((2- methoxyethoxy) methoxy)carbonyl)phenyl)picolinate (3d) (14.3 g, 29.3 mmol) in dichloromethane (150 mL) were added pyridine (12 mL, 146 mmol) and triflic anhydride (7.5 mL g, 44 mmol). After stirring overnight at room temperature under N2. the reaction mixture was poured into ice water and then extracted twice with dichloromethane. After washing the combined organic extracts with water and drying (MgS0 ), the solvent was evaporated in vacuo. The compound was purified by flash chromatography over silica gel column using ethyl acetate: hexane to afford methyl 6-(cyclopropylmethylcarbamoyl)-3-(5-methoxy-2-(((2- methoxyethoxy)methoxy)-carbonyl)-4-(trifluoromethylsulfonyloxy)phenyl)picolinate (3e) (1 g, 93%); H NMR (300 MHz, CDCy a 8.41 (d, J = 8.0, 1H), 8.17 (s, 1H), 8.03 (s, 1H), 7.79 (d, J = 8.0, 1 H), 6.82 (s, 1H), 5.32 (q, J = 6.1, 2H), 3.97 (s, 3H), 3.74 (s, 3H), 3.67 – 3.57 (m, 2H), 3.55 – 3.45 (m, 2H), 3.41 (dd, J = 8.2, 14.5, 2H), 3.34 (s, 3H), 1.36 – 1.17 (m, 1H), 0.58 (d, J = 7.1 , 2H), 0.33 (d, J = 5.1 , 2H).
Methyl 6-(cvclopropylmethylcarbamoyl)-3-(5-methoxy-2-f((2-methoxyethoxy)- methoxy)carbonvn-4-vinylphenyl)picolinate (3f)

To a solution of methyl 6-(cyclopropylmethylcarbamoyl)-3-(5-methoxy-2-(((2- methoxyethoxy)methoxy)carbonyl)-4-(trifluoromethylsulfonyloxy)phenyl)picolinate (3e) (37.4
g, 60.30 mmol) and potassium vinyltrifluoroborate (16.87 g, 120.6 mmol) in DMF (450 mL) and water (45 mL) was bubbled N2 for 5 min. To this mixture was added NaHC03 (20.26 g, 241.2 mmol) and dichloro-bis(triphenylphosphine)palladium (II) (6.34 g, 9.0 mmol). The reaction mixture was stirred at 70 °C for 20 h under N2(reaction progress was checked by 1H N R because product and starting material had same Rf in TLC). The reaction mixture was cooled down to room temperature and diluted with ethyl acetate. The organic layer was separated, washed with water, brine, dried ( gS04) and filtered. The filtrate was concentrated under vacuum to yield crude methyl 6-(cyclopropylmethyl-carbamoyl)-3-(5-methoxy-2-(((2-methoxyethoxy)methoxy)carbonyl)-4-vinylphenyl)-picolinate (3f). The crude product was purified by flash column chromatography (silica gel, 1 kg, eluting with 0-100% ethyl acetate in hexane) to afford methyl 6-(cyclopropylmethylcarbamoyl)-3-(5-methoxy-2-(((2-methoxyethoxy)methoxy) carbonyl)-4-vinylphenyl)picolinate [31) (26.54 g, 88%) as an amber gum; H NMR (300 MHz, DMSO-c¾ δ 8.70 (t, J = 6.1 Hz, 1H), 8.23 (d, J = 8.0 Hz, 1 H), 8.12 (s, 1 H), 8.00 (d, J = 8.0 Hz, 1 H), 6.98 (m, 2H), 5.94 (dd, J = 1.2, 17.8 Hz, 1H), 5.43 (d, J = 12.5 Hz, 1 H), 5.21 (d, J = 6.5 Hz, 2H), 3.88 (s, 3H), 3.64 (s, 3H), 3.48 (d, J = 3.1 Hz, 2H), 3.35 (m, 5H), 3.22 (m, 2H), 1.11 (s, 1H), 0.44 (dt, J = 4.9, 5.5 Hz, 2H), 0.28 (q, J = 4.8 Hz, 2H). IR (KBr); 1732, 1670 cm“1. MS (ES+) 499.1 (M+1).
2-(6-(cvclopropylmethylcarbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzolc acid (3g)

A mixture of methyl 6-(cyclopropylmethylcarbamoyl)-3-(5-methoxy-2-(((2-methoxyethoxy)methoxy) carbonyl)-4-vinylphenyl)picolinate (3f) (27.4 mmol) in DME (160 mL) and 6N HCI (40 mL) was stirred at room temperature for 6 h or till TLC showed complete conversion. The solvent was removed under vacuum. The residue obtained was suspended in water, the solid separated out was collected by filtration, washed with water and dried under vacuum to give 2-(6-(cyclopropylmethylcarbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (3g) (7.0 g, 63%) as a white
solid MP 40 – 42 °C; H NMR (300 MHz, DMSO-de) δ 8.69 (t, J= 6.0 Hz, 1H, NH), 8.20 (d, J= 7.9 Hz, 1H), 8.09 (s, 1 H), 7.95 (d, J= 8.1 Hz, 1H), 6.97 (dd, J= 18.0, 11.3 Hz, 1H), 6.88 (s, 1H), 5.92 (d, J= 7.9 Hz, 1H), 5.38 (d, J= 11.1 Hz, 1H), 3.85 (s, 3H), 3.63 (s, 3H), 3.27-3.17 (m, 2H), 1.15-1.05 (m, 1 H), 0.48-0.40 (m, 2H), 0.31-0.24 (m, 2H); IR (KBr): 3084, 1728, 1650, 1533, 1212, 1143 cm-1; MS (ES+) 433.26 (M+Na); (ES-): 409.28 (M-1); Analysis calculated for θ22Η22Ν2Ο6.0.25Η2Ο; C, 63.68; H, 5.47; N, 6.75; Found C, 63.75; H, 5.56; N, 6.65
Methyl-3-(2-(4-carbamimidoylprienylcarbamoyl)-5-metrioxy-4-vinylphenyl)-6- (cvclopropylmethylcarbamoyl)picolinate (3h)

To a solution of 2-(6-(cyclopropylmethylcarbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (3g) (2.35 g, 5.7 mmol) and 4-aminobenzimidamide dihydrochloride (3j) (1.79 g, 8.6 mmol) in DMF (20 mL) and pyridine (30 mL) at 0 °C was added EDCI (1.65 g, 8.6 mmol) and allowed to warm to room temperature overnight. The reaction mixture was quenched with 6N HCI (60 mL) and extracted with chloroform (3 x 60 mL). The organic layer was dried over MgS04, filtered and purified by flash column chromatography (silica gel, 110 g, eluting with 0 to 100% chloroform in CMA 80 in CMA 50) yielding methyl-3-(2-(4-carbamimidoylphenyl-carbamoyl)-5-methoxy-4-vinylphenyl)-6-(cyclopropylmethylcarbamoyl)picolinate (3h) (2.2 g, 65%) as a white solid MP 266 °C; 1H NMR (300 MHz, DMSO-c/6) δ 10.78 (s, 1 H), 9.26 (s, 2H), 9.03 (s, 2H), 8.67 (t, J = 6.1 , 1 H), 8.22 (d, J = 8.0, 1 H), 8.06 (d, J = 8.0, 1 H), 7.96 (s, 1 H), 7.89 – 7.74 (m, 4H), 7.13 – 6.96 (m, 2H), 6.07 (d, J = 17.7, 1H), 5.45 (d, J = 12.4, 1 H), 3.91 (s, 3H), 3.61 (s, 3H), 3.20 (s, 2H), 1.09 (dd, J = 4.7, 8.2, 1H), 0.43 (dt, J = 4.9, 5.4, 2H), 0.34 – 0.21 (m, 2H); MS (ES+) 528.1 (M+1); Analysis calculated for 
C, 58.93; H, 5.63; N,11.85; Found: C, 58.75; H, 5.65; N, 11.92.
46578
159
3-r2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy -vinyl-phenyll-6-(cvclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid (3i)

3h 3i
To a solution of methyl-3-(2-(4-carbamirriidoylphenylcarbarnoyl)-5-methoxy-4-vinylphenyl)-6-(cyclopropylmethylcarbamoyl)picolinate (3h) (1 g, 1.9 mmol) in methanol (10 mL) and THF
(10 mL) was added 2 N NaOH (10 mL). The reaction mixture was stirred at room
temperature for 3 h, and concentrated in vacuo to remove methanol and THF. The aqueous layer was acidified with 6N HCI to pH 6-7 and the solid obtained was collected by filtration
washed with water and ether to furnish on drying 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid
(3i)(0.775 g, 80%) as the hydrochloride salt as an off white solid.
1H NMR (300 MHz, DMSO-d6) δ 12.67 (s, 1 H), 9.11 (s, 2H), 8.97 (s, 2H), 8.74 (s, 1 H), 7.90
(d, J = 7.8, 1 H), 7.80 (s, 1 H), 7.72 – 7.58 (m, 4H), 6.99 (dd, J = 11.3, 17.7, 1 H), 6.78 (s, 1H),
5.95 (d, J = 17.2, 1H), 5.38 (d, J = 11.9, 1H), 3.82 (s, 3H), 3.18 (s, 2H), 1.06 (s, 1 H), 0.43 (d,
J = 7.9, 2H), 0.25 (d, J = 4.7, 2H); MS (ES+) 514.0 (M+1 ); Analysis calculated for
C2eH27N5O5.HCI.H2O: C, 59.21; H, 5.32; N, 12.33; Found: C, 59.43; H, 5.21; N, 12.06.
Example 1A- Preparation of 3-f2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyll-6-(cvclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride in Form
C

The jacket of a 10 L glass reactor was set to -5 °C. To the reactor was charged 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)-pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d) prepared in Step (11) of Example 1 (500 g, 1.22 mol), 4-amino-benzamidine-2HCI (280 g, 1.34 mol), and 2-propanol (4.05 kg). The mixture was cooled to 0.3 °C, and pyridine (210 g, 2.62 mol) followed by EDCI HCI (310 g, 1.61 mol) was added. The mixture was stirred at -1.1 to -0.3 °C for 22 hrs followed by addition of the second portion of EDCI HCI (58 g, 0.30 mol). The temperature of jacket was set to 14.0 °C, and the mixture was stirred for 89 hrs. The precipitate was filtered, and washed with 1.32 kg of 2-propanol.
The wet product (8a) was recharged to the reactor followed by addition of acetonitrile (1.6 kg) and water (0.57 kg). The mixture was heated to 46 °C. Smopex-234 (21 g) and Acticarbone 2SW (10 g) were added and the mixture was stirred at this temperature for 1 hr. The solution was filtered, and filtrate was returned back to the reactor. The jacket of the reactor was set to -5 °C, and the mixture was cooled to -0.2 “C. NaOH solution (256 g 46% NaOH, 2.95 mol, in 960 g water) was added in 25 min keeping the temperature ❤ °C. The mixture was stirred at 0.2-2.0 °C for 1 hr 40 min and then quenched with cone, acetic acid (40 g, 0.66 mol). Diluted acetic acid (80 g, 1.33 mol AcOH in 1000 g water) was added during 1 hr 20 min (temperature 1.7-3.0 °C), followed by 1250 g water (30 min). The
suspension was stirred at 0-3.0 “for 1 hr, and filtered at 0-5 °C (ice mantle around the filter). The reactor and product (8d) was rinsed with 3.5 kg water.
The wet product (8d) was recharged to the reactor followed by 0.65 kg water and 1.69 kg acetonitrile. The mixture was heated to 57-60 °C, and stirred at this temperature for 14.5 hrs. The mixture was cooled to -2.2 °C (Tjackel= -5 °C), and a solution of NaOH (163 g 46%, 1.87 mol, in 580 g water) was added during 15 min. The temperature rose to -0.4 °C. Hydrochloric acid (407 g 37% HCI, 4 mol) was added in 10 min, the temperature rose to 7.5 °C. The suspension was agitated at -3 – 0 °C for 19 hrs. The product was filtered and the filter cake was rinsed with 2.87 kg water, compressed and pulled dry. The wet product (1.30 kg) was dried at 40-43 °C and 50 mbar for 11 hrs to furnish 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6- (cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) (484 g) as Form C.
Example-1 B: Preparation of 3-f2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyll-6-(cvclopropylmethylcarbartiovQpyridine-2-carboxylic acid hydrochloride in Form A
The procedure was carried out in an identical manner to Example 1 A, with the exception that after the final filtration the filter cake was rinsed with 2.87 kg methyl ierf-butyl ether instead of 2.87 kg water, and pulled dry. The product was dried at 40-43 °C and 50 mbar to furnish 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) as Form A.
PATENT
Methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)picolinate (compound 6a) is (I) (pages 85 and 86). Avoralstat hydrochloride (compound of formula XVIII) is (II) (claim 40, page 109). A Markush structures is presented (claim 1, page 99).
The synthesis of (II) via intermediate (I) is described (example 1, pages 80-93).
A synthesis of the compound 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid (Compound 3i) is described in Schemes A-C.
O y OHCk n Br^ ^OCH3
B Brr22,, AAccOOHH Y^ V”“ \ \ tt–BBuuOOKK
OHC^^^O ” Br^\^0 MeOH ” OHC
1a 1b 66%

1d 95% 1 e

1f
Scheme A


3h 31
Scheme C
Examples. In this section, the following abbreviations are used:



Example-1 : Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b)

7b
Step (1): Preparation of 6-Bromobenzo 1 ,3]dioxole-5-carbaldehyde (1 b):

1b
A solution of bromine (33.0 kg, 206.49 mol) in acetic acid (27.5 L) was added slowly to a solution of piperonal (1a) (29.9 kg, 199.16 mol) in acetic acid (105 L) at room
temperature over a period of 50 min and the reaction mixture was stirred at room temperature for 14.2 h. Additional solution of bromine (33 kg, 206.49 mol) in acetic acid (27.5 L) was added slowly to the reaction mixture over a period of 2 h and the reaction mixture was stirred for 22 h. The reaction mixture was quenched by addition of ice water (500 L) with stirring over a period of 6 h and continued stirring for additional 1.25 h. The mixture was allowed to settle and most of the supernatant liquid was decanted to a waste container using nitrogen pressure. Water (600 L) was added to the solid, stirred, mixture was allowed to settle and then most of the supernatant liquid was decanted to a waste container using nitrogen pressure. Water (100 L) was added to the decanted mixture, stirred for 15 min and the solid obtained was collected by filtration using a centrifuge. The solid was washed with water (2 x 100 L) and air-dried in a tray drier for 3.75 h to afford the crude product 1 b (52 kg). The crude product (51.2 kg) was stirred in n-hexane (178 L) for 3 h, collected by filtration, washed with n-hexane (25 L) and dried to afford 6-bromobenzo[1 ,3]dioxole-5-carbaldehyde (1b) (40.1 1 kg, 87.9%) as a light brown solid. MP: 109-112°C. 1H NMR (300 MHz, CDCI3) δ 10.21 (s, 1 H), 7.37 (s, 1 H), 7.07 (s, 1 H), 6.10 (s, 2H); HNMR (DMSO-cf6): δ 10.06 (s, 1 H), 7.42 (s, 1 H), 7.29 (s, 1 H), 6.20 (d, J =12.3 Hz, 2H)
The process is also illustrated in Fig. 1.
Average yield of isolated 1 b from step-1 is 78 – 88%.
Step (2): Preparation of 2-Bromo-5-hydroxy-4-methoxy-benzaldehyde (1c)

A solution of potassium terf-butoxide (10.7 kg, 95.36 mol) in DMSO (49 L) was stirred at 50 °C for 30 min. Methanol (49 L) was added slowly over a period of 4.25 h and stirred at 50 °C for 30 min. 6-Bromobenzo[1 ,3]dioxole-5-carbaldehyde (1 b) (9.91 kg, 43.27 mol) was added to the reaction mixture in small portions over a period of 45 min and stirred at 50 °C for 1 h. The reaction mixture was cooled to room temperature and split into two equal portions. Each portion was quenched with water (50.9 L) and basified with 50% aqueous NaOH solution (2.4 L). Each portion was extracted with MTBE (4 x 36 L) to remove impurities. The aqueous layer was acidified with cone. HCI to pH ~ 3 to obtain
product as a yellow solid. The solid was collected by filtration using a centrifuge, washed with water (2 x 35 L) and air-dried to afford 2-Bromo-5-hydroxy-4-methoxy-benzaldehyde (1c) (4.37 kg, 40.7%, contains 7 % water); Mp: 100-102°C; 1HNMR (300MHz, DMSO-d6): δ 10.00 (s, 1 H), 9.92 (s,1 H), 7.27 (s, 1 H), 7.26 (s, 1 H), 3.93 (s, 3H).
The process is also illustrated in Fig. 2.
Average yield of isolated product 2-Bromo-5-hydroxy-4-methoxy-benzaldehyde (1c) from step-2 is 40-50%.
Step (3): 5-Hydroxy-4-methoxy-2-(4,4,5,5-tetramethyl-[1 ,3,2]dioxaborolan-2-y benzaldehyde (4a)

2-Bromo-5-hydroxy-4-methoxy-benzaldehyde (1c) [1.3 kg (93%, 7% water content), 5.25 mol] was dissolved in toluene (13 L) in a reaction flask equipped with a Dean Stark apparatus. The solution was heated at reflux with stirring to distil off about 25% of the toluene along with water (90 ml_). The solution was cooled to 90 °C then
bis(pinacolato)diboron (1.5 kg, 5.82 mol), KOAc (772.6 g, 7.87 mol) and Pd(PPh3) (24.3 g, 0.02 mol) were added and the reaction mixture was heated at reflux for 10h. After confirming the completion of reaction by TLC (mobile phase: 100% DCM), the reaction mixture was cooled to room temperature and was kept standing overnight. The reaction mixture was filtered through celite and the celite cake was washed with toluene (4 L). The filtrate of this batch was mixed with the filtrate of another batch (batch size 1.3 kg obtained from an identical reaction). The mixed filtrate was washed with water (17.5 L), brine (17.5 L), dried over Na2S04, filtered and the solution was passed through a pad of silica gel (2 kg, mesh size 230-400). The silica gel pad was washed with toluene. The combined filtrate and washing was concentrated under reduced pressure and the residual crude product was stirred with n-hexane (23 L) for 1 h to obtain a solid product. The solid was collected by filtration, washed with n-hexane (5 L) and dried to afford 5-hydroxy-4-methoxy-2-(4,4,5,5-tetramethyl-[1 ,3,2]dioxaborolan-2-yl)benzaldehyde (4a) (2.47 kg, 84.6%). H NMR (300 MHz, CDCI3) δ 10.54 (s, 1 H), 7.57 (s, 1 H), 7.33 (s, 1 H), 5.89 (s, 1 H), 4.01 (s, 3H), 1.37 (s, 12H); 1H NMR (300 MHz, DMSO-d6) δ 10.35 (s, 1 H), 9.95 (s, 1 H), 7.33 (s, 1 H), 7.23 (s, 1 H), 3.87 (s, 3H), 1.33 (s, 12H); MS (ES+) 301.1 (M+Na); 579.1 (2M+Na); Analysis calculated for C14H19B05: C, 60.46; H, 6.89; Found: C, 60.60; H, 6.87
The average yield of 5-hydroxy-4-methoxy-2-(4,4,5,5-tetramethyl-[1 ,3,2]dioxa-borolan-2-yl)benzaldehyde (4a) from step (3) is 78 – 90%.
The process is also illustrated in Fig. 3.
Step (4): Preparation of 3-Bromo-2,6-dimethylpyridine (5b)

2,6-lutidine (5a) (115 kg, 1073.3 mol) was added into pre-chilled oleum (20-23%, 1015 kg, 2276.7 mol) at 0 °C over a period of 4.5 h (temperature r6ached 14 °C during the addition). Bromine (88.18 kg, 1103.6 mol) was then added at 5-10 °C over a period of 1 h. The reaction mixture was slowly heated to 150 °C over a period of 12h. TLC analysis indicated about 40-50% conversion to product and the formation of a dimer by-product (5%). The reaction mixture was cooled to room temperature and then additional bromine (88.18 kg, 1103.6 mol) was added slowly. The reaction mixture was slowly heated to maintain a temperature of 65-75 °C over a period of 15h. TLC analysis indicated a 65-70 % conversion to product and the formation of 5% dimer by product. The reaction mixture was quenched by addition of water (500L) while maintaining the reaction temperature below 20 °C. The mixture was basified with 6.6 M NaOH (3800 L) while maintain the temperature at < 40 °C. EtOAc (220 L) was added and the mixture was stirred for 1 h then allowed to settle over a period of 2 h. The layers were separated and the aqueous layer was treated with NaOH (10 kg) in water (10 L) and extracted with EtOAc (160 L). The organic extracts were combined washed with brine (100 L), dried over Na2S04 (50.0 kg), filtered and the solvent was evaporated under atmospheric pressure. The residue was vacuum distilled and the desired product 3-bromo-2,6-dimethylpyridine (5b) was collected at 58-60 °C, 2 mmHg (98.45 kg, 49.2 %) as a colorless liquid.
The process is also illustrated in Fig. 4.
Step (5): Preparation of 3-Bromopyridine-2,6-dicarboxylic acid (5c)

5b 5c
To a stirred solution of 3-bromo-2,6-dimethylpyridine (5b) (98 kg, 5326 mol) in water (1310 L) was added KMn0 (225 kg, 1423.6 mol) in 5 equal portions in 1 h intervals at 70 °C. After stirring for 1 h at 70 °C, additional KMn04 (225 Kg, 1423.6 mol) was added in 5 equal portion in 1 h intervals at 90 °C. The reaction mixture was stirred for 12 h at 90 °C. The suspension was filtered hot through celite to obtain a clear solution. The solvent was distilled off to remove about 30% of the total volume. The remaining concentrated solution was chilled to 0 °C and made acidic (to pH 3-4) by the addition of cone. HCI (120 L). The white precipitate obtained was collected by filtration and dried at 70 °C to afford 3-bromopyridine-2,6-dicarboxylic acid (5c) as a white solid (109 kg, 84%).
The process is also illustrated in Fig. 5.
Step (6): Preparation of Dimethyl 3-Bromopyridine-2,6-dicarboxylate (5d)

To a stirred solution of 3-bromopyridine-2,6-dicarboxylic acid (5c) (20.0 kg, 81.29 mol) in methanol (100 L) was added cone. H2S04 (4.4 L) over a period of 30 min. The reaction mixture was heated to 65 °C and maintained at that temperature for 5 h (the reaction was monitored by TLC analysis to determine completion of reaction). The reaction mixture was cooled to room temperature basified by careful addition of aqueous NaHC03 solution (prepared from 10 kg NaHC03 in 120 L of water) and further diluted with water (120 L). The white solid obtained was collected by filtration, washed with plenty of water and then oven-dried at 40 °C to obtain dimethyl 3-bromopyridine-2,6-dicarboxylate (5d) (9.2 kg, 41.3%) as a white solid; 1HNMR (300 MHz, DMSO-cf6) δ 8.47 (d, J = 8.4, 1 H), 8.08 (dd, J = 4.5, 8.4, 1 H), 3.95 (s, 3H), 3.91 (s, 3H); MS (ES+) 570.6 (2M+Na); Analysis calculated for C9H8BrN04: C, 39.44; H, 2.94; Br, 29.15 N, 5. 1 ;
Found: C, 39.52; H, 2.92; Br, 29.28; N, 5.03.
The process is also illustrated in Fig. 6.
6582
Step (7): Preparation of Methyl 3-bromo-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylate (

To a stirred solution of dimethyl 3-bromopyridine-2,6-dicarboxylate (5d) (27 kg, 98.52 mol) in ierf-butanol (135 L) was added at room temperature cyclopropylmethanamine (7.83 kg, 110.1 mol). The reaction mixture was heated at 65 °C for 17 h. The progress of reaction was monitored by TLC and HPLC (HPLC analysis showed the formation of 74% of the product 5e after 17 h. The reaction mixture was cooled to room temperature and then cone. HCI (2.7 L) was added slowly and the mixture was stirred for 15 min. The reaction mixture was concentrated under reduced pressure to obtain the crude product. The crude product was dissolved in hot /-PrOH (54 L) filtered through a celite pad. The filtrate was cooled with stirring to 10 °C to obtain a white precipitate. The solid obtained was collected by filtration, washed with cold
i-PrOH (13 kg), n-hexane (15 L) and dried to provide pure methyl 3-bromo-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylate (5e) (15.7 kg, 50.9%). The filtrate was concentrated under reduced pressure and the crude product can be purified by silica gel column chromatography eluting with tert-butanol in hexanes to furnish additional 10% methyl 3-bromo-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylate (5e). HNMR (300 MHz, DMSO-cf6) δ 8.83 (t, J = 5.9, 1 H), 8.47 – 8.41 (m, 1 H), 8.06 (d, J = 8.4, 1 H), 3.96 (s, 3H), 3.16 (t, J = 6.5, 2H), 1.14 – 0.99 (m, 1 H), 0.42 (m, 2H), 0.30 -0.19 (m, 2H); MS (ES+) 337.0 (M+23), 650.8 (2M+23); Analysis calculated for
C12H13BrN203: C, 46.03; H, 4.18; N, 8.95; Br, 25.52; Found: C, 46.15; H, 4.17; N, 8.72; Br, 25.26.
The average isolated yield for step (7) is 50% to 60%.
The process is also illustrated in Fig. 7.
Step (8): Preparation of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)picolinate (6a)
2

6a
THF (37.5 L) was charged to a 100 L reactor followed by ethyl 3-bromo-6- (cyclopropylmethyl-carbamoyl)pyridine-2-carboxylate (5e) (2.5 kg, 7.98 mol) under a nitrogen atmosphere. The reaction mixture was degassed twice by applying alternate vacuum and nitrogen. 5-Hydroxy-4-methoxy-2-(4,4,5,5-tetramethyl-[1 ,3,2]dioxa-borolan-2-yl)benzaldehyde (4a) (2.88 kg, 10.36 mol) was added, followed by the addition of PPh3 (53.13 g, 0.20 mol), PdCI2(PPh3)2 (120.4 g, 0.17 mol) and a solution of Na2C03(2.12 kg, 20.00 mol) in demineralized water (10.0 L) under nitrogen atmosphere. The reaction mixture was degassed again two times by applying alternate vacuum and nitrogen. The reaction mixture was heated at reflux for 6.5 h, cooled to room temperature and filtered through a Celite bed. Water (75 L) was added to the filtrate and the product was extracted with ethyl acetate (75 L). The aqueous layer was back extracted with ethyl acetate (2 χ 60 L). The combined ethyl acetate extract was divided into two equal portions and each portion was washed with brine (37 L), dried over Na2S04, filtered and concentrated under reduced pressure to give crude methyl 6- ((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)picolinate (6a) as a reddish viscous material (-4.5 Kg) which was used as such for the next step without further purification. An analytical sample was prepared by purification of a small sample by flash column chromatography (silica gel, eluting with 0-100% ethyl acetate in hexane) to furnish methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)-picolinate (6a) as an off-white solid; HNMR (300 MHz, DMSO-d6) δ 9.89 (s, 1 H), 9.52 (s, 1 H), 8.79 (t, J = 6.1 Hz, 1 H), 8.23 (d, J = 8.0 Hz, 1 H), 8.09 (d, J = 8.0 Hz, 1 H), 7.34 (s, 1 H), 6.90 (s, 1 H), 3.85 (s, 3H), 3.62 (s, 3H), 3.22 (m, 2H), 1.16 -1.02 (m, 1 H), 0.49 – 0.38 (m, 2H), 0.32 – 0.22 (m, 2H); MS (ES+) 791.0 (2M+Na), (ES-) 382.7 (M-1), 767.3 (2M-1); Analysis calculated for C20H20N2O6.0.25 H20: C, 61.77; H, 5.31 ; N, 7.20; Found: C, 61.54; H, 5.13; N, 7.05.
The process is also illustrated in Fig. 8.
46582
Step (9): Preparation of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-(((trifluoromethyl)sulfonyl)oxy)phenyl)picolinate (6b)

6a 6b
A solution of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)picolinate (6a) (2.11 kg, estimated about 3.83 mol from step-8) in dichloromethane (16.0 L) and pyridine (1.4 L, 17.4 mol) cooled to -10°C and maintained at that temperature for 1 h was added a solution of triflic anhydride (980.0 ml_, 5.8 mol) in dichloromethane (6.0 L) drop wise over a period of 3 h at -10 °C. The reaction mixture was stirred at -5°C for 1.3 h, quenched with saturated aqueous NaHCO3(10.4 L) and stirred for 30 mins. The organic layer was separated, washed successively with saturated aqueous NaHC03 (10.4 L), 1 HCI (2 x 16.6 L), water (13.2 L), brine (13.2 L), dried over MgS04, filtered and concentrated under reduced pressure to give the crude product. The crude product was stirred with 15% ethyl acetate in n-hexane (7.0 L) for 1 h. The solid obtained was collected by filtration washed with 15% ethyl acetate in n-hexane (3.0 L). The solid was stirred again with 15% ethyl acetate in n-hexane (7.0 L) for 1 h, was collected by filtration and washed with 15% ethyl acetate in n-hexane (3.0 L). The solid was stirred again with 15% ethyl acetate in n-hexane (8.0 L) for 1 h, collected by filtration washed with 15% ethyl acetate in n-hexane (3.0 L). The solid was dried to afford methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-(((trifluoromethyl)sulfonyl)-oxy)phenyl)picolinate (6b) as a light brown solid (1.7 kg, 86% yield, for combined steps 8 & 9). Average isolated yield for combined steps 8 and 9 was 70% to 86%; Ή NMR (300 MHz, DMSO-cf6): δ 9.64 (s, 1 H), 8.78 (t, J = 6.1 , 1 H), 8.29 (d, J = 8.0, 1 H), 8.16 (d, J = 8.0, 1 H), 8.03 (s, 1H), 7.39 (s, 1 H), 4.00 (s, 3H), 3.63 (s, 3H), 3.22 (m, 2H), 1.11 (m, 1 H), 0.52 – 0.39 (m, 2H), 0.28 (m, 2H); MS (ES+) 538.9 (M+Na). The process is also illustrated in Fig. 9.
Step (10): Preparation of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-vinylphenyl)picolinate (6c)

A solution of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4- (((trifluoromethyl)sulfonyl)oxy)phenyl)picolinate (6b) (12 kg, 23.24 mol) in DME (106 L) was charged into reactor under nitrogen. The reaction mixture was degassed twice by applying alternate vacuum and nitrogen. Potassium trifluoro(vinyl)borate (3.9 kg, 29.1 1 mol), PdCI2(PPh3)2 (815 g, 1.13 mol), KHC03 (4.65 g, 46.44 mol) and demineralized water (12 L) was then added under a N2 atmosphere. The reaction mixture was degassed by applying alternate vacuum and nitrogen. The reaction mixture was heated at reflux for 5 h. The reaction mixture was cooled to room temperature and then filtered through a Celite bed. Demineralized water (118 L) was added to the filtrate followed by ethyl acetate (124 L). The mixture was stirred for 20 min and then the organic layer was separated. The aqueous layer was back-extracted with ethyl acetate (2 x 95 L). The combined organic extract was washed with brine (95 L), dried over Na2S04, and filtered. The solvent was evaporated under reduced pressure to give the crude product. The crude product was purified by column chromatography (silica gel, 120 kg, 230-400 mesh size, eluting with ethyl acetate in n-hexane) to obtain methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-vinylphenyl)picolinate (6c) (6 kg, 72%). 1H NMR (300 MHz, CDCI3): δ (ppm) 9.64 (s, 1 H), 8.35 (d, J = 7.8 Hz, 1 H), 8.06-8.03 (m, 2H), 7.78(d, J = 7.8 Hz, 1 H), 7.02-6.92 (m, 1 H), 6.61 (s, 1 H), 5.86 (d, J = 17.7 Hz, 1 H), 5.38 (d, J = 1 1.4 Hz, 1 H), 3.84 (s, 3H), 3.67 (s, 3H), 3.35-3.29 (m, 2H),1.08-1.03 (m, 1H), 0.55-0.49 (m, 2H), 0.29-0.2 4(m, 2H). 1HNMR (300 MHz, DMSO-d6) 6 9.68 (s, 1 H), 8.77 (t, J = 6.1 , 1 H), 8.35 – 8.21 (m, 1 H), 8.16 – 8.01 (m, 2H), 7.14 -6.87 (m, 2H), 6.01 (dd, J = 1.2, 17.8, 1 H), 5.45 (dd, J = 1.1 , 1 1.3, 1 H), 3.91 (s, 3H), 3.64 (s, 3H), 3.23 (m, 2H), 1.21 – 1.01 (m, 1H), 0.51 – 0.40 (m, 2H), 0.34 – 0.20 (m, 2H). MS
(ES+) 417.0 (M+Na); Analysis calculated for C22H22N205: C, 66.99; H, 5.62; N, 7.10;
Found: C, 66.75; H, 5.52; N, 7.06.
The process is also illustrated in Fig. 10.
Step (1 1): Preparation of 2-(6-((cyclopropylmethyl)carbamoyl)-2- (methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d)

To a stirred solution of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-vinylphenyl)picolinate (6c) (1.57 kg, 3.80 mol) in acetonitrile (15.4 L) was added ferf-butyl alcohol (22.2 L), demineralized water (3.2 L) and sodium dihydrogen phosphate monohydrate (323.74 g, 2.346 mol). The reaction mixture was cooled to 0 °C and added 2-methyl-2-butene (5.3 L, 50.0 mol) and stirred at 0 °C for 30 min. A solution of 80% sodium chlorite (1.36 kg, 12.0 mol) in demineralized water (5.2 L) was added to the reaction mixture over a period of 2.5 h at 0 °C [temperature rises to 7 °C during the addition]. The reaction mixture was stirred at 0 °C for 2 h, diluted with water (40 L) and ethyl acetate (24 L). After stirring the mixture, it was allowed to settle and the organic layer was separated. The aqueous layer was back-extracted with ethyl acetate (2 x 20 L) then acidified with 5.9 % aqueous acetic acid (2 L) and extracted once with ethyl acetate (10 L). The organic extracts were combined washed with water (2 x 20 L), a solution of acetic acid (125 mL) in water (20.0 L), brine (2 χ 20 L), dried over Na2S04, filtered and concentrated under reduced pressure (vapor temperature below 40 °C). The residue obtained was dissolved in acetone (7 L) (residue didn’t dissolve completely). The solution was poured slowly into a reactor containing stirred n-hexane (70.0 L) to precipitate the solid product and the mixture was stirred for 2 h. The solid obtained was collected by filtration, washed with 10% acetone in n-hexane (6.3 L), AJ-hexane (6.3 L), dried to afford 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4- methoxy-5-vinylbenzoic acid (6d) as an off-white solid (1.29 Kg, yield: 79.0%). Average isolated yield for step 1 1 is 74% to 84%. 1H NMR (300 MHz, DMSO-d6): δ (ppm) 12.50 (brs, 1 H), 8.69(t, J= 6.0 Hz, 1 H, NH), 8.20 (d, J= 7.9 Hz, 1 H), 8.09 (s, 1 H), 7.95 (d, J= 8.1 Hz, 1 H), 6.97 (dd, J= 18.0, 1 1.3 Hz, 1 H), 6.88 (s, 1 H), 5.92 (d, J= 7.9 Hz, 1 H), 5.38 (d, J= 1 1.1 Hz, 1 H), 3.85 (s, 3H), 3.63 (s, 3H), 3.27-3.17 (m, 2H), 1.15-1.05 (m, 1 H), 0.48-0.40 (m, 2H), 0.31-0.24 (m, 2H); MS (ES+) 433.26, (M+Na); (ES-) 409.28 (M-1). The process is also illustrated in Fig. 1 1.
Step (12): Preparation of Methyl 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylate methanesulfonate (7a

Pyridine (3.8 L, 47.17 mol) and EDCI (5.31 kg, 27.66 mol) were sequentially added to a cooled solution (0 °C) of 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)-pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d) (9 kg, 21.92 mol) and 4-aminobenzamidine dihydrochloride (5.13 kg, 24.65 mol) in /-PrOH (90 L). The reaction mixture was allowed to warm to room temperature and stirred for 2 h. TLC analysis indicated incomplete reaction. Additional EDCI (1.08 kg, 5.6 mol) was added and the reaction mixture was stirred for 8 h. The reaction was still incomplete as indicated by TLC analysis, additional EDCI (0.54 kg, 2.8 mol) was added and the reaction mixture was stirred for 5 h. TLC analysis indicated there was trace amount of unreacted starting material remaining. The reaction mixture was cooled to 0 °C and a solution of
methanesulfonic acid (MSA) (9.13 kg, 95 mol) in MeOH (38.7 L) was added to the cooled mixture over a period of 4 h. The reaction mixture was allowed to warm to room temperature and stirred for 15 h. The product was collected by filtration, washed with a mixture of /‘-PrOH and MeOH (4:1 , 45 L). The wet cake was slurried in a mixture of /-PrOH and MeOH (2:1 , 135 L) stirred for 1 h and the product was collected by filtration and washed with a mixture of /‘-PrOH and MeOH (4:1 , 46.8 L). The product was dried in
2015/046582
a vacuum oven at 45 °C to afford methyl 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethyl-carbamoyl)pyridine-2-carboxylate methanesulfonate (7a) as a pink-colored solid (12.71 kg, 93%). Average isolated yield for this step: >90%.
1H NMR (300 MHz, DMSO-c/6) δ 10.71 (s, 1 H), 9.16 (s, 2H), 8.80 (s, 2H), 8.68 (t, J = 6.1 Hz, 1 H), 8.22 (d, J = 8.0 Hz, 1H), 8.06 (d, J = 8.1 Hz, 1 H), 7.93 (s, 1H), 7.84 – 7.72 (m, 4H), 7.12 – 6.97 (m, 2H), 6.04 (dd, J = 17.8, 1.3 Hz, 1 H), 5.45 (d, J = 12.6 Hz, 1H), 3.91 (s, 3H), 3.60 (s, 3H), 3.25 – 3.16 (m, 2H), 2.32 (s, 3H), 1.10 – 1.01 (m, 1 H), 0.48 – 0.37 (m, 2H), 0.30 – 0.22 (m, 2H); MS (ES+) 528.0 (M+1); Analysis calculated for
C29H29N5O5.CH3SO3H.2H2O. C, 54.62; H, 5.65; N, 10.62; S, 4.86; Found: C, 54.95; H, 5.55; N, 10.61 ; S, 4.87.
The process is also illustrated in Fig. 12.
Step (13): Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-rnethoxy-4- vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrate

(3i) ,a 3i
A pre-cooled (0-5 °C) aq. NaOH solution [prepared from solid NaOH (4 kg, 100 mol) in water (86 L)] was added to a suspension of methyl 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethyl-carbamoyl)pyridine-2-carboxylate methanesulfonate (7a) (28.7 kg, 46 mol) in acetonitrile (86 L) cooled to 0 to 5 °C over a period of 25 mins. The reaction mixture was stirred at 0 to 5 °C for 2.5 h (TLC analysis showed the reaction was complete). The reaction mixture was filtered through a sparkler filter, washed with a mixture of 1 :1 CH3CN / H20 ( 57.4 L). Acetic acid (3.2 L, 55.9 mol) in water (56 L) was added to the filtrate at room temperature over a period of 25 mins and the resulting mixture was stirred at room temperature for 2.5 h. The solid product obtained was collected by filtration, washed with a 1 :4 mixture of CH3CN / H20 (57.5 L). The solid was dried at 45°C in a vacuum oven to afford 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6- (cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrate (3i) as an off-white solid (12,77 kg, 54.1%). Average yield for this step is 50% to 75%. Mp: >200°C; H NMR (300 MHz, DMSO-d6): δ 13.49 (s, 1 H), 8.94 (bs, 4H), 8.56 (t, 1 H), 7.82 – 7.71 (m, 2H), 7.67 -7.56 (m, 4H), 7.51 (d, J = 7.8, 1 H), 6.98 (dd, J = 11.3, 17.8, 1 H), 6.68 (s, 1 H), 5.92 (d, J = 16.6, 1 H), 5.36 (d, J = 12.4, 1 H), 3.80 (s, 3H), 3.16 (m, 2H), 1.05 (m, 1 H), 0.43 (m, 2H), 0.24 (m, 2H); MS (ES+) 514.1 (M+1), 536.1 (M+Na), (ES-) 512.1 ; Analysis calculated for C28H27N5O5.3H2O: C, 59.25; H, 5.86; N, 12.34; Found C, 59.50; H,
5.75; N, 12.05. If needed this material can be crystallized from a mixture of acetone and water.
The process is also illustrated in Fig. 13.
Step 14: Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b

A pre-cooled (5-8 °C) aqueous NaOH solution (prepared from solid NaOH (1.97 kg, 49.25 mol) in demineralized water (41 L) was added to a pre-cooled (0-5 °C) suspension of (3i) (13.8 kg, 26.9 mol) in acetonitrile (41 L). The reaction mixture was stirred at 0-5 °C for 30 min (until the reaction mixture becomes homogeneous). The reaction mixture was filtered through a sparkler filter washed with 50% acetonitrile in demineralized water (4.4 L). The filtrate was charged into a reactor and cooled to 0-5 °C. Aqueous HCI [prepared from cone. HCI (9.3 L) in demineralized water (36 L)] was added slowly with stirring to keep the reaction temperature at or below 15 °C, the resulting mixture was stirred at 10-15 °C for 13 h. The reaction mixture was cooled to 0-5 °C and stirred for 1 h. The solid obtained was collected by filtration and washed with demineralized water (36 L). The solid product was suspended in water (69 L) stirred for 30 mins and collected by filtration washed twice with water (20 L each). The solid product was dried in a vacuum oven at 45°C to afford 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-
(cyclopropylmethyl carbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) (1 1.21 Kg, 75.77%). Mp: >200°C; 1H NMR (300 MHz, DMSO-ci6): δ 12.98 (br s, 1 H), 10.86 (s, 1 H), 9.24 (s, 3H), 9.04 (s, 2H), 8.22 (d, J = 7.8 Hz, 1 H), 7.96 (d, J = 5.7 Hz, 2H), 7.78 (s, 4H), 7.09-6.99 (m, 2H), 6.07 (d, J = 17.7 Hz, 1 H), 5.45(d, J = 11.4 Hz, 1 H), 3.88 (s, 3H), 3.26-3.24 (m, 2H), 1.09 (m, 1 H), 0.47 (m, 2H), 0.28 (m, 2H).
Average isolated yield for this step varies from 63% to 80%.
The process is also illustrated in Fig. 14.
Example-2: Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid sulfate salt (8b)

6d 8a
To a solution of 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d) (2.35 g, 5.7 mmol) and 4-aminobenzamidine dihydrochloride (1.79 g, 8.6 mmol) in DMF (20 mL) and pyridine (30 ml_) at 0 °C was added EDCI (1.65 g, 8.6 mmol) and allowed to warm to room temperature overnight. The
reaction mixture was quenched with 6N HCI (60 mL) and extracted with chloroform (3 x 60 mL). The organic layer was dried over MgS04, filtered and concentrated in vacuum. The residue obtained was purified by flash column chromatography (silica gel, 110 g, eluting with 0 to 100% chloroform in CMA 80 and 0-100% chloroform in CMA 50) to furnish methyl 3-(2-((4-carbamimidoylphenyl)carbamoyl)-5-methoxy-4-vinylphenyl)-6-((cyclopropylmethyl)-carbamoyl)picolinate hydrochloride (8a) (2.2 g, 65%) as a white solid; MP 266 °C; 1HNMR (300 MHz, DMSO-d6) δ 10.78 (s, 1 H), 9.26 (s, 2H), 9.03 (s, 2H), 8.67 (t, J = 6.1 , 1 H), 8.22 (d, J = 8.0, 1 H), 8.06 (d, J = 8.0, 1 H), 7.96 (s, 1 H), 7.89 -7.74 (m, 4H), 7.13 – 6.96 (m, 2H), 6.07 (d, J = 17.7, 1 H), 5.45 (d, J = 12.4, 1 H), 3.91 (s, 3H), 3.61 (s, 3H), 3.20 (s, 2H), 1.09 (dd, J = 4.7, 8.2, 1 H), 0.43 (dt, J = 4.9, 5.4, 2H), 0.34 – 0.21 (m, 2H); MS (ES+) 528.1 (M+1); Analysis calculated for C29H29N505 (H20)1 5 (HCI): C, 58.93; H, 5.63; N, 1 1.85; Found: C, 58.75; H, 5.65; N, 1 1.92.
Step-2: preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid sulfate salt (8b)

8a 8b j0 a solution of methyl 3-(2-((4-carbamimidoylphenyl)carbamoyl)-5-methoxy-4-vinylphenyl)-6-((cyclopropylmethyl)carbamoyl)picolinate hydrochloride (8a) (1.128 g, 2 mmol) in acetonitrile (5 ml), was added 1 N aqueous sodium hydroxide (5.00 ml, 5.00 mmol) and stirred at room temperature for 2 h, TLC [CMA80/CMA50 (7/3)] shows reaction was complete. The reaction mixture was neutralized with a solution of sulfuric acid (0.483 ml, 9.00 mmol) in water (5 mL) and stirred for 10 min at room temperature. To this cold water (5 ml) was added and stirred at room temperature until product crystallized out. Cold water (5 mL) was added to the slurry and stir for additional 20 min, additional cold water (5 mL) was added prior to filtration of solid. The solid obtained was collected by filtration washed with water (5 mL and 2.5 mL), dried under vacuum overnight to afford 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-
(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid sulfate salt (8b) (1.103 g, 90 % yield) as a white solid; MP 221.7 °C; H NMR (300 MHz, DMSO-d6) δ 12.30 – 10.91 (bs, 1 H, D20 exchangeable), 10.69 (bs, 1 H, D20 exchangeable), 9.24 (t, J = 6.0 Hz, 1 H), 9.16 (s, 2H, D2O exchangeable), 8.78 (s, 2H, D2O exchangeable), 8.24 (d, J = 8.0 Hz, 1 H), 8.04 – 7.91 (m, 2H), 7.84 – 7.67 (m, 4H), 7.13 – 6.94 (m, 2H), 6.03 (dd, J = 17.8, 1 .4 Hz, 1 H), 5.51 – 5.37 (m, 1 H), 3.88 (s, 3H), 3.24 (t, J = 6.4 Hz, 2H), 1.16 – 1.01 (m, 1 H), 0.52 – 0.41 (m, 2H), 0.32 – 0.22 (m, 2H); MS (ES+) 514.0 (M+1); Analysis calculated for: C28H27N605 1.0H2SO4 1.5H20: C, 52.66; H, 5.05; N, 10.97; S, 5.02; Found: C, 52.81 ; H, 4.95; N, 10.94; S, 4.64.
Example-3: Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid methane s

To a solution of methyl 3-(2-((4-carbamimidoylphenyl)carbamoyl)-5-methoxy-4-vinylphenyl)-6-((cyclopropylmethyl)carbamoyl)picolinate hydrochloride (8a) (1.128 g, 2 mmol) in acetonitrile (5 ml) was added 1 N aqueous sodium hydroxide (5.00 ml, 5.00 mmol) and stirred at room temperature for 2 h, TLC [CMA80/CMA50 (7/3)] shows reaction was complete. The reaction mixture was neutralized with methanesulfonic acid (0.584 ml, 9.00 mmol) and stirred for 1 h at room temperature. Cold water (5.00 ml) was added to the reaction mixture and stirred at room temperature until product crystallized out. To the slurry was added water (5 ml) of water stirred for additional 20 min, followed by the addition of water (5 ml) prior to filtration. The solid obtained was collected by filtration washed with water (5 ml and 2.5 ml), dried under vacuum to afford 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6- (cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid methane sulfonate salt (8c)
(1 .138 g, 1.867 mmol, 93 % yield) as a white solid; MP 221.2 °C; 1 H NMR (300 MHz,
DMSO-d6) δ 12.89 (s, 1 H, D2O exchangeable), 10.69 (s, 1 H, D2O exchangeable), 9.24
(t, J = 6.0 Hz, 1 H), 9.16 (s, 2H,), 8.85 (s, 2H), 8.24 (d, J = 8.0 Hz, 1 H), 8.06 – 7.91 (m, 2H), 7.86 – 7.70 (m, 4H), 7.15 – 6.96 (m, 2H), 6.03 (dd, J = 17.8, 1.4 Hz, 1 H), 5.52 – 5.35 (m, 1 H), 3.88 (s, 3H), 3.25 (t, J = 6.3 Hz, 2H), 2.34 (s, 3H), 1.17 – 1.01 (m, 1 H), 0.53 -0.43 (m, 2H), 0.32 – 0.23 (m, 2H); MS (ES+) 514.0 (M+1); Analysis calculated for:
CzeH^NsOsCHsSOsH 1.5H20: C, 54.71 ; H, 5.38; N, 11.00; S, 5.04; Found: C, 54.80; H, 5.14; N, 10.94; S, 4.90.
Example-4: Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) in Form C (Compound XX)

The jacket of a 10 L glass reactor was set to -5 °C. To the reactor was charged 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)-pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d) prepared in Step (11) of Example 1 (500 g, 1.22 mol), 4-amino-benzamidine-2HCI (280 g, 1.34 mol), and 2-propanol (4.05 kg). The mixture was cooled
46582
to 0.3 °C, and pyridine (210 g, 2.62 mol) followed by EDCI HCI (310 g, 1.61 mol) was added. The mixture was stirred at -1.1 – -0.3 °C for 22 hrs followed by addition of the second portion of EDCI HCI (58 g, 0.30 mol). The temperature of jacket was set to 14.0 °C, and the mixture was stirred for 89 hrs. The precipitate was filtered, and washed with 1.32 kg of 2-propanol.
The wet product (8a) was recharged to the reactor followed by addition of acetonitrile (1 .6 kg) and 0.57 kg water. The mixture was heated to 46 °C. 21 g of Smopex-234 and 10 g Acticarbone 2SW were added and the mixture was stirred at this temperature for 1 hr. The solution was filtered, and filtrate was returned back to the reactor. The jacket of the reactor was set to -5 °C, and the mixture was cooled to -0.2 °C. NaOH solution (256 g 46% NaOH, 2.95 mol, in 960 g water) was added in 25 min keeping the temperature ❤ °C. The mixture was stirred at 0.2-2.0 °C for 1 hr 40 min and then quenched with cone, acetic acid (40 g, 0.66 mol). Diluted acetic acid (80 g, 1.33 mol AcOH in 1000 g water) was added during 1 hr 20 min (temperature 1.7-3.0 °C), followed by 1250 g water (30 min). The suspension was stirred at 0-3.0 °for 1 hr, and filtered at 0-5 °C (ice mantle around the filter). The reactor and product (8d) was rinsed with 3.5 kg water.
The wet product (8d) was recharged to the reactor followed by 0.65 kg water and 1.69 kg acetonitrile. The mixture was heated to 57-60 °C, and stirred at this temperature for 14.5 hrs. The mixture was cooled to -2.2 °C (Tjacke,= -5 °C), and a solution of NaOH (163 g 46%, 1.87 mol, in 580 g water) was added during 15 min. The temperature rose to -0.4 °C. Hydrochloric acid (407 g 37% HCI, 4 mol) was added in 10 min, the temperature rose to 7.5 °C. The suspension was agitated at -3 – 0 °C for 19 hrs. The product was filtered and the filter cake was rinsed with 2.87 kg water, compressed and pulled dry. The wet product (1.30 kg) was dried at 40-43 °C and 50 mbar for 1 17 hrs to furnish 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) (484 g) as Form C (Compound XX).
/////avoralstat, BCX4161, Fast Track, Treat hereditary angioedema (HAE), Orphan Drug, PRECLINICAL
COc1cc(c(cc1C=C)C(=O)Nc2ccc(cc2)C(=N)N)c3cc(ncc3C(=O)O)C(=O)NCC4CC4

Avoralstat, BCX4161,
CAS 918407-35-9
UNII: UX17773O15
513.5513, C28-H27-N5-O5
Hereditary angioedema (HAE)
Kallikrein inhibitor
BioCryst Pharmaceuticals
![]()
BioCryst is also investigating second-generation plasma kallikrein inhibitors to avoralstat, for treating HAE (in February 2016, this program was listed as being in preclinical development).
Prevent acute attacks in patients with hereditary angioedema (HAE); Treat hereditary angioedema (HAE)
U.S. – Fast Track (Treat hereditary angioedema (HAE));
U.S. – Orphan Drug (Prevent acute attacks in patients with hereditary angioedema (HAE))
26 Feb 2016Clinical trials in Hereditary angioedema (Prevention) in USA (PO, Hard-gelatin capsule) before February 2016
24 Feb 2016Discontinued – Phase-III for Hereditary angioedema (Prevention) in France (PO, Soft-gelatin capsule)
24 Feb 2016Discontinued – Phase-III for Hereditary angioedema (Prevention) in Germany (PO, Soft-gelatin capsule)
| Conditions | Interventions | Phases | Recruitment | Sponsor/Collaborators |
|---|---|---|---|---|
| Hereditary Angioedema|HAE | Drug: BCX4161|Drug: Placebo | Phase 2|Phase 3 | Recruiting | BioCryst Pharmaceuticals |
| Hereditary Angioedema | Drug: BCX4161|Drug: Placebo | Phase 2 | Completed | BioCryst Pharmaceuticals |
| Hereditary Angioedema | Drug: BCX4161 | Phase 1 | Completed | BioCryst Pharmaceuticals |
| Hereditary Angioedema | Drug: BCX4161 | Phase 1 | Completed | BioCryst Pharmaceuticals |
Avoralstat, also known as BCX-4161, is a potent and orally active Kallikrein inhibitor and Bradykinin inhibitor. Avoralstat may be potentially useful for treatment for Hereditary angioedema. Avoralstat inhibits plasma kallikrein and suppresses bradykinin production. Bradykinin is the mediator of acute swelling attacks in HAE patients.
Selective inhibitor of plasma kallikrein that subsequently suppresses bradykinin production
Hereditary angioedema (HAE) is a serious and potentially life-threatening rare genetic illness, caused by mutations in the C1-esterase inhibitor (C1 INH) gene, located on chromosome 11q. HAE is inherited as an autosomal dominant condition, although one quarter of diagnosed cases arise from a new mutation. HAE has been classed as an orphan disease in Europe, with an estimated prevalence of 1 in 50,000. Individuals with HAE experience recurrent acute attacks of painful subcutaneous or submucosal edema of the face, larynx, gastrointestinal tract, limbs or genitalia which, if untreated, may last up to 5 days. Attacks vary in frequency, severity and location and can be life-threatening. Laryngeal attacks, with the potential for asphyxiation, pose the greatest risk. Abdominal attacks are especially painful, and often result in exploratory procedures or unnecessary surgery. Facial and peripheral attacks are disfiguring and debilitating.
HAE has a number of subtypes. HAE type I is defined by C1 INH gene mutations which produce low levels of C1 -inhibitor, whereas HAE type II is defined by mutations which produce normal levels of ineffective C1 protein. HAE type III has separate pathogenesis, being caused by mutations in the F12 gene which codes for the serine protease known as Factor XII. Diagnostic criteria for distinguishing the subtypes of HAE, and distinguishing HAE from other angioedemas, can be found in Ann Allergy Asthma Immunol 2008; 100(Suppl 2): S30-S40 and J Allergy Clin Immunol 2004; 114: 629-37, incorporated herin by reference.
Current treatments for HAE fall into two main types. Older non-specific treatments including androgens and antifibrinolytics are associated with significant side effects, particularly in females. Newer treatments are based on an understanding of the molecular pathology of the disease, namely that C1 INH is the most important inhibitor of kallikrein in human plasma and that C1 INH deficiency leads to unopposed activation of the kallikrein-bradykinin cascade, with bradykinin the most important mediator of the locally increased vascular permeability that is the hallmark of an attack.
Approved therapies include purified plasma-derived C1 INH (Cinryze®, Berinert), the recombinant peptide kallikrein inhibitor ecallantide (Kalbitor®), and the bradykinin receptor B2 inhibitor iticabant (Firazyr®). All of the currently available targeted therapies are administered by intravenous or subcutaneous injection. There is currently no specific targeted oral chronic therapy for HAE.
There are many delivery routes for active pharmaceutical ingredients (APIs). Generally, the oral route of administration is favored. Oral administration provides a number of advantages, such as, but not limited to, patient convenience, flexibility of timing of administration, location of administration and non-invasiveness. Oral administration also provides more prolonged drug exposure compared with intermittent intravenous infusion, which may be important for drugs with schedule-dependent efficacy. For example, a drug with a short half-life can achieve a greater exposure time by either continuous infusion or by continuous oral dosing. The use of oral therapy further has the potential to reduce the cost of healthcare resources for inpatient and ambulatory patient care services.
In the pharmaceutical arts, it is known that a number of APIs cannot be administered effectively by the oral route. The main reasons why these compounds cannot be administered by the oral route are: a) rapid enzymatic and metabolic degradation; b) chemical and/or biological instability; c) low solubility in aqueous medium; and/or d) limited permeability in the gastrointestinal tract. For such compounds, non-oral routes of delivery, such as parenteral administration, mainly via intramuscular or subcutaneous injections, may be developed. However, non-oral administration poses a disadvantage for the patient as well as healthcare providers, and for this reason, it is important to develop alternative routes of administration for such compounds, such as oral routes of administration.
While the oral route of administration is the most convenient for the patient and the most economical, designing formulations for administration by the oral route involves many complications. Several methods are available to predict the ease by which an API may be formulated into a formulation suitable for administration by the oral route. Such methods include, but are not limited to, and Lipinski rule (also referred to as the Rule of Five) and the Biopharmaceutical Drug Disposition Classification System (BDDCS).
The BDDCS divides APIs into four classifications, depending on their solubility and permeability. Class I APIs have high solubility and high permeability; Class II APIs have low solubility and high permeability; Class III APIs have high solubility and low permeability; and Class IV APIs have low solubility and low permeability. APIs in higher classes in the BDDCS face greater challenges in formulating into an effective, pharmaceutically acceptable product than those in lower classes. Of the four classes, APIs falling into Class IV are the most difficult to formulate into a formulation for administration by the oral route that is capable of delivering an effective amount of the API as problems of both solubility and permeability must be addressed (note the BDDCS does not inherently address chemical stability). The role of BDDCS in drug development is described generally in L.Z. Benet J Pharm Sci. 2013, 102(1), 34-42.
Lipinski’s rule (described in Lipinski et al. Adv. Drug Deliv. Rev. 46 (1-3): 3-26) states, in general, that in order to develop a successful formulation for administration by the oral route, an API can have no more than one violation of the following criteria:
i) not more than 5 hydrogen bond donors (nitrogen or oxygen atoms with one or more hydrogen atoms)
ii) not more than 10 hydrogen bond acceptors (nitrogen or oxygen atoms) iii) a molecular mass less than 500 daltons
iv) an octanol-water partition coefficient log P not greater than 5.
J. Zhang et al. Medicinal Chemistry, 2006, 2, 545-553, describes a number of small molecule amidine compounds which have activity as inhibitors of kallikrein. The molecules described in this document fall into Class IV of the BDDCS as described above. The compounds are poorly soluble in aqueous and physiological fluids, and are poorly permeable as demonstrated by oral dosing in rats and in vitro experiments with Caco-2 cells.
Furthermore, 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid, one of the compounds described in Zhang et al., is a Class IV API and violates criteria iii) and iv) as set forth in the Lipinski Rule.
Furthermore, the compounds described in Zhang et al., including 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid, exhibit poor stability with respect to oxidation in air, to light
(photodegradation) and in aqueous and physiological fluids, as well as to elevated temperatures.
Therefore, the compounds described by Zhang et al. including, but not limited to, 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid, not only exhibit poor solubility and permeability characteristics, but also poor stability characteristics. As a result, such compounds are predicted to be especially difficult to formulate into an effective, orally deliverable
pharmaceutical composition that is capable of delivering an effective amount of the compound to a subject.
Polymorphism, the occurrence of different crystal forms, is a property of some molecules. A single molecule may give rise to a variety of polymorphs having distinct crystal structures and physical properties, such as, but not limited to, melting point, thermal behaviors (e.g. measured by thermogravimetric analysis (TGA), or differential scanning calorimetry (DSC), x-ray diffraction pattern, infrared absorption fingerprint, and solid state NMR spectrum. One or more of these techniques may be used to distinguish different polymorphic forms of a compound.
Discovering new polymorphic forms and solvates of a pharmaceutical product can provide alternate forms of the compound that display a number of desirable and advantageous properties, such as, but not limited to, ease of handling, ease of processing, ease of formulation, storage stability, and/or ease of purification. Further, new polymorphic forms and solvates of a pharmaceutically useful compound or salts thereof may further provide for improved pharmaceutical products, by providing compounds that are more soluble in a set of pharmaceutical excipients. Still further, the provision of new polymorphic forms and solvates of a pharmaceutically useful compound or salts thereof enlarges the repertoire of compounds that a formulation scientist has available for formulation optimization, for example by providing a pharmaceutical product with different properties, such as, but not limited to, improved processing characteristics, improved handling characteristics, improved solubility profiles, improved dissolution profile and/or improved shelf-life. Therefore, there is a need for additional polymorphs of pharmaceutically useful compounds, such as, but not limited to, 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6- (cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid and the compounds disclosed herein.
In one aspect, the present invention provides an oral formulation that is capable of delivering an effective amount of the amidine compounds described by Zhang et al. to a subject. In particular, the present invention provides an oral formulation that is capable of delivering an effective amount of 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid to a subject. In one specific aspect, the 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid is present in a particular crystal form designated Form A. In light of the art suggesting the difficulties in formulating such an oral formulation, this result was unexpected.
As described herein, the amidine compounds described in Zhang et al., including, but not limited to, 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6- (cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid (specifically including particular crystal Form A), may now be conveniently used in oral administration and further used in oral administration for the treatment of a number of diseases and conditions in a subject, such as, but not limited to, HAE as described herein.
![]()
May 16 is HAE awareness day
See BioCryst’s video regarding HAE to learn more
Avoralstat is being developed as an oral prophylactic treatment for patients suffering from Hereditary Angioedema (HAE). Avoralstat inhibits plasma kallikrein and suppresses bradykinin production. Bradykinin is the mediator of acute swelling attacks in HAE patients.
In May 2014 BioCryst, announced that the OPuS-1 (OralProphylaxiS-1) Phase 2a proof of concept clinical trial met its primary efficacy endpoint, several secondary endpoints and all other objectives established for the trial. OpuS-1 enrolled 24 HAE patients with a history of HAE attack frequency of at least 1 per week. Treatment with avoralstat demonstrated a statistically significant mean attack rate reduction of 0.45 attacks per week versus placebo, p<0.001. The mean attack rate per week was 0.82 on BCX4161 treatment, compared to 1.27 on placebo.
In December 2014, BioCryst initiated enrollment in OPuS-2 (Oral ProphylaxiS-2). OPuS-2 is a blinded, randomized, 12-week, three-arm, parallel cohort design trial evaluating the efficacy and safety of two different dose regimens of avoralstat administered three-times daily, 300 mg and 500 mg, compared with placebo. The primary efficacy endpoint for the trial will be the mean angioedema attack rate, which will be reported for each avoralstat dose group compared to placebo. The trial is being conducted in the U.S., Canada and Europe. On October 8, 2015, announced that it has completed enrollment of approximately 100 HAE patients with a history of moderately frequent to very frequent attacks in OPuS-2. BioCryst expects to report the OPuS-2 trial results in early 2016.
PATENT
WO200234711
http://www.google.com/patents/WO2002034711A1?cl=en
PATENT
PATENT
Examples
Example 1 – Synthesis of 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl- phenyll-6-(cvclopropylmethyl-carbarnoyl)-pyridine-2-carboxylic acid
The synthesis of the above compound and intermediates is described below. In this section, the following abbreviations are used:
The synthesis of starting material, (4-(benzyloxy)-2-formyl-5-methoxyphenyl)boronic acid (1f) is described in Scheme 1.
f 0HCY ° ΒΓΥΥ°
Preparation of 6-bromobenzofdl[1,3ldioxole-5-carbaldehvde (1b)
1a 1b
To a mixture of piperonal (1a) (498 g, 3.32 mol) in glacial acetic acid (1000 mL) was added a solution of bromine (200 mL, 3.89 mol) in glacial acetic acid (500 mL) over a period of 30 min and stirred at room temperature for 24h. The reaction mixture was poured into water (2000 mL) and the solid that separated was collected by filtration. The solid was dissolved in boiling ethanol (4000 mL) and cooled to room temperature. The solid obtained on cooling was collected by filtration to furnish 6-bromobenzo[d][1 ,3]dioxole-5-carbaldehyde (lb) (365 g, 48 %) as a white solid, MP 126 °C; HNMR (300 MHz, DMSO-d6): δ 10.06 (s, 1 H), 7.42 (s,1 H), 7.29 (s, 1 H), 6.20 (d, J=12.3, 2H); IR (KBr) 3434, 2866, 1673,1489, 1413, 259, 1112, 1031 , 925 cm“1; Analysis calculated for CeH5BrO3.O 25H C, 41.15; H, 2.37; Found: C, 41.07; H, 2.11.
Preparation of 2-bromo-5-hvdroxy-4-methoxybenzaldehyde (1c)
1c
A solution of potassium tert-butoxide (397 g, 3.36 mol) in DMSO (1.5 L) was heated at 50 °C for 30 min. Methanol (1.5 L) was added to it and continued heating at 50 °C for additional 30 min. To the hot reaction mixture was added 6-bromo-benzo[d][1,3]dioxole-5-carbaldehyde (1 b) (350g, 1.53 mol) and continued heating at 50 °C for 30 min. The reaction mixture was cooled to room temperature and quenched with water (2.3 L) and sodium hydroxide (61.2 g, 1.53 mol). The reaction mixture was washed with ether (2 x 1.5 L), acidified to pH 2 using cone. HCI and extracted with ethyl acetate ( 1 L). The ethyl acetate layers were combined and concentrated under vacuum to dryness. The residue obtained was treated with water (1.5 L) and ethyl acetate (1 L). The solid obtained was collected by filtration to furnish 2-bromo-5-hydroxy-4-methoxybenzaldehyde (1c) (97 g, 27.5% as a first crop). The layers from the filtrate were separated and aqueous layer was extracted with ethyl acetate (200 ml_). The ethyl acetate layers were combined dried over MgS04 and concentrated under vacuum to dryness to furnish 2-bromo-5-hydroxy-4-methoxybenzaldehyde (1c) (192 g, 54.4%, second crop) as an orange solid, MP 108 °C; ‘HNMR (300MHz, DMSO-cfe): S 10.00 (s, 1 H), 9.92 (s,1 H), 7.27 (s, 1 H), 7.26 (s, 1 H), 3.93 (s, 3H); IR (KBr) 3477, 2967, 2917,
2837, 2767, 2740, 1657, 1595, 1428, 1270, 1210, 1164, 1022 cm“‘; Analysis calculated for C8H7Br03.H20: C, 38.58; H, 3.64: Found: C, 38.60; H, 3.60.
Preparation of 5-(benzyloxy)-2-bromo-4-methoxybenzaldehvde ( d)
To a solution 2-bromo-5-hydroxy-4-methoxybenzaldehyde (1c) (120 g, 520 mmol) in DMF (1000 mL) was added potassium carbonate (79 g, 572 mmol) and benzyl bromide (68 mL, 572 mmol). The reaction mixture was stirred at room temperature overnight and quenched with water (3000 mL). The solid obtained was collected by filtration, washed with ether and dried under vacuum to furnish 5-(benzyloxy)-2-bromo-4-methoxybenzaldehyde (1d) (113.19 g, 67.9%) as a white solid, MP 144 °C;1HNMR (300 MHz, DMSO-c/6): δ 10.06 (s, 1H), 7.47-7.34 (m, 7H), 5.17 (s, 2H), 3.92 (s, 3H); IR (KBr) 2898, 2851 , 1673, 1592, 1502, 1437, 1402, 1264, 1210, 1158, 1017, 754 cm“1; Analysis calculated for C 5H13Br03: C, 56.10; H, 4.08; Found: C, 55.44; H, 4.08.
Preparation of 1-(benzyloxy)-4-bromo-5-(diethoxymethyl)-2-methoxybenzene (1e)
15 046578
146
1d 1e
To a solution of 5-(benzyloxy)-2-bromo-4-methoxybenzaldehyde (1d) (100 g, 311 mmol) in
ethanol (1500 mL) was added triethyl orthoformate (103 mL, 622 mmol), ammonium nitrate
(7.5 g, 93.3 mmol) and stirred at room temperature overnight. The reaction mixture was
treated with ether (1200 mL) and stirred for 15 min before filtration. The filtrate was
concentrated under vacuum to dryness to give 1-(benzyloxy)-4-bromo-5-(diethoxymethyl)-2-methoxybenzene (1e) (134 g) as a brown syrup; The product was used in the next step
without further purification; 1H N R (300 MHz, DMSO-cf6) δ 7.45 – 7.37 (m, 4H), 7.36 – 7.33
(m, 1 H), 7.17 – 7.14 (m, 1 H), 7.10 (s, 1 H), 5.10 (s, 2H), 3.80 (s, 3H), 3.58 – 3.33 (m, 5H),
1.13 – 1.07 (m, 6H); IR (KBr) 2974, 2879, 1601 , 1503, 1377, 1260, 1163, 1060 cm“1;
Analysis calculated for C19H23Br04: C, 57.73; H, 5.86; Found: C, 57.21 ; H, 5.94.
acid (1fi
To a solution of 1-(benzyloxy)-4-bromo-5-(diethoxymethyl)-2-methoxybenzene (1e) (120 g,
300 mmol) in dry ether (1000 mL) at -78 °C was added n-butyllithium (1.6 M solution in
hexanes, 244 mL, 390 mmol) over a period of 30 min and further stirred at -78 °C for 30 min.
A solution of tri-n-butylborate (110 mL, 405 mmol) in dry ether (300 mL) was added to this
solution at -78 °C over a period of 30 min. The reaction mixture was further stirred for 2 h at -78 °C and warmed to 0 °C. The reaction mixture was quenched with 3N HCI (300 mL) at 0
°C and heated at reflux for 1 h. After cooling to room temperature, the solid obtained was
collected by filtration washed with water (250 mL) dried in vaccum to afford (4-(benzyloxy)-2-formyl-5-methoxyphenyl)boronic acid (1f) (30.85 gm, 37.6% as a white solid. The organic
layer from above filtrate was extracted with 1.5 N NaOH (3 x 200 mL). The combined basic
extracts were acidified with cone. HCI (pH about 4). The solid obtained was collected by
filtration, washed with water and dried under vacuum to furnish a second crop of (4-(benzyloxy)-2-formyl-5-methoxyphenyl)boronic acid (1f) (22.3 g, 26%) as a light orange solid
MP 158 °C; 1H NMR (300 MHz, DMSO-cfe) δ 10.08 (s, 1 H), 7.52 (s, 1 H), 7.48 – 7.33 (m, 5H),
7.24 (s, 1H), 5.18 (s, 2H), 3.89 (s, 3H); 1H NMR (300 MHz, DMSO-d6/D20) δ 10.06 (s, 1H),
7.52 (s, 1H), 7.49 – 7.32 (m, 5H), 7.23 (s, 1 H), 5.18 (s, 2H), 3.89 (s, 3H); MS (ES+) 309.1 (M+Na); IR (KBr) 3335, 2937, 1647, 1545, 1388, 1348, 1268, 1146, 1095 cm-1; Analysis calculated for C15H15BO5.0.25H2O: C, 62.00; H, 5.38; Found: C, 61.77; H, 5.19.
Synthesis of methyl-6-(cvclopropylmethylcarbamoyl¾-3-ftrifluoromethylsulfonyloxyVpicolinate
The synthesis of the intermediate methyl 6-(cyclopropylmethylcarbamoyl)-3-(trifluoromethyl sulfonyloxy)picolinate (2h) is described in Scheme 2.
Preparation of 2-bromo-3-hvdroxy-6-methylpyridine (2b)
![]()
H3C N Br
2a 2b
To a solution of 3-hydroxy-6-methylpyridine (2a) (3000 g, 27.5 mol) in pyridine (24 L) cooled to 15 °C was added a solution of bromine (4.83 kg, 1.55 L, 30.2 mol) in pyridine (3 L) over a period of 50 min maintaining the internal temperature between 20 to 25 DC. After stirring for 19 h at room temperature the solvent was removed under vacuum and the residue was triturated with water. The solid separated was collected by filtration, washed with water and dried under vacuum to give 2-bromo-3-hydroxy-6-methylpyridine (2b) (3502 g, 67.7 %) as a light brown solid which was used as such without further purification; 1H NMR (300 MHz, DMSO-d6) δ 10.43 (s, 1H), 7.18 (d, J = 8.0 Hz, 1 H), 7.08 (d, J
MS (ES+) 188.35, 186.36 (M+1).
(2c)
![]()
2b 2c
A mixture of 2-bromo-3-hydroxy-6-methylpyridine (2b) (3000 g, 15.96 mol), anhydrous potassium carbonate (3308 g, 23.94 mol), and iodomethane (2.491 kg, 1.09 L, 17.556 mol) in 30 L of acetone was heated at 40 °C overnight. The reaction mixture was cooled to room temperature and filtered through Celite. Evaporation of the solvent followed by silica gel chromatography (Hexane: ethyl acetate = 7:3) afforded the desired compound, 2-bromo-3-methoxy-6-methylpyridine (2c) which was used as such for the next step; 1H NMR (300 MHz, DMSO-cfe) δ 7.42 (dd, J = 8.3, 1.5 Hz, 1H), 7.29 – 7.19 (m, 1H), 3.84 (d, J = 1.6 Hz, 3H), 2.37 (d, J = 1.7 Hz, 3H).
2c
2d
To a solution of 2-bromo-3-methoxy-6-methylpyridine (2c) (310 g, 1.53 mol) in 6000 mL of water at 60 °C was added KMnO, (725 g, 4.59 mol) in small portions over a 90 min period with vigorous mechanical stirring. A dark purple solution resulted. This solution was kept at 90 °C for a further 3 h and filtered through Celite while still hot to give a colourless filtrate.
After cooling, the aqueous solution was acidified to pH 1-2 by adding 6 N HCI. The white solid obtained was collected by filtration to give on drying 6-bromo-5-methoxy-2-pyridinecarboxylic acid (2d) (302g, 85%) of product, which was used as such in the next reaction without further purification. An analytical sample was obtained by recrystallization from methanol to give 6-bromo-5-methoxy-2-pyridinecarboxylic acid; 1H NMR (300 MHz, DMSO-tfe) δ 7.40 – 7.28 (m, 1H), 7.17 (d, J = 8.3 Hz, 1 H), 3.83 (d, J = 1.7 Hz, 3H).
Preparation of 6-bromo-N-(cvclopropylmethyl)-5-methoxypicolinamide (2e)
To a solution of 6-bromo-5-methoxy-2-pyridinecarboxylic acid (2d) (12 g, 52 mol) in pyridine (70 mL) was added EDCI (11.5 g, 59 mmol) and cyclopropylmethylamine (3.6 g, 52 mmol). The reaction mixture was stirred at room temperature overnight and then concentrated under vacuum. The reaction mixture was diluted with water (100 mL) and ethyl acetate (100 mL). The organic layer was separated and the water layer was extracted with ethyl acetate (2 x 100 mL). The organic layers were combined and washed with water (2 x 50 mL), brine (500 mL), dried over magnesium sulphate, filtered and concentrated under vacuum to furnish 10.43g of crude product. The crude product was converted into a slurry (silica gel 20 g) and purified by flash column chromatography (silica gel 230 g, eluting with 0-100% ethyl acetate in hexane) to yield compound 6-bromo-N-(cyclopropylmethyl)-5-methoxypicolinamide (2e) (8.02 g, 54%) as off white solid, mp 67-70 °C; 1HNMR (300 MHz, DMSO-d6) δ 8.51 (t, J = 5.8, 1 H), 8.02 (d, J = 8.4, 1 H), 7.65 (d, J = 8.5, 1 H), 3.96 (s, 3H), 3.14 (t, J = 6.5, 2H), 1.11 -0.99 (m, 1 H), 0.47 – 0.36 (m, 2H), 0.27 – 0.20 (m, 2H); MS (ES+) 307.0, 309.0 (100%
M+Na)
Preparation of methyl 6-(cvclopropylmethylcarbamoyl)-3-methoxypicolinate (2f)
To a solution of 6-bromo-N-(cyclopropylmethyl)-5-methoxypicolinamide (2e) (7.5 g, 27.6 mol) in methanol (300 mL) in a 2-L stainless steel bomb was added Pd(OAc)2(750 mg), 1 ,1-bis(diphenylphosphino)-ferrocene (750 mg), and triethylamine (3.9 mL, 27.6 mmol). The reaction mixture was vacuum flushed and charged with CO gas to 150 psi. The reaction mixture was and heated with stirring at 150°C overnight and cooled to room temperature. The catalyst was filtered through a pad of celite, and concentrated to dryness to furnish crude product. The crude was purified by flash column chromatography (silica gel 150 g,
eluting with, 0%, 5%, 10%, 20%, 30%, 50% ethyl acetate/hexanes (250 mL each) as eluents to give methyl 6-(cyclopropylmethyl-carbamoyl)-3-methoxypicolinate (2f) (6.29 g, 86.1 %) as a salmon coloured solid, MP 107 °C; 1HNMR (300 MHz, DMSO-cfe) δ 8.28 (t, J = 6.0, 1H), 7.91 (d, J = 8.8, 1H), 7.55 (d, J = 8.8, 1 H), 3.68 (s, 3H), 3.64 (s, 3H), 2.90 (t, J = 6.5, 2H), 0.89 – 0.68 (m, 1 H), 0.26 – 0.09 (m, 2H), 0.08 – 0.00 (m, 2H); MS (ES+) 287.1 (M+Na); IR (KBr) 3316, 2921 , 1730, 1659, 1534, 1472, 1432, 1315, 1272, 1228, 1189, 1099, 1003, 929, 846, 680 cm“1; Analysis calculated for C13H16 204: C, 59.08; H, 6.10; N, 10.60; Found: C, 58.70; H, 5.97; N, 10.23.
Preparation of 6-(cvclopropylmethylcarbamoyl 3-hvdroxypicolinic acid (2q)
2f 2g
Aluminium chloride method:
To a solution of methyl 6-(cyclopropylmethylcarbamoyl)-3-methoxypicolinate (2f) (0.16 mmol) in dichloromethane (840 mL) was added AICI3 (193 g, 1.5 mol). The reaction mixture was heated at reflux for 12 h under nitrogen. After slowly adding ~2L of 1 N HCI, the organic layer was separated. The aqueous layer was re-extracted several times with ethyl acetate/DME. The combined organic layer was washed with brine, dried (MgSO.4), and evaporated in vacuo to furnish crude 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid. To a solution of 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid was added a solution of acetyl chloride (1 10 mL) in methanol (1.1 L). The reaction mixture was stirred for 12 h at room temperature and then concentrated to dryness in vacuo. After co-evaporating once with methanol, the compound was purified by flash-column chromatography (silica gel, 500 g, eluted with chloroform and 3% methanol in chloroform) to furnish 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid (2g).
Boron tribromide method:
To a stirring solution of methyl 6-(cyclopropylmethylcarbamoyl)-3-ethoxypicolinate (2f) (58.0 g, 208 mmol) was added BBr3 (79 mL, 834 mmol) in CH2CI2 (1.3 L) at 0-5 °C. The reaction mixture was allowed to warm to room temperature and stirred for 18h. The reaction mixture was evaporated to dryness and anhydrous methanol (1 L) was added to the light yellowish solid residue. Insoluble solid was collected by filtration (36 g). Mother liquor was evaporated and co-evaporated with MeOH (2 x 200 mL). The insoluble solid (36 g) was treated with MeOH (500 mL) and acetyl chloride (50 mL) and stirred at room temperature for 18 h (at this point reaction mixture was clear). The mixture was evaporated to dryness and diluted with water and extracted with EtOAc. White solid that separated out from EtOAc layer was collected by filtration, washed with water (2 x 20 mL), dried in vacuo at 50 °C to afford 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid (2g) (5.36 g, 10 %) as a white solid, MP 92-95 °C. 1HNMR (DMSO-cfe) δ 11.04 (s, 1 H, exchangeable with D20), 8.37 (t, J = 6.0, 1 H, exchangeable with D20), 8.12 (d, J = 8.7 Hz, 1 H), 7.57 (d, J = 8.7 Hz, 1 H), 3.90 (m, 3 H), 3.15 (m, 2 H), 1.04 ( m, 1 H), 0.41 (m, 2 H), 0.24 (m, 2 H). IR (KBr): 3346, 3205, 1684 cm“1; MS (ES+): 251.1 (M+1); Analysis calculated for C12H14N2O4.0.1 H2O: C, 57.18; H, 5.67; N, 11.14; Found: C, 57.11 ; H, 5.61; N, 11.09.
Preparation of methyl-6-(cvclopropylmethylcarbamoyl)-3-(trifluoromethylsulfonyloxy) picolinate (2h
To a solution of 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid (2g) (28 mmol) in DMF (200 mL) were added triethylamine (12 mL, 84 mmol) and N-phenyl-bis(trifluoromethanesulfonimide) (12 g, 34 mmol). The reaction mixture was stirred for 1.5 h at room temperature and then poured into ice. After diluting with water and extracting with ethyl acetate, the aqueous phase was re-extracted, and then the combined organic layer was washed with water and concentrated under vacuum to give methyl-6-(cyclopropylmethylcarbamoyl)-3-(trifluoromethylsulfonyloxy)picolinate (2h), which was used in the next step without purification.
1H NMR (300 MHz, CDCI3) δ 8.50 (d, J = 8.6, 1 H), 8.07 (s, 1 H), 7.88 (d, J = 8.6, 1 H), 4.09 (d, J = 12.6, 3H), 3.48 – 3.24 (m, 2H), 1.18 – 1.01 (m, 1 H), 0.69 – 0.44 (m, 2H), 0.42 – 0.20 (m, 2H). MS (ES*): 405.17, 100%, M+Na.
Synthesis of 3-f2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyll-6-(cvclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid:
The synthesis of 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid (3i) is described as shown in Scheme 3.
3-f4-Benzyloxy-2-formyl-5-methoxy-phenylV6-(cvcloDroDvlmethvl-carbarnovn-pyridine-2-carboxylic acid methyl ester (3a)
5 046578
153
3a
To a solution of methyl-6-(cyclopropylmethylcarbamoyl)-3-(trifluoromethylsulfonyloxy)
picolinate (2h) (24.3g, 63 mmol) in DME (225 mL) were added water (25 mL), (4- (benzyloxy)-2-formyl-5-methoxyphenyl)boronic acid (1f) (27.3 g, 95 mmol), NaHC03(15.9 g,
5 189 mmol), and bis(triphenylphosphine)palladium(ll) chloride (0.885 g). The reaction
mixture was stirred at 70°C overnight under nitrogen. After extracting with ethyl acetate, the organic layer was washed with water and brine and dried (MgSO^), and then concentrated
under vacuum. The compound was purified by flash-column chromatography (silica gel, 300 g, eluting with 10%, 20%, 30% and 40% ethyl acetate in hexane) to furnish 3-(4-benzyloxy- 10 2-formyl-5-methoxy-phenyl)-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid
methyl ester (3a) (25 g, 83%) as off white solid, MP 48-50°C: 1H NMR (300 MHz, DMSO-cfe) δ 9.61(s, 1 H), 8.40 (d, J= 7.9 Hz, 1H), 8.14 (t, J= 5.0 Hz, 1H), 7.87 (d, J= 8.1 Hz, 1 H), 7.58
(s, 1H), 7.54-7.30 (m, 5H), 6.71 (s, 1 H), 5.24 (s, 2H), 3.93 (s, 3H), 3.70 (s, 3H), 3.45-3.34 (m,
2H), 1.19-1.05 (m, 1 H), 0.64-0.54 (m, 2H), 0.37-0.30 (m, 2H); IR ( Br) 1735, 1678, 1594,
15 1513, 1437, 1283, 1217, 1141, 1092 cm“1; MS (ES+) 497.29 (M+Na); Analysis calculated for
C27H2eN206: C, 68.34; H, 5.52; N, 5.90; Found; C, 68.16; H, 5.62; N, 5.80.
2-(6-(Cvclopropylmethylcarbamoyl)-2-(methoxycarbonyl)pyridin-3-vn-4-methoxy-5- vinylbenzoic acid (3b)
To a solution of 3-(4-benzyloxy-2-formyl-5-methoxy-phenyl)-6-(cyclopropylmethyl- carbamoyl)-pyridine-2-carboxylic acid methyl ester (3a) (24g, 50.6 mmol) in acetonitrile (50
mL), 2-methyl-2-propanol (350 mL), and water (125 mL) were added sodium dihydrogen
phosphate (12.5 g) and 2-methyl-2-butene (55 mL, 519 mmol). The reaction mixture was cooled in an ice bath and then sodium chlorite (28 g) was added. After stirring for 1 h, the reaction mixture was extracted with ethyl acetate and washed with water. The aqueous layer was re-extracted and then the combined organic layers were dried (MgS04). The solvent was evaporated in vacuo to furnish 5-(benzyloxy)-2-(6- ((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxybenzoic acid (3b) (29 g) which was used for the next step. MS (ES+): 513.24, (M+Na(; (ES ): 489.26, M-1.
Methyl 3-(4-(benzyloxy)-5-methoxy-2-(((2-methoxyethoxy)methoxytoarbonyltohenyl)-6-(cvclopropylmethylcarbamovnpicolinate (3c)
To a mixture of 5-(benzyloxy)-2-(6-(cyclopropylmethylcarbamoyl)-2-(methoxy-carbonyl)pyridin-3-yl)-4-methoxybenzoic acid (3b) (31 g, 63.2 mmol), and triethylamine (17.7 mL, 126.4 mmol) in dichloromethane (300 mL), was added MEM-chloride (9.03 mL, 79 mmol), and stirred at room temperature overnight. The reaction mixture was diluted with water and extracted with ethyl acetate. The organic layer was washed with water and dried over MgS04, filtered and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, 40 g) to furnish methyl 3-(4-(benzyloxy)-5-methoxy-2-(((2-methoxyethoxy)methoxy)carbonyl)phenyl)-6-(cyclopropylmethylcarbamoyl)picolinate (3c) (32.8 g, 89%) as a thick gum; H NMR (300 MHz, CDCI3) δ 8.35 (d, J = 8.0 Hz, 1 H), 8.15 (t, J = 5.7 Hz, 1 H), 7.78 (d, J = 8.0 Hz, 1H), 7.71 (s, 1H), 7.49 (d, J = 6.8 Hz, 2H), 7.36 (ddd, J = 7.5, 14.8, 22.4 Hz, 3H), 6.66 (s, 1 H), 5.37-5.13 (m, 4H), 3.90 (s, 3H), 3.69 (s, 3H), 3.60-3.49 (m, 2H), 3.49 (s, 2H), 3.39 (dd, J = 4.4, 8.4 Hz, 2H), 3.34 (s, 3H), 1.19-1.00 (m, 1H), 0.57 (q, J = 5.8 Hz, 2H), 0.38-0.25 (m, 2H). MS (ES+): 601.24 (M+Na); (ES“): 577.27 (M-1);1H NMR (300 MHz, DMSO-cfe) δ 8.69 (t, 7 = 6.1 Hz, 1H), 8.20 (d, J = 8.0 Hz, 1H), 7.97 (d, J = 8.0 Hz, 1 H), 7.63 (s, 1H), 7.41 (m, 5H), 6.92 (s, 1 H), 5.20 (m, 4H), 3.83 (s, 3H), 3.57 (s, 3H), 3.44 (m, 2H), 3:33 (m, 2H), 3.21 (m, 5H), 1.14 (m, 1H), 0.44 (m, 2H), 0.27 (m, 2H). IR (KBr):
1732, 1671 cm“1. MS (ES+): 601.1(M+Na); Analysis calculated for C31H 2Oe: C, 64.35; H, 5.92; N, 4.84; Found: C, 64.27; H, 6.04; N, 4.79.
Methyl 6-(cvclopropylmethylcarbamoyl)-3-(4-hvdroxy-5-methoxy-2-(((2-methoxyethoxy¾methoxy)carbonyl)phenyl)picolinate (3d)
3c 3d
To a solution of methyl 3-(4-(benzyloxy)-5-methoxy-2-(((2-methoxyethoxy)methoxy)-carbonyl)phenyl)-6-(cyclopropylmethylcarbamoyl)picolinate (3c) (32.8 g, 56.68 mmol) in ethanol (650 mL) was added 10% Pd/C (4 g) and hydrogenated at 45 psi for 5 h. The catalyst was removed by filtration through Celite and the filtrate was concentrated under vacuum to yield methyl 6-(cyclopropylmethylcarbamoyl)-3-(4-hydroxy-5-methoxy-2-(((2-methoxyethoxy)methoxy)carbonyl)phenyl)picolinate (3d) (31.87 g, 86%), which was pure enough to be used as such for the next step. An analytical sample of methyl 6-(cyclopropylmethylcarbamoyl)-3-(4-hydroxy-5-methoxy-2-(((2-methoxyethoxy) methoxy)carbonyl)phenyl)picolinate (3d) was obtained by purification of 350 mg of above crude using flash column chromatography (silica gel, eluting with ethyl acetate in hexane) to afford methyl 6-(cyclopropylmethyl-carbamoyl)-3-(4-hydroxy-5-methoxy-2-(((2-methoxyethoxy)methoxy)carbonyl)-phenyl)picolinate (3d) as a clear gum; 1HNMR (300 MHz, DMSO-d6) δ 9.74 (s, 1 H), 8.68 (t, J = 6.1 Hz, 1H), 8.18 (d, J = 8.0 Hz, 1 H), 7.95 (d, J = 8.0 Hz, 1H), 7.47 (s, 1H), 6.83 (s, 1H), 5.19 (s, 2H), 3.77 (m, 3H), 3.58 (s, 3H), 3.44 (m, 2H), 3.34 (m, 2H), 3.21 (m, 5H), 1.04 (m, 1 H), 0.44 (m, 2H), 0.27 (m, 2H); IR (KBr): 1731 , 1664 cm‘1. MS (ES*): 489.0 (M+1); Analysis calculated for C^e^O,,: C, 59.01; H, 5.78; N, 5.73; Found: C, 58.92; H, 6.15; N, 5.29.
6-(Cvclopropylmethylcarbamovn-3-(5-methoxy-2-(((2-methoxyethoxy^methoxy)-carbonyl)-4- (trifluoromethylsulfonyloxy)phenyl)picolinate (3e)
To a solution of methyl 6-(cyclopropylmethylcarbamoyl)-3-(4-hydroxy-5-methoxy-2-(((2- methoxyethoxy) methoxy)carbonyl)phenyl)picolinate (3d) (14.3 g, 29.3 mmol) in dichloromethane (150 mL) were added pyridine (12 mL, 146 mmol) and triflic anhydride (7.5 mL g, 44 mmol). After stirring overnight at room temperature under N2. the reaction mixture was poured into ice water and then extracted twice with dichloromethane. After washing the combined organic extracts with water and drying (MgS0 ), the solvent was evaporated in vacuo. The compound was purified by flash chromatography over silica gel column using ethyl acetate: hexane to afford methyl 6-(cyclopropylmethylcarbamoyl)-3-(5-methoxy-2-(((2- methoxyethoxy)methoxy)-carbonyl)-4-(trifluoromethylsulfonyloxy)phenyl)picolinate (3e) (1 g, 93%); H NMR (300 MHz, CDCy a 8.41 (d, J = 8.0, 1H), 8.17 (s, 1H), 8.03 (s, 1H), 7.79 (d, J = 8.0, 1 H), 6.82 (s, 1H), 5.32 (q, J = 6.1, 2H), 3.97 (s, 3H), 3.74 (s, 3H), 3.67 – 3.57 (m, 2H), 3.55 – 3.45 (m, 2H), 3.41 (dd, J = 8.2, 14.5, 2H), 3.34 (s, 3H), 1.36 – 1.17 (m, 1H), 0.58 (d, J = 7.1 , 2H), 0.33 (d, J = 5.1 , 2H).
Methyl 6-(cvclopropylmethylcarbamoyl)-3-(5-methoxy-2-f((2-methoxyethoxy)- methoxy)carbonvn-4-vinylphenyl)picolinate (3f)
To a solution of methyl 6-(cyclopropylmethylcarbamoyl)-3-(5-methoxy-2-(((2- methoxyethoxy)methoxy)carbonyl)-4-(trifluoromethylsulfonyloxy)phenyl)picolinate (3e) (37.4
g, 60.30 mmol) and potassium vinyltrifluoroborate (16.87 g, 120.6 mmol) in DMF (450 mL) and water (45 mL) was bubbled N2 for 5 min. To this mixture was added NaHC03 (20.26 g, 241.2 mmol) and dichloro-bis(triphenylphosphine)palladium (II) (6.34 g, 9.0 mmol). The reaction mixture was stirred at 70 °C for 20 h under N2(reaction progress was checked by 1H N R because product and starting material had same Rf in TLC). The reaction mixture was cooled down to room temperature and diluted with ethyl acetate. The organic layer was separated, washed with water, brine, dried ( gS04) and filtered. The filtrate was concentrated under vacuum to yield crude methyl 6-(cyclopropylmethyl-carbamoyl)-3-(5-methoxy-2-(((2-methoxyethoxy)methoxy)carbonyl)-4-vinylphenyl)-picolinate (3f). The crude product was purified by flash column chromatography (silica gel, 1 kg, eluting with 0-100% ethyl acetate in hexane) to afford methyl 6-(cyclopropylmethylcarbamoyl)-3-(5-methoxy-2-(((2-methoxyethoxy)methoxy) carbonyl)-4-vinylphenyl)picolinate [31) (26.54 g, 88%) as an amber gum; H NMR (300 MHz, DMSO-c¾ δ 8.70 (t, J = 6.1 Hz, 1H), 8.23 (d, J = 8.0 Hz, 1 H), 8.12 (s, 1 H), 8.00 (d, J = 8.0 Hz, 1 H), 6.98 (m, 2H), 5.94 (dd, J = 1.2, 17.8 Hz, 1H), 5.43 (d, J = 12.5 Hz, 1 H), 5.21 (d, J = 6.5 Hz, 2H), 3.88 (s, 3H), 3.64 (s, 3H), 3.48 (d, J = 3.1 Hz, 2H), 3.35 (m, 5H), 3.22 (m, 2H), 1.11 (s, 1H), 0.44 (dt, J = 4.9, 5.5 Hz, 2H), 0.28 (q, J = 4.8 Hz, 2H). IR (KBr); 1732, 1670 cm“1. MS (ES+) 499.1 (M+1).
2-(6-(cvclopropylmethylcarbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzolc acid (3g)
A mixture of methyl 6-(cyclopropylmethylcarbamoyl)-3-(5-methoxy-2-(((2-methoxyethoxy)methoxy) carbonyl)-4-vinylphenyl)picolinate (3f) (27.4 mmol) in DME (160 mL) and 6N HCI (40 mL) was stirred at room temperature for 6 h or till TLC showed complete conversion. The solvent was removed under vacuum. The residue obtained was suspended in water, the solid separated out was collected by filtration, washed with water and dried under vacuum to give 2-(6-(cyclopropylmethylcarbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (3g) (7.0 g, 63%) as a white
solid MP 40 – 42 °C; H NMR (300 MHz, DMSO-de) δ 8.69 (t, J= 6.0 Hz, 1H, NH), 8.20 (d, J= 7.9 Hz, 1H), 8.09 (s, 1 H), 7.95 (d, J= 8.1 Hz, 1H), 6.97 (dd, J= 18.0, 11.3 Hz, 1H), 6.88 (s, 1H), 5.92 (d, J= 7.9 Hz, 1H), 5.38 (d, J= 11.1 Hz, 1H), 3.85 (s, 3H), 3.63 (s, 3H), 3.27-3.17 (m, 2H), 1.15-1.05 (m, 1 H), 0.48-0.40 (m, 2H), 0.31-0.24 (m, 2H); IR (KBr): 3084, 1728, 1650, 1533, 1212, 1143 cm-1; MS (ES+) 433.26 (M+Na); (ES-): 409.28 (M-1); Analysis calculated for θ22Η22Ν2Ο6.0.25Η2Ο; C, 63.68; H, 5.47; N, 6.75; Found C, 63.75; H, 5.56; N, 6.65
Methyl-3-(2-(4-carbamimidoylprienylcarbamoyl)-5-metrioxy-4-vinylphenyl)-6- (cvclopropylmethylcarbamoyl)picolinate (3h)
To a solution of 2-(6-(cyclopropylmethylcarbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (3g) (2.35 g, 5.7 mmol) and 4-aminobenzimidamide dihydrochloride (3j) (1.79 g, 8.6 mmol) in DMF (20 mL) and pyridine (30 mL) at 0 °C was added EDCI (1.65 g, 8.6 mmol) and allowed to warm to room temperature overnight. The reaction mixture was quenched with 6N HCI (60 mL) and extracted with chloroform (3 x 60 mL). The organic layer was dried over MgS04, filtered and purified by flash column chromatography (silica gel, 110 g, eluting with 0 to 100% chloroform in CMA 80 in CMA 50) yielding methyl-3-(2-(4-carbamimidoylphenyl-carbamoyl)-5-methoxy-4-vinylphenyl)-6-(cyclopropylmethylcarbamoyl)picolinate (3h) (2.2 g, 65%) as a white solid MP 266 °C; 1H NMR (300 MHz, DMSO-c/6) δ 10.78 (s, 1 H), 9.26 (s, 2H), 9.03 (s, 2H), 8.67 (t, J = 6.1 , 1 H), 8.22 (d, J = 8.0, 1 H), 8.06 (d, J = 8.0, 1 H), 7.96 (s, 1 H), 7.89 – 7.74 (m, 4H), 7.13 – 6.96 (m, 2H), 6.07 (d, J = 17.7, 1H), 5.45 (d, J = 12.4, 1 H), 3.91 (s, 3H), 3.61 (s, 3H), 3.20 (s, 2H), 1.09 (dd, J = 4.7, 8.2, 1H), 0.43 (dt, J = 4.9, 5.4, 2H), 0.34 – 0.21 (m, 2H); MS (ES+) 528.1 (M+1); Analysis calculated for ![]()
C, 58.93; H, 5.63; N,11.85; Found: C, 58.75; H, 5.65; N, 11.92.
46578
159
3-r2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy -vinyl-phenyll-6-(cvclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid (3i)
3h 3i
To a solution of methyl-3-(2-(4-carbamirriidoylphenylcarbarnoyl)-5-methoxy-4-vinylphenyl)-6-(cyclopropylmethylcarbamoyl)picolinate (3h) (1 g, 1.9 mmol) in methanol (10 mL) and THF
(10 mL) was added 2 N NaOH (10 mL). The reaction mixture was stirred at room
temperature for 3 h, and concentrated in vacuo to remove methanol and THF. The aqueous layer was acidified with 6N HCI to pH 6-7 and the solid obtained was collected by filtration
washed with water and ether to furnish on drying 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid
(3i)(0.775 g, 80%) as the hydrochloride salt as an off white solid.
1H NMR (300 MHz, DMSO-d6) δ 12.67 (s, 1 H), 9.11 (s, 2H), 8.97 (s, 2H), 8.74 (s, 1 H), 7.90
(d, J = 7.8, 1 H), 7.80 (s, 1 H), 7.72 – 7.58 (m, 4H), 6.99 (dd, J = 11.3, 17.7, 1 H), 6.78 (s, 1H),
5.95 (d, J = 17.2, 1H), 5.38 (d, J = 11.9, 1H), 3.82 (s, 3H), 3.18 (s, 2H), 1.06 (s, 1 H), 0.43 (d,
J = 7.9, 2H), 0.25 (d, J = 4.7, 2H); MS (ES+) 514.0 (M+1 ); Analysis calculated for
C2eH27N5O5.HCI.H2O: C, 59.21; H, 5.32; N, 12.33; Found: C, 59.43; H, 5.21; N, 12.06.
Example 1A- Preparation of 3-f2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyll-6-(cvclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride in Form
C
The jacket of a 10 L glass reactor was set to -5 °C. To the reactor was charged 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)-pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d) prepared in Step (11) of Example 1 (500 g, 1.22 mol), 4-amino-benzamidine-2HCI (280 g, 1.34 mol), and 2-propanol (4.05 kg). The mixture was cooled to 0.3 °C, and pyridine (210 g, 2.62 mol) followed by EDCI HCI (310 g, 1.61 mol) was added. The mixture was stirred at -1.1 to -0.3 °C for 22 hrs followed by addition of the second portion of EDCI HCI (58 g, 0.30 mol). The temperature of jacket was set to 14.0 °C, and the mixture was stirred for 89 hrs. The precipitate was filtered, and washed with 1.32 kg of 2-propanol.
The wet product (8a) was recharged to the reactor followed by addition of acetonitrile (1.6 kg) and water (0.57 kg). The mixture was heated to 46 °C. Smopex-234 (21 g) and Acticarbone 2SW (10 g) were added and the mixture was stirred at this temperature for 1 hr. The solution was filtered, and filtrate was returned back to the reactor. The jacket of the reactor was set to -5 °C, and the mixture was cooled to -0.2 “C. NaOH solution (256 g 46% NaOH, 2.95 mol, in 960 g water) was added in 25 min keeping the temperature ❤ °C. The mixture was stirred at 0.2-2.0 °C for 1 hr 40 min and then quenched with cone, acetic acid (40 g, 0.66 mol). Diluted acetic acid (80 g, 1.33 mol AcOH in 1000 g water) was added during 1 hr 20 min (temperature 1.7-3.0 °C), followed by 1250 g water (30 min). The
suspension was stirred at 0-3.0 “for 1 hr, and filtered at 0-5 °C (ice mantle around the filter). The reactor and product (8d) was rinsed with 3.5 kg water.
The wet product (8d) was recharged to the reactor followed by 0.65 kg water and 1.69 kg acetonitrile. The mixture was heated to 57-60 °C, and stirred at this temperature for 14.5 hrs. The mixture was cooled to -2.2 °C (Tjackel= -5 °C), and a solution of NaOH (163 g 46%, 1.87 mol, in 580 g water) was added during 15 min. The temperature rose to -0.4 °C. Hydrochloric acid (407 g 37% HCI, 4 mol) was added in 10 min, the temperature rose to 7.5 °C. The suspension was agitated at -3 – 0 °C for 19 hrs. The product was filtered and the filter cake was rinsed with 2.87 kg water, compressed and pulled dry. The wet product (1.30 kg) was dried at 40-43 °C and 50 mbar for 11 hrs to furnish 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6- (cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) (484 g) as Form C.
Example-1 B: Preparation of 3-f2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyll-6-(cvclopropylmethylcarbartiovQpyridine-2-carboxylic acid hydrochloride in Form A
The procedure was carried out in an identical manner to Example 1 A, with the exception that after the final filtration the filter cake was rinsed with 2.87 kg methyl ierf-butyl ether instead of 2.87 kg water, and pulled dry. The product was dried at 40-43 °C and 50 mbar to furnish 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) as Form A.
PATENT
Methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)picolinate (compound 6a) is (I) (pages 85 and 86). Avoralstat hydrochloride (compound of formula XVIII) is (II) (claim 40, page 109). A Markush structures is presented (claim 1, page 99).
The synthesis of (II) via intermediate (I) is described (example 1, pages 80-93).
A synthesis of the compound 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid (Compound 3i) is described in Schemes A-C.
O y OHCk n Br^ ^OCH3
B Brr22,, AAccOOHH Y^ V”“ \ \ tt–BBuuOOKK
OHC^^^O ” Br^\^0 MeOH ” OHC
1a 1b 66%
1d 95% 1 e
![]()
1f
Scheme A
3h 31
Scheme C
Examples. In this section, the following abbreviations are used:
Example-1 : Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b)
7b
Step (1): Preparation of 6-Bromobenzo 1 ,3]dioxole-5-carbaldehyde (1 b):
1b
A solution of bromine (33.0 kg, 206.49 mol) in acetic acid (27.5 L) was added slowly to a solution of piperonal (1a) (29.9 kg, 199.16 mol) in acetic acid (105 L) at room
temperature over a period of 50 min and the reaction mixture was stirred at room temperature for 14.2 h. Additional solution of bromine (33 kg, 206.49 mol) in acetic acid (27.5 L) was added slowly to the reaction mixture over a period of 2 h and the reaction mixture was stirred for 22 h. The reaction mixture was quenched by addition of ice water (500 L) with stirring over a period of 6 h and continued stirring for additional 1.25 h. The mixture was allowed to settle and most of the supernatant liquid was decanted to a waste container using nitrogen pressure. Water (600 L) was added to the solid, stirred, mixture was allowed to settle and then most of the supernatant liquid was decanted to a waste container using nitrogen pressure. Water (100 L) was added to the decanted mixture, stirred for 15 min and the solid obtained was collected by filtration using a centrifuge. The solid was washed with water (2 x 100 L) and air-dried in a tray drier for 3.75 h to afford the crude product 1 b (52 kg). The crude product (51.2 kg) was stirred in n-hexane (178 L) for 3 h, collected by filtration, washed with n-hexane (25 L) and dried to afford 6-bromobenzo[1 ,3]dioxole-5-carbaldehyde (1b) (40.1 1 kg, 87.9%) as a light brown solid. MP: 109-112°C. 1H NMR (300 MHz, CDCI3) δ 10.21 (s, 1 H), 7.37 (s, 1 H), 7.07 (s, 1 H), 6.10 (s, 2H); HNMR (DMSO-cf6): δ 10.06 (s, 1 H), 7.42 (s, 1 H), 7.29 (s, 1 H), 6.20 (d, J =12.3 Hz, 2H)
The process is also illustrated in Fig. 1.
Average yield of isolated 1 b from step-1 is 78 – 88%.
Step (2): Preparation of 2-Bromo-5-hydroxy-4-methoxy-benzaldehyde (1c)
A solution of potassium terf-butoxide (10.7 kg, 95.36 mol) in DMSO (49 L) was stirred at 50 °C for 30 min. Methanol (49 L) was added slowly over a period of 4.25 h and stirred at 50 °C for 30 min. 6-Bromobenzo[1 ,3]dioxole-5-carbaldehyde (1 b) (9.91 kg, 43.27 mol) was added to the reaction mixture in small portions over a period of 45 min and stirred at 50 °C for 1 h. The reaction mixture was cooled to room temperature and split into two equal portions. Each portion was quenched with water (50.9 L) and basified with 50% aqueous NaOH solution (2.4 L). Each portion was extracted with MTBE (4 x 36 L) to remove impurities. The aqueous layer was acidified with cone. HCI to pH ~ 3 to obtain
product as a yellow solid. The solid was collected by filtration using a centrifuge, washed with water (2 x 35 L) and air-dried to afford 2-Bromo-5-hydroxy-4-methoxy-benzaldehyde (1c) (4.37 kg, 40.7%, contains 7 % water); Mp: 100-102°C; 1HNMR (300MHz, DMSO-d6): δ 10.00 (s, 1 H), 9.92 (s,1 H), 7.27 (s, 1 H), 7.26 (s, 1 H), 3.93 (s, 3H).
The process is also illustrated in Fig. 2.
Average yield of isolated product 2-Bromo-5-hydroxy-4-methoxy-benzaldehyde (1c) from step-2 is 40-50%.
Step (3): 5-Hydroxy-4-methoxy-2-(4,4,5,5-tetramethyl-[1 ,3,2]dioxaborolan-2-y benzaldehyde (4a)
2-Bromo-5-hydroxy-4-methoxy-benzaldehyde (1c) [1.3 kg (93%, 7% water content), 5.25 mol] was dissolved in toluene (13 L) in a reaction flask equipped with a Dean Stark apparatus. The solution was heated at reflux with stirring to distil off about 25% of the toluene along with water (90 ml_). The solution was cooled to 90 °C then
bis(pinacolato)diboron (1.5 kg, 5.82 mol), KOAc (772.6 g, 7.87 mol) and Pd(PPh3) (24.3 g, 0.02 mol) were added and the reaction mixture was heated at reflux for 10h. After confirming the completion of reaction by TLC (mobile phase: 100% DCM), the reaction mixture was cooled to room temperature and was kept standing overnight. The reaction mixture was filtered through celite and the celite cake was washed with toluene (4 L). The filtrate of this batch was mixed with the filtrate of another batch (batch size 1.3 kg obtained from an identical reaction). The mixed filtrate was washed with water (17.5 L), brine (17.5 L), dried over Na2S04, filtered and the solution was passed through a pad of silica gel (2 kg, mesh size 230-400). The silica gel pad was washed with toluene. The combined filtrate and washing was concentrated under reduced pressure and the residual crude product was stirred with n-hexane (23 L) for 1 h to obtain a solid product. The solid was collected by filtration, washed with n-hexane (5 L) and dried to afford 5-hydroxy-4-methoxy-2-(4,4,5,5-tetramethyl-[1 ,3,2]dioxaborolan-2-yl)benzaldehyde (4a) (2.47 kg, 84.6%). H NMR (300 MHz, CDCI3) δ 10.54 (s, 1 H), 7.57 (s, 1 H), 7.33 (s, 1 H), 5.89 (s, 1 H), 4.01 (s, 3H), 1.37 (s, 12H); 1H NMR (300 MHz, DMSO-d6) δ 10.35 (s, 1 H), 9.95 (s, 1 H), 7.33 (s, 1 H), 7.23 (s, 1 H), 3.87 (s, 3H), 1.33 (s, 12H); MS (ES+) 301.1 (M+Na); 579.1 (2M+Na); Analysis calculated for C14H19B05: C, 60.46; H, 6.89; Found: C, 60.60; H, 6.87
The average yield of 5-hydroxy-4-methoxy-2-(4,4,5,5-tetramethyl-[1 ,3,2]dioxa-borolan-2-yl)benzaldehyde (4a) from step (3) is 78 – 90%.
The process is also illustrated in Fig. 3.
Step (4): Preparation of 3-Bromo-2,6-dimethylpyridine (5b)
2,6-lutidine (5a) (115 kg, 1073.3 mol) was added into pre-chilled oleum (20-23%, 1015 kg, 2276.7 mol) at 0 °C over a period of 4.5 h (temperature r6ached 14 °C during the addition). Bromine (88.18 kg, 1103.6 mol) was then added at 5-10 °C over a period of 1 h. The reaction mixture was slowly heated to 150 °C over a period of 12h. TLC analysis indicated about 40-50% conversion to product and the formation of a dimer by-product (5%). The reaction mixture was cooled to room temperature and then additional bromine (88.18 kg, 1103.6 mol) was added slowly. The reaction mixture was slowly heated to maintain a temperature of 65-75 °C over a period of 15h. TLC analysis indicated a 65-70 % conversion to product and the formation of 5% dimer by product. The reaction mixture was quenched by addition of water (500L) while maintaining the reaction temperature below 20 °C. The mixture was basified with 6.6 M NaOH (3800 L) while maintain the temperature at < 40 °C. EtOAc (220 L) was added and the mixture was stirred for 1 h then allowed to settle over a period of 2 h. The layers were separated and the aqueous layer was treated with NaOH (10 kg) in water (10 L) and extracted with EtOAc (160 L). The organic extracts were combined washed with brine (100 L), dried over Na2S04 (50.0 kg), filtered and the solvent was evaporated under atmospheric pressure. The residue was vacuum distilled and the desired product 3-bromo-2,6-dimethylpyridine (5b) was collected at 58-60 °C, 2 mmHg (98.45 kg, 49.2 %) as a colorless liquid.
The process is also illustrated in Fig. 4.
Step (5): Preparation of 3-Bromopyridine-2,6-dicarboxylic acid (5c)
![]()
5b 5c
To a stirred solution of 3-bromo-2,6-dimethylpyridine (5b) (98 kg, 5326 mol) in water (1310 L) was added KMn0 (225 kg, 1423.6 mol) in 5 equal portions in 1 h intervals at 70 °C. After stirring for 1 h at 70 °C, additional KMn04 (225 Kg, 1423.6 mol) was added in 5 equal portion in 1 h intervals at 90 °C. The reaction mixture was stirred for 12 h at 90 °C. The suspension was filtered hot through celite to obtain a clear solution. The solvent was distilled off to remove about 30% of the total volume. The remaining concentrated solution was chilled to 0 °C and made acidic (to pH 3-4) by the addition of cone. HCI (120 L). The white precipitate obtained was collected by filtration and dried at 70 °C to afford 3-bromopyridine-2,6-dicarboxylic acid (5c) as a white solid (109 kg, 84%).
The process is also illustrated in Fig. 5.
Step (6): Preparation of Dimethyl 3-Bromopyridine-2,6-dicarboxylate (5d)
To a stirred solution of 3-bromopyridine-2,6-dicarboxylic acid (5c) (20.0 kg, 81.29 mol) in methanol (100 L) was added cone. H2S04 (4.4 L) over a period of 30 min. The reaction mixture was heated to 65 °C and maintained at that temperature for 5 h (the reaction was monitored by TLC analysis to determine completion of reaction). The reaction mixture was cooled to room temperature basified by careful addition of aqueous NaHC03 solution (prepared from 10 kg NaHC03 in 120 L of water) and further diluted with water (120 L). The white solid obtained was collected by filtration, washed with plenty of water and then oven-dried at 40 °C to obtain dimethyl 3-bromopyridine-2,6-dicarboxylate (5d) (9.2 kg, 41.3%) as a white solid; 1HNMR (300 MHz, DMSO-cf6) δ 8.47 (d, J = 8.4, 1 H), 8.08 (dd, J = 4.5, 8.4, 1 H), 3.95 (s, 3H), 3.91 (s, 3H); MS (ES+) 570.6 (2M+Na); Analysis calculated for C9H8BrN04: C, 39.44; H, 2.94; Br, 29.15 N, 5. 1 ;
Found: C, 39.52; H, 2.92; Br, 29.28; N, 5.03.
The process is also illustrated in Fig. 6.
6582
Step (7): Preparation of Methyl 3-bromo-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylate (
To a stirred solution of dimethyl 3-bromopyridine-2,6-dicarboxylate (5d) (27 kg, 98.52 mol) in ierf-butanol (135 L) was added at room temperature cyclopropylmethanamine (7.83 kg, 110.1 mol). The reaction mixture was heated at 65 °C for 17 h. The progress of reaction was monitored by TLC and HPLC (HPLC analysis showed the formation of 74% of the product 5e after 17 h. The reaction mixture was cooled to room temperature and then cone. HCI (2.7 L) was added slowly and the mixture was stirred for 15 min. The reaction mixture was concentrated under reduced pressure to obtain the crude product. The crude product was dissolved in hot /-PrOH (54 L) filtered through a celite pad. The filtrate was cooled with stirring to 10 °C to obtain a white precipitate. The solid obtained was collected by filtration, washed with cold
i-PrOH (13 kg), n-hexane (15 L) and dried to provide pure methyl 3-bromo-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylate (5e) (15.7 kg, 50.9%). The filtrate was concentrated under reduced pressure and the crude product can be purified by silica gel column chromatography eluting with tert-butanol in hexanes to furnish additional 10% methyl 3-bromo-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylate (5e). HNMR (300 MHz, DMSO-cf6) δ 8.83 (t, J = 5.9, 1 H), 8.47 – 8.41 (m, 1 H), 8.06 (d, J = 8.4, 1 H), 3.96 (s, 3H), 3.16 (t, J = 6.5, 2H), 1.14 – 0.99 (m, 1 H), 0.42 (m, 2H), 0.30 -0.19 (m, 2H); MS (ES+) 337.0 (M+23), 650.8 (2M+23); Analysis calculated for
C12H13BrN203: C, 46.03; H, 4.18; N, 8.95; Br, 25.52; Found: C, 46.15; H, 4.17; N, 8.72; Br, 25.26.
The average isolated yield for step (7) is 50% to 60%.
The process is also illustrated in Fig. 7.
Step (8): Preparation of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)picolinate (6a)
2
6a
THF (37.5 L) was charged to a 100 L reactor followed by ethyl 3-bromo-6- (cyclopropylmethyl-carbamoyl)pyridine-2-carboxylate (5e) (2.5 kg, 7.98 mol) under a nitrogen atmosphere. The reaction mixture was degassed twice by applying alternate vacuum and nitrogen. 5-Hydroxy-4-methoxy-2-(4,4,5,5-tetramethyl-[1 ,3,2]dioxa-borolan-2-yl)benzaldehyde (4a) (2.88 kg, 10.36 mol) was added, followed by the addition of PPh3 (53.13 g, 0.20 mol), PdCI2(PPh3)2 (120.4 g, 0.17 mol) and a solution of Na2C03(2.12 kg, 20.00 mol) in demineralized water (10.0 L) under nitrogen atmosphere. The reaction mixture was degassed again two times by applying alternate vacuum and nitrogen. The reaction mixture was heated at reflux for 6.5 h, cooled to room temperature and filtered through a Celite bed. Water (75 L) was added to the filtrate and the product was extracted with ethyl acetate (75 L). The aqueous layer was back extracted with ethyl acetate (2 χ 60 L). The combined ethyl acetate extract was divided into two equal portions and each portion was washed with brine (37 L), dried over Na2S04, filtered and concentrated under reduced pressure to give crude methyl 6- ((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)picolinate (6a) as a reddish viscous material (-4.5 Kg) which was used as such for the next step without further purification. An analytical sample was prepared by purification of a small sample by flash column chromatography (silica gel, eluting with 0-100% ethyl acetate in hexane) to furnish methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)-picolinate (6a) as an off-white solid; HNMR (300 MHz, DMSO-d6) δ 9.89 (s, 1 H), 9.52 (s, 1 H), 8.79 (t, J = 6.1 Hz, 1 H), 8.23 (d, J = 8.0 Hz, 1 H), 8.09 (d, J = 8.0 Hz, 1 H), 7.34 (s, 1 H), 6.90 (s, 1 H), 3.85 (s, 3H), 3.62 (s, 3H), 3.22 (m, 2H), 1.16 -1.02 (m, 1 H), 0.49 – 0.38 (m, 2H), 0.32 – 0.22 (m, 2H); MS (ES+) 791.0 (2M+Na), (ES-) 382.7 (M-1), 767.3 (2M-1); Analysis calculated for C20H20N2O6.0.25 H20: C, 61.77; H, 5.31 ; N, 7.20; Found: C, 61.54; H, 5.13; N, 7.05.
The process is also illustrated in Fig. 8.
46582
Step (9): Preparation of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-(((trifluoromethyl)sulfonyl)oxy)phenyl)picolinate (6b)
6a 6b
A solution of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)picolinate (6a) (2.11 kg, estimated about 3.83 mol from step-8) in dichloromethane (16.0 L) and pyridine (1.4 L, 17.4 mol) cooled to -10°C and maintained at that temperature for 1 h was added a solution of triflic anhydride (980.0 ml_, 5.8 mol) in dichloromethane (6.0 L) drop wise over a period of 3 h at -10 °C. The reaction mixture was stirred at -5°C for 1.3 h, quenched with saturated aqueous NaHCO3(10.4 L) and stirred for 30 mins. The organic layer was separated, washed successively with saturated aqueous NaHC03 (10.4 L), 1 HCI (2 x 16.6 L), water (13.2 L), brine (13.2 L), dried over MgS04, filtered and concentrated under reduced pressure to give the crude product. The crude product was stirred with 15% ethyl acetate in n-hexane (7.0 L) for 1 h. The solid obtained was collected by filtration washed with 15% ethyl acetate in n-hexane (3.0 L). The solid was stirred again with 15% ethyl acetate in n-hexane (7.0 L) for 1 h, was collected by filtration and washed with 15% ethyl acetate in n-hexane (3.0 L). The solid was stirred again with 15% ethyl acetate in n-hexane (8.0 L) for 1 h, collected by filtration washed with 15% ethyl acetate in n-hexane (3.0 L). The solid was dried to afford methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-(((trifluoromethyl)sulfonyl)-oxy)phenyl)picolinate (6b) as a light brown solid (1.7 kg, 86% yield, for combined steps 8 & 9). Average isolated yield for combined steps 8 and 9 was 70% to 86%; Ή NMR (300 MHz, DMSO-cf6): δ 9.64 (s, 1 H), 8.78 (t, J = 6.1 , 1 H), 8.29 (d, J = 8.0, 1 H), 8.16 (d, J = 8.0, 1 H), 8.03 (s, 1H), 7.39 (s, 1 H), 4.00 (s, 3H), 3.63 (s, 3H), 3.22 (m, 2H), 1.11 (m, 1 H), 0.52 – 0.39 (m, 2H), 0.28 (m, 2H); MS (ES+) 538.9 (M+Na). The process is also illustrated in Fig. 9.
Step (10): Preparation of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-vinylphenyl)picolinate (6c)
A solution of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4- (((trifluoromethyl)sulfonyl)oxy)phenyl)picolinate (6b) (12 kg, 23.24 mol) in DME (106 L) was charged into reactor under nitrogen. The reaction mixture was degassed twice by applying alternate vacuum and nitrogen. Potassium trifluoro(vinyl)borate (3.9 kg, 29.1 1 mol), PdCI2(PPh3)2 (815 g, 1.13 mol), KHC03 (4.65 g, 46.44 mol) and demineralized water (12 L) was then added under a N2 atmosphere. The reaction mixture was degassed by applying alternate vacuum and nitrogen. The reaction mixture was heated at reflux for 5 h. The reaction mixture was cooled to room temperature and then filtered through a Celite bed. Demineralized water (118 L) was added to the filtrate followed by ethyl acetate (124 L). The mixture was stirred for 20 min and then the organic layer was separated. The aqueous layer was back-extracted with ethyl acetate (2 x 95 L). The combined organic extract was washed with brine (95 L), dried over Na2S04, and filtered. The solvent was evaporated under reduced pressure to give the crude product. The crude product was purified by column chromatography (silica gel, 120 kg, 230-400 mesh size, eluting with ethyl acetate in n-hexane) to obtain methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-vinylphenyl)picolinate (6c) (6 kg, 72%). 1H NMR (300 MHz, CDCI3): δ (ppm) 9.64 (s, 1 H), 8.35 (d, J = 7.8 Hz, 1 H), 8.06-8.03 (m, 2H), 7.78(d, J = 7.8 Hz, 1 H), 7.02-6.92 (m, 1 H), 6.61 (s, 1 H), 5.86 (d, J = 17.7 Hz, 1 H), 5.38 (d, J = 1 1.4 Hz, 1 H), 3.84 (s, 3H), 3.67 (s, 3H), 3.35-3.29 (m, 2H),1.08-1.03 (m, 1H), 0.55-0.49 (m, 2H), 0.29-0.2 4(m, 2H). 1HNMR (300 MHz, DMSO-d6) 6 9.68 (s, 1 H), 8.77 (t, J = 6.1 , 1 H), 8.35 – 8.21 (m, 1 H), 8.16 – 8.01 (m, 2H), 7.14 -6.87 (m, 2H), 6.01 (dd, J = 1.2, 17.8, 1 H), 5.45 (dd, J = 1.1 , 1 1.3, 1 H), 3.91 (s, 3H), 3.64 (s, 3H), 3.23 (m, 2H), 1.21 – 1.01 (m, 1H), 0.51 – 0.40 (m, 2H), 0.34 – 0.20 (m, 2H). MS
(ES+) 417.0 (M+Na); Analysis calculated for C22H22N205: C, 66.99; H, 5.62; N, 7.10;
Found: C, 66.75; H, 5.52; N, 7.06.
The process is also illustrated in Fig. 10.
Step (1 1): Preparation of 2-(6-((cyclopropylmethyl)carbamoyl)-2- (methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d)
To a stirred solution of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-vinylphenyl)picolinate (6c) (1.57 kg, 3.80 mol) in acetonitrile (15.4 L) was added ferf-butyl alcohol (22.2 L), demineralized water (3.2 L) and sodium dihydrogen phosphate monohydrate (323.74 g, 2.346 mol). The reaction mixture was cooled to 0 °C and added 2-methyl-2-butene (5.3 L, 50.0 mol) and stirred at 0 °C for 30 min. A solution of 80% sodium chlorite (1.36 kg, 12.0 mol) in demineralized water (5.2 L) was added to the reaction mixture over a period of 2.5 h at 0 °C [temperature rises to 7 °C during the addition]. The reaction mixture was stirred at 0 °C for 2 h, diluted with water (40 L) and ethyl acetate (24 L). After stirring the mixture, it was allowed to settle and the organic layer was separated. The aqueous layer was back-extracted with ethyl acetate (2 x 20 L) then acidified with 5.9 % aqueous acetic acid (2 L) and extracted once with ethyl acetate (10 L). The organic extracts were combined washed with water (2 x 20 L), a solution of acetic acid (125 mL) in water (20.0 L), brine (2 χ 20 L), dried over Na2S04, filtered and concentrated under reduced pressure (vapor temperature below 40 °C). The residue obtained was dissolved in acetone (7 L) (residue didn’t dissolve completely). The solution was poured slowly into a reactor containing stirred n-hexane (70.0 L) to precipitate the solid product and the mixture was stirred for 2 h. The solid obtained was collected by filtration, washed with 10% acetone in n-hexane (6.3 L), AJ-hexane (6.3 L), dried to afford 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4- methoxy-5-vinylbenzoic acid (6d) as an off-white solid (1.29 Kg, yield: 79.0%). Average isolated yield for step 1 1 is 74% to 84%. 1H NMR (300 MHz, DMSO-d6): δ (ppm) 12.50 (brs, 1 H), 8.69(t, J= 6.0 Hz, 1 H, NH), 8.20 (d, J= 7.9 Hz, 1 H), 8.09 (s, 1 H), 7.95 (d, J= 8.1 Hz, 1 H), 6.97 (dd, J= 18.0, 1 1.3 Hz, 1 H), 6.88 (s, 1 H), 5.92 (d, J= 7.9 Hz, 1 H), 5.38 (d, J= 1 1.1 Hz, 1 H), 3.85 (s, 3H), 3.63 (s, 3H), 3.27-3.17 (m, 2H), 1.15-1.05 (m, 1 H), 0.48-0.40 (m, 2H), 0.31-0.24 (m, 2H); MS (ES+) 433.26, (M+Na); (ES-) 409.28 (M-1). The process is also illustrated in Fig. 1 1.
Step (12): Preparation of Methyl 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylate methanesulfonate (7a
Pyridine (3.8 L, 47.17 mol) and EDCI (5.31 kg, 27.66 mol) were sequentially added to a cooled solution (0 °C) of 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)-pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d) (9 kg, 21.92 mol) and 4-aminobenzamidine dihydrochloride (5.13 kg, 24.65 mol) in /-PrOH (90 L). The reaction mixture was allowed to warm to room temperature and stirred for 2 h. TLC analysis indicated incomplete reaction. Additional EDCI (1.08 kg, 5.6 mol) was added and the reaction mixture was stirred for 8 h. The reaction was still incomplete as indicated by TLC analysis, additional EDCI (0.54 kg, 2.8 mol) was added and the reaction mixture was stirred for 5 h. TLC analysis indicated there was trace amount of unreacted starting material remaining. The reaction mixture was cooled to 0 °C and a solution of
methanesulfonic acid (MSA) (9.13 kg, 95 mol) in MeOH (38.7 L) was added to the cooled mixture over a period of 4 h. The reaction mixture was allowed to warm to room temperature and stirred for 15 h. The product was collected by filtration, washed with a mixture of /‘-PrOH and MeOH (4:1 , 45 L). The wet cake was slurried in a mixture of /-PrOH and MeOH (2:1 , 135 L) stirred for 1 h and the product was collected by filtration and washed with a mixture of /‘-PrOH and MeOH (4:1 , 46.8 L). The product was dried in
2015/046582
a vacuum oven at 45 °C to afford methyl 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethyl-carbamoyl)pyridine-2-carboxylate methanesulfonate (7a) as a pink-colored solid (12.71 kg, 93%). Average isolated yield for this step: >90%.
1H NMR (300 MHz, DMSO-c/6) δ 10.71 (s, 1 H), 9.16 (s, 2H), 8.80 (s, 2H), 8.68 (t, J = 6.1 Hz, 1 H), 8.22 (d, J = 8.0 Hz, 1H), 8.06 (d, J = 8.1 Hz, 1 H), 7.93 (s, 1H), 7.84 – 7.72 (m, 4H), 7.12 – 6.97 (m, 2H), 6.04 (dd, J = 17.8, 1.3 Hz, 1 H), 5.45 (d, J = 12.6 Hz, 1H), 3.91 (s, 3H), 3.60 (s, 3H), 3.25 – 3.16 (m, 2H), 2.32 (s, 3H), 1.10 – 1.01 (m, 1 H), 0.48 – 0.37 (m, 2H), 0.30 – 0.22 (m, 2H); MS (ES+) 528.0 (M+1); Analysis calculated for
C29H29N5O5.CH3SO3H.2H2O. C, 54.62; H, 5.65; N, 10.62; S, 4.86; Found: C, 54.95; H, 5.55; N, 10.61 ; S, 4.87.
The process is also illustrated in Fig. 12.
Step (13): Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-rnethoxy-4- vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrate
(3i) ,a 3i
A pre-cooled (0-5 °C) aq. NaOH solution [prepared from solid NaOH (4 kg, 100 mol) in water (86 L)] was added to a suspension of methyl 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethyl-carbamoyl)pyridine-2-carboxylate methanesulfonate (7a) (28.7 kg, 46 mol) in acetonitrile (86 L) cooled to 0 to 5 °C over a period of 25 mins. The reaction mixture was stirred at 0 to 5 °C for 2.5 h (TLC analysis showed the reaction was complete). The reaction mixture was filtered through a sparkler filter, washed with a mixture of 1 :1 CH3CN / H20 ( 57.4 L). Acetic acid (3.2 L, 55.9 mol) in water (56 L) was added to the filtrate at room temperature over a period of 25 mins and the resulting mixture was stirred at room temperature for 2.5 h. The solid product obtained was collected by filtration, washed with a 1 :4 mixture of CH3CN / H20 (57.5 L). The solid was dried at 45°C in a vacuum oven to afford 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6- (cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrate (3i) as an off-white solid (12,77 kg, 54.1%). Average yield for this step is 50% to 75%. Mp: >200°C; H NMR (300 MHz, DMSO-d6): δ 13.49 (s, 1 H), 8.94 (bs, 4H), 8.56 (t, 1 H), 7.82 – 7.71 (m, 2H), 7.67 -7.56 (m, 4H), 7.51 (d, J = 7.8, 1 H), 6.98 (dd, J = 11.3, 17.8, 1 H), 6.68 (s, 1 H), 5.92 (d, J = 16.6, 1 H), 5.36 (d, J = 12.4, 1 H), 3.80 (s, 3H), 3.16 (m, 2H), 1.05 (m, 1 H), 0.43 (m, 2H), 0.24 (m, 2H); MS (ES+) 514.1 (M+1), 536.1 (M+Na), (ES-) 512.1 ; Analysis calculated for C28H27N5O5.3H2O: C, 59.25; H, 5.86; N, 12.34; Found C, 59.50; H,
5.75; N, 12.05. If needed this material can be crystallized from a mixture of acetone and water.
The process is also illustrated in Fig. 13.
Step 14: Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b
A pre-cooled (5-8 °C) aqueous NaOH solution (prepared from solid NaOH (1.97 kg, 49.25 mol) in demineralized water (41 L) was added to a pre-cooled (0-5 °C) suspension of (3i) (13.8 kg, 26.9 mol) in acetonitrile (41 L). The reaction mixture was stirred at 0-5 °C for 30 min (until the reaction mixture becomes homogeneous). The reaction mixture was filtered through a sparkler filter washed with 50% acetonitrile in demineralized water (4.4 L). The filtrate was charged into a reactor and cooled to 0-5 °C. Aqueous HCI [prepared from cone. HCI (9.3 L) in demineralized water (36 L)] was added slowly with stirring to keep the reaction temperature at or below 15 °C, the resulting mixture was stirred at 10-15 °C for 13 h. The reaction mixture was cooled to 0-5 °C and stirred for 1 h. The solid obtained was collected by filtration and washed with demineralized water (36 L). The solid product was suspended in water (69 L) stirred for 30 mins and collected by filtration washed twice with water (20 L each). The solid product was dried in a vacuum oven at 45°C to afford 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-
(cyclopropylmethyl carbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) (1 1.21 Kg, 75.77%). Mp: >200°C; 1H NMR (300 MHz, DMSO-ci6): δ 12.98 (br s, 1 H), 10.86 (s, 1 H), 9.24 (s, 3H), 9.04 (s, 2H), 8.22 (d, J = 7.8 Hz, 1 H), 7.96 (d, J = 5.7 Hz, 2H), 7.78 (s, 4H), 7.09-6.99 (m, 2H), 6.07 (d, J = 17.7 Hz, 1 H), 5.45(d, J = 11.4 Hz, 1 H), 3.88 (s, 3H), 3.26-3.24 (m, 2H), 1.09 (m, 1 H), 0.47 (m, 2H), 0.28 (m, 2H).
Average isolated yield for this step varies from 63% to 80%.
The process is also illustrated in Fig. 14.
Example-2: Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid sulfate salt (8b)
6d 8a
To a solution of 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d) (2.35 g, 5.7 mmol) and 4-aminobenzamidine dihydrochloride (1.79 g, 8.6 mmol) in DMF (20 mL) and pyridine (30 ml_) at 0 °C was added EDCI (1.65 g, 8.6 mmol) and allowed to warm to room temperature overnight. The
reaction mixture was quenched with 6N HCI (60 mL) and extracted with chloroform (3 x 60 mL). The organic layer was dried over MgS04, filtered and concentrated in vacuum. The residue obtained was purified by flash column chromatography (silica gel, 110 g, eluting with 0 to 100% chloroform in CMA 80 and 0-100% chloroform in CMA 50) to furnish methyl 3-(2-((4-carbamimidoylphenyl)carbamoyl)-5-methoxy-4-vinylphenyl)-6-((cyclopropylmethyl)-carbamoyl)picolinate hydrochloride (8a) (2.2 g, 65%) as a white solid; MP 266 °C; 1HNMR (300 MHz, DMSO-d6) δ 10.78 (s, 1 H), 9.26 (s, 2H), 9.03 (s, 2H), 8.67 (t, J = 6.1 , 1 H), 8.22 (d, J = 8.0, 1 H), 8.06 (d, J = 8.0, 1 H), 7.96 (s, 1 H), 7.89 -7.74 (m, 4H), 7.13 – 6.96 (m, 2H), 6.07 (d, J = 17.7, 1 H), 5.45 (d, J = 12.4, 1 H), 3.91 (s, 3H), 3.61 (s, 3H), 3.20 (s, 2H), 1.09 (dd, J = 4.7, 8.2, 1 H), 0.43 (dt, J = 4.9, 5.4, 2H), 0.34 – 0.21 (m, 2H); MS (ES+) 528.1 (M+1); Analysis calculated for C29H29N505 (H20)1 5 (HCI): C, 58.93; H, 5.63; N, 1 1.85; Found: C, 58.75; H, 5.65; N, 1 1.92.
Step-2: preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid sulfate salt (8b)
8a 8b j0 a solution of methyl 3-(2-((4-carbamimidoylphenyl)carbamoyl)-5-methoxy-4-vinylphenyl)-6-((cyclopropylmethyl)carbamoyl)picolinate hydrochloride (8a) (1.128 g, 2 mmol) in acetonitrile (5 ml), was added 1 N aqueous sodium hydroxide (5.00 ml, 5.00 mmol) and stirred at room temperature for 2 h, TLC [CMA80/CMA50 (7/3)] shows reaction was complete. The reaction mixture was neutralized with a solution of sulfuric acid (0.483 ml, 9.00 mmol) in water (5 mL) and stirred for 10 min at room temperature. To this cold water (5 ml) was added and stirred at room temperature until product crystallized out. Cold water (5 mL) was added to the slurry and stir for additional 20 min, additional cold water (5 mL) was added prior to filtration of solid. The solid obtained was collected by filtration washed with water (5 mL and 2.5 mL), dried under vacuum overnight to afford 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-
(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid sulfate salt (8b) (1.103 g, 90 % yield) as a white solid; MP 221.7 °C; H NMR (300 MHz, DMSO-d6) δ 12.30 – 10.91 (bs, 1 H, D20 exchangeable), 10.69 (bs, 1 H, D20 exchangeable), 9.24 (t, J = 6.0 Hz, 1 H), 9.16 (s, 2H, D2O exchangeable), 8.78 (s, 2H, D2O exchangeable), 8.24 (d, J = 8.0 Hz, 1 H), 8.04 – 7.91 (m, 2H), 7.84 – 7.67 (m, 4H), 7.13 – 6.94 (m, 2H), 6.03 (dd, J = 17.8, 1 .4 Hz, 1 H), 5.51 – 5.37 (m, 1 H), 3.88 (s, 3H), 3.24 (t, J = 6.4 Hz, 2H), 1.16 – 1.01 (m, 1 H), 0.52 – 0.41 (m, 2H), 0.32 – 0.22 (m, 2H); MS (ES+) 514.0 (M+1); Analysis calculated for: C28H27N605 1.0H2SO4 1.5H20: C, 52.66; H, 5.05; N, 10.97; S, 5.02; Found: C, 52.81 ; H, 4.95; N, 10.94; S, 4.64.
Example-3: Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid methane s
To a solution of methyl 3-(2-((4-carbamimidoylphenyl)carbamoyl)-5-methoxy-4-vinylphenyl)-6-((cyclopropylmethyl)carbamoyl)picolinate hydrochloride (8a) (1.128 g, 2 mmol) in acetonitrile (5 ml) was added 1 N aqueous sodium hydroxide (5.00 ml, 5.00 mmol) and stirred at room temperature for 2 h, TLC [CMA80/CMA50 (7/3)] shows reaction was complete. The reaction mixture was neutralized with methanesulfonic acid (0.584 ml, 9.00 mmol) and stirred for 1 h at room temperature. Cold water (5.00 ml) was added to the reaction mixture and stirred at room temperature until product crystallized out. To the slurry was added water (5 ml) of water stirred for additional 20 min, followed by the addition of water (5 ml) prior to filtration. The solid obtained was collected by filtration washed with water (5 ml and 2.5 ml), dried under vacuum to afford 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6- (cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid methane sulfonate salt (8c)
(1 .138 g, 1.867 mmol, 93 % yield) as a white solid; MP 221.2 °C; 1 H NMR (300 MHz,
DMSO-d6) δ 12.89 (s, 1 H, D2O exchangeable), 10.69 (s, 1 H, D2O exchangeable), 9.24
(t, J = 6.0 Hz, 1 H), 9.16 (s, 2H,), 8.85 (s, 2H), 8.24 (d, J = 8.0 Hz, 1 H), 8.06 – 7.91 (m, 2H), 7.86 – 7.70 (m, 4H), 7.15 – 6.96 (m, 2H), 6.03 (dd, J = 17.8, 1.4 Hz, 1 H), 5.52 – 5.35 (m, 1 H), 3.88 (s, 3H), 3.25 (t, J = 6.3 Hz, 2H), 2.34 (s, 3H), 1.17 – 1.01 (m, 1 H), 0.53 -0.43 (m, 2H), 0.32 – 0.23 (m, 2H); MS (ES+) 514.0 (M+1); Analysis calculated for:
CzeH^NsOsCHsSOsH 1.5H20: C, 54.71 ; H, 5.38; N, 11.00; S, 5.04; Found: C, 54.80; H, 5.14; N, 10.94; S, 4.90.
Example-4: Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) in Form C (Compound XX)
The jacket of a 10 L glass reactor was set to -5 °C. To the reactor was charged 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)-pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d) prepared in Step (11) of Example 1 (500 g, 1.22 mol), 4-amino-benzamidine-2HCI (280 g, 1.34 mol), and 2-propanol (4.05 kg). The mixture was cooled
46582
to 0.3 °C, and pyridine (210 g, 2.62 mol) followed by EDCI HCI (310 g, 1.61 mol) was added. The mixture was stirred at -1.1 – -0.3 °C for 22 hrs followed by addition of the second portion of EDCI HCI (58 g, 0.30 mol). The temperature of jacket was set to 14.0 °C, and the mixture was stirred for 89 hrs. The precipitate was filtered, and washed with 1.32 kg of 2-propanol.
The wet product (8a) was recharged to the reactor followed by addition of acetonitrile (1 .6 kg) and 0.57 kg water. The mixture was heated to 46 °C. 21 g of Smopex-234 and 10 g Acticarbone 2SW were added and the mixture was stirred at this temperature for 1 hr. The solution was filtered, and filtrate was returned back to the reactor. The jacket of the reactor was set to -5 °C, and the mixture was cooled to -0.2 °C. NaOH solution (256 g 46% NaOH, 2.95 mol, in 960 g water) was added in 25 min keeping the temperature ❤ °C. The mixture was stirred at 0.2-2.0 °C for 1 hr 40 min and then quenched with cone, acetic acid (40 g, 0.66 mol). Diluted acetic acid (80 g, 1.33 mol AcOH in 1000 g water) was added during 1 hr 20 min (temperature 1.7-3.0 °C), followed by 1250 g water (30 min). The suspension was stirred at 0-3.0 °for 1 hr, and filtered at 0-5 °C (ice mantle around the filter). The reactor and product (8d) was rinsed with 3.5 kg water.
The wet product (8d) was recharged to the reactor followed by 0.65 kg water and 1.69 kg acetonitrile. The mixture was heated to 57-60 °C, and stirred at this temperature for 14.5 hrs. The mixture was cooled to -2.2 °C (Tjacke,= -5 °C), and a solution of NaOH (163 g 46%, 1.87 mol, in 580 g water) was added during 15 min. The temperature rose to -0.4 °C. Hydrochloric acid (407 g 37% HCI, 4 mol) was added in 10 min, the temperature rose to 7.5 °C. The suspension was agitated at -3 – 0 °C for 19 hrs. The product was filtered and the filter cake was rinsed with 2.87 kg water, compressed and pulled dry. The wet product (1.30 kg) was dried at 40-43 °C and 50 mbar for 1 17 hrs to furnish 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) (484 g) as Form C (Compound XX).
/////avoralstat, BCX4161, Fast Track, Treat hereditary angioedema (HAE), Orphan Drug, PRECLINICAL
COc1cc(c(cc1C=C)C(=O)Nc2ccc(cc2)C(=N)N)c3cc(ncc3C(=O)O)C(=O)NCC4CC4

Pacritinib
パクリチニブ;
| Formula |
C28H32N4O3
|
|---|---|
| CAS |
937272-79-2
|
| Mol weight |
472.5787
|
UPDATE FDA APPROVED 2/28/2022, Vonjo
To treat intermediate or high-risk primary or secondary myelofibrosis in adults with low platelets
A Jak2 inhibitor potentially for the treatment of acute myeloid Leukemia and myelofibrosis.
ONX-0803; SB-1518
CAS No. 937272-79-2
472.57868 g/mol, C28H32N4O3
S*Bio Pte Ltd. and concert innovator
11-(2-pyrrolidin-1-ylethoxy)-14,19-dioxa-5,7,26-triazatetracyclo(19.3.1.1(2,6).1(8,12))heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene
| Pacritinib (SB1518) is a potent and selective inhibitor of Janus Kinase 2 (JAK2) and Fms-Like Tyrosine Kinase-3 (FLT3) with IC50s of 23 and 22 nM, respectively. | ||||||
UPDATED
Pacritinib, sold under the brand name Vonjo, is an anti-cancer medication used to treat myelofibrosis.[1][2] It is a macrocyclic Janus kinase inhibitor. It mainly inhibits Janus kinase 2 (JAK2) and Fms-like tyrosine kinase 3 (FLT3).
Common side effects include diarrhea, low platelet counts, nausea, anemia, and swelling in legs.[2]
Pacritinib in indicated to treat adults who have a rare form of a bone marrow disorder known as intermediate or high-risk primary or secondary myelofibrosis and who have platelet (blood clotting cells) levels below 50,000/µL.[1][2]
The effectiveness and safety of pacritinib were demonstrated in a study that included 63 participants with intermediate or high-risk primary or secondary myelofibrosis and low platelets who received pacritinib 200 mg twice daily or standard treatment.[2] Effectiveness was determined based upon the proportion of participants who had a 35% or greater spleen volume reduction from baseline to week 24.[2] Nine participants (29%) in the pacritinib treatment group had a 35% or greater spleen volume reduction, compared to one participant (3%) in the standard treatment group.[2]
The U.S. Food and Drug Administration (FDA) granted the application for pacritinib priority review, fast track, and orphan drug designations.[2]
Pacritinib is the International nonproprietary name (INN).[3][4]
OLD—
Pacritinib (INN[1]) is a macrocyclic Janus kinase inhibitor that is being developed for the treatment of myelofibrosis. It mainly inhibits Janus kinase 2 (JAK2). The drug is in Phase III clinical trials as of 2013.[2] The drug was discovered in Singapore at the labs of S*BIO Pte Ltd. It is a potent JAK2 inhibitor with activity of IC50 = 23 nM for the JAK2WT variant and 19 nM for JAK2V617F with very good selectivity against JAK1 and JAK3 (IC50 = 1280 and 520 nM, respectively).[3][4] The drug is acquired by Cell Therapeutics, Inc. (CTI) and Baxter international and could effectively address an unmet medical need for patients living with myelofibrosis who face treatment-emergent thrombocytopenia on marketed JAK inhibitors.[5]
Pacritinib is an orally bioavailable inhibitor of Janus kinase 2 (JAK2) and the JAK2 mutant JAK2V617F with potential antineoplastic activity. Oral JAK2 inhibitor SB1518 competes with JAK2 for ATP binding, which may result in inhibition of JAK2 activation, inhibition of the JAK-STAT signaling pathway, and so caspase-dependent apoptosis. JAK2 is the most common mutated gene in bcr-abl-negative myeloproliferative disorders; the JAK2V617F gain-of-function mutation involves a valine-to-phenylalanine modification at position 617. The JAK-STAT signaling pathway is a major mediator of cytokine activity.
Synthesis Reference
A245943 — William AD, Lee AC, Blanchard S, Poulsen A, Teo EL, Nagaraj H, Tan E, Chen D, Williams M, Sun ET, Goh KC, Ong WC, Goh SK, Hart S, Jayaraman R, Pasha MK, Ethirajulu K, Wood JM, Dymock BW: Discovery of the macrocycle 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6). 1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a potent Janus kinase 2/fms-like tyrosine kinase-3 (JAK2/FLT3) inhibitor for the treatment of myelofibrosis and lymphoma. J Med Chem. 2011 Jul 14;54(13):4638-58. doi: 10.1021/jm200326p. Epub 2011 Jun 15.
Pacritinib is an orally bioavailable inhibitor of Janus kinase 2 (JAK2) and the JAK2 mutant JAK2V617F with potential antineoplastic activity. Oral JAK2 inhibitor SB1518 competes with JAK2 for ATP binding, which may result in inhibition of JAK2 activation, inhibition of the JAK-STAT signaling pathway, and so caspase-dependent apoptosis. JAK2 is the most common mutated gene in bcr-abl-negative myeloproliferative disorders; the JAK2V617F gain-of-function mutation involves a valine-to-phenylalanine modification at position 617. The JAK-STAT signaling pathway is a major mediator of cytokine activity.

The compound 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (Compound I) was first described in PCT/SG2006/000352 and shows significant promise as a pharmaceutically active agent for the treatment of a number of medical conditions and clinical development of this compound is underway based on the activity profiles demonstrated by the compound.


PATENT
http://www.google.com/patents/US8415338
Representative Procedure for the Synthesis of Compounds Type (XVIIId) [3-(2-Chloro-pyrimidin-4-yl)-phenyl]-methanol (XIIIa2)

Compound (XIIIa2) was obtained using the same procedure described for compound (XIIIa1); LC-MS (ESI positive mode) m/z 221 ([M+H]+).
4-(3-Allyloxymethyl-phenyl)-2-chloro-pyrimidine (XVa2)

Compound (XVa2) was obtained using the same procedure described for compound (XVa1); LC-MS (ESI positive mode) m/z 271 ([M+H]+).
[4-(3-Allyloxymethyl-phenyl)-pyrimidin-2-yl]-[3-allyloxymethyl-4-(2-pyrrolidin-1-yl-ethoxy)-phenyl]-amine (XVIId1)

Compound (XVIId1) was obtained using the same procedure described for compound (XVIIb1); LC-MS (ESI positive mode) m/z 501.
Macrocycle Example 3 Compound 13

Compound (13) was obtained using the same procedure described for compound (1) HPLC purity at 254 nm: 99%; LC-MS (ESI positive mode) m/z 473 ([M+H]+); 1H NMR (MeOD-d4) δ 8.79 (d, 1H), 8.46 (d, 1H), 8.34-8.31 (m, 1H), 7.98-7.96 (m, 1H), 7.62-7.49 (m, 2H), 7.35 (d, 1H), 7.15-7.10 (m, 1H), 7.07-7.02 (m, 1H), 5.98-5.75 (m, 2H, 2×=CH), 4.67 (s, 2H), 4.67 (s, 2H), 4.39-4.36 (m, 2H), 4.17 (d, 2H), 4.08 (d, 2H), 3.88-3.82 (m, 2H), 3.70 (t, 2H), 2.23-2.21 (m, 2H), 2.10-2.07 (m, 2H).
J MC 2011, 54 4638
http://pubs.acs.org/doi/abs/10.1021/jm200326p

Discovery of the activating mutation V617F in Janus Kinase 2 (JAK2V617F), a tyrosine kinase critically involved in receptor signaling, recently ignited interest in JAK2 inhibitor therapy as a treatment for myelofibrosis (MF). Herein, we describe the design and synthesis of a series of small molecule 4-aryl-2-aminopyrimidine macrocycles and their biological evaluation against the JAK family of kinase enzymes and FLT3. The most promising leads were assessed for their in vitro ADME properties culminating in the discovery of 21c, a potent JAK2 (IC50 = 23 and 19 nM for JAK2WT and JAK2V617F, respectively) and FLT3 (IC50 = 22 nM) inhibitor with selectivity against JAK1 and JAK3 (IC50 = 1280 and 520 nM, respectively). Further profiling of 21c in preclinical species and mouse xenograft and allograft models is described. Compound 21c(SB1518) was selected as a development candidate and progressed into clinical trials where it is currently in phase 2 for MF and lymphoma.
 Discovery of the Macrocycle 11-(2-Pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a Potent Janus Kinase 2/Fms-Like Tyrosine Kinase-3 (JAK2/FLT3) Inhibitor for the Treatment of Myelofibrosis and Lymphoma
Discovery of the Macrocycle 11-(2-Pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a Potent Janus Kinase 2/Fms-Like Tyrosine Kinase-3 (JAK2/FLT3) Inhibitor for the Treatment of Myelofibrosis and Lymphoma(21c)
2“JAK-Inhibitoren: Neue Wirkstoffe für viele Indikationen”. Pharmazeutische Zeitung (in German) (21). 2013.
3William, A. D.; Lee, A. C. -H.; Blanchard, S. P.; Poulsen, A.; Teo, E. L.; Nagaraj, H.; Tan, E.; Chen, D.; Williams, M.; Sun, E. T.; Goh, K. C.; Ong, W. C.; Goh, S. K.; Hart, S.; Jayaraman, R.; Pasha, M. K.; Ethirajulu, K.; Wood, J. M.; Dymock, B. W. (2011). “Discovery of the Macrocycle 11-(2-Pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a Potent Janus Kinase 2/Fms-Like Tyrosine Kinase-3 (JAK2/FLT3) Inhibitor for the Treatment of Myelofibrosis and Lymphoma”. Journal of Medicinal Chemistry 54 (13): 4638–58. doi:10.1021/jm200326p. PMID 21604762.
4Poulsen, A.; William, A.; Blanchard, S. P.; Lee, A.; Nagaraj, H.; Wang, H.; Teo, E.; Tan, E.; Goh, K. C.; Dymock, B. (2012). “Structure-based design of oxygen-linked macrocyclic kinase inhibitors: Discovery of SB1518 and SB1578, potent inhibitors of Janus kinase 2 (JAK2) and Fms-like tyrosine kinase-3 (FLT3)”. Journal of Computer-Aided Molecular Design 26 (4): 437–50. doi:10.1007/s10822-012-9572-z. PMID 22527961.
5http://www.pmlive.com/pharma_news/baxter_licenses_cancer_drug_from_cti_in_$172m_deal_519143
| US8153632 * | Nov 15, 2006 | Apr 10, 2012 | S*Bio Pte Ltd. | Oxygen linked pyrimidine derivatives |
| US8415338 * | Apr 4, 2012 | Apr 9, 2013 | Cell Therapeutics, Inc. | Oxygen linked pyrimidine derivatives |
| US20110294831 * | Dec 9, 2009 | Dec 1, 2011 | S*Bio Pte Ltd. | 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene citrate salt |
| Patent | Submitted | Granted |
|---|---|---|
| OXYGEN LINKED PYRIMIDINE DERIVATIVES [US8153632] | 2009-03-19 | 2012-04-10 |
| ANTIVIRAL JAK INHIBITORS USEFUL IN TREATING OR PREVENTING RETROVIRAL AND OTHER VIRAL INFECTIONS [US2014328793] | 2012-11-30 | 2014-11-06 |
| OXYGEN LINKED PYRIMIDINE DERIVATIVES [US2013172338] | 2013-02-20 | 2013-07-04 |
| METHOD OF SELECTING THERAPEUTIC INDICATIONS [US2014170157] | 2012-06-15 | 2014-06-19 |
| CYCLODEXTRIN-BASED POLYMERS FOR THERAPEUTIC DELIVERY [US2014357557] | 2014-05-30 | 2014-12-04 |
| 11-(2-PYRROLIDIN-1-YL-ETHOXY)-14,19-DIOXA-5,7,26-TRIAZA-TETRACYCLO[19.3.1.1(2,6).1(8,12)]HEPTACOSA-1(25),2(26),3,5,8,10,12(27),16,21,23-DECAENE MALEATE SALT [US2011263616] | 2011-10-27 | |
| 11-(2-PYRROLIDIN-1-YL-ETHOXY)-14,19-DIOXA-5,7,26-TRIAZA-TETRACYCLO[19.3.1.1(2,6).1(8,12)]HEPTACOSA-1(25),2(26),3,5,8,10,12(27),16,21,23-DECAENE CITRATE SALT [US2011294831] | 2011-12-01 | |
| BIOMARKERS AND COMBINATION THERAPIES USING ONCOLYTIC VIRUS AND IMMUNOMODULATION [US2014377221] | 2013-01-25 | 2014-12-25 |
| Oxygen linked pyrimidine derivatives [US8415338] | 2012-04-04 | 2013-04-09 |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(16E)-11-[2-(1-Pyrrolidinyl)ethoxy]-14,19-dioxa-5,7,26-triazatetracyclo[19.3.1.12,6.18,12]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene
|
|
| Clinical data | |
| Legal status |
|
| Routes of administration |
Oral |
| Identifiers | |
| ATC code | None |
| PubChem | CID: 46216796 |
| ChemSpider | 28518965 |
| ChEMBL | CHEMBL2035187 |
| Synonyms | SB1518 |
| Chemical data | |
| Formula | C28H32N4O3 |
| Molecular mass | 472.58 g/mol |



SEE……..http://apisynthesisint.blogspot.in/2016/01/pacritinib.html


join me on Linkedin
join me on Researchgate

join me on Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
|
|
| Clinical data | |
|---|---|
| Trade names | Vonjo |
| Other names | SB1518 |
| License data |
|
| Routes of administration |
By mouth |
| ATC code | |
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| PDB ligand | |
| Chemical and physical data | |
| Formula | C28H32N4O3 |
| Molar mass | 472.589 g·mol−1 |
| 3D model (JSmol) | |
///////Vonjo, FDA APPTOVESD 2022, APPROVALS 2022, PACRITINIB, パクリチニブ, priority review, fast track, orphan drug, UNII-G22N65IL3O, пакритиниб , باكريتينيب , 帕瑞替尼 , SB 1518
c1cc2cc(c1)-c3ccnc(n3)Nc4ccc(c(c4)COC/C=C/COC2)OCCN5CCCC5
C1CCN(C1)CCOC2=C3COCC=CCOCC4=CC=CC(=C4)C5=NC(=NC=C5)NC(=C3)C=C2
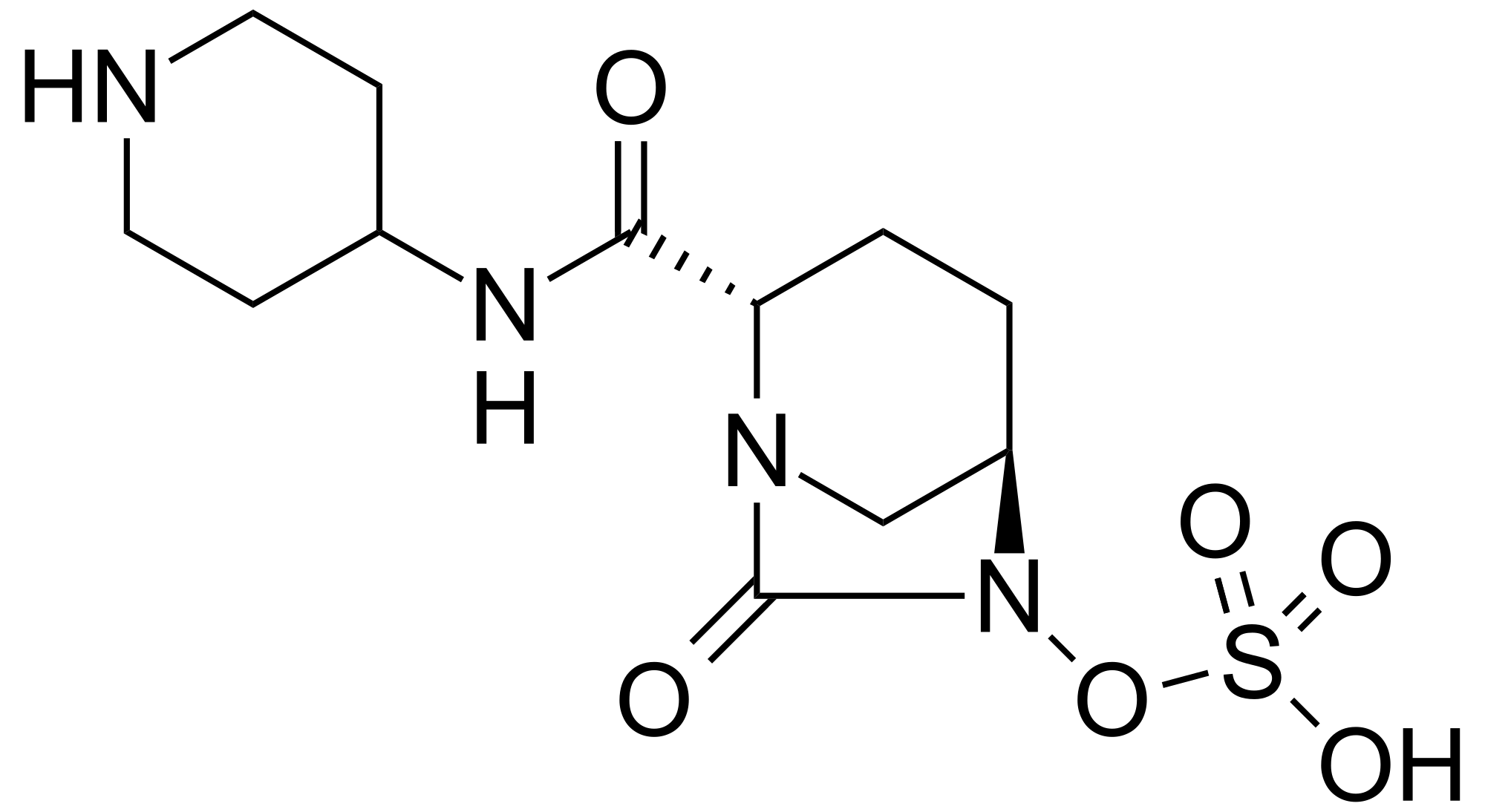
MK 7655, RELEBACTAM
(1R,2S,5R)-7-Oxo-N-(4-piperidinyl)-6-(sulfooxy)-1,6-diazabicyclo[3.2.1]octane-2-carboxamide
| MF C12H22N4O7S | |
| MW | 366.39068 g/mol |
|---|
CAS 1174020-13-3
β-Lactamase inhibitor
MK-7655 is a beta-lactamase inhibitor in phase III clinical studies at Merck & Co for the treatment of serious bacterial infections…….See clinicaltrials.gov, trial identifier numbers NCT01505634 and NCT01506271.
In 2014, Qualified Infectious Disease Product (QIDP) and Fast Track designations were assigned by the FDA for the treatment of complicated urinary tract infections, complicated intra-abdominal infections and hospital-acquired bacterial pneumonia/ventilator-associated bacterial pneumonia.

A concise synthesis of a beta-lactamase inhibitor
Org Lett 2011, 13(20): 5480
http://pubs.acs.org/doi/abs/10.1021/ol202195n
http://pubs.acs.org/doi/suppl/10.1021/ol202195n/suppl_file/ol202195n_si_001.pdf

MK-7655 (1) is a β-lactamase inhibitor in clinical trials as a combination therapy for the treatment of bacterial infection resistant to β-lactam antibiotics. Its unusual structural challenges have inspired a rapid synthesis featuring an iridium-catalyzed N–H insertion and a series of late stage transformations designed around the reactivity of the labile bicyclo[3.2.1]urea at the core of the target.
H NMR (400 MHz, DMSO-d6): δ 8.30 (br s, 2H), 8.20 (d, J = 7.8 Hz, 1H), 4.01 (s, 1H), 3.97-3.85 (m, 1H), 3.75 (d, J = 6.5 Hz, 1H), 3.28 (dd, J = 12.9, 2.5 Hz, 2H), 3.05-2.93 (m, 4H), 2.08-1.97 (m, 1H), 1.95-1.79 (m, 3H), 1.73-1.59 (m, 4H);
13C NMR (DMSO-d6, 100 MHz) δ 169.7, 166.9, 59.8, 58.3, 46.9, 44.3, 42.9, 28.5, 28.3, 20.8, 18.9;
HRMS calculated for C12H20N4O6S (M+H): 349.1182, found: 349.1183.
[α]D 25 = -23.3 (c = 1.0, CHCl3)


WO 2009091856
http://www.google.com/patents/WO2009091856A2?cl=en
EXAMPLE IA
(2S ,5 R)-7-Oxo-N-piperidin-4-yl-6-(sulfooxy)- 1 ,6-diazabicyclo [3.2.1 ]octane-2-carboxamide

Step 1 : tert-butyl 4-({[(2S,5R)-6-(benzyloxy)-7-oxo-l,6-diazabicyclo[3.2.1]oct-2- yljcarbonyl } amino)piperidine- 1 -carboxylate : To a solution of (2S,5R)-6-(phenylmethoxy)-7-oxo-l,6-diazabicyclot3.2.1]octane-
2-carboxylic acid (1.484 g, 5.37 mmol) in dry dichloromethane (60 ml) was added triethylamine (1.88 ml, 13.49 mmol), 2-chloro-l-methylpyridinium iodide (1.60 g, 6.26 mmol), and 4-amino-l- BOC-piperidine (1.30 g, 6.49 mmol) sequentially at room temperature under nitrogen. The reaction was then heated to 500C for 1 hour. The reaction mixture was concentrated under vacuum and purified by silica gel chromatography on an Isco Combiflash (40 g silica gel, 40 mL/min, 254 nM, 15% to 100% EtOAc/hexane over 14 column volumes then 100% EtOAc for 4 column volumes; title compuond eluted at 65% ethyl acetate/hexane) to afford the title compound as a pale orange solid.
Step 2: tert-butyl 4-({[(2S,5R)-6-hydroxy-7-oxo-l ,6-diazabicyclo[3.2.1]oct-2- yl] carbonyl } amino)piρeridine- 1 -carboxylate:
Palladium on carbon (394 mg; 10% Pd/C) was added to a solution of the product of step 1 (1.81 g, 3.95 mmol) in methanol (50.6 mL) and the resulting mixture was stirred under hydrogen (balloon) overnight. LC/MS analysis indicated the reaction was not complete. Acetic acid (6 drops) and additional catalyst (159 mg of 10% Pd/C) were added to the reaction and the resulting mixture was stirred under hydrogen (balloon) for an additional 90 minutes. Additional catalyst (0.2085 g of 10% Pd/C) was added to the reaction and stirring under hydrogen was continued for an additional 2.5 hours at which time the reaction was judged complete by LC-MS analysis. The reaction was filtered through a celite pad and the collected solid was washed well wtih MeOH. The filtrate was concentrated under vacuum to afford the title compound as a colorless oil which was used without purification in the next step.
Step 3 : tert-butyl-4-({ [(2S,5R)-7-oxo-6-(sulfooxy)- 1 ,6-diazabicyclo[3.2.1 ]oct-2- yl] carbonyl } amino)ρiperidine- 1 -carboxylate:
To a solution of the product of step 2 (1.455 g, 3.95 mmol; theoretical yield of step 2) in dry pyridine (30 mL) was added sulfur trioxide pyridine complex (3.2 g, 20.11 mmol) at room temperature under nitrogen. The resulting thick mixture was stirred over the weekend.
The reaction was filtered and the white insoluble solids were washed well with dichloromethane. The filtrate was concentrated in vacuo. The residue was further azeotroped with toluene to remove excess pyridine to afford the title compound which was used without purification in the next step.
Step 4: (2S,5R)-7-oxo-N-piperidin-4-yl-6-(sulfooxy)-l,6-diazabicyclo[3.2.1]octane-2- carboxamide:
To a mixture of the product of step 3 (1.772 g, 3.95 mmol; theoretical yield of step 3) in dry dichloromethane (30 ml) at 00C under nitrogen was slowly added trifluoroacetic acid (6.1 ml, 79 mmol). Immediately the reaction became a solution. After 1 hour, additional trifluoroacetic acid (8 ml) was added to the reaction. The reaction was stirred at 00C until judged complete by LC-MS analysis then concentrated in vacuo. The residue was triturated with ether (3X) to remove excess TFA and organic impurities. The resulting white insoluble solid was collected via centrifugation, dried in vacuo, then purified by preparative HPLC (250X21.2 mm Phenomenex Synergi Polar-RP 80A column; 10 micron; 35 mL/min.; 210 nM; 0% to 30% methanol/water over 15 minutes; title compound eluted at 10% methanol/water). Fractions containing the title compound were combined and Iyophilized overnight to afford the title compound as a white solid. LC-MS (negative ionization mode) m/e 347 (M-H).
Discovery of MK-7655, a beta-lactamase inhibitor for combination with Primaxin
Bioorg Med Chem Lett 2014, 24(3): 780
http://www.sciencedirect.com/science/article/pii/S0960894X13014856

PATENT
WO 2014200786
http://www.google.dj/patents/WO2014200786A1?cl=en


![]()




Exemplary Scheme

– 50% isolated yield overall from 1 to 5

O via crystallization
XAMPLE 1
(2S,5R)-7-oxo-N-piperidin-4-yl-6-(sulfooxy)- 1 ,6-diazabicyclo[3.2.1 ]octane-2-carboxamide 
Preparation of (15′,45)-5-((2-nitrophenyl)sulfonyl)-2-oxa-5-azabicyclo[2.2.2]octan-3 one (2)
To a reactor (R-1) equipped with an additional funnel, nitrogen inlet and agitator was charged (2S,5S)-5-hydroxypiperidine-2-carboxylic acid (77.3 wt%) (50.0 g, 344 mmol), and water (150 mL). Agitation was begun, the pH adjusted to 10-11 by addition of 10 N NaOH (~ 46.5 mL) and the reactor charged with acetone (50.0 mL).
In a separate reactor (R-2) equipped with an agitator and nitrogen inlet was charged 2-nitrobenzene-l-sulfonyl chloride (97%) (106.0 g, 478 mmol) and acetone (80 mL). The contents of R-2 were transferred to R-1 at 23-30 °C while the pH of the solution was maintained at 10-11 by simultaneously addition of 10 N NaOH. After 15 to 30 min, the pH was adjusted to about 6 by addition of 12 N HC1. The solution was charged with EtOAc (500 mL) and the pH adjusted to 3.0 by addition of 12 N HC1. The layers were separated and the aqueous back-extracted with EtOAc (150 mL x 2).
To a separate reactor (R-3) was charged product la in the combined organic layers, 2-nitrobenzene-l-sulfonyl chloride (73.0 g, 329 mmol), and triethylamine (130 mL). The batch in R-3 was agitated at 20-28°C for 30 min. The solution was charged with water (100 mL), the layers separated, and the aqueous back extracted with EtOAc (150 mL x 2). The combined EtOAc layer was washed with 10% NaHC03 (100 mL) and brine (100 mL). The organic phase was concentrated to 150 mL upon which a crystalline slurry was formed. The concentrated solution was agitated at 13-18°C for 2-3 hours followed by filtration of crystalline solids. The resulting wet cake was washed with EtOAc (60 mL) and then dried under vacuum oven at 25-30°C to afford 2 (65.6 g, 79% yield), m.p. 126.0-126.7 °C. 1H NMR (CDC13, 400 MHz) δ: 8.02 (m, 1 H), 7.80-7.71 (m, 2 H), 7.66 (m, 1 H), 4.88 (m, 1 H), 4.55 (dd, J= 3.8, 2.7 Hz, 1 H), 3.78 (dt, J= 11.2, 3.0 Hz, 1 H), 3.66 (dd, J = 11.2, 1.1 Hz, 1 H), 2.44 (m, 1 H), 2.11 (m, 2 H), 1.91 (m, 1 H); 13C NMR (CDC13, 100 MHz) δ: 168.4, 148.3, 134.4, 132.1, 131.0, 130.7, 124.2, 73.5, 51.4, 48.0, 25.1, 23.2
Preparation oftert-butyl 4-((25*,55)-l-((2-nitrophenyl)sulfonyl)-5-(((2- nitrophenyl)sulfony l)oxy)piperidine-2-carboxamido)piperidine- 1 -carboxylate (3)
To a reactor (R-l) was charged lactone 2 (65.5 g, 210 mmol), THF (131 mL) and tert-butyl 4-aminopiperidine-l -carboxylate (44.5 g, 222 mmol). The stirred solution was heated to reflux (typical temperature 72 °C) for ~18 hr. The reaction was cooled to 25-35 °C and then charged with THF (325 mL) and 4-dimethylaminopyridine (40.1 g, 328 mmol) followed by agitation for 30 minutes.
To a separate reactor (R-2) was charged 2-nitrobenzene-l-sulfonyl chloride (60.9 g,
275 mmol) and THF (200 mL). The contents of R-2 were added to R-l over the course of 45 to 75 minutes maintaining batch temperature of 20 to 30°C. The batch in R-l was agitated for 2 to 4 hours at a temperature of 20 to 30°C.
To a separate reactor (R-3) was charged water (600 mL) and methanol (600 mL). The contents of R-3 were charged to the main batch over the course of 45 to 75 minutes with agitation while maintaining temperature of 20 to 30°C. The batch was cooled to 5 to -5°C and then agitated at 5 to -5°C for at least 4 hours. The solids were filtered and then washed twice with methanol (130 mL x 2). The wet cake was dried in a vacuum oven at 40 to 50°C to afford 3 (144.0 g, 98% yield), m.p. 131.8-133.1 °C. 1H NMR (CDC13, 400 MHz) δ: 8.14 (m, 2 H), 7.83-7.74 (m, 6 H), 6.50 (d, J= 7.9 Hz, 1 H), 4.69 (m, 1 H), 4.43 (s, 1H), 4.11 (dd, , J= 13.7, 4.9 Hz, 1H), 3.95 (m, 2H), 3.83 (m, 1H), 3.47 (s, 1H), 3.10 (dd, J= 13.7, 11.0 Hz, 1H), 2.81 (m, 2H), 2.51 (m, 1H), 2.12 (m, 1H), 1.85-1.72 (m, 4H), 1.45 (s, 9H), 1.26 (m, 1H); 13C NMR (CDC13, 100 MHz) δ: 166.9, 154.6, 148.2, 147.6, 135.2, 134.8, 132.6, 132.5, 131.9, 131.6, 131.4, 129.7, 124.9, 124.7, 79.8, 76.5, 55.0, 47.1, 46.0, 31.8, 31.5, 28.4, 27.3, 24.4.
Preparation of N-4-nitrobenzene sulfonyl-O-benzylhydroxylamine
![]()
To a reactor (R-l) was charged O-benzylhydroxylamine hydrochloride (61.0g, 382 mmol) and pyridine (400 mL). The solution cooled to 5 to -5°C.
To a separate reactor (R-2) was charged 4-nitrobenzenesulfonyl chloride (89.0 g, 402 mmol) and pyridine (200 mL). The contents of R-2 were transferred to R-l at a rate to maintain temperature range of -5 to -5°C. The batch in R-l was agitated at 5 to -5 °C for 15 to 45 minutes then warmed to 20 to 30°C for 45 to 75 minutes. Water (250 mL) was then added at a rate to maintain 20 to 30°C and agitated 5 to 15 minutes. The solids were filtered and the wet cake washed with water (100 mL x 3). The wet cake was dried in vacuum oven at 50°C to afford N-4-nitrobenzenesulfonyl-O-benzylhydroxylamine (113.3 g, 96% yield), m.p. 128.4-130.0 °C. 1H NMR (CDCls, 400 MHz) δ: 8.36 (d, J = 8.9 Hz, 2 H), 8.11 (d, J = 8.9 Hz, 2 H), 7.36 (m, 5H), 7.11 (s, 1H), 5.02 (s, 2H); 13C NMR (CDC13, 100 MHz) δ: 151.0, 142.5, 134.9, 130.2, 129.7, 129.3, 128.9, 124.5, 80.2.
Step C. Preparation of tert-butyl 4-((2S,5R)-5-((benzyloxy)amino)piperidine -2-carboxamido)piperidine- 1 -carboxylate (4)
Boc
To a reactor (R-l) was charged tert-butyl 4-((2R,5R)-l-((2-nitrophenyl)sulfonyl)-5-(((2-nitrophenyl)sulfonyl)oxy)piperidine-2-carboxamido)piperidine-l -carboxylate (3) (110 g, 158 mmol), N-4-nitrobenzene sulfonyl-O-benzylhydroxylamine (58 g, 188 mmol), potassium carbonate (25.9 g, 187 mmol) and dimethylacetamide (440 mL). The stirred solution was heated to 60 to 70°C for 24 – 32 hours. The batch was cooled to 20 to 30°C and charged with toluene (660 mL). The batch was extracted with 1 N sodium hydroxide (3×220 mL) then washed with water (220 mL).
The toluene solution was azotropically distilled at ~50°C to about 1/3 volume. The solution was solvent-switched to MeOH at 45-55°C, adjusted to 237 mL.
The batch was cooled to 20-25°C, charged with thioglycolic acid (57.9 g, 629 mmol) at 10 °C, and then charged with K2CO3 anhydrous (172.0 g, 1225 mmol). The batch was agitated at 10-15°C for 0.5 h, warmed to 20-25°C, agitated at 20-25°C for 10-15 h, and heated at 48-53°C for 3-6 h.
The batch was charged with 10 wt% sodium chloride (1.10 L) and toluene (880 mL) at about 40°C. The layers were separated and the aq. layer back-extracted with toluene (3 x440 mL). The combined organic layer was washed with 10% NaHC03 (2 x220 mL). The batch was concentrated at 40-50°C to 165 mL, then cooled to 35-40°C. The batch was charged with seed (50 mg) and agitated for 1 h at 35-40°C. The batch was charged with heptanes (110 mL) at 35-40°C over 1 h, then slowly cooled to 15-20°C over 1 h. The batch was agitated for 3 h and the solids filtered. The wet cake was washed with toluene/heptanes (137.5 mL) then dried in vacuum oven at 30 °C for 3-8 h to affored 4. (47.3 g, 70% overall yield from 3), m.p. 117.5-118.0 °C. 1H NMR (CDC13, 500 MHz) δ: 7.37-7.29 (m, 5 H), 6.64 (d, J= 8.2 Hz, 1 H), 5.36 (brs, 1 H), 4.67 (s, 2 H), 4.00 (m, 2 H), 3.90 (m, 1 H), 3.28 (ddd, J= 11.8, 4.0, 1.7 Hz, 1 H), 3.12 (dd, J= 10.2, 3.2 Hz, 1 H), 2.95 (m, 1 H), 2.86 (m, 2 H), 2.46 (dd, J= 11.8, 9.5 Hz, 1 H), 2.10 (m, 1 H), 1.93-1.83 (m, 3 H), 1.58 (brs, 1 H), 1.45 (s, 9 H), 1.41 (m, 1 H), 1.35-1.23 (m, 3 H); 13C NMR (CDC13, 125 MHz) δ: 172.8, 154.7, 137.7, 128.4 (4 C), 127.9, 79.6, 76.9, 59.8, 57.0, 49.2, 46.1, 42.8 (br, 2 C), 32.0 (2 C), 28.4 (3 C), 28.3, 27.2.
Step D: Preparation of tert-butyl 4-((lR,2S,5R)-6-(benzyloxy)-7-oxo-l,6-diazabicyclo[3.2.1 ]octane-2-carboxamido)piperidine- 1 -carboxylate (5)
To a reactor (R-l) was charged tert-butyl 4-((2S,5R)-5-((benzyloxy)amino)piperidine-2-carboxamido)piperidine-l-carboxylate (4) (46.3 g, 107 mmol), dichloromethane (463 mL), and Hunig’s base (58.0 mL). The batch was cooled to -18°C and then charged with triphosgene in four portions (25.1 g total; 85 mmol) at <-8°C. The batch was agitated at -5 to 0°C for 0.5 h then charged with 11.4 wt% aqueous H3P04 at -5 to 0 °C (347 g, 3541 mmol). The batch was agitated at 20-25°C for 15-20 h then phase cut. The aqueous layer was back-extracted with dichloromethane (138 mL). The combined organic layer was washed with 10% NaHC03 (115 mL), then water (115 mL). The organic solution was concentrated at atmospheric pressure to ~80
mL, then charged with MTBE (347 mL) at 35-45 °C over 0.5 h, then concentrated at 35-45 °C to 231 mL two times to form a slurry.
The slurry was charged with heptanes (139 mL) at 35-45 °C over 2 h, then slowly cooled to 15-20°C over 1 h. The batch was agitated at 15-20°C for 6-8 h. Solids were filtered and the wet cake washed with MTBE/heptanes (1.4 : 1 , 185 mL) then dried under vacuum at 25-30°C for 5-10 hours to afford 5 (43.7 g, 92% yield), m.p. 161.3-161.8 °C. 1H NMR (CDC13, 500 MHz) δ: 7.45-7.32 (m, 5 H), 6.55 (d, J= 8.2 Hz, 1 H), 5.05 (d, J= 11.6 Hz, 1 H), 4.90 (d, J= 11.6 Hz, 1 H), 4.02 (m, 2 H), 3.90 (m, 2 H), 3.30 (m, 1 H), 2.99 (dt, J= 11.7, 1.1 Hz, 1 H), 2.86 (m, 2 H), 2.64 (d, J = 11.7 Hz, 1 H), 2.37 (dd, J= 14.6, 6.9 Hz, 1 H), 2.04-1.82 (m, 4 H), 1.58 (m, 1 H), 1.45 (s, 9 H), 1.30 (m, 2 H); 13C NMR (CDC13, 125 MHz) δ: 168.3, 167.5, 154.7, 135.6, 129.2 (2 C), 128.8, 128.6 (2 C), 79.7, 78.3, 60.4, 57.8, 47.5, 46.8, 42.5 (br, 2 C), 32.0, 31.7, 28.4 (3 C), 20.8, 17.2.
Step E: Preparation of tert-butyl 4-((2S,5R)-6-hydroxy-7-oxo-l,6-diazabicyclo[3.2.1“|octane- 2-carboxamido) iperidine- 1 -carboxylate
tert-butyl 4-((2S,5R)-6-hydroxy-7-oxo-l,6-diazabicyclo[3.2.1]octane-2-carboxamido)piperidine-l -carboxylate (9.2 g, 20.1 mmol) was charged to a glass bottle, and the solids were dissolved in THF (150 mL). The solution was then charged to a hydrogenation reactor along with Pd/Al203 (10 wt%, 1.5 g). The reaction was purged three times with hydrogen and then set to a hydrogen pressure of 50 psi. The reaction temperature was adjusted to 25°C and the reaction was allowed to agitate for 22 hours. After the reaction was complete as determined by HPLC analysis, the solution was filtered through SOLKA-FLOC® (Interational Fiber Corporation, North Tonawanda, NY) to remove the catalyst and the filter cake was washed with THF. The filtrate and washes were then solvent switched by vacuum distillation to iPrOAc to a final volume of 40 mL. The resulting iPrOAc slurry was aged at room temperature for 1 hour. The solids were then filtered and washed with iPrOAc (20 mL) and dried under vacuum and N2 at 40°C to afford the title product (6.62 g., 17.97 mmol, 90% isolated yield). Spectral data matched the reference compound.
Preparation of (2S,5R)-7-oxo-N-piperidin-4-yl-6-(sulfooxy)- 1 ,6-diazabicyclo[3.2.1 ]octane-2-carboxamide
tert-butyl 4-((2S,5R)-6-hydroxy-7-oxo-l,6-diazabicyclo[3.2.1]octane-2-carboxamido)piperidine-l-carboxylate (20 g, 54.3 mmol), THF (200 mL), 2-picoline (10.9 mL, 309 mmol) and pyridine-S03 complex (30.2 g, 190 mmol) were charged to a flask under nitrogen. The heterogeneous mixture was allowed to stir overnight (~15 h). The reaction mixture was cooled to -10°C then DCM (200 mL) was added. 0.5 M K2HP04 (168 mL, 84 mmol) was added over 10 minutes. Bu4NHS04 (19.4 g, 57 mmol) was then added over 10 minutes. The biphasic mixture was stirred for 30 minutes, phase cut and the water layer was back extracted with 40 ml of DCM. The combined DCM solution was washed with water (120 ml), phase cut and the organic solution was solvent-switched to MeCN (320 ml) by vacuum distillation with 3 bed volumes of MeCN (total 1.0 L) and used as is in the next step. The solution of Bu4N+ OSO3 salt 7 in MeCN solution was used with an assumed yield of 100% (37.5 g, 54.3 mmol). The reaction mixture was cooled in an ice bath, and TMSI (10.26 ml, 70.7 mmol) was added via addition funnel over 30 minutes between 0°C and 5°C. The resulting mixture was agitated for 1-2 h and then quenched with H20:MeCN (1 :1, 6 ml) to afford a slurry. The slurry was warmed to room temperature and agitated for 12 h and after this time the pH of the supernatant was about 3.0. Tetrabutylammonium acetate (13.6 ml, 13.59 mmol) was slowly added over 30 min. The slurry was agitated for 1 h and pH of the supernatant was about 4.0. Solids were collected by filtration. The solid was washed with 60 mL of aqueous MeCN to afford 19.5 g of the crude product 8 in a 93% isolated yield from compound 6 .
At this stage, all byproducts (including hydro lyzation products of TMS-carbonate) and impurities were soluble in the organic phase.
The product was dissolved back into 140 ml of MeCN:H20 (1 :2) at room temperature. 1-Butanol (390 ml) as antisolvent was slowly added into the solution to afford a slurry. The slurry was agitated overnight. The white crystalline solid was filtered and washed with 3:1 IPA: water (40 ml) and dried under vacuum and nitrogen at room temperature to afford the title product in the form of a crystalline hydrate. (Yield = 16.3 g, 82%). Spectral data matched reference compound.
Preparation of (2S,5R)-7-oxo-2-(piperidin- 1 -ium-4-ylcarbamoyl)- 1 ,6-diazabicyclo[3.2.1 ]octan-6-yl sulfate (1).
tert-Butyl 4-( {[(25*,5i?)-6-hydroxy-7-oxo- 1 ,6-diazabicyclo[3.2.1 ]oct-2-yl]carbonyl}amino)piperidine-l-carboxylate 16 (0.54 g, 1.5 mmol), THF (5.4 mL), 2-picoline (0.29 mL, 2.9 mmol) and pyridine-S03 complex (0.70 g, 4.4 mmol) were charged to a vial under nitrogen. The heterogeneous mixture was allowed to stir overnight (~15 hr). The reaction mixture was cooled to -10°C then dichloromethane (5.4 mL) was added. 0.5 M K2HPO4 (4.5 mL, 2.3 mmol) was added over 10 minutes. BU4NHSO4 (0.53 g, 1.54 mmol) was then added over 10 min. The biphasic mixture was stirred for 30 min, phase cut and the water layer was back extracted with 1 ml of DCM. The combined DCM solution was washed with water (2.0 mL), phase cut and the organic solution was solvent-switched to MeCN (3.2 mL) by vacuum distillation with 3 bed volumes of MeCN. The product was used as is in the next step (water content less than 1000 ppm).
The solution of Bu4N+S04~~ salt 8 in MeCN solution was used with an assumed yield of 100% (1.0 g, 1.47 mmol). The reaction mixture was cooled in an ice bath, and Ν,Ο-bis(trimethylsilyl)trifluoroacetamide (BSTFA) (0.4 lg, 1.59 mmol) was added into the reaction and was allowed to stir for 10 min. TMSI (0.06g, 0.27 mmol) was added between 0°C and 5°C. The resulting mixture was allowed to agitate for 2 hr and then quenched with H2O (0.07g, 4.1 mmol) and acetic acid (0.08g, 1.5 mmol) to afford a slurry. The slurry was warmed to room temperature and agitated for 12 hr. Filter to collect the solid. The solid was washed with MeCN/water (94:6, 1 mL X 4) to afford the crystalline product 1 (0.38 g) in a 75% yield.
If NO-bis(trimethylsilyl)acetamide (BSA) (0.32g, 1.59 mmol) was applied, the reaction needed 24 hr to achieve full conversion.
Patent
WO2015033191
Scheme 1.

Formula (V)
Formula (VI)

Formula (I)
Scheme – 1
Example -1
Preparation of (2S, 5R)-Sulfuric acid mono-{2-[N’-(4-aminopiperidinyl)-carbonyl]-7-oxo- l,6-diaza-bicyclo[3.2.1]oct-6-yl} ester (I).
Step-1: Preparation of (2S, 5R)-tert-butyl { (6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate (IV):
To a 250 ml round bottom flask equipped with magnetic stirrer was charged a solution of (2S, 5R)-sodium 6-benzyloxy-7-oxo-l,6-diaza-bicyclo [3.2.1] octane-2-carboxylate (11.1 gm, 0.037 mol, prepared using a method disclosed in Indian Patent Application No 699/MUM/2013) in water (180 ml) followed by l-tert-butoxycarbonyl-4-amino-piperidine (7.8 gm, 0.039 mol), EDC hydrochloride (11 gm, 0.055 mol) and 1 -hydro ybenzotriazole (4.8 gm, 0.037 mol) at 30°C successively under stirring. The reaction mixture was stirred for 24 hours at 30°C to provide a suspension. The suspension was filtered under suction and washed with 45°C warm water (40 ml) to provide (2S, 5R)-tert-butyl { (6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate in 12.7 gm quantity in 74% yield after drying under vacuum.
Analysis
NMR: (CDC13,) = 7.36-7.44 (m, 5H), 6.56 (d,lH), 5.06 (d,lH), 4.91 (d, 1H), 4.03 (br s, 1H), 3.88-3.97 (m, 2H), 3.29 (s, 1H), 3.00 (d, 1H), 2.86 (t, 2H), 2.64 (d, 1H), 2.37 (dd, 1H), 1.85-2.01 (m, 4H), 1.54-1.62 (m, 2H), 1.45 (s, 9H), 1.25-1.36 (m, 2H).
MS (ES+) C24H34N405 = 459.5 (M+l).
Step-2: Preparation of (2S, 5R)-tert-butyl { (6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate (V):
To a 100 ml single neck round bottom flask equipped with magnetic stirrer was charged a solution of (2S, 5R)-tert-butyl { (6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate (9 g, 19.5 mmol) in methanol (90 ml) followed by 10% palladium on carbon (2.7 g) at 35°C. The reaction mixture was stirred under 1 atm hydrogen pressure at 35°C for 2 hours. The catalyst was removed by filtering the reaction mixture under suction over a celite bed. The celite bed was washed with dichloromethane (50 ml). The combined filtrate was evaporated under vacuum below 35°C to provide (2S, 5R)-tert-butyl {(6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate in 8.45 g quantity; it was used as such for the next reaction.
Analysis
NMR: (CDC13,) = 6.60 (d, 1H), 3.88-4.10 (m, 4H), 3.78 (s, 1H), 3.20 (d, 1H), 3.90 (t, 2H), 2.80 (d, 1H), 2.46 (dd, 1H), 2.1-2.2 (m, 1H), 2.85-2.20 (m, 4H), 1.70-1.80 (m, 1H), 2.47 (s, 9H), 1.30-1.41 (m, 3H).
MS (ES+) C17H28N405 = 369.4 (M+l).
Step-3: Preparation of Tetrabutyl ammonium salt of (2S, 5R)-tert-butyl {(6-sulfooxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate (VI):
To a 100 ml single neck round bottom flask equipped with magnetic stirrer was charged a solution of (2S, 5R)-tert-butyl {(6-hydroxy-7-oxo-l,6-diaza-bicyclo [3.2.1 ]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate (6.40 g, 7.6 mmol) in dichloromethane (90 ml), triethyl amine (9.3 ml), followed by pyridine – sulfur trioxide complex (5.4 g, 34.2 mmol) at 35°C under stirring. The reaction mixture was stirred for additional 4 hours at 35°C. The solvent was evaporated under vacuum below 40°C to provide a residue. The residue was stirred with 0.5N aqueous potassium dihydrogen phosphate solution (90 ml) for 1 hour. The resulting solution was extracted with dichloromethane (2 x 100 ml) to remove impurities. To the aqueous layer was added tetrabutyl ammonium hydrogen sulfate (6.9 g, 20.52 mmol) and the reaction mixture was stirred for 14 hours at 35°C. It was extracted with dichloromethane (3 x 30 ml). Combined organic layer was dried over sodium sulfate and evaporated under vacuum to provide tetrabutyl ammonium salt of (2S, 5R)-tert-butyl {(6-sulfooxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate in 8.0 g quantity in 62% yield.
Analysis
NMR: (CDC13,) – 6.64 (d, 1H), 4.36 (br s, 1H), 4.05(br s, 2H), 3.90-4.00 (m, 1H), 3.87 (d, 1H), 2.28-3.34 (m, 10H), 3.80-3.95 (m, 2H), 3.74 (d, 1H), 2.42 (dd, 1H), 2.15-2.24 (m, 1H), 1.82-1.97 (m, 4H), 1.61-1.74 (m, 14 H), 1.41-1.52 (m, 10 H), 1.02 (t, 12H).
MS (ES-) C17H27N408S. N(C4H9)4 = 447.4 (M-l) as a free sulfonic acid.
Step-4: Synthesis of (2S, 5R)- Sulfuric acid mono-{ [(4-aminopiperidin-4-yl) carbonyl]-7-oxo-l,6-diaza-bicyclo[3.2.1]-oct-6-yl} ester (I):
To a 100 ml round bottom flask equipped with magnetic stirrer was charged a solution of tetrabutyl ammonium salt of (2S, 5R)-tert-butyl {(6-sulfooxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate (6.0 g) in dichloromethane (15 ml). The solution was cooled to -10°C under stirring and to it was added trifluoro acetic acid (15 ml) drop wise. The reaction mixture was stirred at -10°C for 1 hour. Solvents were evaporated under vacuum below 30°C to its 1/3 volume to provide a thick residue. The thick residue was stirred twice with diethyl ether (60 ml each time) to provide a precipitation. The solid obtained was filtered at suction and suspended in acetone (90 ml). To the suspension was added 10% solution of sodium-2-ethyl-hexanoate in acetone to adjust pH between 4.5 to 5.5. The suspension was stirred for 10 minutes and filtered under suction. The wet cake was washed with acetone and dried under vacuum below 40°C to provide 3 gm crude compound. The crude compound was stirred with aqueous isopropanol (3ml water: 21 ml iospropanol) for overnight to purify further. The resulting suspension was filtered under suction and washed with aqueous isopropanol (1 ml water: 7 ml IPA mixture). Finally the cake was dried under vacuum below 40°C to provide the title compound as a off-white solid in 1.8 g quantity in 65% yield.
Analysis
H1NMR (DMSO-d6, D20 exchange) = 8.19 (d, exchanges with D20), 3.99 (s, 1H), 3.82-3.92 (m, 1H), 3.72 (d, 1H), 2.24 (br d, 3H), 2.90-3.04 (m, 5H), 1.96-2.06 (m, 1H), 1.80-1.94 (m, 3H), 1.58-1.72 (m, 4H).
MS (ES+) C12H20N4O6S = 349.2 (M+l) as a free sulfonic acid;
Purity by HPLC: 99.2%
Specific rotation: [a] D -45.25 °, (c 0.3%, water)
SEE BACTAM SERIES…………..http://apisynthesisint.blogspot.in/p/bactam-series.html
//////
C1CC(N2CC1N(C2=O)OS(=O)(=O)O)C(=O)NC3CCNCC3.O
UPDATE,,,,,,,,,,
 , Jinchu Liu, Debra J. Wallace, Feng Xu , Benjamin D. Sherry, and Nobuyoshi Yasuda*
, Jinchu Liu, Debra J. Wallace, Feng Xu , Benjamin D. Sherry, and Nobuyoshi Yasuda* 
Previous methods to prepare a bicyclic N-hydroxyl urea intermediate in the synthesis of the potent β-lactamase inhibitor relebactam were effective, but deemed unsuitable for long-term use. Therefore, we developed an in situ protection protocol during hydrogenolysis and a robust deprotection/isolation sequence of this unstable intermediate employing a reactive crystallization. During the hydrogenation studies, we discovered a significant rate enhancement of O-benzyl ether hydrogenolysis in the presence of organic amine bases, especially DABCO. The broader utility of the application of organic bases on the hydrogenolysis of a range of O– and N-benzyl-containing substrates was demonstrated.

5 could be isolated by concentrating the filtrate and storing the solution at 5 °C overnight. 1H NMR (500 MHz, CDCl3): δ 6.58 (d, J = 7.9 Hz, 1H), 4.10–3.86 (m, 4H), 3.55 (bs, 1H), 3.14 (bd, J = 11.5 Hz, 1H), 2.86 (bt, J = 12.0 Hz, 2H), 2.76 (d, J = 11.5 Hz, 1H), 2.36 (dd, J = 15.1, 7.1 Hz, 1H), 2.12 (m, 1H), 2.00–1.82 (m, 3H), 1.66 (m, 1H), 1.44 (s, 9H), 1.31 (m, 2H), 0.25 (S, 9H). 13C NMR (125 MHz, CDCl3): δ 169.2, 168.3, 154.8, 79.8, 60.7, 60.0, 47.3, 46.9, 42.6 (br, 2C), 32.2, 31.9, 28.5 (3C), 20.5, 17.5, −0.75 (3C). (+)-ESI HRMS: calcd for C20H36N4NaO3Si (M + Na)+, 463.2347; found, 463.2348.

FINAFLOXACIN
(S-cyano-1-cyclopropyl-ό-fluoro-T-^aS, 7aS)-hexahydropyrrolo [3,4- b]-1,4-oxazin-6(2H)-yl]-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid)
7-[(4aS,7aS)-3,4,4a,5,7,7a-hexahydro-2H-pyrrolo[3,4-b][1,4]oxazin-6-yl]-8-cyano-1-cyclopropyl-6-fluoro-4-oxoquinoline-3-carboxylic acid |
BAY-35-3377
BY-377
CAS Registry Number: 209342-40-5
HYD SALT
(-)-(4aS,7aS)-8-Cyano-1-cyclopropyl-6-fluoro-4-oxo-7-(perhydropyrrolo[3,4-b]-1,4-oxazin-6-yl)-1,4-dihydroquinoline-3-carboxylic acid hydrochloride
209342-41-6,
| C20 H19 F N4 O4 . Cl H | |
| MW | 434.849 |
Synonyms: Finafloxacin, UNII-D26OSN9Q4R,
MerLion Pharmaceuticals (Singapore)…POSTER…….http://www.merlionpharma.com/sites/default/files/file/PPS/F1-2036_Wohlert.pdf
H. pylori, Broad-Spectrum
Finafloxacin is a novel fluoroquinolone being developed by MerLion Pharmaceuticals. Under neutral pH conditions (pH 7.2–7.4), the compound has shown in vitro activity equivalent to that of ciprofloxacin. However, under slightly acidic pH5.8 the compound shows enhanced potency.
Other marketed fluoroquinolones, such as ciprofloxacin, levofloxacin and moxifloxacin, exhibit reduced activity at slightly acidic pH 5.0–6.5. This feature of finafloxacin makes the compound suitable for use in the treatment of infections in acidic foci of infections such as urinary tract infections
Finafloxacin hydrochloride, a novel highly potent antibiotic, is in phase III clinical trials at Alcon for the treatment of ear infections. MerLion Pharmaceuticals is evaluating the product in phase II clinical trials at for the treatment of Helicobacter pylori infection and for the treatment of lower uncomplicated urinary tract infections in females.
A quinolone, finafloxacin holds potential for the treatment of Helicobacter pylori infection and urinary tract infection. Unlike existing antibiotics, finafloxacin demonstrates a unique acid activated activity whereby it becomes increasingly active under acidic conditions.
In 2009, a codevelopment agreement was signed between Chaperone Technologies and MerLion Pharmaceuticals. In 2011, finafloxacin hydrochloride was licensed to Alcon by MerLion Pharmaceuticals in North America for the treatment of ear infections.
MerLion Pharmaceuticals has announced that the FDA has granted a Qualified Infectious Disease Product Designation and Fast Track Status for finafloxacin. The company is currently recruiting patients for the Phase II clinical trial of the compound for the treatment of complicated urinary tract infections (cUTI) and/or acute pyelonephritis compared to ciprofloxacin
Finafloxacin and derivatives thereof can be synthesized according to the methods described in U.S. Patent No. 6,133,260 to Matzke et al., the contents of which are herein incorporated by reference in their entirety. The compositions of the invention are particularly directed toward treating mammalian and human subjects having or at risk of having a microbial tissue infection. Microbial tissue infections that may be treated or prevented in accord with the method of the present invention are referred to in J. P. Sanford et al., “The Sanford Guide to Antimicrobial Therapy 2007” 37 Edition (Antimicrobial Therapy, Inc.). Particular microbial tissue infections that may be treatable by embodiments of the present invention include those infections caused by bacteria, protozoa, fungi, yeast, spores, and parasites.
SYNTHESIS
WO1998026779A1
http://www.google.sc/patents/WO1998026779A1 COPY PASTE ON BROWSER
8-cyano-l-cyclopropyl-6-fluoro-7-((lS, 6S)-2-oxa-5 ,8-di-azabicyclo [4.3.0] non-8-yl)-l, 4-dihydro-4-oxo-3-quinolinecarboxylic acid.
The compounds, which are suitable for use in the invention are known already to some extent in EP-A-0350733, EP-A-0550903 as well as from DE-A-4329600 or can be prepared according to the processes described in .
If, for example 9,10-difluoro-3 ,8-dimethyl-7-oxo-2 ,3-dihydro-7H-pyrido [l ,2,3-d, e] [l, 3,4] benzoxadiazine-6 -carboxylic acid and 2-oxa-5 ,8-diazabicyclo [4.3.0] nonane, the reaction can be represented by the following equation:

The 7-halo-quinolonecarboxylic acid derivatives used for preparing the compounds of Fomel (I) of the invention are known or can be prepared by known methods. Thus, the 7-chloro-8-cyano-l-cyclopropyl-6-fluoro-1 ,4-dihydro-4-oxo-3-quinolinecarboxylic acid, or of the 7-chloro-8-cyano-l-cyclopropyl-6-fluoro- l been ,4-dihydro-4-oxo-3-quinolinecarboxylic acid ethyl ester described in EP-A-0 276 700th The corresponding 7-fluoro derivatives can be, for example, via the following reaction sequence to build:

An alternative process for preparing the intermediate compound 2,4-dichloro-3-cyano-5-fluoro-benzoyl chloride as the starting material for the preparation of 7-chloro-
8-cyano-1-cyclopropyl-6-fluoro-1 ,4-dihydro-4-oxo-3-quinolinecarboxylic acid is used (EP-A-0276700) and in the 3-cyano-2 ,4,5-trifluoro- benzoyl can be converted, is based on 5-fluoro-l ,3-xylene, 5-fluoro-l ,3-xylene, in the presence of a catalyst under ionic conditions in the nucleus disubstituted to 2,4-dichloro-5-fluoro-l ,3-dimethylbenzene, and this is subsequently chlorinated chlorinated under free radical conditions in the side chains of 2,4-dichloro-5-fluoro-3-dichloromethyl-l-trichloro-methylbenzene. This is the 2,4-dichloro-5-fluoro-3-dichloromethyl-benzoic acid to give 2,4-dichloro-5-fluoro-3-formyl-benzoic acid, and then hydrolyzed to 2,4-dichloro-5-fluoro-3 N-hydroxyiminomethyl acid implemented. By treatment with thionyl chloride, 2,4-dichloro-3-cyano-5-fluoro-benzoyl chloride is obtained, which can still be ,4,5-trifluoro-ben-zoylfluorid converted by a chlorine / fluorine exchange on-3-cyano-2 .



The amines used for the preparation of compounds of formula (I) according to the invention are known from EP-A-0550903, EP-A-0551653 as well as from DE-A-4 309 964th
An alternative to the synthesis of lS, 6S-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane-dihydro-drobromid or the free base 1 S, 6S-2-oxa-5 ,8-diazabicyclo [4.3.0 ] nonane and the corresponding IR, 6R enantiomer provides the following path represents:
Starting material for this synthesis is the cis-l ,4-dihydroxy-2-butene, which is converted to the bis-mesylate with mesylation tosylamide for 1-tosylpyrrolidine. This is converted into the epoxide m-chloroperbenzoic. The ring opening of the epoxide by heating in isopropanol with ethanolamine to trans-3-hydroxy-4 – (2-hydroxy-ethylamino)-l-(toluene-4-sulfonyl)-pyrrolidine in 80% yield. Tetrahydrofuran is then in pyridine / reacted with tosyl chloride, with cooling to Tris-tosylate, which as a crude product in a mixture with some tetra-tosyl derivative with basichen reaction conditions to give the racemic trans-5 ,8-bis-tosyl-2-oxa-5, 6 – diazabicyclo [4.3.0] nonane is cylisiert. At this stage occurs with high selectivity of a chromatographic resolution kieselgelgebundenem poly (N-methacryloyl-L-leucine-d menthylamide) as the stationary phase. The desired enantiomer, (lS, 6S) -5,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo [4.3.0] nonane, is of a purity of
> 99% ee. Cleavage of the p-tosyl protecting groups is carried out with HBr-acetic acid to the lS, 6S-2-Oxa-5 ,8-diazabicyclo [4.3.0] nonane dihydrobromide, the one with a base such as sodium or potassium hydroxide or with the aid of ion exchanger can be converted into the free base. The analogous sequence may be used for the preparation of lR, 6R-2-Oxa-5 ,8-diazabicyclo [4.3.0] nonane dihydrobromide.

HBr / AcOH

Synthesis of lS, 6S-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane
Examples of compounds of the invention are mentioned in addition to the compounds listed in the preparation examples, the compounds listed in Table 1 below, which can be used both in racemic form as well as enantiomerically pure or diastereomerically pure compounds. Table 1:


Example 1 Z
8-cyano-1-cyclopropyl-6 ,7-difluoro-1 ,4-dihydro-4-oxo-3-quinoline-carboxylic acid ethyl ester

a 3-bromo-2 ,4,5-trifluoro-benzoate
To a mixture of 1460 ml of methanol and 340 g of triethylamine, 772 g of 3-bromo-2 ,4,5-trifluoro-benzoyl fluoride was added dropwise under ice cooling. There is one
Stirred for an hour at room temperature. The Reaktionsgemsich is concentrated, the residue dissolved in water and methylene chloride, and the aqueous phase was extracted with methylene chloride. After drying the organic phase over sodium sulfate, concentrated, and the residue was distilled in vacuum. This gives 752.4 g of 3-bromo-2 ,4,5-trifluoro-benzoic acid methyl ester of boiling point 122 ° C/20 mbar.
b. 3-Cyano-2 ,4,5-trifluoro-benzoic acid methyl ester:
269 g of 3-bromo-2 ,4,5-trifluoro-benzoic acid methyl ester and 108 g of copper cyanide are heated to reflux in 400 ml of dimethylformamide for 5 hours. , All volatile components of the reaction mixture are then distilled off in vacuo. The distillate was then fractionated on a column. This gives 133 g of 3-cyano-2 ,4,5-trifluoro-benzoate of boiling point 88-89 ° C / 0.01 mbar.
c. 3-Cyano-2 ,4,5-trifluoro-benzoic acid
A solution of 156 g of 3-cyano-2 ,4,5-trifluoro-benzoate in 960 ml of glacial acetic acid, 140 ml of water and 69 ml concentrated sulfuric acid is heated for 8 hours under reflux. Then the acetic acid is distilled off under vacuum and the residue treated with water. Of failed-ne solid is filtered off, washed with water and dried. Obtained
118.6 g of 3-cyano-2 ,4,5-trifluoro-benzoic acid as a white solid, mp 187-190 ° C.
d 3-cyano-2 ,4,5-trifluoro-benzoyl chloride:
111 g of 3-cyano-2 ,4,5-trifluoro-benzoic acid and 84 g of oxalyl chloride are stirred in 930 ml of dry methylene chloride with the addition of a few drops of dimethylformamide for 5 hours at room temperature. The methylene chloride is evaporated and the residue distilled in vacuo. This gives 117.6 g of 3-cyano-2 ,4,5-trifluoro-benzoyl chloride as a yellow oil.
e 2 – (3-cyano-2 ,4,5-trifluoro-benzoyl)-3-dimethylamino-acrylic acid ethyl ester:
To a solution of 36.5 g of 3-dimethylamino-acrylate and 26.5 g of triethylamine in 140 ml toluene, a solution of 55 g 3-cyano-2, 4,5 – trifluoro-benzoyl chloride are added dropwise in 50 ml of toluene so that the temperature 50-55 ° C remains. Then stirred for 2 hours at 50 ° C.
The reaction mixture is concentrated in vacuo and used without further
Processing used in the next step. f 2 – (3-cyano-2 ,4,5-trifluoro-benzoyl)-3-cyclopropylamino-acrylic acid ethyl ester:
To the reaction product of step e 30 g of glacial acetic acid are added dropwise at 20 ° C. A solution of 15.75 g of cyclopropyl amine in 30 ml of toluene is added dropwise. The mixture is stirred at 30 ° C for 1 hour. Are then added 200 ml of water, stirred 15 minutes, the organic phase is separated off and shakes it again with 100 ml of water. The organic phase is dried over sodium sulfate and concentrated in vacuo. The crude product thus obtained is a set-without further purification in the next step.
g 8-cyano-l-cyclopropyl-6 ,7-difluoro-l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid ethyl ester:
The reaction product from stage f and 27.6 g of potassium carbonate are stirred in 80 ml dimethylformamide for 16 hours at room temperature. The reaction mixture is then poured into 750 ml ice water, the solid filtered off with suction and washed with 80 ml cold methanol. After drying, 47 g of 8 – cyano-l-cyclopropyl-6 ,7-difluoro-l ,4-dihydro-4-oxo-3-quinoline carboxylic acid ethyl ester, mp 209-211 ° C.
Example 2 Z
2,4-dichloro-5-fluoro-l ,3-dimethylbenzene

a solvent-free
In 124 g of 3,5-dimethyl-fluorobenzene 1 g of anhydrous iron (III) chloride are pre-loaded and launched with the speed of chlorine (about 4 h), with which the reaction. This is initially slightly exothermic (temperature increase from 24 to 32 ° C) and is maintained by cooling below 30 ° C. After addition of 120 g of chlorine, the mixture is determined. According to GC analysis are 33.4% monochloro compound, formed 58.4% desired product and 5%> overchlorinated connections. The hydrogen chloride is removed and the reaction mixture is then distilled in a column in a water jet vacuum:
In the run 49 g of 2-chloro-5-fluoro-l ,3-dimethylbenzene obtained at 72-74 ° C/22 mbar. After 5 g of an intermediate fraction proceed at 105 ° C/22 mbar 75 g of 2,4 – dichloro-5-fluoro-l ,3-dimethylbenzene via, Melting range: 64 – 65 ° C.
b in 1,2-dichloroethane
1 kg of 3,5-dimethyl-fluorobenzene and 15 g of anhydrous iron (III) chloride are placed in 1 1 1 ,2-dichloroethane and chlorine is introduced in the same extent as the reaction proceeds (about 4 h). The reaction is initially exothermic (temperature rise from 24 to 32 ° C) and is kept below 30 ° C by cooling. After the introduction of 1200 g of chlorine are according to GC analysis 4% monochloro compound, 81.1% and 13.3% desired product overchlorinated connections emerged. After distilling off the solvent and the hydrogen chloride is distilled in a column in a water jet vacuum:
In the run 40 g of 2-chloro-5-fluoro-l ,3-dimethylbenzene receive. After some intermediate run going at 127-128 ° C/50 mbar 1115 g of 2,4-dichloro-5-fTuor-l ,3-dimethyl-ethylbenzene over.
Example 3 Z
2,4-dichloro-5-fluoro-3-dichloromethyl-l-trichloromethylbenzene

In a photochlorination using chlorine inlet and outlet for the hydrogen chloride to a scrubber and a light source in the vicinity of the chlorine inlet tube, 1890 g of 2,4-dichloro-5-fluoro-l ,3-dimethylbenzene pre-loaded and at 140 to 150 ° C. Chlorine metered. Within 30 hours 3850 g of chlorine are introduced. The content of the desired product according to GC analysis is 71.1% and the proportion of connections minderchlorierten 27.7%. The DestiUaton a 60 cm column with Wilson spirals provides a flow of 1142 g, which can be reused in the chlorination. The main fraction at 160-168 ° C / 0.2 mbar gives 2200 g of 2,4-dichloro-5-fluoro-3-dichloromethyl-l-trichloro-methyl benzene having a melting range of 74-76 ° C. After one recrystallization
Sample from methanol, the melting point 81-82 ° C.
Example Z 4
2,4-dichloro-5-fluoro-3-formyl-benzoic acid

In a 2500 ml stirred apparatus with gas discharge are presented 95% sulfuric acid at 70 ° C. and under stirring, 500 g of molten added dropwise 2,4-dichloro-5-fluoro-3-dichloromethyl-1 trichloromethylbenzene. It is after a short while hydrochloric development. Is metered during a 2 h and stirred until the evolution of gas after. After cooling to 20 ° C., the mixture is discharged ice to 4 kg and the precipitated solid is filtered off with suction. The product is after-washed with water and dried.
Yield: 310 g, melting range: 172-174 ° C
Example Z 5
2,4-dichloro-5-fluoro-3-N-hydroxyiminomethyl-benzoic acid

In a stirred reactor 80 g of hydroxylamine hydrochloride in 500 ml of ethanol are charged and added dropwise 200 ml of 45% strength sodium hydroxide solution and then with 40 – 200 g of 2,4-dichloro-5-fluoro-3-formyl-benzoic acid added 45.degree.The reaction is slightly exothermic and it is stirred for 5 h at 60 ° C. After cooling to
Room temperature is provided by the dropwise addition of hydrochloric acid to pH <3, the product taken up in tert-butyl methyl ether, the organic phase separated and the solvent distilled off. The residue obtained 185 g of 2,4-dichloro-5-fluoro-3-N-hydroxyiminomethyl benzoic acid, melting range: 190 – 194 ° C.
Example No. 6
2,4-dichloro-3-cyano-5-benzoyl-fιuor

In a stirred vessel with metering and gas outlet via a reflux condenser to a scrubber 600 ml of thionyl chloride are introduced and registered at 20 ° C. 210 g of 2,4-dichloro-5-fluoro-3-N-hydroxyiminomethyl benzoic acid in the proportion as hydrochloric developed and sulfur dioxide. After the addition the mixture is heated until the gas evolution under reflux. Mixture is then distilled, and boiling in the range of 142-145 ° C/10 mbar, 149 g of 2,4-dichloro-3-cyano-5-fluoro-benzoyl chloride (98.1% purity by GC) Melting range: 73-75 ° C.
Example No. 7
3-Cyano-2 ,4,5-trifluoro-benzoyl

50 g of potassium fluoride are suspended in 120 ml of tetramethylene sulfone and at 15 mbar for drying distilled (ca. 20 mL).Then, 50.4 g of 2,4 – dichloro-3-cyano-5-fluoro-benzoyl chloride was added and stirred at an internal temperature with exclusion of moisture for 12 hours at 180 ° C. Are removed by vacuum distillation to 32.9 g of 3-cyano-2 ,4,5-trifluoro-benzoyl fluoride in the boiling range of 98 –
Obtain 100 ° C/12 mbar.
Example No. 8
3-Cyano-2 ,4,5-trifluoro-benzoyl chloride

76.6 g of 3-cyano-2 ,4,5-trifluoro-benzoyl fluoride together with 1 g of anhydrous
Aluminum chloride introduced at 60-65 ° C and then added dropwise 25 g of silicon tetrachloride gas in the course of development. After the evolution of gas at 65 ° C is distilled in a vacuum. Boiling range 120-122 ° C/14 mbar, 73.2 g of 3 – cyano-2 ,4,5-trifluoro-benzoyl chloride over.
Example No. 9
1 – (toluene-4-sulfonyl-pyrroline

In a 20 1 HC4-HWS boilers are 2.016 kg (17.6 mol)
Submitted methanesulfonyl chloride in dichloromethane and 12 1 at -10 ° C internal temperature under strong cooling (-34 ° C) solution of 705 g (8.0 mol) of 2-butene-l ,4-diol in 1.944 kg (2.68 1 , 19.2 mol) of triethylamine was added dropwise over 30 minutes. A yellow suspension stirred for 1 hour at -10 ° C and then treated with 4 1 of water, the temperature rises to 0 ° C.The suspension is warmed to room temperature, stirred for 10 minutes at room temperature and then fed in a 30 1 separating funnel. The phases are stirred separately (good phase separation) and the aqueous phase extracted with 2 1 of dichloromethane. The combined dichloromethane phases are presented in a pre-cooled 20 1 HC4 vessel and kept at 0 ° C.
In another 20-1 HC4 boiler distillation 1.37 kg (8.0 mol) toluenesulfonamide be submitted in 6 1 toluene. It is mixed with 3.2 kg of 45% sodium hydroxide solution, 0.8 1 of water and 130.5 g Tetrabutylammomiimhydrogensulfat, heated to 40 ° C maximum temperature inside and creates a vacuum. Then, the previously obtained
Dichloromethane (15.2 1) was added dropwise over 1.5 hours while the dichloromethane was removed by distillation at 450 mbar (bath temperature: 60 ° C). During the distillation is foaming. In the end, a solution is available at an internal temperature of 33-40 ° C. After the addition of dichloromethane is distilled off, until barely distillate is (duration: about 85 minutes; internal temperature 40 ° C at 60 ° C bath temperature at the end). The vessel contents will be warm transferred to a separating funnel and rinsed the tank with water and 5 1 2 1 toluene at 50 ° C. Before phase separation, the solids are extracted in the intermediate phase and washed with 0.5 1 of toluene. The organic phase is extracted with 2.4 1 of water, separated and evaporated to dryness on a rotary evaporator. The solid residue (1758 g) is suspended in 50 ° C bath temperature in 1.6 1 of methanol, the suspension is transferred into a 10 1-flanged flask and the flask rinsed with diisopropyl 2,4 1. The mixture is heated to reflux temperature (59 ° C) and stirred for 30 minutes under reflux. The suspension is cooled to 0 ° C., stirred at 0 ° C for 1 hour and extracted with 0.8 1 of a cold mixture of ether Methanol/Diisopropyl-: washed (1 1.5). The crystals are dried under a nitrogen atmosphere at 50 ° C/400 mbar.
Yield: 1456 g (81.5% of theory)
Example Z 10
3 – (toluene-4-sulfonylV6-oxa-3-aza-bicvclo [3.1.0] hexane
o “|” h “CH3
334.5 g (1.5 mol) of l-(toluene-4-sulphonyl)-pyrroline are dissolved in 1.5 1 of dichloromethane at room temperature and over 15 minutes with a suspension of 408 g (approx. 1.65 to 1, 77 mol) of 70-75% m-chloroperbenzoic acid in 900 ml of dichloromethane (cools added in manufacturing from). The mixture is heated under reflux for 16 hr (test for
Peroxide with KI / starch paper shows yet to peroxide), the suspension was cooled to 5 ° C, sucks the precipitated m-chlorobenzoic acid and washed with 300 ml of dichloromethane (peroxide with Precipitation: negative; precipitate was discarded). The filtrate is to destroy excess peroxide with 300 ml of 10% sodium sulfite solution, washed twice (test for peroxide runs now negative), extracted with 300 ml of saturated sodium bicarbonate solution, washed with water, dried with sodium sulfate and about a quarter of the volume evaporated. Again on test peroxide: negative. The mixture is concentrated and the solid residue is stirred with ice cooling, 400 ml of isopropanol, the precipitate filtered off and dried at 70 ° C in vacuum.
Yield: 295 g (82.3%),
Mp: 136-139 ° C,
TLC (dichloromethane methanol 98:2): 1 HK (Jodkammer)
Example CLOSED
trans-3-Hydroxy-4-(2-hydroxy-ethylamino-l-(‘toluene-4-sulfonyl’) pyrrolidine

643.7 g (2.65 mol) 3 – (Toluoι-4-sulfonyl)-6-oxa-3-aza-bicyclo [3.1.0] hexane to 318.5 ml with ethanolamine in 4 1 of isopropanol at reflux for 16 hours cooked. After TLC monitoring, further 35.1 ml (total 5.86 mol) of ethanolamine added to the mixture and boiled again until the next morning. The mixture is filtered hot with suction and the filtrate concentrated on a rotary evaporator to 3.5 ltr. After seeding and stirring at room temperature for 3.5 1 diisopropyl ether are added, and stirred at 0 ° C for 6 hours. The precipitated crystals are filtered off, with 250 ml of a mixture of isopropanol / diisopropyl ether (1: 1) and washed 2 times with 300 ml of diisopropyl ether and dried overnight under high vacuum.
Yield: 663.7 g (83% of theory), content: 96.1% (area% by HPLC). Example Z 12
trans-toluene-4-sulfonic acid {2 – [[4-hydroxy-l-(toluene-4-sulfonyl)-pyrrolidin-3-yl] – ftoluol-4-sulfonyl)-amino]-ethyl ester)

552 g (1.837 mol) of trans-3-hydroxy-4-(2-hydroxy-ethylamino)-l-(toluene-4-sulfonyl) – pyrrolidine are dissolved under argon in 1.65 1 tetrahydrofuran and 0.8 1 of pyridine dissolved and at -10 ° C in portions 700 g (3.675 mol) p-toluenesulfonyl chloride are added thereto. The mixture is then stirred at this temperature for 16 hours. The work is done by adding 4.3 18.5 1% aqueous hydrochloric acid, extraction twice with dichloromethane (3 1, 2 1), washing the combined organic phases with saturated Natriurnhydrogencarbonatlösung (3 1, 2 1), drying over sodium sulfate, extracting and distilling off the solvent in vacuo. The residue is dried overnight at the oil pump and crude in the next reaction. There were 1093 g as a hard foam (content [area% by HPLC]: 80% Tris-tosyl-product and 13% tetra-tosyl-product, yield see next step). Example Z 13
rac. trans-5 ,8-bis-tosyl-2-oxa-5 .6-diazabicyclor4 .3.01 nonane

1092 g of crude trans-toluene-4-sulfonic acid {2 – [[4-hydroxy-l-(toluene-4-sulfonyl) – pyrrolidin-3-yl] – (toluene-4-sulfonyl)-amino]-ethyl} were dissolved in tetrahydrofuran and 9.4 1 at 0-3 ° C with 1.4 1 of a 1.43 molar solution of sodium hydroxide in
Methanol reacted. After half an hour at this temperature, 2.1 1 of water and 430 ml of diluted (2:1) was added to the mixture and acetic acid with previously isolated crystals of trans-toluene-4-sulfonic acid {2 – [[4-hydroxy-l – (toluene-4-sulfo-phenyl)-pyrrolidin-3-yl] – (toluene-4-sulfonyl)-amino] ethyl}-seeded. The suspension is stirred overnight at 0 to -4 ° C. The next morning, the crystals are filtered off, washed twice with 400 ml of cold mixture of tetrahydrofuran / water (4:1) and dried at 3 mbar at 50 ° C overnight.
Yield: 503 g of white crystals (62.7%> of theory over 2 steps), content: 99.7% (area% by HPLC). Example Z 14
Preparative chromatographic resolution of racemic rac. trans-5.8-bis-tosyl-2-oxa-5.6-diazabicyclor4.3.0] nonane
The chromatography of the racemate at room temperature in a column (inner diameter 75 mm), which with 870 g of a chiral stationary phase (kie-selgelgebundenes poly (N-methacryloyl-L-leucine-d menthylamide) based on the mer captomodifizierten silica Polygosil 100 , 10 microns; see EP-A 0 379 917) is filled (bed height: 38 cm). Detection is carried out using a UV detector at 254 nm
For the sample application using a solution of a concentration of 100 g of rac. trans-5 ,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo [4.3.0] nonane in 3000 ml of tetrahydrofuran. A Trenncyclus is carried out under the following conditions: with the aid of a pump is required for 2 min at a flow of 50 ml / min, a part of the sample solution and the same time at a flow rate of 50 ml / min, pure n-heptane to the column.
Thereafter eluted at a flow rate of 100 ml / min 18 minutes with a mixture of n-Heptan/Tetrahydrofuran (3/2 vol / vol). This is followed for 3 minutes at a flow of 100 ml / min elution with pure tetrahydrofuran. Thereafter, further eluted with n-Heptan/Tetrahydro-furan (3/2 vol / vol). This cycle is repeated several times.
The first eluted enantiomer is the (lS, 6R) -5,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo-[4.3.0] nonane, which is isolated by concentration. The eluate of the more retarding enantiomers is largely evaporated in vacuo, and the precipitated crystals are filtered off with suction and dried. From the separation of 179 g of racemate in this
As 86.1 g (96.2% of theory) of the enantiomer (lS, 6S) -5,8-bis-tosyl-2-oxa-5, 6 – diazabicyclo [4.3.0] nonane having a purity of> 99 % ee. Example Z 15
(LR, 6R-2-oxa-5.6-diazabicvclo [4.3.0] nonane dihydrobromide

38.3 g (87 mmol) of (lS, 6R) -5,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo [4.3.0] nonane in 500 ml of 33 -% HBr / glacial acetic acid 10 g added anisole and heated for 4 hours at 60 ° C (bath). After standing overnight, the suspension is cooled, the precipitate filtered, with
100 ml of abs. Ethanol and dried at 70 ° C under high vacuum.
Yield: 23.5 g (93%) of white solid product, mp 309-310 ° C (dec.), DC (dichloromethane/methanol/17% aq ammonia 30:8:1.): 1 HK
[Α] D: + 0.6 ° (c = 0.53, H 2 O) (fluctuating).
Example Z 16
(LS.6S-2-oxa-5.6-diazabicvclor4.3.01nonan-Dihvdrobromid

Z is analogous to Example 15 from (lS, 6S) -5,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo [4.3.0] no-nan (1S, 6S)-2-oxa-5, 6-diazabicyclo [4.3.0] nonane dihydrobromide receive. Example Z 17
(1 R.6R-2-oxa-5.8-diazabicvclo [4.3.Olnonan

1 Method: 5,8 g (20 mmol) of (lS, 6R)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane dihydro-drobromid are suspended in 100 ml of isopropanol at room temperature with 2.4 g ( 42.9 mmol) and powdered potassium hydroxide while leaving about 1 hour in an ultrasonic bath. The suspension is cooled in an ice bath, filtered, washed with isopropanol and the undissolved salt, the filtrate was concentrated and distilled in a Kugelrohr oven at 150-230 ° C oven temperature and 0.7 mbar. Obtained 2.25 g (87.9% of theory) of a viscous oil which crystallizes. [Α] D -21.3 ° (c = 0.92, CHC1 3) Accordingly, this reaction can be carried out in ethanol.
2 Method: A homosexual genie catalyzed mixture of (lR, 6R)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane dihydrobromide and 620 mg (11 mmol) of powdered potassium hydroxide is dry in a Kugelrohr apparatus at 0.2 mbar and increasing oven temperature to 250 ° C distilled. Obtained 490 mg (76.6% of theory) of (lR, 6R) -2 – oxa-5 ,8-diazabicyclo [4.3.0] nonane as a viscous oil which slowly crystallized.
3 Method: 100 g of moist, pretreated cation exchanger (Dowex 50WX, H + – form, 100-200 mesh, capacity: 5.1 meq / g of dry or 1.7 meq / mL) are charged into a column with about 200 ml 1 N HC1 activated and washed neutral with water 3 1. A solution of 2.9 g (10 mmol) of (lS, 6R)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane
Dihydrobromide in 15 ml of water is added to the ion exchanger, and then washed with 2 1 water, and eluted with approximately 1 1 1 N ammonia solution. The eluate is evaporated. concentrated. Yield: 1.3 g of a viscous oil (quantitative), DC (dichloromethane/methanol/17% NH 3 30:8:1): 1 HK, GC: 99.6% (area).
Example Z 18
(LS.6SV2-oxa-5.8-diazabicvclor4.3.01nonan

Z is analogous to Example 17 from (lS, 6S)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane-di-hydrobromide the free base (lS, 6S)-2-oxa-5 ,8-diazabicyclo [ 4.3.0] nonane made.
Example Z 19
2 – (2,4-dichloro-3-cyano-5-fluoro-benzoyl)-3-dimethylamino-acrylic acid ethyl ester

To a solution of 626 g (4.372 mol) of 3-dimethylamino-acrylate and 591 g (4.572 mol) of ethyl-diisopropyl-amine (Hunigs base) in 1060 ml of dichloromethane, a solution of 1075 g starting at room temperature 2,4-dichloro -3-cyano-5-fluoro-benzoyl chloride (94% pure, corresponding to 1010.5 g = 4.00 mol) was dropped in 850 ml of dichloromethane. The temperature rises to 50-55 ° C (dropwise addition about 90 minutes). Then stirred for 2 hours at 50 ° C and the reaction mixture was used without further purification in the next step.
Example Z 20
2 – (2,4-dichloro-3-Cyano-5-fluoro-benzoyl-3-cvclopropylamino-acrylate

To the reaction mixture from the above step 306 g (5.1 mol) of glacial acetic acid are added dropwise under cooling at about 15 ° C. Then, with further cooling at 10-15 ° C. 267.3 g (4.68 mol) of cyclopropyl amine is added dropwise. Immediately after which the reaction mixture is mixed with 1300 ml of water under ice-cooling and 15 minutes stirred well. The dichloromethane layer was separated and used in the next step.
Example 21 Z
7-chloro-8-cyano-1-cyclopropyl-6-fluoro-1.4-dihydro-4-oxo-3-chinolincarbonsäureethyl ester

To a heated to 60-70 ° C suspension of 353 g (2.554 mol) of potassium carbonate in 850 ml of N-methylpyrrolidone, the dichloromethane phase is dropped from the precursor (about 90 minutes). During the addition of the dichloromethane at the same time
Reaction mixture was distilled off. Then the reaction mixture for 5 Vz hours at 60-70 ° C is well stirred. The mixture is cooled to about 50 ° C. and distilled under a vacuum of about 250 mbar residual dichloromethane from. At room temperature is added dropwise 107 ml 30% hydrochloric acid under ice cooling, then to obtain a pH of 5-6 is set. Then, 2,200 ml of water are added under ice cooling. The reaction mixture is thoroughly stirred for 15 minutes, the solid was then filtered off and washed on the filter twice with 1000 ml of water and extracted three times with 1000 ml of ethanol and then dried in a vacuum oven at 60 ° C.
Yield: 1200 g (89.6% of theory).
This product can be purified, if desired by, the solid is stirred in 2000 ml of ethanol for 30 minutes at reflux. You filtered hot with suction, washed with 500 ml of ethanol and dried at 60 ° C in vacuum. Melting point: 180-182 ° C.
Η-NMR (400 MHz, CDC1 3): d = 1.2 to 1.27 (m, 2H), 1.41 (t, 3H), 1.5-1.56 (m, 2H), 4, 1 to 4.8 (m, 1H), 4.40 (q, 2H), 8.44 (d, J = 8.2 Hz, H), 8.64 (s, 1H) ppm.
Example Z 22
7-chloro-8-cyano-1-cvclopropyl-6-fluoro-1 ,4-dihydro-4-oxo-3-quinolinecarboxylic acid

33.8 g (0.1 mol) of 7-chloro-8-cyano-l-cyclopropyl-6-fluoro-l ,4-dihydro-4-oxo-3-quinolinecarboxylate dissolved in a mixture of 100 ml of acetic acid, 20 ml water and 10 ml concentrated sulfuric acid was heated for 3 hours under reflux. After cooling, the mixture is poured onto 100 ml of ice water, the precipitate filtered off, washed with water and ethanol and dried at 60 ° C in vacuum.
Yield: 29.6 g (96% of theory),
Mp 216-21 C. (with decomposition)
Example 1

A 8-Cyano-l-cvclopropyl-6-fluoro-7-((lS.6S-2-oxa-5.8-diazabicvclo [4.3.0] non-8-yl – 1 ,4-dihydro-4-oxo-3 -quinoline carboxylic acid
1.00 g (3.26 mmol) of 7-chloro-8-cyano-l-cyclopropyl-6-fluoro-l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid are heated with 501 mg (3.91 mmol) of ( lS, 6S)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane and 0.9 ml of triethylamine in 30 ml of acetonitrile was stirred at 40-45 ° C under argon for 25 hours. All volatile components in vacuo. removed and the residue recrystallized from ethanol. Yield: 1.22 g (94%)
Melting point: 294 ° C. (with decomposition)
B) 8-Cyano-l-cyclopropyl-6-fluoro-7-(‘(lS.6S-2-oxa-5 ,8-diazabicvclo [4.3.01nonan-8-YLV 1.4-dihydro-4-oxo-3- quinoline carboxylic acid Hvdrochlorid
200 mg (0.63 mmol) of 8-cyano-l-cyclopropyl-6 ,7-difluoro-l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid ethyl ester to be 97 mg (0.75 mmol) of (lS, 6S)-2-oxa-5, 8 – diazabicyclo [4.3.0] nonane and 0.17 ml of triethylamine in 3 ml of acetonitrile was stirred at 40-45 ° C for 2 hours under argon. All volatile components in vacuo. removed, the residue treated with water, insolubles filtered off and the filtrate was extracted with dichloromethane. The organic phase is dried over sodium sulfate and then concentrated under reduced pressure. a. The resulting residue is dissolved in 6 ml of tetrahydrofuran and 2 ml of water and 30 mg (0.72 mmol) of lithium hydroxide monohydrate was added. After 16 hours of stirring at room temperature, acidified with dilute hydrochloric acid and the resulting precipitate was filtered off with suction and dried. Yield: 155 mg (57%) Melting point:> 300 ° C
C) 8-Cyano-l-cvclopropyl-6-fluoro-7-((lS, 6S-2-oxa-5.8-diazabicvclo [4.3.01non-8 yiyi.4-dihydro-4-oxo-3-quinolinecarboxylic acid hydrochloride
1 g (2.5 mmol) of 8-cyano-l-cyclopropyl-6-fluoro-7-((lS, 6S)-2-oxa-5 ,8-diazabicyclo [4.3.0] non-8-yl )-l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid is suspended in 20 ml of water was added to the suspension, 10 ml hydrochloric acid and stirred for In at room temperature for 3 hours. The resulting precipitate is filtered off, washed with ethanol and dried at 80 ° C under high vacuum.
Yield: 987 mg (90.6% of theory), Melting point: 314-316 ° C. (with decomposition).
D) 8-Cyano-l-cvclopropyl-6-fluoro-7-(iS, 6S)-2-oxa-5.8-diazabicyclo [4.3.0] non-8-YLV 1 ,4-dihydro-4-oxo-3 -quinoline carboxylic acid hydrochloride
86.4 g (217 mmol) of 8-cyano-l-cyclopropyl-6-fluoro-7-((lS, 6S)-2-oxa-5, 8 – diazabicyclo [4.3.0] non-8-yl) – l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid are dissolved at room temperature in 963 ml of water and 239 ml of 1 N aqueous sodium hydroxide solution. After filtration and washing with 200 ml of water is added to 477 ml in aqueous hydrochloric acid and the precipitated crystals placed at 95 ° C to 100 ° C in solution. The solution is cooled overnight, the precipitated crystals are filtered off with suction and washed three times with 500 ml of water and dried in vacuum.
Yield 90 g (94.7% of theory), content:> 99% (area% by HPLC) 99.6% ee. [] D 23: -112 ° (c = 0.29, N NaOH).
……………….
Tetrahedron Lett 2009, 50(21): 2525
A novel approach to Finafloxacin hydrochloride (BAY35-3377)Pages 2525-2528 |
||
Finafloxacin hydrochloride, an important clinical compound was synthesized by a novel synthetic approach. An active intermediate ethyl 7-chloro-8-cyano-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate 19 was prepared by a new route. The chiral (S,S′)-N-Boc 10 was derived from protected pyrrolidine and the absolute stereochemistry was established by X-ray analysis.
http://www.sciencedirect.com/science/article/pii/S0040403909005875
……………….



| WO2011003091A1 * | 2 Jul 2010 | 6 Jan 2011 | Alcon Research, Ltd. | Compositions comprising finafloxacin and methods for treating ophthalmic, otic, or nasal infections |
| US7723524 | 29 Sep 2004 | 25 May 2010 | Daiichi Pharmaceutical Co., Ltd. | 8-cyanoquinolonecarboxylic acid derivative |
| US8536167 | 2 Jul 2010 | 17 Sep 2013 | Alcon Research, Ltd. | Methods for treating ophthalmic, otic, or nasal infections |
| DE4329600A1 * | 2 Sep 1993 | 9 Mar 1995 | Bayer Ag | Pyrido [1,2,3-d,e] [1,3,4] benzoxadiazinderivate |
| EP0276700A1 * | 15 Jan 1988 | 3 Aug 1988 | Bayer Ag | 8-Cyano-1-cyclopropyl-1,4-dihydro-4-oxo-3-quinolinecarboxylic acids, process for their preparation, and antibacterial agents containing them |
| EP0350733A2 * | 30 Jun 1989 | 17 Jan 1990 | Bayer Ag | 7-(1-Pyrrolidinyl)-3-quinolone- and -naphthyridone-carboxylic-acid derivatives, method for their preparation and for substituted mono- and bi-cyclic pyrrolidine intermediates, and their antibacterial and feed additive compositions |
| EP0550903A1 * | 28 Dec 1992 | 14 Jul 1993 | Bayer Ag | Quinolone- and naphthyridone carboxylic acid derivatives as antibacterial agents |
| EP0603887A2 * | 23 Dec 1993 | 29 Jun 1994 | Daiichi Pharmaceutical Co., Ltd. | Bicyclic amine derivatives |
| EP0676199A1 * | 23 Mar 1995 | 11 Oct 1995 | Pfizer Inc. | Use of trovafloxacin or derivatives thereof for the manufacture of a medicament for the treatment of H. pylori infections |
| GB2289674A * | Title not available |


Vatiquinone
バチキノン
Vatiquinone; Alpha-Tocotrienol quinone; EPI-743; UNII-6O85FK9I0X; 1213269-98-7; Vincerenone
| Molecular Formula: | C29H44O3 |
|---|---|
| Molecular Weight: | 440.668 g/mol |
2-[(3R,6E,10E)-3-hydroxy-3,7,11,15-tetramethylhexadeca-6,10,14-trienyl]-3,5,6-trimethylcyclohexa-2,5-diene-1,4-dione
2-((R,6E,10E)-3-hydroxy-3,7,11,15-tetramethylhexadeca-6,10,14-trien-1-yl)-3,5,6-trimethylcyclohexa-2,5-diene-1,4-dione
Vatiquinone is in phase II/III clinical trials for the treatment of leigh syndrome in JP. Phase II clinical trials is also ongoing for Friedreich’s ataxia, Parkinson’s disease, Pearson syndrome, cobalamin C deficiency syndrome, hearing loss and Rett’s syndrome.
Vatiquinone was originally developed by Edison Pharmaceuticals, then licensed to Sumitomo Dainippon Pharma in Japan in 2013.
Orphan drug designations for the treatment of Friedreich’s, Leigh syndrome and Rett’s syndrome were granted to the compound by FDA in 2014.
In 2013, the compound was licensed to Sumitomo Dainippon Pharma by Edison Pharmaceuticals in Japan for development and commercialization for the treatment of pediatric orphan inherited mitochondrial and adult central nervous system diseases.
EU
On 17 January 2018, orphan designation (EU/3/17/1971) was granted by the European Commission to Edison Orphan Pharma BV, The Netherlands, for vatiquinone (also known as alpha-tocotrienol quinone) for the treatment of RARS2 syndrome.
Vatiquinone, also known as EPI 743, is an orally bioavailable para-benzoquinone being developed for inherited mitochondrial diseases. The mechanism of action of EPI-743 involves augmenting the synthesis of glutathione, optimizing metabolic control, enhancing the expression of genetic elements critical for cellular management of oxidative stress, and acting at the mitochondria to regulate electron transport.
Vatiquinone has been investigated for the treatment and prevention of Retinopathy, Rett Syndrome, Genetic Disease, Noise-induced Hearing Loss, and Methylmalonic Aciduria and Homocystinuria,Cblc Type.
EPI-743 (vatiquinone) is a compound being developed by BioElectron (previously known as Edison Pharmaceuticals) to treat Friedreich’s ataxia (FA), a rare, autosomal recessive genetic disorder. The disorder is caused by mutations in the FXN gene, which encodes for a protein called frataxin. Frataxin is required for the normal functioning of mitochondria, or the energy factories of the cells. Decreased levels of frataxin, as observed in patients with FA, disrupts the normal function of mitochondria and leads to the gradual development of symptoms associated with the disease: impairment of muscle coordination, loss of muscle strength and sensation, and impaired speech, vision, and hearing.
Currently, there are no drugs available that could cure or help to effectively manage the condition, although a large number of potential treatments are in the pipeline.
EPI-743 is a drug belonging to the class of para-benzoquinones, a group of potent antioxidants. The regulation of oxidative stress is disturbed in people with FA. EPI-743 targets an enzyme called NADPH quinone oxidoreductase 1 (NQO1), helping to increase the biosynthesis of glutathione, a compound essential for the control of oxidative stress. The drug does not target any FA-specific biochemical pathways directly, but helps to improve the regulation of cellular energy metabolism in general. Due to its non-specific mechanism, the drug can be used in a variety of disorders where mitochondrial function is affected.
In December 2012, Edison Pharmaceuticals started a placebo-controlled Phase 2 study (NCT01728064) to examine the safety and efficacy of EPI-743 on visual and neurological function in FA patients. The study was completed in February 2016. The results indicated no significant differences in visual function at six months between patients treated with EPI-743 and those who received a placebo. However, researchers reported a trend toward improvement in neurological function.
In October 2013, the University of South Florida started a small Phase 2 study (NCT01962363) to evaluate the effects of EPI-743 in patients with rare point mutations leading to FA. The study investigated whether treatment with EPI-743 has a discernible impact on neurological function. The results announced in April 2016 demonstrated significant improvements in neurological functions over 18 months. However, the trial only included three participants.
Currently, no further trials testing EPI-743 in FA patients is taking place. However, the drug is in clinical trials for several other disorders that affect the functions of mitochondria, including Leigh syndrome, mitochondrial respiratory chain disease, Pearson syndrome, and others.
In February 2014, the U.S. Food and Drug Administration (FDA) granted orphan drug status to EPI-743, which allows a more expedited drug approval process. The FDA also granted fast track status to EPI-743 for the treatment of FA in March 2014.
Edison Pharmaceuticals is developing vatiquinone, which was awarded Fast Track status for Friedreich’s ataxia in March 2014.
Reference
Bioorg. Med. Chem. Lett. 2011, 21, 3693-3698.
https://www.sciencedirect.com/science/article/pii/S0960894X11005440

Reference
WO2013041676A1 / US9045402B2.

It is known that a-tocotrienol quinones are pharmaceutically active.
US 201 1 /0172312 A1 discloses that tocotrienol quinones are used in treating Leight Syndrome. WO 2010/126909 A1 and US 2006/0281809 A1 disclose that tocotrienol quinones can be used for treating ophthalmic diseases and mitochondrial diseases. US 5,318,993 discloses the activity of tocotrienol quinones as cholesterol suppression. W.D. Shrader et al., Bioorganic & Medical Chemistry Letters 21 (201 1 ), 3693-3698 disclose that the R-isomer of a-tocotrienol quinone is a metabolite of α-tocotrienol and is a potent cellular protectant against oxidative stress and ageing. The R-isomer of α-tocotrienol used for this study has been extracted from Elaeis guineensis. All these documents either use tocotrienol from natural sources or do not disclose the source of tocotrienol respectively tocotrienol quinones or disclose very specific complex synthesis thereof. These methods are very expensive and limited in producing industrial amounts of the desired products.
It is well known that from vitamin E the tocopherols and tocotrienols having the R-configuration have a significantly higher bioactivity (biopotency) than the corresponding S-isomer. This is also the case for the corresponding R-isomers of tocotrienol quinones.
Synthetic pathways to produce the R-isomer of tocotrienol quinones in a stereospecific way are very expensive and therefore only of limited interest.
The synthesis of a-tocotrienol is known from Kabbe and Heitzer, Synthesis 1978, 888-889, however, no indication of chirality whatsoever is indicated.
The synthesis of tocotrienol from the corresponding 4-oxo-chromanol-derivative is known from US 6,096,907, however, no indication of chirality is indicated.
J. Org. Chem. 1981 , 46, 2445-2450 and CH 356754 disclose the chemical transformation of a-tocopherol to a-tocopheryl quinone and to a-tocopherylhydro-quinone, however, neither tocotrienols nor tocotrienol quinones are mentioned.
Separation of chiral compounds by chromatography is principally known. However, it is also known that the quantitative separation is very often very difficult to achieve.
Due to the importance of these substances, there exists a high interest in a process which would produce R-tocotrienol quinones in a large scale in an easy and economic way.
Examples
The present invention is further illustrated by the following experiments.
1 . Chromatographic separation
Starting materials:
Solvents and reagents used as received were heptane (Fluka, 51750), ethanol (Merck, 1 .00983), isopropanol (Sigma-Aldrich, 59300) and acetic acid (Fluka, 45730).
Chromatography:
Preparative separations were performed on an Agilent 1 100 series hplc system consisting of an Agilent 1 100 degasser, Agilent 1 100 preparative pump, Agilent 1 100 diode array detector, Agilent 1 100 MPS G2250A autosampler/fraction collector controlled by chemstation/CC-mode software package.
HPLC conditions for preparative separation:
Column: Daicel Chiracel® OD-H, 250 mm x 20 mm; eluent 0.5% isopropanol, 0.2 % acetic acid in n-heptane; flow 13 ml/min; detection 220 nm, 400 μΙ injection.
Separation of (R)-6-hydroxy-2,5,7,8-tetramethyl-2-((3E,7E)-4,8, 12-trimethyl-trideca-3,7, 11-trienyl) chroman-4-one and (S)-6-hydroxy-2,5,7,8-tetramethyl-2-((3E, 7E)-4,8, 12-trimethyltrideca-3, 7, 11-trienyl) chroman-4-one
Example 1 :
6-Hydroxy-2,5,7,8-tetramethyl-2-((3E,7E)-4,8,12-trimethyltrideca-3,7,1 1 -trienyl) chroman-4-one was prepared according to the example 6a in Kabbe and Heitzer, Synthesis 1978, 888-889.
The product was analyzed by HPLC (Column: Daicel Chiracel® OD-H, 250 mm x 4.6 mm; eluent 1 % ethanol in n-hexane; flow 1 ml/min; detection 220 nm, 2 μΙ injection). Figure 9 b) shows this chromatogram. It shows that the product is a 49.5 : 50.5 mixture (Retention time 13.2 and 14.2 min.)
87.5 mg of this product in heptane was injected and the two peaks with retention time at maximum 35.4 min. (1 ) (50.9%) resp. 43.5 min. (2) (49.1 %) were se-parated by the preparative HPLC separation. Figure 9 a) shows the chromatogram of the preparative HPLC separation.
After evaporation to dryness and dissolution the two collected fractions have been reanalysis on an analytical column (Daicel Chiracel® OD-H, 250 mm x 4.6 mm; eluent 1 % ethanol in n-hexane; flow 1 ml/min; detection 220 nm, 2 μΙ injection). Figure 9 c), respectively Figure 9 d), show the chromatogram of the first fraction, respectively the second fraction. The separation of the two isomers (Retention time 13.2 min, resp. 14.2 min) in the two fraction shows to be 94.9 : 5.1 (Figure 9 c)) resp. 7.1 : 92.9 (Figure 9 d)). Hence, the two isomers have been separation by preparative chromatography almost completely.
Patent
The active component of the formulation of the present invention is selected from alpha- tocotrienol quinone, beta-tocotrienol quinone, gamma-tocotrienol quinone, delta-tocotrienol quinone, and mixtures thereof. In one embodiment, the formulation of the present invention comprises alpha-tocotrienol quinone as the active component. In other embodiments, the formulations of the present invention comprise one or more tocotrienol quinones of Formula I or mixtures thereof, in a pharmaceutically acceptable vehicle, and in other embodiments, the formulations of the present invention comprise alpha-tocotrienol quinone in a pharmaceutically acceptable vehicle. In other particular embodiments, the formulations are administered orally. In other embodiments, the formulations of the present invention comprise one or more tocotrienol quinones of Formula I or mixtures thereof, in an ophthalmically acceptable vehicle for topical, periocular, or intraocular administration, and in other embodiments, the formulations of the present invention comprise alpha-tocotrienol quinone in an ophthalmically acceptable vehicle.
[0120] The formulations of the present invention comprise tocotrienol quinones which can be produced synthetically from the respective tocotrienol by oxidation with suitable oxidizing agents, as for example eerie ammonium nitrate (CAN). Particularly, the formulations of the present invention comprise alpha-tocotrienol quinone (CAS Reg. No. 1401-66-7) produced by oxidation of alpha-tocotrienol. A preferred process for the production of alpha-tocotrienol has been described in co-owned US provisional application USAN 61/197,585 titled “Process for Enrichment and Isolation of alpha-Tocotrienol from Natural Extracts”.
[0121] Syntheses of various members of the tocotrienol family in the d,l- or (RS)-form have been published, see for example Schudel et al, HeIv. Chim. Acta (1963) 46, 2517-2526; H. Mayer et al, HeIv. Chim. Acta (1967) 50, 1376-11393; H.-J. Kabbe et al, Synthesis (1978), 888-889; M. Kajiwara et al, Heterocycles (1980) 14, 1995-1998; S. Urano et al, Chem. Pharm. Bull. (1983) 31, 4341-4345, Pearce et al, J. Med Chem. (1992), 35, 3595-3606 and Pearce et al, J. Med. Chem. (1994). 37, 526-541. None of these reported processes lead to the natural form of the tocotrienols, but rather produces racemic mixtures. Syntheses of natural form d-tocotrienols have been published. See for example. J. Scott et al, HeIv. CMm. Acta (1976) 59, 290-306, Sato et al. (Japanese Patent 63063674); Sato et al. (Japanese Patent NoJP 01233278) and Couladouros et al. (US Patent No. 7,038,067).
[0122] While synthetic and natural tocopherols are readily available in the market, the natural tocotrienol supply is limited, and generally comprises a mixture of tocotrienols. Crude palm oil which is rich in tocotrienols (800-1500 ppm) offers a potential source of natural tocotrienols. Carotech, Malaysia is able to extract and concentrate tocotrienols from crude palm oil, by a process patented in U.S. Pat. No. 5,157,132. Tocomin®-50 typically comprises about 25.32% mixed tocotrienols (7.00% alpha-tocotrienol, 14.42% gamma-tocotrienol, 3.30% delta-tocotrienol and 0.6% beta-tocotrienol ), 6.90% alpha-tocopherol and other phytonutrients such as plant squalene, phytosterols, co-enzyme QlO and mixed carotenoids.
[0123] Other methods for isolation or enrichment of tocotrienol from certain plant oils and plant oil by-products have been described in the literature. For some examples of such isolation and purification processes, see for instance Top A. G. et al, U.S. Pat. No. 5,190,618; Lane R et al, U.S. Pat No. 6,239,171; Bellafiore, L. et al. U.S. Pat. No.6,395,915; May, CY et al, U.S. Pat. No.6,656,358; Jacobs, L et al, U.S. Pat. No. 6,838,104; Sumner, C et al. Int. Pat. Pub. WO 99/38860, or Jacobs, L, Int. Pat. Pub. WO 02/500054. The compounds for use in the present invention and the other therapeutically active agents can be administered at the recommended maximum clinical dosage or at lower doses. Dosage levels of the active compounds in the compositions for use in the present invention may be varied so as to obtain a desired therapeutic response depending on the route of administration, severity of the disease and the response of the patient. When administered in combination with other therapeutic agents, the therapeutic agents can be formulated as separate compositions that are given at the same time or different times, or the therapeutic agents can be given as a single composition.
1: Peragallo JH, Newman NJ. Is there treatment for Leber hereditary optic neuropathy? Curr Opin Ophthalmol. 2015 Nov;26(6):450-7. doi: 10.1097/ICU.0000000000000212. PubMed PMID: 26448041; PubMed Central PMCID: PMC4618295.
2: Miller DK, Menezes MJ, Simons C, Riley LG, Cooper ST, Grimmond SM, Thorburn DR, Christodoulou J, Taft RJ. Rapid identification of a novel complex I MT-ND3 m.10134C>A mutation in a Leigh syndrome patient. PLoS One. 2014 Aug 12;9(8):e104879. doi: 10.1371/journal.pone.0104879. eCollection 2014. PubMed PMID: 25118196; PubMed Central PMCID: PMC4130626.
3: Strawser CJ, Schadt KA, Lynch DR. Therapeutic approaches for the treatment of Friedreich’s ataxia. Expert Rev Neurother. 2014 Aug;14(8):949-57. doi: 10.1586/14737175.2014.939173. Epub 2014 Jul 18. PubMed PMID: 25034024.
4: Enns GM. Treatment of mitochondrial disorders: antioxidants and beyond. J Child Neurol. 2014 Sep;29(9):1235-40. doi: 10.1177/0883073814538509. Epub 2014 Jun 30. PubMed PMID: 24985754.
5: Avula S, Parikh S, Demarest S, Kurz J, Gropman A. Treatment of mitochondrial disorders. Curr Treat Options Neurol. 2014 Jun;16(6):292. doi: 10.1007/s11940-014-0292-7. PubMed PMID: 24700433; PubMed Central PMCID: PMC4067597.
6: Hargreaves IP. Coenzyme Q10 as a therapy for mitochondrial disease. Int J Biochem Cell Biol. 2014 Apr;49:105-11. doi: 10.1016/j.biocel.2014.01.020. Epub 2014 Feb 2. Review. PubMed PMID: 24495877.
7: Chicani CF, Chu ER, Miller G, Kelman SE, Sadun AA. Comparing EPI-743 treatment in siblings with Leber’s hereditary optic neuropathy mt14484 mutation. Can J Ophthalmol. 2013 Oct;48(5):e130-3. doi: 10.1016/j.jcjo.2013.05.011. PubMed PMID: 24093206.
8: Pastore A, Petrillo S, Tozzi G, Carrozzo R, Martinelli D, Dionisi-Vici C, Di Giovamberardino G, Ceravolo F, Klein MB, Miller G, Enns GM, Bertini E, Piemonte F. Glutathione: a redox signature in monitoring EPI-743 therapy in children with mitochondrial encephalomyopathies. Mol Genet Metab. 2013 Jun;109(2):208-14. doi: 10.1016/j.ymgme.2013.03.011. Epub 2013 Mar 24. PubMed PMID: 23583222.
9: Sadun AA, La Morgia C, Carelli V. Mitochondrial optic neuropathies: our travels from bench to bedside and back again. Clin Experiment Ophthalmol. 2013 Sep-Oct;41(7):702-12. doi: 10.1111/ceo.12086. Epub 2013 Apr 11. Review. PubMed PMID: 23433229.
10: Kerr DS. Review of clinical trials for mitochondrial disorders: 1997-2012. Neurotherapeutics. 2013 Apr;10(2):307-19. doi: 10.1007/s13311-013-0176-7. Review. PubMed PMID: 23361264; PubMed Central PMCID: PMC3625388.
11: Blankenberg FG, Kinsman SL, Cohen BH, Goris ML, Spicer KM, Perlman SL, Krane EJ, Kheifets V, Thoolen M, Miller G, Enns GM. Brain uptake of Tc99m-HMPAO correlates with clinical response to the novel redox modulating agent EPI-743 in patients with mitochondrial disease. Mol Genet Metab. 2012 Dec;107(4):690-9. doi: 10.1016/j.ymgme.2012.09.023. Epub 2012 Sep 28. PubMed PMID: 23084792.
12: Martinelli D, Catteruccia M, Piemonte F, Pastore A, Tozzi G, Dionisi-Vici C, Pontrelli G, Corsetti T, Livadiotti S, Kheifets V, Hinman A, Shrader WD, Thoolen M, Klein MB, Bertini E, Miller G. EPI-743 reverses the progression of the pediatric mitochondrial disease–genetically defined Leigh Syndrome. Mol Genet Metab. 2012 Nov;107(3):383-8. doi: 10.1016/j.ymgme.2012.09.007. Epub 2012 Sep 10. PubMed PMID: 23010433.
13: Büsing A, Drotleff AM, Ternes W. Identification of α-tocotrienolquinone epoxides and development of an efficient molecular distillation procedure for quantitation of α-tocotrienol oxidation products in food matrices by high-performance liquid chromatography with diode array and fluorescence detection. J Agric Food Chem. 2012 Aug 29;60(34):8302-13. doi: 10.1021/jf301137b. Epub 2012 Aug 16. PubMed PMID: 22747466.
14: Sadun AA, Chicani CF, Ross-Cisneros FN, Barboni P, Thoolen M, Shrader WD, Kubis K, Carelli V, Miller G. Effect of EPI-743 on the clinical course of the mitochondrial disease Leber hereditary optic neuropathy. Arch Neurol. 2012 Mar;69(3):331-8. doi: 10.1001/archneurol.2011.2972. PubMed PMID: 22410442.
15: Enns GM, Kinsman SL, Perlman SL, Spicer KM, Abdenur JE, Cohen BH, Amagata A, Barnes A, Kheifets V, Shrader WD, Thoolen M, Blankenberg F, Miller G. Initial experience in the treatment of inherited mitochondrial disease with EPI-743. Mol Genet Metab. 2012 Jan;105(1):91-102. doi: 10.1016/j.ymgme.2011.10.009. Epub 2011 Oct 21. PubMed PMID: 22115768.
16: Shrader WD, Amagata A, Barnes A, Enns GM, Hinman A, Jankowski O, Kheifets V, Komatsuzaki R, Lee E, Mollard P, Murase K, Sadun AA, Thoolen M, Wesson K, Miller G. α-Tocotrienol quinone modulates oxidative stress response and the biochemistry of aging. Bioorg Med Chem Lett. 2011 Jun 15;21(12):3693-8. doi: 10.1016/j.bmcl.2011.04.085. Epub 2011 Apr 24. PubMed PMID: 21600768.
17: Gagnon KT. HD Therapeutics – CHDI Fifth Annual Conference. IDrugs. 2010 Apr;13(4):219-23. PubMed PMID: 20373247.
18: Bidichandani SI, Delatycki MB. Friedreich Ataxia. 1998 Dec 18 [updated 2014 Jul 24]. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2016. Available from http://www.ncbi.nlm.nih.gov/books/NBK1281/ PubMed PMID: 20301458.
19: Yu-Wai-Man P, Chinnery PF. Leber Hereditary Optic Neuropathy. 2000 Oct 26 [updated 2013 Sep 19]. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2016. Available from http://www.ncbi.nlm.nih.gov/books/NBK1174/ PubMed PMID: 20301353.

| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US9162957 | METHODS FOR SELECTIVE OXIDATION OF ALPHA TOCOTRIENOL IN THE PRESENCE OF NON-ALPHA TOCOTRIENOLS |
2012-07-19
|
2014-09-04
|
| US9670545 | METHODS AND KITS FOR TREATING AND CLASSIFYING INDIVIDUALS AT RISK OF OR SUFFERING FROM TRAP1 CHANGE-OF-FUNCTION |
2014-06-11
|
2016-06-30
|
| US2017297991 | METHODS FOR SELECTIVE OXIDATION OF ALPHA TOCOTRIENOL IN THE PRESENCE OF NON-ALPHA TOCOTRIENOLS |
2017-01-20
|
|
| US2014221674 | PROCESS FOR THE PRODUCTION OF ALPHA-TOCOTRIENOL AND DERIVATIVES |
2013-09-26
|
2014-08-07
|
| US8575369 | Process for the production of alpha-tocotrienol and derivatives |
2012-01-25
|
2013-11-05
|
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2017037023 | PROCESS FOR THE PRODUCTION OF ALPHA-TOCOTRIENOL AND DERIVATIVES |
2016-03-11
|
|
| US9670170 | RESORUFIN DERIVATIVES FOR TREATMENT OF OXIDATIVE STRESS DISORDERS |
2014-03-14
|
2016-02-11
|
| US9296712 | RESORUFIN DERIVATIVES FOR TREATMENT OF OXIDATIVE STRESS DISORDERS |
2013-03-15
|
2014-09-18
|
| US8106223 | PROCESS FOR THE PRODUCTION OF ALPHA-TOCOTRIENOL AND DERIVATIVES |
2010-04-29
|
2012-01-31
|
| US9567279 | METHODS FOR SELECTIVE OXIDATION OF ALPHA TOCOTRIENOL IN THE PRESENCE OF NON-ALPHA TOCOTRIENOLS |
2015-09-10
|
2016-01-07
|
////////////orphan drug status, EPI-743, fast track, EPI743, EPI-743, EPI 743, Vatiquinone; alpha-Tocotrienol quinone, Vincerenone, バチキノン , BioE-743
CC1=C(C(=O)C(=C(C1=O)C)CCC(C)(CCC=C(C)CCC=C(C)CCC=C(C)C)O)C
19 February 2013 EPI-743 Vatiquinone is a new drug that is based on vitamin E. Tests have shown that it can help improve the function of cells with mitochondrial problems. It may be able to treat people with genetic disorders that affect metabolism and mitochondria Edison Pharmaceuticals and Bambino Gesu Children’s Hospital have announced the commencement of EPI-743 Phase 2 cobalamin C deficiency syndrome trial. EPI-743 is an orally bioavailable small molecule and a member of the para-benzoquinone class of drugs. The trial’s principal investigator, Bambino Gesu Children’s Hospital, division of metabolism Professor Carlo Dionisi-Vici said, “Given the central role of glutathione in cellular redox balance and antioxidant defense systems, we are eager to explore whether a therapeutic that increases glutathione such as EPI-743 will provide clinical benefit.” Improvement in visual function is the primary endpoint of the placebo-controlled study while secondary outcome measurements assess neurologic and neuromuscular function, glutathione biomarkers, quality of life, in addition to safety parameters. The investigation is aimed at assessing the efficacy of EPI-743 in disorders of intermediary metabolism that also result in redox disturbances. EPI-743 is an orally absorbed small molecule that readily crosses into the central nervous system. It works by targeting the enzyme NADPH quinone oxidoreductase 1 (NQO1). Its mode of action is to synchronize energy generation in mitochondria with the need to counter cellular redox stress Friedreich’s ataxia (FRDA) is an autosomal recessive neurodegenerative and cardiodegenerative disorder caused by decreased levels of the protein frataxin. The disease causes the progressive loss of voluntary motor coordination (ataxia) and cardiac complications. Symptoms typically begin in childhood, and the disease progressively worsens as the patient grows older; patients eventually become wheelchair-bound due to motor disabilities. Patients with Friedreich’s ataxia develop loss of visual acuity or changes in color vision. Most have jerky eye movements (nystagmus), but these movements by themselves do not necessarily interfere with vision. ……………… Bioorg Med Chem Lett 2011, 21(12): 3693 http://www.sciencedirect.com/science/article/pii/S0960894X11005440We report that α-tocotrienol quinone (ATQ3) is a metabolite of α-tocotrienol, and that ATQ3 is a potent cellular protectant against oxidative stress and aging. ATQ3 is orally bioavailable, crosses the blood–brain barrier, and has demonstrated clinical response in inherited mitochondrial disease in open label studies. ATQ3 activity is dependent upon reversible 2e-redox-cycling. ATQ3 may represent a broader class of unappreciated dietary-derived phytomolecular redox motifs that digitally encode biochemical data using redox state as a means to sense and transfer information essential for cellular function. 
Figure 1.
The conversion of α-tocotrienol to α-tocotrienol quinone.

Figure 1.
The conversion of α-tocotrienol to α-tocotrienol quinone.

Belinostat (PXD101)
PHASE 2, FAST TRACK FDA , ORPHAN STATUS
Belinostat (PXD101) is a novel HDAC inhibitor with IC50 of 27 nM, with activity demonstrated in cisplatin-resistant tumors.
CLINICAL TRIALS…http://clinicaltrials.gov/search/intervention=Belinostat+OR+PXD101
Belinostat inhibits the growth of tumor cells (A2780, HCT116, HT29, WIL, CALU-3, MCF7, PC3 and HS852) with IC50 from 0.2-0.66 μM. PD101 shows low activity in A2780/cp70 and 2780AD cells. Belinostat inhibits bladder cancer cell growth, especially in 5637 cells, which shows accumulation of G0-G1 phase, decrease in S phase, and increase in G2-M phase. Belinostat also shows enhanced tubulin acetylation in ovarian cancer cell lines. A recent study shows that Belinostat activates protein kinase A in a TGF-β signaling-dependent mechanism and decreases survivin mRNA.
| MW 318.07 | |
| MF | C15H14N2O4S |
414864-00-9 cas no
866323-14-0
(2E)-N-hydroxy-3-[3-(phenylsulfamoyl)phenyl]acrylamide
A novel HDAC inhibitor
…………………………
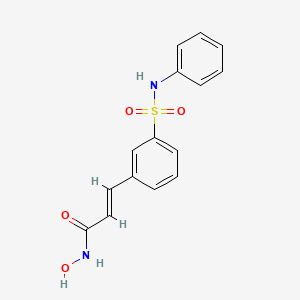
Belinostat (PXD101) is experimental drug candidate under development byTopoTarget for the treatment of hematological malignancies and solid tumors. It is a histone deacetylase inhibitor.[1]
A hydroxamate-type inhibitor of histone deacetylase.
NCI: A novel hydroxamic acid-type histone deacetylase (HDAC) inhibitor with antineoplastic activity. Belinostat targets HDAC enzymes, thereby inhibiting tumor cell proliferation, inducing apoptosis, promoting cellular differentiation, and inhibiting angiogenesis. This agent may sensitize drug-resistant tumor cells to other antineoplastic agents, possibly through a mechanism involving the down-regulation of thymidylate synthase
In 2007 preliminary results were released from the Phase II clinical trial of intravenous belinostat in combination with carboplatin and paclitaxel for relapsedovarian cancer.[2] Final results in late 2009 of a phase II trial for T cell lymphomawere encouraging.[3] Belinostat has been granted orphan drug and fast trackdesignation by the FDA.[4]

The study of inhibitors of histone deacetylases indicates that these enzymes play an important role in cell proliferation and differentiation. The inhibitor Trichostatin A (TSA) (Yoshida et al., 1990a) causes cell cycle arrest at both G1 and G2 phases (Yoshida and Beppu, 1988), reverts the transformed phenotype of different cell lines, and induces differentiation of Friend leukaemia cells and others (Yoshida et al., 1990b). TSA (and SAHA) have been reported to inhibit cell growth, induce terminal differentiation, and prevent the formation of tumours in mice (Finnin et al., 1999).
Trichostatin A (TSA)

Suberoylanilide Hydroxamic Acid (SAHA)

Cell cycle arrest by TSA correlates with an increased expression of gelsolin (Hoshikawa et al., 1994), an actin regulatory protein that is down regulated in malignant breast cancer (Mielnicki et al., 1999). Similar effects on cell cycle and differentiation have been observed with a number of deacetylase inhibitors (Kim et al., 1999). Trichostatin A has also been reported to be useful in the treatment of fibrosis, e.g., liver fibrosis and liver cirrhosis. See, e.g., Geerts et al., 1998.
Recently, certain compounds that induce differentiation have been reported to inhibit histone deacetylases. Several experimental antitumour compounds, such as trichostatin A (TSA), trapoxin, suberoylanilide hydroxamic acid (SAHA), and phenylbutyrate have been reported to act, at least in part, by inhibiting histone deacetylase (see, e.g., Yoshida et al., 1990; Richon et al., 1998; Kijima et al., 1993). Additionally, diallyl sulfide and related molecules (see, e.g., Lea et al., 1999), oxamflatin (see, e.g., Kim et al., 1999), MS-27-275, a synthetic benzamide derivative (see, e.g., Saito et al., 1999; Suzuki et al., 1999; note that MS-27-275 was later re-named as MS-275), butyrate derivatives (see, e.g., Lea and Tulsyan, 1995), FR901228 (see, e.g., Nokajima et al., 1998), depudecin (see, e.g., Kwon et al., 1998), and m-carboxycinnamic acid bishydroxamide (see, e.g., Richon et al., 1998) have been reported to inhibit histone deacetylases. In vitro, some of these compounds are reported to inhibit the growth of fibroblast cells by causing cell cycle arrest in the G1 and G2 phases, and can lead to the terminal differentiation and loss of transforming potential of a variety of transformed cell lines (see, e.g., Richon et al, 1996; Kim et al., 1999; Yoshida et al., 1995; Yoshida & Beppu, 1988). In vivo, phenybutyrate is reported to be effective in the treatment of acute promyelocytic leukemia in conjunction with retinoic acid (see, e.g., Warrell et al., 1998). SAHA is reported to be effective in preventing the formation of mammary tumours in rats, and lung tumours in mice (see, e.g., Desai et al., 1999).
The clear involvement of HDACs in the control of cell proliferation and differentiation suggest that aberrant HDAC activity may play a role in cancer. The most direct demonstration that deacetylases contribute to cancer development comes from the analysis of different acute promyelocytic leukaemias (APL). In most APL patients, a translocation of chromosomes 15 and 17 (t(15;17)) results in the expression of a fusion protein containing the N-terminal portion of PML gene product linked to most of RARσ (retinoic acid receptor). In some cases, a different translocation (t(11 ;17)) causes the fusion between the zinc finger protein PLZF and RARα. In the absence of ligand, the wild type RARα represses target genes by tethering HDAC repressor complexes to the promoter DNA. During normal hematopoiesis, retinoic acid (RA) binds RARα and displaces the repressor complex, allowing expression of genes implicated in myeloid differentiation. The RARα fusion proteins occurring in APL patients are no longer responsive to physiological levels of RA and they interfere with the expression of the RA- inducible genes that promote myeloid differentiation. This results in a clonal expansion of promyelocytic cells and development of leukaemia. In vitro experiments have shown that TSA is capable of restoring RA-responsiveness to the fusion RARα proteins and of allowing myeloid differentiation. These results establish a link between HDACs and oncogenesis and suggest that HDACs are potential targets for pharmaceutical intervention in APL patients. (See, for example, Kitamura et al., 2000; David et al., 1998; Lin et al., 1998).
BELINOSTAT
Furthermore, different lines of evidence suggest that HDACs may be important therapeutic targets in other types of cancer. Cell lines derived from many different cancers (prostate, coloreetal, breast, neuronal, hepatic) are induced to differentiate by HDAC inhibitors (Yoshida and Horinouchi, 1999). A number of HDAC inhibitors have been studied in animal models of cancer. They reduce tumour growth and prolong the lifespan of mice bearing different types of transplanted tumours, including melanoma, leukaemia, colon, lung and gastric carcinomas, etc. (Ueda et al., 1994; Kim et al., 1999).
Psoriasis is a common chronic disfiguring skin disease which is characterised by well-demarcated, red, hardened scaly plaques: these may be limited or widespread. The prevalence rate of psoriasis is approximately 2%, i.e., 12.5 million sufferers in the triad countries (US/Europe/Japan). While the disease is rarely fatal, it clearly has serious detrimental effects upon the quality of life of the patient: this is further compounded by the lack of effective therapies. Present treatments are either ineffective, cosmetically unacceptable, or possess undesired side effects. There is therefore a large unmet clinical need for effective and safe drugs for this condition. Psoriasis is a disease of complex etiology. Whilst there is clearly a genetic component, with a number of gene loci being involved, there are also undefined environmental triggers. Whatever the ultimate cause of psoriasis, at the cellular level, it is characterised by local T-cell mediated inflammation, by keratinocyte hyperproliferation, and by localised angiogenesis. These are all processes in which histone deacetylases have been implicated (see, e.g., Saunders et al., 1999; Bernhard et al, 1999; Takahashi et al, 1996; Kim et al , 2001 ). Therefore HDAC inhibitors may be of use in therapy for psoriasis. Candidate drugs may be screened, for example, using proliferation assays with T-cells and/or keratinocytes.
………………………………………………………………………..
PXD101/Belinostat®
(E)-N-hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide, also known as PXD101 and Belinostat®, shown below, is a well known histone deacetylate (HDAC) inhibitor. It is being developed for treatment of a range of disorders mediated by HDAC, including proliferative conditions (such as cancer and psoriasis), malaria, etc.

PXD101 was first described in WO 02/30879 A2. That document describes a multi-step method of synthesis which may conveniently be illustrated by the following scheme.


…………………………………..
GENERAL SYNTHESIS
IGNORE 10

ENTRY 45 IS BELINOSTAT
Scheme 1

By using amines instead of aniline, the corresponding products may be obtained. The use of aniline, 4-methoxyaniline, 4-methylaniline, 4-bromoaniline, 4-chloroaniline, 4-benzylamine, and 4-phenethyamine, among others, is described in the Examples below.
In another method, a suitable amino acid (e.g., ω-amino acid) having a protected carboxylic acid (e.g., as an ester) and an unprotected amino group is reacted with a sulfonyl chloride compound (e.g., RSO2CI) to give the corresponding sulfonamide having a protected carboxylic acid. The protected carboxylic acid is then deprotected using base to give the free carboxylic acid, which is then reacted with, for example, hydroxylamine 2-chlorotrityl resin followed by acid (e.g., trifluoroacetic acid), to give the desired carbamic acid.
One example of this approach is illustrated below, in Scheme 2, wherein the reaction conditions are as follows: (i) RSO2CI, pyridine, DCM, room temperature, 12 hours; (ii) 1 M LiOH or 1 M NaOH, dioxane, room temperature, 3-48 hours; (iii) hydroxylamine 2-chlorotrityl resin, HOAt, HATU, DIPEA, DCM, room temperature, 16 hours; and (iv) TFA/DCM (5:95, v/v), room temperature, 1.5 hours.
Scheme 2

Additional methods for the synthesis of compounds of the present invention are illustrated below and are exemplified in the examples below.
Scheme 3A

Scheme 3B

Scheme 4


Scheme 8

Scheme 9

……………………………………………………………………..
SYNTHESIS
Example 1
3-Formylbenzenesulfonic acid, sodium salt (1)

Oleum (5 ml) was placed in a reaction vessel and benzaldehyde (2.00 g, 18.84 mmol) was slowly added not exceeding the temperature of the reaction mixture more than 30°C. The obtained solution was stirred at 40°C for ten hours and at ambient temperature overnight. The reaction mixture was poured into ice and extracted with ethyl acetate. The aqueous phase was treated with CaC03 until the evolution of C02 ceased (pH~6-7), then the precipitated CaSO4was filtered off and washed with water. The filtrate was treated with Na2CO3 until the pH of the reaction medium increased to pH 8, obtained CaCO3 was filtered off and water solution was evaporated in vacuum. The residue was washed with methanol, the washings were evaporated and the residue was dried in desiccator over P2Oβ affording the title compound (2.00 g, 51%). 1H NMR (D20), δ: 7.56-8.40 (4H, m); 10.04 ppm (1 H, s).
Example 2 3-(3-Sulfophenyl)acrylic acid methyl ester, sodium salt (2)

Sodium salt of 3-formylbenzenesulfonic acid (1) (1.00 g, 4.80 mmol), potassium carbonate (1.32 g, 9.56 mmol), trimethyl phosphonoacetate (1.05 g, 5.77 mmol) and water (2 ml) were stirred at ambient temperature for 30 min., precipitated solid was filtered and washed with methanol. The filtrate was evaporated and the title compound (2) was obtained as a white solid (0.70 g, 55%). 1H NMR (DMSO- dβl HMDSO), δ: 3.68 (3H, s); 6.51 (1 H, d, J=16.0 Hz); 7.30-7.88 (5H, m).
Example 3 3-(3-Chlorosulfonylphenyl)acrylic acid methyl ester (3)

To the sodium salt of 3-(3-sulfophenyl)acrylic acid methyl ester (2) (0.670 g, 2.53 mmol) benzene (2 ml), thionyl chloride (1.508 g, 0.9 ml, 12.67 mmol) and 3 drops of dimethylformamide were added and the resultant suspension was stirred at reflux for one hour. The reaction mixture was evaporated, the residue was dissolved in benzene (3 ml), filtered and the filtrate was evaporated to give the title compound (0.6’40 g, 97%).
Example 4 3-(3-Phenylsulfamoylphenyl)acrylic acid methyl ester (4a)

A solution of 3-(3-chlorosulfonylphenyl)acrylic acid methyl ester (3) (0.640 g, 2.45 mmol) in dichloromethane (2 ml) was added to a mixture of aniline (0.465 g, 4.99 mmol) and pyridine (1 ml), and the resultant solution was stirred at 50°C for one hour. The reaction mixture was evaporated and the residue was partitioned between ethyl acetate and 10% HCI. The organic layer was washed successively with water, saturated NaCl, and dried (Na2S0 ). The solvent was removed and the residue was chromatographed on silica gel with chloroform-ethyl acetate (7:1 , v/v) as eluent. The obtained product was washed with diethyl ether to give the title compound (0.226 g, 29%). 1H NMR (CDCI3, HMDSO), δ: 3.72 (3H, s); 6.34 (1H, d, J=16.0 Hz); 6.68 (1 H, br s); 6.92-7.89 (10H, m).
Example 5 3-(3-Phenylsulfamoylphenyl)acrylic acid (5a)

3-(3-Phenylsulfamoylphenyl)acrylic acid methyl ester (4a) (0.220 g, 0.69 mmol) was dissolved in methanol (3 ml), 1N NaOH (2.08 ml, 2.08 mmol) was added and the resultant solution was stirred at ambient temperature overnight. The reaction mixture was partitioned between ethyl acetate and water. The aqueous layer was acidified with 10% HCI and stirred for 30 min. The precipitated solid was filtered, washed with water and dried in desiccator over P2Os to give the title compound as a white solid (0.173 g, 82%). Example 6 3-(3-Phenylsulfamoylphenyl)acryloyl chloride (6a)

To a suspension of 3-(3-phenylsulfamoylphenyl)acrylic acid (5a) (0.173 g, 0.57 mmol) in dichloromethane (2.3 ml) oxalyl chloride (0.17 ml, 1.95 mmol) and one drop of dimethylformamide were added. The reaction mixture was stirred at 40°C for one hour and concentrated under reduced pressure to give crude title compound (0.185 g).
Example 7
N-Hydroxy-3-(3-phenylsulfamoylphenyl)acrylamide (7a) (PX105684) BELINOSTAT

To a suspension of hydroxylamine hydrochloride (0.200 g, 2.87 mmol) in tetrahydrofuran (3.5 ml) a saturated NaHCOβ solution (2.5 ml) was added and the resultant mixture was stirred at ambient temperature for 10 min. To the reaction mixture a 3-(3-phenylsulfamoylphenyl)acryloyl chloride (6a) (0.185 g) solution in tetrahydrofuran (2.3 ml) was added and stirred at ambient temperature for one hour. The reaction mixture was partitioned between ethyl acetate and 2N HCI. The organic layer was washed successively with water and saturated NaCl, the solvent was removed and the residue was washed with acetonitrile and diethyl ether.
The title compound was obtained as a white solid (0.066 g, 36%), m.p. 172°C. BELINOSTAT

1H NMR (DMSO-d6, HMDSO), δ: 6.49 (1 H, d, J=16.0 Hz); 7.18-8.05 (10H, m); 9.16 (1 H, br s); 10.34 (1 H, s); 10.85 ppm (1 H, br s).
HPLC analysis on Symmetry C18column: impurities 4% (column size 3.9×150 mm; mobile phase acetonitrile – 0.1 M phosphate buffer (pH 2.5), 40:60; sample concentration 1 mg/ml; flow rate 0.8 ml/ min; detector UV 220 nm).
Anal. Calcd for C15Hι4N204S, %: C 56.59, H 4.43, N 8.80. Found, %: C 56.28, H 4.44, N 8.56.
……………………………………………………………………….
SYNTHESIS
US20100286279

…………………………………………………….
SYNTHESIS AND SPECTRAL DATA
Journal of Medicinal Chemistry, 2011 , vol. 54, 13 pg. 4694 – 4720
(E)-N-Hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide (28, belinostat, PXD101).
http://pubs.acs.org/doi/full/10.1021/jm2003552
http://pubs.acs.org/doi/suppl/10.1021/jm2003552/suppl_file/jm2003552_si_001.pdf
The methyl ester (27) (8.0 g) was prepared according to reported synthetic route,
(Watkins, C. J.; Romero-Martin, M.-R.; Moore, K. G.; Ritchie, J.; Finn, P. W.; Kalvinsh, I.;
Loza, E.; Dikvoska, K.; Gailite, V.; Vorona, M.; Piskunova, I.; Starchenkov, I.; Harris, C. J.;
Duffy, J. E. S. Carbamic acid compounds comprising a sulfonamide linkage as HDAC
inhibitors. PCT Int. Appl. WO200230879A2, April 18, 2002.)
but using procedure D (Experimental Section) or method described for 26 to convert the methyl ester to crude
hydroxamic acid which was further purified by chromatography (silica, MeOH/DCM = 1:10) to
afford 28 (PXD101) as off-white or pale yellow powder (2.5 g, 31%).
LC–MS m/z 319.0 ([M +H]+).
1H NMR (DMSO-d6) 12–9 (very broad, 2H), 7.90 (s, 1H), 7.76 (d, J = 7.7 Hz, 1H), 7.70 (d, J
= 7.8 Hz, 1H), 7.56 (t, J = 7.8 Hz, 1H), 7.44 (d, J = 15.8 Hz, 1H), 7.22 (t, J = 7.8 Hz, 2H), 7.08 (d,
J = 7.8 Hz, 2H), 7.01 (t, J = 7.3 Hz, 1H), 6.50 (d, J = 15.8 Hz, 1H);
13C NMR (DMSO-d6) 162.1,
140.6, 138.0, 136.5, 135.9, 131.8, 130.0, 129.2, 127.1, 124.8, 124.1, 121.3, 120.4.
Anal.
(C15H14N2O4S) C, H, N
………………………………………………..
SYNTHESIS
PXDIOI / Belinostat®
(E)-N-hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide, also known as PXD101 and Belinostat®, shown below, is a well known histone deacetylate (HDAC) inhibitor. It is being developed for treatment of a range of disorders mediated by HDAC, including proliferative conditions (such as cancer and psoriasis), malaria, etc.

PXD101 was first described in WO 02/30879 A2. That document describes a multi-step method of synthesis which may conveniently be illustrated by the following scheme.
Scheme 1
Not isolated

ed on (A)
on (D)

d on (H)

There is a need for alternative methods for the synthesis of PXD101 and related compounds for example, methods which are simpler and/or employ fewer steps and/or permit higher yields and/or higher purity product.
Scheme 5

DMAP, toluene



Synthesis 1 3-Bromo-N-phenyl-benzenesulfonamide (3)

To a 30 gallon (-136 L) reactor was charged aniline (2) (4.01 kg; 93.13 g/mol; 43 mol), toluene (25 L), and 4-(dimethylamino)pyridine (DMAP) (12 g), and the mixture was heated to 50-600C. 3-Bromobenzenesulfonyl chloride (1) (5 kg; 255.52 g/mol; 19.6 mol) was charged into the reactor over 30 minutes at 50-600C and progress of the reaction was monitored by HPLC. After 19 hours, toluene (5 L) was added due to losses overnight through the vent line and the reaction was deemed to be complete with no compound (1) being detected by HPLC. The reaction mixture was diluted with toluene (10 L) and then quenched with 2 M aqueous hydrochloric acid (20 L). The organic and aqueous layers were separated, the aqueous layer was discarded, and the organic layer was washed with water (20 L), and then 5% (w/w) sodium bicarbonate solution (20 L), while maintaining the batch temperature at 45-55°C. The batch was then used in the next synthesis.
Synthesis 2 (E)-3-(3-Phenylsulfamoyl-phenyl)-acrylic acid ethyl ester (5)

To the batch containing 3-bromo-N-phenyl-benzenesulfonamide (3) (the treated organic layer obtained in the previous synthesis) was added triethylamine (2.97 kg; 101.19 g/mol; 29.4 mol), tri(o-tolyl)phosphine (119 g; 304.37 g/mol; 0.4 mol), and palladium (II) acetate (44 g; 224.51 g/mol; 0.2 mol), and the resulting mixture was degassed four times with a vacuum/nitrogen purge at 45-55°C. Catalytic palladium (0) was formed in situ. The batch was then heated to 80-900C and ethyl acrylate (4) (2.16 kg; 100.12 g/mol; 21.6 mol) was slowly added over 2.75 hours. The batch was sampled after a further 2 hours and was deemed to be complete with no compound (3) being detected by HPLC. The batch was cooled to 45-55°C and for convenience was left at this temperature overnight.
The batch was then reduced in volume under vacuum to 20-25 L, at a batch temperature of 45-55°C, and ethyl acetate (20 L) was added. The batch was filtered and the residue washed with ethyl acetate (3.5 L). The residue was discarded and the filtrates were sent to a 100 gallon (-454 L) reactor, which had been pre-heated to 600C. The 30 gallon (-136 L) reactor was then cleaned to remove any residual Pd, while the batch in the 100 gallon (-454 L) reactor was washed with 2 M aqueous hydrochloric acid and water at 45-55°C. Once the washes were complete and the 30 gallon (-136 L) reactor was clean, the batch was transferred from the 100 gallon (-454 L) reactor back to the 30 gallon (-136 L) reactor and the solvent was swapped under vacuum from ethyl acetate/toluene to toluene while maintaining a batch temperature of 45-55°C (the volume was reduced to 20-25 L). At this point, the batch had precipitated and heptanes (10 L) were added to re-dissolve it. The batch was then cooled to 0-100C and held at this temperature over the weekend in order to precipitate the product. The batch was filtered and the residue was washed with heptanes (5 L). A sample of the wet-cake was taken for Pd analysis. The Pd content of the crude product (5) was determined to be 12.9 ppm.
The wet-cake was then charged back into the 30 gallon (-136 L) reactor along with ethyl acetate (50 L) and heated to 40-500C in order to obtain a solution. A sparkler filter loaded with 12 impregnated Darco G60® carbon pads was then connected to the reactor and the solution was pumped around in a loop through the sparkler filter. After 1 hour, a sample was taken and evaporated to dryness and analysed for Pd content. The amount of Pd was found to be 1.4 ppm. A second sample was taken after 2 hours and evaporated to dryness and analysed for Pd content. The amount of Pd had been reduced to 0.6 ppm. The batch was blown back into the reactor and held at 40-500C overnight before the solvent was swapped under vacuum from ethyl acetate to toluene while maintaining a batch temperature of 45-55°C (the volume was reduced to 20-25 L). At this point, the batch had precipitated and heptanes (10 L) were added to re-dissolve it and the batch was cooled to 0-100C and held at this temperature overnight in order to precipitate the product. The batch was filtered and the residue was washed with heptanes (5 L). The filtrate was discarded and the residue was dried at 45-55°C under vacuum for 25 hours. A first lot of the title compound (5) was obtained as an off-white solid (4.48 kg, 69% overall yield from 3-bromobenzenesulfonyl chloride (1)) with a Pd content of 0.4 ppm and a purity of 99.22% (AUC) by HPLC.
Synthesis 3 (E)-3-(3-Phenylsulfamoyl-phenyl)-acrvlic acid (6)

To the 30 gallon (-136 L) reactor was charged the (E)-3-(3-phenylsulfamoyl-phenyl)- acrylic acid ethyl ester (5) (4.48 kg; 331.39 g/mol; 13.5 mol) along with 2 M aqueous sodium hydroxide (17.76 L; -35 mol). The mixture was heated to 40-50°C and held at this temperature for 2 hours before sampling, at which point the reaction was deemed to be complete with no compound (5) being detected by HPLC. The batch was adjusted to pH 2.2 using 1 M aqueous hydrochloric acid while maintaining the batch temperature between 40-500C. The product had precipitated and the batch was cooled to 20-300C and held at this temperature for 1 hour before filtering and washing the cake with water (8.9 L). The filtrate was discarded. The batch was allowed to condition on the filter overnight before being charged back into the reactor and slurried in water (44.4 L) at 40-500C for 2 hours. The batch was cooled to 15-20°C, held for 1 hour, and then filtered and the residue washed with water (8.9 L). The filtrate was discarded. The crude title compound (6) was transferred to an oven for drying at 45-55°C under vacuum with a slight nitrogen bleed for 5 days (this was done for convenience) to give a white solid (3.93 kg, 97% yield). The moisture content of the crude material was measured using Karl Fischer (KF) titration and found to be <0.1% (w/w). To the 30 gallon (-136 L) reactor was charged the crude compound (6) along with acetonitrile (47.2 L). The batch was heated to reflux (about 80°C) and held at reflux for 2 hours before cooling to 0-10°C and holding at this temperature overnight in order to precipitate the product. The batch was filtered and the residue was washed with cold acetonitrile (7.9 L). The filtrate was discarded and the residue was dried under vacuum at 45-55°C for 21.5 hours. The title compound (6) was obtained as a fluffy white solid (3.37 kg, 84% yield with respect to compound (5)) with a purity of 99.89% (AUC) by HPLC.
Synthesis 4 (E)-N-Hvdroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide (PXD101) BELINOSTAT

To the 30 gallon (-136 L) reactor was charged (E)-3-(3-phenylsulfamoyl-phenyl)-acrylic acid (6) (3.37 kg; 303.34 g/mol; 11.1 mol) and a pre-mixed solution of 1 ,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in isopropyl acetate (IPAc) (27 g in 30 L; 152.24 g/mol; 0.18 mol). The slurry was stirred and thionyl chloride (SOCI2) (960 mL; density ~1.631 g/mL; 118.97 g/mol; -13 mol) was added to the reaction mixture and the batch was stirred at 20-300C overnight. After 18.5 hours, the batch was sampled and deemed to be complete with no compound (6) being detected by HPLC. The resulting solution was transferred to a 100 L Schott reactor for temporary storage while the
30 gallon (-136 L) reactor was rinsed with isopropyl acetate (IPAc) and water. Deionized water (28.9 L) was then added to the 30 gallon (-136 L) reactor followed by 50% (w/w) hydroxylamine (6.57 L; -1.078 g/mL; 33.03 g/mol; -214 mol) and another charge of deionized water (1.66 L) to rinse the lines free of hydroxylamine to make a 10% (w/w) hydroxylamine solution. Tetrahydrofuran (THF) (6.64 L) was then charged to the
30 gallon (-136 L) reactor and the mixture was stirred and cooled to 0-100C. The acid chloride solution (from the 100 L Schott reactor) was then slowly charged into the hydroxylamine solution over 1 hour maintaining a batch temperature of 0-10°C during the addition. The batch was then allowed to warm to 20-300C. The aqueous layer was separated and discarded. The organic layer was then reduced in volume under vacuum while maintaining a batch temperature of less than 300C. The intention was to distill out 10-13 L of solvent, but this level was overshot. A larger volume of isopropyl acetate (IPAc) (16.6 L) was added and about 6 L of solvent was distilled out. The batch had precipitated and heptanes (24.9 L) were added and the batch was held at 20-30°C overnight. The batch was filtered and the residue was washed with heptanes (6.64 L). The filtrate was discarded and the residue was dried at 45-55°C under vacuum with a slight nitrogen bleed over the weekend. The title compound (PXD101) was obtained as a light orange solid (3.11 kg, 89% yield with respect to compound (6)) with a purity of 99.25% (AUC) by HPLC.
The title compound (PXD101) (1.2 kg, 3.77 mol) was dissolved in 8 volumes of 1:1 (EtOH/water) at 600C. Sodium bicarbonate (15.8 g, 5 mol%) was added to the solution. Water (HPLC grade) was then added at a rate of 65 mL/min while keeping the internal temperature >57°C. After water (6.6 L) had been added, crystals started to form and the water addition was stopped. The reaction mixture was then cooled at a rate of 10°C/90 min to a temperature of 0-10cC and then stirred at ambient temperature overnight. The crystals were then filtered and collected. The filter cake was washed by slurrying in water (2 x 1.2 L) and then dried in an oven at 45°C for 60 hours with a slight nitrogen bleed. 1.048 kg (87% recovery) of a light orange solid was recovered. Microscopy and XRPD data showed a conglomerate of irregularly shaped birefringant crystalline particles. The compound was found to contain 0.02% water.
As discussed above: the yield of compound (5) with respect to compound (1) was 69%. the yield of compound (6) with respect to compound (5) was 84%. the yield of PXD101 with respect to compound (6) was 89%.
……………….
FORMULATION
Formulation Studies
These studies demonstrate a substantial enhancement of HDACi solubility (on the order of a 500-fold increase for PXD-101) using one or more of: cyclodextrin, arginine, and meglumine. The resulting compositions are stable and can be diluted to the desired target concentration without the risk of precipitation. Furthermore, the compositions have a pH that, while higher than ideal, is acceptable for use.

UV Absorbance
The ultraviolet (UV absorbance E\ value for PXD-101 was determined by plotting a calibration curve of PXD-101 concentration in 50:50 methanol/water at the λmax for the material, 269 nm. Using this method, the E1i value was determined as 715.7.
Methanol/water was selected as the subsequent diluting medium for solubility studies rather than neat methanol (or other organic solvent) to reduce the risk of precipitation of the cyclodextrin.
Solubility in Demineralised Water
The solubility of PXD-101 was determined to be 0.14 mg/mL for demineralised water. Solubility Enhancement with Cvclodextrins
Saturated samples of PXD-101 were prepared in aqueous solutions of two natural cyclodextrins (α-CD and γ-CD) and hydroxypropyl derivatives of the α, β and Y cyclodextrins (HP-α-CD, HP-β-CD and HP-γ-CD). All experiments were completed with cyclodextrin concentrations of 250 mg/mL, except for α-CD, where the solubility of the cyclodextrin was not sufficient to achieve this concentration. The data are summarised in the following table. HP-β-CD offers the best solubility enhancement for PXD-101.

Phase Solubility Determination of HP-β-CD
The phase solubility diagram for HP-β-CD was prepared for concentrations of cyclodextrin between 50 and 500 mg/mL (5-50% w/v). The calculated saturated solubilities of the complexed HDACi were plotted against the concentration of cyclodextrin. See Figure 1.
………………………..

| US2008274120 | 11-7-2008 | Histone Deacetylase (Hdac) Inhibitors (Pxd101) for the Treatment of Cancer Alone or in Combination With Chemotherapeutic Agent |
| US2008227845 | 9-19-2008 | CYCLOOXYGENASE-2 INHIBITOR/HISTONE DEACETYLASE INHIBITOR COMBINATION |
| US2008213399 | 9-5-2008 | Combination Therapies Using Hdac Inhibitors |
| US2008194690 | 8-15-2008 | Pharmaceutical Formulations Of Hdac Inhibitors |
| US7407988 | 8-6-2008 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US7402603 | 7-23-2008 | Cyclooxygenase-2 inhibitor/histone deacetylase inhibitor combination |
| US7183298 | 2-28-2007 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US2005107445 | 5-20-2005 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US6888027 | 5-4-2005 | Carbamic acid compounds comprising a sulfonamide linkage as hdac inhibitors |
| WO2002030879A2 | Sep 27, 2001 | Apr 18, 2002 | Prolifix Ltd | Carbamic acid compounds comprising asulfonamide linkage as hdac inhibitors |
| US7973181 | 7-6-2011 | HYDROXAMIC ACID DERIVATIVES AS INHIBITORS OF HDAC ENZYMATIC ACTIVITY |
| US7928081 | 4-20-2011 | Combined Use of Prame Inhibitors and Hdac Inhibitors |
| US2011077305 | 3-32-2011 | 5-LIPOXYGENASE INHIBITORS |
| US2011003777 | 1-7-2011 | Methods of Treatment Employing Prolonged Continuous Infusion of Belinostat |
| US2010286279 | 11-12-2010 | Methods of Synthesis of Certain Hydroxamic Acid Compounds |
| US2010190694 | 7-30-2010 | Methods for identifying patients who will respond well to cancer treatment |
| US2010010010 | 1-15-2010 | HDAC INHIBITORS |
| US2009312311 | 12-18-2009 | COMBINATION OF ORGANIC COMPOUNDS |
| US2009192211 | 7-31-2009 | CYCLOOXYGENASE-2 INHIBITOR/HISTONE DEACETYLASE INHIBITOR COMBINATION |
| US7557140 | 7-8-2009 | CARBAMIC ACID COMPOUNDS COMPRISING A SULFONAMIDE LINKAGE AS HDAC INHIBITORS |
| WO1998038859A1 * | Mar 4, 1998 | Sep 11, 1998 | Thomas E Barta | Sulfonyl divalent aryl or heteroaryl hydroxamic acid compounds |
| WO1999024399A1 * | Nov 12, 1998 | May 20, 1999 | Darwin Discovery Ltd | Hydroxamic and carboxylic acid derivatives having mmp and tnf inhibitory activity |
| WO2000056704A1 * | Mar 22, 2000 | Sep 28, 2000 | Duncan Batty | Hydroxamic and carboxylic acid derivatives |
| WO2000069819A1 * | May 12, 2000 | Nov 23, 2000 | Thomas E Barta | Hydroxamic acid derivatives as matrix metalloprotease inhibitors |
| WO2001038322A1 * | Nov 22, 2000 | May 31, 2001 | Methylgene Inc | Inhibitors of histone deacetylase |
| EP0570594A1 * | Dec 7, 1992 | Nov 24, 1993 | SHIONOGI & CO., LTD. | Hydroxamic acid derivative based on aromatic sulfonamide |
| EP0931788A2 * | Dec 16, 1998 | Jul 28, 1999 | Pfizer Inc. | Metalloprotease inhibitors |
| GB2312674A * | Title not available |
| WO2002030879A2 | Sep 27, 2001 | Apr 18, 2002 | Prolifix Ltd | Carbamic acid compounds comprising a sulfonamide linkage as hdac inhibitors |
| WO2005063806A1 | Dec 30, 2003 | Jul 14, 2005 | Council Scient Ind Res | Arginine hydrochloride enhances chaperone-like activity of alpha crystallin |
| US4642316 | May 20, 1985 | Feb 10, 1987 | Warner-Lambert Company | Parenteral phenytoin preparations |
| WO2008090585A2 * | Jan 25, 2008 | Jul 31, 2008 | Univ Roma | Soluble forms of inclusion complexes of histone deacetylase inhibitors and cyclodextrins, their preparation processes and uses in the pharmaceutical field |
| WO2009109861A1 * | Mar 6, 2009 | Sep 11, 2009 | Topotarget A/S | Methods of treatment employing prolonged continuous infusion of belinostat |
| WO2010048332A2 * | Oct 21, 2009 | Apr 29, 2010 | Acucela, Inc. | Compounds for treating ophthalmic diseases and disorders |
| WO2011064663A1 | Nov 24, 2010 | Jun 3, 2011 | Festuccia, Claudio | Combination treatment employing belinostat and bicalutamide |
| US20110003777 * | Mar 6, 2009 | Jan 6, 2011 | Topotarget A/S | Methods of Treatment Employing Prolonged Continuous Infusion of Belinostat |
………………………..
SPECTRUM
Tiny Biotech With Three Cancer Drugs Is More Alluring Takeover Bet Now
Forbes
The drug is one of Spectrum’s two drugs undergoing phase 3 clinical trials. Allergan paid Spectrum $41.5 million and will make additional payments of up to $304 million based on achieving certain milestones. So far, Raj Shrotriya, Spectrum’s chairman, …
……………………………..


CAS NO 878592-87-1 of base
7-[3-[2-Amino-1(E)-fluoroethylidene]piperidin-1-yl]-1-cyclopropyl-6-fluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid

Furiex Pharmaceuticals Inc. announced that the FDA has granted Qualified Infectious Disease Product (QIDP) and Fast Track designation for avarofloxacin (JNJ-Q2). Avarofloxacin is a Phase 3-ready broad-spectrum fluoroquinolone antibiotic for the treatment of acute bacterial skin and skin-structure (ABSSSI) infections, community-acquired pneumonia and has proven to be effective in treating methicillin-resistant Staphylococcus aureus (MRSA) infections.
Avarofloxacin is an investigational novel fluoroquinolone antibiotic that has been shown to be effective in a Phase 2 study of ABSSSI infections. In this study, avarofloxacin demonstrated favorable efficacy for both early clinical response endpoints as well as all clinical cure endpoints for the intent to treat population.

Avarofloxacin has a low tendency for development of drug resistance and exhibits a broad range of antibacterial activities in vitro, including MRSA, fluoroquinolone-resistant Staphylococcus aureus, Streptococcus pneumoniae (including multi-drug resistant strains), gram positive, gram negative, atypical respiratory pathogens (such as legionella and mycoplasma) and anaerobic bacteria, which are often associated with abscesses of the skin and other organs.
The availability of IV and oral formulations for avarofloxacin differentiates it from a number of other products for MRSA infections which are only available for intravenous administration.
For more information call (919) 456-7800or visit http://www.furiex.com/
About Methicillin-Resistant Staphylococcus aureus (MRSA)
MRSA is a strain of the bacteria Staphylococcus aureus (staph) which commonly causes skin and soft tissue infections and is resistant to many antibiotics. Although MRSA had previously been primarily a hospital-acquired pathogen, its incidence has been rising in the community, and it has become the most frequent cause of skin and soft tissue infections presenting to emergency departments in the United States. There are a limited number of antibiotics approved to treat MRSA, and their frequent usage has led to emergence of multi-drug resistant bacteria. Thus, we believe there is significant unmet medical need for new antibiotics such as avarofloxacin that provide flexible (hospital and outpatient) treatment options for MRSA.

WO-2006/101603 describes 7-amino alkylidenyl- heterocyclic quinolones as antimicrobial compounds and the synthesis of 7-[(3E)-3-(2-amino-l-fluoroethylidene)-l- piperidinyl]- 1 -cyclopropyl-6-fluoro- 1 ,4-dihydro-8-methoxy-4-oxo 3-quinolinecarboxylic acid is disclosed as compound (303) in Table 1 on page 20. This compound is conveniently referred to as compound ‘A’ hereafter.
compound ‘A’

7-[(3E)-3-(2-amino-1 -fluoroethylidene)-1 -piperidinyl]-1 -cyclopropyl-6-fluoro- 1 ,4-dihydro-8-methoxy-4-oxo 3-quinolinecarboxylic acid
The in vitro antibacterial properties of compound ‘A’ are described by Morrow B.J. et al. in Antimicrobial Agents and Chemotherapy, vol. 54, pp. 1995 – 1964 (2010).
WO-2008/005670 discloses one-pot methods for the production of substituted allylic alcohols as well as extractive methods for the separation of certain isomeric alcohol products which are useful for preparing quinolones such as the antimicrobial compound 7-[(3E)-3-(2-amino- 1 -fluoroethylidene)- 1 -piperidinyl]- 1 -cyclopropyl-6-fluoro- 1 ,4- dihydro-8-methoxy-4-oxo 3-quinolinecarboxylic acid (i.e. compound ‘Α’). An important intermediate in the overall synthesis route of said antimicrobial compound ‘A’ is 2-[(2E)-2-fluoro-2-(3-piperidinylidene)ethyl]-lH-isoindole-l,3(2H)- dione and its hydrochloric acid salt thereof : compound (1 )

2-[(2E)-2-fluoro-2-(3-piperidinylidene)ethyl]-1 H-isoindole-1 ,3(2H)-dione compound (1 ) .HCI

Compound (1) introduces the desired E- stereochemistry into the overall synthesis route for the antimicrobial compound ‘Α’.
WO-2008/005670 discloses a synthesis route for compound (1) on page 38 as depicted below :

– highly enriched (E)

(Step 3a) O
The detailed reaction procedure for compound (1) is disclosed in WO-2008/005670 in Example 1 on pages 37 to 44 affording compound (1) in Method A with a E:Z ratio of 97:3 in an approximate overall yield of 18 % in Method A (step 1 for the first 3 heptane layers has a yield of 34 % with a ratio E:Z of 71 :29, step 2a has a yield of 53.4% with a ratio E:Z of 97:3, and step 3 has quantitave yield), or affording compound (1) in Method B with an approximate overall yield of 15% with a ratio E:Z of 94.4 : 5.6. WO-2008/005670 discloses a synthesis route for the hydrochloric acid addition salt of compound (1) on page 15 in Scheme 2 as depicted below :

into n-butanol,

1 ) 5/6 N HCl in IPA
2) heat to distill
3) add IPA
enriched E-isomer

compound (1 ) .HCl
The detailed reaction procedure to prepare the HCl salt of compound (1) is disclosed in WO-2008/005670 in Example 4 on pages 49 to 52 affording >95% of desired E-isomer with an overall yield of 18 – 22% starting from N-boc-3-piperidone.
The reaction procedures described in WO-2008/005670 for the preparation of compound (1) or its HCl salt are characterized by lack of selectivity of the Wadsworth- Emmons-Horner reaction which produces the undesired Z-isomer in large quantities. This undesired Z-isomer requires additional time consuming separation steps.
Hence there is a need for a more efficient and less waste-producing procedure for the preparation of compound (1) or its HCl salt. WO-2010/056633 discloses a synthesis scheme XIV on page 87 to prepare tert-butyl 4- (2-ethoxy-2-oxoethylidene)piperidinyl-l-carboxylate and a synthesis scheme XXVI on page 111 to prepare (l-benzyl-piperidin-4-ylidene)bromoacetic acid ethyl ester.
In a first embodiment the present invention relates to an improved process for preparing compounds of formula (III) having an improved ratio of the desired (E)-isomer over the undesired (Z)-isomer.

(I)
In a further embodiment the compound (E)-(III) is then converted in to compound (1) or its hydrochloric acid addition salt thereof.

…………………………..
WO 2008005670
http://www.google.com/patents/WO2008005670A2?cl=en
Scheme 1

3 eq NaBH4 OH
30 – 4O0C

2a 2b 2a

2b shows the preparation of alcohol 2
Scheme 2

1 ) HCI (5 eq )

aqueous layer to enriched E isomer pH 9-10 then extract into n-Butanol, discard aqueous layer
Preparation of
7-[3-(2-Amino-l-fluoroethylidene)piperidin-l-yl]-l-cyclopropyl- 6-fluoro-8-methoxy-4-oxo-l,4-dihydroquinoline-3-carboxylic acid (10) and its HCl salt (12)

7-[3-(2-Amino-1-fluoro-ethyhdene)-piperidin-1-yl]-1-cyclopropyl–fluoro-8-mΘthoxy-4-oxo-1 ,4-dιhydro-quιnolιne-3-carboxylιc acid (10)
Step 1: Preparation of 3-(l-fluoro-2-hydroxyethylidene)piperidine-l-carboxylic acid tert-butyl ester (2a)

A 22-L 4-neck round bottom flask, equipped with a thermocouple controller, overhead mechanical stirrer, condenser, nitrogen inlet adapter, and stopper, was charged with N-Boc-3-piperidone (663.36 g, 3.34 mol), 2-methoxyethanol (6.0 L) and 2-fluorotriethylphosphonoacetate (843.54 g, 3.49 mol). The mixture was stirred to obtain a homogeneous solution and then CS2CO3 was added in portions over 1.5 h. After the CS2CO3 addition was complete, NaBH4 was added in portions over 6 h; during most of this addition the reaction temperature was maintained between 35 0C to 40 0C. After the addition was complete, the reaction was allowed to stir overnight after which time HPLC analysis indicated that the reaction was complete. This run was combined with two additional runs of equal size and transferred to a stirred 100-L Hastalloy® reactor containing water (90 L). The aqueous mixture was extracted with heptane (4 x 20 L) followed by extraction with MTBE (methyl tert-butyl ether) (20 L). The first three heptane extracts provided 842 g of the allylic alcohol as 71:29 (E: Z) mixture (HPLC and NMR). The product mixture from the first three heptane extractions was carried on to the next step without any additional purification. The fourth heptane extract gave 114 g of product that was a 67:33 mixture of is: Z alcohols (NMR). MTBE extraction and concentration gave 1.1 Kg of product as a 33:67 mixture of E:Z alcohols (HPLC). The total overall yield for both isomers was 2.06 Kg (83%). 1H NMR of 2a (400 MHz, CDCl3): £ 1.45 (s, 9 H), 1.52 (m, 2 H), 2.40 (m, 2 H), 3.45 (m, 2 H), 3.90 (s, 2 H), 4.25 (d, 2 H). 1H NMR of 2b (400 MHz, CDCl3): δ 1.46 (s, 9 H), 1.65 (m, 2 H), 2.27 (m, 2 H), 3.45 (m, 2 H), 4.1 (s, 2 H), 4.25 (d, 2 H).
Step 2, Method A: Preparation of 3-is-[2-(l,3-dioxo-l,3-dihydroisoindol-2-yl)-l- fluoroethylidene]-piperidine-l-carboxylic acid tert-butyl ester (3-ϋ)

A 22-L 4-neck round bottom flask, equipped with a thermocouple controller, overhead mechanical stirrer, condenser, pressure-equalizing addition funnel, nitrogen inlet adapter, and stopper, was charged with E:Z alcohol mixture 2a and 2b (377.5 g, 1.296 mol corrected), 2-MeTHF (3.31 L), phthalimide (232.8 g, 1.581 mol), and Ph3P (411.3 g, 1.568 mol). The white suspension was stirred under N2 and cooled to -12 0C in an acetone/Dry-Ice bath, DIAD (309 mL, 1.49 mol) was added via the addition funnel over a 36-min period, while the reaction temperature was maintained at -15 0C to -10 0C. After the addition, the reaction was warmed to 20 0C in a water bath and stirred for 2 h. The reaction was cooled to 0 0C in an ice/water bath and quenched with cold 1.0 M HCl (950 mL). The aqueous phase was separated and EtOAc (1.70 L) was added to the organic phase. This phase was washed with cold 1.0 M HCl (0.95 L) (the aqueous phase was pH < 2) and then separated. The organic phase was next washed with cold 4 NNaOH (1.70 L), the alkaline aqueous phase (pH > 13) was separated and the EtOAc layer washed with brine (1.70 L). Concentration of the organic phase at 60 0C under house vacuum (-120 mm Hg) afforded 1,442.0 g of crude 3. This run was repeated on the same scale to provide an additional 1,431.0 g of crude material for a combined yield of 2,873 g (159%). HPLC analysis (area%) indicated crude 3 was a mixture of 3-E (29.4%), 3-Z (10.4 %), Ph3PO (51.0 %), and phthalimide (1.1 %). This was purified by recrystallization as described in step 2a.
Step 2a, Method A: Purification of 3-is-[2-(l,3-dioxo-l,3-dihydroisoindol-2-yl)-l- fluoroethylidene]-piperidine-l-carboxylic acid tert-butyl ester

A 22-L 4-neck round bottom flask equipped with a thermocouple controller, overhead mechanical stirrer, condenser, pressure-equalizing addition funnel, nitrogen inlet adapter and stopper was charged with the combined crude 3 (2,873 g) and MeOH (9.0 L). The solution was stirred under nitrogen and heated to 65 0C, while hot (60 0C) D.I. water (7.8 L) was added over a 15-min period. The solution was stirred at 65 0C for 5 min, and then the heating mantle was replaced with a water bath, and the mixture was gradually cooled to 0 0C over a 4-h period, and continued stirring for 1 h at 0 0C. The off-white solid was collected by filtration, and dried by air-suction at 60 0C for 20 h, this provided 1,172.6 g of a mixture of 3-E and 3-Z.
The partially purified product above was recrystallized a second time in the same manner using hot MeOH (7.2 L) and hot water (5.0 L) except that the water was added over a 10-min period to afford 515.6 g (53.4%) of 3-E as a 97:3 mixture of E:Z geometric isomers. This material was used in the next step without additional purification. . 1H NMR of 3-E (400 MHz, CDCl3): δ 1.48 (s, 9 H), 1.52-1.66 (m, 2 H), 2.28-2.38 (m, 2 H), 3.40-3.51 (m, 2 H), 4.18 (s, 2 H), 4.55 (d, J= 21.0 Hz, 2 H), 7.68- 7.77 (m, 2 H), 7.80-7.89 (m, 2 H). MS: 397 (M+Na)+, 771 (2M+Na)+.
3 -E-[2-( 1 ,3 -dioxo- 1 ,3-dihydroisoindol-2-yl)- 1 -fluoroethylidene]-piperidine- 1 – carboxylic acid tert-butyl ester was also prepared with Method B below: Step 2, Method B: Preparation of 3-£-[2-(l,3-dioxo-l,3-dihydroisoindol-2-yl)-l- fluoroethylidene]-piperidine-l-carboxylic acid tert-butyl ester (3-E)
Preparation of the methanesulfonate and chloride derivatives

2a
A 12-L 4-neck round bottom flask equipped with an overhead stirrer, thermocouple, pressure-equalizing addition funnel, and a nitrogen inlet adapter was charged with 2a (297.0 g, 1.21 mol) and CH2Cl2 (3.9 L). The solution was cooled to 0 0C under N2 and EtsN (320 mL, 2.30 mol) was added via the addition funnel over a 10- min period. This was followed by methanesulfonyl chloride (115 mL, 1.49 mol) added over a 60-min period then the reaction was stirred for an additional 60-min at 0 0C. The mixture was poured into a mixture of deionized water (4.4 L) and saturated NaHCθ3 (0.78 L), the layers were separated, the aqueous layer was extracted with CH2Cl2 (2 x 2 L). All the CH2Cl2 layers were combined and washed with saturated NaHCθ3 (2 L). The CH2Cl2 was removed under vacuum at 40 0C to afford a mixture of the mesylate and chloride (342.3 g). This mixture was taken on to the next step without any purification.
Conversion of the methanesulfonate/chloride to phthalimide 3

A 5-L 4-neck round bottom flask equipped with an overhead stirrer, thermocouple, pressure-equalizing addition funnel, and a nitrogen inlet adapter was charged with the mixture of the mesylate and chloride from above (342.2 g, 1.21 mol) and DMF (2.0 L) followed by potassium phthalimide (224.9 g, 1.21 mol). The mixture was stirred at 60 0C for 1-h then at 20 0C for 18 h. The mixture was poured into ice- water, allowed to stand for 30-min and filtered. The liquors from the filtration were allowed to stand at 0 0C over the weekend and filtered again. The combined solids were dissolved in acetone (4 L) and concentrated on the rotary evaporator, this process was repeated a second time to give the phthalimide derivative 3 as a mixture oiEIZ (79/31) isomers (263.2 g, 58.1 %).
Step 2a, Method B: Purification of 3-£-[2-(l,3-dioxo-l,3-dihydroisoindol-2-yl)-l- fluoroethylidene]-piperidine-l-carboxylic acid tert-butyl ester

A 12-L 4-neck round bottom flask equipped with an overhead stirrer, thermocouple, pressure-equalizing addition funnel, and a nitrogen inlet adapter was charged with the crude phthalimide derivative 3 (263.1 g) and MeOH (2.74 L). The mixture was heated to 66 – 68 0C while water (2.1 L) was added over 20-min, the mixture was stirred at 68 0C for 5-min, then gradually cooled to 20 0C for 18-h. While the crystallization mixture was cooling it was seeded at 60 0C, 56 0C and 530C. This crystallization gave a white solid that was filtered and dried under vacuum at 50 0C to afford 3-E (118.8 g, 45.2%) as a mixture containing 94.4% E and 5.6% Z isomers (NMR analysis).
Step 3: Preparation of 2-[2-fluoro-2-(3-piperidinylidene)ethyl]-lH-isoindole-l,3)- dione (4)

A 12-L 4-neck round bottom flask equipped with an overhead stirrer, thermocouple, pressure-equalizing addition funnel, and a nitrogen inlet adapter was charged with 3-E (578.0 g, 1.544 mol) and CH2Cl2 (4.5 L). The solution was stirred at 200C under N2 and TFA (476 mL, 6.18 mol) was added via the addition funnel over a 10-min period. The mixture was gently heated to 38 0C and stirred for 3 h. The solvent was removed under vacuum to give the TFA salt of 4 (962.6 g). This material was dissolved in CH2Cl2 (4.0 L) and washed with 2.5 NNa2CO3 (4.6 L)-followed by saturated NaHCO3 (4.6 L). The organic phase was dried (MgSO4), filtered, and condensed in vacuo. The off-white solid was dried at 40 0C under vacuum (20 mm Hg) for 20 h to afford 464.3 g of the free base of 4 as slightly yellowish foamy substance. 1H NMR of 4 TFA salt (400 MHz, CDCl3): δ 1.87-1.98 (m, 2 H), 2.42-2.55 (m, 2 H), 3.38-3.50 (m, 2 H), 4.08-4.18 (br s, 2 H), 4.50 (d, J= 21.0 Hz, 2 H), 7.69-7.78 (m, 2 H), 7.79-7.87 (m, 2 H), 7.98-8.23 (br s, 1 H), 12.48 (s, 1 H). MS: 275 (MH)+, 549 (2M+H)+.
Step 4: Preparation of l-Cyclopropyl-ό^-difluoro-S-methoxy^-oxo-l^- dihydroquinoline-3-carboxylic acid difluoroborate ester (6)

A 22-L 4-neck round bottom flask equipped with an overhead stirrer, thermocouple, condenser, pressure equalizing addition funnel, and a nitrogen inlet adapter was charged with quinoline-3-carboxylic acid 5 (450.0 g, 1.524 mol), THF (5.40 L) and K2CO3 (247.2 g, 1.753 mol). This suspension was first stirred at 20 0C under N2 for 5 min, and BF3 »Et20 (259 mL, 2.04 mol) was added dropwise via the addition funnel to the stirred mixture over a 5-min period. After the addition, the mixture was heated to reflux (66 0C) for 6 h. The reaction was cooled to 10 0C, diluted with Et2O (9.0 L) and stirred for 10 min. The solid was filtered and washed with Et2O (200 mL x 2) and then dried at 50 0C under house vacuum (-160 mm Hg) for 20 h to afford 771.O g of crude difluoroborate ester 6. After this, the crude material was suspended in MeCN (8.0 L) and stirred at 20 0C for 20 min; the solid was collected by filtration. The filter cake was re-suspended and stirred in MeCN four more times (2.0 L x 4), and all filtrates were combined and concentrated at 60 0C under hi-vac (~10 mmHg). The resulting off- white solid was dried at 50 0C under house vacuum (-160 mmHg) for 20 h to afford 508.66 g (97.2% isolated yield, HPLC = 99.2% by area) of pure difluoroborate ester 6. 1H NMR of 6 (400 MHz, CD3CN): «51.17-1.28 (m, 2 H), 1.29-1.40 (m, 2 H), 4.19 (s, 3 H), 4.40-4.52 (m, 1 H), 8.16 (dd, J= 6.9, 7.0 Hz, 1 H), 9.17 (s, 1 H). MS: 344 (MH)+, 667 (2M-F)+.
Step 5: Preparation of intermediate 8


A 5-L 4-neck round bottom flask equipped with an overhead stirrer, thermocouple, condenser, pressure-equalizing addition funnel and a nitrogen inlet adapter was charged with difluoroborate ester 6 (320.0 g, 0.933 mol), DMF (1.10 L) and piperidine 4 (289.0 g, 1.053 mole). This suspension was stirred at 20 0C under N2 for 5 min, EtsN (299 mL, 2.15 mol) was added to the stirred mixture via the addition funnel over an additional 5-min period. After this addition, the mixture was heated to 60 0C and stirred for 3 h, to give crude intermediate 7. HPLC analysis (area%) indicated crude 7 is a mixture of 7 (40.5%), 8 (1.7 %), 6 (24.1%), and the rest of unknowns (33.7%). MS: 598 (MH)+. The coupled crude product 7 was carried on to the next step without isolation.
Removal of the Fluoroborate Ester The above stirred reaction mixture containing 7 was treated in the same flask with EtOH (6.80 L) and Et3N (299 mL, 2.147 mol) under N2 at 60 0C. The amber solution was heated to reflux at 72 0C for 2 h and cooled to 20 0C. The reaction mixture was poured into a rapidly stirred 22-L 4-neck round bottom flask containing a 1 : 1 (v/v) ice-water mixture (8.0 L) over a 10-min period; stirring was continued for -10 min. Cold 1 NHCl (4.0 L) was added to the solution over 20 min to adjust the pH from 9-10 to 3; stirring was continued for an additional 20 min at 0 0C. The yellow solid was isolated by filtration and dried in a filter funnel by air-suction using house vacuum (-160 mm Hg) at 20 0C for 20 h to afford 1,889.0 g of crude 8 as a damp solid (HPLC = 33.6%, area%).
Purification of Intermediate 8
To a 22-L 4-neck round bottom flask equipped with an overhead stirrer, thermocouple, pressure-equalizing addition funnel, and a nitrogen inlet adapter was charged with crude 8 (1889.0 g), MeCN (3.6 L) and EtOH (3.2 L). The suspension was heated to reflux (76 0C), while D.I. H2O (500 mL) was added over 10 min. The solution was stirred at 76 0C for 5 min, and then gradually cooled to 10 0C over 1 h; stirred for an additional hour. The yellow solid was collected by filtration, dried in a vacuum oven under house vacuum (-160 mm Hg) at 60 0C for 20 h to afford 229. Ig (45%) of 8, which was used in next step without further purification. 1H ΝMR of 8 (400 MHz, DMSO-d6): £ 1.02-1.10 (m, 2 H), 1.11-1.19 (m, 2 H), 1.67-1.79 (m, 2 H), 2.34-2.45 (m, 2 H), 3.38-3.49 (m, 2 H), 3.78 (s, 3 H), 4.10 (s, 2 H), 4.15-4.26 (m, 1 H), 4.54 (d, J= 21.0 Hz, 1 H), 7.72 (d, J= 9.1 Hz, 1 H), 7.81 (s, 4 H), 8.71 (s, I H), 14.98 (s, 1 H). MS: 550 (MH)+.
Step 6: Preparation of 7-[3-(2-amino-l-fluoro-ethylidene)-piperidin-l-yl]-l- cyclopropyl-6-fluoro-8-methoxy-4-oxo-l,4-dihydro-quinoline-3-carboxylic acid (10)

8 10 A
22-L 4-neck round bottom flask equipped with an overhead stirrer, thermocouple, condenser, pressure-equalizing addition funnel and a nitrogen inlet adapter was charged with 8 (253.6 g, 0.462 mol) and MeOH (5.10 L). This suspension was stirred at 20 0C under N2 and H2NNH2 (86.9 mL, 2.796 mol) was added over a 5-min period. The yellow suspension was heated to 65 0C and refluxed for 1 h. The reaction was cooled to 60 0C and MeCN (3.84 L) was added. The mixture was heated to reflux for 5 min, and then cooled to 20 0C in a water bath. The light-yellow solid was collected by filtration and the filter cake was washed with MeCN (150 mL x 2). The combined filtrate was concentrated at 60 0C affording 322.0 g of crude product 10. This product was recrystallized from a mixture of MeOH (1.0 L) and water (1.195 L) to give 176.6 g (91.2%) of pure product 10 as a light yellow solid.
BASE FORM
1H NMR of 10 (400 MHz, DMSO- d6): £ 1.0-1.09 (m, 2 H), 1.10-1.19 (m, 2 H), 1.66-1.78 (m, 2 H), 2.30-2.41 (m, 2 H), 3.17 (s, 2 H), 3.35 (s, 1 H), 3.36-3. 47 (m, 2 H), 3.74 (s, 3 H), 3.89 (s, 2 H), 4.13-4.22 (m, 1 H), 5.35-6.18 (br, 2 H), 7.74 (d, J= 8.9 Hz, 1 H), 8.69 (s, 1 H).
MS: 420 (MH)+.
Example 3
Preparation of7-[3-(2-Amino-l-fluoro-ethylidene)-piperidin-l-yl]-l-cyclopropyl-
6-fluoro-8-methoxy-4-oxo-l,4-dihydro-quinoline-3-carboxylic acid hydrogen chloride salt (12)

7-[3-(2-amino-l-fluoro-ethylidene)-piperidin-l-yl]-l-cyclopropyl-6-fluoro-8- methoxy-4-oxo-l,4-dihydro-quinoline-3-carboxylic acid (10) was prepared as described in Step 6 of Example 1.
A 5-L 4-neck round bottom flask equipped with an overhead stirrer, thermocouple, condenser, pressure-equalizing addition funnel, and a nitrogen inlet adapter was charged with compound 10 (176.0 g, 0.4196 mol) and EtOH (2.40 L). The suspension was stirred under N2 and cooled to 10 0C with an ice/water bath. A solution of HCl in EtOH (1.25 M, 350 mL) was added via the addition funnel over a 20-min period. After the addition, the reaction was stirred at 10 0C for 5 min. The water bath was replaced with a heating mantle and the solution was heated to 76 0C and stirred for 5 min. The heating mantle was replaced with the water bath, the solution was cooled to 0 0C over 1 h and stirred at this temperature for an additional 1 h. The solid was collected by filtration, washed with ice-cold EtOH (100 mL x 2) and dried at 60 0C under vacuum (~4 mmHg) for 60 h. There was obtained 88.9 g (82%) of HCl salt 12 as an off-white to very light-yellow solid.
HCl SALT
1H NMR of HCl salt 12 (400 MHz, CD3CO2D): £ 1.10-1.19 (m, 2 H), 1.29-1.38 (m, 2 H), 1.81-1.93 (m, 2 H), 2.51-2.60 (m, 2 H), 3.48- 3.60 (m, 2 H), 3.86 (s, 3 H), 4.08 (s, 2 H), 4.18 (s, 1 H), 4.19-4.30 (m, 2 H), 7.92 (d, J= 8.6 Hz, 1 H), 8.98 (s, 1 H) 11.65 (s, 1 H).
MS: 420 (MH)+

………………………………………
http://www.google.com/patents/WO2013045599A1?cl=en
Experiment 6

(VI) compound (1 ) .HCI
Compound (1) .HCI salt from Compound (Via) :
9.8 ml (90.6 mmol) of 1-chloroethyl chloro formate are added slowly to a solution of 30 g (82.3 mmol) of compound (E)-(Va) in 165 ml of toluene kept at 0°C. The reaction mixture is stirred 1 hour at room temperature than 1 hour at 80°C and filtered. 24 ml of ethanol and 15.35 ml (90.6 mmol) of 6M HCI solution in isopropanol are added to the filtrate and the resulting mixture is refluxed for 4 hours then cooled to 0°C. The precipitate is filtered, washed with 16 ml of acetone and 16 ml of toluene and dried under vacuum to give 21.94 g of compound (1) . HCI salt. Yield: 86%>.
NMR and MS data are identical to those of the literature.
http://www.ama-assn.org/resources/doc/usan/avarofloxacin.pdf
November 28, 2012. N12/130. STATEMENT ON A NONPROPRIETARY NAME ADOPTED BY THE USAN COUNCIL. USAN (ZZ-145). AVAROFLOXACIN.
http://www.ama-assn.org/resources/doc/usan/avarofloxacin-hydrochloride.pdf

| WO2006101603A1 | Feb 2, 2006 | Sep 28, 2006 | Janssen Pharmaceutica Nv | 7-amino alkylidenyl-heterocyclic quinolones and naphthyridones |
| WO2008005670A2 | Jun 14, 2007 | Jan 10, 2008 | Janssen Pharmaceutica Nv | One-pot condensation reduction methods for preparing substituted allylic alcohols |
| WO2010056633A2 | Nov 10, 2009 | May 20, 2010 | Janssen Pharmaceutica Nv | 7-amino alkylidenyl-heterocyclic quinolones and naphthyridones |