


| EudraCT | Title | Phase | Status | Date |
|---|---|---|---|---|
| 2019-003807-37 | A Double-Blind, Randomized, Placebo-Controlled Study to Evaluate the Efficacy and Safety of Odevixibat (A4250) in Children with Biliary Atresia Who Have Undergone a Kasai Hepatoportoenterostomy (BOLD) | Phase 3 | Ongoing | 2020-07-29 |
| 2015-001157-32 | An Exploratory Phase II Study to demonstrate the Safety and Efficacy of A4250 | Phase 2 | Completed | 2015-05-13 |
| 2014-004070-42 | An Exploratory, Phase IIa Cross-Over Study to Demonstrate the Efficacy | Phase 2 | Ongoing | 2014-12-09 |
| 2017-002325-38 | An Open-label Extension Study to Evaluate Long-term Efficacy and Safety of A4250 in Children with Progressive Familial Intrahepatic Cholestasis Types 1 and 2 (PEDFIC 2) | Phase 3 | Ongoing | |
| 2017-002338-21 | A Double-Blind, Randomized, Placebo-Controlled, Phase 3 Study to Demonstrate Efficacy and Safety of A4250 in Children with Progressive Familial Intrahepatic Cholestasis Types 1 and 2 (PEDFIC 1) | Phase 3 | Ongoing, Completed |
Home » Posts tagged 'EU 2021'
Tag Archives: EU 2021
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Regdanvimab

| (Heavy chain) QITLKESGPT LVKPTQTLTL TCSFSGFSLS TSGVGVGWIR QPPGKALEWL ALIDWDDNKY HTTSLKTRLT ISKDTSKNQV VLTMTNMDPV DTATYYCARI PGFLRYRNRY YYYGMDVWGQ GTTVTVSSAS TKGPSVFPLA PSSKSTSGGT AALGCLVKDY FPEPVTVSWN SGALTSGVHT FPAVLQSSGL YSLSSVVTVP SSSLGTQTYI CNVNHKPSNT KVDKRVEPKS CDKTHTCPPC PAPELLGGPS VFLFPPKPKD TLMISRTPEV TCVVVDVSHE DPEVKFNWYV DGVEVHNAKT KPREEQYNST YRVVSVLTVL HQDWLNGKEY KCKVSNKALP APIEKTISKA KGQPREPQVY TLPPSRDELT KNQVSLTCLV KGFYPSDIAV EWESNGQPEN NYKTTPPVLD SDGSFFLYSK LTVDKSRWQQ GNVFSCSVMH EALHNHYTQK SLSLSPGK (Light chain) ELVLTQPPSV SAAPGQKVTI SCSGSSSNIG NNYVSWYQQL PGTAPKLLIY DNNKRPSGIP DRFSGSKSGT SATLGITGLQ TGDEADYYCG TWDSSLSAGV FGGGTELTVL GQPKAAPSVT LFPPSSEELQ ANKATLVCLI SDFYPGAVTV AWKADGSPVK AGVETTKPSK QSNNKYAASS YLSLTPEQWK SHRSYSCQVT HEGSTVEKTV APTECS (Disulfide bridge: H22-H97, H155-H211, H231-L215, H237-H’237, H240-H’240, H272-H332, H378-H436, H’22-H’97, H’155-H’211, H’231-L’215, H’272-H’332, H’378-H’436, L22-L89, L138-L197, L’22-L’89, L’138-L’197) |
>Regdanvimab light chain: ELVLTQPPSVSAAPGQKVTISCSGSSSNIGNNYVSWYQQLPGTAPKLLIYDNNKRPSGIP DRFSGSKSGTSATLGITGLQTGDEADYYCGTWDSSLSAGVFGGGTELTVLGQPKAAPSVT LFPPSSEELQANKATLVCLISDFYPGAVTVAWKADGSPVKAGVETTKPSKQSNNKYAASS YLSLTPEQWKSHRSYSCQVTHEGSTVEKTVAPTECS
>Regdanvimab heavy chain: QITLKESGPTLVKPTQTLTLTCSFSGFSLSTSGVGVGWIRQPPGKALEWLALIDWDDNKY HTTSLKTRLTISKDTSKNQVVLTMTNMDPVDTATYYCARIPGFLRYRNRYYYYGMDVWGQ GTTVTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHT FPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKRVEPKSCDKTHTCPPC PAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKT KPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVY TLPPSRDELTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSK LTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK
Regdanvimab
レグダンビマブ;
EMA APPROVED, 2021/11/12, Regkirona
Treatment of adults with coronavirus disease 2019 (COVID-19)
MONOCLONAL ANTIBODY, ANTI VIRAL, PEPTIDE
CAS: 2444308-95-4, CT-P59
Regdanvimab, sold under the brand name Regkirona, is a human monoclonal antibody used for the treatment of COVID-19.[1] The antibody is directed against the spike protein of SARS-CoV-2. It is developed by Celltrion.[2][3] The medicine is given by infusion (drip) into a vein.[1][4]
The most common side effects include infusion-related reactions, including allergic reactions and anaphylaxis.[1]
Regdanvimab was approved for medical use in the European Union in November 2021.[1]
Regdanvimab is a monoclonal antibody targeted against the SARS-CoV-2 spike protein used to treat patients with COVID-19 who are at risk of progressing to severe COVID-19.
Regdanvimab (CT-P59) is a recombinant human IgG1 monoclonal antibody directed at the receptor binding domain (RBD) of the SARS-CoV-2 spike protein.4 It blocks the interaction between viral spike proteins and angiotensin-converting enzyme 2 (ACE2) that allows for viral entry into the cell, thereby inhibiting the virus’ ability to replicate. Trials investigating the use of regdanvimab as a therapeutic candidate for the treatment of COVID-19 began in mid-2020.1,3 It received its first full approval in South Korea in September 2021,3 followed by the EU in November 2021.5

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Synthesis Reference
Kim C, Ryu DK, Lee J, Kim YI, Seo JM, Kim YG, Jeong JH, Kim M, Kim JI, Kim P, Bae JS, Shim EY, Lee MS, Kim MS, Noh H, Park GS, Park JS, Son D, An Y, Lee JN, Kwon KS, Lee JY, Lee H, Yang JS, Kim KC, Kim SS, Woo HM, Kim JW, Park MS, Yu KM, Kim SM, Kim EH, Park SJ, Jeong ST, Yu CH, Song Y, Gu SH, Oh H, Koo BS, Hong JJ, Ryu CM, Park WB, Oh MD, Choi YK, Lee SY: A therapeutic neutralizing antibody targeting receptor binding domain of SARS-CoV-2 spike protein. Nat Commun. 2021 Jan 12;12(1):288. doi: 10.1038/s41467-020-20602-5.
Celltrion’s Monoclonal Antibody Treatment regdanvimab, Approved by the European Commission for the Treatment of COVID-19
- The European Commission (EC) granted marketing authorisation for Celltrion’s regdanvimab following positive opinion by the European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) last week (11/11/2021)
- Celltrion continues to discuss supply agreements with regulatory agencies and contractors in more than 30 countries in Europe, Asia and LATAM to accelerate global access to regdanvimab
- The use of regdanvimab across the Republic of Korea is rapidly increasing to address the ongoing outbreaks
November 14, 2021 08:04 PM Eastern Standard Time
INCHEON, South Korea–(BUSINESS WIRE)–Celltrion Group announced today that the European Commission (EC) has approved Regkirona (regdanvimab, CT-P59), one of the first monoclonal antibody treatments granted marketing authorisation from the European Medicines Agency (EMA). The EC granted marketing authorisation for adults with COVID-19 who do not require supplemental oxygen and who are at increased risk of progressing to severe COVID-19. The decision from the EC follows a positive opinion by the European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) on November 11th, 2021.1
“Today’s achievement, coupled with CHMP positive opinion for regdanvimab, underscores our ongoing commitment to addressing the world’s greatest health challenges,” said Dr. HoUng Kim, Ph.D., Head of Medical and Marketing Division at Celltrion Healthcare. “Typically, the recommendations from the CHMP are passed on to the EC for rapid legally binding decisions within a month or two, however, given the unprecedented times, we have received the EC approval within a day. As part of our global efforts to accelerate access, we have been communicating with the governments and contractors in 30 countries in Europe, Asia and LATAM. We will continue working with all key stakeholders to ensure COVID-19 patients around the world have access to safe and effective treatments.”
Monoclonal antibodies are proteins designed to attach to a specific target, in this case the spike protein of SARS-CoV-2, which works to block the path the virus uses to enter human cells. The EC approval is based on the global Phase III clinical trial involving more than 1,315 people to evaluate the efficacy and safety of regdanvimab in 13 countries including the U.S., Spain, and Romania. Data showed regdanvimab significantly reduced the risk of COVID-19 related hospitalisation or death by 72% for patients at high-risk of progressing to severe COVID-19.
Emergency use authorisations are currently in place in Indonesia and Brazil, and the monoclonal antibody treatment is fully approved in the Republic of Korea. In the U.S., regdanvimab has not yet been approved by the Food and Drug Administration (FDA), but the company is in discussion with the FDA to submit applications for an Emergency Use Authorisation (EUA).
As of November 12th, 2021, more than 22,587 people have been treated with regdanvimab in 129 hospitals in the Republic of Korea.
Notes to Editors:
About Celltrion Healthcare
Celltrion Healthcare is committed to delivering innovative and affordable medications to promote patients’ access to advanced therapies. Its products are manufactured at state-of-the-art mammalian cell culture facilities, designed and built to comply with the US FDA cGMP and the EU GMP guidelines. Celltrion Healthcare endeavours to offer high-quality cost-effective solutions through an extensive global network that spans more than 110 different countries. For more information please visit: https://www.celltrionhealthcare.com/en-us.
About regdanvimab (CT-P59)
CT-P59 was identified as a potential treatment for COVID-19 through screening of antibody candidates and selecting those that showed the highest potency in neutralising the SARS-CoV-2 virus. In vitro and in vivo pre- clinical studies showed that CT-P59 strongly binds to SARS-CoV-2 RBD and significantly neutralise the wild type and mutant variants of concern. In in vivo models, CT-P59 effectively reduced the viral load of SARS-CoV-2 and inflammation in lung. Results from the global Phase I and Phase II/III clinical trials of CT-P59 demonstrated a promising safety, tolerability, antiviral effect and efficacy profile in patients with mild-to-moderate symptoms of COVID-19.2 Celltrion also has recently commenced the development of a neutralising antibody cocktail with CT-P59 against new emerging variants of SARS-CoV-2.
Medical uses
In the European Union, regdanvimab is indicated for the treatment of adults with COVID-19 who do not require supplemental oxygen and who are at increased risk of progressing to severe COVID-19.[1]
Society and culture
Names
Regdanvimab is the proposed international nonproprietary name (pINN).[5]
Legal status
In March 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) started a rolling review of data on regdanvimab.[6][7] In October 2021, the EMA started evaluating an application for marketing authorization for the monoclonal antibody regdanvimab (Regkirona) to treat adults with COVID-19 who do not require supplemental oxygen therapy and who are at increased risk of progressing to severe COVID 19.[8] The applicant is Celltrion Healthcare Hungary Kft.[8] The European Medicines Agency (EMA) concluded that regdanvimab can be used for the treatment of confirmed COVID-19 in adults who do not require supplemental oxygen therapy and who are at high risk of progressing to severe COVID-19.[4]
In November 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) recommended granting a marketing authorization in the European Union for regdanvimab (Regkirona) for the treatment of COVID-19.[9][10] The company that applied for authorization of Regkirona is Celltrion Healthcare Hungary Kft.[10] Regdanvimab was approved for medical use in the European Union in November 2021.[1]
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | Spike protein of SARS-CoV-2 |
| Clinical data | |
| Trade names | Regkirona |
| Other names | CT-P59 |
| License data | EU EMA: by INN |
| Routes of administration | Intravenous infusion |
| ATC code | None |
| Legal status | |
| Legal status | EU: Rx-only [1] |
| Identifiers | |
| CAS Number | 2444308-95-4 |
| DrugBank | DB16405 |
| UNII | I0BGE6P6I6 |
| KEGG | D12241 |
- Tuccori M, Ferraro S, Convertino I, Cappello E, Valdiserra G, Blandizzi C, Maggi F, Focosi D: Anti-SARS-CoV-2 neutralizing monoclonal antibodies: clinical pipeline. MAbs. 2020 Jan-Dec;12(1):1854149. doi: 10.1080/19420862.2020.1854149. [Article]
- Kim C, Ryu DK, Lee J, Kim YI, Seo JM, Kim YG, Jeong JH, Kim M, Kim JI, Kim P, Bae JS, Shim EY, Lee MS, Kim MS, Noh H, Park GS, Park JS, Son D, An Y, Lee JN, Kwon KS, Lee JY, Lee H, Yang JS, Kim KC, Kim SS, Woo HM, Kim JW, Park MS, Yu KM, Kim SM, Kim EH, Park SJ, Jeong ST, Yu CH, Song Y, Gu SH, Oh H, Koo BS, Hong JJ, Ryu CM, Park WB, Oh MD, Choi YK, Lee SY: A therapeutic neutralizing antibody targeting receptor binding domain of SARS-CoV-2 spike protein. Nat Commun. 2021 Jan 12;12(1):288. doi: 10.1038/s41467-020-20602-5. [Article]
- Syed YY: Regdanvimab: First Approval. Drugs. 2021 Nov 1. pii: 10.1007/s40265-021-01626-7. doi: 10.1007/s40265-021-01626-7. [Article]
- EMA Summary of Product Characteristics: Regkirona (regdanvimab) concentrate for solution for intravenous infusion [Link]
- EMA COVID-19 News: EMA recommends authorisation of two monoclonal antibody medicines [Link]
- EMA CHMP Assessment Report: Celltrion use of regdanvimab for the treatment of COVID-19 [Link]
- Protein Data Bank: Crystal Structure of COVID-19 virus spike receptor-binding domain complexed with a neutralizing antibody CT-P59 [Link]
References
- ^ Jump up to:a b c d e f g “Regkirona EPAR”. European Medicines Agency. Retrieved 12 November 2021. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Celltrion Develops Tailored Neutralising Antibody Cocktail Treatment with CT-P59 to Tackle COVID-19 Variant Spread Using Its Antibody Development Platform” (Press release). Celltrion. 11 February 2021. Retrieved 4 March 2021 – via Business Wire.
- ^ “Celltrion Group announces positive top-line efficacy and safety data from global Phase II/III clinical trial of COVID-19 treatment candidate CT-P59” (Press release). Celltrion. 13 January 2021. Retrieved 4 March 2021 – via Business Wire.
- ^ Jump up to:a b “EMA issues advice on use of regdanvimab for treating COVID-19”. European Medicines Agency. 26 March 2021. Retrieved 15 October 2021.
- ^ World Health Organization (2020). “International Nonproprietary Names for Pharmaceutical Substances (INN). Proposed INN: List 124 – COVID-19 (special edition)” (PDF). WHO Drug Information. 34 (3): 660–1.
- ^ “EMA starts rolling review of Celltrion antibody regdanvimab for COVID-19” (Press release). European Medicines Agency (EMA). 24 February 2021. Retrieved 4 March 2021.
- ^ “EMA review of regdanvimab for COVID-19 to support national decisions on early use” (Press release). European Medicines Agency (EMA). 2 March 2021. Retrieved 4 March 2021.
- ^ Jump up to:a b “EMA receives application for marketing authorisation Regkirona (regdanvimab) treating patients with COVID-19”. European Medicines Agency. 4 October 2021. Retrieved 15 October 2021.
- ^ “Regkirona: Pending EC decision”. European Medicines Agency. 11 November 2021. Retrieved 11 November 2021.
- ^ Jump up to:a b “COVID-19: EMA recommends authorisation of two monoclonal antibody medicines”. European Medicines Agency (EMA) (Press release). 11 November 2021. Retrieved 11 November 2021.
Further reading
- Kim C, Ryu DK, Lee J, Kim YI, Seo JM, Kim YG, et al. (January 2021). “A therapeutic neutralizing antibody targeting receptor binding domain of SARS-CoV-2 spike protein”. Nature Communications. 12 (1): 288. doi:10.1038/s41467-020-20602-5. PMC 7803729. PMID 33436577.
External links
- “Regdanvimab”. Drug Information Portal. U.S. National Library of Medicine.
///////////Regdanvimab, Regkirona, MONOCLONAL ANTIBODY, ANTI VIRAL, EU 2021, APPROVALS 2021, EMA 2021, COVID 19, CORONAVIRUS, PEPTIDE, レグダンビマブ , CT-P59, CT P59

NEW DRUG APPROVALS
ONE TIME
$10.00
Diroximel fumarate

Diroximel fumarate
ジロキシメルフマル酸エステル;
| Formula |
C11H13NO6
|
|---|---|
| CAS | 1577222-14-0 |
| Mol weight |
255.224
|
2021/11/15 EMA APPROVED, VUMERITY
Treatment of multiple sclerosis
10356
1577222-14-0 [RN]
2-(2,5-Dioxo-1-pyrrolidinyl)ethyl methyl (2E)-2-butenedioate
K0N0Z40J3W
RDC-5108
дироксимела фумарат [Russian] [INN]
ديروكسيميل فومارات [Arabic] [INN]
富马地罗昔美 [Chinese] [INN]
Diroximel fumarate, sold under the brand name Vumerity, is a medication used for the treatment of relapsing forms of multiple sclerosis (MS).[1][3][4]
Diroximel fumarate was approved for medical use in the United States in October 2019,[5] and in the European Union in November 2021.[2]
History
This drug was formulated by Alkermes in collaboration with Biogen.[6]
Society and culture
Legal status
Diroximel fumarate was approved for medical use in the United States in October 2019.[5]
On 16 September 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Vumerity, intended for the treatment of adults with relapsing remitting multiple sclerosis.[7] The applicant for this medicinal product is Biogen Netherlands B.V.[7] It was approved for medical use in the European Union in November 2021.[2]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
PATENT
US 8669281
https://patents.google.com/patent/US8669281B1/en
PATENT
WO 2014152494
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014152494
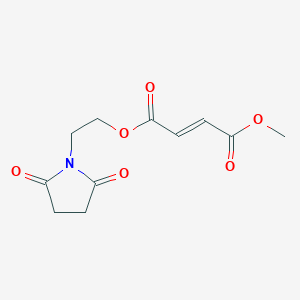
2-(2,5-dioxopyrrolidin-1-yl)ethyl methyl fumarate (14)
2-(2,5-dioxopyrrolidin-1-yl)ethyl methyl fumarate 14 was synthesized following general procedure 1 (1.03 g, 35 %).
1H NMR (400 MHz, DMSO): δ 6.81 (2H, dd, J = 15.8 Hz); 4.36 (2H, t, J = 5.3 Hz); 3.84 (2H, t, J = 5.1 Hz); 3.80 (3H, s); 2.73 (4H, s). [M+H]+ = 256.07.
General Procedure 1
To a mixture of monomethyl fumarate (MMF) (1.0 equivalent) and HBTU (1.5 equivalents) in DMF (25 ml per g of MMF) was added Hünigs base (2.0 equivalents). The dark brown solution was stirred for 10 minutes, where turned into a brown suspension, before addition of the alcohol (1.0 – 1.5 equivalents). The reaction was stirred for 18 hours at room temperature. Water was added and the product extracted into ethyl acetate three times. The combined organic layers were washed with water three times, dried with magnesium sulphate, filtered and concentrated in vacuo at 45 ºC to give the crude product. The crude product was purified by silica chromatography and in some cases further purified by trituration with diethyl ether to give the clean desired ester product. All alcohols were either commercially available or made following known literature procedures.
As an alternative to HBTU (N,N,N’,N’-Tetramethyl-O-(1H-benzotriazol-1 -yl)uronium hexafluorophosphate), any one of the following coupling reagents can be used: EDCI/HOBt (N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride/hydroxybenzotriazole hydrate); COMU ((1-cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium hexafluorophosphate); TBTU (O-(benzotriazol-1 -yl)-N,N,N’,N’-tetramethyluronium tetrafluoroborate); TATU (O-(7-azabenzotriazole-1-yl)-1,1 ,3,3-tetramethyluronium tetrafluoroborate); Oxyma (ethyl (hydroxyimino)cyanoacetate); PyBOP ((benzotriazol-1 -yloxy)tripyrrolidinophosphonium hexafluorophosphate); HOTT (5-(1-oxido-2-pyridyl)-N,N,N’,N’-tetramethylthiuronium hexafluorophosphate); FDPP (pentafluorophenyl diphenylphosphinate); T3P (propylphosphonic anhydride); DMTMM (4-(4,6-dimethoxy-1,3,5-triazin-2-y1)-4-methylmorpholinium tetrafluoroborate); PyOxim ([ethyl
cyano(hydroxyimino)acetato-O2]tri-1-pyrrolidinylphosphonium hexafluorophosphate); TSTU (N,N,N’,N’-tetramethyl-O-(N-succinimidyl)uronium tetrafluoroborate); TDBTU (O-(3,4-dihydro-4-oxo-1,2,3-benzotriazin-3-yl)-N,N,N’,N’-tetramethyluronium tetrafluoroborate); TPTU (O-(2-oxo-1(2H)pyridyl)-N,N,N’,N’-tetramethyluronium tetrafluoroborate); TOTU (O-[(ethoxycarbonyl)cyanomethylenamino]-N,N,N’,N’-tetramethyluronium tetrafluoroborate); IIDQ (isobutyl 1,2-dihydro-2-isobutoxy- 1-quinolinecarboxylate); or PyCIU
(chlorodipyrrolidinocarbenium hexafluorophosphate),
As an alternative to Hünig’s base (diisopropylethylamine), any one of the following amine bases can be used: triethylamine; tributylamine; triphenylamine; pyridine; lutidine (2,6-dimethylpyridine); collidine (2,4,6-trimethylpyridine); imidazole; DMAP (4-(dimethylamino)pyridine); DABCO (1 ,4-diazabicyclo[2.2.2]octane); DBU (1 ,8-
diazabicyclo[5.4.0]undec-7-ene); DBN (1,5-diazabicyclo[4.3.0]non-5-ene); or proton sponge® (N,N,N’,N’-tetramethyl-1 ,8-naphthalenediamine).

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
PATENT
WO 2016124960
PATENT
WO 2017108960
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017108960
Example 3b: Synthesis of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester
Procedure A:
Distilled 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (3 g; 20.96 mmol) and maleic acid anhydride (2.26 g; 23.1 mmol) in toluene (10 mL) were heated to 60°C under stirring for 29 hours. The temperature was raised to 80°C and heated for another 19 hours. Acetyl chloride (0.3 mL; 4.2 mmol) was added and heating (80°C) was continued for 24 hours. The reaction mixture was cooled to RT. The biphasic system was separated, the upper layer was discarded. The lower layer (viscous oil) crystallized. The crystallized compound was suspended in acetone (50 mL) and stirred for 15 minutes before being filtrated off. The product was dried at 50°C for 5 hours and 8 mbar to yield the 1st crop (1.65 g). The mother liquor was evaporated and the obtained oil/solid was suspended in acetone (5 mL) and stirred overnight at RT. The product was filtrated off and dried at 50°C for 5 hours and 8 mbar to yield the 2nd crop (1.41 g). The mother liquor was evaporated and the obtained oil/solid was suspended in a mixture of diethylether/acetone (5 mL/1 mL) and stirred overnight at RT. The product was filtrated off and dried at 8mbar/50°C for 3 hours (3rd crop, 0.37 g).<a name=”
Yield: 3.43 g (68% of theory)
Purity: 1st crop 96.8 area%; 2nd crop 96.0 area-%; 3rd crop 85.4 area-% (HPLC/UV, method A, λ=200nm; tr: 3.8 min.)
1H NMR (400 MHz, DMSO-d6) δ ppm: 2.61 (s, 4 H) 3.66 (t, J=5.47 Hz, 2 H) 4.23 (t, J=5.47 Hz, 2 H) 6.51 – 6.72 (m, 2 H) 6.60 (s, 1 H) 6.63 (s, 1 H) 13.21 (br s, 1 H)
Procedure B:
Reaction performed in a reactor (Mettler Toledo, Optimax):
Distilled 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (20 g; 0.14 mol) and maleic acid anhydride (15 g; 0.15 mol) in toluene (70 mL) were heated to 80°C under stirring (150 rpm) for 29 hours. Acetyl chloride (2 mL; 0.03 mol) was added and heating (80°C) was continued overnight. Stirring speed was raised to 200 rpm) after 15.5 hours (at 80°C) (product precipitated upon raising stirring speed. The reaction mixture was cooled to 20°C within 1 hour, directly after highering stirring speed. The reaction mixture was stirred for 4 hours, before being filtrated off. The filtrated precipitate was washed with toluene (30 mL) and then with heptane (70 mL), the product was dried at 60°C and 18 mbar. The crude product (26.26 g) with -90% purity was suspended in a mixture of acetone (30 mL)/heptane (30 mL) and stirred at RT for 2 days. The product was filtrated off, washed with heptane (30 mL) and dried at 50°C and 7 mbar.
Yield: 24.12 g (72% of theory)
Purity: 97.4 area-% at 200 nm
Procedure C:
a) Ethylene carbonate (8.89 g; 0.1 mol), succinimide (10 g; 0.1 mol) and sodium carbonate (0.53 g, 5 mmol) were heated to 100°C, the temperature was hold overnight. The product was cooled down yielding a brownish solid (13.73 g) which was grinded in a mortar.
b) 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (10 g, 69.9 mmol) from sequence a) and maleic acid anhydride (6.85 g; 69.9 mmol) in toluene (33 mL) were heated to 80°C under stirring for 23 hours. Acetyl chloride (0.5 mL; 7 mmol) was added and heating<a name=”
(80°C) was continued overnight. Heating was stopped and after stirring for another 2 hours the product was filtered off. The product was dried for 2 hours at 60°C and 8 mbar, yielding 15.82 g of crude product.
purity: 63 area-% at 200nm; 80 area-% at 220 nm
Procedure D
a) Ethylene carbonate (44.43 g; 0.5 mol), succinimide (50 g; 0.5 mol) and sodium carbonate (2.67 g; 25 mmol) were heated to 100°C. The reaction mixture was stirred at 100°C for overnight. The mixture was cooled to RT, yielding 72.4 g of the raw product.
40 g of the raw product were suspended in ethylacetate (40 mL) and heated to reflux for 30 minutes. The turbid mixture was cooled to RT and left stirring O/N. The product was filtrated off and dried under vacuum at RT to yield 29.19 g.
b) 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (10 g; 69.9 mmol) from sequence a) and maleic acid anhydride (6.85 g; 69.9 mmol) in toluene (30 mL) were heated to 80°C under stirring. Acetyl chloride (0.5 mL; 7 mmol) was added after 19 hours and heating (80°C) was continued overnight. Heating was stopped and stirring was continued for 2 days. The product was filtrated off and dried at 23 mbar and 60°C.
purity: 82 area% at 200 nm; 91 area-% at 220 nm
Procedure E:
a) Succinimide (500 g; 5.0 mol), ethylene carbonate (444.34 g; 5.0 mol) and sodium carbonate (26.74 g; 0.25 mol) were mixed and slowly heated to 130°C under stirring for 7 hours. The product was distilled via vacuum distillation to yield the product as colourless substance (628.14 g; 87% of theory)
b) The distilled 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (150 g; 1.05 mol) from sequence a) and maleic acid anhydride (102.76 g; 1.05 mol) in toluene (350 mL) were heated to 80°C under stirring for 23 hours. Acetyl chloride (7 mL; 0.01 mol) was added and heating (80°C) was continued. After 6 hours, the reaction mixture was cooled to 20°C within 30 minutes. The product was filtrated off and washed with toluene (200 mL), yielding 221.8 g of a white crystalline product (crude product).
purity: 91 area% at 200 nm; 92 area-% at 220 nm<a name=”
Procedure F:
a) Ethylene carbonate (9.78 g; 0.11 mol), succinimide (10 g; 0.10 mol) and triethylamine (0.7 mL; 5mmol) were heated to 98°C. The reaction mixture was stirred at this temperature overnight. The mixture was cooled to RT, yielding a colourless liquid, which crystallizes upon standing at RT to a colorless solid (14.89 g).
b) The crude 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione from sequence a) (5 g; 35 mmol) and maleic acid anhydride (3.43 g; 35 mmol) in toluene (25 mL) were heated to 80°C under stirring for 24 hours. Acetyl chloride (0.25 mL; 3.5 mmol) was added and heating (80°C) was continued for ~4 hours. The reaction mixture was cooled to RT. The product was filtrated off washed with toluene and dried at 50°C and 8 mbar for 3 hours. Yield: 6.52 g (77%)purity: 93 area% at 200 nm; 94 area-% at 220 nm
Procedure F’
Ethylene carbonate (161.50 g, 1.834 mol) was melted at 50°C in a reactor, succinimide (173.07 g, 1.747 mol) and Et3N (12.2 mL, 87.350 mmol) were added and the reaction mixture was warmed up to 90°C and stirred for 24h. Reaction mixture was cooled to 50°C, 500 mL of acetone was added, followed by addition of maleic anhydride (164.19 g, 1.674 mol) and Et3N (10.15 mL, 72.772 mmol). Reaction mixture was stirred at 50-55°C for 4h, cooled to 0°C and stirred for 20h. Resulting white suspension was filtered off and solid was washed with cold acetone (2×50 mL) and dried for 6h at 50°C and 30 mbar to afford crystalline (Z)-4-(2-(2,5-dioxopyrrolidin-1-yl)ethoxy)-4-oxobut-2-enoic acid.
Yield: 274 g (65%)
Purity: 97.23 area % at 200 nm
Procedure F”
(Z)-4-(2-(2,5-dioxopyrrolidin-1-yl)ethoxy)-4-oxobut-2-enoic acid (250 g, 1.036 mol) was suspended in acetone (500 mL) in 1-L reactor, acetyl chloride (5.53 mL, 77.736 mmol) was added drop wise at 20-25°C and reaction mixture was warmed up to 50-55°C and stirred for 20h. Reaction mixture was cooled to 0°C and stirred for 3h. Resulting white suspension was filtered off and solid was washed with cold acetone <a name=”(2×50 mL) and dried for 6h at 50°C and 30 mbar to afford crystalline (E)-4-(2-(2,5-dioxopyrrolidin-1-yl)ethoxy)-4-oxobut-2-enoic acid (Formula II).
Yield: 231.3 g (92.5%)
Purity: 99.47 area % at 200 nm
Summary:
Procedure B and E, using distilled 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione, showed purities of -90-91 area-% of the crude product, ongoing crystallization of the target compound could improve the purity to -97% also shown in procedure A. Distillation of 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione needs harsh conditions (Ex. 3a; procedure A). Using the crude 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione, produced with Na2CO3 lead to low product purities of 63 area-% (procedure C).
Crystallization of 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (procedure D) lead to product purities comparable to procedure A, B and E with distilled 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione, but crystallization is compounded by a significant product loss of – 25%.
The raw 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione could be used without any disadvantageous impact on product quality by substituting Na2CO3 with triethylamine as shown in procedure F with a purity of 93 area-%.
Procedure G
Two experiments were performed in parallel:
Each with 1 g (7 mmol) 1-(2-hydroxy-ethyl)-pyrrolidine-2,5-dione and 0.75 g (7.7 mmol) maleic acid anhydride in 6 mL acetonitrile in screw capped vials. To one of the reaction mixtures was given 0.1 mL triethylamine. Both mixtures were stirred at RT. Samples were taken and investigated by NMR (in DMSO).
product formation after 1 hour (quantified by NMR):
mixture without triethylamine: 0%
mixture with triethylamine: 55%<a name=”
product formation after 2 hours:
mixture without triethylamine: 0%
mixture with triethylamine: 71 %
Procedure H (isolation of cis intermediate):
1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (5 g; 35 mmol) and maleic acid anhydride (3.43 g; 35 mmol) in toluene (30 mL) were heated to 80°C under stirring for -24 hours. The reaction was cooled to RT, first a biphasic layer was observed, then the product solidified (sticking to glass wall and stirrer). The product was filtrated off after 2.5 hours of stirring, washed with toluene (50 mL) and dried under vacuum. The dried product was milled and suspended again in toluene (60 mL) at RT, after 30 minutes the product was filtrated off and dried under atmospheric conditions to yield 7.24 g of the cis intermediate (86% of theory). The intermediate product was suspended in toluene (30 mL) and heated to 80°C, acetyl chloride (0.25 mL; 3.5 mmol) was added and heating (80°C) was continued for 5 hours. The reaction mixture was cooled to RT and stirred for 2 hours. The product was filtrated off, washed with toluene (30 mL) and dried at 50°C and 8 mbar O/N.
purity: 95.6 area-% at 200nm; (0.2% of Impurity I)
Procedure H (without isolation of cis intermediate):
1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (5 g; 35 mmol) and maleic acid anhydride (3.43 g; 35 mmol) in toluene (30 mL) were heated to 80°C under stirring for 24 hours. Acetyl chloride (0.25 mL; 3.5 mmol) was added and heating (80°C) was continued for ~4 hours. The reaction mixture was cooled to RT. The product was filtrated off washed with toluene (30 mL) and dried at 50°C and 8 mbar for 3 hours.
purity: 93.2 area-% at 200nm; (1.3% of Impurity I)
Procedure I (scale-up without cis isolation)
Maleic acid (959.09 g; 9.8 mol) was added to a reactor under stirring, which was already loaded with toluene (7 L), then 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione<a name=”
(1400 g; 9.8 mol) was added. Then the mixture was heated to 76°C within ~1 h (up to ~50°C the mixture is a suspension with the tendency of conglomeration of solids, very difficult consistency) at 50°C a turbid solution resulted. Stirring was continued at 80°C for 2 days. Acetyl chloride (138 mL; 1.96 mol) was added under enhanced stirring at 80°C. After -5-10 minutes a crystalline precipitate was formed, which transformed into a pasty/syrupy solid, sticking to reactor walls (difficult handling). Heating was continued overnight (reaction completed after 5 hours as IPC showed). Mixture is still an emulsion, seeding was added and the product precipitated. Stirring at 80°C was continued for ~2 hours then the mixture was cooled to RT. The solid was filtrated off and dried at 50°C and 12 mbar overnight to yield 1818.74 g of the product.
purity: 96.34 area-% at 218 nm; (1.5% of Impurity I)
Procedure J:
2L flask (reaction volume ~1 L): Succinimide (460 g; 4.6 mol), ethylene carbonate (450 g; 5.1 mol) and triethylamine (32 mL; 0.23 mol) were heated to 85°C under stirring overnight. Temperature was raised to 95°-97C and heating was continued O/N. The mixture was cooled to 50°C. Acetonitrile (1600 mL) was charged into a 10 L reactor. To the reaction mixture was added acetonitrile (1000 mL) at 50°C and the solution was transferred to the reactor (reactor T ~22°C), triethylamine (35 mL) was added, then maleic acid anhydride (500.81 g; 5.1 mol). The mixture was heated to 55°C for 5.5 hours. A part of the solvent was distilled off (~1200 mL). Then toluene (1200 mL) was added. The mixture was heated to 90°C. The mixture was cooled to 50°C. At 60°C (clear solution), seeding was added ~300 mg, after -3 minutes a suspension resulted. The mixture was further cooled down to 20°C within 10 hours and kept on stirring O/N. The white crystalline product was filtrated off, washed with toluene (1000 mL) and dried at 55°C and 9 mbar for 2 h to yield 908.99 g (81% yield).
905 g of the isolated, crystallized product was suspended in acetonitrile (2.9 L). Acetyl chloride (23 mL) was added and the mixture was heated to 80°C (clear, colorless solution) for 4 hours. Toluene (1000 mL) was added and the mixture was cooled to RT within 2 hours (linear). The mixture was further cooled to 0°C within 60 minutes. The <a name=”product was filtrated off and washed with toluene (1000 mL). The product was dried overnight at 9 mbar and 50°C.
Yield: 742.06 g (66%)
purity: 99.9 area% at 200 nm
Summary:
Isolation of the cis intermediate leads to a significantly lower content of impurities, in particular of Impurity I. Toluene as solvent leads to disadvantageous conditions regarding consistency of the reaction mixture (procedure H). The use of acetonitrile or acetone (procedure I/F) leads to improved reaction conditions and product quality.
Example 3c
Preparation of (E)-4-(2-(2,5-dioxopyrrolidin-1-yl)ethoxy)-4-oxobut-2-enoic acid (Formula II) from ethylene carbonate and succinimide (without isolation of
intermediates)
Procedure
Ethylene carbonate (161.50 g, 1.834 mol) was melted at 50°C in an 1-L reactor, succinimide (173.07 g, 1.747 mol) and Et3N (24.4 mL, 0.175 mol) were added and the reaction mixture was warmed up to 90-92°C and stirred for 24h. Distillation column<a name=”
was set up on the reactor and the remaining Et3N was distilled off. Reaction mixture was cooled to 40-45°C, 500 mL of acetone was added, followed by addition of maleic anhydride (184 g, 1.878 mol) and Et3N (10.96 mL, 78.615 mmol). Reaction was stirred at 40°C for 6h (precipitation occurred after 3h), cooled to 20-25°C and acetyl chloride (20.86 mL, 0.293 mol) was added drop wise. Reaction mixture was then warmed up to 50-55°C and stirred for 20h. Orange solution crystallized upon seeding. Reaction mixture was cooled to 0°C and stirred for 3h. Resulting white suspension was filtered off and solid was washed with cold acetone (2×200 mL) and dried for 6h at 50°C and 30 mbar to afford (E)-4-(2-(2,5-dioxopyrrolidin-1-yl)ethoxy)-4-oxobut-2-enoic acid. Yield: 352.8 g (83.7%)
Purity: 99.69 area % at 200 nm
Example 4: Synthesis of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester
Procedure A:
The starting material (E)-But-2-enedioic acid mono-[2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl] ester (5 g; 20 mmol) was suspended in dichloromethane (60 mL) and cooled to 0°C, triethylamine (3.16 mL; 22.8 mmol) was added, resulting a clear solution. To this solution methylchloroformate (3.3 mL; 20.7 mmol) was carefully added within 30 minutes via syringe (reaction very exothermic). After 15 min of stirring at 0°C, DMAP (0.25 g; 2.1 mmol) was added into the reaction mixture at 0°C, stirring was continued for 3 hours at 0°C. The reaction mixture was poured into water (200 mL) and additional dichloromethane (100 mL) was added. The organic layer was separated and the aqueous layer was extracted once again with dichloromethane (50 mL). The combined organic layers were washed with brine (50 mL). The solvent was evaporated at 52°C. To the brown oil, which solidified, was added acetone (20 mL) and the mixture was stirred overnight. The product was filtrated off (white solid, part I) (2.73 g) and to the mother <a name=”liquor silica was added, the mixture was evaporated. Acetone (50 mL) was added and silica was filtrated off. The solvent was evaporated and diethylether (30 mL) was added to the solid, the mixture was stirred for ~1 hour. The product was filtrated off (part II) (1.6 g).
Overall yield: 4.33 g (82%)
Purity: part I 100 area-% at 200 nm; part II 97.96 area-% at 200 nm
Procedure A’
(E)-4-(2-(2,5-dioxopyrrolidin-1-yl)ethoxy)-4-oxobut-2-enoic acid (Formula II) (200 g, 0.829 mol) was suspended in acetone (2000 mL) in 3-L reactor at 20-25°C and cooled to 0°C. Et3N (150.31 mL, 1.078 mol) was added drop wise at 0-5°C. Into resulting solution, methyl chloroformate (83.27 mL, 1.072 mol) was added drop wise at 0-5°C. Reaction mixture was warmed up to 45°C and stirred for 2h. Upon completion, reaction mixture was cooled to 20-25°C and water (600 mL) was added drop wise with maintaining the temperature at 20-25°C resulting with off white to yellowish solution. pH was adjusted to 7 with 1M HCl. One more volume of water was added and pH corrected if needed. Part of acetone from the reaction mixture (5 volumes or 1000 mL) was distilled off under diminished pressure and reactor walls were washed with 1 more volume of water (200 mL), thus resulting in a solution of acetone/water mixture 1:1 (total 10 volumes). Reaction mixture was gradually cooled to 0°C and stirred for 20h. Resulting white suspension was filtered off and solid was washed with cold water (2×200 mL) and dried for 6h at 50°C and 30 mbar to afford crude 2-(2,5-dioxopyrrolidin-1-yl)ethyl methyl fumarate (Formula I).
Yield: 183.7 g (86.8%)
Purity: 100.00 area % at 200 nm
Crude 2-(2,5-dioxopyrrolidin-1-yl)ethyl methyl fumarate (170 g) was suspended in acetone (850 mL) at 20-25°C and warmed up to 50°C resulting with colorless solution. Water (850 mL) was added in portions at 50°C and solution was cooled gradually. Crystallization started at 32°C. Reaction mixture was stirred at crystallization temperature for 30 minutes and cooled further to 0°C, stirred at 0°C for 2h and resulting <a name=”white suspension was filtered off and solid was washed with cold water (2×170 mL) and dried for 6h at 50°C and 30 mbar to afford crystalline 2-(2,5-dioxopyrrolidin-1-yl)ethyl methyl fumarate.
Yield: 152.5 g (89.7%)
Purity: 100.00 area % at 200 nm
Procedure B:
The starting material (5 g, 20 mmol) was suspended in toluene (25 mL). Acetyl chloride (0.29 mL) and methanol (2.5 mL) were added, the reaction mixture was heated to 55°C and stirred for 3 hours. The reaction mixture was poured into water (100 mL) and extracted with ethylacetate (100 mL). The organic layer was separated and dried over sodium sulfate. The solvent was evaporated (crude product 4.7 g, main impurities dimetylfumarate (13%) and fumaric acid (1%) (HPLC at 200 nm)).
Yield: 4.7 g (88%)
Purity: 82.1 area-% at 200 nm
Procedure C:
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester in polymorphic form A; short: (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form A
The starting material (without isolation of (Z)-But-2-enedioic acid mono-[2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl] ester) (30 g; 0.12 mol) was suspended in dichloromethane (DCM, 160 mL) and cooled to 0°C, triethylamine (TEA, 19 mL; 0.14 mol) was added, resulting a clear solution. To this solution methyl chloroformate (19.74 mL; 0.12 mol) was added carefully within 30 minutes via syringe. Stirring was continued for ~2 hours. Water (200 mL) was added to the reaction mixture and stirring was continued for 5-10 minutes. The organic layer was separated and the aqueous layer was washed with another portion of DCM (100 mL). The combined organic layers were dried over sodium sulfate, before being evaporated. To the crude product was added acetone (50 mL) and the mixture was stirred for 3 hours before being filtered off. The product was washed with heptane (50 mL) and dried at 50°C and 21 mbar for 1 h.<a name=”
Yield: 20.52 g (65%)
Purity: 98.7 area-% at 220 nm; (0.3% of Impurity I)
XRPD diffraction peaks: 7.1, 11.6, 13.5, 13.7, 16.3, 16.7, 18.0, 18.4, 21.1, 22.1, 23.1, 23.9, 24.4, 25.5, 27.0, 27.5, 28.0, 28.6, 30.8, 31.2, 31.9, 32.3, 33.7, 34.2, 34.4, 34.9, 35.1, 35.7, 36.0, 36.8, 38.3, 40.1, 40.5, 41.7, 42.4, 43.0, 43.4, 45.0, 45.3, 46.2, 46.4, 47.0, 48.6, 49.4, 49.9, 52.0 + 0.2 degrees two theta.
The Form A according to Procedure C showed a habitus as depicted in Figure 7a
Procedure D:
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form A The starting material (10 g; 41.5 mmol) was suspended in toluene (70 mL) at 23°C, triethylamine (TEA; 6.3 mL; 45.6 mmol) was added. Methyl chloroformate (6.58 mL; 41.5 mmol) was slowly added within -30 minutes. After stirring for 2 hours water (40 mL) was added and shortly after acetone (110 mL), stirring was continued for ~2 minutes. The organic layer was separated and washed with brine (15 mL). After drying over sodium sulfate, the solvent was evaporated, yielding a slightly grey solid as crude product (9.42 g). The raw product was suspended in acetone (20 mL) and heptane (20 mL). The mixture was heated to reflux for 15 minutes resulting in a clear solution with just a small amount of solid. The mixture was cooled to RT and stirred overnight (precipitation started at 45°C, cooling: flask left in cooling oil bath ~lh to RT). The resulting product showed polymorphic form A.
Yield: 7.83 g (74%)
Purity: 99.4 area-% at 200 nm
The form A according to Procedure D showed a habitus as depicted in Figure 7b<a name=”
Procedure E:
The starting material (1 g; 4.15 mmol) was suspended in dichloromethane (50 mL) at RT. Methyl chloroformate (0.64 mL; 8.3 mmol) was added and stirring was continued overnight, in process control by HPLC showed no conversion.
Procedure F:
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form A
The starting material (7 g; 0.03 mol) and Na2CO3 were suspended in ethylacetate (50 mL). To the suspension was added methyl chloroformate (3.37 mL; 0.04 mol) in one portion. The reaction mixture was heated to 70°C. The temperature was kept for 15.5 h. The reaction mixture was cooled to 20°C and ethyl acetate (70 mL) was added to the white suspension. The solids were filtrated off and the ethyl acetate layer was washed with water (40 mL), dried over Na2S04 and evaporated to yield 6.4 g of the white crystalline crude product.
The crude product was suspended in a mixture of ethylacetate (10 mL) and heptane (10 mL). The suspension was heated to reflux for 30 minutes, then cooled to 23°C and stirred overnight. The product was filtrated off and dried at 8 mbar and 50°C overnight.
Yield: 5.62 g (75%)
Purity: 99.4 area-% at 200 nm
The form A according to Procedure E showed a habitus as depicted in Figure 7c
Procedure G:
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester in polymorphic form B; short: (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form B
(A) 9 g of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methylester was heated to 115°C. The melted compound was stirred for -20 minutes and then dropped into a precooled mortar (0°C).<a name=”
Purity of form B: 98.8 area-% at 200nm
XRPD-pattern: (Figure 4)
(B) 3.00 g of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester (form A) was suspended in 150 mL of dibutyl ether. Suspension was heated to 120° C while mixing. Solution was left at 25°C for 2 days. Crystallized material was filtered and dried at 23 °C at 12 mbar.
XRPD-pattern: (Figure 4′)
A measure of the relative volume change of a solid as a response to pressure change is called compressibility. An API should exhibit good compressibility which is dependent on the polymorphic state.
Experimental data:
The compressibility of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester form B and form A was assessed using a die and a flat-faced punch fitted on a TA-XT2 Texture analyser (Stable Micro Systems Ltd., Godalming, UK). 200 mg of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester sample is compressed in a steel mould (with the rate of displacement 0.03 mm/s). Cyclic procedure (similar to tapping) was performed: compressing, then retracting, relaxation for 15 s and then repeated compressive steps (altogether 10 steps). Each step exerts 0.2 MPa pressure on to the sample. Sample density is calculated by dividing the weight by the sample volume for each cycle. Maximum density is reached within 10 steps. Measurements were performed in duplicates for each sample, results are expressed as an average of duplicate measurements.
Results:
<a name=”
Form B of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester exhibits a higher density at compression, indicating superior compressibility compared to form A.
Procedure H:
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester in polymorphic form C; short: (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form C
(E)-But-2-enedioic acid mono-[2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl] ester (10 g; 41.5 mmol) was suspended in dichloromethane (DCM; 100 mL) and cooled to 0°C, triethylamine (TEA; 6.3 mL; 45.6 mmol) was added, resulting a clear solution. To the reaction mixture was added methyl chloroformate (6.58 mL; 41.5 mmol) within 30 minutes via a syringe pump. After 15 min of stirring at 0°C, DMAP (0.51 g; 4 mmol) was added into the reaction mixture at 0°C. The resulting solution was stirred at 0°C for 2.5 hours, then the cold suspension was poured into water (70 mL), the reactor was washed with further DCM (20 mL), which was added also to the DCM/water mixture. The organic layer was separated and washed with HCl (32% aq) (5 mL) in water (60 mL), then with water (50 mL) and finally with brine (50 mL). To the obtained deep red to brown solution was added silica (40-63 um) and the mixture was stirred for 5 minutes, before being filtered off to yield a colorless solution, which was evaporated to yield a colorless oil (crude product). The obtained oil was dissolved in a mixture of ethyl acetate/heptane (1/4) (20 mL). The mixture was stirred for 2 days before being filtered off. The product was dried under vacuum.
Yield: 2.87 g (26%)
Purity: 90.9 area-% at 200 nm
XRPD diffraction peaks: 11.2, 11.8, 13.0, 13.6, 13.6, 16.8, 18.1, 19.6, 20.6, 21.2, 21.5, 22.3, 23.2, 23.7, 24.3, 24.4, 25.2, 25.6, 26.5, 27.6, 28.4, 29.1, 30.3, 31.1, 32.0, 33.1, 33.8, 36.1, 36.7, 37.5, 38.4, 38.9, 41.6, 42.5, 43.2, 44.8, 46.5, 48.7, 49.6, 49.9 + 0.2 degrees two theta.<a name=”
Procedure I:
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester in polymorphic form D; short: (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form D
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form B (1 g) was suspended in acetonitrile (3 mL). The suspension was stirred for 7 days in a closed screw cap vials followed by slow evaporation of the solvent under ambient conditions within 3 days.
Purity of form D: 96.3 area-% at 200 nm
XRPD diffraction peaks: 6.9, 11.7, 13.6, 13.9, 16.4, 16.9, 18.2, 20.9, 21.3, 22.3, 23.3, 24.0, 24.6, 25.7, 27.5, 27.7, 31.0, 31.3, 32.1, 32.4, 33.9, 35.3, 35.7, 38.4, 41.9, 42.7, 43.1, 43.6, 44.4, 46.5, 48.9 + 0.2 degrees two theta.
Procedure J:
The starting material (obtained via isolation of (Z)-But-2-enedioic acid mono-[2-(2, 5-dioxo-pyrrolidin-1-yl)-ethyl] ester) (400 g; 1.7 mol) and Na2CO3 (264 g; 2.5 mol) were suspended in ethylacetate (2.7 L). To the suspension was added methyl chloroformate (193 mL; 2.5 mol) at 20°C. The reaction mixture was heated to 45°C within 90 minutes (linear heated). The mixture was kept on stirring for 5.5 hours. Ethylacetate (4 L) was added to the white suspension (at 45 °C). The suspension was stirred for 15 minutes before being filtrated off (45 °C suspension). The reactor was rinsed with another portion of ethylacetate (1 L). The filtrated solids were discarded. To the ethylacetate solution was added a mixture of HClaq (32%) (50 mL) and water (1 L) and the mixture was vigorously stirred for 10 minutes (at 35°C). Then the ethylacetate layer was separated (at ~35°C). The ethylacetate layer was transferred back to the reactor and stirred over sodium sulfate for 30 minutes, sodium sulfate was filtrated off and the ethylacetate layer was reduced to 900 mL. The suspension was transferred into a 3 L flask, equipped with a KPG stirrer and reflux condenser. The mixture was heated to reflux (stirring speed 160 rpm), the suspension was stirred until a clear solution was obtained (-30 minutes). Then heptane (550 mL) was added dropwise within 30 minutes<a name=”
under reflux conditions. Then the mixture (still solution) was slowly cooled to RT. The mixture was stirred O/N. The product was filtrated off and the filter cake was rinsed with heptane (500 mL) to yield the crystalline product (362.56 g; 86%).
purity: 99.8 area% at 218 nm (no Impurity I).
Alternative Procedure: Synthesis of (E)-But-2-enedioic acid 2-(2,5-dioxo- pyrrolidin-1-yl) -ethyl ester methyl ester
Procedure A
Monomethylfumarate (20 g, 0.15 mmol) was suspended in dry dichloromethane (400 mL) at RT, 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimid hydrochloride (32.42 g, 0.17 mol), N-(2-hydroxyethyl)succinimide (21.57 g, 0.15 mol) and dimethylaminopyridine (0.94 g, 7.7 mmol) were added. The solution was stirred O/N at RT. The formed yellow solution was diluted with dichloromethane (300 mL) and washed twice with water (2×500 mL). The organic layer was dried over sodium sulfate and concentrated under reduced pressure. To the crude product was added methyl tert. butyl ether (850 mL) and the reaction mixture was refluxed for 2.5 hours, cooled to RT, then filtrated and heated to reflux again for ~2 hours. After cooling to RT, the mixture was stored at ~5°C for 4 days. The white precipitate was filtrated off and washed with isopropylacetate (25 mL). The crystalline product was dried at 50°C and 7 mbar.
Yield: 10.8 g (28%)
Procedure B
Monomethylfumarate (1.5 g; 11.5 mmol) was suspended in dry DCM (30 mL) at 0°C. 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimid hydrochloride (2.47 g; 12.8 mmol), N- (2-hydroxyethyl)succinimide (1.62 g; 11.3 mmol) and DMAP (0.07 g; 0.6 mmol) were added. The solution was stirred overnight at RT. The formed yellow solution was diluted with DCM (50 mL) and washed with water twice (2×35 mL). The organic layer<a name=”
was dried over sodium sulfate and concentrated under reduced pressure. The crude product was purified by flash chromatography (n-heptane:ethyl acetate 1:1->1:2). The final product showed polymorphic form A. The form A according to alternative Procedure B showed a prismatic habitus as depicted in Figure 7d
Yield: 2.3 g (78%)
Purity: 99.5 area-% at 200 nm
Example 5: Kinetic investigations
Monomethyl maleate was prepared in analogy to WO 2014/197860. Samples of 13.2 grams of monomethyl maleate in 50 mL of toluene and 0.1 equivalents of the isomerization catalyst were reacted at 80°C. Samples were taken after the given times and analyzed by HPLC at 200 nm. The absorbance ratio of monomethyl fumarate (3.8 min.) to monomethyl maleate (2.8 min.) was taken as conversion parameter. The results are shown in Figure 1. As it can be seen from Figure 1 the conversion of monomethyl maleate to monomethyl fumarate in the presence of is TMS (trimethylsilylchloride) is advantageously enhanced compared to the one in the presence of AcCl (acetyl chloride).
Example 6: Yield determination
Six samples of 13.2 g (0.1 mol) monomethyl maleate were diluted with toluene (50 mL) and 0.1 eq of the isomerization catalyst (trimethylsilylchloride or acetyl chloride) were added, three samples with trimethylsilylchloride and three samples with acetyl chloride. The resulting reaction mixtures were heated to the temperatures of 45 °C, 51°C and 80°C. After 22 hours the reaction mixtures were cooled to room temperature, the product was filtrated off and dried at 50°C/8-16 mbar overnight. The results are shown in Figure 2 As it can be seen from Figure 2 the isolated yields of the conversion of monomethyl maleate to monomethyl fumarate in the presence of TMS (trimethylsilylchloride) is at any
PATENT
CN 110698442
PATENT
WO 2021053476
PATENT
IN 201921037120
PATENT
WO 2021074842
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021074842
The drug compound having the adopted name “Diroximel fumarate” has chemical name: 2-(2,5-Dioxopyrrolidin-l-yl)ethyl methyl fumarate as below.
Diroximel fumarate is an investigational, novel oral fumarate with a distinct chemical structure and developed by Alkermes pic, for the treatment of relapsing-remitting multiple sclerosis (RRMS) and is currently under review by U.S. Food and Drug Administration. Biogen, under an exclusive license from Alkermes, intends to market Diroximel fumarate under the brand name VUMERITY™.
US 8669281 B1 first disclosed Diroximel fumarate, its preparation, composition and use thereof for treating multiple sclerosis. US 10080733 B2 further discloses the crystalline solid form of Diroximel fumarate having an X-ray powder diffraction pattern comprising 2Q peaks at 11.6, 21.0, 24.3, 27.4, and 27.9 ±0.2 2Q.
WO 2017/108960 A1 also discloses various alternative synthetic approaches to make Diroximel fumarate and crystalline solid forms thereof, designated as Polymorphic forms A to D.
Hence, there remains a need for alternate solid forms of Diroximel fumarate and preparative processes thereof, exhibiting desired bioavailability and stability. Hence, it is desirable to provide a viable solid form of Diroximel fumarate. The known processes for the preparation of Diroximel fumarate are not viable at industrial scale due to the use of expensive reagents and catalyst such as coupling agents disclosed in US 8669281 Bl, with very low yields. Hence, there remains a need for the improved process to make Diroximel fumarate.
In another aspect, the present application provides a process for the preparation of Diroximel fumarate, comprising the step of esterification of monomethyl fumarate with (2,5-dioxopyrrolidin-l-yl)ethanol in the presence of an acid halide.
n another aspect, the present application provides a process for the preparation of Diroximel fumarate, comprising the step of esterification of (E)-4-(2-(2,5-dioxopyrrolidin-l-yl)ethoxy)-4-oxobut-2-enoic acid with methylation agent selected from the group consisting of 2,2-dimethoxypropane, trimethyl orthoformate and dimethyl carbonate.
Diroximel Fumarate
Example-7: Preparation of l-(2-hydroxyethyl)pyrrolidine-2,5-dione
A mixture of succinimide (100 g), ethylene carbonate (70.6 mL) and triethylamine (14 mL) was heated to 90 °C and stirred at the same temperature for 24 hours. The reaction mixture was cooled to 0 °C; methyl /ert-butyl ether (300 mL) was added and the resulting mixture was stirred for 30 minutes at the same temperature. The solid was filtered and dried under vacuum for 5 minutes. The solid was combined with ethyl acetate (100 mL) at 0 °C and stirred at the same temperature for 30 minutes. The solid was filtered and dried in rotatory vacuum dryer at 40 °C for 30 minutes to obtain 142.5 g of the title compound as off-white solid with HPLC purity of 99.6%.
Example-8: Preparation of Diroximel fumarate
To a mixture of (E)-4-methoxy-4-oxobut-2-enoic acid (4.0 g) and dichloromethane (40 mL) at 5 °C, Oxalyl chloride (5.85 g) was added slowly in 10 minutes, then a drop of DMF was added at the same temperature and allowed the reaction mixture to warm up to 27 °C. After complete evolution of the gas, solvent was evaporated from the reaction mixture. To a mixture of l-(2-hydroxyethyl)pyrrolidine-2,5-dione (5.06 g) and dichloromethane (35 mL), diisopropylethylamine (DIPEA) (9.93 g) was added and cooled the reaction mixture to 5 °C. The former mixture of (E)-4-methoxy-4-oxobut-2-enoic acid chloride in dichloromethane was slowly added to this later mixture at 5 °C for 20 minutes and stirred at the same temperature for 1 hour. The reaction mixture was quenched with saturated ammonium chloride solution and the organic layer was separated. Organic layer was washed with 10% citric acid solution and then with brine solution. The solvent from the separated organic layer was evaporated completely at 30 °C and the resultant solid was combined with acetone (15 mL) at 27 °C and stirred for 8 hours at the same temperature. The solid was filtered and the cake was washed with chilled acetone (3 mL) and then with cyclohexane (4 mL). The wet solid was dried at 40 °C under vacuum to obtain 3.3 g of the title compound with HPLC purity of 99.95 %
Example-9: Preparation of Diroximel fumarate
To a mixture of (E)-4-methoxy-4-oxobut-2-enoic acid (100.0 g) and dichloromethane (1000 mL) at 5 °C, Oxalyl chloride (117 g) was added slowly in 15 minutes, then catalytic DMF (1 mL) was added slowly at the same temperature and allowed the reaction mixture to warm up to 27 °C. After complete evolution of the gas, solvent was evaporated from the reaction mixture. To a mixture of l-(2-hydroxyethyl)pyrrolidine-2,5-dione (110 g) and dichloromethane (900 mL), diisopropylethylamine (DIPEA) (139 g) was added and cooled the reaction mixture to -5 °C. The former mixture of (E)-4-methoxy-4-oxobut-2-enoic acid chloride in dichloromethane (100 mL) was slowly added to this later mixture at – 5 °C for 60 minutes and stirred at the same temperature for 1 hour. The reaction mixture was quenched with water and the organic layer was separated. Organic layer was washed with 10% citric acid solution, 10% NaHC03 solution and then with brine solution. The solvent from the separated organic layer was evaporated completely at 30 °C and the resultant solid was combined with acetone (400 mL) at 27 °C. The reaction mixture was heated to 45 °C and stirred at the same temperature for 1 hour. The mixture was cooled to 27 °C and stirred for 8 hours at the same temperature. The solid was filtered and the cake was washed with methanol (200 mL). The wet solid was dried at 45 °C under vacuum to obtain 120 g of the title compound with HPLC purity of 99.97 %
Example-10: Preparation of (E)-4-(2-(2,5-dioxopyrrolidin-l-yl)ethoxy)-4-oxobut-2-enoic acid
A mixture of succinimide (100 g), ethylene carbonate (70.6 mL) and triethylamine (14 mL) was heated to 90 °C and stirred at the same temperature for 24 hours. The reaction mixture was cooled to 50 °C and triethylamine was removed by evaporation under vacuum. The reaction mixture was heated to 90 °C to distill out the traces of triethylamine under vacuum. The reaction mixture was cooled to 40 °C. Acetone (300 mL), maleic anhydride (106.2 g) and triethylamine (6.31 mL) were added. The resulting mixture was stirred at 40 °C for 6 hours. The mixture was cooled to 20 °C and acetyl chloride (12 mL) was added slowly over a period of 30 minutes. The mixture was slowly heated to 50 °C and stirred for 20 hours at the same temperature followed 2 hours at 0 °C. The solid was filtered and washed with cold acetone (2 x 120 mL).The wet solid was dried at 40 °C for 2 hours to obtain 194.2 g of the title compound as white solid with HPLC purity of 99.55%
Example-11: Preparation of Diroximel fumarate
Diroximel Fumarate
To a mixture of (E)-4-(2-(2,5-dioxopyrrolidin-l-yl)ethoxy)-4-oxobut-2-enoic acid (5 g) and 2,2-dimethoxypropane (50 mL) at 29 °C, concentrated hydrochloric acid (1 mL) and water (5 mL) were added and stirred at the same temperature for 17 hours at the same temperature. The pH of the reaction mixture was adjusted to 7 with a saturated aqueous solution of NaHCCh and the solvent was evaporated completely at 40 °C. To the resultant solid, water (50 mL) was added at 29 °C and stirred for 15 minutes. The solid was filtered and dried under vacuum at 29 °C for 5 hours. The resultant solid was combined with methyl /er/-butyl ether (50 mL) at 29 °C and stirred for 20 hours at the same temperature. The solid obtained was filtered and washed with diethyl ether (20 mL). The wet solid was dried under vacuum for 3 hours at 29 °C to obtain 3.3 g of the title compound as white solid with HPLC purity of 98.11%
Example-12: Preparation of Amorphous solid dispersion of Diroximel fumarate with Copovidone
Diroximel fumarate (100 mg) and Copovidone (500 mg) were dissolved in acetone (30 mL) at 30 °C. The clear solution was filtered to make it particle free and the solvent was evaporated in a rotavapor at 45 °C under reduced pressure to obtain the title amorphous solid dispersion. The solid dispersion (100 mg) obtained was combined with Syloid (500 mg) and ground for 20 minutes to obtain the admixture of title compound. XRPD: Amorphous.
Example-13: Crystallization of Diroximel fumarate
Diroximel fumarate (20 g) was dissolved in acetone (80 mL) at 43 °C and methyl tert. butyl ether (30 mL) was added to the clear solution. A suspension of crystalline Diroximel fumarate seed (0.25 g) in methyl tert. butyl ether (10 m) was added at 40 °C and stirred the mixture at the same temperature for 30 minutes. Methyl tert. butyl ether (280 mL) was added slowly for 2 hours at 41 °C. The mixture was cooled to 25 °C in 3
hours and then to 0 °C in 1 hour. The mixture was stirred at 0 °C for 1 hour and the solid was filtered. The wet solid was washed with methyl tert. butyl ether (40 mL) and dried at 42 °C for 6 hours to obtain the title compound.
PXRD: Crystalline; Malvern particle size: Dv (10) 7.776 pm, Dv (50) 31.292 pm & Dv (90) 133.437 pm
Example-14: Crystallization of Diroximel fumarate
Diroximel fumarate (20 g) was dissolved in acetone (80 mL) at 43 °C and DM water (100 mL) was added to the clear solution. A crystalline Diroximel fumarate seed (0.20 g) was added at 42 °C and stirred the mixture at the same temperature for 10 minutes. DM water (100 mL) was added slowly at 41 °C. The mixture was cooled to 28 °C in 1 hour. The mixture was stirred at 28 °C for 2 hour and the solid was filtered to obtain the title compound.
PXRD: Crystalline; Malvern particle size: Dv (10) 8.59 pm, Dv (50) 61.08 pm & Dv (90) 187.07 pm
Example-15: Crystallization of Diroximel fumarate
Diroximel fumarate (20 g) was dissolved in acetone (80 mL) at 45 °C and DM water (400 mL) was added to the clear solution. The mixture was cooled to 30 °C in 1 hour and the solid was filtered to obtain the title compound.
PXRD: Crystalline; Malvern particle size: Dv (10) 7.22 pm, Dv (50) 45.5 pm &
Dv (90) 136.7 pm
Example-16: Crystallization of Diroximel fumarate
Diroximel fumarate (20 g) was dissolved in Isopropyl acetate (360 mL) at 55 °C and cooled to 28 °C. A crystalline Diroximel fumarate seed (0.25 g) was added at 28 °C and cool to 5 °C. The mixture was stirred for 1 hour at the same temperature and the solid was filtered to obtain the title compound.
PXRD: Crystalline; Malvern particle size: Dv (10) 7.3 pm, Dv (50) 43.18 pm & Dv (90) 133.56 pm
PATENT
IN 201941042131
References
- ^ Jump up to:a b “Vumerity- diroximel fumarate capsule”. DailyMed. Retrieved 1 February 2021.
- ^ Jump up to:a b c “Vumerity EPAR”. European Medicines Agency. 14 September 2021. Retrieved 24 November 2021.
- ^ Wang Y, Bhargava P (July 2020). “Diroximel fumarate to treat multiple sclerosis”. Drugs of Today. 56 (7): 431–437. doi:10.1358/dot.2020.56.7.3151521. PMID 32648853. S2CID 220471534.
- ^ Kourakis S, Timpani CA, de Haan JB, Gueven N, Fischer D, Rybalka E (October 2020). “Dimethyl Fumarate and Its Esters: A Drug with Broad Clinical Utility?”. Pharmaceuticals (Basel, Switzerland). 13 (10): 306. doi:10.3390/ph13100306. PMC 7602023. PMID 33066228.
- ^ Jump up to:a b “Drug Approval Package: Vumerity”. U.S. Food and Drug Administration (FDA). 21 April 2020. Retrieved 1 February 2021.
- ^ “Diroximel fumarate”.
- ^ Jump up to:a b “Vumerity: Pending EC decision”. European Medicines Agency. 15 September 2021. Retrieved 17 September 2021. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
External links
- “Diroximel fumarate”. Drug Information Portal. U.S. National Library of Medicine.
 |
|
| Clinical data | |
|---|---|
| Trade names | Vumerity |
| Other names | ALKS-8700 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a620002 |
| License data |
|
| Routes of administration |
By mouth |
| ATC code |
|
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C11H13NO6 |
| Molar mass | 255.226 g·mol−1 |
| 3D model (JSmol) | |
/////////Diroximel fumarate, EU 2021, EMA 2021, APPROVALS 2021, VUMERITY, ジロキシメルフマル酸エステル , K0N0Z40J3W, RDC-5108, дироксимела фумарат , ديروكسيميل فومارات , 富马地罗昔美 ,

NEW DRUG APPROVALS
ONE TIME
$10.00
Vosoritide
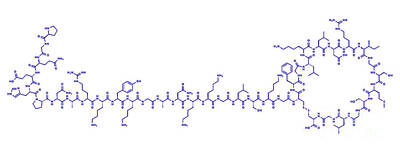
| PGQEHPNARK YKGANKKGLS KGCFGLKLDR IGSMSGLGC (Disulfide bridge: 23-39) |
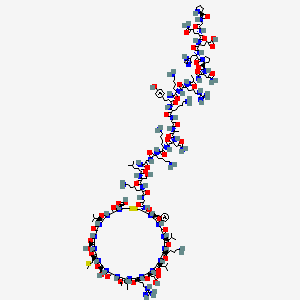

H-Pro-Gly-Gln-Glu-His-Pro-Asn-Ala-Arg-Lys-Tyr-Lys-Gly-Ala-Asn-Lys-Lys-Gly-Leu-Ser-Lys-Gly-Cys(1)-Phe-Gly-Leu-Lys-Leu-Asp-Arg-Ile-Gly-Ser-Met-Ser-Gly-Leu-Gly-Cys(1)-OH
PGQEHPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC
H-PGQEHPNARKYKGANKKGLSKGC(1)FGLKLDRIGSMSGLGC(1)-OH
PEPTIDE1{P.G.Q.E.H.P.N.A.R.K.Y.K.G.A.N.K.K.G.L.S.K.G.C.F.G.L.K.L.D.R.I.G.S.M.S.G.L.G.C}$PEPTIDE1,PEPTIDE1,23:R3-39:R3$$$
L-prolyl-glycyl-L-glutaminyl-L-alpha-glutamyl-L-histidyl-L-prolyl-L-asparagyl-L-alanyl-L-arginyl-L-lysyl-L-tyrosyl-L-lysyl-glycyl-L-alanyl-L-asparagyl-L-lysyl-L-lysyl-glycyl-L-leucyl-L-seryl-L-lysyl-glycyl-L-cysteinyl-L-phenylalanyl-glycyl-L-leucyl-L-lysyl-L-leucyl-L-alpha-aspartyl-L-arginyl-L-isoleucyl-glycyl-L-seryl-L-methionyl-L-seryl-glycyl-L-leucyl-glycyl-L-cysteine (23->39)-disulfide
(4R,10S,16S,19S,22S,28S,31S,34S,37S,40S,43S,49S,52R)-52-[[2-[[(2S)-6-amino-2-[[(2S)-2-[[(2S)-2-[[2-[[(2S)-6-amino-2-[[(2S)-6-amino-2-[[(2S)-4-amino-2-[[(2S)-2-[[2-[[(2S)-6-amino-2-[[(2S)-2-[[(2S)-6-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-4-amino-2-[[(2S)-1-[(2S)-2-[[(2S)-2-[[(2S)-5-amino-5-oxo-2-[[2-[[(2S)-pyrrolidine-2-carbonyl]amino]acetyl]amino]pentanoyl]amino]-4-carboxybutanoyl]amino]-3-(1H-imidazol-5-yl)propanoyl]pyrrolidine-2-carbonyl]amino]-4-oxobutanoyl]amino]propanoyl]amino]-5-carbamimidamidopentanoyl]amino]hexanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]hexanoyl]amino]acetyl]amino]propanoyl]amino]-4-oxobutanoyl]amino]hexanoyl]amino]hexanoyl]amino]acetyl]amino]-4-methylpentanoyl]amino]-3-hydroxypropanoyl]amino]hexanoyl]amino]acetyl]amino]-40-(4-aminobutyl)-49-benzyl-28-[(2S)-butan-2-yl]-31-(3-carbamimidamidopropyl)-34-(carboxymethyl)-16,22-bis(hydroxymethyl)-10,37,43-tris(2-methylpropyl)-19-(2-methylsulfanylethyl)-6,9,12,15,18,21,24,27,30,33,36,39,42,45,48,51-hexadecaoxo-1,2-dithia-5,8,11,14,17,20,23,26,29,32,35,38,41,44,47,50-hexadecazacyclotripentacontane-4-carboxylic acid
Vosoritide
| Formula | C176H290N56O51S3 |
|---|---|
| CAS | 1480724-61-5 |
| Mol weight | 4102.7254 |
1480724-61-5[RN]BMN 111L-Cysteine, L-prolylglycyl-L-glutaminyl-L-α-glutamyl-L-histidyl-L-prolyl-L-asparaginyl-L-alanyl-L-arginyl-L-lysyl-L-tyrosyl-L-lysylglycyl-L-alanyl-L-asparaginyl-L-lysyl-L-lysylglycyl-L-leucyl-L-seryl-L-lysylglycyl-L-cysteinyl-L-phenylalanylglycyl-L-leucyl-L-lysyl-L-leucyl-L-α-aspartyl-L-arginyl-L-isoleucylglycyl-L-seryl-L-methionyl-L-serylglycyl-L-leucylglycyl-, cyclic (23→39)-disulfideL-prolylglycyl-(human C-type natriuretic peptide-(17-53)-peptide (CNP-37)), cyclic-(23-39)-disulfideUNII:7SE5582Q2Pвосоритид [Russian] [INN]فوسوريتيد [Arabic] [INN]伏索利肽 [Chinese] [INN]
Voxzogo, 2021/8/26 EU APPROVED
| Product details | |
|---|---|
| Name | Voxzogo |
| Agency product number | EMEA/H/C/005475 |
| Active substance | Vosoritide |
| International non-proprietary name (INN) or common name | vosoritide |
| Therapeutic area (MeSH) | Achondroplasia |
| Anatomical therapeutic chemical (ATC) code | M05BX |
| Orphan |
This medicine was designated an orphan medicine. This means that it was developed for use against a rare, life-threatening or chronically debilitating condition or, for economic reasons, it would be unlikely to have been developed without incentives. For more information, see Orphan designation. |
| Publication details | |
|---|---|
| Marketing-authorisation holder | BioMarin International Limited |
| Date of issue of marketing authorisation valid throughout the European Union | 26/08/2021 |
On 24 January 2013, orphan designation (EU/3/12/1094) was granted by the European Commission to BioMarin Europe Ltd, United Kingdom, for modified recombinant human C-type natriuretic peptide for the treatment of achondroplasia.
The sponsorship was transferred to BioMarin International Limited, Ireland, in February 2019.
This medicine is now known as Vosoritide.
The medicinal product has been authorised in the EU as Voxzogo since 26 August 2021.
PEPTIDE
| Treatment of Achondroplasia modified recombinant human C-type natriuretic peptide (CNP) |
Vosoritide, sold under the brand name Voxzogo, is a medication used for the treatment of achondroplasia.[1]
The most common side effects include injection site reactions (such as swelling, redness, itching or pain), vomiting and decreased blood pressure.[1]
Vosoritide was approved for medical use in the European Union in August 2021.[1][2]
Voxzogo is a medicine for treating achondroplasia in patients aged 2 years and older whose bones are still growing.
Achondroplasia is an inherited disease caused by a mutation (change) in a gene called fibroblast growth-factor receptor 3 (FGFR3). The mutation affects growth of almost all bones in the body including the skull, spine, arms and legs resulting in very short stature with a characteristic appearance.
Achondroplasia is rare, and Voxzogo was designated an ‘orphan medicine’ (a medicine used in rare diseases) on 24 January 2013. Further information on the orphan designation can be found here: ema.europa.eu/medicines/human/orphan-designations/EU3121094.
Voxzogo contains the active substance vosoritide.
Medical uses
Vosoritide is indicated for the treatment of achondroplasia in people two years of age and older whose epiphyses are not closed.[1]
Mechanism of action

A: Chondrocyte with constitutionally active FGFR3 that down-regulates its development via the MAPK/ERK pathway
B: Vosoritide (BMN 111) blocks this mechanism by binding to the atrial natriuretic peptide receptor B (NPR-B), which subsequently inhibits the MAPK/ERK pathway at the RAF-1 protein.[3]
Vosoritide works by binding to a receptor (target) called natriuretic peptide receptor type B (NPR-B), which reduces the activity of fibroblast growth factor receptor 3 (FGFR3).[1] FGFR3 is a receptor that normally down-regulates cartilage and bone growth when activated by one of the proteins known as acidic and basic fibroblast growth factor. It does so by inhibiting the development (cell proliferation and differentiation) of chondrocytes, the cells that produce and maintain the cartilaginous matrix which is also necessary for bone growth. Children with achondroplasia have one of several possible FGFR3 mutations resulting in constitutive (permanent) activity of this receptor, resulting in overall reduced chondrocyte activity and thus bone growth.[3]
The protein C-type natriuretic peptide (CNP), naturally found in humans, reduces the effects of over-active FGFR3. Vosoritide is a CNP analogue with the same effect but prolonged half-life,[3] allowing for once-daily administration.[4]
Chemistry
Vosoritide is an analogue of CNP. It is a peptide consisting of the amino acids proline and glycine plus the 37 C-terminal amino acids from natural human CNP. The complete peptide sequence isPGQEHPNARKYKGANKKGLS KGCFGLKLDR IGSMSGLGC
with a disulfide bridge between positions 23 and 39 (underlined).[5] The drug must be administered by injection as it would be rendered ineffective by the digestive system if taken by mouth.
History
Vosoritide is being developed by BioMarin Pharmaceutical and, being the only available causal treatment for this condition, has orphan drug status in the US as well as the European Union.[1][2][6] As of September 2015, it is in Phase II clinical trials.[7][4]
Society and culture
Controversy
Some people with achondroplasia, as well as parents of children with this condition, have reacted to vosoritide’s study results by saying that dwarfism is not a disease and consequently does not need treatment.[8]
Research
Vosoritide has resulted in increased growth in a clinical trial with 26 children. The ten children receiving the highest dose grew 6.1 centimetres (2.4 in) in six months, compared to 4.0 centimetres (1.6 in) in the six months before the treatment (p=0.01).[9] The body proportions, more specifically the ratio of leg length to upper body length – which is lower in achondroplasia patients than in the average population – was not improved by vosoritide, but not worsened either.[7][10]
As of September 2015, it is not known whether the effect of the drug will last long enough to result in normal body heights,[10] or whether it will reduce the occurrence of achondroplasia associated problems such as ear infections, sleep apnea or hydrocephalus. This, together with the safety of higher doses, is to be determined in further studies.[4]
References
- ^ Jump up to:a b c d e f g “Voxzogo EPAR”. European Medicines Agency. 23 June 2021. Retrieved 9 September 2021. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b “European Commission Approves BioMarin’s Voxzogo (vosoritide) for the Treatment of Children with Achondroplasia from Age 2 Until Growth Plates Close”. BioMarin Pharmaceutical Inc. (Press release). 27 August 2021. Retrieved 9 September 2021.
- ^ Jump up to:a b c Lorget F, Kaci N, Peng J, Benoist-Lasselin C, Mugniery E, Oppeneer T, et al. (December 2012). “Evaluation of the therapeutic potential of a CNP analog in a Fgfr3 mouse model recapitulating achondroplasia”. American Journal of Human Genetics. 91 (6): 1108–14. doi:10.1016/j.ajhg.2012.10.014. PMC 3516592. PMID 23200862.
- ^ Jump up to:a b c Clinical trial number NCT02055157 for “A Phase 2 Study of BMN 111 to Evaluate Safety, Tolerability, and Efficacy in Children With Achondroplasia (ACH)” at ClinicalTrials.gov
- ^ “International Nonproprietary Names for Pharmaceutical Substances (INN): List 112” (PDF). WHO Drug Information. 28 (4): 539. 2014.
- ^ “Food and Drug Administration Accepts BioMarin’s New Drug Application for Vosoritide to Treat Children with Achondroplasia” (Press release). BioMarin Pharmaceutical. 2 November 2020. Retrieved 9 September 2021 – via PR Newswire.
- ^ Jump up to:a b Spreitzer H (6 July 2015). “Neue Wirkstoffe – Vosoritid”. Österreichische Apothekerzeitung (in German) (14/2015): 28.
- ^ Pollack A (17 June 2015). “Drug Accelerated Growth in Children With Dwarfism, Pharmaceutical Firm Says”. The New York Times.
- ^ “BMN 111 (vosoritide) Improves Growth Velocity in Children With Achondroplasia in Phase 2 Study”. BioMarin. 17 June 2015.
- ^ Jump up to:a b “Vosoritid” (in German). Arznei-News.de. 20 June 2015.
External links
- “Vosoritide”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Voxzogo |
| Other names | BMN-111 |
| Routes of administration |
Subcutaneous injection |
| ATC code | None |
| Legal status | |
| Legal status | EU: Rx-only [1] |
| Identifiers | |
| CAS Number | 1480724-61-5 |
| DrugBank | DB11928 |
| ChemSpider | 44210446 |
| UNII | 7SE5582Q2P |
| KEGG | D11190 |
| Chemical and physical data | |
| Formula | C176H290N56O51S3 |
| Molar mass | 4102.78 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
/////////Vosoritide, Voxzogo, PEPTIDE, ボソリチド (遺伝子組換え) , восоритид , فوسوريتيد , 伏索利肽 , APPROVALS 2021, EU 2021, BMN 111, ORPHAN DRUG
CCC(C)C1C(=O)NCC(=O)NC(C(=O)NC(C(=O)NC(C(=O)NCC(=O)NC(C(=O)NCC(=O)NC(CSSCC(C(=O)NC(C(=O)NCC(=O)NC(C(=O)NC(C(=O)NC(C(=O)NC(C(=O)NC(C(=O)N1)CCCNC(=N)N)CC(=O)O)CC(C)C)CCCCN)CC(C)C)CC2=CC=CC=C2)NC(=O)CNC(=O)C(CCCCN)NC(=O)C(CO)NC(=O)C(CC(C)C)NC(=O)CNC(=O)C(CCCCN)NC(=O)C(CCCCN)NC(=O)C(CC(=O)N)NC(=O)C(C)NC(=O)CNC(=O)C(CCCCN)NC(=O)C(CC3=CC=C(C=C3)O)NC(=O)C(CCCCN)NC(=O)C(CCCNC(=N)N)NC(=O)C(C)NC(=O)C(CC(=O)N)NC(=O)C4CCCN4C(=O)C(CC5=CN=CN5)NC(=O)C(CCC(=O)O)NC(=O)C(CCC(=O)N)NC(=O)CNC(=O)C6CCCN6)C(=O)O)CC(C)C)CO)CCSC)CO
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter a
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
NEW DRUG APPROVALS
ONE TIME TO MAINTAIN THIS BLOG
$10.00
Saquinavir
Saquinavir,
Ro 31 8959, Ro 31-8959, RO 31-8959/000, Ro 318959, RO-31-8959/000, Sch 52852, SCH-52852
(2S)-N-[(2S,3R)-4-[(3S,4aS,8aS)-3-(tert-butylcarbamoyl)-decahydroisoquinolin-2-yl]-3-hydroxy-1-phenylbutan-2-yl]-2-[(quinolin-2-yl)formamido]butanediamide
(2S)-N-[(2S,3R)-4-[(3S,4aS,8aS)-3-(tert-butylcarbamoyl)-3,4,4a,5,6,7,8,8a-octahydro-1H-isoquinolin-2-yl]-3-hydroxy-1-phenylbutan-2-yl]-2-(quinoline-2-carbonylamino)butanediamide
(-)-cis-N-tert-butyldecahydro-2-{(2R,3S)-2-hydroxy-4-phenyl-3-{[N-(2-quinolylcarbonyl)-L-asparaginyl]amino}butyl}-(3S,4aS,8aS)-isoquinoline-3 carboxamide monomethanesulfonate
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Saquinavir mesylate | UHB9Z3841A | 149845-06-7 | IRHXGOXEBNJUSN-YOXDLBRISA-N |
CAS Registry Number: 127779-20-8
CAS Name: (2S)-N1[(1S,2R)-3-[(3S,4aS,8aS)-3-[[(1,1-Dimethylethyl)amino]carbonyl]octahydro-2(1H)-isoquinolinyl]-2-hydroxy-1-(phenylmethyl)propyl]-2-[(2-quinolinylcarbonyl)amino]butanediamide
Additional Names: (S)-N-[(aS)-a-[(1R)-2-[(3S,4aS,8aS)-3-(tert-butylcarbamoyl)octahydro-2(1H)-isoquinolyl]-1-hydroxyethyl]phenethyl]-2-quinaldamido succinamide; N-tert-butyldecahydro-2-[2(R)-hydroxy-4-phenyl-3(S)-[[N-(2-quinolylcarbonyl)-L-asparaginyl]amino]butyl](4aS,8aS)-isoquinoline-3(S)-carboxamide
Manufacturers’ Codes: Ro-31-8959Molecular Formula: C38H50N6O5Molecular Weight: 670.84Percent Composition: C 68.04%, H 7.51%, N 12.53%, O 11.92%
Literature References: Selective HIV protease inhibitor.Prepn: J. A. Martin, S. Redshaw, EP432695; eidem,US5196438 (1991, 1993 both to Hoffmann-LaRoche); K. E. B. Parkes et al.,J. Org. Chem.59, 3656 (1994).In vitro HIV proteinase inhibition: N. A. Roberts et al.,Science248, 358 (1990). Antiviral properties: J. C. Craig et al.,Antiviral Res.16, 295 (1991); S. Galpin et al.,Antiviral Chem. Chemother.5, 43-45 (1994).Clinical evaluation of tolerability and activity: V. S. Kitchen et al.,Lancet345, 952 (1995). Review of pharmacology and clinical experience: S. Kravcik, Expert Opin. Pharmacother.2 303-315 (2001).
Properties: White crystalline solid. [a]D20 -55.9° (c = 0.5 in methanol). Soly (21°): 0.22 g/100 ml water.Optical Rotation: [a]D20 -55.9° (c = 0.5 in methanol)
Derivative Type: Methanesulfonate saltCAS Registry Number: 149845-06-7Additional Names: Saquinavir mesylateManufacturers’ Codes: Ro-31-8959/003Trademarks: Fortovase (Roche); Invirase (Roche)Molecular Formula: C38H50N6O5.CH3SO3HMolecular Weight: 766.95Percent Composition: C 61.08%, H 7.10%, N 10.96%, O 16.69%, S 4.18%
Therap-Cat: Antiviral.Keywords: Antiviral; Peptidomimetics; HIV Protease Inhibitor.
Saquinavir mesylate was first approved by the U.S. Food and Drug Administration (FDA) on Dec 6, 1995, then approved by European Medicine Agency (EMA) on Oct 4, 1996, and approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) on Sep 5, 1997. It was developed by Roche, then marketed as Invirase® by Roche in the US and EU and by Chugai in JP.
Saquinavir mesylate is an inhibitor of HIV-1 protease. It is a peptide-like substrate analogue that binds to the protease active site and inhibits the activity of HIV-1 protease that required for the proteolytic cleavage of viral polyprotein precursors into individual functional proteins found in HIV-1 particles. It is indicated for the treatment of HIV-1 infection in combination with ritonavir and other antiretroviral agents in adults (over the age of 16 years).
Invirase® is available as capsule for oral use, containing 200 mg of free Saquinavir. The recommended dose is 1000 mg twice daily in combination with ritonavir 100 mg twice daily for adults.
Human medicines European public assessment report (EPAR): Invirase, saquinavir, HIV Infections, 03/10/1996, 47, Authorised (updated)
EU 08/09/2021
Invirase is an antiviral medicine used to treat adults infected with the human immunodeficiency virus type 1 (HIV 1), a virus that causes acquired immune deficiency syndrome (AIDS). Invirase should only be used in combination with ritonavir (another antiviral medicine) and other antiviral medicines.
Invirase contains the active substance saquinavir.
| Product details | |
|---|---|
| Name | Invirase |
| Agency product number | EMEA/H/C/000113 |
| Active substance | saquinavir |
| International non-proprietary name (INN) or common name | saquinavir |
| Therapeutic area (MeSH) | HIV Infections |
| Anatomical therapeutic chemical (ATC) code | J05AE01 |
| Publication details | |
|---|---|
| Marketing-authorisation holder | Roche Registration GmbH |
| Date of issue of marketing authorisation valid throughout the European Union | 03/10/1996 |
Invirase can only be obtained with a prescription and treatment should be started by a doctor who has experience in the treatment of HIV infection.
Invirase is available as capsules (200 mg) and tablets (500 mg). For patients already taking HIV medicines, the recommended dose of Invirase is 1,000 mg with 100 mg ritonavir twice daily. For patients who are not taking HIV medicines, Invirase is started at 500 mg twice daily with ritonavir 100 mg twice daily for the first 7 days of treatment, given in combination with other HIV medicines. After 7 days, the recommended dose of Invirase is 1,000 mg twice daily with ritonavir 100 mg twice daily in combination with other HIV medicines.
For more information about using Invirase, see the package leaflet or contact a doctor or pharmacist.
The active substance in Invirase, saquinavir, is a ‘protease inhibitor’. It blocks protease, an enzyme involved in the reproduction of HIV. When the enzyme is blocked, the virus does not reproduce normally, slowing down the spread of infection. Ritonavir is another protease inhibitor that is used as a ‘booster’. It slows the breakdown of saquinavir, increasing the levels of saquinavir in the blood. This allows effective treatment while avoiding a higher dose of saquinavir. Invirase, taken in combination with other HIV medicines, reduces the viral load (the amount of HIV in the blood) and keeps it at a low level. Invirase does not cure HIV infection or AIDS, but it may hold off the damage to the immune system and the development of infections and diseases associated with AIDS.
Invirase received a marketing authorisation valid throughout the EU on 4 October 1996.
Drug Name:Saquinavir MesylateResearch Code:Ro-31-8959; Sch-52852Trade Name:Invirase®MOA:HIV-1 protease inhibitorIndication:HIV infectionStatus:ApprovedCompany:Roche (Originator) , ChugaiSales:ATC Code:J05AE01
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2004-12-17 | New dosage form | Invirase | HIV infection | Tablet | Eq. 500 mg Saquinavir | Roche | Priority |
| 1995-12-06 | First approval | Invirase | HIV infection | Capsule | Eq. 200 mg Saquinavir | Roche | Priority |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 1996-10-04 | First approval | Invirase | HIV infection | Capsule | 200 mg | Roche | |
| 1996-10-04 | First approval | Invirase | HIV infection | Tablet, Film coated | 500 mg | Roche |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2006-09-01 | New dosage form | Invirase | HIV infection | Tablet | 500 mg | Chugai | |
| 1997-09-05 | First approval | Invirase | HIV infection | Capsule | 200 mg | Chugai |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2014-03-13 | Marketing approval | 因服雷/Invirase | HIV infection | Tablet | Eq. 500 mg Saquinavir | Roche | |
| 2009-07-01 | Marketing approval | 因服雷/Invirase | HIV infection | Capsule | Eq. 200 mg Saquinavir | Roche |
Reference:1. US5196438A.Route 2
Reference:1. J. Org. Chem. 1994, 59, 3656-3664.Route 3
Reference:1. WO2006134612A1.
SYN
English: DOI: 10.1021/jo00393a034
DOI: 10.1021/jo00092a026
DOI: 10.1016/S0040-4039(00)77633-7

SYN
In the following, a possible route for the synthesis of Saquinavir is presented. Since Diazomethane is used, the synthesis is not suitable for a scaled up process. Roche has solved this problem with another reaction mechanism. The mechanism for laboratories starts with a ring opening substitution of an epoxid derivative of Phenylalanine with decaisohydroquinoline in dry iso-propanol with nitrogen atmosphere. The intermediate is purified by flash chromatography. In the second step of synthesis, the protection group is removed with gaseous hydrogen and a carbon/palladium catalyst. Furthermore, the new product reacts with N-Benzyloxycarbonyl-Lasparagine(Cbz AsnOH) in the solvents Cbz Asparagine L(Cbz Asn L) and 1- Hydroxybenzotriazolehydrat(HBOT). Afterwards, the protecting group of the former Asparagine is removed with another mixture of gaseous hydrogen and carbon/palladium catalyst. The final intermediate gets stirred in the last step of synthesis together with the solvents Tetrahydrofuran, HBOT and DCC. The mechanism formulated in detail can be found in the Appendix (VIII).7
Kevin E. B. Parkes; David J. Bushnell; et al. Studies toward the Large-Scale Synthesis of the HIV Proteinase Inhibitor Ro 31-8959. J. Org. Chem. 1994, 59, 3656–3664.
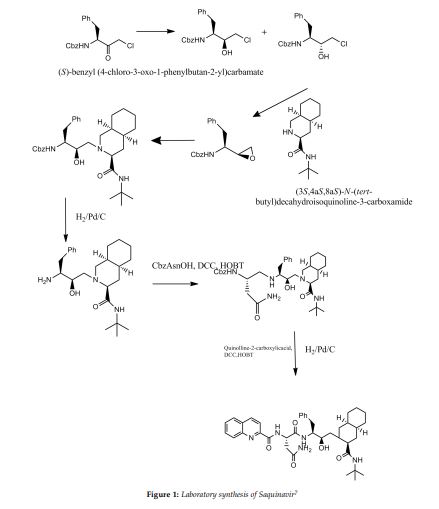
SYN
he synthesis of Ro-31-8959/003 (X) was carried out as follows: Condensation of L-phenylalanine (I) with formaldehyde in concentrated hydrochloric acid gave the tetrahydroisoquinoline (II), which was hydrogenated in 90% acetic acid over rhodium on carbon to yield the decahydroisoquinoline (III) as a mixture of diastereoisomers. Treatment of (III) with benzyl chloroformate in aqueous sodium hydroxide solution gave a mixture of N-protected amino acids which was separated by fractional crystallization of the cyclohexylamine salts to give the (S,S,S)-isomer. Reaction with dicyclohexylcarbodiimide and N-hydroxysuccinimide in dimethoxyethane, followed by treatment of the activated ester with tert-butylamine in dichloromethane and subsequent hydrogenolysis of the benzyloxycarbonyl protecting group gave the decahydroisoquinoline (IV). In the other branch of the synthesis L-phenylalanine was treated with benzyl chloroformate in aqueous sodium hydroxide solution to give the N-protected amino acid. This was converted to the corresponding mixed anhydride with isobutyl chloroformate and N-ethylmorpholine in tetrahydrofuran and immediately reacted with diazomethane in diethyl ether to give the diazomethyl ketone (V). Treatment of (V) with ethereal hydrogen chloride gave the chloromethyl ketone (VI), which on reduction with sodium borohydride in aqueous tetrahydrofuran gave a mixture of diastereoisomeric chlorohydrins. Solvent extraction with boiling n-hexane followed by recrystallization of the less soluble isomer from isopropanol gave pure chlorohydrin (VII), which on treatment with ethanolic potassium hydroxide gave the epoxide (VIII). Condensation of (VIII) with (IV) in ethanol gave the hydroxyethylamine (IX). Hydrogenolysis of (IX) was followed by condensation with N-benzyloxycarbonyl-L-asparagine in tetrahydrofuran in the presence of 1-hydroxybenzotriazole and dicyclohexylcarbodiimide. Hydrogenolysis in ethanol over palladium on charcoal, followed by condensation with quinoline-2-carboxylic acid in tetrahydrofuran in the presence of dicyclohexylcarbodiimide and 1-hydroxybenzotriazole, gave the free base, Ro-31-8959/000. Treatment with methanesulfonic acid in aqueous ethanol then afforded the mesylate salt (X), Ro-31-8959/003.
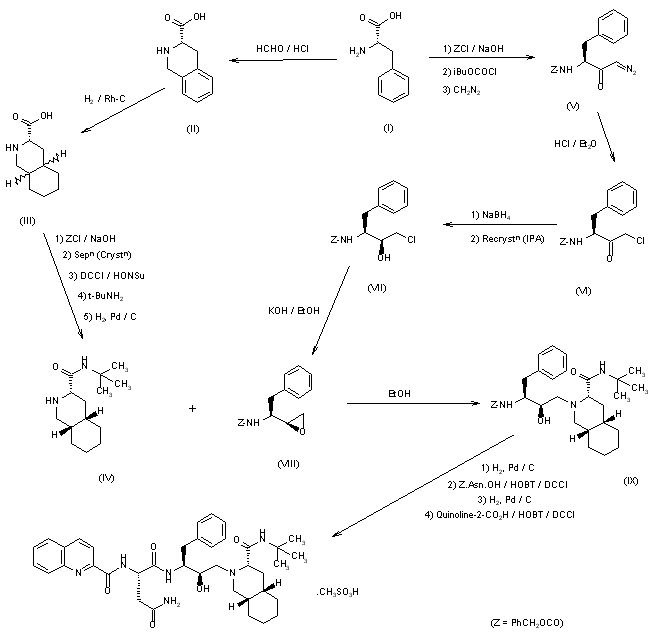
SYN
J Org Chem 1994,59(13),3656
Various new routes for the large-scale synthesis of Ro-31-8959 have been described: 1) The condensation of N-protected-L-phenylalanine (I) with the Mg salt of malonic acid monoethyl ester (II) gives the keto ester (III), which is enantioselectively reduced with NaBH4 to yield the hydroxy ester (IV). The reaction of (IV) with 2,2-dimethoxypropane (V) by means of p-toluenesulfonic acid affords the oxazolidine (VI), which is hydrolyzed with NaOH in ethanol/water to the corresponding acid (VII). The treatment of (VII) with oxalyl chloride, mercaptopyridine-N-oxide (MPO) and bromotrichloromethane affords the bromomethyloxazolidine (VIII), which, without isolation, is treated with acetic acid to give the N-protected 3(S)-amino-2-bromo-4-phenyl-2(S)-butanol (IX). The reaction of (IX) with KOH in methanol yields the epoxide (X), which is condensed with (3S,4aS,8aS)-N-tert-butyldecahydroisoquinoline-3-carboxamide (XI), yielding the protected condensation product (XII). The deprotection of the amino group of (XII) by hydrogenation with H2 over Pd/C affords the amino derivative (XIII), which is condensed with N-benzyloxycarbonyl-asparagine (XIV) in the usual way, giving the protected peptide (XV). The deprotection of (XV) as before yields compound (XVI), with a free amino group that is finally condensed with quinoline-2-carboxylic acid (XVII) by means of dicyclohexylcarbodiimide and hydroxybenzotriazole.
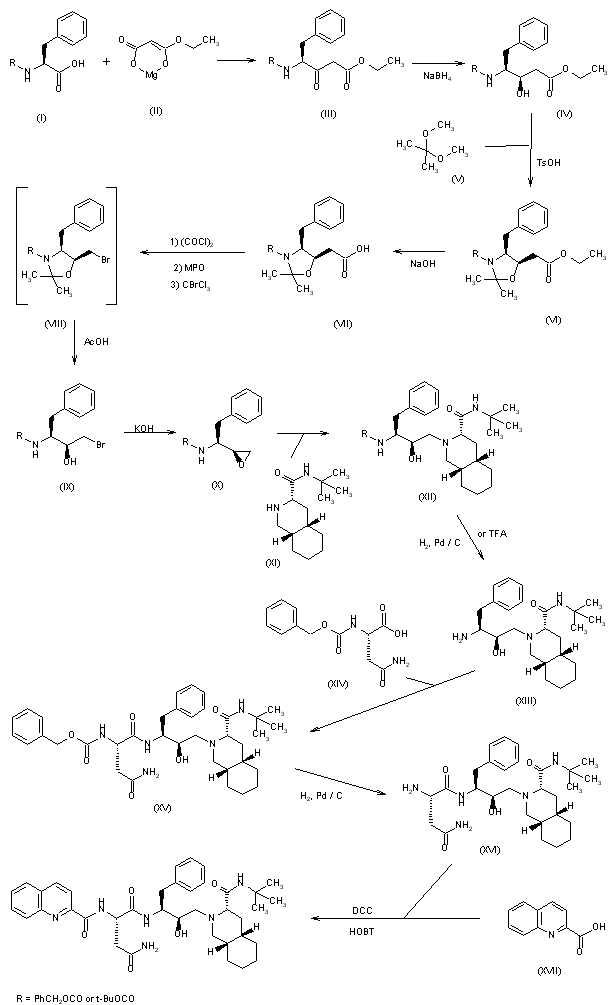
SYN
2) The condensation of N-phthaloyl-L-phenylalaninyl chloride (XVIII) with 1,1,2-tris(trimethylsilyloxy)ethylene (TMS) (XIX) at 90-100 C followed by acidic hydrolysis with HCl gives the acid (XX), which, without isolation, is decarboxylated, yielding 1-hydroxy-3(S)-phthalimido-4-phenyl-2-butanone (XXI). Sequential protection of the OH- group with dihydropyran, reduction of the CO group with NaBH4, mesylation of the resulting OH group with methanesulfonyl chloride and deprotection of the primary OH group gives 2(R)-(methanesulfonyloxy)-4-phenyl-3(S)-phthalimido-1-butanol (XXII). The epoxidation of (XXII) with potassium tert-butoxide yields the epoxide (XXIII), which is condensed with the decahydroisoquinoline (XI) as before, affording the protected condensation product (XXIV). The elimination of the phthalimido group of (XXIV) with methylamine and HCl gives the amino derivative (XIII), already obtained in scheme 16810301a.
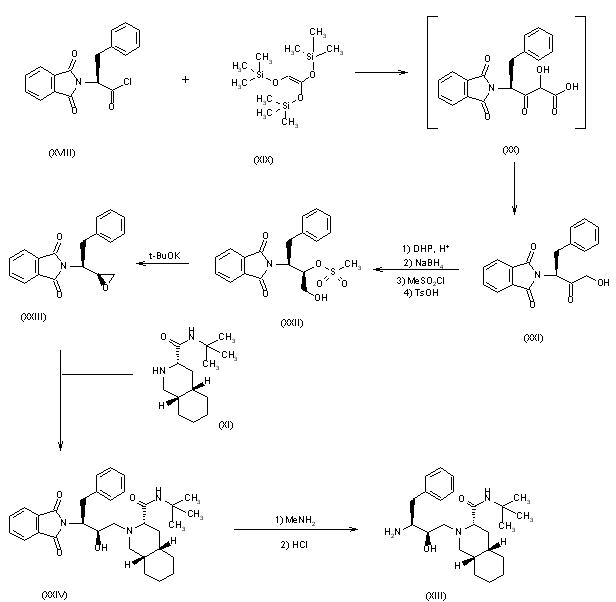
SYN
3) The condensation of N-(tert-butoxycarbonyl)-L-phenylalaninal (XXV) with 2-(trimethylsilyl)thiazole (XXVI) by means of tetrabutylammonium fluoride gives the thiazole derivative (XXVII), which is cleaved by reaction with methyl iodide (formation of the thiazolium derivative) and treated with NaBH4 and HgCl2 to afford the protected 3(S)-amino-2(S)-hydroxy-4-phenylbutanal (XXVIII). Finally, this compound is reductocondensed with isoquinoline (XI) by means of sodium cyanoborohydride to yield the protected condensation product (XII), already obtained in scheme 16810301a.
SYN
4) The selective esterification of 3(S)-azido-4-phenylbutane-1,2(S)-diol (XXIX) with 2,4,6-triiosopropylbenzenesulfonyl chloride (XXX) gives the sulfonate ester (XXXI), which by treatment with KOH is converted to the azido epoxide (XXXII). The condensation of (XXXII) with decahydroisoquinoline (XI) affords the azido condensation product (XXXIII), which is finally hydrogenated with H2 over Pd/C to the amino condensation product (XIII), already obtained in scheme 16810301a. 5) The reaction of (XXIX) with SOCl2 and RuCl3 gives the dioxathiole dioxide (XXXIV), which is condensed with decahydroisoquinoline (XI) to afford the azido condensation product (XXXIII), already obtained.
SYN
The intermediate (3R,4S)-4-[N-(tert-butoxycarbonyl)-N-methylamino]-5-phenyl-3-(tert-butyldimethylsilyloxy)pentanoic acid (VII) has been obtained as follows: The condensation of N-(tert-butoxycarbonyl)-L-phenylalanine (I) with the Mg salt of malonic acid monoethyl ester (II) by means of CDI gives the beta-ketoester (III), which is reduced with NaBH4 to yield (3R,4S)-4-(tert-butoxycarbonylamino)-3-hydroxy-5-phenylpentanoic acid ethyl ester (IV). The protection of the OH group of (IV) with Tbdms-Cl and imidazole in DMF affords the silylated ester (V), which is hydrolyzed with NaOH to provide the corresponding carboxylic acid (VI). Finally, this compound is N-methylated by means of Me-I and NaH in THF to obtain the target intermediate (VII).
SYN
J Label Compd Radiopharm 1998,41(12),1103
[14C]-Saquinavir: The cyclization of [ring-14C]-aniline (I) with crotonic aldehyde (II) by means of HCl and acetic anhydride gives labeled 2-methylquinoline (III), which is brominated with Br2 in acetic acid yielding the tribromo derivative (IV). The hydrolysis of (IV) with hot sulfuric acid afforded labeled quinoline-2-carboxylic acid (V), which is finally condensed with Ro-32-0445 (VI) by means of hydroxybenzotriazole (HOBT) and dicyclohexylcarbodiimide (DCC) in THF.
SYN
Pentadeuterated saquinavir: The nitration of hexadeuterobenzene (VII) with HNO3/H2SO4 gives pentadeuteronitrobenzene (VIII), which is hydrogenated with deuterium/Pt in D1-methanol yielding heptadeuteroaniline (IX). The cyclization of (IX) with crotonic aldehyde (II) by means of DCI/D2O and acetic anhydride as before affords hexadeuterated quinoline (X), which is brominated with Br2 as before giving the tribromo derivative (XI). The hydrolysis of (XI) with sulfuric acid as before yields the acid (XII), which is finally condensed with Ro-32-0445 (VI) as before.
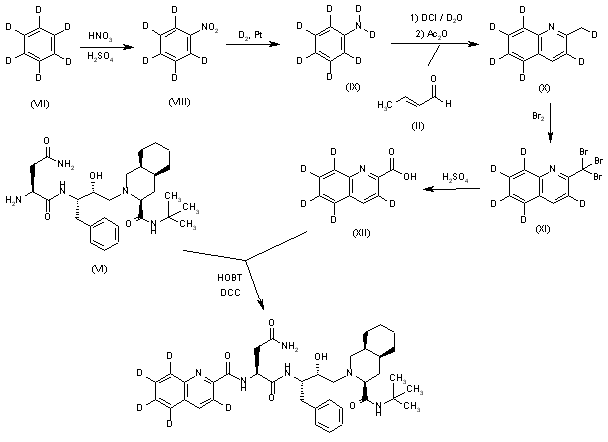
SYN
Tetradeuterated saquinavir: The cyclization of heptadeuteroaniline (IX) with crotonic aldehyde (II) by means of HCl and acetic anhydride as before gives the tetradeuteroquinoline (XIII), which is brominated as described yielding the tribromo derivative (XIV). The hydrolysis of (XIV) with sulfuric acid affords tetradeuterated acid (XV), which is finally condensed with Ro-32-0445 (VI) as indicated.
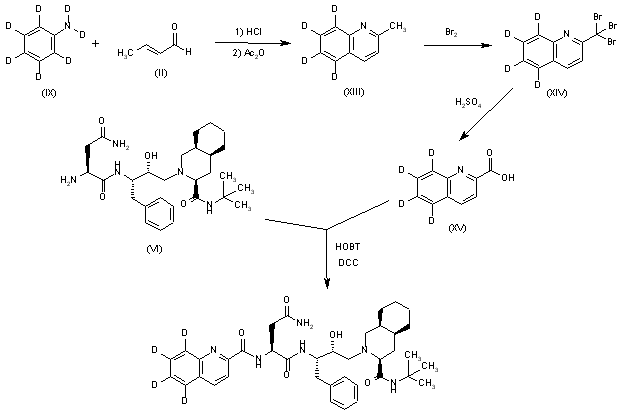
SYN
Tritiated saquinavir: The cyclization of 4-bromoaniline (XVI) with crotonic aldehyde (II) by means of ZnCl2/HCl gives 6-bromo-4-methylquinoline (XVII), which is brominated as before giving tetrabromo derivative (XVIII). The hydrolysis of (XVIII) with sulfuric cid affords 6-bromoquinoline-2-carboxylic acid (XIX), which is condensed with Ro-32-0445 (VI) by means of HOBT and DCC as indicated giving the bromo derivative of saquinavir (XX). Finally, this compound is tritiated with T2 over Pd/C in ethanol.
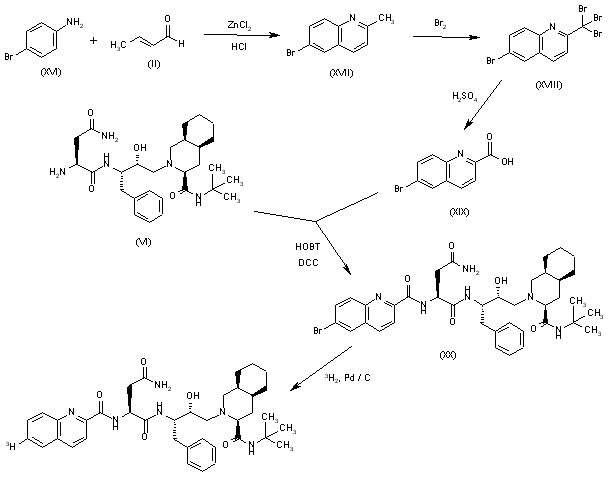
SYN
5)[15N,13C,2H]-Saquinavir: The nitration of [13C6]-benzene (XXI) with [15N]-nitric acid gives the corresponding nitrobenzene (XXII), which is reduced with Sn/HCl to the aniline (XXIII). The cyclization of (XXIII) with crotonic aldehyde (II) by means of ClD/D2O and acetic ahydride yields the tetradeuterated quinoline (XXIV), which is brominated as before givig the tribromo derivative (XXV). The hydrolysis of (XXV) with sulfuric acid as usual affords the [15N,13C6,2H3]-labeled quinoline-2-carboxylic acid (XXVI), which is finally condensed with Ro-32-0445 (VI) by means of HOBT and CDI as indicated.
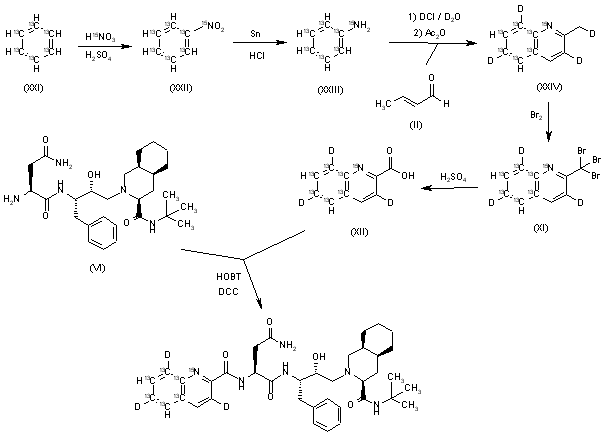
Saquinavir (SQV), sold under the brand names Invirase and Fortovase, is an antiretroviral drug used together with other medications to treat or prevent HIV/AIDS.[3] Typically it is used with ritonavir or lopinavir/ritonavir to increase its effect.[3] It is taken by mouth.[3]
Common side effects include nausea, vomiting, diarrhea, and feeling tired.[3] More serious side effects include problems with QT prolongation, heart block, high blood lipids, and liver problems.[3] It appears to be safe in pregnancy.[3] It is in the protease inhibitor class and works by blocking the HIV protease.[3]
Saquinavir was patented in 1988 and first sold in 1995.[4][5]
Medical uses
Saquinavir is used together with other medications to treat or prevent HIV/AIDS.[3] Typically it is used with ritonavir or lopinavir/ritonavir to increase its effect.[3]
Side effects
The most frequent adverse events with saquinavir in either formulation are mild gastrointestinal symptoms, including diarrhoea, nausea, loose stools and abdominal discomfort. Invirase is better tolerated than Fortovase.[medical citation needed]
Bioavailability and drug interactions
Saquinavir, in the Invirase formulation, has a low and variable oral bioavailability, when given alone. The Fortovase formulation at the standard dosage delivers approximately eightfold more active drug than Invirase, also at the standard dosage.[6]
In the clinic, it was found that the oral bioavailability of saquinavir in both formulations significantly increases when patients also receive the PI ritonavir. For patients, this has the major benefit that they can take less saquinavir, while maintaining sufficient saquinavir blood plasma levels to efficiently suppress the replication of HIV.[medical citation needed]
The mechanism behind this welcome observation was not directly known, but later it was determined that ritonavir inhibits the cytochrome P450 3A4 isozyme. Normally, this enzyme metabolizes saquinavir to an inactive form, but with the ritonavir inhibiting this enzyme, the saquinavir blood plasma levels increased considerably. Additionally, ritonavir also inhibits multidrug transporters, although to a much lower extent.[medical citation needed]
Unlike other protease inhibitors, the absorption of saquinavir seems to be improved by omeprazole.[7]
Mechanism of action
Saquinavir is a protease inhibitor. Proteases are enzymes that cleave protein molecules into smaller fragments. HIV protease is vital for both viral replication within the cell and release of mature viral particles from an infected cell. Saquinavir binds to the active site of the viral protease and prevents cleavage of viral polyproteins, preventing maturation of the virus. Saquinavir inhibits both HIV-1 and HIV-2 proteases.[8]
History

New HIV infections and deaths, before and after the FDA approval of “highly active antiretroviral therapy”,[9] of which saquinavir, ritonavir and indinavir were key as the first three protease inhibitors.Cully, Megan (28 November 2018). “Protease inhibitors give wings to combination therapy”. nature. Open Publishing. Retrieved 28 October 2020. As a result of the new therapies, HIV deaths in the United States fell dramatically within two years.}}[9]
Saquinavir was developed by the pharmaceutical company Roche.[10] Saquinavir was the sixth antiretroviral and the first protease inhibitor approved by the US Food and Drug Administration (FDA), leading ritonavir and indinavir by a few months.[11] This new class of antiretrovirals played a critical role in the development of highly active antiretroviral therapy (HAART), which helped significantly lower the risk of death from AIDS-related causes, as seen by a reduction of the annual U.S. HIV-associated death rate, from over 50,000 to about 18,000 over a period of two years.[9][12]
Roche requested and received approval of Invirase via the FDA’s “Accelerated Approval” program—a process designed to speed drugs to market for the treatment of serious diseases—a decision that was controversial, as AIDS activists disagreed over the benefits of thorough testing versus early access to new drugs.[13][better source needed] It was approved again on November 7, 1997, as Fortovase,[14] a soft gel capsule reformulated for improved bioavailability. Roche announced in May 2005 that, given reduced demand, Fortovase would cease being marketed early in 2006, in favor of Invirase boosted with ritonavir,[15] owing to the ability of the latter co-formulated drug to inhibit the enzyme that metabolizes the AIDS drugs.[citation needed]
Society and culture
Economics
As of 2015, it is not available as a generic medication.[16]
Formulations
Two formulations have been marketed:
- a hard-gel capsule formulation of the mesylate, with trade name Invirase, which requires combination with ritonavir to increase the saquinavir bioavailability;
- a soft-gel capsule formulation of saquinavir (microemulsion,[17] orally-administered formulation), with trade name Fortovase, which was discontinued worldwide in 2006.[18]
References
- ^ “Saquinavir Use During Pregnancy”. Drugs.com. 20 March 2018. Retrieved 28 January 2020.
- ^ “Invirase- saquinavir mesylate capsule INVIRASE- saquinavir mesylate tablet, film coated”. DailyMed. 26 December 2019. Retrieved 28 January 2020.
- ^ Jump up to:a b c d e f g h i “Saquinavir”. The American Society of Health-System Pharmacists. Archived from the original on 8 September 2015. Retrieved 5 September 2015.
- ^ Minor, Lisa K. (2006). Handbook of Assay Development in Drug Discovery. Hoboken: CRC Press. p. 117. ISBN 9781420015706. Archived from the original on 31 March 2016.
- ^ Fischer, Jnos; Ganellin, C. Robin (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 509. ISBN 9783527607495.
- ^ “Fortovase”. Drugs.com. 22 March 2019. Retrieved 28 January2020.
- ^ Winston A, Back D, Fletcher C, et al. (2006). “Effect of omeprazole on the pharmacokinetics of saquinavir-500 mg formulation with ritonavir in healthy male and female volunteers”. AIDS. 20 (10): 1401–6. doi:10.1097/01.aids.0000233573.41597.8a. PMID 16791014. S2CID 44506039.
- ^ Raphael Dolin, Henry Masur, Michael S. Saag. “AIDS Therapy“, Churchill Livingstone, (1999), p. 129.
- ^ Jump up to:a b c “HIV Surveillance—United States, 1981-2008”. Archivedfrom the original on 9 November 2013. Retrieved 8 November 2013.
- ^ J. Hilts, Philip (8 December 1995). “MF.D.A. Backs A New Drug To Fight AIDS”. New York Times. Retrieved 28 October 2020.
- ^ “Antiretroviral Drug Discovery and Development”. NIH. 26 November 2018. Retrieved 29 October 2020.
- ^ The CDC, in its Morbidity and Mortality Weekly Report, ascribes this to “highly active antiretroviral therapy”, without mention of either of these drugs, see the preceding citation. A further citation is needed to make this accurate connection between this drop and the introduction of the protease inhibitors.
- ^ “Drugs! Drugs! Drugs! An Overview of the Approved Anti-HIV Medications”. The Body. Archived from the original on 9 November 2013. Retrieved 20 February 2013.
- ^ “Drug Approval Package: Fortovase/Saquinavir NDA 20828”. U.S. Food and Drug Administration (FDA). 24 December 1999. Retrieved 28 January 2020.
- ^ Withdrawal of Fortovase (PDF) Archived 2006-05-14 at the Wayback Machine
- ^ “Generic Invirase Availability”. Drugs.com. Retrieved 9 July2020.
- ^ Gibaud S, Attivi D (August 2012). “Microemulsions for oral administration and their therapeutic applications” (PDF). Expert Opinion on Drug Delivery. 9 (8): 937–51. doi:10.1517/17425247.2012.694865. PMID 22663249. S2CID 28468973.
- ^ News-Medical.Net. May 18, 2005 Roche to discontinue the sale and distribution of Fortovase (saquinavir) Archived 2015-02-22 at the Wayback Machine
External links
- “Saquinavir”. Drug Information Portal. U.S. National Library of Medicine.
links
| Clinical data | |
|---|---|
| Trade names | Invirase, Fortovase |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a696001 |
| License data | EU EMA: by INNUS DailyMed: Saquinavir |
| Pregnancy category | AU: B1[1] |
| ATC code | J05AE01 (WHO) |
| Legal status | |
| Legal status | US: ℞-only |
| Pharmacokinetic data | |
| Bioavailability | ~4% (without ritonavir boosting)[2] |
| Protein binding | 98% |
| Metabolism | Liver, mainly by CYP3A4 |
| Elimination half-life | 9–15 hours |
| Excretion | feces (81%) and urine (3%) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 127779-20-8 |
| PubChem CID | 441243 |
| IUPHAR/BPS | 4813 |
| DrugBank | DB01232 |
| ChemSpider | 390016 |
| UNII | L3JE09KZ2F |
| KEGG | D00429 |
| ChEMBL | ChEMBL114 |
| NIAID ChemDB | 000640 |
| PDB ligand | ROC (PDBe, RCSB PDB) |
| CompTox Dashboard (EPA) | DTXSID6044012 |
| Chemical and physical data | |
| Formula | C38H50N6O5 |
| Molar mass | 670.855 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
///////////////saquinavir, Antiviral, Peptidomimetics, HIV Protease Inhibitor, Ro-31-8959, EU 2021, APPROVALS 2021, Invirase, Ro 31 8959, Ro 31-8959, RO 31-8959/000, Ro 318959, RO-31-8959/000, Sch 52852, SCH-52852
[H][C@@]12CCCC[C@]1([H])CN(C[C@@H](O)[C@H](CC1=CC=CC=C1)NC(=O)[C@H](CC(N)=O)NC(=O)C1=NC3=C(C=CC=C3)C=C1)[C@@H](C2)C(=O)NC(C)(C)C


NEW DRUG APPROVALS
ONE TIME
$10.00
Bimekizumab
Heavy chain)
EVQLVESGGG LVQPGGSLRL SCAASGFTFS DYNMAWVRQA PGKGLEWVAT ITYEGRNTYY
RDSVKGRFTI SRDNAKNSLY LQMNSLRAED TAVYYCASPP QYYEGSIYRL WFAHWGQGTL
VTVSSASTKG PSVFPLAPSS KSTSGGTAAL GCLVKDYFPE PVTVSWNSGA LTSGVHTFPA
VLQSSGLYSL SSVVTVPSSS LGTQTYICNV NHKPSNTKVD KKVEPKSCDK THTCPPCPAP
ELLGGPSVFL FPPKPKDTLM ISRTPEVTCV VVDVSHEDPE VKFNWYVDGV EVHNAKTKPR
EEQYNSTYRV VSVLTVLHQD WLNGKEYKCK VSNKALPAPI EKTISKAKGQ PREPQVYTLP
PSRDELTKNQ VSLTCLVKGF YPSDIAVEWE SNGQPENNYK TTPPVLDSDG SFFLYSKLTV
DKSRWQQGNV FSCSVMHEAL HNHYTQKSLS LSPGK
(Light chain)
AIQLTQSPSS LSASVGDRVT ITCRADESVR TLMHWYQQKP GKAPKLLIYL VSNSEIGVPD
RFSGSGSGTD FRLTISSLQP EDFATYYCQQ TWSDPWTFGQ GTKVEIKRTV AAPSVFIFPP
SDEQLKSGTA SVVCLLNNFY PREAKVQWKV DNALQSGNSQ ESVTEQDSKD STYSLSSTLT
LSKADYEKHK VYACEVTHQG LSSPVTKSFN RGEC
(Disulfide bridge: H22-H96, H152-H208, H228-L214, H234-H’234, H237-H’237, H269-H329, H375-H433, H’22-H’96, H’152-H’208, H’228-L’214, H’269-H’329, H’375-H’433, L23-L88, L134-L194, L’23-L’88, L’134-L’194)
Bimekizumab
ビメキズマブ (遺伝子組換え)
UCB 4940
| Formula | C6552H10132N1750O2029S42 |
|---|---|
| CAS | 1418205-77-2 |
| Mol weight | 147227.7921 |
EU APPROVED, 2021/8/20, Bimzelx
Immunoglobulin G1, anti-(human interleukin 17A/interleukin 17F) (human-Rattus norvegicus monoclonal UCB4940 heavy chain), disulfide with human-Rattus norvegicus monoclonal UCB4940 light chain, dimer
Protein Sequence
Sequence Length: 1338, 455, 455, 214, 214multichain; modified (modifications unspecified)
| Product details | |
|---|---|
| Name | Bimzelx |
| Agency product number | EMEA/H/C/005316 |
| Active substance | Bimekizumab |
| International non-proprietary name (INN) or common name | bimekizumab |
| Therapeutic area (MeSH) | Psoriasis |
| Anatomical therapeutic chemical (ATC) code | L04AC |
Bimzelx 160 mg solution for injection in pre-filled syringe Bimzelx 160 mg solution for injection in pre-filled pen
The active substance in Bimzelx, bimekizumab, is a monoclonal antibody, a protein designed to attach to interleukins IL-17A, IL-17F and IL-17AF, which are messenger molecules in the body’s immune system (the body’s natural defences). High levels of these interleukins have been shown to be involved in developing inflammatory diseases caused by the immune system, such as plaque psoriasis. By attaching to these interleukins, bimekizumab prevents them from interacting with their receptors (targets) on the surface of the epidermis (outer layer of the skin), which reduces inflammation and improves the symptoms related to plaque psoriasis.,,, https://www.ema.europa.eu/en/documents/overview/bimzelx-epar-medicine-overview_en.pdf
Antipsoriatic, Anti-IL-17A/IL-17F antibody, Monoclonal antibody
Treatment of moderate to severe plaque psoriasis
Bimekizumab, sold under the brand name Bimzelx, is a humanized anti-IL17A, anti-IL-17F, and anti-IL17AF monoclonal antibody[1][2] that is used to treat plaque psoriasis.[1]
The most common side effects include upper respiratory tract infections (nose and throat infection) and oral candidiasis (thrush, a fungal infection in the mouth or throat).[1]
Bimekizumab was approved for medical use in the European Union in August 2021.[1][3]
Drug: bimekizumab
Company: UCB
Used for: psoriasis
Est. 2026 sales: $1.63 billion
Monoclonal antibody treatments for psoriasis are stacking up—but UCB hopes to muscle into the market with bimekizumab this year. The anti-IL-17A and IL-17F injection showed up both Johnson & Johnson’s Stelara and Novartis blockbuster Cosentyx in trials.
UCB’s Stelara head-to-head, the Be Vivid study presented in June at the American Academy of Dermatology and later published in The Lancet, found 85% of bimekizumab patients had a 90% or greater reduction in the area and severity of their psoriasis symptoms at 16 weeks. Complete skin clearance, indicated by a score of PASI 100, happened in 59% of patients.
Stelara, for its part, helped just half of patients reach PASI 90 and 21% achieve complete skin clearance over the same time period.
That Be Vivid readout raised expectations of a potentially favorable outcome in UCB’s head-to-head study with Novartis blockbuster Cosentyx (secukinumab), called Be Radiant.
RELATED: UCB’s bimekizumab blows J&J’s Stelara away in phase 3, raising expectations for Cosentyx showdown
In July, UCB announced that in that phase 3 study, its candidate had “demonstrate(d) superiority to secukinumab for complete skin clearance at both weeks 16 and 48.” The full study results will be presented “in due course,” UCB promised.
The data from the Cosentyx trial could be worth a lot to UCB, Evaluate wrote in June, adding that Jefferies analysts at the time expected annual sales of bimekizumab to top out around $1.5 billion. If bimekizumab beats Cosentyx, the sales forecast could rise to above $2 billion, it said at the time.
Without specific Cosentyx-topping data from the Be Radiant study in hand, Evaluate pegs consensus sales estimates at $1.63 billion in 2026.
One concern for UCB is whether the smaller pharma will be able to compete with the big marketing budgets in psoriasis. AbbVie’s Skyrizi and Humira, Novartis’ Cosentyx, Eli Lilly’s Taltz and Amgen’s Otezla are just a handful of the psoriasis drugs that have spent millions on mainstream TV ads to build brand names.
RELATED: DiCE scores $80M to roll oral IL-17 psoriasis med into the clinic
In September, the FDA and EMA accepted UCB’s biologics license application (BLA) for bimekizumab for adults with moderate to severe plaque psoriasis, the company reported. Ongoing phase 3 trials are evaluating the drug to treat a variety of other conditions, including psoriatic arthritis, ankylosing spondylitis, non-radiographic axial spondyloarthritis and hidradenitis suppurativa.
In the meantime, more competition is on the way. South San Francisco biotech DiCE Molecules, for its part, last month nabbed new funding to the tune of $80 million to roll its oral small molecule IL-17 program into a clinical trial in psoriasis and build out preclinical programs.
In addition to IL-17 rivals, others are also looking to get in on the action—particularly, several TYK2 inhibitors. Bristol Myers Squibb’s deucravacitinib recently bested Otezla in a study, while both Pfizer and Nimbus Therapeutics are in phase 2 studies with prospects of their own.
Psoriatic arthritis (PsA) is a complex and heterogeneous inflammatory disease that affects 20% to 30% of patients with psoriasis and is associated with substantial disability, impaired quality of life (QoL), and several comorbidities.1–3 It involves diverse clinical domains that extend beyond musculoskeletal manifestations (peripheral and axial arthritis, enthesitis and dactylitis): eg, nails, gut, and eyes, in addition to latent or manifest psoriasis.
Although there is still a huge gap in knowledge on the pathophysiology of PsA, what is known has fortunately turned into new treatment approaches that have improved symptoms and outcomes for PsA patients over the last two decades. Pro-inflammatory cytokines have been recognized as potential treatment targets in inflammatory diseases and have led to the creation of a number of anti-cytokine monoclonal antibodies that have revolutionized its treatment, such as TNFα and IL-12/23 inhibitors.4 More recently, the IL-17 pathway has been shown to play an important role in the pathophysiology of psoriatic disease and its blockage has shown to be clinically beneficial, as demonstrated with IL-17A inhibitors secukinumab and ixekizumab.4 Some patients, however, still do not respond, stop responding over time or suffer from side effects, leading to drug discontinuation, and other times combination strategies are required to control all PsA’s disease domains. Thus, there is still a great need for novel therapeutic options.5
Dual inhibitor antibodies target two different cytokines simultaneously potentially offering a better disease control. Interleukin (IL)-17A and IL-17F share structural homology and have a similar biologic function. IL-17A is classically considered to be the most biologically active, but recent studies have shown that IL-17F is also increased in psoriatic skin and synovial cell in psoriatic arthritis, supporting the rationale for targeting both IL-17A and IL-17F in psoriatic disease. Bimekizumab is the first-in-class monoclonal antibody designed to simultaneously target IL-17A and IL-17F.
Medical uses
Bimekizumab is indicated for the treatment of moderate to severe plaque psoriasis in adults who are candidates for systemic therapy.[1]
History
This drug is being developed by Belgian pharmaceutical UCB. Phase III trials have demonstrated that bimekizumab is superior to not only adalimumab[4] but also secukinumab[5] for the treatment of plaque psoriasis.
Names
Bimekizumab is the international nonproprietary name (INN).[6]
The Role of Interleukin (IL)‑17A and IL‑17F in Psoriatic Arthritis
The IL-17 cytokine family comprises six different members (from A to F), of which IL-17A is the most studied. Known to be produced by a wide range of immune cells, IL-17A is involved in the pathophysiology of several inflammatory diseases including spondyloarthritis.6–8
Most non-hematopoietic cells possess IL-17 receptors, including fibroblasts, epithelial cells and synoviocytes,8 but despite this ubiquitous presence, IL-17 seems to have only moderate inflammatory capability per se, rather recruiting and amplifying other pathways, such as IL-6, IL-8, TNF and inflammatory-cell attracting chemokines.6,7,9,10
Still, evidence supporting the centrality of the IL-17 pathway in both PsO and PsA is available from a wide range of data.11 Th17 cells, IL-17 protein and related genes are elevated in both skin, blood and synovial fluid of PsO and PsA patients.11,12 In PsA, increased levels of IL-17+ CD4 and CD813,14, as well as IL-17A+Tγδ cells, have been found in the synovial fluid compared with peripheral blood. Specifically, the levels of IL-17+CD8+ cells in the synovial fluid distinguish PsA from rheumatoid arthritis (RA) and correlate with increased DAS28 scores, C-reactive protein levels, power-doppler findings of activity and prevalence of erosions.13 Inhibition of this pathway is capable of normalizing almost four times more disease-related genes than anti-TNFα treatments.11,15
Within the entire IL-17 family, IL-17F is the most structurally homologous (~50%) to IL-17A8 (Figure 1). They can both be secreted as homodimers (ie IL-17A/A or IL-17F/F) or as heterodimers of IL-17A/IL-17F,9 sharing signaling pathways through the same heterodimeric complex of IL-17 receptors A and C (IL-RA/RC) and biologic function.7–9
| Figure 1 Summarized schematic of inhibition of the IL-17 cytokine family. *Not approved for psoriatic arthritis. Notes: Reprinted by permission from Springer Nature Customer Service Centre GmbH: Springer Nature, BioDrugs, Reis J, Vender R, Torres T. Bimekizumab: the first dual inhibitor of interleukin (IL)-17A and IL-17F for the treatment of psoriatic disease and ankylosing spondylitis, COPYRIGHT 2019.6Abbreviations: IL, interleukin; IL-17RA, IL-17 receptor A; IL-17RB, IL-17 receptor B; IL-17RC, IL-17 receptor C; IL-17RE, IL-17 receptor E. |
The role of both IL-17A and F in psoriasis pathogenesis has been previously addressed.6,9,16
In enthesitis, a central pathologic process in PsA, Tγδ cells have recently been described that are capable of producing both IL-17A and IL-17F even independently of IL-23 stimulation.17 IL-17A and F had already been shown to promote osteogenic differentiation in in vitro models of human periosteum activated through the use of Th17 and Tγδ cells or through culture with serum from patients with ankylosing spondylitis,18 a mechanism potentially implied in the development of enthesitis. Importantly, both cytokines seem to be equipotent in this role, unlike in inflammatory processes where IL-17F seems to be less potent.18
Both IL-17A and IL-17F, when synergized with TNF, lead to increased production of pro-inflammatory cytokines, such as IL-8 and IL-6 in synoviocytes of PsA patients.9 IL-17A seems to be the most pro-inflammatory of the two cytokines.9,19 However, despite some inconsistencies in the literature regarding IL-17F detection levels which might be attributable to differences in methodology,19 IL-17F levels have been reported to be 30–50 times higher in some cytokine microenvironments, such as in psoriatic skin lesions of PsA patients20 or the synovium,21 which might dilute differences in relative potency. Additionally, IL-17F seems to be significantly increased in the synovium of PsA compared to osteoarthritis (OA) patients, unlike IL-17A.21 Dual neutralization of both IL-17A and IL-17F (using bimekizumab) resulted in greater downregulation of pro-inflammatory cytokine production than a single blockade in synovial fibroblasts.9,19 Critically, in in vitro models, anti-TNF blockade alone did not reduce the production of IL-8 as much as both IL-17A and F neutralization or even just anti-IL17A alone.9,19 In in vitro models of human periosteum dual blockade of IL-17A and F was also more effective in suppressing osteogenic differentiation than the blockade of either cytokine individually.18
Interestingly, in Tγδ cells, the predominant IL-17 production seems to be the F subtype.18 Also of note is the recent description that the IL-17receptorC (IL-17RC) competes with IL-17RA for IL-17F, IL-17A and IL-17A/F heterodimers,22 suggesting the possibility of IL-17RA-independent signaling pathways (and thus not targeted by brodalumab, an anti-IL17RA monoclonal antibody).
Bimekizumab
Bimekizumab is a humanized monoclonal IgG1 antibody that selectively neutralizes both IL-17A and IL-17F. In in vitro models, bimekizumab appears to be as potent as ixekizumab at inhibiting IL-17A (also more potent than secukinumab)8 but, unlike those drugs, also possesses the unique ability to inhibit IL-17F as well, functioning as a dual inhibitor. Unlike brodalumab, an IL-17 receptor A blocker – which targets not only IL-17A and F signaling but also IL-17 C, D and E – bimekizumab spares IL-17E (also known as IL-25), for example, which is believed to have anti-inflammatory properties.6
Bimekizumab demonstrates dose-proportional linear pharmacokinetics, with a half-life ranging from 17 to 26 days, and its distribution is restricted to the extravascular compartment.23 Currently, bimekizumab is in advanced clinical development for psoriasis, but also for psoriatic arthritis, and ankylosing spondylitis (both currently in phase III).
Bimekizumab in PsA – Efficacy
Phase I
The first bimekizumab clinical trial in PsA was a phase Ib randomized, double-blind, placebo-controlled clinical trial that included 53 patients (39 treated with bimekizumab, 14 with placebo) with active psoriatic arthritis who had failed conventional disease-modifying antirheumatic drugs (DMARDs) and/or one biologic DMARD. Patients in the active treatment arm were randomized to four different treatment regimens of varying loading doses (ranging from 80 to 560 mg) and maintenance doses (from 40 to 320 mg) at weeks 0, 3 and 6. Patients were followed for up to 20 weeks.9
Patients treated with bimekizumab had a faster response, compared to placebo. This was first detected at week two, with maximal or near-maximal responses maintained up to week 20, for both arthritis and skin psoriasis. ACR20, 50 and 70 responses were maximal at week 8 (80%), week 12 (57%) and week 16 (37%), respectively. For patients with skin involvement, PASI75 and PASI100 responses at week 8 were 100% and 87%, respectively (Table 1).
Phase II
BE ACTIVE10 was a 48-week multicentric, international, phase 2b dose-ranging, randomized, double-blind placebo-controlled trial to assess the efficacy and safety of bimekizumab. Two hundred and six adult patients (out of 308 screened) with active (tender and swollen count >3) PsA (diagnosed according to CASPAR criteria) were enrolled in 5 treatment arms (placebo, 16 mg, 160 mg with single 320 mg loading dose, 160 mg, 320 mg bimekizumab dose, with SC injections every 4 weeks). Concurrent use of TNF inhibitors was not permitted but conventional DMARDs (if on a stable dose and kept throughout the study), corticosteroids (equal or less 10mg/day) and NSAIDs were allowed. Sixteen-milligram bimekizumab (a much lower dose than other treatment arms) was tested with a programmed re-randomization at week 12 to either 160 or 320 mg dosing (meaning no placebo arm after 12 weeks). All patients received treatment up to week 48.
The primary outcome was ACR50 response at 12 weeks, a much more stringent outcome than used for other IL-17 inhibitors. The prespecified analysis was not possible due to the absence of a statistically significant difference versus placebo for the 320 mg group at week 12. All other outcomes were thus considered exploratory, rendering this a failed primary endpoint with no active comparator group.
At 12 weeks, significant ACR50 responses were present for every bimekizumab group, although lower in both the 16 mg and 320 mg dose group (Table 1 reports average values for all bimekizumab treatment groups). The 160 mg dosing had the greatest ACR and PASI response rates. These were confirmed to be increasing response rates up to week 24 and stability thereafter up to week 48, where the results of both 160 and 320 mg were similar. There were also responses in PASI scores, enthesitis, HAQ-DI and SF-36 across all bimekizumab doses. There was no loss of efficacy by week 48.
At the recent American College of Rheumatology (ACR) congress, additional data on BE ACTIVE were reported. BASDAI scoring was improved on the 93 patients in the treatment arm (160–320 mg bimekizumab) who had a baseline score >4 (mean 6.2 ± 1.42). BASDAI50 response rates were 43% and 56% at week 12 and 48, respectively.24
Regarding patient-reported outcomes (PROs), the Health assessment questionnaire Disability Index (HAQ-DI) and the psoriatic arthritis impact of disease-9 (PsAID-9) questionnaire developed specifically to assess health-related quality of life (QoL) in PsA were used on 206 patients from the BE ACTIVE trial. Rapid improvement was registered by week 12 and this response was sustained up to 48 weeks. Better QoL was associated with the better clinical outcomes reported in that study.25,26
Open-Label Extension Study (OLE)
Results from the 108 weeks of follow-up in the open-label extension study of BE ACTIVE (BE ACTIVE2, NCT03347110) have been recently presented.27,28 All patients who completed all 48 weeks of the BE ACTIVE trial were enrolled and switched to the 160 mg dosing regardless of previous treatment dose regimen. Over 108 weeks (an additional 60 weeks of OLE study over the 48 of the original BE ACTIVE trial) there was a 66.7% and 75.4% ACR 50 and body surface area (BSA) 0% response, respectively. Dactylitis and enthesitis were also significantly improved completely resolving in 65.9% and 77.9% of patients, respectively.27 Regarding week 12 responders, ACR20/50/70 and BSA 0% responses were maintained until week 108 in 80/78/81% and 72%, respectively.27 MDA/VLDA responses and DAPSA remission were maintained by 81/72/76% of Week 12 responders, respectively, to Week 120 (MDA/VLDA), and Week 108 (DAPSA remission).
Bimekizumab in PsA – Safety
Phase I
Over 90% of reported adverse events, in both arms, were mild or moderate. In the treatment arm, two fungal infections (oropharyngeal and vulvovaginal candidiasis) were reported, both treated with oral medication. There was no increased incidence of other infections. There were no deaths or severe adverse events resulting from treatment, and no patient discontinued bimekizumab.9
Phase II
No difference was found in the frequency of adverse events between placebo and treatment arms by week 12 in the BE ACTIVE trial. After reallocation (after week 12) and up to the 48 weeks of the trial 151 (74%) of the total 204 patients who ever received bimekizumab reported some AE (exposure adjusted incidence rate 166.8/100 patient-years). Most AE were mild or moderate (the most frequently reported were nasopharyngitis and upper respiratory tract infections) and there was no direct association with bimekizumab dose.
Nine patients (8 of which received bimekizumab) had serious adverse effects. These included one patient with drug-induced liver injury. Another patient also had severe liver enzyme elevation. Both had been given the 320 mg dosing. From the hepatic point of view, the other 11 patients were noted to have increased liver enzymes (>3x ULN). There was no relation with bimekizumab dose, and most were on DMARDs and one was on TB prophylaxis. At least two serious adverse events were related to infections across the entire study period (28 weeks) – 1 hepatitis E infection, 1 cellulitis (both with the 160 mg dosing). Non-severe Candida infection was reported in 7% of the patients, none led to treatment discontinuation. Other serious AEs reported were melanoma in situ (160 mg), suicidal ideation (160 mg loading dose), and neutropenia (320 mg dosing) (only in one patient each).10 In summary, this safety profile overlaps with those of other anti-IL17 therapies.29
In the OLE study, at week 108, serious adverse events occurred in 9.3% of patients (no deaths or major adverse cardiac events) and a total of 8.8% of patients withdrew from the study due to side effects. Full publication is still pending but the authors share that the safety profile observed in the OLE study reflected previous observations.27
Discussion
Dual inhibitor antibodies represent a novel therapeutic strategy, and a logical extension of the success monoclonal antibodies has had over the last couple of decades.
Here we review the most recent information on IL-17A and F inhibition in psoriatic arthritis through the first-of-its-class bimekizumab, a dual inhibitor of both cytokines.
The importance of the IL-17 pathway in psoriatic arthritis, already suggested by preclinical data, was reinforced by the excellent results obtained by secukinumab30 or ixekizumab31 in the control of the disease in the last few years.
Indeed, IL-17 seems to be involved in all of the clinical domains of psoriatic arthritis. In preclinical trials, it has been shown that both IL-17A and F are capable of inducing pro-inflammatory cytokines, like IL-8 or IL-6, in synoviocytes, periosteum and the skin,23 and that this activation was greatly suppressed by blocking both these cytokines simultaneously. Research is expanding on the differential role of IL-17F in different environments,18,21 compared with the more studied IL-17A, as well as possible alternative signaling pathways.22 Taken together these findings could potentially explain different clinical phenotypes in PsA and treatment responses to anti-IL17A (secukinumab, ixekizumab) and IL-17RA (brodalumab) inhibitors furthering support for the use of dual cytokine blockade such as with bimekizumab (Figure 1).
Phase II trials, specifically BE ACTIVE results, have been encouraging. Bimekizumab has shown to be relatively fast-acting, with initial improvements detected by week 8 and well established by week 12. Additionally, at a dose of 160 mg every 4 weeks, bimekizumab has shown to be capable of retaining this level of response in a high percentage of patients for at least 2 years. These results are independent of prior exposure to anti-TNF therapy.10
As with all new drugs, there are still pending questions regarding its optimal use. In BE ACTIVE,10 in which patients received four different dosages through the first 12 weeks, the 160 mg seemed most effective. The initial lower response in the 320 mg group might have been produced by a higher proportion of refractory patients in which bimekizumab took longer to work. This impression is reinforced, in the author’s opinion, by the fact that response rates were different as early as week 4 in both 160 mg (loading dose) and 320 mg dose groups although by that time period both groups had received the same dose. Co-medication was balanced between both groups.
Whichever dose proves best, these results were achieved with mostly mild side-effects that did not lead to treatment discontinuation – most commonly nasopharyngitis, upper respiratory infections and candidiasis. Overall the available data have not revealed any unexpected adverse events. Nonetheless, the number of patients included in the trials is still small. Thirteen out of the 204 patients (6,4%) receiving any dose of bimekizumab in the BE ACTIVE trial had some hepatic adverse effect, raising the need for attentive monitoring by treating physicians. Co-medication needs to be well pondered in this setting as well, but if real-world outcomes of bimekizumab prove as beneficial as in the trials there might be a reduced need for concomitant use of other DMARDs. Although IL-17F has been shown to be associated with increased susceptibility in many forms of human cancer, it has shown a protective role in colon tumorigenesis in mice,32,33 mainly by regulating tumor angiogenesis.6 Longer and bigger trials will be needed to fully ascertain the safety of bimekizumab.
Overall the available results for this new therapeutic option in psoriatic arthritis are encouraging, although it is still early to completely understand the added value offered by bimekizumab. As of yet, however, there are no head-to-head trials directly comparing it to other treatment options in PsA. Anti-IL17A monoclonal antibodies have been evaluated against other therapies, such as anti-TNF inhibitors in the treatment of PsA with mixed results (using different endpoints).34,35
Right now we can only look to early reports from the more advanced Phase 3 trials in psoriasis, where bimekizumab was first studied, which already encompass hundreds of patients and compare bimekizumab with other biologics. A head-to-head comparison with ustekinumab was recently published36 involving 567 patients (321 randomized to bimekizumab, 163 to ustekinumab and 83 to a placebo arm that was switched to bimekizumab at week 16). Using a 320 mg dose of bimekizumab every 4 weeks (and not the 160 mg shown in BE ACTIVE to be the most efficacious in PsA) bimekizumab was superior to ustekinumab (85% vs 49.7% PASI 90 responses at week 16, p<0.001). This response was also sustained throughout the 52-week duration of the study (81.6% vs 55.8%, p<0.001). Similar responses (86.2% vs 47.2% PASI 90 at week 16, p<0.001) in the BE SURE trial comparing bimekizumab (320 mg every 4 weeks or 320 mg until week 16 and then every 8 weeks) and adalimumab (80 mg week 0, 40 mg week 1 and every 2 weeks) were recently presented.37 Switching adalimumab patients to bimekizumab resulted in increased response rates, comparable to rates in bimekizumab-randomized patients at week 56. UCB, the company developing bimekizumab, have also reported the superiority of bimekizumab against secukinumab.38
If nothing else, bimekizumab is a proof-of-concept for a novel avenue in treating inflammatory diseases. Up until now the clinical practice in inflammatory diseases has been to steer clear of the combination of monoclonal antibodies. The results of the trials reported here using bimekizumab to simultaneously inhibit two cytokines, even if related ones, are an important reminder of the redundant and overlapping nature of the immune system and of the multiple pathways through which one arrives at inflammatory disease.
As of yet, however, there are no head-to-head trials directly comparing bimekizumab to conventional DMARDS or other bDMARDs in PsA although the results reported here seem encouraging. Upcoming trials (see Table 2) will hopefully fill this gap in knowledge.
Conclusion
Psoriatic arthritis can be a severe and disabling disease. Although improvements in its treatment have been achieved in the past decade, its pathogenesis is not completely known, and its treatment is still difficult particularly throughout all disease domains.
The IL-17 pathway has been implicated in disease pathogenesis and targeting IL-17A with secukinumab and ixekizumab has shown good results, although there is still a large proportion of patients that respond only partially. The simultaneous blockade of both IL-17A and IL-17F seems to have a synergistic benefit, with IL-17F inhibition contributing with a differentiated role in both osteogenesis and skin inflammation, important domains of PsA.
Bimekizumab uses a novel approach to biologic treatment in psoriatic arthritis through dual cytokine blockade. Mounting evidence from early trials has shown a good safety and efficacy profile, with rapid onset and sustained response, with results now extending to 108 weeks of follow-up. Moreover, clinical trials in skin psoriasis have also shown that bimekizumab is highly effective, confirming the importance of inhibiting these two cytokines in psoriatic disease.
In the near future, phase III trials will help to better understand the potential of bimekizumab in the treatment of psoriatic arthritis.
References
- ^ Jump up to:a b c d e f “Bimzelx EPAR”. European Medicines Agency (EMA). 23 June 2021. Retrieved 24 August 2021. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Lim SY, Oon HH (2019-05-13). “Systematic review of immunomodulatory therapies for hidradenitis suppurativa”. Biologics. 13: 53–78. doi:10.2147/BTT.S199862. PMC 6526329. PMID 31190730.
- ^ “UCB Announces European Commission Approval of Bimzelx (bimekizumab) for the Treatment of Adults with Moderate to Severe Plaque Psoriasis”. UCB (Press release). 24 August 2021. Retrieved 24 August 2021.
- ^ Warren, Richard B.; Blauvelt, Andrew; Bagel, Jerry; Papp, Kim A.; Yamauchi, Paul; Armstrong, April; Langley, Richard G.; Vanvoorden, Veerle; De Cuyper, Dirk; Cioffi, Christopher; Peterson, Luke (2021-07-08). “Bimekizumab versus Adalimumab in Plaque Psoriasis”. New England Journal of Medicine. 385 (2): 130–141. doi:10.1056/NEJMoa2102388. ISSN 0028-4793. PMID 33891379.
- ^ Reich, Kristian; Warren, Richard B.; Lebwohl, Mark; Gooderham, Melinda; Strober, Bruce; Langley, Richard G.; Paul, Carle; De Cuyper, Dirk; Vanvoorden, Veerle; Madden, Cynthia; Cioffi, Christopher (2021-07-08). “Bimekizumab versus Secukinumab in Plaque Psoriasis”. New England Journal of Medicine. 385 (2): 142–152. doi:10.1056/NEJMoa2102383. ISSN 0028-4793. PMID 33891380.
- ^ World Health Organization (2014). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 72”. WHO Drug Information. 28 (3). hdl:10665/331112.
Further reading
- Reis J, Vender R, Torres T (August 2019). “Bimekizumab: The First Dual Inhibitor of Interleukin (IL)-17A and IL-17F for the Treatment of Psoriatic Disease and Ankylosing Spondylitis”. BioDrugs. 33 (4): 391–9. doi:10.1007/s40259-019-00361-6. PMID 31172372. S2CID 174812750.
External links
- “Bimekizumab”. Drug Information Portal. U.S. National Library of Medicine.
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized |
| Target | IL17A, IL17F, IL17AF |
| Clinical data | |
| Trade names | Bimzelx |
| License data | EU EMA: by INN |
| ATC code | None |
| Legal status | |
| Legal status | EU: Rx-only [1] |
| Identifiers | |
| CAS Number | 1418205-77-2 |
| UNII | 09495UIM6V |
| KEGG | D11550 |
//////////Bimekizumab, Bimzelx, EU 2021, APPROVALS 2021, Monoclonal antibody
, plaque psoriasis,ビメキズマブ (遺伝子組換え) , UCB 4940
NEW DRUF APPROVALS
ONE TIME ANTHONY CRASTO +919321316780 amcrasto@gmail.com
$10.00
Tralokinumab
(Heavy chain)
QVQLVQSGAE VKKPGASVKV SCKASGYTFT NYGLSWVRQA PGQGLEWMGW ISANNGDTNY
GQEFQGRVTM TTDTSTSTAY MELRSLRSDD TAVYYCARDS SSSWARWFFD LWGRGTLVTV
SSASTKGPSV FPLAPCSRST SESTAALGCL VKDYFPEPVT VSWNSGALTS GVHTFPAVLQ
SSGLYSLSSV VTVPSSSLGT KTYTCNVDHK PSNTKVDKRV ESKYGPPCPS CPAPEFLGGP
SVFLFPPKPK DTLMISRTPE VTCVVVDVSQ EDPEVQFNWY VDGVEVHNAK TKPREEQFNS
TYRVVSVLTV LHQDWLNGKE YKCKVSNKGL PSSIEKTISK AKGQPREPQV YTLPPSQEEM
TKNQVSLTCL VKGFYPSDIA VEWESNGQPE NNYKTTPPVL DSDGSFFLYS RLTVDKSRWQ
EGNVFSCSVM HEALHNHYTQ KSLSLSLGK
(Light chain)
SYVLTQPPSV SVAPGKTARI TCGGNIIGSK LVHWYQQKPG QAPVLVIYDD GDRPSGIPER
FSGSNSGNTA TLTISRVEAG DEADYYCQVW DTGSDPVVFG GGTKLTVLGQ PKAAPSVTLF
PPSSEELQAN KATLVCLISD FYPGAVTVAW KADSSPVKAG VETTTPSKQS NNKYAASSYL
SLTPEQWKSH RSYSCQVTHE GSTVEKTVAP TECS
(Disulfide bridge: H22-H96, H149-H205, H263-H323, H369-H427, H228-H’228, H231-H’231, L22-L87, L136-L195, H136-L213)
Tralokinumab
トラロキヌマブ (遺伝子組換え)
| Formula | C6374H9822N1698O2014S44 |
|---|---|
| CAS | 1044515-88-9 |
| Mol weight | 143873.2167 |
EU APPROVED, Adtralza, 2021/6/17
Antiasthmatic, Anti-inflammatory, Anti-IL-13 antibody
Tralokinumab is a human monoclonal antibody which targets the cytokine interleukin 13,[1] and is designed for the treatment of asthma and other inflammatory diseases.[2] Tralokinumab was discovered by Cambridge Antibody Technology scientists, using Ribosome Display, as CAT-354[3] and taken through pre-clinical and early clinical development.[4] After 2007 it has been developed by MedImmune, a member of the AstraZeneca group, where it is currently in Ph3 testing for asthma and Ph2b testing for atopic dermatitis.[5][6] This makes it one of the few fully internally discovered and developed drug candidates in AstraZeneca’s late stage development pipeline.
Discovery and development
Tralokinumab (CAT-354) was discovered by Cambridge Antibody Technology scientists[7] using protein optimization based on Ribosome Display.[8] They used the extensive data sets from ribosome display to patent protect CAT-354 in a world-first of sequence-activity-relationship claims.[7] In 2004, clinical development of CAT-354 was initiated with this first study completing in 2005.[9] On 21 July 2011, MedImmune LLC initiated a Ph2b, randomized, double-blind study to evaluate the efficacy of tralokinumab in adults with asthma.[10]
In 2016, MedImmune and AstraZeneca were developing tralokinumab for asthma (Ph3) and atopic dermatitis (Ph2b) while clinical development for moderate-to-severe ulcerative colitis and idiopathic pulmonary fibrosis (IPF) have been discontinued.[9] In July of that year AstraZeneca licensed Tralokinumab to LEO Pharma for skin diseases.[11]
A phase IIb study of Tralokinumab found that treatment was associated with early and sustained improvements in atopic dermatitis symptoms and tralokinumab had an acceptable safety and tolerability profile, thereby providing evidence for targeting IL-13 in patients with atopic dermatitis.[12]
On 15 June 2017, Leo Pharma announced that they were starting phase III clinical trials with tralokinumab in atopic dermatitis.[13]
Society and culture
Legal status
On 22 April 2021, the Committee for Medicinal Products for Human Use (CHMP) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Adtralza, intended for the treatment of moderate‑to‑severe atopic dermatitis.[14]
The applicant for this medicinal product is LEO Pharma A/S.
References
- ^ Kopf M, Bachmann MF, Marsland BJ (September 2010). “Averting inflammation by targeting the cytokine environment”. Nature Reviews. Drug Discovery. 9 (9): 703–18. doi:10.1038/nrd2805. PMID 20811382. S2CID 23769909.
- ^ “Statement On A Nonproprietary Name Adopted By The USAN Council: Tralokinumab” (PDF). American Medical Association.
- ^ Thom G, Cockroft AC, Buchanan AG, Candotti CJ, Cohen ES, Lowne D, et al. (May 2006). “Probing a protein-protein interaction by in vitro evolution” [P]. Proceedings of the National Academy of Sciences of the United States of America. 103 (20): 7619–24. Bibcode:2006PNAS..103.7619T. doi:10.1073/pnas.0602341103. PMC 1458619. PMID 16684878.
- ^ May RD, Monk PD, Cohen ES, Manuel D, Dempsey F, Davis NH, et al. (May 2012). “Preclinical development of CAT-354, an IL-13 neutralizing antibody, for the treatment of severe uncontrolled asthma”. British Journal of Pharmacology. 166 (1): 177–93. doi:10.1111/j.1476-5381.2011.01659.x. PMC 3415647. PMID 21895629.
- ^ “Pipeline”. MedImmune. Retrieved 11 June 2013.
- ^ “Studies found for CAT-354”. ClinicalTrials.gov. Retrieved 11 June 2013.
- ^ Jump up to:a b Human Antibody Molecules for Il-13, retrieved 2015-07-26
- ^ Jermutus L, Honegger A, Schwesinger F, Hanes J, Plückthun A (January 2001). “Tailoring in vitro evolution for protein affinity or stability”. Proceedings of the National Academy of Sciences of the United States of America. 98 (1): 75–80. Bibcode:2001PNAS…98…75J. doi:10.1073/pnas.98.1.75. PMC 14547. PMID 11134506.
- ^ Jump up to:a b “Tralokinumab”. Adis Insight. Springer Nature Switzerland AG.
- ^ Clinical trial number NCT01402986 for “A Phase 2b, Randomized, Double-blind Study to Evaluate the Efficacy of Tralokinumab in Adults With Asthma” at ClinicalTrials.gov
- ^ “AstraZeneca enters licensing agreements with LEO Pharma in skin diseases”.
- ^ Wollenberg A, Howell MD, Guttman-Yassky E, Silverberg JI, Kell C, Ranade K, et al. (January 2019). “Treatment of atopic dermatitis with tralokinumab, an anti-IL-13 mAb”. The Journal of Allergy and Clinical Immunology. 143 (1): 135–141. doi:10.1016/j.jaci.2018.05.029. PMID 29906525.
- ^ “LEO Pharma starts phase 3 clinical study for tralokinumab in atopic dermatitis”. leo-pharma.com. AstraZeneca. 1 July 2016.
- ^ “Adtralza: Pending EC decision”. European Medicines Agency. 23 April 2021. Retrieved 23 April 2021.
| Tralokinumab Fab fragment bound to IL-13. From PDB 5L6Y. | |
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | IL-13 |
| Clinical data | |
| ATC code | D11AH07 (WHO) |
| Identifiers | |
| CAS Number | 1044515-88-9 |
| ChemSpider | none |
| UNII | GK1LYB375A |
| KEGG | D09979 |
| Chemical and physical data | |
| Formula | C6374H9822N1698O2014S44 |
| Molar mass | 143875.20 g·mol−1 |
| (what is this?) (verify) |
/////////Tralokinumab, Adtralza, EU 2021, APPROVALS 2021, Antiasthmatic, Anti-inflammatory, Anti-IL-13 antibody, MONOCLONAL ANTIBODY, PEPTIDE, トラロキヌマブ (遺伝子組換え) ,
NEW DRUG APPROVALS
ONE TIME
$10.00
Dostarlimab
(Heavy chain)
EVQLLESGGG LVQPGGSLRL SCAASGFTFS SYDMSWVRQA PGKGLEWVST ISGGGSYTYY
QDSVKGRFTI SRDNSKNTLY LQMNSLRAED TAVYYCASPY YAMDYWGQGT TVTVSSASTK
GPSVFPLAPC SRSTSESTAA LGCLVKDYFP EPVTVSWNSG ALTSGVHTFP AVLQSSGLYS
LSSVVTVPSS SLGTKTYTCN VDHKPSNTKV DKRVESKYGP PCPPCPAPEF LGGPSVFLFP
PKPKDTLMIS RTPEVTCVVV DVSQEDPEVQ FNWYVDGVEV HNAKTKPREE QFNSTYRVVS
VLTVLHQDWL NGKEYKCKVS NKGLPSSIEK TISKAKGQPR EPQVYTLPPS QEEMTKNQVS
LTCLVKGFYP SDIAVEWESN GQPENNYKTT PPVLDSDGSF FLYSRLTVDK SRWQEGNVFS
CSVMHEALHN HYTQKSLSLS LGK
(Light chain)
DIQLTQSPSF LSAYVGDRVT ITCKASQDVG TAVAWYQQKP GKAPKLLIYW ASTLHTGVPS
RFSGSGSGTE FTLTISSLQP EDFATYYCQH YSSYPWTFGQ GTKLEIKRTV AAPSVFIFPP
SDEQLKSGTA SVVCLLNNFY PREAKVQWKV DNALQSGNSQ ESVTEQDSKD STYSLSSTLT
LSKADYEKHK VYACEVTHQG LSSPVTKSFN RGEC
(Disulfide bridge: H22-H96, H130-L214, H143-H199, H222-H’222, H225-H’225, H257-H317, H363-H421, H’22-H’96, H’130-L’214, H’143-H’199, H’257-H’317, H’363-H’421, L23-L88, L134-L194, L’23-L’88, L’194-L’134)
>Heavy Chain EVQLLESGGGLVQPGGSLRLSCAASGFTFSSYDMSWVRQAPGKGLEWVSTISGGGSYTYY QDSVKGRFTISRDNSKNTLYLQMNSLRAEDTAVYYCASPYYAMDYWGQGTTVTVSSASTK GPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYS LSSVVTVPSSSLGTKTYTCNVDHKPSNTKVDKRVESKYGPPCPPCPAPEFLGGPSVFLFP PKPKDTLMISRTPEVTCVVVDVSQEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTYRVVS VLTVLHQDWLNGKEYKCKVSNKGLPSSIEKTISKAKGQPREPQVYTLPPSQEEMTKNQVS LTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSRLTVDKSRWQEGNVFS CSVMHEALHNHYTQKSLSLSLGK
>Light Chain DIQLTQSPSFLSAYVGDRVTITCKASQDVGTAVAWYQQKPGKAPKLLIYWASTLHTGVPS RFSGSGSGTEFTLTISSLQPEDFATYYCQHYSSYPWTFGQGTKLEIKRTVAAPSVFIFPP SDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLT LSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC
References:
- Statement on a Nonproprietary Name Adopted by the USAN Council: Dostarlimab [Link]
Dostarlimab
Immunoglobulin G4, anti-(programmed cell death protein 1 (PDCD1)) (humanized clone ABT1 γ4-chain), disulfide with humanized clone ABT1 κ-chain, dimer
Protein Sequence
Sequence Length: 1314, 443, 443, 214, 214multichain; modified (modifications unspecified)
- GSK-4057190
- GSK4057190
- TSR 042
- TSR-042
- WBP-285
- ANB 011
| Formula | C6420H9832N1680O2014S44 |
|---|---|
| CAS | 2022215-59-2 |
| Mol weight | 144183.6677 |
Jemperli FDA 2021/4/22 AND EMA 2021/4/21
NEW DRUG APPROVALS
ONE TIME
$10.00
Dostarlimab, sold under the brand name Jemperli, is a monoclonal antibody medication used for the treatment of endometrial cancer.[1][2][3][4]
The most common adverse reactions (≥20%) were fatigue/asthenia, nausea, diarrhea, anemia, and constipation.[1][2] The most common grade 3 or 4 adverse reactions (≥2%) were anemia and transaminases increased.[1][2]
Dostarlimab is a programmed death receptor-1 (PD-1)–blocking antibody.[1][2]
Dostarlimab was approved for medical use in the United States in April 2021.[1][2][5]
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Jemperli | Injection | 50 mg/1mL | Intravenous | GlaxoSmithKline LLC | 2021-04-22 | Not applicable |  |
Medical uses
Dostarlimab is indicated for the treatment of adults with mismatch repair deficient (dMMR) recurrent or advanced endometrial cancer, as determined by an FDA-approved test, that has progressed on or following prior treatment with a platinum-containing regimen.[1][2]
On April 22, 2021, the Food and Drug Administration granted accelerated approval to dostarlimab-gxly (Jemperli, GlaxoSmithKline LLC) for adult patients with mismatch repair deficient (dMMR) recurrent or advanced endometrial cancer, as determined by an FDA-approved test, that has progressed on or following a prior platinum-containing regimen.
Efficacy was evaluated based on cohort (A1) in GARNET Trial (NCT02715284), a multicenter, multicohort, open-label trial in patients with advanced solid tumors. The efficacy population consisted of 71 patients with dMMR recurrent or advanced endometrial cancer who progressed on or after a platinum-containing regimen. Patients received dostarlimab-gxly, 500 mg intravenously, every 3 weeks for 4 doses followed by 1,000 mg intravenously every 6 weeks.
The main efficacy endpoints were overall response rate (ORR) and duration of response (DOR), as assessed by blinded independent central review (BICR) according to RECIST 1.1. Confirmed ORR was 42.3% (95% CI: 30.6%, 54.6%). The complete response rate was 12.7% and partial response rate was 29.6%. Median DOR was not reached, with 93.3% of patients having durations ≥6 months (range: 2.6 to 22.4 months, ongoing at last assessment).
Serious adverse reactions occurred in 34% of patients receiving dostarlimab-gxly. Serious adverse reactions in >2% of patients included sepsis , acute kidney injury , urinary tract infection , abdominal pain , and pyrexia . The most common adverse reactions (≥20%) were fatigue/asthenia, nausea, diarrhea, anemia, and constipation. The most common grade 3 or 4 adverse reactions (≥2%) were anemia and transaminases increased. Immune-mediated adverse reactions can occur including pneumonitis, colitis, hepatitis, endocrinopathies, and nephritis.
The recommended dostarlimab-gxly dose and schedule (doses 1 through 4) is 500 mg every 3 weeks. Subsequent dosing, beginning 3 weeks after dose 4, is 1,000 mg every 6 weeks until disease progression or unacceptable toxicity. Dostarlimab-gxly should be administered as an intravenous infusion over 30 minutes.
View full prescribing information for Jemperli.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
FDA also approved the VENTANA MMR RxDx Panel as a companion diagnostic device for selecting endometrial cancer patients for treatment with dostarlimab-gxly.
This review used the Real-Time Oncology Review (RTOR) pilot program, which streamlined data submission prior to the filing of the entire clinical application, and the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment.
This application was granted priority review, and breakthrough therapy designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
Side effects
Serious adverse reactions in >2% of patients included sepsis, acute kidney injury, urinary tract infection, abdominal pain, and pyrexia.[1][2]
Immune-mediated adverse reactions can occur including pneumonitis, colitis, hepatitis, endocrinopathies, and nephritis.[1][2]
History
Like several other available and experimental monoclonal antibodies, it is a PD-1 inhibitor. As of 2020, it is undergoing Phase I/II and Phase III clinical trials.[6][7][8] The manufacturer, Tesaro, announced prelimary successful results from the Phase I/II GARNET study.[6][9][10]
In 2020, the GARNET study announced that Dostarlimab was demonstrating potential to treat a subset of women with recurrent or advanced endometrial cancer.[11]
April 2021, Dostarlimab is approved for the treatment of recurrent or advanced endometrial cancer with deficient mismatch repair (dMMR), which are genetic anomalies abnormalities that disrupt DNA repair.[12]
On April 22, 2021, the Food and Drug Administration granted accelerated approval to dostarlimab-gxly (Jemperli, GlaxoSmithKline LLC).[1] Efficacy was evaluated based on cohort (A1) in GARNET Trial (NCT02715284), a multicenter, multicohort, open-label trial in patients with advanced solid tumors.[1]
Society and culture
Legal status
On 25 February 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a conditional marketing authorization for the medicinal product Jemperli, intended for the treatment of certain types of recurrent or advanced endometrial cancer.[13] The applicant for this medicinal product is GlaxoSmithKline (Ireland) Limited.[13]
References[
- ^ Jump up to:a b c d e f g h i j k “FDA grants accelerated approval to dostarlimab-gxly for dMMR endometri”. U.S. Food and Drug Administration(FDA) (Press release). 22 April 2021. Retrieved 22 April 2021.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c d e f g h i “Jemperli- dostarlimab injection”. DailyMed. Retrieved 28 April 2021.
- ^ Statement On A Nonproprietary Name Adopted By The USAN Council – Dostarlimab, American Medical Association.
- ^ World Health Organization (2018). “International Nonproprietary Names for Pharmaceutical Substances (INN). Proposed INN: List 119” (PDF). WHO Drug Information. 32 (2).
- ^ “FDA grants accelerated approval for GSK’s Jemperli (dostarlimab-gxly) for women with recurrent or advanced dMMR endometrial cancer” (Press release). GlaxoSmithKline. 22 April 2021. Retrieved 22 April 2021 – via PR Newswire.
- ^ Jump up to:a b Clinical trial number NCT02715284 for “A Phase 1 Dose Escalation and Cohort Expansion Study of TSR-042, an Anti-PD-1 Monoclonal Antibody, in Patients With Advanced Solid Tumors (GARNET)” at ClinicalTrials.gov
- ^ Clinical trial number NCT03981796 for “A Study of Dostarlimab (TSR-042) Plus Carboplatin-paclitaxel Versus Placebo Plus Carboplatin-paclitaxel in Patients With Recurrent or Primary Advanced Endometrial Cancer (RUBY)” at ClinicalTrials.gov
- ^ Clinical trial number NCT03602859 for “A Phase 3 Comparison of Platinum-Based Therapy With TSR-042 and Niraparib Versus Standard of Care Platinum-Based Therapy as First-Line Treatment of Stage III or IV Nonmucinous Epithelial Ovarian Cancer (FIRST)” at ClinicalTrials.gov
- ^ “Data from GARNET study indicates robust activity of dostarlimab in patients with advanced or recurrent endometrial cancer”. Tesaro (Press release). Retrieved 1 January 2020.
- ^ Scalea B (28 May 2019). “Dostarlimab Effective in Endometrial Cancer Regardless of MSI Status”. Targeted Oncology. Retrieved 1 January 2020.
- ^ “GSK Presents New Data from the GARNET Study Demonstrating Potential of Dostarlimab to Treat a Subset of Women with Recurrent or Advanced Endometrial Cancer – Drugs.com MedNews”. Drugs.com. Retrieved 29 April 2020.
- ^ “FDA Approves New Immunotherapy for Endometrial Cancer”. Medscape. Retrieved 23 April 2021.
- ^ Jump up to:a b “Jemperli: Pending EC decision”. European Medicines Agency (EMA) (Press release). 25 February 2021. Retrieved 22 April 2021.
External links
- “Dostarlimab”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT02715284 for “Study of TSR-042, an Anti-programmed Cell Death-1 Receptor (PD-1) Monoclonal Antibody, in Participants With Advanced Solid Tumors (GARNET)” at ClinicalTrials.gov
- Kaplon H, Muralidharan M, Schneider Z, Reichert JM: Antibodies to watch in 2020. MAbs. 2020 Jan-Dec;12(1):1703531. doi: 10.1080/19420862.2019.1703531. [Article]
- Temrikar ZH, Suryawanshi S, Meibohm B: Pharmacokinetics and Clinical Pharmacology of Monoclonal Antibodies in Pediatric Patients. Paediatr Drugs. 2020 Apr;22(2):199-216. doi: 10.1007/s40272-020-00382-7. [Article]
- Green AK, Feinberg J, Makker V: A Review of Immune Checkpoint Blockade Therapy in Endometrial Cancer. Am Soc Clin Oncol Educ Book. 2020 Mar;40:1-7. doi: 10.1200/EDBK_280503. [Article]
- Deshpande M, Romanski PA, Rosenwaks Z, Gerhardt J: Gynecological Cancers Caused by Deficient Mismatch Repair and Microsatellite Instability. Cancers (Basel). 2020 Nov 10;12(11). pii: cancers12113319. doi: 10.3390/cancers12113319. [Article]
- FDA Approved Drug Products: Jemperli (dostarlimab-gxly) for intravenous injection [Link]
- FDA News Release: FDA grants accelerated approval to dostarlimab-gxly for dMMR endometrial cancer [Link]
- Statement on a Nonproprietary Name Adopted by the USAN Council: Dostarlimab [Link]
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized |
| Target | PCDP1 |
| Clinical data | |
| Trade names | Jemperli |
| Other names | TSR-042, WBP-285, dostarlimab-gxly |
| License data | US DailyMed: Dostarlimab |
| Routes of administration | Intravenous |
| Drug class | Antineoplastic |
| ATC code | L01XC40 (WHO) |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| CAS Number | 2022215-59-2 |
| PubChem SID | 384585344 |
| DrugBank | DB15627 |
| UNII | P0GVQ9A4S5 |
| KEGG | D11366 |
| Chemical and physical data | |
| Formula | C6420H9832N1690O2014S44 |
| Molar mass | 144325.73 g·mol−1 |
/////////Dostarlimab, PEPTIDE, ANTINEOPLASTIC, CANCER, ドスタルリマブ , GSK 4057190, GSK4057190, TSR 042, TSR-042, WBP-285, FDA 2021, EU 2021
Lenalidomide hydrate,


Lenalidomide hydrate
レナリドミド水和物
An immunomodulator.
CC-5013 hemihydrate
2,6-Piperidinedione, 3-(4-amino-1,3-dihydro-1-oxo-2H-isoindol-2-yl)-, hydrate (2:1)
(+/-)-2,6-Piperidinedione, 3-(4-amino-1,3-dihydro-1-oxo-2H-isoindol-2-yl)-, hydrate (2:1)
| Formula | (C13H13N3O3)2. H2O |
|---|---|
| CAS | 847871-99-2 |
| Mol weight | 536.5365 |
EMA APPROVED 2021/2/11, Lenalidomide KRKA
Research Code:CDC-501; CC-5013
Trade Name:Revlimid®
MOA:Angiogenesis inhibitor
Indication:Myelodysplastic syndrome (MDS); Mantle cell lymphoma (MCL); Multiple myeloma (MM)
Status:Approved
Company:Celgene (Originator)
Sales:$5,801.1 Million (Y2015); 
$4,980 Million (Y2014);;
$4280 Million (Y2013);;
$3766.6 Million (Y2012);;
$3208.2 Million (Y2011);ATC Code:L04AX04
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2005-12-27 | Marketing approval | Revlimid | Multiple myeloma (MM),Myelodysplastic syndrome (MDS),Mantle cell lymphoma (MCL) | Capsule | 2.5 mg/5 mg/10 mg/15 mg/20 mg/25 mg | Celgene | Priority; Orphan |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2007-06-14 | Marketing approval | Revlimid | Multiple myeloma (MM),Myelodysplastic syndrome (MDS) | Capsule | 2.5 mg/5 mg/7.5 mg/10 mg/15 mg/20 mg/25 mg | Celgene | Orphan |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2010-08-20 | New indication | Revlimid | Myelodysplastic syndrome (MDS) | Capsule | 5 mg | Celgene | |
| 2010-06-25 | Marketing approval | Revlimid | Multiple myeloma (MM) | Capsule | 5 mg | Celgene |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2013-01-23 | Marketing approval | 瑞复美/Revlimid | Multiple myeloma (MM) | Capsule | 5 mg | Celgene | |
| 2013-01-23 | Marketing approval | 瑞复美/Revlimid | Multiple myeloma (MM) | Capsule | 10 mg | Celgene | |
| 2013-01-23 | Marketing approval | 瑞复美/Revlimid | Multiple myeloma (MM) | Capsule | 15 mg | Celgene | |
| 2013-01-23 | Marketing approval | 瑞复美/Revlimid | Multiple myeloma (MM) | Capsule | 25 mg | Celgene |
| Molecular Weight | 259.26 |
| Formula | C13H13N3O3 |
| CAS No. | 191732-72-6 (Lenalidomide); |
| Chemical Name | 3(4-amino-1-oxo 1,3-dihydro-2H-isoindol-2-yl) piperidine-2,6-dione |
Lenalidomide was first approved by the U.S. Food and Drug Administration (FDA) on Dec 27, 2005, then approved by European Medicine Agency (EMA) on June 14, 2007, and approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) on June 25, 2010. It was developed and marketed as Revlimid® by Celgene.
Lenalidomide is an analogue of thalidomide with immunomodulatory, antiangiogenic, and antineoplastic properties. In multiple myeloma cells, the combination of lenalidomide and dexamethasone synergizes the inhibition of cell proliferation and the induction of apoptosis. Revlimid® is indicated for the treatment of multiple myeloma (MM), in combination with dexamethasone, in patients who have received at least one prior therapy, transfusion-dependent anemia due to low-or intermediate-1-risk myelodysplastic syndromes (MDS) associated with a deletion 5q abnormality with or without additional cytogenetic abnormalities and mantle cell lymphoma (MCL) whose disease has relapsed or progressed after two prior therapies, one of which included bortezomib.
Revlimid® is available as capsule for oral use, containing 2.5, 5, 10, 15, 20 or 25 mg of free Lenalidomide. The recommended dose is 25 mg once daily for multiple myeloma (MM), in combination with 40 mg dexamethasone once daily, 10 mg once daily for myelodysplastic syndromes (MDS) and 25 mg once daily for mantle cell lymphoma (MCL).
Lenalidomide, sold under the trade name Revlimid among others, is a medication used to treat multiple myeloma (MM) and myelodysplastic syndromes (MDS).[2] For MM it is used after at least one other treatment and generally together with dexamethasone.[2] It is taken by mouth.[2]
Common side effects include diarrhea, itchiness, joint pain, fever, headache, and trouble sleeping.[2] Severe side effects may include low blood platelets, low white blood cells, and blood clots.[2] Use during pregnancy may harm the baby.[2] The dose may need to be adjusted in people with kidney problems.[2] It has a chemical structure similar to thalidomide but has a different mechanism of action.[3][2] How it works is not entirely clear as of 2019.[2]
Lenalidomide was approved for medical use in the United States in 2005.[2] It is on the World Health Organization’s List of Essential Medicines.[4]
Medical uses
Multiple myeloma
Lenalidomide is used to treat multiple myeloma.[5] It is a more potent molecular analog of thalidomide, which inhibits tumor angiogenesis, tumor-secreted cytokines, and tumor proliferation through induction of apoptosis.[6][7][8]
Lenalidomide is effective at inducing a complete or “very good partial” response and improves progression-free survival. Adverse events more common in people receiving lenalidomide for myeloma include neutropenia, deep vein thrombosis, infections, and an increased risk of other hematological malignancies.[9] The risk of second primary hematological malignancies does not outweigh the benefit of using lenalidomide in relapsed or refractory multiple myeloma.[10] It may be more difficult to mobilize stem cells for autograft in people who have received lenalidomide.[6]
In 2006, lenalidomide received U.S. Food and Drug Administration (FDA) clearance for use in combination with dexamethasone in people with multiple myeloma who have received at least one prior therapy.[11] In 2017, the FDA approved lenalidomide as standalone maintenance therapy (without dexamethasone) for people with multiple myeloma following autologous stem cell transplant.[12]
In 2009, The National Institute for Health and Clinical Excellence issued a final appraisal determination approving lenalidomide in combination with dexamethasone as an option to treat people with multiple myeloma who have received two or more prior therapies in England and Wales.[13]
The use of lenalidomide combined with other drugs was evaluated. It was seen that the drug combinations of lenalidomide plus dexamethasone and continuous bortezomib plus lenalidomide plus dexamethasone probably result in an increase of the overall survival.[14]
Myelodysplastic syndromes
Lenalidomide was approved by the FDA on 27 December 2005 for patients with low- or intermediate-1-risk myelodysplastic syndromes who have chromosome 5q deletion syndrome (5q- syndrome) with or without additional cytogenetic abnormalities.[15][16][17] It was approved on 17 June 2013 by the European Medicines Agency for use in patients with low- or intermediate-1-risk myelodysplastic syndromes who have 5q- deletion syndrome but no other cytogenetic abnormalities and are dependent on red blood cell transfusions, for whom other treatment options have been found to be insufficient or inadequate.[18]
Mantle cell lymphoma
Lenalidomide is approved by FDA as a specialty drug requiring a specialty pharmacy distribution for mantle cell lymphoma in patients whose disease has relapsed or progressed after at least two prior therapies, one of which must have included the medicine bortezomib.[3]
Amyloidosis
Although not specifically approved by the FDA for use in treating amyloidosis, Lenalidomide is widely used in the treatment of that condition, often in combination with dexamethasone. [19]
Adverse effects
In addition to embryo-fetal toxicity, lenalidomide carries black box warnings for hematologic toxicity (including neutropenia and thrombocytopenia) and thromboembolism.[3] Serious potential side effects include thrombosis, pulmonary embolus, hepatotoxicity, and bone marrow toxicity resulting in neutropenia and thrombocytopenia. Myelosuppression is the major dose-limiting toxicity, which is not the case with thalidomide.[20]
Lenalidomide may be associated with such adverse effects as second primary malignancy, severe cutaneous reactions, hypersensitivity reactions, tumor lysis syndrome, tumor flare reaction, hypothyroidism, and hyperthyroidism.[3]
Teratogenicity
Lenalidomide is related to thalidomide, which is known to be teratogenic. Tests in monkeys suggest that lenalidomide is likewise teratogenic.[21] It cannot be prescribed for women who are pregnant or who may become pregnant during therapy.[1] For this reason, the drug is only available in the United States through a restricted distribution system in conjunction with a risk evaluation and mitigation strategy. Females who may become pregnant must use at least two forms of reliable contraception during treatment and for at least four weeks after discontinuing treatment with lenalidomide.[3][22]
Venous thromboembolism
Lenalidomide, like its parent compound thalidomide, may cause venous thromboembolism (VTE), a potentially serious complication with their use. High rates of VTE have been found in patients with multiple myeloma who received thalidomide or lenalidomide in conjunction with dexamethasone, melphalan, or doxorubicin.[23]
Stevens-Johnson syndrome
In March 2008, the U.S. Food and Drug Administration (FDA) included lenalidomide on a list of twenty prescription drugs under investigation for potential safety problems. The drug was investigated for possibly increasing the risk of developing Stevens–Johnson syndrome, a life-threatening skin condition.[24]
FDA ongoing safety review
In 2011, the FDA initiated an ongoing review of clinical trials that found an increased risk of developing cancers such as acute myelogenous leukemia and B-cell lymphoma,[25] though it did not advise patients to discontinue treatment with lenalidomide.[26]
Mechanism of action
Lenalidomide has been used to successfully treat both inflammatory disorders and cancers in the past ten years.[when?] There are multiple mechanisms of action, and they can be simplified by organizing them as mechanisms of action in vitro and in vivo.[27] In vitro, lenalidomide has three main activities: direct anti-tumor effect, inhibition of angiogenesis, and immunomodulation. In vivo, lenalidomide induces tumor cell apoptosis directly and indirectly by inhibition of bone marrow stromal cell support, by anti-angiogenic and anti-osteoclastogenic effects, and by immunomodulatory activity. Lenalidomide has a broad range of activities that can be exploited to treat many hematologic and solid cancers.
On a molecular level, lenalidomide has been shown to interact with the ubiquitin E3 ligase cereblon[28] and target this enzyme to degrade the Ikaros transcription factors IKZF1 and IKZF3.[29] This mechanism was unexpected as it suggests that the major action of lenalidomide is to re-target the activity of an enzyme rather than block the activity of an enzyme or signaling process, and thereby represents a novel mode of drug action. A more specific implication of this mechanism is that the teratogenic and anti-neoplastic properties of lenalidomide, and perhaps other thalidomide derivatives, could be disassociated.
History
See also: Development of analogs of thalidomide
Lenalidomide was approved for medical use in the United States in 2005.[2]
Society and culture
Economics
Lenalidomide costs US$163,381 per year for the average person in the United States as of 2012.[25] Lenalidomide made almost $9.7bn for Celgene in 2018.[30]
In 2013, the UK National Institute for Health and Care Excellence (NICE) rejected lenalidomide for “use in the treatment of people with a specific type of the bone marrow disorder myelodysplastic syndrome (MDS)” in England and Scotland, arguing that Celgene “did not provide enough evidence to justify the GB£3,780 per month (US$5,746.73) price-tag of lenalidomide for use in the treatment of people with a specific type of the bone marrow disorder myelodysplastic syndrome (MDS)”.[31]
Research
Lenalidomide is undergoing clinical trial as a treatment for Hodgkin’s lymphoma,[32] as well as non-Hodgkin’s lymphoma, chronic lymphocytic leukemia and solid tumor cancers, such as carcinoma of the pancreas.[33] One Phase III clinical trial being conducted by Celgene in elderly patients with B-cell chronic lymphocytic leukemia was halted in July 2013, when a disproportionate number of cancer deaths were observed during treatment with lenalidomide versus patients treated with chlorambucil.[34]
1. WO9803502A1 / US2002173658A1.
2. Bioorg. Med. Chem. Lett. 1999, 9, 1625-1630.Route 2
Reference:
1. WO2010139266A1 / US2012077982A1.Route 3
Reference:
1. CN103497175A.Route 4
Reference:
1. WO2010139266A1 / US2012077982A1.Route 5
Reference:
1. CN103554082A.
Clip
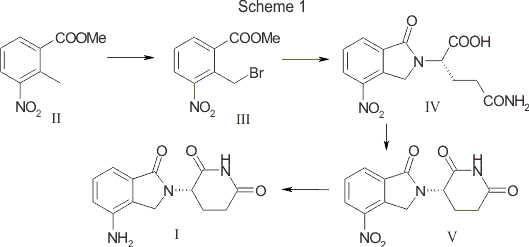
SYN

SCALABLE AND GREEN PROCESS FOR THE SYNTHESIS OF ANTICANCER DRUG LENALIDOMIDE
Yuri Ponomaryov, Valeria Krasikova, Anton Lebedev, Dmitri Chernyak, Larisa Varacheva, Alexandr Chernobroviy

Abstract
A new process for the synthesis of anticancer drug lenalidomide was developed, using platinum group metal-free and efficient reduction of nitro group with the iron powder and ammonium chloride. It was found that the bromination of the key raw material, methyl 2-methyl-3-nitrobenzoate, could be carried out in chlorine-free solvent methyl acetate without forming significant amounts of hazardous by-products. We also have compared the known synthetic methods for cyclization of methyl 2-(bromomethyl)-3-nitrobenzoate and 3-aminopiperidinedione to form lenalidomide nitro precursor.
How to Cite
Ponomaryov, Y.; Krasikova, V.; Lebedev, A.; Chernyak, D.; Varacheva, L.; Chernobroviy, A. Chem. Heterocycl. Compd. 2015, 51, 133. [Khim. Geterotsikl. Soedin. 2015, 51, 133.]
For this article in the English edition see DOI 10.1007/s10593-015-1670-0
SYN
https://link.springer.com/article/10.1007/s10593-015-1670-0

A new process for the synthesis of anticancer drug lenalidomide was developed, using platinum group metal-free and efficient reduction of nitro group with the iron powder and ammonium chloride. It was found that the bromination of the key raw material, methyl 2-methyl-3-nitrobenzoate, could be carried out in chlorine-free solvent methyl acetate without forming significant amounts of hazardous by-products. We also have compared the known synthetic methods for cyclization of methyl 2-(bromomethyl)-3-nitrobenzoate and 3-aminopiperidinedione to form lenalidomide nitro precursor.
SYN

SYN
EP 0925294; US 5635517; WO 9803502
Cyclization of N-(benzyloxycarbonyl)glutamine (I) by means of CDI in refluxing THF gives 3-(benzyloxycarbonylamino)piperidine-2,6-dione (II), which is deprotected with H2 over Pd/C in ethyl acetate/4N HCl to yield 3-aminopiperidine-2,6-dione hydrochloride (III). Bromination of 2-methyl-3-nitrobenzoic acid methyl ester (IV) with NBS in CCl4 provides 2-(bromomethyl)-3-nitrobenzoic acid methyl ester (V), which is cyclized with the aminopiperidine (III) by means of triethylamine in hot DMF to afford 3-(4-nitro-1-oxoisoindolin-2-yl)piperidine-2,6-dione (VI). Finally, the nitro group of compound (VI) is reduced with H2 over Pd/C in methanol (1, 2).
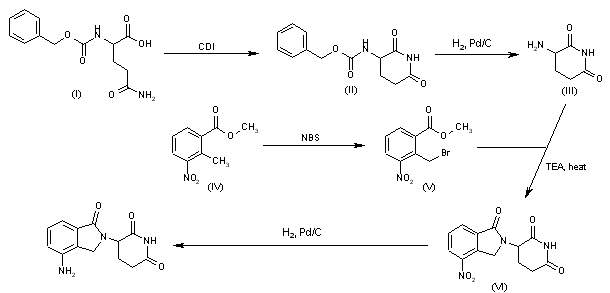
SYN
Bioorg Med Chem Lett 1999,9(11),1625
Treatment of 3-nitrophthalimide (I) with ethyl chloroformate and triethylamine produced 3-nitro-N-(ethoxycarbonyl)phthalimide (II), which was condensed with L-glutamine tert-butyl ester hydrochloride (III) to afford the phthaloyl glutamine derivative (IV). Acidic cleavage of the tert-butyl ester of (IV) provided the corresponding carboxylic acid (V). This was cyclized to the required glutarimide (VI) upon treatment with thionyl chloride and then with triethylamine. The nitro group of (VI) was finally reduced to amine by hydrogenation over Pd/C.
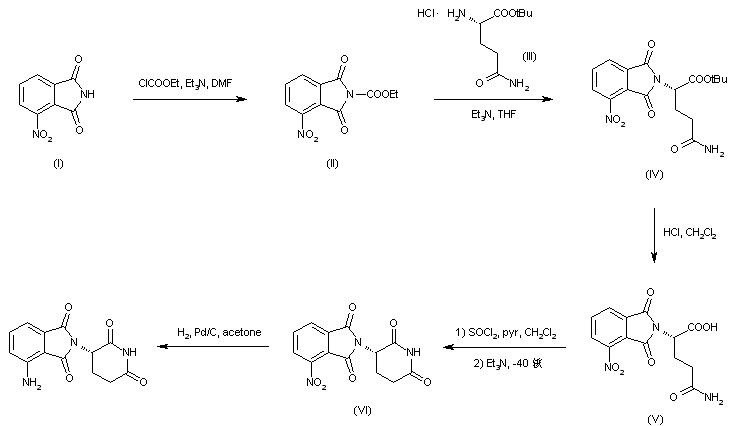
Lenalidomide
- Synonyms:CC-5013, CDC 501
- ATC:L04AX04
- MW:259.27 g/mol
- CAS-RN:191732-72-6
- InChI Key:GOTYRUGSSMKFNF-JTQLQIEISA-N
- InChI:InChI=1S/C13H13N3O3/c14-9-3-1-2-7-8(9)6-16(13(7)19)10-4-5-11(17)15-12(10)18/h1-3,10H,4-6,14H2,(H,15,17,18)/t10-/m0/s1
Synthesis
References
- ^ Jump up to:a b c “Lenalidomide (Revlimid) Use During Pregnancy”. Drugs.com. 13 March 2020. Retrieved 13 August 2020.
- ^ Jump up to:a b c d e f g h i j k “Lenalidomide Monograph for Professionals”. Drugs.com. Retrieved 27 October 2019.
- ^ Jump up to:a b c d e “DailyMed – Revlimid- lenalidomide capsule”. dailymed.nlm.nih.gov. Retrieved 27 October 2019.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ Armoiry X, Aulagner G, Facon T (June 2008). “Lenalidomide in the treatment of multiple myeloma: a review”. Journal of Clinical Pharmacy and Therapeutics. 33 (3): 219–26. doi:10.1111/j.1365-2710.2008.00920.x. PMID 18452408. S2CID 1228171.
- ^ Jump up to:a b Li S, Gill N, Lentzsch S (November 2010). “Recent advances of IMiDs in cancer therapy”. Current Opinion in Oncology. 22 (6): 579–85. doi:10.1097/CCO.0b013e32833d752c. PMID 20689431. S2CID 205547603.
- ^ Tageja N (March 2011). “Lenalidomide – current understanding of mechanistic properties”. Anti-Cancer Agents in Medicinal Chemistry. 11 (3): 315–26. doi:10.2174/187152011795347487. PMID 21426296.
- ^ Kotla V, Goel S, Nischal S, Heuck C, Vivek K, Das B, Verma A (August 2009). “Mechanism of action of lenalidomide in hematological malignancies”. Journal of Hematology & Oncology. 2: 36. doi:10.1186/1756-8722-2-36. PMC 2736171. PMID 19674465.
- ^ Yang B, Yu RL, Chi XH, Lu XC (2013). “Lenalidomide treatment for multiple myeloma: systematic review and meta-analysis of randomized controlled trials”. PLOS ONE. 8 (5): e64354. Bibcode:2013PLoSO…864354Y. doi:10.1371/journal.pone.0064354. PMC 3653900. PMID 23691202.
- ^ Dimopoulos MA, Richardson PG, Brandenburg N, Yu Z, Weber DM, Niesvizky R, Morgan GJ (March 2012). “A review of second primary malignancy in patients with relapsed or refractory multiple myeloma treated with lenalidomide”. Blood. 119 (12): 2764–7. doi:10.1182/blood-2011-08-373514. PMID 22323483.
- ^ “FDA approves lenalidomide oral capsules (Revlimid) for use in combination with dexamethasone in patients with multiple myeloma”. Food and Drug Administration (FDA). 29 June 2006. Retrieved 15 October 2015.[dead link]
- ^ “Lenalidomide (Revlimid)”. Food and Drug Administration(FDA). 22 February 2017.
- ^ “REVLIMID Receives Positive Final Appraisal Determination from National Institute for Health and Clinical Excellence (NICE) for Use in the National Health Service (NHS) in England and Wales”. Reuters. 23 April 2009.
- ^ Piechotta V, Jakob T, Langer P, Monsef I, Scheid C, Estcourt LJ, et al. (Cochrane Haematology Group) (November 2019). “Multiple drug combinations of bortezomib, lenalidomide, and thalidomide for first-line treatment in adults with transplant-ineligible multiple myeloma: a network meta-analysis”. The Cochrane Database of Systematic Reviews. 2019 (11). doi:10.1002/14651858.CD013487. PMC 6876545. PMID 31765002.
- ^ List A, Kurtin S, Roe DJ, Buresh A, Mahadevan D, Fuchs D, et al. (February 2005). “Efficacy of lenalidomide in myelodysplastic syndromes”. The New England Journal of Medicine. 352 (6): 549–57. doi:10.1056/NEJMoa041668. PMID 15703420.
- ^ List AF (August 2005). “Emerging data on IMiDs in the treatment of myelodysplastic syndromes (MDS)”. Seminars in Oncology. 32 (4 Suppl 5): S31-5. doi:10.1053/j.seminoncol.2005.06.020. PMID 16085015.
- ^ List A, Dewald G, Bennett J, Giagounidis A, Raza A, Feldman E, et al. (October 2006). “Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion”. The New England Journal of Medicine. 355 (14): 1456–65. doi:10.1056/NEJMoa061292. PMID 17021321.
- ^ “Revlimid Approved In Europe For Use In Myelodysplastic Syndromes”. The MDS Beacon. Retrieved 17 June 2013.
- ^ “Revlimid and Amyloidosis AL” (PDF). MyelomaUK. Retrieved 3 October 2020.
- ^ Rao KV (September 2007). “Lenalidomide in the treatment of multiple myeloma”. American Journal of Health-System Pharmacy. 64 (17): 1799–807. doi:10.2146/ajhp070029. PMID 17724360.
- ^ “Revlimid Summary of Product Characteristics. Annex I” (PDF). European Medicines Agency. 2012. p. 6.
- ^ Ness, Stacey (13 March 2014). “New Specialty Drugs”. Pharmacy Times. Retrieved 5 November 2015.
- ^ Bennett CL, Angelotta C, Yarnold PR, Evens AM, Zonder JA, Raisch DW, Richardson P (December 2006). “Thalidomide- and lenalidomide-associated thromboembolism among patients with cancer”. JAMA. 296 (21): 2558–60. doi:10.1001/jama.296.21.2558-c. PMID 17148721.
- ^ “Potential Signals of Serious Risks/New Safety Information Identified from the Adverse Event Reporting System (AERS) between January – March 2008”. Food and Drug Administration(FDA). March 2008. Archived from the original on 19 April 2014. Retrieved 16 December 2019.
- ^ Jump up to:a b Badros AZ (May 2012). “Lenalidomide in myeloma–a high-maintenance friend”. The New England Journal of Medicine. 366(19): 1836–8. doi:10.1056/NEJMe1202819. PMID 22571206.
- ^ “FDA Drug Safety Communication: Ongoing safety review of Revlimid (lenalidomide) and possible increased risk of developing new malignancies”. Food and Drug Administration (FDA). April 2011.
- ^ Vallet S, Palumbo A, Raje N, Boccadoro M, Anderson KC (July 2008). “Thalidomide and lenalidomide: Mechanism-based potential drug combinations”. Leukemia & Lymphoma. 49 (7): 1238–45. doi:10.1080/10428190802005191. PMID 18452080. S2CID 43350339.
- ^ Zhu YX, Braggio E, Shi CX, Bruins LA, Schmidt JE, Van Wier S, et al. (November 2011). “Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide”. Blood. 118 (18): 4771–9. doi:10.1182/blood-2011-05-356063. PMC 3208291. PMID 21860026.
- ^ Stewart AK (January 2014). “Medicine. How thalidomide works against cancer”. Science. 343 (6168): 256–7. doi:10.1126/science.1249543. PMC 4084783. PMID 24436409.
- ^ “Top 10 Best-Selling Cancer Drugs of 2018”. Genetic Engineering and Biotechnology News. 22 April 2019. Retrieved 25 April 2019.
- ^ “Revlimid faces NICE rejection for use in rare blood cancer Watchdog’s draft guidance does not recommend Celgene’s drug for NHS use in England and Wales”. Pharma News. 11 July 2013. Retrieved 5 November 2015.
- ^ “Phase II Study of Lenalidomide for the Treatment of Relapsed or Refractory Hodgkin’s Lymphoma”. ClinicalTrials.gov. US National Institutes of Health. February 2009.
- ^ “276 current clinical trials world-wide, both recruiting and fully enrolled, as of 27 February 2009”. ClinicalTrials.gov. US National Institutes of Health. February 2009.
- ^ “Celgene Discontinues Phase 3 Revlimid Study after ‘Imbalance’ of Deaths”. Nasdaq. 18 July 2013.
External links[edit]
- “Lenalidomide”. Drug Information Portal. U.S. National Library of Medicine.
//////////Lenalidomide hydrate, Lenalidomide KRKA, EU 2021, APPROVALS 2021, レナリドミド水和物 , CC-5013 hemihydrate,
#Lenalidomide hydrate, #Lenalidomide KRKA, #EU 2021, #APPROVALS 2021, #レナリドミド水和物 , #CC-5013 hemihydrate,
O.Nc1cccc2C(=O)N(Cc12)C3CCC(=O)NC3=O.Nc4cccc5C(=O)N(Cc45)C6CCC(=O)NC6=O
Setmelanotide

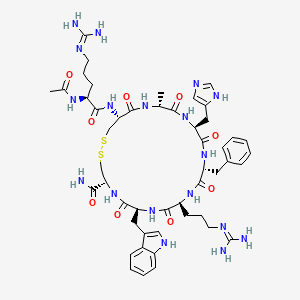

Setmelanotide
Ac-Arg-Cys(1)-D-Ala-His-D-Phe-Arg-Trp-Cys(1)-NH2
- Molecular FormulaC49H68N18O9S2
- Average mass1117.309 Da
- N-acetyl-L-arginyl-L-cysteinyl-D-alanyl-L-histidyl-D-phenylalanyl-L-arginyl-L-tryptophyl-L-cysteinamide (2->8)-disulfide
1,2-Dithia-5,8,11,14,17,20-hexaazacyclotricosane-4-carboxamide, 22-[[(2S)-2-(acetylamino)-5-[(diaminomethylene)amino]-1-oxopentyl]amino]-10-[3-[(diaminomethylene)amino]propyl]-16-(1H-imidazol-5-ylmeth yl)-7-(1H-indol-3-ylmethyl)-19-methyl-6,9,12,15,18,21-hexaoxo-13-(phenylmethyl)-, (4R,7S,10S,13R,16S,19R,22R)- [ACD/Index Name]10011920014-72-8[RN]Imcivree [Trade name]N2-acetyl-L-arginyl-L-cysteinyl-D-alanyl-L-histidyl-D-phenylalanyl-L-arginyl-Ltryptophyl- L-cysteinamide, cyclic (2-8)-disulfideN7T15V1FUYRM-493, BIM-22493UNII-N7T15V1FUYсетмеланотид [Russian] [INN]سيتميلانوتيد [Arabic] [INN]司美诺肽 [Chinese] [INN](4R,7S,10S,13R,16S,19R,22R)-22-[[(2S)-2-acetamido-5-(diaminomethylideneamino)pentanoyl]amino]-13-benzyl-10-[3-(diaminomethylideneamino)propyl]-16-(1H-imidazol-5-ylmethyl)-7-(1H-indol-3-ylmethyl)-19-methyl-6,9,12,15,18,21-hexaoxo-1,2-dithia-5,8,11,14,17,20-hexazacyclotricosane-4-carboxamide
FDA 11/25/2020, Imcivree, To treat obesity and the control of hunger associated with pro-opiomelanocortin deficiency, a rare disorder that causes severe obesity that begins at an early age
Drug Trials Snapshot, 10MG/ML, SOLUTION;SUBCUTANEOUS, Orphan


update Imcivree EMA APPROVED 2021/7/16
DESCRIPTION
IMCIVREE contains setmelanotide acetate, a melanocortin 4 (MC4) receptor agonist. Setmelanotide is an 8 amino acid cyclic peptide analog of endogenous melanocortin peptide α-MSH (alpha-melanocyte stimulating hormone).
The chemical name for setmelanotide acetate is acetyl-L-arginyl-L-cysteinyl-D-alanyl-Lhistidinyl-D-phenylalanyl-L-arginyl-L-tryptophanyl-L-cysteinamide cyclic (2→8)-disulfide acetate. Its molecular formula is C49H68N18O9S2 (anhydrous, free-base), and molecular mass is 1117.3 Daltons (anhydrous, free-base).
The chemical structure of setmelanotide is:
 |
IMCIVREE injection is a sterile clear to slightly opalescent, colorless to slightly yellow solution. Each 1 mL of IMCIVREE contains 10 mg of setmelanotide provided as setmelanotide acetate, which is a salt with 2 to 4 molar equivalents of acetate, and the following inactive ingredients: 100 mg N-(carbonyl-methoxypolyethylene glycol 2000)-1,2-distearoyl-glycero-3phosphoethanolamine sodium salt, 8 mg carboxymethylcellulose sodium (average MWt 90,500), 11 mg mannitol, 5 mg phenol, 10 mg benzyl alcohol, 1 mg edetate disodium dihydrate, and Water for Injection. The pH of IMCIVREE is 5 to 6.
Setmelanotide is a peptide drug and investigational anti-obesity medication which acts as a selective agonist of the MC4 receptor. Setmelanotide binds to and activates MC4 receptors in the paraventricular nucleus (PVN) of the hypothalamus and in the lateral hypothalamic area (LHA), areas involved in the regulation of appetite, and this action is thought to underlie its appetite suppressant effects. Setmelanotide increases resting energy expenditure in both obese animals and humans. Setmelanotide has been reported to possess the following activity profile (cAMP, EC50): MC4 (0.27 nM) > MC3 (5.3 nM) ≈ MC1 (5.8 nM) > MC5 (1600 nM) ≟ MC2 (>1000 nM).
Setmelanotide, sold under the brand name Imcivree, is a medication for the treatment of obesity.[1]
The most common side effects include injection site reactions, skin hyperpigmentation (skin patches that are darker than surrounding skin), headache and gastrointestinal side effects (such as nausea, diarrhea, and abdominal pain), among others.[1] Spontaneous penile erections in males and adverse sexual reactions in females have occurred with treatment.[1] Depression and suicidal ideation have also occurred with setmelanotide.[1]
SYN
WO 2011060355



Medical uses
Setmelanotide is indicated for chronic weight management (weight loss and weight maintenance for at least one year) in people six years and older with obesity due to three rare genetic conditions: pro-opiomelanocortin (POMC) deficiency, proprotein subtilisin/kexin type 1 (PCSK1) deficiency, and leptin receptor (LEPR) deficiency confirmed by genetic testing demonstrating variants in POMC, PCSK1, or LEPR genes considered pathogenic (causing disease), likely pathogenic, or of uncertain significance.[1] Setmelanotide is the first FDA-approved treatment for these genetic conditions.[1]
Setmelanotide is not approved for obesity due to suspected POMC, PCSK1, or LEPR deficiency with variants classified as benign (not causing disease) or likely benign or other types of obesity, including obesity associated with other genetic syndromes and general (polygenic) obesity.[1]
Setmelanotide binds to and activates MC4 receptors in the paraventricular nucleus (PVN) of the hypothalamus and in the lateral hypothalamic area (LHA), areas involved in the regulation of appetite, and this action is thought to underlie its appetite suppressant effects.[2] In addition to reducing appetite, setmelanotide increases resting energy expenditure in both obese animals and humans.[3] Importantly, unlike certain other MC4 receptor agonists, such as LY-2112688, setmelanotide has not been found to produce increases in heart rate or blood pressure.[4]
Setmelanotide has been reported to possess the following activity profile (cAMP, EC50): MC4 (0.27 nM) > MC3 (5.3 nM) ≈ MC1 (5.8 nM) > MC5 (1600 nM) ≟ MC2 (>1000 nM).[5] (19.6-fold selectivity for MC4 over MC3, the second target of highest activity.)
History
Setmelanotide was evaluated in two one-year studies.[1] The first study enrolled participants with obesity and confirmed or suspected POMC or PCSK1 deficiency while the second study enrolled participants with obesity and confirmed or suspected LEPR deficiency; all participants were six years or older.[1] The effectiveness of setmelanotide was determined by the number of participants who lost more than ten percent of their body weight after a year of treatment.[1]
The effectiveness of setmelanotide was assessed in 21 participants, ten in the first study and eleven in the second.[1] In the first study, 80 percent of participants with POMC or PCSK1 deficiency lost ten percent or more of their body weight.[1] In the second study, 46 percent of participants with LEPR deficiency lost ten percent or more of their body weight.[1]
The study also assessed the maximal (greatest) hunger in sixteen participants over the previous 24 hours using an eleven-point scale in participants twelve years and older.[1] In both studies, some, but not all, of participants’ weekly average maximal hunger scores decreased substantially from their scores at the beginning of the study.[1] The degree of change was highly variable among participants.[1]
The U.S. Food and Drug Administration (FDA) granted the application for setmelanotide orphan disease designation, breakthrough therapy designation, and priority review.[1] The FDA granted the approval of Imcivree to Rhythm Pharmaceutical, Inc.[1]
Research
Setmelanotide is a peptide drug and investigational anti-obesity medication which acts as a selective agonist of the MC4 receptor.[6][4] Its peptide sequence is Ac-Arg-Cys(1)-D-Ala-His-D-Phe-Arg-Trp-Cys(1)-NH2. It was first discovered at Ipsen and is being developed by Rhythm Pharmaceuticals for the treatment of obesity and diabetes.[6] In addition, Rhythm Pharmaceuticals is conducting trials of setmelanotide for the treatment of Prader–Willi syndrome (PWS), a genetic disorder which includes MC4 receptor deficiency and associated symptoms such as excessive appetite and obesity.[7] As of December 2014, the drug is in phase II clinical trials for obesity and PWS.[6][8][9][needs update] So far, preliminary data has shown no benefit of Setmelanotide in Prader-Willi syndrome.[10]
PATENT
WO 2007008704
WO 2011060355
WO 2011060352
US 20120225816
PAPER
Journal of Medicinal Chemistry, 61(8), 3674-3684; 2018
PATENT
https://patents.google.com/patent/US9314509
Synthesis of Example 1i.e., Ac-Arg-cyclo(Cys-D-Ala-His-D-Phe-Arg-Trp-Cys)-NH2

The title peptide having the above structure was assembled using Fmoc chemistry on an Apex peptide synthesizer (Aapptec; Louisville, Ky., USA). 220 mg of 0.91 mmol/g (0.20 mmoles) Rink Amide MBHA resin (Polymer Laboratories; Amherst, Mass., USA) was placed in a reaction well and pre-swollen in 3.0 mL of DMF prior to synthesis. For cycle 1, the resin was treated with two 3-mL portions of 25% piperidine in DMF for 5 and 10 minutes respectively, followed by 4 washes of 3-mL DMF—each wash consisting of adding 3 mL of solvent, mixing for 1 minute, and emptying for 1 minute. Amino acids stocks were prepared in NMP as 0.45M solutions containing 0.45M HOBT. HBTU was prepared as a 0.45M solution in NMP and DIPEA was prepared as a 2.73M solution in NMP. To the resin, 2 mL of the first amino acid (0 9 mmoles, Fmoc-Cys(Trt)-OH) (Novabiochem; San Diego, Calif., USA) was added along with 2 mL (0.9 mmoles) of HBTU and 1.5 mL (4.1 mmoles) of DIPEA. After one hour of constant mixing, the coupling reagents were drained from the resin and the coupling step was repeated. Following amino acid acylation, the resin was washed with two 3-mL aliquots of DMF for 1 minute. The process of assembling the peptide (deblock/wash/acylate/wash) was repeated for cycles 2-9 identical to that as described for cycle 1. The following amino acids were used: cycle 2) Fmoc-Trp(Boc)-OH (Genzyme; Cambridge, Mass., USA); cycle 3) Fmoc-Arg(Pbf)-OH (Novabiochem); cycle 4) Fmoc-DPhe-OH (Genzyme); cycle 5) Fmoc-His(Trt)-OH (Novabiochem); cycle 6) Fmoc-D-Ala-OH (Genzyme); cycle 7) Fmoc-Cys(Trt)-OH, (Novabiochem); and cycle 8) Fmoc-Arg(Pbf)-OH (Genzyme). The N-terminal Fmoc was removed with 25% piperidine in DMF as described above, followed by four 3-mL DMF washes for 1 minute. Acetylation of the N-terminus was performed by adding 0.5 mL of 3M DIPEA in NMP to the resin along with 1.45 mL of 0.45M acetic anhydride in NMP. The resin was mixed for 30 minutes and acetylation was repeated. The resin was washed with 3 mL of DMF for a total of 5 times followed with 5 washes with 5 mL of DCM each.
To cleave and deprotect the peptide, 5mL of the following reagent was added to the resin: 2% TIS/5% water/5% (w/v) DTT/88% TFA. The solution was allowed to mix for 3.5 hours. The filtrate was collected into 40 mL of cold anhydrous ethyl ether. The precipitate was pelleted for 10 minutes at 3500 rpm in a refrigerated centrifuge. The ether was decanted and the peptide was re-suspended in fresh ether. The ether workup was performed three times. Following the last ether wash, the peptide was allowed to air dry to remove residual ether.
The peptide was dissolved in 10% acetonitrile and analyzed by mass spectrometry and reverse-phase HPLC employing a 30×4.6 cm C18 column (Vydac; Hesperia, Calif., USA) with a gradient of 2-60% acetonitrile (0.1% TFA) over 30 minutes. This analysis identified a product with ˜53% purity. Mass analysis employing electrospray ionization identified a main product containing a mass of 1118.4 corresponding to the desired linear product. The crude product (˜100 mg) was diluted to a concentration of 2 mg/mL in 5% acetic acid. To this solution, 0.5M iodine/methanol was added dropwise with vigorous stirring until a pale yellow color was achieved. The solution was vigorously stirred for another 10 minutes. Excess iodine was then quenched by adding 1.0M sodium thiosulfate under continuous mixing until the mixture was rendered colorless. The peptide was re-examined by mass spectrometry analysis and HPLC. Mass spectrometry analysis identified a main species with a mass of 1116.4 which indicated successful oxidation to form the cyclic peptide. The peptide solution was purified on a preparative HPLC equipped with a C18 column using a similar elution gradient. The purified product was re-analyzed by HPLC for purity (>95%) and mass spectrometry (1116.9 which is in agreement with the expected mass of 1117.3) and subsequently lyophilized. Following lyophilization, 28 mg of purified product was obtained representing a 24% yield.
The other exemplified peptides were synthesized substantially according to the procedure described for the above-described synthetic process. Physical data for select exemplified peptides are given in Table 1.
TABLE 1 Example Mol. Wt. Mol. Wt. Purity Number (calculated) (ES-MS) (HPLC) 1 1117.3 1116.9 95.1% 2 1117.3 1116.8 99.2% 3 1280.5 1280.6 98.0% 5 1216.37 1216.20 99.9%
Preparation of Pamoate Salt of Example 1
The acetate salt of Example 1 (200 mg, 0.18 mmole) was dissolved in 10 mL of water. Sodium pamoate (155 mg, 0.36 mmole) was dissolved in 10 mL of water. The two solutions were combined and mixed well. The precipitates were collected by centrifugation at 3000 rpm for 20 minutes, washed for three times with water, and dried by lyophilization.
References
- ^ Jump up to:a b c d e f g h i j k l m n o p q r “FDA approves first treatment for weight management for people with certain rare genetic conditions”. U.S. Food and Drug Administration (FDA) (Press release). 27 November 2020. Retrieved 27 November 2020. This article incorporates text from this source, which is in the public domain.
- ^ Kim GW, Lin JE, Blomain ES, Waldman SA (January 2014). “Antiobesity pharmacotherapy: new drugs and emerging targets”. Clinical Pharmacology and Therapeutics. 95 (1): 53–66. doi:10.1038/clpt.2013.204. PMC 4054704. PMID 24105257.
- ^ Chen KY, Muniyappa R, Abel BS, Mullins KP, Staker P, Brychta RJ, et al. (April 2015). “RM-493, a melanocortin-4 receptor (MC4R) agonist, increases resting energy expenditure in obese individuals”. The Journal of Clinical Endocrinology and Metabolism. 100 (4): 1639–45. doi:10.1210/jc.2014-4024. PMC 4399297. PMID 25675384.
- ^ Jump up to:a b Kievit P, Halem H, Marks DL, Dong JZ, Glavas MM, Sinnayah P, et al. (February 2013). “Chronic treatment with a melanocortin-4 receptor agonist causes weight loss, reduces insulin resistance, and improves cardiovascular function in diet-induced obese rhesus macaques”. Diabetes. 62 (2): 490–7. doi:10.2337/db12-0598. PMC 3554387. PMID 23048186.
- ^ Muniyappa R, Chen K, Brychta R, Abel B, Mullins K, Staker P, et al. (June 2014). “A Randomized, Double-Blind, Placebo-Controlled, Crossover Study to Evaluate the Effect of a Melanocortin Receptor 4 (MC4R) Agonist, RM-493, on Resting Energy Expenditure (REE) in Obese Subjects” (PDF). Endocrine Reviews. Rhythm Pharmaceuticals. 35 (3). Retrieved 2015-05-21.
- ^ Jump up to:a b c Lee EC, Carpino PA (2015). “Melanocortin-4 receptor modulators for the treatment of obesity: a patent analysis (2008-2014)”. Pharmaceutical Patent Analyst. 4 (2): 95–107. doi:10.4155/ppa.15.1. PMID 25853469.
- ^ “Obesity and Diabetes Caused by Genetic Deficiencies in the MC4 Pathway”. Rhythm Pharmaceuticals. Retrieved 2015-05-21.
- ^ Jackson VM, Price DA, Carpino PA (August 2014). “Investigational drugs in Phase II clinical trials for the treatment of obesity: implications for future development of novel therapies”. Expert Opinion on Investigational Drugs. 23 (8): 1055–66. doi:10.1517/13543784.2014.918952. PMID 25000213. S2CID 23198484.
- ^ “RM-493: A First-in-Class, Phase 2-Ready MC4 Agonist: A New Drug Class for the Treatment of Obesity and Diabetes”. Rhythm Pharmaceuticals. Archived from the original on 2015-06-14. Retrieved 2015-05-21.
- ^ Duis J, van Wattum PJ, Scheimann A, Salehi P, Brokamp E, Fairbrother L, et al. (March 2019). “A multidisciplinary approach to the clinical management of Prader-Willi syndrome”. Molecular Genetics & Genomic Medicine. 7 (3): e514. doi:10.1002/mgg3.514. PMC 6418440. PMID 30697974.
ADDITIONAL INFORMATION
The peptide sequence is Ac-Arg-Cys(1)-D-Ala-His-D-Phe-Arg-Trp-Cys(1)-NH2. It is being researched by Rhythm Pharmaceuticals for the treatment of obesity and diabetes. In addition, Rhythm Pharmaceuticals is conducting trials of setmelanotide for the treatment of Prader–Willi syndrome (PWS), a genetic disorder which includes MC4 receptor deficiency and associated symptoms such as excessive appetite and obesity. As of December 2014, the drug is in phase II clinical trials for obesity and PWS.
L-Cysteinamide, N2-acetyl-L-arginyl-L-cysteinyl-D-alanyl-L-histidyl-D-phenylalanyl-L-arginyl-L-tryptophyl-, cyclic (2->8)-disulfide
Ac-Arg-Cys(1)-D-Ala-His-D-Phe-Arg-Trp-Cys(1)-NH2
REFERENCES
1: Lee EC, Carpino PA. Melanocortin-4 receptor modulators for the treatment of obesity: a patent analysis (2008-2014). Pharm Pat Anal. 2015;4(2):95-107. doi: 10.4155/ppa.15.1. PubMed PMID: 25853469.
2: Chen KY, Muniyappa R, Abel BS, Mullins KP, Staker P, Brychta RJ, Zhao X, Ring M, Psota TL, Cone RD, Panaro BL, Gottesdiener KM, Van der Ploeg LH, Reitman ML, Skarulis MC. RM-493, a melanocortin-4 receptor (MC4R) agonist, increases resting energy expenditure in obese individuals. J Clin Endocrinol Metab. 2015 Apr;100(4):1639-45. doi: 10.1210/jc.2014-4024. Epub 2015 Feb 12. PubMed PMID: 25675384; PubMed Central PMCID: PMC4399297.
3: Clemmensen C, Finan B, Fischer K, Tom RZ, Legutko B, Sehrer L, Heine D, Grassl N, Meyer CW, Henderson B, Hofmann SM, Tschöp MH, Van der Ploeg LH, Müller TD. Dual melanocortin-4 receptor and GLP-1 receptor agonism amplifies metabolic benefits in diet-induced obese mice. EMBO Mol Med. 2015 Feb 4;7(3):288-98. doi: 10.15252/emmm.201404508. PubMed PMID: 25652173; PubMed Central PMCID: PMC4364946.
4: Jackson VM, Price DA, Carpino PA. Investigational drugs in Phase II clinical trials for the treatment of obesity: implications for future development of novel therapies. Expert Opin Investig Drugs. 2014 Aug;23(8):1055-66. doi: 10.1517/13543784.2014.918952. Epub 2014 Jul 7. Review. PubMed PMID: 25000213.
5: Kievit P, Halem H, Marks DL, Dong JZ, Glavas MM, Sinnayah P, Pranger L, Cowley MA, Grove KL, Culler MD. Chronic treatment with a melanocortin-4 receptor agonist causes weight loss, reduces insulin resistance, and improves cardiovascular function in diet-induced obese rhesus macaques. Diabetes. 2013 Feb;62(2):490-7. doi: 10.2337/db12-0598. Epub 2012 Oct 9. PubMed PMID: 23048186; PubMed Central PMCID: PMC3554387.
6: Kumar KG, Sutton GM, Dong JZ, Roubert P, Plas P, Halem HA, Culler MD, Yang H, Dixit VD, Butler AA. Analysis of the therapeutic functions of novel melanocortin receptor agonists in MC3R- and MC4R-deficient C57BL/6J mice. Peptides. 2009 Oct;30(10):1892-900. doi: 10.1016/j.peptides.2009.07.012. Epub 2009 Jul 29. PubMed PMID: 19646498; PubMed Central PMCID: PMC2755620.
External links
- “Setmelanotide”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Imcivree |
| Other names | RM-493; BIM-22493; IRC-022493; N2-Acetyl-L-arginyl-L-cysteinyl-D-alanyl-L-histidyl-D-phenylalanyl-L-arginyl-L-tryptophyl-L-cysteinamide, cyclic (2-8)-disulfide |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 920014-72-8 |
| PubChem CID | 11993702 |
| ChemSpider | 10166169 |
| UNII | N7T15V1FUY |
| KEGG | D11927 |
| Chemical and physical data | |
| Formula | C49H68N18O9S2 |
| Molar mass | 1117.32 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]C[C@@H]1C(=O)N[C@H](C(=O)N[C@@H](C(=O)N[C@H](C(=O)N[C@H](C(=O)N[C@@H](CSSC[C@@H](C(=O)N1)NC(=O)[C@H](CCCN=C(N)N)NC(=O)C)C(=O)N)Cc2c[nH]c3c2cccc3)CCCN=C(N)N)Cc4ccccc4)Cc5cnc[nH]5 | |
| InChI[hide]InChI=1S/C49H68N18O9S2/c1-26-41(70)63-37(20-30-22-55-25-59-30)46(75)64-35(18-28-10-4-3-5-11-28)44(73)62-34(15-9-17-57-49(53)54)43(72)65-36(19-29-21-58-32-13-7-6-12-31(29)32)45(74)66-38(40(50)69)23-77-78-24-39(47(76)60-26)67-42(71)33(61-27(2)68)14-8-16-56-48(51)52/h3-7,10-13,21-22,25-26,33-39,58H,8-9,14-20,23-24H2,1-2H3,(H2,50,69)(H,55,59)(H,60,76)(H,61,68)(H,62,73)(H,63,70)(H,64,75)(H,65,72)(H,66,74)(H,67,71)(H4,51,52,56)(H4,53,54,57)/t26-,33+,34+,35-,36+,37+,38+,39+/m1/s1Key:HDHDTKMUACZDAA-PHNIDTBTSA-N |
///////////Setmelanotide, FDA 2020, 2020 APPROVALS, Imcivree, Orphan, PEPTIDE, ANTIOBESITY, UNII-N7T15V1FUY, сетмеланотид , سيتميلانوتيد , 司美诺肽 , BIM 22493, RM 493
CC1C(=O)NC(C(=O)NC(C(=O)NC(C(=O)NC(C(=O)NC(CSSCC(C(=O)N1)NC(=O)C(CCCN=C(N)N)NC(=O)C)C(=O)N)CC2=CNC3=CC=CC=C32)CCCN=C(N)N)CC4=CC=CC=C4)CC5=CN=CN5
Odevixibat

Odevixibat
A-4250, AR-H 064974
CAS 501692-44-0
BUTANOIC ACID, 2-(((2R)-2-((2-((3,3-DIBUTYL-2,3,4,5-TETRAHYDRO-7-(METHYLTHIO)-1,1-DIOXIDO-5-PHENYL-1,2,5-BENZOTHIADIAZEPIN-8-YL)OXY)ACETYL)AMINO)-2-(4-HYDROXYPHENYL)ACETYL)AMINO)-, (2S)-
(2S)-2-[[(2R)-2-[[2-[(3,3-dibutyl-7-methylsulfanyl-1,1-dioxo-5-phenyl-2,4-dihydro-1λ6,2,5-benzothiadiazepin-8-yl)oxy]acetyl]amino]-2-(4-hydroxyphenyl)acetyl]amino]butanoic acid
| Molecular Formula | C37H48N4O8S2 |
| Molecular Weight | 740.929 |
-
-
-
- UPDATE 7/20/2021FDA APPROVED, To treat pruritus,
-
- New Drug Application (NDA): 215498
Company: ALBIREO PHARMA INC
-
- AZD8294WHO 10706AR-H064974HY-109120CS-0078340D11716US9694018, 5Originator Albireo AB
- Developer Albireo AB; Albireo Pharma
- ClassAcetamides; Butyric acids; Hepatoprotectants; Small molecules; Sulfones; Thiazepines
- Mechanism of Action Sodium-bile acid cotransporter inhibitors
- Orphan Drug Status Yes – Primary biliary cirrhosis; Biliary atresia; Intrahepatic cholestasis; Alagille syndrome
- New Molecular Entity Yes
- Phase III Biliary atresia; Intrahepatic cholestasis
- Phase II Alagille syndrome; Cholestasis; Primary biliary cirrhosis
- No development reported Non-alcoholic steatohepatitis
- 22 Jul 2020 Albireo initiates an expanded-access programme for Intrahepatic cholestasis in USA, Canada, Australia and Europe
- 14 Jul 2020 Phase-III clinical trials in Biliary atresia (In infants, In neonates) in Belgium (PO) after July 2020 (EudraCT2019-003807-37)
- 14 Jul 2020 Phase-III clinical trials in Biliary atresia (In infants, In neonates) in Germany, France, United Kingdom, Hungary (PO) (EudraCT2019-003807-37)
UPDATE Bylvay, FDA APPROVED2021/7/20 AND EMA 2021/7/16
Odevixibat, sold under the trade name Bylvay, is a medication for the treatment of progressive familial intrahepatic cholestasis (PFIC).[1]
The most common side effects include diarrhea, abdominal pain, hemorrhagic diarrhea, soft feces, and hepatomegaly (enlarged liver).[1]
Odevixibat is a reversible, potent, selective inhibitor of the ileal bile acid transporter (IBAT).[1][2]
In May 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) recommended granting a marketing authorization in the European Union for odevixibat for the treatment of PFIC in people aged six months or older.[1][3]
A-4250 (odevixibat) is a selective inhibitor of the ileal bile acid transporter (IBAT) that acts locally in the gut. Ileum absorbs glyco-and taurine-conjugated forms of the bile salts. IBAT is the first step in absorption at the brush-border membrane. A-4250 works by decreasing the re-absorption of bile acids from the small intestine to the liver, whichreduces the toxic levels of bile acids during the progression of the disease. It exhibits therapeutic intervention by checking the transport of bile acids. Studies show that A-4250 has the potential to decrease the damage in the liver cells and the development of fibrosis/cirrhosis of the liver known to occur in progressive familial intrahepatic cholestasis. A-4250 is a designated orphan drug in the USA for October 2012. A-4250 is a designated orphan drug in the EU for October 2016. A-4250 was awarded PRIME status for PFIC by EMA in October 2016. A-4250 is in phase II clinical trials by Albireo for the treatment of primary biliary cirrhosis (PBC) and cholestatic pruritus. In an open label Phase 2 study in children with cholestatic liver disease and pruritus, odevixibat showed reductions in serum bile acids and pruritus in most patients and exhibited a favorable overall tolerability profile.
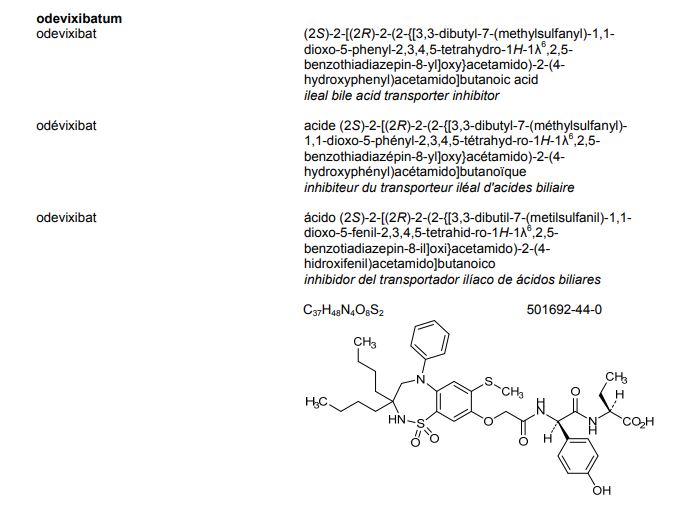
![]()
Odevixibat is a highly potent, non-systemic ileal bile acid transport inhibitor (IBATi) that has has minimal systemic exposure and acts locally in the small intestine. Albireo is developing odevixibat to treat rare pediatric cholestatic liver diseases, including progressive familial intrahepatic cholestasis, biliary atresia and Alagille syndrome.
With normal function, approximately 95 percent of bile acids released from the liver into the bile ducts to aid in liver function are recirculated to the liver via the IBAT in a process called enterohepatic circulation. In people with cholestatic liver diseases, the bile flow is interrupted, resulting in elevated levels of toxic bile acids accumulating in the liver and serum. Accordingly, a product capable of inhibiting the IBAT could lead to a reduction in bile acids returning to the liver and may represent a promising approach for treating cholestatic liver diseases.
The randomized, double-blind, placebo-controlled, global multicenter PEDFIC 1 Phase 3 clinical trial of odevixibat in 62 patients, ages 6 months to 15.9 years, with PFIC type 1 or type 2 met its two primary endpoints demonstrating that odevixibat reduced serum bile acids (sBAs) (p=0.003) and improved pruritus (p=0.004), and was well tolerated with a low single digit diarrhea rate. These topline data substantiate the potential for odevixibat to be first drug for PFIC patients. The Company intends to complete regulatory filings in the EU and U.S. no later than early 2021, in anticipation of regulatory approval, issuance of a rare pediatric disease priority review voucher and launch in the second half of 2021.
Odevixibat is being evaluated in the ongoing PEDFIC 2 open-label trial (NCT03659916) designed to assess long-term safety and durability of response in a cohort of patients rolled over from PEDFIC 1 and a second cohort of PFIC patients who are not eligible for PEDFIC 1.
Odevixibat is also currently being evaluated in a second Phase 3 clinical trial, BOLD (NCT04336722), in patients with biliary atresia. BOLD, the largest prospective intervention trial ever conducted in biliary atresia, is a double-blind, randomized, placebo-controlled trial which will enroll approximately 200 patients at up to 75 sites globally to evaluate the efficacy and safety of odevixibat in children with biliary atresia who have undergone a Kasai procedure before age three months. The company also anticipates initiating a pivotal trial of odevixibat for Alagille syndrome by the end of 2020.
For more information about the PEDFIC 2 or BOLD studies, please visit ClinicalTrials.gov or contact medinfo@albireopharma.com.
The odevixibat PFIC program, or elements of it, have received fast track, rare pediatric disease and orphan drug designations in the United States. In addition, the FDA has granted orphan drug designation to odevixibat for the treatment of Alagille syndrome, biliary atresia and primary biliary cholangitis. The EMA has granted odevixibat orphan designation, as well as access to the PRIority MEdicines (PRIME) scheme for the treatment of PFIC. Its Paediatric Committee has agreed to Albireo’s odevixibat Pediatric Investigation Plan for PFIC. EMA has also granted orphan designation to odevixibat for the treatment of biliary atresia, Alagille syndrome and primary biliary cholangitis.

PATENT
https://patents.google.com/patent/US9694018B1/en
Example 5
1,1-Dioxo-3,3-dibutyl-5-phenyl-7-methylthio-8-(N—{(R)-α-[N—((S)-1-carboxypropyl) carbamoyl]-4-hydroxybenzyl}carbamoylmethoxy)-2,3,4,5-tetrahydro-1,2,5-benzothiadiazepine, Mw. 740.94.
This compound is prepared as described in Example 29 of WO3022286.
PATENT
https://patents.google.com/patent/WO2003022286A1/sv
Example 29
1,1-Dioxo-3,3-dibutyl-5-phenyl-7-methylthio-8-(N-((R)-α-[N-((S)- 1-carboxypropyl) carbamoyl]-4-hydroxybenzyl}carbamoylmethoxy)-2,3,4,5-tetrahydro-1,2,5-benzothiadiazepine
A solution of 1,1-dioxo-3,3-dibutyl-5-phenyl-7-methylthio-8-[N-((R)-α-carboxy-4-hydroxybenzyl)carbamoylmethoxy]-2,3,4,5-tetrahydro-1,2,5-benzothiadiazepine (Example 18; 0.075 g, 0.114 mmol), butanoic acid, 2-amino-, 1,1-dimethylethyl ester, hydrochloride, (2S)-(0.031 g, 0.160 mmol) and Ν-methylmorpholine (0.050 ml, 0.457 mmol) in DMF (4 ml) was stirred at RT for 10 min, after which TBTU (0.048 g, 0.149 mmol) was added. After 1h, the conversion to the ester was complete. M/z: 797.4. The solution was diluted with toluene and then concentrated. The residue was dissolved in a mixture of DCM (5 ml) and TFA (2 ml) and the mixture was stirred for 7h. The solvent was removed under reduced pressure. The residue was purified by preparative HPLC using a gradient of 20-60% MeCΝ in 0.1M ammonium acetate buffer as eluent. The title compound was obtained in 0.056 g (66 %) as a white solid. ΝMR (400 MHz, DMSO-d6): 0.70 (3H, t), 0.70-0.80 (6H, m), 0.85-1.75 (14H, m), 2.10 (3H, s), 3.80 (2H, brs), 4.00-4.15 (1H, m), 4.65 (1H, d(AB)), 4.70 (1H, d(AB)), 5.50 (1H, d), 6.60 (1H, s), 6.65-7.40 (11H, m), 8.35 (1H, d), 8.50 (1H, d) 9.40 (1H, brs).
PATENT
https://patents.google.com/patent/US20140323412A1/en
PATENT
https://patents.google.com/patent/WO2013063526A1/e
PATENT
https://patents.google.com/patent/WO2019245448A1/en
The compound l,l-dioxo-3,3-dibutyl-5-phenyl-7-methylthio-8-(A/-{(R)-a-[A/-((S)-l-carboxypropyl) carbamoyl]-4-hydroxybenzyl}carbamoylmethoxy)-2,3,4,5-tetrahydro-l,2,5-benzothiadiazepine (odevixibat; also known as A4250) is disclosed in WO 03/022286. The structure of odevixibat is shown below.

As an inhibitor of the ileal bile acid transporter (IBAT) mechanism, odevixibat inhibits the natural reabsorption of bile acids from the ileum into the hepatic portal circulation. Bile acids that are not reabsorbed from the ileum are instead excreted into the faeces. The overall removal of bile acids from the enterohepatic circulation leads to a decrease in the level of bile acids in serum and the liver. Odevixibat, or a pharmaceutically acceptable salt thereof, is therefore useful in the treatment or prevention of diseases such as dyslipidemia, constipation, diabetes and liver diseases, and especially liver diseases that are associated with elevated bile acid levels.
According to the experimental section of WO 03/022286, the last step in the preparation of odevixibat involves the hydrolysis of a tert-butyl ester under acidic conditions. The crude compound was obtained by evaporation of the solvent under reduced pressure followed by purification of the residue by preparative HPLC (Example 29). No crystalline material was identified.
Amorphous materials may contain high levels of residual solvents, which is highly undesirable for materials that should be used as pharmaceuticals. Also, because of their lower chemical and physical stability, as compared with crystalline material, amorphous materials may display faster
decomposition and may spontaneously form crystals with a variable degree of crystallinity. This may result in unreproducible solubility rates and difficulties in storing and handling the material. In pharmaceutical preparations, the active pharmaceutical ingredient (API) is for that reason preferably used in a highly crystalline state. Thus, there is a need for crystal modifications of odevixibat having improved properties with respect to stability, bulk handling and solubility. In particular, it is an object of the present invention to provide a stable crystal modification of odevixibat that does not contain high levels of residual solvents, that has improved chemical stability and can be obtained in high levels of crystallinity.
Example 1
Preparation of crystal modification 1
Absolute alcohol (100.42 kg) and crude odevixibat (18.16 kg) were charged to a 250-L GLR with stirring under nitrogen atmosphere. Purified water (12.71 kg) was added and the reaction mass was stirred under nitrogen atmosphere at 25 ± 5 °C for 15 minutes. Stirring was continued at 25 ± 5 °C for 3 to 60 minutes, until a clear solution had formed. The solution was filtered through a 5.0 m SS cartridge filter, followed by a 0.2 m PP cartridge filter and then transferred to a clean reactor.
Purified water (63.56 kg) was added slowly over a period of 2 to 3 hours at 25 ± 5 °C, and the solution was seeded with crystal modification 1 of odevixibat. The solution was stirred at 25 ± 5 °C for 12 hours. During this time, the solution turned turbid. The precipitated solids were filtered through centrifuge and the material was spin dried for 30 minutes. The material was thereafter vacuum dried in a Nutsche filter for 12 hours. The material was then dried in a vacuum tray drier at 25 ± 5 °C under vacuum (550 mm Hg) for 10 hours and then at 30 ± 5 °C under vacuum (550 mm Hg) for 16 hours. The material was isolated as an off-white crystalline solid. The isolated crystalline material was milled and stored in LDPE bags.
An overhydrated sample was analyzed with XRPD and the diffractogram is shown in Figure 2.
Another sample was dried at 50 °C in vacuum and thereafter analysed with XRPD. The diffractogram of the dried sample is shown in Figure 1.
The diffractograms for the drying of the sample are shown in Figures 3 and 4 for 2Q ranges 5 – 13 ° and 18 – 25 °, respectively (overhydrated sample at the bottom and dry sample at the top).
References
- ^ Jump up to:a b c d “First treatment for rare liver disease”. European Medicines Agency (EMA) (Press release). 21 May 2021. Retrieved 21 May 2021. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Odevixibat”. Albireo Pharma. Retrieved 21 May 2021.
- ^ “Bylvay: Pending EC decision”. European Medicines Agency (EMA). 19 May 2021. Retrieved 21 May 2021.
External links
- “Odevixibat”. Drug Information Portal. U.S. National Library of Medicine.
ClinicalTrials.gov
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| NCT04336722 | Efficacy and Safety of Odevixibat in Children With Biliary Atresia Who Have Undergone a Kasai HPE (BOLD) | Phase 3 | Recruiting | 2020-09-02 |
| NCT04483531 | Odevixibat for the Treatment of Progressive Familial Intrahepatic Cholestasis | Available | 2020-08-25 | |
| NCT03566238 | This Study Will Investigate the Efficacy and Safety of A4250 in Children With PFIC 1 or 2 | Phase 3 | Active, not recruiting | 2020-03-05 |
| NCT03659916 | Long Term Safety & Efficacy Study Evaluating The Effect of A4250 in Children With PFIC | Phase 3 | Recruiting | 2020-01-21 |
| NCT03608319 | Study of A4250 in Healthy Volunteers Under Fasting, Fed and Sprinkled Conditions | Phase 1 | Completed | 2018-09-19 |
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| NCT02630875 | A4250, an IBAT Inhibitor in Pediatric Cholestasis | Phase 2 | Completed | 2018-03-29 |
| NCT02360852 | IBAT Inhibitor A4250 for Cholestatic Pruritus | Phase 2 | Terminated | 2017-02-23 |
| NCT02963077 | A Safety and Pharmakokinetic Study of A4250 Alone or in Combination With A3384 | Phase 1 | Completed | 2016-11-16 |
EU Clinical Trials Register
.
 |
|
| Clinical data | |
|---|---|
| Trade names | Bylvay |
| Routes of administration |
By mouth |
| ATC code |
|
| Identifiers | |
| CAS Number |
|
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C37H48N4O8S2 |
| Molar mass | 740.93 g·mol−1 |
| 3D model (JSmol) | |
////////////odevixibat, Orphan Drug Status, phase 3, Albireo, A-4250, A 4250, AR-H 064974
CCCCC1(CN(C2=CC(=C(C=C2S(=O)(=O)N1)OCC(=O)NC(C3=CC=C(C=C3)O)C(=O)NC(CC)C(=O)O)SC)C4=CC=CC=C4)CCCC
| publicationnumber | |||||||
| US-2020046635-A1 | |||||||
| US-2020046636-A1 | US-2020046757-A1 | US-2020046758-A1 | US-2020002299-A1 | WO-2019245448-A1 | WO-2019245449-A1 | US-2019046451-A1 | US-2019070217-A1 |
| US-10441605-B2 | |||||||
| US-2017224720-A1 | |||||||
| US-2017224721-A1 | |||||||
| US-2018264029-A1 | |||||||
| US-2018360869-A1 | |||||||
| US-2018360870-A1 | |||||||
| US-2018360871-A1 | |||||||
| WO-2017133517-A1 | |||||||
| US-2017143738-A1 | |||||||
| US-2017143783-A1 | |||||||
| EP-2968230-A2 | |||||||
| EP-2968262-A1 | |||||||
| US-2014271734-A1 | |||||||
| US-2014275090-A1 | |||||||
| WO-2014144485-A1 | |||||||
| WO-2014144485-A9 | |||||||
| WO-2014144650-A2 | |||||||
| EP-2770990-A1 | |||||||
| EP-2771003-A1 | |||||||
| EP-2771003-B1 | |||||||
| EP-3266457-A1 | |||||||
| EP-3278796-A1 | |||||||
| US-10512657-B2 | |||||||
| US-2013108573-A1 | |||||||
| US-2013109671-A1 | |||||||
| US-2013338093-A1 | |||||||
| US-2014243281-A1 | |||||||
| US-2014323412-A1 | |||||||
| US-2016310518-A1 | |||||||
| US-2017368085-A1 | |||||||
| US-2019169217-A1 | |||||||
| US-2020069715-A1 | |||||||
| WO-2013063512-A1 | |||||||
| WO-2013063526-A1 | |||||||
| EP-2739286-A2 | |||||||
| WO-2013020108-A2 | |||||||
| EP-2637646-B1 | |||||||
| EP-2637668-B1 | |||||||
| EP-3023102-A1 | |||||||
| EP-3023102-B1 | |||||||
| EP-3400944-A1 | |||||||
| US-10000528-B2 | |||||||
| US-10011633-B2 | |||||||
| US-10093697-B2 | |||||||
| US-10221212-B2 | |||||||
| US-2012114588-A1 | |||||||
| US-2013225511-A1 | |||||||
| US-2013236541-A1 | |||||||
| US-2015031636-A1 | |||||||
| US-2015031637-A1 | |||||||
| US-2016193277-A1 | |||||||
| US-2016194353-A1 | |||||||
| US-2017182059-A1 | |||||||
| US-2017182115-A1 | |||||||
| US-2018022776-A1 | |||||||
| US-2018030088-A1 | |||||||
| US-2018030089-A1 | |||||||
| US-2018162904-A1 | |||||||
| US-2018362577-A1 | |||||||
| US-9688720-B2 | |||||||
| US-9694018-B1 | |||||||
| US-10555950-B2 | |||||||
| US-2016220577-A1 | |||||||
| US-9339480-B2 | |||||||
| WO-2008039829-A2 | |||||||
| US-2009069285-A1 | |||||||
| US-7842684-B2 | |||||||
| WO-2007051995-A2 | |||||||
| EP-1896408-A1 | |||||||
| EP-1896409-A1 | |||||||
| EP-1896457-A1 | |||||||
| US-2010048529-A1 | |||||||
| US-2010048530-A1 | |||||||
| US-2010137273-A1 | |||||||
| US-2010152156-A1 | |||||||
| US-2010168039-A1 | |||||||
| US-2010168075-A1 | |||||||
| US-7893048-B2 | |||||||
| US-7906502-B2 | |||||||
| WO-2006137792-A1 | |||||||
| WO-2006137794-A1 | |||||||
| WO-2006137795-A1 | |||||||
| US-2010216759-A1 | |||||||
| US-7863265-B2 | |||||||
| US-2008194494-A1 | |||||||
| US-2009186834-A1 | |||||||
| WO-2006102674-A2 | |||||||
| US-2009005321-A1 | |||||||
| EP-1831151-A1 | |||||||
| US-2008114064-A1 | |||||||
| WO-2006065214-A1 | |||||||
| EP-1699759-A1 | |||||||
| US-2007142304-A1 | |||||||
| US-2008064676-A1 | |||||||
| US-2010099657-A2 | |||||||
| US-7871998-B2 | |||||||
| WO-2005061452-A1 | |||||||
| EP-1638922-A1 | |||||||
| EP-1638926-A1 | |||||||
| EP-1638930-A1 | |||||||
| EP-1675820-A2 | |||||||
| EP-1676833-A1 | |||||||
| US-2005148656-A1 | |||||||
| US-2006142389-A1 | |||||||
| US-2006178432-A1 | |||||||
| US-2006194879-A1 | |||||||
| US-2006258866-A1 | |||||||
| US-2007099928-A1 | |||||||
| US-2007099997-A1 | |||||||
| US-2007244198-A1 | |||||||
| US-7309720-B2 | |||||||
| WO-2004110984-A1 | |||||||
| WO-2004113270-A2 | |||||||
| WO-2004113276-A1 | |||||||
| WO-2004113283-A1 | |||||||
| EP-1610770-A1 | |||||||
| EP-1610770-B1 | |||||||
| EP-1894564-A2 | |||||||
| US-2006199797-A1 | |||||||
| US-7514421-B2 | |||||||
| WO-2004089350-A1 | |||||||
| EP-1572626-A1 | |||||||
| US-2005131068-A1 | |||||||
| WO-2004056748-A1 | |||||||
| EP-1539120-A1 | |||||||
| US-2006083790-A1 | |||||||
| WO-2004006899-A1 | |||||||
| EP-1521742-A1 | |||||||
| US-2005239766-A1 | |||||||
| US-7470678-B2 | |||||||
| WO-2004005247-A1 | |||||||
| EP-1517679-A1 | |||||||
| EP-1517679-B1 | |||||||
| EP-1517883-A1 | |||||||
| EP-1517883-B1 | |||||||
| EP-1517883-B8 | |||||||
| US-2005222261-A1 | |||||||
| US-2005256198-A1 | |||||||
| US-2005267149-A1 | |||||||
| US-7351858-B2 | |||||||
| US-7355069-B2 | |||||||
| US-7521461-B2 | |||||||
| WO-2004000294-A1 | |||||||
| WO-2004000790-A1 | |||||||
| EP-1478368-A1 | |||||||
| US-2005124557-A1 | |||||||
| WO-03061663-A1 | |||||||
| EP-1458672-A1 | |||||||
| EP-1458672-B1 | |||||||
| EP-1458673-A1 | |||||||
| EP-1458673-B1 | |||||||
| EP-1458677-A1 | |||||||
| EP-1458677-B1 | |||||||
| US-2005113362-A1 | |||||||
| US-2005171204-A1 | |||||||
| US-2005215630-A1 | |||||||
| US-2005282822-A1 | |||||||
| US-7256307-B2 | |||||||
| US-7276539-B2 | |||||||
| US-7488844-B2 | |||||||
| US-7514471-B2 | |||||||
| WO-03051821-A1 | |||||||
| WO-03051822-A1 | |||||||
| WO-03051826-A1 | |||||||
| EP-1427423-B1 | |||||||
| EP-1427423-B9 | |||||||
| US-2005038009-A1 | |||||||
| US-7132416-B2 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}