
Home » Posts tagged 'Daiichi Sankyo'
Tag Archives: Daiichi Sankyo
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
DS 2330 by Daiichi Sankyo
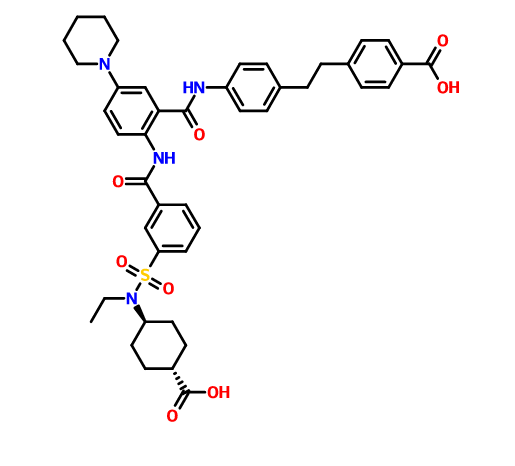
DS 2330
a trans compd
4-[2-(4-{[2-({3-[(trans-4-carboxy-cyclohexyl)(ethyl)sulfocarbamoyl]benzoyl}amino)-5-(piperidin-1-yl)benzoyl]amino}phenyl)ethyl]benzoic acid,
4- [2- (4 – {[2 – ({3 – [(trans-4-carboxy-cyclohexyl) (ethyl) sulfur carbamoyl] benzoyl} amino) -5- (piperidin-1-yl) benzoyl] amino} phenyl) ethyl] benzoate
- Originator Daiichi Sankyo Inc
- Class Hyperphosphataemia therapies
useful for treating hyperphosphatemia, DS-2330, a phosphorous lowering agent, being developed by Daiichi Sankyo, for treating hyperphosphatemia in chronic kidney disease. In April 2016, DS-2330 was reported to be in phase 1 clinical development.
- Phase IHyperphosphataemia
- 31 Oct 2015Phase-I clinical trials in Hyperphosphataemia in USA (unspecified route)
![]()
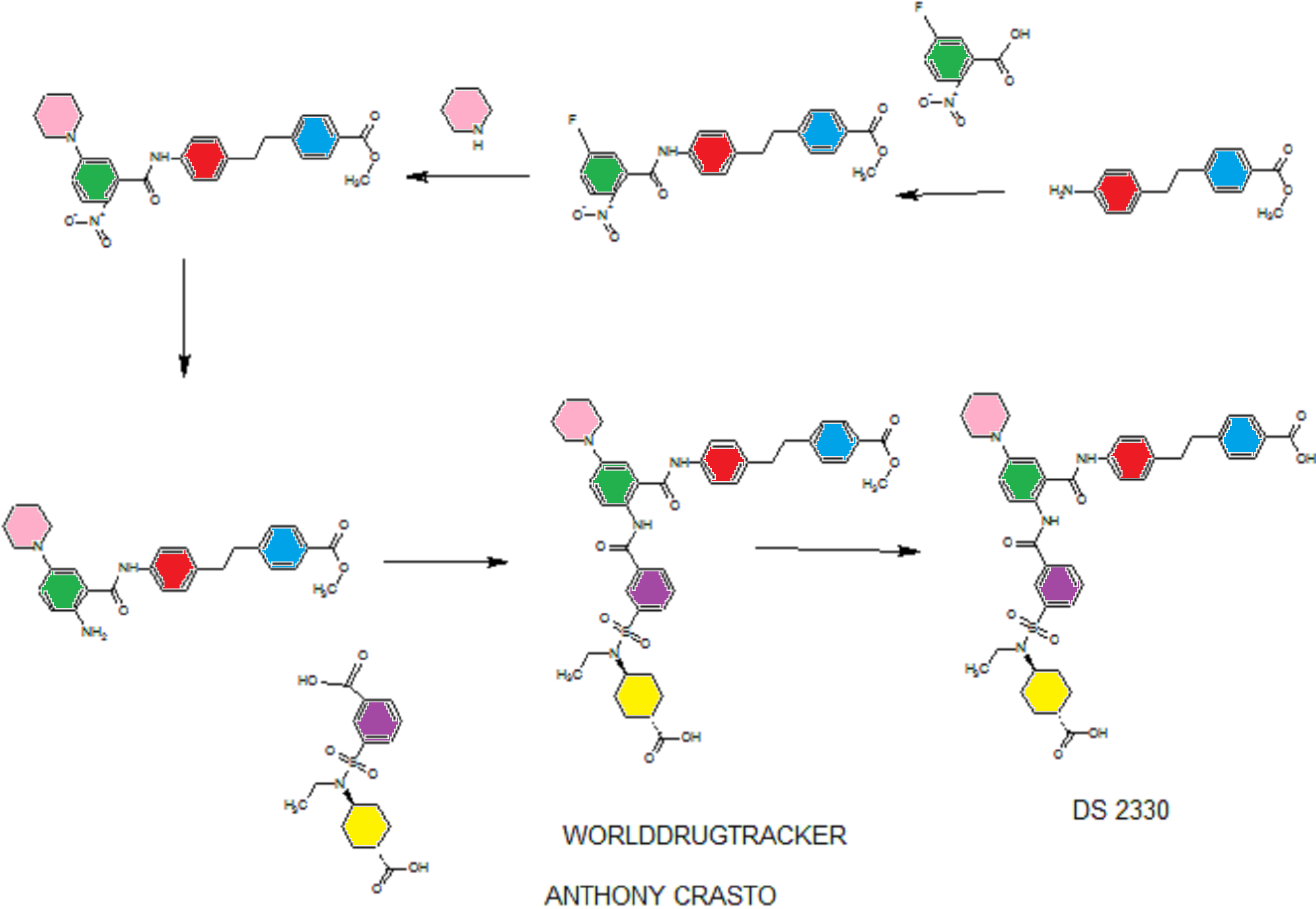
SEE WO2015108038,
PATENT
WO2014175317
http://www.google.com/patents/EP2990400A1?cl=en
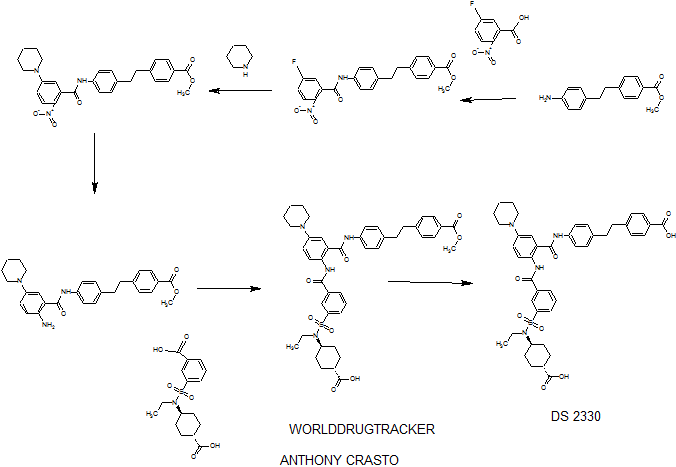
PATENT
he problem is to provide a pharmaceutical for the prevention or treatment of hyperphosphatemia. The solution is a salt of a compound including formula (I), or a crystal of a hydrate thereof.
disodium 4- [2- (4 – {[2 – ({3 – [(trans-4-carboxy-cyclohexyl) (ethyl) sulfur carbamoyl] benzoyl} amino) -5- (piperidin-1-yl ) benzoyl] amino} phenyl) ethyl] benzoic acid trihydrate
Disodium 4- [2- (4 – { [2 – ({3 – [(trans-4-carboxylatocyclohexyl) (ethyl) sulfamoyl] benzoyl} amino) – 5- (piperidin-1-yl) benzoyl] amino} phenyl) ethyl] benzoate trihydrate
of α crystal
4- [2- (4 – {[2 – ({3 – [(trans-4-carboxy-cyclohexyl) (ethyl) sulfur carbamoyl] benzoyl} amino) -5- (piperidin-1-yl) benzoyl] amino} phenyl) ethyl] 1 mol / L NaOH aqueous solution to benzoic acid (1.2 g) (3.1 mL) was added and dissolved completely. After stirring at room temperature for 1 day was added acetonitrile (60 mL), at 40 ° C.
and stirred for further 1 day. The precipitated solid was collected by filtration, and 3 hours drying under reduced pressure at room temperature to give the title compound 1.1 g (85%).
in water (46.4 mL), 1-PrOH (72 mL), 4 mol / L NaOH aqueous solution (25.54 mL) was added, then filtered after stirring insolubles at room temperature, water / 1-PrOH: was washed with (3 7, 80 mL). The filtrate was heated up to 40 ℃, 1-PrOH the (160 mL) was added, and further seed crystal (α crystals, 0.2g) was added. Then the temperature was raised to 50 ℃, 1-PrOH (96 ml) was added, and the mixture was stirred overnight.Thereafter, 1-PrOH (480 ml) was added and after overnight stirring, was collected by filtration the precipitated solid was cooled to room temperature.Thereafter, and vacuum dried overnight at 40 ° C., to give the title compound 39.4 g (96%).
REFERENCES
////////////DS 2330, DS-2330, DAIICHI SANKYO, phase 1
O=C(O)[C@@H]1CC[C@H](CC1)N(CC)S(=O)(=O)c2cccc(c2)C(=O)Nc5ccc(cc5C(=O)Nc4ccc(CCc3ccc(cc3)C(=O)O)cc4)N6CCCCC6
OR
O=C(O)[C@@H]1CC[C@H](CC1)N(CC)S(=O)(=O)c2cccc(c2)C(=O)Nc5ccc(cc5C(=O)Nc4ccc(CCc3ccc(cc3)C(=O)O)cc4)N6CCCCC6
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on Facebook 

 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
CS 3150, angiotensin II receptor antagonist, for the treatment or prevention of such hypertension and heart disease
CS-3150, (XL550)
CS 3150, angiotensin II receptor antagonist, for the treatment or prevention of such hypertension and heart disease similar to olmesartan , losartan, candesartan , valsartan, irbesartan, telmisartan, eprosartan,
Cas name 1H-Pyrrole-3-carboxamide, 1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-, (5S)-
CAS 1632006-28-0 for S conf
MF C22 H21 F3 N2 O4 S
MW 466.47
(S)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide
CAS 1632006-28-0 for S configuration
1- (2-hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxamide
(S) -1- (2- hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxamide
(+/-)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide, CAS 880780-76-7
(+)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide..1072195-82-4
(-)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide..1072195-83-5
WO2008 / 126831 (US Publication US2010-0093826)http://www.google.co.in/patents/EP2133330A1?cl=en
WO 2006012642..compound A;..http://www.google.com/patents/WO2006012642A2?cl=en
WO2006 / 012642 (US Publication US2008-0234270)
WO 2015030010…http://www.google.com/patents/WO2015030010A1?cl=en
- Originator Exelixis
- Developer Daiichi Sankyo Company..
Daiichi Sankyo Company,Limited, 第一三共株式会社 - Class Antihypertensives; Small molecules
- Mechanism of Action Mineralocorticoid receptor antagonists

JAPAN PHASE 2……….Phase 2 Study to Evaluate Efficacy and Safety of CS-3150 in Patients with Essential Hypertension
http://www.clinicaltrials.jp/user/showCteDetailE.jsp?japicId=JapicCTI-121921
Phase II Diabetic nephropathies; Hypertension
- 01 Jan 2015 Daiichi Sankyo initiates a phase IIb trial for Diabetic nephropathies in Japan (NCT02345057)
- 01 Jan 2015 Daiichi Sankyo initiates a phase IIb trial for Hypertension in Japan (NCT02345044)
- 01 May 2013 Phase-II clinical trials in Diabetic nephropathies in Japan (PO)
-
Currently, angiotensin II receptor antagonists and calcium antagonists are widely used as a medicament for the treatment or prevention of such hypertension or heart disease.Mineralocorticoid receptor (MR) (aldosterone receptor) has been known to play an important role in the control of body electrolyte balance and blood pressure, spironolactone having a steroid structure, MR antagonists such as eplerenone, are known to be useful in the treatment of hypertension-heart failure.Renin – angiotensin II receptor antagonists are inhibitors of angiotensin system is particularly effective in renin-dependent hypertension, and show a protective effect against cardiovascular and renal failure. Also, the calcium antagonists, and by the function of the calcium channel antagonizes (inhibits), since it has a natriuretic action in addition to the vasodilating action, is effective for hypertension fluid retention properties (renin-independent) .Therefore, the MR antagonist, when combined angiotensin II receptor antagonists or calcium antagonists, it is possible to suppress the genesis of multiple hypertension simultaneously, therapeutic or prophylactic effect of the stable and sufficient hypertension irrespective of the etiology is expected to exhibit.Also, diuretics are widely used as a medicament for the treatment or prevention of such hypertension or heart disease. Diuretic agent is effective in the treatment of hypertension from its diuretic effect. Therefore, if used in combination MR antagonists and diuretics, the diuretic effect of diuretics, it is possible to suppress the genesis of multiple blood pressure at the same time, shows a therapeutic or prophylactic effect of the stable and sufficient hypertension irrespective of the etiology it is expected.1- (2-hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxamide (hereinafter, compound ( I)) is, it is disclosed in Patent Documents 1 and 2, hypertension, for the treatment of such diabetic nephropathy are known to be useful.
CS-3150 (XL550) is a small-molecule antagonist of the mineralocorticoid receptor (MR), a nuclear hormone receptor implicated in a variety of cardiovascular and metabolic diseases. MR antagonists can be used to treat hypertension and congestive heart failure due to their vascular protective effects. Recent studies have also shown beneficial effects of adding MR antagonists to the treatment regimen for Type II diabetic patients with nephropathy. CS-3150 is a non-steroidal, selective MR antagonist that has the potential for the treatment of hypertension, congestive heart failure, or end organ protection due to vascular damage.
Useful as a mineralocorticoid receptor (MR) antagonist, for treating hypertension, cardiac failure and diabetic nephropathy. It is likely to be CS-3150, a non-steroidal MR antagonist, being developed by Daiichi Sankyo (formerly Sankyo), under license from Exelixis, for treating hypertension and diabetic nephropathy (phase 2 clinical, as of March 2015). In January 2015, a phase II trial for type 2 diabetes mellitus and microalbuminuria was planned to be initiated later that month (NCT02345057).
Exelixis discovered CS-3150 and out-licensed the compound to Daiichi-Sankyo. Two phase 2a clinical trials, one in hypertensive patients and the other in type 2 diabetes with albuminuria, are currently being conducted in Japan by Daiichi-Sankyo.


Mineralocorticoid receptor (MR) (aldosterone receptor) has been known to play an important role in the control of body electrolyte balance and blood pressure, spironolactone having a steroid structure, MR antagonists such as eplerenone, are known to be useful in the treatment of hypertension-heart failure.
CS-3150 (XL550) is a small-molecule antagonist of the mineralocorticoid receptor (MR), a nuclear hormone receptor implicated in a variety of cardiovascular and metabolic diseases. MR antagonists can be used to treat hypertension and congestive heart failure due to their vascular protective effects. Recent studies have also shown beneficial effects of adding MR antagonists to the treatment regimen for Type II diabetic patients with nephropathy. CS-3150 is a non-steroidal, selective MR antagonist that has the potential for the treatment of hypertension, congestive heart failure, or end organ protection due to vascular damage.
Exelixis discovered CS-3150 and out-licensed the compound to Daiichi-Sankyo. Two phase 2a clinical trials, one in hypertensive patients and the other in type 2 diabetes with albuminuria, are currently being conducted in Japan by Daiichi-Sankyo.
Daiichi Sankyo (formerly Sankyo), under license from Exelixis, is developing CS-3150 (XL-550), a non-steroidal mineralocorticoid receptor (MR) antagonist, for the potential oral treatment of hypertension and diabetic nephropathy, microalbuminuria , By October 2012, phase II development had begun ; in May 2014, the drug was listed as being in phase IIb development . In January 2015, a phase II trial for type 2 diabetes mellitus and microalbuminuria was planned to be initiated later that month. At that time, the trial was expected to complete in March 2017 .
Exelixis, following its acquisition of X-Ceptor Therapeutics in October 2004 , was investigating the agent for the potential treatment of metabolic disorders and cardiovascular diseases, such as hypertension and congestive heart failure . In September 2004, Exelixis expected to file an IND in 2006. However, it appears that the company had fully outlicensed the agent to Sankyo since March 2006 .
| Description | Small molecule antagonist of the mineralocorticoid receptor (MR) |
| Molecular Target | Mineralocorticoid receptor |
| Mechanism of Action | Mineralocorticoid receptor antagonist |
| Therapeutic Modality | Small molecule |
In January 2015, a multi-center, placebo-controlled, randomized, 5-parallel group, double-blind, phase II trial (JapicCTI-152774; NCT02345057; CS3150-B-J204) was planned to be initiated later that month in Japan, in patients with type 2 diabetes mellitus and microalbuminuria, to assess the efficacy and safety of different doses of CS-3150 compared to placebo. At that time, the trial was expected to complete in March 2017; later that month, the trial was initiated in the Japan
By October 2012, phase II development had begun in patients with essential hypertension
By January 2011, phase I trials had commenced in Japan
Several patents WO-2014168103,
WO-2015012205 and WO-2015030010
XL-550, claimed in WO-2006012642,
………………………………………………………………….
http://www.google.co.in/patents/EP2133330A1?cl=en
(Example 3)(+/-)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide
-
After methyl 4-methyl-5-[2-(trifluoromethyl) phenyl]-1H-pyrrole-3-carboxylate was obtained by the method described in Example 16 of WO 2006/012642 , the following reaction was performed using this compound as a raw material.
-
Methyl 4-methyl-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxylate (1.4 g, 4.9 mmol) was dissolved in methanol (12 mL), and a 5 M aqueous sodium hydroxide solution (10 mL) was added thereto, and the resulting mixture was heated under reflux for 3 hours. After the mixture was cooled to room temperature, formic acid (5 mL) was added thereto to stop the reaction. After the mixture was concentrated under reduced pressure, water (10 mL) was added thereto to suspend the resulting residue. The precipitated solid was collected by filtration and washed 3 times with water. The obtained solid was dried under reduced pressure, whereby 4-methyl-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxylic acid (1.1 g, 83%) was obtained as a solid. The thus obtained solid was suspended in dichloromethane (10 mL), oxalyl chloride (0.86 mL, 10 mmol) was added thereto, and the resulting mixture was stirred at room temperature for 2 hours. After the mixture was concentrated under reduced pressure, the residue was dissolved in tetrahydrofuran (10 mL), and 4-(methylsulfonyl)aniline hydrochloride (1.0 g, 4.9 mmol) and N,N-diisopropylethylamine (2.8 mL, 16 mmol) were sequentially added to the solution, and the resulting mixture was heated under reflux for 18 hours. After the mixture was cooled to room temperature, the solvent was distilled off under reduced pressure, and acetonitrile (10 mL) and 3 M hydrochloric acid (100 mL) were added to the residue. A precipitated solid was triturated, collected by filtration and washed with water, and then, dried under reduced pressure, whereby 4-methyl-N-[4-(methylsulfonyl) phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide (1.4 g, 89%) was obtained as a solid.
1H-NMR (400 MHz, DMSO-d6) δ11.34 (1H, brs,), 9.89 (1H, s), 7.97 (2H, d, J = 6.6 Hz), 7.87-7.81 (3H, m), 7.73 (1H, t, J = 7.4 Hz), 7.65-7.61 (2H, m), 7.44 (1H, d, J = 7.8 Hz), 3.15 (3H, s), 2.01 (3H, s). -
Sodium hydride (0.12 g, 3 mmol, 60% dispersion in mineral oil) was dissolved in N,N-dimethylformamide (1.5 mL), and 4-methyl -N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide (0.47 g, 1.1 mmol) was added thereto, and then, the resulting mixture was stirred at room temperature for 30 minutes. Then, 1,3,2-dioxathiolane-2,2-dioxide (0.14 g, 1.2 mmol) was added thereto, and the resulting mixture was stirred at room temperature. After 1 hour, sodium hydride (40 mg, 1.0 mmol, oily, 60%) was added thereto again, and the resulting mixture was stirred for 30 minutes. Then, 1,3,2-dioxathiolane-2,2-dioxide (12 mg, 0.11 mmol) was added thereto, and the resulting mixture was stirred at room temperature for 1 hour. After the mixture was concentrated under reduced pressure, methanol (5 mL) was added to the residue and insoluble substances were removed by filtration, and the filtrate was concentrated again. To the residue, tetrahydrofuran (2 mL) and 6 M hydrochloric acid (2 mL) were added, and the resulting mixture was stirred at 60°C for 16 hours. The reaction was cooled to room temperature, and then dissolved in ethyl acetate, and washed with water and saturated saline. The organic layer was dried over anhydrous sodium sulfate and filtered. Then, the filtrate was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (ethyl acetate), whereby the objective compound (0.25 g, 48%) was obtained.
1H-NMR (400 MHz, CDCl3) δ: 7.89-7.79 (m, 6H), 7.66-7.58 (m, 2H), 7.49 (s, 1H), 7.36 (d, 1H, J = 7.4Hz), 3.81-3.63 (m, 4H), 3.05 (s, 3H), 2.08 (s, 3H).
HR-MS (ESI) calcd for C22H22F3N2O4S [M+H]+, required m/z: 467.1252, found: 467.1246.
Anal. calcd for C22H21F3N2O4S: C, 56.65; H, 4.54; N, 6.01; F, 12.22; S, 6.87. found: C, 56.39; H, 4.58; N, 5.99; F, 12.72; S, 6.92.
(Example 4)
-
Resolution was performed 4 times in the same manner as in Example 2, whereby 74 mg of Isomer C was obtained as a solid from a fraction containing Isomer C (tR = 10 min), and 71 mg of Isomer D was obtained as a solid from a fraction containing Isomer D (tR = 11 min).
-
Isomer C: (+)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide
[α]D 21: +7.1° (c = 1.0, EtOH) .
1H-NMR (400 MHz, CDCl3) δ: 7.91 (s, 1H), 7.87-7.79 (m, 5H), 7.67-7.58 (m, 2H), 7.51 (s, 1H), 7.35 (d, 1H, J = 7.0 Hz), 3.78-3.65 (m, 4H), 3.05 (s, 3H), 2.07 (s, 3H).
HR-MS (ESI) calcd for C22H22F3N2O4S [M+H]+, required m/z: 467.1252, found: 467.1260.
Retention time: 4.0 min. -
Isomer D: (-)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide
[α]D 21: -7.2° (c = 1.1, EtOH) .
1H-NMR (400 MHz, CDCl3) δ: 7.88-7.79 (m, 6H), 7.67-7.58 (m, 2H), 7.50 (s, 1H), 7.36 (d, 1H, J = 7.5 Hz), 3.79-3.65 (m, 4H), 3.05 (s, 3H), 2.08 (s, 3H).
HR-MS (ESI) calcd for C22H22F3N2O4S [M+H]+, required m/z: 467.1252, found: 467.1257.
Retention time: 4.5 min.
……………………………………………….
WO 2014168103
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014168103
(7-1) (S) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid quinine salt
obtained by the method of Example 6 the (RS) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid 50.00 g (160 mmol), N, N- dimethylacetamide (25 mL), ethyl acetate (85 mL) was added and dissolved at room temperature (solution 1).
mobile phase A: 0.02mol / L phosphorus vinegar buffer solution (pH 3)
mobile phase B: acetonitrile
solution sending of mobile phase: mobile phase A and I indicates the mixing ratio of mobile phase B in Table 1 below.
flow rate: about 0.8 mL / min
column temperature: 30 ℃ constant temperature in the vicinity of
measuring time: about 20 min
Injection volume: 5 μL
diastereomeric excess (% de), the title compound (retention time about 12 min), was calculated by the following equation using a peak area ratio of R-isomer (retention time of about 13 min).
% De = {[(the title compound (S body) peak area ratio) – (R body peak area ratio)] ÷ [(the title compound (S body) peak area ratio) + (R body peak area ratio)]} × 100
(8-1) (S)-1-(2-hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole -3 – carboxylic acid
obtained by the method of Example 7 (S) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid (8α, 9R) -6′- methoxycinnamate Conan-9-ol 40.00 g (63 mmol) in ethyl acetate (400 mL), was added 2N aqueous hydrochloric acid (100 mL) was stirred at room temperature and separated . The resulting organic layer was concentrated under reduced pressure (120 mL), and added ethyl acetate (200 mL), and further concentrated under reduced pressure to obtain a solution containing the title compound (120 mL).
ethyl acetate (240 mL), was mixed tetrahydrofuran (80 mL) and oxalyl chloride 20.72 g (163 mmol), and cooled to 10 ~ 15 ℃ was. Then the resulting solution was added while keeping the 10 ~ 15 ℃ Example (8-1) and stirred for about 1 hour by heating to 15 ~ 20 ℃. After stirring, acetonitrile (120 mL) and pyridine 2.46 g (31 mmol) was added and the reaction mixture was concentrated under reduced pressure (120 mL), acetonitrile (200 mL) was added and further concentrated under reduced pressure (120 mL).
obtained by the method of Example 7 (S) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole 3-carboxylic acid (8α, 9R) -6′- methoxycinnamate Conan-9-ol 10.00 g (16 mmol) in t- butyl methyl ether (90 mL), water (10 mL) 36w / w% aqueous hydrochloric acid ( 5 mL) was added and stirring at room temperature and separated. The resulting organic layer was concentrated under reduced pressure (30 mL), was added ethyl acetate (50 mL), and further concentrated under reduced pressure to obtain a solution containing the title compound (30 mL).
ethyl acetate (50 mL), was mixed with tetrahydrofuran (20 mL) and oxalyl chloride 5.18 g (41 mmol), and cooled to 0 ~ 5 ℃ was.Then the resulting solution was added in Examples while maintaining the 0 ~ 5 ℃ (12-1), and the mixture was stirred for 6 hours at 0 ~ 10 ℃. After stirring, acetonitrile (30 mL) and pyridine 0.62 g (8 mmol) was added and the reaction mixture was concentrated under reduced pressure (30 mL), acetonitrile (50 mL) was added, and further concentrated under reduced pressure (30 mL).
………………………………………………
Patent literature
Patent Document 2: International Publication WO2008 / 056907 (US Publication US2010-0093826)
Patent Document 3: Pat. No. 2,082,519 JP (US Patent No. 5,616,599 JP)
Patent Document 4: Pat. No. 1,401,088 JP (US Pat. No. 4,572,909)
Patent Document 5: US Pat. No. 3,025,292
Angiotensin II receptor 桔抗 agent
is described in Japanese or the like, its chemical name is (5-methyl-2-oxo-1,3-dioxolen-4-yl ) methyl 4- (1-hydroxy-1-methylethyl) -2-propyl-1 – in [2 ‘(1H- tetrazol-5-yl) biphenyl-4-ylmethyl] imidazole-5-carboxylate, Yes, olmesartan medoxomil of the present application includes its pharmacologically acceptable salt.
 OLMESARTAN
OLMESARTAN
Daiichi Sankyo receives FDA approval for anti-clotting drug Savaysa , EDOXABAN

Edoxaban, DU-176b
Edoxaban (DU-176b, trade names Savaysa, Lixiana) is an anticoagulant drug which acts as a direct factor Xa inhibitor. It was developed by Daiichi Sankyo and approved in July 2011 in Japan for prevention of venous thromboembolisms (VTE) following lower-limb orthopedic surgery.[1] It was also approved by the FDA in January 2015 for the prevention of stroke and non–central-nervous-system systemic embolism.[2]
Daiichi Sankyo receives FDA approval for anti-clotting drug Savaysa
Japanese drug-maker Daiichi Sankyo has obtained approval from US Food and Drug Administration (FDA) for its anti-clotting drug Savaysa (edoxaban tablets)..8 JAN 2015

Daiichi Sankyo, APPROVED IN JAPAN as tosylate monohydrate salt in 2011 for the prevention of venous embolism in patients undergoing total hip replacement surgery



for synthesis see….http://www.sciencedirect.com/science/article/pii/S0968089613002642 Bioorganic & Medicinal Chemistry 21 (2013) 2795–2825, see s[pecific page 2808 for description ie 14/31 of pdf
WO 2010071121, http://www.google.com/patents/WO2010071121A1
WO 2007032498
N’-(5-chloropyridin-2-yl)-N-[(1S,2R,4S)-4-(dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[1,3]thiazolo[5,4-c]pyridine-2-carbonyl)amino]cyclohexyl]oxamide
- N1-(5-Chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-{[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide

Edoxaban (INN, codenamed DU-176b, trade name Lixiana) is an anticoagulant drug which acts as a direct factor Xa inhibitor. It is being developed by Daiichi Sankyo. It was approved in July 2011 in Japan for prevention of venous thromboembolisms (VTE) following lower-limb orthopedic surgery.[1]

In animal studies, edoxaban is potent, selective for factor Xa and has good oral bioavailability.[2]
Daichi Sankyo’s edoxaban tosilate is an orally administered
coagulation factor Xa inhibitor that was approved and launched
in Japan for the preventive treatment of venous thromboembolic
events (VTE) in patients undergoing total knee arthroplasty, total
hip arthroplasty, or hip fracture surgery. Edoxaban has been
shown to have a rapid onset of anticoagulant effect due to short
Tmax (1–2 h) after dosing and sustained for up to 24 h post-dose.
Marketed under the brand name Lixiana, it is currently in phase
III studies in the US for the prevention of stroke and systemic embolic
events in patients with atrial fibrillation (AF) and venous
thromboembolism (VTE).
Several Phase II clinical trials have been conducted, for example for thromboprophylaxis after total hip replacement[3] (phase III early results compare well to enoxaparin[4]), and for stroke prevention in patients with atrial fibrillation[5][6].Those papers follow similar recent major trials showing similar results for the other new factor Xa inhibitors, rivaroxaban and apixaban.
A large phase III trial showed that edoxaban was non inferior to warfarin in preventing recurrent venous thromboembolic events with fewer episodes of major bleeding.[7]


……………..
PATENT
http://www.google.com/patents/WO2014081047A1?cl=en
Chemically, edoxaban is
N1– (5-chloropyridin-2-yl) -N2– ( (IS, 2R/4S) -4- [ (dimethylamino) carbo nyl] -2- { [ ( 5-methyl-4 , 5,6, 7-tetrahydrothiazolo [5 , 4-c] pyridin-2-yl ) carbonyl] amino}eyelohexyl) ethanediamide , represented by the following formula (A) :

(A) The p-toluenesulfonic acid monohydrate salt of compound A is represented b the following formula (B) :

(B)
Edoxaban is known as a compound that exhibits an inhibitory effect on activated blood coagulation factor X (also referred to as activated factor X or FXa) , and is useful as a preventive and/or therapeutic drug for thrombotic diseases.
Several processes are known in the literature for preparing edoxaban for example, U.S. Patent No. 7365205; U.S. Publication No . 20090105491.
U.S. Patent No. 7365205 provides a process for the preparation of edoxaban, wherein the process involves the use of
(IS, 4S, 5S) -4-iodo-6-oxabicyclo [3.2.1] octan-7-one, represented by the following formula (C) :

(C)
as an intermediate.
The present inventors have identified that
(IS, 4S, 5S) -4-bromo-6-oxabicyclo [3.2.1] octan-7-one, represented by the following formula (I) :

( I )
could also be used as an intermediate for the preparation of FXa inhibitory compounds like edoxaban. The present inventors have found that replacement of
(IS, 4S, 5S) -4-iodo-6-oxabicyclo [3.2.1] octan-7-one (C) with
(IS, 4S, 5S) -4-bromo-6-oxabicyclo [3.2.1] octan-7-one (I) has a better atom economy and also an impact on cost.
A method for the synthesis of the
(IS, 4S, 5S) -4 -bromo- 6 -oxabicyclo [3.2.1] octan-7-one (I) was reported in Tetrahedron Letters, 51, (2010) Pages 3433-3435 which involves the reaction of ( IS) -cyclohex-3 -ene- 1-carboxylic acid represented by the following formula (II) :

( Π )
with N-bromosuccinimide in the presence of molecular sieves using dichloromethane as a solvent. However, this reaction is carried out in dark over a period of 7 hours and does not provide a pure product .
Tetrahedron, Vol. 28, Pages 3393 -3399 , 1972 provides a process for the preparation of 4 -bromo- 6 -oxabicyclo [3.2.1] octan-7-one which involves the addition of 20% excess of a 2M solution of bromine in chloroform to a stirred solution of cyclohex- 3 -ene- 1-carboxylic acid (0.04 mol) in chloroform (250 mL) in the absence of a base . Extraction with aqueous sodium bicarbonate followed by acidification gave, after extraction with ether and evaporation of the extract, a mixture of cis & trans 3 , 4-dibromocyclohexanecarboxylic acid (6.7 g) and evaporation of the chloroform layer afforded the bromolactone (0.59 g) . It further provides a process for the preparation of
4 -bromo-6 -oxabicyclo [3.2.1] octan-7-one which involves the treating of cyclohex-3-ene-l-carboxylic acid (0.08 mol) dissolved in chloroform (450 mL) with 20% excess bromine in the presence of an equimolar amount of triethylamine (8.1 g) . After extraction of the amine with 2N hydrochloric acid, and work-up, bromolactone (10.7 g) and a mixture of cis & trans 3 , 4 -dibromocyclohexanecarboxylic acid (6.6 g) were obtained.
Tetrahedron Vol. 48, No. 3, Pages 539-544, 1992 provides a process for the preparation of
(IS, 4S, 5S) -4-bromo-6-oxabicyclo [3.2.1] octan-7-one (I) which involves the addition of 1M solution of bromine in chloroform (30 mL) at 0°C to a solution of ( IS) -cyclohex-3 -ene- 1-carboxylic acid (0.024 mol) of formula (II) in chloroform (600 mL) in the presence of an equimolar amount of triethylamine (3.33 mL) . After work-up, the crude bromolactone obtained was recrystallized from petroleum ether.
However, bromination using bromine does not provide a pure product in good yield.
Heterocycles, Vol. 23, No. 8, Pages 2035-2039, 1985 provides a process for the 4-bromo-6-oxabicyclo [3.2.1] octan-7-one which involves the addition of cyclohex-3-ene-l-carboxylic acid (1.0 mM) in 1 , 2 -dimethoxyethane (2 mL) to a stirred solution of 90% Lead (IV) acetate (1.1 or 2.2 mM) in 1 , 2 -dimethoxyethane (4 mL) followed by the addition of Zinc bromide (2.2 mM) in 1 , 2 -dimethoxyethane (4 mL) and continuing the stirring for 10-30 minutes at 0°C . The reaction mixture was poured into a solution of ice-cold water (30 mL) and 10% hydrochloric acid (10 mL) , and extracted with ether (50 mL X 3) . The combined ether extract was washed successively with saturated sodium hydrogen carbonate solution (20 mL) , 10% sodium thiosulphate solution (5 mL) , and brine (10 mL) , and dried over sodium sulphate. Evaporation of the solvent gave crude lactone which were separated and purified (42% yield) . However, this reaction does not provide a pure product in good yield.
Heterocycles, Vol. 31, No. 6, Pages 987-991, 1990 provides a method for bromolactonization using a
dimethylsulfoxide-trimethylsilyl bromide-amine system. The bromolactonization is carried out for 10 to 72 hours using different solvents and triethylamine or diisopropylethyl amine as base. However, this process does not provide a product in high yield. Further the process afforded the cis isomer exclusively. Journal of the Chemical Society, Perkin Transactions 1:
Organic and Bio-Organic Chemistry (1972-1999) (1994) , (7) , Pages 847-851 provides a method for bromolactonization using a
dimethylsulfoxide-trimethylsilyl bromide-amine system. The bromolactonization is carried out for 12 hours using
dimethylsulfoxide and chloroform solvent system and triethylamine or diisopropylethyl amine as base. However, this process resulted in a low yield of about 55%. Citation List
Patent Literature
PTLl: U.S. Patent No. 7365205
PTL2: U.S. Publication No. 20090105491.
Non Patent Reference
NPLl: Feng Chen et al . , Tetrahedron Letters, 51, (2010) Pages 3433-3435.
NPL2 : G. Belluci et al . , Tetrahedron, Vol. 28, No. 13, Pages 3393-3399, 1972.
NPL3 : Marco Chini et al ., Tetrahedron Vol .48, No. 3, Pages 539-544 , 1992.
NPL4 : Y. Fujimoto et al . , Heterocycles , Vol. 23, No. 8, Pages 2035-2039, 1985.
NPL5: C. Iwata et al . , Heterocycles, Vol. 31, No. 6, Pages 987-991, 1990. –
NPL6 : K. Miyashita et al . , Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999) (1994) , (7) , Pages 847-851.
Summary of Invention
Technical Problem
It is an object of the present invention to solve the problems associated with the prior art, and to provide an improved and efficient method for the preparation of
(IS, 4S, 5S) -4-bromo-6-oxabicyclo [3.2.1] octan-7-one of formula (I).
Solution to Problem As a result of conducting diligent studies to attain the object, the present inventors have found that: surprisingly, the use of N-bromosuccinimide or bromohydantoin (representative is
1, 3-dibromo-5, 5-dimethylhydantoin) as brominating agent in the presence of a base selected from calcium oxide or calcium hydroxide, in specific mole ratios in a solvent selected from the group consisting of dichloromethane , toluene, tetrahydrofuran, ethyl acetate, hexanes, cyclopentyl methyl ether (CPME) or a mixture thereof can efficiently produce a pure
( IS , 4S , 5S) -4 -bromo- 6 -oxabicyclo [3.2.1] octan- 7 -one (I) in better yields. The process provides obvious benefits with respect to economics, convenience to operate at a commercial scale.
…………………………..
SEE
http://www.google.co.ug/patents/US20090105491
………………………….
PATENT
http://www.google.com/patents/EP2589590A1?cl=en
FREE BASE
- (Reference Example 6) N1-(5-Chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-{[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide (X) (production method described in the pamphlet of International Publication No. WO 2007/032498)
-

-
Methanesulfonic acid (66 ml) was added to a suspension of tert-butyl [(1R,2,S,5S)-2-({[(5-chloropyridin-2-yl)amino](oxo)acetyl}amino)-5-(dimethylaminocarbonyl)cyclohexyl]carbamate (5) (95.1 g) in acetonitrile (1900 ml) at room temperature, and the mixture was stirred at this temperature for 2 hours. To the reaction solution, triethylamine (155 ml), 5-methyl-4,5,6,7-tetrahydro[1,3]thiazolo[5,4-c]pyrzdine-2-carboxylic acid hydrochloride (8) (52.5 g), 1-hydroxybenzotriazole (33.0 g), and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (46.8 g) were added under ice cooling, and the mixture was stirred at room temperature for 16 hours. Triethylamine and water were added thereto, and the mixture was stirred for 1 hour under ice cooling. Then, crystals were collected by filtration to obtain the title compound (X) (103.2 g). 1H-NMR (CDCl3) δ : 1.60-1.98 (3H, m), 2.00-2.16 (3H, m), 2.52 (3H, s), 2.78-2.90 (3H, m), 2.92-2.98 (2H, m), 2.95 (3H, s), 3.06 (3H, s), 3.69 (1H, d, J = 15.4 Hz), 3.75 (1H, d, J = 15.4 Hz), 4.07-4.15 (1H, m), 4.66-4.72 (1H, m), 7.40 (1H, dd, J = 8.8, 0.6 Hz), 7. 68 (1H, dd, J = 8.8, 2.4 Hz), 8.03 (1H, d, J = 7.8 Hz), 8.16 (1H, dd, J = 8.8, 0.6 Hz), 8.30 (1H, dd, J = 2. 4, 0.6 Hz), 9.72 (1H, s). MS (ESI) m/z: 548 (M+H)+.

TOSYLATE
- (Reference Example 7) N1-(5-Chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-{[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide mono-p-toluenesulfonate monohydrate (X-a) (production method described in the pamphlet of International Publication No. WO 2007/032498)
-

-
N1-(5-Chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-{[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide (X) (6.2 g) was dissolved in methylene chloride (120 ml). To the solution, a 1 mol/L solution of p-toluenesulfonic acid in ethanol (11.28 ml) was added, and the solvent was distilled off. To the residue, 15% hydrous ethanol (95 ml) was added, and the mixture was dissolved by stirring at 60°C. Then, the mixture was cooled to room temperature and stirred for 1 day. The precipitated crystals were collected by filtration, washed with ethanol, and then dried under reduced pressure at room temperature for 2 hours to obtain the title compound (X-a) (7.4 g).
1H-NMR (DMSO-d6) δ : 1. 45-1. 54 (1H, m), 1.66-1.78 (3H, m), 2.03-2.10 (2H, m), 2.28 (3H, s), 2.79 (3H, s), 2.91-3.02 (1H, m), 2.93 (3H, s), 2.99 (3H, s), 3.13-3.24 (2H, m), 3.46-3.82 (2H, m), 3.98-4.04 (1H, m), 4.43-4.80 (3H, m), 7.11 (2H, d, J = 7.8 Hz), 7.46 (2H, d, J = 8.2 Hz), 8.01 (2H, d, J = 1.8 Hz), 8.46 (1H, t, J = 1.8 Hz), 8.75 (1H, d, J = 6.9 Hz), 9.10-9.28 (1H, br), 10.18 (1H, br), 10.29 (1H, s).
MS (ESI) m/z: 548 (M+H)+.
Anal.: C24H30ClN7O4S·C7H8O3S·H2O
Theoretical: C; 50.43, H; 5.46, N; 13.28, Cl; 4.80, S; 8.69.
Found: C; 50.25, H; 5.36, N; 13.32, Cl; 4.93, S; 8.79. mp (dec.): 245-248°C.

……………………………………………..
PATENT
http://www.google.com/patents/EP2589590A1?cl=en
-
A compound represented by the following formula (X) [hereinafter, also referred to as compound (X)] or a pharmacologically acceptable salt thereof, or a hydrate thereof is a compound that exhibits an FXa inhibitory effect, as disclosed in Patent Literatures 1 to 3, and is useful as a preventive and/or therapeutic drug for thrombotic and/or embolic diseases:
-

-
The pamphlet of International Publication No. WO 2007/032498discloses a process for preparing an FXa inhibitor compound (X) or a pharmacologically acceptable salt thereof, or a hydrate thereof. The process for producing compound (X) disclosed therein involves, as shown in [Scheme A] below, azidifying compound (2) to produce azide compound (3), subsequently reducing compound (3) into amino compound (1a), subsequently treating compound (1a) with anhydrous oxalic acid to obtain compound (1), which is then treated with compound (4) (ethyl[5-chloropyridin-2-yl]amino](oxo)acetate hydrochloride) in the presence of a base to produce compound (5), followed by several steps from compound (5). This pamphlet also discloses crystals of the oxalate of compound (1) as a production intermediate.
-

-
wherein Boc represents a tert-butoxycarbonyl group.


Citation ListPatent Literatures
-
- Patent Literature 1: International Publication No.WO 2004/058715
- Patent Literature 2: International Publication No.WO 2003/016302
- Patent Literature 3: International Publication No.WO 2003/000680
- Patent Literature 4: International Publication No.WO 2007/032498

“First market approval in Japan for LIXIANA (Edoxaban)”. Press Release. Daiichi Sankyo Europe GmbH. 2011-04-22.
- Furugohri T, Isobe K, Honda Y, Kamisato-Matsumoto C, Sugiyama N, Nagahara T, Morishima Y, Shibano T (September 2008). “DU-176b, a potent and orally active factor Xa inhibitor: in vitro and in vivo pharmacological profiles”. J. Thromb. Haemost.6 (9): 1542–9. doi:10.1111/j.1538-7836.2008.03064.x. PMID18624979.
- Raskob, G.; Cohen, A. T.; Eriksson, B. I.; Puskas, D.; Shi, M.; Bocanegra, T.; Weitz, J. I. (2010). “Oral direct factor Xa inhibition with edoxaban for thromboprophylaxis after elective total hip replacement”. Thrombosis and Haemostasis104 (3): 642–649. doi:10.1160/TH10-02-0142.PMID20589317. edit
- “Phase III Trial Finds Edoxaban Outclasses Enoxaparin in Preventing Venous Thromboembolic Events”. 8 Dec 2010.
- Weitz JI, Connolly SJ, Patel I, Salazar D, Rohatagi S, Mendell J, Kastrissios H, Jin J, Kunitada S (September 2010). “Randomised, parallel-group, multicentre, multinational phase 2 study comparing edoxaban, an oral factor Xa inhibitor, with warfarin for stroke prevention in patients with atrial fibrillation”. Thromb. Haemost.104 (3): 633–41. doi:10.1160/TH10-01-0066.
- Edoxaban versus Warfarin in Patients with Atrial Fibrillation Robert P. Giugliano, M.D., Christian T. Ruff, M.D., M.P.H., Eugene Braunwald, M.D., Sabina A. Murphy, M.P.H., Stephen D. Wiviott, M.D., Jonathan L. Halperin, M.D., Albert L. Waldo, M.D., Michael D. Ezekowitz, M.D., D.Phil., Jeffrey I. Weitz, M.D., Jindřich Špinar, M.D., Witold Ruzyllo, M.D., Mikhail Ruda, M.D., Yukihiro Koretsune, M.D., Joshua Betcher, Ph.D., Minggao Shi, Ph.D., Laura T. Grip, A.B., Shirali P. Patel, B.S., Indravadan Patel, M.D., James J. Hanyok, Pharm.D., Michele Mercuri, M.D., and Elliott M. Antman, M.D. for the ENGAGE AF-TIMI 48 InvestigatorsDOI: 10.1056/NEJMoa1310907
- “Edoxaban versus Warfarin for the Treatment of Symptomatic Venous Thromboembolism”. N. Engl. J. Med. August 2013. doi:10.1056/NEJMoa1306638. PMID23991658.
- WO 03/000657 pamphlet WO 03/000680 pamphlet WO 03/016302 pamphlet WO 04/058715 pamphlet WO 05/047296 pamphlet WO 07/032498 pamphlet WO 08/129846 pamphlet WO 08/156159 pamphlet
- J Am Chem Soc 1978, 100(16): 5199
| [1] | 王利华, 赵丽嘉, 李文利, 等. 直接抑制凝血因子Xa 的口服抗凝药物Edoxaban Tosilate Hydrate [J]. 药物评价研究, 2011, 34(6): 478-481. |
| [2] | Ohta T, Komoriya S, Yoshino T, et al. Preparation of N,N’-bis( heterocyclicacyl) cycloalkanediamine and heterocyclediamine derivatives as inhibitors of activated blood coagulation factor X (factor Xa): WO, 2003 000657 [P]. 2003-01-03. (CA 2003, 138: 73271) |
| [3] | Ohta T, Komoriya S, Yoshino T, et al. Preparation of heterocyclic moiety-containing diamine derivatives as FXa inhibitors: WO, 2003 000680 [P]. 2003-01-03. (CA 2003, 138: 89801) |
| [4] | Mochizuki A, Nagata T. Triamine derivative: WO, 2006106963 [P]. 2005-03-31. (CA 2006, 145: 419128) |
| [5] | Kawanami K, Ishikawa H, Shoji M. Process for preparation of optically active (1S,3R,4R)-3-amino-4-hydroxy-N,Ndimethylcyclohexanecarboxamide derivative salt: WO, 2012002538 [P]. 2012-01-05. (CA 2012, 156: 122056) |
| [6] | Sato K, Kubota K. Process for producing optically active carboxylic acid: WO, 2010067824 [P]. 2010-06-17. (CA 2010, 153: 36882) |
| [7] | Yoshikawa K, Yokomizo A, Naito H, et al. Design, synthesis, and SAR of cis-1,2-diaminocyclohexane derivatives as potent factor Xa inhibitors. Part I: Exploration of 5-6fused rings |
| [8] | as alternative S1 moieties [J]. Bioorg Med Chem, 2009, 17(24): 8206-8220. |
| [9] | Sato K, Kawanami K, Yagi T. Process for the preparation of optically active cyclohexane-1,2-diamine derivative from 7-oxabicyclo[4.1.0]heptane compound: WO, 2007032498 |
| [10] | 2007-03-22. (CA 2007, 146: 358502) |
| [11] | Kawanami K. Method for the preparation of optically active diamine derivative: WO, 2010104106 [P]. 2010-09-16. (CA 2010, 153: 406061) |
| [12] | Koyama T, Kondo S. Process for the preparation of diamine derivative: WO, 2010104078 [P]. 2010-09-16. (CA 2010, 153: 382938) |
| [13] | Suzuki T, Ono M. Crystal of diamine derivative and method of producing same: WO, 2011115066 [P]. 2011-09-22. (CA 2011, 155: 467954) |
| US8357808 | 9 Sep 2011 | 22 Jan 2013 | Daiichi Sankyo Company, Limited | Process for producing diamine derivative |
| US8394821 | 13 Jul 2011 | 12 Mar 2013 | Daiichi Sankyo Company, Limited | Activated blood coagulation factor inhibitor |
| US8404847 | 17 Jun 2011 | 26 Mar 2013 | Daiichi Sankyo Company, Limited | Method for producing diamine derivative |
| US8449896 | 16 Dec 2011 | 28 May 2013 | Daiichi Sankyo Company, Limited | Pharmaceutical composition having improved solubility |
| US8541443 | 19 Sep 2012 | 24 Sep 2013 | Daiichi Sankyo Company, Limited | Crystal of diamine derivative and method of producing same |
| US20130004550 * | 22 Aug 2012 | 3 Jan 2013 | Daiichi Sankyo Company, Limited | Sustained-release solid preparation for oral use |
| WO2014081047A1 | 22 Nov 2013 | 30 May 2014 | Daiichi Sankyo Company,Limited | Process for the preparation of (1s,4s,5s)-4-bromo-6-oxabicyclo[3.2.1] octan-7-one |

| Molecular Formula | C24H30ClN7O4S.C7H7HSO3 |
| Molecular Weight | 720.26 |
| CAS Registry Number | 480449-71-6 (912273-65-5) |
Drug formulation , lixiana, edoxaban tosylate monohydrate, CAS 912273-65-5, C24 H30 Cl N7 O4 S . C7 H8 O3 S . H2 O, 738.274
-
N1-(5-chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-{[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide p-toluenesulfonic acid monohydrate represented by the following formula (A) (hereinafter, also referred to as compound A) :
-


-
is known as a compound that exhibits an inhibitory effect on activated blood coagulation factor X (FXa), and is useful as a preventive and/or therapeutic drug for thrombotic diseases (Patent Literature 1 to 8).
-
For example, a method comprising mixing the free form of compound A represented by the following formula (B) (hereinafter, also referred to as compound B):
-

-
with p-toluenesulfonic acid or p-toluenesulfonic acid monohydrate, followed by crystallization from aqueous ethanol, is known as a method for obtaining compound A (Patent Literature 1 to 8). These literature documents do not make any mention about adding p-toluenesulfonic acid or p-toluenesulfonic acid monohydrate in a stepwise manner in the step of obtaining compound A from compound B.
Citation ListPatent Literature
-
- Patent Literature 1: International Publication No. WO 03/000657
- Patent Literature 2: International Publication No. WO 03/000680
- Patent Literature 3: International Publication No. WO 03/016302
- Patent Literature 4: International Publication No. WO 04/058715
- Patent Literature 5: International Publication No. WO 05/047296
- Patent Literature 6: International Publication No. WO 07/032498
- Patent Literature 7: International Publication No. WO 08/129846
- Patent Literature 8: International Publication No. WO 08/156159
SIMILAR

OTHER SALTS

Edoxaban hydrochloride
CAS Number: 480448-29-1
Molecular Formula: C24H30ClN7O4S · HCl
Molecular Weight: 584.52 g.mol-1
Edoxaban is reported to be a member of the so-called “Xaban-group” and as such to be a low molecular inhibitor of the enzyme factor Xa, participating in the blood coagulation system. Therefore, edoxaban is classified as an antithrombotic drug and its possible medical indications are reported to be treatment of thrombosis and thrombosis prophylaxis after orthopaedic operations, such as total hip replacement, as well as for stroke prevention in patients with atrial fibrillation, the prophylaxis of the acute coronary syndrome and the prophylaxis after thrombosis and pulmonary embolism.
The IUPAC name for edoxaban is N’-(5-chloropyridin-2-yl)-N-[(15,2^,4S)-4- (dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[l ,3]thiazolo[5,4-c]pyridine-2- carbonyl)amino]cyclohexyl]oxamide. The chemical structure of edoxaban is shown in the formula (1) below:

formula ( 1 ) While Edoxaban is reported to be soluble in strongly acidic aqueous solutions, its solubility is considered to be very low in neutral or alkaline aqueous media. EP 2 140 867 A 1 claims an edoxaban-containing pharmaceutical composition comprising a water-swelling additive and/or a sugar alcohol. Further, it is alleged that compositions comprising lactose or cornstarch do not have good dissolution properties. The claimed pharmaceutical compositions in EP 2 140 867 Al are considered to show good dissolution properties in a neutral aqueous medium as well. Tablets comprising said composition were produced by wet granulation. However, it turned out that prior art pharmaceutical formulations comprising edoxaban being suitable for oral administration are still improvable with regards to dissolution rate and bioavailability. Further, stability and content uniformity of the known formulations could be improved. Further, due to the intolerance of many people to sugar alcohol(s), such as sorbitol, the use of sugar alcohol(s) should be avoided.
UPDATE
2-amino-5-methyl-4,5,6,7-tetrahydro thiazolone [5,4-c] pyridine
MS (FAB) M / z: 170 (M + H) +
elemental analysis: C 7 H 11 N 3 as S,
theoretical value: C, 49.68; H, 6.55; N, 24.83; S, 18.95
measured value: C, 49.70; H, 6.39; N, 24.91; S, 19.00.
UPDATE

Edoxaban, DU-176b
1H NMR PREDICTION

………….
13 C NMR

FREE BASE
- (Reference Example 6) N1-(5-Chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-{[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide (X) (production method described in the pamphlet of International Publication No. WO 2007/032498)
-

-
Methanesulfonic acid (66 ml) was added to a suspension of tert-butyl [(1R,2,S,5S)-2-({[(5-chloropyridin-2-yl)amino](oxo)acetyl}amino)-5-(dimethylaminocarbonyl)cyclohexyl]carbamate (5) (95.1 g) in acetonitrile (1900 ml) at room temperature, and the mixture was stirred at this temperature for 2 hours. To the reaction solution, triethylamine (155 ml), 5-methyl-4,5,6,7-tetrahydro[1,3]thiazolo[5,4-c]pyrzdine-2-carboxylic acid hydrochloride (8) (52.5 g), 1-hydroxybenzotriazole (33.0 g), and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (46.8 g) were added under ice cooling, and the mixture was stirred at room temperature for 16 hours. Triethylamine and water were added thereto, and the mixture was stirred for 1 hour under ice cooling. Then, crystals were collected by filtration to obtain the title compound (X) (103.2 g).
-
1H-NMR (CDCl3) δ : 1.60-1.98 (3H, m), 2.00-2.16 (3H, m), 2.52 (3H, s), 2.78-2.90 (3H, m), 2.92-2.98 (2H, m), 2.95 (3H, s), 3.06 (3H, s), 3.69 (1H, d, J = 15.4 Hz), 3.75 (1H, d, J = 15.4 Hz), 4.07-4.15 (1H, m), 4.66-4.72 (1H, m), 7.40 (1H, dd, J = 8.8, 0.6 Hz), 7. 68 (1H, dd, J = 8.8, 2.4 Hz), 8.03 (1H, d, J = 7.8 Hz), 8.16 (1H, dd, J = 8.8, 0.6 Hz), 8.30 (1H, dd, J = 2. 4, 0.6 Hz), 9.72 (1H, s).
-
MS (ESI) m/z: 548 (M+H)+.

| Molecular Formula | C24H30ClN7O4S.C7H7HSO3 | |
| Molecular Weight | 720.26 | |
| CAS Registry Number | 480449-71-6 (912273-65-5) |
TOSYLATE
- (Reference Example 7) N1-(5-Chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-{[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide mono-p-toluenesulfonate monohydrate (X-a) (production method described in the pamphlet of International Publication No. WO 2007/032498)
-

-
N1-(5-Chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-{[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide (X) (6.2 g) was dissolved in methylene chloride (120 ml). To the solution, a 1 mol/L solution of p-toluenesulfonic acid in ethanol (11.28 ml) was added, and the solvent was distilled off. To the residue, 15% hydrous ethanol (95 ml) was added, and the mixture was dissolved by stirring at 60°C. Then, the mixture was cooled to room temperature and stirred for 1 day. The precipitated crystals were collected by filtration, washed with ethanol, and then dried under reduced pressure at room temperature for 2 hours to obtain the title compound (X-a) (7.4 g).
-
1H-NMR (DMSO-d6) δ : 1. 45-1. 54 (1H, m), 1.66-1.78 (3H, m), 2.03-2.10 (2H, m), 2.28 (3H, s), 2.79 (3H, s), 2.91-3.02 (1H, m), 2.93 (3H, s), 2.99 (3H, s), 3.13-3.24 (2H, m), 3.46-3.82 (2H, m), 3.98-4.04 (1H, m), 4.43-4.80 (3H, m), 7.11 (2H, d, J = 7.8 Hz), 7.46 (2H, d, J = 8.2 Hz), 8.01 (2H, d, J = 1.8 Hz), 8.46 (1H, t, J = 1.8 Hz), 8.75 (1H, d, J = 6.9 Hz), 9.10-9.28 (1H, br), 10.18 (1H, br), 10.29 (1H, s).
MS (ESI) m/z: 548 (M+H)+.
Anal.: C24H30ClN7O4S·C7H8O3S·H2O
Theoretical: C; 50.43, H; 5.46, N; 13.28, Cl; 4.80, S; 8.69.
Found: C; 50.25, H; 5.36, N; 13.32, Cl; 4.93, S; 8.79. mp (dec.): 245-248°C.
1H NMR PREDICTION, TOSYLATE
CAS NO. 1229194-11-9, N’-(5-chloropyridin-2-yl)-N-[(1S,2R,4S)-4-(dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[1,3]thiazolo[5,4-c]pyridine-2-carbonyl)amino]cyclohexyl]oxamide,4-methylbenzenesulfonic acid,hydrate H-NMR spectral analysis
![N'-(5-chloropyridin-2-yl)-N-[(1S,2R,4S)-4-(dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[1,3]thiazolo[5,4-c]pyridine-2-carbonyl)amino]cyclohexyl]oxamide,4-methylbenzenesulfonic acid,hydrate NMR spectra analysis, Chemical CAS NO. 1229194-11-9 NMR spectral analysis, N'-(5-chloropyridin-2-yl)-N-[(1S,2R,4S)-4-(dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[1,3]thiazolo[5,4-c]pyridine-2-carbonyl)amino]cyclohexyl]oxamide,4-methylbenzenesulfonic acid,hydrate H-NMR spectrum](https://images-blogger-opensocial.googleusercontent.com/gadgets/proxy?url=http%3A%2F%2Fpic11.molbase.net%2Fnmr%2Fnmr_image%2F2015-01-11%2F001%2F717%2F1717235_1h.png&container=blogger&gadget=a&rewriteMime=image%2F*)
13 CNMR PREDICTION, TOSYLATE
CAS NO. 1229194-11-9, N’-(5-chloropyridin-2-yl)-N-[(1S,2R,4S)-4-(dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[1,3]thiazolo[5,4-c]pyridine-2-carbonyl)amino]cyclohexyl]oxamide,4-methylbenzenesulfonic acid,hydrate C-NMR spectral analysis
![N'-(5-chloropyridin-2-yl)-N-[(1S,2R,4S)-4-(dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[1,3]thiazolo[5,4-c]pyridine-2-carbonyl)amino]cyclohexyl]oxamide,4-methylbenzenesulfonic acid,hydrate NMR spectra analysis, Chemical CAS NO. 1229194-11-9 NMR spectral analysis, N'-(5-chloropyridin-2-yl)-N-[(1S,2R,4S)-4-(dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[1,3]thiazolo[5,4-c]pyridine-2-carbonyl)amino]cyclohexyl]oxamide,4-methylbenzenesulfonic acid,hydrate C-NMR spectrum](https://images-blogger-opensocial.googleusercontent.com/gadgets/proxy?url=http%3A%2F%2Fpic11.molbase.net%2Fnmr%2Fnmr_image%2F2015-01-11%2F001%2F717%2F1717235_13c.png&container=blogger&gadget=a&rewriteMime=image%2F*)
………………
(Example 11) N 1 – (5-Chloro-2-yl) -N 2 – [(1S, 2R, 4S)-4-(dimethylcarbamoyl) -2 – {[(5-methyl-4,5 , 6,7-tetrahydro [1,3] thiazolo [5,4-c] pyridin-2-yl) carbonyl] amino} cyclohexyl] Etanjiamido (X) [Production method via Compound (1-p2)]
[0137]
In 10 mL test tube, compound (5-ms) (the compound of Reference Example 8) (100 mg, 0.216 mmol), Compound (1-p2) (81.4 mg, 0.216 mmol), K 3 PO 4 (91.7 mg, 0.432 mmol) and DMF (1 mL) was added, and the mixture was stirred at room temperature conditions for 3 hours. H To the reaction mixture 2 O (2 mL) was added and the resulting slurry was stirred at room temperature overnight, the solid was filtered. The resulting solid H 2 was washed with O (1 mL), was obtained by drying under reduced pressure the title compound (110.0 mg, 92.9%) as a solid.
[0138]
1 H-NMR (500 Hz, CDCl 3 ) delta: 9.72 (s, 1H), 8.30 (dd, 1H, J = 2.5, 0.5 Hz), 8.17 (dd, 1H, J = 9.0, 0.5 Hz), 8.03 (D , 1H, J = 8.5 Hz), 7.68 (dd, 1H, J = 9.0, 2.5 Hz), 7.39 (d, 1H, J = 8.5 Hz), 4.70-4.67 (m, 1H), 4.13-4.09 (m, 1H), 3.73 (d, 1H, J = 16.0 Hz), 3.70 (d, 1H, J = 16.0 Hz), 3.06 (s, 3H), 2.96-2.93 (m, 2H), 2.95 (s, 3H), 2.89-2.79 (m, 3H), 2.52 (s, 3H), 2.14-2.06 (m, 3H), 1.96-1.90 (m, 1H), 1.84-1.78 (m, 1H), 1.69-1.62 (m, 1H ).
References
- “First market approval in Japan for LIXIANA (Edoxaban)”. Press Release. Daiichi Sankyo Europe GmbH. 2011-04-22.
- O’Riordan, Michael (9 January 2015). “FDA Approves Edoxaban for Stroke Prevention in AF and DVT/PE Prevention”. Medscape. Retrieved 10 January 2015.
- http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/206316lbl.pdf
- lexicomp.com
- Savaysa (edoxaban) [prescribing information]. Parsippany, NJ: Daiichi Sankyo; January 2015.
- http://www.drugs.com/cons/edoxaban.html
- Yoshiyuki, I., et al. “Biochemical and pharmalogical profile of darexaban, an oral direct Xa inhibitor.” European Journal of Pharmacology (2011): 49-55
- Katsung, B., S. Masters and A. Trevor. Basic and Clinical Pharmacology 11th Edition. United States of America: McGraw-Hill, 2009
- Turpie AG (January 2008). “New oral anticoagulants in atrial fibrillation”. European Heart Journal 29 (2): 155–65. doi:10.1093/eurheartj/ehm575. PMID 18096568.
Edoxaban, a factor Xa inhibitor, is supplied as edoxaban tosylate monohydrate. The chemical name is N-(5-Chloropyridin-2-yl)-N’-[(1S,2R,4S)-4-(N,N-dimethylcarbamoyl)-2-(5-methyl- 4,5,6,7-tetrahydro[1,3]thiazolo[5,4-c]pyridine-2-carboxamido)cyclohexyl] oxamide mono (4- methylbenzenesulfonate) monohydrate. Edoxaban tosylate monohydrate has the empirical formula C24H30ClN7O4S•C7H8O3S•H2O representing a molecular weight of 738.27. The chemical structure of edoxaban tosylate monohydrate is:
 |
It is a white to pale yellowish-white crystalline powder. The solubility of edoxaban tosylate (pKa 6.7) decreases with increasing pH. It is slightly soluble in water, pH 3 to 5 buffer, very slightly soluble at pH 6 to 7; and practically insoluble at pH 8 to 9.
SAVAYSA is available for oral administration as a 60 mg, 30 mg, or 15 mg round shaped, film-coated tablet, debossed with product identification markings. Each 60 mg tablet contains 80.82 mg edoxaban tosylate monohydrate equivalent to 60 mg of edoxaban. Each 30 mg tablet contains 40.41 mg edoxaban tosylate monohydrate equivalent to 30 mg of edoxaban. Each 15 tablet contains 20.20 mg edoxaban tosylate monohydrate equivalent to 15 mg of edoxaban.
The inactive ingredients are: mannitol, pregelatinized starch, crospovidone, hydroxypropyl cellulose, magnesium stearate, talc, and carnauba wax. The color coatings contain hypromellose, titanium dioxide, talc, polyethylene glycol 8000, iron oxide yellow (60 mg tablets and 15 mg tablets), and iron oxide red (30 mg tablets and 15 mg tablets).
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N’-(5-chloropyridin-2-yl)-N-[(1S,2R,4S)-4-(dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[1,3]thiazolo[5,4-c]pyridine-2-carbonyl)amino]cyclohexyl]oxamide
|
|
| Clinical data | |
| Trade names | Lixiana, Savaysa |
| AHFS/Drugs.com | Monograph |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | 62%; Tmax 1–2 hours |
| Protein binding | 55% |
| Metabolism | Minimal hepatic |
| Biological half-life | 10–14 hours |
| Excretion | 50% renal; <50% bile |
| Identifiers | |
| CAS Registry Number | 912273-65-5 |
| ATC code | None |
| PubChem | CID: 25022378 |
| IUPHAR/BPS | 7575 |
| ChemSpider | 8456212 |
| UNII | NDU3J18APO |
| KEGG | D09710 |
| ChEBI | CHEBI:85973 |
| Chemical data | |
| Formula | C24H30ClN7O4S |
| Molecular mass | 548.056 g/mol |
/////////
SEE ABAN SERIES AT…………http://organicsynthesisinternational.blogspot.in/p/aban-series.html
MHLW, sNDA, JAPAN, Daiichi Sankyo has received approval for Anticancer Agent irinotecan hydrochloride hydrate

Irinotecan (Camptosar, Pfizer; Campto, Yakult Honsha) is a drug used for the treatment of cancer.
Irinotecan prevents DNA from unwinding by inhibition of topoisomerase 1. In chemical terms, it is a semisynthetic analogue of the natural alkaloid camptothecin.
Its main use is in colon cancer, in particular, in combination with other chemotherapy agents. This includes the regimen FOLFIRI, which consists of infusional 5-fluorouracil,leucovorin, and irinotecan.
Irinotecan received accelerated approval by the U.S. Food and Drug Administration (FDA) in 1996[1] and full approval in 1998.[2] During development, it was known as CPT-11.
