
Home » Posts tagged 'chronic kidney disease'
Tag Archives: chronic kidney disease
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Desidustat


Ranjit Desai
DESIDUSTAT
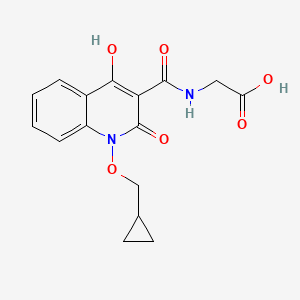
2-(1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carboxamido)acetic acid
desidustat
Glycine, N-((1-(cyclopropylmethoxy)-1,2-dihydro-4-hydroxy-2-oxo-3-quinolinyl)carbonyl)-
N-(1-(Cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carbonyl)glycine
ZYAN1 compound
(1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carbonyl) glycine in 98% yield, as a solid. MS (ESI-MS): m/z 333.05 (M+H) +. 1H NMR (DMSO-d 6): 0.44-0.38 (m, 2H), 0.62-0.53 (m, 2H), 1.34-1.24 (m, 1H), 4.06-4.04 (d, 2H), 4.14-4.13 (d, 2H), 7.43-7.39 (t, 1H), 7.72-7.70 (d, 1H), 7.89-7.85 (m, 1H), 8.11-8.09 (dd, 1H), 10.27-10.24 (t, 1H), 12.97 (bs, 1H), 16.99 (s, 1H). HPLC Purity: 99.85%
Oxemia (Desidustat) has received approval from the Drug Controller General of India. This was an incredible team effort by Zydans across the organization and I am so proud of what we have accomplished. Oxemia is a breakthrough treatment for Anemia associated with Chronic Kidney Disease in Patients either on Dialysis or Not on Dialysis, and will help improve quality of life for CKD patients. Team #zydus , on to our next effort!

Desidustat (INN, also known as ZYAN1) is a drug for the treatment of anemia of chronic kidney disease. This drug with the brand name Oxemia is discovered and developed by Zydus Life Sciences.[1] The subject expert committee of CDSCO has recommended the grant of permission for manufacturing and marketing of Desidustat 25 mg and 50 mg tablets in India,based on some conditions related to package insert, phase 4 protocols, prescription details, and GCP.[2] Clinical trials on desidustat have been done in India and Australia.[3] In a Phase 2, randomized, double-blind, 6-week, placebo-controlled, dose-ranging, safety and efficacy study, a mean hemoglobin increase of 1.57, 2.22, and 2.92 g/dL in desidustat 100, 150, and 200 mg arms, respectively, was observed.[4] The Phase 3 clinical trials were conducted at additional lower doses as of 2019.[5] Desidustat is developed for the treatment of anemia as an oral tablet, where currently injections of erythropoietin and its analogues are drugs of choice. Desidustat is a HIF prolyl-hydroxylase inhibitor. In preclinical studies, effects of desidustat was assessed in normal and nephrectomized rats, and in chemotherapy-induced anemia. Desidustat demonstrated hematinic potential by combined effects on endogenous erythropoietin release and efficient iron utilization.[6][7] Desidustat can also be useful in treatment of anemia of inflammation since it causes efficient erythropoiesis and hepcidin downregulation.[8] In January 2020, Zydus entered into licensing agreement with China Medical System (CMS) Holdings for development and commercialization of desidustat in Greater China. Under the license agreement, CMS will pay Zydus an initial upfront payment, regulatory milestones, sales milestones and royalties on net sales of the product. CMS will be responsible for development, registration and commercialization of desidustat in Greater China.[9] It has been observed that desidustat protects against acute and chronic kidney injury by reducing inflammatory cytokines like IL-6 and oxidative stress [10] A clinical trial to evaluate the efficacy and safety of desidustat tablet for the management of Covid-19 patients is ongoing in Mexico, wherein desidustat has shown to prevent acute respiratory distress syndrome (ARDS) by inhibiting IL-6.[11] Zydus has also received approval from the US FDA to initiate clinical trials of desidustat in chemotherapy Induced anemia (CIA).[12]. Desidustat has met the primary endpoints in the phase 3 clinical trials and Zydus had filed the New Drug Application (NDA) to DCGI in November, 2021.[13]\
CLIP
Zydus receives DCGI approval for new drug Oxemia; what you need to know
The new drug is an oral, small molecule hypoxia-inducible factor-prolyl hydroxylase (HIF-PH) inhibitor, Zydus said in a statement.
Gujarat-based pharma company Zydus Lifesciences on Monday received the Drugs Controller General of India (DCGI) approval for its new drug application for a first-of-its-kind oral treatment for anemia associated with Chronic Kidney Disease (CKD) – Oxemia (Desidustat).
The new drug is an oral, small molecule hypoxia-inducible factor-prolyl hydroxylase (HIF-PH) inhibitor, the drug firm said in a statement.
Desidustat showed good safety profile, improved iron mobilization and LDL-C reduction in CKD patients in DREAM-D and DREAM-ND Phase III clinical trials, conducted in approximately 1,200 subjects. Desidustat provides CKD patients with an oral convenient therapeutic option for the treatment of anemia. The pharma major did not, however, declare the cost per dose if the drug is available in the market.
“After more than a decade of research and development into the science of HIF-PH inhibitors, results have demonstrated that Oxemia addresses this unmet need and additionally reduces hepcidin, inflammation and enables better iron mobilization. This advancement offers ease of convenience for the patient and will also reduce the disease burden by providing treatment at an affordable cost, thereby improving the quality of life for patients suffering from Chronic Kidney Disease,” Chairman of Zydus Lifesciences Pankaj Patel said.
Chronic Kidney Disease (CKD) is a progressive medical condition characterised by a gradual loss of kidney function and is accompanied by comorbidities like anemia, cardiovascular diseases (hypertension, heart failure and stroke), diabetes mellitus, eventually leading to kidney failure.
PATENT
|
Scheme 3:
|
Step 1′a Process for Preparation of ethyl 2-iodobenzoate (XI-a)
Step-2 Process for the Preparation of ethyl 2-((tert-butoxycarbonyl)(cyclopropylmethoxy)aminolbenzoate (XII-a)
Step 3 Process for the Preparation of ethyl 2-((cyclopropylmethoxy)amino)benzoate (XIII-a)
Step 4 Process for the Preparation of ethyl 24N-(cyclopropylinethoxy)-3-ethoxy-3-oxopropanamido)benzoate (XIV-a)
Step 5: Process for the Preparation of ethyl 1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2 dihydroquinolline-3-carboxylate (XY-a)
Purification
Step 6 Process for the Preparation of ethyl (1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carbonyl)glycinate (XVI-a)
Purification
Step 7: Process for the Preparation of (1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carbonyl)glycine (I-a)
Polymorphic Data (XRPD):
References[edit]
- ^ “Zydus receives DCGI approval for new drug Oxemia; what you need to know”.
- ^ CDSCO, SEC Committee. “SEC meeting to examine IND proposals, dated 29.12.2021”. CDSCO website Govt of India. CDSCO. Retrieved 19 January 2022.
- ^ Kansagra KA, Parmar D, Jani RH, Srinivas NR, Lickliter J, Patel HV, et al. (January 2018). “Phase I Clinical Study of ZYAN1, A Novel Prolyl-Hydroxylase (PHD) Inhibitor to Evaluate the Safety, Tolerability, and Pharmacokinetics Following Oral Administration in Healthy Volunteers”. Clinical Pharmacokinetics. 57 (1): 87–102. doi:10.1007/s40262-017-0551-3. PMC 5766731. PMID 28508936.
- ^ Parmar DV, Kansagra KA, Patel JC, Joshi SN, Sharma NS, Shelat AD, Patel NB, Nakrani VB, Shaikh FA, Patel HV; on behalf of the ZYAN1 Trial Investigators. Outcomes of Desidustat Treatment in People with Anemia and Chronic Kidney Disease: A Phase 2 Study. Am J Nephrol. 2019 May 21;49(6):470-478. doi: 10.1159/000500232.
- ^ “Zydus Cadila announces phase III clinical trials of Desidustat”. 17 April 2019. Retrieved 20 April 2019 – via The Hindu BusinessLine.
- ^ Jain MR, Joharapurkar AA, Pandya V, Patel V, Joshi J, Kshirsagar S, et al. (February 2016). “Pharmacological Characterization of ZYAN1, a Novel Prolyl Hydroxylase Inhibitor for the Treatment of Anemia”. Drug Research. 66 (2): 107–12. doi:10.1055/s-0035-1554630. PMID 26367279.
- ^ Joharapurkar AA, Pandya VB, Patel VJ, Desai RC, Jain MR (August 2018). “Prolyl Hydroxylase Inhibitors: A Breakthrough in the Therapy of Anemia Associated with Chronic Diseases”. Journal of Medicinal Chemistry. 61 (16): 6964–6982. doi:10.1021/acs.jmedchem.7b01686. PMID 29712435.
- ^ Jain M, Joharapurkar A, Patel V, Kshirsagar S, Sutariya B, Patel M, et al. (January 2019). “Pharmacological inhibition of prolyl hydroxylase protects against inflammation-induced anemia via efficient erythropoiesis and hepcidin downregulation”. European Journal of Pharmacology. 843: 113–120. doi:10.1016/j.ejphar.2018.11.023. PMID 30458168. S2CID 53943666.
- ^ Market, Capital (20 January 2020). “Zydus enters into licensing agreement with China Medical System Holdings”. Business Standard India. Retrieved 20 January 2020 – via Business Standard.
- ^ Joharapurkar, Amit; Patel, Vishal; Kshirsagar, Samadhan; Patel, Maulik; Savsani, Hardikkumar; Jain, Mukul (22 January 2021). “Prolyl hydroxylase inhibitor desidustat protects against acute and chronic kidney injury by reducing inflammatory cytokines and oxidative stress”. Drug Development Research. 82 (6): 852–860. doi:10.1002/ddr.21792. PMID 33480036. S2CID 231680317.
- ^ “Zydus’ trials of Desidustat shows positive results for Covid-19 management”. The Hindu Business Line. The Hindu. Retrieved 25 January 2021.
- ^ “Zydus receives approval from USFDA to initiate clinical trials of Desidustat in cancer patients receiving chemotherapy”. PipelineReview.com. La Merie Publishing. Retrieved 22 January 2021.
- ^ “Stock Share Price | Get Quote | BSE”.
|
WO – 30.04.2020
|
|
2.WO/2020/058882METHODS OF PRODUCING VENOUS ANGIOBLASTS AND SINUSOIDAL ENDOTHELIAL CELL-LIKE CELLS AND COMPOSITIONS THEREOF
WO – 26.03.2020
|
|
3.110876806APPLICATION OF HIF2ALPHA AGONIST AND ACER2 AGONIST IN PREPARATION OF MEDICINE FOR TREATING ATHEROSCLEROSIS
CN – 13.03.2020
|
|
US – 28.11.2019
|
|
WO – 06.09.2019
|
|
EA – 30.10.2015
Настоящее изобретение относится к новым соединениям общей формулы (I), фармацевтическим композициям, содержащим указанные соединения, применению этих соединений для лечения состояний, опосредованных пролилгидроксилазой HIF, и к способу лечения анемии, включающему введение заявленных соединений |
|
EP – 28.10.2015
|
|
US – 22.10.2015
The present invention relates to novel compounds of the general formula (I), their tautomeric forms, their stereoisomers, their pharmaceutically acceptable salts, pharmaceutical compositions containing them, methods for their preparation, use of these compounds in medicine and the intermediates involved in their preparation. |
|
WO – 03.07.2014
|
 |
|
| Clinical data | |
|---|---|
| Other names | ZYAN1 |
| Identifiers | |
| CAS Number | |
| UNII | |
| Chemical and physical data | |
| Formula | C16H16N2O6 |
| Molar mass | 332.312 g·mol−1 |
| 3D model (JSmol) | |
Date
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| NCT04215120 | Desidustat in the Treatment of Anemia in CKD on Dialysis Patients | Phase 3 | Recruiting | 2020-01-02 |
| NCT04012957 | Desidustat in the Treatment of Anemia in CKD | Phase 3 | Recruiting | 2019-12-24 |
////////// DESIDUSTAT, ZYDUS CADILA, COVID 19, CORONA VIRUS, PHASE 3, ZYAN 1, OXEMIA, APPROVALS 2022, INDIA 2022

TENAPANOR
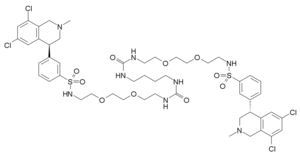
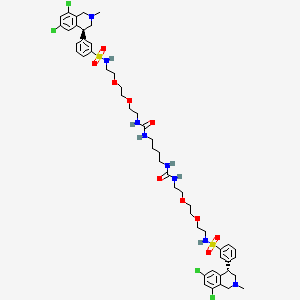


Tenapanor
Molecular FormulaC50H66Cl4N8O10S2
Average mass1145.049 Da
1234423-95-0 [RN]
1234423-95-0 (free base) 1234365-97-9 (2HCl)
9652
3-((S)-6,8-dichloro-2-methyl-1,2,3,4-tetrahydroisoquinolin-4-yl)-N-(26-((3-((S)-6,8-dichloro-2-methyl-1,2,3,4-tetrahydroisoquinolin-4-yl)phenyl)sulfonamido)-10,17-dioxo-3,6,21,24-tetraoxa-9,11,16,18-tetraazahexacosyl)benzenesulfonamide
Benzenesulfonamide, N,N’-(10,17-dioxo-3,6,21,24-tetraoxa-9,11,16,18-tetraazahexacosane-1,26-diyl)bis[3-[(4S)-6,8-dichloro-1,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]-
12,15-Dioxa-2,7,9-triazaheptadecanamide, 17-[[[3-[(4S)-6,8-dichloro-1,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulfonyl]amino]-N-[2-[2-[2-[[[3-[(4S)-6,8-dichloro-1,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulfonyl]amino]ethoxy]ethoxy]ethyl]-8-oxo-
1-[2-[2-[2-[[3-[(4S)-6,8-dichloro-2-methyl-3,4-dihydro-1H-isoquinolin-4-yl]phenyl]sulfonylamino]ethoxy]ethoxy]ethyl]-3-[4-[2-[2-[2-[[3-[(4S)-6,8-dichloro-2-methyl-3,4-dihydro-1H-isoquinolin-4-yl]phenyl]sulfonylamino]ethoxy]ethoxy]ethylcarbamoylamino]butyl]urea
17-[[[3-[(4S)-6,8-Dichloro-1,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulfonyl]amino]-N-[2-[2-[2-[[[3-[(4S)-6,8-dichloro-1,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulfonyl]amino]ethoxy]ethoxy]ethyl]-8-oxo-12,15-dioxa-2,7,9-triazaheptadecanamide
Tenapanor, also known as AZD-1722 and RDX 5791, is an inhibitor of the sodium-proton (Na(+)/H(+)) exchanger NHE3, which plays a prominent role in sodium handling in the gastrointestinal tract and kidney. Tenapanor possesses an excellent preclinical safety profile and, as of now, there are no serious concerns about its side effects.
Tenapanor is a drug developed by Ardelyx, which acts as an inhibitor of the sodium-proton exchanger NHE3. This antiporterprotein is found in the kidney and intestines, and normally acts to regulate the levels of sodium absorbed and secreted by the body. When administered orally, tenapanor selectively inhibits sodium uptake in the intestines, limiting the amount absorbed from food, and thereby reduces levels of sodium in the body.[1] This may make it useful in the treatment of chronic kidney disease and hypertension, both of which are exacerbated by excess sodium in the diet.[2]
Ardelyx and licensees Kyowa Hakko Kirin and Fosun Pharma are developing tenapanor, an NHE3 (Na+/H+ exchange-3) inhibitor that increases fluid content in the GI tract and which also reduces GI tract pain via an unknown TRPV-1-dependent pathway, for treating constipation-predominant irritable bowel syndrome (IBS-C) and hyperphosphatemia in patients with end stage renal disease (ESRD).
Syn

PATENT
WO2010078449
PATENT
WO-2019091503
A novel crystalline form of tenapanor free base, process for its preparation, composition comprising it and its use for the preparation of tenapanor with chemical purity >98.8% is claimed. Also claimed are salt forms of tenapanor, preferably tenapanor phosphate and their use for treating irritable bowel syndrome, constipation, hyperphosphatemia, final stage renal failure, chronic kidney disease and preventing excess sodium in patients with kidney and heart conditions. Further claimed are processes for the preparation of tenapanor comprising the steps of reaction of a diamine compound with 1,4-diisocyanatobutane, followed by deprotection and condensation to obtain tenapanor. Novel intermediates of tenapanor and their use for the preparation of tenapanor are claimed. Tenapanor is known to be a sodium hydrogen exchanger 3 inhibitor and analgesic.
enapanor, having the chemical name 17-[[[3-[(4S)-6,8-dichloro-l,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulphonyl]amino]-N-[2-[2-[2-[[[3-[(4S)-6,8-dichloro-l,2,3,4-tetrahydro-2-methyl -4-isoquinolinyl] phenyl] sulphonyl] amino] ethoxy] ethoxy ] ethyl] – 8 -oxo- 12,15 -dioxa-2 ,7,9-triazaheptadecaneamide, is a selective inhibitor of the sodium protonic NHE3 antiporter. Orally administered tenapanor selectively inhibits the absorption of sodium in the intestine. This leads to an increase of water content in the digestive tract, improved bowel flow and normalization of the frequency of bowel movement and stool consistency. At the same time it exhibits antinociceptive activity and ability to lower serum phosphate levels. Because of these properties, it is clinically tested for the treatment of irritable bowel syndrome, especially when accompanied by constipation, treatment of hyperphosphatemia, especially in patients with dialysis with final stage renal failure, treatment of chronic kidney disease, and prevention of excess sodium in patients with kidney and heart conditions. The tenapanor molecule, which was first described in the international patent application WO 2010/078449, has the following structural formula:
In this document, tenapanor was prepared as bishydrochloride salt. The bishydrochloride salt was prepared only in the form of an amorphous foam, which, after solidification, required grinding for further processing. However, the thus obtained particles are of varying sizes, while a narrow particle size distribution is required for pharmaceutical use in order to ensure uniform behavior. The amorphous foam obtained in the said document is essentially a thickened reaction mixture or a slightly purified reaction mixture containing, in addition to tenapanor, various impurities. The possibilities to purify the reaction mixtures are limited. Moreover, amorphous foams tend to adsorb solvents, and it is usually difficult to remove (or dry out) the residual solvents from the amorphous foam. This is undesirable for pharmaceutical use. A typical feature of amorphous foams is a large specific surface, resulting in a greater interaction of the substance with the surrounding environment. This significantly increases the risk of decomposition of the substance, for example through air oxygen, moisture or light. The present invention aims at overcoming these problems.
It would be advantageous to provide tenapanor solid forms (tenapanor free base or tenapanor salts) which are precipitated in solid forms, thus allowing to filter off the liquid reaction mixture containing the impurities. This results in a significantly improved purity.
The process used in WO 2010/078449 for the preparation of bishydrochloride salt of tenapanor was based on the preparation of 3-(6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinolin-4-yl)benzene-l-sulfonyl chloride of formula III from 4-(3-bromophenyl)-6,8-dichloro-2-methyl-l, 2,3,4-te
Scheme 1
The said document also discloses resolution of the starting tetrahydroisoquinoline of formula II by L-or D-dibenzoylt
(II) (S-II) (R-II)
Scheme 2
WO 2010/078449 discloses further steps of preparation of tenapanor, as shown in Scheme 3.
(V) (I)
Scheme 3
Individual synthetic steps described in Scheme 3 result in low yields: 42% for the reaction of the chloride of formula III with 2-(2-(2-aminoethoxy)ethoxy)ethylamine of formula IV, and 59% for the subsequent reaction with 1,4-diisocyanatobutane of formula V. The products of both synthetic steps are isolated by preparative chromatography which is technologically an unsuitable isolation and purification technique. The low yields and the need to use preparative chromatography for the isolation are caused by an abundance of side products and impurities and by the inability of the intermediates as well as of the product to provide a crystalline form.
Thus present invention thus further aims at providing a method of preparation of tenapanor which would be economically effective, in particular in relation to the expensive starting compound 4-(3-bromophenyl)-6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinoline, and which would also enable industrial scale production, in particular by removing steps which cannot be scaled up effectively or which cannot be scaled up at all. Furthermore, the method of preparation of tenapanor should provide tenapanor in a form which is useful for use in pharmaceutical forms and does not have the disadvantages of an amorphous foam.
Tenapanor free base in the form of an amorphous solid foam was prepared by the procedure disclosed in patent application WO 2010/078449, Example 202. The chemical purity of the tenapanor prepared by this procedure was 96.5% (HPLC). The structure of tenapanor was verified by MS and H and 13C NMR spectra.
Step A
Preparation of (5)- -(3-(benzylthio)phenyl)-6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinoline
Potassium carbonate (9.30 g) and anhydrous xylene (500 ml) were added to the reaction vessel. Benzyl mercaptane (25 g) was added dropwise to the stirred mixture under ice -cooling. The resulting mixture was stirred at 25 °C for lh.
(S)-4-(3-bromophenyl)-6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinoline 50 g in anhydrous xylene (500 ml), Pd2(dba)3 (3 g) and Xantphos (3 g). The resulting solution was stirred at 25 °C for 30 minutes and then added to a solution of benzyl mercaptane. The resulting reaction mixture was maintained at 140 °C for 16 h. The mixture was then concentrated and the residue was subjected to preparative chromatography on silica gel with the mobile phase ethyl acetate / petroleum ether (1: 100-1 :50). 20 g of product are obtained as a yellow oil (36% yield).
Ste B
Preparation of (5) -3 -(6 , 8 -dichloro-2 -methyl- 1,2,3 ,4-tetr ahydroisoquinolin-4-yl)benzenesulf onyl chloride hydrochloride
(S)-4-(3-(benzylthio)phenyl)-6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinoline (16 g) was dissolved in the reaction vessel in acetic acid/water (160 mL: 16 mL) mixture. The mixture was cooled in an ice bath and then gaseous Cl2 was introduced into the well stirred mixture. After disappearance of the starting material, the reaction mixture was purged with nitrogen and concentrated in vacuo. A product (10 g, 66.6%) was obtained as a colorless substance.
Step C
Preparation of (S)-N-(2-(2-(2-aminoethoxy)ethoxy)ethyl)-3-(6,8-dichloro-2-methyl-l, 2,3,4-tetrahydroisoquinolin-4-yl)benzenesulfonamide
2-(2-(2-Aminoethoxy)ethoxy)ethylamine HC1 (30 g; 0.2 mol) and triethylamine (5.2 g; 52 mmol) were dissolved in dichloromethane (500 ml) and the mixture was chilled in an ice bath. (S)-3-(6,8-Dichloro-2-methyl-l,2,3,4-tetrahydroisoquinolin-4-yl)benzenesulfonyl chloride hydrochloride (10 g; 26 mmol) was added in parts during 40 minutes to the chilled reaction mixture. The ice bath was removed and the reaction mixture was stirred at laboratory temperature for additional 30 minutes.
The dichloromethane solution was extracted three times by brine (2x 250 ml), dried over sodium sulphate, and concentrated in vacuo. The residue was purified using preparative chromatography on silica gel with dichloromethane-methanol mobile phase.
Yield 7.2 g. HRMS 502.1247 [M+H]+, C22H29CI2N3O4S.
Step D
Preparation of 17-[[[3-[(4S)-6,8-dichloro-l,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulphonyl]amino]-N-[2-[2-[2-[[[3-[(4S)-6,8-dichloro-l,2,3,4-tetrahydro-2-methyl -4-isoquinolinyl] phenyl] sulphonyl] amino] ethoxy] ethoxy ] ethyl] – 8 -oxo- 12,15 -dioxa-2 ,7,9-triazah
(S)-N-(2-(2-(2-aminoethoxy)ethoxy)ethyl)-3-(6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinolin-4-yl)benzenesulfonamide (5g; 10 mmol) prepared in step A was dissolved in dichloromethane (50 ml). Triethylamine (1.5 g; 14.9 mmol) and 1 ,4-diisocyanatobutane (0.48 g; 3.4 mmol) were added to the solution. The reaction mixture was cooled using ice and stirred overnight. The resulting fine suspension was filtered off, the filtrate was concentrated and the obtained product was purified by preparative chromatography on on silica gel with dichloromethane-methanol mixture as a mobile phase
Yield: 2 g of tenapanor in the form of amorphous solid foam. HPLC purity 96.5 %.
HRMS 1143.3186 [M+H]+, C5oH66Cl4N8010S2. *H NMR (500MHz, DMSO, ppm):7.69-7.66 (m, 6H), 7.54-7.50 (m, 6H), 6.89 (bs, 2H), 5.9 (t, 2H), 5.79 (t, 2H), 4.4 (dd, 2H), 3.7 (dd, 4H), 3.44-3.44 (m, 8H), 3.35 (dd, 8H), 3.12 (dd, 4H), 2.96-2.64 (m, 12H), 2.37 (s, 6H), 1.31 (bs, 4H).
Ste E
Preparation of bishydrochloride salt of tenapanor
Tenapanor free base (1 g; 0.85 mmol) prepared in step B was dissolved in a mixture of methanol (10 ml) and 4M aqueous HCl (0.5 ml; 2 mmol) under mild reflux. The solution was concentrated on rotary vacuum evaporator, and the title product was obtained in the yield of 1 g of amorphous solid foam.
Example 1
Preparation of tenapanor, crystalline form I
Tenapanor free base (200 mg, 0.17 mmol), prepared as in step D of the comparative example, was dissolved in 0.4 ml acetonitrile under mild reflux. The clear solution was cooled at the rate of 1 °C/min with stirring to laboratory temperature (i.e., range from 22 °C to 26 °C) and then stirred for additional 2 hours at this temperature. The resulting crystals were isolated by filtration on sintered glass filter and dried for 6 hours in a vacuum oven at 40 °C. Crystallization yield was 170 mg of crystalline form I of tenapanor. HPLC showed a purity of 99.5%.
Examples 4 to 9 illustrate the inventive method of preparation of crystalline tenapanor.
Example 4
Preparation of (5)- -(3-(benzylthio)phenyl)-6,8-dichloro-2-methyl-l ,2,3,4-tetrahydroisoquinoline
DIPEA (9.6 mL) and anhydrous dioxane (100 mL) were added to a reaction vessel. Benzyl mercaptan (8.1 ml) was added dropwise to the stirred mixture under ice -cooling. The resulting mixture was stirred at 25 °C for lh.
In a second reaction vessel, (S)-4-(3-bromophenyl)-6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinoline (21.2 g) in anhydrous dioxane (140 mL), Pd2(dba)3 (835 mg)and Xantphos (835 mg) were mixed. The resulting solution was stirred at 25 °C for 30 minutes and then added to the solution of benzyl mercaptan. The resulting reaction mixture was maintained at gentle reflux for 3 hours.
After cooling, the suspension obtained was filtered through a thin layer of celite. HC1 was added to the filtrate. The precipitated hydrochloride was isolated by filtration, washed well and dried. 21 g of pinkish product were obtained (81.6% yield).
Example 5
Preparation of (5) -3 -(6 , 8 -dichloro-2 -methyl- 1,2,3 ,4-tetr ahydroisoquinolin-4-yl)benzenesulf onyl chloride hydrochlorid
(S)-4-(3-(benzylthio)phenyl)-6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinoline hydrochloride (11.1 g) was stirred in DCM/2M HC1 (70 mL:6 mL) mixture in a reaction vessel. The mixture was cooled in an ice bath and then gaseous Cl2 was introduced into the vigorously stirred mixture. After disappearance of the starting material, the resulting suspension was bubbled through by nitrogen and the product was filtered off and washed with DCM. 9.2 g of white product was obtained (82.7% yield).
Example 6
In the reaction vessel, t-butyl 2-(2-(2-amionoethoxy)ethoxy)ethylcarbamate (21.8 g) was stirred in DCM. The mixture was cooled in an ice bath under an inert atmosphere. To the cooled solution was
added 1 ,4-diisocyanatobutane (6.14 g) and TEA (0.1 mL). The cooling bath was removed and the reaction mixture was further stirred for 2 h.
35% HCl was added to the reaction mixture and the mixture was stirred under gentle reflux overnight.
After cooling, the precipitated product was filtered off and washed with DCM.
The product was recrystallized from propan-2-ol. 22.3 g of white product was obtained (80% yield).
Example 7
Preparation of (5)-N-(2-(2-(2-aminoethoxy)ethoxy)ethyl)-3-(6,8-dichloro-2-methyl-l , 2,3,4-tetrahydroisoquinolin-4-yl)benzenesulfonamide
(S)-3-(6,8-dichloro-2-methyl-l ,2,3,4-tetrahydroisoquinolin-4-yl)benzenesulfonyl chloride hydrochloride (11.7 g) prepared in Example 2 was stirred in dichloromethane (100 ml) and the suspension was cooled in an ice bath. To the cooled suspension was added a solution of t-butyl 2-(2-(2-amionoethoxy)ethoxy)ethylcarbamate (6.8 g) and DIPEA (14 ml) in DCM (50 ml). The resulting solution was stirred for 2 hours in an ice bath. The reaction mixture was extracted twice with water. Concentrated HCl (15 mL) was added to the dichloromethane solution and the mixture heated at gentle reflux for 2 h.
The precipitated product, after cooling, was extracted into water. The aqueous phase was separated and basified with Na2C03. The product as the free base was extracted into DCM and the dichloromethane solution was dried over sodium sulfate and concentrated in vacuo. 12.9 g of product were obtained.
Yield 93.4%. HRMS 502.1247 [M+H]+, C22H29CI2N3O4S.
Example 8
Preparation of 17-[[[3-[(45)-6,8-dichloro-l ,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl] sulfonyl]amino]-N-[2-[2-[2-[[[3-[(45)-6,8-dichloro-l ,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl] phenyl] sulf onyl] amino] ethoxy ] ethoxy ] ethyl] – 8 -oxo- 12,15 -dioxa-2 ,7 ,9-triazaheptadecanamide (tenapanor free base)
(S)-N-(2-(2-(2-aminoethoxy)ethoxy)ethyl)-3-(6,8-dichloro-2-methyl-l,2,3,4- tetrahydroisoquinolin- 4-yl)benzenesulfonamide (12.9 g) prepared in Example 4 was dissolved in dichloromethane (150 ml). To the solution was added triethylamine (0.3 ml) and 1,4-diisocyanatobutane (1.7 g). The reaction mixture was stirred at 25 °C for 2 h. The resulting reaction mixture was extracted with water and aqueous Na2C03. The dichloromethane solution of the product was dried over sodium sulfate and concentrated to a solid foam. Yield 13.9 g. The crude product was taken up in acetone (100 ml) and then recrystallized from methanol (80 ml). 7.3 g of white crystalline product was obtained. Yield 49.8%.
HRMS 1143.3186 [M+H]+, C5oH66Cl4N8010S2. !H NMR (500MHz, DMSO, ppm):7.69-7.66 (m, 6H), 7.54-7.50 (m, 6H), 6.89 (bs, 2H), 5.9 (t, 2H), 5.79 (t, 2H), 4.4 (dd, 2H), 3.7 (dd, 4H), 3.44-3.44 (m, 8H), 3.35 (dd, 8H), 3.12 (dd, 4H), 2.96-2.64 (m, 12H), 2.37 (s, 6H), 1.31 (bs, 4H)
Example 9
Preparation of 17-[[[3-[(45)-6,8-dichloro-l,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl] sulfonyl]amino]-N-[2-[2-[2-[[[3-[(45)-6,8-dichloro-l,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl] phenyl] sulf onyl] amino] ethoxy ] ethoxy ] ethyl] – 8 -oxo- 12,15 -dioxa-2 ,7 ,9-
(S)-3-(6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinolin-4-yl)benzenesulfonyl chloride hydrochloride (0.81 g) prepared in Example 2 and l,l’-(butane-l,4-diyl)bis(3-(2-(2-(2-aminoethoxy)ethoxy)ethyl)urea) dihydrochloride prepared according to Example 3 (0.48 g) were stirred in anhydrous ΝΜΡ (10 ml). To the suspension was added DIPEA (2 mL) and the resulting solution was stirred at 60 °C for 1.5 h. Water (10 mL) was added dropwise to the reaction mixture and the mixture was cooled to 5 °C. The precipitated product was isolated and stirred in acetone at 5 °C overnight. The beige product was filtered off (0.67 g) and recrystallized from methanol (12 ml).
0.53 g of a colorless crystalline product was obtained.
Yield 78.7 %. HRMS 502.1247 [M+H]+, C22H29CI2N3O4S. DSC analysis showed the melting temperature of 130.5 °C.
Example 10
Tenapanor (1.48 g, 1.3 mmol) is dissolved in 10 ml of tetrahydrofurane (THF). From the thus prepared solution, 1 ml is taken and phosphoric acid (0.4 mmol) is added. The mixture is stirred at room temperature for 24 hours. Salt of tenapanor with phosphoric acid precipitated from the solution in solid stable form, the salt was filtered off, washed with THF and dried by stream of inert gas. XRPD confirmed amorphousness of the product.
Example 11
Tenapanor (1.48 g, 1.3 mmol) is dissolved in 10 ml of tetrahydrofurane (THF). From the thus prepared solution, 1 ml is taken and hydrobromic acid (0.4 mmol) is added. The mixture is stirred at room temperature for 24 hours. Salt of tenapanor with hydrobromic acid precipitated from the solution in solid stable form, the salt was filtered off, washed with THF and dried by stream of inert gas. XRPD confirmed amorphousness of the product.
Example 12
Tenapanor (1.48 g, 1.3 mmol) is dissolved in 10 ml of acetone. From the thus prepared solution, 1 ml is taken and phosphoric acid (0.4 mmol) is added. The mixture is stirred at room temperature for 24 hours. Salt of tenapanor with phosphoric acid precipitated from the solution in solid stable form, the salt was filtered off, washed with acetone and dried by stream of inert gas. XRPD confirmed amorphousness of the product.
Example 13
Tenapanor (1.48 g, 1.3 mmol) is dissolved in 10 ml of acetone. From the thus prepared solution, 1 ml is taken and citric acid (0.4 mmol) is added. The mixture is stirred at room temperature for 24 hours. Salt of tenapanor with citric acid precipitated from the solution in solid stable form, the salt was filtered off, washed with acetone and dried by stream of inert gas. XRPD confirmed amorphousness of the product.
Other pharmaceutically acceptable acids were tested by the procedures shown in Examples 10-13, but did not yield salts which would precipitate in amorphous stable solid form from the solution. The tested acids were: methanesulfonic acid, benzenesulfonic acid, oxalic acid, maleinic acid, tartaric acid, fumaric acid, trichloroacetic acid.
Example 14
Tenapanor (500 mg, 0.44 mmol) is dissolved in 20 ml of THF at 45 °C. To this clear solution, a solution of phosphoric acid in THF (50 μ1/5 ml) is added dropwise during 10 minutes. The resulting suspension is stirred at room temperature for 30 minutes. The precipitated salt of tenapanor with phosph (79 %) oric is filtered off, washed with 3 ml of THF and dried by stream of inert gas. Yield: 430 mg of colourless salt of tenapanor with phosphoric acid. XRPD showed amorphousness of the product.
Example 15
Tenapanor (500 mg, 0.44 mmol) is dissolved in 20 ml of THF at 45 °C. To this clear solution, hydrobromic acid (48%; 100 μΐ) is added dropwise during 10 minutes. A fine precipitate forms already during the dropwise addition of HBr, and the suspension is stirred at room temperature for 30 minutes. The precipitated salt of tenapanor with HBr is filtered off, washed with 3 ml of THF and dried by stream of inert gas. Yield: 397 mg (69 %) of colourless salt of tenapanor with HBr (1 :2). XRPD showed amorphousness of the product.
References
- ^ Spencer AG, Labonte ED, Rosenbaum DP, Plato CF, Carreras CW, Leadbetter MR, Kozuka K, Kohler J, Koo-McCoy S, He L, Bell N, Tabora J, Joly KM, Navre M, Jacobs JW, Charmot D (2014). “Intestinal inhibition of the na+/h+ exchanger 3 prevents cardiorenal damage in rats and inhibits na+ uptake in humans”. Sci Transl Med. 6 (227): 227ra36. doi:10.1126/scitranslmed.3007790. PMID 24622516.
- ^ Salt-buster drug cuts sodium absorbed from food. New Scientist, 14 March 2014
REFERENCES
1: Johansson SA, Knutsson M, Leonsson-Zachrisson M, Rosenbaum DP. Effect of Food Intake on the Pharmacodynamics of Tenapanor: A Phase 1 Study. Clin Pharmacol Drug Dev. 2017 Mar 24. doi: 10.1002/cpdd.341. [Epub ahead of print] PubMed PMID: 28339149.
2: Johansson S, Rosenbaum DP, Ahlqvist M, Rollison H, Knutsson M, Stefansson B, Elebring M. Effects of Tenapanor on Cytochrome P450-Mediated Drug-Drug Interactions. Clin Pharmacol Drug Dev. 2017 Mar 16. doi: 10.1002/cpdd.346. [Epub ahead of print] PubMed PMID: 28301096.
3: Chey WD, Lembo AJ, Rosenbaum DP. Tenapanor Treatment of Patients With Constipation-Predominant Irritable Bowel Syndrome: A Phase 2, Randomized, Placebo-Controlled Efficacy and Safety Trial. Am J Gastroenterol. 2017 Feb 28. doi: 10.1038/ajg.2017.41. [Epub ahead of print] PubMed PMID: 28244495.
4: Carney EF. Dialysis: Efficacy of tenapanor in hyperphosphataemia. Nat Rev Nephrol. 2017 Apr;13(4):194. doi: 10.1038/nrneph.2017.27. PubMed PMID: 28239171.
5: Block GA, Rosenbaum DP, Leonsson-Zachrisson M, Åstrand M, Johansson S, Knutsson M, Langkilde AM, Chertow GM. Effect of Tenapanor on Serum Phosphate in Patients Receiving Hemodialysis. J Am Soc Nephrol. 2017 Feb 3. pii: ASN.2016080855. doi: 10.1681/ASN.2016080855. [Epub ahead of print] PubMed PMID: 28159782.
6: Koliani-Pace J, Lacy BE. Update on the Management of Chronic Constipation. Curr Treat Options Gastroenterol. 2017 Mar;15(1):126-134. doi: 10.1007/s11938-017-0118-2. Review. PubMed PMID: 28116695.
7: Charoenphandhu N, Kraidith K, Lertsuwan K, Sripong C, Suntornsaratoon P, Svasti S, Krishnamra N, Wongdee K. Na(+)/H(+) exchanger 3 inhibitor diminishes hepcidin-enhanced duodenal calcium transport in hemizygous β-globin knockout thalassemic mice. Mol Cell Biochem. 2017 Mar;427(1-2):201-208. doi: 10.1007/s11010-016-2911-y. PubMed PMID: 27995414.
8: Thammayon N, Wongdee K, Lertsuwan K, Suntornsaratoon P, Thongbunchoo J, Krishnamra N, Charoenphandhu N. Na(+)/H(+) exchanger 3 inhibitor diminishes the amino-acid-enhanced transepithelial calcium transport across the rat duodenum. Amino Acids. 2017 Apr;49(4):725-734. doi: 10.1007/s00726-016-2374-1. PubMed PMID: 27981415.
9: Afsar B, Vaziri ND, Aslan G, Tarim K, Kanbay M. Gut hormones and gut microbiota: implications for kidney function and hypertension. J Am Soc Hypertens. 2016 Dec;10(12):954-961. doi: 10.1016/j.jash.2016.10.007. Review. PubMed PMID: 27865823.
10: Johansson S, Leonsson-Zachrisson M, Knutsson M, Spencer AG, Labonté ED, Deshpande D, Kohler J, Kozuka K, Charmot D, Rosenbaum DP. Preclinical and Healthy Volunteer Studies of Potential Drug-Drug Interactions Between Tenapanor and Phosphate Binders. Clin Pharmacol Drug Dev. 2016 Sep 22. doi: 10.1002/cpdd.307. [Epub ahead of print] PubMed PMID: 27654985.
11: Ketteler M, Liangos O, Biggar PH. Treating hyperphosphatemia – current and advancing drugs. Expert Opin Pharmacother. 2016 Oct;17(14):1873-9. doi: 10.1080/14656566.2016.1220538. Review. PubMed PMID: 27643443.
12: Johansson S, Rosenbaum DP, Knutsson M, Leonsson-Zachrisson M. A phase 1 study of the safety, tolerability, pharmacodynamics, and pharmacokinetics of tenapanor in healthy Japanese volunteers. Clin Exp Nephrol. 2016 Jul 1. [Epub ahead of print] PubMed PMID: 27368672.
13: Block GA, Rosenbaum DP, Leonsson-Zachrisson M, Stefansson BV, Rydén-Bergsten T, Greasley PJ, Johansson SA, Knutsson M, Carlsson BC. Effect of Tenapanor on Interdialytic Weight Gain in Patients on Hemodialysis. Clin J Am Soc Nephrol. 2016 Sep 7;11(9):1597-605. doi: 10.2215/CJN.09050815. PubMed PMID: 27340281; PubMed Central PMCID: PMC5012484.
14: Nusrat S, Miner PB Jr. New pharmacological treatment options for irritable bowel syndrome with constipation. Expert Opin Emerg Drugs. 2015;20(4):625-36. doi: 10.1517/14728214.2015.1105215. Review. PubMed PMID: 26548544.
15: Spencer AG, Greasley PJ. Pharmacologic inhibition of intestinal sodium uptake: a gut centric approach to sodium management. Curr Opin Nephrol Hypertens. 2015 Sep;24(5):410-6. doi: 10.1097/MNH.0000000000000154. Review. PubMed PMID: 26197202.
16: Zielińska M, Wasilewski A, Fichna J. Tenapanor hydrochloride for the treatment of constipation-predominant irritable bowel syndrome. Expert Opin Investig Drugs. 2015;24(8):1093-9. doi: 10.1517/13543784.2015.1054480. Review. PubMed PMID: 26065434.
17: Thomas RH, Luthin DR. Current and emerging treatments for irritable bowel syndrome with constipation and chronic idiopathic constipation: focus on prosecretory agents. Pharmacotherapy. 2015 Jun;35(6):613-30. doi: 10.1002/phar.1594. Review. PubMed PMID: 26016701.
18: Gerritsen KG, Boer WH, Joles JA. The importance of intake: a gut feeling. Ann Transl Med. 2015 Mar;3(4):49. doi: 10.3978/j.issn.2305-5839.2015.03.21. PubMed PMID: 25861604; PubMed Central PMCID: PMC4381464.
19: Labonté ED, Carreras CW, Leadbetter MR, Kozuka K, Kohler J, Koo-McCoy S, He L, Dy E, Black D, Zhong Z, Langsetmo I, Spencer AG, Bell N, Deshpande D, Navre M, Lewis JG, Jacobs JW, Charmot D. Gastrointestinal Inhibition of Sodium-Hydrogen Exchanger 3 Reduces Phosphorus Absorption and Protects against Vascular Calcification in CKD. J Am Soc Nephrol. 2015 May;26(5):1138-49. doi: 10.1681/ASN.2014030317. PubMed PMID: 25404658; PubMed Central PMCID: PMC4413764.
20: Spencer AG, Labonte ED, Rosenbaum DP, Plato CF, Carreras CW, Leadbetter MR, Kozuka K, Kohler J, Koo-McCoy S, He L, Bell N, Tabora J, Joly KM, Navre M, Jacobs JW, Charmot D. Intestinal inhibition of the Na+/H+ exchanger 3 prevents cardiorenal damage in rats and inhibits Na+ uptake in humans. Sci Transl Med. 2014 Mar 12;6(227):227ra36. doi: 10.1126/scitranslmed.3007790. PubMed PMID: 24622516.
 |
|
| Clinical data | |
|---|---|
| Routes of administration |
Oral |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.243.471 |
| Chemical and physical data | |
| Formula | C50H66Cl4N8O10S2 |
| Molar mass | 1145.046 g/mol g·mol−1 |
| 3D model (JSmol) | |
//////////////Tenapanor, AZD 1722, RDX 5791, chronic kidney disease, hypertension
CN1CC(C2=CC(=CC(=C2C1)Cl)Cl)C3=CC(=CC=C3)S(=O)(=O)NCCOCCOCCNC(=O)NCCCCNC(=O)NCCOCCOCCNS(=O)(=O)C4=CC=CC(=C4)C5CN(CC6=C(C=C(C=C56)Cl)Cl)C
Atrasentan



Atrasentan
- 173937-91-2

- as HCl: 195733-43-8
A-147627, (+)-A-127722, ABT-627,173937-91-2,
(2R,3R,4S)-4-(1,3-benzodioxol-5-yl)-1-[2-(dibutylamino)-2-oxoethyl]-2-(4-methoxyphenyl)pyrrolidine-3-carboxylic acid
Endothelin ET-A antagonist
Diabetic nephropathy; End stage renal disease; Renal disease
FDA APPROVED 4/02/2025, Vanrafia, To reduce proteinuria in adults with primary immunoglobulin A nephropathy at risk of rapid disease progression
- (2R,3R,4S)-4-(Benzo[d][1,3]dioxol-5-yl)-1-(2-(dibutylamino)-2-oxoethyl)-2-(4-methoxyphenyl)pyrrolidine-3-carboxylic acid
- (2R,3R,4S)-4-Benzo[1,3]dioxol-5-yl-1-dibutylcarbamoylmethyl-2-(4-methoxy-phenyl)-pyrrolidine-3-carboxylic acid
- 3-Pyrrolidinecarboxylic acid, 4-(1,3-benzodioxol-5-yl)-1-[2-(dibutylamino)-2-oxoethyl]-2-(4-methoxyphenyl)-, (2alpha,3beta,4alpha)-
- 3-Pyrrolidinecarboxylic acid, 4-(1,3-benzodioxol-5-yl)-1-[2-(dibutylamino)-2-oxoethyl]-2-(4-methoxyphenyl)-, (2R,3R,4S)-rel-
- 4-(1,3-Benzodioxol-5-yl)-1-[2-(dibutylamino)-2-oxoethyl]-2-(4-methoxyphenyl)-3-pyrrolidinecarboxylic acid, (2R,3R,4S)-rel-
- rel-(2R,3R,4S)-4-(Benzo[d][1,3]dioxol-5-yl)-1-(2-(dibutylamino)-2-oxoethyl)-2-(4-methoxyphenyl)pyrrolidine-3-carboxylic acid
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Atrasentan hydrochloride | E4G31X93ZA | 195733-43-8 | IJFUJIFSUKPWCZ-SQMFDTLJSA-N |
Atrasentan is an experimental drug that is being studied for the treatment of various types of cancer,[1] including non-small cell lung cancer.[2] It is also being investigated as a therapy for diabetic kidney disease.
Atrasentan failed a phase 3 trial for prostate cancer in patients unresponsive to hormone therapy.[3] A second trial confirmed this finding.[4]
It is an endothelin receptor antagonist selective for subtype A (ETA). While other drugs of this type (sitaxentan, ambrisentan) exploit the vasoconstrictive properties of endothelin and are mainly used for the treatment of pulmonary arterial hypertension, atrasentan blocks endothelin induced cell proliferation.
In April 2014, de Zeeuw et al. showed that 0.5 mg and 1.25 mg of atrasentan reduced urinary albumin by 35 and 38% respectively with modest side effects. Patients also had decreased home blood pressures (but no change in office readings) decrease total cholesterol and LDL. Patients in the 1.25 mg dose group had increased weight gain which was presumably due to increased edema and had to withdraw from the study more than the placebo or 0.5 mg dose group.[5] Reductions in proteinuria have been associated with beneficial patient outcomes in diabetic kidney disease with other interventions but is not an accepted end-point by the FDA.
The recently initiated SONAR trial[6] will determine if atrasentan reduces kidney failure in diabetic kidney disease.
Useful for treating nephropathy and chronic kidney disease associated with Type II diabetes. For a prior filing see WO2015006219 , claiming the stable solid composition in the form of a tablet comprising atrasentan and an anti-oxidant. AbbVie (following its spin-out from Abbott), is developing atrasentan (phase III; February 2015) for treating chronic kidney disease, including diabetic nephropathy.
PAPER
European Journal of Organic Chemistry
Enantioselective Synthesis of the Pyrrolidine Core of Endothelin Antagonist ABT-627 (Atrasentan) via 1,2-Oxazines
Year:2003
Volume:2003
Issue:18
page:3524-3533
PATENT
http://www.google.com/patents/US20080132710
EXAMPLE 1
A mixture of bromoacetyl bromide (72.3 mL) in toluene (500 mL) at 0° C. was treated with dibutylamine (280 mL) in toluene (220 mL) while keeping the solution temperature below 10° C., stirred at 0° C. for 15 minutes, treated with 2.5% aqueous phosphoric acid (500 mL) and warmed to 25° C. The organic layer was isolated, washed with water (500 mL) and concentrated to provide the product as a solution in toluene.
EXAMPLE 25-((E)-2-nitroethenyl)-1,3-benzodioxole
3,4-methylenedioxybenzaldehyde (15.55 Kg) was treated sequentially with ammonium acetate (13.4 Kg,), acetic acid (45.2 Kg) and nitromethane (18.4 Kg), warmed to 70° C., stirred for 30 minutes, warmed to 80° C., stirred for 10 hours, cooled to 10° C. and filtered. The filtrant was washed with acetic acid (2×8 Kg) and water (2×90 Kg) and dried under a nitrogen stream then in under vacuum at 50° C. for 2 days.
EXAMPLE 3ethyl 3-(4-methoxyphenyl)-3-oxopropanoate
A mixture of potassium tert-amylate (50.8 Kg) in toluene (15.2 Kg) at 5° C. was treated with 4-methoxyacetophenone (6.755 Kg) and diethyl carbonate (6.4 Kg) in toluene over 1 hour while keeping the solution temperature below 10° C., warmed to 60° C. for 8 hours, cooled to 20° C. and treated with acetic acid (8 Kg) and water (90 Kg) over 30 minutes while keeping the solution temperature below 20° C. The organic layer was isolated, washed with 5% aqueous sodium bicarbonate (41 Kg) and concentrated at 50° C. to 14.65 Kg.
EXAMPLE 4ethyl 2-(4-methoxybenzoyl)-4-nitromethyl-3-(1,3-benzodioxol-5-yl)butyrate
A mixture of EXAMPLE 3 (7.5 Kg) in THF (56 Kg) was treated with EXAMPLE 3 (8.4 Kg), cooled to 17° C., treated with sodium ethoxide (6.4 g), stirred for 30 minutes, treated with more sodium ethoxide (6.4 g), stirred at 25° C. until HPLC shows less than 1 area % ketoester remaining and concentrated to 32.2 Kg.
EXAMPLE 5ethyl cis,cis-2-(4-methoxyphenyl)-4-(1,3-benzodioxol-5-yl)pyrrolidine-3-carboxylate
Raney nickel (20 g), from which the water had been decanted, was treated sequentially with THF (20 mL), EXAMPLE 4 (40.82 g), and acetic acid (2.75 mL). The mixture was stirred under hydrogen (60 psi) until hydrogen uptake slowed, treated with trifluoroacetic acid, stirred under hydrogen (200 psi) until HPLC shows no residual imine and less than 2% nitrone and filtered with a methanol (100 mL) wash. The filtrate, which contained 13.3 g of EXAMPLE 5, was concentrated with THF (200 mL) addition to 100 mL, neutralized with 2N aqueous NaOH (50 mL), diluted with water (200 mL), and extracted with ethyl acetate (2×100 mL). The extract was used in the next step.
EXAMPLE 6ethyl trans,trans-2-(4-methoxyphenyl)-4-(1,3 -benzodioxol-5 -yl)pyrrolidine-3-carboxylate
Example 501E (38.1 g) was concentrated with ethanol (200 mL) addition to 100 mL, treated with sodium ethoxide (3.4 g), heated to 75° C., cooled to 25° C. when HPLC showed less than 3% of EXAMPLE 1E and concentrated. The concentrate was mixed with isopropyl acetate (400 mL), washed with water (2×150 mL) and extracted with 0.25 M phosphoric acid (2×400 mL). The extract was mixed with ethyl acetate (200 mL) and neutralized to pH 7 with sodium bicarbonate (21 g), and the organic layer was isolated.
EXAMPLE 7ethyl (2R,3R,4S)-(+)-2-(4-methoxyphenyl)-4-(1,3-benzodioxol-5-yl)pyrrolidine-3-carboxylate, (S)-(+) mandelate
EXAMPLE 501F was concentrated with acetonitrile (100 mL) addition to 50 mL, treated with (S)-(+)-mandelic acid (2.06 g), stirred until a solution formed, stirred for 16 hours, cooled to 0° C., stirred for 5 hours and filtered. The filtrant was dried at 50° C. under a nitrogen stream for 1 day. The purity of the product was determined by chiral HPLC using Chiralpak AS with 95:5:0.05 hexane/ethanol/diethylamine, a flow rate of 1 mL/min. and UV detection at 227 nm. Retention times were 15.5 minutes for the (+)-enantiomer and 21.0 minutes for the (−)-enantiomer.
EXAMPLE 8(2R,3R,4S)-(+)-2-(4-methoxyphenyl)-4-(1,3-benzodioxol-5-yl)-1-(N,N-di(n-butyl)aminocarbonylmethyl)pyrrolidine-3-carboxylic acid
A mixture of EXAMPLE 7 (20 g) in ethyl acetate (150 mL) and 5% aqueous sodium bicarbonate was stirred at 25° C. until the salt dissolved and gas evolution stopped. The organic layer was isolated and concentrated. The concentrate was treated with acetonitrile (200 mL), concentrated to 100 mL, cooled to 10° C., treated with diisopropylethylamine (11.8 mL) and EXAMPLE 1 (10.5 g), stirred for 12 hours and concentrated. The concentrate was treated with ethanol (200 mL), concentrated to 100 mL, treated with 40% aqueous NaOH (20 mL), stirred at 60° C. for 4 hours, cooled, poured into water (400 mL), washed with hexanes (2×50 mL then 2×20 mL), treated with ethyl acetate (400 mL) and adjusted to pH 5 with concentrated HCl (12 mL). The organic layer was isolated and concentrated.
………………….



SYN
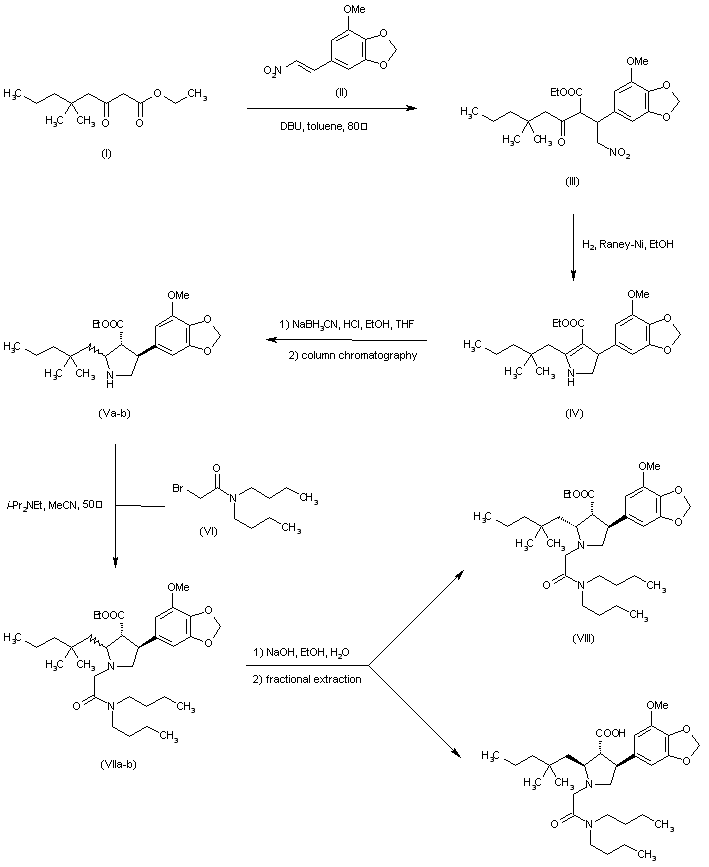
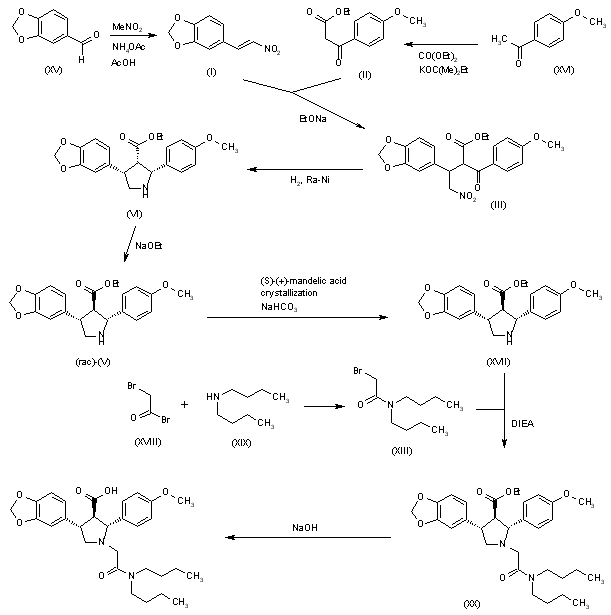
Condensation of ketoester (I) with nitrovinyl benzodioxole (II) in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene gave adduct (III). Hydrogenation of the nitro group of (III) over Raney Nickel with concomitant cyclization yielded dihydropyrrole (IV). Further reduction of (IV) with sodium cyanoborohydride provided a mixture of diastereomeric pyrrolidines. Chromatographic separation removed the cis,cis isomer, affording a mixture of trans,trans and cis,trans products (V). N-Alkylation of the pyrrolidine (V) with N,N-dibutyl bromoacetamide (VI) furnished (VIIa-b). Finally, selective hydrolysis of the ester group from the trans,trans isomer produced a mixture of cis,trans ester (VIII) and the target trans,trans acid, which were readily separated by fractional extraction.
SYN
SYN
J Med Chem 1996,39(5),1039
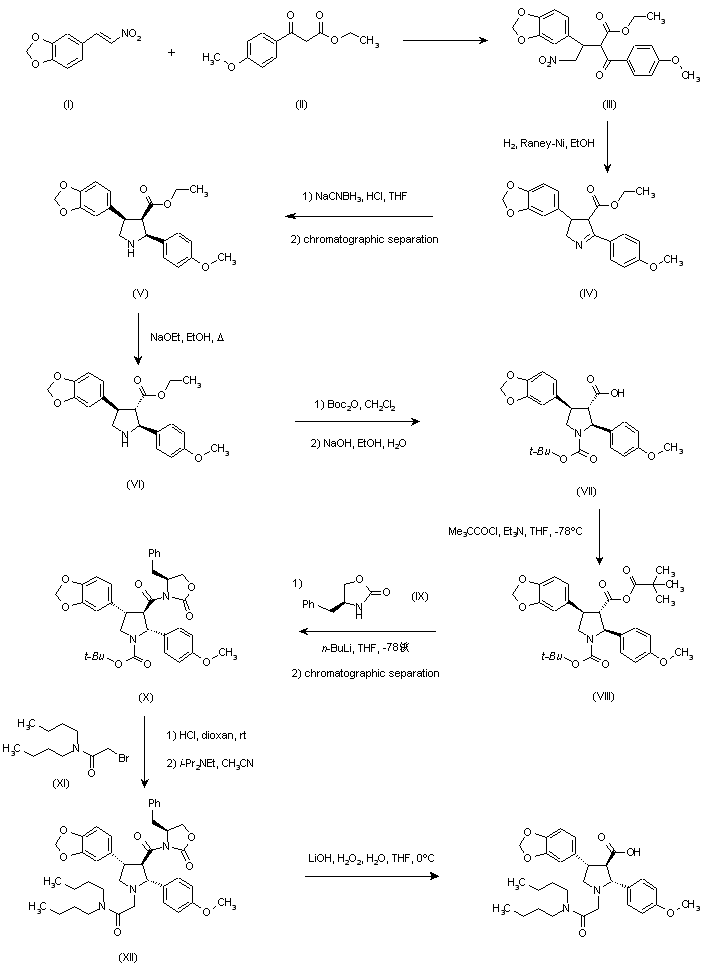
The Michael reaction between 3,4-(methylenedioxy)-beta-nitrostyrene (I) and ethyl (4-methoxybenzoyl)acetate (II) in the presence of DBU gave adduct (III) as a mixture of isomers. Hydrogenation of this nitro ketone over Raney-Ni afforded, after spontaneous cyclization of the resulting amino ketone, the pyrroline (IV). Further reduction of the imine with NaBH3CN yielded a mixture of three pyrrolidine isomers. The desired trans-trans isomer (VI) could not be separated from the cis-trans isomer by column chromatography. However, the pure cis-cis compound (V) was isomerized to (VI) with NaOEt in refluxing EtOH. The protection of the amine as the tert-butyl carbamate with Boc2O, and saponification of the ester function provided the racemic acid (VII). Resolution of (VII) was achieved by conversion to the mixed anhydride (VIII) with pivaloyl chloride, followed by condensation with the lithium salt of (S)-4-benzyl-2-oxazolidinone (IX), and chromatographic separation of the resulting diastereomeric imides. Alternatively, racemic (VII) could be resolved by crystallization of its salt with (R)-a-methylbenzylamine. Removal of the Boc group from the appropriate isomer (X) with HCl in dioxan, followed by alkylation with N,N-dibutylbromoacetamide (XI) in the presence of i-Pr2NEt furnished the pyrrolidinylacetamide (XII). Finally, hydrolysis of the imide with lithium hydroperoxide provided the target acid.
SYN
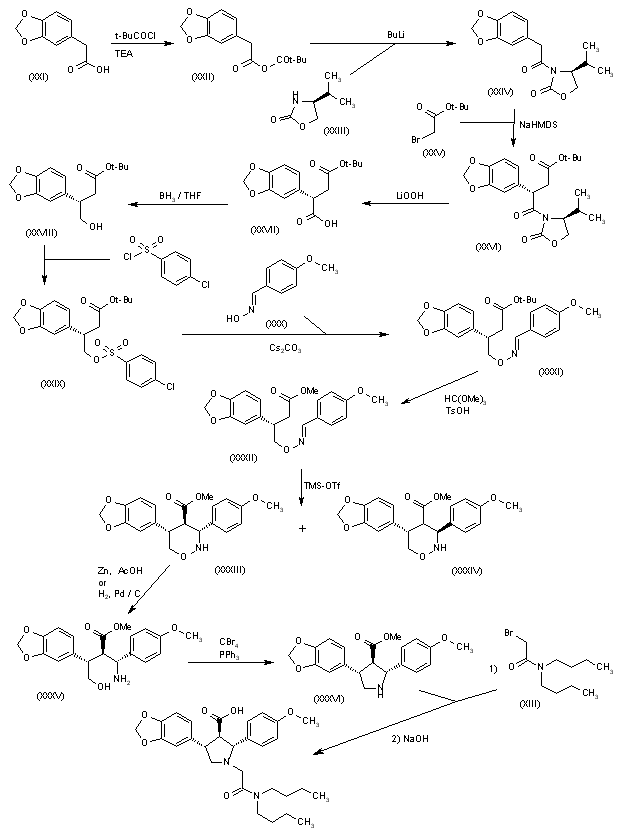
Reaction of 2-(1,3-dioxol-5-yl)acetic acid (XXI) with pivaloyl chloride and TEA gives the corresponding anhydride (XXII), which is condensed with the chiral oxazolidinone (XXIII) by means of n-BuLi in THF to yield the amide (XXIV). Condensation of (XXIV) with 2-bromoacetic acid tert-butyl ester (XXV) by means of NaHMDS in THF affords the adduct (XXVI). Elimination of the chiral auxiliary of (XXVI) by means of LiOOH in THF/water provides the chiral succinic acid hemiester (XXVII) (93% ee), which is selectively reduced with BH3璗HF complex to give the 4-hydroxysuccinate (XXVIII). Reaction of succinate (XXVIII) with 4-chlorophenylsulfonyl chloride, TEA and DMAP in dichloromethane yields the sulfonate (XXIX), which is condensed with 4-methoxybenzaldoxime (XXX) by means of Cs2CO3 in hot acetonitrile to afford the oxime ether (XXXI). Transesterification of the tert-butyl ester of (XXXI) with trimethyl orthoformate and p-toluenesulfonic acid in hot methanol provides the methyl ester (XXXII), which is cyclized by means of trimethylsilyl triflate and tributylamine in dichloroethane to afford a 9:1 diastereomeric mixture of perhydro-1,2-oxazines (XXXIII) and (XXXIV) which is easily separated. The reductive N-O-bond cleavage of the major oxazine diastereomer (XXXIII) by means of Zn/HOAc or H2 over Pd/C gives the trisubstituted 4-aminobutanol (XXXV), which is cyclized by means of CBr4, PPh3 and TEA to yield chiral pyrrolidine (XXXVI) (4). Finally, pyrrolidine (XXXVI) is alkylated with N,N-dibutyl-2-bromoacetamide (XIII) followed by ester hydrolysis as before.
References
1
- “Atrasentan”. NCI Dictionary of Cancer Terms. National Institute of Cancer.
- 2
- Chiappori, Alberto A.; Haura, Eric; Rodriguez, Francisco A.; Boulware, David; Kapoor, Rachna; Neuger, Anthony M.; Lush, Richard; Padilla, Barbara; Burton, Michelle; Williams, Charles; Simon, George; Antonia, Scott; Sullivan, Daniel M.; Bepler, Gerold (March 2008). “Phase I/II Study of Atrasentan, an Endothelin A Receptor Antagonist, in Combination with Paclitaxel and Carboplatin as First-Line Therapy in Advanced Non–Small Cell Lung Cancer”. Clinical Cancer Research 14 (5): 1464–9. doi:10.1158/1078-0432.CCR-07-1508. PMID 18316570.
- 3
- “Addition of experimental drug to standard chemotherapy for advanced prostate cancer shows no benefit in phase 3 clinical trial” (Press release). National Cancer Institute. April 21, 2011. Retrieved October 18, 2014.
- 4
- Quinn, David I; Tangen, Catherine M; Hussain, Maha; Lara, Primo N; Goldkorn, Amir; Moinpour, Carol M; Garzotto, Mark G; Mack, Philip C; Carducci, Michael A; Monk, J Paul; Twardowski, Przemyslaw W; Van Veldhuizen, Peter J; Agarwal, Neeraj; Higano, Celestia S; Vogelzang, Nicholas J; Thompson, Ian M (August 2013). “Docetaxel and atrasentan versus docetaxel and placebo for men with advanced castration-resistant prostate cancer (SWOG S0421): a randomised phase 3 trial”. The Lancet Oncology 14 (9): 893–900. doi:10.1016/S1470-2045(13)70294-8. PMID 23871417.
- 5
- de Zeeuw, Dick; Coll, Blai; Andress, Dennis; Brennan, John J.; Tang, Hui; Houser, Mark; Correa-Rotter, Ricardo; Kohan, Donald; Lambers Heerspink, Hiddo J.; Makino, Hirofumi; Perkovic, Vlado; Pritchett, Yili; Remuzzi, Giuseppe; Tobe, Sheldon W.; Toto, Robert; Viberti, Giancarlo; Parving, Hans-Henrik (May 2014). “The endothelin antagonist atrasentan lowers residual albuminuria in patients with type 2 diabetic nephropathy”. Journal of the American Society of Nephrology 25 (5): 1083–93. doi:10.1681/ASN.2013080830. PMID 24722445.
- 6
Clinical trial number NCT01858532 for “Study Of Diabetic Nephropathy With Atrasentan (SONAR)” at ClinicalTrials.gov
Granted in February 2015, this patent claims novel crystalline anhydrous S-mandelate salt of atrasentan. Useful for treating nephropathy and chronic kidney disease associated with Type II diabetes.
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (2R,3R,4S)-4-(1,3-Benzodioxol-5-yl)-1-[2-(dibutylamino)-2-oxoethyl]-2-(4-methoxyphenyl)pyrrolidine-3-carboxylic acid | |
| Clinical data | |
| Legal status |
?
|
| Identifiers | |
| CAS number | 173937-91-2 |
| ATC code | None |
| PubChem | CID 159594 |
| ChemSpider | 140321 |
| UNII | V6D7VK2215 |
| ChEMBL | CHEMBL9194 |
| Chemical data | |
| Formula | C29H38N2O6 |
| Molecular mass | 510.621 g/mol |
READ MORE ON SENTAN SERIES………..http://medcheminternational.blogspot.in/p/sentan-series.html
- Szczepankiewicz BG, Bal RB, von Geldern TW, Wu-Wong JR, Chiou WJ, Dixon DB, Opgenorth TJ, Hoffman DJ, Borre AJ, Marsh KC, Nguyen BN: The effects of diminishing albumin binding to some Endothelin receptor antagonists. Life Sci. 1998;63(21):1905-12. doi: 10.1016/s0024-3205(98)00466-4. [Article]
- Rajasekaran A, Julian BA, Rizk DV: IgA Nephropathy: An Interesting Autoimmune Kidney Disease. Am J Med Sci. 2021 Feb;361(2):176-194. doi: 10.1016/j.amjms.2020.10.003. Epub 2020 Oct 8. [Article]
- FDA Approved Drug Products: Vanrafia (atrasentan) tablets for oral use (April 2025) [Link]
- Novartis Media Release: Novartis receives FDA accelerated approval for Vanrafia® (atrasentan), the first and only selective endothelin A receptor antagonist for proteinuria reduction in primary IgA nephropathy (IgAN) [Link]
- StatPearls [Internet]: IgA Nephropathy (Berger Disease) [Link]
- ResearchGate: Total Synthesis of Atrasentan (Craig S. Harris, Reims Symposium, October 2002) [Link]
//////////ATRASENTAN, FDA 2025, APPROVALS 2025, Vanrafia, A 147627, (+)-A-127722, ABT 627, UNII-V6D7VK2215
