
Home » Posts tagged 'Boehringer Ingelheim'
Tag Archives: Boehringer Ingelheim
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
BI-882370

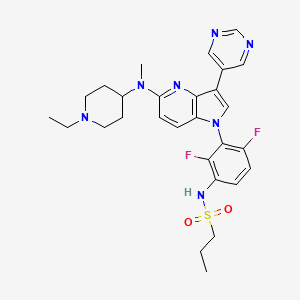
BI-882370
XP-102
N-(3-(5-((1-ethylpiperidin-4-yl)(methyl)amino)-3-(pyrimidin-5-yl)-1H-pyrrolo[3,2-b]pyridin-1-yl)-2,4-difluorophenyl)propane-1-sulfonamide
CAS 1392429-79-6
Chemical Formula: C28H33F2N7O2S
Molecular Weight: 569.68
Elemental Analysis: C, 59.03; H, 5.84; F, 6.67; N, 17.21; O, 5.62; S, 5.63
BI 882370 is a highly potent and selective RAF inhibitor that binds to the DFG-out (inactive) conformation of the BRAF kinase. BI 882370 inhibits proliferation of human BRAF-mutant melanoma cells with 100× higher potency (1-10 nmol/L) than vemurafenib.
Xynomic, under license from Boehringer Ingelheim , is investigating for treating BRAF mutant cancers, including colorectal cancer and melanoma; in October 2017, preclinical data were reported in the melanoma and colorectal cancer settings.
- Originator Boehringer Ingelheim
- Developer Boehringer Ingelheim; Xynomic Pharmaceuticals
- Class Antineoplastics; Piperidines; Pyridines; Pyrimidines; Pyrroles; Small molecules
- Mechanism of Action Proto oncogene protein b raf inhibitors
- Preclinical Colorectal cancer; Malignant melanoma
- 20 Dec 2018 Xynomic Pharma plans a phase Ib trial for Colorectal cancer (in combination with BI 860585) in third quarter of 2019
- 01 Jun 2018 Xynomic Pharmaceuticals plans a phase I trial for Colorectal cancer and Malignant melanoma in 2018 or 2019
- 06 Nov 2017 Chemical structure information added
-
US8889684
PATENT
WO2012104388

PATENT
Novel crystalline salts (monosuccinate salt), designated as Form A, of BI-882370 and their substantially anhydrous and non-solvated, processes for their preparation and compositions comprising them. Also claimed are their use as a RAF kinase Inhibitor, for the treatment of cancers and other diseases, such as infections, inflammations and autoimmune diseases.
The compound N-(3-(5-((l -ethylpiperidin-4-yl)(methyl)andno)-3-(pyrimidin-5-yl)-lH-pyrrolo [3, 2-Z>]pyri din- l-yl)-2,4-difluorophenyl)propane-l -sulfonamide (BI 882370), having Formula I:
I
is a RAF kinase inhibitor useful in the treatment of various diseases including cancer. The compound of Formula I, as well as its preparation and use, have been described in
WO/2012/104388, which is incorporated herein by reference in its entirety.
The RAS-RAF-MAPK (mitogen-activated protein kinase) signaling pathway plays a critical role in transmitting proliferation signals generated by the cell surface receptors and cytoplasmic signaling elements to the nucleus. Constitutive activation of this pathway is involved in malignant transformation by several oncogenes. Activating mutations in RAS
occur in approximately 15 % of cancers, and recent data has shown that B-RAF is mutated in about 7% of cancers (Wellbrock et al, “The RAF proteins take centre stage”, Nature Rev. Mol. Cell Biol., 2004, 5, 875-885), identifying it as another important oncogene in this pathway. In mammals, the RAF family of serine/threonine kinases comprises three members: A-RAF, B-RAF and C-RAF. However, activating mutations have so far been only identified in B-RAF underlining the importance of this isoform. It is believed that B-RAF is the main isoform that couples RAS to MEK, and that C-RAF and A-RAF signal to ERK only to fine-tune cellular responses (Wellbrock et al. Nature Rev. Mol. Cell Biol, 2004, 5, 875-885). The most common cancer mutation in B-RAF results in a valine to glutamic acid exchange at position 600 of the protein (V600E), which dramatically enhances B-RAF activity, presumably because its negative charge mimics activation loop phosphorylation (Wan et al , “Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF”, Cell, 2004, 116, 855-867). The highest incidence of B-RAF V600 mutations occurs in malignant melanoma (39%), thyroid cancer (46%), colorectal cancer (10%), biliary tract cancer (10%), prostate cancer (4%), ovary cancer (3%) and non-small cell lung cancer (2%), but they also occur at a low frequency in a wide variety of other cancers (frequencies of mutations according to COSMIC (Catalogue Of Somatic Mutations In Cancer; Wellcome Trust Sanger Institute) release v.53, 15th May 2011 ;
http://www.sanger.ac.uk/genetics/CGP/cosmic/). Literature supported the hypothesis that B-RA 600E mutated tumor cells seem to rely heavily on the continued activation of this pathway – a phenomenon termed “oncogene addiction” – whereas normal B-RAFwt cells use a broader range of signals. This provides an Achilles’ heel that can be exploited
therapeutically by treating patients with somatically mutated B-RAFV600E using orally available B-RAF inhibitors.
The key role of B-RAF V600E in aberrant ERK signaling and consequently oncogenesis has been demonstrated in several independent experimental approaches such as
overexpression of oncogenic/mutated B-RAF in vitro and in vivo (Wan et al., Cell, 2004, 116, 855-867; Wellbrock et al, Cancer Res. 2004, 64: 2338-2342), siRNA knock-down in vitro (Karasarides et al., Oncogene, “V599EB-RAF is an oncogene in melanocytes”, 2004, 23, 6292-6298) or in inducible short-hairpin RNA xenograft models where gain-of-function B-RAF signaling was found to be strongly associated with in vivo tumorigenicity (Hoeflich et al, “Oncogenic BRAF is required for tumor growth and maintenance in melanoma models”, Cancer Res., 2006, 66, 999-1006).
Treatment of B-RAFV600E mutated melanoma or colon carcinoma cells induces a B-RAF inhibition phenotype (e.g. reduction of phospho-MEK and phospho-ERK levels, reduction of cyclin D expression and induction of p27 expression). Consequently, these cells are locked in the Gl -phase of the cell cycle and do not proliferate.
Clinical proof of mechanism and proof of concept has been established for treating in cancer in B-RAFV600E mutated melanoma patients treated with Zelboraf®, B-RAF inhibitor (PLX-4032, vemurafenib, from Plexxikon/Daiichi Sankyo/Roche. Bollag et al., “Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma”, Nature, 2010, 467(7315), 596-9.; Flaherty et al, New Engl. J. Med., “Inhibition of Mutated, Activated BRAF in Metastatic Melanoma”, 2010, 363, 809-819; Chapman et al. “Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation”, New Engl. J. Med, 2011, 364:2507-2516. Favorable response rates were observed in both Phase I and Phase III clinical trials. It was reported, that melanoma patients carrying a B-RAFV600K mutation also do respond to therapy (Rubinstein et al, “Incidence of the V600K mutation among melanoma patients with BRAF mutations, and potential therapeutic response to the specific BRAF inhibitor PLX4032”, J. Transl. Med , 2010, 8, 67).
The most frequent B-RAF mutation is the exchange at amino acid position 600 from valine to glutamate with more than 90% frequency of all B-RAF mutations (Wellbrock et al. Nature Rev. Mol. Cell Biol, 2004, 5, 875-885), the second most frequent mutation is an alteration from valine to lysine, other mutations were found with lower frequency at that position (Wellbrock et al. Nature Rev. Mol. Cell Biol, 2004, 5, 875-885 and frequencies of mutations according to COSMIC (Catalogue Of Somatic Mutations In Cancer; Wellcome Trust Sanger Institute) release v53, 15th May 2011 ;
http://www.sanger.ac.uk/genetics/CGP/cosmic/). Additional mutations were found at e.g. the glycine rich loop (Wellbrock et al. Nature Rev. Mol. Cell Biol, 2004, 5, 875-885). Not all of these rather rare mutations seem to lead to direct activation of B-RAF (Wan et al. ,
“Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF”, Cell, 2004, 116, 855-867).
The compound of Formula I is a highly potent and selective RAF inhibitor that binds to the DFG-out (inactive) conformation of the B-RAF kinase. The compound inhibited proliferation of human B-RAF-mutant melanoma cells with 100 times higher potency (1-10 nmol/L) than vemurafenib, whereas wild-type cells were not affected at 1,000 nmol/L. A solution of the compound administered orally was efficacious in mouse models of B-RAF-mutant melanomas and colorectal carcinomas, and at 25 mg/kg twice daily showed superior efficacy compared with vemurafenib, dabrafenib, or trametinib. The compound was also active in A375 melanoma-bearing mice that were resistant to vemurafenib, particularly when dosed in combination with trametinib. Mice treated with the compound did not show any body weight loss or clinical signs of intolerability, and no pathologic changes were observed in several major organs investigated, including skin. Furthermore, in a pilot study in rats (up to 60 mg/kg daily for 2 weeks), the compound lacked toxicity in terms of clinical chemistry, hematology, pathology, and toxicogenomics. These results are described in Waizenegger et al., Mol. Cancer Ther., 2016, 75(3); 354-65, which is incorporated herein by reference in its entirety.
For the manufacture, purification, and formulation of a drug, it may be advantageous to employ a form of the drug having superior stability or other desirable formulation property exhibited by, for example, one or more salt or crystalline forms of the drug. Formation of salts of basic or acidic drugs can sometimes provide forms of the drug that have
advantageous properties such as solubility, non-hygroscopicity, crystallinity, and other physical properties that advantageous for formulating the drug. On the other hand, discovering a suitable salt or other crystalline form that is suitable for formulation is difficult, since there are numerous variables in the formation of a salt or crystalline form. These include the existence of numerous possible acids and bases that might be used as a counter-ion, various stoichiometric ratios that may be possible for combining a given basic or acid drug with an acid or base counter-ion, a wide variety of solvents and solvent systems
(including combinations of solvents) that potentially can be used to attempt to form salts or crystalline forms, and a variety of conditions (such as temperature or heating or cooling conditions) under which salts or crystalline forms may be generated. All of these variables of which may affect the properties of the salts or crystalline forms that might be obtained. Salts or solid forms may also have a variety of properties that render them unsuitable for drug development and formulation such as lack of crystallinity (amorphous forms), the presence or formation of multiple crystalline forms, which may interconvert and/or have different properties (polymorphism), lack of aqueous solubility, hygroscopicity, or stickiness of the solid. Furthermore, the formation of salts and crystalline forms and their properties are generally very unpredictable.
Accordingly, the crystalline salt forms of the compound of Formula I provided herein help satisfy the ongoing need for the development of a RAF kinase inhibitor for the treatment of serious diseases.
Preparation of A^-(3-(5-((l-ethylpiperidin-4-yl)(methyl)amino)-3-(pyrimidin-5-yl)-lH-pyrrolo[3,2-Z>]pyridin-l- amide (BI 882370)
Step 1. 4-(6-Methyl-5-nitro-pyridin-2-yl)-piperazine-l-carboxylic acid tert-butyi ester
(3)
1 2 3
DIPEA (62.82 mL, 0.435 mol) is added to the solution of 6-chloro-3-nitro-2-methylpyridine (1) (50 g, 290 mmol) and N-Boc-piperazine (2) (53.95 g, 290 mmol) in dry MeCN (200 mL) and stirred for 4 h at 50 °C. After the reaction is finished the reaction mixture is diluted with MeCN and water and stirred for 30 min. The precipitated product is collected by filtration, washed with water and the solid is dried in vacuo.
Step 2. 4- [6-((£’)-2-Dimethylamino-vinyl)-5-nitro-pyridin-2-yl] -piperazine- 1-carboxylic acid
To a stirred solution of 4-(6-methyl-5-nitro-pyridin-2-yl)-piperazine- 1-carboxylic acid tert-butyl ester (3) (13 g, 40.3 mmol) in DMF (35 mL) is added N,N-dimethylformamide dimethylacetal (14.47 g, 121 mmol) and stirred in argon atmosphere for 36 h at 90 °C.
Additional 1.5 eq. of N^V-dimethylformamide dimethylacetal is added and stirred for 12 h at 90 °C. The reaction mixture is poured into water and extracted with DCM. The combined organic layers are washed with water, dried over anhydrous Na2S04 and concentrated in vacuo. The residue is used without further purification for the next step.
Step -(lH-pyrrolo[3,2-Z>]pyridin-5-yl)piperazine-l-carboxylic acid tert-butyl ester (5)
4 5
4-[6-((i?)-2-Dimethylairdno-vinyl)-5-nitro-pyridin-2-yl]-piperazine-l-carboxylic acid tert-butyl ester (36.4 g, 96 mmol) is taken up in MeOH, Pd/C (0.56 g, 10 %) is added and the mixture is hydrogenated in an autoclave at 60 psi for 16 h. The reaction mixture is filtered and concentrated under reduced pressure. The residue is purified by column chromatography viaNP MPLC. The product containing fractions of compound (5) (HPLC-MS method B: tRet. = 1.55 min.; MS (M+H)+ = 303) are combined and evaporated in vacuo.
Step 4. N- -Amino-2,6-difluorophenyl)acetamide (7)
6 7
Compound (6) (55.0 g, 254 mmol) is taken-up in MeOH (1.0 L). Pd/C (10.0 g, 10 %) is added and the mixture is hydrogenated in an autoclave at 200 psi for 3 h. The reaction mixture is filtered and concentrated under reduced pressure. The residue is purified by NP-MPLC on silica gel using DCM/MeOH (96:4) as eluent. The product containing fractions of the aniline intermediate (HPLC-MS method B: tRet. = 0.25 min.; MS (M-H)“ = 185) are combined and evaporated.
Step 5. N- -Difluoro-3-(propylsulfonamido)phenyl)acetamide (9)
To the aniline intermediate (35.0 g, 188 mmol) in DCM (100 mL) pyridine (6.6 mL, 75 mmol) and ^-propane sulfonyl chloride (8) (29.5 mL, 263 mmol) are added and the mixture is stirred at rt for 16 h. The reaction mixture is diluted with EtOAc (200 mL), washed with H2O and HC1 (aq., 1 N) and the layers are separated, dried over MgS04 and evaporated to yield the sulfonamide (9) which was used without further purification.
Step 6. N-
9 10
The sulfonylated aniline (9) (38.0 g, 130 mmol) is taken-up in EtOH (250 mL), H2O (200 mL) and concentrated hydrochloric acid (200 mL) and heated to 80 °C for 2 h. The reaction mixture is concentrated under reduced pressure, aqueous NaOH (4 N) is added until pH = 6 is reached and the mixture is extracted 2 x with DCM. The combined organic layer is washed with brine, dried over MgS04, filtered and evaporated to yield the deacylated aniline (10) (HPLC-MS method B: tRet. = 0.22 min.; MS (M-H)“ = 249) as a hydrochloride which was used without further purification.
Step 7. N-(2 -Difluoro-3-iodophenyl)propane-l-sulfonamide (11)
10 11
The hydrochloride of compound (10) is taken-up in DCM and extracted with NaHCCb solution. The organic layer is dried over MgSCn, filtered and evaporated. To the free base (10) (3.55 g, 14.21 mmol) in TFA (80 mL) at 0 °C is added NaNC (1.96 g, 28.4 mmol) in small portions and the mixture is stirred for 30 min. KI (23.83 g, 142 mmol) is added and stirring is continued for additional 15 min. The reaction mixture is diluted with Et^O and stirred for 1 h. Na2S203 solution (semiconc.) is added and the mixture is extracted 3 x with Et20. The combined organic layer is dried over MgSCn, filtered and concentrated in vacuo. The residue is purified by column chromatography via NP-MPLC. The product containing fractions of compound (11) (HPLC-MS method A: tRet. = 1.58 min.; MS (M-H)“ = 360) are combined and evaporated in vacuo.
Step 8. 4-((l-(2,6-Difluoro-3-(propylsulfonamido)phenyl)-lH-pyrrolo [3,2-b] pyridin-5-yl)
12
The lH-pyrrolo [3,2-*] pyridine (5) (10.0 g, 30.27 mmol), sulfonamide (11) (16.4 g,
45.4 mmol), Cul (576 mg, 3.03 mmol), ^^-(l ^^^-^N’-bismethyl-l^-cyclohexandiamine
(1.91 mL, 12.1 mmol) and CS2CO3 (29.6 g, 90.85 mmol) are taken-up in dry toluene (3 mL) and the resulting mixture is flushed with argon and stirred for 16 h at 120 °C. After the addition of further Cul (576 mg, 3.03 mmol), trans-(\R,2R)-N,N’-bismet y 1-1,2-cyclohexandiamine (1.91 mL, 12.1 mmol) and CS2CO3 (20.0 g, 60.0 mmol) the reaction mixture is stirred for further 24 h. The solvent is removed in vacuo, the residue is taken up in DCM and extracted with NaHCC solution (semiconc). The organic layer is dried over MgS04, filtered, the solvent is removed in vacuo and the residue is purified viaNP-MPLC. The product containing fractions of (12) (HPLC-MS method C: teet. = 1.62 mia; MS (M+H)+ = 564) are combined and the solvent is removed in vacuo.
Step 9. 4-((l-(2,6-Difluoro-3-(propylsulfonamido)phenyl)-3-iodo-lH-pyrrolo[3,2-b]pyridin-5 3)
To a solution of sulfonamide (12) (1.078 g, 1.9 mmol) in DMF (4 mL)/THF (100 μί) is added NIS (474 mg, 2.1 mmol) and the mixture is stirred for 1 h at rt. The reaction mixture is diluted with 30 mL DCM and extracted with NaHCCb solution (semiconc). The combined organic layer is dried over MgSCn, filtered and concentrated under reduced pressure. The residue is purified by column chromatography via RP HPLC. The product containing fractions of (13) (HPLC-MS method B: tRet. = 2.035 mia; MS (M+H)+ = 688) are freeze dried.
Step 10. 4-((l-(2,6-Difluoro-3-(propylsulfonamido)phenyl)-3-(pyrimidin-5-yl)-lH-pyrrolo[3,2-b]pyridin-5-yl)(methyl)amino)piperidine-l-carboxylic acid tert-butyi ester (15)
13 15
Sulfonamide (13) (770 mg, 1.12 mmol), pyrimidin-5-yl-boronic acid (14) (194 mg, 1.57 mmol), Pd(dppf)Cl2 (82 mg, 0.11 mmol), LiCl (142 mg, 3.35 mmol) and Na2C03 (294 mg, 2.8 mmol) are taken-up in dioxane/LhO (2: 1 mixture, 12 mL), and the resulting mixture is flushed with argon and stirred for 1 h at 100 °C. The reaction mixture is diluted with DCM and extracted with NaHCCb solution (semi-concentrated). The organic layer is dried over MgS04, filtered, Isolute® is added, the solvent is removed in vacuo and the residue is purified via RP HPLC. The product containing fractions of (15) (HPLC-MS method C: tRet. = 2.149 min.; MS (M+H)+ = 642) are freeze dried.
Step 11. N-(2,4-Difluoro-3-(5-(methyl(piperidin-4-yl)amino)-3-(pyrimidin-5-yl)- 1H-pyrrolo[3,2-b]pyridin-l-yl)phenyl)propane-l-sulfonamide
15 16
To a solution of example compound (15) (154 mg, 0.24 mmol) in DCM/MeOH (1 : 1, 4 mL) is added HC1 (in dioxane, 4 N, 2 mL) and the mixture is stirred for 3 h at rt. The solvent is removed in vacuo. Obtained compound (16) (HPLC-MS method B: tRet. = 1.02 min.; MS (M+H)+ = 542) is used without further purification.
Step 12. ^-(3-(5-((l-Ethylpiperidin-4-yl)(methyl)amino)-3-(pyrimidin-5-yl)-lH-pyrrolo [3,2-Z>] pyridin- l-yl)-2,4-diflu
Compound I was obtained from compound (16) by reductive alkylation with acetaldehyde (40% in iPrOH) in the presence of 1.5 eq. sodium acetoxyborohydride in iPrOH. The crude product was recrystallized from ethanol to obtain the title compound in 84% yield.
Scale-Up Synthesis of A/-(3-(5-((l-ethylpiperidin-4-yl)(methyl)amino)-3-(pyrimidin-5-yl)-lH-pyrrolo[3,2-Z>]pyridin-l-yl)-2,4-difluorophenyl)propane- 1-sulfonamide (BI 882370)
Step 1. N-(2,4-Difluoro-3-(5-(methyl(piperidin-4-yl)amino)-3-(pyrimidin-5-yl)-lH-pyrrolo[
15 16
Isopropanol (8.83 kg) and compound (15) (1.80 kg, 2.8 mol) were added into a reactor, and the mixture was stirred and heated to 55-60 °C. Concentrated hydrochloric acid (2.76 kg, 28 mol) was dropped into the reactor over than 20 min. at 60-65 °C. Then, the reaction mass was heated to 60-70 °C and held for 1 h. The conversion was monitored by HPLC, and reached about 99.5% after about 1 h.
The reaction mass was cooled and the isopropanol was removed by distillation under reduced pressure at not more than 50 °C. A brown oil was obtained, dissolved into water (6.75 kg) and washed by extraction with ethyl acetate (2.02 kg) at 20-30 °C. The water-phase was cooled to 15-20 °C. The pH was adjusted to 8.0-8.5 with 10% aqueous NaOH solution (-8.0 kg) at 20-30°C. The mixture was stirred for 3-4h at 20-30°C with the pH adjusted to 8.0-8.5 by addition of 10% NaOH solution every half-hour. The product was isolated by filtration and the cake washed with water (3.6 kg). The solid was dried under vacuum at 45-50 until the water content was not more than 5.5%. This provided about 1.64 kg of crude compound (16) (yield 108% of theoretical; the crude product containing water and NaCl detected). The crude product was used directly).
Step 12. ^-(3-(5-((l-Ethylpiperidin-4-yl)(methyl)amino)-3-(pyrimidin-5-yl)-lH-pyrr -Z>] pyridin- l-yl)-2,4-difluorophenyl)propane- 1-sulfonamide (I)
Bl 878426 Bl 882370
Process:
Dichloromethane (19.88 kg) and compound (16) (1.5kg, 2.77mol) were added into a reactor, and the mixture was stirred and cooled to 0-10°C under a nitrogen atmosphere. Sodium triacetoxyborohydride (95%, 0.93 kg, 4.16 mol) was added into the mixture at 0-10°C. The mixture was stirred for 20-30 min. at 0- 10°C. Acetaldehyde in DCM (40%,
1.07 kg, 9.71 mol) added into the mixture slowly over 2 h at 0-10 °C. The reaction mixture was stirred at 0-10 °C under a nitrogen atmosphere for 0.5-lh. The conversion was monitored by HPLC, and reached about 99.5% after about 0.5-1 h.
Water (15 kg) was added into the reaction mass at a temperature below 15 °C. The mixture was stirred at 15-30 °C for 20-30 min. Aqueous ammonia (25%, 1.13 kg, 16.61 mol) was added into the mixture and the mixture was then stirred for 0.5 h. The organic phase was separated and then washed by extraction with water (15 kg) at 20-25 °C. Activated charcoal (0.15 kg) was added into the organic phase. The mixture was stirred for 1 h and then filtered. The filtrate was concentrated under reduced pressure at not more than 40°C, and compound (I) (1.58 kg, 100% yield) was obtained as a foamy solid.
Investigation of the Crystallinity of iV-(3-(5-((l-Ethylpiperidin-4-yl)(methyl)amino)-3-(pyrimidin-5-yl)- lH-pyrrolo [3,2-Z>] pyridin- l-yl)-2,4-difluorophenyl)propane- 1-sulfonamide Free Base
Investigation of the crystallinity of N-(3-(5-((l-ethylpiperidin-4-yl)(methyl)amino)-3-(py rimidin-5-y 1)- lH-pyrrolo[3 ,2-b] pyridin- 1 -y l)-2,4-difluoropheny l)propane- 1 -sulfonamide free base, obtained by recrystallization from aqueous ethanol, which was used as a starting material to investigate salt formation showed that the compound had low crystallinity, as seen in FIG. 1.
Investigation of Salt forms of iV-(3-(5-((l-Ethylpiperidin-4-yl)(methyl)amino)-3-(pyrimidin-5-yl)- lH-pyrrolo [3,2-Z>] pyridin- l-yl)-2,4-difluorophenyl)propane- 1-sulfonamide
The compound N-(3-(5-((l-ethylpiperidin-4-yl)(methyl)andno)-3-(pyrimidin-5-yl)-lH-pyrrolo [3 ,2-Z>]pyri din- l-yl)-2,4-difluorophenyl)propane-l -sulfonamide was combined with various acids in various solvent systems.
A 96-well master plate was charged by dosing compound in MeOH (stock solution) with a concentration of approx. 40 mg/mL. This plate was placed in a vacuum oven for liquid removal to obtain the same amount of solid material in each well. Subsequently different solvents/solvent mixtures and the acids were added to the solid material in each well (approx. 500μί) and the whole plate was heated up to 50 °C for 2 hours while stirring (using a small stirring bar added to each well).
The acids used were as shown in Table 1. The solvents used were as shown in Table 2. Crystallinity of salts obtained either by the slurry experiment or crystallization by evaporation.
To investigate crystal formation by a slurry experiment, the plate was allowed to cool and the crystallinity of the resulting salts was investigated by XRPD. An image of the master plate showing the salts obtained is shown in FIG. 2A and images of XRPD performed on the salt from each of the master plate wells, showing the crystallinity of the salts formed, is shown in FIG. 2B.
To investigate crystal formation by an evaporation experiment, after the heating period, the solutions were filtered at the same temperature (50 °C) using a preheated filter plate to ensure that no non-dissolved material can be transferred into the other crystallization plates. The filtrate was dispensed into an evaporation plate (approx.. 200μί). The solvents were allowed to evaporate, and the crystallinity of the resulting salts was investigated by XRPD. An image of the master plate showing the salts obtained is shown in FIG. 3A and images of XRPD performed on the salt from each of the evaporation plate wells, showing the crystallinity of the salts formed, is shown in FIG. 3B.
Table 1. Salts Used for Salt Form Investigation
Table 2. Solvents Used for Salt Form Investigation
REFERENCES
1: Waizenegger IC, Baum A, Steurer S, Stadtmüller H, Bader G, Schaaf O, Garin-Chesa P, Schlattl A, Schweifer N, Haslinger C, Colbatzky F, Mousa S, Kalkuhl A, Kraut N, Adolf GR. A Novel RAF Kinase Inhibitor with DFG-Out-Binding Mode: High Efficacy in BRAF-Mutant Tumor Xenograft Models in the Absence of Normal Tissue Hyperproliferation. Mol Cancer Ther. 2016 Mar;15(3):354-65. doi: 10.1158/1535-7163.MCT-15-0617. Epub 2016 Feb 25. PubMed PMID: 26916115.
/////////////// BI-882370, BI 882370, BI882370, XP-102, Boehringer Ingelheim, Xynomic Pharmaceuticals, Preclinical, Colorectal cancer, Malignant melanoma
CCN1CCC(CC1)N(C)c3ccc4n(cc(c2cncnc2)c4n3)c5c(F)ccc(NS(=O)(=O)CCC)c5F
FDA Approves Spiriva Respimat (tiotropium) for the Maintenance Treatment of COPD

Ridgefield, Conn., September 25, 2014 – Boehringer Ingelheim Pharmaceuticals, Inc. announced today that the U.S. Food and Drug Administration (FDA) approved Spiriva Respimat (tiotropium bromide) inhalation spray for the long-term, once-daily maintenance treatment of bronchospasm associated with chronic obstructive pulmonary disease (COPD), including chronic bronchitis and emphysema and to reduce exacerbations in COPD patients. Boehringer Ingelheim anticipates Spiriva Respimat to be available in January 2015.

Spiriva Respimat provides a pre-measured amount of medicine in a slow-moving mist that helps patients inhale the medicine. Spiriva Respimat was developed to actively deliver medication in a way that does not depend of how fast air is breathed in from the inhaler.
READ AT




MAKE IN INDIA
FDA Approves Striverdi Respimat, Olodaterol to Treat Chronic Obstructive Pulmonary Disease

FDA Approves Striverdi Respimat to Treat Chronic Obstructive Pulmonary Disease
July 31, 2014 — Today, the U.S. Food and Drug Administration approved
Striverdi Respimat (olodaterol) inhalation spray to treat patients with chronic
obstructive pulmonary disease (COPD), including chronic bronchitis and/or emphysema
that are experiencing airflow obstruction. Striverdi Respimat can be used once daily
over a long period of time.
read at
http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm407465.htm
See my old post cut paste here
BI launches COPD drug Striverdi, olodaterol in UK and Ireland
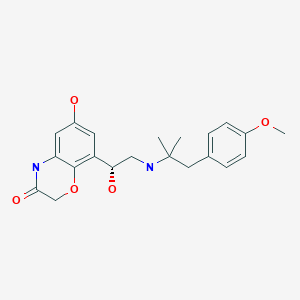
Olodaterol
オロダテロール
BI-1744
BI-1744-CL (hydrochloride) marketed as drug
Boehringer Ingelheim Pharma innovator
synthesis…..http://wendang.baidu.com/view/d4f95541e518964bcf847c22.html
Olodaterol (trade name Striverdi) is a long acting beta-adrenoceptor agonist used as an inhalation for treating patients with chronic obstructive pulmonary disease (COPD), manufactured by Boehringer-Ingelheim.[1]
see……….https://www.thieme-connect.de/DOI/DOI?10.1055/s-0029-1219649 ……… synfacts
Olodaterol is a potent agonist of the human β2-adrenoceptor with a high β1/β2 selectivity. Its crystalline hydrochloride salt is suitable for inhalation and is currently undergoing clinical trials in man for the treatment of asthma. Olodaterol has a duration of action that exceeds 24 hours in two preclinical animal models of bronchoprotection and it has a better safety margin compared with formoterol.
Olodaterol hydrochloride [USAN]
Bi 1744 cl
Bi-1744-cl
Olodaterol hydrochloride
Olodaterol hydrochloride [usan]
UNII-65R445W3V9
868049-49-4 [RN] FREE FORM
CAS 869477-96-3 HCL SALT
R ENANTIOMER
2H-1,4-Benzoxazin-3(4H)-one, 6-hydroxy-8-((1R)-1-hydroxy-2-((2-(4-methoxyphenyl)- 1,1-dimethylethyl)amino)ethyl)-, hydrochloride (1:1)
2H-1,4-benzoxazin-3(4H)-one, 6-hydroxy-8-((1R)-1-hydroxy-2-((2-(4-methoxyphenyl)- 1,1-dimethylethyl)amino)ethyl)-, hydrochloride (1:1)
6-Hydroxy-8-((1R)-1-hydroxy-2-((2-(4-methoxyphenyl)-1,1-dimethylethyl)amino)ethyl)- 2H-1,4-benzoxazin-3(4H)-one hydrochloride
clinical trialshttp://clinicaltrials.gov/search/intervention=Olodaterol+OR+BI+1744
Boehringer Ingelheim has launched a new chronic obstructive pulmonary disease drug, Striverdi in the UK and Ireland.
Striverdi (olodaterol) is the second molecule to be licenced for delivery via the company’s Respimat Soft Mist inhaler, following the COPD blockbuster Spiriva (tiotropium). The drug was approved in Europe in November based on results from a Phase III programme that included more than 3,000 patients with moderate to very severe disease.http://www.pharmatimes.com/Article/14-07-01/BI_launches_COPD_drug_Striverdi_in_UK_and_Ireland.aspx
Olodaterol hydrochloride is a drug candidate originated by Boehringer Ingelheim. The product, delivered once-daily by the Respimat Soft Mist Inhaler, was first launched in Denmark and the Netherlands in March 2014 for the use as maintenance treatment of chronic obstructive pulmonary disease (COPD), including chronic bronchitis and/or emphysema. In 2013, approval was obtained in Russia and Canada for the same indication, and in the U.S, the product was recommended for approval. Phase III clinical trials for the treatment of COPD are ongoing in Japan.
| Systematic (IUPAC) name | |
|---|---|
| 6-hydroxy-8-{(1R)-1-hydroxy-2-{[1-(4-methoxyphenyl)-2-methylpropan-2-yl]amino}ethyl}-4H-1,4-benzoxazin-3-one | |
| Clinical data | |
| Trade names | Striverdi |
| AHFS/Drugs.com | UK Drug Information |
| Pregnancy cat. | No experience |
| Legal status | POM (UK) |
| Routes | Inhalation |
| Identifiers | |
| CAS number | 868049-49-4; 869477-96-3 (hydrochloride) |
| ATC code | R03AC19 |
| PubChem | CID 11504295 |
| ChemSpider | 9679097 |
| UNII | VD2YSN1AFD |
| ChEMBL | CHEMBL605846 |
| Synonyms | BI 1744 CL |
| Chemical data | |
| Formula | C21H26N2O5 free form C21 H26 N2 O5 . Cl H; of hcl salt |
| Mol. mass | 386.44 g/mol free form; 422.902 as hyd salt |
Medical uses
Olodaterol is a once-daily maintenance bronchodilator treatment of airflow obstruction in patients with COPD including chronic bronchitis and/or emphysema, and is administered in an inhaler called Respimat Soft Mist Inhaler.[2][3][4][5][6][7]
As of December 2013, olodaterol is not approved for the treatment of asthma. Olodaterol monotherapy was previously evaluated in four Phase 2 studies in asthma patients. However, currently there are no Phase 3 studies planned for olodaterol monotherapy in patients with asthma.
In late January 2013, Olodaterol CAS# 868049-49-4 was the focus of an FDA committee reviewing data for the drug’s approval as a once-daily maintenance bronchodilator to treat chronic obstructive pulmonary disease (COPD), as well as chronic bronchitis and emphysema. The FDA Pulmonary-Allergy Drugs Advisory Committee recommended that the clinical data from the Boehringer Ingelheim Phase III studies be included in their NDA.
Also known as the trade name Striverdi Respimat, Olodaterol is efficacious as a long-acting beta-agonist, which patients self-administer via an easy to use metered dose inhaler. While early statistics from clinical trials of Olodaterol were encouraging, a new set of data was released earlier this week, which only further solidified the effectual and tolerable benefits of this COPD drug.
On September 10, 2013 results from two Phase 3 studies of Olodaterol revealed additional positive results from this formidable COPD treatment. The conclusion from these two 48 week studies, which included over 3,000 patients, showed sizable and significant improvements in the lung function of patients who were dosed with Olodaterol. Patients in the aforementioned studies were administered either a once a day dosage of Olodaterol via the appropriate metered-dose inhaler or “usual care”. The “usual care” included a variety of treatment options, such as inhaled corticosteroids (not Olodaterol), short and long acting anticholinergics, xanthines and beta agonists, which were short acting. The clinical trial participants who were dosed with Olodaterol displayed a rapid onset of action from this drug, oftentimes within the first five minutes after taking this medication. Additionally, patients dispensed the Olodaterol inhaler were successfully able to maintain optimum lung function for longer than a full 24 hour period. The participants who were given Olodaterol experienced such an obvious clinical improvement in their COPD symptoms, and it quickly became apparent that the “usual care” protocol was lacking in efficacy and reliability.
A staggering 24 million patients in the United States suffer from chronic obstructive pulmonary disease, and this patient population is in need of an effectual, safe and tolerable solution. Olodaterol is shaping up to be that much needed solution. Not only have the results from studies of Olodaterol been encouraging, the studies themselves have actually been forward thinking and wellness centered. Boehringer Ingelheim is the first company to included studies to evaluate exercise tolerance in patients with COPD, and compare the data to those patients who were dosed with Olodaterol. By including exercise tolerance as an important benchmark in pertinent data for Olodaterol, Boehringer Ingelheim has created a standard for COPD treatment expectations. The impaired lung function for patients with COPD contributes greatly to their inability to exercise and stay healthy. Patients who find treatments and management techniques to combat the lung hyperinflation that develops during exercise have a distinct advantage to attaining overall good health.
– See more at: http://www.lgmpharma.com/blog/olodaterol-offers-encouraging-results-patients-copd/#sthash.DOjcrGxc.dpuf
Data has demonstrated that Striverdi, a once-daily long-acting beta2 agonist, significantly improved lung function versus placebo and is comparable to improvements shown with the older LABA formoterol. The NHS price for the drug is £26.35 for a 30-day supply.
Boehringer cited Richard Russell at Wexham Park Hospital as saying that the licensing of Stirverdi will be welcomed by clinicians as it provides another option. He added that the trial results showing improvements in lung function “are particularly impressive considering the study design, which allowed participants to continue their usual treatment regimen. This reflects more closely the real-world patient population”.
Significantly, the company is also developing olodaterol in combination with Spiriva, a long-acting muscarinic antagonist. LAMA/LABA combinations provide the convenience of delivering the two major bronchodilator classes.
 Olodaterol is a novel, long-acting beta2-adrenergic agonist (LABA) that exerts its pharmacological effect by binding and activating beta2-adrenergic receptors located primarily in the lungs. Beta2-adrenergic receptors are membrane-bound receptors that are normally activated by endogenous epinephrine whose signalling, via a downstream L-type calcium channel interaction, mediates smooth muscle relaxation and bronchodilation. Activation of the receptor stimulates an associated G protein which then activates adenylate cyclase, catalyzing the formation of cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA). Elevation of these two molecules induces bronchodilation by relaxation of airway smooth muscles. It is by this mechanism that olodaterol is used for the treatment of chronic obstructive pulmonary disease (COPD) and the progressive airflow obstruction that is characteristic of it. Treatment with bronchodilators helps to mitigate associated symptoms such as shortness of breath, cough, and sputum production. Single doses of olodaterol have been shown to improve forced expiratory volume in 1 sec (FEV1) for 24 h in patients with COPD, allowing once daily dosing. A once-a-day treatment with a LABA has several advantages over short-acting bronchodilators and twice-daily LABAs including improved convenience and compliance and improved airflow over a 24-hour period. Despite similarities in symptoms, olodaterol is not indicated for the treatment of acute exacerbations of COPD or for the treatment of asthma.
Olodaterol is a novel, long-acting beta2-adrenergic agonist (LABA) that exerts its pharmacological effect by binding and activating beta2-adrenergic receptors located primarily in the lungs. Beta2-adrenergic receptors are membrane-bound receptors that are normally activated by endogenous epinephrine whose signalling, via a downstream L-type calcium channel interaction, mediates smooth muscle relaxation and bronchodilation. Activation of the receptor stimulates an associated G protein which then activates adenylate cyclase, catalyzing the formation of cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA). Elevation of these two molecules induces bronchodilation by relaxation of airway smooth muscles. It is by this mechanism that olodaterol is used for the treatment of chronic obstructive pulmonary disease (COPD) and the progressive airflow obstruction that is characteristic of it. Treatment with bronchodilators helps to mitigate associated symptoms such as shortness of breath, cough, and sputum production. Single doses of olodaterol have been shown to improve forced expiratory volume in 1 sec (FEV1) for 24 h in patients with COPD, allowing once daily dosing. A once-a-day treatment with a LABA has several advantages over short-acting bronchodilators and twice-daily LABAs including improved convenience and compliance and improved airflow over a 24-hour period. Despite similarities in symptoms, olodaterol is not indicated for the treatment of acute exacerbations of COPD or for the treatment of asthma.Adverse effects
Adverse effects generally were rare and mild in clinical studies. Most common, but still affecting no more than 1% of patients, were nasopharyngitis (running nose), dizziness and rash. To judge from the drug’s mechanism of action and from experiences with related drugs, hypertension (high blood pressure), tachycardia (fast heartbeat), hypokalaemia (low blood levels of potassium), shaking, etc., might occur in some patients, but these effects have rarely, if at all, been observed in studies.[1]
Interactions
Based on theoretical considerations, co-application of other beta-adrenoceptor agonists, potassium lowering drugs (e. g. corticoids, many diuretics, and theophylline), tricyclic antidepressants, and monoamine oxidase inhibitors could increase the likelihood of adverse effects to occur. Beta blockers, a group of drugs for the treatment of hypertension (high blood pressure) and various conditions of the heart, could reduce the efficacy of olodaterol.[1] Clinical data on the relevance of such interactions are very limited.
Pharmacology
Mechanism of action
Like all beta-adrenoceptor agonists, olodaterol mimics the effect of epinephrine at beta-2 receptors (β₂-receptors) in the lung, which causes the bronchi to relax and reduces their resistance to airflow.[3]
Olodaterol is a nearly full β₂-agonist, having 88% intrinsic activity compared to the gold standard isoprenaline. Its half maximal effective concentration (EC50) is 0.1 nM. It has a higher in vitro selectivity for β₂-receptors than the related drugs formoterol and salmeterol: 241-fold versus β₁- and 2299-fold versus β₃-receptors.[2] The high β₂/β₁ selectivity may account for the apparent lack of tachycardia in clinical trials, which is mediated by β₁-receptors on the heart.
Pharmacokinetics
Once bound to a β₂-receptor, an olodaterol molecule stays there for hours – its dissociation half-life is 17.8 hours –, which allows for once-a-day application of the drug[3] like with indacaterol. Other related compounds generally have a shorter duration of action and have to be applied twice daily (e.g. formoterol, salmeterol). Still others (e. g. salbutamol, fenoterol) have to be applied three or four times a day for continuous action, which can also be an advantage for patients who need to apply β₂-agonists only occasionally, for example in an asthma attack.[8]
History
On 29 January 2013 the U.S. Food and Drug Administration (FDA) Pulmonary-Allergy Drugs Advisory Committee (PADAC) recommended that the clinical data included in the new drug application (NDA) for olodaterol provide substantial evidence of safety and efficacy to support the approval of olodaterol as a once-daily maintenance bronchodilator treatment for airflow obstruction in patients with COPD.[9]
On 18 October 2013 approval of olodaterol in the first three European countries – the United Kingdom, Denmark and Iceland – was announced by the manufacturer.[10]

Figure Chemical structures of salmeterol, formoterol, inda- caterol, and emerging once-daily long-acting β2-agonists
CLIP
Synthetic approaches to the 2013 new drugs – ScienceDirect

Olodaterol hydrochloride was approved for long-term, once-daily maintenance treatment of chronic
obstructive pulmonary disease (COPD) in 2013 in the following countries: Canada, Russia, United
Kingdom, Denmark, and Iceland.142, 143 The drug has been recommended by a federal advisory panel for
approval by the FDA.142, 143 Developed and marketed by Boehringer Ingelheim, olodaterol is a longacting
β2-adrenergic receptor agonist with high selectivity over the β1- and β3-receptors (219- and 1622-fold, respectively).144 Upon binding to and activating the β2-adrenergic receptor in the airway, olodaterol
stimulates adenyl cyclase to synthesize cAMP, leading to the relaxation of smooth muscle cells in the
airway. Administered by inhalation using the Respimat®
Soft Mist inhaler, it delivers significant
bronchodilator effects within five minutes of the first dose and provides sustained improvement in
forced expiratory volume (FEV1) for over 24 hours.143 While several routes have been reported in the
patent and published literature,144-146 the manufacturing route for olodaterol hydrochloride disclosed in
2011 is summarized in Scheme 19 below.147
Commercial 2’,5’-dihydroxyacetophenone (122) was treated with one equivalent of benzyl bromide
and potassium carbonate in methylisobutylketone (MIBK) to give the 5’-monobenzylated product in
76% yield. Subsequent nitration occurred at the 4’-position to provide nitrophenol 123 in 87% yield.
Reduction of the nitro group followed by subjection to chloroacetyl chloride resulted in the construction
of benzoxazine 124 in 82% yield. Next, monobromination through the use of tetrabutylammonium
tribromide occurred at the acetophenone carbon to provide bromoketone 125, and this was followed by
asymmetric reduction of the ketone employing (−)-DIP chloride to afford an intermediate bromohydrin,
which underwent conversion to the corresponding epoxide 126 in situ upon treatment with aqueous
NaOH. This epoxide was efficiently formed in 85% yield and 98.3% enantiomeric excess. Epoxide
126 underwent ring-opening upon subjection to amine 127 to provide amino-alcohol 128 in in 84-90%
yield and 89.5-99.5% enantiomeric purity following salt formation with HCl. Tertiary amine 127 was
itself prepared in three steps by reaction of ketone 129 with methylmagnesium chloride, Ritter reaction
of the tertiary alcohol with acetonitrile, and hydrolysis of the resultant acetamide with ethanolic
potassium hydroxide. Hydrogenative removal of the benzyl ether within 128 followed by
recrystallization with methanolic isopropanol furnished olodaterol hydrochloride (XVI) in 63-70%
yield. Overall, the synthesis of olodaterol hydrochloride required 10 total steps (7 linear) from
commercially available acetophenone 122.
142. Gibb, A.; Yang, L. P. H. Drugs 2013, 73, 1841.
143. http://www.boehringeringelheim.com/news/news_releases/press_releases/2013/18_october_2013_olodaterol.html.
144. Bouyssou, T.; Hoenke, C.; Rudolf, K.; Lustenberger, P.; Pestel, S.; Sieger, P.; Lotz, R.; Heine,
C.; Buettner, F. H.; Schnapp, A.; Konetzki, I. Bioorg. Med. Chem. Lett. 2010, 20, 1410.
145. Trunk, M. J. F.; Schiewe, J. US Patent 20050255050A1, 2005.
146. Lustenberger, P.; Konetzki, I.; Sieger, P. US Patent 20090137578A1, 2009.
147. Krueger, T.; Ries, U.; Schnaubelt, J.; Rall, W.; Leuter, Z. A.; Duran, A.; Soyka, R. US Patent
20110124859A1, 2011.
PATENT
WO 2004045618 or
http://www.google.com/patents/EP1562603B1?cl=en
Example

a)
To a solution of 3.6 g 1,1-dimethyl-2-(4-methoxyphenyl)-ethylamine in 100 mL of ethanol at 70 ° C. 7.5 g of (6-benzyloxy-4H-benzo [1,4] oxazin-3-one )-glyoxal added and allowed to stir for 15 minutes. Then within 30 minutes at 10 to 20 ° C. 1 g of sodium borohydride added. It is stirred for one hour, with 10 mL of acetone and stirred for another 30 minutes. The reaction mixture is diluted with 150 mL ethyl acetate, washed with water, dried with sodium sulfate and concentrated. The residue is dissolved in 50 mL of methanol and 100 mL ethyl acetate and acidified with conc. Hydrochloric acid. After addition of 100 mL of diethyl ether, the product precipitates. The crystals are filtered, washed and recrystallized from 50 mL of ethanol. Yield: 7 g (68%; hydrochloride), mp = 232-234 ° C.
b)
6.8 g of the above obtained benzyl compound in 125 mL of methanol with the addition of 1 g of palladium on carbon (5%) was hydrogenated at room temperature and normal pressure. The catalyst is filtered and the filtrate was freed from solvent. Recrystallization of the residue in 50 mL of acetone and a little water, a solid is obtained, which is filtered and washed.
Yield: 5.0 g (89%; hydrochloride), mp = 155-160 ° C.
The (R) – and (S)-enantiomers of Example 3 can be obtained from the racemate, for example, by chiral HPLC (for example, column: Chirobiotic T, 250 x 1.22 mm from the company Astec). As the mobile phase, methanol with 0.05% triethylamine and 0.05% acetic acid. Silica gel with a grain size of 5 microns, to which is covalently bound the glycoprotein teicoplanin can reach as column material used. Retention time (R enantiomer) = 40.1 min, retention time (S-enantiomer) = 45.9 min. The two enantiomers can be obtained by this method in the form of free bases. According to the invention of paramount importance is the R enantiomer of Example 3
PATENT
WO 2005111005
http://www.google.fm/patents/WO2005111005A1?cl=en
Scheme 1.




Scheme 1:
Example 1 6-Hydroxy-8-{(1-hydroxy-2-r2-(4-methoxy-phenyl) – 1, 1-dimethyl-ethylamino]-ethyl)-4H-benzor 41oxazin-3-one – Hvdrochlorid

a) l-(5-benzyloxy-2-hydroxy-3-nitro-phenyl)-ethanone
To a solution of 81.5 g (0.34 mol) l-(5-benzyloxy-2-hydroxy-phenyl)-ethanone in 700 ml of acetic acid are added dropwise under cooling with ice bath, 18 mL of fuming nitric acid, the temperature does not exceed 20 ° C. increases. The reaction mixture is stirred for two hours at room temperature, poured onto ice water and filtered. The product is recrystallized from isopropanol, filtered off and washed with isopropanol and diisopropyl ether. Yield: 69.6 g (72%), mass spectroscopy [M + H] + = 288
b) l-(3-Amino-5-benzyloxy-2-hydroxy-phenyl)-ethanone
69.5 g (242 mmol) of l-(5-benzyloxy-2-hydroxy-3-nitro-phenyl)-ethanone are dissolved in 1.4 L of methanol and in the presence of 14 g of rhodium on carbon (10%) as catalyst at 3 bar room temperature and hydrogenated. Then the catalyst is filtered off and the filtrate concentrated. The residue is reacted further without additional purification. Yield: 60.0 g (96%), R f value = 0.45 (silica gel, dichloromethane).
c) 8-acetyl-6-benzyloxy-4H-benzoπ .4] oxazin-3-one
To 60.0 g (233 mmol) of l-(3-Amino-5-benzyloxy-2-hydroxy-phenyl)-ethanone and 70.0 g (506 mmol) of potassium carbonate while cooling with ice bath, 21.0 ml (258 mmol) of chloroacetyl chloride added dropwise. Then stirred overnight at room temperature and then for 6 hours under reflux. The hot reaction mixture is filtered and then concentrated to about 400 mL and treated with ice water. The precipitate is filtered off, dried and purified by chromatography on a short silica gel column (dichloromethane: methanol = 99:1). The product-containing fractions are concentrated, suspended in isopropanol, diisopropyl ether, and extracted with
Diisopropyl ether. Yield: 34.6 g (50%), mass spectroscopy [M + H] + = 298
d) 6-Benzyloxy-8-(2-chloro-acetyl)-4H-benzoFl, 4] oxazin-3-one 13.8 g (46.0 mmol) of 8-benzyloxy-6-Acetyl-4H-benzo [l, 4] oxazin -3-one and 35.3 g (101.5 mmol) of benzyltrimethylammonium dichloriodat are stirred in 250 mL dichloroethane, 84 mL glacial acetic acid and 14 mL water for 5 hours at 65 ° C. After cooling to room temperature, treated with 5% aqueous sodium hydrogen sulfite solution and stirred for 30 minutes. The precipitated solid is filtered off, washed with water and diethyl ether and dried. Yield: 13.2 g (86%), mass spectroscopy [M + H] + = 330/32.
e) 6-Benzyloxy-8-((R-2-chloro-l-hydroxy-ethyl)-4H-benzori ,41-oxazin-3-one The procedure is analogous to a procedure described in the literature (Org. Lett ., 2002, 4, 4373-4376).
To 13:15 g (39.6 mmol) of 6-benzyloxy-8-(2-chloro-acetyl)-4H-benzo [l, 4] oxazin-3-one and 25.5 mg (0:04 mmol) Cρ * RhCl [(S, S) -TsDPEN] (Cp * = pentamethylcyclopentadienyl and TsDPEN = (lS, 2S)-Np-toluenesulfonyl-l ,2-diphenylethylenediamine) in 40 mL of dimethylformamide at -15 ° C and 8 mL of a mixture of formic acid and triethylamine (molar ratio = 5: 2) dropwise. It is allowed for 5 hours at this temperature, stirring, then 25 mg of catalyst and stirred overnight at -15 ° C. The reaction mixture is mixed with ice water and filtered. The filter residue is dissolved in dichloromethane, dried with sodium sulfate and the solvent evaporated. The residue is recrystallized gel (dichloromethane / methanol gradient) and the product in diethyl ether / diisopropyl ether. Yield: 10.08 g (76%), R f value = 00:28 (on silica gel, dichloromethane ethanol = 50:1).
f) 6-Benzyloxy-8-(R-oxiranyl-4H-benzo [“L4] oxazin-3-one 6.10 g (30.1 mmol) of 6-benzyloxy-8-((R)-2-chloro-l-hydroxy- ethyl)-4H-benzo [l, 4] oxazin-3-one are dissolved in 200 mL of dimethylformamide. added to the solution at 0 ° C with 40 mL of a 2 molar sodium hydroxide solution and stirred at this temperature for 4 hours. the reaction mixture is poured onto ice water, stirred for 15 minutes, and then filtered The solid is washed with water and dried to give 8.60 g (96%), mass spectroscopy [M + H] + = 298..
g) 6-Benyloxy-8-{(R-l-hydroxy-2-r2-(4-methoxy-phenyl)-dimethyl-ll-ethvIaminol-ethyl)-4H-benzo-3-Tl A1oxazin
5.25 g (17.7 mmol) of 6-benzyloxy-8-(R)-oxiranyl-4H-benzo [l, 4] oxazin-3-one and 6.30 g (35.1 mmol) of 2 – (4-methoxy-phenyl 1, 1 – dimethyl-ethyl to be with 21 mL
Of isopropanol and stirred at 135 ° C for 30 minutes under microwave irradiation in a sealed reaction vessel. The solvent is distilled off and the residue chromatographed (alumina, ethyl acetate / methanol gradient). The product thus obtained is purified by recrystallization from a mixture further Diethylether/Diisopropylether-. Yield: 5:33 g (63%), mass spectroscopy [M + H] + = 477 h) 6-Hydroxy-8-{(R)-l-hydroxy-2-[2 – (4-methoxy-phenyl)-l, l-dimethyl-ethylamino] – ethyl}-4H-benzo [1, 4, 1 oxazin-3-one hydrochloride
A suspension of 5:33 g (11.2 mmol) of 6-Benyloxy-8-{(R)-l-hydroxy-2-[2 – (4-methoxy-phenyl)-l, l-dimethyl-ethylamino]-ethyl}-4H -benzo [l, 4] oxazin-3-one in 120 mL of methanol with 0.8 g of palladium on carbon (10%), heated to 50 ° C and hydrogenated at 3 bar hydrogen pressure. Then the catalyst is filtered off and the filtrate concentrated. The residue is dissolved in 20 mL of isopropanol, and 2.5 mL of 5 molar hydrochloric acid in isopropanol. The product is precipitated with 200 mL of diethyl ether, filtered off and dried. Yield: 4.50 g (95%, hydrochloride), mass spectroscopy [M + H] + = 387
PATENT
WO 2007020227
http://www.google.com.ar/patents/WO2007020227A1?cl=en
PATENT
WO 2008090193
or
http://www.google.com/patents/EP2125759B1?cl=en
PAPER
Discovery of olodaterol, a novel inhaled beta(2)-adrenoceptor agonist with a 24h bronchodilatory efficacy
Bioorg Med Chem Lett 2010, 20(4): 1410
http://www.sciencedirect.com/science/article/pii/S0960894X09018101
The discovery of the β2-adrenoceptor agonist (R)-4p designated olodaterol is described. The preclinical profile of the compound suggests a bronchoprotective effect over 24 h in humans.

CLIP
Australia
http://www.tga.gov.au/pdf/auspar/auspar-olodaterol-140327-pi.pdf
CLIP
DUTCH
FDA
Click to access 203108Orig1s000ChemR.pdf
NDA 203108
Striverdi® Respimat® (olodaterol) Inhalation Spray
Boehringer Ingelheim Pharmaceuticals, Inc.
References
- Striverdi UK Drug Information
- Bouyssou, T.; Casarosa, P.; Naline, E.; Pestel, S.; Konetzki, I.; Devillier, P.; Schnapp, A. (2010). “Pharmacological Characterization of Olodaterol, a Novel Inhaled 2-Adrenoceptor Agonist Exerting a 24-Hour-Long Duration of Action in Preclinical Models”. Journal of Pharmacology and Experimental Therapeutics334 (1): 53–62. doi:10.1124/jpet.110.167007. PMID20371707.
- Casarosa, P.; Kollak, I.; Kiechle, T.; Ostermann, A.; Schnapp, A.; Kiesling, R.; Pieper, M.; Sieger, P.; Gantner, F. (2011). “Functional and Biochemical Rationales for the 24-Hour-Long Duration of Action of Olodaterol”. Journal of Pharmacology and Experimental Therapeutics337 (3): 600–609. doi:10.1124/jpet.111.179259. PMID21357659.
- Bouyssou, T.; Hoenke, C.; Rudolf, K.; Lustenberger, P.; Pestel, S.; Sieger, P.; Lotz, R.; Heine, C.; Büttner, F. H.; Schnapp, A.; Konetzki, I. (2010). “Discovery of olodaterol, a novel inhaled β2-adrenoceptor agonist with a 24h bronchodilatory efficacy”. Bioorganic & Medicinal Chemistry Letters20 (4): 1410–1414. doi:10.1016/j.bmcl.2009.12.087. PMID20096576.
- Joos G, Aumann JL, Coeck C, et al. ATS 2012 Abstract: Comparison of 24-Hour FEV1 Profile for Once-Daily versus Twice-Daily Treatment with Olodaterol, A Novel Long-Acting ß2-Agonist, in Patients with COPD[dead link]
- Van Noord, J. A.; Smeets, J. J.; Drenth, B. M.; Rascher, J.; Pivovarova, A.; Hamilton, A. L.; Cornelissen, P. J. G. (2011). “24-hour Bronchodilation following a single dose of the novel β2-agonist olodaterol in COPD”. Pulmonary Pharmacology & Therapeutics24 (6): 666–672. doi:10.1016/j.pupt.2011.07.006. PMID21839850.
- van Noord JA, Korducki L, Hamilton AL and Koker P. Four Weeks Once Daily Treatment with BI 1744 CL, a Novel Long-Acting ß2-Agonist, is Effective in COPD Patients. Am. J. Respir. Crit. Care Med. 2009; 179: A6183[dead link]
- Haberfeld, H, ed. (2009). Austria-Codex (in German) (2009/2010 ed.). Vienna: Österreichischer Apothekerverlag. ISBN3-85200-196-X.
- Hollis A (31 January 2013). “Panel Overwhelmingly Supports Boehringer COPD Drug Striverdi”. FDA News/Drug Industry Daily.
- “New once-daily Striverdi (olodaterol) Respimat gains approval in first EU countries”. Boehringer-Ingelheim. 18 October 2013.
External links
The active moiety olodaterol is a selective beta2-adrenergic bronchodilator. The drug substance, olodaterol hydrochloride, is chemically described as 2H-1,4- Benzoxazin-3H(4H)-one, 6-hydroxy-8-[(1R)-1-hydroxy-2-[[2-(4-methoxyphenyl)-1,1-dimethylethyl]-amino]ethyl]-, monohydrochloride. Olodaterol hydrochloride is a white to off-white powder that is sparingly-slightly soluble in water and slightly soluble in ethanol. The molecular weight is 422.9 g/mole (salt): 386.5 g/mole (base), and the molecular formula is C21H26N2O5 x HCl as a hydrochloride. The conversion factor from salt to free base is 1.094.
The structural formula is:
 |
The drug product, STRIVERDI RESPIMAT, is composed of a sterile, aqueous solution of olodaterol hydrochloride filled into a 4.5 mL plastic container crimped into an aluminum cylinder (STRIVERDI RESPIMAT cartridge) for use with the STRIVERDI RESPIMAT inhaler.
Excipients include water for injection, benzalkonium chloride, edetate disodium, and anhydrous citric acid. The STRIVERDI RESPIMAT cartridge is only intended for use with the STRIVERDI RESPIMAT inhaler. The STRIVERDI RESPIMAT inhaler is a hand held, pocket sized oral inhalation device that uses mechanical energy to generate a slow-moving aerosol cloud of medication from a metered volume of the drug solution. The STRIVERDI RESPIMAT inhaler has a yellow-colored cap.
When used with the STRIVERDI RESPIMAT inhaler, each cartridge containing a minimum of 4 grams of a sterile aqueous solution delivers the labeled number of metered actuations after preparation for use. Each dose (1 dose equals 2 actuations) from the STRIVERDI RESPIMAT inhaler delivers 5 mcg olodaterol in 22.1 mcL of solution from the mouthpiece. As with all inhaled drugs, the actual amount of drug delivered to the lung may depend on patient factors, such as the coordination between the actuation of the inhaler and inspiration through the delivery system. The duration of inspiration should be at least as long as the spray duration (1.5 seconds).
| WO2002030928A1 | 28 Sep 2001 | 11 Apr 2003 | Boehringer Ingelheim Pharma | Crystalline monohydrate, method for producing the same and the use thereof in the production of a medicament |
| WO2003000265A1 | 8 Jun 2002 | 3 Jan 2003 | Boehringer Ingelheim Pharma | Crystalline anticholinergic, method for its production, and use thereof in the production of a drug |
| WO2004045618A2 * | 11 Nov 2003 | 3 Jun 2004 | Boehringer Ingelheim Pharma | Novel medicaments for the treatment of chronic obstructive pulmonary diseases |
| EP0073505A1 * | 28 Aug 1982 | 9 Mar 1983 | Boehringer Ingelheim Kg | Benzo-heterocycles |
| EP0321864A2 * | 15 Dec 1988 | 28 Jun 1989 | Boehringer Ingelheim Kg | Ammonium compounds, their preparation and use |
| US4460581 | 12 Oct 1982 | 17 Jul 1984 | Boehringer Ingelheim Kg | Antispasmodic agents, antiallergens |
| US4656168 * | 13 Oct 1983 | 7 Apr 1987 | Merck & Co., Inc. | Vision defects; adrenergic blocking and hypotensive agents |
Organic spectroscopy should be brushed up and you get confidence
read my blog
ORGANIC SPECTROSCOPY INTERNATIONAL is the blog
Organic chemists from Industry and academics to interact on Spectroscopy techniques for Organic compounds ie NMR, MASS, IR, UV Etc. email me ……….. amcrasto@gmail.com
http://orgspectroscopyint.blogspot.in/ is the link
 amcrasto@gmail.com
amcrasto@gmail.com
Glenmark Generics receives final ANDA approval for Telmisartan Tablets

Glenmark Generics receives final ANDA approval for Telmisartan Tablets
Mumbai, India, July 8, 2014
Glenmark Generics Inc., USA a subsidiary of Glenmark Generics Limited has been granted final abbreviated new drug approval (ANDA) from the United States Food and Drug Administration (US FDA) for Telmisartan Tablets. Glenmark will commence distribution of the product immediately.
Telmisartan Tablets are Glenmark’s generic version of Boehringer Ingelheim’s Micardis®. Telmisartan is indicated for the treatment of hypertension.
The approval is for the 20mg, 40mg and 80mg tablets. For the 12 month period ending March 2014, Telmisartan garnered annual sales of USD 250 Million according to IMS Health.
Glenmark receives USFDA approval for telmisartan tablets
Telmisartan, which is the generic version of Boehringer Ingelheim’s Micardis, garnered annual sales of $ 250 million for the 12 month period ending March 2014
BI launches COPD drug Striverdi, olodaterol in UK and Ireland
Olodaterol
BI-1744
BI-1744-CL (hydrochloride) marketed as drug
Boehringer Ingelheim Pharma innovator
synthesis…..http://wendang.baidu.com/view/d4f95541e518964bcf847c22.html
Olodaterol (trade name Striverdi) is a long acting beta-adrenoceptor agonist used as an inhalation for treating patients with chronic obstructive pulmonary disease (COPD), manufactured by Boehringer-Ingelheim.[1]
see……….https://www.thieme-connect.de/DOI/DOI?10.1055/s-0029-1219649 ……… synfacts
Olodaterol is a potent agonist of the human β2-adrenoceptor with a high β1/β2 selectivity. Its crystalline hydrochloride salt is suitable for inhalation and is currently undergoing clinical trials in man for the treatment of asthma. Olodaterol has a duration of action that exceeds 24 hours in two preclinical animal models of bronchoprotection and it has a better safety margin compared with formoterol.
Olodaterol hydrochloride [USAN]
Bi 1744 cl
Bi-1744-cl
Olodaterol hydrochloride
Olodaterol hydrochloride [usan]
UNII-65R445W3V9
868049-49-4 [RN] FREE FORM
CAS 869477-96-3 HCL SALT
R ENANTIOMER
2H-1,4-Benzoxazin-3(4H)-one, 6-hydroxy-8-((1R)-1-hydroxy-2-((2-(4-methoxyphenyl)- 1,1-dimethylethyl)amino)ethyl)-, hydrochloride (1:1)
2H-1,4-benzoxazin-3(4H)-one, 6-hydroxy-8-((1R)-1-hydroxy-2-((2-(4-methoxyphenyl)- 1,1-dimethylethyl)amino)ethyl)-, hydrochloride (1:1)
6-Hydroxy-8-((1R)-1-hydroxy-2-((2-(4-methoxyphenyl)-1,1-dimethylethyl)amino)ethyl)- 2H-1,4-benzoxazin-3(4H)-one hydrochloride
clinical trialshttp://clinicaltrials.gov/search/intervention=Olodaterol+OR+BI+1744
Boehringer Ingelheim has launched a new chronic obstructive pulmonary disease drug, Striverdi in the UK and Ireland.
Striverdi (olodaterol) is the second molecule to be licenced for delivery via the company’s Respimat Soft Mist inhaler, following the COPD blockbuster Spiriva (tiotropium). The drug was approved in Europe in November based on results from a Phase III programme that included more than 3,000 patients with moderate to very severe disease.http://www.pharmatimes.com/Article/14-07-01/BI_launches_COPD_drug_Striverdi_in_UK_and_Ireland.aspx
Olodaterol hydrochloride is a drug candidate originated by Boehringer Ingelheim. The product, delivered once-daily by the Respimat Soft Mist Inhaler, was first launched in Denmark and the Netherlands in March 2014 for the use as maintenance treatment of chronic obstructive pulmonary disease (COPD), including chronic bronchitis and/or emphysema. In 2013, approval was obtained in Russia and Canada for the same indication, and in the U.S, the product was recommended for approval. Phase III clinical trials for the treatment of COPD are ongoing in Japan.
| Systematic (IUPAC) name | |
|---|---|
| 6-hydroxy-8-{(1R)-1-hydroxy-2-{[1-(4-methoxyphenyl)-2-methylpropan-2-yl]amino}ethyl}-4H-1,4-benzoxazin-3-one | |
| Clinical data | |
| Trade names | Striverdi |
| AHFS/Drugs.com | UK Drug Information |
| Pregnancy cat. | No experience |
| Legal status | POM (UK) |
| Routes | Inhalation |
| Identifiers | |
| CAS number | 868049-49-4; 869477-96-3 (hydrochloride) |
| ATC code | R03AC19 |
| PubChem | CID 11504295 |
| ChemSpider | 9679097 |
| UNII | VD2YSN1AFD |
| ChEMBL | CHEMBL605846 |
| Synonyms | BI 1744 CL |
| Chemical data | |
| Formula | C21H26N2O5 free form C21 H26 N2 O5 . Cl H; of hcl salt |
| Mol. mass | 386.44 g/mol free form; 422.902 as hyd salt |
Medical uses
Olodaterol is a once-daily maintenance bronchodilator treatment of airflow obstruction in patients with COPD including chronic bronchitis and/or emphysema, and is administered in an inhaler called Respimat Soft Mist Inhaler.[2][3][4][5][6][7]
As of December 2013, olodaterol is not approved for the treatment of asthma. Olodaterol monotherapy was previously evaluated in four Phase 2 studies in asthma patients. However, currently there are no Phase 3 studies planned for olodaterol monotherapy in patients with asthma.
In late January 2013, Olodaterol CAS# 868049-49-4 was the focus of an FDA committee reviewing data for the drug’s approval as a once-daily maintenance bronchodilator to treat chronic obstructive pulmonary disease (COPD), as well as chronic bronchitis and emphysema. The FDA Pulmonary-Allergy Drugs Advisory Committee recommended that the clinical data from the Boehringer Ingelheim Phase III studies be included in their NDA.
Also known as the trade name Striverdi Respimat, Olodaterol is efficacious as a long-acting beta-agonist, which patients self-administer via an easy to use metered dose inhaler. While early statistics from clinical trials of Olodaterol were encouraging, a new set of data was released earlier this week, which only further solidified the effectual and tolerable benefits of this COPD drug.
On September 10, 2013 results from two Phase 3 studies of Olodaterol revealed additional positive results from this formidable COPD treatment. The conclusion from these two 48 week studies, which included over 3,000 patients, showed sizable and significant improvements in the lung function of patients who were dosed with Olodaterol. Patients in the aforementioned studies were administered either a once a day dosage of Olodaterol via the appropriate metered-dose inhaler or “usual care”. The “usual care” included a variety of treatment options, such as inhaled corticosteroids (not Olodaterol), short and long acting anticholinergics, xanthines and beta agonists, which were short acting. The clinical trial participants who were dosed with Olodaterol displayed a rapid onset of action from this drug, oftentimes within the first five minutes after taking this medication. Additionally, patients dispensed the Olodaterol inhaler were successfully able to maintain optimum lung function for longer than a full 24 hour period. The participants who were given Olodaterol experienced such an obvious clinical improvement in their COPD symptoms, and it quickly became apparent that the “usual care” protocol was lacking in efficacy and reliability.
A staggering 24 million patients in the United States suffer from chronic obstructive pulmonary disease, and this patient population is in need of an effectual, safe and tolerable solution. Olodaterol is shaping up to be that much needed solution. Not only have the results from studies of Olodaterol been encouraging, the studies themselves have actually been forward thinking and wellness centered. Boehringer Ingelheim is the first company to included studies to evaluate exercise tolerance in patients with COPD, and compare the data to those patients who were dosed with Olodaterol. By including exercise tolerance as an important benchmark in pertinent data for Olodaterol, Boehringer Ingelheim has created a standard for COPD treatment expectations. The impaired lung function for patients with COPD contributes greatly to their inability to exercise and stay healthy. Patients who find treatments and management techniques to combat the lung hyperinflation that develops during exercise have a distinct advantage to attaining overall good health.
– See more at: http://www.lgmpharma.com/blog/olodaterol-offers-encouraging-results-patients-copd/#sthash.DOjcrGxc.dpuf
Data has demonstrated that Striverdi, a once-daily long-acting beta2 agonist, significantly improved lung function versus placebo and is comparable to improvements shown with the older LABA formoterol. The NHS price for the drug is £26.35 for a 30-day supply.
Boehringer cited Richard Russell at Wexham Park Hospital as saying that the licensing of Stirverdi will be welcomed by clinicians as it provides another option. He added that the trial results showing improvements in lung function “are particularly impressive considering the study design, which allowed participants to continue their usual treatment regimen. This reflects more closely the real-world patient population”.
Significantly, the company is also developing olodaterol in combination with Spiriva, a long-acting muscarinic antagonist. LAMA/LABA combinations provide the convenience of delivering the two major bronchodilator classes.
Olodaterol is a novel, long-acting beta2-adrenergic agonist (LABA) that exerts its pharmacological effect by binding and activating beta2-adrenergic receptors located primarily in the lungs. Beta2-adrenergic receptors are membrane-bound receptors that are normally activated by endogenous epinephrine whose signalling, via a downstream L-type calcium channel interaction, mediates smooth muscle relaxation and bronchodilation. Activation of the receptor stimulates an associated G protein which then activates adenylate cyclase, catalyzing the formation of cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA). Elevation of these two molecules induces bronchodilation by relaxation of airway smooth muscles. It is by this mechanism that olodaterol is used for the treatment of chronic obstructive pulmonary disease (COPD) and the progressive airflow obstruction that is characteristic of it. Treatment with bronchodilators helps to mitigate associated symptoms such as shortness of breath, cough, and sputum production. Single doses of olodaterol have been shown to improve forced expiratory volume in 1 sec (FEV1) for 24 h in patients with COPD, allowing once daily dosing. A once-a-day treatment with a LABA has several advantages over short-acting bronchodilators and twice-daily LABAs including improved convenience and compliance and improved airflow over a 24-hour period. Despite similarities in symptoms, olodaterol is not indicated for the treatment of acute exacerbations of COPD or for the treatment of asthma.Adverse effects
Adverse effects generally were rare and mild in clinical studies. Most common, but still affecting no more than 1% of patients, were nasopharyngitis (running nose), dizziness and rash. To judge from the drug’s mechanism of action and from experiences with related drugs, hypertension (high blood pressure), tachycardia (fast heartbeat), hypokalaemia (low blood levels of potassium), shaking, etc., might occur in some patients, but these effects have rarely, if at all, been observed in studies.[1]
Interactions
Based on theoretical considerations, co-application of other beta-adrenoceptor agonists, potassium lowering drugs (e. g. corticoids, many diuretics, and theophylline), tricyclic antidepressants, and monoamine oxidase inhibitors could increase the likelihood of adverse effects to occur. Beta blockers, a group of drugs for the treatment of hypertension (high blood pressure) and various conditions of the heart, could reduce the efficacy of olodaterol.[1] Clinical data on the relevance of such interactions are very limited.
Pharmacology
Mechanism of action
Like all beta-adrenoceptor agonists, olodaterol mimics the effect of epinephrine at beta-2 receptors (β₂-receptors) in the lung, which causes the bronchi to relax and reduces their resistance to airflow.[3]
Olodaterol is a nearly full β₂-agonist, having 88% intrinsic activity compared to the gold standard isoprenaline. Its half maximal effective concentration (EC50) is 0.1 nM. It has a higher in vitro selectivity for β₂-receptors than the related drugs formoterol and salmeterol: 241-fold versus β₁- and 2299-fold versus β₃-receptors.[2] The high β₂/β₁ selectivity may account for the apparent lack of tachycardia in clinical trials, which is mediated by β₁-receptors on the heart.
Pharmacokinetics
Once bound to a β₂-receptor, an olodaterol molecule stays there for hours – its dissociation half-life is 17.8 hours –, which allows for once-a-day application of the drug[3] like with indacaterol. Other related compounds generally have a shorter duration of action and have to be applied twice daily (e.g. formoterol, salmeterol). Still others (e. g. salbutamol, fenoterol) have to be applied three or four times a day for continuous action, which can also be an advantage for patients who need to apply β₂-agonists only occasionally, for example in an asthma attack.[8]
History
On 29 January 2013 the U.S. Food and Drug Administration (FDA) Pulmonary-Allergy Drugs Advisory Committee (PADAC) recommended that the clinical data included in the new drug application (NDA) for olodaterol provide substantial evidence of safety and efficacy to support the approval of olodaterol as a once-daily maintenance bronchodilator treatment for airflow obstruction in patients with COPD.[9]
On 18 October 2013 approval of olodaterol in the first three European countries – the United Kingdom, Denmark and Iceland – was announced by the manufacturer.[10]
Figure Chemical structures of salmeterol, formoterol, inda- caterol, and emerging once-daily long-acting β2-agonists
CLIP
Synthetic approaches to the 2013 new drugs – ScienceDirect
Olodaterol hydrochloride was approved for long-term, once-daily maintenance treatment of chronic
obstructive pulmonary disease (COPD) in 2013 in the following countries: Canada, Russia, United
Kingdom, Denmark, and Iceland.142, 143 The drug has been recommended by a federal advisory panel for
approval by the FDA.142, 143 Developed and marketed by Boehringer Ingelheim, olodaterol is a longacting
β2-adrenergic receptor agonist with high selectivity over the β1- and β3-receptors (219- and 1622-fold, respectively).144 Upon binding to and activating the β2-adrenergic receptor in the airway, olodaterol
stimulates adenyl cyclase to synthesize cAMP, leading to the relaxation of smooth muscle cells in the
airway. Administered by inhalation using the Respimat®
Soft Mist inhaler, it delivers significant
bronchodilator effects within five minutes of the first dose and provides sustained improvement in
forced expiratory volume (FEV1) for over 24 hours.143 While several routes have been reported in the
patent and published literature,144-146 the manufacturing route for olodaterol hydrochloride disclosed in
2011 is summarized in Scheme 19 below.147
Commercial 2’,5’-dihydroxyacetophenone (122) was treated with one equivalent of benzyl bromide
and potassium carbonate in methylisobutylketone (MIBK) to give the 5’-monobenzylated product in
76% yield. Subsequent nitration occurred at the 4’-position to provide nitrophenol 123 in 87% yield.
Reduction of the nitro group followed by subjection to chloroacetyl chloride resulted in the construction
of benzoxazine 124 in 82% yield. Next, monobromination through the use of tetrabutylammonium
tribromide occurred at the acetophenone carbon to provide bromoketone 125, and this was followed by
asymmetric reduction of the ketone employing (−)-DIP chloride to afford an intermediate bromohydrin,
which underwent conversion to the corresponding epoxide 126 in situ upon treatment with aqueous
NaOH. This epoxide was efficiently formed in 85% yield and 98.3% enantiomeric excess. Epoxide
126 underwent ring-opening upon subjection to amine 127 to provide amino-alcohol 128 in in 84-90%
yield and 89.5-99.5% enantiomeric purity following salt formation with HCl. Tertiary amine 127 was
itself prepared in three steps by reaction of ketone 129 with methylmagnesium chloride, Ritter reaction
of the tertiary alcohol with acetonitrile, and hydrolysis of the resultant acetamide with ethanolic
potassium hydroxide. Hydrogenative removal of the benzyl ether within 128 followed by
recrystallization with methanolic isopropanol furnished olodaterol hydrochloride (XVI) in 63-70%
yield. Overall, the synthesis of olodaterol hydrochloride required 10 total steps (7 linear) from
commercially available acetophenone 122.
142. Gibb, A.; Yang, L. P. H. Drugs 2013, 73, 1841.
143. http://www.boehringeringelheim.com/news/news_releases/press_releases/2013/18_october_2013_olodaterol.html.
144. Bouyssou, T.; Hoenke, C.; Rudolf, K.; Lustenberger, P.; Pestel, S.; Sieger, P.; Lotz, R.; Heine,
C.; Buettner, F. H.; Schnapp, A.; Konetzki, I. Bioorg. Med. Chem. Lett. 2010, 20, 1410.
145. Trunk, M. J. F.; Schiewe, J. US Patent 20050255050A1, 2005.
146. Lustenberger, P.; Konetzki, I.; Sieger, P. US Patent 20090137578A1, 2009.
147. Krueger, T.; Ries, U.; Schnaubelt, J.; Rall, W.; Leuter, Z. A.; Duran, A.; Soyka, R. US Patent
20110124859A1, 2011.
PATENT
WO 2004045618 or
http://www.google.com/patents/EP1562603B1?cl=en
Example
a)
To a solution of 3.6 g 1,1-dimethyl-2-(4-methoxyphenyl)-ethylamine in 100 mL of ethanol at 70 ° C. 7.5 g of (6-benzyloxy-4H-benzo [1,4] oxazin-3-one )-glyoxal added and allowed to stir for 15 minutes. Then within 30 minutes at 10 to 20 ° C. 1 g of sodium borohydride added. It is stirred for one hour, with 10 mL of acetone and stirred for another 30 minutes. The reaction mixture is diluted with 150 mL ethyl acetate, washed with water, dried with sodium sulfate and concentrated. The residue is dissolved in 50 mL of methanol and 100 mL ethyl acetate and acidified with conc. Hydrochloric acid. After addition of 100 mL of diethyl ether, the product precipitates. The crystals are filtered, washed and recrystallized from 50 mL of ethanol. Yield: 7 g (68%; hydrochloride), mp = 232-234 ° C.
b)
6.8 g of the above obtained benzyl compound in 125 mL of methanol with the addition of 1 g of palladium on carbon (5%) was hydrogenated at room temperature and normal pressure. The catalyst is filtered and the filtrate was freed from solvent. Recrystallization of the residue in 50 mL of acetone and a little water, a solid is obtained, which is filtered and washed.
Yield: 5.0 g (89%; hydrochloride), mp = 155-160 ° C.
The (R) – and (S)-enantiomers of Example 3 can be obtained from the racemate, for example, by chiral HPLC (for example, column: Chirobiotic T, 250 x 1.22 mm from the company Astec). As the mobile phase, methanol with 0.05% triethylamine and 0.05% acetic acid. Silica gel with a grain size of 5 microns, to which is covalently bound the glycoprotein teicoplanin can reach as column material used. Retention time (R enantiomer) = 40.1 min, retention time (S-enantiomer) = 45.9 min. The two enantiomers can be obtained by this method in the form of free bases. According to the invention of paramount importance is the R enantiomer of Example 3
PATENT
WO 2005111005
http://www.google.fm/patents/WO2005111005A1?cl=en
Scheme 1.
Scheme 1:
Example 1 6-Hydroxy-8-{(1-hydroxy-2-r2-(4-methoxy-phenyl) – 1, 1-dimethyl-ethylamino]-ethyl)-4H-benzor 41oxazin-3-one – Hvdrochlorid
a) l-(5-benzyloxy-2-hydroxy-3-nitro-phenyl)-ethanone
To a solution of 81.5 g (0.34 mol) l-(5-benzyloxy-2-hydroxy-phenyl)-ethanone in 700 ml of acetic acid are added dropwise under cooling with ice bath, 18 mL of fuming nitric acid, the temperature does not exceed 20 ° C. increases. The reaction mixture is stirred for two hours at room temperature, poured onto ice water and filtered. The product is recrystallized from isopropanol, filtered off and washed with isopropanol and diisopropyl ether. Yield: 69.6 g (72%), mass spectroscopy [M + H] + = 288
b) l-(3-Amino-5-benzyloxy-2-hydroxy-phenyl)-ethanone
69.5 g (242 mmol) of l-(5-benzyloxy-2-hydroxy-3-nitro-phenyl)-ethanone are dissolved in 1.4 L of methanol and in the presence of 14 g of rhodium on carbon (10%) as catalyst at 3 bar room temperature and hydrogenated. Then the catalyst is filtered off and the filtrate concentrated. The residue is reacted further without additional purification. Yield: 60.0 g (96%), R f value = 0.45 (silica gel, dichloromethane).
c) 8-acetyl-6-benzyloxy-4H-benzoπ .4] oxazin-3-one
To 60.0 g (233 mmol) of l-(3-Amino-5-benzyloxy-2-hydroxy-phenyl)-ethanone and 70.0 g (506 mmol) of potassium carbonate while cooling with ice bath, 21.0 ml (258 mmol) of chloroacetyl chloride added dropwise. Then stirred overnight at room temperature and then for 6 hours under reflux. The hot reaction mixture is filtered and then concentrated to about 400 mL and treated with ice water. The precipitate is filtered off, dried and purified by chromatography on a short silica gel column (dichloromethane: methanol = 99:1). The product-containing fractions are concentrated, suspended in isopropanol, diisopropyl ether, and extracted with
Diisopropyl ether. Yield: 34.6 g (50%), mass spectroscopy [M + H] + = 298
d) 6-Benzyloxy-8-(2-chloro-acetyl)-4H-benzoFl, 4] oxazin-3-one 13.8 g (46.0 mmol) of 8-benzyloxy-6-Acetyl-4H-benzo [l, 4] oxazin -3-one and 35.3 g (101.5 mmol) of benzyltrimethylammonium dichloriodat are stirred in 250 mL dichloroethane, 84 mL glacial acetic acid and 14 mL water for 5 hours at 65 ° C. After cooling to room temperature, treated with 5% aqueous sodium hydrogen sulfite solution and stirred for 30 minutes. The precipitated solid is filtered off, washed with water and diethyl ether and dried. Yield: 13.2 g (86%), mass spectroscopy [M + H] + = 330/32.
e) 6-Benzyloxy-8-((R-2-chloro-l-hydroxy-ethyl)-4H-benzori ,41-oxazin-3-one The procedure is analogous to a procedure described in the literature (Org. Lett ., 2002, 4, 4373-4376).
To 13:15 g (39.6 mmol) of 6-benzyloxy-8-(2-chloro-acetyl)-4H-benzo [l, 4] oxazin-3-one and 25.5 mg (0:04 mmol) Cρ * RhCl [(S, S) -TsDPEN] (Cp * = pentamethylcyclopentadienyl and TsDPEN = (lS, 2S)-Np-toluenesulfonyl-l ,2-diphenylethylenediamine) in 40 mL of dimethylformamide at -15 ° C and 8 mL of a mixture of formic acid and triethylamine (molar ratio = 5: 2) dropwise. It is allowed for 5 hours at this temperature, stirring, then 25 mg of catalyst and stirred overnight at -15 ° C. The reaction mixture is mixed with ice water and filtered. The filter residue is dissolved in dichloromethane, dried with sodium sulfate and the solvent evaporated. The residue is recrystallized gel (dichloromethane / methanol gradient) and the product in diethyl ether / diisopropyl ether. Yield: 10.08 g (76%), R f value = 00:28 (on silica gel, dichloromethane ethanol = 50:1).
f) 6-Benzyloxy-8-(R-oxiranyl-4H-benzo [“L4] oxazin-3-one 6.10 g (30.1 mmol) of 6-benzyloxy-8-((R)-2-chloro-l-hydroxy- ethyl)-4H-benzo [l, 4] oxazin-3-one are dissolved in 200 mL of dimethylformamide. added to the solution at 0 ° C with 40 mL of a 2 molar sodium hydroxide solution and stirred at this temperature for 4 hours. the reaction mixture is poured onto ice water, stirred for 15 minutes, and then filtered The solid is washed with water and dried to give 8.60 g (96%), mass spectroscopy [M + H] + = 298..
g) 6-Benyloxy-8-{(R-l-hydroxy-2-r2-(4-methoxy-phenyl)-dimethyl-ll-ethvIaminol-ethyl)-4H-benzo-3-Tl A1oxazin
5.25 g (17.7 mmol) of 6-benzyloxy-8-(R)-oxiranyl-4H-benzo [l, 4] oxazin-3-one and 6.30 g (35.1 mmol) of 2 – (4-methoxy-phenyl 1, 1 – dimethyl-ethyl to be with 21 mL
Of isopropanol and stirred at 135 ° C for 30 minutes under microwave irradiation in a sealed reaction vessel. The solvent is distilled off and the residue chromatographed (alumina, ethyl acetate / methanol gradient). The product thus obtained is purified by recrystallization from a mixture further Diethylether/Diisopropylether-. Yield: 5:33 g (63%), mass spectroscopy [M + H] + = 477 h) 6-Hydroxy-8-{(R)-l-hydroxy-2-[2 – (4-methoxy-phenyl)-l, l-dimethyl-ethylamino] – ethyl}-4H-benzo [1, 4, 1 oxazin-3-one hydrochloride
A suspension of 5:33 g (11.2 mmol) of 6-Benyloxy-8-{(R)-l-hydroxy-2-[2 – (4-methoxy-phenyl)-l, l-dimethyl-ethylamino]-ethyl}-4H -benzo [l, 4] oxazin-3-one in 120 mL of methanol with 0.8 g of palladium on carbon (10%), heated to 50 ° C and hydrogenated at 3 bar hydrogen pressure. Then the catalyst is filtered off and the filtrate concentrated. The residue is dissolved in 20 mL of isopropanol, and 2.5 mL of 5 molar hydrochloric acid in isopropanol. The product is precipitated with 200 mL of diethyl ether, filtered off and dried. Yield: 4.50 g (95%, hydrochloride), mass spectroscopy [M + H] + = 387
PATENT
WO 2007020227
http://www.google.com.ar/patents/WO2007020227A1?cl=en
PATENT
WO 2008090193
or
http://www.google.com/patents/EP2125759B1?cl=en
PAPER
Discovery of olodaterol, a novel inhaled beta(2)-adrenoceptor agonist with a 24h bronchodilatory efficacy
Bioorg Med Chem Lett 2010, 20(4): 1410
http://www.sciencedirect.com/science/article/pii/S0960894X09018101
The discovery of the β2-adrenoceptor agonist (R)-4p designated olodaterol is described. The preclinical profile of the compound suggests a bronchoprotective effect over 24 h in humans.
CLIP
Australia
http://www.tga.gov.au/pdf/auspar/auspar-olodaterol-140327-pi.pdf
CLIP
DUTCH
http://mri.medagencies.org/download/NL_H_2498_001_PAR.pdf
FDA
Click to access 203108Orig1s000ChemR.pdf
NDA 203108
Striverdi® Respimat® (olodaterol) Inhalation Spray
Boehringer Ingelheim Pharmaceuticals, Inc.
References
- Striverdi UK Drug Information
- Bouyssou, T.; Casarosa, P.; Naline, E.; Pestel, S.; Konetzki, I.; Devillier, P.; Schnapp, A. (2010). “Pharmacological Characterization of Olodaterol, a Novel Inhaled 2-Adrenoceptor Agonist Exerting a 24-Hour-Long Duration of Action in Preclinical Models”. Journal of Pharmacology and Experimental Therapeutics 334 (1): 53–62. doi:10.1124/jpet.110.167007. PMID 20371707.
- Casarosa, P.; Kollak, I.; Kiechle, T.; Ostermann, A.; Schnapp, A.; Kiesling, R.; Pieper, M.; Sieger, P.; Gantner, F. (2011). “Functional and Biochemical Rationales for the 24-Hour-Long Duration of Action of Olodaterol”. Journal of Pharmacology and Experimental Therapeutics 337 (3): 600–609. doi:10.1124/jpet.111.179259. PMID 21357659.
- Bouyssou, T.; Hoenke, C.; Rudolf, K.; Lustenberger, P.; Pestel, S.; Sieger, P.; Lotz, R.; Heine, C.; Büttner, F. H.; Schnapp, A.; Konetzki, I. (2010). “Discovery of olodaterol, a novel inhaled β2-adrenoceptor agonist with a 24h bronchodilatory efficacy”. Bioorganic & Medicinal Chemistry Letters 20 (4): 1410–1414. doi:10.1016/j.bmcl.2009.12.087. PMID 20096576.
- Joos G, Aumann JL, Coeck C, et al. ATS 2012 Abstract: Comparison of 24-Hour FEV1 Profile for Once-Daily versus Twice-Daily Treatment with Olodaterol, A Novel Long-Acting ß2-Agonist, in Patients with COPD[dead link]
- Van Noord, J. A.; Smeets, J. J.; Drenth, B. M.; Rascher, J.; Pivovarova, A.; Hamilton, A. L.; Cornelissen, P. J. G. (2011). “24-hour Bronchodilation following a single dose of the novel β2-agonist olodaterol in COPD”. Pulmonary Pharmacology & Therapeutics 24 (6): 666–672. doi:10.1016/j.pupt.2011.07.006. PMID 21839850.
- van Noord JA, Korducki L, Hamilton AL and Koker P. Four Weeks Once Daily Treatment with BI 1744 CL, a Novel Long-Acting ß2-Agonist, is Effective in COPD Patients. Am. J. Respir. Crit. Care Med. 2009; 179: A6183[dead link]
- Haberfeld, H, ed. (2009). Austria-Codex (in German) (2009/2010 ed.). Vienna: Österreichischer Apothekerverlag. ISBN 3-85200-196-X.
- Hollis A (31 January 2013). “Panel Overwhelmingly Supports Boehringer COPD Drug Striverdi”. FDA News/Drug Industry Daily.
- “New once-daily Striverdi (olodaterol) Respimat gains approval in first EU countries”. Boehringer-Ingelheim. 18 October 2013.
External links
The active moiety olodaterol is a selective beta2-adrenergic bronchodilator. The drug substance, olodaterol hydrochloride, is chemically described as 2H-1,4- Benzoxazin-3H(4H)-one, 6-hydroxy-8-[(1R)-1-hydroxy-2-[[2-(4-methoxyphenyl)-1,1-dimethylethyl]-amino]ethyl]-, monohydrochloride. Olodaterol hydrochloride is a white to off-white powder that is sparingly-slightly soluble in water and slightly soluble in ethanol. The molecular weight is 422.9 g/mole (salt): 386.5 g/mole (base), and the molecular formula is C21H26N2O5 x HCl as a hydrochloride. The conversion factor from salt to free base is 1.094.
The structural formula is:
|
The drug product, STRIVERDI RESPIMAT, is composed of a sterile, aqueous solution of olodaterol hydrochloride filled into a 4.5 mL plastic container crimped into an aluminum cylinder (STRIVERDI RESPIMAT cartridge) for use with the STRIVERDI RESPIMAT inhaler.
Excipients include water for injection, benzalkonium chloride, edetate disodium, and anhydrous citric acid. The STRIVERDI RESPIMAT cartridge is only intended for use with the STRIVERDI RESPIMAT inhaler. The STRIVERDI RESPIMAT inhaler is a hand held, pocket sized oral inhalation device that uses mechanical energy to generate a slow-moving aerosol cloud of medication from a metered volume of the drug solution. The STRIVERDI RESPIMAT inhaler has a yellow-colored cap.
When used with the STRIVERDI RESPIMAT inhaler, each cartridge containing a minimum of 4 grams of a sterile aqueous solution delivers the labeled number of metered actuations after preparation for use. Each dose (1 dose equals 2 actuations) from the STRIVERDI RESPIMAT inhaler delivers 5 mcg olodaterol in 22.1 mcL of solution from the mouthpiece. As with all inhaled drugs, the actual amount of drug delivered to the lung may depend on patient factors, such as the coordination between the actuation of the inhaler and inspiration through the delivery system. The duration of inspiration should be at least as long as the spray duration (1.5 seconds).
| WO2002030928A1 | 28 Sep 2001 | 11 Apr 2003 | Boehringer Ingelheim Pharma | Crystalline monohydrate, method for producing the same and the use thereof in the production of a medicament |
| WO2003000265A1 | 8 Jun 2002 | 3 Jan 2003 | Boehringer Ingelheim Pharma | Crystalline anticholinergic, method for its production, and use thereof in the production of a drug |
| WO2004045618A2 * | 11 Nov 2003 | 3 Jun 2004 | Boehringer Ingelheim Pharma | Novel medicaments for the treatment of chronic obstructive pulmonary diseases |
| EP0073505A1 * | 28 Aug 1982 | 9 Mar 1983 | Boehringer Ingelheim Kg | Benzo-heterocycles |
| EP0321864A2 * | 15 Dec 1988 | 28 Jun 1989 | Boehringer Ingelheim Kg | Ammonium compounds, their preparation and use |
| US4460581 | 12 Oct 1982 | 17 Jul 1984 | Boehringer Ingelheim Kg | Antispasmodic agents, antiallergens |
| US4656168 * | 13 Oct 1983 | 7 Apr 1987 | Merck & Co., Inc. | Vision defects; adrenergic blocking and hypotensive agents |
Organic spectroscopy should be brushed up and you get confidence
read my blog
ORGANIC SPECTROSCOPY INTERNATIONAL is the blog
Organic chemists from Industry and academics to interact on Spectroscopy techniques for Organic compounds ie NMR, MASS, IR, UV Etc. email me ……….. amcrasto@gmail.com
http://orgspectroscopyint.blogspot.in/ is the link
amcrasto@gmail.com
BI-836845 a fully human mAb targeting IGF-1 created using HuCAL technology from Morphosys, for the potential iv infusion treatment of cancer, including solid tumors and breast cancer.

BI-836845
Human monoclonal IgG1 lambda antibody against IGF-1 (insulin growth factor-1) and IGF-2
| IGF pathway modulator (iv, cancer), Boehringer Ingelheim; |
Phase 2 Clinical
Anticancer protein kinase inhibitor; Anticancer monoclonal antibody
WO-2008155387
Boehringer Ingelheim International Gmbh

Boehringer Ingelheim is developing BI-836845, a fully human mAb targeting IGF-1 created using HuCAL technology from Morphosys, for the potential iv infusion treatment of cancer, including solid tumors and breast cancer.
In April 2011, a phase I trial was initiated in the UK . In October 2011, another phase I trial was initiated in Taiwan. In February 2014, recruitment was ongoing. At that time, the trial was expected to be completed in March 2015 In June 2014, the drug was listed as being in phase I development for solid tumors in Japan and for breast cancer
In May 2014, an open-label, randomized, parallel-assigned, phase II trial (NCT02123823; 1280.4; 2013-001110-15) to evaluate the safety and efficacy of BI-836845 and everolimus in combination with exemestane in women with breast cancer (expected n = 198) was planned to be initiated in Belgium, France and the Netherlands. At that time, the trial was expected to complete in December 2017
In June 2014, an open-label, single-group assigned, phase I trial (NCT02145741; 1280.15) to evaluate BI-836845 in Japanese patients (expected n = 18) with advanced solid tumors was planned to be initiated in Japan. At that time, the trial was expected to complete in June 2015
In March 2011, a non-randomized, open-label, phase I study (NCT01317420; 1280.2; 2010-021714-29) was planned to begin later that month in patients with solid tumors (expected n = 70) in the UK, to assess the safety, efficacy, pharmacokinetics, pharmacodynamics and pharmacogenomics of BI-836845. The study began in April 2011; at that time, completion was expected in March 2013 .
In June 2012, preclinical data were presented at the 48th ASCO meeting in Chicago, IL. In the study, the combination of BI-836845 plus rapamycin was more effective than single agent therapy at inhibiting Ewing’s sarcoma cell proliferation in vitro and in a nude mouse xenograft model .

In November 2011, preclinical data were presented at the 23rd AACR-NCI-EORTC International Conference in San Francisco, CA. BI-836845 potently inhibited proliferation of the multiple myeloma cell line LP-1 with an EC50 of 0.4 nM.
BI-836845 is a human monoclonal IgG1 lambda antibody against IGF-1 (insulin growth factor-1) and IGF-2 (insulin growth factor-2). Phase II clinical trials are ongoing at Boehringer Ingelheim for the treatment of patients with breast cancer, and phase I clinical trials are ongoing with patients with advanced solid tumors.
Insulin-like growth factor-1 (IGF-1; a 70 amino-acid polypeptide) and insulin-like growth factor-2 (IGF-2; a 67 amino-acid polypeptide) are 7.5-kD soluble factors present in serum that can potently stimulate the growth of many mammalian cells (reviewed by Pollack et al., 2004). Although IGFs can be detectable in a number of tissues the main source of circulating IGFs is the liver which secretes the IGFs and IGF binding proteins (IGFBPs) in response to a complex signaling pathway that is initiated in the pituitary gland and transduced via growth hormone. On secretion into the bloodstream the IGFs form complexes with the IGFBPs which not only protects them from proteolytic degradation in the serum en route to their target tissues but also prevents their association with the IGF receptors. In addition to this endocrine source of IGFs they are also known to be secreted in an autocrine or paracrine manner in target tissues themselves. This is known to occur during normal fetal development where the IGFs play a key role in the growth of tissues, bone and organs. It is also seen in many cancer tissues where there is thought to be paracrine signaling between tumour cells and stromal cells or autocrine IGF production by the tumour cells themselves (reviewed by LeRoith D, 2003).
30 May 2014
MEDIA ALERT
ASCO 2014: Boehringer Ingelheim to present latest oncology research, including overall survival results
• Highly anticipated new overall survival data for Giotrif® (afatinib*) to be presented on June 2nd (3:00 – 6:00 PM, E Hall D2 [Abstract #8004 scheduled for 4:00 – 4:12 PM])
• 7 total abstracts accepted for Giotrif® (afatinib*), nintedanib** and BI 836845**: 1 for oral presentation and 6 posters
| BI 836845 (IGF ligand antibody)** | ||
| A Phase I dose escalation study of weekly BI 836845, a fully human, affinity-optimized, insulin-like growth factor (IGF) ligand neutralizing antibody, in patients with advanced solid cancers | Chia-Chi Lin, Kwang-Yu Chang, Dennis Chin-Lun Huang, Vicky Marriott, Ludy van Beijsterveldt, Li-Tzong Chen, Ann-Lii Cheng | Sunday, June 1 8:00 – 11:45 AM S Hall A2 (Abstract #2617 Poster #80) |
| Phase I dose escalation study of 3-weekly BI 836845, a fully human, affinity optimized, insulin-like growth factor (IGF) ligand neutralizing antibody, in patients with advanced solid tumours | Rihawi K, Ong M, Michalarea V, Bent L, Buschke S4, Bogenrieder T, Anthoney A, de Bono J, Twelves CJ | Sunday, June 1 8:00 – 11:45 AM S Hall A2 (Abstract #2622 Poster #85) |
The activity of the IGFs is thought to be regulated by a complex and relatively poorly understood interaction involving seven different IGFBPs and other serum proteins. Activation of the IGFs involves their release from this ternary complex after proteolytic release of the serum binding protein and IGFBPs, this is thought to occur in close proximity to cell surfaces where the IGFs are then free to bind to their receptors and transduce intracellular signals that ultimately leads to cellular proliferation and the inhibition of apoptosis. IGF-1 and IGF-2 are able to bind to the IGF-1 receptor (IGF-1R) expressed on many normal tissues, which functionally is a 460 kD heterotetramer consisting of a dimerised alpha- and beta-subunit, with similar affinities (Rubin et al., 1995). IGF-2 can also bind to the IGF-2 receptor (also know as the mannose-6-phosphate receptor) which does not have any known signaling function, rather it is thought to act as a sink for IGF-2 and prevent it from binding and signaling through the IGF-1R. In this respect the IGF-2R has been demonstrated to be a tumour suppressor protein. The IGF-1R is structurally similar to the insulin receptor which exists in two forms, IR-A and IR-B, which differ by an alternatively spliced 12 amino acid exon deletion in the extracellular domain of IR-A. IR-B is the predominant IR isoform expressed in most normal adult tissues where it acts to mediate the effects of insulin on metabolism. IR-A on the other hand is known to be highly expressed in developing fetal tissues but not in adult normal tissues. Recent studies have also shown that IR-A, but not IR-B, is highly expressed in some cancers. The exon deletion in IR-A has no impact on insulin binding but does cause a small conformational change that allows IGF-2 to bind with much higher affinity than for IR-B (Frasca et al., 1999; Pandini et al., 2002). Thus, because of it’s expression in cancer tissues and increase propensity for IGF-2 binding, IR-A may be as important as IGF1-R in mediating the mitogenic effects of IGF-2 in cancer.
Binding of the IGFs to IGF-1R triggers a complex intracellular signaling cascade which results in activation of proteins that stimulate growth and inhibit apoptosis (reviewed by Pollack et al., 2004). In terms of growth, upregulated translation is induced by the activation of p70 S6 kinase, which in turn phosphorylates the S6 ribosomal protein (Dufner and Thomas, 1999). Thus, IGF-stimulated cell growth can be measured by the rapid increase in phosphorylated S6 ribosomal protein.
Unlike the EGFR and Her2neu receptors there is no known amplification of the IGF1-R or IR-A receptors in cancers indicating that receptor activation is controlled by the presence of active ligand. There is a very large body of scientific, epidemiological and clinical literature implicating a role for the IGFs in the development, progression and metastasis of many different cancer types (reviewed by Jerome et al., 2003; and Pollack et al., 2004).
For example, in colorectal cancer the expression of IGF-2 mRNA and protein is elevated in clinical colorectal tumour specimens compared with adjacent normal tissue (Freier et al., 1999; Li et al., 2004). There is also a positive correlation of elevated IGF serum levels with proliferating cell index in patients with colorectal neoplasia (Zhao et al., 2005). In addition, elevated circulating levels of IGF-2 correlate with an increased risk of developing colorectal cancers and adenomas (Renehan et al., 2000a) and b); Hassan et al., 2000). Loss of parental imprinting (LOI) of the IGF-2 gene, an epigenetic alteration that results in elevated IGF-2 expression, is a heritable molecular trait that has recently been identified in patients with colorectal and other tumour types. Loss of IGF-2 imprinting has been shown to be associated with a five-fold risk of colorectal neoplasia (Cui et al., 2003; Cruz-Correa et al., 2004) and adenomas (Woodson et al., 2004). Antibodies targeting the alpha-subunit of the IGF-1R which block IGF binding and internalize the receptor have been shown to delay the growth of the xenografted colon cancer-derived cell lines such as COLO 205 (Burtrum et al., 2003).
Elevated levels of IGFs are associated with a poor prognosis in human pulmonary adenocarcinomas (Takanami et al., 1996) and IGFs are expressed and secreted by many SCLC— and NSCLC-derived cell lines (Quinn et al., 1996). Transgenic over-expression of IGF-2 induces spontaneous lung tumours in a murine model (Moorhead et al., 2003). In terms of hepatocellular carcinoma (HCC), human clinical specimens and animal models of HCC express higher levels of IGF mRNA and protein than corresponding normal tissues and this has been correlated with increased tumour growth (Wang et al., 2003; Ng et al., 1998). IGF-2 has also been shown to be a serological marker of HCC with elevated levels in the serum of HCC patients compared with controls (Tsai et al., 2005). An orthotopic xenograft tumour model of HCC was established using Hep 3B cells, and used to demonstrate that inhibition of IGF-2 expression using a methylated oligonucleotide enhances survival (Yao et al., 2003a) and b).
Many childhood solid tumours such as Ewing sarcoma and rhabdomyosarcoma appear to be particularly dependent on the IGF signaling pathway for their growth (Scotlandi et al., 1996). LOI of the IGF-2 gene has been implicated as a primary genetic event in the development for embryonal rhabdomyosarcoma (Fukuzawa et al., 1999). Autocrine IGF signaling is also thought to strongly influence the growth of Ewing sarcoma in cases where the type-1 EWS-FLI1 chimeric transcription factor is expressed through a chromosomal translocation resulting in elevated expression of target genes including the IGF ligands and IGF-1R, and reduced expression of IGFBP-3. Antibodies and small molecule compounds targeting the IGF-1R have been shown to reduce the growth of xenografted pediatric solid tumour derived cell lines (Kolb et al., 2008; Manara et al., 2007).
Using IGF ligand-specific antibodies it has been demonstrated that the growth of human prostate cancer cells in adult human bone implanted into SCID mice can be inhibited (Goya et al., 2004). In addition, it was demonstrated that the same IGF ligand antibodies could block the paracrine supply of IGF and suppress the liver metastasis of human colorectal cancer cells in a murine xenograft system (Miyamoto et al., 2005).
There is also considerable evidence suggesting that the IGF signaling system reduces the sensitivity of cancers to chemotherapeutic agents and radiation. One of the earliest findings in this respect was the demonstration that IGF-1R knock-out mouse embryos are refractory to transformation by viruses, oncogenes and over-expressed growth factor receptors (Sell et al., 1993; Sell et al., 1994) and that over-expression of IGF-1R protects cells from UV irradiation and gamma radiation-induced apoptosis (Kulik et al., 1997). Furthermore, using liver tumour cell lines that secrete large amounts of IGF-2, it was found that neutralization of IGF-2 significantly increased response to chemotherapeutic agents such as cisplatin and etoposide in vitro, especially at lower, cytostatic doses, suggesting that IGF-2 can reduce the susceptibility to chemotherapeutic agents (Lund et al., 2004). Consistent with these findings it has been demonstrated that antibodies targeting the IGF-1R increase the susceptibility of tumour xenografts to growth inhibition by chemotherapeutic drugs and radiation (Goetsch et al., 2005).
A number of antibodies that show cross-reactive binding to human IGF-1 and human IGF-2 have been reported. Antibody sm1. was raised against human IGF-1 and shows 40% cross-reactivity to human IGF-2 and was shown to inhibit the proliferation of a mouse fibroblast cell line BALB/c3T3 which was stimulated with 20 ng/ml human IGF-1 (Russell et al., 1984). In a study designed to functionally epitope map IGF-1 by raising monoclonal antibodies to whole IGF-1 protein and portions of the protein a number of antibodies where identified that cross reacted with IGF-2 (Manes et al., 1997). The percent cross-reactivity with IGF-2 ranged from 0 to 800% and several antibodies were identified which were equally IGF-1 and IGF-2 reactive. KM1486 is a rat monoclonal antibody that cross-reacts with human IGF-1 and IGF-2 and it was demonstrated that KM1486 can inhibit growth of human prostate cancer cells in human adult bone implanted into nonobese diabetic/severe combined immunodeficient mice (Goya et al., 2004). In addition, it was demonstrated that KM1486 suppresses the liver metastasis of human colorectal cancers (Miyamoto et al., 2005). KM1486 has also been described in WO 03/093317, JP 2003-310275, WO 2005/018671, WO 2005/028515, and WO 2005/027970.
For the treatment of human disease an antibody with a fully human sequence is highly desirable in order to minimize the risk of generating a human anti-antibody reaction and neutralizing antibodies that will rapidly eliminate the administered antibody from the body and thereby reduce the therapeutic effect. As such, and given the roles of IGF-1 and IGF-2 dependent signaling in the development and progression of cancers it would be desirable to obtain high affinity fully human antibodies that co-neutralise the mitogenic effects of both ligands.
In addition, to maximize the therapeutic potential of such an antibody, it is important to have a suitably long terminal half life (T1/2). Prior to terminal half life determination in human subjects, the most accurate estimation of an antibody’s human terminal half life can be obtained from administration to non-human primates such as cynomolgus monkeys. For example, bevacizumab, a registered humanized monoclonal antibody against vascular endothelial growth factor (VEGF) used for the treatment of several human cancers, has a terminal half-life in cynomolgus monkeys of 8.57±0.38 days (Lin et al., 1999), which translates to a terminal half life in humans of approximately 20 days allowing for a single administration once every two weeks (Lu et al., 2008).

It was a further object of the invention to obtain an antibody that does not affect binding of insulin to its receptor.
The clinical development of therapeutic agents is supported by pharmacodynamic biomarkers of drug activity. Clinical studies with antibodies targeting the IGF-1R have demonstrated that an increase in total serum IGF-1 levels may be a useful pharmacodynamic marker for these agents (Pollack et al., 2007). The reason for the increase in total serum IGF-1 levels is likely due to a feedback mechanism involving pituitary growth hormone (GH) secretion which releases both IGF-1 and IGFBPs from the liver. Indeed, in humans it has been demonstrated that free or bioactive IGF-1, which represents only around 1% of total IGF-1 levels, determines the feedback response (Chen et al., 2005). The inventors thus sought to confirm whether total serum IGF-1 levels are also a useful pharmacodynamic marker for the activity of a therapeutic anti-IGF antibody. In this case it would be desirable for such antibody to be cross-reactive with IGFs from a suitable animal species, e.g. mouse or rat, such that a pharmacodynamic effect can already be tested pre-clinically.

Boehringer Ingelheim
The Boehringer Ingelheim group is one of the world’s 20 leading pharmaceutical companies. Headquartered in Ingelheim, Germany, Boehringer Ingelheim operates globally with 142 affiliates and a total of more than 47,400 employees. The focus of the family-owned company, founded in 1885, is researching, developing, manufacturing and marketing new medications of high therapeutic value for human and veterinary medicine.
Taking social responsibility is an important element of the corporate culture at Boehringer Ingelheim. This includes worldwide involvement in social projects, such as the initiative “Making more Health” and caring for the employees. Respect, equal opportunities and reconciling career and family form the foundation of the mutual cooperation. In everything it does, the company focuses on environmental protection and sustainability.
In 2013, Boehringer Ingelheim achieved net sales of about 14.1 billion euros. R&D expenditure corresponds to 19.5% of its net sales.

Fig.1 Production of MAb
| Adam, P.J.; Friedbichler, K.; Hofmann, M.H.; Bogenrieder, T.; Borges, E.; Adolf, G.R. BI 836845, a fully human IGF ligand neutralizing antibody, to improve the efficacy of rapamycin by blocking rapamycin-induced AKT activation 48th Annu Meet Am Soc Clin Oncol (ASCO) (June 1-5, Chicago) 2012, Abst 3092 |
| Lin, C.-C.; Chang, K.-Y.; Huang, D.C.; Marriott, V.; Van Beijsterveldt, L.; Chen, L.-T.; Cheng, A.-L. A phase I dose escalation study of weekly BI 836845, a fully human, affinity-optimized, insulin-like growth factor (IGF) ligand neutralizing antibody, in patients with advanced solid cancers 50th Annu Meet Am Soc Clin Oncol (ASCO) (May 30-June 3, Chicago) 2014, Abst 2617 |
Adam, P.J.; Ostermann, E.; Lamche, H.R.; Hofmann, M.H.; Kroez, M.; Borges, E.; Adolf, G.R.
Pharmacodynamic properties and anti-tumor efficacy of BI 836845, a fully human IGF ligand neutralizing antibody
AACR-NCI-EORTC Int Conf Mol Targets Cancer Ther (November 12-16, San Francisco) 2011, Abst A208
| Rihawi, K.; Ong, M.; Michalarea, V.; et al. Phase I dose escalation study of 3-weekly BI 836845, a fully human, affinity optimized, insulin-like growth factor (IGF) ligand neutralizing antibody, in patients with advanced solid tumors 50th Annu Meet Am Soc Clin Oncol (ASCO) (May 30-June 3, Chicago) 2014, Abst 2622 |
Boehringer-Ingelheim …A Well-Balanced Pipeline
Promising Drugs in Boehringer-Ingelheim Pipeline
| Compound* | Clinical phase | Indication | Therapeutic principle | Mode of action |
|---|---|---|---|---|
| Olodaterol | Submitted | Chronic obstructive pulmonary Disease (COPD) | Long-acting beta-agonist | Bronchodilation |
| Tiotropium | Submitted | Cystic fibrosis (CF) | Bronchodilatator | Long Acting Muscarinic Antagonist |
| Afatinib | Phase III | Breast cancer | Signal transduction inhibition | Novel irreversible ErbB Family blocker |
| Afatinib | Phase III | Head and neck cancer | Signal transduction inhibition | Novel irreversible ErbB Family blocker |
| Deleobuvir (BI 207127) |
Phase III | Hepatitis C | Direct acting antiviral small molecule | Oral NS5B RNA-dependent polymerase inhibitor |
| Empagliflozin | Phase III | Diabetes mellitus type II |
SGLT-2-inhibitor | Inhibition of glucose transporter-2 |
| Faldaprevir (BI 201335) |
Phase III | Hepatitis C | Direct acting antiviral small molecule | Oral HCV NS3/4A protease inhibitor |
| Nintedanib | Phase III | Non-small cell lung cancer (NSCLC) | Angiogenesis inhibition | Triple angiokinase inhibitor, simultaneously blocks VEGFR, FGFR, PDGFR |
| Nintedanib | Phase III | Ovarian cancer | Angiogenesis inhibition | Triple angiokinase inhibitor, simultaneously blocks VEGFR, FGFR, PDGFR |
| Nintedanib | Phase III | Idiopathic pulmonary fibrosis (IPF) | Anti-fibrotic kinase inhibition | Anti-fibrotic kinase inhibitor |
| Tiotropium | Phase III | Asthma | Bronchodilatator | Long Acting Muscarinic Antagonist |
| Volasertib | Phase III | Various cancer types | Cell-cycle kinase inhibition | PLK-1 antagonist |
* These are investigational agents; their safety and efficacy have not yet been established.
Status: April 2013
Successful Products from our Boehringer-Ingelheim Research & Development
| Product name | First launch | Active ingredient | Indication |
|---|---|---|---|
| Gilotrif™ | 2013 | Afatinib | Non-small cell lung cancer (NSCLC) |
| Trajenta® | 2011 | Linagliptin | Diabetes mellitus type II |
| Pradaxa® | 2010 2008 |
Dabigatran etexilate | Stroke prevention in atrial fibrillationPrevention of venous thromboembolic events (VTE) in adults |
| Spiriva® Respimat Soft Mist™ InhalerSpiriva® |
2007 2002 |
Tiotropium | COPD |
| Micardis® | 1998 | Telmisartan | Essential hypertension |
| Sifrol® / Mirapex® /Mirapexin® | 20061997 | Pramipexole | Restless legs syndrome (RLS) Parkinson’s disease (PD) |
| Viramune® | 1996 | Nevirapine | HIV/AIDS |


Oncology Websites
No flash player detected. For optimized usage of this website your browser should support shockwave flash. For downloading see Macromedia Flash Player
US FDA grants breakthrough therapy designation to Boehringer Ingelheim’s volasertib to treat patients with AML

Volasertib
755038-65-4
CHEMICAL NAMES
1. Benzamide, N-[trans-4-[4-(cyclopropylmethyl)-1-piperazinyl]cyclohexyl]-4-[[(7R)-7-
ethyl-5,6,7,8-tetrahydro-5-methyl-8-(1-methylethyl)-6-oxo-2-pteridinyl]amino]-3-
methoxy-
2. N-{trans-4-[4-(cyclopropylmethyl)piperazin-1-yl]cyclohexyl}-4-{[(7R)-7-ethyl-5-methyl-8-
(1-methylethyl)-6-oxo-5,6,7,8-tetrahydropteridin-2-yl]amino}-3-methoxybenzamide
CODE DESIGNATION BI 6727
| Ingelheim, Germany Thursday, September 19, 2013, 16:00 Hrs [IST] |
|
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation to Boehringer Ingelheim’s volasertib, a selective and potent polo-like kinase (Plk) inhibitor, for the treatment of patients with acute myeloid leukaemia (AML), a type of blood cancer. |
http://www.pharmabiz.com/NewsDetails.aspx?aid=77733&sid=2

Volasertib (also known as BI 6727) is a small molecule inhibitor of the PLK1 (polo-like kinase 1) protein being developed byBoehringer Ingelheim for use as an anti-cancer agent. Volasertib is the second in a novel class of drugs called dihydropteridinone derivatives.[1]
Mechanism of action
Volasertib is a novel small-molecule targeted therapy that blocks cell division by competitively binding to the ATP-binding pocket of the PLK1 protein. PLK1 proteins are found in the nuclei of all dividing cells and control multiple stages of the cell cycle and cell division.[2] [3] [4] The levels of the PLK1 protein are tightly controlled and are raised in normal cells that are dividing. Raised levels of the PLK1 protein are also found in many cancers including; breast, non-small cell lung, colorectal, prostate, pancreatic, papillary thyroid, ovarian, head and neck and Non-Hodgkin’s Lymphoma.[5] [3] [6] [4] [7] [8] Raised levels of PLK1 increase the probability of improper segregation of chromosomes which is a critical stage in the development of many cancers. Raised levels of PLK1 have been associated with a poorer prognosis and overall survival in some cancers[4][9] [10] In addition to its role in cell division, there is evidence that PLK1 also interacts with components of other pathways involved in cancer development including the K-Ras oncogene and the retinoblastoma and p53 tumour suppressors[11] These observations have led to PLK1 being recognised as an important target in the treatment of cancer.
Volasertib can be taken either orally or via intravenous infusion, once circulating in the blood stream it is distributed throughout the body, crosses the cell membrane and enters the nucleus of cells where it binds to its target; PLK1. Volasertib inhibits PLK1 preventing its roles in the cell-cycle and cell division which leads to cell arrest and programmed cell death.[2] Volasertib binds to and inhibits PLK1 at nanomolar doses however, it has also been shown to inhibit other PLK family members; PLK2 and PLK3 at higher; micromolar doses. The roles of PLK2 and PLK3 are less well understood; however they are known to be active during the cell cycle and cell division.[12]
Volasertib inhibits PLK1 in both cancer and normal cells; however it only causes irreversible inhibition and cell death in cancer cells, because inhibition of PLK1 in cancer cells arrests the cell cycle at a different point to normal, non-cancer cells. In cancer cells PLK1 inhibition results in G2/M cell cycle arrest followed by programmed cell death, however, in normal cells inhibition of PLK1 only causes temporary, reversible G1 and G2 arrest without programmed cell death.[13] This specificity for cancer cells improves the efficacy of the drug and minimizes the drug related toxicity.
Clinical uses
Volasertib is currently undergoing investigation in phase 1 and 2 trials and has yet to be licensed by the FDA. Volasertib may be effective in several malignancies evidenced by the fact that its target PLK1 is overexpressed in up to 80% of malignancies, where it has been associated with a poorer treatment outcome and reduced overall survival.[1][4][9]Further phase 1 and 2 trials are active, investigating the effects of Volasertib both as a single agent and in combination with other agents in solid tumours and haematological malignancies including; ovarian cancer, urothelial cancer and acute myeloid leukaemia.[14]
Studies
Preclinical studies on volasertib have demonstrated that it is highly effective at binding to and blocking PLK1 function and causing programmed cell death in colon and non-small cell lung cancer cells both in vitro and in vivo. Volasertib can also cause cell death in cancer cells that have are no longer sensitive to existing anti-mitotic drugs such as vinca alkaloids and taxanes.[13] This suggests that volasertib may be effective when used as a second line treatment in patients who have developed resistance to vinca alkaloid and taxane chemotherapeutics.
A first in man trial of volasertib in 65 patients with solid cancers reported that the drug is safe to administer to patients and is stable in the bloodstream. This study also reported favourable anti-cancer activity of the drug; three patients achieved a partial response, 48% of patients achieved stable disease and 6 patients achieved progression free survival of greater than 6 months.[15] A further phase 1 trial of volasertib in combination with cytarabine in patients with relapsed / refractory acute myeloid leukaemiareported that 5 of 28 patients underwent a complete response, 2 achieved a partial response and a further 6 patients no worsening of their disease.[16]
- Schoffski, P. (2009). “Polo-like kinase (PLK) inhibitors in preclinical and early clinical development in oncology”. Oncologist 14 (6): 559–70. ISSN (Electronic) 1083-7159 (Linking) 1549-490X (Electronic) 1083-7159 (Linking).
- Barr, F. A.; H. H. Sillje, E. A. Nigg (2004). “Polo-like kinases and the orchestration of cell division”. Nat Rev Mol Cell Biol 5 (6): 429–40. ISSN (Print) 1471-0072 (Linking) 1471-0072 (Print) 1471-0072 (Linking).
- Garland, L. L.; C. Taylor, D. L. Pilkington, J. L. Cohen, D. D. Von Hoff (2006). “A phase I pharmacokinetic study of HMN-214, a novel oral stilbene derivative with polo-like kinase-1-interacting properties, in patients with advanced solid tumors”. Clin Cancer Res 12 (17): 5182–9. ISSN (Print) 1078-0432 (Linking) 1078-0432 (Print) 1078-0432 (Linking).
- Santamaria, A.; R. Neef, U. Eberspacher, K. Eis, M. Husemann, D. Mumberg, S. Prechtl, V. Schulze, G. Siemeister, L. Wortmann, F. A. Barr, E. A. Nigg (2007). “Use of the novel Plk1 inhibitor ZK-thiazolidinone to elucidate functions of Plk1 in early and late stages of mitosis”. Mol Biol Cell 18 (10): 4024–36. ISSN (Print) 1059-1524 (Linking) 1059-1524 (Print) 1059-1524 (Linking).
- Fisher, R.A.H.; D.K. Ferris (2002). “The functions of Polo-like kinases and their relevance to human disease.”. Curr Med Chem 2: 125–134.
- Holtrich, U.; G. Wolf, A. Brauninger, T. Karn, B. Bohme, H. Rubsamen-Waigmann, K. Strebhardt (1994). “Induction and down-regulation of PLK, a human serine/threonine kinase expressed in proliferating cells and tumors”. Proc Natl Acad Sci U S A 91 (5): 1736–40. doi:10.1073/pnas.91.5.1736. ISSN (Print) 0027-8424 (Linking) 0027-8424 (Print) 0027-8424 (Linking). PMC 43238. PMID 8127874.
- Steegmaier, M.; M. Hoffmann, A. Baum, P. Lenart, M. Petronczki, M. Krssak, U. Gurtler, P. Garin-Chesa, S. Lieb, J. Quant, M. Grauert, G. R. Adolf, N. Kraut, J. M. Peters, W. J. Rettig (2007). “BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo”. Curr Biol 17 (4): 316–22. doi:10.1016/j.cub.2006.12.037. ISSN (Print) 0960-9822 (Linking) 0960-9822 (Print) 0960-9822 (Linking). PMID 17291758.
- Winkles, J. A.; G. F. Alberts (2005). “Differential regulation of polo-like kinase 1, 2, 3, and 4 gene expression in mammalian cells and tissues”. Oncogene 24 (2): 260–6.doi:10.1038/sj.onc.1208219. ISSN (Print) 0950-9232 (Linking) 0950-9232 (Print) 0950-9232 (Linking). PMID 15640841.
- Eckerdt, F.; J. Yuan, K. Strebhardt (2005). “Polo-like kinases and oncogenesis”. Oncogene 24 (2): 267–76. doi:10.1038/sj.onc.1208273. ISSN (Print) 0950-9232 (Linking) 0950-9232 (Print) 0950-9232 (Linking). PMID 15640842.
- Weichert, W.; A. Ullrich, M. Schmidt, V. Gekeler, A. Noske, S. Niesporek, A. C. Buckendahl, M. Dietel, C. Denkert (2006). “Expression patterns of polo-like kinase 1 in human gastric cancer”. Cancer Sci 97 (4): 271–6. ISSN (Print) 1347-9032 (Linking) 1347-9032 (Print) 1347-9032 (Linking).
- Liu, X.; R. L. Erikson (2003). “Polo-like kinase (Plk)1 depletion induces apoptosis in cancer cells”. Proc Natl Acad Sci U S A 100 (10): 5789–94. doi:10.1073/pnas.1031523100.ISSN (Print) 0027-8424 (Linking) 0027-8424 (Print) 0027-8424 (Linking). PMC 156279. PMID 12732729.
- Schmit, T. L.; N. Ahmad (2007). “Regulation of mitosis via mitotic kinases: new opportunities for cancer management”. Mol Cancer Ther 6 (7): 1920–31. ISSN (Print) 1535-7163 (Linking) 1535-7163 (Print) 1535-7163 (Linking).
- Rudolph, D.; M. Steegmaier, M. Hoffmann, M. Grauert, A. Baum, J. Quant, C. Haslinger, P. Garin-Chesa, G. R. Adolf (2009). “BI 6727, a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity”. Clin Cancer Res 15 (9): 3094–102. ISSN (Print) 1078-0432 (Linking) 1078-0432 (Print) 1078-0432 (Linking).
- ClinicalTrials.gov (2011). “Clinical Trials.gov Search of: Volasertib”. Missing or empty
|url=(help) - Gil, T.; P. Schöffski, A. Awada, H. Dumez, S. Bartholomeus, J. Selleslach, M. Taton, H. Fritsch, P. Glomb, Munzert G.M. (2010). “Final analysis of a phase I single dose-escalation study of the novel polo-like kinase 1 inhibitor BI 6727 in patients with advanced solid tumors”. J Clin Oncol 28.
- Bug, G.; R. F. Schlenk, C. Müller-Tidow, M. Lübbert, A. Krämer, F. Fleischer, T. Taube, O. G. Ottmann, H. Doehner (2010). “Phase I/II Study of BI 6727 (volasertib), An Intravenous Polo-Like Kinase-1 (Plk1) Inhibitor, In Patients with Acute Myeloid Leukemia (AML): Results of the Dose Finding for BI 6727 In Combination with Low-Dose Cytarabine”. 52nd ASH Annual Meeting and Exposition. Orange County Convention Centre, Florida: American Society of Haematology.
VOLASERTIB TRIHYDROCHLORIDE
CHEMICAL NAMES
1. Benzamide, N-[trans-4-[4-(cyclopropylmethyl)-1-piperazinyl]cyclohexyl]-4-[[(7R)-7-
ethyl-5,6,7,8-tetrahydro-5-methyl-8-(1-methylethyl)-6-oxo-2-pteridinyl]amino]-3-
methoxy-, hydrochloride (1:3)
2. N-{trans-4-[4-(cyclopropylmethyl)piperazin-1-yl]cyclohexyl}-4-{[(7R)-7-ethyl-5-methyl-8-
(1-methylethyl)-6-oxo-5,6,7,8-tetrahydropteridin-2-yl]amino}-3-methoxybenzamide
trihydrochloride
MOLECULAR FORMULA C34H50N8O3 . 3 HCl
MOLECULAR WEIGHT 728.2
SPONSOR Boehringer Ingelheim Pharmaceuticals, Inc.
CODE DESIGNATION BI 6727 CL3
CAS REGISTRY NUMBER 946161-17-7
Volasertib is a highly potent and selective inhibitor of the serine-threonine Polo like kinase 1 (Plk1), a key regulator of cell-cycle progression. Volasertib is a dihydropteridinone derivative with distinct pharmacokinetic (PK) properties. The problem underlying this invention was to develop improved dosage schedules for combination therapy of advanced and/or metastatic solid tumours.
Volasertib (I) is known as the compound N-[trans-4-[4-(cyclopropylmethyl)-1-piperazinyl]cyclohexyl]-4-[[(7R)-7-ethyl-5,6,7,8-tetrahydro-5-methyl-8-(1-methylethyl)-6-oxo-2-pteridinyl]amino]-3-methoxy-benzamide,

This compound is disclosed in WO 04/076454. Furthermore, trihydrochloride salt forms and hydrates thereof are known from WO 07/090844. They possess properties which make those forms especially suitable for pharmaceutical use. The above mentioned patent applications further disclose the use of this compound or its monoethanesulfonate salt for the preparation of pharmaceutical compositions intended especially for the treatment of diseases characterized by excessive or abnormal cell proliferation.
U.S. 8,188,086
Several dihydropteridione derivatives effectively prevent cell proliferation. G. Linz and co-inventors report a comprehensive method for preparing pharmacologically active crystalline and anhydrous forms of compound 1 (Figure 1) that are suitable for drug formulations.

The inventors list several criteria for the properties of 1 and its manufacturing procedure:
- favorable bulk characteristics such as drying times, filterability, solubility in biologically acceptable solvents, and thermal stability;
- purity of the pharmaceutical composition;
- low hygroscopicity;
- no or low tendency toward polymorphism; and
- scalability to a convenient commercial process.
They describe their finding that the tri-HCl salt of 1 satisfies these criteria as “surprising”.
Free base 1 is prepared by condensing cyclopropylmethylpiperazine derivative 2 with pteridinone 3 in the presence of p-toluenesulfonic acid (TsOH), as shown in Figure 1. After the reaction is complete, the crude free base 1 is recovered as a viscous oil. It is then treated with HCl in an organic solvent to form 1·3HCl, isolated in 91% yield. Alternatively, the free base is not isolated; instead, concd HCl is added to the reaction mixture, followed by acetone. The crude salt is recovered in 92% yield.
The salt is purified by crystallization from refluxing EtOH, adding water, and cooling to precipitate the crystals. The inventors do not report the purity of this or any other reaction product.
The inventors obtained a hydrated form of the tri-HCl salt by dissolving the free base in EtOH at room temperature, followed by adding concd HCl and cooling to 2 °C. An anhydrous form can be recovered by drying the hydrate at 130 °C. The solubility of the hydrated salt in aqueous and organic media is reported, as are X-ray diffraction data for the hydrated form. The hydrated salt has good solid-state stability.
The patent also contains the syntheses of reactants 2 and 3 (Figures 2 and 3). The preparation of 2 begins with the formation of amide 7. Acid 4 is treated with SOCl2–DMF to form acid chloride 5; the crude product is added to a suspension of chiral difunctionalized cyclohexane 6 in THF and aq K2CO3 to produce 7. The crude product is recovered in 98% yield and oxidized to 8 with RuCl3 and N-methylmorpholine N-oxide (NMMO) in 91% yield.

Amide 8 reacts with cyclopropylmethylpiperazine 9 in the presence of methanesulfonic acid (MsOH). The solvent is evaporated, and the reaction mixture is treated with NaBH4. After further workup, product 10 is isolated in 46% yield. The nitro group is then hydrogenated over Raney Ni to give 2 in 90% yield. An alternative method for preparing10 is also described.
To prepare 3, readily available amino acid 11 is esterified and alkylated to form 12. In a multistep, one-pot procedure, 11 is first treated with HC(OMe)3 and SOCl2. Further reaction with NaBH(OAc)3, acetone, and NH4OH produces 12 as its HCl salt in 90% yield. The salt is treated with aq NaOH to form the free base, which reacts with pyrimidine 13 in the presence of NaHCO3 to form 14 in 79% isolated yield.

The pteridinone system is formed by hydrogenating 14 over a Pt/C catalyst in the presence of V(acac)3. Precursor 15 is recovered in 90% yield and methylated with (MeO)2CO and K2CO3 to give 3 in 82% isolated yield.
The inventors succeeded in developing a route for making a crystalline salt that is suitable for preparing pharmaceutical formulations. The many synthetic steps, however, use a large number of solvents that are frequently evaporated to dryness. [This observation implies that the processes have a significant environmental burden. —Ed.] (Boehringer Ingelheim International [Ingelheim am Rhein, Germany]. US Patent U.S. 8,188,086,
Phase 3 Boehringer Ingelheim Announces Interim Results Evaluating Virologic Response Rates in HCV/HIV Co-Infected Patients Treated with Faldaprevir

Faldaprevir
BI 201335
http://clinicaltrials.gov/ct2/show/NCT01343888
CAS Number: 801283-95-4
Molecular Formula: C40H49BrN6O9S
Molecular Weight: 869.82 g.mol
RIDGEFIELD, Conn., March 4, 2013
Boehringer Ingelheim Pharmaceuticals, Inc. announced the first interim results in HCV/HIV co-infected patients from the company’s ongoing hepatitis C (HCV) clinical trial program, HCVerso™. These results, from the Phase 3 trial STARTVerso™ 4, were presented today at the 20th annual Conference on Retroviruses and Opportunistic Infections (CROI) in Atlanta, GA.
The interim results showed that 80% of HCV/HIV co-infected patients achieved early treatment success (ETS)*, as defined by the study protocol, when given an investigational HCV regimen that included faldaprevir (BI 201335). Results were consistent across patients regardless of HIV therapy or prior HCV treatment status, including patients who were HCV treatment-naive or had previously relapsed during HCV treatment with pegylated interferon and ribavirin (PegIFN/RBV). Patients who achieved ETS were eligible for randomization to a shortened duration of treatment (24 weeks versus 48 weeks). Investigators also reported on-treatment virologic response at week 12, which showed that 84% of all study patients had undetectable levels of hepatitis C virus.
For more information, please visit http://us.boehringer-ingelheim.com

Faldaprevir, also known as BI 201335, is an investigational, oral protease inhibitor that is specifically designed to target viral replication in the liver. The ongoing multi-study Phase 3 STARTVerso™ trial program, evaluating faldaprevir combined with PegIFN/RBV in treatment-naive, treatment-experienced and HIV co-infected patients with chronic genotype-1 HCV, is near clinical completion. BI 207127 is an investigational NS5B non-nucleoside polymerase inhibitor that has shown the potential to eliminate interferon from HCV treatment when combined in a regimen with faldaprevir and RBV. Phase 2 trials of this interferon-free regimen have been completed and Phase 3 HCVerso™ trials investigating this regimen are now underway.
Faldaprevir and BI 207127 are investigational compounds and not approved by the FDA. Their safety and efficacy have not been established.
Hepatitis C is a blood-born infectious disease and a leading cause of chronic liver disease,transplant and failure that affects as many as 150 million people globally. In the United States, an estimated 4.1 million Americans have been infected with HCV, of which approximately 3.2 million have chronic HCV infection. Since 1999 there has been a significant increase in deaths due to chronic HCV, accounting for 15,000 deaths in the United States in 2007.
