PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
BMS-986115 has been used in trials studying the treatment of Various Advanced Cancer.
Varegacestat is an orally bioavailable, gamma secretase (GS) and pan-Notch inhibitor, with potential antineoplastic activity. Upon administration, varegacestat binds to GS and blocks the proteolytic cleavage and release of the Notch intracellular domain (NICD), which would normally follow ligand binding to the extracellular domain of the Notch receptor. This prevents both the subsequent translocation of NICD to the nucleus to form a transcription factor complex and the expression of Notch-regulated genes. This results in the induction of apoptosis and the inhibition of growth of tumor cells that overexpress Notch. Overexpression of the Notch signaling pathway plays an important role in tumor cell proliferation and survival. The integral membrane protein GS is a multi-subunit protease complex that cleaves single-pass transmembrane proteins, such as Notch receptors, at residues within their transmembrane domains and leads to their activation
AL 102 (previously known as BMS 986115), was developed as an orally active a gamma-secretase and pan-Notch inhibitor. The drug participated in phase I clinical trials in solid tumor patients. The drug was safe and well-tolerated and stabilized disease for more than six months in 14% of patients, however, Bristol-Myers Squibb terminated the study because of the changes in the business objectives. Ayala, an Israeli biotech company, licensed rights for the development of AL 102 from Bristol-Myers Squibb. In December 2018, Ayala in collaborating with Novartis decided to investigate AL102 for treatment of multiple myeloma. Ayala studied AL102, an inhibitor of the Notch pathway, in blood cancers. It is known that the pathway regulates cell-fate determination during development and maintains adult tissue balance. Cumulative evidence indicates that Notch is overactive in multiple myeloma and participates in its onset and progression.
In a 100 mL round-bottomed flask, a solution of Intermediate B-1 (1683 mg, 5.94 mmol), Et3N (1.656 mL, 11.88 mmol), and Intermediate S-1 in DMF (20 mL) was treated with o-benzotriazol-1-yl-N,N,N′,N′-tetramethyluronium tetrafluoroborate (3815 mg, 11.88 mmol) and stirred at room temperature for 1 hour. The reaction mixture was diluted with water and saturated aqueous NaHCO3. An off white precipitate formed and was filtered and washed with water. The resulting solid was dried on the filter under a stream of nitrogen to give Intermediate 1A (3.7 g, 99% yield). MS (ES): m/z=632.4[M+H+]; HPLC: RT=3.635 min Purity=98%. (H2O/MeOH with TFA, CHROMOLITH® ODS S5 4.6×50 mm, gradient=4 min, wavelength=220 nm). 1H NMR (400 MHz, methanol-d4) δ 7.53 (t, J=4.5 Hz, 1H), 7.46-7.30 (m, 3H), 7.28-7.23 (m, 1H), 7.23-7.18 (m, 2H), 5.37 (s, 1H), 2.88 (td, J=10.4, 3.4Hz, 1H), 2.60 (td, J=10.2, 4.1 Hz, 1H), 2.54-2.40 (m, 1H), 2.47 (s, 3H), 2.33-2.12 (m, 3H), 1.98-1.69 (m, 4H), 1.51 (s, 9H).
In a 250 mL round-bottomed flask, a solution of Intermediate 1A (3.7 g, 5.86 mmol) in DCM (25 mL) was treated with TFA (25 mL) and the resulting pale orange solution was stirred at room temperature for 1.5 hours. The reaction mixture was then concentrated to give Intermediate 1B. HPLC: RT=3.12 min (H2O/MeOH with TFA, CHROMOLITH® ODS S5 4.6×50 mm, gradient=4 min, wavelength=220 nm). MS (ES): m/z=576.3 (M+H)+. 1H NMR (400 MHz, methanol-d4) δ 7.54 (t, J=4.5 Hz, 1H), 7.49-7.29 (m, 3H), 7.28-7.15 (m, 3H), 5.38 (br. s., 1H), 2.89 (td, J=10.3, 3.7 Hz, 1H), 2.67 (td, J=9.9, 4.2Hz, 1H), 2.56-2.38 (m, 1H), 2.48 (s, 3H), 2.34-2.13 (m, 3H), 2.00-1.71 (m, 4H).
Example 1
In a 250 mL round-bottomed flask, a solution of Intermediate 1B (4.04 g, 5.86 mmol) in THF (50 mL) was treated with ammonia (2M in iPrOH) (26.4 mL, 52.7 mmol), followed by HOBT (1.795 g, 11.72 mmol) and EDC (2.246 g, 11.72 mmol). The resulting white suspension was stirred at room temperature overnight. The reaction mixture was diluted with water and saturated aqueous NaHCO3. The resulting solid was filtered, rinsed with water and then dried on the filter under a stream of nitrogen. The crude product was suspended in 20 mL of iPrOH and stirred at room temperature for 20 min and then filtered and washed with iPrOH and dried under vacuum to give 2.83 g of solid. The solid was dissolved in refluxing EtOH (100 mL) and slowly treated with 200 mg activated charcoal added in small portions. The hot mixture was filtered through CELITE® and rinsed with hot EtOH. The filtrate was reduced to half volume, allowed to cool and the white precipitate formed was filtered and rinsed with EtOH to give 2.57 g of white solid. A second recrystallization from EtOH (70 mL) afforded Example 1 (2.39 g, 70% yield) as a white solid. HPLC: RT=10.859 min (H2O/CH3CN with TFA, Sunfire C18 3.5 μm, 3.0×150 mm, gradient=15 min, wavelength=220 and 254 nm); MS (ES): m/z=575.3 [M+H+]; 1H NMR (400 MHz, methanol-d4) δ 7.57-7.50 (m, 1H), 7.47-7.30 (m, 3H), 7.29-7.15 (m, 3H), 5.38 (s, 1H), 2.85-2.75 (m, 1H), 2.59 (td, J=10.5, 4.0 Hz, 1H), 2.53-2.41 (m, 4H), 2.31-2.10 (m, 3H), 1.96-1.70 (m, 4H).
[00180] To a cold (-25 °C) stirred solution of 2,6-lutidine (18.38 mL, 158 mmol) in DCM (120 mL) was added Tf20 (24.88 mL, 147 mmol) over 3 min, and the mixture was stirred for 5 min. To the reaction mixture was added 3,3,3-trifluoropropan-l-ol (12 g, 105 mmol) over an interval of 3 min. After 2 hr, the reaction mixture was warmed to room temperature and stirred for 1 hr. The reaction mixture was concentrated to half its volume, then purified by loading directly on a silica gel column (330g ISCO) and the product was eluted with DCM to afford Intermediate S-IA (13.74 g, 53%) as a colorless oil. 1H NMR (400 MHz, CDC13) δ ppm 4.71 (2 H, t, J= 6.15 Hz), 2.49-2.86 (2 H, m).
[00181] To a stirring solution of 5,5,5-trifluoropentanoic acid (14.76 g, 95 mmol) and DMF (0.146 rriL) in DCM (50 mL) was slowly added oxalyl chloride (8.27 mL, 95 mmol). After 2h, the mixture was concentrated to dryness. A separate flask was changed with (S)-4-benzyloxazolidin-2-one (16.75 g, 95 mmol) in THF (100 mL) and then cooled to -78 °C. To the solution was slowly added n-BuLi (2.5M, 37.8 mL, 95 mmol) over 10 min, stirred for 10 min, and then a solution of the above acid chloride in THF (50 mL) was slowly added over 5 min. The mixture was stirred for 30 min, and then warmed to room temperature. The reaction was quenched with sat aq NH4C1. Next, 10% aq LiCl was then added to the mixture, and the mixture was extracted with Et20. The organic layer was washed with sat aq NaHC03 then with brine, dried (MgSC^), filtered and concentrated to dryness. The residue was purified by Si02 chromatography (ISCO, 330 g column, eluting with a gradient from 100% hexane to 100% EtOAc) to afford the product Intermediate S-IB; (25.25 g, 85%): 1H NMR (400 MHz, CDC13) δ ppm 7.32-7.39 (2 H, m), 7.30 (1 H, d, J= 7.05 Hz), 7.18-7.25 (2 H, m), 4.64-4.74 (1 H, m), 4.17-4.27 (2 H, m), 3.31 (1 H, dd, J= 13.35, 3.27 Hz), 3.00-3.11 (2 H, m), 2.79 (1 H, dd, J= 13.35, 9.57 Hz), 2.16-2.28 (2 H, m), 1.93-2.04 (2 H, m).
[00182] To a cold (-78 °C), stirred solution of Intermediate S-IB (3.03 g, 9.61 mmol) in THF (20 mL) was added NaHMDS (1.0M in THF) (10.6 mL, 10.60 mmol) under a nitrogen atmosphere. After 2 hours, tert-butyl 2-bromoacetate (5.62 g, 28.8 mmol) was added neat via syringe at -78 °C and stirring was maintained at the same temperature. After 6 hours, the reaction mixture was warmed to room temperature. The reaction mixture was partitioned between saturated NH4C1 and EtOAc. The organic phase was separated, and the aqueous phase was extracted with EtOAc (3x). The combined organics were washed with brine, dried (Na2s04), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (Teledyne ISCO
CombiFlash Rf, 5% to 100% solvent A/B = hexanes/EtOAc, REDISEP® Si02 120g). Concentration of the appropriate fractions provided Intermediate S-1C (2.79 g, 67.6%) as a colorless viscous oil: 1H NMR (400 MHz, CDC13) δ ppm 7.34 (2 H, d, J= 7.30 Hz), 7.24-7.32 (3 H, m), 4.62-4.75 (1 H, m, J= 10.17, 6.89, 3.43, 3.43 Hz), 4.15-4.25 (3 H, m), 3.35 (1 H, dd, J= 13.60, 3.27 Hz), 2.84 (1 H, dd, J= 16.62, 9.57 Hz), 2.75 (1 H, dd, J = 13.35, 10.07 Hz), 2.47 (1 H, dd, J= 16.62, 4.78 Hz), 2.11-2.23 (2 H, m), 1.90-2.02 (1 H, m), 1.72-1.84 (1 H, m), 1.44 (9 H, s).
[00183] To a cool (0 °C), stirred solution of Intermediate S-1C (2.17 g, 5.05 mmol) in THF (50 mL) and water (15 mL) was added a solution of LiOH (0.242 g, 10.11 mmol) and H202 (2.065 mL, 20.21 mmol) in H20 (2 mL). After 10 min, the reaction mixture was removed from the ice bath, stirred for lh, and then cooled to 0 °C. Saturated aqueous NaHCC”3 (25 mL) and saturated aqueous Na2s03 (25 mL) were added to the reaction mixture, and the mixture was stirred for 10 min, and then partially concentrated. The resulting mixture was extracted with DCM (2x), cooled with ice and made acidic with cone. HC1 to pH 3. The mixture was saturated with solid NaCl, extracted with EtOAc (3x), and then dried over MgS04, filtered and concentrated to a colorless oil to afford Intermediate S-ID, 1.2514g, 92%): 1H NMR (400 MHz, CDCI3) δ ppm 2.83-2.95 (1 H, m), 2.62-2.74 (1 H, m), 2.45 (1 H, dd, J= 16.62, 5.79 Hz), 2.15-2.27 (2 H, m), 1.88-2.00 (1 H, m), 1.75-1.88 (1 H, m), 1.45 (9 H, s). Intermediate S-l : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and Intermediate S-1E: (2R,3R)-3-(tert-butoxycarbonyl)- 6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid
(S-1E)
[00184] To a cold (-78 °C) stirred solution of Intermediate S-1D (5 g, 18.50 mmol) in THF (60 mL) was slowly added LDA (22.2 mL, 44.4 mmol, 2.0M) over 7 min. After stirring for 2 hr, Intermediate S- 1 A (6.38 g, 25.9 mmol) was added to the reaction mixture over 3 min. After 60 min, the reaction mixture was warmed to -25 °C
(ice/MeOH/dry ice) and stirred for an additional 60 min at which time sat aq NH4C1 was added. The separated aqueous phase was acidified with IN HC1 to pH 3, and then extracted with Et20. The combined organic layers were washed with brine (2x), dried over MgS04, filtered and concentrated to provide a 1 :4 (II :I1E) mixture (as determined by 1H NMR) of Intermediate S-l and Intermediate S-1E (6.00 g, 89%) as a pale yellow solid. 1H NMR (500 MHz, CDC13) δ ppm 2.81 (1 H, ddd, J = 10.17, 6.32, 3.85 Hz), 2.63- 2.76 (1 H, m), 2.02-2.33 (4 H, m), 1.86-1.99 (2 H, m), 1.68-1.85 (2 H, m), 1.47 (9 H, s).
[00185] To a cold (-78 °C), stirred solution of a mixture of Intermediate S-l and Intermediate S-1E (5.97 g, 16.30 mmol) in THF (91 mL) was added LDA (19 mL, 38.0 mmol, 2.0M in THF/hexane/ethyl benzene) dropwise via syringe over 10 min (internal temperature never exceeded -65 °C, J-KEM® probe in reaction solution). The mixture was stirred for 15 min, and then warmed to room temperature (24 °C water bath), stirred for 15 min, and then cooled to -78 °C for 15 min. To the reaction mixture was added Et2AlCl (41 mL, 41.0 mmol, 1M in hexane) via syringe (internal temperature never exceeded -55 °C), and the mixture was stirred for 10 min, and then warmed to room temperature (24 °C bath) for 15 min and then back to -78 °C for 15 min. Meanwhile, a 1000 mL round bottom flask was charged with MeOH (145 mL) and precooled to -78 °C. With vigorous stirring the reaction mixture was transferred via cannula over 5 min to the MeOH. The flask was removed from the bath, ice was added followed by the slow addition of IN HC1 (147 mL, 147 mmol). Gas evolution was observed as the HC1 was added. The reaction mixture was allowed to warm to room temperature during which the gas evolution subsided. The reaction mixture was diluted with EtOAc (750 mL), saturated with NaCl, and the organic phase was separated, washed with a solution of potassium fluoride (8.52 g, 147 mmol) and IN HC1 (41 mL, 41.0 mmol) in water (291 mL), brine (100 mL), and then dried (Na2s04), filtered and concentrated under vacuum. 1H NMR showed the product was a 9: 1 mixture of Intermediate S-l and Intermediate S- 1E. The enriched mixture of Intermediate S-l and Intermediate S-1E (6.12 g, >99% yield) was obtained as a dark amber solid: 1H NMR (400 MHz, CDC13) δ ppm 2.64-2.76 (2 H, m), 2.04-2.35 (4 H, m), 1.88-2.00 (2 H, m), 1.71-1.83 (2 H, m), 1.48 (9 H, s).
[00186] To a stirred solution of a 9: 1 enriched mixture of Intermediate S-l and Intermediate S-1E (5.98 g, 16.33 mmol) in DMF (63 mL) were added potassium carbonate (4.06 g, 29.4 mmol) and benzyl bromide (2.9 mL, 24.38 mmol), the mixture was then stirred overnight at room temperature. The reaction mixture was diluted with EtOAc (1000 mL), washed with 10% LiCl (3×200 mL), brine (200 mL), dried (Na2S04), filtered, concentrated, and then dried under vacuum. The residue was purified by Si02 chromatography using a toluene:hexane gradient. Diastereomerically purified
[00187] To a solution of Intermediate S-1F (4.81 g, 10.54 mmol) in MeOH (100 mL) was added 10% palladium on carbon (wet, Degussa type, 568.0 mg, 0.534 mmol) in a H2– pressure flask. The vessel was purged with N2 (4x), then purged with H2 (2x), and finally, pressurized to 50 psi and shaken overnight. The reaction vessel was
depressurized and purged with nitrogen. The mixture was filtered through CELITE®, washed with MeOH and then concentrated and dried under vacuum. Intermediate S-1 (3.81 g, 99% yield)) was obtained as a colorless solid: 1H NMR (400 MHz, chloroform-d) δ 2.62-2.79 (m, 2H), 2.02-2.40 (m, 4H), 1.87-2.00 (m, 2H), 1.67-1.84 (m, 2H), 1.48 (s, 9H).
[00188] Intermediate S-1 as a mixture with Intermediate S-IE was prepared in a similar procedure as above from Intermediate S-1D to afford a 1 :2.2 mixture of
Intermediate S-1 and Intermediate S-IE (8.60 g, 23.48 mmol), which was enriched using LDA (2.0 M solution in THF, ethyl benzene and heptane, 28.2 mL, 56.4 mmol) and diethyl aluminum chloride (1.0 M solution in hexane, 59 mL, 59.0 mmol) in THF (91 mL). After workup as described above, the resulting residue was found to be a 13.2: 1 (by 1H NMR) mixture of Intermediate S-1 and Intermediate S-IE, which was treated as follows: The crude material was dissolved in MTBE (43 mL). Hexanes (26 mL) were slowly charged to the reaction mixture while maintaining a temperature below 30 °C. The reaction mixture was stirred for 10 min. Next, tert-butylamine (2.7 mL, 1.1 eq) was charged slowly over a period of 20 minutes while maintaining a temperature below 30 °C. This addition was observed to be exothermic. The reaction mixture was stirred for 2 hrs below 30 °C and then filtered. The solid material was washed with 5:3 MTBE: hexane (80 mL), and the filtrate was concentrated and set aside. The filtered solid was dissolved in dichloromethane (300 mL), washed with IN HC1 (lOOmL), and the organic layer was washed with brine (100 mL x 2), and then concentrated under reduced pressure below 45 °C to afford Intermediate S-l (5.46 g, 64%).
A second alternate procedure for preparing Intermediate S-l :
[00189] To a stirred solution of 5,5,5-trifluoropentanoic acid (5 g, 32.0 mmol) in THF (30 mL) and hexane (30 mL) at 0 °C, was added tert-butyl 2,2,2-trichloroacetimidate (11.46 mL, 64.1 mmol). The mixture was stirred for 15 min at 0 °C. Boron trifluoride etherate (0.406 mL, 3.20 mmol) was added and the reaction mixture was allowed to warm to room temperature overnight. To the clear reaction mixture was added solid NaHC03 (5 g) and stirred for 30 min. The mixture was filtered through MgSC^ and washed with hexanes (200 mL). The solution was allowed to rest for 45 min, and the resulting solid material was removed by filtering on the same MgSC^ filter again, washed with hexanes (100 mL) and concentrated under reduced pressure without heat. The volume was reduced to about 30 mL, filtered through a clean fritted funnel, washed with hexane (5 mL), and then concentrated under reduced pressure without heat. The resulting neat oil was filtered through a 0.45μιη nylon membrane filter disk to provide Intermediate S-1G (6.6 g, 31.4 mmol 98% yield) as a colorless oil: 1H NMR (400 MHz, CDC13) δ ppm 1.38 (s, 9 H) 1.74-1.83 (m, 2 H) 2.00-2.13 (m, 2 H) 2.24 (t, J= 7.28 Hz, 2 H). Intermediate S-1H: (4S)-4-(Propan-2-yl)-3-(5,5,5-trifluoropentanoyl)-l,3-oxazolidin-2- one
[00190] To a stirred solution of 5,5,5-trifluoropentanoic acid (5.04 g, 32.3 mmol) in DCM (50 mL) and DMF (3 drops) was added oxalyl chloride (3.4 mL, 38.8 mmol) dropwise over 5 min. The solution was stirred until all bubbling subsided. The reaction mixture was concentrated under reduced pressure to give pale yellow oil. To a separate flask charged with a solution of (4S)-4-(propan-2-yl)-l,3-oxazolidin-2-one (4.18 g, 32.4 mmol) in THF (100 mL) at -78 °C was added n-BuLi (2.5M in hexane) (13.0 mL, 32.5 mmol) dropwise via syringe over 5 min. After stirring for 10 min, the above acid chloride, dissolved in THF (20 mL), was added via cannula over 15 min. The reaction mixture was warmed to 0 °C, and was allowed to warm to room temperature as the bath warmed and stirred overnight. To the reaction mixture was added saturated NH4C1, and the mixture was extracted with EtOAc (2x). The combined organics were washed with brine, dried (Na2s04), filtered and concentrated under reduced pressure. The crude material was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 5% to 60% solvent A/B = hexanes/EtOAc, REDISEP® Si02 120g). Concentration of the appropriate fractions provided Intermediate S-1H (7.39 g, 86%) as a colorless oil: 1H NMR (400 MHz, CDC13) δ ppm 4.44 (1 H, dt, J= 8.31, 3.53 Hz), 4.30 (1 H, t, J= 8.69 Hz), 4.23 (1 H, dd, J= 9.06, 3.02 Hz), 2.98-3.08 (2 H, m), 2.32-2.44 (1 H, m, J= 13.91, 7.02, 7.02, 4.03 Hz), 2.13-2.25 (2 H, m), 1.88-2.00 (2 H, m), 0.93 (3 H, d, J= 7.05 Hz), 0.88 (3 H, d, J= 6.80 Hz).
[00191] To a cold (-78 °C), stirred solution of diisopropylamine (5.3 mL, 37.2 mmol) in THF (59 mL) under a nitrogen atmosphere was added n-BuLi (2.5M in hexane) (14.7 mL, 36.8 mmol). The mixture was then warmed to 0 °C to give a 0.5M solution of LDA. A separate vessel was charged with Intermediate S-1H (2.45 g, 9.17 mmol). The material was azeotroped twice with benzene (the RotoVap air inlet was fitted with a nitrogen inlet to completely exclude humidity), and then toluene (15.3 mL) was added. This solution was added to a flask containing dry lithium chloride (1.96 g, 46.2 mmol). To the resultant mixture, cooled to -78 °C, was added the LDA solution (21.0 mL, 10.5 mmol) and the mixture was stirred at -78 °C for 10 min, then warmed to 0 °C for 10 min., and then cooled to -78 °C. To a separate reaction vessel containing Intermediate S-1G (3.41 g, 16.07 mmol), also azeotroped twice with benzene, was added toluene (15.3 mL), cooled to -78 °C and LDA (37.0 mL, 18.5 mmol) was added. The resulting solution was stirred at -78 °C for 25 min. At this time the enolate derived from the ester was transferred via cannula into the solution of the oxazolidinone enolate and stirred at -78 °C for an additional 5 min, at which time the septum was removed and solid powdered bis(2- ethylhexanoyloxy)copper (9.02 g, 25.8 mmol) was rapidly added to the reaction vessel and the septum was replaced. The vessel was immediately removed from the cold bath and immersed into a warm water bath (40 °C) with rapid swirling and with a concomitant color change from the initial turquoise to brown. The reaction mixture was stirred for 20 min, was then poured into 5% aqueous NH4OH (360 mL) and extracted with EtOAc (2x). The combined organics were washed with brine, dried (Na2s04), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 0% to 60% solvent A/B = hexanes/EtOAc, REDISEP® Si02 120g). Concentration of the appropriate fractions provided a mixture of Intermediate S- II and Intermediate S-1J (2.87 g, 66%) as a pale yellow viscous oil. 1H NMR showed the product was a 1.6: 1 mixture of diastereomers S-1LS-1J as determined by the integration of the multiplets at 2.74 and 2.84 ppm: 1H NMR (400 MHz, CDC13) δ ppm 4.43-4.54 (2 H, m), 4.23-4.35 (5 H, m), 4.01 (1 H, ddd, J= 9.54, 6.27, 3.51 Hz), 2.84 (1 H, ddd, J = 9.41, 7.28, 3.64 Hz), 2.74 (1 H, ddd, J= 10.29, 6.27, 4.02 Hz), 2.37-2.48 (2 H, m, J = 10.38, 6.98, 6.98, 3.51, 3.51 Hz), 2.20-2.37 (3 H, m), 1.92-2.20 (8 H, m), 1.64-1.91 (5 H, m), 1.47 (18 H, s), 0.88-0.98 (12 H, m). Intermediate S-1 : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and Intermediate S-IE: (2R,3R)-3-(tert-Butoxycarbonyl)- 6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid
(S-IE)
[00192] To a cool (0 °C), stirred solution of Intermediate S-1I and Intermediate S-1 J (4.54 g, 9.51 mmol) in THF (140 mL) and water (42 mL) were sequentially added hydrogen peroxide (30% in water) (10.3 g, 91 mmol) and LiOH (685.3 mg, 28.6 mmol). The mixture was stirred for 1 hr. At this time the reaction vessel was removed from the cold bath and then stirred for 1.5 hr. To the reaction mixture were added saturated NaHC03 (45 mL) and saturated Na2s03 (15 mL), and then the mixture was partially concentrated under reduced pressure. The resulting crude solution was extracted with DCM (3x). The aqueous phase was acidified to pH~l-2 with IN HC1, extracted with DCM (3x) and then EtOAc (lx). The combined organics were washed with brine, dried (Na2s04), filtered and concentrated under reduced pressure to provide a mixture of Intermediates S-1 and S-IE (3.00 g, 86%) as a colorless oil: 1H NMR (400 MHz, CDC13) δ ppm 2.76-2.84 (1 H, m, diastereomer 2), 2.64-2.76 (3 H, m), 2.04-2.35 (8 H, m), 1.88- 2.00 (4 H, m), 1.71-1.83 (4 H, m), 1.48 (9 H, s, diastereomer 1), 1.46 (9 H, s,
diastereomer 2); 1H NMR showed a 1.7: 1 mixture of S-1E:S-1F by integration of the peaks for the t-butyl groups. Intermediate S-1 : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and Intermediate S-IF: (2R,3R)-3-(fert-Butoxycarbonyl)- 6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid
[00193] To a cold (-78 °C) stirred solution of diisopropylamine (1.7 mL, 11.93 mmol) in THF (19 mL) under a nitrogen atmosphere was added n-BuLi (2.5M in hexanes) (4.8 mL, 12.00 mmol). The mixture was stirred for 5 min and then warmed to 0 °C. In a separate vessel, to a cold (-78 °C) stirred solution of the mixture of Intermediates S-1 and S-1E (1.99 g, 5.43 mmol) in THF (18 mL) was added the LDA solution prepared above via cannula slowly over 25 min. The mixture was stirred for 15 min, then warmed to room temperature (placed in a 24 °C water bath) for 15 min, and then again cooled to -78 °C for 15 min. To the reaction mixture was added Et2AlCl (1M in hexane) (11.4 mL, 11.40 mmol) via syringe. The mixture was stirred for 10 min, warmed to room
temperature for 15 min and then cooled back to -78 °C for 15 min. Methanol (25 mL) was rapidly added, swirled vigorously while warming to room temperature, and then concentrated to ~l/4 the original volume. The mixture was dissolved in EtOAc and washed with IN HC1 (50 mL) and ice (75 g). The aqueous phase was separated and extracted with EtOAc (2x). The combined organics were washed with a mixture of KF (2.85g in 75 mL water) and IN HC1 (13 mL) [resulting solution pH 3-4], then with brine, dried (Na2s04), filtered and concentrated under reduced pressure to give a 9: 1 (S-LS-1E) enriched diastereomeric mixture (as determined by 1H NMR) of Intermediate S-1 and Intermediate S-1E (2.13 g, >99%) as a pale yellow viscous oil: 1H NMR (400 MHz, CDC13) δ ppm 2.64-2.76 (2 H, m), 2.04-2.35 (4 H, m), 1.88-2.00 (2 H, m), 1.71-1.83 (2 H, m), 1.48 (9 H, s).
[00194] To a cold (-78 °C), stirred solution of Intermediate S-1D (1.72 g, 6.36 mmol) in THF (30 mL) was slowly added LDA (7.32 mL, 14.6 mmol) over 7 min. After stirring for 1 h, 4,4,4-trifluorobutyltrifluoromethanesulfonate (2.11 g, 8.11 mmol) was added to the reaction mixture over 2 min. After 15 min, the reaction mixture was warmed to -25 °C (ice/MeOH/dry ice) for lh, and then cooled to -78 °C. After 80 min, the reaction was quenched with a saturated aqueous NH4C1 solution (10 mL). The reaction mixture was further diluted with brine and the solution was adjusted to pH 3 with IN HC1. The aqueous layer was extracted with ether. The combined organics were washed with brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure to provide a mixture of Intermediates S-2 and S-2A (2.29 g, 95%) as a colorless oil. 1H NMR (400MHz, chloroform-d) δ 2.83-2.75 (m, 1H), 2.64 (ddd, J = 9.9, 6.7, 3.6 Hz, 1H), 2.32-2.03 (m, 5H), 1.98-1.70 (m, 3H), 1.69-1.52 (m, 3H), 1.50-1.42 (m, 9H). 1H NMR showed a 1 :4.5 mixture (S-2:S-2A) of diastereomers by integration of the peaks for the t- Bu groups.
[00195] A mixture of Intermediate S-2 and Intermediate S-2A (2.29 g, 6.02 mmol) was dissolved in THF (38 mL) to give a colorless solution which was cooled to -78 °C. Then, LDA (7.23 mL, 14.5 mmol) (2.0M in heptane/THF/ethylbenzene) was slowly added to the reaction mixture over 3 min. After stirring for 15 min, the reaction mixture was placed in a room temperature water bath. After 15 min the reaction mixture was placed back in a -78 °C bath and then diethylaluminum chloride (14.5 mL, 14.5 mmol) (1M in hexane) was added slowly over 5 min. The reaction mixture was stirred at -78 °C. After 15 min, the reaction mixture was placed in a room temperature water bath for 10 min, and then cooled back to -78 °C. After 15 min, the reaction was quenched with MeOH (30.0 mL, 741 mmol), removed from the -78 °C bath and concentrated. To the reaction mixture was added ice and HC1 (60.8 mL, 60.8 mmol) and the resulting mixture was extracted with EtOAc (2x 200 mL). The organic layer was washed with potassium fluoride (3.50g, 60.3 mmol) in 55 mL H20 and 17.0 mL of IN HC1. The organics were dried over anhydrous magnesium sulfate and concentrated under reduced pressure to provide an enriched mixture of Intermediate S-2 and Intermediate S-2A (2.25g, 98% yield) as a light yellow oil. 1H NMR (400MHz, chloroform-d) δ 2.83-2.75 (m, 1H), 2.64 (ddd, J= 9.9, 6.7, 3.6 Hz, 1H), 2.32-2.03 (m, 5H), 1.98-1.70 (m, 3H), 1.69-1.52 (m, 3H), 1.50-1.42 (m, 9H). 1H NMR showed a 9: 1 ratio in favor of the desired diastereomer Intermediate S-2.
[00196] To a stirred 9: 1 mixture of Intermediate S-2 and Intermediate S-2A (2.24 g, 5.89 mmoL) and potassium carbonate (1.60 g, 11.58 mmoL) in DMF (30 mL) was added benzyl bromide (1.20 mL, 10.1 mmoL)). The reaction mixture was stirred at room temperature for 19 h. The reaction mixture was diluted with ethyl acetate (400 mL) and washed with 10% LiCl solution (3 x 100 mL), brine (50 mL), and then dried over anhydrous magnesium sulfate, filtered and concentrated to dryness under vacuum. The residue was purified by flash chromatography (Teledyne ISCO CombiFlash 0%> to 100% solvent A/B = hexane/EtOAc, REDISEP® Si02 220 g, detecting at 254 nm, and monitoring at 220 nm). Concentration of the appropriate fractions provided Intermediate S-2B (1.59 g, 57.5%). HPLC: RT = 3.863 min (CHROMOLITH® SpeedROD column 4.6 x 50 mm, 10-90% aqueous methanol over 4 minutes containing 0.1% TFA, 4 mL/min, monitoring at 220 nm), 1H NMR (400MHz, chloroform-d) δ 7.40-7.34 (m, 5H), 5.17 (d, J= 1.8 Hz, 2H), 2.73-2.64 (m, 1H), 2.55 (td, J= 10.0, 3.9 Hz, 1H), 2.16-1.82 (m, 5H), 1.79-1.57 (m, 3H), 1.53-1.49 (m, 1H), 1.45 (s, 9H), 1.37-1.24 (m, 1H).
[00197] To a stirred solution of Intermediate S-2B (1.59 g, 3.37 mmoL) in MeOH (10 mL) and EtOAc (10 mL) under nitrogen was added 10%> Pd/C (510 mg). The atmosphere was replaced with hydrogen and the reaction mixture was stirred at room temperature for 2.5 h. The palladium catalyst was filtered off through a 4 μΜ polycarbonate film and rinsed with MeOH. The filtrate was concentrated under reduced pressure to give intermediate S-2 (1.28 g, 99%). 1H NMR (400MHz, chloroform-d) δ 2.76-2.67 (m, 1H), 2.65-2.56 (m, 1H), 2.33-2.21 (m, 1H), 2.17-2.08 (m, 3H), 1.93 (dtd, J= 14.5, 9.9, 5.2 Hz, 1H), 1.84-1.74 (m, 2H), 1.70-1.52 (m, 3H), 1.48 (s, 9H).
[00198] In a 1 L round-bottomed flask was added 2-amino-3-methylbenzoic acid (11.2 g, 74.1 mmol) and Ν,Ο-dimethylhydroxylamine hydrochloride (14.45 g, 148 mmol) in DCM (500 mL) to give a pale brown suspension. The reaction mixture was treated with Et3N (35 mL), HOBT (11.35 g, 74.1 mmol) and EDC (14.20 g, 74.1 mmol) and then stirred at room temperature for 24 hours. The mixture was then washed with 10% LiCl, and then acidified with IN HCl. The organic layer was washed successively with 10%> LiCl and aq NaHC03. The organic layer was decolorized with charcoal, filtered, and the filtrate was dried over MgSC^. The mixture was filtered and concentrated to give 13.22 g (92% yield) of Intermediate A-1A. MS(ES): m/z = 195.1 [M+H+]; HPLC: RT = 1.118 min. (H20/MeOH with TFA, CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm); 1H NMR (500MHz, chloroform-d) δ 7.22 (dd, J= 7.8, 0.8 Hz, 1H), 7.12-7.06 (m, 1H), 6.63 (t, J= 7.5 Hz, 1H), 4.63 (br. s., 2H), 3.61 (s, 3H), 3.34 (s, 3H), 2.17 (s, 3H).
[00199] In a 500 mL round-bottomed flask, a solution of l-fluoro-3-iodobenzene (13.61 mL, 116 mmol) in THF (120 mL) was cooled in a -78 °C bath. A solution of n- BuLi, (2.5M in hexane, 46.3 mL, 116 mmol) was added dropwise over 10 minutes. The solution was stirred at -78 °C for 30 minutes and then treated with a solution of
Intermediate A-1 A (6.43 g, 33.1 mmol) in THF (30 mL). After 1.5 hours, the reaction mixture was added to a mixture of ice and IN HCl (149 mL, 149 mmol) and the reaction flask was rinsed with THF (5 ml) and combined with the aqueous mixture. The resulting mixture was diluted with 10% aq LiCl and the pH was adjusted to 4 with IN NaOH. The mixture was then extracted with Et20, washed with brine, dried over MgS04, filtered and concentrated. The resulting residue was purified by silica gel chromatography (220g ISCO) eluting with a gradient from 10% EtOAc/hexane to 30% EtOAc/hexane to afford Intermediate A-l (7.11 g, 94% yield) as an oil. MS(ES): m/z = 230.1 [M+H+]; HPLC: RT = 2.820 min Purity = 99%. (H20/MeOH with TFA, CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm).
[00225] In a 1 L round-bottomed flask, a solution of 2-(lH-benzo[d][l,2,3]triazol-l- yl)-2-((phenoxycarbonyl)amino)acetic acid (J. Org. Chem., 55:2206-2214 (1990)) (19.37 g, 62.0 mmol) in THF (135 mL) was cooled in an ice/water bath and treated with oxalyl chloride (5.43 mL, 62.0 mmol) and 4 drops of DMF. The reaction mixture was stirred for 4 hours. Next, a solution of Intermediate A- 1 (7.11 g, 31.0 mmol) in THF (35 mL) was added and the resulting solution was removed from the ice/water bath and stirred at room temperature for 1.5 hours. The mixture was then treated with a solution of ammonia, (7M in MeOH) (19.94 mL, 140 mmol). After 15 mins, another portion of ammonia, (7M in MeOH) (19.94 mL, 140 mmol) was added and the resulting mixture was sealed under N2 and stirred overnight at room temperature. The reaction mixture was then concentrated to ~l/2 volume and then diluted with AcOH (63 mL) and stir at room temperature for 4 hours. The reaction mixture was then concentrated, and the residue was diluted with 500 mL water to give a precipitate. Hexane and Et20 were added and the mixture was stirred at room temperature for 1 hour to form an orange solid. Et20 was removed under a stream of nitrogen and the aqueous layer was decanted. The residue was triturated with 40 mL of iPrOH and stirred at room temperature to give a white precipitate. The solid was filtered and washed with iPrOH, then dried on a filter under a stream of nitrogen to give racemic Intermediate B-1A (5.4 g, 41.7%yield).
[00226] Racemic Intermediate B-1A (5.9 g, 14.3 mmol) was resolved using the Chiral SFC conditions described below. The desired stereoisomer was collected as the second peak in the elution order: Instrument: Berger SFC MGIII, Column: CHIRALPAK® IC 25 x 3 cm, 5 cm; column temp: 45 °C; Mobile Phase: C02/MeOH (45/55); Flow rate: 160 mL/min; Detection at 220 nm.
[00227] After evaporation of the solvent, Intermediate B-1A (2.73 g, 46% yield) was obtained as a white solid. HPLC: RT = 3.075 min. (H20/MeOH with TFA,
CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm).
[00228] In a 100 mL round-bottomed flask, a solution of Intermediate B-1A (2.73 g, 6.54 mmol) in acetic acid (12 mL) was treated with HBr, 33% in HOAc (10.76 mL, 65.4 mmol) and the mixture was stirred at room temperature for 1 hour. The solution was diluted with Et20 to give a yellow precipitate. The yellow solid was filtered and rinsed with Et20 under nitrogen. The solid was transferred to 100 mL round bottom flask and water was added (white precipitate formed). The slurry was slowly made basic with saturated NaHC03. The resulting tacky precipitate was extracted with EtOAc. The organic layer was washed with water, dried over MgS04, and then filtered and
concentrated to dryness to give Intermediate B-l (1.68 g, 91% yield) as a white foam solid. MS(ES): m/z = 284.2 [M+H+]; HPLC: RT = 1.72 min (H20/MeOH with TFA, CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm). 1H NMR (400MHz, DMSO-d6) δ 10.01 (br. s., 1H), 7.56-7.44 (m, 2H), 7.41-7.26 (m, 3H), 7.22-7.11 (m, 2H), 4.24 (s, 1H), 2.55 (br. s., 2H), 2.41 (s, 3H). [00229] The compounds listed below in Table 6 (Intermediates B-2 to B-3) were prepared according to the general synthetic procedure described for Intermediate B-l , using the starting materials Intermediate A- 10 and Intermediate A-4, respectively.
[00240] In a 100 mL round-bottomed flask, a solution of Intermediate B-l (1683 mg, 5.94 mmol), Et3N (1.656 mL, 11.88 mmol), and Intermediate S-l in DMF (20 mL) was treated with o-benzotriazol-l-yl-A .A .N’.N’-tetramethyluronium tetrafluoroborate (3815 mg, 11.88 mmol) and stirred at room temperature for 1 hour. The reaction mixture was diluted with water and saturated aqueous NaHC03. An off white precipitate formed and was filtered and washed with water. The resulting solid was dried on the filter under a stream of nitrogen to give Intermediate 1A (3.7 g, 99% yield). MS(ES): m/z =
632.4[M+H+]; HPLC: RT = 3.635 min Purity = 98%. (H20/MeOH with TFA,
[00241] In a 250 mL round-bottomed flask, a solution of Intermediate 1A (3.7 g, 5.86 mmol) in DCM (25 mL) was treated with TFA (25 mL) and the resulting pale orange solution was stirred at room temperature for 1.5 hours. The reaction mixture was then concentrated to give Intermediate IB. HPLC: RT = 3.12 min (H20/MeOH with TFA, CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm).
[00242] In a 250 mL round-bottomed flask, a solution of Intermediate IB (4.04 g, 5.86 mmol) in THF (50 mL) was treated with ammonia (2M in iPrOH) (26.4 mL, 52.7 mmol), followed by HOBT (1.795 g, 11.72 mmol) and EDC (2.246 g, 11.72 mmol). The resulting white suspension was stirred at room temperature overnight. The reaction mixture was diluted with water and saturated aqueous NaHC03. The resulting solid was filtered, rinsed with water and then dried on the filter under a stream of nitrogen. The crude product was suspended in 20 mL of iPrOH and stirred at room temperature for 20 min and then filtered and washed with iPrOH and dried under vacuum to give 2.83 g of solid. The solid was dissolved in re fluxing EtOH(100 mL) and slowly treated with 200 mg activated charcoal added in small portions. The hot mixture was filtered through CELITE® and rinsed with hot EtOH. The filtrate was reduced to half volume, allowed to cool and the white precipitate formed was filtered and rinsed with EtOH to give 2.57 g of white solid. A second recrystallization from EtOH (70 mL) afforded Example 1 (2.39 g, 70% yield) as a white solid. HPLC: RT = 10.859 min (H20/CH3CN with TFA, Sunfire C18 3.5μπι, 3.0x150mm, gradient = 15 min, wavelength = 220 and 254 nm); MS(ES): m/z = 575.3 [M+H+]; 1H NMR (400MHz, methanol-d4) δ 7.57-7.50 (m, 1H), 7.47-7.30 (m, 3H), 7.29-7.15 (m, 3H), 5.38 (s, 1H), 2.85-2.75 (m, 1H), 2.59 (td, J= 10.5, 4.0 Hz, 1H), 2.53-2.41 (m, 4H), 2.31-2.10 (m, 3H), 1.96-1.70 (m, 4H).
Structure–activity relationships in a series of (2-oxo-1,4-benzodiazepin-3-yl)-succinamides identified highly potent inhibitors of γ-secretase mediated signaling of Notch1/2/3/4 receptors. On the basis of its robust in vivo efficacy at tolerated doses in Notch driven leukemia and solid tumor xenograft models, 12 (BMS-906024) was selected as a candidate for clinical evaluation.
Discovery of Clinical Candidate BMS-906024: A Potent Pan-Notch Inhibitor for the Treatment of Leukemia and Solid Tumors
An enantioselective synthesis of (S)-7-amino-5H,7H-dibenzo[b,d]azepin-6-one (S–1) is described. The key step in the sequence involved crystallization-induced dynamic resolution (CIDR) of compound 7 using Boc-d-phenylalanine as a chiral resolving agent and 3,5-dichlorosalicylaldehyde as a racemization catalyst to afford S–1 in 81% overall yield with 98.5% enantiomeric excess.
Crystallization-Induced Dynamic Resolution toward the Synthesis of (S)-7-Amino-5H,7H-dibenzo[b,d]-azepin-6-one: An Important Scaffold for γ-Secretase Inhibitors
† Department of Discovery Synthesis, Biocon Bristol-Myers Squibb Research Centre, Biocon Park, Bommasandra IV Phase, Jigani Link Road, Bengaluru 560099, India
‡Bristol-Myers Squibb Company, P.O Box 4000, Princeton, New Jersey 08543-4000, United StatesOrg. Process Res. Dev.
ZAVZPRET is indicated for the acute treatment of migraine with or without aura in adults.
The recommended dose of ZAVZPRET is 10 mg given as a single spray in one nostril, as needed. The maximum dose that may be given in a 24-hour period is 10 mg (one spray). The safety of treating more than 8 migraines in a 30-day period has not been established, Nasal spray: 10 mg of zavegepant per device. Each unit-dose nasal spray device delivers a single spray containing 10 mg of zavegepant.
ZAVZPRET (zavegepant) nasal spray contains zavegepant hydrochloride, a calcitonin generelated peptide receptor antagonist. Zavegepant hydrochloride is described chemically as (R)-N- (3-(7-methyl-1H-indazol-5-yl)-1-(4-(1-methylpiperidin-4-yl) piperazin-1-yl)-1-oxopropan-2-yl)- 4-(2-oxo-1,2-dihydroquinolin-3-yl) piperidine-1-carboxamide hydrochloride and its structural formula is:
Its molecular formula is C36H46N8O3․HCl, representing a molecular weight of 675. 28 g/mol. Zavegepant free base has a molecular weight of 638.82 g/mol. Zavegepant hydrochloride is a white to off-white powder, freely soluble in water, and has pKa values of 4.8 and 8.8. Each unit-dose ZAVZPRET device for nasal administration delivers 10 mg of zavegepant (equivalent to 10.6 mg of zavegepant hydrochloride) in a buffered aqueous solution containing dextrose, hydrochloric acid, sodium hydroxide, and succinic acid in water for injection. The solution has a pH of 5.3 to 6.7.
Active ingredients in ZAVZPRET: zavegepant Inactive ingredients in ZAVZPRET: dextrose, hydrochloric acid, sodium hydroxide, and succinic acid in water for injection.
The most common adverse reactions include taste disorders, nausea, nasal discomfort, and vomiting.[1]
Zavegepant was approved for medical use in the United States in March 2023.[1][2][3]
Medical usesZavegepant is a Calcitonin Gene-related Peptide Receptor Antagonist. The mechanism of action of zavegepant is as a Calcitonin Gene-related Peptide Receptor Antagonist.
Zavegepant is indicated for the acute treatment of migraine with or without aura in adults.[1]
Zavegepant is an antagonist of the calcitonin gene-related peptide (CGRP) receptor currently in phase 3 trials in an intranasal formulation for the treatment of migraine. If FDA approved, it will join other previously-approved “-gepant” drugs [rimegepant] and [ubrogepant] as an additional treatment alternative for patients with migraine, particularly those for whom traditional triptan therapy has proven ineffective. On April 15th, 2020, a phase 2 clinical trial (NCT04346615: Safety and Efficacy Trial of Vazegepant Intranasal for Hospitalized Patients With COVID-19 Requiring Supplemental Oxygen) began to investigate the use of intranasally administered zavegepant to combat the acute respiratory distress syndrome (ARDS) sometimes seen in patients with COVID-19. Acute lung injury activates the release of CGRP, which plays a role in the development of ARDS – CGRP antagonists, then, may help to blunt the significant inflammation associated with COVID-19. The clinical trial is expected to complete in September 2020.
Zavegepant is a highly soluble small molecule calcitonin gene related peptide (CGRP) receptor antagonist, with potential analgesic and immunomodulating activities. Upon administration, zavegepant targets, binds to and inhibits the activity of CGRP receptors located on mast cells in the brain. This may inhibit neurogenic inflammation caused by trigeminal nerve release of CGRP. In addition, by blocking the CGRP receptors located in smooth muscle cells within vessel walls, zavegepant inhibits the pathologic dilation of intracranial arteries. Zavegepant, by blocking the CGRP receptors, also suppresses the transmission of pain by inhibiting the central relay of pain signals from the trigeminal nerve to the caudal trigeminal nucleus. Altogether, this may relieve migraine. As CGRP receptors induce the release of pro-inflammatory mediators, such as interleukin-6 (IL-6), from inflammatory cells, zavegepant may prevent an IL-6-mediated inflammatory response. Zavegepant may also inhibit the CGRP-mediated induction of eosinophil migration and the stimulation of beta-integrin-mediated T cell adhesion to fibronectin at the site of inflammation, and may abrogate the CGRP-mediated polarization of the T cell response towards the pro-inflammatory state characterized by Th17 and IL-17. This may improve lung inflammation and oxygenation, prevent edema, and further lung injury. CGRP, a 37 amino-acid peptide expressed in and released from a subset of polymodal primary sensory neurons of the trigeminal ganglion and nerve fibers projecting to the airways and by pulmonary neuroendocrine cells, plays an important role in pain transmission, inflammation, and neurogenic vasodilatation. It is released upon acute lung injury and upregulation of transient receptor potential (TRP) channels.
Azepino-indazoles as calcitonin gene-related peptide (CGRP) receptor antagonists
PMID: 33096162Publication Date: 2021-01-01Journal: Bioorganic & medicinal chemistry lettersDiscovery of (R)-N-(3-(7-methyl-1H-indazol-5-yl)-1-(4-(1-methylpiperidin-4-yl)-1-oxopropan-2-yl)-4-(2-oxo-1,2-dihydroquinolin-3-yl)piperidine-1-carboxamide (BMS-742413): a potent human CGRP antagonist with superior safety profile for the treatment of migraine through intranasal delivery PMID: 23632269Publication Date: 2013-06-01Journal: Bioorganic & medicinal chemistry letters
////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Migraine is a chronic and debilitating disorder characterized by recurrent attacks lasting four to 72 hours with multiple symptoms, including typically one-sided, pulsating headaches of moderate to severe pain intensity that are associated with nausea or vomiting, and/or sensitivity to sound (phonophobia) and sensitivity to light (photophobia). Migraines are often preceded by transient neurological warning symptoms, known as auras, which typically involve visual disturbances such as flashing lights, but may also involve numbness or tingling in parts of the body. Migraine is both widespread and disabling. The Migraine Research Foundation ranks migraine as the world’s third most prevalent illness, and the Global Burden of Disease Study 2015 rates migraine as the seventh highest specific cause of disability worldwide. According to the Migraine Research Foundation, in the United States, approximately 36 million individuals suffer from migraine attacks. While most sufferers experience migraine attacks once or twice per month, more than 4 million people have chronic migraine, defined as experiencing at least 15 headache days per month, of which at least eight are migraine, for more than three months. Others have episodic migraine, which is characterized by experiencing less than 15 migraine days per month. People with episodic migraine may progress to chronic migraine over time. Migraine attacks can last four hours or up to three days. More than 90% of individuals suffering from migraine attacks are unable to work or function normally during a migraine attack, with many experiencing comorbid conditions such as depression, anxiety and insomnia. Also, those suffering from migraine often have accompanying nausea and have an aversion to consuming food or liquids during an attack.
CGRP (calcitonin gene-related peptide) is a 37 amino acid neuropeptide, which belongs to a family of peptides that includes calcitonin, adrenomedullin and amylin. In humans, two forms of CGRP (a-CGRP and 0-CGRP) exist and have similar activities. They vary by three amino acids and exhibit differential distribution. At least two CGRP receptor subtypes may also account for differential activities. The CGRP receptor is located within pain-signaling pathways, intracranial arteries and mast cells and its activation is thought to play a causal role in migraine pathophysiology. For example, research and clinical studies have shown: serum levels of CGRP are elevated during migraine attacks, infusion of intravenous CGRP produces persistent pain in migraine sufferers and non-migraine sufferers, and treatment with anti-migraine drugs normalizes CGRP activity.
Currently, clinicians use a number of pharmacologic agents for the acute treatment of migraine. A study published by the American Headache Society in 2015 concluded that the medications deemed effective for the acute treatment of migraine fell into the following classes: triptans, ergotamine derivatives, non-steroidal anti-inflammatory drugs (“NSAIDs”), opioids and combination medications. The current standard of care for the acute treatment of migraine is prescription of triptans, which are serotonin 5-HT IB/ID receptor agonists. Triptans have been developed and approved for the acute treatment of migraine over the past two decades. The initial introduction of triptans represented a shift toward drugs more selectively targeting the suspected pathophysiology of migraine. While triptans account for almost 80% of anti-migraine therapies prescribed at office visits by healthcare providers, issues such as an incomplete effect or headache recurrence remain important clinical limitations. In fact, only about 30% of patients from clinical trials are pain free at two hours after taking triptans. In addition, triptans are contraindicated in patients with cardiovascular disease, cerebrovascular disease, or significant risk factors for either because of potential systemic and cerebrovascular vasoconstriction from the 5-HT IB -mediated effects. Also, according to a January 2017 study published in the journal Headache, an estimated 2.6 million migraine sufferers in the United States have a cardiovascular event, condition or procedure that limits the potential of triptans as a treatment option.
Accordingly, there remains a significant unmet medical need for a novel migraine-specific medication that provides enhanced patient benefits compared to existing therapies.
Possible CGRP involvement in migraine has been the basis for the development and clinical testing of a number of compounds, including for example, advanced clinical candidates rimegepant (BHV-3000) and zavegepant (BHV-3500), which are developed by Biohaven Pharmaceutical Holding Company Ltd., New Haven, CT.
Zavegepant (also known as vazegepant) is a third generation, high affinity, selective and structurally unique small molecule CGRP receptor antagonist having the following formula I:
I
Zavegepant is described, for example, in WO 03/104236 published December 18, 2003 and US 8,481,546 issued July 9, 2013, which are incorporated herein in their entireties by reference.
While zavegepant is a highly soluble molecule, its bioavailability characteristics may render it challenging to prepare the drug in an oral dosage form. Enhancing the bioavailability of zavegepant and other CGRP inhibitors by different administration routes would therefore be desirable.
Calcitonin gene-related peptide (CGRP) is widely distributed in nociceptive pathways in human peripheral and central nervous system and its receptors are also expressed in pain pathways. While CGRP is involved in migraine pathophysiology, its role in non-headache pain has not been quite clear. There remains a need for new medicines to treat various pain disorders in patients in need thereof.
Scheme 1
Scheme 3
Scheme 4
tert-butyl 4-(2-methoxy-2-oxoethylidene)piperidine-l -carboxylate. Sodium hydride in mineral oil (60%, 7.92 g, 198.02 mmoles) was washed with hexanes then suspended in dimethylformamide (220 mL). The mixture was cooled to 0°C. Trimethyl phosphonoacetate (29.0 mL, 189.82 mmoles) was added dropwise to the stirred reaction mixture. After 20 min at 0°C, a solution of A-/c/7-butoxycarbonyl-4-pi peri done (30.41 g, 152.62 mmoles) in dimethylformamide (80 mL) was added to the mixture dropwise. The reaction was stirred at room temperature for 3 h and then diluted with diethyl ether (650 mL). The mixture was washed once with water and the aqueous layer was extracted once with diethyl ether. The combined organic layers were washed 4 times with water and the aqueous phase was discarded. The organic phase was washed with brine and dried over magnesium sulfate, filtered, and concentrated to dryness. The title compound was obtained as a white solid in 92% yield. 1 H- NMR (300 MHz, CDCh): 5 = 5.68 (s, 1 H), 3.66 (s, 3 H), 3.40-3.51 (m, 4 H), 2.90 (t, J= 5.49, 2 H), 2.25 (t, J= 5.49, 2 H), 1.44 (s, 9 H).
ed-butyl 4-(2-methoxy-2-oxoethyl)piperidine-l -carboxylate. A solution of tert-butyl 4- (2-methoxy-2-oxoethylidene)piperidine-l -carboxylate (35.71 g, 140 mmoles) in a mixture of 1 : 1 ethyl acetate/methanol (220 mL) was carefully treated with 50% wet 10% palladium on carbon (3.3 g). The reaction vessel was charged with 55 psi of hydrogen gas and the mixture was shaken on a Parr apparatus at room temperature for 16 h. The reaction mixture was then filtered to remove the catalyst and the filtrate concentrated in vacuo. The title compound was obtained as a clear colorless oil in 97% yield. ‘H-NMR (300 MHz, CDCh): 5 = 4.04 (d, J= 10.25, 2 H), 3.64 (s, 3 H), 2.68 (t, J= 12.44, 2 H), 2.21 (d, J= 6.95, 2 H), 1.98-1.77 (m, 1 H), 1.64 (d, J= 13.54, 2 H), 1.41 (s, 9 H), 1.25-0.99 (m, 2 H).
4-[2-Hydroxy-l-methoxycarbonyl-2-(2-nitro-phenyl)-ethyl]-piperidine-l-carboxylic acid tert-butyl ester. A A-diisopropylamine (4.40 mL, 31.3 mmoles) was dissolved in tetrahydrofuran (50 mL). The mixture was cooled to -78°C. Butyllithium (2.5 M in hexanes, 12.4 mL, 31 mmoles) was added dropwise to the stirred solution. After stirring at -78°C for 30 min, a solution of tert-butyl 4-(2-methoxy-2-oxoethyl)piperidine-l -carboxylate (6.65 g, 25.8 mmoles) in tetrahydrofuran (15 mL) was added dropwise to the mixture. Stirring was continued at -78°C for 1 h. A solution of 2-nitrobenzaldehyde (3.90 g, 25.8 mmoles) in tetrahydrofuran (20 mL) was then added to the mixture dropwise, and then stirring was continued at -78°C for a further 2.5 h. The reaction was quenched with cold aqueous ammonium chloride and then diluted with water. The mixture was extracted twice with ethyl acetate and the aqueous phase was discarded. The material was dried (magnesium sulfate) filtered, and concentrated to dryness. Silica gel chromatography afforded the desired product in 94% yield as light yellow foam. MS m/e (M- C4H8+H)+= 353.1.
4-(4-Hydroxy-2-oxo-l , 2, 3, 4-tetrahydro-quinolin-3-yl)-piperidine-l -carboxylic acid tertbutyl ester. In a 3 neck flask fitted with a nitrogen inlet, thermometer, and a mechanical stirrer, 4-[2-hydroxy-l -methoxy carbonyl-2-(2-nitro-phenyl)-ethyl]-piperidine-l -carboxylic acid tertbutyl ester (9.93 g, 24.3 mmoles) was dissolved in acetic acid (1.75 moles, 100 mL). Iron powder (8.90 g, 159 mmoles) was added to the vessel with stirring. The stirred mixture was slowly heated to 80°C for 30 min and then cooled to room temperature. It was then diluted with ethyl acetate and filtered through a pad of celite. Solids were washed with 20% methanol/ethyl acetate, and then with methanol. The filtrate was concentrated and the residue partitioned between ethyl acetate and aqueous sodium bicarbonate. The layers were separated. The resulting aqueous phase was extracted twice with ethyl acetate. The organic layers were combined. The mixture was washed twice with water and the aqueous phase was discarded. The material was dried (magnesium sulfate) filtered, and concentrated to dryness. Silica gel chromatography afforded the title compound as light yellow foam in 77% yield. MS m/e (M-H)’ = 345.1.
3-(Piperidin-4-yl)quinolin-2(lH) hydrochloride . A stirred solution of 4-(4-hydroxy-2- oxo-l,2,3,4-tetrahydro-quinolin-3-yl)-piperidine-l-carboxylic acid tert-butyl ester (5.60 g, 16.2 mmoles) in ethyl acetate (70 mL) was treated with HC1 in dioxane (4N, 40 mmoles, 10 mL). The mixture was stirred at room temperature for 45 min. More HC1 in dioxane (4N, 120 mmoles, 30 mL) was then added and stirring was continued at room temperature for 16 h. The resulting solid was collected by filtration and washed with ethyl acetate. It was then suspended in 5% water-isopropanol (100 mL) and the mixture was warmed to reflux and stirred for 20 min. The mixture was cooled to room temperature and stirred at room temperature for 16 h. The solid was collected by filtration, washed with isopropanol, and dried under high vacuum. The title compound was obtained as white solid in 75% yield. ‘H-NMR (DMSO-de) 5 11.85 (s, 1 H), 9.02 (bs, 1 H), 8.88 (bs, 1 H), 7.70 (t, J= 3.81 Hz, 2 H), 7.53 – 7.30 (d, J= 8.24 Hz, 1 H), 7.17 (t, J= 7.48 Hz, 2 H), 3.36 (d, J= 12.51 Hz, 2 H), 3.10 – 2.94 (m, 3 H), 2.01 (d, J= 13.43 Hz, 2 H), 1.87 – 1.73 (m, 2 H); MS m/e (M+H)+ = 229.0.
4-Iodo-2,6-dimethylbenzenamine hydrochloride . To a suspension of sodium bicarbonate (126 g, 1.5 moles) and 2,6-dimethylaniline (61.5 mL, 500 mmoles) in methanol (700 mL) was added iodine monochloride (1.0 M in dichloromethane, 550 mL, 550 mmoles) at room temperature over 1 h. After addition was complete, stirring was continued for 3 h. The reaction was filtered to remove excess sodium bicarbonate and the solvent removed in vacuo. The residue was re-dissolved in diethyl ether (1.5 L) and treated with hydrochloric acid (2M in ether, 375 mL, 750 mmoles). The resulting suspension was stored in the freezer (-15°C) overnight. The solid was filtered and washed with diethyl ether until it became colorless, to give 126.5 g (89%) as a grey-green powder. ‘H-NMR (DMSO-de) 5 2.33 (s, 6 H), 7.48 (s, 2 H), 9.05 (bs, 3 H); 13C-NMR (DMSO-de) 5 17.4, 91.5, 133.1, 131.2, 136.9.
Methyl 2 -(benzyloxy carbonyl) acrylate . To a flame dried three-neck round bottom flask, fitted with a mechanical stirrer, was added (S)-methyl 2-(benzyloxycarbonyl)-3- hydroxypropanoate (129 g, 509 mmoles), anhydrous dichloromethane (2 L), and methanesulfonyl chloride (49.3 mL, 636 mmoles). The mixture was cooled to -15°C, and treated with tri ethylamine (213 mL, 1527 mmoles), dropwise, to ensure the temperature of the reaction mixture did not exceed 0°C. The addition of the first equivalent of triethylamine was exothermic. After addition of tri ethylamine, the mixture was stirred at 0°C for 30 min. The cooling bath was removed and the mixture stirred at room temperature for 1.5 h. The reaction was quenched by addition of methanol (21 mL). The mixture was washed with 0.5% aqueous potassium bisulfate until the washings were pH 5, then saturated sodium bicarbonate, and brine, dried over sodium sulfate, and concentrated. Flash chromatography (silica gel, 1 :9 ethyl acetate/hexanes) gave I l l g (92%) as a viscous colorless oil, which crystallized upon standing. ’H-NMR (DMSO-de) 5 3.71 (s, 3 H), 5.10 (s, 2 H), 5.60 (s, 1 H), 5.76 (s, 1 H), 7.39-7.35 (m, 5 H), 8.96 (s, 1 H); 13C-NMR (DMSO-de) 5 52.3, 65.9, 127.8, 128.1, 128.3, 128.8, 133.3, 136.3, 153.5, 163.7.
(Z)-Methyl 3-(4-amino-3,5-dimethylphenyl)-2-(benzyloxycarbonyl) acrylate. A 2 L round bottom flask was charged 4-iodo-2,6-dimethylbenzenamine hydrochloride salt (55 g, 194 mmoles), methyl 2-(benzyloxycarbonyl)acrylate (59.2 g, 252 mmoles), tetrabutylammonium chloride (59.2 g, 213 mmoles), palladium (II) acetate (4.34 g, 19.4 mmoles), and tetrahydrofuran (1.2 L, degassed by a flow of nitrogen for 30 min). The mixture was stirred so that a suspension was formed and then degassed by a flow of nitrogen for 30 min. Triethylamine (110 mL, 789 mmoles) was added and the resulting mixture was heated at reflux for 3 h. After cooling to room temperature, the reaction mixture was filtered through a pad of celite, washed with tetrahydrofuran (2 x 100 mL), and concentrated. The residue was dissolved in di chloromethane, washed with water (3X) and brine (2X), dried over sodium sulfate, and concentrated. Flash chromatography (silica gel, using 1 :9 ethyl acetate/dichloromethane) gave a tan solid. The solid was recrystallized from warm methanol (210 mL) and water (100 mL). The mixture was held at room temperature overnight, then at 0°C for 2 h, and finally at -15°C for 2 h. The resulting solid was filtered, washed with ice cold 1 : 1 methanol/water, and dried under high vacuum overnight to give 44.7 g (65%) as a light tan solid which was a mixture of ZZE isomers (73 :27). ’H-NMR (DMSO-de) 5, 2.05 (s, 6 H), 3.61 (s, 0.8 H), 3.68 (s, 2.2 H), 5.00 (s, 0.54 H), 5.13 (s, 1.46 H), 5.24 (s, 2 H), 7.40-7.21 (m, 8 H), 8.51 (s, 0.27 H), 8.79 (s, 0.73 H); 13C-NMR (DMSO-de) 5 17.8, 51.7, 65.3, 119.4, 120.0, 120.3, 127.3, 127.7, 128.3, 130.9, 135.8, 137.2, 146.9, 154.7, 166.0.
(R)-Methyl 3-(4-amino-3,5-dimethylphenyl)-2-(benzyloxycarbonyl)propanoate. A flame- dried 2 L Parr hydrogenation bottle was charged with (Z)-methyl 3-(4-amino-3,5- dimethylphenyl)-2-(benzyloxycarbonyl)acrylate (84.5 g, 239 mmoles), di chloromethane (300 mL), and methanol (300 mL). The bottle was swirled so that a light brown suspension was formed. The mixture was degassed using a flow of nitrogen for 30 min. To this was quickly added (-)-l,2-bis((2A,5A)-2,5-diethylphospholano)-bezene(cyclooctadiene) rhodium (I) tetrafluoroborate ([(2A,5A)-Et-DuPhosRh]BF4) (2.11 g, 3.20 mmoles). The bottle was immediately attached to a Parr Hydrogenator. After 5 cycles of hydrogen (60 psi) and vacuum, the bottle was pressurized to 65 psi and the suspension was agitated at room temperature for 16 h. The reaction had become homogeneous. The reaction mixture was concentrated, and the resulting residue purified by flash chromatography (silica gel, 1 :9 ethyl acetate/dichloromethane) to give 82.9 g (98%). ‘H-NMR (DMSO-de) 5 2.04 (s, 6 H), 2.65 (dd, J= 13.4, 9.8 Hz, 1H), 2.82 (dd, J= 13.7, 5.2 Hz, 1 H), 3.62 (s, 3 H), 4.15-4.10 (m, 1H), 4.41 (s, 2 H), 5.00 (s, 2 H), 6.68 (s, 2 H), 7.37-7.28 (m, 5 H), 7.70 (d, J= 7.9 Hz, 1 H); 13C-NMR (DMSO-de) 5 17.7, 35.9, 51.7, 56.1, 65.3, 120.4, 124.0, 127.5, 127.7, 128.2, 128.3, 136.9, 142.6, 155.9, 172.5.
(R)-Methyl 2-(benzyloxycarbonyl)-3-(7-methyl-lH-indazol-5-yl)propanoate. (R)-Methyl 3-(4-amino-3,5-dimethylphenyl)-2-(benzyloxycarbonyl)propanoate (50.0 g, 140 mmoles) was weighed into a flame-dried 5 L three neck round bottom flask, followed by the addition of toluene (2.4 L) and glacial acetic acid (120 mL, 2.1 moles). The mixture was mechanically stirred to form a clear solution, and then potassium acetate (103 g, 1.05 moles) was added. To the resulting white suspension, z.w-amyl nitrite (20.7 mL, 154 mmoles) was added dropwise at room temperature, and the resulting mixture was stirred at room temperature for 16 h. Saturated sodium bicarbonate (I L) was added, followed by the careful addition of solid sodium bicarbonate to neutralize the acetic acid. The mixture was extracted with a mixture of di chloromethane (2 L) and brine (1.5 L). After separation, the aqueous layer was extracted with di chloromethane (500 mL). The combined organic layers were dried over anhydrous sodium sulfate and filtered. Solvents were removed to afford a tan solid, which was washed with hexanes (2 L) and toluene (150 mL). The solid was recrystallized from hot acetone (260 mL) and hexanes (700 mL). The slightly cloudy mixture was allowed to cool to room temperature slowly, then to 0°C for 1.5 h, and finally to -15°C for 1.5 h. The resulting solid was filtered and washed with ice-cold acetone/hexanes (1 : 1, 200 mL) to afford 39.1 g (76% yield). Analytical HPLC showed >98% UV purity. The enantiomeric excess (ee) was determined to be 99.8% (conditions: Chiralpak AD column, 4.6 x 250 mm, 10 pm; A = ethanol, B = 0.05% diethylamine/heptane; 85%B @1.0 mL/min. for 55 min. The retention times for R was 44.6 min and for S was 28.8 min). ‘H-NMR (DMSO-de) 5 2.48 (s, 3 H), 2.93 (dd, J= 13.4, 10.7 Hz, 1H), 3.10 (dd, J= 13.7, 4.9 Hz, 1H), 3.63 (s, 3H), 4.32-4.27 (m, 1 H), 4.97 (s, 2 H), 7.03 (s, 1 H), 7.24-7.22 (m, 2 H), 7.29 -7.27 (m, 3 H), 7.41 (s, 1 H), 7.83 (d, J= 8.2 Hz, 1H), 7.99 (s, 1H), 13.1 (s, 1 H); 13C-NMR (DMSO-de) 5 16.7, 36.5, 51.8, 56.0, 65.3, 117.6, 119.6, 122.7, 127.2, 127.4, 127.6, 128.2, 129.3, 133.4, 136.8, 139.2, 155.9, 172.4. Mass spec.: 368.16 (MH)+.
(R)-Methyl 2-amino-3-(7-methyl-lH-indazol-5-yl)propanoate. A Parr hydrogenation bottle was charged with (R)-methyl 2-(benzyloxycarbonyl)-3-(7-methyl-lH-indazol-5- yl)propanoate (11.0 g, 29.9 mmoles) and methanol (75 mL). The suspension was purged with nitrogen and treated with palladium (10% on charcoal, 700 mg). The bottle was shaken under hydrogen (15 psi) overnight. The mixture was filtered through a pad of celite to remove the catalyst. Concentration of the eluent gave 7.7 g (quant.) as an oil which was used without further purification. XH-NMR (CD3OD) 5 2.54 (s, 3 H), 2.98 (dd, J= 13.5, 7.0 Hz, 1 H), 3.09 (dd, J= 13.5, 5.9 Hz, 1 H), 3.68 (s, 3 H), 3.75 (dd, J= 7.0, 6.2 Hz, 1 H), 7.01 (s, 1 H), 7.39 (s, 1 H), 7.98 (s, 1 H). Mass spec.: 232.34 (M-H)’.
(R)-methyl 3-(7-methyl-lH-indazol-5-yl)-2-(4-(2-oxo-l,2-dihydroquinolin-3- yl)piperidine-l-carboxamido)propanoate. To a solution of (R)-methyl 2-amino-3-(7-methyl-lH- indazol-5-yl)propanoate hydrochloride (7.26 g, 27.0 mmoles) in dimethylformamide (50 mL) at room temperature was added N, A’-disuccinimidyl carbonate (7.60 g, 29.7 mmoles) followed by triethylamine (11.29 mL, 81 mmoles). The resulting mixture was stirred for 30 min and treated with 3-(piperidin-4-yl)quinolin-2(lH)-one (6.77 g, 29.9 mmoles) in portions. The reaction was allowed to stir for 24 h. The mixture was concentrated, dissolved in ethyl acetate, and washed sequentially with water, brine, and 0.5 N HC1 (2X). The organic phase was dried over magnesium sulfate, filtered, and concentrated. The resulting residue was purified by flash chromatography (silica gel, 20: 1 ethyl acetate/methanol) to give 11.9 g (78%). 1 H-NMR (CD3OD) 5 13.0 (s, 1 H), 11.8 (s, 1 H), 7.98 (s, 1 H), 7.63 (d, J= 7.6 Hz, 1 H), 7.57 (s, 1 H), 7.45 – 7.41 (m, 2 H), 7.27 (d, J= 8.2Hz, 1 H), 7.16 (t, J= 7.9 Hz, 1 H), 7.03 (s, 1 H), 6.85 (d, J= 7.9 Hz, 1 H), 4.31 – 4.26 (m, 1 H), 4.10 – 4.08 (m, 2 H), 3.60 (s, 3 H), 3.07 – 3.01 (m, 2 H), 2.93 – 2.88 (m, 1 H), 2.77 – 2.67 (m, 2 H), 2.48 (s, 3 H), 1.78 – 1.72 (m, 2 H), 1.34 – 1.26 (m, 2 H). Mass spec.: 488.52 (MH)+.
(R)-3-(7-methyl-lH-indazol-5-yl)-2-(4-(2-oxo-l,2-dihydroquinolin-3-yl)piperidine-l- carboxamido)propanoic acid. A solution of (R)-methyl 3-(7-methyl-lH-indazol-5-yl)-2-(4-(2- oxo-1, 2-dihydroquinolin-3-yl)piperidine-l-carboxamido)propanoate_(5.50 g, 11.3 mmoles) in tetrahydrofuran (50 mL) and methanol (10 mL) was cooled to 0°C. To this was added a cold (0°C) solution of lithium hydroxide monohydrate (0.95 g, 22.6 mmoles) in water (20 mL), dropwise over 15 min. The reaction was stirred at room temperature for additional 3 h. The mixture was concentrated to remove the organic solvents. The resulting residue was dissolved in a minimum amount of water, cooled to 0°C, and treated with cold (0°C) IN HC1 until pH 2 was attained. The resulting solid was collected by filtration, washed with cold water and ether, and then dried overnight under high vacuum to give 5.0 g (94%) as a white solid. ’H-NMR (DMSO- d6) 5 13.05 (bs, 1 H), 11.77 (s, 1 H), 7.98 (s, 1 H), 7.62 (d, J= 8.0 Hz, 1 H), 7.55 (s, 1 H), 7.44 (d, J= 8.2Hz, 1 H), 7.42 (s, 1 H), 7.27 (d, J= 8.2 Hz, 1 H), 7.16 (t, J= 7.6 Hz, 1 H), 7.05 (s, 1 H), 6.65 (d, J= 7.9 Hz, 1 H), 4.27 – 4.22 (m, 1 H), 4.10 – 4.07 (m, 2 H), 3.12 – 3.07 (m, 1 H), 3.03 – 2.99 (m, 1 H), 2.93 – 2.88 (m, 1 H), 2.77 – 2.66 (m, 2 H), 2.47 (s, 3 H), 1.77 – 1.74 (m, 2 H), 1.34 – 1.27 (m, 2 H). Mass spec.: 474.30 (MH)+.
(R)-N-(3-(7-methyl-lH-indazol-5-yl)-l-(4-(l-methylpiperidin-4-yl)piperazin-l-yl)-l- oxopropan-2-yl)-4-(2-oxo-l,2-dihydroquinolin-3-yl)piperidine-l-carboxamide (I). A flask was charged with (R)-3-(7-methyl-lH-indazol-5-yl)-2-(4-(2-oxo-l,2-dihydroquinolin-3- yl)piperidine-l-carboxamido)propanoic acid (2.9 g, 6.11 mmoles), triethylamine (3.00 mL, 21.5 mmoles), l-(l-methylpiperidin-4-yl)piperazine (1.23 g, 6.72 mmoles), and dimethylformamide (10 mL). The resulting solution was treated with 2-(lH-benzotriazole-l-yl)-l, 1,3,3- tetramethyluronium tetrafluoroborate (2.26 g, 7.03 mmoles) in portions. The reaction was allowed to stir at room temperature overnight. The mixture was concentrated under vacuum to remove dimethylformamide. The crude product was dissolved in 7% methanol in di chloromethane and purified by flash chromatography using 7% methanol in di chloromethane containing 2% of aqueous ammonium hydroxide as eluent. The pure fractions were collected and solvent was removed under vacuum. The desired product was crystallized from hot acetone to give the compound having Formula I in 77% yield. Analytical HPLC showed 99.0 % UV purity at 230 nm. The enantiomeric excess (ee) was determined to be >99.9% (conditions: Chiralpak AD column, 4.6 x 250 mm, 10 pm; eluent: 70% (0.05% diethylamine)/heptane/30%ethanol; @1.0 mL/min. for 45 min. The retention times were 18.7 min for R and 28.1 min for S). ‘H-NMR (500 MHz, DMSO-de) 5 ppm 13.01 (s, 1 H), 11.76 (s, 1 H), 7.96 (s, 1 H), 7.62 (d, J= 7.10 Hz, 1 H), 7.60 (s, 1 H), 7.42 (m, 1 H), 7.36 (s, 1 H), 7.26 (d, J = 8.25 Hz, 1 H), 7.14 (m, 1 H), 7.00 (s, 1 H), 6.69 (d, J= 8.25 Hz, 1 H), 4.78 (q, J= 7.79 Hz, 1 H), 4.14 (d, J= 12.37 Hz, 2 H), 3.54 (dd, J= 9.16, 4.58 Hz, 1 H), 3.24 (m, 1 H), 3.11 (m, 1 H), 2.97 (m, 1 H), 2.89 (m, 2 H), 2.69 (m, 4 H), 2.32 (m, 1 H), 2.21 (m, 1 H), 2.07 (m, 4 H), 1.95 (t, J= 8.25 Hz, 1 H), 1.87 (m, J= 11.28, 11.28, 3.55, 3.44 Hz, 1 H), 1.76 (t, J= 12.03 Hz, 2 H), 1.68 (t, J= 11.11 Hz, 2 H), 1.53 (t, J= 8.25 Hz, 1 H), 1.32 (m, 4 H), 1.16 (m, 2 H); 13C-NMR (DMSO-de) 5 16.80, 27.30, 30.51, 30.51, 30.67, 35.50, 38.04, 41.74, 44.00, 44.16, 45.35, 45.78, 48.14, 48.39, 51.45, 54.76, 54.76, 60.61, 114.53, 117.79, 119.29, 119.34, 121.57, 122.78, 127.46, 127.79, 129.29, 129.79, 133.31, 133.72, 136.98, 137.41, 139.12, 156.50, 161.50, 170.42.
Accurate mass analysis: m/z 639.3770, [MH]+, A = -0.2 ppm. Optical rotation: -27.36° @ 589 nm, concentration = 4.71 mg/mL in methanol. DESCRIPTION AND DOSAGE FORM
The physical and chemical properties of zavegepant (BHV-3500) drug substance mono-hydrochloride salt form are provided in Table 1.
A pyrimidine and thiazole derived ANTINEOPLASTIC AGENT and PROTEIN KINASE INHIBITOR of BCR-ABL KINASE. It is used in the treatment of patients with CHRONIC MYELOID LEUKEMIA who are resistant or intolerant to IMATINIB.
An orally bioavailable synthetic small molecule-inhibitor of SRC-family protein-tyrosine kinases. Dasatinib binds to and inhibits the growth-promoting activities of these kinases. Apparently because of its less stringent binding affinity for the BCR-ABL kinase, dasatinib has been shown to overcome the resistance to imatinib of chronic myeloid leukemia (CML) cells harboring BCR-ABL kinase domain point mutations. SRC-family protein-tyrosine kinases interact with a variety of cell-surface receptors and participate in intracellular signal transduction pathways; tumorigenic forms can occur through altered regulation or expression of the endogenous protein and by way of virally-encoded kinase genes. (NCI Thesaurus)
SPRYCEL (dasatinib) is an inhibitor of multiple tyrosine kinases.
The chemical name for dasatinib is N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2- methyl-4-pyrimidinyl]amino]-5-thiazolecarboxamide, monohydrate. The molecular formula is C22H26ClN7O2S • H2O, which corresponds to a formula weight of 506.02 (monohydrate).
The anhydrous free base has a molecular weight of 488.01. Dasatinib has the following chemical structure: Dasatinib is a white to off-white powder and has a melting point of 280°–286° C.
The drug substance is insoluble in water and slightly soluble in ethanol and methanol. SPRYCEL tablets are white to off-white, biconvex, film-coated tablets containing dasatinib, with the following inactive ingredients: lactose monohydrate, microcrystalline cellulose, croscarmellose sodium, hydroxypropyl cellulose, and magnesium stearate. The tablet coating consists of hypromellose, titanium dioxide, and polyethylene glycol
Drug Name:Dasatinib HydrateResearch Code:BMS-354825Trade Name:Sprycel®MOA:Kinase inhibitorIndication:Acute lymphoblastic leukaemia (ALL); Chronic myeloid leukemia (CML )Status:ApprovedCompany:Bristol-Myers Squibb (Originator)Sales:$1,620 Million (Y2015); $1,493 Million (Y2014); $1,280 Million (Y2013); $1,019 Million (Y2012); $803 Million (Y2011);ATC Code:L01XE06Approved Countries or Area
SPRYCEL (dasatinib) is a kinase inhibitor. The chemical name for dasatinib is N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-5-thiazolecarboxamide, monohydrate. The molecular formula is C22H26ClN7O2S • H2O, which corresponds to a formula weight of 506.02 (monohydrate). The anhydrous free base has a molecular weight of 488.01. Dasatinib has the following chemical structure:
Dasatinib is a white to off-white powder. The drug substance is insoluble in water and slightly soluble in ethanol and methanol.
SPRYCEL tablets are white to off-white, biconvex, film-coated tablets containing dasatinib, with the following inactive ingredients: lactose monohydrate, microcrystalline cellulose, croscarmellose sodium, hydroxypropyl cellulose, and magnesium stearate. The tablet coating consists of hypromellose, titanium dioxide, and polyethylene glycol.
Dasatinib hydrate was first approved by the U.S. Food and Drug Administration (FDA) on June 28, 2006, then approved by European Medicine Agency (EMA) on Nov 20, 2006, and approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) on Jan 21, 2009. It was developed and marketed as Sprycel® by Bristol Myers Squibb in the US.
Dasatinibhydrate is a kinase inhibitor.It is indicated for the treatment ofchronic myeloid leukemia and acutelymphoblastic leukemia.
Sprycel® is available as film-coatedtabletfor oral use, containing 20, 50, 70, 80, 100 or 140 mg offreeDasatinib. The recommended dose is 100 mg once daily forchronic myeloid leukemia. Another dose is 140 mg once daily for accelerated phase chronic myeloid leukemia, myeloid or lymphoid blast phase chronic myeloid leukemia, or Ph+ acutelymphoblastic leukemia.
Dasatinib, also known as BMS-354825, is an orally bioavailable synthetic small molecule-inhibitor of SRC-family protein-tyrosine kinases. Dasatinib binds to and inhibits the growth-promoting activities of these kinases. Apparently because of its less stringent binding affinity for the BCR-ABL kinase, dasatinib has been shown to overcome the resistance to imatinib of chronic myeloid leukemia (CML) cells harboring BCR-ABL kinase domain point mutations.
In the EU dasatinib is indicated for children with
newly diagnosed Philadelphia chromosome-positive chronic myelogenous leukaemia in chronic phase (Ph+ CML CP) or Ph+ CML CP resistant or intolerant to prior therapy including imatinib.[2]
newly diagnosed Ph+ acute lymphoblastic leukaemia (ALL) in combination with chemotherapy.[2]
newly diagnosed Ph+ CML in chronic phase (Ph+ CML-CP) or Ph+ CML-CP resistant or intolerant to prior therapy including imatinib.[2]
and adults with
newly diagnosed Philadelphia-chromosome-positive (Ph+) chronic myelogenous leukaemia (CML) in the chronic phase;[2]
chronic, accelerated or blast phase CML with resistance or intolerance to prior therapy including imatinib mesilate;[2]
Ph+ acute lymphoblastic leukaemia (ALL) and lymphoid blast CML with resistance or intolerance to prior therapy.[2]
On October 11, 2011, the U.S. Food and Drug Administration (FDA) announced that dasatinib may increase the risk of a rare but serious condition in which there is abnormally high blood pressure in the arteries of the lungs (pulmonary hypertension, PAH).[9] Symptoms of PAH may include shortness of breath, fatigue, and swelling of the body (such as the ankles and legs).[9] In reported cases, people developed PAH after starting dasatinib, including after more than one year of treatment.[9] Information about the risk was added to the Warnings and Precautions section of the Sprycel drug label.[9]
Dasatinib is an ATP-competitive protein tyrosine kinase inhibitor. The main targets of dasatinib are BCR/Abl (the “Philadelphia chromosome”), Src, c-Kit, ephrin receptors, and several other tyrosine kinases.[11] Strong inhibition of the activated BCR-ABL kinase distinguishes dasatinib from other CML treatments, such as imatinib and nilotinib.[11][12] Although dasatinib only has a plasma half-life of three to five hours, the strong binding to BCR-ABL1 results in a longer duration of action.[12]
Dasatinib was developed by collaboration of Bristol-Myers Squibb and Otsuka Pharmaceutical Co., Ltd,[13][14][15] and named for Bristol-Myers Squibb research fellow Jagabandhu Das, whose program leader says that the drug would not have come into existence had he not challenged some of the medicinal chemists‘ underlying assumptions at a time when progress in the development of the molecule had stalled.[16]
Society and culture
Legal status
Dasatinib was approved for used in the United States in June 2006 and in the European Union in November 2006[17][2]
In October 2010, dasatinib was approved in the United States for the treatment of newly diagnosed adults with Philadelphia chromosome positive chronic myeloid leukemia in chronic phase (CP-CML).[18]
In November 2017, dasatinib was approved in the United States for the treatment of children with Philadelphia chromosome-positive (Ph+) chronic myeloid leukemia (CML) in the chronic phase.[19]
Approval was based on data from 97 pediatric participants with chronic phase CML evaluated in two trials—a Phase I, open-label, non-randomized, dose-ranging trial and a Phase II, open-label, non-randomized trial.[19] Fifty-one participants exclusively from the Phase II trial were newly diagnosed with chronic phase CML and 46 participants (17 from the Phase I trial and 29 from the Phase II trial) were resistant or intolerant to previous treatment with imatinib.[19] The majority of participants were treated with dasatinib tablets 60 mg/m2body surface area once daily.[19] Participants were treated until disease progression or unacceptable toxicity.[19]
Economics
The Union for Affordable Cancer Treatment objected to the price of dasatinib, in a letter to the U.S. trade representative. The average wholesale price in the U.S. is $367 per day, twice the price in other high income countries. The price in India, where the average annual per capita income is $1,570, and where most people pay out of pocket, is Rs6627 ($108) a day. Indian manufacturers offered to supply generic versions for $4 a day, but, under pressure from the U.S., the Indian Department of Industrial Policy and Promotion refused to issue a compulsory license.[20]
Bristol-Myers Squibb justified the high prices of cancer drugs with the high R&D costs, but the Union of Affordable Cancer Treatment said that most of the R&D costs came from the U.S. government, including National Institutes of Health funded research and clinical trials, and a 50% tax credit. In England and Wales, the National Institute for Health and Care Excellence recommended against dasatinib because of the high cost-benefit ratio.[20]
The Union for Affordable Cancer Treatment said that “the dasatinib dispute illustrates the shortcomings of US trade policy and its impact on cancer patients”[20]
Dasatinib has been shown to eliminate senescent cells in cultured adipocyte progenitor cells.[21] Dasatinib has been shown to induce apoptosis in senescent cells by inhibiting Src kinase, whereas quercetin inhibits the anti-apoptotic protein Bcl-xL.[21] Administration of dasatinib along with quercetin to mice improved cardiovascular function and eliminated senescent cells.[22] Aged mice given dasatinib with quercetin showed improved health and survival.[22]
Giving dasatinib and quercetin to mice eliminated senescent cells and caused a long-term resolution of frailty.[23] A study of fourteen human patients suffering from idiopathic pulmonary fibrosis (a disease characterized by increased numbers of senescent cells) given dasatinib and quercetin showed improved physical function and evidence of reduced senescent cells.[21]Route 1
Balaji, N.; Sultana, Sayeeda. Trace level determination and quantification of potential genotoxic impurities in dasatinib drug substance by UHPLC/infinity LC. International Journal of Pharmacy and Pharmaceutical Sciences. Department of Chemistry. St. Peter’s University. Tamil Nadu, India 600054. Volume 8. Issue 10. Pages 209-216. 2016
SYN 2
Reference
Zhang, Shaoning; Wei, Hongtao; Ji, Min. Synthesis of dasatinib. Zhongguo Yiyao Gongye Zazhi. Dept. of Pharmaceutical Engineering, School of Chemistry & Chemical Engineering. Southeast University. Nanjing, Jiangsu Province, Peop. Rep. China 210096. Volume 41. Issue 3. Pages 161-163. 2010
SYN 3
Reference
Suresh, Garbapu; Nadh, Ratnakaram Venkata; Srinivasu, Navuluri; Yennity, Durgaprasad. A convenient new and efficient commercial synthetic route for dasatinib (Sprycel). Synthetic Communications. Division of Chemistry, Department of Science and Humanities. Vignan’s Foundation for Science Technology and Research University. Guntur, India. Volume 47. Issue 17. Pages 1610-1621. 2017
SYN 4
Reference
Chen, Bang-Chi; Zhao, Rulin; Wang, Bei; Droghini, Roberto; Lajeunesse, Jean; Sirard, Pierre; Endo, Masaki; Balasubramanian, Balu; Barrish, Joel C. A new and efficient preparation of 2-aminothiazole-5-carbamides: applications to the synthesis of the anticancer drug dasatinib. ARKIVOC (Gainesville, FL, United States). Discovery Chemistry. Bristol-Myers Squibb Research and Development. Princeton, USA 08543. Issue 6.Pages 32-38. 2010
SYN 5
Reference
An, Kang; Guan, Jianning; Yang, Hao; Hou, Wen; Wan, Rong. Improvement on the synthesis of Dasatinib. Jingxi Huagong Zhongjianti. College of Science. Nanjing University of Technology. Nanjing, Jiangsu Province, Peop. Rep. China 211816. Volume 41. Issue 2. Pages 42-44. 2011
To a solution of S— sec-butyl-amine (7.31 g, 0.1 mol) in chloroform (80 mL) at 0° C. was slowly added benzoyl isothiocyanate (13.44 mL, 0.1 mol). The mixture was allowed to warm to 10° C. and stirred for 10 min. The solvent was then removed under reduced pressure, and the residue was dissolved in MeOH (80 mL). An aqueous solution (10 mL) of NaOH (4 g, 0.1 mol) was added to this solution, and the mixture was stirred at 60° C. for another 2 h. The MeOH was then removed under reduced pressure, and the residue was stirred in water (50 mL). The precipitate was collected by vacuum filtration and dried to provide S-1-sec-butyl-thiourea (12.2 g, 92% yield). mp 133-134° C.; 1H NMR (500 MHz, DMSO-D6) δ 7.40 (s, 1H), 7.20 (br s, 1H), 6.76 (s, 1H), 4.04 (s, 1H), 1.41 (m, 2H), 1.03 (d, J=6.1 Hz, 3H), 0.81 (d, J=7.7 Hz, 3H); 13C NMR (125 MHz, DMSO-D6) δ 182.5, 50.8, 28.8, 19.9, 10.3; LRMS m/z 133.2 (M+H); Anal. Calcd for C5H12N2S: C, 45.41; H, 9.14; N, 21.18; S, 24.25. Found: C, 45.49; H, 8.88; N, 21.32; S, 24.27.
Example 2Preparation of Intermediate:
(R)-1-sec-Butylthiourea
(R)-1-sec-Butylthiourea was prepared in 92% yield according to the general method outlined for Example 1. mp 133-134° C.; 1H NMR(500 MHz, DMSO) δ 0.80(m, 3H, J=7.7), 1.02(d, 3H, J=6.1), 1.41(m, 2H), (3.40, 4.04)(s, 1H), 6.76(s, 1H), 7.20(s, br, 1H), 7.39(d, 1H, J=7.2); 13C NMR (500 MHz, DMSO) δ: 10.00, 19.56, 28.50, 50.20, 182.00; m/z 133.23 (M+H); Anal. Calcd for C5H12N2S: C, 45.41; H, 9.14; N, 21.18; S, 24.25. Found: C, 45.32; H, 9.15; N, 21.14; S, 24.38.
Example 3Preparation of:
To a solution of 3-amino-N-methyl-4-methylbenzamide hydrochloride (1.0 g, 5 mmol) in acetone (10 mL) at 0° C. was added pyridine (1.2 mL, 15 mmol) dropwise via syringe. 3-Methoxyacryloyl chloride (0.72 mL. 6.5 mmol) was added and the reaction stirred at room temperature for 1 h. The solution was cooled again to 0° C. and 1N HCl (1.5 mL) was added dropwise via pipet. The reaction mixture was stirred for 5 min, then water (8.5 mL) was added via an addition funnel. The acetone was removed in vacuo and the resulting solution stirred for 4h. Crystallization began within 15 min. After stirring for 4 h, the vessel was cooled in an ice bath for 30 min, filtered, and rinsed with ice cold water (2×3 mL) to give compound 3A (0.99 g, 78% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.95 (s, 1H), 8.12 (br s, 1H), 7.76 (s, 1H), 7.29 (m, 2H), 7.05 (d, J=7.9 Hz, 1H), 5.47 (d, J=12.3 Hz, 1H), 3.48 (s, 3H), 2.54 (d, J=4.7 Hz, 3H), 2.03 (s, 3H); HPLC rt 2.28 min (Condition A).
3B. Example 3To a 50 mL RBF containing the above compound 3A (0.5 g, 2.0 mmol) was added THF (2.5 mL) and water (2 mL), followed by NBS (0.40 g, 2.22 mmol), and the solution was stirred for 90 min. R-sec-butylthiourea (Ex. 2) (267 mg), was added, and the solution was heated to 75° C. for 8 h. Conc. NH4OH was added to adjust the pH to 10 followed by the addition of EtOH (15 mL). Water (15 mL) was added and the slurry stirred for 16 h, filtered, and washed with water to give Example 3 as a light brown solid (0.48 g, 69% yield, 98% purity). MS 347.1; HPLC 2.59.
Example 4Preparation of:
Example 4 is prepared following the methods of Example 3 but using the appropriate acryl benzamide and Example 1.
Example 5Preparation of:
N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (The Compound of Formula (IV))
5A. 1-(6-Chloro-2-methylpyrimidin-4-yl)thiourea
To a stirring slurry of 4-amino-5-chloro-2-methylpyrimidine (6.13 g, 42.7 mmol) in THF (24 mL) was added ethyl isothiocyanatoformate (7.5 mL, 63.6 mmol), and the mixture heated to reflux. After 5h, another portion of ethyl isothiocyanato formate (1.0 mL, 8.5 mmol) was added and after 10h, a final portion (1.5 mL, 12.7 mmol) was added and the mixture stirred 6h more. The slurry was evaporated under vacuum to remove most of the solvent and heptane (6 mL) added to the residue. The solid was collected by vacuum filtration and washed with heptane (2×5 mL) giving 8.01 g (68% yield) of the intermediate ethyl 6-chloro-2-methylpyrimidin-4-ylcarbamothioylcarbamate.A solution of ethyl 6-chloro-2-methylpyrimidin-4-ylcarbamothioylcarbamate (275 mg, 1.0 mmol) and 1N sodium hydroxide (3.5 eq) was heated and stirred at 50° C. for 2h. The resulting slurry was cooled to 20-22° C. The solid was collected by vacuum filtration, washed with water, and dried to give 185 mg of 1-(6-chloro-2-methylpyrimidin-4-yl)thiourea (91% yield). 1H NMR (400 MHz, DMSO-d6): δ2.51 (S, 3H), 7.05 (s, 1H), 9.35 (s,1H), 10.07 (s, 1H), 10.91 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ: 25.25, 104.56, 159.19, 159.33, 167.36, 180.91.
To a cold stirring solution of 2-chloro-6-methylaniline (59.5 g 0.42 mol) and pyridine (68 ml, 0.63 mol) in THF (600 mL) was added 3-ethoxyacryloyl chloride (84.7 g, 0.63 mol) slowly keeping the temp at 0-5° C. The mixture was then warmed and stirred for 2 h. at 20° C. Hydrochloric acid (1N, 115 mL) was added at 0-10° C. The mixture was diluted with water (310 mL) and the resulting solution was concentrated under vacuum to a thick slurry. The slurry was diluted with toluene (275 mL) and stirred for 15 min. at 20-22° C. then 1 h. at 0° C. The solid was collected by vacuum filtration, washed with water (2×75 mL) and dried to give 74.1 g (73.6% yield) of (E)-N-(2-chloro-6-methylphenyl)-3-ethoxyacrylamide). 1H NMR (400 Hz, DMSO-d6) δ 1.26 (t, 3H, J=7 Hz), 2.15 (s, 3H), 3.94 (q, 2H, J=7 Hz), 5.58 (d, 1H, J=12.4 Hz), 7.10-7.27 (m, 2H, J=7.5 Hz), 7.27-7.37 (d, 1H, J=7.5 Hz), 7.45(d, 1H, J=12.4 Hz), 9.28 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 14.57, 18.96, 67.17, 97.99, 126.80, 127.44, 129.07, 131.32, 132.89, 138.25, 161.09, 165.36.
To a mixture of compound 5B (5.00 g, 20.86 mmol) in 1,4-dioxane (27 mL) and water (27 mL) was added NBS (4.08 g, 22.9 mmol) at −10 to 0° C. The slurry was warmed and stirred at 20-22° C. for 3h. Thiourea (1.60 g, 21 mmol) was added and the mixture heated to 80° C. After 2h, the resulting solution was cooled to 20-22° and conc. ammonium hydroxide (4.2 mL) was added dropwise. The resulting slurry was concentrated under vacuum to about half volume and cooled to 0-5° C. The solid was collected by vacuum filtration, washed with cold water (10 mL), and dried to give 5.3 g (94.9% yield) of 2-amino-N-(2-chloro-6-methylphenyl)thiazole-5-carboxamide. 1H NMR (400 MHz, DMSO-d6) δ δ 2.19 (s, 3H), 7.09-7.29 (m, 2H, J=7.5), 7.29-7.43 (d, 1H, J=7.5), 7.61 (s, 2H), 7.85 (s, 1H), 9.63 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ: 18.18, 120.63, 126.84, 127.90, 128.86, 132.41, 133.63, 138.76, 142.88, 159.45, 172.02.
To a stirring solution of compound 5C (5.00 g, 18.67 mmol) and 4,6-dichloro-2-methylpyrimidine (3.65 g 22.4/mmol) in THF (65 mL) was added a 30% wt. solution of sodium t-butoxide in THF (21.1 g, 65.36 mmol) slowly with cooling to keep the temperature at 10-20° C. The mixture was stirred at room temperature for 1.5 h and cooled to 0-5° C. Hydrochloric acid, 2N (21.5 mL) was added slowly and the mixture stirred 1.75 h at 0-5° C. The solid was collected by vacuum filtration, washed with water (15 mL) and dried to give 6.63 g (86.4% yield) of compound 5D. 1H NMR (400 MHz, DMSO-d6) δ 2.23 (s, 3H), 2.58 (s, 3H), 6.94 (s, 1H), 7.18-7.34, (m, 2H, J=7.5), 7.34-7.46 (d, 1H, J=7.5), 8.31 (s, 1H), 10.02 (s, 1H), 12.25 (s, 1H).
5E. Example 5To a mixture of compound 5D (4.00 g, 10.14 mmol) and hydroxyethylpiperazine (6.60 g, 50.69 mmol) in n-butanol (40 mL) was added DIPEA (3.53 mL, 20.26 mmol). The slurry was heated at 118° C. for 4.5 h, then cooled slowly to room temperature. The solid was collected by vacuum filtration, washed with n-butanol (5 mL), and dried. The product (5.11 g) was dissolved in hot 80% EtOH—H2O (80 mL), and the solution was clarified by filtration. The hot solution was slowly diluted with water (15 mL) and cooled slowly to room temperature. The solid was collected by vacuum filtration, washed with 50% ethanol-water (5 mL) and dried affording 4.27 g (83.2% yield) of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide as monohydrate. 1H NMR (400 MHz, DMSO-d6) δ 2.23 (s, 3H), 2.40 (s, 3H), 2.42 (t, 2H, J=6), 2.48 (t, 4H, J=6.3), 3.50 (m, 4H), 3.53 (q, 2H, J=6), 4.45 (t, 1H, J=5.3), 6.04 (s, 1H), 7.25 (t, 1H, J=7.6), 7.27 (dd, 1H, J=7.6, 1.7), 7.40 (dd, 1H, J=7.6, 1.7), 8.21 (s, 1H), 9.87 (s, 1H), 11.47.
To a slurry of (E)-N-(2-chloro-6-methylphenyl)-3-ethoxyacrylamide 5B (120 mg, 0.50 mmol) in THF (0.75 ml) and water (0.5 mL) was added NBS (98 mg, 0.55 mmol) at 0° C. The mixture was warmed and stirred at 20-22° C. for 3h. To this was added 1-(6-chloro-2-methylpyrimidin-4-yl)thiourea 5A (100 mg, 0.49 mmol), and the slurry heated and stirred at reflux for 2h. The slurry was cooled to 20-22° C. and the solid collected by vacuum filtration giving 140 mg (71% yield) of 2-(6-chloro-2-methylpyrimidin-4-ylamino)-N-(2-chloro-6-methylphenyl)thiazole-5-carboxamide 5D. 1H NMR (400 MHz, DMSO-d6) δ 2.23 (s, 3H), 2.58 (s, 3H), 6.94 (s, 1H), 7.18-7.34, (m, 2H, J=7.5), 7.34-7.46 (d, 1H, J=7.5), 8.31 (s, 1H), 10.02 (s, 1H), 12.25 (s, 1H).Compound 5D was elaborated to N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide, following Step 5E.
2-piperazin-1-yl-ethanol (8.2 g, 63.1 mmol) was added to a solution of 4,6-dichloro-2-methylpyrimidine (5.2 g, 31.9 mmol) in dichloromethane (80 ml) at rt. The mixture was stirred for two hours and triethylamine (0.9 ml) was added. The mixture was stirred at rt for 20h. The resultant solid was filtered. The cake was washed with dichloromethane (20 ml). The filtrate was concentrated to give an oil. This oil was dried under high vacuum for 20h to give a solid. This solid was stirred with heptane (50 ml) at rt for 5h. Filtration gave 7C (8.13 g) as a white solid
7B. Example 7
To a 250 ml of round bottom flask were charged compound 5C (1.9 g, 7.1 mmol), compound 7C (1.5 g, 5.9 mmol), K2CO3 (16 g, 115.7 mmol), Pd (OAc)2 (52 mg, 0.23 mmol) and BINAP (291 mg, 0.46 mmol). The flask was placed under vacuum and flushed with nitrogen. Toluene was added (60 ml). The suspension was heated to 100-110° C. and stirred at this temperature for 20h. After cooling to room temperature, the mixture was applied to a silica gel column. The column was first eluted with EtOAC, and then with 10% of MeOH in EtOAC. Finally, the column was washed with 10% 2M ammonia solution in MeOH/90% EtOAC. The fractions which contained the desired product were collected and concentrated to give compound IV as a yellow solid (2.3 g).
Analytical MethodsSolid State Nuclear Magnetic Resonance (SSNMR)All solid-state C-13 NMR measurements were made with a Bruker DSX-400, 400 MHz NMR spectrometer. High resolution spectra were obtained using high-power proton decoupling and the TPPM pulse sequence and ramp amplitude cross-polarization (RAMP-CP) with magic-angle spinning (MAS) at approximately 12 kHz (A. E. Bennett et al, J. Chem. Phys., 1995, 103, 6951), (G. Metz, X. Wu and S. O. Smith, J. Magn. Reson. A., 1994, 110, 219-227). Approximately 70 mg of sample, packed into a canister-design zirconia rotor was used for each experiment. Chemical shifts (δ) were referenced to external adamantane with the high frequency resonance being set to 38.56 ppm (W. L. Earl and D. L. VanderHart, J. Magn. Reson., 1982, 48, 35-54).X-Ray Powder DiffractionOne of ordinary skill in the art will appreciate that an X-ray diffraction pattern may be obtained with a measurement error that is dependent upon the measurement conditions employed. In particular, it is generally known that intensities in a X-ray diffraction pattern may fluctuate depending upon measurement conditions employed. It should be further understood that relative intensities may also vary depending upon experimental conditions and, accordingly, the exact order of intensity should not be taken into account. Additionally, a measurement error of diffraction angle for a conventional X-ray diffraction pattern is typically about 5% or less, and such degree of measurement error should be taken into account as pertaining to the aforementioned diffraction angles. Consequently, it is to be understood that the crystal forms of the instant invention are not limited to the crystal forms that provide X-ray diffraction patterns completely identical to the X-ray diffraction patterns depicted in the accompanying Figures disclosed herein. Any crystal forms that provide X-ray diffraction patterns substantially identical to those disclosed in the accompanying Figures fall within the scope of the present invention. The ability to ascertain substantial identities of X-ray diffraction patterns is within the purview of one of ordinary skill in the art.X-Ray powder diffraction data for the crystalline forms of Compound (IV) were obtained using a Bruker GADDS (BRUKER AXS, Inc., 5465 East Cheryl Parkway Madison, Wis. 53711 USA) (General Area Detector Diffraction System) manual chi platform goniometer. Powder samples were placed in thin walled glass capillaries of 1 mm or less in diameter; the capillary was rotated during data collection. The sample-detector distance was 17 cm. The radiation was Cu Kα (45 kV 111 mA, λ=1.5418 Å). Data were collected for 3<2θ<35° with a sample exposure time of at least 300 seconds.Single Crystal X-RayAll single crystal data were collected on a Bruker-Nonius (BRUKER AXS, Inc., 5465 East Cheryl Parkway Madison, Wis. 53711 USA) Kappa CCD 2000 system using Cu Kα radiation (λ=1.5418 Å) and were corrected only for the Lorentz-polarization factors. Indexing and processing of the measured intensity data were carried out with the HKL2000 software package (Otwinowski, Z. & Minor, W. (1997) in Macromolecular Crystallography, eds. Carter, W. C. Jr & Sweet, R. M. (Academic, NY), Vol. 276, pp. 307-326) in the Collect program suite (Data collection and processing user interface: Collect: Data collection software, R. Hooft, Nonius B. V., 1998).The structures were solved by direct methods and refined on the basis of observed reflections using either the SDP (SDP, Structure Determination Package, Enraf-Nonius, Bohemia NY 11716 Scattering factors, including f′ and f″, in the SDP software were taken from the “International Tables for Crystallography”, Kynoch Press, Birmingham, England, 1974; Vol IV, Tables 2.2A and 2.3.1) software package with minor local modifications or the crystallographic package, MAXUS (maXus solution and refinement software suite: S. Mackay, C. J. Gilmore, C. Edwards, M. Tremayne, N. Stewart, K. Shankland. maXus: a computer program for the solution and refinement of crystal structures from diffraction data).The derived atomic parameters (coordinates and temperature factors) were refined through full matrix least-squares. The function minimized in the refinements was Σw(|Fo|−|Fc|)2. R is defined as Σ∥Fo|−|Fc∥/Σ|Fo| while Rw=[Σw(|Fo|−|Fc|)2/Σw|Fo|2]1/2 where w is an appropriate weighting function based on errors in the observed intensities. Difference maps were examined at all stages of refinement. Hydrogens were introduced in idealized positions with isotropic temperature factors, but no hydrogen parameters were varied.The derived atomic parameters (coordinates and temperature factors) were refined through full matrix least-squares. The function minimized in the refinements was Σw(|Fo|−|Fc|)2. R is defined as Σ∥Fo|−|Fc∥/Σ|Fo| while Rw=[Σw(|Fo|−|Fc|)2/Σw|Fo|2]1/2 where w is an appropriate weighting function based on errors in the observed intensities. Difference maps were examined at all stages of refinement. Hydrogens were introduced in idealized positions with isotropic temperature factors, but no hydrogen parameters were variedDifferential Scanning CalorimetryThe DSC instrument used to test the crystalline forms was a TA Instruments® model Q1000. The DSC cell/sample chamber was purged with 100 ml/min of ultra-high purity nitrogen gas. The instrument was calibrated with high purity indium. The accuracy of the measured sample temperature with this method is within about +/−1° C., and the heat of fusion can be measured within a relative error of about +/−5%. The sample was placed into an open aluminum DSC pan and measured against an empty reference pan. At least 2 mg of sample powder was placed into the bottom of the pan and lightly tapped down to ensure good contact with the pan. The weight of the sample was measured accurately and recorded to a hundredth of a milligram. The instrument was programmed to heat at 10° C. per minute in the temperature range between 25 and 350° C.The heat flow, which was normalized by a sample weight, was plotted versus the measured sample temperature. The data were reported in units of watts/gram (“W/g”). The plot was made with the endothermic peaks pointing down. The endothermic melt peak was evaluated for extrapolated onset temperature, peak temperature, and heat of fusion in this analysis.Thermogravimetric Analysis (TGA)The TGA instrument used to test the crystalline forms was a TAInstruments® model Q500. Samples of at least 10 milligrams were analyzed at a heating rate of 10° C. per minute in the temperature range between 25° C. and about 350° C.
Example 8Preparation of:
crystalline monohydrate of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV)An example of the crystallization procedure to obtain the crystalline monohydrate form is shown here:
Charge 48 g of the compound of formula (IV).
Charge approximately 1056 mL (22 mL/g) of ethyl alcohol, or other suitable alcohol.
Charge approximately 144 mL of water.
Dissolve the suspension by heating to approximately 75° C.
Optional: Polish filter by transfer the compound of formula (IV) solution at 75° C. through the preheated filter and into the receiver.
Rinse the dissolution reactor and transfer lines with a mixture of 43 mL of ethanol and 5 mL of water.
Heat the contents in the receiver to 75-80° C. and maintain 75-80° C. to achieve complete dissolution.Charge approximately 384 mL of water at a rate such that the batch temperature is maintained between 75-80° C.Cool to 75° C., and, optionally, charge monohydrate seed crystals. Seed crystals are not essential to obtaining monohydrate, but provide better control of the crystallization.
Cool to 70° C. and maintain 70° C. for ca. 1 h.
Cool from 70 to 5 C over 2 h, and maintain the temperature between 0 at 5° C. for at least 2 h.
Filter the crystal slurry.
Wash the filter cake with a mixture of 96 mL of ethanol and 96 mL of water.
Dry the material at ≦50° C. under reduced pressure until the water content is 3.4 to 4.1% by KF to afford 41 g (85 M %). Alternately, the monohydrate can be obtained by:
1) An aqueous solution of the acetate salt of compound IV was seeded with monohydrate and heated at 80° C. to give bulk monohydrate.
2) An aqueous solution of the acetate salt of compound IV was seeded with monohydrate. On standing several days at room temperature, bulk monohydrate had formed.
3) An aqueous suspension of compound IV was seeded with monohydrate and heated at 70° C. for 4 hours to give bulk monohydrate. In the absence of seeding, an aqueous slurry of compound IV was unchanged after 82 days at room temperature.
4) A solution of compound IV in a solvent such as NMP or DMA was treated with water until the solution became cloudy and was held at 75-85° C. for several hours. Monohydrate was isolated after cooling and filtering.
5) A solution of compound IV in ethanol, butanol, and water was heated. Seeds of monohydrate were added to the hot solution and then cooled. Monohydrate was isolated upon cooling and filtration.
One of ordinary skill in the art will appreciate that the monohydrate of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 1 or by a representative sampling of peaks as shown in Table 1.Representative peaks taken from the XRPD of the monohydrate of the compound of formula (IV) are shown in Table 1.TABLE 1 2-Theta d(Å) Height 17.994 4.9257 915 18.440 4.8075 338 19.153 4.6301 644 19.599 4.5258 361 21.252 4.1774 148 24.462 3.6359 250 25.901 3.4371 133 28.052 3.1782 153The XRPD is also characterized by the following list comprising 2θ values selected from the group consisting of: 4.6±0.2, 11.2±0.2, 13.8±0.2, 15.2±0.2, 17.9±0.2, 19.1±0.2, 19.6±0.2, 23.2±0.2, 23.6±0.2. The XRPD is also characterized by the list of 2θ values selected from the group consisting of: 18.0±0.2, 18.4±0.2, 19.2±0.2, 19.6±0.2, 21.2±0.2, 24.5±0.2, 25.9±0.2, and 28.0±0.2.Single crystal x-ray data was obtained at room temperature (+25° C.). The molecular structure was confirmed as a monohydrate form of the compound of Formula (IV).The following unit cell parameters were obtained for the monohydrate of the compound of formula (IV) from the x-ray analysis at 25° C.:a(Å)=13.8632(7); b(Å)=9.3307(3); c(Å)=38.390(2);V(Å3) 4965.9(4); Z′=1; Vm=621Space group PbcaMolecules/unit cell 8Density (calculated) (g/cm3) 1.354Wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).Single crystal x-ray data was also obtained at −50° C. The monohydrate form of the compound of Formula (IV) is characterized by unit cell parameters approximately equal to the following:Cell dimensions:
a(Å)=13.862(1);
b(Å)=9.286(1);
c(Å)=38.143(2);
Volume=4910(1) Å3Space group PbcaMolecules/unit cell 8Density (calculated) (g/cm3) 1.369wherein the compound is at a temperature of about −50° C.The simulated XRPD was calculated from the refined atomic parameters at room temperature.The monohydrate of the compound of formula (IV) is represented by the DSC as shown in FIG. 2. The DSC is characterized by a broad peak between approximately 95° C. and 130° C. This peak is broad and variable and corresponds to the loss of one water of hydration as seen in the TGA graph. The DSC also has a characteristic peak at approximately 287° C. which corresponds to the melt of the dehydrated form of the compound of formula (IV).The TGA for the monohydrate of the compound of Formula (IV) is shown in FIG. 2 along with the DSC. The TGA shows a 3.48% weight loss from 50° C. to 175° C. The weight loss corresponds to a loss of one water of hydration from the compound of Formula (IV).The monohydrate may also be prepared by crystallizing from alcoholic solvents, such as methanol, ethanol, propanol, i-propanol, butanol, pentanol, and water.
Example 9Preparation of:
crystalline n-butanol solvate of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV)The crystalline butanol solvate of the compound of formula (IV) is prepared by dissolving compound (IV) in 1-butanol at reflux (116-118° C.) at a concentration of approximately 1 g/25 mL of solvent. Upon cooling, the butanol solvate crystallizes out of solution. Filter, wash with butanol, and dry.The following unit cell parameters were obtained from the x-ray analysis for the crystalline butanol solvate, obtained at room temperature:a(Å)=22.8102(6); b(Å)=8.4691(3); c(Å)=15.1436(5); β=95.794(2);V(Å3) 2910.5(2); Z′=1; Vm=728Space group P21/aMolecules/unit cell 4Density (calculated) (g/cm3) 1.283Wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the butanol solvate of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 3 or by a representative sampling of peaks. Representative peaks for the crystalline butanol solvate are 2θ values of: 5.9±0.2, 12.0±0.2, 13.0±0.2, 17.7±0.2, 24.1±0.2, and 24.6±0.2.
Example 10Preparation of:
crystalline ethanol solvate of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV)
To a 100-mL round bottom flask was charged 4.00 g (10.1 mmol) of 5D (contained 2.3 Area % 5C) 6.60 g (50.7 mmol) of 7B, 80 mL of n-butanol and 2.61 g (20.2 mmol) of DIPEA. The resulting slurry was heated to 120° C. and maintained at 120° C. for 4.5 h whereby HPLC analysis showed 0.19 relative Area % of residual 5D to compound IV. The homogeneous mixture was cooled to 20° C. and left stirring overnight. The resulting crystals were filtered. The wet cake was washed twice with 10-mL portions of n-butanol to afford a white crystalline product. HPLC analysis showed this material to contain 99.7 Area % compound IV and 0.3 Area % 5C.The resulting wet cake was returned to the 100-mL reactor, and charged with 56 mL (12 mL/g) of 200 proof ethanol. At 80° C. an additional 25 mL of ethanol was added. To this mixture was added 10 mL of water resulting in rapid dissolution. Heat was removed and crystallization was observed at 75-77° C. The crystal slurry was further cooled to 20° C. and filtered. The wet cake was washed once with 10 mL of 1:1 ethanol: water and once with 10 mL of n-heptane. The wet cake contained 1.0% water by KF and 8.10% volatiles by LOD. The material was dried at 60° C./30 in Hg for 17 h to afford 3.55 g (70 M %) of material containing only 0.19% water by KF, 99.87 Area % by HPLC. The 1H NMR spectrum, however revealed that the ethanol solvate had been formed.The following unit cell parameters were obtained from the x-ray analysis for the crystalline ethanol solvate (di-ethanolate, E2-1), obtained at −40° C.:a(Å)=22.076(1); b(Å)=8.9612(2); c(Å)=16.8764(3); β=114.783(1);V(Å3) 3031.1(1); Z′=1; Vm=758Space group P21/aMolecules/unit cell 4Density (calculated) (g/cm3) 1.271Wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the ethanol solvate (E2-1) of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 4 or by a representative sampling of peaks. Representative peaks for the crystalline ethanol solvate are 2θ values of: 5.8±0.2, 11.3±0.2, 15.8±0.2, 17.2±0.2, 19.5±0.2, 24.1±0.2, 25.3±0.2, and 26.2±0.2.In addition, during the process to form the ethanolate (diethanolate) the formation of another ethanol solvate (½ ethanolate, T1E2-1) has been observed. To date this additional ethaonol solvate is known strictly as a partial desolvation product of the original diethanolate form E2-1, and has only been observed on occasion during crystallization of E2-1The following unit cell parameters were obtained from the x-ray analysis for the crystalline ½ ethanol solvate T1E2-1, obtained at −10° C.:a(Å)=22.03(2); b(Å)=9.20(1); c(Å)=12.31(1);β=93.49(6)V(Å3) 2491(4)); Z′=1; Vm=623;Space group P21/aMolecules/unit cell 4Density (calculated) (g/cm3) 1.363Wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the ethanol solvate (T1E2-1) of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 7 or by a representative sampling of peaks. Representative peaks for the crystalline ethanol solvate are 2θ values of: 7.20±0.2, 12.01±0.2, 12.81±0.2, 18.06±0.2, 19.30±0.2, and 25.24±0.2.
Example 11Preparation of:
crystalline N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV) (Neat form N-6)To a mixture of compound 5D (175.45 g, 0.445 mol) and hydroxyethylpiperazine (289.67 g, 2.225 mol) in NMP (1168 mL) was added DIPEA (155 mL, 0.89 mol). The suspension was heated at 110° C. (solution obtained) for 25 min., then cooled to about 90° C. The resulting hot solution was added dropwise into hot (80° C.) water (8010) mL, keeping the temperature at about 80° C. The resulting suspension was stirred 15 min at 80° C. then cooled slowly to room temperature. The solid was collected by vacuum filtration, washed with water (2×1600 mL) and dried in vacuo at 55-60° C. affording 192.45 g (88.7% yield) of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide. 1H NMR (400 MHz, DMSO-d6): δ 2.24 (s, 3H), 2.41 (s, 3H), 2.43 (t, 2H, J=6), 2.49 (t, 4H, J=6.3), 3.51 (m, 4H), 3.54 (q, 2H, J=6), 4.46 (t, 1H, J=5.3), 6.05 (s, 1H), 7.26 (t, 1H, J=7.6), 7.28 (dd, 1H, J=7.6, 1.7), 7.41 (dd, 1H, J=7.6, 1.7), 8.23 (s, 1H), 9.89 (s, 1H), 11.48. KF0.84; DSC: 285.25° C. (onset), 286.28° C. (max).The following unit cell parameters were obtained from the x-ray analysis for the neat crystalline compound IV, obtained at 23° C.:a(Å)=22.957(1); b(Å)=8.5830(5); c(Å)=13.803(3); β=112.039(6);V(Å3)=2521.0(5); Z′=1; Vm=630Space group P21/aMolecules/unit cell 4Density (calculated) (g/cm3) 1.286Wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the crystalline form of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 5 or by a representative sampling of peaks. Representative peaks for the crystalline neat form (N-6) are 2θ values of: 6.8±0.2, 11.1±0.2, 12.3±0.2, 13.2±0.2, 13.7±0.2, 16.7±0.2, 21.0±0.2, 24.3±0.2, and 24.8±0.2.
Example 12Preparation of:
crystalline N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV) (neatform T1H1-7)The title neat form may be prepared by heating the monohydrate form of the compound of formula (IV) above the dehydration temperature.The following unit cell parameters were obtained from the x-ray analysis for the neat crystalline (T1H1-7) compound IV, obtained at 25° C.:a(Å)=13.4916; b(Å)=9.3992(2); c(Å)=38.817(1);V(Å3)=4922.4(3); Z′=1; Vm=615Space group PbcaDensity (calculated) (g/cm3) 1.317Wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the neat crystalline form (T1H1-7) of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 6 or by a representative sampling of peaks. Representative peaks for the crystalline neat form (T1H1-7)) are 2θ values of: 8.0±0.2, 9.7±0.2, 11.2±0.2, 13.3±0.2, 17.5±0.2, 18.9±0.2, 21.0±0.2, 22.0±0.2.Obviously, numerous modifications and variations of the present invention are possible in light of the above teachings. It is therefore to be understood that within the scope of the appended claims, the invention may be practiced otherwise than as specifically described herein.PATENThttps://patents.google.com/patent/US8680103B2/enAminothiazole-aromatic amides of formula I
wherein Ar is aryl or heteroaryl, L is an optional alkylene linker, and R2, R3, R4, and R5, are as defined in the specification herein, are useful as kinase inhibitors, in particular, inhibitors of protein tyrosine kinase and p38 kinase. They are expected to be useful in the treatment of protein tyrosine kinase-associated disorders such as immunologic and oncological disorders [see, U.S. Pat. No. 6,596,746 (the ‘746 patent), assigned to the present assignee and incorporated herein by reference], and p38 kinase-associated conditions such as inflammatory and immune conditions, as described in U.S. patent application Ser. No. 10/773,790, filed Feb. 6, 2004, claiming priority to U.S. Provisional application Ser. No. 60/445,410, filed Feb. 6, 2003 (hereinafter the ‘410 application), both of which are also assigned to the present assignee and incorporated herein by reference.The compound of formula (IV), ′N-(2-Chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-5-thiazolecarboxamide, is an inhibitor of SRC/ABL and is useful in the treatment of oncological diseases.
Other approaches to preparing 2-aminothiazole-5-carboxamides are described in the ‘746 patent and in the ‘410 application. The ‘746 patent describes a process involving treatment of chlorothiazole with n-BuLi followed by reaction with phenyl isocyanates to give chlorothiazole-benzamides, which are further elaborated to aminothiazole-benzamide final products after protection, chloro-to-amino substitution, and deprotection, e.g.,
The ‘410 application describes a multi-step process involving first, converting N-unsubstituted aminothiazole carboxylic acid methyl or ethyl esters to bromothiazole carboxylic acid esters via diazotization with tert-butyl nitrite and subsequent CuBr2 treatment, e.g.,
then, hydrolyzing the resulting bromothiazole esters to the corresponding carboxylic acids and converting the acids to the corresponding acyl chlorides, e.g.,
then finally, coupling the acyl chlorides with anilines to afford bromothiazole-benzamide intermediates which were further elaborated to aminothiazole-benzamide final products, e.g.,
Other approaches for making 2-aminothiazole-5-carboxamides include coupling of 2-aminothiazole-5-carboxylic acids with amines using various coupling conditions such as DCC [Roberts et al, J. Med. Chem. (1972), 15, at p. 1310], and DPPA [Marsham et al., J. Med. Chem. (1991), 34, at p. 1594)].The above methods present drawbacks with respect to the production of side products, the use of expensive coupling reagents, less than desirable yields, and the need for multiple reaction steps to achieve the 2-aminothiazole-5-carboxamide compounds.Reaction of N,N-dimethyl-N′-(aminothiocarbonyl)-formamidines with α-haloketones and esters to give 5-carbonyl-2-aminothiazoles has been reported. See Lin, Y. et al, J. Heterocycl. Chem. (1979), 16, at 1377; Hartmann, H. et al, J. Chem. Soc. Perkin Trans. (2000), 1, at 4316; Noack, A. et al; Tetrahedron (2002), 58, at 2137; Noack, A.; et al. Angew. Chem. (2001), 113, at 3097; and Kantlehner, W. et al., J. Prakt. Chem./Chem.-Ztg. (1996), 338, at 403. Reaction of β-ethoxy acrylates and thioureas to prepare 2-aminothiazole-5-carboxylates also has been reported. See Zhao, R., et al., Tetrahedron Lett. (2001), 42, at 2101. However, electrophilic bromination of acrylanilide and crotonanilide has been known to undergo both aromatic bromination and addition to the α,β-unsaturated carbon-carbon double bonds. See Autenrieth, Chem. Ber. (1905), 38, at 2550; Eremeev et al., Chem. Heterocycl. Compd. Engl. Transl. (1984), 20, at 1102.New and efficient processes for preparing 2-aminothiazole-5-carboxamides are desired.
SUMMARY OF THE INVENTION
This invention is related to processes for the preparation of 2-aminothiazole-5-aromatic amides having the formula (I),
wherein L, Ar, R2, R3, R4, R5, and m are as defined below, comprising reacting a compound having the formula (II),
wherein Q is the group —O—P*, wherein P* is selected so that, when considered together with the oxygen atom to which P* is attached, Q is a leaving group, and Ar, L, R2, R3, and m are as defined below, with a halogenating reagent in the presence of water followed by a thiourea compound having the formula (III),
wherein, R4 and R5 are as defined below, to provide the compound of formula (I),
wherein,Ar is the same in formulae (I) and (II) and is aryl or heteroaryl;L is the same in formulae (I) and (II) and is optionally-substituted alkylene;R2 is the same in formulae (I) and (II), and is selected from hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, heteroaryl, cycloalkyl, and heterocyclo;R3 is the same in formulae (I) and (II), and is selected from hydrogen, halogen, cyano, haloalkyl, alkyl, substituted alkyl, alkenyl, substituted alkenyl, aryl, heteroaryl, cycloalkyl, and heterocyclo;R4 is (i) the same in each of formulae (I) and (III), and (ii) is independently selected from hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, heteroaryl, cycloalkyl, and heterocyclo, or alternatively, R4 is taken together with R5, to form heteroaryl or heterocyclo;R5 is (i) the same in each of formulae (I) and (III), and (ii) is independently selected from hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, heteroaryl, cycloalkyl, and heterocyclo, or alternatively, R5 is taken together with R4, to form heteroaryl or heterocyclo; andm is 0 or 1.Applicants have surprisingly discovered said process for converting β-(P*)oxy acryl aromatic amides and thioureas to 2-aminothiazole derivatives, wherein the aromatic amides are not subject to further halogenation producing other side products. Aminothiazole-aromatic amides, particularly, 2-aminothiazole-5-benzamides, can thus be efficiently prepared with this process in high yield.In another aspect, the present invention is directed to crystalline forms of the compound of formula (IV).
EXAMPLESExample 1Preparation of Intermediate:
(S)-1-sec-Butylthiourea
To a solution of S-sec-butyl-amine (7.31 g, 0.1 mol) in chloroform (80 mL) at 0° C. was slowly added benzoyl isothiocyanate (13.44 mL, 0.1 mol). The mixture was allowed to warm to 10° C. and stirred for 10 min. The solvent was then removed under reduced pressure, and the residue was dissolved in MeOH (80 mL). An aqueous solution (10 mL) of NaOH (4 g, 0.1 mol) was added to this solution, and the mixture was stirred at 60° C. for another 2 h. The MeOH was then removed under reduced pressure, and the residue was stirred in water (50 mL). The precipitate was collected by vacuum filtration and dried to provide S-1-sec-butyl-thiourea (12.2 g, 92% yield). mp 133-134° C.; 1H NMR (500 MHz, DMSO-D6) δ 7.40 (s, 1H), 7.20 (br s, 1H), 6.76 (s, 1H), 4.04 (s, 1H), 1.41 (m, 2H), 1.03 (d, J=6.1 Hz, 3H), 0.81 (d, J=7.7 Hz, 3H); 13C NMR (125 MHz, DMSO-D6) δ 182.5, 50.8, 28.8, 19.9, 10.3; LRMS m/z 133.2 (M+H); Anal. Calcd for C5H12N2S: C, 45.41; H, 9.14; N, 21.18; S, 24.25. Found: C, 45.49; H, 8.88; N, 21.32; S, 24.27.
Example 2Preparation of Intermediate:
(R)-1-sec-Butylthiourea
(R)-1-sec-Butylthiourea was prepared in 92% yield according to the general method outlined for Example 1. mp 133-134° C.; 1H NMR (500 MHz, DMSO) δ 0.80 (m, 3H, J=7.7), 1.02 (d, 3H, J=6.1), 1.41 (m, 2H), (3.40, 4.04) (s, 1H), 6.76 (s, 1H), 7.20 (s, br, 1H), 7.39 (d, 1H, J=7.2); 13C NMR (500 MHz, DMSO) δ: 10.00, 19.56, 28.50, 50.20, 182.00; m/z 133.23 (M+H); Anal. Calcd for C5H12N2S: C, 45.41; H, 9.14; N, 21.18; S, 24.25. Found: C, 45.32; H, 9.15; N, 21.14; S, 24.38.
Example 3Preparation of:
To a solution of 3-amino-N-methyl-4-methylbenzamide hydrochloride (1.0 g, 5 mmol) in acetone (10 mL) at 0° C. was added pyridine (1.2 mL, 15 mmol) dropwise via syringe. 3-Methoxyacryloyl chloride (0.72 mL 6.5 mmol) was added and the reaction stirred at room temperature for 1 h. The solution was cooled again to 0° C. and 1N HCl (1.5 mL) was added dropwise via pipette. The reaction mixture was stirred for 5 min, then water (8.5 mL) was added via an addition funnel. The acetone was removed in vacuo and the resulting solution stirred for 4 h. Crystallization began within 15 min. After stirring for 4 h, the vessel was cooled in an ice bath for 30 min, filtered, and rinsed with ice cold water (2×3 mL) to give compound 3A (0.99 g, 78% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.95 (s, 1H), 8.12 (br s, 1H), 7.76 (s, 1H), 7.29 (m, 2H), 7.05 (d, J=7.9 Hz, 1H), 5.47 (d, J=12.3 Hz, 1H), 3.48 (s, 3H), 2.54 (d, J=4.7 Hz, 3H), 2.03 (s, 3H); HPLC rt 2.28 min (Condition A).
3B. Example 3To a 50 mL RBF containing the above compound 3A (0.5 g, 2.0 mmol) was added THF (2.5 mL) and water (2 mL), followed by NBS (0.40 g, 2.22 mmol), and the solution was stirred for 90 min. R-sec-butylthiourea (Ex. 2) (267 mg), was added, and the solution was heated to 75° C. for 8 h. Conc. NH4OH was added to adjust the pH to 10 followed by the addition of EtOH (15 mL). Water (15 mL) was added and the slurry stirred for 16 h, filtered, and washed with water to give Example 3 as a light brown solid (0.48 g, 69% yield, 98% purity). MS 347.1; HPLC 2.59.
Example 4Preparation of:
Example 4 is prepared following the methods of Example 3 but using the appropriate acryl benzamide and Example 1.
Example 5Preparation of:
N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (The compound of Formula (IV))
5A. 1-(6-Chloro-2-methylpyrimidin-4-yl)thiourea
To a stirring slurry of 4-amino-5-chloro-2-methylpyrimidine (6.13 g, 42.7 mmol) in THF (24 mL) was added ethyl isothiocyanatoformate (7.5 mL, 63.6 mmol), and the mixture heated to reflux. After 5 h, another portion of ethyl isothiocyanato formate (1.0 mL, 8.5 mmol) was added and after 10 h, a final portion (1.5 mL, 12.7 mmol) was added and the mixture stirred 6 h more. The slurry was evaporated under vacuum to remove most of the solvent and heptane (6 mL) added to the residue. The solid was collected by vacuum filtration and washed with heptane (2×5 mL) giving 8.01 g (68% yield) of the intermediate ethyl 6-chloro-2-methylpyrimidin-4-ylcarbamothioylcarbamate.A solution of ethyl 6-chloro-2-methylpyrimidin-4-ylcarbamothioylcarbamate (275 mg, 1.0 mmol) and 1N sodium hydroxide (3.5 eq) was heated and stirred at 50° C. for 2 h. The resulting slurry was cooled to 20-22° C. The solid was collected by vacuum filtration, washed with water, and dried to give 185 mg of 1-(6-chloro-2-methylpyrimidin-4-yl)thiourea (91% yield). 1H NMR (400 MHz, DMSO-d6): δ2.51 (S, 3H), 7.05 (s, 1H), 9.35 (s, 1H), 10.07 (s, 1H), 10.91 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ: 25.25, 104.56, 159.19, 159.33, 167.36, 180.91.
To a cold stirring solution of 2-chloro-6-methylaniline (59.5 g 0.42 mol) and pyridine (68 ml, 0.63 mol) in THF (600 mL) was added 3-ethoxyacryloyl chloride (84.7 g, 0.63 mol) slowly keeping the temp at 0-5° C. The mixture was then warmed and stirred for 2 h. at 20° C. Hydrochloric acid (1N, 115 mL) was added at 0-10° C. The mixture was diluted with water (310 mL) and the resulting solution was concentrated under vacuum to a thick slurry. The slurry was diluted with toluene (275 mL) and stirred for 15 min. at 20-22° C. then 1 h. at 0° C. The solid was collected by vacuum filtration, washed with water (2×75 mL) and dried to give 74.1 g (73.6% yield) of (E)-N-(2-chloro-6-methylphenyl)-3-ethoxyacrylamide). 1H NMR (400 Hz, DMSO-d6) δ 1.26 (t, 3H, J=7 Hz), 2.15 (s, 3H), 3.94 (q, 2H, J=7 Hz), 5.58 (d, 1H, J=12.4 Hz), 7.10-7.27 (m, 2H, J=7.5 Hz), 7.27-7.37 (d, 1H, J=7.5 Hz), 7.45 (d, 1H, J=12.4 Hz), 9.28 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 14.57, 18.96, 67.17, 97.99, 126.80, 127.44, 129.07, 131.32, 132.89, 138.25, 161.09, 165.36.
To a mixture of compound 5B (5.00 g, 20.86 mmol) in 1,4-dioxane (27 mL) and water (27 mL) was added NBS (4.08 g, 22.9 mmol) at −10 to 0° C. The slurry was warmed and stirred at 20-22° C. for 3 h. Thiourea (1.60 g, 21 mmol) was added and the mixture heated to 80° C. After 2 h, the resulting solution was cooled to 20-22° and conc. ammonium hydroxide (4.2 mL) was added dropwise. The resulting slurry was concentrated under vacuum to about half volume and cooled to 0-5° C. The solid was collected by vacuum filtration, washed with cold water (10 mL), and dried to give 5.3 g (94.9% yield) of 2-amino-N-(2-chloro-6-methylphenyl)thiazole-5-carboxamide. 1H NMR (400 MHz, DMSO-d6) δ δ 2.19 (s, 3H), 7.09-7.29 (m, 2H, J=7.5), 7.29-7.43 (d, 1H, J=7.5), 7.61 (s, 2H), 7.85 (s, 1H), 9.63 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ: 18.18, 120.63, 126.84, 127.90, 128.86, 132.41, 133.63, 138.76, 142.88, 159.45, 172.02.
To a stirring solution of compound 5C (5.00 g, 18.67 mmol) and 4,6-dichloro-2-methylpyrimidine (3.65 g 22.4/mmol) in THF (65 mL) was added a 30% wt. solution of sodium t-butoxide in THF (21.1 g, 65.36 mmol) slowly with cooling to keep the temperature at 10-20° C. The mixture was stirred at room temperature for 1.5 h and cooled to 0-5° C. Hydrochloric acid, 2N (21.5 mL) was added slowly and the mixture stirred 1.75 h at 0-5° C. The solid was collected by vacuum filtration, washed with water (15 mL) and dried to give 6.63 g (86.4% yield) of compound 5D. 1H NMR (400 MHz, DMSO-d6) δ 2.23 (s, 3H), 2.58 (s, 3H), 6.94 (s, 1H), 7.18-7.34, (m, 2H, J=7.5), 7.34-7.46 (d, 1H, J=7.5), 8.31 (s, 1H), 10.02 (s, 1H), 12.25 (s, 1H).
5E. Example 5To a mixture of compound 5D (4.00 g, 10.14 mmol) and hydroxyethylpiperazine (6.60 g, 50.69 mmol) in n-butanol (40 mL) was added DIPEA (3.53 mL, 20.26 mmol). The slurry was heated at 118° C. for 4.5 h, then cooled slowly to room temperature. The solid was collected by vacuum filtration, washed with n-butanol (5 mL), and dried. The product (5.11 g) was dissolved in hot 80% EtOH—H2O (80 mL), and the solution was clarified by filtration. The hot solution was slowly diluted with water (15 mL) and cooled slowly to room temperature. The solid was collected by vacuum filtration, washed with 50% ethanol-water (5 mL) and dried affording 4.27 g (83.2% yield) of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide as monohydrate. 1H NMR (400 MHz, DMSO-d6) δ 2.23 (s, 3H), 2.40 (s, 3H), 2.42 (t, 2H, J=6), 2.48 (t, 4H, J=6.3), 3.50 (m, 4H), 3.53 (q, 2H, J=6), 4.45 (t, 1H, J=5.3), 6.04 (s, 1H), 7.25 (t, 1H, J=7.6), 7.27 (dd, 1H, J=7.6, 1.7), 7.40 (dd, 1H, J=7.6, 1.7), 8.21 (s, 1H), 9.87 (s, 1H), 11.47.
To a slurry of (E)-N-(2-chloro-6-methylphenyl)-3-ethoxyacrylamide 5B (120 mg, 0.50 mmol) in THF (0.75 ml) and water (0.5 mL) was added NBS (98 mg, 0.55 mmol) at 0° C. The mixture was warmed and stirred at 20-22° C. for 3 h. To this was added 1-(6-chloro-2-methylpyrimidin-4-yl)thiourea 5A (100 mg, 0.49 mmol), and the slurry heated and stirred at reflux for 2 h. The slurry was cooled to 20-22° C. and the solid collected by vacuum filtration giving 140 mg (71% yield) of 2-(6-chloro-2-methylpyrimidin-4-ylamino)-N-(2-chloro-6-methylphenyl)thiazole-5-carboxamide 5D. 1H NMR (400 MHz, DMSO-d6) δ 2.23 (s, 3H), 2.58 (s, 3H), 6.94 (s, 1H), 7.18-7.34, (m, 2H, J=7.5), 7.34-7.46 (d, 1H, J=7.5), 8.31 (s, 1H), 10.02 (s, 1H), 12.25 (s, 1H).Compound 5D was elaborated to N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide, following Step 5E.
2-Piperazin-1-yl-ethanol (8.2 g, 63.1 mmol) was added to a solution of 4,6-dichloro-2-methylpyrimidine (5.2 g, 31.9 mmol) in dichloromethane (80 ml) at rt. The mixture was stirred for two hours and triethylamine (0.9 ml) was added. The mixture was stirred at rt for 20 h. The resultant solid was filtered. The cake was washed with dichloromethane (20 ml). The filtrate was concentrated to give an oil. This oil was dried under high vacuum for 20 h to give a solid. This solid was stirred with heptane (50 ml) at rt for 5 h. Filtration gave 7C (8.13 g) as a white solid
7B. Example 7
To a 250 ml of round bottom flask were charged compound 5C (1.9 g, 7.1 mmol), compound 7C (1.5 g, 5.9 mmol), K2CO3 (16 g, 115.7 mmol), Pd (OAc)2 (52 mg, 0.23 mmol) and BINAP (291 mg, 0.46 mmol). The flask was placed under vacuum and flushed with nitrogen. Toluene was added (60 ml). The suspension was heated to 100-110° C. and stirred at this temperature for 20 h. After cooling to room temperature, the mixture was applied to a silica gel column. The column was first eluted with EtOAC, and then with 10% of MeOH in EtOAC. Finally, the column was washed with 10% 2M ammonia solution in MeOH/90% EtOAC. The fractions which contained the desired product were collected and concentrated to give compound IV as a yellow solid (2.3 g).
Analytical MethodsSolid State Nuclear Magnetic Resonance (SSNMR)All solid-state C-13 NMR measurements were made with a Bruker DSX-400, 400 MHz NMR spectrometer. High resolution spectra were obtained using high-power proton decoupling and the TPPM pulse sequence and ramp amplitude cross-polarization (RAMP-CP) with magic-angle spinning (MAS) at approximately 12 kHz (A. E. Bennett et al, J. Chem. Phys., 1995, 103, 6951), (G. Metz, X. Wu and S. O, Smith, J. Magn. Reson. A, 1994, 110, 219-227). Approximately 70 mg of sample, packed into a canister-design zirconia rotor was used for each experiment. Chemical shifts (6) were referenced to external adamantane with the high frequency resonance being set to 38.56 ppm (W. L. Earl and D. L. VanderHart, J. Magn. Reson., 1982, 48, 35-54).X-Ray Powder DiffractionOne of ordinary skill in the art will appreciate that an X-ray diffraction pattern may be obtained with a measurement error that is dependent upon the measurement conditions employed. In particular, it is generally known that intensities in a X-ray diffraction pattern may fluctuate depending upon measurement conditions employed. It should be further understood that relative intensities may also vary depending upon experimental conditions and, accordingly, the exact order of intensity should not be taken into account. Additionally, a measurement error of diffraction angle for a conventional X-ray diffraction pattern is typically about 5% or less, and such degree of measurement error should be taken into account as pertaining to the aforementioned diffraction angles. Consequently, it is to be understood that the crystal forms of the instant invention are not limited to the crystal forms that provide X-ray diffraction patterns completely identical to the X-ray diffraction patterns depicted in the accompanying Figures disclosed herein. Any crystal forms that provide X-ray diffraction patterns substantially identical to those disclosed in the accompanying Figures fall within the scope of the present invention. The ability to ascertain substantial identities of X-ray diffraction patterns is within the purview of one of ordinary skill in the art.X-Ray powder diffraction data for the crystalline forms of Compound (IV) were obtained using a Bruker GADDS (BRUKER AXS, Inc., 5465 East Cheryl Parkway Madison, Wis. 53711 USA) (General Area Detector Diffraction System) manual chi platform goniometer. Powder samples were placed in thin walled glass capillaries of 1 mm or less in diameter; the capillary was rotated during data collection. The sample-detector distance was 17 cm. The radiation was Cu Kα (45 kV 111 mA, λ=1.5418 Å). Data were collected for 3<2θ<35° with a sample exposure time of at least 300 seconds.Single Crystal X-RayAll single crystal data were collected on a Bruker-Nonius (BRUKER AXS, Inc., 5465 East Cheryl Parkway Madison, Wis. 53711 USA) Kappa CCD 2000 system using Cu Kα radiation (λ=1.5418 Å) and were corrected only for the Lorentz-polarization factors. Indexing and processing of the measured intensity data were carried out with the HKL2000 software package (Otwinowski, Z. & Minor, W. (1997) in Macromolecular Crystallography, eds. Carter, W. C. Jr. & Sweet, R. M. (Academic, NY), Vol. 276, pp. 307-326) in the Collect program suite (Data collection and processing user interface: Collect: Data collection software, R. Hooft, Nonius B. V., 1998).The structures were solved by direct methods and refined on the basis of observed reflections using either the SDP (SDP, Structure Determination Package, Enraf-Nonius, Bohemia N.Y. 11716 Scattering factors, including f′ and f″, in the SDP software were taken from the “International Tables for Crystallography”, Kynoch Press, Birmingham, England, 1974; Vol IV, Tables 2.2A and 2.3.1) software package with minor local modifications or the crystallographic package, MAXUS (maXus solution and refinement software suite: S. Mackay, C. J. Gilmore, C. Edwards, M. Tremayne, N. Stewart, K. Shankland. maXus: a computer program for the solution and refinement of crystal structures from diffraction data).The derived atomic parameters (coordinates and temperature factors) were refined through full matrix least-squares. The function minimized in the refinements was Σw(|Fo|−|Fc|)2. R is defined as Σ∥Fo|−|Fc∥/Σ|Fo| while Rw=[Σw(|Fo|−|Fc|)2/Σw|Fo|2]1/2 where w is an appropriate weighting function based on errors in the observed intensities. Difference maps were examined at all stages of refinement. Hydrogens were introduced in idealized positions with isotropic temperature factors, but no hydrogen parameters were varied.The derived atomic parameters (coordinates and temperature factors) were refined through full matrix least-squares. The function minimized in the refinements was Σw(|Fo|−|Fc|)2. R is defined as Σ∥Fo|−|Fc∥/Σ|Fo| while Rw=[Σw(|Fo|−|Fc|)2/Σw|Fo|2]1/2 where w is an appropriate weighting function based on errors in the observed intensities. Difference maps were examined at all stages of refinement. Hydrogens were introduced in idealized positions with isotropic temperature factors, but no hydrogen parameters were variedDifferential Scanning CalorimetryThe DSC instrument used to test the crystalline forms was a TA INSTRUMENTS° model Q1000. The DSC cell/sample chamber was purged with 100 ml/min of ultra-high purity nitrogen gas. The instrument was calibrated with high purity indium. The accuracy of the measured sample temperature with this method is within about +/−1° C., and the heat of fusion can be measured within a relative error of about +/−5%. The sample was placed into an open aluminum DSC pan and measured against an empty reference pan. At least 2 mg of sample powder was placed into the bottom of the pan and lightly tapped down to ensure good contact with the pan. The weight of the sample was measured accurately and recorded to a hundredth of a milligram. The instrument was programmed to heat at 10° C. per minute in the temperature range between 25 and 350° C.The heat flow, which was normalized by a sample weight, was plotted versus the measured sample temperature. The data were reported in units of watts/gram (“W/g”). The plot was made with the endothermic peaks pointing down. The endothermic melt peak was evaluated for extrapolated onset temperature, peak temperature, and heat of fusion in this analysis.Thermogravimetric Analysis (TGA)The TGA instrument used to test the crystalline forms was a TA INSTRUMENTS® model Q500. Samples of at least 10 milligrams were analyzed at a heating rate of 10° C. per minute in the temperature range between 25° C. and about 350° C.
Example 8Preparation of:
Crystalline monohydrate of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV)An example of the crystallization procedure to obtain the crystalline monohydrate form is shown here:Charge 48 g of the compound of formula (IV).Charge approximately 1056 mL (22 mL/g) of ethyl alcohol, or other suitable alcohol.Charge approximately 144 mL of water.Dissolve the suspension by heating to approximately 75° C.Optional: Polish filter by transfer the compound of formula (IV) solution at 75° C. through the preheated filter and into the receiver.Rinse the dissolution reactor and transfer lines with a mixture of 43 mL of ethanol and 5 mL of water.Heat the contents in the receiver to 75-80° C. and maintain 75-80° C. to achieve complete dissolution.Charge approximately 384 mL of water at a rate such that the batch temperature is maintained between 75-80° C.Cool to 75° C., and, optionally, charge monohydrate seed crystals. Seed crystals are not essential to obtaining monohydrate, but provide better control of the crystallization.Cool to 70° C. and maintain 70° C. for ca. 1 h.Cool from 70 to 5 C over 2 h, and maintain the temperature between 0 at 5° C. for at least 2 h.Filter the crystal slurry.Wash the filter cake with a mixture of 96 mL of ethanol and 96 mL of water.Dry the material at ≦50° C. under reduced pressure until the water content is 3.4 to 4.1% by KF to afford 41 g (85 M %).Alternately, the monohydrate can be obtained by:1) An aqueous solution of the acetate salt of compound IV was seeded with monohydrate and heated at 80° C. to give bulk monohydrate.2) An aqueous solution of the acetate salt of compound IV was seeded with monohydrate. On standing several days at room temperature, bulk monohydrate had formed.3) An aqueous suspension of compound IV was seeded with monohydrate and heated at 70° C. for 4 hours to give bulk monohydrate. In the absence of seeding, an aqueous slurry of compound IV was unchanged after 82 days at room temperature.4) A solution of compound IV in a solvent such as NMP or DMA was treated with water until the solution became cloudy and was held at 75-85° C. for several hours. Monohydrate was isolated after cooling and filtering.5) A solution of compound IV in ethanol, butanol, and water was heated. Seeds of monohydrate were added to the hot solution and then cooled. Monohydrate was isolated upon cooling and filtration.One of ordinary skill in the art will appreciate that the monohydrate of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 1 or by a representative sampling of peaks as shown in Table 1.Representative peaks taken from the XRPD of the monohydrate of the compound of formula (IV) are shown in Table 1.TABLE 1 2-Theta d(Å) Height 17.994 4.9257 915 18.440 4.8075 338 19.153 4.6301 644 19.599 4.5258 361 21.252 4.1774 148 24.462 3.6359 250 25.901 3.4371 133 28.052 3.1782 153The XRPD is also characterized by the following list comprising 2θ values selected from the group consisting of: 4.6±0.2, 11.2±0.2, 13.8±0.2, 15.2±0.2, 17.9±0.2, 19.1±0.2, 19.6±0.2, 23.2±0.2, 23.6±0.2. The XRPD is also characterized by the list of 2θ values selected from the group consisting of: 18.0±0.2, 18.4±0.2, 19.2±0.2, 19.6±0.2, 21.2±0.2, 24.5±0.2, 25.9±0.2, and 28.0±0.2.Single crystal x-ray data was obtained at room temperature (+25° C.). The molecular structure was confirmed as a monohydrate form of the compound of Formula (IV).The following unit cell parameters were obtained for the monohydrate of the compound of formula (IV) from the x-ray analysis at 25° C.:a(Å)=13.8632(7); b(Å)=9.3307(3); c(Å)=38.390(2);V(Å3) 4965.9(4); Z′=1; Vm=621Space group PbcaMolecules/unit cell 8Density (calculated) (g/cm3) 1.354wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).Single crystal x-ray data was also obtained at −50° C. The monohydrate form of the compound of Formula (IV) is characterized by unit cell parameters approximately equal to the following:Cell dimensions: a(Å)=13.862(1);
b(Å)=9.286(1);
c(Å)=38.143(2);
Volume=4910(1) Å3Space group PbcaMolecules/unit cell 8Density (calculated) (g/cm3) 1.369wherein the compound is at a temperature of about −50° C.The simulated XRPD was calculated from the refined atomic parameters at room temperature.The monohydrate of the compound of formula (IV) is represented by the DSC as shown in FIG. 2. The DSC is characterized by a broad peak between approximately 95° C. and 130° C. This peak is broad and variable and corresponds to the loss of one water of hydration as seen in the TGA graph. The DSC also has a characteristic peak at approximately 287° C. which corresponds to the melt of the dehydrated form of the compound of formula (IV).The TGA for the monohydrate of the compound of Formula (IV) is shown in FIG. 2 along with the DSC. The TGA shows a 3.48% weight loss from 50° C. to 175° C. The weight loss corresponds to a loss of one water of hydration from the compound of Formula (IV).The monohydrate may also be prepared by crystallizing from alcoholic solvents, such as methanol, ethanol, propanol, i-propanol, butanol, pentanol, and water.
Example 9Preparation of:
Crystalline n-butanol solvate of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV)The crystalline butanol solvate of the compound of formula (IV) is prepared by dissolving compound (IV) in 1-butanol at reflux (116-118° C.) at a concentration of approximately 1 g/25 mL of solvent. Upon cooling, the butanol solvate crystallizes out of solution. Filter, wash with butanol, and dry.The following unit cell parameters were obtained from the x-ray analysis for the crystalline butanol solvate, obtained at room temperature:a(Å)=22.8102(6); b(Å)=8.4691(3); c(Å)=15.1436(5); β=95.794(2);V(Å3) 2910.5(2); Z′=1; Vm=728Space group P21/aMolecules/unit cell 4Density (calculated) (g/cm3) 1.283wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the butanol solvate of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 3 or by a representative sampling of peaks. Representative peaks for the crystalline butanol solvate are 2θ values of: 5.9±0.2, 12.0±0.2, 13.0±0.2, 17.7±0.2, 24.1±0.2, and 24.6±0.2.
Example 10Preparation of:
Crystalline ethanol solvate of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV)
To a 100-mL round bottom flask was charged 4.00 g (10.1 mmol) of 5D (contained 2.3 Area % 5C) 6.60 g (50.7 mmol) of 7B, 80 mL of n-butanol and 2.61 g (20.2 mmol) of DIPEA. The resulting slurry was heated to 120° C. and maintained at 120° C. for 4.5 h whereby HPLC analysis showed 0.19 relative Area % of residual 5D to compound IV. The homogeneous mixture was cooled to 20° C. and left stirring overnight. The resulting crystals were filtered. The wet cake was washed twice with 10-mL portions of n-butanol to afford a white crystalline product. HPLC analysis showed this material to contain 99.7 Area % compound IV and 0.3 Area % 5C.The resulting wet cake was returned to the 100-mL reactor, and charged with 56 mL (12 mL/g) of 200 proof ethanol. At 80° C. an additional 25 mL of ethanol was added. To this mixture was added 10 mL of water resulting in rapid dissolution. Heat was removed and crystallization was observed at 75-77° C. The crystal slurry was further cooled to 20° C. and filtered. The wet cake was washed once with 10 mL of 1:1 ethanol:water and once with 10 mL of n-heptane. The wet cake contained 1.0% water by KF and 8.10% volatiles by LOD. The material was dried at 60° C./30 in Hg for 17 h to afford 3.55 g (70 M %) of material containing only 0.19% water by KF, 99.87 Area % by HPLC. The 1H NMR spectrum, however revealed that the ethanol solvate had been formed.The following unit cell parameters were obtained from the x-ray analysis for the crystalline ethanol solvate (di-ethanolate, E2-1), obtained at −40° C.:a(Å)=22.076(1); b(Å)=8.9612(2); c(Å)=16.8764(3); β=114.783(1);V(Å3) 3031.1(1); Z′=1; Vm=758Space group P21/aMolecules/unit cell 4Density (calculated) (g/cm3) 1.271wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the ethanol solvate (E2-1) of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 4 or by a representative sampling of peaks. Representative peaks for the crystalline ethanol solvate are 2θ values of: 5.8±0.2, 11.3±0.2, 15.8±0.2, 17.2±0.2, 19.5±0.2, 24.1±0.2, 25.3±0.2, and 26.2±0.2.In addition, during the process to form the ethanolate (diethanolate) the formation of another ethanol solvate (½ ethanolate, T1E2-1) has been observed. To date this additional ethanol solvate is known strictly as a partial desolvation product of the original diethanolate form E2-1, and has only been observed on occasion during crystallization of E2-1The following unit cell parameters were obtained from the x-ray analysis for the crystalline ½ ethanol solvate T1E2-1, obtained at −10° C.:a(Å)=22.03(2); b(Å)=9.20(1); c(Å)=12.31(1);β=93.49(6)V(Å3) 2491(4)); Z′=1; Vm=623;Space group P21/aMolecules/unit cell 4Density (calculated) (g/cm3) 1.363wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the ethanol solvate (T1E2-1) of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 7 or by a representative sampling of peaks. Representative peaks for the crystalline ethanol solvate are 2θ values of: 7.20±0.2, 12.01±0.2, 12.81±0.2, 18.06±0.2, 19.30±0.2, and 25.24±0.2.
Example 11Preparation of:
Crystalline N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV) (Neat form N-6)To a mixture of compound 5D (175.45 g, 0.445 mol) and hydroxyethylpiperazine (289.67 g, 2.225 mol) in NMP (1168 mL) was added DIPEA (155 mL, 0.89 mol). The suspension was heated at 110° C. (solution obtained) for 25 min., then cooled to about 90° C. The resulting hot solution was added dropwise into hot (80° C.) water (8010) mL, keeping the temperature at about 80° C. The resulting suspension was stirred 15 min at 80° C. then cooled slowly to room temperature. The solid was collected by vacuum filtration, washed with water (2×1600 mL) and dried in vacuo at 55-60° C. affording 192.45 g (88.7% yield) of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide. 1H NMR (400 MHz, DMSO-d6): δ 2.24 (s, 3H), 2.41 (s, 3H), 2.43 (t, 2H, J=6), 2.49 (t, 4H, J=6.3), 3.51 (m, 4H), 3.54 (q, 2H, J=6), 4.46 (t, 1H, J=5.3), 6.05 (s, 1H), 7.26 (t, 1H, J=7.6), 7.28 (dd, 1H, J=7.6, 1.7), 7.41 (dd, 1H, J=7.6, 1.7), 8.23 (s, 1H), 9.89 (s, 1H), 11.48. KF0.84; DSC: 285.25° C. (onset), 286.28° C. (max).The following unit cell parameters were obtained from the x-ray analysis for the neat crystalline compound IV, obtained at 23° C.:a(Å)=22.957(1); b(Å)=8.5830(5); c(Å)=13.803(3); β=112.039(6);V(Å3)=2521.0(5); Z′=1; Vm=630Space group P21/aMolecules/unit cell 4Density (calculated) (g/cm3) 1.286wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the crystalline form of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 5 or by a representative sampling of peaks. Representative peaks for the crystalline neat form (N-6) are 2θ values of: 6.8±0.2, 11.1±0.2, 12.3±0.2, 13.2±0.2, 13.7±0.2, 16.7±0.2, 21.0±0.2, 24.3±0.2, and 24.8±0.2.
Example 12Preparation of:
Crystalline N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV) (neat form T1H1-7)The title neat form may be prepared by heating the monohydrate form of the compound of formula (IV) above the dehydration temperature.The following unit cell parameters were obtained from the x-ray analysis for the neat crystalline (T1H1-7) compound IV, obtained at 25° C.:a(Å)=13.4916; b(Å)=9.3992(2); c(Å)=38.817(1);V(Å3)=4922.4(3); Z′=1; Vm=615Space group PbcaDensity (calculated) (g/cm3) 1.317wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the neat crystalline form (T1H1-7) of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 6 or by a representative sampling of peaks. Representative peaks for the crystalline neat form (T1H1-7)) are 2θ values of: 8.0±0.2, 9.7±0.2, 11.2±0.2, 13.3±0.2, 17.5±0.2, 18.9±0.2, 21.0±0.2, 22.0±0.2.Obviously, numerous modifications and variations of the present invention are possible in light of the above teachings. It is therefore to be understood that within the scope of the appended claims, the invention may be practiced otherwise than as specifically described herein. PAPERhttps://pubs.acs.org/doi/abs/10.1021/jm060727j
2-Aminothiazole (1) was discovered as a novel Src family kinase inhibitor template through screening of our internal compound collection. Optimization through successive structure−activity relationship iterations identified analogs 2 (Dasatinib, BMS-354825) and 12m as pan-Src inhibitors with nanomolar to subnanomolar potencies in biochemical and cellular assays. Molecular modeling was used to construct a putative binding model for Lck inhibition by this class of compounds. The framework of key hydrogen-bond interactions proposed by this model was in agreement with the subsequent, published crystal structure of 2 bound to structurally similar Abl kinase. The oral efficacy of this class of inhibitors was demonstrated with 12m in inhibiting the proinflammatory cytokine IL-2 ex vivo in mice (ED50 ∼ 5 mg/kg) and in reducing TNF levels in an acute murine model of inflammation (90% inhibition in LPS-induced TNFα production when dosed orally at 60 mg/kg, 2 h prior to LPS administration). The oral efficacy of 12m was further demonstrated in a chronic model of adjuvant arthritis in rats with established disease when administered orally at 0.3 and 3 mg/kg twice daily. Dasatinib (2) is currently in clinical trials for the treatment of chronic myelogenous leukemia.
PATENT
https://patents.google.com/patent/WO2019209908A1/enDasatinib (DAS), having the chemical designation N-(2-chloro-6-methylphenyl)-2- [[6-[4-(2-hydroxyethyl)-l-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-5- thiazolecarboxamide, monohydrate, is an orally bioavailable inhibitor of the receptor tyrosine kinase (RTK) epidermal growth factor receptor (ErbB; EGFR) family, with antineoplastic activity. Dasatinib has the following structure:
Dasatinib is commercially marketed under the name SPRY CEL® and is indicated for the treatment of patients with newly diagnosed Philadelphia chromosome-positive chronic myeloid leukemia in chronic phase, for the treatment of patients chronic, accelerated, or myeloid or lymphoid blast phase Philadelphia chromosome-positive chronic myeloid leukemia with resistance or intolerance to prior therapy and for the treatment of patients with Philadelphia chromosome-positive acute lymphoblastic leukemia with resistance or intolerance to prior therapy.Solid forms of dasatinib are described in U.S. Patent Nos. 7491725 (butanol solvate, monohydrate, diethanolate, hemi-ethanolate, anhydrous), 8680103 (butanol solvate, monohydrate, diethanolate, hemi-ethanolate, anhydrous), 7973045 (anhydrous), 8067423 (isopropyl alcohol solvate), 8242270 (butanol solvate, monohydrate, diethanolate, hemi- ethanolate, anhydrous), 8884013 (monohydrates), 9249134 (amorphous), 9456992 (solid dispersion nanoparticles), 9556164 (saccharin salt crystal) and 9884857 (saccharinate, glutarate, nicotinate); in U.S. Publication Nos. 20160250153 (solid dispersion nanoparticles), 20160264565 (Form-SDI), 20160361313 (solid dispersion nanoparticles), 20170183334 (salts) and 20140031352 (anti-oxidative acid); in International Publication Nos.W02010067374 (solvated forms and Form I), W02010139980, W02010139981,W02013065063 (anhydrous), W02017103057, W02017108605 (solid dispersion),WO2017134617 (amorphous), WO2014086326 (NMP, isoamyl-OH, 1, 3-propanediol process), WO2015107545, WO2015181573, WO2017134615 (PG solvate), W02010062715 (isosorbide dimethyl ether, N,N’-dimethylethylene urea, N,N’-dimethyl-N,N’-propylene urea), WO2010139979 (DCM, DMSP, monohydrate), WO2011095588 (anhydrate, hydrochloride, hemi-ethanol), W02012014149 (N-methylformamide) and W02017002131 (propandiol, monohydrate); and in Chinese Patent Nos. CN102643275, CN103059013, CN103819469, CN104341410. None of the references describe an ethyl formate solvate of dasatinib.Dasatinib co-crystals are described in U.S. Patent No. 9,340,536 (co-crystals selected from methyl-4-hydroxybenzoate, nicotinamide, ethyl gallate, methyl gallate, propyl gallate, ethyl maltol, vanillin, menthol, and (lR,2S,5R)-(-)-menthol) and International Publication No. W02016001025 (co-crystal selected from menthol or vanillin). None of the references describe dasatinib co-crystal comprising dasatinib and a second compound, as a co-crystal former, wherein the second compound is selected from butyl paraben, propyl paraben and ethyl vanillin.Dasatinib (DAS), having the chemical designation N-(2-chloro-6-methylphenyl)-2- [[6-[4-(2-hydroxyethyl)-l-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-5- thiazolecarboxamide, monohydrate, is an orally bioavailable inhibitor of the receptor tyrosine kinase (RTK) epidermal growth factor receptor (ErbB; EGFR) family, with antineoplastic activity. Dasatinib has the following structure:
Dasatinib is commercially marketed under the name SPRY CEL® and is indicated for the treatment of patients with newly diagnosed Philadelphia chromosome-positive chronic myeloid leukemia in chronic phase, for the treatment of patients chronic, accelerated, or myeloid or lymphoid blast phase Philadelphia chromosome-positive chronic myeloid leukemia with resistance or intolerance to prior therapy and for the treatment of patients with Philadelphia chromosome-positive acute lymphoblastic leukemia with resistance or intolerance to prior therapy.Solid forms of dasatinib are described in U.S. Patent Nos. 7491725 (butanol solvate, monohydrate, diethanolate, hemi-ethanolate, anhydrous), 8680103 (butanol solvate, monohydrate, diethanolate, hemi-ethanolate, anhydrous), 7973045 (anhydrous), 8067423 (isopropyl alcohol solvate), 8242270 (butanol solvate, monohydrate, diethanolate, hemi- ethanolate, anhydrous), 8884013 (monohydrates), 9249134 (amorphous), 9456992 (solid dispersion nanoparticles), 9556164 (saccharin salt crystal) and 9884857 (saccharinate, glutarate, nicotinate); in U.S. Publication Nos. 20160250153 (solid dispersion nanoparticles), 20160264565 (Form-SDI), 20160361313 (solid dispersion nanoparticles), 20170183334 (salts) and 20140031352 (anti-oxidative acid); in International Publication Nos.W02010067374 (solvated forms and Form I), W02010139980, W02010139981,W02013065063 (anhydrous), W02017103057, W02017108605 (solid dispersion),WO2017134617 (amorphous), WO2014086326 (NMP, isoamyl-OH, 1, 3-propanediol process), WO2015107545, WO2015181573, WO2017134615 (PG solvate), W02010062715 (isosorbide dimethyl ether, N,N’-dimethylethylene urea, N,N’-dimethyl-N,N’-propylene urea), WO2010139979 (DCM, DMSP, monohydrate), WO2011095588 (anhydrate, hydrochloride, hemi-ethanol), W02012014149 (N-methylformamide) and W02017002131 (propandiol, monohydrate); and in Chinese Patent Nos. CN102643275, CN103059013, CN103819469, CN104341410. None of the references describe an ethyl formate solvate of dasatinib.Dasatinib co-crystals are described in U.S. Patent No. 9,340,536 (co-crystals selected from methyl-4-hydroxybenzoate, nicotinamide, ethyl gallate, methyl gallate, propyl gallate, ethyl maltol, vanillin, menthol, and (lR,2S,5R)-(-)-menthol) and International Publication No. W02016001025 (co-crystal selected from menthol or vanillin). None of the references describe dasatinib co-crystal comprising dasatinib and a second compound, as a co-crystal former, wherein the second compound is selected from butyl paraben, propyl paraben and ethyl vanillin. hereafter. ClaimsHide Dependent What is claimed is:1. A dasatinib co-crystal comprising dasatinib and a second compound, wherein the second compound is selected from butyl paraben, propyl paraben and ethyl vanillin.2. The dasatinib co-crystal according to claim 1, wherein a molar ratio of the dasatinib to the second compound is about 1: 1.3. The dasatinib co-crystal according to claim 1, wherein the second compound is butyl paraben.4. The dasatinib co-crystal according to claim 3, wherein a molar ratio of the dasatinib to the butyl paraben is about 1 : 1.5. The dasatinib co-crystal according to claim 1, which is Form I co-crystal of dasatinib and butyl paraben.6. The dasatinib co-crystal according to claim 5, characterized by having at least 2 or more X-ray powder diffraction peaks selected from about 4.9, 9.8, 11.3, 14.9, 17.5, 20.8, 21.6, 22.6 and 25.4° 2Q degrees.7. The dasatinib co-crystal according to claim 5, characterized by a thermal event at about 287.3 °C, as measured by differential scanning calorimetry.8. The dasatinib co-crystal according to claim 5, characterized by a weight loss of 8.1% from about 70 °C through about 165 °C, as measured by thermal gravimetric analysis.9. The dasatinib co-crystal of claim 5 monoclinic, P2i/C.10. The dasatinib co-crystal d of claim 5 which has single crystal parametersa = 18.630 (2) Ab = 8.725 (1) Ac = 22.331 (2) Aa = g = 90°, b = 104.575 (8)°.11. The dasatinib co-crystal of claim 5 which has a cell volume is about 3512.9 A3.12. The dasatinib co-crystal according to claim 1, wherein the second compound is ethyl vanillin.13. The dasatinib co-crystal according to claim 9, wherein a molar ratio of the dasatinib to the ethyl vanillin is about 1 : 1.14. The dasatinib co-crystal according to claim 1, which is Form II co-crystal of dasatinib and ethyl vanillin.15. The dasatinib co-crystal according to claim 14, characterized by having at least 2 or more X-ray powder diffraction peaks selected from about 5.7, 10.9, 13.5, 17.1, 18.4, 19.4, 23.7 and 26.3° 2Q degrees.16. The dasatinib co-crystal according to claim 14, characterized by one or more thermal events selected from about 140 °C, about 181 °C, and about 293 °C, as measured by differential scanning calorimetry.17. The dasatinib co-crystal according to claim 14, characterized by a weight loss of 24.3% from about 120 through 250 °C, as measured by thermal gravimetric analysis.18. The dasatinib co-crystal of claim 14 monoclinic, P2i/n.19. The dasatinib co-crystal d of claim 14 which has single crystal parametersa = 18.452 (1) Ab = 9.441 (6) Ac = 19.377 (1) Aa = g = 90°, b = 108.78 (1)°.20. The dasatinib co-crystal of claim 5 which has a cell volume is about 3195.71 A3.21. The dasatinib co-crystal according to claim 1, wherein the second compound is propyl paraben.22. The dasatinib co-crystal according to claim 21, wherein a molar ratio of the dasatinib to the propyl paraben is about 1 : 1.23. The dasatinib co-crystal according to claim 1, which is Form III co-crystal ofdasatinib and propyl paraben.24. The dasatinib co-crystal according to claim 23, characterized by having at least 2 or more X-ray powder diffraction peaks selected from about 4.8, 9.6, 11.9, 14.8, 18.4, 22.2, 23.9 and 26.1° 2Q degrees.25. The dasatinib co-crystal of claim 23 monoclinic, P2i/n.26. The dasatinib co-crystal of claim 23 which has single crystal parametersa = 18.859 (9) Ab = 8.131 (6) Ac = 22.473 (1) Aa = g = 90°, b = 103.87(1)°.27. The dasatinib co-crystal of claim 23 which has a cell volume is about 3345.51 A3.28. An ethyl formate solvate of dasatinib.29. The ethyl formate solvate of dasatinib according to claim 28, wherein a molar ratio of the dasatinib to the ethyl formate is about 1 : 1.30. The ethyl formate solvate of dasatinib according to claim 1, which is Form I of ethyl formate solvate of dasatinib.31. The ethyl formate solvate of dasatinib according to claim 30, characterized by having at least 2 or more X-ray powder diffraction peaks selected from about 6.0, 12.1, 15.1, 18.0, 23.8 and 24.8° 2Q degrees.32. The ethyl formate solvate of dasatinib according to claim 30, characterized by athermal event at about 287.3 °C, as measured by differential scanning calorimetry.33. The ethyl formate solvate of dasatinib according to claim 30, characterized by aweight loss of 8.1% from about 70 °C through about 165 °C, as measured by thermal gravimetric analysis.34. The ethyl formate solvate of dasatinib of claim 23 orthorhombic, P2i/c.35. The ethyl formate solvate of dasatinib of claim 23 which has single crystal parameters a = 14.8928 (5) Ab = 8.3299 (3) Ac = 22.18990 (6) Aa = g =b = 90°.36. The ethyl formate solvate of dasatinib of claim 23 which has a cell volume is about 2731.9 A3.37. A pharmaceutical composition comprising a pharmaceutically effective amount of the dasatinib co-crystal according to claim 1 and pharmaceutically acceptable excipient.38. A method of treating disease in a patient comprising administering a pharmaceutical formulation according to claim 37 to the patient in need thereof.39. A method of treating disease according to claim 38, wherein the disease ismyelogenous leukemia.40. A method of treating disease according to claim 38, wherein the disease isPhiladelphia chromosome-positive (Ph+) chronic myeloid leukemia (CML) in chronic phase.41. A method of treating disease according to claim 38, wherein the disease Ph+ acute lymphoblastic leukemia (Ph+ ALL).42. A method of making the dasatinib co-crystal according to claim 1, comprisingdissolving dasatinib and a second compound, wherein the second compound is selected from the group consisting of butyl paraben, propyl paraben and ethyl vanillin, in heated methanol (-10: 1 – wt(mg)DAs:v(mL)MeOH and molD,\s:mohnci compound is 1 : 1.1) to form a clear solution, heating the solution under vacuum for about l8-20h to yield the dasatinib co-crystal.43. A process for the preparation Form II co-crystal of dasatinib and ethyl vanillin,according to claim 14, comprising: (g) dissolving Form I of ethyl formate solvate of dasatinib and ethyl vanillin in N-methyl-2-pyrrolidone to form a solution;(h) adding water to the solution;(i) stirring the solution for about 12-24 hours to form a slurry;(j) filtering the slurry to yield a precipitate;(k) washing the precipitate with water; and(l) drying the precipitate under vacuum with warming to yield Form II co crystal of dasatinib and ethyl vanillin.44. A process for the preparation of Form I of ethyl formate solvate of dasatinib,according to claim 30, comprising:(d) dissolving dasatinib in ethyl formate to form a solution;(e) stirring the solution for about 12-24 hours form a slurry;(f) filtering the slurry to yield Form I of ethyl formate solvate of dasatinib.45. A process for the preparation of Form I of ethyl formate solvate of dasatinib,according to claim 30, comprising:(g) dissolving dasatinib in N-Methyl-2-pyrrolidone to form a solution;(h) adding ethyl formate to the solution to form a slurry;(i) adding additional ethyl formate to the slurry;(j) stirring the slurry for about 2 hours;(k) filtering the slurry to yield a precipitate; and(l) washing the precipitate with ethyl formate to yield Form I of ethyl formate solvate of dasatinib.
ATENThttps://patents.google.com/patent/WO2013065063A1/en Dasatinib, N-(2-chloro-6-methylphenyl)-2- [(6-[4-(2-hydroxyl)- 1 -piperazinyl]-2-methyl-4-pyrimidinyl]amino]-5- thiazolecarboxamide compound having the following chemical structure of Formula (I)
Formula IAlso known as BMS-354825, it is a drug produced by Bristol Myers Squibb and sold under the trade name Sprycel. Dasatinib is an oral dual BCR/ABL and SRC family tyrosine kinase inhibitor approved for use in patients with chronic myelogenous leukemia (CML) after Imatinib treatment has failed and Philadelphia chromosome- positive acute lymphoblastic leukemia (Ph + ALL). It is also being assessed for use in metastatic melanoma.A preparation of Dasatinib is described in US patent No. 6596746 (B l ), where the process is done by reacting compound of the following formula III with N-(2- hydroxyethyl) piperazine at 80° C.
Formula IIIThe compound of Formula (I) and its preparation is described in US Patent No. 6596746, US patent application No. 2005/0176965 Al , and US patent application No. 2006/0004067 Al .l Polymorphism is defined as “the ability of a substance to exist as two or more crystalline phases that have different arrangement and /or conformations of the molecules in the crystal Lattice. Thus, in the strict sense, polymorphs are different crystalline forms of the same pure substance in which the molecules have different arrangements and / or different configurations of the molecules”. Different polymorphs may differ in their physical properties such as melting point, solubility, X-ray diffraction patterns, 1R etc. Polymorphic forms of a compound can be distinguished in the laboratory by analytical methods such as X-ray diffraction (XRD), Differential Scanning Calorimetry (DSC) and Infrared spectrometry (IR). Solvent medium and mode of crystallization play very important role in obtaining a crystalline form.The discovery of new polymorphic forms is a continuing goal of formulators. The new polymorphs may be advantageous for dosage form development and enhancing bioavailability owing to the altered physiochemical properties. Some form may turn out to be more efficacious. Discovering novel processes to prepare known polymorphic forms is also a primary goal of the pharmaceutical development scientists. New processes can provide novel intermediates or synthetic pathways that result in product with increased chemical and polymorphic purity in addition to providing cost and other advantages. There is thus a need to provide novel synthetic routes and intermediates that can realize these goals.Several crystalline forms of Dasatinib are described in the literature; these are designated as HI -7, BU-2, E2-1 , N-6, T1 H1 -7 and TIE2-1. Crystalline Dasatinib monohydrate (H I -7) and butanol solvate (BU-2) along with the processes for their preparation are described in WO 2005077945. In addition US 2006/0004067, which is continuation of US 2005215795 also describe two ethanol solvates (E2-1 ; TIE2-1) and two anhydrous forms (N-6 and T1 H1 -7).WO 2009053854 discloses various Dasatinib solvates including their crystalline form, amorphous form and anhydrous form.US patent No. 7973045 discloses the anhydrous form of Dasatinib and process for preparation thereof. The anhydrous form disclosed therein have typical characteristic XRD peaks at about 7.2, 1 1.9, 14.4, 16.5, 17.3, 19.1 , 20.8, 22.4, 23.8, 25.3 and 29.1 on the 2- theta value. WO 2010062715 discloses isosorbide dimethyl ether solvate, Ν,Ν’- dimethylethylene urea solvate and N,N’-dimethyl-N,N’-propylene urea solvate of Dasatinib.WO 2010067374 discloses novel crystalline form I, solvates of DMF, DMSO, toluene, isopropyl acetate and processes for their preparation.WO 2010139979 discloses MDC solvate and process of preparation, for use in the manufacture of pure Dasatinib.WO 2010139980 discloses a process for the preparation of crystalline Dasatinib monohydrate.The present invention is a step forward in this direction and provides a novel anhydrous form and process for its preparation, which can be used for the preparation of pure Dasatinib, in particularly Dasatinib monohydrate.The process for preparing Dasatinib monohydrate is described in US 2006/0004067. Further studies by the inventors have shown that the preparation of Dasatinib by using the method, which is disclosed in US 2006/0004067 yields the monohydrate with ~ 90% purity. Therefore the present invention provides a novel anhydrous form which can be used to get Dasatinib monohydrate with high yield and purity.Preparing API with increased purity is always an aim of the pharmaceutical development team. The inventors of the present invention have found that preparingDasatinib monohydrate using the novel anhydrous form of the present invention resulted in a highly pure product with a good yield.Scheme 1 shows a general process for the preparation of Dasatinib as disclosed in US 2006/0004067. Intermediate 3 and N-(2-hydroxyethyl) piperazine are heated together in a solvent system comprising n-butanol as a solvent and diisopropyl ethylamine (DIPEA) as a base. On cooling of the reaction mixture, Dasatinib precipitates out which is isolated by filtration.
DasatinibScheme 1Example – 1In a reaction vessel, N-(2-chloro-6-methylphenyl)-2-[(6-chloro-2-methyl-4- pyrimidinyl) amino] -5-thiazolecarboxamide (1 gm, 2.54 mmol) and N-(2- hydroxyethyl) piperazine (5.3 gm, 40.70 mmol) was added under stirring. The reaction mixture was heated at 80 °C for 2H. Acetonitrile was added into reaction mixture at 80 °C and stirred for 30 min. Cooled the suspension to room temperature and stirred for 30 min. Filtered, washed with acetonitrile and dried at 60 °C under vacuum to get 950 mg anhydrous N-(2-chloro-6-methylphenyl)-2-[(6-[4-(2-hydroxy 1)- 1 -piperaziny l]-2- methyl-4-pyrimidinyl]amino]-5-thiazole carboxamide (76.73 % Yield).HPLC Purity 99.90 %M/C by KF 0.12 %DSC 278.17 °CTGA 2.05 %XRD as provided in Fig. 2
Patent
Publication numberPriority datePublication dateAssigneeTitleUS7491725B22004-02-062009-02-17Bristol-Myers Squibb CompanyProcess for preparing 2-aminothiazole-5-aromatic carboxamides as kinase inhibitorsWO2009147238A12008-06-062009-12-10Boehringer Ingelheim International GmbhSolid pharmaceutical formulations comprising bibw 2992WO2010062715A22008-11-032010-06-03Teva Pharmaceutical Industries Ltd.Polymorphs of dasatinib and process for preparation thereofWO2010067374A22008-12-082010-06-17Hetero Research FoundationPolymorphs of dasatinibWO2010139980A12009-06-032010-12-09Generics [Uk] LimitedProcess for preparing crystalline dasatinib monohydrateWO2010139979A22009-06-032010-12-09Generics [Uk] LimitedProcesses for preparing crystalline formsWO2010139981A22009-06-032010-12-09Generics [Uk] LimitedProcesses for preparing crystalline formsWO2011003853A22009-07-062011-01-13Boehringer Ingelheim International GmbhProcess for drying of bibw2992, of its salts and of solid pharmaceutical formulations comprising this active ingredientUS7973045B22007-10-232011-07-05Teva Pharmaceutical Industries Ltd.Anhydrous form of dasatinib and process for preparation thereofWO2011095588A12010-02-042011-08-11Ratiopharm GmbhPharmaceutical composition comprising n-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-5-thiazolecarboxamidWO2012014149A12010-07-302012-02-02Ranbaxy Laboratories LimitedN-methylformamide solvate of dasatinibCN102643275A2011-02-212012-08-22江苏先声药物研究有限公司A new preparation method for Dasatinib N-6 crystal formCN103059013A2011-10-182013-04-24北京本草天源药物研究院New crystal of Dasatinib monohydrate and preparation method thereofWO2013065063A12011-11-032013-05-10Cadila Healthcare LimitedAnhydrous form of dasatinib, process for its preparation and its useUS20140031352A12012-07-242014-01-30Laurus Labs Private LimitedSolid forms of tyrosine kinase inhibitors, process for the preparation and their pharmaceutical composition thereofCN103819469A2012-11-162014-05-28重庆医药工业研究院有限责任公司Crystal form of dasatinib and preparation method for crystal form of dasatinibWO2014086326A12012-12-062014-06-12Zentiva, K.S.A method for the preparation and purification of new and known polymorphs and solvates of dasatinibUS8884013B22010-02-082014-11-11Nan Jing Cavendish Bio-Engineering Technology Co., Ltd.Polymorphs of Dasatinib, preparation methods and pharmaceutical compositions thereofCN104341410A2013-08-092015-02-11上海科胜药物研发有限公司New Dasatinib crystal form and preparation method thereofWO2015107545A12013-12-182015-07-23Dharmesh Mahendrabhai ShahWater soluble salts of dasatinib hydrateWO2015181573A12014-05-262015-12-03Egis Gyógyszergyár Zrt.Dasatinib saltsWO2016001025A12014-06-302016-01-07Basf SeMulticomponent crystals of dasatinib with menthol or vanillinUS9249134B22013-03-262016-02-02Cadila Healthcare LimitedProcess for preparation of amorphous form of dasatinibUS9340536B22012-06-152016-05-17Basf SeMulticomponent crystals comprising dasatinib and selected co-crystal formersUS20160250153A12012-01-132016-09-01Xspray Microparticles AbNovel methodsUS20160264565A12013-11-082016-09-15Shilpa Medicare LimitedCrystalline dasatinib processWO2017002131A12015-06-292017-01-05Msn Laboratories Private LimitedCrystalline forms of n-(2-chloro-6-methy]phenvn-2-[f6-[4-(2-hvdroxvethvl)-l- piperazinvil-2-methvl-4-pvrimidinvllaminol-5-thiazolecarboxamide and their process thereofUS9556164B22013-07-252017-01-31Basf SeSalts of Dasatinib in crystalline formWO2017103057A12015-12-162017-06-22Synthon B.V.Pharmaceutical composition comprising anhydrous dasatinibWO2017108605A12015-12-222017-06-29Synthon B.V.Pharmaceutical composition comprising amorphous dasatinibWO2017134615A12016-02-032017-08-10Dr. Reddy’s Laboratories LimitedSolid state forms of dasatinib and processes for their preparationWO2017134617A12016-02-032017-08-10Dr. Reddy’s Laboratories LimitedProcess for the preparation of amorphous dasatinibUS9884857B22013-07-252018-02-06Basf SeSalts of dasatinib in amorphous form
PATENTSPublication numberPriority datePublication dateAssigneeTitleUS20060079563A1 *1999-04-152006-04-13Jagabandhu DasCyclic protein tyrosine kinase inhibitorsUS20070219370A1 *2006-03-152007-09-20Bristol-Myers Squibb CompanyProcess for preparing n-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl]amino] -5-thiazolecarboxamide and related metabolites thereofUS20080275009A1 *2005-09-212008-11-06Bristol-Myers Squibb CompanyOral administration of n-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-1,3-thiazole-5-carboxamide and salts thereofUS20090030203A1 *2005-08-052009-01-29Bristol-Myers Squibb CompanyPreparation of 2-amino-thiazole-5-carboxylic-acid derivativesUS20090118297A1 *2007-10-232009-05-07Ondrej SimoPolymorphs of dasatinib and process for preparation thereofWO2012014149A12010-07-302012-02-02Ranbaxy Laboratories LimitedN-methylformamide solvate of dasatinibUS20120309968A1 *2010-02-082012-12-06Nan Jing Cavendish Bio-Engineering Technology Co., Ltd.Polymorphs of dasatinib, preparation methods and pharmaceutical compositions thereofUS8530492B22009-04-172013-09-10Nektar TherapeuticsOligomer-protein tyrosine kinase inhibitor conjugatesUS8680103B22004-02-062014-03-25Bristol-Myers Squibb CompanyProcess for preparing 2-aminothiazole-5-aromatic carboxamides as kinase inhibitorsWO2014102759A22012-12-312014-07-03Ranbaxy Laboratories LimitedProcess for the preparation of dasatinib and its intermediatesUS8816077B22009-04-172014-08-26Nektar TherapeuticsOligomer-protein tyrosine kinase inhibitor conjugatesUS20150057446A1 *2012-04-202015-02-26Shilpa Medicare LimitedProcess for preparing dasatinib monohydrateWO2016001025A12014-06-302016-01-07Basf SeMulticomponent crystals of dasatinib with menthol or vanillinUS9340536B22012-06-152016-05-17Basf SeMulticomponent crystals comprising dasatinib and selected co-crystal formersUS9556164B22013-07-252017-01-31Basf SeSalts of Dasatinib in crystalline formUS9884857B22013-07-252018-02-06Basf SeSalts of dasatinib in amorphous formWO2018078392A12016-10-292018-05-03Cipla LimitedPolymorphs of dasatinibWO2018100585A12016-12-012018-06-07Natco Pharma LimitedAn improved process for the preparation of dasatinib polymorphWO2018134189A12017-01-202018-07-26Cerbios-Pharma SaCo-crystal of an antitumoral compoundWO2018134190A12017-01-202018-07-26Cerbios-Pharma SaCo-crystals of an antitumoral compoundUS10174018B22016-12-132019-01-08Princeton Drug Discovery IncProtein kinase inhibitorsWO2019209908A12018-04-252019-10-31Johnson Matthey Public Limited CompanyCrystalline forms of dasatinibUS10722484B22016-03-092020-07-28K-Gen, Inc.Methods of cancer treatmentUS10799459B12019-05-172020-10-13Xspray Microparticles AbRapidly disintegrating solid oral dosage forms containing dasatinibFamily To Family CitationsUS7396935B22003-05-012008-07-08Bristol-Myers Squibb CompanyAryl-substituted pyrazole-amide compounds useful as kinase inhibitorsUS7652146B2 *2004-02-062010-01-26Bristol-Myers Squibb CompanyProcess for preparing 2-aminothiazole-5-carboxamides useful as kinase inhibitorsTW200600513A *2004-06-302006-01-01Squibb Bristol Myers CoA method for preparing pyrrolotriazine compoundsPE20061394A1 *2005-03-152006-12-15Squibb Bristol Myers CoMetabolites of n- (2-chloro-6-methylphenyl) -2 – [[6- [4- (2-hydroxyethyl) -1-piperazinyl] -2-methyl-4-pyrimidinyl] amino] -5-thiazolecarboxamidesUS20060235006A1 *2005-04-132006-10-19Lee Francis YCombinations, methods and compositions for treating cancerPL1885339T32005-05-052015-12-31Bristol Myers Squibb Holdings IrelandFormulations of a src/abl inhibitorWO2008076883A22006-12-152008-06-26Abraxis Bioscience, Inc.Triazine derivatives and their therapeutical applicationsWO2010139979A22009-06-032010-12-09Generics [Uk] LimitedProcesses for preparing crystalline formsWO2010139980A1 *2009-06-032010-12-09Generics [Uk] LimitedProcess for preparing crystalline dasatinib monohydrateEP2359813A12010-02-042011-08-24Ratiopharm GmbHPharmaceutical composition comprising N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-5-thiazolecarboxamidCN102250084A *2010-02-082011-11-23南京卡文迪许生物工程技术有限公司Dasatinib polymorphic substance as well as preparation method and pharmaceutical composition thereofCN102643275B *2011-02-212016-04-20江苏先声药物研究有限公司The preparation method that a kind of Dasatinib N-6 crystal formation is newWO2013065063A12011-11-032013-05-10Cadila Healthcare LimitedAnhydrous form of dasatinib, process for its preparation and its useUS20150087687A12012-03-232015-03-26Dennis BrownCompositions and methods to improve the therapeutic benefit of indirubin and analogs thereof, including meisoindigoSG10201610869TA2012-06-262017-02-27Del Mar PharmaceuticalsMethods for treating tyrosine-kinase-inhibitor-resistant malignancies in patients with genetic polymorphisms or ahi1 dysregulations or mutations employing dianhydrogalactitol, diacetyldianhydrogalactiCN103664929B *2012-08-302016-08-03石药集团中奇制药技术(石家庄)有限公司Dasatinib polycrystalline form medicament and preparation methodCN102838595B *2012-09-132014-09-24江苏奥赛康药业股份有限公司Preparation method of high-purity dasatinib and by-product of dasatinibCN103819469A *2012-11-162014-05-28重庆医药工业研究院有限责任公司Crystal form of dasatinib and preparation method for crystal form of dasatinibCZ306598B62012-12-062017-03-22Zentiva, K.S.A method of preparation and purification of new and known polymorphs and dasatinib solvatesCN105764502A2013-07-262016-07-13现代化制药公司Combinatorial methods to improve the therapeutic benefit of bisantrene and analogs and derivatives thereofCN103408542B *2013-08-132016-06-29南京优科生物医药研究有限公司A kind of preparation method of highly purified Dasatinib anhydrideWO2015049645A2 *2013-10-042015-04-09Alembic Pharmaceuticals LimitedAn improved process for the preparation of dasatinibCZ306732B62013-12-192017-05-31Zentiva, K.S.A method of preparation of the anhydrous polymorphic form of N-6 DasatinibCN104788445B *2015-04-102017-06-23山东新时代药业有限公司A kind of synthetic method of Dasatinib intermediateCN106668022B *2015-11-052020-09-15武汉应内药业有限公司Application of aminothiazole MyD88 specific inhibitor TJM2010-5* Cited by examiner, † Cited by third party, ‡ Family to family citation
^World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
https://patents.google.com/patent/US8884013B2/enDasatinib, with the trade name SPRYCEL™, is a oral tyrosine kinase inhibitor and developed by BMS Company. It is used to cure adult chronic myelogenous leukemia (CML), acute lymphatic leukemia (ALL) with positive Philadelphia chromosome, etc. Its chemical name is N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidyl]amino]-5-thiazolformamide and its chemical structure is as following:
Five polymorphs of Dasatinib and the preparation methods thereof were described by Bristol-Myers Squibb in the Chinese Patent Application No. CN200580011916.6 (publication date is 13 Jun. 2007). The preparation methods instructed in this document are:Monohydrate: Dasatinib (48 g) was added into ethanol (1056 mL 22 ml/g) and water (144 mL), and dissolved by heating to 75° C.; the mixture was purified, filtrated and transferred to the receiver. The solution reactor and transferring pipes were washed with the mixture of ethanol (43 mL) and water (5 mL). The solution was heated to 75˜80° C. to be soluble completely and water (384 mL) was heated and the temperature of the solution was kept between 75° C. and 80° C. The seed crystal of monohydrate (preferable) was added when cooling to 75° C., and keep the temperature at 70° C. for 1 h; cooling to 5° C. within 2 h and keeping the temperature at 0˜5° C. for 2 h. The slurry was filtrated and the filter cake was washed by the mixture of ethanol (96 mL) and water (96 mL); after being dried under vacuum≦50° C. 41 g of solid was obtained.Butanol solvate: under refluxing (116° C.˜118° C.), Dasatinib was dissolved in 1-butanol (about 1 g/25 mL) to yield crystalline butanol solvate of Dasatinib. When cooling, this butanol solvate was recrystallized from solution. The mixture was filtrated and the filter cake was dried after being washed with butanol.Ethanol solvate: 5D (4 g, 10.1 mmol), 7B (6.6 g, 50.7 mmol), n-bubanol (80 mL) and DIPEA (2.61 g, 20.2 mmol)) were added into a 100 ml round flask. The obtained slurry was heated to 120° C. and kept the temperature for 4.5 h, and then cooled to 20° C. and stirred over night. The mixture was filtrate, and the wet filter cake was washed with n-butanol (2×10 mL) to yield white crystal product. The obtained wet filter cake was put back to the 100 ml reactor and 56 mL (12 mL/g) of 200 proof ethanol was added. Then additional ethanol (25 mL) was added at 80° C., and water (10 mL) was added into the mixture to make it dissolved rapidly. Heat was removed and crystallization was observed at 75° C.˜77° C. The crystal slurry was further cooled to 20° C. and filtrated. The wet filter cake was washed with ethanol:water (1:1, 10 mL) once and then washed with n-heptane (10 mL) once. After that it was dried under the condition of 60° C./30 in Hg for 17 h to yield 3.55 g of substance only containing 0.19% water.Neat form of N-6: DIPEA (155 mL, 0.89 mmol) was added into the mixture of compound 5D (175.45 g, 0.445 mol) and hydroxyethylpiperazine (289.67 g, 2.225 mol) in NMP (1168 mL). The suspension was heated at 110° C. for 25 min to be solution, which was then cooled down to about 90° C. The obtained solution was added dropwise into hot water (80° C., 8010 mL), and the mixture was stirred at 80° C. with heat preservation for 15 min and cooled to room temperature slowly. The solid was filtrated under vacuum and collected, washed by water (2×1600 mL) and dried under vacuum at 55° C.˜60° C. to give 192.45 of compound.Neat form of T1H1-7 (neat form and pharmaceutically acceptable carrier): monohydrate of Dasatinib was heated over dehydrate temperature to yield.Because Dasatinib is practically insoluble in water or organic solvent (e.g. methanol, ethanol, propanol, isopropanol, butanol, pentanol, etc.), even in the condition of heating, a large amount (over 100 times) of solvent is needed, which is disadvantageous in industrial production; in addition, with the method described in the Patent document of CN200580011916.6, the related substances in products can not be lowed effectively during the process of crystal preparation to improve the products quality.In terms of polymorphs of drug, each polymorph has different chemical and physical characteristics, including melting point, chemical stability, apparent solubility, rate of dissolution, optical and mechanical properties, vapor pressure as well as density. Such characteristics can directly influence the work-up or manufacture of bulk drug and formulation, and also affect the stability, solubility and bioavailability of formulation. Consequently, polymorph of drug is of great importance to quality, safety and efficacy of pharmaceutical preparation. When it comes to Dasatinib, there are still needs in the art for new polymorphs suitable for industrial production and with excellent physical and chemical properties as well.Example 1Preparation of the Polymorph IA. Dasatinib (10 g) and DMSO (40 ml) were added into a flask and heated up to 60˜70° C. by stirring, after dissolving, the mixture (120 mL) of water and acetone (1:1) was added under heat preservation. When crystal was precipitated, cooled it down to 0° C. to grow the grains for 10 minutes. Filtrate it and the cake was washed by water and then by the mixture of water and acetone (1:1). After that it was dried under −0.095 MPa at about 50° C. using phosphorus pentoxide as drying aid to give 7.7 g of white solid. Yield was 77%.Contrasts Index of raw material Items before transformation Index of Polymorph I Appearance off-white powder White crystal powder Related substance 0.85% 0.07% KF moisture 0.67% 3.59% 70~150 0.72% 3.63% TGA weight loss The following items of products prepared by Method A were detected: microscope-crystal form (See. FIG. 1); XRPD Test (See. FIG. 2), IR Test (See. FIG. 3), DSC-TGA Test (See. FIG. 4-1, 4–2), 13C Solid-state NMR Test (See. FIG. 5).B. Dasatinib (10 g) and DMSO (40 ml) were added into a flask and heated slowly up to 60˜70° C. by stirring, after dissolving, the mixture (160 mL) of ethanol and water (1:1) was added under heat preservation. When crystal was precipitated, cooled it down to 0° C. to grow the grains for 10 minutes. Filtrate it and the cake was washed by the mixture of ethanol and water (1:1) and dried under −0.095 MPa at about 50° C. using phosphorus pentoxide as drying aid to give 7.7 g of white solid. Yield was 87%.Contrasts Index of raw material Items before transformation Index of Polymorph I Appearance off-white powder White crystal powder Related substance 0.85% 0.08% KF moisture 0.67% 3.58% 70~150 0.72% 3.67% TGA weight lossHPLC.Related Substances DeterminationHPLC conditions and system applicability: octadecylsilane bonded silica as the filler; 0.05 mol/L of potassium dihydrogen phosphate (adjusted to pH 2.5 by phosphoric acid, 0.2% triethylamine)-methanol (45:55) as the mobile phase; detection wavelength was 230 nm; the number of theoretical plates should be not less than 2000, calculated according to the peak of Dasatinib. The resolution of the peak of Dasatinib from the peaks of adjacent impurities should meet requirements.Determination method: sample was dissolved in mobile phase to be the solution containing 0.5 mg per milliliter. 20 μL of such solution was injected into liquid chromatograph, and chromatogram was recorded until the sixfold retention time of major component peak. If there were impurities peaks in the chromatogram of sample solution, total impurities and any single impurity were calculated by normalization method on the basis of peak area.Stability of Polymorph in the FormulationsThe XRPD patterns of capsules and tablets respectively prepared in the Example 3 and Example 4 have been tested, and compared with XRPD characteristic peaks of Polymorph I of Dasatinib prepared by the Method A in the Example 1 in the present invention, as listed in the following table:Bulk Drug Capsules 1 Capsules 2 Tablets 2 (Polymorph (Polymorph (Polymorph Tablets 1 (Polymorph I) I) I) (Polymorph I) I) 2θ 2θ 2θ 2θ 2θ 9.060 9.080 9.070 9.060 9.070 11.100 11.120 11.110 11.100 11.110 13.640 13.670 13.650 13.640 13.650 15.100 15.120 15.110 15.100 15.110 17.820 17.840 17.830 17.820 17.820 19.380 19.400 19.390 19.380 19.390 22.940 22.970 22.950 22.950 22.950The results in the above-mentioned comparative table have shown that the crystal form had substantially no change after Polymorph I of Dasatinib in the invention were prepared into capsules or tablets by the formulation process.In addition, The relative substances of capsules and tablets respectively prepared in the Example 3 and Example 4 have been tested, and compared with those of Polymorph I of Dasatinib prepared by the Method A in the Example 1 in the present invention, as listed in the following table:Bulk Drug (Polymorph I) Capsules 1 Capsules 2 Tablets 1 Tablets 2 0.07% 0.08% 0.08% 0.07% 0.08%The results in the above-mentioned comparative table have shown that the Polymorph I of Dasatinib was stable, and there were no significantly changes in respect to the relative substances, after Polymorph I of Dasatinib in the invention were prepared into capsules or tablets by the formulation process.INDUSTRIAL APPLICATIONThe present invention provides novel polymorphs of Dasatinib, preparing methods, and pharmaceutical composition comprising them. These polymorphs have better physicochemical properties, are more stable and are more suitable for industrial scale production, furthermore, are suitable for long-term storage, and are advantageous to meet the requirements of formulation process and long-term storage of formulations. The preparation technique of this invention was simple, quite easy for operation and convenient for industrial production, and the quality of the products was controllable with paralleled yields. In addition, by the methods of polymorph preparation in this invention, the amount of organic solvent used in crystal transformation could be reduced greatly, which led to reduced cost of products; organic solvents in Class III with low toxicity could be used selectively to prepare the polymorphs of this invention, reducing the toxic effects of the organic solvents potentially on human body to some extent.PATENThttps://patents.google.com/patent/WO2010067374A2/enDasatinib are antineoplastic agents, which were disclosed in WO Patent Publication No. 00/62778 and U.S. Patent No. 6,596,746. Dasatinib, chemically N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4- pyrimidinyl]amino]-5-thiazolecarboxamide, is represented by the following structure:
Polymorphism is defined as “the ability of a substance to exist as two or more crystalline phases that have different arrangement and /or conformations of the molecules in the crystal Lattice. Thus, in the strict sense, polymorphs are different crystalline forms of the same pure substance in which the molecules have different arrangements and / or different configurations of the molecules”. Different polymorphs may differ in their physical properties such as melting point, solubility, X-ray diffraction patterns, etc. Although those differences disappear once the compound is dissolved, they can appreciably influence pharmaceutically relevant properties of the solid form, such as handling properties, dissolution rate and stability. Such properties can significantly influence the processing, shelf life, and commercial acceptance of a polymorph. It is therefore important to investigate all solid forms of a drug, including all polymorphic forms, and to determine the stability, dissolution and flow properties of each polymorphic form. Polymorphic forms of a compound can be distinguished in the laboratory by analytical methods such as X-ray diffraction (XRD), Differential Scanning Calorimetry (DSC) and Infrared spectrometry (IR).Solvent medium and mode of crystallization play very important role in obtaining a crystalline form over the other. Dasatinib can exist in different polymorphic forms, which differ from each other in terms of stability, physical properties, spectral data and methods of preparation.U.S. Patent Application No. 2005/0215795 A1 (herein after referred to as the 795 patent application) described five crystalline forms of dasatinib (monohydrate, butanol solvate, ethanol solvate, neat form (N-6) and neat form (T1H1-7)), characterized by powder X-ray diffraction (P-XRD) pattern.According to the ‘795 patent application, dasatinib monohydrate is characterized by an X-ray powder diffraction pattern having peaks expressed as 2Θ at approximately 18.0, 18.4, 19.2, 19.6, 21.2, 24.5, 25.9 and 28.0 ± 0.2 degrees. As per the process exemplified in the ‘795 patent application, dasatinb monohydrate can be obtained in dasatinib, by heating and dissolving the dasatinib in an ethanol and water mixture. Crystallizing the monohydrate from the ethanol and water mixture and cooled to get dasatinib monohydrate.According to the ‘795 patent application, dasatinib crystalline butanol solvate is characterized by an X-ray powder diffraction pattern having peaks expressed as 2Θ at approximately 5.9, 12.0, 13.0, 17.7, 24.1 and 24.6 ± 0.2 degrees.According to the 795 patent application, dasatinib crystalline ethanol solvate is characterized by an X-ray powder diffraction pattern having peaks expressed as 2Θ at approximately 5.8, 11.3, 15.8, 17.2, 19.5, 24.1, 25.3 and 26.2 ± 0.2 degrees.According to the 795 patent application, dasatinib crystalline neat form (N-6) is characterized by an X-ray powder diffraction pattern having peaks expressed as 2Θ at approximately 6.8, 11.1, 12.3, 13.2, 13.7, 16.7, 21.0, 24.3 and 24.8 ± 0.2 degrees.According to the 795 patent application, dasatinib crystalline neat form (T1H1-7) is characterized by an X-ray powder diffraction pattern having peaks expressed as 2Θ at approximately 8.0, 9.7, 11.2, 13.3, 17.5, 18.9, 21.0 and 22.0 ± 0.2 degrees.U.S. Patent application No. 2006/0094728 disclosed ethanolate form (T1E2-1) of dasatinib, characterized by an X-ray powder diffraction pattern having peaks expressed as 2Θ at approximately 7.2, 12.0, 12.8, 18.0, 19.3 and 25.2 ± 0.2 degrees. We have discovered novel crystalline form of dasatinib, dasatinib dimethylformamide solvate, dasatinib dimethyl sulfoxide solvate, dasatinib toluene solvate and dasatinib isopropyl acetate solvate.Another object of the present invention is to provide process for preparing the novel crystalline form of dasatinib, dasatinib dimethylformamide solvate, dasatinib dimethyl sulfoxide solvate, dasatinib toluene solvate, dasatinib isopropyl acetate solvate and known crystalline dasatinib monohydrate.Still another object of the present invention is to provide pharmaceutical compositions containing the novel crystalline form of dasatinib.Reference Example2-(6-Cholro-2-methylpyrimidin-4-yl-amino)-N-(2-chloro-6-methylphenyl) thiazole-5-carboxamide (15 gm) was added to 1-(2-hydroxyethyl)piperazine at 250C and heated to 850C, stirred for 2 hours 30 minutes at 850C. To the solution was added water (500 ml) at 800C and slowly cooled to 250C, stirred for 1 hour at 250C. The solid was collected by filtration and the solid was washed with water (50 ml), and then dried the solid at 550C under vacuum to obtain 15 gm of dasatinib.Example 1Dasatinib (5 gm) obtained according to reference example was dissolved in ethyl acetate (300 ml) at 250C and heated to reflux temperature. To the solution was added methanol (100 ml) and stirred for 30 minutes at reflux temperature to form clear solution. The solution was slowly cooled to room temperature and then cooled to O0C, stirred for 1 hour at O0C. The solid was collected by filtration and the solid was washed with mixture of ethyl acetate and methanol (20 ml, 3:1), and then dried the solid at 500C under vacuum to obtain 3.5 gm of crystalline dasatinib form I.Example 2Dasatinib (5 gm, HPLC purity: 99.2%) was dissolved in acetone (100 ml) and methanol (250 ml) and heated to reflux temperature, stirred for 30 minutes at reflux temperature to form clear solution. The solution was cooled to room temperature and then cooled to 200C, stirred for 1 hour at 200C. The solid was collected by filtration and the solid was washed with mixture of acetone (10 ml) and methanol (25 ml), and then dried the solid at 500C under vacuum to obtain 4 gm of crystalline dasatinib form I (HPLC purity: 99.85%).Example 3Dasatinib (5 gm, HPLC purity: 99.2%) was dissolved in dimethylformamide (25 ml) at 250C and heated to 650C to form clear solution. To the solution was slowly added acetone (50 ml) at 650C and stirred for 1 hour at 650C. The solution was slowly cooled to 250C and stirred for 1 hour at 250C. The contents are filtered and the solid obtained was washed with mixture of dimethylformamide and acetone (15 ml, 1:2), and then dried the solid at 500C under vacuum to obtain 4 gm of dasatinib dimethylformamide solvate (HPLC purity: 99.94%).Example 4Dasatinib (5 gm) was dissolved in dimethylformamide (25 ml) at 250C and heated to 650C to form clear solution. Ethyl acetate (50 ml) was added slowly to the solution at 650C and stirred for 1 hour at 650C. The solution was slowly cooled to 250C, stirred for 1 hour at 250C and filtered. The solid obtained was washed with mixture of dimethylformamide and ethyl acetate (30 ml, 1:2), and then dried the solid at 500C under vacuum to obtain 4 gm of dasatinib dimethylformamide solvate.Example 5Dasatinib (5 gm, HPLC purity: 99.2%) was dissolved in dimethylformamide (25 ml) and heated to 650C to form a clear solution. The solution was cooled to 250C and then cooled to 50C, stirred for 4 hour at 50C. The solid was collected by filtration and the solid was washed with chilled dimethylformamide (10 ml), and then dried the solid at 500C under vacuum to obtain 4 gm of dasatinib dimethylformamide solvate (HPLC purity: 99.9%).Example 6Dasatinib (5 gm, HPLC purity: 99.2%) was dissolved in dimethylformamide (25 ml) and heated to 650C to form a clear solution. Water (50 ml) was added slowly to the solution at 650C and stirred for 1 hour at 650C. The solution was cooled to 250C and stirred for 30 minutes at 250C. The solid was collected by filtration and the solid was washed with mixture of dimethylformamide and water (15 ml, 1 :2), and then dried the solid at 500C under vacuum to obtain 4.7 gm of dasatinib dimethylformamide solvate (HPLC purity: 99.93%).Example 7Dasatinib dimethylformamide solvate (4.7 gm) obtained as in example 6 was dissolved in water (50 ml) and heated to 750C, stirred for 4 hours at 750C. The solution was cooled to 250C, stirred for 30 minutes at 250C and filtered. The solid obtained was washed with water (15 ml), and then dried at 500C under vacuum to obtain 4.7 gm of dasatinib monohydrate.Example 8Dasatinib (20 gm) was dissolved in dimethyl sulfoxide (100 ml) at 250C and heated to 650C to form clear solution. To the solution was slowly added water (200 ml) at 650C and stirred for 1 hour at 650C. The solution was slowly cooled to 250C and stirred for 30 minutes at 250C. The solid was collected by filtration and the solid was washed with mixture of dimethyl sulfoxide and water (30 ml, 1 :2), and then dried the solid at 500C under vacuum to obtain 19.5 gm of dasatinib monohydrate.Example 9Dasatinib (5 gm) was dissolved in isopropyl acetate (65 ml) and heated to 800C, stirred for 1 hour at 800C to form a clear solution. The solution was cooled to 250C, stirred for 1 hour at 250C and filtered. The solid obtained was washed with isopropyl acetate (15 ml) to obtain 5 gm of dasatinib isopropyl acetate solvate.Example 10Dasatinib (6 gm) was dissolved in toluene (100 ml) and heated to reflux temperature, stirred for 2 hours at reflux temperature to form a clear solution. The solution was slowly cooled to 250C. The contents are filtered and the solid obtained was washed with toluene (20 ml) to obtain 5.5 gm of dasatinib toluene solvate.Example 11Dasatinib (5 gm) was dissolved in dimethyl sulfoxide (20 ml) at 250C and heated to 650C. To the solution was slowly added ethyl acetate (200 ml) at 650C and the solution was slowly cooled to O0C, stirred for 2 hours at O0C. The solid was collected by filtration and the solid was washed with mixture of dimethyl sulfoxide and ethyl acetate (55 ml, 1 :10), and then dried the solid at 500C under vacuum to obtain 4 gm of dasatinib dimethyl sulfoxide solvate. PATENThttps://patents.google.com/patent/WO2014086326A1/enDasatinib, N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)- 1 -piperazinyl]-2- methyl-4-pyrimidmyl]amino]-5-thiazole carboxamide of formula I, also known as BMS- 354825, is a cancer treatment drug developed by Bristol-Myers Squibb and sold under the trade name Sprycel®. Dasatinib is a multi- BCR/ABL and Src family tyrosine kinase inhibitor and it is used for treatment of chronic myelogenous leukaemia (CML) as a secondary drug after primary treatment with imatinib (Gleevec®). It is also used for treatment of acute lymphoblastic leukaemia caused by mutation/translocation of chromosomes and development of the so-called Philadelphia chromosome (Ph+ ALL). However, its potential is so wide that the possibility of using it for treatment of other types of cancer, including advanced stages of prostate cancer, is still being investigated.
(I)In accordance with the basic patent WO2000062778A1, dasatinib is prepared by reaction of the key intermediate of formula II with l-(2-hydroxyethyl)piperazine in the presence of a base and a suitable solvent (Scheme 1). A similar preparation method was later used in a number of other process patents, only varying the corresponding base or solvent. Through the selection of a suitable solvent or procedure a great number of solvates or polymorphs can be prepared. Polymorphs have been one of the most frequently studied physical characteristics of active pharmaceutical substances (API) recently. Thus, different polymorphs of one API may have entirely different physical-chemical properties such as solubility, melting point, mechanical resistance of crystals but they may also influence the chemical and physical stability. Then, these properties may have an impact on further processes such as handling of the particular API, grinding or formulation method. These various physical-chemical characteristics of polymorphs influence the resulting bioavailability of the solid dosage form. Therefore, looking for new polymorphs and solvates is becoming an important tool for obtaining a polymorph form with the desired physical-chemical characteristics.
The process patent WO2005077945A2 describes preparation of the following solvates of dasatinib: monohydrate, butanol solvate, as well as two anhydrous forms (N-6 and T1H1- 7). A related patent also mentions two ethanol solvates, the hemi-ethanol and diethanol solvates (US 8 242 270 B2). Salts, various combinations of salts and their solvates have been described in detail in the patent application WO2007035874A1.Another process patent, WO2009053854A2, dealt with the preparation of a number of solvates or mixed solvates out of which especially the isopropanol and mixed isopropanol/dimethyl sulfoxide solvates, as well as a new solid form B, another anhydrous polymorph of dasatinib, are worth mentioning. Other patent applications have also dealt with the preparation of other solvates/mixed solvates (WO2010067374A2), or processes for the preparation and purification of the monohydrate/anhydrous form (WO2010139981A2) and its polymorphs (WO2011095059 Al).API solvates or salts are used in drug formulations in many cases. In the case of solvates the limits for individual solvents, their contents or maximum daily doses have to be strictly observed. Then, these limits can dramatically restrict their effective use. Thus, the clearly most convenient option is the use of sufficiently stable polymorphs of API that do not contain any solvents bound in the crystalline structure.Some of the above mentioned patent documents describe preparation of a stable anhydrous form of dasatinib (N-6). In accordance with individual patent documents the main disadvantages of the preparation of N-6 is the necessity of desolvation of the solvated form of the API at high temperatures (WO2009053854A2), or application of an increased temperature (50°C and more) and vacuum for a relatively long time (8-12h; WO2010139981A2 and WO2005077945A2). These procedures are very demanding from the point of view of general technology, energy and time, to say nothing of the necessity to work under an inert atmosphere to prevent possible oxidation-degradation reactions of the API. This is because dasatinib may be oxidized by atmospheric oxygen to the corresponding N-oxide (oxidation occurs in the piperazine ring), which may undergo the Cope elimination at increased temperatures. This secondary reaction may subsequently impair the purity of the prepared API.With a view to the above mentioned facts it is obvious that completely new methods and processes have to be developed even for polymorphs or solvates that are already well- known. Generally, the development of technologically and economically more efficient procedures is the main decisive parameter in their industrial utilization for the preparation of the API.Dasatinib of formula I is prepared by a reaction of the intermediate of formula II with l-(2- hydroxyethyl)piperazine in the presence of diisopropylethylamine (DIPEA) in an organic solvent from the group of dipolar aprotic solvents, higher alcohols or diols.If a dipolar aprotic solvent from the group of N-methyl-2-pyrrolidone (NMP), N^iV-dimethyl formamide (DMF), AyV-dimethyl acetamide (DMA), dimethyl sulfoxide (DMSO), formamide (FA), N,N -dimethyl propylene urea (DMPU) and l,3-dimethyl-2-imidazolidinone (DMI) is used, the reaction is carried out at 50-110°C under an inert atmosphere for 1/2-6 hours. In a preferable embodiment, NMP, DMSO, DMPU or DMI is used and the reaction is carried out at 90°C for 1-3 hours. The result of the reaction is crude dasatinib in the form of a solution in the corresponding solvent.If an alcohol from the group of isoamyl alcohol or 1,3-propanediol is used as a solvent for preparation of the crude dasatinib, the reaction mixture is heated at 120-160°C for 2-12 hours, in a preferable embodiment at 135°C for 3-6 hours.If dipolar aprotic solvents (NMP, DMF, DMA, DMSO, FA, DMPU and DMI) are used, in step a) a precipitant is added to the hot solution (90°C) under continuous stirring in an inert atmosphere in a 2- 15 fold, most preferably 4-10fold (by volume) amount with respect to the dipolar aprotic solvent. Suitable precipitants comprise especially acetonitrile, propionitrile, most preferably acetonitrile.After addition of the precipitant the obtained solution is withdrawn from the heating bath and is slowly left to cool down to 22°C under continuous stirring in an inert atmosphere. Crystallization occurs within 1-120 minutes (depending on the volume, until complete cooling). After having cooled down to 22°C (laboratory temperature), the suspension is stirred for another hour. The corresponding solvate of dasatinib is aspirated by well-known techniques in an inert atmosphere at 10-35 °C, most preferably at 22°C, and washed with the respective co-solvent.The solvate of dasatinib obtained this way can be directly used in the next step – recrystallization, without the necessity of drying. If necessary, the product may be dried at 10- 35°C, most preferably at 25°C, and at the pressure of 10-200 kPa, most preferably 50 kPa, for 6-24 hours, most preferably 12 hours.If NMP is used as the solvent in step a), the corresponding NMP solvate is isolated. The obtained dried crystalline NMP solvate (NM) of dasatinib has a characteristic XRPD pattern, which is presented in Figure no. 1. The NMP solvate (NM) has the following characteristic peaks: 5.88; 6.73; 10.73; 11.92; 13.39; 14.97; 16.72; 18.95; 20.17; 21.46; 22.81; 24.65; 25.18; 26.02 and 28.06 ± 0.2° 2-theta.If isoamyl alcohol or 1,3-propanediol are used as the solvents in step a), the reaction mixture is left to cool down to 22°C after expiration of the reaction time (3-6 h). Crystallization generally begins when the inner temperature of the reaction mixture drops to 100°C. After cooling down to 22°C (laboratory temperature), the suspension is further stirred for another 1 hour. Crystalline dasatinib is aspirated by well-known techniques in an inert atmosphere at 10-35°C, most preferably at 22°C, and washed with the corresponding solvent.The obtained product is dried at 10-35°C, most preferably at 25°C, and at the pressure of 10-200 kPa, most preferably 50 kPa, for 6-24 hours, most preferably 12 hours.The obtained crystalline isoamyl alcohol solvate (SI) of dasatinib has a characteristic XRPD pattern, which is shown in Figure no. 2. The solvate (SI) has the following characteristic peaks: 5.72; 10.35; 11.42; 12.61; 13.14; 14.27; 15.33; 17.18; 17.44; 17.97; 19.12; 19.95; 20.38; 22.05; 22.42; 23.01; 23.46; 23.68; 25.26; 26.20; 26.45; 26.62 and 27.78 ± 0.2° 2-theta.The obtained crystalline 1,3-propanediol solvate (SP) of dasatinib has a characteristic XRPD pattern, which is shown in Figure no. 3. The solvate (SP) has the following characteristic peaks: 6.04; 12.01; 15.10; 17.95; 18.35; 18.77; 21.25; 21.51; 22.96; 24.08; 24.62; 25.80; 26.16; 28.16 and 33.6578 ± 0.2° 2-theta.These solvates (or polymorph forms) are then easily converted to the desired anhydrous polymorph N-6 or another solvate in steps b) and c). All the forms prepared this way are sufficiently stable and can easily be isolated in the chemical purities of 99% and higher (in accordance with HPLC).The anhydrous polymorph form N-6 is prepared in the following way: any solvate or another polymorph is dissolved under an inert atmosphere at 90°C (reflux) in a 10-30 times, most preferably 20 times, the (weight) amount of the crystallization solvent. Suitable crystallization solvents include especially methanol, ethanol, isopropanol, most preferably methanol.A co-solvent is added in 0.1-10 times, most preferably ½-l times, the volume of the crystallization solvent used in an inert atmosphere at 90°C. The co-solvent can be, e.g., acetonitrile, propionitrile and their mixtures, most preferably acetonitrile. After addition of the co-solvent the obtained solution is withdrawn from the heating bath and is slowly left to cool down to 22°C under continuous stirring in an inert atmosphere. Crystallization occurs during 1-120 minutes (depending on the volume, until complete cooling). After having cooled down to 22°C (laboratory temperature), the suspension is stirred for another hour. Crystalline dasatinib is aspirated by well-known techniques in an inert atmosphere at 10-35°C, most preferably at 22°C, and washed with the corresponding co-solvent. The chemical purity of the obtained product is 99% (in accordance with HPLC); it is the polymorph form N-6 and its XRPD pattern is shown in Figure no. 4. The polymorph form N-6 has the following characteristic peaks: 6.77; 12.31; 13.16; 13.75; 16.70; 17.20; 18.54; 19.34; 20.25; 20.95; 21.94; 24.28; 24.82; and 27.80 ± 0.2° 2-theta.Brief Description of Drawings:Figure 1: shows an X-ray powder diffraction pattern of the crystalline solvate NM. Individual axes: independently variable: reflection angle 2Θ, dependently variable: intensity of detected radiation.Figure 2: shows an X-ray powder diffraction pattern of the isoamyl alcohol crystalline solvate SI. Individual axes: independently variable: reflection angle 2Θ, dependently variable: intensity of detected radiation. Figure 3: shows an X-ray powder diffraction pattern of the 1,3 propanediol crystalline solvate SP. Individual axes: independently variable: reflection angle 2Θ, dependently variable: intensity of detected radiation.Figure 4: shows an X-ray powder diffraction pattern of the crystalline anhydrous form N-6. Individual axes: independently variable: reflection angle 2Θ, dependently variable: intensity of detected radiation.Examples: The following working examples illustrate methods for the preparation of dasatinib of formula I, its polymorph form N-6 and its solvates NM, SI, SP.The polymorph forms and solvates of dasatinib were characterized with X-ray powder diffraction using the following methods:The diffraction patterns were measured using an X’PERT PRO MPD PANalytical diffractometer with a graphite monochromator, radiation used CuKa (λ=1.542 A), excitation voltage: 45 kV, anode current: 40 mA, measured range: 2 – 40° 2Θ, increment: 0.01° 2Θ. The measurement was carried out using a flat powder sample that was placed on a Si plate. For the primary optic setting programmable divergence diaphragms with the irradiated sample area of 10 mm, Soller diaphragms 0.02 rad and an anti-dispersion diaphragm ¼ were used. For the secondary optic setting an X’Celerator detector with the maximum opening of the detection slot, Soller diaphragms 0.02 rad and an anti-dispersion diaphragm 5.0 mm were used. HPLC method:Stock solution of samples: dissolve 5.0 mg of the sample in 10.0 ml of 50% acetonitrile R with water.Dimensions of the chromatographic HPLC column: / = 0.10 m, d= 3 mm- stationary phase: Zorbax Eclipse Plus Phenyl-Hexyl RRHD 1.8 μιη; temperature: 35 °C. Mobile phase: A: phosphate buffer (0.01 M sodium dihydrogen phosphate, pH treated by addition of sodium hydroxide to 7.00 ± 0.05); B: acetonitrile R.Gradient (A/B; flow 0.6 ml/min): 0 min 80/20; 10 min 50/50; 11 min 50/50; 12 min 80/20. Detection at the wavelength of 220 nm.Feed: 2 μΐ of the sample stock solution Example 1.Preparation of the NMP solvate (NM) of dasatinib:The intermediate of formula II (1.00 g; 2.54 mmol) and l-(2-hydroxyethyl)piperazine (1.66 g; 12.77 mmol) were dissolved in N-methylpyrrolidone (5 ml) under an inert atmosphere and diisopropylethylamine (0.9 ml, 5.18 mmol) was added to the reaction mixture. The reaction mixture was stirred and heated up to 90°C for 70 minutes and then acetonitrile (30 ml) was added to the reaction. The mixture was withdrawn from the heating bath and stirred intensively. Crystallization started after 5 minutes, the suspension was left to cool down under continuous stirring. After achieving the laboratory temperature it was stirred for another 2 hours. The crystalline substance was aspirated on frit S3, washed with acetonitrile (5 ml) and dried by suctioning under an inert nitrogen atmosphere for 15 minutes. The XRPD pattern of the sample obtained this way corresponds to the NMP solvate (NM) and can be used in the subsequent steps without the necessity of drying. Drying after 6 hours in an exsiccator at the laboratory temperature in vacuo (50 kPa) provided 1.2 g of crystalline dasatinib; 80% of the theoretical yield. HPLC purity 99.12%. The 1H NMR and 13C NMR spectra correspond to the data known from the literature. The XRPD pattern of the dried product corresponds to the NMP solvate (NM). The NM solvate is characterized by the reflections presented in Table 1 :Table 1 – NM forminterplanarpos. distance[°2Th.] [nm] rel. int. [%]5.88 1.5024 81.86.73 1.3131 100.010.73 0.8236 10.611.92 0.7420 59.213.39 0.6606 19.614.97 0.5915 38.416.72 0.5298 45.018.95 0.4679 10.920.17 0.4399 13.921.46 0.4138 13.422.81 0.3895 21.024.65 0.3608 13.325.18 0.3534 14.426.02 0.3422 11.928.06 0.3177 5.8
BuspironeCAS Registry Number: 36505-84-7CAS Name: 8-[4-[4-(2-Pyrimidinyl)-1-piperazinyl]butyl]-8-azaspiro[4.5]decane-7,9-dioneMolecular Formula: C21H31N5O2Molecular Weight: 385.50Percent Composition: C 65.43%, H 8.11%, N 18.17%, O 8.30%Literature References: Non-benzodiazepine anxiolytic; 5-hydroxytryptamine (5-HT1) receptor agonist. Prepn: Y. H. Wu et al.,J. Med. Chem.15, 477 (1972); Y. H. Wu, J. W. Rayburn, DE2057845 (1971 to Bristol-Myers); eidem,US3717634 (1973 to Mead-Johnson). Pharmacology: L. E. Allen et al.,Arzneim.-Forsch.24, 917 (1974). Comparison with diazepam in treatment of anxiety: H. L. Goldberg, R. J. Finnerty, Am. J. Psychiatry136, 1184 (1979); A. F. Jacobson et al.,Pharmacotherapy5, 290 (1985). Nonsynergistic effect with alcohol: T. Seppala et al.,Clin. Pharmacol. Ther.32, 201 (1982). Disposition and metabolism: S. Caccia et al.,Xenobiotica13, 147 (1983). Series of articles on chemistry, pharmacology, addictive potential, and clinical trials: J. Clin. Psychiatry43, pp 1-116 (1982); on pharmacology, safety and clinical comparison with clorazepate: Am. J. Med.80, Suppl. 3B, 1-51 (1986). Review of pharmacology and therapeutic efficacy: K. L. Goa, A. Ward, Drugs32, 114-129 (1986). Review: M. W. Jann, Pharmacotherapy8, 100-116 (1988); D. P. Taylor, FASEB J.2, 2445-2452 (1988). Derivative Type: HydrochlorideCAS Registry Number: 33386-08-2Trademarks: Ansial (Vita); Ansiced (Abello); Axoren (Glaxo Wellcome); Bespar (BMS); Buspar (BMS); Buspimen (Menarini); Buspinol (Zdravlje); Buspisal (Lesvi); Narol (Almirall)Molecular Formula: C21H31N5O2.HClMolecular Weight: 421.96Percent Composition: C 59.77%, H 7.64%, N 16.60%, O 7.58%, Cl 8.40%Properties: Crystals from abs ethanol, mp 201.5-202.5°. LD50 i.p. in rats: 136 mg/kg (Allen).Melting point: mp 201.5-202.5°Toxicity data: LD50 i.p. in rats: 136 mg/kg (Allen) Therap-Cat: Anxiolytic.Keywords: Anxiolytic; Arylpiperazines; Serotonin Receptor Agonist.
Buspirone, sold under the brand name Buspar, among others, is a medication primarily used to treat anxiety disorders, particularly generalized anxiety disorder.[9][10] Benefits support its short term use.[11] It has not been found to be effective in treating psychosis.[9] It is taken by mouth, and it may take up to four weeks to have an effect.[9][10]
Buspirone was first made in 1968 and approved for medical use in the United States in 1986.[9][10] It is available as a generic medication.[11] In 2018, it was the 92nd most-commonly prescribed medication in the United States, with more than 8 million prescriptions.[13][14]
Buspirone has no immediate anxiolytic effects, and hence has a delayed onset of action; its full clinical effectiveness may require 2–4 weeks to manifest itself.[20] The drug has been shown to be similarly effective in the treatment of generalized anxiety disorder (GAD) to benzodiazepines including diazepam, alprazolam, lorazepam, and clorazepate.[2] Buspirone is not known to be effective in the treatment of other anxiety disorders besides GAD,[21] although there is some limited evidence that it may be useful in the treatment of social phobia as an adjunct to selective serotonin reuptake inhibitors (SSRIs).[2][22]
SSRI and SNRI antidepressants such as paroxetine and venlafaxine may cause jaw pain/jaw spasm reversible syndrome (although it is not common), and buspirone appears to be successful in treating bruxism on SSRI/SNRI-induced jaw clenching.[25][26]
It is unclear if there is a risk of tardive dyskinesia or other movement disorders with buspirone.[9]
Overdose
Buspirone appears to be relatively benign in cases of single-drug overdose, although no definitive data on this subject appear to be available.[29] In one clinical trial, buspirone was administered to healthy male volunteers at a dosage of 375 mg/day, and produced side effects including nausea, vomiting, dizziness, drowsiness, miosis, and gastric distress.[15][16][18] In early clinical trials, buspirone was given at dosages even as high as 2,400 mg/day, with akathisia, tremor, and muscle rigidity observed.[30] Deliberate overdoses with 250 mg and up to 300 mg buspirone have resulted in drowsiness in about 50% of individuals.[30] One death has been reported in association with 450 mg buspirone together with alprazolam, diltiazem, alcohol, cocaine.[30]
Interactions
Buspirone has been shown in vitro to be metabolized by the enzymeCYP3A4.[8] This finding is consistent with the in vivo interactions observed between buspirone and these inhibitors or inducers of cytochrome P450 3A4 (CYP3A4), among others:[27]
In addition to binding to serotonin receptors, buspirone is an antagonist of the dopamineD2 receptor with weak affinity.[2][35] It preferentially blocks inhibitory presynaptic D2 autoreceptors, and antagonizes postsynaptic D2 receptors only at higher doses.[2] In accordance, buspirone has been found to increase dopaminergicneurotransmission in the nigrostriatal pathway at low doses, whereas at higher doses, postsynaptic D2 receptors are blocked and antidopaminergic effects such as hypoactivity and reduced stereotypy, though notably not catalepsy, are observed in animals.[2] Buspirone has also been found to bind with much higher affinity to the dopamine D3 and D4 receptors, where it is similarly an antagonist.[45]
A major metabolite of buspirone, 1-(2-pyrimidinyl)piperazine (1-PP), occurs at higher circulating levels than buspirone itself and is known to act as a potent α2-adrenergic receptor antagonist.[44][46][47] This metabolite may be responsible for the increased noradrenergic and dopaminergic activity observed with buspirone in animals.[46][48] In addition, 1-PP may play an important role in the antidepressant effects of buspirone.[48] Buspirone also has very weak and probably clinically unimportant affinity for the α1-adrenergic receptor.[35][49] However, buspirone has been reported to have shown “significant and selective intrinsic efficacy” at the α1-adrenergic receptor expressed in a “tissue- and species-dependent manner”.[49]
Unlike benzodiazepines, buspirone does not interact with the GABAA receptor complex.[2][50]
Pharmacokinetics
Buspirone has a low oralbioavailability of 3.9% relative to intravenous injection due to extensive first-pass metabolism.[2] The time to peak plasma levels following ingestion is 0.9 to 1.5 hours.[2] It is reported to have an elimination half-life of 2.8 hours,[2] although a review of 14 studies found that the mean terminal half-life ranged between 2 and 11 hours, and one study even reported a terminal half-life of 33 hours.[4] Buspirone is metabolized primarily by CYP3A4, and prominent drug interactions with inhibitors and inducers of this enzyme have been observed.[7][8] Major metabolites of buspirone include 5-hydroxybuspirone, 6-hydroxybuspirone, 8-hydroxybuspirone, and 1-PP.[4][5][6] 6-Hydroxybuspirone has been identified as the predominant hepatic metabolite of buspirone, with plasma levels that are 40-fold greater than those of buspirone after oral administration of buspirone to humans.[5] The metabolite is a high-affinity partial agonist of the 5-HT1A receptor (Ki = 25 nM) similarly to buspirone, and has demonstrated occupancy of the 5-HT1A receptor in vivo.[5] As such, it is likely to play an important role in the therapeutic effects of buspirone.[5] 1-PP has also been found to circulate at higher levels than those of buspirone itself and may similarly play a significant role in the clinical effects of buspirone.[46][48]
Buspirone was first synthesized, by a team at Mead Johnson, in 1968,[21] but was not patented until 1975.[54][55] It was initially developed as an antipsychoticdrug acting on the D2 receptor, but was found to be ineffective in the treatment of psychosis; it was then used as an anxiolytic instead.[2] In 1986, Bristol-Myers Squibb gained FDA approval for buspirone in the treatment of GAD.[21][56] The patent placed on buspirone expired in 2001 and it is now available as a generic drug.
Buspirone was primarily sold under the brand name Buspar.[57][59] Buspar is currently listed as discontinued by the US Federal Drug Administration.[60] In 2010, in response to a citizen petition, the US FDA determined that Buspar was not withdrawn for sale because of reasons of safety or effectiveness.[61]
Alkylation of 1-(2-pyrimidyl)piperazine (1) with 3-chloro-1-cyanopropane (2, 4-chlorobutyronitrile) gives 3, which is reduced either by hydrogenation over Raney nickel catalyst, or with LAH. The resulting 1° amine (4) from the previous step is then reacted with 3,3-tetramethyleneglutaric anhydride (5, 8-Oxaspiro[4.5]decane-7,9-dione) in order to yield buspirone (6).
A continuous flow method for the direct conversion of alcohols to amines via a hydrogen borrowing approach is reported. The method utilises a low loading (0.5%) of a commercial catalyst system ([Ru(p-cymene)Cl2]2 and DPEPhos), reagent grade solvent and is selective for primary alcohols. Successful methylation of amines using methanol and the direct dimethylamination of alcohols using commercial dimethylamine solution are reported. The synthesis of two pharmaceutical agents Piribedil (5) and Buspirone (25) were accomplished in good yields employing these new methods.
http://www.rsc.org/suppdata/c8/gc/c8gc03328e/c8gc03328e2.pdf 8-(4-hydroxybutyl)-8-azaspiro[4.5]decane-7,9-dione (23): A solution of 3,3-tetramethyleneglutaric anhydride (0.25 mol/L in THF) was combined in a tee piece with a solution of 4-amino-1-butanol (0.25 mol/L in THF) and reacted in a 20 mL reactor coil (stainless steel, 20 min residence time) heated at 250 °C. The output was concentrated in vacuo and the residue purified by column chromatography on silica gel to afford the product in 84% yield (Rf = 0.31, 63% DCM/AcOEt). 1H NMR (400 MHz, CDCl3) δ = 3.78 (t, J = 7.2 Hz, 2H), 3.65 (t, J = 6.0 Hz, 2H), 2.58 (s, 4H), 1.77 – 1.64 (m, 4H), 1.64 – 1.53 (m, 4H), 1.53 – 1.43 (m, 4H). 13C NMR (100 MHz, CDCl3) δ = 172.33, 62.28, 44.87, 39.47, 39.14, 37.54, 29.81, 24.35, 24.17. HRMS for [C13H22NO3] + calculated 240.1594 found 240.1605.
8-(4-(4-(pyrimidin-2-yl)piperazin-1-yl)butyl)-8-azaspiro[4.5]decane-7,9-dione (Buspirone, 25): The flow system was flushed with THF, the back-pressure regulator was set to 50 bar, and the coil reactor heated to 250 °C. Then a solution (10 mL overall volume) containing 1-(2-pyrimidyl)piperazine (2 mmol), 8-(4-hydroxybutyl)- 8-azaspiro[4.5]decane-7,9-dione (23) (2 mmol), dichloro(p-cymene)ruthenium(II) dimer (0.08 mmol) and bis[(2- diphenylphosphino)phenyl] ether (DPEPhos, 0.17 mmol) was pumped at 0.8 ml/min through a heated coil (8 mL, Phoenix reactor). The output solution obtained in steady state (monitored using the FlowUV) was concentrated in vacuo and purified by column chromatography on silica gel to afford the desired product in 76% yield (Rf = 0.29, 5% MeOH/DCM). 1H NMR (400 MHz, CDCl3) δ = 8.31 (d, J = 4.7 Hz, 2H), 6.48 (t, J = 4.7 Hz, 1H), 3.84 (t, J = 5.1 Hz, 4H), 3.79 (t, J = 6.8 Hz, 2H), 2.60 (s, 4H), 2.50 (t, J = 5.1 Hz, 4H), 2.40 (t, J = 6.8 Hz, 2H), 1.79 – 1.65 (m, 4H), 1.65 – 1.42 (m, 8H). 13C NMR (100 MHz, CDCl3) δ = 172.19, 161.63, 157.68, 109.77, 58.31, 53.06, 44.92, 43.60, 39.48, 39.35, 37.56, 26.04, 24.19, 24.19. HRMS for [C21H32N5O2] + calculated 386.2551 found 386.2570.
PAPER
Organic Preparations and Procedures International, 40(4), 391-394; 2008
The condensation of 1-(2-pyrimidinyl)piperazine (I) with 3-chloro-1-cyanopropane (II) by means of Na2CO3 in n-butanol gives 4-(2-pyrimidinyl)-1-(3-cyanopropyl)piperazine (III). This product is reduced with LiAlH4 or with H2 and Raney-Ni yielding 4-(2-pyrimidinyl)-1-(4-aminobutyl)piperazine (IV), which is finally condensed with 8-oxaspiro[4.5]decane-7,9-dione-(3,3-tetramethylene-glutaric anhydride) (V) in pyridine.
Buspirone, 8-[4-[4-(2-pyrimidyl)-1-piperazinyl]butyl]-8-azaspiro [4,5] decan-7,9-dione (5.2.6), is synthesized by the reaction of 1-(2-pyrimidyl)-4-(4-aminobutyl)piperazine (5.2.4) with 8-oxaspiro[4,5]decan-7,9-dione (5.2.5). In turn, 1-(2-pyrimidyl)-4-(4-aminobutyl)piperazine (5.2.4) is synthesized by the reaction of 1-(2-pyrimidyl)piperazine with 4-chlorobutyronitrile, giving 4-(2-pyrimidyl)-1-(3-cyanopropyl)piperazine (5.2.3), which is hydrogenated with Raney nickel into buspirone (5.2.4) [51–55].
Buspirone is an extremely specific drug that could possibly represent a new chemical class of anxiolytics—azaspirones. As an anxiolytic, its activity is equal to that of benzodiazepines; however, it is devoid of anticonvulsant and muscle relaxant properties, which are characteristic of benzodiazepines. It does not cause dependence or addiction. The mechanism of its action is not conclusively known. It does not act on the GABA receptors, which occurs in benzodiazepine use; however, it has a high affinity for seratonin (5-HT) receptors and a moderate affinity for dopamine (D2) receptors. Buspirone is effective as an anxiolytic. A few side effects of buspirone include dizziness, drowsiness, headaches, nervousness, fatigue, and weakness. This drug is intended for treatment of conditions of anxiety in which stress, muscle pain, rapid heart rate, dizziness, fear, etc. are observed; in other words, conditions of anxiety not associated with somewhat common, usual, and everyday stress. Synonyms for buspirone are anizal, axoren, buspar, buspimen, buspinol, narol, travin, and others.
Buspirone (Buspar®, 59, Figure 11.17) is a drug used for the treatment of anxiety and depression, thought to produce its effects by binding to the serotonin 5HT1A receptor [114–116]. Mainly as a result of hydroxylation reactions, it is extensively converted to various metabolites and blood concentrations return to low levels a few hours after dosing [117]. A major metabolite, 6-hydroxybuspirone, produced by the action of liver cytochrome P450 CYP3A4, was present at much higher concentrations in human blood than buspirone itself. For development of 6-hydroxybuspirone as a potential antianxiety drug, preparation and testing of the two enantiomers as well as the racemate was of interest. An enantioselective microbial reduction process was developed for the reduction of 6-oxobuspirone 60 to (R)-6-hydroxybuspirone 61a or (S)-6-hydroxybuspitone 61b. About 150 microbial cultures were screened for the enantioselective reduction of 60. Rhizopus stolonifer SC 13898, Neurospora crassa SC 13816, Mucor racemosus SC 16198, and Pseudomonas putida SC 13817 gave >50% reaction yields and >95% ee of (S)-6-hydroxybuspirone 61a. The yeast strains Hansenula polymorpha SC 13845 and Candida maltosa SC 16112 gave (R)-6-hydroxybuspirone in >60% reaction yield and >97% ee [118]. The NADPH-dependent (R)-reductase (RHBR) from H. polymorpha SC 13845 was purified to homogeneity, its N-terminal and internal amino acid sequences were determined and the corresponding gene was cloned and expressed in E. coli. To regenerate the NADPH required for reduction, glucose-6-phosphate dehydrogenase gene from Saccharomyces cerevisiae was cloned and coexpressed in the same E. coli strain. Recombinant cultures coexpressing (R)-reductase (RHBR) and glucose 6-phosphate dehydrogenase catalyzed the reduction of 6-ketobuspirone to (R)-6-hydroxybuspirone 61a in 99% yield and 99.9% ee at 50 g/L substrate input [119].
The NADH-dependent (S)-reductase (SHBR) from P. putida SC 16269 was also purified to homogeneity, its N-terminal and internal amino acid sequences were determined and the corresponding gene was cloned and expressed in E. coli. To regenerate the NADH required for reduction, the NAD+ dependent formate dehydrogenase gene from Pichia pastoris was also cloned and co-expressed in the same E. coli strain. Recombinant E. coli coexpressing (S)-reductase and formate dehydrogenase was used to catalyze the reduction of 6-ketobuspirone to (S)-6-hydroxybuspirone 61b, in >98% yield and >99.8% ee at 50 g/L substrate input [119].
The present invention relates to methods of treating anxiety and depression using R-6-hydroxy-buspirone and pharmaceutical compositions containing R-6-hydroxy-buspirone.
Buspirone, chemically: 8-[4-[4-(2-pyrimidinyl)1-piperazinyl]butyl-8-azaspiro(4,5)-decane-7,9-dione, is approved for the treatment of anxiety disorders and depression by the United States Food and Drug Administration. It is available under the trade name BUSPAR® from Bristol-Myers Squibb Company.
Studies have shown that buspirone is extensively metabolized in the body. (See, for example, Mayol, et al., Clin. Pharmacol. Ther., 37, p. 210, 1985). One of the metabolites is 6-hydroxy-8-[4-[4-(2-pyrimidinyl)1-piperazinyl]butyl-8-azaspiro(4,5)-decane-7,9-dione having Formula I. This metabolite is also known as BMS 28674, BMS 442608, or
as 6-hydroxy-buspirone. This compound is believed to be the active metabolite of buspirone and its use in treating anxiety disorders and depression is disclosed in U.S. Pat. No. 6,150,365. The specific stereochemistry of 6-hydroxy-buspirone has not been described previously. Neither racemic 6-hydroxy-buspirone nor its enantiomers are commercially available at the present time.
Preclinical studies demonstrate that 6-hydroxy-buspirone, like buspirone, demonstrates a strong affinity for the human 5-HT1A receptor. In functional testing, 6-hydroxy-buspirone produced a dose-dependent anxiolytic response in the rat pup ultrasonic vocalization test, a sensitive method for assessment of anxiolytic and anxiogenic effects (Winslow and Insel, 1991, Psychopharmacology, 105:513-520).
Clinical studies in volunteers orally dosed with buspirone demonstrate that 6-hydroxy-buspirone blood plasma levels were not only 30 to 40 times higher but were sustained compared to buspirone blood plasma levels. The time course of 6-hydroxy-buspirone blood plasma levels, unlike buspirone blood plasma levels, correlate more closely with the sustained anxiolytic effect seen following once or twice a day oral dosing with buspirone.
Although buspirone is an effective treatment for anxiety disorders and depression symptomatology in a significant number of patients treated, about a third of patients get little to no relief from their anxiety and responders often require a week or more of buspirone treatment before experiencing relief from their anxiety symptomatology. Further, certain adverse effects are reported across the patient population. The most commonly observed adverse effects associated with the use of buspirone include dizziness, nausea, headache, nervousness, lightheadedness, and excitement. Also, since buspirone can bind to central dopamine receptors, concern has been raised about its potential to cause unwanted changes in dopamine-mediated neurological functions and a syndrome of restlessness, appearing shortly after initiation of oral buspirone treatment, has been reported in small numbers of patients. While buspirone lacks the prominent sedative effects seen in more typical anxiolytics such as the benzodiazepines, patients are nonetheless advised against operating potentially dangerous machinery until they experience how they are affected by buspirone.
It can be seen that it is desirable to find a medicament with buspirone’s advantages but which demonstrates more robust anxiolytic potency with a lack of the above described adverse effects.
Formation of 6-hydroxy-buspirone occurs in the liver by action of enzymes of the P450 system, specifically CYP3A4. Many substances such as grapefruit juice and certain other drugs; e.g. erythromycin, ketoconazole, cimetidine, etc., are inhibitors of the CYP3A4 isozyme and may interfere with the formation of this active metabolite from buspirone. For this reason it would be desirable to find a compound with the advantages of buspirone but without the drug—drug interactions when coadministered with agents affecting the activity level of the CYP3A4 isozyme.
EXAMPLE 3One-Step Synthesis of 6-Hydroxy-buspirone (I)
Buspirone (19.3 g, 50 mmole) was dissolved in dry THF (400 mL) and the resulting solution was cooled to −78° C. A solution of KN(SiMe3)2 in toluene (100 mL, 1 M) was added slowly. After the reaction mixture was stirred at −78° C. for 1 h, a solution of 2-(phenylsulfonyl)-3-phenyloxaziridine (Davis reagent, prepared according to literature method: F. A. Davis, et al., Org. Synth., 1988, 66, 203) (17.0 g, 65 mmole) in dry THF (150 mL, precooled to −78° C.) was added quickly via a cannular. After stirred for 30 mins at −78° C., the reaction was quenched with 1 N HCl solution (500 mL). It was extracted with EtOAc (3×500 mL). The aqueous layer was separated, neutralized with saturated sodium bicarbonate solution, and extracted with EtOAc (3×500 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under reduced pressure to give a white solid residue which was subjected to column chromatography using CH2Cl2/MeOH/NH4OH (200:10:1) as the eluent to give pure 6-hydroxy-buspirone (I, 7.2 g) and a mixture of buspirone and 6-hydroxy-buspirone (I). The mixture was purified by above column chromatography to afford another 3.3 g of pure 6-hydroxy-buspirone (I).
^ Jump up to:ab Wong H, Dockens RC, Pajor L, Yeola S, Grace JE, Stark AD, et al. (August 2007). “6-Hydroxybuspirone is a major active metabolite of buspirone: assessment of pharmacokinetics and 5-hydroxytryptamine1A receptor occupancy in rats”. Drug Metabolism and Disposition. 35 (8): 1387–92. doi:10.1124/dmd.107.015768. PMID17494642. S2CID25558546.
^ Jump up to:abcd Zhu M, Zhao W, Jimenez H, Zhang D, Yeola S, Dai R, et al. (April 2005). “Cytochrome P450 3A-mediated metabolism of buspirone in human liver microsomes”. Drug Metabolism and Disposition. 33 (4): 500–7. doi:10.1124/dmd.104.000836. PMID15640381. S2CID10142905.
^ Sontheimer DL, Ables AZ (March 2001). “Is imipramine or buspirone treatment effective in patients wishing to discontinue long-term benzodiazepine use?”. The Journal of Family Practice. 50(3): 203. PMID11252203.
^ Prisco V, Iannaccone T, Di Grezia G (2017-04-01). “Use of buspirone in selective serotonin reuptake inhibitor-induced sleep bruxism”. European Psychiatry. Abstract of the 25th European Congress of Psychiatry. 41: S855. doi:10.1016/j.eurpsy.2017.01.1701.
^ Lamberg TS, Kivistö KT, Laitila J, Mårtensson K, Neuvonen PJ (1998). “The effect of fluvoxamine on the pharmacokinetics and pharmacodynamics of buspirone”. European Journal of Clinical Pharmacology. 54 (9–10): 761–6. doi:10.1007/s002280050548. PMID9923581. S2CID21939719.
^ Jump up to:abc Roth BL, Driscol J. “PDSP Ki Database”. Psychoactive Drug Screening Program (PDSP). University of North Carolina at Chapel Hill and the United States National Institute of Mental Health. Retrieved 14 August 2017.
^ Nelson DR, Thomas DR (May 1989). “[3H]-BRL 43694 (Granisetron), a specific ligand for 5-HT3 binding sites in rat brain cortical membranes”. Biochemical Pharmacology. 38 (10): 1693–5. doi:10.1016/0006-2952(89)90319-5. PMID2543418.
^ Jump up to:ab Borsini F, Giraldo E, Monferini E, Antonini G, Parenti M, Bietti G, Donetti A (September 1995). “BIMT 17, a 5-HT2A receptor antagonist and 5-HT1A receptor full agonist in rat cerebral cortex”. Naunyn-Schmiedeberg’s Archives of Pharmacology. 352 (3): 276–82. doi:10.1007/bf00168557. PMID8584042. S2CID19340842.
^ Plassat JL, Amlaiky N, Hen R (August 1993). “Molecular cloning of a mammalian serotonin receptor that activates adenylate cyclase”. Molecular Pharmacology. 44 (2): 229–36. PMID8394987.
^ Lovenberg TW, Baron BM, de Lecea L, Miller JD, Prosser RA, Rea MA, et al. (September 1993). “A novel adenylyl cyclase-activating serotonin receptor (5-HT7) implicated in the regulation of mammalian circadian rhythms”. Neuron. 11 (3): 449–58. doi:10.1016/0896-6273(93)90149-l. PMID8398139. S2CID28729004.
^ Jump up to:ab Blier P, Curet O, Chaput Y, de Montigny C (July 1991). “Tandospirone and its metabolite, 1-(2-pyrimidinyl)-piperazine–II. Effects of acute administration of 1-PP and long-term administration of tandospirone on noradrenergic neurotransmission”. Neuropharmacology. 30 (7): 691–701. doi:10.1016/0028-3908(91)90176-c. PMID1681447. S2CID44297577.
^ Zuideveld KP, Rusiç-Pavletiç J, Maas HJ, Peletier LA, Van der Graaf PH, Danhof M (December 2002). “Pharmacokinetic-pharmacodynamic modeling of buspirone and its metabolite 1-(2-pyrimidinyl)-piperazine in rats”. The Journal of Pharmacology and Experimental Therapeutics. 303 (3): 1130–7. doi:10.1124/jpet.102.036798. PMID12438536. S2CID14139919.
^ Dockens RC, Salazar DE, Fulmor IE, Wehling M, Arnold ME, Croop R (November 2006). “Pharmacokinetics of a newly identified active metabolite of buspirone after administration of buspirone over its therapeutic dose range”. Journal of Clinical Pharmacology. 46(11): 1308–12. doi:10.1177/0091270006292250. PMID17050795.
^ Jajoo HK, Mayol RF, LaBudde JA, Blair IA (1989). “Metabolism of the antianxiety drug buspirone in human subjects”. Drug Metabolism and Disposition. 17 (6): 634–40. PMID2575499.
Tasimelteon is a white to off-white crystalline powder, it is non hygroscopic, soluble in water across relevant pH values and freely soluble in alcohols, cyclohexane, and acetonitrile. Conducted in vivo studies demonstrate that tasimelteon is highly permeable substance. Photostability testing and testing on stress conditions demonstrated that the active substance degrades in light.
Tasimelteon exhibits stereoisomerism due to the presence of two chiral centres. Active substance is manufactured as a single, trans-1R,2R isomer. Enantiomeric purity is controlled routinely during manufacture of active substance intermediates by chiral HPLC/specific optical rotation and additionally controlled in the active substance. Stability data indicates tasimelteon is isomerically stable.
Polymorphism has been observed in polymorphic screening studies for tasimelteon and two forms have been identified. The thermodynamically more stable form has been chosen for development and the manufacturing process consistently yields active substance of single, desired polymorphic form. It was demonstrated that milling of the active substance does not affect polymorphic form. Polymorphism is additionally controlled in active substance release and shelf-life specifications using X-ray powder diffraction analysis.
Tasimelteon is synthesized in nine main steps using linear synthesis and using commercially available well-defined starting materials with acceptable specifications. Three intermediates are isolated for control of active substance quality including stereochemical control. The active substance is isolated by slow recrystallisation or precipitation of tasimelteon from an ethanol/water mixture which ensures the formation of desired polymorphic form. Up to two additional, optional recrystallisations may be performed for unmilled tasimelteon to ensure that milled tasimelteon active substance is of high purity. Seed crystals complying with active substance specifications can be used optionally. Active substance is jet milled (micronised) to reduce and control particle size, which is critical in finished product performance with regards to content uniformity and dissolution…….http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/003870/WC500190309.pdf
launched in 2014 in the U.S. by Vanda Pharmaceuticals for the treatment of non-24-hour sleep-wake disorder in totally blind subjects. In 2015, the European Committee for Medicinal Products of the European Medicines Agency granted approval for the same indication. In 2010 and 2011, orphan drug designations were assigned for the treatment of non-24 hour sleep/wake disorder in blind individuals without light perception in the U.S. and the E.U., respectively.
Tasimelteon (trade name Hetlioz) is a drug approved by the U.S. Food and Drug Administration (FDA)[2] in January 2014 for the treatment of non-24-hour sleep–wake disorder (also called Non-24, N24 and N24HSWD).[3] In June 2014, the European Medicines Agency accepted an EU filing application for tasimelteon[4] and in July 2015, the drug was approved in Europe for the treatment of non-24-hour sleep-wake rhythm disorder in totally blind adults,[5] but not in the rarer case of non-24 in sighted people.
Tasimelteon is a selective agonist for the melatonin receptors MT1 and MT2, similar to other members of the melatonin receptor agonistclass of which ramelteon (2005) and agomelatine (2009) were the first approved.[6] As a treatment for N24HSWD, as with melatonin or other melatonin derivatives, the patient may experience improved sleep timing while taking the drug. Reversion to baseline sleep performance occurs within a month of discontinuation.[7]
Development
Tasimelteon (previously known as BMS-214,778) was developed for the treatment of insomnia and other sleep disorders. A phase II trial on circadian rhythm sleep disorders was concluded in March 2005.[8] A phase III insomnia trial was conducted in 2006.[9] A second phase III trial on insomnia, this time concerning primary insomnia, was completed in June 2008.[10] In 2010, the FDA granted orphan drug status to tasimelteon, then regarded as an investigational medication, for use in totally blind adults with N24HSWD.[11] (Through mechanisms such as easing the approval process and extending exclusivity periods, orphan drug status encourages development of drugs for rare conditions that otherwise might lack sufficient commercial incentive.)
On completion of Phase III trials, interpretations of the clinical trials by the research team concluded that the drug may have therapeutic potential for transient insomnia in circadian rhythm sleep disorders.[12] A year-long (2011–2012) study at Harvard tested the use of tasimelteon in blind subjects with non-24-hour sleep-wake disorder. The drug has not been tested in children nor in any non-blind people.
FDA approval
In May 2013 Vanda Pharmaceuticals submitted a New Drug Application to the Food and Drug Administration for tasimelteon for the treatment of non-24-hour sleep–wake disorder in totally blind people. It was approved by the FDA on January 31, 2014 under the brand name Hetlioz.[3] In the opinion of Public Citizen, an advocacy group, the FDA erroneously allowed it to be labelled without stating that it is only approved for use by totally blind people.[13] However, FDA updated its press release on Oct. 2, 2014 to clarify the approved use of Hetlioz, which includes both sighted and blind individuals. The update did not change the drug labeling (prescribing information).[14]
Toxicity
Experiments with rodents revealed fertility impairments, an increase in certain cancers, and serious adverse events during pregnancy at dosages in excess of what is considered the “human dose”.[15][16]
The master body clock controls the timing of many aspects of physiology, behavior and metabolism that show daily rhythms, including the sleep-wake cycles, body temperature, alertness and performance, metabolic rhythms and certain hormones which exhibit circadian variation. Outputs from the
suprachiasmatic nucleus (SCN) control many endocrine rhythms including those of melatonin secretion by the pineal gland as well as the control of Cortisol secretion via effects on the hypothalamus, the pituitary and the adrenal glands. This master body clock, located in the SCN, spontaneously generates rhythms of approximately 24.5 hours. These non-24-hour rhythms are synchronized each day to the 24-hour day-night cycle by light, the primary environmental time cue which is detected by specialized cells in the retina and transmitted to the SCN via the retino-hypothalamic tract. Inability to detect this light signal, as occurs in most totally blind individuals, leads to the inability of the master body clock to be reset daily and maintain entrainment to a 24-hour day.
Non-24-Hour Disorder, Non-24, also referred to as Non-24-Hour Sleep-Wake Disorder, (N24HSWD) or Non-24-Hour Disorder, is an orphan indication affecting approximately 65,000 to 95,000 people in the U.S. and 140,000 in Europe. Non- 24 occurs when individuals, primarily blind with no light perception, are unable to synchronize their endogenous circadian pacemaker to the 24-hour light/dark cycle. Without light as a synchronizer, and because the period of the internal clock is typically a little longer than 24 hours, individuals with Non-24 experience their circadian drive to initiate sleep drifting later and later each day. Individuals with Non-24 have abnormal night sleep patterns, accompanied by difficulty staying awake during the day. Non-24 leads to significant impairment, with chronic effects impacting the social and occupational functioning of these individuals.
In addition to problems sleeping at the desired time, individuals with Non-24 experience excessive daytime sleepiness that often results in daytime napping.
The severity of nighttime sleep complaints and/or daytime sleepiness complaints varies depending on where in the cycle the individual’s body clock is with respect to their social, work, or sleep schedule. The “free running” of the clock results in approximately a 1-4 month repeating cycle, the circadian cycle, where the circadian drive to initiate sleep continually shifts a little each day (about 15 minutes on average) until the cycle repeats itself. Initially, when the circadian cycle becomes desynchronous with the 24h day-night cycle, individuals with Non-24 have difficulty initiating sleep. As time progresses, the internal circadian rhythms of these individuals becomes 180 degrees out of synchrony with the 24h day-night cycle, which gradually makes sleeping at night virtually impossible, and leads to extreme sleepiness during daytime hours.
Eventually, the individual’s sleep-wake cycle becomes aligned with the night, and “free-running” individuals are able to sleep well during a conventional or socially acceptable time. However, the alignment between the internal circadian rhythm and the 24-hour day-night cycle is only temporary.
In addition to cyclical nighttime sleep and daytime sleepiness problems, this condition can cause deleterious daily shifts in body temperature and hormone secretion, may cause metabolic disruption and is sometimes associated with depressive symptoms and mood disorders.
It is estimated that 50-75% of totally blind people in the United States (approximately 65,000 to 95,000) have Non-24. This condition can also affect sighted people. However, cases are rarely reported in this population, and the true rate of Non-24 in the general population is not known.
The ultimate treatment goal for individuals with Non-24 is to entrain or synchronize their circadian rhythms into an appropriate phase relationship with the 24-hour day so that they will have increased sleepiness during the night and increased wakefulness during the daytime. Tasimelteon
Tasimelteon is a circadian regulator which binds specifically to two high affinity melatonin receptors, Mella (MT1R) and Mellb (MT2R). These receptors are found in high density in the suprachiasmatic nucleus of the brain (SCN), which is responsible for synchronizing our sleep/wake cycle. Tasimelteon has been shown to improve sleep parameters in prior clinical studies, which simulated a desynchronization of the circadian clock. Tasimelteon has so far been studied in hundreds of individuals and has shown a good tolerability profile.
Tasimelteon has the chemical name: tr ns-N-[[2-(2,3-dihydrobenzofuran- 4-yl)cycloprop-lyl] methyl] propanamide, has the structure of Formula I:
Formula I
and is disclosed in US 5856529 and in US 20090105333, both of which are incorporated herein by reference as though fully set forth.
Tasimelteon is a white to off-white powder with a melting point of about 78°C (DSC) and is very soluble or freely soluble in 95% ethanol, methanol, acetonitrile, ethyl acetate, isopropanol, polyethylene glycols (PEG-300 and PEG- 400), and only slightly soluble in water. The native pH of a saturated solution of tasimelteon in water is 8.5 and its aqueous solubility is practically unaffected by pH. Tasimelteon has 2-4 times greater affinity for MT2R relative to MTIR. It’s affinity (¾) for MTIR is 0.3 to 0.4 and for MT2R, 0.1 to 0.2. Tasimelteon is useful in the practice of this invention because it is a melatonin agonist that has been demonstrated, among other activities, to entrain patients suffering from Non-24.
Metabolites of tasimelteon include, for example, those described in “Preclinical Pharmacokinetics and Metabolism of BMS-214778, a Novel
Melatonin Receptor Agonist” by Vachharajani et al., J. Pharmaceutical Sci., 92(4):760-772, which is hereby incorporated herein by reference. The active metabolites of tasimelteon can also be used in the method of this invention, as can pharmaceutically acceptable salts of tasimelteon or of its active metabolites. For example, in addition to metabolites of Formula II and III, above, metabolites of tasimelteon also include the monohydroxylated analogs M13 of Formula IV, M12 of Formula V, and M14 of Formula VI.
Formula IV
Formula V
MO
Formula VI
Thus, it is apparent that this invention contemplates entrainment of patients suffering free running circadian rhythm to a 24 hour circadian rhythm by administration of a circadian rhythm regulator (i.e., circadian rhythm modifier) capable of phase advancing and/or entraining circadian rhythms, such as a melatonin agonist like tasimelteon or an active metabolite oftasimelteon or a pharmaceutically acceptable salt thereof. Other MT1R and MT2R agonists, i.e., melatonin agonists, can have similar effects on the master body clock. So, for example, this invention further contemplates the use of melatonin agonists such as but not limited to melatonin, N-[l-(2,3-dihydrobenzofuran-4- yl)pyrrolidin-3-yl]-N-ethylurea and structurally related compounds as disclosed in US 6,211,225, LY-156735 ((R)-N-(2-(6-chloro-5-methoxy-lH-indol- 3yl) propyl) acetamide) (disclosed in U.S. Patent No. 4,997,845), agomelatine (N- [2-(7-methoxy-l-naphthyl)ethyl]acetamide) (disclosed in U.S. Patent No.
Additional melatonin agonists include, without limitation, those listed in U.S. Patent Application Publication No. 20050164987, which is incorporated herein by reference, specifically: TAK-375 (see Kato, K. et al. Int. J.
Neuropsychopharmacol. 2000, 3 (Suppl. 1): Abst P.03.130; see also abstracts P.03.125 and P.03.127), CGP 52608 (l-(3-allyl-4-oxothiazolidine-2-ylidene)-4- met- hylthiosemicarbazone) (See Missbach et al., J. Biol. Chem. 1996, 271, 13515-22), GR196429 (N-[2-[2,3,7,8-tetrahydro-lH-fur-o(2,3-g)indol-l- yl] ethyl] acetamide) (see Beresford et al., J. Pharmacol. Exp. Ther. 1998, 285, 1239-1245), S20242 (N-[2-(7-methoxy napth-l-yl) ethyl] propionamide) (see Depres-Brummer et al., Eur. J. Pharmacol. 1998, 347, 57-66), S-23478 (see Neuropharmacology July 2000), S24268 (see Naunyn Schmiedebergs Arch. June 2003), S25150 (see Naunyn Schmiedebergs Arch. June 2003), GW-290569, luzindole (2-benzyl-N-acetyltryptamine) (see U.S. Patent No. 5,093,352), GR135531 (5-methoxycarbonylamino-N-acetyltrypt- amine) (see U.S. Patent Application Publication No. 20010047016), Melatonin Research Compound A, Melatonin Agonist A (see IMSWorld R&D Focus August 2002), Melatonin
Analogue B (see Pharmaprojects August 1998), Melatonin Agonist C (see Chem. Pharm. Bull. (Tokyo) January 2002), Melatonin Agonist D (see J. Pineal Research November 2000), Melatonin Agonist E (see Chem. Pharm. Bull. (Tokyo) Febrary 2002), Melatonin Agonist F (see Reprod. Nutr. Dev. May 1999), Melatonin Agonist G (see J. Med. Chem. October 1993), Melatonin Agonist H (see Famaco March 2000), Melatonin Agonist I (see J. Med. Chem. March 2000), Melatonin Analog J (see Bioorg. Med. Chem. Lett. March 2003), Melatonin Analog K (see MedAd News September 2001), Melatonin Analog L, AH-001 (2-acetamido-8- methoxytetralin) (see U.S. Patent No. 5,151,446), GG-012 (4-methoxy-2- (methylene propylamide)indan) (see Drijfhout et al., Eur. J. Pharmacol. 1999, 382, 157-66), Enol-3-IPA, ML-23 (N-2,4-dinitrophenyl-5-methoxy-tryptamine ) (see U.S. Patent No. 4,880,826), SL-18.1616, IP-100-9 (US 5580878), Sleep Inducing Peptide A, AH-017 (see U.S. Patent No. 5,151,446), AH-002 (8-methoxy- 2-propionamido-tetralin) (see U.S. Patent No. 5,151,446), and IP-101.
Metabolites, prodrugs, stereoisomers, polymorphs, hydrates, solvates, and salts of the above compounds that are directly or indirectly active can, of course, also be used in the practice of this invention.
Melatonin agonists with a MT1R and MT2R binding profile similar to that of tasimelteon, which has 2 to 4 time greater specificity for MT2R, are preferred.
Tasimelteon can be synthesized by procedures known in the art. The preparation of a 4-vinyl-2,3-dihydrobenzofuran cyclopropyl intermediate can be carried out as described in US7754902, which is incorporated herein by reference as though fully set forth.
Pro-drugs, e.g., esters, and pharmaceutically acceptable salts can be prepared by exercise of routine skill in the art.
In patients suffering a Non-24, the melatonin and Cortisol circadian rhythms and the natural day/night cycle become desynchronized. For example, in patients suffering from a free-running circadian rhythm, melatonin and Cortisol acrophases occur more than 24 hours, e.g., >24.1 hours, prior to each previous day’s melatonin and Cortisol acrophase, respectively, resulting in desynchronization for days, weeks, or even months, depending upon the length of a patient’s circadian rhythm, before the melatonin, Cortisol, and day /night cycles are again temporarily synchronized.
Chronic misalignment of Cortisol has been associated with metabolic, cardiac, cognitive, neurologic, neoplastic, and hormonal disorders. Such disorders include, e.g., obesity, depression, neurological impairments.
Figure : Melatonin receptor agonists. The applied colors indicate the mutual properties with the general melatonin receptor agonists pharmacophore.
INTRODUCTION
Tasimelteon has the chemical name: trans-N-[[2-(2,3-dihydrobenzofuran-4-yl)cycloprop-1yl]methyl]propanamide, has the structure of Formula I:
and is disclosed in U.S. Pat. No. 5,856,529 and in US 20090105333, both of which are incorporated herein by reference as though fully set forth.
Tasimelteon is a white to off-white powder with a melting point of about 78° C. (DSC) and is very soluble or freely soluble in 95% ethanol, methanol, acetonitrile, ethyl acetate, isopropanol, polyethylene glycols (PEG-300 and PEG-400), and only slightly soluble in water. The native pH of a saturated solution of tasimelteon in water is 8.5 and its aqueous solubility is practically unaffected by pH. Tasimelteon has 2-4 times greater affinity for MT2R relative to MT1R. It’s affinity (Ki) for MT1R is 0.3 to 0.4 and for MT2R, 0.1 to 0.2. Tasimelteon is useful in the practice of this invention because it is a melatonin agonist that has been demonstrated, among other activities, to entrain patients suffering from Non-24.
IV 3-hydroxy-2-(2-propenyl) benzoic acid methyl ester
V 2,3-dihydro-4-hydroxy-2-benzofurancarboxylic acid methyl ester
VI benzofuran-4-carboxylic acid methyl ester
VII benzofuran-4-carboxylic acid
VIII 2,3-dihydro-4-benzofurancarboxylic acid
IX 2,3-dihydro-4-benzofuranmethanol
X 2,3-dihydro-4-benzofurancarboxaldehyde
XI Propanedioic acid
XII (E) -3 – (2,3-dihydro-4-benzofuranyl) propenoic acid
XIII thionyl chloride
XIV (E) -3 – (2,3-dihydro-4-benzofuranyl) propenoyl chloride
XV (3aS, 6R, 7aR)-hexahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide
XVI (3aS,6R,7aR)-1-[(E)-3-(2,3-dihydro-4-benzofuranyl)-1-oxo-2-propenyl]hexahydro-8,8-dimethyl-3H-3a,6-methano-2,1-benzisothiazole-2,2-dioxide
XVII (3aS,6R,7aR)-1-[[(1R,2R)-2-(2,3-dihydro-4-benzofuranyl)cyclopropyl]carbonyl]hexahydro-8,8-dimethyl-3H-3a,6-methano-2,1-benzisothiazole-2,2-dioxide
XVIII [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanemethanol
XIX [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanecarboxaldehyde
XX hydroxylamine hydrochloride
XXI [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanecarbaldehyde oxime
XXII [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanemethanamine
– Preparation of VII (aminoalkylindols): Structure-Activity Relationships of Novel Cannabinoid Mimetics Eissenstat et al, J.. Med. Chem. 1995, 38, 3094-3105
– G. Birznieks et al. Melatonin agonist VEC-162 Improves sleep onset and maintenance in a model of transient insomnia. Sleep 2007, 30, 0773 Abstract.
-. Rajaratnam SM et al, The melatonin agonist VEC-162 Phase time immediately advances the human circadian system, Sleep 2006, 29, 0159 Abstract.
-. AK Singh et al, Evolution of a manufacturing route for a highly potent drug candidate, 229th ACS Natl Meet, March 13-17, 2005, San Diego, Abstract MEDI 576.
– Vachharajani NN et al, Preclinical pharmacokinetics and metabolism of BMS-214778, a novel melatonin receptor agonist, J Pharm Sci. 2003 Apr; 92 (4) :760-72.
. – JW Scott et al, Catalytic Asymmetric Synthesis of a melotonin antagonist; synthesis and process optimization. 223rd ACS Natl Meet, April 7-11, Orlando, 2002, Abstract ORGN 186.
The syntheses of the 4-aryl-propenoic acid derivatives, 2 and 3, are shown in Reaction Scheme 1. The starting aldehydes, 1 , can be prepared by methods well known to those skilled in the art. Condensation of malonic acid with the aldehydes, 1, in solvents such as pyridine with catalysts such as piperidine or pyrrolidine, gives the 4-aryl- propenoic acid, 2. Subsequent conversion of the acid to the acid chloride using reagents such as thionyl chloride, phosphoryl chloride, or the like, followed by reaction with N,0-dimethyl hydroxylamine gives the amide intermediate 3 in good yields. Alternatively, aldehyde 1 can be converted directly to amide 3 using reagents such as diethyl (N-methoxy- N-methyl-carbamoylmethyl)phosphonate with a strong base such as sodium hydride.
Reaction Scheme 2
The conversion of the amide intermediate 3 to the racemic, trans- cyclopropane carboxaldehyde intermediate, 4, is shown in Reaction Scheme 2. Intermediate 3 was allowed to react with cyclopropanating reagents such as trimethylsulfoxonium iodide and sodium hydride in solvents such as DMF, THF, or the like. Subsequent reduction using reagents such as LAH in solvents such as THF, ethyl ether, or the like, gives the racemic, trans-cyclopropane carboxaldehyde intermediates, 4.
Reaction Scheme 3
Racemic cyclopropane intermediate 5 (R = halogen) can be prepared from intermediate 2 as shown in Reaction Scheme 3. Intermediate 2 was converted to the corresponding allylic alcohol by treatment with reducing agents such as sodium borohydride plus iodine in solvents such as THF. Subsequent acylation using reagents such as acetic anhydride in pyridine or acetyl chloride gave the allylic acetate which was allowed to react with cyclopropanating reagents such as sodium chloro-difluoroacetate in diglyme to provide the racemic, trans- cyclopropane acetate intermediates, 5. Reaction Scheme 4
The conversion of the acid 2 to the chiral cyclopropane carboxaldehyde intermediate, (-)-(trans)-4, is shown in Reaction Scheme 4. Intermediate 2 is condensed with (-)-2,10-camphorsultam under standard conditions, and then cyclopropanated in the presence of catalysts such as palladium acetate using diazomethane generated from reagents such as 1-methyl-3-nitro-1-nitrosoguanidine. Subsequent reduction using reagents such as LAH in solvents such as THF, followed by oxidation of the alcohol intermediates using reagents such as DMSO/oxalyl chloride, or PCC, gives the cyclopropane carboxaldehyde intermediate, (-)-(trans)-4, in good yields. The enantiomer, (+)-(trans)-4, can also be obtained employing a similar procedure using (+)-2,10- camphorsultam in place of (-)-2,10-camphorsultam.
When it is desired to prepare compounds of Formula I wherein m = 2, the alcohol intermediate may be activated in the conventional manner such as with mesyl chloride and treated with sodium cyanide followed by reduction of the nitrile group with a reducing agent such as LAH to produce the amine intermediate 6.
Reaction Scheme 5
Reaction Scheme 5 shows the conversion of intermediates 4 and 5 to the amine intermediate, 7, and the subsequent conversion of 6. or 7 to compounds of Formula I. The carboxaldehyde intermediate, 4, is condensed with hydroxylamine and then reduced with reagents such as LAH to give the amine intermediate, 7. The acetate intermediate 5 is hydrolyzed with potassium hydroxide to the alcohol, converted to the mesylate with methane sulfonyl chloride and triethyl amine in CH2CI2and then converted to the azide by treatment with sodium azide in solvents such as DMF. Subsequent reduction of the azide group with a reducing agent such as LAH produced the amine intermediate 7. Further reaction of 6 or 7 with acylating reagents gives compounds of Formula I. Suitable acylating agents include carboxylic acid halides, anhydrides, acyl imidazoles, alkyl isocyanates, alkyl isothiocyanates, and carboxylic acids in the presence of condensing agents, such as carbonyl imidazole, carbodiimides, and the like. Reaction Scheme 6
Reaction Scheme 6 shows the alkylation of secondary amides of Formula I (R2 = H) to give tertiary amides of Formula I (R2 = alkyl). The secondary amide is reacted with a base such as sodium hydride, potassium tert-butoxide, or the like, and then reacted with an alkylating reagent such as alkyl halides, alkyl sulfonate esters, or the like to produce tertiary amides of Formula I.
Reaction Scheme 7
Reaction Scheme 7 shows the halogenation of compounds of Formula I. The carboxamides, i (Q1 = Q2 = H), are reacted with excess amounts of halogenating agents such as iodine, N-bromosuccinimide, or the like to give the dihalo-compounds of Formula I (Q1 = Q2 = halogen). Alternatively, a stoichiometric amount of these halogenating agents can be used to give the monohalo-compounds of Formula I (Q1 = H, Q2 = halogen; or Q1 = halogen, Q2 = H). In both cases, additives such as lead IV tetraacetate can be used to facilitate the reaction. Biological Activity of the Compounds
The compounds of the invention are melatonergic agents. They have been found to bind human melatonergic receptors expressed in a stable cell line with good affinity. Further, the compounds are agonists as determined by their ability, like melatonin, to block the forskolin- stimulated accumulation of cAMP in certain cells. Due to these properties, the compounds and compositions of the invention should be useful as sedatives, chronobiotic agents, anxiolytics, antipsychotics, analgesics, and the like. Specifically, these agents should find use in the treatment of stress, sleep disorders, seasonal depression, appetite regulation, shifts in circadian cycles, melancholia, benign prostatic hyperplasia and related conditions
A mixture of benzofuran-4-carboxylic acid [Eissenstat, et al.. J. Medicinal Chemistry, 38 (16) 3094-3105 (1995)] (2.8 g, 17.4 mmol) and thionyl chloride (25 mL) was heated to reflux for 2 h and then concentrated in vacuo. The solid residue was dissolved in ethyl acetate (50 mL) and a solution of N,O-dimethylhydroxylamine hydrochloride (2.8 g) in saturated NaHC03(60 mL) was added with stirring. After stirring for 1.5 h, the ethyl acetate layer was separated. The aqueous layer was extracted with ethyl acetate. The ethyl acetate extracts were combined, washed with saturated NaHCO3 and concentrated in vacuo to give an oil (3.2 g, 95.4%).
Step 2: Benzofuran-4-carboxaldehyde
A solution of N-methoxy-N-methyl-benzofuran-4-carboxamide (3.2 g, 16.6 mmol) in THF (100 mL) was cooled to -45°C and then LAH (0.7 g, 18.7 mmol) was added. The mixture was stirred for 15 min, allowed to warm to -5°C, and then recooled to -45°C. Saturated KHS04 (25 mL) was added with vigorous stirring, and the mixture was allowed to warm to room temperature. The precipitate was filtered and washed with acetone. The filtrate was concentrated in vacuo to give an oil (2.3 g, 94%). Preparation 2
2,3-Dihydrobenzofuran-4-carboxaldehyde
Step 1 : 2,3-Dihydrobenzofuran-4-carboxylic acid
Benzofuran-4-carboxylic acid (10.0 g, 61 .7 mmol) was hydrogenated (60 psi) in acetic acid (100 mL) over 10% Pd/C (2 g) for 12 hr. The mixture was filtered and the filtrate was diluted with water (500 mL) to give 2,3- dihydrobenzofuran-4-carboxylic acid as a white powder (8.4 g, 83%). A sample was recrystallized from isopropanol to give fine white needles (mp: 185.5-187.5°C).
Step 2: (2,3-Dihydrobenzofuran-4-yl)methanol
A solution of 2,3-dihydrobenzofuran-4-carboxylic acid (10 g, 61 mmol) in THF (100 mL) was stirred as LAH (4.64 g, 122 mmol) was slowly added. The mixture was heated to reflux for 30 min. The mixture was cooled and quenched cautiously with ethyl acetate and then with 1 N HCI (150 mL). The mixture was then made acidic with 12 N HCI until all the inorganic precipitate dissolved. The organic layer was separated, and the inorganic layer was extracted twice with ethyl acetate. The organic layers were combined, washed twice with brine, and then concentrated in vacuo. This oil was Kϋgelrohr distilled to a clear oil that crystallized upon cooling (8.53 g, 87.6%).
Step 3: 2.3-Dihydrobenzofuran-4-carboxaldehyde
DMSO (8.10 mL, 1 14 mmol) was added at -78°C to a stirred solution of oxalyl chloride in CH2CI2 (40 mL of a 2M solution). A solution of (2,3- dihydrobenzofuran-4-yl)methanol (8.53 g, 56.9 mmol) in CH2CI2 (35 mL) was added dropwise, and the solution stirred at -78°C for 30 min. Triethyl amine (33 mL, 228 mmol) was added cautiously to quench the reaction. The resulting suspension was stirred at room temperature for 30 min and diluted with CH2CI2 (100 mL). The organic layer was washed three times with water, and twice with brine, and then concentrated in vacuo to an oil (8.42 g, 100%) that was used without purification.
Trimethylsulfoxonium iodide (9.9 g, 45 mmol) was added in small portions to a suspension of sodium hydride (1 .8 g, 45 mmol) in DMF (120 mL). After the foaming had subsided (10 min), a solution of (trans)- N-methoxy-N-methyl-3-(2,3-dihydrobenzofuran-4-yl)propenamide (3.5 g, 15 mmol) in DMF (60 mL) was added dropwise, with the temperature maintained between 35-40°C. The mixture was stirred for 3 h at room temperature. Saturated NH4CI (50 mL) was added dropwise and the mixture was extracted three times with ethyl acetate. The organic extracts were combined, washed with H2O and brine, dried over K2CO3, and concentrated in vacuo to give a white wax (3.7 g, 100%).
A solution of (±)-(trans)-N-methoxy-N-methyl-2-(2,3-dihydrobenzofuran- 4-yl)cyclopropanecarboxamide (3.7 g, 15 mmol) in THF (10 mL) was added dropwise to a rapidly stirred suspension of LAH (683 mg, 18 mmol) in THF (50 mL) at -45°C, maintaining the temperature below -40°C throughout. The cooling bath was removed, the reaction was allowed to warm to 5°C, and then the reaction was immediately recooled to -45°C. Potassium hydrogen sulfate (3.4 g, 25.5 mmol) in H20 (50 mL) was cautiously added dropwise, the temperature maintained below – 30°C throughout. The cooling bath was removed and the suspension was stirred at room temperature for 30 min. The mixture was filtered through Celite and the filter cake was washed with ether. The combined filtrates were then washed with cold 1 N HCI, 1 N NaOH, and brine. The filtrates were dried over MgSO4, and concentrated in vacuo to give a clear oil (2.6 g, 99%).
To a solution of (-)-2,10-camphorsultam (8.15 g, 37.9 mmol) in 50 mL toluene at 0°C was added sodium hydride (1.67 g, 41.7 mmol). After stirring for 0.33 h at 0°C and 0.5 h at 20°C and recooling to 0°C, a solution of 3-(2,3-dihydrobenzofuran-4-yl)-2-propenoyl chloride
(37.9 mmol), prepared in situ from the corresponding acid and thionyl chloride (75 mL), in toluene (50 mL), was added dropwise. After stirring for 18 h at 20°C, the mixture was diluted with ethyl acetate and washed with water, 1 N HCI, and 1 N NaOH. The organic solution was dried and concentrated in vacuo to give 15.8 g of crude product. Recrystallization form ethanol-methanol (600 mL, 1 :1) gave the product (13.5 g, 92%, mp 199.5-200°C).
1 -Methyl-3-nitro-1 -nitrosoguanidine (23.88g 163 mmol) was added in portions to a mixture of 10 N sodium hydroxide (60 mL) and ether (200 mL) at 0°C. The mixture was shaken vigorously for 0.25 h and the ether layer carefully decanted into a solution of (-)-N-[3-(2,3-dihydrobenzofuran-4-yl)-2-propenoyl]-2,10-camphorsultam (9.67 g, 25 mmol) and palladium acetate (35 mg) in methylene chloride (200 mL). After stirring for 18 h, acetic acid (5 mL) was added to the reaction and the mixture stirred for 0.5 h. The mixture was washed with 1 N HCI, 1 N NaOH and brine. The solution was dried, concentrated in vacuo and the residue crystallized twice from ethanol to give the product (6.67 g, 66.5%, mp 157-159°C).
A solution of (-)-N-[(trans)-2-(2,3-dihydrobenzofuran-4-yl)cyclo-propanecarbonylj-2,10-camphorsultam (4.3 g, 10.7 mmol) in THF (50 mL) was added dropwise to a mixture of LAH (0.81 g, 21.4 mmol) in THF (50 mL) at -45°C. The mixture was stirred for 2 hr while it warmed to 10°C. The mixture was recooled to -40°C and hydrolyzed by the addition of saturated KHS0 (20 mL). The mixture was stirred at room temperature for 30 minutes and filtered. The precipitate was washed twice with acetone. The combined filtrate and acetone washes were concentrated in vacuo. The gummy residue was dissolved in ether, washed with 1 N NaOH and 1 N HCI, and then dried in vacuo to give the product (2.0 g, 98.4%).
Step 4: (-)-(trans)-2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane- carboxaldehyde DMSO (1.6 g, 21 mmol) was added to oxalyl chloride in CH2CI2(7.4 mL of 2 M solution, 14.8 mmole) at -78°C. The (-)-(trans)-2-(2,3-dihydrobenzofuran-4-yl)-cyclopropylmethanol (2.0 g, 10.5 mmol) in CH2CI2(15 mL) was added. The mixture was stirred for 20 min and then triethylamine (4.24 g, 42 mmol) was added. The mixture was warmed to room temperature and stirred for 30 min. The mixture was diluted with CH2CI2 and washed with water, 1 N HCI, and then 1 N NaOH. The organic layer was dried and concentrated iι> vacuo to give the aldehyde product (1.98 g, 100%).
Preparation 24
(-)-(trans)-2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane-methanamine A mixture of (-)-(trans)-2-(2,3-dihydrobenzofuran-4-yl)cyclopropane-carboxaldehyde (1.98 g, 10.5 mmol), hydroxylamine hydrochloride (2.29 g, 33 mmol), and 30% NaOH (3.5 mL, 35 mmol), in 5:1
ethanol/water (50 mL) was heated on a steam bath for 2 h. The solution was concentrated in vacuo. and the residue mixed with water. The mixture was extracted with CH2CI2. The organic extracts were dried and concentrated in vacuo to give a solid which NMR analysis showed to be a mixture of the cis and trans oximes. This material was dissolved in THF (20 mL) and added to solution of alane in THF [prepared from LAH (1.14 g, 30 mmol) and H2S04 (1.47 g, 15 mmol) at 0°Cj. The reaction was stirred for 18 h, and quenched successively with water (1.15 mL), 15% NaOH (1.15 mL), and then water (3.45 mL). The mixture was filtered and the filtrate was concentrated in vacuo. The residue was mixed with ether and washed with water and then 1 N HCI. The acid washes were made basic and extracted with CH2CI . The extracts were dried and concentrated in vacuo to give the amine product (1.4 g, 70.5%). The amine was converted to the fumarate salt in ethanol (mp: 197-198°C).
Anal. Calc’d for C12H15NO • C4H404: C, 62.94; H, 6.27; N, 4.59.
Found: C, 62.87; H, 6.31 ; N, 4.52.
This compound was prepared similar to the above procedure using propionyl chloride and (-)-(trans)-2-(2,3-dihydrobenzofuran-4-yl)- cyclopropanemethanamine to give an oil that solidified upon standing to an off-white solid (61 %, mp: 71-72°C). IR (NaCI Film): 3298, 1645, 1548, 1459, 1235 cm“1.
Mo5 : -17.3°
Anal. Calc’d for C15H19N02: C, 73.44; H, 7.87; N, 5.71 . Found: C, 73.28; H, 7.68; N, 5.58
SYNTHESIS
Synthesis Path
SYN
Tasimelteon (Hetlioz)Tasimelteon, which is marketed by Vanda Pharmaceuticals as Hetlioz and developed in partnership with Bristol-Myers Squibb,is a drug that was approved by the US FDA in January 2014 for the treatment of non-24-hour sleep–wake disorder (also called Non-24, N24 and N24HSWD).234 Tasimelteon is a melatonin MT1
and MT2 receptor agonist; because it exhibits a greater affinity to the MT2 receptor than MT1, is also known as Dual Melatonin
Receptor Agonist.234 Two randomized controlled trials (phases II
and III) demonstrated that tasimelteon improved sleep latency
and maintenance of sleep with a shift in circadian rhythms, and
therefore has the potential to treat patients with transient insomnia
associated with circadian rhythm sleep disorders.235 Preclinical
studies showed that the drug has similar phase-shifting properties
to melatonin, but with less vasoconstrictive effects.236 The most
likely scale preparation of the drug, much of which has been published
in the chemical literature, is described below in Scheme 44.
Activation of commercial bis-ethanol 250 with 2.5 equivalents
of the Vilsmeier salt 251 followed by treatment with base resulted
an intramolecular cyclization reaction with the proximal phenol
and concomitant elimination of the remaining imidate to deliver
the vinylated dihydrobenzofuran 252 in 76% yield.237 Interestingly,
this reaction could be performed on multi-kilogram scale, required
no chromatographic purification, and generated environmentallyfriendly
DMF and HCl as byproducts.237 Sharpless asymmetric
dihydroxylation of olefin 252 delivered diol 253 in 86% yield and
impressive enantioselectivity (>99% ee). This diol was then activated
with trimethylsilyl chloride and then treated with base to generate epoxide 254.238 Next, a modified Horner–Wadsworth–
Emmons reaction involving triethylphosphonoacetate (TEPA, 255)
was employed to convert epoxide 254 to cyclopropane 256.239
The reaction presumably proceeds through removal of the acidic
TEPA proton followed by nucleophilic attack at the terminal epoxide
carbon. The resulting alkoxide undergoes an intramolecular
phosphoryl transfer reaction resulting in an enolate, which then attacked the newly formed phosphonate ester in an SN2 fashion
resulting in the trans-cyclopropane ester, which was ultimately
saponified and re-acidified to furnish cyclopropane acid 256.239
Conversion of this acid to the corresponding primary amide preceded
carbonyl reduction with sodium borohydride. The resulting
amine was acylated with propionyl chloride to furnish tasimelteon
(XXXI) as the final product in 86% yield across the four-step
sequence.
Submitted by Michael C. Weismiller, James C. Towson, and Franklin A. Davis1.
Checked by David I. Magee and Robert K. Boeckman, Jr..
1. Procedure
(−)-2,10-Camphorsultam. A dry, 2-L, three-necked, round-bottomed flask is equipped with a 1.5-in egg-shaped Teflon stirring bar, a 250-mL addition funnel, and a 300-mL Soxhlet extraction apparatus equipped with a mineral oil bubbler connected to an inert-gas source. The flask is charged with 600 mL of dry tetrahydrofuran (THF) (Note 1) and6.2 g (0.16 mol) of lithium aluminum hydride (Note 2). Into the 50-mL Soxhlet extraction thimble is placed 35.0 g (0.16 mol) of (−)-(camphorsulfonyl)imine (Note 3) and the reaction mixture is stirred and heated at reflux. After all of the(camphorsulfonyl)imine has been siphoned into the reaction flask (3–4 hr), the mixture is allowed to cool to room temperature. The unreacted lithium aluminum hydride is cautiously hydrolyzed by dropwise addition of 200 mL of 1 Nhydrochloric acid via the addition funnel (Note 4). After the hydrolysis is complete the contents of the flask are transferred to a 1-L separatory funnel, the lower, silver-colored aqueous layer is separated, and the upper layer placed in a 1-L Erlenmeyer flask. The aqueous phase is returned to the separatory funnel and washed with methylene chloride (3 × 100 mL). After the reaction flask is rinsed with methylene chloride (50 mL), the organic washings are combined with the THF phase and dried over anhydrous magnesium sulfate for 10–15 min. Filtration through a 300-mL sintered-glass funnel of coarse porosity into a 1-L round-bottomed flask followed by removal of the solvent on arotary evaporator gives 33.5 g (95%) of the crude (−)-2,10-camphorsultam. The crude sultam is placed in a 250-mL Erlenmeyer flask and crystallized from approximately 60 mL of absolute ethanol. The product is collected on a 150-mL sintered-glass funnel of coarse porosity and dried in a vacuum desiccator to give 31.1 g (88%) of the pure sultam. A second crop of crystals can be gained by evaporating approximately half the filtrate; the residue is crystallized as above to give 1.4 g (4%). The combined yield of white crystalline solid, mp 183–184°C, [α]D −30.7° (CHCl3, c 2.3) is92% (Note 5) and (Note 6).
2. Notes
1. Tetrahydrofuran (Aldrich Chemical Company, Inc.) was distilled from sodium benzophenone.
2. Lithium aluminum hydride was purchased from Aldrich Chemical Company, Inc.
3. (−)-(Camphorsulfonyl)imine, [(7S)-(−)-10,10-dimethyl-5-thia-4-azatricyclo[5.2.1.03,7]dec-3-ene 5,5-dioxide] was prepared by the procedure of Towson, Weismiller, Lal, Sheppard, and Davis, Org. Synth., Coll. Vol. VIII, 1993, 104.
4. The addition must be very slow at first (1 drop/5 sec) until the vigorous reaction has subsided.
6. Checkers obtained material having the same mp (183–184°C) and [α]D − 31.8° (CHCl3, c 2.3).
3. Discussion
(−)-2,10-Camphorsultam was first prepared by the catalytic hydrogenation of (−)-(camphorsulfonyl)imine overRaney nickel.2 Lithium aluminum hydride reduction was used by Oppolzer and co-workers in their synthesis of the sultam.3,4 However, because of the low solubility of the sultam in tetrahydrofuran, a large amount of solvent was required.4 In the procedure described here the amount of solvent is significantly reduced by using a Soxhlet extractor to convey the imine slowly into the reducing medium.5
Oppolzer’s chiral auxiliary,6 (−)-2,10-camphorsultam, is useful in the asymmetric Diels–Alder reaction,3,4 and for the preparation of enantiomerically pure β-substituted carboxylic acids7 and diols,8 in the stereoselective synthesis of Δ2-isoxazolines,9 and in the preparation of N-fluoro-(−)-2,10-camphorsultam, an enantioselective fluorinating reagent.10
References and Notes
Department of Chemistry, Drexel University, Philadelphia, PA 19104.
Shriner, R. L.; Shotton, J. A.; Sutherland, H. J. Am. Chem. Soc.1938, 60, 2794.
Submitted by James C. Towson, Michael C. Weismiller, G. Sankar Lal, Aurelia C. Sheppard, Anil Kumar, and Franklin A. Davis1.
Checked by David I. Magee and Robert K. Boeckman, Jr..
1. Procedure
A. (+)-(1S)-10-Camphorsulfonamide. Into a 2-L, two-necked, round-bottomed flask, equipped with a 250-mL dropping funnel, a magnetic stirring bar, and a reflux condenser fitted with an outlet connected to a disposable pipettedipped in 2 mL of chloroform in a test tube for monitoring gas evolution, were placed 116 g (0.5 mol) ofcamphorsulfonic acid (Note 1) and 750 mL of reagent-grade chloroform. The suspension of camphorsulfonic acid was heated to reflux and 71.4 g (43.77 mL, 0.6 mol, 1.2 equiv) of freshly distilled thionyl chloride was added dropwise over a 1-hr period. Heating was continued until gas evolution (sulfur dioxide and hydrogen chloride) had ceased (approximately 9–10 hr). The resultant solution of camphorsulfonyl chloride in chloroform was converted tocamphorsulfonamide without further purification.
In a 5-L, two-necked, round-bottomed flask fitted with a 250-mL dropping funnel and a mechanical stirrer was placed a solution of 1.6 L of reagent-grade ammonium hydroxide solution and the flask was cooled to 0°C in an ice bath. The solution of the crude camphorsulfonyl chloride, prepared in the preceding section, was added dropwise to the ammonium hydroxide solution at 0–10°C over a period of 1 hr. The reaction mixture was warmed to room temperature, stirred for 4 hr, the organic layer separated, and the aqueous layer was extracted with methylene chloride (3 × 250 mL). The combined organic layers were washed with brine (250 mL) and dried over anhydrousmagnesium sulfate. Removal of the solvent on the rotary evaporator gave 104.0 g (90%) of the crudecamphorsulfonamide (Note 2) and (Note 3).
B. (−)-(Camphorsulfonyl)imine. A 1-L, round-bottomed flask is equipped with a 2-in. egg-shaped magnetic stirring bar, a Dean–Stark water separator, and a double-walled condenser containing a mineral oil bubbler connected to an inert gas source. Into the flask are placed 5 g of Amberlyst 15 ion-exchange resin (Note 4) and 41.5 g of the crude(+)-(1S)-camphorsulfonamide in 500 mL of toluene. The reaction mixture is heated at reflux for 4 hr. After the reaction flask is cooled, but while it is still warm (40–50°C), 200 mL of methylene chloride is slowly added to dissolve any(camphorsulfonyl)imine that crystallizes. The solution is filtered through a 150-mL sintered glass funnel of coarse porosity an the reaction flask and filter funnel are washed with an additional 75 mL of methylene chloride.
Isolation of the (−)-(camphorsulfonyl)imine is accomplished by removal of the toluene on the rotary evaporator. The resulting solid is recrystallized from absolute ethanol (750 mL) to give white crystals, 34.5–36.4 g (90–95%), mp225–228°C; [α]D −32.7° (CHCl3, c 1.9) (Note 5).
C. (+)-(2R, 8aS)-10-Camphorylsulfonyloxaziridine. A 5-L, three-necked, round-bottomed Morton flask is equipped with an efficient mechanical stirrer, a 125-mm Teflon stirring blade, a Safe Lab stirring bearing (Note 6), and a 500-mL addition funnel. Into the flask are placed the toluene solution of (−)-(camphorsulfonyl)imine (39.9 g, 0.187 mol)prepared in Step B and a room-temperature solution of 543 g (3.93 mol, 7 equiv based on oxone) of anhydrouspotassium carbonate dissolved in 750 mL of water. The reaction mixture is stirred vigorously and a solution of 345 g (0.56 mol, 6 equiv of KHSO5) of oxone dissolved in 1250 mL of water is added dropwise in three portions over 45 min(Note 7) and (Note 8). Completion of the oxidation is determined by TLC (Note 9) and the reaction mixture is filtered through a 150-mL sintered-glass funnel of coarse porosity to remove solids. The filtrate is transferred to a 3-L separatory funnel, the toluene phase is separated and the aqueous phase is washed with methylene chloride (3 × 100 mL). The filtered solids and any solids remaining in the Morton flask are washed with an additional 200 mL of methylene chloride. The organic extracts are combined and washed with 100 mL of saturated sodium sulfite, dried over anhydrousmagnesium sulfate for 15–20 min, filtered, and concentrated on the rotary evaporator. The resulting white solid is crystallized from approximately 500 mL of hot 2-propanol to afford, after drying under vacuum in a desiccator, 35.9 g(84%) of white needles, mp 165–167°C, [α]D +44.6° (CHCl3, c 2.2) (Note 10) and (Note 11).
(−)-(2S,8aR)-10-(camphorylsulfonyl)oxaziridine is prepared in a similar manner starting from (−)-10-camphorsulfonic acid; mp 166–167°C, [α]D +43.6° (CHCl3, c 2.2).
2. Notes
1. (1S)-(+)-10-Camphorsulfonic acid was purchased from Aldrich Chemical Company, Inc.
2. The crude sulfonamide is contaminated with 5–10% of the (camphorsulfonyl)imine, the yield of which increases on standing.
3. The 1H NMR spectrum of (+)-(1S)-10-camphorsulfonamide is as follows: (CDCl3) δ: 0.93 (s, 3 H, CH3), 1.07 (s, 3 H, CH3), 1.40–2.50 (m, 7 H), 3.14 and 3.53 (AB quartet, 2 H, CH2-SO2, J = 15.1), 5.54 (br s, 2 H, NH2).
4. Amberlyst 15 ion-exchange resin is a strongly acidic, macroreticular resin purchased from Aldrich Chemical Company, Inc.
5. The spectral properties of (−)-(camphorsulfonyl)imine are as follows: 1H NMR (CDCl3) δ: 1.03 (s, 3 H, CH3), 1.18 (s, 3 H, CH3), 1.45–2.18 (m, 6 H), 2.65 (m, 1 H), 3.10 and 3.28 (AB quartet, 2 H, CH2-SO2, J = 14.0); 13C NMR (CDCl3) δ: 19.01 (q, CH3), 19.45 (q, CH3), 26.64 (t), 28.44 (t), 35.92 (t), 44.64 (d), 48.00 (s), 49.46 (t), 64.52 (s), 195.52 (s); IR (CHCl3) cm−1: 3030, 2967, 1366. Checkers obtained material having identical melting point and [α]D−32.3° (CHCl3, c 1.8).
6. The SafeLab Teflon bearing can be purchased from Aldrich Chemical Company, Inc. A glass stirring bearing lubricated with silicone grease is unsatisfactory because the dissolved salts solidify in the shaft, causing freezing.
7. Efficient stirring is important and indicated by a milky white appearance of the solution.
8. Occasionally batches of oxone purchased from Aldrich Chemical Company, Inc., have exhibited reduced reactivity in this oxidation. Oxone exposed to moisture prior to use also gives reduced reactivity in this oxidation. If this occurs, oxone is added until oxidation is complete as determined by TLC (Note 9). Potassium carbonate is added as needed to maintain the pH at approximately 9.0. Oxone stored in the refrigerator under an inert atmosphere has shown no loss in reactivity for up to 6 months.
9. Oxidation is generally complete after addition of the oxone solution. The oxidation is monitored by TLC as follows. Remove approximately 0.5 mL of the toluene solution from the nonstirring solution, spot a 250-μm TLC silica gel plate, elute with methylene chloride, and develop with 10% molybdophosphoric acid in ethanol and heating(camphorsulfonyl)imine Rf = 0.28 and (camphorylsulfonyl)oxaziridine Rf = 0.62. If (camphorsulfonyl)imine is detected, stirring is continued at room temperature until the reaction is complete (see (Note 8)). If the reaction mixture takes on a brownish color after addition of oxone and has not gone to completion after 30 min, the reaction mixture is filtered through a 150-mL sintered-glass funnel of coarse porosity, and the solids are washed with 50 mL of methylene chloride. The aqueous/organic extracts are returned to the 5-L Morton flask and stirred vigorously and 52 g (0.08 mol, 1 equiv KHSO5) of oxone is added over 5 min and stirring continued until oxidation is complete (approximately 10–15 min).
10. The submitters employed a toluene solution of crude imine prepared in Part B and obtained somewhat higher yields (90–95%). However, the checkers obtained yields in this range on one half the scale using isolatedsulfonylimine.
11. The spectral properties of (+)-(camphorsulfonyl)oxaziridine are as follows: 1H NMR (CDCl3) δ: 1.03 (s, 3 H, CH3), 1.18 (s, 3 H, CH3), 1.45–2.18 (m, 6 H), 2.65 (d, 1 H), 3.10 and 3.28 (AB quartet, 2 H, CH2-SO2, J = 14.0); 13C NMR (CDCl3) δ: 19.45 (q, CH3), 20.42 (q, CH3), 26.55 (t), 28.39 (t), 33.64 (t), 45.78 (d), 48.16 (s), 48.32 (t), 54.07 (s), 98.76 (s). The checkers obtained material (mp 165–167°C) having [α]D +44.7° (CHCl3, c 2.2).
3. Discussion
Camphorsulfonamide, required for the preparation of the (camphorsulfonyl)imine, was previously prepared in two steps. The first step involved conversion of camphorsulfonic acid to the sulfonyl chloride with PCl5 or SOCl2. The isolated sulfonyl chloride was converted in a second step to the sulfonamide by reaction with ammonium hydroxide. This modified procedure is more efficient because it transforms camphorsulfonic acid directly to camphorsulfonamide, avoiding isolation of the camphorsulfonyl chloride.
(Camphorsulfonyl)imine has been reported as a by-product of reactions involving the camphorsulfonamide.2,3,4,5Reychler in 1898 isolated two isomeric camphorsulfonamides,2 one of which was shown to be the(camphorsulfonyl)imine by Armstrong and Lowry in 1902.3 Vandewalle, Van der Eycken, Oppolzer, and Vullioud described the preparation of (camphorsulfonyl)imine in 74% overall yield from 0.42 mol of the camphorsulfonyl chloride.6 The advantage of the procedure described here is that, by using ammonium hydroxide, the camphorsulfonyl chloride is converted to the sulfonamide in >95% yield.7 The sulfonamide is of sufficient purity that it can be used directly in the cyclization step, which, under acidic conditions, is quantitative in less than 4 hr. These modifications result in production of the (camphorsulfonyl)imine in 86% overall yield from the sulfonyl chloride.
In addition to the synthesis of enantiomerically pure (camphorylsulfonyl)oxaziridine7 and its derivatives,8 the(camphorsulfonyl)imine has been used in the preparation of (−)-2,10-camphorsultam (Oppolzers’ auxiliary),6,9 (+)-(3-oxocamphorysulfonyl) oxaziridine,10 and the N-fluoro-2,10-camphorsultam, an enantioselective fluorinating reagent.11
The N-sulfonyloxaziridines are an important class of selective, aprotic oxidizing reagents.121314 Enantiomerically pure N-sulfonyloxaziridines have been used in the asymmetric oxidation of sulfides to sulfoxides (30–91% ee),15selenides to selenoxides (8–9% ee).16 disulfides to thiosulfinates (2–13% ee),5 and in the asymmetric epoxidation of alkenes (19–65% ee).17,18 Oxidation of optically active sulfonimines (R*SO2N=CHAr) affords mixtures of N-sulfonyloxaziridine diastereoisomers requiring separation by crystallization and/or chromatography.3
(+)-(Camphorylsulfonyl)oxaziridine described here is prepared in four steps from inexpensive (1S)-(+)- or (1R)-(+)-10-camphorsulfonic acid in 77% overall yield.7 Separation of the oxaziridine diastereoisomers is not required because oxidation is sterically blocked from the exo face of the C-N double bond in the (camphorsulfonyl)imine. In general, (camphorsulfonyl)oxaziridine exhibits reduced reactivity compared to other N-sulfonyloxaziridines. For example, while sulfides are asymmetrically oxidized to sulfoxides (3–77% ee), this oxaziridine does not react with amines or alkenes.7 However, this oxaziridine is the reagent of choice for the hydroxylation of lithium and Grignard reagents to give alcohols and phenols because yields are good to excellent and side reactions are minimized.19 This reagent has also been used for the stereoselective oxidation of vinyllithiums to enolates.20
The most important synthetic application of the (camphorylsulfonyl)oxaziridines is the asymmetric oxidation of enolates to optically active α-hydroxy carbonyl compounds.14,21,22,23,24 Chiral, nonracemic α-hydroxy carbonylcompounds have been used extensively in asymmetric synthesis, for example, as chiral synthons, chiral auxiliaries, and chiral ligands. This structural array is also featured in many biologically active natural products. This oxidizing reagent gives uniformly high chemical yields regardless of the counterion, and stereoselectivities are good to excellent (50–95% ee).9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24 Since the configuration of the oxaziridine three-membered ring controls the stereochemistry, both α-hydroxy carbonyl optical isomers are readily available. Representative examples of the asymmetric oxidation of prochiral enolates by (+)-(2R,8aS)-camphorylsulfonyl)oxaziridine are given in Tables I and II.
Davis, F. A.; Jenkins, Jr., R. H.; Awad, S. B.; Stringer, O. D.; Watson, W. H.; Galloy, J. J. Am. Chem. Soc.1982, 104, 5412.
Vandewalle, M.; Van der Eycken, J.; Oppolzer, W.; Vullioud, C. Tetrahedron, 1986, 42, 4035.
Davis, F. A.; Towson, J. C.; Weismiller, M. C.; Lal, S.; Carroll, P. J. J. Am. Chem. Soc.1988, 110, 8477.
Davis, F. A.; Weismiller, M. C.; Lal, G. S.; Chen, B. C.; Przeslawski, R. M. Tetrahedron Lett., 1989, 30, 1613.
Oppolzer, W. Tetrahedron1987, 43, 1969.
Glahsl, G.; Herrmann, R. J. Chem. Soc., Perkin Trans. I1988, 1753.
Differding, E.; Lang, R. W. Tetrahedron Lett.1988, 29, 6087.
For recent reviews on the chemistry of N-sulfonyloxaziridines, see: (a) Davis, F. A.; Jenkins, Jr., R. H. in “Asymmetric Synthesis,” Morrison, J. D., Ed.; Academic Press: Orlando, FL, 1984, Vol. 4, Chapter 4;
Davis, F. A.; Haque, S. M. in “Advances in Oxygenated Processes,” Baumstark, A. L., Ed.; JAI Press: London, Vol. 2;
Davis, F. A.; Sheppard, A. C. Tetrahedron1989, 45, 5703.
Davis, F. A.; McCauley, Jr., J. P.; Chattopadhyay, S.; Harakal, M. E.; Towson, J. C.; Watson, W. H.; Tavanaiepour, I. J. Am. Chem. Soc.1987, 109, 3370.
Davis, F. A.; Stringer, O. D.; McCauley, Jr., J. M. Tetrahedron1985, 41, 4747.
Davis, F. A.; Chattopadhyay, S. Tetrahedron Lett.1986, 27, 5079.
Davis, F. A.; Harakal, M. E.; Awad, S. B. J. Am. Chem. Soc.1983, 105, 3123.
Davis, F. A.; Wei, J.; Sheppard, A. C.; Gubernick S. Tetrahedron Lett.1987, 28, 5115.
Davis, F. A.; Lal, G. S.; Wei, J. Tetrahedron Lett.1988, 29, 4269.
Davis, F. A.; Haque, M. S.; Ulatowski, T. G.; Towson, J. C. J. Org. Chem.1986, 51, 2402.
Davis, F. A.; Haque, M. S. J. Org. Chem.1986, 51, 4083; Davis, F. A.; Haque, M. S.; Przeslawski, R. M. J. Org. Chem.1989, 54, 2021.
Davis, F. A.; Ulatowski, T. G.; Haque, M. S. J. Org. Chem.1987, 52, 5288.
Davis, F. A.; Sheppard, A. C., Lal, G. S. Tetrahedron Lett.1989, 30, 779.
Davis, F. A.; Sheppard, A. C.; Chen, B. C.; Haque, M. S. J. Am. Chem. Soc.1990, 112, 6679.
a US 5 856 529 (Bristol-Myers Squibb; 5.1.1999; appl. 9.12.1997; USA-prior. 10.12.1996).
b US 7 754 902 (Vanda Pharms.; 13.7.2010; appl. 18.5.2006).
Jump up^Vachharajani, Nimish N.; Yeleswaram, Krishnaswamy; Boulton, David W. (April 2003). “Preclinical pharmacokinetics and metabolism of BMS-214778, a novel melatonin receptor agonist”. Journal of Pharmaceutical Sciences. 92 (4): 760–72. doi:10.1002/jps.10348. PMID12661062.
Jump up^Sack, R. L.; Brandes, R. W.; Kendall, A. R.; Lewy, A. J. (2000). “Entrainment of Free-Running Circadian Rhythms by Melatonin in Blind People”. New England Journal of Medicine. 343 (15): 1070–7. doi:10.1056/NEJM200010123431503. PMID11027741.
Jump up^“Side Effects Drug Center: Hetlioz Warnings and Precautions”. RxList. February 10, 2014. In animal studies, administration of tasimelteon during pregnancy resulted in developmental toxicity (embryofetal mortality, neurobehavioral impairment, and decreased growth and development in offspring) at doses greater than those used clinically.
//////////////BMS-214778, VEC-162, Tasimelteon, Hetlioz, FDA 2014, 609799-22-6 , BMS-214778, VEC-162, ATC N05CH03, タシメルテオン , EU 2015, VANDA, BMS, orphan drug designations
CCC(=O)NCC1CC1C2=C3CCOC3=CC=C2
Chemical and physical properties
Tasimelteon has two stereogenic centers. Besides the medically used trans-1 R , 2 R isomer (in the picture above left), there are thus three further stereoisomers that do not arise in the synthesis.
Tasimelteon is a white to off-white crystalline non-hygroscopic substance, soluble in water at physiologically relevant pH levels and readily soluble in alcohols, cyclohexane and acetonitrile. The compound occurs in two crystal forms. It is an anhydrate melting at 74 ° C and a hemihydrate . [4] The hemihydrate is from about 35 ° C the water of hydration and converts thereby in the anhydrate form to. [4] The anhydrate crystallizes in a monoclinic lattice with the space group P 2 1 , and the hemihydrate crystallizes in a tetragonal lattice with the space group P 4 3 21 2. [4]
4 Kaihang Liu, Zhou Xinbo, Zhejing Xu, Bai Hongzhen, Jianrong Zhu Jianming Gu, Guping Tang, Liu Xingang, Hu Xiurong: anhydrate and hemihydrate of Tasimelteon: Synthesis, structure, and pharmacokinetic study in J. Pharm. Biomed. Anal. 151 (2018) 235-243, doi : 10.1016 / j.jpba.2017.12.035 .
BMS compd for treatment for type 2 diabetes( GPR40 agonists with a dual mechanism of action, promoting both glucose-dependent insulin and incretin secretion)
BMS-986118 is a GPR40 full agonist. GPR40 is a G-protein-coupled receptor expressed primarily in pancreatic islets and intestinal L-cells that has been a target of significant recent therapeutic interest for type II diabetes. Activation of GPR40 by partial agonists elicits insulin secretion only in the presence of elevated blood glucose levels, minimizing the risk of hypoglycemia
NOTE CAS OF , 1H-Pyrazole-5-acetic acid, 1-[4-[[(3S,4S)-1-(5-chloro-2-methoxy-4-pyridinyl)-3-methyl-4-piperidinyl]oxy]phenyl]-4,5-dihydro-4-methyl-3-(trifluoromethyl)-, (4S,5S)- IS 1610562-73-6
G protein-coupled receptor 40 (GPR40) has become an attractive target for the treatment of diabetes since it was shown clinically to promote glucose-stimulated insulin secretion. Herein, we report our efforts to develop highly selective and potent GPR40 agonists with a dual mechanism of action, promoting both glucose-dependent insulin and incretin secretion. Employing strategies to increase polarity and the ratio of sp3/sp2 character of the chemotype, we identified BMS-986118 (compound 4), which showed potent and selective GPR40 agonist activity in vitro. In vivo, compound 4 demonstrated insulinotropic efficacy and GLP-1 secretory effects resulting in improved glucose control in acute animal models.
To a stirred solution of methyl 2-((4S,5S)-1-(4-(((3R,4R)-1-(5-chloro-2-methoxypyridin-4-yl)-3-methylpiperidin-4-yl)oxy)phenyl)-4-methyl-3-(trifluoromethyl)-4,5-dihydro-1H-pyrazol-5-yl)acetate (5.5 g, 9.9 mmol) in THF (90 mL) and water (9 mL) at room temperature was added 2 N LiOH solution (12 mL, 24 mmol). The reaction mixture was stirred at room temperature for 16 h, and 1 N HCl (25 mL, 25 mmol) was added at 0 °C to pH = 4–5. The solvent was evaporated, and the residue was extracted three times with EtOAc. The organic extracts were dried over Na2SO4; the solution was filtered and concentrated. The residue was recrystallized from isopropanol to give 4(neutral form) as white solid (4.3 g, 7.7 mmol, 78% yield).
The final two steps used to prepare greater than 1 kg of a compound evaluated as a treatment for type 2 diabetes are reported. The application of a palladium-catalyzed C–O coupling presented significant challenges due to the nature of the reactants, impurities produced, and noncrystalline coupling intermediate. Process development was able to address these limitations and enable production of kilogram quantities of the active pharmaceutical ingredient (API) in greater efficiency than a Mitsunobu reaction for formation of the key bond. The development of a sequence that telescopes the coupling with the subsequent ester hydrolysis to yield the API and the workup and final product crystallization necessary to produce high-quality drug substance without the need of column chromatography are discussed.
Bruce Ellsworth, Director, Head of Fibrosis Discovery Chemistry at Bristol-Myers Squibb
Rick Ewing, Head, External Partnerships, Discovery Chemistry and Molecular Technologies at Bristol-Myers Squibb
1: Li Z, Qiu Q, Geng X, Yang J, Huang W, Qian H. Free fatty acid receptor
agonists for the treatment of type 2 diabetes: drugs in preclinical to phase II
clinical development. Expert Opin Investig Drugs. 2016 Aug;25(8):871-90. doi:
10.1080/13543784.2016.1189530. PubMed PMID: 27171154.
Discovery of BMS-986118, a dual MOA GPR40 agonist that produces glucose-dependent insulin and GLP-1 secretion Bruce A Ellsworth, Jun Shi, Elizabeth A Jurica, Laura L Nielsen, Ximao Wu, Andres H Hernandez, Zhenghua Wang, Zhengxiang Gu, Kristin N Williams, Bin Chen, Emily C Cherney, Xiang-Yang Ye, Ying Wang, Min Zhou, Gary Cao, Chunshan Xie, Jason J Wilkes, Heng Liu, Lori K Kunselman, Arun Kumar Gupta, Ramya Jayarama, Thangeswaran Ramar, J. Prasada Rao, Bradley A Zinker, Qin Sun, Elizabeth A Dierks, Kimberly A Foster, Tao Wang, Mary Ellen Cvijic, Jean M Whaley, Jeffrey A Robl, William R Ewing.
Mechanism of Action Agammaglobulinaemia tyrosine kinase inhibitors
Highest Development Phases
Phase I Rheumatoid arthritis
Most Recent Events
30 Jan 2018 Bristol-Myers Squibb completes a phase I trial in Rheumatoid arthritis (In volunteers, In adults, Combination therapy) in USA (PO) (NCT03262740)
10 Nov 2017 Bristol-Myers Squibb completes a phase I drug-drug interaction trial in Healthy volunteers (NCT03131973)
03 Nov 2017 Safety, pharmacokinetic, and pharmacodynamic data from a pharmacokinetic trial in healthy volunteers presented at the 81st American College of Rheumatology and the 52nd Association of Rheumatology Health Professionals Annual Scientific Meeting (ACR/ARHP-2017)
BMS-986195 is a potent, covalent, irreversible inhibitor of Bruton’s tyrosine kinase (BTK), a member of the Tec family of non-receptor tyrosine kinases essential in antigen-dependent B-cell signaling and function. BMS-986195 is more than 5000-fold selective for BTK over all kinases outside of the Tec family, and selectivity ranges from 9- to 1010-fold within the Tec family. BMS-986195 inactivated BTK in human whole blood with a rapid rate of inactivation (3.5×10-4 nM-1·min-1) and potently inhibited antigen-dependent interleukin-6 production, CD86 expression and proliferation in B cells (IC50 <1 nM) without effect on antigen-independent measures in the same cells.
Bristol-Myers Squibb is developing BMS-986195, an oral candidate for the treatment of rheumatoid arthritis. A phase I clinical trial in healthy adult volunteers is ongoing.
Credit: Tien Nguyen/C&EN
Presented by: Scott H. Watterson, principal scientist at Bristol-Myers Squibb
Target: Bruton’s tyrosine kinase (BTK)
Disease: Autoimmune diseases such as rheumatoid arthritis
Reporter’s notes: Completing another set of back-to-back presentations on the same target, Watterson revealed another BTK inhibitor also in Phase II clinical trials. Chemists made BMS-986195 in seven steps, and the molecule showed high levels of BTK inactivation in mice. The team aimed to develop an effective compound that required low doses and that had low metabolic degradation.
otein kinases, the largest family of human enzymes, encompass well over 500 proteins. Btk is a member of the Tec family of tyrosine kinases, and is a regulator of early B-cell development, as well as mature B-cell activation, signaling, and survival.
B-cell signaling through the B-cell receptor (BCR) leads to a wide range of biological outputs, which in turn depend on the developmental stage of the B-cell. The magnitude and duration of BCR signals must be precisely regulated. Aberrant BCR-mediated signaling can cause dysregulated B-cell activation and/or the formation of pathogenic auto-antibodies leading to multiple autoimmune and/or inflammatory diseases. Mutation of Btk in humans results in X-linked agammaglobulinaemia (XLA). This disease is associated with the impaired maturation of B-cells, diminished immunoglobulin production, compromised T-cell-independent immune responses and marked attenuation of the sustained calcium signal upon BCR stimulation.
Evidence for the role of Btk in allergic disorders and/or autoimmune disease and/or inflammatory disease has been established in Btk-deficient mouse models. For example, in standard murine preclinical models of systemic lupus erythematosus (SLE), Btk deficiency has been shown to result in a marked amelioration of disease progression. Moreover, Btk deficient mice are also resistant to developing collagen-induced arthritis and are less susceptible to Staphylococcus-induced arthritis.
A large body of evidence supports the role of B-cells and the humoral immune system in the pathogenesis of autoimmune and/or inflammatory diseases. Protein-based therapeutics (such as Rituxan) developed to deplete B-cells, represent an important approach to the treatment of a number of autoimmune and/or inflammatory diseases.
Because of Btk’s role in B-cell activation, inhibitors of Btk can be useful as inhibitors of B-cell mediated pathogenic activity (such as autoantibody production).
Btk is also expressed in mast cells and monocytes and has been shown to be important for the function of these cells. For example, Btk deficiency in mice is associated with impaired IgE -mediated mast cell activation (marked diminution of T F-alpha and other inflammatory cytokine release), and Btk deficiency in humans is associated with greatly reduced TNF-alpha production by activated monocytes.
Thus, inhibition of Btk activity can be useful for the treatment of allergic disorders and/or autoimmune and/or inflammatory diseases including, but not limited to: SLE, rheumatoid arthritis, multiple vasculitides, idiopathic thrombocytopenic purpura (ITP), myasthenia gravis, allergic rhinitis, multiple sclerosis (MS), transplant rejection, type I diabetes, membranous nephritis, inflammatory bowel disease, autoimmune hemolytic anemia, autoimmune thyroiditis, cold and warm agglutinin diseases, Evan’s syndrome, hemolytic uremic syndrome/thrombotic thrombocytopenic purpura (HUS/TTP), sarcoidosis, Sjogren’s syndrome, peripheral neuropathies (e.g., Guillain-Barre syndrome), pemphigus vulgaris, and asthma.
In addition, Btk has been reported to play a role in controlling B-cell survival in certain B-cell cancers. For example, Btk has been shown to be important for the survival of BCR-Abl-positive B-cell acute lymphoblastic leukemia cells. Thus inhibition of Btk activity can be useful for the treatment of B-cell lymphoma and leukemia.
In view of the numerous conditions that are contemplated to benefit by treatment involving modulation of protein kinases, it is immediately apparent that new compounds capable of modulating protein kinases such as Btk and methods of using these compounds should provide substantial therapeutic benefits to a wide variety of patients.
WO 2016/065226 discloses indole carboxamide compounds useful as Btk inhibitors, including (S)-4-(3-(but-2-ynamido)piperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide (Example 223), which has the structure:
Also disclosed is multistep synthesis process for preparing (S)-4-(3-(but-2-ynamido) piperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide.
There are difficulties associated with the adaptation of the multistep synthesis disclosed in WO 2016/065226 to larger scale synthesis, such as production in a pilot plant or a manufacturing plant for commercial production. Further, there is a continuing need to find a process that has few synthesis steps, provides higher yields, and/or generates less waste.
Applicants have discovered a new synthesis process for the preparation of (S)-4-(3-(but-2-ynamido)piperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide that has fewer synthesis steps and/or provides higher yields than the process disclosed in WO 2016/065226. Furthermore, this process contains no metal-catalyzed steps, no genotoxic intermediates, and is adaptable to large scale manufacturing.
Step 1 : Preparation of Methyl (S)-2-amino-4-(3-((tert-butoxycarbonyl)amino)piperidin-l-yl)-5-fluorobenz
To a 250 mL ChemGlass reactor were charged methyl 2-amino-4,5-difluoro-benzoate (11.21 g, 59.90 mmol), tert-butyl N-[(3S)-3-piperidyl]carbamate (10 g, 49.930 mmol), potassium phosphate, dibasic (10.44 g, 59.94 mmol), and dimethyl sulfoxide (100 mL, 1400 mmol). The resulting thin slurry was heated to 95 to 100 °C and agitated at this temperature for 25 hours. The mixture was cooled to 50 °C. Methanol (100 mL) was added and followed by slow addition of water (50 mL). The mixture was aged at 50 °C for 30 minutes to result in a thick white slurry. Additional water (150 mL) was slowly charged to the above mixture and agitated at 50 °C for 1 hour. The slurry was cooled to 20 °C in 1 hour and aged at this temperature for 4 hours. The slurry was filtrated. The wet cake washed with 25% MeOH in water (30 mL), water (100 mL) and dried under vacuum at 60 °C for 24 h. Methyl (S)-2-amino-4-(3-((tert-butoxycarbonyl)amino) piperidin-l-yl)-5-fluorobenzoate was obtained as a white solid (7 g, yield: 72.5%). ¾ MR (400MHz, METHANOLS) δ 7.34 (d, J=14.6 Hz, 1H), 6.27 (d, J=7.3 Hz, 1H), 3.83-3.71 (s, 3H), 3.68-3.57 (m., 1H), 3.50 -3.40 (m 1H), 3.39 -3.31 (m, 1H), 3.31-3.26 (m, 1H), 2.86-2.70 (m, 1H), 2.64 (t, J=10.0 Hz, 1H), 1.97-1.84 (m, 1H), 1.84-1.74 (m, 1H), 1.73-1.61 (m, 1H), 1.44 (s, 9H), 1.38 (m, 1H). LC-MS [M+H] 368.
Step 2: Preparation of Methyl (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxylate
To a reactor were charged methyl (S)-2-amino-4-(3-((tert-butoxycarbonyl)amino) piperidin-l-yl)-5-fluorobenzoate (5.0 g), DPPOH (diphenyl phosphate, 6.81 g, 2 eq) and 3-hydroxybutanone (1.2 eq, 1.44 g), followed by addition of isopropyl acetate (100 mL, 20 mL/g). The mixture was allowed to warm up to 70 to 75 °C, resulting in a yellow solution. The solution was stirred at 70 to 75 °C for 30 h to complete the cyclization.
Water (2 mL) was added and the mixture was aged at 70 °C over 24 h to remove the Boc group. The mixture was cooled to room temperature. Next, aqueous 20% K3PO4 solution (50 mL) was added and the mixture was stirred for 15 min. The organic layer was separated and washed with water (50 mL). The organic layer was then concentrated under vacuum (200 Torr) to -50 mL. The resulting slurry was stirred at 50 °C for 2 h and then heptane (100 mL) was added over 1 h. The mixture was cooled to room
temperature, stirred for 20 h, and then filtered. The cake was washed with heptane (50 mL). Methyl (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxylate, DPPOH salt was obtained as a light yellow solid. The wet-cake was added to a reactor. Isopropyl acetate (100 mL) was added, followed by addition of aqueous K3PO4 solution (4 g in water 50 mL). The mixture was stirred at room temperature for -half-hour, resulting in a two phase clear solution (pH >10 for aqueous). The organic layer was separated and washed with water (50 mL), and then concentrated under vacuum to a volume of 15 mL. The resulting slurry was stirred at room temperature for 4 h, then heptane (75 mL) was added over 1 h. The mixture was aged at room temperature for 24 h, then concentrated to a volume to -50 mL. The slurry was filtered. The cake was washed with heptane 20 mL and dried under vacuum at 50 °C for 24 h. Methyl (S)-4-(3- aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxylate was obtained as a light yellow solid (2.76 g, yield: 69%). ¾ NMR (400MHz, DMSO-d6) δ 10.64 (s, 1H), 7.33 (d, J=13.7 Hz, 1H), 3.89 (s, 3H), 3.14 (br. m., 1H), 3.07-2.90 (m, 2H), 2.84 (br. m., 1H), 2.70 (br. m., 1H), 2.35 (s, 3H), 2.33 (s, 3H), 1.87 (br. m., 1H), 1.67 (br. m., 3H). LC-MS: M+H= 320.
Alternative Preparation
Step 2: Preparation of ethyl (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxylate trifluoroacetic acid salt
To a reactor were charged ethyl (S)-2-amino-4-(3-((tert-butoxycarbonyl)amino) piperidin-l-yl)-5-fluorobenzoate (1.0 g, limiting reagent), DPPOH (diphenyl phosphate, 1.97 g, 3.0 eq) and 3-hydroxybutanone (1.4 eq, 0.32 g), followed by addition of toluene (20 mL, 20 mL/g). The mixture was allowed to warm up to 80-90 °C, resulting in a yellow solution. The solution was stirred at 80-90 °C for 10 h to complete the
cyclization. Water (0.4 mL, 0.4 ml/g) was added and the mixture was aged at 80-90 °C for 8 hours. The mixture was cooled to room temperature. Next, aqueous 20% K3PO4 solution (15 mL, 15 mL/g) was added and the mixture was stirred for 0.5 hour. The organic layer was separated and the aqueous layer was washed with toluene (7.5 mL, 7.5 mL/g). To combined organic layers water (10 mL, 10 mL/g) was added and the mixture was stirred for 0.5 hour. The organic layer was separated. To the organic layer water (10 mL, 10 mL/g) was added and the mixture was stirred for 0.5 hour. The organic layer was separated. The organic layer was concentrated under vacuum (100 Torr) to 8 mL (8 ml/g). Following concentration the reaction mixture was cooled to 20-25 °C and MTBE (20 mL, 20 mL/g) was added. Trifluoroacetic acid (1.2 eq., 0.36 g) was slowly added to make the salt maintaining temperature at 20-25 °C. The resulting slurry was aged for 4 hours and then filtered. The filtered solids are washed with MTBE (8 mL, 8 mL/g) and the cake
was dried under vacuum at 50 °C. (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxylate trifluoroacetic acid salt was obtained as a white to tan crystalline material (85% yield, 1.0 g). ¾ NMR (400 MHz, DMSO-d6) δ 10.74 (s, 1H), 8.16-7.88 (m, 2H), 7.37 (d, 7=13.6 Hz, 1H), 4.38 (q, 7=7.1 Hz, 2H), 3.18-3.01 (m, 3H), 2.96 (br s, 1H), 2.35 (s, 6H), 2.30 (s, 1H), 2.12 (br d, 7=9.3 Hz, 1H), 1.78 (br s, 2H), 1.45-1.31 (m, 4H), 1.10 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ 165.1, 165.1, 158.4, 158.1, 135.4, 134.7, 134.6, 132.2, 128.8, 128.2, 126.9, 126.8, 118.7, 115.7, 110.6, 110.3,108.7, 108.6, 106.6, 106.5, 83.5, 79.8, 60.5, 54.9, 51.7, 48.7, 47.2, 28.4, 26.8, 23.6, 14.2, 11.1, 10.2
Step 3A: Preparation of (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide
A 40 mL vial was charged with methyl (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxylate (1.5 g, 4.70 mmol), followed by the addition of N,N-dimethylformamide (12.0 mL, 8.0 mL/g). The vial was purged with N2. Formamide (1.49 mL, 37.6 mmol) was added followed by sodium methoxide solution in methanol (35 wt%, 1.29 mL, 3.76 mmol). The resulting solution was heated at 50 °C over 8 hours. The reaction mixture was cooled down to room temperature and the reaction was quenched with water (12.0 mL, 8.0 mL/g). 2-methyltetrahydrofuran (30 mL, 20 mL/g) was added to the mixture. The mixture was shaken vigorously. The layers were separated and the aqueous layer was extracted with 2-methyltetrahydrofuran (15 mL, 10 mL/g) two more times. Organic extracts were then washed with brine and water (15 mL each, 10 mL/g). The organic layer was evaporated. Solids were dried in vacuo at 60 °C to afford (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide as a yellow solid (1.04 g, 69% yield). ¾ NMR (500MHz, DMSO-d6) δ 10.60 (br. s.,
Step 3B: Alternative Preparation of (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide
A 100 mL Hastelloy high pressure EasyMax reactor was charged with methyl (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxylate (1.5 g, 4.70 mmol), followed by addition of 7 N ammonia solution in methanol (45.0 mL, 30.0 mL/g) followed by addition of l,3,4,6,7,8-hexahydro-2H-pyrimido[l,2-a]pyrimidine (1.33 g, 9.39 mmol). The reactor was sealed and purged with N2 three times. The reactor was then heated to 80 °C for 24 hrs. The reaction mixture was cooled to room temperature and the vessel contents were purged with N2 three times. Volatiles were concentrated to ~6 mL (4 mL/g) and water (24 mL, 16 mL/g) was added. The yellow precipitate was collected and filtered. The precipitate was washed with methanol/water mixture (20:80 v/v, 6 mL, 4 mL/g), and then water (18 mL, 12 mL/g). The solids were dried in vacuo at 60 °C to afford (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide as a yellow crystalline material (0.93 g, 62% yield). ¾ MR (500MHz, DMSO-de) δ 10.60 (br. s., 1H), 7.91 (br. s., 1H), 7.40 (d, J=14.0 Hz, 1H), 7.32 (br. s., 1H), 3.10 (br. s., 1H), 2.98 (br. s., 2H), 2.82 (br. s., 1H), 2.68 (br. s., 1H), 2.34 (br. s., 3H), 2.30 (br. s., 3H), 1.88 (br. s., 1H), 1.67 (br. s., 2H), 1.45 (br. s., 2H), 1.05 (br. s., 1H). LCMS [M+H] 305.24.
Alternative Preparation:
Step 3C: Preparation of (,S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide 2-butynoic acid salt
Ethyl (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxylate trifluoroacetic acid salt (1.0 g, limiting reagent) and formamide (5 mL, 5 mL/g) were added to a nitrogen inerted reactor. The temperature was maintained at 20-25 °C. To the reactor was added a solution of 20 wt% potassium t-butoxide in THF. The reaction mixture was allowed to sit for 6 hours. To reaction mixture was added Me-THF (15 mL, 15 mL/g) and 12.5 wt % aqueous NaCl (5 mL, 5 mL/g). The reaction mixture was stirred for 0.5 hour. The organic layer was separated, 5 wt% aqueous NaCl (1 mL, 1 mL/g) and 0.25 N aqueous NaOH (4 mL, 4 mL/g) were added, and then stirred for 0.5 hour. The organic layer was separated and 5 wt% aqueous NaCl (5 mL, 5 mL/g) was added, the mixture was stirred for 0.5 hour, and organic phase was separated. The rich organic phase was dried distillation at a pressure of 100 mtorr with Me-THF to obtain KF in 1.5-4wt% range at 5 mL Me-THF volume. The volume was adjusted to 15 mL Me-THF by adding Me-THF (10 mL, 10 mL/g) and EtOH (4 mL, 4 mL/g). Next, 2-butynoic acid (1.0 eq., 0.19 g) was added and the mixture was agitated for 10 hrs. The resulting slurry was filtered. The cake was washed with Me-THF (10 mL, 10 mL/g) and dried under vacuum at 75 °C to afford (,S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide 2-butynoic acid salt (0.7 g, 80% yield) as white crystalline powder. ¾ NMR (400 MHz, DMSO-d6) δ 10.68 (s, 1H), 7.98 (br s, 1H), 7.50-7.32 (m, 2H), 3.32 (br d, J=8.6 Hz, 2H), 3.21 (br t, J=10.5 Hz, 1H), 3.13-2.89 (m, 3H), 2.32 (d, J=5.1 Hz, 5H), 2.11 (br d, J=10.9 Hz, 1H), 1.81-1.67 (m, 4H), 1.55-1.28 (m, 1H).
Step 4A: Preparation of (S)-4-(3-(but-2-ynamido)piperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide
To Reactor-1 was charged N,N-dimethylformamide (DMF, 12.77 kg, 13.5 L). Reactor-1 was purged with N2 to inert. (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide (3.0 kg, 1.0 equiv) was charged followed by 2-butynoic acid (0.854 kg, 1.04 equiv). Reactor-1 was rinsed with DMF (1.42 kg, 1.5 L). The mixture was sparged with N2 for 20 min. Triethylamine (2.99 kg, 3.0 equiv) was charged followed by a DMF rinse (1.42 kg, 1.5 L). TBTU (O-(Benzotriazol-l-yl)-N,N,N’,N’-tetramethyluronium tetrafluorob orate, 3.256 kg, 1.04 equiv) was charged followed by a DMF rinse (1.42 kg, 1.5 L). The reaction mixture was agitated for 1.5 h at 20 °C. MeTHF (46.44 kg, 60 L) was charged to the batch. The reaction was quenched with LiCl (20 wt%, 26.76 kg, 24 L) at 20 °C. The bottom aqueous layer was discharged as waste. The organic layer was washed with 2N HCl solution (24.48 kg, 24 L), 10 wt% sodium bicarbonate solution (25.44 kg, 24 L) and deionized water (24.0 kg, 24 L). THF (26.61 kg, 30 L) was charged into Reactor-1. The rich organic stream in MeTHF/TFIF was polish filtered. The stream was distilled down to 15 L at 75-100 Torn Constant volume distillation was carried out at 15 L with THF feed (39.92 kg, 45 L). The stream was heated to 60 °C for 1 hr and cooled to 50 °C. MTBE (33.30 kg, 45 L) was charged slowly over 2 h. The slurry was aged at 50 °C for 4 h and cooled to 20 °C over 2 h, and aged at 20 °C for >2 h. The 1st drop slurry was filtered and was rinsed with MTBE (8.88 kg, 12 L) twice. Wet cake was dried under vacuum 60 to 70 °C at 25 mbar overnight (>15 h). Reactor-1 was thoroughly cleaned with IPA. The dry cake was charged into Reactor-1 followed by the charge of IPA (47.10 kg, 60 L). The batch was heated to 60 °C to achieve full dissolution and cooled to 40 °C. Rich organic (24 L) was transferred to Reactor-2 for crystallization. The stream was distilled at 24 L constant volume and 100 mbar using remaining rich organic from reactor-1 as distillation feed. Following distillation completion, the batch was heated to 60 °C, aged at 60 °C for 2 h, cooled to 20 °C over 2 h, and aged at 20 °C over 2 h. The slurry was filtered. IPA (1.18 kg) was used to rinse the reactor and washed the cake. The wet cake was dried under vacuum at 70 °C and 25 mbar for >15 h. The dry cake (2.196 kg, 63.2% yield) was discharged as an off-white crystalline solid. ¾ NMR (400MHz, DMSO-d6): δ 10.62 (s, 1H), 8.48 (d, J= 7.1 Hz, 1H), 7.91 (s, 1H), 7.39 (d, J=7.4 Hz, 1H), 7.33 (s, 1H), 3.88 (m, 1H), 3.11 (t, J= 8.0 Hz, 1H), 3.0 (m, 1H), 2.96 (m, 1H), 2.78 (t, J= 10.0 Hz, 1H), 2.35 (s, 3H), 2.30 (s, 3H), 1.92 (s, 3H), 1.86 (m, 1H), 1.31 (m, 1H), 1.70 (m, 2H); 13C NMR (400 MHz, DMSO-d6): δ 168.2, 153.2, 151.9, 134.4, 133.2, 132.1, 126.5, 112.3, 108.4, 106.0, 82.3, 75.7, 56.9, 51.9, 46.3, 29.7, 24.4, 11.1, 10.2, 3.0; LC-MS: M+H= 371.2.
Step 4B: Alternative preparation of (S)-4-(3-(but-2-ynamido)piperidin-l-yl)-5-fluoro-2,3-dimeth -lH-indole-7-carboxamide
To Reactor-1 was charged N,N-dimethylformamide (DMF 4.5 mL, 4.5 mL/g). Reactor-1 was purged with N2 to inert. (,S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide 2-butynoic acid salt (1.0 g, limiting reagent) was charged followed by 2-butynoic acid (0.065g, 0.3 equiv.). The mixture was inerted with N2 for 20 min. N-methylmorpholine (0.78 g, 3.0 equiv) was charged. Next,
diphenylphosphinic chloride (0.79 g, 1.3 equiv) was charged over 0.5 h while maintaining the reaction temperature at 20-25 °C. The reaction mixture was agitated for 1.5 hour at 20 °C. Me-THF (14 mL, 14 mL/g) was charged to the reaction mixture. The reaction was quenched with the addition of aqueous NaCl (12.5 wt%, 6 mL, 6 mL/g) at 20 °C. The bottom aqueous layer was discharged as waste. Aqueous NaCl (12.5 wt%, 6 mL, 6 mL/g) at 20 °C was added to the organic layer, stirred for 0.5 hour and the bottom aqueous layer was discharged to waste. Deionized water (6 mL, 6 mL/g) was charged to the organic layer, stirred for 0.5 hour and the bottom aqueous layer was discharged to waste. THF (8 mL, 8 mL/g) was charged into Reactor-1 and the mixture was
concentrated under vacuum to remove Me-THF and water, and reconstituted in 4 L/kg of THF. The mixture was heated to 60 °C and stirred for 1 hour; the temperature was reduced to 50 °C and MTBE (12 mL, 12 mL/g) was added. The mixture was aged for 4 hours while maintaining the temperature of 50 °C and then cooled to room temperature. The solids were filtered and washed with MTBE (6.5 mL, 6.5 mL/g). The solids of crude were dried at 70 °C under vacuum for 12 hours.
Crude (S)-4-(3-(but-2-ynamido)piperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide was charged to Reactor-2, followed by THF (12 mL, 12 mL/g). The mixture was stirred for 0.5 hour. The solution was polish filtered. The solution was concentrated under vaccuum to remove THF and reconstituted in EtOH (7 mL, 7 mL/g). (S)-4-(3-(but-2-ynamido)piperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide seeds (0.01 g, 0.01 g/g) were added, the mixture was heated to 60 °C and aged for 2 hours, n-heptane (21 mL, 21 mL/g) was added slowly over 4 hours. The mixture was aged for additional 2 hours at 60 °C, followed by cooldown to room temperature. The slurry was filtered, washed with n-heptane (6 mL, 6 mL/g), and dried under vacuum at 70 °C for 12 hours. The dry cake (0.68 g, 71% yield) was discharged as an off-white crystalline solid. ¾ NMR (400MHz, DMSO-d6): δ 10.62 (s, 1H), 8.48 (d, J= 7.1 Hz, 1H), 7.91 (s, 1H), 7.39 (d, J=7.4 Hz, 1H), 7.33 (s, 1H), 3.88 (m, 1H), 3.11 (t, J= 8.0 Hz, 1H), 3.0 (m, 1H), 2.96 (m, 1H), 2.78 (t, J= 10.0 Hz, 1H), 2.35 (s, 3H), 2.30 (s, 3H), 1.92 (s, 3H), 1.86 (m, 1H), 1.31 (m, 1H), 1.70 (m, 2H); 13C MR (400 MHz, DMSO-d6): δ 168.2, 153.2, 151.9, 134.4, 133.2, 132.1, 126.5, 112.3, 108.4, 106.0, 82.3, 75.7, 56.9, 51.9, 46.3, 29.7, 24.4, 11.1, 10.2, 3.0; LC-MS: M+H= 371.2.
Applicants have discovered a new synthesis process for the preparation of (S)-4- (3-(but-2-ynamido)piperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide which offers significant advantages.
The new synthesis process utilizes fewer synthesis steps (4 vs 8) than the process disclosed in WO 2016/065226.
Additionally, the process of the present invention provided (S)-4-(3-(but-2-ynamido)piperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide at an overall
yield of 22% (step 1 : 73.%, step 2: 69%, step 3 : 69%, step 4: 63%). In comparison, (S)-4-(3-(but-2-ynamido)piperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide was prepared according to the process of WO 2016/065226, which provided (S)-4-(3-(but-2-ynamido)piperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide at an overall yield of 2.9% yield (step 1 : 91%, step 2: 71%, step 3 : 35%, step 4: 88%, step 5: 80%, step 6: 29%, step 7: 99%, step 8: 63%).
Furthermore, the process of the present invention does not include any transition metal-catalyzed steps, no genotoxic intermediates, and is adaptable to large scale manufacturing. In comparison, the process disclosed in WO 2016/065226 employed lead (Pb) in process step (8) and included a potentially genotoxic hydrazine intermediate in process step 8.
The process of the present invention has an estimated manufacturing cycle time of approximately 6 months versus a estimated manufacturing cycle time of approximately 12 months for the process disclosed in WO 2016/065226.
For treatment for pulmonary fibrosis, phase 2, The lysophosphatidic acid receptor, LPA1, has been implicated as a therapeutic target for fibrotic disorders
Lysophospholipids (LPs), including lysophosphatidic acid (LPA), sphingosine 1-phospate (S1P), lysophosphatidylinositol (LPI), and lysophosphatidylserine (LysoPS), are bioactive lipids that transduce signals through their specific cell-surface G protein-coupled receptors, LPA1-6, S1P1-5, LPI1, and LysoPS1-3, respectively. These LPs and their receptors have been implicated in both physiological and pathophysiological processes such as autoimmune diseases, neurodegenerative diseases, fibrosis, pain, cancer, inflammation, metabolic syndrome, bone formation, fertility, organismal development, and other effects on most organ systems.
Originator Amira Pharmaceuticals
DeveloperB ristol-Myers Squibb; Duke University
Class Antifibrotics; Azabicyclo compounds; Carboxylic acids; Small molecules; Tetrazoles
Mechanism of Action Lysophosphatidic acid receptor antagonists
Orphan Drug Status Yes – Fibrosis
Phase II Idiopathic pulmonary fibrosis
Phase IPsoriasis
Most Recent Events
05 May 2016 Bristol-Myers Squibb plans a phase I trial for Psoriasis in Australia (PO, Capsule, Liquid) (NCT02763969)
01 May 2016 Preclinical trials in Psoriasis in USA (PO) before May 2016
14 Mar 2016 Bristol-Myers Squibb withdraws a phase II trial for Systemic scleroderma in USA, Canada, Poland and United Kingdom (PO) (NCT02588625)
BMS-986020, also known as AM152 and AP-3152 free acid, is a potent and selective LPA1 antagonist. BMS-986020 is in Phase 2 clinical development for treating idiopathic pulmonary fibrosis. BMS-986020 selectively inhibits the LPA receptor, which is involved in binding of the signaling molecule lysophosphatidic acid, which in turn is involved in a host of diverse biological functions like cell proliferation, platelet aggregation, smooth muscle contraction, chemotaxis, and tumor cell invasion, among others
PRODUCT PATENT
GB 2470833, US 20100311799, WO 2010141761
Hutchinson, John Howard; Seiders, Thomas Jon; Wang, Bowei; Arruda, Jeannie M.; Roppe, Jeffrey Roger; Parr, Timothy
[0562] Compound XLV-1 was prepared by the same method as described in the synthesis of compound 1-4 (Scheme 1-A).
[0563] To a solution of compound XLV-1 (8 g, 28.08 mmol) in dry toluene (150 mL) was added compound XLV-2 (1.58 g, 10.1 mmol), triethylamine (8.0 mL) and DPPA (9.2 g, 33.6 mmol). The reaction mixture was heated to 80 °C for 3 hours. The mixture was diluted with EtOAc (50 mL), washed with brine, dried over Na2S04, filtered and concentrated. The residue was purified by column chromatography (PE/EA = 10 IX) to give compound XLV-3 (9.4 g, yield: 83 %). MS (ESI) m/z (M+H)+402.0.
[0564] Compound 74 was prepared analogously to the procedure described in the synthesis of Compound 28 and was carried through without further characterization.
[0565] Compound 74a was prepared analogously to the procedure described in the synthesis of Compound 44a. Compound 74a: 1HNMR (DMSO-d6 400MHz) δ 7.81 (d, J = 8.4 Hz, 2H), 7.41 (d, J = 8.4 Hz, 2H), 7.52 (d, J = 8.4 Hz, 2H), 7.29-7.32 (m, 7 H), 5.78 (q, 1 H), 2.15 (s, 3 H), 1.52 (d, J = 6.0 Hz, 3H), 1.28 (br, 2 H), 0.74 (br, 2 H). MS (ESI) m/z (M+H)+ 483.1.
Paper
Development of a Concise Multikilogram Synthesis of LPA-1 Antagonist BMS-986020 via a Tandem Borylation–Suzuki Procedure
The process development for the synthesis of BMS-986020 (1) via a palladium catalyzed tandem borylation/Suzuki reaction is described. Evaluation of conditions culminated in an efficient borylation procedure using tetrahydroxydiboron followed by a tandem Suzuki reaction employing the same commercially available palladium catalyst for both steps. This methodology addressed shortcomings of early synthetic routes and was ultimately used for the multikilogram scale synthesis of the active pharmaceutical ingredient 1. Further evaluation of the borylation reaction showed useful reactivity with a range of substituted aryl bromides and iodides as coupling partners. These findings represent a practical, efficient, mild, and scalable method for borylation.
1: Kihara Y, Mizuno H, Chun J. Lysophospholipid receptors in drug discovery. Exp
Cell Res. 2015 May 1;333(2):171-7. doi: 10.1016/j.yexcr.2014.11.020. Epub 2014
Dec 8. Review. PubMed PMID: 25499971; PubMed Central PMCID: PMC4408218.
Mechanism of Action Amyloid precursor protein secretase inhibitors; Notch signalling pathway inhibitors
Phase I Solid tumours
Most Recent Events
30 Aug 2016Bristol-Myers Squibb terminates a phase I trial for Solid tumours (late-stage disease, second-line therapy or greater) in USA, Australia and Canada (NCT01986218)
25 Jan 2016Bristol-Myers Squibb completes enrolment in its phase I trial for Solid tumours in USA, Australia and Canada (NCT01986218)
31 Dec 2013Phase-I clinical trials in Solid tumours (late-stage disease) in Canada & Australia (Oral)
DETAILS WILL BE UPDATED SOON………….
BMS-986115 is an orally bioavailable, gamma secretase (GS) and pan-Notch inhibitor, with potential antineoplastic activity. Upon administration, GS/pan-Notch inhibitor BMS 986115 binds to GS and blocks the proteolytic cleavage and release of the Notch intracellular domain (NICD), which would normally follow ligand binding to the extracellular domain of the Notch receptor. This prevents both the subsequent translocation of NICD to the nucleus to form a transcription factor complex and the expression of Notch-regulated genes. This results in the induction of apoptosis and the inhibition of growth of tumor cells that overexpress Notch. Overexpression of the Notch signaling pathway plays an important role in tumor cell proliferation and survival
Dan O’Malley (Rice University) Currently: Bristol-Myers Squibb
PICTURES WILL BE UPDATED………….
Useful for the treatment of conditions related to the Notch pathway, such as cancer and other proliferative diseases.
Notch signaling has been implicated in a variety of cellular processes, such as cell fate specification, differentiation, proliferation, apoptosis, and angiogenesis. (Bray, Nature Reviews Molecular Cell Biology, 7:678-689 (2006); Fortini, Developmental Cell 16:633-647 (2009)). The Notch proteins are single-pass heterodimeric transmembrane molecules. The Notch family includes 4 receptors, NOTCH 1-4, which become activated upon binding to ligands from the DSL family (Delta-like 1, 3, 4 and Jagged 1 and 2).
The activation and maturation of NOTCH requires a series of processing steps, including a proteolytic cleavage step mediated by gamma secretase, a multiprotein complex containing Presenilin 1 or Presenilin 2, nicastrin, APH1, and PEN2. Once NOTCH is cleaved, NOTCH intracellular domain (NICD) is released from the membrane. The released NICD translocates to the nucleus, where it functions as a transcriptional activator in concert with CSL family members (RBPSUH, “suppressor of hairless”, and LAG1). NOTCH target genes include HES family members, such as HES- 1. HES- 1 functions as transcriptional repressors of genes such as HERP 1 (also known as HEY2), HERP2 (also known as HEY1), and HATH1 (also known as ATOH1).
The aberrant activation of the Notch pathway contributes to tumorigenesis. Activation of Notch signaling has been implicated in the pathogenesis of various solid tumors including ovarian, pancreatic, as well as breast cancer and hematologic tumors such as leukemias, lymphomas, and multiple myeloma. The role of Notch inhibition and its utility in the treatment of various solid and hematological tumors are described in Miele, L. et al, Current Cancer Drug Targets, 6:313-323 (2006); Bolos, V. et al, Endocrine Reviews, 28:339-363 (2007); Shih, I.-M. et al, Cancer Research, 67: 1879- 1882 (2007); Yamaguchi, N. et al., Cancer Research, 68: 1881-1888 (2008); Miele, L., Expert Review Anti-cancer Therapy, 8: 1 197-1201 (2008); Purow, B., Current Pharmaceutical Biotechnology, 10: 154-160 (2009); Nefedova, Y. et al, Drug Resistance Updates, 1 1 :210-218 (2008); Dufraine, J. et al, Oncogene, 27:5132-5137 (2008); and Jun, H.T. et al, Drug Development Research, 69:319-328 (2008).
There remains a need for compounds that are useful as Notch inhibitors and that have sufficient metabolic stability to provide efficacious levels of drug exposure. Further, there remains a need for compounds useful as Notch inhibitors that can be orally or intravenously administered to a patient.
U.S. Patent No. 7,053,084 Bl discloses succinoylamino benzodiazepine compounds useful for treating neurological disorders such as Alzheimer’s Disease. The reference discloses that these succinoylamino benzodiazepine compounds inhibit gamma secretase activity and the processing of amyloid precursor protein linked to the formation of neurological deposits of amyloid protein. The reference does not disclose the use of these compounds in the treatment of proliferative diseases such as cancer.
Applicants have found potent compounds that have activity as Notch inhibitors and have sufficient metabolic stability to provide efficacious levels of drug exposure upon intravenous or oral administration. These compounds are provided to be useful as pharmaceuticals with desirable stability, bioavailability, therapeutic index, and toxicity values that are important to their drugability.
In a 100 mL round-bottomed flask, a solution of Intermediate B-1 (1683 mg, 5.94 mmol), Et3N (1.656 mL, 11.88 mmol), and Intermediate S-1 in DMF (20 mL) was treated with o-benzotriazol-1-yl-N,N,N′,N′-tetramethyluronium tetrafluoroborate (3815 mg, 11.88 mmol) and stirred at room temperature for 1 hour. The reaction mixture was diluted with water and saturated aqueous NaHCO3. An off white precipitate formed and was filtered and washed with water. The resulting solid was dried on the filter under a stream of nitrogen to give Intermediate 1A (3.7 g, 99% yield). MS (ES): m/z=632.4[M+H+]; HPLC: RT=3.635 min Purity=98%. (H2O/MeOH with TFA, CHROMOLITH® ODS S5 4.6×50 mm, gradient=4 min, wavelength=220 nm). 1H NMR (400 MHz, methanol-d4) δ 7.53 (t, J=4.5 Hz, 1H), 7.46-7.30 (m, 3H), 7.28-7.23 (m, 1H), 7.23-7.18 (m, 2H), 5.37 (s, 1H), 2.88 (td, J=10.4, 3.4Hz, 1H), 2.60 (td, J=10.2, 4.1 Hz, 1H), 2.54-2.40 (m, 1H), 2.47 (s, 3H), 2.33-2.12 (m, 3H), 1.98-1.69 (m, 4H), 1.51 (s, 9H).
In a 250 mL round-bottomed flask, a solution of Intermediate 1A (3.7 g, 5.86 mmol) in DCM (25 mL) was treated with TFA (25 mL) and the resulting pale orange solution was stirred at room temperature for 1.5 hours. The reaction mixture was then concentrated to give Intermediate 1B. HPLC: RT=3.12 min (H2O/MeOH with TFA, CHROMOLITH® ODS S5 4.6×50 mm, gradient=4 min, wavelength=220 nm). MS (ES): m/z=576.3 (M+H)+. 1H NMR (400 MHz, methanol-d4) δ 7.54 (t, J=4.5 Hz, 1H), 7.49-7.29 (m, 3H), 7.28-7.15 (m, 3H), 5.38 (br. s., 1H), 2.89 (td, J=10.3, 3.7 Hz, 1H), 2.67 (td, J=9.9, 4.2Hz, 1H), 2.56-2.38 (m, 1H), 2.48 (s, 3H), 2.34-2.13 (m, 3H), 2.00-1.71 (m, 4H).
Example 1
In a 250 mL round-bottomed flask, a solution of Intermediate 1B (4.04 g, 5.86 mmol) in THF (50 mL) was treated with ammonia (2M in iPrOH) (26.4 mL, 52.7 mmol), followed by HOBT (1.795 g, 11.72 mmol) and EDC (2.246 g, 11.72 mmol). The resulting white suspension was stirred at room temperature overnight. The reaction mixture was diluted with water and saturated aqueous NaHCO3. The resulting solid was filtered, rinsed with water and then dried on the filter under a stream of nitrogen. The crude product was suspended in 20 mL of iPrOH and stirred at room temperature for 20 min and then filtered and washed with iPrOH and dried under vacuum to give 2.83 g of solid. The solid was dissolved in refluxing EtOH (100 mL) and slowly treated with 200 mg activated charcoal added in small portions. The hot mixture was filtered through CELITE® and rinsed with hot EtOH. The filtrate was reduced to half volume, allowed to cool and the white precipitate formed was filtered and rinsed with EtOH to give 2.57 g of white solid. A second recrystallization from EtOH (70 mL) afforded Example 1 (2.39 g, 70% yield) as a white solid. HPLC: RT=10.859 min (H2O/CH3CN with TFA, Sunfire C18 3.5 μm, 3.0×150 mm, gradient=15 min, wavelength=220 and 254 nm); MS (ES): m/z=575.3 [M+H+]; 1H NMR (400 MHz, methanol-d4) δ 7.57-7.50 (m, 1H), 7.47-7.30 (m, 3H), 7.29-7.15 (m, 3H), 5.38 (s, 1H), 2.85-2.75 (m, 1H), 2.59 (td, J=10.5, 4.0 Hz, 1H), 2.53-2.41 (m, 4H), 2.31-2.10 (m, 3H), 1.96-1.70 (m, 4H).
[00180] To a cold (-25 °C) stirred solution of 2,6-lutidine (18.38 mL, 158 mmol) in DCM (120 mL) was added Tf20 (24.88 mL, 147 mmol) over 3 min, and the mixture was stirred for 5 min. To the reaction mixture was added 3,3,3-trifluoropropan-l-ol (12 g, 105 mmol) over an interval of 3 min. After 2 hr, the reaction mixture was warmed to room temperature and stirred for 1 hr. The reaction mixture was concentrated to half its volume, then purified by loading directly on a silica gel column (330g ISCO) and the product was eluted with DCM to afford Intermediate S-IA (13.74 g, 53%) as a colorless oil. 1H NMR (400 MHz, CDC13) δ ppm 4.71 (2 H, t, J= 6.15 Hz), 2.49-2.86 (2 H, m).
[00181] To a stirring solution of 5,5,5-trifluoropentanoic acid (14.76 g, 95 mmol) and DMF (0.146 rriL) in DCM (50 mL) was slowly added oxalyl chloride (8.27 mL, 95 mmol). After 2h, the mixture was concentrated to dryness. A separate flask was changed with (S)-4-benzyloxazolidin-2-one (16.75 g, 95 mmol) in THF (100 mL) and then cooled to -78 °C. To the solution was slowly added n-BuLi (2.5M, 37.8 mL, 95 mmol) over 10 min, stirred for 10 min, and then a solution of the above acid chloride in THF (50 mL) was slowly added over 5 min. The mixture was stirred for 30 min, and then warmed to room temperature. The reaction was quenched with sat aq NH4C1. Next, 10% aq LiCl was then added to the mixture, and the mixture was extracted with Et20. The organic layer was washed with sat aq NaHC03 then with brine, dried (MgSC^), filtered and concentrated to dryness. The residue was purified by Si02 chromatography (ISCO, 330 g column, eluting with a gradient from 100% hexane to 100% EtOAc) to afford the product Intermediate S-IB; (25.25 g, 85%): 1H NMR (400 MHz, CDC13) δ ppm 7.32-7.39 (2 H, m), 7.30 (1 H, d, J= 7.05 Hz), 7.18-7.25 (2 H, m), 4.64-4.74 (1 H, m), 4.17-4.27 (2 H, m), 3.31 (1 H, dd, J= 13.35, 3.27 Hz), 3.00-3.11 (2 H, m), 2.79 (1 H, dd, J= 13.35, 9.57 Hz), 2.16-2.28 (2 H, m), 1.93-2.04 (2 H, m).
[00182] To a cold (-78 °C), stirred solution of Intermediate S-IB (3.03 g, 9.61 mmol) in THF (20 mL) was added NaHMDS (1.0M in THF) (10.6 mL, 10.60 mmol) under a nitrogen atmosphere. After 2 hours, tert-butyl 2-bromoacetate (5.62 g, 28.8 mmol) was added neat via syringe at -78 °C and stirring was maintained at the same temperature. After 6 hours, the reaction mixture was warmed to room temperature. The reaction mixture was partitioned between saturated NH4C1 and EtOAc. The organic phase was separated, and the aqueous phase was extracted with EtOAc (3x). The combined organics were washed with brine, dried (Na2s04), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (Teledyne ISCO
CombiFlash Rf, 5% to 100% solvent A/B = hexanes/EtOAc, REDISEP® Si02 120g). Concentration of the appropriate fractions provided Intermediate S-1C (2.79 g, 67.6%) as a colorless viscous oil: 1H NMR (400 MHz, CDC13) δ ppm 7.34 (2 H, d, J= 7.30 Hz), 7.24-7.32 (3 H, m), 4.62-4.75 (1 H, m, J= 10.17, 6.89, 3.43, 3.43 Hz), 4.15-4.25 (3 H, m), 3.35 (1 H, dd, J= 13.60, 3.27 Hz), 2.84 (1 H, dd, J= 16.62, 9.57 Hz), 2.75 (1 H, dd, J = 13.35, 10.07 Hz), 2.47 (1 H, dd, J= 16.62, 4.78 Hz), 2.11-2.23 (2 H, m), 1.90-2.02 (1 H, m), 1.72-1.84 (1 H, m), 1.44 (9 H, s).
[00183] To a cool (0 °C), stirred solution of Intermediate S-1C (2.17 g, 5.05 mmol) in THF (50 mL) and water (15 mL) was added a solution of LiOH (0.242 g, 10.11 mmol) and H202 (2.065 mL, 20.21 mmol) in H20 (2 mL). After 10 min, the reaction mixture was removed from the ice bath, stirred for lh, and then cooled to 0 °C. Saturated aqueous NaHCC”3 (25 mL) and saturated aqueous Na2s03 (25 mL) were added to the reaction mixture, and the mixture was stirred for 10 min, and then partially concentrated. The resulting mixture was extracted with DCM (2x), cooled with ice and made acidic with cone. HC1 to pH 3. The mixture was saturated with solid NaCl, extracted with EtOAc (3x), and then dried over MgS04, filtered and concentrated to a colorless oil to afford Intermediate S-ID, 1.2514g, 92%): 1H NMR (400 MHz, CDCI3) δ ppm 2.83-2.95 (1 H, m), 2.62-2.74 (1 H, m), 2.45 (1 H, dd, J= 16.62, 5.79 Hz), 2.15-2.27 (2 H, m), 1.88-2.00 (1 H, m), 1.75-1.88 (1 H, m), 1.45 (9 H, s). Intermediate S-l : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and Intermediate S-1E: (2R,3R)-3-(tert-butoxycarbonyl)- 6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid
(S-1E)
[00184] To a cold (-78 °C) stirred solution of Intermediate S-1D (5 g, 18.50 mmol) in THF (60 mL) was slowly added LDA (22.2 mL, 44.4 mmol, 2.0M) over 7 min. After stirring for 2 hr, Intermediate S- 1 A (6.38 g, 25.9 mmol) was added to the reaction mixture over 3 min. After 60 min, the reaction mixture was warmed to -25 °C
(ice/MeOH/dry ice) and stirred for an additional 60 min at which time sat aq NH4C1 was added. The separated aqueous phase was acidified with IN HC1 to pH 3, and then extracted with Et20. The combined organic layers were washed with brine (2x), dried over MgS04, filtered and concentrated to provide a 1 :4 (II :I1E) mixture (as determined by 1H NMR) of Intermediate S-l and Intermediate S-1E (6.00 g, 89%) as a pale yellow solid. 1H NMR (500 MHz, CDC13) δ ppm 2.81 (1 H, ddd, J = 10.17, 6.32, 3.85 Hz), 2.63- 2.76 (1 H, m), 2.02-2.33 (4 H, m), 1.86-1.99 (2 H, m), 1.68-1.85 (2 H, m), 1.47 (9 H, s).
[00185] To a cold (-78 °C), stirred solution of a mixture of Intermediate S-l and Intermediate S-1E (5.97 g, 16.30 mmol) in THF (91 mL) was added LDA (19 mL, 38.0 mmol, 2.0M in THF/hexane/ethyl benzene) dropwise via syringe over 10 min (internal temperature never exceeded -65 °C, J-KEM® probe in reaction solution). The mixture was stirred for 15 min, and then warmed to room temperature (24 °C water bath), stirred for 15 min, and then cooled to -78 °C for 15 min. To the reaction mixture was added Et2AlCl (41 mL, 41.0 mmol, 1M in hexane) via syringe (internal temperature never exceeded -55 °C), and the mixture was stirred for 10 min, and then warmed to room temperature (24 °C bath) for 15 min and then back to -78 °C for 15 min. Meanwhile, a 1000 mL round bottom flask was charged with MeOH (145 mL) and precooled to -78 °C. With vigorous stirring the reaction mixture was transferred via cannula over 5 min to the MeOH. The flask was removed from the bath, ice was added followed by the slow addition of IN HC1 (147 mL, 147 mmol). Gas evolution was observed as the HC1 was added. The reaction mixture was allowed to warm to room temperature during which the gas evolution subsided. The reaction mixture was diluted with EtOAc (750 mL), saturated with NaCl, and the organic phase was separated, washed with a solution of potassium fluoride (8.52 g, 147 mmol) and IN HC1 (41 mL, 41.0 mmol) in water (291 mL), brine (100 mL), and then dried (Na2s04), filtered and concentrated under vacuum. 1H NMR showed the product was a 9: 1 mixture of Intermediate S-l and Intermediate S- 1E. The enriched mixture of Intermediate S-l and Intermediate S-1E (6.12 g, >99% yield) was obtained as a dark amber solid: 1H NMR (400 MHz, CDC13) δ ppm 2.64-2.76 (2 H, m), 2.04-2.35 (4 H, m), 1.88-2.00 (2 H, m), 1.71-1.83 (2 H, m), 1.48 (9 H, s).
[00186] To a stirred solution of a 9: 1 enriched mixture of Intermediate S-l and Intermediate S-1E (5.98 g, 16.33 mmol) in DMF (63 mL) were added potassium carbonate (4.06 g, 29.4 mmol) and benzyl bromide (2.9 mL, 24.38 mmol), the mixture was then stirred overnight at room temperature. The reaction mixture was diluted with EtOAc (1000 mL), washed with 10% LiCl (3×200 mL), brine (200 mL), dried (Na2S04), filtered, concentrated, and then dried under vacuum. The residue was purified by Si02 chromatography using a toluene:hexane gradient. Diastereomerically purified
[00187] To a solution of Intermediate S-1F (4.81 g, 10.54 mmol) in MeOH (100 mL) was added 10% palladium on carbon (wet, Degussa type, 568.0 mg, 0.534 mmol) in a H2– pressure flask. The vessel was purged with N2 (4x), then purged with H2 (2x), and finally, pressurized to 50 psi and shaken overnight. The reaction vessel was
depressurized and purged with nitrogen. The mixture was filtered through CELITE®, washed with MeOH and then concentrated and dried under vacuum. Intermediate S-1 (3.81 g, 99% yield)) was obtained as a colorless solid: 1H NMR (400 MHz, chloroform-d) δ 2.62-2.79 (m, 2H), 2.02-2.40 (m, 4H), 1.87-2.00 (m, 2H), 1.67-1.84 (m, 2H), 1.48 (s, 9H).
[00188] Intermediate S-1 as a mixture with Intermediate S-IE was prepared in a similar procedure as above from Intermediate S-1D to afford a 1 :2.2 mixture of
Intermediate S-1 and Intermediate S-IE (8.60 g, 23.48 mmol), which was enriched using LDA (2.0 M solution in THF, ethyl benzene and heptane, 28.2 mL, 56.4 mmol) and diethyl aluminum chloride (1.0 M solution in hexane, 59 mL, 59.0 mmol) in THF (91 mL). After workup as described above, the resulting residue was found to be a 13.2: 1 (by 1H NMR) mixture of Intermediate S-1 and Intermediate S-IE, which was treated as follows: The crude material was dissolved in MTBE (43 mL). Hexanes (26 mL) were slowly charged to the reaction mixture while maintaining a temperature below 30 °C. The reaction mixture was stirred for 10 min. Next, tert-butylamine (2.7 mL, 1.1 eq) was charged slowly over a period of 20 minutes while maintaining a temperature below 30 °C. This addition was observed to be exothermic. The reaction mixture was stirred for 2 hrs below 30 °C and then filtered. The solid material was washed with 5:3 MTBE: hexane (80 mL), and the filtrate was concentrated and set aside. The filtered solid was dissolved in dichloromethane (300 mL), washed with IN HC1 (lOOmL), and the organic layer was washed with brine (100 mL x 2), and then concentrated under reduced pressure below 45 °C to afford Intermediate S-l (5.46 g, 64%).
A second alternate procedure for preparing Intermediate S-l :
[00189] To a stirred solution of 5,5,5-trifluoropentanoic acid (5 g, 32.0 mmol) in THF (30 mL) and hexane (30 mL) at 0 °C, was added tert-butyl 2,2,2-trichloroacetimidate (11.46 mL, 64.1 mmol). The mixture was stirred for 15 min at 0 °C. Boron trifluoride etherate (0.406 mL, 3.20 mmol) was added and the reaction mixture was allowed to warm to room temperature overnight. To the clear reaction mixture was added solid NaHC03 (5 g) and stirred for 30 min. The mixture was filtered through MgSC^ and washed with hexanes (200 mL). The solution was allowed to rest for 45 min, and the resulting solid material was removed by filtering on the same MgSC^ filter again, washed with hexanes (100 mL) and concentrated under reduced pressure without heat. The volume was reduced to about 30 mL, filtered through a clean fritted funnel, washed with hexane (5 mL), and then concentrated under reduced pressure without heat. The resulting neat oil was filtered through a 0.45μιη nylon membrane filter disk to provide Intermediate S-1G (6.6 g, 31.4 mmol 98% yield) as a colorless oil: 1H NMR (400 MHz, CDC13) δ ppm 1.38 (s, 9 H) 1.74-1.83 (m, 2 H) 2.00-2.13 (m, 2 H) 2.24 (t, J= 7.28 Hz, 2 H). Intermediate S-1H: (4S)-4-(Propan-2-yl)-3-(5,5,5-trifluoropentanoyl)-l,3-oxazolidin-2- one
[00190] To a stirred solution of 5,5,5-trifluoropentanoic acid (5.04 g, 32.3 mmol) in DCM (50 mL) and DMF (3 drops) was added oxalyl chloride (3.4 mL, 38.8 mmol) dropwise over 5 min. The solution was stirred until all bubbling subsided. The reaction mixture was concentrated under reduced pressure to give pale yellow oil. To a separate flask charged with a solution of (4S)-4-(propan-2-yl)-l,3-oxazolidin-2-one (4.18 g, 32.4 mmol) in THF (100 mL) at -78 °C was added n-BuLi (2.5M in hexane) (13.0 mL, 32.5 mmol) dropwise via syringe over 5 min. After stirring for 10 min, the above acid chloride, dissolved in THF (20 mL), was added via cannula over 15 min. The reaction mixture was warmed to 0 °C, and was allowed to warm to room temperature as the bath warmed and stirred overnight. To the reaction mixture was added saturated NH4C1, and the mixture was extracted with EtOAc (2x). The combined organics were washed with brine, dried (Na2s04), filtered and concentrated under reduced pressure. The crude material was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 5% to 60% solvent A/B = hexanes/EtOAc, REDISEP® Si02 120g). Concentration of the appropriate fractions provided Intermediate S-1H (7.39 g, 86%) as a colorless oil: 1H NMR (400 MHz, CDC13) δ ppm 4.44 (1 H, dt, J= 8.31, 3.53 Hz), 4.30 (1 H, t, J= 8.69 Hz), 4.23 (1 H, dd, J= 9.06, 3.02 Hz), 2.98-3.08 (2 H, m), 2.32-2.44 (1 H, m, J= 13.91, 7.02, 7.02, 4.03 Hz), 2.13-2.25 (2 H, m), 1.88-2.00 (2 H, m), 0.93 (3 H, d, J= 7.05 Hz), 0.88 (3 H, d, J= 6.80 Hz).
[00191] To a cold (-78 °C), stirred solution of diisopropylamine (5.3 mL, 37.2 mmol) in THF (59 mL) under a nitrogen atmosphere was added n-BuLi (2.5M in hexane) (14.7 mL, 36.8 mmol). The mixture was then warmed to 0 °C to give a 0.5M solution of LDA. A separate vessel was charged with Intermediate S-1H (2.45 g, 9.17 mmol). The material was azeotroped twice with benzene (the RotoVap air inlet was fitted with a nitrogen inlet to completely exclude humidity), and then toluene (15.3 mL) was added. This solution was added to a flask containing dry lithium chloride (1.96 g, 46.2 mmol). To the resultant mixture, cooled to -78 °C, was added the LDA solution (21.0 mL, 10.5 mmol) and the mixture was stirred at -78 °C for 10 min, then warmed to 0 °C for 10 min., and then cooled to -78 °C. To a separate reaction vessel containing Intermediate S-1G (3.41 g, 16.07 mmol), also azeotroped twice with benzene, was added toluene (15.3 mL), cooled to -78 °C and LDA (37.0 mL, 18.5 mmol) was added. The resulting solution was stirred at -78 °C for 25 min. At this time the enolate derived from the ester was transferred via cannula into the solution of the oxazolidinone enolate and stirred at -78 °C for an additional 5 min, at which time the septum was removed and solid powdered bis(2- ethylhexanoyloxy)copper (9.02 g, 25.8 mmol) was rapidly added to the reaction vessel and the septum was replaced. The vessel was immediately removed from the cold bath and immersed into a warm water bath (40 °C) with rapid swirling and with a concomitant color change from the initial turquoise to brown. The reaction mixture was stirred for 20 min, was then poured into 5% aqueous NH4OH (360 mL) and extracted with EtOAc (2x). The combined organics were washed with brine, dried (Na2s04), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 0% to 60% solvent A/B = hexanes/EtOAc, REDISEP® Si02 120g). Concentration of the appropriate fractions provided a mixture of Intermediate S- II and Intermediate S-1J (2.87 g, 66%) as a pale yellow viscous oil. 1H NMR showed the product was a 1.6: 1 mixture of diastereomers S-1LS-1J as determined by the integration of the multiplets at 2.74 and 2.84 ppm: 1H NMR (400 MHz, CDC13) δ ppm 4.43-4.54 (2 H, m), 4.23-4.35 (5 H, m), 4.01 (1 H, ddd, J= 9.54, 6.27, 3.51 Hz), 2.84 (1 H, ddd, J = 9.41, 7.28, 3.64 Hz), 2.74 (1 H, ddd, J= 10.29, 6.27, 4.02 Hz), 2.37-2.48 (2 H, m, J = 10.38, 6.98, 6.98, 3.51, 3.51 Hz), 2.20-2.37 (3 H, m), 1.92-2.20 (8 H, m), 1.64-1.91 (5 H, m), 1.47 (18 H, s), 0.88-0.98 (12 H, m). Intermediate S-1 : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and Intermediate S-IE: (2R,3R)-3-(tert-Butoxycarbonyl)- 6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid
(S-IE)
[00192] To a cool (0 °C), stirred solution of Intermediate S-1I and Intermediate S-1 J (4.54 g, 9.51 mmol) in THF (140 mL) and water (42 mL) were sequentially added hydrogen peroxide (30% in water) (10.3 g, 91 mmol) and LiOH (685.3 mg, 28.6 mmol). The mixture was stirred for 1 hr. At this time the reaction vessel was removed from the cold bath and then stirred for 1.5 hr. To the reaction mixture were added saturated NaHC03 (45 mL) and saturated Na2s03 (15 mL), and then the mixture was partially concentrated under reduced pressure. The resulting crude solution was extracted with DCM (3x). The aqueous phase was acidified to pH~l-2 with IN HC1, extracted with DCM (3x) and then EtOAc (lx). The combined organics were washed with brine, dried (Na2s04), filtered and concentrated under reduced pressure to provide a mixture of Intermediates S-1 and S-IE (3.00 g, 86%) as a colorless oil: 1H NMR (400 MHz, CDC13) δ ppm 2.76-2.84 (1 H, m, diastereomer 2), 2.64-2.76 (3 H, m), 2.04-2.35 (8 H, m), 1.88- 2.00 (4 H, m), 1.71-1.83 (4 H, m), 1.48 (9 H, s, diastereomer 1), 1.46 (9 H, s,
diastereomer 2); 1H NMR showed a 1.7: 1 mixture of S-1E:S-1F by integration of the peaks for the t-butyl groups. Intermediate S-1 : (2R,3S)-3-(fert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and Intermediate S-IF: (2R,3R)-3-(fert-Butoxycarbonyl)- 6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid
[00193] To a cold (-78 °C) stirred solution of diisopropylamine (1.7 mL, 11.93 mmol) in THF (19 mL) under a nitrogen atmosphere was added n-BuLi (2.5M in hexanes) (4.8 mL, 12.00 mmol). The mixture was stirred for 5 min and then warmed to 0 °C. In a separate vessel, to a cold (-78 °C) stirred solution of the mixture of Intermediates S-1 and S-1E (1.99 g, 5.43 mmol) in THF (18 mL) was added the LDA solution prepared above via cannula slowly over 25 min. The mixture was stirred for 15 min, then warmed to room temperature (placed in a 24 °C water bath) for 15 min, and then again cooled to -78 °C for 15 min. To the reaction mixture was added Et2AlCl (1M in hexane) (11.4 mL, 11.40 mmol) via syringe. The mixture was stirred for 10 min, warmed to room
temperature for 15 min and then cooled back to -78 °C for 15 min. Methanol (25 mL) was rapidly added, swirled vigorously while warming to room temperature, and then concentrated to ~l/4 the original volume. The mixture was dissolved in EtOAc and washed with IN HC1 (50 mL) and ice (75 g). The aqueous phase was separated and extracted with EtOAc (2x). The combined organics were washed with a mixture of KF (2.85g in 75 mL water) and IN HC1 (13 mL) [resulting solution pH 3-4], then with brine, dried (Na2s04), filtered and concentrated under reduced pressure to give a 9: 1 (S-LS-1E) enriched diastereomeric mixture (as determined by 1H NMR) of Intermediate S-1 and Intermediate S-1E (2.13 g, >99%) as a pale yellow viscous oil: 1H NMR (400 MHz, CDC13) δ ppm 2.64-2.76 (2 H, m), 2.04-2.35 (4 H, m), 1.88-2.00 (2 H, m), 1.71-1.83 (2 H, m), 1.48 (9 H, s).
[00194] To a cold (-78 °C), stirred solution of Intermediate S-1D (1.72 g, 6.36 mmol) in THF (30 mL) was slowly added LDA (7.32 mL, 14.6 mmol) over 7 min. After stirring for 1 h, 4,4,4-trifluorobutyltrifluoromethanesulfonate (2.11 g, 8.11 mmol) was added to the reaction mixture over 2 min. After 15 min, the reaction mixture was warmed to -25 °C (ice/MeOH/dry ice) for lh, and then cooled to -78 °C. After 80 min, the reaction was quenched with a saturated aqueous NH4C1 solution (10 mL). The reaction mixture was further diluted with brine and the solution was adjusted to pH 3 with IN HC1. The aqueous layer was extracted with ether. The combined organics were washed with brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure to provide a mixture of Intermediates S-2 and S-2A (2.29 g, 95%) as a colorless oil. 1H NMR (400MHz, chloroform-d) δ 2.83-2.75 (m, 1H), 2.64 (ddd, J = 9.9, 6.7, 3.6 Hz, 1H), 2.32-2.03 (m, 5H), 1.98-1.70 (m, 3H), 1.69-1.52 (m, 3H), 1.50-1.42 (m, 9H). 1H NMR showed a 1 :4.5 mixture (S-2:S-2A) of diastereomers by integration of the peaks for the t- Bu groups.
[00195] A mixture of Intermediate S-2 and Intermediate S-2A (2.29 g, 6.02 mmol) was dissolved in THF (38 mL) to give a colorless solution which was cooled to -78 °C. Then, LDA (7.23 mL, 14.5 mmol) (2.0M in heptane/THF/ethylbenzene) was slowly added to the reaction mixture over 3 min. After stirring for 15 min, the reaction mixture was placed in a room temperature water bath. After 15 min the reaction mixture was placed back in a -78 °C bath and then diethylaluminum chloride (14.5 mL, 14.5 mmol) (1M in hexane) was added slowly over 5 min. The reaction mixture was stirred at -78 °C. After 15 min, the reaction mixture was placed in a room temperature water bath for 10 min, and then cooled back to -78 °C. After 15 min, the reaction was quenched with MeOH (30.0 mL, 741 mmol), removed from the -78 °C bath and concentrated. To the reaction mixture was added ice and HC1 (60.8 mL, 60.8 mmol) and the resulting mixture was extracted with EtOAc (2x 200 mL). The organic layer was washed with potassium fluoride (3.50g, 60.3 mmol) in 55 mL H20 and 17.0 mL of IN HC1. The organics were dried over anhydrous magnesium sulfate and concentrated under reduced pressure to provide an enriched mixture of Intermediate S-2 and Intermediate S-2A (2.25g, 98% yield) as a light yellow oil. 1H NMR (400MHz, chloroform-d) δ 2.83-2.75 (m, 1H), 2.64 (ddd, J= 9.9, 6.7, 3.6 Hz, 1H), 2.32-2.03 (m, 5H), 1.98-1.70 (m, 3H), 1.69-1.52 (m, 3H), 1.50-1.42 (m, 9H). 1H NMR showed a 9: 1 ratio in favor of the desired diastereomer Intermediate S-2.
[00196] To a stirred 9: 1 mixture of Intermediate S-2 and Intermediate S-2A (2.24 g, 5.89 mmoL) and potassium carbonate (1.60 g, 11.58 mmoL) in DMF (30 mL) was added benzyl bromide (1.20 mL, 10.1 mmoL)). The reaction mixture was stirred at room temperature for 19 h. The reaction mixture was diluted with ethyl acetate (400 mL) and washed with 10% LiCl solution (3 x 100 mL), brine (50 mL), and then dried over anhydrous magnesium sulfate, filtered and concentrated to dryness under vacuum. The residue was purified by flash chromatography (Teledyne ISCO CombiFlash 0%> to 100% solvent A/B = hexane/EtOAc, REDISEP® Si02 220 g, detecting at 254 nm, and monitoring at 220 nm). Concentration of the appropriate fractions provided Intermediate S-2B (1.59 g, 57.5%). HPLC: RT = 3.863 min (CHROMOLITH® SpeedROD column 4.6 x 50 mm, 10-90% aqueous methanol over 4 minutes containing 0.1% TFA, 4 mL/min, monitoring at 220 nm), 1H NMR (400MHz, chloroform-d) δ 7.40-7.34 (m, 5H), 5.17 (d, J= 1.8 Hz, 2H), 2.73-2.64 (m, 1H), 2.55 (td, J= 10.0, 3.9 Hz, 1H), 2.16-1.82 (m, 5H), 1.79-1.57 (m, 3H), 1.53-1.49 (m, 1H), 1.45 (s, 9H), 1.37-1.24 (m, 1H).
[00197] To a stirred solution of Intermediate S-2B (1.59 g, 3.37 mmoL) in MeOH (10 mL) and EtOAc (10 mL) under nitrogen was added 10%> Pd/C (510 mg). The atmosphere was replaced with hydrogen and the reaction mixture was stirred at room temperature for 2.5 h. The palladium catalyst was filtered off through a 4 μΜ polycarbonate film and rinsed with MeOH. The filtrate was concentrated under reduced pressure to give intermediate S-2 (1.28 g, 99%). 1H NMR (400MHz, chloroform-d) δ 2.76-2.67 (m, 1H), 2.65-2.56 (m, 1H), 2.33-2.21 (m, 1H), 2.17-2.08 (m, 3H), 1.93 (dtd, J= 14.5, 9.9, 5.2 Hz, 1H), 1.84-1.74 (m, 2H), 1.70-1.52 (m, 3H), 1.48 (s, 9H).
[00198] In a 1 L round-bottomed flask was added 2-amino-3-methylbenzoic acid (11.2 g, 74.1 mmol) and Ν,Ο-dimethylhydroxylamine hydrochloride (14.45 g, 148 mmol) in DCM (500 mL) to give a pale brown suspension. The reaction mixture was treated with Et3N (35 mL), HOBT (11.35 g, 74.1 mmol) and EDC (14.20 g, 74.1 mmol) and then stirred at room temperature for 24 hours. The mixture was then washed with 10% LiCl, and then acidified with IN HCl. The organic layer was washed successively with 10%> LiCl and aq NaHC03. The organic layer was decolorized with charcoal, filtered, and the filtrate was dried over MgSC^. The mixture was filtered and concentrated to give 13.22 g (92% yield) of Intermediate A-1A. MS(ES): m/z = 195.1 [M+H+]; HPLC: RT = 1.118 min. (H20/MeOH with TFA, CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm); 1H NMR (500MHz, chloroform-d) δ 7.22 (dd, J= 7.8, 0.8 Hz, 1H), 7.12-7.06 (m, 1H), 6.63 (t, J= 7.5 Hz, 1H), 4.63 (br. s., 2H), 3.61 (s, 3H), 3.34 (s, 3H), 2.17 (s, 3H).
[00199] In a 500 mL round-bottomed flask, a solution of l-fluoro-3-iodobenzene (13.61 mL, 116 mmol) in THF (120 mL) was cooled in a -78 °C bath. A solution of n- BuLi, (2.5M in hexane, 46.3 mL, 116 mmol) was added dropwise over 10 minutes. The solution was stirred at -78 °C for 30 minutes and then treated with a solution of
Intermediate A-1 A (6.43 g, 33.1 mmol) in THF (30 mL). After 1.5 hours, the reaction mixture was added to a mixture of ice and IN HCl (149 mL, 149 mmol) and the reaction flask was rinsed with THF (5 ml) and combined with the aqueous mixture. The resulting mixture was diluted with 10% aq LiCl and the pH was adjusted to 4 with IN NaOH. The mixture was then extracted with Et20, washed with brine, dried over MgS04, filtered and concentrated. The resulting residue was purified by silica gel chromatography (220g ISCO) eluting with a gradient from 10% EtOAc/hexane to 30% EtOAc/hexane to afford Intermediate A-l (7.11 g, 94% yield) as an oil. MS(ES): m/z = 230.1 [M+H+]; HPLC: RT = 2.820 min Purity = 99%. (H20/MeOH with TFA, CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm).
[00225] In a 1 L round-bottomed flask, a solution of 2-(lH-benzo[d][l,2,3]triazol-l- yl)-2-((phenoxycarbonyl)amino)acetic acid (J. Org. Chem., 55:2206-2214 (1990)) (19.37 g, 62.0 mmol) in THF (135 mL) was cooled in an ice/water bath and treated with oxalyl chloride (5.43 mL, 62.0 mmol) and 4 drops of DMF. The reaction mixture was stirred for 4 hours. Next, a solution of Intermediate A- 1 (7.11 g, 31.0 mmol) in THF (35 mL) was added and the resulting solution was removed from the ice/water bath and stirred at room temperature for 1.5 hours. The mixture was then treated with a solution of ammonia, (7M in MeOH) (19.94 mL, 140 mmol). After 15 mins, another portion of ammonia, (7M in MeOH) (19.94 mL, 140 mmol) was added and the resulting mixture was sealed under N2 and stirred overnight at room temperature. The reaction mixture was then concentrated to ~l/2 volume and then diluted with AcOH (63 mL) and stir at room temperature for 4 hours. The reaction mixture was then concentrated, and the residue was diluted with 500 mL water to give a precipitate. Hexane and Et20 were added and the mixture was stirred at room temperature for 1 hour to form an orange solid. Et20 was removed under a stream of nitrogen and the aqueous layer was decanted. The residue was triturated with 40 mL of iPrOH and stirred at room temperature to give a white precipitate. The solid was filtered and washed with iPrOH, then dried on a filter under a stream of nitrogen to give racemic Intermediate B-1A (5.4 g, 41.7%yield).
[00226] Racemic Intermediate B-1A (5.9 g, 14.3 mmol) was resolved using the Chiral SFC conditions described below. The desired stereoisomer was collected as the second peak in the elution order: Instrument: Berger SFC MGIII, Column: CHIRALPAK® IC 25 x 3 cm, 5 cm; column temp: 45 °C; Mobile Phase: C02/MeOH (45/55); Flow rate: 160 mL/min; Detection at 220 nm.
[00227] After evaporation of the solvent, Intermediate B-1A (2.73 g, 46% yield) was obtained as a white solid. HPLC: RT = 3.075 min. (H20/MeOH with TFA,
CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm).
[00228] In a 100 mL round-bottomed flask, a solution of Intermediate B-1A (2.73 g, 6.54 mmol) in acetic acid (12 mL) was treated with HBr, 33% in HOAc (10.76 mL, 65.4 mmol) and the mixture was stirred at room temperature for 1 hour. The solution was diluted with Et20 to give a yellow precipitate. The yellow solid was filtered and rinsed with Et20 under nitrogen. The solid was transferred to 100 mL round bottom flask and water was added (white precipitate formed). The slurry was slowly made basic with saturated NaHC03. The resulting tacky precipitate was extracted with EtOAc. The organic layer was washed with water, dried over MgS04, and then filtered and
concentrated to dryness to give Intermediate B-l (1.68 g, 91% yield) as a white foam solid. MS(ES): m/z = 284.2 [M+H+]; HPLC: RT = 1.72 min (H20/MeOH with TFA, CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm). 1H NMR (400MHz, DMSO-d6) δ 10.01 (br. s., 1H), 7.56-7.44 (m, 2H), 7.41-7.26 (m, 3H), 7.22-7.11 (m, 2H), 4.24 (s, 1H), 2.55 (br. s., 2H), 2.41 (s, 3H). [00229] The compounds listed below in Table 6 (Intermediates B-2 to B-3) were prepared according to the general synthetic procedure described for Intermediate B-l , using the starting materials Intermediate A- 10 and Intermediate A-4, respectively.
[00240] In a 100 mL round-bottomed flask, a solution of Intermediate B-l (1683 mg, 5.94 mmol), Et3N (1.656 mL, 11.88 mmol), and Intermediate S-l in DMF (20 mL) was treated with o-benzotriazol-l-yl-A .A .N’.N’-tetramethyluronium tetrafluoroborate (3815 mg, 11.88 mmol) and stirred at room temperature for 1 hour. The reaction mixture was diluted with water and saturated aqueous NaHC03. An off white precipitate formed and was filtered and washed with water. The resulting solid was dried on the filter under a stream of nitrogen to give Intermediate 1A (3.7 g, 99% yield). MS(ES): m/z =
632.4[M+H+]; HPLC: RT = 3.635 min Purity = 98%. (H20/MeOH with TFA,
[00241] In a 250 mL round-bottomed flask, a solution of Intermediate 1A (3.7 g, 5.86 mmol) in DCM (25 mL) was treated with TFA (25 mL) and the resulting pale orange solution was stirred at room temperature for 1.5 hours. The reaction mixture was then concentrated to give Intermediate IB. HPLC: RT = 3.12 min (H20/MeOH with TFA, CHROMOLITH® ODS S5 4.6 x 50 mm, gradient = 4 min, wavelength = 220 nm).
[00242] In a 250 mL round-bottomed flask, a solution of Intermediate IB (4.04 g, 5.86 mmol) in THF (50 mL) was treated with ammonia (2M in iPrOH) (26.4 mL, 52.7 mmol), followed by HOBT (1.795 g, 11.72 mmol) and EDC (2.246 g, 11.72 mmol). The resulting white suspension was stirred at room temperature overnight. The reaction mixture was diluted with water and saturated aqueous NaHC03. The resulting solid was filtered, rinsed with water and then dried on the filter under a stream of nitrogen. The crude product was suspended in 20 mL of iPrOH and stirred at room temperature for 20 min and then filtered and washed with iPrOH and dried under vacuum to give 2.83 g of solid. The solid was dissolved in re fluxing EtOH(100 mL) and slowly treated with 200 mg activated charcoal added in small portions. The hot mixture was filtered through CELITE® and rinsed with hot EtOH. The filtrate was reduced to half volume, allowed to cool and the white precipitate formed was filtered and rinsed with EtOH to give 2.57 g of white solid. A second recrystallization from EtOH (70 mL) afforded Example 1 (2.39 g, 70% yield) as a white solid. HPLC: RT = 10.859 min (H20/CH3CN with TFA, Sunfire C18 3.5μπι, 3.0x150mm, gradient = 15 min, wavelength = 220 and 254 nm); MS(ES): m/z = 575.3 [M+H+]; 1H NMR (400MHz, methanol-d4) δ 7.57-7.50 (m, 1H), 7.47-7.30 (m, 3H), 7.29-7.15 (m, 3H), 5.38 (s, 1H), 2.85-2.75 (m, 1H), 2.59 (td, J= 10.5, 4.0 Hz, 1H), 2.53-2.41 (m, 4H), 2.31-2.10 (m, 3H), 1.96-1.70 (m, 4H).
Structure–activity relationships in a series of (2-oxo-1,4-benzodiazepin-3-yl)-succinamides identified highly potent inhibitors of γ-secretase mediated signaling of Notch1/2/3/4 receptors. On the basis of its robust in vivo efficacy at tolerated doses in Notch driven leukemia and solid tumor xenograft models, 12 (BMS-906024) was selected as a candidate for clinical evaluation.
Discovery of Clinical Candidate BMS-906024: A Potent Pan-Notch Inhibitor for the Treatment of Leukemia and Solid Tumors
For some disease targets, an indirect approach may be best. Or so Ashvinikumar V. Gavai and his colleagues atBristol-Myers Squibbfound in their quest toward a potential cancer drug. Gavai unveiled BMS-906024, which is an experimental—and slightly roundabout—treatment for a number of cancers, including breast, lung, and colon cancers, and leukemia.
Cancers have a tendency to relapse or to become resistant to treatments that once worked. Research at BMS and elsewhere had suggested that a family of proteins called Notch is implicated in that resistance and in cancer progression more generally. Gavai, director of oncology chemistry at BMS in Princeton, N.J., and his team set out to block Notch family signaling.
Notch family members lack enzymatic activity, so blocking them directly is difficult. Instead, BMS developed inhibitors of an enzyme that is essential for activating Notch signaling—γ-secretase.
Company: Bristol-Myers Squibb
Target: pan-Notch
Disease: breast, lung, colon cancer; leukemia
Interfering with Notch, even in this indirect way, can have detrimental effects on the gastrointestinal tract. Only two of the four Notch family members are linked to that side effect, Gavai says. But he and his team think their drug will be most effective if it acts on all four family members roughly equally—a so-called pan-Notch inhibitor. By selecting a molecule that’s well tolerated in animals and carefully scheduling doses of the drug in humans, it could be possible to minimize side effects, he says.
The BMS team relied on Notch signaling assays in leukemia and breast cancer cell lines to find leads. They soon learned that for their molecules to work, three chiral centers had to be in the S,R,Sconfiguration. After that, they strove to make the molecules last in the bloodstream. They removed an isobutyl group and tweaked some other parts of their candidate’s succinamide side chain. It was tough to retain both a long half-life and activity against Notch, Gavai told C&EN. “You’d optimize one and lose the other.”
His team threaded the needle with BMS-906024. Their studies with mice suggest that a dose of 4–6 mg once a week could be effective in people. That’s lower than doses being tested for other Notch-targeted agents, according to the website clinicaltrials.gov. The mouse studies also back the idea that Notch is involved in cancer drug resistance and suggest that Notch could be a target for taking on cancer stem cells, which are notoriously resistant to chemotherapy.
BMS-906024 is in Phase I clinical trials, both alone and in combination with other agents. Patients with colon, lung, breast, and other cancers are receiving intravenous doses of the compound to determine its safety and optimum dose ranges.
(From left, front row) Gavai, Weifeng Shan, (second row) Aaron Balog, Patrice Gill, Gregory Vite, (third row) Francis Lee, Claude Quesnelle, (rear row) Wen-Ching Han, Richard Westhouse.
An enantioselective synthesis of (S)-7-amino-5H,7H-dibenzo[b,d]azepin-6-one (S–1) is described. The key step in the sequence involved crystallization-induced dynamic resolution (CIDR) of compound 7 using Boc-d-phenylalanine as a chiral resolving agent and 3,5-dichlorosalicylaldehyde as a racemization catalyst to afford S–1 in 81% overall yield with 98.5% enantiomeric excess.
Crystallization-Induced Dynamic Resolution toward the Synthesis of (S)-7-Amino-5H,7H-dibenzo[b,d]-azepin-6-one: An Important Scaffold for γ-Secretase Inhibitors
† Department of Discovery Synthesis, Biocon Bristol-Myers Squibb Research Centre, Biocon Park, Bommasandra IV Phase, Jigani Link Road, Bengaluru 560099, India
‡Bristol-Myers Squibb Company, P.O Box 4000, Princeton, New Jersey 08543-4000, United States
Claude Quesnelle
Senior Research Investigator/Chemist at Bristol-Myers Squibb
RICHARD LEE
BMS-906024 is a novel, potent Notch receptor inhibitor . Cancers have a tendency to relapse or to become resistant to treatments that once worked. A family of proteins called Notch is implicated in that resistance and in cancer progression more generally. BMS-906024 is in Phase I clinical trials, both alone and in combination with other agents. Patients with colon, lung, breast, and other cancers are receiving intravenous doses of the compound to determine its safety and optimum dose ranges.
New Phase I drug structure by Bristol-Myers Squibb disclosed at the spring 2013 American Chemical Society meeting in New Orleans to treat breast, lung, and colon cancers and leukemia.[1] The drug works as an pan-Notch inhibitor. The structure is one of a set patented in 2012,[2] and it currently being studied in clinical trials.[3][4]
useful for the treatment of conditions related to the Notch pathway, such as cancer and other proliferative diseases.
Notch signaling has been implicated in a variety of cellular processes, such as cell fate specification, differentiation, proliferation, apoptosis, and angiogenesis. (Bray, Nature Reviews Molecular Cell Biology, 7:678-689 (2006); Fortini, Developmental Cell 16:633-647 (2009)). The Notch proteins are single-pass heterodimeric transmembrane molecules. The Notch family includes 4 receptors, NOTCH 1-4, which become activated upon binding to ligands from the DSL family (Delta-like 1, 3, 4 and Jagged 1 and 2).
The activation and maturation of NOTCH requires a series of processing steps, including a proteolytic cleavage step mediated by gamma secretase, a multiprotein complex containing Presenilin 1 or Presenilin 2, nicastrin, APH1, and PEN2. Once NOTCH is cleaved, NOTCH intracellular domain (NICD) is released from the membrane. The released NICD translocates to the nucleus, where it functions as a transcriptional activator in concert with CSL family members (RBPSUH, “suppressor of hairless”, and LAG1). NOTCH target genes include HES family members, such as HES- 1. HES- 1 functions as transcriptional repressors of genes such as HERP 1 (also known as HEY2), HERP2 (also known as HEY1), and HATH1 (also known as ATOH1).
The aberrant activation of the Notch pathway contributes to tumorigenesis. Activation of Notch signaling has been implicated in the pathogenesis of various solid tumors including ovarian, pancreatic, as well as breast cancer and hematologic tumors such as leukemias, lymphomas, and multiple myeloma. The role of Notch inhibition and its utility in the treatment of various solid and hematological tumors are described in Miele, L. et al, Current Cancer Drug Targets, 6:313-323 (2006); Bolos, V. et al, Endocrine Reviews, 28:339-363 (2007); Shih, I.-M. et al, Cancer Research, 67: 1879- 1882 (2007); Yamaguchi, N. et al., Cancer Research, 68: 1881-1888 (2008); Miele, L., Expert Review Anti-cancer Therapy, 8: 1 197-1201 (2008); Purow, B., Current Pharmaceutical Biotechnology, 10: 154-160 (2009); Nefedova, Y. et al, Drug Resistance Updates, 1 1 :210-218 (2008); Dufraine, J. et al, Oncogene, 27:5132-5137 (2008); and Jun, H.T. et al, Drug Development Research, 69:319-328 (2008).
There remains a need for compounds that are useful as Notch inhibitors and that have sufficient metabolic stability to provide efficacious levels of drug exposure. Further, there remains a need for compounds useful as Notch inhibitors that can be orally or intravenously administered to a patient.
U.S. Patent No. 7,053,084 Bl discloses succinoylamino benzodiazepine compounds useful for treating neurological disorders such as Alzheimer’s Disease. The reference discloses that these succinoylamino benzodiazepine compounds inhibit gamma secretase activity and the processing of amyloid precursor protein linked to the formation of neurological deposits of amyloid protein. The reference does not disclose the use of these compounds in the treatment of proliferative diseases such as cancer.
Applicants have found potent compounds that have activity as Notch inhibitors and have sufficient metabolic stability to provide efficacious levels of drug exposure upon intravenous or oral administration. These compounds are provided to be useful as pharmaceuticals with desirable stability, bioavailability, therapeutic index, and toxicity values that are important to their drugability.
PAPER
Structure–activity relationships in a series of (2-oxo-1,4-benzodiazepin-3-yl)-succinamides identified highly potent inhibitors of γ-secretase mediated signaling of Notch1/2/3/4 receptors. On the basis of its robust in vivo efficacy at tolerated doses in Notch driven leukemia and solid tumor xenograft models, 12 (BMS-906024) was selected as a candidate for clinical evaluation.
Discovery of Clinical Candidate BMS-906024: A Potent Pan-Notch Inhibitor for the Treatment of Leukemia and Solid Tumors
[00219] To a stirred solution of 5,5,5-trifluoropentanoic acid (5 g, 32.0 mmol) in THF (30 mL) and hexane (30 mL) at 0 °C, was added tert-butyl 2,2,2-trichloroacetimidate (11.46 mL, 64.1 mmol). The mixture was stirred for 15 min at 0 °C. Boron trifluoride etherate (0.406 mL, 3.20 mmol) was added and the reaction mixture was allowed to warm to room temperature overnight. To the clear reaction mixture was added solid aHC03 (5 g) and stirred for 30 min. The mixture was filtered through MgS04 and washed with hexanes (200 mL). The solution was allowed to rest for 45 min, and the resulting solid material was removed by filtering on the same MgS04 filter again, washed with hexanes (100 mL) and concentrated under reduced pressure without heat. The volume was reduced to about 30 mL, filtered through a clean fritted funnel, washed with hexane (5 mL), and then concentrated under reduced pressure without heat. The resulting neat oil was filtered through a 0.45μηι nylon membrane filter disk to provide tert-butyl 5,5,5- trifluoropentanoate (6.6 g, 31.4 mmol 98% yield) as a colorless oil: XH NMR (400 MHz, CDC13) δ ppm 1.38 (s, 9 H) 1.74-1.83 (m, 2 H) 2.00-2.13 (m, 2 H) 2.24 (t, J=7.28 Hz, 2 H).
[00220] To a stirred solution of 5,5,5-trifluoropentanoic acid (5.04 g, 32.3 mmol) in DCM (50 mL) and DMF (3 drops) was added oxalyl chloride (3.4 mL, 38.8 mmol) dropwise over 5 min and the solution was stirred until all bubbling subsided. The reaction mixture was concentrated under reduced pressure to give pale yellow oil. To a separate flask charged with a solution of (45)-4-(propan-2-yl)-l,3-oxazolidin-2-one (4.18 g, 32.4 mmol) in THF (100 mL) at -78 °C was added n-BuLi (2.5M in hexane) (13.0 mL, 32.5 mmol) dropwise via syringe over 5 min. After stirring for 10 min, the above acid chloride dissolved in THF (20 mL) was added via cannula over 15 min. The reaction mixture was warmed to 0 °C, and was allowed to warm to room temperature as the bath warmed and stirred overnight. To the reaction mixture was added saturated NH4CI, and then extracted with EtOAc (2x). The combined organics were washed with brine, dried (Na2S04), filtered and concentrated under reduced pressure. The crude material was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 5% to 60% solvent A/B=hexanes/EtOAc, REDISEP® S1O2 120g). Concentration of appropriate fractions provided Preparation IB (7.39 g, 86%) as a colorless oil: XH NMR (400 MHz, CDC13) δ ppm 4.44 (1 H, dt, J=8.31, 3.53 Hz), 4.30 (1 H, t, J=8.69 Hz), 4.23 (1 H, dd, J=9.06, 3.02 Hz), 2.98-3.08 (2 H, m), 2.32-2.44 (1 H, m, J=13.91, 7.02, 7.02, 4.03 Hz), 2.13-2.25 (2 H, m), 1.88-2.00 (2 H, m), 0.93 (3 H, d, J=7.05 Hz), 0.88 (3 H, d, J=6.80 Hz). Preparation 1C: (25′,3R)-tert-Butyl 6,6,6-trifluoro-3-((5)-4-isopropyl-2-oxooxazolidine- 3 -carbonyl)-2-(3 ,3,3 -trifluoropropyl)hexanoate, and
[00221] To a cold (-78 °C), stirred solution of diisopropylamine (5.3 mL, 37.2 mmol) in THF (59 mL) under nitrogen atmosphere was added n-BuLi (2.5M in hexane) (14.7 mL, 36.8 mmol), then warmed to 0 °C to give a 0.5M solution of LDA. A separate vessel was charged with Preparation IB (2.45 g, 9.17 mmol), the material was azeotroped twice with benzene (the RotoVap air inlet was fitted with nitrogen inlet to completely exclude humidity) then toluene (15.3 mL) was added. This solution was added to a flask containing dry lithium chloride (1.96 g, 46.2 mmol). To the resultant mixture, cooled to -78 °C, was added LDA solution (21.0 mL, 10.5 mmol) and stirred at -78 °C for 10 min, warmed to 0 °C for 10 min then recooled to -78 °C. To a separate reaction vessel containing Preparation 1A (3.41 g, 16.07 mmol), also azeotroped twice with benzene, was added toluene (15.3 mL), cooled to -78 °C and LDA (37.0 mL, 18.5 mmol) was added, the resulting solution was stirred at -78° for 25 min. At this time the enolate derived from the ester was transferred via cannula into the solution of the oxazolidinone enolate, stirred at -78 °C for an additional 5 min at which time the septum was removed and solid powdered bis(2-ethylhexanoyloxy)copper (9.02 g, 25.8 mmol) was rapidly added to the reaction vessel and the septum replaced. The vessel was immediately removed from the cold bath and immersed into a warm water bath (40 °C) with rapid swirling with a concomitant color change from the initial turquoise to brown. The reaction mixture was stirred for 20 min, was poured into 5% aqueous NH4OH (360 mL) and extracted with EtOAc (2x). The combined organics were washed with brine, dried (Na2S04), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 0% to 60% solvent A/B=hexanes/EtOAc, REDISEP® S1O2 120g). Concentration of appropriate fractions provided Preparation 1C (2.87 g, 66%) as pale yellow viscous oil. XH NMR showed the product was a 1.6: 1 mixture of diastereoisomers 1C: 1D as determined by the integration of the multiplets at 2.74 & 2.84 ppm: XH NMR (400 MHz, CDC13) δ ppm 4.43-4.54 (2 H, m), 4.23-4.35 (5 H, m), 4.01 (1 H, ddd, J=9.54, 6.27, 3.51 Hz), 2.84 (1 H, ddd, J=9.41, 7.28, 3.64 Hz), 2.74 (1 H, ddd, J=10.29, 6.27, 4.02 Hz), 2.37-2.48 (2 H, m, J=10.38, 6.98, 6.98, 3.51, 3.51 Hz), 2.20-2.37 (3 H, m), 1.92-2.20 (8 H, m), 1.64-1.91 (5 H, m), 1.47 (18 H, s), 0.88-0.98 (12 H, m). Preparation IE: (2R,35)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and
[00222] To a cool (0 °C), stirred solution of Preparation 1C and ID (4.54 g, 9.51 mmol) in THF (140 mL) and water (42 mL) was sequentially added hydrogen peroxide (30% in water) (10.3 g, 91 mmol) and LiOH (685.3 mg, 28.6 mmol) and the mixture was stirred for 1 hr. At this time the reaction vessel was removed from the cold bath and then stirred for 1.5 hr. The reaction was judged complete by HPLC. To the reaction mixture was added saturated NaHC03 (45 mL) and saturated a2S03(15 mL), and then partially concentrated under reduced pressure. The resulting crude solution was extracted with DCM (3x). The aqueous phase was acidified to pH~l-2 with IN HC1, extracted with DCM (3x) and EtOAc (lx). The combined organics were washed with brine, dried (Na2S04), filtered and concentrated under reduced pressure to provide a mixture of Preparation IE and IF (3.00 g, 86%) as colorless oil: XH NMR (400 MHz, CDC13) δ ppm 2.76-2.84 (1 H, m, diastereoisomer 2), 2.64-2.76 (3 H, m), 2.04-2.35 (8 H, m), 1.88-2.00 (4 H, m), 1.71-1.83 (4 H, m), 1.48 (9 H, s, diastereoisomer 1), 1.46 (9 H, s, diastereoisomer 2); XH NMR showed a 1.7: 1 mixture of 1E: 1F by integration of the peaks for the ?-butyl groups.
Preparation IE: (2R,35)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and
[00223] To a cold (-78 °C), stirred solution of diisopropylamine (1.7 mL, 11.93 mmol) in THF (19 mL) under nitrogen atmosphere was added n-BuLi (2.5M in hexanes) (4.8 mL, 12.00 mmol). The mixture was stirred for 5 min and then warmed to 0 °C. In a separate vessel, to a cold (-78 °C) stirred solution of the mixture of Preparation IE and IF (1.99 g, 5.43 mmol) in THF (18 mL) was added the LDA solution prepared above via cannula slowly over 25 min. The mixture was stirred for 15 min, then warmed to room temperature (placed in a 24 °C water bath) for 15 min, and then again cooled to -78 °C for 15 min. To the reaction mixture was added Et2AlCl (1M in hexane) (11.4 mL, 1 1.40 mmol) via syringe, stirred for 10 min, warmed to room temperature for 15 min and then cooled back to -78 °C for 15 min. Methanol (25 mL) was rapidly added, swirled vigorously while warming to room temperature, then concentrated to ~l/4 original volume. The mixture was dissolved in EtOAc and washed with IN HCl (50 mL) and ice (75 g). The aqueous phase was separated, extracted with EtOAc (2x). The combined organics were washed with a mixture of KF (2.85g in 75 mL water) and IN HCl (13 mL) [resulting solution pH 3-4], then with brine, dried (Na2S04), filtered and concentrated under reduced pressure to give a 9: 1 (IE: IF) enriched diastereoisomeric mixture (as determined by XH NMR) of Preparation IE and Preparation IF (2.13 g, >99%) as a pale yellow viscous oil: XH NMR (400 MHz, CDC13) δ ppm 2.64-2.76 (2 H, m), 2.04-2.35 (4 H, m), 1.88-2.00 (2 H, m), 1.71-1.83 (2 H, m), 1.48 (9 H, s). Preparation 1 G: (35)-3 -Amino- 1 -methyl-5-phenyl- 1 ,3 -dihydro-2H- 1 ,4-benzodiazepin-2- one, and
[00224] Racemic 3-amino-l-methyl-5-phenyl-l,3-dihydro-2H-l,4-benzodiazepin-2- one (Rittle, K.E. et al, Tetrahedron Letters, 28(5):521-522 (1987)) was prepared according to the literature procedure. The enantiomers were separated under chiral-SFC conditions using the following method: CHIRALPAK® AS-H 5×25; Mobile phase: 30% MeOH+ 0.1% DEA in C02; Flow rate: 280 mL/min; Pressure: 100 bar; Temperature: 35 °C.
[00225] Obtained the S-enantiomer (Preparation 1G): HPLC: RT=1.75 min (30% MeOH + 0.1% DEA in C02 on CHIRALPAK® AS-H 4.6×250 mm, 3 mL/min, 35 °C, 100 bar, 230 nm, ΙΟμΙ injection); ¾ NMR (400 MHz, CDC13) δ ppm 7.58-7.63 (2 H, m), 7.55 (1 H, ddd, J=8.50, 7.1 1, 1.76 Hz), 7.40-7.47 (1 H, m), 7.34-7.40 (3 H, m), 7.31 (1 H, dd, J=7.81, 1.51 Hz), 7.14-7.22 (1 H, m), 4.46 (1 H, s), 3.44 (3 H, s), 3.42 (2 H, s); [a]D= -155° (c=1.9, MeOH) (Lit. Rittle, K.E. et al, Tetrahedron Letters, 28(5):521-522 (1987): [a]D=-236°).
[00226] Also obtained the R-enantiomer (Preparation 1H): HPLC: RT=1.71 min; [a]D=+165° (c=2.1, MeOH) (Lit [a]D= +227°).
Alternate procedure to make Preparation 1 G:
Preparation 1G»CSA salt: (35)-3-Amino-l-methyl-5-phenyl-l,3-dihydro-2H-l,4- benzodiazepin-2-one, (15)-(+)-10-camphorsulfonic acid salt
[00227] Preparation lG’CSA was prepared from racemic 3-amino-l-methyl-5-phenyl- l,3-dihydro-2H-l,4-benzodiazepin-2-one (9.98g, 37.6 mmol) (prepared according to the literature as shown above) according to the literature procedure (Reider, P.J. et al, J. Org. Chem., 52:955-957 (1987)). Preparation lG’CSA (16.91g, 99%) was obtained as a colorless solid: Optical Rotation: [a]D = -26.99° (c=l, H20) (Lit. [a]D = -27.8° (c=l,
[00228] To a stirred solution of Preparation 1G (1.45 g, 5.47 mmol) and a 9: 1 mixture of Preparation IE and IF (1.989 g, 5.43 mmol) in DMF (19 mL) was added O- benzotriazol-l-yl-N,N,N’,N’-tetra-methyluronium tetrafluoroborate (1.79 g, 5.57 mmol) and triethylamine (3.0 mL, 21.52 mmol) and stirred overnight. The reaction was judged complete by LCMS. The reaction mixture was poured into water (125 mL) and the precipitated solid was collected by filtration, washed with water and air dried to provide an 8: 1 mixture of Preparation II and Preparation 1J (2.95 g, 89%) as a cream solid: MS (ES): m/z= 614 [M+H]+;XH NMR (400 MHz, CDC13) δ ppm 7.55-7.65 (3 H, m), 7.44- 7.52 (2 H, m), 7.35-7.45 (4 H, m), 5.52 (1 H, d, J=8.03 Hz), 3.48 (3 H, s), 2.63 (2 H, ddd, J=9.35, 3.95, 3.76 Hz), 2.14-2.25 (4 H, m), 1.90-2.03 (3 H, m), 1.69-1.82 (1 H, m), 1.51 (9 H, s).
Preparation IK: (25,,3R)-6,6,6-Trifluoro-3-(((35)-l-methyl-2-oxo-5-phenyl-2,3-dihydro- lH-l,4-benzodiazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoic acid, and
[00229] To a cool (0 °C), stirred solution of the above mixture of Preparation II and Preparation 1 J (2.95 g, 4.81 mmol) in DCM (20 mL) was added TFA (20 mL, 260 mmol). The reaction mixture was stirred for lhr, then allowed to warm to room temperature and stirred for 2.5 hr. The reaction was judged complete by LCMS. The reaction mixture was diluted with toluene (50 mL) and concentrated under reduced pressure. The residue mixture was redissolved in toluene (50 mL) and concentrated under reduced pressure then dried under high vacuum. The crude product was dissolved in DCM, S1O2 (15g) was added, concentrated, then was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 0% to 45% solvent A/B=DCM/EtOAc, REDISEP® S1O2 80g). Concentration of appropriate fractions provided a mixture of Preparation IK and Preparation 1L (2.00 g, 75%) as a cream solid: HPLC: RT=2.770 min
(CHROMOLITH® SpeedROD 4.6 x 50 mm (4 min grad) eluting with 10-90% aqueous MeOH over 4 minutes containing 0.1% TFA, 4 mL/min, monitoring at 254 nm); MS (ES): m/z= 558 [M+H]+; XH NMR (400 MHz, CDC13) δ ppm 8.32 (1 H, d, J=8.03 Hz), 7.65-7.71 (1 H, m), 7.50-7.60 (3 H, m), 7.41-7.49 (2 H, m), 7.39 (1 H, dd, J=7.91, 1.63 Hz), 7.23-7.35 (2 H, m), 5.59 (1 H, d, J=8.03 Hz), 3.51 (3 H, s), 2.81 (1 H, ddd, J=10.54, 6.90, 3.64 Hz), 2.67-2.76 (1 H, m), 2.22-2.33 (3 H, m), 1.99-2.12 (3 H, m), 1.85-1.94 (1 H, m), 1.79 (1 H, ddd, J=13.87, 7.84, 3.64 Hz). Example 1 :
[00230] To a stirred solution of an 8: 1 mixture of Preparation IK and Preparation 1L (3.46 g, 6.21 mmol) in DMF (25 mL) under nitrogen atmosphere was added ammonium chloride (3.32 g, 62.1 mmol), EDC (3.55 g, 18.52 mmol), HOBT (2.85 g, 18.61 mmol), and triethyl amine (16 mL, 1 15 mmol) and stirred overnight. The reaction was judged complete by LCMS. The reaction mixture was poured into water (200 mL) with vigorous swirling and then allowed to sit. The solid was collected by filtration, washed with water, allowed to dry to afford 3.6 g colorless solid. The solid was purified by preparative SFC chromatography (Lux-Cellulose-2 (3x25cm), 8% methanol in CO2, 140ml/min @220nm and 35 °C; Sample: 3.6g in 50cc methanol, conc.=70mg/ml, Stack injection:
0.5cc/9.2min). Fractions containing product were concentrated, dried overnight under vacuum. Obtained Example 1 (2.74 g, 79%) as a colorless solid (Crystal Form -1): HPLC: RT=9.601 min (H20/CH3CN with TFA, Sunfire CI 8 3.5um, 4.6x150mm, 4.6x150mm, gradient = 15 min, wavelength = 220 and 254 nm). MS (ES): m/z= 557 [M+H]+; XH NMR (400 MHz, DMSO-d6) δ ppm 9.54 (1 H, d, J=7.28 Hz), 7.71-7.80 (1 H, m), 7.68 (2 H, d, J=8.78 Hz), 7.50-7.62 (3 H, m), 7.45 (2 H, t, J=7.28 Hz), 7.29-7.40 (2 H, m), 7.15 (1 H, br. s.), 5.30 (1 H, d, J=7.28 Hz), 3.39 (3 H, s), 2.74-2.86 (1 H, m), 2.02-2.32 (3 H, m), 1.45-1.79 (4 H, m); [a]D = -107.0° (5.73 mg/mL, DMSO).
[00231] Crystal Form A-2 was prepared by adding approximately 1 mg of Example 1 to approximately 0.7 mL of acetone/acetonitrile/water solution (2:2: 1). A mixture of colorless needles and thin blades crystals were obtained after one day of slow evaporation of the solution at room temperature. The thin blade crystals were separated to provide crystal Form A-2.
[00232] Crystal Form EA-3 was prepared by adding approximately 1 mg of Example 1 to approximately 0.7 mL of ethyl acetate/heptane solution (1 : 1). Colorless blade crystals were obtained after three days of slow evaporation of the solution at room temperature.
[00233] Crystal Form THF-2 was obtained by adding approximately 5 mg of Example 1 to approximately 0.7 mL of THF/water solution (4: 1). Colorless blade-like crystals were obtained after one day of solvent evaporation at room temperature.
Alternate Procedure to Make Example 1 : Preparation 1M: 3,3,3-Trifluoropropyl trifluoromethanesulfonate
[00234] To a cold (-25 °C), stirred solution of 2,6-lutidine (18.38 mL, 158 mmol) in CH2CI2 (120 mL) was added Tf20 (24.88 mL, 147 mmol) over 3 min, and stirred for 5 min. To the reaction mixture was added 3,3,3-trifluoropropan-l-ol (12 g, 105 mmol) over an interval of 3 min. After 2 hr, the reaction mixture was warmed to room temperature and stirred for 1 hr. The reaction mixture was concentrated to half volume, then purified by loading directly on silica gel column (330g ISCO) and eluted with CH2C12. Obtained Preparation 1M (13.74 g, 53%) as a colorless oil. XH NMR (400 MHz, CDCI3) δ ppm 4.71 (2 H, t, J=6.15 Hz), 2.49-2.86 (2 H, m).
[00235] Preparation IN was prepared from 5,5,5-trifluoropentanoic acid (3.35 g, 21.46 mmol) and (45)-4-benzyl-l,3-oxazolidin-2-one (3.80 g, 21.46 mmol) by the general methods shown for Preparation IB. Preparation IN (5.67 g, 84%) was obtained as a colorless viscous oil: XH NMR (400 MHz, CDC13) δ ppm 7.32-7.39 (2 H, m), 7.30 (1 H, d, J=7.05 Hz), 7.18-7.25 (2 H, m), 4.64-4.74 (1 H, m), 4.17-4.27 (2 H, m), 3.31 (1 H, dd, J=13.35, 3.27 Hz), 3.00-3.1 1 (2 H, m), 2.79 (1 H, dd, J=13.35, 9.57 Hz), 2.16-2.28 (2 H, m), 1.93-2.04 (2 H, m).
[00236] To a cold (-78 °C), stirred solution of Preparation IN (3.03 g, 9.61 mmol) in THF (20 mL) was added NaHMDS (1.0M in THF) (10.6 mL, 10.60 mmol) under nitrogen atmosphere. After 2 hours, tert-butyl 2-bromoacetate (5.62 g, 28.8 mmol) was added neat via syringe at -78 °C and stirring was maintained at the same temperature. After 6 hours, the reaction mixture was warmed to room temperature. The reaction mixture was partitioned between saturated NH4C1 and EtOAc. The organic phase was separated, and the aqueous was extracted with EtOAc (3x). The combined organics were washed with brine, dried (Na2S04), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 5% to 100% solvent A/B=hexanes/EtO Ac, REDISEP® Si02 120g). Concentration of appropriate fractions provided Preparation 10 (2.79 g, 67.6%) as a colorless viscous oil: XH NMR (400 MHz, CDC13) δ ppm 7.34 (2 H, d, J=7.30 Hz), 7.24-7.32 (3 H, m), 4.62- 4.75 (1 H, m, J=10.17, 6.89, 3.43, 3.43 Hz), 4.15-4.25 (3 H, m), 3.35 (1 H, dd, J=13.60, 3.27 Hz), 2.84 (1 H, dd, J=16.62, 9.57 Hz), 2.75 (1 H, dd, J=13.35, 10.07 Hz), 2.47 (1 H, dd, J=16.62, 4.78 Hz), 2.1 1-2.23 (2 H, m), 1.90-2.02 (1 H, m), 1.72-1.84 (1 H, m), 1.44 (9 H, s). -2-(2-tert-Butoxy-2-oxoethyl)-5,5,5-trifluoropentanoic acid
[00237] Preparation IP was prepared from Preparation 10 (2.79 g, 6.50 mmol) by the general methods shown for Preparation IE. Preparation IP (1.45 g, 83%) was obtained as a colorless oil: XH NMR (400 MHz, CDC13) δ ppm 2.83-2.95 (1 H, m), 2.62-2.74 (1 H, m), 2.45 (1 H, dd, J=16.62, 5.79 Hz), 2.15-2.27 (2 H, m), 1.88-2.00 (1 H, m), 1.75-1.88 (1 H, m), 1.45 (9 H, s). Preparation IE: (2R,35′)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and
[00238] To a cold (-78 °C), stirred solution of Preparation IP (5.44 g, 20.13 mmol) in THF (60 mL) was slowly added LDA (24.60 mL, 44.3 mmol) over 7 min. After stirring for 2 hr, Preparation 1M (6.44 g, 26.2 mmol) was added to the reaction mixture over 3 min. After 45 min, the reaction mixture was warmed to -25 °C bath (ice/MeOH/dry ice) for 1 hr, and then warmed to 0 °C. After 45 min, Preparation 1M (lg) was added and the reaction mixture was stirred for 20 min. The reaction was quenched with water and IN NaOH and was extracted with (¾(¾. The organic layer was again extracted with IN NaOH (2x) and the aqueous layers were combined. The aqueous layer was cooled in ice/water bath and then acidified with concentrated HCl to pH 2. Next, the aqueous layer was extracted with EtOAc. The combined organics were washed with brine, dried over anhydrous sodium sulphate, and concentrated under reduced pressure. The residue was dried under high vacuum to provide a 1 :5 (IE: IF) mixture (as determined by XH NMR) of Preparation IE and Preparation IF (5.925 g, 80%) as a pale yellow solid. XH NMR (500 MHz, CDC13) 8 ppm 2.81 (1 H, ddd, J=10.17, 6.32, 3.85 Hz), 2.63-2.76 (1 H, m), 2.02- 2.33 (4 H, m), 1.86-1.99 (2 H, m), 1.68-1.85 (2 H, m), 1.47 (9 H, s).
Preparation IE: (2R,35)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and
[00239] A mixture of Preparation IE and Preparation IF (64 mg, 1.758 mmol) was taken in THF (6 mL) to give a colorless solution which was cooled to -78 °C. Then, LDA (2.149 mL, 3.87 mmol) (1.8M in heptane/THF/ethylbenzene) was slowly added to the reaction mixture over 10 min. After stirring for 15 min the reaction mixture was placed in a room temperature water bath. After 15 min the reaction mixture was placed back in -78 °C bath and then diethylaluminum chloride (3.87 mL, 3.87 mmol) (1M in hexane) was added slowly over 5 min. The reaction mixture was stirred at -78 °C. After 15 min the reaction mixture was placed in a room temperature water bath for 10 min and then cooled back to -78 °C bath. After 15 min the reaction was quenched with MeOH (8 mL, 198 mmol), removed from the -78 °C bath and concentrated. To the reaction mixture was added ice and HC1 (16 mL, 16.00 mmol), followed by extraction with EtOAc (2x). The organic layer was washed with potassium fluoride (920 mg, 15.84 mmol) (in 25 mL FLO) and HC1 (4.5 mL, 4.50 mmol). The organics were dried over anhydrous magnesium sulphate and concentrated under reduced pressure to provide a 9: 1 (IE: IF) enriched mixture of Preparation IE and Preparation IF (540 mg, 1.583 mmol, 90% yield) as light yellow/orange solid. ¾ NMR (400 MHz, CDC13) δ ppm 2.64-2.76 (2 H, m), 2.04-2.35 (4 H, m), 1.88-2.00 (2 H, m), 1.71-1.83 (2 H, m), 1.48 (9 H, s). It was converted to Example 1 by the sequence of reactions as outlined above.
(1Q) [00240] A clean and dry 5 L four neck round bottom flask equipped with mechanical stirring, thermometer socket and nitrogen bubbler at room temperature was charged with Ν,Ν-dimethyl formamide (2.07 L), a 1.2: 1 mixture of Preparation IE and Preparation IF (207 g, 0.5651 moles), potassium carbonate (1 17.1 g, 0.8476 moles) followed by benzyl bromide (116 g, 0.6781 moles) over 15-20 min. The reaction mixture was stirred for 2-3 hr. After completion of the reaction, the reaction mixture was concentrated to dryness at 50-55 °C under vacuum. Ethyl acetate (3.1 L, 30 Vol.) was charged into the concentrated reaction mass and then washed with water (2.07 L), brine (0.6 L) then dried over anhydrous sodium sulfate (207 g), filtered and concentrated to dryness at 40-45 °C under vacuum. The residue was dissolved in dichloromethane (1.035 L, 5 vol.) and then absorbed onto silica gel (60-120) (607 g, 3.0 w/w), then was purified with column chromatography using petroleum ether and ethyl acetate as solvents. After pooling several batches, Preparation 1Q (235 g) was obtained. HPLC purity: 99.77%, Preparation IE: (2R,35)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid
[00241] A clean and dry 2 L autoclave was charged with methanol (540 mL) and was purged with nitrogen for 5-10 minutes. To the autoclave was added 10% palladium on carbon (12 g, 20%), purged with nitrogen once again for 5-10 min then was charged with Preparation 1Q (60g, 0.1315 moles), the autoclave was flushed with methanol (60mL) and stirred for 4-6 hr at 20-25 °C under 5Kg hydrogen pressure. After completion of the reaction, the reaction mass was filtered through CELITE®, washed with methanol (180 mL), dried with anhydrous sodium sulfate (60 g), filtered and concentrated to dryness at 45-50 °C under vacuum. Obtained Preparation IE (45.8 g, 95%) as a colorless solid: HPLC purity: 98.9%.
Alternate procedure to make Preparation IE: Preparation IE: (2R,35)-3-(te^Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid
[00242] Preparation IE was prepared in a procedure identical as above from a mixture of Preparations IE and IF (200g, 0.5460 moles) using LDA (1.8 M solution in THF, ethyl benzene and heptane) (698mL, 2.3equiv.) and diethyl aluminum chloride (1.0 M solution in hexane) (1256mL, 2.3equiv) in THF (2.0L). After workup as explained above, the resulting residue was treated as follows: The crude material was added to a 2L four neck round bottom flask, followed by the addition of MTBE (1.0L) charged below 30 °C. The resulting mixture was stirred for 5-10 minutes to obtain a clear solution.
Hexanes (600mL) was charged to the reaction mixture at a temperature below 30 °C. The reaction mixture was stirred for 10 min. Next, tert-butylamine (43.8g, l. leq) was charged slowly over a period of 15 minutes below 30 °C. This addition was observed to be exothermic. The reaction mixture was stirred for 2 hrs below 30 °C and filtered. The solid material was washed with 5:3 MTBE: hexane (200mL), the filtrate was
concentrated and transferred to an amber color bottle. The filtered solid was dissolved in dichloromethane (2.0L), washed with IN HC1 (2.0), the organic layer was washed with brine (1.0L x 2), then was concentrated under reduced pressure below 45 °C. This material was found to be 91.12% pure. The material was repurified by the above t- butylamine crystallization purification procedure. Obtained Preparation IE (78 g, 39%): HPLC purity: 99.54%.
[00243] A clean and dry 2 L four neck round bottom flask equipped with mechanical stirring, thermometer socket and nitrogen bubbler was charged with N,N- dimethylformamide (457 mL), Preparation IE (45.7g, 0.1248moles) and Preparation lG’CSA (62.08g, 0.1248moles) under nitrogen atmosphere at 20-25 °C. The reaction mixture was stirred for 15-20 minutes to make clear solution at 20-25 °C. To the reaction mixture was added TBTU (48.16g, 0.1498 moles) at 20-25 °C followed by triethylamine (50.51g, 0.4992 moles) over 15-20 minutes at 20-25 °C. The reaction mixture was stirred for 60-120 minutes at 20-25 °C under nitrogen atmosphere. After completion of the reaction, the reaction was quenched into water (1.37L, 30 Vol.) at 20-25 °C under stirring. The reaction mixture was stirred for 30 minutes at 20-25 °C. The reaction mixture was filtered and washed with water (228 mL). The resulting solid material was dissolved in ethyl acetate (457 mL), washed with water (2×137 mL), brine (137 mL), and then dried with anhydrous sodium sulfate (45.7g). Activated charcoal (9.14 g, 20%) was charged into the reaction mixture and stirred for 30 minutes. The mixture was filtered through CELITE® bed and 1 micron filter cloth, washed charcoal bed with ethyl acetate (137 mL), concentrated to 1.0 Vol. stage and then petroleum ether (457 mL, 10 Vol.) was charged and stirred for 30 minutes at 20-25 °C. The solid was collected by filtration, washed with petroleum ether (137 mL) and then dried under vacuum at 40-45 °C for 8 hr until loss on drying was less than 3.0%. Obtained Preparation II (65.2 g, 85%): HPLC purity: 98.26%.
[00244] A clean and dry 3 L four neck round bottom flask equipped with mechanical stirring, thermometer socket and nitrogen bubbler was charged with dichloromethane (980 mL) under nitrogen atmosphere followed by Preparation II (140 g, 0.2282 moles) at 20-25 °C. The reaction mixture was cooled to 0-5 °C and trifluoroacetic acid (980 mL) was charged slowly for 30-40 minutes. The resulting mixture was stirred for 2 hr at 0-5 °C under nitrogen atmosphere. The reaction temperature was raised to 20 to 25 °C, and the reaction mixture was stirred for 1-2 hr at 20 to 25 °C. After completion of the reaction, the reaction mixture was concentrated to dryness at 50 to 55 °C under vacuum. Toluene (3×700 mL,) was charged into the concentrated reaction mass, and then distilled off at 50 to 55 °C under vacuum. After complete concentration from toluene, ethyl acetate (280 mL) was charged into the reaction mass at 20 to 25 °C, stirred for 60 minutes, then the solid was collected by filtration, washed with ethyl acetate (140 mL), dried under vacuum at 50 to 55 °C for 12 hr until loss on drying was less than 2.0%. Obtained Preparation IK (106 g, 84%): HPLC purity: 98.43%.
Example 1 :
[00245] A reaction vessel was charged with Preparation IK (30 g, 53.81 mmol), HOBt (8.7g, 64.38 mmol), and THF (150 mL) at room temperature. To the homogeneous solution was added EDCI (12.4g, 64.68 mmol), stirred for 15 min, then cooled to 8 °C. To the reaction mixture was added ammonia (2M in IP A) (81 mL, 162 mmol) over 5 min so as to maintain a temperature below 10 °C. The resulting heavy slurry was stirred for 10 min, warmed to room temperature over 30 min, then stirred for 4 hr. At the completion of the reaction, water (230 mL) was slowly added over 15 min to maintain a temperature below 20 °C, and then stirred for 2 hr. The solid was collected by filtration, washed with water (3X60 mL), then dried under vacuum 48 hr at 55 °C. The above crude product was charged into a 1 L 3 -necked round flask. IP A (200 mL) was added, then heated to 80 °C resulting in a homogeneous solution. Water (170 mL) was slowly added (15 min) to maintain an internal temperature >75 °C. The resulting slurry was stirred and cooled to room temperature for 2 hr. The solid was collected by filtration, washed with water (2 X 50 mL), then dried under vacuum (55 °C for 24 h, and 30 °C for 48 h).
Obtained Example 1 (23.4 g, 78% yield): HPLC purity: 99.43%.
Preparation 2A: (35)-3-Amino-5-phenyl-l,3-dihydro-2H-l,4-benzodiazepin-2-one, and Preparation 2B: -3-Amino-5-phenyl-l,3-dihydro-2H-l,4-benzodiazepin-2-one
(2A) (2B)
[00246] Racemic 3-amino-5-phenyl-l,3-dihydro-2H-l,4-benzodiazepin-2-one (J. Med. Chem., 49:231 1-2319 (2006), compound# 5) was prepared according to the literature procedure. The enantiomers were separated on Berger SFC MGIII Column: Lux 25X3 cm, 5cm; Mobile phase: 30% MeOH+ 0.1% DEA in C02; Flow rate: 150 mL/min;
Temperature: 40 °C; Detector wavelength: 250 nM. Obtained the S-enantiomer
Preparation 2A as a white solid: XH NMR (400 MHz, DMSO-d6) δ ppm 10.67 (1 H, br. s.), 7.58 (1 H, td, J=7.65, 1.76 Hz), 7.37-7.53 (5 H, m), 7.23-7.30 (2 H, m), 7.14-7.22 (1 H, m), 4.23 (1 H, s), 2.60 (2 H, br. s.); HPLC: RT=3.0625 min (30% MeOH + 0.1% DEA in C02 on OD-H Column, 3 mL/min, 35 °C, 96 bar, 230 nm, ΙΟμΙ inj); [a]D = -208.3° (5.05 mg/niL, MeOH). Also obtained the R-enantiomer Preparation 2B as an off white solid: HPLC: RT=3.970 min; [a]D = 182.1° (2.01 mg/mL, MeOH).
[00247] Preparation 2C was prepared from Preparation 2A (564 mg, 2.244 mmol) and a mixture of Preparation IE and Preparation IF (822 mg, 2.244 mmol) according to the general procedure shown for Preparation II. Obtained Preparation 2C and Preparation 2D (1.31 g, 97%): HPLC: RT=3.443 min (CHROMOLITH® ODS 4.6 x 50 mm (4 min grad) eluting with 10-90% aqueous MeOH over 4 minutes containing 0.% TFA, 4 mL/min, monitoring at 220 nm); MS (ES): m/z= 600.3 [M+H]+.
Preparation 2E: (25′,3R)-6,6,6-Trifluoro-3-(((35)-2-oxo-5-phenyl-2,3-dihydro-lH-l,4- benzodiazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoic acid, and
(2E) (2F) [00248] A mixture of Preparation 2E and Preparation 2F was prepared from a mixture of Preparation 2C and Preparation 2D (1.3 lg, 2.185 mmol) by the general methods shown for Preparation IK. Obtained a mixture of Preparation 2E and Preparation 2F (1.18 g, 99%): HPLC: RT=2.885 min (CHROMOLITH® ODS 4.6 x 50 mm (4 min grad) eluting with 10-90% aqueous MeOH over 4 minutes containing 0.% TFA, 4 mL/min, monitoring at 220 nm). MS (ES): m/z= 544.2 [M+H]+.
Example 2:
[00249] Example 2 was prepared from a mixture of Preparation 2E and Preparation 2F (354 mg, 0.651 mmol) by the general methods shown for Example 1. After separation of the diastereoisomers, Example 2 was obtained (188 mg, 52%) as a white solid: HPLC: RT=9.063 min (H20/CH3CN with TFA, Sunfire C18 3.5um, 4.6x150mm, 4.6x150mm, gradient = 15 min, wavelength = 220 and 254 nm); MS (ES): m/z= 543 [M+H]+; XH NMR (400 MHz, DMSO-d6) δ ppm 10.87 (1 H, br. s.), 9.50-9.55 (1 H, m), 7.62-7.69 (2 H, m), 7.40-7.57 (5 H, m), 7.29-7.36 (2 H, m), 7.22-7.28 (1 H, m), 7.16 (1 H, br. s.), 5.25 (1 H, d), 3.30-3.32 (1 H, m), 2.75-2.86 (1 H, m), 2.44-2.48 (1 H, m), 2.06-2.34 (3 H, m), 1.51- 1.77 (4 H, m); [a]D = -114.4° (8.04 mg/mL, DMSO).
[00250] Crystal Form M2- 1 was prepared by adding approximately 1 mg of Example 2 to approximately 0.7 mL of MeOH/fluorobenzene solution (3 : 1). Colorless plate-like crystals were obtained after 2 days of solvent evaporation at room temperature.
To a stirred solution of 5,5,5-trifluoropentanoic acid (5 g, 32.0 mmol) in THF (30 mL) and hexane (30 mL) at 0° C., was added tert-butyl 2,2,2-trichloroacetimidate (11.46 mL, 64.1 mmol). The mixture was stirred for 15 min at 0° C. Boron trifluoride etherate (0.406 mL, 3.20 mmol) was added and the reaction mixture was allowed to warm to room temperature overnight. To the clear reaction mixture was added solid NaHCO3 (5 g) and stirred for 30 min. The mixture was filtered through MgSO4 and washed with hexanes (200 mL). The solution was allowed to rest for 45 min, and the resulting solid material was removed by filtering on the same MgSO4 filter again, washed with hexanes (100 mL) and concentrated under reduced pressure without heat. The volume was reduced to about 30 mL, filtered through a clean fitted funnel, washed with hexane (5 mL), and then concentrated under reduced pressure without heat. The resulting neat oil was filtered through a 0.45 μm nylon membrane filter disk to provide tert-butyl 5,5,5-trifluoropentanoate (6.6 g, 31.4 mmol 98% yield) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ ppm 1.38 (s, 9H) 1.74-1.83 (m, 2H) 2.00-2.13 (m, 2H) 2.24 (t, J=7.28 Hz, 2H).
To a stirred solution of 5,5,5-trifluoropentanoic acid (5.04 g, 32.3 mmol) in DCM (50 mL) and DMF (3 drops) was added oxalyl chloride (3.4 mL, 38.8 mmol) dropwise over 5 min and the solution was stirred until all bubbling subsided. The reaction mixture was concentrated under reduced pressure to give pale yellow oil. To a separate flask charged with a solution of (4S)-4-(propan-2-yl)-1,3-oxazolidin-2-one (4.18 g, 32.4 mmol) in THF (100 mL) at −78° C. was added n-BuLi (2.5M in hexane) (13.0 mL, 32.5 mmol) dropwise via syringe over 5 min. After stirring for 10 min, the above acid chloride dissolved in THF (20 mL) was added via cannula over 15 min. The reaction mixture was warmed to 0° C., and was allowed to warm to room temperature as the bath warmed and stirred overnight. To the reaction mixture was added saturated NH4Cl, and then extracted with EtOAc (2×). The combined organics were washed with brine, dried (Na2SO4), filtered and concentrated under reduced pressure. The crude material was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 5% to 60% solvent A/B=hexanes/EtOAc, REDISEP® SiO2 120 g). Concentration of appropriate fractions provided Preparation 1B (7.39 g, 86%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ ppm 4.44 (1H, dt, J=8.31, 3.53 Hz), 4.30 (1H, t, J=8.69 Hz), 4.23 (1H, dd, J=9.06, 3.02 Hz), 2.98-3.08 (2H, m), 2.32-2.44 (1H, m, J=13.91, 7.02, 7.02, 4.03 Hz), 2.13-2.25 (2H, m), 1.88-2.00 (2H, m), 0.93 (3H, d, J=7.05 Hz), 0.88 (3H, d, J=6.80 Hz).
Preparation 1C: (2S,3R)-tert-Butyl 6,6,6-trifluoro-3-((S)-4-isopropyl-2-oxooxazolidine-3-carbonyl)-2-(3,3,3-trifluoropropyl)hexanoate, and
To a cold (−78° C.), stirred solution of diisopropylamine (5.3 mL, 37.2 mmol) in THF (59 mL) under nitrogen atmosphere was added n-BuLi (2.5M in hexane) (14.7 mL, 36.8 mmol), then warmed to 0° C. to give a 0.5M solution of LDA. A separate vessel was charged with Preparation 1B (2.45 g, 9.17 mmol), the material was azeotroped twice with benzene (the RotoVap air inlet was fitted with nitrogen inlet to completely exclude humidity) then toluene (15.3 mL) was added. This solution was added to a flask containing dry lithium chloride (1.96 g, 46.2 mmol). To the resultant mixture, cooled to −78° C., was added LDA solution (21.0 mL, 10.5 mmol) and stirred at −78° C. for 10 min, warmed to 0° C. for 10 min then recooled to −78° C. To a separate reaction vessel containing Preparation 1A (3.41 g, 16.07 mmol), also azeotroped twice with benzene, was added toluene (15.3 mL), cooled to −78° C. and LDA (37.0 mL, 18.5 mmol) was added, the resulting solution was stirred at −78° for 25 min. At this time the enolate derived from the ester was transferred via cannula into the solution of the oxazolidinone enolate, stirred at −78° C. for an additional 5 min at which time the septum was removed and solid powdered bis(2-ethylhexanoyloxy)copper (9.02 g, 25.8 mmol) was rapidly added to the reaction vessel and the septum replaced. The vessel was immediately removed from the cold bath and immersed into a warm water bath (40° C.) with rapid swirling with a concomitant color change from the initial turquoise to brown. The reaction mixture was stirred for 20 min, was poured into 5% aqueous NH4OH (360 mL) and extracted with EtOAc (2×). The combined organics were washed with brine, dried (Na2SO4), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 0% to 60% solvent A/B=hexanes/EtOAc, REDISEP® SiO2 120 g). Concentration of appropriate fractions provided Preparation 1C (2.87 g, 66%) as pale yellow viscous oil. 1H NMR showed the product was a 1.6:1 mixture of diastereoisomers 1C:1D as determined by the integration of the multiplets at 2.74 & 2.84 ppm: 1H NMR (400 MHz, CDCl3) δ ppm 4.43-4.54 (2H, m), 4.23-4.35 (5H, m), 4.01 (1H, ddd, J=9.54, 6.27, 3.51 Hz), 2.84 (1H, ddd, J=9.41, 7.28, 3.64 Hz), 2.74 (1H, ddd, J=10.29, 6.27, 4.02 Hz), 2.37-2.48 (2H, m, J=10.38, 6.98, 6.98, 3.51, 3.51 Hz), 2.20-2.37 (3H, m), 1.92-2.20 (8H, m), 1.64-1.91 (5H, m), 1.47 (18H, s), 0.88-0.98 (12H, m).
Preparation 1E: (2R,3S)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid, and
To a cool (0° C.), stirred solution of Preparation 1C and 1D (4.54 g, 9.51 mmol) in THF (140 mL) and water (42 mL) was sequentially added hydrogen peroxide (30% in water) (10.3 g, 91 mmol) and LiOH (685.3 mg, 28.6 mmol) and the mixture was stirred for 1 hr. At this time the reaction vessel was removed from the cold bath and then stirred for 1.5 hr. The reaction was judged complete by HPLC. To the reaction mixture was added saturated NaHCO3(45 mL) and saturated Na2SO3 (15 mL), and then partially concentrated under reduced pressure. The resulting crude solution was extracted with DCM (3×). The aqueous phase was acidified to pH-1-2 with 1N HCl, extracted with DCM (3×) and EtOAc (1×). The combined organics were washed with brine, dried (Na2SO4), filtered and concentrated under reduced pressure to provide a mixture of Preparation 1E and 1F (3.00 g, 86%) as colorless oil: 1H NMR (400 MHz, CDCl3) δ ppm 2.76-2.84 (1H, m, diastereoisomer 2), 2.64-2.76 (3H, m), 2.04-2.35 (8H, m), 1.88-2.00 (4H, m), 1.71-1.83 (4H, m), 1.48 (9H, s, diastereoisomer 1), 1.46 (9H, s, diastereoisomer 2); 1H NMR showed a 1.7:1 mixture of 1E:1F by integration of the peaks for the t-butyl groups.
Preparation 1E: (2R,3S)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid, and
To a cold (−78° C.), stirred solution of diisopropylamine (1.7 mL, 11.93 mmol) in THF (19 mL) under nitrogen atmosphere was added n-BuLi (2.5M in hexanes) (4.8 mL, 12.00 mmol). The mixture was stirred for 5 min and then warmed to 0° C. In a separate vessel, to a cold (−78° C.) stirred solution of the mixture of Preparation 1E and 1F (1.99 g, 5.43 mmol) in THF (18 mL) was added the LDA solution prepared above via cannula slowly over 25 min. The mixture was stirred for 15 min, then warmed to room temperature (placed in a 24° C. water bath) for 15 min, and then again cooled to −78° C. for 15 min. To the reaction mixture was added Et2AlCl (1M in hexane) (11.4 mL, 11.40 mmol) via syringe, stirred for 10 min, warmed to room temperature for 15 min and then cooled back to −78° C. for 15 min. Methanol (25 mL) was rapidly added, swirled vigorously while warming to room temperature, then concentrated to ˜¼ original volume. The mixture was dissolved in EtOAc and washed with 1N HCl (50 mL) and ice (75 g). The aqueous phase was separated, extracted with EtOAc (2×). The combined organics were washed with a mixture of KF (2.85 g in 75 mL water) and 1N HCl (13 mL) [resulting solution pH 3-4], then with brine, dried (Na2SO4), filtered and concentrated under reduced pressure to give a 9:1 (1E:1F) enriched diastereoisomeric mixture (as determined by 1H NMR) of Preparation 1E and Preparation 1F (2.13 g, >99%) as a pale yellow viscous oil: 1H NMR (400 MHz, CDCl3) δ ppm 2.64-2.76 (2H, m), 2.04-2.35 (4H, m), 1.88-2.00 (2H, m), 1.71-1.83 (2H, m), 1.48 (9H, s).
Preparation 1G: (3S)-3-Amino-1-methyl-5-phenyl-1,3-dihydro-2H-1,4-benzodiazepin-2-one, and
Racemic 3-amino-1-methyl-5-phenyl-1,3-dihydro-2H-1,4-benzodiazepin-2-one (Rittle, K. E. et al., Tetrahedron Letters, 28(5):521-522 (1987)) was prepared according to the literature procedure. The enantiomers were separated under chiral-SFC conditions using the following method: CHIRALPAK® AS-H 5×25; Mobile phase: 30% MeOH+0.1% DEA in CO2; Flow rate: 280 mL/min; Pressure: 100 bar; Temperature: 35° C.
Obtained the S-enantiomer (Preparation 1G): HPLC: RT=1.75 min (30% MeOH+0.1% DEA in CO2 on CHIRALPAK® AS-H 4.6×250 mm, 3 mL/min, 35° C., 100 bar, 230 nm, 10 μl injection); 1H NMR (400 MHz, CDCl3) δ ppm 7.58-7.63 (2H, m), 7.55 (1H, ddd, J=8.50, 7.11, 1.76 Hz), 7.40-7.47 (1H, m), 7.34-7.40 (3H, m), 7.31 (1H, dd, J=7.81, 1.51 Hz), 7.14-7.22 (1H, m), 4.46 (1H, s), 3.44 (3H, s), 3.42 (2H, s); [α]D=−155° (c=1.9, MeOH) (Lit. Rittle, K. E. et al.,Tetrahedron Letters, 28(5):521-522 (1987): [α]D=−236°).
Also obtained the R-enantiomer (Preparation 1H): HPLC: RT=1.71 min; [α]D=+165° (c=2.1, MeOH) (Lit [α]D=+227°).
Alternate Procedure to Make Preparation 1G
Preparation 1G•CSA salt: (3S)-3-Amino-1-methyl-5-phenyl-1,3-dihydro-2H-1,4-benzodiazepin-2-one, (1 S)-(+)-10-camphorsulfonic acid salt
Preparation 1G•CSA was prepared from racemic 3-amino-1-methyl-5-phenyl-1,3-dihydro-2H-1,4-benzodiazepin-2-one (9.98 g, 37.6 mmol) (prepared according to the literature as shown above) according to the literature procedure (Reider, P. J. et al., J. Org. Chem., 52:955-957 (1987)). Preparation 1G•CSA (16.91 g, 99%) was obtained as a colorless solid: Optical Rotation: [α]D=−26.99° (c=1, H2O) (Lit. [α]D=−27.8° (c=1, H2O))
Preparation 1I: tert-Butyl (2S,3R)-6,6,6-trifluoro-3-(((3S)-1-methyl-2-oxo-5-phenyl-2,3-dihydro-1H-1,4-benzodiazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoate, and
To a stirred solution of Preparation 1G (1.45 g, 5.47 mmol) and a 9:1 mixture of Preparation 1E and 1F (1.989 g, 5.43 mmol) in DMF (19 mL) was added 0-benzotriazol-1-yl-N,N,N′,N′-tetra-methyluronium tetrafluoroborate (1.79 g, 5.57 mmol) and triethylamine (3.0 mL, 21.52 mmol) and stirred overnight. The reaction was judged complete by LCMS. The reaction mixture was poured into water (125 mL) and the precipitated solid was collected by filtration, washed with water and air dried to provide an 8:1 mixture of Preparation 1I and Preparation 1J (2.95 g, 89%) as a cream solid: MS (ES): m/z=614 [M+H]+; 1H NMR (400 MHz, CDCl3) δ ppm 7.55-7.65 (3H, m), 7.44-7.52 (2H, m), 7.35-7.45 (4H, m), 5.52 (1H, d, J=8.03 Hz), 3.48 (3H, s), 2.63 (2H, ddd, J=9.35, 3.95, 3.76 Hz), 2.14-2.25 (4H, m), 1.90-2.03 (3H, m), 1.69-1.82 (1H, m), 1.51 (9H, s).
Preparation 1K: (2S,3R)-6,6,6-Trifluoro-3-(((3S)-1-methyl-2-oxo-5-phenyl-2,3-dihydro-1H-1,4-benzodiazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoic acid, and
To a cool (0° C.), stirred solution of the above mixture of Preparation 1I and Preparation 1J (2.95 g, 4.81 mmol) in DCM (20 mL) was added TFA (20 mL, 260 mmol). The reaction mixture was stirred for 1 hr, then allowed to warm to room temperature and stirred for 2.5 hr. The reaction was judged complete by LCMS. The reaction mixture was diluted with toluene (50 mL) and concentrated under reduced pressure. The residue mixture was redissolved in toluene (50 mL) and concentrated under reduced pressure then dried under high vacuum. The crude product was dissolved in DCM, SiO2(15 g) was added, concentrated, then was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 0% to 45% solvent A/B=DCM/EtOAc, REDISEP® SiO2 80 g). Concentration of appropriate fractions provided a mixture of Preparation 1K and Preparation 1L (2.00 g, 75%) as a cream solid: HPLC: RT=2.770 min (CHROMOLITH® SpeedROD 4.6×50 mm (4 min grad) eluting with 10-90% aqueous MeOH over 4 minutes containing 0.1% TFA, 4 mL/min, monitoring at 254 nm); MS (ES): m/z=558 [M+H]+; 1H NMR (400 MHz, CDCl3) δ ppm 8.32 (1H, d, J=8.03 Hz), 7.65-7.71 (1H, m), 7.50-7.60 (3H, m), 7.41-7.49 (2H, m), 7.39 (1H, dd, J=7.91, 1.63 Hz), 7.23-7.35 (2H, m), 5.59 (1H, d, J=8.03 Hz), 3.51 (3H, s), 2.81 (1H, ddd, J=10.54, 6.90, 3.64 Hz), 2.67-2.76 (1H, m), 2.22-2.33 (3H, m), 1.99-2.12 (3H, m), 1.85-1.94 (1H, m), 1.79 (1H, ddd, J=13.87, 7.84, 3.64 Hz).
Example 1
To a stirred solution of an 8:1 mixture of Preparation 1K and Preparation 1L (3.46 g, 6.21 mmol) in DMF (25 mL) under nitrogen atmosphere was added ammonium chloride (3.32 g, 62.1 mmol), EDC (3.55 g, 18.52 mmol), HOBT (2.85 g, 18.61 mmol), and triethyl amine (16 mL, 115 mmol) and stirred overnight. The reaction was judged complete by LCMS. The reaction mixture was poured into water (200 mL) with vigorous swirling and then allowed to sit. The solid was collected by filtration, washed with water, allowed to dry to afford 3.6 g colorless solid. The solid was purified by preparative SFC chromatography (Lux-Cellulose-2 (3×25 cm), 8% methanol in CO2, 140 ml/min @220 nm and 35° C.; Sample: 3.6 g in 50 cc methanol, conc.=70 mg/ml, Stack injection: 0.5 cc/9.2 min). Fractions containing product were concentrated, dried overnight under vacuum. Obtained Example 1 (2.74 g, 79%) as a colorless solid (Crystal Form N-1): HPLC: RT=9.601 min (H2O/CH3CN with TFA, Sunfire C18 3.5 um, 4.6×150 mm, 4.6×150 mm, gradient=15 min, wavelength=220 and 254 nm). MS (ES): m/z=557 [M+H]+; 1H NMR (400 MHz, DMSO-d6) δ ppm 9.54 (1H, d, J=7.28 Hz), 7.71-7.80 (1H, m), 7.68 (2H, d, J=8.78 Hz), 7.50-7.62 (3H, m), 7.45 (2H, t, J=7.28 Hz), 7.29-7.40 (2H, m), 7.15 (1H, br. s.), 5.30 (1H, d, J=7.28 Hz), 3.39 (3H, s), 2.74-2.86 (1H, m), 2.02-2.32 (3H, m), 1.45-1.79 (4H, m); [α]D=−107.0° (5.73 mg/mL, DMSO).
Crystal Form A-2 was prepared by adding approximately 1 mg of Example 1 to approximately 0.7 mL of acetone/acetonitrile/water solution (2:2:1). A mixture of colorless needles and thin blades crystals were obtained after one day of slow evaporation of the solution at room temperature. The thin blade crystals were separated to provide crystal Form A-2.
Crystal Form EA-3 was prepared by adding approximately 1 mg of Example 1 to approximately 0.7 mL of ethyl acetate/heptane solution (1:1). Colorless blade crystals were obtained after three days of slow evaporation of the solution at room temperature.
Crystal Form THF-2 was obtained by adding approximately 5 mg of Example 1 to approximately 0.7 mL of THF/water solution (4:1). Colorless blade-like crystals were obtained after one day of solvent evaporation at room temperature.
To a cold (−25° C.), stirred solution of 2,6-lutidine (18.38 mL, 158 mmol) in CH2Cl2 (120 mL) was added Tf2O (24.88 mL, 147 mmol) over 3 min, and stirred for 5 min. To the reaction mixture was added 3,3,3-trifluoropropan-1-ol (12 g, 105 mmol) over an interval of 3 min. After 2 hr, the reaction mixture was warmed to room temperature and stirred for 1 hr. The reaction mixture was concentrated to half volume, then purified by loading directly on silica gel column (330 g ISCO) and eluted with CH2Cl2. Obtained Preparation 1M (13.74 g, 53%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ ppm 4.71 (2H, t, J=6.15 Hz), 2.49-2.86 (2H, m).
Preparation 1N was prepared from 5,5,5-trifluoropentanoic acid (3.35 g, 21.46 mmol) and (4S)-4-benzyl-1,3-oxazolidin-2-one (3.80 g, 21.46 mmol) by the general methods shown for Preparation 1B. Preparation 1N (5.67 g, 84%) was obtained as a colorless viscous oil: 1H NMR (400 MHz, CDCl3) δ ppm 7.32-7.39 (2H, m), 7.30 (1H, d, J=7.05 Hz), 7.18-7.25 (2H, m), 4.64-4.74 (1H, m), 4.17-4.27 (2H, m), 3.31 (1H, dd, J=13.35, 3.27 Hz), 3.00-3.11 (2H, m), 2.79 (1H, dd, J=13.35, 9.57 Hz), 2.16-2.28 (2H, m), 1.93-2.04 (2H, m).
To a cold (−78° C.), stirred solution of Preparation 1N (3.03 g, 9.61 mmol) in THF (20 mL) was added NaHMDS (1.0M in THF) (10.6 mL, 10.60 mmol) under nitrogen atmosphere. After 2 hours, tert-butyl 2-bromoacetate (5.62 g, 28.8 mmol) was added neat via syringe at −78° C. and stirring was maintained at the same temperature. After 6 hours, the reaction mixture was warmed to room temperature. The reaction mixture was partitioned between saturated NH4Cl and EtOAc. The organic phase was separated, and the aqueous was extracted with EtOAc (3×). The combined organics were washed with brine, dried (Na2SO4), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 5% to 100% solvent A/B=hexanes/EtOAc, REDISEP® SiO2 120 g). Concentration of appropriate fractions provided Preparation 10 (2.79 g, 67.6%) as a colorless viscous oil: 1H NMR (400 MHz, CDCl3) δ ppm 7.34 (2H, d, J=7.30 Hz), 7.24-7.32 (3H, m), 4.62-4.75 (1H, m, J=10.17, 6.89, 3.43, 3.43 Hz), 4.15-4.25 (3H, m), 3.35 (1H, dd, J=13.60, 3.27 Hz), 2.84 (1H, dd, J=16.62, 9.57 Hz), 2.75 (1H, dd, J=13.35, 10.07 Hz), 2.47 (1H, dd, J=16.62, 4.78 Hz), 2.11-2.23 (2H, m), 1.90-2.02 (1H, m), 1.72-1.84 (1H, m), 1.44 (9H, s).
Preparation 1P was prepared from Preparation 1O (2.79 g, 6.50 mmol) by the general methods shown for Preparation 1E. Preparation 1P (1.45 g, 83%) was obtained as a colorless oil: 1H NMR (400 MHz, CDCl3) δ ppm 2.83-2.95 (1H, m), 2.62-2.74 (1H, m), 2.45 (1H, dd, J=16.62, 5.79 Hz), 2.15-2.27 (2H, m), 1.88-2.00 (1H, m), 1.75-1.88 (1H, m), 1.45 (9H, s).
Preparation 1E: (2R,3S)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid, and
To a cold (−78° C.), stirred solution of Preparation 1P (5.44 g, 20.13 mmol) in THF (60 mL) was slowly added LDA (24.60 mL, 44.3 mmol) over 7 min. After stirring for 2 hr, Preparation 1M (6.44 g, 26.2 mmol) was added to the reaction mixture over 3 min. After 45 min, the reaction mixture was warmed to −25° C. bath (ice/MeOH/dry ice) for 1 hr, and then warmed to 0° C. After 45 min, Preparation 1M (1 g) was added and the reaction mixture was stirred for 20 min. The reaction was quenched with water and 1N NaOH and was extracted with CH2Cl2. The organic layer was again extracted with 1N NaOH (2×) and the aqueous layers were combined. The aqueous layer was cooled in ice/water bath and then acidified with concentrated HCl to pH 2. Next, the aqueous layer was extracted with EtOAc. The combined organics were washed with brine, dried over anhydrous sodium sulphate, and concentrated under reduced pressure. The residue was dried under high vacuum to provide a 1:5 (1E:1F) mixture (as determined by 1H NMR) of Preparation 1E and Preparation 1F (5.925 g, 80%) as a pale yellow solid. 1H NMR (500 MHz, CDCl3) δ ppm 2.81 (1H, ddd, J=10.17, 6.32, 3.85 Hz), 2.63-2.76 (1H, m), 2.02-2.33 (4H, m), 1.86-1.99 (2H, m), 1.68-1.85 (2H, m), 1.47 (9H, s).
Preparation 1E: (2R,3S)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid, and
A mixture of Preparation 1E and Preparation 1F (64 mg, 1.758 mmol) was taken in THF (6 mL) to give a colorless solution which was cooled to −78° C. Then, LDA (2.149 mL, 3.87 mmol) (1.8M in heptane/THF/ethylbenzene) was slowly added to the reaction mixture over 10 min. After stirring for 15 min the reaction mixture was placed in a room temperature water bath. After 15 min the reaction mixture was placed back in −78° C. bath and then diethylaluminum chloride (3.87 mL, 3.87 mmol) (1M in hexane) was added slowly over 5 min. The reaction mixture was stirred at −78° C. After 15 min the reaction mixture was placed in a room temperature water bath for 10 min and then cooled back to −78° C. bath. After 15 min the reaction was quenched with MeOH (8 mL, 198 mmol), removed from the −78° C. bath and concentrated. To the reaction mixture was added ice and HCl (16 mL, 16.00 mmol), followed by extraction with EtOAc (2×). The organic layer was washed with potassium fluoride (920 mg, 15.84 mmol) (in 25 mL H2O) and HCl (4.5 mL, 4.50 mmol). The organics were dried over anhydrous magnesium sulphate and concentrated under reduced pressure to provide a 9:1 (1E:1F) enriched mixture of Preparation 1E and Preparation 1F (540 mg, 1.583 mmol, 90% yield) as light yellow/orange solid. 1H NMR (400 MHz, CDCl3) δ ppm 2.64-2.76 (2H, m), 2.04-2.35 (4H, m), 1.88-2.00 (2H, m), 1.71-1.83 (2H, m), 1.48 (9H, s). It was converted to Example 1 by the sequence of reactions as outlined above.
A clean and dry 5 L four neck round bottom flask equipped with mechanical stirring, thermometer socket and nitrogen bubbler at room temperature was charged with N,N-dimethyl formamide (2.07 L), a 1.2:1 mixture of Preparation 1E and Preparation 1F (207 g, 0.5651 moles), potassium carbonate (117.1 g, 0.8476 moles) followed by benzyl bromide (116 g, 0.6781 moles) over 15-20 min. The reaction mixture was stirred for 2-3 hr. After completion of the reaction, the reaction mixture was concentrated to dryness at 50-55° C. under vacuum. Ethyl acetate (3.1 L, 30 Vol.) was charged into the concentrated reaction mass and then washed with water (2.07 L), brine (0.6 L) then dried over anhydrous sodium sulfate (207 g), filtered and concentrated to dryness at 40-45° C. under vacuum. The residue was dissolved in dichloromethane (1.035 L, 5 vol.) and then absorbed onto silica gel (60-120) (607 g, 3.0 w/w), then was purified with column chromatography using petroleum ether and ethyl acetate as solvents. After pooling several batches, Preparation 1Q (235 g) was obtained. HPLC purity: 99.77%,
A clean and dry 2 L autoclave was charged with methanol (540 mL) and was purged with nitrogen for 5-10 minutes. To the autoclave was added 10% palladium on carbon (12 g, 20%), purged with nitrogen once again for 5-10 min then was charged with Preparation 1Q (60 g, 0.1315 moles), the autoclave was flushed with methanol (60 mL) and stirred for 4-6 hr at 20-25° C. under 5 Kg hydrogen pressure. After completion of the reaction, the reaction mass was filtered through CELITE®, washed with methanol (180 mL), dried with anhydrous sodium sulfate (60 g), filtered and concentrated to dryness at 45-50° C. under vacuum. Obtained Preparation 1E (45.8 g, 95%) as a colorless solid: HPLC purity: 98.9%.
Preparation 1E was prepared in a procedure identical as above from a mixture of Preparations 1E and 1F (200 g, 0.5460 moles) using LDA (1.8 M solution in THF, ethyl benzene and heptane) (698 mL, 2.3 equiv.) and diethyl aluminum chloride (1.0 M solution in hexane) (1256 mL, 2.3 equiv) in THF (2.0 L). After workup as explained above, the resulting residue was treated as follows: The crude material was added to a 2 L four neck round bottom flask, followed by the addition of MTBE (1.0 L) charged below 30° C. The resulting mixture was stirred for 5-10 minutes to obtain a clear solution. Hexanes (600 mL) was charged to the reaction mixture at a temperature below 30° C. The reaction mixture was stirred for 10 min. Next, tert-butylamine (43.8 g, 1.1 eq) was charged slowly over a period of 15 minutes below 30° C. This addition was observed to be exothermic. The reaction mixture was stirred for 2 hrs below 30° C. and filtered. The solid material was washed with 5:3 MTBE: hexane (200 mL), the filtrate was concentrated and transferred to an amber color bottle. The filtered solid was dissolved in dichloromethane (2.0 L), washed with 1N HCl (2.0), the organic layer was washed with brine (1.0 L×2), then was concentrated under reduced pressure below 45° C. This material was found to be 91.12% pure. The material was repurified by the above t-butylamine crystallization purification procedure. Obtained Preparation 1E (78 g, 39%): HPLC purity: 99.54%.
A clean and dry 2 L four neck round bottom flask equipped with mechanical stirring, thermometer socket and nitrogen bubbler was charged with N,N-dimethylformamide (457 mL), Preparation 1E (45.7 g, 0.1248 moles) and Preparation 1G•CSA (62.08 g, 0.1248 moles) under nitrogen atmosphere at 20-25° C. The reaction mixture was stirred for 15-20 minutes to make clear solution at 20-25° C. To the reaction mixture was added TBTU (48.16 g, 0.1498 moles) at 20-25° C. followed by triethylamine (50.51 g, 0.4992 moles) over 15-20 minutes at 20-25° C. The reaction mixture was stirred for 60-120 minutes at 20-25° C. under nitrogen atmosphere. After completion of the reaction, the reaction was quenched into water (1.37L, 30 Vol.) at 20-25° C. under stirring. The reaction mixture was stirred for 30 minutes at 20-25° C. The reaction mixture was filtered and washed with water (228 mL). The resulting solid material was dissolved in ethyl acetate (457 mL), washed with water (2×137 mL), brine (137 mL), and then dried with anhydrous sodium sulfate (45.7 g). Activated charcoal (9.14 g, 20%) was charged into the reaction mixture and stirred for 30 minutes. The mixture was filtered through CELITE® bed and 1 micron filter cloth, washed charcoal bed with ethyl acetate (137 mL), concentrated to 1.0 Vol. stage and then petroleum ether (457 mL, 10 Vol.) was charged and stirred for 30 minutes at 20-25° C. The solid was collected by filtration, washed with petroleum ether (137 mL) and then dried under vacuum at 40-45° C. for 8 hr until loss on drying was less than 3.0%. Obtained Preparation 11 (65.2 g, 85%): HPLC purity: 98.26%.
A clean and dry 3 L four neck round bottom flask equipped with mechanical stirring, thermometer socket and nitrogen bubbler was charged with dichloromethane (980 mL) under nitrogen atmosphere followed by Preparation 1I (140 g, 0.2282 moles) at 20-25° C. The reaction mixture was cooled to 0-5° C. and trifluoroacetic acid (980 mL) was charged slowly for 30-40 minutes. The resulting mixture was stirred for 2 hr at 0-5° C. under nitrogen atmosphere. The reaction temperature was raised to 20 to 25° C., and the reaction mixture was stirred for 1-2 hr at 20 to 25° C. After completion of the reaction, the reaction mixture was concentrated to dryness at 50 to 55° C. under vacuum. Toluene (3×700 mL,) was charged into the concentrated reaction mass, and then distilled off at 50 to 55° C. under vacuum. After complete concentration from toluene, ethyl acetate (280 mL) was charged into the reaction mass at 20 to 25° C., stirred for 60 minutes, then the solid was collected by filtration, washed with ethyl acetate (140 mL), dried under vacuum at 50 to 55° C. for 12 hr until loss on drying was less than 2.0%. Obtained Preparation 1K (106 g, 84%): HPLC purity: 98.43%.
Example 1
A reaction vessel was charged with Preparation 1K (30 g, 53.81 mmol), HOBt (8.7 g, 64.38 mmol), and THF (150 mL) at room temperature. To the homogeneous solution was added EDCI (12.4 g, 64.68 mmol), stirred for 15 min, then cooled to 8° C. To the reaction mixture was added ammonia (2M in IPA) (81 mL, 162 mmol) over 5 min so as to maintain a temperature below 10° C. The resulting heavy slurry was stirred for 10 min, warmed to room temperature over 30 min, then stirred for 4 hr. At the completion of the reaction, water (230 mL) was slowly added over 15 min to maintain a temperature below 20° C., and then stirred for 2 hr. The solid was collected by filtration, washed with water (3×60 mL), then dried under vacuum 48 hr at 55° C. The above crude product was charged into a 1 L 3-necked round flask. IPA (200 mL) was added, then heated to 80° C. resulting in a homogeneous solution. Water (170 mL) was slowly added (15 min) to maintain an internal temperature>75° C. The resulting slurry was stirred and cooled to room temperature for 2 hr. The solid was collected by filtration, washed with water (2×50 mL), then dried under vacuum (55° C. for 24 h, and 30° C. for 48 h). Obtained Example 1 (23.4 g, 78% yield): HPLC purity: 99.43%.
For some disease targets, an indirect approach may be best. Or so Ashvinikumar V. Gavai and his colleagues atBristol-Myers Squibbfound in their quest toward a potential cancer drug. Gavai unveiled BMS-906024, which is an experimental—and slightly roundabout—treatment for a number of cancers, including breast, lung, and colon cancers, and leukemia.
Cancers have a tendency to relapse or to become resistant to treatments that once worked. Research at BMS and elsewhere had suggested that a family of proteins called Notch is implicated in that resistance and in cancer progression more generally. Gavai, director of oncology chemistry at BMS in Princeton, N.J., and his team set out to block Notch family signaling.
Notch family members lack enzymatic activity, so blocking them directly is difficult. Instead, BMS developed inhibitors of an enzyme that is essential for activating Notch signaling—γ-secretase.
Company: Bristol-Myers Squibb
Target: pan-Notch
Disease: breast, lung, colon cancer; leukemia
Interfering with Notch, even in this indirect way, can have detrimental effects on the gastrointestinal tract. Only two of the four Notch family members are linked to that side effect, Gavai says. But he and his team think their drug will be most effective if it acts on all four family members roughly equally—a so-called pan-Notch inhibitor. By selecting a molecule that’s well tolerated in animals and carefully scheduling doses of the drug in humans, it could be possible to minimize side effects, he says.
The BMS team relied on Notch signaling assays in leukemia and breast cancer cell lines to find leads. They soon learned that for their molecules to work, three chiral centers had to be in the S,R,Sconfiguration. After that, they strove to make the molecules last in the bloodstream. They removed an isobutyl group and tweaked some other parts of their candidate’s succinamide side chain. It was tough to retain both a long half-life and activity against Notch, Gavai told C&EN. “You’d optimize one and lose the other.”
His team threaded the needle with BMS-906024. Their studies with mice suggest that a dose of 4–6 mg once a week could be effective in people. That’s lower than doses being tested for other Notch-targeted agents, according to the website clinicaltrials.gov. The mouse studies also back the idea that Notch is involved in cancer drug resistance and suggest that Notch could be a target for taking on cancer stem cells, which are notoriously resistant to chemotherapy.
BMS-906024 is in Phase I clinical trials, both alone and in combination with other agents. Patients with colon, lung, breast, and other cancers are receiving intravenous doses of the compound to determine its safety and optimum dose ranges.
(From left, front row) Gavai, Weifeng Shan, (second row) Aaron Balog, Patrice Gill, Gregory Vite, (third row) Francis Lee, Claude Quesnelle, (rear row) Wen-Ching Han, Richard Westhouse.
BMS-906024
Company: Bristol-Myers Squibb
Meant to treat: cancers including breast, lung, colon, and leukemia
Mode of action: pan-Notch inhibitor
Medicinal chemistry tidbit: The BMS team used an oxidative enolate heterocoupling en route to the candidate– a procedure from Phil Baran’s lab at Scripps Research Institute. JACS 130, 11546
Status in the pipeline: Phase I
Relevant documents: WO 2012/129353
PAPER
An enantioselective synthesis of (S)-7-amino-5H,7H-dibenzo[b,d]azepin-6-one (S–1) is described. The key step in the sequence involved crystallization-induced dynamic resolution (CIDR) of compound 7 using Boc-d-phenylalanine as a chiral resolving agent and 3,5-dichlorosalicylaldehyde as a racemization catalyst to afford S–1 in 81% overall yield with 98.5% enantiomeric excess.
Crystallization-Induced Dynamic Resolution toward the Synthesis of (S)-7-Amino-5H,7H-dibenzo[b,d]-azepin-6-one: An Important Scaffold for γ-Secretase Inhibitors
† Department of Discovery Synthesis, Biocon Bristol-Myers Squibb Research Centre, Biocon Park, Bommasandra IV Phase, Jigani Link Road, Bengaluru 560099, India
‡Bristol-Myers Squibb Company, P.O Box 4000, Princeton, New Jersey 08543-4000, United States
1. Quesnelle, Claude; Kim, Soong-Hoon; Lee, Francis; Gavai, Ashvinikumar. Bis(fluoroalkyl)-1,4-benzodiazepinone compounds as Notch receptor inhibitors and their preparation and use in the treatment of cancer. PCT Int. Appl. (2012), WO 2012129353 A1 20120927.
USE OF DIANHYDROGALACTITOL AND ANALOGS OR DERIVATIVES THEREOF IN COMBINATION WITH PLATINUM-CONTAINING ANTINEOPLASTIC AGENTS TO TREAT NON-SMALL-CELL CARCINOMA OF THE LUNG AND BRAIN METASTASES

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....












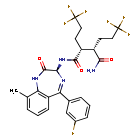
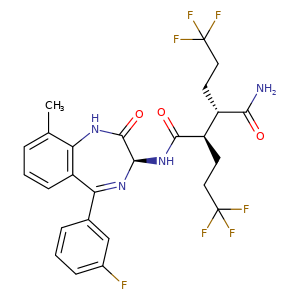
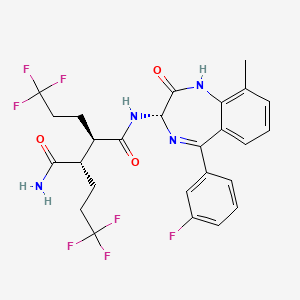
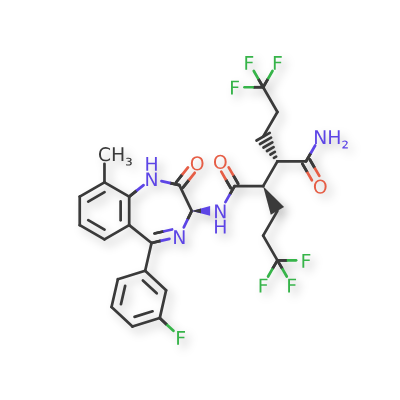












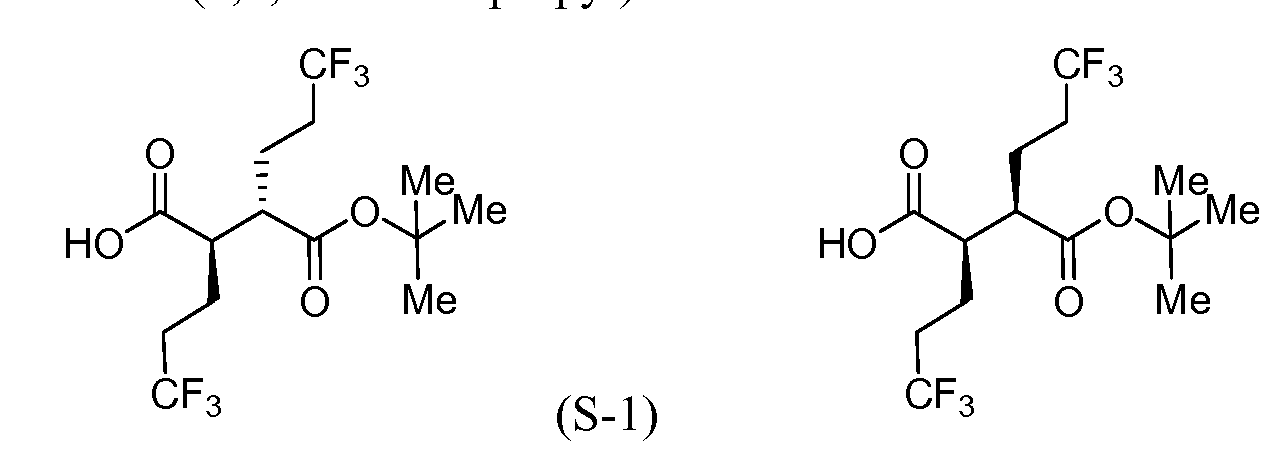





























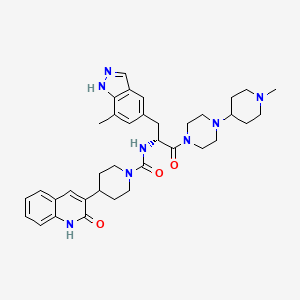
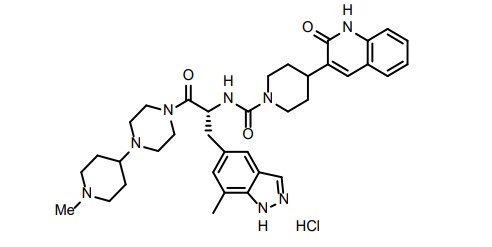



















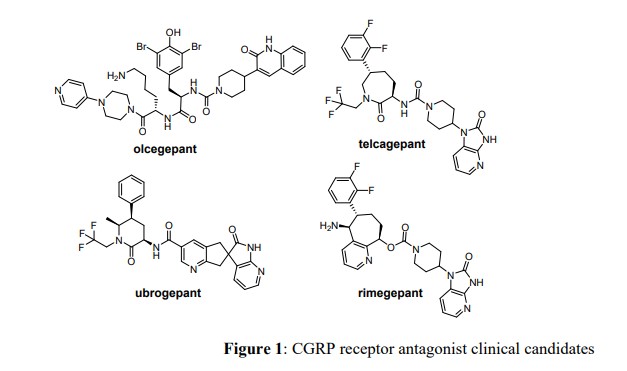






















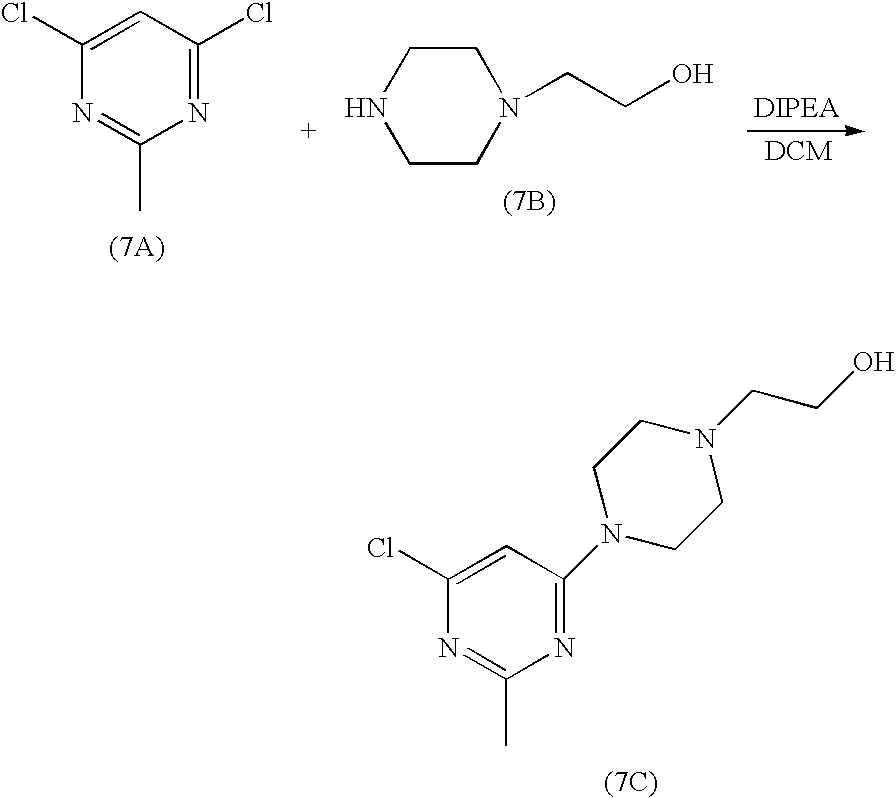




































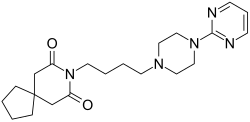
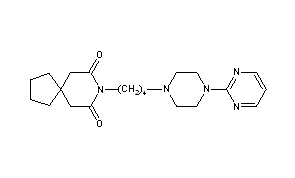










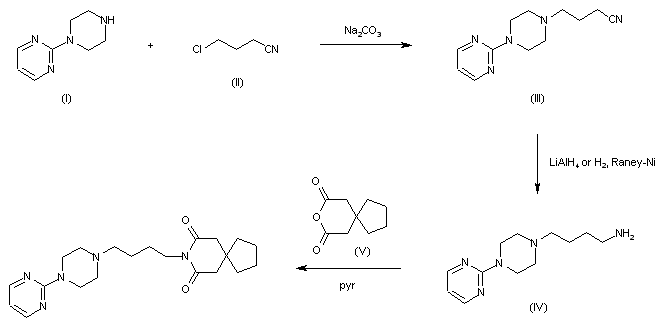




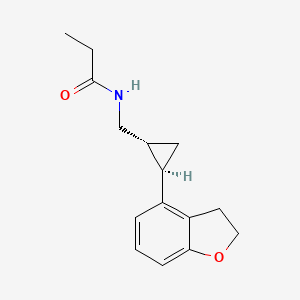





























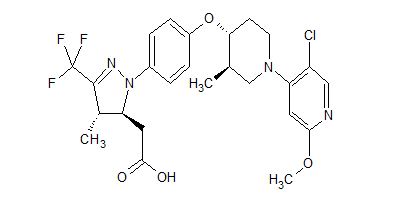
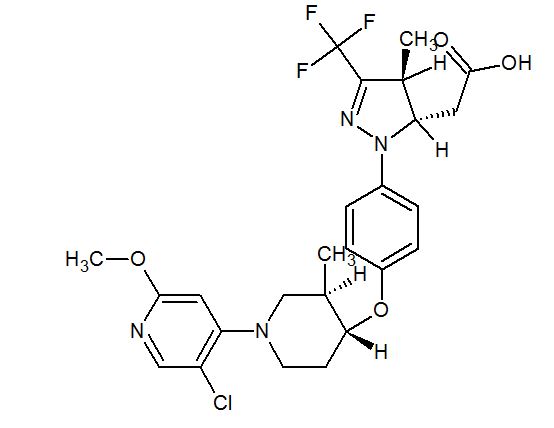
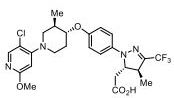
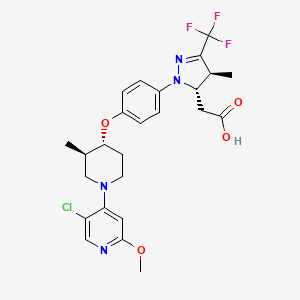



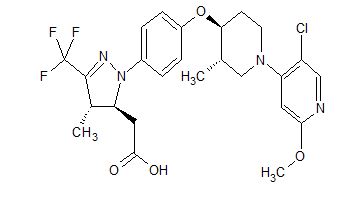






 Rick Ewing, Head, External Partnerships, Discovery Chemistry and Molecular Technologies at Bristol-Myers Squibb
Rick Ewing, Head, External Partnerships, Discovery Chemistry and Molecular Technologies at Bristol-Myers Squibb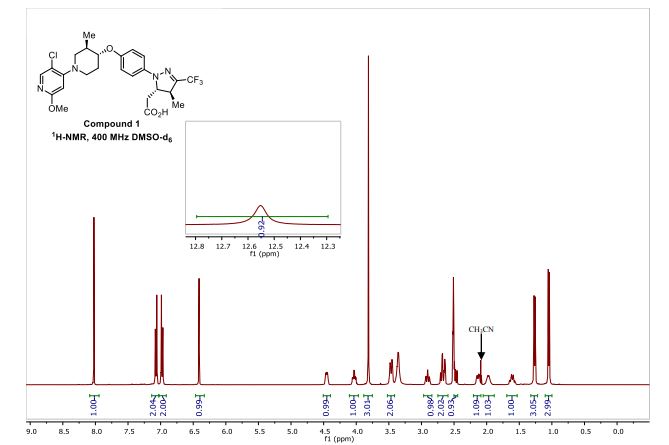
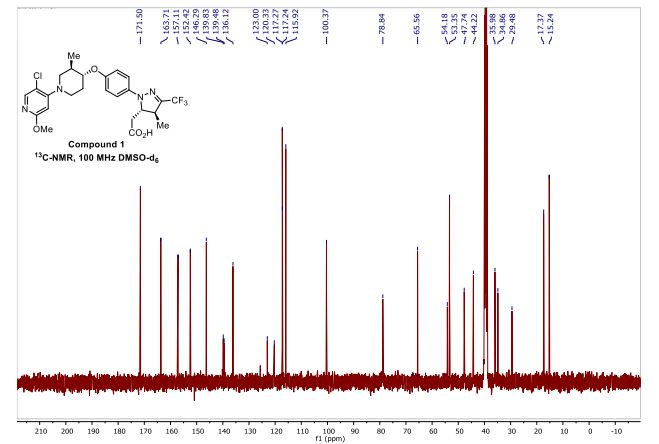










































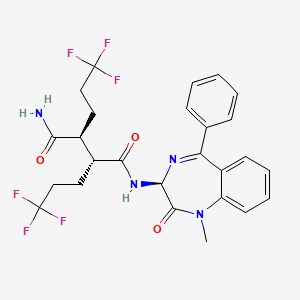

























































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}