
Home » Posts tagged 'APPROVALS 2025'
Tag Archives: APPROVALS 2025
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Luvometinib
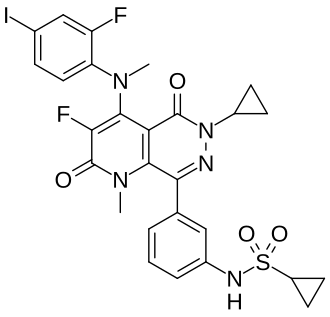
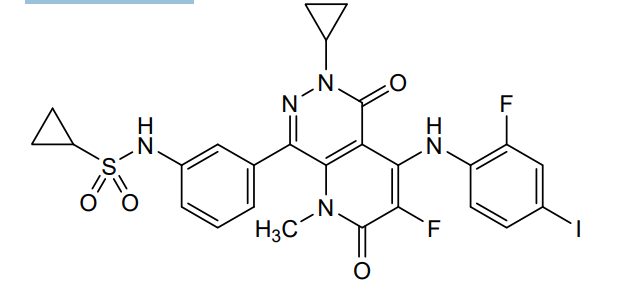
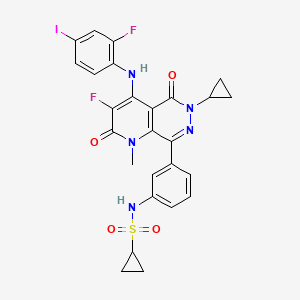
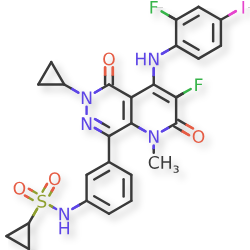
Luvometinib
CAS 2739690-43-6
MF C26H22F2IN5O4S MW665.5 g/mol
CHINA 2025, APPROVALS 2025
N-[3-[6-cyclopropyl-3-fluoro-4-(2-fluoro-4-iodoanilino)-1-methyl-2,5-dioxopyrido[2,3-d]pyridazin-8-yl]phenyl]cyclopropanesulfonamide
N-{3-[6-cyclopropyl-3-fluoro-4-(2-fluoro-4-iodoanilino)-1-methyl-2,5-dioxo-1,2,5,6-tetrahydropyrido[2,3-
d]pyridazin-8-yl]phenyl}cyclopropanesulfonamide
mitogen-activated protein kinase (MEK) inhibitor, antineoplastic, FCN 159, FCN-159, B2DYT4V89X
Luvometinib is a drug for the treatment of various types of cancer. It is a selective, orally administered inhibitor of mitogen-activated protein kinase kinases 1 and 2 (MEK1/MEK2), developed by Fosun Pharma for the treatment of rare malignancies, especially those driven by abnormal abnormal mitogen-activated protein kinase (MAPK) activation.[1][2]
In May 2025, it was approved in China for the treatment of histiocytic neoplasms such as Langerhans cell histiocytosis (LCH) and the genetic disease neurofibromatosis type 1 (NF1).[2]
Luvometinib is an orally bioavailable inhibitor of mitogen-activated protein kinase kinase (MAP2K, MAPK/ERK kinase, or MEK) 1 and 2, with potential antineoplastic activity. Upon administration, luvometinib selectively binds to and inhibits the activity of MEK1 and MEK2, preventing the activation of MEK1/2-dependent effector proteins and transcription factors, which may result in the inhibition of growth factor-mediated cell signaling and tumor cell proliferation. MEK1/2 are dual-specificity threonine/tyrosine kinases that play key roles in the activation of the RAS/RAF/MEK/ERK pathway that regulates cell growth. This pathway is often dysregulated in a variety of tumor cell types through BRAF, KRAS and NRAS mutations.
Luvometinib is a small molecule drug. The usage of the INN stem ‘-tinib’ in the name indicates that Luvometinib is a tyrosine kinase inhibitor. Luvometinib is under investigation in clinical trial NCT07004075 (FCN-159 Monotherapy Versus Chemotherapy by Investigator’s Choice in Pediatric Low-grade Glioma Patients With BRAF Alteration). Luvometinib has a monoisotopic molecular weight of 665.04 Da.
SYN
Example 8
N-(3-(6-allyl-3-ƒluoro-4-(2-ƒluoro-4-iodophenylamino)-1-methyl-2,5-dioxo-1,2,5,6- tetrahydropyrido[2,3-d]pyridazin-8-yl)phenyl)cyclopropanesulƒonamide (8)
The title compound 8 was prepared following the same procedure as described for Example 5 by substituting methanesulfonyl chloride with cyclopropanesulfonyl chloride. MS-ESI (m/z): 666 [M + 1]+.
PAT
Example 8
N-(3-(6-cyclopropyl-3-fluoro-4-(2-fluoro-4-iodophenylamino)-1-methyl-2,5-dioxo-1,2,5,6-tetrahydropyrido[2,3-d]pyridazin-8-yl)phenyl)cyclopropanesulfonamide (8)
[0136] The title compound 8 was prepared following the same procedure as described for Example 5 by substituting methanesulfonyl chloride with cyclopropanesulfonyl chloride. MS-ESI (m/z): 666 [M + 1] +.
PAT
- Protein kinase inhibitorsPublication Number: CN-106905316-BPriority Date: 2013-04-18Grant Date: 2021-06-01
- Certain protein kinase inhibitorsPublication Number: EP-2986611-B1Priority Date: 2013-04-18Grant Date: 2019-02-06
- Certain protein kinase inhibitorsPublication Number: US-10022374-B2Priority Date: 2013-04-18Grant Date: 2018-07-17



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

References
- Cheng Y, Tian H (2017). “Current Development Status of MEK Inhibitors”. Molecules. 22 (10). Basel, Switzerland: 1551. doi:10.3390/molecules22101551. PMC 6151813. PMID 28954413.
- Keam SJ (2025). “Luvometinib: First Approval”. Drugs. 85 (9): 1177–1183. doi:10.1007/s40265-025-02217-6. PMID 40751881.
| Clinical data | |
|---|---|
| Trade names | 复迈宁 (Fu Mainin) |
| Other names | FCN-159 |
| Routes of administration | Oral |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2739690-43-6 |
| PubChem CID | 135210935 |
| IUPHAR/BPS | 13495 |
| UNII | B2DYT4V89X |
| Chemical and physical data | |
| Formula | C26H22F2IN5O4S |
| Molar mass | 665.45 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
//////////luvometinib, CHINA 2025, APPROVALS 2025, antineoplastic, FCN 159, FCN-159, B2DYT4V89X, ANAX, PTFEON, ADVECT, BLUE JET
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
ADVERTISEMENT
Advect Process Systems Ltd.
ADVECT PROCESS SYSTEMS CANADA LTD
51 Beechwood Rd., Cambridge, ON Canada N1S 3S1, Call Now +1 306 850 6737, Mail Now, ask@advectprocess.com
ADVERTISEMENT
BLUE JET HEALTHCARE LTD, https://bluejethealthcare.com
Looking for a Reliable SNAC Manufacturer? Let’s Talk.
At Blue Jet Healthcare Ltd, we specialize in the scalable, high-purity production of SNAC—a critical excipient powering the next generation of oral peptide therapeutics.
With increasing demand for SNAC across global pharma pipelines, choosing the right manufacturing partner is essential. Quality, timelines, and consistency matter.

Phone No. +91 (22) 22075307 / +91 (22) 22071691
Business Development/ Contract Manufacturing: marketing1@bluejethealthcare.com, madhu.gautam71@gmail.com
Aficamten


Aficamten
C18H19N5O2, 337.4 g/mol
FDA 2025, APPROVALS 2025, Myqorzo, 12/19/2025, To treat symptomatic obstructive hypertrophic cardiomyopathy
CK-3773274, B1I77MH6K1, BAY-3723113; CK 3773274; CK 274; MYQORZO
N-[(1R)-5-(5-ethyl-1,2,4-oxadiazol-3-yl)-2,3-dihydro-1H-inden-1-yl]-1-methylpyrazole-4-carboxamide
- (R)-N-(5-(5-ethyl-1,2,4-oxadiazol-3-yl)-2,3-dihydro-1H-inden-1-yl)-1-methyl-1H-pyrazole-4-carboxamide
- N-((1R)-5-(5-Ethyl-1,2,4-oxadiazol-3-yl)-2,3-dihydro-1H-inden-1-yl)- 1-methyl-1H-pyrazole-4-carboxamide
- OriginatorCytokinetics
- DeveloperBayer; Cytokinetics; Sanofi
- ClassAmides; Cardiovascular therapies; Heart failure therapies; Indenes; Oxadiazoles; Pyrazoles; Small molecules
- Mechanism of ActionCardiac myosin inhibitors
- Orphan Drug StatusYes – Hypertrophic cardiomyopathy
- RegisteredHypertrophic cardiomyopathy
- 20 Dec 2025Cytokinetics plans to launch aficamten in the USA in second half of January 2026
- 19 Dec 2025Registered for Hypertrophic cardiomyopathy in USA (PO)
- 19 Dec 2025Aficamten carries a black box warning for the risk of heart failure
Aficamten, sold under the brand name Myqorzo, is a medication used for the treatment of symptomatic obstructive hypertrophic cardiomyopathy.[1] It is a cardiac myosin inhibitor[2] developed by Cytokinetics.[3][4]
Aficamten binds directly to the motor domain of cardiac myosin and prevents it from entering the force-producing state.[5] This lowers cardiac contractility, leading to reduced left ventricular outflow tract obstruction in people with hypertrophic cardiomyopathy.[5]
Aficamten was approved for medical use in the United States in December 2025.[6]
Medical uses
Aficamten is indicated for the treatment of adults with symptomatic obstructive hypertrophic cardiomyopathy to improve functional capacity and symptoms.[1][6]
Symptomatic obstructive hypertrophic cardiomyopathy is an inherited condition where people have thickened heart muscle and reduced blood flow from the left side of the heart to the rest of the body, causing symptoms such as shortness of breath, fatigue, and potentially life-threatening cardiac events.[6]
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019144041&_cid=P10-MJP428-30255-1

Example 15
Synthesis of Compound 184
1. Synthesis of Intermediate 15-2:

[0262] To a solution of tert-butyl N-[(1R)-5-(N-hydroxycarbamimidoyl)-2,3-dihydro-1H-inden-1-yl] carbamate (16 g, 54.9 mmol, 1.0 equiv) in dioxane (300 mL) was added propanoyl propanoate (8.4 g, 64.5 mmol, 1.2 equiv). The mixture was stirred at 105 oC for 8 h, cooled to r.t., concentrated under reduced pressure, and purified by silica gel
chromatography (EA/PE, 1/9) to give 17.5 g (97%) of tert-butyl N-[(1R)-5-(5-ethyl-1,2,4-oxadiazol-3-yl)-2,3-dihydro-1H-inden-1-yl]carbamate as a white solid.
2. Synthesis of Intermediate 15-3:

[0263] To a solution of tert-butyl N-[(1R)-5-(5-ethyl-1,2,4-oxadiazol-3-yl)-2,3-dihydro-1H-inden-1-yl]carbamate (17.6 g, 53.4 mmol, 1.0 equiv) in DCM (120 mL) was added TFA (24 mL). The mixture was stirred at room temperature overnight and concentrated under reduced pressure. The mixture was then poured into ethanol (50 mL) and water (5 mL) and the pH was adjusted to 12 with sodium hydroxide solution (2 N). The mixture was then extracted with dichloromethane (200 mL) three times. The combined organic layers were dried over anhydrous sodium sulfate and concentrated under reduced pressure to give 11.2 g of (1R)-5-(5-ethyl-1,2,4-oxadiazol-3-yl)-2,3-dihydro-1H-inden-1-amine as a brown oil. 3. Synthesis of Compound 184:

[0264] To a solution of 1-methyl-1H-pyrazole-4-carboxylic acid (6.1 g, 48.4 mmol, 1.0 equiv) in DMF (300 mL) were added DIEA (12.6 g, 97.5 mmol, 2.0 equiv), HOAt (19.8 g, 145.8 mmol, 3.0 equiv), and EDCI (28 g, 146.1 mmol, 3.0 equiv). The mixture was stirred for 15 min, and (1R)-5-(5-ethyl-1,2,4-oxadiazol-3-yl)-2,3-dihydro-1H-inden-1-amine (11.2 g, 48.9 mmol, 1.0 equiv) was then added. The mixture was then stirred for 3 h, diluted with DCM, washed with NH4Cl solution three times, dried over sodium sulfate, concentrated under reduced pressure, and purified by silica gel chromatography (EA/PE, 74/26) to give an intermediate product. The intermediate product was triturated with a mixture of EA and PE (1/10) to afford 14.5 g (88%) of (R)-N-(5-(5-ethyl-1,2,4-oxadiazol-3-yl)-2,3-dihydro-1H-inden-1-yl)-1-methyl-1H-pyrazole-4-carboxamide (Compound 184) as a white solid. LRMS (ES) m/z 338 (M+H). 1H-NMR: (DMSO, 300MHz, ppm): į 8.41 (1H, d, J = 8.4 Hz), 8.16 (1H, s), 7.91-7.79 (3H, m), 7.34 (1H, d, J = 7.9 Hz), 5.53 (1H, q, J = 8.3 Hz), 3.84 (3H, s), 3.13-2.81 (4H, m), 2.44 (1H, dd, J = 7.9, 4.7 Hz), 1.95 (1H, m), 1.33 (3H, t, J = 7.5 Hz).



PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021011807&_cid=P10-MJP428-30255-1

(R)-N-(5-(5-ethyl- 1,2,4-oxadiazol-3-yl)-2,3-dihydro-1H-inden-l-yl)-1-methyl-1H-pyrazole-4-carboxamide,
SYN
https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c01290



PAT
- Cardiac sarcomere inhibitorsPublication Number: US-10836755-B2Priority Date: 2018-01-19Grant Date: 2020-11-17
- Cardiac sarcomere inhibitorsPublication Number: US-12065436-B2Priority Date: 2018-01-19Grant Date: 2024-08-20
- Cardiac sarcomere inhibitorsPublication Number: US-2023119665-A1Priority Date: 2018-01-19
- Cardiac sarcomere inhibitorsPublication Number: US-11472796-B2Priority Date: 2018-01-19Grant Date: 2022-10-18
- Cardiac sarcomere inhibitorsPublication Number: US-2025059173-A1Priority Date: 2018-01-19
- Dihydrobenzofuran and indene analogs as myocardial inhibitorsPublication Number: CN-117964573-APriority Date: 2018-01-19
- Cardiac sarcomere inhibitorsPublication Number: TW-202436291-APriority Date: 2018-01-19
- Dihydrobenzofurans and indene analogs as cardiomyome inhibitorsPublication Number: CN-111757875-BPriority Date: 2018-01-19Grant Date: 2024-01-09
- Dihydrobenzofuran and indene analogs as myocardial inhibitorsPublication Number: CN-117924208-APriority Date: 2018-01-19
- Dihydrobenzofuran and indene analogs as inotropic agentsPublication Number: CN-111757875-APriority Date: 2018-01-19
- Cardiac sarcomere inhibitorsPublication Number: TW-I835770-BPriority Date: 2018-01-19Grant Date: 2024-03-21
- Dihydrobenzofuran and inden analogs as cardiac sarcomere inhibitorsPublication Number: EP-3740481-A1Priority Date: 2018-01-19
- Dihydrobenzofuran and inden analogs as cardiac sarcomere inhibitorsPublication Number: EP-3740481-B9Priority Date: 2018-01-19Grant Date: 2024-10-23
- Cardiac sarcomere inhibitorsPublication Number: US-2021147399-A1Priority Date: 2018-01-19
- Cardiac sarcomere inhibitorsPublication Number: EP-4491622-A2Priority Date: 2018-01-19
REF
- Clinical Evaluation of the Effect of Aficamten on <scp>QT</scp>/<scp>QTc</scp> Interval in Healthy ParticipantsPublication Name: Clinical and Translational SciencePublication Date: 2025-04PMCID: PMC11979292PMID: 40200648DOI: 10.1111/cts.70218
- Effect of Hepatic Impairment or Renal Impairment on the Pharmacokinetics of AficamtenPublication Name: Clinical PharmacokineticsPublication Date: 2025-02-05PMCID: PMC11954688PMID: 39907965DOI: 10.1007/s40262-025-01481-9
- The clinical utility of cardiac myosin inhibitors for the management of hypertrophic cardiomyopathy: a scoping reviewPublication Name: Heart Failure ReviewsPublication Date: 2024-12-17PMCID: PMC11802616PMID: 39690360DOI: 10.1007/s10741-024-10476-w
- Obstructive Hypertrophic Cardiomyopathy: A Review of New TherapiesPublication Name: Future CardiologyPublication Date: 2023-10PMID: 37933625DOI: 10.2217/fca-2023-0056
- Aficamten: A Breakthrough Therapy for Symptomatic Obstructive Hypertrophic CardiomyopathyPublication Name: American journal of cardiovascular drugs : drugs, devices, and other interventionsPublication Date: 2023-08-01PMID: 37526885DOI: 10.1007/s40256-023-00599-0
- Synthesis of AficamtenPublication Name: SynfactsPublication Date: 2021-11-17DOI: 10.1055/s-0041-1737088
- Discovery of Aficamten (CK-274), a Next-Generation Cardiac Myosin Inhibitor for the Treatment of Hypertrophic CardiomyopathyPublication Name: Journal of Medicinal ChemistryPublication Date: 2021-10-04PMID: 34606259DOI: 10.1021/acs.jmedchem.1c01290
- Emerging Medical Treatment for Hypertrophic CardiomyopathyPublication Name: Journal of Clinical MedicinePublication Date: 2021-03-01PMCID: PMC7957690PMID: 33804412DOI: 10.3390/jcm10050951
- Small Molecules Acting on Myofilaments as Treatments for Heart and Skeletal Muscle DiseasesPublication Name: International Journal of Molecular SciencesPublication Date: 2020-12-16PMCID: PMC7767104PMID: 33339418DOI: 10.3390/ijms21249599
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Contraindiations
Use with rifampin is contraindicated.[1]
Adverse effects
The US prescription label for aficamten contains a boxed warning that it reduces left ventricular ejection fraction and can cause heart failure due to systolic dysfunction.[1]
History
The effectiveness and safety of aficamten were studied in 282 adults with symptomatic obstructive hypertrophic cardiomyopathy randomly assigned to receive aficamten or placebo for 24 weeks.[6] At the end of the study, participants receiving aficamten had an increase in exercise capacity measured by peak oxygen uptake compared to no change in exercise capacity among those receiving placebo.[6] Also, 59 percent of participants receiving aficamten experienced an improvement in physical activity limitations (measured using the New York Heart Association Classification system) compared to 24 percent of individuals receiving placebo.[6]
Society and culture
Legal status
Aficamten was approved for medical use in the United States in December 2025.[6][7] The US Food and Drug Administration granted the application for aficamten orphan drug and breakthrough therapy designations.[6]
In December 2025, the Committee for Medicinal Products for Human Use of the European Medicines Agency adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Myqorzo, intended for the treatment of adults with obstructive hypertrophic cardiomyopathy.[5] The applicant for this medicinal product is Cytokinetics (Ireland) Limited.[5]
Names
Aficamten is the international nonproprietary name.[8]
Aficamten is sold under the brand name Myqorzo.[6]
References
- https://www.accessdata.fda.gov/drugsatfda_docs/label/2025/219083s000lbl.pdf [bare URL PDF]
- Chuang, Chihyuan; Collibee, Scott; Ashcraft, Luke; Wang, Wenyue; Vander Wal, Mark; Wang, Xiaolin; et al. (October 2021). “Discovery of Aficamten (CK-274), a Next-Generation Cardiac Myosin Inhibitor for the Treatment of Hypertrophic Cardiomyopathy”. Journal of Medicinal Chemistry. 64 (19): 14142–14152. doi:10.1021/acs.jmedchem.1c01290. ISSN 0022-2623. PMID 34606259. S2CID 238355647.
- Zhao, Xue; Liu, Hongzhong; Tian, Wei; Fang, Ligang; Yu, Mengyang; Wu, Xiaofei; et al. (2023). “Safety, tolerability, pharmacokinetics, and pharmacodynamics of single and multiple doses of aficamten in healthy Chinese participants: a randomized, double-blind, placebo-controlled, phase 1 study”. Frontiers in Pharmacology. 14 1227470. doi:10.3389/fphar.2023.1227470. PMC 10482267. PMID 37680714.
- Sebastian, Sneha Annie; Padda, Inderbir; Lehr, Eric J.; Johal, Gurpreet (September 2023). “Aficamten: A Breakthrough Therapy for Symptomatic Obstructive Hypertrophic Cardiomyopathy”. American Journal of Cardiovascular Drugs: Drugs, Devices, and Other Interventions. 23 (5): 519–532. doi:10.1007/s40256-023-00599-0. ISSN 1179-187X. PMID 37526885. S2CID 260348901.
- “Myqorzo EPAR”. European Medicines Agency (EMA). 12 December 2025. Retrieved 22 December 2025. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- “FDA approves drug to improve functional capacity and symptoms in adults with rare inherited heart condition”. U.S. Food and Drug Administration (FDA) (Press release). 22 December 2025. Retrieved 22 December 2025.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - “Cytokinetics Announces FDA Approval of Myqorzo (aficamten) for the Treatment of Adults with Symptomatic Obstructive Hypertrophic Cardiomyopathy to Improve Functional Capacity and Symptoms” (Press release). Cytokinetics. 19 December 2025. Retrieved 22 December 2025 – via GlobeNewswire News Room.
- World Health Organization (2021). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 86”. WHO Drug Information. 35 (3). hdl:10665/346562.
Further reading
- Maron, Martin S.; Masri, Ahmad; Choudhury, Lubna; Olivotto, Iacopo; Saberi, Sara; Wang, Andrew; et al. (January 2023). “Phase 2 Study of Aficamten in Patients With Obstructive Hypertrophic Cardiomyopathy”. Journal of the American College of Cardiology. 81 (1): 34–45. doi:10.1016/j.jacc.2022.10.020. hdl:2158/1295661. PMID 36599608. S2CID 255472935.
External links
- Clinical trial number NCT05186818 for “Aficamten vs Placebo in Adults With Symptomatic Obstructive Hypertrophic Cardiomyopathy (SEQUOIA-HCM) (SEQUOIA-HCM)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Myqorzo |
| Other names | CK-3773274 |
| License data | US DailyMed: Aficamten |
| Routes of administration | By mouth |
| Drug class | Cardiac myosin inhibitor |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2364554-48-1 |
| PubChem CID | 139331495 |
| DrugBank | DB18490 |
| ChemSpider | 114935503 |
| UNII | B1I77MH6K1 |
| KEGG | D12253 |
| ChEMBL | ChEMBL4847050 |
| PDB ligand | 6I6 (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C18H19N5O2 |
| Molar mass | 337.383 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
//////////Aficamten, FDA 2025, APPROVALS 2025, Myqorzo, CK-3773274, CK 3773274, B1I77MH6K1, BAY 3723113; CK 3773274; CK 274, MYQORZO
Zoliflodacin



Zoliflodacin
- CAS 1620458-09-4
- AZD-0914
- AZD0914
- FWL2263R77
- ETX0914
MF C22H22FN5O7 MW 487.4 g/mol
FDA 2025, APPROVALS 2025, 12/12/2025, Nuzolvence
(4′R,6′S,7′S)-17′-fluoro-4′,6′-dimethyl-13′-[(4S)-4-methyl-2-oxo-1,3-oxazolidin-3-yl]spiro[1,3-diazinane-5,8′-5,15-dioxa-2,14-diazatetracyclo[8.7.0.02,7.012,16]heptadeca-1(17),10,12(16),13-tetraene]-2,4,6-trione
Spiro[isoxazolo[4,5-g][1,4]oxazino[4,3-a]quinoline-5(6H),5′(2′H)-pyrimidine]-2′,4′,6′(1′H,3′H)-trione, 11-fluoro-1,2,4,4a-tetrahydro-2,4-dimethyl-8-[(4S)-4-methyl-2-oxo-3-oxazolidinyl]-, (2R,4S,4aS)-
(2R,4S,4aS)-11-Fluoro-2,4-dimethyl-8-[(4S)-4-methyl-2-oxo-1,3-oxazolidin-3-yl]-1,2,4,4a-tetrahydro-2′H,6H-spiro[1,4-oxazino[4,3-a][1,2]oxazolo[4,5-g]quinoline-5,5′-pyrimidine]-2′,4′,6′(1′H,3′H)-trione
To treat uncomplicated urogenital gonorrhea due to Neisseria gonorrhoeae
Zoliflodacin, sold under the brand name Nuzolvence, is an antibiotic used for the treatment of antibiotic-resistant Neisseria gonorrhoeae (gonorrhea).[2] Zoliflodacin is being developed as part of a public-private partnership between Innoviva Specialty Therapeutics and the Global Antibiotic Research & Development Partnership (GARDP).[3] Zoliflodacin is taken by mouth.[2]
The most common side effects include low white blood cell counts, headache, dizziness, nausea, and diarrhea.[2]
Zoliflodacin was approved for medical use in the United States in December 2025.[2]
SYN
- Facile Synthesis of Spirocyclic Tetrahydroquinolines via C(sp3)–H Functionalization in a Cascade Redox ProcessDOI: 10.1055/s-0040-1720890Publication Date: 2022Publication Name: Synthesis
- Synthesis of ZoliflodacinDOI: 10.1055/s-0040-1707088Publication Date: 2020Publication Name: Synfacts
SYN

SYN
SYN
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US106042502&_cid=P11-MJMADN-82597-1
(2R,4S,4aS)-11-Fluoro-2,4-dimethyl-8-[(4S)-4-methyl-2-oxo-1,3-oxazolidin-3-yl]-1,2,4,4a-tetrahydro-2′H,6H-spiro[1,4-oxazino[4,3-a][1,2]oxazolo[4,5-g]quinoline-5,5′-pyrimidine]-2′,4′,6′(1′H,3′H)-trione


| 1H NMR (400 MHz, DMSO-d 6) δ: 0.9 (d, 3H), 1.15 (d, 3H), 1.4 (d, 3H), 2.9 (d, 1H), 3.1 (t, 1H), 3.6-3.7 (m, 2H), 3.8-4.0 (m, 1H), 3.9 (d, 1H), 4.1 (d, 1H), 4.2 (q, 1H), 4.6-4.7 (m, 2H), 7.6 (s, 1H), 11.5 (s, 1H), 11.8 (s, 1H). MS (ES) MH +: 488.4 for C 22H 22FN 5O 7, [α] D 20=+224 (c=1; MeOH). |
Alternative Synthesis of Example 5

1H NMR (300 MHz, DMSO-d 6) δ: 1.0 (d, 3H), 1.3 (d, 3H), 1.4 (d, 3H), 3.1 (d, 1H), 3.5-4.3 (m, 7H), 4.5-4.8 (m, 2H), 7.6 (s, 1H), 11.5 (br. s., 1H), 11.7 (br. s., 1H). MS (ES) MH +: 488 for C 22H 22FN 5O 7.
SYN
https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/mic90
2.3.2 Chemical synthesis
The synthesis of zoliflodacin described below was reported in 2015 [47]. The first step, starting from 2,3,4-trifluorobenzaldehyde, consists of the protection of the aldehyde function to an acetal group. After deprotonation using n-BuLi, formylation is performed with DMF to introduce an aldehyde group, which is then converted to oxime using hydroxylamine. Chlorination with N-chlorosuccinimide (NCS), followed by reaction with L-alaninol and intramolecular SNAr allows the formation of the benzisoxazole ring. The oxazolidinone moiety is obtained using 1,1′-carbonyldiimidazole (CDI). The deprotection of the aldehyde is then performed in acidic conditions followed by another SNAr at the ortho position of the aldehyde using (2R,6S)-2,6-dimethylmorpholine. Finally, a Knoevenagel condensation between the aldehyde and hexahydropyrimidine-2,4,6-trione is performed, followed by an intramolecular rearrangement consisting in an [1-5] hydride shift and then intramolecular cyclization leading to zoliflodacin (Fig. 5).

PAT
- High throughput screening assay to identify DNA topoisomerase inhibitorsPublication Number: US-12234504-B1Priority Date: 2023-10-16Grant Date: 2025-02-25
- Treatment of pathogenic neisseria sp. infection with triazole antifungal agentsPublication Number: US-2025281464-A1Priority Date: 2022-04-29
- Methods and materials for treatment of neisseria gonorrhoeae infectionPublication Number: WO-2022204231-A2Priority Date: 2021-03-26
- Methods and materials for treatment of neisseria gonorrhoeae infectionPublication Number: EP-4313040-A2Priority Date: 2021-03-26
- Methods and materials for treatment of neisseria gonorrhoeae infectionPublication Number: EP-4313040-A2Priority Date: 2021-03-26
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Medical uses
Zoliflodacin is indicated for the treatment of uncomplicated urogenital gonorrhea in people who weigh at least 77 pounds (35 kg).[2]
Susceptible bacteria
Zoliflodacin has shown in vitro activity against the following species of bacteria:[4] Staphylococcus aureus, Streptococcus pneumoniae, Haemophilus influenzae, Moraxella catarrhalis, Neisseria gonorrhoeae, and Chlamydia trachomatis
Adverse effects
Animal studies showed that zoliflodacin might cause birth defects, pregnancy loss, or male fertility problems.[2]
Mechanism of action
It has a mechanism of action which involves inhibition of bacterial type II topoisomerases.[4][5][6]
History

A high throughput screening campaign aimed at identifying compounds with whole cell antibacterial activity performed at Pharmacia & Upjohn identified compound PNU-286607, a progenitor of Zoliflodacin, as having the desired activity.[7]
Subsequent research at AstraZeneca led to the discovery that the nitroaromatic in PNU-286607 could be replaced with a fused benzisoxazole ring,[8] which allowed for an exploration of different groups at the 3-position of the heterocycle. This work was continued at Entasis Pharmaceuticals where extensive optimization resulted in the discovery of ETX0914.[4]
Researchers tested zoliflodacin in a study with 930 participants who had uncomplicated urogenital gonorrhea.[2] Two-thirds of participants received a single 3-gram dose of zoliflodacin dissolved in water.[2] The other third received the standard treatment of ceftriaxone shot plus azithromycin pill.[2] The study measured how well the medicines cleared the bacteria 4 to 8 days after treatment.[2] The study showed 91% of participants who took zoliflodacin were cured and 96% of participants who received the standard treatment were cured.[2]
Society and culture
Legal status
Zoliflodacin was approved for medical use in the United States in December 2025.[3]
The US Food and Drug Administration (FDA) granted the application for zoliflodacin fast track, qualified infectious disease product, and priority review designations for the uncomplicated urogenital gonorrhea indication.[2] The FDA approval for zoliflodacin was granted to Entasis Therapeutics.[2]
Names
Zoliflodacin is the international nonproprietary name.[9]
Zoliflodacin is sold under the brand name Nuzolvence.[3]
References
- https://innovivaspecialtytherapeutics.com/wp-content/uploads/2025/12/NUZOLVENCE-zoliflodacin-Full-Prescribing-Information-December-2025.pdf [bare URL PDF]
- “FDA Approves Two Oral Therapies to Treat Gonorrhea”. U.S. Food and Drug Administration (FDA) (Press release). 12 December 2025. Retrieved 13 December 2025. This article incorporates text from this source, which is in the public domain.
- Pierre G (12 December 2025). “Nuzolvence (Zoliflodacin) Receives U.S. FDA Approval”. Global Antibiotic Research & Development Partnership (GARDP). Retrieved 13 December 2025.
- Basarab GS, Kern GH, McNulty J, Mueller JP, Lawrence K, Vishwanathan K, et al. (July 2015). “Responding to the challenge of untreatable gonorrhea: ETX0914, a first-in-class agent with a distinct mechanism-of-action against bacterial Type II topoisomerases”. Scientific Reports. 5 (1) 11827. Bibcode:2015NatSR…511827B. doi:10.1038/srep11827. PMC 4501059. PMID 26168713.
- Bradford PA, Miller AA, O’Donnell J, Mueller JP (June 2020). “Zoliflodacin: An Oral Spiropyrimidinetrione Antibiotic for the Treatment of Neisseria gonorrheae, Including Multi-Drug-Resistant Isolates”. ACS Infectious Diseases. 6 (6): 1332–1345. doi:10.1021/acsinfecdis.0c00021. PMID 32329999.
- Pisano L, Giovannuzzi S, Supuran CT (June 2024). “Management of Neisseria gonorrhoeae infection: from drug resistance to drug repurposing”. Expert Opinion on Therapeutic Patents. 34 (6): 511–524. doi:10.1080/13543776.2024.2367005. PMID 38856987.
- Miller AA, Bundy GL, Mott JE, Skepner JE, Boyle TP, Harris DW, et al. (August 2008). “Discovery and characterization of QPT-1, the progenitor of a new class of bacterial topoisomerase inhibitors”. Antimicrobial Agents and Chemotherapy. 52 (8): 2806–2812. doi:10.1128/AAC.00247-08. PMC 2493097. PMID 18519725.
- Basarab GS, Brassil P, Doig P, Galullo V, Haimes HB, Kern G, et al. (November 2014). “Novel DNA gyrase inhibiting spiropyrimidinetriones with a benzisoxazole scaffold: SAR and in vivo characterization”. Journal of Medicinal Chemistry. 57 (21): 9078–9095. doi:10.1021/jm501174m. PMID 25286019.
- World Health Organization (2016). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 76”. WHO Drug Information. 30 (3). hdl:10665/331020.
Further reading
- Luckey A, Balasegaram M, Barbee LA, Batteiger TA, Broadhurst H, Cohen SE, et al. (2025). “Zoliflodacin versus ceftriaxone plus azithromycin for treatment of uncomplicated urogenital gonorrhoea: an international, randomised, controlled, open-label, phase 3, non-inferiority clinical trial”. The Lancet. doi:10.1016/S0140-6736(25)01953-1.
- Taylor SN, Marrazzo J, Batteiger BE, Hook EW, Seña AC, Long J, et al. (November 2018). “Single-Dose Zoliflodacin (ETX0914) for Treatment of Urogenital Gonorrhea”. The New England Journal of Medicine. 379 (19): 1835–1845. doi:10.1056/NEJMoa1706988. hdl:1805/19865. PMID 30403954.
External links
- Clinical trial number NCT03959527 for “Zoliflodacin in Uncomplicated Gonorrhoea” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Nuzolvence |
| Other names | AZD0914; ETX0914 |
| AHFS/Drugs.com | Nuzolvence |
| License data | US DailyMed: Zoliflodacin |
| Routes of administration | By mouth |
| Drug class | Antibacterial |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1][2] |
| Pharmacokinetic data | |
| Bioavailability | 97.8% |
| Metabolism | Liver |
| Onset of action | Fasted: 1.5–2.3 hFed: 4 h |
| Elimination half-life | 5.3–6.3 h |
| Excretion | Feces (79.6%)Urine (18.2%) |
| Identifiers | |
| IUPAC name | |
| PubChem CID | 76685216 |
| DrugBank | 12817 |
| UNII | FWL2263R77 |
| KEGG | D11726 |
| Chemical and physical data | |
| Formula | C22H22FN5O7 |
| Molar mass | 487.444 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
//////////Zoliflodacin, FDA 2025, APPROVALS 2025, Nuzolvence, AZD-0914, AZD 0914, FWL2263R77, ETX 0914
Sevabertinib
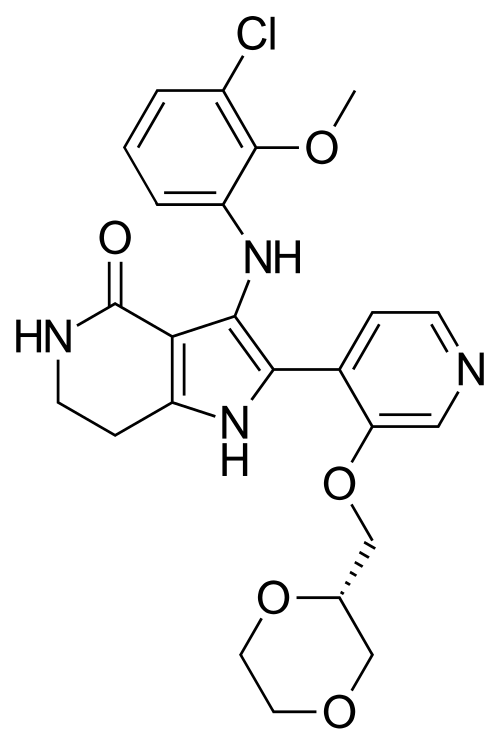
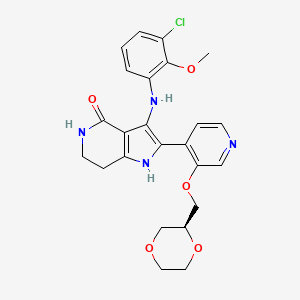
Sevabertinib
CAS 2521285-05-0
MF C24H25ClN4O5, 484.9 g/mol
3-(3-chloro-2-methoxyanilino)-2-[3-[[(2S)-1,4-dioxan-2-yl]methoxy]-4-pyridinyl]-1,5,6,7-tetrahydropyrrolo[3,2-c]pyridin-4-one
11/19/2025, FDA 2025, APPROVALS 2025, Hyrnuo, 2A7VPM5RWH, BAY-2927088, BAY 2927088
To treat locally advanced or metastatic non-squamous non-small cell lung cancer with tumors that have activating HER2 tyrosine kinase domain activating mutations in patients who received a systemic therapy
Sevabertinib, sold under the brand name Hyrnuo, is an anti-cancer medication used for the treatment of non-small cell lung cancer.[1] Sevabertinib is a kinase inhibitor.[1] It is taken by mouth.[1]
Sevabertinib was approved for medical use in the United States in November 2025.[2]
SYN
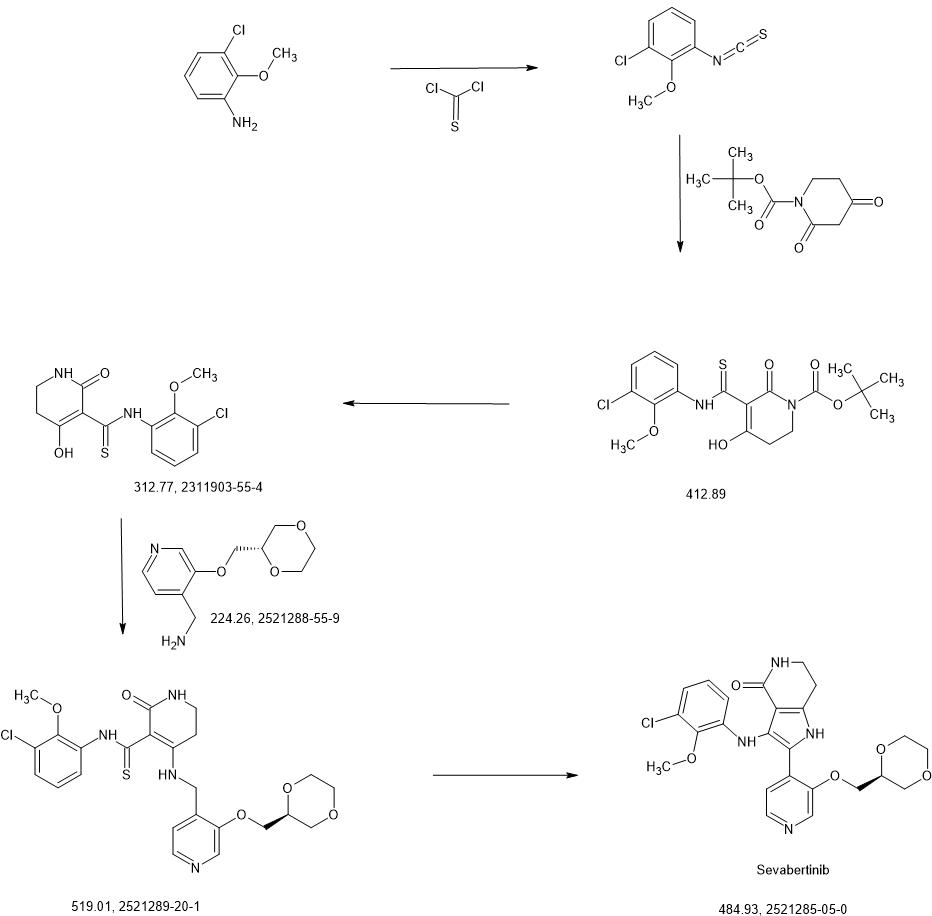
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020216781&_cid=P22-MICIVF-33261-1
Intermediate 3-1
1-chloro-3-isothiocyanato-2-methoxybenzene
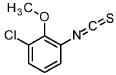
3-chloro-2-methoxyaniline (CAS 511 14-68-2, 8.4 ml, 63 mmol) was solved in DCM (100 ml) and sat. sodium bicarbonate solution (100 ml) was added. To the ice cooled mixture was slowly added thiophosgene (5.4 ml, 70 mmol). The reaction was stirred at 0°C for 2 h. At RT the DCM layer was separated and washed with sat. sodium bicarbonate solution, filtered through a hydrophobic filter and concentrated under reduced pressure to give the title compound (12.97 g, 100 % yield) which was used directly in the next step.
1H-NMR (400MHz, DMSO-de): d [ppm]= 7.51 (dd, 1 H), 7.35 (dd, 1 H), 7.20 (t, 1 H), 3.85 -3.91 (m, 3H).
Intermediate 4-1
tert- butyl 5-[(3-chloro-2-methoxyphenyl)carbamothioyl]-4-hydroxy-6-oxo-3,6-dihydropyridine-1(2/-/)-carboxylate
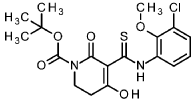
To an ice-cooled solution of 1-chloro-3-isothiocyanato-2-methoxybenzene (intermediate 3-1 , 4.00 g, 20.0 mmol) and tert- butyl 2,4-dioxopiperidine-1-carboxylate (CAS 845267-78-9, 4.27 g, 20.0 mmol) in acetonitrile (92 ml) was added dropwise DBU (4.5 ml, 30 mmol). The reaction was stirred at RT overnight. To the reaction mixture was added ice-water (200 ml_) and cone. HCI (2 ml_). The mixture was stirred for 20 min. and extracted with DCM. The organic phase was filtered over a water-repellent filter, conentrated under reduced pressure and purified by flash chromatography (silica, hexane / EtOAc gradient 0-50 %) to give 6.54 g of the title compound (71 % yield).
1H-NMR (400MHz, DMSO-de): d [ppm]= 13.36 (br s, 1 H), 7.73 (d, 1 H), 7.47 (dd, 1 H), 7.22 (t, 1 H), 3.76 – 3.82 (m, 5H), 2.88 (t, 2H), 1.48 (s, 9H).
LC-MS (method 1): Rt = 1.49 min; MS (ESIpos): m/z = 413.1 [M+H]+
Intermediate 5-1
A/-(3-chloro-2-methoxyphenyl)-4-hydroxy-2-oxo-1 ,2,5,6-tetrahydropyridine-3-carbothioamide
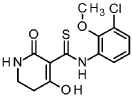
To a solution of tert- butyl 5-[(3-chloro-2-methoxyphenyl)carbamothioyl]-4-hydroxy-6-oxo-3,6-dihydropyridine-1 (2/-/)-carboxylate (intermediate 4-1 , 6.54 g, 15.8 mmol) in dichloromethane (94 ml) was added TFA (12 ml, 160 mmol) and the mixture was stirred 1.5 h at RT. The reaction mixture was concentrated under reduced pressure and the residue was solved in EtOAc and washed with sat. sodium bicarbonate solution and brine. The organic layer was filtered through a hydrophobic filter and the filtrate was dried to dryness. The residue was purified by flash chromatography (silica, hexane / EtOAc gradient 20-100 %) to give 4.06 g of the title compound (78 % yield).
1H-NMR (400 MHz, DMSO-de): d [ppm]= 16.45 (d, 1 H), 14.69 (s, 1 H), 14.33 (s, 1 H), 9.37 (br s, 1 H), 8.18 (br s, 1 H), 7.76 – 7.87 (m, 1 H), 7.37 – 7.45 (m, 1 H), 7.15 – 7.23 (m, 1 H), 3.73 – 3.76 (m, 3H), 3.43 (td, 1 H), 3.27 – 3.32 (m, 1 H), 2.79 (t, 1 H), 2.59 – 2.69 (m, 1 H).
LC-MS (method 1): Rt = 1.19 min; MS (ESIpos): m/z = 313 [M+H]+
ntermediate 6-2
A/-(3-chloro-2-methoxyphenyl)-4-{[(3-{[(2S)-1 ,4-dioxan-2-yl]methoxy}pyridin-4-yl)methyl]amino}-2-oxo-1 ,2,5,6-tetrahydropyridine-3-carbothioamide
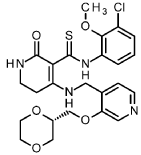
A mixture of A/-(3-chloro-2-methoxyphenyl)-4-hydroxy-2-oxo-1 ,2,5,6-tetrahydropyridine-3-carbothioa ide (intermediate 5-1 , 866 mg, 2.77 mmol) and 1-(3-{[(2S)-1 ,4-dioxan-2-yl]methoxy}pyridin-4-yl)methanamine (intermediate 2-8, 776 mg, 80% purity, 2.77 mmol) in ACN (22 ml) was treated with A/,0-bis(trimethylsilyl)acetamide (2.05 ml, 8.6 mmol, CAS 10416-59-8) and stirred at 80°C for 4 h. The reaction mixture was concentrated under reduced pressure and purified by flash chromatography (silica, DCM / EtOH gradient 0-20%) to give 1.23 g (95% purity, 81 % yield) of the title compound.
1H-NMR (400MHz, DMSO-d6): d [ppm]= 2.78 (t, 2H), 3.16 (td, 2H), 3.40 – 3.54 (m, 3H), 3.59 – 3.69 (m, 2H), 3.71 (s, 3H), 3.73 – 3.79 (m, 1 H), 3.83 – 3.95 (m, 2H), 4.16 (t, 2H), 4.67 (d, 2H), 7.11 (t, 1 H), 7.27 – 7.33 (m, 2H), 7.73 (br s, 1 H), 7.81 (dd, 1 H), 8.24 (d, 1 H), 8.39 (s, 1 H), 13.69 (s, 1 H), 14.79 (s, 1 H).
LC-MS (method 2): Rt = 1.09 min; MS (ESIpos): m/z = 519 [M+H]+
Example 2
3-(3-chloro-2-methoxyanilino)-2-(3-{[(2S)-1 ,4-dioxan-2-yl]methoxy}pyridin-4-yl)-1 ,5,6,7-tetrahydro-4H-pyrrolo[3,2-c]pyridin-4-one (Stereoisomer 1)

The title compound from example 1 (140 mg) was separated into enantiomers by preparative chiral HPLC to give title compound (enantiomer 1 , 27 mg at Rt = 14.0 – 17.0 min) and enantiomer 2 (25 mg at Rt = 20.0 – 24.8 min, see example 3).
Preparative chiral HPLC method:
Instrument: Labomatic HD5000, Labocord-5000; Gilson GX-241 , Labcol Vario 4000; column: Cellulose SB 5m, 250×30 mm; eluent A: hexane + 0.1 vol. % diethylamine (99 %); eluent B: 2-propanol; isocratic: 50 % A + 50 % B; flow 50 ml/min; UV: 254 nm.
Analytical chiral HPLC method:
Instrument: Agilent HPLC 1260; column: Cellulose SB 3m, 100×4.6 mm; eluent A: hexane + 0.1 vol. % diethylamine (99 %); eluent B: 2-propanol; isocratic: 50 % A + 50 % B, flow 1.4 ml/min; temperature: 25°C; UV: 254 nm
Analytical chiral HPLC: Rt = 4.49 min.
Optical rotation:[a]D = 1.7° +/- 0.98° (c = 3.6 mg/2 ml, methanol)
Enantioselective synthesis confirmed the title compound as 3-(3-chloro-2-methoxyanilino)-2-(3-{[(2S)-1 ,4-dioxan-2-yl]methoxy}pyridin-4-yl)-1 ,5,6,7-tetrahydro-4/-/-pyrrolo[3,2-c]pyridin-4-one. 872 mg (95% purity, 72% yield) of the title compound were prepared in analogy to example 1 using A/-(3-chloro-2-methoxyphenyl)-4-{[(3-{[(2S)-1 ,4-dioxan-2-yl]methoxy}pyridin-4-yl)methyl]amino}-2-oxo-1 ,2,5,6-tetrahydropyridine-3-carbothioamide (intermediate 6-2, 1.23 g, 2.36 mmol) as starting material, followed by purification with preparative HPLC (method 10, gradient: 0.00-0.50 min 15% B, 0.50-6.00 min 15-55% B).
1H-NMR (400MHz, DMSO-d6): d [ppm]= 2.86 (t, 2H), 3.38 – 3.47 (m, 3H), 3.53 (td, 1 H), 3.69
– 3.78 (m, 2H), 3.83 (dd, 1 H), 3.88 (s, 3H), 3.90 (m, 1 H), 3.98 – 4.08 (m, 1 H), 4.12 – 4.18 (m, 1 H), 4.28 (dd, 1 H), 6.12 – 6.17 (quin, 1 H), 6.66 – 6.71 (m, 2H), 7.16 (s, 1 H), 7.28 (d, 1 H),
7.52 (s, 1 H), 8.04 (d, 1 H), 8.39 (s, 1 H), 11.07 (s, 1 H).
Analytical chiral HPLC: Rt = 4.46 min.
Optical rotation:[a]D = -12.5° +/- 0.52° (c = 5.6 mg/ l, chloroform)
PAT
- 4H-pyrrolo[3,2-c]pyridin-4-one compoundPublication Number: CN-114127064-BPriority Date: 2019-04-24Grant Date: 2023-12-26
- 4H-Pyrrolo[3,2-c]pyridin-4-one compoundsPublication Number: CN-117946100-APriority Date: 2019-04-24
- 4H-pyrrolo [3,2-c ] pyridin-4-one compoundsPublication Number: CN-117986251-APriority Date: 2019-04-24
- 4h-pyrrolo[3,2-c]pyridin-4-one compoundsPublication Number: TW-I849114-BPriority Date: 2019-04-24Grant Date: 2024-07-21
- 4H-pyrrolo[3,2-c]pyridin-4-one compoundPublication Number: KR-20220004103-APriority Date: 2019-04-24
- 4H-PYRROLO[3,2-C]PYRIDIN-4-ONE COMPOUNDSPublication Number: PE-20220254-A1Priority Date: 2019-04-24
- 4H-Pyrrolo [3,2-C] Pyridine-4-one CompoundPublication Number: JP-2022532850-APriority Date: 2019-04-24
- 4h-pyrrolo[3,2-c]pyridin-4-one compoundsPublication Number: US-2022298157-A1Priority Date: 2019-04-24
- 4H-PYRROL[3,2-C]PYRIDIN-4-ONE, ITS USES, PHARMACEUTICAL COMPOSITION, AND KIT OF PARTSPublication Number: BR-112021019998-B1Priority Date: 2019-04-24
- 4h-pyrrolo[3,2-c]pyridin-4-one compoundsPublication Number: WO-2020216781-A1Priority Date: 2019-04-24
- 4h-pyrrolo[3,2-c]pyridin-4-one compoundsPublication Number: TW-202106683-APriority Date: 2019-04-24
- 4H-pyrrolo(3,2-c)pyridin-4-one compoundsPublication Number: AU-2020262221-A1Priority Date: 2019-04-24
- 4H-pyrrolo [3,2-c ] pyridin-4-one compoundsPublication Number: CN-114127064-APriority Date: 2019-04-24
- 4h-pyrrolo[3,2-c]pyridin-4-one compoundsPublication Number: EP-3959211-A1Priority Date: 2019-04-24
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Medical uses
Sevabertinib is indicated for the treatment of adults with locally advanced or metastatic non-squamous non-small cell lung cancer whose tumors have HER2 (ERBB2) tyrosine kinase domain activating mutations.[1][2]
Adverse effects
The US prescribing information includes warnings and precautions for diarrhea, hepatotoxicity, interstitial lung disease/pneumonitis, ocular toxicity, pancreatic enzyme elevation, and embryo-fetal toxicity.[2]
History
Efficacy was evaluated in people with unresectable or metastatic, non-squamous non-small cell lung cancer with HER2 (ERBB2) tyrosine kinase domain activating mutations who had received prior systemic therapy and received sevabertinib in SOHO-01 (NCT05099172), an open-label, single-arm, multi-center, multi-cohort clinical trial.[2] HER2 (ERBB2) activating mutations were determined in tumor tissue or plasma by local laboratories prior to enrollment.[2]
The US Food and Drug Administration granted the application for sevabertinib priority review, breakthrough therapy, and orphan drug designations.[2]
Society and culture
Legal status
Sevabertinib was approved for medical use in the United States in November 2025.[3][4]
Names
Sevabertinib is the international nonproprietary name.[5]
Sevabertinib is sold under the brand name Hyrnuo.[1][3]
References
- “HYRNUO (sevabertinib) tablets, for oral use” (PDF). Bayer HealthCare Pharmaceuticals Inc. U.S. Food and Drug Administration.
- “FDA grants accelerated approval to sevabertinib for non-squamous non-small cell lung cancer”. U.S. Food and Drug Administration (FDA). 19 November 2025. Retrieved 21 November 2025. This article incorporates text from this source, which is in the public domain.
- “U.S. FDA Approves Hyrnuo (sevabertinib) for Previously Treated Patients with HER2-Mutated Locally Advanced or Metastatic Non-Squamous Non-Small Cell Lung Cancer” (Press release). Bayer. 20 November 2025. Retrieved 21 November 2025 – via Business Wire.
- “U.S. FDA grants accelerated approval to Bayer’s Hyrnuo (sevabertinib) for patients with previously treated advanced HER2-mutant non-small cell lung cancer”. Bayer (Press release). 20 November 2025. Retrieved 21 November 2025.
- World Health Organization (2025). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 93”. WHO Drug Information. 39 (1). hdl:10665/381075.
Further reading
- Le X, Kim TM, Loong HH, Prelaj A, Goh BC, Li L, et al. (November 2025). “Sevabertinib in Advanced HER2-Mutant Non-Small-Cell Lung Cancer”. The New England Journal of Medicine. 393 (18): 1819–1832. doi:10.1056/NEJMoa2511065. PMID 41104928.
- Siegel F, Siegel S, Kotýnková K, Karsli Uzunbas G, Korr D, Tomono H, et al. (October 2025). “Sevabertinib, a Reversible HER2 Inhibitor with Activity in Lung Cancer”. Cancer Discovery: OF1 – OF14. doi:10.1158/2159-8290.CD-25-0605. PMID 41090369.
External links
- “Sevabertinib”. NCI Drug Dictionary.
- “Sevabertinib ( Code – C185187 )”. EVS Explore.
- Clinical trial number NCT05099172 for “First in Human Study of BAY2927088 in Participants Who Have Advanced Non-small Cell Lung Cancer (NSCLC) With Mutations in the Genes of Epidermal Growth Factor Receptor (EGFR) and/or Human Epidermal Growth Factor Receptor 2 (HER2)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Hyrnuo |
| Other names | BAY2927088, sevabertinib hydrate (JAN JP) |
| License data | US DailyMed: Sevabertinib |
| Routes of administration | By mouth |
| Drug class | Antineoplastic |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| CAS Number | 2521285-05-0 |
| PubChem CID | 155234713 |
| DrugBank | DB21667 |
| ChemSpider | 129786615 |
| UNII | 2A7VPM5RWH |
| KEGG | D13098 |
| Chemical and physical data | |
| Formula | C24H25ClN4O5 |
| Molar mass | 484.94 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
/////sevabertinib, FDA 2025, APPROVALS 2025, Hyrnuo, 2A7VPM5RWH, BAY-2927088, BAY 2927088
Ziftomenib

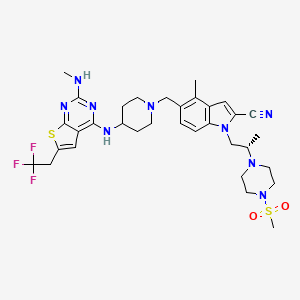
Ziftomenib
CAS 2134675-36-6
4MOD1F4ENC, KO 539
717.9 g/mol, C33H42F3N9O2S2
APPROVALS 2025, FDA 2025, 11/13/2025, Komzifti
4-methyl-5-[[4-[[2-(methylamino)-6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl]amino]piperidin-1-yl]methyl]-1-[(2S)-2-(4-methylsulfonylpiperazin-1-yl)propyl]indole-2-carbonitrile
To treat adults with relapsed or refractory acute myeloid leukemia with a susceptible nucleophosmin 1 mutation who have no satisfactory alternative treatment options
Ziftomenib, sold under the brand name Komzifti, is an anti-cancer medication used for the treatment of acute myeloid leukemia.[1] Ziftomenib is a menin inhibitor.[1] It is taken by mouth.[1]
Ziftomenib blocks the interaction between two proteins, menin (MEN1) and KMT2A (also known as mixed lineage leukemia protein, MLL).[2][3]
Ziftomenib was approved for medical use in the United States in November 2025.[4][5]
Ziftomenib, also known as KO539, is an orally bioavailable inhibitor of the menin-mixed lineage leukemia (MLL; myeloid/lymphoid leukemia; KMT2A) fusion protein, with potential antineoplastic activity. Upon oral administration, ziftomenib prevents the interaction between the two proteins menin and MLL, and thus the formation of the menin-MLL complex. This reduces the expression of downstream target genes and results in an inhibition of the proliferation of MLL-rearranged leukemic cells. The menin-MLL complex plays a key role in the survival, growth and proliferation of certain kinds of leukemia cells
SYN

syn
WO2022086986
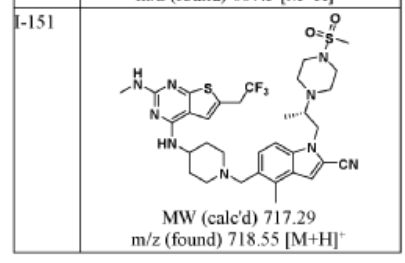
above similar not same
pat
WO2020069027
WO2018175746
WO2017161028
WO2018106820
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US239825810&_cid=P20-MI88RV-91969-1

PAT
- Inhibitors of the myst family of lysine acetyl transferasesPublication Number: US-2025051343-A1
- Small molecule inhibitors of dyrk/clk and uses thereofPublication Number: US-2025051325-A1
- VCP/p97 INHIBITOR FOR THE TREATMENT OF CANCERPublication Number: US-2025049800-A1
- RNAi agents for inhibiting expression of HIF-2 alpha (EPAS1), compositions thereof, and methods of usePublication Number: US-12221610-B2Grant Date: 2025-02-11
- Inhibitors of the myst family of lysine acetyl transferasesPublication Number: US-2025051343-A1
- Small molecule inhibitors of dyrk/clk and uses thereofPublication Number: US-2025051325-A1
- VCP/p97 INHIBITOR FOR THE TREATMENT OF CANCERPublication Number: US-2025049800-A1
- RNAi agents for inhibiting expression of HIF-2 alpha (EPAS1), compositions thereof, and methods of usePublication Number: US-12221610-B2Grant Date: 2025-02-11
- N-(3-hydroxy-4-piperidinyl)benzamide derivatives and pharmaceutical compositionsPublication Number: NZ-201856-APriority Date: 1981-10-01
- Novel n-(3-hydroxy-4-piperidinyl)benzamide derivativesPublication Number: EP-0076530-B1Priority Date: 1981-10-01Grant Date: 1985-12-11
- Novel N-(3-hydroxy-4-piperidinyl)benzamide derivativesPublication Number: EP-0076530-A2Priority Date: 1981-10-01
- Boron-containing polyphosphonates for the treatment of calcogenic tumorsPublication Number: JP-S5817120-APriority Date: 1981-06-30
- Novel n-aryl-piperazinealkanamidesPublication Number: IE-53465-B1Priority Date: 1981-06-23
- Novel((bis(aryl)methylene)-1-piperidinyl)-alkyl-pyrimidinonesPublication Number: IE-56180-B1Priority Date: 1982-11-01
- Novel ((bis(aryl)methylene)-1-piperidinyl)alkyl-pyrimidinonesPublication Number: EP-0110435-B1Priority Date: 1982-11-01Grant Date: 1989-01-04
- Novel ((bis(aryl)methylene)-1-piperidinyl)alkyl-pyrimidinonesPublication Number: EP-0110435-A1Priority Date: 1982-11-01
- NEW // BIS (ARYL) METHYLENE / -1-PIPERIDINYL / -ALKYL-PyrimidinonesPublication Number: BG-60538-B2Priority Date: 1982-11-01
- Process for preparing n-(3-hydroxy-4-piperidinyl)benzamide derivativesPublication Number: KR-860001584-B1Priority Date: 1982-07-30Grant Date: 1986-10-10
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Medical uses
Ziftomenib is indicated for the treatment of adults with relapsed or refractory acute myeloid leukemia with a susceptible nucleophosmin 1 mutation who have no satisfactory alternative treatment options.[1]
Adverse effects
The US prescribing information includes warnings and precautions for differentiation syndrome, QTc interval prolongation, and embryo-fetal toxicity.[4]
History
Efficacy was evaluated in KO-MEN-001 (NCT04067336), an open-label, single, arm, multi-center trial in 112 adults with relapsed or refractory acute myeloid leukemia with an nucleophosmin 1 mutation identified using next-generation sequencing or polymerase chain reaction.[4] Participants with nucleophosmin 1 mutations, including type A, B, and D mutations and other nucleophosmin 1 mutations likely to result in cytoplasmic localization of the nucleophosmin 1 protein, were enrolled.[4]
The US Food and Drug Administration granted the application for ziftomenib priority review, breakthrough therapy, and orphan drug designations.[4]
Society and culture
Legal status
Ziftomenib was approved for medical use in the United States in November 2025.[6]
Names
Ziftomenib is the international nonproprietary name.[7][8]
Ziftomenib is sold under the brand name Komzifti.[6]
References
- https://kuraoncology.com/wp-content/uploads/prescribinginformation.pdf
- “Ziftomenib”. NCI Cancer Dictionary. National Cancer Institute.
- Rausch J, Dzama MM, Dolgikh N, Stiller HL, Bohl SR, Lahrmann C, et al. (October 2023). “Menin inhibitor ziftomenib (KO-539) synergizes with drugs targeting chromatin regulation or apoptosis and sensitizes acute myeloid leukemia with MLL rearrangement or NPM1 mutation to venetoclax”. Haematologica. 108 (10): 2837–2843. doi:10.3324/haematol.2022.282160. PMC 10543165. PMID 37102614.
- “FDA approves ziftomenib for relapsed or refractory acute myeloid leukemia with a NPM1 mutation”. U.S. Food and Drug Administration (FDA). 13 November 2025. Retrieved 14 November 2025. This article incorporates text from this source, which is in the public domain.
- “Novel Drug Approvals for 2025”. U.S. Food and Drug Administration (FDA). 13 November 2025. Retrieved 14 November 2025.
- “Kura Oncology and Kyowa Kirin Announce FDA Approval of Komzifti (ziftomenib), the First and Only Once-Daily Targeted Therapy for Adults with Relapsed or Refractory NPM1-Mutated Acute Myeloid Leukemia” (Press release). Kura Oncology. 13 November 2025. Retrieved 14 November 2025 – via GlobeNewswire News Room.
- World Health Organization (2022). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 87”. WHO Drug Information. 36 (1). hdl:10665/352794.
- World Health Organization (2022). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 88”. WHO Drug Information. 36 (3). hdl:10665/363551.
Further reading
- Wang ES, Issa GC, Erba HP, Altman JK, Montesinos P, DeBotton S, et al. (October 2024). “Ziftomenib in relapsed or refractory acute myeloid leukaemia (KOMET-001): a multicentre, open-label, multi-cohort, phase 1 trial”. The Lancet. Oncology. 25 (10): 1310–1324. doi:10.1016/S1470-2045(24)00386-3. PMID 39362248.
External links
- Clinical trial number NCT04067336 for “First in Human Study of Ziftomenib in Relapsed or Refractory Acute Myeloid Leukemia” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Komzifti |
| Other names | KO-539; KO539 |
| AHFS/Drugs.com | Komzifti |
| License data | US DailyMed: Ziftomenib |
| Routes of administration | By mouth |
| Drug class | Antineoplastic |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2134675-36-6 |
| PubChem CID | 138497449 |
| IUPHAR/BPS | 11680 |
| DrugBank | DB17171 |
| ChemSpider | 115009296 |
| UNII | 4MOD1F4ENC |
| KEGG | D12419 |
| ChEMBL | ChEMBL5095038 |
| PDB ligand | K5O (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C33H42F3N9O2S2 |
| Molar mass | 717.88 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
////////Ziftomenib, APPROVALS 2025, FDA 2025, 4MOD1F4ENC, Komzifti
Elinzanetant
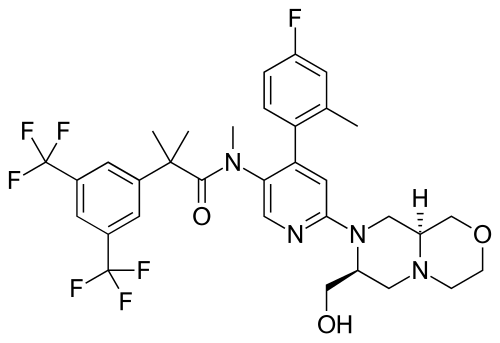
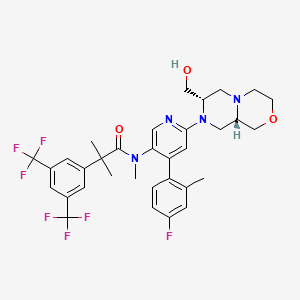
Elinzanetant
CAS 929046-33-3
MW 668.6 g/mol MF C33H35F7N4O3
N-[6-[(7S,9aS)-7-(hydroxymethyl)-3,4,6,7,9,9a-hexahydro-1H-pyrazino[2,1-c][1,4]oxazin-8-yl]-4-(4-fluoro-2-methylphenyl)-3-pyridinyl]-2-[3,5-bis(trifluoromethyl)phenyl]-N,2-dimethylpropanamide
FDA 10/24/2025, Lynkuet, To treat moderate-to-severe vasomotor symptoms due to menopause
BAY-3427080; GSK-1144814; NT-814, UNII-NZW2BOW35N
Elinzanetant, sold under the brand name Lynkuet, is a medication used for the treatment of moderate to severe vasomotor symptoms due to menopause.[4] It is an neurokinin 1 and neurokinin 3 receptor antagonist.[4] It was developed by Bayer Healthcare.[4] It is taken by mouth.[4]
Elinzanetant is a non-hormonal, selective, neurokinin 1 (NK-1) and neurokinin 3 (NK-3) receptor antagonist.[5] By blocking NK-1 and NK-3 receptors signaling, elinzanetant is postulated to normalize neuronal activity involved in thermo- and sleep regulation in the hypothalamus.[5]
Elinzanetant is an orally bioavailable neurokinin/tachykinin 1 receptor (NK1-receptor; NK1R; NK-1R) and NK3 receptor (NK-3R; NK3R) antagonist, that may be used to treat vasomotor symptoms in menopausal woman. Upon oral administration, elinzanetant targets, competitively binds to and blocks the activity of the NK1R and NK3R in the central nervous system (CNS), thereby inhibiting the binding of the endogenous ligands and neuropeptides substance P (SP; neurokinin-1; NK1) and neurokinin B (NKB). This inhibits NK1R/NK3R-mediated signal transduction and may prevent certain menopausal symptoms such as hot flashes. Neurokinin-mediated signaling may increase during hormone deficiency and may cause hot flashes.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021094247&_cid=P20-MHLSZY-53200-1
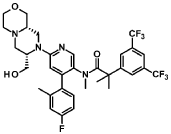
“Compound A” refers to 2-[3,5-Bis(trifluoromethyl)phenyl]-N-{4-(4-fluoro-2-methylphenyl)-6-[(7S,9aS)-7-(hydroxymethyl)hexahydropyrazino[2,l-c][l,4]oxazin-8(lH)-yl]-3-pyridi-nyl}-N,2-dimethylpropanamide, and has the chemical structure depicted below.
(Compound A).
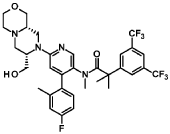
Example 8
2-[3.5-Bis(trifluoromethyl¾phenyl1-N-{4-(4-fluoro-2-methylphenyl¾-6-[(7S.9aS¾-7-(hvdroxymethyl¾hexa-hvdropyrazino[2,l-c1[l,41oxazin-8(lH)-yl1-3-pyridinyl}-N, 2-dimethyl propanamide as anhydrous crys talline form (Compound A)
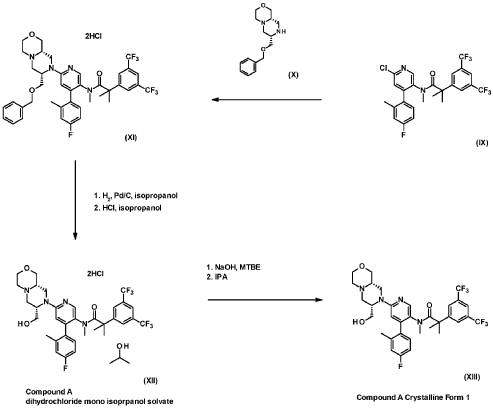
Example 7 (2-[3,5-Bis(trifluoromethyl)phenyl]-N-{4-(4-fluoro-2-methylphenyl)-6-[(7S,9aS)-7-(hy-droxymethyl)hexahydropyrazino[2,l-c][l,4]oxazin-8(lH)-yl]-3-pyridinyl}-N,2-dimethylpropanamide dihydrochloride salt mono-isopropanol solvate (Compound XII)) (3.4 kg), methyl-f-butyl ether (from now on, MTBE) (15.0 L/kg of Example 7) and NaOH 2.5N (4.9 L/kg of Example 7) were loaded, heated to 40QC and stirred for 10 to 30 min. The layers were settled for not less than 30 min at 40QC and the bottom aqueous layer discarded.
An aqueous solution of L-cysteine 9 wt% (5.0 L water per kg of Example 7+ 0.5 w/w L-cysteine per Ex ample 7) was added over the organic layer and stirred at 40QC for not less than 60 min. The layers were settled for not less than 30 min at 40QC and the bottom aqueous layer discarded.
Water (5.0 L/kg of Example 7) was added over the organic layer and stirred at 40QC for not less than 15 min. The layers were settled for not less than 60 min at 40QC and the bottom aqueous layer dis carded.
Water (5.0 L/kg of Example 7) was added over the organic layer and stirred at 40QC for not less than 15 min. The layers were settled for not less than 60 min at 40QC and the bottom aqueous layer dis carded.
The organic layer was concentrated at atmospheric pressure to 2.5 L/kg of Example 7. Iso-octane (8.3 L/kg of Example 7) was added at 50/55QC in not less than lh and the solution distilled under light vac uum to 4.0 L/kg of Example 7. A sample was taken for controlling the water and MTBE removal.
Isopropanol (0.8 L/kg of Example 7) was added and stirred at 65/75QC until total dissolution. The solu tion was cooled down to 45/55QC and filtered to remove any foreign matters. Iso-octane (4.5 L/kg of Example 7) was added and the batch heated to 70QC for not less than 30 min. The solution was cooled down to 50QC and seeded with a slurry of 2-[3,5-Bis(trifluoromethyl)phenyl]-N-{4-(4-fluoro-2-methylphenyl)-6-[(7S,9aS)-7-(hydroxymethyl)hexahydropyrazino[2,l-c][l,4]oxazin-8(lH)-yl]-3-pyridi-nyl}-N,2-dimethylpropanamide(0.008% w/w of Example 7) in iso-octane (0.07 L/kg of Example 7) and isopropanol (0.01 L/kg of Example 7). The seeds were aged at 50QC for not less than 3h and additional iso-octane (4.2 L/kg of Example 7) was added in not less than 3h keeping the temperature at 50/55QC. The slurry was held at 50QC for not less than 8h, cooled down to 0QC in not less than 5h and aged for not less than 3h before proceeding with the centrifugation step.
The slurry was centrifuged and the cake washed with iso-octane (2 x 3.3 L/kg of Example 7).
The wet product was dried under vacuum at 50QC to obtain 2.34 kg of the title compound (yield = 82.7%). This product was sieved for delumping to obtain 2.26kg of the title compound with a 99.8% purity as a white powder.
NMR spectrometer: Varian Agilent Mercury Vx 400 (16 scans, sw 6400 Hz, 25 °C).
*H NMR (400 MHz, DMSO-ds): d 8.02 (s, 1 H), 7.85 (s, 1 H), 7.74 (bd, 2 H), 7.22-6.92 (m, 3 H), 6.61 (s, 1 H), 4.70 (m, 1 H), 4.21 (bd, 1 H), 4.09 (bd, 1 H), 3.75 (m, 3 H), 3.55 (td, 11.3 Hz, 2.2 Hz, 1 H), 3.40 (bd, 1 H), 3.15 (t, 10.5 Hz, 1 H), 3.02 (d, 11.3 Hz, 1 H), 2.63 (d, 11.3 Hz, 1 H), ca. 2.5 (bd, 2 H), 2.31-2.00 (m, 7 H), 1.58-1.10 (m, 6 H).
SYN
- WO2007028654
- https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2007028654&_cid=P20-MHLT4M-58180-1
Example 34
2-[3,5-Bis(trifluoromethyl)phenyl]-yV-{4-(4-fluoro-2-methylphenyl)-6-[(7S,9aS)-7-(hydroxymethyl)hexahydropyrazino[2,1 -c][1 ,4]oxazin-8(1 H)-yl]-3-pyridinyl}-A/,2-dimethylpropanamide (E34)
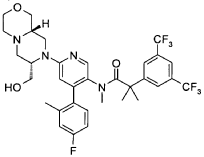
2-[3,5-bis(trifluoromethyl)phenyl]-Λ/-[6-[(7S,9aS)-7-({[(1 , 1 -dimethylethyl)(dimethyl)silyl]oxy}methyl)hexahydropyrazino[2,1-c][1 ,4]oxazin-8(1H)-yl]-4-(4-fluoro-2-methylphenyl)-3-pyridinyl]-Λ/,2-dimethylpropanamide (D24) (390 mg, 0.498 mmol) was dissolved 17 ml. of methanol. To this solution was added concentrated HCI (0.9 mL) at 00C, and stirring was continued at room temperature for 3h (complete conversion). The reaction mixture was loaded on a SCX cartridge and washed with MeOH. The product was eluted with 0.5 M methanolic ammonia. The product-containing fractions were evaporated, leaving the target compound as a white solid: 310 mg, 0.464 mmol, 93%.
UPLC/MS: m/z= 669 (M+1 ).
1H-NMR (DMSO-d6): δ (ppm) 8.07-7.97 (s, 1 H), 7.88-7.81 (s, 1 H), 7.79-7.69 (br. s, 2H), 7.19-7.11 (d, 1 H), 7.14-7.06 (br. s, 2H) 6.64-6.56 (s, 1 H), 4.75-4.65 (m, 1 H), 4.31-4.13 (br. S, 1 H), 4.15-4.01 (br. s, 1 H), 3.80-3.68 (m, 3H), 3.58-3.49 (t, 1 H); 3.43-3.34 (m, 1 H); 3.18-3.09 (t, 1 H); 3.04-2.98 (d, 1 H); 2.68-2.58 (d, 1 H); 2.51-2.45 (s, 3H); 2.20-2.13 (s, 3H); 2.29-2.00 (m, 4H); 1.54-1.39 (s, 3H); 1.39-1.28 (s, 3H).
SYN
Crystalline forms of a pyridine derivative
Publication Number: WO-2010015626-A1
Priority Date: 2008-08-05
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2010015626&_cid=P20-MHLT79-60873-1
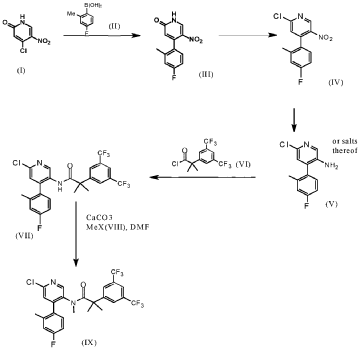
ntermediate 7
2-r3.5-bis(trifluoromethyl)phenvn-Λ/-f4-(4-fluoro-2-methylphenyl)-6-r(7S,9aS)-7-(hvdroxymethyl)hexahvdro-pyrazinoF2,1-c1f1 ,41oxazin-8(1/-/)-vn-3-pyridinyl}-Λ/.2-dimethylpropanamide
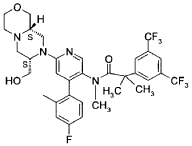
16.90 g of bis(trifluoromethyl)phenyl]-Λ/-[6-chloro-4-(4-fluoro-2-methylphenyl)-3-pyridinyl]- Λ/,2-dimethylpropanamide (WO 2005/002577) 4.58 g sodium tert-butoxide and 2.1O g Bis-(tri-terf-butylphposphine-palladium(O) catalyst was loaded into the vessel under nitrogen.
10.00 g intermediate 6 dissolved in 338 mL toluene was charged to afford a dark brown solution. The solution was heated to 800C and stirred for at least 16 h (thin suspension obtained).
The reaction mixture was cooled down to 20-25°C and 16.90 g celite was added to give a brown suspension. The suspension was filtered over 16.90 g celite and washed with 33.8 mL toluene. 338 mL sat. sodium bicarbonate solution was added and the biphasic system was stirred for 5 min. at 20-250C. After phase separation, the aqueous layer was extracted twice with 118 mL toluene. The combined organic layers were treated with 90 mL of a 10% aqueous cysteine solution and stirred for 1 h at 25°C. After phase separation the organic layer was treated again with 90 mL of a 10% cysteine-solution and stirred for a further 1 h at 25°C. After phase separation , the organic layer was washed with 85 mL half saturated sodium bicarbonate solution then solvent exchanged to dioxane. The dioxane solution was cooled down to 10-150C. 63.5 mL of 4M hydrogen chloride in dioxane was added at 10-150C over at least 10 min. The solution was warmed to 20-25°C and stirred for 2 h.
Dioxane was concentrated down to 85 mL at 45°C under reduced pressure. 85 mL water and 254 mL dichloromethane were added to the residue to give a thin suspension. The biphasic system was stirred for 5 min. at 20-250C. The layers were separated and the organic phase was washed with 33.8 mL saturated sodium bicarbonate solution at 20-25°C (pH adjusted to 7-8). The biphasic system was stirred for 5 min. at 20-250C and the organic layer separated and concentrated under reduced pressure at 500C to afford crude title compound as a pale brown solid. 8.00 g of the title compound (78.8% a/a HPLC) was dissolved in 16 mL ethyl acetate. The filter was loaded with 80 to 104 g silica gel and conditioned with ethyl acetate. The product solution was loaded on top of the column and chromatography was started using ethyl acetate as solvent. The product fractions were combined and partially concentrated at 45-50°C under reduced pressure. To the mixture was added 2.64 g to 4.00 g silicycle (Si-Thiol, 1.2 mmol/g) at rt and stirred for 2 h. Filtration over 8.00 g celite and washing with 32 mL ethyl acetate gave the filtrate which was concentrated to dryness at 45°C afford the title compound as a light brown solid. ( Weight yield 72%) 1H-NMR [ppm, CDCI3]: 8.04-7.91 , (m, 1 H); 7.77, (s, 1 H); 7.72-7.60, (m, 2H); 7.59-7.16, (m, 1H); 7.06-6.74, (m, 2H); 6.44, (s, 1 H); 4.64-4.43, (m, 1 H); 4.38-4.18, (m, 1 H); 4.07-3.96, (m, 2H); 3.95-3.76, (m, 3H); 3.76-3.61 , (m, 1 H); 3.37-3.27, (m, 1 H); 3.16-2.98, (m, 2H); 2.84-2.70, (m, 1 H); 2.67-2.51 , (m, 2H); 2.49-2.22, (m, 5H); 2.19-2.06, (m, 2H); 1.64-1.31 , (m, 5H), OH broad and not observed
LIT
- Elinzanetant: a phase III therapy for postmenopausal patients with vasomotor symptomsPublication Name: Expert Opinion on Investigational DrugsPublication Date: 2024-01-02PMID: 38224099DOI: 10.1080/13543784.2024.2305122
- Elinzanetant (NT-814), a Neurokinin 1,3 Receptor Antagonist, Reduces Estradiol and Progesterone in Healthy WomenPublication Name: The Journal of clinical endocrinology and metabolismPublication Date: 2021-02-24PMCID: PMC8277204PMID: 33624806DOI: 10.1210/clinem/dgab108
- Pleiotypic responses of regenerating liverPublication Name: Advances in Enzyme RegulationPublication Date: 1976PMID: 9791DOI: 10.1016/0065-2571(76)90023-6
PAT
Pyridine Derivatives and Their Use in the Treatment of Psychotic Disorders
Publication Number: US-2008269208-A1
Priority Date: 2005-06-06
- Pyridine Derivatives And Their Use In The Treatment Of Psychotic DisordersPublication Number: US-2011190276-A1Priority Date: 2005-09-09
- Pyridine derivatives and their use in the treatment of psychotic disordersPublication Number: US-7683056-B2Priority Date: 2005-09-09Grant Date: 2010-03-23
- Pyridine derivatives and their use in the treatment of psychotic disordersPublication Number: US-7919491-B2Priority Date: 2005-09-09Grant Date: 2011-04-05
- Pyridine derivatives and their use in the treatment of psychotic disordersPublication Number: US-8097618-B2Priority Date: 2005-09-09Grant Date: 2012-01-17
- Pyridine derivatives and their use in the treatment of psychotic disordersPublication Number: WO-2007028654-A1Priority Date: 2005-09-09
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Medical uses
Elinzanetant is indicated for the treatment of moderate to severe vasomotor symptoms associated with menopause.[4]
Society and culture
Legal status
In September 2025, the Committee for Medicinal Products for Human Use of the European Medicines Agency adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Lynkuet, intended for the treatment of moderate to severe vasomotor symptoms (hot flushes).[5] The applicant for this medicinal product is Bayer AG.[5]
Lynkuet was approved for medical use in the United States in October 2025.[6]
Names
Elinzanetant is the international nonproprietary name.[7]
Elinzanetant is sold under the brand name Lynkuet.[8]
References
- “Details for: Lynkuet”. Drug and Health Products Portal. 23 July 2025. Retrieved 28 September 2025.
- “Lynkuet product information”. Lynkuet. 23 July 2025. Retrieved 28 September 2025.
- “MHRA approves elinzanetant to treat moderate to severe vasomotor symptoms (hot flushes) caused by menopause”. Medicines and Healthcare products Regulatory Agency (Press release). 8 July 2025. Retrieved 28 September 2025.
- https://www.accessdata.fda.gov/drugsatfda_docs/label/2025/219469s000lbl.pdf
- “Lynkuet EPAR”. European Medicines Agency (EMA). 19 September 2025. Retrieved 27 September 2025. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- “Novel Drug Approvals for 2025”. U.S. Food and Drug Administration (FDA). 24 October 2025. Retrieved 29 October 2025.
- World Health Organization (2020). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 84”. WHO Drug Information. 34 (3). hdl:10665/340680.
- “Bayer’s Lynkuet (elinzanetant), the First and Only Neurokinin 1 and Neurokinin 3 Receptor Antagonist, Receives FDA Approval for Moderate to Severe Hot Flashes Due to Menopause” (Press release). Bayer. 24 October 2025. Retrieved 29 October 2025 – via Business Wire.
External links
- Clinical trial number NCT05042362 for “A Study to Learn More About How Well Elinzanetant Works and How Safe it is for the Treatment of Vasomotor Symptoms (Hot Flashes) That Are Caused by Hormonal Changes Over 26 Weeks in Women Who Have Been Through the Menopause (OASIS-1)” at ClinicalTrials.gov
- Clinical trial number NCT05099159 for “A Study to Learn More About How Well Elinzanetant Works and How Safe it is for the Treatment of Vasomotor Symptoms (Hot Flashes) That Are Caused by Hormonal Changes Over 26 Weeks in Women Who Have Been Through the Menopause (OASIS-2)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Lynkuet |
| Other names | BAY-3427080; GSK-1144814; NT-814 |
| License data | US DailyMed: Elinzanetant |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | CA: ℞-only[1][2]UK: POM (Prescription only)[3]US: ℞-only[4] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 929046-33-3 |
| PubChem CID | 16063568 |
| ChemSpider | 17223178 |
| UNII | NZW2BOW35N |
| KEGG | D12123 |
| CompTox Dashboard (EPA) | DTXSID101337049 |
| Chemical and physical data | |
| Formula | C33H35F7N4O3 |
| Molar mass | 668.657 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
///////elinzanetant, Lynkuet, FDA 2025, APPROVALS 2025, BAY 3427080, GSK 1144814, NT 814
Remibrutinib
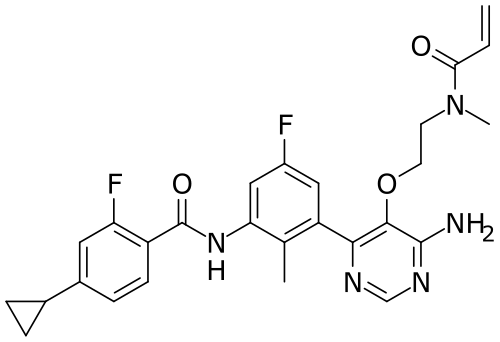
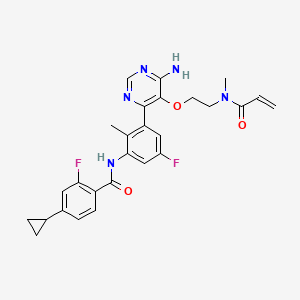
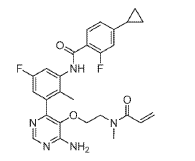
Remibrutinib
CAS 1787294-07-8
N-[3-[6-amino-5-[2-[methyl(prop-2-enoyl)amino]ethoxy]pyrimidin-4-yl]-5-fluoro-2-methylphenyl]-4-cyclopropyl-2-fluorobenzamide
MW 507.5 g/mol, MF C27H27F2N5O3
- LOU064
- NVP-LOU064-NXA
- LOU064-NXA
- I7MVZ8HDNU
- WHO 11062
APPROVALS 2025, FDA 2025, 9/30/2025, To treat chronic spontaneous urticaria in adults who remain symptomatic despite H1 antihistamine treatment
Remibrutinib, sold under the brand name Rhapsido, is a medication used for the treatment of chronic spontaneous urticaria.[1] Remibrutinib is an oral, small molecule kinase inhibitor that inhibits Bruton’s tyrosine kinase (BTK).[1] It is taken by mouth.[1]
SYN
Discovery of LOU064 (Remibrutinib), a Potent and Highly Selective Covalent Inhibitor of Bruton’s Tyrosine Kinase
https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b01916
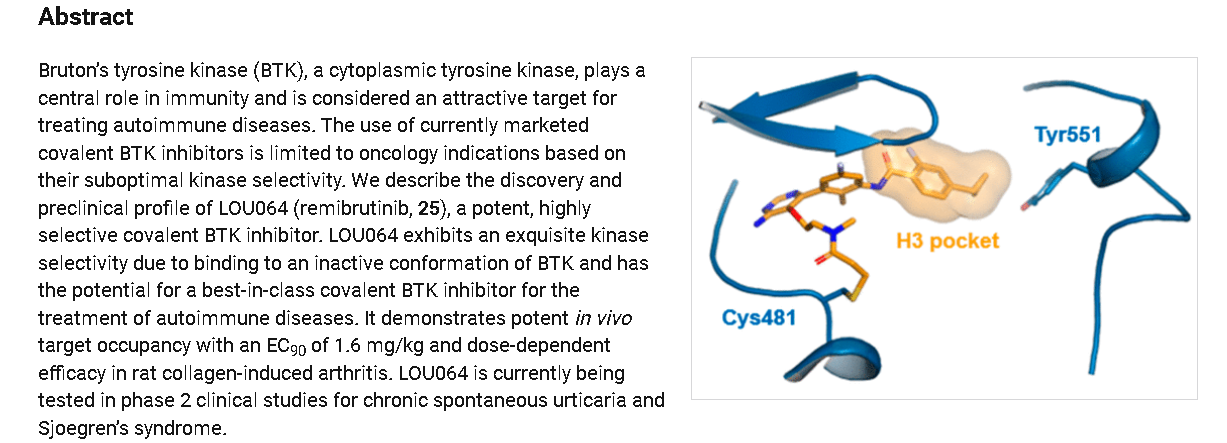
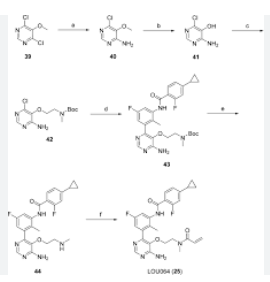
SYN
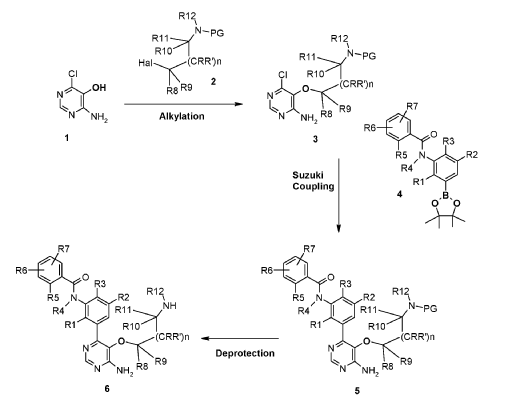
- WO2015079417
- https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015079417&_cid=P12-MH1R14-25744-1
Example 6
N-(3-(6-Amino-5-(2-(N-methylacrylamido)ethoxy)pyrimidin-4-yl)-5-fluoro-2- methylphenyl)-4-cyclopropyl-2-fluorobenzamide
(1) tert-Butyl (2-((4-amino-6-chloropyrimidin-5-yl)oxy)ethyl)(methyl)carbamate, INT 8
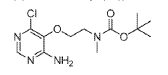
To a solution of 4-amino-6-chloropyrimidin-5-ol (content 90%, 2.00 g, 12.37 mmol) in THF (120 mL) was added N-Boc-N-methyl-2-hydroxyethylamine (6.07 g, 34.64 mmol) followed by SMOPEX-301 (1 mmol/g, 30.90 g, 30.90 mmol). Then, a solution of DIAD (6.01 mL, 30.52 mmol) in THF (20 mL) was added slowly. The reaction mixture was stirred at 60 °C for 3 hr. The mixture was filtered through a pad of Celite. The filtrate was concentrated to afford an oil which was triturated with EtOAc and a white precipitate was formed. The solid was filtered off to afford INT 8. The mother liquor was concentrated and the residue was purified by flash chromatography (silica; DCM/EtOAc gradient, 0- 100%) to afford more INT 8 as a beige solid.
UPLC-MS: MS (ESI): [M+H]+ 303.1, rt = 0.86 min. 1H NMR (DMSO-d6): δ (ppm) 7.97 (s, 1H), 7.26 (s, br, 2H), 4.02-3.93 (m, 2H), 3.54 (t, 2H), 2.89 (s, br, 3H), 1.39 (s, 9H).
(2) tert-Butyl (2-((4-amino-6-(3-(4-cyclopropyl-2-fluorobenzamido)-5-fluoro-2- methylphenyl)pyrimidin-5-yl)oxy)ethyl)(methyl)carbamate, INT 9
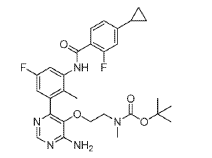
To a solution of INT 8 (447 mg, 1.48 mmol) in DME (7.0 mL) and water (1.0 mL) was added INT 5 (638 mg, 1.54 mmol) followed by aqueous sodium carbonate solution (1 M, 4.21 mL, 4.21 mmol). The mixture was degassed with argon for 10 min and bis(triphenylphosphine)palladium(II) dichloride (49.2 mg, 0.070 mmol) was added. The reaction mixture was stirred at 110 °C for 10 min in a microwave reactor. More INT 5 (232 mg, 0.56 mmol) was added and the reaction mixture was stirred at 110 °C for an additional 15 min in a microwave reactor. The mixture was partitioned between saturated aqueous sodium hydrogen carbonate solution and EtOAc. The organic layer was washed with water and brine, dried over magnesium sulfate, filtered and concentrated. The residue was purified by flash chromatography (silica; DCM/EtOAc gradient, 0-100%) to afford INT 9 as an off-white solid.
UPLC-MS: MS (ESI): [M+H]+ 554.3, rt = 1.21 min. 1H NMR (DMSO-d6): δ (ppm) rotamers 9.76 (s, 1H), 8.19 (s, 1H), 7.74-7.53 (m, 2H) 7.20-6.85 (m, 5H), 3.57-3.48 (m, 2H), 3.29- 3.15 (m, 2H), 2.58 (s, 3H), 2.08-1.99 (overlapping s, 3H and m, 1H), 1.34 and 1.28 (s, 9H), 1.10-1.02 (m, 2H), 0.84-0.77 (m, 2H).
(3) N-(3-(6-Amino-5-(2-(methylamino)ethoxy)pyrimidin-4-yl)-5-fluoro-2- methylphenyl)-4-cyclopropyl-2-fluorobenzamide, INT 10
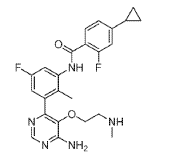
To a solution of INT 9 (335 mg, 0.61 mmol) in DCM (5.0 mL) was added TFA (0.47 mL, 6.05 mmol). The reaction mixture was stirred at RT for 15 hr. The mixture was concentrated under reduced pressure. The residue was dried in vacuum to afford INT 10 as theTFA salt as a brown oil.
UPLC-MS: MS (ESI): [M+H]+ 454.3, rt = 0.73 min. 1H NMR (DMSO-d6): δ (ppm) 10.02 (s, 1H), 9.07-8.13 (s, v br, number of H cannot be assigned), 8.58 (s, 1H), 8.51 (s, br, 2H), 7.71-7.61 (m, 2H), 7.29-7.22 (m, 1H), 7.14-7.05 (m, 2H), 3.75-3.65 (m, 2H), 3.16-3.07 (m, 2H), 2.48 (s, 3H, overlapping with solvent peak), 2.12 (s, 3H), 2.10-1.99 (m, 1H), 1.11-1.03 (m, 2H), 0.83-0.76 (m, 2H).
(4) N-(3-(6-Amino-5-(2-(N-methylacrylamido)ethoxy)pyrimidin-4-yl)-5-fluoro-2-methylphenyl)-4-cyclopropyl-2-fluorobenzamide
To a solution of acrylic acid (62 mg, 0.87 mmol) in DMF (4.0 mL) was added DIPEA (0.302 mL, 1.73 mmol) followed by T3P solution (50% in DMF) (0.438 mL, 0.750 mmol). The mixture was stirred at RT for 30 min. To a solution of INT 10 (containing 3.0 eq TFA, content 90%, 510 mg, 0.577 mmol) and DIPEA (0.302 mL, 1.731 mmol) in DMF (2.0 mL) at 0 °C was added dropwise the above solution. The reaction mixture was stirred at 0 °C for 30 min. The mixture was diluted with water and extracted with EtOAc. The organic layer was washed with water (2x) and brine (2x), dried over magnesium sulfate, filtered and concentrated. The residue was purified by flash chromatography (silica;
DCM/(MeOH with 2% aqueous ammonium hydroxide) gradient, 0-9%) to afford the title compound Example 6 as a white solid.
UPLC-MS: MS (ESI): [M+H]+ 508.3, rt = 0.95 min. 1H NMR (DMSO-d6): δ (ppm) rotamers 9.77 and 9.56 (s, total 1H), 8.25-8.14 (m, 1H), 7.79-7.50 (m, 2H), 7.17-6.93 (m, 5H), 6.70-6.55 (m, 1H), 6.06 (t, 1H), 5.59 (d, 1H), 3.63-3.40 (m, 4H), 2.80 and 2.49 (s, total 3H, peak at 2.49 overlapping with solvent peak), 2.09-1.93 (m, 4H), 1.11-1.00 (m, 2H), 0.85-0.76 (m, 2H).
PAT
- US9512084,
- https://patentscope.wipo.int/search/en/detail.jsf?docId=US133778840&_cid=P12-MH1R60-30520-1
SYN
Publication Date: 2020
Publication Name: Synfacts
PAT
- Amino Pyrimidine DerivativesPublication Number: US-2023312483-A1Priority Date: 2013-11-29
- A complex of bisphenol and a phosphorus compound and heat-developable image-recording material containing the samePublication Number: DE-60121375-T2Priority Date: 2000-01-11Grant Date: 2007-07-05
- Photothermographic materialPublication Number: DE-60011207-T2Priority Date: 1999-10-26Grant Date: 2005-06-23
- CD19 binding molecules and uses thereofPublication Number: US-12221481-B2Grant Date: 2025-02-11

AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Medical uses
Remibrutinib is indicated for the treatment of chronic spontaneous urticaria in adults who remain symptomatic despite H1 antihistamine treatment[1]
Society and culture
Legal status
Remibrutinib was approved for medical use in the United States in September 2025.[2]
Names
Remibrutinib is the international nonproprietary name.[3]
Remibrutinib is sold under the brand name Rhapsido.[2]
References
- https://www.novartis.com/us-en/sites/novartis_us/files/rhapsido.pdf
- “Novartis receives FDA approval for Rhapsido (remibrutinib), the only oral, targeted BTKi treatment for chronic spontaneous urticaria (CSU)” (Press release). Novartis Pharmaceuticals. 30 September 2025. Retrieved 1 October 2025 – via PR Newswire.
- World Health Organization (2020). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 83”. WHO Drug Information. 34 (1). hdl:10665/339768.
Further reading
- Maurer, Marcus; Berger, William; Giménez-Arnau, Ana; Hayama, Koremasa; Jain, Vipul; Reich, Adam; et al. (December 2022). “Remibrutinib, a novel BTK inhibitor, demonstrates promising efficacy and safety in chronic spontaneous urticaria”. The Journal of Allergy and Clinical Immunology. 150 (6): 1498–1506.e2. doi:10.1016/j.jaci.2022.08.027. hdl:10230/55511. ISSN 1097-6825. PMID 36096203.
- Maurer, Marcus; Giménez-Arnau, Ana; Jain, Vipul; Tillinghast, Jeffrey; Tolcachier, Alberto; Nigen, Simon; et al. (February 2022). “Remibrutinib Treatment Improves Quality of Life in Patients with Chronic Spontaneous Urticaria”. Journal of Allergy and Clinical Immunology. 149 (2): AB179. doi:10.1016/j.jaci.2021.12.589. S2CID 246522006.
External links
- Clinical trial number NCT05030311 for “A Phase 3 Study of Efficacy and Safety of Remibrutinib in the Treatment of CSU in Adults Inadequately Controlled by H1 Antihistamines (REMIX-1)” at ClinicalTrials.gov
- Clinical trial number NCT05032157 for “A Phase 3 Study of Efficacy and Safety of Remibrutinib in the Treatment of CSU in Adults Inadequately Controlled by H1-antihistamines (REMIX-2)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Rhapsido |
| License data | US DailyMed: Remibrutinib |
| Routes of administration | By mouth |
| ATC code | L04AA60 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1787294-07-8 |
| PubChem CID | 118107483 |
| IUPHAR/BPS | 10457 |
| DrugBank | DB16852 |
| ChemSpider | 78317000 |
| UNII | I7MVZ8HDNU |
| KEGG | D12285 |
| ChEMBL | ChEMBL4483575 |
| PDB ligand | N6Z (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C27H27F2N5O3 |
| Molar mass | 507.542 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
- Maurer M, Berger W, Gimenez-Arnau A, Hayama K, Jain V, Reich A, Haemmerle S, Lheritier K, Walsh P, Xia S, Storim J: Remibrutinib, a novel BTK inhibitor, demonstrates promising efficacy and safety in chronic spontaneous urticaria. J Allergy Clin Immunol. 2022 Dec;150(6):1498-1506.e2. doi: 10.1016/j.jaci.2022.08.027. Epub 2022 Sep 9. [Article]
- Nuesslein-Hildesheim B, Ferrero E, Schmid C, Huck C, Smith P, Tisserand S, Rubert J, Bornancin F, Eichlisberger D, Cenni B: Remibrutinib (LOU064) inhibits neuroinflammation driven by B cells and myeloid cells in preclinical models of multiple sclerosis. J Neuroinflammation. 2023 Aug 26;20(1):194. doi: 10.1186/s12974-023-02877-9. [Article]
- Bozek A, Reich A: Evaluating remibrutinib in the treatment of chronic spontaneous urticaria. Immunotherapy. 2025 May;17(7):479-484. doi: 10.1080/1750743X.2025.2510892. Epub 2025 Jun 2. [Article]
- Kaul M, End P, Cabanski M, Schuhler C, Jakab A, Kistowska M, Kinhikar A, Maiolica A, Sinn A, Fuhr R, Cenni B: Remibrutinib (LOU064): A selective potent oral BTK inhibitor with promising clinical safety and pharmacodynamics in a randomized phase I trial. Clin Transl Sci. 2021 Sep;14(5):1756-1768. doi: 10.1111/cts.13005. Epub 2021 Apr 9. [Article]
- Gimeno R, Ribas-Llaurado C, Pesque D, Andrades E, Cenni B, Ambros B, Pujol R, Gimenez-Arnau AM: Remibrutinib inhibits hives effector cells stimulated by serum from chronic urticaria patients independently of FcepsilonR1 expression level and omalizumab clinical response. Clin Transl Allergy. 2023 Mar;13(3):e12227. doi: 10.1002/clt2.12227. [Article]
- Dorner T, Kaul M, Szanto A, Tseng JC, Papas AS, Pylvaenaeinen I, Hanser M, Abdallah N, Grioni A, Santos Da Costa A, Ferrero E, Gergely P, Hillenbrand R, Avrameas A, Cenni B, Siegel RM: Efficacy and safety of remibrutinib, a selective potent oral BTK inhibitor, in Sjogren’s syndrome: results from a randomised, double-blind, placebo-controlled phase 2 trial. Ann Rheum Dis. 2024 Feb 15;83(3):360-371. doi: 10.1136/ard-2023-224691. [Article]
- FDA Approved Drug Products: RHAPSIDO (remibrutinib) tablets, for oral use [Link]
- Novartis: Novartis receives FDA approval for Rhapsido® (remibrutinib), the only oral, targeted BTKi treatment for chronic spontaneous urticaria (CSU) [Link]
//////////Remibrutinib, APPROVALS 2025, FDA 2025, Rhapsido, LOU064, NVP-LOU064-NXA, LOU064-NXA, I7MVZ8HDNU, WHO 11062
Fovinaciclib
Fovinaciclib
CAS 2146171-49-3
MF C29H40N8OS
Exact Mass: 548.3046
Molecular Weight: 548.75
7-cyclopentyl-N,N-dimethyl-2-({5-[4-(4-methylpiperazin-1-yl)piperidin-1-yl]pyridin-2-yl}amino) thieno[3,2-d]pyrimidine-6-carboxamide
7-cyclopentyl-N,N-dimethyl-2-((5-(4-(4-methylpiperazin-1-yl)piperidin-1-yl)pyridin-2-yl)amino)thieno[3,2-d]pyrimidine-6-carboxamide
7-cyclopentyl-N, N-dimethyl-2- ( (5- (4- (4-methylpiperazin-1-yl) piperidin-1-yl) pyridin-2-yl) amino) thieno [3, 2-d] pyrimidine-6-carboxamide
7-Cyclopentyl-N,N-dimethyl-2-((5-(4-(1-methylpiperidin-4-yl)piperazin-1-yl)pyridin-2-yl)amino Thieno[3,2-d]pyrimidine-6-carboxamide
cyclin dependent kinase inhibitor, antineoplastic, Fovinaciclibum, LPW3H579X8, inzhou Aohong Pharmaceutical Co
- OriginatorChongqing Fochon Pharmaceutical
- DeveloperAhon Pharmaceutical; Chongqing Fochon Pharmaceutical; Shanghai Fosun Pharmaceutical
- Class2 ring heterocyclic compounds; Amides; Amines; Antineoplastics; Cyclopentanes; Piperazines; Piperidines; Pyridines; Pyrimidines; Small molecules; Thiophenes
- Mechanism of ActionCyclin-dependent kinase 4 inhibitors; Cyclin-dependent kinase 6 inhibitors
- MarketedHER2 negative breast cancer
- No development reportedSolid tumours
- 04 Sep 2025Chemical structure information added.
- 02 Sep 2025Launched for HER2-negative-breast-cancer (Late-stage disease, Second-line therapy or greater) in China (PO) (Shanghai Henlius Biotech pipeline, September 2025)
- 26 Aug 2025Registered for HER2-negative-breast-cancer (Late-stage disease, Second-line therapy or greater) in China (PO) prior to August 2025
Fovinaciclib is an orally bioavailable inhibitor of cyclin-dependent kinase (CDK) types 4 (CDK4) and 6 (CDK6), with potential antineoplastic activity. Upon administration, fovinaciclib selectively inhibits CDK4 and CDK6, which inhibits the phosphorylation of retinoblastoma protein (Rb) early in the G1 phase, prevents CDK-mediated G1/S transition and leads to cell cycle arrest. This suppresses DNA replication and decreases tumor cell proliferation. CDK4 and 6 are serine/threonine kinases that are upregulated in many tumor cell types and play key roles in the regulation of both cell cycle progression from the G1-phase into the S-phase and cell proliferation.
On May 29, 2025, China’s National Medical Products Administration (NMPA) approved the Class 1 innovative drug Fovinaciclib (CDK4&6 inhibitor), developed by Jinzhou Aohong Pharmaceutical Co., Ltd. This medication, in combination with fulvestrant, is indicated for the treatment of adult patients with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative recurrent or metastatic breast cancer, who have experienced disease progression following prior endocrine therapy.
Notably, Fovinaciclib represents an excellent example of scaffold hopping—its design replaces the pyrrolo-pyrimidine core of Ribociclib (first approved on March 13, 2017) with a thieno-pyrimidine ring.
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN236278427&_cid=P21-MGRD95-18783-1
| Example 3 |
| 7-Cyclopentyl-N,N-dimethyl-2-((5-(4-(1-methylpiperidin-4-yl)piperazin-1-yl)pyridin-2-yl)amino Thieno[3,2-d]pyrimidine-6-carboxamide (3) |
According to the synthesis method of Example 2, CH
3 CHO replaced by CH
2 O, to prepare the title compound 7-cyclopentyl-N,N-dimethyl-2-((5-(4-(1-methylpiperidin-4-yl)piperazin-1-yl)pyridin-2-yl)amino)thieno[3,2-d]pyrimidine-6-carboxamide (3). MS-ESI (m/z): 549 [M+1] + .
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017193872&_cid=P21-MGRDEF-24321-1
[0266]
7-cyclopentyl-N, N-dimethyl-2- ( (5- (4- (4-methylpiperazin-1-yl) piperidin-1-yl) pyridi n-2-yl) amino) thieno [3, 2-d] pyrimidine-6-carboxamide (5)
[0267]
To a solution of 7-cyclopentyl-N, N-dimethyl-2- ( (5- (4- (piperazin-1-yl) piperidin-1-yl) pyridin-2-yl) amino) thieno [3, 2-d] pyrimidine-6-carboxamide (4) (1.5 g, 2.8 mmol) in DCM (45 mL) was added NaBH (OAc) 3(3.56 mg, 16.8 mmol) followed by CH 2O (40%in water, 252 mg, 3.4 mmol) . The mixture was stirred at r.t. for 30 min. The mixture was diluted with saturated aqueous NaHCO 3(100 mL) and extracted with DCM (2 × 30 mL) . The extracts were dried over Na 2SO 4. Solvents were evaporated under reduced pressure. The residue was purified by column chromatography on silica gel eluting with 96: 3: 1 DCM/methanol/ammonia to give 7-cyclopentyl-N, N-dimethyl-2- ( (5- (4- (4-methylpiperazin-1-yl) piperidin-1-yl) pyridin-2-yl) amino) thieno [3, 2-d] pyrimidine-6-carboxamide (5) . MS-ESI (m/z) : 549 [M + 1] +.
PAT
- Certain protein kinase inhibitorsPublication Number: JP-2019516790-APriority Date: 2016-05-07
- Certain protein kinase inhibitorsPublication Number: US-2019209566-A1Priority Date: 2016-05-07
- Certain protein kinase inhibitorsPublication Number: WO-2017193872-A1Priority Date: 2016-05-07
- Certain protein kinase inhibitorsPublication Number: US-10835535-B2Priority Date: 2016-05-07Grant Date: 2020-11-17
- A class of protein kinase inhibitorsPublication Number: CN-109153686-BPriority Date: 2016-05-07Grant Date: 2021-04-30
- specific protein kinase inhibitorsPublication Number: KR-102374033-B1Priority Date: 2016-05-07Grant Date: 2022-03-14
- Certain protein kinase inhibitorsPublication Number: EP-3452484-B1Priority Date: 2016-05-07Grant Date: 2023-07-05
- Certain protein kinase inhibitorsPublication Number: ES-2954148-T3Priority Date: 2016-05-07Grant Date: 2023-11-20
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////////Fovinaciclib, CHINA 2025, APPROVALS 2025, cyclin dependent kinase inhibitor, antineoplastic, Fovinaciclibum, LPW3H579X8, inzhou Aohong Pharmaceutical Co
Imlunestrant

Imlunestrant
CAS 2408840-26-4
as tosylate: 2408840-41-3
(5R)-5-[4-[2-[3-(fluoromethyl)azetidin-1-yl]ethoxy]phenyl]-8-(trifluoromethyl)-5H-chromeno[4,3-c]quinolin-2-ol
- (5r)-5-(4-(2-(3-(fluoromethyl)azetidin-1-yl)ethoxy)phenyl)-8-(trifluoromethyl)-5h-(1)benzopyrano(4,3-c)quinolin-2-ol
- 5h-(1)benzopyrano(4,3-c)quinolin-2-ol, 5-(4-(2-(3-(fluoromethyl)-1-azetidinyl)ethoxy)phenyl)-8-(trifluoromethyl)-, (5r)-
MF C29H24F4N2O3 MW 524.516
FDA 9/25/2025, Inluriyo, LY3484356, LY-3484356, To treat estrogen receptor-positive, human epidermal growth factor receptor 2-negative, estrogen receptor-1-mutated advanced or metastatic breast cancer with disease progression following at least one line of endocrine therapy
Imlunestrant, sold under the brand name Inluriyo, is an anti-cancer medication used for the treatment of breast cancer.[1] It is an is an estrogen receptor antagonist.[1] It is used as the salt, imlunestrant tosylate.[2] It is taken by mouth.[1] It was developed by Eli Lilly and Company.[2]
The most common adverse events and laboratory abnormalities include decreased hemoglobin, musculoskeletal pain, decreased calcium, decreased neutrophils, increased AST, fatigue, diarrhea, increased ALT, increased triglycerides, nausea, decreased platelets, constipation, increased cholesterol, and abdominal pain.[2]
Imlunestrant was approved for medical use in the United States in September 2025.[2]
SYN
- Imlunestrant with or without Abemaciclib in Advanced Breast CancerPublication Name: The New England journal of medicinePublication Date: 2025-03-27PMID: 39660834DOI: 10.1056/nejmoa2410858
- Targeting the Estrogen Receptor for the Treatment of Breast Cancer: Recent Advances and ChallengesPublication Name: Journal of Medicinal ChemistryPublication Date: 2023-06-28PMID: 37377342DOI: 10.1021/acs.jmedchem.3c00136
- Novel endocrine therapies: What is next in estrogen receptor positive, HER2 negative breast cancer?Publication Name: Cancer Treatment ReviewsPublication Date: 2023-06PMID: 37146385DOI: 10.1016/j.ctrv.2023.102569
- Oral Selective Estrogen Receptor Degraders (SERDs) as a Novel Breast Cancer Therapy: Present and Future from a Clinical PerspectivePublication Name: International Journal of Molecular SciencesPublication Date: 2021-07-22PMCID: PMC8345926PMID: 34360578DOI: 10.3390/ijms22157812
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US281655517&_cid=P12-MG7DCV-14904-1
Example 1A
5-(4-{2-[3-(Fluoromethyl)azetidin-1-yl]ethoxy}phenyl)-8-(trifluoromethyl)-5H-[1]benzopyrano[4,3-c]quinolin-2-ol, Isomer 1Separate the two enantiomers of 5-(4-{2-[3-(fluoromethyl)azetidin-1-yl]ethoxy}phenyl)-8-(trifluoromethyl)-5H-[1]benzopyrano[4,3-c]quinolin-2-ol by chiral SFC with the following conditions: Column: LUX® Cellulose-1, 5×25 cm; eluting with a mobile phase of 30% iPrOH (with 0.5% DMEA) in CO 2; column temperature: 40° C.; flow rate: 300 g/minute; UV detection wavelength: 270 nm to give Example 1A as the first eluting enantiomer (Isomer 1). ES/MS (m/z): 525.2 (M+H). Confirm enantiomeric enrichment of Isomer 1 by chiral analytical SFC, >99% ee, t (R): 1.30 minutes; column: CHIRALCEL® OD-H, 4.6×150 mm; eluting with a mobile phase of 30% MeOH (0.2% IPA) in CO 2; column temperature: 40° C.; flow rate: 5 mL/minute; UV detection wavelength: 225 nm. Isolate the title compound of Example 1B to give the second eluting enantiomer (Isomer 2). ES/MS (m/z): 525.2 (M+H). Confirm enantiomeric enrichment of Isomer 2 by chiral analytical SFC, 98% ee, t (R): 2.03 minutes; column: CHIRALCEL® OD-H, 4.6×150 mm; eluting with a mobile phase of 30% MeOH (0.2% IPA) in CO 2; column temperature: 40° C.; flow rate: 5 mL/minute; UV detection wavelength: 225 nm.
Alternate Preparation Example 1B
Crystalline 5-(4-{2-[3-(Fluoromethyl)azetidin-1-yl]ethoxy}phenyl)-8-(trifluoromethyl)-5H-[1]benzopyrano[4,3-c]quinolin-2-ol, Isomer 2
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020014435&_cid=P12-MG7DHN-18354-1
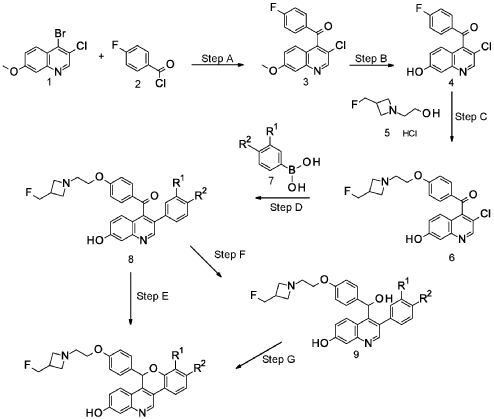
EXAMPLE 1
Racemic 5-(4-{2-[3-(Fluoromethyl)azetidin-l-yl]ethoxy}phenyl)-8-(trifluoromethyl)-5H- [ 1 ]benzopyrano[4,3 -c]quinolin-2-ol
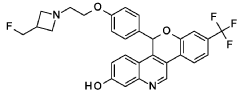
Cool a solution of (4-{2-[3-(fluoromethyl)azetidin-l-yl]ethoxy}phenyl){3-[2-fluoro-4-(trifluoromethyl)phenyl]-7-hydroxyquinolin-4-yl}methanone (5.27 g, 9.71 mmol) in 1,4-dioxane (100 mL) to 5 °C. Add lithium triethylborohydride (1 M in THF, 30.0 mL, 30.0 mmol). Remove the cooling bath and stir for 1.5 hours at room temperature. Quench the mixture with water. Add saturated NH4Cl solution and EtOAc. Separate the layers and extract the aqueous layer with EtOAc. Combine the organic extracts, dry over anhydrous MgS04, filter, and concentrate the filtrate. Dissolve the crude residue in THF (100 mL).
Add sodium hydride (60% in mineral oil, 1.94 g, 48.5 mmol). Reflux the solution for 1.5 hours. Add additional sodium hydride (60% in mineral oil, 1.94 g, 48.5 mmol), then reflux for an additional 30 minutes. Cool the solution to room temperature and quench with water. Add EtOAc and saturated NH4Cl solution. Separate the layers and extract the aqueous layer with EtOAc. Combine the organic extract, dry over anhydrous MgS04, filter, and concentrate the filtrate. Purify the residue by silica gel column chromatography eluting with a gradient of 5-7% MeOH in DCM to give the title compound (3.70 g, 72%) as a light yellow foam. ES/MS (m/z): 525.2 (M+H).
Prepare the following compounds in a manner essentially analogous to the method of Example 1, with the following variations in procedure. For the reduction, use 3 to 5 equivalents of lithium triethylborohydride with reaction times from 30 minutes to one hour and drying of the organic layers over magnesium sulfate or sodium sulfate. ETse the crude residue directly or purify by silica gel column chromatography eluting with a gradient of 0-5-7.5-10% MeOH in DCM before cyclization. Complete the cyclization by refluxing in THF for up to 16 hours, or in DMF, from 2 hours at room temperature for Ex 2, to 2 hours at 85 °C for Ex 8. Extract with DCM or EtOAc and dry organic layers over magnesium sulfate or sodium sulfate. Purify by silica gel column chromatography using up to 10% (MeOH or 7 M ammoniated MeOH) in DCM (Ex 2: gradient 0-10% MeOH in DCM; Ex 5: gradient 4-10% 7 M ammoniated MeOH in DCM; Ex 8: gradient 5-7.5% 7 M ammoniated MeOH in DCM) or by high pH reversed phase HPLC as noted.
EXAMPLE 1A
-(4-{2-[3-(Fluoromethyl)azetidin-l-yl]ethoxy}phenyl)-8-(trifluoromethyl)-5H- [l]benzopyrano[4,3-c]quinolin-2-ol, Isomer 1
and
EXAMPLE 1B
5-(4-{2-[3-(Fluoromethyl)azetidin-l-yl]ethoxy}phenyl)-8-(trifluoromethyl)-5H- [l]benzopyrano[4,3-c]quinolin-2-ol, Isomer 2
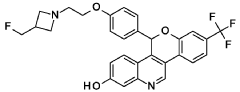
Separate the two enantiomers of 5-(4-{2-[3-(fluoromethyl)azetidin-l-yl]ethoxy}phenyl)-8-(trifluoromethyl)-5H-[l]benzopyrano[4,3-c]quinolin-2-ol by chiral SFC with the following conditions: Column: LUX® Cellulose-l, 5 x 25 cm; eluting with a mobile phase of 30% iPrOH (with 0.5% DMEA) in C02; column temperature: 40 °C; flow rate: 300 g/minute; UV detection wavelength: 270 nm to give Example 1 A as the first eluting enantiomer (Isomer 1). ES/MS (m/z): 525.2 (M+H). Confirm enantiomeric enrichment of Isomer 1 by chiral analytical SFC, >99% ee, /(R>: 1.30 minutes; column: CHFRALCEL® OD-H, 4.6 x 150 mm; eluting with a mobile phase of 30% MeOH (0.2% IP A) in C02; column temperature: 40 °C; flow rate: 5 mL/minute; UV detection wavelength: 225 nm. Isolate the title compound of Example 1B to give the second eluting enantiomer (Isomer 2). ES/MS (m/z): 525.2 (M+H). Confirm enantiomeric enrichment of Isomer 2 by chiral analytical SFC, 98% ee, /(R>: 2.03 minutes; column: CHIRALCEL® OD-H, 4.6 x 150 mm; eluting with a mobile phase of 30% MeOH (0.2% IP A) in C02; column temperature: 40 °C; flow rate: 5 mL/minute; UV detection wavelength: 225 nm.
Alternate Preparation EXAMPLE 1B
Crystalline 5-(4-{2-[3-(Fluoromethyl)azetidin-l-yl]ethoxy}phenyl)-8-(trifluoromethyl)-5H- [l]benzopyrano[4,3-c]quinolin-2-ol, Isomer 2
Stir 5-(4-{2-[3-(fluoromethyl)azetidin-l-yl]ethoxy}phenyl)-8-(trifluoromethyl)-5H-[l]benzopyrano[4,3-c]quinolin-2-ol, 4-methylbenzenesulfonic acid, Isomer 2 (23.8 g, 0.034 mol) in water (250 mL) at 1000 rpm. Add NaOH (76 pL) and stir the solution for 2 hours. Add DCM (600 mL). Separate the mixture, dry the DCM extract with magnesium sulfate, filter the material through a syringe filter (0.45 pm), and concentrate to dryness. Allow the material to sit under a N2 stream over a weekend. Add 1 : 1 EtOH/water (80 mL) and stir the mixture with sonication. Collect a tan solid by filtration on a nylon membrane to give the title compound (10.47 g, 0.02 mol, 59%).
PAT
- Selective estrogen receptor degradersPublication Number: US-2023234960-A1Priority Date: 2018-07-12
- Selective estrogen receptor degraderPublication Number: CN-112424205-BPriority Date: 2018-07-12Grant Date: 2023-10-31
- selective estrogen receptor degraderPublication Number: CN-117379428-APriority Date: 2018-07-12
- Selective estrogen receptor degradersPublication Number: US-11993608-B2Priority Date: 2018-07-12Grant Date: 2024-05-28
- Selective estrogen receptor degradersPublication Number: US-12128040-B2Priority Date: 2018-07-12Grant Date: 2024-10-29
PAT
https://patents.google.com/patent/US11926634B2/en
Selective estrogen receptor degraders (SERDs) bind to the estrogen receptor (ER) and downregulate ER-mediated transcriptional activity. The degradation and downregulation caused by SERDs can be useful in the treatment of various proliferative immune mediated disorders, cell proliferation disorders, including cancers such as breast cancer, ovarian cancer, endometrial cancer, prostate cancer, uterine cancer, gastric cancer, and lung cancer as well as mutations due to emerging resistance. Some small molecule examples of SERDs have been disclosed in the literature (see, e.g., WO2005073204, WO2014205136, and WO2016097071). Nonetheless, there is a need for new SERDs to treat ER-positive cancers, such as breast cancer, gastric cancer, and/or lung cancer.
As described in U.S. Pat. No. 10,654,866 (the ‘866 patent) a series of SERDs of the following formula have been discovered, along with pharmaceutically acceptable salts thereof:
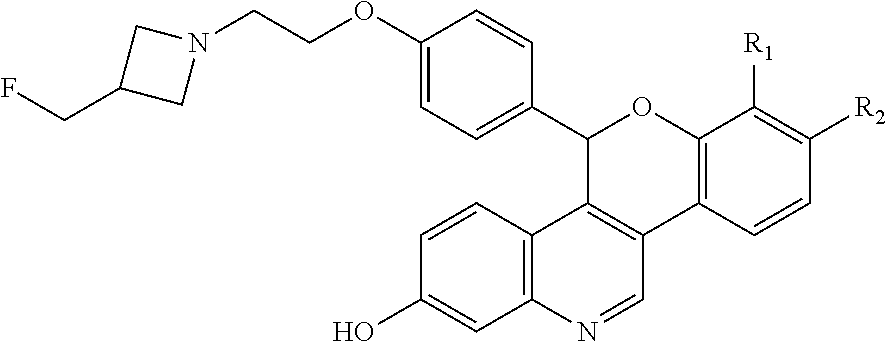
wherein one of R1 and R2 are independently Cl, F, —CF3, or —CH3, and the other is H.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Trade names | Inluriyo |
| Other names | LY3484356, LY-3484356 |
| AHFS/Drugs.com | Inluriyo |
| License data | US DailyMed: Imlunestrant |
| Routes of administration | By mouth |
| Drug class | Estrogen receptor antagonist |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2408840-26-4as tosylate: 2408840-41-3 |
| PubChem CID | 146603228 |
| DrugBank | DB19043 |
| ChemSpider | 115010421 |
| UNII | 9CXQ3PF69Uas tosylate: F7UDT90EW5 |
| KEGG | D12216as tosylate: D12217 |
| ChEMBL | ChEMBL5095183 |
| Chemical and physical data | |
| Formula | C29H24F4N2O3 |
| Molar mass | 524.516 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- https://www.accessdata.fda.gov/drugsatfda_docs/label/2025/218881s000lbl.pdf
- “FDA approves imlunestrant for ER-positive, HER2-negative, ESR1-mutated advanced or metastatic breast cancer”. U.S. Food and Drug Administration (FDA). 25 September 2025. Retrieved 27 September 2025. This article incorporates text from this source, which is in the public domain.
- “U.S. FDA approves Inluriyo (imlunestrant) for adults with ER+, HER2-, ESR1-mutated advanced or metastatic breast cancer” (Press release). Eli Lilly. 25 September 2025. Retrieved 27 September 2025 – via PR Newswire.
- World Health Organization (2022). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 88”. WHO Drug Information. 36 (3). hdl:10665/363551.
Further reading
- Jhaveri, Komal L.; Jeselsohn, Rinath; Lim, Elgene; Hamilton, Erika P.; Yonemori, Kan; Beck, J. Thaddeus; et al. (June 2022). “A phase 1a/b trial of imlunestrant (LY3484356), an oral selective estrogen receptor degrader (SERD) in ER-positive (ER+) advanced breast cancer (aBC) and endometrial endometrioid cancer (EEC): Monotherapy results from EMBER”. Journal of Clinical Oncology. 40 (16_suppl): 1021. doi:10.1200/JCO.2022.40.16_suppl.1021. S2CID 249445691.
- Jhaveri, Komal; O’Shaughnessy, Joyce; Andre, Fabrice; Goetz, Matthew P.; Harbeck, Nadia; Martín, Miguel; et al. (March 2023). “Abstract OT1-01-02: EMBER-4: A phase 3 adjuvant trial of imlunestrant vs standard endocrine therapy (ET) in patients with ER+, HER2- early breast cancer (EBC) with an increased risk of recurrence who have previously received 2 to 5 years of adjuvant ET”. Cancer Research. 83 (5_Supplement): OT1–01–02-OT1-01–02. doi:10.1158/1538-7445.SABCS22-OT1-01-02.
- Neven, P.; Stahl, N.; Losada, M.J. Vidal; Jimenez, M. Martin; Kaufman, P.A.; Harbeck, N.; et al. (October 2023). “273P A preoperative window-of-opportunity (WOO) study of imlunestrant in ER+, HER2- early breast cancer (EBC): Final analysis from EMBER-2”. Annals of Oncology. 34: S292 – S293. doi:10.1016/j.annonc.2023.09.470. S2CID 264385454.
External links
- Clinical trial number NCT04975308 for “A Study of Imlunestrant, Investigator’s Choice of Endocrine Therapy, and Imlunestrant Plus Abemaciclib in Participants With ER+, HER2- Advanced Breast Cancer (EMBER-3)” at ClinicalTrials.gov
/////////Imlunestrant, FDA 2025, APPROVALS 2025, Inluriyo, CANCER, LY3484356, LY 3484356, 9CXQ3PF69U
Donidalorsen
Donidalorsen
CAS 2304692-48-4
| 분자량 Mw | 8672.64 |
|---|---|
| 화학식Mf | C296H435N83O151P20S15 |
ISIS 721744, ISIS-721744
FDA 8/21/2025, Dawnzera, To prevent attacks of hereditary angioedema
DNA, D((2′-O-(2-METHOXYETHYL))M5RU-SP-(2′-O-(2-METHOXYETHYL))RG-SP-(2′-O-(2-METHOXYETHYL))M5RC-(2′-O-(2-METHOXYETHYL))RA-(2′-O-(2-METHOXYETHYL))RA-SP-G-SP-T-SP-M5C-SP-T-SP-M5C-SP-T-SP-T-SP-G-SP-G-SP-M5C-SP-(2′-O-(2-METHOXYETHYL))RA-(2′-O-(2-METHOXYETHYL)
- WHO 11653
- DNA, d((2′-O-(2-methoxyethyl))m5rU-sp-(2′-O-(2-methoxyethyl)(rG-sp-(2′-O-(2- methoxyethyl))m5rC-(2′-O-(2-methoxyethyl))rA-(2′-O-(2-methoxyethyl))rA-spG-sp-T-sp-m5C-sp-T-sp-m5C-sp-T-sp-T-sp-G-sp-G-sp-m5C-sp-(2′-O-(2-methoxyethyl))rA-(2′-O-(2-methoxyethyl))rA-(2′-O-(2-methoxyethyl))rA-sp-(2′-O-(2-methoxyethyl))m5rC-sp-(2′-O-(2-methoxyethyl))rA), 5′-(26-((2-(acetylamino)-2-deoxy-beta-D-galactopyranosyl)oxy)-14,14-bis((3-((6-((2-(acetylamino)-2-deoxybeta-D-galactopyranosyl)oxy)hexyl)amino)-3-oxopropoxy)methyl)-8,12,19-trioxo16-oxa-7,13,20-triazahexacos-1-yl hydrogen phosphate)
- UNII-ZD4D8M32TL
| Ingredient | UNII | CAS | . |
|---|---|---|---|
| Donidalorsen sodium | Y30VEG5PH1 | 2304701-45-7 |
Donidalorsen, sold under the brand name Dawnzera, is a medication used to prevent attacks of hereditary angioedema.[1] Donidalorsen is a prekallikrein-directed antisense oligonucleotide.[1] It is given by injection under the skin (subcutaneous).[1]
Donidalorsen was approved for medical use in the United States in August 2025.[2]
Donidalorsen is under investigation in clinical trial NCT05392114 to assess the long-term safety and efficacy of donidalorsen in the prophylactic treatment of hereditary angioedema (HAE)
Donidalorsen is an antisense oligonucleotide designed to reduce the production of prekallikrein (PKK). PKK plays an important role in the activation of inflammatory mediators associated with acute attacks of Hereditary angioedema (HAE).
AWNZERA™ (donidalorsen) approved in the U.S. as first and only RNA-targeted prophylactic treatment for hereditary angioedema
August 21, 2025
– DAWNZERA demonstrated significant and sustained HAE attack rate reduction and long-term disease control –
– Offers longest dosing option for HAE, with dosing every 4 or 8 weeks –
– Compelling profile supported by recently published switch data –
– Ionis’ second independent launch in just nine months, with potential for two additional launches next year –
– Ionis to host webcast today at 12:15pm ET –
CARLSBAD, Calif.–(BUSINESS WIRE)–Aug. 21, 2025– Ionis Pharmaceuticals, Inc. (Nasdaq: IONS) announced today that the U.S. Food and Drug Administration (FDA) has approved DAWNZERA™ (donidalorsen) for prophylaxis to prevent attacks of hereditary angioedema (HAE) in adult and pediatric patients 12 years of age and older. DAWNZERA is the first and only RNA-targeted medicine approved for HAE, designed to target plasma prekallikrein (PKK), a key protein that activates inflammatory mediators associated with acute attacks of HAE. DAWNZERA 80mg is self-administered via subcutaneous autoinjector once every four (Q4W) or eight weeks (Q8W).
This press release features multimedia. View the full release here: https://www.businesswire.com/news/home/20250818615141/en/
![]()
DAWNZERA (donidalorsen) logo
HAE is a rare and potentially life-threatening genetic condition that involves recurrent attacks of severe swelling (angioedema) in various parts of the body, including the hands, feet, genitals, stomach, face and/or throat. HAE is estimated to affect approximately 7,000 people in the U.S.
“DAWNZERA represents a significant advance for people living with HAE who need improved treatment options. With strong and durable efficacy, convenient administration and the longest dosing option available, we believe DAWNZERA will be the prophylactic treatment of choice for many people living with HAE. Importantly, the recently published switch data empowers patients and physicians with a roadmap for switching to DAWNZERA from other prophylactic therapies,” said Brett P. Monia, Ph.D., chief executive officer, Ionis. “At Ionis, we are dedicated to turning groundbreaking science into life-changing medicines. With the early success of our first independent launch of TRYNGOLZA® for familial chylomicronemia syndrome (FCS), and now with DAWNZERA, our second independent medicine approved in less than nine months, we are proudly delivering on that vision. To the patients, families, advocacy partners and investigators who helped make this moment a reality, we express our deepest gratitude.”
The approval of DAWNZERA was based on positive results from the Phase 3 global, multicenter, randomized, double-blind, placebo-controlled OASIS-HAE study in patients with HAE. The study met its primary endpoint, with DAWNZERA Q4W significantly reducing monthly HAE attack rate by 81% compared to placebo over 24 weeks. Mean attack rate reduction increased to 87% when measured from the second dose, a key secondary endpoint. Additionally, DAWNZERA Q4W reduced moderate-to-severe HAE attacks by ~90% over 24 weeks when measured from the second dose.
These results are bolstered by the ongoing OASISplus open-label extension (OLE) study, in which DAWNZERA Q8W had a similar effect as Q4W over time. DAWNZERA demonstrated 94% total mean attack rate reduction from baseline across both dosing groups after one year in the OLE.
The OASISplus study also includes a switch cohort evaluating DAWNZERA Q4W in patients previously treated with lanadelumab, C1-esterase inhibitor or berotralstat for at least 12 weeks. Switching to DAWNZERA reduced mean HAE attack rate by 62% from prior prophylactic treatment over 16 weeks, with no mean increase in breakthrough attacks observed during the switch. A total of 84% of patients surveyed preferred DAWNZERA over their prior prophylactic treatment, citing better disease control, less time to administer and less injection site pain or reactions.
Across clinical studies, DAWNZERA demonstrated a favorable safety and tolerability profile. The most common adverse reactions (incidence ≥ 5%) were injection site reactions, upper respiratory tract infection, urinary tract infection and abdominal discomfort.
“As the first FDA-approved RNA-targeted therapy for HAE, DAWNZERA represents a welcome advance in therapeutic options for preventing attacks. Today’s approval gives people living with HAE and their physicians another important choice for aligning treatment with individual needs,” said Anthony J. Castaldo, CEO & chairman of the board, U.S. Hereditary Angioedema Association (HAEA) and Hereditary Angioedema International (HAEi).
“People living with HAE manage this condition for all their lives, and many continue to face unpredictable, painful and dangerous breakthrough attacks even with current treatments. Durable efficacy is essential in maintaining long-term disease control,” said Marc Riedl, M.D., M.S., clinical director, U.S. HAEA Angioedema Center; University of California, San Diego; OASIS-HAE and OASISplus trial investigator. “DAWNZERA is positioned to help meet patient needs, providing substantial and sustained reduction of HAE attacks, continued improvement over time and reduced burden of treatment.”
DAWNZERA will be available in the U.S. in the coming days.
Ionis is committed to helping people access the medicines they are prescribed and will offer a suite of services designed to meet the unique needs of the HAE community through Ionis Every Step™. As part of Ionis Every Step, patients and healthcare providers will have access to a wide range of support and resources including dedicated support from a Patient Education Manager, assistance with the insurance approval process, information on affordability programs, access to the DAWNZERA Direct digital companion and other ongoing services and resources to help patients stay on track. Visit DAWNZERA.com for more information.

AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- “DAWNZERA (donidalorsen) injection, for subcutaneous use” (PDF). Highlights of Prescribing Information. Ionis Pharmaceuticals, Inc.
- “Dawnzera (donidalorsen) approved in the U.S. as first and only RNA-targeted prophylactic treatment for hereditary angioedema” (Press release). Ionis Pharmaceuticals, Inc. 21 August 2025. Retrieved 22 August 2025 – via Business Wire.
- “Donidalorsen: An Investigational RNA-targeted Medicine” (PDF). Ionis Pharmaceuticals, Inc.
- Farkas H, Balla Z (March 2024). “Kallikrein inhibitors for angioedema: the progress of preclinical and early phase studies”. Expert Opinion on Investigational Drugs. 33 (3): 191–200. doi:10.1080/13543784.2024.2320700. PMID 38366937.
- “Dawnzera: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 22 August 2025.
- World Health Organization (2021). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 86”. WHO Drug Information. 35 (3). hdl:10665/346562.
Further reading
- Raja A, Shuja MH, Raja S, Qammar A, Kumar S, Khurram L, et al. (December 2024). “Efficacy and safety of Donidalorsen in Hereditary Angioedema with C1 inhibitor deficiency: a systematic review and a meta analysis”. Archives of Dermatological Research. 317 (1): 110. doi:10.1007/s00403-024-03652-3. PMID 39666085.
External links
- Clinical trial number NCT05139810 for “OASIS-HAE: A Study to Evaluate the Safety and Efficacy of Donidalorsen (ISIS 721744 or IONIS-PKK-LRx) in Participants With Hereditary Angioedema (HAE)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Dawnzera |
| Other names | ISIS 721744, ISIS-721744 |
| AHFS/Drugs.com | Dawnzera |
| License data | US DailyMed: Donidalorsen |
| Routes of administration | Subcutaneous |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| CAS Number | 2304692-48-42304701-45-7 |
| DrugBank | DB18751DBSALT003520 |
| UNII | ZD4D8M32TLY30VEG5PH1 |
- [1]. Fijen LM, Riedl MA, Bordone L, et al. Inhibition of Prekallikrein for Hereditary Angioedema. N Engl J Med. 2022;386(11):1026-1033. [Content Brief][2]. Valerieva A, Longhurst HJ. Treatment of hereditary angioedema-single or multiple pathways to the rescue. Front Allergy. 2022;3:952233. [Content Brief]
//////////Donidalorsen, FDA 2025, APPROVALS 2025, Dawnzera, ISIS-721744 FREE ACID, ISIS 721744

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}