WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ]
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
Palopegteriparatide is a human parathyroid hormone analogue corresponding to amino acid residues 1 – 34 of human parathyroid hormone, to which a methoxy polyethylene glycol (molecular weight: ca. 43,000) is bound via a cleavable linker (pegylation site: S1). Palopegteriparatide is a pegylated synthetic peptide (molecular weight: ca. 48,000) consisting of 34 amino acid residues.
Palopegteriparatide was approved for medical use in the European Union in November 2023,[2] and in the United States in August 2024.[1][5]
Medical uses
Palopegteriparatide is indicated for the treatment of adults with hypoparathyroidism.[1][2]
Adverse effects
The US Food and Drug Administration (FDA) prescription label for palopegteriparatide includes warnings for a potential risk of risk of unintended changes in serum calcium levels related to number of daily injections and total delivered dose, serious hypocalcemia and hypercalcemia (blood calcium levels that are too high), osteosarcoma (a rare bone cancer) based on findings in rats, orthostatic hypotension (dizziness when standing), and a risk of a drug interaction with digoxin (a medicine for certain heart conditions).[5]
History
The effectiveness of palopegteriparatide was evaluated in a 26-week, randomized, double-blind, placebo-controlled trial that enrolled 82 adults with hypoparathyroidism.[5] Prior to randomization, all participants underwent an approximate four-week screening period in which calcium and active vitamin D supplements were adjusted to achieve an albumin-corrected serum calcium concentration between 7.8 and 10.6 mg/dL, a magnesium concentration ≥1.3 mg/dL and below the upper limit of the reference range, and a 25(OH) vitamin D concentration between 20 to 80 ng/mL.[5] During the double-blind period, participants were randomized to either palopegteriparatide (N = 61) or placebo (N= 21), at a starting dose of 18 mcg/day, co-administered with conventional therapy (calcium and active vitamin D).[5] Study drug and conventional therapy were subsequently adjusted according to the albumin-corrected serum calcium levels.[5] At the end of the trial, 69% of the participants in the palopegteriparatide group compared to 5% of the participants in the placebo group were able to maintain their calcium level in the normal range, without needing active vitamin D and high doses of calcium (calcium dose ≤ 600 mg/day).[5]
In September 2023, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Yorvipath, intended for the treatment of chronic hypoparathyroidism in adults.[4][6] The applicant for this medicinal product is Ascendis Pharma Bone Diseases A/S.[4] Palopegteriparatide was approved for medical use in the European Union in November 2023.[2]
^ Jump up to:abcdef“Yorvipath EPAR”. European Medicines Agency. 19 October 2020. Archived from the original on 10 December 2023. Retrieved 11 December 2023. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
^“Yorvipath Product information”. Union Register of medicinal products. 20 November 2023. Archived from the original on 26 November 2023. Retrieved 11 December 2023.
^ Jump up to:abc“Yorvipath: Pending EC decision”. European Medicines Agency. 15 September 2023. Archived from the original on 24 September 2023. Retrieved 24 September 2023. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
^“EU/3/20/2350”. European Medicines Agency. 15 September 2023. Archived from the original on 24 September 2023. Retrieved 24 September 2023.
^World Health Organization (2021). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 86”. WHO Drug Information. 35 (3). hdl:10665/346562.
^World Health Organization (2023). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 89”. WHO Drug Information. 37 (1). hdl:10665/366661.
Clinical trial number NCT04701203 for “A Trial Investigating the Safety, Tolerability and Efficacy of TransCon PTH Administered Daily in Adults With Hypoparathyroidism (PaTHway)” at ClinicalTrials.gov
Seladelpar was approved for medical use in the United States in August 2024.[1][5]
Seladelpar is a peroxisome proliferator-activated receptor (PPAR)-delta (δ) agonist. Seladelpar is a single enantiomer of the R-configuration.5 On August 14, 2024, seladelpar was granted accelerated approval by the FDA for the treatment of primary biliary cholangitis,6 which is a condition associated with aberrant bile acid metabolism. Seladelpar works to block bile acid synthesis.1
Medical uses
Seladelpar is indicated for the treatment of primary biliary cholangitis in combination with ursodeoxycholic acid in adults who have an inadequate response to ursodeoxycholic acid, or as monotherapy in people unable to tolerate ursodeoxycholic acid.[1]
Clinically, Seladelpar reduces pruritus and IL-31 in patients with primary biliary cholangitis.[6]
^ Billin AN (October 2008). “PPAR-beta/delta agonists for Type 2 diabetes and dyslipidemia: an adopted orphan still looking for a home”. Expert Opinion on Investigational Drugs. 17 (10): 1465–1471. doi:10.1517/13543784.17.10.1465. PMID18808307. S2CID86564263.
Lazertinib is an oral, third-generation, epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI).2,3 Lazertinib was first approved in South Korea on January 18, 2021, for the treatment of EGFR T790M mutation-positive non-small cell lung cancer (NSCLC) with EGFR mutations.1 It was approved by the FDA on August 19, 2024.5 Lazertinib is used alone or in combination with other chemotherapeutic agents.4
The most common adverse reactions include rash, nail toxicity, infusion-related reactions (amivantamab), musculoskeletal pain, edema, stomatitis, venous thromboembolism, paresthesia, fatigue, diarrhea, constipation, COVID-19 infection, hemorrhage, dry skin, decreased appetite, pruritus, nausea, and ocular toxicity.[2]
Lazertinib was approved for medical use in South Korea in January 2021,[4][5] and in the United States in August 2024.[2][6]
Medical uses
Lazertinib is indicated in combination with amivantamab for the first-line treatment of adults with locally advanced or metastatic non-small cell lung cancer with epidermal growth factor receptor exon 19 deletions or exon 21 L858R substitution mutations.[2
History
Efficacy was evaluated in MARIPOSA (NCT04487080), a randomized, active-controlled, multicenter trial of 1074 participants with exon 19 deletion or exon 21 L858R substitution mutation-positive locally advanced or metastatic non-small cell lung cancer and no prior systemic therapy for advanced disease.[2] Participants were randomized (2:2:1) to receive lazertinib in combination with amivantamab, osimertinib monotherapy, or lazertinib monotherapy (an unapproved regimen for non-small cell lung cancer) until disease progression or unacceptable toxicity.[2]
Society and culture
Legal status
Lazertinib was approved for medical use in the United States in August 2024.[2]Names
^World Health Organization (2018). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 79”. WHO Drug Information. 32 (1). hdl:10665/330941.
External links
Clinical trial number NCT04487080 for “A Study of Amivantamab and Lazertinib Combination Therapy Versus Osimertinib in Locally Advanced or Metastatic Non-Small Cell Lung Cancer (MARIPOSA)” at ClinicalTrials.gov
Aprocitentan, sold under the brand name Tryvio, is a medication used to treat hypertension (high blood pressure).[1] It is developed by Idorsia.[2] It is taken by mouth.[1]
Aprocitentan is indicated for the treatment of hypertension in combination with other antihypertensive drugs, to lower blood pressure in adults who are not adequately controlled on other medications.[1]
Data from animal reproductive toxicity studies with other endothelin-receptor agonists indicate that use is contraindicated in pregnant women.[1]
Mechanism of action
Aprocitentan is an endothelin receptor agonist that inhibits the protein endothelin-1 from binding to endothelin A and endothelin B receptors.[1][4] Endothelin-1 mediates various adverse effects via its receptors, such as inflammation, cell proliferation, fibrosis, and vasoconstriction.[1]
Society and culture
Economics
Aprocitentan is developed by Idorsia, which sold it to Janssen and purchased the rights back in 2023, for US$343 million.[6]
Legal status
Aprocitentan was approved for medical use in the United States in March 2024.[1]
In April 2024, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Jeraygo, intended for the treatment of resistant hypertension in adults.[7] The applicant for this medicinal product is Idorsia Pharmaceuticals Deutschland GmbH.[7]
The following example was prepared according to the procedures described below. All compounds were characterized by 1H-NMR (300 MHz) and occasionally by 13C-NMR (75 MHz) (Varian Oxford, 300 MHz; chemical shifts are given in ppm relative to the solvent used; multiplicities: s=singlet, d=doublet, t=triplet; m=multiplet), by LC-MS (Finnigan Navigator with HP 1100 Binary Pump and DAD, column: 4.6×50 mm, Develosil RP Aqueous, 5 μm, 120 A, gradient: 5-95% acetonitrile in water, 1 min, with 0.04% trifluoroacetic acid, flow: 4.5 ml/min), t R is given in min; by TLC (TLC-plates from Merck, Silica gel 60 F 254) and occasionally by melting point.
Preparation A: Benzylsulfamide Potassium Salt
A.i. Benzylsulfamide
Chlorosulfonylisocyanate (14.14 g) was dissolved in DCM (50 mL) and cooled to 0° C. A solution of t-BuOH (9.6 mL) in DCM (50 mL) was added within 30 min. Stirring was continued for additional 30 min at rt. The solution thus obtained was then added at 0° C. within 1 h to a solution of benzylamine (10.7 g) and TEA (15.32 mL) in DCM (200 mL). Stirring is continued for 10 h at rt. The mixture was concentrated in vacuo, taken up in EA (500 mL) and washed with water (2×40 mL) and brine (30 mL), dried over MgSO 4, filtered. The filtrate was concentrated in vacuo and the crude material was crystallized from EA and dried under HV to give N-benzyl-N′-tert-butoxycarbonyl sulfamide (13.68 g).
This material was dissolved in dioxane (20 ml) and 4 M HCl in dioxane (120 mL) was added within 1 h at rt. The mixture was stirred for 8 h before the solvent was evaporated and the residue dried under HV to give benzylsulfamide as an off-white powder (9.47 g).
To a solution of benzylsulfamide (17.98 g) in McOH (300 mL) was carefully added potassium tert-butylate (10.8 g). The mixture was stirred at rt for 15 min before the solvent was evaporated. The remaining residue was dried under HV to give benzylsulfamide potassium salt as an off-white powder (21.73 g).
To a solution of 4-bromophenylacetic acid (50 g) in methanol (250 ml) was added dropwise thionyl chloride (34.2 mL) while the temperature of the reaction mixture was kept at 0-5° C. Upon complete addition cooling was stopped and the mixture was allowed to warm to rt. Stirring was continued for 75 min before the solvent was removed in vacuo. The yellow oil was dissolved in benzene and again concentrated. The residue was dissolved in EA, washed with water, brine, 2 N aq. Na 2CO 3, and again brine. The org. extract was dried over MgSO 4, filtered, concentrated and dried under HV at 85° C. for 30 min to give the expected product as a yellow oil (52.4 g).
At 40° C., a solution of intermediate B.i (52 g) in THF (100 mL) was carefully added over a period of 40 min to a suspension of NaH (15.6 g) in dry THF (450 mL). Stirring was continued for 70 min without heating and the temperature dropped to 27° C. The evolution of gas stopped before dimethylcarbonate (76.42 mL) was added dropwise while the temperature of the mixture was maintained at 29-31° C. Stirring was continued for 22 h at rt. The mixture was cooled to −10° C. and then carefully neutralized to pH 6-7 with aq. HCl before bulk of the THF was removed in vacuo. The residue was dissolved in EA (700 mL), washed 3 times with 1 N aq. HCl-solution and once with brine, dried over MgSO 4. Most of the EA was evaporated before Hex was added. The product crystallised overnight at 4° C. The crystals were collected, washed with Hex and dried to give the expected product as pale yellow crystals (45.9 g).
A solution of intermediate B.ii (11.73 g) in MeOH (100 mL) was added at 0° C. to a solution of sodium (2.83 g) in MeOH (100 mL). The mixture was stirred for 18 h at rt before formamidine hydrochloride (4.10 g) was added. The suspension was stirred at rt for 4 h. The solvent was removed and the residue was suspended in 10% aq. citric acid (100 mL) and stirred for 10 min. The white precipitate was collected, washed with 10% aq. citric acid, water, evaporated three times from cyclohexane and dried under HV at 40° C. to give 5-(4-bromophenyl)-pyrimidine-4,6-diol as a pale beige powder (9.90 g).
LC-MS: t R=0.62 min, [M+H] +=266.89/268.89 (Br-isotopes).
B.iv. 5-(4-bromo-phenyl)-4,6-dichloro-pyrimidine
To a suspension of 5-(4-bromophenyl)-pyrimidine-4,6-diol (9.90 g) in POCl 3 (130 mL) was carefully added N,N-dimethylaniline (13.5 mL). The mixture is heated to 130° C. for 2 h. The dark brown solution is concentrated in vacuo and the residue was poured into ice/water. The suspension is diluted with 2 N HCl and water and stirred for 20 min. The precipitate that formed is collected and washed with water. The solid material is dissolved in EA, washed with 1 N aq. HCl and brine. The org. phase is dried over MgSO 4 and evaporated. The material is further purified by column chromatography on silica gel eluting with Hex:EA 95:5 to 1:1 followed by crystallisation from Hex/EA at −20° C. to give 4,6-dichloro-5-(4-bromophenyl)-pyrimidine as pale yellow crystals (8.3 g).
A solution of 5-(4-bromophenyl)-4,6-dichloro-pyrimidine (4.00 g, 13.2 mmol) and benzylsulfamide potassium salt (7.38 g, 32.9 mmol) in DMSO (30 mL) was stirred at it for 24 h before being diluted with a 10% aq. citric acid solution (200 mL). The suspension that formed was filtered. The collected solid was washed well with water and dried under HV at 40° C. for 48 h to give the expected product as a white powder (6.15 g).
t-BuOK (18.5 g, 164.5 mmol) was added portionwise to a suspension of intermediate 1.i (7.46 g, 16.4 mmol) in ethylene glycol (50 mL). The mixture became warm and thick and was diluted with DME (75 mL). The mixture was stirred at 95° C. for 24 h before it was cooled to rt, diluted with water (50 mL) and a 10% aq. citric acid solution (250 mL). The milky suspension was extracted with EA (2×300 mL). The combined org. extracts were dried over MgSO 4, filtered and the filtrate was concentrated. The remaining crystalline solid was suspended in MeOH, collected, washed well with MeOH and dried under HV to give the expected product as a white crystalline powder (6.49 g).
To a solution of intermediate 1.ii (6.49 g, 13.5 mmol) in THF (120 mL) was added carefully NaH (1.77 g, 40.6 mmol, 55% dispersion in mineral oil). The mixture was stirred for 10 min before 2-chloro-5-bromo-pyrimidine (3.93 g, 20.3 mmol) was added. The mixture was diluted with DMF (15 mL) and then stirred at rt for 20 min. The mixture was heated to 60° C. and stirred for 3 h before being again cooled to rt. The reaction was quenched with water and 10% aq. citric acid solution (250 mL) and the mixture was extracted with EA (2×300 mL). The org. extracts were washed with water, combined, dried over MgSO 4, filtered and the solvent of the filtrate was evaporated. The crude product was crystallised from MeOH/ether. The crystalline material was collected, washed with additional MeOH/ether and dried under HV to give the expected product as a white powder (6.47 g).
A solution of borontribromide (25.5 mL, 1 M in DCM) was slowly added to a solution of intermediate 1.iii (6.50 g, 10.2 mmol) in chloroform (250 mL). The mixture became turbid and an oily residue separated. The mixture was stirred at rt. Another portion of BBr 3 solution (5 mL) was added after 6, 24, and 33 h. After the last addition of BBr 3, the beige suspension was stirred vigorously for additional 2 h before being carefully quenched with MeOH. The mixture became slightly warm and clear. The solution was washed with cold water (0° C., 2×150 mL). The washings were extracted back with DCM. The combined org. extracts were again washed with water, dried over MgSO 4, filtered and concentrated. The crude product was purified by CC on silica gel eluting with heptane:EA 1:1 followed by crystallisation from DCM. The purified crystalline product was dried under HIV at 45° C. for 48 h to give the expected product as a white, crystalline powder (1.62 g).
^ Jump up to:ab“Jeraygo EPAR”. European Medicines Agency. 25 April 2024. Archived from the original on 30 April 2024. Retrieved 27 April 2024. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
Further reading
Mahfooz K, Najeed S, Tun HN, Khamosh M, Grewal D, Hussain A, et al. (July 2023). “New Dual Endothelin Receptor Antagonist Aprocitentan in Hypertension: A Systematic Review and Meta-Analysis”. Current Problems in Cardiology. 48 (7): 101686. doi:10.1016/j.cpcardiol.2023.101686. PMID36893968.
CAS Name: 1-[[(6R,7R)-7-[[(2Z)-(2-Amino-4-thiazolyl)(methoxyimino)acetyl]amino]-2-carboxy-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-3-yl]methyl]-1-methylpyrrolidinium inner salt
Additional Names: 1-[[(6R,7R)-7-[2-(2-amino-4-thiazolyl)glyoxylamido]-2-carboxy-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-3-yl]methyl]-1-methylpyrrolidinium hydroxide inner salt 72-(Z)-2-(O-methyloxime); 7-[(Z)-2-(2-aminothiazol-4-yl)-2-methoxyiminoacetamido]-3-(1-methylpyrrolidinio)methyl-3-cephem-4-carboxylate
Manufacturers’ Codes: BMY-28142
Molecular Formula: C19H24N6O5S2
Molecular Weight: 480.56
Percent Composition: C 47.49%, H 5.03%, N 17.49%, O 16.65%, S 13.34%
Literature References: Semisynthetic, fourth generation cephalosporin antibiotic. Prepn: S. Aburaki et al.,DE3307550; eidem,US4406899 (both 1983 to Bristol-Myers); and antibacterial activity: T. Naito et al.,J. Antibiot.39, 1092 (1986). In vitro comparative antimicrobial spectrum: N. J. Khan et al.,Antimicrob. Agents Chemother.26, 585 (1984); and b-lactamase stability: H. C. Neu et al.,J. Antimicrob. Chemother.17, 441 (1986). HPLC determn in plasma and urine: R. H. Barbhaiya et al.,Antimicrob. Agents Chemother.31, 55 (1987). Clinical evaluations in infection: N. Clynes et al.,Diagn. Microbiol. Infect. Dis.12, 257 (1989); S. Oster et al.,Antimicrob. Agents Chemother.34, 954 (1990). Review of clinical pharmacokinetics: M. P. Okamoto et al.,Clin. Pharmacokinet.25, 88-102 (1993).
Properties: Colorless powder, mp 150° (dec). uv max (pH 7 phosphate buffer): 235, 257 nm (e 16700, 16100).
Melting point: mp 150° (dec)
Absorption maximum: uv max (pH 7 phosphate buffer): 235, 257 nm (e 16700, 16100)
Derivative Type: Sulfate
Molecular Formula: C19H24N6O5S2.H2SO4
Molecular Weight: 578.64
Percent Composition: C 39.44%, H 4.53%, N 14.52%, O 24.89%, S 16.62%
Properties: mp 210° (dec). uv max (pH 7 phosphate buffer): 236, 258 nm (e 17200, 16900).
Melting point: mp 210° (dec)
Absorption maximum: uv max (pH 7 phosphate buffer): 236, 258 nm (e 17200, 16900)
Derivative Type: Hydrochloride monohydrate
CAS Registry Number: 123171-59-5
CAS Name: 1-[[(6R,7R)-7-[[(2Z)-(2-Amino-4-thiazolyl)(methoxyimino)acetyl]amino]-2-carboxy-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-3-yl]methyl]-1-methylpyrrolidinium chloride monohydrochloride monohydrate
FDA APPROVED 2/22/2024, To treat complicated urinary tract infections, Exblifep
BMY 28142
BMY-28142
Cefepime is a fourth-generation cephalosporinantibiotic. Cefepime has an extended spectrum of activity against Gram-positive and Gram-negativebacteria, with greater activity against both types of organism than third-generation agents. A 2007 meta-analysis suggested when data of trials were combined, mortality was increased in people treated with cefepime compared with other β-lactam antibiotics.[1] In response, the U.S. Food and Drug Administration (FDA) performed their own meta-analysis which found no mortality difference.[2]
Cefepime is a broad-spectrum cephalosporin antibiotic and has been used to treat bacteria responsible for causing pneumonia and infections of the skin and urinary tract. Some of these bacteria include Pseudomonas, Escherichia, and Streptococcus species. The following represents MIC susceptibility data for a few medically significant microorganisms:[7]
Escherichia coli: ≤0.007 – 128 μg/ml
Pseudomonas aeruginosa: 0.06 – >256 μg/ml
Streptococcus pneumoniae: ≤0.007 – >8 μg/ml
Chemistry
The combination of the syn-configuration of the methoxyiminomoiety and the aminothiazole moiety confers extra stability to β-lactamase enzymes produced by many bacteria. The N–methylpyrrolidine moiety increases penetration into Gram-negative bacteria. These factors increase the activity of cefepime against otherwise resistant organisms including Pseudomonas aeruginosa and Staphylococcus aureus.
Semisynthetic, fourth generation cephalosporin antibiotic. Prepn: S. Aburaki et al., DE 3307550; eidem, US 4406899 (both 1983 to Bristol-Myers); and antibacterial activity: T. Naito et al., J. Antibiot. 39, 1092 (1986).
Trade names
Following expiration of the Bristol-Myers Squibb patent,[] cefepime became available as a generic and is now] marketed by numerous companies worldwide under tradenames including Neopime (Neomed), Maxipime, Cepimax, Cepimex, and Axepim.
^ Yahav D, Paul M, Fraser A, Sarid N, Leibovici L (May 2007). “Efficacy and safety of cefepime: a systematic review and meta-analysis”. The Lancet. Infectious Diseases. 7 (5): 338–348. doi:10.1016/S1473-3099(07)70109-3. PMID17448937.
^ “Cefepime (maxipime), large spectrum 4th generation cephalosporin, resistant to beta-lactamases]”.
^World Health Organization (2019). Executive summary: the selection and use of essential medicines 2019: report of the 22nd WHO Expert Committee on the selection and use of essential medicines. Geneva: World Health Organization. hdl:10665/325773. WHO/MVP/EMP/IAU/2019.05. License: CC BY-NC-SA 3.0 IGO.
^ Chapman TM, Perry CM (2003). “Cefepime: a review of its use in the management of hospitalized patients with pneumonia”. American Journal of Respiratory Medicine. 2 (1): 75–107. doi:10.1007/bf03256641. PMID14720024.
Berdazimer sodium, sold under the brand name Zelsuvmi, is a medication used for the treatment for molluscum contagiosum.[1] Berdazimer sodium is a nitric oxide releasing agent.[1] It is a polymer formed from sodium 1-hydroxy-3-methyl-3-(3-(trimethoxysilyl)propyl)-1-triazene-2-oxide and tetraethyl silicate.[2]
Berdazimer sodium was approved for medical use in the United States in January 2024.[3][4][5]
Medical uses
Berdazimer sodium is indicated for the topical treatment of molluscum contagiosum.[1]
Pharmacology
Mechanism of action
Berdazimer sodium is a nitric oxide releasing agent.[1] The mechanism of action for the treatment of molluscum contagiosum is unknown.[1]
Pharmacodynamics
The pharmacodynamics of berdazimer sodium are unknown.[1]
Society and culture
Legal status
Berdazimer sodium was approved for medical use in the United States in January 2024.[4]
Berdazimer is a polymeric substance consisting of a polysiloxane backbone (Si-O-Si bonds) with covalently bound N-diazeniumdiolate nitric oxide (NO) donors. It releases NO through exposure to proton donors like water, which will degrade the N-diazeniumdiolate entity.2 Berdazimer was previously investigated as a potential treatment for molluscum contagiosum, a viral cutaneous infection mainly affecting children, sexually active adults, and immunocompromised patients. It is one of the 5 most prevalent skin diseases in the world and the third-most common viral skin infection in children.3 Previously, the first line treatment for molluscum contagiosum was surgical excision, although it poses challenges such as repeated doctor visits, post-surgical scarring and skin discoloration, and fear and anxiety in the pediatric population.3
On Jan 05, 2024, the FDA approved berdazimer under the brand name ZELSUVMI for the treatment of adult and pediatric molluscum contagiosum, and it is the first drug to be approved for this condition. This decision is based on positive results demonstrated in 2 Phase 3 trials, B-SIMPLE 4 and B-SIMPLE 2, where reduced lesion counts were observed with once-a-day uses of berdazimer.5
^World Health Organization (2018). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 79”. WHO Drug Information. 32 (1). hdl:10665/330941.
Further reading
Pera Calvi I, R Marques I, Cruz SA, Mesquita YL, Padrao EM, Souza RM, et al. (2023). “Safety and efficacy of topical nitric oxide-releasing berdazimer gel for molluscum contagiosum clearance: A systematic review and meta-analysis of randomized controlled trials”. Pediatric Dermatology. 40 (6): 1060–1063. doi:10.1111/pde.15419. PMID37721050. S2CID262045499.
Han H, Smythe C, Yousefian F, Berman B (February 2023). “Molluscum Contagiosum Virus Evasion of Immune Surveillance: A Review”. Journal of Drugs in Dermatology. 22 (2): 182–189. doi:10.36849/JDD.7230. PMID36745361. S2CID256613906.
Clinical trial number NCT04535531 for “A Phase 3 Molluscum Contagiosum Efficacy and Safety Study (B-SIMPLE4)” at ClinicalTrials.gov
Clinical trial number NCT03927703 for “A Phase 3 Efficacy & Safety of SB206 & Vehicle Gel for the Treatment of MC (B-SIMPLE2)” at ClinicalTrials.gov
Clinical trial number NCT03927716 for “A Phase 3 Randomized Parallel Group Study Comparing the Efficacy & Safety of SB206 & Vehicle Gel in the Treatment of MC (B-SIMPLE1)” at ClinicalTrials.gov
fda approved 4/3/2024, To treat certain bloodstream infections, bacterial skin and associated tissue infections, and community-acquired bacterial pneumonia Press Release zevtera
Ceftobiprole, sold under the brand name Zevtera among others, is a fifth-generation[5]cephalosporin antibacterial used for the treatment of hospital-acquired pneumonia (excluding ventilator-associated pneumonia) and community-acquired pneumonia. It is marketed by Basilea Pharmaceutica under the brand names Zevtera and Mabelio.[6][7][8][9][10][11] Like other cephalosporins, ceftobiprole exerts its antibacterial activity by binding to important penicillin-binding proteins and inhibiting their transpeptidase activity which is essential for the synthesis of bacterial cell walls. Ceftobiprole has high affinity for penicillin-binding protein 2a of methicillin-resistant Staphylococcus aureus strains and retains its activity against strains that express divergent mecA gene homologues (mecC or mecALGA251). Ceftobiprole also binds to penicillin-binding protein 2b in Streptococcus pneumoniae (penicillin-intermediate), to penicillin-binding protein 2x in Streptococcus pneumoniae (penicillin-resistant), and to penicillin-binding protein 5 in Enterococcus faecalis.[12]
Medical uses
In the US, ceftobiprole is indicated for the treatment of adults with Staphylococcus aureus bloodstream infections (bacteremia) including those with right-sided infective endocarditis;[4] adults with acute bacterial skin and skin structure infections;[4] and people with community-acquired bacterial pneumonia.[4]
Microbiology
Ceftobiprole has shown in vitro antimicrobial activity against a broad range of Gram-positive and Gram-negative pathogens. Among the Gram-positive pathogens, ceftobiprole has demonstrated good in vitro activity against methicillin-resistant Staphylococcus aureus, methicillin-susceptible Staphylococcus aureus and coagulase-negative staphylococci, as well as against strains of methicillin-resistant Staphylococcus aureus with reduced susceptibility to linezolid, daptomycin or vancomycin.[13] Ceftobiprole has also displayed potent activity against Streptococcus pneumoniae (including penicillin-sensitive, penicillin-resistant and ceftriaxone-resistant strains) and Enterococcus faecalis, but not against Enterococcus faecium. For Gram-negative pathogens, ceftobiprole has shown good in vitro activity against Haemophilus influenzae (including both ampicillin-susceptible and ampicillin-non-susceptible isolates), Pseudomonas aeruginosa and strains of Escherichia coli, Klebsiella pneumoniae and Proteus mirabilis that do not produce extended-spectrum β-lactamases (ESBL). Like all other cephalosporins, ceftobiprole was inactive against strains that produce extended-spectrum β-lactamases.[14]
The efficacy of ceftobiprole has been demonstrated in two large randomized, double-blind, phase 3 clinical trials in patients with hospital-acquired and community-acquired pneumonia. Ceftobiprole was non-inferior to ceftazidime plus linezolid in the treatment of hospital-acquired pneumonia (excluding ventilator-acquired pneumonia) and non-inferior to ceftriaxone with or without linezolid in the treatment of community-acquired pneumonia.[15][16]
Pharmacology
Ceftobiprole medocaril
Ceftobiprole is the active moiety of the prodrug ceftobiprole medocaril and is available for intravenous treatment only. It is mainly excreted via the kidney.[17]
Society and culture
Legal status
500 mg powder
Ceftobiprole has been approved for the treatment of adults with hospital acquired pneumonia (excluding ventilator-acquired pneumonia) and community-acquired pneumonia in twelve European countries, Canada, and Switzerland.[18]
In February 2010, the Committee for Medicinal Products for Human Use of the European Medicines Agency adopted a negative opinion, recommending the refusal of the marketing authorization for the medicinal product Zeftera, intended for treatment of complicated skin and soft-tissue infections in adults. The company that applied for authorization is Janssen-Cilag International N.V. The applicant requested a re-examination of the opinion. After considering the grounds for this request, the CHMP re-examined the opinion, and confirmed the refusal of the marketing authorization in June 2010.[19]
Processes for producing ceftobiprole medocaril are known per se. What the processes known from the prior art have in common is that, starting from 7-aminocephalosporanic acid, a large number of intermediates have to be produced, isolated and purified in order to obtain ceftobiprole medocaril of the general formula (1) in sufficient purity.
The compound of the general formula (1) is known per se and is described, for example, in WO 99/65920. It can be used for the treatment and prophylaxis of bacterial infectious diseases, especially infectious diseases caused by methicillin-resistant Staphylococcus Aureus strains.
WO 99/65920 describes, as the last step in the production process of ceftobiprole Medocaril, a reaction in which the Medocaril prodrug unit is introduced into a compound of the general formula (2).
The compound of the general formula (2) is also known per se and has been described, for example, in EP 0 849 269 A1. The compound of the general formula (2) is prepared according to EP 0 849 269 A1 starting from (2R,6R,7R)-te rt. B u toxyc abonylamin o-3-formyl-8-oxo-5-thia-1 -azabicyclo[4.2.0]oct-3-ene-2-carboxylic acid benzhydryl ester by Wittig reaction with (1 ‘-allyloxycarbonyl-2- oxo-[1,3’]bipyrrolidinyl-3-yl)-triphenylphosphonium bromide. The resulting Δ2 reaction product is reisomerized to the desired Δ3 isomer by sulfoxidation and subsequent reduction and then deprotected from the benzhydryl ester with trifluoroacetic acid. The acylation in position 7 occurs by reaction with (Z)-(5-amino-[1,2,4]-thiadiazol-3-yl)-trityloxyiminothioacetic acid S-benzothiazol-2-yl ester. The compound of the general formula (2) is then obtained by removing the protective groups.
In EP 1 067 131 A1 the formation of the ylide in toluene or a mixture of toluene and dichloromethane is tert by adding alkali. Butylate in tetrahydrofuran, which allows the base to be added as a solution. The reaction of the ylide with the corresponding aldehyde is described at a reaction temperature of -70 0 C.
EP 0 841 339 A1 relates to cephalosporin derivatives and processes for their production. WO 95/29182 also discloses intermediates for the production of cephalosporins.
WO 01/90111 describes a further production of ceftobiprole Medocaril in several stages starting from desacetyl-7-aminocephalosporanic acid by acylation with (Z)-(5-amino-[1,2,4]-thiadiazol-3-yl)- trityloxyiminothioacetic acid S-benzothiazol-2-yl ester in N,N-dimethylformamide, followed by in situ esterification with diphenyldiazomethane in dichloromethane to give the corresponding benzohydryl ester, which is precipitated and isolated by adding hexane. In the next step, this product is oxidized to the corresponding aldehyde using TEMPO/NaOCI in dichloromethane/water or with Braunstein in tetrahydrofuran/dichloromethane. The next reaction step involves the Wittig reaction to the 3-vinyl-substituted derivative, in which the reaction takes place in dichloromethane/toluene/tetrahydrofuran at -78°C. The crude product is stirred with ethanol and made from dichloromethane/tert. Butyl methyl ether recrystallized or purified chromatographically. According to the method disclosed in WO 01/901 11, the Wittig reaction is carried out at low temperatures of -80 to -70 0 C in a complex solvent mixture of dichloromethane, toluene and tetrahydrofuran. This leads to significant disadvantages when carrying out the reaction on a production scale, since regeneration of the process solvents is difficult.
5 , 1 4 g of 7-amino-3-formyl-ceph-3-em-4-carboxy I at was dissolved in 2 7 , 8 m bis(trimethylsilyl)acetamide and 50 ml propylene oxide. 16.8 g of (1 R/S,3’R)-(1′-tert-butyloxycarbonyl-2-oxo-[1,3′]bipyrrolidinyl-3-yl)-triphenylphosphonium bromide (EP1067131, WO02/14332) slowly added in portions. Stirring was continued at 1 ° C until the starting material had reacted and then the crystalline precipitate was added
Nitrogen atmosphere filtered off and washed with 50 ml cyclohexane/bis(tirmethylsilyl)acetamide 99.5/0.5. After drying under vacuum, the desired product was obtained in silylated form.
The material was dissolved in 100ml dichloromethane and at 0 0 C with 50ml 3%
NaHCC> 3 solution added. The phases were separated, the organic phase was washed with 30 ml of water and the combined water phases were adjusted to pH 3.5 with 3% H 3 PO 4 after activated carbon treatment. The crystalline precipitate was filtered, washed with water and dried under vacuum.
10.28 g of 7-amino-3-formyl-ceph-3-em-4-carboxylate were dissolved in 55.6 ml of bis(trimethylsilyl)acetamide and 100 ml of propylene oxide. 33.6 g of (1R/S, 3’R)-(1′-tert. Butyloxycarbonyl-2-oxo-[1,3′]bipyrrolidinyl-3-yl)-triphenylphosphonium bromide (EP1067131, WO02.) were then added at 0 0 C /14332) slowly added in portions over 22 hours. The mixture was stirred at 1° C. until the starting material had reacted and then the reaction mixture was cooled to -20 ° C. The crystalline precipitate was filtered off under a nitrogen atmosphere and washed in portions with 180 ml of cyclohexane/bis(trimethylsilyl)acetamide 99.5/0.5. After drying under vacuum, the bissilylated
1.0g (6R,7R)-7-Trimethylsilylamino-3[E-(R)-1′-(5-tert.butyloxycarbonyl)-2-oxo-[I.Slbipyrrolidinyl-S-ylidenemethyO-δ-oxo-δ -thia-i-aza-bicyclo^^.Oloct^-ene^- carboxylic acid trimethylsilyl esters were dissolved in 10ml dichloromethane and a solution of 300mg dicyclohexylamine in 1ml EtOH and 10ml ethyl acetate was added. The precipitate was filtered off, washed with ethyl acetate and dried in vacuo.
3.0g (6R,7R)-7-Amino-3[E-(R)-1′-(5-tert-butyloxycarbonyl)-2-oxo-[1,3′]bipyrrolidinyl-3-ylidenemethyl]-8 -oxo-5-thia-1-aza-bicyclo[4.2.0]oct-2-ene-2-carboxylic acid in silylated form was dissolved in 150 ml of dichloromethane at 0°. 600 μl of DMF/water 5/1 and 1.8 ml of bis(trimethylsilyl) acetamide and 2.29 g of 2-trityloxyimino-2-(5-amino-1,2,4-thiadiazol-3-yl) acetic acid chloride were then added Hydrochoride (J. Antibiotics 37:557 – 571, 1984) was added in portions.
After 3 hours at 0°, the mixture was poured into 30 ml MeOH/120 ml water and the methylene chloride phase was separated off. The organic phase was concentrated to 66g and 25ml of trifluoroacetic acid was added. After 10 minutes, 1.5 ml triethylsilane and 10 ml water were added and the mixture was cooled to -15 ° C. The organic phase was separated off and again with 6 ml
Washed trifluoroacetic acid/water 1/1. The combined aqueous phases were diluted to 150 ml with water and filtered through an adsorber resin column with XAD-1600. After washing out the column with water, elution was carried out with water/acetonitrile 85/15. The product-containing fractions were concentrated in vacuo and allowed to stand at 0° for post-crystallization. The crystalline
Product was filtered off, washed with water and dried under vacuum.
Auswaage: 2,66g
4.2 Variant B:
7.4g (6R,7R)-7-Amino-3[E-(R)-1′-(5-tert-butyloxycarbonyl)-2-oxo-[1,3′]bipyrrolidinyl-3-ylidenemethyl]-8 -oxo-5-thia-1-aza-bicyclo[4.2.0]oct-2-ene-2-carboxylic acid was dissolved at 0° in 781 ml of dichloromethane with the addition of 6.7 ml of triethylamine. 8.65 g of 2-trityloxyimino-2-(5-amino-1,2,4-thiadiazol-3-yl)-acetic acid chloride hydrochloride were then added in portions. After the starting material had reacted, the mixture was poured into 500 ml of water and the methylene chloride phase was separated off. The organic phase was dried over Na 2 SC> 4 and concentrated in vacuo.
The residue was dissolved in 148 ml of dichloromethane and 4.5 ml of triethylsilane and 74 ml of trifluoroacetic acid were added at room temperature. After 30 minutes, 222 ml of dichloromethane and 222 ml of water were added and the mixture was cooled to -20 0 C. The organic phase was separated off and washed again with a mixture of 37 ml of trifluoroacetic acid and 148 ml of water. The combined aqueous phases were diluted with water to 364 ml, filtered through an adsorber resin and eluted with acetonitrile/water 15/85.
The filtrate was concentrated to 35g on a Rotavapor, filtered and washed with water.
After drying in a vacuum, 4.5 g of the sample was obtained.
6.0g (6R,7R)-7-Amino-3[E-(R)-1′-(5-tert-butyloxycarbonyl)-2-oxo-[1,3′]bipyrrolidinyl-3-ylidenemethyl]-8 -oxo-5-thia-1-aza-bicyclo[4.2.0]oct-2-ene-2-carboxylic acid in silylated form was dissolved in 300 ml of dichloromethane at 0°. 1200 μl of DMF/water 5/1 and 8.1 ml of bis(tirmethylsilyl)acetamide were then added as well as 5.3g of 2-trityloxyimino-2-(5-amino-1,2,4-thiadiazol-3-yl)acetic acid chloride Hydrochoride (J. Antibiotics 37:557 – 571, 1984) added in portions. The mixture was then poured into 60 ml MeOH/240 ml water and the methylene chloride phase was separated off. The organic phase was concentrated to 48g and 1.5 ml of triethylsilane was added. After adding 50ml
Trifluoroacetic acid was stirred at room temperature for 60 min, 20 ml of water was added and the mixture was cooled to -15°C. The organic phase was separated off and washed again with 20 ml trifluoroacetic acid/water 1/1. The combined aqueous phases were diluted to 500 ml with water and treated with 2.0 g of activated carbon. After filtration, the solution was concentrated in vacuo.
The residue was diluted to 50 ml with water and adjusted to pH 6.9 with saturated NaHCO 3 solution. The mixture was stirred at 0 0 C for 2 hours, filtered and the precipitate washed with water.
0, 5 3 g ( 6 R , 7 R )-7-[(Z)-2-(5-amino-[1,2,4]thiadiazol-3-yl)-2-hydroxyimino-acetylamino]-8- oxo-3-[(E)-(R)-2-oxo-[1,3′]bipyrrolidinyl-3-ylidenemethyl]-5-thia-1 – aza-bicyclo[4.2.0]oct-2-ene- 2 carboxylic acid were dissolved in 5 ml of dimethyl sulfoxide and 0.27 g of carbonic acid (5-methyl-2-oxo-[1,3]dioxol-4-ylmethyl)-4-nitrophenyl ester were added and stirred at room temperature. A solution of sodium ethyl hexanoate in 30 ml of acetone was added for precipitation. The precipitate was filtered and washed with acetone.
^ Zhanel GG, Lam A, Schweizer F, Thomson K, Walkty A, Rubinstein E, et al. (2008). “Ceftobiprole: a review of a broad-spectrum and anti-MRSA cephalosporin”. American Journal of Clinical Dermatology. 9 (4): 245–254. doi:10.2165/00128071-200809040-00004. PMID18572975. S2CID24357533.
^ Farrell DJ, Flamm RK, Sader HS, Jones RN (April 2014). “Activity of ceftobiprole against methicillin-resistant Staphylococcus aureus strains with reduced susceptibility to daptomycin, linezolid or vancomycin, and strains with defined SCCmec types”. International Journal of Antimicrobial Agents. 43 (4): 323–327. doi:10.1016/j.ijantimicag.2013.11.005. PMID24411474.
^ Nicholson SC, Welte T, File TM, Strauss RS, Michiels B, Kaul P, et al. (March 2012). “A randomised, double-blind trial comparing ceftobiprole medocaril with ceftriaxone with or without linezolid for the treatment of patients with community-acquired pneumonia requiring hospitalisation”. International Journal of Antimicrobial Agents. 39 (3): 240–246. doi:10.1016/j.ijantimicag.2011.11.005. PMID22230331.
^“Zeftera (previously Zevtera) EPAR”. European Medicines Agency (EMA). 18 February 2010. Retrieved 6 April 2024. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
External links
Clinical trial number NCT03138733 for “Ceftobiprole in the Treatment of Patients With Staphylococcus Aureus Bacteremia” at ClinicalTrials.gov
Clinical trial number NCT03137173 for “Ceftobiprole in the Treatment of Patients With Acute Bacterial Skin and Skin Structure Infections” at ClinicalTrials.gov
Clinical trial number NCT00326287 for “Ceftobiprole in the Treatment of Patients With Community-Acquired Pneumonia” at ClinicalTrials.gov
Clinical trial number NCT03439124 for “Ceftobiprole in the Treatment of Pediatric Patients With Pneumonia” at ClinicalTrials.gov
////////Ceftobiprole, BAL-9141, BAL-9141-000, BAL-9141000, BAL9141-000, RO 63-9141, RO-63-9141, RO-639141, fda 2024, zevtera, approvals 2024, Ceftobiprole medocaril sodium salt
Fda approved, To treat paroxysmal nocturnal hemoglobinuria, 12/5/2023, Fabhalta ‘CHINA 2024
Iptacopan is a small-molecule factor B inhibitor previously investigated as a potential treatment for the rare blood disease paroxysmal nocturnal hemoglobinuria (PNH) by inhibiting the complement factor B.1 Factor B is a positive regulator of the alternative complement pathway, where it activates C3 convertase and subsequently C5 convertase.2 This is of particular importance to PNH, where one of the disease hallmarks is the mutation of the PIGA gene. Due to this mutation, all progeny erythrocytes will lack the glycosyl phosphatidylinositol–anchored proteins that normally anchor 2 membrane proteins, CD55 and CD59, that protect blood cells against the alternative complement pathway.3 Additionally, iptacopan has the benefit of targeting factor B, which only affect the alternative complement pathway, leaving the classic and lectin pathway untouched for the body to still mount adequate immune responses against pathogens.2
On December 6th, 2023, Iptacopan under the brand name Fabhalta was approved by the FDA for the treatment of adults with PNH. This approval was based on favorable results obtained from the phase III APPL-PNH and APPOINT-PNH studies, where 82.3% and 77.5% of patients experienced a sustained hemoglobin improvement without transfusions respectively.5
Iptacopan was approved by the US Food and Drug Administration (FDA) for the treatment of adults with paroxysmal nocturnal hemoglobinuria in December 2023.[2][3]
Medical uses
Iptacopan is indicated for the treatment of adults with paroxysmal nocturnal hemoglobinuria.[1][4]
In a clinical study with twelve participants, iptacopan as a single drug led to the normalization of hemolytic markers in most patients, and no serious adverse events occurred during the 12-week study.[5][6]
Iptacopan is also investigated as a drug in other complement-mediated diseases, like age-related macular degeneration and some types of glomerulopathies.[7]
To a 3 L three-necked flask were successively added tetrahydrofuran (150 mL) and 4-bromoxynil (50 g). Isopropylmagnesium chloride lithium chloride coordination complex (1.3 M, 210 mL) was slowly added to the reaction system under nitrogen atmosphere. After the reaction was carried out at room temperature for 2 h, the reaction system was diluted with anhydrous tetrahydrofuran (500 mL) for dilution. The reaction system was cooled to −5° C., and 4-methoxypyridine (25 mL) was added, followed by slowly dropwise addition of benzyl chloroformate (35 mL) (the system temperature was maintained below 0° C.). After the dropwise addition was completed, the reaction system was successively reacted at 0° C. for 2 h, and then warmed to room temperature and reacted at that temperature for 16 h. After the reaction was completed, hydrochloric acid solution (6 M, 150 mL) was added. The mixture was stirred at room temperature for half an hour, added with water (1000 mL) for dilution, and extracted twice with ethyl acetate (500 mL). The extract phase was washed with saturated brine (50 mL), then dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated, and the resulting crude product was separated and purified by a silica gel column (petroleum ether:ethyl acetate=3:1 to 1:1) to give intermediate 1 (23 g, yield: 23%). MS m/z (ESI): 333.0 [M+H].
Intermediate 2:
To a 500 mL single-neck flask were successively added intermediate 1 (28 g), zinc powder (55 g) and acetic acid (200 mL). The reaction mixture was heated to 100° C. and reacted at that temperature for 16 h. After the reaction was completed, the reaction mixture was filtered. The filtrate was added with water (500 mL) for dilution and extracted with ethyl acetate (500 mL). The extract phase was washed twice with saturated aqueous sodium bicarbonate solution (500 mL), washed once with saturated brine (100 mL), dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated under reduced pressure to give intermediate 2 (26 g, yield: 73%). MS m/z (ESI): 334.8 [M+H].
Intermediate 3:
To a 1 L single-neck flask were successively added tetrahydrofuran (100 mL), ethanol (100 mL) and intermediate 2 (26 g), and sodium borohydride (2 g) was added in batches. The mixture was reacted at room temperature for 2 h. After the reaction was completed, the system was cooled to 0° C., and saturated aqueous ammonium chloride solution (100 mL) was added until the temperature did not increase any more. Water (300 mL) was added for dilution, followed by extraction with ethyl acetate (200 mL×2). The extract phase was washed with saturated brine (500 mL), dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated under reduced pressure to give intermediate 3 (25 g, yield: 76%). MS m/z (ESI): 336.9 [M+H].
Intermediate 4:
Dichloromethane (200 mL) was added to a 500 mL single-neck flask, and then intermediate 3 (25 g), imidazole (6.6 g) and tert-butyldiphenylchlorosilane (25 g) were successively added. The mixture was reacted at room temperature for 2 h. After the reaction was completed, water (500 mL) was added for dilution, followed by the extraction with dichloromethane (200 mL). The extract phase was washed with water (50 mL), dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated under reduced pressure. The residue was separated and purified by a silica gel column (petroleum ether:ethyl acetate=10:1) to give intermediate 4 (5.7 g, yield: 13%, R f=0.55; isomer R f=0.50). MS m/z (ESI): 597.0 [M+23].
Intermediate 5:
To a 250 mL single-neck flask were successively added a solution of tetrabutylammonium fluoride in tetrahydrofuran (1 M, 30 mL) and intermediate 4 (5 g). The mixture was reacted at room temperature for 2 h. After the reaction was completed, water (100 mL) was added for dilution, followed by the extraction with ethyl acetate (50 mL×3). The extract phase was washed with saturated brine (100 mL), dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated under reduced pressure. The residue was separated and purified by a silica gel column (petroleum ether:ethyl acetate=3:1 to 0:1) to give a racemic intermediate. The intermediate was subjected to SFC chiral resolution (apparatus: SFC Thar prep 80; column: CHIRALPAK AD-H, 250 mm×20 mm, 5 m; modifier: 35% methanol (0.2% aqueous ammonia); column temperature: 40° C.; column pressure: 60 bar; wavelength: 214/254 nm; flow rate: 40 g/min; Rt=4.78 min) to give intermediate 5 (1.2 g, yield: 41%). MS m/z (ESI): 358.8 [M+23].
Intermediate 6:
To a 100 mL single-neck flask were successively added N,N-dimethylformamide (15 mL) as a solvent, intermediate 5 (1.2 g) and iodoethane (1.1 g). After the reaction system was cooled to 0° C., sodium hydrogen (60%, 243 mg) was added. Then the system was warmed to room temperature and reacted at that temperature for 2 h. After the reaction was completed, water (30 mL) was added for dilution, followed by the extraction with ethyl acetate (50 mL). The extract phase was washed with saturated brine (10 mL), dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated under reduced pressure to give intermediate 6 (1.2 g, yield: 83%). MS m/z (ESI): 386.9 [M+23].
Intermediate 7:
To a 100 mL single-neck flask were successively added methanol (10 mL), water (10 mL), concentrated sulfuric acid (10 mL) and intermediate 6 (1.2 g). The mixture was heated to 80° C. and reacted at that temperature for 48 h. After the reaction was completed, the reaction mixture was concentrated to remove methanol. The residue was made neutral with saturated aqueous sodium hydroxide solution and extracted three times with ethyl acetate (10 mL). The extract phase was washed with saturated brine (5 mL), dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated under reduced pressure to give intermediate 7 (850 mg, yield: 81%). MS m/z (ESI): 264.1 [M+H]. 1H NMR (400 MHz, CDCl 3) δ 8.01 (d, J=8.3 Hz, 2H), 7.49 (d, J=8.3 Hz, 2H), 4.13 (dd, J=11.7, 2.4 Hz, 1H), 3.92 (s, 3H), 3.82-3.70 (m, 1H), 3.62-3.47 (m, 2H), 3.27-3.10 (m, 1H), 3.02-2.88 (m, 1H), 2.07-1.97 (m, 1H), 1.95-1.85 (m, 1H), 1.82-1.62 (m, 2H), 1.27 (t, J=7.0 Hz, 3H).
Intermediate 8:
To a 250 mL single-neck flask were successively added dichloromethane (50 mL), 5-methoxy-7-methyl-1H-indole (3 g), BOC anhydride (5.68 g), 4-dimethylaminopyridine (227 mg) and triethylamine (2.26 g). The mixture was reacted at room temperature for 16 h. After the reaction was completed, the reaction mixture was quenched by adding saturated ammonium chloride solution (5 mL) and extracted three times with dichloromethane (20 mL). The combined organic phases were washed with water (5 mL), dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated. The residue was purified by column chromatography on silica gel (petroleum ether:ethyl acetate=10:1) to give intermediate 8 (4.6 g, yield: 94%). MS m/z (ESI): 262.0 [M+H].
Intermediate 9:
To a 250 mL single-neck flask were successively added dichloromethane (80 mL), N-methylformanilide (3.8 g) and oxalyl chloride (3.6 g). The mixture was stirred at room temperature for 3 h. Then the reaction temperature was lowered to −14° C., and intermediate 8 (2.5 g) was added. The reaction system was naturally warmed to room temperature and stirred for 1 h. After the reaction was completed, the reaction liquid was poured into ice water and extracted three times with dichloromethane (100 mL). The combined extract phases were washed twice with water (10 mL), dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated. The residue was separated and purified by a silica gel column (petroleum ether:ethyl acetate=20:1) to give intermediate 9 (1.3 g, yield: 47%). MS m/z (ESI): 290.0 [M+H]. 1H NMR (400 MHz, CDCl 3) δ 10.65 (s, 1H), 7.65 (d, J=3.4 Hz, 1H), 7.49 (d, J=3.4 Hz, 1H), 6.76 (s, 1H), 3.98 (s, 3H), 2.70 (s, 3H), 1.65 (s, 9H).
Intermediate 10:
To a 50 mL three-necked flask were successively added 1,2-dichloroethane (5 mL), intermediate 7 (127 mg) and intermediate 9 (130 mg). The mixture was reacted at room temperature for 18 h. Then sodium triacetoxyborohydride (438.72 mg) was added, and the system was successively reacted at room temperature for 18 h. After the reaction was completed, dichloromethane (10 mL) was added for dilution, followed by a wash with 10 mL of water. The organic phase was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated. The residue was separated and purified by a silica gel column (methanol:dichloromethane=1:10) to give intermediate 10 (50 mg, yield: 14.58%). MS m/z (ESI): 437.3 [M+H], RT=1.142 min.
Intermediate 11:
To a 50 mL three-necked flask were successively added tetrahydrofuran (0.5 mL), methanol (0.5 mL), water (0.5 mL), sodium hydroxide (44 mg) and intermediate 10 (50 mg). The mixture was reacted at room temperature for 18 h. After the reaction was completed, the reaction liquid was directly concentrated under reduced pressure and lyophilized to give intermediate 11 (50 mg, yield: 92%). MS m/z (ESI): 423.1 [M+H].
The alternative pathway (AP) of the complement system is a key contributor to the pathogenesis of several human diseases including age-related macular degeneration, paroxysmal nocturnal hemoglobinuria (PNH), atypical hemolytic uremic syndrome (aHUS), and various glomerular diseases. The serine protease factor B (FB) is a key node in the AP and is integral to the formation of C3 and C5 convertase. Despite the prominent role of FB in the AP, selective orally bioavailable inhibitors, beyond our own efforts, have not been reported previously. Herein we describe in more detail our efforts to identify FB inhibitors by high-throughput screening (HTS) and leveraging insights from several X-ray cocrystal structures during optimization efforts. This work culminated in the discovery of LNP023 (41), which is currently being evaluated clinically in several diverse AP mediated indications.
a Reagents and conditions: (a) i PrMgCl·LiCl, Cbz-Cl, THF; (b) Zn, AcOH; (c) LiBH4, THF; (d) TBDPS-Cl, imidazole, DMF; (e) separation of diastereomers by flash chromatography; (f) TBAF, THF; (g) NaH, EtI, DMF; (h) Ba(OH)2, i PrOH, H2O; (i) K2CO3, MeI, DMF; (j) H2, Pd/C, MeOH; (k) (±)-50, DIPEA, DMA; (l) K2CO3, MeOH; then TMS-diazomethane, toluene, MeOH; (m) chiral SFC; (n) LiOH, H2O, MeOH, THF; (o) (2S,4S)-50, NaBH(OAc)3, DCE.
4-((2S,4S)-(4-Ethoxy-1-((5-methoxy-7-methyl-1H-indol-4- yl)methyl)piperidin-2-yl))benzoic Acid (41, LNP023). Step 1: tert-Butyl 4-(((2S,4S)-4-Ethoxy-2-(4-(methoxycarbonyl)phenyl)- piperidin-1-yl)methyl)-5-methoxy-7-methyl-1H-indole-1-carboxylate (58). To a solution of tert-butyl 4-formyl-5-methoxy-7-methyl1H-indole-1-carboxylate (57) (1.5 g, 5.18 mmol) and methyl 4- ((2S,4S)-4-ethoxypiperidin-2-yl)benzoate ((2S,4S)-50) (1.185 g, 4.50 mmol) in DCE (20 mL) was added NaBH(OAc)3 (3 g, 14.1 mmol), and this was stirred at rt for 21.5h. Additional tert-butyl 4-formyl-5- methoxy-7-methyl-1H-indole-1-carboxylate (57) (500 mg, 1.90 mmol) was added, and this was stirred for 20 h. The reaction was diluted with EtOAc, washed successively with 5% aqueous NaHCO3, H2O, and brine, dried over Na2SO4, filtered, and concentrated to provide the title compound (2.415 g, quant) which was used without further purification. MS (ESI+) m/z 537.4 (M + H). The absolutestereochemistry was ultimately determined via cocrystallization of 41 with the catalytic domain of FB. Step 2: 4-((2S,4S)-(4-Ethoxy-1-((5-methoxy-7-methyl-1H-indol-4- yl)methyl)piperidin-2-yl))benzoic Acid (41, LNP023). To a solution of tert-butyl 4-(((2S,4S)-4-ethoxy-2-(4-(methoxycarbonyl)phenyl)- piperidin-1-yl)methyl)-5-methoxy-7-methyl-1H-indole-1-carboxylate (58) (2.415 g, 4.50 mmol) in THF (10 mL) and MeOH (20 mL) was added 1 M LiOH in H2O (15 mL, 15 mmol), and this was stirred at 70 °C for 8 h. The reaction was cooled to rt, diluted with H2O, half saturated aqueous KHSO4 and citric acid, saturated with sodium chloride, then extracted with 9:1 DCM/TFE, dried with Na2SO4, filtered, and concentrated. RP-HPLC-B purification provided the title compound (730 mg, 38% for 2 steps). 1 H NMR (400 MHz, D2O) δ 7.96 (d, J = 8.0 Hz, 2H), 7.58 (d, J = 8.1 Hz, 2H), 7.30 (d, J = 3.2 Hz, 1H), 6.66 (s, 1H), 6.20 (s, 1H), 4.62−4.47 (m, 1H), 4.06 (d, J = 13.2 Hz, 1H), 3.97−3.76 (m, 2H), 3.66−3.48 (m, 5H), 3.43−3.29 (m, 1H), 3.26−3.15 (m, 1H), 2.35 (s, 3H), 2.31−2.11 (m, 2H), 2.00 (d, J = 15.4 Hz, 1H), 1.93−1.74 (m, 1H), 1.25−1.07 (m, 3H). HRMS calcd for C25H31N2O4 (M + H)+ 423.2284, found 423.2263. 4-((2S,4S)-(4-Ethoxy-1-((5-methoxy-7-methyl-1H-indol-4- yl)methyl)piperidin-2-yl))benzoic Acid Hydrochloride (41· HCl). To a solution of 41 (620 mg, 1.47 mmol) in H2O (10 mL) and acetonitrile (3 mL) was added 5 M aqueous HCl (0.5 mL, 2.5 mmol). The mixture was then lyophilized, and the resulting solid was suspended in i PrOH and heated to 70 °C. The mixture turned into a solution after 1.5 h and was then cooled to rt with stirring. After about 5 h, the mixture turned into a suspension and the solid was collected by filtration and dried under high vacuum at 50 °C to provide the title compound as the hydrochloride salt (450 mg, 65%). 1 H NMR (400 MHz, methanol-d4) δ 10.73 (s, 1H), 8.23 (d, J = 8.2 Hz, 2H), 7.74 (d, J = 8.3 Hz, 2H), 7.36−7.31 (m, 1H), 6.77 (s, 1H), 6.42−6.31 (m, 1H), 4.40−4.19 (m, 2H), 3.87−3.80 (m, 1H), 3.76 (s, 3H), 3.68− 3.50 (m, 4H), 3.45−3.38 (m, 1H), 2.51 (s, 3H), 2.30−2.18 (m, 2H), 2.13−1.89 (m, 2H), 1.31 (t, J = 7.0 Hz, 3H). MS (ESI+) m/z 423.3 (M + H).
SYN European Journal of Medicinal Chemistry 291 (2025) 117643
Iptacopan (Fabhalta®), a first-in-class oral therapeutic agent discovered by Novartis, specifically targets the complement Factor B protein within the alternative complement system. NMPA granted marketing authorization in 2024, indicated for complement inhibitor-naïve adult patients diagnosed with paroxysmal nocturnal hemoglobinuria (PNH) [75]. By competitively binding to the catalytic domain of Factor B, the drug effectively blocks C3 convertase assembly, thereby suppressing downstream cleavage of C3 into its active fragments. This dual inhibitory action addresses both intravascular erythrocyte destruction and extravascular hemolytic processes characteristic of PNHpathogenesis [76]. Clinical validation emerged from the multinational APPOINT-PNH study (ClinicalTrials.gov identifier NCT04820530), where treatment-naïve participants exhibited sustained hemoglobin stabilization (≥12 g/dL) in 79.6 % of cases, achieving transfusion in dependence over 24 weeks. Secondary endpoints revealed significant improvements in fatigue scores and health-related quality metrics [77]. Safety monitoring identified encapsulated bacterial infection as critical risks, necessitating mandatory vaccination ≥2 weeks pre-treatment. Common treatment-emergent adverse events comprised transient gastrointestinal disturbances (nausea 18.3 %, diarrhea 14.7 %) and mild cephalgia (22.1 %), with resolution typically occurring within 4 weeks [78]. The synthetic pathway of Iptacopan, delineated in Scheme 18, initiates with nucleophilic substitution between Ipta-001 and Ipta-002, followed by Grignard coupling yielding Ipta-003 [79]. This intermedi ate undergoes NaBH4-mediated reduction and TMSCl-induced silanization to afford Ipta-004. Acid-catalyzed TMS deprotection (HCl/MeOH) delivers Ipta-005, which progresses through sequential alkylation (methyl iodide/K2CO3 catalytic hydrogenation (H)/Pd–C), transesterification (EtONa), and to construct Ipta-006. Condensation with Ipta-007 and subsequent reduction forms Ipta-008. Strategic TFA-mediated Boc cleavage in DCM followed by HCl-induced salt formation in dioxane ultimately furnishes Iptacopan hydrochloride.
75-79
[75] Iptacopan, Drugs and Lactation Database (Lactmed®), National Institute of Child Health and Human Development, Bethesda (MD), 2006. [76] J.H. Jang, L. Wong, B.S. Ko, S.S. Yoon, K. Li, I. Baltcheva, P.K. Nidamarthy, R. Chawla, G. Junge, E.S. Yap, Iptacopan monotherapy in patients with paroxysmal nocturnal hemoglobinuria: a 2-cohort open-label proof-of-concept study, Blood Adv 6 (2022) 4450–4460. [77] A.M. Risitano, C. de Castro, B. Han, A.G. Kulasekararaj, J.P. Maciejewski, P. Scheinberg, Y. Ueda, S. Vallow, G. Bermann, M. Dahlke, R. Kumar, R. Peffault de Latour, Patient-reported improvements in patients with PNH treated with iptacopan from two phase 3 studies, Blood Adv 9 (2025) 1816–1826. [78] C.M. de Castro, B.J. Patel, Iptacopan for the treatment of paroxysmal nocturnal hemoglobinuria, Expert Opin Pharmacother 25 (2024) 2331–2339. [79] N. Mainolfi, T. Ehara, R.G. Karki, K. Anderson, A. Mac Sweeney, S.M. Liao, U. A. Argikar, K. Jendza, C. Zhang, J. Powers, D.W. Klosowski, M. Crowley, T. Kawanami, J. Ding, M. April, C. Forster, M. Serrano-Wu, M. Capparelli, R. Ramqaj, C. Solovay, F. Cumin, T.M. Smith, L. Ferrara, W. Lee, D. Long, M. Prentiss, A. De Erkenez, L. Yang, F. Liu, H. Sellner, F. Sirockin, E. Valeur, P. Erbel, D. Ostermeier, P. Ramage, B. Gerhartz, A. Schubart, S. Flohr, N. Gradoux, R. Feifel, B. Vogg, C. Wiesmann, J. Maibaum, J. Eder, R. Sedrani, R.A. Harrison, M. Mogi, B.D. Jaffee, C.M. Adams, Discovery of 4-((2S,4S)-4-Ethoxy-1-((5- methoxy-7-methyl-1H-indol-4-yl)methyl)piperidin-2-yl)benzoic acid (LNP023), a factor B inhibitor specifically designed to be applicable to treating a diverse array of complement mediated diseases, J. Med. Chem. 63 (2020) 5697–5722.
.
//////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Clinical trial number NCT04558918 for “Study of Efficacy and Safety of Twice Daily Oral LNP023 in Adult PNH Patients With Residual Anemia Despite Anti-C5 Antibody Treatment (APPLY-PNH)” at ClinicalTrials.gov
Clinical trial number NCT04820530 for “Study of Efficacy and Safety of Twice Daily Oral Iptacopan (LNP023) in Adult PNH Patients Who Are Naive to Complement Inhibitor Therapy (APPOINT-PNH)” at ClinicalTrials.gov
fda approved 4/26/2024, To treat WHIM syndrome (warts, hypogammaglobulinemia, infections and myelokathexis), Xolremdi
Mavorixafor (AMD-070) is a potent, selective and orally available CXCR4 antagonist, with an IC50 value of 13 nM against CXCR4125I-SDF binding, and also inhibits the replication of T-tropic HIV-1 (NL4.3 strain) in MT-4 cells and PBMCs with an IC50 of 1 and 9 nM, respectively.
AMD-070 is a small molecule drug candidate that belongs to a new investigational class of anti-HIV drugs known as entry (fusion) inhibitors. Approximately 76% of HIV-patients with measurable viral load are infected with a strain of virus that is resistant to one or more classes of antiretroviral agents, thus reducing treatment options. Unlike many existing HIV drugs that target the virus after it has infected a healthy cell, AMD-070 blocks the virus from entering a healthy cell, thus preventing the replication process. AMD-070 targets the CXCR4 receptor on HIV and prevents the virus from entering and infecting healthy cells. AMD-070 is specific for the CXCR4 receptor and does not interact with any other chemokine receptors in vitro. AMD-070 strongly inhibits viral infection by all CXCR4 using virus (including virus using CXCR4 alone and/or virus using CXCR4 and CCR5) in vitro. AMD-070 is orally bioavailable in animals, it has suitable PK and toxicity profile for oral dosing. AMD-070 shows additive or synergistic effects in vitro in combination with other known anti-HIV agents. AMD-070 is active against CXCR4 using HIV strains that are resistant to existing antiretroviral therapies in vitro, reveals potent anti-HIV activity against CXCR4-using laboratory strains and clinical isolates. MD-070 had been in phase II clinical trials by Genzyme for the treatment of HIV infection. However, this research has been discontinued. AMD-070 has been studied in Phase I/II clinical trials for the treatment of Renal cell carcinoma and Phase I clinical trials for the treatment of malignant melanoma and solid tumours.
A novel and practical synthesis of mavorixafor (1) is reported. The novelty of this synthetic route is the use of 8-chloro-5,6,7,8-tetrahydroquinoline (9) and 1,4-diaminobutane as the materials, instead of 8-amino-5,6,7,8-tetrahydroquinoline (4) and N,N-diprotected aminobutyraldehyde (6a or 6b). The preparation of (S)-8-(4-aminobutylamino)-5,6,7,8-tetrahydroquinoline (13) by resolution with N-acetyl-l-leucine was first achieved. Then the one-pot synthesis of 1 from 13 involving protection, condensation, and subsequent hydrolysis was successfully developed. In addition, the final product with a satisfactory purity (>99.5%, detected by both achiral and chiral HPLC) was obtained by a simple operation (salification) without column chromatographic purification.
Charge diethyl-4-aminobutyl acetal (E) (1.00 wt, 1.00 eq) to vessel A. Charge acetonitrile (10.0 vol, 7.8 wt) and adjust temperature to 20°C. Heat the mixture to 40°C. Concentrate the reaction mixture to 6.0 vol under reduced pressure at 35 to 45°C.
[0098] Acetonitrile filler (5.0 vol, 3.9wt) at 35 to 45°C. Concentrate the reaction mixture to 6.0 vol under reduced pressure 35 to 45°C. This step is repeated once as described below.
[0099] Acetonitrile filler (5.0 vol, 3.9wt) at 35 to 45°C. Concentrate the reaction mixture to 6.0 vol under reduced pressure at 35 to 45°C. Cool to 20°C.
[00100] Charge di-tert-butyl dicarbonate (1.1 eq, 1.5 wt) to a drum, followed by acetonitrile (0.4 vol, 0.3 wt) and agitate until fully dissolved. Concentrate the reaction mixture to 6.0 vol under reduced pressure at 35 to 45°C.
[00101] Charge this di-tert-butyl dicarbonate solution in acetonitrile to vessel A maintaining 20°C. Charge acetonitrile (1.5 vol, 1.1 wt) to the solution as a line rinse and stir at 20°C for 30 to 60 min..
[00102] Charge 4-dimethylaminopyridine (0.076 wt, 0.10 eq) to the vessel A at 20°C. Heat the solution to 40°C. Concentrate the reaction mixture to 5.0 vol under reduced pressure. Charge acetonitrile (5.0 vol, 3.9 wt) to the solution. Concentrate the reaction mixture to 5.0 vol under reduced pressure.
[00103] Take the resulting solution of Dl into next reaction without isolation.
Step IB: Preparation of Cl
[00104] Charge acetonitrile (2.0 vol, 1.6 wt) at 35 to 45°C to vessel A containing solution of D-1 from Step 1A.
[00105] Charge di-tert-butyl dicarbonate (1.4 eq, 1.9 wt) to a drum, followed by acetonitrile (10.0 vol, 7.8 wt) and agitate until fully dissolved. Charge this di-tert-butyl dicarbonate solution to vessel A, 2 to 6 h while distilling under vacuum at 35 to 45°C maintaining the volume of the reaction at 7.0 vol. Load acetonitrile (3.0 vol, 2.4 wt) over 20 to 40 min. as a line rinse while distilling under vacuum at 35 to 45°C, maintaining the volume of the reaction at 7.0 vol.
[00106] Charge di-tert-butyl dicarbonate, (0.14 eq, 0.19 wt) to a drum, followed by acetonitrile (1.0 vol, 0.74 wt) and agitate until fully dissolved. Charge this di-tert-butyl dicarbonate solution to vessel A over 20 to 40 min.. Charge acetonitrile (0.3 vol, 0.24 wt) over 10 to 20 min as a line rinse while distilling under vacuum at 35 to 45°C, maintaining the volume of the reaction at 7.0 vol.
[00107] Concentrate the reaction mixture to 5.0 vol distilling under vacuum at 35 to 45°C.
[00108] Charge n-heptane, (7.5 vol, 5.1 wt) to the reaction mixture, and concentrate the reaction mixture to 5.0 vol under reduced pressure at 40°C. This step is repeated once as described below.
[00109] Charge n-heptane, (7.5 vol, 5.1 wt) to the reaction mixture, and concentrate the reaction mixture to 5.0 vol under reduced pressure at 40°C.
[00110] Charge decolorizing, activated charcoal (0.2 wt) to the solution and stir for 1 to 2 h at 40°C. Filter the reaction mixture at 40°C. Charge n-heptane, (2.0 vol, 1.4 wt) to the reactor vessel and stir for 5 to 15 min. at 20°C before charging to the filter as a line rinse. Combine the filtrate and wash, and as required adjust to 20°C.
[00111] Take the resulting solution of Cl into next reaction without isolation.
Step 1C: Preparation of Bl
[00112] Charge 15% v/v acetic acid (2.0 vol) caution gas evolution, to vessel A containing solution of Cl from Step IB, maintaining the temperature at 20°C and stir for 10 min. at 20°C. Allow the phases to separate for 15 min. at 20°C. Discharge the aqueous phase to waste, retaining the organic phase in vessel A. This step is repeated once as described below.
[00113] Charge 15% v/v acetic acid (2.0 vol) maintaining 20°C and stir for 10 min. at 20°C. Allow the phases to separate for 15 min. at 20°C. Discharge the aqueous phase to waste, retaining the organic phase in vessel A.
[00114] Adjust the reaction to 30°C. Charge 4% w/w sodium chloride solution (2.1 vol) to the vessel maintaining the temperature at 30°C. Charge glacial acetic acid (4.1 vol, 4.3 wt) to the vessel maintaining 30°C. Stir the reaction mixture for 2 h maintaining the temperature at 30°C.
[00115] Charge purified water, (6.0 vol) at 30°C. Stir the contents for 5 to 10 min. at 30°C, and separate the phases, retaining the upper organic phase in vessel A. Charge the lower aqueous phase to vessel B.
[00116] Charge purified water (4.0 vol) at 30°C and stir for 5 to 10 min. maintaining the temperature at 30°C. Separate the phases at 30°C, retaining the upper organic phase in vessel A. Charge the lower aqueous phase to vessel B.
[00117] Adjust the temperature to 30°C of vessel B containing combined aqueous phases. Charge n-heptane, (2.0 vol, 1.4 wt) to vessel B and stir for 5 to 10 min. maintaining the temperature at 30°C. Separate the phases at 30°C, over 15 min.. Charge the upper organic phase to vessel A and recharge the lower aqueous phase to vessel B. This step is repeated two additional times as described below.
[00118] Charge n-heptane, (2.0 vol, 1.4 wt) to vessel B and stir for 5 to 10 min. maintaining the temperature at 30°C. Separate the phases at 30°C, over 15 min.. Charge the upper organic phase to vessel A and recharge the lower aqueous phase to vessel B.
[00119] Charge n-heptane, (2.0 vol, 1.4 wt) to vessel B and stir for 5 to 10 min. maintaining the temperature at 30°C. Separate the phases at 30°C, over 15 min., discharge the lower aqueous phase to waste and charge the upper organic layer to vessel A.
[00120] Concentrate the combined organic phases in vessel A to 3.0 vol at 10 to 20°C under reduced pressure. Offload the solution to new HDPE drum(s) and line rinse with n-heptane (0.5
vol, 0.4 wt) at 20°C. Homogenize the drum and store as “Bl solution in n-heptane,” and take into next reaction without isolation.
Step ID: Preparation of F-2d
[00121] Calculate a new 1.00 wt based on the above assay.
[00122] Charge “Bl solution in n-heptane” from Step 1C (1.00 wt, 1.00 eq, corrected for w/w assay, ca. 3.0 vol), into an appropriate vessel. THF load (3.0 vol, 2.7 wt). Heat the reaction mixture to 40°C.
[00123] Charge purified water, (0.02 vol, 0.02 wt) followed by sodium metabisulphite, (0.125 eq, 0.08 wt) as a solid via the charge hole at 40°C. Stir the resulting mixture for 30 to 35 min. at 40°C. This step was repeated four additional times to add the reagent in five portions total, as detailed below.
[00124] Charge purified water, (0.02 vol, 0.02 wt) followed by sodium metabisulphite, (0.125 eq, 0.08 wt) as a solid via the charge hole at 40°C. Stir the resulting mixture for 30 to 35 min. at 40°C.
[00125] Charge purified water, (0.02 vol, 0.02 wt) followed by sodium metabisulphite, (0.125 eq, 0.08 wt) as a solid via the charge hole at 40°C. Stir the resulting mixture for 30 to 35 min. at 40°C.
[00126] Charge purified water, (0.02 vol, 0.02 wt) followed by sodium metabisulphite, (0.125 eq, 0.08 wt) as a solid via the charge hole at 40°C. Stir the resulting mixture for 30 to 35 min. at 40°C.
[00127] Charge purified water, (0.02 vol, 0.02 wt) followed by sodium metabisulphite, (0.125 eq, 0.08 wt) as a solid via the charge hole at 40°C. Stir the resulting mixture for 36 hours at 40°C.
[00128] Cool the reaction mixture to 20°C over 3 to 4 h at a target constant rate. Filter the reaction mixture at 20°C on a 1-2 pm cloth.
[00129] Wash the solid with a pre-mixed mixture of THF (0.5 vol, 0.5 wt) and n-heptane (0.5 vol, 0.3 wt) maintaining the temperature at 20°C. This step was repeated an additional three times, as detailed below.
[00130] Wash the solid with n-heptane, (2.0 vol, 1.4 wt) as a line rinse and apply to the filtercake at 20°C.
[00131] Wash the solid with n-heptane, (2.0 vol, 1.4 wt) as a line rinse and apply to the filtercake at 20°C.
[00132] Wash the solid with acetonitrile, (2.0 vol, 1.6 wt) as a line rinse and apply to the filtercake at 20°C.
[00133] Dry the solid at 38°C under a flow of nitrogen for 12 h.
[00137] Charge J, (1.00 wt, 1.00 eq) to vessel A. Charge purified water, (1.0 vol, 1.0 wt) to vessel A and as necessary adjust the temperature to 20°C. Charge concentrated hydrochloric acid, (4.0 eq, 3.0 vol, 3.6 wt) to vessel A maintaining the temperature at 20°C. Line rinse with purified water, (0.5 vol, 0.5 wt) maintaining the contents of vessel A at 15 to 25°C.
[00138] Charge chloroacetic acid, (1.3 wt, 1.5 eq) and purified water, (1.0 vol, 1.0 wt) to vessel B and as necessary, adjust the temperature to 20°C. Stir until fully dissolved, expected 10 to 20 min.
[00139] Charge the chloroacetic acid solution to vessel A maintaining the temperature of vessel A at 20°C. Line rinse vessel A with purified water, (0.5 vol, 0.5 wt) at 15 to 25°C and charge to vessel B at 20°C. Heat the reaction mixture to 80°C. Stir the reaction mixture at 80°C for 20 h.
[00140] Cool the reaction mixture to 10°C over 1.5 h. Load 47% w/w potassium phosphate solution (6.0 vol) over 60 min. targeting a constant rate maintaining 10°C. Adjust the pH of the reaction mixture by charging 47% w/w potassium phosphate solution to pH 7.0 maintaining the reaction temperature at 10°C. Expected charge is 2.0 to 3.5 vol 47% w/w potassium phosphate solution.
[00141] Stir the slurry for >30 min. maintaining 10°C and rechecking the pH, pass criterion pH 7.0. Filter the reaction mixture through 20 pm cloth at 10°C. Wash the filter-cake with purified water, (1.0 vol, 1.0 wt) at 10°C. This step is repeated additional three times as described below.
[00142] Slurry wash the filter-cake in the reactor vessel with purified water, (10.0 vol, 10.0 wt) for 45 min. to 90 min. at 10°C. Filter the mixture through 20 pm cloth at 10°C.
[00143] Slurry wash the filter-cake in the reactor vessel with purified water, (10.0 vol, 10.0 wt) for 45 min. to 90 min. at 10°C. Filter the mixture through 20 pm cloth at 10°C.
[00144] Slurry wash the filter-cake in the reactor vessel with purified water, (10.0 vol, 10.0 wt) for 45 min. to 90 min. at 10°C. Filter the mixture through 20 pm cloth at 10°C.
[00145] Wash the filter-cake with purified water, (1.0 vol, 1.0 wt) at 10°C. The filter-cake was washed with purified water additional five times as described below.
[00146] Wash the filter-cake with purified water, (1.0 vol, 1.0 wt) at 10°C.
[00147] Wash the filter-cake with acetonitrile, (2×1.3 vol, 2×1.0 wt) at 10°C.
[00148] Dry the filter-cake on the filter under vacuum and strong nitrogen flow through the filter cake at 20°C until the water content is <15.0% w/w by Karl-Fisher analysis.
[00149] Dry the filter-cake on the filter under vacuum and strong nitrogen flow through the filter cake at 30°C until the water content is <5.0% w/w by Karl-Fisher analysis.
[00150] Dry the filter-cake on the filter under vacuum and strong nitrogen flow through the filter cake at 50°C until the water content is <1.0% w/w by Karl-Fisher analysis.
[00151] Yield of compound Gl: about 75%.
Step 2B: Preparation of F-3a
Charge di-/c/7-butyl dicarbonate, (1.85 wt, 1.4 eq) to vessel A followed by N,N-dimethylformamide, (2.6 wt, 2.7 vol) and stir at 20°C for 20 min. until dissolution achieved. Add A,A-diisopropylethylamine, (0.08 wt, 0.11 vol, 0.1 eq) to contents of vessel A at 20°C. Heat the contents of vessel A to 40°C.
[00153] Charge Gl, (1.00 wt) to vessel B followed by YW-di methyl form am ide, (5.2 wt, 5.5 vol) and adjust to 14°C.
[00154] Charge the Gl/DMF solution from vessel B to vessel A over 5 h at 40°C, at an approximately constant rate. Line rinse with Y,Y-di methyl form am ide, (0.4 wt, 0.4 vol), maintaining vessel A at 40°C. Stir the resulting reaction mixture at 40°C for 16 h.
[00155] Charge decolorizing charcoal activated, (0.20 wt). Adjust the mixture to 40°C and stir at 40°C for 60 to 90 min..
[00156] Clarify (filter) the reaction mixture into vessel B at 40°C. Charge N,N-dimethylformamide, (0.9 wt, 1.0 vol) via vessel A and filter at 40°C. Charge purified water, (3.5 vol) to the combined filtrates, over 60 min., maintaining the temperature at 40°C. As required, cool the mixture to 35°C over 30 to 60 min..
[00157] Filler F-3a, (0.02 wt) as seed material at 35°C. Stir at 34°C for 1.5 h then check for crystallization. Cool slurry to 30°C over 40 min.
[00158] Filler F-3a, (0.02 wt) as seed material at 30°C. Stir at 30°C for 1.5 h then check for crystallization.
[00159] Cool slurry at 20°C over 3.5 h at a targeted constant rate. Stir at 20°C for 3 hours. Charge purified water, (1.0 vol), maintaining the temperature at 20°C over 60 min..
Stir at 20°C for 3 hours.
[00160] Cool slurry to 2°C over 2.5 h. Stir at 2°C for 2.5 hours. Filter through 20 pm cloth and pull dry until no further filtrate passes. Wash the solid with pre-mixed Y,Y-di methyl form am ide / purified water, (2.0 vol, 1:2 v:v) at 2°C. Wash the solid with purified water, (2 x 3.0 vol) at 2°C. Dry under vacuum at 28°C until KF <0.2% w/w, and Y,Y-di methyl form am ide <0.4% w/w.
[00161] Yield of compound F-3a: 62-70%.
Example 3: Synthesis of Mavorixafor:
Scheme VI:
nce
Step 3A: Preparation of imine Q-1
[00162] To vessel A charge purified water, (8.7 vol, 8.7 wt) followed by potassium phosphate, (5.52 eq, 5.3 wt) portion-wise and cool to 15°C. Charge tetrahydrofuran, (4.3 vol, 3.8 wt) and n-heptane, (2.2 vol, 1.5 wt) to vessel A and cool the biphasic mixture to 0°C. Charge Fl, (1.00 eq, 1.00 wt) to the vessel in 2 portions maintaining 0°C.
[00163] Charge F-2d, (1.10 eq, 1.95 wt) to the vessel in 4 portions maintaining 0°C, ensuring portions are spaced by 10 min.. Stir the resulting biphasic mixture for 1.5 h at 0°C. Allow the layers to separate for 45 min. at 0°C before separating the layers. Retain the upper organic phase within vessel A.
[00164] Take the resulting solution of Ql into next reaction without isolation.
Step 3B: Preparation of amine P-1
[00165] To vessel B, charge tetrahydrofuran, (6.0 vol, 5.3 wt) and adjust to 15°C. Charge zinc chloride, (1.5 eq, 0.92 wt) to vessel B in 4 portions, maintaining 10 to 30°C. Adjust the reaction mixture in vessel B to 15°C. Stir the mixture at 15°C for 1 hour. Charge sodium borohydride,(1.0 eq, 0.17 wt) to vessel B in 2 portions maintaining 15°C. Cool the reaction mixture in vessel B to 15°C. Stir the mixture for 1 hour maintaining 15°C. Cool the reaction mixture in vessel B to -5°C.
[00166] Cool the retained organic solution of Ql in vessel A, from Step 3A, to -5°C.
[00167] Charge the organic solution in vessel A into vessel B over 1 to 2 h maintaining -5°C. Charge tetrahydrofuran, (1.0 vol, 0.9 wt) to vessel A as a line rinse and adjust to -5°C. Transfer the contents of vessel A to vessel B maintaining -5°C.
[00168] Stir the resulting reaction mixture in vessel B for 1.5 h maintaining -5°C.
[00169] Charge purified water, (4.5 vol, 4.5 wt) and glacial acetic acid, (1.0 eq, 0.27 wt, 0.26 vol) to the cleaned vessel A and cool to 0°C. Charge the contents of vessel B to vessel A over 1 to 2 h maintaining 0°C. Charge tetrahydrofuran, (1.0 vol, 0.9 wt) to vessel B as a vessel rinse, cool to 0°C and transfer to vessel A maintaining 0°C.
[00170] Warm the resulting mixture in vessel A to 30°C. Stir the resulting mixture in vessel A at 30°C for 1 h. Allow the layers to settle for 15 min. at 30°C before separating the layers. Retain the upper organic phase.
[00171] Cool the retained organic phase to 15°C. Charge to the vessel 25% w/w ammonia solution (3.0 vol) at 10 to 30°C. Cool the reaction mixture to 20°C. Charge to the vessel 25% w/w ammonium chloride solution (3.0 vol) at 20°C and stir for 1 h. Separate the layers for 15 min. at 20°C, retain the upper organic phase. Wash the retained organic phase with 10% w/w sodium chloride solution (3.0 vol) at 20°C for 10 min.. Allow the layers to settle for 10 min. at 20°C before separating and retaining the upper organic phase within the vessel.
[00172] Charge tert-butyl methyl ether, (0.5 vol, 0.4 wt) to the organic phase. Cool the mixture to 5°C. Adjust the pH of the reaction mixture to pH 5 with hydrochloric acid aqueous solution (expected ca. 9.0 vol) over 1 h at a targeted constant rate at 5°C. Stir the mixture at 5°C for 45 min.. Measure the pH of the aqueous phase to confirm the value is pH 5.
[00173] Charge sodium chloride, (2.1 wt) to the reaction mixture at 5°C and stir the mixture until everything is dissolved. Adjust the temperature of the reaction mixture to 20°C. Separate the layers at 20°C and retain the organic phase within the vessel. Tetrahydrofuran charge, (1.5 vol, 1.3 wt) maintaining 20°C.
[00174] Charge to the vessel 24% w/w sodium chloride solution (7.5 vol) at 20°C and stir for 10 min.. Separate the layers at 20°C and retain the organic phase in the vessel. This step is repeated additional one more time as described below.
[00175] Charge to the vessel 24% w/w sodium chloride solution (7.5 vol) at 20°C and stir for 10 min.. Separate the layers at 20°C and retain the organic phase in the vessel.
[00176] Heat the retained organic phase to 35°C and concentrate the mixture to 6.0 vol under reduced pressure maintaining 35°C.
[00177] Tetrahydrofuran charge, (15.0 vol, 13.2 wt) maintaining 35°C. Concentrate the mixture to 6.0 vol under reduced pressure maintaining 35°C.
[00178] Tetrahydrofuran charge, (15.0 vol, 13.2 wt) maintaining 35°C. Concentrate the mixture to 11.0 vol under reduced pressure maintaining 35°C.
[00179] Cool the mixture to -5°C. Load tert-butyl methyl ether, (10.0 vol, 7.4 wt) over 1 h maintaining -5°C. Stir the mixture at -5°C for 1.5 hours. Filter the solid on 1 to 2 pm filter cloth at -5°C. Wash the solid with pre-mixed tetrahydrofuran, (1.9 vol, 1.7 wt) and tert-butyl methyl ether, (3.1 vol, 1.9 wt) at -5°C as a displacement wash.
[00180] Wash the solid with tert-butyl methyl ether, (5.0 vol, 3.7 wt) at -5°C.
[00181] Dry the solid on the filter under a flow of nitrogen at 23°C.
[00182] Yield of compound P-1: 76-87%.
Step 3C: Preparation of compound 0-1
[00183] Charge purified water, (2.0 vol, 2.0 wt) followed by potassium phosphate, (3.3 eq, 1.54 wt), carefully portion-wise, maintaining <15°C, to vessel A. Charge toluene, (4.5 vol, 3.9 wt) to the vessel maintaining <15°C. As necessary, adjust the temperature to 10°C.
[00184] Charge P-1, (1.00 eq, 1.00 wt) to the vessel in two portions maintaining 10°C. Stir the reaction mixture at 10°C for 15 min..
[00185] Load F-3a, (1.1 eq, 0.64 wt) in 4 equal portions ensuring portions are spaced by 10 min. at 10°C.
[00186] Tetrabutylammonium iodide (TBAI) filler (0.20 eq, 0.16 wt). Heat the reaction mixture to 40°C. Stir the reaction mixture at 40°C for 30 h.
[00187] Charge pre-mixed 2-mercaptoacetic acid, (0.40 eq, 0.08 wt, 0.06 vol), and toluene, (0.5 vol, 0.4 wt) over 20 min. to Vessel A at 40°C. Line rinse with toluene, (0.5 vol, 0.4 wt) at 40°C. Adjust the temperature of the reaction mixture to 50°C. Stir the mixture at 50°C for 2.5 hours.
[00188] Adjust the temperature of Vessel A to 20°C. Charge purified water, (3.0 vol, 3.0 wt) maintaining 20°C. Stir the reaction mixture at 20°C for 15 min. and transfer to a new, clean HDPE container. Line/vessel rinse with toluene, (0.5 vol, 0.4 wt) at 20°C. Clarify (filter) the reaction mixture via a 1 pm filter at 20°C into clean Vessel A. Wash the vessel and the filter with toluene, (0.5 vol, 0.4 wt) at 20°C. Allow the layers to separate for 15 min. at 20°C, retaining the upper organic layer (organic layer 1).
[00189] Wash the aqueous layer with toluene, (2.5 vol, 2.2 wt) at 20°C for 15 min.. Allow the layers to separate for 15 min. at 20°C. Retain the upper organic layer (organic layer 2).
[00190] Combine the organic layer 1 and organic layer 2 and adjust the temperature to 20°C. Wash the combined organic layers with 10% w/w sodium chloride solution (5.0 vol) at 20°C for 15 min.. Allow the layers to settle for 15 min. at 20°C. Retain the upper organic layer.
[00191] Take the resulting solution of Ol into next reaction without isolation.
Step 3D: Preparation of compound Kl
[00192] Charge n-butanol, (2.4 wt, 3.0 vol) to vessel B and adjust to 5°C. Charge concentrated sulfuric acid, (1.1 wt, 5.0 eq, 0.6 vol) slowly to Vessel B maintaining <15°C. Line rinse with toluene, (0.4 wt, 0.5 vol) maintaining <15°C. Adjust the temperature of Vessel B to 25°C.
[00193] Heat the n-butanol/ sulfuric acid solution in Vessel B to 55°C. Charge the organic layer from Vessel A (from Step 3C) to the butanol/ sulfuric acid solution in Vessel B over 60 to 90 min. maintaining 55°C. Charge toluene, (1.3 wt, 1.5 vol) to Vessel A as a line rinse and transfer to Vessel B maintaining 55°C. Stir the contents of Vessel B at 55°C for 1.5 h.
[00194] Stir the mixture in Vessel B for 4.5 h at 55°C. Cool the contents of Vessel B to 20°C over 10 h. Filter the slurry over 1-2 pm filter cloth under nitrogen at 20°C. Wash the filter cake with pre-mixed toluene, (3.5 wt, 4.0 vol) and n-butanol, (1.0 vol, 0.8 wt) at 20°C. Wash the filter cake with toluene, (4.3 wt, 5.0 vol) at 20°C. Dry the solid at 30°C under vacuum.
[00195] Correct the output weight for assay. Expected 50-55% w/w.
[00196] Yield of compound K1: 89-92%.
Step 3E: Preparation of Mavorixafor Drug Substance
[00197] Charge Kl, (1.00 eq, 1.00 wt, corrected for HPLC assay) in vessel A followed by nitrogen-purged purified water, (2.0 wt, 2.0 vol) and if necessary, adjust the temperature to 20°C. Charge nitrogen-purged toluene, (12.0 wt, 14.0 vol) to the solution maintaining 20°C. Charge nitrogen-purged n-butanol, (0.8 wt, 1.0 vol) to the solution maintaining 20°C. Heat the biphasic mixture to 30°C. Charge nitrogen-purged 3.0 M aqueous sodium hydroxide solution (6.2 eq, 5.9 vol) maintaining 30°C. Check the pH (expected 12 to 13). Adjust the pH of the aqueous layer to pH 10.0 with nitrogen-purged 0.3 M sulfuric acid solution (expected up to 2.5 vol) maintaining 30°C. Stir the mixture at 30°C for 45 min..
[00198] Measure the pH to confirm the value is pH 10.0.
[00199] Allow the layers to settle at 30°C for 30 min. and separate the layers retaining the organic phase in the vessel, and discharge the aqueous layer into a separate container (container C).
[00200] Charge pre-mixed toluene, (4.1 wt, 4.7 vol) and n-butanol, (0.24 wt, 0.3 vol) to a separate vessel; heat the contents to 30°C and charge the aqueous layer from container C. As required adjust the temperature to 30°C and stir for 5 to 10 min. at 30°C. Allow the phases to separate for 10 to 15 min. at 30°C. Discharge the aqueous phase to waste and combine the organic phase to the organic phase in vessel A.
[00201] Charge nitrogen-purged purified water, (2.0 wt, 2.0 vol) to the organic layer maintaining the temperature at 30°C and stir for 5 to 10 min. at 30°C. Allow the phases to separate for 10 to 15 min. at 30°C. Discharge the aqueous phase to waste retaining the organic phase in the vessel. Heat the retained organic solution to 40°C. Concentrate the resulting organic phase to 7.0 vol by vacuum distillation at 40°C.
[00202] Charge nitrogen -purged toluene, (13.0 wt, 15.0 vol) to the mixture and concentrate the solution 7.0 vol by vacuum distillation at 40°C. This step is repeated additional one time as described below.
[00203] Charge nitrogen -purged toluene, (13.0 wt, 15.0 vol) to the mixture and concentrate the solution 7.0 vol by vacuum distillation at 40°C.
[00204] Charge nitrogen-purged toluene, (7.0 wt, 8.0 vol) to the mixture at 40°C, heat to 55°C and clarify the hot reaction mixture under nitrogen via a 1 pm filter.
[00205] Charge clarified nitrogen-purged toluene, (1.7 wt, 2.0 vol) to the mixture as a line and vessel rinse at 40°C. Concentrate the solution to 7.0 vol by vacuum distillation at 40°C. At the end of the distillation the product is expected to have precipitated. Heat the mixture to 63°C.
[00206] Adjust the temperature to 60.5°C. This batch will be referred to as the main batch.
[00207] Load seed material, (0.02 wt) to a new clean container. Charge clarified nitrogen-purged toluene, (0.09 wt, 0.10 vol) to this seed material and gently shake.
[00208] Seed the main batch with the slurry maintaining the temperature at 60.5 ± 2°C. Stir the reaction at the 60.5± 2°C for 1 hour.
[00209] Cool to 40°C for 2.5 h. Stir the reaction at 40°C for 1 hour.
[00210] Cool to 30°C over 2 h.. Stir the reaction at 30°C for 1 h.
[00211] Cool to 25°C 50 min. Stir the reaction at 25°C over 2 hours.
[00212] Cool to 2°C over 4 h. Stir the mixture for 12 hours at 2°C.
[00213] Filter the mixture at 2°C over 1 to 2 pm cloth. Wash the filter cake with clarified nitrogen-purged toluene, (2.0 vol, 1.7 wt) at 2°C. Dry the filter cake under vacuum and a flow of nitrogen for 1.5 h.
[00214] Dry the solid at 40°C under vacuum and a flow of nitrogen until drying specification is achieved.
[00215] Yield of the final compound mavorixafor: 72%.
[00216] When toluene is used as the recrystallization solvent, optionally with a dissolution aid such butanol or methanol, for maxorixa for recrystallization, advantages were found compared to using dichloromethane and isopropyl acetate. We have found that these solvents do not react with the API, and accordingly we believe that this change has caused the significant reduction of impurities A (imine), B (N-formyl) and C (acetamide) that we have observed.
[00217] In some embodiments, the mavorixafor composition included 7000, 6000, 5000, 4500, 4450, 4000, 3500, 3000, 2500, 2000, 1750, 1700, 1650, 1600, 1550, 1500, 1450, 1400, 1400, 1400, 1400 gold 50 ppm of toluene or less. In some embodiments, the mavorixafor composition comprises a detectable amount of toluene. In some embodiments, the mavorixafor composition comprises from a detectable amount of toluene to 1350 ppm of toluene.
////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Mavorixafor (AMD-070) is a potent, selective and orally available CXCR4 antagonist, with an IC50 value of 13 nM against CXCR4125I-SDF binding, and also inhibits the replication of T-tropic HIV-1 (NL4.3 strain) in MT-4 cells and PBMCs with an IC50 of 1 and 9 nM, respectively.
125I-SDF-CXCR413 nM (IC50)HIV-1 (NL4.3 strain)1 nM (IC50, in MT-4 cells)HIV-1 (NL4.3 strain)9 nM (IC50, in PBMCs)HIV-1 (NL4.3 strain)3 nM (IC90, in MT-4 cells)HIV-1 (NL4.3 strain)26 nM (IC90, in PBMCs)
In Vitro
Mavorixafor (AMD-070) is a potent and orally available CXCR4 antagonist, with an IC50 value of 13 nM against CXCR4 125I-SDF binding, and also inhibits the replication of T-tropic HIV-1 (NL4.3 strain) in MT-4 cells and PBMCs with an IC50 of 1 and 9 nM, respectively. Mavorixafor (AMD-070) shows no effect on other chemokine receptors (CCR1, CCR2b, CCR4, CCR5, CXCR1, and CXCR2)[1]. Mavorixafor (AMD-070) (6.6 µM) significantly suppresses the anchorage-dependent growth, the migration and matrigel invasion of the B88-SDF-1 cells[2].MCE has not independently confirmed the accuracy of these methods. They are for reference only.
In Vivo
Mavorixafor (AMD-070) (2 mg/kg, p.o.) significantly reduces the number of metastatic lung nodules in mice, and lowers the expression of human Alu DNA in mice, without body weight loss[2].MCE has not independently confirmed the accuracy of these methods. They are for reference only.
Arimoclomol maleate is in a phase III clinical trials by Orphazyme for the treatment of Niemann-Pick disease type C (NP-C). It is also in phase II clinical studies for the treatment of amyotrophic lateral sclerosis (ALS).
Arimoclomol (INN; originally codenamed BRX-345, which is a citrate salt formulation of BRX-220) is an experimental drug developed by CytRx Corporation, a biopharmaceutical company based in Los Angeles, California. In 2011 the worldwide rights to arimoclomol were bought by Danish biotech company Orphazyme ApS.[1] The European Medicines Agency (EMA) and U.S. Food & Drug Administration (FDA) granted orphan drug designation to arimoclomol as a potential treatment for Niemann-Pick type C in 2014 and 2015 respectively.[2][3]
Fig. 1 Structures of (±)-bimoclomol (1) and (R)-(+)-arimoclomol (2).
The present disclosure provides an optimized four-step process for preparing an ultra-pure composition comprising arimoclomol citrate, i.e. N-{[(2R)-2-hydroxy-3-piperidin-l-ylpropyl]oxy}pyridine-3-carboximidoyl chloride 1-oxide citrate. The optimized process comprises a plurality of optimized sub-steps, each contributing to an overall improved process, providing the ultra-pure composition comprising arimoclomol citrate. The ultra-pure composition comprising arimoclomol citrate meets the medicines agencies’ high regulatory requirements. An overview of the four-steps process is outlined below:
Step 1: Overview of process for preparing ORZY-01
Step 2: Overview of process for preparing ORZY-03
Step 4: Overview of process for preparing BRX-345 (ORZY-05)
The previously reported two-step synthesis of ORZY-01 as shown below includes a 2 hour reflux in step 1A, followed by purification of intermediate compound (V) to increase the batch quality.
(R,Z)-3-(N’-(2-hydroxy-3-(piperidin-1-yl)propoxy)carboximidoyl chloride)pyridine-1-oxide1 – (R)-(+)-Arimoclomol – 2 A solution of (R,Z)-3-(N’-(2-hydroxy-3-(piperidin-1-yl)propoxy)carbamimidoyl)pyridine-1-oxide 12 (205 mg, 0.70 mmol) in conc. hydrochloric acid (1.1 mL, 13.9 mmol) and water (3 mL) was cooled to -5 °C for 15 minutes. Sodium nitrite (63 mg, 0.91 mmol) in water (0.5 mL) was then added dropwise to the reaction mixture and the reaction was stirred at -5 °C for 2.5 hours. The reaction mixture was made alkaline with NaOH (7 M, 3 mL). An additional 10 mL of water was added followed by DCM (30 mL) containing EtOAc (5 mL) and the organics were dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by FCC on Biotage Isolera using Biotage SNAP 10 g Si cartridge eluting with gradient elution 0-30% MeOH:DCM both containing 0.1% Et3N to afford the title compound (160 mg, 73% yield) as a colourless semi-solid. Analytical data was consistent with literature values. See ESI section SFC traces for specific enantiomeric ratios of 2 synthesised under the various methodologies quoted in the text. Optical rotation was not determined as it was determined in the ultimate product of this 2·citrate and comparative run times on SFC. 1H NMR (600 MHz, CDCl3) δ: 8.63 (t, J = 1.4 Hz, 1H), 8.16 (ddd, J = 6.4, 1.6, 0.9 Hz, 1H), 7.66 – 7.62 (m, 1H), 7.25 (dd, J = 8.0, 6.6 Hz, 1H), 4.26 (qd, J = 11.3, 5.2 Hz, 2H), 4.07 (dd, J = 9.2, 4.7 Hz, 1H), 2.62 (s, 2H), 2.47 – 2.31 (m, 4H), 1.65 – 1.51 (m, 4H), 1.42 (s, 2H); 13C NMR (151 MHz, CDCl3) δ: 140.3, 137.7, 133.1, 132.5, 125.7, 123.9, 78.7, 64.9, 60.9, 54.8, 25.8, 24.0.
(R)-(+)- Arimoclomol citrate – 2·citrate (R,Z)-3-(N’-(2-hydroxy-3-(piperidin-1-yl)propoxy)carboximidoyl chloride)pyridine-1-oxide (159 mg, 0.51 mmol) was dissolved in acetone (3 mL) and citric acid (97 mg, 0.51 mmol) was added. The reaction mixture was left to stir at room temperature for 18 hours. After this time the mixture was sonicated and the precipitate was filtered, rinsed with cold acetone (1 mL) and dried under vacuum to afford the title compound (165 mg, 64% yield) as a white amorphous solid. Analytical data was consistent with literature values. m.p. 161-162 °C, Acetone (lit. 163-165 °C, EtOH); [α]D 20 +8.0 (c=1, H2O); IR νmax (neat): 3423, 3228, 2949, 2868, 1722, 1589, 1483, 1433, 1307, 1128, 972, 829 cm-1; 1H NMR (600 MHz, d6-DMSO) δ: 8.54 (t, J = 1.5 Hz, 1H), 8.39 – 8.35 (m, 1H), 7.72 – 7.68 (m, 1H), 7.55 (dd, J = 8.0, 6.5 Hz, 1H), 4.28 (ddd, J = 17.6, 13.3, 7.4 Hz, 3H), 3.35 (br. s, 2H), 3.13 – 2.74 (m, 6H), 2.59 (d, J = 15.2 Hz, 2H), 2.56 – 2.51 (m, 2H), 1.77 – 1.61 (m, 4H), 1.48 (s, 2H); 13C NMR (151 MHz, d6-DMSO) δ: 176.6, 171.3, 140.5, 136.4, 132.7, 131.5, 126.8, 123.3, 77.8, 71.4, 63.8, 58.7, 53.1, 44.0, 30.7, 23.0, 21.9; HRMS (m/z TOF MS ES+) for C14H20ClN3O3 [M+H]+ calc. 314.1271, observed 314.1263; SFC er purity R:S >99:1
Procedure for the conversion of (R)-(+)-Bimoclomol 1 into (R)-(+)-Arimoclomol 2 To a solution of (R)-(+)-bimoclomol (61 mg, 0.21 mmol) in acetone (2 mL) was added benzenesulfonic acid (33 mg, 0.21 mmol). The reaction mixture was stirred at room temperature for 1.5 hours. The reaction mixture was concentrated in vacuo. Separately to a suspension of hydrogen peroxide-urea adduct (39 mg, 0.41 mmol) in acetonitrile (6 mL) at -5°C (ice-salt bath) was added trifluoroacetic anhydride (58 μL, 0.41 mmol) dropwise. A suspension of (R)-(+)-bimoclomol, 1, benzenesulfonic acid salt, as made above, in acetonitrile (3 mL) was then added dropwise to this solution. The reaction mixture was stirred for 18 hours, whilst slowly warming to room temperature. Aqueous Na2S2O5 solution (0.5 M, 1 mL) was added and the reaction mixture stirred for 1 hour. The reaction mixture was made alkaline with NaOH (7 M) and extracted with DCM (2 x 30 mL). The combined organics were washed with brine, dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by FCC on a Biotage Isolera using Biotage SNAP 10g Si cartridge eluting with gradient elution 0-35% MeOH in DCM to afford the title compound (35 mg, 55% yield) as a colourless semi-solid. Analytical data of the products was consistent with literature and/or previous samples synthesised above.
Arimoclomol is believed to function by stimulating a normal cellular protein repair pathway through the activation of molecular chaperones. Since damaged proteins, called aggregates, are thought to play a role in many diseases, CytRx believes that arimoclomol could treat a broad range of diseases.
Arimoclomol has been shown to extend life in an animal model of ALS[11] and was well tolerated in healthy human volunteers in a Phase I study. CytRx is currently conducting a Phase II clinical trial.[12]
Arimoclomol also has been shown to be an effective treatment in an animal model of Spinal Bulbar Muscular Atrophy (SBMA, also known as Kennedy’s Disease).[13]
Arimoclomol was discovered by Hungarian researchers, as a drug candidate to treat insulin resistance[14][15] and diabetic complications such as retinopathy, neuropathy and nephropathy. Later, the compound, along with other small molecules, was screened for further development by Hungarian firm Biorex, which was sold to CytRx Corporation, who developed it toward a different direction from 2003.
^ Kieran D, Kalmar B, Dick JR, Riddoch-Contreras J, Burnstock G, Greensmith L (April 2004). “Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice”. Nat. Med. 10 (4): 402–5. doi:10.1038/nm1021. PMID15034571. S2CID2311751.
^ Kalmar B, Greensmith L, Malcangio M, McMahon SB, Csermely P, Burnstock G (December 2003). “The effect of treatment with BRX-220, a co-inducer of heat shock proteins, on sensory fibers of the rat following peripheral nerve injury”. Exp. Neurol. 184 (2): 636–47. doi:10.1016/S0014-4886(03)00343-1. PMID14769355. S2CID5316222.
^ Rakonczay Z, Iványi B, Varga I, et al. (June 2002). “Nontoxic heat shock protein coinducer BRX-220 protects against acute pancreatitis in rats”. Free Radic. Biol. Med. 32 (12): 1283–92. doi:10.1016/S0891-5849(02)00833-X. PMID12057766.
^ Kalmar B, Burnstock G, Vrbová G, Urbanics R, Csermely P, Greensmith L (July 2002). “Upregulation of heat shock proteins rescues motoneurones from axotomy-induced cell death in neonatal rats”. Exp. Neurol. 176 (1): 87–97. doi:10.1006/exnr.2002.7945. PMID12093085. S2CID16071543.

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE

 Subscribe in a reader
Subscribe in a reader












 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain.

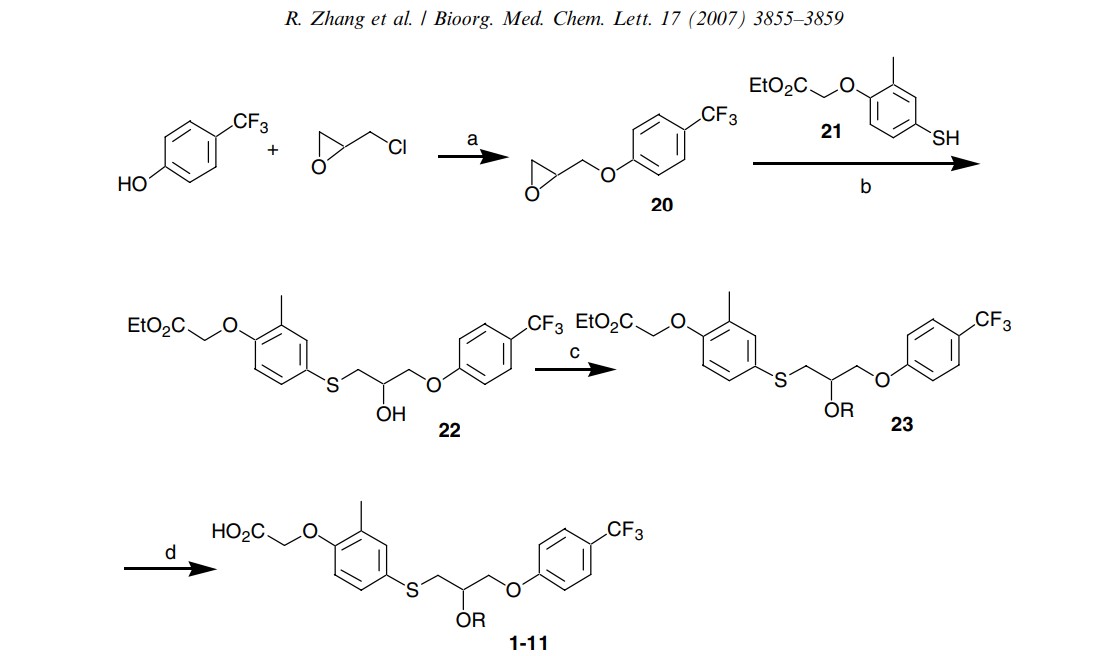
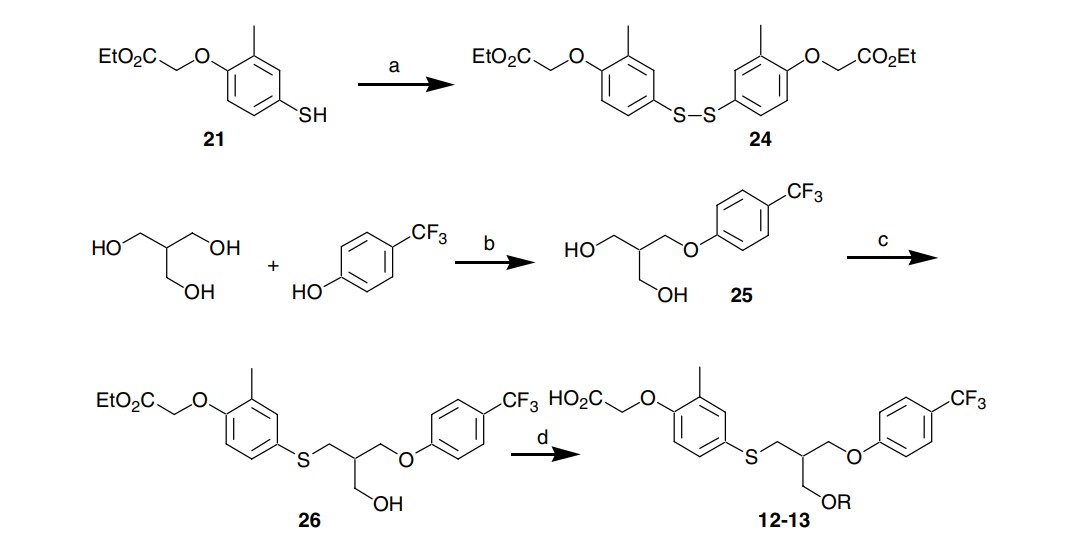


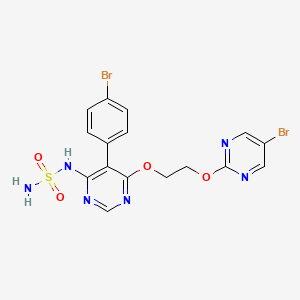



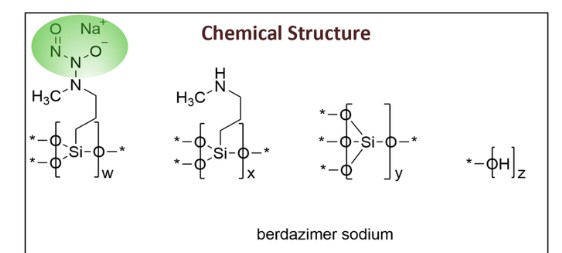


 References
References
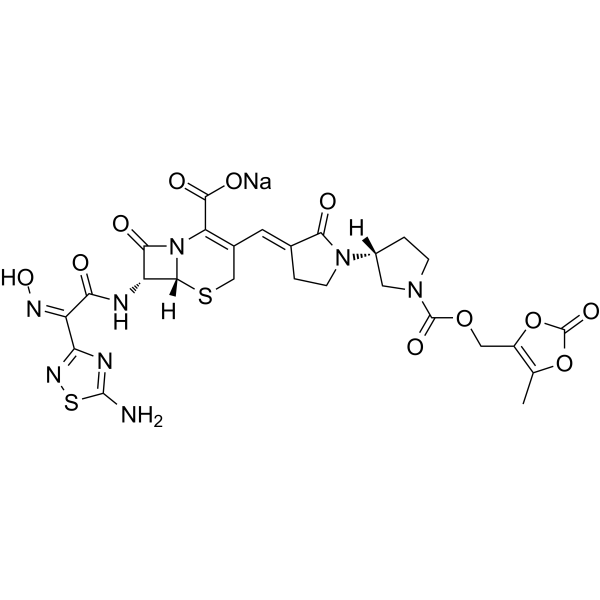
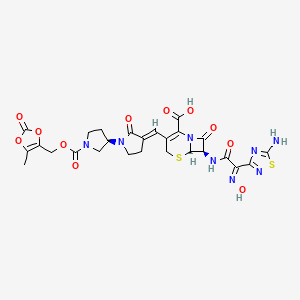

(1)_(30204271666).jpg)
(1)_(29608654394).jpg)

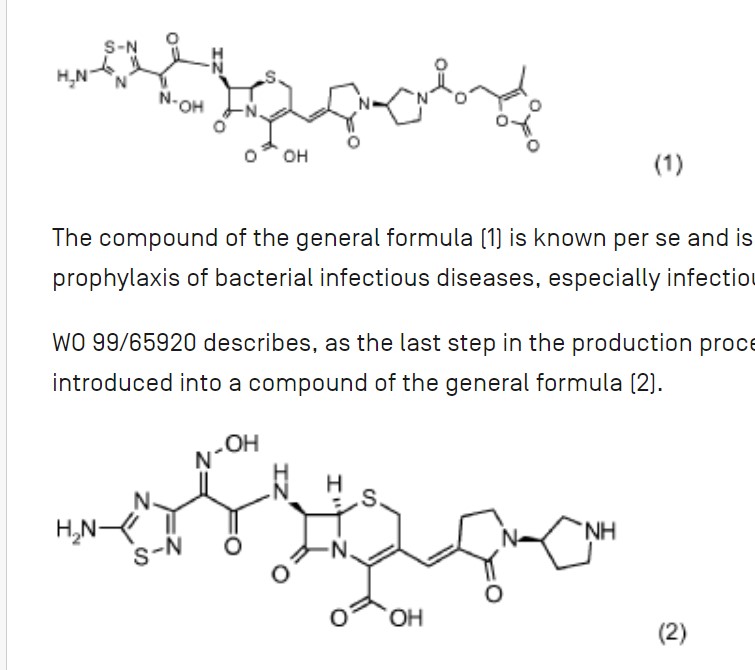






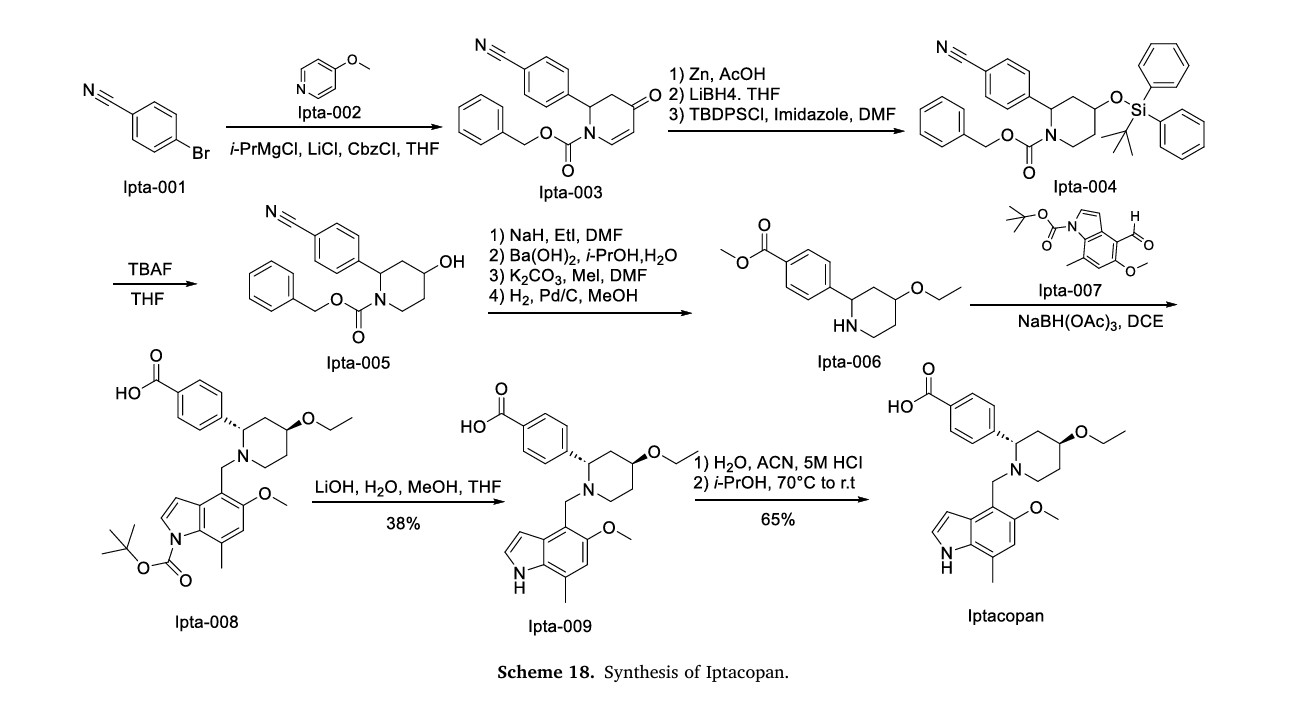










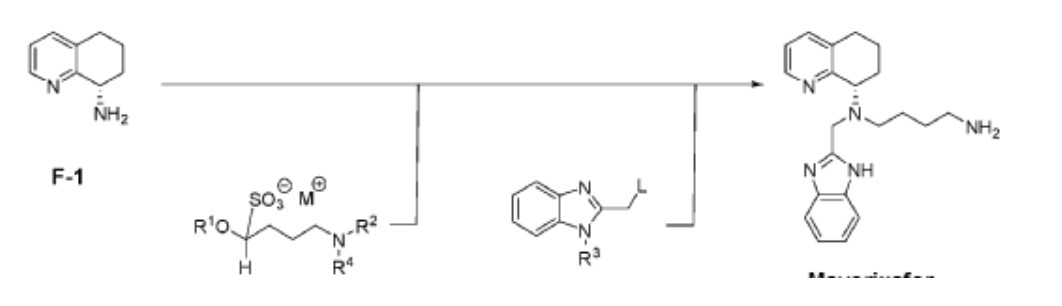

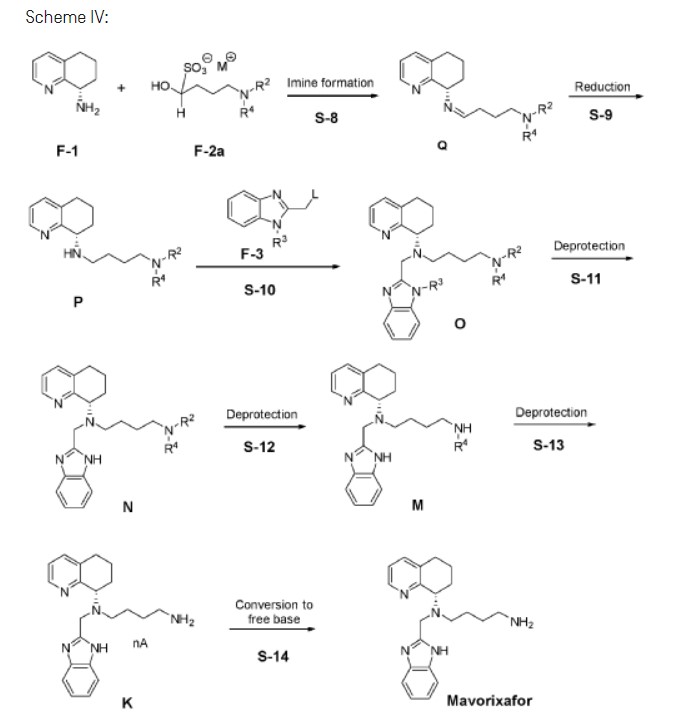

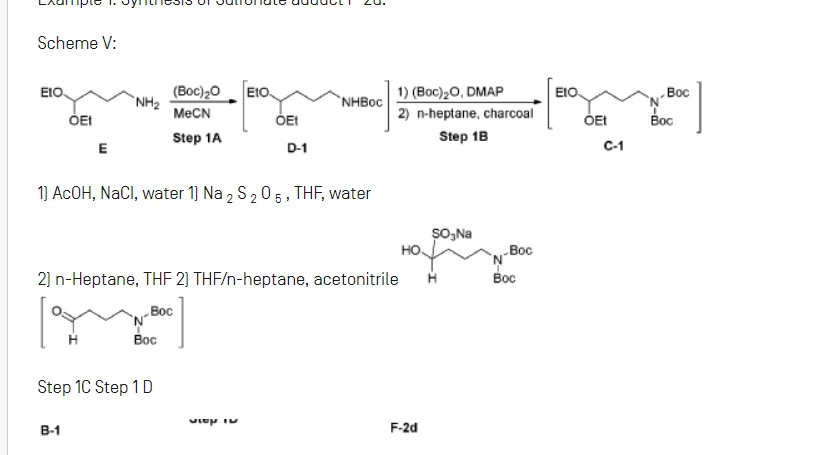

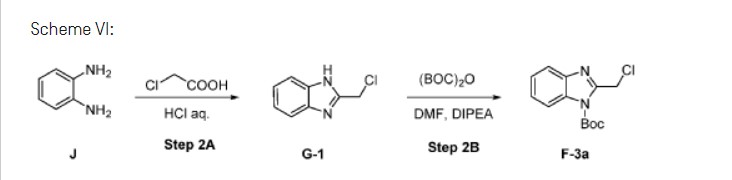

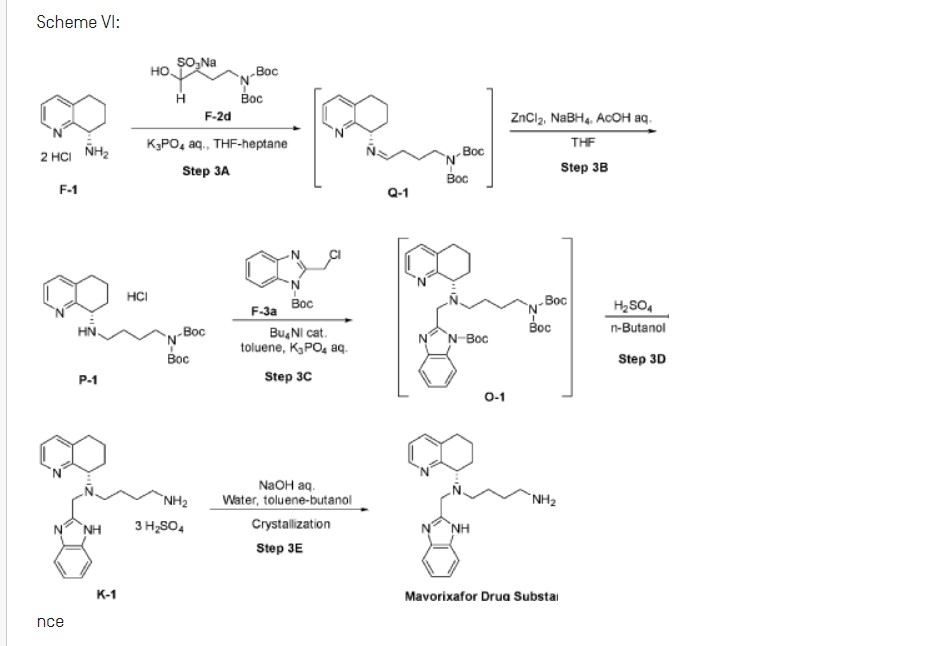








![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
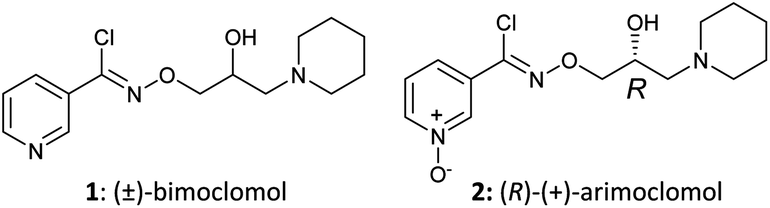{kind=link}
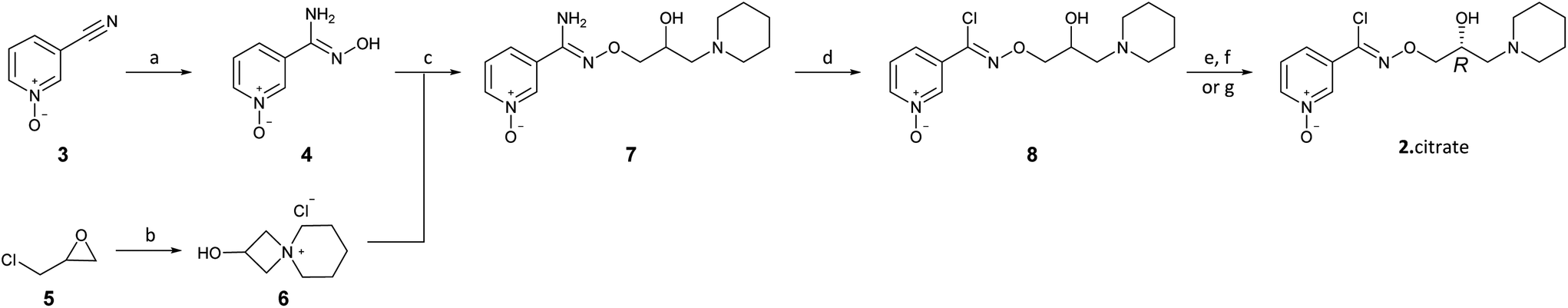{kind=link}
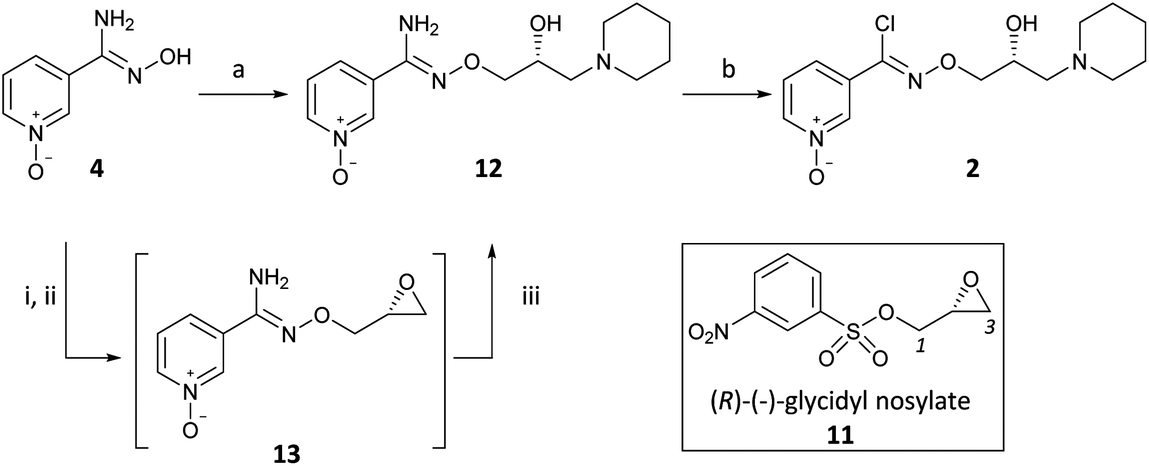{kind=link}
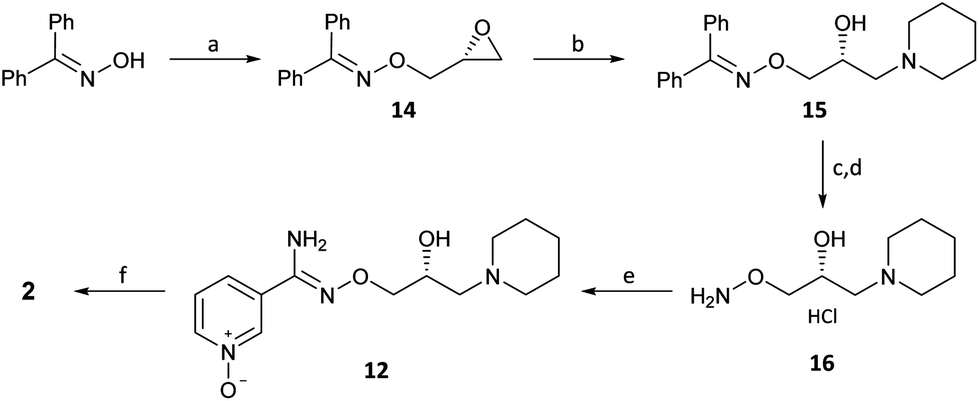{kind=link}
{kind=link}