
Home » Posts tagged 'approvals 2024' (Page 3)
Tag Archives: approvals 2024
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Sulopenem



Sulopenem
- 120788-07-0
- CP-70429
- 349.5 g/mol, C12H15NO5S3
- XX514BJ1XW
- PF-03709270
- PF03709270
(5R,6S)-6-[(1R)-1-hydroxyethyl]-7-oxo-3-[(1R,3S)-1-oxothiolan-3-yl]sulfanyl-4-thia-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid
- (5R,6S)-6-((1R)-1-HYDROXYETHYL)-7-OXO-3-(((1R,3S)-1-OXOTETRAHYDRO-1H-1.LAMBA.(SUP 4)-THIOPHEN-3-YL)SULFANYL)-4-THIA-1-AZABICYCLO(3.2.0)HEPT-2-ENE-2-CARBOXYLIC ACID
- (5R,6S)-6-((1R)-1-Hydroxyethyl)-7-oxo-3-(((3S)-tetrahydro-3-thienyl)thio)-4-thia-1-azabicyclo(3.2.0)hept-2-ene-2-carboxylic acid, (R)-S-oxide
- 4-THIA-1-AZABICYCLO(3.2.0)HEPT-2-ENE-2-CARBOXYLIC ACID, 6-((1R)-1-HYDROXYETHYL)-7-OXO-3-(((1R,3S)-TETRAHYDRO-1-OXIDO-3-THIENYL)THIO)-, (5R,6S)-
- 4-THIA-1-AZABICYCLO(3.2.0)HEPT-2-ENE-2-CARBOXYLIC ACID, 6-(1-HYDROXYETHYL)-7-OXO-3-((TETRAHYDRO-3-THIENYL)THIO)-, S-OXIDE, (5R-(3(1R*,3S*),5.ALPHA.,6.ALPHA.(R*)))-
FDA APPROVED sulopenem etzadroxil, probenecid, 10/25/2024, To treat uncomplicated urinary tract infections (uUTI)
Drug Trial Snapshot
Sulopenem (CP-70,429) is a thiopenem antibiotic derivative from the penem family, which unlike most related drugs is orally active. It was developed in Japan in the 1990s, and has been approved to treat uncomplicated urinary tract infections in combination with probenecid (Brand name: Orlynvah). It has reached Phase III clinical trials on several occasions and continues to be the subject of ongoing research into potential applications, especially in the treatment of multiple drug resistant urinary tract infections.[1][2][3][4][5]
In October 2024, the US Food and Drug Administration approved sulopenem etzadroxil with probenecid combination for the treatment of urinary tract infections caused by Escherichia coli, Klebsiella pneumoniae, or Proteus mirabilis in adult women with limited alternative oral antibiotic options. The combination was developed by Iterum Therapeutics under the trade name ORLYNVAH™.[6]


JP 1995278137; US 5013729; WO 8808845, J Org Chem 1992,57(16),4352-61
1) The reaction of L-aspartic acid (I) with NaNO2, NaBr and H2SO4 gives 2(S)-bromosuccinic acid (II), which is reduced with methyl sulfide borane complex in THF, yielding 2(S)-bromobutane-1,4-diol (III). The cyclization of (III) with Cs2CO3 in methylene chloride affords (R)-(2-hydroxyethyl)oxirane (IV), which is acylated with methanesulfonyl chloride to the corresponding mesylate (V). The cyclization of (V) with Na2S in acetonitrile/water gives 3(R)-hydroxythiolane (VI), which is acylated with p-toluenesulfonyl chloride, affording the corresponding tosylate (VII). The controlled oxidation of (VII) with potassium peroxymonosulfate (oxone) gives 3(R)-(p-toluenesulfonyloxy)thiolane-1(R)-oxide (VIII), which by reaction with potassium thioacetate in acetone is converted to 3(S)-(acetylthio)thiolane 1(R)-oxide (IX). The reaction of (IX) with NaOEt and CS2 in ethanol yields the trithiocarbonate (X), which is condensed with the chloroazetidinone (XI), yielding the trithiocarbonate ester (XII). The condensation of (XII) with 2-chloroallyloxalyl fluoride (XIII) by means of diisopropylethylamine in methylene chloride affords the substituted oxalamic ester (XIV), which is cyclized by means of triethyl phosphite in refluxing chloroform to the fully protected penem derivative (XV). The reaction of (XV) with tetrabutylammonium fluoride (TBAF) in THF eliminates the protecting tert-butyldimethylsilyl group, yielding the chloroallyl ester (XVI), which is treated with triphenylphosphine and sodium 2-ethylhexanoate in dichloromethane to obtain the corresponding sodium salt (XVII). Finally, this compound is treated with HCl in cool water.

US 4921972
2) The intermediate 3(R)-(p-toluenesulfonyloxy)thiolane (VII) can be obtained by two other synthetic pathways: a) The racemic 2-hydroxy-4-(methylsulfanyl)butyric acid ethyl ester (XVIII) is submitted to optical resolution with Pseudomonas fluorescens lipase in toluene/water, yielding the corresponding 2(R)-hydroxy ester (XIX), which is reduced with NaBH4 in THF/water to afford 4-(methylsulfanyl)butane-1,2(R)-diol (XX). The acylation of (XX) with p-toluenesulfonyl chloride and pyridine yields the ditosylate (XXI), which is cyclized in refluxing benzene to give 1(R)-methyl-3(R)-(p-toluenesulfonyloxy)thiolanium p-toluenesulfonate (XXII). Finally, this compound is treated with trifluoroacetic acid in pyridine to afford the thiolane (VII), already described. b) The reduction of 4-chloro-3(R)-hydroxybutyric acid methyl ester (XXIII) with lithium borohydride in THF gives 4-chlorobutane-1,3(R)-diol (XXIV), which is tosylated as before, yielding the bis(tosyloxy) derivative (XXV). Finally, this compound is cyclized with Na2S in hot acetonitrile/water to afford the thiolane (VII), already described.

https://pubsapp.acs.org/cen/coverstory/88/8836cover.html
References
- ^ Minamimura M, Taniyama Y, Inoue E, Mitsuhashi S (July 1993). “In vitro antibacterial activity and beta-lactamase stability of CP-70,429 a new penem antibiotic”. Antimicrobial Agents and Chemotherapy. 37 (7): 1547–1551. doi:10.1128/AAC.37.7.1547. PMC 188011. PMID 8363389.
- ^ Hamilton-Miller JM (November 2003). “Chemical and microbiologic aspects of penems, a distinct class of beta-lactams: focus on faropenem”. Pharmacotherapy. 23 (11): 1497–1507. doi:10.1592/phco.23.14.1497.31937. PMID 14620395. S2CID 43705118.
- ^ Ednie LM, Appelbaum PC (May 2009). “Antianaerobic activity of sulopenem compared to six other agents”. Antimicrobial Agents and Chemotherapy. 53 (5): 2163–2170. doi:10.1128/AAC.01557-08. PMC 2681565. PMID 19223615.
- ^ Bader MS, Loeb M, Leto D, Brooks AA (April 2020). “Treatment of urinary tract infections in the era of antimicrobial resistance and new antimicrobial agents”. Postgraduate Medicine. 132 (3): 234–250. doi:10.1080/00325481.2019.1680052. PMID 31608743. S2CID 204545734.
- ^ Veeraraghavan B, Bakthavatchalam YD, Sahni RD (December 2021). “Oral Antibiotics in Clinical Development for Community-Acquired Urinary Tract Infections”. Infectious Diseases and Therapy. 10 (4): 1815–1835. doi:10.1007/s40121-021-00509-4. PMC 8572892. PMID 34357517.
- ^ “Iterum Therapeutics Receives U.S. FDA Approval of ORLYNVAH™ (Oral Sulopenem) for the Treatment of Uncomplicated Urinary Tract Infections”. Iterum Therapeutics plc. 2024-10-25. Retrieved 2024-10-25.
| Clinical data | |
|---|---|
| ATC code | None |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 120788-07-0 |
| PubChem CID | 9950244 |
| DrugBank | DB15284 |
| ChemSpider | 8125855 |
| UNII | XX514BJ1XW |
| KEGG | D05969 |
| CompTox Dashboard (EPA) | DTXSID20869656 |
| Chemical and physical data | |
| Formula | C12H15NO5S3 |
| Molar mass | 349.43 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
FDA Approved Drug Products: Orlynvah (sulopenem etzadroxil and probenecid) tablets for oral use (October 2024) [Link]
FDA News Release: FDA approves new treatment for uncomplicated urinary tract infections in adult women who have limited or no alternative oral antibiotic treatment options [Link]
//////Sulopenem, Orlynvah, FDA 2024, APPROVALS 2024, CP-70,429, 120788-07-0, CP-70429, XX514BJ1XW, PF-03709270, PF03709270
#Sulopenem, #Orlynvah, #FDA 2024, #APPROVALS 2024, #CP-70,429, #120788-07-0, #CP-70429, #XX514BJ1XW, #PF-03709270, #PF03709270
Revumenib
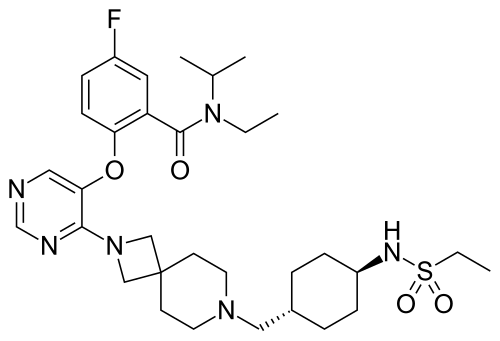
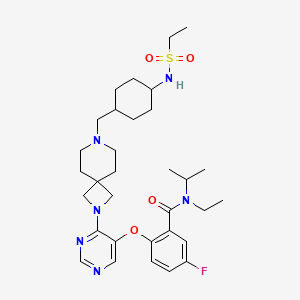
Revumenib
- SNDX-5613
- 2169919-21-3
- LZ0M43NNF2
- SNDX5613
- C32H47FN6O4S, 630.82
- SNDX-50613 free base
- SNDX-5613 free base
- SNDX50613 free base
- SNDX5613 free base
FDA APPROVED 11/15/2024, Revuforj, To treat relapsed or refractory acute leukemia
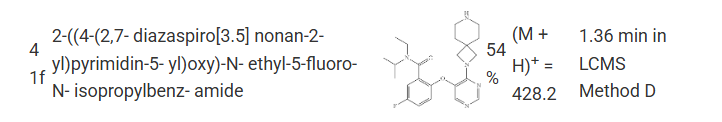
N-ethyl-2-[4-[7-[[4-(ethylsulfonylamino)cyclohexyl]methyl]-2,7-diazaspiro[3.5]nonan-2-yl]pyrimidin-5-yl]oxy-5-fluoro-N-propan-2-ylbenzamide
- N-Ethyl-2-[4-[7-[[4-(ethylsulfonylamino)cyclohexyl]methyl]-2,7-diazaspiro[3.5]nonan-2-yl]pyrimidin-5-yl]oxy-5-fluoro-N-propan-2-ylbenzamide
- 2-({4-[7-({(1r,4r)-4-[(ethanesulfonyl)amino]cyclohexyl}methyl)-2,7-diazaspiro[3.5]nonan-2-yl]pyrimidin-5-yl}oxy)-N-ethyl-5-fluoro-N-(propan-2-yl)benzamide
- N-ethyl-2-((4-(7-(((1r,4r)-4-(ethylsulfonamido)cyclohexyl)methyl)-2,7-diazaspiro[3.5]nonan-2-yl)pyrimidin-5-yl)oxy)-5-fluoro-N-isopropylbenzamide
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Revumenib citrate | YL4RYN734D | 2761046-45-9 | UBXFWTFPYATLBT-SGBGZXBGSA-N |
| Revumenib sesquifumarate | 75HI05N8HS | 2169919-22-4 | AXNUWYROYVRYIM-OQIJCFCCSA-N |
Revumenib, sold under the brand name Revuforj, is an anti-cancer medication used for the treatment of acute leukemias harboring lysine methyltransferase 2A gene (KMT2A) rearrangements.[1] It is designed to disrupt the interaction between menin and KMT2A (also known as MLL), which plays a role in the pathogenesis of these leukemias.[2] It is taken by mouth.[1]
The most common adverse reactions include hemorrhage, nausea, increased phosphate, musculoskeletal pain, infection, increased aspartate aminotransferase, febrile neutropenia, increased alanine aminotransferase, increased intact parathyroid hormone, bacterial infection, diarrhea, differentiation syndrome, electrocardiogram QT prolonged, decreased phosphate, increased triglycerides, decreased potassium, decreased appetite, constipation, edema, viral infection, fatigue, and increased alkaline phosphatase.[3]
Revumenib was approved for medical use in the United States in November 2024.[1][3] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication.[4]\
PATENT
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US11479557 | No | 2022-10-25 | 2037-06-08 |  |
| US10683302 | No | 2020-06-16 | 2037-06-08 | |
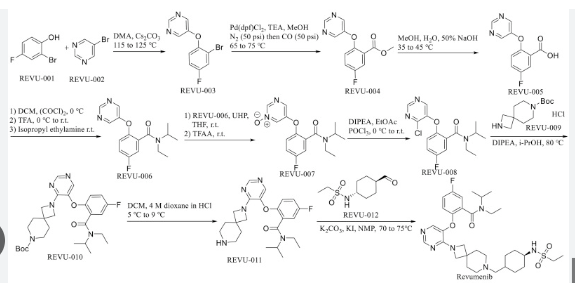
PATENT US11479557
https://patents.google.com/patent/US11479557B2/en
Intermediate 50((1r,4r)-4-(Ethylsulfonamido)cyclohexyl)methyl 4-methylbenzenesulfonate

Step 1: Methyl (1r,4r)-4-(ethylsulfonamido)cyclohexane-1-carboxylate

A solution of methyl (1r,4r)-4-aminocyclohexane-1-carboxylate hydrochloride (120 g, 0.62 mol) and Et3N (346 mL, 2.48 mol) in anhydrous DCM (2.5 L) was stirred at RT for 30 min. Ethanesulfonyl chloride (80.6 g, 0.63 mol) was added dropwise over 30 min to the reaction mixture at 0-5° C. After addition, the mixture was stirred at 0° C. for 3 h. The mixture was quenched with water (250 mL) at 0° C. After partition, the organic layer was washed with H2O (600 mL, 5 volumes) and 1 N HCl (2×600 mL, 2×5 volumes), H2O (600 mL, 5 volumes) and brine (600 mL, 5 volumes), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to afford crude methyl (1r,4r)-4-(ethylsulfonamido)cyclohexane-1-carboxylate (117.6 g, 76%) as alight yellow solid, which was used for the next step without further purification. 1H NMR (CDCl3 400 MHz): δ 4.36 (d, J=8.0 Hz, 1H), 3.67 (s, 3H), 3.29-3.22 (m, 1H), 3.04 (q, J=7.6 Hz, 2H), 2.25-2.21 (m, 1H), 2.15-2.09 (m, 2H), 2.08-2.01 (m, 2H), 1.58-1.51 (m, 2H), 1.39-1.25 (m, 5H).Step 2. N-((1r,4r)-4-(hydroxymethyl)cyclohexyl)ethanesulfonamide

To a solution of crude methyl (1r,4r)-4-(ethylsulfonamido)cyclohexane-1-carboxylate (100 g, 402 mmol) in anhydrous THF (1 L) was added LiAlH4 (403 mL, 403 mmol, 1 M in THF) dropwise at 0-5° C. under N2 over about 1 h. The mixture was then stirred at 0° C. for 2 h under N2. Additional LiAlH4 (40 mL, 40 mmol, 1 M in THF) was then added to the reaction mixture. The mixture was stirred at 0° C. for 1 h under N2. The mixture was quenched with 20% NaCl solution (20 mL) slowly at 0° C. and diluted with THF (500 mL, 5 volumes). The mixture was warmed to 15° C. and stirred for 15 min. The mixture was filtered and rinsed with THF (2×200 mL). The filter cake was suspended within THF (1 L, 10 volumes) for 30 min. The suspension was filtered and rinsed with THF (2×200 mL). The filter cake suspension and filtration was repeated twice in THF (1 L, 10 volumes), and was then rinsed with THF (2×200 mL). The combined filtrate was dried over anhydrous Na2SO4, concentrated under reduced pressure to afford crude N-((1r,4r)-4-(hydroxymethyl)cyclohexyl)ethanesulfonamide (72 g, 81%) as a white solid, which was used for the next step without further purification; 1H NMR (CDCl3 400 MHz): δ 4.23 (d, J=8.0 Hz, 1H), 3.46 (t, J=6.4 Hz, 2H), 3.25-3.18 (m, 1 H), 3.04 (q, J=7.6 Hz, 2H), 2.11-2.07 (m, 2H), 1.88-1.84 (m, 2H), 1.46-1.35 (m, 4H), 1.29-1.24 (m, 2H), 1.09-1.00 (m, 2H).Step 3: ((1r,4r)-4-(Ethylsulfonamido)cyclohexyl)methyl 4-methylbenzenesulfonate
To a solution of crude N-((1r,4r)-4-(hydroxymethyl)cyclohexyl)ethanesulfonamide (30 g, 136 mmol) in anhydrous DCM (300 mL) was added TsCl (25.84 g, 136 mmol), DMAP (1.66 g, 13.6 mmol) and Et3N (41.2 g, 408 mmol). The mixture was stirred at 10° C. for 6 h under N2. The mixture was then quenched with H2O (200 mL). After partition, the organic layer was washed with H2O (2×150 mL) and brine (150 mL), dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel eluting with petroleum ether/ethyl acetate=1/0˜2/1 to give ((1r,4r)-4-(ethylsulfonamido)cyclohexyl)methyl 4-methylbenzenesulfonate (37 g, 73%) as a white solid; 1H NMR (CDCl3 400 MHz): δ 7.78 (d, J=8.4 Hz, 2H), 7.35 (d, J=8.8 Hz, 2H), 4.23 (d, J=7.6 Hz, 1H), 3.81 (d, J=6.4 Hz, 2H), 3.19-3.14 (m, 1H), 3.01 (q, J=7.6 Hz, 2H), 2.46 (s, 3H), 2.09-2.03 (m, 2H), 1.79-1.74 (m, 2H), 1.66-1.56 (m, 1H), 1.35 (t, J=7.6 Hz, 3H), 1.28-1.18 (m, 2H), 1.09-1.01 (m, 2H).
Medical uses
Revumenib is indicated for the treatment of relapsed or refractory acute leukemia with a lysine methyltransferase 2A gene (KMT2A) translocation.[1][3]
History
Efficacy was evaluated in a single-arm cohort of an open-label, multicenter trial (SNDX-5613-0700, NCT04065399; AUGMENT-101) in 104 adult and pediatric participants (at least 30 days old) with relapsed or refractory (R/R) acute leukemia with a lysine methyltransferase 2A gene translocation.[3] Participants with an 11q23 partial tandem duplication were excluded.[3] Revumenib was administered until disease progression, unacceptable toxicity, failure to achieve morphological leukemia-free state by four cycles of treatment, or hematopoietic stem cell transplantation.[3]
The US Food and Drug Administration (FDA) granted the application for revumenib priority review, breakthrough therapy, and orphan drug designations.[3]
Society and culture
Legal status
Revumenib was approved for medical use in the United States in November 2024.[3][5][6]
Names
Revumenib is the international nonproprietary name.[7]
It is sold under the brand name Revuforj.[1][3]
References
- ^ Jump up to:a b c d e f “Revuforj- revumenib tablet, film coated”. DailyMed. 19 November 2024. Retrieved 28 November 2024.
- ^ Hussain H, Zaidi SM, Hasan SM, Jahan AS, Rangwala BS, Rangwala HS, et al. (May 2024). “Revumenib (SNDX-5613): a promising menin inhibitor for the management of relapsed and refractory acute myeloid leukaemia (AML)”. Annals of Medicine and Surgery (2012). 86 (5): 2379–2381. doi:10.1097/MS9.0000000000001888. PMC 11060303. PMID 38694289.
- ^ Jump up to:a b c d e f g h i “FDA approves revumenib for relapsed or refractory acute leukemia with a KMT2A translocation”. U.S. Food and Drug Administration (FDA) (Press release). 15 November 2024. Archived from the original on 20 November 2024. Retrieved 20 November 2024.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ New Drug Therapy Approvals 2024 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2025. Archived from the original on 21 January 2025. Retrieved 21 January 2025.
- ^ “Novel Drug Approvals for 2024”. U.S. Food and Drug Administration (FDA). 1 October 2024. Retrieved 29 November 2024.
- ^ “Syndax Announces FDA Approval of Revuforj (revumenib), the First and Only Menin Inhibitor to Treat Adult and Pediatric Patients with Relapsed or Refractory Acute Leukemia with a KMT2A Translocation” (Press release). Syndax Pharmaceuticals. 15 November 2024. Retrieved 20 November 2024 – via PR Newswire.
- ^ World Health Organization (2022). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 88”. WHO Drug Information. 36 (3). hdl:10665/363551.
Further reading
- Nadiminti KV, Sahasrabudhe KD, Liu H (November 2024). “Menin inhibitors for the treatment of acute myeloid leukemia: challenges and opportunities ahead”. Journal of Hematology & Oncology. 17 (1): 113. doi:10.1186/s13045-024-01632-8. PMC 11575055. PMID 39558390.
External links
- “Revumenib (Code C165776)”. NCI Thesaurus.
- “Revumenib citrate”. NCI Drug Dictionary.
- Clinical trial number NCT04065399 for “A Study of Revumenib in R/R Leukemias Including Those With an MLL/KMT2A Gene Rearrangement or NPM1 Mutation (AUGMENT-101)” at ClinicalTrials.gov
- Clinical trial number NCT05918913 for “Expanded Access Program for Revumenib” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Revuforj |
| Other names | SNDX-5613 |
| AHFS/Drugs.com | revuforj |
| License data | US DailyMed: Revumenib |
| Routes of administration | By mouth |
| Drug class | Antineoplastic; menin inhibitor |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2169919-21-32169919-22-4 |
| PubChem CID | 132212657 |
| DrugBank | DB18515DBSALT003460 |
| ChemSpider | 95502909 |
| UNII | LZ0M43NNF275HI05N8HS |
| KEGG | D12728D12729 |
| ChEMBL | ChEMBL4650827ChEMBL4650278 |
| PDB ligand | OQ4 (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C32H47FN6O4S |
| Molar mass | 630.82 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
Salman MY, Stein EM: Revumenib for patients with acute leukemia: a new tool for differentiation therapy. Haematologica. 2024 Nov 1;109(11):3488-3495. doi: 10.3324/haematol.2022.282621. [Article]- FDA Approved Drug Products: Revuforj (revumenib) tablets for oral use (November 2024) [Link]
- FDA News Release: FDA approves revumenib for relapsed or refractory acute leukemia with a KMT2A translocation [Link]
- Syndax Pharmaceuticals: Revumenib Physiologically Based Pharmacokinetic (PBPK) Model for Evaluation of Age Effect and CYP3A4-Mediated Drug–Drug Interaction (DDI) in Relapsed/Refractory Acute Leukemias [Link]
- Syndax Pharmaceuticals: Syndax Announces FDA Approval of Revuforj® (revumenib), the First and Only Menin Inhibitor to Treat Adult and Pediatric Patients with Relapsed or Refractory Acute Leukemia with a KMT2A Translocation [Link]
///////////Revumenib, APPROVALS 2024, FDA 2024, Revuforj, SNDX-5613, 2169919-21-3, revumenib, Z0M43NNF2, SNDX5613, SNDX-50613 free base, SNDX-5613 free base, SNDX50613 free base, SNDX5613 free base
Acoramidis



Acoramidis
- AG-10
- 1446711-81-4
- AG10
- Acorami
292.30 g/mol,
C15H17FN2O3
3-[3-(3,5-dimethyl-1H-pyrazol-4-yl)propoxy]-4-fluorobenzoic acid
FDA APPROVED 11/22/2024, Attruby To treat cardiomyopathy of wild-type or variant transthyretin-mediated amyloidosis
Drug Trials Snapshot
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Acoramidis hydrochloride | VY9C88C2NV | 2242751-53-5 | MGFZEARHINUOMX-UHFFFAOYSA-N |
Acoramidis, sold under the brand name Attruby, is a medication used for the treatment of cardiomyopathy.[1] It is a near-complete (>90%) transthyretin stabilizer, developed to mimic the protective properties of the naturally-occurring T119M mutation,[4][5] to treat transthyretin amyloid cardiomyopathy. It is taken by mouth.[1]
The most common adverse reactions include diarrhea and upper abdominal pain.[6]
Acoramidis was approved for medical use in the United States in November 2024,[6][7][8][9] and in the European Union in February 2025.[2][3]
PATENTS
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US9169214 | No | 2015-10-27 | 2031-05-05 | |
| US9913826 | No | 2018-03-13 | 2033-03-14 | |
| US11058668 | No | 2021-07-13 | 2039-03-22 | |
| US10842777 | No | 2020-11-24 | 2031-05-05 | |
| US11919865 | No | 2024-03-05 | 2038-02-16 | |
| US12070449 | No | 2024-08-27 | 2039-03-22 | |
| US12005043 | No | 2024-06-11 | 2039-08-16 | |
| US10398681 | No | 2019-09-03 | 2031-05-05 | |
| US9642838 | No | 2017-05-09 | 2031-05-05 | |
| US8877795 | No | 2014-11-04 | 2031-05-05 | |
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US10513497 | No | 2019-12-24 | 2038-02-16 | |
| US11260047 | No | 2022-03-01 | 2039-08-16 | |
SYN


Penchala SC, Connelly S, Wang Y, Park MS, Zhao L, Baranczak A, Rappley I, Vogel H, Liedtke M, Witteles RM, Powers ET, Reixach N, Chan WK, Wilson IA, Kelly JW, Graef IA, Alhamadsheh MM: AG10 inhibits amyloidogenesis and cellular toxicity of the familial amyloid cardiomyopathy-associated V122I transthyretin. Proc Natl Acad Sci U S A. 2013 Jun 11;110(24):9992-7. doi: 10.1073/pnas.1300761110. Epub 2013 May 28
PATENT
https://patents.google.com/patent/US9913826B2/en
Chemical Synthesis

Methyl 3-(3-bromopropoxy)-4-fluorobenzoate (Compound 2)
To a solution of methyl 4-fluoro-3-hydroxybenzoate 1 (3.0 g, 17.6 mmol, 1 equiv) and 1,3-dibromopropane (9.0 ml, 88.2 mmol, 5 equiv) in DMF (40 ml) was added K2CO3 (2.93 g, 21.2 mmol, 1.2 equiv). The reaction mixture was stirred at room temperature for 16 hours. The mixture was diluted with EtOAc (1.5 L), washed with brine (3×0.5 L) and dried with Na2SO4. The solution was filtered and concentrated. The residue was purified by flash column chromatography (silica gel, 1-10% EtOAc/hexanes) to afford compound 2 (4.21 g, 82% yield); 1H NMR (CD3OD, 600 MHz) δ 7.67-7.61 (m, 2H), 7.14-7.07 (m, 1H), 4.21 (t, 2H, J=5.89 Hz), 3.89 (s, 3H), 3.62 (t, 2H, J=6.38 Hz), 2.38-2.31 (m, 2H); (ESI+) m/z: calcd for C11H12BrFO3+H+290.00; found 290.01 (M+H+).Methyl 3-(3-(3,5-dimethyl-1H-pyrazol-4-yl)propoxy)-4-fluorobenzoate (Compound 4)
A solution of 2 (780 mg, 2.69 mmol, 1 equiv) in benzene (3 ml) was added dropwise to a solution of acetyl acetone (0.552 ml, 5.38 mmol, 2 equiv) and DBU (0.804 ml, 5.38 mmol, 2 equiv) in benzene (7 ml). The reaction mixture was stirred at room temperature for 3 days. The mixture was filtered and concentrated. The residue was purified by flash column chromatography (silica gel, 1-10% EtOAc/hexanes) to afford compound 3 which was used in the next step directly. Hydrazine hydrate (0.36 ml, 6.73 mmol, 2.5 equiv) was added to a solution 3 in ethanol (5 ml) and the reaction was heated under reflux for 4 hours. The reaction was concentrated and purified by flash column chromatography (silica gel, 1-20% MeOH/CH2Cl2) to afford compound 4 (288 mg, 35% yield) in two steps; 1H NMR (CD3OD, 600 MHz) δ 7.64-7.58 (m, 2H), 7.20-7.15 (m, 1H), 4.01 (t, 2H, J=6.0 Hz), 3.86 (s, 3H), 2.58 (t, 2H, J=7.2 Hz), 2.12 (s, 6H), 1.97-1.92 (m, 2H); HRMS (DART) m/z: calcd for C16H19FN2O+H+307.1458; found 307.1452 (M+H+).3-(3-(3,5-Dimethyl-1H-pyrazol-4-yl)propoxy)-4-fluorobenzoic acid (Compound VIIc)
To a suspension of 4 (100 mg, 0.33 mmol, 1 equiv) in a mixture of THF (3 ml) and water (3 ml) was added LiOH.H2O (27.5 mg, 0.66 mmol, 2 equiv). The reaction mixture was stirred at room temperature for 14 hr after which it was cooled to 0° C. and carefully acidified to pH 2-3 with IN aqueous HCl. The mixture was extracted with EtOAc (3×30 ml) and the combined organic extracts were dried over anhydrous sodium sulfate and concentrated in vacuo. The crude product was subjected to flash column chromatography (silica gel, 10-50% MeOH/CH2Cl2) to give Compound VIIc (68 mg, 71% yield) as a white solid (>98% purity by HPLC); 1H NMR (CD3OD, 600 MHz) δ 7.65-7.58 (m, 2H), 7.20-7.14 (m, 1H), 4.00 (t, 2H, J=6.0 Hz), 2.58 (t, 2H, J=5.8 Hz), 2.12 (s, 6H), 1.97-1.92 (m, 2H); HRMS (DART) m/z: calcd for C15H17FN2O3+H+ 293.1301; found 293.1293 (M+H+).3-(3-(3,5-Dimethyl-1H-pyrazol-4-yl)propoxy)-4-fluorobenzamide
To a suspension of 4 (100 mg, 0.33 mmol, 1 equiv) in a mixture of THF (3 ml) and water (3 ml) is added (23.1 mg, 0.66 mmol, 2 equiv) of NH4OH. The reaction mixture is stirred at room temperature for 14 hr after which it is cooled to 0° C. and carefully adjusted to pH 7 with IN aqueous HCl. The mixture is extracted with EtOAc (3×30 ml) and the combined organic extracts are dried over anhydrous sodium sulfate and concentrated in vacuo. The crude product is subjected to flash column chromatography (silica gel, 10-50% MeOH/CH2Cl2) to give 3-(3-(3,5-Dimethyl-1H-pyrazol-4-yl)propoxy)-4-fluorobenzamide.N-ethyl 3-(3-(3,5-Dimethyl-1H-pyrazol-4-yl)propoxy)-4-fluorobenzamide
To a suspension of 4 (100 mg, 0.33 mmol, 1 equiv) in a mixture of THF (3 ml) and water (3 ml) is added (27.1 mg, 0.66 mmol, 2 equiv) of C2H5NH2. The reaction mixture is adjusted to pH 9.0 with 0.5N NaOH, then stirred at room temperature for 14 hr after which it is cooled to 0° C. and carefully adjusted to pH 7 with IN aqueous HCl. The mixture is extracted with EtOAc (3×30 ml) and the combined organic extracts are dried over anhydrous sodium sulfate and concentrated in vacuo. The crude product is subjected to flash column chromatography (silica gel, 10-50% MeOH/CH2Cl2) to give N-ethyl 3-(3-(3,5-Dimethyl-1H-pyrazol-4-yl)propoxy)-4-fluorobenzamide.
PATENT
https://patents.google.com/patent/US10513497B2/en
Example 1Preparation of 3-(3-Hydroxy-propyl)-pentane-2, 4-dione (a Compound of Formula IV)

A compound of Formula IIIa (100 g, 495 mmol 1.0 equiv.) was dissolved in acetone (1 L). A compound of Formula II (49.59 g, 495 mmol, 1.0 equiv.) was added to above solution, followed by addition of K2CO3 (82.14 g, 594.38 mmol, 1.2 equiv.) and KI (41.11 g, 247 mmol, 0.5 equiv.) at room temperature with stirring. The reaction mixture was heated to 60±5° C. and stirred for 40 h at this temperature. The reaction mixture was filtered and then concentrated under reduced pressure to afford a compound of Formula IV (102 g) as viscous orange liquid.Example 2Preparation of 3(3, 5-Dimethyl-1H-pyrazol-4-yl) propane-1-ol (a compound of Formula V)

A compound of Formula IV (100 g, 632 mmol, 1.0 equiv.) was dissolved in ethanol (1 L). Hydrazine hydrate (87 g, 1738 mmol, 2.75 equiv.) and conc. HCl (4.6 mL, 0.2 equiv.) were added to above solution at room temperature. The reaction mixture was heated to 75±5° C. and stirred for 3 h at this temperature. After completion of reaction by TLC (70% ethyl acetate: n-hexane, visible in iodine) and observation of product peak in mass spectrum, the reaction mixture was concentrated under reduce pressure to afford a compound of Formula V (70 g) as a colorless liquid syrup which was used as such for next step.Example 3Preparation of 4-(3-Bromo-propyl)-3, 5-dimethyl-1H-pyrazole (a compound of formula VIa)

A compound of Formula V (35 g, 227 mmol, 1.0 equiv.) was dissolved in 1,2-dichloroethane (525 mL). PBr3 (64.67mL, 681 mmol, 3 equiv.) was added in small portions at room temperature over 30 minutes. The reaction mixture was heated up to 75±5° C. and stirred for 3 h at this temperature. After completion of reaction by TLC (50% ethyl acetate: n-hexane, visible in iodine) and observation of product peak in Mass spectrum, the reaction mixture was diluted with dichloromethane (350 mL) and quenched with saturated solution of NaHCO3 till pH=7 to 8. Both organic and aqueous layers were separated and collected. The organic layer was dried over MgSO4 and filtered. Filtrate was concentrated under reduce pressure to afford a compound of Formula VIa (38 g) as a viscous orange liquid.Example 4Preparation of 3-[3-(3, 5-Dimethyl-1H-pyrazol-4-yl)-propoxy]-4-fluoro-benzoic acid methyl ester (a compound of Formula VIIIa)

A compound of Formula VIIa (19 g, 111 mmol, 1.0 equiv.) was dissolved in DMF (190 mL). A compound of Formula VIa (31.5 g, 145.14 mmol, 1.3 equiv.) was added followed by K2CO3 (38.6 g, 279.18 mmol, 2.5 equiv.) at room temperature under stirred conditions. The reaction mixture was stirred for 16 to 18 h at room temperature. After completion of reaction in TLC (50% ethyl acetate: n- hexane), the reaction mixture was diluted with water (190 mL) and ethyl acetate (95 mL). Both organic layer and aqueous layer were separated and collected. Aqueous layer was extracted with ethyl acetate (190 mL). The combined organic extract was washed with water (95 mL), brine (95 mL), dried over Na2SO4 and filtered. The filtered organic layer was concentrated under reduce pressure to afford a crude viscous orange liquid (40 g). The crude was further purified by column chromatography using silica gel (285 g) and eluted with varying quantity of ethyl acetate in hexane to afford pure product, a compound of Formula VIIIa (25 g) as an off white solid.Example 5Preparation of 3-[3-(3,5-Dimethyl-1H-pyrazol-4-yl)-propoxy]-4-fluoro-benzoic acid methyl ester (a compound of Formula VIIIa)

4-(3-Bromopropyl)-3,5-dimethyl-1H-pyrazole hydrobromide (VIa) and DMSO were charged into vessel and agitated at 20±10° C. for 10 minutes. The mixture was then heated to 55±5° C. with stirring. To this mixture was transferred a stirred solution containing 4-fluoro-3-hydroxy-benzoic acid methyl ester (VIIa), potassium carbonate and anhydrous DMSO. The DMSO solution of the alkyl bromide were slowly transferred in order to maintaining an internal temperature of 55.0±5° C. Addition was complete after 6 hours and the mixture was agitated at 55.0±5° C. for an additional hour at 55.0±5° C. The mixture was cooled to 25±5° C. over the course of 30 minutes and water added while maintaining a temperature below 25° C. The mixture was extracted with ethyl acetate and the aqueous layer back extracted with ethyl acetate. The pooled ethyl acetate solutions were washed brine. The combined ethyl acetate washes were concentrated under vacuum to a minimal volume and heptane was added, which precipitates VIIIa. The mixture was heated to 75±5° C. and aged with stirring for 1 hour. The mixture was cooled to 25±5° C. over the course of two hours and the resulting solids collected by filtration. The filter cake was washed with ethyl acetate in heptane (30%). Isolated solids were dried with a nitrogen flow. Solids are charged to vessel and combined with ethyl acetate and heptane. The resulting mixture is heated to 75±5° C. to dissolve solids. The solution was cooled to 25±5° C. over the course of two hours and the resulting solids collected by filtration. The solids were washed with a 30% ethyl acetate/heptane solvent mixture and dried in vacuum oven at 55° C. to give VIIIa in >99.5% purity.Example 6Preparation of 3-[3-(3, 5-Dimethyl-1H-pyrazol-4-yl)-propoxy]-4-fluoro-benzoic acid (a compound of Formula IX)

A compound of Formula VIIIa (19 g, 62 mmol, 1 equiv.) was dissolved in methanol (95 mL, 5 vol.) at room temperature. A solution of LiOH.H2O (6.5 g, 155 mmol, 2.5 equiv.) in water (57 mL) was added in small portions at room temperature over 10 to 15 minutes. The reaction mixture was stirred for 2 h at room temperature. After completion of reaction by TLC (70% ethyl acetate: n-hexane), the reaction mixture is concentrated below 45° C. under reduced pressure to afford a solid residue of Formula IX.Example 7Preparation of a Pharmaceutically Acceptable Salt of Formula I
The solid residue of Formula IX was dissolved in water (57 mL) and stirred for 10 min and cooled to 0±5° C. The aqueous solution was acidified with conc. HCl (20-25 mL) to pH=2 and stirred for 30 minutes at 0±5° C. Precipitation was observed which was filtered and dried at room temperature to afford pure product, a compound of Formula Ia (17.5 g) as an off-white solid.Example 8Additional Preparation of a Pharmaceutically Acceptable Salt of Formula I

Water and concentrated HCl were charged to a vessel and cooled with stirring to 10±5° C. Compound of Formula IX and water were charged to a second vessel and cooled with stirring to 10±5° C. The HCl solution in vessel
1 was transferred to a vessel containing compound of Formula IX mixture over not less than 15 minutes, while maintaining a temperature of <25° C. The resulting slurry was aged with stirring at 20±5° C. for 44 hours. The solids were collected by filtration, washed with 0.2 N HCl (3 ×) and dried under vacuum at ≥55° C. to provide Ia as white solid, >99.8% purity.Example 9Preparation of 3-[3-(3,5-dimethyl-1H-pyrazol-4-yl)propoxy]-4-fluorobenzoic acid hydrochloride salt (Compound Ia) from VIIIa

A jacketed glass vessel is charged with compound of formula VIIIa (1.0 equiv.) and methanol. The mixture is cooled with stirring to 10±5° C. and over the course of 20 minutes an aqueous solution of sodium hydroxide (3 equiv.) is charged. The mixture is aged with stirring at 20±5° C. for NLT
2 hours at which point the reaction is complete. Stirring is stopped and water is added. Methanol is then removed by vacuum distillation at an internal temperature of NMT
35° C. The resulting concentrated, clear aqueous solution is cooled to 10° C. and concentrated HCl is added until the pH was lowered to between 1.4-1.6 (pH meter) to precipitate the HCl salt. The solids are collected by filtration, washed with 0.2 N HCl and dried under vacuum at 50° C. to give a compound of Formula Ia in NLT 99.5% purity.Example 10Preparation of 3-[3-(3,5-dimethyl-1H-pyrazol-4-yl)propoxy]-4-fluorobenzoic acid (compound of formula IX) from VIIIa

Methyl 3-(3-(3,5-dimethyl-1H-pyrazol-4-yl)propoxy)-4-fluorobenzoate (Compound of formula VIIIa) and methanol were charged into a vessel and the resulting mixture was agitated at 20±5° C. until dissolved. The solution was cooled to 10±5° C. and over the course of 20 minutes a sodium hydroxide solution was added while maintaining a temperature ≤25° C. The mixture temperature was adjusted to 25±5° C. and aged with stirring for 18 hours. The reaction mixture was filtered. Water was added to filtrate and the resulting mixture concentrated under vacuum until volume of the mixture was reduced to minimal volume. Water was again added and the resulting mixture concentrated under vacuum until volume of the mixture was reduced to minimal volume. The pH of the aqueous mixture was adjusted to 5.5±0.5 by addition of concentrated hydrochloric acid then 0.5N HCl. The temperature of the mixture was adjusted to 7±5° C. and aged with stirring for an additional hour. The solids were collected by filtration, washed with water and partially dried under vacuum at ≥55° C. to provide compound of Formula IX as white solids with >99.5% HPLC purity.Example 11Conversion of the Hydrochloride Salt to Free Base
3-[3-(3,5-Dimethyl-1H-pyrazol-4-yl)-propoxy]-4-fluorobenzoic acid hydrochloride (10.0 g, 30.4 mmol, 1.0 equiv.) was taken in deionized water (30.0 mL) at room temperature and was cooled to 10±5° C. To this mixture was added saturated sodium bicarbonate to pH≅6-7 and stirred for 30 minute at this temperature. The off white precipitate obtained was filtered and washed with deionized water (20 mL). Solid compound was dried at room temperature to afford 3-[3-(3,5-dimethyl-1H-pyrazol-4-yl)-propoxy]-4-fluorobenzoic acid (the compound of Formula IX) (7.40 g, 83.2%) as an off-white solid.
Medical uses
Acoramidis is indicated for the treatment of the cardiomyopathy of wild-type or variant transthyretin-mediated amyloidosis (ATTR-CM) in adults to reduce cardiovascular death and cardiovascular-related hospitalization.[1][6][10]
ATTR-CM is a rare and serious disease that affects the heart muscle.[6] In people with ATTR-CM, there is a build-up of protein deposits in the heart, causing the walls of the heart to become stiff, and making the left ventricle unable to properly relax and fill with blood (called cardiomyopathy).[6] As the condition progresses, the heart can become unable to pump blood out adequately, causing heart failure.[6] There are two types of ATTR-CM, hereditary ATTR-CM (hATTR-CM) and wild-type ATTR-CM (wATTR-CM).[6] In hATTR-CM, which can run in families, there’s a variant in the transthyretin gene, which results in protein deposits in the heart. In wATTR-CM, there is no variant in the transthyretin gene.[6]
Side effects
The most common side effects are diarrhea and abdominal pain.[11]
History
The efficacy and safety of acoramidis were evaluated in a multicenter, international, randomized, double-blind, placebo-controlled study in 611 adult participants with wild-type or hereditary (variant) ATTR-CM (NCT03860935).[6]
Clinical trials
Phase I data indicated acoramidis achieved near-complete (>90%) TTR stabilization across the entire dosing interval at steady state.[12]
Phase II and the Open-Label Extension (OLE) data indicated after a median of 38 months, long-term treatment with acoramidis was generally well tolerated and resulted in a median decline in NT-proBNP levels, normalization of serum TTR, and sustained stabilization of TTR in individuals with ATTR-CM. [13]
Phase III data from ATTRibute-CM indicated acoramidis resulted in a significantly better four-step primary hierarchical outcome containing components of mortality, morbidity, and function than placebo at 30 months in participants with ATTR-CM. Adverse events were similar in the two groups.[14]
Other analyses from ATTRibute-CM indicated a 50% reduction in cumulative cardiovascular hospitalizations (CVH), a 42% reduction in all-cause mortality (ACM) and recurrent CVH, and a 3-month time-to-separation of the Kaplan Meier curves for ACM or CVH. No other treatment has demonstrated this degree of treatment effect this quickly in participants with ATTR-CM.[15][16][17]
In vitro data indicated acoramidis exhibits near-complete (>90%) TTR stabilization at therapeutic trough concentrations, and its TTR stabilization exceeds that of tafamidis’ across a range of destabilizing TTR mutations.[18]
Society and culture
Legal status
Acoramidis was approved for medical use in the United States in November 2024.[6][7][19] The approval was granted to BridgeBio Pharma.[10]
In December 2024, the Committee for Medicinal Products for Human Use of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Beyonttra, intended for the treatment of transthyretin amyloidosis in adults with cardiomyopathy.[2] The applicant for this medicinal product is BridgeBio Europe B.V.[2] Acoramidis was designated an orphan medicine by the EMA.[2] Acoramidis was authorized for medical use in the European Union in February 2025.[2][3]
Names
During development, acoramidis was known as AG10 (the Alhamadsheh-Graef molecule 10).[20]
Acoramidis is the international nonproprietary name.[21]
Acoramidis is sold under the brand names Attruby[1][6] and Beyonttra.[2][3]
References
- ^ Jump up to:a b c d e “Attruby- acoramidis hydrochloride tablet, film coated”. DailyMed. 26 November 2024. Retrieved 28 November 2024.
- ^ Jump up to:a b c d e f g “Beyonttra EPAR”. European Medicines Agency (EMA). 12 December 2024. Retrieved 15 December 2024. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b c d “Beyonttra PI”. Union Register of medicinal products. 11 February 2025. Retrieved 16 February 2025.
- ^ Penchala SC, Connelly S, Wang Y, Park MS, Zhao L, Baranczak A, et al. (June 2013). “AG10 inhibits amyloidogenesis and cellular toxicity of the familial amyloid cardiomyopathy-associated V122I transthyretin”. Proceedings of the National Academy of Sciences of the United States of America. 110 (24): 9992–9997. Bibcode:2013PNAS..110.9992P. doi:10.1073/pnas.1300761110. PMC 3683741. PMID 23716704.
- ^ Miller M, Pal A, Albusairi W, Joo H, Pappas B, Haque Tuhin MT, et al. (September 2018). “Enthalpy-Driven Stabilization of Transthyretin by AG10 Mimics a Naturally Occurring Genetic Variant That Protects from Transthyretin Amyloidosis”. Journal of Medicinal Chemistry. 61 (17): 7862–7876. doi:10.1021/acs.jmedchem.8b00817. PMC 6276790. PMID 30133284.
- ^ Jump up to:a b c d e f g h i j k “FDA approves drug for heart disorder caused by transthyretin-mediated”. U.S. Food and Drug Administration. 1 October 2024. Retrieved 27 November 2024. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b “Novel Drug Approvals for 2024”. U.S. Food and Drug Administration (FDA). 1 October 2024. Retrieved 20 December 2024.
- ^ “FDA approves BridgeBio Pharma’s Attruby to treat rare heart disease ATTR-CM”. PMLiVE. 25 November 2024. Retrieved 25 November 2024.
- ^ New Drug Therapy Approvals 2024 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2025. Archived from the original on 21 January 2025. Retrieved 21 January 2025.
- ^ Jump up to:a b LeMieux J (25 November 2024). “Bridgebio’s Attruby, to Treat Heart Condition ATTR-CM, Receives FDA Approval”. Genetic Engineering and Biotechnology News. Retrieved 25 November 2024.
- ^ “FDA approves BridgeBio’s Attruby for ATTR-CM treatment”. Pharmaceutical Technology. 25 November 2024. Retrieved 25 November 2024.
- ^ Fox JC, Hellawell JL, Rao S, O’Reilly T, Lumpkin R, Jernelius J, et al. (January 2020). “First-in-Human Study of AG10, a Novel, Oral, Specific, Selective, and Potent Transthyretin Stabilizer for the Treatment of Transthyretin Amyloidosis: A Phase 1 Safety, Tolerability, Pharmacokinetic, and Pharmacodynamic Study in Healthy Adult Volunteers”. Clinical Pharmacology in Drug Development. 9 (1): 115–129. doi:10.1002/cpdd.700. PMC 7003869. PMID 31172685.
- ^ Masri A, Aras M, Falk RH, Grogan M, Jacoby D, Judge DP, et al. (March 2022). “Long-Term Safety and Tolerability of Acoramidis (Ag10) in Symptomatic Transthyretin Amyloid Cardiomyopathy: Updated Analysis from an Ongoing Phase 2 Open-Label Extension Study”. Journal of the American College of Cardiology. 79 (9): 227. doi:10.1016/S0735-1097(22)01218-9.
- ^ Gillmore JD, Judge DP, Cappelli F, Fontana M, Garcia-Pavia P, Gibbs S, et al. (January 2024). “Efficacy and Safety of Acoramidis in Transthyretin Amyloid Cardiomyopathy”. The New England Journal of Medicine. 390 (2): 132–142. doi:10.1056/NEJMoa2305434. PMID 38197816.
- ^ “Program Planner”. http://www.abstractsonline.com. Archived from the original on 6 February 2021. Retrieved 19 October 2024.
- ^ Alexander K, Judge D, Cappelli F, Fontana M, Garcia-Pavia P, Grogan M, et al. (6 May 2024). Acoramidis Achieves Early Reduction in Cardiovascular Death or Hospitalization in Transthyretin Amyloid Cardiomyopathy (ATTR-CM): Results from the ATTRibute-CM Clinical Trial OC7 (#281) (Report). doi:10.26226/m.65f9bf8ae6f73964e1d4f069.
- ^ “BridgeBio Shares Recurrent Event Analysis of ATTRibute-CM, Demonstrating a 42% Reduction by Acoramidis on the Composite Endpoint of All-Cause Mortality and Recurrent Cardiovascular-related Hospitalization Events”. HFSA. Retrieved 19 October 2024.
- ^ Ji A, Wong P, Judge DP, Graef IA, Fox J, Sinha U (November 2023). “Acoramidis produces near-complete TTR stabilization in blood samples from patients with variant transthyretin amyloidosis that is greater than that achieved with tafamidis”. European Heart Journal. 44 (Supplement_2). doi:10.1093/eurheartj/ehad655.989. ISSN 0195-668X.
- ^ “Attruby (acoramidis), a Near Complete TTR Stabilizer (≥90%), approved by FDA to Reduce Cardiovascular Death and Cardiovascular-related Hospitalization in ATTR-CM Patients” (Press release). BridgeBio Pharma. 23 November 2024. Archived from the original on 25 November 2024. Retrieved 28 November 2024 – via GlobeNewswire.
- ^ “FDA approves Stanford Medicine-developed drug that treats rare heart disease”. Stanford. 27 November 2024. Retrieved 29 November 2024.
- ^ World Health Organization (2024). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 83”. WHO Drug Information. 38 (1). hdl:10665/378096.
Further reading
- Penchala SC, Connelly S, Wang Y, Park MS, Zhao L, Baranczak A, et al. (June 2013). “AG10 inhibits amyloidogenesis and cellular toxicity of the familial amyloid cardiomyopathy-associated V122I transthyretin”. Proceedings of the National Academy of Sciences of the United States of America. 110 (24): 9992–9997. Bibcode:2013PNAS..110.9992P. doi:10.1073/pnas.1300761110. PMC 3683741. PMID 23716704.
External links
- Clinical trial number NCT03860935 for “Efficacy and Safety of AG10 in Subjects With Transthyretin Amyloid Cardiomyopathy (ATTRibute-CM)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Pronunciation | ə-corAM-i-dis |
| Trade names | Attruby, others |
| Other names | AG10 |
| AHFS/Drugs.com | Monograph |
| License data | US DailyMed: Acoramidis |
| Routes of administration | By mouth |
| Drug class | Amyloidogenesis suppressant |
| ATC code | C01EB25 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1]EU: Rx-only[2][3] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1446711-81-42242751-53-5 |
| PubChem CID | 7146471371464713 |
| IUPHAR/BPS | 135307127 |
| DrugBank | DB17999 |
| ChemSpider | 35033544 |
| UNII | T12B44A1OEVY9C88C2NV |
| KEGG | D11972D11973 |
| ChEMBL | ChEMBL3940890ChEMBL4650226 |
| PDB ligand | 16V (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C15H17FN2O3 |
| Molar mass | 292.310 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
- Nuvolone M, Girelli M, Merlini G: Oral Therapy for the Treatment of Transthyretin-Related Amyloid Cardiomyopathy. Int J Mol Sci. 2022 Dec 18;23(24):16145. doi: 10.3390/ijms232416145. [Article]
- Penchala SC, Connelly S, Wang Y, Park MS, Zhao L, Baranczak A, Rappley I, Vogel H, Liedtke M, Witteles RM, Powers ET, Reixach N, Chan WK, Wilson IA, Kelly JW, Graef IA, Alhamadsheh MM: AG10 inhibits amyloidogenesis and cellular toxicity of the familial amyloid cardiomyopathy-associated V122I transthyretin. Proc Natl Acad Sci U S A. 2013 Jun 11;110(24):9992-7. doi: 10.1073/pnas.1300761110. Epub 2013 May 28. [Article]
- Fox JC, Hellawell JL, Rao S, O’Reilly T, Lumpkin R, Jernelius J, Gretler D, Sinha U: First-in-Human Study of AG10, a Novel, Oral, Specific, Selective, and Potent Transthyretin Stabilizer for the Treatment of Transthyretin Amyloidosis: A Phase 1 Safety, Tolerability, Pharmacokinetic, and Pharmacodynamic Study in Healthy Adult Volunteers. Clin Pharmacol Drug Dev. 2020 Jan;9(1):115-129. doi: 10.1002/cpdd.700. Epub 2019 Jun 6. [Article]
- FDA Approved Drug Products: Attruby (acoramidis) tablets for oral administration (November 2024) [Link]
- FDA News Release: FDA approves drug for heart disorder caused by transthyretin-mediated amyloidosis [Link]
/////////Acoramidis, Attruby, AG 10, AG10. AG-10, WHO 11205, APPROVALS 2024, FDA 2024
Landiolol


Landiolol
- 133242-30-5
- ONO-1101
- Ono 1101
- WHO 7516
FDA APPROVED 11/22/2024, Rapiblyk, To treat supraventricular tachycardia
C25H39N3O8
509.6 g/mol
[(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]methyl 3-[4-[(2S)-2-hydroxy-3-[2-(morpholine-4-carbonylamino)ethylamino]propoxy]phenyl]propanoate
- [(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]methyl 3-[4-[(2S)-2-hydroxy-3-[2-(morpholine-4-carbonylamino)ethylamino]propoxy]phenyl]propanoate
- UNII-62NWQ924LH
- (S-(R*,R*))-(2,2-Dimethyl-1,3-dioxolan-4-yl)methyl 4-(2-hydroxy-3-((2-((4-morpholinylcarbonyl)amino)ethyl)amino)propoxy)benzenepropanoate
- Benzenepropanoic acid, 4-((2S)-2-hydroxy-3-((2-((4-morpholinylcarbonyl)amino)ethyl)amino)propoxy)-, ((4S)-2,2-dimethyl-1,3-dioxolan-4-yl)methyl ester
- (-)-((S)-2,2-Dimethyl-1,3-dioxolan-4-yl)methyl p-((S)-2-hydroxy-3-((2-(4-morpholinecarboxamido)ethyl)amino)propoxy)hydrocinnamate
- Benzenepropanoic acid, 4-(2-hydroxy-3-((2-((4-morpholinylcarbonyl)amino)ethyl)amino)propoxy)-, (2,2-dimethyl-1,3-dioxolan-4-yl)methyl ester, (S-(R*,R*))-

Landiolol hydrochloride- 144481-98-1
- Landiolol HCl
- ONO 1101 hydrochloride
- Onoact
Landiolol, sold under the brand name Onoact among others, is a medication used for the treatment of tachycardia, atrial fibrillation, and atrial flutter.[1][4] It is a beta-adrenergic blocker;[4] an ultra short-acting, β1-superselective intravenous adrenergic antagonist, which decreases the heart rate effectively with less negative effect on blood pressure or myocardial contractility.[6][7] In comparison to other beta blockers, landiolol has the shortest elimination half-life (3 to 4 minutes), ultra-rapid onset of effect (heart rate begins to decrease immediately after completion of administration), and predictable effectiveness with inactive metabolites (heart rate returns to baseline levels at 30 min after completion of landiolol hydrochloride administration).[8] The pure S-enantiomer structure of landiolol is believed to develop less hypotensive side effects in comparison to other β-blockers. This has a positive impact on the treatment of patients when reduction of heart rate without decrease in arterial blood pressure is desired.[9] It is used as landiolol hydrochloride.
Landiolol was approved for medical use in Japan in 2002,[10][11] in Canada in November 2023,[1] and in the United States in November 2024.[12][13][14]
Syn
- Landiolol 1 is a potent cardioselective beta-blocker with ultrarapid action, used as an arrhythmic agent in the form of the hydrochloride salt.
- [0004]The synthesis of Landiolol 1 is disclosed in US 5013734 , JP 3302647 , CN 100506814 , JP 2539734 and Chemical & Pharmaceutical Bulletin 1992, 40 (6) 1462-1469. The main synthetic route for the preparation of Landiolol is reported in the following scheme:

The synthesis of landiolol appeared in an earlier patent in 1990. Esterification of 3- (4-hydroxyphenyl)propionic acid (141) with 2,2-dimethyl- 1,3-dioxolan-4-ylmethyl chloride (142) in DMSO gave desired ester 143 in 57% yield. Treatment of phenol 143 with bromo epoxide 144 in the present of K2CO3 afforded ether 145 in 76% yield. Epoxide 145 was then reacted with free amine 146 via a neucleophilic ring opening process to provide landiolol (14).

Yield:144481-98-1 95.9%
Reaction Conditions:
with hydrogenchloride in ethyl acetate at 5 – 10; for 2 h;
Steps:
1.6 Preparation of Lantilolol Hydrochloride
Add Lantilolol (10g, 19.62mmol) and 100mL of ethyl acetate to the reaction flask. The temperature of the ice-water bath is lowered below 5 ° C, and a temperature of 10-18 ° C is added dropwise to a 15-18% HCl-ethyl acetate solution 4.63g A large amount of solid was gradually precipitated, dripped, stirred below 10 ° C for 2h, filtered, washed with ethyl acetate, and dried under vacuum at 50 ° C to obtain 10.28 g of a white solid with a yield of 95.9% and an HPLC purity of 99.85%.
References:
CN110483470,2019,A Location in patent:Paragraph 0031; 0045-0047
EP2687521,2014,A1
https://patents.google.com/patent/EP2687521B1/en

- [0025]Typically, to activate the salen catalyst, preferably (R,R)-N,N’-bis(3,5-di-tert-butylsalicylidene)-1,2-ciclohexanediamino cobalt 16 is reacted with 1.0 ÷ 3.0 equivalents of a carboxylic acid, preferably 4-nitrobenzoic acid 17, preferably 1.5 ÷ 2.5 equivalents. The reaction is carried out in a polar aprotic solvent, preferably dichloromethane, at a temperature of 10 ÷ 40°C, preferably at a temperature of 20 ÷ 30°C. 4 ÷ 15 Volumes of solvent are used, preferably 7 ÷ 12 volumes with respect to the amount of (R,R)-N,N’-bis(3,5-di-tert-butylsalicylidene)-1,2-ciclohexanediamino cobalt 16. After a dark brown color appears, the solvent is removed thereby obtaining the catalyst the in active form. This is then added with 10 ÷ 100 equivalents of starting product (S)-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl 3-(4-hydroxyphenyl)propanoate 3, preferably 20 ÷ 50 equivalents, then with a polar aprotic solvent, preferably methyl tert-butyl ether (MTBE). 1 ÷ 5 Volumes of solvent are used, preferably 2 ÷ 3 volumes with respect to the amount of (S)-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl 3-(4-hydroxyphenyl)propanoate 3. Afterwards, 2.0 ÷ 3.0 equivalents of a compound of formula 4, typically epichlorohydrin, are added, preferably 2.0 ÷ 2,5 equivalents. The reaction is carried out at a temperature of 10 ÷ 40°C, preferably at a temperature of 20 ÷ 30°C. The reaction is monitored by UPLC analysis using a C18 column and water / acetonitrile containing 1% formic acid as the eluent phase. After completion of the reaction, water and toluene are added and phases are separated. The organic phase is then distilled to recover (S)-epichlorohydrin and washed with dilute sodium hydroxide. The organic phase is then concentrated to small volume, added with a polar solvent, acetonitrile or methanol, preferably acetonitrile, concentrated again to small volume to remove toluene and finally added with 5 ÷ 30 volumes of a polar solvent, such as acetonitrile or methanol, preferably acetonitrile. The suspension is filtered thus recovering the catalyst and the resulting solution can be directly used in step b, or (S)-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl 3-(4-((2R)-3-chloro-2-hydroxypropoxy)phenyl)-propanoate 5 can be isolated as an oil that can be stored at room temperature for some days. In order to obtain (S)-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl 3-(4-((2R)-3-chloro-2-hydroxypropoxy)phenyl)propanoate 5 as an oil, the solution in the polar solvent is added with decolorizing filter aid, the obtained suspension is filtered and the resulting solution is evaporated to dryness. (S)-(2,2-Dimethyl-1,3-dioxolan-4-yl)methyl 3-(4-((2R)-3-chloro-2-hydroxy-propoxy)phenyl)propanoate 5 is obtained as an oil.
- [0026]Typically, (S)-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl 3-(4-((2R)-3-chloro-2-hydroxypropoxy)phenyl)propanoate 5 obtained in step a, either isolated as an oil or directly from the polar solvent solution, is reacted with an inorganic base, preferably potassium carbonate, in an amount of 1.0 ÷ 6.0 equivalents, 3.0 ÷ 4.0 equivalents, in the presence of a ionic inorganic catalyst, preferably potassium iodide, in catalytic amounts (0.05 ÷ 0.20 eq). Thereafter, 2-(morpholine-4-carboxamido)ethanamine as base or a salt thereof, such as the oxalate or the hydrochloride, preferably the oxalate, is added in an amount of 1.0 ÷ 4.0 equivalents, preferably 2.0 ÷ 3.0 equivalents. The reaction is carried out in a polar solvent, preferably acetonitrile, at a temperature 20 ÷ 85°C, preferably 60 ÷ 85°C. 5 ÷ 30 Volumes of solvent are used, preferably 10 ÷ 20 volumes with respect to the amount of (S)-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl 3-(4-((2R)-3-chloro-2-hydroxypropoxy)-phenyl)propanoate 5. The reaction is monitored by UPLC analysis using a C 18 column and water / acetonitrile containing 1% formic acid as the eluent. After completion of the reaction, ethyl acetate and water are added and the phases separated. The organic phase is then extracted with water at pH 2 ÷ 5, preferably 3 ÷ 4. The phases are separated and the aqueous phase is extracted again with ethyl acetate at pH 8 ÷ 13, preferably 9 ÷ 12. The solvent of the organic phase is then replaced with 2 ÷ 20 volumes of a polar solvent, such as isopropanol, and the resulting solution can be directly used in step c, or Landiolol 1 can be isolated. For this purpose, the solvent is removed or replaced with a polar solvent, for example diisopropyl ether, to promote solidification, then the solvent is stripped from the resulting suspension thereby obtaining Landiolol 1 as an oil which solidifies in time.
- [0027]Typically, Landiolol 1 obtained in step b directly from the polar solvent solution or by dissolution of the isolated product is directly salified to give Landiolol hydrochloride 2, preferably with hydrochloric acid. Salification is carried out in a polar solvent, preferably isopropanol, in amounts of 2 ÷ 20 volumes of solvent, preferably 5 ÷ 10 volumes with respect to the amount of Landiolol 1. After addition of the acid, the solvent is evaporated off and the product is crystallized by adding 1 ÷ 20 volumes of a polar solvent, preferably acetone. The suspension is filtered and the solid is dried at 25 ÷ 35°C under vacuum for 12 hours to obtain Landiolol hydrochloride 2. The enantiomeric excess of the final product is analyzed using a Chiralcel OD column and hexane / ethanol as the eluent phase containing diethylamine.
- [0028]The process of the invention is particularly advantageous in that is effected without isolating any intermediates. Intermediate 5 is obtained with high purity in very high yields under very mild reactions conditions. Furthermore, the starting material 4 in which X is chlorine (epichlorohydrin), is very inexpensive and easily commercially available. The catalysts used are commercially available at low costs and can be easily recovered by simple filtration. Surprisingly, the reaction to give Landiolol 1 starting from the novel intermediate 5 in which X is chlorine provides a markedly higher yield than those obtained with most processes mentioned in the background of the invention, which conversely start from intermediate 7. The resulting Landiolol 1 can be directly converted to Landiolol hydrochloride 2 in good overall yields, with no further purifications neither intermediate steps. The resulting Landiolol hydrochloride 2 has very high enantiomeric purity.
- [0029]Furthermore, the process of the invention allows to recover (S)-epichlorohydrin 12, which is a high added value product that can also be used in the synthesis of Landiolol 1 according to the following scheme, to prepare compound 3:
- [0030]The synthesis of intermediate 15 from 12 in very high yields is described in literature in a number of publications. Some publications which the disclose it are the following: Catalysis Communications, 8(12), 2087-2095; 2007; CN100506814 ; Journal of Molecular Catalysis A: Chemical, 236(1-2), 72-76; 2005; Chinese Journal of Chemistry, 23(9), 1275-1277; 2005; Synthetic Communications, 35(11), 1441-1445; 2005; Synthetic Communications, 31(22), 3411-3416; 2001; Chemistry Letters, (11), 2019-22; 1990; Khimiya Geterotsiklicheskikh Soedinenii, (1), 33-6; 1991. The synthesis of 3 in high yields starting from 15 and 19 is described in CN100506814 . A further publication disclosing it is US5013734 . Both publications have already been mentioned in the background of the invention for the synthesis of Landiolol 1.
- [0031]The invention is illustrated in detail by the following examples.
- [0032]
- [0033]A suspension of (R,R)-N,N’-bis(3,5-di-tert-butylsalicylidene)-1,2-ciclohexanediamino cobalt (16) (50 mg, 0.0828 mmol) in MTBE (1 ml) is added with acetic acid (10 mg, 0.166 mmol). The mixture is left under stirring for 1 h at 20-25°C until a dark color appears. Afterwards, (S)-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl 3-(4-hydroxyphenyl)propanoate (3), (500 mg, 1.78 mmol), then epichlorohydrin (compound of formula 4 in which X is chlorine) (340 mg, 3.56 mmol) are added thereto. The mixture is left under stirring at 20-25°C, monitoring by UPLC. After completion of the reaction, water (5 ml) and toluene (5 ml) are added, the phases are separated and the solvent and (S)-epichlorohydrin are removed from the organic phase under reduced pressure to obtain 600 mg (90.4%) of a dark oil.
- [0034]
- [0035]A suspension of (R,R)-N,N’-bis(3,5-di-tert-butylsalicylidene)-1,2-ciclohexanediamino cobalt (16) (1,9 g, 3.19 mmol) in dichloromethane (20 ml) is added with 4-nitrobenzoic acid 17 (1.1 g, 6.38 mmol). The mixture is left under stirring for 1 h at 20-25°C until a dark color appears. The solvent is replaced with MTBE (30 ml), subsequently (S)-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl 3-(4-hydroxyphenyl)propanoate (3), (18 g, 63,8 mmol) and then epichlorohydrin (compound of formula 4 in which X is chlorine) (13,4 g, 140 mmol) are added. The mixture is left under stirring at 20-25°C, monitoring by UPLC. After completion of the reaction, toluene (300 ml) and water (150 ml) are added and the phases are separated. The organic phase is evaporated to dryness thereby recovering the enriched (S)-epichlorohydrin. Toluene (300 ml) and 10% NaOH (100 ml) are added. The phases are separated, the resulting solution is concentrated to a volume of about 50 ml, added with 100 ml of acetonitrile, concentrated to a volume of 50 ml and finally added with 250 ml of acetonitrile. Decolorizing filter aid (2.5 g) is added, the mixture is left under stirring for 15′ and the suspension is filtered. The filtrate is evaporated to dryness to obtain 23.7 g (99,6%) of a red-brownish oil.
- [0036]A suspension of (R,R)-N,N’-bis(3,5-di-tert-butylsalicylidene)-1,2-ciclohexanediamino cobalt (16) (470 mg, 0.780 mmol) in dichloromethane (5 ml) is added with 4-nitrobenzoic acid 17 (270 mg, 1.56 mmol). The mixture is left under stirring for 45′ at 20-25°C until a dark color appears. The resulting solution is concentrated to a volume of about 2 ml, added with 5 ml of MTBE, concentrated to a volume of 2 ml and finally added with 6 ml of MTBE, subsequently with (S)-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl 3-(4-hydroxyphenyl)propanoate (3), (5 g, 17.7 mmol) and then with epichlorohydrin (compound of formula 4 in which X is chlorine) (3.7 g, 38.9 mmol). The mixture is left under stirring at 20-25°C, monitoring by UPLC. After completion of the reaction, toluene (25 ml) and water (25 ml) are added and the phases are separated. The organic phase is evaporated to dryness thereby recovering the enriched (S)-epichlorohydrin. Acetonitrile (25 ml) is added and the suspension is filtered thereby recovering the catalyst. The resulting solution is concentrated to a volume of about 5 ml, added with 15 ml of toluene, then concentrated to a volume of 5 ml and finally added with 20 ml of toluene and decolorizing filter aid (20.0 g). The mixture is left under stirring for 15′ and the suspension is filtered. The filtrate is evaporated to dryness to obtain 6.1 g (92.4%) of a yellow oil.
LC-MS (ESI+) [M+H]+ = 373
1H-NMR (CDCl3) (chemical shifts expressed in ppm with respect to TMS): 1,37 (3H, s, CH3); 1,43 (3H, s, CH3); 2,65 (2H, t, J = 7 Hz, CH2-Ar); 2,83 (1H, bs, OH); 2,91 (2H, t, J = 7 Hz, CH2-CO); 3,66 – 3,81 (3H, m, CH in 4 oxolane and CH2-Cl); 4.00 – 4,25 (6H, m, CH in 4 oxolane, CH2-OCO, CH2-OAr and CH in 5 oxolane); 4,25 (1H, m, CH-OH); 6,84 and 7,13 (4H, system AA’XX’, aromatics).
13C-NMR (CDCl3) (ppm): 25,3 (CH3); 26,6 (CH3); 29,9 (CH2); 35,8 (CH2); 45,9 (CH2-Cl); 64,6 (CH2); 66,2 (CH2); 68,5 (CH2); 69,7 (CH); 73,4 (CH); 109,7; 114,5 (CH); 129,3 (CH); 133,1; 156,7; 172,6 (COOR).
Elemental analysis: C, 58.3%; H, 6.9%; Cl, 9.3%; O, 25.5%. (% calculated: C, 58.0; H, 6.8; Cl, 9.5; O, 25.7).
FT-IR (UATR, cm-1): 3456, 2987, 2936, 1733, 1612, 1512, 1372, 1241, 1154, 1041,828,741,720. - [0037]
- [0038]A suspension of (S)-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl 3-(4-((2R)-3-chloro-2-hydroxypropoxy)phenyl)propanoate (5) prepared according Example 3 (0.50 g, 0.00134 mol) in isopropanol (10 ml) is added with 2-(morpholine-4-carboxamido)ethanamino hydrochloride (18) (1.4 g, 0.00670 mol), heated to 30-35°C and dropwise added with 30% NaOH, keeping pH at 10-11. The mixture is left under stirring at 35-40°C, monitoring by UPLC. After completion of the reaction, ethyl acetate (20 ml) and water (20 ml) are added and the phases are separated. The organic phase is added with water (20 ml) and adjusted to pH 3-4 with hydrochloric acid. The phases are separated and the resulting aqueous phase is then adjusted to pH 10-11 with sodium hydroxide and re-extracted with ethyl acetate (20 ml). The solvent is then evaporated off under reduced pressure to obtain 0.38 g (55.6%) of a pale yellow oil which solidifies in time to a pale yellow solid.
- [0039]
- [0040]A solution of (S)-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl 3-(4-((2R)-3-chloro-2-hydroxypropoxy)phenyl)propanoate (5) prepared according to Example 3 (0.30 g, 0.805 mmol) in acetonitrile (6.0 ml) is added with potassium carbonate 0.45 g (3.22 mmol), and KI 0.013 g (0.0805 mmol), then refluxed for 2 h and added with 2-(morpholine-4-carboxamido)ethanamino oxalate (6) (0.64 g, 2.42 mmol). The mixture is refluxed under stirring, monitoring by UPLC. After completion of the reaction, ethyl acetate (10 ml) and water (10 ml) are added and the phases are separated. The organic phase is added with water (10 ml) and adjusted to pH 4-5 with hydrochloric acid, the phases are separated and the resulting aqueous phase is then adjusted to pH 11-12 with sodium hydroxide and re-extracted with ethyl acetate (10 ml). Then the solvent is evaporated off under reduced pressure to obtain 0.29 g (70.7%) of a pale yellow oil which solidifies in time to a pale yellow solid.
LC-MS (ESI+) [M+H]+ = 510
1H-NMR (CDCl3) (chemical shifts expressed in ppm with respect to TMS) (assigned based on the hetero correlation HSQC spectrum): 1.36 (3H, s, CH3); 1.42 (3H, s, CH3); 2.63 (2H, t, J = 7 Hz, CH2-Ar); 2.75 – 2.93 (8H, m, CH2-CO, CH-CH 2 -NH, CH2-CH 2 -NH, NH and OH); 3.35 (6H, m, 2CH2-N morpholine and CH 2 -NH); 3.65 (4H, m, 2CH2-O morpholine), 3.68 (1H, m, CH in 4 oxolane); 3.94 (2H, bd, CH2-OAr); 4.00 – 4.20 (4H, m, CH in 4 oxolane, CH2-OCO and CH in 5 oxolane); 4.25 (1H, m, CH-OH); 5.21 (1H, bt, NH carbamate); 6.83 and 7.11 (4H, system AA’XX’, aromatics).
13C-NMR (CDCl3) (ppm) (multiplicity was assigned by DEPT-135): 25.3 (CH3); 26.6 (CH3); 29.9 (CH2); 35.8 (CH2); 40.2 (CH2); 43.8 (CH2-N morpholine); 49.2 (CH2); 51.5 (CH2); 64.6 (CH2); 66.2 (CH2); 66.4 (CH2-O morpholine); 68.3 (CH); 70.3 (CH2); 73.4 (CH); 109.7; 114.4 (CH); 129.2 (CH); 132.8; 157.0; 158.0; 172.5 (COOR).
FT-IR (UATR, cm-1): 3350. 2858, 1735, 1626, 1512, 1454, 1371, 1244, 1153, 1115, 1040. 829, 733. - [0041]
- [0042]A solution of Landiolol (1) prepared according to Example 5 (100 mg, 0.196 mmol) in isopropanol (6.0 ml) is added with 18% isopropanol hydrochloric acid (40 mg, 0.197 mmol). The solvent is then evaporated off under reduced pressure and the residue is crystallized from acetone (2 ml). The suspension is filtered and the crystal is dried at 25°C for 12 h to obtain 80 mg (74.7%) of a white solid.
- [0043]
- [0044]A suspension of (R,R)-N,N’-bis(3,5-di-tert-butylsalicylidene)-1,2-ciclohexanediamino cobalt (16) (47 mg, 0.0780 mmol) in dichloromethane (1 ml) is added with 4-nitrobenzoic acid 17 (27 mg, 0.156 mmol). The mixture is left under stirring for 45′ at 20-25°C until a dark color appears. The resulting solution is concentrated to a volume of about 0.5 ml, added with 0.5 ml of MTBE, concentrated to a volume of 0.5 ml and finally added with 0.5 ml of MTBE, then with (S)-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl 3-(4-hydroxyphenyl)propanoate (3), (0.5 g, 1,77 mmol) and then with epichlorohydrin (compound of formula 4 in which X is chlorine) (0.37 g, 3,89 mmol). The mixture is left under stirring at 20-25°C, monitoring by UPLC. After completion of the reaction, toluene (10 ml) and water (10 ml) are added and the phases are separated. The organic phase is evaporated recovering the enriched (S)-epichlorohydrin, then added again with toluene (10 ml) and washed with 10% NaOH (10 ml). The resulting solution is concentrated to a volume of about 2 ml, added with 5 ml of acetonitrile, concentrated to a volume of 2 ml and finally added with 10 ml of acetonitrile). The suspension is filtered thus recovering the catalyst and the solution is added with potassium carbonate 0.79 g (5,64 mmol), and KI 0.026 g (0.161 mmol), refluxed for 2 h, then added with 2-(morpholine-4-carboxamido)ethanamino oxalate (6) (1.07 g, 4.03 mmol). The mixture is refluxed under stirring, monitoring by UPLC. After completion of the reaction, ethyl acetate (20.0 ml) and water (20 ml) are added and the phases are separated. The organic phase is then adjusted to pH 4-5 with hydrochloric acid and extracted with water (20 ml). The phases are separated and the resulting aqueous phase is then adjusted to pH 11-12 with sodium hydroxide and re-extracted with ethyl acetate (20 ml). The resulting solution is concentrated to a volume of about 5 ml, added with 20 ml of isopropanol, concentrated to a volume of 5 ml and finally added with 30 ml of isopropanol, then 18% isopropanol hydrochloric acid (0.24 g, 1.18 mmol). The solvent is then evaporated off under reduced pressure and the residue is crystallized from acetone (10 ml). The suspension is filtered and the crystal is dried at 25°C for 12 h to obtain 0.48 g (49.7% total, enantiomeric purity: 99.8%) of a white solid.
m.p.: 126°C (from literature 123-127°C)
LC-MS (ESI+) [M+H]+ = 510
FT-IR (UATR, cm-1): 3265, 2941, 2789, 2419, 1723, 1615, 1538, 1515, 1435, 1371, 1260. 1242, 1196, 1118, 1047, 887, 838, 821, 771.
Medical uses
Landiolol is indicated as an antiarrhythmic agent to treat
- Supraventricular tachycardia and for the rapid control of ventricular rate in patients with atrial fibrillation or atrial flutter in perioperative, postoperative, or other circumstances where short-term control of the ventricular rate with a short acting agent is desirable.
- Non-compensatory sinus tachycardia where, in the physician’s judgment the rapid heart rate requires specific intervention.
Landiolol has been approved for the treatment of ventricular fibrillation or ventricular tachycardia in Japan.
In the United States, landiolol is indicated for the short-term reduction of ventricular rate in adults with supraventricular tachycardia including atrial fibrillation and atrial flutter.[4]
Landiolol can be used as first-line treatment for acute ventricular rate control in patients with atrial fibrillation (Level I recommendation- 2020 Guidelines of the European Society of Cardiology[15]).
Mode of action
The drug acts as an ultra-short-acting β1-selective blocking agent. It is rapidly hydrolyzed to an inactive form by both carboxylesterase in the liver and pseudocholinesterase in the plasma, resulting in an elimination half-life of about four minutes.[16] Landiolol is a highly selective beta-1-adrenoreceptor antagonist (the selectivity for beta-1-receptor blockade is 255 times higher than for beta-2-receptor blockade) that inhibits the positive chronotropic effects of the catecholamines adrenaline and noradrenaline on the heart, where beta-1-receptors are predominantly located. Landiolol, as other beta-blockers, is thought to reduce the sympathetic drive, resulting in reduction in heart rate, decrease in spontaneous firing of ectopic pacemakers, slowing the conduction and increase the refractory period of the AV node. Landiolol does not exhibit any membrane-stabilizing activity or intrinsic sympathomimetic activity in vitro. In preclinical and clinical studies, landiolol controlled tachycardia in an ultra-short acting manner with a fast onset and offset of action and further demonstrated anti-ischaemic and cardioprotective effects.[17] To date, landiolol has the shortest plasma half-time and the highest cardio-selectivity among β-blockers in clinical use. The selectivity of landiolol for β1-receptor blockade is 255 times higher than for β2-receptor blockade. In comparison, Metoprolol, has a much less cardioselectivity (landiolol is 100 times more cardioselective than metoprolol,[18] and 8 times more cardioselectove than esmolol[19]), and sixty times longer half-life (3–4 hours comparing to 3–4 minutes in case of landiolol). FDA points out that CYP2D6 poor metabolizers will have decreased cardioselectivity for metoprolol due to increased metoprolol blood levels, since the gene variation reduces the conversion of metoprolol to inactive metabolites leading to almost 5-fold higher plasma concentrations of metoprolol.[20]
Activation of β2 adrenergic receptors contributes to bronchial dilation and acceleration of alveolar fluid clearance in the pulmonary airway system. Consequently, a cardio-selective β1-blocker with limited effect on β2-receptor decreases the heart rate without the pulmonary adverse effects in patients with COPD or Asthma. Pharmacological stimulation of β2 receptors increases coronary blood flow in healthy humans and in patients with mildly atherosclerotic coronary arteries. Thus, not only does a cardio-selective β1-blocker reduce myocardial oxygen demand during exercise, but it also unveils β2-receptor-mediated coronary exercise hyperemia, while reducing the heart rate selectively. Interestingly, landiolol does not possess any sodium and calcium antagonistic properties, which makes it a more suitable cardio-selective β-blocker for patients with heart failure due to its lesser potency for negative inotropy, while offering higher potency for heart rate reduction. Contrary to landiolol, exposure to other β-blockers such as esmolol amplifies the re-expression of β-receptors which explains the drug tolerance effect seen during long-term esmolol infusion. Long term exposure of cells to betablockers which act as pharmacochaperones will raise the total surface level of β1-adrenergic receptors, resulting in exaggerating responses to endogenous agonists such as catecholamines, if the treatment is suddenly stopped. This phenomenon has been described as the betablocker withdrawal rebound. However, landiolol lacks appreciable pharmacochaperoning activity, as landiolol can hardly permeate cell membranes due to its large polar surface area.
Biotransformation
Landiolol is metabolised via hydrolysis of the ester moiety. In vitro and in vivo data suggest that landiolol is mainly metabolised in the plasma by pseudocholinesterases and carboxylesterases. Hydrolysis releases a ketal (the alcoholic component) that is further cleaved to yield glycerol and acetone, and the carboxylic acid component (metabolite M1), which subsequently undergoes beta-oxidation to form metabolite M2 (a substituted benzoic acid). The beta-1-adrenoreceptor blocking activity of landiolol metabolites M1 and M2 is 1/200 or less of the parent compound indicating a negligible effect on pharmacodynamics taking into account the maximum recommended landiolol dose and infusion duration.
Neither landiolol nor the metabolites M1 and M2 showed inhibitory effects on the metabolic activity of different cytochrome P450 molecular species (CYP1A2, 2C9, 2C19, 2D6 and 3A4) in vitro. The cytochrome P450 content was not affected in rats after repeated intravenous administration of landiolol. There are no data on a potential effect of landiolol or its metabolites on CYP P450 induction or time dependent inhibition available.
| IV β-Blocker | max. elimination half-life (min) | cardio-selectivity (β1/β2) | metabilization |
|---|---|---|---|
| Landiolol | 4 | 250 | pseudocholinesterases |
| Esmolol | 9 | 30 | ery-esterases |
| Metoprolol | 420 | 3 | cytochrom P2D6 (Leber) |
History
The beneficial effects of landiolol have been demonstrated in over sixty clinical trials (pubmed search -August 2018). Landiolol was generally well tolerated, with a relatively low risk of hypotension and bradycardia. Most clinical trials with landiolol have been conducted in peri-operative settings for the treatment or prophylaxis of supraventricular tachycardia or tachyarrhythmia before or after cardiac and non-cardiac surgeries. Randomized clinical trials have been published to compare landiolol with placebo<[21][22][23] diltiazem,[24] and amiodaron[25] in patients with or without heart failure. Case reports on the use of landiolol after myocardial infarction,[26] refractory electrical storm[27] have been published. The fast turnover of landiolol will diminish most adverse events due to self-limiting administration. Landiolol may be cardio-protective in septic rats by normalizing coronary microcirculation through blockage of sepsis-induced decrease in expression of VEGF signaling system but independent of inflammatory cytokines.
The efficacy and safety of landiolol in septic shock has been investigated in a multi-center prospective randomized controlled trial, and the results of the study have been published in the renown Journal Lancet Respiratory in 2020, demonstrating clinical impact of landiolol in sepsis patients through significant reduction of new-onset arrhythmia and keeping the patients within the target heart rate range.
Furthermore, landiolol demonstrated a positive clinical impact regarding ventilation-free days, ICU-free days and hospital-free days. Patients in the landiolol group had a survival rate of 88% by day 28, in contrast to a mortality rate of 20% in the control group by day 28. These are very important findings which may include landiolol in the standard of care for sepsis patients, since tachycardia and atrial fibrillation are key prognostic factors for sepsis. Additionally, tachycardia exceeding 100 beats per min (bpm) on admission to an intensive care unit (ICU) is a risk factor for worsening prognosis.[28]
A publication in the Journal of Cardiology illustrated in a prospective real-world setting, the safety and effectiveness of landiolol for the treatment of atrial fibrillation or atrial flutter in chronic heart failure (over one thousand patients at 209 medical institutions throughout Japan). In this survey, which is one of the largest studies ever performed in patients with chronic heart failure requiring intravenous rate control, report of serious hypotension was in less than 1% of patients, which highlights the cardio-selectivity of landiolol with limited effect on blood pressure. Noteworthy, over 70% of patients were in the NYHA class III or IV (35% NYHA IV), and close to 50% had a LVEF below 40%. The median time to first return to sinus rhythm after administration of landiolol was 14 hours, and the median highest infusion rate was 3 μg/kg/min.[29]
The excellent tolerance of landiolol at lower dosage (3–5 μg/kg/min) allows to initiate prophylactic use during surgery and post-operatively. Landiolol prophylaxis is associated with reduced incidence of postoperative atrial fibrillation without triggering adverse events related to a beta-blockade. Optimized infusion scheme with continuing landiolol infusion in the post-operative period seems to be associated with better response, while infusion limited to the intraoperative period may not be sufficient[30]
Society and culture
Legal status
Landiolol was approved for medical use in Japan in 2002,[10] in Canada in November 2023,[1] and in the United States in November 2024.[13]
Brand names
It is sold under various brand names including Rapibloc, Raploc, Runrapiq, Landibloc, Onoact, Corbeta, and Rapiblyk.
References
- ^ Jump up to:a b c d “Summary Basis of Decision for Sibboran”. Health Canada. 2 July 2024. Retrieved 12 October 2024.
- ^ “Details for: Sibboran”. Health Canada. 20 November 2023. Retrieved 3 March 2024.
- ^ “Regulatory Decision Summary for Sibboran”. Drug and Health Products Portal. 21 December 2022. Retrieved 2 April 2024.
- ^ Jump up to:a b c d https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/217202s000lbl.pdf
- ^ “List of nationally authorised medicinal products Active substance: landiolol” (PDF). Procedure no.: PSUSA/00010570/201802. Retrieved 12 October 2024.
- ^ Ikeshita K, Nishikawa K, Toriyama S, Yamashita T, Tani Y, Yamada T, et al. (2008). “Landiolol has a less potent negative inotropic effect than esmolol in isolated rabbit hearts”. Journal of Anesthesia. 22 (4): 361–6. doi:10.1007/s00540-008-0640-4. PMID 19011773. S2CID 5731527.
- ^ Wada Y, Aiba T, Tsujita Y, Itoh H, Wada M, Nakajima I, et al. (April 2016). “Practical applicability of landiolol, an ultra-short-acting β1-selective blocker, for rapid atrial and ventricular tachyarrhythmias with left ventricular dysfunction”. Journal of Arrhythmia. 32 (2): 82–8. doi:10.1016/j.joa.2015.09.002. PMC 4823575. PMID 27092187.
- ^ Atarashi H, Kuruma A, Yashima M, Saitoh H, Ino T, Endoh Y, et al. (August 2000). “Pharmacokinetics of landiolol hydrochloride, a new ultra-short-acting beta-blocker, in patients with cardiac arrhythmias”. Clinical Pharmacology and Therapeutics. 68 (2): 143–50. doi:10.1067/mcp.2000.108733. PMID 10976545. S2CID 46146913.
- ^ Iguchi S, Iwamura H, Nishizaki M, Hayashi A, Senokuchi K, Kobayashi K, et al. (June 1992). “Development of a highly cardioselective ultra short-acting beta-blocker, ONO-1101”. Chemical & Pharmaceutical Bulletin. 40 (6): 1462–9. doi:10.1248/cpb.40.1462. PMID 1356643.
- ^ Jump up to:a b “Ono Submits an Application of Onoact for Intravenous Infusion 50mg/150mg, a Short-Acting Selective β1 Blocker, in Japan for Additional Indication of Tachyarrhythmia in Pediatric Patients with Low Cardiac Function for a Partial Change in Approved Items of”. Ono Pharmaceutical. 28 October 2021. Retrieved 29 November 2024.
- ^ “A Short-Acting Selective β1 Blocker, Onoact for Intravenous Infusion 50mg/150mg Approved for Additional Indication of Tachyarrhythmia in Pediatric Patients with Low Cardiac Function in Japan”. Ono Pharmaceutical (Press release). 24 August 2022. Retrieved 28 November 2024.
- ^ “U.S. FDA Approves AOP Health’s Rapiblyk (landiolol) for Atrial Fibrillation and Atrial Flutter in the Critical Care Setting” (Press release). AOP Health. 27 November 2024. Retrieved 28 November 2024 – via Business Wire.
- ^ Jump up to:a b “Novel Drug Approvals for 2024”. U.S. Food and Drug Administration (FDA). 1 October 2024. Retrieved 29 November 2024.
- ^ New Drug Therapy Approvals 2024 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2025. Archived from the original on 21 January 2025. Retrieved 21 January 2025.
- ^ Hindricks G, Potpara T, Dagres N, Arbelo E, Bax JJ, Blomström-Lundqvist C, et al. (February 2021). “2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS): The Task Force for the diagnosis and management of atrial fibrillation of the European Society of Cardiology (ESC) Developed with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC”. European Heart Journal. 42 (5): 373–498. doi:10.1093/eurheartj/ehaa612. hdl:1887/3279676. PMID 32860505.
- ^ Circ J. 2016 Apr 25;80(5):1106-7
- ^ “Rapibloc Summary of Product Characteristics” (PDF). Archived from the original (PDF) on 16 June 2019. Retrieved 11 September 2018.
- ^ Baker JG (February 2005). “The selectivity of beta-adrenoceptor antagonists at the human beta1, beta2 and beta3 adrenoceptors”. British Journal of Pharmacology. 144 (3): 317–22. doi:10.1038/sj.bjp.0706048. PMC 1576008. PMID 15655528.
- ^ Okajima M, Takamura M, Taniguchi T (August 2015). “Landiolol, an ultra-short-acting β1-blocker, is useful for managing supraventricular tachyarrhythmias in sepsis”. World Journal of Critical Care Medicine. 4 (3): 251–7. doi:10.5492/wjccm.v4.i3.251. PMC 4524822. PMID 26261777.
- ^ Dean L (2017). “Metoprolol Therapy and CYP2D6 Genotype”. In Pratt VM, McLeod HL, Rubinstein WS, et al. (eds.). Medical Genetics Summaries. National Center for Biotechnology Information (NCBI). PMID 28520381. Bookshelf ID: NBK425389.
- ^ Ojima T, Nakamori M, Nakamura M, Katsuda M, Hayata K, Kato T, et al. (July 2017). “Randomized clinical trial of landiolol hydrochloride for the prevention of atrial fibrillation and postoperative complications after oesophagectomy for cancer”. The British Journal of Surgery. 104 (8): 1003–1009. doi:10.1002/bjs.10548. PMID 28444964. S2CID 1079409.
- ^ Xiao J, He P, Zou Q, Zhao Y, Xue Z, Deng X, et al. (March 2015). “Landiolol in the treatment of the intraoperative supraventricular tachycardia: a multicenter, randomized, double-blind, placebo-controlled study”. Journal of Clinical Anesthesia. 27 (2): 120–8. doi:10.1016/j.jclinane.2014.07.003. PMID 25434501.
- ^ Sezai A, Minami K, Nakai T, Hata M, Yoshitake I, Wakui S, et al. (June 2011). “Landiolol hydrochloride for prevention of atrial fibrillation after coronary artery bypass grafting: new evidence from the PASCAL trial”. The Journal of Thoracic and Cardiovascular Surgery. 141 (6): 1478–87. doi:10.1016/j.jtcvs.2010.10.045. PMID 21269646.
- ^ Sakamoto A, Kitakaze M, Takamoto S, Namiki A, Kasanuki H, Hosoda S (2012). “Landiolol, an ultra-short-acting β₁-blocker, more effectively terminates atrial fibrillation than diltiazem after open heart surgery: prospective, multicenter, randomized, open-label study (JL-KNIGHT study)”. Circulation Journal. 76 (5): 1097–101. doi:10.1253/circj.CJ-11-1332. PMID 22361918.
- ^ Shibata SC, Uchiyama A, Ohta N, Fujino Y (April 2016). “Efficacy and Safety of Landiolol Compared to Amiodarone for the Management of Postoperative Atrial Fibrillation in Intensive Care Patients”. Journal of Cardiothoracic and Vascular Anesthesia. 30 (2): 418–22. doi:10.1053/j.jvca.2015.09.007. PMID 26703973.
- ^ Kiyokuni M, Konishi M, Sakamaki K, Kawashima C, Narikawa M, Doi H, et al. (October 2016). “Beneficial effect of early infusion of landiolol, a very short-acting beta-1 adrenergic receptor blocker, on reperfusion status in acute myocardial infarction”. International Journal of Cardiology. 221: 321–6. doi:10.1016/j.ijcard.2016.07.076. PMID 27404699.
- ^ Kanamori K, Aoyagi T, Mikamo T, Tsutsui K, Kunishima T, Inaba H, et al. (2015). “Successful Treatment of Refractory Electrical Storm With Landiolol After More Than 100 Electrical Defibrillations”. International Heart Journal. 56 (5): 555–7. doi:10.1536/ihj.15-102. PMID 26346519.
- ^ Kakihana Y, Nishida O, Taniguchi T, Okajima M, Morimatsu H, Ogura H, et al. (2020). “Efficacy and safety of landiolol, an ultra-short-acting β1-selective antagonist, for treatment of sepsis-related tachyarrhythmia (J-Land 3S): A multicentre, open-label, randomised controlled trial”. The Lancet Respiratory Medicine. 8 (9): 863–872. doi:10.1016/S2213-2600(20)30037-0. PMID 32243865.
- ^ Yamashita T, Nakasu Y, Mizutani H, Sumitani K (2019). “A prospective observational survey on landiolol in atrial fibrillation/Atrial flutter patients with chronic heart failure – AF-CHF landiolol survey”. Journal of Cardiology. 74 (5): 418–425. doi:10.1016/j.jjcc.2019.05.012. PMID 31255463.
- ^ Balik M, Sander M, Trimmel H, Heinz G (2018). “Landiolol for managing post-operative atrial fibrillation”. European Heart Journal Supplements. 20 (Suppl A): A10 – A14. doi:10.1093/eurheartj/sux036. PMC 5909769. PMID 30188958.
Further reading
Shiga T (June 2022). “Benefits and safety of landiolol for rapid rate control in patients with atrial tachyarrhythmias and acute decompensated heart failure”. European Heart Journal Supplements. 24 (Suppl D): D11 – D21. doi:10.1093/eurheartjsupp/suac023. PMC 9190747. PMID 35706898.
- Rao SJ, Kanwal A, Kanwal A, Danilov A, Frishman WH (2024). “Landiolol: An Ultra-Short-Acting β-Blocker”. Cardiology in Review. 32 (5): 468–472. doi:10.1097/CRD.0000000000000555. PMID 37185629.
| showvteBeta blockers (C07) |
|---|
| Clinical data | |
|---|---|
| Trade names | Onoact, others |
| Other names | ONO-1101 |
| AHFS/Drugs.com | Rapiblyk |
| License data | US DailyMed: Landiolol |
| Routes of administration | Intravenous |
| Drug class | Antiarrhythmic |
| ATC code | C07AB14 (WHO) |
| Legal status | |
| Legal status | CA: ℞-only[1][2][3]US: ℞-only[4]Rx-only[5] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 133242-30-5 144481-98-1 |
| PubChem CID | 114905164457 |
| ChemSpider | 102855 |
| UNII | 62NWQ924LHG8HQ634Y17 |
| KEGG | D12410 D01847 |
| CompTox Dashboard (EPA) | DTXSID10158026 |
| Chemical and physical data | |
| Formula | C25H39N3O8 |
| Molar mass | 509.600 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
/////Landiolol, Rapiblyk, FDA 2024, APPROVALS 2024, supraventricular tachycardia, 133242-30-5, ONO-1101, Ono 1101, WHO 7516
Iomeprol



Iomeprol
- 78649-41-9
- Iomeprolum
- Iomeron
- Iomeprolo
WeightAverage: 777.089
Monoisotopic: 776.8541
Chemical FormulaC17H22I3N3O8
FDA APPROVED, 11/27/2024, Iomervu, For use as a radiographic contrast agent
1-N,3-N-bis(2,3-dihydroxypropyl)-5-[(2-hydroxyacetyl)-methylamino]-2,4,6-triiodobenzene-1,3-dicarboxamide
- N1,N3-Bis(2,3-dihydroxypropyl)-5-(2-hydroxy-N-methylacetamido)-2,4,6-triiodoisophthalamide
- 1-N,3-N-bis(2,3-dihydroxypropyl)-5-[(2-hydroxyacetyl)-methylamino]-2,4,6-triiodobenzene-1,3-dicarboxamide
- DTXCID2028987
- E7337
- N,N’-bis(2,3-dihydroxypropyl)-5-[glycoloyl(methyl)amino]-2,4,6-triiodoisophthalamide
Iomeprol, sold under the brand name Imeron among others, is a medication used as a radiocontrast agent in X-ray imaging.[1][2][3]
Iomeprol was approved for medical use in the United States in November 2024.[1][4][5]
Experimental Properties
| Property | Value | Source |
|---|---|---|
| melting point (°C) | 285-291 | https://www.chemos.de/import/data/msds/GB_en/78649-41-9-A0017152-GB-en.pdf |
| boiling point (°C) | 198 | https://datasheets.scbt.com/sds/eghs/en/sc-211652.pdf |
the first synthesis method is to take 5-amino-2, 4, 6-triiodoisophthalic acid as a starting material, methylate amino to chlorinate the starting material to prepare diacyl chloride, then use acetoxy acetyl chloride to carry out acylation reaction, then carry out amidation reaction with 3-amino-1, 2-propylene glycol, and finally use sodium hydroxide to hydrolyze the product to obtain iomeprol, as shown in the synthesis scheme 1
U.S. Patent No. 4,364,921 discloses three preparation processes of iopromide. One of them is shown in the following reaction scheme 1.
[Reaction scheme 1]

U.S. patent No. 4,364,921 are those basically under the same concept as shown in the following reaction schemes 3 and 4.
[Reaction scheme 3]

[Reaction scheme 4]

References:

M F C Co., Ltd.;Park Yung-ho;Hwang Seong-gwan;Park Jang-ha;Seo Rak-seok;Kim Gyeong-deok KR2020/77762, 2020, A Location in patent:Paragraph 0133-0136
Yield:78649-41-9 95%
Reaction Conditions:
with sodium hydroxide in N,N-dimethyl acetamide at 10 – 60; for 16 h;
Steps:
6 Synthesis of Iomeprole
Prepared N,N-bis(2,3-dihydroxypropyl)-5-(2-hydroxyacetamido)-2,4,6-triiodoisophthalamide 20 g (0.026 mol) in a reactor and 60 g of N,N-dimethylacetamide was added, followed by heating and stirring at 60 °C to dissolve.1.3 g (0.033 mol) of caustic soda was added to the reactor and stirred to dissolve. The reactor was cooled to 10 °C or less, and 4.5 g (0.032 mol) of iodomethane was added to the reactor, followed by stirring at 25 °C for 14 to 16 hours. After confirming the completion of the reaction, acetic acid was added to neutralize the reaction solution.The solvent was removed through concentration under reduced pressure at 60 to 65 °C and 60 g of ethanol was added to crystallize. The wet body was obtained by filtration and dried under reduced pressure in an oven at 60 °C for 12 hours to obtain iomeprol (yield 95%, purity 99.8%).
Iomeprol (CAS NO.: ), with its systematic name of , N,N’-bis(2,3-dihydroxypropyl)-5-((hydroxyacetyl)methylamino)-2,4,6-triiodo-, could be produced through many synthetic methods.
Following is one of the synthesis routes:
Firstly, 5-Amino-2,4,6-triiodo-1,3-benzenedicarboxylic acid (I) is treated with chloride to produce the dichloride (II). Secondly, compound (II) is treated with acetoxyacetyl chloride to afford compound (III), which is methylated with iodomethane. Lastly, condensation with 3-amino-1,2-propanediol of the N-methyl derivative (IV) thus obtained, followed by deacetylation with alkali metal hydroxide, yields iomeprol.

PATENT
https://patents.google.com/patent/WO2009134030A1/en
The process for preparing iopromide of formula (1) according to the present invention is shown in the following reaction scheme 5.
[Reaction scheme 5]

Step 1
5-amino-2,4,6-triiodoisophthalic acid dichloride of formula (2) is used as a starting material. The compound of formula (2) is reacted with methoxyacetyl chloride in dimethylacetamide solvent to produce 5-methoxyacetylamino-2,4,6-triiodoisophthalic acid dichloride of formula (3) which is then used without an additional purification procedure for the next step. The compound of formula (3) is reacted with 2,3-dihydroxypropylamine in dimethylacetamide solvent in the presence of triethylamine to form 5-methoxyacetylamino-2,4,6-triiodoisophthalic acid (2,3-dihydroxypropyl)amide chloride of formula (4), as shown in the following reaction scheme 6.
[Reaction scheme 6]

In the above reaction scheme, if 2,3-dihydroxypropylamine is used preferably in 0.6 to 1 equivalents, more preferably in 0.7 equivalents, the compound of formula (4) can be obtained in reasonable yield with minimizing the generation of the compound of formula (5) which is a bismer by-product.
In addition, since the unreacted compound of formula (3) existing in the filtrate obtained along with the compound of formula (4) can be recycled to the next batch without an additional recovery procedure, the loss of the yield of iopromide which occurs during the removal procedure of the bismer by-product generated in a large amount in conventional process can be prevented ultimately.
Step 2
The compound of formula (4) is reacted with acetic anhydride in acetic acid solvent in the presence of sulfuric acid as a catalyst to convert into the compound of formula (19). Sulfuric acid of preferably 0.01 to 0.2 moles, more preferably 0.05 to 0.1 moles per 1 molar reaction is added at a temperature of preferably from 0 to 30°C, more preferably from 5 to 25°C. Acetic anhydride of preferably 0.19 to 1 L, more preferably 0.35 to 0.7 L per 1 molar reaction is used.
5-methoxyacetylamino-2,4,6-triiodoisophthalic acid-N,N’-bis-(2,3-diacetoxypropyl) diamide of the compound of following formula (21), which is generated by a simultaneous conversion of the bismer by-product of formula (5) already produced in the step 1 with the conversion of the compound of formula (4) into the compound of formula (19), is easily removed by the simple crystallization procedure of the compound of formula (19). That is, the by-product of the compound of formula (21) can be removed even without an additional removal procedure. It consequently means that the bismer by-product of the compound of formula (5) which is hard to remove according to conventional process can be effectively removed in the present invention even without any additional purification procedures.
[Formula 5]

[Formula 21]

Step 3
The compound of formula (19) is reacted with 2,3-dihydroxy-N-methypropylamine in dimethylacetamide solvent in the presence of triethylamine as a base to convert into the compound of formula (20). By the hydrolysis of the compound of formula (20) in aqueous NaOH solution without further purification therefor, iopromide of formula (1) can be obtained.
The present invention will be explained more specifically by the following examples. However, the examples are not intended to limit the scope of the present invention thereto.
Example 1: Synthesis of 5-methoxyacetylamino-2,4,6-triiodoisophthalic acid (2,3-dihydroxypropyl)amide chloride (Formula 4)
5-amino-2,4,6-triiodoisophthalic acid dichloride (13.7 kg, 23 mol) was dissolved in dimethylacetamide (17.2 kg) and the mixture was cooled to 15 ℃ . Methoxyacetyl chloride (3.74 kg, 34.5 mol) was added dropwise thereto for 2 hours, and then the mixture was stirred for 15 hours. After confirming the disappearance of the starting material by HPLC analysis for reaction, methylene chloride (45.7 kg) and water (11.5 kg) were subsequently added to the reaction mixture upon being stirred, and then the stirring was stopped and the layers became separated. The obtained organic layer was washed with aqueous sodium bicarbonate solution and concentrated by distillation under reduced pressure. To a solution of the obtained concentrate dissolved in dimethylacetamide (43.1 kg), triethylamine (1.95 kg, 19.32 mol) was added and then a solution of 2,3-dihydroxypropylamine (1.47 kg, 16.13 mol) dissolved in dimethylacetamide (10.78 kg) was added dropwise thereto for 5 hours with maintaining 0 to 5 ℃ . After additional 3 hours with stirring, the reaction mixture was concentrated by distillation under reduced pressure and to the concentrate, methylene dichloride (213.33 kg) was added dropwise for 5 hours to form solid. The solid was filtered and the title compound was obtained as a white solid (10.98 kg, yield 66.1 %).
1 H NMR(dmso-d6, 500MHz) 10.2,10.06(2s, 1H); 8.79, 8.71, 8.63 (3t, 1H); 4.5~4.0(br, 2H); 4.04, 4.00(2s, 2H); 3.71~3.66(m, 1H); 3.48, 3.47(2s, 3H); 3.40~3.36(m, 2H); 3.36~3.27(m, 1H); 3.2~3.09 (m, 1H)
Example 2: Synthesis of 5-methoxyacetylamino-2,4,6-triiodoisophthalic acid (2,3-diacetoxypropyl)amide chloride (Formula 19)
5-methoxyacetylamino-2,4,6-triiodoisophthalic acid (2,3-dihydroxypropyl)amide chloride (9.97 kg, 13.8 mol) was dispersed in acetic acid, and then anhydrous acetic acid (7.45 kg) was added thereto and the mixture was cooled to 5 ℃ . Sulfuric acid (135 g) was slowly added thereto and the mixture was stirred for 1 hour. To the obtained clear solution, sodium acetate trihydrate (376 g) was added and dissolved at 0 to 5 ℃ . And then water (96.6 kg) was added for 3 hours with maintaining 0 to 10 ℃ to produce solid. The produced solid was filtered and the title compound was obtained as a white solid (10.04 kg, yield 90.2 %).
1 H NMR(dmso-d6, 500MHz) 10.12, 9.99(2s, 1H); 8.91, 8.80(2t, 1H); 5.10~5.06(m, 1H); 4.32~4.27(m, 1H); 4.20~4.16(m, 1H); 4.01, 4.00(2s, 2H); 3.52~3.37(m, 2H); 3.47, 3.46(2s, 3H); 2.02(s, 6H)
Example 3: Synthesis of 5-methoxyacetylamino-2,4,6-triiodoisophthalic acid [(2,3-dihydroxy-N-methylpropyl)-(2,3-dihydroxypropyl)]diamide (iopromide)
5-methoxyacetylamino-2,4,6-triiodoisophthalic acid (2,3-diacetoxypropyl)amide chloride (7.23 kg, 8.97 mol) was dissolved in dimethylacetamide (12.6 kg), and triethylamine (1.95 kg, 19.32 mol) was added thereto and a solution of 2,3-dihydroxy-N-methylpropylamine (943 g, 8.97 mol) dissolved in dimethylacetamide (4.2 kg) was added dropwise thereto at room temperature. After additional 2 hours with stirring, the solution was concentrated by distillation under reduced pressure. To an aqueous solution of the obtained 5-methoxyacetylamino-2,4,6-triiodoisophthalic acid [(2,3-dihydroxy-N-methylpropyl)-(2,3-diacetoxypropyl)]diamide dissolved in water, a solution of sodium hydroxide (897 g, 22.43 mol) dissolved in water was added and the reaction mixture was stirred for 10 hours with maintaining 20 to 25 ℃ . The reaction solution was passed through cation exchange resin column and anion exchange resin column to produce a colorless and transparent aqueous solution. The obtained aqueous solution was distilled under reduced pressure to remove water completely, crystallized in ethanol and filtered to obtain a white crystalline iopromide (6.032 kg, yield 85 %).
1 H NMR(dmso-d6, 500MHz) 10.07, 10.03, 9.97, 9.90(4s,1H); 8.66, 8.57, 8.52(3t, 1H); 4.76~4.74(m, 1H); 4.72, 4.67(2t, 1H); 4.59~4.58(m, 1H); 4.54~4.44(m, 1H); 4.00(s, 2H); 3.89~3.88(m, 1H); 3.69~3.68(m, 2H); 3.47(s, 3H); 3.44~3.38(m, 4H); 3.23~3.17(m, 3H), 2.85~2.83(4s, 3H)
Example 4: Synthesis of 5-methoxyacetylamino-2,4,6-triiodoisophthalic acid [(2,3-dihydroxy-N-methylpropyl)-(2,3-diacetoxypropyl)]diamide (Formula 20)
5-methoxyacetylamino-2,4,6-triiodoisophthalic acid (2,3-diacetoxypropyl)amide chloride (80.65 g, 0.1 mol) was dissolved in dimethylacetamide (140.5 g), and then triethylamine (11.13 g, 0.11 mol) was added thereto and a solution of 2,3-dihydroxy-N-methylpropylamine (10.5 g, 0.1 mol) dissolved in dimethylacetamide (46.9 g) was added dropwise thereto at room temperature. After additional 2 hours with stirring, the produced solid was filtered and then the filtrate was concentrated by distillation under reduced pressure. To the obtained concentrate, diethyl ether was added dropwise to form solid. The formed solid was filtered and the title compound was obtained as a white solid (85.8 g, yield 98 %).
1 H NMR(dmso-d6, 500MHz) 10.10, 10.06, 10.00, 9.92(4s,1H); 8.93, 8.83, 8.78(3m, 1H); 5.09(br, 1H); 4.78~4.74(m, 1H); 4.62~4.58(m, 1H); 4.34~4.26(m, 1H); 4.22~4.16(m, 1H); 4.00(s, 2H); 3.89(br, 1H); 3.72~3.65,(m, 1H); 3.49~3.40(br, 2H);3.47(s, 3H); 3.48~3.38(m, 2H); 3.22~3.12,(m, 1H); 3.07~3.04(m, 1H); 2.87~2.82(m, 2H); 2.03(s, 3H); 2.02(s, 3H)
PATENT
https://patents.google.com/patent/RU2563645C2/ru
Scheme 3

Example 1Preparation of a compound of formula 2 in which both substituents R and R’ are -NH-CH
2 -CH(OH)CH
2 OH groups using a starting solution heated to 60
° C.In a 2 L four-necked jacketed reactor equipped with a mechanical stirrer, reflux condenser and combination pH/temperature electrode, an aqueous solution of the sodium salt of 3,5-disubstituted phenol 1 corresponding to a phenol concentration of 22.8% (w/w) (1175 g solution; 0.816 mol; pH 9.6) was heated to 60 °C and then solid I
2 (250.6 g; 0.988 mol) was added in one portion. When the pH spontaneously dropped to 5, a 50% (w/w) aqueous solution of HIO
3 (173.6 g; 0.494 mol) was slowly added over 2 h. The reaction mixture was maintained at 60 °C for an additional 4 h, during which time the pH spontaneously remained in the range of 5-5.5. The red solution was cooled to 25°C and quenched by adding 18% (w/w) aqueous sodium bisulfite solution until the color was lost and the oxidation-reduction potential, measured with a suitable electrode, reached a stable negative value in the range from 0 to -20 mV.During quenching of the reaction mixture, the pH was maintained at 5 by adding minimal amounts of 30% (w/w) aqueous NaOH solution.HPLC analysis (the results of which are shown in Fig. 1) showed the degree of conversion of the starting compound to 3,5-disubstituted-2,4,6-triiodophenol 2b to be >98% (by area % on the HPLC chromatogram), and the resulting solution was used in the next stage of the synthesis without any further processing.Example 2Preparation of a compound of formula 2 in which both substituents R and R’ are -NH-CH
2 -CH(OH)CH
2 OH groups using a starting solution heated to 40
° CIn a 2 L jacketed four-neck reactor equipped with a mechanical stirrer, reflux condenser and combination pH/temperature electrode, solid I
2 (250.6 g, 0.988 mol) was added in one portion to an aqueous solution of the sodium salt of 3,5-disubstituted phenol 1 corresponding to a phenol concentration of 22.8% (w/w) (1175 g solution, 0.816 mol; pH 9.6) heated to 40 °C. When the pH spontaneously dropped to 5, a 50% (w/w) aqueous solution of HIO
3 (173.6 g, 0.494 mol) was slowly added over 3 h. The reaction mixture was then heated for 2 h at 40°C, 1 h at 50°C and 1 h at 60°C, during which time the pH spontaneously remained in the range of 5-5.5. The resulting red solution was cooled to 25°C, the pH was adjusted to 7 and maintained at this level by adding 30% (w/w) aqueous NaOH solution during quenching, which was carried out by adding 18% (w/w) aqueous sodium bisulfite solution until the color was lost and the oxidation-reduction potential, measured with a suitable electrode, reached stable negative values in the range of -20 to -50 mV.HPLC analysis showed the conversion of the starting compound to 3,5-disubstituted-2,4,6-triiodophenol 2b to be >98% (based on area % of the HPLC chromatogram), and the resulting solution was used in the next step of the synthesis without any further treatment.Example 3Preparation of a compound of formula 2 in which both substituents R and R’ are -NH-CH
2 -CH(OH)CH
2 OH groups using a stock solution heated to 30
° C and quenching the reaction mixture with bisulfite at pH5 in the final stepIn a 4 L four-necked jacketed reactor equipped with a mechanical stirrer, reflux condenser and combination pH/temperature electrode, an aqueous solution of the sodium salt of 3,5-disubstituted phenol 1 corresponding to a phenol concentration of 22.8% (w/w) (1175 g of solution; 0.816 mol; pH 9.6) was diluted with H
2 O (1054 g), heated to 30 °C and then solid I
2 (250.6 g; 0.988 mol) was added in one portion. When the pH spontaneously dropped to 5, a 50% (w/w) aqueous solution of HIO
3 (173.6 g; 0.494 mol) was slowly added over 4 h. The temperature of the reaction mixture was raised to 60°C and maintained at this temperature for a further 4 h, with the pH spontaneously remaining in the range of 5-5.5. The resulting red solution was cooled to 25°C and quenched by adding 18% (w/w) aqueous sodium bisulfite solution, maintaining the pH at 5 by adding 30% (w/w) aqueous NaOH solution until the color was lost and the oxidation-reduction potential, measured with a suitable electrode, reached stable negative values in the range of 0 to -20 mV.HPLC analysis showed the conversion of the starting compound to 3,5-disubstituted-2,4,6-triiodophenol 2b to be >98% (based on area % of the HPLC chromatogram), and the resulting solution was used in the next step of the synthesis without any further treatment.Example 4Preparation of a compound of formula 2 in which both substituents R and R’ are -NH-CH
2 -CH(OH)CH
2 OH groups using a starting solution heated to 30
° C and quenching the reaction mixture with bisulfite at pH7 in the final stepIn a 4 L four-necked jacketed reactor equipped with a mechanical stirrer, reflux condenser and combination pH/temperature electrode, an aqueous solution of the sodium salt of 3,5-disubstituted phenol 1 corresponding to a phenol concentration of 22.8% (w/w) (1175 g solution; 0.816 mol; pH 9.6) was diluted with H
2 O (1054 g), heated to 30 °C and then solid I
2 (250.6 g; 0.988 mol) was added in one portion. When the pH spontaneously dropped to 5, a 50% (w/w) aqueous solution of HIO
3 (173.6 g; 0.494 mol) was slowly added over 4 h. The temperature of the reaction mixture was raised to 60°C and maintained at this temperature for a further 4 h, during which time the pH spontaneously remained in the range of 5-5.5. The resulting red solution was cooled to 25°C, the pH was adjusted to 7 and maintained at this level by adding 30% (w/w) aqueous NaOH solution during quenching, which was carried out by adding 18% (w/w) aqueous sodium bisulfite solution until the color was lost and the oxidation-reduction potential, measured with a suitable electrode, reached stable negative values in the range of -20 to -50 mV.HPLC analysis showed the conversion of the starting compound to 3,5-disubstituted-2,4,6-triiodophenol 2b to be >98% (based on area % of the HPLC chromatogram), and the resulting solution was used in the next step of the synthesis without any further treatment.Example 5Preparation of a compound of formula 2, in which both substituents R and R’ are -NH-CH
2 -CH(OH)CH
2 OH groups, using a starting solution at room temperature (approximately 20
° C)In a 4 L four-necked jacketed reactor equipped with a mechanical stirrer, reflux condenser and combination pH/temperature electrode, an aqueous solution of 3,5-disubstituted phenol 1 sodium salt corresponding to a phenol concentration of 22.8% (w/w) (1175 g solution; 0.816 mol; pH 9.6) was first diluted with H
2 O (1054 g) maintaining the temperature at 20 °C, and then solid I
2 (250.6 g; 0.988 mol) was added in one portion. The resulting solution was then heated to 40 °C and when the pH spontaneously dropped to 5, a 50% (w/w) aqueous solution of HIO
3 (173.6 g; 0.494 mol) was slowly added over 4 h . The temperature of the reaction mixture was raised to 60°C over 2 h and maintained at this temperature for a further 3 h, during which time the pH spontaneously remained in the range of 5-5.5. The red solution was then cooled to 25°C, the pH was adjusted to 7 and maintained at this level by adding 30% (w/w) aqueous NaOH solution, and quenched with sodium bisulfite (18% (w/w) aqueous solution) until color loss and stable negative values (in the range of -20 to -50 mV) of the oxidation-reduction potential were reached, measured with suitable redox electrodes.HPLC analysis showed the conversion of the starting compound to 3,5-disubstituted-2,4,6-triiodophenol 2b to be >98% (based on area % of the HPLC chromatogram), and the resulting solution was used in the next step of the synthesis without any further treatment.Example 6Preparation of a compound of formula 2 in which the substituent R is a -NH-CH
2 -CH(OH)CH
2 OH group and R’ is -NH-CH(CH
2 OH)
2 using a starting solution heated to 60
° C.In a 1 L jacketed four-neck reactor equipped with a mechanical stirrer, reflux condenser, and combination pH/temperature electrode, N-(2,3-dihydroxypropyl)-N’-[2-hydroxy-1-(hydroxymethyl)ethyl]-5-hydroxy-1,3-benzenedicarboxamide (100.3 g, 0.305 mol) was dissolved in H
2 O (430 g) and converted to the corresponding sodium salt by adding 30% (w/w) NaOH (40.6 g, 0.305 mol) (pH 9.5). The solution was heated to 60 °C and solid I
2 (93.1 g, 0.367 mol) was added in one portion; When the pH spontaneously dropped to 5, 50% (w/w) aqueous HIO3 (64.5 g, 0.183 mol) was added slowly over 2 h
. The reaction temperature was maintained at 60 °C for a further 4 h, during which time the pH of the reaction mixture spontaneously remained in the range 5-5.5. The resulting red solution was cooled to 25 °C and quenched by adding 18% (w/w) aqueous sodium bisulfite solution, maintaining the pH at 5, by adding 30% (w/w) aqueous NaOH until colourlessness and a stable negative oxidation-reduction potential, measured with a suitable oxidation-reduction electrode, in the range 0 to -20 mV.HPLC analysis (Fig. 2) showed the degree of conversion of the starting compound to N-(2,3-dihydroxypropyl)-N’-[2-hydroxy-1-(hydroxymethyl)ethyl]-5-hydroxy-2,4,6-triiodo-1,3-benzenedicarboxamide >98% (by area % on the HPLC chromatogram), and the resulting solution was used in the next step of the synthesis without any additional processing.Example 7Preparation of a compound of formula 2 in which both substituents R and R’ are -NH-CH(CH
2 OH)
2 groups using a starting solution heated to 60
° C.In a 0.5 L jacketed four-neck reactor equipped with a mechanical stirrer, reflux condenser, and combination pH/temperature electrode, N,N’-bis[2-hydroxy-1-(hydroxymethyl)ethyl]-5-hydroxy-1,3-benzenedicarboxamide (50 g, 0.152 mol) was dissolved in H
2 O (215 g) and converted to the corresponding sodium salt by adding 30% (w/w) NaOH (20.3 g, 0.152 mol) (pH 9.5). The solution was heated to 60 °C and solid I
2 (46.4 g, 0.183 mol) was added in one portion; When the pH spontaneously dropped to 5, 50% (w/w) aqueous HIO3 (32.2 g, 0.091 mol) was added slowly over 2 h
. The reaction temperature was maintained at 60 °C for an additional 4 h, during which time the pH of the reaction mixture spontaneously remained in the range 5-5.5. The resulting red solution was cooled to 25 °C and quenched by adding 18% (w/w) aqueous sodium bisulfite solution, maintaining the pH at 5, with 30% (w/w) aqueous NaOH until colorlessness and a stable negative oxidation-reduction potential (in the range of -20 to -50 mV), as measured by a suitable oxidation-reduction electrode.HPLC analysis showed the conversion of the starting compound to N,N’-bis[2-hydroxy-1-(hydroxymethyl)ethyl]-5-hydroxy-2,4,6-triiodo-1,3-benzenedicarboxamide to be >98% (based on area % of the HPLC chromatogram), and the resulting solution was used in the next step of the synthesis without any further processing.Comparative example 1This test was carried out to evaluate the efficiency of the iodination reaction disclosed by Patil et al., in ARKIVOC, 2006, 104 and Tetrahedron Lett., 2005, 46, 7179.In a 50 mL three-necked round-bottomed flask equipped with a thermometer and a reflux condenser, solid 3,5-disubstituted phenol 1 (16.4 g, 50 mmol) was suspended in ethanol (30 mL). Then, to the resulting suspension, heated to 38-40 °C, solid I
2 (15.2 g, 60 mmol) was added in one portion and a solution of HIO
3 (5.3 g, 30 mmol) in H
2 O (3 mL) was added in the indicated order over 5 min. The resulting dark brown mixture was stirred at 38-40 °C for about 1 h and then the change in appearance of the reaction mixture was recorded, which turned into a clear dark brown solution. The reaction mixture was maintained at the above temperature for a total of 3.5 h, then cooled to room temperature, which caused crystallization of the pale yellow solid product. After 15 h at room temperature, the solid was isolated by filtration and dried to give the desired 3,5-disubstituted-2,4,6-triiodophenol (12.1 g, 17 mmol). Yield 34.3%.During the iodination reaction, the reaction mixture was analyzed by HPLC. In particular, the first analysis was performed 1.5 hours after the start of iodination (the reaction time was chosen based on the literature sources cited), and its results are shown in Fig. 3, and the second analysis was performed after another 2 hours (the total reaction time was 3.5 hours), and its results are shown in Fig. 4. The results show that even after 3.5 hours, the conversion is not complete and a significant amount (13% based on the area on the HPLC chromatogram) of the original substrate is still present. On the other hand, a longer reaction time leads to the formation of a significant amount of impurities, which are decay products, which are easily detected after 3.5 hours of reaction (Fig. 4). This is certainly a factor that adversely affects the reaction yields. However, the low reaction yields can also be attributed to the solubility of 3,5-disubstituted-2,4,6-triiodophenol 2b in an alcohol medium, as confirmed by the analysis of the mother liquor shown in Fig. 5, which interferes with the quantitative isolation of the iodination product.In this regard, the increase in both the reaction yield and the product purity due to the use of an aqueous medium and reaction conditions, expressed in the above results, is evident from a comparison of Figs. 3-5 with Figs. 1 and 2, which show chromatograms (HPLC) of crude reaction solutions (Examples 1 and 6, respectively) obtained using the method of the present invention.
PATENT
https://patents.google.com/patent/KR20200032280A/en
[Scheme 3]

Example One.
Iomeproletic Produce
5- (2-hydroxyacetamido) -N, N’-bis (2, 3-dihydroxypropyl) -2, 4, 6-triiodoisophthalamide (1b) 5g (1 equivalent) and 3.6 g (5 equivalents) of calcium chloride was added to 25 g of methanol together, and dissolved at reflux at room temperature or 70 ° C. for 60 minutes.
After cooling the temperature of the solution to 10 ℃ to 15 ℃ 0.3g (0.62 equivalents) of calcium hydroxide was added and stirred at the same temperature for 1 hour.
2.48 g (3 equivalents) of dimethyl sulfate was added to the reaction solution and stirred for 3 hours at the same temperature until the reaction was completed.
After completion of the reaction, 1 mL of HCl (35%) was added to acidify, 25 mg of 2-butanol was added, stirred at a temperature of 70 to 80 ° C. for 2 hours, cooled, filtered and washed with 2-butanol to obtain an iomeprole crude product.
The above prepared omeprolol was added to a mixture of 25 mL of methanol and 10 mL of water, heated to 50 ° C. to dissolve, put 20 mL of 2-butanol, refluxed at 90 ° C. for 3 hours, cooled to room temperature, and the resulting crystal was filtered.
After washing with 2-butanol and drying under reduced pressure at 90 ° C. for 12 hours, 4.17 g of Iomeprole (HPLC: 99.3%) was obtained.
Example 2.
Preparation of Iomeprole
5- (2-hydroxyacetamido) -N, N’-bis (2, 3-dihydroxypropyl) -2, 4, 6-triiodoisophthalamide (1b) 5g (1 equivalent) and 3.6 g (5 equivalents) of calcium chloride was added to 25 g of methanol together and refluxed at 70 ° C for 30 minutes to dissolve.
After cooling the solution to 0-5 ° C., 0.3 g (0.62 equivalents) of calcium hydroxide was added and stirred at the same temperature for 1 hour.
2.48 g (3 equivalents) of dimethyl sulfate was added to the reaction solution and stirred for 7 hours at the same temperature until the reaction was completed.
After completion of the reaction, 1 mL of HCl (35%) was added to acidify, 30 mL of 2-butanol was added, refluxed at a temperature of 70-80 ° C. for 2 hours, stirred, filtered and washed with 2-butanol to obtain an iomeprole crude product.
After adding the above prepared omeprolol to a mixture of 25 mL of methanol and 10 mL of water, the temperature was raised to 50 ° C. to dissolve, 20 mL of 2-butanol was added, refluxed at 90 ° C. for 3 hours, cooled to room temperature, and the resulting crystal was filtered. .
After washing with 2-butanol and drying under reduced pressure at 90 ° C. for 12 hours, 4.21 g of Iomeprole (HPLC: 99.1%) was obtained.
Comparative example 1 (Inorganic chloride Non-addition )
After adding 5- (2-hydroxyacetamido) -N, N’-bis (2, 3-dihydroxypropyl) -2, 4, 6-triiodoisophthalamide (1b) to 25 g of methanol It was refluxed for 30 minutes.
After the temperature of the turbid solution was cooled to 10 ° C to 15 ° C, 0.3 g (0.62 equivalent) of calcium hydroxide was added and stirred at the same temperature for 1 hour.
2.48 g (3 equivalents) of dimethyl sulfate was added to the reaction solution and stirred at the same temperature for 5 hours.
As a result of reactivity review by HPLC, synthesis of iomeprole progressed 5%, 5- (2-hydroxyacetamido) -N, N’-bis (2, 3-dihydroxypropyl) -2, 4, 6- Triiodoisophthalamide (1b) was found to be 90% or more remaining, so that the reactivity was very low.
Comparative example 2 (Inorganic base Non-addition )
5- (2-hydroxyacetamido) -N, N’-bis (2, 3-dihydroxypropyl) -2, 4, 6-triiodoisophthalamide (1b) and 3.6 g of calcium chloride are methanol After adding to 25 g, the mixture was refluxed for 30 minutes to dissolve.
After the temperature of the solution was cooled from 10 ° C to 15 ° C, 2.48 g (3 equivalents) of dimethyl sulfate was added to the reaction solution and stirred at the same temperature for 5 hours.
As a result of reactivity review by HPLC, synthesis of iomeprole proceeds 0.5%, 5- (2-hydroxyacetamido) -N, N’-bis (2, 3-dihydroxypropyl) -2, 4, 6- Triiodoisophthalamide (1b) was found to be very low reactivity with more than 99% remaining.
PATENT
https://patents.google.com/patent/CN102363600B/en
.Synthetic route is seen formula 1:

Embodiment
Embodiment 1
5-methylamino-2,4,6-triiodo m-phthaloyl chloride synthetic:
In the there-necked flask that agitator and reflux condensing tube are housed, 76g(0.133mol under the room temperature) 5-methylamino-2,4,6-triiodo m-phthalic acid is dissolved in 250mL(2.53mol) in the ethyl acetate, after waiting to stir, add 29mL(0.4mol) sulfur oxychloride, be warming up to 50 ℃ of acyl chloride reaction temperature then, stir and finished reaction in 6 hours.After treating that ethyl acetate and sulfur oxychloride boil off under the decompression, residue boils off solvent and washs through frozen water after adding the 100mL ethyl acetate, dry product 68.9g, yield is that 84.9%(is with 5-methylamino-2,4,6-triiodo m-phthalic acid meter), m.p.167 ~ 169 ℃.
Embodiment 2
5-methylamino-2,4,6-triiodo m-phthaloyl chloride synthetic:
The acyl chloride reaction temperature is 80 ℃, and in 3 hours reaction times, all the other are operated with embodiment 1.
Embodiment 3
5-[N-methyl-2-chloracetyl amido]-2,4,6-triiodo m-phthaloyl chloride synthetic:
In reaction flask, add 36.6g(0.06mol under the room temperature) 5-methylamino-2,4,6-triiodo m-phthaloyl chloride and 80mL N,N-dimethylacetamide, heating in water bath to 50 ℃, after the stirring and dissolving, be cooled to 10 ℃, begin to drip 10.2g(0.09mol) chloroacetyl chloride, dropwising the back heats up 50 ℃, stirred 3 hours, and be cooled to 10 ℃, be added dropwise to the 150mL frozen water.Filter, through the frozen water washing, dry product 38.7g, yield be 94%(with 5-methylamino-2,4,6-triiodo m-phthaloyl chloride meter), m.p.194 ~ 196 ℃.
Embodiment 4
5-[N-methyl-2-chloracetyl amido]-2,4,6-triiodo m-phthaloyl chloride synthetic:
The chlorine acylation temperature is 90 ℃, and in 1 hour reaction times, all the other are operated with embodiment 3.
Embodiment 5
5-[N-methyl-2-chloracetyl amido]-N, N ‘-two (2,3-dihydroxypropyl)-2,4,6-three iodo-1,3-benzenedicarboxamide synthetic:
In reaction flask, add 41.2g(0.06mol under the room temperature) 5-[N-methyl-2-chloracetyl amido]-2,4,6-triiodo m-phthaloyl chloride and 80mL N, the N-N,N-DIMETHYLACETAMIDE after the stirring and dissolving, is cooled to 10 ℃, add 16g(0.16mol) triethylamine and 14.6g(0.16mol) 3-amino-1, the 2-propylene glycol is heated to 20 ℃ then, stirs and finishes reaction in 15 hours, be chilled to after-filtration below 10 ℃, after evaporated under reduced pressure, residue is dissolved in the 160mL methyl alcohol with filtrate, adds 200mL water and stirs evenly, leave standstill, with the solid filtering of separating out, the washing, dry product 43.4g, yield is that 91%(is with 5-[N-methyl-2-chloracetyl amido]-2,4,6-triiodo m-phthaloyl chloride meter), m.p.204 ~ 207 ℃.
Embodiment 6
5-[N-methyl-2-chloracetyl amido]-N, N ‘-two (2,3-dihydroxypropyl)-2,4,6-three iodo-1,3-benzenedicarboxamide synthetic:
The amidate action temperature is 50 ℃, and in 8 hours reaction times, all the other are operated with embodiment 5.
Embodiment 7
5-[N-methyl-glycolamide base]-N, N ‘-two (2,3-dihydroxypropyl)-2,4,6-three iodo-1,3-benzenedicarboxamide synthetic:
In reaction flask, add 47.7g(0.06mol) 5-[N-methyl-2-chloracetyl amido]-N, N ‘-two (2,3-dihydroxypropyl)-2,4,6-three iodo-1,3-benzenedicarboxamide, 250mL water, stir and to add 26g(0.32mol down) sodium acetate, back flow reaction is after 24 hours, the pressure reducing and steaming solvent.The residue dissolve with methanol filters, and filtrate boils off methyl alcohol, and vacuum-drying gets solid, is dissolved in the 150mL water, adds gac 1.8g, and reflux 30min filters.Filtrate is successively respectively by 732 Zeo-karbs and 717 anionite-exchange resin, and pressure reducing and steaming solvent again is after the resistates vacuum-drying, add 180mL ethanol and carry out recrystallization, get white solid 40.1g, HPLC detects purity greater than 99.0%, and yield is that 86%(is with 5-[N-methyl-2-chloracetyl amido]-N, N ‘-two (2, the 3-dihydroxypropyl)-2,4,6-three iodo-1,3-benzenedicarboxamide meter), m.p.〉280 ℃. 1H-NMR?(DMSO-D 6)δ(ppm):2.91(s,4H),3.21~3.36(m,4H),?3.41~3.65(m,4H),?3.85(s,2H),?3.98(m,2H),?4.1~?4.5(m,5H),?8.2~?8.4(d,?2H); 13C-NMR(D 2O)δ(ppm)33.2,44.7,60.7,64.6,71.2,90.1,99.8,99.9,145.5,150.5,150.6,171.3,171.4,173.9。
PATENT
https://patents.google.com/patent/CN114213273A/en
synthetic route is as follows:

Example 1
The synthesis process of iomeprol with 5-amido-N, N’ -bis (2, 3-dihydroxypropyl) -2,4, 6-triiodo-1, 3-benzenedicarboxamide as initial material includes successive chloroacetylation, methylation, hydrolysis and hydroxylation to synthesize iomeprol product, and includes the following steps:

the specific process comprises 4 reaction steps:
s1, chloroacetylation (preparation of 5- (2-chloroacetamido) -2,4, 6-triiodo-N, N’ -bis (2, 3-bis (2-chloroacetoxy) propyl) -isophthalamide):
12g of 5-amino-N.N’ -bis (2, 3-dihydroxypropyl) -2,4, 6-triiodoisophthalamide was dissolved in 24g N, N-dimethylacetamide and stirred at room temperature. 12g of chloroacetyl chloride were added while controlling the temperature below 60 ℃. After the addition, the temperature is kept between 50 and 60 ℃, the stirring is carried out for 4 hours, and after the reaction is finished, the vacuum concentration is carried out below 65 ℃ until the reaction is dried. 36ml of ethyl acetate and 36ml of a 5% aqueous solution of sodium hydrogencarbonate were added to the concentrate to conduct extraction. The aqueous layer after separation was extracted twice with 10ml of ethyl acetate. The organic solutions thus extracted were mixed, 15ml of 5% saline was added thereto, and the mixture was washed, and after recovering the organic solution layer, magnesium sulfate was added to remove water, followed by filtration and distillation under reduced pressure to obtain 18.5g of an oily substance.
S2, methylation reaction (preparation of 5- (N-methyl-2-chloroacetamido) -2,4, 6-triiodo-N, N’ -bis (2, 3-bis (2-chloroacetoxy) propyl) -isophthalamide):
dissolving 18.5g of the oily substance in the previous step by using 37ml of acetone, cooling to 0 ℃, adding 3.8g of potassium carbonate, keeping the temperature and stirring, dropwise adding 5.8g of dimethyl sulfate while stirring, keeping the temperature and reacting for 8 hours, after the reaction is finished, carrying out vacuum concentration to dryness at 60 ℃, dissolving the concentrated solution in 50ml of ethyl acetate and 50ml of water, and adding 10ml of ethyl acetate into the separated water layer for secondary extraction. The organic solutions thus extracted were mixed, 15ml of 5% saline was added thereto, and the mixture was washed, and after recovering the organic solution layer, magnesium sulfate was added to remove water, followed by filtration and distillation under reduced pressure to obtain 19.3g of an oily substance.
S3, ester hydrolysis reaction (preparation of 5- (N-methyl-2-chloroacetamido) -2,4, 6-triiodo-N, N’ -bis (2, 3-dihydroxypropyl) -isophthalamide):
the above oily substance was dissolved in 20ml of methanol, 6.0g of water was added, 30.0g of 30% aqueous sodium hydroxide solution was added, and the reaction was carried out at 15 ℃ for 4 hours, and after the completion of the reaction, methanol was distilled off under reduced pressure to obtain an aqueous solution containing the objective compound.
S4 hydroxylation reaction (preparation of 5- (N-methyl-2-hydroxyacetamido) -2,4, 6-triiodo-N, N’ -bis (2, 3-dihydroxypropyl) -isophthalamide, i.e. iomeprol):
adding 6.0g of sodium acetate into the aqueous solution, heating to reflux for 24 hours, distilling under reduced pressure to remove the solvent, adding methanol into the residue to dissolve, filtering, evaporating the methanol from the filtrate, dissolving the residue in pure water, adding activated carbon, heating to reflux for 30 minutes, filtering, sequentially passing the filtrate through cation resin and anion resin, evaporating to remove the solvent, adding 70ml of ethanol to recrystallize, filtering, drying under reduced pressure at 50 ℃ to obtain 8.5g of white solid, wherein the HPLC purity is 99.2%, and the molar yield is 64%.
Example 2
S1, chloroacetylation (preparation of 5- (2-chloroacetamido) -2,4, 6-triiodo-N, N’ -bis (2, 3-bis (2-chloroacetoxy) propyl) -isophthalamide):
500g of 5-amino-N.N’ -bis (2, 3-dihydroxypropyl) -2,4, 6-triiodoisophthalamide was dissolved in 1000g N, N-dimethylacetamide and stirred at room temperature. 500g of chloroacetyl chloride was added while controlling the temperature below 60 ℃. After the addition, the temperature is kept between 50 and 60 ℃, the stirring is carried out for 4 hours, and after the reaction is finished, the vacuum concentration is carried out below 65 ℃ until the reaction is dried. 1500ml of ethyl acetate and 1500ml of 5% aqueous sodium bicarbonate solution were added to the concentrate to extract. The aqueous layer after separation was extracted twice with 480ml of ethyl acetate. The organic solutions thus extracted were mixed, and 650ml of 5% brine was added thereto, followed by washing, and after recovering the organic solution layer, magnesium sulfate was added to remove water, followed by filtration and distillation under reduced pressure to obtain 772.6g of an oil.
S2, methylation reaction (preparation of 5- (N-methyl-2-chloroacetamido) -2,4, 6-triiodo-N, N’ -bis (2, 3-bis (2-chloroacetoxy) propyl) -isophthalamide):
dissolving 772.6g of the oily matter in the previous step by 1545ml of acetone, cooling to 0 ℃, adding 158.5g of potassium carbonate, keeping the temperature and stirring, dropwise adding 241.5g of dimethyl sulfate while stirring, keeping the temperature and reacting for 8 hours, after the reaction is finished, vacuum concentrating at 60 ℃ to dryness, dissolving the concentrated solution in 1500ml of ethyl acetate and 1500ml of water, and adding 480ml of ethyl acetate into the separated water layer for secondary extraction. The organic solutions thus extracted were mixed, and then 650ml of 5% saline was added thereto, followed by washing, and after recovering the organic solution layer, magnesium sulfate was added thereto to remove water, followed by filtration and distillation under reduced pressure to obtain 805.7g of an oily substance.
S3, ester hydrolysis reaction (preparation of 5- (N-methyl-2-chloroacetamido) -2,4, 6-triiodo-N, N’ -bis (2, 3-dihydroxypropyl) -isophthalamide):
the above oil was dissolved in 800ml of methanol, 250g of water was added, 1250g of a 30% aqueous solution of sodium hydroxide was added, and the reaction was carried out at 15 ℃ for 4 hours, and after the completion of the reaction, methanol was distilled off under reduced pressure to obtain an aqueous solution containing the objective compound.
S4 hydroxylation reaction (preparation of 5- (N-methyl-2-hydroxyacetamido) -2,4, 6-triiodo-N, N’ -bis (2, 3-dihydroxypropyl) -isophthalamide, i.e. iomeprol):
adding 250g of sodium acetate into the aqueous solution, heating to reflux for 24 hours, distilling under reduced pressure to remove the solvent, adding methanol into the residue to dissolve, filtering, evaporating the filtrate to remove the methanol, dissolving the residue in pure water, adding activated carbon, heating to reflux for 60 minutes, filtering, sequentially passing the filtrate through cation resin and anion resin, evaporating to remove the solvent, adding 3000ml of ethanol to recrystallize, filtering, drying under reduced pressure at 50 ℃ to obtain 365.0g of white solid, wherein the HPLC purity is 99.1%, and the molar yield is 66%.
Compared with two synthesis methods in patent EP0026281A1, the synthesis method of iomeprol developed by the invention does not use thionyl chloride, reduces the difficulty of waste gas treatment, does not use acetoxy acetyl chloride, and ensures the process stability. Meanwhile, the used starting material (compound II) is a common intermediate of other contrast agents iohexol and ioversol, so that corresponding supporting construction is reduced.
Therefore, the synthetic route designed by the invention has the advantages of mild reaction conditions, stable quality, high yield, low cost, environmental protection and suitability for industrial production.
The embodiments are described in a progressive manner, each embodiment focuses on differences from other embodiments, and the same or similar parts among the embodiments are referred to each other. The device disclosed by the embodiment corresponds to the method disclosed by the embodiment, so that the description is simple, and the relevant points can be referred to the method part for description.
Side effects
It is classified as a water-soluble, nephrotrophic, low osmolar X-ray contrast medium.[2] Low osmolar non-ionic agents are better tolerated and less likely to cause side effects than the high osmolar ionic agents.[2]
Society and culture
Iomeprol is not metabolized in the human body but excreted in unchanged form.[medical citation needed] It is decomposed slowly and can therefore accumulate in the environment.[6]
Legal status
Iomeprol was approved for medical use in the United States in November 2024.[1][4]
Brand names
Iomeprol is sold under the brand name Iomervu.[1]
References
- ^ Jump up to:a b c d e https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/216017s000,216017s000lbl.pdf
- ^ Jump up to:a b c Rossiter D (2014). South African medicines formulary (11th ed.). Rondebosch, South Africa: Health and Medical Pub. Group .of the South African Medical Association. ISBN 978-1-875098-30-9. OCLC 869772940.
- ^ Haberfeld H, ed. (2020). Austria-Codex (in German). Vienna: Österreichischer Apothekerverlag. Iomeron 300 mg J/ml-Infusionsflasche.
- ^ Jump up to:a b “Novel Drug Approvals for 2024”. U.S. Food and Drug Administration (FDA). 1 October 2024. Retrieved 29 November 2024.
- ^ New Drug Therapy Approvals 2024 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2025. Archived from the original on 21 January 2025. Retrieved 21 January 2025.
- ^ Pfundstein P, Martin C, Schulz W, Seitz W, Ruth KM, Wille A, et al. (January 2015). “IC-ICP/MS-Analytik”. GIT Labor-Fachzeitschrift (in German): 29–31.
- Dooley M, Jarvis B: Iomeprol: a review of its use as a contrast medium. Drugs. 2000 May;59(5):1169-86. doi: 10.2165/00003495-200059050-00013. [Article]
- Katayama H, Spinazzi A, Fouillet X, Kirchin MA, Taroni P, Davies A: Iomeprol: current and future profile of a radiocontrast agent. Invest Radiol. 2001 Feb;36(2):87-96. doi: 10.1097/00004424-200102000-00004. [Article]
- Rosati G: Clinical pharmacology of iomeprol. Eur J Radiol. 1994 May;18 Suppl 1:S51-60. doi: 10.1016/0720-048x(94)90094-9. [Article]
- EMC Summary of Product Characteristics: Iomeron (iomeprol) solution for injection [Link]
- FDA Approved Drug Products: IOMERVU (iomeprol) injection, for intra-arterial or intravenous use [Link]
- Medsafe NZ: IOMERON® (iomeprol) safety data sheet [Link]
| showvteContrast media (V08) |
|---|
| Clinical data | |
|---|---|
| Trade names | Iomervu, others |
| License data | US DailyMed: Iomeprol |
| Routes of administration | Intravenous, intra-arterial |
| ATC code | V08AB10 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1]In general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Metabolism | none |
| Elimination half-life | 109±20 min |
| Excretion | Kidney |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 78649-41-9 |
| PubChem CID | 3731 |
| DrugBank | DB11705 |
| ChemSpider | 3600 |
| UNII | 17E17JBP8L |
| KEGG | D01719 |
| ChEBI | CHEBI:31710 |
| CompTox Dashboard (EPA) | DTXSID1049061 |
| Chemical and physical data | |
| Formula | C17H22I3N3O8 |
| Molar mass | 777.089 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
////////////Iomeprol, Iomervu, FDA 2024, APPROVALS 2024, 78649-41-9, Iomeprolum, Iomeron, Iomeprolo, UNII-17E17JBP8L, E-7337, E 7337, E-7337, E7337, XRAY CONTRAST AGENT
Crinecerfont



Crinecerfont
CAS 752253-39-7
SSR125543
SSR 125543
SSR-125543, WHO 10958, UNII-MFT24BX55I, 06-RORI,NBI-74788
- (S)-4-(2-chloro-4-methoxy-5-methylphenyl)-N-(2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl)-5-methyl-N-(prop-2-yn-1-yl)thiazol-2-amine
- 2-Thiazolamine, 4-(2-chloro-4-methoxy-5-methylphenyl)-N-((1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl)-5-methyl-N-2-propynyl-
- 2-Thiazolamine, 4-(2-chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]-5-methyl-N-2-propyn-1-yl
FDA APPROVED 12/13/2024, Crenessity, To treat classic congenital adrenal hyperplasia
Press Release
4-(2-chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]-5-methyl-N-prop-2-ynyl-1,3-thiazol-2-amine
WeightAverage: 483.04
Monoisotopic: 482.1594906
Chemical FormulaC27H28ClFN2OS

CAS No. : 321839-75-2
| Molecular Weight | 519.50 |
|---|---|
| Formula | C27H29Cl2FN2OS |
Crinecerfont, sold under the brand name Crenessity, is a medication used for the treatment of congenital adrenal hyperplasia.[1] It is a corticotropin-releasing factor type 1 receptor (CRF1R) antagonist developed to treat classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency (21OHD).[1] It is taken by mouth.[1]
The most common side effects of crinecerfont in adults include fatigue, dizziness, and arthralgia (joint pain).[2] For children, the most common side effects include headache, abdominal pain, and fatigue.[2]
Crinecerfont was approved for medical use in the United States in December 2024.[2][3] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication.[4]
A medication used to reduce the amount of steroid replacement required in patients with a genetic disease that causes, amongst other symptoms, a steroid deficiency.
- OriginatorSanofi
- DeveloperNeurocrine Biosciences; Sanofi
- ClassAmines; Antidepressants; Anxiolytics; Chlorobenzenes; Cyclopropanes; Fluorobenzenes; Halogenated hydrocarbons; Phenyl ethers; Small molecules; Thiazines; Thiazoles
- Mechanism of ActionCorticotropin releasing factor receptor 1 antagonists
- Orphan Drug StatusYes – Congenital adrenal hyperplasia
- MarketedCongenital adrenal hyperplasia
- DiscontinuedMajor depressive disorder; Post-traumatic stress disorders
20 Dec 2024Launched for Congenital adrenal hyperplasia (Adjunctive treatment, In adolescents, In children) in USA (PO)
- 20 Dec 2024Launched for Congenital adrenal hyperplasia (Adjunctive treatment, In adolescents, In children, In the elderly, In adults) in USA (PO)
- 20 Dec 2024Launched for Congenital adrenal hyperplasia (Adjunctive treatment, In the elderly, In adults) in USA (PO)



SYN

https://patents.google.com/patent/US12128033
Example processes and certain intermediates of the present invention are shown in Scheme I to Scheme VII below.
A representative Coupling-Step of 2-cyclopropylacetic acid (Compound
1A) with N,O-dimethylhydroxylamine or a salt thereof in the presence of a coupling-step reagent (e.g., 1,1′-carbonyldiimidazole), a coupling-step base (e.g., triethylamine), and a coupling-step solvent (e.g., dichloromethane) to prepare 2-cyclopropyl-N-methoxy-N-methylacetamide (Compound
2A) is provided below in Scheme I.

A representative Reacting-Step between 2-cyclopropyl-N-methoxy-N-methylacetamide (Compound
2A) with an organomagnesium reagent of 4-bromo-2-fluoro-1-methylbenzene in the presence of a reacting-step solvent (e.g., tetrahydrofuran (THF)) to prepare 2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethan-1-one (Compound
3A) is provided below in Scheme II.

A representative Condensing-Step of 2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethan-1-one (Compound
3A) with a Compound of Formula (Ic) or a salt thereof, in the presence of a condensing-step acid (e.g., p-toluenesulfonic acid) and a condensing-step solvent (e.g., toluene) to prepare a Compound of Formula (Ie) is provided below in Scheme III.

-
- wherein:
- R1c, R2c, and R3c are each independently selected from: H, C1-C6 alkoxy, C1-C6 alkyl, C1-C6 haloalkyl, and halogen.
A representative Reducing-Step of a Compound of Formula (Ie) in the presence of a reducing-catalyst (e.g., sponge nickel and Pd/Cu—C), hydrogen, and a reducing-step solvent (e.g., ethanol) to prepare a Compound of Formula (Ig) is provided below in Scheme IV.

-
- wherein:
- R1c, R2c, and R3c are each independently selected from: H, C1-C6 alkoxy, C1-C6 alkyl, C1-C6 haloalkyl, and halogen.
A representative Deprotecting-Step of a Compound of Formula (Ig), or a salt thereof, in the presence of a deprotecting-catalyst (e.g., Pd), hydrogen, and a deprotecting-step solvent (e.g., ethanol) to prepare (S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethan-1-amine (Compound
6A) or a salt thereof is provided below in Scheme V.

-
- wherein:
- R1c, R2c, and R3c are each independently selected from: H, C1-C6 alkoxy, C1-C6 alkyl, C1-C6 haloalkyl, and halogen.
A representative Cyclizing-Step of (S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethan-1-amine (Compound
6A) or a salt thereof, with 1-(2-chloro-4-methoxy-5-methylphenyl)-2-thiocyanatopropan-1-one (Compound
8A) or a tautomeric form thereof, in the presence of a cyclizing-step solvent (e.g., n-heptane) to prepare (S)-4-(2-chloro-4-methoxy-5-methylphenyl)-N-(2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl)-5-methylthiazol-2-amine (Compound
9A) or a salt thereof is provided below in Scheme VI.

A representative Alkylating-Step of (S)-4-(2-chloro-4-methoxy-5-methylphenyl)-N-(2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl)-5-methylthiazol-2-amine (Compound
9A) or a salt thereof, with a Compound of Formula (Ii), wherein LG is suitable leaving group (e.g., Br), in the presence of an alkylating-step solvent (e.g., methyl tert-butyl ether (MTBE), toluene, and mixtures thereof), a phase-transfer catalyst (e.g., tetra-n-butylammonium bromide (TBAB)), an alkylating-step base (e.g., potassium hydroxide), and water to prepare 4-(2-chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]-5-methyl-N-prop-2-ynyl-1,3-thiazol-2-amine (Compound 1) or a pharmaceutically acceptable salt thereof is provided below in Scheme VII.

One aspect of the present invention includes every combination of one or more process steps and intermediates related thereto used in the preparation of 4-(2-chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]-5-methyl-N-prop-2-ynyl-1,3-thiazol-2-amine (Compound 1), and/or pharmaceutically acceptable salts, and crystalline forms thereof, such as those processes exemplified by Schemes I, II, III, IV, V, VI, VII, and VII (supra) and Compounds contained therein.
8A was previously described in International Publication Number WO2010/125414 by Sanofi-Aventis.Example 1: Preparation of 4-(2-chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]-5-methyl-N-prop-2-ynyl-1,3-thiazol-2-amine (Compound 1), See FIG. 5 for a general synthetic schemeStep1A: Preparation of 2-Cyclopropyl-N-methoxy-N-methylacetamide (Compound 2A)

A suspension of 1,1′-carbonyldiimidazole (CDI, 152.6 kg, 1.01 eq.) in DCM (682 kg, 513 L, 7.3 w/w relative to 2-cyclopropylacetic acid) was treated with a solution of 2-cyclopropylacetic acid (Compound
1A, 93.6 kg, 1 eq.) in DCM (248 kg, 186 L, 2.65 w/w) over at least 1 h, keeping the temperature ≤25° C. and compensating for significant effervescence. The resulting mixture was stirred for 15 min at 22° C. and then N,O-dimethylhydroxylamine-HCl (93.6 kg, 1.03 eq.) was added in portions, keeping the temperature ≤30° C. Subsequently, triethylamine (46.4 kg, 0.49 eq.) was added to the stirring mixture at 20-25° C. The resulting mixture was stirred at 22° C. at least 1 h. The mixture was washed once with KHSO4 solution (0.24 M, 357.1 kg, 0.09 eq.), once with KHSO4 solution (0.40 M, 365.4 kg, 0.15 eq.), once with KHSO4 solution (0.80 M, 384.5 kg, 0.30 eq.), and once with NaHCO3 solution (0.60 M, 393.1 kg, 0.24 eq.). Residual DCM was removed by two put-and-takes of THF (166.6 kg, 1.78 w/w) and vacuum distillation (50-60° C., to minimum volume/until distillation stops) to provide Compound
2A. THF (333.2 kg. 3.56 w/w) was added and the yield was determined by correcting for the LOD and GC-FID purity of the sample (131.5 kg, 98.2% corrected). 1H-NMR (400 MHz, DMSO-d6) δ (ppm) −0.01-0.03 (m, 2H), 0.32-0.36 (m, 2H), 0.81-0.90 (br m, 1H), 2.18 (d, J=6.80 Hz, 2H), 2.97 (s, 3H), 3.53 (s, 3H). ESI-MS: 144.0 [M+H]+.Step 1B: Preparation of 2-Cyclopropyl-1-(3-fluoro-4-methylphenyl)ethan-1-one (Compound 3A)

Mg (turnings, 28.6 kg, 1.37 eq.) were suspended in THF (244.7 kg, 2.0 w/w) and DIBAL-H (1 M in n-heptane, 18.9 kg, 0.03 eq.) was added dropwise at 30° C. The resulting mixture was stirred at 30° C. for at least 10 min and then 4-bromo-2-fluoro-1-methylbenzene (neat, 21.1 kg, 0.13 eq.) was added over at least 30 min at 30-50° C. Subsequently, the mixture was treated with a solution of 4-bromo-2-fluoro-1-methylbenzene (191.6 kg, 1.18 eq.) in THF (414.5 kg, 3.37 w/w) at 30-50° C. over 3 h or less. The mixture was stirred at 30° C. for at least 1 h. The mixture was cooled to 12-18° C. and subsequently treated with 2-cyclopropyl-N-methoxy-N-methylacetamide (Compound
2A, 123.0 kg, 1 eq., 25.9% w/w solution in THF) over at least 1 h at 15-25° C. The resulting mixture was stirred at 20-25° C. for at least 1 h. The stirring mixture was then treated with aqueous HCl (3 M, 10.3% w/w, 668.9 kg, 2.24 eq.) at 10-25° C. and the resulting mixture was stirred at least 2 h until no Mg turnings were observed (check pH 3.0-3.5). The layers were separated, and the aqueous layer discarded. The organic layer was distilled at 55-65° C. and 400 mbar until distillation halts. Heptane (290.3 kg, 2.36 w/w) was added. The layers were separated, and the organic layer was washed once with NaHCO3 solution (0.63 M, 211.6 kg, 0.15 eq.) and once with NaCl solution (2.57 M, 213.0 kg, 0.55 eq.). The residual solvents were removed by vacuum distillation at 58-62° C. until distillation stops and then one put-and-take of toluene (275.5 kg, 2.24 w/w) at 107-117° C. until distillation stops. Toluene (275.5 kg, 2.24 w/w) was added and the yield was determined by correcting for the LOD and GC-FID purity of the sample (150.7 kg, 91.3% corrected). 1H NMR (400 MHz, DMSO-d6) β (ppm) 0.07-0.21 (m, 2H), 0.40-0.54 (m, 2H), 1.02 (ttt, J=8.16, 8.16, 6.68, 6.68, 4.86, 4.86 Hz, 1H), 2.30 (d, J=1.77 Hz, 3H), 2.91 (d, J=6.57 Hz, 2H), 7.44 (t, J=7.83 Hz, 1H), 7.57-7.78 (m, 2H). ESI-MS: 193.1 [M+H]+.Step 1C: Preparation of (S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)-N-(1-phenylethyl)ethan-1-imine (Compound 4A)

A mixture of 2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethan-1-one (Compound
3A, 150.7 kg, 1 eq., as a 27.6% w/w solution in toluene), (S)-(−)-1-phenylethylamine (112.9 kg, 1.19 eq.), and p-toluenesulfonic acid (7.4 kg, 0.05 eq.) was heated to reflux at 110-120° C. for 23-25 h in a reactor set up in a Dean-Stark configuration. The solvent was then removed at 125-135° C. under atmospheric pressure until distillation halts and a portion of toluene (275 kg, 2.24 w/w) was added to afford a suspension. The suspension was heated to reflux at 110-120° C. for 23-25 h. The mixture was cooled to 22° C. and washed twice with aqueous NH4Cl (10%, 301.2 kg, 0.72 eq.) and once with aqueous NaHCO3 (5%, 301.2 kg, 0.23 eq., check pH 8-9). The solvent was removed at 125-135° C. and atmospheric pressure to a target volume of 256 L, the mixture was filtered over CELITE®, and the cake was washed with toluene (25 kg). The resulting mixture containing Compound 4A was used directly in the next step without further isolation. The yield was determined by correcting for the LOD and GC-FID purity of the sample (208.4 kg, 90.0% corrected). EL-MS: 294.1 [M−H]*, 190.1 [M-C6H5CH(CH3)]+, 105.1 [C6H5CH(CH3)]+.Step 1D: Preparation of (S)-2-Cyclopropyl-1-(3-fluoro-4-methylphenyl)-N—((S)-1-phenylethyl)ethan-1-amine (Compound 5A) as the Hydrochloride Salt

Sponge nickel catalyst (144 kg, 0.70 w/w, shipped as a 50% w/w suspension in water) was added to a hydrogenation reactor, equipped with a dip tube capable of removing material from the top of the mass inside, minimizing the amount of water introduced. The supernatant was discarded, ethanol (329.3 kg, 1.58 w/w, anhydrous) was added, the suspension was stirred and then allowed to settle. This process was repeated four more times and the supernatant is checked; ≤1% H2O w/w (Karl Fisher (KF)). Compound 4A (208.4 kg, 1 eq., as a 62.6% solution in toluene) was added to the mixture in the hydrogenation reactor. Ethanol (389.4 kg, 1.86 w/w) was used to rinse the addition flask into the hydrogenation reactor. The hydrogenation reactor was pressurized/depressurized twice with nitrogen (2 bar), twice with hydrogen (5 bar), and then pressurized with hydrogen (9.8-10.2 bar). The resulting mixture was heated to 33-37° C. and stirred for 17-19 h. The system was depressurized/pressurized three times with nitrogen (1 bar). The suspension was filtered and washed three times with ethanol (total amount, 493.8 kg, 2.37 w/w). The filtrate was combined with HCl (concentrated, 83.4 kg, 1.07 eq.) and the resulting mixture stirred 25-35 min at 20-24° C. The mixture was concentrated by distillation at 78-80° C. and atmospheric pressure to remove water with a distillate target volume of 1167 L (5.6 L/kg based on imine Compound 4A) and the KF of the solution checked (≤1.5% H2O w/w). The mixture was stirred at 48-52° C. for 55-65 min, then 68-72° C. for 55-65 min, then cooled to 20-24° C. at a rate of 12° C./h and stirred for 25-35 min, then cooled to 0-4° C. at a rate of 10° C./h and stirred for 55-65 min. The suspension was filtered, the cake was washed twice with precooled ethanol (total amount, 329.2 kg, 1.58 w/w, 0° C.), and the collected solid was dried at 40° C. to afford Compound
5A as the HCl salt (156.5 kg, 66.4% uncorrected). 1H NMR (400 MHz, DMSO-d6) δ (ppm) −0.33–0.06 (m, 2H), 0.11-0.31 (m, 3H), 1.57 (d, J=6.57 Hz, 3H), 1.95 (br t, J=7.07 Hz, 2H), 2.26 (d, J=1.26 Hz, 3H), 3.68 (br d, J=7.83 Hz, 1H), 3.92 (br t, J=6.44 Hz, 1H), 6.98 (dd, J=7.71, 1.14 Hz, 1H), 7.28-7.36 (m, 2H), 7.37-7.50 (m, 5H). EST-MS: 298.2 m/z [M+H]+.Step 1E: Preparation of (S)-2-Cyclopropyl-1-(3-fluoro-4-methylphenyl)ethan-1-amine (Compound 6A) as the Hydrochloride Salt

5A (HCl salt, 156.5 kg, 1.00 eq.) and Pd/C (7.8 kg, 10% Pd basis) were added to an inerted hydrogenation reactor. The reactor was then pressurized/depressurized twice with nitrogen (2 bar) and then methanol (494.5 kg, 3.16 w/w) was added. The reactor was depressurized/pressurized three times with nitrogen (2 bar) then three times with hydrogen (5 bar), pressurized with hydrogen (9.8-10.2 bar), heated to 58-62° C. and stirred for 7-9 h. The reaction mixture was cooled to 20-24° C. The reactor was depressurized/pressurized three times with nitrogen (1 bar) and the suspension was filtered and washed three times with methanol (total amount, 492.9 kg, 3.15 w/w). The solution was concentrated at 63-67° C. and atmospheric pressure to a distillate target volume of 1408 L (9.0 L/kg Compound
6A), n-Heptane (1173.8 kg, 7.5 w/w) was added and the resulting mixture was heated to reflux at 65-80° C. and atmospheric pressure in Dean-Stark configuration to remove methanol. The suspension was cooled to 31-35° C. and filtered, the cake washed with n-heptane (147.1 kg, 0.94 w/w), and the solid dried at 40° C. to provide Compound
6A as the HCl salt (101.0 kg, 93.8% uncorrected, 99.6% ee). 1H NMR (400 MHz, DMSO-d6) δ (ppm) −0.12-0.14 (m, 2H), 0.26-0.42 (m, 2H), 0.44-0.55 (m, 1H), 1.70-1.83 (m, 2H), 2.23 (d, J=1.52 Hz, 3H), 4.24 (t, J=7.33 Hz, 1H), 7.22-7.29 (m, 1H), 7.29-7.36 (m, 1H), 7.40 (dd, J=10.99, 1.39 Hz, 1H). ESI-MS: 194.2 [M+H]+, 177.0 [M-NH2]+.Step 1F: Preparation of (S)-4-(2-chloro-4-methoxy-5-methylphenyl)-N-(2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl)-5-methylthiazol-2-amine (Compound 9A)

A mixture of n-heptane (146 kg), water (142 kg), Compound
6A (HCl salt, 57.4 kg), and aqueous sodium hydroxide (30% w/w, 41.0 kg) was stirred together. The layers were partitioned, and the aqueous layer removed. The organic layer was washed with water (170 kg) and the layers partitioned. The organic layer was set aside, n-Heptane (145 kg) and 1-(2-chloro-4-methoxy-5-methylphenyl)-2-thiocyanatopropan-1-one (Compound
8A, 66.1 kg, the preparation of Compound
8A has been previously described in International Publication Number WO2010/125414) were added to the reactor and heated to 85° C. The previously set aside organic layer containing the free base of Compound
6A was added at 84-85° C. to the reactor and rinsed with n-heptane (20 kg). The resulting mixture was stirred for 2 h at 83° C. Subsequently, the solvent was switched to methanol by four put-and-take additions/vacuum distillations of methanol (180 kg) at 55° C. with the target volume being 287 L remaining in the reactor. The suspension was cooled to 5° C. and water (570 kg) was added over 4 h at 5-10° C., with the first 60 kg added very slowly. The suspension was aged 2 h at 5° C. and then isolated by filtration, washed with a mixture of methanol/water (91/115 kg) and then a mixture of methanol/water (134/57 kg). The yellow solid was dried at 25° C. and 1 mbar for 17 h then 40° C. and 1 mbar for 22 h to afford Compound
9A (97.4 kg, 87.5% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm −0.01-0.14 (m, 2H), 0.29-0.42 (m, 2H), 0.61-0.73 (m, 1H), 1.47 (dt, J=13.83, 6.85 Hz, 1H), 1.76 (dt, J=13.89, 7.20 Hz, 1H), 2.00 (s, 3H), 2.11 (s, 3H), 2.19 (d, J=1.01 Hz, 3H), 3.82 (s, 3H), 4.54 (q, J=7.58 Hz, 1H), 7.00 (s, 1H), 7.06 (d, J=0.76 Hz, 1H), 7.08-7.14 (m, 2H), 7.18-7.23 (m, 1H), 7.89 (d, 1=8.08 Hz, 1H). ESI-MS: 445.3 m/z [M+H]+.Step 1G: Preparation of 4-(2-chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]-5-methyl-N-prop-2-ynyl-1,3-thiazol-2-amine (Compound 1)

A mixture of MTBE (279 kg), tetra-n-butylammonium bromide (10.5 kg), and Compound
9A (95.4 kg) were heated at 60° C. external temperature for 30 min and then cooled to 0° C. Aqueous potassium hydroxide (52.4% w/w, 364 kg) and propargyl bromide (39.4 kg as an 80% w/w solution in toluene, 1.19 eq.) were added at 0-5° C. The propargyl bromide additional funnel was washed with MTBE (25 kg) and the biphasic mixture was aged 14.5 h at 4-6° C. with vigorous stirring. Subsequently, water (191 kg) was added and the aqueous layer was discharged at 20° C. The organic layer was washed twice with water (382 kg) and once with aqueous acetic acid (5.26% w/w, 190 kg) at 20° C. The mixture is polish filtered, rinsed with ethanol (11 kg) and then the solvent switched to ethanol by 3 put-and-take additions/vacuum distillations of ethanol (300 kg) at 25-30° C. for the first cycle and then 35-40° C. with the target volume of each cycle being 250 L remaining in the reactor. Ethanol (164 kg) was added and the mixture heated at 60° C. external for 0.5 h before it was cooled to 25° C. in 1 h and seeded with authentic Form I (free base) of Compound 1 (0.340 kg) which can be prepared as described below in Example 2 and Example 3. The suspension was aged for 5 h, cooled to 0° C. in 2 h, aged 12 h, filtered, and washed twice with ethanol (24 kg each) pre-cooled to 0° C. The white solid was dried at 40° C. and 1 mbar for 19 h to yield 80.15 kg of Compound 1 (77.2% yield). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 0.14 (qt, J=8.59, 4.42 Hz, 2H), 0.29-0.48 (m, 2H), 0.61-0.82 (m, 1H), 1.89 (dt, J=14.08, 6.98 Hz, 1H), 2.07 (br d, J=7.83 Hz, 1H), 2.10 (s, 3H), 2.14 (s, 3H), 2.20 (d, J=1.01 Hz, 3H), 3.11 (t, J=2.27 Hz, 1H), 3.83 (s, 3H), 3.94-4.22 (m, 2H), 5.26 (t, J=7.58 Hz, 1H), 7.05 (s, 1H), 7.10-7.36 (m, 4H). ESI-MS: 483.2 m/z [M+H]+.Example 2: Preparation of 4-(2-chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]-5-methyl-N-prop-2-ynyl-1,3-thiazol-2-amine (Compound 1)
A mixture of MTBE (2 mL), tetra-n-butylammonium bromide (110 mg), and Compound
9A (1.003 g) at 0° C. was treated with aqueous potassium hydroxide (52.4% w/w, 1.80 mL, 2.73 g) and propargyl bromide (405 mg as an 80% w/w solution in toluene) maintaining the temperature at 0-5° C. The resulting biphasic mixture was aged 23 h at 4-6° C. Subsequently, water (2 mL) and MTBE (2 mL) were added and the aqueous layer was discharged. The organic layer was washed twice with water (4 mL) and once with aqueous acetic acid (5% w/w, 2 mL) at 20° C. Ethanol (4 mL) was added and then the solvent was switched to ethanol by 3 put-and-take additions/vacuum distillations of ethanol (6 mL) at 35-40° C. with the target volume of each cycle being 2 mL remaining in the vessel, except for the third cycle where the mixture was concentrated to dryness. Ethanol (4 mL) was added to the vessel and the mixture heated at 60° C. (external) for 0.5 h before it was cooled to 20° C. in 1 h and aged 18 h. The resulting suspension was cooled to 0° C., aged 6 h, filtered, and washed twice with ethanol (2 mL each) pre-cooled to 0° C. to afford a solid. The solid was dried at 40° C. under vacuum to afford Compound 1 (506 mg, 46% yield) as Form I. The 1H NMR and ESI-MS data matches that as described above in Example 1, Step 1G.Example 3: Preparation of 4-(2-chloro-4-methoxy-5-methylphenyl)-N-1(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]-5-methyl-N-prop-2-ynyl-1,3-thiazol-2-amine (Compound 1)
A mixture of MTBE (40 mL), tetra-n-butylammonium bromide (1.1 g), and Compound
9A (10.0 g) was heated to 45° C., aged for 10 min, then cooled to 0° C. The solution was treated with aqueous potassium hydroxide (52.4% w/w, 38.2 g) and propargyl bromide (3.36 g as an 80% w/w solution in toluene) maintaining the temperature at 0-5° C. The resulting biphasic mixture stirred vigorously for 16 h at 4-6° C. Subsequently, water (20 mL) was added and the aqueous layer was discharged. The organic layer was washed twice with water (40 mL) and once with aqueous acetic acid (5.2% w/w, 20 mL) at 20° C. The solvent was switched to ethanol by 4 put-and-take additions/vacuum distillations of ethanol (15 mL) at 35-40° C. with the target volume of each cycle being 15 mL remaining in the vessel. The solution was weighed to approximate the amount of ethanol remaining, and ethanol (26 mL) was added to the vessel to bring the total amount of ethanol to 40 mL. The solution was cooled to 4° C. and stirred for 45 min to afford a suspension. The suspension was heated to 38° C. in 15 min, aged 10 min, then cooled to 20° C. over 14 h. The suspension was cooled to 0° C., aged 1.5 h, filtered, and the solids washed twice with ethanol (7.5 mL each) pre-cooled to 0° C. The solid was dried at 40° C. under vacuum to afford Compound 1 (8.27 g, 76% yield) as Form I. The 1H NMR and ESL-MS data matches that as described above in Example 1, Step 1G.
The crystalline free base Compound
1, Form I was characterized by X-ray powder diffraction (XRPD) (FIG. 1
, Table 2) and DSC (FIG. 2
). The DSC indicated the crystalline Compound
1, Form I has an onset of melt (temperature) at about 83.7° C. (76.6 J/g). The Thermogravimetric Analysis (TGA) (FIG. 2
) of the crystalline free base exhibited substantially no weight loss (about 0.2%) from room temperature to ˜125° C. indicating Form I for the free base of Compound
1 is anhydrous.
Medical uses
Crinecerfont is indicated as adjunctive treatment to glucocorticoid replacement to control androgens in people four years of age and older with classic congenital adrenal hyperplasia.[1][2]
Adverse effects
The US Food and Drug Administration prescription label for crinecerfont has a warning for acute adrenal insufficiency or adrenal crisis.[2]
History
Crinecerfont’s approval is based on two randomized, double-blind, placebo-controlled trials in 182 adults and 103 children with classic congenital adrenal hyperplasia.[2] In the first trial, 122 adults received crinecerfont twice daily and 60 received placebo twice daily for 24 weeks.[2] After the first four weeks of the trial, the glucocorticoid dose was reduced to replacement levels, then adjusted based on levels of androstenedione, an androgen hormone.[2] The primary measure of efficacy was the change from baseline in the total glucocorticoid daily dose while maintaining androstenedione control at the end of the trial.[2] The group that received crinecerfont reduced their daily glucocorticoid dose by 27% while maintaining control of androstenedione levels, compared to a 10% daily glucocorticoid dose reduction in the group that received placebo.[2]
In the second trial, 69 children received crinecerfont twice daily and 34 received placebo twice daily for 28 weeks.[2] The primary measure of efficacy was the change from baseline in serum androstenedione at week four.[2] The group that received crinecerfont experienced a statistically significant reduction from baseline in serum androstenedione, compared to an average increase from baseline in the placebo group.[2] At the end of the trial, children assigned to crinecerfont were able to reduce their daily glucocorticoid dose by 18% while maintaining control of androstenedione levels compared to an almost 6% daily glucocorticoid dose increase in children assigned to placebo.[2]
The US Food and Drug Administration (FDA) granted the application for crinecerfont fast track, breakthrough therapy, orphan drug, and priority review designations.[2] The FDA granted the approval of Crenessity to Neurocrine Biosciences, Inc.[2]
Society and culture
Legal status
Crinecerfont was approved for medical use in the United States in December 2024.[1][2][5]
Names
Crinecerfont is the international nonproprietary name.[6]
Crinecerfont is sold under the brand name Crenessity.[1]
References
- ^ Jump up to:a b c d e f g “Crenessity- crinecerfont; capsule Crenessity- crinecerfont solution”. DailyMed. 1 December 2024. Retrieved 25 January 2025.
- ^ Jump up to:a b c d e f g h i j k l m n o p q “FDA Approves New Treatment for Congenital Adrenal Hyperplasia”. U.S. Food and Drug Administration (FDA) (Press release). 1 October 2024. Retrieved 16 December 2024. This article incorporates text from this source, which is in the public domain.
- ^ “Novel Drug Approvals for 2024”. U.S. Food and Drug Administration (FDA). 1 October 2024. Retrieved 20 December 2024.
- ^ New Drug Therapy Approvals 2024 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2025. Archived from the original on 21 January 2025. Retrieved 21 January 2025.
- ^ “Neurocrine Biosciences Announces FDA Approval of Crenessity (crinecerfont), a First-in-Class Treatment for Children and Adults With Classic Congenital Adrenal Hyperplasia” (Press release). Neurocrine Biosciences. 13 December 2024. Retrieved 16 December 2024 – via PR Newswire.
- ^ World Health Organization (2019). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 82”. WHO Drug Information. 33 (3). hdl:10665/330879.
Further reading
- Auchus, Richard; Chan, Jean; Farber, Robert; Fechner, Patricia; Giri, Nagdeep; Nokoff, Natalie; et al. (1 November 2022). “OR18-4 Crinecerfont (NBI-74788), a Novel CRF1 Receptor Antagonist, Lowers Adrenal Androgens and Precursors in Adolescents with Classic Congenital Adrenal Hyperplasia”. Journal of the Endocrine Society. 6 (Supplement_1): A618. doi:10.1210/jendso/bvac150.1281. PMC 9625506.
- Auchus, Richard J; Sarafoglou, Kyriakie; Fechner, Patricia Y; Vogiatzi, Maria; Giri, Nagdeep; Roberts, Eiry; et al. (8 May 2020). “OR25-03 The Effects of Crinecerfont (NBI-74788), a Novel CRF1 Receptor Antagonist, on Adrenal Androgens and Precursors in Patients with Classic Congenital Adrenal Hyperplasia: Results from A Multiple-Dose Phase 2 Study”. Journal of the Endocrine Society. 4 (Supplement_1): OR25-03. doi:10.1210/jendso/bvaa046.221. PMC 7209526.
- Auchus, Richard J; Sarafoglou, Kyriakie; Fechner, Patricia Y; Vogiatzi, Maria G; Imel, Erik A; Davis, Shanlee M; et al. (17 February 2022). “Crinecerfont Lowers Elevated Hormone Markers in Adults With 21-Hydroxylase Deficiency Congenital Adrenal Hyperplasia”. The Journal of Clinical Endocrinology & Metabolism. 107 (3): 801–812. doi:10.1210/clinem/dgab749. PMC 8851935. PMID 34653252.
- Newfield, Ron S; Sarafoglou, Kyriakie; Fechner, Patricia Y; Nokoff, Natalie J; Auchus, Richard J; Vogiatzi, Maria G; et al. (18 October 2023). “Crinecerfont, a CRF1 Receptor Antagonist, Lowers Adrenal Androgens in Adolescents With Congenital Adrenal Hyperplasia”. The Journal of Clinical Endocrinology & Metabolism. 108 (11): 2871–2878. doi:10.1210/clinem/dgad270. PMC 10583973. PMID 37216921.
External links
- “Crinecerfont (Code C174708)”. NCI Thesaurus.
- Clinical trial number NCT03525886 for “Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of NBI-74788 in Adults With Congenital Adrenal Hyperplasia” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Crenessity |
| Other names | SSR-125543, NBI-74788 |
| AHFS/Drugs.com | Crenessity |
| License data | US DailyMed: Crinecerfont |
| Routes of administration | By mouth |
| Drug class | Corticotropin-releasing factor type 1 receptor antagonist |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 752253-39-7 |
| PubChem CID | 5282340 |
| DrugBank | DB18518 |
| ChemSpider | 4445507 |
| UNII | MFT24BX55I |
| KEGG | D12366 |
| ChEBI | CHEBI:34969 |
| ChEMBL | ChEMBL291657 |
| CompTox Dashboard (EPA) | DTXSID10996687 |
| Chemical and physical data | |
| Formula | C27H28ClFN2OS |
| Molar mass | 483.04 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
- Prete A, Auchus RJ, Ross RJ: Clinical advances in the pharmacotherapy of congenital adrenal hyperplasia. Eur J Endocrinol. 2021 Nov 30;186(1):R1-R14. doi: 10.1530/EJE-21-0794. [Article]
- Yogi A, Kashimada K: Current and future perspectives on clinical management of classic 21-hydroxylase deficiency. Endocr J. 2023 Oct 30;70(10):945-957. doi: 10.1507/endocrj.EJ23-0075. Epub 2023 Jun 29. [Article]
- FDA Approved Drug Products: Crenessity (crinecerfont) capsules/solution for oral administration (December 2024) [Link]
- FDA News Release: FDA Approves New Treatment for Congenital Adrenal Hyperplasia [Link]
///////Crinecerfont, Crenessity, FDA 2024, APPROVALS 2024, 752253-39-7, SSR125543, SSR 125543, SSR-125543, WHO 10958, 06-RORI, NBI-74788, ORPHAN DRUG
Ensartinib



Ensartinib
X396, X 396
- 1370651-20-9
- C26H27Cl2FN6O3,
561.4 g/mol - SMA5ZS5B22
6-amino-5-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-N-[4-[(3R,5S)-3,5-dimethylpiperazine-1-carbonyl]phenyl]pyridazine-3-carboxamide
- 3-Pyridazinecarboxamide, 6-amino-5-((1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-N-(4-(((3R,5S)-3,5-dimethyl-1-piperazinyl)carbonyl)phenyl)
- 6-amino-5-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-N-[4-[(3R,5S)-3,5-dimethylpiperazine-1-carbonyl]phenyl]pyridazine-3-carboxamide
- 6-Amino-5-((1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)- N-(4-((3R,5S)-3,5-dimethylpiperazine- 1-carbonyl)phenyl)pyridazine-3-carboxamide
FDA 12/18/2024, Ensacove, To treat non-small cell lung cancer
Ensartinib, sold under the brand name Ensacove, is an anti-cancer medication used for the treatment of non-small cell lung cancer.[1] Ensartinib is an Anaplastic lymphoma kinase (ALK) inhibitor used as the salt ensartinib hydrochloride.[1] It is taken by mouth.[1]
The most common adverse reactions include rash, musculoskeletal pain, constipation, cough, pruritis, nausea, edema, pyrexia, and fatigue.[2]
Ensartinib was approved for medical use in the United States in December 2024.[1][2][3][4]
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US90227390&_cid=P11-M9JBTT-36001-1
Synthesis of 6-[bis(tert-butoxycarbonyl)amino]-5-[(1R)-1-(2,6-dichloro-3-fluoro-phenyl)ethoxy]pyridazine-3-carboxylic acid (B)


Synthesis of 6-[bis(tert-butoxycarbonyl)amino]-5-[(1S)-1-(2,6-dichloro-3-fluoro-phenyl)ethoxy]pyridazine-3-carboxylic acid (C)

| Step 1: To a solution of A5 (41.8 g, 200 mmol) in 1,2-dichloroethane (800 mL) was added Boc-L-Pro (26.9 g, 125 mmol) followed by EDCI (31.1 g, 163 mmol) and DMAP (4.12 g, 33.8 mmol) at 0° C. The resulting mixture was stirred at r.t. overnight and then water (350 mL) was added and separated, the water phase was extracted with DCM(150 mL×3), dried over MgSO 4, concentrated and purified by column chromatography to (PE:EA=30:1) to give C1 (13.72 g, yield: 65.6%). |
| Step 2: The procedure from C1 to C was similar to that of B1 to B (9.46 g, yield: 26.4% from C1). |

Medical uses
Ensartinib is indicated for the treatment of adults with anaplastic lymphoma kinase (ALK)-positive locally advanced or metastatic non-small cell lung cancer who have not previously received an ALK-inhibitor.[1][2]
History
Efficacy was evaluated in eXALT3 (NCT02767804), an open-label, randomized, active-controlled, multicenter trial in 290 participants with locally advanced or metastatic ALK-positive non-small cell lung cancer who had not previously received an ALK-targeted therapy.[2] Participants were randomized 1:1 to receive ensartinib or crizotinib.[2]
Society and culture
Legal status
Ensartinib was approved for medical use in the United States in December 2024.[2][3][5]
Name
Ensartinib is the international nonproprietary name.[6]
Ensartinib is sold under the brand name Ensacove.[1][2][3]
References
- ^ Jump up to:a b c d e f g “Ensacove (ensartinib) capsules, for oral use” (PDF). Xcovery Holdings, Inc. U.S. Food and Drug Administration. December 2024.
- ^ Jump up to:a b c d e f g “FDA approves ensartinib for ALK-positive locally advanced or metastatic non-small cell lung cancer”. U.S. Food and Drug Administration (FDA). 18 December 2024. Retrieved 20 December 2024. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c “Novel Drug Approvals for 2024”. U.S. Food and Drug Administration (FDA). 1 October 2024. Retrieved 20 December 2024.
- ^ New Drug Therapy Approvals 2024 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2025. Archived from the original on 21 January 2025. Retrieved 21 January 2025.
- ^ “FDA Approval of Ensartinib for ALK-Positive Locally Advanced or Metastatic Non-Small Cell Lung Cancer (NSCLC)” (Press release). Xcovery Holdings. 19 December 2024. Retrieved 20 December 2024 – via Business Wire.
- ^ World Health Organization (2017). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 77”. WHO Drug Information. 31 (1). hdl:10665/330984.
External links
- “Ensartinib (Code C102754)”. NCI Thesaurus.
- Clinical trial number NCT02767804 for “eXalt3: Study Comparing X-396 (Ensartinib) to Crizotinib in ALK Positive Non-Small Cell Lung Cancer (NSCLC) Patients” at ClinicalTrials.gov
Horn L, Infante JR, Reckamp KL, Blumenschein GR, Leal TA, Waqar SN, Gitlitz BJ, Sanborn RE, Whisenant JG, Du L, Neal JW, Gockerman JP, Dukart G, Harrow K, Liang C, Gibbons JJ, Holzhausen A, Lovly CM, Wakelee HA: Ensartinib (X-396) in ALK-Positive Non-Small Cell Lung Cancer: Results from a First-in-Human Phase I/II, Multicenter Study. Clin Cancer Res. 2018 Jun 15;24(12):2771-2779. doi: 10.1158/1078-0432.CCR-17-2398. Epub 2018 Mar 21. [Article]- FDA Approved Drug Products: ENSACOVETM (ensartinib) capsules, for oral use (Dec 2024) [Link]
- NCI Formulary: Ensartinib (X-396) [Link]
| Clinical data | |
|---|---|
| Trade names | Ensacove |
| Other names | X-396 |
| License data | US DailyMed: Ensartinib |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| CAS Number | 1370651-20-9 |
| PubChem CID | 56960363 |
| DrugBank | DB14860 |
| ChemSpider | 58828042 |
| UNII | SMA5ZS5B22 |
| KEGG | D11346 |
| ChEMBL | ChEMBL4113131 |
| ECHA InfoCard | 100.306.918 |
| Chemical and physical data | |
| Formula | C26H27Cl2FN6O3 |
| Molar mass | 561.44 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
\/////////Ensartinib, FDA 2024, APPROVALS 2024, Ensacove, X396, X 396, GLXC-15836, BCP26265, EX-A2941, NSC793150, s8230
BENZGALANTAMINE


BENZGALANTAMINE
CAS 224169-27-1
Benzgalantamine gluconate, 1542321-58-3
- 6H-Benzofuro[3a,3,2-ef][2]benzazepin-6-ol, 4a,5,9,10,11,12-hexahydro-3-methoxy-11-methyl-, benzoate (ester), (4aS,6R,8aS)- (9CI)
- Alpha 1062
- GLN 1062
- Memogain
6h-benzofuro(3a,3,2-ef)(2)benzazepin-6-ol, 4a,5,9,10,11,12-hexahydro-3-methoxy-11-methyl-, benzoate (ester), (4as,6r,8as)-
| Formula | C24H25NO4 |
|---|---|
| Molar mass | 391.467 g·mol−1 |


External IDs GLN-1062 gluconate
UNIILN7PMJ4P57
CAS Number1542321-58-3
WeightAverage: 587.622
Monoisotopic: 587.236661015
Chemical FormulaC30H37NO11
Benzgalantamine, sold under the brand name Zunveyl, is a medication used for the treatment of mild to moderate dementia of the Alzheimer’s type.[1] It is a cholinesterase inhibitor.[1] Benzgalantamine is a prodrug of galantamine.[1]
The most common side effects include nausea, vomiting, diarrhea, dizziness, headache, and decreased appetite.[1]
Benzgalantamine was approved for medical use in the United States in July 2024.[1][2][3]
compounds that, in addition to enhancing the sensitivity to acetylcholine and choline, and to their agonists, of neuronal cholinergic receptors, and/or acting as cholinesterase inhibitors and/or neuroprotective agents, have enhanced blood-brain barrier permeability in comparison to their parent compounds. The compounds are derived (either formally by their chemical structure or directly by chemical synthesis) from natural compounds belonging to the class of amaryllidaceae alkaloids e.g., Galantamine, Narwedine and Lycoramine, or from metabolites of said compounds. The compounds of the present invention can either interact as such with their target molecules, or they can act as “pro-drugs”, in the sense that after reaching their target regions in the body, they are converted by hydrolysis or enzymatic attack to the original parent compound and react as such with their target molecules, or both. The compounds of this disclosure may be used as medicaments for the treatment of human brain diseases associated with a cholinergic deficit, including the neurodegenerative diseases Alzheimer’s and Parkinson’s disease and the neurological/psychiatric diseases vascular dementia, schizophrenia and epilepsy. Galantamine derivatives disclosed herein have higher efficacy and lower levels of adverse side effects in comparison to galantamine, in treatment of human brain diseases.
Benzgalantamine is a prodrug of galantamine. Gastrointestinal adverse effects are the most frequently reported side effects in patients undergoing treatment with cholinesterase inhibitors, including galantamine, and are often a reason for treatment discontinuation.2 As a prodrug, benzagalantamine remains inert as it passes through the stomach, thereby avoiding many of the gastrointestinal effects associated with peripheral cholinesterase inhibition.4
Benzgalantamine was approved by the FDA in July 2024 for the treatment of mild-to-moderate dementia in Alzheimer’s patients.3,4
SCHEME


US20090253654
https://patentscope.wipo.int/search/en/detail.jsf?docId=US42863485&_cid=P12-M8ZQT3-74791-1

| O-Benzoyl-galantamine(=(4aS,6R,8aS)-4a,5,9,10,11,12-Hexahydro-3-methoxy-11-methyl-6H-benzofuro[3a,3,2-ef][2]benzazepin-6-ol, benzoate (ester)); yield: 78% |
| O-3,4-Dichlorobenzoyl-galantamine(=(4aS,6R,8aS)-4a,5,9,10,11,12-Hexahydro-3-methoxy-11-methyl-6H-benzofuro[3a,3,2-ef][2]benzazepin-6-ol, 3,4-dichlorobenzoate (ester)); off-white solid; mp. 69-70° C. |
WO2009127218
US20220220121
https://patentscope.wipo.int/search/en/detail.jsf?docId=US368470159&_cid=P12-M8ZR8V-88578-1
Experiment 1
The gluconate salt of Alpha-1062 was created according to the following previously established general scheme:

AND
US20090253654
Medical uses
Benzgalantamine is indicated for the treatment of mild to moderate dementia of the Alzheimer’s type in adults.[1][2]
Side effects
The most common side effects include nausea, vomiting, diarrhea, dizziness, headache, and decreased appetite.[1]
Society and culture
Legal status
Benzgalantamine was approved for medical use in the United States in July 2024.[1][2]
Names
Benzgalantamine is the international nonproprietary name.[4]
References
- ^ Jump up to:a b c d e f g h i “Zunveyl- benzgalantamine tablet, delayed release”. DailyMed. 8 August 2024. Retrieved 15 August 2024.
- ^ Jump up to:a b c https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2024/218549Orig1s000ltr.pdf
- ^ “Alpha Cognition’s Oral Therapy Zunveyl Receives FDA Approval to Treat Alzheimer’s Disease” (Press release). Alpha Cognition. 29 July 2024. Archived from the original on 4 August 2024. Retrieved 4 August 2024 – via Business Wire.
- ^ World Health Organization (2022). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 88”. WHO Drug Information. 36 (3). hdl:10665/363551.
- Baakman AC, ‘t Hart E, Kay DG, Stevens J, Klaassen ES, Maelicke A, Groeneveld GJ: First in human study with a prodrug of galantamine: Improved benefit-risk ratio? Alzheimers Dement (N Y). 2016 Jan 20;2(1):13-22. doi: 10.1016/j.trci.2015.12.003. eCollection 2016 Jan. [Article]
- Bakker C, van der Aart J, Hart EP, Klaassen ES, Bergmann KR, van Esdonk MJ, Kay DG, Groeneveld GJ: Safety, pharmacokinetics, and pharmacodynamics of Gln-1062, a prodrug of galantamine. Alzheimers Dement (N Y). 2020 Oct 13;6(1):e12093. doi: 10.1002/trc2.12093. eCollection 2020. [Article]
- FDA Approved Drug Products: Zunveyl (benzgalantamine) delayed-release tablets for oral use [Link]
- Fierce Pharma: Alpha Cognition’s delayed-release Alzheimer’s drug Zunveyl passes muster with FDA [Link]
- Alpha Cognition: Corporate Presentation Oct 2024 [Link]
External links
- “Benzgalantamine (Code C188656)”. NCI Thesaurus.
| Clinical data | |
|---|---|
| Trade names | Zunveyl |
| Other names | ALPHA-1062 |
| AHFS/Drugs.com | Zunveyl |
| License data | US DailyMed: Benzgalantamine |
| Routes of administration | By mouth |
| Drug class | Cholinesterase inhibitor |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| CAS Number | 224169-27-11542321-58-3 |
| DrugBank | DB19353 |
| UNII | XOI2Q0ZF7GLN7PMJ4P57 |
| KEGG | D12930D12931 |
| ChEMBL | ChEMBL5095056 |
| Chemical and physical data | |
| Formula | C24H25NO4 |
| Molar mass | 391.467 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
//////////BENZGALANTAMINE, Alpha 1062, GLN 1062, Memogain, FDA 2024, APPROVALS 2024, Zunveyl
Deuruxolitinib


Deuruxolitinib
C17H18N6, 314.422
Fda approved Leqselvi, 7/25/2024, To treat severe alopecia areata
C-21543, CTP 543, CTP-543, CTP543
(3r)-3-(2,2,3,3,4,4,5,5-d8)cyclopentyl-3-(4-(7h-pyrrolo(2,3-d)pyrimidin-4-yl)-1h-pyrazol-1-yl)propanenitrile
1h-pyrazole-1-propanenitrile, .beta.-(cyclopentyl-2,2,3,3,4,4,5,5-d8)-4-(7h-pyrrolo(2,3-d)pyrimidin-4-yl)-, (.beta.r)-D8-ruxolitinib
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Deuruxolitinib phosphate | 8VJ43S4LCM | 2147706-60-1 | JFMWPOCYMYGEDM-NTVOUFPTSA-N |
unii
0CA0VSF91Y
Deuruxolitinib, sold under the brand name Leqselvi, is a medication used for the treatment of alopecia areata.[1] It is a Janus kinase inhibitor selective for JAK1 and JAK2.[2] Although the relative effectiveness of deuruxolitinib and another Janus kinase inhibitor—baricitinib—for alopecia areata may vary depending on the population studied, both drugs are more effective than alternative treatments.[3]
Deuruxolitinib was approved for medical use in the United States in July 2024.[1][4]
Medical uses
Deuruxolitinib is indicated for the treatment of adults with severe alopecia areata.[1]
Side effects
The FDA prescribing label for deuruxolitinib contains a boxed warning for serious infections; malignancies; cardiovascular death, myocardial infarction, and stroke; and thrombosis.[5]
Society and culture
Names
Deuruxolitinib is the international nonproprietary name[6] and the United States Adopted Name.[7]
SYN
20240108633METHOD FOR PREVENTING OR TREATING DISEASE OR CONDITION ASSOCIATED WITH ANTITUMOR AGENT
20240058345TREATMENT OF HAIR LOSS DISORDERS WITH DEUTERATED JAK INHIBITORS
2023553253重水素化JAK阻害剤による脱毛障害の治療のためのレジメン
20230390292REGIMENS FOR THE TREATMENT OF HAIR LOSS DISORDERS WITH DEUTERATED JAK INHIBITORS
20230322787PROCESS FOR PREPARING ENANTIOMERICALLY ENRICHED JAK INHIBITORS
1020230093504중수소화된 JAK 억제제를 이용한 탈모 장애의 치료를 위한 요법
WO/2023/018954TREATMENT OF JAK-INHIBITION-RESPONSIVE DISORDERS WITH PRODRUGS OF JAK INHIBITORS
2022171838TREATMENT OF ALOPECIA CAUSED BY DEUTERATED JAK INHIBITOR
2022171838TREATMENT OF ALOPECIA CAUSED BY DEUTERATED JAK INHIBITOR
20220226327Combination therapy comprising JAK pathway inhibitor and rock inhibitor
20220213105PROCESS FOR PREPARING ENANTIOMERICALLY ENRICHED JAK INHIBITORS
20220202834JAK inhibitor with a vitamin D analog for treatment of skin diseases
20210387991Deuterated JAK inhibitor and uses thereof SUN
WO/2020/163653PROCESS FOR PREPARING ENANTIOMERICALLY ENRICHED JAK INHIBITORS CONCERT
20200222408TREATMENT OF HAIR LOSS DISORDERS WITH DEUTERATED JAK INHIBITORS
2019516684Treatment of Hair Loss Disorders with Deuterated JAK Inhibitors
PATENT
US20210387991
USE OF COMPD NOT SYNTHESIS
https://patentscope.wipo.int/search/en/detail.jsf?docId=US344953814&_cid=P12-M0XGHQ-19840-2
Example 1
Synthesis of Compound 10

| HRMS: Agilent 6530 Q-TOF LC/MS system with electrospray ionization in positive mode. The measured time-of-flight mass-to-charge ratio (m/z) is 333.22839 (theoretical value=333.22735). |
| Clinical data | |
|---|---|
| Trade names | Leqselvi |
| Other names | CTP-543 |
| License data | US DailyMed: Deuruxolitinib |
| Routes of administration | By mouth |
| Drug class | Janus kinase inhibitor |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1513883-39-0as phosphate: 2147706-60-1 |
| PubChem CID | 72704611as phosphate: 154572727 |
| DrugBank | DB18847 |
| ChemSpider | 115010950 |
| UNII | 0CA0VSF91Yas phosphate: 8VJ43S4LCM |
| KEGG | D11866as phosphate: D11867 |
| ChEMBL | ChEMBL4594381 |
| Chemical and physical data | |
| Formula | C17H18N6 |
| Molar mass | 306.373 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
References
King B, Mesinkovska N, Mirmirani P, Bruce S, Kempers S, Guttman-Yassky E, Roberts JL, McMichael A, Colavincenzo M, Hamilton C, Braman V, Cassella JV: Phase 2 randomized, dose-ranging trial of CTP-543, a selective Janus Kinase inhibitor, in moderate-to-severe alopecia areata. J Am Acad Dermatol. 2022 Aug;87(2):306-313. doi: 10.1016/j.jaad.2022.03.045. Epub 2022 Mar 29. [Article]Yan T, Wang T, Tang M, Liu N: Comparative efficacy and safety of JAK inhibitors in the treatment of moderate-to-severe alopecia areata: a systematic review and network meta-analysis. Front Pharmacol. 2024 Apr 10;15:1372810. doi: 10.3389/fphar.2024.1372810. eCollection 2024. [Article]Barati Sedeh F, Michaelsdottir TE, Henning MAS, Jemec GBE, Ibler KS: Comparative Efficacy and Safety of Janus Kinase Inhibitors Used in Alopecia Areata: A Systematic Review and Meta-analysis. Acta Derm Venereol. 2023 Jan 25;103:adv00855. doi: 10.2340/actadv.v103.4536. [Article]Sardana K, Bathula S, Khurana A: Which is the Ideal JAK Inhibitor for Alopecia Areata – Baricitinib, Tofacitinib, Ritlecitinib or Ifidancitinib – Revisiting the Immunomechanisms of the JAK Pathway. Indian Dermatol Online J. 2023 Jun 28;14(4):465-474. doi: 10.4103/idoj.idoj_452_22. eCollection 2023 Jul-Aug. [Article]FDA Approved Drug Products: LEQSELVI (deuruxolitinib) tablets, for oral use [Link]AJMC: FDA Approves Deuruxolitinib for Alopecia Areata [Link]
^ Jump up to:a b c d “Archived copy” (PDF). Archived (PDF) from the original on 29 July 2024. Retrieved 26 July 2024.
- ^ King, Brett; Mesinkovska, Natasha; Mirmirani, Paradi; Bruce, Suzanne; Kempers, Steve; Guttman-Yassky, Emma; et al. (August 2022). “Phase 2 randomized, dose-ranging trial of CTP-543, a selective Janus Kinase inhibitor, in moderate-to-severe alopecia areata”. Journal of the American Academy of Dermatology. 87 (2): 306–313. doi:10.1016/j.jaad.2022.03.045. ISSN 1097-6787. PMID 35364216. S2CID 247866262.
- ^ SEDEH, Farnam Barati; MICHAELSDÓTTIR, Thorunn Elísabet; HENNING, Mattias Arvid Simon; JEMEC, Gregor Borut Ernst; IBLER, Kristina Sophie (25 January 2023). “Comparative Efficacy and Safety of Janus Kinase Inhibitors Used in Alopecia Areata: A Systematic Review and Meta-analysis”. Acta Dermato-Venereologica. 103: 4536. doi:10.2340/actadv.v103.4536. ISSN 0001-5555. PMC 10391778. PMID 36695751.
- ^ “U.S. FDA Approves Leqselvi (deuruxolitinib), an Oral JAK Inhibitor for the Treatment of Severe Alopecia Areata” (Press release). Sun Pharmaceutical. 25 July 2024. Archived from the original on 26 July 2024. Retrieved 26 July 2024 – via PR Newswire.
- ^ http://www.leqselvi.com/&a=Prescribing Information
- ^ World Health Organization (2021). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 86”. WHO Drug Information. 35 (3). hdl:10665/346562.
- ^ “Deuruxolitinib”. American Medical Association. Retrieved 27 July 2024.
Further reading
Passeron T, King B, Seneschal J, Steinhoff M, Jabbari A, Ohyama M, et al. (2023). “Inhibition of T-cell activity in alopecia areata: recent developments and new directions”. Frontiers in Immunology. 14: 1243556. doi:10.3389/fimmu.2023.1243556. PMC 10657858. PMID 38022501.
External links
- “Deuruxolitinib (Code C175770)”. NCI Thesaurus.
- “Deuruxolitinib Phosphate (Code C175771)”. NCI Thesaurus.
- Clinical trial number NCT04518995 for “Study to Evaluate the Efficacy and Safety of CTP-543 in Adults With Moderate to Severe Alopecia Areata (THRIVE-AA1) (THRIVE-AA1)” at ClinicalTrials.gov
- Clinical trial number NCT04797650 for “Study to Evaluate the Efficacy and Safety of CTP-543 in Adults With Moderate to Severe Alopecia Areata (THRIVE-AA2) (THRIVE-AA2)” at ClinicalTrials.gov
////Deuruxolitinib, alopecia areata, Leqselvi , approvals 2024, fda 2024, C-21543, CTP 543, CTP-543, CTP543, UNII-0CA0VSF91Y, WHO 11622
Vorasidenib


Vorasidenib
6-(6-chloropyridin-2-yl)-N2,N4-bis[(2R)-1,1,1-trifluoropropan-2-yl]-1,3,5-triazine-2,4-diamine
CAS 1644545-52-7, C14H13ClF6N6, 414.74
FDA APPROVED, 8/6/2024, Voranigo, To treat Grade 2 astrocytoma or oligodendroglioma
UNII 789Q85GA8P
- AG 881
- AG-881
- AG881
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Vorasidenib citrate | X478M962XG | 2316810-02-1 | YOUTVRFNJAAFNS-DLVAHKFUSA-N |
| Vorasidenib citrate anhydrous | W4XG3EQK7B | 2316810-00-9 | OCEHQNOYRLHJCI-WPRTUUMNSA-N |
Vorasidenib, sold under the brand name Voranigo, is an anti-cancer medication used for the treatment of certain forms of glioma.[1][2] Vorasidenib acts to inhibit the enzymes isocitrate dehydrogenase-1 (IDH1) and isocitrate dehydrogenase-2 (IDH2).[1][2]
The most common adverse reactions include fatigue, headache, increased risk of COVID-19 infection, musculoskeletal pain, diarrhea, nausea, and seizures.[2]
Vorasidenib was approved for medical use in the United States in August 2024.[2][3] It is the first approval by the US Food and Drug Administration (FDA) of a systemic therapy for people with grade 2 astrocytoma or oligodendroglioma with a susceptible isocitrate dehydrogenase-1 or isocitrate dehydrogenase-2 mutation.[2]
Medical uses
Vorasidenib is indicated for the treatment of people aged twelve years of age and older with grade 2 astrocytoma or oligodendroglioma with a susceptible isocitrate dehydrogenase-1 or isocitrate dehydrogenase-2 mutation, following surgery including biopsy, sub-total resection, or gross total resection.[2]
Side effects
The most common adverse reactions include fatigue, headache, increased risk of COVID-19 infection, musculoskeletal pain, diarrhea, nausea, and seizures.[2] The most common grade 3 or 4 laboratory abnormalities include increased alanine aminotransferase, increased aspartate aminotransferase, GGT increased, and decreased neutrophils.[2]
History
Efficacy was evaluated in 331 participants with grade 2 astrocytoma or oligodendroglioma with a susceptible isocitrate dehydrogenase-1 or isocitrate dehydrogenase-2 mutation following surgery enrolled in INDIGO (NCT04164901), a randomized, multicenter, double-blind, placebo-controlled trial.[2] Participants were randomized 1:1 to receive vorasidenib 40 mg orally once daily or placebo orally once daily until disease progression or unacceptable toxicity.[2] Isocitrate dehydrogenase-1 or isocitrate dehydrogenase-2 mutation status was prospectively determined by the Life Technologies Corporation Oncomine Dx Target Test.[2] Participants randomized to placebo were allowed to cross over to vorasidenib after documented radiographic disease progression.[2] Participants who received prior anti-cancer treatment, including chemotherapy or radiation therapy, were excluded.[2]
Society and culture
Legal status
Vorasidenib was approved for medical use in the United States in August 2024.[2]
The FDA granted the application for vorasidenib priority review, fast track, breakthrough therapy, and orphan drug designations.[2]
SYN
WO/2024/161041NOVEL COMPOUNDS THAT CAN BE USED AS THERAPEUTIC AGENTS
20240254118PRMT5 INHIBITORS AND USES THEREOF
118359585共晶体、其药物组合物以及涉及其的治疗方法
WO/2024/148437USE OF PCLX-001 OR PCLX-002 AS A RADIOSENSITIZER
20240238424HETEROBIFUNCTIONAL COMPOUNDS AND METHODS OF TREATING DISEASE
1020240097895CD73 화합물
WO/2024/137852PRMT5 INHIBITORS AND USES THEREOF
2024057088THERAPEUTICALLY ACTIVE COMPOUNDS AND THEIR METHODS OF USE
20240116928CD73 COMPOUNDS
117586228Preparation method of triazine medicine
20240041892THERAPEUTICALLY ACTIVE COMPOUNDS AND THEIR METHODS OF USE
117529323Therapeutically active compounds and methods of use thereof
WO/2024/006929CD73 COMPOUNDS
PATENT
https://patents.google.com/patent/US10028961B2/en
Step 3: Preparation of 6-(6-chloropyridin-2-yl)-N2,N4-bis((R)-1,1,1-trifluoro propan-2-yl)-1,3,5-triazine-2,4-diamine
A mixture of 2,4-dichloro-6-(6-chloro-pyridin-2-yl)-1,3,5-triazine (0.27 g, 1.04 mol), (R)-1,1,1-trifluoropropan-2-amine hydrochloride (0.39 g, 2.6 mol), and potassium carbonate (0.43 g, 3.1 mol) in dry 1,4-dioxane (2.5 mL) was stirred under the atmosphere of N2 at 50° C. for 36 hr then at 100° C. for another 36 hr until the reaction was complete. The resulting mixture was filtered through Celite and the cake was washed with EtOAc. The filtrate was concentrated and the residue was purified by standard methods to give the desired product.

1H NMR (400 MHz, CDCl3) δ 8.32 (m, 1H), 7.80 (m, 1H), 7.48 (d, J=7.9 Hz, 1H), 5.61 (m, 1.5H), 5.25 (m, 0.5H), 5.09 (m, 0.5H), 4.88 (m, 1.5H), 1.54-1.26 (m, 6H). LC-MS: m/z 415 (M+H)+.
The procedure set forth in Example 10 was used to produce the following compounds using the appropriate starting materials.Compound 6-(6-Chloropyridin-2-yl)-N2,N4-bis((S)-1,1,1-trifluoropropan-2-yl)-1,3,5-triazine-2,4-diamine

1H NMR (400 MHz, CDCl3) δ 8.29-8.16 (m, 1H), 7.72 (d, J=7.6 Hz, 1H), 7.41 (d, J=7.9 Hz, 1H), 5.70-5.13 (m, 2H), 5.09-4.71 (m, 2H), 1.34 (m, 6H). LC-MS: m/z 415 (M+H)+.Compound 6-(6-Chloropyridin-2-yl)-N2—((R)-1,1,1-trifluoropropan-2-yl)-N4—((S)-1,1,1-trifluoropropan-2-yl)-1,3,5-triazine-2,4-diamine

1H NMR (400 MHz, CDCl3) δ 8.41-8.23 (m, 1H), 7.83 (s, 1H), 7.51 (d, J=6.2 Hz, 1H), 5.68-5.20 (m, 2H), 5.18-4.81 (m, 2H), 1.48-1.39 (m, 6H). LC-MS: m/z 415 (M+H)+.Compound 6-(6-Chloropyridin-2-yl)-N2,N4-bis(1,1,1-trifluoropropan-2-yl)-1,3,5-triazine-2,4-diamine

1H NMR (400 MHz, CDCl3) δ 8.29-8.16 (m, 1H), 7.72 (d, J=7.6 Hz, 1H), 7.41 (d, J=7.9 Hz, 1H), 5.70-5.13 (m, 2H), 5.09-4.71 (m, 2H), 1.34 (m, 6H). LC-MS: m/z 415 (M+H)+.

| Clinical data | |
|---|---|
| Trade names | Voranigo |
| License data | US DailyMed: Vorasidenib |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1644545-52-7 |
| PubChem CID | 117817422 |
| IUPHAR/BPS | 10663 |
| DrugBank | DB17097 |
| ChemSpider | 64835242 |
| UNII | 789Q85GA8P |
| KEGG | D11834 |
| ChEMBL | ChEMBL4279047 |
| Chemical and physical data | |
| Formula | C14H13ClF6N6 |
| Molar mass | 414.74 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
References
^ Jump up to:a b c “Voranigo- vorasidenib citrate tablet, film coated”. DailyMed. 9 August 2024. Retrieved 15 August 2024.
- ^ Jump up to:a b c d e f g h i j k l m n o “FDA approves vorasidenib for Grade 2 astrocytoma or oligodendroglioma with a susceptible IDH1 or IDH2 mutation”. U.S. Food and Drug Administration (FDA). 6 August 2024. Archived from the original on 7 August 2024. Retrieved 7 August 2024.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Servier’s Voranigo (vorasidenib) Tablets Receives FDA Approval as First Targeted Therapy for Grade 2 IDH-mutant Glioma” (Press release). Servier Pharmaceuticals. 6 August 2024. Archived from the original on 7 August 2024. Retrieved 7 August 2024 – via PR Newswire.
Further reading
- Mellinghoff IK, Lu M, Wen PY, Taylor JW, Maher EA, Arrillaga-Romany I, et al. (March 2023). “Vorasidenib and ivosidenib in IDH1-mutant low-grade glioma: a randomized, perioperative phase 1 trial”. Nature Medicine. 29 (3): 615–622. doi:10.1038/s41591-022-02141-2. PMC 10313524. PMID 36823302.
- Mellinghoff IK, Penas-Prado M, Peters KB, Burris HA, Maher EA, Janku F, et al. (August 2021). “Vorasidenib, a Dual Inhibitor of Mutant IDH1/2, in Recurrent or Progressive Glioma; Results of a First-in-Human Phase I Trial”. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research. 27 (16): 4491–4499. doi:10.1158/1078-0432.CCR-21-0611. PMC 8364866. PMID 34078652.
- Mellinghoff IK, van den Bent MJ, Blumenthal DT, Touat M, Peters KB, Clarke J, et al. (August 2023). “Vorasidenib in IDH1- or IDH2-Mutant Low-Grade Glioma”. The New England Journal of Medicine. 389 (7): 589–601. doi:10.1056/NEJMoa2304194. PMID 37272516.
- Popovici-Muller J, Lemieux RM, Artin E, Saunders JO, Salituro FG, Travins J, et al. (April 2018). “Discovery of AG-120 (Ivosidenib): A First-in-Class Mutant IDH1 Inhibitor for the Treatment of IDH1 Mutant Cancers”. ACS Medicinal Chemistry Letters. 9 (4): 300–305. doi:10.1021/acsmedchemlett.7b00421. PMC 5900343. PMID 29670690.
External links
Clinical trial number NCT04164901 for “Study of Vorasidenib (AG-881) in Participants With Residual or Recurrent Grade 2 Glioma With an IDH1 or IDH2 Mutation (INDIGO)” at ClinicalTrials.gov
- Clinical trial number NCT02481154 for “Study of Orally Administered AG-881 in Patients With Advanced Solid Tumors, Including Gliomas, With an IDH1 and/or IDH2 Mutation” at ClinicalTrials.gov
- Clinical trial number NCT03343197 for “Study of AG-120 and AG-881 in Subjects With Low Grade Glioma” at ClinicalTrials.gov
////////Vorasidenib, Voranigo, FDA 2024, APPROVALS 2024, AG 881, AG-881, AG881

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}