
Home » Posts tagged 'approvals 2024' (Page 2)
Tag Archives: approvals 2024
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Envonalkib
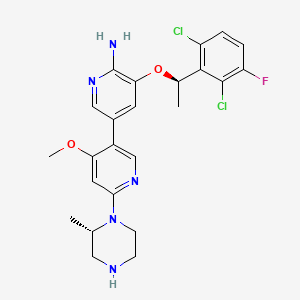
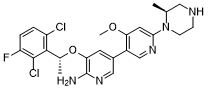
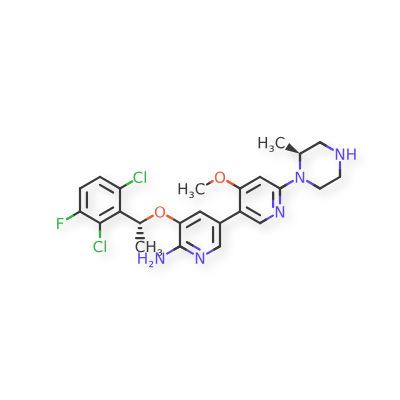
Envonalkib
- CAS 1621519-26-3
- QB7KTQ7VW9
- 5-((1R)-1-(2,6-Dichloro-3-fluorophenyl)ethoxy)-4′-methoxy-6′-((2S)-2-methyl-1-piperazinyl)(3,3′-bipyridin)-6-amine
- 506.4 g/mol, C24H26Cl2FN5O2
TQ-B3139, Chia Tai Tianqing, Anluoqing, cancer
ENVONALKIB is a small molecule drug with a maximum clinical trial phase of II and has 1 investigational indication.
SYN
https://patentscope.wipo.int/search/en/WO2014117718
Example 27: 5-[(2,6-dichloro-3-fluorophenyl)ethoxy-4′-methoxy-6′ …
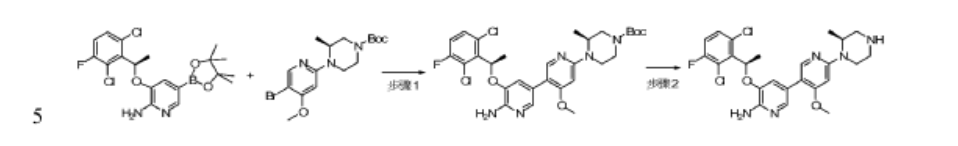
Step 1: 5-((R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-4′-methoxy-6′-((S)-2-methyl-4-tert-butoxycarbonylpiperazin-1-yl)-3,3′-bipyridin-6-amine
To dioxane (10 mL) and water (1.5 mL) were added tert-butyl (S)-4-(5-bromo-4-methoxypyridin-2-yl)-3-methylpiperidin-1-carboxylate (106 mg, 0.275 mmol), (R)-3-(1-(2,6-dichloro-3-fluorophenyl)ethoxy)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2-aminopyridine (140 mg, 0.33 mmol), tetrakis(triphenylphosphine)palladium (32 mg, 0.0275 mmol) and cesium carbonate (179 mg, 0.55 mmol), the atmosphere was replaced with nitrogen, and the reaction was carried out at 100 ° C. overnight. After cooling, the mixture was separated by silica gel column chromatography to give 5-(2,6-dichloro-3-fluorophenyl)ethoxy)-4′-methoxy-6-(5-(2-methyl-4-tert-butoxycarbonylpiperidin-1-yl)-3,3′-bipyridin-6-amine) (70 mg) in a yield of 42%. MS m/z [ESI]: 606.2 [M+1].
Step 2: 5-((R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-4′-methoxy-6′-((S)-2-methylpiperazin-1-yl)-3,3′-bipyridin-6-amine
To a stirred dichloromethane solution (10 mL) of 5-((R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-4′-methoxy-6′-((S)-2-methyl-4-tert-butoxycarbonylpiperidin-1-yl)-3,3′-bipyridin-6-amine (67 mg, 0.11 mmol) was added trifluoroacetic acid (1 mL) and stirred for 1 hour. The pH was adjusted to greater than 13 with sodium hydroxide solution, and the mixture was extracted with dichloromethane. The organic phase was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated. The product was separated and purified by column chromatography (with dichloromethane:methanol = 8:1 as eluent) to give 5-((R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-4′-methoxy-6′-((S)-2-methylpiperidin-1-yl)-3,3′-bipyridin-6-amine (30 mg). Yield: 55%, MS m/z [ESI]: 506.1[M+1]. 1H-NM (400 MHz, CDC1 3 ):5= 7.94(1H, s), 7.71(1H, s), 7.28-7.32(lH, m), 7.07(1H, t, J=8.4Hz), 6.97(1H, s), 6.04-6.13(2H, m), 4.86 (2H : s), 4.57-4.59(lH, m), 4.03 (1H, d, J=14Hz), 3.76(3H, s), 3.07-3.33(4H, m), 2.88-3.00(lH, m), 1.84(3H, d, J=6.8Hz), 1.34 (3H, d, J=6.8Hz).
SYN
CN107949560
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US154015806&_cid=P11-MEF9W1-27198-1
Example 27: 5-((R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-4′-methoxy-6′-((S)-2-methylpiperazin-1-yl)-[3,3′-bipyridin]-6-amine
General Synthetic Methods:
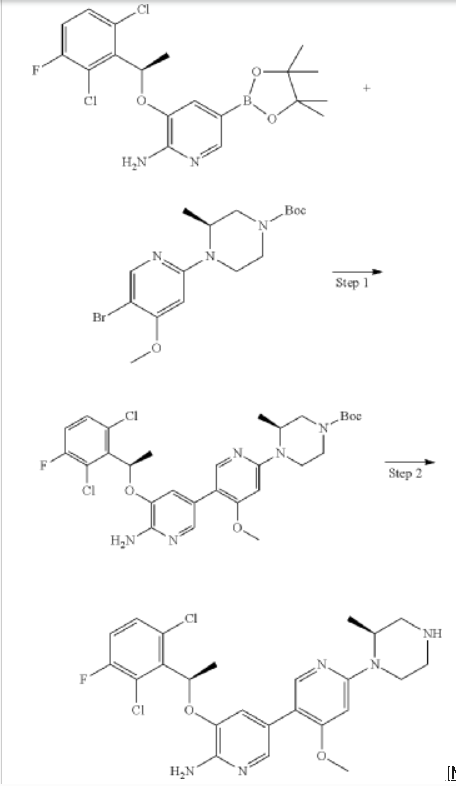
Step 1: (S)-tert-butyl 4-(6′-amino-5′-((R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-4-methoxy-[3,3′-bipyridin]-6-yl)-3-methylpiperazine-1-carboxylate
Step 2: 5-((R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-4′-methoxy-6′-((S)-2-methylpiperazin-1-yl)-[3,3′-bipyridin]-6-amine
SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Envonalkib, also known as TQ-B3139, is a novel small-molecule TKI, developed by Chia Tai Tianqing Pharmaceutical Group. It targets ALK, ROS1, and c-Met kinases, exhibiting potent antitumor activity against cancers harboring these genetic alterations. In 2024, the NMPA approved Envonalkib under the brand name Anluoqing for the treatment of adult patients with ALK-positive locally advanced or metastatic NSCLC who have not received prior ALK inhibitor therapy [24]. Envonalkib exerts its therapeutic effects through selective inhibition of the kinase activities of ALK, ROS1, and c-Met, thereby interrupting the downstream signaling pathways that are crucial for tumor cell proliferation and survival [25]. The inhibition of these targets results in cell cycle arrest and apoptosis in cancer cells。The clinical efficacy of Envonalkib was evidenced in a Phase III randomized, open-label, multicenter clinical trial (NCT04009317), which compared Envonalkib with crizotinib in treatment-naïve patients with ALK-positive advanced NSCLC [25,26]. In the reported study, Envonalkib demonstrated a me dian PFS of 24.87 months, which was markedly superior to the 11.60 months achieved with crizotinib (hazard ratio [HR] = 0.47, p < 0.0001). Notably, in patients harboring brain metastases, Envonalkib exhibited a
central nervous system objective response rate (CNS-ORR) of 78.95 %, a substantial improvement over the 23.81 % observed with crizotinib. In terms of safety profile, Envonalkib was generally well-tolerated. Treat ment-related adverse events (TRAEs) of Grade ≥3 were noted in 55.73 % of patients receiving Envonalkib, contrasting with the 42.86 % incidence in the crizotinib cohort. The predominant TRAEs encompassed elevated liver enzymes, neutropenia, and gastrointestinal symptoms, all of which
were amenable to effective management through appropriate support ive care measures. The regulatory approval of Envonalkib thus in troduces a novel therapeutic modality for patients with ALK-positive NSCLC, effectively addressing a significant unmet medical need within this patient population [25].
The synthesis of Envonalkib, illustrated in Scheme 6, initiates with Mitsunobu coupling of Envo-001 and Envo-002, affording Envo-003 [27]. Sequential reduction and NBS-bromination converts Envo-003 to
Envo-005 via Envo-004. Miyaura borylation of Envo-005 constructs Envo-006, which undergoes Suzuki-Miyaura cross-coupling with Envo-007 followed by deprotection to deliver Envonalkib. In parallel,
Envo-009 reacts with Envo-010 through Buchwald-Hartwig cross coupling to form Envo-011. This intermediate is brominated to produce Envo-007, which is used in the Suzuki-Miyaura coupling with Envo-006
[24] X. Li, Y. Xia, C. Wang, S. Huang, Q. Chu, Efficacy of ALK inhibitors in Asian
patients with ALK inhibitor-naïve advanced ALK-Positive non-small cell lung
cancer: a systematic review and network meta-analysis, Transl. Lung Cancer Res.
13 (2024) 2015–2022.
[25] Y. Yang, J. Min, N. Yang, Q. Yu, Y. Cheng, Y. Zhao, M. Li, H. Chen, S. Ren, J. Zhou,
W. Zhuang, X. Qin, L. Cao, Y. Yu, J. Zhang, J. He, J. Feng, H. Yu, L. Zhang, W. Fang,
Envonalkib versus crizotinib for treatment-naive ALK-Positive non-small cell lung
cancer: a randomized, multicenter, open-label, phase III trial, Signal Transduct
Target Ther 8 (2023) 301.
[26] R. Garcia-Carbonero, A. Carnero, L. Paz-Ares, Inhibition of HSP90 molecular
chaperones: moving into the clinic, Lancet Oncol. 14 (2013) e358–e369.
[27] F. Gong, X. Li, R. Zhao, X. Zhang, X. Xu, X. Liu, D. Xiao, Y. Han, Process for
Preparation of Pyridine Substituted 2-aminopyridine Protein Kinase Inhibitor
Crystal, 2017. CN107949560B.




AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
- New drugs approved by the NMPA in 2024: Synthesis and clinical applicationsPublication Name: European Journal of Medicinal ChemistryPublication Date: 2025-07-05PMID: 40262297DOI: 10.1016/j.ejmech.2025.117643
- Efficacy of ALK inhibitors in Asian patients with ALK inhibitor-naïve advanced ALK-positive non-small cell lung cancer: a systematic review and network meta-analysisPublication Name: Translational Lung Cancer ResearchPublication Date: 2024-08-31PMCID: PMC11384493PMID: 39263024DOI: 10.21037/tlcr-24-604
- Envonalkib versus crizotinib for treatment-naive ALK-positive non-small cell lung cancer: a randomized, multicenter, open-label, phase III trialPublication Name: Signal Transduction and Targeted TherapyPublication Date: 2023-08-14PMCID: PMC10423717PMID: 37574511DOI: 10.1038/s41392-023-01538-w
- Pharmacokinetic, pharmacodynamic, and behavioural studies of deschloroketamine in Wistar ratsPublication Name: British Journal of PharmacologyPublication Date: 2021-10-31PMID: 34519023DOI: 10.1111/bph.15680
//////////Envonalkib, china 2024, approvals 2024, TQ-B3139, TQ B3139, Chia Tai Tianqing, Anluoqing, cancer, QB7KTQ7VW9
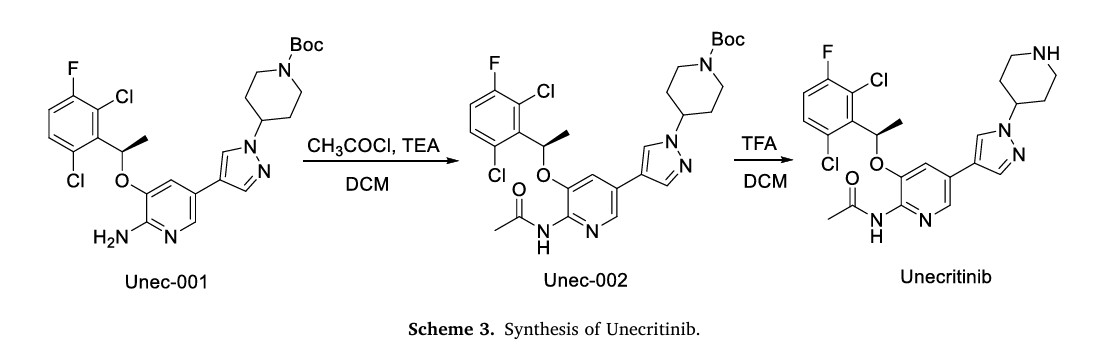
Unecritinib
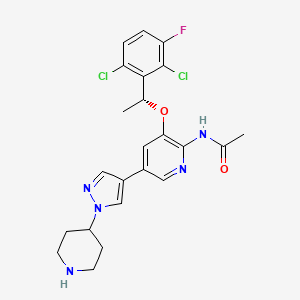
Unecritinib
- CAS 1418026-92-2
- 4T3Z98RR86
- TQ-B3101
492.4 g/mol, C23H24Cl2FN5O2
N-[3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-(1-piperidin-4-ylpyrazol-4-yl)pyridin-2-yl]acetamide
- Acetamide, N-[3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-[1-(4-piperidinyl)-1H-pyrazol-4-yl]-2-pyridinyl]-
- N-{3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-[1- (piperidin-4-yl)-1H-pyrazol-4-yl]pyridin-2-yl}acetamide
Chia Tai Tianqing Pharmaceutical Group
Unecritinib is an orally available, small molecule inhibitor of the receptor tyrosine kinases anaplastic lymphoma kinase (ALK), C-ros oncogene 1 (ROS1) and Met (hepatocyte growth factor receptor; HGFR; c-Met), with potential antineoplastic activity. Upon oral administration,unecritinib targets, binds to and inhibits the activity of ALK, ROS1 and c-Met, which leads to the disruption of ALK-, ROS1- and c-Met-mediated signaling and the inhibition of cell growth in ALK-, ROS1- and c-Met-expressing tumor cells. ALK, ROS1 and c-Met, overexpressed or mutated in many tumor cell types, play key roles in tumor cell proliferation, survival, invasion and metastasis.
UNECRITINIB is a small molecule drug with a maximum clinical trial phase of II (across all indications) and has 3 investigational indications.
- OriginatorChia Tai Tianqing Pharmaceutical Group
- ClassAcetamides; Antineoplastics; Benzofurans; Chlorobenzenes; Esters; Ethers; Fluorobenzenes; Ketones; Morpholines; Piperidines; Pyrazoles; Pyridines; Small molecules
- Mechanism of ActionAnaplastic lymphoma kinase inhibitors; Proto-oncogene protein c-met inhibitors; ROS1 protein inhibitors
- RegisteredNon-small cell lung cancer
- No development reportedAnaplastic large cell lymphoma
- 07 Sep 2024Efficacy and adverse events data from a phase II trial in Non-small cell lung cancer presented at the 25th World Conference on Lung Cancer (WCLC-2024)
- 17 May 2024Chemical structure information added
- 17 May 2024No development reported – Phase-II for Anaplastic large cell lymphoma (In adolescents, In children, Late-stage disease, Refractory metastatic disease, Second-line therapy or greater, In adults) in China (PO)
PATENT
https://patentscope.wipo.int/search/en/WO2013041038
Example 11: Synthesis of
(R)-N-(3-(l-(2,6-dichloro-3-fluorophenyl)ethoxy)- 5-(l -(piperidin-4-yl)-lH-pyrazol-4-yl)pyridin-2-yl)acetamide (Compound 18)
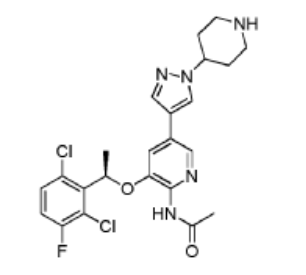
Step 1. To a solution of (R)-tert-butyl 4-(4-(6-amino-5-(l-(2,6-dichloro-3-fluorophenyl)ethoxy)pyridin-3 -yl)- 1 H-pyrazol- 1 -yl)piperidine- 1 -carboxylate ( 4g, 7.27 mmol, 1.0 eq) and pyridine ( 2.3g, 29.1 mmol, 4.0 eq) in 50 ml DCM was added acetyl chloride (0.86g, 10.9 mmol, 1.5 eq) in an ice bath. The reaction mixture was stirred at room temperature for overnight. The resulting mixture was washed with H20 (3×20 mL). The organic layer was dried and concentrated. The crude product was purified on silica gel column to give (R)-tert-butyl 4-(4-(6-acetamido-5-(l-(2,6-dichloro-3-fluorophenyl)ethoxy)pyridin-3-yl)-lH-pyrazol-l-yl)piperidine-l-carboxylatel .66g (38.6% yield).
Step 2. To a solution of (R)-tert-butyl 4-(4-(6-acetamido-5-(l-(2,6-dichloro-3 -fluorophenyl)ethoxy)pyridin-3 -yl)- 1 H-pyrazol- 1 -yl)piperidine- 1 -carboxylate (500 mg, 0.84 mmol, 1.0 eq) in DCM (5 mL) was added trifluoroacetic acid (2 ml) in an ice bath. The reaction mixture was stirred at room temperature for 2 hours. The pH of the reaction mixture was adjusted to 9 by saturated bicarbonate sodium in an ice bath. The aqueous solution was extracted with ethyl acetate (3×20 mL), the combined organic layers were washed with brine, dried over (MgSC^), filtered, and concentrated. The crude product was purified by silica gel column to give (R)-N-(3 -( 1 -(2,6-dichloro-3 -fluorophenyl)ethoxy)-5-( 1 -(piperidin-4-yl)- 1 H-pyrazol-4-yl)pyridin-2-yl)acetamide 250 mg (60.2% yield).
^-NMR^DC , 400Hz): 51.88(d, J=6.4Hz, 3H), 51.90-1.94(m, 2H), 52.16-2.20(m, 2H), 52.48(s, 3H), 52.76-2.824(m, 2H), 53.25-3.28(m, 2H), 53.69-3.74(m, 1H), 54.22-4.26 (m, 1H), 56.10-6.15(m, 1H), 57.05-7.07 (m, 1H), 57.09(s, 1H), 57.30-7.33 (m, 1H), 57.59(s, 1H), 57.62(s, 1H), 58.06(s, 1H),
58.12(s, 1H). MS m/z 493 [M+l]
PATENT
CN102850328
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN85774618&_cid=P12-MECPSG-91316-1
SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Unecritinib, developed by Chia Tai Tianqing Pharmaceutical Group, is a novel small-molecule tyrosine kinase inhibitor. It targets c-rosoncogene 1 (ROS1), anaplastic lymphoma kinase (ALK), and c-mesen
chymal-epithelial transition factor (c-MET) kinases, exhibiting potent antitumor activity against cancers harboring these genetic alterations. In 2024, the NMPA approved Unecritinib under the brand name Anbaini for the treatment of adult patients with ROS1-positive locally advanced or metastatic non-small cell lung cancer (NSCLC). Unecritinib exerts its therapeutic effects through selective inhibition of the kinase activities of ROS1, ALK, and c-MET, which effectively disrupts the downstream signaling pathways that are crucial for the proliferation and survival of tumor cells. Consequently, this inhibition induces cell cycle arrest and apoptosis in cancer cells that express these specific targets [13]. The clinical efficacy of Unecritinib was established in a Phase II single-arm, multicenter clinical trial (NCT03750739) enrolling patients with ROS1-positive advanced NSCLC. Among 111 evaluable patients, an ORR of 80.2 % was achieved, along with a median PFS of 16.5 months. These findings underscore the robust antitumor activity of Unecritinib in this specific patient cohort. In terms of safety, Unecritinib exhibited a
favorable tolerability profile. The most frequently reported treatment-related adverse events were neutropenia, leukopenia, vomit ing, and nausea, which were predominantly of mild (Grade 1) or mod
erate (Grade 2) severity. Importantly, no dose-limiting toxicities were observed, and the maximum tolerated dose was not established, further supporting its favorable safety profile. The approval of Unecritinib represents a novel therapeutic strategy for patients with ROS1-positive NSCLC, effectively addressing a significant unmet medical need within this population [13].
The synthesis of Unecritinib, depicted in Scheme 3, initiates with acetylation of Unec-001 to yield Unec-002, which undergoes deprotection to afford Unecritinib [14]
[13] S. Lu, H. Pan, L. Wu, Y. Yao, J. He, Y. Wang, X. Wang, Y. Fang, Z. Zhou, X. Wang,
X. Cai, Y. Yu, Z. Ma, X. Min, Z. Yang, L. Cao, H. Yang, Y. Shu, W. Zhuang, S. Cang,
J. Fang, K. Li, Z. Yu, J. Cui, Y. Zhang, M. Li, X. Wen, J. Zhang, W. Li, J. Shi, X. Xu,
D. Zhong, T. Wang, J. Zhu, Efficacy, safety and pharmacokinetics of unecritinib
(TQ-B3101) for patients with ROS1 positive advanced non-small cell lung cancer: a
phase I/II trial, Signal Transduct Target Ther 8 (2023) 249.
[14] A. Zhang, M. Geng, Y. Wang, J. Ai, X. Peng, Preparation of Pyridine Compounds as
Inhibitors of c-Met And/Or ALK Kinases, Shanghai Institute of Materia Medica,
2013 CN102850328A.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
/////////Unecritinib, Chia Tai Tianqing Pharmaceutical Group, 1418026-92-2, 4T3Z98RR86, TQ B3101, APPROVALS 2024, CHINA 2024
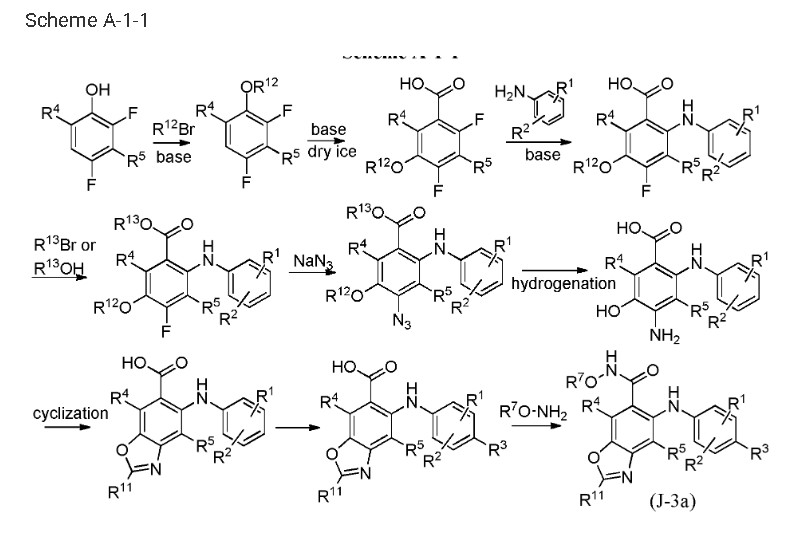
Tunlametinib
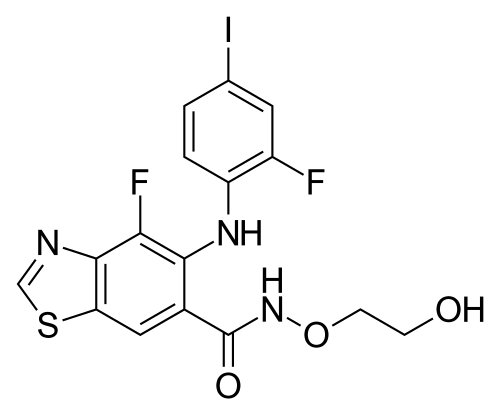
Tunlametinib
- CAS 1801756-06-8
- IF25NR1PV3
- HL085
- C16H12F2IN3O3S
491.3 g/mol
4-fluoro-5-(2-fluoro-4-iodoanilino)-N-(2-hydroxyethoxy)-1,3-benzothiazole-6-carboxamide
- 4-Fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2-hydroxyethoxy)-6-benzothiazolecarboxamide
- 4-fluoro-5-(2-fluoro-4-iodoanilino)-N-(2-hydroxyethoxy)-1,3-benzothiazole-6-carboxamide
- 6-Benzothiazolecarboxamide, 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2-hydroxyethoxy)-
Tunlametinib, an oral selective inhibitor of mitogen-activated protein kinase kinase 1 and 2 (MEK1/2), was developed by Shanghai KeChow Pharmaceuticals Co., Ltd. Marketed under the brand name
Keluping,
Tunlametinib is a pharmaceutical drug for the treatment of cancer. It is an inhbitor of mitogen-activated protein kinase kinase.[1]
In China, tunlametinib was approved in 2024 for the treatment of patients with NRAS-mutated advanced melanoma who were previously treated with a PD-1/PD-L1 targeting agent.[2][3]
It is also being studied for use in combination with vemurafenib in patients with advanced BRAF V600-mutant solid tumors.[4]
PAT
US9937158
PAT
https://patents.google.com/patent/WO2013107283A1/en
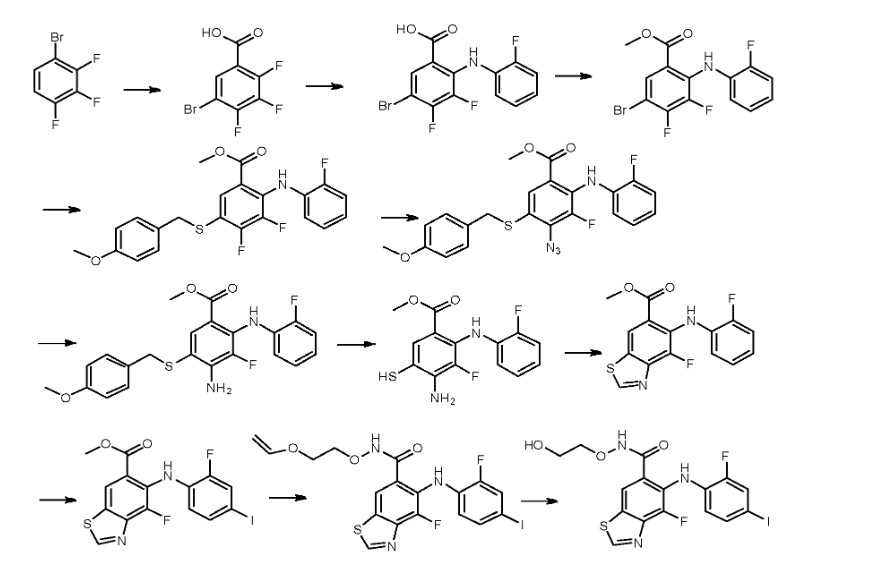
Step 1:

[0435] To a solution of 2,3,4-trifluorobromobenzene in appropriate solvent (include aliphatic and aromatic hydrocarbon(such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), sulfolane, HMPA, DMPU, prefer anhydrous THF, ethyl ether and dioxane) was added strong base (such as LDA, nBuLi,
LiHDMS) at low temperature (-50 °C 80 °C, prefer -78 °C) under nitrogen atmosphere. The reaction is kept stirring for some time (0.5-12 h, prefer 0.5-2 h) and is added dry ice. After several hours (3-12 h, prefer 5-10 h), 5-bromo-2,3,4-trifluorobenzoic acid is obtained after conventional workup.
Step 2:

[0436] 5-Bromo-2,3,4-trifluorobenzoic acid can be reacted with halogenated aniline (such as o-fluoroaniline, o-chloroaniline, o-bromoaniline, o-iodoaniline) in the presence of base (such as LDA, n-BuLi, LiHDMS) in appropriate solvent (include aliphatic and aromatic
hydrocarbon(such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2- methoxyethyl ether, tetrahydrofuran, dioxane), sulfolane, HMPA, DMPU, prefer anhydrous THF, ethyl ether and dioxane) at low temperature (-50 °C— -80 °C, prefer -78 °C) for some time (such as 3-12 h, prefer 5-10 h). 5-Bromo-3,4-difluoro-2-((2-fluorophenyl)amino)benzoic acid is obtained after conventional workup.
Step 3:

[0437] 5-Bromo-3,4-difluoro-2-((2-fluorophenyl)amino)benzoic acid can be reacted with MeOH in the presence of SOCl2 in appropriate solvent (include aliphatic and aromatic hydrocarbon(such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), aliphatic and aromatic halo-hydrocarbon (such as dichloromethane, 1,2-dichloroethane, chloroform, phenixin, chlorobenzene, o-dichlorobenzene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), ketone(such as acetone, methyl ethyl ketone, methyl isopropyl ketone, methyl isobutyl ketone), ester(such as ethyl acetate, methyl acetate), nitrile(such as acetonitrile, propiononitrile), amide(such as N,N-dimethylformamide, N,N-dimethylacetamide and N-methylpyrrolidin-2-one), DMSO, sulfolane, HMPA, DMPU, prefer methanol and ethanol). The reaction proceeds for several hours (3-12 h, prefer 5-10 h). Methyl 5-bromo-3,4-difluoro-2-((2-fluorophenyl) amino)benzoate is obtained after conventional workup.
Step 4:

[0438] To a solution of methyl 5-bromo-3,4-difluoro-2-((2-fluorophenyl) amino)benzoate in appropriate solvent (include aliphatic and aromatic hydrocarbon(such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), ester(such as ethyl acetate, methyl acetate), nitrile(such as acetonitrile, propiononitrile), amide(such as N,N-dimethylformamide, N,N-dimethylacetamide and N-methylpyrrolidin-2-one), DMSO, sulfolane, HMPA, DMPU, prefer dioxane) was added base (such as aliphatic and aromatic amine(such as, but not limited to, N-ethyl-N-isopropylpropan-2-amine, triethylamine, diethylamine, DBU, t-butylamine, cyclopropanamine, dibutylamine, diisopropylamine, 1,2- dimethylpropanamine), inorganic base(such as Na2C03, K2C03, NaHC03, KHC03, t-BuONa, t- BuOK), prefer N-ethyl-N-isopropylpropan-2-amine) at ambient temperature under nitrogen atmosphere, followed by Pd catalyst (such as tris(dibenzylideneacetone)dipalladium,
bis(dibenzylideneacetone) palladium, bis(triphenylphosphine)palladium(II) chloride, palladium diacetate, tetrakis(triphenylphosphine)palladium, bis(triphenylphosphinepalladium)acetate, prefer tris(dibenzylideneacetone) dipalladium) and phosphine ligand (such as
dimethylbisdiphenylphosphinoxanthene, tri-tert-butylphosphine, tri-p-tolylphosphine, tris(4- chlorophenyl)phosphine, triisopropylphosphine, tris(2,6-dimethoxyphenyl)phosphine, 1, 1 ‘- bis(diphenylphosphino)ferrocene, prefer dimethylbisdiphenylphosphinoxanthene). The reaction is kept stirring at high temperature (80-130 °C, prefer 90-110 °C) for some time (8-24 h, prefer 12-18 h). Methyl 3,4-difluoro-2- ((2-fluorophenyl)amino)-5-((4-methoxybenzyl)thio)benzoate is obtained after conventional workup. Step 5:

[0439] Methyl 3,4-difluoro-2-((2-fluorophenyl)amino)-5-((4-methoxy benzyl)thio)benzoate can be reacted with azide (such as NaN3, KN3) at high temperature (60-120 °C, prefer 80-100 °C) in appropriate solvent (include aliphatic and aromatic hydrocarbon(such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), aliphatic and aromatic halo-hydrocarbon (such as dichloromethane, 1,2-dichloroethane, chloroform, phenixin, chlorobenzene, o-dichlorobenzene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), ketone(such as acetone, methyl ethyl ketone, methyl isopropyl ketone, methyl isobutyl ketone), ester(such as ethyl acetate, methyl acetate), nitrile (such as acetonitrile, propiononitrile), amide (such as N,N-dimethylformamide, N,N-dimethylacetamide and N-methylpyrrolidin-2-one), DMSO, sulfolane, HMPA, DMPU, prefer N,N-dimethylformamide and N,N-dimethylacetamide) for some time (1-12 h, prefer 3-10 h). Methyl 4-azido-3-fluoro-2-((2-fluorophenyl) amino)-5-((4-methoxybenzyl)thio)benzoate is obtained after conventional workup.
Step 6:

[0440] Methyl 4-azido-3-fluoro-2-((2-fluorophenyl)amino)-5-((4-methoxy
benzyl)thio)benzoate can be hydrogenated catalyzed by appropriate catalyst (such as Pd/C, Pt, Ni) in the solvent (include aliphatic and aromatic hydrocarbon(such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), ester(such as ethyl acetate, methyl acetate), amide (such as N,N-dimethylformamide, N,N- dimethylacetamide and N-methylpyrrolidin-2-one), DMSO, sulfolane, HMPA, DMPU, prefer methanol, ethanol, propan-l-ol and water) for some time (1-12 h, prefer 3-10 h). Methyl 4- amino-3-fluoro-2-((2-fluorophenyl)amino)-5-((4-methoxybenzyl)thio)benzoate is obtained after conventional workup. Step 7:

[0441] 4-Amino-3-fluoro-2-((2-fluorophenyl)amino)-5-((4-methoxybenzyl)thio)benzoate can be deprotected in the presence of acid (such as CF3COOH, HCOOH, CH3COOH and n- C5H11COOH, prefer CF3COOH) at certain temperature (20-75 °C, prefer 25-75 °C) in
appropriate aromatic aliphatic ether (such as anisole and phenetole, prefer anisole) for some time (1-12 h, prefer 3-10 h). Methyl 4-amino-3-fluoro-2-((2-fluorophenyl)amino)-5- mercaptobenzoate is obtained after conventional workup.
Step 8:

[0442] Methyl 4-amino-3-fluoro-2-((2-fluorophenyl)amino)-5-mercapto benzoate can be cyclized in the presence of acid (such as ^-toluenesulfonic acid, pyridinium toluene-4- sulphonate, formic acid, acetic acid, sulfuric acid) in appropriate solvent (include aliphatic and aromatic hydrocarbon (such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), aliphatic and aromatic halo-hydrocarbon (such as
dichloromethane, 1,2-dichloroethane, chloroform, phenixin, chlorobenzene, o-dichlorobenzene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), ketone(such as acetone, methyl ethyl ketone, methyl isopropyl ketone, methyl isobutyl ketone), ester(such as ethyl acetate, methyl acetate), nitrile (such as acetonitrile, propiononitrile), amide (such as N,N-dimethylformamide, N,N-dimethylacetamide and N- methylpyrrolidin-2-one), DMSO, sulfolane, HMPA, DMPU, prefer methyl acetate, ethyl acetate and trimethoxymethane) for some time (0.2-12 h, prefer 0.5-10 h). Methyl 4-fluoro-5-((2- fluorophenyl)amino) benzo[d]thiazole-6-carboxylate is obtained after conventional workup. Step 9:

[0443] Methyl 4-fluoro-5-((2-fluorophenyl)amino)benzo[d]thiazole-6- carboxylate can be reacted with halogenations reagent (such as NIS) in the presence of acid (such as trifluoroacetic acid, trifluoromethanesulfonic acid, methanesulfonic acid, formic acid, acetic acid) at ambient temperature in appropriate solvent (include aliphatic and aromatic hydrocarbon(such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), aliphatic and aromatic halo-hydrocarbon (such as dichloromethane, 1,2-dichloroethane, chloroform, phenixin, chlorobenzene, o-dichlorobenzene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), ketone(such as acetone, methyl ethyl ketone, methyl isopropyl ketone, methyl isobutyl ketone), ester(such as ethyl acetate, methyl acetate), nitrile (such as acetonitrile, propiononitrile), amide (such as N,N- dimethylformamide, N,N-dimethylacetamide and N-methylpyrrolidin-2-one), DMSO, sulfolane, HMPA, DMPU, prefer N,N-dimethylformamide and N,N-dimethylacetamide) for some time (1- 12 h, prefer 3-10 h). Methyl 4-fluoro-5-((2-fluoro-4-iodophenyl) amino)benzo[d]thiazole-6- carboxylate is obtained after conventional workup.
Step 10:

[0444] 4-Fluoro-5-((2-fluoro-4-iodophenyl)amino)benzo[d]thiazole-6-carboxylic acid can be reacted with O-(2-(vinyloxy)ethyl)hydroxylamine in the presence of coupling reagent(such as HOBt, EDCI, HATU, TBTU) at ambient temperature in appropriate solvent(include aliphatic and aromatic hydrocarbon(such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), aliphatic and aromatic halo-hydrocarbon (such as dichloromethane, 1,2-dichloroethane, chloroform, phenixin, chlorobenzene, o-dichlorobenzene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), ketone(such as acetone, methyl ethyl ketone, methyl isopropyl ketone, methyl isobutyl ketone), ester(such as ethyl acetate, methyl acetate), nitrile (such as acetonitrile, propiononitrile), amide (such as N,N-dimethylformamide, N,N-dimethylacetamide and N- methylpyrrolidin-2-one), DMSO, sulfolane, HMPA, DMPU, prefer dichloromethane, 1,2- dichloroethane and N,N-dimethylformamide) for some time (1-12 h, prefer 3-10 h). 4-Fluoro-5- ((2-fluoro-4-iodophenyl) amino)-N-(2-(vinyloxy)ethoxy)benzo[d]thiazole-6-carboxamide is obtained after conventional workup. Step 11:

[0445] 4-Fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2-(vinyloxy)ethoxy)benzo[d]thiazole- 6-carboxamide can be reacted in the presence of acid (such as HCl, H2S04, trifluoroacetic acid) in appropriate solvent (include aliphatic and aromatic hydrocarbon (such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), aliphatic and aromatic halo-hydrocarbon (such as dichloromethane, 1,2-dichloroethane, chloroform, phenixin, chlorobenzene, o-dichlorobenzene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), ketone(such as acetone, methyl ethyl ketone, methyl isopropyl ketone, methyl isobutyl ketone), ester(such as ethyl acetate, methyl acetate), nitrile (such as acetonitrile, propiononitrile), amide (such as N,N-dimethylformamide, N,N-dimethylacetamide and N-methylpyrrolidin-2-one), DMSO, sulfolane, HMPA, DMPU, prefer dichloromethane and 1,2-dichloroethane) for some time (1-12 h, prefer 3-10 h). 4-Fluoro- 5-((2-fluoro-4-iodophenyl)amino)-N-(2-hydroxy ethoxy)benzo[d]oxazole-6-carboxamide is obtained after conventional workup.
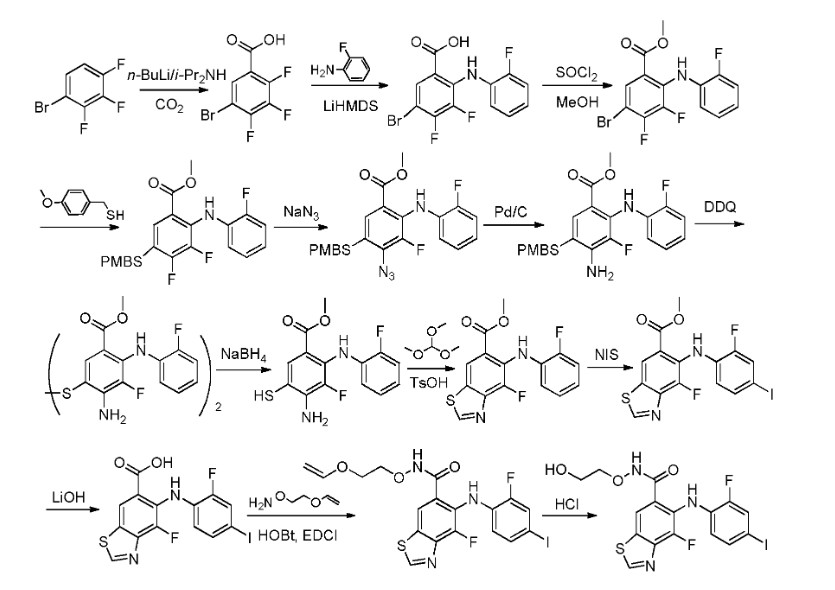
Example 9: Preparation of 4-fluoro-5-((2-fluoro-4-iodophenyDamino)-N-(2- hydroxyethoxy)benzo[d]thiazole-6-carboxamide (Compound 9)

Step 1: 5-bromo-2,3,4-trifluorobenzoic acid
[0510] To a solution of diisopropylamine (10.14 g, 100.20 mmol) in THF (100 mL) was added «-BuLi (40.08 mL, 2.5 M in hexane, 100.20 mmol) at -78 °C under nitrogen atmosphere. The stirring was maintained at this temperature for 1 h. Then a solution of l-bromo-2,3,4- trifluorobenzene (17.62 g, 83.50 mmol) in THF (120 mL) was added. After stirring for 1 h at -78 °C, the mixture was transferred to a bottle with dry ice. The mixture was stirred overnight at room temperature. The reaction was quenched with 10% aqueous HCl and pH was adjusted to 1- 2. The mixture was extracted with ethyl acetate (100 mL x 3). The combined organic extracts were washed with water (100 mL) and brine (100 mL) sequentially, dried over Na2S04, filtered and concentrated under reduced pressure to afford the desired product (20.12 g, 94.5% yield). 1H NMR (400 MHz, DMSO-d6): δ 13.95 (s, 1H), 7.97 (m, 1H).
Step 2: 5-bromo-3,4-difluoro-2-((2-fluorophenyl)amino)benzoic acid
[0511] To a solution of 2-fluoroaniline (17.54 g, 157.80 mmol) and 5-bromo-2,3,4- trifluorobenzoic acid (20.12 g, 78.90 mmol) in THF (120 mL) was added LiHMDS (236.7 mL, 1 M in THF, 236.7 mmol) dropwisely at -78 °C under nitrogen atmosphere. The mixture was allowed to slowly warm to room temperature and stirred at this temperature overnight. The reaction was quenched with water (100 mL) and acidified to pH 2-3 with 10% HCl (aq.). The mixture was extracted with ethyl acetate (100 mL χ 3). The combined organic extracts were washed with water (100 mL) and brine (100 mL) sequentially, dried over Na2S04, filtered and concentrated in vacuo to afford the desired product (pale yellow solid, 24.24 g, 88.8% yield). 1H NMR (400 MHz, DMSO-d6): δ 9.22 (s, 1H), 8.01 (dd, J= 7.4, 2.1 Hz, 1H), 7.25 (m, 1H), 7.10 (m, 3H).
Step 3: methyl 5-bromo-3,4-difluoro-2-((2-fluorophenyl)amino)benzoate
[0512] To a solution of 5-bromo-3,4-difluoro-2-((2-fluorophenyl)amino) benzoic acid (24.24 g, 70.04 mmol) in MeOH (300 mL) was added thionyl chloride (20 mL). After stirring at 85 °C overnight, most MeOH was removed in vacuo. The residue was neutralized with saturated sodium bicarbonate (aq.) and extracted with ethyl acetate (100 mL χ 3). The combined organic layer was washed with water (100 mL) and brine (100 mL) sequentially, dried over Na2S04, filtered and concentrated. After purification by column chromatography on silica gel (petroleum ether/ethyl acetate, 50: 1, v/v), the corresponding product was obtained as a white solid (22.33 g, 88.5% yield). 1H NMR (400 MHz, CDC13): δ 9.06 (s, 1H), 8.01 (dd, J= 7.1, 2.3 Hz, 1H), 7.04 (m, 4H), 3.92 (s, 3H).
Step 4: methyl 3,4-difluoro-2-((2-fluorophenyl)amino)-5-((4-methoxybenzyl)thio)benzoate
[0513] To a solution of methyl 5-bromo-3,4-difluoro-2-((2-fluorophenyl) amino)benzoate (22.33 g, 62.01 mmol) in anhydrous 1,4-dioxane (200 mL) was added N,N- diisopropylethylamine (16.03 g, 124.04 mmol). Then Pd2(dba)3 (2.84 g, 3.10 mmol) followed by Xantphos (3.59 g, 6.20 mmol) and 4-methoxy-a-toluenethiol (10.27 g, 65.11 mmol) was added under nitrogen atmosphere. The mixture was stirred overnight at 100 °C under N2 atmosphere and then allowed to warm to ambient temperature. The insoluble matter was filtered off and the filter cake was washed ethyl acetate. The filtrate was diluted with water (300 mL) and extracted with ethyl acetate (100 mL x 3). The combined organic layers were washed with water (100 mL) and brine (100 mL) sequentially, dried over Na2S04, filtered and concentrated. The crude product was purified by column chromatography on silica gel (petroleum ether/ethyl acetate, 50: 1, v/v) to give the desired product (pale yellow solid, 24.35 g, 90.6% yield). 1H NMR (400 MHz, CDC13): δ 9.12 (s, 1H), 7.78 (d, 1H), 7.25 (m, 6H), 6.85 (m, 2H), 4.03 (s, 2H), 3.90 (s, 3H), 3.80 (s, 3H). Step 5: methyl 4-azido-5-(4-methoxybenzylthio)-3-fluoro-2-((2-fluorophenyl)amino)benzoate
[0514] To a solution of methyl 5-(4-methoxybenzylthio)-3,4-difluoro-2- ((2- fluorophenyl)amino)benzoate (24.35 g, 56.18 mmol) in DMF (200 mL) was added NaN3 (4.38 g, 67.41 mmol) at ambient temperature. The mixture was stirred at 90 °C for 3 h. Then water (200 mL) was added. The solution was extracted with ethyl acetate (100 mL χ 3). The combined organic extracts were washed with water (100 mL) and brine (100 mL), dried over Na2S04 and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (petroleum ether/ethyl acetate, 10: 1, v/v) and gave the desired product (white solid, 21.04 g, 82.1% yield). 1H NMR (400 MHz, CDC13): δ 8.98 (s, 1H), 7.75 (s, 1H), 7.10 (m, 6H), 6.84 (m, 2H), 4.03 (s, 2H), 3.92 (s, 3H), 3.81 (s, 3H). Step 6: methyl 4-amino-5-(4-methoxybenzylthio)-3-fluoro-2-((2-fluorophenyl)amino)benzoate To a solution of methyl 4-azido-5-(4-methoxybenzylthio)-3-fluoro-2-((2- fluorophenyl)amino)benzoate (21.04 g, 46.09 mmol) in MeOH (500 mL) was added and 10% palladium on carbon (3.40 g) under nitrogen atmosphere. Then the nitrogen atmosphere was completely changed to hydrogen atmosphere. The mixture was stirred for 2 h at ambient temperature. After the insoluble matter was filtered off, the solvent was evaporated in vacuo to give the desired product (19.46 g, 98.1% yield). 1H NMR (400 MHz, CDC13): δ 9.07 (s, 1H), 7.77 (s, 1H), 7.06 (m, 4H), 6.95 (m, 2H), 6.81 (d, J = 8.3 Hz, 2H), 4.68 (s, 2H), 3.85 (s, 5H), 3.81 (s, 3H).
Step 7: dimethyl 5,5′-disulfanediylbis(4-amino-3-fluoro-2-((2-fluorophenyl)amino)benzoate)
[0515] To a solution of methyl 4-amino-5-(4-methoxybenzylthio)-3-fluoro-2-((2- fluorophenyl)amino)benzoate (19.46 g, 45.21 mmol) in CH2C12 (180 mL) was added DDQ (11.29 g, 49.73 mmol) followed by water (20 mL). After stirring at ambient temperature for 10 h, the reaction was quenched by saturated sodium bicarbonate (aq., 100 mL). The aqueous layer was extracted by CH2C12 (100 mL χ 3). The combined organic phase was washed with water (100 mL) and brine (100 mL) sequentially, dried over Na2S04, filtered and concentrated. The crude product was purified by column chromatography on silica gel (petroleum ether/ethyl acetate, 5: 1, v/v) to give the desired product (pale yellow solid, 9.81 g, 35.1% yield). 1H NMR (400 MHz, CDC13): δ 9.34 (s, 2H), 7.46 (s, 2H), 7.06 (m, 8H), 4.89 (br, 4H), 3.75 (s, 6H). Step 8: methyl 4-amino-3-fluoro-2-((2-fluorophenyl)amino)-5-mercaptobenzoate
[0516] To a solution of dimethyl 5,5′-disulfanediylbis(4-amino-3-fluoro-2-((2- fluorophenyl)amino)benzoate) (9.81 g, 15.86 mmol) in THF/MeOH (100 mL, 10: 1, v/v) was added NaBH4 (3.00 g, 79.29 mmol) portion-wise in 1 h. After stirring at ambient temperature for 1 h, the reaction was quenched with 10% HCl (aq.) and pH was adjusted to 1-2. The aqueous layer was extracted with CH2C12 (50 mL χ 3). The combined organic phase was washed with water (50 mL) and brine (50 mL) sequentially, dried over Na2S04, filtered and concentrated in vacuo. The crude product was used directly in the next step without further purification.
Step 9: methyl 4-fluoro-5-((2-fluorophenyl)amino)benzofdJthiazole-6-carboxylate
[0517] To a solution of methyl 4-amino-3-fluoro-2-((2-fluorophenyl)amino)-5- mercaptobenzoate in trimethyl orthoformate (50 mL) was added p-TsOU (0.61 g, 3.17 mmol). The reaction mixture was stirred for 1 h and treated with water (100 mL). The precipitate was filtered off and the filter cake was washed with water to afford the desired product (pale yellow solid, 8.64 g, 85.1% yield for two steps). 1H MR (400 MHz, CDC13): δ 9.13 (s, 1H), 8.68 (s, 1H), 8.46 (s, 1H), 7.10 (m, 1H), 7.01 (m, 1H), 6.92 (s, 2H), 3.97 (s, 3H).
Step 10: methyl 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)benzofdJthiazole-6-carboxylate
[0518] To a solution of methyl 4-fluoro-5-((2-fluorophenyl)amino)benzo[d]thiazole-6- carboxylate (8.64 g, 26.97 mmol) in DMF (100 mL) was added NIS (6.68 g, 29.67 mmol) followed by trifluoroacetic acid (0.5 mL). After stirring for 5 h at ambient temperature, the reaction was treated by water (150 mL). The precipitate was filtered off and the filter cake was washed with water. The desired product was obtained as a yellow solid (10.34 g, 86.0% yield). 1H NMR (400 MHz, CDC13): δ 9.14 (s, 1H), 8.66 (s, 1H), 8.46 (s, 1H), 7.42 (d, J= 10.4 Hz, 1H), 7.31 (d, J= 8.8 Hz, 1H), 6.63 (dd, J= 15.0, 8.7 Hz, 1H), 3.97 (s, 3H).
Step 11: 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)benzo[d]thiazole-6-carboxylic acid
[0519] To a solution of methyl 4-fluoro-5-((2-fluoro-4-iodophenyl)amino) benzo[d]thiazole-6- carboxylate (10.34 g, 23.17 mmol) in THF and MeOH (20 mL, 4: 1, v/v) was added 5.0 M LiOH (aq., 2 mL, 10 mmol). After stirring at ambient temperature for 2 h, the reaction was treated with 1.0 M HCl (aq.) till the solution was acidic. The aqueous layer was extracted with ethyl acetate (50 mL x 3). The combined organic phase was washed with water (100 mL) and brine (100 mL) sequentially, dried over Na2S04, filtered and concentrated to give the desired product (9.51 g, 95.0% yield). 1H NMR (400 MHz, DMSO-d6): δ 11.10 (s, 1H), 9.18 (s, 1H), 8.68 (s, 1H), 8.45 (s, 1H), 7.41 (m, 1H), 7.30 (m, 1H), 6.65 (m, 1H). Step 12: 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2-(vinyloxy)etho
carboxamide
[0520] To a solution of 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)benzo[d]thiazole-6- carboxylic acid (519 mg, 1.20 mmol) in CH2C12 (10 mL) was added HOBt (254 mg, 1.63 mmol) and EDCI (314 mg, 1.63 mmol). The mixture was stirred for 1 h and O-(2-
(vinyloxy)ethyl)hydroxyl -amine (172 mg, 1.62 mmol) was added. After stirring for 4 h at ambient temperature, the reaction was treated with saturated H4C1 (aq.). The resultant mixture was extracted with CH2C12 (30 mL χ 3). The combined organic extracts were washed with water (30 mL) and brine (30 mL), dried over Na2S04 filtered, and concentrated in vacuo. The crude product (492 mg) was used directly in the next step without further purification.
Step 13: 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2-hydroxyethoxy)benzo[d]thiazole-6- carboxamide
[0521] To a solution of 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2- (vinyloxy)ethoxy)benzo[d]thiazole-6-carboxamide (492 mg, 1.00 mmol) in CH2C12 (10 mL) was added 1.0 N HCl (aq., 5 mL, 5 mmol). After stirring for 1 h, the reaction mixture was neutralized with saturated NaHC03 (aq.). The aqueous layer was washed with CH2C12 (30 mL). The combined organic layer was washed with water (30 mL x 2) and brine (30 mL), dried over Na2S04, filtered and concentrated in vacuo. The crude product was purified by column chromatography on silica gel (CH2Cl2/MeOH, 50: 1, v/v) and gave the desired product as a white solid (446 mg, 75.9% yield for the two steps). 1H MR (400 MHz, DMSO-d6): δ 11.80 (s, 1H), 9.55 (s, 1H), 8.22 (s, 1H), 8.12 (s, 1H), 7.55 (d, J= 11.0 Hz, 1H), 7.31 (d, J= 8.5 Hz, 1H), 6.48 (d, J= 9.2 Hz, 1H), 4.72 (s, 1H), 3.84 (m, 2H), 3.57 (m, 2H). MS APCI(+)m/z: 491.8, [M+H].
Example 9A: Preparation of 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2- hydroxyethoxy)benzo[d]thiazole-6-carboxamide (Compound 9)

Step 1: 5-bromo-2,3,4-trifluorobenzoic aci

[0522] To a solution of l-bromo-2,3,4-trifluorobenzene (13.64 g, 64.6 mmol) in THF (120 mL) was added lithium diisopropylamide (2.0 M in THF, 33.9 mL, 67.8 mmol) at -78 °C under nitrogen atmosphere. After stirring for 1 h at -78 °C, the mixture was transferred to a bottle with dry ice. The mixture was stirred overnight at room temperature. The reaction was quenched with 10% aqueous HC1 (300 mL) and extracted with ethyl acetate (200 mL x 3). The combined organic extracts were washed with 5% sodium hydroxide (300 mL). The aqueous layer was acidized to pH 1 and extracted with ethyl acetate (200 mL χ 3). The combined organic extract was dried over Na2S04, filtered and concentrated under reduced pressure to afford the desired product (white solid, 13.51 g, 82% yield). 1H MR (400 MHz, CDC13): δ 13.94 (s, 1H), 7.95 (m,
1H).
Step 2: 5-bromo-3,4-difluoro-2-((2-fluorophenyl)amino)benzoic

[0523] To a solution of 2-fluoroaniline (10.2 mL, 105.8 mmol) and 5-bromo-2,3,4- trifluorobenzoic acid (13.51 g, 52.9 mmol) in THF (120 mL) was added LiHMDS (158.7 mL, 1 M in THF, 158.7 mmol) dropwisely at -78 °C under nitrogen atmosphere. The mixture was allowed to slowly warm to room temperature and stirred at this temperature overnight. The reaction was quenched with 10% HC1 (aq., 100 mL) and extracted with ethyl acetate (200 mL x 3). The combined organic extracts were washed with water (200 mL x 3) and brine (200 mL) sequentially, dried over Na2S04, filtered and concentrated in vacuo to afford the desired product (pale yellow solid, 13.73 g, 75% yield). 1H MR (400 MHz, DMSO-d6): δ 9.21 (s, 1H), 8.01 (d, 1H), 7.26 (m, 1H), 7.01-7.16 (m, 3H).
Step 3: methyl 5-bromo-3,4-difluoro-2- -fluorophenyl)amino)benzoate

[0524] To a solution of 5-bromo-3,4-difluoro-2-((2-fluorophenyl)amino)benzoic acid (13.73 g, 39.6 mmol) in MeOH (300 mL) was added SOCl2 (60 mL). After stirring at 85 °C overnight, most MeOH was removed in vacuo. The residue was neutralized with saturated sodium bicarbonate (aq.) and extracted with ethyl acetate (300 mL χ 3). The combined organic extract was washed with water (200 mL x 3) and brine (200 mL) sequentially, dried over Na2S04, filtered and concentrated in vacuo to afford the corresponding product (gray solid, 12.58 g, 90% yield). 1H MR (400 MHz, CDC13): δ 9.09 (s, 1H), 8.05 (d, 1H), 7.00-7.14 (m, 4H), 3.94 (s, 3H).
Step 4: methyl 3,4-difluoro-2-((2-fluorophenyl)amino)-5-((4-methoxybenzyl)thio)benzoate

[0525] To a solution of methyl 5-bromo-3,4-difluoro-2-((2-fluorophenyl)amino)benzoate (12.85 g, 35.6 mmol) in anhydrous 1,4-dioxane (30 mL) was added N,N-diisopropylethylamine (9.21 g, 71.2 mmol). Then Pd2(dba)3 (1.63 g, 1.78 mmol) followed by Xantphos (2.06 g, 3.56 mmol) and 4-methoxy-a-toluenethiol (5.48 g, 35.6 mmol) was added under nitrogen atmosphere. The mixture was stirred overnight at 100 °C under N2 atmosphere and then allowed to cool to ambient temperature. The reaction was quenched with water (150 mL) and extracted with ethyl acetate (200 mL χ 3). The combined organic extract was washed with water (200 mL χ 3) and brine (200 mL) sequentially, dried over Na2S04, filtered and concentrated. The crude product was purified by column chromatography on silica gel (petroleum ether/ethyl acetate, 50: 1, v/v) to give the desired product (pale yellow solid, 12.64 g, 82% yield). 1H NMR (400 MHz, CDC13): δ 9.12 (s, 1H), 7.78 (d, 1H), 7.06-7.44 (m, 6H), 6.82-6.88 (m, 2H), 4.03 (s, 2H), 3.90 (s, 3H), 3.80 (s, 3H).
Step 5: methyl 4-azido-5-(4-methoxybenzylthio)-3-fluoro-2-((2-fluorophenyl)amino)benzoate

[0526] To a solution of methyl 5-(4-methoxybenzylthio)-3,4-difluoro-2-((2- fluorophenyl)amino)benzoate (12.64 g, 29.2 mmol) in DMF (30 mL) was added NaN3 (2.28 g, 35.0 mmol) at ambient temperature. The mixture was stirred at 90 °C for 3 h. Then water (150 mL) was added. The solution was extracted with ethyl acetate (100 mL χ 3). The combined organic extracts were washed with water (100 mL χ 3) and brine (100 mL), dried over Na2S04 and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (petroleum ether/ethyl acetate, 10: 1, v/v) and gave the desired product (white solid, 10.38 g, 78% yield). 1H NMR (400 MHz, CDC13): δ 8.98 (s, 1H), 7.75 (s, 1H), 7.02-7.28 (m, 6H), 6.83- 6.85 (m, 2H), 4.03 (s, 2H), 3.92 (s, 3H), 3.81 (s, 3H).
Step 6: methyl 4-amino-5-(4-methoxybenzylthio)-3-fluoro-2-((2-fluorophenyl)amino)benzoate

[0527] To a solution of methyl 4-azido-5-(4-methoxybenzylthio)-3-fluoro-2-((2- fluorophenyl)amino)benzoate (10.38 g, 22.7 mmol) in MeOH (100 mL) was added and 10% palladium on carbon (1.55 g) under nitrogen atmosphere. Then the nitrogen atmosphere was completely changed to hydrogen atmosphere. The mixture was stirred at ambient temperature for 6 h. After the insoluble matter was filtered off, the solvent was evaporated in vacuo to give the desired product (9.79 g, 100% yield).1H MR (400 MHz, CDC13): δ 9.08 (s, 1H), 7.78 (s, 1H), 6.93-7.28 (m, 8H), 4.65 (s, 2H), 4.00 (s, 2H), 3.89 (s, 3H), 3.75 (s, 3H).
Step 7: methyl 4-amino-3-fluoro-2-((2-fluorophenyl)amino)-5-mercaptobenzoate

[0528] To a solution of methyl 4-amino-3-fluoro-2-((2-fluorophenyl)amino)-5-((4- methoxybenzyl)thio)benzoate (9.79 g, 22.7 mmol) in anisole (12 mL) was added CF3COOH (20 mL). After stirring at ambient temperature for 23 h, the solvent was removed in vacuo. To the residue was added water (30 mL). The mixture was neutralized with 25% aqueous ammonia and extracted with ethyl acetate (100 mL χ 3). The combined organic layer was washed with water (100 mL x 3) and brine (100 mL) sequentially, dried over Na2S04, filtered and concentrated to give the desired product (white solid, 5.28 g, 75% yield). The product was used directly in the next step without further purification.
Step 8: methyl 4-fluoro-5-((2-fluorophenyl)amino)benzofdJthiazole-6-carboxylate

[0529] To a solution of methyl 4-amino-3-fluoro-2-((2-fluorophenyl)amino)-5- mercaptobenzoate (2.07 g, 6.67 mmol) in trimethyl orthoformate (20 mL) was added p-TsOU (166 mg, 0.65 mmol). The reaction mixture was stirred for 1 h and treated with water (100 mL). The precipitate was filtered off and the filter cake was washed with water to afford the desired product (white solid, 1.963 g, 92% yield for two steps). 1H NMR (400 MHz, DMSO-d6): δ 9.01 (s, 1H), 8.08 (s, 1H), 7.90 (s, 1H), 7.15-6.78 (m, 4H), 3.91 (s, 3H).
Step 9: methyl 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)benzofdJthiazole-6-carboxylate

[0530] To a solution of methyl 4-fluoro-5-((2-fluorophenyl)amino)benzo[d]thiazole-6- carboxylate (1.963 g, 6.14 mmol) in DMF (10 mL) was added NIS (1.5 g, 6.5 mmol) followed by trifluoroacetic acid (0.5 mL). After stirring for 4 h at ambient temperature, the reaction was treated by saturated H4C1 (aq.). The aqueous layer was extracted with ethyl acetate (150 mL χ 3). The combined organic layer was washed with water (100 mL x 3) and brine (100 mL) sequentially, dried over Na2S04, filtered and concentrated in vacuo. After purification by flash column chromatography on silica gel (petroleum ether/ethyl acetate, 10: 1, v/v), the desired product was obtained as white solid (1.889 g, 69% yield). 1H NMR (400 MHz, DMSO-d6): δ 9.03 (s, 1H), 8.10 (s, 1H), 7.93 (s, 1H), 7.18-6.72 (m, 3H), 3.91 (s, 3H).
Step 10: 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2-(vinyloxy
carboxamide

[0531] To a solution of O-(2-(vinyloxy)ethyl)hydroxyl-amine (172 mg, 1.62 mmol) in THF (6 mL) was added LiHMDS (2.5 mL, 1 M in THF, 2.5 mmol) at -78 °C. After stirring at this temperature for 10 min, a solution of methyl 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)benzo[d] thiazole-6-carboxylate (360 mg, 0.81 mmol) in THF was syringed dropwisely. Then the mixture was allowed to warm to ambient temperature, quenched with saturated NH4C1 (aq., 20 mL) and extracted with ethyl acetate (15 mL χ 3). The combined organic extract was washed with water (10 mL x 3) and brine (10 mL), dried over Na2S04, filtered and concentrated in vacuo. After purification by flash chromatography (petroleum ether/ethyl acetate, 10: 1, v/v), the desired product was obtained (410 mg, 98% yield). 1H NMR (400 MHz, DMSO-d6): δ 11.85 (s, 1H),
8.98 (s, 1H), 8.04 (s, 1H), 7.89 (s, 1H), 7.55 (d, J= 10.8 Hz, 1H), 7.31 (d, J = 8.1 Hz, 1H), 6.53 (dd, J= 13.9, 6.6 Hz, 1H), 6.42 (d, J= 6.0 Hz, 1H), 4.21 (d, J= 14.5 Hz, 1H), 4.01 (m, 3H), 3.83 (m, 2H).
Step 11: 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2-hydroxyethoxy)benzofdJthiazole-6- carboxamide

[0532] To a solution of 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2- (vinyloxy)ethoxy)benzo[d]thiazole-6-carboxamide (410 mg, 0.8 mmol) in CH2C12 (5 mL) was added 1.0 N HCl (aq., 5 mL, 5 mmol) dropwise. After stirring for 1 h, the reaction mixture was neutralized with saturated NaHC03 (aq.). The organic layer was separated, washed with water (30 mL x 2) and brine (30 mL) sequentially, dried over Na2S04, filtered and concentrated in vacuo. The crude product was purified by column chromatography on silica gel (CH2Cl2/MeOH, 15: 1, v/v) and the desired product was obtained as a white solid (290 mg, 52 % yield). 1H MR (400 MHz, DMSO-de): δ 11.83 (s, 1H), 8.92 (s, 1H), 8.03 (s, 1H), 7.90 (s, 1H), 7.56 (d, J= 9.4 Hz, 1H), 7.30 (d, J= 8.7 Hz, 1H), 6.41 (m, 1H), 4.72 (m, 1H), 3.85 (m, 2H), 3.59 (m, 2H). MS (ES+): m/z 492.35 [MH+].
SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Tunlametinib, an oral selective inhibitor of mitogen-activated protein kinase kinase 1 and 2 (MEK1/2), was developed by Shanghai KeChow Pharmaceuticals Co., Ltd. Marketed under the brand name
Keluping, it received conditional approval from the NMP in 2024 for the treatment of patients with advanced melanoma harboring NRAS mutations, particularly those who have not responded to anti-PD-1/PD-L1therapies [1]. Tunlametinib exerts its antitumor effects by targeting the MEK1/2 kinases within the RAS-RAF-MEK-ERK signaling pathway, thereby disrupting downstream signaling cascades and inhibiting tumor cell growth and proliferation [2]. Its clinical efficacy was demonstrated in a Phase II pivotal registration study (NCT05217303) involving patients with advanced NRAS-mutant melanoma [3]. The study reported a confirmed objective response rate (ORR) of 34.8 % and a median progression-free survival (mPFS) of 4.2 months. These findings suggest that Tunlametinib holds promise as a treatment option for NRAS-mutant melanoma, including in patients who have failed immunotherapy. In terms of safety, Tunlametinib has been generally well-tolerated [4]. Adverse events frequently encountered during treatment primarily consist of increased blood creatine phosphokinase (CPK) levels, diarrhea, and edema. Additionally, warnings and precautions pertinent to Tunlametinib therapy encompass decreased left ventricular ejection fraction (LVEF), skin toxicity, ocular toxicity, interstitial lung disease,
gastrointestinal reactions, and elevated CPK levels [5].
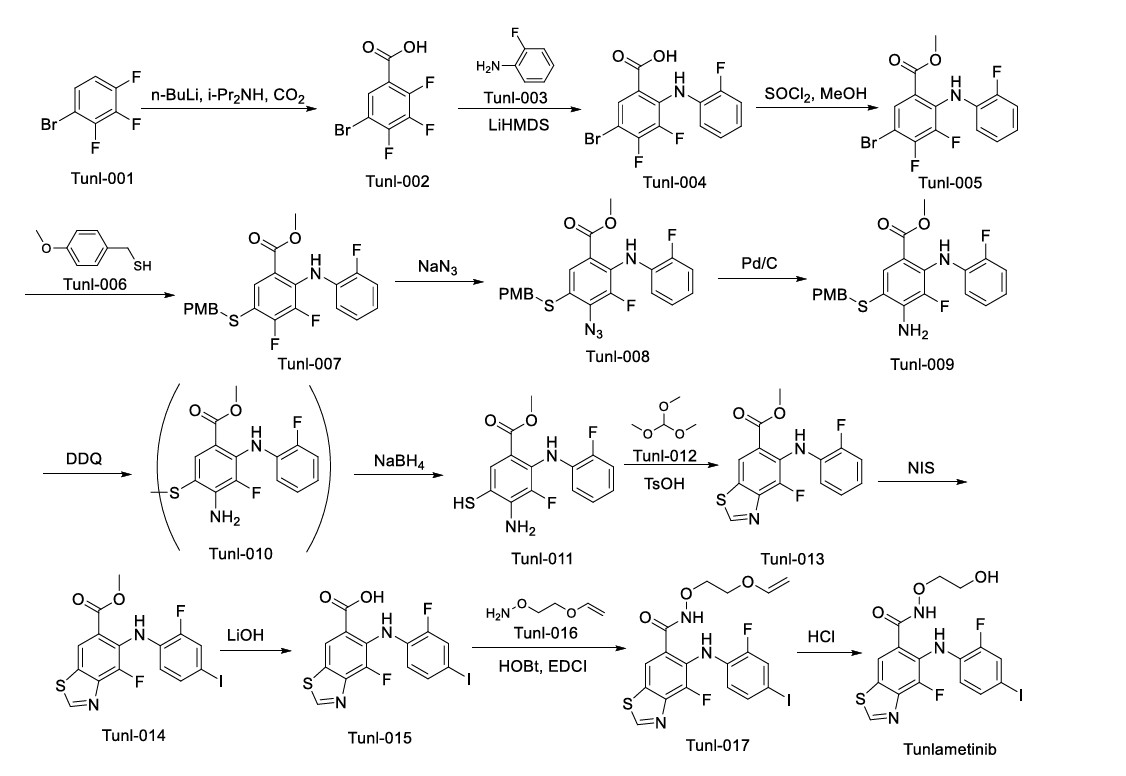
The synthetic pathway of Tunlametinib, illustrated in Scheme 1, begins with carboxylation of Tunl-001 to yield Tunl-002 [6]. Nucleophilic substitution of Tunl-002 with Tunl-003 then produces Tunl-004,
which undergoes esterification to form Tunl-005. Subsequent nucleophilic substitution between Tunl-05 and Tunl-006 generates Tunl-007. This intermediate undergoes azidation to afford Tunl-008, followed by
reduction to Tunl-009. Treatment of Tunl-009 with DDQ converts it to Tunl-010, which is deprotected to yield Tunl-011. Cycloaddition of Tunl-011 with Tunl-012 forms Tunl-013. Iodination of Tunl-013 gives
Tunl-014, which is hydrolyzed to produce Tunl-015. Amidation of Tunl-015 with Tunl-016 yields Tunl-017, and its subsequent acidolysis completes the synthesis of Tunlametinib.
[1] Y. Liu, Y. Cheng, G. Huang, X. Xia, X. Wang, H. Tian, Preclinical characterization of
tunlametinib, a novel, potent, and selective MEK inhibitor, Front. Pharmacol. 14
(2023) 1271268.
[2] S.J. Keam, Tunlametinib: first approval, Drugs 84 (2024) 1005–1010.
[3] X. Wei, Z. Zou, W. Zhang, M. Fang, X. Zhang, Z. Luo, J. Chen, G. Huang, P. Zhang,
Y. Cheng, J. Liu, J. Liu, J. Zhang, D. Wu, Y. Chen, X. Ma, H. Pan, R. Jiang, X. Liu,
X. Ren, H. Tian, Z. Jia, J. Guo, L. Si, A phase II study of efficacy and safety of the MEK inhibitor tunlametinib in patients with advanced NRAS-Mutant melanoma,
Eur. J. Cancer 202 (2024) 114008.
[4] Q. Zhao, T. Wang, H. Wang, C. Cui, W. Zhong, D. Fu, W. Xi, L. Si, J. Guo, Y. Cheng,
H. Tian, P. Hu, Phase I pharmacokinetic study of an oral, small-molecule MEK
inhibitor tunlametinib in patients with advanced NRAS mutant melanoma, Front.
Pharmacol. 13 (2022) 1039416.
[5] Y. Shi, X. Han, Q. Zhao, Y. Zheng, J. Chen, X. Yu, J. Fang, Y. Liu, D. Huang, T. Liu,
H. Shen, S. Luo, H. Yu, Y. Cao, X. Zhang, P. Hu, Tunlametinib (HL-085) plus
vemurafenib in patients with advanced BRAF V600-mutant solid tumors: an open-
label, single-arm, multicenter, phase I study, Exp. Hematol. Oncol. 13 (2024) 60.
[6] H. Tian, C. Ji, C. Liu, L. Kong, Y. Cheng, G. Huang, Benzoheterocyclic Compounds
and Use Thereof, 2014. US9937158B2.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- “Tunlametinib”. NCI Drug Dictionary. National Cancer Institute.
- “Tunlametinib Wins Approval in China for NRAS+ Advanced Melanoma After PD-1/PD-L1 Therapy”. 18 March 2024.
- Keam SJ (2024). “Tunlametinib: First Approval”. Drugs. 84 (8): 1005–1010. doi:10.1007/s40265-024-02072-x. PMID 39034326.
- Shi Y, Han X, Zhao Q, Zheng Y, Chen J, Yu X, et al. (2024). “Tunlametinib (HL-085) plus vemurafenib in patients with advanced BRAF V600-mutant solid tumors: An open-label, single-arm, multicenter, phase I study”. Experimental Hematology & Oncology. 13 (1): 60. doi:10.1186/s40164-024-00528-0. PMC 11167782. PMID 38867257.
| Clinical data | |
|---|---|
| Other names | HL-085 |
| ATC code | None |
| Legal status | |
| Legal status | Rx in China |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1801756-06-8 |
| PubChem CID | 71621329 |
| ChemSpider | 115006753 |
| UNII | IF25NR1PV3 |
| ChEMBL | ChEMBL5095241 |
| Chemical and physical data | |
| Formula | C16H12F2IN3O3S |
| Molar mass | 491.25 g·mol−1 |
/////////Tunlametinib, CHINA 2024, APPROVALS 2024, Shanghai KeChow, Keluping,1801756-06-8, IF25NR1PV3, HL 085
Taletrectinib
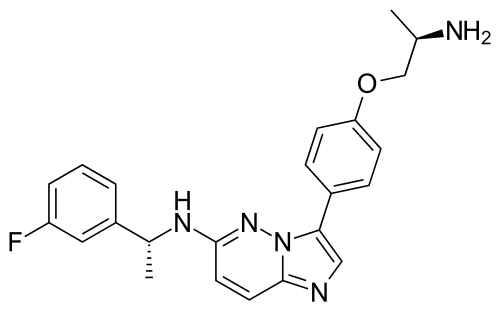
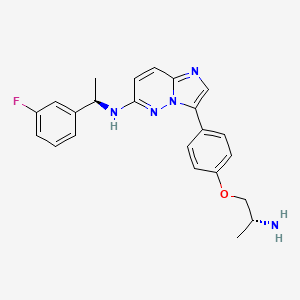
Taletrectinib
CAS 1505514-27-1
as salt: 1505515-69-4, Taletrectinib adipate
FDA 6/11/2025, Ibtrozi, To treat locally advanced or metastatic ROS1-positive non-small cell lung cancer ALSO CHINA 2024 APPROVED |
405.5 g/mol, C23H24FN5O, UNII-W4141180YD
3-[4-[(2R)-2-aminopropoxy]phenyl]-N-[(1R)-1-(3-fluorophenyl)ethyl]imidazo[1,2-b]pyridazin-6-amine
Taletrectinib adipate
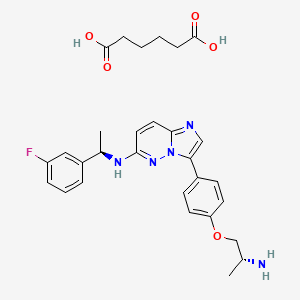
WeightAverage: 551.619
Monoisotopic: 551.254397378
Chemical FormulaC29H34FN5O5
DS-6051B, CAS 1505515-69-4,
6KLL51GNBG, 3-{4-[(2R)-2-aminopropoxy]phenyl}-N-[(1R)-1-(3-fluorophenyl)ethyl]imidazo[1,2-b]pyridazin-6-amine; hexanedioic acid
Taletrectinib, sold under the brand name Ibtrozi, is an anti-cancer medication used for the treatment of non-small cell lung cancer.[1][2] It is used as the salt, taletrectinib adipate.[1] Taletrectinib is a kinase inhibitor.[1] It is taken by mouth.[1]
Taletrectinib was approved for medical use in the United States in June 2025.[3]
SYN
US20200062765
https://patentscope.wipo.int/search/en/detail.jsf?docId=US289038418&_cid=P12-MCIHV1-02369-1
Example 1
tert-Butyl [(2R)-1-(4-bromophenoxy)propan-2-yl]carbamate (1)
Example 2
6-Fluoroimidazo[1,2-b]pyridazine methanesulfonate (2)
Example 3
tert-Butyl {(2R)-1-[4-(6-fluoroimidazo[1,2-b]pyridazin-3-yl)phenoxy]propan-2-yl}carbamate (3)
Example 4
tert-Butyl {(2R)-1-[4-(6-{[(1R)-1-(3-fluorophenyl)ethyl]amino}imidazo[1,2-b]pyridazin-3-yl)phenoxy]propan-2-yl}carbamate hydrochloride (4)
Example 5
3-{4-[(2R)-2-Aminopropoxy]phenyl}-N-[(1R)-1-(3-fluorophenyl)ethylimidazo[1,2-b]pyridazin-6-amine dihydrochloride (5)
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023272701&_cid=P12-MCIHPU-95869-1
The NMR data for the crystalline form A of Compound 1 adipate are as follows: 1H NMR (500 MHz, DMSO) δ 1.13-1.14 (d, J=5.0 Hz, 3H) , 1.47-1.48 (d, J=5.0 Hz, 7H) , 2.15-2.18 (t, J=5.0 Hz, J=10.0 Hz, 4H) , 3.25-3.29 (m, 1H) , 3.79-3.83 (m, 2H) , 4.80-4.85 (m, 1H) , 6.76-6.77 (d, J=5.0 Hz, 1H) , 6.92-6.94 (d, J=10.0 Hz, 2H) , 7.01-7.05 (t, J=10.0 Hz, 1H) , 7.23-7.28 (m, 2H) , 7.37-7.42 (m, 1H) , 7.64-7.65 (d, J=5.0 Hz, 1H) , 7.72-7.76 (t, J=10.0 Hz, 4H) .
[0148]
The IR data for the crystalline form A of Compound 1 adipate are as follows: IR (cm -1) : 1701, 1628, 1612, 1586, 1463, 1333, 1246, 1110, 829, 821.
Example 5: Preparation and Characterization of Crystalline Form A of Compound 1 Free Base
[0212]
Compound 1 HCl (75.5 g) (e.g., obtained by using the method described in Example 5 of U.S. Application Publication No. 2020/0062765) was dissolved in ethanol (604 mL) at 50℃. Sodium hydroxide (68.1 g) was added to the above solution. The mixture was cooled to 1℃ in 1.5 hours and stirred for 18.5 hours. The mixture was then filtered, and the solid thus obtained was washed with a cooled mixture of ethanol (151 mL) and water (151 mL) and dried. The solid thus obtained was confirmed to be the crystalline form A of Compound 1 free base.
[0213]
The NMR data for the crystalline form A of Compound 1 free base are as follows: 1H NMR (500 MHz, DMSO) δ 1.09-1.10 (d, J=5.0 Hz, 3H) , 1.48-1.49 (d, J=5.0 Hz, 3H) , 3.16-3.20 (m, 1H) , 3.75-3.79 (m, 2H) , 4.82-4.86 (m, 1H) , 6.76-6.78 (d, J=10.0 Hz, 1H) , 6.92-6.94 (m, 2H) , 7.01-7.05 (m, 1H) , 7.23-7.28 (m, 2H) , 7.37-7.42 (m, 1H) , 7.62-7.63 (d, J=5.0 Hz, 1H) , 7.72-7.75 (m, 4H) .
[0214]
The IR data for the crystalline form A of Compound 1 free base are as follows: IR (cm -1) : 3350, 3247, 3055, 2961, 2923, 2864, 1611, 1586, 1349, 829, 819.
SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Taletrectinib is an oral, next-generation ROS1 TKI developed by Nuvation Bio Inc. for the treatment of ROS1-positive NSCLC. In 2024, the NMPA approved taletrectinib for adult patients with locally advanced or metastatic ROS1-positive NSCLC, regardless of prior ROS1TKI treatment [47]. Under an exclusive license agreement, Innovent Biologics will commercialize taletrectinib in China under the brand
name DOVBLERON®. Taletrectinib exerts its pharmacological action through the mechanism of selectively impeding the ROS1 receptor tyrosine kinase, which effectively disrupts the signaling cascades which are responsible for facilitating the growth and survival of cancer cells in ROS1-positive NSCLC. This inhibition of the ROS1 receptor tyrosine kinase is a key event in the drug’s mode of action, as it specifically targets the molecular processes that drive the progression of the disease in ROS1-positive NSCLC cases [48]. The NMPA granted approval founded on the data sourced from the crucial Phase 2 TRUST – I study. This study substantiated that patients administered with taletrectinib achieved sustained responses and extended PFS. Regarding safety, taletrectinib boasted a generally good tolerability. It presented an advantageous safety profile and favorable tolerability characteristics, as evidenced by the low incidences of dose reduction and treatment discontinuation triggered by adverse effects. [49]. Overall, taletrectinib represents a promising therapeutic option for patients with advanced ROS1-positive NSCLC, offering efficacy in both TKI-naïve and TKI-pretreated populations, including those with CNS metastases [50–52].
The synthesis of Taletrectinib, illustrated in Scheme 12, commences with Mitsunobu coupling of Tale-001 and Tale-002 to afford Tale-003, which then undergoes Suzuki coupling with Tale-004 constructing
Tale-005 [53]. Sequential acidolysis/deprotection of Tale-005 ultimately delivers Taletrectinib
[47] M. P´ erol, N. Yang, C.M. Choi, Y. Ohe, S. Sugawara, N. Yanagitani, G. Liu, F.G.M.
D. Braud, J. Nieva, M. Nagasaka, 1373P efficacy and safety of taletrectinib in
patients (pts) with ROS1+ non-small cell lung cancer (NSCLC): interim analysis of
global TRUST-II study, Ann. Oncol. 34 (2023) S788–S789.
[48] G. Harada, F.C. Santini, C. Wilhelm, A. Drilon, NTRK fusions in lung cancer: from
biology to therapy, Lung Cancer 161 (2021) 108–113.
[49] W. Li, A. Xiong, N. Yang, H. Fan, Q. Yu, Y. Zhao, Y. Wang, X. Meng, J. Wu, Z. Wang,
Y. Liu, X. Wang, X. Qin, K. Lu, W. Zhuang, Y. Ren, X. Zhang, B. Yan, C.M. Lovly,
C. Zhou, Efficacy and safety of taletrectinib in Chinese patients with ROS1+ non-
small cell lung cancer: the phase II TRUST-I study, J. Clin. Oncol. 42 (2024)
2660–2670.
[50] M. Nagasaka, D. Brazel, S.I. Ou, Taletrectinib for the treatment of ROS-1 positive
non-small cell lung cancer: a drug evaluation of phase I and II data, Expert Opin
Investig Drugs 33 (2024) 79–84.
[51] S. Waliany, J.J. Lin, Taletrectinib: TRUST in the continued evolution of treatments
for ROS1 fusion-positive lung cancer, J. Clin. Oncol. 42 (2024) 2622–2627.
[52] M. Nagasaka, Y. Ohe, C. Zhou, C.M. Choi, N. Yang, G. Liu, E. Felip, M. P´ erol,
B. Besse, J. Nieva, L. Raez, N.A. Pennell, A. Dimou, F. Marinis, F. Ciardiello,
T. Seto, Z. Hu, M. Pan, W. Wang, S. Li, S.I. Ou, TRUST-II: a global phase II study of
taletrectinib in ROS1-positive non-small-cell lung cancer and other solid tumors,
Future Oncol. 19 (2023) 123–135.
[53] Y. Takeda, K. Yoshikawa, Y. Kagoshima, Y. Yamamoto, R. Tanaka, Y. Tominaga,
M. Kiga, Y. Hamada, Preparation of imidazo[1,2-b]pyridazine Derivatives as
Potent Inhibitors of ROS1 Kinase and NTRK Kinase, 2013. WO2013183578A1.

Medical uses
Taletrectinib is indicated for the treatment of adults with locally advanced or metastatic ROS1-positive non-small cell lung cancer.[1][2]
Adverse effects
The FDA prescribing information for taletrectinib includes warnings and precautions for hepatotoxicity, interstitial lung disease/pneumonitis, QTc interval prolongation, hyperuricemia, myalgia with creatine phosphokinase elevation, skeletal fractures, and embryo-fetal toxicity.[1][3]
History
The efficacy of taletrectinib to treat ROS1-positive non-small cell lung cancer was evaluated in participants with locally advanced or metastatic, ROS1-positive non-small cell lung cancer enrolled in two multi-center, single-arm, open-label clinical trials, TRUST-I (NCT04395677) and TRUST-II (NCT04919811).[3] The efficacy population included 157 participants (103 in TRUST-I; 54 in TRUST-II) who were naïve to treatment with a ROS1 tyrosine kinase inhibitor (TKI) and 113 participants (66 in TRUST-I; 47 in TRUST-II) who had received one prior ROS1 tyrosine kinase inhibitor.[3] Participants may have received prior chemotherapy for advanced disease.[3] The US Food and Drug Administration (FDA) granted the application for taletrectinib priority review, breakthrough therapy, and orphan drug designations.[3]
Society and culture
Legal status
Taletrectinib was approved for medical use in the United States in June 2025.[3][4]
Names
Taletrectinib is the international nonproprietary name.[5]
Taletrectinib is sold under the brand name Ibtrozi.[3][4]
References
- ^ Jump up to:a b c d e f g “Prescribing Information for NDA 219713, Supplement 000” (PDF). Drugs@FDA. U.S. Food and Drug Administration. April 2025. Retrieved 14 June 2025.
- ^ Jump up to:a b Khan I, Sahar A, Numra S, Saha N, Nidhi, Parveen R (April 2025). “Efficacy and safety of taletrectinib for treatment of ROS1 positive non-small cell lung cancer: A systematic review”. Expert Opinion on Pharmacotherapy. 26 (6): 765–772. doi:10.1080/14656566.2025.2487150. PMID 40170301.
- ^ Jump up to:a b c d e f g h “FDA approves taletrectinib for ROS1-positive non-small cell lung cancer”. U.S. Food and Drug Administration (FDA). 11 June 2025. Retrieved 13 June 2025.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b “U.S. Food and Drug Administration Approves Nuvation Bio’s Ibtrozi (taletrectinib), a Next-Generation Oral Treatment for Advanced ROS1-Positive Non-Small Cell Lung Cancer”. Nuvation Bio (Press release). 12 June 2025. Retrieved 13 June 2025.
- ^ World Health Organization (2021). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 85”. WHO Drug Information. 35 (1). hdl:10665/340684.
External links
- Clinical trial number NCT04395677 for “A Study of AB-106 in Subjects With Advanced NSCLC Harboring ROS1 Fusion Gene” at ClinicalTrials.gov
- Clinical trial number NCT04919811 for “Taletrectinib Phase 2 Global Study in ROS1 Positive NSCLC (TRUST-II)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Ibtrozi |
| License data | US DailyMed: Taletrectinib |
| Routes of administration | By mouth |
| Drug class | Antineoplastic |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| CAS Number | 1505514-27-1as salt: 1505515-69-4 |
| PubChem CID | 72202474as salt: 72694302 |
| DrugBank | DB18711 |
| ChemSpider | 114934673as salt: 88297530 |
| UNII | W4141180YDas salt: 6KLL51GNBG |
| KEGG | D12363as salt: D12364 |
| ChEMBL | ChEMBL4650989as salt: ChEMBL4650361 |
| Chemical and physical data | |
| Formula | C23H24FN5O |
| Molar mass | 405.477 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
/////////Taletrectinib, FDA 2025, APPROVALS 2025, Ibtrozi, CANCER, AB-106, DS-6051a, UNII-W4141180YD, DS 6051B, APPROVALS 2024, CHINA 2024, Nuvation Bio Inc
Levacetylleucine
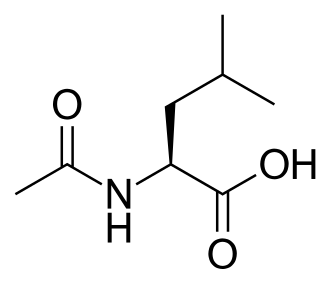
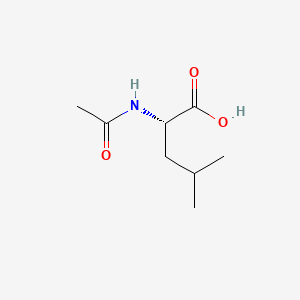

Levacetylleucine
WeightAverage: 173.212
Monoisotopic: 173.105193347
Chemical FormulaC8H15NO3
- N-Acetyl-L-leucine
- CAS 1188-21-2
- acetyl-L-leucine
- Ac-Leu-OH
- N-Acetylleucine
- NSC 206316
- UNII-E915HL7K2O
NSC-206316
(2S)-2-acetamido-4-methylpentanoic acid
FDA APPROVED 9/24/2024, To treat Niemann-Pick disease type C
Press Release
Drug Trials Snapshot
- Originator University of Munich; University of Oxford
- Developer IntraBio
- Class Acetamides; Amino acids; Esters; Neuroprotectants; Pentanoic acids; Small molecules; Vestibular disorder therapies
- Mechanism of Action Calcium channel modulators
- Orphan Drug StatusYes – Tay-Sachs disease; Niemann-Pick disease type C; Ataxia telangiectasia
Registered Niemann-Pick disease type C
- Phase IIIAtaxia telangiectasia
- Phase IISandhoff disease; Tay-Sachs disease
18 Mar 2025Phase-III clinical trials in Ataxia telangiectasia (In adolescents, In children, In the elderly, In adults) in Switzerland, Slovakia, Spain, Germany, USA, United Kingdom (PO) (NCT06673056)
- 04 Nov 2024IntraBio plans a phase III trial for Ataxia telangiectasia (In children, In adolescents, In adults, In elderly) in the US, Germany, Slovakia, Spain and Switzerland (PO, Suspension) in March 2025 (NCT06673056)
- 24 Sep 2024Registered for Niemann-Pick disease type C (In adolescents, In children, In adults) in USA (PO)
Levacetylleucine (N-acetyl-L-leucine), sold under the brand name Aqneursa, is a medication used for the treatment of neurological manifestations of Niemann-Pick disease type C.[1][2] Levacetylleucine is a modified version of the amino acid leucine.[1] It is the L-form of acetylleucine. It is taken by mouth.[1]
The most common side effects include abdominal pain, difficulty swallowing, upper respiratory tract infections, and vomiting.[1][2]
Levacetylleucine was approved for medical use in the United States in September 2024.[1][2][3] Levacetylleucine is the second medication approved by the US Food and Drug Administration (FDA) for the treatment of Niemann-Pick disease type C.[2] The FDA considers it to be a first-in-class medication.[4]
DATA
N-acetyl-D, L-leucine is the active ingredient of Tanganil ® which helps treat vertigo attacks.

N-Acetyl-D, L-leucine
Unlike the majority of chemical syntheses of active principles where it is desirable to separate the enanti omers and / or to retain the selective stereo information during the synthesis steps, the synthesis of N-acetyl-D, L-leucine is carried out from L-leucine and therefore involves a racemization step. This racemization takes place before the acetylation step, via a Schiff base formed in situ with salicylic aldehyde (Yamada et al., J. Org. Chem., 1983 48, 843- 846).

Two competitive reactions are then involved: the acetylation of leucine, the main reaction, where acetic anhydride reacts with the amine function of leucinate of sodium to give N-acetyleucinate and the hydrolysis of acetic anhydride to acetic acid, a side reaction described below.

This synthesis has a molar yield of 70%. The limiting steps are essentially the secondary reaction of hydrolysis of acetic anhydride and the step of isolation of the racemized leucine before the acetylation reaction. Indeed, on an industrial scale, the quantities of products brought into play for isolations prove to be very restrictive.
There is therefore a real need to develop a new process for the preparation of N-actéyl-D, L-leucine which is faster and more economical.
The inventors thus discovered that the racemization step could be carried out after the L-leucine acetylation step making it possible to avoid a step of isolating the intermediate product and that this process could be carried out in continuous flow. Du Vigneaud & Meyer (J. Biol Chem, 1932, 98, 295-308) had already shown that it was possible to racemize different acetylated amino acids by bringing them into the presence of acetic anhydride for several hours. However, no examples had been made with acetyl leucine. By attempting to reproduce this process with acetyl-leucine, the inventors have thus found that this racemization reaction did not give satisfactory results with acetyl-leucine because of a competitive hydrolysis reaction of acetic anhydride. used. The inventors have also surprisingly discovered that the racemization reaction of N-acetyl-L-leucine could be improved by producing it in a continuous flow. It seems indeed that the realization of this continuous flow process allows better control of the mixing of the reagents and therefore to better control the reaction. The inventors have also shown that the racemization of N-acetyl-L-Leucine in continuous flow was obtained in a very short time of the order of a few minutes.
Furthermore, there is also a need to develop a new method of acetylation of leucine for the preparation of N-actyle-leucine which is faster and more economical. The inventors have discovered that the acetylation reaction of leucine can be improved by making it in a continuous flow. The process according to the invention gives good yields, in a very short time and using fewer reagents compared to the method known hitherto.
Indeed, DeWitt et al. (J Am Chem Soc (1951) 73 (7) 3359-60) described the preparation of N-acetyl-L-Leucine by reacting L-Leucine with 3 molar equivalents of acetic anhydride and sodium hydroxide for 2 hours 20 minutes. . N-acetyl-L-leucine is then obtained in a yield of only 70-80%. In addition, the authors of this publication clearly indicated that a molar ratio between L-Leucine and acetic anhydride below 2 resulted in much lower yields.
SYNTHESIS
H. D. DeWitt and A. W. Ingersoll. The Preparation of Pure N-Acetyl-L-leucine and L-Leucine. Journal of the American Chemical Society 1951 73 (7), 3359-3360. DOI: 10.1021/ja01151a108
PATENT
https://patents.google.com/patent/WO2012038515A1/en
EXAMPLES
A. Acetylation of L-Leucine in Continuous Flow

A. L. Study of the molar ratio of acetic anhydride to leucine
The objective of this study is to define the necessary molar ratio of acetic anhydride so that the acetylation reaction with acetic anhydride is complete and is not disadvantageous by competition with the acetic anhydride hydrolysis reaction. In this study, the residence time in the reactor / exchanger (1 process plate) was set at 9 seconds, for a temperature of the reaction medium of between 25 and 30 ° C.
The ratio range studied is between 0.9 and 2.0 molar equivalents. The optimum is obtained for a ratio between 1.20 and 2.00, more particularly between 1.30 and 1.60. Below this ratio, the acetylation reaction is disadvantageous compared to the acetic hydrolysis reaction. Beyond this, the drop in pH (acid instead of base) also disadvantages the acetylation reaction.
EXAMPLES 1-10:
A solution of sodium L-leucinate, for passage in continuous flow reactor, is prepared in the following manner: 700 g of L-leucine are dissolved in a solution of 576 g of sodium hydroxide and 3.5 liters of Demineralized Water. This solution is the main fluid process. The reaction between this solution and the acetic anhydride is carried out in a continuous flow in a Boostec® reactor, made of silicon carbide. The reactor / exchanger is configured with an injection-type process plate comprised between two utility plates. The volume of the process plate is 10 mL. The temperature in the reactor is maintained by the circulation of a coolant heated by a thermostatic bath. The transformation of L-leucine to N-acetyl-L-leucine is monitored online by quantitative Raman spectroscopy. This method of analysis is calibrated beforehand with solutions of known concentration prepared with pure L-leucine and N-acetyl-L-leucine.
Example 1
The temperature of the thermostated bath is set at 25 ° C. The sodium leucinate solution and pure acetic anhydride are introduced into the reactor at respective flow rates set at 4.06 kg.h -1 and 0.42 kg h -1 . These flow rates correspond to a molar ratio of acetic anhydride to leucine of 0.91 equivalents. The total flow rate is therefore 4.48 kg.h -1 , which corresponds to a residence time (equivalent to the reaction time) of 8.7 s The yield of acetyl-L-leucinate determined by Raman spectroscopy online at the outlet of the reactor is 40% Example 2:
The temperature of the thermostated bath is set at 25 ° C. The sodium leucinate solution and pure acetic anhydride are introduced into the reactor at respective flow rates set at 3.95 kg · h -1 and 0.45 kg · h -1 . These flow rates correspond to a molar ratio of acetic anhydride to leucine of 1.01 equivalents. The total flow rate is therefore 4.40 kg.h -1 , which corresponds to a residence time of 8.9 S. The yield of acetyl-L-leucinate determined by in-line Raman spectroscopy at the outlet of the reactor is 52.degree. %.
Example 3
The temperature of the thermostated bath is set at 25 ° C. The sodium leucinate solution and pure acetic anhydride are introduced into the reactor at respective flow rates set at 3.89 kg · h -1 and 0.52 kg · h -1 . These flow rates correspond to a molar ratio of acetic anhydride to leucine of 1.18 equivalents. The total flow rate is therefore 4.41 kg.h -1 , which corresponds to a residence time of 8.9 S. The yield of acetyl-L-leucinate determined by in-line Raman spectroscopy at the outlet of the reactor is 57.degree. %. Example 4
The temperature of the thermostated bath is set at 25 ° C. The sodium leucinate solution and pure acetic anhydride are introduced into the reactor at respective flow rates set at 3.82 kg. h -1 and 0.57 kg h -1 . These flow rates correspond to a molar ratio of acetic anhydride to leucine of 1.32 equivalents. The total flow is therefore 4.39 kg. h “1 , which corresponds to a residence time of 8.9 S. The yield of acetyl-L-leucinate determined by in-line Raman spectroscopy at the outlet of the reactor is 83%.
Example 5
The temperature of the thermostated bath is set at 25 ° C. The sodium leucinate solution and pure acetic anhydride are introduced into the reactor at respective rates set at 3.64 kg. h -1 and 0.55 kg h -1 . These flow rates correspond to a molar ratio of acetic anhydride to leucine of 1.34 equivalents. The total flow is therefore 4, 19 kg. h “1 , which corresponds to a residence time of 9.4 s The yield of acetyl-L-leucinate determined by in-line Raman spectroscopy at the outlet of the reactor is 98%.
Example 6
The temperature of the thermostated bath is set at 25 ° C. The sodium leucinate solution and pure acetic anhydride are introduced into the reactor at respective rates set at 3.66 kg. h 1 and 0.62 kg h -1 . These flow rates correspond to a molar ratio of acetic anhydride to leucine of 1.50 equivalents. The total flow is therefore 4.28 kg. h “1 , which corresponds to a residence time of 9.2 s The yield of acetyl-L-leucinate determined by in-line Raman spectroscopy at the outlet of the reactor is 96%.
The temperature of the thermostated bath is set at 25 ° C. The sodium leucinate solution and pure acetic anhydride are introduced into the reactor at respective flow rates fixed at 3.67 kg. h -1 and 0.64 kg h -1 . These flow rates correspond to a molar ratio of acetic anhydride to leucine of 1.54 equivalents. The total flow is therefore 4.31 kg. h “1 , which corresponds to a residence time of 9.1 sec The yield of acetyl-L-leucinate determined by in-line Raman spectroscopy at the outlet of the reactor is 100%. Example 8
The temperature of the thermostated bath is set at 25 ° C. The sodium leucinate solution and pure acetic anhydride are introduced into the reactor at respective flow rates set at 3.63 kg. h -1 and 0.73 kg h -1 . These flow rates correspond to a molar ratio of acetic anhydride to leucine of 1.78 equivalents. The total flow is therefore 4.36 kg. h “1 , which corresponds to a residence time of 9.0 s The yield of acetyl-L-leucinate determined by in-line Raman spectroscopy at the outlet of the reactor is 90%.
PATENT
https://patents.google.com/patent/CN104592052A/en
Example 1:
100gL-leucine adds 1000ML2NNaOH rising temperature for dissolving, adds 1ML salicylic aldehyde, 95 degree of insulations of intensification 3 hours, recording optically-active is 0, be cooled to 5 degree and keep, dripping 80ML diacetyl oxide, dropwise maintenance 0.5 hour, be warmed up to 60 degree, add proper amount of active carbon decolouring, add 160ML HCl and adjust PH 2.5, be cooled to 4 degree, suction filtration, the 118g. of oven dry
Example 2:
100gL-leucine adds 1200ML 2NNaOH rising temperature for dissolving, adds 3ML salicylic aldehyde, 95 degree of insulations of intensification 3 hours, recording optically-active is 0, be cooled to 5 degree and keep, dripping 80ML diacetyl oxide, dropwise maintenance 0.5 hour, be warmed up to 60 degree, add proper amount of active carbon decolouring, add the 3.0. that 180ML HCl adjusts PH, be cooled to 4 degree, suction filtration, the 110g. of oven dry
Example 3:
100gL-leucine adds 1000ML 2NNaOH rising temperature for dissolving, adds 2ML salicylic aldehyde, 95 degree of insulations of intensification 3 hours, recording optically-active is 0, be cooled to 5 degree and keep, dripping 80ML diacetyl oxide, dropwise maintenance 0.5 hour, be warmed up to 60 degree, add proper amount of active carbon decolouring, add 180ML HCl and adjust PH 3.0, be cooled to 4 degree, suction filtration, the 120g. of oven dry
Medical uses
Levacetylleucine is indicated for the treatment of neurological manifestations of Niemann-Pick disease type C in people weighing at least 15 kilograms (33 lb).[1][2]
Adverse effects
The most common side effects include abdominal pain, difficulty swallowing, upper respiratory tract infections, and vomiting.[2]
Levacetylleucine may cause embryo-fetal harm if used during pregnancy.[1][2]
History
The safety and efficacy of levacetylleucine for the treatment of Niemann-Pick disease type C were evaluated in a randomized, double-blind, placebo-controlled, two-period, 24-week crossover study.[2] The duration was twelve weeks for each treatment period.[2] The study enrolled 60 participants.[2] To be eligible for the study participants had to be four years of age or older with a confirmed diagnosis of Niemann-Pick disease type C and at least mild disease-related neurological symptoms.[2] Participants could receive miglustat, an enzyme inhibitor, as background treatment in the study.[2]
The US Food and Drug Administration (FDA) granted the application for levacetylleucine priority review, fast track, orphan drug, and rare pediatric disease designations.[2] The FDA granted approval of Aqneursa to IntraBio Inc.[2]
Society and culture
Legal status
Levacetylleucine was approved for medical use in the United States in September 2024.[1][2][5]
Names
Levacetylleucine is the international nonproprietary name.[6]
Research
Levacetylleucine is being studied for the treatment of GM2 gangliosidoses (Tay-Sachs and Sandhoff diseases),[7] ataxia-telangiectasia,[8] Lewy body dementia,[9] amyotrophic lateral sclerosis, restless legs syndrome, multiple sclerosis, and migraine.[10]
References
- ^ Jump up to:a b c d e f g h i “Aqneursa- levacetylleucine granule, for suspension”. DailyMed. 24 September 2024. Retrieved 5 October 2024.
- ^ Jump up to:a b c d e f g h i j k l m n o “FDA Approves New Drug to Treat Niemann-Pick Disease, Type C”. U.S. Food and Drug Administration (Press release). 24 September 2024. Retrieved 25 September 2024. This article incorporates text from this source, which is in the public domain.
- ^ “IntraBio Announces U.S. FDA Approval of Aqneursa for the Treatment of Niemann-Pick Disease Type C”. IntraBio (Press release). 25 September 2024. Retrieved 26 September 2024.
- ^ New Drug Therapy Approvals 2024 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2025. Archived from the original on 21 January 2025. Retrieved 21 January 2025.
- ^ “Novel Drug Approvals for 2024”. U.S. Food and Drug Administration (FDA). 1 October 2024. Retrieved 29 November 2024.
- ^ World Health Organization (2024). “International nonproprietary names for pharmaceutical substances (INN): proposed INN: list 131”. WHO Drug Information. 38 (2). hdl:10665/378367. ISBN 9789240098558.
- ^ Martakis K, Claassen J, Gascon-Bayari J, Goldschagg N, Hahn A, Hassan A, et al. (March 2023). “Efficacy and Safety of N-Acetyl-l-Leucine in Children and Adults With GM2 Gangliosidoses”. Neurology. 100 (10): e1072 – e1083. doi:10.1212/WNL.0000000000201660. PMC 9990862. PMID 36456200.
- ^ Fields T, Patterson M, Bremova-Ertl T, Belcher G, Billington I, Churchill GC, et al. (January 2021). “A master protocol to investigate a novel therapy acetyl-L-leucine for three ultra-rare neurodegenerative diseases: Niemann-Pick type C, the GM2 gangliosidoses, and ataxia telangiectasia”. Trials. 22 (1): 84. doi:10.1186/s13063-020-05009-3. PMC 7821839. PMID 33482890.
- ^ Passmore P (15 April 2014). A clinical trial to test amlodipine as a new treatment for vascular dementia. ISRCTN registry (Report). doi:10.1186/isrctn31208535.
- ^ Strupp M, Bayer O, Feil K, Straube A (February 2019). “Prophylactic treatment of migraine with and without aura with acetyl-DL-leucine: a case series”. Journal of Neurology. 266 (2): 525–529. doi:10.1007/s00415-018-9155-6. PMID 30547273. S2CID 56148131.
Further reading
- Churchill GC, Strupp M, Factor C, Bremova-Ertl T, Factor M, Patterson MC, et al. (August 2021). “Acetylation turns leucine into a drug by membrane transporter switching”. Scientific Reports. 11 (1): 15812. Bibcode:2021NatSR..1115812C. doi:10.1038/s41598-021-95255-5. PMC 8338929. PMID 34349180.
- Bremova-Ertl T, Ramaswami U, Brands M, Foltan T, Gautschi M, Gissen P, et al. (February 2024). “Trial of N-Acetyl-l-Leucine in Niemann-Pick Disease Type C”. The New England Journal of Medicine. 390 (5): 421–431. doi:10.1056/NEJMoa2310151. PMID 38294974.
- Tifft CJ (February 2024). “N-Acetyl-l-Leucine and Neurodegenerative Disease”. The New England Journal of Medicine. 390 (5): 467–470. doi:10.1056/NEJMe2313791. PMID 38294981.
External links
- Clinical trial number NCT05163288 for “A Pivotal Study of N-Acetyl-L-Leucine on Niemann-Pick Disease Type C” at ClinicalTrials.gov
- Bremova-Ertl T, Ramaswami U, Brands M, Foltan T, Gautschi M, Gissen P, Gowing F, Hahn A, Jones S, Kay R, Kolnikova M, Arash-Kaps L, Marquardt T, Mengel E, Park JH, Reichmannova S, Schneider SA, Sivananthan S, Walterfang M, Wibawa P, Strupp M, Martakis K: Trial of N-Acetyl-l-Leucine in Niemann-Pick Disease Type C. N Engl J Med. 2024 Feb 1;390(5):421-431. doi: 10.1056/NEJMoa2310151. [Article]
- Fields T, M Bremova T, Billington I, Churchill GC, Evans W, Fields C, Galione A, Kay R, Mathieson T, Martakis K, Patterson M, Platt F, Factor M, Strupp M: N-acetyl-L-leucine for Niemann-Pick type C: a multinational double-blind randomized placebo-controlled crossover study. Trials. 2023 May 29;24(1):361. doi: 10.1186/s13063-023-07399-6. [Article]
- FDA Approved Drug Products: Aqneursa (levacetylleucine) for oral suspension (September 2024) [Link]
- FDA News Release: FDA Approves New Drug to Treat Niemann-Pick Disease, Type C [Link]
| Clinical data | |
|---|---|
| Trade names | Aqneursa |
| Other names | IB1001 |
| AHFS/Drugs.com | Aqneursa |
| License data | US DailyMed: Levacetylleucine |
| Pregnancy category | Not recommended |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1188-21-2 |
| PubChem CID | 70912 |
| DrugBank | DB16956 |
| ChemSpider | 1918 |
| UNII | E915HL7K2O |
| KEGG | D12967 |
| ChEBI | CHEBI:17786 |
| ChEMBL | ChEMBL56021 |
| PDB ligand | LAY (PDBe, RCSB PDB) |
| CompTox Dashboard (EPA) | DTXSID6045870 |
| ECHA InfoCard | 100.013.370 |
| Chemical and physical data | |
| Formula | C8H15NO3 |
| Molar mass | 173.212 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
/////////Levacetylleucine, Aqneursa, Niemann-Pick disease type C, FDA 2024, APPROVALS 2024, N-Acetyl-L-leucine, 1188-21-2, acetyl-L-leucine, Ac-Leu-OH, N-Acetylleucine, NSC 206316, UNII-E915HL7K2O, ORPHAN DRUG, NSC-206316, NSC 206316
Trospium chloride



Trospium chloride
CAS
47608-32-2
10405-02-4
WeightAverage: 392.518
Monoisotopic: 392.22202025
Chemical FormulaC25H30NO3
T4Y8ORK057
- 73954-17-3
- 8-Benziloyloxy-6,10-ethano-5-azoniaspiro(4.5)decane chloride
- 3-[(2-hydroxy-2,2-diphenylacetyl)oxy]-8lambda5-azaspiro[bicyclo[3.2.1]octane-8,1′-pyrrolidin]-8-yliumchloride
- spiro[8-azoniabicyclo[3.2.1]octane-8,1′-azolidin-1-ium]-3-yl 2-hydroxy-2,2-diphenylacetate;chloride
- SMR002533165
- spiro[8-azoniabicyclo[3.2.1]octane-8,1′-azolidin-1-ium]-3-yl 2-hydroxy-2,2-diphenylacetate;chloride
FDA 2024, Cobenfy 9/26/2024, To treat schizophrenia
Press Release
Drug Trials Snapshot
Trospium chloride is a muscarinic antagonist used to treat overactive bladder.[3] It has side effects typical of this class of drugs, namely dry mouth, stomach upset, and constipation; these side effects cause problems with people taking their medicine as directed. However it doesn’t cause central nervous system side effects like some other muscarinic antagonists.[4]
Chemically it is a quaternary ammonium cation which causes it to stay in periphery rather than crossing the blood–brain barrier.[5] It works by causing the smooth muscle in the bladder to relax.[3]
It was patented in 1966 and approved for medical use in 1974.[6] It was first approved in the US in 2004, and an extended release version was brought to market in 2007. It became generic in the EU in 2009, and the first extended-release generic was approved in the US in 2012.
SYN

Tropium chloride is one of the azoniaspironortropine derivatives and is used for the treatment of urinary bladder dysfunction due to bladder dysfunction, night urination, overactive bladder, and urinary incontinence. Useful compounds. The chemical name of the tropium chloride is (1R, 3R, 5S) -3-[(hydroxydiphenylacetyl) oxy] spiro [8-azoniabiscyclo [3,2,1] octane-8,1 ‘ -Pyrrolidinium] chloride ((1R, 3R, 5S) -3-[(Hydroxydiphenylacetyl) oxy] spiro [8-azoniabicyclo [3,2,1] octane-8,1’-pyrrolidinium] chloride) It is represented by Formula (1).

As a method for preparing the thromium chloride, US Patent No. 3,480,626 (1969) is prepared in the form of a free base of nortropine benzilate represented by the formula (2) as an intermediate, as shown in Scheme 1 below Thereafter, it is reacted with 1,4-dichlorobutane of the formula (3) to synthesize a thromium chloride, which is then recrystallized in ethanol-ether to disclose a two-step process for obtaining the thromium chloride. However, the method does not use a base, has a long reaction time, a low yield (about 46%), and instead of intramolecular cyclization, positions 1 and 4 of butane represented by the formula (4) as side reactants. There is a disadvantage in that a large amount of the compound in the form of substituted 1,4-nortropin benzylate is produced.

PATENT
https://patents.google.com/patent/KR20090076081A/en

Example 1 Preparation of Tropium Chloride
In a 1 L reactor equipped with a stirrer, 100 g of nortropin benzylate hydrochloride, 59 ml of 1,4-dichlorobutane, 89 ml of 1,8-diazabicyclo and 5 ml of 1,8-diazabicyclo [5,4,0] undec-7-ene and 500 ml of acetonitrile The reaction was carried out at 60 ° C. for 2 hours. Thin-Layer Chromatography (TLC) confirmed the completion of the reaction, when the reaction was complete, cooled to 5 ℃, stirred for 1 hour at the same temperature, the resulting crystals were filtered, dried at 60 ℃, white 92.6 g (yield: 81%) of the target compound were obtained. The 1 H-NMR (D 2 O, 400 MHz) data of the obtained compound are as follows: δ 1.34 to 1.36 (2H, d), 1.80 to 1.87 (2H, m), 1.98 (4H, s), 2.44 to 2.48 (2H , d), 3.21-3.24 (2H, t), 3.43-3.46 (2H, t), 3.56 (2H, s), 5.12-5.13 (1H, t), 7.31-7.74 (10H, m).
PATENT
https://patents.google.com/patent/CN102718760B/en


Medical uses
Trospium chloride is used for the treatment of overactive bladder with symptoms of urge incontinence and frequent urination.[3][4][2]
It should not be used with people who retain urine, who have severe digestive conditions, myasthenia gravis, narrow-angle glaucoma, or tachyarrhythmia.[3]
It should be used with caution in people who have problems with their autonomous nervous system (dysautonomia) or who have gastroesophageal reflux disease, or in whom fast heart rates are undesirable, such as people with hyperthyroidism, coronary artery disease and congestive heart failure.[3]
There are no adequate and well-controlled studies of trospium chloride in pregnant women and there are signs of harm to the fetus in animal studies. The drug was excreted somewhat in the milk of nursing mothers.[3] The drug was studied in children.[3]
Side effects
Side effects are typical of gastrointestinal effects of anticholinergic drugs, and include dry mouth, indigestion, and constipation. These side effects lead to problems with adherence, especially for older people.[4] The only CNS side effect is headache, which was very rare. Tachycardia is a rare side effect.[3]
Pharmacology
Mechanism of action
| Target | Affinity (Ki, nM) | Species |
|---|---|---|
| M1 | 3.5 | Human |
| M2 | 1.1 | Human |
| M3 | 1.0 | Human |
| M4 | 1.4 | Human |
| M5 | 6.0 | Human |
| Notes: Values are Ki, unless otherwise specified. The smaller the value, the more strongly the drug binds to the site. | ||
Trospium chloride is a muscarinic antagonist. Trospium chloride blocks the effect of acetylcholine on muscarinic receptors organs that are responsive to the compounds, including the bladder.[3] Its parasympatholytic action relaxes the smooth muscle in the bladder.[4] Receptor assays showed that trospium chloride has negligible affinity for nicotinic receptors as compared to muscarinic receptors at concentrations obtained from therapeutic doses.[3] The drug has high and similar affinity for all five of the muscarinic acetylcholine receptor subtypes, including the M1, M2, M3, M4, and M5 receptors.[9][10][11]
Pharmacokinetics
After oral administration, less than 10% of the dose is absorbed. Mean absolute bioavailability of a 20 mg dose is 9.6% (range: 4.0 to 16.1%). Peak plasma concentrations (Cmax) occur between 5 and 6 hours post-dose. Mean Cmax increases greater than dose-proportionally; a 3-fold and 4-fold increase in Cmax was observed for dose increases from 20 mg to 40 mg and from 20 mg to 60 mg, respectively. AUC exhibits dose linearity for single doses up to 60 mg. Trospium chloride exhibits diurnal variability in exposure with a decrease in Cmax and AUC of up to 59% and 33%, respectively, for evening relative to morning doses.[12]
Administration with a high fat meal resulted in reduced absorption, with AUC and Cmax values 70 to 80% lower than those obtained when trospium chloride was administered while fasting. Therefore, it is recommended that trospium chloride should be taken at least one hour prior to meals or on an empty stomach.[12]
Protein binding ranged from 50 to 85% when concentration levels of trospium chloride (0.5 to 50 ng/mL) were incubated with human serum in vitro. The 3H-trospium chloride ratio of plasma to whole blood was 1.6:1. This ratio indicates that the majority of 3H-trospium chloride is distributed in plasma. The apparent volume of distribution for a 20 mg oral dose is 395 (± 140) liters.[12]
The metabolic pathway of trospium in humans has not been fully defined. Of the 10% of the dose absorbed, metabolites account for approximately 40% of the excreted dose following oral administration. The major metabolic pathway is hypothesized as ester hydrolysis with subsequent conjugation of benzylic acid to form azoniaspironortropanol with glucuronic acid. Cytochrome P450 is not expected to contribute significantly to the elimination of trospium. Data taken from in vitro human liver microsomes investigating the inhibitory effect of trospium on seven cytochrome P450 isoenzyme substrates (CYP1A2, 2A6, 2C9, 2C19, 2D6, 2E1, and 3A4) suggest a lack of inhibition at clinically relevant concentrations.[12]
The plasma half-life for trospium chloride following oral administration is approximately 20 hours. After oral administration of an immediate-release formulation of 14C-trospium chloride, the majority of the dose (85.2%) was recovered in feces and a smaller amount (5.8% of the dose) was recovered in urine; 60% of the radioactivity excreted in urine was unchanged trospium. The mean renal clearance for trospium (29 L/hour) is 4-fold higher than average glomerular filtration rate, indicating that active tubular secretion is a major route of elimination for trospium. There may be competition for elimination with other compounds that are also renally eliminated.[12]
Chemistry
Anticholinergic drugs used to treat overactive bladder were all amines as of 2003. Quaternary ammonium cations in general are more hydrophilic than other amines and don’t cross membranes well, so they tend to be poorly absorbed from the digestive system, and to not cross the blood–brain barrier. Oxybutynin, tolterodine, darifenacin, and solifenacin are tertiary amines while trospium chloride and propantheline are quaternary amines.[5]
History
The synthesis of trospium was described by scientists from Dr. Robert Pfleger Chemische Fabrik GmbH, Heinz Bertholdt, Robert Pfleger, and Wolfram Schulz, in US. Pat. No. 3,480,626 (the US equivalent to DE119442), and its activity was first published in the literature in 1967.[13][14]
The first regulatory approval was granted in Germany in August 1999 to Madaus AG for Regurin 20 mg Tablets.[15]: 13 Madaus is considered the originator for regulatory filings worldwide.[16] The German filing was recognized throughout Europe under the Mutual Recognition Procedure.[15]: 13
Madaus licensed the US rights to trospium chloride to Interneuron in 1999 and Interneuron ran clinical trials in the US to win FDA approval.[17][18] Interneuron changed its name to Indevus in 2002[19] Indevus entered into a partnership with Odyssey Pharmaceuticals, a subsidiary of Pliva, to market the drug in April 2004,[20] and won FDA approval for the drug, which it branded as Sanctura, in May 2004.[21][22] The approval earned Indevus a milestone payment of $120M from Pliva, which had already paid Indevus $30 million at signing; the market for overactive bladder therapies was estimated to be worth $1.1 billion in 2004.[23] In 2005 Pliva exited the relationship, selling its rights to Esprit Pharma,[24] and in September 2007 Allergan acquired Esprit, and negotiated a new agreement with Indevus under which Allergan would completely take over the US manufacturing, regulatory approvals, and marketing.[25] A month before, Indevus had received FDA approval for an extended release formulation that allowed once a day dosing, Sanctura XR.[26] Indevus had developed intellectual property around the extended release formulation which it licensed to Madaus for most of the world.[25]
In 2012 the FDA approved the first generic version of the extended release formulation, granting approval to the ANDA that Watson Pharmaceuticals had filed in 2009.[27] Annual sales in the US at that time were $67M.[28] European patents had expired in 2009.[29]
As of 2016, the drug is available worldwide under many brand names and formulations, including oral, extended release, suppositories, and injections.[1]
Society and culture
Marketing rights to the drug became subject to parallel import litigation in Europe in the case of Speciality European Pharma Ltd v Doncaster Pharmaceuticals Group Ltd / Madaus GmbH (Case No. A3/2014/0205) which was resolved in March 2015. Madaus had exclusively licensed the right to use the Regurin trademark to Speciality European Pharma Ltd. In 2009, when European patents expired on the drug, Doncaster Pharmaceuticals Group, a well known parallel importer, which had been selling the drug in the UK under another label, Ceris, which was used in France, began to put stickers on their packaging with the Regurin name. Speciality and Madaus sued and initially won based on the argument that 90% of prescriptions were already generic, but Doncaster appealed and won the appeal based on the argument that it could not charge a premium with a generic label. The case has broad implications for trade in the EU.[29][30]
Research
In 2007 Indevus partnered with Alkermes to develop and test an inhaled form of trospium chloride as a treatment for COPD; it was in Phase II trials at that time.[31]
Reference
- ^ Jump up to:a b “International brands of trospium”. Drugs.com. Retrieved 13 May 2016.
- ^ Jump up to:a b FDA “Trospium chloride label” (PDF). U.S. Food and Drug Administration. January 2011.
- ^ Jump up to:a b c d e f g h i j “Regurin XL 60mg”. UK eMC. 3 July 2015.
- ^ Jump up to:a b c d Biastre K, Burnakis T (February 2009). “Trospium chloride treatment of overactive bladder”. Ann Pharmacother. 43 (2): 283–95. doi:10.1345/aph.1L160. PMID 19193592. S2CID 20102756.
- ^ Jump up to:a b Pak RW, Petrou SP, Staskin DR (December 2003). “Trospium chloride : a quaternary amine with unique pharmacologic properties”. Curr Urol Rep. 4 (6): 436–40. doi:10.1007/s11934-003-0023-1. PMID 14622495. S2CID 4512769.
- ^ Fischer J, Ganellin CR (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 446. ISBN 9783527607495.
- ^ Liu T (2020). “BindingDB BDBM50540489 Flotros::IP-631::IP631::Regurin::Regurin xl::Sanctura::Sanctura xr::Spasmo-lyt::Trospium chloride::Uraplex”. Journal of Medicinal Chemistry. 63 (11): 5763–5782. doi:10.1021/acs.jmedchem.9b02100. PMC 8007111. PMID 32374602. Retrieved 28 October 2024.
- ^ Del Bello F, Bonifazi A, Giorgioni G, Piergentili A, Sabbieti MG, Agas D, et al. (June 2020). “Novel Potent Muscarinic Receptor Antagonists: Investigation on the Nature of Lipophilic Substituents in the 5- and/or 6-Positions of the 1,4-Dioxane Nucleus”. J Med Chem. 63 (11): 5763–5782. doi:10.1021/acs.jmedchem.9b02100. PMC 8007111. PMID 32374602.
- ^ Peretto I, Petrillo P, Imbimbo BP (November 2009). “Medicinal chemistry and therapeutic potential of muscarinic M3 antagonists”. Med Res Rev. 29 (6): 867–902. doi:10.1002/med.20158. PMID 19399831.
- ^ Pak RW, Petrou SP, Staskin DR (December 2003). “Trospium chloride: a quaternary amine with unique pharmacologic properties”. Curr Urol Rep. 4 (6): 436–440. doi:10.1007/s11934-003-0023-1. PMID 14622495.
- ^ Rosa GM, Bauckneht M, Scala C, Tafi E, Leone Roberti Maggiore U, Ferrero S, et al. (November 2013). “Cardiovascular effects of antimuscarinic agents in overactive bladder”. Expert Opin Drug Saf. 12 (6): 815–827. doi:10.1517/14740338.2013.813016. PMID 23800037.
- ^ Jump up to:a b c d e Doroshyenko O, Jetter A, Odenthal KP, Fuhr U (2005). “Clinical pharmacokinetics of trospium chloride”. Clin Pharmacokinet. 44 (7): 701–20. doi:10.2165/00003088-200544070-00003. PMID 15966754. S2CID 10968270.
- ^ US 6974820 which cites US 3480626 and Bertholdt H, Pfleger R, Schulz W (1967). “[On azoniaspire-compounds. 2. Preparation of esterified azoniaspire-compounds of nortropan-3-alpha- or 3-beta-ol (1)]”. Arzneimittelforschung. 17 (6): 719–26. PMID 5632538.
- ^ DE patent 1194422, Bertholdt H, Pfleger R, Schulz W, “[Verfahren zur Herstellung von Azoniaspironortropanderivaten] (A process for preparing azonia-spirono-tropane derivatives)”, issued 10 June 1965, assigned to Dr. Robert Pfleger Chemische Fabrik GmbH
- ^ Jump up to:a b “Trospium Chloride 20mg Film-Coated Tablets, Public Assessment Report” (PDF). Medicines and Healthcare products Regulatory Agency. 7 April 2011.
- ^ “Trospium chloride”. AdisInsight. Springer Nature Switzerland AG.
- ^ Miller J (23 September 2002). “Indevus to apply for new drug status for incontinence drug”. Boston Business Journal.
- ^ Herper M (25 September 2002). “A Biotech Phoenix Could Be Rising”. Forbes.
- ^ “Indevus Pharmaceuticals, Inc., Formerly Interneuron, to Begin Trading on Nasdaq”. Indevus Press Release. 2 April 2002.
- ^ “Indevus and PLIVA Sign Co-Promotion and Licensing Agreement for SANCTURA -Trospium Chloride”. Indevus Press Release. 7 April 2004. Archived from the original on 27 August 2021. Retrieved 14 May 2016.
- ^ “Sanctura (trospium chloride)”. CenterWatch. Archived from the original on 5 August 2019. Retrieved 13 May 2016.
- ^ “Indevus Announces FDA Approval Of Sanctura”. Indevus Press Release. 28 May 2004.
- ^ Osterweil N (28 May 2004). “FDA approves Indevus’ Sanctura”. for First Word Pharma.
- ^ “Novartis, P&G enter agreement for OAB drug”. Urology Times. 21 July 2005.
- ^ Jump up to:a b “Indevus Announces Allergan as New Partner for Sanctura Brand”. Indevus Press Release. 19 September 2007.
- ^ “Indevus’ Sanctura XR approved by US FDA”. The Pharma Letter. 13 August 2007.
- ^ “ANDA 091289 approval letter” (PDF). U.S. Food and Drug Administration. 12 October 2012.
- ^ “Watson’s Generic Sanctura XR Receives FDA Approval”. Watson Press Release. 12 October 2012.
- ^ Jump up to:a b “Court takes a permissive approach to parallel importers within the EU”. Lexology. 6 March 2015.
- ^ R.P.C. (2015) 132 (7): 521-540. doi: 10.1093/rpc/rcv039
- ^ “Alkermes, Indevus testing COPD drug”. UPI. 25 April 2007.
External links
Trospium chloride at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
| Clinical data | |
|---|---|
| Pronunciation | /ˈtroʊspiəm/ TROHS-pee-əm |
| Trade names | Regurin, Sanctura, others[1] |
| AHFS/Drugs.com | Monograph |
| Routes of administration | By mouth |
| Drug class | Antimuscarinic (peripherally selective) |
| ATC code | G04BD09 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[2]In general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Protein binding | 50–85% |
| Elimination half-life | 20 hours |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 10405-02-4 |
| PubChem CID | 107979 |
| DrugBank | DB00209 |
| ChemSpider | 10482307 |
| UNII | 1E6682427E |
| ChEBI | CHEBI:32270 |
| ChEMBL | ChEMBL1201344 |
| CompTox Dashboard (EPA) | DTXSID7023724 |
| ECHA InfoCard | 100.030.784 |
| Chemical and physical data | |
| Formula | C25H30ClNO3 |
| Molar mass | 427.97 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
- Trospium [Link]
- FDA drug approval: Trospium [Link]
- FDA Approved Drug Products: Cobenfy (xanomeline tartrate/trospium chloride) capsules for oral use (September 2024) [Link]
- DailyMed Label: TROSPIUM CHLORIDE oral capsule, extended release [Link]
///////Trospium chloride, Cobenfy, APPROVALS 2024, FDA 2024, SMR002533165
Flurpiridaz F 18



Flurpiridaz F 18
WeightAverage: 367.84
Monoisotopic: 367.1328329
Chemical FormulaC18H22ClFN2O3
- 863887-89-2
- Bms 747158-02
2-tert-butyl-4-chloro-5-[[4-(2-(18F)fluoranylethoxymethyl)phenyl]methoxy]pyridazin-3-one
- 2-tert-butyl-4-chloro-5-[[4-(2-(18F)fluoranylethoxymethyl)phenyl]methoxy]pyridazin-3-one
- 2-TERT-BUTYL-4-CHLORO-5-((4-((2-(18F)FLUOROETHOXY)METHYL)PHENYL)METHOXY)PYRIDAZIN-3(2H)-ONE
- 3(2H)-PYRIDAZINONE, 4-CHLORO-2-(1,1-DIMETHYLETHYL)-5-((4-((2-(FLUORO-18F)ETHOXY)METHYL)PHENYL)METHOXY)-
- UNII-TY3V24C029
FDA APPROVED 9/27/2024, Flyrcado, A radioactive diagnostic drug to evaluate for myocardial ischemia and infarction
Flurpiridaz (18F), sold under the brand name Flyrcado, is a cyclotron-produced radioactive diagnostic agent for use with positron emission tomography (PET) myocardial perfusion imaging under rest or stress (pharmacologic or exercise).[3] Flurpiridaz (18F) It is given by intravenous injection.[3]
The most common adverse reactions include dyspnea (shortness of breath), headache, angina pectoris (severe pain in the chest), chest pain, fatigue, ST segment changes, flushing, nausea, abdominal pain, dizziness, and arrhythmia (irregular heartbeat).[3]
Flurpiridaz (18F) was approved for medical use in the United States in September 2024.[3][4][5][6]
PATENT
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US9687571 | No | 2017-06-27 | 2032-11-01 |  |
| US9603951 | No | 2017-03-28 | 2031-05-02 | |
| US9161997 | No | 2015-10-20 | 2026-02-04 | |
| US8936777 | No | 2015-01-20 | 2031-06-30 | |
| US8226929 | No | 2012-07-24 | 2028-06-21 | |
| US7344702 | No | 2008-03-18 | 2026-05-26 | |
SYN
https://ejnmmipharmchem.springeropen.com/articles/10.1186/s41181-022-00182-z


Chemistry
Synthesis of precursor of [18F]Flurpiridaz (7) [2-(4-((1-tert-Butyl-5-chloro-6-oxo-1,6-dihydropyridazine-4-yloxy)methyl)benzyloxy)ethyl-4- methylbenzensulfonate] (6)
Precursor 6 was synthesized according to the literature procedures with few changes (Purohit et al. 2008; Nagel 2014) Briefly, to a mixture of mucochloric acid (1) (1.18 g, 6.98 mmol) and Na2CO3 (0.33 g, 3.11 mmol) in 15 ml of distilled water was added tert-butylhydrazine hydrochloride (0.86 g, 6.90 mmol) in ice-water bath and reaction mixture was stirred for about 4 h. White precipitate was washed by water and dried under reduced vacuum after filtration. Then, 13.2 ml of benzene and acetic acid (1.86 g, 30,95 mmol) were added and reaction was kept at 40 °C for 4 h. Organic phase was extracted with 10 ml of water and washed by 5 ml of 1.25 M NaOH(aq), 5 ml of 5 M HCl(aq) and 10 ml of water respectively. 0.83 g of DCP (2) was obtained as an orange solid. 1.0 g of DCP (2) (4,53 mmol) was dissolved in 15 ml of dry DMF, 1,4-phenylene dimethanol (3.2 g, 23.16 mmol) and Cs2CO3 (6.0 g, 18.41 mmol) were slowly added to the solution and reaction was stirred at 68 °C under nitrogen atmosphere for about 6 h and allowed to be cooled down to room temperature. Crude product was extracted with CHCl3/water several times and evaporated under vacuum. Residue was subjected to flash column chromatography (silica gel 40 g, EtOAc/Hexane 3:2) and 0.91 g of compound 3 was obtained as white solid. Then, 0.91 g of an alcoholic compound 3 was dissolved in 15 ml of freshly distilled dichloromethane and 0.14 ml of PBr3 was slowly added to the solution. The reaction was carried out at room temperature for about 2 h under nitrogen atmosphere. Crude product was extracted with 30 ml of water and dried under vacuum. White solid product 4 was successfully obtained in a quantitative yield without further purification for next step. KOtBu (0.28 g, 2.49 mmol) and 11.2 ml of ethylene glycol were stirred at room temperature under nitrogen atmosphere. Then, 0.95 g of bromide compound 4 dissolved in 8 ml of dry THF was added slowly into the reaction mixture and the reaction was stirred at 60 °C for overnight. After cooling to room temperature, THF was evaporated and residue was extracted with CHCl3/water several times. Organic phase was evaporated under vacuum and residue was submitted to flash column chromatograpy (silica gel 40 g, EtOAc/Hexane 2:1) and 0.86 g of compound 5 was obtained as colorless oil in quantitative yield. Finally, to a mixture of 0.85 g of compound 5 and tosyl chloride (690 mg, 3.62 mmol) in 6 ml of dry dichloromethane, 0.64 ml of DIPEA and 4-(dimethylamino) pyridine (445 mg, 3.64 mmol) were added and reaction was carried out at room temperature for 2.5 h under nitrogen atmosphere. Dichloromethane was evaporated and crude product was directly subjected to flash column chromatograpy (silica gel 45 g, EtOAc/Hexane 2:1). 0.9 g of pure tosylate 6 (precursor of [18F]Flurpiridaz) was obtained by recrystallisation in dichloromethane at + 4 °C. Tosylate 6 was further purified through semipreparative HPLC for an accurate spectroscopic characterization (Fig. 1). Anal. Calcd for C25H29ClN2O6S: C, 57.63; H, 5.61; Cl, 6.80; N, 5.38; S, 6.15. Found: C, 57.86; H, 5.84; Cl, 7.03; N, 5.66; S, 6.34.
1H NMR ((CDCl3, 400 MHz) δ (ppm)): 7,80 (d, J = 9.1 Hz, 2H); 7,73 (s, 1H); 7,39 (d, J = 9.1 Hz, 2H); 5,29 (s, 2H,); 4,49 (s, 2H,); 4,20–4,19 (m, 2H); 3,70–3,65 (m, 2H); 2,42 (s, 3H); 1,60 (s, 9H).
Synthesis of [18F]Flurpiridaz (7)
Preliminary studies & synthesis of [19F]Flurpiridaz (7) (Cold runs)
Materials KF, Ethanol, and Acetonitrile were obtained from Sigma Aldrich. Kryptofix K2.2.2./K2CO3 (22 mg Kryptofix K2.2.2., 7 mg K2CO3, 300 µl acetonitrile and 300 µl pure water), TBA-HCO3 (0.075 M) solution and QMA Cartridges were from ABX. Sep-Pak C18 Plus Light Cartridge was from Waters.
Methods
Firstly, consecutive cold syntheses of [19F]Flurpiridaz (7) were performed using stable isotope fluorine-19 and optimum reaction parameters were tried to be determined.
Eluent solution-I (Kryptofix K2.2.2./K2CO3)
50 mg of KF was dissolved in 2 mL of ultrapure water and directly passed through the preconditioned QMA cartridge. The QMA cartridge was rinsed with 5 mL of ultrapure water and dried with N2. [19F]F trapped on the QMA cartridge was eluted into the reaction vial with 600 µL of Kryptofix K2.2.2./K2CO3 solution. Solvents in the reaction vial were removed at 100 °C, [19F]F and Kryptofix K2.2.2./K2CO3 were dried gently. Then, 10 mg of precursor 6 dissolved in 2 mL of anhydrous acetonitrile was added to the reaction vial and the mixture was sealed and heated at 95 °C for 10 min. The reaction solution was diluted with 5 ml of ultrapure water and directly passed through a preconditioned C-18 cartridge. C-18 cartridge was rinsed with 5 mL of ultrapure water and dried with air. Finally, C-18 cartridge was eluted with 5 mL of ethanol and transferred into the final product vial. The final product was diluted with 5 mL of ultrapure water (n = 3) and analyzed by HPLC (described in HPLC analysis of precursor 6) to determine its composition.
Chromatogram analysis indicated that four different separate peaks were observed. The unreacted precursor 6 was detected around 11.55 min. The other two peaks were detected between 8 and 9 min. Another major peak around 5.5 min was detected. It was concluded that the chemical yield of product 7 was low due to the majority of side-product formations
Medical uses
Flurpiridaz (18F) is indicated for positron emission tomography myocardial perfusion imaging under rest or stress (pharmacologic or exercise) in adults with known or suspected coronary artery disease to evaluate for myocardial ischemia and infarction.[2][3]
History
Flurpiridaz F-18 is a fluorine 18-labeled agent developed by Lantheus Medical Imaging for the diagnosis of coronary artery disease.[7]
The efficacy and safety of flurpiridaz (18F) were evaluated in two prospective, multicenter, open-label clinical studies in adults with either suspected CAD (Study 1: NCT03354273) or known or suspected CAD (Study 2: NCT01347710).[3] Study 1 evaluated the sensitivity (ability to designate an imaged patient with disease as positive) and specificity (ability to designate an imaged patient without disease as negative) of flurpiridaz (18F) for the detection of significant CAD in subjects with suspected CAD who were scheduled for invasive coronary angiography (ICA).[3] Across three flurpiridaz (18F) imaging readers, estimates of sensitivity ranged from 74% to 89% and estimates of specificity ranged from 53% to 70% for CAD defined as at least 50% narrowing of an artery.[3]
Study 2 evaluated the sensitivity and specificity of flurpiridaz (18F) for the detection of significant CAD in subjects with known or suspected CAD who had ICA without intervention within 60 days prior to imaging or were scheduled for ICA.[3] Across three flurpiridaz (18F) imaging readers, estimates of sensitivity ranged from 63% to 77% and estimates of specificity ranged from 66% to 86% for CAD defined as at least 50% narrowing of an artery.[3]
Society and culture
Legal status
Flurpiridaz (18F) was approved for medical use in the United States in September 2024.[2][3]
Names
Flurpiridaz (18F) is the international nonproprietary name.[8]
References
- ^ “Flurpiridaz F 18”. AMA Finder. Retrieved 27 September 2024.
- ^ Jump up to:a b c d e “Flyrcado (flurpiridaz F 18) injection, for intravenous use” (PDF). U.S. Food and Drug Administration (FDA). Retrieved 27 September 2024.
- ^ Jump up to:a b c d e f g h i j k “FDA approves imaging drug for evaluation of myocardial ischemia”. U.S. Food and Drug Administration (FDA). 27 September 2024. Retrieved 27 September 2024. This article incorporates text from this source, which is in the public domain.
- ^ “Drug Approval Package: Flyrcado Injection”. U.S. Food and Drug Administration (FDA). 25 October 2024. Retrieved 21 January 2025.
- ^ “Novel Drug Approvals for 2024”. U.S. Food and Drug Administration. 1 October 2024. Retrieved 8 November 2024.
- ^ New Drug Therapy Approvals 2024 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2025. Archived from the original on 21 January 2025. Retrieved 21 January 2025.
- ^ “Flurpiridaz F-18”. Inxight Drugs. Retrieved 27 September 2024.
- ^ World Health Organization (2011). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 65”. WHO Drug Information. 25 (1). hdl:10665/74623.
Further reading
- Maddahi J, Agostini D, Bateman TM, Bax JJ, Beanlands RS, Berman DS, et al. (October 2023). “Flurpiridaz F-18 PET Myocardial Perfusion Imaging in Patients With Suspected Coronary Artery Disease”. Journal of the American College of Cardiology. 82 (16): 1598–1610. doi:10.1016/j.jacc.2023.08.016. PMID 37821170.
- Matsumoto N (2023). “Progress of 18F-flurpiridaz in Clinical Trials”. Annals of Nuclear Cardiology. 9 (1): 91–93. doi:10.17996/anc.23-00011. PMC 10696143. PMID 38058576.
External links
- Clinical trial number NCT03354273 for “An International Study to Evaluate Diagnostic Efficacy of Flurpiridaz (18F) Injection PET MPI in the Detection of Coronary Artery Disease (CAD)” at ClinicalTrials.gov
- Clinical trial number NCT01347710 for “A Phase 3 Multi-center Study to Assess PET Imaging of Flurpiridaz F 18 Injection in Patients With CAD” at ClinicalTrials.gov
- Maddahi J, Agostini D, Bateman TM, Bax JJ, Beanlands RSB, Berman DS, Dorbala S, Garcia EV, Feldman J, Heller GV, Knuuti JM, Martinez-Clark P, Pelletier-Galarneau M, Shepple B, Tamaki N, Tranquart F, Udelson JE: Flurpiridaz F-18 PET Myocardial Perfusion Imaging in Patients With Suspected Coronary Artery Disease. J Am Coll Cardiol. 2023 Oct 17;82(16):1598-1610. doi: 10.1016/j.jacc.2023.08.016. [Article]
- Berman DS, Maddahi J, Tamarappoo BK, Czernin J, Taillefer R, Udelson JE, Gibson CM, Devine M, Lazewatsky J, Bhat G, Washburn D: Phase II safety and clinical comparison with single-photon emission computed tomography myocardial perfusion imaging for detection of coronary artery disease: flurpiridaz F 18 positron emission tomography. J Am Coll Cardiol. 2013 Jan 29;61(4):469-477. doi: 10.1016/j.jacc.2012.11.022. Epub 2012 Dec 19. [Article]
- Maddahi J, Lazewatsky J, Udelson JE, Berman DS, Beanlands RSB, Heller GV, Bateman TM, Knuuti J, Orlandi C: Phase-III Clinical Trial of Fluorine-18 Flurpiridaz Positron Emission Tomography for Evaluation of Coronary Artery Disease. J Am Coll Cardiol. 2020 Jul 28;76(4):391-401. doi: 10.1016/j.jacc.2020.05.063. [Article]
- Patel KK, Singh A, Bateman TM: The Potential of F-18 Flurpiridaz PET/CT Myocardial Perfusion Imaging for Precision Imaging. Curr Cardiol Rep. 2022 Aug;24(8):987-994. doi: 10.1007/s11886-022-01713-5. Epub 2022 May 26. [Article]
- FDA Approved Drug Products: FLYRCADO (flurpiridaz F 18) injection, for intravenous use [Link]
/////////////Flurpiridaz F 18, Flyrcado, APPROVALS 2024, FDA 2024, Bms 747158-02, BMS 747158-02, BM-747158-02, BMS747158-02
| Clinical data | |
|---|---|
| Trade names | Flyrcado |
| Other names | NMB58, BMS-747158-02, flurpiridaz F-18, flurpiridaz F 18[1] (USAN US) |
| AHFS/Drugs.com | Flyrcado |
| License data | US DailyMed: Flurpiridaz |
| Routes of administration | Intravenous |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 863887-89-2 |
| PubChem CID | 11405965 |
| DrugBank | DB18773 |
| ChemSpider | 9580860 |
| UNII | TY3V24C029 |
| KEGG | D10009 |
| CompTox Dashboard (EPA) | DTXSID00235517 |
| Chemical and physical data | |
| Formula | C18H22Cl[18F]N2O3[2] |
| Molar mass | 367.8 [2] |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
Inavolisib



Inavolisib
WeightAverage: 407.378
Monoisotopic: 407.140510438
Chemical FormulaC18H19F2N5O4
- GDC-0077
- CAS 2060571-02-8
- GDC0077
- RG6114
- WHO 11204
- GDC 0077
- GDC-0077
- RG-6114
- RG6114
- RO-7113755
- RO7113755
FDA APPROVED, 10/10/2024, Itovebi, To treat locally advanced or metastatic breast cancer
Drug Trials Snapshot
(2S)-2-[[2-[(4S)-4-(difluoromethyl)-2-oxo-1,3-oxazolidin-3-yl]-5,6-dihydroimidazo[1,2-d][1,4]benzoxazepin-9-yl]amino]propanamide
- (2S)-2-((2-((4S)-4-(difluoromethyl)-2-oxo-3-oxazolidinyl)-5,6-dihydroimidazo(1,2-D)(1,4)benzoxazepin-9-yl)amino)propanamide
- propanamide, 2-((2-((4S)-4-(difluoromethyl)-2-oxo-3-oxazolidinyl)-5,6-dihydroimidazo(1,2-D)(1,4)benzoxazepin-9-yl)amino)-, (2S)-
Inavolisib, sold under the brand name Itovebi, is an anti-cancer medication used for the treatment of breast cancer.[2][3] It is an inhibitor and degrader of mutant phosphatidylinositol 3-kinase (PI3K) alpha.[4] The PI3K-mediated signalling pathway has shown to play an important role in the development of tumours as dysregulation is commonly associated with tumour growth and resistance to antineoplastic agents and radiotherapy.[5]
The most common adverse reactions include decreased neutrophils, decreased hemoglobin, increased fasting glucose, decreased platelets, decreased lymphocytes, stomatitis, diarrhea, decreased calcium, fatigue, decreased potassium, increased creatinine, increased ALT, nausea, decreased sodium, decreased magnesium, rash, decreased appetite, COVID-19 infection, and headache.[3]
Inavolisib was approved for medical use in the United States in October 2024.[3][6][7]
SYN
Hanan EJ, Braun MG, Heald RA, MacLeod C, Chan C, Clausen S, Edgar KA, Eigenbrot C, Elliott R, Endres N, Friedman LS, Gogol E, Gu XH, Thibodeau RH, Jackson PS, Kiefer JR, Knight JD, Nannini M, Narukulla R, Pace A, Pang J, Purkey HE, Salphati L, Sampath D, Schmidt S, Sideris S, Song K, Sujatha-Bhaskar S, Ultsch M, Wallweber H, Xin J, Yeap S, Young A, Zhong Y, Staben ST: Discovery of GDC-0077 (Inavolisib), a Highly Selective Inhibitor and Degrader of Mutant PI3Kalpha. J Med Chem. 2022 Dec 22;65(24):16589-16621. doi: 10.1021/acs.jmedchem.2c01422. Epub 2022 Dec 1.





PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US215633239&_cid=P11-M9XU5W-08686-1


Example 101 (S)-2-((2-((S)-4-(Difluoromethyl)-2-oxooxazolidin-3-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl)amino)propanamide 101
Step 1: 4-Bromo-2-hydroxybenzaldehyde
Step 2: 5-Bromo-2-(1H-imidazol-2-yl)phenol
Step 3: 9-Bromo-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepine
Step 4: 9-Bromo-2,3-diiodo-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepine
Step 5: 9-Bromo-2-iodo-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepine
Step 6: (R)-2,2-Dimethyl-[1,3]dioxolane-4-carbaldehyde
Step 7: (R)-4-Difluoromethyl-2,2-dimethyl-[1,3]dioxolane
Step 8: (R)-3-(tert-Butyldimethylsilanyloxy)-1,1-difluoropropan-2-ol
Step 9: ((S)-2-Azido-3,3-difluoropropoxy)-tert-butyldimethylsilane
Step 10: (S)-1-(tert-Butyldimethylsilanyloxymethyl)-2,2-difluoroethylamine
Step 12: (S)-3-(9-Bromo-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-2-yl)-4-(difluoromethyl)oxazolidin-2-one
Step 13: (S)-2-((2-((S)-4-(Difluoromethyl)-2-oxooxazolidin-3-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl)amino)propanamide
Medical uses
Inavolisib is indicated in combination with palbociclib and fulvestrant for the treatment of adults with endocrine-resistant, PIK3CA-mutated, hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative, locally advanced or metastatic breast cancer, as detected by an FDA-approved test, following recurrence on or after completing adjuvant endocrine therapy.[3]
Side effects
The most common adverse reactions include decreased neutrophils, decreased hemoglobin, increased fasting glucose, decreased platelets, decreased lymphocytes, stomatitis, diarrhea, decreased calcium, fatigue, decreased potassium, increased creatinine, increased ALT, nausea, decreased sodium, decreased magnesium, rash, decreased appetite, COVID-19 infection, and headache.[3]
History
Efficacy was evaluated in INAVO120 (NCT04191499), a randomized, double-blind, placebo-controlled, multicenter trial in 325 participants with endocrine-resistant, PIK3CA-mutated HR-positive, HER2-negative locally advanced or metastatic breast cancer whose disease progressed during or within twelve months of completing adjuvant endocrine therapy and who had not received prior systemic therapy for locally advanced or metastatic disease.[3] Primary endocrine resistance was defined as relapse while on the first two years of adjuvant endocrine therapy (ET) and secondary endocrine resistance was defined as relapse while on adjuvant ET after at least two years or relapse within twelve months of completing adjuvant ET.[3]
Structure, reactivity, and synthesis
Inavolisib is a synthetic, organic, small compound (the full structure can be seen here).[8] When binding to phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha (p110α), inavolisib’s carbonyl group can accept a hydrogen bond from the Tyr836 (conserved) in p110α. The difluoromethyl group can interact with the hydroxyl group presented on Ser774 (conserved) in p110α, which is 3.2Å nearer than of which on the equivalent residue Ser754 in p110δ. Additionally, the amide group can interact with Gln859 (non-conserved). This results in a very high selectivity regarding PI3Kα isoforms.[4][9]
Compared to similar PI3K inhibiting compounds, inavolisib has a higher thermodynamic aqueous solubility that proved advantageous in the formulation process and aiding greater consistency in predictions of absorption.[4]
Inavolisibcan be developed as a derivative of 1,3-oxazole[10] or by means of stereo-controlled N-arylation of alpha-amino acids.[11]
Metabolism and biotransformation
Inavolisib is orally administered, though there is little knowledge about its metabolism.[12]However, absorption, metabolism, and excretion data of taselisib, a molecule with a related chemical scaffold, suggest moderately slow absorption into the systemic circulation, metabolism to play a minor role in drug clearance, and biliary excretion to be the main route of excretion.[13]
Molecular mechanisms of action
Inavolisib is a selective PI3K-p110α (PIK3CA) inhibitor, which may offer antineoplastic functionality.[8] Therefore, it may serve as a new addition to combination therapy with conventional cancer treatment, such as chemotherapy. Combining inavolisib with palbociclib and fulvestrant might improve treatment of breast cancer.[14]
Next to its inhibitory enzymatic ability, it is suggested that inavolisib binds to – and activates degradation of – mutated forms of p110α. Members of the PI3K family regulate cellular processes such as cell growth and proliferation, survival, remodelling, and intracellular transport of organelles.[15] PI3K also plays an essential role for the immune system.
The class I isoform PI3K alpha (PI3Kα) is often times expressed in solid tumours through gene amplification or activated mutations.[4] Mutations in PI3Kα can often be found in cancer cells, especially HR+ breast cancer, which causes a disruption of the PI3K pathway. This leads to increased tumour growth and metastasis. One of the most common mutations can be found in PIK3CA, which plays a significant role in tumour cell proliferation.
In preclinical studies, inavolisib has shown to specifically initiate the degradation of this p110α oncogene with the help of proteasomes.[16] After binding to the mutant PI3Kα, inavolisib blocks phosphorylation of PIP2 to PIP3, thereby stopping downstream signalling.[17]
Consequently, biomarkers in the PI3K pathway are reduced, cell proliferation inhibited, and the rate of PIK3CA-mutant breast cancer apoptosis increased (in comparison to the wild type). The exact mechanism of action of inhibitors like inavolisib on mutated PI3Kα and the inhibitors’ influence on mutant structures are still unknown.[18]
Toxicity
Inavolisib is able to induce a cytotoxic response but this is directed towards tumour cells that contain the PI3K mutation, thereby inhibiting further tumour growth and leading to cell loss.[19]
Society and culture
Legal status
In October 2024, the US Food and Drug Administration (FDA) approved inavolisib for the treatment of PIK3CA-mutant breast cancer based on the results from the INAVO120 trial.[3][6][20][21] The drug application was granted priority review and breakthrough therapy designations by the FDA.[3]
Names
Inavolisib is the international nonproprietary name.[22][23]
Inavolisib is sold under the brand name Itovebi.[3]
Research
Due to inavolisib’s ability to inhibit the PI3K pathway through HER2-dependent degradation, it is undergoing clinical trials to potentially make use of it as an antineoplastic (anti-cancer) drug to treat breast cancer.[4][24][17]
References
- ^ “Register of Innovative Drugs”. Health Canada. 3 November 2006. Retrieved 17 April 2025.
- ^ Jump up to:a b “Itovebi- inavolisib tablet, film coated”. DailyMed. 11 October 2024. Retrieved 11 November 2024.
- ^ Jump up to:a b c d e f g h i j “FDA approves inavolisib with palbociclib and fulvestrant for endocrine-resistant, PIK3CA-mutated, HR-positive, HER2-negative, advanced breast cancer”. U.S. Food and Drug Administration (FDA). 10 October 2024. Retrieved 11 October 2024. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d e Hanan EJ, Braun MG, Heald RA, MacLeod C, Chan C, Clausen S, et al. (December 2022). “Discovery of GDC-0077 (Inavolisib), a Highly Selective Inhibitor and Degrader of Mutant PI3Kα”. Journal of Medicinal Chemistry. 65 (24). American Chemical Society (ACS): 16589–16621. doi:10.1021/acs.jmedchem.2c01422. PMID 36455032. S2CID 254149451.
- ^ “CID 124173720, Inavolisib”. PubChem. National Center for Biotechnology Information, U.S. National Library of Medicine. Retrieved 21 September 2023.
- ^ Jump up to:a b “Novel Drug Approvals for 2024”. U.S. Food and Drug Administration (FDA). 1 October 2024. Retrieved 29 November 2024.
- ^ New Drug Therapy Approvals 2024 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2025. Archived from the original on 21 January 2025. Retrieved 21 January 2025.
- ^ Jump up to:a b “inavolisib — Ligand page”. IUPHAR/BPS Guide to Pharmacology. Retrieved 21 September 2023.
- ^ Vanhaesebroeck B, Perry MW, Brown JR, André F, Okkenhaug K (October 2021). “PI3K inhibitors are finally coming of age”. Nature Reviews. Drug Discovery. 20 (10). Springer Science and Business Media LLC: 741–769. doi:10.1038/s41573-021-00209-1. PMC 9297732. PMID 34127844.
- ^ Chen J, Lv S, Liu J, Yu Y, Wang H, Zhang H (December 2021). “An Overview of Bioactive 1,3-Oxazole-Containing Alkaloids from Marine Organisms”. Pharmaceuticals. 14 (12). MDPI AG: 1274. doi:10.3390/ph14121274. PMC 8706051. PMID 34959674.
- ^ Han C, Kelly SM, Cravillion T, Savage SJ, Nguyen T, Gosselin F (2019). “Synthesis of PI3K inhibitor GDC-0077 via a stereocontrolled N-arylation of α-amino acids”. Tetrahedron. 75 (32). Elsevier BV: 4351–4357. doi:10.1016/j.tet.2019.04.057. ISSN 0040-4020. S2CID 150262658.
- ^ “Inavolisib: Uses, Interactions, Mechanism of Action”. DrugBank. 20 May 2019. DB15275. Retrieved 21 September 2023.
- ^ Ma S, Cho S, Sahasranaman S, Zhao W, Pang J, Ding X, et al. (April 2023). “Absorption, Metabolism, and Excretion of Taselisib (GDC-0032), a Potent β-Sparing PI3K Inhibitor in Rats, Dogs, and Humans”. Drug Metabolism and Disposition. 51 (4): 436–450. doi:10.1124/dmd.122.001096. PMID 36623882.
- ^ “A trial looking at a new drug called inavolisib for breast cancer that has spread (WO41554)”. Cancer Research UK. 22 June 2021. Retrieved 21 September 2023.
- ^ Koyasu S (April 2003). “The role of PI3K in immune cells”. Nature Immunology. 4 (4). Springer Science and Business Media LLC: 313–319. doi:10.1038/ni0403-313. PMID 12660731. S2CID 9951653.
- ^ Hong R, Edgar K, Song K, Steven S, Young A, Hamilton P, et al. (15 February 2018). “Abstract PD4-14: GDC-0077 is a selective PI3Kalpha inhibitor that demonstrates robust efficacy in PIK3CA mutant breast cancer models as a single agent and in combination with standard of care therapies”. Cancer Research. 78 (4_Supplement). American Association for Cancer Research (AACR): PD4–14–PD4–14. doi:10.1158/1538-7445.sabcs17-pd4-14. ISSN 0008-5472.
- ^ Jump up to:a b “Inavolisib (PI3K alpha inhibitor)”. Genentech. Retrieved 21 September 2023.
- ^ Menteş M, Karakuzulu BB, Uçar GB, Yandım C (August 2022). “Comparative molecular dynamics analyses on PIK3CA hotspot mutations with PI3Kα specific inhibitors and ATP”. Computational Biology and Chemistry. 99. Elsevier BV: 107726. doi:10.1016/j.compbiolchem.2022.107726. PMID 35842959. S2CID 250404770.
- ^ Song KW, Edgar KA, Hanan EJ, Hafner M, Oeh J, Merchant M, et al. (January 2022). “RTK-Dependent Inducible Degradation of Mutant PI3Kα Drives GDC-0077 (Inavolisib) Efficacy”. Cancer Discovery. 12 (1). American Association for Cancer Research (AACR): 204–219. doi:10.1158/2159-8290.cd-21-0072. PMC 9762331. PMID 34544753.
- ^ “FDA Approves Genentech’s Itovebi, a Targeted Treatment for Advanced Hormone Receptor-Positive, HER2-Negative Breast Cancer With a PIK3CA Mutation” (Press release). Genentech. 10 October 2024. Retrieved 11 October 2024 – via Business Wire.
- ^ “U.S. Food and Drug Administration Approves FoundationOne Liquid CDx as a Companion Diagnostic for Itovebi (inavolisib) to Identify Patients with Hormone Receptor-Positive, HER2-Negative Breast Cancer with a PIK3CA Mutation” (Press release). Foundation Medicine. 11 October 2024. Retrieved 11 October 2024 – via Business Wire.
- ^ World Health Organization (2020). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 84”. WHO Drug Information. 34 (3). hdl:10665/340680.
- ^ World Health Organization (2023). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 90”. WHO Drug Information. 37 (3). hdl:10665/373341.
- ^ Vanhaesebroeck B, Burke JE, Madsen RR (January 2022). “Precision Targeting of Mutant PI3Kα in Cancer by Selective Degradation”. Cancer Discovery. 12 (1). American Association for Cancer Research (AACR): 20–22. doi:10.1158/2159-8290.cd-21-1411. PMC 7612218. PMID 35022207.
External links
- Clinical trial number NCT04191499 for “A Study Evaluating the Efficacy and Safety of Inavolisib + Palbociclib + Fulvestrant vs Placebo + Palbociclib + Fulvestrant in Patients With PIK3CA-Mutant, Hormone Receptor-Positive, Her2-Negative, Locally Advanced or Metastatic Breast Cancer (INAVO120)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Itovebi |
| Other names | GDC-0077, RG6114, Ro7113755 |
| AHFS/Drugs.com | Itovebi |
| License data | US DailyMed: Inavolisib |
| Routes of administration | By mouth |
| Drug class | PI3K inhibitor |
| ATC code | None |
| Legal status | |
| Legal status | CA: ℞-only[1]US: ℞-only[2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2060571-02-8 |
| PubChem CID | 124173720 |
| IUPHAR/BPS | 9636 |
| DrugBank | DB15275 |
| ChemSpider | 59718498 |
| UNII | L4C1UY2NYH |
| KEGG | D11942 |
| ChEMBL | ChEMBL4650215 |
| PDB ligand | X3N (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C18H19F2N5O4 |
| Molar mass | 407.378 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
//////////Inavolisib, FDA 2024, APPROVALS 2024, GDC-0077, 2060571-02-8, GDC0077, RG6114, WHO 11204, GDC 0077, GDC-0077, RG-6114, RG6114, RO-7113755, RO7113755
Vanzacaftor



Vanzacaftor
- CAS 2374124-49-7
- COM1POP492
- VX-121
- 617.8 g/mol, C32H39N7O4S
FDA APPROVED vanzacaftor, tezacaftor, and deutivacaftor, 12/20/2024, Alyftrek , To treat cystic fibrosis
(14S)-8-[3-(2-dispiro[2.0.24.13]heptan-7-ylethoxy)pyrazol-1-yl]-12,12-dimethyl-2,2-dioxo-2λ6-thia-3,9,11,18,23-pentazatetracyclo[17.3.1.111,14.05,10]tetracosa-1(22),5(10),6,8,19(23),20-hexaen-4-one
Vanzacaftor (VX-121) is an orally active noval corrector of Cystic fibrosis transmembrane conductance regulator (CFTR). Vanzacaftor improves processing and trafficking of CFTR protein as well as increases chloride transport in triple combined with Tezacaftor (HY-15448) and Deutivacaftor. Vanzacaftor-Tezacaftor-Deutivacaftor is safe and well tolerated, improving lung function, respiratory symptoms, and CFTR function with cystic fibrosis, which is promising for research in the field of cystic fibrosis diseases.
| Cystic fibrosis (CF) is a recessive genetic disease that affects approximately 70,000 children and adults worldwide. Despite progress in the treatment of CF, there is no cure. |
PATENTS
https://patentscope.wipo.int/search/en/detail.jsf?docId=US356967369&_cid=P12-M9W6P5-06241-1
Example 104: Preparation of (14S)-8-[3-(2-{dispiro[2.0.2.1]heptan-7-yl}ethoxy)-1H-pyrazol-1-yl]-12,12-dimethyl(20-deuterio)-2λ6-thia-3,9,11,18,23-pentaazatetracyclo[17.3.1.111,14.05,10]tetracosa-1(23),5,7,9,19,21-hexaene-2,2,4-trione (Compound 300)

Step 1: (14S)-8-[3-(2-{Dispiro[2.0.2.1]heptan-7-yl}ethoxy)-1H-pyrazol-1-yl]-12,12-dimethyl-2,2,4-trioxo-2λ6-thia-3,9,11,18,23-pentaazatetracyclo[17.3.1.111,14.05,10]tetracosa-1(23),5,7,9,19,21-hexaen-20-yl 4-methylbenzene-1-sulfonate

To a stirred solution of (14S)-8-[3-(2-{dispiro[2.0.2.1]heptan-7-yl}ethoxy)-1H-pyrazol-1-yl]-20-hydroxy-12,12-dimethyl-2λ 6-thia-3,9,11,18,23-pentaazatetracyclo[17.3.1.111,14.05,10]tetracosa-1 (23),5,7,9,19,21-hexaene-2,2,4-trione (150 mg, 0.2367 mmol) in anhydrous dichloromethane (3.000 mL) was added 4-methylbenzenesulfonyl chloride (58 mg, 0.3042 mmol), triethylamine (80 μL, 0.5740 mmol) and catalytic amount of N,N-dimethylpyridin-4-amine (10 mg, 0.08185 mmol). The reaction mixture was stirred at room temperature overnight. The reaction mixture was quenched with saturated aqueous ammonium chloride solution and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated. The resultant brown residue was purified by silica gel column chromatography using a shallow gradient 100% hexanes to 100% ethyl acetate to afford (14S)-8-[3-(2-{dispiro[2.0.2.1]heptan-7-yl}ethoxy)-1H-pyrazol-1-yl]-12,12-dimethyl-2,2,4-trioxo-2λ 6-thia-3,9,11,18,23-pentaazatetracyclo[17.3.1.111,14.05,10]tetracosa-1(23),5,7,9,19,21-hexaen-20-yl 4-methylbenzene-1-sulfonate (120 mg, 51%) as a white solid. ESI-MS m/z calc. 787.28217, found 788.42 (M+1) +; Retention time: 1.39 min (LC Method J).
Step 2: (14S)-8-[3-(2-{Dispiro[2.0.2.1]heptan-7-yl}ethoxy)-1H-pyrazol-1-yl]-12,12-dimethyl(20-deuterio)-2λ6-thia-3,9,11,18,23-pentaazatetracyclo[17.3.1.111,14.05,10]tetracosa-1(23),5,7,9,19,21-hexaene-2,2,4-trione (Compound 300)

A solution of (14S)-8-[3-(2-{dispiro[2.0.2.1]heptan-7-yl}ethoxy)-1H-pyrazol-1-yl]-12,12-dimethyl-2,2,4-trioxo-2λ 6-thia-3,9,11,18,23-pentaazatetracyclo[17.3.1.111,14.05,10]tetracosa-1(23),5,7,9,19,21-hexaen-20-yl 4-methylbenzene-1-sulfonate (120 mg, 0.1523 mmol) in dry N,N-dimethylformamide (1 mL) was purged with nitrogen for 5 min using a balloon. Then, dichloronickel; triphenyl-phosphane (30 mg, 0.04586 mmol) and tricyclohexylphosphane (34 mg, 0.1212 mmol) were added. The resultant green solution was stirred for 5 min under nitrogen atmosphere and tetradeuterioboranuide (sodium salt) (87 mg, 2.079 mmol) was added in one portion. The resultant dark reddish brown mixture was stirred at room temperature for 1 h. Additional dichloronickel; triphenylphosphane (30 mg, 0.04586 mmol), tricyclohexylphosphane (34 mg, 0.1212 mmol) and tetradeuterioboranuide (sodium salt) (87 mg, 2.079 mmol) were added and the mixture was stirred at room temperature under nitrogen overnight. The reaction mixture was diluted with water and extracted with ethyl acetate. The organic layer was dried over magnesium sulfate, filtered and evaporated. The resultant residue was dissolved in dimethyl sulfoxide and filtered through a Whatman filter disc (puradisc 25 TF) and the filtrate was purified by reverse phase HPLC-MS using a dual gradient run from 50%-99% mobile phase B over 15.0 min (mobile phase A=water (5 mM hydrochloric acid), mobile phase B=acetonitrile) to afford (14S)-8-[3-(2-{dispiro[2.0.2.1]heptan-7-yl}ethoxy)-1H-pyrazol-1-yl]-12,12-dimethyl(20-deuterio)-2λ 6-thia-3,9,11,18,23-pentaazatetracyclo[17.3.1.111,14.05,10]tetracosa-1(23),5,7,9,19,21-hexaene-2,2,4-trione (Compound 300) (35 mg, 37%) as a white solid. 1H NMR (400 MHz, dimethyl sulfoxide-d 6) δ 12.52 (s, 1H), 8.20 (d, J=2.8 Hz, 1H), 7.81 (d, J=8.2 Hz, 1H), 7.56 (d, J=7.1 Hz, 1H), 7.04 (d, J=7.2 Hz, 1H), 6.98 (s, 1H), 6.90 (d, J=8.1 Hz, 1H), 6.08 (d, J=2.7 Hz, 1H), 4.25-4.17 (m, 2H), 3.92 (d, J=12.5 Hz, 1H), 3.17 (s, 1H), 2.94 (d, J=13.2 Hz, 1H), 2.72 (s, 1H), 2.20-2.06 (m, 1H), 1.81 (q, J=6.6 Hz, 4H), 1.60 (s, 3H), 1.56 (d, J=13.5 Hz, 2H), 1.51 (s, 3H), 1.46 (d, J=6.5 Hz, 1H), 1.36-1.26 (m, 1H), 1.23 (s, 1H), 0.87-0.76 (m, 4H), 0.70-0.59 (m, 2H), 0.50 (dd, J=8.0, 4.3 Hz, 2H). ESI-MS m/z calc. 618.2847, found 619.25 (M+1) +; Retention time: 1.28 min (LC Method J).
- US11866450, Compound 195
- US11866450, Compound 252
- US11866450, Compound 253
- US11866450, Compound 274
- US11866450, Compound 298
- US11866450, Compound 301
- [1]. Uluer AZ, et al. Safety and efficacy of vanzacaftor-tezacaftor-deutivacaftor in adults with cystic fibrosis: randomised, double-blind, controlled, phase 2 trials[J]. Lancet Respir Med. 2023 Jun;11(6):550-562. [Content Brief][2]. Kolski-Andreaco A, et al. Potentiation of BKCa channels by cystic fibrosis transmembrane conductance regulator (CFTR) correctors VX-445 and VX-121[J]. J Clin Invest. 2024 Jul 2:e176328. [Content Brief]
//////Vanzacaftor, Alyftrek , cystic fibrosis, COM1POP492, VX-121, FDA 2024, APPROVALS 2024
#Vanzacaftor, #Alyftrek , #cystic fibrosis, #COM1POP492, #VX-121, #FDA 2024, #APPROVALS 2024
Probenecid



Probenecid
- 57-66-9
- 4-(Dipropylsulfamoyl)benzoic acid
- Probenecid acid
- Benemid
4-(dipropylsulfamoyl)benzoic acid
C13H19NO4S, 285.359
HC 5006- NSC-18786
FDA APPROVED, 10/25/2024, sulopenem etzadroxil, probenecid, Orlynvah, To treat uncomplicated urinary tract infections (uUTI)
Drug Trial Snapshot
Probenecid, also sold under the brand name Probalan, is a medication that increases uric acid excretion in the urine. It is primarily used in treating gout and hyperuricemia.
Probenecid was developed as an alternative to caronamide[1] to competitively inhibit renal excretion of some drugs, thereby increasing their plasma concentration and prolonging their effects.
Experimental Properties
| Property | Value | Source |
|---|---|---|
| melting point (°C) | 195 °C | PhysProp |
| water solubility | 27.1 mg/L | Not Available |
| logP | 3.21 | HANSCH,C ET AL. (1995) |
| pKa | 3.4 | SANGSTER (1994) |
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US12109197 | No | 2024-10-08 | 2039-04-01 | |
| US11554112 | No | 2023-01-17 | 2039-04-01 | |
| US11478428 | No | 2022-10-25 | 2039-12-23 | |
| US7795243 | No | 2010-09-14 | 2029-06-03 | |

PATENT
https://patents.google.com/patent/CN103613521A/en
At present, the production technique of probenecid mainly contains two kinds:
(1) p-methyl benzenesulfonic acid-dipropyl amine method
Take p-methyl benzenesulfonic acid as raw material, through potassium bichromate or potassium permanganate oxidation, then react generation with chlorsulfonic acid generation sulfonating chlorinating to carboxyl benzene sulfonyl chloride, amidate action occurs then in organic solvent and obtain the finished product probenecid.Reaction process route is as follows:

This technique in a large number with an organic solvent, seriously polluted; Heavy metal recovery and treatment cost are high; Chlorsulfonic acid transportation, storage and use are dangerous large, and acid mist is obvious.Along with the increasing of environmental protection pressure, people increase severely day by day to the concern of environment, and this route is substantially in end-of-life state.
(2) to methyl benzenesulfonamide-Halopropane method
To methyl benzenesulfonamide, through potassium bichromate or potassium permanganate oxidation, be P―Carboxybenzenesulfonamide, under the effect of alkali, with Halopropane generation alkylated reaction, after acidifying, obtain probenecid.Reaction process route is as follows:

This process using sodium dichromate 99 or potassium permanganate oxidation are to methyl benzenesulfonamide, and yield is on the low side (lower than 50%).In addition, the waste water that contains chromium or manganese is difficult to dispose, and these have all seriously restricted further developing of this technique.
Reaction scheme of the present invention is as follows:

embodiment 1
(1) diazotization reaction
Get 68.6g para-amino benzoic acid (0.5mol), 250g water and 127.4ml hydrochloric acid (31%, 1.25mol) join in 2000ml there-necked flask, in ice-water bath, stir, be cooled to 0-5 ℃, drip sodium nitrite solution (34.5g Sodium Nitrite, 0.5mol, be dissolved in 190g water), control temperature at 10-20 ℃, it is 4 hours that time for adding is controlled, after dropping finishes, at this temperature, continue reaction 1 hour, obtain diazotization reaction liquid.
(2) sulfonating chlorinating reaction
In 5000ml there-necked flask, add 250g water, 765ml hydrochloric acid (31%, 7.5mol), in ice-water bath, stir, be cooled to-5 ℃, start to pass into liquid sulfur dioxide, control temperature at-3–1 ℃, when passing into 64g sulfurous gas (1mol), sulfurous gas absorbs complete, obtains sulfonating chlorinating reagent.
In sulfonating chlorinating reagent, add diazotization reaction liquid, adding the time control of diazotization reaction liquid is 5 hours, is warming up to gradually 5-10 ℃, continues reaction 8 hours at this temperature; Filtration obtains 121g to carboxyl benzene sulfonyl chloride.
(3) synthetic probenecid reaction
In 1000ml there-necked flask, add 350g water, 152g dipropyl amine (1.5mol), open and stir, when temperature is greater than 15 ℃, start to divide gradually 40 batches add step (2) gained to carboxyl benzene sulfonyl chloride, temperature control 40-50 ℃, adds and at this temperature, stirs 3 hours continuing after carboxyl benzene sulfonyl chloride.Drip hydrochloric acid (31%), regulate pH value to 2-3, continue to stir 1 hour.Filter, obtain 135g probenecid crude product, put in 500ml pure water, agitator treating 1 hour, heavy 122.8g after filtering, being dried, yield 86.2%(is in para-amino benzoic acid), purity 98.2%.
embodiment 2
(1) diazotization reaction
Get 68.6g para-amino benzoic acid (0.5mol), 250g water and 152.9ml hydrochloric acid (31%, 1.5mol) join in 2000ml there-necked flask, in ice-water bath, stir, be cooled to 0-5 ℃, drip sodium nitrite solution (36.0g Sodium Nitrite, 0.52mol, be dissolved in 190g water), control temperature at 0-10 ℃, it is 3 hours that time for adding is controlled, after dropping finishes, at this temperature, continue reaction 1 hour, obtain diazotization reaction liquid.
(2) sulfonating chlorinating reaction
In 5000ml there-necked flask, add 250g water, 887ml hydrochloric acid (31%, 8.7mol), in ice-water bath, stir, be cooled to-5 ℃, start to pass into liquid sulfur dioxide, control temperature at 0-5 ℃, when passing into 112g sulfurous gas (1.75mol), sulfurous gas absorbs complete, obtains sulfonating chlorinating reagent.
In sulfonating chlorinating reagent, add diazotization reaction liquid, adding the time control of diazotization reaction liquid is 4 hours, is warming up to gradually 5-15 ℃, continues reaction 5 hours at this temperature; Filtration obtains 150g to carboxyl benzene sulfonyl chloride.
(3) synthetic probenecid reaction
In 1000ml there-necked flask, add 350g water, 192g dipropyl amine (1.9mol), open and stir, when temperature is greater than 15 ℃, start to divide gradually 35 batches add step (2) gained to carboxyl benzene sulfonyl chloride, temperature control 40-50 ℃, adds and at this temperature, stirs 2 hours continuing after carboxyl benzene sulfonyl chloride.Drip hydrochloric acid (31%), regulate pH value to 2-3, continue to stir 1 hour.Filter, obtain 155.4g probenecid crude product, put in 500ml pure water, agitator treating 1 hour, heavy 129.5g after filtering, being dried, yield 90.9%(is in para-amino benzoic acid), purity 98.7%.
embodiment 3
(1) diazotization reaction
Get 68.6g para-amino benzoic acid (0.5mol), 250g water and 203.9ml hydrochloric acid (31%, 2mol) join in 2000ml there-necked flask, in ice-water bath, stir, be cooled to-10–5 ℃, drip sodium nitrite solution (38.0g Sodium Nitrite, 0.55mol, be dissolved in 190g water), control temperature at 0-10 ℃, it is 5 hours that time for adding is controlled, after dropping finishes, at this temperature, continue reaction 1 hour, obtain diazotization reaction liquid.
(2) sulfonating chlorinating reaction
In 5000ml there-necked flask, add 250g water, 968ml hydrochloric acid (31%, 9.5mol), in ice-water bath, stir, be cooled to-5 ℃, start to pass into liquid sulfur dioxide, control temperature at 5-10 ℃, when passing into 160g sulfurous gas (2.5mol), sulfurous gas absorbs complete, obtains sulfonating chlorinating reagent.
In sulfonating chlorinating reagent, add diazotization reaction liquid, adding the time control of diazotization reaction liquid is 3 hours, is warming up to gradually 10-15 ℃, continues reaction 20 hours at this temperature; Filtration obtains 146.7g to carboxyl benzene sulfonyl chloride, needn’t be dried, and directly enters next step reaction.
(3) synthetic probenecid reaction
In 1000ml there-necked flask, add 350g water, 202g dipropyl amine (2mol), open to stir, when temperature is greater than 30 ℃, start to divide gradually 30 batches add step (2) gained to carboxyl benzene sulfonyl chloride, temperature control 40-50 ℃, adds and at this temperature, stirs 4 hours continuing after carboxyl benzene sulfonyl chloride.Drip hydrochloric acid (31%), regulate pH value to 2-3, continue to stir 1 hour.Filtration obtains 151.7g probenecid crude product, puts in 500ml pure water, and agitator treating 1 hour, heavy 128.5g after filtering, being dried, yield 90.2%(is in para-amino benzoic acid), purity 98.8%.Medical uses
Probenecid is primarily used to treat gout and hyperuricemia.
Probenecid is sometimes used to increase the concentration of some antibiotics and to protect the kidneys when given with cidofovir. Specifically, a small amount of evidence supports the use of intravenous cefazolin once rather than three times a day when it is combined with probenecid.[2]
It has also found use as a masking agent,[3] potentially helping athletes using performance-enhancing substances to avoid detection by drug tests.
Adverse effects
Mild symptoms such as nausea, loss of appetite, dizziness, vomiting, headache, sore gums, or frequent urination are common with this medication. Life-threatening side effects such as thrombocytopenia, hemolytic anemia, leukemia and encephalopathy are extremely rare.[4] Theoretically probenecid can increase the risk of uric acid kidney stones.
Drug interactions
Some of the important clinical interactions of probenecid include those with captopril, indomethacin, ketoprofen, ketorolac, naproxen, cephalosporins, quinolones, penicillins, methotrexate, zidovudine, ganciclovir, lorazepam, and acyclovir. In all these interactions, the excretion of these drugs is reduced due to probenecid, which in turn can lead to increased concentrations of these.[5]
Pharmacology
Pharmacodynamics
In gout, probenecid competitively inhibits the reabsorption of uric acid through the organic anion transporter (OAT) at the proximal tubules. This leads to preferential reabsorption of probenecid back into plasma and excretion of uric acid in urine,[6] thus reducing blood uric acid levels and reducing its deposition in various tissues.
Probenecid also inhibits pannexin 1.[7] Pannexin 1 is involved in the activation of inflammasomes and subsequent release of interleukin-1β causing inflammation. Inhibition of pannexin 1 thus reduces inflammation, which is the core pathology of gout.[7]
Pharmacokinetics
In the kidneys, probenecid is filtered at the glomerulus, secreted in the proximal tubule and reabsorbed in the distal tubule. Probenicid lowers the concentration of certain drugs in urine drug screens by reducing renal excretion of these drugs.
Historically, probenecid has been used to increase the duration of action of drugs such as penicillin and other beta-lactam antibiotics. Penicillins are excreted in the urine at proximal and distal convoluted tubules through the same organic anion transporter (OAT) as seen in gout. Probenecid competes with penicillin for excretion at the OAT, which in turn increases the plasma concentration of penicillin.[8]
History
During World War II, probenecid was used to extend limited supplies of penicillin. This use exploited probenecid’s interference with drug elimination (via urinary excretion) in the kidneys and allowed lower doses of penicillin to be used.[9]
Probenecid was added to the International Olympic Committee‘s list of banned substances in January 1988, due to its use as a masking agent.[10]
References
- ^ Mason RM (June 1954). “Studies on the effect of probenecid (benemid) in gout”. Annals of the Rheumatic Diseases. 13 (2): 120–130. doi:10.1136/ard.13.2.120. PMC 1030399. PMID 13171805.
- ^ Cox VC, Zed PJ (March 2004). “Once-daily cefazolin and probenecid for skin and soft tissue infections”. The Annals of Pharmacotherapy. 38 (3): 458–463. doi:10.1345/aph.1d251. PMID 14970368. S2CID 11449580.
- ^ Morra V, Davit P, Capra P, Vincenti M, Di Stilo A, Botrè F (December 2006). “Fast gas chromatographic/mass spectrometric determination of diuretics and masking agents in human urine: Development and validation of a productive screening protocol for antidoping analysis”. Journal of Chromatography A. 1135 (2): 219–229. doi:10.1016/j.chroma.2006.09.034. hdl:2318/40201. PMID 17027009. S2CID 20282106.
- ^ Kydd AS, Seth R, Buchbinder R, Edwards CJ, Bombardier C (November 2014). “Uricosuric medications for chronic gout”. The Cochrane Database of Systematic Reviews (11): CD010457. doi:10.1002/14651858.CD010457.pub2. PMC 11262558. PMID 25392987.
- ^ Cunningham RF, Israili ZH, Dayton PG (March–April 1981). “Clinical pharmacokinetics of probenecid”. Clinical Pharmacokinetics. 6 (2): 135–151. doi:10.2165/00003088-198106020-00004. PMID 7011657. S2CID 24497865.
- ^ “Probenecid”. PubChem. U.S. National Library of Medicine. Retrieved 2022-06-12.
- ^ Jump up to:a b Silverman W, Locovei S, Dahl G (September 2008). “Probenecid, a gout remedy, inhibits pannexin 1 channels”. American Journal of Physiology. Cell Physiology. 295 (3): C761 – C767. doi:10.1152/ajpcell.00227.2008. PMC 2544448. PMID 18596212.
- ^ Ho RH (January 2010). “4.25 – Uptake Transporters”. In McQueen CA, Kim RB (eds.). Comprehensive Toxicology (Second ed.). Oxford: Elsevier. pp. 519–556. doi:10.1016/B978-0-08-046884-6.00425-5. ISBN 978-0-08-046884-6.
- ^ Butler D (November 2005). “Wartime tactic doubles power of scarce bird-flu drug”. Nature. 438 (7064): 6. Bibcode:2005Natur.438….6B. doi:10.1038/438006a. PMID 16267514.
- ^ Wilson W, Derse E, eds. (2001). Doping in Elite Sport: The Politics of Drugs in the Olympic Movement. Human Kinetics. p. 86. ISBN 0-7360-0329-0.
| Clinical data | |
|---|---|
| Trade names | Probalan |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a682395 |
| Routes of administration | By mouth |
| ATC code | M04AB01 (WHO) |
| Legal status | |
| Legal status | In general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Protein binding | 75-95% |
| Elimination half-life | 2-6 hours (dose: 0.5-1 g) |
| Excretion | kidney (77-88%) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 57-66-9 |
| PubChem CID | 4911 |
| IUPHAR/BPS | 4357 |
| DrugBank | DB01032 |
| ChemSpider | 4742 |
| UNII | PO572Z7917 |
| KEGG | D00475 |
| ChEMBL | ChEMBL897 |
| CompTox Dashboard (EPA) | DTXSID9021188 |
| ECHA InfoCard | 100.000.313 |
| Chemical and physical data | |
| Formula | C13H19NO4S |
| Molar mass | 285.36 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
/////////probenecid, APPROVALS 2024, FDA 2024, Orlynvah, HC 5006, NSC-18786
#probenecid, #APPROVALS 2024, #FDA 2024, #Orlynvah, #HC 5006, #NSC-18786

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}