
Home » Posts tagged 'Antineoplastic' (Page 5)
Tag Archives: Antineoplastic
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Lunbotinib
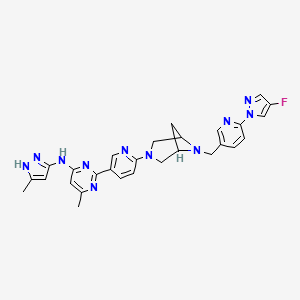
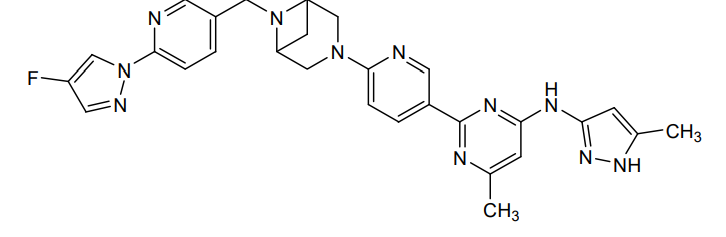
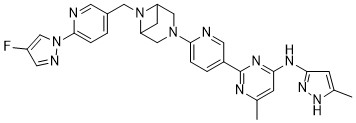
Lunbotinib
CAS 2479961-46-9
MF C28H28FN11 MW537.6 g/mol
2-[6-(6-{[6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl]methyl}-3,6-diazabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl]-6-methyl-N-(5-methyl1H-pyrazol-3-yl)pyrimidin-4-amine
tyrosine kinase inhibitor, antineoplastic, KL3T9ZU6HQ
- 2-(6-(6-((6-(4-fluoropyrazol-1-yl)pyridin-3-yl)methyl)-3,6-diazabicyclo(3.1.1)heptan-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine
- 2-[6-[6-[[6-(4-fluoropyrazol-1-yl)pyridin-3-yl]methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine
Lunbotinib is an orally bioavailable selective inhibitor of the proto-oncogene receptor tyrosine kinase rearranged during transfection (RET), with potential antineoplastic activity. Upon oral administration, lunbotinib selectively binds to various RET fusions and mutations, including solvent front resistance mutations, and inhibits the activity of RET. This results in an inhibition of cell growth of tumors that exhibit increased RET activity due to these fusions and mutations. RET overexpression, activating mutations, and fusions result in the upregulation and/or overactivation of RET tyrosine kinase activity in various cancer cell types. Dysregulated RET activity plays a key role in the development and progression of certain cancers. Lunbotinib is able to penetrate the blood-brain barrier (BBB) and may also be able to overcome resistance mechanisms to first generation selective RET inhibitors (SRIs).
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020168939&_cid=P12-MHKH7H-14851-1
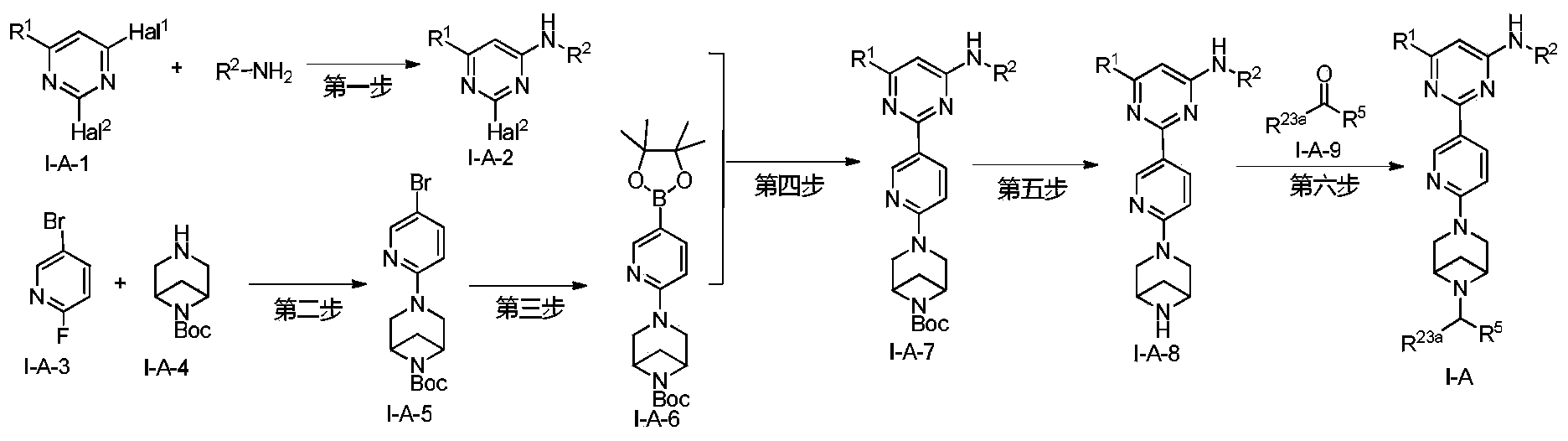
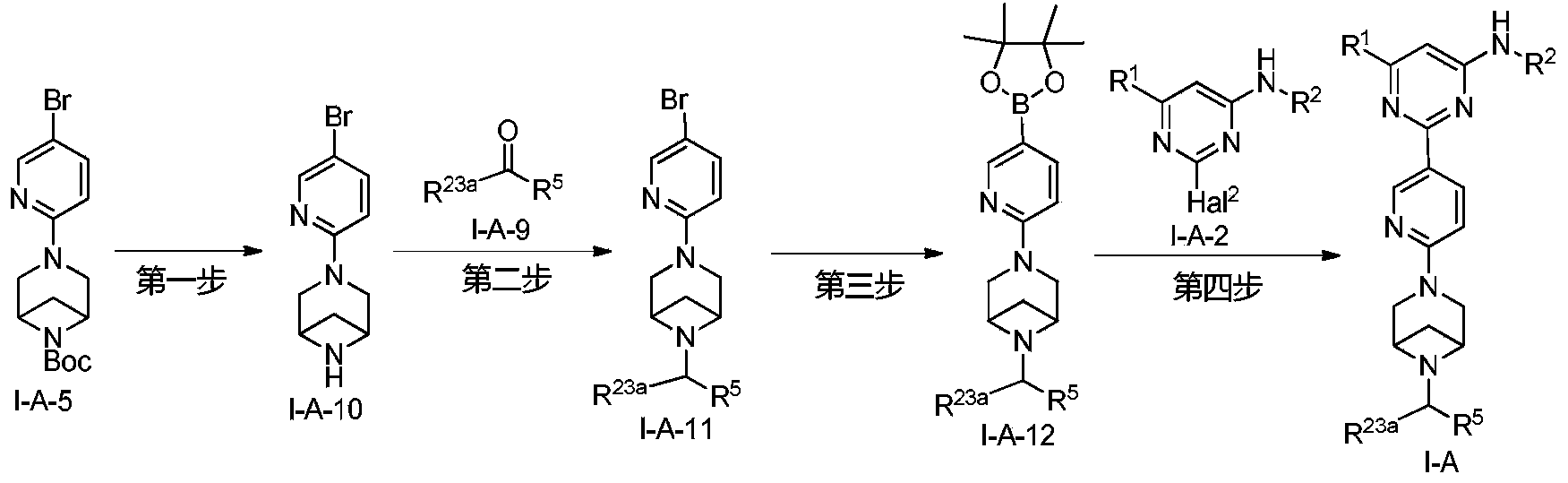
Example 6: 2-(6-(6-((6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)methyl)-3,6-diazabicyclo[3.1.1]heptane-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (Compound 17)
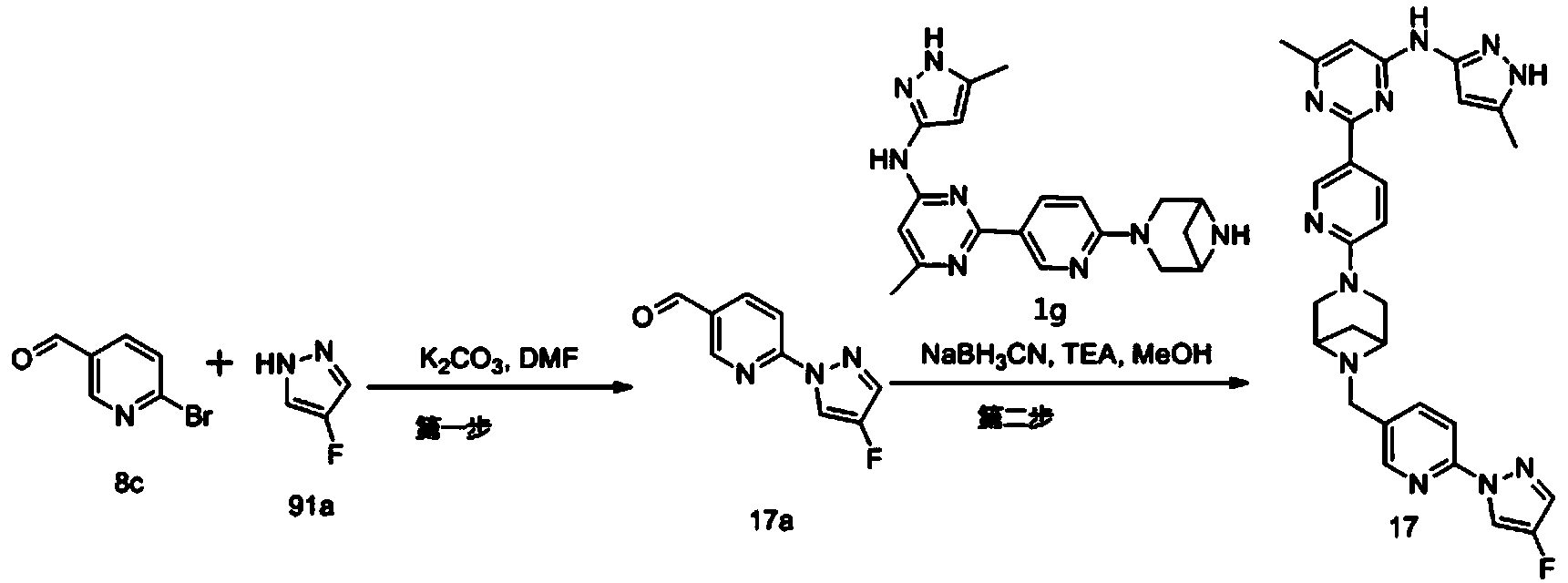
Step 1: Preparation of 6-(4-fluoro-1H-pyrazol-1-yl)nicotinaldehyde (compound 17a)
[0396]Compound 8c (2.0 g), 91a hydrochloride (1.58 g), and potassium carbonate (4.45 g) were sequentially added to DMF (15 mL), and the mixture was heated to 80 °C and stirred for 14 h. The reaction mixture was cooled to room temperature, diluted with water (100 mL), and extracted with DCM (50 mL x 2). The organic phases were combined, washed with water and saturated brine, dried over anhydrous sodium sulfate, filtered, concentrated under reduced pressure, and purified by silica gel column chromatography (PE:EA = 10:1) to give compound 17a (0.81 g). MS m/z (ESI): 192.1 [M+H]
[0397]Step 2: Preparation of 2-(6-(6-((6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)methyl)-3,6-diazabicyclo[3.1.1]heptane-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (compound 17)
[0398]1 g of trifluoroacetate (22.82 mg) and compound 17a (27.47 mg) were added to methanol (1.0 mL), followed by the sequential addition of triethylamine (4.45 mg) and sodium cyanoborohydride (13.86 mg), and the reaction was carried out at room temperature for 14 h. After the reaction was completed, the reaction solution was concentrated to dryness under reduced pressure and purified by Prep-HPLC to obtain compound 17 (7.0 mg). MS m/z (ESI): 538.3 [M+H]
[0399]
1H NMR(400MHz,DMSO-d 6)δ11.98(s,1H),9.66(s,1H),9.12(d,J=2.16Hz,1H),8.67(dd,J=4.54,0.64Hz,1H),8.43(dd,J=8.94,2.28Hz,1H),8.41(d,J=1.68,1H),7.98(dd,J=8.48Hz,2.12 1H),7.92(d,J=4.28,1H),7.87(d,J=8.4,1H),6.78(d,J=9.0Hz,2H),6.31(br,1H),3.78-3.71(m,4H),3.68-3.52(m,4H),2.59-2.52(m,1H),2.33(s,3H),2.25(s,3H),1.60(d,J=8.36Hz,1H).
PAT
- Heterocyclic compound, pharmaceutical composition comprising same, preparation method therefor, and use thereofPublication Number: US-2022144847-A1Priority Date: 2019-02-19
- Heterocyclic compounds, pharmaceutical compositions containing the same and preparation methods and uses thereofPublication Number: CN-113316578-BPriority Date: 2019-02-19Grant Date: 2023-10-31
- Heterocyclic compounds, pharmaceutical compositions containing the same and preparation methods and uses thereofPublication Number: CN-117263945-APriority Date: 2019-02-19
- Heterocyclic compounds, pharmaceutical compositions containing the same and preparation methods and uses thereofPublication Number: CN-117327078-APriority Date: 2019-02-19
- Heterocyclic compounds, pharmaceutical compositions containing same, methods for their preparation and usePublication Number: JP-7615056-B2Priority Date: 2019-02-19Grant Date: 2025-01-16
- Salt and crystal form of pyrimidine compound, and preparation methods thereforPublication Number: US-2023295174-A1Priority Date: 2020-07-28
- Heterocyclic compound, pharmaceutical composition comprising same, preparation method therefor, and use thereofPublication Number: WO-2020168939-A1Priority Date: 2019-02-19
- Heterocyclic compounds, pharmaceutical compositions containing the same, and preparation methods and uses thereofPublication Number: CN-113316578-APriority Date: 2019-02-19
- Heterocyclic compound, pharmaceutical composition comprising same, preparation method therefor, and use thereofPublication Number: EP-3929198-A1Priority Date: 2019-02-19
- Heterocyclic compounds, drug compositions containing them, methods of their manufacture and usePublication Number: JP-2022521859-APriority Date: 2019-02-19
- Use of heterocyclic compound for treating diseases related to ret genetic change and method thereforPublication Number: WO-2024240017-A1Priority Date: 2023-05-19
- Uses and methods of heterocyclic compounds for treating diseases associated with kinase resistance mutationsPublication Number: CN-116801882-APriority Date: 2021-03-24
- Use of heterocyclic compound in treating diseases related to kinase drug-resistant mutation and method thereforPublication Number: EP-4316490-A1Priority Date: 2021-03-24
- Salt and crystal form of pyrimidine compound, and preparation methods thereforPublication Number: EP-4190781-A1Priority Date: 2020-07-28
- Salts, crystal forms of pyrimidine compounds and methods for their preparationPublication Number: JP-2023535361-APriority Date: 2020-07-28



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
//////////Lunbotinib, tyrosine kinase inhibitor, antineoplastic, KL3T9ZU6HQ
Lirodegimod
Lirodegimod
CAS 2502186-79-8
MF C60H74ClN10O14PS, MW 1257.79

[2-[[(5S,8S,10aR)-3-acetyl-8-[[(2S)-5-amino-1-[2-chloro-3-[4-[[(2S)-1-[(2S,4R)-4-hydroxy-2-[[(1S)-1-[4-(4-methyl-1,3-thiazol-5-yl)phenyl]ethyl]carbamoyl]pyrrolidin-1-yl]-3,3-dimethyl-1-oxobutan-2-yl]amino]-4-oxobutyl]phenoxy]-5-oxopentan-2-yl]carbamoyl]-6-oxo-1,2,4,5,8,9,10,10a-octahydropyrrolo[1,2-a][1,5]diazocin-5-yl]carbamoyl]-1H-indole-5-carbonyl]phosphonic acid
KT 333, KT333, ANTINEOPLASTIC, Fast Track (United States), Orphan Drug (United States), 4Q6ZHJ2MNA
Lirodegimod is a small molecule drug. The usage of the INN stem ‘-imod’ in the name indicates that Lirodegimod is a immunomodulator, both stimulant/suppressive and stimulant. Lirodegimod has a monoisotopic molecular weight of 1256.45 Da.
Safety, PK, PD, Clinical Activity of KT-333 in Adult Patients With Refractory Lymphoma, Large Granular Lymphocytic Leukemia, Solid Tumors
CTID: NCT05225584
Phase: Phase 1
Status: Completed
Date: 2025-03-19
PAT
- Stat3 degraders and uses thereofPublication Number: US-2023212201-A1Priority Date: 2021-12-11
- Stat3 degraders and uses thereofPublication Number: US-2025019388-A1Priority Date: 2021-12-11
- Stat degraders and uses thereofPublication Number: US-2024016942-A1Priority Date: 2020-03-17
- Stat degraders and uses thereofPublication Number: WO-2020206424-A1Priority Date: 2019-04-05
- Stat degraders and uses thereofPublication Number: US-11746120-B2Priority Date: 2019-04-05Grant Date: 2023-09-05
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////Lirodegimod, KT 333, KT333, ANTINEOPLASTIC, Fast Track, Orphan Drug, 4Q6ZHJ2MNA
Iodofalan (131I)
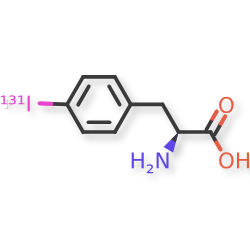
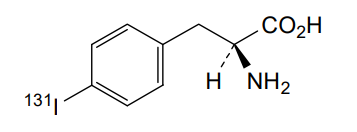
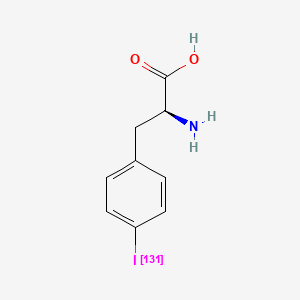
Iodofalan (131I)
CAS 76641-05-9
MFC9H10131INO2
Molecular FormulaC9H10INO2
Molecular Weight295.09
4-(131I)iodo-L-phenylalanine
(2S)-2-amino-3-(4-iodophenyl)propanoic acid
radiopharmaceutical, antineoplastic, Phase 2, Glioblastoma, 606VTF676Y, 131I-TLX-101, ACD 101
- 4-Iodophenylalanine I-131
- 4-(131I)Iodo-L-phenylalanine
- 4-Iodo-L-phenylalanine-131I
- ACD-101
- L-Phenylalanine, 4-(iodo-131I)-
- OriginatorTherapeia
- DeveloperTelix Pharmaceuticals; Therapeia
- ClassAmino acids; Antineoplastics; Radioisotopes; Radiopharmaceutical diagnostics; Radiopharmaceuticals; Small molecules
- Mechanism of ActionApoptosis stimulants; Positron-emission tomography enhancers
- Orphan Drug StatusYes – Glioblastoma
- Phase IIGlioblastoma
- 14 Oct 2025Telix Pharmaceuticals receives IND approval for TLX 101 in Glioblastoma
- 27 Jul 2025Telix Pharmaceuticals plans a phase III IPAX BrIGHT trial for Glioblastoma (Monotherapy, Combination therapy, Recurrent, Second-line therapy or greater) in Australia(IV) (NCT07100730)(EudraCT2025-521785-10) in September 2025
- 16 Apr 2025Telix has submitted for ethics approval a registration-enabling study of TLX101 in recurrent glioblastoma.
Iodofalan (131I) is a radiopharmaceutical that has garnered significant attention in oncological research due to its targeted therapeutic potential. This compound, which includes the radioactive isotope Iodine-131, has been explored for its efficacy in treating certain types of cancers, particularly those associated with the thyroid. Various research institutions worldwide have been studying Iodofalan (131I) to better understand its clinical benefits, optimize its usage, and minimize potential side effects. As a drug type, Iodofalan (131I) is categorized as a targeted radiopharmaceutical therapy, which leverages the properties of radioactive isotopes to destroy cancer cells with precision. Currently, its primary indications include differentiated thyroid cancer and non-resectable metastatic thyroid cancer, among other investigational uses.
Iodofalan (131I) Mechanism of Action
The mechanism of action for Iodofalan (131I) centers on the properties of Iodine-131, a beta-emitting isotope. When administered, Iodofalan (131I) is selectively absorbed by thyroid cells. This selectivity is due to the thyroid gland’s natural ability to uptake iodine, a key element required for the production of thyroid hormones. Cancerous thyroid tissues retain this ability, making them ideal targets for Iodofalan (131I) therapy.
Once absorbed by the thyroid cancer cells, the radioactive decay of Iodine-131 begins. This decay process emits beta particles, which possess sufficient energy to destroy nearby cells. The radiation from these beta particles causes direct DNA damage, leading to cell death. Additionally, the gamma radiation emitted by Iodine-131 can be used diagnostically to track the distribution and uptake of the compound in the body via imaging techniques such as SPECT (Single Photon Emission Computed Tomography).
The dual role of Iodofalan (131I) in both treatment and diagnostic contexts underscores its importance in managing thyroid cancers. By delivering a localized radiation dose to thyroid cancer cells, Iodofalan (131I) minimizes damage to surrounding healthy tissues, which is a significant advantage over traditional external beam radiotherapy.
What is the indication of Iodofalan (131I)?
The primary indication for Iodofalan (131I) is the treatment of differentiated thyroid cancer, a category that includes papillary and follicular thyroid cancers. These subtypes are characterized by their ability to absorb iodine, making them particularly amenable to radioiodine therapy. Iodofalan (131I) is typically used in cases where the thyroid cancer is not amenable to surgical removal or has metastasized to other parts of the body. In such scenarios, the radiopharmaceutical offers a non-invasive therapeutic option that can target and destroy cancer cells even in distant metastatic sites.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US42129729&_cid=P21-MHE8B5-15309-1
EXAMPLE 1
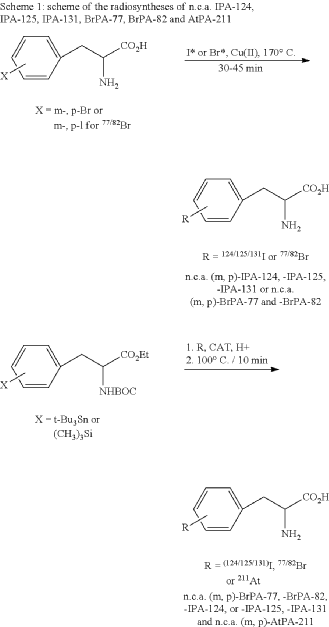
EXAMPLE 2
General synthesis of 3,4-[124I]iodo-L-phenylalanine (m, p-IPA-124), 3,4-[125I]iodo-L-phenylalanine (m,p-IPA-125) and 3,4-[131I]iodo-L-phenylalanine (m,p-IPA-131) by non-isotopic radioiodo-debromination
PAT
- Pharmaceutical combinations and uses thereofPublication Number: US-2024197715-A1Priority Date: 2022-11-18
- Pharmaceutical combinations and uses thereofPublication Number: WO-2024105610-A1Priority Date: 2022-11-18
- Iodine-labeled homoglutamic acid and glutamic acid derivativesPublication Number: US-2013034497-A1Priority Date: 2009-11-17
- MALIGNAS NEOPLASIAS THERAPY.Publication Number: ES-2341575-T3Priority Date: 2005-11-25Grant Date: 2010-06-22
- Therapy of malignant neoplasiasPublication Number: US-2007128108-A1Priority Date: 2005-11-18
- Therapy of malignant neoplasias
- Publication Number: US-9682158-B2
- Priority Date: 2005-11-18
- Grant Date: 2017-06-20
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////////Iodofalan (131I), radiopharmaceutical, antineoplastic, Phase 2, Glioblastoma, 606VTF676Y, 131I-TLX-101, ACD 101
Inlexisertib
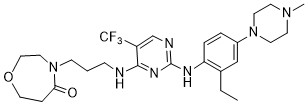
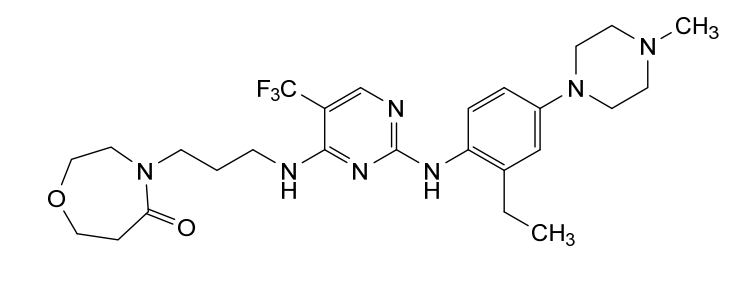
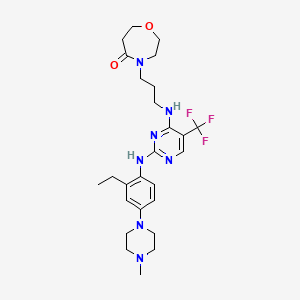
Inlexisertib
CAS 2543673-19-2
MF C26H36F3N7O2, 535.62
4-(3-((2-((2-ethyl-4-(4-methylpiperazin-1-yl)phenyl)amino)-5-(trifluoromethyl)pyrimidin-4-yl)amino)propyl)-1,4-oxazepan-5-one
4-[3-[[2-[2-ethyl-4-(4-methylpiperazin-1-yl)anilino]-5-(trifluoromethyl)pyrimidin-4-yl]amino]propyl]-1,4-oxazepan-5-one

serine/ threonine kinase inhibitor, antineoplastic, DCC 3116, JM2ZTM8S7S
Inlexisertib is an orally bioavailable inhibitor of the serine/threonine-protein kinase ULK 1 and 2, with potential antineoplastic activity. Upon oral administration, inlexisertib targets and binds to ULK1/2. This inhibits cancer autophagy, which mutant RAS cancer cells use for their survival, and results in tumor cell death. ULK1/2 mediates the autophagocytotic process and is often upregulated in cancers, especially in mutant RAS cancers. Autophagy plays a key role in a tumor cell proliferation and survival, and mediates tumor cell resistance.
- A Study of Inlexisertib (DCC-3116) in Combination With Anticancer Therapies in Participants With Advanced MalignanciesCTID: NCT05957367Phase: Phase 1/Phase 2Status: RecruitingDate: 2025-06-05
- A Phase 1/2 Study of Inlexisertib (DCC-3116) in Patients With RAS/MAPK Pathway Mutant Solid TumorsCTID: NCT04892017Phase: Phase 1/Phase 2Status: RecruitingDate: 2025-05-06
SYN
https://patents.google.com/patent/US11530206B2/en
PAT
Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereof
Publication Number: JP-7593947-B2
Priority Date: 2019-05-10
Grant Date: 2024-12-03
- PHENYLAMINOPYRIMIDINE AMIDE INHIBITORS OF AUTOPHAGY AND METHODS OF THEIR APPLICATIONPublication Number: HR-P20231730-T1Priority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: US-12071432-B2Priority Date: 2019-05-10Grant Date: 2024-08-27
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: CN-118878519-APriority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: CN-118878520-APriority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: CN-118930524-APriority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: AU-2020275392-B2Priority Date: 2019-05-10Grant Date: 2023-09-14
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: CN-114127057-BPriority Date: 2019-05-10Grant Date: 2024-07-12
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: EP-3966207-B1Priority Date: 2019-05-10Grant Date: 2023-11-01
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: EP-4342469-A2Priority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: ES-2966807-T3Priority Date: 2019-05-10Grant Date: 2024-04-24
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: EP-3966207-A1Priority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: KR-20220008873-APriority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitor and how to use itPublication Number: JP-2022531801-APriority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: US-11530206-B2Priority Date: 2019-05-10Grant Date: 2022-12-20
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: US-2023039712-A1Priority Date: 2019-05-10
- Combination of dcc-3116 and mapkap pathway inhibitors for use in the treatment of cancerPublication Number: WO-2024050351-A1Priority Date: 2022-09-02
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: US-2020354352-A1Priority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: WO-2020231806-A1Priority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: AU-2020275392-A1Priority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitor and method of usePublication Number: CN-114127057-APriority Date: 2019-05-10
PAT
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020231806&_cid=P12-MHCSWS-98394-1

AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
/////////Inlexisertib, serine/ threonine kinase inhibitor, antineoplastic, DCC 3116, JM2ZTM8S7S
Icovamenib
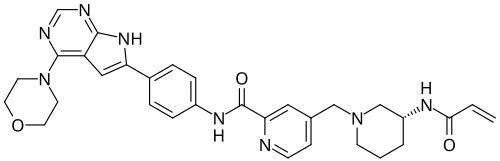
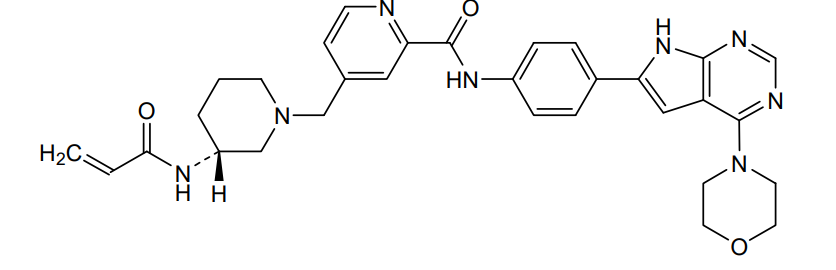
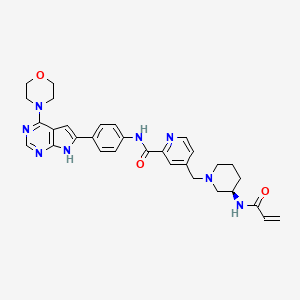
Icovamenib
CAS 2448172-22-1
MF C31H34N8O3 MW 566.7 g/mol
N-{4-[4-(morpholin-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl}-4-{[(3R)-3-(prop-2-enamido) piperidin-1-yl]methyl}pyridine-2-carboxamide
N-[4-(4-morpholin-4-yl-7H-pyrrolo[2,3-d]pyrimidin-6-yl)phenyl]-4-[[(3R)-3-(prop-2-enoylamino)piperidin-1-yl]methyl]pyridine-2-carboxamide
menin-MLL (mixed-lineage leukemia) protein interaction inhibitor,
antineoplastic, BMF-219, BMF 219, 2Z737MY35A, Menin-MLL inhibitor 21
Icovamenib is an investigational irreversible covalent inhibitor of menin. It is developed by Biomea Fusion for diabetes, lymphoma, leukemia, and multiple myeloma.[1][2][3]
Icovamenib is an orally bioavailable, irriversible inhibitor of menin, an essential co-factor of oncogenic menin-mixed lineage leukemia (MLL; myeloid/lymphoid leukemia; KMT2A) fusion proteins, with potential antineoplastic activity. Upon oral administration, icovamenib specifically targets and binds to menin, thereby preventing the interaction between the two proteins menin and MLL and the formation of the menin-MLL complex. This reduces the expression of downstream target genes, such as MYC and Bcl2, and results in an inhibition of the proliferation of MLL-rearranged tumor cells. Menin, an essential transcriptional regulator, plays a key role in oncogenic signaling in cancers driven by oncogenic MLL-fusions.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US299042443&_cid=P20-MH9YDY-31032-1
Example 9
Synthesis of Compound 10
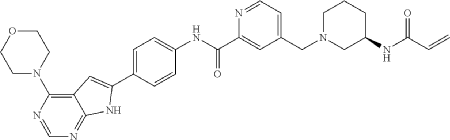
Compound 10
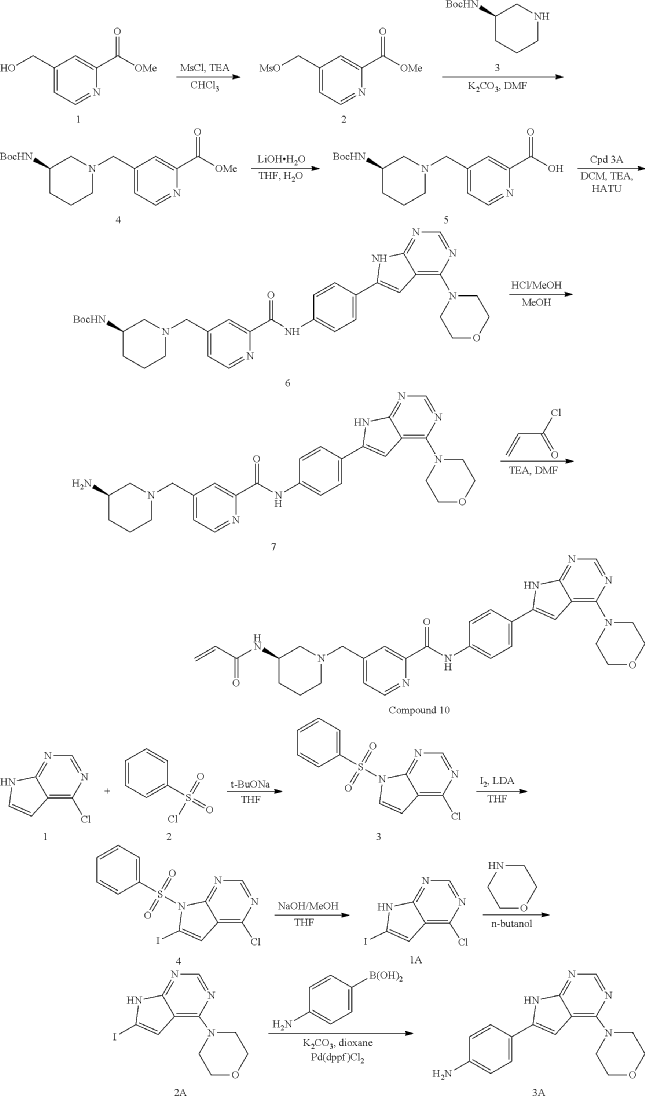
General Procedure for Preparation of Intermediate 2
| 1H NMR: CDCl 3 400 MHz 8.80 (d, J=4.85 Hz, 1H), 8.15 (d, J=0.66 Hz, 1H), 7.53 (dt, J=4.91, 0.85 Hz, 1H), 5.27-5.34 (m, 2H), 4.00-4.08 (m, 3H), 3.11 (s, 3H) |
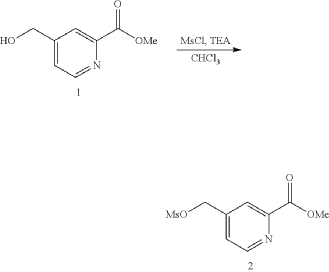
General Procedure for Preparation of Intermediate 5—
| To a solution of Intermediate 4 (1.50 g, 4.29 mmol, 1 eq) in THF (7.00 mL) was added LiOH.H 2O (540.3 mg, 12.8 mmol, 3 eq) in H 2O (7.00 mL). The mixture was stirred at 25° C. for 3 h. TLC (Dichloromethane:Methanol=10:1, R f=0) showed the reaction was complete. The mixture was poured into H 2O (20.0 mL) and extracted with DCM (10.0 mL×3). Then the organic phases dried over Na 2SO 4, filtered and concentrated under vacuum. The crude without purification. Give the Intermediate 5 (1.20 g, crude) as a yellow solid. |
General Procedure for Preparation of Intermediate 6—
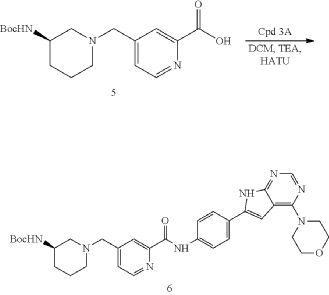
| To a solution of Intermediate 5 (0.80 g, 2.39 mmol, 1 eq), Intermediate 3A (704.4 mg, 2.39 mmol, 1 eq), TEA (1.69 g, 16.7 mmol, 2.32 mL, 7 eq) in DCM (10.0 mL) was added HATU (1.36 g, 3.58 mmol, 1.5 eq). The mixture was stirred at 20° C. for 12 h. LCMS showed the reaction was complete. The mixture was poured into H 2O (40.0 mL) and extracted with DCM (20.0 mL×3). Then the organic phases were washed with brine (50.0 mL) dried over Na 2SO 4, filtered and concentrated under vacuum. The crude for next step without purification. Give the Intermediate 6 (0.60 g, crude) as a yellow solid. |
General Procedure for Preparation of Intermediate 7—
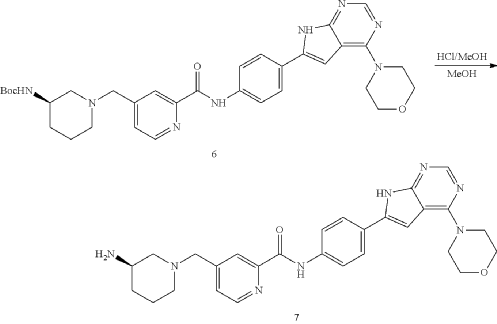
| To a solution of Intermediate 6 (0.50 g, 816.0 umol, 1 eq) in MeOH (5.00 mL) was added HCl/MeOH (4 M, 5.00 mL, 24.51 eq). The mixture was stirred at 20° C. for 12 h. LCMS showed the reaction was complete. The mixture was concentrated under vacuum. The crude for next step without purification. Give the Intermediate 7 (0.50 g, crude, HCl) as a yellow solid. |
General Procedure for Preparation of Compound 10—
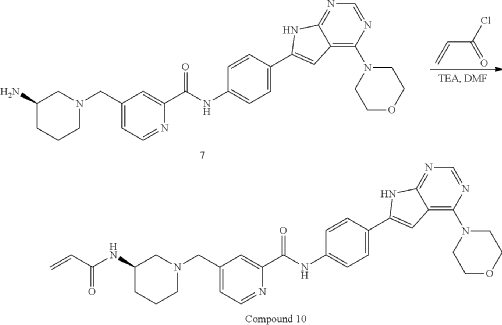
| To a solution of Intermediate 3 (0.50 g, 910.6 umol, 1 eq, HCl) in DMF (10.0 mL) was added TEA (645.0 mg, 6.37 mmol, 887.2 uL, 7 eq) and prop-2-enoyl chloride (82.4 mg, 910.6 umol, 74.2 uL, 1 eq). Then the mixture was stirred at 20° C. for 12 h. LCMS showed the reaction was complete. The mixture was poured into H 2O (50.0 mL), then was filtered and filter cake was concentrated in vacuum. The crude product was purified by reversed-phase HPLC (column: Phenomenex Luna C18 200*40 mm*10 um; mobile phase: [water(0.05% HCl)-ACN]; B %: 10%-30%, 10 min) and (column: Xtimate C18 150*25 mm*5 um; mobile phase: [water(10 mM NH 4HCO 3)-ACN]; B %: 30%-60%, 10 min). Give the Intermediate Compound 10 (20.0 mg, 35.0 umol, 3.85% yield, 99.3% purity) as a yellow solid. |
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2024172911&_cid=P20-MH9YNT-37455-1
PAT
- Substituted pyridines as irreversible inhibitors of menin-MLL interactionPublication Number: US-11702421-B2Priority Date: 2018-12-31Grant Date: 2023-07-18
- Irreversible inhibitors of menin-mll interactionPublication Number: US-2023227458-A1Priority Date: 2018-12-31
- N-[4-[4-(4-MORPHOLINYL)-7H-PYRROLO[2,3-d]PYRIMIDIN-6-YL]PHENYL]-4-[[3(R)-[(1-OXO-2-PROPEN-1-YL)AMINO]-1-PIPERIDINYL]METHYL]-2-PYRIDINE CARBOXAMIDE AND USES THEREOFPublication Number: US-2024376112-A1Priority Date: 2018-12-31
- Covalent inhibitors of menin-mll interaction for diabetes mellitusPublication Number: WO-2023018825-A1Priority Date: 2021-08-11
- Covalent inhibitors of Mennin-MLL interaction for diabetesPublication Number: CN-118076357-APriority Date: 2021-08-11
- Irreversible inhibitors of menin-mll interactionPublication Number: US-2020223853-A1Priority Date: 2018-12-31
- Substituted pyridines as irreversible inhibitors of menin-MLL interactionPublication Number: US-11084825-B2Priority Date: 2018-12-31Grant Date: 2021-08-10
- Irreversible inhibitors of menin-mll interactionPublication Number: US-2022169627-A1Priority Date: 2018-12-31
- Crystalline forms of an irreversible inhibitor of menin-mll interactionPublication Number: US-2023086137-A1Priority Date: 2021-08-20
- Crystalline form of n-[4-[4-(4-morpholinyl)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]-4-[[3(r)-[(1-oxo -2-propen-1-yl)amino]-1-piperidinyl]methyl]-2-pyridinecarboxamide, an irreversible menin-mll inhibitor for the treatment of cancerPublication Number: EP-4387972-A1Priority Date: 2021-08-20
- Crystalline forms of N-[4-[4-(4-morpholinyl)-7H-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]-4-[[3(r)-[(1-oxo-2-propen-1-yl)amino]-1-piperidinyl]methyl]-2-pyridinecarboxamide as an irreversible inhibitor of menin-MLL interactionPublication Number: US-12018032-B2Priority Date: 2021-08-20Grant Date: 2024-06-25
- Crystalline form of n-[4-[4-(4-morpholinyl)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]-4-[[3(r)-[(1-oxo -2-propen-1-yl)amino]-1-piperidinyl]methyl]-2-pyridinecarboxamide, an irreversible menin-mll inhibitor for the treatment of cancerPublication Number: WO-2023022912-A1Priority Date: 2021-08-20
- Crystalline forms of an irreversible inhibitor of menin-mll interactionPublication Number: US-2024343731-A1Priority Date: 2021-08-20
- Synthetic methods for preparing a pyridinecarboxamide compoundPublication Number: WO-2024011450-A1Priority Date: 2022-07-13
- Menin-mll inhibitors and compositions for proliferation of beta cellsPublication Number: WO-2024006391-A1Priority Date: 2022-06-28
- Flt3 combination therapy for cancer and compositions thereforPublication Number: WO-2023225005-A1Priority Date: 2022-05-17
- Treatment of cancer with menin inhibitors and immuno-oncology agentsPublication Number: WO-2023172925-A1Priority Date: 2022-03-08
- Treatment of hematological malignancies with menin inhibitors and p-glycoprotein inhibitorsPublication Number: WO-2023150635-A1Priority Date: 2022-02-04
- Crystalline forms of N[4[4-(4-Morpholinyl)-7H-Pyrrolo[2-3-D]Pyrimidin-6-yl]Phenyl]-4-[[3(R)-[(1-Oxo-2-Protein-1-yl)Amino]-1-Piperidinyl]Methyl]2-Pyridinecarboxamide]Publication Number: US-12215113-B2Priority Date: 2023-01-18Grant Date: 2025-02-04
- CRYSTALLINE FORMS OF N-[4-[4-(4-MORPHOLINYL)-7H-PYRROLO[2,3-d]PYRIMIDIN-6-YL]PHENYL]-4-[[3(R)-[(1-OXO-2-PROPEN-1-YL)AMINO]-1-PIPERIDINYL]METHYL]-2-PYRIDINECARBOXAMIDE AS IRREVERSIBLE INHIBITORS OF MENIN-MLL INTERACTIONPublication Number: US-2024417404-A1Priority Date: 2023-01-18
- Crystalline forms of n-[4-[4-(4-morpholinyl)-7h-pyrrolo[2,3-d]pyrimidin-6- yl]phenyl]-4-[[3(r)-[(l-oxo-2-propen-l-yl)amino]-l-piperidinyl]methyl]-2-pyridinecarboxamide as a covalent inhibitor of menin-mll interactionPublication Number: WO-2024155710-A1Priority Date: 2023-01-18
- Crystalline forms of n-[4-[4-(4-morpholinyl)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]-4-[[3(r)-[(l-oxo-2-propen-l-yl)amino]-l-piperidinyl]methyl]-2- pyridinecarboxamide as a covalentinhibitor of menin-mll interactionPublication Number: WO-2024155719-A1Priority Date: 2023-01-18
- Combinations of lsd1 inhibitors and menin inhibitors for treating cancerPublication Number: WO-2024110649-A1Priority Date: 2022-11-24
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Rodriguez, Jose E.; Abitbol, Alexander; Abuzgaya, Fathi; Perez, Cesar; Mourya, Sanchita; Munneke, Brian; Morris, Stephan W.; Butler, Thomas (20 June 2023). “91-LB: COVALENT-111, a Phase 1/2 Trial of BMF-219, a Covalent Menin Inhibitor, in Patients with Type 2 Diabetes Mellitus—Preliminary Results”. Diabetes. 72 (Supplement_1) 91-LB. doi:10.2337/db23-91-LB. S2CID 259444592.
- Ravandi-Kashani, F.; Kishtagari, A.; Carraway, H.; Schiller, G.; Curran, E.; Yadav, B.; Cacovean, A.; Morris, S.; Butler, T.; Lancet, J. (23 June 2022). “P587: Covalent-101: A Phase 1 Study of BMF-219, A Novel Oral Irreversible Menin Inhibitor, in Patients with Relapsed/Refractory Acute Leukemia, Diffuse Large B-Cell Lymphoma, and Multiple Myeloma”. HemaSphere. 6: 486–487. doi:10.1097/01.HS9.0000845236.32931.83.
- Somanath, Priyanka; Lu, Daniel; Law, Brian; Archer, Tenley C.; Cacovean, Alexandru; Palmer, James T.; Kinoshita, Taisei; Butler, Thomas (5 November 2021). “Novel Irreversible Menin Inhibitor, BMF-219, Shows Potent Single Agent Activity in Clinically Relevant DLBCL Cells”. Blood. 138 (Supplement 1): 4318. doi:10.1182/blood-2021-148045.
| Clinical data | |
|---|---|
| Other names | BMF-219 |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2448172-22-1 |
| PubChem CID | 154988914 |
| ChemSpider | 115037287 |
| UNII | 2Z737MY35A |
| Chemical and physical data | |
| Formula | C31H34N8O3 |
| Molar mass | 566.666 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
/////////Icovamenib, antineoplastic, BMF-219, BMF 219, 2Z737MY35A, Menin-MLL inhibitor 21
Ibrilatazar
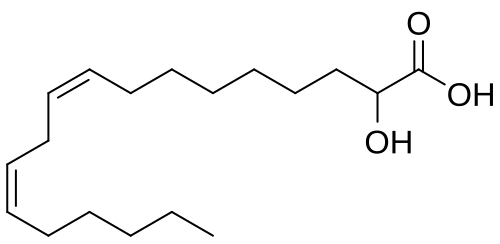
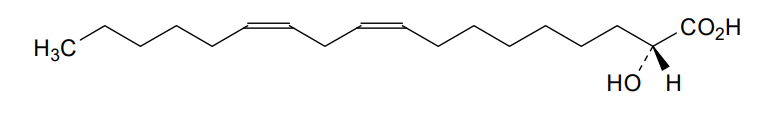
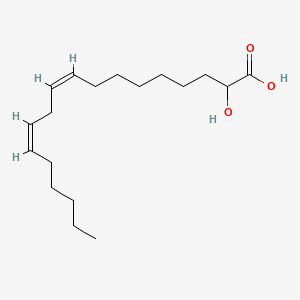
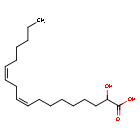
Ibrilatazar
CAS 57818-44-7
MF C18H32O3 MW 296.4 g/mol
rac-(2R)-(9Z,12Z)-2-hydroxyoctadeca-9,12-dienoic acid
(9Z,12Z)-2-hydroxyoctadeca-9,12-dienoic acid
peroxisome proliferator activated receptor (PPAR) alpha and gamma agonist, antineoplastic, ABILITY PHARMA, ABTL 0812, alpha-Hydroxylinoleic acid, ABTL0812
- alpha-Hydroxylinoleic acid
- ABTL0812
- 2-hydroxylinoleic acid
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| ABTL-0812 Sodium | X1840C8161 | Not Available | VFXKYDDSDQXKLC-NBTZWHCOSA-M |
Ibrilatazar also known as α-hydroxylinoleic acid is a small-molecule, experimental cancer drug being developed by Ability Pharmaceuticals.[1]
Ibrilatazar is an orally bioavailable, lipid analogue and inhibitor of raptor-mammalian target of rapamycin (mTOR) (mTOR complex 1; mTORC1), rictor-mTOR (mTOR complex 2; mTORC2) and dihydrofolate reductase (DHFR) with potential antineoplastic activity. Upon oral administration, ibrilatazar binds to and inhibits both mTORC1 and mTORC2, which may result in apoptosis and a decrease in proliferation in mTORC1/2-expressing tumor cells. mTOR is a serine/threonine kinase that is upregulated in some tumors; it plays an important role in the PI3K/Akt/mTOR signaling pathway which is often deregulated in cancer cells. In addition, ibrilatazar inhibits DHFR, an enzyme that reduces dihydrofolic acid to tetrahydrofolic acid, thereby blocking tetrahydrofolate synthesis, and resulting in both the depletion of nucleotide precursors and the inhibition of DNA, RNA and protein synthesis. This induces autophagy-induced cell death and further inhibition of cell proliferation.
- A Study of ABTL0812 in Pancreatic CancerCTID: NCT03417921Phase: Phase 1/Phase 2Status: SuspendedDate: 2024-07-31
- ABTL0812 in Combination With FOLFIRINOX for First-line Treatment of Metastatic Pancreatic StudyCTID: NCT04431258Phase: Phase 1/Phase 2Status: CompletedDate: 2024-03-18
- Phase I/Ib Clinical Trial of ABTL0812 in Advanced Cancer PatientsCTID: NCT02201823Phase: Phase 1Status: CompletedDate: 2015-07-02
- Microbiological production method of γ- and δ-lactonesPublication Number: JP-H03187387-APriority Date: 1989-08-04
- Process for the microbiological production of gamma- and delta-lactonesPublication Number: US-5168054-APriority Date: 1989-08-04Grant Date: 1992-12-01
- Degradation control of environmentally degradable disposable materialsPublication Number: US-2002123546-A1Priority Date: 1988-08-08
- Degradation control of environmentally degradable disposable materialsPublication Number: US-6323307-B1Priority Date: 1988-08-08Grant Date: 2001-11-27
- Degradation control of environmentally degradable disposable materialsPublication Number: US-6740731-B2Priority Date: 1988-08-08Grant Date: 2004-05-25
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US38087288&_cid=P12-MH8IQK-97634-1
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
History
In 2015, Ability announced that it had received orphan drug designation (ODD) for pediatric cancer neuroblastoma from the European Medical Agency (EMA) and the US Food and Drug Administration (FDA).[1] Also in 2016 a preclinical study confirmed that ABTL0812 was well tolerated.[2] In December 2016 the company announced Ibrilatazar has received an Orphan Drug Designation for the treatment of pancreatic cancer.[1]
Mechanism of action
One mechanism of action is the activation of the PPAR-alpha and PPAR-gamma receptors which in turn up-regulate the expression of the TRIB3 gene, leading to inhibition of the PI3K/AKT/mTOR pathway. This pathway is excessively activated in most human cancers, supporting tumor growth. It is a principal target of various new anti-tumour drugs. Tumor cells are killed via autophagic cell death, rather than apoptosis.[3][4]
ABTL0812 activates the PPAR receptors, inducing TRIB3 over-expression. TRIB3 binds to the Akt oncogene and inhibits the Akt/mTOR axis.[3]
Clinical trials
ABTL0812 showed efficacy in Phase I clinical trials in patients with advanced cancer, with low toxicity and high tolerability.[3]
References
- “Ability Pharmaceuticals Announces Orphan Drug Designation in the US for ABTL0812 in Pancreatic Cancer”. Ability Pharmaceuticals SL.
- “Ability Pharmaceuticals Announces Positive Phase 1 1b Study Results Of ABTL0812 In Cancer Patients With Advanced Solid Tumors”. http://www.biospace.com.
- “New mechanism of antitumor action identified”. Medical Xpress. 25 January 2016.
- Erazo T, Lorente M, López-Plana A, Muñoz-Guardiola P, Fernández-Nogueira P, García-Martínez JA, et al. (May 2016). “The New Antitumor Drug ABTL0812 Inhibits the Akt/mTORC1 Axis by Upregulating Tribbles-3 Pseudokinase”. Clinical Cancer Research. 22 (10): 2508–19. doi:10.1158/1078-0432.ccr-15-1808. hdl:2445/207600. PMID 26671995.
| Clinical data | |
|---|---|
| Other names | α-Hydroxylinoleic acid; 2-Hydroxylinoleic acid; ABTL-0812 |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| IUPAC name | |
| CAS Number | 57818-44-7 |
| PubChem CID | 21158511 |
| ChemSpider | 20118100 |
| UNII | 0DE74TJ7EZ |
| ChEBI | CHEBI:136927 |
| CompTox Dashboard (EPA) | DTXSID301258077 |
| Chemical and physical data | |
| Formula | C18H32O3 |
| Molar mass | 296.451 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
/////////Ibrilatazar, peroxisome proliferator activated receptor (PPAR) alpha and gamma agonist, antineoplastic, ABILITY PHARMA, ABTL 0812, alpha-Hydroxylinoleic acid, ABTL0812
Fovinaciclib
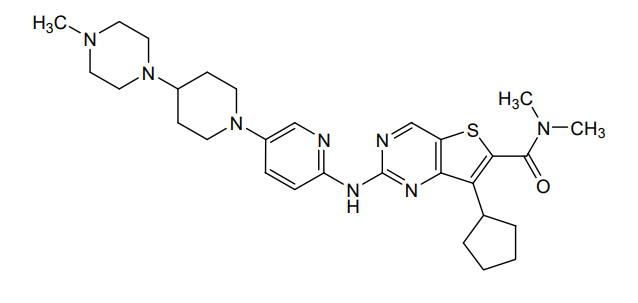
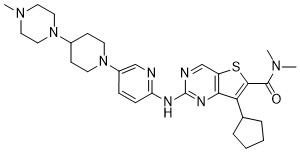
Fovinaciclib
CAS 2146171-49-3
MF C29H40N8OS
Exact Mass: 548.3046
Molecular Weight: 548.75
7-cyclopentyl-N,N-dimethyl-2-({5-[4-(4-methylpiperazin-1-yl)piperidin-1-yl]pyridin-2-yl}amino) thieno[3,2-d]pyrimidine-6-carboxamide
7-cyclopentyl-N,N-dimethyl-2-((5-(4-(4-methylpiperazin-1-yl)piperidin-1-yl)pyridin-2-yl)amino)thieno[3,2-d]pyrimidine-6-carboxamide
7-cyclopentyl-N, N-dimethyl-2- ( (5- (4- (4-methylpiperazin-1-yl) piperidin-1-yl) pyridin-2-yl) amino) thieno [3, 2-d] pyrimidine-6-carboxamide
7-Cyclopentyl-N,N-dimethyl-2-((5-(4-(1-methylpiperidin-4-yl)piperazin-1-yl)pyridin-2-yl)amino Thieno[3,2-d]pyrimidine-6-carboxamide
cyclin dependent kinase inhibitor, antineoplastic, Fovinaciclibum, LPW3H579X8, inzhou Aohong Pharmaceutical Co
- OriginatorChongqing Fochon Pharmaceutical
- DeveloperAhon Pharmaceutical; Chongqing Fochon Pharmaceutical; Shanghai Fosun Pharmaceutical
- Class2 ring heterocyclic compounds; Amides; Amines; Antineoplastics; Cyclopentanes; Piperazines; Piperidines; Pyridines; Pyrimidines; Small molecules; Thiophenes
- Mechanism of ActionCyclin-dependent kinase 4 inhibitors; Cyclin-dependent kinase 6 inhibitors
- MarketedHER2 negative breast cancer
- No development reportedSolid tumours
- 04 Sep 2025Chemical structure information added.
- 02 Sep 2025Launched for HER2-negative-breast-cancer (Late-stage disease, Second-line therapy or greater) in China (PO) (Shanghai Henlius Biotech pipeline, September 2025)
- 26 Aug 2025Registered for HER2-negative-breast-cancer (Late-stage disease, Second-line therapy or greater) in China (PO) prior to August 2025
Fovinaciclib is an orally bioavailable inhibitor of cyclin-dependent kinase (CDK) types 4 (CDK4) and 6 (CDK6), with potential antineoplastic activity. Upon administration, fovinaciclib selectively inhibits CDK4 and CDK6, which inhibits the phosphorylation of retinoblastoma protein (Rb) early in the G1 phase, prevents CDK-mediated G1/S transition and leads to cell cycle arrest. This suppresses DNA replication and decreases tumor cell proliferation. CDK4 and 6 are serine/threonine kinases that are upregulated in many tumor cell types and play key roles in the regulation of both cell cycle progression from the G1-phase into the S-phase and cell proliferation.
On May 29, 2025, China’s National Medical Products Administration (NMPA) approved the Class 1 innovative drug Fovinaciclib (CDK4&6 inhibitor), developed by Jinzhou Aohong Pharmaceutical Co., Ltd. This medication, in combination with fulvestrant, is indicated for the treatment of adult patients with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative recurrent or metastatic breast cancer, who have experienced disease progression following prior endocrine therapy.
Notably, Fovinaciclib represents an excellent example of scaffold hopping—its design replaces the pyrrolo-pyrimidine core of Ribociclib (first approved on March 13, 2017) with a thieno-pyrimidine ring.
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN236278427&_cid=P21-MGRD95-18783-1
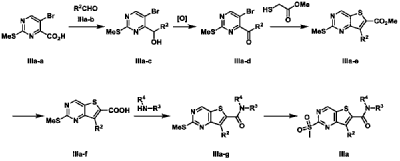
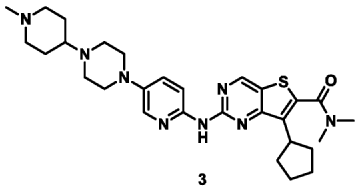
| Example 3 |
| 7-Cyclopentyl-N,N-dimethyl-2-((5-(4-(1-methylpiperidin-4-yl)piperazin-1-yl)pyridin-2-yl)amino Thieno[3,2-d]pyrimidine-6-carboxamide (3) |
According to the synthesis method of Example 2, CH
3 CHO replaced by CH
2 O, to prepare the title compound 7-cyclopentyl-N,N-dimethyl-2-((5-(4-(1-methylpiperidin-4-yl)piperazin-1-yl)pyridin-2-yl)amino)thieno[3,2-d]pyrimidine-6-carboxamide (3). MS-ESI (m/z): 549 [M+1] + .
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017193872&_cid=P21-MGRDEF-24321-1
[0266]
7-cyclopentyl-N, N-dimethyl-2- ( (5- (4- (4-methylpiperazin-1-yl) piperidin-1-yl) pyridi n-2-yl) amino) thieno [3, 2-d] pyrimidine-6-carboxamide (5)
[0267]
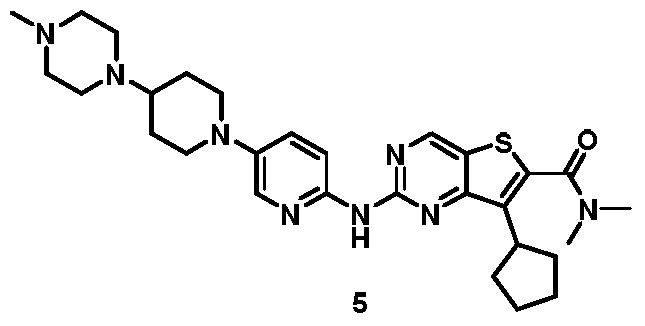
To a solution of 7-cyclopentyl-N, N-dimethyl-2- ( (5- (4- (piperazin-1-yl) piperidin-1-yl) pyridin-2-yl) amino) thieno [3, 2-d] pyrimidine-6-carboxamide (4) (1.5 g, 2.8 mmol) in DCM (45 mL) was added NaBH (OAc) 3(3.56 mg, 16.8 mmol) followed by CH 2O (40%in water, 252 mg, 3.4 mmol) . The mixture was stirred at r.t. for 30 min. The mixture was diluted with saturated aqueous NaHCO 3(100 mL) and extracted with DCM (2 × 30 mL) . The extracts were dried over Na 2SO 4. Solvents were evaporated under reduced pressure. The residue was purified by column chromatography on silica gel eluting with 96: 3: 1 DCM/methanol/ammonia to give 7-cyclopentyl-N, N-dimethyl-2- ( (5- (4- (4-methylpiperazin-1-yl) piperidin-1-yl) pyridin-2-yl) amino) thieno [3, 2-d] pyrimidine-6-carboxamide (5) . MS-ESI (m/z) : 549 [M + 1] +.
PAT
- Certain protein kinase inhibitorsPublication Number: JP-2019516790-APriority Date: 2016-05-07
- Certain protein kinase inhibitorsPublication Number: US-2019209566-A1Priority Date: 2016-05-07
- Certain protein kinase inhibitorsPublication Number: WO-2017193872-A1Priority Date: 2016-05-07
- Certain protein kinase inhibitorsPublication Number: US-10835535-B2Priority Date: 2016-05-07Grant Date: 2020-11-17
- A class of protein kinase inhibitorsPublication Number: CN-109153686-BPriority Date: 2016-05-07Grant Date: 2021-04-30
- specific protein kinase inhibitorsPublication Number: KR-102374033-B1Priority Date: 2016-05-07Grant Date: 2022-03-14
- Certain protein kinase inhibitorsPublication Number: EP-3452484-B1Priority Date: 2016-05-07Grant Date: 2023-07-05
- Certain protein kinase inhibitorsPublication Number: ES-2954148-T3Priority Date: 2016-05-07Grant Date: 2023-11-20
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////////Fovinaciclib, CHINA 2025, APPROVALS 2025, cyclin dependent kinase inhibitor, antineoplastic, Fovinaciclibum, LPW3H579X8, inzhou Aohong Pharmaceutical Co
Foselutoclax
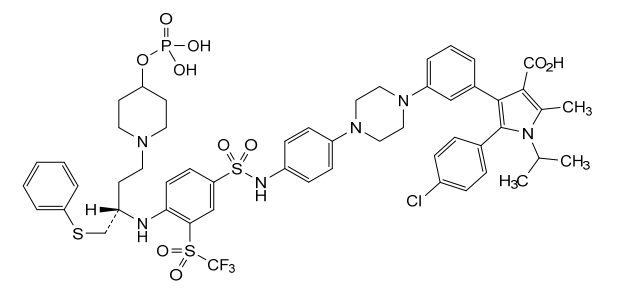
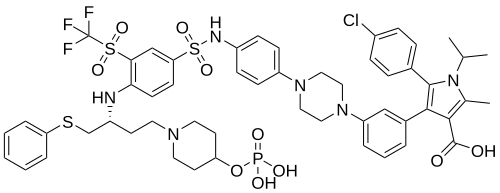
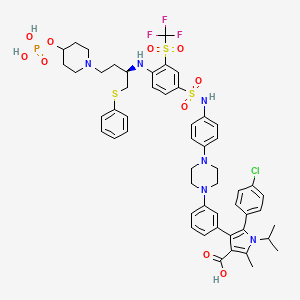
Foselutoclax
CAS 2271269-01-1
MF C53H59ClF3N6O10PS3 MW 1159.7 g/mol
(10R)-14-chloro-25-methyl-7,7-dioxo-10-[(phenylsulfanyl)methyl]-134-(phosphonooxy)-21-(propan-2-yl)-83-(trifluoromethanesulfonyl)-21H-7λ6-thia-6,9-diaza-4(1,4)-piperazina-13(1)-piperidina-2(2,3)-pyrrola-1(1),3(1,3),5,8(1,4)-tetrabenzenatridecaphane-24-carboxylic acid
5-(4-chlorophenyl)-2-methyl-4-[3-[4-[4-[[4-[[(2R)-1-phenylsulfanyl-4-(4-phosphonooxypiperidin-1-yl)butan-2-yl]amino]-3-(trifluoromethylsulfonyl)phenyl]sulfonylamino]phenyl]piperazin-1-yl]phenyl]-1-propan-2-ylpyrrole-3-carboxylic acid
B-cell lymphoma 2 (Bcl-2) inhibitor, antineoplastic, VT53CL5GES, UBX 1325
Foselutoclax is an investigational new drug that is being evaluated for the treatment of age-related eye diseases, particularly diabetic macular edema (DME) and wet age-related macular degeneration (AMD). Developed by Unity Biotechnology, this senolytic compound acts as a potent inhibitor of Bcl-xL, a protein that senescent cells rely on for survival.[1] Foselutoclax is designed to selectively eliminate senescent cells in the retina, potentially addressing the underlying causes of vision loss in these conditions.[2]
- Assess the Efficacy and Safety of Repeat Intravitreal Injections of Foselutoclax (UBX1325) in Patients With DME (ASPIRE)CTID: NCT06011798Phase: Phase 2Status: CompletedDate: 2025-08-05
- Safety, Tolerability and Evidence of Activity Study of UBX1325 in Patients With Diabetic Macular Edema (BEHOLD)CTID: NCT04857996Phase: Phase 2Status: CompletedDate: 2024-05-16
- Safety and Tolerability Study of UBX1325 in Patients With Diabetic Macular Edema or Neovascular Age-Related Macular DegenerationCTID: NCT04537884Phase: Phase 1Status: CompletedDate: 2022-03-10
REF
- Therapeutic targeting of cellular senescence in diabetic macular edema: preclinical and phase 1 trial resultsPublication Name: Nature MedicinePublication Date: 2024-02PMID: 38321220DOI: 10.1038/s41591-024-02802-4
- Senolytics in the treatment of diabetic retinopathyPublication Name: Frontiers in PharmacologyPublication Date: 2022-08-26PMCID: PMC9462063PMID: 36091769DOI: 10.3389/fphar.2022.896907
- Senolytic drugs: from discovery to translationPublication Name: Journal of Internal MedicinePublication Date: 2020-08-04PMCID: PMC7405395PMID: 32686219DOI: 10.1111/joim.13141
- bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell deathPublication Name: CellPublication Date: 1993-08-27PMID: 8358789DOI: 10.1016/0092-8674(93)90508-n
PAT
Treatment of Lung Diseases Using Pharmaceutical Agents that Eliminate Senescent Cells
Publication Number: US-2020354336-A9
Priority Date: 2017-08-11
- Senescent Cells and for Treating CancerPublication Number: US-2022017485-A1Priority Date: 2018-06-13
- Acyl sulfonamides that are bcl family antagonists for use in clinical management of conditions caused or mediated by senescent cells and for treating cancerPublication Number: EP-4335516-A2Priority Date: 2018-06-13
- Methods of Inhibiting Pathological AngiogenesisPublication Number: US-2020253991-A1Priority Date: 2017-10-31
- Methods of inhibiting pathological angiogenesisPublication Number: US-11129838-B2Priority Date: 2017-10-31Grant Date: 2021-09-28
- Treatment of Lung Diseases Using Pharmaceutical Agents that Eliminate Senescent CellsPublication Number: US-2020199103-A1Priority Date: 2017-08-11
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US279621490&_cid=P21-MGPXU3-15237-1
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US421382898&_cid=P21-MGPXWE-19244-1
A crystalline solid meglumine salt of of (R)-5-(4-chlorophenyl)-1-isopropyl-2-methyl-4-(3-(4-(4-((4-((1-(phenylthio)-4-(4-((phosphonooxy)methyl)piperidin-1-yl)butan-2-yl)amino)-3-((trifluoromethyl)sulfonyl)phenyl)sulfonamido)phenyl)piperazin-1-yl)phenyl)-1H-pyrrole-3-carboxylic acid, the compound of Formula I:
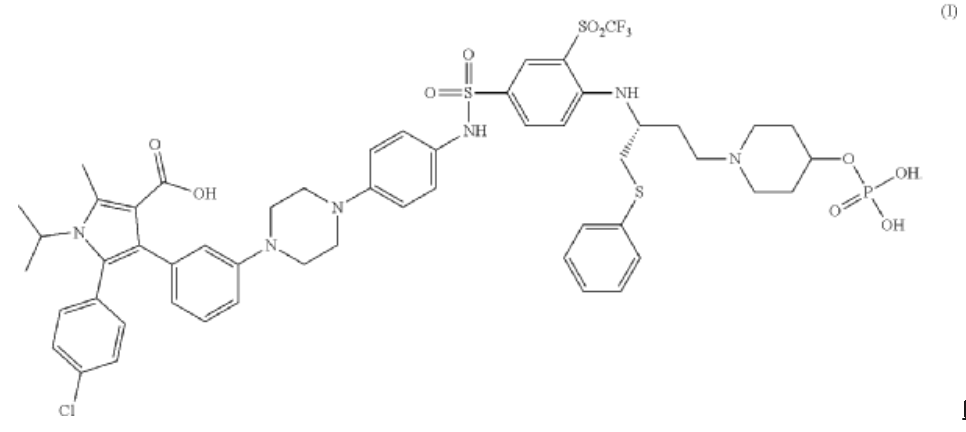
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US348024244&_cid=P21-MGPXWE-19244-1
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | UBX1325 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2271269-01-1 |
| PubChem CID | 147562879 |
| IUPHAR/BPS | 13366 |
| ChemSpider | 115277082 |
| UNII | VT53CL5GES |
| Chemical and physical data | |
| Formula | C53H59ClF3N6O10PS3 |
| Molar mass | 1159.69 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Crago SM (22 June 2023). “Design for Phase 2B ASPIRE Study of UBX1325 for DME announced by UNITY”. Modern Retina. Archived from the original on 13 August 2024.
- Macha N, Yu M, Sapieha P, Klier S, Ghosh A, White L, et al. (September 2024). “Multifocal Electroretinography Changes after UBX1325 (Foselutoclax) Treatment in Neovascular Age-Related Macular Degeneration”. Journal of Clinical Medicine. 13 (18): 5540. doi:10.3390/jcm13185540. PMC 11433175. PMID 39337030.
//////////foselutoclax, antineoplastic, VT53CL5GES, UBX 1325
Fosdesdenosine sipalabenamide
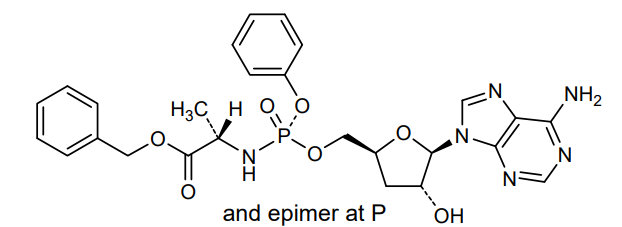
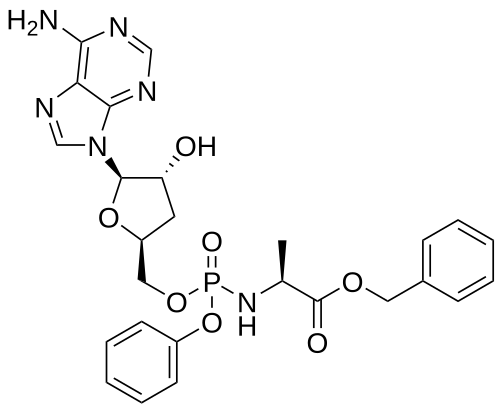
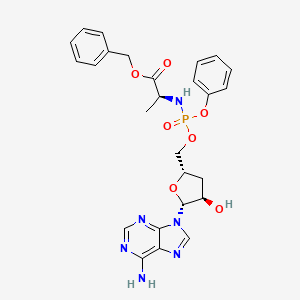
Fosdesdenosine sipalabenamide
CAS 2348493-39-8
MF C26H29N6O7P, MW=568.5 g/mol
benzyl N-(P-ambo-3′-deoxy-OP-phenyl-5′-adenylyl)-Lalaninate
benzyl (2S)-2-[[[(2S,4R,5R)-5-(6-aminopurin-9-yl)-4-hydroxyoxolan-2-yl]methoxy-phenoxyphosphoryl]amino]propanoate
3′-Deoxyadenosine 5′-O-phenyl-(benzoxy-L-alaninyl)-phosphatenucleoside analogue, antineoplastic, NUC 7738, Y7BFN2M72F
Fosdesdenosine sipalabenamide is an investigational new drug that is being evaluated for the treatment of advanced solid tumors and lymphoma.[1] This compound is a phosphoramidate derivative of cordycepin (3′-deoxyadenosine), an adenosine analog originally isolated from the fungus Cordyceps.[2][3] As a nucleoside analog with potential antineoplastic properties, Fosdesdenosine sipalabenamide is designed to inhibit RNA synthesis and act as an RNA inhibitor.[1] The drug is being developed by NuCana Plc.[1]
Fosdesdenosine Sipalabenamide is a phosphoramidate derivative of the monophosphate form of cordycepin (3′-deoxyadenosine; 3′-dA), an adenosine derivative first isolated from Cordyceps sinensis, with potential antineoplastic, antioxidant, and anti-inflammatory activities. Upon administration and cellular uptake of fosdesdenosine sipalabenamide by passive diffusion, cordycepin monophosphate (3′-dAMP) is converted into its active anti-cancer metabolite 3′-deoxyadenosine triphosphate (3′-dATP). 3′-dATP functions as a ribonucleoside analogue and competes with ATP during transcription. Therefore, this agent causes RNA synthesis inhibition, inhibits cellular proliferation, and induces apoptosis. Also, 3′-dAMP activates AMP-activated protein kinase (AMPK) and reduces mammalian target of rapamycin (mTOR) signaling. This prevents the hyperphosphorylation of the translation repressor protein 4E-BP1. This results in the induction of tumor cell apoptosis and a decrease in tumor cell proliferation. mTOR, a serine/threonine kinase belonging to the phosphatidylinositol 3-kinase (PI3K)-related kinase (PIKK) family, plays an important role in the PI3K/AKT/mTOR signaling pathway that regulates cell growth and proliferation, and its expression or activity is frequently dysregulated in human cancers. Compared to cordycepin alone, the addition of the phosphoramidate moiety may overcome cancer resistance and allow for greater cytotoxicity as fosdesdenosine sipalabenamide does not require a nucleoside transporter for cellular uptake, is independent of enzymatic activation by adenosine kinase (AK) and is not susceptible to enzymatic degradation by adenosine deaminase (ADA). Altogether, this may help overcome cancer resistance to cordycepin.
SYN
Publication Name: Journal of Medicinal Chemistry
Publication Date: 2022-11-23
PMCID: PMC9743095
PMID: 36417756
DOI: 10.1021/acs.jmedchem.2c01348
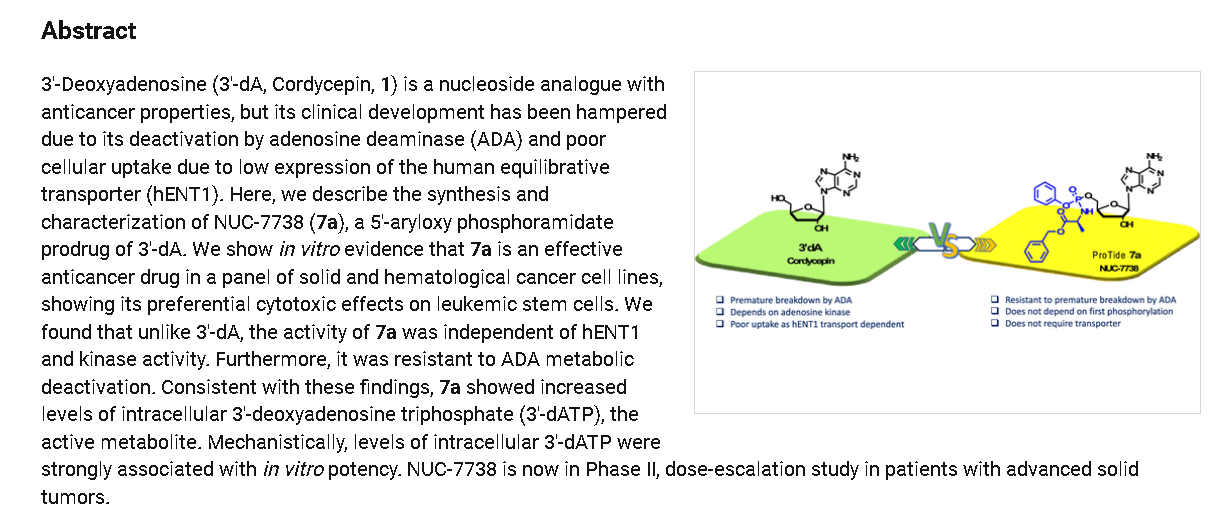
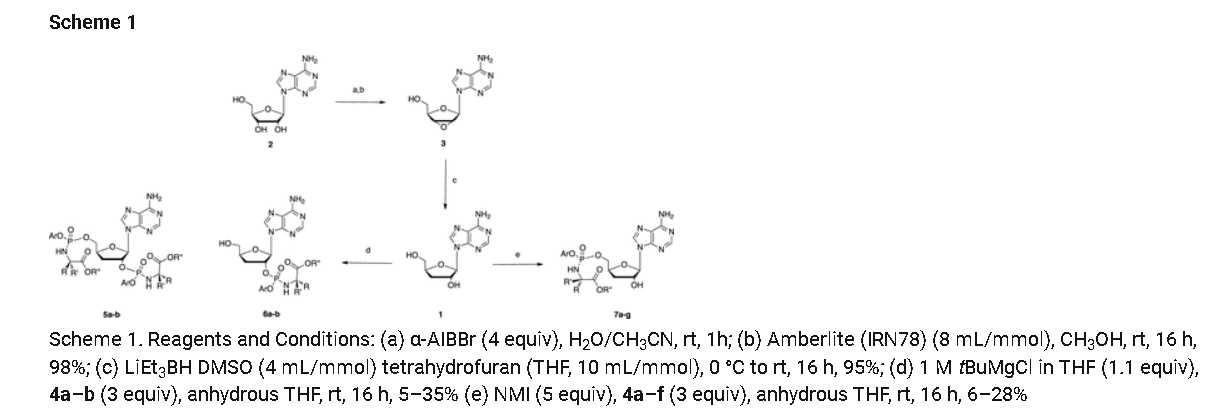
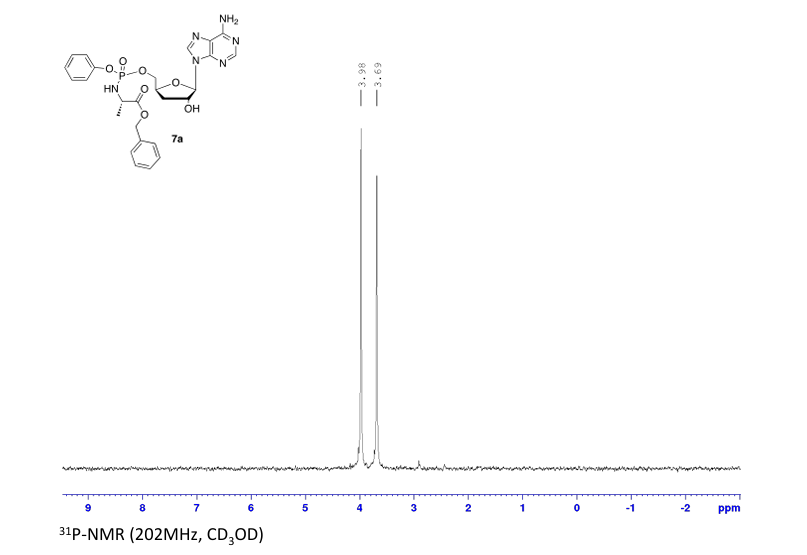
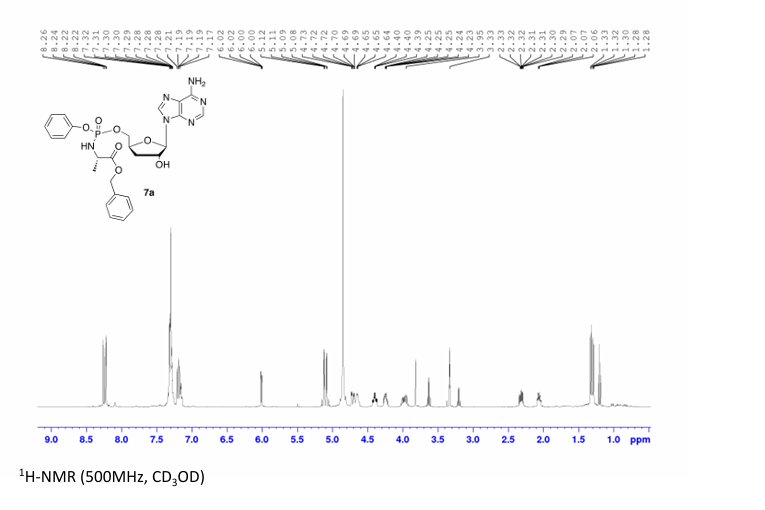
Rp)- and (Sp)-3′-Deoxyadenosine 5′-O-phenyl-(benzoxy-l-alaninyl)-phosphate (7a)
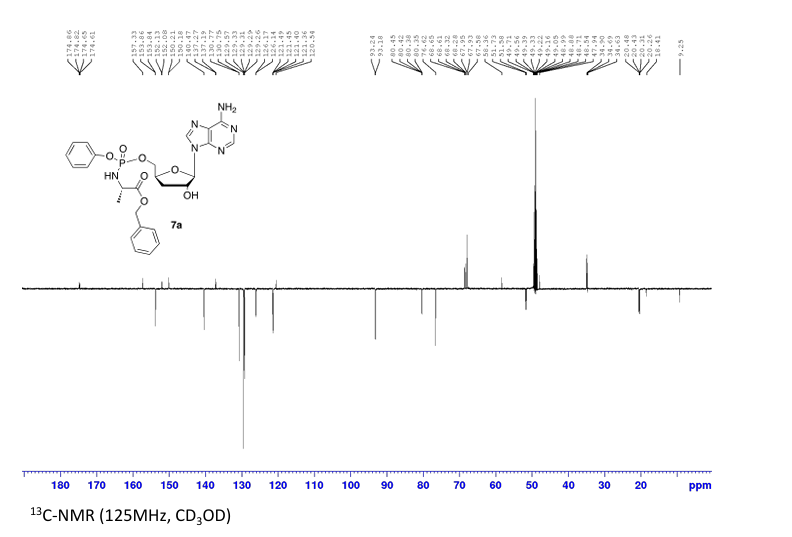
Prepared according to general procedure C using 3′-deoxyadenosine (1) (0.05 g, 0.20 mmol) in anhydrous THF (4 mL), N-methyl imidazole (0.080 μL, 1.0 mmol), and phenyl(benzyloxy-l-alaninyl) phosphorochloridate (4a) (0.021 g, 0.6 mmol) in THF (2.4 mL) Purification by Biotage Isolera One (cartridge SNAP 25 g, 25 mL/min, CH3OH/CH2Cl2 1–8% 10 CV, 8% 5 CV) and preparative TLC (1000 μM, eluent system CH3OH/CH2Cl2 5/95) afforded the title compound 7a as a white solid (0.032 g, 28%). 31P NMR (202 MHz, CD3OD) δP 3.91, 3.73. 1H NMR (500 MHz, CDCl3) δH 8.26 (s, 0.5H, H-8), 8.24 (s, 0.5H, H-8), 8.22 (s, 0.5H, H-2), 8.21 (s, 0.5H, H-2), 7.34–7.25 (m, 7H, Ar), 7.21–7.13 (m, 3H, Ar), 6.01 (d, J = 1.5 Hz, 0.5H, H-1′), 6.00 (d, J = 1.5 Hz, 0.5H, H-1′), 5.15–5.04 (m, 2H, CH2Ph), 4.73–4.63 (m, 2H, H-2′, H-4′), 4.43–4.35 (m, 1H, H-5′), 4.27–4.20 (m, 1H, H-5′), 4.03–3.91 (m, 1H, CHCH3), 2.35–2.28 (m, 1H, H-3′), 2.09–2.02 (m, 1H, H-3′), 1.32 (d, J = 7.4 Hz, 1.5 H, CHCH3), 1.28 (d, J = 7.4 Hz, 1.5 H, CHCH3). 13C NMR (125 MHz, CD3OD) δC 174.84 (d, 3JC-P = 4.5 Hz, C=O), 174.63 (d, 3JC-P = 4.5 Hz, C═O), 157.32 (C-6), 157.31 (C-6), 153.86 (C-2), 153.84 (C-2), 152.13 (C-4), 152.07 (C-4), 150.20 (C-Ar), 150.18 (C-Ar), 140.47 (C-8), 137.26 (C-Ar), 137.19 (C-Ar), 130.76 (CH-Ar), 130.74 (CH-Ar), 129.57 (CH-Ar), 129.32 (CH-Ar), 129.31 (CH-Ar), 129.29 (CH-Ar), 129.26 (CH-Ar), 126.16 (CH-Ar), 126.14 (CH-Ar), 121.46 (d, 3JC-P = 4.7 Hz, CH-Ar), 121.38 (d, 3JC-P = 4.7 Hz, CH-Ar) 120.54 (C-5), 120.53 (C-5), 93.24 (C-1′), 93.18 (C-1′), 80.43 (d, 3JC-P = 3.6 Hz, C-4′), 80.36 (d, 3JC-P = 3.6 Hz, C-4′), 76.62 (C-2′), 68.62 (d, 2JC-P = 5.3 Hz, C-5′), 68.30 (d, 2JC-P = 5.3 Hz, C-5′), 67.95 (CH2Ph), 67.92 (CH2Ph), 51.74 (CHCH3), 51.60 (CHCH3), 34.91 (C-3′), 34.70 (C-3′), 20.45 (d, 3JC-P = 7.0 Hz, CHCH3), 20.28 (d, 3JC-P = 7.0 Hz, CHCH3). Reversed-phase HPLC eluting with H2O/CH3CN from 100/10 to 0/100 in 30 min, F = 1 mL/min, λ = 254 nm, tR 13.56 and 13.75 min. C26H29N6O7P required m/z 568.2 [M]. MS (ES+) found m/z 569.2 [M + H]+, 591.2 [M + Na]+, 1159.4 [2M+Na]+.
The two diastereoisomers 7a-Rp and 7a-Sp were separated via Biotage Isolera One (cartridge SNAP-Ultra C18 12 g, F: 12 mL/min, isocratic eluent system: H2O/CH3OH 45/55 in 30 min, 150 mg sample) to obtain:
7a-Rp as Fast Eluting Isomer (76 mg)
31P NMR (202 MHz, CD3OD) δP 3.91. 1H NMR (500 MHz, CDCl3) δH 8.26 (s, 1H, H-8), 8.22 (s, 1H, H-2), 7.37–7.25 (m, 7H, Ar), 7.22–7.12 (m, 3H, Ar), 6.01 (d, J = 1.5 Hz, 1H, H-1′), 5.12 (AB q, JAB = 12.0 Hz, 2H, CH2Ph), 4.74–4.70 (m, 1H, H-2′), 4.69–4.62 (m, 1H, H-4′), 4.44–4.38 (m, 1H, H-5′), 4.28–4.21 (m, 1H, H-5′), 3.99–3.90 (m, 1H, CHCH3), 2.35–2.27 (m, 1H, H-3′), 2.09–2.02 (m, 1H, H-3′), 1.29 (d, J = 7.0 Hz, 3H, CHCH3). HPLC reversed-phase HPLC eluting with H2O/CH3CN from 90/10 to 0/100 in 30 min, F = 1 mL/min, λ = 254 nm, showed one peak with tR 13.56 min.
7a-Sp as Slow-Eluting Isomer (61 mg)
31P NMR (202 MHz, CD3OD) δP 3.73. 1H NMR (500 MHz, CDCl3) δH 8.24 (s, 1H, H-8), 8.22 (s, 1H, H-2), 7.36–7.26 (m, 7H, Ar), 7.22–7.13 (m, 3H, Ar), 6.01 (d, J = 1.5 Hz, 1H, H-1′), 5.08 (AB q, JAB = 12.0 Hz, 2H, CH2Ph), 4.70–4.67 (m, 1H, H-2′), 4.66–4.60 (m, 1H, H-4′), 4.41–4.35 (m, 1H, H-5′), 4.26–4.19 (m, 1H, H-5′), 4.02–3.94 (m, 1H, CHCH3), 2.36–2.27 (m, 1H, H-3′), 2.08–2.01 (m, 1H, H-3′), 1.34–1.30 (m, 3H, CHCH3). HPLC reversed-phase HPLC eluting with H2O/CH3CN from 90/10 to 0/100 in 30 min, F = 1 mL/min, λ = 254 nm, tR 13.75 min.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
……
| Clinical data | |
|---|---|
| Other names | NUC-7738 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2348493-39-8 |
| PubChem CID | 166177279 |
| DrugBank | DB19148 |
| UNII | Y7BFN2M72F |
| ChEMBL | ChEMBL5277528 |
| Chemical and physical data | |
| Formula | C26H29N6O7P |
| Molar mass | 568.527 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- “Fosdesdenosine sipalabenamide”. PatSnap.
- “Fosdesdenosine Sipalabenamide”. PubChem. U.S. National Library of Medicine.
- Serpi M, Ferrari V, McGuigan C, Ghazaly E, Pepper C (December 2022). “Synthesis and Characterization of NUC-7738, an Aryloxy Phosphoramidate of 3′-Deoxyadenosine, as a Potential Anticancer Agent”. Journal of Medicinal Chemistry. 65 (23): 15789–15804. doi:10.1021/acs.jmedchem.2c01348. PMC 9743095. PMID 36417756.
….///////Fosdesdenosine sipalabenamide, antineoplastic, NUC 7738, Y7BFN2M72F
Flezurafenib
Flezurafenib
CAS 2760321-00-2
MF C26H21FN4O3 MW456.5 g/mol, P26TTM6U27
5-({(3S)-3-[4-(4-fluorophenyl)-1H-imidazol-2-yl]-3,4-dihydro-2H-1-benzopyran-6-yl}oxy)-3,4-dihydro-1,8-naphthyridin-2(1H)-one
5-[[(3S)-3-[5-(4-fluorophenyl)-1H-imidazol-2-yl]-3,4-dihydro-2H-chromen-6-yl]oxy]-3,4-dihydro-1H-1,8-naphthyridin-2-one
rapidly accelerated fibrosarcoma (Raf) kinase inhibitor,
antineoplastic
Flezurafenib is an investigational new drug designed as a rapidly accelerated fibrosarcoma (RAF) kinase inhibitor which is being evaluated for the treatment of cancer. Developed by Jazz Pharmaceuticals, this novel therapeutic agent is currently being explored for its efficacy against solid tumors and hematological malignancies harboring oncogenic mutations that activate the RAS-RAF-MAPK signaling pathway.[1][2] As of January 2025, flezurafenib has reached Phase 1 clinical trials, where it is being evaluated for the treatment of advanced cancers and advanced malignant solid neoplasms.[1]
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022023450&_cid=P11-MGN3DV-58095-1
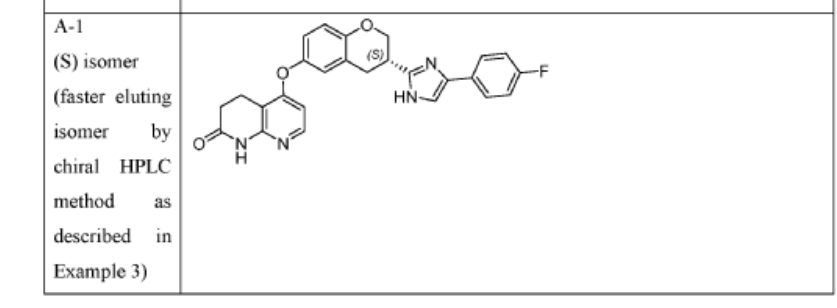
[0402] Example 3. Chiral Synthesis of Compounds A-l and A-2
[0403] A. Synthesis of P2
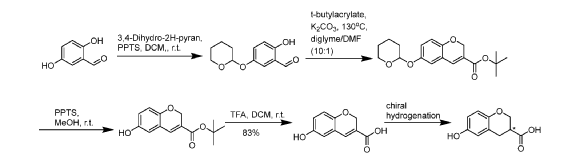
[0404] Step 1: To a solution of 2,5-dihydroxybenzaldehyde (200 g, 1448 mmol) and pyridinium p-toluenesulfonate (18.2 g, 72.4 mmol) in DCM (3.75 L) was added 3,4-dihydro-2H-pyran (165 mL, 1810 mmol) dropwise over 10 minutes and the reaction temperature warmed to 30 °C. The reaction was stirred for 2 hours and checked by UPLC-MS which indicated the reaction was 92% complete (~5% starting material and ~3% later running unknown). The reaction was stopped. The reaction was washed with water (1.5 L) and the DCM solution was passed through a 750g silica pad and followed through by DCM (2.5 L). The DCM solution was reduced in-vacuo and the crude product was then slowly diluted with Pet. Ether to ~1L total volume, stirred and cooled to -10° C to afford a thick yellow slurry. The product was filtered and washed with Pet. Ether (2 x 150 mL) and pulled dry for 3 hours to afford 2-hydroxy-5-tetrahydropyran-2-yloxy-benzaldehyde (265g, 1192 mmol, 82% yield) as a bright yellow solid. ¾ NMR (400 MHz, DMSO-d6) d/ppm: 10.35 (s, 1H), 10.23 (s, 1H), 7.32 – 7.19 (m, 2H), 6.94 (d, J = 8.9 Hz, 1H), 5.36 (t, J = 3.3 Hz, 1H), 3.77 (ddd, J = 11.2, 8.8, 3.6 Hz, 1H), 3.59 – 3.49 (m, 1H), 1.94 – 1.45 (m, 6H). UPLC-MS (ES+, Short acidic): 1.64 min, m/z 223.0 [M+H]+ (100%).
[0405] Step 2: 2-hydroxy-5-tetrahydropyran-2-yloxy-benzaldehyde (107 g, 481 mmol) was dissolved in diglyme (750 mL) and K2CO3 (133 g, 963 mmol) was added on one portion with stirring to afford a bright yellow suspension. The reaction was then heated to 140°C and tert-butyl acrylate (155 mL, 1059 mmol) in DMF (75 mL) was added over 10 minutes starting at ~110°C and up to 130°C. Maintained this temperature for a further 1 hour. UPLC-MS indicated that the
reaction had progressed 75%. After a further hour this showed clean conversion to 85% product and little or no side-products. After another 3 hours UPLC-MS showed 88% product (previous reactions had showed that further heating did not afford more conversion). The dark brown reaction was cooled to room temperature overnight and filtered to remove inorganics. The reaction was suspended in EtOAc (2.5 L) and water (2.5 L) and the phases separated. The aqueous was re-extracted with EtOAc (2.5 L) and the combined organics were washed with brine (2 x 1.5 L) and the organics were reduced in-vacuo. The crude product was then purified on silica (2Kg) loading in a minimum volume of DCM. A gradient of EtOAc in Pet. Ether (10 – 25%) was run and clean product fractions combined and reduced in-vacuo to afford tert-butyl 6-tetrahydropyran-2-yloxy-2H-chromene-3-carboxylate (93.5 g, 281 mmol, 58% yield) as a yellow solid. ¾ NMR (400 MHz, DMSO-d6) d/ppm: 7.37 (q, J = 1.2 Hz, 1H), 7.05 (d, J = 2.9 Hz, 1H), 6.94 (dd, J = 8.8, 2.9 Hz, 1H), 6.79 (dd, J = 8.7, 0.7 Hz, 1H), 5.35 (t, J = 3.3 Hz, 1H), 4.82 (d, J = 1.4 Hz, 2H), 3.77 (ddt, J = 13.3, 8.3, 4.2 Hz, 1H), 3.59 – 3.48 (m, 1H), 1.93 – 1.49 (m, 6H), 1.49 (s, 9H). UPLC-MS (ES+, Short acidic): 2.18 min, m/z ([M+H]+) not detected (100%).
[0406] Step 3: tert-butyl 6-tetrahydropyran-2-yloxy-2H-chromene-3-carboxylate (215 g, 647 mmol) was suspended in MeOH (1.6 L) at room temperature (did not dissolve immediately) and pyridinium p-toluenesulfonate (16.3 g, 64.7 mmol) added. The reaction was warmed to 40°C with a hot water bath and checked by UPLC-MS for progress after 1 hour which indicated the reaction was complete and was a clear orange solution. The reaction was reduced in-vacuo and the crude product dissolved in DCM (2 L) and washed with water (1 L). The organic layer was dried (MgSC>4), filtered and reduced in-vacuo to afford the crude product as a yellow solid. This was suspended in Pet. Ether and stirred in an ice bath before filtering, to afford a bright yellow solid. This was dried under high vac at 50°C for 2 hours to afford tert-butyl 6-hydroxy-2H-chromene-3-carboxylate (144.4 g, 582 mmol, 90% yield). ¾ NMR (400 MHz, DMSO-d6) d/ppm: 9.17 (s, 1H), 7.33 (s, 1H), 6.76 – 6.64 (m, 3H), 4.77 (d, J = 1.4 Hz, 2H), 1.49 (s, 9H). UPLC-MS (ES+, Short acidic): 1.71 min, m/z 247.2 [M-H]- (100%).
[0407] Step 4: tert-Butyl 6-hydroxy-2H-chromene-3-carboxylate (84. g, 338.34mmol) was dissolved in DCM (500mL) and trifluoroacetic acid (177.72mL, 2320.9mmol) added at room temperature and the reaction stirred to give a brown solution. Initially gas evolution was noted and the reaction was stirred over several days at room temperature. DCM and TFA were removed in-vacuo and finally azeotroped with 200ml of toluene before slurrying with diethyl ether and filtering to give the crude product 6-hydroxy-2H-chromene-3-carboxylic acid (53.15g, 276.58mmol, 81.745% yield) as a cream solid. ¾ NMR (400 MHz, DMSO-d6) d/ppm: 12.77 (s, 1H), 9.14 (s, 1H), 7.37 (t, J = 1.4 Hz, 1H), 6.72 (dd, J = 2.4, 0.9 Hz, 1H), 6.70 – 6.64 (m, 2H), 4.78 (d, J = 1.4 Hz, 2H).
[0408] Step 5: (R)-Phanephos and [RuCl2(p-cym)]2 (1.2: 1 eq., 6.6 mg, 3.0 mg respectively) were weighed into a 50 mL glass lined Parr vessel followed by the substrate (1.845 g, 9.6 mmol). Methanol (16 mL, 0.6 M substrate concentration) was added to the vessel followed by triethylamine (135 μL, 0.96 mmol, 0.1 eq.). A PTFE stirrer bar was added and the thermocouple was covered with PTFE tape. The vessel was sealed and purged with nitrogen 5 times (at ~2 bar) and 5 times with stirring (~500 rpm). The vessel was then purged with hydrogen 5 times (at -10 bar) and 5 times with stirring (~500 rpm). The vessel was then pressurised to 5 bar hydrogen pressure and heated to 40 °C (with 1500 rpm stirring speed). The pressure was kept constant but with venting and refilling to 5 bar after sampling. After 21.5 hours, the vessel was allowed to cool. After 22.5 hours, the vessel was vented and purged with nitrogen. Each -0.1 mL sample was diluted to -1 mL with MeOH for SFC analysis. Work-up procedure: MeOH removed by concentrating under vacuum, followed by addition of EtOAc (10 mL) and 1 M HC1 (10 mL). The layers were mixed before separating. The EtOAc layer was washed with a further portion of 1 M HC1 (4 mL) before removing the aqueous layer to leave the EtOAc organic phase. The aqueous layer was then washed with a further portion of EtOAc (4 mL) and the organic layers were combined. EtOAc was then removed under vacuum to leave behind the product as a greyish solid (See Table 29). P2 is the first eluting product with a retention time of 5.8 min and PI is the second eluting product with a retention time of 6.1 min using the SFC method as described in Example 1.
[0409] B. Synthesis of 5-fluoro-3,4-dihydro-l,8-naphthyridin-2(lH)-one
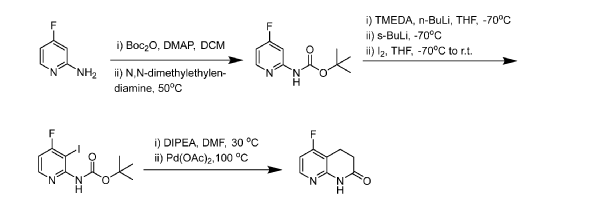
0410] Step 1: 2-Amino-4-fluoropyridine (400 g, 3568 mmol) was charged into a 10 L fixed reactor vessel and then taken up in DCM (4 L) as a slurry under nitrogen atmosphere. To this was added DMAP (43.6 g, 357 mmol) and cooled to 10°C. Di-tert-butyldicarbonate (934 g, 4282 mmol) was added, as a solution in DCM (1 L), over the space of 1.5 hours. The reaction was stirred at room temperature for 2 hours after which time the complete consumption of the starting material was evident by NMR. To the reaction was added N,N-dimethylethylenediamine (390 mL, 3568 mmol) and the reaction warmed to 40°C overnight (converting any di-BOC material back to the mono-BOC desired product). Allowed to cool to room temperature and then diluted with further DCM (2 L) and washed with water (2 L). Extracted with further DCM (2 L), washed with water
(1 L), brine (1.2 L) and dried (MgSO4) before filtering. The solvents were removed in-vacuo and the resultant product was slurried in DCM/Pet. Ether (1:1) (500 mL). Filtered, washed with further Pet. Ether and pulled dry to afford tert- butyl N-(4-fluoro-2-pyridyl)carbamate (505 g, 2380 mmol, 67% yield) as a cream solid product. A second crop of material was isolated from the mother liquors after passing through a short pad of silica followed by trituration with DCM/Pet. Ether (1:1) (-200 mL) to afford tert-butyl N-(4-fluoro-2-pyridyl)carbamate (46.7 g, 220 mmol, 6% yield). ¾ NMR (400 MHz, DMSO-d6) d/ppm: 10.13 (d, J = 1.7 Hz, 1H), 8.26 (dd, J = 9.4, 5.7 Hz, 1H), 7.60 (dd, J = 12.3, 2.4 Hz, 1H), 6.94 (ddd, J = 8.2, 5.7, 2.4 Hz, 1H), 1.47 (s, 9H). UPLC-MS (ES+, Short acidic): 1.64 min, m/z 213.1 [M+H]+ (98%).
[0411] Step 2: tert-butyl-N-(4-fluoro-2-pyridyl)carbamate (126 g, 594 mmol) and TMEDA (223 mL, 1484 mmol) were taken up in dry THF (1.7 L) and then cooled to -78°C under nitrogen atmosphere. To this solution was added n-butyllithium solution (2.5M solution in hexanes) (285 mL, 713 mmol) and then allowed to stir for a further 10 minutes. sec-Butyllithium solution (1.2M in cyclohexane) (509 mL, 713 mmol) was added keeping the reaction temperature below -70°C whilst stirred for 1 hour. After this time, Iodine (226 g, 891 mmol) in THF (300 mL) was added slowly and dropwise over 30 minutes to keep the temp below -65°C. Stirred at -70°C for another 10 minutes and then quenched by the addition of sat. aq. NH4CI solution (400 mL) and then a solution of sodium thiosulphate (134 g, 848 mmol) dissolved in water (600 mL). This addition raised the temperature to — 25°C. The reaction was warmed to room temperature then transferred to the 5L separator and extracted with EtOAc (2 x 1.5 L) and then washed with brine (500 mL), dried (MgSCL) and then evaporated in vacuo to afford crude material (~200g). This was taken up in hot DCM (500 mL) (slurry added to the silica pad) and then passed through a 2Kg silica pad. Washed through with DCM (10 x 1 L fractions) and then the product was eluted from the column with EtOAc in Pet. Ether (10% to 100%), (1 L at each 10% increase, with 1 L fractions). This gave 2 mixed fractions and clean product containing fractions, which were combined and evaporated in vacuo to afford tert-butyl N-(4-fluoro-3-iodo-2-pyridyl)carbamate (113.4 g, 335.4 mmol, 57% yield) as a white solid. Clean by UPLC-MS and NMR. The mixed fractions were combined with previous crude material to afford 190g in total of a cream solid that was composed of -50% of the desired product. This was re-columned as above to afford a combined second crop from all 4 batches as a cream solid tert-butyl N-(4-fluoro-3-iodo-2-pyridyl) carbamate (107.5 g, 318 mmol, 54% yield). ¾ NMR (400 MHz, DMSO-d6) d/ppm: 9.47 (s, 1H), 8.33 (dd, J = 8.7, 5.5 Hz, 1H), 7.19 (dd, J = 7.3, 5.5 Hz, 1H), 1.46 (s, 9H). UPLC-MS (ES+, Short acidic): 1.60 mm, m/z 339.1 [M+H]+ (100%).
[0412] Step 3: tert-butyl N-(4-fluoro-3-iodo-2-pyridyl)carbamate (300 g, 887 mmol), 3,3-dimethoxyprop- 1 -ene (137 mL, 1153 mmol) and DIPEA (325 mL, 1863 mmol) were suspended in DMF (2 L) and water (440 mL) to give a yellow slurry. This was degassed for 20 minutes at 30°C. To this mixture was then added Palladium (II) acetate (19.92 g, 89 mmol) in one portion and degassed again for a further 15mins. The reaction was slowly and carefully heated to 100°C. Gas evolution at around 85°C (large volumes of off gassing, presumably due to the loss of Boc group as CO2 and isobutylene). The reaction became darker once off gassing finished and full solubility achieved. The reaction was then heated at 100°C for 3 hours and checked by UPLC-MS (70% desired product, 18% un-cyclised intermediate and 7% des-iodo BOC). The reaction was heated for a further 2 hours and this showed 81% desired product, 12% un-cyclised intermediate and 8% des-iodo BOC. After 7 hours the reaction showed 89% desired product, 4% un-cyclised
intermediate and 7% des-iodo BOC. The reaction was heated overnight. The reaction solution was cooled and filtered through celite and evaporated in-vacuo to a thick dark orange slurry which was then suspended in water (1 L) and acidified to pH~l-2 with aq. HC1 (4N) solution. This was then basified to pH~9 with sat. aq. Na2CO3 solution. Extracted with DCM (2 x 2L) and washed with brine and dried (MgS04). EtOAc (2 L) was added to the solution and then the organics were passed through a 500g silica plug. This was then followed by DCM/EtOAc (1 : 1) (2 L) and finally EtOAc (2 L) (the final wash through contained only baseline). The product containing fractions were combined and reduced in-vacuo to give an orange slurry and then suspended in hot diethyl ether (300 mL), cooled back to ~10°C in an ice bath with stirring before being filtered and washed with 150 mL of ice cold diethyl ether. Pulled dry to afford 5-fluoro-3,4-dihydro-lH-l,8-naphthyridin- 2-one (58.4 g, 351.5 mmol, 39.6 % yield) as a cream fluffy solid. ¾ NMR (400 MHz, DMSO-d6) d/ppm: 10.69 (s, 1H), 8.29 – 7.90 (m, 1H), 6.92 (dd, J = 8.8, 5.7 Hz, 1H), 2.88 (dd, J= 8.3, 7.1 Hz, 2H), 2.57 – 2.47 (m, 2H). UPLC-MS (ES+, Short acidic): 1.04 mm, m/z 167.0 [M+H]+ (100%).
[0413] C. Synthesis of Compounds A-l and A-2
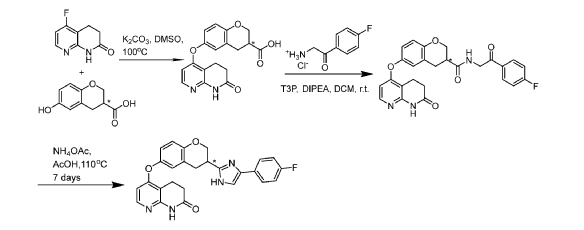
[0414] Step 1: Potassium carbonate (832mg, 6.02mmol) was added to a stirred solution of 5- fluoro-3,4-dihydro-lH-l,8-naphthyridin-2-one (250mg, 1.5mmol), P2 (see step A, 292mg, 1.5mmol; 85% ee) and DMSO (2mL) at room temperature. The reaction was degassed and flushed with nitrogen 3 times before being stirred under a nitrogen atmosphere for 18 hours at 100°C. The reaction mixture was cooled to room temperature and diluted with water (20mL) and the resulting mixture extracted with EtOAc (20mL). A solution of citric acid (1156.3mg, 6.02mmol) in water (lOmL) was then added to the aqueous layer resulting in a solid precipitate which was filtered and dried in vacuo to give (S)- or (R)-6-[(7-oxo-6, 8-dihydro- 5H-1 ,8-naphthyridin-4-yl)oxy]chromane-3 -carboxylic acid (345mg, 1.01 mmol, 67% yield) as a white solid. UPLC-MS (ES+, Short acidic): 1.29 mm, m/z 341.1 [M+H]+. ¾ NMR (400 MHz, DMSO-d6) d/ppm: 12.71 (lH, br s), 10.47 (1H, s), 7.95 (1H, d, J = 6.0Hz), 6.97 (1H, d, J = 2.4Hz), 6.89 (1H, dd, J = 8.4Hz, 2.4Hz), 6.83 (1H, d, J = 8.4Hz), 6.24 (1H, d, J = 6.0Hz), 4.33 (1H, dd, J = 11.2Hz, 3.2Hz), 4.15 (1H, dd, J = 11.2Hz, 7.2Hz), 3.05-2.89 (5H, m), 2.53 (2H, t, J = 7.6Hz).
[0415] Step 2: Propylphosphonic anhydride (0.91mL, 1.52mmol) was added to a stirred solution of (S)-6-[(7-oxo-6,8-dihydro-5H-l,8-naphthyridin-4-yl)oxy]chromane-3-carboxylic acid (345mg, 1.01 mmol), 2-amino- l-(4-fluorophenyl)ethanone hydrochloride (288mg, 1.52mmol), N,N-diisopropylethylamine (0.88mL, 5.07mmol) andDCM (lOmL) at room temperature. After stirring for 2 hours the reaction was complete by LCMS. Water (50mL) and DCM (50mL) were added and the organic layer separated and washed with sat. aq. Na2CO3 (50mL). The organic layer was dried over sodium sulfate and solvent removed in vacuo. The residue was purified by column chromatography using an eluent of 0-5% MeOH in DCM to give (S)- or (R)-N-[2-(4-fluorophenyl)-2-oxo-ethyl]-6-[(7-oxo-6,8-dihydro-5H-l,8-naphthyridin-4-yl)oxy]chromane-3-carboxamide (300mg, 0.63mmol, 62% yield) as a yellow solid. UPLC-MS (ES+, Short acidic): 1.52 mm, m/z 476.4 [M+H]+. ¾ NMR (400 MHz, DMSO-d6) d/ppm: 10.47 (1H, s), 8.60-8.54 (1H, m), 8.08 (1H, dd, J = 8.8Hz, 5.6Hz), 7.95 (1H, d, J = 5.6Hz), 7.41-7.37 (2H, m), 7.01-6.97 (1H, m), 6.90 (1H, dd, J = 8.8Hz, 3.2Hz), 6.86 (1H, d, J = 8.8Hz), 6.25 (1H, d, J = 5.6Hz), 4.65 (2H, d, J = 6.0Hz), 4.42-4.35 (1H, m), 3.96 (1H, t, J = 9.6Hz), 3.03-2.87 (5H, m), 2.55-2.52 (2H, m), 1 exchangeable proton not seen.
[0416] Step 3: (S)- or (R)-N-[2-(4-fluorophenyl)-2-oxo-ethyl]-6-[(7-oxo-6, 8-dihydro- 5H-1, 8-naphthyridin-4-yl)oxy]chromane-3 -carboxamide (300mg, 0.63mmol), ammonium acetate
(1216mg, 15.77mmol) and acetic acid (5mL) were combined in a sealable vial, the vial sealed and the reaction stirred and heated to 130°C for 18 hours after which time the reaction was complete by LCMS. The reaction was cooled to room temperature and AcOH removed in vacuo. DCM (50mL) was added to the residue and sat. aq. Na2CO3 (50mL) added. The organic layer was separated and washed with brine, dried over sodium sulfate and solvent removed in vacuo. The residue was purified by column chromatography using an eluent of 0-10% MeOH in DCM to give (R)- or (S)-5 – [3 – [4-(4-fluorophenyl)- 1 H-imidazol-2-y 1] chroman-6-yl] oxy-3 ,4-dihydro- 1 H- 1 , 8-naphthyridin-2-one (141mg, 0.31mmol, 49% yield) as a yellow solid.
[0417] Chiral LCMS of the product, together with chiral LCMS’s of Compounds A-l and A-2 showed that this product is predominantly Compounds A-l (Fig. 7), with a similar ee to that of the starting acid (85% ee), however accurate analysis cannot be done due to overlap of the peaks. UPLC-MS (ES+, Short acidic): 1.36 mm, m/z 457.2 [M+H]+. Ή NMR (400 MHz, DMSO-d6) d/ppm: 12.31 (0.2H, s), 12.10 (0.8H, s), 10.47 (1H, s), 7.96 (1H, d, J = 6.0Hz), 7.80-7.75 (1.8H, m), 7.69-7.65 (0.2H, m), 7.59-7.78 (0.8H, m), 7.29-7.23 (0.4H, m), 7.19-7.13 (1.8H, m), 7.03-7.00 (1H, m), 6.92 (1H, dd, J = 8.8Hz, 2.8Hz), 6.89 (1H, d, J = 8.8Hz), 6.27 (1H, d, J = 6.0Hz), 4.55-4.48 (1H, m), 4.16-4.09 (1H, m), 3.44-3.36 (1H, m), 3.30-3.21 (1H, m), 3.16-3.09 (1H, m), 2.94 (2H, t, J = 7.2Hz), 2.54 (2H, t, J = 7.2Hz).
[0439] A. Synthesis of P2
[0440] Step 1: 2,5-Dihydroxybenzaldehyde (13.6 kg, 98.18 mol) was dried using 2 x azeotropic concentrations with 2 x 125-130 kg of THF at up to 35 °C, concentrating under vacuum to 27-41 kg each time. The THF was then removed using 4 x azeotropic concentrations with 4 x 179-187 kg of DCM at up to 35 °C, concentrating under vacuum to 27-41 kg each time. The concentrate was diluted with DCM (284 kg) and pyridine p-toluenesulfonate (PPTS; 1.25 kg, 4.97 mol) was added. 3,4-dihydro-2H-pyran (10.4 kg, 123.63 mol) was added slowly at between 25-35 °C and the reaction was stirred at 30 °C for 90 minutes. The mixture was added to a solution of Na2CO3 (7.1 kg) in water (138 kg) at -15 °C and allowed to warm to 25 °C and then stirred for 6 h. The mixture was filtered through Celite® (33 kg), washing with DCM (92.5 kg). The filtrate was allowed to stand for 1 h and then the organic phase was separated and concentrated to 27-41 kg.
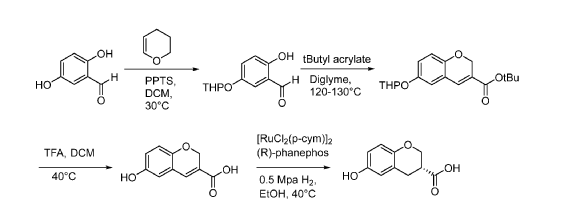
The DCM was then removed using 3 x azeotropic concentrations with 3 x 105 kg n-heptane at up to 35 °C, concentrating under vacuum to 27-41 kg each time. The concentrate was diluted with n- heptane (210 kg) and the heated to 30-40 °C and stirred for 6 h. The solution was then cooled to – 5 to -15 °C over 4 h, stirred for 9 h and filtered, washing the filter cake with n-heptane (39.5 kg).
The wet cake was dried at 30-40 °C for 24 h in vacuo to give 2-hydroxy-5-(oxan-2- yloxy)benzaldehyde (9.38 kg, 40.6%). Additional product (8.00 kg, 34.3%) was recovered by dissolving solid attached to the walls of the reaction vessel with 42 kg DCM and concentrating the resultant solution in vacuo to give a further 8.00 kg (34.3% yield ) of product to give a total yield of 74.9% (17.38 kg). LCMS (ES-): 15.18 mm, m/z 221.12 [M-H]-.
[0441] Step 2: To a stirring solution of 2-hydroxy-5-(oxan-2-yloxy)benzaldehyde (16.95 kg, 76.27 mol) in diglyme (113.4 kg) was added K2CO3 (21.4 kg, 154.83 mol) and the mixture was heated to between 80-90 °C. Tert-butyl prop-2-enoate (20.0 kg, 156.04 mol) was added, and the mixture was heated to between 120-130 °C and stirred for 18 hr. The mixture was cooled and
filtered, and the filter cake washed with EtOAc (80.0 kg). The filtrate was diluted with EtOAc (238.0 kg) and water (338.0 kg) and stirred for 1 hr at 20-30 °C, then stood for 2 hr. The mixture was filtered through Celite® (40.0 kg), and the filter cake washed with EtOAc (84.0 kg). The filtrate was left to stand for 2 hr and the aqueous layer was extracted with EtOAc (312.0 kg), stirring for 1 hr at 0-30 °C and standing for 2 hr. The organic layers were combined and washed with 2 x 345 kg water, stirring at between 20-30 °C for 1 hr and standing for 2 hr for each wash. The combined organics were then concentrated to 182.4 kg maintaining the temperature below 50 °C under vacuum. This gave the product tert-butyl 6-(oxan-2-yloxy)-2H-chromene-3-carboxylate as a 9.3% solution in diglyme/EtOAc (66.9% yield) and was used in the next stage without further isolation. LCMS (ES-): 20.26 mm, m/z 247.12 [M-THP]-.
[0442] Step 3: Tert-butyl 6-(oxan-2-yloxy)-2H-chromene-3-carboxylate (16.9 kg, 50.84 mol) as a 181.8 kg solution in diglyme/EtOAc was concentrated to 68 kg under vacuum at 50 °C. TFA (110.3 kg, 1002.46 mol) was added and the reaction was warmed to 40 °C under nitrogen flow and then stirred for 8 hrs. The mixture was then diluted with DCM (222.0 kg) and cooled to between -5 and -15 °C, and then stirred for 7 hrs. The solid was filtered and the filter cake washed with DCM (67.0 kg). The wet cake was dried for 24 hr under vacuum at between 30-40 °C to give 6-hydroxy-2H-chromene-3-carboxylic acid (8.75 kg, 78.5% yield). LCMS (ES-): 0.85 min, m/z 191.11 [M-H]-.
[0443] Step 4: To a stirring solution of 6-hydroxy-2H-chromene-3-carboxylic acid (7.19 kg, 37.4 mol) in N2-degassed EtOH (60 kg) was added (R)-Phanephos (131 g, 0.227 mol), [RuCl2(p-cym)]2 (70 g, 0.114 mol), and Et3N (5.6 kg, 55.3 mol). The reaction atmosphere was replaced with 3 x N2 and then 3 x H2, adjusting the H2 pressure to between 0.5-0.6 MPa, and then stirred for 18 hrs at 40 °C. The atmosphere was then replaced with 3 x N2 and then 3 x H2, adjusting the H2 pressure to between 0.5-0.6 MPa again and the mixture was stirred for a further 18 hrs.
[0444] The mixture was concentrated in vacuo to ca. 30 kg at no more than 40 °C. The reaction was diluted with MTBE (53 kg) and cooled to between 15-25 °C. 5% Na2CO3 (80 kg) was added dropwise, and the mixture was stirred for 2 hrs and stood for 2 hrs at between 15-25 °C. The aqueous layer was collected and 5% Na2CO3 (48 kg) was added to the organic layer, then stirred for 2 hrs at 15-25 °C and filtered through Celite® (10.0 kg). The wet cake was washed with water (20 kg) and the combined aqueous filtrate and aqueous layer were diluted with IP Ac (129.0 kg). The pH of the mixture was adjusted to 1-3 with dropwise addition of 6 N HC1 (29 kg) at 15-25 °C and stirred for 2 hrs. The mixture was filtered through Celite® (10 kg), washing the filter cake with IP Ac (34 kg) and the filtrate was left to stand for 2 hrs at 15-25 °C. The aqueous layer was then extracted with IP Ac (34 kg) and the combined organic layers were concentrated to ca. 35 kg under vacuum at no more than 40 °C. Me-cyclohexane (21 kg) was added dropwise at 15-25 °C and concentrated to ca. 35 kg under vacuum at no more than 40 °C. Further Me-cyclohexane (20 kg) was added dropwise at 15-25 °C and stirred for 3 hrs. The mixture was then stirred at 40-50 °C for 4 hrs and cooled to 15-25 °C over 3 hrs and then stirred for a further 2 hrs.
[0445] The mixture was then filtered, washing the filter cake with 16.4 kg of IPAc/Me-cyclohexane (1/4, v/v). The wet cake was dried for 24 hrs at 35-45 °C under vacuum to give (3R)-6-hydroxy-3,4-dihydro-2H-l-benzopyran-3-carboxylic acid (5.2 kg, 68.6% yield, chiral purity 95.5%). Further product was isolated by rinsing solid from the reaction vessel wall with EtOH (42 kg) and concentrating to dryness. The resulting solid was suspended in IP Ac (875mL) and Me-cyclohexane (2625mL) and stirred for 5 h at 40 °C and then cooled to 20 °C over 2 h and stirred for 16 h and filtered. The filter cake was then split into 2 equal batches and each batch suspended in IP Ac (912mL) and Me-cyclohexane (2737mL). The resulting mixtures were stirred at 45 °C for 18 h and then filtered and the filter cake dried at 45 °C to give (3R)-6-hydroxy-3,4-dihydro-2H-l-benzopyran-3 -carboxylic acid (1.27 kg, 17% yield, chiral purity 96.2%). LCMS (ES-): 1.74 min, m/z 193.03 [M-H]-.
[0446] Chiral resolution to improve chiral purity:
[0447] (3R)-6-Hydroxy-3,4-dihydro-2H-l-benzopyran-3-carboxylicacid (P2; 5.94 kg, 30.59 mol) (chiral purity =95.5%) was dissolved in IP Ac (138.2 kg) and stirred for 2 hrs at 20-30 °C. The solution obtained was filtered through Celite® (12 kg), washing through with IP Ac (25 kg). In a separate vessel, (S)-(+)-2-phenylglycinol (4.4 kg, 32.07 mol) was dissolved in IP Ac (56 kg), stirring for 1 hr at 40-50 °C. The filtrate was added to this solution over 4 hrs at 40-50 °C, and stirred for 1 hr. The mixture was then stirred for 1 hr at 15-25 °C, and concentrated to ca. 120 kg under vacuum at no more than 40 °C. The concentrate was stirred for 3 hrs at 15-25 °C and filtered, washing through with IP Ac (12 kg) (chiral purity = 96.2%).
[0448] The wet cake was redissolved in EtOH (29 kg), heated to 40-50 °C and diluted with IP Ac (64 kg). 30 g of dry product was added and stirred for 30 min at 15-25 °C. The mixture was concentrated to ca. 42 kg under vacuum at no more than 40 °C, and rediluted with IP Ac (64 kg). This step was repeated two additional times, then stirred at 40-50 °C for 8 hrs. The mixture was filtered, washing through with IP Ac (13 kg) (chiral purity = 97.7%). This recrystallisation process was repeated two further times, for a total of 3 recrystallisation rounds to give material with 98.9% chiral purity.
[0449] The wet cake (10.7 kg) was then dissolved in IN HC1 (45.4 kg) and stirred for 1 hr at 20-30 °C. The mixture was filtered through Celite® (11.5 kg), washing through with IP Ac (28 kg). The aqueous layer was extracted with IP Ac (28.8 kg) and the combined organic layers were washed with water (30 kg), then concentrated to ca. 24 kg at 40 °C under vacuum. Me-cyclohexane (19 kg) was added at 20 °C and the mixture was concentrated to ca. 24 kg at 40 °C under vacuum. This step was repeated twice more. The concentrate was diluted with Me-cyclohexane (29 kg) and stirred for 1 hr at 15-25 °C. The mixture was filtered, and the wet cake was rinsed with Me-Cyclohexane (59 kg). The wet cake was dried under vacuum at 35-45 °C for 16 hrs to give (3R)-6-hydroxy-3,4-dihydro-2H-l-benzopyran-3-carboxylic acid (3.02 kg, 50.2% yield).
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US350349340&_cid=P11-MGN37Z-55206-1
PAT
- Methods of treating solid tumors with mitogen activated protein kinase (mapk) pathway alterationsPublication Number: WO-2024173931-A2Priority Date: 2023-02-17
- Crystalline forms of (s)-5-((3-(4-(4-fluorophenyl)-1h-imidazol-2-yl)chroman-6-yl)oxy)-3,4-dihydro-1,8-naphthyridin-2(1h)-onePublication Number: TW-202421110-APriority Date: 2022-11-29
- Crystalline forms of (s)-5-((3-(4-(4-fluorophenyl)-1h-imidazol-2-yl)chroman-6-yl)oxy)-3,4-dihydro-1,8-naphthyridin-2(1h)-onePublication Number: WO-2024115583-A1Priority Date: 2022-11-29
- Chiral synthesis of fused bicyclic raf inhibitorsPublication Number: US-2022041595-A1Priority Date: 2020-07-28
- Chiral synthesis of fused bicyclic raf inhibitorsPublication Number: WO-2022023450-A1Priority Date: 2020-07-28
- Chiral synthesis of fused bicyclic raf inhibitorsPublication Number: EP-4188923-A1Priority Date: 2020-07-28
- Chiral synthesis of fused bicyclic RAF inhibitorsPublication Number: KR-20230058630-APriority Date: 2020-07-28
- Chiral Synthesis of Fused Bicyclic RAF InhibitorsPublication Number: CN-116348465-APriority Date: 2020-07-28
- Chiral Synthesis of Fused Bicyclic RAF InhibitorsPublication Number: JP-2023535595-APriority Date: 2020-07-28
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | JZP-815 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2760321-00-2 |
| PubChem CID | 162772363 |
| IUPHAR/BPS | 13233 |
| UNII | P26TTM6U27 |
| KEGG | D13132 |
| Chemical and physical data | |
| Formula | C26H21FN4O3 |
| Molar mass | 456.477 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- “JZP-815”. PatSnap.
- Riaud M, Maxwell J, Soria-Bretones I, Dankner M, Li M, Rose AA (February 2024). “The role of CRAF in cancer progression: from molecular mechanisms to precision therapies”. Nature Reviews. Cancer. 24 (2): 105–122. doi:10.1038/s41568-023-00650-x. PMID 38195917.
///////////flezurafenib, JZP-815, JZP 815, P26TTM6U27, ANTINEOPLASTIC, CANCER

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}