
Home » Posts tagged 'Anti-inflammatory'
Tag Archives: Anti-inflammatory
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Navepdekinra
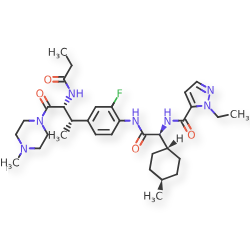
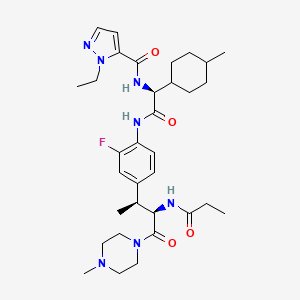
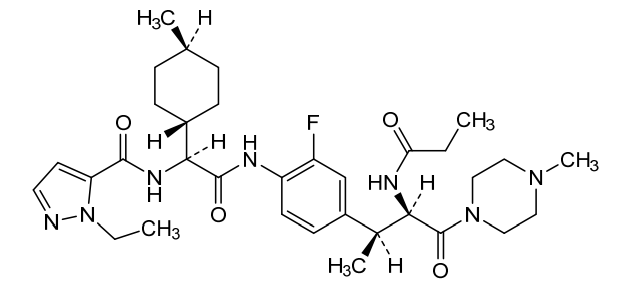
Navepdekinra
CAS 2467732-66-5
MF C33H48FN7O4 MW625.78
1H-Pyrazole-5-carboxamide, 1-ethyl-N-[(1S)-2-[[2-fluoro-4-[(1S,2R)-1-methyl-3-(4-methyl-1-piperazinyl)-3-oxo-2-[(1-oxopropyl)amino]propyl]phenyl]amino]-1-(trans-4-methylcyclohexyl)-2-oxoethyl]-
1-ethyl-N-{(1S)-2-{2-fluoro-4-[(2S,3R)-4-(4-methylpiperazin-1-yl)-4-oxo-3-propanamidobutan-2-yl]anilino}-1-[(1r,4S)-4-methylcyclohexyl]-2-oxoethyl}-1H-pyrazole-5-carboxamide
1-ethyl-N-{(1S)-2-{2-fluoro-4-[(2S,3R)-4-(4-methylpiperazin-1-yl)-4-oxo-3-propanamidobutan-2-
yl]anilino}-1-[(1r,4S)-4-methylcyclohexyl]-2-oxoethyl}-1H-pyrazole-5-carboxamide
interleukin-17A (IL-17A) inhibitor, anti-inflammatory, DC-806, LY4100504, DC 806, LY 4100504, Y64F9MC2QM
Navepdekinra (also known as DC-806 or LY4100504) is an experimental, orally active small-molecule inhibitor of interleukin-17A (IL-17A). It was primarily developed to treat autoimmune and inflammatory conditions, such as psoriasis, by disrupting the interaction between IL-17A and its receptor.
Key Properties and Development
- Mechanism: It is a potent inhibitor with an IC50 of 10.81 nM, designed to provide an oral alternative to existing injectable IL-17 biologic therapies.
- Acquisition: The drug was originally developed by DICE Therapeutics, which was acquired by Eli Lilly and Company in 2023 for approximately $2.4 billion to bolster their immunology pipeline.
Navepdekinra (DC-806) is an orally active, potent interleukin-17A (IL-17A) inhibitor (IC50 = 10.81 nM). Navepdekinra disrupts the IL-17A protein-receptor interaction, suppressing the downstream pro-inflammatory signaling pathway. Navepdekinra inhibits arthritis in a collage-induced arthritis (CIA) rat model. Navepdekinra can be used for psoriasis, psoriatic arthritis, and ankylosing spondylitis
SYN
Example 210: N-[(2R,3S)-3-{4-[(2S)-2-[(1-ethyl-1H-pyrazol-5-yl)formamido]-2-[(1r,4S)-4-methylcyclo hexyl]acetamido]-3-fluorophenyl}-1-(4-methylpiperazin-1-yl)-1-oxobutan-2-yl]propanamide) (234)
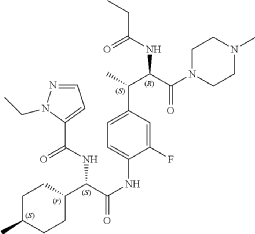
Following General Procedure R, 0.227 g, 0.310 mmol, 1.0 eq) of 82d in DMF (1 mL) were added 1-ethyl-1H-pyrazole-5-carboxylic acid (0.052 g, 0.372 mmol, 1.2 eq), DIPEA (0.43 mL, 2.482 mmol, 8.0 eq) and then HATU (0.177 g, 0.465 mmol, 1.5 eq.) and the resulting mixture was stirred at RT for 1 h. The mixture was concentrated to dryness and the residue was purified via reverse phase column chromatography on a 120 g C18 cartridge eluting with a 5-95% H 2O:MeCN eluent (0.1% ammonia) to afford 234 (0.025 g) as a white solid. 1H NMR (400 MHz, DMSO-d 6) δ 9.86 (s, 1H), 8.46 (d, J=8.3 Hz, 1H), 8.26 (d, J=8.7 Hz, 1H), 7.75 (t, J=8.3 Hz, 1H), 7.47 (d, J=2.1 Hz, 1H), 7.15-7.07 (m, 1H), 7.05-6.97 (m, 2H), 4.86 (t, J=9.4 Hz, 1H), 4.53 (t, J=8.4 Hz, 1H), 4.47 (q, J=7.2 Hz, 2H), 3.46-3.38 (m, 2H), 3.29-3.14 (m, 2H), 3.12-2.99 (m, 2H), 2.25-2.03 (m, 5H), 1.98 (s, 3H), 1.81 (ddt, J=15.0, 9.9, 5.6 Hz, 2H), 1.74-1.60 (m, 4H), 1.58-1.47 (m, 1H), 1.28 (t, J=7.1 Hz, 4H), 1.20 (d, J=7.0 Hz, 3H), 1.14-1.02 (m, 1H), 0.99 (t, J=7.6 Hz, 3H), 0.93-0.87 (m, 1H), 0.86 (d, J=6.5 Hz, 3H). UPLC-MS (basic 4 min): rt=1.76 min; m/z=626.4 for [M+H] +.
PAT
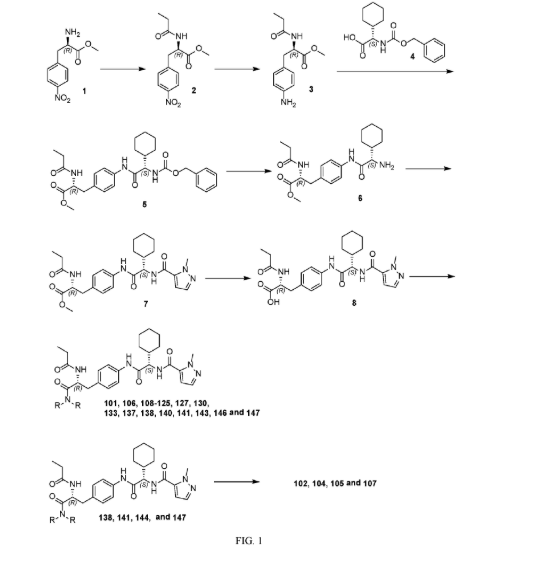
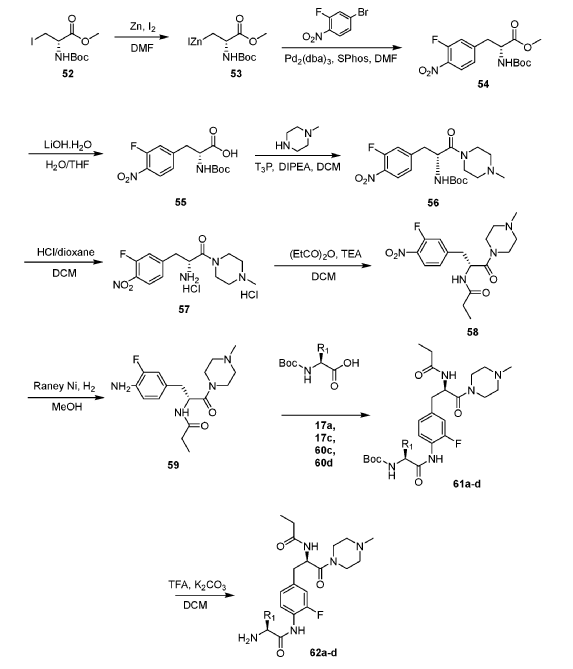
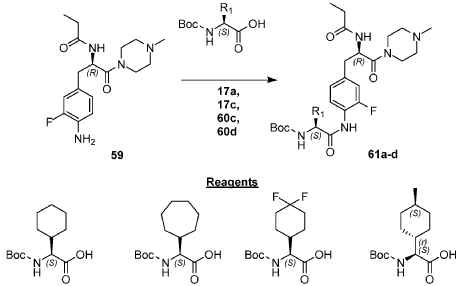
Example 1: Exemplary Scheme—Synthesis of Intermediate Compounds 62a-62d
PAT
IL-17 Ligands And Uses Thereof
Publication Number: US-2020247785-A1
Priority Date: 2019-02-06
- Substituted benzenecarboxamides as IL-17A modulatorsPublication Number: US-11274094-B2Priority Date: 2019-09-16Grant Date: 2022-03-15
- Il-17a modulators and uses thereofPublication Number: US-2021101886-A1Priority Date: 2019-09-16
- IL-17 ligands and uses thereofPublication Number: US-11447468-B2Priority Date: 2019-02-06Grant Date: 2022-09-20
- Il-17 ligands and uses thereofPublication Number: US-2023053746-A1Priority Date: 2019-02-06
- IL-17 ligands and uses thereofPublication Number: US-12234225-B2Priority Date: 2019-02-06Grant Date: 2025-02-25
- Mannose 6-phosphate or asgpr receptor binding compounds for the degradation of extracellular proteinsPublication Number: WO-2023028338-A2Priority Date: 2021-08-27
- Potent asgpr-binding compounds for the degradation of immunoglobulins and other proteinsPublication Number: WO-2022235699-A2Priority Date: 2021-05-03
- Il-17a modulators and uses thereofPublication Number: WO-2021055376-A1Priority Date: 2019-09-16
- Substituted benzenecarboxamides as il-17a modulatorsPublication Number: US-2023141212-A1Priority Date: 2019-09-16
- Substituted benzenecarboxamides as il-17a modulatorsPublication Number: US-2023145481-A1Priority Date: 2019-09-16



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

- [1]. Paul R. Fatheree, et al. IL-17 Ligands And Uses Thereof. US20200247785A1.[2]. Kim D, et al. Next-Generation Anti-IL-17 Agents for Psoriatic Disease: A Pipeline Review. Am J Clin Dermatol. 2025 May;26(3):307-320. [Content Brief][3]. Xiaobing Deng, et al. The Critical and Unexpected Role of a Methyl Group in Interleukin-17A Inhibitors. bioRxiv 2025.10.02.680113
//////////navepdekinra, interleukin-17A (IL-17A) inhibitor, anti-inflammatory, DC-806, LY4100504, DC 806, LY 4100504, Y64F9MC2QM
Glasmacinal



Glasmacinal
CAS 2097822-02-9
MF C37H62N2O10 MW694.90
[(2S,3R,4S,6R)-4-(dimethylamino)-2-[[(2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-2-ethyl-3,4,10,13-tetrahydroxy-3,5,6,8,10,12,14-heptamethyl-15-oxo-1-oxa-6-azacyclopentadec-11-yl]oxy]-6-methyloxan-3-yl] benzoate
- (2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-11-[[2-O-Benzoyl-3,4,6-trideoxy-3-(dimethylamino)-beta-D-xylo-hexopyranosyl]oxy]-2-ethyl-3,4,10,13-tetrahydroxy-3,5,6,8,10,12,14-heptamethyl-1-oxa-6-azacyclopentadecan-15-one
- 1-Oxa-6-azacyclopentadecan-15-one, 11-[[2-O-benzoyl-3,4,6-trideoxy-3-(dimethylamino)-beta-D-xylo-hexopyranosyl]oxy]-2-ethyl-3,4,10,13-tetrahydroxy-3,5,6,8,10,12,14-heptamethyl-, (2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-
(2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-11-{[2-O-benzoyl-3,4,6-trideoxy-3-(dimethylamino) -β-D-xylo-hexopyranosyl]oxy}-2-ethyl3,4,10,13-tetrahydroxy-3,5,6,8,10,12,14-heptamethyl-1-oxa-6-
azacyclopentadecan-15-one
non-antibacterial macrolide, anti-inflammatory, EP 395, M3T8D3P634
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US234729681&_cid=P12-MKVZ26-57135-1
Example 2: (2S,3R,4S,6R)-4-(dimethylamino)-2-[[(2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-2-ethyl-3,4,10,13-tetrahydroxy-3,5,6,8,10,12,14-heptamethyl-15-oxo-1-oxa-6-azacyclopentadec-11-yl]oxy]-6-methyl-tetrahydropyran-3-yl] benzoate)

To a mixture of (2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-11-[(2S,3R,4S,6R)-4-(dimethylamino)-3-hydroxy-6-methyl-tetrahydropyran-2-yl]oxy-2-ethyl-3,4,10,13-tetrahydroxy-3,5,6,8,10,12,14-heptamethyl-1-oxa-6-azacyclopentadecan-15-one (Example 1) (0.5 g, 0.8500 mmol) and Triethylamine (428.2 mg, 4.23 mmol) in DCM (5 ml), cooled on ice, was added Benzoyl chloride (356.9 mg, 2.54 mmol). The reaction mixture was allowed to reach room temperature. After 3 days good conversion to the desired benzoylated product was obtained and the mixture was portioned between DCM and saturated sodium hydrogen carbonate solution. The organic phase was dried over magnesium sulphate and concentrated to a white foam. The product was purified using reversed phase chromatography (see general information)
PAT
- Azithromycin Derivatives With Epithelial Barrier Enhancement PropertiesPublication Number: US-2018354981-A1Priority Date: 2015-11-19
- Azithromycin derivatives with epithelial barrier enhancement propertiesPublication Number: US-10723752-B2Priority Date: 2015-11-19Grant Date: 2020-07-28
- Azithromycin Derivatives With Epithelial Barrier Enhancement PropertiesPublication Number: US-2020317710-A1Priority Date: 2015-11-19
- Azithromycin derivatives with epithelial barrier enhancement propertiesPublication Number: US-12049477-B2Priority Date: 2015-11-19Grant Date: 2024-07-30
- Azithromycin derivatives with epithelial barrier enhancement propertiesPublication Number: US-11236120-B2Priority Date: 2015-11-19Grant Date: 2022-02-01
- Compounds
- Publication Number: US-2022106349-A1
- Priority Date: 2015-11-19
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
ADVERTISEMENT
ANAX LABORATORIES, WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
ADVERTISEMENT
Advect Process Systems Ltd. https://advectprocess.com

ADVECT PROCESS SYSTEMS CANADA LTD
51 Beechwood Rd., Cambridge, ON Canada N1S 3S1, Call Now +1 306 850 6737, Mail Now, ask@advectprocess.com
///////glasmacinal, ANAX, ADVECT, non-antibacterial macrolide, anti-inflammatory, EP 395, M3T8D3P634
Cenacitinib



Cenacitinib
CAS 2641636-52-2
MF C19H19F2N7O3 MW431.4
Urea, N-[(1R,2S)-2-fluorocyclopropyl]-N′-[5-[(7-fluoro-2,3-dihydro-1,4-benzodioxin-5-yl)amino]-7-(methylamino)pyrazolo[1,5-a]pyrimidin-3-yl]-
N-{5-[(7-fluoro-2,3-dihydro-1,4-benzodioxin-5-yl)amino]-7-(methylamino)pyrazolo[1,5-a]pyrimidin-3-yl}-N′-[(1R,2S)-2-fluorocyclopropyl]urea
Janus kinase inhibitor, anti-inflammatory, VTX958, VTX 958, SB88R8KGL3
VTX958 for the Treatment of Moderately to Severely Active Crohn’s Disease
CTID: NCT05688852
Phase: Phase 2
Status: Terminated
Date: 2025-07-03
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US323750705&_cid=P22-MKEUDK-45432-1
Example 4: Synthesis of 1-(5-((7-fluoro-2,3-dihydrobenzo[b][1,4]dioxin-5-yl)amino)-7-(methylamino)pyrazolo[1,5-a]pyrimidin-3-yl)-3-((1R,2S)-2-fluorocyclopropyl)urea (5)


| Step 1: To a solution of 1E (100 g, 288 mmol) and 2E (57 g, 345 mmol) in dry 1,4-dioxane (3000 mL) under N 2 atmosphere was added Cs 2CO 3 (141 g, 432 mmol), Pd(OAc) 2 (5.2 g, 23.3 mmol) and BINAP (28.6 g, 46.6 mmol). After stirring at 115° C. overnight, the reaction mixture was cooled to rt. and diluted with hexane (3000 mL). The solid was collected by filtration and washed with 2×1500 mL (50% hexane in DCM). The solid was suspended into 5000 mL water and stirred for 1 h. The solid was collected by filtration and dried under vacuum to afford compound 2 (90 g, 65%) as a brown solid. |
PAT
Publication Number: US-2021139486-A1
Priority Date: 2019-11-08
- Tyk2 pseudokinase ligandsPublication Number: EP-4054581-A1Priority Date: 2019-11-08
- Tyk2 pseudokinase ligandsPublication Number: US-2023348478-A1Priority Date: 2019-11-08
- Substituted pyrazolo[1,5-a]pyrimidines as TYK2 pseudokinase ligandsPublication Number: US-11753411-B2Priority Date: 2019-11-08Grant Date: 2023-09-12
- TYK2 pseudokinase ligandsPublication Number: CN-114929226-BPriority Date: 2019-11-08Grant Date: 2024-09-27
- TYK2 pseudokinase ligandPublication Number: CN-114929226-APriority Date: 2019-11-08
- Preparation of a tyk2 inhibitorPublication Number: WO-2024151992-A1Priority Date: 2023-01-13
- Crystalline forms of a tyk2 inhibitorPublication Number: US-2024010654-A1Priority Date: 2022-07-06
- Crystalline forms of TYK2 inhibitorsPublication Number: CN-119816502-APriority Date: 2022-07-06
- Crystalline forms of a tyk2 inhibitorPublication Number: EP-4551576-A1Priority Date: 2022-07-06
- Crystalline forms of a tyk2 inhibitorPublication Number: WO-2024011136-A1Priority Date: 2022-07-06
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions,Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////cenacitinib, cenacitinib, Janus kinase inhibitor, anti-inflammatory, VTX958, VTX 958, SB88R8KGL3
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
Zemprocitinib


Zemprocitinib
CAS 2417414-44-7
MF C16H19N5O2S MW 345.4 g/mol
N-[3-(3,5,8,10-tetrazatricyclo[7.3.0.02,6]dodeca-1,4,6,8,11-pentaen-3-yl)-1-bicyclo[1.1.1]pentanyl]propane-1-sulfonamide
N-[3-(imidazo[4,5-d]pyrrolo[2,3-b]pyridin-1(6H)-yl)bicyclo[1.1.1]pentan-1-yl]propane-1-sulfonamide
Janus kinase inhibitor, anti-inflammatory, LNK 01001, LG6MM3RP86
Zemprocitinib (also known as LNK01001) is a selective Janus kinase (JAK) 1 inhibitor, a type of small molecule drug being developed for inflammatory and autoimmune conditions like rheumatoid arthritis, atopic dermatitis, and ankylosing spondylitis. It works by blocking the JAK1 enzyme, reducing the inflammatory signals that cause these diseases, and has shown promising results in clinical trials, with development reaching Phase 3.
Key Aspects:
- Drug Class: JAK1 Inhibitor.
- Mechanism: Blocks Janus Kinase 1, a key enzyme in inflammatory pathways.
- Developer: Initially Lynk Pharmaceuticals.
- Potential Uses: Rheumatoid Arthritis, Atopic Dermatitis, Ankylosing Spondylitis, Psoriasis, Alopecia Areata.
- Development Stage: Reached Phase 3 clinical trials for several indications.
- Chemical Info: CAS: 2417414-44-7; Formula: C16H19N5O2S.
In Summary:
Zemprocitinib is an investigational drug targeting inflammation by inhibiting JAK1, with potential to treat various autoimmune disorders, showing strong efficacy in early clinical trials for conditions like rheumatoid arthritis.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US347660217&_cid=P21-MJDP3D-82397-1
Example 1



Step 1. 4-Chloro-1-tosyl-1H-pyrrolo[2,3-b]pyridine (1b)
Step 2. 4-Chloro-5-nitro-1-tosyl-1H-pyrrolo[2,3-b]pyridine (1c)
Step 3. Tert-butyl 3-((5-nitro-1-tosyl-1H-pyrrolo[2,3-b]pyridin-4-yl)amino)bicyclo[1.1.1]pentane-1-carboxylate (Id)
Step 4. Tert-butyl 3-((5-amino-1-tosyl-1H-pyrrolo[2,3-b]pyridin-4-yl)amino)bicyclo[1.1.1]pentane-1-carboxylate (le)
Step 5. Tert-butyl 3-(6-tosylimidazo[4,5-d]pyrrolo[2,3-b]pyridin-1(6H)-yl)bicyclo[1.1.1]pentane-1-carboxylate (1f)
Step 6. 3-(6-Tosylimidazo[4,5-d]pyrrolo[2,3-b]pyridin-1(6LF)-yl)bicyclo[1.1.1]pentane-1-carboxylic acid (1g)
Step 7. Tert-butyl (3-(6-tosylimidazo[4,5-d]pyrrolo[2,3-b]pyridin-1(6LF)-yl)bicyclo[1.1.1]pentan-1-yl)carbamate (1h)
Step 8. Tert-butyl (3-(imidazo[4,5-d]pyrrolo[2,3-b]pyridin-1(6H)-yl)bicyclo[1.1.1]pentan-1-yl)carbamate (1i)
Step 9. 3-(Imidazo[4,5-d]pyrrolo[2,3-b]pyridin-1(6H)-yl)bicyclo[1.1.1]pentan-1-amine 2,2,2-trifluoroacetate (1j)
Step 10. N-(3-(Imidazo[4,5-d]pyrrolo[2,3-b]pyridin-1(6H)-yl)bicyclo[1.1.1]pentan-1-yl)propane-1-sulfonamide (1)
PAT
- Tricyclic janus kinase 1 inhibitors, and compositions and methods thereofPublication Number: ES-2993867-T3Priority Date: 2018-11-01Grant Date: 2025-01-10
- Tricyclic janus kinase 1 inhibitors, and compositions and methods thereofPublication Number: JP-2024147699-APriority Date: 2018-11-01
- Tricyclic janus kinase 1 inhibitors, and compositions and methods thereofPublication Number: EP-3856742-B1Priority Date: 2018-11-01Grant Date: 2024-10-02
- Tricyclic janus kinase 1 inhibitors, and compositions and methods thereofPublication Number: US-2022009927-A1Priority Date: 2018-11-01
- Tricyclic janus kinase 1 inhibitors, and compositions and methods thereofPublication Number: US-2023357247-A1Priority Date: 2018-11-01
- Tricyclic janus kinase 1 inhibitors, and compositions and methods thereofPublication Number: US-2023339950-A1Priority Date: 2018-11-01
- Tricyclic Janus kinase 1 inhibitors, and compositions and methods thereofPublication Number: AU-2019372677-B2Priority Date: 2018-11-01Grant Date: 2024-05-30
- Tricyclic janus kinase 1 inhibitors, and compositions and methods thereofPublication Number: TW-202432555-APriority Date: 2018-11-01
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////Zemprocitinib, Janus kinase inhibitor, anti-inflammatory, LNK 01001, LG6MM3RP86
Nibrozetone
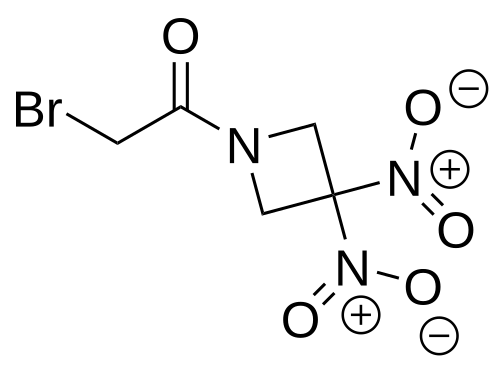
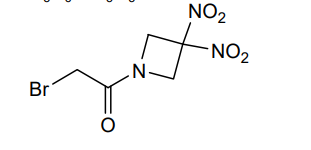
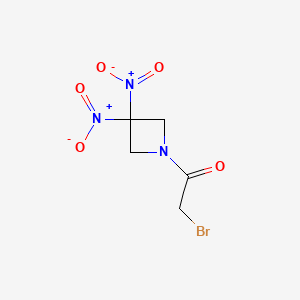
Nibrozetone
CAS 925206-65-1
MF C5H6BrN3O5 MW268.02 g/mol
2-bromo-1-(3,3-dinitroazetidin-1-yl)ethan-1-one
2-Bromo-1-(3,3-dinitroazetidin-1-yl)ethanone
2-BROMO-1-(3,3-DINITROAZETIDIN-1-YL)ETHAN-1-ONE
anti-inflammatory, RRx-001, RRx 001, ABDNAZ
Nibrozetone is an investigational new drug that is being evaluated by EpicentRx for the treatment of oral mucositis in head and neck cancer patients. It is a small molecule that combines direct inhibition of the NLRP3 inflammasome, induction of NRF2, and release of nitric oxide under hypoxic conditions.[1][2] It has received Fast Track designation from the FDA for severe oral mucositis in head and neck cancer patients.[3]
Nibrozetone (RRx-001) is an investigational, multi-action small molecule drug that is being developed by EpicentRx for a range of conditions, including head and neck cancers, small cell lung cancer, and neurodegenerative diseases like Parkinson’s and ALS. Its mechanism involves inhibiting the NLRP3 inflammasome, activating the Nrf2 pathway, and releasing nitric oxide in hypoxic tumor environments, while also protecting healthy tissues. It is being evaluated for its potential to reduce the side effects of cancer treatments and as a disease-modifying therapy itself.
How it works
- Anti-inflammatory: Nibrozetone inhibits the NLRP3 inflammasome, which is a key driver of inflammation in several diseases.
- Antioxidant: It activates the Nrf2 pathway, a cellular defense mechanism that protects against oxidative stress.
- Tumor-specific delivery: It acts as a “hypoxia-activated” drug, releasing a nitric oxide-releasing radical only in the low-oxygen environment of tumors, which can be toxic to cancer cells.
- Protective to normal tissue: The drug’s protective mechanisms are thought to keep it from causing harm to healthy tissues outside of the tumor environment.
Current and potential uses
- Oral mucositis: It is being studied to prevent and treat severe mouth sores that can be a side effect of head and neck cancer radiation therapy.
- Small cell lung cancer (SCLC): It is being investigated in a Phase 3 trial for the treatment of SCLC.
- Neurodegenerative diseases: Animal studies have shown promising neuroprotective effects in models of Parkinson’s and ALS.
- Other potential applications: Research is ongoing for its use as a treatment for other conditions, including endometriosis, toxic exposures, and various types of cancers.
- RRx-001 in Lung Cancer, Ovarian Cancer and Neuroendocrine Tumors Prior to Re-administration of Platinum Based Doublet Regimens (QUADRUPLE THREAT)CTID: NCT02489903Phase: Phase 2Status: CompletedDate: 2025-03-17
- RRx-001 for Reducing Oral Mucositis in Patients Receiving Chemotherapy and Radiation for Head and Neck CancerCTID: NCT05966194Phase: Phase 2Status: RecruitingDate: 2024-11-15
- Safety and Efficacy of RRx-001 in the Attenuation of Oral Mucositis in Patients Receiving Chemoradiation for the Treatment of Oral CancersCTID: NCT03515538Phase: Phase 2Status: CompletedDate: 2024-11-04
- Safety and Pharmacokinetic Study of RRx-001 in Cancer SubjectsCTID: NCT01359982Phase: Phase 1Status: CompletedDate: 2024-11-01
- RRx-001 Given With Irinotecan and Temozolomide for Pediatric Patients With Recurrent or Progressive Malignant Solid and Central Nervous System TumorsCTID: NCT04525014Phase: Phase 1Status: TerminatedDate: 2024-10-31
REF
- Dinitroazetidines Are a Novel Class of Anticancer Agents and Hypoxia-Activated Radiation Sensitizers Developed from Highly Energetic MaterialsPublication Name: Cancer ResearchPublication Date: 2012-05-14PMID: 22589277DOI: 10.1158/0008-5472.can-11-2303
- Properties of delta5-3beta-hydroxysteroid oxidoreductase isolated from Streptomyces griseocarneusPublication Name: Acta microbiologica Academiae Scientiarum HungaricaePublication Date: 1975PMID: 5856
PAT
- Dinitroazetidines Are a Novel Class of Anticancer Agents and Hypoxia-Activated Radiation Sensitizers Developed from Highly Energetic MaterialsPublication Name: Cancer ResearchPublication Date: 2012-05-14PMID: 22589277DOI: 10.1158/0008-5472.can-11-2303
- Properties of delta5-3beta-hydroxysteroid oxidoreductase isolated from Streptomyces griseocarneusPublication Name: Acta microbiologica Academiae Scientiarum HungaricaePublication Date: 1975PMID: 5856
- Cyclic nitro compounds, pharmaceutical compositions thereof and uses thereofPublication Number: US-8927527-B2Priority Date: 2005-08-12Grant Date: 2015-01-06
- Cyclic nitro compounds, pharmaceutical compositions thereof and uses thereofPublication Number: US-9226915-B2Priority Date: 2005-08-12Grant Date: 2016-01-05
- Cyclic nitro compounds, pharmaceutical compositions thereof and uses thereofPublication Number: WO-2007022225-A2Priority Date: 2005-08-12
- Cyclic nitro compounds, pharmaceutical compositions thereof and uses thereofPublication Number: US-2022016077-A1Priority Date: 2005-08-12
- Cyclic nitro compounds, pharmaceutical compositions thereof and uses thereofPublication Number: US-11925617-B2Priority Date: 2005-08-12Grant Date: 2024-03-12
- Methods of synthesizing and isolating N-(bromoacetyl)-3,3-dinitroazetidine and a composition including the samePublication Number: US-8471041-B2Priority Date: 2010-02-09Grant Date: 2013-06-25
- Methods of synthesizing and isolating n-(bromoacetyl)-3,3-dinitroazetidine and a composition including the samePublication Number: WO-2011100090-A1Priority Date: 2010-02-09
- Methods of synthesizing and isolating n-(bromoacetyl)-3,3-dinitroazetidine and a composition including the samePublication Number: IL-221141-A0Priority Date: 2010-02-09
- Cyclic nitro compounds, pharmaceutical compositions thereof and uses thereofPublication Number: EP-1924253-A2Priority Date: 2005-08-12
- Cyclic nitro compounds, pharmaceutical compositions thereof and uses thereofPublication Number: EP-1924253-B1Priority Date: 2005-08-12Grant Date: 2014-12-10
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2011100090&_cid=P11-MHTYGA-61308-1
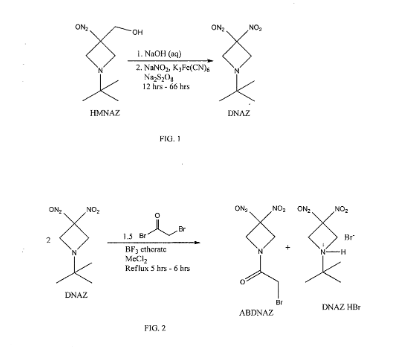
Cyclic nitro compounds, such as ABDNAZ, are being investigated for their potential use in treating cancer. Methods of synthesizing ABDNAZ have been described, such as in United States Patent No. 7,507,842 to Bednarski et al.
(“Bednarski”). In Bednarski, ABDNAZ is synthesized by reacting
l-½rt-butyl-3,3-dinitroazetidine (DNAZ) with bromoacetyl bromide and boron trifluoride etherate. For every mole of ABDNAZ produced, a mole of a hydrogen bromide salt of DNAZ (DNAZ HBr) is also produced as a coproduct. The ABDNAZ is isolated from the DNAZ HBr by cooling the reaction mixture, adding
dichloromethane, and filtering the DNAZ HBr. Solid DNAZ HBr is sensitive to impact, friction, and other external stimuli and, therefore, must be handled carefully. The dichloromethane filtrate is washed with water, dried, and then the dichloromethane is evaporated, producing a crude ABDNAZ mixture. The product is washed sequentially with diethyl ether and dried under vacuum, yielding ABDNAZ that is approximately 98% pure and at a yield of approximately 75% (based on bromoacetyl bromide). The 2% of impurities remaining in the ABDNAZ are believed to include
bromoacetic acid, unreacted DNAZ, and DNAZ HBr. This method of producing ABDNAZ is referred to herein as the Bednarski process. While the Bednarski process provides ABDNAZ at a reasonable purity and yield, the purity is not sufficient for pharmaceutical uses. In addition, solid DNAZ HBr produced during the Bednarski process is an explosive compound, which adds to the complexity of producing
Example 2
Synthesis of ABDNAZ from DNAZ
A three neck round bottom flask (3 L) equipped with a magnetic stir bar and a water jacketed reflux condenser was charged with the dichloromethane solution of DNAZ (produced as described in Example 1). A nitrogen gas purge of the apparatus was initiated and, after ten minutes, boron trifluoride diethyletherate (6.37 mL, 52 mmol) was added, followed by bromoacetyl bromide (33.77 mL, 388 mmol). The flask was sealed, except for a small vent at the top of the condenser, and the solution was heated to a mild reflux. After six hours (± 0.5 hour), heating was stopped and dichloromethane (1000 mL) and distilled water (800 mL) were added, in that order, to the heterogeneous mixture. The two-phase system was stirred vigorously for sixteen hours, until all solids (DNAZ HBr) were dissolved. The two-phase system was then transferred to a separatory funnel. The aqueous phase was removed and the organic phase was washed with additional distilled water (4 x 500 mL). The organic phase was dried with sodium sulfate (100 g – 150 g) and then transferred to a single neck, round bottom flask. The solution was concentrated on a rotary evaporator to approximately half of its initial volume and then ethanol (250 mL) was added. The remaining dichloromethane was removed by a rotary evaporator, causing precipitation of clear, colorless crystals. The flask was chilled in an ice bath for thirty minutes. The precipitate was isolated by vacuum filtration, rinsed with additional cold ethanol (5 x 150 mL), and dried to afford pure ABDNAZ (56.04 g, 81% yield): Ή NMR
(d6-acetone) δ 4.02 (s, 2H, -CH2Br ), 4.96 (br s, 2H, ring -CH2), 5.36 (br s, 2H, ring -CH2); 13C NMR (d6-acetone) δ 25.58, 58.58, 60.53, 107.69, 167.48.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2007022225&_cid=P11-MHTYDP-59218-1
Example 5: Synthesis of ABDNAZ
[00139] A 25 ml, three-neck, round bottom flask was charged with 7 ml of methylene chloride and 2.50 g (12.3 mmol) of t-BuDNAZ prepared as described in Archibald et at, Journal of Organic Chemistry, 1990, 2920. Under nitrogen, 0.16 ml (1.23 mmol) of boron trifluoride etherate was added. After stirring 5 min. at ambient temperature, 0.54 ml (6.15 mol) of bromoacetyl bromide was added. The solution was heated between 50-600C for 2 h. The darkened reaction mixture was cooled to ambient temperature, diluted with 50 ml methylene chloride, and filtered. The solid was identified as the HBr salt of t-BuDNAZ. The methylene chloride filtrate was washed with two 20 ml portions of water, dried over sodium sulfate, filtered, and evaporated under reduced pressure. The resultant solid was washed with three 20 ml portions of ethyl ether and dried under vacuum to yield 1.24 g (75.2% based on bromoacetyl bromide) of BrADNAZ as a white solid (mp = 124-1250C). 1H NMR (CDCl3): δ 3.76 (s, 2H), 4.88 (br s, 2H), 5.14 (br s, 2H); 13C NMR (CDCl3): δ 165.2, 105.0, 59.72, 57.79, 23.90. CaIc. for C5H6BrN3O5: %C 22.41, %H 2.26, %N 15.68; Found: %C 22.61, %H 2.36, %N 15.58.
HPLC/MS C-8 reverse phase column with acetonitrile/water mobile phase – m/e 266.95 (100%), 268.95 (98.3%). FT-IR 3014.24 (weak), 1677.66, 1586.30, 1567.65, 1445.55 (NO2), 1367.80, 1338.00, 1251.27 cm‘1.
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Oronsky B, Takahashi L, Gordon R, Cabrales P, Caroen S, Reid T (2023). “RRx-001: a chimeric triple action NLRP3 inhibitor, Nrf2 inducer, and nitric oxide superagonist”. Frontiers in Oncology. 13 1204143. doi:10.3389/fonc.2023.1204143. PMC 10258348. PMID 37313460.
- Jayabalan N, Oronsky B, Cabrales P, Reid T, Caroen S, Johnson AM, et al. (April 2023). “A Review of RRx-001: A Late-Stage Multi-Indication Inhibitor of NLRP3 Activation and Chronic Inflammation”. Drugs. 83 (5): 389–402. doi:10.1007/s40265-023-01838-z. PMC 10015535. PMID 36920652.
- Ryan C (30 March 2023). “FDA Grants Fast Track Designation to RRx-001 for Severe Oral Mucositis in Head and Neck Cancer”. OncLive.
| Clinical data | |
|---|---|
| Other names | Rrx-001 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 925206-65-1 |
| PubChem CID | 15950826 |
| DrugBank | DB12060 |
| ChemSpider | 13092644 |
| UNII | 7RPW6SU9SC |
| KEGG | D12720 |
| ChEMBL | ChEMBL3526802 |
| Chemical and physical data | |
| Formula | C5H6BrN3O5 |
| Molar mass | 268.023 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
/////////Nibrozetone, anti-inflammatory, RRx-001, RRx 001, ABDNAZ
Lomedeucitinib
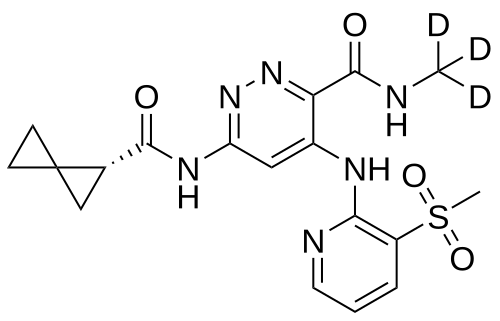
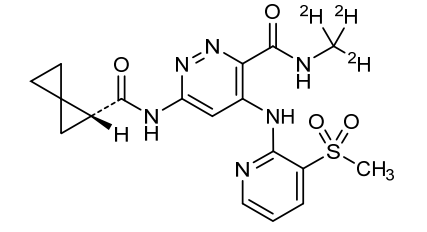
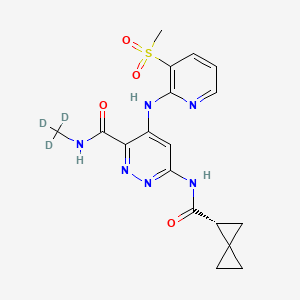
Lomedeucitinib
CAS 2328068-29-5
MF C18H172H3N6O4S
MW 419.5 g/mol

4-{[3-(methanesulfonyl)pyridin-2-yl]amino}-N-(2H3)methyl-6-[(1R)-spiro[2.2]pentane-1-carboxamido]pyridazine-3-carboxamide
4-[(3-methylsulfonyl-2-pyridinyl)amino]-6-[[(2R)-spiro[2.2]pentane-2-carbonyl]amino]-N-(trideuteriomethyl)pyridazine-3-carboxamide
Janus kinase inhibitor, anti-inflammatory, BMS-986322, BMS 986322, EYQ7KA55XA
Lomedeucitinib is an investigational new drug that is being evaluated for the treatment of psoriasis and psoriatic arthritis. It is a tyrosine kinase 2 (TYK2) inhibitor.[1]
- A Study to Evaluate Effectiveness and Safety of BMS-986322 in Participants With Moderate-to-Severe PsoriasisCTID: NCT05730725Phase: Phase 2Status: CompletedDate: 2024-09-19
- A Study to Evaluate the Drug Levels, Metabolism, and Removal of BMS-986322 in Healthy Adult Male ParticipantsCTID: NCT06088264Phase: Phase 1Status: CompletedDate: 2024-03-29
- A Study Investigating Interactions Between BMS-986322 and Rosuvastatin, Metformin and Methotrexate in Healthy ParticipantsCTID: NCT05615012Phase: Phase 1Status: CompletedDate: 2024-03-27
- A Study to Investigate the Interaction of BMS-986322 and a Combined Oral Hormonal Contraceptive (Ethinyl Estradiol [EE]/Norethindrone [NET]) in Healthy Female ParticipantsCTID: NCT05579574Phase: Phase 1Status: CompletedDate: 2023-08-18
- A Study to Assess the Safety and Tolerability of BMS-986322 in Healthy Participants of Japanese DescentCTID: NCT05546151Phase: Phase 1Status: CompletedDate: 2023-06-22
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US333829535&_cid=P10-MHIXWK-98212-1
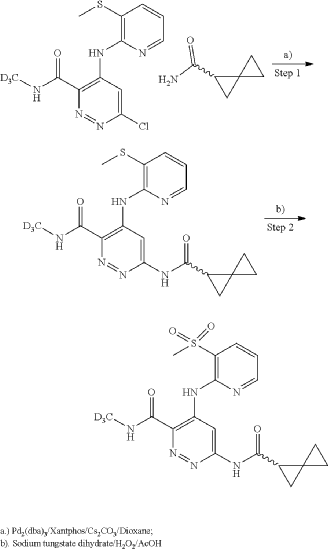
General Scheme for Examples 252 and 253:
Example 252
Step 1
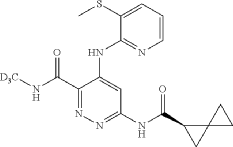
| A mixture of cesium carbonate (149 mg, 0.457 mmol), Xantphos (14.43 mg, 0.025 mmol), Pd 2(dba) 3 (11.42 mg, 0.012 mmol), 6-chloro-N-(methyl-d3)-4-((3-(methylthio)pyridin-2-yl)amino)pyridazine-3-carboxamide (65 mg, 0.208 mmol), and (R)-spiro[2.2]pentane-1-carboxamide (50.8 mg, 0.457 mmol) in dioxane (3 mL) was degassed using a vacuum/N2 fill cycle three times. The reaction was heated at 110° C. for 16 hours. The reaction was diluted with water and DCM. The DCM layer was separated and washed two more times with water and then dried (Na 2SO 4), filtered and concentrated. Purification via automated flash chromatography, eluting with methanol in DCM from 0 to 10%, gave the title compound (R)—N-(methyl-d3)-4-((3-(methylthio)pyridin-2-yl)amino)-6-(spiro[2.2]pentane-1-carboxamido)pyridazine-3-carboxamide (54 mg, 67% yield). 1H NMR (400 MHz, CHLOROFORM-d) δ 12.15 (br s, 1H), 9.88 (s, 1H), 8.68 (br s, 1H), 8.36 (br d, J=3.5 Hz, 1H), 8.25 (br s, 1H), 7.72 (br d, J=7.4 Hz, 1H), 6.97 (br dd, J=7.0, 5.1 Hz, 1H), 2.51 (s, 3H), 2.21-2.09 (m, 1H), 1.58-1.10 (m, 6H), 1.08-0.93 (m, 5H). |
| LCMS (ESI) m/e 388.1 [(M+H) +, calc’d C 18H 18D 3N 6O 2S 1, 388.1]; LC/MS retention time (method D): t R=0.80 min. |
Step 2
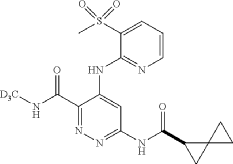
To a suspension of hydrogen peroxide (30% solution in water, 0.258 mL, 2.52 mmol) and (R)—N-(methyl-d3)-4-((3-(methylthio)pyridin-2-yl)amino)-6-(spiro[2.2]pentane-1-carboxamido)pyridazine-3-carboxamide (0.0489 g, 0.126 mmol) in AcOH (1 mL) was added sodium tungstate dihydrate (0.042 g, 0.126 mmol) at room temperature. After stirring at room temperature for 1 hour, the reaction was diluted with water, basified with Na 2CO 3 powder and extracted three times with DCM. The DCM layers were combined, washed with Na 2S 2O 3 (5% solution), dried (Na 2SO 4), filtered and concentrated. The crude product was purified using reverse phase prepHPLC to give the title compound (R)—N-(methyl-d3)-4-((3-(methylsulfonyl)pyridin-2-yl)amino)-6-(spiro[2.2]pentane-1-carboxamido)pyridazine-3-carboxamide (16.2 mg, 31%) as a colorless solid. 1H NMR (500 MHz, DMSO-d 6) δ 12.07 (s, 1H), 11.22 (s, 1H), 9.49 (s, 1H), 9.16 (s, 1H), 8.63 (dd, J=4.6, 1.5 Hz, 1H), 8.29 (dd, 0.1=7.8, 1.4 Hz, 1H), 7.34 (dd, 0.1=7.8, 4.7 Hz, 1H), 2.48-2.43 (m, 1H), 1.46-1.41 (m, 1H), 1.42-1.36 (m, 1H), 0.95-0.82 (m, 3H), 0.80-0.73 (m, 1H). (3H methyl sulfone was buried under DMSO peak). LCMS (ESI) m/e 420.0 [(M+H) +, calc’d C 18H 18D 3N 6O 4S, 420.1]; LC/MS retention time (method E): t R=1.38 min; OR: −205.39 (20° C.).
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US242383764&_cid=P10-MHIXVD-97150-1
PAT
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: US-11787779-B2Priority Date: 2017-11-21Grant Date: 2023-10-17
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: US-2024002364-A1Priority Date: 2017-11-21
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: KR-102702228-B1Priority Date: 2017-11-21Grant Date: 2024-09-02
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: NZ-805343-APriority Date: 2017-11-21
- Sulfonepyridine alkylamide-substituted heteroaryl compoundsPublication Number: JP-2023098942-APriority Date: 2017-11-21
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: AU-2023255024-A1Priority Date: 2017-11-21
- Heteroaryl compounds substituted with sulfone pyridinylalkylamidesPublication Number: CN-111315737-BPriority Date: 2017-11-21Grant Date: 2024-06-18
- The heteroaryl compounds are substituted with sulfone-pyridine alkyl amidesPublication Number: IL-274816-B2Priority Date: 2017-11-21
- Sulfonepyridine alkylamide substituted heteroaryl compoundsPublication Number: JP-7490107-B2Priority Date: 2017-11-21Grant Date: 2024-05-24
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: AU-2022228101-A1Priority Date: 2017-11-21
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: TW-I776994-BPriority Date: 2017-11-21Grant Date: 2022-09-11
- Sulfonepyridine alkylamide-substituted heteroaryl compoundsPublication Number: JP-7258903-B2Priority Date: 2017-11-21Grant Date: 2023-04-17
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: AU-2022228101-B2Priority Date: 2017-11-21Grant Date: 2023-08-03
- The heteroaryl compounds are substituted with sulfone-pyridine alkyl amidesPublication Number: IL-274816-B1Priority Date: 2017-11-21
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: US-2019152948-A1Priority Date: 2017-11-21
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: CA-3083122-A1Priority Date: 2017-11-21
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: KR-20200089706-APriority Date: 2017-11-21
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: US-11021462-B2Priority Date: 2017-11-21Grant Date: 2021-06-01
- Sulfone pyridine alkyl amide-substituted heteroaryl compoundsPublication Number: US-2021253554-A1Priority Date: 2017-11-21
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | BMS-986322 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2328068-29-5 |
| PubChem CID | 138620496 |
| IUPHAR/BPS | 13210 |
| UNII | EYQ7KA55XA |
| KEGG | D12725 |
| ChEMBL | ChEMBL5314608 |
| Chemical and physical data | |
| Formula | C18H17D3N6O4S |
| Molar mass | 419.47 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Ahsan S, Degener R, Schlamp M (2024). “Non-Invasive Treatments Invade the Psoriasis Pipeline”. Drugs in Context. 13: 2024–5–6. doi:10.7573/dic.2024-5-6. PMC 11313207. PMID 39131603.
////////lomedeucitinib, Janus kinase inhibitor, anti-inflammatory, BMS-986322, BMS 986322, EYQ7KA55XA
Girocitinib
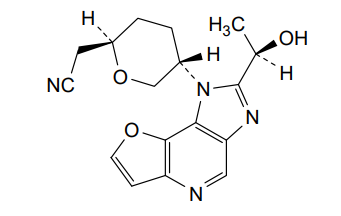
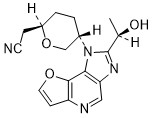
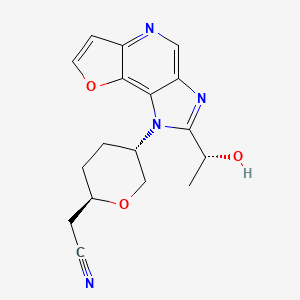
Girocitinib
CAS 2222137-79-1
MFC17H18N4O3 MW 326.36
2-[(2R,5S)-5-[4-[(1R)-1-hydroxyethyl]-12-oxa-3,5,8-triazatricyclo[7.3.0.02,6]dodeca-1,4,6,8,10-pentaen-3-yl]oxan-2-yl]acetonitrile
[(2R,5S)-5-{2-[(1R)-1-hydroxyethyl]-1H-furo[3,2-b]imidazo[4,5-d]pyridin-1-yl}oxan-2-yl]acetonitrile
2-((2R,5S)-5-(2-((R)-1-hydroxyethyl)-1H-furo[3,2-b]imidazo[4,5-d]pyridin-1-yl)tetrahydro-2H-pyran-2-yl)acetonitrile
Janus kinase inhibitor, anti-inflammatory, A0IES9T8GO
In an era where targeted therapies are redefining the landscape of medical treatment, Girocitinib emerges as a beacon of hope for many. This innovative drug, developed by leading pharmaceutical research institutions, primarily targets specific proteins involved in disease progression. Classified as a tyrosine kinase inhibitor (TKI), Girocitinib has shown significant promise in the treatment of various cancers, particularly non-small cell lung cancer (NSCLC). The drug is currently in the advanced stages of clinical trials, with researchers optimistic about its potential to provide a more effective and less toxic treatment option compared to conventional therapies.
Girocitinib is designed to interfere with the signaling pathways that promote cancer cell growth and survival. It does this by inhibiting the activity of tyrosine kinases, enzymes that play a key role in the activation of many proteins by signaling pathways within the cell. Tyrosine kinases are often overactive in cancer cells, leading to unchecked proliferation and survival. By targeting these enzymes, Girocitinib effectively disrupts these malign processes, thereby slowing down or even halting the progression of the disease.
The primary indication for Girocitinib is non-small cell lung cancer (NSCLC), which accounts for approximately 85% of all lung cancer cases. NSCLC is notoriously difficult to treat, especially in its advanced stages, and current treatments often come with significant side effects. Clinical trials have shown that Girocitinib can significantly improve progression-free survival in patients with specific genetic mutations that make them more responsive to TKI therapy. These mutations can be identified through genetic testing, allowing for a more personalized treatment approach that increases the likelihood of success.
In addition to NSCLC, researchers are exploring the potential of Girocitinib to treat other types of cancer, including colorectal cancer and certain forms of leukemia. Early-stage trials have shown encouraging results, suggesting that Girocitinib could become a versatile tool in the oncology arsenal. Its ability to target specific molecular pathways makes it a promising candidate for combination therapies, which aim to enhance treatment efficacy while minimizing resistance and adverse effects.
The development of Girocitinib is a testament to the power of modern science and technology in addressing some of the most challenging health issues of our time. The drug’s journey from the laboratory to clinical trials has been marked by rigorous research and collaboration among scientists, healthcare professionals, and patients. As we await the results of ongoing studies, there is a palpable sense of anticipation in the medical community, as Girocitinib holds the promise of transforming cancer treatment for many patients.
In conclusion, Girocitinib represents a significant advancement in the field of targeted cancer therapy. Its mechanism of action, which involves the inhibition of tyrosine kinases, offers a more precise and potentially less harmful treatment option for patients with NSCLC and possibly other cancers. As research progresses, Girocitinib may well become a cornerstone in the fight against cancer, providing hope and improved outcomes for countless individuals around the world.
PDT PAT
WO2018067422
SYN
https://patents.google.com/patent/US10738060B2/en?oq=US10738060
Example 4: Synthesis of 2-[(2R,5S)-5-[2-[(R)-1-Hydroxyethyl]furo[3,2-b]imidazo[4,5-d]pyridin-1-yl]tetrahydropyran-2-yl] acetonitrile (4)
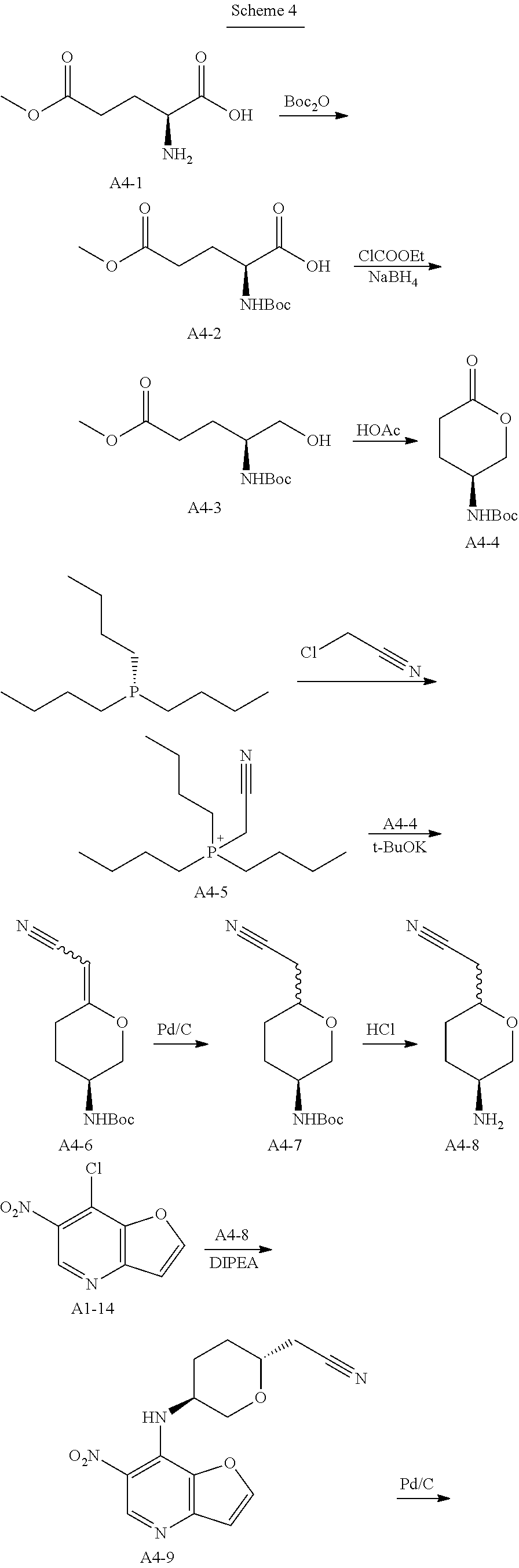
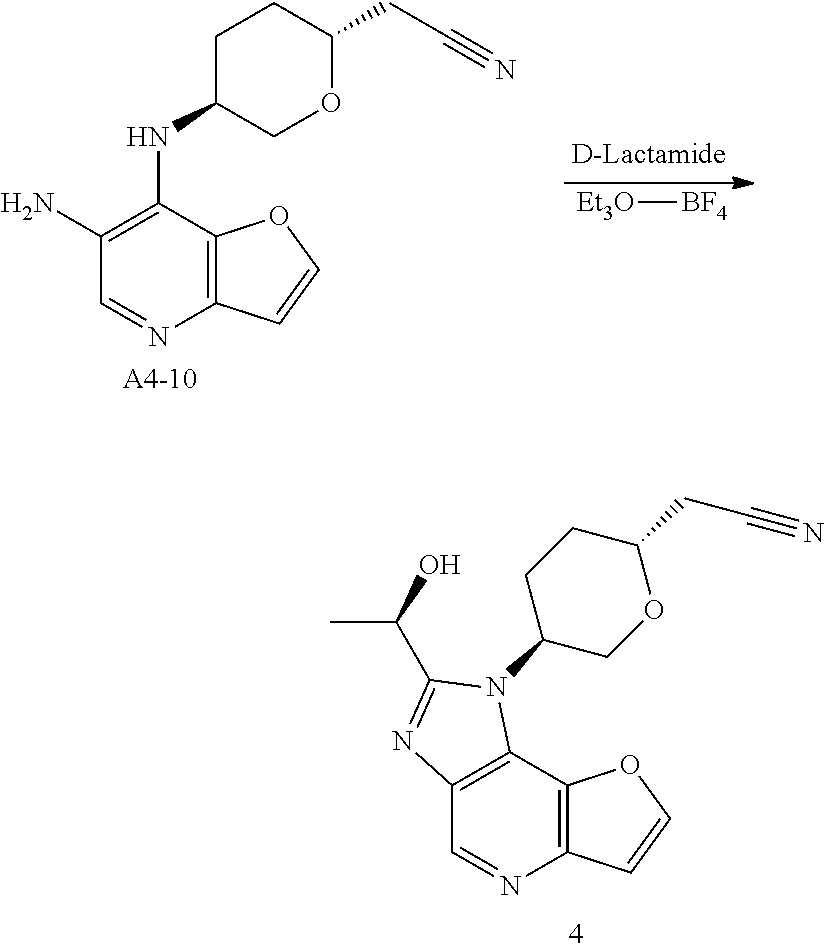
Step 1. In a round bottom flask, triethylamine (188 g, 1.86 mol, 1.0 eq) was added dropwise to a stirred solution of di-tert-butyl dicarbonate (162 g, 0.744 mol, 1.2 eq) and compound A4-1 (100 g, 0.62 mol, 1.0 eq) in water (500 mL) and 1,4-dioxane (500 mL). After stirring for 18 hrs at room temperature, the solution was extracted with MTBE (500 mL*2) and the aqueous phase was cooled on ice and carefully acidified to pH 3 by slow addition of 10% citric acid solution. The urethane was then extracted twice with ethyl acetate, and the combined extracts was washed with brine, dried over anhydrous sodium sulfate, and concentrated to give compound A4-2 as clear viscous oil (180 g, yield 100%). MS-ESI:[M+1]+: 262.1
Step 2. A solution of compound A4-2 (40 g, 0.153 mmol, 1.0 eq) in THF (600 mL) was treated with 4-methylmorpholine (17 g, 0.168, 1.1 eq) at room temperature. The resulting mixture was cooled to 0° C. before being treated with isobutyl chloroformate (22.7 g, 0.166 mmol, 1.08 eq) dropwise. The resulting reaction mixture was stirred at 0° C. for an addition 20 mins before being filtered and washed with THF. Then the clear filtrate solution was cooed to 0° C., and treated with a solution of NaBH4 (11.2 g, 0.295 mol, 1.93 eq) in water (100 mL). The resulting mixture was stirred overnight at room temperature, and then quenched with an aqueous HCl solution (1.0 mol/L,200 mL) dropwise, The mixture was extracted with ethyl acetate, and the combined extracts was washed with brine, dried over anhydrous sodium sulfate, concentrated to give compound A4-3 as a yellow oil (25 g, yield 66%). MS-ESI:[M+1]+: 248.1
Step 3. A solution of compound of A4-3 (25 g, 0.1 mol, 1.0 eq) in toluene (300 mL) and acetic acid (150 mL) was heated to reflux for 5 hrs and then cooled, concentrated under vacuum. The residual was added saturated sodium bicarbonate solution to pH 7-8 in ice-bath. Then the mixture was extracted three times with ethyl acetate, and the combined extracts was washed with brine, dried over anhydrous sodium sulfate, concentrated and recrystallized by ethyl acetate and PE to give compound A4-4 as a white powder (8.0 g, yield 37.2%). GC-MS: 215
Step 4. A solution of tributyl phosphine (72.9 g, 0.36 mol, 1.0 eq) in nitromethane (500 mL), was added dropwise chloroacetonitrile (27.2 g, 0.36 mol, 1.0 eq) in nitrogen atmosphere. The resulting reaction mixture was stirred for 16 hrs at room temperature, then concentrated. The residual oil solidified when a small amount of ethyl acetate was added. The solid was recrystallized by ethyl acetate and DCM to afford compound A4-5 as a white powder (95 g, yield 95%).
Step 5. To a solution of dry compound A4-5 (8.3 g, 30 mmol, 3.0 eq) in N,N-dimethylacetamide (30 mL) in nitrogen atmosphere, was added solid Potassium tert-butoxide (3.1 g, 28 mmol, 2.8 eq) in portions at 0° C. The resulting mixture was gradually warmed to 30° C. and stirred for 2 hrs. The resulting ylide solution was then treated with compound A4-4 (2.15 g, 10 mmol, 1.0 eq), and stirred overnight at 70° C. After cooled to room temperature, the resulting slurry was poured into the mixture of ice-water (100 mL) and saturated sodium bicarbonate solution (100 mL). The mixture was extracted twice with ethyl acetate, and the combined extracts was washed three times with brine, dried over anhydrous sodium sulfate, concentrated to give compound A4-6 as yellow oil without purification (7.5 g, yield 100%). MS-ESI:[M+1]+: 239.1
Step 6. To a solution of compound A4-6 (7.5 g, 10 mmol, 1.0 eq) in methanol (200 mL), was added 10% Pd/C (0.5 g,50% wet). Hydrogenation was carried out under atmospheric pressure at room temperature until hydrogen uptake ceased. The catalyst was filtered and washed by methanol. The filtrates was concentrated under vacuum, and purified by silica gel column chromatography to give compound A4-7 as off-white powder (1.6 g, yield 66.7%). MS-ESI:[M+1]+: 241.1
Step 7. To a solution of compound A4-7 (1.6 g, 6.67 mmol, 1.0 eq) in DCM (20 mL), was added TFA (10 g, 88.5 mmol, 13.2 eq). The reaction mixture was stirred for 2 hrs at room temperature until TLC showed the reaction was complete, then concentrated under vacuum. Water (20 mL) was added and the solution was treated with aqueous sodium hydroxide solution (4 mol/L) to pH 10. Then the aqueous phase was extracted six times with DCM/methanol (10/1). The combined extracts was dried over anhydrous sodium sulfate, concentrated to give compound A4-8 as light-brown oil (950 mg, yield 100%). MS-ESI:[M+1]+: 141.1
Step 8. To a solution of compound A1-14 (prepared as step 4 to 12 in example 1) (600 mg, 3.0 mmol, 1.0 eq) in n-butanol (15 mL), was added compound A4-8 (950 mg, 6.7 mmol, 2.26 eq) and DIPEA (1.36 g, 10.5 mmol, 3.5 eq). The reaction mixture was stirred for 1 hr at 135° C., concentrated and purified by silica gel column chromatography to give compound A4-9 (2R,5S) as light-yellow powder (254 mg, yield 28.0%).MS-ESI: [M+1]+: 303.1.
1H NMR (300 MHz, d6-DMSO): 9.063 (s, 1H), 8.503 (d, 1H), 9.326 (d, 1H), 7.176 (d, 1H), 4.431-4.513 (m, 1H), 4.128-4.156 (m, 1H), 3.633-3.659 (m, 1H), 3.448-3.518 (m, 1H), 2.775-2.841 (m, 2H), 2.205-2.312 (m, 1H), 1.829-1.859 (m, 2H), 1.501-1.521 (m, 1H).
Step 9. To a solution of compound A4-9 (254 g, 0.84 mmol, 1.0 eq) in methanol (20 mL), was added 10% Pd/C (0.15 g,50% wet). Hydrogenation was carried out under atmospheric pressure at room temperature until hydrogen uptake ceased. The catalyst was filtered and washed by methanol. The filtrates was concentrated under vacuum, and compound A4-10 was obtained as yellow oil (230 mg, yield 100%). MS-ESI:[M+1]+: 273.1
Step 10. A solution of D-Lactamide (388 mg, 4.2 mmol, 5.0 eq) and Et3O—BF4 (1.3 g, 6.72 mmol, 8.0 eq) in THF (10 mL) was stirred for 30 mins at room temperature in nitrogen atmosphere. Then the above solution was added to the mixture of compound A4-10 (230 mg, 0.84 mmol, 1.0 eq) in ethanol (10 mL). After stirring for 3 hrs at 85° C. until HPLC showed the reaction was complete, the mixture was concentrated, added water and extracted four times with ethyl acetate. The organic phases was discarded and the aqueous phase was treated with saturated sodium bicarbonate solution to pH 8, extracted twice with ethyl acetate. The second organic phases was dried over anhydrous sodium sulfate, concentrated and purified by silica gel column chromatography to give the title compound as light-yellow powder (120 mg, yield 43.8%). MS-ESI: [M+1]+: 327.6,
1H NMR (300 MHz, CDCl3): 9.039 (s, 1H), 7.939 (d, 1H), 7.196 (d, 1H), 5.235-5.336 (m, 1H), 4.806-4.973 (m, 1H), 4.403-4.483 (t, 1H), 4.096-6.116 (m, 2H), 2.700-2.807 (m, 4H), 2.105-2.312 (m, 2H), 1.830-1.852 (d, 3H).
SYN
US2022227777
https://patents.google.com/patent/US20220227777A1
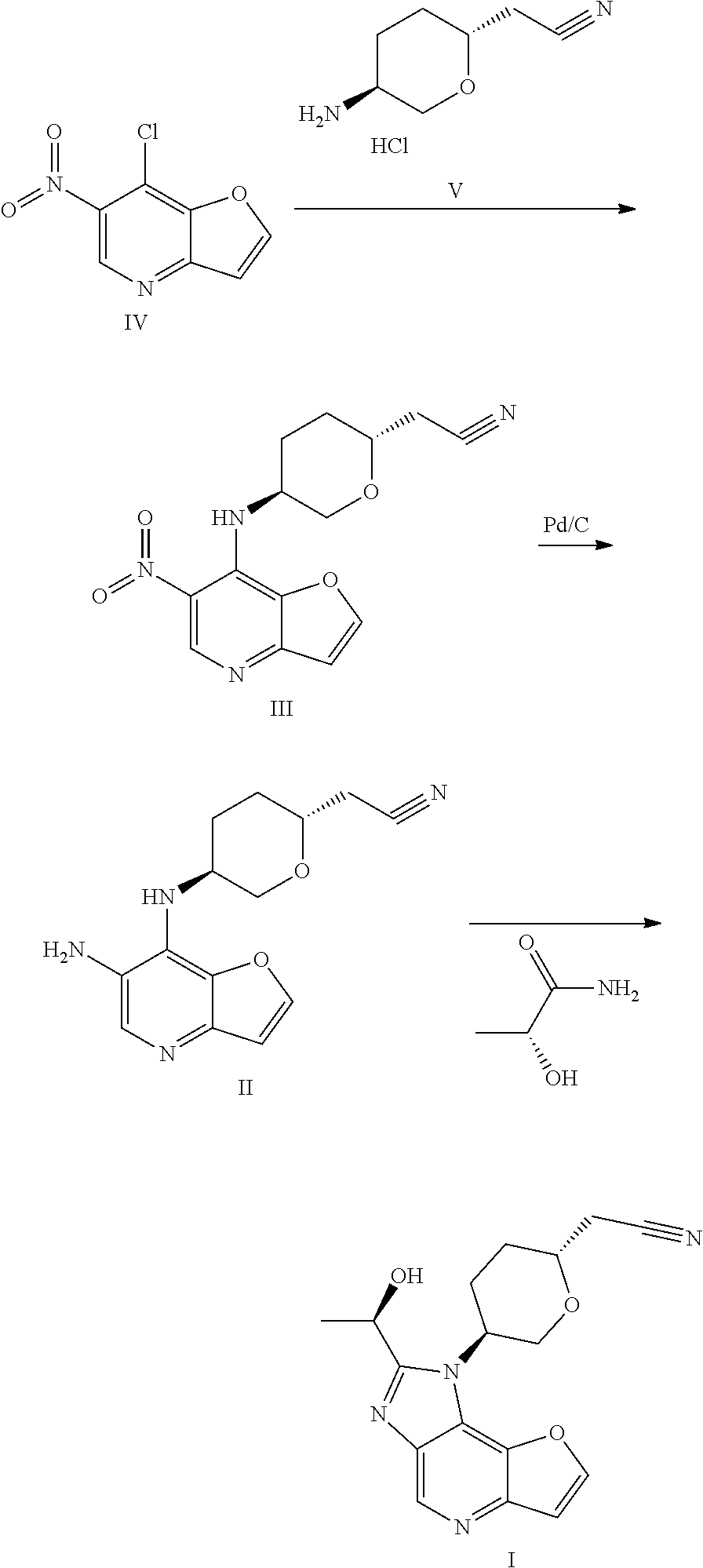
International patent application WO2018067422A1 discloses 1H-furo[3,2-b]imidazo[4,5-d]pyridine derivatives as selective JAK1 kinase inhibitors and preparation methods thereof, wherein compound I and its preparation method is disclosed.
Preparation of a Compound of Formula I
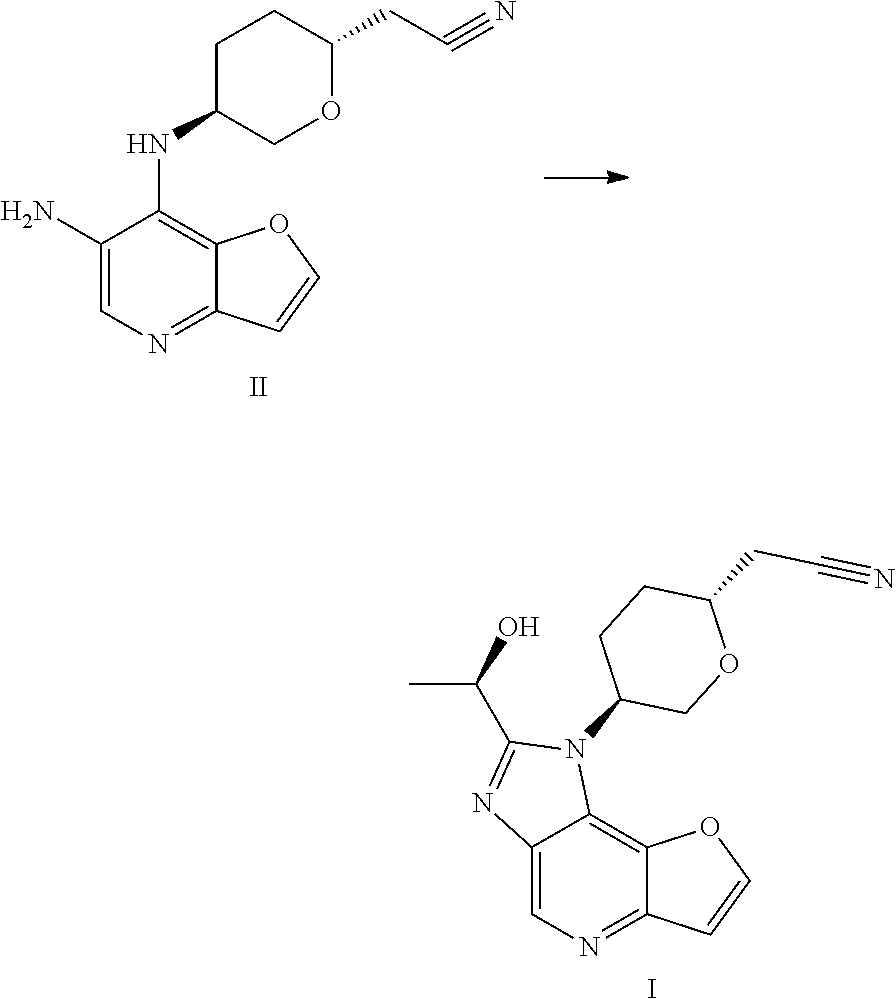
- [0204]THF (60 mL, 12 V), (R)-lactamide (6.6 g, 4.0 eq) and Et3O—BF4 (13.9 g, 4.0 eq) were added to a 250 mL three-necked flask #1, the system was stirred; the materials in three-necked flask #1 were stirred under nitrogen protection for later use; a compound of formula II (5.0 g, 1.0 eq) and ethanol (80 mL, 16 V) were added to another 250 mL three-necked flask #2; the system was heated to 70±5° C. under nitrogen protection; the materials in three-necked flask #1 were added to three-necked flask #2 with a syringe dropwise within 10-20 minutes; the system was heated to 85±5° C. (internal temperature was in the range of 72-75° C.) under nitrogen protection for reacting for 2 hours; the system was cooled to room temperature; the reaction liquid was concentrated with a rotary evaporator until there was basically no fraction flowing out; 1M HCl (80 mL) was added to the residual concentrated liquid, the pH was about 1 (determined with a pH test paper); the system was extracted four times with DCM (50 mL×4); the pH of the aqueous phase was adjusted to 7-8 with saturated sodium bicarbonate solution; the system was stirred at room temperature for 0.5 hour, then was filtered, the filter cake was washed with water (60 mL) and EA (10 mL), respectively; the filter cake was dried under vacuum at 50° C. for 16 hours; 4.3 g of faint yellow solid was obtained, with a purity of 95.0%; the solid was dissolved with methanol (30 mL); 4.1 g of silicon based metal eliminator and 1.0 g of activated carbon were added, the system was heated to 50° C. and stirred for 1 hour, then was cooled, filtered, washed with methanol (30 mL); the filtrate was concentrated with rotary evaporator until there was basically no fraction flowing out; methanol (10 mL) and MTBE (25 mL) were added to the residue, the system was heated to 50° C., and was stirred for 0.5 hour, then was cooled, the system was cooled to 10±5° C. and stirred for 0.5 hour; filtered, the filter cake was washed with MTBE (25 mL); the filter cake was dried under vacuum at 50° C. for 16 hours, 3.2 g of faint yellow solid was obtained, with a purity of 97.9%.
- [0205]MS-ESI: [M+1]+: 327.6
- [0206]1H NMR (400 MHz, CDCl3): 8.988 (s, 1H), 7.922 (d, 1H), 7.175 (d, 1H), 5.200-5.265 (m, 1H), 4.859-4.942 (m, 1H), 4.350-4.406 (t, 1H), 4.020-4.108 (m, 2H), 3.067 (d, 1H), 2.619-2.779 (m, 3H), 2.108-2.269 (m, 2H), 1.790-1.895 (m, 3H).
- [0207]THF (650 mL, 12 V), (R)-lactamide (70.6 g, 4.0 eq) and Et3O—BF4 (150.6 g, 4.0 eq) were added to a 1000 mL three-necked flask #1, the system was stirred; the materials in three-necked flask #1 were stirred under nitrogen protection for later use; a compound of formula II (54 g, 1.0 eq) and ethanol (860 mL, 16 V) were added to another 2000 mL three-necked flask #2; the system was heated to 70±5° C. under nitrogen protection; the materials in three-necked flask #1 were slowly added to three-necked flask #2 dropwise within 1 hour; the system was heated to 85±5° C. (internal temperature was in the range of 72-75° C.) under nitrogen protection for reacting for 2 hours; the system was cooled to room temperature; the reaction liquid was concentrated with a rotary evaporator until there was basically no fraction flowing out; 1M HCl (450 mL) was added to the residual concentrated liquid, the pH was about 1 (determined with a pH test paper); the system was extracted four times with DCM (270 mL×4); the pH of the aqueous phase was adjusted to 7-8 with saturated sodium bicarbonate solution; the system was stirred at room temperature for 0.5 hour, then was filtered, the filter cake was washed with water (540 mL); MTBE (270 mL) was added to the filter cake, the system was stirred at room temperature for 0.5 hour, filtered, the filter cake was washed with MTBE (108 mL); the filter cake was dried under vacuum at 50° C. for 16 hours; 49.2 g of light yellow solid was obtained, with an HPLC purity of 94.2%; the solid was dissolved with methanol (380 mL); silicon based metal eliminator (44 g) and activated carbon (5.4 g) were added, the system was heated to 50° C. and stirred for 1 hour, then was cooled, filtered, washed with methanol (430 mL); the filtrate was concentrated with a rotary evaporator to (80-110 mL, 1.5 V-2 V); MTBE (540 mL) was added to the residue, the system was heated to 50° C., and was stirred for 1 hour, then was cooled to 10±5° C. and stirred for 0.5 hour; filtered, the filter cake was washed with MTBE (270 mL); 42.4 g of filter cake was obtained, with an HPLC purity of 96.9%; the filter cake was dried under vacuum at 50° C. for 16 hours, 41.0 g of light yellow solid was obtained, with an HPLC purity of 96.7%, a yield of 63.3%.
- [0208]Purification of a Compound of Formula I:
- [0209]A compound of formula I (41 g) was dissolved with methanol; silica gel (50 g) was added to the solution, the system was concentrated to dryness for later use; silica gel (200 g) was added to the chromatographic column, the column was compacted with an air pump; a compound of formula I mixed with silica gel was added to the chromatographic column, the column was compacted with an air pump; the chromatographic column was eluted with an eluent (VMeOH:VDCM=1:100-1:30); qualified components were collected, concentrated to dryness; the product was dried under vacuum at 50° C. for 16 hours; 36 g of off-white solid was obtained, with an HPLC purity of 98.5%.
- [0210]The MS-ESI and 1H NMR data are consistent with example 21.
- [0211]THF (60 mL, 6 V), (R)-lactamide (13.2 g, 4.0 eq) and Et3O—BF4 (27.9 g, 4.0 eq) were added to a 100 mL three-necked flask #1, the system was stirred; the materials in #1 were stirred under nitrogen protection for later use; a compound of formula II (10 g, 1.0 eq) and ethanol (100 mL, 10 V) were added to another 250 mL three-necked flask #2; the system was heated to 70±5° C. under nitrogen protection; the materials in three-necked flask #1 were slowly added to three-necked flask #2 dropwise within 20 minutes; the system was heated to 80±5° C. (internal temperature was in the range of 72-75° C.) under nitrogen protection for reacting for 0.5 hour; the system was cooled to room temperature 20-30° C.; the reaction liquid was concentrated to about 50-80 mL with a rotary evaporator between 30-40° C.; water (100 mL, 10 V) was added to the system, then the system was concentrated with a rotary evaporator between 30-40° C. until there was basically no fraction flowing out; the system was cooled to 20-30° C.; the temperature of the system was controlled at 20-30° C., 12M HCl (5.5 g) was used to adjust the pH of the system to 2-3, the system was extracted with ethyl acetate (50 mL×2, 5V×2); the organic phase was discarded, and the aqueous phase was transferred to a flask; the temperature of the system was controlled at 20-30° C., the pH of the system was adjusted to 8-9 with saturated potassium carbonate solution (23 g); the temperature of the system was controlled at 20-25° C., the system was stirred for 2 hours, then was filtered, the filter cake was washed with water (50 mL) and MTBE (50 mL); the filter cake was dried with an air blower at 50° C. for 24 hours, 18 g of earth yellow solid was obtained, with an HPLC purity of 93.5%.
- [0212]The MS-ESI and 1H NMR data are consistent with example 21.
- [0213]THF (120 mL, 12 V), (R)-lactamide (13.2 g, 4.0 eq) and Et3O—BF4 (27.8 g, 4.0 eq) were added to a 250 mL three-necked flask #1, the system was stirred; the materials in #1 were stirred under nitrogen protection for later use; a compound of formula II (10 g, 1.0 eq) and ethanol (140 mL, 14 V) were added to another 500 mL three-necked flask #2; the system was heated to 40-45° C. (internal temperature) under nitrogen protection; the materials in three-necked flask #1 were added to three-necked flask #2 dropwise within 1 hour; the system was maintained at 40-45° C. (internal temperature) under nitrogen protection for reacting for 4.5 hours; the system was cooled to room temperature, and water (20 mL, 2V) was added; the system was concentrated with a rotary evaporator at 30-40° C. until there was basically no fraction flowing out; the system was cooled to 20-30° C.; the temperature of the system was controlled at 20-30° C., 12M HCl (3 mL) was used to adjust the pH of the system to 2-3, the system was extracted with ethyl acetate (50 mL×2, 5V×2); the organic phase was discarded, and the aqueous phase was transferred to a flask; the temperature of the system was controlled at 20-30° C., the pH of the system was adjusted to 8-9 with 50% potassium carbonate solution (15 mL); the temperature of the system was controlled at 20-25° C., the system was stirred for 2 hours, then was filtered, the filter cake was washed with water (50 mL) and acetone (50 mL); the crude product was triturated and stirred with water (50 mL) at 20-25° C. for 1 hour; the system was filtered, the filter cake was washed with water (50 mL) and acetone (50 mL); the filter cake was dried with an air blower at 50° C. for 24 hours, 17.8 g of khaki solid was obtained, with an HPLC purity of 95.3%.
- [0214]The MS-ESI and 1H NMR data are consistent with example 21.
- [0215]THF (60 mL, 12 V), (R)-lactamide (6.6 g, 4.0 eq) and Et3O—BF4 (13.9 g, 4.0 eq) were added to a 250 mL three-necked flask #1, the system was stirred; the materials in three-necked flask #1 were stirred under nitrogen protection for later use; a compound of formula II (5 g, 1.0 eq) and ethanol (70 mL, 14 V) were added to another 250 mL three-necked flask #2; the system was heated to 40-45° C. (internal temperature) under nitrogen protection; the materials in three-necked flask #1 were added to three-necked flask #2 dropwise within 20 minutes; the system was maintained at 40-45° C. (internal temperature) under nitrogen protection for reacting for 3 hours; the system was cooled to room temperature and was filtered, the filter cake was washed with THF (10 mL); water (10 mL, 2V) was added to the filtrate; the filtrate was concentrated with a rotary evaporator to 10-20 mL (2V-4V), the concentrated residue was exchanged with ethyl acetate (25 mL×2) and concentrated to 10-20 mL (2V-4V); water (50 mL, 10V) was added to the concentrated residue; the internal temperature was controlled at 20-25° C., 12M HCl (4.1 g) was used to adjust the pH of the system to 1-2; activated carbon (0.5 g) was added to the system, and the system was stirred at room temperature for 2 hours, and was filtered, the filter cake was washed with water (10 mL) and 1M HCl (10 mL); the combined filtrate was extracted with ethyl acetate (25 mL×2), the organic phase was discarded; the internal temperature was controlled at 20-25° C., the pH of the system was adjusted to 9-10 with saturated potassium carbonate solution (15 g); the internal temperature was controlled at 15-20° C., the system was stirred for 1 hour, and was filtered, the filter cake was washed with water (10 mL); the filter cake was triturated with acetone aqueous solution (50 mL, V/V=1:1) for 1 hour; the system was filtered, the filter cake was washed with acetone aqueous solution (10 mL, V/V=1:1); the filter cake was dried with an air blower at 50° C. for 24 hours; 5.0 g of pale gray solid was obtained, with an HPLC purity of 95.6%, and a yield of 83.5%;
- [0216]Purification of a Compound of Formula I:
- [0217]5.0 g of the obtained solid and methanol (40 mL) were added to a flask, and were stirred for 10 minutes at room temperature, the materials were basically dissolved and the solution was clear; activated carbon (0.5 g) and silica gel (4.0 g) were added to the system; the system was heated to 50-55° C., the temperature was maintained and the system was stirred for 2 hours, then was filtered with silica gel (5 g), the filter cake was washed with methanol (50 mL); the filtrate was concentrated with a rotary evaporator to 5-10 mL; MTBE (50 mL) was added to the concentrated residue; the system was heated to reflux, and was allowed for reflux for 1 hour; the system was cooled to 5-10° C., the temperature was maintained and the system was stirred for 1 hour and was filtered, the filter cake was washed with MTBE; the filter cake was dried with a drying oven under vacuum at 50° C. for 16 hours; 3.0 g of off-white solid was obtained, with a yield of 60% and a purity of 97.9%; the filtrate was concentrated to dryness to obtain 1.4 g of yellow solid.
- [0218]The MS-ESI and 1H NMR data are consistent with example 21.
PAT
- NEW SELECTIVE JAK1 INHIBITORS AND THEIR USEPublication Number: HR-P20211965-T1Priority Date: 2016-10-03
- Novel JAK1 selective inhibitors and uses thereofPublication Number: KR-102399848-B1Priority Date: 2016-10-03Grant Date: 2022-05-19
- Novel Jak1-selective inhibitors and their usesPublication Number: CN-113214278-BPriority Date: 2016-10-03Grant Date: 2022-10-28
- Jak1 selective inhibitors and uses thereofPublication Number: US-RE49834-EPriority Date: 2016-10-03Grant Date: 2024-02-13
- Novel jak1 selective inhibitors and uses thereofPublication Number: US-2019256523-A1Priority Date: 2016-10-03
- JAK1 selective inhibitors and uses thereofPublication Number: US-10738060-B2Priority Date: 2016-10-03Grant Date: 2020-08-11
- Novel Jak1-selective inhibitors and their usesPublication Number: CN-108366994-BPriority Date: 2016-10-03Grant Date: 2021-10-01
- Novel Jak1-selective inhibitors and their usesPublication Number: CN-113214278-APriority Date: 2016-10-03
- Novel jak1 selective inhibitors and uses thereofPublication Number: EP-3509591-B1Priority Date: 2016-10-03Grant Date: 2021-11-17
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereof.Publication Number: MX-2024006688-APriority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: US-12195476-B2Priority Date: 2019-06-06Grant Date: 2025-01-14
- Novel jak1 selective inhibitors and uses thereofPublication Number: CA-3039178-A1Priority Date: 2016-10-03
- Novel jak1 selective inhibitors and uses thereofPublication Number: EP-3509591-A1Priority Date: 2016-10-03
- Novel JAK1 selective inhibitors and uses thereofPublication Number: JP-2019537559-APriority Date: 2016-10-03
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: EP-4248967-A3Priority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: JP-2023089169-APriority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compounds, crystal forms of furoimidazopyridine compounds and crystal forms of their saltsPublication Number: CN-113906035-BPriority Date: 2019-06-06Grant Date: 2023-11-10
- Synthesis method of furoimidazopyridine compounds, crystal forms of furoimidazopyridine compounds and crystal forms of their saltsPublication Number: CN-117327083-APriority Date: 2019-06-06
- METHOD OF SYNTHESIS OF FUROIMIDAZOPYRIDINE COMPOUND, CRYSTAL FORM OF FUROIMIDAZOPYRIDINE COMPOUND, AND CRYSTAL FORM OF ITS SALT.Publication Number: MX-2024004146-APriority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: US-2022227777-A1Priority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: AU-2020289149-B2Priority Date: 2019-06-06Grant Date: 2023-05-11
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: EP-4248966-A2Priority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: EP-4248966-A3Priority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: EP-4248967-A2Priority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: WO-2020244348-A1Priority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: AU-2020289149-A1Priority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound and crystal form of salt thereofPublication Number: CN-113906035-APriority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: EP-3981771-A1Priority Date: 2019-06-06

AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////Girocitinib, Janus kinase inhibitor, anti-inflammatory, A0IES9T8GO
Frevecitinib
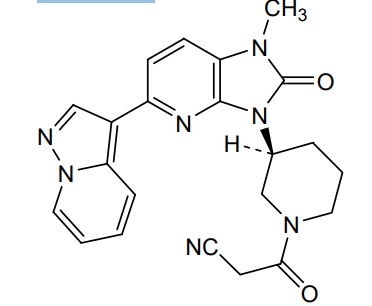
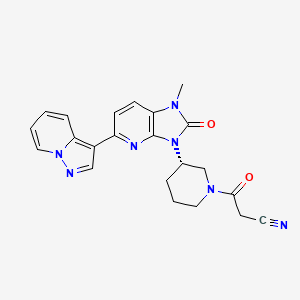
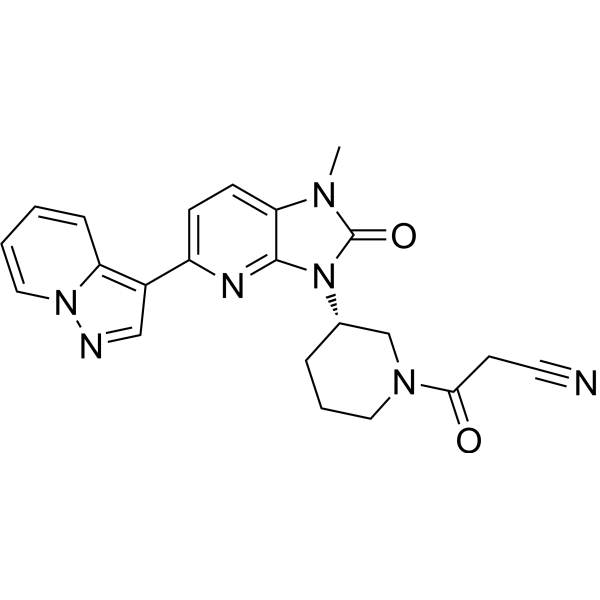
Frevecitinib
CAS 1299417-07-4
MF C22H21N7O2 MW 415.4 g/mol
3-[(3S)-3-(1-methyl-2-oxo-5-pyrazolo[1,5-a]pyridin-3-ylimidazo[4,5-b]pyridin-3-yl)piperidin-1-yl]-3-oxopropanenitrile
3-{(3S)-3-[1-methyl-2-oxo-5-(pyrazolo[1,5-a]pyridin-3-
yl)-1,2-dihydro-3H-imidazo[4,5-b]pyridin-3-yl]piperidin1-yl}-3-oxopropanenitrile
Janus kinase inhibitor, anti-inflammatory, 5N5L287M8T, KN 002, KN-002
Single and Multiple Ascending Dose Study of KN-002
CTID: NCT05006521
Phase: Phase 1
Status: Completed
Date: 2024-08-07
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2011157397&_cid=P11-MH2TVG-48083-1
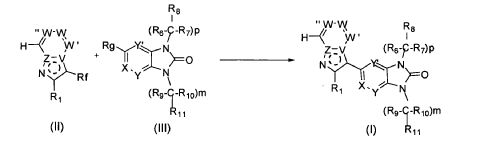
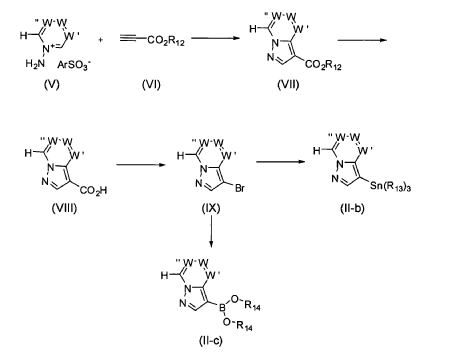
SYN
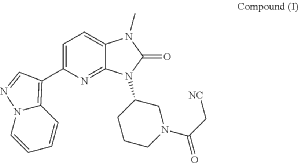
It has now been found that a drug substance disclosed in WO2011/051452, namely the compound (S)-3-(3-(1-methyl-2-oxo-5-(pyrazolo[1,5-a]pyridine-3-yl)-1H-imidazo[4,5-b]pyridine-3(2H)-yl)piperidin-1-yl)-3-oxopropanenitrile having the structure shown below and known herein as compound (I) can be prepared in different polymorphic forms. Surprisingly one form exists as a polymorph with particularly advantageous stability properties. Compound (I) as prepared following the process in WO2011/051452 is known as Form I herein.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US76222175&_cid=P11-MH2U0A-51623-1
PAT
- N-containing heteroaryl derivatives as jak3 kinase inhibitorsPublication Number: KR-101675614-B1Priority Date: 2009-10-29Grant Date: 2016-11-11
- N-containing heteroaryl derivatives as jak3 kinase inhibitorsPublication Number: US-2012245140-A1Priority Date: 2009-10-29
- N-containing heteroaryl derivatives as jak3 kinase inhibitorsPublication Number: US-2013131038-A9Priority Date: 2009-10-29
- N-containing heteroaryl derivatives as JAK3 kinase inhibitorsPublication Number: US-8501735-B2Priority Date: 2009-10-29Grant Date: 2013-08-06
- N-containing heteroaryl derivatives as JAK3 kinase inhibitorsPublication Number: US-8946257-B2Priority Date: 2009-10-29Grant Date: 2015-02-03
- Heteroaryl imidazolone derivatives as jak inhibitorsPublication Number: EP-2582703-A1Priority Date: 2010-06-15
- Heteroaryl Imidazolone Derivatives as Jap InhibitorsPublication Number: KR-20130113331-APriority Date: 2010-06-15
- Heteroaryl imidazolone derivatives as jak inhibitorsPublication Number: US-2013089512-A1Priority Date: 2010-06-15
- Heteroaryl imidazolone derivatives as jak inhibitorsPublication Number: WO-2011157397-A1Priority Date: 2010-06-15
- N-containing heteroaryl derivatives as jak3 kinase inhibitorsPublication Number: EP-2493895-B1Priority Date: 2009-10-29Grant Date: 2017-04-26
- Novel polymorphsPublication Number: US-2018016284-A1Priority Date: 2015-02-05
- Novel polymorphsPublication Number: US-2019031687-A1Priority Date: 2015-02-05
- Novel polymorphsPublication Number: WO-2016124464-A1Priority Date: 2015-02-05
- Heteroaryl imidazolone derivatives as jak inhibitorsPublication Number: CA-2802588-A1Priority Date: 2010-06-15
- Heteroaryl imidazolone derivatives as JAK inhibitorsPublication Number: CN-102933583-APriority Date: 2010-06-15
- Novel polymorphsPublication Number: EP-3053927-A1Priority Date: 2015-02-05
- Novel polymorphsPublication Number: EP-3253769-B1Priority Date: 2015-02-05Grant Date: 2019-03-13
- New polymorphPublication Number: JP-2018502929-APriority Date: 2015-02-05
- New polymorphPublication Number: JP-6685326-B2Priority Date: 2015-02-05Grant Date: 2020-04-22
- PolymorphsPublication Number: US-10087196-B2Priority Date: 2015-02-05Grant Date: 2018-10-02
- Crystalline form of a JAK3 kinase inhibitorPublication Number: US-10155757-B2Priority Date: 2015-03-10Grant Date: 2018-12-18
- Crystalline form of a jak3 kinase inhibitorPublication Number: US-2018044336-A1Priority Date: 2015-03-10
- Crystalline form of a jak3 kinase inhibitorPublication Number: WO-2016142201-A1Priority Date: 2015-03-10
- Polymorphic forms of (s)-3-(3(1-methyl-2-oxo-5-(pyrazolo[1,5-a]pyridine-3(2h)-yl)piperidin-1-yl)-3-oxopropanenitrilePublication Number: CA-2972977-CPriority Date: 2015-02-05Grant Date: 2019-04-09
- polymorphPublication Number: CN-107207533-BPriority Date: 2015-02-05Grant Date: 2019-04-16
- Formulation of a pan-jak inhibitorPublication Number: TW-202440105-APriority Date: 2022-12-02
- Formulation of a pan-jak inhibitorPublication Number: US-2024261224-A1Priority Date: 2022-12-02
- Formulation of a pan-jak inhibitorPublication Number: WO-2024119058-A2Priority Date: 2022-12-02
- Formulation of a pan-jak inhibitorPublication Number: WO-2024119058-A3Priority Date: 2022-12-02
- Crystalline form of a jak3 kinase inhibitorPublication Number: EP-3268364-A1Priority Date: 2015-03-10
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////Frevecitinib, Janus kinase inhibitor, anti-inflammatory, 5N5L287M8T, KN 002, KN-002
Envudeucitinib
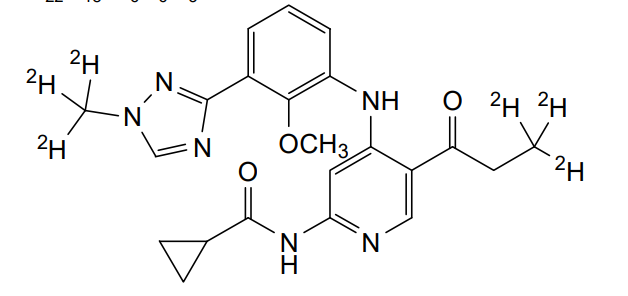
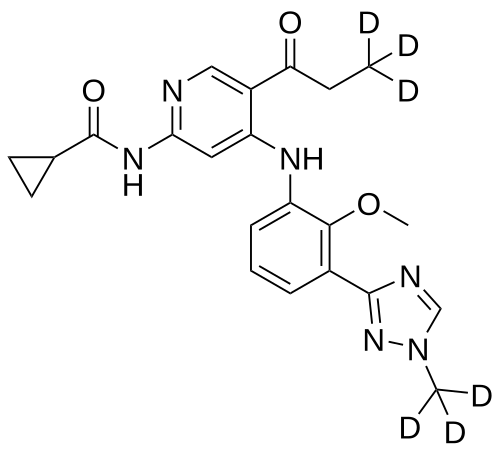
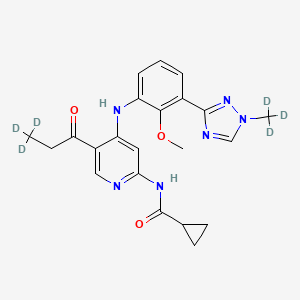
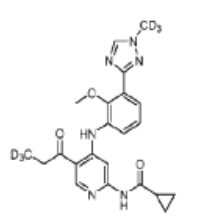
Envudeucitinib
CAS 2417135-66-9
MF C22H18[2]H6N6O3 MW426.5 g/mol
N-[4-{2-methoxy-3-[1-(2H3)methyl-1H-1,2,4-triazol-3-yl]anilino}-5-(3,3,3-2H3)propanoylpyridin-2-yl] cyclopropanecarboxamide
N-(4-(2-methoxy-3-(1-(trideuteriomethyl)-1,2,4-triazol-3-yl)anilino)-5-(3,3,3-trideuteriopropanoyl)pyridin-2-yl)cyclopropanecarboxamide
N-[4-[2-methoxy-3-[1-(trideuteriomethyl)-1,2,4-triazol-3-yl]anilino]-5-(3,3,3-trideuteriopropanoyl)pyridin-2-yl]cyclopropanecarboxamide
Janus kinase inhibitor, anti-inflammatory, Fronthera U.S. Pharmaceuticals, psoriasis, FTP 637
Envudeucitinib is an investigational new drug that is being evaluated for the treatment of psoriasis. It is a selective tyrosine kinase 2 (TYK2) inhibitor developed by Fronthera U.S. Pharmaceuticals LLC and now owned by Alumis, Inc. for the treatment of autoimmune diseases. Envudeucitinib targets the TYK2 signaling pathway, which plays a crucial role in regulating multiple pro-inflammatory cytokines such as IL-12, IL-23, and type I interferons.[1][2]
PAT
- Crystalline forms of a tyk2 inhibitor and uses thereofPublication Number: WO-2024081603-A1Priority Date: 2022-10-10
- Crystalline forms of a tyk2 inhibitor and uses thereofPublication Number: WO-2024059529-A1Priority Date: 2022-09-12
- Tyk2 inhibitors and uses thereofPublication Number: WO-2023227946-A1Priority Date: 2022-05-27
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2024081603&_cid=P11-MGGDZU-88200-1
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023227946&_cid=P11-MGGE36-91523-1
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | FTP-637 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2417135-66-9 |
| PubChem CID | 158715582 |
| IUPHAR/BPS | 13205 |
| UNII | KD2MDJ4GAB |
| KEGG | D13123 |
| Chemical and physical data | |
| Formula | C22H18D6N6O3 |
| Molar mass | 426.506 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Deng L, Wan L, Liao T, Wang L, Wang J, Wu X, et al. (August 2023). “Recent progress on tyrosine kinase 2 JH2 inhibitors”. International Immunopharmacology. 121 110434. doi:10.1016/j.intimp.2023.110434. PMID 37315371.
- Loo WJ, Turchin I, Prajapati VH, Gooderham MJ, Grewal P, Hong CH, et al. (2023). “Clinical Implications of Targeting the JAK-STAT Pathway in Psoriatic Disease: Emphasis on the TYK2 Pathway”. Journal of Cutaneous Medicine and Surgery. 27 (1_suppl): 3S – 24S. doi:10.1177/12034754221141680. PMID 36519621.
////////Envudeucitinib, Janus kinase inhibitor, anti-inflammatory, Fronthera U.S. Pharmaceuticals, psoriasis, FTP 637
CLOBETASOL
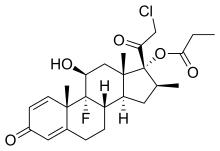
Clobetasol propionate
- Molecular FormulaC25H32ClFO5
- Average mass466.970 Da
CCI 4725, CCI-4725, GR 2/925, GR-2/925,(8S,9R,10S,11S,13S,14S,16S,17R)-17-(chloroacetyl)-9-fluoro-11-hydroxy-10,13,16-trimethyl-3-oxo-6,7,8,9,10,11,12,13,14,15,16,17-dodecahydro-3H-cyclopenta[a]phenanthren-17-yl propanoate
246-634-3[EINECS], 25122-46-7[RN]
(11β,16β)-21-chloro-9-fluoro-11-hydroxy-16-methyl-3,20-dioxopregna-1,4-dien-17-yl propanoate
Active Moieties
| NAME | KIND | UNII | CAS | INCHI KEY |
|---|---|---|---|---|
| Clobetasol | prodrug | ADN79D536H | 25122-41-2 | FCSHDIVRCWTZOX-DVTGEIKXSA-N |
Clobetasol
CAS Registry Number: 25122-41-2
CAS Name: (11b,16b)-21-Chloro-9-fluoro-11,17-dihydroxy-16-methylpregna-1,4-diene-3,20-dione
Molecular Formula: C22H28ClFO4, Molecular Weight: 410.91
Percent Composition: C 64.30%, H 6.87%, Cl 8.63%, F 4.62%, O 15.57%
Literature References: Topical corticosteroid. Prepn: Elks et al.,DE1902340; eidem,US3721687 (1969, 1973 both to Glaxo). Review of pharmacology and clinical efficacy in skin disorders: E. A. Olsen, R. C. Cornell, J. Am. Acad. Dermatol.15, 246-255 (1986).
Derivative Type: 17-Propionate
CAS Registry Number: 25122-46-7
Manufacturers’ Codes: GR-2/925
Trademarks: Clobesol (GSK); Dermovate (GSK); Olux (Connetics); Psorex (GSK); Temovate (GSK)
Molecular Formula: C25H32ClFO5
Molecular Weight: 466.97
Percent Composition: C 64.30%, H 6.91%, Cl 7.59%, F 4.07%, O 17.13%
Properties: White or almost white colorless, crystalline powder, mp 195.5-197°. [a]D +103.8° (c = 1.04 in dioxane). uv max (ethanol): 237 nm (e 15000). Insol in water.
Melting point: mp 195.5-197°
Optical Rotation: [a]D +103.8° (c = 1.04 in dioxane)
Absorption maximum: uv max (ethanol): 237 nm (e 15000)
Therap-Cat: Glucocorticoid; anti-inflammatory.
Keywords: Glucocorticoid.
Clobetasol propionate is a corticosteroid used to treat corticosteroid-responsive dermatoses and plaque psoriasis.
Clobetasol propionate is a corticosteroid used to treat skin conditions such as eczema, contact dermatitis, seborrheic dermatitis, and psoriasis.[2] It is applied to the skin as a cream, ointment, or shampoo.[2][3] Use should be short term and only if other weaker corticosteroids are not effective.[3] Use is not recommended in rosacea or perioral dermatitis.[2]
Common side effects include skin irritation, dry skin, redness, pimples, and telangiectasia.[2] Serious side effects may include adrenal suppression, allergic reactions, cellulitis, and Cushing’s syndrome.[2] Use in pregnancy and breastfeeding is of unclear safety.[4] Clobetasol is believed to work by activating steroid receptors.[2] It is a US Class I (Europe: class IV) corticosteroid, making it one of the strongest available.
Clobetasol propionate was patented in 1968 and came into medical use in 1978.[5] It is available as a generic medication.[3] In 2019, it was the 180th most commonly prescribed medication in the United States, with more than 3 million prescriptions.[6][7]
SYNTHESIS OF KEY INTERMEDIATE
SYN

DE 1902340
US 3992422
DE 2613875
EP 72200
WO 2012122452
CN 112110972
PATENT
IN 201821008147
Clobetasol propionate (C25H32ClFO5); CAS Registry No.[25112-46-7]; IUPAC name: 17-(2′- Chloroacetyl)-9-fluoro-l l-hydroxy-10,13,16-trimethyl-3-oxo-6,7,8,l 1,12,14,15,16-octahydrocyclopenta[a]phenanthren-17-yl] propionate is a potent halogen adrenal corticosteroid of the gluco-corticoid class used to treat various skin disorders including eczema and psoriasis. It is also highly effective for contact dermatitis caused by exposure to poison ivy/oak.In the US 3721687Apatentshow use of methanesulfonyl chloride and Pyridine as base to protect alcohol and at the time of LiCI reaction results with 10-15%ene impurity and less yield.In the methanesulfonyl chloride step used with pyridine as base which is a hazardous.Mesyl compound converted to Clobetasol propionate by using LiCl in Dimethylformamide reaction at IOO-IlO0C forms 10-15% with ene impurity. The synthesis of Clobetasol propionate results in small quantities of the eneimpurity. Clobetasol propionate desired compound to be with impurities which must be minimized. Ene impurity can be reduced to very low levels by reaction itself. However, if used recrystallization reduce ene impurity it is time consuming and very expensive. Further, because recrystallizations have high losses, unacceptably low yields.


Example I: Betamethasone to betamethasone 17- propionate To a 100 ml 4-neck round bottom flask (RBF) equipped with halfmoon stirrer, thermowelland addition funnel, mounted in a tub bath, was charged betamethasone (5.0g, 0.0127mole), Dimethylformamide (20ml). Cooled the reaction mass to 10-15°C. Slowly added trimethyl ortho propionate (3.42g, 0.0255mole) and p-toluenesulfonic acid (PTSA)(0.30g, 0.00174 mole) to the reaction mass at 10-15°C. Stirred the contents 10-15°C for 4 hr. The reaction was monitored for completion by TLC. Further continued stirring at the same temperature for Ihr till reaction complies by TLC. After reaction completion, added H2SO4UP to pH=1.0-2.0 in to reaction mass.Reaction mass was quenched in Purified water (25ml) at 25-30°C. Cooled reaction mass temperature to 0-5°C. Stirred for I hr and filtered and washed with Purified water (10mlX2). Suck dried under vacuum completely to get cream coloured solid. Dried in tray drier at 50-55°C.Dry weight-5.40g(94.50%); HPLC: 98.5%;mp215-218°C. IR (KBr, on’):3454.90, 3370.99 (-OH); 1719.86, 1659.10 (C=O)iC25H33FO6;
MS 448.52m/z 449.2255 [M+H]; 1HNMR (300MHz, CDCl3S ppm): Spectrum is recorded on Varian, and Tetra Methyl Silane (TMS) as internal standard. 1H-NMR Spectrum shows Aromatic-HK-7.17-7.22(d,lH); Hj-6.37-6.38 (d,lH);Hr6.14 (s,lH); HF-4.04-4.06(s,2H);HE2.23-2.28 (q,2H);Hc-l.39-1.43 (d,3H); HB-1.14 (t,3H);HA-0.96-2.96 (m,20H). 13CMR (300MHz, CDCl3Sppm): 8.692 (CH2-CH3); 16.693; 19.664; 21.353; 23.042; 27.568; 30.387; 36.456; 43.400; 46.547; 47.422; 71.760; 93.547; 124.307; 125.728; 127.903; 129.222; 130.443; 132.472; 145.632; 153.044; 167.424; 175.031 (O-C=O); 185.772 (Cyclic C=O); 196.732 (CH-CO-CH2-OH).
Example 2: Betamethasone 17- propionateto betamethasone 21-tosylate 100ml 4-neck RBF equipped with halfmoon stirrer, thermowell, reflux condenser mountained in water bath, was charged Stage-1 (5.0g, O.Olllmole), Dimethylformamide (20ml). Added 4-Dimethylaminopyridine as base (4.10g, 0.0335mole) and p-toluenesulfonyl chloride (4.24.Og, 0.0222mole)slowly, Stirredfor2-3 hr at 25-30°C. Stirred reaction mass at 25-30°C till reaction complies by TLC.As such reaction mass used insitue for next step. Reaction mass aliquot taken (2ml) and quenched in DM water (20ml), precipited material fdtered and washed with DM water (20ml). Suck dried well. Dried in tray drier at 50-55°C to get dry white solid. Dry weight-0.598g, (89.0%); HPLC: 98.5%; mp-170-175°C (dec). IR (KBr,cm”1):3291.91, 2980.39 (-OH); 1739.15, 1661.99 (C=O); C32H39FO8S; MS 602.71mA 603.2317 [M+H]; 1HNMR (300MHz, CDCl3Sppm): Spectrum is recorded on Varian, and Tetra Methyl Silane (TMS) as internal standard. 1H-NMR Spectrum shows Aromatic-Ηκ7.17-7.22(d,lH); Hj-6.37-6.38 (d,lH);Hr6.14 (s,lH); HG-4.334-4.393 (m,lH);HF-3.846- 4.007(d,2H);HE-2.273-2.349 (q,2H);HD-l.671-1.688 (s,lH); Hc-I-306-1.331 (d,3H); Hb1.055-1.105 (t,3H);HA-0.941 -2.634 (m,18H).13CMR (300MHz, CDCl3Sppm): 9.055 (CH2- CH3); 17.244; 20.002; 21.353; 23.168; 27.901; 30.622; 33.508; 34.881; 36.783; 43.642; 46.637; 47.330; 48.113; 48.417; 66.613; 70.902; 93.801; 102.732; 124.415; 129.324; 130.443; 132.472; 145.632; 153.583; 168.042; 174.853 (O-C=O); 186.208 (Cyclic C=O); 205.491 (CH-CO-CH2-OAr).
Example 3:Betamethasone 21-tosylateto Clobetasol propionate As such reaction mass used insitue for next step. Added. lithium chloride (LiCl)1.04 gm (0.0245mole). Stirred the reaction mass at 60-65°C for 5-6 hr.Reaction completion checked by TLC.After reaction completion, Added DM water (200ml). Stirred the reaction mass at 10-15°C for Ihr and Filtered washed with DM water (30mlx2).Dried in oven at 50-55°C to get white crystalline powder. Dry weight-4.42gm, (85.0%); HPLC:99.70%;mp-158-161°C. IR (KBr, cm_1):3299.62, 2976.53 (-OH); 1734.32, (C=0);1662.95 (C=C);C25H32C1F05; MSΑβ6.9Ίτη/ζ 467 [M+H];’HNMR (300MHz, CDCl35ppm): Spectrum is recorded on Varian, and Tetra Methyl Silane (TMS) as internal standard. 1H-NMR Spectrum shows AromaticHk-7.094-7. 128(d,IH); Hj-6.267-6.307 (d,lH); Hr6.066-6.076 (s,IH); H0-4.334-4.393 (m,lH); Hf-3.846-4.007 (d,2H); HE-2.273-2.349 (q,2H); Hd-I .671-1.688 (s,lH); Hc-1.306- 1.331 (d,3H); Hb-I .055-1.105 (t,3H); HA-0.941-2.634 (m,17H).13CMR (300MHz, CDCl35ppm): 8.692 (CH2-CH3); 16.693; 19.664; 21.353; 23.042; 27.568; 30.387; 36.456; 41.104; 46.547; 47.422; 71.760; 93.547; 124.307; 125.728; 127.903; 129.222; 130.443; 132.472; 145.632; 153.044; 168.312; 173.101 (O-C=O); 185.802 (Cyclic C=O); 204.602(CH-CO-CH2-C1)
SYN
Ruben Vardanyan, Victor Hruby, in Synthesis of Best-Seller Drugs, 2016
Synthesis of clobetasol propionate (27.1.13) starts from the known betamethasone 17-propionate (27.1.26), a potent glucocorticoid steroid with antiinflammatory and immunosuppressive properties, which was mesylated with methanesulfonyl chloride in pyridine to produce 9α-fluoro-11β-hydroxy-21-methylsulfonyloxy-16β-methyl 17-propionyloxypregna-1,4-diene-3,20-dione (27.1.27). The obtained product was refluxed in acetone, DMF, and dry LiCl mixture to produce the desired clobetasol propionate (27.1.13) [34] (Scheme 27.2.).

Clobetasol propionate, its structural formula (formula (I)), is a potent halogen-containing adrenocorticoid drug, has strong anti-inflammatory, anti-pruritic and vasoconstrictive effects, and its anti-inflammatory effect is approximately hydrogenated It is 112 times that of cortisone, and it is also used to treat neurodermatitis, contact dermatitis, eczema, discoid lupus erythematosus and other symptoms. It is currently widely used in clinical practice. It has been very popular in the international market and ranks among the top hormones. At present, there are only a few domestic companies in normal production, and the total yield is about 88%.[0003]

[0004] Formula (I).[0005] The process route for the production of synthetic clobetasol propionate is complex, technically difficult, and product quality requirements are strict. This is due to the complex structure of corticosteroids. The chemical structure of this type of drug is composed of three six-membered rings and one five-membered ring fused together to form a special molecular structure composed of 21 carbon atoms, with special molecular configuration steric effects and steric barriers. Group role. The functional groups on the drug structure interfere with each other, which makes the chemical reaction very complicated. It is manifested in many synthetic process steps, low raw material utilization rate, large amount of auxiliary materials, long production cycle, and many side reactions. The reaction process has various problems such as a large amount of solvents, a large amount of waste water and waste gas, and difficulty in recycling. Low technical indicators, low cost and other aspects.[0006] US patent, patent number 3721687, discloses two synthetic processes.[0007] Process method (1) adopts 9a-fluoro-113-hydroxy-16a-methyl-17 oxopropyl-1,4-diene-3,20-dione to synthesize clobetasol propionate, 9a -Fluoro-11-hydroxy-16 a -methyl-17oxopropyl-1,4-diene-3,20-dione and lithium chloride mixture, mixed with dimethylformamide (DMF) in acetone The solution is refluxed for four days, the solution is moved to a vacuum, ethanol, methanol, and acetone are added, and the mixture is refluxed for another 4 days. Most of the solution is moved to a vacuum, water is added to the residue, the crude product is put into the ether solution, and the mixture is passed through with chloroform. The aluminum is purified by filtration and recrystallized with ethanol to produce clobetasol propionate as a raw material. Method I uses too much acetone, and there is a certain risk of operation.[0008] Process method 2 adopts 21-chloro-9a-fluoro-1I@ -hydroxy-16a-methyl-17_oxopropyl-4ene-3,20-dione to synthesize clobetasol propionate Cable. Dissolve 21-chloro-9 a -fluoro-11 P -hydroxy-16 a -methyl-17oxopropyl_4ene-3,20-dione in acetone, cool in an ice bath, and add slowly while stirring Chromic acid (prepared by chromic acid: add 53.3ml of concentrated sulfuric acid to 250ml of water and add 66.7g of chromium trioxide); 4 hours later, the mixture reaches room temperature, ether is added, and it is left for another 20 minutes. The mixture is washed with water, and then the solution is moved to a vacuum ; The residue is recrystallized with acetone-petroleum ether, which pollutes the environment. In the past, organic solvents were not safe for production operations. [0009] Chinese patent, application number 200610053511.5, provides a method of mixing betamethasone 17-propionate sulfonate and anhydrous lithium chloride in a ratio of 1:1 to 2 and dissolving in dimethylformamide ( DMF), the chlorination reaction is carried out; second, after the chlorination reaction is complete, it is separated by ice water, and then centrifuged to dry, after drying, the crude clobetasol propionate is obtained; third, the clobetasol propionate is crude The crude tasol is dissolved in methanol or ethanol, activated carbon is added, decolorized, filtered, and the activated carbon is recovered; fourth, the filtrate is concentrated under reduced pressure, crystallized, dehydrated, and dried to obtain the raw material of clobetasol propionate. It has the characteristics of easy availability of starting materials, simple reaction steps, less dangerous and harmful solvents, mature technology, and convenient industrial production.[0010] The process route is as follows:[0011]

[0012] Clobetasol propionate uses betamethasone as the starting material, goes through the steps of cyclic ester-hydrolysis-sulfonation-chlorination, and then undergoes rough refinement to obtain clobetasol propionate-a refined substance, and then undergoes dissolution , Filtration, concentration, cooling, centrifugation, and drying to obtain clobetasol propionate. But its process route is longer, there are many influencing factors, and there are many side reactions. Moreover, the solvents used are very polluting and difficult to recycle.

Example 1[0034] 20g of Betamethasone 17-ester obtained by cyclic ester hydrolysis reaction was dissolved in 150ml of acetone, and after fully stirring and dissolving, 6g of ZnCl2 was added, and the temperature was raised to 35°C, and then 30g of BTC was introduced, After the BTC is passed, the reaction is kept warm for 3 hours. After the reaction is completed, the temperature is 40°C, and the concentration is reduced under reduced pressure until the solution contains 30ml of acetone. Then 300ml of drinking water is added for water separation and filtration. After drying for 16 hours at °C, 19.64 g of crude clobetasol propionate was obtained. The yield was 98.2%, and the crude clobetasol propionate content was 96.9% after analysis.Example 2[0036] 20g of betamethasone 17-ester compound obtained by cyclic ester hydrolysis reaction was dissolved in 150ml of acetone, and after fully stirring and dissolving, 7.2g of FeCl3 was added, heated to 30°C, and then 24g of BTC was introduced After the BTC is passed, the reaction is kept warm for 2 hours. After the reaction is completed, the solution is concentrated under reduced pressure at a temperature of 35°C until the solution contains 20ml of acetone, and then 300ml of drinking water is added for water precipitation, filtered, and finally at the temperature After drying for 10 hours at 85°C, 19.5 g of crude clobetasol propionate was obtained. The yield was 97.5%, and the crude clobetasol propionate content was 95.6% after analysis.Example 3[0038] 20g of Betamethasone 17-ester obtained by the cyclic ester hydrolysis reaction was dissolved in 150ml of acetone, and after fully stirring and dissolving, 4g of AlCl3 was added and the temperature was raised to 35°C, and then 28g of BTC was introduced, After the BTC is passed through, the reaction is kept warm for 4 hours. After the reaction is completed, the temperature is 30°C, and concentrated under reduced pressure until the solution contains 20ml of acetone. Then 300ml of drinking water is added for water precipitation, filtered, and finally at a temperature of 75 After drying for 18 hours at °C, 19.62g crude clobetasol propionate was obtained. The yield was 98.1%, and the crude clobetasol propionate content was 95.8% after analysis.Example 4[0040] The betamethasone 17-ester 20g obtained by the cyclic ester hydrolysis reaction was dissolved in 100ml of acetone, and after being fully stirred to dissolve, 4g of ZnCl3 was added, heated to 40°C, and then passed into 25g of BTC, After the BTC is passed, the reaction is kept for 5 hours. After the reaction is completed, the temperature is 40°C, and the concentration is reduced under reduced pressure until the solution contains 20ml of acetone. Then 200ml of drinking water is added for water precipitation, filtered, and finally at a temperature of 80°C. After drying for 18 hours at °C, 19.54 g of crude clobetasol propionate was obtained. The yield was 97.7%, and the crude clobetasol propionate content was 96.2% after analysis.[0041] Example 5 [0042] 20g of Betamethasone 17-ester obtained by cyclic ester hydrolysis reaction was dissolved in 200ml of acetone, and after fully stirring and dissolving, 8g of ZnCl3 was added and the temperature was raised to 50°C. Then pass in 40g BTC. After passing the BTC, keep it warm and react for 3 hours. After the reaction is completed, perform vacuum concentration at a temperature of 40°C until the solution contains 40ml of acetone, and then add 400ml of drinking water for hydrolysis. Filter, and finally dry at 85°C for 18 hours to obtain 19.58 g of crude clobetasol propionate. The yield was 97.9%, and the crude clobetasol propionate content was 96.9% after analysis.Example 6[0044] 20g of betamethasone 17-ester compound obtained by cyclic ester hydrolysis reaction was dissolved in 80ml of acetone, and after fully stirring and dissolving, 6g of ZnCl3 was added, and after the temperature was raised to 40°C, 25g of BTC was introduced, After the BTC is passed, the reaction is kept for 3 hours. After the reaction is completed, it is concentrated under reduced pressure at a temperature of 40°C, and concentrated until the solution contains 10ml of acetone. Then 150ml of drinking water is added for water precipitation, filtered, and finally at a temperature of 85°C. After drying for 18 hours at °C, 19.3g crude clobetasol propionate was obtained. The yield was 96.5%, and the crude clobetasol propionate content was 96.5% after analysis.
Publication numberPriority datePublication dateAssigneeTitleUS3721687A *1968-01-191973-03-20Glaxo Lab Ltd3-keto-delta 4-9alpha-halo-11-oxygenated-16-methyl or methylene-17alpha-acyloxy-20-keto-21-halo pregnenesCN1923842A *2006-09-112007-03-07Zhejiang Dingtai Pharmaceutical Co., Ltd.Manufacturing method of clobetasol propionate
Publication numberPriority datePublication dateAssigneeTitleFamily To Family CitationsCN105646630A *2015-08-102016-06-08Shandong Taihua Biological Technology Co., Ltd.One-pot Preparation of Clobetasol Propionate IntermediateCN112110972A *2019-06-212020-12-22Henan Lihua Pharmaceutical Co., Ltd.A kind of preparation method of clobetasol propionateCN112028957A *2020-07-292020-12-04Henan Lihua Pharmaceutical Co., Ltd.A kind of clobetasol propionate intermediate and preparation method
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Medical uses
Clobetasol propionate is used for the treatment of various skin disorders including eczema, herpes labialis,[8] psoriasis, and lichen sclerosus. It is also used to treat several auto-immune diseases including alopecia areata, lichen planus (auto immune skin nodules), and mycosis fungoides (T-cell skin lymphoma). It is used as first-line treatment for both acute and chronic GVHD of the skin.[9]
Clobetasol propionate is used cosmetically by dark-skinned women for skin whitening, although this use is controversial. The U.S. Food and Drug Administration has not approved it for that purpose, and sales without a prescription are illegal in the U.S. Nonetheless, skin-whitening creams containing this ingredient can sometimes be found in ethnic beauty supply stores in New York City and on the internet. It is also sold internationally, and does not require a prescription in some countries. Whitening creams with clobetasol propionate, such as Hyprogel, can make skin thin and easily bruised, with visible capillaries, and acne. It can also lead to hypertension, elevated blood sugar, suppression of the body’s natural steroids, and stretch marks, which may be permanent.[10]
Clobetasol propionate is, along with mercury and hydroquinone, “amongst the most toxic and most used agents in lightening products.” Many products sold illegally have higher concentrations of clobetasol propionate than is permitted for prescription drugs.[11]
Contraindications
According to the California Environmental Protection Agency, clobetasol propionate should not be used by pregnant women, or women expecting to become pregnant soon, as studies with rats shows a risk of birth defects:[12]
“Studies in the rat following oral administration at dosage levels up to 50 mcg/kg per day revealed that the females exhibited an increase in the number of resorbed embryos and a decrease in the number of living fetuses at the highest dose. Pregnancy: Teratogenic Effects (i.e., possibility of causing abnormalities in fetuses): Pregnancy Category C: Clobetasol propionate has not been tested for teratogenicity when applied topically; however, it is absorbed percutaneously, and when administered subcutaneously it was a significant teratogen in both the rabbit and mouse. Clobetasol propionate has greater teratogenic potential than steroids that are less potent. There are no adequate and well-controlled studies of the teratogenic effects of clobetasol propionate in pregnant women. Temovate Cream and Ointment should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.”
Forms
Clobetasol propionate is marketed and sold worldwide under numerous names, including Clobex, Clob-x (Colombia), Clovate, Clobet (Biolab Thailand) Clonovate (T.O. Chemicals, Thailand), Cormax (Watson, US), Haloderm (Switzerland, by ELKO Org), Pentasol (Colombia), Cosvate, Clop (Cadila Healthcare, India), Propysalic (India), Temovate (US), Dermovate[13] (GlaxoSmithKline, Canada, Estonia, Pakistan, Switzerland, Portugal, Romania, Israel), Olux, ClobaDerm, Tenovate, Dermatovate, Butavate, Movate, Novate, Salac (Argentina), and Powercort, Lotasbat and Kloderma (Indonesia), Lemonvate (Italy), Delor (Ethiopia), Psovate (Turkey).
References
- ^ “Clobetasol Propionate Topical Ointment 0.05% Information – Drug Encyclopedia”. Kaiser Permanente.
- ^ Jump up to:a b c d e f “Clobetasol Propionate Monograph for Professionals”. Drugs.com. American Society of Health-System Pharmacists. Retrieved 13 April 2019.
- ^ Jump up to:a b c British national formulary : BNF 76 (76 ed.). Pharmaceutical Press. 2018. p. 1210. ISBN 9780857113382.
- ^ “Clobetasol topical Use During Pregnancy”. Drugs.com. Retrieved 13 April 2019.
- ^ Fischer J, Ganellin CR (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 487. ISBN 9783527607495.
- ^ “The Top 300 of 2019”. ClinCalc. Retrieved 16 October 2021.
- ^ “Clobetasol – Drug Usage Statistics”. ClinCalc. Retrieved 16 October 2021.
- ^ Hull C, McKeough M, Sebastian K, Kriesel J, Spruance S (March 2009). “Valacyclovir and topical clobetasol gel for the episodic treatment of herpes labialis: a patient-initiated, double-blind, placebo-controlled pilot trial”. Journal of the European Academy of Dermatology and Venereology. 23 (3): 263–7. doi:10.1111/j.1468-3083.2008.03047.x. PMID 19143902. S2CID 205588376.
- ^ E. Fougera and Co. “CLOBETASOL PROPIONATE CREAM USP, 0.05% CLOBETASOL PROPIONATE OINTMENT USP, 0.05%<“. NIH Daily Med.
- ^ Saint Louis C (January 15, 2010). “Creams Offering Lighter Skin May Bring Risks”. New York Times.
- ^ Gbetoh MH, Amyot M (October 2016). “Mercury, hydroquinone and clobetasol propionate in skin lightening products in West Africa and Canada”. Environmental Research. 150: 403–410. Bibcode:2016ER….150..403G. doi:10.1016/j.envres.2016.06.030. hdl:1866/19006. PMID 27372064.
- ^ Office of Environmental Health Hazard Assessment (August 22, 1997). “Chemicals Under Consideration For Possible Listing Via The “Formally Required To Be Labeled Or Identified” Mechanism”. California Environmental Protection Agency. Archived from the original on 2001-07-20. Retrieved 2007-05-06.
- ^ “DERMOVATE 0.05% W/V OINTMENT – Clobetasol Topical(0.05% w/v) Glaxo SmithKline Pharmaceuticals Ltd”. GNH. Retrieved 2021-07-16.
External links
- “Clobetasol propionate”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Pronunciation | /kloʊˈbeɪtəsɒl/[1] |
| Trade names | Temovate, Clobex, Cormax, others |
| AHFS/Drugs.com | Monograph |
| License data | US DailyMed: Clobetasol_propionate |
| Pregnancy category | AU: B3 |
| Routes of administration | Topical |
| ATC code | D07AD01 (WHO) |
| Legal status | |
| Legal status | In general: ℞ (Prescription only) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 25122-46-7 |
| PubChem CID | 32798 |
| IUPHAR/BPS | 7062 |
| DrugBank | DB01013 |
| ChemSpider | 30399 |
| UNII | 779619577M |
| KEGG | D01272 |
| ChEBI | CHEBI:31414 |
| ChEMBL | ChEMBL1159650 |
| CompTox Dashboard (EPA) | DTXSID6045907 |
| ECHA InfoCard | 100.042.380 |
| Chemical and physical data | |
| Formula | C25H32ClFO5 |
| Molar mass | 466.97 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
//////////////////Clobetasol propionate, CCI 4725, CCI-4725, GR 2/925, GR-2/925, Glucocorticoid, anti-inflammatory
[H][C@@]12C[C@H](C)[C@](OC(=O)CC)(C(=O)CCl)[C@@]1(C)C[C@H](O)[C@@]1(F)[C@@]2([H])CCC2=CC(=O)C=C[C@]12C

NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}
{kind=link}
{kind=link}
{kind=link}