

Roche is investing $775 million in cash and equity for access to Blueprint Medicines’ oncology drug candidate pralsetinib, which is under review by the US Food and Drug Administration.
Home » Posts tagged '2020 APPROVALS' (Page 3)
Tag Archives: 2020 APPROVALS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Bulevirtide acetate

Bulevirtide acetate
(N-Myristoyl-glycyl-L-threonyl-L-asparaginyl-L-leucyl-L-seryl-L-valyl-Lprolyl-L-asparaginyl-L-prolyl-L-leucyl-glycyl-L-phenylalanyl-L-phenylalanyl-L-prolyl-L-aspartyl-L-histidyl-Lglutaminyl-L-leucyl-L-aspartyl-L-prolyl-L-alanyl-L-phenylalanyl-glycyl-L-alanyl-L-asparaginyl-L-seryl-Lasparaginyl-L-asparaginyl-Lprolyl-L-aspartyl-L-tryptophanyl-L-aspartyl-L-phenylalanyl-L-asparaginyl-L-prolylL-asparaginyl-L-lysyl-L-aspartyl-L-histidyl-L-tryptophanyl-L-prolyl-L-glutamyl-L-alanyl-L-asparaginyl-L-lysylL-valylglycinamide, acetate salt.
molecular formula C248H355N65O72,
molecular mass is 5398.9 g/mol
ブレビルチド酢酸塩;
APROVED 2020/7/31, EU, Hepcludex
MYR GmbH
|
Antiviral, Entry inhibitor
|
|
| Disease |
Hepatitis delta virus infection
|
|---|
Bulevirtide is a 47-amino acid peptide with a fatty acid, a myristoyl residue, at the N-terminus and an amidated C-terminus. The active substance is available as acetate salt. The counter ion acetate is bound in ionic form to basic groups of the peptide molecule and is present in a non-stoichiometric ratio. The chemical name of bulevirtide is (N-Myristoyl-glycyl-L-threonyl-L-asparaginyl-L-leucyl-L-seryl-L-valyl-Lprolyl-L-asparaginyl-L-prolyl-L-leucyl-glycyl-L-phenylalanyl-L-phenylalanyl-L-prolyl-L-aspartyl-L-histidyl-Lglutaminyl-L-leucyl-L-aspartyl-L-prolyl-L-alanyl-L-phenylalanyl-glycyl-L-alanyl-L-asparaginyl-L-seryl-Lasparaginyl-L-asparaginyl-Lprolyl-L-aspartyl-L-tryptophanyl-L-aspartyl-L-phenylalanyl-L-asparaginyl-L-prolylL-asparaginyl-L-lysyl-L-aspartyl-L-histidyl-L-tryptophanyl-L-prolyl-L-glutamyl-L-alanyl-L-asparaginyl-L-lysylL-valylglycinamide, acetate salt. It corresponds to the molecular formula C248H355N65O72, its relative molecular mass is 5398.9 g/mol
Bulevirtide appears as a white or off-white hygroscopic powder. It is practically insoluble in water and soluble at concentrations of 1 mg/ml in 50% acetic acid and about 7 mg/ml in carbonate buffer solution at pH 8.8, respectively. The structure of the active substance (AS) was elucidated by a combination of infrared spectroscopy (IR), mass spectrometry (MS), amino acid analysis and sequence analysis Other characteristics studied included ultraviolet (UV) spectrum, higher order structure (1D- and 2D- nuclear magnetic resonance spectroscopy (NMR)) and aggregation (Dynamic Light Scattering). Neither tertiary structure nor aggregation states of bulevirtide have been identified. With regard to enantiomeric purity, all amino acids are used in L-configuration except glycine, which is achiral by nature. Two batches of bulevirtide acetate were evaluated for enanatiomeric purity and no relevant change in configuration during synthesis was detected.
Bulevirtide is manufactured by a single manufacturer. It is a chemically synthesised linear peptide containing only naturally occurring amino acids. The manufacturing of this peptide is achieved using standard solidphase peptide synthesis (SPPS) on a 4-methylbenzhydrylamine resin (MBHA resin) derivatised with Rink amide linker in order to obtain a crude peptide mixture. This crude mixture is purified through a series of washing and preparative chromatography steps. Finally, the purified peptide is freeze-dried prior to final packaging and storage. The process involves further four main steps: synthesis of the protected peptide on the resin while side-chain functional groups are protected as applicable; cleavage of the peptide from the resin, together with the removal of the side chain protecting groups to obtain the crude peptide; purification; and lyophilisation. Two chromatographic systems are used for purification. No design space is claimed. Resin, Linker Fmoc protected amino acids and myristic acid are starting materials in line with ICH Q11. Sufficient information is provided on the source and the synthetic route of the starting materials. The active substance is obtained as a nonsterile, lyophilised powder. All critical steps and parameters were presented and clearly indicated in the description of the manufacturing process. The process description includes also sufficient information on the type of equipment for the SPPS, in-process controls (IPCs). The circumstances under which reprocessing might be performed were clearly presented. No holding times are proposed. Overall the process is sufficiently described.
The finished product is a white to off white lyophilised powder for solution for injection supplied in single-use vials. Each vial contains bulevirtide acetate equivalent to 2 mg bulevirtide. The composition of the finished product was presented. The powder is intended to be dissolved in 1 ml of water for injection per vial. After reconstitution the concentration of bulevirtide net peptide solution in the vial is 2 mg/ml. The components of the formulation were selected by literature review and knowledge of compositions of similar products available on the market at that time, containing HCl, water, mannitol, sodium carbonate, sodium hydrogen carbonate and sodium hydroxide. All excipients are normally used in the manufacture of lyophilisates. The quality of the excipients complies with their respective Ph. Eur monographs. The intrinsic properties of the active substance and the compounding formulation do not support microbiological growth as demonstrated by the stability data. No additional preservatives are therefore needed.
Hepcludex is an antiviral medicine used to treat chronic (long-term) hepatitis delta virus (HDV) infection in adults with compensated liver disease (when the liver is damaged but is still able to work), when the presence of viral RNA (genetic material) has been confirmed by blood tests.
HDV is an ‘incomplete’ virus, because it cannot replicate in cells without the help of another virus, the hepatitis B virus. Because of this, patients infected with the virus always also have hepatitis B.
HDV infection is rare, and Hepcludex was designated an ‘orphan medicine’ (a medicine used in rare diseases) on 19 June 2015. For further information on the orphan designation, see EU/3/15/1500.
Hepcludex contains the active substance bulevirtide.
Bulevirtide, sold under the brand name Hepcludex, is an antiviral medication for the treatment of chronic hepatitis D (in the presence of hepatitis B).[2]
The most common side effects include raised levels of bile salts in the blood and reactions at the site of injection.[2]
Bulevirtide works by attaching to and blocking a receptor (target) through which the hepatitis delta and hepatitis B viruses enter liver cells.[2] By blocking the entry of the virus into the cells, it limits the ability of HDV to replicate and its effects in the body, reducing symptoms of the disease.[2]
Bulevirtide was approved for medical use in the European Union in July 2020.[2]
Medical uses
Bulevirtide is indicated for the treatment of chronic hepatitis delta virus (HDV) infection in plasma (or serum) HDV-RNA positive adult patients with compensated liver disease.[2][3]
Pharmacology
Mechanism of action
Bulevirtide binds and inactivates the sodium/bile acid cotransporter, blocking both viruses from entering hepatocytes.[4]
The hepatitis B virus uses its surface lipopeptide pre-S1 for docking to mature liver cells via their sodium/bile acid cotransporter (NTCP) and subsequently entering the cells. Myrcludex B is a synthetic N-acylated pre-S1[5][6] that can also dock to NTCP, blocking the virus’s entry mechanism.[7]
The drug is also effective against hepatitis D because the hepatitis D virus is only infective in the presence of a hepatitis B virus infection.[7]
References
- ^ Deterding, K.; Wedemeyer, H. (2019). “Beyond Pegylated Interferon-Alpha: New Treatments for Hepatitis Delta”. Aids Reviews. 21 (3): 126–134. doi:10.24875/AIDSRev.19000080. PMID 31532397.
- ^ Jump up to:a b c d e f g “Hepcludex EPAR”. European Medicines Agency (EMA). 26 May 2020. Retrieved 12 August 2020. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Summary of opinion: Hepcludex” (PDF). European Medicines Agency. 28 May 2020.
- ^ Francisco, Estela Miranda (29 May 2020). “Hepcludex”. European Medicines Agency. Retrieved 6 August 2020.
- ^ Volz T, Allweiss L, Ben MBarek M, Warlich M, Lohse AW, Pollok JM, et al. (May 2013). “The entry inhibitor Myrcludex-B efficiently blocks intrahepatic virus spreading in humanized mice previously infected with hepatitis B virus”. Journal of Hepatology. 58 (5): 861–7. doi:10.1016/j.jhep.2012.12.008. PMID 23246506.
- ^ Abbas Z, Abbas M (August 2015). “Management of hepatitis delta: Need for novel therapeutic options”. World Journal of Gastroenterology. 21 (32): 9461–5. doi:10.3748/wjg.v21.i32.9461. PMC 4548107. PMID 26327754.
- ^ Jump up to:a b Spreitzer H (14 September 2015). “Neue Wirkstoffe – Myrcludex B”. Österreichische Apothekerzeitung (in German) (19/2015): 12.
External links
- “Bulevirtide”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Hepcludex |
| Other names | MyrB, Myrcludex-B[1] |
| License data | |
| Routes of administration |
Subcutaneous injection |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| DrugBank | |
| UNII | |
| KEGG | |
| ChEMBL | |
/////////Bulevirtide acetate, ブレビルチド酢酸塩 , orphan designation, MYR GmbH, PEPTIDE, EU 2020, 2020 APPROVALS
Nifurtimox

Nifurtimox
| Formula |
C10H13N3O5S
|
|---|---|
| CAS |
23256-30-6
|
| Mol weight |
287.2923
|
FDA APPROVED, 2020/8/6, LAMPIT
|
Antiprotozoal
|
|
| Disease |
Chagas disease
|
|---|
IUPAC Name
3-methyl-4-[(E)-[(5-nitrofuran-2-yl)methylidene]amino]-1lambda6-thiomorpholine-1,1-dione
SMILES
CC1CS(=O)(=O)CCN1\N=C\C1=CC=C(O1)[N+]([O-])=O
SYN
Danong Chen, Glenn Rice. 2013. Novel formulations of nitrofurans including nifurtimox with enhanced activity with lower toxicity.US20150140089A1
| melting point (°C) | 177-183 | https://www.chemicalbook.com/ChemicalProductProperty_US_CB3903922.aspx |
- OriginatorBayer
- ClassAntiprotozoals; Nitrofurans; Small molecules; Thiamorpholines; Thiazines
- Mechanism of ActionDNA damage modulators
- RegisteredChagas disease
- 07 Aug 2020Registered for Chagas disease (In adolescents, In children, In infants) in USA (PO)
- 31 Jan 2020Preregistration for Chagas disease (In infants, In children, In adolescents) in USA (PO)
- 29 Jan 2020Bayer completes a phase I trial in Chagas disease in Argentina (PO) (NCT03334838)
Title: Nifurtimox
CAS Registry Number: 23256-30-6
CAS Name: 3-Methyl-N-[(5-nitro-2-furanyl)methylene]-4-thiomorpholinamine 1,1-dioxide
Additional Names: 4-[(5-nitrofurfurylidene)amino]-3-methylthiomorpholine-1,1-dioxide; tetrahydro-3-methyl-4-[(5-nitrofurfurylidene)amino]-2H-1,4-thiazine 1,1-dioxide; 1-[(5-nitrofurfurylidene)amino]-2-methyltetrahydro-1,4-thiazine 4,4-dioxide
Manufacturers’ Codes: Bay 2502
Trademarks: Lampit (Bayer)
Molecular Formula: C10H13N3O5S
Molecular Weight: 287.29
Percent Composition: C 41.81%, H 4.56%, N 14.63%, O 27.85%, S 11.16%
Literature References: Prepn from 5-nitrofurfural and 4-amino-3-methyltetrahydro-1,4-thiazine 1,1-dioxide: Herlinger et al., DE 1170957 corresp to US 3262930 (1964 and 1966 to Bayer). Series of articles on pharmacology and clinical findings: Arzneim.-Forsch. 22, 1563-1642 (1972). Toxicity data: K. Hoffmann, ibid. 1590.
Properties: Orange-red crystals from dil acetic acid, mp 180-182°. LD50 in mice, rats (mg/kg): 3720, 4050 by gavage (Hoffmann).
Melting point: mp 180-182°
Toxicity data: LD50 in mice, rats (mg/kg): 3720, 4050 by gavage (Hoffmann)
Therap-Cat: Antiprotozoal (Trypanosoma).
Keywords: Antiprotozoal (Trypanosoma).
Nifurtimox, sold under the brand name Lampit, is a medication used to treat Chagas disease and sleeping sickness.[1][4] For sleeping sickness it is used together with eflornithine in nifurtimox-eflornithine combination treatment.[4] In Chagas disease it is a second-line option to benznidazole.[5] It is given by mouth.[1]
Common side effects include abdominal pain, headache, nausea, and weight loss.[1] There are concerns from animal studies that it may increase the risk of cancer but these concerns have not be found in human trials.[5] Nifurtimox is not recommended in pregnancy or in those with significant kidney or liver problems.[5] It is a type of nitrofuran.[5]
Nifurtimox came into medication use in 1965.[5] It is on the World Health Organization’s List of Essential Medicines.[4] It is not available commercially in Canada.[1] It was approved for medical use in the United States in August 2020.[3] In regions of the world where the disease is common nifurtimox is provided for free by the World Health Organization (WHO).[6]
Chagas disease, caused by a parasite known as Trypanosoma cruzi (T.cruzi), is a vector-transmitted disease affecting animals and humans in the Americas. It is commonly known as American Trypanosomiasis.11
The CDC estimates that approximately 8 million people in Central America, South America, and Mexico are infected with T. cruzi, without symptoms. If Chagas disease is left untreated, life-threatening sequelae may result.11
Nifurtimox, developed by Bayer, is a nitrofuran antiprotozoal drug used in the treatment of Chagas disease. On August 6 2020, accelerated FDA approval was granted for its use in pediatric patients in response to promising results from phase III clinical trials. Continued approval will be contingent upon confirmatory data.10 A convenient feature of Bayer’s formulation is the ability to divide the scored tablets manually without the need for pill-cutting devices.10
Medical uses
Nifurtimox has been used to treat Chagas disease, when it is given for 30 to 60 days.[7][8] However, long-term use of nifurtimox does increase chances of adverse events like gastrointestinal and neurological side effects.[8][9] Due to the low tolerance and completion rate of nifurtimox, benznidazole is now being more considered for those who have Chagas disease and require long-term treatment.[5][9]
In the United States nifurtimox is indicated in children and adolescents (birth to less than 18 years of age and weighing at least 2.5 kilograms (5.5 lb) for the treatment of Chagas disease (American Trypanosomiasis), caused by Trypanosoma cruzi.[2]
Nifurtimox has also been used to treat African trypanosomiasis (sleeping sickness), and is active in the second stage of the disease (central nervous system involvement). When nifurtimox is given on its own, about half of all patients will relapse,[10] but the combination of melarsoprol with nifurtimox appears to be efficacious.[11] Trials are awaited comparing melarsoprol/nifurtimox against melarsoprol alone for African sleeping sickness.[12]
Combination therapy with eflornithine and nifurtimox is safer and easier than treatment with eflornithine alone, and appears to be equally or more effective. It has been recommended as first-line treatment for second-stage African trypanosomiasis.[13]
Pregnancy and breastfeeding
Use of nifurtimox should be avoided in pregnant women due to limited use.[5][8][14] There is limited data shown that nifurtimox doses up to 15 mg/kg daily can cause adverse effects in breastfed infants.[15] Other authors do not consider breastfeeding a contraindication during nifurtimox use.[15]
Side effects
Side effects occur following chronic administration, particularly in elderly people. Major toxicities include immediate hypersensitivity such as anaphylaxis and delayed hypersensitivity reaction involving icterus and dermatitis. Central nervous system disturbances and peripheral neuropathy may also occur.[8]
Contraindications
Nifurtimox is contraindicated in people with severe liver or kidney disease, as well as people with a background of neurological or psychiatric disorders.[5][16][20]
Mechanism of action
Nifurtimox forms a nitro-anion radical metabolite that reacts with nucleic acids of the parasite causing significant breakdown of DNA.[8] Its mechanism is similar to that proposed for the antibacterial action of metronidazole. Nifurtimox undergoes reduction and creates oxygen radicals such as superoxide. These radicals are toxic to T. cruzi. Mammalian cells are protected by presence of catalase, glutathione, peroxidases, and superoxide dismutase. Accumulation of hydrogen peroxide to cytotoxic levels results in parasite death.[8]
Manufacturing and availability
.jpg)
A bottle of nifurtimox
Nifurtimox is sold under the brand name Lampit by Bayer.[3] It was previously known as Bayer 2502.
Nifurtimox is only licensed for use in Argentina and Germany,[citation needed] where it is sold as 120-mg tablets. It was approved for medical use in the United States in August 2020.[3]
Research
Nifurtimox is in a phase-II clinical trial for the treatment of pediatric neuroblastoma and medulloblastoma.[21]
SYN
Nifurtimox
2006, Pages 559-582
Nifurtimox, 1,1-dioxide 4-[(5-nitrofuryliden)amino]-3-methylthiomorpholine (37.4.7), is made by the following scheme. Interaction of 2-mercaptoethanol with propylene oxide in the presence of potassium hydroxide gives (2-hydroxyethyl)-(2-hydroxypropylsul-fide) (37.4.3), which undergoes intramolecular dehydration using potassium bisulfate to make 2-methyl-1,4-oxithiane (37.4.4). Oxidation of this using hydrogen peroxide gives 2-methyl-1,4-oxithian-4,4-dioxide (37.4.5), which when reacted with hydrazine transforms to 4-amino-3-methyltetrahydro-1,4-thiazin-1,1-dioxide (37.4.6). Reacting this with 5-nitrofurfurol gives the corresponding hydrazone—the desired nifurtimox [58,59].
58. H. Herlinger, K.H. Heinz, S. Petersen, M.Bock, Ger. Pat. 1.170.957 (1964).
59. H. Herlinger, K.H. Heinz, S. Petersen, M. Bock, U.S. Pat. 3.262.930 (1966)

References
- ^ Jump up to:a b c d e f “Nifurtimox (Systemic)”. Drugs.com. 1995. Archived from the original on 20 December 2016. Retrieved 3 December 2016.
- ^ Jump up to:a b “Lampit (nifurtimox) tablets, for oral use” (PDF). U.S. Food and Drug Administration(FDA). Bayer HealthCare Pharmaceuticals Inc. Retrieved 6 August 2020.
- ^ Jump up to:a b c d “Lampit: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 6 August 2020.
- ^ Jump up to:a b c World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ Jump up to:a b c d e f g h Bern, Caryn; Montgomery, Susan P.; Herwaldt, Barbara L.; Rassi, Anis; Marin-Neto, Jose Antonio; Dantas, Roberto O.; Maguire, James H.; Acquatella, Harry; Morillo, Carlos (2007-11-14). “Evaluation and Treatment of Chagas Disease in the United States”. JAMA. 298 (18): 2171–81. doi:10.1001/jama.298.18.2171. ISSN 0098-7484. PMID 18000201.
- ^ “Trypanosomiasis, human African (sleeping sickness)”. World Health Organization. February 2016. Archived from the original on 4 December 2016. Retrieved 7 December2016.
- ^ Coura JR, de Castro SL (2002). “A critical review of Chagas disease chemotherapy”. Mem Inst Oswaldo Cruz. 97 (1): 3–24. doi:10.1590/S0074-02762002000100001. PMID 11992141.
- ^ Jump up to:a b c d e f g h “Nifurtimox Drug Information, Professional”. http://www.drugs.com. Archivedfrom the original on 2016-11-08. Retrieved 2016-11-09.
- ^ Jump up to:a b Jackson, Yves; Alirol, Emilie; Getaz, Laurent; Wolff, Hans; Combescure, Christophe; Chappuis, François (2010-11-15). “Tolerance and Safety of Nifurtimox in Patients with Chronic Chagas Disease”. Clinical Infectious Diseases. 51 (10): e69–e75. doi:10.1086/656917. ISSN 1058-4838. PMID 20932171.
- ^ Pepin J, Milord F, Mpia B, et al. (1989). “An open clinical trial of nifurtimox for arseno-resistant T. b. gambiense sleeping sickness in central Zaire”. Trans R Soc Trop Med Hyg. 83(4): 514–7. doi:10.1016/0035-9203(89)90270-8. PMID 2694491.
- ^ Bisser S, N’Siesi FX, Lejon V, et al. (2007). “Equivalence Trial of Melarsoprol and Nifurtimox Monotherapy and Combination Therapy for the Treatment of Second-Stage Trypanosoma brucei gambiense Sleeping Sickness”. J Infect Dis. 195 (3): 322–329. doi:10.1086/510534. PMID 17205469.
- ^ Pepin J (2007). “Combination Therapy for Sleeping Sickness: A Wake-Up Call”. J Infect Dis. 195 (3): 311–13. doi:10.1086/510540. PMID 17205466.
- ^ Priotto G, Kasparian S, Mutombo W, et al. (July 2009). “Nifurtimox-eflornithine combination therapy for second-stage African Trypanosoma brucei gambiensetrypanosomiasis: a multicentre, randomised, phase III, non-inferiority trial”. Lancet. 374(9683): 56–64. doi:10.1016/S0140-6736(09)61117-X. hdl:10144/72797. PMID 19559476.
- ^ Schaefer, Christof; Peters, Paul W. J.; Miller, Richard K. (2014-09-17). Drugs During Pregnancy and Lactation: Treatment Options and Risk Assessment. Academic Press. ISBN 9780124079014. Archived from the original on 2017-09-08.
- ^ Jump up to:a b “Nifurtimox use while Breastfeeding | Drugs.com”. http://www.drugs.com. Archived from the original on 2016-11-08. Retrieved 2016-11-07.
- ^ Jump up to:a b c “Parasites – American Trypanosomiasis (also known as Chagas Disease)”. U.S. Centers for Disease Control and Prevention (CDC). Archived from the original on 2016-11-06. Retrieved 2016-11-09.
- ^ Jump up to:a b Forsyth, Colin J.; Hernandez, Salvador; Olmedo, Wilman; Abuhamidah, Adieb; Traina, Mahmoud I.; Sanchez, Daniel R.; Soverow, Jonathan; Meymandi, Sheba K. (2016-10-15). “Safety Profile of Nifurtimox for Treatment of Chagas Disease in the United States”. Clinical Infectious Diseases. 63 (8): 1056–1062. doi:10.1093/cid/ciw477. ISSN 1537-6591. PMC 5036918. PMID 27432838.
- ^ Castro, José A.; de Mecca, Maria Montalto; Bartel, Laura C. (2006-08-01). “Toxic side effects of drugs used to treat Chagas’ disease (American trypanosomiasis)”. Human & Experimental Toxicology. 25 (8): 471–479. doi:10.1191/0960327106het653oa. ISSN 0960-3271. PMID 16937919.
- ^ Jump up to:a b Estani, Sergio Sosa; Segura, Elsa Leonor (1999-09-01). “Treatment of Trypanosoma cruzi infection in the undetermined phase. Experience and current guidelines of treatment in Argentina”. Memórias do Instituto Oswaldo Cruz. 94: 363–365. doi:10.1590/S0074-02761999000700070. ISSN 0074-0276. PMID 10677756.
- ^ “Chagas disease”. World Health Organization. Archived from the original on 2014-02-27. Retrieved 2016-11-08.
- ^ Clinical trial number NCT00601003 for “Study of Nifurtimox to Treat Refractory or Relapsed Neuroblastoma or Medulloblastoma” at ClinicalTrials.gov. Retrieved on July 10, 2009.
External links
- “Nifurtimox”. Drug Information Portal. U.S. National Library of Medicine.
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Lampit[1] |
| Other names | Bayer 2502[1] |
| AHFS/Drugs.com | Drugs.com archive Lampit |
| License data |
|
| Routes of administration |
By mouth |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | Low |
| Metabolism | Liver (Cytochrome P450 oxidase (CYP) involved) |
| Elimination half-life | 2.95 ± 1.19 hours |
| Excretion | Kidney, very low |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.041.377 |
| Chemical and physical data | |
| Formula | C10H13N3O5S |
| Molar mass | 287.29 g·mol−1 |
| 3D model (JSmol) | |
| Chirality | Racemic mixture |
| Melting point | 180 to 182 °C (356 to 360 °F) |
///////////Nifurtimox, LAMPIT, 2020 APPROVALS, FDA 2020, ニフルチモックス, CHAGAS DISEASE, ANTI PROTOZOAL
Imlifidase
MDSFSANQEI RYSEVTPYHV TSVWTKGVTP PANFTQGEDV FHAPYVANQG WYDITKTFNG
KDDLLCGAAT AGNMLHWWFD QNKDQIKRYL EEHPEKQKIN FNGEQMFDVK EAIDTKNHQL
DSKLFEYFKE KAFPYLSTKH LGVFPDHVID MFINGYRLSL TNHGPTPVKE GSKDPRGGIF
DAVFTRGDQS KLLTSRHDFK EKNLKEISDL IKKELTEGKA LGLSHTYANV RINHVINLWG
ADFDSNGNLK AIYVTDSDSN ASIGMKKYFV GVNSAGKVAI SAKEIKEDNI GAQVLGLFTL
STGQDSWNQT N
Imlifidase
イムリフィダーゼ;
| Formula |
C1575H2400N422O477S6
|
|---|---|
| CAS |
1947415-68-0
|
| Mol weight |
35070.8397
|
EMA APPROVED, 2020/8/25, Idefirix
Pre-transplant treatment to make patients with donor specific IgG eligible for kidney transplantation
Immunosuppressant, Immunoglobulin modulator (enzyme)
Imlifidase is under investigation in clinical trial NCT02854059 (IdeS in Asymptomatic Asymptomatic Antibody-Mediated Thrombotic Thrombocytopenic Purpura (TTP) Patients).
Imlifidase, brand name Idefirix, is a medication for the desensitization of highly sensitized adults needing kidney transplantation, but unlikely to receive a compatible transplant.[1]
Imlifidase is a cysteine protease derived from the immunoglobulin G (IgG)‑degrading enzyme of Streptococcus pyogenes.[1] It cleaves the heavy chains of all human IgG subclasses (but no other immunoglobulins), eliminating Fc-dependent effector functions, including CDC and antibody-dependent cell-mediated cytotoxicity (ADCC).[1] Thus, imlifidase reduces the level of donor specific antibodies, enabling transplantation.[1]
The benefits with imlifidase are its ability to convert a positive crossmatch to a negative one in highly sensitized people to allow renal transplantation.[1] The most common side effects are infections and infusion related reactions.[1]
In June 2020, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) recommended the approval of Imlifidase.[1][2]
Medical uses
Per the CHMP recommendation, imlifidase will be indicated for desensitization treatment of highly sensitized adult kidney transplant people with positive crossmatch against an available deceased donor.[1] The use of imlifidase should be reserved for people unlikely to be transplanted under the available kidney allocation system including prioritization programmes for highly sensitized people.[1]
History
Imlifidase was granted orphan drug designations by the European Commission in January 2017, and November 2018,[3][4] and by the U.S. Food and Drug Administration (FDA) in both February and July 2018.[5][6]
In February 2019, Hansa Medical AB changed its name to Hansa Biopharma AB.[4]
| PHASE | STATUS | PURPOSE | CONDITIONS | COUNT |
|---|---|---|---|---|
| 2 | Recruiting | Treatment | Anti-Glomerular Basement Membrane Disease | 1 |
| 2 | Recruiting | Treatment | Guillain-Barré Syndrome (GBS) | 1 |
| 2 | Recruiting | Treatment | Kidney Transplant Rejection | 1 |
| 2 | Terminated | Treatment | Thrombotic Thrombocytopenic Purpura (TTP) | 1 |
| Not Available | Recruiting | Not Available | Kidney Transplant Failure and Rejection | 1 |
References
- ^ Jump up to:a b c d e f g h i “Imlifidase: Pending EC decision”. European Medicines Agency (EMA). 25 June 2020. Retrieved 26 June 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “New treatment to enable kidney transplant in highly sensitised patients”. European Medicines Agency (Press release). 26 June 2020. Retrieved 26 June 2020. This article incorporates text from this source, which is in the public domain.
- ^ “EU/3/16/1826”. European Medicines Agency (EMA). 12 January 2017. Retrieved 27 June 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b “EU/3/18/2096”. European Medicines Agency (EMA). 13 February 2019. Retrieved 27 June 2020. This article incorporates text from this source, which is in the public domain.
- ^ “Imlifidase Orphan Drug Designation and Approval”. U.S. Food and Drug Administration (FDA). 3 July 2018. Retrieved 27 June 2020.
- ^ “Imlifidase Orphan Drug Designation and Approval”. U.S. Food and Drug Administration (FDA). 14 February 2018. Retrieved 27 June 2020.
Further reading
- Ge S, Chu M, Choi J, Louie S, Vo A, Jordan SC, et al. (October 2019). “Imlifidase Inhibits HLA Antibody-Mediated NK Cell Activation and Antibody-Dependent Cell-Mediated Cytotoxicity (ADCC) In Vitro”. Transplantation. doi:10.1097/TP.0000000000003023. PMID 31644495.
- Lin J, Boon L, Bockermann R, Robertson AK, Kjellman C, Anderson CC (March 2020). “Desensitization using imlifidase and EndoS enables chimerism induction in allosensitized recipient mice”. Am. J. Transplant. doi:10.1111/ajt.15851. PMID 32185855.
- Lonze BE, Tatapudi VS, Weldon EP, Min ES, Ali NM, Deterville CL, et al. (September 2018). “IdeS (Imlifidase): A Novel Agent That Cleaves Human IgG and Permits Successful Kidney Transplantation Across High-strength Donor-specific Antibody”. Ann. Surg. 268 (3): 488–496. doi:10.1097/SLA.0000000000002924. PMID 30004918.
- Lorant T, Bengtsson M, Eich T, Eriksson BM, Winstedt L, Järnum S, et al. (November 2018). “Safety, immunogenicity, pharmacokinetics, and efficacy of degradation of anti-HLA antibodies by IdeS (imlifidase) in chronic kidney disease patients”. Am. J. Transplant. 18 (11): 2752–2762. doi:10.1111/ajt.14733. PMC 6221156. PMID 29561066.
External links
- “Imlifidase”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Pronunciation | im lif’ i dase |
| Trade names | Idefirix |
| Other names | HMED-IdeS |
| Routes of administration |
Intravenous |
| ATC code | |
| Identifiers | |
| CAS Number | |
| DrugBank | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C1575H2400N422O477S6 |
| Molar mass | 35071.36 g·mol−1 |
//////////Imlifidase, Idefirix, PEPTIDE, イムリフィダーゼ , 2020 APPROVALS, EMA 2020, EU 2020
Pralsetinib
Pralsetinib
| Formula |
C27H32FN9O2
|
|---|---|
| CAS |
2097132-94-8
|
| Mol weight |
533.6005
|
Cyclohexanecarboxamide, N-[(1S)-1-[6-(4-fluoro-1H-pyrazol-1-yl)-3-pyridinyl]ethyl]-1-methoxy-4-[4-methyl-6-[(5-methyl-1H-pyrazol-3-yl)amino]-2-pyrimidinyl]-, cis–
2097132-94-8 [RN]
BLU-667
BS-15942
Other Names
- cis-N-[(1S)-1-[6-(4-Fluoro-1H-pyrazol-1-yl)-3-pyridinyl]ethyl]-1-methoxy-4-[4-methyl-6-[(5-methyl-1H-pyrazol-3-yl)amino]-2-pyrimidinyl]cyclohexanecarboxamide
- BLU 123244
- BLU 667
- Pralsetinib
- X 581238
- cis-N-{(1S)-1-[6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl]ethyl}-1-methoxy-4-{4-methyl-6-[(5-methyl-1H-pyrazol-3-yl)amino]pyrimidin-2-yl}cyclohexane-1-carboxamide
N-[(1S)-1-[6-(4-fluoropyrazol-1-yl)pyridin-3-yl]ethyl]-1-methoxy-4-[4-methyl-6-[(5-methyl-1H-pyrazol-3-yl)amino]pyrimidin-2-yl]cyclohexane-1-carboxamide
FDA APPROVED GAVRETO, 2020/9/4
Pralsetinib, sold under the brand name Gavreto, is a medication for the treatment of metastatic RET fusion-positive non-small cell lung cancer (NSCLC).[1] Pralsetinib is a tyrosine kinase inhibitor. It is taken by mouth.[1]
The most common adverse reactions include increased aspartate aminotransferase (AST), decreased hemoglobin, decreased lymphocytes, decreased neutrophils, increased alanine aminotransferase (ALT), increased creatinine, increased alkaline phosphatase, fatigue, constipation, musculoskeletal pain, decreased calcium, hypertension, decreased sodium, decreased phosphate, and decreased platelets.[1]
Pralsetinib was approved for medical use in the United States in September 2020.[1][2][3][4]
Medical uses
Pralsetinib is indicated for the treatment of adults with metastatic RET fusion-positive non-small cell lung cancer (NSCLC) as detected by an FDA approved test.[1][4]
History
Efficacy was investigated in a multicenter, open-label, multi-cohort clinical trial (ARROW, NCT03037385) with 220 participants aged 26-87 whose tumors had RET alterations.[1][4] Identification of RET gene alterations was prospectively determined in local laboratories using either next generation sequencing, fluorescence in situ hybridization, or other tests.[1] The main efficacy outcome measures were overall response rate (ORR) and response duration determined by a blinded independent review committee using RECIST 1.1.[1] The trial was conducted at sites in the United States, Europe and Asia.[4]
Efficacy for RET fusion-positive NSCLC was evaluated in 87 participants previously treated with platinum chemotherapy.[1] The ORR was 57% (95% CI: 46%, 68%); 80% of responding participants had responses lasting 6 months or longer.[1] Efficacy was also evaluated in 27 participants who never received systemic treatment.[1] The ORR for these participants was 70% (95% CI: 50%, 86%); 58% of responding participants had responses lasting 6 months or longer.[1]
The US Food and Drug Administration (FDA) granted the application for pralsetinib priority review, orphan drug, and breakthrough therapy designations[1]and granted approval of Gavreto to Blueprint Medicines.[1]
PATENT
US 20170121312

https://patents.google.com/patent/US20170121312A1/en
-
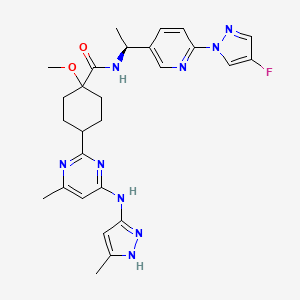
- Step 7: Synthesis of (1R,4S)—N—((S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-1-methoxy-4-(4-methyl-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)cyclohexane-carboxamide (Compound 129) and (1S,4R)—N—((S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-1-methoxy-4-(4-methyl-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)cyclohexanecarboxamide (Compound 130)
- [0194]

- [0195]
The title compounds were prepared from methyl 1-methoxy-4-(4-methyl-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)cyclohexanecarboxylate (192 mg, 0.53 mmol) using the same two-step procedure (hydrolysis and amide coupling) outlined in Synthetic Protocols 1 and 2, with PyBOP as the amide coupling reagent instead of HATU. The products were initially isolated as a mixture of diastereomers (190 mg), which was then dissolved in 6 mL methanol and purified by SFC (ChiralPak AD-H 21×250 mm, 40% MeOH containing 0.25% DEA in CO2, 2.5 mL injections, 70 mL/min). Peak 1 was concentrated to give (1R,4S)—N—((S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-1-methoxy-4-(4-methyl-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)cyclohexanecarboxamide (29 mg, 10%) as a white solid. Peak 2 was concentrated to give (1s,4R)—N—((S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-1-methoxy-4-(4-methyl-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)cyclohexane-carboxamide (130 mg, 46%) as a white solid.
- [0194]
Example 6. Synthesis of Compound 149Step 1: Synthesis of Methyl 4-(2-chloro-6-methylpyrimidin-4-yl)-1-methoxycyclohexane-1-carboxylate
-
- [0196]

- [0197]
Methyl 4-iodo-1-methoxycyclohexanecarboxylate (3.37 g, 11.3 mmol) was dissolved in dimethylacetamide (38 mL) in a pressure vessel under a stream of N2. Rieke Zinc (17.7 mL of a 50 mg/mL suspension in THF, 13.6 mmol) was added quickly via syringe, and the vessel was capped and stirred at ambient temperature for 15 minutes. The vessel was opened under a stream of N2 and 2,4-dichloro-6-methylpyrimidine (1.84 g, 11.3 mmol) was added followed by PdCl2dppf (826 mg, 1.13 mmol). The vessel was capped and heated to 80° C. for one hour, then cooled to room temperature. The reaction mixture was diluted with EtOAc, filtered through celite, and the filtrate was washed with H2O (3×), brine, dried over sodium sulfate, filtered, and concentrated. The resulting residue was purified by flash-column chromatography on silica gel (gradient elution, 0 to 50% EtOAc-hexanes) to give methyl 4-(2-chloro-6-methylpyrimidin-4-yl)-1-methoxycyclohexane-1-carboxylate (74 mg, 2.2%) as a colorless oil. MS (ES+) C14H19ClN2O3 requires: 298, found: 299 [M+H]+.
- [0196]
Step 2: Synthesis of tert-Butyl 3-((4-(4-methoxy-4-(methoxycarbonyl)cyclohexyl)-6-methylpyrimidin-2-yl)amino)-5-methyl-1H-pyrazole-1-carboxylate
-
- [0198]

- [0199]
Methyl 4-(2-chloro-6-methylpyrimidin-4-yl)-1-methoxycyclohexane-1-carboxylate (70.5 mg, 0.236 mmol), tert-butyl 3-amino-5-methyl-1H-pyrazole-1-carboxylate (69.8 mg, 0.354 mmol), di-tert-butyl(2′,4′,6′-triisopropyl-[1,1′-biphenyl]-2-yl)phosphine (20.0 mg, 0.2 equiv.), Pd2(dba)3 (21.6 mg, 0.1 equiv.), and potassium acetate (70 mg, 0.71 mmol) were combined in a vial under nitrogen and 0.98 mL dioxane was added. The reaction mixture was heated to 115° C. for 2 h, then cooled to ambient temperature. The reaction mixture was diluted with EtOAc, filtered through celite, concentrated onto silica gel, and the resulting residue was purified by flash-column chromatography on silica gel (gradient elution, 0 to 100% ethyl acetate-hexanes) to give tert-butyl 3-((4-(4-methoxy-4-(methoxycarbonyl)cyclohexyl)-6-methylpyrimidin-2-yl)amino)-5-methyl-1H-pyrazole-1-carboxylate (48 mg, 44%) as a yellow oil. MS (ES+) C23H33N5O5 requires: 459, found: 460 [M+H]+.
- [0198]
Step 3: Synthesis of 1-Methoxy-4-(6-methyl-2-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-4-yl)cyclohexane-1-carboxylic acid
-
- [0200]

- [0201]
Lithium hydroxide monohydrate (13 mg, 0.31 mmol) was added to a solution of tert-butyl 3-((4-(4-methoxy-4-(methoxycarbonyl)cyclohexyl)-6-methylpyrimidin-2-yl)amino)-5-methyl-1H-pyrazole-1-carboxylate (47.7 mg, 0.104 mmol) in THF/MeOH/H2O (17:1:1, 1.8 mL). The reaction mixture was heated to 60° C. and stirred for 16 h. The reaction mixture was then cooled to ambient temperature and concentrated to give crude 1-methoxy-4-(6-methyl-2-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-4-yl)cyclohexane-1-carboxylic acid (57 mg, crude) which was used in the subsequent amide coupling without any further purification. MS (ES+) C17H23N5O3 requires: 345, found: 346 [M+H]+.
- [0200]
Step 4: Synthesis of (1s,4R)—N—((S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-1-methoxy-4-(6-methyl-2-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-4-yl)cyclohexane-1-carboxamide (Compound 149)
-
- [0202]

- [0203]
The title compound was prepared from 1-methoxy-4-(6-methyl-2-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-4-yl)cyclohexane-1-carboxylic acid (57 mg, 0.104 mmol) using the same procedured (amide coupling) outlined in Synthetic Protocols 1 and 2, with PyBOP as the amide coupling reagent instead of HATU. The products were initially isolated as a mixture of diastereomers (36 mg), which was then dissolved in 6 mL methanol-DCM (1:1) and purified by SFC (ChiralPak IC-H 21×250 mm, 40% MeOH containing 0.25% DEA in CO2, 1.0 mL injections, 70 mL/min). Peak 1 was an undesired isomer, and Peak 2 was concentrated to give (1 s,4R)—N—((S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-1-methoxy-4-(6-methyl-2-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-4-yl)cyclohexane-1-carboxamide (13.4 mg, 24%) as a white solid.
- [0202]
Synthesis of IntermediatesExample 7. Synthesis of Ketone and Boronate IntermediatesA. Methyl 1-methoxy-4-oxocyclohexane-1-carboxylate
-
- [0204]

- [0205]
The title compound was prepared as described in WO 2014/130810 A1 page 86.
- [0204]
B. Ethyl 1-ethoxy-4-oxocyclohexane-1-carboxylate
-
- [0206]

- [0206]
Step 1: Synthesis of ethyl 8-ethoxy-1,4-dioxaspiro[4.5]decane-8-carboxylate
-
- [0207]
A solution of 1,4-dioxaspiro[4.5]decan-8-one (20.0 g, 128 mmol) in CHBr3 (3234 g, 1280 mmol) was cooled to 0° C. and potassium hydroxide (57.5 g, 1024 mmol) in EtOH (300 mL) was added dropwise over 2.5 hrs. After stirring the mixture for 23 h, the mixture was concentrated, and the residue was partitioned between EtOAc and H2O. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure to give crude product, which was purified by flash column chromatography on silica gel (gradient elution, PE:EA=15:1 to 10:1) to obtain the title compound (18.0 g).
- [0207]
Step 2: Synthesis of ethyl 1-ethoxy-4-oxocyclohexane-1-carboxylate
-
- [0208]
To a solution of ethyl 8-ethoxy-1,4-dioxaspiro[4.5]decane-8-carboxylate (10 g, 43 mmol) in 1,4-dioxane (250 mL) was added aqueous HCl (6 M, 92.5 mL), and the mixture was stirred for 23 h at ambient temperature. The mixture was then diluted with H2O and extracted with EtOAc.
- [0209]
The organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure to give a crude residue, which was purified by flash column chromatography on silica gel (PE:EA=15:1) to obtain the product (8.0 g). 1H NMR (400 MHz, DMSO) δ 4.20-4.13 (m, 2H), 3.43 (q, J=6.9 Hz, 1H), 2.48-2.39 (m, 1H), 2.24-2.12 (m, 2H), 2.10-2.01 (m, 1H), 1.22 (t, J=7.1 Hz, 2H), 1.17 (t, J=7.0 Hz, 2H).
- [0208]
C. Ethyl 6,6-dimethyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)cyclohex-3-ene-1-carboxylate
-
- [0210]

- [0210]
Step 1: Synthesis of ethyl 2,2-dimethyl-4-oxocyclohexane-1-carboxylate
-
- [0211]
A solution of methylmagnesium bromide (3M, 109.8 mL, 329.4 mmol) was added dropwise to a suspension of CuCN (14.75 g, 164.7 mmol) in diethyl ether (50 mL) at 0° C. The mixture was stirred for 30 min at 0° C. and then cooled to −78° C. The solution of ethyl 2-methyl-4-oxocyclohex-2-ene-1-carboxylate (10 g, 54.9 mmol) in diethyl ether (10 mL) was then added dropwise. The mixture was stirred between −40° C. to −20° C. for 2 h, then was warmed to ambient temperature for 16 h. The reaction mixture was carefully added to a saturated solution of ammonium chloride. The aqueous layer was extracted twice with diethyl ether, and the organic layers were combined. The combined organic layer was washed with brine, dried over sodium sulfate, filtered and concentrated. The residue was purified by flash column chromatography on silica gel (PE:EA=10:1) to give ethyl 2,2-dimethyl-4-oxocyclohexane-1-carboxylate (1.16 g).
- [0211]
Step 2: Synthesis of ethyl 6,6-dimethyl-4-(((trifluoromethyl)sulfonyl)oxy)cyclohex-3-ene-1-carboxylate
-
- [0212]
Ethyl 2,2-dimethyl-4-oxocyclohexane-1-carboxylate (1.16 g, 5.85 mmol) and DIPEA (3.03 g, 23.4 mmol) were dissolved in dry toluene (2 mL) and heated at 45° C. for 10 minutes. Trifluoromethanesulfonic anhydride (6.61 g, 23.4 mmol) in DCM (20 mL) was added dropwise over 10 min and the mixture was heated at 45° C. for 2 h. The mixture was allowed to cool to room temperature, concentrated, diluted with water (60 mL) and extracted with DCM (2×40 mL). The organic layer was washed with saturated sodium bicarbonate solution (20 mL) and brine (20 mL), dried over sodium sulfate, filtered, and concentrated. The crude product was purified by flash column chromatography on silica gel (gradient elution, 0 to 100% ethyl acetate-petroleum ether) to afford ethyl 6,6-dimethyl-4-(((trifluoromethyl)sulfonyl)oxy)cyclohex-3-ene-1-carboxylate (1 g).
- [0212]
Step 3: Synthesis of ethyl 6,6-dimethyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)cyclohex-3-ene-1-carboxylate
-
- [0213]
Ethyl 6,6-dimethyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)cyclohex-3-ene-1-carboxylate (1 g, 3.03 mmol), 4,4,5,5-tetramethyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (1.15 g, 4.54 mmol), Pd(dppf)Cl2 (73.5 mg, 0.09 mmol) and potassium acetate (891 mg, 9.08 mmol) were suspended in 1,4-dioxane (20 mL). The reaction mixture was flushed with nitrogen, then heated to 100° C. for 2 h. The mixture was cooled to room temperature, filtered, and concentrated, and the resulting brown oil was purified by flash column chromatography on silica gel (gradient elution, 0 to 100% ethyl acetate-petroleum ether) to afford ethyl 6,6-dimethyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)cyclohex-3-ene-1-carboxylate (618 mg).
- [0213]
D. Ethyl 6-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)cyclohex-3-ene-1-carboxylate
-
- [0214]

- [0215]
Ethyl 6-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)cyclohex-3-ene-1-carboxylate was prepared using the same synthetic protocol as described above using ethyl 2-methyl-4-oxocyclohexane-1-carboxylate as the starting material.
- [0214]
E. Methyl 2-methyl-5-oxotetrahydro-2H-pyran-2-carboxylate
-
- [0216]

- [0216]
Step 1: Synthesis of methyl 2-methyl-3,4-dihydro-2H-pyran-2-carboxylate
-
- [0217]
A mixture of acrylaldehyde (120 g, 2.14 mol), methyl methacrylate (200 g, 2.00 mol) and hydroquinone (2.2 g, 20 mmol) were heated in a sealed steel vessel at 180° C. for one h. The mixture was then cooled to ambient temperature and concentrated. The residue was purified by silica gel column chromatography (gradient elution, petroleum ether:ethyl acetate=100:1 to 80:1) to give methyl 2-methyl-3,4-dihydro-2H-pyran-2-carboxylate (70 g, 22% yield) as a pale yellow oil. 1H-NMR (400 MHz, CDCl3): δ 6.38 (d, J=6.4 Hz, 1H), 4.73-4.70 (m, 1H), 3.76 (s, 3H), 2.25-2.22 (m, 1H), 1.99-1.96 (m, 2H), 1.79-1.77 (m, 1H), 1.49 (s, 3H).
- [0217]
Step 2: Synthesis of methyl 5-hydroxy-2-methyltetrahydro-2H-pyran-2-carboxylate
-
- [0218]
To a solution of methyl 2-methyl-3,4-dihydro-2H-pyran-2-carboxylate (20.0 g, 128 mmol) in anhydrous tetrahydrofuran (200 mL) was added borane (67 mL, 1 M in tetrahydrofuran) dropwise at −5° C. The reaction mixture was stirred at 0° C. for 3 hours. This reaction was monitored by TLC. The mixture was quenched by a solution of sodium acetate (10.5 g, 128 mmol) in water (15 mL). Then the mixture was treated with 30% hydrogen peroxide solution (23.6 g, 208.2 mmol) slowly at 0° C. and stirred at 30° C. for 3 h. The mixture was then partitioned between saturated sodium sulfite solution and tetrahydrofuran. The aqueous layer was further extracted with tetrahydrofuran (2×). The combined organic layers were washed with saturated brine, dried over sodium sulfate and concentrated in vacuo. The residue was purified by a silica gel column chromatography (gradient elution, petroleum ether:ethyl acetate=10:1 to 1:1) to give crude methyl 5-hydroxy-2-methyltetrahydro-2H-pyran-2-carboxylate (18 g, crude) as a pale yellow oil, which used directly for next step.
- [0218]
Step 3: Synthesis of methyl 2-methyl-5-oxotetrahydro-2H-pyran-2-carboxylate
-
- [0219]
To a solution of methyl 5-hydroxy-2-methyltetrahydro-2H-pyran-2-carboxylate (18.0 g, 103 mmol) in anhydrous dichloromethane (200 mL) was added PCC (45.0 g, 209 mmol) in portions. The reaction mixture was stirred at ambient temperature until TLC indicated the reaction was completed. Petroleum ether (500 mL) was then added and the mixture was filtered. The filter cake was washed with petroleum ether (100 mL), and the filtrate was concentrated under vacuum to give methyl 2-methyl-5-oxotetrahydro-2H-pyran-2-carboxylate (15 g, 84% yield) as a pale yellow oil. 1H-NMR (400 MHz, CDCl3): δ 4.25 (d, J=17.6 Hz, 1H), 4.07 (d, J=17.6 Hz, 1H), 3.81 (s, 3H), 2.52-2.44 (m, 3H), 2.11-2.04 (m, 1H), 1.53 (s, 3H).
- [0219]
Example 8. Synthesis of Iodide IntermediatesA. Methyl 1-methoxy-4-iodocyclohexane-1-carboxylate
-
- [0220]

- [0220]
Step 1: Synthesis of methyl 1-methoxy-4-hydroxycyclohexane-1-carboxylate
-
- [0221]
Methyl 1-methoxy-4-oxocyclohexanecarboxylate (4.00 g, 21.5 mmol) was dissolved in methanol (100 mL) and the solution was cooled to 0° C. Sodium borohydride (2.03 g, 53.7 mmol) was added in portions over 20 min. The reaction mixture was stirred for 30 min, then was quenched by addition of aqueous saturated NH4Cl solution. The quenched reaction mixture was evaporated to remove the MeOH, then the aqueous suspension was extracted with DCM (3×). The combined organic layers were dried over sodium sulfate, filtered, and concentrated to yield a residue that was purified by flash-column chromatography on silica gel (gradient elution, 5% to 100% ethyl acetate-hexanes) to afford methyl 1-methoxy-4-hydroxycyclohexane-1-carboxylate (2.00 g, 49.5%) as a colorless oil. MS (ES+) C9H16O4 requires: 188, found: 211 [M+Na]+.
- [0221]
Step 2: Synthesis of methyl 1-methoxy-4-iodocyclohexane-1-carboxylate
-
- [0222]
Methyl 1-methoxy-4-hydroxycyclohexane-1-carboxylate (2.00 g, 10.6 mmol) was dissolved in THF (20 mL) and imidazole (723 mg, 10.6 mmol) and triphenylphosphine (3.34 g, 12.8 mmol) were added. The mixture was cooled to 0° C., and then a solution of iodine (3.24 g, 12.8 mmol) in THF (10 mL) was added dropwise over 15 min. The reaction mixture was allowed to warm to ambient temperature and was then stirred for 2 days, after which it was poured over saturated sodium thiosulfate solution and extracted with EtOAc. The organic layer was dried over sodium sulfate, filtered, concentrated, and the residue was triturated with hexane (40 mL, stir for 20 min). The mixture was filtered, and the filtrate was evaporated to provide a residue that was purified by flash-column chromatography on silica gel (gradient elution, 0 to 30% ethyl acetate-hexanes) to give the title compound (2.37 g, 75%) as a pale yellow oil. MS (ES+) C9H15IO3 requires: 298, found: 299 [M+H]+.
- [0222]
B. Ethyl 1-ethoxy-4-iodocyclohexane-1-carboxylate
-
- [0223]

- [0224]
The title compound was prepared as described above using ethyl 1-ethoxy-4-oxocyclohexane-1-carboxylate as a starting material. C11H19IO3 requires: 326, found: 327 [M+H]−.
- [0223]
Example 9. Synthesis of Amine IntermediatesA. (S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethan-1-amine
-
- [0225]

- [0225]
Step 1: Synthesis of 1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethan-1-one
-
- [0226]
4-Fluoro-1H-pyrazole (4.73 g, 55 mmol) and potassium carbonate (17.27 g, 125 mmol) were combined and stirred in N,N-dimethylformamide (41.7 mL) for 10 minutes in an open sealed tube before addition of 2-bromo-5-acetylpyridine (10 g, 50 mmol). The reaction tube was sealed and stirred for 20 hours at 100° C. The reaction mixture was then cooled to room temperature and poured into water (˜700 mL). The mixture was sonicated and stirred for 20 minutes, after which a beige solid was isolated by filtration, washed with small amounts of water, and dried to yield 1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethan-1-one (9.81 g, 96% yield). MS: M+1=206.0.
- [0226]
Step 2: Synthesis of (R)—N—((S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-2-methylpropane-2-sulfinamide
-
- [0227]
To a stirred room temperature solution of 1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethan-1-one (9.806 g, 47.8 mmol) in THF (96 mL) was added (R)-(+)-t-Butylsulfinamide (5.79 g, 47.8 mmol) followed by titanium (IV) ethoxide (21.8 g, 96 mmol). The solution was stirred at 75° C. on an oil bath for 15 hours. The reaction solution was cooled to room temperature and then to −78° C. (external temperature) before the next step. To the −78° C. solution was added dropwise over nearly 55 minutes L-Selectride (143 mL of 1N in THF, 143 mmol). During addition, some bubbling was observed. The reaction was then stirred after the addition was completed for 15 minutes at −78° C. before warming to room temperature. LC-MS of sample taken during removal from cold bath showed reaction was completed. The reaction was cooled to −50° C. and quenched slowly with methanol (˜10 mL), then poured into water (600 mL) and stirred. An off-white precipitate was removed by filtration, with ethyl acetate used for washes. The filtrate was diluted with ethyl acetate (800 mL), the layers were separated, and the organic layer was dried over sodium sulfate, filtered, and concentrated down. The crude was purified by silica gel chromatography to yield (R)—N—((S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-2-methylpropane-2-sulfinamide (10.5 g, 99% purity, 70.3% yield) as a light yellow solid. MS: M+1=311.1.
- [0227]
Step 3: Synthesis of (S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethan-1-amine
- [0228]
A solution of (R)—N—((S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-2-methylpropane-2-sulfinamide (10.53 g, 33.9 mmol)) in methanol (79 mmol) and 4N HCl/dioxane (85 mL, 339 mmol) was stirred for 2.5 hours, at which point LC-MS showed reaction was complete. The reaction solution was poured into diethyl ether (300 mL) and a sticky solid was formed. The mixture was treated with ethyl acetate (200 mL) and sonicated. The solvents were decanted, and the sticky solid was treated with more ethyl acetate (˜200 mL), sonicated and stirred. The bulk of the sticky solid was converted to a suspension. A light yellow solid was isolated by filtration, washed with smaller amounts of ethyl acetate, and dried to yield (S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethan-1-amine (7.419 g, 78% yield). LC-MS confirmed desired product in high purity. MS: M+1=207.1.
PATENT
CN 111440151
PATENT
CN 111362923
References
- ^ Jump up to:a b c d e f g h i j k l m n “FDA approves pralsetinib for lung cancer with RET gene fusions”. U.S. Food and Drug Administration (FDA). 4 September 2020. Retrieved 8 September 2020. This article incorporates text from this source, which is in the public domain.
- ^ “Blueprint Medicines Announces FDA Approval of Gavreto (pralsetinib) for the Treatment of Adults with Metastatic RET Fusion-Positive Non-Small Cell Lung Cancer” (Press release). Blueprint Medicines. 4 September 2020. Retrieved 8 September 2020 – via PR Newswire.
- ^ “Roche announces FDA approval of Gavreto (pralsetinib) for the treatment of adults with metastatic RET fusion-positive non-small cell lung cancer”. Roche (Press release). 7 September 2020. Retrieved 8 September 2020.
- ^ Jump up to:a b c d “Drug Trial Snapshot: Gavreto”. U.S. Food and Drug Administration. 4 September 2020. Retrieved 16 September 2020. This article incorporates text from this source, which is in the public domain.
External links
- “Pralsetinib”. Drug Information Portal. U.S. National Library of Medicine.
- “Pralsetinib”. NCI Drug Dictionary. National Cancer Institute.
- Clinical trial number NCT03037385 for “Phase 1/2 Study of the Highly-selective RET Inhibitor, Pralsetinib (BLU-667), in Patients With Thyroid Cancer, Non-Small Cell Lung Cancer, and Other Advanced Solid Tumors (ARROW)” at ClinicalTrials.gov
- “Understanding Metastatic RET Fusion-Positive Non-Small Cell Lung Cancer (NSCLC)” (PDF).
- “Understanding Metastatic RET-Driven Thyroid Cancers” (PDF).
| Clinical data | |
|---|---|
| Trade names | Gavreto |
| Other names | BLU-667 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
By mouth |
| Drug class | Tyrosine kinase inhibitor |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C27H32FN9O2 |
| Molar mass | 533.612 g·mol−1 |
| 3D model (JSmol) | |
Roche buys into Blueprint’s RET inhibitor
The deal positions pralsetinib to compete against Lilly’s Retevmo
by Lisa M. Jarvis
JULY 18, 2020 | APPEARED IN VOLUME 98, ISSUE 28

Pralsetinib is a small-molecule inhibitor of RET alterations—rare genetic fusions or mutations that occur at low levels across lung, thyroid, and many other cancers.
The drug will go up against Eli Lilly and Company’s Retevmo, an RET inhibitor that received FDA approval in May for certain lung and thyroid cancers. Lilly acquired Retevmo in its $8 billion purchase of Loxo Oncology in 2019, a deal to obtain Loxo’s pipeline of small molecules for genetically defined tumors.
But SVB Leerink analyst Andrew Berens points out that Retevmo has side effects: it can cause an irregular heart rhythm called QT prolongation and hemorrhagic events. That leaves room for pralsetinib, which Roche will be better able to get in front of oncologists, Berens argues. In addition to a vast commercial network, Roche brings diagnostic tools to help identify cancer patients whose tumors feature RET alterations.
The FDA has a deadline of Nov. 23 to decide on approving the drug for lung cancer.
Roche’s move lowers the likelihood of a takeover of Blueprint, which had appeared on many investors’ short lists of acquisition targets. “We were surprised by the profuse language framing this deal as ensuring Blueprint’s independence,” Piper Sandler stock analyst Christopher J. Raymond told investors in a note.
//////////Pralsetinib, GAVRETO, 2020 APPROVALS, FDA 2020
CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3CCC(CC3)(C(=O)NC(C)C4=CN=C(C=C4)N5C=C(C=N5)F)OC
Clascoterone
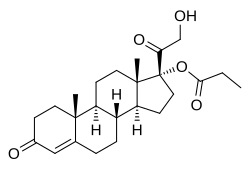
Clascoterone
(1R,3aS,3bR,9aR,9bS,11aS)-1-(2-hydroxyacetyl)-9a,11a-dimethyl-7-oxo-1H,2H,3H,3aH,3bH,4H,5H,7H,8H,9H,9aH,9bH,10H,11H,11aH-cyclopenta[a]phenanthren-1-yl propanoate
| Formula |
C24H34O5
|
|---|---|
| CAS |
19608-29-8
|
| Mol weight |
402.5238
|
FDA APPROVED, 2020/8/26, Winlevi
|
クラスコステロン;
|
Anti-acne, Androgen receptor antagonist
Clascoterone, sold under the brand name Winlevi, is an antiandrogen medication which is used topically in the treatment of acne.[1][2][3] It is also under development for the treatment of androgen-dependent scalp hair loss.[2] The medication is used as a cream by application to the skin, for instance the face and scalp.[3]
Clascoterone is an antiandrogen, or antagonist of the androgen receptor (AR), the biological target of androgens such as testosterone and dihydrotestosterone.[4][5] It shows no systemic absorption when applied to skin.[3]
The medication, developed by Cassiopea and Intrepid Therapeutics,[2] was approved by the US Food and Drug Administration (FDA) for acne in August 2020.[6][7]
Medical uses
Clascoterone is indicated for the topical treatment of acne vulgaris in females and males age 12 years and older.[1][8] It is applied to the affected skin area in a dose of 1 mg cream (or 10 mg clascoterone) twice per day, once in the morning and once in the evening.[1] The medication should not be used ophthalmically, orally, or vaginally.[1]
Available forms
Clascoterone is available in the form of a 1% (10 mg/g) cream for topical use.[1]
Contraindications
Clascoterone has no contraindications.[1]
Side effects
The incidences of local skin reactions with clascoterone were similar to placebo in two large phase 3 randomized controlled trials.[1][9] Suppression of the hypothalamic–pituitary–adrenal axis (HPA axis) may occur during clascoterone therapy in some individuals due to its cortexolone metabolite.[1][8] HPA axis suppression as measured by the cosyntropin stimulation test was observed to occur in 3 of 42 (7%) of adolescents and adults using clascoterone for acne.[1][8] HPA axis function returned to normal within 4 weeks following discontinuation of clascoterone.[1][8] Hyperkalemia (elevated potassium levels) occurred in 5% of clascoterone-treated individuals and 4% of placebo-treated individuals.[1]
Pharmacology
Pharmacodynamics
Clascoterone is an steroidal antiandrogen, or antagonist of the androgen receptor (AR), the biological target of androgens such as testosterone and dihydrotestosterone (DHT).[1][4][5] In a bioassay, the topical potency of the medication was greater than that of progesterone, flutamide, and finasteride and was equivalent to that of cyproterone acetate.[10] Likewise, it is significantly more efficacious as an antiandrogen than other AR antagonists such as enzalutamide and spironolactone in scalp dermal papilla cells and sebocytes in vitro.[5]\
Pharmacokinetics
Steady-state levels of clascoterone occur within 5 days of twice daily administration.[1] At a dosage of 6 g clascoterone cream applied twice daily, maximal circulating levels of clascoterone were 4.5 ± 2.9 ng/mL, area-under-the-curve levels over the dosing interval were 37.1 ± 22.3 h*ng/mL, and average circulating levels of clascoterone were 3.1 ± 1.9 ng/mL.[1] In rodents, clascoterone has been found to possess strong local antiandrogenic activity, but negligible systemic antiandrogenic activity when administered via subcutaneous injection.[10] Along these lines, the medication is not progonadotropic in animals.[10]
The plasma protein binding of clascoterone is 84 to 89% regardless of concentration.[1]
Clascoterone is rapidly hydrolyzed into cortexolone (11-deoxycortisol) and this compound is a possible primary metabolite of clascoterone based on in-vitro studies in human liver cells.[1][8] During treatment with clascoterone, cortexolone levels were detectable and generally below or near the low limit of quantification (0.5 ng/mL).[1] Clascoterone may also produce other metabolites, including conjugates.[1]
The elimination of clascoterone has not been fully characterized in humans.[1]
Chemistry
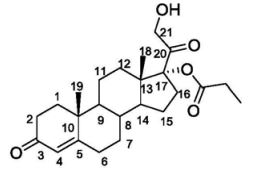
Clascoterone, also known as cortexolone 17α-propionate or 11-deoxycortisol 17α-propionate, as well as 17α,21-dihydroxyprogesterone 17α-propionate or 17α,21-dihydroxypregn-4-en-3,20-dione 17α-propionate, is a synthetic pregnane steroid and a derivative of progesterone and 11-deoxycortisol (cortexolone).[11] It is specifically the C17α propionate ester of 11-deoxycortisol.[10]
An analogue of clascoterone is 9,11-dehydrocortexolone 17α-butyrate (CB-03-04).[12]
History
C17α esters of 11-deoxycortisol were unexpectedly found to possess antiandrogenic activity.[10] Clascoterone, also known as cortexolone 17α-propionate, was selected for development based on its optimal drug profile.[10] The medication was approved by the US Food and Drug Administration (FDA) for the treatment of acne in August 2020.[6]
Two large phase 3 randomized controlled trials evaluated the effectiveness of clascoterone for the treatment of acne over a period of 12 weeks.[1][8][9] Clascoterone decreased acne symptoms by about 8 to 18% more than placebo.[1][9] The defined treatment success endpoint was achieved in about 18 to 20% of individuals with clascoterone relative to about 7 to 9% of individuals with placebo.[1][8][9] The comparative effectiveness of clascoterone between males and females was not described.[1][9]
A small pilot randomized controlled trial in 2011, found that clascoterone cream decreased acne symptoms to a similar or significantly greater extent than tretinoin 0.05% cream.[8][13] No active comparator was used in the phase III clinical trials of clascoterone for acne.[8] Hence, it’s unclear how clascoterone compares to other therapies used in the treatment of acne.[8]
The FDA approved clascoterone based on evidence from two clinical trials (Trial 1/NCT02608450 and Trial 2/NCT02608476) of 1440 participants 9 to 58 years of age with acne vulgaris.[14] The trials were conducted at 99 sites in the United States, Poland, Romania, Bulgaria, Ukraine, Georgia, and Serbia.[14]
Participants applied clascoterone or vehicle (placebo) cream twice daily for 12 weeks.[14] Neither the participants nor the health care providers knew which treatment was being given until after the trial was completed.[14] The benefit of clascoterone in comparison to placebo was assessed after 12 weeks of treatment using the Investigator’s Global Assessment (IGA) score that measures the severity of disease (on a scale from 0 to 4) and a decrease in the number of acne lesions.[14]
Society and culture
Names
Clascoterone is the generic name of the drug and its INN and USAN.[11][15]
Research
Clascoterone has been suggested as a possible treatment for hidradenitis suppurativa (acne inversa), an androgen-dependent skin condition.[16]
………………………………………………………………………….
PATENT
CN 112028956
https://patents.google.com/patent/CN112028956A/en

Example 1
Preparation of intermediate I
Wherein R is DMTr
Dissolving the compound 11-deoxycortisol (1.04g, 3.0mmol, 1eq.) in 10mL of anhydrous pyridine, dissolving dried DMTrCl (1.2-1.5eq) in 5mL of anhydrous dichloromethane, dropwise adding a dichloromethane solution of DMTrCl into the reactant solution at room temperature, and reacting for 4 hours at room temperature; the reaction was quenched with methanol and the solvent was evaporated to dryness with an oil pump to give intermediate I in 85% yield (the next reaction was carried out without work-up, the solvent environment and catalyst were similar to the reaction of this step).
1 H NMR (600MHz, CDCl 3 ) (ppm) 7.25-7.31 (m, 5H, H-DMTr), 7.15-7.18 (m, 4H, H-DMTr), 6.81-6.84 (m, 4H, H-DMTr), 5.73(1H,s,H-4),4.65(1H,dd,J=19.8,4.8Hz,H-21),4.30(1H,dd,J=19.8,4.8Hz,H-21),3.80(6H ,s),2.71(s,1H,17-OH),2.66-2.71(m,1H,H-16β),2.27-2.45(m,4H),1.19(3H,s,H-19),0.96- 1.87(m, 14H), 0.72(s, 3H, H-18).
MS + 303(DMTr protecting group fragment), 649[M + H] +
Melting point: 95-97 deg.C
Example 2:
preparation of intermediate II
Wherein R is DMTr
Under the protection of nitrogen, dissolving the intermediate product I (1eq.) in 5mL of anhydrous dichloromethane, adding DMAP (0.1eq.) into the solution, dropwise adding triethylamine (1.2eq.) and propionic anhydride or propionyl chloride (1.2eq. ), reacting at 40 ℃ for 12 hours after dropwise adding, and evaporating the solvent to obtain an intermediate product II.
Or under the protection of nitrogen, dissolving the intermediate product I (1eq.) in 5mL of anhydrous pyridine, adding DMAP (0.1eq.) into the solution, dropwise adding triethylamine (1.2eq.) and propionic anhydride or propionyl chloride (1.2eq .), reacting at 80 ℃ for 4 hours after dropwise adding, and evaporating the solvent to obtain an intermediate product II. (the reaction in the step can be directly carried out for the next step of removing DMTr protecting group to obtain the reaction after solvent evaporation without strict purification post-treatment)
1 H NMR (600MHz, CDCl 3 ) (ppm) 7.26-7.32 (m, 5H, H-DMTr), 7.14-7.18 (m, 4H, H-DMTr), 6.81-6.84 (m, 4H, H-DMTr), 5.72(1H,s,H-4),4.65(1H,dd,J=19.8,4.8Hz,H-21),4.30(1H,dd,J=19.8,4.8Hz,H-21),3.81(6H ,s),2.66-2.71(m,1H,H-16β),2.35(m,2H,-CH 2 CH 3 ),2.27-2.45(m,4H),1.19(3H,s,H-19), 1.15 (t, 3H, J=7.8Hz, -CH 2 CH 3 ), 0.96-1.87 (m, 14H), 0.72 (s, 3H, H-18);
MS + :303(DMTr protecting group fragment), 727[ M + Na [)] + ,768[M+Na+CH 3 CN] + .
Example 3:
preparation of target Compound 1 (21-hydroxy-17- (1-oxopropoxy) pregn-4-ene-3, 20-dione)
Dissolving the concentrated intermediate product II in an ethyl acetate solution, slowly dropwise adding 0.5M hydrochloric acid solution or 2% trifluoroacetic acid-ethyl acetate solution at 0 ℃, reacting for 5 minutes at 0 ℃, removing DMTr protective groups, adding 5% sodium bicarbonate aqueous solution at 0 ℃, stirring, neutralizing acid in a reaction system, washing an ethyl acetate organic layer twice by using 5% sodium bicarbonate aqueous solution, removing acid and other water-soluble impurities in the ethyl acetate organic layer, drying the ethyl acetate organic layer by anhydrous sodium sulfate, evaporating to remove part of ethyl acetate solvent, adding petroleum ether into the remaining small amount of ethyl acetate solution, and recrystallizing in a system with 10 times of solvent amount of ethyl acetate-petroleum ether (5:1) to obtain a target product with high purity of 90%. The total yield from 11-deoxycortisol is up to 70%. The final product was free of isomerized by-products by HPLC and was not found by LCMS.
1 H NMR (600 MHz, CDCl 3 ) (ppm): 5.75 (s, 1H, H-4), 4.28 (d, 1H, J=18.0 Hz, H-21), 4.23 (d, 1H, J=18.0 Hz, H-21), 3.05(s, 1H, 21-OH), 2.81-2.86(m, 1H, H-16β), 2.34-2.46(m, 3H), 2.35(m, 2H, -CH 2 CH 3 ) ,2.28-2.33(m,1H),2.03-2.07(m,1H),1.86-1.94(m,2H),1.67-1.77(m,3H),1.55-1.64(m,3H),1.35-1.46( m, 3H), 1.19(s, 3H, H-19), 1.15(t, 3H, J=7.8Hz, -CH 2 CH 3 ), 1.08-1.11(m, 1H), 1.00-1.05(m, 1H ),0.69(s,3H,H-18);
MS + : 403[M+H] + , 444[M+H+CH 3 CN] +
Melting point: 128-130 ℃.
PATENT
WO 2009019138,
EXAMPLES Example 1
Alcoho lysis with CCL of cortexolone 17α, 21-dipropionate
Add butanol (0.4g, 5.45 mmoles) and CCL (17.4g, 3.86 U/mg, FLUKA) to a solution of cortexolone- 17α,21-dipropionate (0.5g, 1.09 mmoles) in toluene (50ml). Maintain the mixture under stirring, at 30 0C, following the progress of the reaction in TLC (Toluene/ethyl acetate 6/4) until the initial material is dissolved (24h). Remove the enzyme by means of filtration using a Celite layer. Recover the cortexolone 17α-propionate (0.437, 99%) after evaporation under low pressure. Through crystallisation, from diisopropyl ether you obtain a product with a purity >99% in HPLC.
1 H-NMR (500MHz, CDCl3) relevant signals δ (ppm) 5.78 (br s, 1 H, H-4), 4.32 (dd, 1 H, H-21), 4.25 (dd, IH, H-21), 1.22 (s, 3H, CH3-19), 1.17 (t, 3H, CH3), 0.72 (s, 3H5 CH3-18). P.f. 114 0C Example 2
According to the method described in example 1 prepare cortexolone- 17α- butanoate.
1H-NMR relevant signals δ (ppm) 5.78 (br s, IH, H-4), 4.32 (dd, IH, H-21), 4.26 (dd, IH, H-21), 1.23 (s, 3H, CH3-19), 0.97 (t, 3H, CH3), 0.73 (s, 3H, CH3-18). P.F. 134-136 0C
Example 3
According to the method described in the example prepare cortexolone- 17α- valerate.
1H-NMR relevant signals δ (ppm) 5.77 (br s, IH, H-4), 4.32 (dd, IH, H-21), 4.26
(dd, IH, H-21), 1.22 (s, 3H, CH3-19), 0.95 (t, 3H, CH3), 0.72 (s, 3H, CH3-18). P.f.
114 0C (diisopropyl ether).
Example 4
According to the method described in the example prepare 9,11-dehydro- cortexolone- 17α-butanoate.
1 H-NMR relevant signals δ (ppm) 5.77 (br s, IH, H-4), 5.54 (m, IH, H-9), 4.29
(dd, IH, H-21), 4.24 (dd, IH, H-21), 1.32 (s, 3H, CH3-19), 0.94(t, 3H, CH3), 0.68
(s, 3H, CH3– 18). P.f. 135-136 0C (acetone/hexane).
Example 5
Alcoho lysis with CALB of cortexolone- 17α, 21-dipropionate
Dissolve cortexolone, 17α, 2-dipropionate (0.5g, 1 .09 mmoles) in acetonitrile
(40ml), add CALB (2.3g, 2.5 U/mg Fluka) and octanol (0.875ml). Leave the mixture under stirring, at 30 0C, for 76 hrs. Remove the enzyme by means of filtration using a paper filter. Once the solvents evaporate, recover a solid
(0.4758) which upon analysis 1H-NMR shall appear made up of cortexolone- 17α- propionate at 91%.
Example 6
Crystallisation
Add the solvent (t-butylmethylether or diisopropylether) to the sample according to the ratios indicated in Table 3. Heat the mixture to the boiling temperature of the solvent, under stirring, until the sample dissolves completely. Cool to room temperature and leave it at this temperature, under stirring, for 6 hours. Filter using a buchner funnel and maintain the solid obtained, under low pressure, at a room temperature for 15 hours and then, at 400C, for 5 hours.
Example 7
Precipitation Disslove the sample in the suitable solvent (dichloromethane, acetone, ethyl acetate or ethanol) according to the ratios indicated in table 3 and then add the solvent, hexane or water, according to the ratios indicated in table 3, maintaining the mixture, under stirring, at room temperature. Recover the precipitate by filtration using a buchner funnel and desiccate as in example 6. Example 8.
Obtaining a pharmaceutical form containing the medication in a defined crystalline form.
Prepare a fluid cream containing 2 % cetylic alcohol, 16% glyceryl monostearate, 10% vaseline oil, 13 % propylene glycol, 10% poly ethylengly col with low polymerization 1.5% polysorbate 80 and 47.5 % purified water. Add 1 g of cortexolone 17α-propionate of crystalline form III to 100 g of this cream and subject the mixture to homogenisation by means of a turbine agitator until you obtain homogeneity. You obtain a cream containing a fraction of an active ingredient dissolved in the formulation vehicle and a non-dissolved fraction of an active ingredient, present as a crystal of crystalline form III. This preparation is suitable for use as a formulation vehicle for skin penetration tests on Franz cells, where a coefficient of penetration in the range of 0.04 to 0.03 cm/h is observed on the preparation. Example 9.
Obtaining the pharmaceutical form containing the medication in solvate form IV for replacing the solvent during the galenic formulation procedure Dissolve lOOg of cortexolone 17α-propionate of crystalline form III in 2500 g of propylene glycol under stirring at room temperature. Separately prepare, by using a turbo emulsifϊer raising the temperature up to about 700C, an emulsion with 250 g of Cetylic alcohol, 1500 g of glyceryl monostearate, 1000 g of liquid paraffin, 5 g of mixed tocopherols, 100 g of polysorbate 80 and 4650 g of water. After cooling the emulsion up to about 300C, add – under stirring and under negative pressure – the cortexolone 17α-propionate solution in propylene glycol. Maintain the emulsioned cream under stirring until you obtain homogeneity, making sure the temperature remains low by means the circulation of a coolant. The cream contains a dispersed crystalline fraction, made up of an active ingredient in solvate crystalline form IV, formed due to the precipitation of the active ingredient itself from the glycolic solution which contained it when the latter was added to the predominantly aqueous formulation. The DRX spectra of the crystalline form present in the cream are indicated in Fig. 30.
PAPER
Tetrahedron Letters, 49(31), 4610-4612; 2008
Abstract
Several 17α-monoesters of cortexolone and its Δ9-derivative are endowed with antiandrogenic activity. Their synthesis can be accomplished by means of a lipase-catalyzed chemoselective alcoholysis of the corresponding 17α,21-diesters.
Graphical abstract

1H NMR (500 MHz, CDCl3): selected data δ 5.78 (br s, 1H, H-4), 4.32 (dd, 1H, H-21, J18.3 and 4.9 Hz), 4.25 (dd, 1H, H-21, J18.3 and 4.9 Hz), 1.22 (s, 3H, CH3-19), 1.17 (t, 3H, CH3, J7.6 Hz), 0.72 (s, 3H, CH3-18) MP 133 °C (t-butylmethylether)
…………………………………………………………………..
PATENT
https://patents.google.com/patent/EP2503005B1/en
-
Cortexolone derivatives in which the hydroxyl group at position C-17α is esterified with short chain aliphatic or aromatic acids and the derivatives of the corresponding 9,11-dehydro derivative, are known to have an antiandrogenic effect.
- [0002]
EP 1421099 describes cortexolone 17α-propionate and 9,11-dehydro-cortexolone-17-α-butanoate regarding a high antiandrogenic biological activity demonstrated both “in vitro” and “in vivo” on the animal.
- [0003]
US3530038 discloses the preparation of a crystalline form of cortexolone-17α-propionate having a melting point of 126-129 °C and an IR spectrum with bands at (cm-1): 3500, 1732, 1713, 1655 and 1617.
- [0004]
A method for obtaining the above mentioned derivatives is described by Gardi et al. (Gazz. Chim. It. 63, 43 1,1963) and in the United States patent US3152154 providing for the transformation of cortexolone, or transformation of 9,11-dehydrocortexolone, in the intermediate orthoester using orthoesters available in the market as a mixture of aprotic solvents such as cyclohexane and DMF, in presence of acid catalysis (ex. PTSA.H20). The intermediate orthoester thus obtained can be used as is or upon purification by suspension in a solvent capable of solubilising impurities, preferably in alcohols. The subsequent hydrolysis in a hydroalcoholic solution, buffered to pH 4-5 preferably in acetate buffer, provides the desired monoester.
- [0005]
Such synthesis is indicated in the diagram 1 below

- [0006]
However, the monoesters thus obtained were, in the reaction conditions, unstable and, consequently hard to manipulate and isolate (R. Gardi et al Tetrahedron Letters, 448, 1961). The instability is above all due to the secondary reaction of migration of the esterifying acyl group from position 17 to position 21.
- [0007]
It is thus known that in order to obtain the above mentioned monoesters with a chemical purity in such a manner to be able to proceed to the biological tests, it is necessary to use, at the end of the synthesis, a purification process which is generally performed by means of column chromatography.
- [0008]
Furthermore, US3152154 describes how the hydrolysis of the diester in a basic environment is not convenient due to the formation of a mixture of 17α,21-diol, of 17- and 21 -monoesters, alongside the initial non-reacted product.
- [0009]
Now, it has been surprisingly discovered that an alcoholysis reaction using a lipase from Candida as a biocatalyst can be usefully applied during the preparation of 17α monoesters of cortexolone, or its 9,11-dehydroderivatives.
- [0010]
As a matter of fact, it has been discovered that such enzymatic alcoholysis of the 17,21-diester of the cortexolone, or of its derivative 9,11-dehydro, selectively occurs in position 21 moving to the corresponding monoester in position 17, as shown in diagram 2 below:

- [0011]
The chemoselectivity of the special enzymatic reaction in alcoholysis conditions, according to the present invention, opens new perspectives for preparation, at industrial level with higher yields, of 17α-monoesters with respect to the methods already indicated in literature.
- [0012]
The diesters serving as a substrate for the reaction of the invention can be prepared according to the prior art, for example following the one described in B.Turner, (Journal of American Chemical Society, 75, 3489, 1953) which provides for the esterification of corticosteroids with a linear carboxylic acid in presence of its anhydride and PTSA monohydrate.
EXAMPLES
-
- Example 1
Alcoholysis with CCL of cortexolone 17α, 21-dipropionate
-
-
- [0055]
Add butanol (0.4g, 5.45 mmoles) and CCL (17.4g, 3.86 U/mg, FLUKA) to a solution of cortexolone-17α,21-dipropionate (0.5g, 1.09 mmoles) in toluene (50ml). Maintain the mixture under stirring, at 30 °C, following the progress of the reaction in TLC (Toluene/ethyl acetate 6/4) until the initial material is dissolved (24h). Remove the enzyme by means of filtration using a Celite layer. Recover the cortexolone 17α-propionate (0.437, 99%) after evaporation under low pressure. Through crystallisation, from diisopropyl ether you obtain a product with a purity >99% in HPLC.
- [0056]
1H-NMR (500MHz, CDCl3) relevant signals δ (ppm) 5.78 (br s, 1 H, H-4), 4.32 (dd, 1 H, H-21), 4.25 (dd, 1H, H-21), 1.22 (s, 3H, CH3-19), 1.17 (t, 3H, CH3), 0.72 (s, 3H, CH3-18). P.f. 114 °C
- [0055]
-
Example 2 (comparative)
-
-
- [0057]
According to the method described in example 1 prepare cortexolone-17α-butanoate.
- [0058]
1H-NMR relevant signals δ (ppm) 5.78 (br s, 1H, H-4), 4.32 (dd, 1H, H-21), 4.26 (dd, 1H, H-21), 1.23 (s, 3H, CH3-19), 0.97 (t, 3H, CH3), 0.73 (s, 3H. CH3-18). P.F. 134-136 °C
- [0057]
-
Example 3 (comparative)
According to the method described in the example prepare cortexolone-17α-valerate.
-
-
- [0059]
1H-NMR relevant signals δ (ppm) 5.77 (br s, 1H, H-4), 4.32 (dd, 1H, H-21), 4.26 (dd, 1H, H-21), 1.22 (s, 3H, CH3-19), 0.95 (t, 3H, CH3), 0.72 (s, 3H, CH3-18). P.f. 114 °C (diisopropyl ether).
- [0059]
-
Example 4 (comparative)
According to the method described in the example prepare 9, 11-dehydro-cortexolone-17α-butanoate.
-
-
- [0060]
1H-NMR relevant signals δ (ppm) 5.77 (br s, 1H, H-4), 5.54 (m, 1H, H-9), 4.29 (dd, 1H, H-21), 4.24 (dd, 1H, H-21), 1.32 (s, 3H, CH3-19), 0.94(t, 3H, CH3), 0.68 (s, 3H, CH3-18). P.f. 135-136 °C (acetone/hexane).
- [0060]
-
Example 5
Alcoholysis with CALB of cartexolone-17α, 21-dipropionate
-
-
- [0061]
Dissolve cortexolone, 17α, 2-dipropionate (0.5g, 1.09 mmoles) in acetonitrile (40ml), add CALB (2.3g, 2.5 U/mg Fluka) and octanol (0.875ml). Leave the mixture under stirring, at 30 °C, for 76 hrs. Remove the enzyme by means of filtration using a paper filter. Once the solvents evaporate, recover a solid (0.4758) which upon analysis 1H-NMR shall appear made up of cortexolone-17α-propionate at 91%.
- [0061]
-
Example 6
Crystallisation
-
-
- [0062]
Add the solvent (t-butylmethylether or diisopropylether) to the sample according to the ratios indicated in Table 3. Heat the mixture to the boiling temperature of the solvent, under stirring, until the sample dissolves completely. Cool to room temperature and leave it at this temperature, under stirring, for 6 hours. Filter using a buchner funnel and maintain the solid obtained, under low pressure, at a room temperature for 15 hours and then, at 40°C, for 5 hours.
- [0062]
-
Example 7 (comparative)
Precipitation
-
-
- [0063]
Disslove the sample in the suitable solvent (dichloromethane, acetone, ethyl acetate or ethanol) according to the ratios indicated in table 3 and then add the solvent, hexane or water, according to the ratios indicated in table 3, maintaining the mixture, under stirring, at room temperature. Recover the precipitate by filtration using a buchner funnel and desiccate as in example 6.
- [0063]
-
Example 8.
Obtaining a pharmaceutical form containing the medication in a defined crystalline form.
- [0064]
Prepare a fluid cream containing 2 % cetylic alcohol, 16% glyceryl monostearate, 10% vaseline oil, 13 % propylene glycol, 10% polyethylenglycol with low polymerization 1.5% polysorbate 80 and 47.5 % purified water. Add 1 g of cortexolone 17α-propionate of crystalline form III to 100 g of this cream and subject the mixture to homogenisation by means of a turbine agitator until you obtain homogeneity. You obtain a cream containing a fraction of an active ingredient dissolved in the formulation vehicle and a non-dissolved fraction of an active ingredient, present as a crystal of crystalline form III. This preparation is suitable for use as a formulation vehicle for skin penetration tests on Franz cells, where a coefficient of penetration in the range of 0.04 to 0.03 cm/h is observed on the preparation.
References
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t u v w “Winlevi (clascoterone) cream, for topical use”(PDF). Cassiopea. Retrieved 9 September 2020.
- ^ Jump up to:a b c http://adisinsight.springer.com/drugs/800026561
- ^ Jump up to:a b c Kircik LH (July 2019). “What’s new in the management of acne vulgaris”. Cutis. 104(1): 48–52. PMID 31487336.
- ^ Jump up to:a b Rosette C, Rosette N, Mazzetti A, Moro L, Gerloni M (February 2019). “Cortexolone 17α-Propionate (Clascoterone) is an Androgen Receptor Antagonist in Dermal Papilla Cells In Vitro”. J Drugs Dermatol. 18 (2): 197–201. PMID 30811143.
- ^ Jump up to:a b c Rosette C, Agan FJ, Mazzetti A, Moro L, Gerloni M (May 2019). “Cortexolone 17α-propionate (Clascoterone) Is a Novel Androgen Receptor Antagonist that Inhibits Production of Lipids and Inflammatory Cytokines from Sebocytes In Vitro”. J Drugs Dermatol. 18 (5): 412–418. PMID 31141847.
- ^ Jump up to:a b “Cassiopea Receives FDA Approval for Winlevi (clascoterone cream 1%), First-in-Class Topical Acne Treatment Targeting the Androgen Receptor”. Cassiopea (Press release). Retrieved 2020-08-30.
- ^ “Winlevi: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 9 September 2020.
- ^ Jump up to:a b c d e f g h i j Barbieri, John S. (2020). “A New Class of Topical Acne Treatment Addressing the Hormonal Pathogenesis of Acne”. JAMA Dermatology. 156 (6): 619–620. doi:10.1001/jamadermatol.2020.0464. ISSN 2168-6068. PMID 32320045.
- ^ Jump up to:a b c d e Hebert A, Thiboutot D, Stein Gold L, Cartwright M, Gerloni M, Fragasso E, Mazzetti A (April 2020). “Efficacy and Safety of Topical Clascoterone Cream, 1%, for Treatment in Patients With Facial Acne: Two Phase 3 Randomized Clinical Trials”. JAMA Dermatol. 156 (6): 621–630. doi:10.1001/jamadermatol.2020.0465. PMC 7177662. PMID 32320027.
- ^ Jump up to:a b c d e f Celasco G, Moro L, Bozzella R, Ferraboschi P, Bartorelli L, Quattrocchi C, Nicoletti F (2004). “Biological profile of cortexolone 17alpha-propionate (CB-03-01), a new topical and peripherally selective androgen antagonist”. Arzneimittelforschung. 54 (12): 881–6. doi:10.1055/s-0031-1297043. PMID 15646372.
- ^ Jump up to:a b https://chem.nlm.nih.gov/chemidplus/rn/19608-29-8
- ^ Celasco G, Moroa L, Bozzella R, Ferraboschi P, Bartorelli L, Di Marco R, Quattrocchi C, Nicoletti F (2005). “Pharmacological profile of 9,11-dehydrocortexolone 17alpha-butyrate (CB-03-04), a new androgen antagonist with antigonadotropic activity”. Arzneimittelforschung. 55 (10): 581–7. doi:10.1055/s-0031-1296908. PMID 16294504.
- ^ Trifu V, Tiplica GS, Naumescu E, Zalupca L, Moro L, Celasco G (2011). “Cortexolone 17α-propionate 1% cream, a new potent antiandrogen for topical treatment of acne vulgaris. A pilot randomized, double-blind comparative study vs. placebo and tretinoin 0·05% cream”. Br. J. Dermatol. 165 (1): 177–83. doi:10.1111/j.1365-2133.2011.10332.x. PMID 21428978. S2CID 38404925.
- ^ Jump up to:a b c d e “Drug Trial Snapshot: Winlevi”. U.S. Food and Drug Administration (FDA). 26 August 2020. Retrieved 10 September 2020. This article incorporates text from this source, which is in the public domain.
- ^ World Health Organization (2019). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 82”. WHO Drug Information. 33 (3): 106. hdl:10665/330879.
- ^ Der Sarkissian SA, Sun HY, Sebaratnam DF (August 2020). “Cortexolone 17 α-proprionate for hidradenitis suppurativa”. Dermatol Ther: e14142. doi:10.1111/dth.14142. PMID 32761708.
External links
- “Clascoterone”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT02608450 for “A Study to Evaluate the Safety and Efficacy of CB-03-01 Cream, 1% in Subjects With Facial Acne Vulgaris (25)” at ClinicalTrials.gov
- Clinical trial number NCT02608476 for “A Study to Evaluate the Safety and Efficacy of CB-03-01 Cream, 1% in Subjects With Facial Acne Vulgaris (26)” at ClinicalTrials.gov
 |
|
| Clinical data | |
|---|---|
| Trade names | Winlevi |
| Other names | CB-03-01; Breezula; 11-Deoxycortisol 17α-propionate; 17α-(Propionyloxy)- deoxycorticosterone; 21-Hydroxy-3,20-dioxopregn-4-en-17-yl propionate |
| License data |
|
| Routes of administration |
Topical (cream) |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.210.810 |
| Chemical and physical data | |
| Formula | C24H34O5 |
| Molar mass | 402.531 g·mol−1 |
| 3D model (JSmol) | |
/////////Clascoterone, クラスコステロン , FDA 2020, 2020 APPROVALS, ANTI ACNE
[H][C@@]12CC[C@](OC(=O)CC)(C(=O)CO)[C@@]1(C)CC[C@@]1([H])[C@@]2([H])CCC2=CC(=O)CC[C@]12C
Copper Cu 64 dotatate, 銅(Cu64)ドータテート;

Copper Cu 64 dotatate
銅(Cu64)ドータテート;
UNII-N3858377KC
N3858377KC
Copper 64-DOTA-tate
Copper Cu-64 dotatate
Copper dotatate Cu-64
Diagnostic (neuroendocrine tumors), Radioactive agent
| Formula |
C65H86CuN14O19S2. 2H
|
|---|---|
| CAS: |
1426155-87-4
|
| Mol weight |
1497.1526
|
FDA APPROVED 2020. 2020/9/3. Detectnet
2-[4-[2-[[(2R)-1-[[(4R,7S,10S,13R,16S,19R)-10-(4-aminobutyl)-4-[[(1S,2R)-1-carboxy-2-hydroxypropyl]carbamoyl]-7-[(1R)-1-hydroxyethyl]-16-[(4-hydroxyphenyl)methyl]-13-(1H-indol-3-ylmethyl)-6,9,12,15,18-pentaoxo-1,2-dithia-5,8,11,14,17-pentazacycloicos-19-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-2-oxoethyl]-10-(carboxylatomethyl)-7-(carboxymethyl)-1,4,7,10-tetrazacyclododec-1-yl]acetate;copper-64(2+)
Copper Cu 64 dotatate, sold under the brand name Detectnet, is a radioactive diagnostic agent indicated for use with positron emission tomography (PET) for localization of somatostatin receptor positive neuroendocrine tumors (NETs) in adults.[1]
Common side effects include nausea, vomiting and flushing.[2]
It was approved for medical use in the United States in September 2020.[1][2]
History
The U.S. Food and Drug Administration (FDA) approved copper Cu 64 dotatate based on data from two trials that evaluated 175 adults.[3]
Trial 1 evaluated adults, some of whom had known or suspected NETs and some of whom were healthy volunteers.[3] The trial was conducted at one site in the United States (Houston, TX).[3] Both groups received copper Cu 64 dotatate and underwent PET scan imaging.[3] Trial 2 data came from the literature-reported trial of 112 adults, all of whom had history of NETs and underwent PET scan imaging with copper Cu 64 dotatate.[3] The trial was conducted at one site in Denmark.[3] In both trials, copper Cu 64 dotatate images were compared to either biopsy results or other images taken by different techniques to detect the sites of a tumor.[3] The images were read as either positive or negative for presence of NETs by three independent image readers who did not know participant clinical information.[3]
PATENT
https://patents.google.com/patent/WO2013029616A1/en
PATENT
https://patents.google.com/patent/US20140341807
-
Known imaging techniques with tremendous importance in medical diagnostics are positron emission tomography (PET), computed tomography (CT), magnetic resonance imaging (MRI), single photon computed tomography (SPECT) and ultrasound (US). Although today’s imaging technologies are well developed they rely mostly on non-specific, macroscopic, physical, physiological, or metabolic changes that differentiate pathological from normal tissue.
- [0003]
Targeting molecular imaging (MI) has the potential to reach a new dimension in medical diagnostics. The term “targeting” is related to the selective and highly specific binding of a natural or synthetic ligand (binder) to a molecule of interest (molecular target) in vitro or in vivo.
- [0004]
MI is a rapidly emerging biomedical research discipline that may be defined as the visual representation, characterization and quantification of biological processes at the cellular and sub-cellular levels within intact living organisms. It is a novel multidisciplinary field, in which the images produced reflect cellular and molecular pathways and in vivo mechanism of disease present within the context of physiologically authentic environments rather than identify molecular events responsible for disease.
- [0005]
Several different contrast-enhancing agents are known today and their unspecific or non-targeting forms are already in clinical routine. Some examples listed below are reported in literature.
- [0006]
For example, Gd-complexes could be used as contrast agents for MRI according to “Contrast Agents I” by W. Krause (Springer Verlag 2002, page one and following pages). Furthermore, superparamagnetic particles are another example of contrast-enhancing units, which could also be used as contrast agents for MRI (Textbook of Contrast Media, Superparamagnetic Oxides, Dawson, Cosgrove and Grainger Isis Medical Media Ltd, 1999, page 373 and following pages). As described in Contrast Agent II by W. Krause (Springer Verlag 2002, page 73 and following pages), gas-filled microbubbles could be used in a similar way as contrast agents for ultrasound. Moreover “Contrast Agents II” by W. Krause (Springer Verlag, 2002, page 151 and following pages) reports the use of iodinated liposomes or fatty acids as contrast agents for X-Ray imaging.
- [0007]
Contrast-enhancing agents that can be used in functional imaging are mainly developed for PET and SPECT.
- [0008]
The application of radiolabelled bioactive peptides for diagnostic imaging is gaining importance in nuclear medicine. Biologically active molecules which selectively interact with specific cell types are useful for the delivery of radioactivity to target tissues. For example, radiolabelled peptides have significant potential for the delivery of radionuclides to tumours, infarcts, and infected tissues for diagnostic imaging and radiotherapy.
- [0009]
DOTA (1,4,7,10-tetrakis(carboxymethyl)-1,4,7,10tetraazacyclododecane) and its derivatives constitute an important class of chelators for biomedical applications as they accommodate very stably a variety of di- and trivalent metal ions. An emerging area is the use of chelator conjugated bioactive peptides for labeling with radiometals in different fields of diagnostic and therapeutic nuclear oncology.
- [0010]
There have been several reports in recent years on targeted radiotherapy with radiolabeled somatostatin analogs.
- [0011]
US2007/0025910A1 discloses radiolabled somatostatin analogs primarily based on the ligand DOTA-TOC. The radionucleotide can be (64)Copper and the somatostatin analog may be octreotide, lanreotide, depreotide, vapreotide or derivatives thereof. The compounds of US2007/0025910A1 are useful in radionucleotide therapy of tumours.
- [0012]
US2007/0025910A1 does not disclose (64)Cu-DOTA-TATE. DOTA-TATE and DOTA-TOC differ clearly in affinity for the 5 known somatostatin receptors (SST1-SST2). Accordingly, the DOTA-TATE has a 10-fold higher affinity for the SST2 receptor, the receptor expressed to the highest degree on neuroendocrine tumors. Also the relative affinity for the other receptor subtypes are different. Furthermore, since 177Lu-DOTATATE is used for radionuclide therapy, only 64Cu-DOTATATE and not 64Cu-DOTATOC can be used to predict effect of such treatment by a prior PET scan.
- [0013]
There exists a need for further peptide-based compounds having utility for diagnostic imaging techniques, such as PET.
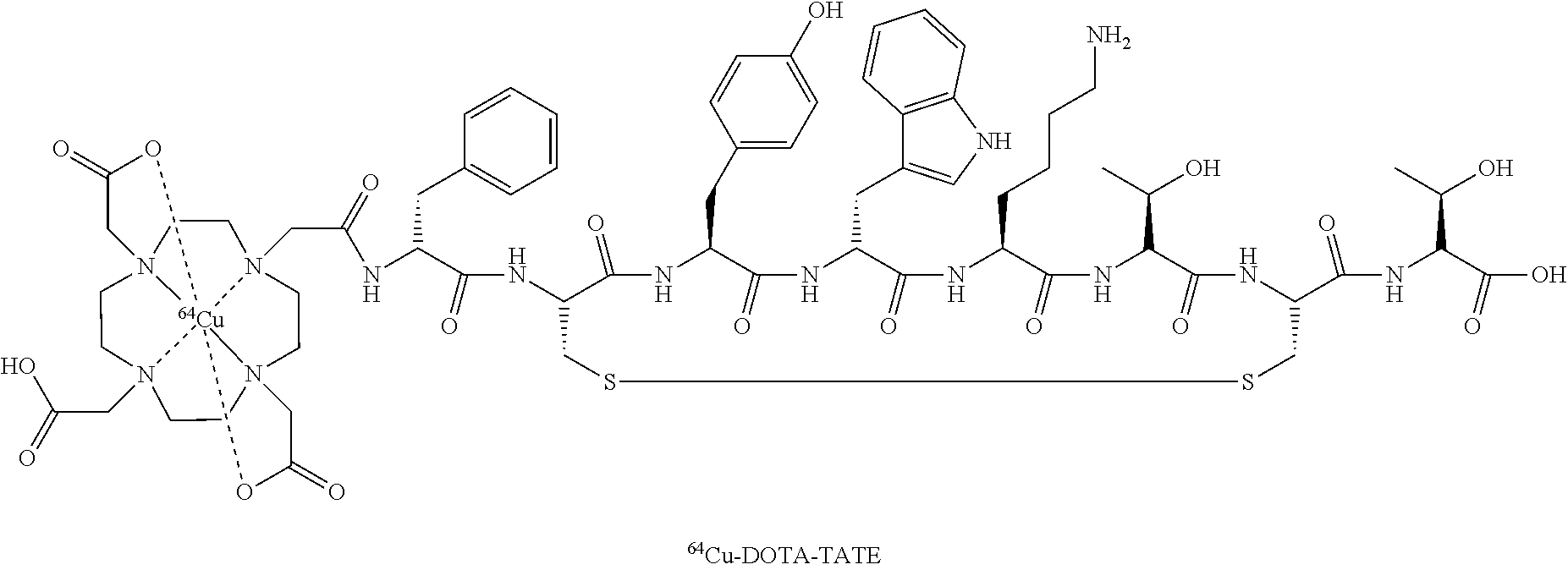
-
- EXAMPLE
- [0033]
Preparation of “Cu-Dotatate-DOTA-TATE
- [0034]
64Cu was produced using a GE PETtrace cyclotron equipped with a beamline. The 64Cu was produced via the 64Ni (p,n) 64Cu reaction using a solid target system consisting of a water cooled target mounted on the beamline. The target consisted of 64Ni metal (enriched to >99%) electroplated on a silver disc backing. For this specific type of production a proton beam with the energy of 16 MeV and a beam current of 20 uA was used. After irradiation the target was transferred to the laboratory for further chemical processing in which the 64Cu was isolated using ion exchange chromatography. Final evaporation from aq. HCl yielded 2-6 GBq of 64Cu as 64CuCl2 (specific activity 300-3000 TBq/mmol; RNP >99%). The labeling of 64Cu to DOTA-TATE was performed by adding a sterile solution of DOTA-TATE (0.3 mg) and Gentisic acid (25 mg) in aq Sodium acetate (1 ml; 0.4M, pH 5.0) to a dry vial containing 64CuCl2 (˜1 GBq). Gentisic acid was added as a scavenger to reduce the effect of radiolysis. The mixture was left at ambient temperature for 10 minutes and then diluted with sterile water (1 ml). Finally, the mixture was passed through a 0.22 μm sterile filter (Millex GP, Millipore). Radiochemical purity was determined by RP-HPLC and the amount of unlabeled 64Cu2+ was determined by thin-layer chromatography. All chemicals were purchased from Sigma-Aldrich unless specified otherwise. DOTA-Tyr3-Octreotate (DOTA-TATE) was purchased from Bachem (Torrance, Calif.). Nickel-64 was purchased in +99% purity from Campro Scientific Gmbh. All solutions were made using Ultra pure water (<0.07 μSimens/cm). Reversed-phase high pressure liquid chromatography was performed on a Waters Alliance 2795 Separations module equipped with at Waters 2489 UV/Visible detector and a Caroll Ramsey model 105 S-1 radioactivity detector—RP-HPLC column was Luna C18, HST, 50×2 mm, 2.5 μm, Phenomenex. The mobile phase was 5% aq. acetonitrile (0.1% TFA) and 95% aq. acetonitrile (0.1% TFA).
- [0035]
Thin layer chromatography was performed with a Raytest MiniGita Star TLC-scanner equipped with a Beta-detector. The eluent was 50% aq methanol and the TLC-plate was a Silica60 on Al foil (Fluka). Ion exchange chromatography was performed on a Dowex 1×8 resin (Chloride-form, 200-400 mesh).
References
- ^ Jump up to:a b “FDA approval letter” (PDF). 3 September 2020. Retrieved 5 September 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b “RadioMedix and Curium Announce FDA Approval of Detectnet (copper Cu 64 dotatate injection) in the U.S.” (Press release). Curium. 8 September 2020. Retrieved 9 September 2020 – via GlobeNewswire.
- ^ Jump up to:a b c d e f g h “Drug Trials Snapshots: Detectnet”. U.S. Food and Drug Administration (FDA). 3 September 2020. Retrieved 10 September 2020. This article incorporates text from this source, which is in the public domain.
External links
- “Copper dotatate Cu-64”. Drug Information Portal. U.S. National Library of Medicine.
- “copper Cu 64 dotatate injection safety data sheet” (PDF). Curium US LLC. 15 March 2020.



The FDA has approved copper Cu 64 dotatate injection (Detectnet) for the localization of somatostatin receptor–positive neuroendocrine tumors (NETs), according to an announcement from RadioMedix Inc. and Curium Pharma.1
The positron emission tomography (PET) diagnostic agent is anticipated to launch immediately, according to Curium. Doses will be accessible through several nuclear pharmacies or through the nuclear medicine company.
“Detectnet brings an exciting advancement in the diagnosis of NETs for healthcare providers, patients, and their caregivers,” Ebrahim Delpassand MD, CEO of RadioMedix, stated in a press release. “The phase 3 results demonstrate the clinical sensitivity and specificity of Detectnet which will provide a great aid to clinicians in developing an accurate treatment approach for their [patients with] NETs.”
Copper Cu 64 dotatate adheres to somatostatin receptors with highest affinity for subtype 2 receptors (SSTR2). Specifically, the agent binds to somatostatin receptor–expressing cells, including malignant neuroendocrine cells; these cells overexpress SSTR2. The agent is a positron-producing radionuclide that possesses an emission yield that permits PET imaging.
“Perhaps most exciting is that the 12.7-hour half-life allows Detectnet to be produced centrally and shipped to sites throughout the United States,” added Delpassand. “This will help alleviate shortages or delays that have been experienced with other somatostatin analogue PET agents.”
Two single-center, open-label studies confirmed the efficacy of the diagnostic agent, according to Curium.2 In Study 1, investigators conducted a prospective analysis of 63 patients, which included 42 patients with known or suspected NETs according to histology, conventional imaging, or clinical evaluations, and 21 healthy volunteers. The majority of the participants, or 88% (n = 37) had a history of NETs at the time that they underwent imaging. Just under half of patients (44%; n = 28) were men and the majority were white (86%). Moreover, patients had a mean age of 54 years.
Images produced by the PET agent were interpreted to be either positive or negative for NET via 3 independent readers who had been blinded to the clinical data and other imaging information. Moreover, the results from the diagnostic agent were compared with a composite reference standard that was comprised of 1 oncologist’s blinded evaluation of patient diagnosis based on available histopathology results, reports of conventional imaging that had been done within 8 weeks before the PET imaging, as well as clinical and laboratory findings, which involved chromogranin A and serotonin levels.
Additionally, the percentage of patients who tested positive for disease via composite reference as well as through PET imaging was used to quantify positive percent agreement. Conversely, the percentage of participants who did not have disease per composite reference and who were determined to be negative for disease per PET imaging was used to quantify negative percent agreement.
Results showed that the percent reader agreement for positive detection in 62 scans was 91% (95% CI, 75-98) and negative detection was 97% (95% CI, 80-99). For reader 2, these percentages were 91% (95% CI, 75-98) and 80% (95% CI, 61-92), respectively, for 63 scans. Lastly, the percent reader agreement for reader 3 in 63 scans was 91% (95% CI, 75-98) positive and 90% (95% CI, 72-97) negative.
Study 2 was a retrospective analysis in which investigators examined published findings collected from 112 patients; 63 patients were male, while 43 were female. The mean age of patients included in the analysis was 62 years. All patients had a known history of NETs. Results demonstrated similar performance with the PET imaging agent.
In both safety and efficacy trials, a total of 71 patients were given a single dose of the diagnostic agent; the majority of these patients had known or suspected NETs and 21 were healthy volunteers. Adverse reactions such as nausea, vomiting, and flushing were reported at a rate of less than 2%. In all clinical experience that has been published, a total of 126 patients with a known history of NETs were given a single dose of the PET diagnostic agent. A total of 4 patients experienced nausea immediately after administration.
“Curium is excited to bring the first commercially available Cu 64 diagnostic agent to the US market,” Dan Brague, CEO of Curium, North America, added in the release. “Our unique production capabilities and distribution network allow us to deliver to any nuclear pharmacy, hospital, or imaging center its full dosing requirements first thing in the morning, to provide scheduling flexibility to the institution and its patients. We look forward to joining with healthcare providers and our nuclear pharmacy partners to bring this highly efficacious agent to the market.”
References
1. RadioMedix and Curium announce FDA approval of Detectnet (copper Cu 64 dotatate injection) in the US. News release. RadioMedix Inc and Curium. September 8, 2020. Accessed September 9, 2020. https://bit.ly/3m6iC0q.
2. Detectnet. Prescribing information. Curium Pharma; 2020. Accessed September 9, 2020. https://bit.ly/32eZxS3.
///////////////Copper Cu 64 dotatate, 銅(Cu64)ドータテート , FDA 2020, 2020 APPROVALS, Diagnostic, neuroendocrine tumors, Radioactive agent,
CC(C1C(=O)NC(CSSCC(C(=O)NC(C(=O)NC(C(=O)NC(C(=O)N1)CCCCN)CC2=CNC3=CC=CC=C32)CC4=CC=C(C=C4)O)NC(=O)C(CC5=CC=CC=C5)NC(=O)CN6CCN(CCN(CCN(CC6)CC(=O)[O-])CC(=O)[O-])CC(=O)O)C(=O)NC(C(C)O)C(=O)O)O.[Cu+2]
Amisulpride, アミスルプリド ,
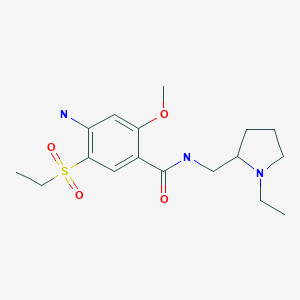
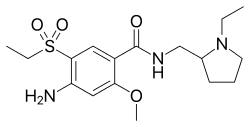
Amisulpride
FDA 2020, Barhemsys APPROVED, 2020/2/27
| Name |
Amisulpride (INN);
Deniban (TN); Solian (TN) アミスルプリド;
|
|---|---|
| Formula |
C17H27N3O4S
|
| CAS |
71675-85-9
|
| Mol weight |
369.479
|
Antipsychotic, Dopamine receptor antagonist, Neuropsychiatric agent
amisulpride(标准品)
275-831-7 [EINECS]
Synthesis ReferenceUS4401822
4-Amino-N-[(1-ethyl-2-pyrrolidinyl)methyl]-5-(ethylsulfonyl)-2-methoxybenzamide
Amisulpride
CAS Registry Number: 71675-85-9
CAS Name: 4-Amino-N-[(1-ethyl-2-pyrrolidinyl)methyl]-5-(ethylsulfonyl)-2-methoxybenzamide
Additional Names: 4-amino-N-[(1-ethyl-2-pyrrolidinyl)methyl]-5-(ethylsulfonyl)-o-anisamide; aminosultopride
Manufacturers’ Codes: DAN-2163
Trademarks: Deniban (Synthelabo); Socian (Synthelabo); Solian (Synthelabo); Sulamid (Baldacci)
Molecular Formula: C17H27N3O4S
Molecular Weight: 369.48
Percent Composition: C 55.26%, H 7.37%, N 11.37%, O 17.32%, S 8.68%
Literature References: Dopamine receptor antagonist. Prepn: M. Thominet et al., BE 872585; eidem, US 4401822 (1979, 1983 both to Soc. d’Etudes Sci. Ind. de l’Ile-de-France).
Crystal structure: H. L. DeWinter et al., Acta Crystallogr. C46, 313 (1990). Psychopharmacology: G. Perrault et al., J. Pharmacol. Exp. Ther. 280, 73 (1997). HPLC determn in plasma and urine: B. Malavasi et al., J. Chromatogr. B 676, 107 (1996). Series of articles on pharmacology and clinical efficacy in schizophrenia: Int. Clin. Psychopharmacol. 12, Suppl. 2, S11-S36 (1997).
Properties: Crystals from acetone, mp 126-127°. LD50 in male mice (mg/kg): 56-60 i.v.; 175-180 i.p.; 224-250 s.c.; 1024-1054 orally (Thominet).
Melting point: mp 126-127°
Toxicity data: LD50 in male mice (mg/kg): 56-60 i.v.; 175-180 i.p.; 224-250 s.c.; 1024-1054 orally (Thominet)
Therap-Cat: Antipsychotic.
Keywords: Antipsychotic; Benzamides; Dopamine Receptor Antagonist.
Amisulpride (trade name Solian) is an antipsychotic drug sold by Sanofi-Aventis. but is approved for use in Europe and Australia for the treatment of psychoses and schizophrenia. Additionally, it is approved in Italy for the treatment of dysthymia (under the brand name Deniban). Amisulpride is a selective dopamine antagonist.
Amisulpride is an antiemetic and antipsychotic medication used at lower doses intravenously to prevent and treat postoperative nausea and vomiting; and at higher doses orally and intramuscularly to treat schizophrenia and acute psychotic episodes. It is sold under the brandnames Barhemsys[6] (as an antiemetic) and Solian, Socian, Deniban and others (as an antipsychotic).[2] It is also used to treat dysthymia.[7]
It is usually classed with the atypical antipsychotics. Chemically it is a benzamide and like other benzamide antipsychotics, such as sulpiride, it is associated with a high risk of elevating blood levels of the lactation hormone, prolactin (thereby potentially causing the absence of the menstrual cycle, breast enlargement, even in males, breast milk secretion not related to breastfeeding, impaired fertility, impotence, breast pain, etc.), and a low risk, relative to the typical antipsychotics, of causing movement disorders.[8][9][10] It has also been found to be modestly more effective in treating schizophrenia than the typical antipsychotics.[9]
Amisulpride is approved for use in the United States in adults for the prevention of postoperative nausea and vomiting (PONV), either alone or in combination with an antiemetic of a different class; and to treat PONV in those who have received antiemetic prophylaxis with an agent of a different class or have not received prophylaxis.[6]
Amisulpride is believed to work by blocking, or antagonizing, the dopamine D2 receptor, reducing its signalling. The effectiveness of amisulpride in treating dysthymia and the negative symptoms of schizophrenia is believed to stem from its blockade of the presynapticdopamine D2 receptors. These presynaptic receptors regulate the release of dopamine into the synapse, so by blocking them amisulpride increases dopamine concentrations in the synapse. This increased dopamine concentration is theorized to act on dopamine D1 receptors to relieve depressive symptoms (in dysthymia) and the negative symptoms of schizophrenia.[7]
It was introduced by Sanofi-Aventis in the 1990s. Its patent expired by 2008, and generic formulations became available.[11] It is marketed in all English-speaking countries except for Canada and the United States.[10] A New York City based company, LB Pharmaceuticals, has announced the ongoing development of LB-102, also known as N-methyl amisulpride, an antipsychotic specifically targeting the United States.[12][13] A poster presentation at European Neuropsychopharmacology[14] seems to suggest that this version of amisulpride, known as LB-102 displays the same binding to D2, D3 and 5HT7 that amisulpride does.[15][16]
Medical uses
Schizophrenia
In a 2013 study in a comparison of 15 antipsychotic drugs in effectiveness in treating schizophrenic symptoms, amisulpride was ranked second and demonstrated high effectiveness. 11% more effective than olanzapine (3rd), 32-35% more effective than haloperidol, quetiapine, and aripiprazole, and 25% less effective than clozapine (1st).[9] Although according to other studies it appears to have comparable efficacy to olanzapine in the treatment of schizophrenia.[17][18] Amisulpride augmentation, similarly to sulpirideaugmentation, has been considered a viable treatment option (although this is based on low-quality evidence) in clozapine-resistant cases of schizophrenia.[19][20] Another recent study concluded that amisulpride is an appropriate first-line treatment for the management of acute psychosis.[21]
Contraindications
Amisulpride’s use is contraindicated in the following disease states[2][22][8]
- Pheochromocytoma
- Concomitant prolactin-dependent tumours e.g. prolactinoma, breast cancer
- Movement disorders (e.g. Parkinson’s disease and dementia with Lewy bodies)
- Lactation
- Children before the onset of puberty
Neither is it recommended to use amisulpride in patients with hypersensitivities to amisulpride or the excipients found in its dosage form.[2]
Adverse effects
- Very Common (≥10% incidence)[1]
- Extrapyramidal side effects (EPS; including dystonia, tremor, akathisia, parkinsonism). Produces a moderate degree of EPS; more than aripiprazole (not significantly, however), clozapine, iloperidone (not significantly), olanzapine (not significantly), quetiapine (not significantly) and sertindole; less than chlorpromazine (not significantly), haloperidol, lurasidone (not significantly), paliperidone (not significantly), risperidone (not significantly), ziprasidone (not significantly) and zotepine (not significantly).[9]
- Insomnia
- Hypersalivation
- Nausea
- Headache
- Hyperactivity
- Anxiety
- Vomiting
- Hyperprolactinaemia (which can lead to galactorrhoea, breast enlargement and tenderness, sexual dysfunction, etc.)
- Weight gain (produces less weight gain than chlorpromazine, clozapine, iloperidone, olanzapine, paliperidone, quetiapine, risperidone, sertindole, zotepine and more (although not statistically significantly) weight gain than haloperidol, lurasidone, ziprasidone and approximately as much weight gain as aripiprazole and asenapine)[9]
- Anticholinergic side effects (although it does not bind to the muscarinic acetylcholine receptors and hence these side effects are usually quite mild) such as
- – constipation
- – dry mouth
- – disorder of accommodation
- – Blurred vision
- Rare (<1% incidence)[1][2][23][22][8]
- Blood dyscrasias such as leucopenia, neutropenia and agranulocytosis
- QT interval prolongation (in a recent meta-analysis of the safety and efficacy of 15 antipsychotic drugs amisulpride was found to have the 2nd highest effect size for causing QT interval prolongation[9])
Hyperprolactinaemia results from antagonism of the D2 receptors located on the lactotrophic cells found in the anterior pituitary gland. Amisulpride has a high propensity for elevating plasma prolactin levels as a result of its poor blood-brain barrier penetrability and hence the resulting greater ratio of peripheral D2 occupancy to central D2 occupancy. This means that to achieve the sufficient occupancy (~60–80%[24]) of the central D2 receptors in order to elicit its therapeutic effects a dose must be given that is enough to saturate peripheral D2receptors including those in the anterior pituitary.[25][26]
- Somnolence. It produces minimal sedation due to its absence of cholinergic, histaminergic and alpha adrenergic receptor antagonism. It is one of the least sedating antipsychotics.[9]
Discontinuation
The British National Formulary recommends a gradual withdrawal when discontinuing antipsychotics to avoid acute withdrawal syndrome or rapid relapse.[27] Symptoms of withdrawal commonly include nausea, vomiting, and loss of appetite.[28] Other symptoms may include restlessness, increased sweating, and trouble sleeping.[28] Less commonly there may be a felling of the world spinning, numbness, or muscle pains.[28] Symptoms generally resolve after a short period of time.[28]
There is tentative evidence that discontinuation of antipsychotics can result in psychosis.[29] It may also result in reoccurrence of the condition that is being treated.[30] Rarely tardive dyskinesia can occur when the medication is stopped.[28]
Overdose
Torsades de pointes is common in overdose.[31][32] Amisulpride is moderately dangerous in overdose (with the TCAs being very dangerous and the SSRIs being modestly dangerous).[33][34]
Interactions
Amisulpride should not be used in conjunction with drugs that prolong the QT interval (such as citalopram, venlafaxine, bupropion, clozapine, tricyclic antidepressants, sertindole, ziprasidone, etc.),[33] reduce heart rate and those that can induce hypokalaemia. Likewise it is imprudent to combine antipsychotics due to the additive risk for tardive dyskinesia and neuroleptic malignant syndrome.[33]
Pharmacology
Pharmacodynamics
Amisulpride and its relatives sulpiride, levosulpiride, and sultopride have been shown to bind to the high-affinity GHB receptor at concentrations that are therapeutically relevant (IC50 = 50 nM for amisulpride).[37]Amisulpride functions primarily as a dopamine D2 and D3 receptor antagonist. It has high affinity for these receptors with dissociation constantsof 3.0 and 3.5 nM, respectively.[36] Although standard doses used to treat psychosis inhibit dopaminergic neurotransmission, low doses preferentially block inhibitory presynaptic autoreceptors. This results in a facilitation of dopamine activity, and for this reason, low-dose amisulpride has also been used to treat dysthymia.[2]
Amisulpride, sultopride and sulpiride respectively present decreasing in vitro affinities for the D2 receptor (IC50 = 27, 120 and 181 nM) and the D3 receptor (IC50 = 3.6, 4.8 and 17.5 nM).[39]
Though it was long widely assumed that dopaminergic modulation is solely responsible for the respective antidepressant and antipsychoticproperties of amisulpride, it was subsequently found that the drug also acts as a potent antagonist of the serotonin 5-HT7 receptor (Ki = 11.5 nM).[36] Several of the other atypical antipsychotics such as risperidone and ziprasidone are potent antagonists at the 5-HT7 receptor as well, and selective antagonists of the receptor show antidepressant properties themselves. To characterize the role of the 5-HT7 receptor in the antidepressant effects of amisulpride, a study prepared 5-HT7 receptor knockout mice.[36] The study found that in two widely used rodent models of depression, the tail suspension test, and the forced swim test, those mice did not exhibit an antidepressant response upon treatment with amisulpride.[36] These results suggest that 5-HT7 receptor antagonism mediates the antidepressant effects of amisulpride.[36]
Amisulpride also appears to bind with high affinity to the serotonin 5-HT2B receptor (Ki = 13 nM), where it acts as an antagonist.[36] The clinical implications of this, if any, are unclear.[36] In any case, there is no evidence that this action mediates any of the therapeutic effects of amisulpride.[36]
Society and culture
Brand names
Brand names include: Amazeo, Amipride (AU), Amival, Solian (AU, IE, RU, UK, ZA), Soltus, Sulpitac (IN), Sulprix (AU), Midora (RO) and Socian (BR).[40][41]
Availability
Amisulpride was not approved by the Food and Drug Administration for use in the United States until February 2020, but it is used in Europe,[41]Israel, Mexico, India, New Zealand and Australia[2] to treat psychosis and schizophrenia.[42][43]
Amisulpride was approved for use in the United States in February 2020.[44][6]
CLIP

Dopamine receptor antagonist. Prepn: M. Thominet et al., BE 872585; eidem, U.S. Patent 4,401,822 (1979, 1983 both to Soc. d’Etudes Sci. Ind. de l’Ile-de-France).
CLIP
4-Amino-N-((1-ethyl-2-pyrrolidinyl)methyl)-5-(ethylsulfonyl)-o-anisamide, could be produced through many synthetic methods.
Following is one of the synthesis routes:
Firstly, the acetylation of 5-aminosalicylic acid (I) with acetic anhydride in hot acetic acid affords 5-acetaminosalicylic acid (II), which is methylated with dimethyl sulfate and K2CO3 in refluxing acetone producing methyl 2-methoxy-5-acetaminobenzoate (III). Secondly, nitration of (III) with HNO3 in acetic acid affords methyl 2-methoxy-4-nitro-5-acetaminobenzoate (IV), which is deacetylated with H2SO4 in refluxing methanol to give methyl 2-methoxy-4-nitro-5-aminobenzoate (V). Next, the diazotation of (V) with NaNO2-HCl, followed by reaction with sodium ethylmercaptide, oxidation with H2O2 and hydrolysis with NaOH in ethanol yields 2-methoxy-4-nitro-5-(ethylsulfonyl)benzoic acid (VI), which is condensed with N-ethyl-2-aminomethylpyrrolidine (VII) in the presence of ethyl chloroformate and triethylamine in dioxane affording 2-methoxy-4-nitro-N-[(1-ethyl-2-pyrrolidinyl) methyl]-5-(ethylsulfonyl)benzamide (VIII). At last, this compound is reduced with H2 over Raney-Ni in ethanol.

CLIP
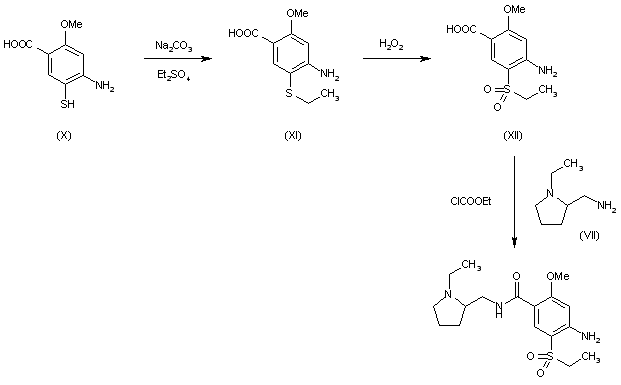
BE 0872585; ES 476755; FR 2415099; GB 2083458; JP 54145658; US 4294828; US 4401822
Alkylation of 2-methoxy-4-amino-5-mercaptobenzoic acid (X) with diethyl sulfate acid Na2CO3 gives 2-methoxy-4-amino-5-ethylthiobenzoic acid (XI), which is oxidized with H2O2 in acetic acid yielding 2-methoxy-4-amino-5-(ethylsulfonyl)benzoic acid (XII). Finally, this compound is condensed with (VII) by means of ethyl chloroformate.
CLIP
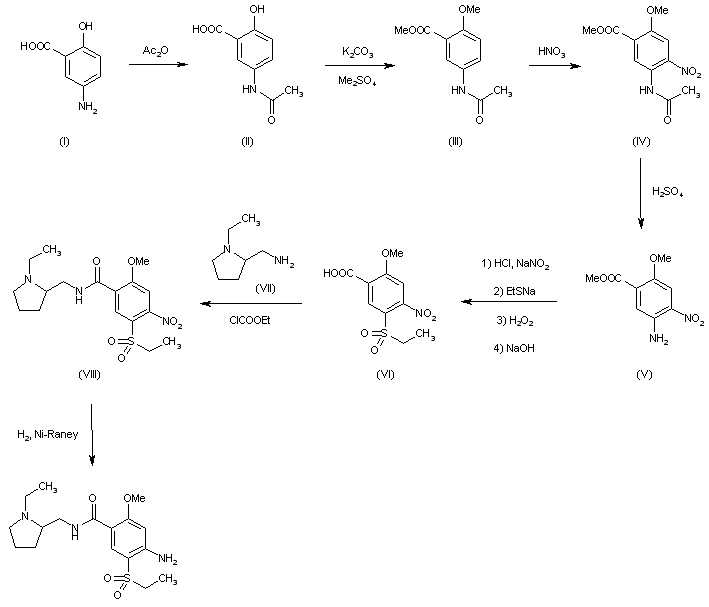
| FR 2460930 |
Acetylation of 5-aminosalicylic acid (I) with acetic anhydride in hot acetic acid gives 5-acetaminosalicylic acid (II), which is methylated with dimethyl sulfate and K2CO3 in refluxing acetone yielding methyl 2-methoxy-5-acetaminobenzoate (III). Nitration of (III) with HNO3 in acetic acid affords methyl 2-methoxy-4-nitro-5-acetaminobenzoate (IV), which is deacetylated with H2SO4 in refluxing methanol to give methyl 2-methoxy-4-nitro-5-aminobenzoate (V). The diazotation of (V) with NaNO2-HCl, followed by reaction with sodium ethylmercaptide, oxidation with H2O2 and hydrolysis with NaOH in ethanol yields 2-methoxy-4-nitro-5-(ethylsulfonyl)benzoic acid (VI), which is condensed with N-ethyl-2-aminomethylpyrrolidine (VII) by means of ethyl chloroformate and triethylamine in dioxane affording 2-methoxy-4-nitro-N-[(1-ethyl-2-pyrrolidinyl) methyl]-5-(ethylsulfonyl)benzamide (VIII). Finally, this compound is reduced with H2 over Raney-Ni in ethanol.
CLIP
Treatment of thiourea (I) with iodomethane provided S-methylthiouronium iodide (II). This was further condensed with N-methylpiperazine (III) to afford the intermediate piperazine-1-carboxamidine (IV)
CLIP
Regioselective lithiation of 1,2,4-trichlorobenzene (V) with n-BuLi at -60 C, followed by quenching of the resultant organolithium compound (VI) with N,N-dimethylformamide yielded 2,3,5-trichlorobenzaldehyde (VII) (1), which was then reduced with NaBH4 to provide alcohol (VIII). Bromination of (VIII) using PBr3 afforded compound (IX), whose bromide atom was displaced with KCN to give the trichlorophenylacetonitrile (X). Claisen condensation of (X) with ethyl formate in the presence of NaOEt furnished the oxo nitrile sodium enolate (XI), which was subsequently O-alkylated with iodomethane yielding the methoxy acrylonitrile (XII). Finally, cyclization of (XII) with the piperazine-1-carboxamidine (IV) in EtOH gave rise to the target pyrimidine derivative
PATENT
https://patents.google.com/patent/US20130096319A1/en
Amisulpride is represented by the formula (I) as given below.

The product patent U.S. Pat. No. 4,401,822 describes preparation of amisulpride as shown in scheme (I)

The synthesis of amisulpride involves oxidation of 2-methoxy-4-amino-5-ethyl-thio benzoic acid (III) using acetic acid and hydrogen peroxide at 40-45° C. for few hours to obtain 2-methoxy-4-amino-5-ethyl-sulfonyl benzoic acid (IV). In our attempt to repeat this reaction, we found that almost 22 hours were required for completion and the purity of compound (IV) was 87.6%.
-
- [0006]
Thus, the product patent method suffers from the disadvantages such as high reaction time, low yield and low purity.
- [0007]
Liu Lie et al, Jingxi Huagong Zhongjianti 2008, 38 (3), 29-32 describes the process for the preparation of 2-methoxy-4-amino-5-ethyl-sulfonyl benzoic acid (IV) as shown in scheme (II).
-

- [0008]
4-amino salicylic acid (VI) is treated with dimethyl sulphate in the presence of potassium hydroxide and acetone to give 4-amino-2-methoxy-methyl benzoate in 4 hours, which is further treated with potassium thiocynate to give compound of formula (VIII). 4-Amino-2-,methoxy-5-thiocyanatobenzoate (VIII) is treated with bromoethane to give 4-amino-5-ethylthio-2-methoxy benzoic acid (IX) which is further converted to 2-methoxy-4-amino-5-ethyl-sulfonyl benzoic acid (IV) via oxidation with hydrogen peroxide and acetic acid.
- [0009]
The yield of conversion of compound (VIII) to compound (IX) is 57% and the overall yield of compound (IV) from compound (VI) is 24% only. Thus, the above process suffers from the disadvantages such as low yield and in that it uses bromoethane which is skin and eye irritant and has carcinogenic effects.
- [0010]
Therefore, there is, an unfulfilled need to provide industrially feasible process for the preparation of 2-methoxy-4-amino-5-ethyl-sulfonyl benzoic acid (IV) and amisulpride (I) with higher purity and yield, since it is one of the key intermediates in the manufacture of amisulpride.
- [0006]
SUMMARY OF THE INVENTION
The present invention is related to a novel process for the preparation of amisulpride (I) that involves: (i) methylation of 4-amino-salicylic-acid (VI) with dimethyl sulphate and base, optionally in presence of TBAB to obtain 4-amino-2-methoxy methyl benzoate (VII) and (ii) oxidation of 4-amino-2-methoxy-5-ethyl thio benzoic acid (IX) or 4-amino-2-methoxy-5-ethyl thio methyl benzoate (X) with oxidizing agent in the presence of sodium tungstate or ammonium molybdate to give 2-methoxy-4-amino-5-ethyl-sulfonyl benzoic acid (IV) or 2-methoxy-4-amino-5-ethyl-sulfonyl methyl benzoate (XI) respectively.
-
- Example 13
- [0097]
Preparation of crude amisulpride
- [0098]
To a stirring mixture of 4-amino-2-methoxy-5-ethyl sulphonyl benzoic acid (IV) and acetone (5.0 L) at 0-5° C., triethyl amine (0.405 Kg) was added and stirred followed by addition of ethyl chloroformate (0.368 Kg). N-ethyl-2-amino methyl pyrrolidine (0.627 Kg) was added to the reaction mass at 5-10° C. Temperature of reaction mass was raised to 25-30° C. and stirred for 120 min. To the same reaction mass triethyl amine (0.405 Kg) and ethyl chloroformate (0.368 Kg) was added with maintaining the temperature. Reaction mass was stirred for 120 min. After completion of reaction, water (4.0 L) was added. Reaction mass was filtered and washed with water (2.0 L). Filtrate was collected and water was added (9.0 L). pH of the reaction mass was adjusted to 10.8-11.2 by using 20% NaOH solution. Reaction mass was stirred for 240-300 min, filtered and washed with water. Solid was dried under vacuum
- [0099]
Yield : 70%
- [0100]
Purity: 98%
- [0097]
Example 14
- [0101]
Purification of amisulpride
- [0102]
Amisulpride (1 kg) was charged in acetone (6 liters) and the reaction mixture was heated till a clear solution was obtained. Slurry of activated carbon (0.1 kg in 1 liter) was added in acetone. The reaction mass was stirred at 50-55 ° C. for 60 minutes and filtered hot. The filtrate was concentrated and further heated to dissolve the solid. The reaction mass was cooled to 0-5° C., stirred and filtered. The precipitated solid was washed with acetone and dried.
- [0103]
Yield: 750 gm (75%)
- [0104]
HPLC purity: 99.8% (quantitative)
- [0105]
M.P.: 125° C.
- [0106]
DSC: shows endotherm at 133° C.
- [0107]
Particle size: d10=0.637, d50=6.0, d90=13.325 microns
CLIP

References
- ^ Jump up to:a b c d “Amisulpride 100 mg Tablets – Summary of Product Characteristics (SmPC)”. (emc). 5 July 2019. Retrieved 26 February 2020.
- ^ Jump up to:a b c d e f g h i j k “Solian tablets and solution product information” (PDF). TGA eBusiness Services. Sanofi-Aventis Australia Pty Ltd. 27 September 2019. Retrieved 26 February2020.
- ^ Jump up to:a b c Rosenzweig, P.; Canal, M.; Patat, A.; Bergougnan, L.; Zieleniuk, I.; Bianchetti, G. (2002). “A review of the pharmacokinetics, tolerability and pharmacodynamics of amisulpride in healthy volunteers”. Human Psychopharmacology. 17 (1): 1–13. doi:10.1002/hup.320. PMID 12404702.
- ^ Caccia, S (May 2000). “Biotransformation of Post-Clozapine Antipsychotics Pharmacological Implications”. Clinical Pharmacokinetics. 38 (5): 393–414. doi:10.2165/00003088-200038050-00002. PMID 10843459.
- ^ Noble, S; Benfield, P (December 1999). “Amisulpride: A Review of its Clinical Potential in Dysthymia”. CNS Drugs. 12 (6): 471–483. doi:10.2165/00023210-199912060-00005.
- ^ Jump up to:a b c “Barhemsys (amisulpride) injection, for intravenous use” (PDF). U.S. Food and Drug Administration (FDA). February 2020. Retrieved 26 February 2020.
- ^ Jump up to:a b Pani L, Gessa GL (2002). “The substituted benzamides and their clinical potential on dysthymia and on the negative symptoms of schizophrenia”. Molecular Psychiatry. 7 (3): 247–53. doi:10.1038/sj.mp.4001040. PMID 11920152.
- ^ Jump up to:a b c d Rossi, S, ed. (2013). Australian Medicines Handbook (2013 ed.). Adelaide: The Australian Medicines Handbook Unit Trust. ISBN 978-0-9805790-9-3.
- ^ Jump up to:a b c d e f g Leucht, S; Cipriani, A; Spineli, L; Mavridis, D; Orey, D; Richter, F; Samara, M; Barbui, C; Engel, RR; Geddes, JR; Kissling, W; Stapf, MP; Lässig, B; Salanti, G; Davis, JM (September 2013). “Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis”. Lancet. 382 (9896): 951–962. doi:10.1016/S0140-6736(13)60733-3. PMID 23810019.
- ^ Jump up to:a b Brayfield, A, ed. (June 2017). “Amisulpride: Martindale: The Complete Drug Reference”. MedicineComplete. Pharmaceutical Press. Retrieved 5 August 2017.
- ^ De Silva, V; Hanwella, R (April 2008). “Pharmaceutical patents and the quality of mental healthcare in low- and middle-income countries”. The Psychiatrist. 32 (4): 121–23. doi:10.1192/pb.bp.107.015651.
- ^ “Pipeline”. LB Pharmaceuticals. Retrieved 29 August 2019.
- ^ “About Us”. LB Pharmaceuticals. Retrieved 26 February 2020.
- ^ “Data presented at 2017 ECNP meeting (European Neuropsychopharmacology, 2017, 27 (S4), S922-S923)”. LB Pharmaceuticals. Retrieved 26 February 2020.
- ^ “Building a translational bridge from animals to man for clinical candidate LB-102, a next-generation benzamide antipsychotic (P.101)” (PDF). LB Pharmaceuticals. Retrieved 29 August 2019.
- ^ Grattan V, Vaino AR, Prensky Z, Hixon MS (August 2019). “Antipsychotic Benzamides Amisulpride and LB-102 Display Polypharmacy as Racemates, S Enantiomers Engage Receptors D2 and D3, while R Enantiomers Engage 5-HT7”. ACS Omega. 4 (9): 14151–4. doi:10.1021/acsomega.9b02144. ISSN 2470-1343. PMC 6714530. PMID 31497735.
- ^ Komossa, K; Rummel-Kluge, C; Hunger, H; Schmid, F; Schwarz, S; Silveira da Mota Neto, JI; Kissling, W; Leucht, S (January 2010). “Amisulpride versus other atypical antipsychotics for schizophrenia”. The Cochrane Database of Systematic Reviews (1): CD006624. doi:10.1002/14651858.CD006624.pub2. PMC 4164462. PMID 20091599.
- ^ Leucht, S; Corves, C; Arbter, D; Engel, RR; Li, C; Davis, JM (January 2009). “Second-generation versus first-generation antipsychotic drugs for schizophrenia: a meta-analysis”. Lancet. 373 (9657): 31–41. doi:10.1016/S0140-6736(08)61764-X. PMID 19058842.
- ^ Solanki, RK; Sing, P; Munshi, D (October–December 2009). “Current perspectives in the treatment of resistant schizophrenia”. Indian Journal of Psychiatry. 51 (4): 254–60. doi:10.4103/0019-5545.58289. PMC 2802371. PMID 20048449.
- ^ Mouaffak, F; Tranulis, C; Gourevitch, R; Poirier, MF; Douki, S; Olié, JP; Lôo, H; Gourion, D (2006). “Augmentation Strategies of Clozapine With Antipsychotics in the Treatment of Ultraresistant Schizophrenia”. Clinical Neuropharmacology. 29 (1): 28–33. doi:10.1097/00002826-200601000-00009. PMID 16518132.
- ^ Nuss, P.; Hummer, M.; Tessier, C. (2007). “The use of amisulpride in the treatment of acute psychosis”. Therapeutics and Clinical Risk Management. 3 (1): 3–11. doi:10.2147/tcrm.2007.3.1.3. PMC 1936283. PMID 18360610.
- ^ Jump up to:a b c Joint Formulary Committee (2013). British National Formulary (BNF) (65 ed.). London, UK: Pharmaceutical Press. ISBN 978-0-85711-084-8.
- ^ Jump up to:a b Truven Health Analytics, Inc. DRUGDEX System (Internet) [cited 2013 Sep 19]. Greenwood Village, CO: Thomsen Healthcare; 2013.
- ^ Brunton, L; Chabner, B; Knollman, B (2010). Goodman and Gilman’s The Pharmacological Basis of Therapeutics (12th ed.). New York: McGraw-Hill Professional. ISBN 978-0-07-162442-8.
- ^ McKeage, K; Plosker, GL (2004). “Amisulpride: a review of its use in the management of schizophrenia”. CNS Drugs. 18 (13): 933–956. doi:10.2165/00023210-200418130-00007. ISSN 1172-7047. PMID 15521794.
- ^ Natesan, S; Reckless, GE; Barlow, KB; Nobrega, JN; Kapur, S (October 2008). “Amisulpride the ‘atypical’ atypical antipsychotic — Comparison to haloperidol, risperidone and clozapine”. Schizophrenia Research. 105 (1–3): 224–235. doi:10.1016/j.schres.2008.07.005. PMID 18710798.
- ^ Joint Formulary Committee, BMJ, ed. (March 2009). “4.2.1”. British National Formulary (57 ed.). United Kingdom: Royal Pharmaceutical Society of Great Britain. p. 192. ISBN 978-0-85369-845-6.
Withdrawal of antipsychotic drugs after long-term therapy should always be gradual and closely monitored to avoid the risk of acute withdrawal syndromes or rapid relapse.
- ^ Jump up to:a b c d e Haddad, Peter; Haddad, Peter M.; Dursun, Serdar; Deakin, Bill (2004). Adverse Syndromes and Psychiatric Drugs: A Clinical Guide. OUP Oxford. p. 207–216. ISBN 9780198527480.
- ^ Moncrieff J (July 2006). “Does antipsychotic withdrawal provoke psychosis? Review of the literature on rapid onset psychosis (supersensitivity psychosis) and withdrawal-related relapse”. Acta Psychiatrica Scandinavica. 114 (1): 3–13. doi:10.1111/j.1600-0447.2006.00787.x. PMID 16774655.
- ^ Sacchetti, Emilio; Vita, Antonio; Siracusano, Alberto; Fleischhacker, Wolfgang (2013). Adherence to Antipsychotics in Schizophrenia. Springer Science & Business Media. p. 85. ISBN 9788847026797.
- ^ Isbister, GK; Balit, CR; Macleod, D; Duffull, SB (August 2010). “Amisulpride overdose is frequently associated with QT prolongation and torsades de pointes”. Journal of Clinical Psychopharmacology. 30 (4): 391–395. doi:10.1097/JCP.0b013e3181e5c14c. PMID 20531221.
- ^ Joy, JP; Coulter, CV; Duffull, SB; Isbister, GK (August 2011). “Prediction of Torsade de Pointes From the QT Interval: Analysis of a Case Series of Amisulpride Overdoses”. Clinical Pharmacology & Therapeutics. 90 (2): 243–245. doi:10.1038/clpt.2011.107. PMID 21716272.
- ^ Jump up to:a b c Taylor, D; Paton, C; Shitij, K (2012). Maudsley Prescribing Guidelines in Psychiatry(11th ed.). West Sussex: Wiley-Blackwell. ISBN 978-0-47-097948-8.
- ^ Levine, M; Ruha, AM (July 2012). “Overdose of atypical antipsychotics: clinical presentation, mechanisms of toxicity and management”. CNS Drugs. 26 (7): 601–611. doi:10.2165/11631640-000000000-00000. PMID 22668123.
- ^ Roth, BL; Driscol, J. “PDSP Ki Database”. Psychoactive Drug Screening Program (PDSP). University of North Carolina at Chapel Hill and the United States National Institute of Mental Health. Retrieved 14 August 2017.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac ad ae af ag ah ai aj ak al am an ao ap aq ar asat au av aw Abbas AI, Hedlund PB, Huang XP, Tran TB, Meltzer HY, Roth BL (2009). “Amisulpride is a potent 5-HT7 antagonist: relevance for antidepressant actions in vivo”. Psychopharmacology. 205 (1): 119–28. doi:10.1007/s00213-009-1521-8. PMC 2821721. PMID 19337725.
- ^ Jump up to:a b Maitre, M.; Ratomponirina, C.; Gobaille, S.; Hodé, Y.; Hechler, V. (April 1994). “Displacement of [3H] gamma-hydroxybutyrate binding by benzamide neuroleptics and prochlorperazine but not by other antipsychotics”. European Journal of Pharmacology. 256(2): 211–214. doi:10.1016/0014-2999(94)90248-8. PMID 7914168.
- ^ Schoemaker H, Claustre Y, Fage D, Rouquier L, Chergui K, Curet O, Oblin A, Gonon F, Carter C, Benavides J, Scatton B (1997). “Neurochemical characteristics of amisulpride, an atypical dopamine D2/D3 receptor antagonist with both presynaptic and limbic selectivity”. J. Pharmacol. Exp. Ther. 280 (1): 83–97. PMID 8996185.
- ^ Blomme, Audrey; Conraux, Laurence; Poirier, Philippe; Olivier, Anne; Koenig, Jean-Jacques; Sevrin, Mireille; Durant, François; George, Pascal (2000), “Amisulpride, Sultopride and Sulpiride: Comparison of Conformational and Physico-Chemical Properties”, Molecular Modeling and Prediction of Bioactivity, Springer US, pp. 404–405, doi:10.1007/978-1-4615-4141-7_97, ISBN 9781461368571
- ^ “Amisulpride international”. Drugs.com. 3 February 2020. Retrieved 26 February 2020.
- ^ Jump up to:a b “Active substance: amisulpride” (PDF). 28 September 2017. EMA/658194/2017; Procedure no.: PSUSA/00000167/201701. Retrieved 26 February 2020.
- ^ Lecrubier, Y.; et al. (2001). “Consensus on the Practical Use of Amisulpride, an Atypical Antipsychotic, in the Treatment of Schizophrenia”. Neuropsychobiology. 44 (1): 41–46. doi:10.1159/000054913. PMID 11408792.
- ^ Kaplan, A. (2004). “Psychotropic Medications Around the World”. Psychiatric Times. 21(5).
- ^ “Barhemsys: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 26 February 2020.
External links
- “Amisulpride”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT01991860 at ClinicalTrials.gov
- Clinical trial number NCT02337062 at ClinicalTrials.gov
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Solian, Barhemsys, others |
| Other names | APD421 |
| AHFS/Drugs.com | International Drug Names |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
By mouth, intravenous |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | 48%[3][2] |
| Protein binding | 16%[2] |
| Metabolism | Hepatic (minimal; most excreted unchanged)[2] |
| Elimination half-life | 12 hours[3] |
| Excretion | Renal[3] (23–46%),[4][5]Faecal[2] |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.068.916 |
| Chemical and physical data | |
| Formula | C17H27N3O4S |
| Molar mass | 369.48 g/mol g·mol−1 |
| 3D model (JSmol) | |
- Rosenzweig P, Canal M, Patat A, Bergougnan L, Zieleniuk I, Bianchetti G: A review of the pharmacokinetics, tolerability and pharmacodynamics of amisulpride in healthy volunteers. Hum Psychopharmacol. 2002 Jan;17(1):1-13. [PubMed:12404702]
- Moller HJ: Amisulpride: limbic specificity and the mechanism of antipsychotic atypicality. Prog Neuropsychopharmacol Biol Psychiatry. 2003 Oct;27(7):1101-11. [PubMed:14642970]
- Weizman T, Pick CG, Backer MM, Rigai T, Bloch M, Schreiber S: The antinociceptive effect of amisulpride in mice is mediated through opioid mechanisms. Eur J Pharmacol. 2003 Oct 8;478(2-3):155-9. [PubMed:14575800]
- Leucht S, Pitschel-Walz G, Engel RR, Kissling W: Amisulpride, an unusual “atypical” antipsychotic: a meta-analysis of randomized controlled trials. Am J Psychiatry. 2002 Feb;159(2):180-90. [PubMed:11823257]
- Rehni AK, Singh TG, Chand P: Amisulpride-induced seizurogenic effect: a potential role of opioid receptor-linked transduction systems. Basic Clin Pharmacol Toxicol. 2011 May;108(5):310-7. doi: 10.1111/j.1742-7843.2010.00655.x. Epub 2010 Dec 22. [PubMed:21176108]
Patent
Publication numberPriority datePublication dateAssigneeTitle
US4052445A *1975-02-011977-10-04Deutsche Gold- Und Silber-Scheideanstalt Vormals RoesslerProcess for the production of alkyl sulfonic acids
Family To Family Citations
US20100105755A1 *2008-09-122010-04-29Auspex Pharmaceuticals, Inc.Substituted benzamide modulators of dopamine receptor
Non-Patent
Title
Jeyakumar et al. (Tetrahedron Letters 47 (2006) 4573-4576) *
Sato et al. (Tetrahedron 57 (2001) 2469-2476) *
WO2019113084A1 *2017-12-052019-06-13Sunovion Pharmaceuticals Inc.Crystal forms and production methods thereof
Family To Family Citations
CN102807516A *2012-08-162012-12-05四川省百草生物药业有限公司Intermediate in amisulpride and method for preparing amisulpride by using intermediate
CN103319385B *2013-06-182015-07-08苏州诚和医药化学有限公司Method for synthesizing 2-methoxy-4-amino-5-ethylsulfonyl benzoic acid
CN103553989B *2013-11-082015-03-11苏州诚和医药化学有限公司Synthetic method of 2-methoxyl-4-amino-5-ethyl sulfuryl methyl benzoate
CN105237422A *2015-09-062016-01-13南京理工大学Synthetic method of 4-amino-5-chloro-2-methoxyl benzoic acid
///////////////Amisulpride, アミスルプリド , 标准品 , FDA 2020, 2020 APPROVALS, Barhemsys, SOLIAN, Antipsychotic, Benzamides, Dopamine Receptor Antagonist,
CCN1CCCC1CNC(=O)C1=CC(=C(N)C=C1OC)S(=O)(=O)CC
Delgocitinib

Delgocitinib
デルゴシチニブ
3-[(3S,4R)-3-methyl-7-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1,7-diazaspiro[3.4]octan-1-yl]-3-oxopropanenitrile
1,6-Diazaspiro(3.4)octane-1-propanenitrile, 3-methyl-beta-oxo-6-(7H-pyrrolo(2,3-d)pyrimidin-4-yl)-, (3S,4R)-
3-((3S,4R)-3-methyl-6-(7H-pyrrolo(2,3-d)pyrimidin-4-yl)-1,6-diazaspiro(3.4)octan-1-yl)-3-oxopropanenitrile
| Formula |
C16H18N6O
|
|---|---|
| CAS |
1263774-59-9
|
| Mol weight |
310.3537
|
Approved, Japan 2020, Corectim, 2020/1/23, atopic dermatitis, Japan Tobacco (JT)
Torii
7/23/2025 fda approved, Anzupgo
| To treat moderate-to-severe chronic hand eczema when topical corticosteroids are not advisable or produce an inadequate response |
UNII-9L0Q8KK220, JTE-052, LP-0133, ROH-201, 9L0Q8KK220, LEO 124249A, LEO 124249, HY-109053
CS-0031558, D11046, GTPL9619, JTE-052A, JTE052
Delgocitinib, also known as LEO-124249 and JTE052, is a potent and selective JAK inhibitor. JTE-052 reduces skin inflammation and ameliorates chronic dermatitis in rodent models: Comparison with conventional therapeutic agents. JTE-052 regulates contact hypersensitivity by downmodulating T cell activation and differentiation.
Delgocitinib is a JAK inhibitor first approved in Japan for the treatment of atopic dermatitis in patients 16 years of age or older. Japan Tobacco is conducting phase III clinical trials for the treatment of atopic dermatitis in pediatric patients. Leo is developing the drug in phase II clinical trials for the treatment of inflammatory skin diseases, such as atopic dermatitis, and chronic hand eczema and for the treatment of discoid lupus erythematosus. Rohto is evaluating the product in early clinical development for ophthalmologic indications.
In 2014, the drug was licensed to Leo by Japan Tobacco for the development, registration and marketing worldwide excluding Japan for treatment of inflammatory skin conditions. In 2016, Japan Tobacco licensed the rights of co-development and commercialization in Japan to Torii. In 2018, Japan Tobacco licensed the Japanese rights of development and commercialization to Rohto for the treatment of ophthalmologic diseases.
Delgocitinib, sold under the brand name Corectim among others, is a medication used for the treatment of autoimmune disorders and hypersensitivity, including inflammatory skin conditions.[3] Delgocitinib was developed by Japan Tobacco and approved in Japan for the treatment of atopic dermatitis.[3] In the United States, delgocitinib is in Phase III clinical trials and the Food and Drug Administration has granted delgocitinib fast track designation for topical treatment of adults with moderate to severe chronic hand eczema.[4]
Delgocitinib works by blocking activation of the JAK-STAT signaling pathway which contributes to the pathogenesis of chronic inflammatory skin diseases.[5]
PATENTS
WO 2018117151
IN 201917029002
IN 201917029003
IN 201917029000
PATENTS
WO 2011013785
https://patents.google.com/patent/WO2011013785A1/en
[Production Example 6]: Synthesis of Compound 6

(1) Optically active substance of 2-benzylaminopropan-1-ol

To a solution of (S)-(+)-2-aminopropan-1-ol (50.0 g) and benzaldehyde (74 ml) in ethanol (500 ml) was added 5% palladium carbon (5.0 g) at room temperature and normal pressure. Hydrogenated for 8 hours. The reaction mixture was filtered through celite and concentrated under reduced pressure to give the title compound (111.2 g).
1 H-NMR (DMSO-D 6 ) δ: 7.34-7.27 (4H, m), 7.23-7.18 (1H, m), 4.53-4.47 (1H, m), 3.76 (1H, d, J = 13.5 Hz) , 3.66 (1H, d, J = 13.5 Hz), 3.29-3.24 (2H, m), 2.65-2.55 (1H, m), 1.99 (1H, br s), 0.93 (3H, d, J = 6.4 Hz) .
(2) Optically active substance of [benzyl- (2-hydroxy-1-methylethyl) -amino] acetic acid tert-butyl ester

To a mixture of optically active 2-benzylaminopropan-1-ol (111.2 g), potassium carbonate (111.6 g) and N, N-dimethylformamide (556 ml) cooled to 0 ° C., tert-butyl bromoacetate was added. Ester (109 ml) was added dropwise over 20 minutes and stirred at room temperature for 19.5 hours. The mixture was acidified to pH 2 by adding 2M aqueous hydrochloric acid and 6M aqueous hydrochloric acid, and washed with toluene (1000 ml). The separated organic layer was extracted with 0.1 M aqueous hydrochloric acid (300 ml). The combined aqueous layer was adjusted to pH 10 with 4M aqueous sodium hydroxide solution and extracted with ethyl acetate (700 ml). The organic layer was washed successively with water (900 ml) and saturated aqueous sodium chloride solution (500 ml). The separated aqueous layer was extracted again with ethyl acetate (400 ml). The combined organic layers were dried over anhydrous sodium sulfate and concentrated under reduced pressure to give the title compound (160.0 g).
1 H-NMR (DMSO-D 6 ) δ: 7.37-7.26 (4H, m), 7.24-7.19 (1H, m), 4.26 (1H, dd, J = 6.9, 3.9 Hz), 3.76 (1H, d, J = 14.1 Hz), 3.68 (1H, d, J = 13.9 Hz), 3.45-3.39 (1H, m), 3.29-3.20 (1H, m), 3.24 (1H, d, J = 17.2 Hz), 3.13 ( 1H, d, J = 17.0 Hz), 2.84-2.74 (1H, m), 1.37 (9H, s), 0.96 (3H, d, J = 6.8 Hz).
(3) Optically active substance of [benzyl- (2-chloropropyl) -amino] acetic acid tert-butyl ester

(3)-(1) Optically active form of [benzyl- (2-chloro-1-methylethyl) -amino] acetic acid tert-butyl ester

To a solution of [benzyl- (2-hydroxy-1-methylethyl) -amino] acetic acid tert-butyl ester optically active substance (160.0 g) cooled to 0 ° C. in chloroform (640 ml) was added thionyl chloride (50.0 ml). Was added dropwise and stirred at 60 ° C. for 2 hours. The reaction mixture was cooled to 0 ° C., saturated aqueous sodium hydrogen carbonate solution (1000 ml) and chloroform (100 ml) were added and stirred. The separated organic layer was washed with a saturated aqueous sodium chloride solution (500 ml), and the aqueous layer was extracted again with chloroform (450 ml). The combined organic layers were dried over anhydrous sodium sulfate and concentrated under reduced pressure to obtain the title compound (172.9 g).
1 H-NMR (CDCl 3 ) δ: 7.40-7.22 (5H, m), 4.05-3.97 (0.4H, m), 3.93-3.81 (2H, m), 3.70-3.65 (0.6H, m), 3.44- 3.38 (0.6H, m), 3.29 (0.8H, s), 3.27 (1.2H, d, J = 2.4 Hz), 3.24-3.15 (0.6H, m), 3.05-2.99 (0.4H, m), 2.94 -2.88 (0.4H, m), 1.50 (1.2H, d, J = 6.4 Hz), 1.48 (3.6H, s), 1.45 (5.4H, s), 1.23 (1.8H, d, J = 6.8 Hz) .
(3)-(2) Optically active form of [benzyl- (2-chloropropyl) -amino] acetic acid tert-butyl ester

[Benzyl- (2-chloro-1-methylethyl) -amino] acetic acid tert-butyl ester optically active substance (172.9 g) was dissolved in N, N-dimethylformamide (520 ml) and stirred at 80 ° C. for 140 minutes. did. The reaction mixture was cooled to 0 ° C., water (1200 ml) was added, and the mixture was extracted with n-hexane / ethyl acetate (2/1, 1000 ml). The organic layer was washed successively with water (700 ml) and saturated aqueous sodium chloride solution (400 ml), and the separated aqueous layer was extracted again with n-hexane / ethyl acetate (2/1, 600 ml). The combined organic layers were concentrated under reduced pressure, and the obtained residue was purified by silica gel column chromatography (eluent: n-hexane / ethyl acetate = 50/1 to 40/1) to give the title compound (127.0 g )
1 H-NMR (CDCl 3 ) δ: 7.37-7.29 (4H, m), 7.28-7.23 (1H, m), 4.05-3.97 (1H, m), 3.91 (1H, d, J = 13.5 Hz), 3.86 (1H, d, J = 13.7 Hz), 3.29 (2H, s), 3.03 (1H, dd, J = 13.9, 6.6 Hz), 2.91 (1H, dd, J = 13.9, 6.8 Hz), 1.50 (3H, d, J = 6.4 Hz), 1.48 (9H, s).
(4) Optically active substance of 1-benzyl-3-methylazetidine-2-carboxylic acid tert-butyl ester

To a solution of [benzyl- (2-chloropropyl) -amino] acetic acid tert-butyl ester optically active substance (60.0 g) cooled to −72 ° C. and hexamethylphosphoramide (36.0 ml) in tetrahydrofuran (360 ml), Lithium hexamethyldisilazide (1.0 M tetrahydrofuran solution, 242 ml) was added dropwise over 18 minutes, and the temperature was raised to 0 ° C. over 80 minutes. A saturated aqueous ammonium chloride solution (300 ml) and water (400 ml) were sequentially added to the reaction mixture, and the mixture was extracted with ethyl acetate (500 ml). The organic layer was washed successively with water (700 ml) and saturated aqueous sodium chloride solution (500 ml), and the separated aqueous layer was extracted again with ethyl acetate (300 ml). The combined organic layers were dried over anhydrous sodium sulfate, concentrated under reduced pressure, and the resulting residue was purified by silica gel column chromatography (developing solvent: n-hexane / ethyl acetate = 50/1 to 4/1). To give the title compound (50.9 g).
1 H-NMR (CDCl 3 ) δ: 7.34-7.21 (5H, m), 3.75 (1H, d, J = 12.6 Hz), 3.70-3.67 (1H, m), 3.58 (1H, d, J = 12.6 Hz ), 3.05-3.01 (1H, m), 2.99-2.95 (1H, m), 2.70-2.59 (1H, m), 1.41 (9H, s), 1.24 (3H, d, J = 7.1 Hz).
(5) Optically active substance of 3-methylazetidine-1,2-dicarboxylic acid di-tert-butyl ester

1-Benzyl-3-methylazetidine-2-carboxylic acid tert-butyl ester optically active substance (43.5 g) and di-tert-butyl dicarbonate (38.2 g) in tetrahydrofuran / methanol (130 ml / 130 ml) solution 20% Palladium hydroxide carbon (3.5 g) was added thereto, and hydrogenated at 4 atm for 2 hours. The mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure to give the title compound (48.0 g).
1 H-NMR (DMSO-D 6 ) δ: 4.44 (1H, d, J = 8.8 Hz), 3.99-3.77 (1H, m), 3.45-3.37 (1H, m), 3.00-2.88 (1H, m) , 1.45 (9H, s), 1.40-1.30 (9H, m), 1.02 (3H, d, J = 7.2 Hz).
(6) Optically active substance of 3-methyl-2- (3-methyl-but-2-enyl) -azetidine-1,2-dicarboxylic acid di-tert-butyl ester

Optically active substance (48.0 g) of 3-methylazetidine-1,2-dicarboxylic acid di-tert-butyl ester cooled to -69 ° C. and 1-bromo-3-methyl-2-butene (25.4 ml) Lithium hexamethyldisilazide (1.0 M tetrahydrofuran solution, 200 ml) was added to a tetrahydrofuran solution (380 ml). The reaction mixture was warmed to −20 ° C. in 40 minutes and further stirred at the same temperature for 20 minutes. A saturated aqueous ammonium chloride solution (200 ml) and water (300 ml) were successively added to the reaction mixture, and the mixture was extracted with n-hexane / ethyl acetate (1 / 1,500 ml). The separated organic layer was washed successively with water (200 ml) and saturated aqueous sodium chloride solution (200 ml), dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (eluent: n-hexane / ethyl acetate = 15/1 to 8/1) to give the titled compound (44.5 g).
1 H-NMR (CDCl 3 ) δ: 5.29-5.21 (1H, m), 3.77-3.72 (1H, m), 3.49-3.44 (1H, m), 2.73-2.52 (3H, m), 1.76-1.74 ( 3H, m), 1.66-1.65 (3H, m), 1.51 (9H, s), 1.43 (9H, s), 1.05 (3H, d, J = 7.3 Hz).
(7) Optically active substance of 3-methyl-2- (2-oxoethyl) azetidine-1,2-dicarboxylic acid di-tert-butyl ester

3-methyl-2- (3-methyl-but-2-enyl) -azetidine-1,2-dicarboxylic acid di-tert-butyl ester optically active substance (44.5 g) in chloroform / cooled to −70 ° C. An ozone stream was passed through the methanol solution (310 ml / 310 ml) for 1 hour. To this reaction mixture, a solution of triphenylphosphine (44.7 g) in chloroform (45 ml) was added little by little, and then the mixture was warmed to room temperature. To this mixture were added saturated aqueous sodium thiosulfate solution (200 ml) and water (300 ml), and the mixture was extracted with chloroform (500 ml). The separated organic layer was washed with a saturated aqueous sodium chloride solution, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure to obtain the title compound (95.0 g). This product was subjected to the next step without further purification.
1 H-NMR (DMSO-D 6 ) δ: 9.65 (1H, t, J = 2.6 Hz), 3.79-3.74 (1H, m), 3.45-3.40 (1H, m), 2.99-2.80 (3H, m) , 1.46 (9H, s), 1.34 (9H, s), 1.06 (3H, d, J = 7.2 Hz).
(8) Optically active substance of 2- (2-benzylaminoethyl) -3-methylazetidine-1,2-dicarboxylic acid di-tert-butyl ester

To a solution of the residue (95.0 g) obtained in (7) in tetrahydrofuran (300 ml) was added benzylamine (34 ml) at room temperature, and the mixture was stirred for 2 hours. The mixture was cooled to 0 ° C., sodium triacetoxyborohydride (83.3 g) was added, and the mixture was stirred at room temperature for 1.5 hours. Water (300 ml) was added to the reaction mixture, and the mixture was extracted with n-hexane / ethyl acetate (1/3, 600 ml). The separated organic layer was washed with water (300 ml) and saturated aqueous sodium chloride solution (200 ml), and then extracted twice with 5% aqueous citric acid solution (300 ml, 200 ml) and three times with 10% aqueous citric acid solution (250 ml × 3). . The combined aqueous layers were basified to pH 10 with 4M aqueous sodium hydroxide solution and extracted with chloroform (300 ml). The organic layer was washed with a saturated aqueous sodium chloride solution (200 ml), dried over anhydrous magnesium sulfate and concentrated under reduced pressure to obtain the title compound (46.9 g).
1 H-NMR (DMSO-D 6 ) δ: 7.34-7.26 (4H, m), 7.22-7.17 (1H, m), 3.74-3.65 (2H, m), 3.61 (1H, t, J = 7.8 Hz) , 3.28 (1H, t, J = 7.5 Hz), 2.76-2.66 (2H, m), 2.57-2.45 (1H, m), 2.15 (1H, br s), 2.05-1.89 (2H, m), 1.42 ( 9H, s), 1.27 (9H, s), 0.96 (3H, d, J = 7.1 Hz).
(9) Optically active substance of 2- (2-benzylaminoethyl) -3-methylazetidine-2-dicarboxylic acid dihydrochloride

2- (2-Benzylaminoethyl) -3-methylazetidine-1,2-dicarboxylic acid di-tert-butyl ester optically active substance (46.5 g), 4M hydrochloric acid 1,4-dioxane (230 ml) and water (4.1 ml) was mixed and stirred at 80 ° C. for 2 hours. The mixture was concentrated under reduced pressure, azeotroped with toluene, and then slurry washed with n-hexane / ethyl acetate (1/1, 440 ml) to give the title compound (30.1 g).
1 H-NMR (DMSO-D 6 ) δ: 10.24 (1H, br s), 9.64 (2H, br s), 8.90 (1H, br s), 7.58-7.53 (2H, m), 7.47-7.41 (3H , m), 4.21-4.10 (2H, m), 4.02-3.94 (1H, m), 3.46-3.37 (1H, m), 3.20-3.10 (1H, m), 2.99-2.85 (2H, m), 2.69 -2.54 (2H, m), 1.10 (3H, d, J = 7.2 Hz).
(10) Optically active substance of 6-benzyl-3-methyl-1,6-diazaspiro [3.4] octan-5-one

To a solution of 2- (2-benzylaminoethyl) -3-methylazetidine-2-dicarboxylic acid dihydrochloride optically active substance (29.1 g) and N, N-diisopropylethylamine (65 ml) in chloroform (290 ml), At room temperature, O- (7-azabenzotriazol-1-yl) -N, N, N ′, N′-tetramethyluronium hexafluorophosphate (41.3 g) was added and stirred for 4 hours. To this reaction mixture were added saturated aqueous sodium hydrogen carbonate solution (200 ml) and water (100 ml), and the mixture was extracted with chloroform (200 ml). The organic layer was washed with a saturated aqueous sodium chloride solution, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (developing solvent: chloroform / methanol = 20/1 to 10/1) to give the titled compound (21.3 g).
1 H-NMR (DMSO-D 6 ) δ: 7.38-7.31 (2H, m), 7.30-7.22 (3H, m), 4.52 (1H, d, J = 14.8 Hz), 4.29 (1H, d, J = 14.8 Hz), 3.35-3.27 (2H, m), 3.22-3.17 (1H, m), 3.05 (2H, dd, J = 9.5, 4.0 Hz), 2.77-2.66 (1H, m), 2.16-2.10 (1H , m), 1.96-1.87 (1H, m), 0.94 (3H, d, J = 7.1 Hz).
(11) Optically active substance of 6-benzyl-3-methyl-1,6-diazaspiro [3.4] octane-1-carboxylic acid tert-butyl ester

Concentrated sulfuric acid (4.8 ml) was slowly added dropwise to a suspension of lithium aluminum hydride (6.8 g) in tetrahydrofuran (300 ml) under ice cooling, and the mixture was stirred for 30 minutes. To this mixture was added dropwise a solution of 6-benzyl-3-methyl-1,6-diazaspiro [3.4] octan-5-one optically active substance (21.3 g) in tetrahydrofuran (100 ml) at the same temperature. Stir for 45 minutes. Water (7.0 ml), 4M aqueous sodium hydroxide solution (7.0 ml) and water (14.0 ml) were sequentially added to the reaction mixture, and the mixture was stirred as it was for 30 minutes. To this mixture was added anhydrous magnesium sulfate and ethyl acetate (100 ml), and the mixture was stirred and filtered through celite. Di-tert-butyl dicarbonate (23.4 g) was added to the filtrate at room temperature and stirred for 3 hours. The mixture was concentrated under reduced pressure to a half volume and washed twice with a saturated aqueous ammonium chloride solution (200 ml × 2). N-Hexane (200 ml) was added to the separated organic layer, and the mixture was extracted 5 times with a 10% aqueous citric acid solution. The separated aqueous layer was basified with 4M aqueous sodium hydroxide solution and extracted with chloroform. The organic layer was washed with a saturated aqueous sodium chloride solution (200 ml), dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (eluent: chloroform / methanol = 40/1 to 20/1) to give the titled compound (15.6 g).
1 H-NMR (DMSO-D 6 ) δ: 7.34-7.27 (4H, m), 7.26-7.21 (1H, m), 3.84-3.69 (1H, m), 3.62-3.47 (2H, m), 3.19- 3.05 (1H, m), 3.02-2.92 (1H, m), 2.76-2.69 (1H, m), 2.47-2.24 (4H, m), 1.95-1.77 (1H, m), 1.36 (9H, s), 1.03 (3H, d, J = 7.0 Hz).
(12) Optically active substance of 3-methyl-1,6-diazaspiro [3.4] octane-1-carboxylic acid tert-butyl ester

20% of optically active form of 6-benzyl-3-methyl-1,6-diazaspiro [3.4] octane-1-carboxylic acid tert-butyl ester (10.0 g) in tetrahydrofuran / methanol (50 ml / 50 ml) solution Palladium hydroxide on carbon (2.0 g) was added and hydrogenated at 4 atm for 24 hours. The mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure to give the title compound (7.3 g).
1 H-NMR (DMSO-D 6 ) δ: 3.88-3.71 (1H, m), 3.44-3.06 (2H, m), 3.02-2.64 (4H, m), 2.55-2.38 (1H, m), 2.31- 2.15 (1H, m), 1.81-1.72 (1H, m), 1.37 (9H, s), 1.07 (3H, d, J = 7.0 Hz).
(13) Optical activity of 3-methyl-6- (7H-pyrrolo [2,3-d] pyrimidin-4-yl) -1,6-diazaspiro [3.4] octane-1-carboxylic acid tert-butyl ester body

The optically active substance (6.9 g) of 3-methyl-1,6-diazaspiro [3.4] octane-1-carboxylic acid tert-butyl ester was converted into 4-chloro-7H-pyrrolo [2,3-d] pyrimidine ( 4.3 g), potassium carbonate (7.7 g) and water (65 ml) and stirred for 4 hours at reflux. The mixture was cooled to room temperature, water (60 ml) was added, and the mixture was extracted with chloroform / methanol (10/1, 120 ml). The organic layer was washed successively with water, saturated aqueous ammonium chloride solution and saturated aqueous sodium chloride solution, and dried over anhydrous sodium sulfate. To this mixture, silica gel (4 g) was added, stirred for 10 minutes, filtered through celite, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (developing solvent: chloroform / ethyl acetate = 1/1, then chloroform / methanol = 50/1 to 20/1) to give the title compound (10.0 g). Obtained.
1 H-NMR (DMSO-D 6 ) δ: 11.59 (1H, br s), 8.09 (1H, s), 7.12-7.09 (1H, m), 6.64-6.59 (1H, m), 4.09-3.66 (5H , m), 3.39-3.21 (1H, m), 2.64-2.44 (2H, m), 2.27-2.06 (1H, m), 1.36 (3H, s), 1.21 (6H, s), 1.11 (3H, d , J = 6.5 Hz).
(14) Optically active form of 4- (3-methyl-1,6-diazaspiro [3.4] oct-6-yl) -7H-pyrrolo [2,3-d] pyrimidine dihydrochloride

Optically active form of 3-methyl-6- (7H-pyrrolo [2,3-d] pyrimidin-4-yl) -1,6-diazaspiro [3.4] octane-1-carboxylic acid tert-butyl ester (9 0.5 g), 4M hydrochloric acid 1,4-dioxane (50 ml), chloroform (50 ml) and methanol (100 ml) were mixed and stirred at 60 ° C. for 30 minutes. The mixture was concentrated under reduced pressure and azeotroped with toluene to give the title compound (9.3 g).
1 H-NMR (DMSO-D 6 ) δ: 12.91 (1H, br s), 9.97-9.64 (2H, m), 8.45-8.35 (1H, m), 7.58-7.47 (1H, m), 7.04-6.92 (1H, m), 4.99-4.65 (1H, m), 4.32-3.21 (7H, m), 3.04-2.90 (1H, m), 2.46-2.31 (1H, m), 1.27 (3H, d, J = 6.0 Hz).
(15) 3- [3-Methyl-6- (7H-pyrrolo [2,3-d] pyrimidin-4-yl) -1,6-diazaspiro [3.4] oct-1-yl] -3-oxo Optically active form of propionitrile

4- (3-Methyl-1,6-diazaspiro [3.4] oct-6-yl) -7H-pyrrolo [2,3-d] pyrimidine dihydrochloride optically active substance (8.8 g) was converted to 1- The mixture was mixed with cyanoacetyl-3,5-dimethylpyrazole (6.8 g), N, N-diisopropylethylamine (20 ml) and 1,4-dioxane (100 ml) and stirred at 100 ° C. for 1 hour. The mixture was cooled to room temperature, saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with chloroform / methanol (10/1). The separated organic layer was washed with a saturated aqueous sodium chloride solution, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (developing solvent: chloroform / methanol = 30/1 to 9/1). The residue obtained by concentration under reduced pressure was slurry washed with n-heptane / ethanol (2/1, 90 ml) to obtain a solid (7.3 g). The solid was slurried again with n-heptane / ethanol (5/1, 90 ml) to give the title compound as crystals 1 (6.1 g).
1 H-NMR (DMSO-D 6 ) δ: 11.60 (1H, br s), 8.08 (1H, s), 7.11 (1H, dd, J = 3.5, 2.4 Hz), 6.58 (1H, dd, J = 3.4 , 1.9 Hz), 4.18-4.14 (1H, m), 4.09-3.93 (3H, m), 3.84-3.73 (1H, m), 3.71 (1H, d, J = 19.0 Hz), 3.66 (1H, d, J = 18.7 Hz), 3.58 (1H, dd, J = 8.2, 6.0 Hz), 2.70-2.58 (2H, m), 2.24-2.12 (1H, m), 1.12 (3H, d, J = 7.1 Hz).
[Α] D = + 47.09 ° (25 ° C., c = 0.55, methanol)
1-Butanol (39 ml) was added to the obtained crystal 1 (2.6 g), and the mixture was heated and stirred at 100 ° C. After complete dissolution, the solution was cooled to room temperature by 10 ° C. every 30 minutes and further stirred at room temperature overnight. The produced crystals were collected by filtration, washed with 1-butanol (6.2 ml), and dried under reduced pressure to give crystals 2 (2.1 g) of the title compound.
PATENTS
WO 2017006968
WO 2018117152
WO 2018117151
PATENT
WO 2018117153
https://patentscope.wipo.int/search/zh/detail.jsf?docId=WO2018117153&tab=FULLTEXT
Janus kinase (JAK) inhibitors are of current interest for the treatment of various diseases including autoimmune diseases, inflammatory diseases, and cancer. To date, two JAK inhibitors have been approved by the U.S. Food & Drug Administration (FDA). Ruxolitinib has been approved for the treatment of primary myelofibrosis and polycythemia vera (PV), and tofacitinib has been approved for the treatment of rheumatoid arthritis. Other JAK inhibitors are in the literature. The compound 3-((3S,4R)-3-methyl-6-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1,6-diazaspiro[3.4]octan-1-yl)-3-oxopropanenitrile (Compound A) (see structure below) is an example of a spirocyclic JAK inhibitor reported in U.S. Pat. Pub. Nos. 2011/0136778 and International Pat. Pub. No. PCT/JP2016/070046.
[Chem. 1]
Step A. Preparation of S-MABB-HC (Compound [5])
[Chem. 2]

Step 1
[Chem. 3]
 S-BAPO [1] (35.0 g, 212 mmol) was added to water (175 mL) at room temperature under nitrogen atmosphere. To the resulting suspension were added toluene (53 mL) and potassium carbonate (32.2 g, 233 mmol) at room temperature. To the resulting solution was added dropwise TBBA (434.4 g, 223 mmol) at room temperature, and then the used dropping funnel was washed with toluene (17 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at 65°C for 21 hours, and then cooled to room temperature. After toluene (105 mL) was added to the reaction mixture and then the mixture was stirred, the organic layer was separated out. The organic layer was washed with water (175 mL), aqueous layer was removed, and then the solvent was removed out of the organic layer in vacuo. Toluene (105 mL) was added to the residue and the toluene solution was concentrated. The operation was repeated two more times to give a toluene solution of S-BBMO [2] (74.0 g, 212 mmol in theory). The given toluene solution of S-BBMO was used in the next step, assuming that the yield was 100 %.
S-BAPO [1] (35.0 g, 212 mmol) was added to water (175 mL) at room temperature under nitrogen atmosphere. To the resulting suspension were added toluene (53 mL) and potassium carbonate (32.2 g, 233 mmol) at room temperature. To the resulting solution was added dropwise TBBA (434.4 g, 223 mmol) at room temperature, and then the used dropping funnel was washed with toluene (17 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at 65°C for 21 hours, and then cooled to room temperature. After toluene (105 mL) was added to the reaction mixture and then the mixture was stirred, the organic layer was separated out. The organic layer was washed with water (175 mL), aqueous layer was removed, and then the solvent was removed out of the organic layer in vacuo. Toluene (105 mL) was added to the residue and the toluene solution was concentrated. The operation was repeated two more times to give a toluene solution of S-BBMO [2] (74.0 g, 212 mmol in theory). The given toluene solution of S-BBMO was used in the next step, assuming that the yield was 100 %.
A crude product of S-BBMO which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 7.36-7.13 (5H, m), 4.26 (1H, dd, J = 6.8, 3.9 Hz), 3.72 (2H, dd, J = 14.2, 6.8 Hz), 3.47-3.38 (1H, m), 3.30-3.08 (3H, m), 2.79 (1H, sext, J = 6.8 Hz), 1.35 (9H, s), 0.96 (3H, d, J = 6.8 Hz).
MS: m/z = 280 [M+H] +
[Chem. 2]
Step 1
[Chem. 3]
A crude product of S-BBMO which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 7.36-7.13 (5H, m), 4.26 (1H, dd, J = 6.8, 3.9 Hz), 3.72 (2H, dd, J = 14.2, 6.8 Hz), 3.47-3.38 (1H, m), 3.30-3.08 (3H, m), 2.79 (1H, sext, J = 6.8 Hz), 1.35 (9H, s), 0.96 (3H, d, J = 6.8 Hz).
MS: m/z = 280 [M+H] +
[0134]
Step 2
[Chem. 4]
 To the toluene solution of S-BBMO [2] (74.0 g, 212 mmol) were added toluene (200 mL), tetrahydrofuran (35 mL), and then triethylamine (25.7 g, 254 mmol) at room temperature under nitrogen atmosphere. To the mixture was added dropwise methanesulfonyl chloride (26.7 g, 233 mmol) at 0°C, and then the used dropping funnel was washed with toluene (10 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at room temperature for 2 hours and further at 65°C for 22 hours, and then cooled to room temperature. After sodium bicarbonate water (105 mL) was added to the reaction mixture and then the mixture was stirred, the organic layer was separated out. The organic layer was washed with water (105 mL), aqueous layer was removed, and then the solvent was removed out of the organic layer in vacuo. Toluene (105 mL) was added to the residue, and the toluene solution was concentrated. The operation was repeated two more times to give a toluene solution of R-BCAB [3] (75.3 g, 212 mmol in theory). The given toluene solution of R-BCAB was used in the next step, assuming that the yield was 100 %.
To the toluene solution of S-BBMO [2] (74.0 g, 212 mmol) were added toluene (200 mL), tetrahydrofuran (35 mL), and then triethylamine (25.7 g, 254 mmol) at room temperature under nitrogen atmosphere. To the mixture was added dropwise methanesulfonyl chloride (26.7 g, 233 mmol) at 0°C, and then the used dropping funnel was washed with toluene (10 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at room temperature for 2 hours and further at 65°C for 22 hours, and then cooled to room temperature. After sodium bicarbonate water (105 mL) was added to the reaction mixture and then the mixture was stirred, the organic layer was separated out. The organic layer was washed with water (105 mL), aqueous layer was removed, and then the solvent was removed out of the organic layer in vacuo. Toluene (105 mL) was added to the residue, and the toluene solution was concentrated. The operation was repeated two more times to give a toluene solution of R-BCAB [3] (75.3 g, 212 mmol in theory). The given toluene solution of R-BCAB was used in the next step, assuming that the yield was 100 %.
A crude product of R-BCAB which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 7.28-7.11 (5H, m), 4.24-4.11 (1H, m), 3.80 (2H, d, J = 3.6 Hz), 3.24 (2H, d, J = 3.6 Hz), 2.98-2.78 (2H, m), 1.46-1.37 (12H, m).
MS: m/z = 298 [M+H] +
[Chem. 4]
A crude product of R-BCAB which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 7.28-7.11 (5H, m), 4.24-4.11 (1H, m), 3.80 (2H, d, J = 3.6 Hz), 3.24 (2H, d, J = 3.6 Hz), 2.98-2.78 (2H, m), 1.46-1.37 (12H, m).
MS: m/z = 298 [M+H] +
[0135]
Step 3
[Chem. 5]
 To the toluene solution of R-BCAB [3] (75.3 g, 212 mmol) were added tetrahydrofuran (88.0 mL) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (42.0 mL) at room temperature under nitrogen atmosphere. To the resulting solution was added dropwise a solution of lithium bis(trimethylsilyl)amide /tetrahydrofuran (195 mL, 233 mmol) at 0°C, and then the used dropping funnel was washed with tetrahydrofuran (17.0 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at 0°C for 1 hour, and then warmed to room temperature. After water (175 mL) and toluene (175 mL) were added to the reaction mixture and then the mixture was stirred, the organic layer was separated out. The resulting organic layer was washed with aqueous ammonium chloride (175 mL) and then water (175 mL), and the solvent was removed out of the organic layer in vacuo. Ethyl acetate (175 mL) was added to the residue and the ethyl acetate solution was concentrated. The operation was repeated two more times to give an ethyl acetate solution of S-MABB [4] (66.5 g, 212 mmol in theory). The given ethyl acetate solution of S-MABB was used in the next step, assuming that the yield was 100 %.
To the toluene solution of R-BCAB [3] (75.3 g, 212 mmol) were added tetrahydrofuran (88.0 mL) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (42.0 mL) at room temperature under nitrogen atmosphere. To the resulting solution was added dropwise a solution of lithium bis(trimethylsilyl)amide /tetrahydrofuran (195 mL, 233 mmol) at 0°C, and then the used dropping funnel was washed with tetrahydrofuran (17.0 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at 0°C for 1 hour, and then warmed to room temperature. After water (175 mL) and toluene (175 mL) were added to the reaction mixture and then the mixture was stirred, the organic layer was separated out. The resulting organic layer was washed with aqueous ammonium chloride (175 mL) and then water (175 mL), and the solvent was removed out of the organic layer in vacuo. Ethyl acetate (175 mL) was added to the residue and the ethyl acetate solution was concentrated. The operation was repeated two more times to give an ethyl acetate solution of S-MABB [4] (66.5 g, 212 mmol in theory). The given ethyl acetate solution of S-MABB was used in the next step, assuming that the yield was 100 %.
A crude product of S-MABB which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 7.28-7.25 (10H, m), 3.75 (1H, d, J = 12.7 Hz), 3.68 (1H, d, J = 1.4 Hz), 3.66 (1H, d, J = 6.7 Hz), 3.46 (2H, d, J = 12.7 Hz), 3.30-3.17 (2H, m), 2.95 (1H, dd, J = 6.2, 1.2 Hz), 2.77 (1H, dd, J = 6.1, 2.2 Hz), 2.65-2.55 (1H, m), 2.48-2.40 (2H, m), 1.35 (9H, s), 1.35 (9H, s), 1.12 (3H, d, J = 7.2 Hz), 1.09 (3H, d, J = 6.2 Hz).
MS: m/z = 262 [M+H] +
[Chem. 5]
A crude product of S-MABB which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 7.28-7.25 (10H, m), 3.75 (1H, d, J = 12.7 Hz), 3.68 (1H, d, J = 1.4 Hz), 3.66 (1H, d, J = 6.7 Hz), 3.46 (2H, d, J = 12.7 Hz), 3.30-3.17 (2H, m), 2.95 (1H, dd, J = 6.2, 1.2 Hz), 2.77 (1H, dd, J = 6.1, 2.2 Hz), 2.65-2.55 (1H, m), 2.48-2.40 (2H, m), 1.35 (9H, s), 1.35 (9H, s), 1.12 (3H, d, J = 7.2 Hz), 1.09 (3H, d, J = 6.2 Hz).
MS: m/z = 262 [M+H] +
[0136]
Step 4
[Chem. 6]
 To the ethyl acetate solution of S-MABB [4] (66.5 g, 212 mmol in theory) were added ethyl acetate (175 mL) and active carbon (3.5 g) under nitrogen atmosphere, and then the mixture was stirred at room temperature for 2 hours. The active carbon was removed by filtration, and the residue on the filter was washed with ethyl acetate (175 mL). The washings were added to the filtrate. To the solution was added S-MABB-HC crystal (17.5 mg) that was prepared according to the method described herein at 0°C, and then 4 M hydrogen chloride/ethyl acetate (53.0 mL, 212 mmol) was dropped thereto at 0°C. The reaction mixture was stirred at 0°C for 17 hours, and then the precipitated solid was collected on a filter, and washed with ethyl acetate (70 mL). The resulting wet solid was dried in vacuo to give S-MABB-HC [5] (48.3 g, 162 mmol, yield: 76.4 %).
To the ethyl acetate solution of S-MABB [4] (66.5 g, 212 mmol in theory) were added ethyl acetate (175 mL) and active carbon (3.5 g) under nitrogen atmosphere, and then the mixture was stirred at room temperature for 2 hours. The active carbon was removed by filtration, and the residue on the filter was washed with ethyl acetate (175 mL). The washings were added to the filtrate. To the solution was added S-MABB-HC crystal (17.5 mg) that was prepared according to the method described herein at 0°C, and then 4 M hydrogen chloride/ethyl acetate (53.0 mL, 212 mmol) was dropped thereto at 0°C. The reaction mixture was stirred at 0°C for 17 hours, and then the precipitated solid was collected on a filter, and washed with ethyl acetate (70 mL). The resulting wet solid was dried in vacuo to give S-MABB-HC [5] (48.3 g, 162 mmol, yield: 76.4 %).
S-MABB-HC which was prepared by the same process was measured about NMR, MS, and Cl-content.
1H-NMR (DMSO-d 6) δ: 11.08 (1H, br s), 10.94 (1H, br s), 7.52-7.42 (10H, m), 5.34 (1H, t, J = 8.4 Hz), 4.90 (1H, br s), 4.45-4.10 (5H, m), 3.92-3.49 (3H, br m), 3.10-2.73 (2H, br m), 1.35 (9H, s), 1.29 (9H, s), 1.24 (3H, d, J = 6.7 Hz), 1.17 (3H, d, J = 7.4 Hz).
MS: m/z = 262 [M+H-HCl] +
Cl content (ion chromatography): 11.9 % (in theory: 11.9 %).
[Chem. 6]
S-MABB-HC which was prepared by the same process was measured about NMR, MS, and Cl-content.
1H-NMR (DMSO-d 6) δ: 11.08 (1H, br s), 10.94 (1H, br s), 7.52-7.42 (10H, m), 5.34 (1H, t, J = 8.4 Hz), 4.90 (1H, br s), 4.45-4.10 (5H, m), 3.92-3.49 (3H, br m), 3.10-2.73 (2H, br m), 1.35 (9H, s), 1.29 (9H, s), 1.24 (3H, d, J = 6.7 Hz), 1.17 (3H, d, J = 7.4 Hz).
MS: m/z = 262 [M+H-HCl] +
Cl content (ion chromatography): 11.9 % (in theory: 11.9 %).
[0137]
Step B. Preparation of S-MACB-HC (Compound [6])
[Chem. 7]
 To a solution of S-MABB-HC [5] (5.0 g, 16.8 mmol) in methanol (15.0 mL) was added 5 % palladium carbon (made by Kawaken Fine Chemicals Co., Ltd., PH type, 54.1 % water-content 1.0 g) at room temperature under nitrogen atmosphere. The reaction vessel was filled with hydrogen, the reaction mixture was stirred at hydrogen pressure of 0.4 MPa at room temperature for 12 hours, the hydrogen in the reaction vessel was replaced with nitrogen, and then the 5 % palladium carbon was removed by filtration. The reaction vessel and the 5 % palladium carbon were washed with methanol (10 mL). The washings were added to the filtrate to give a methanol solution of S-MACB-HC [6] (24.8 g, 16.8 mmol in theory). The given methanol solution of S-MACB-HC was used in the next step, assuming that the yield was 100 %.
To a solution of S-MABB-HC [5] (5.0 g, 16.8 mmol) in methanol (15.0 mL) was added 5 % palladium carbon (made by Kawaken Fine Chemicals Co., Ltd., PH type, 54.1 % water-content 1.0 g) at room temperature under nitrogen atmosphere. The reaction vessel was filled with hydrogen, the reaction mixture was stirred at hydrogen pressure of 0.4 MPa at room temperature for 12 hours, the hydrogen in the reaction vessel was replaced with nitrogen, and then the 5 % palladium carbon was removed by filtration. The reaction vessel and the 5 % palladium carbon were washed with methanol (10 mL). The washings were added to the filtrate to give a methanol solution of S-MACB-HC [6] (24.8 g, 16.8 mmol in theory). The given methanol solution of S-MACB-HC was used in the next step, assuming that the yield was 100 %.
A crude product of S-MACB-HC which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 9.60 (br s, 1H), 4.97 (d, 1H, J = 9.2 Hz), 4.61 (d, 1H, J = 8.4 Hz), 4.01 (dd, 1H, J = 10.0, 8.4 Hz), 3.78-3.74 (m, 1H), 3.54 (dd, 1H, J = 9.6, 8.4 Hz), 3.35 (dd, 1H, J = 10.0, 6.0 Hz), 3.15-3.03 (m, 1H), 3.00-2.88 (m, 1H), 1.49 (s, 9H), 1.47 (s, 9H), 1.22 (d, 3H, J = 6.8 Hz), 1.14 (d, 3H, J = 7.2 Hz).
MS: m/z = 172 [M+H] + (free form)
[Chem. 7]
A crude product of S-MACB-HC which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 9.60 (br s, 1H), 4.97 (d, 1H, J = 9.2 Hz), 4.61 (d, 1H, J = 8.4 Hz), 4.01 (dd, 1H, J = 10.0, 8.4 Hz), 3.78-3.74 (m, 1H), 3.54 (dd, 1H, J = 9.6, 8.4 Hz), 3.35 (dd, 1H, J = 10.0, 6.0 Hz), 3.15-3.03 (m, 1H), 3.00-2.88 (m, 1H), 1.49 (s, 9H), 1.47 (s, 9H), 1.22 (d, 3H, J = 6.8 Hz), 1.14 (d, 3H, J = 7.2 Hz).
MS: m/z = 172 [M+H] + (free form)
[0138]
Step C. Preparation of S-ZMAB (Compound [7])
[Chem. 8]
 To the methanol solution of S-MACB-HC [6] (24.8 g, 16.8 mmol in theory) was added dropwise N,N-diisopropylethylamine (4.8 g, 36.9 mmol) at room temperature under nitrogen atmosphere, and then the used dropping funnel was washed with tetrahydrofuran (2.5 mL) and the washings were added to the reaction mixture. To the resulting reaction mixture was added dropwise benzyl chloroformate (3.0 g, 17.6 mmol) at 0°C, and then the used dropping funnel was washed with tetrahydrofuran (2.5 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at 0°C for 1 hour, and then the solvent was removed in vacuo. After toluene (25.0 mL) and an aqueous solution of citric acid (25.0 mL) was added to the residue and then the mixture was stirred, the organic layer was separated out. The resulting organic layer was washed with sodium bicarbonate water (25.0 mL) and then water (25.0 mL), and the solvent in the organic layer was removed out of the organic layer in vacuo. Toluene (15.0 mL) was added to the residue and the toluene solution was concentrated. The operation was repeated one more time to give a toluene solution of S-ZMAB [7] (6.9 g, 16.8 mmol in theory). The given toluene solution of S-ZMAB was used in the next step, assuming that the yield was 100 %.
To the methanol solution of S-MACB-HC [6] (24.8 g, 16.8 mmol in theory) was added dropwise N,N-diisopropylethylamine (4.8 g, 36.9 mmol) at room temperature under nitrogen atmosphere, and then the used dropping funnel was washed with tetrahydrofuran (2.5 mL) and the washings were added to the reaction mixture. To the resulting reaction mixture was added dropwise benzyl chloroformate (3.0 g, 17.6 mmol) at 0°C, and then the used dropping funnel was washed with tetrahydrofuran (2.5 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at 0°C for 1 hour, and then the solvent was removed in vacuo. After toluene (25.0 mL) and an aqueous solution of citric acid (25.0 mL) was added to the residue and then the mixture was stirred, the organic layer was separated out. The resulting organic layer was washed with sodium bicarbonate water (25.0 mL) and then water (25.0 mL), and the solvent in the organic layer was removed out of the organic layer in vacuo. Toluene (15.0 mL) was added to the residue and the toluene solution was concentrated. The operation was repeated one more time to give a toluene solution of S-ZMAB [7] (6.9 g, 16.8 mmol in theory). The given toluene solution of S-ZMAB was used in the next step, assuming that the yield was 100 %.
A crude product of S-ZMAB which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (CDCl 3) δ: 7.38-7.28 (m, 10H), 5.16-5.04 (m, 4H), 4.60 (d, 1H, J = 9.2 Hz), 4.18-4.12 (m, 2H), 4.04 (t, 1H, J = 8.6 Hz), 3.66 (dd, 1H, J = 7.6, 7.2 Hz), 3.50 (dd, 1H, J = 8.0, 5.2 Hz), 3.05-2.94 (m, 1H), 2.60-2.50 (m, 1H), 1.43 (br s, 18H), 1.33 (d, 3H, J = 6.5 Hz), 1.15 (d, 3H, J = 7.2 Hz).
MS: m/z = 328 [M+Na] +.
[Chem. 8]
A crude product of S-ZMAB which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (CDCl 3) δ: 7.38-7.28 (m, 10H), 5.16-5.04 (m, 4H), 4.60 (d, 1H, J = 9.2 Hz), 4.18-4.12 (m, 2H), 4.04 (t, 1H, J = 8.6 Hz), 3.66 (dd, 1H, J = 7.6, 7.2 Hz), 3.50 (dd, 1H, J = 8.0, 5.2 Hz), 3.05-2.94 (m, 1H), 2.60-2.50 (m, 1H), 1.43 (br s, 18H), 1.33 (d, 3H, J = 6.5 Hz), 1.15 (d, 3H, J = 7.2 Hz).
MS: m/z = 328 [M+Na] +.
[0139]
Step D. Preparation of RS-ZMBB (Compound [8])
[Chem. 9]
 To the toluene solution of S-ZMAB [7] (6.9 g, 16.8 mmol) was added tetrahydrofuran (15.0 mL) at room temperature under nitrogen atmosphere. A solution of lithium bis(trimethylsilyl)amide/tetrahydrofuran (14.7 mL, 17.6 mmol) was added dropwise to the toluene solution at -70°C. The used dropping funnel was washed with tetrahydrofuran (2.5 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at -70°C for 6 hours, and then a solution of TBBA (3.4 g, 17.6 mmol) in tetrahydrofuran (2.5 mL) was added dropwise to the reaction mixture at -70°C. The used dropping funnel was washed with tetrahydrofuran (2.5 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at -70°C for 1 hour, and then warmed to room temperature. To the reaction mixture were added an aqueous ammonium chloride (25 mL) and toluene (25 mL) and then the mixture was stirred, the organic layer was separated out. The resulting organic layer was washed with an aqueous solution of citric acid (25 mL, x 2), sodium bicarbonate water (25 mL), and then water (25 mL), and then the solvent was removed out of the organic layer in vacuo. Acetonitrile (15 mL) was added to the residue and the acetonitrile solution was concentrated. The operation was repeated two more times. Acetonitrile (15 mL) and active carbon (0.25 g) were added to the residue, the mixture was stirred at room temperature for 2 hours. The active carbon was removed by filtration, and the reaction vessel and the residue on the filter was washed with acetonitrile (10 mL). The washings were added to the filtration, and then the filtration was concentrated in vacuo to give an acetonitrile solution of RS-ZMBB [8] (13.2 g, 16.8 mmol in theory). The given acetonitrile solution of RS-ZMBB was used in the next step, assuming that the yield was 100 %.
To the toluene solution of S-ZMAB [7] (6.9 g, 16.8 mmol) was added tetrahydrofuran (15.0 mL) at room temperature under nitrogen atmosphere. A solution of lithium bis(trimethylsilyl)amide/tetrahydrofuran (14.7 mL, 17.6 mmol) was added dropwise to the toluene solution at -70°C. The used dropping funnel was washed with tetrahydrofuran (2.5 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at -70°C for 6 hours, and then a solution of TBBA (3.4 g, 17.6 mmol) in tetrahydrofuran (2.5 mL) was added dropwise to the reaction mixture at -70°C. The used dropping funnel was washed with tetrahydrofuran (2.5 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at -70°C for 1 hour, and then warmed to room temperature. To the reaction mixture were added an aqueous ammonium chloride (25 mL) and toluene (25 mL) and then the mixture was stirred, the organic layer was separated out. The resulting organic layer was washed with an aqueous solution of citric acid (25 mL, x 2), sodium bicarbonate water (25 mL), and then water (25 mL), and then the solvent was removed out of the organic layer in vacuo. Acetonitrile (15 mL) was added to the residue and the acetonitrile solution was concentrated. The operation was repeated two more times. Acetonitrile (15 mL) and active carbon (0.25 g) were added to the residue, the mixture was stirred at room temperature for 2 hours. The active carbon was removed by filtration, and the reaction vessel and the residue on the filter was washed with acetonitrile (10 mL). The washings were added to the filtration, and then the filtration was concentrated in vacuo to give an acetonitrile solution of RS-ZMBB [8] (13.2 g, 16.8 mmol in theory). The given acetonitrile solution of RS-ZMBB was used in the next step, assuming that the yield was 100 %.
A crude product of RS-ZMBB which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 7.38-7.29 (m, 5H), 5.09-4.96 (m, 2H), 3.91 (t, 0.4H, J = 8.0 Hz), 3.79 (t, 0.6H, J = 8.0 Hz), 3.55 (t, 0.4H, J = 7.2 Hz), 3.46 (t, 0.6H, J = 7.5 Hz), 3.14-3.04 (m, 1H), 2.83-2.72 (m, 2H), 1.38 (br s, 9H), 1.37 (br s, 3.6H), 1.34 (br s, 5.4H), 1.12-1.09 (m, 3H).
MS: m/z = 420 [M+H] +.
[Chem. 9]
A crude product of RS-ZMBB which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 7.38-7.29 (m, 5H), 5.09-4.96 (m, 2H), 3.91 (t, 0.4H, J = 8.0 Hz), 3.79 (t, 0.6H, J = 8.0 Hz), 3.55 (t, 0.4H, J = 7.2 Hz), 3.46 (t, 0.6H, J = 7.5 Hz), 3.14-3.04 (m, 1H), 2.83-2.72 (m, 2H), 1.38 (br s, 9H), 1.37 (br s, 3.6H), 1.34 (br s, 5.4H), 1.12-1.09 (m, 3H).
MS: m/z = 420 [M+H] +.
[0140]
Step E. Preparation of RS-ZMAA-DN . 2H 2 O (Compound [9])
[Chem. 10]
 To the acetonitrile solution of RS-ZMBB [8] (13.2 g, 16.8 mmol in theory) was added acetonitrile (15 mL) at room temperature under nitrogen atmosphere. p-Toluenesulfonic acid mono-hydrate (6.4 g, 33.6 mmol) was added to the solution at room temperature. The reaction mixture was stirred at 50°C for 12 hours, and then cooled to room temperature, and water (7.5 mL) was added dropwise to the reaction mixture. The reaction mixture was cooled to 0°C, and then 4 mol/L aqueous sodium hydroxide (17.6 mL, 70.5 mmol) was added dropwise thereto. After stirring the reaction mixture at room temperature for 1 hour, acetonitrile (75 mL) was added dropwise thereto at room temperature, and the reaction mixture was stirred for 3 hours. The precipitated solid was collected on a filter, and washed with a mixture of acetonitrile : water = 4 : 1 (10 mL) and then acetonitrile (10 mL). The resulting wet solid was dried in vacuo to give RS-ZMAA-DN .2H 2O [9] (5.2 g, 13.4 mmol, yield: 85.4 %).
To the acetonitrile solution of RS-ZMBB [8] (13.2 g, 16.8 mmol in theory) was added acetonitrile (15 mL) at room temperature under nitrogen atmosphere. p-Toluenesulfonic acid mono-hydrate (6.4 g, 33.6 mmol) was added to the solution at room temperature. The reaction mixture was stirred at 50°C for 12 hours, and then cooled to room temperature, and water (7.5 mL) was added dropwise to the reaction mixture. The reaction mixture was cooled to 0°C, and then 4 mol/L aqueous sodium hydroxide (17.6 mL, 70.5 mmol) was added dropwise thereto. After stirring the reaction mixture at room temperature for 1 hour, acetonitrile (75 mL) was added dropwise thereto at room temperature, and the reaction mixture was stirred for 3 hours. The precipitated solid was collected on a filter, and washed with a mixture of acetonitrile : water = 4 : 1 (10 mL) and then acetonitrile (10 mL). The resulting wet solid was dried in vacuo to give RS-ZMAA-DN .2H 2O [9] (5.2 g, 13.4 mmol, yield: 85.4 %).
RS-ZMAA-DN .2H 2O which was prepared by the same process was measured about NMR, MS, Na-content, and water-content.
1H-NMR (DMSO-d 6) δ: 7.32-7.22 (m, 5H), 4.97 (d, 1H, J = 12.7 Hz), 4.84 (d, 1H, J = 12.7 Hz), 3.79 (t, 1H, J = 8.0 Hz), 3.29 (d, 1H, J = 14.8 Hz), 3.16-3.12 (m, 1H), 2.17-2.09 (m, 2H), 1.07 (d, 3H, J = 6.9 Hz).
MS: m/z = 352 [M+H] + (anhydrate)
Na content (ion chromatography): 13.3 % (after correction of water content)(13.1 % in theory)
Water content (Karl Fischer’s method): 9.8 % (9.3 % in theory)
[Chem. 10]
RS-ZMAA-DN .2H 2O which was prepared by the same process was measured about NMR, MS, Na-content, and water-content.
1H-NMR (DMSO-d 6) δ: 7.32-7.22 (m, 5H), 4.97 (d, 1H, J = 12.7 Hz), 4.84 (d, 1H, J = 12.7 Hz), 3.79 (t, 1H, J = 8.0 Hz), 3.29 (d, 1H, J = 14.8 Hz), 3.16-3.12 (m, 1H), 2.17-2.09 (m, 2H), 1.07 (d, 3H, J = 6.9 Hz).
MS: m/z = 352 [M+H] + (anhydrate)
Na content (ion chromatography): 13.3 % (after correction of water content)(13.1 % in theory)
Water content (Karl Fischer’s method): 9.8 % (9.3 % in theory)
[0141]
Step F. Preparation of RS-ZMAA (Compound [10])
[Chem. 11]
 To 1 mol/L hydrochloric acid (180 mL) were added RS-ZMAA-DN .2H 2O [9] (30 g, 77.5 mmol) and acetonitrile (60 mL), and the mixture was stirred at room temperature for about 15 minutes. After ethyl acetate (240 mL) was added to the reaction mixture and then the mixture was stirred, the organic layer was separated out. The organic layer was washed with 10 % brine (60 mL x 2). The organic layer was stirred with magnesium sulfate (6 g), the magnesium sulfate was removed by filtration, and the residue on the filter was washed with ethyl acetate (60 mL). The filtrate and the washings are combined, and the solvent was removed out in vacuo. Tetrahydrofuran (240 mL) was added to the residue and the tetrahydrofuran solution was concentrated. The operation was repeated two more times. Tetrahydrofuran (60 mL) was added to the residue to give a tetrahydrofuran solution of RS-ZMAA [10]. The given tetrahydrofuran solution of RS-ZMAA was used in the next step, assuming that the yield was 100 %.
To 1 mol/L hydrochloric acid (180 mL) were added RS-ZMAA-DN .2H 2O [9] (30 g, 77.5 mmol) and acetonitrile (60 mL), and the mixture was stirred at room temperature for about 15 minutes. After ethyl acetate (240 mL) was added to the reaction mixture and then the mixture was stirred, the organic layer was separated out. The organic layer was washed with 10 % brine (60 mL x 2). The organic layer was stirred with magnesium sulfate (6 g), the magnesium sulfate was removed by filtration, and the residue on the filter was washed with ethyl acetate (60 mL). The filtrate and the washings are combined, and the solvent was removed out in vacuo. Tetrahydrofuran (240 mL) was added to the residue and the tetrahydrofuran solution was concentrated. The operation was repeated two more times. Tetrahydrofuran (60 mL) was added to the residue to give a tetrahydrofuran solution of RS-ZMAA [10]. The given tetrahydrofuran solution of RS-ZMAA was used in the next step, assuming that the yield was 100 %.
RS-ZMAA which was prepared by the same process was measured about NMR and MS.
1H-NMR (DMSO-D 6) δ: 7.35-7.28 (m, 5H), 5.06-4.94 (m, 2H), 3.86 (dt, 1H, J = 48.4, 7.9 Hz), 3.50 (dt, 1H, J = 37.9, 7.4 Hz), 3.16-3.02 (br m, 1H), 2.91-2.77 (br m, 2H), 1.08 (d, 3H, J = 6.9 Hz)
MS: m/z = 308 [M+H] +.
[Chem. 11]
RS-ZMAA which was prepared by the same process was measured about NMR and MS.
1H-NMR (DMSO-D 6) δ: 7.35-7.28 (m, 5H), 5.06-4.94 (m, 2H), 3.86 (dt, 1H, J = 48.4, 7.9 Hz), 3.50 (dt, 1H, J = 37.9, 7.4 Hz), 3.16-3.02 (br m, 1H), 2.91-2.77 (br m, 2H), 1.08 (d, 3H, J = 6.9 Hz)
MS: m/z = 308 [M+H] +.
[0142]
Step G. Preparation of RS-ZMOO (Compound [11])
[Chem. 12]
 To the tetrahydrofuran solution of RS-ZMAA [10] (25.8 mmol in theory) was added tetrahydrofuran (50 mL) under nitrogen atmosphere. Boron trifluoride etherate complex (4.40 g) was added dropwise thereto at 0°C to 5°C. The used dropping funnel was washed with tetrahydrofuran (5 mL) and the washings were added to the reaction mixture. To the reaction mixture was added dropwise 1.2 mol/L borane-tetrahydrofuran complex (43.0 mL) at 0°C to 5°C, and the reaction mixture was stirred at 0°C to 5°C for about 30 minutes, and then further stirred at room temperature overnight. To the reaction mixture was added dropwise 1.2 mol/L borane-tetrahydrofuran complex (21.1 mL) at 0°C to 5°C, and then the reaction mixture was stirred at room temperature overnight. After stirring, water (40 mL) was added dropwise to the reaction mixture at 0°C to 15°C. To the reaction mixture was added sodium bicarbonate (5.42 g) at 0°C to 15°C. The sodium bicarbonate left in the vessel was washed with water (10 mL), and the washings were added to the reaction mixture. The reaction mixture was stirred at room temperature for 2 hours, and then toluene (50 mL) was added thereto and the reaction mixture was further stirred. The organic layer was separated out. The resulting organic layer was washed with 10 % brine (20 mL x 1), a mixture (x 3) of 5 % sodium bicarbonate water (20 mL) and 10 % brine (20 mL), a mixture (x 1) of 5 % aqueous potassium hydrogensulfate (10 mL) and 10 % brine (10 mL), and then 10 % brine (20 mL x 2). The organic layer was stirred with magnesium sulfate (8.9 g), the magnesium sulfate was removed by filtration, and the residue on the filter was washed with toluene (20 mL). The washings were added to the filtration, and then the filtrate was concentrated in vacuo. To the concentrated residue was added toluene (80 mL). The solution was concentrated in vacuo, and toluene (15 mL) was added thereto to give a toluene solution of RS-ZMOO [11]. The given toluene solution of RS-ZMOO was used in the next step, assuming that the yield was 100 %.
To the tetrahydrofuran solution of RS-ZMAA [10] (25.8 mmol in theory) was added tetrahydrofuran (50 mL) under nitrogen atmosphere. Boron trifluoride etherate complex (4.40 g) was added dropwise thereto at 0°C to 5°C. The used dropping funnel was washed with tetrahydrofuran (5 mL) and the washings were added to the reaction mixture. To the reaction mixture was added dropwise 1.2 mol/L borane-tetrahydrofuran complex (43.0 mL) at 0°C to 5°C, and the reaction mixture was stirred at 0°C to 5°C for about 30 minutes, and then further stirred at room temperature overnight. To the reaction mixture was added dropwise 1.2 mol/L borane-tetrahydrofuran complex (21.1 mL) at 0°C to 5°C, and then the reaction mixture was stirred at room temperature overnight. After stirring, water (40 mL) was added dropwise to the reaction mixture at 0°C to 15°C. To the reaction mixture was added sodium bicarbonate (5.42 g) at 0°C to 15°C. The sodium bicarbonate left in the vessel was washed with water (10 mL), and the washings were added to the reaction mixture. The reaction mixture was stirred at room temperature for 2 hours, and then toluene (50 mL) was added thereto and the reaction mixture was further stirred. The organic layer was separated out. The resulting organic layer was washed with 10 % brine (20 mL x 1), a mixture (x 3) of 5 % sodium bicarbonate water (20 mL) and 10 % brine (20 mL), a mixture (x 1) of 5 % aqueous potassium hydrogensulfate (10 mL) and 10 % brine (10 mL), and then 10 % brine (20 mL x 2). The organic layer was stirred with magnesium sulfate (8.9 g), the magnesium sulfate was removed by filtration, and the residue on the filter was washed with toluene (20 mL). The washings were added to the filtration, and then the filtrate was concentrated in vacuo. To the concentrated residue was added toluene (80 mL). The solution was concentrated in vacuo, and toluene (15 mL) was added thereto to give a toluene solution of RS-ZMOO [11]. The given toluene solution of RS-ZMOO was used in the next step, assuming that the yield was 100 %.
RS-ZMOO which was prepared by the same process was measured about NMR and MS.
1H-NMR (CDCl 3) δ: 7.39-7.30 (m, 5H), 5.10 (s, 2H), 4.15-4.01 (br m, 2H), 3.83-3.73 (br m, 3H), 3.48 (dd, 1H, J = 8.3, 6.4 Hz), 2.59-2.50 (br m, 1H), 2.46-2.40 (br m, 1H), 2.07-1.99 (m, 1H), 1.14 (d, 3H, J = 7.2 Hz)
MS: m/z = 280 [M+H]+.
[Chem. 12]
RS-ZMOO which was prepared by the same process was measured about NMR and MS.
1H-NMR (CDCl 3) δ: 7.39-7.30 (m, 5H), 5.10 (s, 2H), 4.15-4.01 (br m, 2H), 3.83-3.73 (br m, 3H), 3.48 (dd, 1H, J = 8.3, 6.4 Hz), 2.59-2.50 (br m, 1H), 2.46-2.40 (br m, 1H), 2.07-1.99 (m, 1H), 1.14 (d, 3H, J = 7.2 Hz)
MS: m/z = 280 [M+H]+.
[0143]
Step H. Preparation of RS-ZMSS (Compound [12])
[Chem. 13]
 To the toluene solution of RS-ZMOO [11] (23.7 mmol in theory) was added toluene (55 mL) under nitrogen atmosphere. And, triethylamine (5.27 g) was added dropwise thereto at -10°C to 10°C, and the used dropping funnel was washed with toluene (1.8 mL) and the washings were added to the reaction mixture. To this reaction mixture was added dropwise methanesulfonyl chloride (5.69 g) at -10°C to 10°C, and then the used dropping funnel was washed with toluene (1.8 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at 0°C to 10°C for about 2 hours, and then water (28 mL) was added dropwise thereto at 0°C to 20°C. The reaction mixture was stirred at 0°C to 20°C for about 30 minutes, and then, the organic layer was separated out. The resulting organic layer was washed twice with 10 % brine (18 mL). The organic layer was stirred with magnesium sulfate (2.75 g), the magnesium sulfate was removed by filtration, and the residue on the filter was washed with toluene (18 mL). The washings were added to the filtrate, and then the solvent was removed from the filtrate in vacuo. To the concentrated residue was added toluene up to 18 mL to give a toluene solution of RS-ZMSS [12]. The given toluene solution of RS-ZMSS was used in the next step, assuming that the yield was 100 %.
To the toluene solution of RS-ZMOO [11] (23.7 mmol in theory) was added toluene (55 mL) under nitrogen atmosphere. And, triethylamine (5.27 g) was added dropwise thereto at -10°C to 10°C, and the used dropping funnel was washed with toluene (1.8 mL) and the washings were added to the reaction mixture. To this reaction mixture was added dropwise methanesulfonyl chloride (5.69 g) at -10°C to 10°C, and then the used dropping funnel was washed with toluene (1.8 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at 0°C to 10°C for about 2 hours, and then water (28 mL) was added dropwise thereto at 0°C to 20°C. The reaction mixture was stirred at 0°C to 20°C for about 30 minutes, and then, the organic layer was separated out. The resulting organic layer was washed twice with 10 % brine (18 mL). The organic layer was stirred with magnesium sulfate (2.75 g), the magnesium sulfate was removed by filtration, and the residue on the filter was washed with toluene (18 mL). The washings were added to the filtrate, and then the solvent was removed from the filtrate in vacuo. To the concentrated residue was added toluene up to 18 mL to give a toluene solution of RS-ZMSS [12]. The given toluene solution of RS-ZMSS was used in the next step, assuming that the yield was 100 %.
RS-ZMSS which was prepared by the same process was measured by NMR and MS.
1H-NMR (DMSO-D 6) δ: 7.37-7.27 (br m, 5H), 5.10-4.98 (m, 2H), 4.58-4.22 (br m, 4H), 3.84 (dt, 1H, J = 45.6, 8.1 Hz), 3.48-3.33 (br m, 1H), 3.17-3.10 (m, 6H), 2.81-2.74 (br m, 1H), 2.22-2.12 (m, 2H)
MS: m/z = 436 [M+H] +.
[Chem. 13]
RS-ZMSS which was prepared by the same process was measured by NMR and MS.
1H-NMR (DMSO-D 6) δ: 7.37-7.27 (br m, 5H), 5.10-4.98 (m, 2H), 4.58-4.22 (br m, 4H), 3.84 (dt, 1H, J = 45.6, 8.1 Hz), 3.48-3.33 (br m, 1H), 3.17-3.10 (m, 6H), 2.81-2.74 (br m, 1H), 2.22-2.12 (m, 2H)
MS: m/z = 436 [M+H] +.
[0144]
Step I. Preparation of SR-ZMDB (Compound [13])
[Chem. 14]
 To a toluene solution of RS-ZMSS [12] (23.7 mmol in theory) was added toluene (55 mL) under nitrogen atmosphere. And, benzylamine (17.8 g) was added dropwise thereto at room temperature, and the used dropping funnel was washed with toluene (9.2 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at room temperature for about 1 hour, at 55°C to 65°C for about 3 hours, and then at 70°C to 80°C for 6 hours. After the reaction mixture was cooled to room temperature, 10 % NaCl (28 mL) was added dropwise thereto, and the reaction mixture was stirred at room temperature for about 30 minutes. After toluene (37 mL) was added to the reaction mixture and then the mixture was stirred, the organic layer was separated out. The resulting organic layer was washed with a mixture (x 2) of 10 % brine (18 mL) and acetic acid (2.84 g), and then 10 % brine (11 mL, x 1). The solvent of the organic layer was removed in vacuo to a half volume, and acetic anhydride (1.45 g) was added to the concentrated residue at room temperature. The mixture was stirred for about 3 hours. To the reaction mixture were added dropwise a solution of potassium hydrogensulfate (3.87 g) and water (92 mL) at room temperature. The reaction mixture was stirred, and then the aqueous layer was separated out. The resulting aqueous layer was washed with toluene (18 mL), and toluene (73 mL) and then sodium bicarbonate (6.56 g) were added to the aqueous layer at room temperature, and the mixture was stirred. The organic layer was separated out, and washed with 10 % brine (11 mL). The organic layer was stirred with magnesium sulfate (2.75 g), the magnesium sulfate was removed by filtration. The residue on the filter was washed with toluene (18 mL), and the washings were added to the filtrate, and then the filtrate was concentrated in vacuo. Toluene (44 mL) was added to the concentrated residue to give a toluene solution of SR-ZMDB [13]. The given toluene solution of SR-ZMDB was used in the next step, assuming that the yield was 100 %.
To a toluene solution of RS-ZMSS [12] (23.7 mmol in theory) was added toluene (55 mL) under nitrogen atmosphere. And, benzylamine (17.8 g) was added dropwise thereto at room temperature, and the used dropping funnel was washed with toluene (9.2 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at room temperature for about 1 hour, at 55°C to 65°C for about 3 hours, and then at 70°C to 80°C for 6 hours. After the reaction mixture was cooled to room temperature, 10 % NaCl (28 mL) was added dropwise thereto, and the reaction mixture was stirred at room temperature for about 30 minutes. After toluene (37 mL) was added to the reaction mixture and then the mixture was stirred, the organic layer was separated out. The resulting organic layer was washed with a mixture (x 2) of 10 % brine (18 mL) and acetic acid (2.84 g), and then 10 % brine (11 mL, x 1). The solvent of the organic layer was removed in vacuo to a half volume, and acetic anhydride (1.45 g) was added to the concentrated residue at room temperature. The mixture was stirred for about 3 hours. To the reaction mixture were added dropwise a solution of potassium hydrogensulfate (3.87 g) and water (92 mL) at room temperature. The reaction mixture was stirred, and then the aqueous layer was separated out. The resulting aqueous layer was washed with toluene (18 mL), and toluene (73 mL) and then sodium bicarbonate (6.56 g) were added to the aqueous layer at room temperature, and the mixture was stirred. The organic layer was separated out, and washed with 10 % brine (11 mL). The organic layer was stirred with magnesium sulfate (2.75 g), the magnesium sulfate was removed by filtration. The residue on the filter was washed with toluene (18 mL), and the washings were added to the filtrate, and then the filtrate was concentrated in vacuo. Toluene (44 mL) was added to the concentrated residue to give a toluene solution of SR-ZMDB [13]. The given toluene solution of SR-ZMDB was used in the next step, assuming that the yield was 100 %.
1H-NMR (CDCl 3) δ: 7.35-7.20 (m, 10H), 5.08 (d, 2H, J = 23.6 Hz), 3.94 (q, 1H, J = 7.9 Hz), 3.73-3.42 (br m, 2H), 3.30-3.23 (m, 1H), 3.05 (dd, 1H, J = 19.7, 9.5 Hz), 2.79 (dt, 1H, J = 69.6, 6.1 Hz), 2.57-2.32 (br m, 4H), 1.96-1.89 (m, 1H), 1.09 (d, 3H, J = 6.9 Hz)
MS: m/z = 351 [M+H] +.
[Chem. 14]
1H-NMR (CDCl 3) δ: 7.35-7.20 (m, 10H), 5.08 (d, 2H, J = 23.6 Hz), 3.94 (q, 1H, J = 7.9 Hz), 3.73-3.42 (br m, 2H), 3.30-3.23 (m, 1H), 3.05 (dd, 1H, J = 19.7, 9.5 Hz), 2.79 (dt, 1H, J = 69.6, 6.1 Hz), 2.57-2.32 (br m, 4H), 1.96-1.89 (m, 1H), 1.09 (d, 3H, J = 6.9 Hz)
MS: m/z = 351 [M+H] +.
[0145]
Step J. Preparation of SR-MDOZ (Compound [14])
[Chem. 15]
 To a solution of 1-chloroethyl chloroformate (3.72 g) in toluene (28 mL) was added dropwise the toluene solution of SR-ZMDB [13] (23.7 mmol in theory) at 0°C to 10°C under nitrogen atmosphere, and then the used dropping funnel was washed with toluene (4.6 mL) and the washings were added to the reaction mixture. To the reaction mixture was added triethylamine (718 mg) at 0°C to 10°C, and the reaction mixture was stirred at 15°C to 25°C for about 2 hours. Then, methyl alcohol (46 mL) was added to the reaction mixture, and the mixture was stirred at 50°C to 60°C for additional about 2 hours. The solvent of the reaction mixture was removed in vacuo to a volume of about less than 37 mL. To the concentrated residue was added dropwise 2 mol/L hydrochloric acid (46 mL) at 15°C to 20°C, and the mixture was stirred, and the aqueous layer was separated out. The resulting aqueous layer was washed with toluene (28 mL, x 2). To the aqueous layer were added 20 % brine (46 mL) and tetrahydrofuran (92 mL), and then 8 mol/L aqueous sodium hydroxide (18 mL) was added dropwise thereto at 0°C to 10°C. The organic layer was separated out from the reaction mixture, washed with 20 % brine (18 mL, x 2), and then the solvent of the organic layer was removed in vacuo. To the concentrated residue was added tetrahydrofuran (92 mL), and the solution was concentrated in vacuo. The operation was repeated one more time. The concentrated residue was dissolved in tetrahydrofuran (92 mL). The solution was stirred with magnesium sulfate (2.75 g), and the magnesium sulfate was removed by filtration. The residue on the filter was washed with tetrahydrofuran (28 mL), the washings were added to the filtrate, and the filtrate was concentrated in vacuo. The volume of the concentrated residue was adjusted to about 20 mL with tetrahydrofuran to give a tetrahydrofuran solution of SR-MDOZ [14] (net weight: 4.01 g, 15.4 mol, yield: 65.0 %).
To a solution of 1-chloroethyl chloroformate (3.72 g) in toluene (28 mL) was added dropwise the toluene solution of SR-ZMDB [13] (23.7 mmol in theory) at 0°C to 10°C under nitrogen atmosphere, and then the used dropping funnel was washed with toluene (4.6 mL) and the washings were added to the reaction mixture. To the reaction mixture was added triethylamine (718 mg) at 0°C to 10°C, and the reaction mixture was stirred at 15°C to 25°C for about 2 hours. Then, methyl alcohol (46 mL) was added to the reaction mixture, and the mixture was stirred at 50°C to 60°C for additional about 2 hours. The solvent of the reaction mixture was removed in vacuo to a volume of about less than 37 mL. To the concentrated residue was added dropwise 2 mol/L hydrochloric acid (46 mL) at 15°C to 20°C, and the mixture was stirred, and the aqueous layer was separated out. The resulting aqueous layer was washed with toluene (28 mL, x 2). To the aqueous layer were added 20 % brine (46 mL) and tetrahydrofuran (92 mL), and then 8 mol/L aqueous sodium hydroxide (18 mL) was added dropwise thereto at 0°C to 10°C. The organic layer was separated out from the reaction mixture, washed with 20 % brine (18 mL, x 2), and then the solvent of the organic layer was removed in vacuo. To the concentrated residue was added tetrahydrofuran (92 mL), and the solution was concentrated in vacuo. The operation was repeated one more time. The concentrated residue was dissolved in tetrahydrofuran (92 mL). The solution was stirred with magnesium sulfate (2.75 g), and the magnesium sulfate was removed by filtration. The residue on the filter was washed with tetrahydrofuran (28 mL), the washings were added to the filtrate, and the filtrate was concentrated in vacuo. The volume of the concentrated residue was adjusted to about 20 mL with tetrahydrofuran to give a tetrahydrofuran solution of SR-MDOZ [14] (net weight: 4.01 g, 15.4 mol, yield: 65.0 %).
SR-MDOZ which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (CDCl 3) δ: 7.37-7.28 (m, 5H), 5.08 (dd, 2H, J = 16.8, 12.8 Hz), 4.00 (dd, 1H, J = 17.1, 8.3 Hz), 3.40-3.31 (m, 1H), 3.24 (d, 1H, J = 12.7 Hz), 3.00 (dd, 1H, J = 54.9, 12.4 Hz), 2.87-2.57 (m, 3H), 2.47-2.27 (m, 1H), 1.91-1.80 (m, 1H), 1.14 (d, 3H, J = 7.2 Hz)
MS: m/z = 261 [M+H] +.
[Chem. 15]
SR-MDOZ which was prepared by the same process was evaporated to dryness and then measured about NMR and MS.
1H-NMR (CDCl 3) δ: 7.37-7.28 (m, 5H), 5.08 (dd, 2H, J = 16.8, 12.8 Hz), 4.00 (dd, 1H, J = 17.1, 8.3 Hz), 3.40-3.31 (m, 1H), 3.24 (d, 1H, J = 12.7 Hz), 3.00 (dd, 1H, J = 54.9, 12.4 Hz), 2.87-2.57 (m, 3H), 2.47-2.27 (m, 1H), 1.91-1.80 (m, 1H), 1.14 (d, 3H, J = 7.2 Hz)
MS: m/z = 261 [M+H] +.
[0146]
Step K. Preparation of SR-MDOZ-OX (Compound [15])
[Chem. 16]
 Under nitrogen atmosphere, oxalic acid (761 mg) was dissolved in tetrahydrofuran (40 mL), and the tetrahydrofuran solution of SR-MDOZ [14] (3.84 mmol in theory) was added dropwise to the solution of oxalic acid at room temperature. To the solution was added SR-MDOZ-OX crystal (1 mg) that was prepared according to the method described herein at room temperature, and the mixture was stirred at room temperature for about 3.5 hours to precipitate the crystal. To the slurry solution was added dropwise the tetrahydrofuran solution of SR-MDOZ (3.84 mmol) at room temperature, and the mixture was stirred at room temperature for about 1 hour. The slurry solution was heated, and stirred at 50°C to 60°C for about 2 hours, and then stirred at room temperature overnight. The slurry solution was filtrated, and the wet crystal on the filter was washed with tetrahydrofuran (10 mL), dried in vacuo to give SR-MDOZ-OX [15] (2.32 g, 6.62 mol, yield: 86.2 %).
Under nitrogen atmosphere, oxalic acid (761 mg) was dissolved in tetrahydrofuran (40 mL), and the tetrahydrofuran solution of SR-MDOZ [14] (3.84 mmol in theory) was added dropwise to the solution of oxalic acid at room temperature. To the solution was added SR-MDOZ-OX crystal (1 mg) that was prepared according to the method described herein at room temperature, and the mixture was stirred at room temperature for about 3.5 hours to precipitate the crystal. To the slurry solution was added dropwise the tetrahydrofuran solution of SR-MDOZ (3.84 mmol) at room temperature, and the mixture was stirred at room temperature for about 1 hour. The slurry solution was heated, and stirred at 50°C to 60°C for about 2 hours, and then stirred at room temperature overnight. The slurry solution was filtrated, and the wet crystal on the filter was washed with tetrahydrofuran (10 mL), dried in vacuo to give SR-MDOZ-OX [15] (2.32 g, 6.62 mol, yield: 86.2 %).
SR-MDOZ-OX which was prepared by the same process was measured about NMR, MS, and elementary analysis.
1H-NMR (DMSO-D 6) δ: 7.37-7.30 (m, 5H), 5.15-5.01 (m, 2H), 3.92 (dt, 1H, J = 43.5, 8.4 Hz), 3.48-3.12 (br m, 5H), 2.67-2.56 (m, 1H), 2.46-2.35 (m, 1H), 2.12-2.05 (m, 1H), 1.13 (d, 3H, J = 6.9 Hz)
MS: m/z = 261 [M+H] +
elementary analysis: C 58.4wt % , H 6.4wt % , N 7.9 % wt % (theoretically, C 58.3wt % , H 6.3wt % , N 8.0wt % )
[Chem. 16]
SR-MDOZ-OX which was prepared by the same process was measured about NMR, MS, and elementary analysis.
1H-NMR (DMSO-D 6) δ: 7.37-7.30 (m, 5H), 5.15-5.01 (m, 2H), 3.92 (dt, 1H, J = 43.5, 8.4 Hz), 3.48-3.12 (br m, 5H), 2.67-2.56 (m, 1H), 2.46-2.35 (m, 1H), 2.12-2.05 (m, 1H), 1.13 (d, 3H, J = 6.9 Hz)
MS: m/z = 261 [M+H] +
elementary analysis: C 58.4wt % , H 6.4wt % , N 7.9 % wt % (theoretically, C 58.3wt % , H 6.3wt % , N 8.0wt % )
[0147]
Step L. Preparation of SR-MDPZ (Compound [16])
[Chem. 17]
 To SR-MDOZ-OX [15] (12.0 g, 34.2 mmol) were added ethanol (36 mL), water (72 mL), CPPY [20] (5.36 g, 34.9 mmol), and then K 3PO 4 (21.8 g, 103 mmol) under nitrogen atmosphere. The reaction mixture was stirred at 80°C for 5 hours, and then cooled to 40°C. Toluene (120 mL) was added thereto at 40°C, and the organic layer was separated out. The resulting organic layer was washed with 20 % aqueous potassium carbonate (48 mL), followed by washing twice with water (48 mL). The solvent of the organic layer was then removed in vacuo. tert-butanol (60 mL) was added to the residue and the tert-butanol solution was concentrated. The operation was repeated two more times. tert-Butanol (36 mL) was added to the concentrated residue to give a solution of SR-MDPZ [16] in tert-butanol (61.1 g, 34.2 mmol in theory). The given tert-butanol solution of SR-MDPZ was used in the next step, assuming that the yield was 100 %.
To SR-MDOZ-OX [15] (12.0 g, 34.2 mmol) were added ethanol (36 mL), water (72 mL), CPPY [20] (5.36 g, 34.9 mmol), and then K 3PO 4 (21.8 g, 103 mmol) under nitrogen atmosphere. The reaction mixture was stirred at 80°C for 5 hours, and then cooled to 40°C. Toluene (120 mL) was added thereto at 40°C, and the organic layer was separated out. The resulting organic layer was washed with 20 % aqueous potassium carbonate (48 mL), followed by washing twice with water (48 mL). The solvent of the organic layer was then removed in vacuo. tert-butanol (60 mL) was added to the residue and the tert-butanol solution was concentrated. The operation was repeated two more times. tert-Butanol (36 mL) was added to the concentrated residue to give a solution of SR-MDPZ [16] in tert-butanol (61.1 g, 34.2 mmol in theory). The given tert-butanol solution of SR-MDPZ was used in the next step, assuming that the yield was 100 %.
SR-MDPZ which was prepared by the same process was isolated as a solid from a mixture of ethyl acetate and n-heptane, and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 11.59 (br s, 1H), 8.08 (s, 1H), 7.41-7.26 (br m, 3H), 7.22-7.08 (br m, 3H), 6.64-6.51 (br m, 1H), 5.07-4.91 (br m, 2H), 4.09-3.67 (br m, 5H), 3.47-3.32 (br m, 1H), 2.67-2.55 (br m, 2H), 2.21-2.15 (br m, 1H), 1.11 (d, 3H, J = 6.9 Hz).
MS: m/z = 378 [M+H] +
[Chem. 17]
SR-MDPZ which was prepared by the same process was isolated as a solid from a mixture of ethyl acetate and n-heptane, and then measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 11.59 (br s, 1H), 8.08 (s, 1H), 7.41-7.26 (br m, 3H), 7.22-7.08 (br m, 3H), 6.64-6.51 (br m, 1H), 5.07-4.91 (br m, 2H), 4.09-3.67 (br m, 5H), 3.47-3.32 (br m, 1H), 2.67-2.55 (br m, 2H), 2.21-2.15 (br m, 1H), 1.11 (d, 3H, J = 6.9 Hz).
MS: m/z = 378 [M+H] +
[0148]
Step M. Preparation of SR-MDOP (Compound [17])
[Chem. 18]
 To the solution of SR-MDPZ [16] in tert-butanol (34.2 mmol in theory) were added ammonium formate (10.8 g, 171 mmol), water (60 mL), and 10 % palladium carbon (made by Kawaken Fine Chemicals Co., Ltd., M type, 52.6 % water-content, 1.20 g) under nitrogen atmosphere. The reaction mixture was stirred at 40°C for 13 hours, and then cooled to room temperature, and the resulting precipitate was removed by filtration. The reaction vessel and the residue on the filter were washed with tert-butanol (24 mL), the washings was added to the filtrate, and 8 M aqueous sodium hydroxide (25.7 mL, 205 mmol) and sodium chloride (13.2 g) were added to the filtrate. The reaction mixture was stirred at 50°C for 2 hours, and then toluene (84 mL) was added thereto at room temperature, and the organic layer was separated out. The resulting organic layer was washed with 20 % brine (60 mL), stirred with anhydrous sodium sulfate, and then the sodium sulfate was removed by filtration. The residue on the filter was washed with a mixture of toluene : tert-butanol = 1 : 1 (48 mL), the washings was added to the filtrate, and the filtrate was concentrated in vacuo. To the concentrated residue was added toluene (60 mL), and the solution was stirred at 50°C for 2 hours, and then the solvent was removed in vacuo. To the concentrated residue was added toluene (60 mL) again, and the solution was concentrated. To the concentrated residue was added toluene (48 mL), and the solution was stirred at room temperature for 1 hour, and then at ice temperature for 1 hour. The precipitated solid was collected on a filter, and washed with toluene (24 mL). The resulting wet solid was dried in vacuo to give SR-MDOP [17] (7.07 g, 29.1 mmol, yield: 84.8 %).
To the solution of SR-MDPZ [16] in tert-butanol (34.2 mmol in theory) were added ammonium formate (10.8 g, 171 mmol), water (60 mL), and 10 % palladium carbon (made by Kawaken Fine Chemicals Co., Ltd., M type, 52.6 % water-content, 1.20 g) under nitrogen atmosphere. The reaction mixture was stirred at 40°C for 13 hours, and then cooled to room temperature, and the resulting precipitate was removed by filtration. The reaction vessel and the residue on the filter were washed with tert-butanol (24 mL), the washings was added to the filtrate, and 8 M aqueous sodium hydroxide (25.7 mL, 205 mmol) and sodium chloride (13.2 g) were added to the filtrate. The reaction mixture was stirred at 50°C for 2 hours, and then toluene (84 mL) was added thereto at room temperature, and the organic layer was separated out. The resulting organic layer was washed with 20 % brine (60 mL), stirred with anhydrous sodium sulfate, and then the sodium sulfate was removed by filtration. The residue on the filter was washed with a mixture of toluene : tert-butanol = 1 : 1 (48 mL), the washings was added to the filtrate, and the filtrate was concentrated in vacuo. To the concentrated residue was added toluene (60 mL), and the solution was stirred at 50°C for 2 hours, and then the solvent was removed in vacuo. To the concentrated residue was added toluene (60 mL) again, and the solution was concentrated. To the concentrated residue was added toluene (48 mL), and the solution was stirred at room temperature for 1 hour, and then at ice temperature for 1 hour. The precipitated solid was collected on a filter, and washed with toluene (24 mL). The resulting wet solid was dried in vacuo to give SR-MDOP [17] (7.07 g, 29.1 mmol, yield: 84.8 %).
SR-MDOP which was prepared by the same process was measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 11.57 (br s, 1H), 8.07 (s, 1H), 7.10 (d, 1H, J = 3.2 Hz), 6.58 (d, 1H, J = 3.2 Hz), 3.92-3.59 (br m, 4H), 3.49 (dd, 1H, J = 8.3, 7.2 Hz), 2.93 (dd, 1H, J = 7.2, 6.1 Hz), 2.61-2.53 (m, 2H), 2.12-2.01 (br m, 2H), 1.10 (d, 3H, J = 6.9 Hz).
MS: m/z = 244 [M+H] +.
[Chem. 18]
SR-MDOP which was prepared by the same process was measured about NMR and MS.
1H-NMR (DMSO-d 6) δ: 11.57 (br s, 1H), 8.07 (s, 1H), 7.10 (d, 1H, J = 3.2 Hz), 6.58 (d, 1H, J = 3.2 Hz), 3.92-3.59 (br m, 4H), 3.49 (dd, 1H, J = 8.3, 7.2 Hz), 2.93 (dd, 1H, J = 7.2, 6.1 Hz), 2.61-2.53 (m, 2H), 2.12-2.01 (br m, 2H), 1.10 (d, 3H, J = 6.9 Hz).
MS: m/z = 244 [M+H] +.
[0149]
Step N. Preparation of Compound A mono-ethanolate (Compound [18])
[Chem. 19]
 Under nitrogen atmosphere, acetonitrile (60 mL) and triethylamine (416 mg, 4.11 mmol) were added to SR-MDOP [17] (5.00 g, 20.5 mmol), and to the solution was added dropwise a solution of DPCN [21] (3.69 g, 22.6 mmol) in acetonitrile (35 mL) at 45°C, and then the used dropping funnel was washed with acetonitrile (5.0 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at 45°C for 3 hours, and then cooled to room temperature. After 5 % sodium bicarbonate water (25 mL), 10 % brine (25 mL), and ethyl acetate (50 mL) were added to the reaction mixture and then the mixture was stirred, the organic layer was separated out. The solvent of the organic layer was removed out in vacuo. Tetrahydrofuran (50 mL) was added to the residue and the tetrahydrofuran solution was concentrated. The operation was repeated three more times. To the concentrated residue was added tetrahydrofuran (50 mL), and water was added the solution to adjust the water content to 5.5 %. The resulting precipitate was removed by filtration. The reaction vessel and the residue on the filter were washed with tetrahydrofuran (15 mL), the washings were added to the filtrate, and the solvent was removed out of the filtrate in vacuo. To the concentrated residue were added ethanol (50 mL) and Compound A crystal (5.1 mg) that was prepared according to the method described in the following Example 15. The mixture was stirred at room temperature for 1 hour, and concentrated in vacuo. To the residue was ethanol (50 mL), and the solution was concentrated again. To the concentrated residue was added ethanol (15 mL), and the solution was stirred at room temperature for 1 hour. The precipitated solid was collected on the filter, and washed with ethanol (20 mL). The resulting wet solid was dried in vacuo to give Compound A mono-ethanolate [18] (6.26 g, 17.6 mmol, yield: 85.5 %).
Under nitrogen atmosphere, acetonitrile (60 mL) and triethylamine (416 mg, 4.11 mmol) were added to SR-MDOP [17] (5.00 g, 20.5 mmol), and to the solution was added dropwise a solution of DPCN [21] (3.69 g, 22.6 mmol) in acetonitrile (35 mL) at 45°C, and then the used dropping funnel was washed with acetonitrile (5.0 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at 45°C for 3 hours, and then cooled to room temperature. After 5 % sodium bicarbonate water (25 mL), 10 % brine (25 mL), and ethyl acetate (50 mL) were added to the reaction mixture and then the mixture was stirred, the organic layer was separated out. The solvent of the organic layer was removed out in vacuo. Tetrahydrofuran (50 mL) was added to the residue and the tetrahydrofuran solution was concentrated. The operation was repeated three more times. To the concentrated residue was added tetrahydrofuran (50 mL), and water was added the solution to adjust the water content to 5.5 %. The resulting precipitate was removed by filtration. The reaction vessel and the residue on the filter were washed with tetrahydrofuran (15 mL), the washings were added to the filtrate, and the solvent was removed out of the filtrate in vacuo. To the concentrated residue were added ethanol (50 mL) and Compound A crystal (5.1 mg) that was prepared according to the method described in the following Example 15. The mixture was stirred at room temperature for 1 hour, and concentrated in vacuo. To the residue was ethanol (50 mL), and the solution was concentrated again. To the concentrated residue was added ethanol (15 mL), and the solution was stirred at room temperature for 1 hour. The precipitated solid was collected on the filter, and washed with ethanol (20 mL). The resulting wet solid was dried in vacuo to give Compound A mono-ethanolate [18] (6.26 g, 17.6 mmol, yield: 85.5 %).
Compound A mono-ethanolate which was prepared by the same process was measured by NMR and MS.
1H-NMR (DMSO-d 6) δ: 11.59 (br s, 1H), 8.08 (s, 1H), 7.11 (dd, 1H, J = 3.5, 2.3 Hz), 6.58 (dd, 1H, J = 3.5, 1.8 Hz), 4.34 (t, 1H, J = 5.1 Hz), 4.16 (t, 1H, J = 8.3 Hz), 4.09-3.92 (m, 3H), 3.84-3.73 (m, 1H), 3.71 (d, 1H, J = 19.0 Hz), 3.65 (d, 1H, J = 19.0 Hz), 3.58 (dd, 1H, J = 8.2, 5.9 Hz), 3.44 (dq, 2H, J = 6.7, 5.1 Hz), 2.69-2.60 (m, 2H), 2.23-2.13 (br m, 1H), 1.12 (d, 3H, J = 7.1 Hz), 1.06 (t, 3H, J = 6.7 Hz).
MS: m/z = 311 [M+H] +
[Chem. 19]
Compound A mono-ethanolate which was prepared by the same process was measured by NMR and MS.
1H-NMR (DMSO-d 6) δ: 11.59 (br s, 1H), 8.08 (s, 1H), 7.11 (dd, 1H, J = 3.5, 2.3 Hz), 6.58 (dd, 1H, J = 3.5, 1.8 Hz), 4.34 (t, 1H, J = 5.1 Hz), 4.16 (t, 1H, J = 8.3 Hz), 4.09-3.92 (m, 3H), 3.84-3.73 (m, 1H), 3.71 (d, 1H, J = 19.0 Hz), 3.65 (d, 1H, J = 19.0 Hz), 3.58 (dd, 1H, J = 8.2, 5.9 Hz), 3.44 (dq, 2H, J = 6.7, 5.1 Hz), 2.69-2.60 (m, 2H), 2.23-2.13 (br m, 1H), 1.12 (d, 3H, J = 7.1 Hz), 1.06 (t, 3H, J = 6.7 Hz).
MS: m/z = 311 [M+H] +
[0150]
Step O. Purification of Compound A (Compound [19])
[Chem. 20]
 Compound A mono-ethanolate [18] (4.00 g, 11.2 mmol) and n-butanol (32 mL) were mixed under nitrogen atmosphere, and the mixture was dissolved at 110°C. The mixture was cooled to 85°C, and Compound A crystal (4.0 mg) that was prepared according to the method described herein was added thereto, and the mixture was stirred at 85°C for 2 hours, at 75°C for 1 hour, and then at room temperature for 16 hours. The precipitated solid was collected on a filter, and washed with n-butanol (8.0 mL) and then ethyl acetate (8.0 mL). The resulting wet solid was dried in vacuo to give Compound A [19] (3.18 g, 10.2 mmol, yield: 91.3 %).
Compound A mono-ethanolate [18] (4.00 g, 11.2 mmol) and n-butanol (32 mL) were mixed under nitrogen atmosphere, and the mixture was dissolved at 110°C. The mixture was cooled to 85°C, and Compound A crystal (4.0 mg) that was prepared according to the method described herein was added thereto, and the mixture was stirred at 85°C for 2 hours, at 75°C for 1 hour, and then at room temperature for 16 hours. The precipitated solid was collected on a filter, and washed with n-butanol (8.0 mL) and then ethyl acetate (8.0 mL). The resulting wet solid was dried in vacuo to give Compound A [19] (3.18 g, 10.2 mmol, yield: 91.3 %).
Compound A which was prepared by the same process was measured by NMR and MS.
1H-NMR (DMSO-d 6) δ: 11.59 (br s, 1H), 8.08 (s, 1H), 7.11 (dd, 1H, J = 3.5, 2.5 Hz), 6.58 (dd, 1H, J = 3.5, 1.8 Hz), 4.16 (t, 1H, J = 8.3 Hz), 4.09-3.93 (m, 3H), 3.84-3.73 (m, 1H), 3.71 (d, 1H, J = 19.0 Hz), 3.65 (d, 1H, J = 19.0 Hz), 3.58 (dd, 1H, J = 8.2, 5.9 Hz), 2.69-2.59 (m, 2H), 2.23-2.13 (m, 1H), 1.12 (d, 3H, J = 7.2 Hz).
MS: m/z = 311 [M+H] +
[Chem. 20]
Compound A which was prepared by the same process was measured by NMR and MS.
1H-NMR (DMSO-d 6) δ: 11.59 (br s, 1H), 8.08 (s, 1H), 7.11 (dd, 1H, J = 3.5, 2.5 Hz), 6.58 (dd, 1H, J = 3.5, 1.8 Hz), 4.16 (t, 1H, J = 8.3 Hz), 4.09-3.93 (m, 3H), 3.84-3.73 (m, 1H), 3.71 (d, 1H, J = 19.0 Hz), 3.65 (d, 1H, J = 19.0 Hz), 3.58 (dd, 1H, J = 8.2, 5.9 Hz), 2.69-2.59 (m, 2H), 2.23-2.13 (m, 1H), 1.12 (d, 3H, J = 7.2 Hz).
MS: m/z = 311 [M+H] +
[0151]
Using Compound A, which was prepared by the same method, the single crystal X-ray analysis was carried out.
(1) Preparation of Single crystal
To 10 mg of Compound A in a LaPha ROBO Vial(R) 2.0 mL wide-mouthed vial was added 0.5 mL of chloroform. The vial was covered with a cap, in which Compound A was completely dissolved. In order to evaporate the solvent slowly, a hole was made on the septum attached in the cap with a needle of a TERUMO(R) syringe, and the vial was still stood at room temperature. The resulting single crystal was used in the structural analysis.
(2) Measuring instrument
Beam line: SPring-8 BL32B2
Detector: Rigaku R-AXIS V diffractometer
(3) Measuring method
The radiant light of 0.71068Å was irradiated to the single crystal to measure X-ray diffraction data.
(4) Assay method
Using the X-ray anomalous scattering effect of the chlorine atom in the resulting Compound A chloroform-solvate, the absolute configuration of Compound A was identified as (3S,4R). Based on the obtained absolute configuration of Compound A, the absolute configurations of each process intermediate were identified.
(1) Preparation of Single crystal
To 10 mg of Compound A in a LaPha ROBO Vial(R) 2.0 mL wide-mouthed vial was added 0.5 mL of chloroform. The vial was covered with a cap, in which Compound A was completely dissolved. In order to evaporate the solvent slowly, a hole was made on the septum attached in the cap with a needle of a TERUMO(R) syringe, and the vial was still stood at room temperature. The resulting single crystal was used in the structural analysis.
(2) Measuring instrument
Beam line: SPring-8 BL32B2
Detector: Rigaku R-AXIS V diffractometer
(3) Measuring method
The radiant light of 0.71068Å was irradiated to the single crystal to measure X-ray diffraction data.
(4) Assay method
Using the X-ray anomalous scattering effect of the chlorine atom in the resulting Compound A chloroform-solvate, the absolute configuration of Compound A was identified as (3S,4R). Based on the obtained absolute configuration of Compound A, the absolute configurations of each process intermediate were identified.
REFERENCES
1: Nakagawa H, Nemoto O, Yamada H, Nagata T, Ninomiya N. Phase 1 studies to assess the safety, tolerability and pharmacokinetics of JTE-052 (a novel Janus kinase inhibitor) ointment in Japanese healthy volunteers and patients with atopic dermatitis. J Dermatol. 2018 Jun;45(6):701-709. doi: 10.1111/1346-8138.14322. Epub 2018 Apr 17. PubMed PMID: 29665062; PubMed Central PMCID: PMC6001687.
2: Nakagawa H, Nemoto O, Igarashi A, Nagata T. Efficacy and safety of topical JTE-052, a Janus kinase inhibitor, in Japanese adult patients with moderate-to-severe atopic dermatitis: a phase II, multicentre, randomized, vehicle-controlled clinical study. Br J Dermatol. 2018 Feb;178(2):424-432. doi: 10.1111/bjd.16014. Epub 2018 Jan 15. PubMed PMID: 28960254.
3: Tanimoto A, Shinozaki Y, Yamamoto Y, Katsuda Y, Taniai-Riya E, Toyoda K, Kakimoto K, Kimoto Y, Amano W, Konishi N, Hayashi M. A novel JAK inhibitor JTE-052 reduces skin inflammation and ameliorates chronic dermatitis in rodent models: Comparison with conventional therapeutic agents. Exp Dermatol. 2018 Jan;27(1):22-29. doi: 10.1111/exd.13370. Epub 2017 Jul 3. PubMed PMID: 28423239.
4: Nomura T, Kabashima K. Advances in atopic dermatitis in 2015. J Allergy Clin Immunol. 2016 Dec;138(6):1548-1555. doi: 10.1016/j.jaci.2016.10.004. Review. PubMed PMID: 27931536.
5: Amano W, Nakajima S, Yamamoto Y, Tanimoto A, Matsushita M, Miyachi Y, Kabashima K. JAK inhibitor JTE-052 regulates contact hypersensitivity by downmodulating T cell activation and differentiation. J Dermatol Sci. 2016 Dec;84(3):258-265. doi: 10.1016/j.jdermsci.2016.09.007. Epub 2016 Sep 13. PubMed PMID: 27665390.
6: Tanimoto A, Shinozaki Y, Nozawa K, Kimoto Y, Amano W, Matsuo A, Yamaguchi T, Matsushita M. Improvement of spontaneous locomotor activity with JAK inhibition by JTE-052 in rat adjuvant-induced arthritis. BMC Musculoskelet Disord. 2015 Nov 6;16:339. doi: 10.1186/s12891-015-0802-0. PubMed PMID: 26546348; PubMed Central PMCID: PMC4636776.
7: Amano W, Nakajima S, Kunugi H, Numata Y, Kitoh A, Egawa G, Dainichi T, Honda T, Otsuka A, Kimoto Y, Yamamoto Y, Tanimoto A, Matsushita M, Miyachi Y, Kabashima K. The Janus kinase inhibitor JTE-052 improves skin barrier function through suppressing signal transducer and activator of transcription 3 signaling. J Allergy Clin Immunol. 2015 Sep;136(3):667-677.e7. doi: 10.1016/j.jaci.2015.03.051. Epub 2015 Jun 24. PubMed PMID: 26115905.
8: Tanimoto A, Ogawa Y, Oki C, Kimoto Y, Nozawa K, Amano W, Noji S, Shiozaki M, Matsuo A, Shinozaki Y, Matsushita M. Pharmacological properties of JTE-052: a novel potent JAK inhibitor that suppresses various inflammatory responses in vitro and in vivo. Inflamm Res. 2015 Jan;64(1):41-51. doi: 10.1007/s00011-014-0782-9. Epub 2014 Nov 12. PubMed PMID: 25387665; PubMed Central PMCID: PMC4286029.
References
- “Anzupgo EPAR”. European Medicines Agency. 25 July 2024. Retrieved 25 July 2024. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- “Anzupgo PI”. Union Register of medicinal products. 23 September 2024. Retrieved 27 September 2024.
- Dhillon S (April 2020). “Delgocitinib: First Approval”. Drugs. 80 (6): 609–615. doi:10.1007/s40265-020-01291-2. PMID 32166597. S2CID 212681247.
- Park B (5 August 2020). “Delgocitinib Cream Gets Fast Track Status for Chronic Hand Eczema”. empr.com.
- Szalus K, Trzeciak M, Nowicki RJ (November 2020). “JAK-STAT Inhibitors in Atopic Dermatitis from Pathogenesis to Clinical Trials Results”. Microorganisms. 8 (11): 1743. doi:10.3390/microorganisms8111743. PMC 7694787. PMID 33172122.
- “Meeting highlights from the Committee for Medicinal Products for Human Use (CHMP) 22-25 July 2024”. European Medicines Agency (Press release). 25 July 2024. Retrieved 29 July 2024.
/////////Delgocitinib, デルゴシチニブ , JAPAN 2020, 2020 APPROVALS, Corectim, UNII-9L0Q8KK220, JTE-052, 9L0Q8KK220, LEO 124249A, LEO 124249, HY-109053, CS-0031558, D11046, GTPL9619, JTE-052A, JTE052, LP-0133 , ROH-201, atopic dermatitis
CC1CN(C12CCN(C2)C3=NC=NC4=C3C=CN4)C(=O)CC#N

 |
|
| Clinical data | |
|---|---|
| Trade names | Corectim, others |
| Other names | JTE-052; JTE-052A |
| ATC code | |
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| CompTox Dashboard (EPA) | |
| Chemical and physical data | |
| Formula | C16H18N6O |
| Molar mass | 310.361 g·mol−1 |
| 3D model (JSmol) | |
https://pubs.acs.org/doi/10.1021/acs.oprd.1c00031
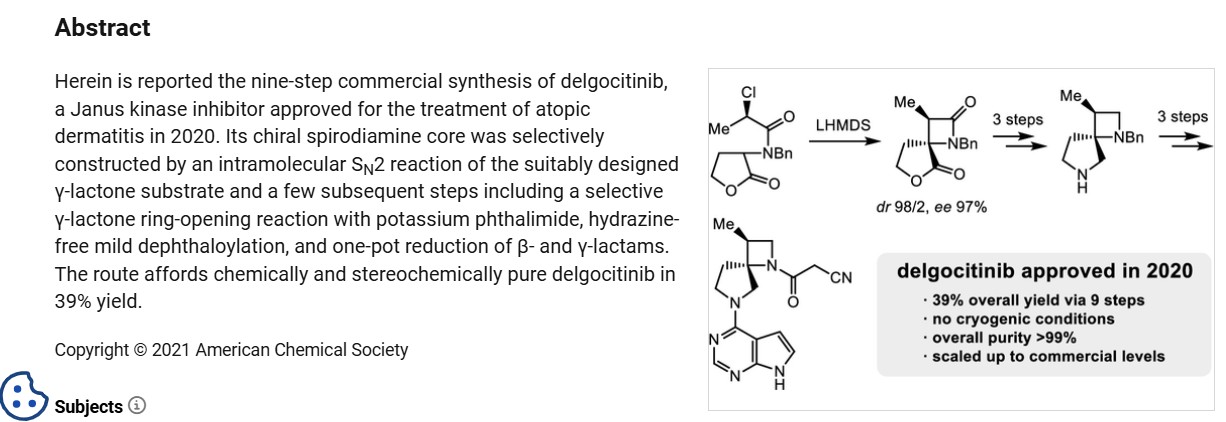
https://www.chemicalbook.com/article/synthesis-of-delgocitinib.htm
Synthesis of Delgocitinib
Delgocitinib is synthesised using bromolactone as raw material by chemical reaction. The specific synthesis steps are as follows:

Synthesis of Delgocitinib
Dec 26,2023
Synthesis of Delgocitinib
Delgocitinib is synthesised using bromolactone as raw material by chemical reaction. The specific synthesis steps are as follows:

A stereocontrolled kilogram scale synthesis of delgocitinib has been disclosed, beginning with an SN2 reaction involving bromolactone 128 and benzyl amine to provide α-amino lactone 129, which was isolated as the HCl salt after precipitation from hydrochloric acid in ethyl acetate. Amine 129 was then acylated with enantiomerically pure acid chloride 131 (prepared by thionyl chloride treatment of commercial acid 130) to furnish lactone 132. In the crucial spirocyclic ring ringforming sequence of the synthesis, lactone 132 was treated with LHMDS to form an enolate that underwent SN2 displacement of the chloride, forming the spirolactone 133 and establishing both stereocenters with 98:2 dr and 96% ee.
The lactone ring of 133 was then opened by an attack of potassium phthalimide on the γ- carbon, and the resulting carboxylic acid was converted to the ethyl ester by treatment with ethyl iodide. Finally, treatment with diethylenetriamine released phthalimide, providing a free amine for subsequent cyclization to spirolactam 134 via the corresponding ethyl ester intermediate. This sequence took place in 80% yield over four steps and provided the spirolactam in >99% de after recrystallization.
The carbonyl groups within spirolactam 134 were then reduced with lithium aluminum hydride and aluminum chloride in THF, and the resulting diamine 135 was crystallized as a succinic acid salt in 86% yield. The SNAr reaction of 135 with chloropyrrolopyrimidine 136 followed by hydrogenative removal of the benzyl protecting group provided amine 137 in 92% yield over 2 steps. Finally, amine 137 was acylated with cyanoacetyl pyrazole 138 and recrystallized from n-butanol with 3 wt % BHT to provide delgocitinib in 86% yield, >99% ee, and >99% de.
Dotinurad ドチヌラド
Dotinurad
ドチヌラド
(3,5-dichloro-4-hydroxyphenyl)-(1,1-dioxo-2H-1,3-benzothiazol-3-yl)methanone
| Formula |
C14H9Cl2NO4S
|
|---|---|
| CAS |
1285572-51-1
|
| Mol weight |
358.1966
|
PMDA, Urece, APROVED JAPAN 2020/1/23, Antihyperuricemic
305EB53128UNII-305EB53128
1285572-51-1,
Dotinurad is a urate transporter inhibitor.
Patents
WO 2011040449

https://patents.google.com/patent/WO2011040449A1/en
Uric acid is produced by metabolizing a purine produced by the degradation of a nucleic acid in the body and adenosine triphosphate (ATP), which is an energy source of the living body, to xanthine, and further undergoes oxidation by xanthine oxidase or xanthine dehydrogenase. In humans, uric acid (dissociation constant pKa = 5.75) is the final metabolite of purines and exists in the body as free forms or salts.
Uric acid is normally excreted in the urine, but when uric acid production exceeds excretion and blood uric acid increases, hyperuricemia occurs. If a state in which the blood level of uric acid exceeds the upper limit of solubility (about 7 mg / dL) continues for a long period of time, crystals of urate (usually sodium salt) precipitate.
In the blood, the precipitated crystals deposit on cartilage tissue and joints, form precipitates and become gouty nodules, causing acute gouty arthritis, and then transition to chronic gouty arthritis.
When uric acid crystals are precipitated in urine, renal disorders such as interstitial nephritis (gouty kidney), urinary calculi, and the like are caused. After the seizures of acute gouty arthritis have subsided, drug therapy is given along with lifestyle improvement guidance to correct hyperuricemia.
Correcting hyperuricemia and appropriately managing uric acid levels are also important in preventing acute gouty arthritis, gouty kidneys, urinary tract stones, and the like.
Hyperuricemia is considered to be associated with a high rate of lifestyle-related diseases such as obesity, hyperlipidemia, impaired glucose tolerance, and hypertension (see Non-Patent Document 1 (pp7-9)). Increased serum uric acid levels are positively related to cardiovascular mortality, and higher serum uric acid levels increase mortality due to ischemic heart disease. It has been suggested that it is associated with the risk of death from disease (see Non-Patent Document 2).
Furthermore, serum uric acid levels have also been shown to be a powerful risk factor for myocardial infarction and stroke (see Non-Patent Document 3). To date, hyperuricemia is obesity, hyperlipidemia, dyslipidemia, impaired glucose tolerance, diabetes, metabolic syndrome, kidney disease (eg, renal failure, urine protein, end-stage renal disease (ESRD), etc.), heart It is known to be associated with vascular diseases (for example, hypertension, coronary artery disease, carotid artery disease, vascular endothelial disorder, arteriosclerosis, cardiac hypertrophy, cerebrovascular disease, etc.) or risk factors of these diseases (Non-Patent Documents 2 to 11) reference). In cerebrovascular dementia, it has also been reported that the concentration of uric acid in the cerebrospinal cord is increased (see Non-Patent Document 12).
Under such circumstances, it has been suggested that the treatment for lowering the blood uric acid level may delay the progression of kidney disease and reduce the risk of cardiovascular disease (Non-Patent Documents 5, 8, 13, 14), it has been reported that it should also be applied to asymptomatic hyperuricemia (see Non-Patent Document 14).
Therefore, reducing the blood uric acid level in the above-mentioned diseases is effective for the treatment or prevention of these diseases, and is considered to be important in terms of preventing recurrence of cardiovascular accidents and maintaining renal function.
The main factors that increase blood uric acid levels include excessive uric acid production and decreased uric acid excretion. Therefore, as a method for lowering blood uric acid level, it is conceivable to suppress the production of uric acid or promote the excretion of uric acid, and allopurinol is a drug having the former mechanism of action (uric acid production inhibitor). Benzbromarone, probenecid, JP-A 2006-176505 (Patent Document 1) and the like are known as drugs having the latter mechanism of action (uric acid excretion promoters).
According to the Japanese guidelines for treatment of hyperuricemia and gout, in principle, uric acid excretion-promoting agents are applied to hyperuricemia-reducing types and uric acid production-inhibiting agents are applied to excessive uric acid production types, respectively. (See Non-Patent Document 1 (pp31-32)).
In Japan, it is said that about 60% of hyperuricemia patients have a reduced uric acid excretion type, and about 25% are a mixed type of reduced uric acid excretion type and excessive uric acid production type (Non-patent Document 15). About 85% of the patients showed a decrease in uric acid excretion, and the average value of uric acid clearance was significantly lower than that of healthy individuals even in patients with excessive uric acid production, and the decrease in uric acid excretion was fundamental in all gout patients. Is also reported (Non-Patent Document 16).
Therefore, in hyperuricemia (especially gout), treatment for patients with reduced uric acid excretion is considered to be important, and the existence significance of uric acid excretion promoters is extremely large.
Among the major uric acid excretion promoters, probenecid is weakly used and is rarely used because of its gastrointestinal tract disorders and interactions with other drugs. On the other hand, severe liver damage has been reported for benzbromarone, which has a strong uric acid excretion promoting action and is widely used in Japan as a uric acid excretion promoting drug (see Non-Patent Document 17).
Benzbromarone or its analogs inhibit mitochondrial respiratory chain enzyme complex activity, uncoupling action, respiration inhibition, fatty acid β oxidation inhibition, mitochondrial membrane potential reduction, apoptosis, generation of reactive oxygen species, etc. Has been suggested to be involved in the development of liver damage (see Non-Patent Documents 18 and 19). Hexahydrate, which is the active body of benzbromarone, is also toxic to mitochondria.
Furthermore, benzbromarone has an inhibitory action on cytochrome P450 (CYP), which is a drug metabolizing enzyme. In particular, the inhibition against CYP2C9 is very strong, suggesting the possibility of causing a pharmacokinetic drug interaction (non-) (See Patent Documents 20 and 21).
Furthermore, although a nitrogen-containing fused ring compound having a URAT1 inhibitory action, which is a kind of uric acid transporter, and having a structure similar to that of the compound of the present invention is described in JP-A-2006-176505 (Patent Document 1), the effect is sufficient. In addition, no practical uric acid excretion promoter has been developed yet.
Recently, it has been found that the uric acid excretion promoting action depends on the urinary concentration of a drug having the same action, that is, the uric acid excretion promoting drug is excreted in the urine and exhibits a medicinal effect (Patent Document 2). Non-Patent Documents 22 and 23).
Therefore, a stronger pharmacological effect is expected if it is a uric acid excretion promoter that is excreted more in the urine, but the above existing uric acid excretion promoters have a very low concentration in urine, and a satisfactory activity can be obtained sufficiently. I can’t say that.
Regarding the urinary excretion of drugs, it is assumed that the administered drug is excreted as it is as an unchanged form or converted into an active metabolite and excreted. In the latter case, the active metabolite is produced. There is a risk that the individual difference in the amount becomes large, and in order to obtain stable drug efficacy and safety, a drug excreted as an unchanged substance is more desirable.
As described above, there is a demand for the development of a highly safe pharmaceutical having a high unchanged body urine concentration and a remarkable uric acid excretion promoting action as compared with existing uric acid excretion promoting drugs.
JP 2006-176505 A WO2005 / 121112
Treatment Guidelines for Hyperuricemia and Gout (1st Edition) pp7-9 and pp31-32, Gout and Nucleic Acid Metabolism, Volume 26, Supplement 1, 2002 Japan Gout and Nucleic Acid Metabolism Society JAMA 283: 2404-2410 (2000) Stroke 37: 1503-1507 (2006) Nephrology 9: 394-399 (2004) Semin. Nephrol. 25: 43-49 (2005)J. Clin. Hypertens. 8: 510-518 (2006) J. Hypertens. 17: 869-872 (1999) Curr. Med. Res. Opin. 20: 369-379 (2004) Curr. Pharm. Des. 11: 4139-4143 (2005)Hypertension 45: 991-996 (2005) Arch. Intern. Med. 169: 342-350 (2009) J. Neural. Transm. Park Dis. Dement. Sect. 6: 119-126 (1993) Am. J. Kidney Dis. 47: 51-59 (2006) Hyperuricemia and gout 9: 61-65 (2001) Japanese clinical trials 54: 3230-3236 (1996) Japanese clinical trial 54: 3248-3255 (1996) J. Hepatol. 20: 376-379 (1994) J. Hepatol. 35: 628-636 (2001) Hepatology 41: 925-935 (2005) Saitama Medical University Journal (J. Saitama. Med. School) 30: 187-194 (2004) Drug Metab. Dispos. 31: 967-971 (2003) 42nd Annual Meeting of the Japanese Gout and Nucleic Acid Metabolism General Assembly Program / Abstracts, p59 (2009) ACR 2008 Annual Scientific Meeting, No. 28





PATENT
JP 2011074017
PATENT
WO 2018199277
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018199277
//////////Dotinurad, Antihyperuricemic, JAPAN 2020, 2020 APPROVALS , ドチヌラド , VOFLAIHEELWYGO-UHFFFAOYSA-N, HY-109031, CS-0030545
C1N(C2=CC=CC=C2S1(=O)=O)C(=O)C3=CC(=C(C(=C3)Cl)O)Cl
Teprotumumab-trbw

Tepezza (teprotumumab-trbw)
Company: Horizon Therapeutics plc
Date of Approval: January 21, 2020
Treatment for: Thyroid Eye Disease
UNIIY64GQ0KC0A
CAS number1036734-93-6
R-1507 / R1507 / RG-1507 / RG1507 / RO-4858696 / RO-4858696-000 / RO-4858696000 / RO4858696 / RO4858696-000 / RV-001 / RV001
Tepezza (teprotumumab-trbw) is a fully human monoclonal antibody (mAb) and a targeted inhibitor of the insulin-like growth factor 1 receptor (IGF-1R) for the treatment of active thyroid eye disease (TED).
FDA Approves Tepezza (teprotumumab-trbw) for the Treatment of Thyroid Eye Disease (TED) – January 21, 2020
Today, the U.S. Food and Drug Administration (FDA) approved Tepezza (teprotumumab-trbw) for the treatment of adults with thyroid eye disease, a rare condition where the muscles and fatty tissues behind the eye become inflamed, causing the eyes to be pushed forward and bulge outwards (proptosis). Today’s approval represents the first drug approved for the treatment of thyroid eye disease.
“Today’s approval marks an important milestone for the treatment of thyroid eye disease. Currently, there are very limited treatment options for this potentially debilitating disease. This treatment has the potential to alter the course of the disease, potentially sparing patients from needing multiple invasive surgeries by providing an alternative, non surgical treatment option,” said Wiley Chambers, M.D., deputy director of the Division of Transplant and Ophthalmology Products in the FDA’s Center for Drug Evaluation and Research. “Additionally, thyroid eye disease is a rare disease that impacts a small percentage of the population, and for a variety of reasons, treatments for rare diseases are often unavailable. This approval represents important progress in the approval of effective treatments for rare diseases, such as thyroid eye disease.”
Thyroid eye disease is associated with the outward bulging of the eye that can cause a variety of symptoms such as eye pain, double vision, light sensitivity or difficulty closing the eye. This disease impacts a relatively small number of Americans, with more women than men affected. Although this condition impacts relatively few individuals, thyroid eye disease can be incapacitating. For example, the troubling ocular symptoms can lead to the progressive inability of people with thyroid eye disease to perform important daily activities, such as driving or working.
Tepezza was approved based on the results of two studies (Study 1 and 2) consisting of a total of 170 patients with active thyroid eye disease who were randomized to either receive Tepezza or a placebo. Of the patients who were administered Tepezza, 71% in Study 1 and 83% in Study 2 demonstrated a greater than 2 millimeter reduction in proptosis (eye protrusion) as compared to 20% and 10% of subjects who received placebo, respectively.
The most common adverse reactions observed in patients treated with Tepezza are muscle spasm, nausea, alopecia (hair loss), diarrhea, fatigue, hyperglycemia (high blood sugar), hearing loss, dry skin, dysgeusia (altered sense of taste) and headache. Tepezza should not be used if pregnant, and women of child-bearing potential should have their pregnancy status verified prior to beginning treatment and should be counseled on pregnancy prevention during treatment and for 6 months following the last dose of Tepezza.
The FDA granted this application Priority Review, in addition to Fast Track and Breakthrough Therapy Designation. Additionally, Tepezza received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases or conditions. Development of this product was also in part supported by the FDA Orphan Products Grants Program, which provides grants for clinical studies on safety and efficacy of products for use in rare diseases or conditions.
The FDA granted the approval of Tepezza to Horizon Therapeutics Ireland DAC.
Teprotumumab (RG-1507), sold under the brand name Tepezza, is a medication used for the treatment of adults with thyroid eye disease, a rare condition where the muscles and fatty tissues behind the eye become inflamed, causing the eyes to be pushed forward and bulge outwards (proptosis).[1]
The most common adverse reactions observed in people treated with teprotumumab-trbw are muscle spasm, nausea, alopecia (hair loss), diarrhea, fatigue, hyperglycemia (high blood sugar), hearing loss, dry skin, dysgeusia (altered sense of taste) and headache.[1] Teprotumumab-trbw should not be used if pregnant, and women of child-bearing potential should have their pregnancy status verified prior to beginning treatment and should be counseled on pregnancy prevention during treatment and for six months following the last dose of teprotumumab-trbw.[1]
It is a human monoclonal antibody developed by Genmab and Roche. It binds to IGF-1R.
Teprotumumab was first investigated for the treatment of solid and hematologic tumors, including breast cancer, Hodgkin’s and non-Hodgkin’s lymphoma, non-small cell lung cancer and sarcoma.[2][3] Although results of phase I and early phase II trials showed promise, research for these indications were discontinued in 2009 by Roche. Phase II trials still in progress were allowed to complete, as the development was halted due to business prioritization rather than safety concerns.
Teprotumumab was subsequently licensed to River Vision Development Corporation in 2012 for research in the treatment of ophthalmic conditions. Horizon Pharma (now Horizon Therapeutics, from hereon Horizon) acquired RVDC in 2017, and will continue clinical trials.[4] It is in phase III trials for Graves’ ophthalmopathy (also known as thyroid eye disease (TED)) and phase I for diabetic macular edema.[5] It was granted Breakthrough Therapy, Orphan Drug Status and Fast Track designations by the FDA for Graves’ ophthalmopathy.[6]
In a multicenter randomized trial in patients with active Graves’ ophthalmopathy Teprotumumab was more effective than placebo in reducing the clinical activity score and proptosis.[7] In February 2019 Horizon announced results from a phase 3 confirmatory trial evaluating teprotumumab for the treatment of active thyroid eye disease (TED). The study met its primary endpoint, showing more patients treated with teprotumumab compared with placebo had a meaningful improvement in proptosis, or bulging of the eye: 82.9 percent of teprotumumab patients compared to 9.5 percent of placebo patients achieved the primary endpoint of a 2 mm or more reduction in proptosis (p<0.001). Proptosis is the main cause of morbidity in TED. All secondary endpoints were also met and the safety profile was consistent with the phase 2 study of teprotumumab in TED.[8] On 10th of July 2019 Horizon submitted a Biologics License Application (BLA) to the FDA for teprotumumab for the Treatment of Active Thyroid Eye Disease (TED). Horizon requested priority review for the application – if so granted (FDA has a 60-day review period to decide) it would result in a max. 6 month review process.[9]
History[edit]
Teprotumumab-trbw was approved for use in the United States in January 2020, for the treatment of adults with thyroid eye disease.[1]
Teprotumumab-trbw was approved based on the results of two studies (Study 1 and 2) consisting of a total of 170 patients with active thyroid eye disease who were randomized to either receive teprotumumab-trbw or a placebo.[1] Of the subjects who were administered Tepezza, 71% in Study 1 and 83% in Study 2 demonstrated a greater than two millimeter reduction in proptosis (eye protrusion) as compared to 20% and 10% of subjects who received placebo, respectively.[1]
The U.S. Food and Drug Administration (FDA) granted the application for teprotumumab-trbw fast track designation, breakthrough therapy designation, priority review designation, and orphan drug designation.[1] The FDA granted the approval of Tepezza to Horizon Therapeutics Ireland DAC.[1]
References
- ^ Jump up to:a b c d e f g h “FDA approves first treatment for thyroid eye disease”. U.S. Food and Drug Administration (FDA) (Press release). 21 January 2020. Retrieved 21 January 2020. This article incorporates text from this source, which is in the public domain.
- ^ https://clinicaltrials.gov/ct2/show/NCT01868997
- ^ http://adisinsight.springer.com/drugs/800015801
- ^ http://www.genmab.com/product-pipeline/products-in-development/teprotumumab
- ^ http://adisinsight.springer.com/drugs/800015801
- ^ http://www.genmab.com/product-pipeline/products-in-development/teprotumumab
- ^ Smith, TJ; Kahaly, GJ; Ezra, DG; Fleming, JC; Dailey, RA; Tang, RA; Harris, GJ; Antonelli, A; Salvi, M; Goldberg, RA; Gigantelli, JW; Couch, SM; Shriver, EM; Hayek, BR; Hink, EM; Woodward, RM; Gabriel, K; Magni, G; Douglas, RS (4 May 2017). “Teprotumumab for Thyroid-Associated Ophthalmopathy”. The New England Journal of Medicine. 376 (18): 1748–1761. doi:10.1056/NEJMoa1614949. PMC 5718164. PMID 28467880.
- ^ “Horizon Pharma plc Announces Phase 3 Confirmatory Trial Evaluating Teprotumumab (OPTIC) for the Treatment of Active Thyroid Eye Disease (TED) Met Primary and All Secondary Endpoints”. Horizon Pharma plc. Retrieved 22 March 2019.
- ^ “Horizon Therapeutics plc Submits Teprotumumab Biologics License Application (BLA) for the Treatment of Active Thyroid Eye Disease (TED)”. Horizon Therapeutics plc. Retrieved 27 August 2019.
External links
- “Teprotumumab”. Drug Information Portal. U.S. National Library of Medicine.
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | IGF-1R |
| Clinical data | |
| Other names | teprotumumab-trbw, RG-1507 |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| DrugBank | |
| ChemSpider |
|
| UNII | |
| KEGG | |
| ChEMBL | |
| ECHA InfoCard | 100.081.384 |
| Chemical and physical data | |
| Formula | C6476H10012N1748O2000S40 |
| Molar mass | 145.6 kg/mol g·mol−1 |
/////////Teprotumumab-trbw, APPROVALS 2020, FDA 2020, ORPHAN, BLA, fast track designation, breakthrough therapy designation, priority review designation, and orphan drug designation, Tepezza, Horizon Therapeutics, MONOCLONAL ANTIBODY, 2020 APPROVALS, active thyroid eye disease, Teprotumumab
https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-thyroid-eye-disease

{kind=link}
{kind=link}