
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

Potential diabetes breakthrough Harvard researchers discover hormone that spurs beta cell production

Betatrophin causes a specific increase in pancreatic β cell replication. Betatrophin is a secreted protein expressed in liver and fat. The increase in β cell replication and mass improves glycemic control. (Credit: Peng Yi, Ji-Sun Park, Douglas A. Melton/Cell)
Potential diabetes breakthrough
Researchers at the Harvard Stem Cell Institute (HSCI) have discovered a hormone that holds promise for a dramatically more effective treatment of type 2 diabetes, a metabolic illness afflicting an estimated 26 million Americans.
The researchers believe that the hormone might also have a role in treating type 1, or juvenile, diabetes.
The work was published by the journal Cell.
The hormone, called betatrophin, causes mice to produce insulin-secreting pancreatic beta cells at up to 30 times the normal rate. The new beta cells only produce insulin when called for by the body, offering the potential for the natural regulation of insulin and a great reduction in the complications associated with diabetes, the leading medical cause of amputations and non-genetic loss of vision.
The researchers who discovered betatrophin, HSCI co-director Doug Melton and postdoctoral fellow Peng Yi, caution that much work remains to be done before it could be used as a treatment in humans. But the results of their work, which was supported in large part by a federal research grant, already have attracted the attention of drug manufacturers.
“If this could be used in people,” said Melton, Harvard’s Xander University Professor and co-chair of the University’s Department of Stem Cell and Regenerative Biology, “it could eventually mean that instead of taking insulin injections three times a day, you might take an injection of this hormone once a week or once a month, or in the best case maybe even once a year.”
Type 2 diabetes, a disease associated with the national obesity epidemic, is usually caused by a combination of excess weight and lack of exercise. It causes patients to slowly lose beta cells and the ability to produce adequate insulin. One recent study has estimated that diabetes treatment and complications cost the United States $218 billion annually, or about 10 percent of the nation’s entire health bill.
“Our idea here is relatively simple,” Melton said. “We would provide this hormone, the type 2 diabetic will make more of their own insulin-producing cells, and this will slow down, if not stop, the progression of their diabetes. I’ve never seen any treatment that causes such an enormous leap in beta cell replication.”
CHINA MARKET-Takeda and Sanofi Sign Co-promotion Agreement to Expand Reach of Diabetes Treatment Alogliptin in China

ALOGLIPTIN
22.04.2013
• Alogliptin is a DPP-4 inhibitor that is designed to slow the inactivation of incretin hormones GLP-1 and GIP
• Agreement is part of Takeda’s strategy to complement capabilities through partnerships
• Agreement complements Sanofi’s diabetes portfolio and expand its offer of innovative diabetes treatment to Chinese patients
• The regulatory approval of alogliptin in China is expected in 2013
Shanghai, China, April 22, 2013 – Takeda and Sanofi today announced that they have entered into an agreement for the co-promotion of alogliptin in China for the treatment of type 2 diabetes. Alogliptin is Takeda’s new type 2 diabetes therapy, which has been filed for marketing authorization in China. It is a dipeptidyl peptidase-4 inhibitor (DPP-4i) that is designed to slow the inactivation of incretin hormones GLP-1 (glucagon-like peptide-1) and GIP (glucose-dependent insulinotropic peptide).
Under the terms of the agreement, Takeda will grant Sanofi the exclusive right to co-promote alogliptin in China. Sanofi will utilize its commercial capabilities and experience to promote the product in defined territories in China. The commercial terms of the agreement were not disclosed.
“Diabetes has become a major public health problem in China with a rapid increase in the prevalence over recent years. China is now the country with the largest number of people with diabetes,” said Haruhiko Hirate, Corporate Officer and Head of North Asia of Takeda. “The collaboration will expand our reach to Chinese physicians treating patients with type 2 diabetes. Both Takeda and Sanofi have a long history and significant experience in diabetes and this makes for a win-win partnership, as we work together to advance patient care and help to meet the needs of this growing patient population.”
“We are pleased to announce the collaboration with Takeda,” said Fabrice Baschiera, General Manager, Pharmaceutical Operations, Sanofi China. “Alogliptin reinforces the strategic focus of Sanofi in the diabetes field. The new addition of alogliptin strengthens our offer of innovative diabetes treatment to Chinese patients, which includes best-in-class oral and insulin drugs. We look forward to working with Takeda to make alogliptin more widely available to patients with type 2 diabetes in China,” added Mr. Baschiera.
Alogliptin was approved and marketed in Japan in 2010 under the brand name of Nesina®, where it is currently the best-selling DPP-4i for type 2 diabetes. It was approved by the U.S. FDA as a monotherapy and also in fixed-dose combination with pioglitazone (Oseni®) and metformin (Kazano®) in January 2013 for the treatment of type 2 diabetes in adults as adjuncts to diet and exercise.
In China, the rapid economic development has brought mass urbanization, changing diets and increasingly sedentary lifestyles. These factors greatly increase the risk of developing type 2 diabetes. China has the largest number of people with diabetes1, with approximately 92.4 million adults suffering from the disease, 60.7% of which are undiagnosed2. Over the next 20 years, an additional 40 million Chinese adults are expected to develop type 2 diabetes, surpassing the overall prevalence rate of the United States3.
Alogliptin is under registration review in China. Takeda is expecting to obtain the regulatory approval in 2013.
Notes
1 International Diabetes Federation. New diabetes figures in China: IDF press statement
2 Diabetes: Wenying Yang et al, N ENGL J MED, March 25, 2010;
3 Kantar Health. The Burden of the Complicated Type 2 Diabetes Patient in China.
About Alogliptin
Alogliptin is a DPP-4i for the treatment of type 2 diabetes as an adjunct to diet and exercise. DPP-4 is designed to slow the inactivation of incretin hormones GLP-1 and GIP. As a result, an increased amount of active incretins enables the pancreas to secrete insulin in a glucose-dependent manner, thereby assisting in the management of blood glucose levels. A New Drug Application (NDA) for NESINA (alogliptin) was approved in April 2010 by the Japanese Ministry of Health, Labour and Welfare for the treatment of type 2 diabetes, and the therapy is available under the same brand name in Japan. NESINA (alogliptin) was approved by the U.S. FDA as a monotherapy and also in fixed-dose combination with pioglitazone (OSENI) and metformin (KAZANO) in January 2013 for the treatment of type 2 diabetes in adults as adjuncts to diet and exercise.
About Type 2 Diabetes
Type 2 diabetes is the most common form of diabetes affecting millions of people globally. Type 2 diabetes is a progressive and chronic condition and patients should work with a health care professional to manage and monitor their disease. In addition to diet and exercise, patients often need to take multiple medications in order to help them manage their blood glucose levels. According to the International Diabetes Federation, the global health care expenditures for diabetes (both type 1 and 2) were estimated at $471.6 billion in 2012. By 2030, this number is projected to exceed $595 billion. China is now the country with the largest number of people with diabetes and 92.4 million adults are suffering from the disease.
About Takeda
Located in Osaka, Japan, Takeda is a research-based global company with its main focus on pharmaceuticals. As the largest pharmaceutical company in Japan and one of the global leaders of the industry, Takeda is committed to strive towards better health for patients worldwide through leading innovation in medicine. Additional information about Takeda is available through its corporate website,www.takeda.com.
About Sanofi
Sanofi, a global and diversified healthcare leader, discovers, develops and distributes therapeutic solutions focused on patients’ needs. Sanofi has core strengths in the field of healthcare with seven growth platforms: diabetes solutions, human vaccines, innovative drugs, consumer healthcare, emerging markets, animal health and the new Genzyme. Sanofi is listed in Paris (EURONEXT: SAN) and in New York (NYSE: SNY).
From a structural point has uracil (Uracil) structure, synthesis of these compounds are usually replaced with urea or urea and 1,3 – parents Electric reagent directly related ring, and substituted ureas from amines and isocyanate obtained. Compound 1 and methyl isocyanate urea derivatives obtained by reacting 2 , 2 and 1,3 – diethyl reaction 3 , 3 chlorination with phosphorus oxychloride to obtain 4 , 4 with a secondary amine 5 reaction of 6 , 6 de-Boc protected with acid reaction and generate benzoate Alogliptin benzoate.
EMA- Committee for Medicinal Products for Human Use (CHMP) adopted a positive opinion, recommending the granting of a marketing authorisation for Spedra, for the treatment of erectile dysfunction in adult men.

AVANAFIL, SPEDRA
On 25 April 2013, the Committee for Medicinal Products for Human Use (CHMP) adopted a positive opinion, recommending the granting of a marketing authorisation for the medicinal product Spedra, 50 mg, 100 mg, 200 mg, tablet intended for the treatment of erectile dysfunction in adult men.
The applicant for this medicinal product is VIVUS BV.
They may request a re-examination of any CHMP opinion, provided they notify the European Medicines Agency in writing of their intention within 15 days of receipt of the opinion. The active substance of Spedra is avanafil, a selective phosphodiesterase (PDE) type 5 inhibitor that leads to higher cyclic guanosine monophosphate (cGMP)-specific PDE5 levels. This enhances smooth muscle relaxation, which results in an inflow of blood into the penile tissues, thereby producing an erection. The benefit with Spedra is its effect on the ability of men with erectile dysfunction to achieve and maintain an erection sufficient for satisfactory sexual activity. It was observed in clinical trials that Spedra increased the percentage of sexual attempts resulting in successful intercourse by roughly 20-30% over placebo in the general population of adult men with erectile dysfunction. The most common side effects are headache, flushing, nasal and sinus congestion, dyspepsia and back pain. A pharmacovigilance plan for Spedra will be implemented as part of the marketing authorisation. The approved indication is: “Treatment of erectile dysfunction in adult men. In order for Spedra to be effective, sexual stimulation is required.” Detailed recommendations for the use of this product will be described in the summary of product characteristics (SmPC), which will be published in the European public assessment report (EPAR) and made available in all official European Union languages after the marketing authorisation has been granted by the European Commission. The CHMP, on the basis of quality, safety and efficacy data submitted, considers there to be a favourable benefit-to-risk balance for Spedra and therefore recommends the granting of the marketing authorisation.
Avanafil is a PDE5 inhibitor approved for erectile dysfunction on April 27, 2012.[1] Avanafil is known by the trademark name Stendra and was developed by Vivus Inc. It acts by inhibiting a specific phosphodiesterase type 5 enzyme which is found in various body tissues, but primarily in the corpus cavernosum penis, as well as the retina. Other similar drugs are sildenafil, tadalafil and vardenafil. The advantage of avanafil is that it has very fast onset of action compared with other PDE5 inhibitors.
Avanafil can be synthesized from a benzylamine derivative and a pyrimidine derivative:

“FDA approves Stendra for erectile dysfunction” (Press release). Food and Drug Administration (FDA). April 27, 2012.
Yamada, K.; Matsuki, K.; Omori, K.; Kikkawa, K.; 2004, U.S. Patent 6,797,709
A cutting that phenanthrene by a methylthio urea ( a ) and ethoxy methylene malonate ( 2 ) cyclization of 3 , chloride, phosphorus oxychloride get 4 , 4 with benzyl amine 5 occurred SNAr the reaction product after oxidation with mCPBA 6 . In pyrimidine, if the 2 – and 4 – positions are active simultaneously the same leaving group in the case, SNAr reaction occurs preferentially at 4 – position, but does not guarantee the 2 – side reaction does not occur. Here is an activity of the poor leaving group sulfide spans 2 – bit, and a good leaving group active chlorine occupy four – position, thus ensuring a high regioselectivity of the reaction. 4 – position after completion of the reaction, then the 2 – position of the group activation, where sulfide sulfoxide better than the leaving group. Amino alcohols 7 and 6 recurrence SNAr reaction 8 , 8 after alkaline hydrolysis and acid alpha amidation get that phenanthrene.
European Medicines Agency recommends approval of first treatment for pseudobulbar affect
26/04/2013

Dextromethorphan

Quinidine
European Medicines Agency recommends approval of first treatment for pseudobulbar affect
Medicine to help curb bouts of uncontrolled emotional expression in patients with certain neurological disorders
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended the granting of a marketing authorisation for Nuedexta, a medicine for the treatment of pseudobulbar affect in adults.
Pseudobulbar affect is a medical condition in which patients experience sudden and uncontrollable bouts of laughing or crying unrelated or disproportionate to their emotional state. It occurs when certain neurological disorders, such as multiple sclerosis (MS) and amyotrophic lateral sclerosis (ALS) or a stroke, damage areas of the brain that are involved in the control of normal expression of emotion. This damage can disrupt brain signalling, resulting in the alteration or loss of control of emotional expression.
Although pseudobulbar affect is a non-life-threatening condition, it can have a significant impact on an individual’s ability to interact normally in society and on their relationships with others. There is currently no treatment approved for pseudobulbar affect in the European Union.
Nuedexta is a combination of two known active substances, dextromethorphan hydrobromide and quinidine sulphate. In studies, treatment with these medicines significantly decreased episodes of involuntary, uncontrollable laughing or crying.
Pseudobulbar affect is observed in a number of neurological conditions. Nuedexta has currently only been studied in patients with MS and ALS. Nuedexta is not suitable for treating episodes of laughing or crying brought on by mood swings and not due to pseudobulbar affect.
The CHMP’s opinion on Nuedexta will now be sent to the European Commission for the adoption of a marketing authorisation.
Note
- The marketing authorisation holder for Nuedexta is Jenson Pharmaceutical Services Ltd.
Dextromethorphan/quinidine (trade name Nuedexta) is a combination drug containing the active ingredients dextromethorphan and quinidine. It was the first FDA-approved drug for the treatment of pseudobulbar affect (PBA).
In a 12 week randomized, double-blind trial, amyotrophic lateral sclerosis and multiple sclerosis patients with significant PBA were given either Nudexta 30/10 mg or placebo. In 326 randomized patients, the PBA-episode daily rate was 46.9% (p < 0.0001) lower for Nudexta than for placebo.
Nuedexta was approved in February 2011 and is marketed in the United States by Avanir Pharmaceuticals.
Celgene makes good start to 2013 as Revlimid hits $1 billion
April 26, 2013
Celgene Corp has posted a healthy set of financials for the first quarter despite a 4.1% decline in net income to $410.2 million, as product sales increased 15% to $1.43 billion. read more at————http://www.pharmatimes.com/Article/13-04-26/Celgene_makes_good_start_to_2013_as_Revlimid_hits_1_billion.aspx

(RS)-3-(4-amino-1-oxo 1,3-dihydro-2H-isoindol- 2-yl)piperidine-2,6-dione
Lenalidomide
REVLIMID® is an oral immunomodulatory drug marketed in the United States and many international markets, in combination with dexamethasone, for treatment of patients with multiple myeloma who have received at least one prior therapy. It is also marketed in the United States and certain international markets for the treatment of transfusion-dependent anemia due to low- or intermediate-1-risk myelodysplastic syndromes, or MDS, associated with a deletion 5q cytogenetic abnormality with or without additional cytogenetic abnormalitie.Revlimid Worldwide annual sales in 2011 was $3.2bLenalidomide (Revlimid) is a derivative of thalidomideintroduced in 2004.It was initially intended as a treatment for multiple myeloma, for which thalidomide is an accepted therapeutic treatment. Lenalidomide has also shown efficacy in the class of hematological disorders known as myelodysplastic syndromes (MDS). Lenalidomide has significantly improved overall survival in myeloma (which generally carries a poor prognosis), although toxicity remains an issue for users. [1]It costs $163,381 per year for the average patient.[2]
Lenalidomide has been used to successfully treat both inflammatory disorders and cancers in the past 10 years. There are multiple mechanisms of action, and they can be simplified by organizing them as mechanisms of action in vitro and in vivo.[3] In vitro, lenalidomide has three main activities: direct anti-tumor effect, inhibition of the microenvironment support for tumor cells, and immunomodulatory role. In vivo, lenalidomide induces tumor cell apoptosis directly and indirectly by inhibition of bone marrow stromal cell support, by anti-angiogenic and anti-osteoclastogenic effects, and by immunomodulatory activity. Lenalidomide has a broad range of activities that can be exploited to treat many hematologic and solid cancers.
- McCarthy; Philip L. McCarthy, Kouros Owzar, Craig C. Hofmeister, et al. (May 10, 2012). “Lenalidomide after Stem-Cell Transplantation for Multiple Myeloma”. N Engl J Med 366 (19): 1770–1781. doi:10.1056/NEJMoa1114083. PMID 22571201.
- Badros, Ashraf Z. Badros (May 10, 2012). “Lenalidomide in Myeloma — A High-Maintenance Friend”. N Engl J Med 366 (19): 1836–1838. doi:10.1056/NEJMe1202819. PMID 22571206.
- Vallet S, Palumbo A, Raje N, Boccadoro M, Anderson KC (July 2008). “Thalidomide and lenalidomide: Mechanism-based potential drug combinations”. Leukemia & Lymphoma 49 (7): 1238–45. doi:10.1080/10428190802005191. PMID 18452080.
Shire move delays Intuniv generic until 2014

Guanfacine (brand name Tenex, and the extended release Intuniv) is a sympatholytic. It is a selective α2A receptor agonist. These receptors are concentrated heavily in the prefrontal cortex and the locus coeruleus, with the potential to improve attention abilities via modulating post-synaptic α2A receptors in the prefrontal cortex. Guanfacine lowers both systolic and diastolic blood pressure by activating the central nervous system α2A norepinephrine autoreceptors, which results in reduced peripheral sympathetic outflow and thus a reduction in peripheral sympathetic tone. Its side-effects are dose dependent, with practically no dryness of the mouth at doses of 2 mg and less
April 26, 2013
Shire has come to an agreement with drugmakers Actavis and Watson that lays to rest all pending litigation over their attempts to launch a generic form of the attention-deficit hyperactivity drug (ADHD) Intuniv (guanfacine hydrochloride) in the US.
Under the settlement, Shire has granted Actavis a license to make and market its version of Intuniv in the US from December 1 next year, in return for a 25% royalty on gross profit during the 180 day period of exclusivity.
read more at—–http://www.pharmatimes.com/Article/13-04-26/Shire_move_delays_Intuniv_generic_until_2014.aspx
TOXINS-Occurrence of ochratoxin A in Korean paprika

ochratoxin A
Occurrence of ochratoxin A in Korean red paprika

http://www.ncbi.nlm.nih.gov/pubmed/23605491
National Agricultural Products Quality Management Service, Seoul, 150-804, Korea, ahnjs@naqs.go.kr.
A large amount-260,000 tons-of red paprika is consumed annually in Korea, where the people prefer hot and pungent to sweet foods.
,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,
Occurrence of ochratoxin A in herbal drugs of Indian origin – a report.
Source
Medicinal Plant Research Laboratory, University Department of Botany, Bhagalpur University, 812 007, Bhagalpur, India.
Abstract
This paper contains a report of occurrence of ochratoxin A in some common herbal medicines collected from different store-houses and shop-keepers of Bihar, India. Of 129 samples of 9 plants, 55 were found to be contaminated with various levels of ochratoxin A. The level of ochratoxin A was found maximal in barks ofHolarrhena antidysenterica (1.14…
View original post 190 more words
TOXINS-Occurrence of ochratoxin A in herbal drugs of Indian origin – a report.

ochratoxin A
Occurrence of ochratoxin A in Korean red paprika

http://www.ncbi.nlm.nih.gov/pubmed/23605491
National Agricultural Products Quality Management Service, Seoul, 150-804, Korea, ahnjs@naqs.go.kr.
A large amount-260,000 tons-of red paprika is consumed annually in Korea, where the people prefer hot and pungent to sweet foods.
,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,,
Occurrence of ochratoxin A in herbal drugs of Indian origin – a report.
Source
Medicinal Plant Research Laboratory, University Department of Botany, Bhagalpur University, 812 007, Bhagalpur, India.
Abstract
This paper contains a report of occurrence of ochratoxin A in some common herbal medicines collected from different store-houses and shop-keepers of Bihar, India. Of 129 samples of 9 plants, 55 were found to be contaminated with various levels of ochratoxin A. The level of ochratoxin A was found maximal in barks ofHolarrhena antidysenterica (1.14 – 2.34 μg/g) whereas it was minimal in rhizomes ofTacca aspera (0.3 – 0.74 μg/g).Aspergillus ochraceus, A sulphureus and Penicillium viridicatum isolates obtained from drug samples were also examined for their toxigenic potentials. 19 isolates ofA ochraceus, 13 ofA sulphureus and 37 isolates ofP viridicatum were found to be toxigenic out of 67, 33, and 107 isolates, respectively. The ochratoxin A produced by Aochraceus was in the range of 0.09 to 2.44 μg/mL, byA sulphureus 0.1 to 1.76 μg/mL, and byP viridicatum 0.14 to 2.78 μg/mL of the culture filtrate.
Ochratoxin A, a toxin produced by Aspergillus ochraceus, Aspergillus carbonarius and Penicillium verrucosum, is one of the most abundant food-contaminating mycotoxins.It is also a frequent contaminant of water-damaged houses and of heating ducts.Human exposure can occur through consumption of contaminated food products, particularly contaminated grain and pork products, as well as coffee, wine grapes and dried grapes.[4][5][6] The toxin has been found in the tissues and organs of animals, including human blood and breast milk. Ochratoxin A, like most toxic substances, has large species- and sex-specific toxicological differences.
Drug Spotlight: Zioptan, Tafluprost, Merck,

 |
|---|
isopropyl (5Z)-7-{(1R,2R,3R,5S)-2-[(1E)-3,3-difluoro-4-phenoxybut-1-en-1-yl]-3,5-dihydroxycyclopentyl}hept-5-enoate,
Drug: Zioptan
Generic molecule: tafluprost
Company: Merck
Approval date: Feb. 10, 2012
The scoop: Merck says this is the first (get ready for a mouthful) preservative-free prostaglandin analog ophthalmic solution and is for treating elevated eye pressure in some patients with the most common form of glaucoma. Merck sells the ointment in the U.S. and most of Europe, while it licensed it to Japanese drugmaker Santen in Japan, Germany and northern Europe.
Tafluprost (trade names Taflotan, marketed by Santen Pharmaceutical Co. and Zioptan, by Merck (U.S.)) is a prostaglandin analogue used topically (as eye drops) to control the progression of glaucoma and in the management of ocular hypertension. It reduces http://en.wikipedia.org/wiki/Intraocular_pressure”; rel=”nofollow”>intraocular pressure by increasing the outflow of aqueous fluid from the eyes.[1][2]
Taflotan contains 15 µg/ml Tafluprost. Taflotan sine is a preservative-free, single-dose formulation containing 0.3 ml per dose.[3]

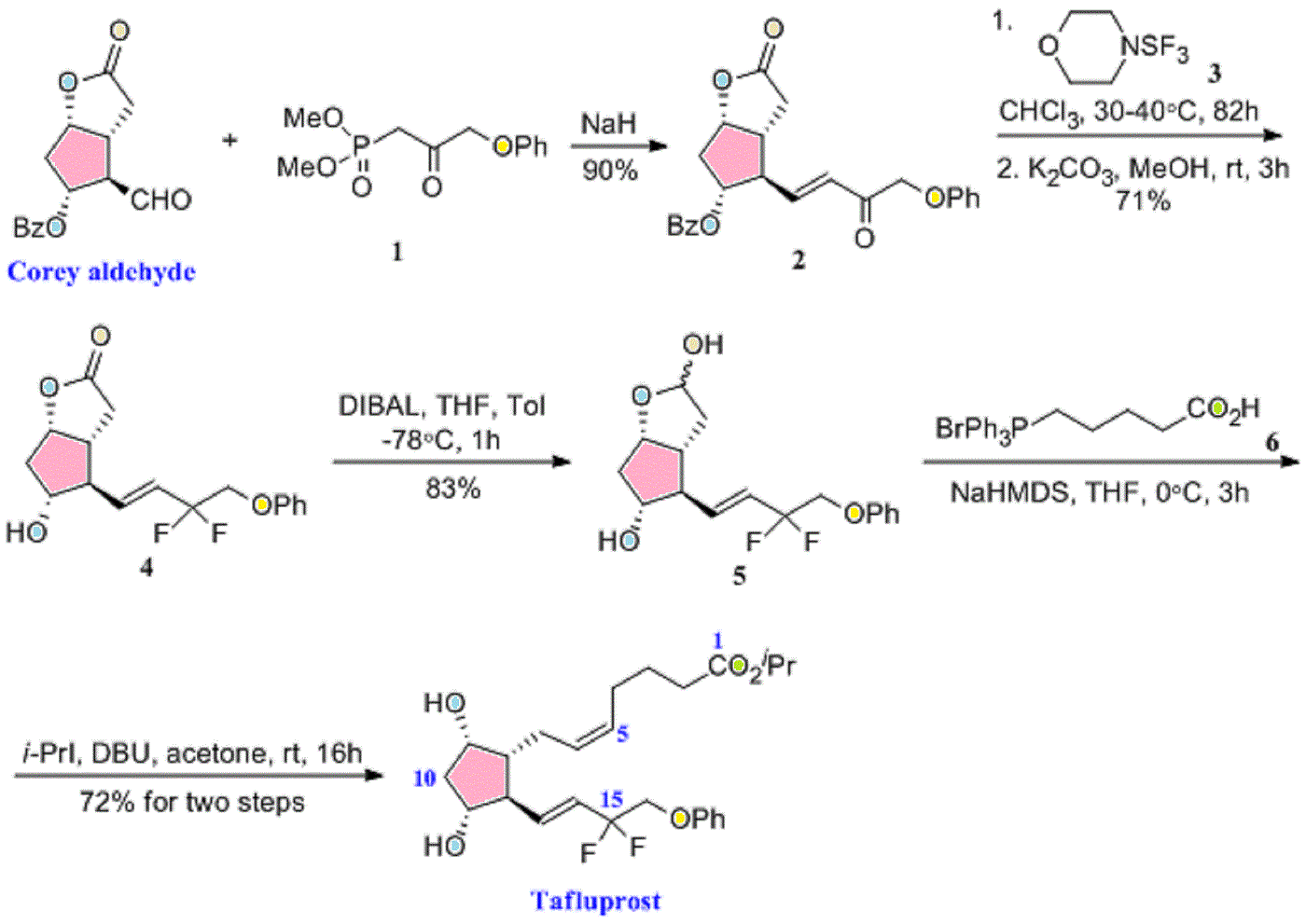
|
|
| Systematic (IUPAC) name | |
|---|---|
| isopropyl (5Z)-7-{(1R,2R,3R,5S)-2-[(1E)-3,3-difluoro-4-phenoxybut-1-en-1-yl]-3,5-dihydroxycyclopentyl}hept-5-enoate | |
| Clinical data | |
| Trade names | Saflutan, Taflotan, Tapros, Zioptan |
| AHFS/Drugs.com | International Drug Names |
| Pregnancy cat. | C (US) |
| Legal status | ℞-only (US) |
| Routes | Topical (eye drops) |
| Identifiers | |
| CAS number | 209860-87-7 |
| ATC code | S01EE05 |
| PubChem | CID 6433101 |
| ChemSpider | 8044182 |
| UNII | 1O6WQ6T7G3 |
| ChEBI | CHEBI:66899 |
| ChEMBL | CHEMBL1963683 |
| Chemical data | |
| Formula | C25H34F2O5 |
| Mol. mass | 452.531266 g/mol |


- Schubert-Zsilavecz, M, Wurglics, M, Neue Arzneimittel 2008/2009
- Santen Home Page
- Gelbe Liste (in German)

European Patent No. 8509621 discloses a process for the preparation of tafluprost. In the first step, (3afl,4fl,5fl,6aS)-4-formyl-2-oxohexahydro-2 — cyclopenta[b]furan-5-ylbenzoate (CTAF 1 (i)) is condensed with dimethyl (2-oxo-3- phenoxypropyl)-phosphonate in the presence of lithium chloride and triethylamine, to provide (3aft,4F?,5F?,6aS)-2-oxo-4-((£)-3-oxo-4-phenoxybut-1 -en-1 -yl)hexahydro-2H- cyclopenta[b]-furan-5-ylbenzoate (CTAF1 ). In the second step, CTAF 1 is reacted with morpholinosulfurtrifluoride to provide (3aH,4H,5H,6aS)-4-((£)-3,3-difluoro-4- phenoxybut-1 -en-1 -yl)-2-oxohexahydro-2 –cyclopenta-[b]furan-5-yl benzoate (CTAF2). CTAF 2 is debenzoylated by potassium carbonate in methanol, to provide (3aH,4H,5H,6aS)-4-((£)-3,3-difluoro-4-phenoxybut-1 -en-1 -yl)-5-hydroxyhexahydro-2H- cyclopenta[b]furan-2-one(CTAF 3), which is further reduced by diisobutyl aluminum hydride (DIBALH) to provide (3af?,4f?,5f?,6aS)-4-((£)-3,3-difluoro-4-phenoxybut-1 -en-1 – yl) hexahydro-2H-cyclopenta[b]furan-2,5-diol (CTAF 4). CTAF 4 is then treated with (4- carboxybutyl)triphenylphosphonium bromide, in the presence of potassium bis(trimethylsilyl)amide in THF, to provide (Z)-7-((1 f?,2f?,3f?,5S)-2-((£)-3,3-difluoro-4- phenoxybut-1 -en-1 -yl)-3,5-dihydroxycyclopentyl)hept-5-enoic acid (“tafluprost free acid,” CTAF5), which is reacted with isopropyl iodide in the presence of DBU to provide (Z)- isopropyl 7-((1 F?,2F?,3F?,5S)-2-((£)-3,3-difluoro-4-phenoxybut-1 -en-1 -yl)-3,5-dihydroxy- cyclopentyl)hept-5-enoate (“tafluprost,” CTAF 6). The reaction sequence is summarized in Scheme 1 .

CTAF 1(i)
CTAF 1 CTAF 2

U.S. Patent Application Publication No. 2010/0105775A1 discloses amino acid salts of prostaglandins. The application also discloses a process for the preparation of prostaglandins, comprising forming an amino acid salt of a prostaglandin and converting the amino acid salt to the prostaglandin.
EXAMPLE 1 : Preparation of CTAF 1

CTAF1(i)
CTAF1
To a stirred suspension of sodium hydride (60% dispersion in mineral oil, 0.217 g, 5.429 mmol) in THF (5 ml_) was added a solution of dimethyl (2-oxo-3- phenoxypropyl)phosphonate(1 .21 g, 4.705 mmol) in THF (2 ml_), over 15 minutes at 0- 5°C under a nitrogen atmosphere. The mixture was warmed to 25-35 , 0.5 M zinc chloride solution in THF (9.4 ml_, 4.705 mmol) was added over 10 minutes, and then the mixture was stirred for 15 minutes at 25-35<€. CTAF1 (i) (3af?,4F?,5F?,6aS)-4-formyl-2- oxohexahydro-2 –cyclopenta[b]furan-5-yl benzoate (1 g) in dichloromethane (10 ml_) was added over 5 minutes at 25-35 °C. The temperature was raised to 35-40 °C and the mixture was stirred for 2hours under a nitrogen atmosphere. The mixture was cooled to 15°C and the reaction was quenched by adding acetic acid (0.2 mL), followed by adding saturated ammonium chloride solution (10 mL), and further stirring for 15 minutes. The organic layer was separated and the aqueous layer was extracted with ethyl acetate (5 mL). The combined organic layers were evaporated under reduced pressure below 50°C. The crude product was purified by column chromatography on silica gel (100-200 mesh) with 30% ethyl acetate in hexane, to afford the title compound (0.9 g, 61 %yield).
EXAMPLE 2: Preparation of CTAF 2

CTAF1 CTAF2
To a stirred solution of CTAF1 (5 g, 0.0123 mol) in dichloromethane (100 mL) was added diethylaminosulfurtrifluoride (13 mL, 0.09841 mol) at 0-5 °C under a nitrogen atmosphere. The temperature was raised to 25-35 °C and maintained for 24 hours under a nitrogen atmosphere at the same temperature. The mass was slowly added into a saturated sodium bicarbonate solution (75 mL) at 0-5 °C. Temperature was raised to 25- 35 °C, the layers were separated, and the aqueous layer was extracted with dichloromethane (2×25 mL). The combined organic layer was washed with water (25mL) and dried over sodium sulfate (5 g). The organic layer was evaporated to dryness under reduced pressure below 40 °C. The crude product was purified by column chromatography on silica gel (100-200 mesh) with 30% ethyl acetate in hexane, to afford the title compound (4.2 g, 79% yield). EXAMPLE 3: Preparation of CTAF 4

CTAF 2 CTAF 4
CTAF 2 (2.30 g, 5.37 mmol) was dissolved in toluene (25 mL) and the solution was cooled to -65 °C under nitrogen. Diisobutyl aluminum hydride (1 .5 M in toluene, 1 1 .8 mL, 17.7mmol) was added over 15 minutes at -61 to -65 . The mixture was stirred for 3hours and then the reaction was quenched by adding methanol (1 .5 mL). Sulfuric acid (1 M, 25 mL) was added and the temperature rose to -20°C during the addition. Methyl t-butyl ether (MTBE) (10 mL) was added and the mixture was allowed to warm to room temperature. The organic phase was separated and the aqueous phase was extracted with MTBE (2x 10 mL). The combined organic phase was washed with water (10 mL), saturated aqueous sodium bicarbonate (10 mL), and then brine (10 mL). The washes were back-extracted with MTBE (10 mL). The combined organic phases were dried with magnesium sulfate, filtered, and evaporated to give a colourless oil (2.20 g). The crude product was chromatographed on silica (60 g), eluting with a mixture of ethyl acetate and heptane (2:1 by volume), and then with ethyl acetate, to give CTAF 4 as a colourless oil (1 .71 g, 97% yield).
EXAMPLE 4: Preparation of CTAF 2

CTAF1 CTAF2
To a stirred solution of CTAF1 (20 g, 0.0492 mol) in dichloromethane(400 mL) was added diethylaminosulfurtrifluoride (52 mL, 0.393 mol) at 0-10°C under a nitrogen atmosphere. The temperature was raised to 25-35 and maintained for 96hours under a nitrogen atmosphere at that temperature. The mass was slowly added to a saturated NaHCOs solution (600 mL) at 0-10°C. The mixture was heated to 25-35 <€ and filtered through aCelite bed. The layers were separated and the aqueous layer was extracted with DCM (2×100 mL). The combined organic layer was washed with 10% NaCI solution (100 mL) and evaporated to dryness under reduced pressure below 40°C. The residue was purified by column chromatography on silica gel (100-200 mesh) with 30% ethyl acetate in hexane.
Column purified material was dissolved in MTBE (80 mL) at 40°C and stirred for 30 minutes at that temperature. Diisopropyl ether (160 mL) was added at 35-40 and stirring continued for 30 minutes at 35-40 . Cooled the mass to 5-15°C and stirred for 30 minutes at that temperature. The solid was filtered, washed with a mixture of MTBE and diisopropyl ether (DIPE) (1 :2 by volume, 60 mL), and dried at 40°C under vacuum, to afford pure CTAF2 (12.0 g, 57% yield).
EXAMPLE 5: Preparation of CTAF 5

(4-Carboxybutyl)triphenylphosphonium bromide (10.32 g, 23.3 mmol, 4 eq) was suspended in THF (20 mL) under a nitrogen atmosphere and cooled to 5°C. NaHMDS solution (1 M in THF, 46.6 mL, 46.6 mmol, 8 eq) was added over 10 minutes. The red/orange mixture was stirred for 30 minutes. A solution of CTAF 4 (1 .90 g, 5.82 mmol) in THF (10 mL) was added over 30 minutes at 0-3 . The mixture was stirred for 1 .5hours and then the reaction was quenched by adding water (30 mL) and the masswas warmed to room temperature. The aqueous phase was separated and the organic phase was washed with water (20 mL). The combined aqueous phases were washed with MTBE (30 mL). The organic phases up to this point were discarded. The aqueous phase was acidified with 2M hydrochloric acid (14 mL, to pH 3-4) and extracted with ethyl acetate (2×30 mL). The combined ethyl acetate layers were washed with brine (20 mL), dried with magnesium sulfate, filtered, and evaporated under reduced pressure to give CTAF 5 asa yellow oil (8.60 g).
A 2.96 g sample was removed and the remainder (5.64 g) was chromatographed on silica (30 g) eluting with ethyl acetate to give purified CTAF 5 (1 .41 g) asa yellow oil. NMR analysis showed approximately 90% purity, remainder triphenyl phosphine oxide.
EXAMPLE 6: Preparation of CTAF 5 DCHA salt

CTAF 5 CTAF 5 DCHA sa t
CTAF5 (1 1 .72 g, 90% purity, 25.7 mmol, containing 1 .4% trans isomer) was dissolved in acetone (60 mL). Dicyclohexylamine (4.66 g, 25.7 mmol) was added and the mixture was stirred at room temperature overnight. The solid was filtered and washed with acetone (6 mL), then dried to give the DCHA salt (12.93 g, 85% yield, 0.29% trans-isomer).
A sample (7.03 g) was further purified by recrystallisation. It was dissolved in hot acetone (30 mL) and cooled to room temperature with stirring. The mixture was stirred for 3 hours, filtered and the solid was washed with acetone (3 mL) and dried to give a white solid (6.41 g, 91 % recovery, 0.1 1 % trans-isomer).
A PXRD pattern of the product is shown as Fig. 1 , obtained using copper Ka radiation. In the drawing, the y-axis is intensity units and the x-axis is the 2-theta angle, in degrees. EXAMPLE 7: Pre aration of CTAF 6

CTAF 5 DCH A sa l ^ I AI- O
CTAF 5 DCHA salt (5.80 g, 9.80 mmol) was suspended in ethyl acetate (20 mL). Sulfuric acid (1 M, 20 mL) was added and the mixture was stirred until a clear solution was obtained. The organic phase was separated and the aqueous phase was extracted with ethyl acetate (2×20 mL). The combined organic layers were washed with water (15 mL) and brine (15 mL), dried with magnesium sulfate, filtered, and evaporated. The residue was dissolved in acetone (40 mL) and charged into a jacketed vessel at 30°C. 1 ,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) (8.95 g, 58.8 mmol) was added, then 2- iodopropane (10.0 g, 58.8 mmol) was added, and the mixture was stirred for 20hours. The mixture was concentrated under reduced pressure and the residue was partitioned between ethyl acetate (30 mL) and aqueous potassium dihydrogen orthophosphate (8 g) in water (50 mL). The organic phase was separated and the aqueous was extracted with ethyl acetate (30 mL). The combined organic phases were washed with brine (20 mL), dried with magnesium sulfate, filtered and evaporated to give a yellow oil (4.83 g). The crude product was chromatographed on silica (130 g), eluting with a mixture of ethyl acetate and heptane (2:1 by volume), to give CTAF 6 (3.98 g, 90% yield) as a colorless oil.
DRUG SPOTLIGHT-Afinitor (everolimus) , Novartis:

Afinitor (everolimus)
40-O-(2-hydroxyethyl)-rapamycin
Company: Novartis
Approval Status: Approved July 2012
Treatment Area: hormone receptor-positive, HER2-negative breast cancer
Everolimus is a derivative of Rapamycin (sirolimus), and works similarly to Rapamycin as an mTOR (mammalian target of rapamycin) inhibitor. It is currently used as an immunosuppressant to prevent rejection of organ transplants. In a similar fashion to other mTOR inhibitors Everolimus’ effect is solely on the mTORC1 protein and not on the mTORC2 protein.
159351-69-6 CAS NO
BRANDS
| Afinitor | Novartis |
| Certican | Novartis |
| VOTUBIA | Novartis |
| Zortress | Novartis |
Afinitor (everolimus), an inhibitor of mTOR (mammalian target of rapamycin), is an antineoplastic agent.
Afinitor is specifically approved for the treatment of postmenopausal women with advanced hormone receptor-positive, HER2-negative breast cancer (advanced HR+ BC) in combination with exemestane, after failure of treatment with letrozole or anastrozole.
Afinitor is supplied as a tablet for oral administration. The recommended dose of Afinitor for breast cancer is 10 mg, to be taken once daily, at the same time every day, either consistently with food or consistently without food.
FDA Approval
The FDA approval of Afinitor for the treatment of advanced hormone receptor-positive, HER2-negative breast cancer was based on a randomized, double-blind, multicenter study in 724 postmenopausal women with estrogen receptor-positive, HER 2/neu-negative advanced breast cancer with recurrence or progression following prior therapy with letrozole or anastrozole.
Everolimus is indicated for the treatment of postmenopausal women with advanced hormone receptor-positive, HER2-negative breast cancer (advanced HR+ BC) in combination with exemestane, after failure of treatment with letrozole or anastrozole. Indicated for the treatment of adult patients with progressive neuroendocrine tumors of pancreatic origin (PNET) with unresectable, locally advanced or metastatic disease. Indicated for the treatment of adult patients with advanced renal cell carcinoma (RCC) after failure of treatment with sunitinib or sorafenib. Indicated for the treatment of adult patients with renal angiomyolipoma and tuberous sclerosis complex (TSC), not requiring immediate surgery. Indicated in pediatric and adult patients with tuberous sclerosis complex (TSC) for the treatment of subependymal giant cell astrocytoma (SEGA) that requires therapeutic intervention but cannot be curatively resected.
Everolimus (RAD-001) is the 40-O-(2-hydroxyethyl) derivative of sirolimus and works similarly to sirolimus as an inhibitor of mammalian target of rapamycin (mTOR).
It is currently used as an immunosuppressant to prevent rejection of organ transplants and treatment of renal cell cancer and other tumours. Much research has also been conducted on everolimus and other mTOR inhibitors for use in a number of cancers.
It is marketed by Novartis under the tradenames Zortress (USA) and Certican (Europe and other countries) in transplantation medicine, and Afinitor in oncology.
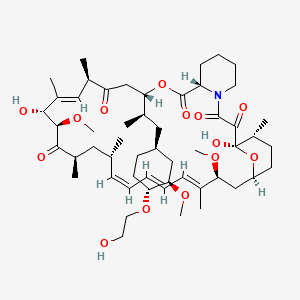
AFINITOR (everolimus), an inhibitor of mTOR, is an antineoplastic agent.
The chemical name of everolimus is (1R,9S,12S,15R,16E,18R,19R,21R,23S,24E,26E,28E,30S,32S,35R)-1,18- dihydroxy-12-{(1R)-2-[(1S,3R,4R)-4-(2-hydroxyethoxy)-3-methoxycyclohexyl]-1-methylethyl}-19,30-dimethoxy15,17,21,23,29,35-hexamethyl-11,36-dioxa-4-aza-tricyclo[30.3.1.04,9]hexatriaconta-16,24,26,28-tetraene-2,3,10,14,20pentaone.
The molecular formula is C53H83NO14 and the molecular weight is 958.2. The structural formula is:
 |
AFINITOR Tablets are supplied for oral administration and contain 2.5 mg, 5 mg, 7.5 mg, or 10 mg of everolimus. The tablets also contain anhydrous lactose, butylated hydroxytoluene, crospovidone, hypromellose, lactose monohydrate, and magnesium stearate as inactive ingredients.
AFINITOR DISPERZ (everolimus tablets for oral suspension) is supplied for oral administration and contains 2 mg, 3 mg, or 5 mg of everolimus. The tablets for oral suspension also contain butylated hydroxytoluene, colloidal silicon dioxide, crospovidone, hypromellose, lactose monohydrate, magnesium stearate, mannitol, and microcrystalline cellulose as inactive ingredients.
- R.N Formica Jra, K.M Lorberb, A.L Friedmanb, M.J Biaa, F Lakkisa, J.D Smitha, M.I Lorber (March 2004). “The evolving experience using everolimus in clinical transplantation”. Elsevier 36 (2): S495–S499.
- “Afinitor approved in US as first treatment for patients with advanced kidney cancer after failure of either sunitinib or sorafenib” (Press release). Novartis. 2009-03-30. Retrieved April 6, 2009.
- “Novartis receives US FDA approval for Zortress (everolimus) to prevent organ rejection in adult kidney transplant recipients” (Press release). Novartis. 2010-04-22. Retrieved April 26, 2010.
- “Novartis’ Afinitor Cleared by FDA for Treating SEGA Tumors in Tuberous Sclerosis”. 1 Nov 2010.
- http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm254350.htm
- “US FDA approves Novartis drug Afinitor for breast cancer”. 20 Jul 2012.
PATENTS
|
Country
|
Patent Number
|
Approved
|
Expires (estimated)
|
|---|---|---|---|
| United States | 6440990 | 1993-09-24 | 2013-09-24 |
| Canada | 2145383 | 2004-11-16 | 2013-09-24 |
| Canada | 2225960 | 2004-05-11 | 2016-07-12 |
| United States | 7297703 | 1999-12-06 | 2019-12-06 |
|
10-28-2011
|
METHODS OF TREATMENT
|
|
|
1-21-2011
|
ANTI-IGF1R
|
|
|
1-14-2011
|
HISTONE H2AX (HH2AX) BIOMARKER FOR FTI SENSITIVITY
|
|
|
3-24-2010
|
Thermal treatment of a drug eluting implantable medical device
|
|
|
1-13-2010
|
Therapeutic phosphonate compounds
|
|
|
10-21-2009
|
Processes for preparing water-soluble polyethylene glycol conjugates of macrolide immunosuppressants
|
|
|
10-16-2009
|
Heparin Prodrugs and Drug Delivery Stents Formed Therefrom
|
|
|
9-11-2009
|
PHOSPHONATE COMPOUNDS HAVING IMMUNO-MODULATORY ACTIVITY
|
|
|
12-31-2008
|
Phosphonate compounds having immuno-modulatory activity
|
|
|
10-8-2008
|
Anti-inflammatory phosphonate compounds
|
|
6-27-2008
|
Genes Involved in Neurodegenerative Conditions
|
|
|
10-24-2007
|
Fluid treatment of a polymeric coating on an implantable medical device
|
|
|
7-11-2007
|
Oxepane isomer of 42-O-(2-hydroxy)ethyl-rapamycin
|
|
|
2-9-2007
|
40-O-(2-hydroxy)ethyl-rapamycin coated stent
|
|
|
1-5-2007
|
Methods for treating neurofibromatosis 1
|
|
|
9-8-2006
|
Anti-inflammatory phosphonate compounds
|
| WO1994009010A1 | Sep 24, 1993 | Apr 28, 1994 | Sandoz Ag | O-alkylated rapamycin derivatives and their use, particularly as immunosuppressants |
| WO2007135397A1 * | May 18, 2007 | Nov 29, 2007 | Christoph Beckmann | 36 -des (3 -methoxy-4 -hydroxycyclohexyl) 36 – (3 -hydroxycycloheptyl) derivatives of rapamycin for the treatment of cancer and other disorders |
| EP0663916A1 | Sep 24, 1993 | Jul 26, 1995 | Novartis AG | O-alkylated rapamycin derivatives and their use, particularly as immunosuppressants |
| US5665772 | Sep 24, 1993 | Sep 9, 1997 | Sandoz Ltd. | O-alkylated rapamycin derivatives and their use, particularly as immunosuppressants |
| US20030125800 | Apr 24, 2002 | Jul 3, 2003 | Shulze John E. | Drug-delivery endovascular stent and method for treating restenosis |
…………………………………….
Rapamycin is a known macrolide antibiotic produced by Streptomvces hvgroscopicus. having the structure depicted in Formula A:

See, e.g., McAlpine, J.B., et al., J. Antibiotics (1991) 44: 688; Schreiber, S.L., et al., J. Am. Chem. Soc. (1991) J_13: 7433‘- US Patent No. 3 929 992. Rapamycin is an extremely potent immunosuppressant and has also been shown to have antitumor and antifungal activity. Its utility as a pharmaceutical, however, is restricted by its very low and variable bioavailabiiity as well as its high toxicity. Moreover, rapamycin is highly insoluble, making it difficult to formulate stable galenic compositions.
Everolimus, 40-O-(2-hydroxyethyl)-rapamycin of formula (1) is a synthetic derivative of rapamycin (sirolimus) of formula (2), which is produced by a certain bacteria strain and is also pharmaceutically active.

(1) (2)
Everolimus is marketed under the brand name Certican for the prevention of rejection episodes following heart and kidney transplantation, and under the brand name Afinitor for treatment of advanced kidney cancer.
Due to its complicated macrolide chemical structure, everolimus is, similarly as the parent rapamycin, an extremely unstable compound. It is sensitive, in particular, towards oxidation, including aerial oxidation. It is also unstable at temperatures higher than 25°C and at alkaline pH.
Everolimus and a process of making it have been disclosed in WO 94/09010
Synthesis

Alkylation of rapamycin (I) with 2-(tert-butyldimethylsilyloxy)ethyl triflate (II) by means of 2,6-lutidine in hot toluene gives the silylated target compound (III), which is deprotected by means of 1N HCl in methanol (1). (Scheme 21042401a) Manufacturer Novartis AG (CH). References 1. Cottens, S., Sedrani, R. (Sandoz-Refindungen VmbH; Sandoz-Patent GmbH; Sandoz Ltd.). O-Alkylated rapamycin derivatives and their use, particularly as immunosuppressants. EP 663916, EP 867438, JP 96502266, US 5665772, WO 9409010.EP 0663916; EP 0867438; JP 1996502266; JP 1999240884; US 5665772; WO 9409010
…………..
SYNTHESIS
https://www.google.com/patents/WO2012103960A1
(US 5,665,772, EP 663916). The process principle is shown in the scheme below, wherein the abbreviation RAP-OH has been used as an abbreviation for the rapamycin structure of formula (2) above, L is a leaving group and P is a trisubstituted silyl group serving as a OH- protective group.
RAP-OH + L-CH2-CH2-0-P — –> RAP-O-CH2-CH2-O-P — – > RAP-O-CH2-CH2-OH
(2) (4) (1)
Specifically, the L- group is a trifluoromethanesulfonate (triflate) group and the protective group P- is typically a tert-butyldimethylsilyloxy- group. Accordingly, the known useful reagent within the above general formula (3) for making everolimus from rapamycin is 2-(tert-butyldimethylsilyloxy)ethyl triflate of formula (3 A):

According to a known synthetic procedure disclosed in Example 8 of WO 94/09010 and in Example 1 of US application 2003/0125800, rapamycin (2) reacts in hot toluene and in the presence of 2,6-lutidine with a molar excess of the compound (3 A), which is charged in several portions, to form the t-butyldimethylsilyl-protected everolimus (4A). This compound is isolated and deprotected by means of IN aqueous HC1 in methanol. Crude everolimus is then purified by column chromatography. Yields were not reported.

(2) (3A) (4A) (1)
In an article of Moenius et al. (J. Labelled Cpd. Radiopharm. 43, 113-120 (2000)), which used the above process for making C14-labelled and tritiated everolimus, a diphenyl- tert.butylsilyloxy -protective group was used as the alkylation agent of formula (3B).

Only 8% yield of the corresponding compound (4B)

and 21% yield of the compound (1) have been reported.
Little is known about the compounds of the general formula (3) and methods of their preparation. The synthesis of the compound (3 A) was disclosed in Example 1 of US application 2003/0125800. It should be noted that specification of the reaction solvent in the key step B of this synthesis was omitted in the disclosure; however, the data about isolation of the product allow for estimation that such solvent is dichloromethane. Similarly also a second article of Moenius et al. (J. Labelled Cpd. Radiopharm.42, 29-41 (1999)) teaches that dichloromethane is the solvent in the reaction.
It appears that the compounds of formula (3) are very reactive, and thus also very unstable compounds. This is reflected by the fact that the yields of the reaction with rapamycine are very low and the compound (3) is charged in high molar extent. Methods how to monitor the reactivity and/or improve the stability of compounds of general formula (3), however, do not exist.
Thus, it would be useful to improve both processes of making compounds of formula (3) and, as well, processes of their application in chemical synthesis.
xample 6: 40-O-[2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl]rapamycin
In a 100 mL flask, Rapamycin (6 g, 6.56 mmol) was dissolved in dimethoxyethane (4.2 ml) and toluene (24 ml) to give a white suspension and the temperature was raised to 70°C. After 20 min, N,N-diisopropylethylamine (4.56 ml, 27.6 mmol) and 2-((2,3-dimethylbutan-2- yl)dimethylsilyloxy)ethyl trifluoromethanesulfonate (8.83 g, 26.3 mmol) were added in 2 portions with a 2 hr interval at 70°C. The mixture was stirred overnight at room temperature, then diluted with EtOAc (40 ml) and washed with sat. NaHC03 (30 ml) and brine (30 ml). The organic layer was dried with Na2S04, filtered and concentrated. The cmde product was chromatographed on a silica gel column (EtOAc/heptane 1/1 ; yield 4.47 g).
Example 7: 40-O-(2-hydroxyethyl)-rapamycin [everolimus]
In a 100 mL flask, 40-O-[2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl]rapamycin (4.47 g, 4.06 mmol) was dissolved in methanol (20 ml) to give a colorless solution. At 0°C, IN aqueous hydrochloric acid (2.0 ml, 2.0 mmol) was added and the mixture was stirred for 90 min. The reaction was followed by TLC (ethyl acetate/n-heptane 3 :2) and HPLC. Then 20 ml of saturated aqueous NaHC03 were added, followed by 20 ml of brine and 80 ml of ethyl acetate. The phases were separated and the organic layer was washed with saturated aqueous NaCl until pH 6/7. The organic layer was dried by Na2S04, filtered and concentrated to yield 3.3 g of the product.
……………………….
SYNTHESIS
https://www.google.co.in/patents/WO1994009010A1
Example 8: 40-O-(2-Hydroxy)ethyl-rapamycin
a) 40-O-[2-(t-Butyldimethylsilyl)oxy]ethyl-rapamycin
A solution of 9.14 g (10 mmol) of rapamycin and 4.70 mL (40 mmol) of 2,6-lutidine in 30 mL of toluene is warmed to 60°C and a solution of 6.17 g (20 mmol) of 2-(t-butyldimethylsilyl)oxyethyl triflate and 2.35 mL (20 mmol) of 2,6-lutidine in 20 mL of toluene is added. This mixture is stirred for 1.5h. Then two batches of a solution of 3.08 g (10 mmol) of triflate and 1.2 mL (10 mmol) of 2,6-lutidine in 10 mL of toluene are added in a 1.5h interval. After addition of the last batch, stirring is continued at 60°C for 2h and the resulting brown suspension is filtered. The filtrate is diluted with ethyl acetate and washed with aq. sodium bicarbonate and brine. The organic solution is dried over anhydrous sodium sulfate, filtered and concentrated. The residue is purified by column chromatography on silica gel (40:60 hexane-ethyl acetate) to afford 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl-rapamycin as a white solid: 1H NMR (CDCl3) δ 0.06 (6H, s), 0.72 (1H, dd), 0.90 (9H, s), 1.65 (3H, s), 1.75 (3H, s), 3.02 (1H, m), 3.63 (3H, m), 3.72 (3H, m); MS (FAB) m/z 1094 ([M+Na]+), 1022 ([M-(OCH3+H2O)]+).
b) 40-O-(2-Hydroxy)ethyl-rapamycin
To a stirred, cooled (0°C) solution of 4.5 g (4.2 mmol) of 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl-rapamycin in 20 mL of methanol is added 2 mL of IN HCl. This solution is stirred for 2h and neutralized with aq. sodium bicarbonate. The mixture is extracted with three portions of ethyl acetate. The organic solution is washed with aq.
sodium bicarbonate and brine, dried over anhydrous sodium sulfate, filtered and
concentrated. Purification by column chromatography on silica gel (ethyl acetate) gave the title compound as a white solid:1H NMR (CDCl3) δ 0.72 (1H, dd), 1.65 (3H, s), 1.75 (3H, s), 3.13 (5H, s and m), 3.52-3.91 (8H, m); MS (FAB) m/z 980 ([M+Na]+), 926 ([M-OCH3]+), 908 ([M-(OCH3+H2O)]+), 890 ([M-(OCH3+2H2O)]+), 876 ([M-(2CH3OH+OH)]+), 858 ([M-(OCH3+CH3OH+2H2O)]+).
MBA (rel. IC50) 2.2
IL-6 dep. prol. (rel. IC50) 2.8
MLR (rel. IC50) 3.4
…………………..
synthesis
Everolimus (Everolimus) was synthesized by the Sirolimus (sirolimus, also known as rapamycin Rapamycin) ether from. Sirolimus is from the soil bacterium Streptomyces hygroscopicus isolated metabolites. Activation end sirolimus (triflate, Tf) the other end of the protection (t-butyldimethylsilyl, TBS) of ethylene glycol 1 reaction of 2 , because the hydroxyl group 42 hydroxyl site over the 31-bit resistance is small, so the reaction only occurs in 42. Compound 2under acidic conditions TBS protection is removed everolimus.
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY

