
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

Balsam for the bones: Chemists develop a nanopaste for the repair of bone defects
Following accidents or cancer surgery surgeons often have to transplant healthy bone tissue or synthetic material to repair the resulting bone defects. Unfortunately, these procedures do not always have the desired effect.
Now a professor for inorganic chemistry, Matthias Epple was attracted to the interface between biology and medical science. “We have been investigating the impact of mineral tissue such as teeth, bone and sea shells for many years and are now using the knowledge we have gained to produce new biomaterials.” To achieve this he has collaborated closely with medical scientists and his current project – carried out with three of his doctoral students – was no exception.
“The repair of bone defects presents a real challenge for surgeons,” he relates. “When possible they collect the patient’s own bone from various locations, such as the iliac crest, and implant it where needed to fill defects.” The researcher explained…
View original post 267 more words
Glenmark Pharmaceuticals Ltd. through its Swiss Subsidiary receives USD 4 Mn. as research fee payment from Forest Laboratories Inc.

Total Payment received for the mpges-1 program from Forest Laboratories is USD 15 million
March 25, 2014: Glenmark Pharmaceuticals Ltd. has informed the Stock Exchange today that the company through its Swiss subsidiary has received
USD 4 million as research fee payment from Forest Laboratories Inc. on a collaboration for the development of novel mPGES-1 inhibitors to treatchronic inflammatory conditions, including pain.
Under the terms of the agreement signed in FY 2012-13, Forest made USD 6 million upfront payment and also provided an additional USD 3 million
to support the next phase of work. In September 2013, Glenmark received an additional amount of USD 2 million as research fee payment from Forest Laboratories Inc.
Hence, the total amount received by Glenmark from Forest Laboratories Inc towards its novel mPEGS-1 inhibitors program is USD15 million.
read at
Zabofloxacin
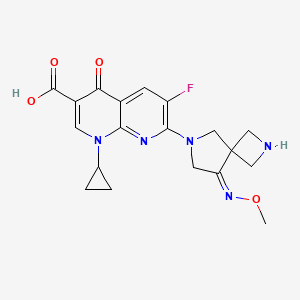
zabofloxacin, 219680-11-2
Example 1. l-Cyclopropyl-6-fluoro-7-[8-(methoxyimino)-2,6-diazaspiro[3,4]oct-6-yl]-4- oxo-l,4-dihydro[l,8]naphthyridine-3-carboxylic acid methanesulfonate
30 350mg of
7-[2-(t-buthoxycarbonyl)-8-(methoxyimino)-2,6-diazaspiro[3.4]oct-6-yl]-l- cyclopropyl-6-fluoro-4-oxo-l,4-dihydro[l,8]naphthyridine-3-carboxylic acid was dissolved in 5ml of dichloromethane and thereto 0.6ml of trifluoroacetic acid was dropped. The mixture was stirred for 5 hours at room temperature and thereto 10ml (if ethylether was added. It was stirred additionally for 1 hour and thus precipitated solid was filtered, dissolved in 5ml of diluted NaOH and neutralized with diluted hydrochloric acid. The precipitate thus obtained was filtered and dried. The resulting solid was added to 5ml of lN-methanesulfonic acid in ethanol and stirred for 1 hour. Thus obtained precipitate was filtered and dried to give 185g of the titled compound(yield : 47.8%). m.p. : 228- 229 °C
1H-NMR(DMSO-dG+CF3COOD, ppm): 0.97(s, 2H), 1.14(d, 2H), 2.48(s, 3H), 3.57(bs, IH), 3.88(s, 3H), 4.06-4.17(m, 411), 4.40(s, 2H), 4.49(s, 2H), 7.88(d, Hi, J=12.67Hz), 8.49(s, IH).
………………………………..
aspartate of 1-cyclopropyl-6-fluoro-7-(8-methoxyimino-2,6-diaza-spiro[3.4]oct-6-yl)-4-oxo-1,4-dihydro-[1,8]naphthyridine-3-carboxylic acid comprises a step of reacting 1-cyclopropyl-6-fluoro-7-(8-methoxyimino-2,6-diaza-spiro[3.4]oct-6-yl)-4-oxo-1,4-dihydro-[1,8]naphthyridine-3-carboxylic acid with aspartic acid in a solvent. The method can be represented by Scheme 1.

Example 1 Preparation of the D-Aspartic Acid Salt of 1-cyclopropyl-6-fluoro-7-(8-methoxyimino-2,6-diaza-spiro[3.4]oct-6-yl)-4-oxo-1,4-dihydro-[1,8]naphthyridine-3-carboxylic acid
1-Cyclopropyl-6-fluoro-7-(8-methoxyimino-2,6-diaza-spiro[3.4]oct-6-yl)-4-oxo-1,4-dihydro-[1,8]naphthyridine-3-carboxylic acid (5.0 g) was added to 50% ethanol (80 mL), and then the mixture was stirred at 50° C. for 10 minutes. D-Aspartic acid (2.0 g) was added and then the mixture was stirred at 50° C. for 1 hour. The mixture was cooled to room temperature, and then the resulting solid was collected by filtration. Ethanol (100 mL) was added to the filtrate, and then the mixture was stirred for 30 minutes. The resulting solid was collected by filtration to obtain a total of 5.55 g of the target compound (yield: 83%). Melting point: 200-201° C. 1H NMR (D2O): δ 0.97 (bs, 2H), 1.27 (d, 2H), 2.00 (dd, 1H, J=8.8, 17.6 Hz), 2.77 (dd, 1H, J=3.3, 17.0 Hz), 3.53 (bs, 1H), 3.84 (dd, 1H, J=3.3, 8.78 Hz), 4.01 (s, 3H), 4.31-4.45 (m, 8H), 7.46 (d, 1H, J=12.2 Hz), 8.42 (s, 1H).
Example 2 Preparation of L-Aspartic Acid Salt of 1-cyclopropyl-6-fluoro-7-(8-methoxyimino-2,6-diaza-spiro[3.4]oct-6-yl)-4-oxo-1,4-dihydro-[1,8]naphthyridine-3-carboxylic acid
1-Cyclopropyl-6-fluoro-7-(8-methoxyimino-2,6-diaza-spiro[3.4]oct-6-yl)-4-oxo-1,4-dihydro-[1,8]naphthyridine-3-carboxylic acid (500 mg) was added to 50% ethanol (20 mL), and then the mixture was stirred at 50° C. for 10 minutes. L-Aspartic acid (174 mg) was added and then the mixture was stirred at 50° C. for 1 hour. The mixture was cooled to room temperature. Ethanol (20 mL) was added to the reaction mixture, and then the mixture was stirred for 30 minutes. The resulting solid was collected by filtration to obtain 550 mg of the target compound (yield: 82%). Melting point: 205-206° C. 1H NMR (d6-DMSO): δ 0.93 (d, 2H, J=3.5 Hz), 1.20 (d, 2H, J=6.8 Hz), 2.42 (dd, 1H, J=9.2, 17.3 Hz), 2.59 (dd, 1H, J=3.3, 17.2 Hz), 3.50 (m, 1H), 3.59 (1H, dd, J=3.1, 9.1 Hz), 3.91 (s, 3H), 4.24 (m, 6H), 4.41 (br, 2H), 7.59 (d, 1H, J=12.4 Hz), 8.41 (s, 1H).
Example 3 Preparation of Hydrochloric Acid Salt, Phosphate Salt, and Formate Salt of 1-cyclopropyl-6-fluoro-7-(8-methoxyimino-2,6-diaza-spiro[3.4]oct-6-yl)-4-oxo-1,4-dihydro-[1,8]naphthyridine-3-carboxylic acid
3-1 Hydrochloric Acid Salt
Ethanol (3 mL) was cooled to 0° C. and acetyl chloride (1.13 mL) was added, and then the mixture was stirred for 30 minutes. 1-Cyclopropyl-6-fluoro-7-(8-methoxyimino-2,6-diaza-spiro[3.4]oct-6-yl)-4-oxo-1,4-dihydro-[1,8]naphthyridine-3-carboxylic acid (800 mg) was added to the reaction mixture, and then stirred at 0° C. for 30 minutes. Tetrahydrofuran (4 mL) was added, and then the mixture was stirred for 30 minutes. The resulting solid was collected by filtration and dried to obtain 776 mg of the target compound (yield: 89%). Melting point: 244-245° C. 1H NMR (d6-DMSO): δ 1.07 (d, 2H, J=4.7 Hz), 1.21 (d, 2H, J=6.8 Hz), 3.68 (m, 1H), 3.94 (s, 3H), 4.17 (m, 2H), 4.40 (s, 2H), 4.53 (s, 2H), 8.03 (d, 1H, J=12.5 Hz), 8.59 (s, 1H).
Han J, Kim JC, Chung MK, Kim B, Choi DR.
Biol Pharm Bull. 2003 Jun;26(6):832-9
Jin HE, Kang IH, Shim CK.
J Pharm Pharm Sci. 2011;14(3):291-305.
Jin HE, Lee KR, Kang IH, Chung SJ, Shim CK.
J Pharm Biomed Anal. 2011 Mar 25;54(4):873-7. doi: 10.1016/j.jpba.2010.11.001. Epub 2010 Nov 9.
Kosowska-Shick, K.; Credito, K.; Pankuch, G.A.; Lin, G.; Bozdogan, B.; McGhee, P.; Dewasse, B.;
Choi, D.-R.; Ryu, J.M.; Appelbaum, P.C. Antipneumococcal activity of DW-224a, a new
quinolone, compared to those of eight other agents. Antimicrob. Agents Chemother. 2006, 50,
2064–2071.
Park, H.-S.; Kim, H.-J.; Seol, M.-J.; Choi, D.-R.; Choi, E.-C.; Kwak, J.-H. In vitro and in vivo
antibacterial activities of DW-224a, a new fluoronaphthyridone. Antimicrob. Agents Chemother.
2006, 50, 2261–2264.
Dong Wha Pharmaceutical Co. Ltd. A study to evaluate efficacy and safety profile of
Zabofloxacin tablet 400 mg and moxifloxacin tablet 400 mg. Available online:
http://www.clinicaltrials.gov/ct2/show/NCT01658020 (accessed on 15 July 2013).
Dong Wha Pharmaceutical Co. Ltd. A new quinolone antibiotic. Available online:
http://www.dong-wha.co.kr/english/rnd/rnd02_03.asp (accessed on 15 April 2013).
| US4957922 * | Mar 29, 1989 | Sep 18, 1990 | Bayer Aktiengesellschaft | Infusion solutions of 1-cyclopropyl-6-fluoro-1,4-di-hydro-4-oxo-7-(1-piperazinyl)-quinoline-3-carboxylic acid |
| US5563149 * | Aug 8, 1994 | Oct 8, 1996 | Cheil Foods & Chemicals, Inc. | Aqueous solutions of pyridone carboxylic acids |
| US6552196 * | Sep 6, 2001 | Apr 22, 2003 | Dong Wha Pharmaceutical Industrial Co., Ltd. | Quinolone carboxylic acid derivatives |
|
7-23-2010
|
ASPARTATE OF 1-CYCLOPROPYL-6-FLUORO-7-(8-METHOXYIMINO-2,6-DIAZA-SPIRO[3.4]OCT-6-YL)-4-OXO-1,4-DIHYDRO-[1,8]NAPHTHYRIDINE-3-CARBOXYLIC ACID, METHOD FOR PREPARING THE SAME, AND ANTIMICROBIAL PHARMACEUTICAL COMPOSITION COMPRISING THE SAME
|
PRINOMASTAT

PRINOMASTAT
Molecular Formula: C18H21N3O5S2
Molecular Weight: 423.5064
CAS No: 192329-42-3
IUPAC Name: 2-[(Hydroxyamino)methyl]-5,6-dimethyl-4-(4-pyridin-4-yloxyphenyl)sulfonylmorpholine-3-thione
3-Thiomorpholinecarboxamide,N-hydroxy-2,2-dimethyl-4-[[4-(4-pyridinyloxy)phenyl]sulfonyl]-, (S)-; AG 3340;KB-R 9896; Prinomastat
Prinomastat, AG-3362(maleate), AG-3354(HCl), AG-3340
Agouron (Originator)
Prinomastat (AG-3340) is a matrix metalloprotease (MMP) inhibitor with specific selectivity for MMPs 2, 3, 9, 13, and 14. Investigations have been carried out to determine whether the inhibition of these MMPs is able to block tumour metastasis by preventing MMP degradation of the extracellular matrix proteins and angiogenesis.
Prinomastat is a synthetic hydroxamic acid derivative with potential antineoplastic activity. Prinomastat inhibits matrix metalloproteinases (MMPs) (specifically, MMP-2, 9, 13, and 14), thereby inducing extracellular matrix degradation, and inhibiting angiogenesis, tumor growth and invasion, and metastasis. As a lipophilic agent, prinomastat crosses the blood-brain barrier.
Prinomastat underwent a Phase III trial to investigate its effectiveness against non-small cell lung cancer (nsclc), in combination with gemcitabine chemotherapy. However, it was discovered that Prinomastat did not improve the outcome of chemotherapy in advanced Non-Small-Cell Lung Cancer. [1] [2]
Matrix metalloproteinases (“MMPs”) are a family of enzymes, including, collagenases, gelatinases, matrilysin, and stromelysins, that are involved in the degradation and remodeling of connective tissues. These enzymes are contained in a number of cell types that are found in or are associated with connective tissue, such as fibroblasts, monocytes, macrophages, endothelial cells and metastatic tumor cells. They also share a number of properties, including zinc and calcium dependence, secretion as zymogens, and, 40-50% amino acid sequence homology.
Matrix metalloproteinases degrade the protein components of the extracellular matrix, i.e., the protein components found in the linings of joints, interstitial connective tissue, basement membranes, cartilage and the like. These proteins include collagen, proteoglycan, fibronectin and lamanin.
In a number of pathological disease conditions, however, deregulation of matrix metalloproteinase activity leads to the uncontrolled breakdown of extracellular matrix. These disease conditions include arthritis (e.g., rheumatoid arthritis and osteoarthritis), periodontal disease, aberrant angiogenesis, tumor metastasis and invasion, tissue ulceration (e.g., comeal ulceration, gastric ulceration or epidermal ulceration), bone disease, HIV-infection and complications from diabetes.
Administration of matrix metalloproteinase inhibitors has been found to reduce the rate of connective tissue degradation, thereby leading to a favorable therapeutic effect. For example, in Cancer Res., 53, 2087 (1993), a synthetic matrix metalloproteinase inhibitor was shown to have in vivo efficacy in a murine model for ovarian cancer with an apparent mode of action consistent with inhibition of matrix remodeling. The design and uses of MMP inhibitors are reviewed, for example, in J. Enzyme Inhibition, 2, 1-22 (1987); Progress in Medicinal Chemistry, 29, 271-334 (1992); Current Medicinal Chemistry, 2, 743-762 (1995); Exp. Opin. Ther. Patents, 5, 12871296 (1995); and Drug Discovery Today, 1, 16-26 (1996).
Matrix metalloproteinase inhibitors are also the subject of numerous patents and patent applications, including: U.S. Pat. Nos. 5,189,178; 5,183,900; 5,506,242; 5,552,419; and 5,455,258; European Patent Application Nos. EP 0 438 223 and EP 0 276 436; International Publication Nos. WO 92/21360; WO 92/06966; WO 92/09563; WO 96/00214; WO 95/35276; and WO 96/27583.
Further, U.S. patent application Ser. Nos. 6,153,757 and 5,753,653 relate to prinomistat and its synthesis, the disclosures of each are incorporated herein by reference in their entireties.
Prinomastat, shown below, is a potent inhibitor of certain metalloproteinases (MMP), particularly matrix metalloproteinases and tumor necrosis factor-α convertase. International Publication No. WO 97/208824 discloses the chemical structure of prinomastat, its pharmaceutical composition, as well as pharmaceutical uses, methods of its preparation and intermediates useful in its synthesis.

Until now, metabolites of prinomastat have not been identified, isolated, purified or synthesized. Further, it is shown that some of these metabolites are potent matrix metalloproteinase inhibitors


The sulfonation of 4-chlorodiphenyl ether (I) with chlorosulfonic acid in dichloromethane gives the 4-(4-chlorophenoxy)benzenesulfonic acid (II), which is treated with oxalyl chloride and DMF in the same solvent yielding the sulfonyl chloride (III).
The reduction of (III) with trimethyl phosphite and KOH in toluene affords the methylsulfanyl derivative (IV), which is chlorinated with SO2Cl2 in dichloromethane to give the chloromethylsulfanyl derivative (V). The condensation of (V) with the silylated enol ether (VI) by means of ZnCl2 and KOH in refluxing dichloromethane yields 4-[4-(4-chlorophenoxy)phenylsulfanylmethyl]tetrahydropyran-4-carboxylic acid (VII), which is treated with oxalyl chloride affording the corresponding acyl chloride (VIII).
The reaction of (VIII) with NH2OH in dichloromethane provides the carbohydroxamic acid (IX), which is finally oxidized with oxone (potassium peroxymonosulfate) in N-methyl-2-pyrrolidone/H2O to furnish the target sulfone.

The cyclization of D-penicillamine (I) with 1,2-dichloroethane by means of DBU and TMS-Cl in DMF gives 2,2-dimethylthiomorpholine-3(S)-carboxylic acid (XV), which is treated with isobutylene (XVI) and sulfuric acid in dioxane to yield the corresponding tert-butyl ester (XVII). The sulfonation of (XVII) with the sulfonyl chloride (VI) as before affords 2,2-dimethyl-4-[4-(4-pyridyloxy)phenylsulfonyl]thiomorpholine-3(S)-carboxylic acid tert-butyl ester (XVIII), which is finally treated with HCl in refluxing dioxane to give the previously reported free acid intermediate (XIV).
The cyclization of D-penicillamine methyl ester (XIX) with 1,2-dibromoethane by means of DBU in DMF gives 2,2-dimethylthiomorpholine-3(S)-carboxylic acid methyl ester (XX), which is sulfonated with the sulfonyl chloride (VI) as before, affording 2,2-dimethyl-4-[4-(4-pyridyloxy)phenylsulfonyl]thiomorpholine-3(S)-carboxylic acid methyl ester (XXI). Finally, this compound is hydrolyzed with refluxing aqueous HCl to yield the previously reported intermediate (XIV).

The silylation of D-penicillamine (I) with dimethylhexylsilyl chloride (Dmhs-Cl) and DBU gives the ester (XI), which is cyclized with 1,2-dichloroethane and DBU in DMF, yielding 2,2-dimethylthiomorpholine-3(S)-carboxylic acid dimethylhexylsilyl ester (XII).
The sulfonation of (XII) with the sulfonyl chloride (VI) as before affords 2,2-dimethyl-4-[4-(4-pyridyloxy)phenylsulfonyl]thiomorpholine-3(S)-carboxylic acid dimethylhexylsilyl ester (XIII), which is desilylated in refluxing methanol to give the free acid (XIV) Finally, this compound is treated with oxalyl chloride and hydroxylamine in dichloromethane.
References
- Hande, Kenneth R; Mary Collier, Linda Paradiso, Jill Stuart-Smith, Mary Dixon, Neil Clendeninn, Geoff Yeun, Donna Alberti, Kim Binger and George Wilding (2004). “Phase I and Pharmacokinetic Study of Prinomastat, a Matrix Metalloprotease Inhibitor”. Journal of Drugs in Dermatology: JDD 3 (4): 393–7. PMID 15303783.
- Bissett, K Donald; en J. O’Byrne, J. von Pawel, Ulrich Gatzemeier, Allan Price, Marianne Nicolson, Richard Mercier, Elva Mazabel, Carol Penning, Min H. Zhang, Mary A. Collier, Frances A. Shepherd (2005). “Phase III Study of Matrix Metalloproteinase Inhibitor Prinomastat in Non–Small-Cell Lung Cancer”.Journal of Clinical Oncology 10: 909. doi:10.1158/1078-0432.CCR-0981-3.
clinical trial results
1. Phase II, prinomastat in patients with esophageal adenocarcinoma.
All patients, regardless of treatment arm, were able to successfully undergo neoadjuvant combined modality therapy and esophagectomy. However, early closure of the study due to unexpected thrombo-embolic events precluded any conclusions regarding clinical activity of prinomastat in locally advanced esophageal cancer patients.
2. Phase III study of prinomastat in non-small-cell lung cancer.
Prinomastat does not improve the outcome of chemotherapy in advanced NSCLC.
Green…Asymmetric hydrogentation of unfunctionalised olefins/enamines/imines
Asymmetric hydrogentation of unfunctionalised olefins/enamines/imines
The reaction survey found that the predominant strategy for the introduction of chirality was through classical chemical resolutions as opposed to introductions through biotransformation or transition metal or organometallic catalytic means.
Asymmetric hydrogenation provides an elegant methodology for the introduction of chirality, meeting many of the goals of green chemistry and is finding increasing application in API synthesis.47
The efficiency of this approach is elegantly exemplified by the Merck second generation synthesis of sitagliptin 5 (Scheme ), where an unprecedented final stage asymmetric hydrogenation of the unprotected enamide 6 resulted in an increase in overall yield of almost 50% and produced 100 kg less waste per kg sitagliptin48 when compared with the first generation approach.49
 |
||
| Scheme The synthesis of sitagliptin. | ||
There are challenging areas remaining within the field, for example, the hydrogenation of enamides and related substrates in the synthesis of amino acids has numerous examples50 but few examples exist for unsubstitued enamines41 and imines. Some classes of alkene offer additional challenges.51 For the pharmaceutical industry, the limited time for synthetic route identification is an issue and access to catalyst and ligand diversity is required to ensure the application of this approach.52
Some pharmaceutical companies have synthesised their own ligands and have found very effective catalysts.53 The majority of academic asymmetric hydrogenation approaches are based on homogeneous catalysis to overcome issues of activation and mass transfer. For pharmaceutical use, efficient catalyst and ligand recovery, and eliminating heavy metal contamination of the API are significant requirements for the industry.
These controls are often easier to achieve with heterogeneous methodology where there are less examples.50 The demonstration of organocatalytic hydride transfer offers the possibility of future access to metal free asymmetric hydrogenations.54
- 47………V. Farina, J. T. Reeves, C. H. Senanayake and J. J. Song, Chem. Rev., 2006, 106, 2734–2793. See also Asymmetric Catalysis on Industrial Scale Challenges, Approaches and Solutions, ed. H.-U. Blaser and E. Schmidt, Wiley-VCH, Weinheim, 2004 Search PubMed .
- 48………..http://www.epa.gov/greenchemistry/pubs/pgcc/winners/gspa06.html .
- 49……K. B. Hansen, J. Balsells, S. Dreher, Y. Hsiao, M. Kubryk, M. Palucki, N. Rivera, D. Steinhuebel, J. D. Armstrong III, D. Askin and E. J. J. Grabowski, Org. Process Res. Dev., 2005, 9, 634–639 Search PubMed .
- 50………..M. Studer, H.-U. Blaser and C. Exner, Adv. Synth. Catal., 2003, 345, 45–65 CrossRef CAS Search PubMed .
- 51……..X. Cui and K. Burgess, Chem. Rev., 2005, 105, 3272–3296 CrossRef CAS Search PubMed
- 52……….I. C. Lennon and C. J. Pilkington, Synthesis, 2003, 1639–1642 CrossRef CAS Search PubMed .
- 53………G. Hoge, H.-P. Wu, W. S. Kissel, D. A. Plum, D. J. Greene and J. Bao, J. Am. Chem. Soc., 2004, 126, 5966–5967 CrossRef CAS Search PubMed .
- 54……..H. Adolfsson, Angew. Chem., Int. Ed., 2005, 44, 3340–3342 CrossRef CAS Search PubMed .
APREMILAST …….FDA approves Celgene’s Otezla for psioratic arthritis

APREMILAST
PDE4 inhibitor
N-{2-[(1S)-1-(3-Ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethyl]-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl}acetamide
(+)-2-[l-(3-ethoxy-4-methoxyphenyl)-2- methanesulfonylethyl]-4-acetylaminoisoindolin-l,3-dione,
(S)—N-{2-[1-(3-ethoxy-4-methoxy-phenyl)-2-methanesulfonylethyl]-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl}acetamide
(S)-N-{2-[1-(3-Ethoxy-4-methoxyphenyl)-2-methanesulfonylethyl]-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl}acetamide
Molecular Formula: C22H24N2O7S Molecular Weight: 460.50016
608141-41-9 CAS NO
Celgene (Originator)
MARCH 22, 2014
Just as the American Academy of Dermatology meeting opens its doors in Denver, Celgene Corp has been boosted by a green light from US regulators for Otezla as a treatment for psoriatic arthritis.
The US Food and Drug Administration has approved Otezla (apremilast), making it the first oral treatment for adults with active PsA. The thumbs-up for the phosphodieasterase-4 (PDE-4) inhibitor is primarily based on three trials involving 1,493 patients where Otezla showed improvement in signs and symptoms of the disease, including tender and swollen joints and physical function, compared to placebo.
Read more at:
COPY PASTE LINK
CC-10004, , Apremilast (USAN), SureCN302992, Apremilast (CC-10004), QCR-202,
- Apremilast
- CC 10004
- CC-10004
- CC10004
- UNII-UP7QBP99PN
- CLINICAL TRIALS….http://clinicaltrials.gov/search/intervention=Apremilast+OR+CC-10004
Apremilast is an orally available small molecule inhibitor of PDE4 being developed byCelgene for ankylosing spondylitis, psoriasis, and psoriatic arthritis.[1][2] The drug is currently in phase III trials for the three indications. Apremilast, an anti-inflammatory drug, specifically inhibits phosphodiesterase 4. In general the drug works on an intra-cellular basis to moderate proinflammatory and anti-inflammatory mediator production.
 APREMILAST
APREMILAST
Apremilast is being tested for its efficacy in treating “psoriasis, psoriatic arthritis and other chronic inflammatory diseases such as ankylosing spondylitis, Behcet’s disease, and rheutmatoid arthritis.
“Apremilast is Celgene’s lead oral phosphodiesterase IV inhibitor and anti-TNF alpha agent in phase III clinical studies at Celgene for the oral treatment of moderate to severe plaque-type psoriasis and for the oral treatment of psoriatic arthritis.
Early clinical development is also ongoing for the treatment of acne, Behcet’s disease, cutaneous sarcoidosis, prurigo nodularis, ankylosing spondylitis, atopic or contact dermatitis and rheumatoid arthritis. No recent development has been reported for research for the treatment of skin inflammation associated with cutaneous lupus erythematosus.
In 2011, Celgene discontinued development of the compound for the management of vision-threatening uveitis refractory to other modes of systemic immunosuppression due to lack of efficacy.
Celgene had been evaluating the potential of the drug for the treatment of asthma; however, no recent development has been reported for this research. The drug candidate is also in phase II clinical development at the William Beaumont Hospital Research Institute for the treatment of chronic prostatitis or chronic pelvic pain syndrome and for the treatment of vulvodynia (vulvar pain).
In 2013, orphan drug designations were assigned to the product in the U.S. and the E.U. for the treatment of Behcet’s disease.
Celgene Corp has been boosted by more impressive late-stage data on apremilast, an oral drug for psoriatic arthritis, this time in previously-untreated patients.
The company is presenting data from the 52-week PALACE 4 Phase III study of apremilast tested in PsA patients who have not taken systemic or biologic disease modifying antirheumatic drugs (DMARDs) at the American College of Rheumatology meeting in San Diego. The results from the 527-patient trial show that at week 16, patients on 20mg of the first-in-class oral inhibitor of phosphodiesterase 4 (PDE4) achieved an ACR20 (ie a 20% improvement in the condition) response of 29.2% and 32.3% for 30mg aapremilast, compared with 16.9% for those on placebo.
After 52 weeks, 53.4% on the lower dose and 58.7% on 30mg achieved an ACR20 response. ACR50 and 70 was reached by 31.9% and 18.1% of patients, respectively, for apremilast 30mg. The compound was generally well-tolerated and discontinuation rates for diarrhoea and nausea were less than 2% over 52 weeks.
Commenting on the data, Alvin Wells, of the Rheumatology and Immunotherapy Center in Franklin, Wisconsin, noted that apremilast demonstrated long-term safety and tolerability and significant clinical benefit in treatment-naive patients. He added that “these encouraging results suggest that apremilast may have the potential to be used alone and as a first-line therapy”. Celgene is also presenting various pooled data from the first three trials in the PALACE programme which, among other things, shows that apremilast significantly improves swollen and tender joints.
Treatment for PSA, which affects about 30% of the 125 million people worldwide who have psoriasis, currently involves injectable tumour necrosis factor (TNF) inhibitors, notably AbbVie’s Humira (adalimumab) and Pfizer/Amgen’s Enbrel (etanercept), once patients have not responded to DMARDs (at least in the UK). While the biologics are effective, the side effect profile can be a concern, due to the risk of infection and tuberculosis and many observers believe that apremilast will prove popular with patients and doctors due to the fact that it is oral, not injectable.
Apremilast was filed for PsA with the US Food and Drug Administration in the first quarter and will be submitted on both sides of the Atlantic for psoriasis before year-end. The European filing will also be for PsA.
Apremilast impresses for Behcet’s disease
Celgene has also presented promising Phase II data on apremilast as a treatment for the rare inflammatory disorder Behcet’s disease. 71% of patients achieved complete response at week 12 in clearing oral ulcers
 APREMILAST
APREMILAST
- “Apremilast Palace Program Demonstrates Robust and Consistent Statistically Significant Clinical Benefit Across Three Pivotal Phase III Studies (PALACE-1, 2 & 3) in Psoriatic Arthritis” (Press release). Celgene Corporation. 6 September 2012. Retrieved 2012-09-10.
- “US HOT STOCKS: OCZ, VeriFone, Men’s Wearhouse, AK Steel, Celgene”. The Wall Street Journal. 6 September 2012. Retrieved 2012-09-06.
- Discovery of (S)-N-[2-[1-(3-ethoxy-4-methoxyphenyl)-2-methanesulfonylethyl]-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl] acetamide (apremilast), a potent and orally active phosphodiesterase 4 and tumor necrosis factor-alpha inhibitor.
Man HW, Schafer P, Wong LM, Patterson RT, Corral LG, Raymon H, Blease K, Leisten J, Shirley MA, Tang Y, Babusis DM, Chen R, Stirling D, Muller GW.
J Med Chem. 2009 Mar 26;52(6):1522-4. doi: 10.1021/jm900210d.
- Therapeutics: Silencing psoriasis.Crow JM.Nature. 2012 Dec 20;492(7429):S58-9. doi: 10.1038/492S58a. No abstract available.
- NMR…http://file.selleckchem.com/downloads/nmr/S803401-Apremilast-HNMR-Selleck.pdf
- WO 2003080049
- WO 2013126495
- WO 2013126360
- WO 2003080049
- WO 2006065814
- US2003/187052 A1 …..MP 144 DEG CENT
- US2007/155791
-
J. Med. Chem., 2008, 51 (18), pp 5471–5489DOI: 10.1021/jm800582j
-
J. Med. Chem., 2011, 54 (9), pp 3331–3347DOI: 10.1021/jm200070e


合成路线:
US2013217918A1
US2014081032A1
…………………………………………
INTRODUCTION
2-[l-(3-ethoxy-4-methoxyphenyl)-2-methylsulfonylethyl]-4- acetylaminoisoindoline-l ,3-dione is a PDE4 inhibitor that is currently under investigation as an anti-inflammatory for the treatment of a variety of conditions, including asthma, chronic obstructive pulmonary disease, psoriasis and other allergic, autoimmune and rheumatologic conditions. S-enantiomer form of 2-[l-(3-ethoxy-4-methoxyphenyl)-2-methylsulfonylethyl]-4- acetylaminoisoindoline-l ,3-dione can be prepared by reacting (5)-aminosulfone 1 with intermediate 2.

Existing methods for synthesizing (S)-aminosulfone 1 involve resolution of the corresponding racemic aminosulfone by techniques known in the art. Examples include the formation and crystallization of chiral salts, and the use of chiral high performance liquid chromatography. See, e.g., Jacques, J., et al., Enantiomers, Racemates and Resolutions (Wiley Interscience, New York, 1981); Wilen, S. H., et al, Tetrahedron 33:2725 (1977); Eliel, E. L., Stereochemistry of Carbon Compounds (McGraw Hill, NY, 1962); and Wilen, S. H., Tables of Resolving Agents and Optical Resolutions p. 268 (E.L. Eliel, Ed., Univ. of Notre Dame Press, Notre Dame, IN, 1972). In one example, as depicted in Scheme 1 below, (5)-aminosulfone 1 is prepared by resolution of racemic aminosulfone 3 with N-Ac-L-Leu. Racemic aminosulfone 3 is prepared by converting 3-ethoxy-4-methoxybenzonitrile 4 to enamine intermediate 5 followed by enamine reduction and borate hydrolysis. This process has been reported in U.S. Patent
Application Publication No. 2010/0168475.

CH2CI2, NaOH

Scheme 1
The procedure for preparing an enantiomerically enriched or enantiomerically pure aminosulfone, such as compound 1, may be inefficient because it involves the resolution of racemic aminosulfone 3. Thus, a need exists as to asymmetric synthetic processes for the preparation of an enantiomerically enriched or enantiomerically pure aminosulfone, particularly for manufacturing scale production. Direct catalytic asymmetric hydrogenation of a suitable enamine or ketone intermediate is of particular interest because it eliminates the need for either classic resolution or the use of stoichiometric amount of chiral auxiliary, and thus, may be synthetically efficient and economical.
……………………………………….
SYNTHESIS OF KEY INTERMEDIATE
Example 1
Synthesis of 1 -(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethenamine

[00232] A slurry of dimethylsulfone (85 g, 903 mmol) in THF (480 ml) was treated with a
1.6M solution of n-butyllithium in hexane (505 ml, 808 mmol) at 0 – 5 °C. The resulting mixture was agitated for 1 hour then a solution of 3-ethoxy-4-methoxybenzonitrile (80 g, 451 mmol) in THF (240 ml) was added at 0 – 5 °C. The mixture was agitated at 0 – 5 °C for 0.5 hour, warmed to 25 – 30 °C over 0.5 hour and then agitated for 1 hour. Water (1.4 L) was added at 25 – 30 °C and the reaction mass was agitated overnight at room temperature (20 – 30 °C). The solid was filtered and subsequently washed with a 2: 1 mixture of water :THF (200 ml), water (200 ml) and heptane (2 x 200 ml). The solid was dried under reduced pressure at 40 – 45 °C to provide the product as a white solid (102 g, 83% yield); 1H NMR (DMSO-d6) δ 1.34 (t, J=7.0 Hz, 3H), 2.99 (s, 3H), 3.80 (s, 3H), 4.08 (q, J=7.0 Hz, 2H), 5.03 (s, 1H), 6.82 (s, 2H), 7.01 (d, J=8.5 Hz, 1H), 7.09 – 7.22 (m, 2H).
Example 2
Synthesis of (R)- 1 -(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethanamine

[00233] A solution of bis(l,5-cyclooctadiene)rhodium(I) trifluoromethanesulfonate (36 mg, 0.074 mmol) and (i?)-l-[(5)-2-(diphenylphosphino)ferrocenyl]ethyldi-tert-butylphosphine (40 mg, 0.074 mmol) in 25 mL of 2,2,2-trifluoroethanol was prepared under nitrogen. To this solution was then charged l-(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethenamine (2.0 g, 7.4 mmol). The resulting mixture was heated to 50 °C and hydrogenated under 90 psig hydrogen pressure. After 18 h, the mixture was cooled to ambient temperature and removed from the hydrogenator. The mixture was evaporated and the residue was purified by chromatography on a CI 8 reverse phase column using a water-acetonitrile gradient. The appropriate fractions were pooled and evaporated to -150 mL. To this solution was added brine (20 mL), and the resulting solution was extracted with EtOAc (3 x 50 mL). The combined organic layers were dried (MgS04) and evaporated to provide the product as a white crystalline solid (1.4 g, 70% yield); achiral HPLC (Hypersil BDS C8, 5.0 μπι, 250 x 4.6 mm, 1.5 mL/min, 278nm, 90/10 gradient to 80/20 0.1% aqueous TFA/MeOH over 10 min then gradient to 10/90 0.1% aqueous TFA/MeOH over the next 15 min): 9.11 (99.6%); chiral HPLC (Chiralpak AD-H 5.0 μιη Daicel, 250 x 4.6 mm, 1.0 mL/min, 280 nm, 70:30:0.1 heptane-z-PrOH-diethylamine): 7.32 (97.5%), 8.26 (2.47%); 1H NMR (DMSO-de) δ 1.32 (t, J= 7.0 Hz, 3H), 2.08 (s, 2H), 2.96 (s, 3H), 3.23 (dd, J= 3.6, 14.4 Hz, 1H), 3.41 (dd, J= 9.4, 14.4 Hz, 1H), 3.73 (s, 3H), 4.02 (q, J= 7.0 Hz, 2H), 4.26 (dd, J= 3.7, 9.3 Hz, 1H), 6.89 (s, 2H), 7.02 (s, 1H); 13C NMR (DMSO-d6) δ 14.77, 41.98, 50.89, 55.54, 62.03, 63.68, 111.48, 111.77, 118.36, 137.30, 147.93, 148.09. Example 3
Synthesis of (6 -l-(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethanamine N-Ac-L-Leu salt

[00234] A solution of bis(l,5-cyclooctadiene)rhodium(I) trifluoromethanesulfonate (17 mg, 0.037 mmol) and (5)-l-[(i?)-2-(diphenylphosphino)ferrocenyl]ethyldi-tert-butylphosphine (20 mg, 0.037 mmol) in 10 mL of 2,2,2-trifluoroethanol was prepared under nitrogen. To this solution was then charged l-(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethenamine (2.0 g, 7.4 mmol). The resulting mixture was heated to 50 °C and hydrogenated under 90 psig hydrogen pressure. After 18 h, the mixture was cooled to ambient temperature and removed from the hydrogenator. Ecosorb C-941 (200 mg) was added and the mixture was stirred at ambient temperature for 3 h. The mixture was filtered through Celite, and the filter was washed with additional trifluoroethanol (2 mL). Then, the mixture was heated to 55 °C, and a solution of N- acetyl-L-leucine (1.3 g, 7.5 mmol) was added dropwise over the course of 1 h. Stirring proceeded at the same temperature for 1 h following completion of the addition, and then the mixture was cooled to 22 °C over 2 h and stirred at this temperature for 16 h. The crystalline product was filtered, rinsed with methanol (2 x 5 mL), and dried under vacuum at 45 °C to provide the product as a white solid (2.6 g, 80% yield); achiral HPLC (Hypersil BDS Cg, 5.0 μιη, 250 x 4.6 mm, 1.5 mL/min, 278nm, 90/10 gradient to 80/20 0.1% aqueous TFA/MeOH over 10 min then gradient to 10/90 0.1% aqueous TFA/MeOH over the next 15 min): 8.57 (99.8%); chiral HPLC (Chiralpak AD-H 5.0 μιη Daicel, 250 x 4.6 mm, 1.0 mL/min, 280 nm, 70:30:0.1 heptane-z-PrOH-diethylamine): 8.35 (99.6%); 1H NMR (DMSO-<¾) δ 0.84 (d, 3H), 0.89 (d, J= 6.6 Hz, 3H), 1.33 (t, J= 7.0 Hz, 3H), 1.41 – 1.52 (m, 2H), 1.62 (dt, J= 6.7, 13.5 Hz, 1H), 1.83 (s, 3H), 2.94 (s, 3H), 3.28 (dd, J= 4.0, 14.4 Hz, 1H), 3.44 (dd, J= 9.1, 14.4 Hz, 1H), 3.73 (s, 3H), 4.02 (q, J= 6.9 Hz, 2H), 4.18 (q, J= 7.7 Hz, 1H), 4.29 (dd, J= 4.0, 9.1 Hz, 1H), 5.46 (br, 3H), 6.90 (s, 2H), 7.04 (s, 1H), 8.04 (d, J= 7.9 Hz, 1H); Anal. (C20H34N2O7S) C, H, N. Calcd C, 53.79; H, 7.67; N 6.27. Found C, 53.78; H, 7.57; N 6.18.
SUBSEQUENT CONVERSION
S-enantiomer form of 2-[l-(3-ethoxy-4-methoxyphenyl)-2-methylsulfonylethyl]-4- acetylaminoisoindoline-l ,3-dione can be prepared by reacting (5)-aminosulfone 1 with intermediate 2.
……………………………………
 APREMILAST
APREMILAST
GENERAL SYNTHESIS AND SYNTHESIS OF APREMILAST




(apremilast)
[0145] Preparation of 3-Ethoxy-4-methoxybenzonitrile (Compound 2). 3-Ethoxy-
4-methoxybenzaldehyde (Compound 1, 10.0 gm, 54.9 mmol, Aldrich) and hydroxylamine hydrochloride (4.67 gm, 65.9 mmol, Aldrich) were charged to a 250 mL three-necked flask at room temperature, followed by the addition of anhydrous acetonitrile (50 mL). The reaction mixture was stirred at room temperature for thirty minutes and then heated to reflux (oil bath at 85 °C). After two hours of reflux, the reaction mixture was cooled to room temperature, and added 50 mL of deionized water. The mixture was concentrated under reduced pressure to remove acetonitrile and then transferred to a separatory funnel with an additional 80 mL of deionized water and 80 mL dichloromethane. The aqueous layer was extracted with dichloromethane (3 x 50 mL). The combined organic layers were washed successively with water (80 mL) and saturated sodium chloride (80 mL). The organic layer was dried over anhydrous sodium sulfate (approximately 20 gm). The organic layer was filtered and concentrated under reduced pressure to give a yellow oil. Purification by silica gel chromatography (0 to 1 % MeOH/DCM ) afforded 3-Ethoxy-4-methoxybenzonitrile
(Compound 2) as a white solid (7.69 gm, 79 % yield). MS (ESI positive ion) m/z 178.1 (M + 1). HPLC indicated >99% purity by peak area. 1H-NMR (500 MHz, DMSO-c¾: δ ppm 1.32 (t, 3H), 3.83 (s, 3H), 4.05 (q, 2H), 7.10 (d, J = 8.0 Hz, 1H), 7.35 (d, J = 2.0 Hz, 1H), 7.40 (dd, J = 2.0 Hz, 1H).
[0146] Preparation of l-(3-Ethoxy-4-methoxyphenyi)-2-
(niethylsulfonyl)ethanamine (Compound 3). Dimethyl sulfone (2.60 gm, 27.1 mmol, Aldrich) and tetrahydrofuran (10 mL, Aldrich) were charged to a 250 mL three-necked flask at room temperature. The mixture was cooled to 0 – 5 °C, and the solution gradually turned white. n-Butyllithium (10.8 mL, 27.1 mmol, 2.5 M solution in hexanes, Aldrich) was added to the flask at a rate such that the reaction mixture was maintained at 5 – 10 °C. The mixture was stirred at 0 – 5 °C for one hour, turning light-yellow. 3-Ethoxy-4-methoxybenzonitrile (Compound 2, 4.01 gm, 22.5 mmol) in tetrahydrofuran (8 mL) was then charged to the flask at a rate such that the reaction mixture was maintained at 0 – 5 °C. The mixture was stirred at 0 – 5 °C for another 15 minutes. After warming to room temperature, the reaction mixture was stirred for another 1.5 hours and then transferred to a second 250 mL three-necked flask containing a suspension of sodium borohydride (1.13 gm, 29.3 mmol, Aldrich) in
tetrahydrofuran (1 1 mL), maintained at – 5 – 0 °C for 30 minutes. Trifluoroacetic acid (“TFA,” 5.26 mL, 68.3 mmol, Aldrich) was charged to the flask at a rate such that the reaction mixture was maintained at 0 – 5 °C. The mixture was stirred at 0 – 5 °C for 40 minutes and an additional 17 hours at room temperature. The reaction mixture was then charged with 2.7 mL of deionized water over five minutes at room temperature. The mxiture was stirred at room temperature for 15 hours. Aqueous NaOH (10 N, 4.9 mL) was charged to the flask over 15 minutes at 45 °C. The mixture was stirred at 45 °C for two hours, at 60 °C for 1.5 hours, and at room temperature overnight. After approximately 17 hours at room temperature the mixture was cooled to 0 °C for thirty minutes and then concentrated under reduced pressure. The residual material was charged with deionized water (3 mL) and absolute ethanol (3 mL) and stirred at 0 – 5 °C for 2 hours. The mixture was filtered under vacuum, and the filtered solid was washed with cold absolute ethanol (3 x 5 mL), followed by deionized water until the pH of the wash was about 8. The solid was air dried overnight, and then in a vacuum oven at 60 °C for 17 hours to afford Compound 3 as a white solid (4.75 gm, 77 %). MS (ESI positive ion) m/z 274.1 (M + 1). Ή-NMR (500 MHz, DMSO-c¾): δ ppm 1.32 (t, J = 7.0 Hz, 3H), 2.08 (bs, 2H), 2.95 (s, 3H), 3.23 (dd, J = 4.0 Hz, 1H), 3.40 (dd, J = 9.5 Hz, 1H), 3.72 (s, 3H), 4.01 (q, J = 7.0 Hz, 2H), 4.25 (dd, J = 3.5 Hz, 1H), 6.88 (s, 2H), 7.02 (s, 1H).
[0147] Preparation of 4-Nitroisobenzofuran-l,3-dione (Compound 5). Into a 250 mL round bottom flask, fitted with a reflux condenser, was placed 3-nitrophthalic acid (21.0 gm, 99 mmol, Aldrich) and acetic anhydride (18.8 mL, 199 mmol, Aldrich). The solid mixture was heated to 85 °C, under nitrogen, with gradual melting of the solids. The yellow mixture was heated at 85 °C for 15 minutes, and there was noticeable thickening of the mixture. After 15 minutes at 85 °C, the hot mixture was poured into a weighing dish, and allowed to cool. The yellow solid was grinded to a powder and then placed on a cintered funnel, under vacuum. The solid was washed with diethyl ether (3 x 15 mL), under vacuum and allowed to air dry overnight, to afford 4-nitroisobenzofuran-l ,3-dione, Compound 5, as a light-yellow solid (15.8 gm, 82 %). MS (ESI positive ion) m/z 194.0 (M + 1). TLC: Rf = 0.37 (10% MeOH/DCM with 2 drops Acetic acid) Ή-NMR (500 MHz, DMSO-i¾: δ ppm 8.21 (dd, J = 7.5 Hz, 1H), 8.39 (dd, J = 7.5 Hz, 1H), 8.50 (dd, J = 7.5 Hz, 1 H).
[0148] Preparation of 2-(l-(3-Ethoxy-4-methoxyphenyI)-2-
(methylsulfonyl)ethyl)-4-nitroisoindoline-l,3-dione (Compound 6). Into a 2 – 5 mL microwave vial was added 4-nitroisobenzofuran-l ,3-dione (Compound 5, 0.35 gm, 1.82 mmol), the amino-sulfone intermediate (Compound 3, 0.50 gm, 1.82 mmol) and 4.0 mL of glacial acetic acid. The mixture was placed in a microwave at 125 °C for 30 minutes. After 30 minutes the acetic acid was removed under reduced pressure. The yellow oil was taken up in ethyl acetate and applied to a 10 gm snap Biotage samplet. Purification by silica gel chromatography (0 to 20 % Ethyl Acetate/Hexanes) afforded Compound 6 as a light-yellow solid (0.67 gm, 82 %). MS (ESI positive ion) m/z 449.0 (M + 1). TLC: Rf = 0.19
(EtOAc:Hexanes, 1 : 1). HPLC indicated 99% purity by peak area. Ή-NMR (500 MHz, DMSO-c¾: δ ppm 1.32 (t, 3H), 2.99 (s, 3H), 3.73 (s, 3H), 4.02 (m, 2H), 4.21 (dd, J = 5.0 Hz, 1H), 4.29 (dd, J = 10.0 Hz, 1H), 5.81 (dd, J = 5.0 Hz, 1H), 6.93 (d, J – 8.5 Hz, 1H), 7.00 (dd, J = 2.0 Hz, 1H), 7.10 (d, J = 2.5 Hz, 1H), 8.07 (t, J = 15.5 Hz, 1H), 8.19 (dd, J = 8.5 Hz, 1H), 8.30 (dd, J = 9.0 Hz, 1H).
[0149] Preparation of 4-Amino-2-(l-(3-ethoxy-4-methoxyphenyl)-2-
(methylsulfonyl)ethyl)isoindoline-l,3-dione (Compound 7). Compound 6 (0.54 gm, 1.20 mmol) was taken up in ethyl acetate / acetone (1 : 1 , 24 mL) and flowed through the H-cube™ hydrogen reactor using a 10 % Pd/C CatCart™ catalyst cartridge system (ThalesNano, Budapest Hungary). After eluting, the yellow solvent was concentrated under reduced pressure to give Compound 7 as a yellow foam solid (0.48 gm, 95 %). MS (ESI positive ion) m/z 419.1 (M + 1). 1H-NMR (500 MHz, DMSO-<¾): δ ppm 1.31 (t, J = 7.0 Hz, 3H), 2.99 (s, 3H), 3.72 (s, 3H), 4.04 (q, J = 7.0 Hz, 2H), 4.09 (m, 1H), 4.34 (m, 1H), 5.71 (dd, J = 5.5 Hz, 1H), 6.52 (bs, 2H), 6.92-6.98 (m, 3H), 7.06 (bs, 1 H), 7.42 (dd, J = 7.0 Hz, 1H).
[0150] Preparation of N-(2-(l-(3-ethoxy-4-methoxyphenyl)-2-
(methylsuIfonyl)ethyl)-l,3-dioxoisoindolin-4-yl)acetamide (Apremilast, Compound 8).
Into a 2-5 mL microwave vial was placed Compound 7 (0.18 gm, 0.43 mmol), acetic anhydride (0.052 mL, 0.53 mmol) and acetic acid (4 mL). The microwave vial was placed into a Biotage microwave and heated to 125 °C for 30 minutes. The solvents were removed under reduced pressure and the residue was purified by silica gel chromatography (0 to 5% MeOH/DCM) to afford apremilast (Compound 8) as a yellow oil (0.14 gm, 71%). HPLC indicated 94.6% purity by peak area.
1H-NMR (500 MHz, DMSO-c 6): δ ppm 1.31 (t, 3H), 2.18 (s, 3H), 3.01 (s, 3H), 3.73 (s, 3H), 4.01 (t, J = 7.0 Hz, 2H), 4,14 (dd, J = 4.0 Hz, 1H), 4.33 (m, 1H), 5.76 (dd, J = 3.0 Hz, 1H), 6.95 (m, 2H), 7.06 (d, J = 1.5 Hz, 1H), 7.56 (d, J = 7.0 Hz, 1H), 7.79 (t, J = 7.7 Hz, 1H), 8.43 (d, J = 8.5 Hz, 1H), 9.72 (bs, 1H).
……………………..
SYNTHESIS
5. EXAMPLES
Certain embodiments provided herein are illustrated by the following non-limiting examples.
5.1 PREPARATION OF (+)-2-[l-(3-ETHOXY-4-METHOXYPHENYL)-2- METHANESULFONYLETHYLJ-4- ACETYL AMINOISOINDOLIN-1,3- DIONE (APREMILAST)

5.1.1 Preparation of 3-aminopthalic acid
10% Pd/C (2.5 g), 3-nitrophthalic acid (75.0 g, 355 mmol) and ethanol (1.5 L) were charged to a 2.5 L Parr hydrogenator under a nitrogen atmosphere. Hydrogen was charged to the reaction vessel for up to 55 psi. The mixture was shaken for 13 hours, maintaining hydrogen pressure between 50 and 55 psi. Hydrogen was released and the mixture was purged with nitrogen 3 times. The suspension was filtered through a celite bed and rinsed with methanol. The filtrate was concentrated in vacuo. The resulting solid was reslurried in ether and isolated by vacuum filtration. The solid was dried in vacua to a constant weight, affording 54 g (84%> yield) of 3-aminopthalic acid as a yellow product. 1H-NMR (DMSO-d6) δ: 3.17 (s, 2H), 6.67 (d, 1H), 6.82 (d, 1H), 7.17 (t, 1H), 8-10 (brs, 2H). 13C-NMR(DMSO-d6) δ: 112.00, 115.32, 118.20, 131.28, 135.86, 148.82, 169.15, 170.09.
5.1.2 Preparation of 3-acetamidopthalic anhydride
A I L 3 -necked round bottom flask was equipped with a mechanical stirrer, thermometer, and condenser and charged with 3-aminophthalic acid (108 g, 596 mmol) and acetic anhydride (550 mL). The reaction mixture was heated to reflux for 3 hours and cooled to ambient temperature and further to 0-5. degree. C. for another 1 hour. The crystalline solid was collected by vacuum filtration and washed with ether. The solid product was dried in vacua at ambient temperature to a constant weight, giving 75 g (61% yield) of 3-acetamidopthalic anhydride as a white product. 1H-NMR (CDCI3) δ: 2.21 (s, 3H), 7.76 (d, 1H), 7.94 (t, 1H), 8.42 (d, 1H), 9.84 (s, 1H).
5.1.3 Resolution of 2-(3-ethoxy-4-methoxyphenyl)-l-(methylsulphonyl)- ethyl-2-amine
A 3 L 3 -necked round bottom flask was equipped with a mechanical stirrer, thermometer, and condenser and charged with 2-(3-ethoxy-4-methoxyphenyl)-l-(methylsulphonyl)-eth-2-ylamine (137.0 g, 500 mmol), N-acetyl-L-leucine (52 g, 300 mmol), and methanol (1.0 L). The stirred slurry was heated to reflux for 1 hour. The stirred mixture was allowed to cool to ambient temperature and stirring was continued for another 3 hours at ambient temperature. The slurry was filtered and washed with methanol (250 mL). The solid was air-dried and then dried in vacuo at ambient temperature to a constant weight, giving 109.5 g (98% yield) of the crude product (85.8% ee). The crude solid (55.0 g) and methanol (440 mL) were brought to reflux for 1 hour, cooled to room temperature and stirred for an additional 3 hours at ambient temperature. The slurry was filtered and the filter cake was washed with methanol (200 mL). The solid was air-dried and then dried in vacuo at 30°C. to a constant weight, yielding 49.6 g (90%> recovery) of (S)-2-(3-ethoxy-4- methoxyphenyl)-l-(methylsulphonyl)-eth-2-ylamine-N-acety 1-L-leucine salt (98.4% ee). Chiral HPLC (1/99 EtOH/20 mM KH2P04 @pH 7.0, Ultron Chiral ES-OVS from Agilent Technologies, 150 mm.times.4.6 mm, 0.5 mL/min., @240 nm): 18.4 min (S-isomer, 99.2%), 25.5 min (R-isomer, 0.8%)
5.1.4 Preparation of (+)-2-[l-(3-ethoxy-4-methoxyphenyl)-2- methanesulfonylethyl] -4-acetylaminoisoindolin- 1 ,3-dione
A 500 mL 3 -necked round bottom flask was equipped with a mechanical stirrer,
thermometer, and condenser. The reaction vessel was charged with (S)-2-(3-ethoxy-4- methoxyphenyl)-l-(methylsulphonyl)-eth-2-yl amine N-acetyl-L-leucine salt (25 g, 56 mmol, 98% ee), 3-acetamidophthalic anhydride (12.1 g, 58.8 mmol), and glacial acetic acid (250 mL). The mixture was refluxed over night and then cooled to <50°C. The solvent was removed in vacuo, and the residue was dissolved in ethyl acetate. The resulting solution was washed with water (250 mL x
2), saturated aqeous NaHC03 (250 mL.times.2), brine (250 mL.times.2), and dried over sodium sulphate. The solvent was evaporated in vacuo, and the residue recrystallized from a binary solvent containing ethanol (150 mL) and acetone (75 mL). The solid was isolated by vacuum filtration and washed with ethanol (100 mL.times.2). The product was dried in vacuo at 60°C. to a constant weight, affording 19.4 g (75% yield) of Compound 3 APREMILAST with 98% ee. Chiral HPLC (15/85 EtOH/20 mM KH2P04 @pH 3.5, Ultron Chiral ES-OVS from Agilent Technology, 150 mm x 4.6 mm, 0.4 mL/min., @240 nm): 25.4 min (S-isomer, 98.7%), 29.5 min (R-isomer, 1.2%).
1H-NMR (CDC13) δ: 1.47 (t, 3H), 2.26 (s, 3H), 2.87 (s, 3H), 3.68-3.75 (dd, 1H), 3.85 (s, 3H), 4.07-4.15 (q, 2H), 4.51-4.61 (dd, 1H), 5.84-5.90 (dd, 1H), 6.82-8.77 (m, 6H), 9.46 (s, 1H).
13C-NMR(DMSO-d6) δ: 14.66, 24.92, 41.61, 48.53, 54.46, 55.91, 64.51, 111.44, 112.40, 115.10, 118.20, 120.28, 124.94, 129.22, 131.02, 136.09, 137.60, 148.62, 149.74, 167.46, 169.14, 169.48.
…………………………………..
NMR
1H-NMR (CDCl3) δ: 1.47 (t, 3H), 2.26 (s, 3H), 2.87 (s, 3H), 3.68-3.75 (dd, 1H), 3.85 (s, 3H), 4.07-4.15 (q, 2H), 4.51-4.61 (dd, 1H), 5.84-5.90 (dd, 1H), 6.82-8.77 (m, 6H), 9.46 (s, 1H). 13C-NMR (DMSO-d6) δ: 14.66, 24.92, 41.61, 48.53, 54.46, 55.91, 64.51, 111.44, 112.40, 115.10, 118.20, 120.28, 124.94, 129.22, 131.02, 136.09, 137.60, 148.62, 149.74, 167.46, 169.14, 169.48.
…………….

aReagents and conditions: (a) LiN(SiMe3)2, then Me2SO2/n-BuLi/BF3Et2O, −78 °C; (b) N-Ac-l-leucine, MeOH; (c) HOAc, reflux.
……………………
SARCOIDOSIS
Sarcoidosis is a disease of unknown cause. Sarcoidosis is characterized by the presence of granulomas in one or more organ systems. The most common sites of involvement are the lungs and the lymph nodes in the mediastinum and hilar regions. However, sarcoidosis is a systemic disease and a variety of organ systems or tissues may be the source of primary or concomitant clinical manifestations and morbidity. The clinical course of sarcoidosis is extremely variable, and ranges from a mild or even asymptomatic disease with spontaneous resolution to a chronic progressive disease leading to organ system failure and, in 1-5% of cases, death. See Cecil
Textbook of Medicine, 21st ed. (Goldman, L., Bennett, J. C. eds), W. B. Saunders Company, Philadelphia, 2000, p. 433-436.
While the cause of sarcoidosis is unknown, a substantial body of information suggests that immune mechanisms are important in disease pathogenesis. For example, sarcoidosis is
characterized by enhanced lymphocyte and macrophage activity. See Thomas, P.D. and
Hunninghake, G.W., Am. Rev. Respir. Dis., 1987, 135: 747-760. As sarcoidosis progresses, skin rashes, erythema nodosum and granulomas may form. Granulomas or fibrosis caused by sarcoidosis can occur throughout the body, and may affect the function of vital organs such as the lungs, heart, nervous system, liver or kidneys. In these cases, the sarcoidosis can be fatal. See
http://www.nlm.nih.gov/medlineplus/sarcoidosis.html (accessed November 12, 2009).
Moreover, a variety of exogenous agents, both infectious and non-infectious, have been hypothesized as a possible cause of sarcoidosis. See Vokurka et ah, Am. J. Respir. Crit. Care Med., 1997, 156: 1000-1003; Popper et al, Hum. Pathol, 1997, 28: 796-800; Almenoff et al, Thorax, 1996, 51 : 530-533; Baughman et al., Lancet, 2003, 361 : 1111-1118. These agents include mycobaceria, fungi, spirochetes, and the agent associated with Whipple’s disease. Id.
Sarcoidosis may be acute or chronic. Specific types of sarcoidosis include, but are not limited to, cardiac sarcoidosis, cutaneous sarcoidosis, hepatic sarcoidosis, oral sarcoidosis, pulmonary sarcoidosis, neurosarcoidosis, sinonasal sarcoidosis, Lofgren’s syndrome, lupus pernio, uveitis or chronic cutaneous sarcoidosis.
As the lung is constantly confronted with airborne substances, including pathogens, many researchers have directed their attention to identification of potential causative transmissible agents and their contribution to the mechanism of pulmonary granuloma formation associated with sarcoidosis. See Conron, M. and Du Bois, R.M., Clin. Exp. Allergy, 2001, 31 : 543-554; Agostini et al, Curr. Opin. Pulm. Med. , 2002, 8: 435-440.
Corticosteroid drugs are the primary treatment for the inflammation and granuloma formation associated with sarcoidosis. Rizatto et al. , Respiratory Medicine, 1997, 91 : 449-460. Prednisone is most often prescribed drug for the treatment of sarcoidosis. Additional drugs used to treat sarcoidosis include methotrexate, azathioprine, hydroxychloroquine, cyclophosphamide, minocycline, doxycycline and chloroquin. TNF-a blockers such as thalidomide and infliximab have been reported to be effective in treating patients with sarcoidosis. Baughman et al, Chest, 2002, 122: 227-232; Doty et al, Chest, 2005, 127: 1064-1071. Antibiotics have also been studied for the treatment of sarcoidosis, such as penicillin antibiotics, cephalosporin antibiotics, macrolide antibiotics, lincomycin antibiotics, and tetracycline antibiotics. Specific examples include minocycline hydrochloride, clindamycin, ampicillin, or clarithromycin. See, e.g., U.S. Patent Publication No. 2007/0111956.
There currently lacks a Food and Drug Administration-approved therapeutic agent for the treatment of sarcoidosis, and many patients are unable to tolerate the side effects of the standard corticosteroid therapy. See Doty et al, Chest, 2005, 127: 1064-1071. Furthermore, many cases of sarcoidosis are refractory to standard therapy. Id. Therefore, a demand exists for new methods and compositions that can be used to treat patients with sarcoidosis.
……………..
合成路线:
US2013217918A1
US2014081032A1
PATENTS
|
8-15-2012
|
PROCESSES FOR THE PREPARATION OF AMINOSULFONE COMPOUNDS
|
|
|
11-4-2011
|
HETEROCYCLIC COMPOUNDS AS PHOSPHODIESTERASE INHIBITORS
|
|
|
5-27-2011
|
Nanosuspension of a Poorly Soluble Drug via Microfluidization Process
|
|
|
5-28-2010
|
METHODS AND COMPOSITIONS USING PDE4 INHIBITORS FOR THE TREATMENT AND MANAGEMENT OF CANCERS
|
Garcinia Cambogia Kills 89% of Pancreatic Cancer Cells and Synergizes with Curcumin

Garcinia Cambogia Extract Explained
The latest in innovation in weight loss supplements is Garcinia Cambogia. It is unparalleled in its ability to help boost your body’s weight loss potential, and help you achieve your perfect weight.
There’s no wonder it’s quickly gained a huge following, with endorsements from celebrities to health experts, with scientifically proven ability to help you increase your fat burning power.
As with all supplements like this, there are questions as to how it works, and just how it can benefit you, with your health and in losing weight. This site’s goal is to hopefully answer some of these questions, and to show you just how you can benefit from this amazing supplement.

What is Garcinia Cambogia?
Garcinia Cambogia is a fruit, that is grown all over Asia, but originating in Indonesia and grows particularly well grows best with tropical conditions. It rose to prominence after appearance on the massively lauded American health show, Doctor Oz. It had recently been subject to a medical trial where the study scientifically proved it was highly effective in increasing burning up fat and aiding in overall weight loss.
Can Garcinia Cambogia Extract Help Me Lose Weight?
Well Garcinia Cambogia contains a useful compound called Hydroxycitric Acid, which I’ll refer to as HCA for ease of reference. Garcinia Cambogia contains one of the highest known concentrations of HCA, and this was why it was noticed as a potential weight loss supplement. HCA has two main mechanisms in which it works to boost your fat burning potential:

Firstly it will reduce the ability for the body to convert carbohydrates into fat cells, meaning that even without a calorific controlled diet; you will be able to aid your body’s ability to burn of existing fat, while not gaining additional fat.
Secondly it will also suppress your appetite, meaning that it will not only help reduce the weight you can put on by stopping putting on additional fat, it will also massively reduce the cravings and hunger that usually lead to breaking a diet and weight loss routine. This means that your body will just be burning off the existing fat, helping you to achieve that perfect weight!
What About Side Effects form Garcinia Cambogia?
The most amazing thing about Garcinia Cambogia is that the side effects of the product are almost non-existent in the all-natural extract. By this I mean an extract that contains purely Garcinia Cambogia extract without any additional additives that some unrepeatable sellers will try to pass off as the quality product. Those extracts that contain additives can cause side effect in users of Garcinia Cambogia, which are related to the different additives and binding agents added.
The cost of Garcinia Cambogia from a supplier, whom ensures a high quality and natural product, will range from $40-50 a bottle. There is however introductory offers from some suppliers, such as Miracle Garcinia Cambogia currently offering a free bottle of Garcinia Cambogia with every order.
This means the overall cost per bottle of this amazing product can drop as low as $28.99. Most of these offers unfortunately do have a limited stock and therefore won’t be around forever.
Garcinia gummi-gutta is a tropical[2] species of Garcinia native to Indonesia. Common names include garcinia cambogia (a former scientific name), as well as gambooge, brindleberry,[3] brindall berry, Malabar tamarind,[2] assam fruit, vadakkan puli (northern tamarind) and kudam puli (pot tamarind).[4] This fruit looks like a small pumpkin and is green to pale yellow in color. It has recently received considerable media attention because of its purported effects on weight loss, although there is no clinical evidence to support this claim.

Cultivation


Ripe fruit
Garcinia gummi-gutta is grown for its fruit in southeast Asia, coastal Karnataka/Kerala, India, and west and central Africa. It thrives in most moist forests.
Garcinia gummi-gutta is one of several closely related Garcinia species from the plant family Guttiferae.[5] With thin skin and deep vertical lobes, the fruit of G. gummi-gutta and related species range from about the size of an orange to that of a grapefruit; G. gummi-gutta looks more like a small yellowish, greenish or sometimes reddish pumpkin.[6] The color can vary considerably. When the rinds are dried and cured in preparation for storage and extraction, they are dark brown or black in color.
Along the west coast of South India, G. gummi-gutta is popularly termed “Malabar tamarind,” and shares culinary uses with the tamarind (Tamarindus indica). The latter is a small and the former a quite large evergreen tree. G. gummi-gutta is also called “goraka” or, in some areas, simply “kattcha puli” (souring fruit).
Uses
Cooking
Garcinia gummi-gutta is used in cooking, including in the preparation of curries. The fruit rind and extracts of Garcinia species are called for in many traditional recipes,[7] and various species of Garcinia are used similarly in food preparation in Assam (India), Thailand, Malaysia, Burma and other Southeast Asian countries. In the Indian Ayurvedic medicine, “sour” flavors are said to activate digestion. The extract and rind of Garcinia gummi-gutta is a curry condiment in India. It is an essential souring ingredient in the Southern Thai variant of kaeng som, a sour curry.
Garcinia gummi-gutta is employed commercially in fish curing, especially in Sri Lanka (Colombo curing) and South India, which makes use of the antibacterial qualities of the fruit.
The trees can be found in forested areas and also are protected in plantations otherwise given over to pepper, spice, and coffee production.
Traditional medicine
Aside from its use in food preparation and preservation, extracts of G. gummi-gutta are sometimes used in traditional medicine aspurgatives. The fruit rind is also used to make medicine.
Weight loss
In late 2012, a United States television personality, Dr. Oz, promoted Garcinia cambogia extract as a “magic” weight-loss aid. Dr. Oz’s previous endorsements have often led to a substantial increase in consumer interest in the promoted products. However, a dearth of scientific evidence and clinical trials do not support claims that Garcinia cambogia is an effective weight-loss aid.[8][9] A meta-analysis found a possible small, short-term weight loss effect (under 1 kilogram).[10] However, side effects—namely hepatotoxicity (chemical-driven liver damage)—led to one preparation being withdrawn from the market.[11][12]
A 1998 randomized controlled trial looked at the effects of hydroxycitric acid, the purported active component in Garcinia gummi-gutta, as a potential antiobesity agent in 135 people. The conclusion from this trial was that “Garcinia cambogia failed to produce significant weight loss and fat mass loss beyond that observed with placebo”.[13]

When the fruit is sun dried for several days, it becomes black with a shrivelled body
References
- “Garcinia gummi-gutta (L.) Roxb.”. The Plant List. Royal Botanic Gardens, Kew and Missouri Botanical Garden. Retrieved 1 June 2013.
- “USDA GRIN Taxonomy”.
- “Potential treatments for insulin resistance in the horse: A comparative multi-species review”. Science Direct. Retrieved 6 October 2013.
- “Meals that heal – Soul curry”. The Hindu. Retrieved 3 October 2013.
- Publications & Information Directorate, Council of Scientific & Industrial Research (1986). G. cambogia Desr. The Useful Plants of India. (New Delhi: Publications & Information Directorate, 1986) 229.
- “Fruit yellowish or reddish, size of an orange having six or eight deep longitudinal grooves in its fleshy pericarp. Pulp acid of a pleasant flavor. It is dried among the Singalese who use it in curries.” Uphof, J.C. Th. (1968).
- “The acid rinds of the ripe fruit are eaten, and in Ceylon are dried, and eaten as a condiment in curries.” Drury, Heber (1873). “Garcinia gambogia(Desrous) N. 0. Clusiaceae”. The Useful Plants of India, second edition. London: William H. Allen & Co. p. 220.
- Belluz, Julia; Hoffman, Steven J. (1 January 2013). “Dr. Oz’s Miraculous Medical Advice; Pay no attention to that man behind the curtain”. Slate. The Slate Group. Retrieved 31 May 2013.
- Márquez F1, Babio N, Bulló M, Salas-Salvadó J (2012). “Evaluation of the safety and efficacy of hydroxycitric acid or Garcinia cambogia extracts in humans”. Crit Rev Food Sci Nutr 52 (7): 585–94. doi:10.1080/10408398.2010.500551. PMID 22530711.
- Hepatotoxicity (from hepatic toxicity) implies driven liver damage.
- Lobb, A. (2009). “Hepatoxicity associated with weight-loss supplements: A case for better post-marketing surveillance”. World Journal of Gastroenterology 15 (14): 1786–1787. doi:10.3748/wjg.15.1786. PMC 2668789. PMID 19360927.
- Kim YJ1, Choi MS, Park YB, Kim SR, Lee MK, Jung UJ (2013). “Garcinia Cambogia attenuates diet-induced adiposity but exacerbates hepatic collagen accumulation and inflammation”. World J Gastroenterol 19 (29): 4689–701. doi:10.3748/wjg.v19.i29.4689. PMID 23922466.
- Heymsfield, S. B.; Allison, D. B.; Vasselli, J. R.; Pietrobelli, A.; Greenfield, D.; Nunez, C. (1998). “Garcinia cambogia (Hydroxycitric Acid) as a Potential Antiobesity Agent: A Randomized Controlled Trial”. JAMA: the Journal of the American Medical Association 280 (18): 1596–1600.doi:10.1001/jama.280.18.1596. PMID 9820262.
When Antibiotics Fail, Are Herbs the Answer?


Complex Compounds in Natural Herbs:
Now looking at the rate of adaptation and mutability of bacteria, it is inevitable that they will form resistance to most forms of simplistic human made antibiotic compounds. And when everything fails we will fall back to the old biblical medicinal herbs such as Ginger, Garlic, Black Pepper, Ashwagandha, etc. These herbs not only contain dozens of mild antibiotic compounds, they are also widely available in abundance.
Brisbane scientists make cancer treatment breakthrough

DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY

