
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

HELP, Need one time help to pay 10 year concessional subscription to this, your favorite blog to WordPress
Just One viewer please come forward
Dear Kind Viewer’s
WordPress is kind to me and negotiated a one time 10 year concessional subscription of 260 US dollars…….https://newdrugapprovals.org/
I need one time help to pay this one time 10 year concessional subscription to our favorite blog.
This is done to keep this blog running even after my death.
Currently I am paying 99 US Dollars per annum
email me
amcrasto@gmail.com
call +919323115463
Paypal will work for me via email request to you by me, Indian govt does not allow automatic transfer via paypal buttons on the blog.
email me at amcrasto@gmail.com and tell me amount, i will request you via paypal

DR ANTHONY CRASTO
LIONEL MY SON, MY MOTIVATION
.
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed…
View original post 226 more words
Tianagliflozin IND filed by Tianjin Institute of Pharmaceutical research


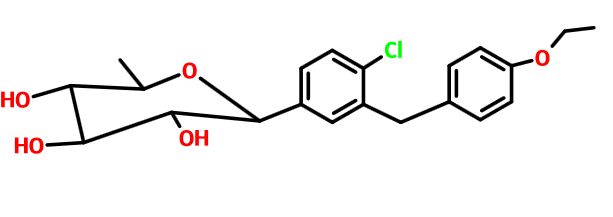
Tianagliflozin,
taigeliejing, 6-deoxydapagliflozin
| Molecular Formula: | C21H25ClO5 |
|---|---|
| Molecular Weight: | 392.8732 g/mol |
IND Filing…Tianjin Institute of Pharmaceutical research
Tianjin Institute Of Pharmaceutical Research,
(3R,4S,5S,6R)-2-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-6-methyloxane-3,4,5-triol
1–[4–Chloro–3–(4–ethoxybenzyl)phenyl]–1,6–dideoxy–β–d–glucopyranose
![]()
CAS N. 1461750-27-5
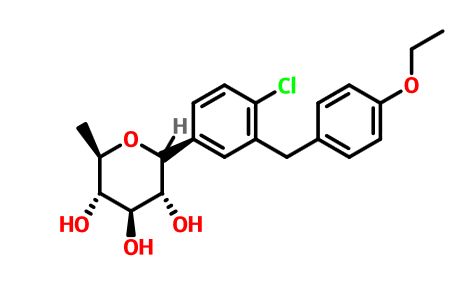

The structures of dapagliflozin and 6-deoxydapagliflozin (1)
,deletion of the 6-OH in the sugar moiety of dapagliflozin led to the discovery of a more potent SGLT2 inhibitor, 6-deoxydapagliflozin (1, ). In an in vitro assay, 1 was a more active SGLT2 inhibitor, with IC 50 = 0.67 nM against human SGLT2 (hSGLT2), as compared with 1.1 nM for dapagliflozin, leading to the identification of 1 as the most active SGLT2 inhibitor discovered so far in this field. Also in an in vivo assay, 1 also introduced more urinary glucose in a rat urinary glucose excretion test (UGE) and exhibited more potent blood glucose inhibitory activity in a rat oral glucose tolerance test (OGTT) than dapagliflozin.
Tianjin Institute Of Pharmaceutical Research,天津药物研究院

SPECTRAL DATA of Tianagliflozin
1 as a white solid (3.65 g, 93 %). R f = 0.35 (EtOAc);
m.p.: 148–149 °C;
1H NMR (400 MHz, DMSO-d 6): δ = 7.35 (d, 1H, J = 8.4 Hz), 7.25 (s, 1H), 7.18 (d, 1H, J = 8.0 Hz), 7.08 (d, 2H, J = 8.4 Hz), 6.81 (d, 2H, J = 8.4 Hz), 4.95 (d, 1H, J = 5.2 Hz, OH), 4.90 (d, 1H, J = 4.4 Hz, OH), 4.79 (d, 1H, J = 5.6 Hz, OH), 3.92–4.01 (m, 5H), 3.24–3.29 (m, 1H), 3.18–3.22 (m, 1H), 3.09–3.15 (m, 1H), 2.89–2.95 (m, 1H), 1.29 (t, 3H, J = 7.0 Hz, CH2 CH 3 ), 1.15 (d, 3H, J = 6.0 Hz, CHCH 3 ) ppm;
13C NMR (100 MHz, DMSO-d 6): δ = 156.85, 139.65, 137.82, 131.83, 131.16, 130.58, 129.52, 128.65, 127.14, 114.26, 80.71, 77.98, 75.77, 75.51, 74.81, 62.84, 37.55, 18.19, 14.62 ppm;
IR (KBr): v¯¯¯ = 3,564 (w), 3,385 (s), 2,981 (s), 2,899 (s), 2,861 (s), 1,613 (m), 1,512 (s), 1,477 (m), 1,247 (s), 1,102 (s), 1,045 (s), 1,012 (s) cm−1;
HR–MS: calcd for C21H29ClNO5 ([M + NH4]+) 410.1729, found 410.1724.
PATENT
CN 103864737
http://www.google.com/patents/CN103864737A?cl=en
PATENT
WO 2014094544
http://www.google.com/patents/WO2014094544A1?cl=en


-27-


1 D1 -6 Optionally, the step (7 ‘) is the step (7’) in place:
LS l- [4 – D (I- Dl- 6)

A.

(DMSO-d 6, 400 MHz), δ 7.35 (d, 1H, J = 8.0 Hz), 7.28 (d, 1H, J ‘. 2.0 Hz), 7.17 (dd, IH, / = 2.0 Hz and 8.4 Hz), 7.05 (d, 2H, J: 8.8 Hz), 6.79 (d, 2H, 8.8 Hz): 4.924,95 (m, 2H), 4,81 (d, IH, 6,0 Hz), 3.93- 3.99 (m, 5H), 3,85 (d, 1H, J = 10,4 Hz), 3,66 (dd, IH, 5,2 Hz and 11,6 Hz), 3.17-3,28 (m, 3H), 3.02-3.08 (m: IH), 1.28 (t, 3H, J = 7,0 Hz), 0,80 (s, 9H), -0.05 (s, 3H), -0.09 (s, 3H) .
PATENT
[0066] The added 100mL dried over anhydrous methanol 0. 5g of sodium metal, nitrogen at room temperature with stirring, until the sodium metal disappeared. Followed by addition of 5. 2g (10mmol) of compound 6, stirring was continued at room temperature for 3 hours. To the reaction system was added 5g strong acid cation exchange resin, stirred at room temperature overnight, the reaction mixture until pH = 7. The resin was removed by suction, and the filtrate evaporated to dryness on a rotary evaporator, the residue was further dried on a vacuum pump to give the product I-D1-6, as a white foamy solid.
PATENT
WO 2014139447
PATENT related
http://www.google.com/patents/WO2013044608A1?cl=en
http://link.springer.com/article/10.1007%2Fs40242-014-4043-9#/page-1
Design of SGLT2 Inhibitors for the Treatment of Type 2 Diabetes: A History Driven by Biology to Chemistry.
Abstract
A brief history of the design of sodium-dependent glucose cotransporter 2 (SGLT2) inhibitors is reviewed. The design of O-glucoside SGLT2 inhibitors by structural modification of phlorizin, a naturally occurring O-glucoside, in the early stage was a process mainly driven by biology with anticipation of improving SGLT2/SGLT1 selectivity and increasing metabolic stability. Discovery of dapagliflozin, a pioneering C-glucoside SGLT2 inhibitor developed by Bristol-Myers Squibb, represents an important milestone in this history. In the second stage, the design of C-glycoside SGLT2 inhibitors by modifications of the aglycone and glucose moiety of dapagliflozin, an original structural template for almost all C-glycoside SGLT2 inhibitors, was mainly driven by synthetic organic chemistry due to the challenge of designing dapagliflozin derivatives that are patentable, biologically active and synthetically accessible. Structure-activity relationships (SAR) of the SGLT2 inhibitors are also discussed.
http://www.ncbi.nlm.nih.gov/pubmed/25557661
Paper

Discovery of 6-Deoxydapagliflozin as a Highly Potent Sodium-dependent Glucose Cotransporter 2 (SGLT2) Inhibitor for the Treatment of Type 2 Diabetes
http://www.ingentaconnect.com/content/ben/mc/2014/00000010/00000003/art00009?crawler=true
CLIP

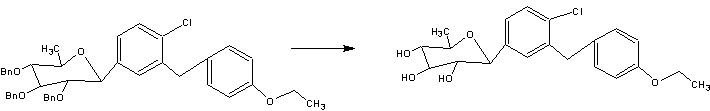
A facile synthesis of 6-deoxydapagliflozin


The synthetic route to the target compound 1 is shown in Scheme 3. The starting material methyl 2,3,4-tri-O-benzyl-6-deoxy-6-iodo-α–d-glucopyranoside (3) was prepared from commercially available methyl α–d-glucopyranoside (2) according to a known method [5, 6].
Iodide 3 was reductively deiodinated to give 4 in 91 % yield under hydrogenolytic conditions using 10 % Pd/C as catalyst in the presence of Et3N as base in THF/MeOH at room temperature.
when the iodide 3 was treated with Barton–McCombie reagent (n-Bu3SnH/AIBN) [7] in toluene at room temperature no reaction occurred; however, when the reaction was carried out at elevated temperatures, such as reflux, a complex mixture formed with only a trace amount (3 %, entry 1) of the desired product 4.
When the iodide 3 was treated with LiAlH4 in THF at 0 °C to room temperature, another complex mixture was produced with only a trace amount (2 %, entry 2) of 4.
When Pd(OH)2 was used as the hydrogenolysis catalyst instead of 10 % Pd/C, the desired 4 was indeed formed (14 %, entry 4), but most of the starting material was converted to a few more polar byproducts, which were believed to result from the cleavage of at least one of the benzyl groups.
pdf available
Monatshefte für Chemie – Chemical Monthly
December 2013, Volume 144, Issue 12, pp 1903-1910
////////IND Filing, SGLT-2 inhibitor, type 2 diabetes, Tianagliflozin, taigeliejing, 6-deoxydapagliflozin, 1461750-27-5
Clc1c(cc(cc1)C2[C@@H]([C@H]([C@@H]([C@H](O2)C)O)O)O)Cc3ccc(cc3)OCC
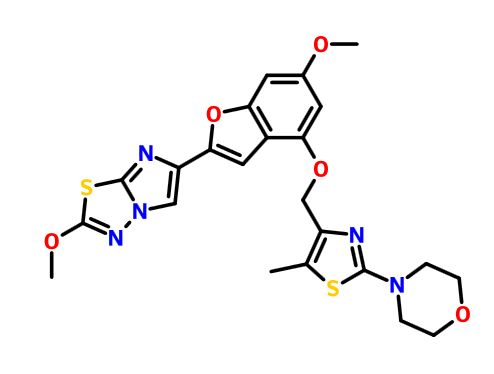
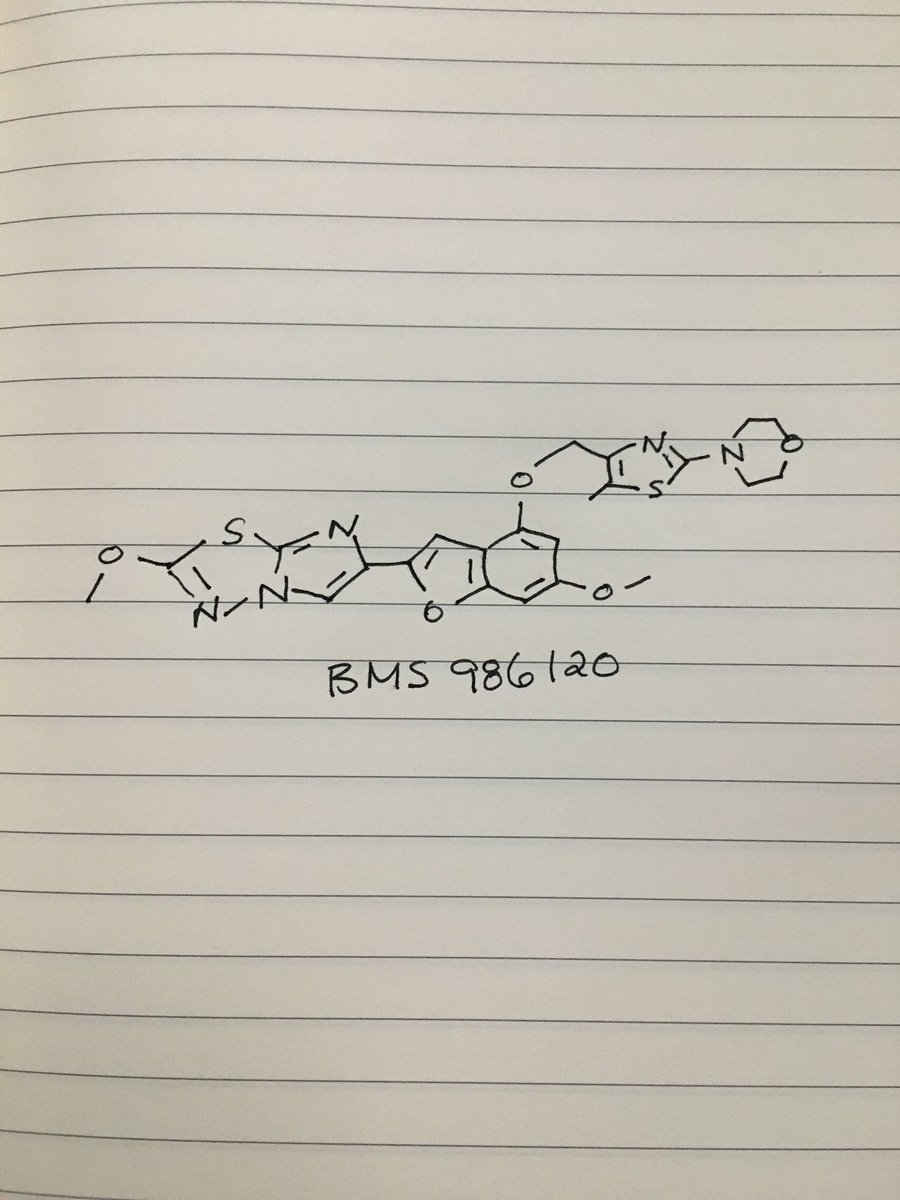
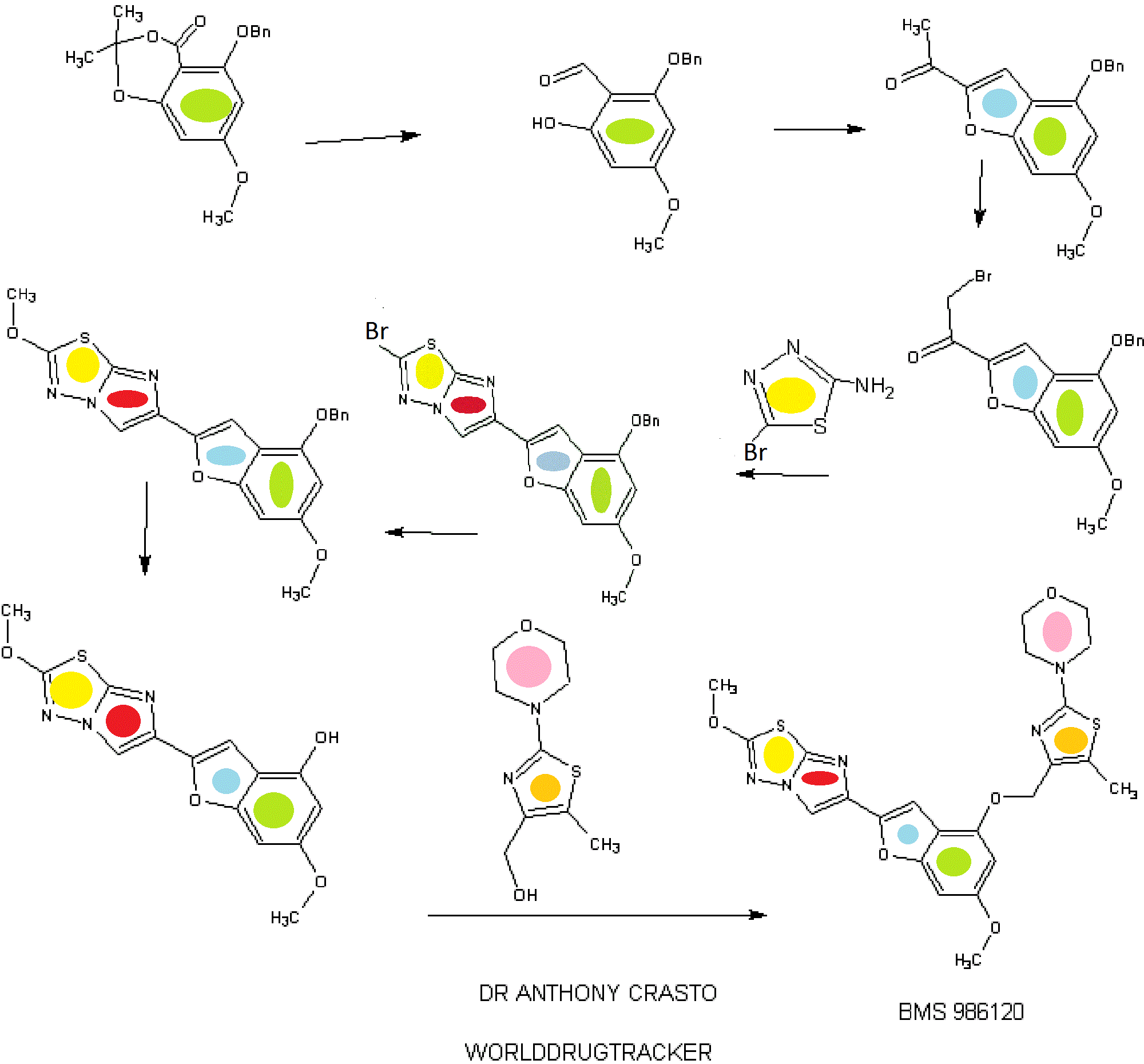
BMS 986120

 .
.
Picture credit….Bethany Halford
BMS 986120
Originator Bristol-Myers Squibb
Bristol-Myers Squibb Company, Université de Montréal
| Molecular Formula: | C23H23N5O5S2 |
|---|---|
| Molecular Weight: | 513.58922 g/mol |
4-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-5-methyl-1,3-thiazol-2-yl]morpholine
4-(4-(((6-Methoxy-2-(2-methoxyimidazo[2,l-b][l,3,4]thiadiazol-6-yl)benzofuran-4-yl) oxy)methyl)-5-methylthiazol-2-yl)morpholine
Imidazo[2,1-b] -1,3,4-thiadiazole, 2-methoxy-6-[6-methoxy-4-[[5-methyl-2-(4-morpholinyl)-4- thiazolyl]methoxy]-2-benzofuranyl]-
CAS 1478712-37-6
Phase I Thrombosis
- 02 Apr 2015 Bristol-Myers Squibb plans a phase I trial in Thrombosis (In volunteers) in United Kingdom (NCT02439190)
- 01 Aug 2014 Preclinical trials in Thrombosis in USA (PO)
https://clinicaltrials.gov/ct2/show/NCT02208882
https://clinicaltrials.gov/ct2/show/NCT02439190
Class Imidazoles; Small molecules; Thiadiazoles
$BMY antithrombic compound
PATENT
http://www.google.com/patents/WO2013163279A1?cl=en
Thromboembolic diseases remain the leading cause of death in developed countries despite the availability of anticoagulants such as warfarin (COUMADIN®), heparin, low molecular weight heparins (LMWH), synthetic pentasaccharides, and antiplatelet agents such as aspirin and clopidogrel (PLAVIX®).
Current anti-platelet therapies have limitations including increased risk of bleeding as well as partial efficacy (relative cardiovascular risk reduction in the 20 to
30% range). Thus, discovering and developing safe and efficacious oral or parenteral antithrombotics for the prevention and treatment of a wide range of thromboembolic disorders remains an important goal.
Alpha-thrombin is the most potent known activator of platelet aggregation and degranulation. Activation of platelets is causally involved in atherothrombotic vascular occlusions. Thrombin activates platelets by cleaving G-protein coupled receptors termed protease activated receptors (PARs). PARs provide their own cryptic ligand present in the N-terminal extracellular domain that is unmasked by proteolytic cleavage, with subsequent intramolecular binding to the receptor to induce signaling (tethered ligand mechanism; Coughlin, S.R., Nature, 407:258-264 (2000)). Synthetic peptides that mimic the sequence of the newly formed N-terminus upon proteolytic activation can induce signaling independent of receptor cleavage. Platelets are a key player in atherothrombotic events. Human platelets express at least two thrombin receptors, commonly referred to as PARI and PAR4. Inhibitors of PARI have been investigated extensively, and several compounds, including vorapaxar and atopaxar have advanced into late stage clinical trials. Recently, in the TRACER phase III trial in ACS patients, vorapaxar did not significantly reduce cardiovascular events, but significantly increased the risk of major bleeding (Tricoci, P. et al, N. Eng. J. Med., 366(l):20-33 (2012). Thus, there remains a need to discover new antiplatelet agents with increased efficacy and reduced bleeding side effects.
There are several early reports of preclinical studies of PAR4 inhibitors. Lee, F-Y. et al., “Synthesis of l-Benzyl-3-(5′-hydroxymethyl-2′-furyl)indazole Analogues as Novel Antiplatelet Agents”, J. Med. Chem., 44(22):3746-3749 (2001) discloses in the abstract that the compound

58
“was found to be a selective and potent inhibitor or protease-activated receptor type 4 (PAR4)-dependent platelet activation. ”
Compound 58 is also referred to as YD-3 in Wu, C-C. et al, “Selective Inhibition of Protease-activated Receptor 4-dependent Platelet Activation by YD-3”, Thromb. Haemost., 87: 1026-1033 (2002). Also, see Chen, H.S. et al, “Synthesis and platelet activity”, J. Bioorg. Med. Chem., 16: 1262-1278 (2008).
EP1166785 Al and EP0667345 disclose various pyrazole derivatives which are useful as inhibitors of platelet aggregation.\
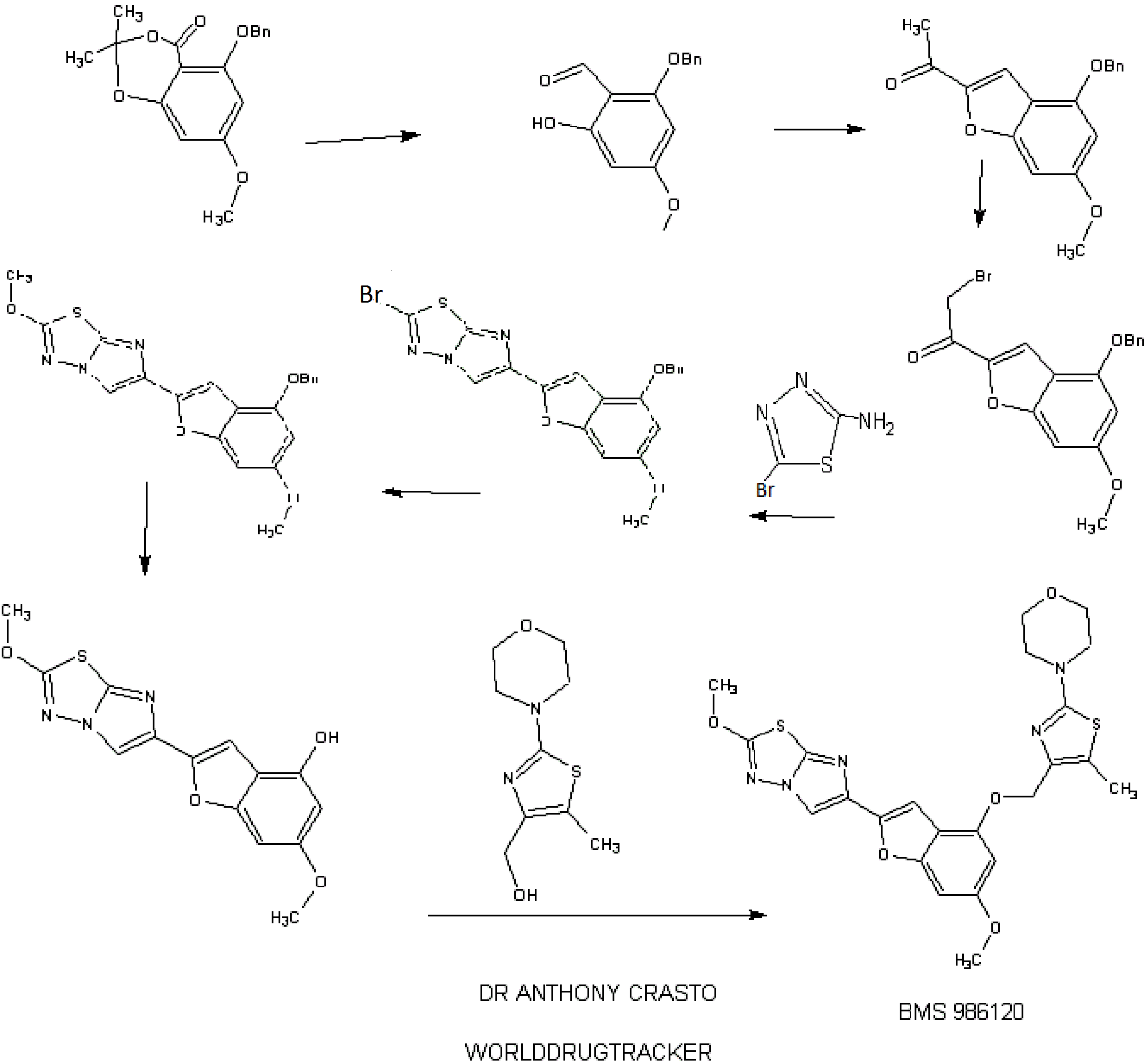
IB. 5-(Benzyloxy)-7-methoxy-2,2-dimethyl-4H-benzo[d][l,3]dioxin-4-one

A solution of 5-hydroxy-7-methoxy-2,2-dimethyl-4H-benzo[d][l,3]dioxin-4- one (30.00 g, 0.134 mol, see Kamisuki, S. et al, Tetrahedron, 60:5695-5700 (2004) for preparation) in N,N-dimethylformamide (400 mL) was treated with powdered anhydrous potassium carbonate (19.41 g, 0.14 mol) added all at once. The resulting mixture was stirred in vacuo for 10 min. and then flushed with nitrogen. The reaction flask was placed in a water bath (22 °C) and treated with benzyl bromide (24.03 g, 0.14 mol) added dropwise over 15 min. The resulting mixture was then stirred at 22 °C for 18 h (no starting material left by tic). The solid was filtered and washed with N,N- dimethylformamide. The filtrate was evaporated in vacuo and the residual oil was diluted with ethyl acetate (500 mL), washed with cold 0.1 N hydrochloric acid, saturated sodium bicarbonate and brine. After drying over anhydrous magnesium sulfate, evaporation of the solvent gave a thick syrup. Crystallization form ethyl acetate (50 mL) and hexane (150 mL) gave 35.17 g of 5-(benzyloxy)-7-methoxy-2,2-dimethyl-4H- benzo[d][l ,3]dioxin-4-one as large colorless prisms. Chromatography of the mother liquors on silica gel (4 x 13 cm, elution toluene – ethyl acetate 0-5%) gave 6.64 g of additional material to afford a total yield of 41.81 g (99%). HRMS(ESI) calcd for
Ci8Hi905 [M+H]+ m/z 315.1227, found 315.1386. 1H NMR (CDC13, 600 MHz) δ 1.68 (s, 6H), 3.77 (s, 3H), 5.19 (s, 2H), 5.19 (s, 2H), 6.04 (d, J = 2.03 Hz, 1H), 6.15 (d, J = 2.03 Hz, 1H), 7.27 (broad t, 1H), 7.36 (broad t, 2H), 7.52 (broad d, 2H).
1 C. 2-(Benzyloxy)-6-hydroxy-4-methoxybenzaldehyde

A solution of 5-(benzyloxy)-7-methoxy-2,2-dimethyl-4H-benzo[d][l ,3]dioxin- 4-one (Example IB, 6.76 g, 21.5 mmol) in dichloromethane (120 mL) was cooled to -78 °C and treated with 43 mL (64.5 mmol) of a 1.5 M solution of diisobutylaluminum hydride in toluene added dropwise over 20 min. The resulting mixture was then stirred at -78 °C for 3 h. The reaction mixture was quenched by the careful addition of methanol (5 mL) added dropwise over 15 min, followed by IN hydrochloric acid (50 mL) added dropwise over 15 min. The cooling bath was then removed and an additional 150 mL of IN hydrochloric acid was added over 20 min. The mixture was then stirred at 22 °C for 2 h and diluted with dichloromethane (400 mL). The organic phase was collected and the aqueous phase (pH ~1) was extracted with dichloromethane (3 x 50 mL). The combined organic extracts were washed with brine, dried over anhydrous magnesium sulfate and concentrated in vacuo. The residual oil was diluted with tetrahydrofuran (70 mL), treated with 10 mL of 0.1N hydrochloric acid and stirred at 20 °C for 2 h. The reaction mixture was diluted with ethyl acetate (300 mL), washed with brine, dried over anhydrous magnesium sulfate, evaporated in vacuo to give a clear oil. Chromatography on silica gel (4 x 13 cm, elution toluene) gave 4.08 g (73% yield) of the title aldehyde as a clear oil which solidified on standing. LC (Method C): 2.237 min. HRMS(ESI) calcd for Ci5Hi504 [M+H]+ m/z 259.0965, found 259.1153. 1H NMR (CDC13, 600 MHz) δ 3.80 (s, 3H), 5.07 (s, 2H), 5.97 (d, J= 2.1 Hz, 1H), 6.01 (d, J= 2.1 Hz, 1H), 7.3 – 7.4 (m, 5 H), 10.15 (s, 1H), 12.49 (s, 1H).
ID. 1 -(4-(Benzyloxy)-6-methoxybenzofuran-2-yl)ethanone

A solution of 2-(benzyloxy)-6-hydroxy-4-methoxybenzaldehyde (Example 1C, 3.46 g, 13.4 mmol) in N,N-dimethylformamide (50 mL) was treated with powdered anhydrous cesium carbonate (4.58 g, 14.05 mmol) added all at once. The resulting mixture was stirred in vacuo for 10 min. and then flushed with nitrogen. The reaction flask was placed in a water bath (22 °C) and treated with chloroacetone (1.74 g, 18.7 mmol) added dropwise over 5 min. The resulting mixture was then stirred at 22 °C for 18 h (no starting aldehyde left by tic and formation of the intermediate alkylated aldehyde). The solid was filtered and washed with N,N-dimethylformamide. The filtrate was evaporated in vacuo and the residual oil was diluted with ethyl acetate (300 mL), washed with cold 0.1 N hydrochloric acid, saturated sodium bicarbonate and brine. After drying over anhydrous magnesium sulfate, evaporation of the solvent gave a thick syrup. This syrup was diluted with tetrahydrofuran (50 mL) and ethyl acetate (50 mL), treated p- toluenesulfonic acid monohydrate (0.2 g) and stirred at 20 °C for 1 h (tic indicated complete cyclization of the intermediate alkylated aldehyde to the benzofuran). The reaction mixture was diluted with ethyl acetate (300 mL), washed with saturated sodium bicarbonate and brine. After drying over anhydrous magnesium sulfate, evaporation of the solvent gave a thick syrup. Chromatography on silica gel (4 x 12 cm, elution toluene – ethyl acetate 2-4%) gave 3.51 g (88% yield) of the title benzofuran as a yellow solid. Recrystallization from ethyl acetate (10 mL) and hexane (20 mL) gave the title material as large yellow prisms (3.15 g). LC (Method D): 2.148 min. HRMS(ESI) calcd for Ci8Hiv04 [M+H]+ m/z 297.1121, found 297.1092. 1H NMR (CDC13, 600 MHz) δ 2.51 (s, 3H), 3.82 (s, 3H), 5.13 (s, 2H), 6.37 (d, J= 1.77 Hz, 1H), 6.63 (broad s, 1H), 7.34 (broad t, 1H), 7.39 (broad t, 2H), 7.44 (broad d, 2H), 7.55 (d, J = 0.7 Ηζ,ΙΗ). IE. l-(4-(Benzyloxy)-6-methoxybenzofuran-2-yl)-2-bromoethanone

A 250-mL, three-necked flask is equipped with a magnetic stirring bar and purged with a nitrogen atmosphere was charged with anhydrous tetrahydrofuran (25 mL) followed by 9.3 mL (9.3 mmol) of a 1M solution of lithium bis(trimethylsilyl)amide in tetrahydrofuran. The mixture was cooled to -78 °C and treated with a solution of l-(4- (benzyloxy)-6-methoxybenzofuran-2-yl)ethanone (Example ID, 2.40 g, 8.1 mmole) in tetrahydrofuran (20 mL) added dropwise over 10 min. The resulting mixture was then stirred at -78 °C for 45 min. Then chlorotrimethylsilane (1.18 mL, 9.31 mmol) was added dropwise over 5 min and the resulting solution was stirred at -78 °C for another 20 min. The cooling bath was then removed and the mixture is allowed to warm to room temperature over 30 min. The reaction mixture was then quenched by addition to a cold solution of ethyl acetate (200 mL), saturated sodium bicarbonate (30 mL) and ice. The organic phase was rapidly dried over anhydrous magnesium sulfate (magnetic stirring) and evaporated in vacuo to give the silyl enol ether as an oil which is co-evaporated with toluene (20 mL). The silyl enol ether was then dissolved in dry tetrahydrofuran (40 mL), cooled to -20 °C and treated with solid sodium bicarbonate (0.10 g) followed by N- bromosuccinimide (1.44 g, 8.1 mmol) added in small portions over 15 min. The reaction mixture was allowed to warm to 0 °C over 2h and then quenched by addition of ethyl acetate (300 mL) and saturated sodium bicarbonate. The organic phase was washed with brine, dried over anhydrous magnesium sulfate and evaporated to give an orange oil. Chromatography on silica gel (4 x 12 cm, elution toluene – ethyl acetate 0-5%) gave 2.62 g (86% yield) of the title bromomethylketone as a yellow solid. Recrystallization from ethyl acetate (10 mL) and hexane (20 mL) gave yellow prisms (2.30 g). LC (Method E): 1.977 min. HRMS(ESI) calcd for Ci8Hi6Br04 [M+H]+ m/z 375.0226, found 375.0277. 1H NMR (CDCls, 600 MHz) δ 3.84 (s, 3H), 4.33 (s, 2H), 5.14 (s, 2H), 6.38 (d, J = 1.76 Hz, 1H), 6.64 (broad s, 1H), 7.35 (broad t, 1H), 7.40 (broad t, 2H), 7.44 (broad d, 2H), 7.70 (s, 1H). 1 EE. 1 -(4-(Benzyloxy)-6-methoxybenzofuran-2-yl)-2-chloroethanone

Benzyltrimethylammonium dichloroiodate (117 g, 169 mmol) was added to a solution of l-(4-(benzyloxy)-6-methoxybenzofuran-2-yl)ethanone (Example ID, 50 g, 170 mmol) in THF (500 mL) in a 1 L multineck round bottom flask under nitrogen atmosphere. The reaction mixture was stirred at RT for 6 h, cooled to 0 °C and quenched with 10% NaHCC”3 solution. The organic layer was washed with 1 M sodium thiosulphate solution, water, and brine, dried over Na2S04, and concentrated in vacuo (bath temperature <45 °C). The residue was triturated with 5% EtOAc in pet. ether and dried to obtain the title chloromethylketone as a pale yellow solid (48 g, 130 mmol, 78%). 1H NMR (300 MHz, DMSO-d6) δ 3.84-3.82 (d, J =4.5Hz, 3H) 4.98 (s, 2H), 5.27(s, 2H), 6.62 -6.61 (d, J = 1.8Hz, 1H), 6.92-6.93 (m, 1H), 7.54-7.36 (m, 5H), 8.10-8.09 (d, J = 3Hz, 1H); MS m/z: [M+H]+ 331.0. IF. 6-(4-(Benzyloxy)-6-methoxybenzofuran-2-yl)-2-bromoimidazo[2, 1 – b] [ 1 ,3 ,4]thiadiazole

A mixture of l-(4-(benzyloxy)-6-methoxybenzofuran-2-yl)-2-bromoethanone (Example IE, 3.00 g, 8.0 mmol) and 5-bromo-l,3,4-thiadiazol-2-amine (1.65 g, 9.16 mmol) in isopropanol (100 mL) was heated in a pressure flask equipped with a magnetic stirring bar at 78-80 °C for 18 h (homogeneous after 20 min and then formation of a precipitate after 2 h). The cooled mixture is then transferred into five 20 mL microwave vials and then heated in a microwave apparatus to 150 °C for 30 min. Each vial was then diluted with dichloromethane (250 mL) washed with saturated sodium bicarbonate (25 mL) and brine (25 mL), dried over anhydrous magnesium sulfate. The fractions were combined and concentrated in vacuo. Chromatography of the orange-brown residual solid on silica gel (4 x 10 cm, slow elution with dichloromethane due to poor solubility) gave 2.96 g of the title imidazothiadiazole contaminated with some l-(4-(benzyloxy)-6- methoxybenzofuran-2-yl)ethanone. The solid material was triturated with ethyl acetate (20 mL), filtered, washed with ethyl acetate (10 ml) and dried in vacuo to give 2.34 g (64% yield) of pure title imidazothiadiazole as an off white solid which is used as such for the next step. LC (Method E): 2.188 min. HRMS(ESI) calcd for C2oHi5BrN303S [M+H]+ m/z 456.00175, found 456.00397. 1H NMR (CDC13, 600 MHz) δ 3.82 (s, 3H), 5.16 (s, 2H), 6.38 (d, J= 1.67 Hz, 1H), 6.66 (broad s, 1H), 7.15 (s, 1H), 7.31 (broad t, 1H), 7.38 (broad t, 2H), 7.45 (broad d, 2H), 8.02 (s, 1H).
Alternatively, Example IF, 6-(4-(benzyloxy)-6-methoxybenzofuran-2-yl)-2- bromoimidazo[2,l-b][l,3,4]thiadiazole, was prepared as follows:
A 1000-mL, three-necked flask equipped with a magnetic stirring bar and purged with a nitrogen atmosphere was charged with dry NMP (200 mL) followed by 1- (4-(benzyloxy)-6-methoxybenzofuran-2-yl)-2-chloroethanone (Example 1EE, 50 g, 150 mmol) and 5-bromo-l,3,4-thiadiazol-2-amine (27.2 g, 151 mmol). The resulting mixture was stirred at 80 °C for 8h. TLC (8:2 dichloromethane/pet. ether) and LC/MS showed intermediate uncyclized material (m/z 476) and the reaction mixture was stirred at 120 °C for 3h. The reaction mixture was cooled to RT, quenched with water and extracted with EtOAc (3X). The combined organic layers were washed with brine, dried over Na2S04, and concentrated in vacuo. The thick brown residue was purified by silica gel chromatography (0 to 100% dichloromethane in pet. ether) to give a brown solid. This material was triturated with EtOAc and dried to obtain the title imidazothiadiazole (24 g, 50 mmol, 33%>) as a light brown solid. (See the procedure set forth above for analytical data).
1 G. 6-(4-(Benzyloxy)-6-methoxybenzofuran-2-yl)-2-methoxyimidazo[2, 1 – b][l,3,4]thiadiazole

A solution of 6-(4-(benzyloxy)-6-methoxybenzofuran-2-yl)-2- bromoimidazo[2,l-b][l,3,4]thiadiazole (Example IF, 2.30 g, 5.04 mmol) in a mixture of dichloromethane (180 mL) and methanol (45 mL) was treated at 22 °C with 4.2 mL of a 25 wt.% solution of sodium methoxide in methanol (0.2 mmol) added in one portion. More methanol (45 mL) was added and the mixture was stirred for 1 h. The reaction mixture was quenched by the addition of 25 mL of IN hydrochloric acid followed by 20 ml of saturated sodium bicarbonate. The solvent was evaporated under reduced pressure and the residue was diluted with dichloromethane (400 mL), washed with brine, dried over anhydrous magnesium sulfate and evaporated in vacuo. Chromatography of the residue on silica gel (3 x 10 cm, elution with dichloromethane – ethyl acetate 0-4%) gave 1.70 g (83% yield) of the title compound as a white solid. This material was recrystallized from ethyl acetate (30 mL per gram, 80% recovery) to give white needles. LC (Method
D): 2.293 min. HRMS(ESI) calcd for C21H18N3O4S [M+H]+ m/z 408.1013, found 408.1024. 1H NMR (CDC13, 600 MHz) δ 3.81 (s, 3H), 4.18 (s, 3H), 5.16 (s, 2H), 6.37 (d, J = 1.75 Hz, 1H), 6.67 (broad s, 1H), 7.07 (s, 1H), 7.31 (broad t, 1H), 7.37 (broad t, 2H), 7.45 (broad d, 2H), 7.81 (s, 1H).
1H. 6-Methoxy-2-(2-methoxyimidazo[2,l-b][l,3,4]thiadiazol-6-yl)benzofuran-4-ol

A mixture of 6-(4-(benzyloxy)-6-methoxybenzofuran-2-yl)-2- methoxyimidazo[2,l-b][l,3,4]thiadiazole (Example 1G, 1.250 g, 3.06 mmol) and pentamethylbenzene (3.17 g, 21.4 mmol) in dichloromethane (200 mL) was cooled to -78 °C under a nitrogen atmosphere and then treated immediately (to avoid crystallization) with 8 mL (8 mmol) of a 1 M solution of boron trichloride in dichloromethane added dropwise over 3 min. The resulting mixture was stirred at -78 °C for 1 h. The reaction mixture was then quenched by the addition of a solution of sodium bicarbonate (6 g) in water (100 mL) added in one portion. The cooling bath was removed and the resulting mixture was stirred at room temperature for 1 h. The solid formed was filtered, washed successively with water (50 m) and dichloromethane (50 mL). The filter cake was allowed to soak with anhydrous ethanol (15 ml) and then sucked dry. The white solid obtained was then dried under vacuum for 24 h to give 0.788 g (80%> yield) of pure title material (> 95% by hplc). The combined filtrate and washings were diluted with dichloromethane (600 mL) and stirred in a warm water bath till the organic phase was clear with no apparent solid in suspension. The organic phase was collected, dried over anhydrous magnesium sulfate and rapidly filtered while still warm. The filtrate was evaporated and the residue (product and pentamethylbenzene) was triturated with toluene (20 mL), the solid collected and washed with toluene (20 mL) to give 0.186 g (19% yield, 99% combined yield) of title material as a tan solid (> 95% by hplc). LC (Method E): 1.444 min. HRMS(ESI) calcd for C14H12N3O4S [M+H]+ m/z 318.0543, found 318.0578. 1H NMR (DMSO-de, 600 MHz) 5 3.71 (s, 3H), 4.16 (s, 3H), 6.21 (d, J = 1.87 Hz, 1H), 6.61 (broad s, 1H), 6.95 (s, 1H), 8.29 (s, 1H), 9.96 (s, 1H).
Example 94
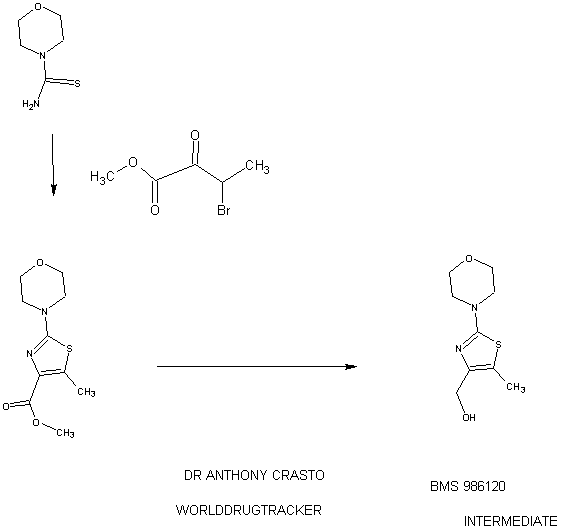
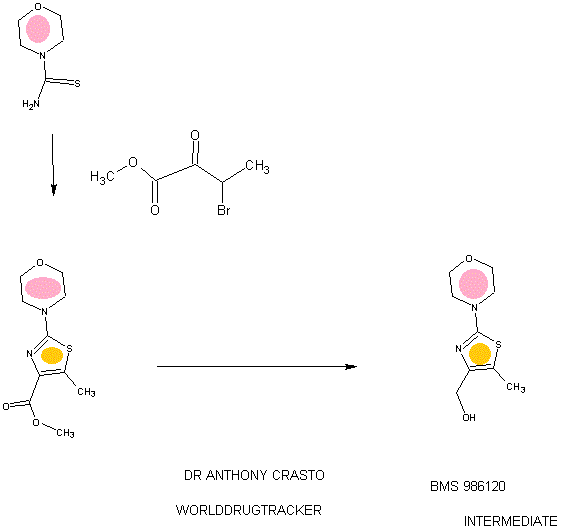
4-(4-(((6-Methoxy-2-(2-methoxyimidazo[2,l-b][l,3,4]thiadiazol-6-yl)benzofuran-4-yl) oxy)methyl)-5-methylthiazol-2-yl)morpholine

94 A. Methyl 5-methyl-2-morpholinothiazole-4-carboxylate [00258] A solution of methyl 2-bromo-5-methylthiazole-4-carboxylate (2.80 g, 11.86 mmol) and morpholine (4.5 mL, 51.7 mmol) in THF (10 mL) was heated at reflux under nitrogen for 18 h. The volatiles were then removed under reduced pressure and the crude product was purified on the ISCO using a REDISEP® 40 g column (0 to 40% EtOAc- DCM), to give the title compound (2.20 g, 77%) as a yellow solid. LCMS (APCI): calcd for CioHisNzOsS [M+H]+ m/z 243.07, found 243.1. 1H NMR (CDC13, 400 MHz) δ ppm: 3.89 (s, 3H), 3.77-3.83 (m, 4H), 3.41-3.47 (m, 4H), 2.64 (s, 3H). [00259] Alternatively, Example 94A, methyl 5-methyl-2-morpholinothiazole-4- carboxylate, was prepared as follows:
94AA. Methyl 3-bromo-2-oxobutanoate

A 5L 4-neck round bottom flask equipped with a mechanical stirrer, temperature thermocouple, condenser and a 1L addition funnel, was charged copper(II) bromide (962 g, 4310 mmol) and ethyl acetate (2 L). A solution of methyl 2-ketobutyrate (250 g, 2150 mmol) in CHC13 (828 mL) was added dropwise. A scrubber (400 mL 1 N NaOH) was connected and the reaction mixture was heated to reflux (75 °C). The reaction started as a dark green color and as heating progressed, it became a light green with a white precipitate forming. NMR after one hour at reflux indicated that the reaction was complete. The reaction was cooled to RT and filtered through a pad of CELITE®. The filtrate was concentrated to an oil, dissolved in methylene chloride (500 mL) and filtered again through CELITE®. The filtrate was then passed through a pad of silica gel and eluted with ethyl acetate. Concentration of the filtrate provided the title bromoketoester (399 g, 2040 mmol, 95%) as a yellow oil. 1H NMR (400MHz, CDC13) δ 5.18 (q, J = 6.7 Hz, 1H), 3.94 (s, 3H), 1.83 (d, J = 6.8 Hz, 3H). 94AAA. Morpholine-4-carbothioamide

To a solution of morpholine (199 g, 2280 mmol) in CHC13 (1 L) was added isothiocyanatotrimethylsilane (150 g, 1140 mmol) dropwise. A white precipitate formed almost immediately, and the reaction was stirred for 1 h at RT. The reaction was then filtered and the resulting solid was washed with additional CHC13 and dried in vacuo to give the title thiourea as a white solid. (137 g, 937 mmol, 82%). 1H NMR (400MHz, DMSO-de) δ 3.81 – 3.71 (m, 2H), 3.17 – 3.08 (m, 2H).
94 A. Methyl 5-methyl-2-morpholinothiazole-4-carboxylate

To a solution of morpholine-4-carbothioamide (Example 94 AAA, 175 g, 1200 mmol) in methanol (500 mL) was charged methyl 3-bromo-2-oxobutanoate (Example 94AA, 233 g, 1200 mmol). The reaction was then heated to reflux for 1 hour, cooled to RT, and filtered. The filtrate was concentrated and the crude product was purified on by silica gel chromatography. The title thiazole (206g, 850 mmol, 71%) was isolated as a yellow oil. (See the procedure set forth above for analytical data).
(5-Methyl-2-morpholinothiaz l-4-yl)methanol

The compound was prepared according to the protocol described for Example 92B. The crude product was purified on the ISCO using a REDISEP® Gold 24 g column (0 to 50% EtOAc-DCM) to give the title compound as a white solid (0.086 g, 51%). LCMS (APCI): calcd for C9Hi5N202S [M+H]+ m/z 215.08, found 215.1. 1H NMR (CDCI3, 400 MHz) δ ppm: 4.48 (d, J= 4.7 Hz, 2H), 3.77-3.83 (m, 4H), 3.37-3.43 (m, 4H), 2.30 (t, J= 4.7 Hz, 1H), 2.28 (s, 3H).
Example 94. 4-(4-(((6-Methoxy-2-(2-methoxyimidazo[2, 1 -b] [ 1 ,3,4]thiadiazol-6-yl) benzofuran-4-yl)oxy)methyl)-5 -methylthiazol-2-yl)morpholine

The title compound was prepared according to the protocol described for Example 86. The crude product was purified on the ISCO using a REDISEP® 4 g column (0 to 40% EtOAc-DCM) and the obtained solid was suspended in MeOH, sonicated, filtered and dried to give the title compound as an off-white solid (0.094 g, 53%). LC (Method C): 2.314 min. HRMS(ESI): calcd for C23H24N505S2 [M+H]+ m/z 514.122, found 514.126. 1H NMR (CDC13, 400 MHz) δ ppm: 7.83 (s, 1H), 7.06 (d, J = 0.8 Hz, 1H), 6.69 (d, J= 0.8 Hz, 1H), 6.50 (d, J= 2.0 Hz, 1H), 5.05 (s, 2H), 4.21 (s, 3H), 3.85 (s, 3H), 3.78- 3.84 (m, 4H), 3.39- 3.46 (m, 4H), 2.37 (s, 3H).
ABSTRACT
251st Am Chem Soc (ACS) Natl Meet (March 13-17, San Diego) 2016, Abst MEDI 263


| Patent ID | Date | Patent Title |
|---|---|---|
| US2015094297 | 2015-04-02 | IMIDAZOTHIADIAZOLE AND IMIDAZOPYRAZINE DERIVATIVES AS PROTEASE ACTIVATED RECEPTOR 4 (PAR4) INHIBITORS FOR TREATING PLATELET AGGREGATION |
////////BMS 986120, phase 1, Bristol-Myers Squibb , Imidazoles, Small molecules, Thiadiazoles, 1478712-37-6
c1(sc2nc(cn2n1)c3cc4c(cc(cc4o3)OC)OCc5nc(sc5C)N6CCOCC6)OC
CC1=C(N=C(S1)N2CCOCC2)COC3=C4C=C(OC4=CC(=C3)OC)C5=CN6C(=N5)SC(=N6)OC
PF 06650808
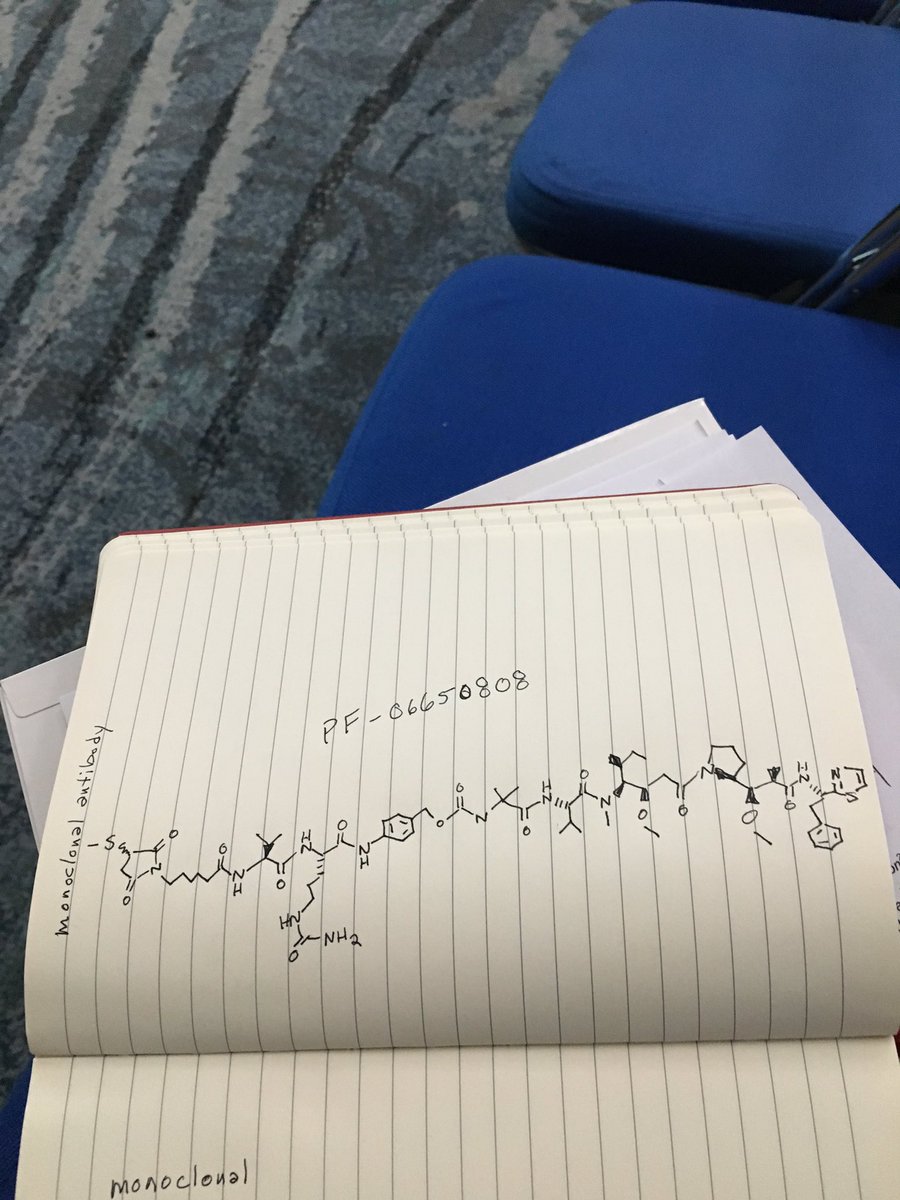 .
.
Picture credit….Bethany Halford

PF 06650808
CAS 1822383-80-1
A biologic for cancer treatment (Pfizer Inc.)
- Originator Pfizer
- Class Antineoplastics
- Mechanism of Action Notch-3 receptor antagonists
- No development reported Solid tumours
- 24 Jun 2018 Biomarkers information updated
- 28 Apr 2018 No recent reports of development identified for phase-I development in Solid-tumours(Late-stage disease) in USA (IV)
- 01 Jul 2017 Pfizer completes a phase I trial in Solid tumours (Late-stage disease) in USA (IV) (NCT02129205)
Company: Pfizer
Target: Neurogenic locus notch homolog protein 3 (NOTCH3): Activation and mutation of the NOTCH signaling pathway can lead to cancer.
Disease: Cancer
Notes: PF06650808 is an antibody-drug conjugate that delivers a cytotoxic payload molecule directly to tumor cells, explained Andreas Maderna, an associate research fellow at Pfizer. The payload molecule in PF06650808 was inspired by the marine natural product dolostatin 10, which is produced by cyanobacteria consumed by a type of sea slug.
https://cen.acs.org/articles/94/i15/New-drug-candidates-shine-San-Diego.html
PATENT
WO 2015171907
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015171907
The present invention relates to stable isotopic identification of biologic products, methods of stable isotopic identification of such biologic products, and stable isotopic methods and systems for correlating biologic products to the processes by which they are made.
PATENT
WO 2018045058
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018045058&tab=PCTDESCRIPTION&maxRec=1000
CLIP
Rosen, L.S.; Wesolowski, R.; Gibson, B.; et al.
A Phase 1 dose escalation, safety, and pharmacokinetic study of PF-06650808, an anti-Notch3 antibody drug conjugate, in adult patients with advanced solid tumors
Eur Cancer Congr (September 25-29, Vienna) 2015, Abst 3OLBA
Maderna, A.
Therapeutic targeting the NOTCH3 receptor with antibody drug conjugates
251st Am Chem Soc (ACS) Natl Meet (March 13-17, San Diego) 2016, Abst MEDI 262
Hurvitz, S.A.; von Euw, E.; O’Brien, N.; et al.
Preclinical evaluation of targeting Notch-3 in breast cancer
107th Annu Meet Am Assoc Cancer Res (AACR) (April 16-20, New Orleans) 2016, Abst 1206
Chen, J.; Geles, K.; Silva, M.; Waterhouse, R.; Ma, D.; Charati, M.; Sapra, P.; Mccarthy, T.
Evaluate the impact of conjugation on targeting capacity, pharmacokinetics and tissue distribution of antibody drug conjugate, PF-06650808, in tumor bearing mice
22nd Int Symp Radiopharm Sci (ISRS) (May 14-19, Dresden) 2017, Abst P 052
///////////
PF 06650808
| Phase 1 |
$PFE compound inspired by auristatins
https://clinicaltrials.gov/ct2/show/NCT02129205
http://www.pfizer.com/sites/default/files/product-pipeline/8_7_2014_Pipeline_Update.pdf
ALL DATA COMING………
Notch-3 receptor antagonists
Neoplasms
Breast
| Pfizer |
Cancer
PF-06650808, is currently being examined in a Ph1 clinical trial (Protocol B7501001).
Notch3
Researchers are also exploring the use of Notch3 targeting. “The Notch pathway plays an important role in the growth of several solid tumours, including breast and ovarian cancer and melanoma,” explained Joerger. “In particular, Notch3 alterations such as gene amplification and upregulation are associated with poor patient survival. Research using Notch3 targeting as an innovative approach to treat solid malignancies included 27 patients unselected for Notch3 who received increasing doses of the anti-Notch3 antibody-drug conjugate PF-06650808. Responses were seen in two breast cancer patients (LBA 30). While preliminary, targeting Notch3 may become a new treatment approach in patients with selected solid tumours.”
The anti-Notch3 antibody-drug conjugate PF-06650808 is being developed by Pfizer.
- 31 Jul 2014 Phase-I clinical trials in Solid tumours (Late-stage disease) in USA (Parenteral)
- 30 Apr 2014 Preclinical trials in Solid tumours in USA (Parenteral)
- 30 Apr 2014 Pfizer plans a phase I trial for Solid tumours (late-stage disease, second-line therapy or greater) in USA (NCT02129205)
251st Am Chem Soc (ACS) Natl Meet (March 13-17, San Diego) 2016, Abst MEDI 262


/////////PF 06650808, PF-06650808, PF-6650808, monoclonal antibody, pfizer, phase 1, Solid tumours , Notch-3 receptor antagonists
C1(C(N(C(C1)=O)CCCCCC(=O)NC([C@H](C)C)C(=O)NC(C(=O)Nc2ccc(cc2)COC(=O)NC(C)(C)C(=O)N[C@@H](C(C)C)C(=O)[N@](C)C(C(CC)C)[C@@H](OC)CC(=O)N3CCC[C@H]3C(OO)C(C)C(=O)N[C@H](c4nccs4)CC)CCCNC(=O)N)=O)SC
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
P.S
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India.
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP
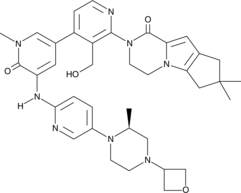
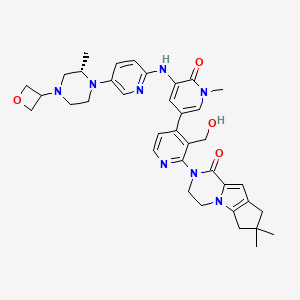
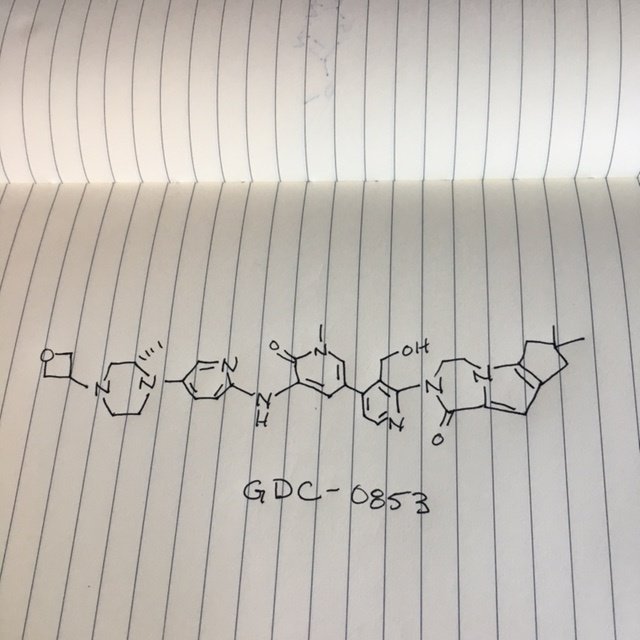
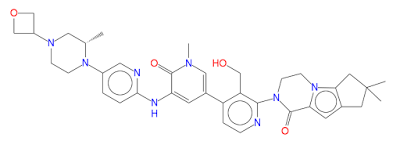
GDC 0853, Fenebrutinib
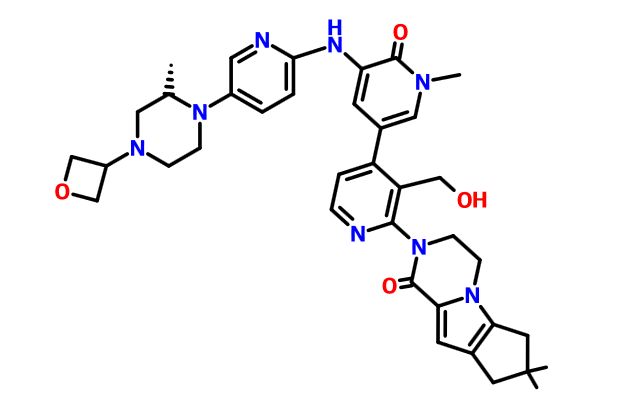
 .
.
Picture credit….Bethany Halford
GDC 0853, Fenebrutinib
GDC-0853; RG 7845
| Molecular Formula: | C37H44N8O4 |
|---|---|
| Molecular Weight: | 664.79646 g/mol |
2-[3-(hydroxymethyl)-4-[1-methyl-5-[(7-methyl-6,8-dihydro-5H-[1,2,4]triazolo[1,5-a]pyrazin-2-yl)amino]-6-oxo-3-pyridyl]-2-pyridyl]-3,4,6,7,8,9-hexahydropyrazino[1,2-a]indol-1-one
3-[3-(hydroxymethyl)-4-[1-methyl-5-[[5-[2-methyl-4-(oxetan-3-yl)piperazin-1-yl]pyridin-2-yl]amino]-6-oxopyridin-3-yl]pyridin-2-yl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one
3-[3-(hydroxymethyl)-4-[5-[[5-[(2S)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]-2-pyridyl]amino]-6-oxo-1H-pyridin-3-yl]-2-pyridyl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one
2H-Cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-1(6H)-one, 2-[1,6-dihydro-3′-(hydroxymethyl)-1-methyl-5-[[5-[(2S) -2-methyl-4-(3-oxetanyl)-1-piperazinyl]-2-pyridinyl]amino] -6-oxo[3,4′-bipyridin]-2′-yl]-3,4,7,8-tetrahydro-7,7- dimethyl-
s ISoMER 1434048-34-6 desired
r iSoMER 1434048-57-3 undesired
Phase 1
Patients with Patients with Resistant B-Cell Lymphoma or Chronic Lymphocytic Leukemia..
@genentech‘s Btk inhibitor
https://clinicaltrials.gov/ct2/show/NCT01991184
Bruton tyrosine kinase inhibitor
- 01 Sep 2015 Phase-I clinical trials in Autoimmune disorders (In volunteers) in USA (PO, Capsule and Tablet) (NCT02699710)
- 16 Oct 2014 Discontinued – Phase-I for Non-Hodgkin’s lymphoma (Second-line therapy or greater) in USA (unspecified route)
- 16 Oct 2014 Discontinued – Phase-I for Chronic lymphocytic leukaemia (Second-line therapy or greater) in USA (unspecified route)

GDC-0853; RG 7845; RO 7010939
2-[1,6-dihydro-3′-(hydroxymethyl)-1-methyl-5-[[5-[(2S)-2-methyl-4-(3-oxetanyl)-1-piperazinyl]-2-pyridinyl]amino]-6-oxo[3,4′-bipyridin]-2′-yl]-3,4,7,8-tetrahydro-7,7-dimethyl-2H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-1(6H)-one
GDC-0853 is an orally bioavailable, selective, and reversible Bruton’s tyrosine kinase (BTK) inhibitor with IC50s ranging from 2-9 nM for basophil activation, B cell receptor activation, and constitutive p-BTK activity in whole blood lysates.1,2 In rats, treatment for longer than 7 days leads to pancreatic toxicity but it does not occur in mice or dogs, even at higher doses.3 Formulations containing GDC-0853 were well-tolerated in Phase I clinical trials and are in additional clinical trials for rheumatoid arthritis, lupus erythematosus, lymphoma, and leukemia.
- Originator Genentech
- Class Antineoplastics; Antirheumatics; Piperazines; Pyrazines; Pyridines
- Mechanism of Action Agammaglobulinaemia tyrosine kinase inhibitors
Highest Development Phases
- Phase II Rheumatoid arthritis; Systemic lupus erythematosus; Urticaria
- Phase I Autoimmune disorders
- Discontinued Chronic lymphocytic leukaemia; Non-Hodgkin’s lymphoma
Most Recent Events
- 01 Jun 2018 Chemical structure information added
- 07 Nov 2017 Genentech initiates enrolment in a phase II extension trial for Systemic Lupus Erythematosus in Spain (EudraCT2017-001764-37)
- 13 Sep 2017 Genentech initiates enrolment in a phase I trial in Healthy volunteers in United Kingdom (NCT03290703)
BTK inhibitor GDC-0853 An orally available inhibitor of Bruton’s tyrosine kinase (BTK) with potential antineoplastic activity. Upon administration, GDC-0853 inhibits the activity of BTK and prevents the activation of the B-cell antigen receptor (BCR) signaling pathway. This prevents both B-cell activation and BTK-mediated activation of downstream survival pathways, which leads to the inhibition of the growth of malignant B-cells that overexpress BTK. BTK, a member of the Src-related BTK/Tec family of cytoplasmic tyrosine kinases, is overexpressed in B-cell malignancies; it plays an important role in B-lymphocyte development, activation, signaling, proliferation and survival.
SCHEME

MAIN

Patent
WO 2013067274
https://www.google.co.in/patents/WO2013067274A1?cl=en
part
Example 271a (S)-tert-Butyl 4-(6-(5-Chloro-2-methoxypyridin-3-ylamino)pyridin-3-yl)-3-methylpiperazine-1-carboxylate 271a
A 100-mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with 1,4-dioxane (40 mL), (S)-tert-butyl 4-(6-amino pyridin-3-yl)-3-methylpiperazine-1-carboxylate 101h (2.04 g, 7.0 mmol), 3-bromo-5-chloro-2-methoxypyridine (2.8 g, 12.6 mmol), Pd2(dba)3 (640 mg, 0.70 mmol), XantPhos (404.6 mg, 0.70 mmol), and cesium carbonate (4.56 g, 14.0 mmol). After three cycles of vacuum/argon flush, the mixture was heated at 100 °C for 4 h. After this time the reaction was cooled to room temperature. It was then filtered and the filtrate was evaporated under reduced pressure. The residue was purified by silica-gel column chromatography eluting with 1:3 ethyl acetate/petroleum ether to afford 271a (1.7 g, 57%) as a yellow solid. MS-ESI: [M+H]+ 434.2
Example 271btert-Butyl (3S)-4-(6-{[5-(2-{4,4-Dimethyl-9-oxo-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-10-yl}-3-(hydroxymethyl)pyridin-4-yl)-2-methoxypyridin-3-yl] amino}pyridin-3-yl)-3-methylpiperazine-1-carboxylate 271b
A 100-mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with 271a (650 mg, 1.50 mmol), {3-[(acetyloxy)methyl]-2-{4,4-dimethyl-9-oxo-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-10-yl}pyridin-4-yl}boronic acid 199e (1.79 g, 4.5 mmol), Pd2(dba)3 (137.2 mg, 0.15 mmol), P(cy)3(167.4 mg, 0.60 mmol), Cs2CO3 (978 mg, 3.0 mmol), dioxane (20 mL), and water (0.5 mL). After three cycles of vacuum/argon flush, the mixture was heated at 110°C for 16 h. After this time the reaction was cooled to room temperature. Lithium hydroxide monohydrate (1.89 g, 45 mmol) and water (2.0 mL) were added. The resulting mixture was stirred at 45°C for 4 h. It was then filtered and the filtrate was evaporated under reduced pressure. The residue was purified by silica-gel column chromatography eluting with 3:1 ethyl acetate/petroleum ether to afford 271b (290 mg, 27%) as a yellow solid. MS-ESI: [M+H]+ 709.3
Example 271c 10-[3-(Hydroxymethyl)-4-[5-({5-[(2S)-2-methylpiperazin-1-yl]pyridin-2-yl}amino)-6-oxo-1,6-dihydropyridin-3-yl]pyridin-2-yl]-4,4-dimethyl-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-9-one 271c
A solution of 271b (286.6 mg, 0.40 mmol) in dioxane/HCl (30 mL) was stirred at 50 °C for 2 h. It was evaporated under reduced pressure to afford 271c (450 mg, crude) as a black solid. MS-ESI: [M+H]+ 595.3
Example 271 3-[3-(hydroxymethyl)-4-[5-[[5-[(2S)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]-2-pyridyl]amino]-6-oxo-1H-pyridin-3-yl]-2-pyridyl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one 271
To a solution of 271c (450 mg, 0.75 mmol) in methanol (10 mL) was added oxetan-3-one (162 mg, 2.25 mmol), NaBH3CN (141.8 mg, 2.25 mmol), and ZnCl2 (306 mg, 2.25 mmol). The reaction was stirred at room temperature for 3 h. The mixture was evaporated under reduced pressure and the residue was diluted with water (5 mL). It was then extracted with dichloromethane (3 X 10 mL) and the combined dichloromethane extract was concentrated under reduced pressure. The residue was purified by reverse-phase prep-HPLC to afford 271 (23.0 mg, 8.8%, over two steps) as a yellow solid. MS-ESI: [M+H]+651.3. 1H NMR (500 MHz, CDCl3) δ 9.76 (s, 1H), 8.74 (d, J = 2.0 Hz, 1H), 8.53 (d, J = 5.0 Hz, 1H), 7.99 (d, J = 3.0 Hz, 1H), 7.84 (s, 1H), 7.73 (s, 1H), 7.41 (d, J = 4.5 Hz, 1H), 7.35 (dd, J = 2.5 Hz, 8.5 Hz, 1H), 6.87 (s, 1H), 6.85 (d, J = 9.0 Hz, 1H), 5.16-5.13 (m, 1H), 4.72-4.69 (m, 5H), 4.54-4.53 (m, 1H), 4.36-4.35 (m, 1H), 4.19-4.17 (m, 2H), 3.89-3.87 (m, 1H), 3.56-3.49 (m, 2H), 3.11-3.09 (m, 2H), 2.60-2.48 (m, overlap, 7H), 2.24-2.21 (m, 1H), 1.29 (s, 6H), 1.02 (d, J = 6.0 Hz, 3H)
271
………………………..
syn of 191 j
is intermediatenot product, is acid
To a mixture of 4-chloro-2-{4,4-dimethyl-9-oxo-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-10-yl}pyridine-3-carbaldehyde 108a (500 mg, 1.46 mmol), tert-butyl alcohol (20 mL), and dichloromethane (5 mL) was added 2-methyl-2-butene (3066 mg, 43.8 mmol). An aqueous solution (8 mL) of NaClO2 (263 mg, 2.92 mmol) and NaH2PO4·2water (683 mg, 4.38 mmol) was added dropwise at -10°C and the reaction mixture was stirred at -10 °C for overnight. It was concentrated under reduced pressure and the residue was extracted with ethyl acetate (4 × 20 mL). The combined organic extract was dried over MgSO4 and concentrated. The residue was purified with reverse-phase prep-HPLC to afford 210a (315 mg, 60%) as a pale yellow solid. MS-ESI: [M+H]+ 360.1
Example 210b 2-{4,4-Dimethyl-9-oxo-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-10-yl} -4-[1-methyl-5-({5-[(2S)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]pyridin-2-yl}amino)-6-oxo-1,6-dihydropyridin-3-yl]pyridine-3-carboxylic Acid 210b
A 25-mL round-bottomed flask equipped with a reflux condenser was charged with 210a (400 mg, 1.1 mmol), (S)-1-methyl-3-(5-(2-methyl-4-(oxetan-3-yl)piperazin-1-yl)pyridin-2-ylamino)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2(1H)-one 191j (536 mg, 1.1 mmol), PdCl2(dppf) (81 mg, 0.11 mmol), K3PO4 (466 mg, 2.2 mmol), sodium acetate (216 mg, 2.2 mmol), acetonitrile (10 mL), and water (0.2 mL). After three cycles of vacuum/argon flush, the mixture was heated at 100°C for 3 h. It was then filtered and the filtrate was evaporated in vacuo. The residue was purified by silica-gel column chromatography eluting with 1:3 petroleum/ethyl acetate to afford 210b as a yellow solid (306 mg, 41%). MS-ESI: [M+H]+ 679.3
construction, use your discretion
Example 130a (3S)-tert- utyl 3-methyl-4-(6-nitropyridin-3-yl)piperazine-l-carboxylate 130a

130a
Following the procedures as described for compound lOlg, reaction of 5-bromo-2-nitropyridine (10.5 g, 50 mmol), and (JS)-tert-butyl-3 -methylpiperazine- 1 -carboxylate (10.0 g, 50 mmol) afforded 130a as a yellow solid (8.05 g, 50%). LCMS: [M+H]+ 323
Example 130b (3 S)-tert-butyl-4-(6-aminopyridin-3 -yl)-3 -methylpiperazine- 1 -carboxylate 130b

130b
Following the procedures as described for compound lOlh, hydrogenation of 130a (5.8 g) afforded 130bas a brown solid (4.9 g, 96%). LCMS: [M+H]+ 293
Example 130c (3 S)-tert-Butyl-4-(6-(5 -bromo- 1 -methyl -2 -oxo- 1,2-dihydropyridin-3 -yl amino) pyridine-3 -yl)-3 -methylpiperazine- 1 -carboxylate 130c
N

Following the procedures as described for compound lOli, reaction of 130b (4.0 g) and 3,5-dibromo-l-methylpyridin-2(lH)-one (5.5 g) afforded 130c as a yellow solid (5.4 g, 83%). LCMS: [M+H]+ 478
Example 130d (3 S)-5 -Bromo- 1 -methyl-3 -(5 -(2-methylpiperazin- 1 -yl)pyridin- 2-ylamino)pyridine-2(lH)-one 130d

Following the procedures as described for compound lOlj, acidic hydrolysis of the Boc group of 130c (3.1 g) afforded 130d as a yellow solid (2.3 g, 95%). LCMS: [M+H]+ 380.
Example 130e (3 S)-5 -Bromo- 1 -methyl-3 -(5 -(2 -methyl-4-(ox etan-3-yl)piperazin-l-yl) pyridine -2-ylamino)pyridin-2(lH)-one 130e

Following the procedures as described for compound 101k, reductive amination of 130d (2.35 g) with oxetan-3-one (0.4 mL) afforded 130e as a yellow solid (2.6 g, 98%). LCMS: [M+H]+ 434.
Example 13 Of (3S)-l-methyl-3-(5-(2-methyl-4-(oxetan-3-yl)piperazin-l-yl)pyridin-2-ylamino) -5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyridin-2(lH)-one 130f
 check pyridine ring position
check pyridine ring position
A 100 mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with 130e (1.0 g, 1.0 eq., 2.3 mmol), Pin2B2 (1.46 g, 2.50 eq., 5.75 mmol), Pd2(dba)3 (105 mg, 0.05 eq., 0.125 mmol), X-Phos (93 mg, 0.1 eq., 0.23 mmol), AcOK (676 mg, 3.0 eq., 6.9 mmol), and dioxane (50 mL). After three cycles of vacuum/argon flush, the mixture was heated at 90 °C for 4 hrs, then cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure and the resulting residue was washed with 3: 1 PE/EA (80 mL) to afford 130f as yellow solid (1.0 g, 90%). MS: [M+H]+ 482.
check pyridine ring position, use your discretion
Example 191h ( 3S)-5 -Bromo- 1 -methyl-3 -(5 -(2-methylpiperazin- 1 -yl)pyridin- -ylamino)pyridine-2(lH)-one 191h

Following the procedure described for compound lOlj and starting with (3S)-tert-butyl 4-(6-(5 -bromo- 1 -methyl-2-oxo- 1 ,2-dihydropyridin-3 -ylamino)pyridine-3 -yl)-3 -methyl-piperazine-l-carboxylate 191g (3.1 g, 6.5 mmol) afforded 191h as a yellow solid (2.3 g, 94%). MS-ESI: [M+H]+ 378.
Example 1 1 i (S)-5 -Bromo- 1 -methyl-3-(5-(2-methyl-4-(oxetan-3-yl)piperazin- 1 -yl)pyridin-2-ylamino)pyridin-2(lH)-one 191i
A mixture of (5)-5-bromo-l-methyl-3-(5-(2-methylpiperazin-l-yl)pyridin-2-ylamino)pyridin-2(lH)-one 191h (40.0 g, 106 mmol), oxetan-3-one (1 1.4 g, 159 mmol), NaBH3CN (10.0 g, 159 mmol), and zinc chloride (21.3 g, 159 mmol) in methanol (700 mL) was stirred at 50°C for 5 hours. The mixture was added to water (100 mL) and concentrated under reduced pressure. The residue was extracted with dichloromethane (200 mL x 3). The combined organic layer was concentrated under reduced pressure and the residue was purified by silica-gel column chromatography eluting with 40: 1 dichloromethane /methanol to afford 191i (35 g, 73%). MS: [M+H]+ 434.
Example 191j (J5)-l-Methyl-3-(5-(2-methyl-4-(oxetan-3-yl)piperazin-l-yl)-pyridin- -ylamino) -5-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)pyridin-2(lH)-one 191j

191 i 191j
A 100-mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with (5)-tert-butyl-4-(6-(5-bromo-l-methyl-2-oxo-l ,2-dihydropyridin-3-ylamino)pyridine-3-yl)-3-methylpiperazine-l-carboxylate 191i (1.0 g, 1.0 eq., 2.3 mmol), Pin2B2 (1.46 g, 2.50 eq., 5.75 mmol), Pd2(dba)3 (105 mg, 0.05 eq., 0.125 mmol), X-Phos (93 mg, 0.1 eq., 0.23 mmol), potassium acetate (676 mg, 3.0 eq., 6.9 mmol), and dioxane (50 mL). After three cycles of vacuum/argon flush, the mixture was heated at 90°C for 4 h. It was then cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure and the resulting residue was washed with 3 : 1 petroleum ether/ethyl acetate (80 mL) to afford 191j as yellow solid (1.0 g, 90%). MS: [M+H]+ 482.
pipeline
http://www.gene.com/medical-professionals/pipeline

Pictrelisib, GDC-0941, RG7321 and GNE0941
| Patent ID | Date | Patent Title |
|---|---|---|
| US8921353 | 2014-12-30 | Heteroaryl pyridone and aza-pyridone compounds |
| US2014378432 | 2014-12-25 | HETEROARYL PYRIDONE AND AZA-PYRIDONE COMPOUNDS |
| US8716274 | 2014-05-06 | Heteroaryl pyridone and aza-pyridone compounds |
Development of an Efficient Manufacturing Process for Reversible Bruton’s Tyrosine Kinase Inhibitor GDC-0853
 , Theresa Cravillion†, Ngiap-Kie Lim†, Qingping Tian†, Danial Beaudry†, Jessica L. Defreese†, Alec Fettes‡, Philippe James¶, David Linder¶, Sushant Malhotra†, Chong Han† , Remy Angelaud†, and Francis Gosselin†
, Theresa Cravillion†, Ngiap-Kie Lim†, Qingping Tian†, Danial Beaudry†, Jessica L. Defreese†, Alec Fettes‡, Philippe James¶, David Linder¶, Sushant Malhotra†, Chong Han† , Remy Angelaud†, and Francis Gosselin† 
Efforts toward the process development of reversible Bruton’s tyrosine kinase (BTK) inhibitor GDC-0853 (1) are described. A practical synthesis of GDC-0853 was accomplished via a key highly regioselective Pd-catalyzed C–N coupling of tricyclic lactam 5 with 2,4-dichloronicotinaldehyde (6) to afford the C–N coupling product 3, a Suzuki–Miyaura cross-coupling of intermediate 3 with boronic ester 4 derived from a Pd-catalyzed borylation of tetracyclic bromide 7, to generate penultimate aldehyde intermediate 2 and subsequent aldehyde reduction and recrystallization. Process development of starting materials 5, 6, and 7 is also discussed.
(S)-2-(3′-(Hydroxymethyl)-1-methyl-5-((5-(2-methyl-4-(oxetan-3-yl)piperazin-1-yl)pyridin-2-yl)amino)-6-oxo-1,6-dihydro-[3,4′-bipyridin]-2′-yl)-7,7-dimethyl-2,3,4,6,7,8-hexahydro-1H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-1-one (crude GDC-0853, 1)
GDC-0853 (1, 196 kg, 81% yield, >99 A%, Pd < 10 ppm): mp 271 °C (DSC);
FTIR (cm–1, neat) 3430, 3313, 2945, 2865, 1606, 1573;
1H NMR (400 MHz, CDCl3) δ 8.65 (d, J = 2.2 Hz, 1H), 8.48 (d, J = 5.1 Hz, 1H), 7.96 (d, J = 2.7 Hz, 1H), 7.83 (d, J = 2.3 Hz, 2H), 7.36 (d, J = 5.1 Hz, 1H), 7.31 (dd, J = 8.9, 2.8 Hz, 1H), 6.87–6.76 (m, 2H), 5.18–4.98 (m, 1H), 4.77–4.58 (m, 5H), 4.50 (m, 1H), 4.33 (m, 1H), 4.16 (m, 2H), 3.86 (m, 1H), 3.71 (s, 3H), 3.61–3.38 (m, 2H), 3.07 (m, 2H), 2.67–2.39 (m, 7H), 2.20 (dd, J = 10.8, 6.3 Hz, 1H), 1.27 (s, 6H), 0.98 (d, J = 6.3 Hz, 3H);
13C NMR (101 MHz, CDCl3) δ 161.7, 157.6, 154.3, 150.3, 148.4, 141.9, 140.0, 131.4, 131.1, 129.7, 128.8, 127.7, 125.8, 123.9, 117.2, 116.3, 112.4, 111.3, 75.5, 75.5, 59.4, 59.1, 56.3, 52.9, 50.0, 49.2, 48.2, 45.9, 42.7, 40.9, 39.6, 38.5, 30.3, 15.3.
HRMS (ESI+) calcd for C37H45N8O4 ([M + H]+), 665.3564; found, 665.3588.
https://pubs.acs.org/doi/suppl/10.1021/acs.oprd.8b00134/suppl_file/op8b00134_si_001.pdf


/////////////
O=C1N(C)C=C(C2=CC=NC(N3CCN4C(C3=O)=CC5=C4CC(C)(C)C5)=C2CO)C=C1NC(N=C6)=CC=C6N7CCN(C8COC8)C[C@@H]7C
//////GDC 0853, genentech, Btk inhibitor, phase 1, Patients with Resistant B-Cell Lymphoma, Chronic Lymphocytic Leukemia, Bruton tyrosine kinase inhibitor, GDC-0853, RG 7845, 1434048-34-6, Fenebrutinib
N1(CCN(CC1C)C2COC2)c3cnc(cc3)NC=4C(N(\C=C(/C=4)c5c(c(ncc5)N6CCn7c(C6=O)cc8CC(Cc78)(C)C)CO)C)=O
CC1CN(CCN1C2=CN=C(C=C2)NC3=CC(=CN(C3=O)C)C4=C(C(=NC=C4)N5CCN6C7=C(CC(C7)(C)C)C=C6C5=O)CO)C8COC8
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Googleplus amcrasto@gmail.com
P.S
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India.
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP
SIDE CHAIN
MAIN
GLPG 1690

Picture credit….Bethany Halford
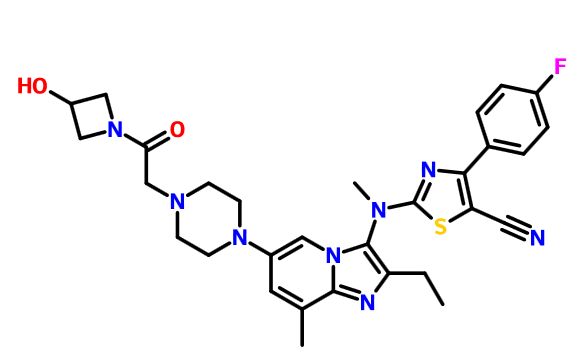
GLPG 1690
2-[[2-ethyl-6-[4-[2-(3-hydroxyazetidin-1-yl)-2-oxoethyl]piperazin-1-yl]-8-methylimidazo[1,2-a]pyridin-3-yl]-methylamino]-4-(4-fluorophenyl)-1,3-thiazole-5-carbonitrile
5- Thiazolecarbonitrile, 2-[[2-ethyl-6-[4-[2-(3-hydroxy-1-azetidinyl)-2-oxoethyl]- 1-piperazinyl]-8-methylimidazo[1,2-a]pyridin-3-yl]methylamino]-4-(4-fluorophenyl)-
CAS 1628260-79-6
![]()
$GLPG compound for treating idiopathic pulmonary fibrosis
| Molecular Formula: | C30H33FN8O2S |
|---|---|
| Molecular Weight: | 588.698823 g/mol |
| Galapagos Nv |
http://files.glpg.com/docs/website_1/Poster_ERS_2015_final.pdf
http://www.glpg.com/docs/view/56b360a81f6b2-en
Phase I Idiopathic pulmonary fibrosis
| Description | Selective autotaxin (ENPP2; ATX) inhibitor |
| Molecular Target | Autotaxin (ENPP2) (ATX) |
- Originator Galapagos NV
- Class Anti-inflammatories; Small molecules
- Mechanism of Action ENPP2 protein inhibitors
- 23 Sep 2015 Pharmacodynamics data from a preclinical trial in Indiopathic pulmonary fibrosis released by Galapagos
- 22 Sep 2015 Pharmacokinetics data from a phase I trial in healthy volunteers released by Galapagos
- 22 Sep 2015 Updated adverse events data from a phase I trial in healthy volunteers released by Galapagos
GLPG1690
GLPG1690 is a selective autotaxin inhibitor discovered by Galapagos, with potential application in idiopathic pulmonary disease (IPF). In a Phase 1 study in healthy human volunteers, GLPG1690 demonstrated favorable safety and tolerability, as well as a strong pharmacodynamic signal implying target engagement. Galapagos is currently preparing a Phase 2 study in IPF, to be filed for approval before the end of 2015. GLPG1690 is fully proprietary to Galapagos.
| Source: Galapagos NV
- Fully owned and proprietary clinical asset for pulmonary fibrosis
- GLPG1690 acts on autotaxin target
- Novel mode of action, originating from Galapagos target discovery engine
- Filing for Phase 2 clinical trial in 2015
MECHELEN, Belgium, March 16, 2015 (GLOBE NEWSWIRE) — Galapagos NV (Euronext: GLPG) announced that Janssen Pharmaceutica NV and Galapagos have mutually agreed to terminate the inflammation alliance and option agreements between the companies. Galapagos views the molecules emerging from the alliance as strong additions to its growing proprietary pipeline. Among others, all rights to candidate drug GLPG1690, a selective autotaxin inhibitor, return to Galapagos. Galapagos has successfully completed a First-in-Human Phase 1 trial for GLPG1690 and is preparing a Phase 2 clinical trial in idiopathic pulmonary fibrosis (IPF).
“We are pleased to regain the rights to GLPG1690 to pursue the most suitable clinical application of autotaxin inhibition. There is a large unmet medical need in IPF, and our pre-clinical data with GLPG1690 supports its potential as a competitive and novel approach in this disease area,” said Dr Piet Wigerinck, Chief Scientific Officer of Galapagos. “The alliance with Janssen has been underway since October 2007 and has generated three clinical molecules, two of which are now proprietary Phase 2 assets of Galapagos: GLPG1205 and GLPG1690. This program is a valuable component of our development portfolio, and regaining the rights is a next step in our transformation into a mature biotech company with a proprietary product pipeline.”
Galapagos identified autotaxin as playing a key role in inflammation, using an inflammation assay in its unique target discovery platform. Pharmacology and translational studies published by other parties in the literature since then suggest autotaxin may play a key role in metabolic disease, arthritic pain, oncology, and lung disease.
GLPG1690 is a potent and selective inhibitor of autotaxin. In a Phase 1 study in healthy human volunteers, GLPG1690 demonstrated favorable safety and tolerability, as well as a strong pharmacodynamic signal implying target engagement. Galapagos is currently preparing a Phase 2 study in IPF, to be filed for approval before the end of 2015.
About IPF
Idiopathic pulmonary fibrosis (IPF) is a chronic and ultimately fatal disease characterized by a progressive decline in lung function. Pulmonary fibrosis involves scarring of lung tissue and is the cause of shortness of breath. Fibrosis is usually associated with a poor prognosis. The term “idiopathic” is used because the cause of pulmonary fibrosis is still unknown. Estimated incidence of IPF is up to 16.3 per 100,000 persons in the US and 7.4 per 100,000 persons in Europe, with approximately 30,000-35,000 new patients diagnosed with IPF worldwide each year. The goals of treatment in IPF are essentially to reduce the symptoms, slow down disease progression, reduce acute exacerbations, and prolong survival. Approved treatments thus far have improved the overall survival of IPF patients, but unwanted side effects with these treatments are common, presenting an unmet need for effective treatments with safer side effect profiles.
| Source: Galapagos NV
MECHELEN, Belgium, Sept. 22, 2015 (GLOBE NEWSWIRE) — Galapagos NV (Euronext & NASDAQ: GLPG) presents pre-clinical and Phase 1 results for autotaxin inhibitor GLPG1690 at the European Respiratory Society Annual Meeting in Amsterdam, Netherlands. Galapagos expects to file an exploratory Phase 2 study in idiopathic pulmonary fibrosis before year end. GLPG1690 has potential application in other pulmonary diseases such as chronic obstructive pulmonary disease (COPD), as supported by the presentation on pre-clinical findings at ERS this year:
“Pharmacological profile and efficacy of GLPG1690, a novel ATX inhibitor for COPD treatment,” poster PA2129 in Poster Discussion Session: “New targets and modalities for the treatment of asthma and COPD” (September 28, 2015; Room D201-202, 10:45 AM – 12:45 PM)
Galapagos is the first to show efficacy of an autotaxin inhibitor in pre-clinical models for COPD and IPF, pointing to novel therapeutic areas for autotaxin inhibition. The poster shows how GLPG1690 acts as a potent inhibitor of mouse and human autotaxin (IC50: 100 -500 nM range). Furthermore, GLPG1690 reduces inflammation in a mouse steroid-resistant tobacco smoke model to a similar extent as a standard therapy for COPD.
Galapagos also presents the topline results with GLPG1690 in Phase 1 in healthy human volunteers: “Favorable human safety, pharmacokinetics and pharmacodynamics of the autotaxin inhibitor GLPG1690, a potential new treatment in COPD,” oral presentation OA484 in session “Advances in the future treatment of COPD” (September 27, 2015; Room 2.1, 10:45 AM – 12:45 PM)
GLPG1690 was safe and well tolerated up to a single oral dose of 1500 mg and up to 1000 mg twice daily for 14 days, with no significant adverse effects on ECGs, vital signs or laboratory parameters. The compound also showed good oral bioavailability with a half-life of 5 hours and a dose-proportional increase in exposure. GLPG1690 showed concentration-dependent reduction of a relevant biomarker (plasma LPA18:2 levels) with a maximum of approximately 90%. At steady state, continuous reduction of this biomarker levels of >60% was observed from 0 to 24 hours. The presentation will also include relevant pre-clinical model data for COPD and IPF with GLPG1690.
Both the presentation and the posters will be made available on the Galapagos website after the conference.
About Galapagos
Galapagos (Euronext & NASDAQ: GLPG) is a clinical-stage biotechnology company specialized in the discovery and development of small molecule medicines with novel modes of action, with a pipeline comprising three Phase 2 programs, two Phase 1 trials, five pre-clinical studies, and 20 discovery small-molecule and antibody programs in cystic fibrosis, inflammation, and other indications. In the field of inflammation, AbbVie and Galapagos signed a collaboration agreement for the development and commercialization of filgotinib. Filgotinib is an orally-available, selective inhibitor of JAK1 for the treatment of rheumatoid arthritis and potentially other inflammatory diseases, currently in Phase 2B studies in RA and in Phase 2 in Crohn’s disease. Galapagos reported good activity and a favorable safety profile in both the DARWIN 1 and 2 trials in RA. AbbVie and Galapagos also signed a collaboration agreement in cystic fibrosis to develop and commercialize molecules that address mutations in the CFTR gene. Potentiator GLPG1837 is currently in a Phase 1 trial, and corrector GLPG2222 is at the pre-clinical candidate stage. GLPG1205, a first-in-class inhibitor of GPR84 and fully-owned by Galapagos, is currently being tested in a Phase 2 proof-of-concept trial in ulcerative colitis patients. GLPG1690, a fully proprietary, first-in-class inhibitor of autotaxin, has shown favorable safety in a Phase 1 trial and is expected to enter Phase 2 in idiopathic pulmonary fibrosis. The Galapagos Group, including fee-for-service subsidiary Fidelta, has approximately 400 employees, operating from its Mechelen, Belgium headquarters and facilities in The Netherlands, France, and Croatia. More info at www.glpg.com
CONTACT
Galapagos NV
Elizabeth Goodwin, Head of Corporate Communications & IR
Tel: +31 6 2291 6240
ir@glpg.com
MECHELEN, Belgium, Feb. 16, 2015 (GLOBE NEWSWIRE) — Galapagos NV (Euronext: GLPG) announced today that GLPG1690, a first-in-class molecule for pulmonary disease, has demonstrated target engagement, a good safety profile, and favorable drug properties in a Phase 1 study. Galapagos is developing GLPG1690 within its alliance with Janssen Pharmaceutica NV.
The aim of the Phase 1 study was to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of oral single and multiple ascending doses of GLPG1690. The randomized, double-blind, placebo-controlled, single center study was conducted in 40 healthy volunteers in Belgium. In the first part of the study, single ascending doses were evaluated. In the second part, the new compound was administered daily for 14 days.
GLPG1690 proved to be safe and well-tolerated over a wide dose range in healthy volunteers. Engagement of the thus far undisclosed novel target was confirmed using a relevant biomarker. GLPG1690 displayed a favorable pharmacokinetic and pharmacodynamic profile. The data shown in Phase 1 encourage Galapagos to explore a Phase 2 study design in pulmonary disease.
“GLPG1690 is the first molecule against this target ever to be evaluated clinically, and we are pleased with the outcome of the Phase 1 study,” said Dr Piet Wigerinck, CSO of Galapagos. “Galapagos continues to deliver novel therapeutics from its unique target and drug discovery engine.”
In 2007, Galapagos announced an alliance agreement with Janssen Pharmaceutica NV providing the option to worldwide, commercial licenses to certain Galapagos internal inflammatory disease programs. These programs are based on novel targets for inflammatory disorders that were identified and validated by Galapagos using its proprietary target discovery engine. Subsequent Galapagos research led to the discovery of GLPG1690, a first-in-class molecule that entered the clinic for inflammatory disorders. Galapagos is responsible for execution of Phase 1 and Phase 2A studies with GLPG1690.
SYNTHESIS
INTRODUCTION
relates to compounds that are inhibitors of autotaxin, also known as ectonucleotide pyrophosphatase/phosphodiesterase 2 (NPP2 or ENPP2), that is involved in fibrotic diseases, proliferative diseases, inflammatory diseases, autoimmune diseases, respiratory diseases, cardiovascular diseases, neurodegenerative diseases, dermatological disorders, and/or abnormal angiogenesis associated diseases. The present invention also provides methods for the production of a compound of the invention, pharmaceutical compositions comprising a compound of the invention, methods for the prophylaxis and/or treatment of diseases involving fibrotic diseases, proliferative diseases, inflammatory diseases, autoimmune diseases, respiratory diseases, cardiovascular diseases, neurodegenerative diseases, dermatological disorders, and/or abnormal angiogenesis associated diseases by administering a compound
STAGE 1

STAGE2

STAGE 3

STAGE4

STAGE 5

FINAL

PATENT
US2014303140
http://www.google.com/patents/US20140303140
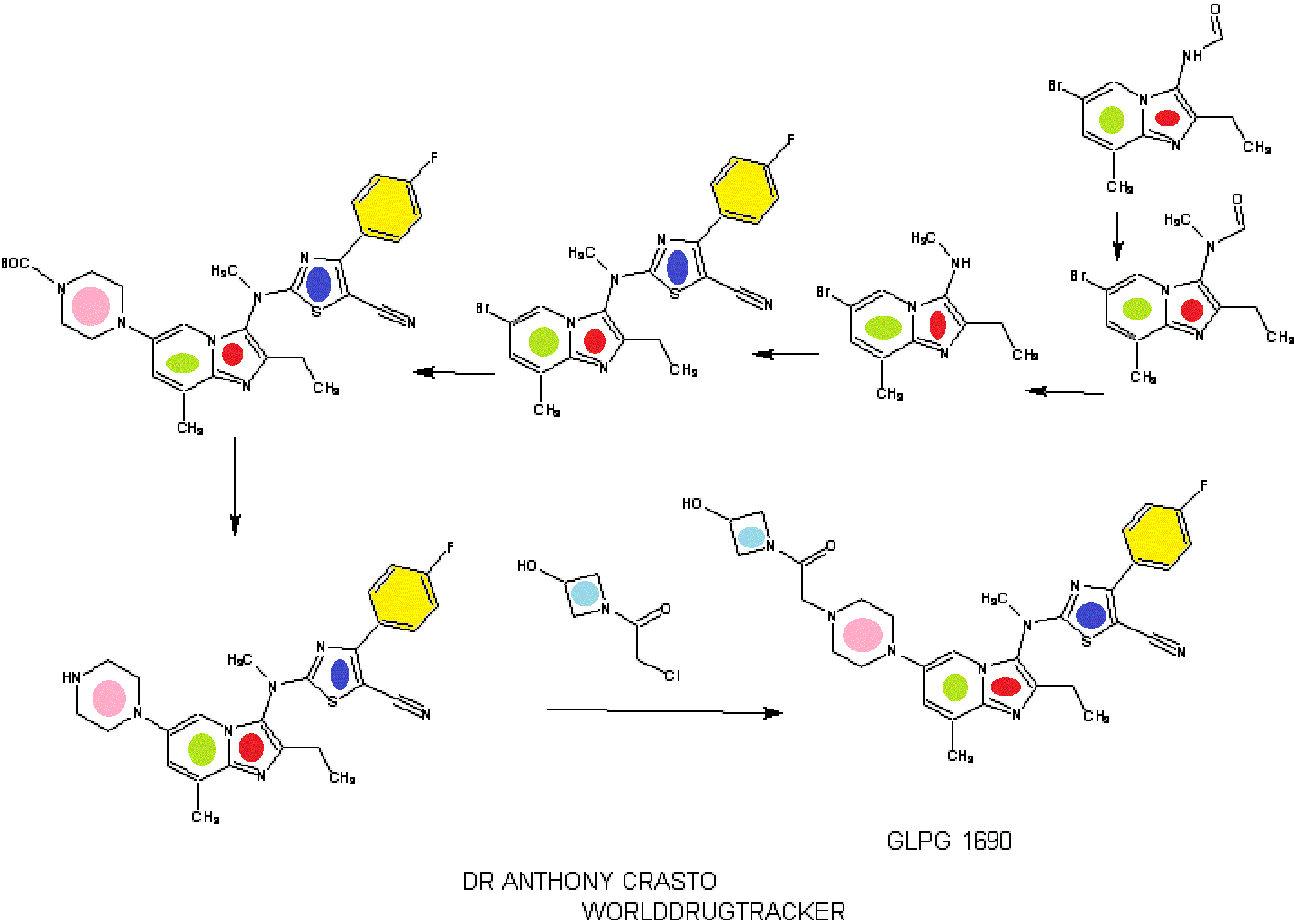
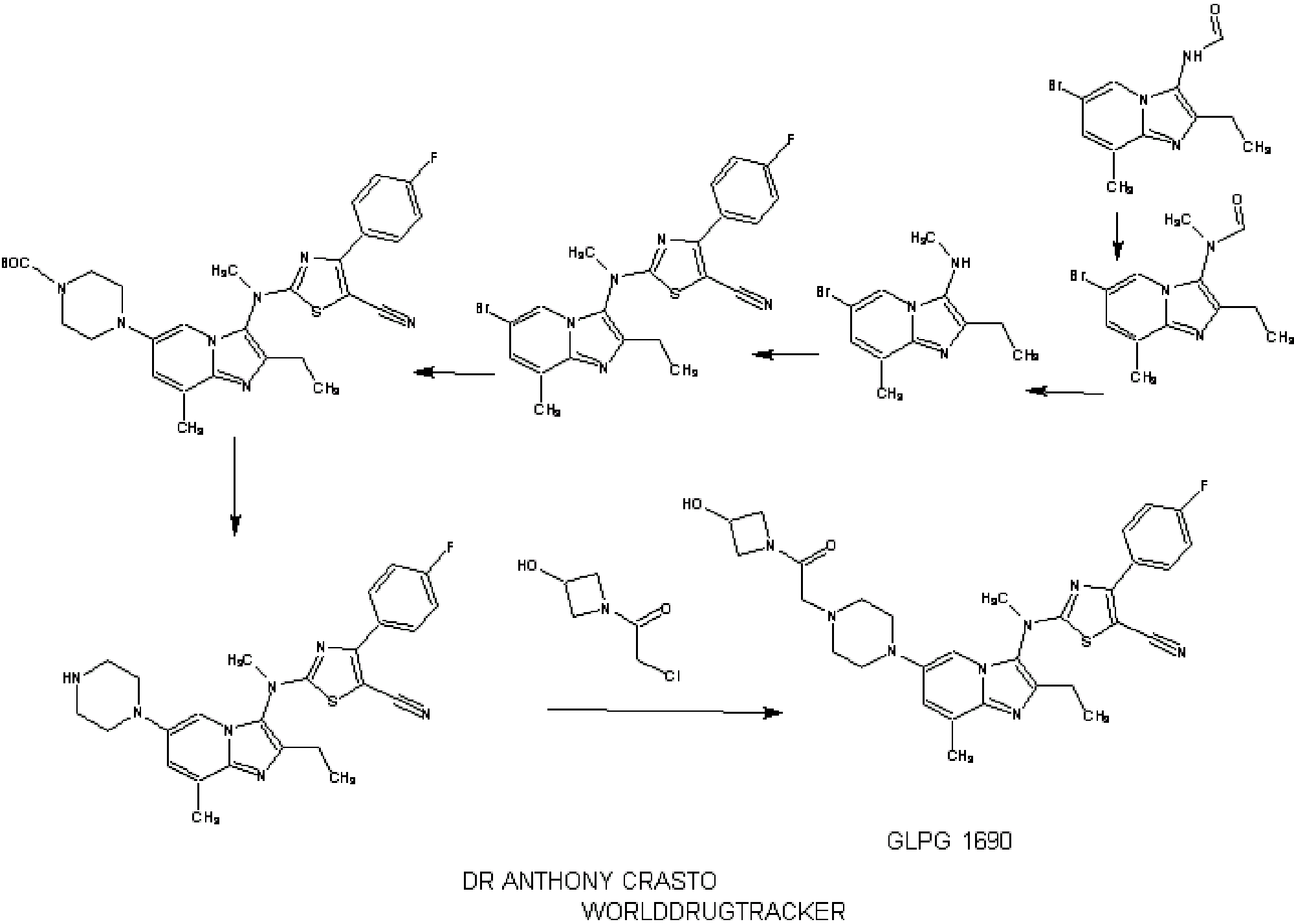
1.2.4.4. Illustrative Synthesis of Intermediate Gen-3-e: N-(6-bromo-2-ethyl-8-methylimidazo[1,2-a]pyridin-3-yl)-N-methylformamide
-

-
To a suspension of formamide Gen-2-d (720 g, 2.55 mol, 1 eq.) in 5 L of acetone were added potassium carbonate (1 kg, 7.66 mol, 3 eq.) and methyl iodide (700 g, 4.93 mol, 1.9 eq.). The reaction mixture was heated to 40° C. overnight. Additional methyl iodide (25 g, 0.18 mol, 0.07 eq.) was then introduced and stirring continued for 1 h at 40° C. The reaction mixture was filtered and washed with acetone (2×300 mL) and DCM (2×300 mL). The filtrate was concentrated in vacuo and the residue was partitioned between DCM (3 L) and water (1 L). The aqueous layer was further extracted with DCM. The combined organic layers were then washed with brine, dried over Na2SO4, filtered and concentrated in vacuo. The solid was triturated with Et2O (1 L) at r.t. for 1 h, filtered off and dried to afford Intermediate Gen-3-e.
-
Rotamer A (Major): 1H NMR δ (ppm) (400 MHz, CDCl3): 8.19 (1H, s), 7.78 (1H, s), 7.15 (1H, s), 3.24 (3H, s), 2.72 (2H, q), 2.59 (3H, s), 1.31 (3H, t)
-
Rotamer B (Minor): 1H NMR δ (ppm) (400 MHz, CDCl3): 8.49 (1H, s), 7.65 (1H, s), 7.08 (1H, s), 3.36 (3H, s), 2.72 (2H, q), 2.59 (3H, s), 1.31 (3H, t)
-
LC-MS: MW (calcd): 295 (79Br), 297 (81Br); m/z MW (obsd): 296 (79Br M+1), 298 (81Br M+1)
1.2.5.2. Illustrative Synthesis of Intermediate Gen-4-d: (6-Bromo-2-ethyl-8-methyl-imidazo[1,2-a]pyridin-3-yl)-methyl-amine
-

-
Intermediate Gen-3-e (80 g, 270 mmol, 1 eq.) was dissolved in a 1.25 M HCl solution in MeOH (540 mL, 2.5 eq.) and the resulting mixture was refluxed overnight. 270 mL of 1.25 M HCl solution in MeOH were added and heating continued overnight. After 48 h, additional 70 mL of the 1.25 M HCl solution in MeOH were introduced in the reaction mixture. Heating was maintained overnight until conversion was complete. The crude mixture was then concentrated in vacuo and the residue was partitioned between EtOAc (300 mL) and water (700 mL). A saturated NaHCO3 solution was added until pH reached 8-9. The aqueous layer was extracted twice with EtOAc (2×300 mL). The combined organic layers were then washed with brine (200 mL), dried over Na2SO4, filtered and concentrated in vacuo to give Intermediate Gen-4-d (6-bromo-2-ethyl-8-methyl-imidazo[1,2-a]pyridin-3-yl)-methyl-amine) as a free base.
-
1H NMR δ (ppm) (400 MHz, CDCl3): 8.05 (1H, s), 7.04 (1H, s), 2.84-2.78 (5H, m), 2.60 (3H, s), 1.35 (3H, t)
-
LC-MS: MW (calcd): 267 (79Br), 269 (81Br); m/z MW (obsd): 268 (79Br M+1), 270 (81Br M+1)
1.2.6.4. Illustrative Synthesis of Intermediate Gen-5-t: 2-[(6-Bromo-2-ethyl-8-methyl-imidazo[1,2-a]pyridin-3-yl)-methyl-amino]-4-(4-fluoro-phenyl)-thiazole-5-carbonitrile
-
-
To a solution of amine Gen-4-d (4.4 g, 16.6 mmol, 1 eq.) in THF (44 mL) under argon was slowly added NaH (60% in oil suspension, 2.0 g, 50.0 mmol, 3 eq.). The reaction mixture was heated at 90° C. for 30 min then cooled to 40° C. before adding the chlorothiazole Gen-12-a (4.74 g, 19.9 mmol, 1.2 eq.). The reaction mixture was stirred at 90° C. overnight. After cooling to r.t. the mixture was slowly quenched by addition of water and then diluted with EtOAc. The organic layer was separated and the aqueous layer extracted with EtOAc. The combined organic layers were then washed with water and brine, dried over Na2SO4, filtered and concentrated in vacuo. The residue was triturated in Et2O, filtered and washed with Et2O and MeCN. Recrystallization was performed in MeCN (180 mL) to afford Intermediate Gen-5-t (2-[(6-Bromo-2-ethyl-8-methyl-imidazo[1,2-a]pyridin-3-yl)-methyl-amino]-4-(4-fluoro-phenyl)-thiazole-5-carbonitrile).
-
1H NMR δ (ppm) (400 MHz, CDCl3): 8.15 (2H, dd), 7.80 (1H, s), 7.22-7.14 (3H, m), 3.62 (3H, s), 2.77 (2H, q), 2.64 (3H, s), 1.35 (3H, t)
-
LC-MS: MW (calcd): 469 (79Br), 471 (81Br); m/z MW (obsd): 470 (79Br M+1), 472 (81Br M+1)

1.2.7.1.4. Illustrative Synthesis of 4-(3-{[5-Cyano-4-(4-fluoro-phenyl)-thiazol-2-yl]-methyl-amino}-2-ethyl-8-methyl-imidazo[1,2-a]pyridin-6-yl)-piperazine-1-carboxylic acid tert-butyl ester
-

-
To a solution of Intermediate Gen-5-t (24.2 g, 51.5 mmol, 1 eq.) in toluene under argon were successively added N-Boc piperazine (14.4 g, 77.3 mmol, 1.5 eq.), sodium tert-butoxide (9.9 g, 103 mmol, 2 eq.), JohnPhos (1.54 g, 5.15 mmol, 0.1 eq.) and Pd2(dba)3 (2.36 g, 2.58 mmol, 0.05 eq.). The reaction mixture was heated at 115° C. for 1 h. After cooling to r.t., the crude product was filtered on Celpure® P65 and the residue dissolved in EtOAc and washed with water. The organic layer was further washed with brine, dried over Na2SO4, filtered and concentrated in vacuo. The crude product was purified by chromatography on silica gel (elution with heptane/EtOAc:90/10 to 20/80) to afford the expected product.
-
1H NMR δ (ppm) (400 MHz, CDCl3): 8.16 (2H, dd), 7.17 (2H, app t), 6.99 (2H, bs), 3.62-3.53 (4H, m), 3.60 (3H, s), 3.04-2.93 (4H, m), 2.74 (2H, q), 2.62 (3H, s), 1.47 (9H, s), 1.33 (3H, t).
-
LC-MS: MW (calcd): 575; m/z MW (obsd): 576 (M+1)
1.2.7.8.4. Illustrative Synthesis of Compound 1: 2-[(2-Ethyl-8-methyl-6-piperazin-1-yl-imidazo[1,2-a]pyridin-3-yl)-methyl-amino]-4-(4-fluoro-phenyl)-thiazole-5-carbonitrile
-

-
4-(3-{[5-Cyano-4-(4-fluoro-phenyl)-thiazol-2-yl]-methyl-amino}-2-ethyl-8-methyl-imidazo[1,2-a]pyridin-6-yl)-piperazine-1-carboxylic acid tert-butyl ester was prepared from intermediate Gen-5-t using Boc-piperazine and method Flb.
-
To a solution of 4-(3-{[5-Cyano-4-(4-fluoro-phenyl)-thiazol-2-yl]-methyl-amino}-2-ethyl-8-methyl-imidazo[1,2-a]pyridin-6-yl)-piperazine-1-carboxylic acid tert-butyl ester (24.4 g, 42 mmol, 1 eq.) in MeOH (100 mL) was added a 2 M HCl solution in Et2O (127 mL, 254 mmol, 6 eq.). The reaction mixture was stirred at r.t. for 3.5 h then concentrated in vacuo. The residue was partitioned between EtOAc and water. The aqueous layer was extracted twice with EtOAc. A 2 M NaOH solution was added to the aqueous layer until pH reached 8-9 and further extraction with EtOAc was performed. The combined organic layers were then washed with brine, dried over Na2SO4, filtered and concentrated in vacuo. The solid was triturated with heptane (100 mL) at r.t. overnight, filtered off, washed with heptane and Et2O, and dried to afford the expected compound.
-
1H NMR δ (ppm) (400 MHz, CDCl3): 8.17 (2H, dd), 7.18 (2H, app t), 6.99 (2H, bs), 3.61 (3H, s), 3.09-2.98 (8H, m), 2.75 (2H, q), 2.61 (3H, s), 1.34 (3H, t).
-
LC-MS: MW (calcd): 475; m/z MW (obsd): 476 (M+1)
1.2.7.14. Illustrative Synthesis of Compound 2: 2-((2-ethyl-6-(4-(2-(3-hydroxyazetidin-1-yl)-2-oxoethyl)piperazin-1-yl)-8-methylimidazo[1,2-a]pyridin-3-yl)(methyl)amino)-4-(4-fluorophenyl)thiazole-5-carbonitrile
-

-
To a solution of amine compound 1 (12.6 g, 27 mmol, 1 eq.) in 100 mL of MeCN were added potassium carbonate (7.3 g, 53 mmol, 2 eq.) and Gen13-a (5.2 g, 34 mmol, 1.3 eq.). The reaction mixture was refluxed for 5.5 h then cooled to r.t. and stirred for 40 h. The crude product was filtered and washed with MeCN. The collected precipitate was then suspended in 300 mL of water, stirred for 1 h, filtered, and finally washed with water and MeCN. The solid obtained was dried in vacuo for 48 h to afford Compound 2.
-
1H NMR (400 MHz, CDCl3) δ ppm 8.20-8.12 (2H, m), 7.22-7.13 (2H, m), 6.99 (2H, s), 4.68 (1H, m), 4.43 (1H, dd), 4.26 (1H, dd), 4.14-4.05 (1H, m), 3.88 (1H, dd), 3.61 (3H, s), 3.58-3.52 (1H, m), 3.14-3.02 (6H, m), 2.74 (2H, q), 2.70-2.62 (4H, m), 2.59 (3H, s), 1.33 (3H, t)
-
LC-MS: MW (calcd): 588; m/z MW (obsd): 589 (M+1)
| US9249141 | Dec 17, 2014 | Feb 2, 2016 | Galapagos Nv | Compounds and pharmaceutical compositions thereof for the treatment of inflammatory disorders |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015111872 | 2015-04-23 | NOVEL COMPOUNDS AND PHARMACEUTICAL COMPOSITIONS THEREOF FOR THE TREATMENT OF INFLAMMATORY DISORDERS |
| US2014303140 | 2014-10-09 | NOVEL COMPOUNDS AND PHARMACEUTICAL COMPOSITIONS THEREOF FOR THE TREATMENT OF INFLAMMATORY DISORDERS |
////////////GLPG 1690, idiopathic pulmonary fibrosis, PHASE 1, GALAPAGOS, 1628260-79-6
n12c(c(nc1c(cc(c2)N3CCN(CC3)CC(=O)N4CC(C4)O)C)CC)N(C)c5nc(c(s5)C#N)c6ccc(cc6)F
CCC1=C(N2C=C(C=C(C2=N1)C)N3CCN(CC3)CC(=O)N4CC(C4)O)N(C)C5=NC(=C(S5)C#N)C6=CC=C(C=C6)F
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Googleplus amcrasto@gmail.com
P.S
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India.
DISCLAIMER
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP
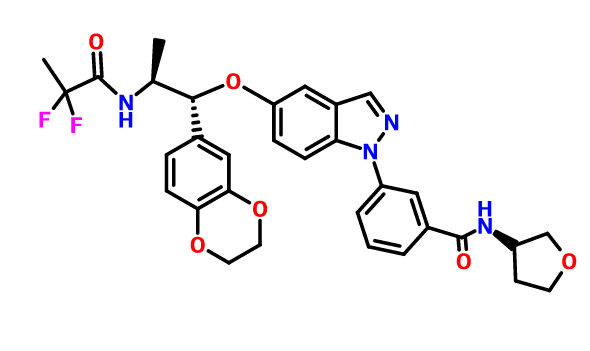
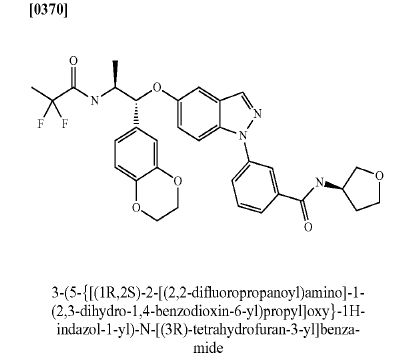
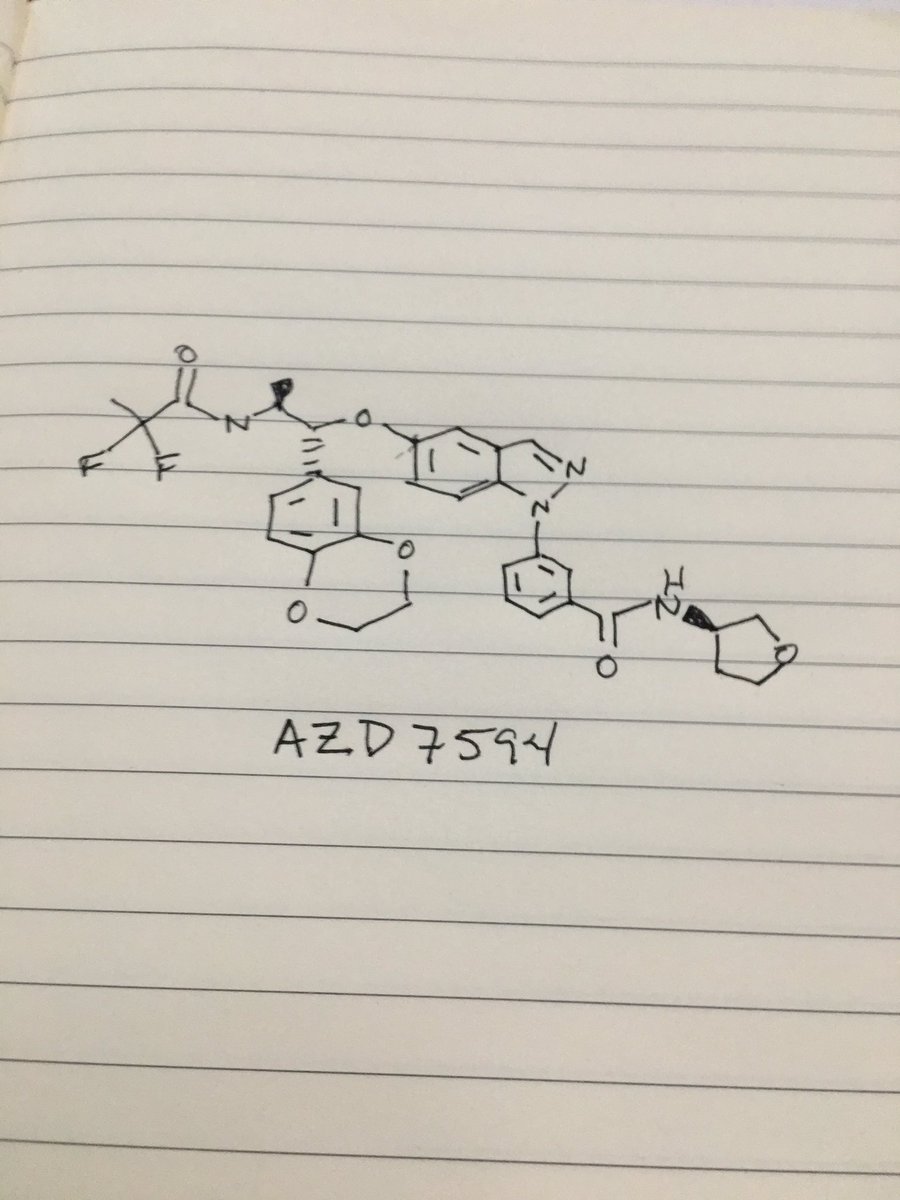
AZD 7594
 .
.
Picture credit….Bethany Halford

AZD 7594
$AZN‘s asthma candidate
AZ13189620; AZD-7594
Bayer Pharma Aktiengesellschaft, Astrazeneca Ab
| Molecular Formula: | C32H32F2N4O6 |
|---|---|
| Molecular Weight: | 606.616486 g/mol |
3-[5-[(1R,2S)-2-(2,2-difluoropropanoylamino)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)propoxy]indazol-1-yl]-N-(oxolan-3-yl)benzamide
- Cas 1196509-60-0
AZD-7594 is in phase II clinical trials by AstraZeneca for the treatment of mild to moderate asthma.
It is also in phase I clinical trials for the treatment of chronic obstructive pulmonary disorder (COPD).
https://clinicaltrials.gov/ct2/show/NCT02479412
| Company | AstraZeneca plc |
| Description | Inhaled selective glucocorticoid receptor (GCCR) modulator |
| Molecular Target | Glucocorticoid receptor (GCCR) |
- Phase II Asthma
- Phase I Chronic obstructive pulmonary disease
- 01 Feb 2016 AstraZeneca completes a phase II trial in Asthma in Bulgaria and Germany (Inhalation) (NCT02479412)
- 09 Jan 2016 AstraZeneca plans to initiate a phase I trial in Healthy volunteers in USA (IV and PO) (NCT02648438)
- 01 Jan 2016 Phase-I clinical trials in Chronic obstructive pulmonary disease (In volunteers) in USA (PO, IV, Inhalation) (NCT02648438)
PATENT
http://www.google.com/patents/WO2009142569A1
PATENT
US20100804345
UNWANTED ISOMER
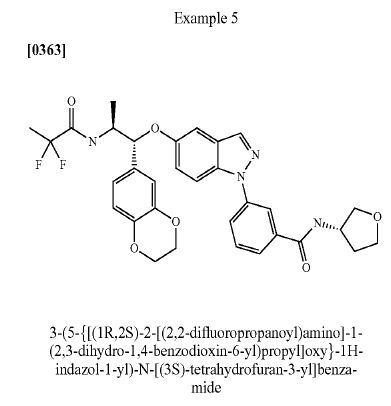

WANTED COMPD


PATENT
Example 6
WANTED ISOMER

3-(5- { TC 1 R,2SV2-r(2,2-difluoropropanoyl)aminol- 1 -(2,3-dihydro-l ,4-benzodioxin-6-5 yDpropylioxy) – 1 H-indazol- 1 -ylVN-[(3R)-tetrahydrofuran-3-vnbenzamide. APCI-MS: m/z 607 [MH+] 1H NMR ^OO MHz, DMSOd6) δ 8.71 (IH, d), 8.65 (IH, d), 8.24 (IH, s), 8.18 (IH, s), 7.90 – 7.84 (2H, m), 7.77 (IH, d), 7.65 (IH, t), 7.21 (IH, dd), 7.13 (IH, d), 6.89 – 6.78 (3H, m), 5.17 (IH, d), 4.48 (IH, m), 4.23 – 4.10 (5H, m), 3.89 – 3.82 (2H, m), 3.72 (IH, td), 3.61 (IH, dd), 2.16 (IH, m), 1.94 (IH, m), 1.55 (3H, t), 1.29 (3H, d). LC (method A) rt = 12.03 min LC (method B) rt = 11.13 min Chiral SFC (method B) rt = 4.71 min M.p. = 177 °C
UNWANTED

o 3-(5- { IY 1 R,2S V2-r(2,2-difluoropropanoyl‘)amino‘|- 1 -(2,3-dihydro- 1 ,4-benzodioxin-6- yl)propyl]oxy } – 1 H-indazol- 1 -yP-N-IO S)-tetrahydrofuran-3 -yl“|benzamide
APCI-MS: m/z 607 [MH+]
1H NMR (400 MHz, DMSO-J6) δ 8.71 (IH, d), 8.65 (IH, d), 8.24 (IH, s), 8.18 (IH, s),
7.90 – 7.84 (2H, m), 7.77 (IH, d), 7.65 (IH, t), 7.21 (IH, dd), 7.13 (IH, d), 6.89 – 6.78 (3H,s m), 5.17 (IH, d), 4.48 (IH, m), 4.24 – 4.11 (5H, m), 3.90 – 3.81 (2H, m), 3.72 (IH, td), 3.61
(IH, dd), 2.16 (IH, m), 1.94 (IH, m), 1.55 (3H, t), 1.29 (3H, d).
LC (Method A) rt = 12.02 min
LC (Method B) rt = 11.12 min
Chiral SFC (method B) rt = 5.10 min o M.p. = 175 0C
PATENT
WO 2011061527
http://www.google.com/patents/WO2011061527A1?cl=en
Intermediate 12
( 1 R,2S)-2-amino- 1 -(2,3 -dihydrobenzo b [ 1 ,41dioxin-6-yl)propan- 1 -ol hydrochloride. (12)

5-6 N HC1 in 2-propanol (8 mL, 40-48 mmol) was added to tert-butyl (lR,2S)-l-(2,3- dihydrobenzo[b][l,4]dioxin-6-yl)-l-hydroxypropan-2-ylcarbamate (I2a) (3.1 g, 10.02 mmol) in ethyl acetate (40 mL) at 40°C and stirred for 3 hours. The reaction mixture was allowed to reach r.t. and was concentrated by evaporation. Ether was added and the salt was filtered off and washed with ether. The salt was found to be hygroscopic. Yield 2.10 g (85%)
APCI-MS: m/z 210 [MH+-HC1]
1H-NMR (300 MHz, DMSO-^): δ 8.01 (brs, 3H), 6.87-6.76 (m, 3H), 5.93 (brd, 1H), 4.79 (brt, 1H), 4.22 (s, 4H), 3.32 (brm, 1H), 0.94 (d, 3H).
tert-butyl (1R,2S)- 1 -(2,3-dihvdrobenzorbl Γ 1 ,41dioxin-6-yl)- 1 -hvdroxypropan-2-ylcarbamate.

The diastereoselective catalytic Meerwein-Ponndorf-Verley reduction was made by the method described by Jingjun Yin et. al. J. Org. Chem. 2006, 71, 840-843.
(S)-tert-butyl 1 -(2,3-dihydrobenzo[b] [ 1 ,4]dioxin-6-yl)- 1 -oxopropan-2-ylcarbamate (I2b) (3.76 g, 12.23 mmol), aluminium isopropoxide (0.5 g, 2.45 mmol) and 2-propanol (12 mL, 157.75 mmol) in toluene (22 mL) were stirred at 50°C under argon for 16 hours. The reaction mixture was poured into 1M HC1 (150 mL) and the mixture was extracted with ethyl acetate (250 mL). The organic phase was washed with water (2×50 mL) and brine (100 mL), dried over Na2SC”4, filtered and concentrated. The crude product was purified by flash- chromatography on silica using ethyl acetate/hexane (1/2) as eluent. Fractions containing product were combined. Solvent was removed by evaporation to give the desired product as a colourless solid. Yield 3.19 g (84%) APCI-MS: m/z 236, 210, 192 [MH -tBu-18, MH -BOC, MH -BOC- 18]
1H NMR (300 MHz, DMSO-^): δ 6.80-6.70 (m, 3H), 6.51 (d, IH), 5.17 (d, IH), 4.36 (t, IH),
4.19 (s, 4H), 3.49 (m, IH), 1.31 (s, 9H), 0.93 (d, 3H).
(S)-tert-butyl 1 -(2,3-dihydrobenzo[bl [ 1 ,41dioxin-6-yD- 1 -oxopropan-2-ylcarbamate. (I2b)

A suspension of (S)-tert-butyl l-(methoxy(methyl)amino)-l-oxopropan-2-ylcarbamate (3 g, 12.92 mmol) in THF (30 mL) was placed under a protective atmosphere of argon and cooled down to -15 to -20°C. Isopropylmagnesium chloride, 2M in THF (6.5 mL, 13.00 mmol), was added keeping the temperature below -10°C. The temperature was allowed to reach 0°C. A freshly prepared solution of (2,3-dihydrobenzo[b][l,4]dioxin-6-yl)magnesium bromide, 0.7M in THF (20 mL, 14.00 mmol) was added. The temperature was allowed to reach r.t. overnight. The reaction mixture was poured into ice cooled IN HC1 (300 mL). TBME (300 mL) was added and the mixture was transferred to a separation funnel. The water phase was back extracted with TBME (200 mL). The ether phases were washed with water, brine and dried (Na2S04). The crude product was purified by flash chromatography using TBME /Heptane 1/2 as eluent. Fractions containing the product were combined and solvents were removed by evaporation to give the subtitle compound as a slightly yellow sticky oil/gum. Yield 3.76g
(95%)
APCI-MS: m/z 208 [MH+ – BOC]
1H NMR (300 MHz, DMSO-^): δ 7.50 (dd, IH), 7.46 (d, IH), 7.24 (d, IH), 6.97 (d, IH), 4.97 (m, IH), 4.30 (m, 4H), 1.36 (s, 9H), 1.19 (d, 3H).
Intermediate 13
(lR,2S)-2-amino-l-(4H-benzo[dl[l,31dioxin-7- l)propan-l-ol hydrochloride (13)

Tert-butyl ( 1 R,2S)- 1 -(4H-benzo[d] [ 1 ,3]dioxin-7-yl)- 1 -hydroxypropan-2-ylcarbamate (I3b) (403 mg, 1.30 mmol) was dissolved in ethyl acetate (5 mL) and 5-6 N HC1 solution in 2- propanol (1.5 mL, 7.5-9 mmol) was added. The mixture was stirred at 50 °C for 1.5 hours. The solvents was removed by evaporation. The residual sticky gum was treated with ethyl acetate and evaporated again to give a solid material that was suspended in acetonitrile and stirred for a few minutes. The solid colourless salt was collected by filtration and was found to be somewhat hygroscopic. The salt was quickly transferred to a dessicator and dried under reduced pressure. Yield 293 mg (92%)
APCI-MS: m/z 210 [MH+ -HC1]
1H NMR (300 MHz, DMSO-^) δ 8.07 (3H, s), 7.05 (IH, d), 6.92 (IH, dd), 6.85 (IH, d), 6.03 (IH, d), 5.25 (2H, s), 4.87 (3H, m), 3.42 – 3.29 (IH, m), 0.94 (3H, d).
(4S.5R -5-(4H-benzordiri.31dioxin-7-vn- -methyloxazolidin-2-one (I3a

A mixture of (lR,2S)-2-amino-l-(4H-benzo[d][l,3]dioxin-7-yl)propan-l-ol hydrochloride (I3b) (120 mg, 0.49 mmol), DIEA (0.100 mL, 0.59 mmol) and CDI (90 mg, 0.56 mmol) in THF (2 mL) was stirred at r.t. for 2 hours. The reaction mixture was concentrated by evaporation and the residual material was partitioned between ethyl acetate and water. The organic phase was washed with 10% NaHS04, dried over MgS04, filtered and evaporated. The crude product was analysed by LC/MS and was considered pure enough for further analysis by NMR. Yield 66 mg (57%)
The relative cis conformation of the product was confirmed by comparing the observed 1H- NMR with the literature values reported for similar cyclised norephedrine (Org. Lett. 2005 (07), 13, 2755-2758 and Terahedron Assym. 1993, (4), 12, 2513-2516). In a 2D NOESY experiment a strong NOE cross-peak was observed for the doublet at 5.64 with the multiplet at 4.19 ppm. This also confirmed the relative czs-conformation.
APCI-MS: m/z 236 [MH+]
1H NMR (400 MHz, CDC13) δ 6.99 (d, J= 8.0 Hz, IH), 6.88 (dd, J= 8.0, 1.4 Hz, IH), 6.83 (s, IH), 5.81 (brs,lH), 5.64 (d, J= 8.0 Hz, IH), 5.26 (s, 2H), 4.91 (s, 2H), 4.19 (m, IH), 0.85 (d, J = 6.4 Hz, 3H). Tert-butyl ( 1 R,2S)- 1 -(4H-benzord1 Γ 1 ,31dioxin-7-yl)- 1 -hvdroxypropan-2-ylcarbamate (I3b)

A mixture (S)-tert-butyl l-(4H-benzo[d][l,3]dioxin-7-yl)-l-oxopropan-2-ylcarbamate (I3c) (680 mg, 2.21 mmol), triisopropoxyaluminum (140 mg, 0.69 mmol) and propan-2-ol (3 mL, 38.9 mmol) in toluene (3 mL) was stirred at 65 °C for 15 hours. The reaction mixture was allowed to cool down, poured into 1M HC1 (50 mL) and extracted with ethyl acetate (2×50 mL). The organic phase was washed with water, brine, dried over MgS04, filtered and solvents were removed by evaporation to afford a colourless solid. The crude product was purified by flash chromatography, (solvent A = Heptane, solvent B = EtOAc + 10% MeOH. A gradient of 10%B to 50%B in A was used). The obtained product was crystallised from DCM / heptane to afford the subtitle compound as colourless needles. Yield 414 mg (60%)
APCI-MS: m/z 210 [MH+ -BOC]
1H NMR (400 MHz, DMSO- ¾ δ 6.97 (1H, d), 6.88 (1H, d), 6.77 (1H, s), 6.56 (1H, d), 5.27 (1H, d), 5.22 (2H, s), 4.83 (2H, s), 4.44 (1H, t), 3.53 (1H, m), 1.32 (9H, s), 0.93 (3H, d). (S)-Tert-butyl 1 -(4H-benzord1 Γ 1 ,31dioxin-7-vD- 1 -oxopropan-2-ylcarbamate (I3c)

7-Bromo-4H-benzo[d][l,3]dioxine (1 g, 4.65 mmol) was dissolved in THF (5 mL) and added to magnesium (0.113 g, 4.65 mmol) under a protective atmosphere of argon. One small iodine crystal was added. The coloured solution was heated with an heat gun in short periods to initiate the Grignard formation. When the iodine colour vanished the reaction was allowed to proceed at r.t. for 1.5 hours.
In a separate reaction tube (S)-tert-butyl l-(methoxy(methyl)amino)-l-oxopropan-2- ylcarbamate (1 g, 4.31 mmol) was suspended in THF (5 mL) and cooled in an ice/acetone bath to below -5 °C. Isopropylmagnesium chloride, 2M solution in THF (2.5 mL, 5.00 mmol) was slowly added to form a solution. To this solution was added the above freshly prepared Grignard reagent. The mixture was allowed to reach r.t. and stirred for 4 hours. The reaction mixture was slowly poured into ice-cold 150 mL 1M HC1. Ethyl acetate (150 mL) was added and the mixture was stirred for a few minutes and transferred to a separation funnel. The organic phase was washed with water and brine, dried over MgS04, filtered and concentrated. The obtained crude product was further purified by flash chromatography using a prepacked 70g silica column with a gradient of 10% TBME to 40% TBME in heptane as eluent. The subtitle compound was obtained as a colourless solid. Yield 790 mg (59%>)
APCI-MS: m/z 208 [MH+ -BOC]
1H NMR (400 MHz, DMSO-^) δ 7.53 (IH, dd), 7.39 (IH, s), 7.30 (IH, d), 7.22 (IH, d), 5.30 (2H, s), 4.98 (IH, m), 4.95 (2H, s), 1.35 (9H, s), 1.20 (3H, d).
Preparation 4
3-(5-([(lR,2S)-2-[(2,2-difluoropropanoyl)aminol-l-(2,3-dihydro-l,4-benzodioxin-6- yl)propyl]oxy| – 1 H-indazol- 1 -yl)-N-[(3R)-tetrahydrofuran-3-yllbenzamide

TEA (2.0 g, 20.65 mmol) was added to a mixture of 3-(5-((lR,2S)-2-(2,2- difluoropropanamido)- 1 -(2,3-dihydrobenzo[b] [ 1 ,4]dioxin-6-yl)propoxy)-l H-indazol-1 – yl)benzoic acid (14) (3.6 g, 6.70 mmol), (R)-tetrahydrofuran-3 -amine hydrochloride (0.99 g, 8.0 mmol) and HBTU (2.65 g, 6.99 mmol) in DCM (15 mL). The reaction was stirred at r.t. for 3h, then quenched by addition of a mixture of water and ethyl acetate. The mixture was shaken and the organic layer was collected. The water phase was extracted twice with ethyl acetate. The combined organic layers were washed with a small portion of water and dried over magnesium sulphate. The product was purified by flash chromatography (silica, eluent: a gradient of ethyl acetate in heptane). The residue was crystallized by dissolving in refluxing acetonitrile (50 mL) and then allowing to cool to r.t. over night. The solid was collected by filtration, washed with a small volume of acetonitrile and dried at 40°C in vaccum to give the title compound (2.5 g, 61%).
APCI-MS: m/z 607 [MH+]
1H NMR (400 MHz, DMSO-d6) δ 8.71 (IH, d), 8.65 (IH, d), 8.24 (IH, s), 8.18 (IH, s), 7.90 – 7.84 (2H, m), 7.77 (IH, d), 7.65 (IH, t), 7.21 (IH, dd), 7.13 (IH, d), 6.89 – 6.78 (3H, m), 5.17 (IH, d), 4.48 (IH, m), 4.23 – 4.10 (5H, m), 3.89 – 3.82 (2H, m), 3.72 (IH, td), 3.61 (IH, dd), 2.16 (IH, m), 1.94 (IH, m), 1.55 (3H, t), 1.29 (3H, d).
LC (method A) rt = 12.03 min
LC (method B) rt = 11.13 min
Chiral SFC (method B) rt = 4.71 min
M.p. = 177 °C
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015080434 | 2015-03-19 | PHENYL AND BENZODIOXINYL SUBSTITUTED INDAZOLES DERIVATIVES |
| US8916600 | 2014-12-23 | Phenyl and benzodioxinyl substituted indazoles derivatives |
| US8211930 | 2012-07-03 | Phenyl and Benzodioxinyl Substituted Indazoles Derivatives |
REFERENCES
https://www.astrazeneca.com/content/dam/az/press-releases/2014/Q2/Pipeline-table.pdf
////////AZD 7594, AZ13189620, AZD-7594 , phase 2, astrazeneca, 1196509-60-0
c21cc(ccc1n(nc2)c3cc(ccc3)C(=O)NC4COCC4)O[C@H](c5cc6c(cc5)OCCO6)[C@@H](NC(=O)C(F)(F)C)C
CC(C(C1=CC2=C(C=C1)OCCO2)OC3=CC4=C(C=C3)N(N=C4)C5=CC=CC(=C5)C(=O)NC6CCOC6)NC(=O)C(C)(F)F
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Googleplus amcrasto@gmail.com
P.S
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India.
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP
PF 06650833
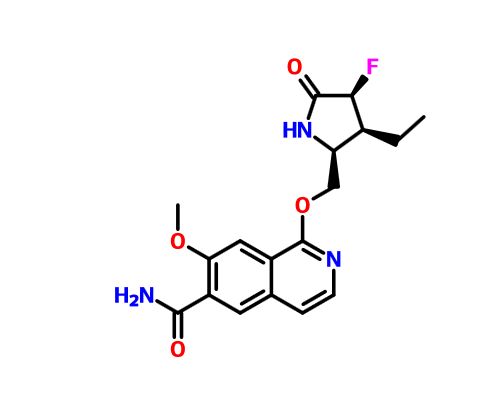
 .
.
Picture credit….Bethany Halford
PF 06650833
MFC18H20FN3O4, MW361.37
1-{[(2S,3S,4S)-3-ethyl-4-fluoro-5-oxopyrrolidin-2-yl]methoxy}-7-methoxyisoquinoline-6-carboxamide
6-Isoquinolinecarboxamide, 1-[[(2S,3S,4S)-3-ethyl-4-fluoro-5-oxo-2-pyrrolidinyl]methoxy]-7-methoxy-
CAS 1817626-54-2
WO 2015150995
1st disclosures is @pfizer‘s on inflammatory disease treatment targeting IRAK4
$PFE IRAK4 inhibitor
Phase I Lupus vulgaris
- 01 Feb 2016 Pfizer completes a phase I pharmacokinetics trial in Healthy volunteers in USA (PO) (NCT02609139)
- 01 Nov 2015 Pfizer initiates a phase I pharmacokinetics trial in Healthy volunteers in USA (PO) (NCT02609139)
- 01 Jun 2015 Pfizer completes a phase I trial for Lupus (In volunteers) in USA (PO) (NCT02224651)
Compounds useful for the treatment of autoimmune and inflammatory diseases associated with lnterleukin-1 Receptor Associated Kinase (IRAK) and more particularly compounds that modulate the function of IRAK4.
Protein kinases are families of enzymes that catalyze the phosphorylation of specific residues in proteins, broadly classified in tyrosine and serine/threonine kinases. Inappropriate activity arising from dysregulation of certain kinases by a variety of mechanisms is believed to underlie the causes of many diseases, including but not limited to, cancer, cardiovascular diseases, allergies, asthma, respiratory diseases, autoimmune diseases, inflammatory diseases, bone diseases, metabolic disorders, and neurological and neurodegenerative diseases. As such, potent and selective inhibitors of kinases are sought as potential treatments for a variety of human diseases.
There is considerable interest in targeting the innate immune system in the treatment of autoimmune diseases and sterile inflammation. Receptors of the innate immune system provide the first line of defense against bacterial and viral insults. These receptors recognize bacterial and viral products as well as pro-inflammatory cytokines and thereby initiate a signaling cascade that ultimately results in the up-regulation of inflammatory cytokines such as TNFa, IL6, and interferons. Recently it has become apparent that self-generated ligands such as nucleic acids and products of inflammation such as high-mobility group protein B1 (HMGB1) and Advanced Glycated End-products (AGE) are ligands for Toll-like receptors (TLRs) which are key receptors of the innate immune system (O’Neill 2003, Kanzler et al 2007, Wagner 2006). This demonstrates the role of TLRs in the initiation and perpetuation of inflammation due to autoimmunity.
lnterleukin-1 receptor associated kinase 4 (I RAK4) is a ubiquitously expressed serine/threonine kinase involved in the regulation of innate immunity (Suzuki & Saito 2006). IRAK4 is responsible for initiating signaling from TLRs and members of the I L- 1/18 receptor family. Kinase-inactive knock-ins and targeted deletions of IRAK4 in mice were reported to cause reductions in TLR and IL-1 induced pro-inflammatory cytokines (Kawagoe et al 2007; Fraczek et al. 2008; Kim et al. 2007). IRAK4 kinase-dead knock-in mice have also been shown to be resistant to induced joint inflammation in the antigen-induced-arthritis (AIA) and serum transfer-induced (K/BxN) arthritis models (Koziczak-Holbro 2009). Likewise, humans deficient in IRAK4 also appear to display the inability to respond to challenge by Toll ligands and IL-1 (Hernandez & Bastian 2006). However, the immunodeficient phenotype of IRAK4-null individuals is narrowly restricted to challenge by gram positive bacteria, but not gram negative bacteria, viruses or fungi. This gram positive sensitivity also lessens with age, implying redundant or compensating mechanisms for innate immunity in the absence of IRAK4 (Lavine et al 2007).
These data indicate that inhibitors of IRAK4 kinase activity should have therapeutic value in treating cytokine driven autoimmune diseases while having minimal immunosuppressive side effects. Additional recent studies suggest that targeting IRAK4 may be useful in other inflammatory pathologies such as atherosclerosis and diffuse large B-cell lymphoma (Rekhter et al 2008; Ngo et al 2011). Therefore, inhibitors of IRAK4 kinase activity are potential therapeutics for a wide variety of diseases including but not limited to autoimmunity, inflammation, cardiovascular diseases, cancer, and metabolic diseases. See the following references for additional information: N. Suzuki and T. Saito, Trends in Immunology, 2006, 27, 566. T. Kawagoe, S. Sato, A. Jung, M. Yamamoto, K. Matsui, H. Kato, S. Uematsu, O. Takeuchi and S. Akira, Journal of Experimental Medicine, 2007, 204, 1013. J. Fraczek, T. W. Kim, H. Xiao, J. Yao, Q. Wen, Y. Li, J.-L. Casanova, J. Pryjma and X. Li, Journal of Biological Chemistry, 2008, 283, 31697. T. W. Kim, K. Staschke, K. Bulek, J. Yao, K. Peters, K.-H. Oh, Y. Vandenburg, H. Xiao, W. Qian, T. Hamilton, B. Min, G. Sen, R. Gilmour and X. Li, Journal of Experimental Medicine, 2007, 204, 1025. M. Koziczak-Holbro, A. Littlewood- Evans,
B. Pollinger, J. Kovarik, J. Dawson, G. Zenke, C. Burkhart, M. Muller and H. Gram, Arthritis & Rheumatism, 2009, 60, 1661. M. Hernandez and J. F. Bastian, Current Allergy and Asthma Reports, 2006, 6, 468. E. Lavine, R. Somech, J. Y. Zhang, A. Puel, X. Bossuyt, C. Picard, J. L. Casanova and C. M. Roifman, Journal of Allergy and Clinical Immunology, 2007, 120, 948. M. Rekhter, K. Staschke, T. Estridge, P. Rutherford, N. Jackson, D. Gifford-Moore, P. Foxworthy,
C. Reidy, X.-d. Huang, M. Kalbfleisch, K. Hui, M.S. Kuo, R. Gilmour and C. J. Vlahos, Biochemical and Biophysical Research Communications, 2008, 367, 642. O’Neill, L. A. (2003). “Therapeutic targeting of Toll-like receptors for inflammatory and infectious diseases.” Curr Opin Pharmacol 3(4): 396. Kanzler, H et al. (2007) “Therapeutic targeting of innate immunity with toll-like receptor agonists and antagonists.” Nature Medicine 13:552. Wagner, H. (2006) “Endogenous TLR ligands and autoimmunity” /Advances in Immunol 91 : 159. Ngo, V. N. et al. (2011) “Oncogenically active MyD88 mutations in human lymphoma” Nature 470: 115.
PATENT
Preparation 1 : 1-chloro-7-methoxyisoquinoline-6-carbonitrile (P1) Step 1. Synthesis of methyl 4-iodo-3-methoxybenzoate (CAS 35387-92-9. CD.
To a solution of 3-hydroxy-4-iodobenzoic acid (CAS 58123-77-6, C12) (10800 g, 40.9 moles) in DMF (65 L) was added K2C03 (25398 g, 184 moles), followed by the slow addition of dimethyl sulfate (11352 g, 90 moles). This mixture was heated to about 50 °C for over night. The reaction mixture was cooled to about 25 °C, diluted with EtOAc (50 L) and filtered through a plug of Celite®. The solid was thoroughly washed with EtOAc (10 L X 3). The combined EtOAc filtrates were poured into water. After stirring for about 30 min, the EtOAc layer was separated and it was further washed sequentially with water, 1 M NaOH and brine. The EtOAc layer was separated, dried over Na2S04, filtered and concentrated to provide the title compound C1. Yield: 11750 g (98%).
Step 2. Synthesis of (4-iodo-3-methoxyphenyl)methanol (CAS 244257-61-2, C2).
To a solution of compound C1 (11750 g, 40.2 moles) in THF (35 L) was added NaBH4 (7645 g, 201.09 moles) and refluxed. While refluxing, MeOH (25 L) was slowly added into the reaction mixture at a rate of about 1 L per hour. After completion of the reaction, it was poured into a solution of cold dilute HCI. Once the excess of NaBH4was quenched, the solution was filtered and extracted with EtOAc (2.5 L X 3). The combined EtOAc extracts were washed sequentially with water, brine and dried over Na2S04. The solvent was evaporated under reduced pressure and the resulting crude material was treated with MTBE. The resulting solid was filtered and filtrate was washed with water, brine, dried over Na2S0 , and filtered. The solvent was evaporated under reduced pressure to provide the title compound C2. Yield: 9900 g (93%).
Step 3. Synthesis of 4-iodo-3-methoxybenzaldehyde (CAS 121404-83-9, C3).
To a solution of compound C2 (9900 g, 34.5 moles) in CHCI3 (186 L), was added manganese dioxide (18000 g, 207 moles) and the resulting mixture was refluxed for about 16 h. The mixture was cooled to about 25 °C and filtered through a Celite pad, which was then washed thoroughly with CHCI3. The CHCI3 was evaporated under reduced pressure to provide the title compound C3. Yield: 9330 g (95%). 1 H NMR (400 MHz, CDCI3): δ 9.95 (s, 1 H), 7.99 (d, 1 H), 7.14 (dd, 1 H), 3.95 (s, 3 H).
Step 3. Synthesis of 6-iodo-7-methoxyisoquinoline (CAS 244257-63-4. C4).
To a solution of compound C3 (9300 g, 35 moles) in toluene (60 L) was added amino acetaldehyde dimethyl acetal (5590 g, 53 moles) and the mixture was refluxed for about 4 h, while removing the liberated water by the use of a Dean – Stark water separator. The reaction mixture was cooled to about 0 °C, after which trifluoroacetic anhydride (22305 g, 106 moles) followed by BF3-Et20 (15080 g, 106 moles) were added, keeping internal temperature below 5 °C. The reaction mixture was stirred at about 25 °C for about 16 h and quenched by pouring into a mixture of ice and ammonium hydroxide. The product was extracted with EtOAc (10 L X 3), and the combined EtOAc extracts were washed sequentially with water and brine. The combined EtOAc extracts were dried over Na2S04, filtered, and concentrated to afford a dark tan colored residue. This was treated with a mixture of MTBE and hexane (1 :1 v/v, 30 L), followed by 6 M HCI (9 L), with stirring. The precipitated solid was filtered and washed with MTBE. The solid was suspended in EtOAc (5 L) and made alkaline with ammonium hydroxide. The EtOAc layer was separated, washed with brine, dried over Na2S04, filtered, and concentrated to afford crude compound C4 as a brown solid. HPLC (230 nm) showed it to be about 83% pure.
The crude material (1000 g) was taken in AcOH (2.5 L) and stirred for about 90 min at about 25 °C. The solid was filtered and washed with AcOH (500 ml_). The filtrate was neutralized with saturated aqueous Na2C03 solution. The resulting precipitated solid was filtered, washed with water (4 L), and oven dried at about 70 – 75 °C for about 5 h to afford about 780 g of pure C4. Similarly, the remaining crude C4 (4 kg) was purified to provide the title compound C4. Yield: 4300 g (42%). 1H NMR (400 MHz, CDCI3): δ 9.15 (s, 1 H), 8.45 (d, 1 H), 8.35 (s, 1 H), 7.45 (d, 1 H), 7.15 (s, 1 H) 4.00 (s, 3 H).
Step 4. Synthesis of 7-methoxyisoquinoline-6-carbonitrile (C5).
To a solution of compound C4 (4300 g , 15 moles) in DMSO (39 L) was added copper(l) cyanide (2954 g, 33 moles) and the mixture was heated to about 120 °C for about 3 h. The reaction mixture was quenched by pouring into a mixture of ice and ammonium hydroxide (40 L) and filtered. The filtrate was extracted with EtOAc (10 L X 2). While stirring, the solid residue was again treated with ammonium hydroxide solution (10 L) and EtOAc (10 L). After filtration, the precipitated material was repeatedly washed with a mixture of MeOH and CHCI3 (1 :9, v/v) several times and the combined extracts were washed with brine. The extracts were dried over Na2S04, filtered, and concentrated under reduced pressure. The resulting crude material was triturated with hexane to provide the title compound C5. Yield: 2250 g (87%). 1H NMR (400 MHz, CDCI3): δ 9.25 (br. s, 1 H), 8.55 (br. s, 1 H), 8.15 (s, 1 H), 7.60 (d, 1 H), 7.30 (s, 1 H), 4.05 (s, 3 H).

A solution of a reactant such as 1-(((2S,3S,4S)-3-ethyl-4-fluoro-5-oxopyrrolidin-2-yl)methoxy)-7-methoxyisoquinoline-6-carbonitrile (200 mg, 0.5 mmol) in concentrated H2SO4 (1.5 ml.) was warmed to about 55 °C for about two hours, then cooled to about 20 °C. The reaction mixture was added dropwise with vigorous stirring to 7.3 ml_ of ice cold concentrated ammonium hydroxide with cooling in ice. The precipitated solid was filtered and washed with water, heptane, ether, and dried under vacuum. The residue may be used directly for subsequent work, or it may be purified by chromatography or HPLC.
ABSTRACTS
251st Am Chem Soc (ACS) Natl Meet (March 13-17, San Diego) 2016, Abst MEDI 261




//////////PF 06650833, IRAK4 inhibitor, inflammatory disease treatment , PFIZER, 1817626-54-2
N1C([C@H](C([C@H]1COc3c2cc(c(cc2ccn3)C(=O)N)OC)CC)F)=O
NC(=O)c2cc3ccnc(OC[C@H]1NC(=O)[C@@H](F)[C@H]1CC)c3cc2OC
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Googleplus amcrasto@gmail.com
P.S
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India.
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP
LY 2922470

LY 2922470

LY 2922470
Picture credit….Bethany Halford
(3S)-3-[4-[[5-[(8-methoxy-3,4-dihydro-2H-quinolin-1-yl)methyl]thiophen-2-yl]methoxy]phenyl]hex-4-ynoic acid
Benzenepropanoic acid, 4-[[5-[(3,4-dihydro-8-methoxy-1(2H)-quinolinyl)methyl]-2-thienyl]methoxy]-β-1-propyn-1-yl-, (βS)-
Glucose Lowering Agents, Signal Transduction Modulators
| CAS | 1423018-12-5 |
|---|---|
| Molecular Formula: | C28H29NO4S |
| Molecular Weight: | 475.59916 g/mol |
|---|
https://clinicaltrials.gov/ct2/show/NCT01867216
- Phase I Type 2 diabetes mellitus
Eli Lilly
Antihyperglycaemics
- 28 Jan 2014 Eli Lilly completes a phase I trial in Type-2 diabetes mellitus in USA (NCT01867216)
- 30 Jun 2013 Phase-I clinical trials in Type-2 diabetes mellitus in USA (PO)
- 14 Jun 2013 Eli Lilly plans a phase I trial for Type-2 diabetes mellitus in USA (NCT01867216)
PATENT
WO 2013025424
https://www.google.com/patents/US20130045990?cl=de
| Also published as | CA2843474A1, CA2843474C, CN103687856A, CN103687856B, EP2744806A1, US8431706, WO2013025424A1, Less « |
| Inventors | Chafiq Hamdouchi |
| Original Assignee | Eli Lilly And Company |



Preparation 18-Methoxyquinoline
Add potassium hydroxide (435 g, 7.76 mol) to a solution of 8-hydroxy quinoline (250 g, 1.724 mol) in THF (10 L) at ambient temperature and stir. Add methyl iodide (435 g, 2.58 mol) dropwise and stir overnight. Filter the reaction mixture and wash the solid with THF (2 L). Concentrate the solution to dryness; add water; extract with dichloromethane (2×3 L); combine the organic layers; and wash with brine. Collect the organic layers and dry over sodium sulfate. Remove the solids by filtration. Collect the filtrate and concentrate under reduced pressure to give a red oil, which solidifies on standing, to give the title compound (281 g, 102%), which can be used without further purification. ESI (m/z) 160(M+H).
Preparation 2
8-Methoxy-1,2,3,4-tetrahydroquinoline
Add sodium cyanoborohydride (505 g, 8.11 mol) in EtOH (1 L) to a solution of 8-methoxy quinoline (425 g, 2.673 mol) in EtOH (9 L), and stir. Cool the reaction mixture to an internal temperature of 0° C. and add HCl (35%, 1.12 L, 10.962 mol) dropwise over 60 min so that the internal temperature did not rise above 20° C. Allow the reaction mixture to warm to ambient temperature and then heat to reflux for 2.5 hours. Cool to ambient temperature and stir overnight. Add ammonium hydroxide (25%, 1 L); dilute with water (15 L); and extract the mixture with dichloromethane (3×10 L). Combine the organic layers and dry over sodium sulfate. Remove the solids by filtration. Collect the filtrate and concentrate under reduced pressure to give a residue. Purify the residue by silica gel flash chromatography, eluting with ethyl acetate: hexane (1:10) to give the title compound (357 g, 82%). ESI (m/z) 164(M+H).
Preparation 3
Methyl-5-methylthiophene-2-carboxylate
Add thionyl chloride (153 ml, 2.1 mol) dropwise over 20 min to a solution of 5-methylthiophene-2-carboxylic acid (100 g, 0.703 mol) in MeOH (1 L) at 0° C. and stir. After the addition is complete, heat the reaction mixture to reflux for 3.5 hours. Cool and concentrate in vacuo to give a thick oil. Dilute the oil with EtOAc (500 ml) and sequentially wash with water (300 ml) then brine (300 ml). Dry the organic layer over sodium sulfate. Remove the solids by filtration. Collect the filtrate and concentrate under reduced pressure to give the title compound (106 g, 97%), which is used without further purification. ESI (m/z) 156(M+H).
Preparation 4
Methyl 5-(bromomethyl)thiophene-2-carboxylate
Add freshly recrystallised NBS (323.8 g, 1.81 mol) to a solution of methyl-5-methylthiophene-2-carboxylate (258 g, 1.65 mol) in chloroform (2.6 L) at room temperature, and stir. Add benzoyl peroxide (3.99 g, 0.016 mol) and heat the reaction mixture to reflux for 7 hours. Cool the reaction mixture to ambient temperature and filter through diatomaceous earth. Wash the filter cake with chloroform (250 ml). Collect the organic layers and remove the solvent to give the title compound (388 g, 100%), which is used without further purification. ESI (m/z) 236(M+H).
Preparation 5
Methyl-5-[8-methoxy-3,4-dihydro-2H-quinolin-1-yl)methyl]thiophene-2-carboxylate
Add methyl-5-(bromoethyl)thiophene-2-carboxylate (432.5 g, 1.84 mol) in EtOH (500 ml) to a solution of 8-methoxy-1,2,3,4-tetrahydroquinoline (300 g 1.84 mol) in EtOH (1 L) and stir. Add DIPEA (641 ml, 3.67 mol) dropwise and stir at room temperature overnight. After completion of the reaction, remove the EtOH in vacuo, and add water (5 L). Extract the aqueous with EtOAc (3×3 L); combine the organic layers; and dry over sodium sulfate. Filter the solution and concentrate under reduced pressure to give a residue. Purify the residue by silica gel flash chromatography eluting with ethyl acetate: hexane (6:94) to give the title compound (325 g, 56%). ESI (m/z) 318(M+H).
Preparation 6
[5-[(8-Methoxy-3,4-dihydro-2H-quinolin-1-yl)methyl]-2-thienyl]methanol
Add DIBAL-H (1 M in toluene 2.7 L, 2.66 mol) slowly via a cannula over a period of 1.5 h to a stirred solution of methyl-5-(8-methoxy-3,4-dihydroquinolin-1(2H)-yl)methyl)thiophene-2-carboxylate (281 g, 0.886 mol) in THF (4 L) at −70° C. Monitor the reaction via thin layer chromatography (TLC) for completion. After completion of the reaction, allow the reaction mixture to warm to 20° C. and add a saturated solution of ammonium chloride. Add a solution of sodium potassium tartrate (1.3 Kg in 5 L of water), and stir overnight. Separate the organic layer; extract the aqueous phase with EtOAc (2×5 L); then combine the organic layers; and dry the combined organic layers over sodium sulfate. Remove the solids by filtration. Remove the solvent from the filtrate under reduced pressure to give the title compound as a white solid (252 g, 98%). ESI (m/z) 290(M+H).
Preparation 7
Ethyl(3S)-3-[4-[[5-[(8-methoxy-3,4-dihydro-2H-quinolin-1-yl)methyl]-2-thienyl]methoxy]phenyl]hex-4-ynoate
Add tributylphosphine (50% solution in EtOAc, 543 ml, 1.34 mol) to a solution of ADDP (282.5 g, 1.5 eq) in THF (3 L) and cool the mixture to an internal temperature of 0° C., then stir for 15 minutes. Add (S)-ethyl 3-(4-hydroxyphenyl)hex-4-ynoate (173.5 g, 0.747 mol) in THF (3 L) dropwise over 15 min; then add 5-((8-methoxy-3,4-dihydroquinolin-1(2H)-yl)methyl)thiophene-2-yl)methanol (216 g, 0747 mol) in THF (5 L) dropwise. Allow the reaction mixture to warm to ambient temperature and stir overnight. Filter the reaction mixture through diatomaceous earth and wash the filter cake with ethyl acetate (2 L). Concentrate the organic filtrate to dryness. Add water (4 L); extract with ethyl acetate (3×5 L); combine the organic layers; and dry the combined organic layers over sodium sulfate. Remove the solids by filtration and concentrate under reduced pressure to give an oil. Purify the residue by silica gel flash chromatography by eluting with ethyl acetate: hexane (6:94) to give the title compound (167 g, 44%). ESI (m/z) 504(M+H).
Example 1
(3S)-3-[4-[[5-[(8-Methoxy-3,4-dihydro-2H-quinolin-1-yl)methyl]-2-thienyl]methoxy]phenyl]hex-4-ynoic acid

Add a solution of potassium hydroxide (49.76 g, 0.88 mol) in water (372 ml) to a solution of (S)-ethyl-3-(4-((5-8-methoxy-3,4-dihydroquinolin-1(2H)-yl)methyl)thiophen-2-yl)methoxy) phenyl)hex-4-ynoate (149 g, 0.296 mol) in EtOH (1.49 L) at room temperature and stir overnight. Concentrate the reaction mixture to dryness and add water (1.3 L). Extract the resulting solution with EtOAc (2×300 ml) and separate. Adjust the pH of the aqueous layer to pH=6 with 2 N HCl. Collect the resulting solids. Recrystallise the solids from hot MeOH (298 ml, 2 vol) to give the title compound (91 g, 65%). ESI (m/z) 476(M+H).
Abstract
GPR40 agonists for the treatment of type 2 diabetes: From the laboratory to the patient
251st Am Chem Soc (ACS) Natl Meet (March 13-17, San Diego) 2016, Abst MEDI 260

Presenter

Chafiq Hamdouchi
Senior Research Advisor at Eli Lilly and Company
https://www.linkedin.com/in/chafiq-hamdouchi-4988126
Summary
Dr. Hamdouchi earned his bachelor’s degree and doctorate in organic chemistry from Louis Pasteur University, Strasbourg-France.
Following two postdoctoral fellowships, sponsored by the National Science Foundation-USA and Ministerio de Educación y Ciencia-Spain, he joined Eli Lilly and Company in 1995.
Throughout his 20 years of career at Lilly, he has contributed to a sustainable drug discovery portfolio from preclinical hypothesis to clinical proof-of-concept that spans the oncology, neuroscience and endocrinology therapeutic areas. He has led multidisciplinary (chemistry, pharmacology, ADMET, PK, medical) scientific teams in USA, Europe and Asia to deliver a number of compounds that achieved first human dose.
He is a co-inventor of six innovative molecules being pursued in clinical development for the treatment of Diabetes, Cancer and Neurodegenerative Diseases.
He has an extensive patent and publication record and deep experience in conducting drug discovery and development in Asia through effective partnership and mentorship.
SEE AT…………ONE ORGANIC CHEMIST ONE DAY BLOG
LINK……http://oneorganichemistoneday.blogspot.in/2016/03/chafiq-hamdouchi-senior-research.html
| Patent ID | Date | Patent Title |
|---|---|---|
| US8431706 | 2013-04-30 | 1,2,3,4-tetrahydroqinoline derivative useful for the treatment of diabetes |
References
GPR40 agonists for the treatment of type 2 diabetes: From the laboratory to the patient
251st Am Chem Soc (ACS) Natl Meet (March 13-17, San Diego) 2016, Abst MEDI 260
//////Phase 1, LY2922470, LY 2922470, Eli Lilly, Type 2 diabetes mellitus, 1423018-12-5, Chafiq Hamdouchi
CC#CC(CC(=O)O)C1=CC=C(C=C1)OCC2=CC=C(S2)CN3CCCC4=C3C(=CC=C4)OC
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Googleplus amcrasto@gmail.com
P.S
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India.
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP
HELP, Need one time help to pay 10 year concessional subscription to this, your favorite blog to WordPress
Just One viewer please come forward
Dear Kind Viewer’s
WordPress is kind to me and negotiated a one time 10 year concessional subscription of 260 US dollars…….https://newdrugapprovals.org/
I need one time help to pay this one time 10 year concessional subscription to our favorite blog.
This is done to keep this blog running even after my death.
Currently I am paying 99 US Dollars per annum
email me
amcrasto@gmail.com
call +919323115463
Paypal will work for me via email request to you by me, Indian govt does not allow automatic transfer via paypal buttons on the blog.
email me at amcrasto@gmail.com and tell me amount, i will request you via paypal
DR ANTHONY CRASTO
LIONEL MY SON, MY MOTIVATION
.
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, He cried bitterly and we had never seen him so depressed
Now I keep Lionel as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son and family happy.

ps
The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent,
///////////
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on Facebook
Googleplus amcrasto@gmail.com
P.S
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India.
DISCLAIMER
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY

