

.
Home » Uncategorized (Page 72)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
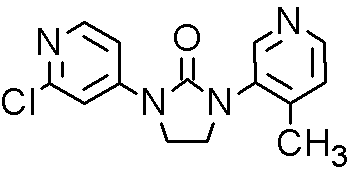
CFG 920, Novartis Scientists team up with Researchers at Aurigene, Bangalore, India,
CFG920,
Inhibitor Of Prostate Cancer With Fewer Cardiac Side Effects
Cas 1260006-20-9
Novartis
Target: CYP17/CYP11B2
Disease: Castration-resistant prostate cancer
MF C14H13ClN4O
MW: 288.0778
Elemental Analysis: C, 58.24; H, 4.54; Cl, 12.28; N, 19.40; O, 5.54
Steroid 17-alpha-hydroxylase inhibitors
CFG920 is a CYP17 inhibitor, is also an orally available inhibitor of the steroid 17-alpha-hydroxylase/C17,20 lyase (CYP17A1 or CYP17), with potential antiandrogen and antineoplastic activities. Upon oral administration, CYP17 inhibitor CFG920 inhibits the enzymatic activity of CYP17A1 in both the testes and adrenal glands, thereby inhibiting androgen production. This may decrease androgen-dependent growth signaling and may inhibit cell proliferation of androgen-dependent tumor cells.
https://clinicaltrials.gov/ct2/show/NCT01647789
NCT01647789: A Study of Oral CFG920 in Patients With Castration Resistant Prostate Cancer2012
- 09 Nov 2015Adverse events, efficacy and pharmacokinetics data from the phase I part of a phase I/II trial in Prostate cancer (Metastatic disease) presented at the 27th AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics (AACR-NCI-EORTC-2015)
- 29 Jan 2013Phase-I clinical trials in Prostate cancer in Spain (PO)
- 10 Dec 2012Phase-I clinical trials in Prostate cancer in Canada (PO)
In August 2015, preclinical data were presented at the 250th ACS meeting in Boston, MA. In monkeys, treatment with CFG-920 (3 mg/kg, po) showed good bioavailability with F value of 93%, Tmax of 0.5 h, Cmax of 1382 nM.dn and AUC of 2364 nM.h, while CFG-920 (10 mg/kg, po) showed F value of 183%, Cmax of 1179 nM.dn and Tmax of 1.04 h
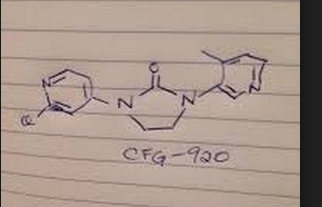
Bethany Halford on Twitter: “CFG920 – @Novartis CMOS for …
Bethany Halford on Twitter: “CFG920 – @Novartis CMOS for castration resistant prostate cancer #ACSBoston MEDI 1st disclosures http://t.co/XJJ3tCvpUk”

Novartis is developing CFG-920 (structure shown), an oral CYP17 inhibitor, for the potential treatment of metastatic castration-resistant prostate cancer. In March 2013, a phase I/II trial was initiated and at that time, the study was expected to complete in January 2015; in August 2015, clinical data were presented
2015 250th (August 19) Abs MEDI 341
Discovery of CFG920, a dual CYP17/CYP11B2 inhibitor, for the treatment of castration resistant prostate cancer
American Chemical Society National Meeting and Exposition
Christoph Gaul, Prakash Mistry, Henrik Moebitz, Mark Perrone, Bjoern Gruenenfelder, Nelson Guerreiro, Wolfgang Hackl, Peter Wessels, Estelle Berger, Mark Bock, Saumitra Sengupta, Venkateshwar Rao, Murali Ramachandra, Thomas Antony, Kishore Narayanan, Samiulla Dodheri, Aravind Basavaraju, Shekar Chelur

CHEMISTRY COLLABORATORS
Novartis-Aurigene team: (from left) Brahma Reddy V, Thomas Antony, Murali Ramachandra, Venkateshwar Rao G, Wesley Roy Balasubramanian, Kishore Narayanan, Samiulla DS, Aravind AB, and Shekar Chelur. Not pictured: Björn Grünenfelder, Saumitra Sengupta, Nelson Guerreiro, Andrea Gerken, Mark Perrone, Mark Bock, Wolfgang Hackl, Henrik Möbitz, Peter Wessels, Christoph Gaul, Prakash Mistry, and Estelle Marrer.
Credit: Aurigene

| Aurigene Discovery Technologies Limited, an independent subsidiary of Dr Reddy’s, CSN Murthy is chief executive officer |
Preclinical and clinical studies were performed to evaluate the efficacy of CFG-920, a dual cytochrome P450 (CYP)17 and CYP11B2 dual inhibitor, for the potential treatment of castration resistant prostate cancer. CFG-920 showed potent activity against human CYP17 and CYP11B2 enzymes with IC50 values of 0.023 and 0.034 microM, respectively. In monkeys, treatment with CFG-920 (3 mg/kg, po) showed good bioavailability (93%), Tmax of 0.5 h, Cmax of 1382 nM.dn and AUC of 2364 nM.h, while CFG-920 (10 mg/kg, po) showed F value of 183%, Cmax of 1179 nM.dn and Tmax of 1.04 h. In a phase I, first-in-man study, patients received continuous po dosing of CFG-920 (50 mg, bid) plus prednisone (5 mg) in 28-day cycles. At the time of presentation, CFG-920 was under phase II development.

CFG920
WO 2010149755

Novartis team: (clockwise from left) Wolfgang Hackl, Henrik Möbitz, Peter Wessels, Christoph Gaul, Prakash Mistry, and Estelle Marrer., Credit: Novartis
Prostate cancer is the most commonly occurring cancer in men. Doctors often treat the metastatic stage of the disease by depriving the patient of sex hormones via chemical or surgical castration. But if it progresses far enough, the cancer can survive this therapy, transforming into the castration-resistant form. “Once the cancer becomes castration-resistant, the prognosis is poor,” said Novartis’s Christoph Gaul.
In recent years, CYP17, a bifunctional 17α-hydroxylase/17,20-lyase cytochrome P450 enzyme, has emerged as a target for treating castration-resistant prostate cancer. The enzyme catalyzes the biosynthesis of sex hormones, including testosterone, and blocking it can starve prostate cancer of the androgens it needs to thrive.
Johnson & Johnson’s CYP17 inhibitor, abiraterone acetate (Zytiga), a steroid that binds irreversibly to CYP17, was approved by the Food & Drug Administration in 2011. But Novartis scientists thought they could make a better CYP17 inhibitor, Gaul told C&EN. They teamed up with researchers at Aurigene, in Bangalore, India, and came up with their clinical candidate, CFG920.
Unlike abiraterone, CFG920 isn’t a steroid, and it inhibits CYP17 reversibly. It also reversibly inhibits another cytochrome P450 enzyme, CYP11B2, which is involved in the synthesis of the mineralocorticoids, hormones that regulate cardiac function.
Treating prostate cancer patients by lowering their androgen levels turns out to have negative cardiac side effects: Patients’ lipid metabolism is thrown off and their mineralocorticoid levels jump, leading to increases in blood pressure. Those changes can be stressful for the heart. “If prostate cancer patients don’t die because of the cancer, a lot of times they die because of cardiac disease,” Gaul said.
Because CFG920 also keeps mineralocorticoid levels in check, Novartis is hoping the drug candidate will ameliorate some of the cardiac side effects of inhibiting CYP17. The compound is currently in Phase I clinical trials.
PATENT
WO 2010149755
Example 58
Prύpιn”ation ofI'(2’ChIoroψ}ri(ibi-^’\l)’3’f4’metMψ}τUin’3’yl)-imiJazoliJin’2’θne (5HA)-

Using the same reaction conditions as in Example 14. 1-(4-methyl-pyridin-3-yl)- itnida/olidin-2-onc ().-.!.4b: 600 mg. 3.3898 mmol) uas reacted with 2-chloro-4-iodo- py.idine (974 mg.4.067 mmol). 1 , 4-dioxane (60 mL). copper iodide (65 mg, 0.3398 mmol), /r<w.v-1.2-diamino cycK)hexane (0.12 ml,, 1.0169 mmol) and potassium phosphate (2.15 g, 10.1694 mmol) to afford 810 mg of the product (83% yield).
1H NMR (C1DCI3. 300 Mi l/): 6 8.5-8.4 (m. 211). 8.3 (d. IH), 7.6-7.5 (m, 2H). 7.2 (S. 111). 4.1-3.9 (ni. 4H), 2.35 <s. 3H)
LCVIS puιϊt>: 90.8%. nι-7 – 289.1 (M M)
HPl C: 97.14%
REFERENCES
1: Gomez L, Kovac JR, Lamb DJ. CYP17A1 inhibitors in castration-resistant prostate cancer. Steroids. 2015 Mar;95:80-7. doi: 10.1016/j.steroids.2014.12.021. Epub 2015 Jan 3. Review. PubMed PMID: 25560485; PubMed Central PMCID: PMC4323677.
2: Yin L, Hu Q, Hartmann RW. Recent progress in pharmaceutical therapies for castration-resistant prostate cancer. Int J Mol Sci. 2013 Jul 4;14(7):13958-78. doi: 10.3390/ijms140713958. Review. PubMed PMID: 23880851; PubMed Central PMCID: PMC3742227.
///////CFG-920, CYP17 inhibitor (prostate cancer), Novartis, CFG 920, Novartis scientists, team up , researchers , Aurigene, Bangalore, India,
Novartis Molecule for functionally liver selective glucokinase activators for the treatment of type 2 diabetes


(R)-3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide
3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide
(3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide)
cas 866772-52-3
NVP-LBX192
LBX-192

54 Discovery and Evaluation of NVP-LBX192, a Liver Targeted Glucokinase Activator
Thursday, October 8, 2009: 10:30 AM
Nathan Hale North (Hilton Third Floor)
Glucokinase (GK) activators are currently under investigation by a number of pharmaceutical companies with only a few reaching clinical evaluation. A GK activator has the promise of potentially affecting both the beta-cell of the pancreas, by improving glucose sensitive insulin secretion, as well as the liver, by reducing uncontrolled glucose output and restoring post prandial glucose uptake and storage as glycogen. We will describe our efforts to generate liver selective GK activators which culminated in the discovery of NVP-LBX192 (3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide). This compound activated the GK enzyme in vitro at low nM concentrations and significantly reduced glucose levels during an oral glucose tolerance test in normal as well as diabetic mice.
https://acs.confex.com/acs/nerm09/webprogram/Paper75087.html
| Molecular Formula: | C26H33N5O4S2 |
|---|---|
| Molecular Weight: | 543.70132 g/mol |
Sulfonamide-Thiazolpyridine Derivatives, Glucokinase Activators, Treatment Of Type 2 Diabetes
2009 52 (19) 6142 – 6152
Investigation of functionally liver selective glucokinase activators for the treatment of type 2 diabetes
Journal of Medicinal Chemistry
Bebernitz GR, Beaulieu V, Dale BA, Deacon R, Duttaroy A, Gao JP, Grondine MS, Gupta RC, Kakmak M, Kavana M, Kirman LC, Liang JS, Maniara WM, Munshi S, Nadkarni SS, Schuster HF, Stams T, Denny IS, Taslimi PM, Vash B, Caplan SL
2010 240th (August 22) Medi-198
Glucokinase activators with improved physicochemicalproperties and off target effects
American Chemical Society National Meeting and Exposition
Kirman LC, Schuster HF, Grondine MS et al
2010 240th (August 22) Medi-197
Investigation of functionally liver selective glucokinase activators
American Chemical Society National Meeting and Exposition
Schuster HF, Kirman LC, Bebernitz GC et al
PATENT
http://www.google.com/patents/US7750020
EXAMPLE 1 3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide

A. Phenylacetic Acid Ethyl Ester
A solution of phenylacetic acid (50 g, 0.36 mol) in ethanol (150 mL) is treated with catalytic amount of sulfuric acid (4 mL). The reaction mixture is refluxed for 4 h. The reaction is then concentrated in vacuo. The residue is dissolved in diethyl ether (300 mL) and washed with saturated aqueous sodium bicarbonate solution (2×50 mL) and water (1×100 mL). The organic layer dried over sodium sulfate filtered and concentrated in vacuo to give phenylacetic acid ethyl ester as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 1.2 (t, J=7.2, 3H), 3.6 (s, 2H), 4.1 (q, J=7.2, 2H), 7.3 (m, 5H); MS 165 [M+1]+.
B. (4-Chlorosulfonyl-phenyl)-acetic acid ethyl ester
To a cooled chlorosulfonic acid (83.83 g, 48 mL, 0.71 mol) under nitrogen is added the title A compound, phenylacetic acid ethyl ester (59 g, 0.35 mol) over a period of 1 h. Reaction temperature is brought to RT (28° C.), then heated to 70° C., maintaining it at this temperature for 1 h while stirring. Reaction is cooled to RT and poured over saturated aqueous sodium chloride solution (200 mL) followed by extraction with DCM (2×200 mL). The organic layer is washed with water (5×100 mL), followed by saturated aqueous sodium chloride solution (1×150 mL). The organic layer dried over sodium sulfate, filtered and concentrated in vacuo to give crude (4-chlorosulfonyl-phenyl)acetic acid ethyl ester. Further column chromatography over silica gel (60-120 mesh), using 100% hexane afforded pure (4-chlorosulfonyl-phenyl)-acetic acid ethyl ester as a colorless oil.
C. [4-(4-Methyl-piperazine-1-sulfonyl)-phenyl]-acetic acid ethyl ester
A solution of N-methylpiperazine (9.23 g, 10.21 ml, 0.092 mol), DIEA (13 g, 17.4 mL, 0.10 mol) and DCM 80 mL is cooled to 0° C., and to this is added a solution of the title B compound, (4-chlorosulfonyl-phenyl)-acetic acid ethyl ester (22 g, 0.083 mol) in 50 mL of DCM within 30 min. Reaction mixture stirred at 0° C. for 2 h, and the reaction mixture is washed with water (100 mL), followed by 0.1 N aqueous hydrochloric acid solution (1×200 mL). The organic layer dried over sodium sulfate, filtered and concentrated under vacuo to give crude [4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-acetic acid ethyl ester. Column chromatography over silicagel (60-120 mesh), using ethyl acetate afforded pure [4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-acetic acid ethyl ester as white crystalline solid: 1H NMR (400 MHz, CDCl3) δ 1.3 (t, J=7.4, 3H), 2.3 (s, 3H), 2.5 (m, 4H), 3.0 (br s, 4H), 3.7 (s, 2H), 4.2 (q, J=7.4, 2H), 7.4 (d, J=8.3, 2H), 7.7 (d, J=7.3, 2H); MS 327 [M+1]+.
D. 3-Cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid ethyl ester
A solution of the title C compound, [4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-acetic acid ethyl ester (15 g, 0.046 mol) in a mixture of THF (60 mL) and DMTP (10 mL) is cooled to −78° C. under nitrogen. The resulting solution is stirred at −78° C. for 45 min and to this is added LDA (25.6 mL, 6.40 g, 0.059 mol, 25% solution in THF/Hexane). A solution of iodomethylcyclopentane (11.60 g, 0.055 mol) in a mixture of DMTP (12 mL) and THF (20 mL) is added over a period of 15 min at −78° C. and reaction mixture stirred at −78° C. for 3 h further, followed by stirring at 25° C. for 12 h. The reaction mixture is then quenched by the dropwise addition of saturated aqueous ammonium chloride solution (50 mL) and is concentrated in vacuo. The residue is diluted with water (50 mL) and extracted with ethyl acetate (3×100 mL). The organic solution is washed with a saturated aqueous sodium chloride (2×150 mL), dried over sodium sulfate, filtered and concentrated in vacuo. Column chromatography over silica gel (60-120 mesh), using 50% ethyl acetate in hexane as an eluent to afford 3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid ethyl ester as a white solid: 1H NMR (400 MHz, CDCl3) δ 0.9-2.1 (m, 11H), 1.2 (t, J=7.1, 3H), 2.3 (s, 3H), 2.5 (br s, 4H), 3.0 (br s, 4H), 3.6 (m, 1H), 4.1 (q, J=7.1, 2H), 7.5 (d, J=8.3, 2H), 7.7 (d, J=8.3, 2H); MS 409 [M+1]+.
E. 3-Cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid
A solution of the title D compound, 3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid ethyl ester (14 g, 0.034 mol) in methanol:water (30 mL:10 mL) and sodium hydroxide (4.11 g, 0.10 mol) is stirred at 60° C. for 8 h in an oil bath. The methanol is then removed in vacuo at 45-50° C. The residue is diluted with water (25 mL) and extracted with ether (1×40 mL). The aqueous layer is acidified to pH 5 with 3 N aqueous hydrochloric acid solution. The precipitated solid is collected by vacuum filtration, washed with water (20 mL), followed by isopropyl alcohol (20 mL). Finally, solid cake is washed with 100 mL of hexane and dried under vacuum at 40° C. for 6 h to give 3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid as a white solid: 1H NMR (400 MHz, CDCl3) δ 1.1-2.0 (m, 11H), 2.4 (s, 3H), 2.7 (br s, 4H), 3.1 (br s, 4H), 3.6 (m, 1H), 7.5 (d, J=8.3, 2H), 7.6 (d, J=8.3, 2H); MS 381 [M+l]+.
F. 5-Methoxy-thiazolo[5,4-b]pyridin-2-ylamine
A solution of 6-methoxy-pyridin-3-ylamine (5.0 g, 0.0403 mol) in 10 mL of acetic acid is added slowly to a solution of potassium thiocyanate (20 g, 0.205 mol) in 100 mL of acetic acid at 0° C. followed by a solution of bromine (2.5 mL, 0.0488 mol) in 5 mL of acetic acid. The reaction is stirred for 2 h at 0° C. and then allowed to warm to RT. The resulting solid is collected by filtration and washed with acetic acid, then partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The insoluble material is removed by filtration and the organic layer is evaporated and dried to afford 5-methoxy-thiazolo[5,4-b]pyridin-2-ylamine as a tan solid.
G. 3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide
A solution of the title E compound, 3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid (5 g, 0.013 mol) in DCM (250 mL) is cooled to 0° C. and then charged HOBt hydrate (2.66 g, 0.019 mol), followed by EDCI hydrochloride (6 g, 0.031 mol). The reaction mixture is stirred at 0° C. for 5 h. After that the solution of the title F compound, 5-methoxy-thiazolo[5,4-b]pyridin-2-ylamine (2.36 g, 0.013 mol) and D1EA (8 mL, 0.046 mol) in a mixture of DCM (60 mL) and DMF (20 mL) is added dropwise over 30 min. Reaction temperature is maintained at 0° C. for 3 h, then at RT (28° C.) for 3 days. Reaction is diluted with (60 mL) of water and the organic layer is separated and washed with saturated sodium bicarbonate solution (2×50 mL) followed by water washing (2×50 mL) and saturated sodium chloride aqueous solution (1×150 mL). Finally the organic layer is dried over sodium sulfate, filtered, and evaporated under vacuo. The crude product is purified using column chromatography over silica gel (60-120 mesh), using 40% ethyl acetate in hexane as an eluent to afford 3-cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide as a white solid: 1H NMR (400 MHz, CDCl3) δ 0.9-2.1 (m, 11H), 2.2 (s, 3H), 2.5 (br s, 4H), 3.1 (br s, 4H), 3.7 (m, 1H), 4.0 (s, 3H), 6.8 (d, J=8.8, 1H), 7.5 (d, J=8.3, 2H), 7.7 (d, J=8.3, 2H), 7.8 (d, J=8.8, 1H), 8.6 (s, 1H); MS 617 [M+1]+.
H. 3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide dihydrochloride
The title G compound, 3-cyclopentyl-2-(4-methyl piperazinyl sulfonyl)phenyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)propionamide (2.8 g, 0.0051 mol) is added to a cooled solution of 10% hydrochloric acid in isopropanol (3.75 mL). The reaction mixture is stirred at 0° C. for 1 h and then at RT for 2 h. The solid is separated, triturated with 10 mL of isopropanol and collected by vacuum filtration and washed with 50 mL of hexane. The solid is dried at 70° C. for 48 h to afford 3-cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide dihydrochloride as an off white solid.
EXAMPLE 2 (R)-3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide

The title compound is obtained analogously to Example 1 by employing the following additional resolution step:
The racemic title E compound of Example 1,3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid (10 g, 0.026 mol) in 1,4-dioxane (500 mL) is treated in a three necked 1 liter flask, equipped with heating mantle, water condenser, calcium chloride guard tube and mechanical stirrer with 3.18 g (0.026 mol) of (R)-(+)-1-phenylethylamine. This reaction mixture is then refluxed at 100° C. for 1 h. The clear reaction solution is cooled to RT (27° C.) and stirred for 10 h. The crystallized salt is collected by filtration under vacuum, washed with 5 mL of hexane and dried under vacuum to afford salt A.
The salt A is dissolved in 1,4-dioxane (500 mL) and heated at 100° C. for 1 h. The clear reaction solution is cooled to RT (27° C.) and stirred for 10 h. The crystallized product is collected by filtration under vacuum, washed with 50 mL of hexane, and dried under vacuum to afford salt B.
The salt B is dissolved in 1,4-dioxane (290 mL) and heated at 100° C. for 1 h. The clear reaction solution is cooled to RT (27° C.) and stirred for 10 h. The crystallized product is collected by filtration under vacuum, washed with 30 mL of hexane, and dried under vacuum to afford salt C.
The salt C is dissolved in 1,4-dioxane (100 mL) and heated at 100° C. for 1 h. The clear reaction solution is cooled to RT (27° C.) and stirred for 10 h. The crystallized product is collected by filtration under vacuum, washed with 30 ml of hexane, and dried under vacuum to afford salt D.
The salt D is treated with aqueous hydrochloric acid solution (20 mL, 1 mL of concentrated hydrochloric acid diluted with 100 mL of water) and stirred for 5 min. The white solid precipitates out and is collected by vacuum filtration, washed with 10 mL of cold water, 5 mL of isopropanol and 20 mL of hexane, and dried under vacuum to yield the hydrochloride salt of (R)-(−)-3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid, salt E.
The salt E is neutralized by stirring with aqueous sodium bicarbonate solution (10 mL, 1 g of sodium bicarbonate dissolved in 120 mL of water) for 5 min. The precipitated solid is collected by filtration, washed with 10 mL of cold water, 100 mL of hexane, and dried to afford (R)-(−)-3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid: m.p. 202.2-203.4° C.
Alternatively, the title compound may be obtained by the resolution of the racemic title compound of Example 1 using the following preparative chiral HPLC method:
- Column: Chiralcel OD-R (250×20 mm) Diacel make, Japan;
- Solvent A: water:methanol:acetonitrile (10:80:10 v/v/v);
- Solvent B: water:methanol:acetonitrile (05:90:05 v/v/v);
- Using gradient elution: gradient program (time, min/% B): 0/0, 20/0, 50/100, 55/0, 70/0;
- Flow rate: 6.0 mL/min; and
- Detection: by UV at 305 nm.
EXAMPLE 3 (S)-3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide

The title compound is prepared analogously to Example 2.
J MED CHEM 2009, 52, 6142-52
Investigation of Functionally Liver Selective Glucokinase Activators for the Treatment of Type 2 Diabetes
† Novartis Institutes for BioMedical Research, Inc., 100 Technology Square, Cambridge, Massachusetts 02139
‡ Torrent Research Centre, Village Bhat, Gujarat, India
J. Med. Chem., 2009, 52 (19), pp 6142–6152
DOI: 10.1021/jm900839k
http://pubs.acs.org/doi/abs/10.1021/jm900839k

Type 2 diabetes is a polygenic disease which afflicts nearly 200 million people worldwide and is expected to increase to near epidemic levels over the next 10−15 years. Glucokinase (GK) activators are currently under investigation by a number of pharmaceutical companies with only a few reaching early clinical evaluation. A GK activator has the promise of potentially affecting both the β-cells of the pancreas, by improving glucose sensitive insulin secretion, as well as the liver, by reducing uncontrolled glucose output and restoring post-prandial glucose uptake and storage as glycogen. Herein, we report our efforts on a sulfonamide chemotype with the aim to generate liver selective GK activators which culminated in the discovery of 3-cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide (17c). This compound activated the GK enzyme (αKa = 39 nM) in vitro at low nanomolar concentrations and significantly reduced glucose levels during an oral glucose tolerance test in normal mice.


PATENT
EP-1735322-B1
Example 2(R)-3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide
The title compound is obtained analogously to Example 1 by employing the following additional resolution step:
The racemic title E compound of Example 1, 3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid (10 g, 0.026 mol) in 1,4-dioxane (500 mL) is treated in a three necked 1 liter flask, equipped with heating mantle, water condenser, calcium chloride guard tube and mechanical stirrer with 3.18 g (0.026 mol) of (R)-(+)-1-phenylethylamine. This reaction mixture is then refluxed at 100°C for 1 h. The clear reaction solution is cooled to RT (27°C) and stirred for 10 h. The crystallized salt is collected by filtration under vacuum, washed with 5 mL of hexane and dried under vacuum to afford salt A.
The salt A is dissolved in 1,4-dioxane (500 mL) and heated at 100°C for 1 h. The clear reaction solution is cooled to RT (27°C) and stirred for 10 h. The crystallized product is collected by filtration under vacuum, washed with 50 mL of hexane, and dried under vacuum to afford salt B.
The salt B is dissolved in 1,4-dioxane (290 mL) and heated at 100°C for 1 h. The clear reaction solution is cooled to RT (27°C) and stirred for 10 h. The crystallized product is collected by filtration under vacuum, washed with 30 mL of hexane, and dried under vacuum to afford salt C.
The salt C is dissolved in 1,4-dioxane (100 mL) and heated at 100°C for 1 h. The clear reaction solution is cooled to RT (27°C) and stirred for 10 h. The crystallized product is collected by filtration under vacuum, washed with 30ml of hexane, and dried under vacuum to afford salt D.
The salt D is treated with aqueous hydrochloric acid solution (20 mL, 1 mL of concentrated hydrochloric acid diluted with 100 mL of water) and stirred for 5 min. The white solid precipitates out and is collected by vacuum filtration, washed with 10 mL of cold water, 5 mL of isopropanol and 20 mL of hexane, and dried under vacuum to yield the hydrochloride salt of (R)-(-)-3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid, salt E.
The salt E is neutralized by stirring with aqueous sodium bicarbonate solution (10 mL, 1 g of sodium bicarbonate dissolved in 120 mL of water) for 5 min. The precipitated solid is collected by filtration, washed with 10 mL of cold water, 100 mL of hexane, and dried to afford (R)-(-)-3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid: m.p. 202.2-203.4°C.
Alternatively, the title compound may be obtained by the resolution of the racemic title compound of Example 1 using the following preparative chiral HPLC method:
- Column: Chiralcel OD-R (250 x 20 mm) Diacel make, Japan;
- Solvent A: water:methanol:acetonitrile (10:80:10 v/v/v);
- Solvent B: water:methanol:acetonitrile (05:90:05 v/v/v);
- Using gradient elution: gradient program (time, min / %B): 0/0, 20/0, 50/100, 55/0, 70/0;
- Flow rate: 6.0 mL/min; and
- Detection: by UV at 305 nm.
REFERENCES
US 7750020
WO-2005095418-A1
US-20080103167-A1
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015218151 | 2015-08-06 | NOVEL PHENYLACETAMIDE COMPOUND AND PHARMACEUTICAL CONTAINING SAME |
| US7750020 | 2010-07-06 | Sulfonamide-Thiazolpyridine Derivatives As Glucokinase Activators Useful The Treatment Of Type 2 Diabetes |
///NOVARTIS, DIABETES, Sulfonamide-Thiazolpyridine Derivatives, Glucokinase Activators, Treatment Of Type 2 Diabetes, 866772-52-3, Novartis Molecule, functionally liver selective glucokinase activators, treatment of type 2 diabetes , NVP-LBX192, LBX-192
c1(sc2nc(ccc2n1)OC)NC(C(c3ccc(cc3)S(=O)(=O)N4CCN(CC4)C)CC5CCCC5)=O
RP 6530, Tenalisib


(S)-2-(l-(9H-purin-6-ylamino)propyl)-3-(3-fluorophenyl)-4H-chromen-4-one (Compound A1 is RP 6530).

RP 6530
RP 6530, RP6530, RP-6530
Tenalisib
RP6530-1401, NCI-2015-01804, 124584, NCT02567656
(S)-2-(l-(9H-purin-6-ylamino)propyl)-3-(3-fluorophenyl)-4H-chromen-4-one
3-(3-fluorophenyl)-2-[(1S)-1-(7H-purin-6-ylamino)propyl]chromen-4-one
MW415.4, C23H18FN5O2
CAS 1639417-53-0, 1693773-94-2
A PI3K inhibitor potentially for the treatment of hematologic malignancies.
An inhibitor of phosphoinositide-3 kinase (PI3K) δ/γ isoforms and anti-cellular proliferation agent for treatment of hematol. malignancies
Rhizen Pharmaceuticals is developing RP-6530, a PI3K delta and gamma dual inhibitor, for the potential oral treatment of cancer and inflammation In November 2013, a phase I trial in patients with hematologic malignancies was initiated in Italy ]. In September 2015, a phase I/Ib study was initiated in the US, in patients with relapsed and refractory T-cell lymphoma. At that time, the study was expected to complete in December 2016
PATENTS……..WO 11/055215 , WO 12/151525.
Inventors
| Inventors | Meyyappan Muthuppalaniappan, Srikant Viswanadha, Govindarajulu Babu, Swaroop Kumar V.S. Vakkalanka, |
| Incozen Therapeutics Pvt. Ltd., Rhizen… |
View original post 3,692 more words
RP 6503, Novartis to develop and commercialize Rhizen’s inhaled dual PI3K-delta gamma inhibitor

RP 6503
phase 1

RP 6503
| Molecular Formula: | C30H24F2N6O5S |
|---|---|
| Molecular Weight: | 618.610566 g/mol |
Mass: 619.1 (M++l). MP: 175-178° C Specific optical rotation (C=l in chloroform, at 25°C) : [a]D = + 147.16.

A1
RP 6503
(S)-N-(5-(4-amino-l-(l-(5-fluoro-3-(3-fluorophenyl)-4-oxo-4H-chromen-2-yl) ethyl)-lH-pyrazolo[3,4-d]pyrimidin-3-yl)-2-methoxyphenyl)methanesulfonamide
(S)-N-[5-[4-amino-1-[1-[5-fluoro-3-(3-fluorophenyl)-4-oxochromen-2-yl]ethyl]pyrazolo[3,4-d]pyrimidin-3-yl]-2-methoxyphenyl]methanesulfonamide

![]()
Novartis to develop and commercialize Rhizen’s inhaled dual PI3K-delta gamma inhibitor and related compounds worldwide
The immune pipeline includes ‘dual PI3K inhibitors for various indications’ licensed to Novartis
‘inhaled dual inhibitor’,
Phosphoinositide-3 kinase delta inhibitor; Phosphoinositide-3 kinase gamma inhibitor
WO2011055215A2 and WO2012151525A1 and U.S. Publication Nos. US20110118257 and US20120289496
Rhizen Pharmaceuticals Sa INNOVATOR
| Incozen Therapeutics Pvt. Ltd., Rhizen Pharmaceuticals Sa |
PATENT
http://www.google.com/patents/WO2011055215A2?cl=en
PATENT
http://www.google.com/patents/WO2012151525A1?cl=en
scheme 1A:
Ste -1

Step-2

Scheme 2

SCHEME 3

SCHEME4

List of Intermediates





Intermediate 27: 2-( l -(4-amino-3-iodo-lH-pyrazolo[3,4-d]pyrimidin- l – yl)ethyl)-5-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one: To a solution of 3-iodo- l H- pyrazolo[3,4-d]pyrimidin-4-amine (0.800 g, 2.88 mmol) in DMF (5 ml), potassium carbonate (0.398 g, 2.88 mmol) was added and stirred at RT for 30 min. To this mixture intermediate 22 (0.500 g, 1.44 mmol) was added and stirred for 12h. The reaction mixture was diluted with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with methanol: dichloromethane to afford the title compound as a off-white solid (0.300 g, 38%). Ή-NMR (5 ppm, DMSO-d63, 400 MHz): 8.02 (s, 1 H), 7.94 (s, 1 H), 7.84 (dt, J = 8.4,5.7 Hz, 1H), 7.47 (d, 7 = 8.6 Hz, 1H), 7.29 (m, 3H), 7.09 (dt, 7 = 8.8,2.3 Hz, 1 H), 6.87 (s, 2H), 5.88 (q, 7 = 7.0 Hz, 1H), 1.82 (d, 7 = 7.0 Hz, 3H).
SYNTHESIS
MAIN PART
PATENT
http://www.google.com/patents/WO2015198289A1?cl=en
Prashant Kashinath Bhavar, Swaroop Kumar Venkata Satya VAKKALANKA
The present invention relates to a selective dual delta (δ) and gamma (γ) PI3K protein kinase modulator (S)-N-(5-(4-amino-1-(1-(5-fluoro-3-(3-fluorophenyl)-4-oxo-4H- chromen-2-yl)ethyl)-1H-pyrazolo[3,4-d]pyrimidin-3-yl)-2-methoxyphenyl) methane sulfonamide, methods of preparing them, pharmaceutical compositions containing them and methods of treatment, prevention and/or amelioration of PI3K kinase mediated diseases or disorders with them.
compound of formula (Al):
(Al).
The process comprises the steps of:
(a) subjecting (R)-5-fluoro-3-(3-fluorophenyl)-2-(l-hydroxyethyl)-4H-chromen-4-one:

to a Mitsunobu reaction with 3-(4-methoxy-3-nitrophenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-amine:

(for example, in the presence of triphenylphosphine and diisopropylazodicarboxylate) to give (S)-2-(l-(4-amino-3-(4-methoxy-3-nitrophenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-5-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (Intermediate 3):

Intermediate 3;
(b) reducing Intermediate 3, for example with a reducing agent such as Raney Ni, to give (S)-2-(l-(4-amino-3-(3-amino-4-methoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin- l-yl)ethyl)-5-fluoro-3-( -fluorophenyl)-4H-chromen-4-one (Intermediate 4):

Intermediate 4;
The intermediates described herein may be prepared by the methods described in International Publication Nos. WO 11/055215 and WO 12/151525, both of which are hereby incorporated by reference.
Intermediate 1: N-(5-bromo-2-methoxyphenyl)methanesulfonamide:
To a solution of 5-bromo-2-methoxyaniline(1.00 g, 4.94 mmol) in dichloromethane (10 ml), pyridine (0.800 ml, 9.89 mmol) was added and cooled to 0°C. Methane sulphonyl chloride (0.40 ml, 5.19 mmol) was added and stirred for 30 min. The reaction mixture was quenched with water, extracted with ethyl acetate, dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude product was chromatographed with ethyl acetate : petroleum ether to afford the title compound as a reddish solid (1.20 g, 87%).
Intermediate 2: N-(2-methoxy-5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)methanesulfonamide: Potassium acetate (0.841 g, 8.57 mmol) and bis(pinacolato)diboron (1.190 g, 4.71 mmol) were added to a solution of intermediate 1 (1.20 g, 4.28 mmol) in dioxane (17.5 ml) and the solution was degassed for 30 min.[l, -Bis(diphenylphosphino)ferrocene]dichloro palladium(II).CH2Ci2 (0.104 g, 0.128 mmol) was added under nitrogen atmosphere and heated to 80°C. After 2h the
reaction mixture was filtered through celite and concentrated. The crude product was purified by column chromatography with ethyl acetate : petroleum ether to afford the title compound as a yellow solid (1.00 g, 71%).JH-NMR (δ ppm, CDCb, 400 MHz): 7. 91 (d, / = 1.2Hz, 1H), 7. 62 (dd, / = 8.1, 1.2Hz, 1H), 6. 92 (d, / = 8.1Hz, 1H), 6.73 (s, 1H), 3.91 (s, 3H), 2.98 (s, 3H), 1.32 (s, 12H).
Intermediate 3: (S)-2-(l-(4-amino-3-(4-methoxy-3-nitrophenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-5-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one: (S)-2-(l-(4-amino-3-(4-methoxy-3-nitrophenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-5-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one: To a solution of (R)-5-fluoro-3-(3-fluorophenyl)-2-(l-hydroxyethyl)-4H-chromen-4-one (0.500 g, 1.64 mmol) in THF (5 ml), 3-(4-methoxy-3-nitrophenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-amine (0.564 g, 1.97 mmol) and triphenylphosphine (0.649 g, 2.47 mmol) were added followed by the addition of diisopropylazodicarboxylate (0.50 ml, 2.47 mmol). ((R)-5-fluoro-3-(3-fluorophenyl)-2-(l-hydroxyethyl)-4H-chromen-4-one can be prepared as described for Intermediates 23, 25, and 26 in International Publication No. WO 2012/0151525.). After 4h at room temperature, the mixture was concentrated and the residue was purified by column chromatography with ethyl acetate : petroleum ether to afford the title compound as a brown solid (0.270 g, 29%). JH-NMR (δ ppm, DMSO-d6, 400 MHz): 8.04 (s, 1H), 7.83 (m, 1H), 7.63-7.50 (m, 3H), 7.29 (m, 2H), 7.06 (dt, J = 8.7,2.2Hz, 1H), 6.94 (m, 2H), 6.75 (dd, J = 8.1,2.1Hz, 1H), 5.95 (q, J = 7.0Hz, 1H), 4.98 (s, 2H), 3.81 (s, 3H), 1.86 (d, J = 7.0 Hz, 3H).
[109] Intermediate 4: (S)-2-(l-(4-amino-3-(3-amino-4-methoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-5-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one:
(S)-2-(l-(4-amino-3-(3-amino-4-methoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-5-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one : To a solution of Intermediate 3 (0.260 g, 0.455 mmol) in ethanol (5 ml), Raney Ni (0.130 g) was added and hydrogeneated at 20psi at 50°C for 24h. The reaction mixture was passed through celitepad and concentrated to afford the title compound as a brown solid (0.150 g, 60%). Mass : 540.8 (M+).
Example A
N-(5-(4-amino-l-(l-(5-fluoro-3-(3-fluorophenyl)-4-oxo-4H-chromen-2-yl)ethyl)-lH- pyrazolo[3,4-d]pyrimidin-3-yl)-2-methoxyphenyl)methanesulfonamide
To a solution of 2-(l-(4-amino-3-iodo-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-5-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (0.200 g, 0.366 mmol) in DME (2.1 ml) and water (0.67 ml), intermediate 2 (0.179 g, 0.550 mmol) and sodium carbonate (0.116 g, 1.10 mmol) were added and the system was degassed for 30 min. (2-(l-(4-amino-3-iodo-lH^yrazolo[3,4-d]pyrimidin-l-yl)ethyl)-5-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one can be prepared as described for Intermediates 23, 25, and 26 in International Publication No. WO 2012/0151525). Bis(diphenylphosphino) ferrocene]dichloropalladium(II) (0.059 g, 0.075 mmol) was added and kept under microwave irradiation (microwave power = 100W, temperature = 100 °C) for 45 min. The reaction mixture was Celite filtered, concentrated and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with methanol: dichloromethane to afford the title compound as a brown solid (0.080 g, 35%). MP: 216-218 °C. ¾-NMR (δ ppm, CDCb, 400 MHz): 8.20 (s, 1H), 7.73 (s, 1H), 7.53 (m, 2H), 7.31 (m, 2H), 7.07-6.73 (m, 6H), 6.07 (q, / = 6.2 Hz, 1H), 3.98 (s, 3H), 3.14 (s, 3H), 2.01 (d, / = 6.0Hz, 3H).
Example Al and A2
Method A
(S)-N-(5-(4-amino-l-(l-(5-fluoro-3-(3-fluorophenyl)-4-oxo-4H-chromen-2-yl)ethyl)- lH-pyrazolo[3,4-d]pyrimidin-3-yl)-2-methoxyphenyl)methanesulfonamide
and (R)-N-(5-(4-amino-l-(l-(5-fluoro-3-(3-fluorophenyl)-4-oxo-4H-chromen-2- yl)ethyl)-lH-p anesulfonamide

The two enantiomerically pure isomers were separated by preparative SFC (supercritical fluid) conditions from N-(5-(4-amino-l-(l-(5-fluoro-3-(3-fluorophenyl)-4-oxo-4H-chromen-2-yl)ethyl)-lH-pyrazolo[3,4-d]pyrimidin-3-yl)-2-methoxyphenyl)methanesulfonamide (0.500 g) on a CHIRALPAK AS-H column (250 x 30 mm; 5μπι) using methanol : CO2 (55:45) as the mobile phase at a flow rate of 80g / min.
Example Al (S-isomer): Brown solid (0.247 g). Enantiomeric excess: 97.4%. Retention time: 2.14 min. Mass: 619.1 (M++l). MP: 156-158° C.
Example A2 (R-isomer): Brown solid (0.182 g). Enantiomeric excess: 99.3%. Retention t: 3.43 min. Mass: 619.1 (M++l). MP: 168-171° C.
Method Al
(S)-N-(5-(4-amino-l-(l-(5-fluoro-3-(3-fluorophenyl)-4-oxo-4H-chromen-2-yl)ethyl)- lH-pyrazolo[3,4-d]pyrimidin-3-yl)-2-methoxyphenyl)methanesulfonamide
and (R)-N-(5-(4-amino-l-(l-(5-fluoro-3-(3-fluorophenyl)-4-oxo-4H-chromen-2- yl)ethyl)-lH-p anesulfonamide

The two enantiomerically pure isomers were separated by preparative SFC (supercritical fluid) conditions from N-(5-(4-amino-l-(l-(5-fluoro-3-(3-fluorophenyl)-4-oxo-4H-chromen-2-yl)ethyl)-lH-pyrazolo[3,4-d]pyrimidin-3-yl)-2-methoxyphenyl) methanesulfonamide (15.0 g) on a CHIRALPAK AS-H column (250 x 20 mm; 5μπι) using methanol : CO2 (45:55) as the mobile phase at a flow rate of 120g / min.
Example Al (S-isomer): Enantiomeric excess: 100 %. Retention time: 2.21 min. Mass: 619.1 (M++l). MP: 175-178° C Specific optical rotation (C=l in chloroform, at 25°C) : [a]D = + 147.16.
Example A2 (R-isomer): Enantiomeric excess: 99.3%. Retention t: 3.72 min. Mass: 619.1 (M++l). MP: 154-157° C. Specific optical rotation (C=l in chloroform, at 25°C) : [a]D = – 159.54.
Method B
Example Al
(S)-N-(5-(4-amino-l-(l-(5-fluoro-3-(3-fluorophenyl)-4-oxo-4H-chromen-2-yl)ethyl)- lH-pyrazolo[3,4-d]pyrimidin-3-yl)-2-methoxyphenyl)methanesulfonamide
To a solution of Intermediate 4 (0.500 g, 0.923 mmol) in dichloromethane (5 ml) cooled to 0°C, pyridine (0.200 ml, 1.84 mmol) was added and stirred for 10 min. Methanesulphonyl chloride (0.100 ml, 0.923 mmol) was added stirred for 30 min. The reaction mixture was quenched with water, extracted with dichloromethane and dried over sodium sulphate. The crude product was column chromatographed with methanol : dichloromethane to afford the title compound as an off-white solid (0.240 g, 42%). MP: 211-213°C. ¾-NMR (δ ppm, DMSO-d6, 400 MHz): 9.15 (s, 1H), 8.06 (s, 1H), 7.83 (m, 1H), 7.49 (m, 4H), 7.28 (m, 4H), 7.08 (dt, / = 8.6, 1.7 Hz, 1H), 6.92 (s, 2H), 5.98 (q, / = 6.9 Hz, 1H), 3.88 (s, 3H), 2.99 (s, 3H), 1.88 (d, / = 7.0 Hz, 3H). Enantiomeric excess: 85.4% as determined by HPLC on a chiralpak AS-3R column, enriched in the fast eluting isomer (retention time = 7.46 min.).

CLIPS
La Chaux-de-Fonds, Switzerland, Sept. 6, 2013 — La Chaux-de-Fonds, Switzerland (6 September 2013): Rhizen Pharmaceuticals S.A. announces a scientific poster presentation on the pre-clinical characterization of its lead calcium release activated channel (CRAC) inhibitor, RP3128, for the treatment of respiratory disorders and an oral presentation on the pharmacological profile of its novel, dual Phosphoinositide-3 kinase (PI3K) delta/gamma inhibitor, RP6503, in the pulmonary disease systems, at the European Respiratory Society Annual Congress (ERS), to be held from 7-11 September 2013, at Barcelona, Spain.
RP6503 is a novel, potent and selective inhibitor of the delta and gamma isoforms of PI3K. It is to be delivered via the inhalation route and has a long duration of action along with excellent PI3K isoform selectivity, which is expected to result in better safety. RP3128 has been optimized with high potency for CRAC channel inhibition, selectivity over the other voltage gated channels and excellent oral bioavailability. Rhizen intends to move both these compounds to the clinic in 2014.
Details of the presentations:
1. Abstract of the Poster Presentation: “Pre-clinical characterization of RP3128, a novel and potent CRAC channel inhibitor for the treatment of respiratory disorders”
Time and Location- 8 September 2013 between 14.45-16.45 in Room 3.6, at Poster Discussion: New drugs in respiratory medicine, at FIRA BARCELONA, Convention Centre de Gran Via, Barcelona, Spain
2. Abstract of Oral Presentation: “In vitro and in vivo pharmacological profile of RP6503, a novel dual PI3K delta/gamma inhibitor, in pulmonary disease systems”
Time and Location- 11 September 2013 at 8.45 in Room 3.9; Session 8.30-10.30, at the Oral Presentation: Emerging new targets for the treatment of respiratory diseases, at FIRA BARCELONA, Convention Centre de Gran Via, Barcelona, Spain
CLIPS
La Chaux-de-Fonds, Switzerland , Dec. 09, 2015 — Rhizen Pharmaceuticals S.A. announced today that they have entered into an exclusive, worldwide license agreement with Novartis for the development and commercialization of Rhizen’s, inhaled dual PI3K-delta gamma inhibitor and its closely related compounds for various indications.
Under the terms of the agreement, Rhizen will receive an upfront payment and is eligible to receive development, regulatory and sales milestones payments. In addition Rhizen is also eligible to receive tiered royalties on annual nets sales.
The lead compound is a novel, potent, and selective dual PI3K-delta gamma inhibitor with demonstrated anti-inflammatory and immuno-modulatory activity in pre-clinical systems and models representative of respiratory diseases. With a favorable ADME and PK profile and high therapeutic index in animals, the inhaled dual PI3K-delta gamma inhibitor holds promise in the treatment of human airway disorders.
About Rhizen Pharmaceuticals S.A.:
Rhizen Pharmaceuticals is an innovative, clinical-stage biopharmaceutical company focused on the discovery and development of novel therapeutics for the treatment of cancer, immune and metabolic disorders. Since its establishment in 2008, Rhizen has created a diverse pipeline of proprietary drug candidates targeting several cancers and immune associated cellular pathways. Rhizen is headquartered in La-Chaux-de-Fonds, Switzerland. For additional information, please visit Rhizen’s website, http://www.rhizen.com.
SEE
http://www.atsjournals.org/doi/abs/10.1164/ajrccm-conference.2013.187.1_MeetingAbstracts.A3880
| WO2011055215A2 | Nov 3, 2010 | May 12, 2011 | Incozen Therapeutics Pvt. Ltd. | Novel kinase modulators |
| WO2012008302A1 | Jun 28, 2011 | Jan 19, 2012 | National University Corporation Tottori University | Method for preparing novel hipsc by means of mirna introduction |
| WO2012121953A1 | Feb 29, 2012 | Sep 13, 2012 | The Trustees Of Columbia University In The City Of New York | Methods and pharmaceutical compositions for treating lymphoid malignancy |
| WO2012151525A1 | May 4, 2012 | Nov 8, 2012 | Rhizen Pharmaceuticals Sa | Novel compounds as modulators of protein kinases |
| WO2013164801A1 | May 3, 2013 | Nov 7, 2013 | Rhizen Pharmaceuticals Sa | Process for preparation of optically pure and optionally substituted 2- (1 -hydroxy- alkyl) – chromen – 4 – one derivatives and their use in preparing pharmaceuticals |
| US20110118257 | May 19, 2011 | Rhizen Pharmaceuticals Sa | Novel kinase modulators | |
| US20120289496 | May 4, 2012 | Nov 15, 2012 | Rhizen Pharmaceuticals Sa | Novel compounds as modulators of protein kinases |
///////RP 6503, Novartis, develop, commercialize, Rhizen, inhaled dual PI3K-delta gamma inhibitor, PHASE 1, RP-6503
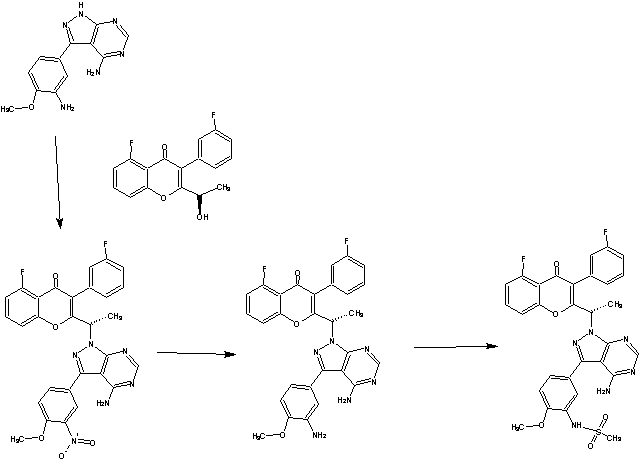
c21c(cccc1O/C(=C(\C2=O)c3cc(ccc3)F)C(C)n4c6ncnc(c6c(n4)c5cc(c(cc5)OC)NS(=O)(=O)C)N)F
CC(C1=C(C(=O)C2=C(O1)C=CC=C2F)C3=CC(=CC=C3)F)N4C5=C(C(=N4)C6=CC(=C(C=C6)OC)NS(=O)(=O)C)C(=NC=N5)N
/////
EDQM adopts revised monograph for WFI allowing non-destillation techniques
DRUG REGULATORY AFFAIRS INTERNATIONAL

In a press release the EDQM has announced that the new monograph draft on Water for Injection (169) had been adopted. Read on to learn more about the production of WFI with membrane systems.
In a press release, the European Pharmacopeia Commission has announced that the revised monograph on Water for Injection (WFI) had been adopted.
According to the revised monograph, it will be allowed in Europe in future to produce WFI with a purification method equivalent to distillation like e.g. reverse osmosis coupled with appropriate techniques. Moreover, the EDQM declares that a notice to the respective supervisory authorities will be required when a “non-distillation” technology is used for the production of WFI. Besides, the EDQM points out that it is not only a matter of equivalence of a specification but rather the robustness of the purification of WFI. Therefore, Annex 1, which is currently under revision, will also include requirements with…
View original post 90 more words
QP Education and Qualification – What is needed?
DRUG REGULATORY AFFAIRS INTERNATIONAL
![]()
We are frequently asked about the educational requirements in order to become a Qualified Person in Europe. Comprehensive educational modules are offered, especially in the UK. These training courses contain different topics like pharmaceutical law, Microbiology, Quality Management etc and require the trainee to take part in multiple courses over an extended period. But is this needed to become a QP in Europe?
Read more about QP education and qualification.
We are frequently asked about the educational requirements in order to become a Qualified Person in Europe. Comprehensive educational modules are offered, especially in the UK. These training courses contain different topics like pharmaceutical law, Microbiology, Quality Management etc and require the trainee to take part in multiple courses over an extended period. But is this needed to become a QP in Europe?
The answer comes in two parts.
First: If you are located in the UK then those…
View original post 445 more words
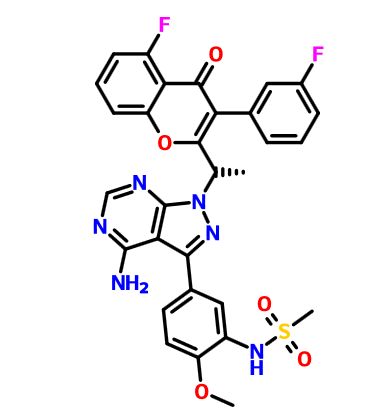
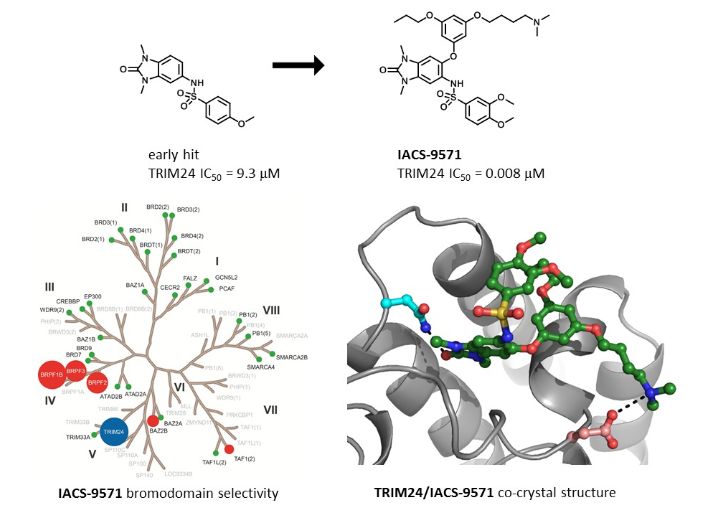
IACS -9571
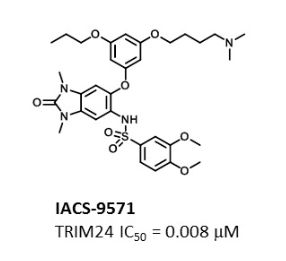

IACS-9571
TRIM24/BRPF1 bromodomain inhibitor
IACS-9571; IACS 9571; IACS9571.
| Molecular Formula: | C32H42N4O8S |
|---|---|
| Molecular Weight: | 642.76288 g/mol |
N-[6-[3-[4-(dimethylamino)butoxy]-5-propoxyphenoxy]-1,3-dimethyl-2-oxobenzimidazol-5-yl]-3,4-dimethoxybenzenesulfonamide
BOARD OF REGENTS, UNIVERSITY OF TEXAS SYSTEM
IACS-9571 is a potent and selective inhibitor TRIM24 and BRPF1. The bromodomain containing proteins TRIM24 (Tripartite motif containing protein 24) and BRPF1 (bromodomain and PHD finger containing protein 1) are involved in the epigenetic regulation of gene expression and have been implicated in human cancer. Overexpression of TRIM24 correlates with poor patient prognosis and BRPF1 is a scaffolding protein required for the assembly of histone acetyltransferase complexes, where the gene of MOZ (monocytic leukemia zinc finger protein) was first identified as a recurrent fusion partner in leukemia patients (8p11 chromosomal rearrangements). IACS-9571 has low nanomolar affinities for TRIM24 and BRPF1 (ITC Kd = 31 nM and 14 nM, respectively). With its excellent cellular potency (EC50 = 50 nM) and favorable pharmacokinetic properties (F = 29%), IACS-9571 is a high-quality chemical probe for the evaluation of TRIM24 and/or BRPF1 bromodomain function in vitro and in vivo. (J Med Chem. 2015 Jun 10. [Epub ahead of print] )

PAPER
http://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.5b00405
Structure-Guided Design of IACS-9571, a Selective High-Affinity Dual TRIM24-BRPF1 Bromodomain Inhibitor
†Institute for Applied Cancer Science, and ‡Core for Biomolecular Structure and Function, The University of Texas MD Anderson Cancer Center, 1881 East Road, Unit 1956, Houston, Texas 77054, United States
§ Department of Epigenetics and Molecular Carcinogenesis, The University of Texas MD Anderson Cancer Center,
1515 Holcombe Boulevard
, Houston, Texas 77030, United States
J. Med. Chem., 2016, 59 (4), pp 1440–1454
DOI: 10.1021/acs.jmedchem.5b00405
Publication Date (Web): June 10, 2015
Copyright © 2015 American Chemical Society

The bromodomain containing proteins TRIM24 (tripartite motif containing protein 24) and BRPF1 (bromodomain and PHD finger containing protein 1) are involved in the epigenetic regulation of gene expression and have been implicated in human cancer. Overexpression of TRIM24 correlates with poor patient prognosis, and BRPF1 is a scaffolding protein required for the assembly of histone acetyltransferase complexes, where the gene of MOZ (monocytic leukemia zinc finger protein) was first identified as a recurrent fusion partner in leukemia patients (8p11 chromosomal rearrangements). Here, we present the structure guided development of a series of N,N-dimethylbenzimidazolone bromodomain inhibitors through the iterative use of X-ray cocrystal structures. A unique binding mode enabled the design of a potent and selective inhibitor 8i (IACS-9571) with low nanomolar affinities for TRIM24 and BRPF1 (ITC Kd = 31 nM and ITC Kd = 14 nM, respectively). With its excellent cellular potency (EC50 = 50 nM) and favorable pharmacokinetic properties (F = 29%), 8i is a high-quality chemical probe for the evaluation of TRIM24 and/or BRPF1 bromodomain function in vitro and in vivo.
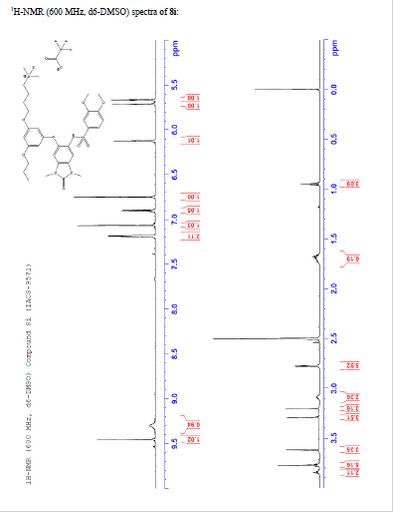
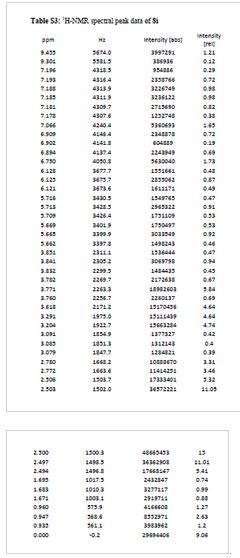
TFA salt of 8i (106 mg, 57%) as a white solid. 1H NMR (600 MHz, DMSO-d6) δ 9.46 (s, 1H), 9.30 (br-s, 1H), 7.19 (m, 2H), 7.07 (s, 1H), 6.90 (d, J = 9.0 Hz, 1H), 6.75 (s, 1H), 6.13 (t, J = 2.2 Hz, 1H), 5.71 (t, J = 2.0 Hz, 1H), 5.67 (t, J = 2.0 Hz, 1H), 3.84 (t, J = 5.9 Hz, 2H), 3.77 (m, 5H), 3.62 (s, 3H), 3.29 (s, 3H), 3.20 (s, 3H), 3.12–3.05 (m, 2H), 2.78 (d, J = 4.7 Hz, 6H), 1.77–1.63 (m, 6H), 0.95 (t, J = 7.3 Hz, 3H). 13C NMR (600 MHz, DMSO-d6) δ 160.3, 160.0, 159.3, 154.1, 152.0, 148.4, 143.9, 131.8, 128.2, 126.0, 121.9, 120.5, 110.4, 109.4, 106.4, 100.6, 95.9, 95.8, 95.2, 68.9, 66.7, 56.3, 55.6, 55.4, 42.1, 27.1, 27.0, 25.6, 21.9, 20.7, 10.4. MS (ESI) m/z 644 [M + H]+.
NMR
IACS -9571
N-(6-(3-(4-(dimethylamino)butoxy)-5- propoxyphenoxy)-l,3-dimethyl-2-oxo-2,3-dihydro-lH-benzo[d]imidazol-5-yl)-3,4- dimethoxybenzenesulfonamide 2,2,2-trifluoroacetate
 CLICK ON IMAGE
CLICK ON IMAGE
ABSTRACT
251st ACS National Meeting & Exposition
13–17 March 2016
San Diego, United States
MEDI 5
Discovery and development of a potent dual TRIM24/BRPF1 bromodomain inhibitor, IACS -9571, using structure- based drug design Wylie S. Palmer 1 , wpalmer@mdanderson.org, Guillaume Poncet -Montagne 1 , Gang Liu 1 , Alessia Petrocchi 1 , N aphtali Reyna 1 , Govindan Subramanian 1 , Jay Theroff 1 , Maria Kost -Alimova 1 , Jennifer Bardenhagen 1 , Elisabetta Leo 1 , Hannah Sheppard 1 , Trang Tieu 1 , Shi Xi 1 , Yanai Zhan 1 , Shuping Zhao 1 , Michelle Barton 2 , Giulio Draetta 1 , Carlo Toniatti 1 , Philip Jones 1 , Mary Ge ck Do 1 , Jannik Andersen 1 . (1) Institute for Applied Cancer Science, The University of Texas, MD Anderson Cancer Center, Houston, Texas, United States (2) Department of Epigenetics and Molecular Carcinogenesis, The University of Texas, MD Anderson Cancer Center, Houston, Texas, United States
Bromodomains are an important class of chromatin remodeling proteins that recognize acetylated lysine residues on histone tails. As epigenetic targets they regulate gene transcription and offer a new way to treat diseas es, particularly in inflammation and oncology. The bromodomain and extra- terminal (BET) family has emerged as an important and druggable example of this class of proteins with the successful entry of small- molecule inhibitors into the clinic. Other families of bromodomains are only starting to be explored, such as the Tripartite Motif -containing 24 protein (TRIM24) and bromodomain- PHD finger protein 1 (BRPF1). Both proteins contain a dual PHD -bromo motif which have a role in recognizing specific histone mar ks. TRIM24 recognizes the dual histone marks of unmodified H3K4 and acetylated- H3K23 within the same histone tail. TRIM24 is a potent co- activator of ER -alpha and overexpression of TRIM24 has been linked to poor survival rates in breast cancer patients.
This presentation will describe the structure guided development of a series of N,N- dimethyl -benzimidazolones through the iterative use of X -ray cocrystal structures. A unique binding mode enabled the design of a potent and selective inhibitor (IACS -9571) with low nanomolar affinities for TRIM24 and BRPF1 (ITC Kd = 31 nM and ITC Kd = 14 nM, respectively). With its excellent cellular potency (EC 50 = 50 nM) and favorable pharmacokinetic properties, IACS -9571 is a high- quality chemical probe for the evaluation of TRIM24 and/or BRPF1 bromodomain function in vitro and in vivo

PATENT
WO-2016033416-A1
Synthesis of Intermediates:
N-(6-bromo-l ,3-dimethyl-2-oxo-2,3-dihydro-lH-benzo[d]imidazol-5-yl)-2,2,2- trifluoroacetamide (Intermediate 1):
Step 1 : 5-nitro-lH-benzo[d]imidazol-2(3H)-one:
To a 0 °C solution of 4-nitrobenzene- 1 ,2-diamine (44 g, 285 mmol) in 80 mL of DMF was added l, l’-carbonyldiimidazole (70 g, 428 mmol). The reaction mixture was stirred at RT for 4 h, then water (250 mL) was added. The resulting suspension was filtered, and the collected solids were washed with water (200 mL) and dried to give 5-nitro-lH- benzo[d]imidazol-2(3H)-one as a yellow solid (45 g, 88%). MS (ES+) C7H5N3O3 requires: 179, found: 180 [M+H]+.
Step 2: l,3-dimethyl-5-nitro-lH-benzo[d]imidazol-2(3H)-one:
To a solution of 5-nitro-lH-benzo[d]imidazol-2(3H)-one (55 g, 309 mmol) in 150 mL of DMF was added K2CO3 (85 g, 618 mmol), the reaction mixture was cooled to 0 °C, then iodomethane (109 g, 772 mmol) was slowly added. The reaction mixture was stirred at RT overnight, then water was added to the reaction mixture. The resulting suspension was filtered and the collected solids were washed with water (200 mL) and dried to give 1,3- dimethyl-5-nitro-lH-benzo[d]imidazol-2(3H)-one as a yellow solid (55 g, 86%). MS (ES+) C9H9N3O3 requires: 207, found: 208 [M+H] +.
Step 3: 5-amino-l,3-dimethyl-lH-benzo[d]imidazol-2(3H)-one:
To a solution of l,3-dimethyl-5-nitro-lH-benzo[d]imidazol-2(3H)-one (50 g, 240 mmol) in 200 mL of EtOAc under an inert atmosphere was added 10% palladium on activated carbon (5 g, 24 mmol). The reaction mixture was then charged with hydrogen and stirred at RT under an ¾ atmosphere overnight. The reaction mixture was filtered through a pad of celite then concentrated to give 5-amino-l,3-dimethyl-lH-benzo[d]imidazol-2(3H)- one as a yellow solid (32 g, 68%). MS (ES+) C9H11N3O requires: 177, found: 178 [M+H]+.
Step 4: 5-amino-6-bromo-l ,3-dimethyl-lH-benzo[d]imidazol-2(3H)-one:
To a 0 °C solution of 5-amino-l ,3-dimethyl-lH-benzo[d]imidazol-2(3H)-one (4 g, 22.6 mmol) in 25 mL of CHCI3 and 25 mL of AcOH was slowly added drop wise bromine (3.5 g, 22.6mmol). The mixture was stirred at RT for 30 min, then concentrated and purified by silica gel chromatography (1 : 1 EtOAc/ hexanes) to afford 5-amino-6-bromo-l ,3-dimethyl- lH-benzo[d]imidazol-2(3H)-one as a yellow solid (3.2 g, 69%). MS (ES+) C9HioBrN30 requires: 256, found: 257 [M+H]+.
Step 5: N-(6-bromo-l ,3-dimethyl-2-oxo-2,3-dihydro-lH-benzo[d]imidazol-5-yl)-2,2,2- trifluoroacetamide:
To a 0 °C solution of 5-amino-6-bromo-l ,3-dimethyl-lH-benzo[d]imidazol- 2(3H)-one (1.50 g, 5.9 mmol) in DCM (45 ml) was added DMAP (72 mg, 0.59 mmol), triethylamine (1.63 ml, 11.7 mmol) and trifluoroacetic anhydride (0.91 ml, 6.4 mmol). The reaction mixture was stirred for 2 h and warmed to RT. The reaction mixture was then quenched with water and the organic phase was washed with brine, dried over sodium sulfate, filtered and concentrated to give N-(6-bromo-l,3-dimethyl-2-oxo-2,3-dihydro-lH- benzo[d]imidazol-5-yl)-2,2,2-trifluoroacetamide (Intermediate 1) as a yellow solid (2.20 g, 100%). MS (ES+) CiiH9BrF3N302 requires: 352, found 353 [M+H]+.
5-amino-6-(3-hydroxyphenoxy)-l,3-dimethyl-lH-benzo[d]imidazol-2(3H)-one (Intermediate 2, Route A):
To a mixture of 5-amino-6-(3-(benzyloxy)phenoxy)-l,3-dimethyl-lH- benzo[d]imidazol-2(3H)-one (400 mg, 1.07 mmol) in DCM (20 mL) at -78 °C was added tribromoborane (5.3 mL, 5.3 mmol). The mixture was warmed up to room temperature gradually, then quenched by methanol dropwise, concentrated, and purified by column chromatography (20-100% EtOAc/hexanes and then 0-40% methanol/EtOAc) to give 5- amino-6-(3-hydroxyphenoxy)-l,3-dimethyl-lH-benzo[d]imidazol-2(3H)-one as a solid (240 mg, 79%). MS (ES+) C15H15N3O3 requires: 285, found: 286 [M+H]+.
5-amino-6-(3-hydroxyphenoxy)-l,3-dimethyl-lH-benzo[d]imidazol-2(3H)-one (Intermediate 2, Route B):
Step 2
Step 1: 3-[(ieri-butyldimethylsilyl)oxy]phenol:
A mixture of lH-imidazole (2.25 g, 33.1 mmol), ieri-butylchlorodimethylsilane (3.83 g, 25.4 mmol) and resorcinol (5.6 g, 51 mmol) in THF (30 ml) was stirred at 80 °C for 5 h. The resulting suspension of the cooled reaction mixture was filtered and the collected filtrate was concentrated and purified by silica-gel chromatography (20:80 to 0:100, EtOAc/hexanes) to give 3-((ieri-butyldimethylsilyl)oxy)phenol (2.78 g, 49%). MS (ES+) C12H20O2S1 requires: 224, found 225 [M+H]+.
Step 2: 5-amino-6-(3-((ier^butyldimethylsilyl)oxy)phenoxy)-l ,3-dimethyl-lH- benzo[d]imidazol-2(3H)-one:
A mixture of 3-((ieri-butyldimethylsilyl)oxy)phenol (1.39 g, 6.20 mmol), quinolin-8-ol (79 mg, 0.55 mmol), copper(I) chloride (20 mg, 0.21 mmol), potassium phosphate (526 mg, 2.48 mmol) and 5-amino-6-bromo-l ,3-dimethyl-lH-benzo[d]imidazol- 2(3H)-one (529 mg, 2.07 mmol) in diglyme (20 ml) in a 100 mL round-bottom flask was degassed under a nitrogen atmosphere and heated to 120 °C for 24 h. To the cooled reaction mixture was added silica gel, stirred for 2 min, then the mixture was filtered through a pad of silica gel. The collected filtrate was concentrated and purified by column chromatography (20:80 to 0: 100, EtOAc/hexanesthen 0: 100 to 40:60, MeOH/EtOAc) to give 5-amino-6-(3- ((ieri-butyldimethylsilyl)oxy)phenoxy)-l,3-dimethyl-lH-benzo[d]imidazol-2(3H)-one (521 mg, 63%). MS (ES+) C21H29N3O3S1 requires: 399, found 400 [M+H]+.
Step 3: 5-amino-6-(3-hydroxyphenoxy)-l,3-dimethyl-lH-benzo[d]imidazol-2(3H)-one:
To a 0 °C solution of 5-amino-6-(3-((ieri-butyldimethylsilyl)oxy)phenoxy)-l,3- dimethyl-lH-benzo[d]imidazol-2(3H)-one (623 mg, 1.56 mmol) in THF was added a solution of ieira-butylammonium fluoride (0.90 mL, 3.1 mmol) in THF, the reaction mixture was allowed to warm up to RT and then stirred for 1-2 h. The reaction mixture was quenched with 1 M hydrogen chloride (0.10 mL, 3.1 mmol) and then partitioned between EtOAc and water. The seperated organic layer was washed with water twice, then concentrated and purified by column chromatography (20-80% EtOAc/hexanes and 0-40% MeOH/DCM) to give 5-amino-6-(3-hydroxyphenoxy)-l ,3-dimethyl-lH-benzo[d]imidazol-2(3H)-one (120 mg, 27%) as a solid. MS (ES+) C15H15N3O3 requires: 285, found 286 [M+H]+.
EXAMPLE 10: N-(6-(3-(4-(dimethylamino)butoxy)-5-propoxyphenoxy)-l,3-dimethyl-2- oxo-2,3-dihydro-lH-benzo[d]imidazol-5-yl)-3,4-dimethoxybenzenesulfonamide 2,2,2-
To a solution of N-(6-(3-(4-aminobutoxy)-5-propoxyphenoxy)-l ,3-dimethyl-2- oxo-2,3-dihydro-lH-benzo[d]imidazol-5-yl)-3,4-dimethoxybenzenesulfonamide 2,2,2- trifluoroacetate (180 mg, 0.247 mmol) in methanol (3.0 ml) was added triethylamine (0.034 ml, 0.25 mmol), acetic acid (0.028 ml, 0.49 mmol), formaldehyde (0.054 ml, 2.0 mmol), and sodium triacetoxyborohydride (131 mg, 0.618 mmol). The reaction mixture was stirred at room temperature and checked by LCMS every 30 minutes. After 3 h the reaction was complete by LCMS. The reaction was quenched with a few drops of TFA and concentrated under reduced pressure. The residue was purified by prep-HPLC using a gradient of 20-60% ACN/water containing 0.1% TFA to afford N-(6-(3-(4-(dimethylamino)butoxy)-5- propoxyphenoxy)-l,3-dimethyl-2-oxo-2,3-dihydro-lH-benzo[d]imidazol-5-yl)-3,4- dimethoxybenzenesulfonamide 2,2,2-trifluoroacetate (106 mg, 57%) as a white solid. MS (ES+) C32H42N4O8S requires: 642, found 643 [M+H]+. ¾ NMR (600 MHz, DMSO-ifc) δ 9.46 (s, 1H), 9.30 (br-s, 1H), 7.19 (m, 2H), 7.07 (s, 1H), 6.90 (d, 7 = 9.0 Hz, 1H), 6.75 (s, 1H), 6.13 (t, 7 = 2.2 Hz, 1H), 5.71 (t, J = 2.0 Hz, 1H), 5.67 (t, J = 2.0 Hz, 1H), 3.84 (t, 7 = 5.9 Hz, 2H), 3.77 (m, 5H), 3.62 (s, 3H), 3.29 (s, 3H), 3.20 (s, 3H), 3.12-3.05 (m, 2H), 2.78 (d, 7 = 4.7 Hz, 6H), 1.77-1.63 (m, 6H), 0.95 (t, 7 = 7.3 Hz, 3H)
References
1: Palmer WS, Poncet-Montange G, Liu G, Petrocchi A, Reyna N, Subramanian G, Theroff J, Yau A, Kost-Alimova M, Bardenhagen JP, Leo E, Shepard HE, Tieu TN, Shi X, Zhan Y, Zhao S, Draetta G, Toniatti C, Jones P, Geck Do M, Andersen JN. Structure-Guided Design of IACS-9571, a Selective High-Affinity Dual TRIM24-BRPF1 Bromodomain Inhibitor. J Med Chem. 2015 Jun 10. [Epub ahead of print] PubMed PMID: 26061247.
US-20160060260-A1
Institute for Applied Cancer Science, The University of Texas, MD Anderson Cancer Center, Houston, Texas, United States

The University of Texas MD Anderson Cancer Center | University of Texas System

The new Institute for Applied Cancer Science will be located at the south campus of M.D.

Draetta arrived at MD Anderson in 2011 to direct the Institute for Applied Cancer Science. He oversees the moon shots platforms
Department of Epigenetics and Molecular Carcinogenesis, The University of Texas, MD Anderson Cancer Center, Houston, Texas, United States

///////IACS-9571, TRIM24, BRPF1 bromodomain inhibitor, IACS-9571, IACS 9571, IACS9571, BOARD OF REGENTS, UNIVERSITY OF TEXAS SYSTEM
CAS BASE 1800477-30-8
CAS OF 1:1 TRIFLUOROACETATE 1883598-69-3
c1(cc(cc(c1)OCCC)Oc3cc2N(C(N(c2cc3NS(=O)(=O)c4cc(c(cc4)OC)OC)C)=O)C)OCCCCN(C)C
CCCOC1=CC(=CC(=C1)OC2=C(C=C3C(=C2)N(C(=O)N3C)C)NS(=O)(=O)C4=CC(=C(C=C4)OC)OC)OCCCCN(C)C
TFA salt of 8i (106 mg, 57%) as a white solid. 1H NMR (600 MHz, DMSO-d6) δ 9.46 (s, 1H), 9.30 (br-s, 1H), 7.19 (m, 2H), 7.07 (s, 1H), 6.90 (d, J = 9.0 Hz, 1H), 6.75 (s, 1H), 6.13 (t, J = 2.2 Hz, 1H), 5.71 (t, J = 2.0 Hz, 1H), 5.67 (t, J = 2.0 Hz, 1H), 3.84 (t, J = 5.9 Hz, 2H), 3.77 (m, 5H), 3.62 (s, 3H), 3.29 (s, 3H), 3.20 (s, 3H), 3.12–3.05 (m, 2H), 2.78 (d, J = 4.7 Hz, 6H), 1.77–1.63 (m, 6H), 0.95 (t, J = 7.3 Hz, 3H). 13C NMR (600 MHz, DMSO-d6) δ 160.3, 160.0, 159.3, 154.1, 152.0, 148.4, 143.9, 131.8, 128.2, 126.0, 121.9, 120.5, 110.4, 109.4, 106.4, 100.6, 95.9, 95.8, 95.2, 68.9, 66.7, 56.3, 55.6, 55.4, 42.1, 27.1, 27.0, 25.6, 21.9, 20.7, 10.4. MS (ESI) m/z 644 [M + H]+.
HELP, Need one time help to pay 10 year concessional subscription to this, your favorite blog to WordPress
Just One viewer please come forward
Dear Kind Viewer’s
WordPress is kind to me and negotiated a one time 10 year concessional subscription of 260 US dollars…….https://newdrugapprovals.org/
I need one time help to pay this one time 10 year concessional subscription to our favorite blog.
This is done to keep this blog running even after my death.
Currently I am paying 99 US Dollars per annum
email me
amcrasto@gmail.com
call +919323115463
Paypal will work for me via email request to you by me, Indian govt does not allow automatic transfer via paypal buttons on the blog.
email me at amcrasto@gmail.com and tell me amount, i will request you via paypal

DR ANTHONY CRASTO
LIONEL MY SON, MY MOTIVATION
.
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed…
View original post 226 more words
Tianagliflozin IND filed by Tianjin Institute of Pharmaceutical research


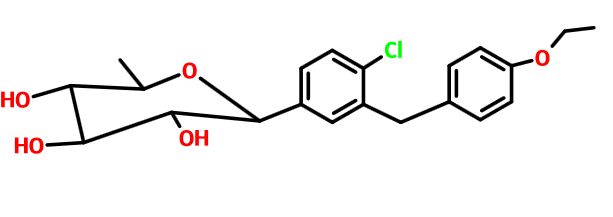
Tianagliflozin,
taigeliejing, 6-deoxydapagliflozin
| Molecular Formula: | C21H25ClO5 |
|---|---|
| Molecular Weight: | 392.8732 g/mol |
IND Filing…Tianjin Institute of Pharmaceutical research
Tianjin Institute Of Pharmaceutical Research,
(3R,4S,5S,6R)-2-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-6-methyloxane-3,4,5-triol
1-[4-Chloro-3-(4-ethoxybenzyl)phenyl]-1,6-dideoxy-b-D-glucopyranose
D-Glucitol, 1,5-anhydro-1-C-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-6-deoxy-, (1S)-
1–[4–Chloro–3–(4–ethoxybenzyl)phenyl]–1,6–dideoxy–β–d–glucopyranose
6-deoxydapagliflozin
A SGLT-2 inhibitor potentially for the treatment of type 2 diabetes.
![]()
CAS N. 1461750-27-5
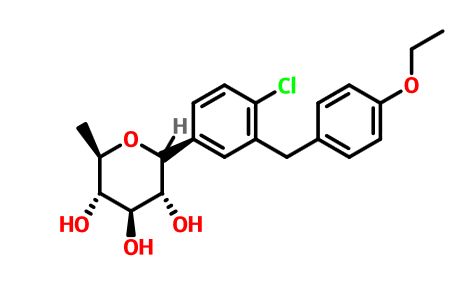

The structures of dapagliflozin and 6-deoxydapagliflozin (1)
,deletion of the 6-OH in the sugar moiety of dapagliflozin led to the discovery of a more potent SGLT2 inhibitor, 6-deoxydapagliflozin (1, ). In an in vitro assay, 1 was a more active SGLT2 inhibitor, with IC 50 = 0.67 nM against human SGLT2 (hSGLT2), as compared with 1.1 nM for dapagliflozin, leading to the identification of 1 as the most active SGLT2 inhibitor discovered so far in this field. Also in an in vivo assay, 1 also introduced more urinary glucose in a rat urinary glucose excretion test (UGE) and exhibited more potent blood glucose inhibitory activity in a rat oral glucose tolerance test (OGTT) than dapagliflozin.
Given the fact that 6-dexoydapagliflozin (1) is a very promising SGLT2 inhibitor that could be used to treat type 2 diabetes, led to preclinical trials
Tianjin Institute Of Pharmaceutical Research,天津药物研究院

SPECTRAL DATA of Tianagliflozin
1 as a white solid (3.65 g, 93 %). R f = 0.35 (EtOAc);
m.p.: 148–149 °C;
1H NMR (400 MHz, DMSO-d 6): δ = 7.35 (d, 1H, J = 8.4 Hz), 7.25 (s, 1H), 7.18 (d, 1H, J = 8.0 Hz), 7.08 (d, 2H, J = 8.4 Hz), 6.81 (d, 2H, J = 8.4 Hz), 4.95 (d, 1H, J = 5.2 Hz, OH), 4.90 (d, 1H, J = 4.4 Hz, OH), 4.79 (d, 1H, J = 5.6 Hz, OH), 3.92–4.01 (m, 5H), 3.24–3.29 (m, 1H), 3.18–3.22 (m, 1H), 3.09–3.15 (m, 1H), 2.89–2.95 (m, 1H), 1.29 (t, 3H, J = 7.0 Hz, CH2 CH 3 ), 1.15 (d, 3H, J = 6.0 Hz, CHCH 3 ) ppm;
13C NMR (100 MHz, DMSO-d 6): δ = 156.85, 139.65, 137.82, 131.83, 131.16, 130.58, 129.52, 128.65, 127.14, 114.26, 80.71, 77.98, 75.77, 75.51, 74.81, 62.84, 37.55, 18.19, 14.62 ppm;
IR (KBr): v¯¯¯ = 3,564 (w), 3,385 (s), 2,981 (s), 2,899 (s), 2,861 (s), 1,613 (m), 1,512 (s), 1,477 (m), 1,247 (s), 1,102 (s), 1,045 (s), 1,012 (s) cm−1;
HR–MS: calcd for C21H29ClNO5 ([M + NH4]+) 410.1729, found 410.1724.
PATENT
CN 103864737
http://www.google.com/patents/CN103864737A?cl=en
PATENT
WO 2014094544
http://www.google.com/patents/WO2014094544A1?cl=en


-27-


1 D1 -6 Optionally, the step (7 ‘) is the step (7’) in place:
LS l- [4 – D (I- Dl- 6)

A.

(DMSO-d 6, 400 MHz), δ 7.35 (d, 1H, J = 8.0 Hz), 7.28 (d, 1H, J ‘. 2.0 Hz), 7.17 (dd, IH, / = 2.0 Hz and 8.4 Hz), 7.05 (d, 2H, J: 8.8 Hz), 6.79 (d, 2H, 8.8 Hz): 4.924,95 (m, 2H), 4,81 (d, IH, 6,0 Hz), 3.93- 3.99 (m, 5H), 3,85 (d, 1H, J = 10,4 Hz), 3,66 (dd, IH, 5,2 Hz and 11,6 Hz), 3.17-3,28 (m, 3H), 3.02-3.08 (m: IH), 1.28 (t, 3H, J = 7,0 Hz), 0,80 (s, 9H), -0.05 (s, 3H), -0.09 (s, 3H) .
PATENT
[0066] The added 100mL dried over anhydrous methanol 0. 5g of sodium metal, nitrogen at room temperature with stirring, until the sodium metal disappeared. Followed by addition of 5. 2g (10mmol) of compound 6, stirring was continued at room temperature for 3 hours. To the reaction system was added 5g strong acid cation exchange resin, stirred at room temperature overnight, the reaction mixture until pH = 7. The resin was removed by suction, and the filtrate evaporated to dryness on a rotary evaporator, the residue was further dried on a vacuum pump to give the product I-D1-6, as a white foamy solid.
PATENT
WO 2014139447
PATENT related
http://www.google.com/patents/WO2013044608A1?cl=en
http://link.springer.com/article/10.1007%2Fs40242-014-4043-9#/page-1
Med Chem. 2015;11(4):317-28.
Design of SGLT2 Inhibitors for the Treatment of Type 2 Diabetes: A History Driven by Biology to Chemistry.
Abstract
A brief history of the design of sodium-dependent glucose cotransporter 2 (SGLT2) inhibitors is reviewed. The design of O-glucoside SGLT2 inhibitors by structural modification of phlorizin, a naturally occurring O-glucoside, in the early stage was a process mainly driven by biology with anticipation of improving SGLT2/SGLT1 selectivity and increasing metabolic stability. Discovery of dapagliflozin, a pioneering C-glucoside SGLT2 inhibitor developed by Bristol-Myers Squibb, represents an important milestone in this history. In the second stage, the design of C-glycoside SGLT2 inhibitors by modifications of the aglycone and glucose moiety of dapagliflozin, an original structural template for almost all C-glycoside SGLT2 inhibitors, was mainly driven by synthetic organic chemistry due to the challenge of designing dapagliflozin derivatives that are patentable, biologically active and synthetically accessible. Structure-activity relationships (SAR) of the SGLT2 inhibitors are also discussed.
http://www.ncbi.nlm.nih.gov/pubmed/25557661
Paper

Discovery of 6-Deoxydapagliflozin as a Highly Potent Sodium-dependent Glucose Cotransporter 2 (SGLT2) Inhibitor for the Treatment of Type 2 Diabetes
http://www.ingentaconnect.com/content/ben/mc/2014/00000010/00000003/art00009?crawler=true
CLIP

A facile synthesis of 6-deoxydapagliflozin
Keywords. Carbohydrates Drug research Hydrogenolysis Dapagliflozin SGLT2 inhibitor


The synthetic route to the target compound 1 is shown in Scheme 3. The starting material methyl 2,3,4-tri-O-benzyl-6-deoxy-6-iodo-α–d-glucopyranoside (3) was prepared from commercially available methyl α–d-glucopyranoside (2) according to a known method [5, 6].
Iodide 3 was reductively deiodinated to give 4 in 91 % yield under hydrogenolytic conditions using 10 % Pd/C as catalyst in the presence of Et3N as base in THF/MeOH at room temperature.
when the iodide 3 was treated with Barton–McCombie reagent (n-Bu3SnH/AIBN) [7] in toluene at room temperature no reaction occurred; however, when the reaction was carried out at elevated temperatures, such as reflux, a complex mixture formed with only a trace amount (3 %, entry 1) of the desired product 4.
When the iodide 3 was treated with LiAlH4 in THF at 0 °C to room temperature, another complex mixture was produced with only a trace amount (2 %, entry 2) of 4.
When Pd(OH)2 was used as the hydrogenolysis catalyst instead of 10 % Pd/C, the desired 4 was indeed formed (14 %, entry 4), but most of the starting material was converted to a few more polar byproducts, which were believed to result from the cleavage of at least one of the benzyl groups.
pdf available
Monatshefte für Chemie – Chemical Monthly
December 2013, Volume 144, Issue 12, pp 1903-1910
////////IND Filing, SGLT-2 inhibitor, type 2 diabetes, Tianagliflozin, taigeliejing, 6-deoxydapagliflozin, 1461750-27-5
Clc1c(cc(cc1)C2[C@@H]([C@H]([C@@H]([C@H](O2)C)O)O)O)Cc3ccc(cc3)OCC
CCOC1=CC=C(C=C1)CC2=C(C=CC(=C2)C3C(C(C(C(O3)C)O)O)O)Cl
c1(c(cc(cc1)C2OC(C(C(C2O)O)O)C)Cc3ccc(cc3)OCC)Cl
BRIVARACETAM
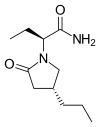
BRIVARACETAM, UCB-34714
(2S)-2-[(4R)-2-oxo-4-propylpyrrolidin-1-yl]butanamide
(2S)-2-[(4R)-2-Oxo-4-propyl-1-pyrrolidinyl]butanamide
1-Pyrrolidineacetamide, α-ethyl-2-oxo-4-propyl-, (αS,4R)-
CAS 357336-20-0
| Molecular Formula: | C11H20N2O2 |
|---|---|
| Molecular Weight: | 212.2887 g/mol |
UNII-U863JGG2IA
UCB; For the treatment of partial onset seizures related to epilepsy, Approved February 2016
Brivaracetam, the 4-n-propyl analog of levetiracetam, is a racetam derivative with anticonvulsant properties.[1][2] Brivaracetam is believed to act by binding to the ubiquitous synaptic vesicle glycoprotein 2A (SV2A).[3] Phase II clinical trials in adult patients with refractory partial seizures were promising. Positive preliminary results from stage III trials have been recorded,[4][5] along with evidence that it is around 10 times more potent[6] for the prevention of certain types of seizure in mouse models than levetiracetam, of which it is an analogue.
On 14 January 2016, the European Commission,[7] and on 18 February 2016, the USFDA[8] approved brivaracetam under the trade name Briviact (by UCB). The launch of this anti-epileptic is scheduled for the first quarter of that year. Currently, brivaracetam is still not approved in other countries like Australia, Canada and Switzerland.
Brivaracetam was approved by European Medicine Agency (EMA) on Jan 14, 2016 and approved by the U.S. Food and Drug Administration (FDA) on Feb 18, 2016. It was developed and marketed as Briviact® by UCB in EU/US.
Brivaracetam is a selective high-affinity synaptic vesicle protein 2A ligand, as an adjunctive therapy in the treatment of partial-onset seizures with or without secondary generalization in adult and adolescent patients from 16 years of age with epilepsy.
Briviact® is available in three formulations, including film-coated tablets, oral solution and solution for injection/infusion. And it will be available as 10 mg, 25 mg, 50 mg, 75 mg and 100 mg film-coated tablets, a 10 mg/ml oral solution, and a 10 mg/ml solution for injection/infusion. The recommended starting dose is either 25 mg twice a day or 50 mg twice a day, depending on the patient’s condition. The dose can then be adjusted according to the patient’s needs up to a maximum of 100 mg twice a day. Briviact can be given by injection or by infusion (drip) into a vein if it cannot be given by mouth.
European Patent No. 0 162 036 Bl discloses compound (S)-α-ethyl-2-oxo-l- pyrrolidine acetamide, which is known under the International Non-proprietary Name of Levetiracetam.

Levetiracetam
Levetiracetam is disclosed as a protective agent for the treatment and prevention of hypoxic and ischemic type aggressions of the central nervous system in European patent EP 0 162 036 Bl. This compound is also effective in the treatment of epilepsy.
The preparation of Levetiracetam has been disclosed in European Patent No. 0 162 036 and in British Patent No. 2 225 322.
International patent application having publication number WO 01/62726 discloses 2-oxo-l -pyrrolidine derivatives and methods for their preparation. It particularly discloses compound (2S)-2-[(4R)-2-oxo-4-propyl-pyrrolidin-l-yl] butanamide known under the international non propriety name of brivaracetam.

Brivaracetam
International patent application having publication number WO 2005/121082 describes a process of preparation of 2-oxo-l -pyrrolidine derivatives and particularly discloses a process of preparation of (2S)-2-[(4S)-4-(2,2-difluorovinyl)-2-oxo-pyrrolidin-l- yl]butanamide known under the international non propriety name of seletracetam.

Seletracetam
Kenda et al., in J. Med. Chem. 2004, 47, 530-549, describe processes of preparation of 2-oxo-l -pyrrolidine derivatives and particularly discloses compound 1-((1S)-I- carbamoyl-propyl)-2-oxo-pyrrolidone-3-carboxylic acid as a synthetic intermediate.
WO2005028435
CLIPS
Find better ways to make old and new epilepsy drugs. J. Surtees and co-inventors disclose alternative processes for making active pharmaceutical ingredients (APIs) that are used to treat epilepsy and seizures. One compound that can be prepared by their processes is the established drug levetiracetam (1, Figure 1), marketed under the trade name Keppra. Because 1 is now off-patent, there is obvious interest in new drugs.

The inventors also claim that seletracetam (2) and brivaracetam (3) (Figure 2) can be prepared by their processes. These drugs are apparently much more active than 1.
All of the drugs are used as single isomers, so a stereoselective synthesis is desirable. The inventors describe two routes for preparing the molecules; the first, shown in Figure 1, is the synthesis of 1 by the reaction between pyrrolidone (4) and chiral bromo amide 5 in the presence of a base. GC analysis showed that the conversion is 40.3% and that the product contains 51% of the (S)-enantiomer and 49% of the (R)-isomer. No details of their separation are given, although the use of chiral HPLC is discussed.
The same reaction is used to prepare derivative 6 of 1. Compound 7 is prepared from the corresponding hydroxy ester and then condensed with 4 to give 6. Chiral HPLC showed that the product is a mixture of 89.3% (S)-enantiomer 6 and 10.7% of its (R)-isomer.
The inventors do not describe the detailed preparation of 2, but they report that acid 8 is prepared in 41% yield from pyrrolidone 9 and acid 10 in the presence of NaH (Figure 2). Ammonolysis of 8 produces 2; no reaction details are provided.

In a reaction similar to the preparation of 8, acid 11 is prepared from 10 and pyrrolidone 12. The product is isolated in 77% yield and can be converted to 3 by ammonolysis. Again, no details are provided for this reaction.
The second route for preparing the substituted pyrrolidones does not start with simple pyrrolidones and is the subject of additional claims. The route involves a cyclization reaction, shown in Figure 3. The preparation of enantiomer 13 begins with the reaction of racemic salt 14 and optically pure bromo ester 15. This step produces intermediate 16, isolated as a yellow oil. The crude material is treated with 2-hydroxypyridine (2-HP) to cyclize it to 17. This ester is hydrolyzed to give acid 18. Conversion to 13 is carried out by adding ClCO2Et, followed by reaction with liquid NH3 in the presence of K2CO3. The overall yield of 13 is 32%.

This route is also used to prepare levetiracetam (1) by treating 5 with the HCl salt of amino ester 19 to give 20, recovered as its HCl salt in 49% yield. The salt is basified with Et3N and treated with 2-HP to cyclize it to 1, initially isolated as an oil. GC analysis showed 100% conversion, and chiral HPLC showed that the product contains 98.6% (S)-isomer and 1.4% (R)-isomer.
The inventors also prepared 1 and its (R)-enantiomer 21 by using a similar reaction scheme with alternative substrates to 5. Figure 4 outlines the route, which starts from protected hydroxy amide 22 and amino ester 23. When the reaction is carried out in the presence of Cs2CO3, the product is (R)-enantiomer24, which is used without purification to prepare 21 by treating it with 2-HP. Chiral HPLC showed that the product is 94% (R) and 6% (S).

When the reaction between 22 and 23 is run with K2CO3, the product is (S)-enantiomer 25. This is used to prepare 1, but the product contains only 79% (S)-isomer.
The inventors do not comment on the apparent stereoselectivity of the carbonate salts in the reaction of 22 with 23. This is an intriguing finding and worthy of investigation. (UCB S.A. [Brussels]. US Patent 8,338,621

SYNTHESIS
PATENT
WO2005028435
Example 1: Synthesis of (2S)-2-((4R)-2-oxo-4-n-propyl-l-pyrrolidinyl)butanamide 1.1 Synthesis of (2S)-2-aminobutyramide free base

1800 ml of isopropanol are introduced in a 5L reactor. 1800 g of (2S)-2- aminobutyramide tartrate are added under stirring at room temperature. 700 ml of a 25% aqueous solution of ammonium hydroxide are slowly added while maintaining the temperature below 25°C. The mixture is stirred for an additional 3 hours and then the reaction is allowed to complete at 18°C for 1 hour. The ammonium tartrate is filtered. Yield : 86%.
1.2 Synthesis of 5-hydroxy-4-n-propyl-furan-2-one

Heptane (394 ml) and morpholine (127.5 ml) are introduced in a reactor. The mixture is cooled to 0°C and glyoxylic acid (195 g, 150 ml, 50w% in water) is added. The mixture is heated at 20°C during 1 hour, and then valeraldehyde (148.8 ml) is added . The reaction mixture is heated at 43°C during 20 hours. After cooling down to 20CC, a 37 % aqueous solution of HCl (196.9 ml) is slowly added to the mixture, which is then stirred during 2 hours.
After removal of the heptane phase, the aqueous phase is washed three times with heptane. Diisopropyl ether is added to the aqueous phase. The organic phase is removed, and the aqueous phase further extracted with diisopropyl ether (2x). The diisopropyl ether phases are combined, washed with brine and then dried by azeotropic distillation. After filtration and evaporation of the solvent, 170g of 5- hydroxy-4-n-propyl-furan-2-one are obtained as a brown oil. Yield: 90.8 %
1.3 Synthesis of (2S)-2-((4R)-2-oxo-4-n-propyl-l-pyrrolidinyl)butanamide and (2S)-2-((4S)-2-oxo-4-n-propyl-l-pyrrolidinyl)butanamide

(S, R) (S, S) The (2S)-2-aιninobutyrarnide solution in isopropanol containing 250 g obtained as described here above is dried by azeotropic distillation under vacuum. To the dried (2S)-2-am obutyraιnide solution is added 5-hydroxy-4-n-propyl-furan-2-one (290 g) between 15°C and 25 °C; the mixture is heated to 30 °C and kept for at least 2 hours at that temperature. Acetic acid (1, 18 eq.), Pd/C catalyst (5 w/w%; Johnson Matthey 5% Pd on carbon – type 87L) are then added and hydrogen introduced into the system under pressure. The temperature is kept at 40 °C maximum and the H2 pressure maintained between 0,2 bar and 0,5 bar followed by stirring for at least 20 hours following the initial reaction. The solution is then cooled to between 15 °C and 25 °C and filtered to remove the catalyst. The solution of product in isopropanol is solvent switched to a solution of product in isopropyl acetate by azeotropic distillation with isopropyl acetate. The organic solution is washed with aqueous sodium bicarbonate followed by a brine wash and then filtered. After recristallisation, 349 g of (2S)-2-((4R)-2- oxo-4-n-propyl-l-pyrrolidinyl)butanamide and (2S)-2-((4S)-2-oxo-4-n-propyl-l- pyιτolidinyl)butanamide are obtained (Yield: 82.5%).
1.4 Preparation of (2S)-2-((4R)-2-oxo-4-n-propyl-l-pyrrolidinyl)butanamide The chromatographic separation of the two diastereoisomers obtained in 1.3 is performed using of (CHIRALPAK AD 20 um) chiral stationary phase and a 45/55 (volume /volume) mixture of n-heptane and ethanol as eluent at a temperature of 25 + 2°C. The crude (2S)-2-((4R)-2-oxo-4-n-propyl-l-pyrrolidinyl)butanamide thus obtained is recristallised in isopropylacetate, yielding pure (2S)-2-((4R)-2-oxo-4-n-propyl-l- pyrrolidinyl)butanamide (Overall yield: 80%) .
Example 2: Synthesis of (2S)-2-((4R)-2-oxo-4-n-propyl-l-pyrrolidinyl)butanamide

Example 1 is repeated except that in step 1.1 a solution of (2S)-2- aminoburyramide.HCl in isopropanol is used (27.72 g, 1.2 equivalent), which is neutralised with a NHs/isopropanol solution (3,4-3,7 mol/L). The resulting ainmonium chloride is removed from this solution by filtration and the solution is directly used for reaction -with 5-hydroxy-4-n-propyl-furan-2-one (23.62 g, 1.0 equivalent) without intermediate drying of the (2S)-2-aminobutyramide solution. Yield after separation of the two diastereoisomers and recristallisation: approximately 84%.

Ref ROUTE1
1. WO0162726A2.
2. WO2005028435A1 / US2007100150A1.
3. J. Med. Chem. 1988, 31, 893-897.
4. J. Org. Chem. 1981, 46, 4889-4894.
PATENT
https://www.google.com/patents/WO2007031263A1?cl=en
Example 3-Synthesis of brivaracetam (I)
3.a. Synthesis of (S) and (R) 2-((R)-2-oxo-4-propyl-pyrrolidin-l-yl)-butyric acid methyl ester fVIaa*) and (Wlab)

(VIaa) (VIab) A slurry of 60% sodium hydride suspension in mineral oil (0.94g, 23.4 mmol) in tetrahydrofuran (30 mL) is cooled at 0°C under a nitrogen atmosphere. A solution of substantially optically pure (R)-4-propyl-pyrrolidin-2-one (Ilia) (2g, 15.7 mmol) in tetrahydrofuran (2 mL) is added over a 15 minutes period. The reaction mixture is stirred 10 min at 0°C then a solution of methyl-2-bromo-butyric acid methyl ester (V) (3.69g, 20.4 mmol) in tetrahydrofuran (2mL) was added over a 20 minutes period. The reaction mixture is stirred at O0C until maximum conversion of starting material and the reaction mixture is then allowed to warm to room temperature and diluted with water (20 mL). Tetrahydrofuran is removed by evaporation and the residue is extracted with isopropyl acetate (20 ml + 10 mL). The combined organic layers are dried on anhydrous magnesium sulfate and evaporated to afford 3g (13.2 mmol, 86 %) of a mixture of epimers of compound (Via), as a mixture respectively of epimer (VIaa) and epimer (VIab). 1H NMR(400 MHz, CDCI3) of the mixture of epimers (VIaa) and (VIab) : δ = 4.68
(dd, J= 10.8, J= 5.1, 2×1 H) ; 3.71 (s, 2x3H); 3.60 (t app, J= 8.2, IH); 3.42 (t app, J= 8.7, IH); 313 (dd, J= 9.2, J = 6.8, IH); 2.95 (dd, J= 9.2, J= 6.8, IH); 2.56 (dd, J= 16.6, J = 8.7, 2xlH); 2.37 (dm, 2xlH); 2.10 (m, 2xlH); 2.00 (m, 2xlH); 1.68 (m, 2xlH); 1.46 (m, 2x2H); 1.36 (m, 2x2H); 0.92 (m, 2x6H).
13C NMR (400 MHz, CDCl3) of the mixture of epimers (VIaa) and (VIab) : δ =
175.9; 175.2; 171.9; 55.3; 52.4; 49.8; 49.5; 38.0; 37.8; 37.3; 36.9; 32.5; 32.2; 22.6; 22.4; 21.0; 14.4; 11.2; 11.1
HPLC (GRAD 90/10) of the mixture of epimers (VIaa) and (VIab): retention time= 9.84 minutes (100 %)
GC of the mixture of epimers (VIaa) and (VIab): retention time = 13.33 minutes (98.9 %)
MS of the mixture of epimers (VIaa) and (VIab) (ESI) : 228 MH+
3.b. Ammonolysis of compound of the mixture of (VIaa) and (VIab)

(VIaa) (VIab) (I) (VII)
A solution of (VIaa) and (VIab) obtained in previous reaction step (1.46g, 6.4 mmol) in aqueous ammonia 50 % w/w (18 mL) at 00C is stirred at room temperature for 5.5hours. A white precipitate that appears during the reaction, is filtered off, is washed with water and is dried to give 0.77g (3.6 mmol, yield = 56 %) of white solid which is a mixture of brivaracetam (I) and of compound (VII) in a 1 :1 ratio.
1H NMR of the mixture (I) and (VII) (400 MHz, CDCI3) : δ = 6.36 (s, broad, IH); 5.66 (s, broad, IH); 4.45 (m, IH); 3.53 (ddd, J= 28.8, J= 9.7, J= 8.1, IH); 3.02 (m, IH); 2.55 (m, IH); 2.35 (m, IH); 2.11 (m, IH); 1.96 (m, IH); 1.68 (m, IH); 1.38 (dm, 4H); 0.92 (m, 6H). 13c NMR of the mixture (I) and (VII) (400 MHz, CDCl3) : δ = 176.0; 175.9; 172.8;
172.5; 56.4; 56.3; 50.0; 49.9; 38.3; 38.1; 37.3; 37.0; 32.3; 32.2; 21.4; 21.3; 21.0; 20.9; 14.4; 10.9; 10.8
HPLC (GRAD 90/10) of the mixture of (I) and (VII) retention time= 7.67 minutes (100 %)
Melting point of the mixture of (I) and (VII) = 104.90C (heat from 400C to 1200C at 10°C/min)
Compounds (I) and (VII) are separated according to conventional techniques known to the skilled person in the art. A typical preparative separation is performed on a 11.7g scale of a 1 :1 mixture of compounds (I) and (VII) : DAICEL CHIRALPAK® AD 20 μm, 100*500 mm column at 300C with a 300 mL/minutes debit, 50 % EtOH – 50 % Heptane. The separation affords 5.28g (45 %) of compound (VII), retention time = 14 minutes and 5.2Og (44 %) of compounds (I), retention time = 23 minutes.
1H NMR of compound (I) (400 MHz, CDCl3): δ = 6.17 (s, broad, IH); 5.32 (s, broad, IH); 4.43 (dd, J= 8.6, J= 7.1, IH); 3.49 (dd, J= 9.8, J= 8.1, IH); 3.01 (dd, J= 9.8, J= 7.1, IH); 2.59 (dd, J= 16.8, J= 8.7, IH); 2.34 (m, IH); 2.08 (dd, J= 16.8, J= 7.9, IH); 1.95 (m, IH); 1.70 (m, IH); 1.47-1.28 (m, 4H); 0.91 (dt, J= 7.2, J= 2.1, 6H)
HPLC (GRAD 90/10) of compound (I) : retention time = 7.78 minutes
1H NMR of compound (VII) (400 MHz, CDCl3): δ = 6.14 (s, broad, IH); 5.27 (s, broad, IH); 4.43 (t app, J = 8.1, IH); 3.53 (t app, J = 9.1, IH); 3.01 (t app, J = 7.8, IH); 2.53 (dd, J = 16.5, J = 8.8, IH); 2.36 (m, IH); 2.14 (dd, J = 16.5, J = 8.1, IH); 1.97 (m, IH); 1.68 (m, IH); 1.43 (m, 2H); 1.34 (m, 2H); 0.92 (m, 6H)
3c. Epimerisation of compound of (2RV2-((R)-2-oxo-4-propyl-pyπOlidin-l-ylV butyramide (VID
Compound (VII) (200 mg, 0.94 mmol) is added to a solution of sodium tert- butoxide (20 mg, 10 % w/w) in isopropanol (2 mL) at room temperature. The reaction mixture is stirred at room temperature for 18h. The solvent is evaporated to afford 200 mg
(0.94 mmol, 100 %) of a white solid. Said white solid is a mixture of brivaracetam (I) and of (VII) in a ratio 49.3 / 50.7.
HPLC (ISO80): retention time= 7.45 min (49.3%) brivaracetam (I); retention time= 8.02 minutes (50.7%) compound (VII).
Route 2

Reference:ROUTE 2
1. WO2007031263A1 / US2009318708A1.
PATENT
http://www.google.com/patents/WO2007065634A1?cl=en
(scheme 3).

Scheme 3
scheme 4.

5h. Synthesis of brivaracetam and (V) A suspension of (Id) and (Ie) (0.6 g, 2.3 mmol) in MIBK (10 mL) is heated at
120°C for 6 hours. The resulting solution is concentrated and separated on chromatography column (Silicagel 600.068-0.200 mm, cyclohexane/EtOAc : 10/90) to give 0.13 g of brivaracetam (0.6 mmol, 26 %, ee = 94 %) and (V).
1H NMR (400 MHz, CDCl3): δ = 6.17 (s, broad, IH); 5.32 (s, broad, IH); 4.43 (dd, J= 8.6, J= 7.1, IH); 3.49 (dd, J= 9.8, J= 8.1, IH); 3.01 (dd, J= 9.8, J= 7.1, IH); 2.59 (dd, J= 16.8, J= 8.7, IH); 2.34 (m, IH); 2.08 (dd, J= 16.8, J= 7.9, IH); 1.95 (m, IH); 1.70 (m, IH); 1.47-1.28 (m, 4H); 0.91 (dt, J= 7.2,J= 2.1, 6H).
HPLC (method 90/10) : Retention time = 7.78 minutes Chiral HPLC : Retention time = 9.66 minutes (97%) MS (ESI): 213 MH+

Route 3
Reference:1. WO2007065634A1 / US2009012313A1.
PAPER
http://pubs.acs.org/doi/abs/10.1021/acs.oprd.6b00094
A Biocatalytic Route to the Novel Antiepileptic Drug Brivaracetam
Chemical Process Research and Development, Pharma Sciences, UCB Pharma S.A., Chemin du Foriest, 1420 Braine l’Alleud, Belgium
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.6b00094
* E-mail: arnaud.schule@ucb.com; Telephone: +32.2.386.6208.


AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
References
- von Rosenstiel P (Jan 2007). “Brivaracetam (UCB 34714)”. Neurotherapeutics 4 (1): 84–7. doi:10.1016/j.nurt.2006.11.004.PMID 17199019.
- Malawska B, Kulig K (Jul 2005). “Brivaracetam UCB”. Current Opinion in Investigational Drugs 6 (7): 740–746. PMID 16044671.
- Rogawski MA, Bazil CW (Jul 2008). “New molecular targets for antiepileptic drugs: alpha(2)delta, SV2A, and K(v)7/KCNQ/M potassium channels”. Current Neurology and Neuroscience Reports 8 (4): 345–352. doi:10.1007/s11910-008-0053-7. PMC 2587091.PMID 18590620.
- Clinical trial number NCT00464269 for “Double-blind, Randomized Study Evaluating the Efficacy and Safety of Brivaracetam in Adults With Partial Onset Seizures” at ClinicalTrials.gov
- Rogawski MA (Aug 2008). “Brivaracetam: a rational drug discovery success story”. British Journal of Pharmacology 154 (8): 1555–7.doi:10.1038/bjp.2008.221. PMC 2518467. PMID 18552880.
- Matagne A, Margineanu DG, Kenda B, Michel P, Klitgaard H (Aug 2008). “Anti-convulsive and anti-epileptic properties of brivaracetam (ucb 34714), a high-affinity ligand for the synaptic vesicle protein, SV2A”. British Journal of Pharmacology 154 (8): 1662.doi:10.1038/bjp.2008.198. PMID 18500360.
- http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/003898/human_med_001945.jsp&mid=WC0b01ac058001d124
- http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm486827.htm
|
|||
| Names | |||
|---|---|---|---|
| IUPAC name
(2S)-2-[(4R)-2-oxo- 4-propylpyrrolidin-1-yl] butanamide
|
|||
| Identifiers | |||
| 357336-20-0 |
|||
| ChEMBL | ChEMBL607400 |
||
| ChemSpider | 8012964 |
||
| Jmol interactive 3D | Image | ||
| PubChem | 9837243 | ||
| UNII | U863JGG2IA |
||
| Properties | |||
| C11H20N2O2 | |||
| Molar mass | 212.15 g/mol | ||
| Pharmacology | |||
| ATC code | N03 | ||
| Legal status |
|
||
| Oral | |||
| Pharmacokinetics: | |||
| Nearly 100% | |||
| <20% | |||
| Hydrolysis, CYP2C8-mediated hydroxylation | |||
| 8 hrs | |||
| >75% renal | |||
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|||


//////BRIVARACETAM, UCB, 2016 FDA, UCB-34714
CCCC1CC(=O)N(C1)C(CC)C(=O)N
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021260721&_cid=P12-KXX1JU-33531-1
Brivaracetam is chemically known as (2S)-2-[(4R)-2-oxo-4-propyltetrahydro-1H-pyrrol-1-yl] butanamide, having the chemical structure of formula 1 as below:
Brivaracetam is basically a chemical analogue of Levetiracetam, marketed under the brand name of BRIVIACT for the treatment as adjunctive therapy in the treatment of partial-onset seizures in patients at 16 years of age and older with epilepsy. Brivaracetam has an advantage over Levetiracetam in that it gets into the brain “much more quickly,” which means that “it could be used for status epilepticus, or acute seizures than cluster, or prolonged seizures”. From the Phase III trials, the self-reported rate of irritability with Brivaracetam was 2% for both drug doses (100 mg and 200 mg) vs 1% for placebo, which compares to as much as 10% for Levetiracetam in some post-marketing studies.
With the improved safety profile and possibility to be used for wider range of epilepsy, Brivaracetam is considered as one of the most promising 3rd generation antiepileptic drugs.
Brivaracetam molecule is first disclosed in patent publication WO2001062726, which describes 2-oxo-1 -pyrrolidine derivatives and methods for their preparation. This patent publication further discloses compound (2S)-2-[(4R)-2-oxo-4-propyl-pyrrolidin-1-yl] butanamide which is known under the international non propriety name as Brivaracetam. As per Biopharmaceutics Classification System, Brivaracetam is a class I drug (high solubility and permeability).
Some prior arts US6784197 and US7629474 disclose a process for synthesizing a diastereomeric mixture of (2S)-2-[(4R)-2-oxo-4-propylpyrrolidin-1-yl]-butanamide and (2S)-2-[(4S)-2-oxo-4-propylpyrrolidin-1-yl]-butanamide (Brivaracetam) which is purified by chiral HPLC (Scheme-I & Scheme-II respectively, as provided below). This process used for chiral resolution makes it difficult for bulk manufacturing as well as it affects the overall yield making the process uneconomical.
Scheme-I
Synthesis of (2S)-2-(2-oxo-4-propyl-1-pyrrolidinyl)butanamide
[As disclosed in columns 37-38 of US 6784197 B2]
Scheme-II
1.1 Synthesis of (2S)-2-aminobutyramide-Free base
1.2 Synthesis of 5-hydroxy-4-n-propylfuran-2-one
1.3 Synthesis of (2S)-2-((4R)-2-oxo-4-n-propyl-1-pyrrolidinyl)butanamide and
(2S)-2-((4S)-2-oxo-4-n-propyl-1-pyrrolidinyl)butanamide
[As disclosed in columns 6-7 of US 7629474 B2]
Moreover, some prior arts such as US7122682B2, US8076493B2, US8338621B2 and US8957226B2 also describe processes for preparing Brivaracetam, wherein, the purifications are reportedly done by chiral HPLC methods resulting into similar shortcomings.
Kenda et al.: Journal of Medicinal Chemistry, 2004, 47, 530-549 further proposes selection of (2S)-2-[(4R)-2-oxo-4-propylpyrrolidin-1-yl]butanamide 83α (ucb 34714; Brivaracetam) as the most interesting candidate showing 10 times more potency than Levetiracetam as an antiseizure agent in audiogenic seizure-prone mice. This article further discloses methods for synthesizing the said compound Brivaracetam. However, here too these compounds are synthesized as mixtures of stereoisomers (racemic or diastereoisomeric mixtures), separated by preparative HPLC on silica gel and/or chiral phases.
All these processes for the preparation of Brivaracetam as described in the above- mentioned prior arts suffer from many disadvantages which includes difficulty in achieving desired chiral purity, tedious and cumbersome work up procedures, high temperature and longer time reaction, multiple crystallizations or isolation steps, use of excess reagents and solvents, column chromatographic separations & purifications etc. All these disadvantages affect the overall yield as well as the quality of the final product Brivaracetam and intermediates produced thereof; further, rendering such processes to be uneconomical and unsuitable for industrial scale-ups.
As a result, enantioselective synthesis of Brivaracetam was perceived to be a possible way of overcoming such problems in view of the large-scale synthesis. However, very few prior arts have been found to report successful reduction of such concept into practice.
WO2016191435A1 (also as IN201717005820A) relates to a process for a scalable synthesis of enantiomerically pure Brivaracetam from an intermediate (4R)-4- Propyldihydrofuran-2(3H)-one (compound IV):
, wherein, R is saturated or unsaturated C1-20 alkyl or C1 alkyl-unsubstituted or substituted aryl, comprising the steps of decarboxylation of
the compound of formula IV
to produce the compound of formula VI
ring-opening of the compound of formula VI to produce the compound of formula VII
, wherein Rl is saturated or unsaturated Cl-20 alkyl or Cl alkyl-unsubstituted or substituted aryl; and X is CI Br I OMs, OTs, ONs; or
the compound of formula X
reacting the compound of formula VI with (S)-2- aminobutanamide or its salt to produce the compound of formula XII in one step; or reacting the compound of formula VI with alkyl (S)-2- aminobutanoate to produce Xll-a
, wherein R in the compound of formula Xll-a is a saturated or unsaturated C1-C20 alkoxyl; and then converting Xll-a to XII that is Brivaracetam by aminolysis and amide formation reaction.
In above mentioned prior arts, the synthesis of chiral lactone which is the key starting material for making Brivaracetam involved Grignard addition, column chromatography and Krapcho decarboxylation techniques at high temperature, all of which are not at all recommendable in view of process perspective at industrial levels. Further the final step of the said reaction often involved cryogenic condition –30°C which is also difficult with respect to industrial scale up activities.
Furthermore, prior art IN201641030239A disclosed a process for the preparation of Brivaracetam of Formula (I) by means of converting enantiomerically pure compound of Formula VII to obtain enantiomerically pure compound of Formula XI:
, wherein X is each independently selected from halogen; alkyl or aryl sulfonyloxy; OR2; R2 is optionally substituted C1-C12 alkyl, aryl, alkyl aryl, aryl alkyl;
such that the said process further comprises steps of:
1) cyclizing compound of formula VII to give enantiomerically pure compound of formula IX:
, wherein R2 is optionally substituted C1-C12 alkyl, aryl, alkyl aryl or aryl alkyl;
2) converting the compound of formula IX to give a enantiomerically pure compound of formula X:
, wherein X is halogen;
3) converting compound of formula X to give a enantiomerically pure compound of formula XI:
wherein X is each independently selected from halogen; alkyl or aryl sulfonyloxy; OR2; R2 as defined above; followed by 4) treating the enantiomerically pure compound of formula XI with (S)- aminobutyramide of formula XII or its salt thereof to obtain Brivaracetam of Formula I.
However, this process suffered from drawbacks of handling acid chloride. Acid chlorides are unstable and storing a large amount of acid chloride is also not recommendable in view of safety and stability in industries. Moreover, this prior art process involves use of HBr in acetic acid, where HBr liberates Br that is hazardous and not recommendable for industrial scale-up activities in view of safety and handling.
Some other prior arts such as CN108503573A, CN105646319A, CN106588740A, CN106588831A, CN108689903B and CN108929289A report various processes of synthesizing Brivaracetam from its lactone intermediate by various ring opening techniques. Among these, in particular CN108929289A discloses a process of reacting a compound represented by the formula IV with (S) -2-aminobutyramide in order to obtain Brivaracetam. The synthetic route is as follows:
Also, CN108689903B relates to a new preparation method of Brivaracetam that comprises steps of: a) subjecting a compound of formula III and (S) -2-aminobutanamide or salt thereof to condensation reaction, in the presence of a condensing agent, in order to obtain a compound shown in a formula IV, wherein the compound has two chiral centres; b) removal of the hydroxy-protecting group R1 to obtain a compound of formula V; and c) carrying out chlorination reaction on the compound shown in the formula V using a chlorination reagent to obtain a compound shown in the formula VI; and d) carrying out substitution reaction on the compound shown in the formula VI in the presence of an
alkaline reagent, and closing a ring to obtain Brivaracetam of formula I having two chiral centres.
It has further been noted that although the above reaction goes through formation of intermediates V and/or VI; however, these intermediates are not essentially formed from the key lactone intermediate of Brivaracetam that is (4R)-4-propyldihydrofuran-2(3H)-one [or (R)-lactone].
Furthermore, CN111196771A relates to a preparation method of Brivaracetam which comprises the steps of: 1) carrying out ring-opening reaction on a compound R-4-propyldihydrofuran-2-ketone in a formula II and a compound S-2-aminobutanamide in a formula III to obtain an intermediate compound in a formula I; 2) condensing the said intermediate compound of formula I is followed by cyclization to produce Brivaracetam
However, the ring-opening reaction in step 1 of this process essentially occurs under acidic conditions, specifically in presence of Lewis acids like tetra-isopropyl titanate, anhydrous aluminium trichloride, anhydrous zinc chloride, boron trifluoride diethyl etherate etc.; and also in presence of organic solvents chosen from one or more of anhydrous tetrahydrofuran, 2-methyltetrahydrofuran, acetone, dimethyl sulfoxide and N, N-dimethylformamide; which makes this process both industrially non-scalable and environmentally unfriendly.
The prior art Org. Process Res. Dev.2016. v 20. no 9. p 1566-1575 in its scheme 4, on page 17 also discloses a scheme for synthesizing Brivaracetam from its lactone intermediate:
Nevertheless, it has been noted that the process reported in this prior art provides Brivaracetam (API) with a very poor yield of ~30% and also having an inferior chiral purity of 95.9% ee, which does not even meet the ICH-specification for the Finished Product (API).
Furthermore, a recently filed patent application WO2020148787A1 (also as IN201931002041) recites a new, improved and economical process for enantioselective synthesis and purification of a key intermediate of Brivaracetam that is the R-lactone, essentially utilizing a low chiral loading and without involving any chiral chromatographic resolution technique. Even though this prior art also discloses a process for the preparation of a chirally pure Brivaracetam of formula I utilizing the said intermediate; however, that process is mostly a conventional one.
Accordingly, there is still a need in the art for a more economical and improved process for the synthesis of Brivaracetam with better purity and yield which overcomes the drawbacks of above prior arts.
Therefore, the present inventors have developed a cost effective, novel and efficient process for the preparation of Brivaracetam which essentially avoids all the drawbacks involved in prior art as mentioned above. The currently developed process is advantageously capable of producing the key lactone intermediate with more than 80% ee applying transfer hydrogenation with a very simple operation in view of process perspective. Further, by means of using such chiral lactone with more than 80% ee, the currently developed process is also capable of delivering >99.9% chirally pure Brivaracetam with excellent yield.
EXAMPLES:
EXAMPLE 1: Synthesis of (3R)-N-[(1S)-1-carbamoylpropyl]-3-(hydroxymethyl) hexanamide [Intermediate 7 of scheme A of the present invention]
Example 1 illustrates one pot process for preparing purified (3R)-N-[(1S)-1- carbamoylpropyl]-3-(hydroxymethyl) hexanamide [Intermediate 7] from Intermediate 3 (80% ee) as developed in step 1 of scheme A of the present invention.
Procedure:
In the first step of scheme A of the present invention, a mixture of (R)/(S)-4-propyldihydrofuran-2(3H)-one (Intermediate 3, R: S isomer = 80: 20) (1 eq), (S)-2-aminobutanamide (1.1 eq), triethylamine (1.5 eq) is refluxed at a temperature of 95±5 °C for 24h. The mixture is then cooled to 60-65 °C, washed with a mixture of dichloromethane and diisopropyl ether (2.5 vol) in order to get Intermediate-7 [(3R)-N-[(1S)-1-carbamoyl-propyl]-3-(hydroxymethyl) hexanamide] (80% yield).
Results:
Formation of Intermediate 7 is confirmed further by following analytical studies: a) The 1H NMR analysis is conducted and the data as illustrated in accompanying figure 1 depicts: (400 MHz, DMSO-d6): δ 0.6 (t, J= Hz, 6H), 1.07-1.18 (m, 1H), 1.21-1.35 (m, 3H), 1.45-1.43 (m, 1H), 1.61-1.72 (m, 1H), 1.75-1.90 (m, 1H), 2.03 (dd, J=6.64 & 14.08 Hz, 1H), 2.18 (dd, J=7.0 & 14.08 Hz, 1H), 3.28 (t, J=5.36 Hz, 2H), 4.07-4.18 (m, 1H), 4.43 (t, J=5.2 Hz, 1H), 6.95 (s, 1H), 7.28 (s, 1H), 7.76 (d, J=8.08 Hz, 1H).; thus, confirming formation of Intermediate 7 of the present invention.
b) The LCMS analysis is further conducted and the data as graphically illustrated in accompanying figure 2 provides a (M+H+) value of 231.0; thus, confirming formation of Intermediate 7 of the present invention.
c) The HPLC study is also conducted and the data as graphically illustrated in accompanying figure 3 confirms formation of Intermediate 7 of the present invention with chiral purity of 97.38%
EXAMPLE 2: Preparation of (3R)-N-(1S)-1-amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide [Intermediate 8A of scheme A of the present invention]: Example 2 illustrates a process for preparing (3R)-N-(1S)-1-Amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide [Intermediate 8A] from Intermediate 7 of example 1 above as developed in the present invention.
Procedure:
In second step of scheme A of the present invention, the said Intermediate 7 of example 1 above that is (3R)-N-(1S)-1-Amino-1-oxobutan-2-yl)-3-(hydroxymethyl) hexanamide (~98% Chemical purity and ~97% Chiral purity) (1736.86 mmol) is dissolved in DCM (1.2 L) at RT into a RBF under N2 atm. Then the solution is cooled to 10-20 C and Oxaloyl chloride (2605.29 mmol) is added to this cooled solution at 10-20 °C. The mixture is stirred for 24 h at 25-40 °C under N2 atm. Completion of the reaction is monitored by TLC. After completion of reaction, the solvent is distilled off and the residual mass is diluted with water (6 L), stirred at 30-50 °C for 4 h. Slurry mass is then filtered and washed with water (2×400 mL) followed by MTBE (800 mL). The solid is dried under vacuum at 50-55 °C for 4-5 h to afford Intermediate 8A that is (3R)-N-(1S)-1-Amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide as a white solid (92% yield).
Results:
Formation of Intermediate 8A is confirmed further by following analytical studies: a) The 1H NMR analysis is conducted and the data as illustrated in accompanying figure 4 depicts: 1H NMR (400 MHz, DMSO-d6) : δ 0.83 (t, J=7.44 Hz, 3H), 0.85 (t, J=6.72 Hz, 3H), 1.20-1.40 (m, 4H), 1.43-1.56 (m, 1H), 2.08-2.18 (m, 1H), 2.20-2.28 (m, 2H), 3.61 (dd, J=4.6 & 10.8 Hz, 1 H), 3.67 (dd, J=4.6 & 10.8 Hz, 1H), 4.07-4.18 (m, 1H), 6.95 (s, 1H), 7.29 (s, 1H), 7.89 (d, J=8.12 Hz, 1H); thus confirming formation of Intermediate 8A of the present invention.
b) The LCMS analysis is further conducted and the data as graphically illustrated in accompanying figure 5 (a, b) provides a (M+H+) value of 249.20; thus, confirming formation of Intermediate 8A of the present invention.
EXAMPLE 3: Process for purification of Intermediate 8A forming Intermediate 8B Example 3 illustrates a process for purifying the said Intermediate 8A of example 2 above of the present invention.
Procedure:
The Intermediate 8A as obtained in example 2 above [that is (3R)-N-(1S)-1-Amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide] is first dissolved in a polar solvent like Acetonitrile raising the temperature to 50 to 60 °C; followed by stirring and then addition of another solvent methyl tert-butyl ether (MTBE) which is less polar in nature. The mixture is then cooled down to 0°C, the filtered mass thus obtained is dried in order to obtain a white solid of Intermediate 8B. The material thus obtained is further dissolved in THF (5 vol) at 60 °C, cooled to 20-30°C, followed by addition of heptane (5 vol), stirred at 10°C to 30 °C for 1 h. The mass obtained is filtered and washed with heptane (2×1 vol), dried under vacuum at 50-55°C in order to afford formation of purer form of Intermediate 8A that is Intermediate 8B that is (3R)-N-(1S)-1-amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide as a white solid having a chemical purity of 99.9% along with a chiral purity of 100% (yield: 390 g).
Results:
The purification of Intermediate 8A is further confirmed by the following analytical test results:
a) Chiral HPLC: A Chiral HPLC as illustrated in accompanying figure 6 confirmed formation of purest form of Intermediate 8B having 100% chiral purity [Peak 1; RT (min) = 6.244; %Area=100%].
b) GLP-HPLC: A GLP-HPLC as illustrated in accompanying figure 7 further confirmed formation of Intermediate 8B having 99.9% chemical purity [Peak 3; BRIV8; RT = 29.278; % Area=99.90%].
EXAMPLE 4: Synthesis of (3R)-N-[(1S)-1-carbamoylpropyl]-3-(hydroxymethyl) hexanamide [Intermediate 7’ of scheme B of the present invention]
Example 4 illustrates one pot process for preparing purified (3R)-N-[(1S)-1-carbamoylpropyl]-3-(hydroxymethyl) hexanamide [Intermediate 7’] from Intermediate 6 (99.99% ee) as developed in step-1 scheme B of the present invention.
Procedure:
In another method, in the first step of scheme B of the present invention, a mixture of (R)/(S)-4-propyldihydrofuran-2(3H)-one (Intermediate 6: S isomer = 99.99% : 0.1%) (1 eq), (S)-2-aminobutanamide (1.7 eq), triethylamine (5 eq) is refluxed at a temperature between 95±5 °C for 24 h. Then, the crude reaction mass is cooled and washed with dichloromethane and diisopropyl ether mixture (2.5 vol) in order to achieve Intermediate-7’ of scheme B of the present invention [(3R)-N-[(1S)-1-carbamoylpropyl]-3-(hydroxymethyl) hexanamide] (90% yield).
Results:
The chiral purity of the formed Intermediate 7’ is analyzed by HPLC method and the data as graphically illustrated in accompanying figure 8 confirms formation of Intermediate 7’ of the present invention with chiral purity of 99.11%
EXAMPLE 5: Preparation of (3R)-N-(1S)-1-amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide [Intermediate 8’ of scheme B of present invention]: Example 5 illustrates a process for preparing purest form of (3R)-N-(1S)-1-amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide [Intermediate 8’] from Intermediate 7’ of example 4 as developed in scheme B (step 2) of the present invention.
Procedure:
In the second step of scheme B of the present invention, the intermediate 7’ of the above example 4 that is (3R)-N-(1S)-1-amino-1-oxobutan-2-yl)-3-(hydroxymethyl) hexanamide (98% Chemical purity and >99%Chiral purity) (1736.86 mmol) is dissolved in DCM (1.2 L) at RT in a round bottomed flask under N2 atm. Then the solution is cooled to 10-30 °C and 1-Chloro-N,N,2-trimethyl-1-propenylamine (2605.29 mmol) is added to this cooled solution at 10-30 °C. The mixture is stirred for 24 h at 25-40 °C under N2 atm. Completion of the reaction is monitored by TLC. After completion of the reaction, the solvent is distilled off and the residual mass is diluted with water (6 L), stirring at 30-50 °C for 4 h. The slurry mass thus obtained is then filtered and washed with water (2×400 mL) followed by methyl tert-buty ether (MTBE) (800 mL). The solid thus obtained is dried under vacuum at 50-55 °C for 4-5 h in order to afford formation of Intermediate 8’ that is (3R)-N-(1S)-1-amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide as a white solid (92% yield).
Results:
Formation of Intermediate 8’ is confirmed further by following analytical studies: a) The 1H NMR analysis is conducted and the data as illustrated in accompanying figure 9 depicts: (400 MHz, DMSO-d6): 1H NMR (400 MHz, DMSO-d6) : δ 0.83 (t, J=7.44 Hz, 3H), 0.85 (t, J=6.72 Hz, 3H), 1.20-1.40 (m, 4H), 1.43-1.56 (m, 1H), 2.08-2.18 (m, 1H), 2.20-2.28 (m, 2H), 3.61 (dd, J=4.6 & 10.8 Hz, 1 H), 3.67 (dd, J=4.6 & 10.8 Hz, 1H), 4.07-4.18 (m, 1H), 6.95 (s, 1H), 7.29 (s, 1H), 7.89 (d, J=8.12 Hz, 1H); thus, confirming formation of Intermediate 8’ of the present invention.
b) The LCMS analysis is further conducted and the data as graphically illustrated in accompanying figure 10 provides a (M+H+) value of 249.1; thus, confirming formation of Intermediate 8’ of the present invention.
c) The HPLC data as illustrated in accompanying figure 11 confirms 100% chiral purity of Intermediate 8’.
EXAMPLE 6: Preparation of (2S)-2-[(4R)-2-oxo-4-propyl-pyrrolidin-1-yl] butanamide [Brivaracetam-API]:
Example 6 illustrates a process for preparing (2S)-2-[(4R)-2-oxo-4-propyl-pyrrolidin-1-yl] butanamide [Brivaracetam API] from Intermediate 8B of example 3 or from Intermediate 8’ of example 5 as developed in step 3 of scheme A or scheme B of the present invention respectively.
Procedure:
In the final step of scheme A or scheme B of the present invention, the intermediate 8B of example 3 or intermediate 8’ of example 5 above that is (3R)-N-((1S)-1-Amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide (1608.04 mmol) is dissolved in dimethyl acetamide (0.5 vol) and isopropylacetate (2 L) into a RBF at 25-30 °C under N2 atm. Then 18-Crown-6 (160.79 mmol) is added into the solution and stirred at RT for 30 min. Reaction mixture is then cooled to 0-10 °C and t-BuOK (1.5 eq) is added portion wise to the cooled solution over 1 h maintaining the temperature from – 0-10 °C to 25 °C under N2 atm. Stirring is then continued for 2 h at -10 °C to 0 °C and then for 12 h at 15-25 °C under N2 atm. Completion of reaction is monitored by TLC. After completion of reaction, the reaction mixture is quenched with addition of 1M HCl solution (pH~6.5-7.0). The resulting mixture is extracted with i-PrOAc (2 L) and MTBE (1 L). Water (0.5 L) is added to the combined organic extract and then filtered through celite bed, washed the bed with MTBE-i-PrOAc (1:1) (400 mL). The organic part is separated and the aqueous part is re-extracted with i-PrOAc-MTBE (1:1) (2 ×0.8 L). The combined organic phases are washed with brine solution (100 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuum under a rotary evaporator to afford crude API. Distillation of dimethylacetamide solvent from the crude is then done at high vacuum pressure (0.05 mm Hg) at 70 °C. Crude product is then dissolved in isopropyl acetate (1.6 L) and treated with activated charcoal (7% w/w) to afford a tech-grade crude of Brivaracetam API as a white solid (yield: 90%) with 97.82% chemical purity.
Results:
Formation of Brivaracetam API is confirmed further by following analytical studies: a) The 1H NMR analysis is conducted and the data as illustrated in accompanying figure 12 depicts: 1H NMR (400 MHz, DMSO-d6) : δ 0.77 (t, J=7.32 Hz, 3H), 0.87 (t, J=7.2 Hz, 3H), 1.21-1.31 (m, 2H), 1.33-1.43 (m, 2H), 1.50-1.62 (m, 1H), 1.73-1.84 (m, 1H), 1.97 (dd, J=8.0 & 16.12 Hz, 1H), 2.18-2.28 (m, 1H), 2.37 (dd, J=8.4 & 16.14 Hz, 1H), 3.11 (dd, J=7.16 & 9.44 Hz, 1H), 3.36 (dd, J=9.2 & 17.5 Hz, 1H), 4.30 dd, J=5.44 & 10.28 Hz, 1H), 6.98 (s, 1H), 7.32 (s, 1H); thus, confirming formation of Brivaracetam API of the present invention.
b) The LCMS analysis is further conducted and the data as graphically illustrated in accompanying figure 13 provides a (M+H+) value of 213.0; thus, confirming formation of Brivaracetam API of the present invention.
^ Purification of Brivaracetam API:
The Brivaracetam thus formed above is further purified by means of dissolving the said material (307 g) in 30% i-PrOAc -MTBE (1 vol) at 55-60 °C, cool to 20-30°C. A mixture of Heptane and MTBE and DIPE (2:2:1) is added, stirred at 10 °C to 30°C for 1 h. The obtained mass is filtered and washed with heptane, which is subsequently dried under vacuum at 40-45 °C to afford (3R)-N-((1S)-1-amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide as a white solid (yield: 80%, chiral purity 99.93% and chemical purity 99.94%).
Results:
a) Chiral HPLC: A Chiral HPLC as illustrated in accompanying figure 14 confirmed formation of purest form of Brivaracetam API having 99.93% chiral purity [Peak 2; RT (min) = 9.45; %Area=99.93%].
b) GLP-HPLC: A GLP-HPLC as illustrated in accompanying figure 15 further confirmed formation of Brivaracetam API having 99.9% chemical purity [Peak 2; RT = 21.138; % Area=99.94%].