
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
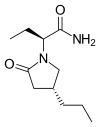
BRIVARACETAM, UCB-34714
(2S)-2-[(4R)-2-oxo-4-propylpyrrolidin-1-yl]butanamide
| Molecular Formula: | C11H20N2O2 |
|---|---|
| Molecular Weight: | 212.2887 g/mol |
UNII-U863JGG2IA
UCB; For the treatment of partial onset seizures related to epilepsy, Approved February 2016
Brivaracetam, the 4-n-propyl analog of levetiracetam, is a racetam derivative with anticonvulsant properties.[1][2] Brivaracetam is believed to act by binding to the ubiquitous synaptic vesicle glycoprotein 2A (SV2A).[3] Phase II clinical trials in adult patients with refractory partial seizures were promising. Positive preliminary results from stage III trials have been recorded,[4][5] along with evidence that it is around 10 times more potent[6] for the prevention of certain types of seizure in mouse models than levetiracetam, of which it is an analogue.
On 14 January 2016, the European Commission,[7] and on 18 February 2016, the USFDA[8] approved brivaracetam under the trade name Briviact (by UCB). The launch of this anti-epileptic is scheduled for the first quarter of that year. Currently, brivaracetam is still not approved in other countries like Australia, Canada and Switzerland.
Brivaracetam was approved by European Medicine Agency (EMA) on Jan 14, 2016 and approved by the U.S. Food and Drug Administration (FDA) on Feb 18, 2016. It was developed and marketed as Briviact® by UCB in EU/US.
Brivaracetam is a selective high-affinity synaptic vesicle protein 2A ligand, as an adjunctive therapy in the treatment of partial-onset seizures with or without secondary generalization in adult and adolescent patients from 16 years of age with epilepsy.
Briviact® is available in three formulations, including film-coated tablets, oral solution and solution for injection/infusion. And it will be available as 10 mg, 25 mg, 50 mg, 75 mg and 100 mg film-coated tablets, a 10 mg/ml oral solution, and a 10 mg/ml solution for injection/infusion. The recommended starting dose is either 25 mg twice a day or 50 mg twice a day, depending on the patient’s condition. The dose can then be adjusted according to the patient’s needs up to a maximum of 100 mg twice a day. Briviact can be given by injection or by infusion (drip) into a vein if it cannot be given by mouth.
European Patent No. 0 162 036 Bl discloses compound (S)-α-ethyl-2-oxo-l- pyrrolidine acetamide, which is known under the International Non-proprietary Name of Levetiracetam.

Levetiracetam
Levetiracetam is disclosed as a protective agent for the treatment and prevention of hypoxic and ischemic type aggressions of the central nervous system in European patent EP 0 162 036 Bl. This compound is also effective in the treatment of epilepsy.
The preparation of Levetiracetam has been disclosed in European Patent No. 0 162 036 and in British Patent No. 2 225 322.
International patent application having publication number WO 01/62726 discloses 2-oxo-l -pyrrolidine derivatives and methods for their preparation. It particularly discloses compound (2S)-2-[(4R)-2-oxo-4-propyl-pyrrolidin-l-yl] butanamide known under the international non propriety name of brivaracetam.

Brivaracetam
International patent application having publication number WO 2005/121082 describes a process of preparation of 2-oxo-l -pyrrolidine derivatives and particularly discloses a process of preparation of (2S)-2-[(4S)-4-(2,2-difluorovinyl)-2-oxo-pyrrolidin-l- yl]butanamide known under the international non propriety name of seletracetam.

Seletracetam
Kenda et al., in J. Med. Chem. 2004, 47, 530-549, describe processes of preparation of 2-oxo-l -pyrrolidine derivatives and particularly discloses compound 1-((1S)-I- carbamoyl-propyl)-2-oxo-pyrrolidone-3-carboxylic acid as a synthetic intermediate.
WO2005028435
CLIPS
Find better ways to make old and new epilepsy drugs. J. Surtees and co-inventors disclose alternative processes for making active pharmaceutical ingredients (APIs) that are used to treat epilepsy and seizures. One compound that can be prepared by their processes is the established drug levetiracetam (1, Figure 1), marketed under the trade name Keppra. Because 1 is now off-patent, there is obvious interest in new drugs.

The inventors also claim that seletracetam (2) and brivaracetam (3) (Figure 2) can be prepared by their processes. These drugs are apparently much more active than 1.
All of the drugs are used as single isomers, so a stereoselective synthesis is desirable. The inventors describe two routes for preparing the molecules; the first, shown in Figure 1, is the synthesis of 1 by the reaction between pyrrolidone (4) and chiral bromo amide 5 in the presence of a base. GC analysis showed that the conversion is 40.3% and that the product contains 51% of the (S)-enantiomer and 49% of the (R)-isomer. No details of their separation are given, although the use of chiral HPLC is discussed.
The same reaction is used to prepare derivative 6 of 1. Compound 7 is prepared from the corresponding hydroxy ester and then condensed with 4 to give 6. Chiral HPLC showed that the product is a mixture of 89.3% (S)-enantiomer 6 and 10.7% of its (R)-isomer.
The inventors do not describe the detailed preparation of 2, but they report that acid 8 is prepared in 41% yield from pyrrolidone 9 and acid 10 in the presence of NaH (Figure 2). Ammonolysis of 8 produces 2; no reaction details are provided.

In a reaction similar to the preparation of 8, acid 11 is prepared from 10 and pyrrolidone 12. The product is isolated in 77% yield and can be converted to 3 by ammonolysis. Again, no details are provided for this reaction.
The second route for preparing the substituted pyrrolidones does not start with simple pyrrolidones and is the subject of additional claims. The route involves a cyclization reaction, shown in Figure 3. The preparation of enantiomer 13 begins with the reaction of racemic salt 14 and optically pure bromo ester 15. This step produces intermediate 16, isolated as a yellow oil. The crude material is treated with 2-hydroxypyridine (2-HP) to cyclize it to 17. This ester is hydrolyzed to give acid 18. Conversion to 13 is carried out by adding ClCO2Et, followed by reaction with liquid NH3 in the presence of K2CO3. The overall yield of 13 is 32%.

This route is also used to prepare levetiracetam (1) by treating 5 with the HCl salt of amino ester 19 to give 20, recovered as its HCl salt in 49% yield. The salt is basified with Et3N and treated with 2-HP to cyclize it to 1, initially isolated as an oil. GC analysis showed 100% conversion, and chiral HPLC showed that the product contains 98.6% (S)-isomer and 1.4% (R)-isomer.
The inventors also prepared 1 and its (R)-enantiomer 21 by using a similar reaction scheme with alternative substrates to 5. Figure 4 outlines the route, which starts from protected hydroxy amide 22 and amino ester 23. When the reaction is carried out in the presence of Cs2CO3, the product is (R)-enantiomer24, which is used without purification to prepare 21 by treating it with 2-HP. Chiral HPLC showed that the product is 94% (R) and 6% (S).

When the reaction between 22 and 23 is run with K2CO3, the product is (S)-enantiomer 25. This is used to prepare 1, but the product contains only 79% (S)-isomer.
The inventors do not comment on the apparent stereoselectivity of the carbonate salts in the reaction of 22 with 23. This is an intriguing finding and worthy of investigation. (UCB S.A. [Brussels]. US Patent 8,338,621

SYNTHESIS
PATENT
WO2005028435
Example 1: Synthesis of (2S)-2-((4R)-2-oxo-4-n-propyl-l-pyrrolidinyl)butanamide 1.1 Synthesis of (2S)-2-aminobutyramide free base

1800 ml of isopropanol are introduced in a 5L reactor. 1800 g of (2S)-2- aminobutyramide tartrate are added under stirring at room temperature. 700 ml of a 25% aqueous solution of ammonium hydroxide are slowly added while maintaining the temperature below 25°C. The mixture is stirred for an additional 3 hours and then the reaction is allowed to complete at 18°C for 1 hour. The ammonium tartrate is filtered. Yield : 86%.
1.2 Synthesis of 5-hydroxy-4-n-propyl-furan-2-one

Heptane (394 ml) and morpholine (127.5 ml) are introduced in a reactor. The mixture is cooled to 0°C and glyoxylic acid (195 g, 150 ml, 50w% in water) is added. The mixture is heated at 20°C during 1 hour, and then valeraldehyde (148.8 ml) is added . The reaction mixture is heated at 43°C during 20 hours. After cooling down to 20CC, a 37 % aqueous solution of HCl (196.9 ml) is slowly added to the mixture, which is then stirred during 2 hours.
After removal of the heptane phase, the aqueous phase is washed three times with heptane. Diisopropyl ether is added to the aqueous phase. The organic phase is removed, and the aqueous phase further extracted with diisopropyl ether (2x). The diisopropyl ether phases are combined, washed with brine and then dried by azeotropic distillation. After filtration and evaporation of the solvent, 170g of 5- hydroxy-4-n-propyl-furan-2-one are obtained as a brown oil. Yield: 90.8 %
1.3 Synthesis of (2S)-2-((4R)-2-oxo-4-n-propyl-l-pyrrolidinyl)butanamide and (2S)-2-((4S)-2-oxo-4-n-propyl-l-pyrrolidinyl)butanamide

(S, R) (S, S) The (2S)-2-aιninobutyrarnide solution in isopropanol containing 250 g obtained as described here above is dried by azeotropic distillation under vacuum. To the dried (2S)-2-am obutyraιnide solution is added 5-hydroxy-4-n-propyl-furan-2-one (290 g) between 15°C and 25 °C; the mixture is heated to 30 °C and kept for at least 2 hours at that temperature. Acetic acid (1, 18 eq.), Pd/C catalyst (5 w/w%; Johnson Matthey 5% Pd on carbon – type 87L) are then added and hydrogen introduced into the system under pressure. The temperature is kept at 40 °C maximum and the H2 pressure maintained between 0,2 bar and 0,5 bar followed by stirring for at least 20 hours following the initial reaction. The solution is then cooled to between 15 °C and 25 °C and filtered to remove the catalyst. The solution of product in isopropanol is solvent switched to a solution of product in isopropyl acetate by azeotropic distillation with isopropyl acetate. The organic solution is washed with aqueous sodium bicarbonate followed by a brine wash and then filtered. After recristallisation, 349 g of (2S)-2-((4R)-2- oxo-4-n-propyl-l-pyrrolidinyl)butanamide and (2S)-2-((4S)-2-oxo-4-n-propyl-l- pyιτolidinyl)butanamide are obtained (Yield: 82.5%).
1.4 Preparation of (2S)-2-((4R)-2-oxo-4-n-propyl-l-pyrrolidinyl)butanamide The chromatographic separation of the two diastereoisomers obtained in 1.3 is performed using of (CHIRALPAK AD 20 um) chiral stationary phase and a 45/55 (volume /volume) mixture of n-heptane and ethanol as eluent at a temperature of 25 + 2°C. The crude (2S)-2-((4R)-2-oxo-4-n-propyl-l-pyrrolidinyl)butanamide thus obtained is recristallised in isopropylacetate, yielding pure (2S)-2-((4R)-2-oxo-4-n-propyl-l- pyrrolidinyl)butanamide (Overall yield: 80%) .
Example 2: Synthesis of (2S)-2-((4R)-2-oxo-4-n-propyl-l-pyrrolidinyl)butanamide

Example 1 is repeated except that in step 1.1 a solution of (2S)-2- aminoburyramide.HCl in isopropanol is used (27.72 g, 1.2 equivalent), which is neutralised with a NHs/isopropanol solution (3,4-3,7 mol/L). The resulting ainmonium chloride is removed from this solution by filtration and the solution is directly used for reaction -with 5-hydroxy-4-n-propyl-furan-2-one (23.62 g, 1.0 equivalent) without intermediate drying of the (2S)-2-aminobutyramide solution. Yield after separation of the two diastereoisomers and recristallisation: approximately 84%.

Ref ROUTE1
1. WO0162726A2.
2. WO2005028435A1 / US2007100150A1.
3. J. Med. Chem. 1988, 31, 893-897.
4. J. Org. Chem. 1981, 46, 4889-4894.
PATENT
https://www.google.com/patents/WO2007031263A1?cl=en
Example 3-Synthesis of brivaracetam (I)
3.a. Synthesis of (S) and (R) 2-((R)-2-oxo-4-propyl-pyrrolidin-l-yl)-butyric acid methyl ester fVIaa*) and (Wlab)

(VIaa) (VIab) A slurry of 60% sodium hydride suspension in mineral oil (0.94g, 23.4 mmol) in tetrahydrofuran (30 mL) is cooled at 0°C under a nitrogen atmosphere. A solution of substantially optically pure (R)-4-propyl-pyrrolidin-2-one (Ilia) (2g, 15.7 mmol) in tetrahydrofuran (2 mL) is added over a 15 minutes period. The reaction mixture is stirred 10 min at 0°C then a solution of methyl-2-bromo-butyric acid methyl ester (V) (3.69g, 20.4 mmol) in tetrahydrofuran (2mL) was added over a 20 minutes period. The reaction mixture is stirred at O0C until maximum conversion of starting material and the reaction mixture is then allowed to warm to room temperature and diluted with water (20 mL). Tetrahydrofuran is removed by evaporation and the residue is extracted with isopropyl acetate (20 ml + 10 mL). The combined organic layers are dried on anhydrous magnesium sulfate and evaporated to afford 3g (13.2 mmol, 86 %) of a mixture of epimers of compound (Via), as a mixture respectively of epimer (VIaa) and epimer (VIab). 1H NMR(400 MHz, CDCI3) of the mixture of epimers (VIaa) and (VIab) : δ = 4.68
(dd, J= 10.8, J= 5.1, 2×1 H) ; 3.71 (s, 2x3H); 3.60 (t app, J= 8.2, IH); 3.42 (t app, J= 8.7, IH); 313 (dd, J= 9.2, J = 6.8, IH); 2.95 (dd, J= 9.2, J= 6.8, IH); 2.56 (dd, J= 16.6, J = 8.7, 2xlH); 2.37 (dm, 2xlH); 2.10 (m, 2xlH); 2.00 (m, 2xlH); 1.68 (m, 2xlH); 1.46 (m, 2x2H); 1.36 (m, 2x2H); 0.92 (m, 2x6H).
13C NMR (400 MHz, CDCl3) of the mixture of epimers (VIaa) and (VIab) : δ =
175.9; 175.2; 171.9; 55.3; 52.4; 49.8; 49.5; 38.0; 37.8; 37.3; 36.9; 32.5; 32.2; 22.6; 22.4; 21.0; 14.4; 11.2; 11.1
HPLC (GRAD 90/10) of the mixture of epimers (VIaa) and (VIab): retention time= 9.84 minutes (100 %)
GC of the mixture of epimers (VIaa) and (VIab): retention time = 13.33 minutes (98.9 %)
MS of the mixture of epimers (VIaa) and (VIab) (ESI) : 228 MH+
3.b. Ammonolysis of compound of the mixture of (VIaa) and (VIab)

(VIaa) (VIab) (I) (VII)
A solution of (VIaa) and (VIab) obtained in previous reaction step (1.46g, 6.4 mmol) in aqueous ammonia 50 % w/w (18 mL) at 00C is stirred at room temperature for 5.5hours. A white precipitate that appears during the reaction, is filtered off, is washed with water and is dried to give 0.77g (3.6 mmol, yield = 56 %) of white solid which is a mixture of brivaracetam (I) and of compound (VII) in a 1 :1 ratio.
1H NMR of the mixture (I) and (VII) (400 MHz, CDCI3) : δ = 6.36 (s, broad, IH); 5.66 (s, broad, IH); 4.45 (m, IH); 3.53 (ddd, J= 28.8, J= 9.7, J= 8.1, IH); 3.02 (m, IH); 2.55 (m, IH); 2.35 (m, IH); 2.11 (m, IH); 1.96 (m, IH); 1.68 (m, IH); 1.38 (dm, 4H); 0.92 (m, 6H). 13c NMR of the mixture (I) and (VII) (400 MHz, CDCl3) : δ = 176.0; 175.9; 172.8;
172.5; 56.4; 56.3; 50.0; 49.9; 38.3; 38.1; 37.3; 37.0; 32.3; 32.2; 21.4; 21.3; 21.0; 20.9; 14.4; 10.9; 10.8
HPLC (GRAD 90/10) of the mixture of (I) and (VII) retention time= 7.67 minutes (100 %)
Melting point of the mixture of (I) and (VII) = 104.90C (heat from 400C to 1200C at 10°C/min)
Compounds (I) and (VII) are separated according to conventional techniques known to the skilled person in the art. A typical preparative separation is performed on a 11.7g scale of a 1 :1 mixture of compounds (I) and (VII) : DAICEL CHIRALPAK® AD 20 μm, 100*500 mm column at 300C with a 300 mL/minutes debit, 50 % EtOH – 50 % Heptane. The separation affords 5.28g (45 %) of compound (VII), retention time = 14 minutes and 5.2Og (44 %) of compounds (I), retention time = 23 minutes.
1H NMR of compound (I) (400 MHz, CDCl3): δ = 6.17 (s, broad, IH); 5.32 (s, broad, IH); 4.43 (dd, J= 8.6, J= 7.1, IH); 3.49 (dd, J= 9.8, J= 8.1, IH); 3.01 (dd, J= 9.8, J= 7.1, IH); 2.59 (dd, J= 16.8, J= 8.7, IH); 2.34 (m, IH); 2.08 (dd, J= 16.8, J= 7.9, IH); 1.95 (m, IH); 1.70 (m, IH); 1.47-1.28 (m, 4H); 0.91 (dt, J= 7.2, J= 2.1, 6H)
HPLC (GRAD 90/10) of compound (I) : retention time = 7.78 minutes
1H NMR of compound (VII) (400 MHz, CDCl3): δ = 6.14 (s, broad, IH); 5.27 (s, broad, IH); 4.43 (t app, J = 8.1, IH); 3.53 (t app, J = 9.1, IH); 3.01 (t app, J = 7.8, IH); 2.53 (dd, J = 16.5, J = 8.8, IH); 2.36 (m, IH); 2.14 (dd, J = 16.5, J = 8.1, IH); 1.97 (m, IH); 1.68 (m, IH); 1.43 (m, 2H); 1.34 (m, 2H); 0.92 (m, 6H)
3c. Epimerisation of compound of (2RV2-((R)-2-oxo-4-propyl-pyπOlidin-l-ylV butyramide (VID
Compound (VII) (200 mg, 0.94 mmol) is added to a solution of sodium tert- butoxide (20 mg, 10 % w/w) in isopropanol (2 mL) at room temperature. The reaction mixture is stirred at room temperature for 18h. The solvent is evaporated to afford 200 mg
(0.94 mmol, 100 %) of a white solid. Said white solid is a mixture of brivaracetam (I) and of (VII) in a ratio 49.3 / 50.7.
HPLC (ISO80): retention time= 7.45 min (49.3%) brivaracetam (I); retention time= 8.02 minutes (50.7%) compound (VII).

Reference:ROUTE 2
1. WO2007031263A1 / US2009318708A1.
PATENT
http://www.google.com/patents/WO2007065634A1?cl=en
(scheme 3).

Scheme 3
scheme 4.

5h. Synthesis of brivaracetam and (V) A suspension of (Id) and (Ie) (0.6 g, 2.3 mmol) in MIBK (10 mL) is heated at
120°C for 6 hours. The resulting solution is concentrated and separated on chromatography column (Silicagel 600.068-0.200 mm, cyclohexane/EtOAc : 10/90) to give 0.13 g of brivaracetam (0.6 mmol, 26 %, ee = 94 %) and (V).
1H NMR (400 MHz, CDCl3): δ = 6.17 (s, broad, IH); 5.32 (s, broad, IH); 4.43 (dd, J= 8.6, J= 7.1, IH); 3.49 (dd, J= 9.8, J= 8.1, IH); 3.01 (dd, J= 9.8, J= 7.1, IH); 2.59 (dd, J= 16.8, J= 8.7, IH); 2.34 (m, IH); 2.08 (dd, J= 16.8, J= 7.9, IH); 1.95 (m, IH); 1.70 (m, IH); 1.47-1.28 (m, 4H); 0.91 (dt, J= 7.2,J= 2.1, 6H).
HPLC (method 90/10) : Retention time = 7.78 minutes Chiral HPLC : Retention time = 9.66 minutes (97%) MS (ESI): 213 MH+

Route 3
Reference:1. WO2007065634A1 / US2009012313A1.
PAPER
http://pubs.acs.org/doi/abs/10.1021/acs.oprd.6b00094
A Biocatalytic Route to the Novel Antiepileptic Drug Brivaracetam


AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
References
- von Rosenstiel P (Jan 2007). “Brivaracetam (UCB 34714)”. Neurotherapeutics 4 (1): 84–7. doi:10.1016/j.nurt.2006.11.004.PMID 17199019.
- Malawska B, Kulig K (Jul 2005). “Brivaracetam UCB”. Current Opinion in Investigational Drugs 6 (7): 740–746. PMID 16044671.
- Rogawski MA, Bazil CW (Jul 2008). “New molecular targets for antiepileptic drugs: alpha(2)delta, SV2A, and K(v)7/KCNQ/M potassium channels”. Current Neurology and Neuroscience Reports 8 (4): 345–352. doi:10.1007/s11910-008-0053-7. PMC 2587091.PMID 18590620.
- Clinical trial number NCT00464269 for “Double-blind, Randomized Study Evaluating the Efficacy and Safety of Brivaracetam in Adults With Partial Onset Seizures” at ClinicalTrials.gov
- Rogawski MA (Aug 2008). “Brivaracetam: a rational drug discovery success story”. British Journal of Pharmacology 154 (8): 1555–7.doi:10.1038/bjp.2008.221. PMC 2518467. PMID 18552880.
- Matagne A, Margineanu DG, Kenda B, Michel P, Klitgaard H (Aug 2008). “Anti-convulsive and anti-epileptic properties of brivaracetam (ucb 34714), a high-affinity ligand for the synaptic vesicle protein, SV2A”. British Journal of Pharmacology 154 (8): 1662.doi:10.1038/bjp.2008.198. PMID 18500360.
- http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/003898/human_med_001945.jsp&mid=WC0b01ac058001d124
- http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm486827.htm
|
|||
| Names | |||
|---|---|---|---|
| IUPAC name
(2S)-2-[(4R)-2-oxo- 4-propylpyrrolidin-1-yl] butanamide
|
|||
| Identifiers | |||
| 357336-20-0 |
|||
| ChEMBL | ChEMBL607400 |
||
| ChemSpider | 8012964 |
||
| Jmol interactive 3D | Image | ||
| PubChem | 9837243 | ||
| UNII | U863JGG2IA |
||
| Properties | |||
| C11H20N2O2 | |||
| Molar mass | 212.15 g/mol | ||
| Pharmacology | |||
| ATC code | N03 | ||
| Legal status |
|
||
| Oral | |||
| Pharmacokinetics: | |||
| Nearly 100% | |||
| <20% | |||
| Hydrolysis, CYP2C8-mediated hydroxylation | |||
| 8 hrs | |||
| >75% renal | |||
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|||


//////BRIVARACETAM, UCB, 2016 FDA, UCB-34714
CCCC1CC(=O)N(C1)C(CC)C(=O)N
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021260721&_cid=P12-KXX1JU-33531-1
Brivaracetam is chemically known as (2S)-2-[(4R)-2-oxo-4-propyltetrahydro-1H-pyrrol-1-yl] butanamide, having the chemical structure of formula 1 as below:
Brivaracetam is basically a chemical analogue of Levetiracetam, marketed under the brand name of BRIVIACT for the treatment as adjunctive therapy in the treatment of partial-onset seizures in patients at 16 years of age and older with epilepsy. Brivaracetam has an advantage over Levetiracetam in that it gets into the brain “much more quickly,” which means that “it could be used for status epilepticus, or acute seizures than cluster, or prolonged seizures”. From the Phase III trials, the self-reported rate of irritability with Brivaracetam was 2% for both drug doses (100 mg and 200 mg) vs 1% for placebo, which compares to as much as 10% for Levetiracetam in some post-marketing studies.
With the improved safety profile and possibility to be used for wider range of epilepsy, Brivaracetam is considered as one of the most promising 3rd generation antiepileptic drugs.
Brivaracetam molecule is first disclosed in patent publication WO2001062726, which describes 2-oxo-1 -pyrrolidine derivatives and methods for their preparation. This patent publication further discloses compound (2S)-2-[(4R)-2-oxo-4-propyl-pyrrolidin-1-yl] butanamide which is known under the international non propriety name as Brivaracetam. As per Biopharmaceutics Classification System, Brivaracetam is a class I drug (high solubility and permeability).
Some prior arts US6784197 and US7629474 disclose a process for synthesizing a diastereomeric mixture of (2S)-2-[(4R)-2-oxo-4-propylpyrrolidin-1-yl]-butanamide and (2S)-2-[(4S)-2-oxo-4-propylpyrrolidin-1-yl]-butanamide (Brivaracetam) which is purified by chiral HPLC (Scheme-I & Scheme-II respectively, as provided below). This process used for chiral resolution makes it difficult for bulk manufacturing as well as it affects the overall yield making the process uneconomical.
Scheme-I
Synthesis of (2S)-2-(2-oxo-4-propyl-1-pyrrolidinyl)butanamide
[As disclosed in columns 37-38 of US 6784197 B2]
Scheme-II
1.1 Synthesis of (2S)-2-aminobutyramide-Free base
1.2 Synthesis of 5-hydroxy-4-n-propylfuran-2-one
1.3 Synthesis of (2S)-2-((4R)-2-oxo-4-n-propyl-1-pyrrolidinyl)butanamide and
(2S)-2-((4S)-2-oxo-4-n-propyl-1-pyrrolidinyl)butanamide
[As disclosed in columns 6-7 of US 7629474 B2]
Moreover, some prior arts such as US7122682B2, US8076493B2, US8338621B2 and US8957226B2 also describe processes for preparing Brivaracetam, wherein, the purifications are reportedly done by chiral HPLC methods resulting into similar shortcomings.
Kenda et al.: Journal of Medicinal Chemistry, 2004, 47, 530-549 further proposes selection of (2S)-2-[(4R)-2-oxo-4-propylpyrrolidin-1-yl]butanamide 83α (ucb 34714; Brivaracetam) as the most interesting candidate showing 10 times more potency than Levetiracetam as an antiseizure agent in audiogenic seizure-prone mice. This article further discloses methods for synthesizing the said compound Brivaracetam. However, here too these compounds are synthesized as mixtures of stereoisomers (racemic or diastereoisomeric mixtures), separated by preparative HPLC on silica gel and/or chiral phases.
All these processes for the preparation of Brivaracetam as described in the above- mentioned prior arts suffer from many disadvantages which includes difficulty in achieving desired chiral purity, tedious and cumbersome work up procedures, high temperature and longer time reaction, multiple crystallizations or isolation steps, use of excess reagents and solvents, column chromatographic separations & purifications etc. All these disadvantages affect the overall yield as well as the quality of the final product Brivaracetam and intermediates produced thereof; further, rendering such processes to be uneconomical and unsuitable for industrial scale-ups.
As a result, enantioselective synthesis of Brivaracetam was perceived to be a possible way of overcoming such problems in view of the large-scale synthesis. However, very few prior arts have been found to report successful reduction of such concept into practice.
WO2016191435A1 (also as IN201717005820A) relates to a process for a scalable synthesis of enantiomerically pure Brivaracetam from an intermediate (4R)-4- Propyldihydrofuran-2(3H)-one (compound IV):
, wherein, R is saturated or unsaturated C1-20 alkyl or C1 alkyl-unsubstituted or substituted aryl, comprising the steps of decarboxylation of
the compound of formula IV
to produce the compound of formula VI
ring-opening of the compound of formula VI to produce the compound of formula VII
, wherein Rl is saturated or unsaturated Cl-20 alkyl or Cl alkyl-unsubstituted or substituted aryl; and X is CI Br I OMs, OTs, ONs; or
the compound of formula X
reacting the compound of formula VI with (S)-2- aminobutanamide or its salt to produce the compound of formula XII in one step; or reacting the compound of formula VI with alkyl (S)-2- aminobutanoate to produce Xll-a
, wherein R in the compound of formula Xll-a is a saturated or unsaturated C1-C20 alkoxyl; and then converting Xll-a to XII that is Brivaracetam by aminolysis and amide formation reaction.
In above mentioned prior arts, the synthesis of chiral lactone which is the key starting material for making Brivaracetam involved Grignard addition, column chromatography and Krapcho decarboxylation techniques at high temperature, all of which are not at all recommendable in view of process perspective at industrial levels. Further the final step of the said reaction often involved cryogenic condition –30°C which is also difficult with respect to industrial scale up activities.
Furthermore, prior art IN201641030239A disclosed a process for the preparation of Brivaracetam of Formula (I) by means of converting enantiomerically pure compound of Formula VII to obtain enantiomerically pure compound of Formula XI:
, wherein X is each independently selected from halogen; alkyl or aryl sulfonyloxy; OR2; R2 is optionally substituted C1-C12 alkyl, aryl, alkyl aryl, aryl alkyl;
such that the said process further comprises steps of:
1) cyclizing compound of formula VII to give enantiomerically pure compound of formula IX:
, wherein R2 is optionally substituted C1-C12 alkyl, aryl, alkyl aryl or aryl alkyl;
2) converting the compound of formula IX to give a enantiomerically pure compound of formula X:
, wherein X is halogen;
3) converting compound of formula X to give a enantiomerically pure compound of formula XI:
wherein X is each independently selected from halogen; alkyl or aryl sulfonyloxy; OR2; R2 as defined above; followed by 4) treating the enantiomerically pure compound of formula XI with (S)- aminobutyramide of formula XII or its salt thereof to obtain Brivaracetam of Formula I.
However, this process suffered from drawbacks of handling acid chloride. Acid chlorides are unstable and storing a large amount of acid chloride is also not recommendable in view of safety and stability in industries. Moreover, this prior art process involves use of HBr in acetic acid, where HBr liberates Br that is hazardous and not recommendable for industrial scale-up activities in view of safety and handling.
Some other prior arts such as CN108503573A, CN105646319A, CN106588740A, CN106588831A, CN108689903B and CN108929289A report various processes of synthesizing Brivaracetam from its lactone intermediate by various ring opening techniques. Among these, in particular CN108929289A discloses a process of reacting a compound represented by the formula IV with (S) -2-aminobutyramide in order to obtain Brivaracetam. The synthetic route is as follows:
Also, CN108689903B relates to a new preparation method of Brivaracetam that comprises steps of: a) subjecting a compound of formula III and (S) -2-aminobutanamide or salt thereof to condensation reaction, in the presence of a condensing agent, in order to obtain a compound shown in a formula IV, wherein the compound has two chiral centres; b) removal of the hydroxy-protecting group R1 to obtain a compound of formula V; and c) carrying out chlorination reaction on the compound shown in the formula V using a chlorination reagent to obtain a compound shown in the formula VI; and d) carrying out substitution reaction on the compound shown in the formula VI in the presence of an
alkaline reagent, and closing a ring to obtain Brivaracetam of formula I having two chiral centres.
It has further been noted that although the above reaction goes through formation of intermediates V and/or VI; however, these intermediates are not essentially formed from the key lactone intermediate of Brivaracetam that is (4R)-4-propyldihydrofuran-2(3H)-one [or (R)-lactone].
Furthermore, CN111196771A relates to a preparation method of Brivaracetam which comprises the steps of: 1) carrying out ring-opening reaction on a compound R-4-propyldihydrofuran-2-ketone in a formula II and a compound S-2-aminobutanamide in a formula III to obtain an intermediate compound in a formula I; 2) condensing the said intermediate compound of formula I is followed by cyclization to produce Brivaracetam
However, the ring-opening reaction in step 1 of this process essentially occurs under acidic conditions, specifically in presence of Lewis acids like tetra-isopropyl titanate, anhydrous aluminium trichloride, anhydrous zinc chloride, boron trifluoride diethyl etherate etc.; and also in presence of organic solvents chosen from one or more of anhydrous tetrahydrofuran, 2-methyltetrahydrofuran, acetone, dimethyl sulfoxide and N, N-dimethylformamide; which makes this process both industrially non-scalable and environmentally unfriendly.
The prior art Org. Process Res. Dev.2016. v 20. no 9. p 1566-1575 in its scheme 4, on page 17 also discloses a scheme for synthesizing Brivaracetam from its lactone intermediate:
Nevertheless, it has been noted that the process reported in this prior art provides Brivaracetam (API) with a very poor yield of ~30% and also having an inferior chiral purity of 95.9% ee, which does not even meet the ICH-specification for the Finished Product (API).
Furthermore, a recently filed patent application WO2020148787A1 (also as IN201931002041) recites a new, improved and economical process for enantioselective synthesis and purification of a key intermediate of Brivaracetam that is the R-lactone, essentially utilizing a low chiral loading and without involving any chiral chromatographic resolution technique. Even though this prior art also discloses a process for the preparation of a chirally pure Brivaracetam of formula I utilizing the said intermediate; however, that process is mostly a conventional one.
Accordingly, there is still a need in the art for a more economical and improved process for the synthesis of Brivaracetam with better purity and yield which overcomes the drawbacks of above prior arts.
Therefore, the present inventors have developed a cost effective, novel and efficient process for the preparation of Brivaracetam which essentially avoids all the drawbacks involved in prior art as mentioned above. The currently developed process is advantageously capable of producing the key lactone intermediate with more than 80% ee applying transfer hydrogenation with a very simple operation in view of process perspective. Further, by means of using such chiral lactone with more than 80% ee, the currently developed process is also capable of delivering >99.9% chirally pure Brivaracetam with excellent yield.
EXAMPLES:
EXAMPLE 1: Synthesis of (3R)-N-[(1S)-1-carbamoylpropyl]-3-(hydroxymethyl) hexanamide [Intermediate 7 of scheme A of the present invention]
Example 1 illustrates one pot process for preparing purified (3R)-N-[(1S)-1- carbamoylpropyl]-3-(hydroxymethyl) hexanamide [Intermediate 7] from Intermediate 3 (80% ee) as developed in step 1 of scheme A of the present invention.
Procedure:
In the first step of scheme A of the present invention, a mixture of (R)/(S)-4-propyldihydrofuran-2(3H)-one (Intermediate 3, R: S isomer = 80: 20) (1 eq), (S)-2-aminobutanamide (1.1 eq), triethylamine (1.5 eq) is refluxed at a temperature of 95±5 °C for 24h. The mixture is then cooled to 60-65 °C, washed with a mixture of dichloromethane and diisopropyl ether (2.5 vol) in order to get Intermediate-7 [(3R)-N-[(1S)-1-carbamoyl-propyl]-3-(hydroxymethyl) hexanamide] (80% yield).
Results:
Formation of Intermediate 7 is confirmed further by following analytical studies: a) The 1H NMR analysis is conducted and the data as illustrated in accompanying figure 1 depicts: (400 MHz, DMSO-d6): δ 0.6 (t, J= Hz, 6H), 1.07-1.18 (m, 1H), 1.21-1.35 (m, 3H), 1.45-1.43 (m, 1H), 1.61-1.72 (m, 1H), 1.75-1.90 (m, 1H), 2.03 (dd, J=6.64 & 14.08 Hz, 1H), 2.18 (dd, J=7.0 & 14.08 Hz, 1H), 3.28 (t, J=5.36 Hz, 2H), 4.07-4.18 (m, 1H), 4.43 (t, J=5.2 Hz, 1H), 6.95 (s, 1H), 7.28 (s, 1H), 7.76 (d, J=8.08 Hz, 1H).; thus, confirming formation of Intermediate 7 of the present invention.
b) The LCMS analysis is further conducted and the data as graphically illustrated in accompanying figure 2 provides a (M+H+) value of 231.0; thus, confirming formation of Intermediate 7 of the present invention.
c) The HPLC study is also conducted and the data as graphically illustrated in accompanying figure 3 confirms formation of Intermediate 7 of the present invention with chiral purity of 97.38%
EXAMPLE 2: Preparation of (3R)-N-(1S)-1-amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide [Intermediate 8A of scheme A of the present invention]: Example 2 illustrates a process for preparing (3R)-N-(1S)-1-Amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide [Intermediate 8A] from Intermediate 7 of example 1 above as developed in the present invention.
Procedure:
In second step of scheme A of the present invention, the said Intermediate 7 of example 1 above that is (3R)-N-(1S)-1-Amino-1-oxobutan-2-yl)-3-(hydroxymethyl) hexanamide (~98% Chemical purity and ~97% Chiral purity) (1736.86 mmol) is dissolved in DCM (1.2 L) at RT into a RBF under N2 atm. Then the solution is cooled to 10-20 C and Oxaloyl chloride (2605.29 mmol) is added to this cooled solution at 10-20 °C. The mixture is stirred for 24 h at 25-40 °C under N2 atm. Completion of the reaction is monitored by TLC. After completion of reaction, the solvent is distilled off and the residual mass is diluted with water (6 L), stirred at 30-50 °C for 4 h. Slurry mass is then filtered and washed with water (2×400 mL) followed by MTBE (800 mL). The solid is dried under vacuum at 50-55 °C for 4-5 h to afford Intermediate 8A that is (3R)-N-(1S)-1-Amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide as a white solid (92% yield).
Results:
Formation of Intermediate 8A is confirmed further by following analytical studies: a) The 1H NMR analysis is conducted and the data as illustrated in accompanying figure 4 depicts: 1H NMR (400 MHz, DMSO-d6) : δ 0.83 (t, J=7.44 Hz, 3H), 0.85 (t, J=6.72 Hz, 3H), 1.20-1.40 (m, 4H), 1.43-1.56 (m, 1H), 2.08-2.18 (m, 1H), 2.20-2.28 (m, 2H), 3.61 (dd, J=4.6 & 10.8 Hz, 1 H), 3.67 (dd, J=4.6 & 10.8 Hz, 1H), 4.07-4.18 (m, 1H), 6.95 (s, 1H), 7.29 (s, 1H), 7.89 (d, J=8.12 Hz, 1H); thus confirming formation of Intermediate 8A of the present invention.
b) The LCMS analysis is further conducted and the data as graphically illustrated in accompanying figure 5 (a, b) provides a (M+H+) value of 249.20; thus, confirming formation of Intermediate 8A of the present invention.
EXAMPLE 3: Process for purification of Intermediate 8A forming Intermediate 8B Example 3 illustrates a process for purifying the said Intermediate 8A of example 2 above of the present invention.
Procedure:
The Intermediate 8A as obtained in example 2 above [that is (3R)-N-(1S)-1-Amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide] is first dissolved in a polar solvent like Acetonitrile raising the temperature to 50 to 60 °C; followed by stirring and then addition of another solvent methyl tert-butyl ether (MTBE) which is less polar in nature. The mixture is then cooled down to 0°C, the filtered mass thus obtained is dried in order to obtain a white solid of Intermediate 8B. The material thus obtained is further dissolved in THF (5 vol) at 60 °C, cooled to 20-30°C, followed by addition of heptane (5 vol), stirred at 10°C to 30 °C for 1 h. The mass obtained is filtered and washed with heptane (2×1 vol), dried under vacuum at 50-55°C in order to afford formation of purer form of Intermediate 8A that is Intermediate 8B that is (3R)-N-(1S)-1-amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide as a white solid having a chemical purity of 99.9% along with a chiral purity of 100% (yield: 390 g).
Results:
The purification of Intermediate 8A is further confirmed by the following analytical test results:
a) Chiral HPLC: A Chiral HPLC as illustrated in accompanying figure 6 confirmed formation of purest form of Intermediate 8B having 100% chiral purity [Peak 1; RT (min) = 6.244; %Area=100%].
b) GLP-HPLC: A GLP-HPLC as illustrated in accompanying figure 7 further confirmed formation of Intermediate 8B having 99.9% chemical purity [Peak 3; BRIV8; RT = 29.278; % Area=99.90%].
EXAMPLE 4: Synthesis of (3R)-N-[(1S)-1-carbamoylpropyl]-3-(hydroxymethyl) hexanamide [Intermediate 7’ of scheme B of the present invention]
Example 4 illustrates one pot process for preparing purified (3R)-N-[(1S)-1-carbamoylpropyl]-3-(hydroxymethyl) hexanamide [Intermediate 7’] from Intermediate 6 (99.99% ee) as developed in step-1 scheme B of the present invention.
Procedure:
In another method, in the first step of scheme B of the present invention, a mixture of (R)/(S)-4-propyldihydrofuran-2(3H)-one (Intermediate 6: S isomer = 99.99% : 0.1%) (1 eq), (S)-2-aminobutanamide (1.7 eq), triethylamine (5 eq) is refluxed at a temperature between 95±5 °C for 24 h. Then, the crude reaction mass is cooled and washed with dichloromethane and diisopropyl ether mixture (2.5 vol) in order to achieve Intermediate-7’ of scheme B of the present invention [(3R)-N-[(1S)-1-carbamoylpropyl]-3-(hydroxymethyl) hexanamide] (90% yield).
Results:
The chiral purity of the formed Intermediate 7’ is analyzed by HPLC method and the data as graphically illustrated in accompanying figure 8 confirms formation of Intermediate 7’ of the present invention with chiral purity of 99.11%
EXAMPLE 5: Preparation of (3R)-N-(1S)-1-amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide [Intermediate 8’ of scheme B of present invention]: Example 5 illustrates a process for preparing purest form of (3R)-N-(1S)-1-amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide [Intermediate 8’] from Intermediate 7’ of example 4 as developed in scheme B (step 2) of the present invention.
Procedure:
In the second step of scheme B of the present invention, the intermediate 7’ of the above example 4 that is (3R)-N-(1S)-1-amino-1-oxobutan-2-yl)-3-(hydroxymethyl) hexanamide (98% Chemical purity and >99%Chiral purity) (1736.86 mmol) is dissolved in DCM (1.2 L) at RT in a round bottomed flask under N2 atm. Then the solution is cooled to 10-30 °C and 1-Chloro-N,N,2-trimethyl-1-propenylamine (2605.29 mmol) is added to this cooled solution at 10-30 °C. The mixture is stirred for 24 h at 25-40 °C under N2 atm. Completion of the reaction is monitored by TLC. After completion of the reaction, the solvent is distilled off and the residual mass is diluted with water (6 L), stirring at 30-50 °C for 4 h. The slurry mass thus obtained is then filtered and washed with water (2×400 mL) followed by methyl tert-buty ether (MTBE) (800 mL). The solid thus obtained is dried under vacuum at 50-55 °C for 4-5 h in order to afford formation of Intermediate 8’ that is (3R)-N-(1S)-1-amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide as a white solid (92% yield).
Results:
Formation of Intermediate 8’ is confirmed further by following analytical studies: a) The 1H NMR analysis is conducted and the data as illustrated in accompanying figure 9 depicts: (400 MHz, DMSO-d6): 1H NMR (400 MHz, DMSO-d6) : δ 0.83 (t, J=7.44 Hz, 3H), 0.85 (t, J=6.72 Hz, 3H), 1.20-1.40 (m, 4H), 1.43-1.56 (m, 1H), 2.08-2.18 (m, 1H), 2.20-2.28 (m, 2H), 3.61 (dd, J=4.6 & 10.8 Hz, 1 H), 3.67 (dd, J=4.6 & 10.8 Hz, 1H), 4.07-4.18 (m, 1H), 6.95 (s, 1H), 7.29 (s, 1H), 7.89 (d, J=8.12 Hz, 1H); thus, confirming formation of Intermediate 8’ of the present invention.
b) The LCMS analysis is further conducted and the data as graphically illustrated in accompanying figure 10 provides a (M+H+) value of 249.1; thus, confirming formation of Intermediate 8’ of the present invention.
c) The HPLC data as illustrated in accompanying figure 11 confirms 100% chiral purity of Intermediate 8’.
EXAMPLE 6: Preparation of (2S)-2-[(4R)-2-oxo-4-propyl-pyrrolidin-1-yl] butanamide [Brivaracetam-API]:
Example 6 illustrates a process for preparing (2S)-2-[(4R)-2-oxo-4-propyl-pyrrolidin-1-yl] butanamide [Brivaracetam API] from Intermediate 8B of example 3 or from Intermediate 8’ of example 5 as developed in step 3 of scheme A or scheme B of the present invention respectively.
Procedure:
In the final step of scheme A or scheme B of the present invention, the intermediate 8B of example 3 or intermediate 8’ of example 5 above that is (3R)-N-((1S)-1-Amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide (1608.04 mmol) is dissolved in dimethyl acetamide (0.5 vol) and isopropylacetate (2 L) into a RBF at 25-30 °C under N2 atm. Then 18-Crown-6 (160.79 mmol) is added into the solution and stirred at RT for 30 min. Reaction mixture is then cooled to 0-10 °C and t-BuOK (1.5 eq) is added portion wise to the cooled solution over 1 h maintaining the temperature from – 0-10 °C to 25 °C under N2 atm. Stirring is then continued for 2 h at -10 °C to 0 °C and then for 12 h at 15-25 °C under N2 atm. Completion of reaction is monitored by TLC. After completion of reaction, the reaction mixture is quenched with addition of 1M HCl solution (pH~6.5-7.0). The resulting mixture is extracted with i-PrOAc (2 L) and MTBE (1 L). Water (0.5 L) is added to the combined organic extract and then filtered through celite bed, washed the bed with MTBE-i-PrOAc (1:1) (400 mL). The organic part is separated and the aqueous part is re-extracted with i-PrOAc-MTBE (1:1) (2 ×0.8 L). The combined organic phases are washed with brine solution (100 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuum under a rotary evaporator to afford crude API. Distillation of dimethylacetamide solvent from the crude is then done at high vacuum pressure (0.05 mm Hg) at 70 °C. Crude product is then dissolved in isopropyl acetate (1.6 L) and treated with activated charcoal (7% w/w) to afford a tech-grade crude of Brivaracetam API as a white solid (yield: 90%) with 97.82% chemical purity.
Results:
Formation of Brivaracetam API is confirmed further by following analytical studies: a) The 1H NMR analysis is conducted and the data as illustrated in accompanying figure 12 depicts: 1H NMR (400 MHz, DMSO-d6) : δ 0.77 (t, J=7.32 Hz, 3H), 0.87 (t, J=7.2 Hz, 3H), 1.21-1.31 (m, 2H), 1.33-1.43 (m, 2H), 1.50-1.62 (m, 1H), 1.73-1.84 (m, 1H), 1.97 (dd, J=8.0 & 16.12 Hz, 1H), 2.18-2.28 (m, 1H), 2.37 (dd, J=8.4 & 16.14 Hz, 1H), 3.11 (dd, J=7.16 & 9.44 Hz, 1H), 3.36 (dd, J=9.2 & 17.5 Hz, 1H), 4.30 dd, J=5.44 & 10.28 Hz, 1H), 6.98 (s, 1H), 7.32 (s, 1H); thus, confirming formation of Brivaracetam API of the present invention.
b) The LCMS analysis is further conducted and the data as graphically illustrated in accompanying figure 13 provides a (M+H+) value of 213.0; thus, confirming formation of Brivaracetam API of the present invention.
^ Purification of Brivaracetam API:
The Brivaracetam thus formed above is further purified by means of dissolving the said material (307 g) in 30% i-PrOAc -MTBE (1 vol) at 55-60 °C, cool to 20-30°C. A mixture of Heptane and MTBE and DIPE (2:2:1) is added, stirred at 10 °C to 30°C for 1 h. The obtained mass is filtered and washed with heptane, which is subsequently dried under vacuum at 40-45 °C to afford (3R)-N-((1S)-1-amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide as a white solid (yield: 80%, chiral purity 99.93% and chemical purity 99.94%).
Results:
a) Chiral HPLC: A Chiral HPLC as illustrated in accompanying figure 14 confirmed formation of purest form of Brivaracetam API having 99.93% chiral purity [Peak 2; RT (min) = 9.45; %Area=99.93%].
b) GLP-HPLC: A GLP-HPLC as illustrated in accompanying figure 15 further confirmed formation of Brivaracetam API having 99.9% chemical purity [Peak 2; RT = 21.138; % Area=99.94%].

