
Home » Uncategorized (Page 54)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Ibipinabant Revisited

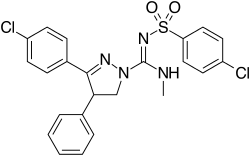
Ibipinabant
| cas 464213-10-3; UNII-O5CSC6WH1T; BMS-646256; SLV-319; |
| Molecular Formula: | C23H20Cl2N4O2S |
|---|---|
| Molecular Weight: | 487.4015 g/mol |
(4S)-5-(4-chlorophenyl)-N-(4-chlorophenyl)sulfonyl-N’-methyl-4-phenyl-3,4-dihydropyrazole-2-carboximidamide
1H-Pyrazole-1-carboximidamide, 3-(4-chlorophenyl)-N’-[(4-chlorophenyl)sulfonyl]-4,5-dihydro-N-methyl-4-phenyl-, (4S)-
(4S)-3-(4-Chlorophenyl)-N-[(4-chlorophenyl)sulfonyl]-4,5-dihydro-N’-methyl-4-phenyl-1H-pyrazole-1-carboximidamide
1H-Pyrazole-1-carboximidamide, 3-(4-chlorophenyl)-N-[(4-chlorophenyl)sulfonyl]-4,5-dihydro-N‘-methyl-4-phenyl-, (4S)-
(-)-(4S)-N-Methyl-N’-((4-chlorophenyl)sulfonyl)-3-(4-chlorophenyl)-4,5-dihydro-4-phenyl-1 H-pyrazole-1 -carboxamidine
4S)-(−)-3-(4-Chlorophenyl)-N-methyl-N‘-[(4-chlorophenyl)sulfonyl]-4-phenyl-4,5-dihydro-1H-pyrazole-1-carboxamidine
It was originally developed by Solvay, which was acquired by Abbott in 2010.
SLV 319, UNII:O5CSC6WH1T, (S)-SLV 319, BMS 646256, JD 5001
- Originator Solvay
- Class Antipsychotics; Imides; Obesity therapies; Pyrazoles; Small molecules; Sulfonamides
- Mechanism of ActionCannabinoid receptor CB1 antagonists
Ibipinabant, also known as BMS-646256, JD-5001 and SLV-319, is a potent and highly selective CB1 antagonist. It has potent anorectic effects in animals, and was researched for the treatment of obesity, although CB1 antagonists as a class have now fallen out of favour as potential anorectics following the problems seen with rimonabant, and so ibipinabant is now only used for laboratory research, especially structure-activity relationship studies into novel CB1 antagonists
Ibipinabant (SLV319, BMS-646,256) is a drug used in scientific research which acts as a potent and highly selective CB1antagonist.[1] It has potent anorectic effects in animals,[2] and was researched for the treatment of obesity, although CB1 antagonists as a class have now fallen out of favour as potential anorectics following the problems seen with rimonabant, and so ibipinabant is now only used for laboratory research, especially structure-activity relationship studies into novel CB1 antagonists.[3][4][5]

| Inventors | Josephus H.M. Lange, Cornelis G Kruse,Jacobus Tipker, Jan Hoogendoorn |
| Applicant | Solvay Pharmaceuticals B.V. |
PATENT
WO 2002076949
https://www.google.com/patents/WO2002076949A1?cl=en
Example IV
(-)-(4S)-N-methyl-N’-((4-chlorophenyl)sulfonyl)-3-(4-chlorophenyl)-4,5- dihydro-4-phenyl-1 H-pyrazole-1 -carboxamidine
(-)-(4S)-N-Methyl-N’-((4-chlorophenyl)sulfonyl)-3-(4-chlorophenyl)-4,5-dihydro-4-phenyl-1 H-pyrazole-1 -carboxamidine (7.16 gram, 0.0147 mol)) ([α25 D] = -150°, c = 0.01 , MeOH) (melting point: 169-170 °C) was obtained via chiral chromatographic separation of racemic N-methyl-N’-((4-chlorophenyl)sulfonyl)-3- (4-chlorophenyl)-4,5-dihydro-4-phenyl-1 H-pyrazole-1 -carboxamidine (18 gram, 0.037 mol) using a Chiralpak AD, 20 μm chiral stationary phase. The mobile phase consisted of a mixture of hexane/ethanol (80/20 (v/v)) and 0.1 % ammonium hydroxide (25 % aqueous solution).
Example III N-Methyl-N’-((4-chlorophenyl)sulfonyl)-3-(4-chlorophenyl)-4,5-dihydro-4- phenyl-1 H-pyrazole-1 -carboxamidine
Part A: To a solution of N-((4-chlorophenyl)sulfonyl)carbamic acid methyl ester (CAS: 34543-04-9) (2.99 gram, 12.0 mmol) and pyridine (4 ml) in 1 ,4-dioxane (20 ml) is added 3-(4-chlorophenyl)-4,5-dihydro-4-phenyl-1 H-pyrazole (3.39 gram, 13.2 mmol) and the resulting mixture is stirred for 4 hours at 100 °C After concentration in vacuo the residue is dissolved in dichloromethane, successively washed with water, 1 N HCI and water, dried over anhydrous Na2SO4, filtered and concentrated in vacuo to a volume of 20 ml. Methyl-tert-butyl ether (60 ml) is added and the resulting solution is concentrated to a volume of 20 ml. The formed crystals are collected by filtration and recrystallised from methyl-te/τ-butyl ether to give 3-(4-chlorophenyl)-N-((4-chlorophenyl)sulfonyl)-4,5-dihydro-4-phenyl-1 H- pyrazole-1-carboxamide (4J5 gram, 76 % yield) Melting point: 211-214 °C
Part B: A mixture of 3-(4-chlorophenyl)-N-((4-chlorophenyl)sulfonyl)-4,5-dihydro- 4-phenyl-1 H-pyrazole-1 -carboxamide (3.67 gram, 7J5 mmol) and phosphorus pentachloride (1.69 gram, 8.14 mmol) in chlorobenzene (40 ml) is heated at reflux for 1 hour. After thorough concentration in vacuo, the formed N-((4- chlorophenyl)sulfonyl)-3-(4-chlorophenyl)-4,5-dihydro-4-phenyl-1 H-pyrazole-1- carboximidoyl chloride is suspended in dichloromethane and reacted with cold methylamine (1.5 ml). After stirring at room temperature for 1 hour, the mixture is concentrated in vacuo. The residue is crystallised from diethyl ether to give N-methyl-N’-((4-chlorophenyl)sulfonyl)-3-(4-chlorophenyl)-4,5-dihydro-4-phenyl- 1 H-pyrazole-1 -carboxamidine (2.29 gram, 61 % yield). Melting point: 96-98 °C(dec).
PATENT
WO 2008074816
https://google.com/patents/WO2008074816A1?cl=en
PAPER
An expedient atom-efficient synthesis of the cannabinoid CB1receptor inverse agonist ibipinabant
- Abbott Healthcare Products B.V., Chemical Design & Synthesis Unit, C.J. van Houtenlaan 36, 1381 CP Weesp, The Netherlands
http://www.sciencedirect.com/science/article/pii/S0040403911000955
http://dx.doi.org/10.1016/j.tetlet.2011.01.068

A novel synthetic route to the highly selective and orally active cannabinoid CB1 receptor inverse agonist ibipinabant is described which combines the use of inexpensive, commercially available reagents and mild reaction conditions with a high degree of atom-efficiency. The method is expected to enable the rapid synthesis of a variety of sulfonylguanidines.
PAPER
JD-5006 and JD-5037: Peripherally restricted (PR) cannabinoid-1 receptor blockers related to SLV-319 (Ibipinabant) as metabolic disorder therapeutics devoid of CNS liabilities
- Jenrin Discovery, 2515 Lori Lane North, Wilmington, DE 19810, USA
http://dx.doi.org/10.1016/j.bmcl.2012.08.004
http://www.sciencedirect.com/science/article/pii/S0960894X12009936

- Scheme 1.
Reagents and conditions: (a) ArylSO2NHCO2CH3, toluene, reflux, 3–6 h, 30–95%; (b) PCl5, chlorobenzene, reflux, 2–4 h, 50–90%; (c) NH2Me.HCl, Et3N, CH2Cl2, ice-bath to rt, 16–20 h, 30–70%; (d) X = CO2Et/X′ = CO2H: LiOH, H2O/THF(1:3), 50%; X′ = CO2H/X′ = CONH2, (1) IBCF, NMM, CH2Cl2, (2) NH3/THF, 60%; X′ = CO2H/X′ = CONHOH, (1) IBCF, NMM, CH2Cl2, (2) NH2OH·HCl, NMM, CH2Cl2, 40%; X = OCH2CO2Et/OCH2CO2H: LiOH, H2O/THF(1:3), rt, 50%; X′ = OCH2CO2H/X′ = OCH2CONH2: (1) IBCF, NMM, CH2Cl2, (2) NH3/THF, 30%; X = NO2:/X′ = NHCOCH3: (1) Fe/HCl, EtOH, H2O, reflux 1–2 h, 90%; (2) Ac2O, pyr, CH2Cl2, rt, 8 h, 90%; X = NO2:/X′ = NHCO2Et: (1) Fe/HCl, EtOH, H2O, reflux 1–2 h, 90%; (2) ClCO2Et, pyr, CH2Cl2, 0 °C to rt, 8 h, 90%
Clip
http://molpharm.aspetjournals.org/content/87/2/197.full.pdf
Paper
Lange et al (2005) Novel 3,4-diarylpyrazolines as potent cannabinoid CB1 receptor antagonists with lower lipophilicity. Bioorg.Med.Chem.Lett. 15 4794. PMID: 16140010.
http://www.sciencedirect.com/science/article/pii/S0960894X05010139
http://dx.doi.org/10.1016/j.bmcl.2005.07.054
Paper
Lange et al (2004) Synthesis, biological properties, and molecular modeling investigations of novel 3,4-diarylpyrazolines as potent and selective CB1 cannabinoid receptor antagonists. J.Med.Chem. 47 627. PMID:14736243.
A series of novel 3,4-diarylpyrazolines was synthesized and evaluated in cannabinoid (hCB1 and hCB2) receptor assays. The 3,4-diarylpyrazolines elicited potent in vitroCB1 antagonistic activities and in general exhibited high CB1 vs CB2 receptor subtype selectivities. Some key representatives showed potent pharmacological in vivo activities after oral dosing in both a CB agonist-induced blood pressure model and a CB agonist-induced hypothermia model. Chiral separation of racemic 67, followed by crystallization and an X-ray diffraction study, elucidated the absolute configuration of the eutomer 80 (SLV319) at its C4 position as 4S. Bioanalytical studies revealed a high CNS−plasma ratio for the development candidate 80. Molecular modeling studies showed a relatively close three-dimensional structural overlap between 80 and the known CB1 receptor antagonist rimonabant (SR141716A). Further analysis of the X-ray diffraction data of 80 revealed the presence of an intramolecular hydrogen bond that was confirmed by computational methods. Computational models and X-ray diffraction data indicated a different intramolecular hydrogen bonding pattern in the in vivo inactive compound 6. In addition, X-ray diffraction studies of 6 revealed a tighter intermolecular packing than 80, which also may contribute to its poorer absorption in vivo. Replacement of the amidine -NH2 moiety with a -NHCH3 group proved to be the key change for gaining oral biovailability in this series of compounds leading to the identification of 80

 ] = −150°, c = 0.01, MeOH; mp 171−172 °C;
] = −150°, c = 0.01, MeOH; mp 171−172 °C;  ] = + 150°, c = 0.01, MeOH; mp 171−172 °C; 1H NMR (400 MHz, DMSO-d6) δ 2.94 (d, J = 4 Hz, 3H), 3.96 (dd, J = 11 and 4 Hz, 1H), 4.46 (t, J = 11 Hz, 1H), 5.05 (dd, J = 11 and 4 Hz, 1H), 7.20−7.35 (m, 5H), 7.45 (dt, J = 8 and 2 Hz, 2H), 7.53 (dt, J = 8 and 2 Hz, 2H), 7.77 (dt,J = 8 and 2 Hz, 2H), 7.82 (dt, J = 8 and 2 Hz, 2H), 8.19 (br d, J = 4 Hz, 1H); HRMS (C23H21Cl2N4O2S) [M+H]+: found m/z 487.0749, calcd 487.0762. Anal. (C23H20Cl2N4O2S) C, H, N.
] = + 150°, c = 0.01, MeOH; mp 171−172 °C; 1H NMR (400 MHz, DMSO-d6) δ 2.94 (d, J = 4 Hz, 3H), 3.96 (dd, J = 11 and 4 Hz, 1H), 4.46 (t, J = 11 Hz, 1H), 5.05 (dd, J = 11 and 4 Hz, 1H), 7.20−7.35 (m, 5H), 7.45 (dt, J = 8 and 2 Hz, 2H), 7.53 (dt, J = 8 and 2 Hz, 2H), 7.77 (dt,J = 8 and 2 Hz, 2H), 7.82 (dt, J = 8 and 2 Hz, 2H), 8.19 (br d, J = 4 Hz, 1H); HRMS (C23H21Cl2N4O2S) [M+H]+: found m/z 487.0749, calcd 487.0762. Anal. (C23H20Cl2N4O2S) C, H, N.Paper

changed to

Process for the second pilot-plant implementation
Process parameter ranges and typical results from approximately 20 lab experiments conducted on the process shown in Scheme



Figure 3. Ishikawa diagram for the API step, highlighting factors that potentially affect the enantiomeric purity of the product. Factors shown in blue were accounted for in the sulfonylation reaction and distillative crystallization models. Factors shown in red were not included in the models
| process parameter | min. value | max. value | # of “levels” |
|---|---|---|---|
| sulfonylation reaction model | |||
| temp. (°C) | 5 | 35 | 7 |
| 4-chlorobenzenesulfonyl chloride (equiv) | 1.0 | 1.2 | 6 |
| conc. (mL/g) | 5 | 10 | 6 |
| reaction time (h) | 2 | 5 | 4 |
| distillative crystallization model | |||
| pressure (mbar) | 300 | 1013 | 6 |
| residual 2(AP) | 0.05 | 2.0 | 6 |
| distillation time (h) | 8 | 48 | 4 |
| distillation end point (wt % EtOH) | 90 | 98 | 3 |
REFERENCES
1: Schirris TJ, Ritschel T, Herma Renkema G, Willems PH, Smeitink JA, Russel FG. Mitochondrial ADP/ATP exchange inhibition: a novel off-target mechanism underlying ibipinabant-induced myotoxicity. Sci Rep. 2015 Sep 29;5:14533. doi: 10.1038/srep14533. PubMed PMID: 26416158; PubMed Central PMCID: PMC4586513.
2: Chorvat RJ, Berbaum J, Seriacki K, McElroy JF. JD-5006 and JD-5037: peripherally restricted (PR) cannabinoid-1 receptor blockers related to SLV-319 (Ibipinabant) as metabolic disorder therapeutics devoid of CNS liabilities. Bioorg Med Chem Lett. 2012 Oct 1;22(19):6173-80. doi: 10.1016/j.bmcl.2012.08.004. Epub 2012 Aug 20. PubMed PMID: 22959249.
3: Tomlinson L, Tirmenstein MA, Janovitz EB, Aranibar N, Ott KH, Kozlosky JC, Patrone LM, Achanzar WE, Augustine KA, Brannen KC, Carlson KE, Charlap JH, Dubrow KM, Kang L, Rosini LT, Panzica-Kelly JM, Flint OP, Moulin FJ, Megill JR, Zhang H, Bennett MJ, Horvath JJ. Cannabinoid receptor antagonist-induced striated muscle toxicity and ethylmalonic-adipic aciduria in beagle dogs. Toxicol Sci. 2012 Oct;129(2):268-79. doi: 10.1093/toxsci/kfs217. Epub 2012 Jul 21. PubMed PMID: 22821849.
4: Dawes J, Allenspach C, Gamble JF, Greenwood R, Robbins P, Tobyn M. Application of external lubrication during the roller compaction of adhesive pharmaceutical formulations. Pharm Dev Technol. 2013 Feb;18(1):246-56. doi: 10.3109/10837450.2012.705299. Epub 2012 Jul 20. PubMed PMID: 22813432.
5: Leane MM, Sinclair W, Qian F, Haddadin R, Brown A, Tobyn M, Dennis AB. Formulation and process design for a solid dosage form containing a spray-dried amorphous dispersion of ibipinabant. Pharm Dev Technol. 2013 Mar-Apr;18(2):359-66. doi: 10.3109/10837450.2011.619544. Epub 2012 Jan 23. PubMed PMID: 22268601.
6: Rohrbach K, Thomas MA, Glick S, Fung EN, Wang V, Watson L, Gregory P, Antel J, Pelleymounter MA. Ibipinabant attenuates β-cell loss in male Zucker diabetic fatty rats independently of its effects on body weight. Diabetes Obes Metab. 2012 Jun;14(6):555-64. doi: 10.1111/j.1463-1326.2012.01563.x. Epub 2012 Feb 24. PubMed PMID: 22268426.
7: Lynch CJ, Zhou Q, Shyng SL, Heal DJ, Cheetham SC, Dickinson K, Gregory P, Firnges M, Nordheim U, Goshorn S, Reiche D, Turski L, Antel J. Some cannabinoid receptor ligands and their distomers are direct-acting openers of SUR1 K(ATP) channels. Am J Physiol Endocrinol Metab. 2012 Mar 1;302(5):E540-51. doi: 10.1152/ajpendo.00258.2011. Epub 2011 Dec 13. PubMed PMID: 22167524; PubMed Central PMCID: PMC3311290.
8: Gamble JF, Leane M, Olusanmi D, Tobyn M, Supuk E, Khoo J, Naderi M. Surface energy analysis as a tool to probe the surface energy characteristics of micronized materials–a comparison with inverse gas chromatography. Int J Pharm. 2012 Jan 17;422(1-2):238-44. doi: 10.1016/j.ijpharm.2011.11.002. Epub 2011 Nov 10. PubMed PMID: 22100516.
9: Sinclair W, Leane M, Clarke G, Dennis A, Tobyn M, Timmins P. Physical stability and recrystallization kinetics of amorphous ibipinabant drug product by fourier transform raman spectroscopy. J Pharm Sci. 2011 Nov;100(11):4687-99. doi: 10.1002/jps.22658. Epub 2011 Jun 16. PubMed PMID: 21681752.
10: Gamble JF, Tobyn M, Dennis AB, Shah T. Roller compaction: application of an in-gap ribbon porosity calculation for the optimization of downstream granule flow and compactability characteristics. Pharm Dev Technol. 2010 Jun;15(3):223-9. doi: 10.3109/10837450903095342. PubMed PMID: 22716462.
11: Zhang H, Patrone L, Kozlosky J, Tomlinson L, Cosma G, Horvath J. Pooled sample strategy in conjunction with high-resolution liquid chromatography-mass spectrometry-based background subtraction to identify toxicological markers in dogs treated with ibipinabant. Anal Chem. 2010 May 1;82(9):3834-9. doi: 10.1021/ac100287a. PubMed PMID: 20387806.
12: Lange JH, van der Neut MA, den Hartog AP, Wals HC, Hoogendoorn J, van Stuivenberg HH, van Vliet BJ, Kruse CG. Synthesis, SAR and intramolecular hydrogen bonding pattern of 1,3,5-trisubstituted 4,5-dihydropyrazoles as potent cannabinoid CB(1) receptor antagonists. Bioorg Med Chem Lett. 2010 Mar 1;20(5):1752-7. doi: 10.1016/j.bmcl.2010.01.049. Epub 2010 Jan 20. PubMed PMID: 20137935.
References
- Lange, JH; Coolen, HK; Van Stuivenberg, HH; Dijksman, JA; Herremans, AH; Ronken, E; Keizer, HG; Tipker, K; et al. (2004). “Synthesis, biological properties, and molecular modeling investigations of novel 3,4-diarylpyrazolines as potent and selective CB(1) cannabinoid receptor antagonists”. Journal of Medicinal Chemistry. 47 (3): 627–43. doi:10.1021/jm031019q. PMID 14736243.
- Need, AB; Davis, RJ; Alexander-Chacko, JT; Eastwood, B; Chernet, E; Phebus, LA; Sindelar, DK; Nomikos, GG (2006). “The relationship of in vivo central CB1 receptor occupancy to changes in cortical monoamine release and feeding elicited by CB1 receptor antagonists in rats”.Psychopharmacology. 184 (1): 26–35. doi:10.1007/s00213-005-0234-x. PMID 16328376.
- Lange, JH; Van Stuivenberg, HH; Veerman, W; Wals, HC; Stork, B; Coolen, HK; McCreary, AC; Adolfs, TJ; Kruse, CG (2005). “Novel 3,4-diarylpyrazolines as potent cannabinoid CB1 receptor antagonists with lower lipophilicity”. Bioorganic & Medicinal Chemistry Letters. 15 (21): 4794–8. doi:10.1016/j.bmcl.2005.07.054. PMID 16140010.
- Srivastava, BK; Joharapurkar, A; Raval, S; Patel, JZ; Soni, R; Raval, P; Gite, A; Goswami, A; et al. (2007). “Diaryl dihydropyrazole-3-carboxamides with significant in vivo antiobesity activity related to CB1 receptor antagonism: synthesis, biological evaluation, and molecular modeling in the homology model”. Journal of Medicinal Chemistry. 50 (24): 5951–66. doi:10.1021/jm061490u. PMID 17979261.
- Srivastava, BK; Soni, R; Joharapurkar, A; Sairam, KV; Patel, JZ; Goswami, A; Shedage, SA; Kar, SS; et al. (2008). “Bioisosteric replacement of dihydropyrazole of 4S-(−)-3-(4-chlorophenyl)-N-methyl-N’-(4-chlorophenyl)-sulfonyl-4-phenyl-4,5-dihydro-1H-pyrazole-1-caboxamidine (SLV-319) a potent CB1 receptor antagonist by imidazole and oxazole”. Bioorganic & Medicinal Chemistry Letters. 18 (3): 963–8. doi:10.1016/j.bmcl.2007.12.036. PMID 18207393.
| Patent ID | Date | Patent Title |
|---|---|---|
| US9174965 | 2015-11-03 | Pyrimidinylpiperidinyloxypyridone analogues as GPR119 modulators |
| US2015133479 | 2015-05-14 | PYRIMIDINYLPIPERIDINYLOXYPYRIDONE ANALOGUES AS GPR119 MODULATORS |
| US8940716 | 2015-01-27 | Bicyclic heteroaryl compounds as GPR119 modulators |
| US8853205 | 2014-10-07 | Heteropyrrole analogs acting on cannabinoid receptors |
| US8729084 | 2014-05-20 | Benzofuranyl analogues as GPR119 modulators |
| US2014080788 | 2014-03-20 | NOVEL BICYCLIC NITROGEN CONTAINING HETEROARYL TGR5 RECEPTOR MODULATORS |
| US8513265 | 2013-08-20 | [6, 6] and [6, 7]-bicyclic GPR119 G protein-coupled receptor agonists |
| US8513424 | 2013-08-20 | Pyridone GPR119 G protein-coupled receptor agonists |
| US8476283 | 2013-07-02 | [6, 5]â??bicyclic GPR119 G protein-coupled receptor agonists |
| US8314095 | 2012-11-20 | [6, 6] and [6, 7]-bicyclic GPR119 G protein-coupled receptor agonists |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
4S-(−)-3-(4-chlorophenyl)-N-methyl-N’-[(4-chlorophenyl)-sulfonyl]-4-phenyl-4,5-dihydro-1H-pyrazole-1-carboxamidine
|
|
| Identifiers | |
| CAS Number | 464213-10-3 |
| ATC code | none |
| PubChem | CID 9826744 |
| ChemSpider | 24765166 |
| UNII | O5CSC6WH1T |
| KEGG | D09349 |
| ChEMBL | CHEMBL158784 |
| Chemical data | |
| Formula | C24H22Cl2N4O2S |
| Molar mass | 501.427 |
///////// 464213-10-3, UNII-O5CSC6WH1T, BMS-646256, SLV-319, Ibipinabant, JD 5001, solvay, abbott
c2cc(Cl)ccc2C1=NN(C(NC)=NCS(=O)(=O)c3ccc(Cl)cc3)CC1c4ccccc4
PF-04745637

PF-04745637
cas 1917294-46-2
MW 509.00, MF C27 H32 Cl F3 N2 O2
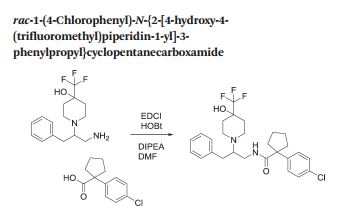
Cyclopentanecarboxamide, 1-(4-chlorophenyl)-N-[2-[4-hydroxy-4-(trifluoromethyl)-1-piperidinyl]-3-phenylpropyl]-
rac-1-(4-Chlorophenyl)-N-f2-r4-hvdroxy-4-(trifluoromethyl)piperidin-1-vn-3-phenylpropyDcyclopentanecarboxamide
|
PRODUCT PATENT WO-2016067143-A1
|
| Applicants: | PFIZER INC. [US/US]; 235 East 42nd Street New York, New York 10017 (US) |
| Inventors: | SWAIN, Nigel Alan; (GB). PRYDE, David Cameron; (GB). RAWSON, David James; (GB). RYCKMANS, Thomas; (GB). SKERRATT, Sarah Elizabeth; (GB). AMATO, George Salvatore; (US). MARRON, Brian Edward; (US). REISTER, Steven Michael; (US). |

TrpA1 is a member of the Transient Receptor Potential (Trp) family of ion channels. It was first described as being activated in response to noxious cold. It is activated by a number of exogenous chemical compounds and some endogenous inflammatory mediators. It has also been reported to be activated in response to mechanical stress.
There is substantial evidence for the involvement of TrpA1 in the physiology of pain, including neuropathic and inflammatory pain, and in pruritus (itch). For example, see:
Bautista, D.M. et al., “TRPA 1: A Gatekeeper for Inflammation” , Annu. Rev. Physiol.2013, 75, 181-200;
Bishnoi, M. & Premkumar, L.S., “Changes in TRP Channels Expression in Painful
Conditions”, Open Pain Journal 2013, 6(Suppl. 1), 10-22;Brederson, J.-D. et al., “Targeting TRP channels for pain relief, Eur. J. Pharmacol.2013, 716, 61-76;
Radresa, O. et al., “Roles of TRPAI in Pain Pathophysiology and Implications for the Development of a New Class of Analgesic Drugs”, Open Pain Journal 2013, 6(Suppl. 1), 137-153; and Toth, B.I. & Biro, T., “TRP Channels and Pruritus” , Open Pain Journal 2013, 6(Suppl.1), 62-80.
There is a continuing interest in finding new compounds that interact with TrpA1.
 Nigel Swain
Nigel Swain
PATENT
E8 that is 1-(4-chlorophenyl)-/V-[2-(4-hydroxy-4-(trifluoromethyl)piperidin-1-yl)-3-phenylpropyl]-cyclopentanecarboxamide, or a pharmaceutically acceptable salt thereof. This compound is represented by formula (lE).

Example 1
rac-1-(4-Chlorophenyl)-N-f2-r4-hvdroxy-4-(trifluoromethyl)piperidin-1-vn-3-phenylpropyDcyclopentanecarboxamide

Method 1
To a solution of rac-1-(1-amino-3-phenylpropan-2-yl)-4-(trifluoromethyl)piperidin-4-ol (Preparation 2, 50 mg, 0.214 mmol) in DMF (1 mL) was added 1-(4-chlorophenyl)cyclopentanecarboxylic acid (37 mg, 0.165 mmol), DIPEA (0.035 mL, 0.198 mmol) and EDCI (38 mg, 0.198 mmol), followed by HOBt (30 mg, 0.198 mmol) and the reaction was stirred at room temperature for 18 hours. Water was added and the reaction stirred for a further 2 hours. DCM was added with further stirring for 1 hour followed by elution through a phase separation cartridge. The organic filtrate was concentrated in vacuo. The residue was dissolved in MeOH and treated with ethereal HCI with standing for 18 hours. The resulting suspension was filtered and triturated with EtOAc, heptanes and TBME to afford the title compound as the hydrochloride salt (69 mg, 82%).
1H NMR (400MHz, DMSO-d6): δ ppm 1.50-1.60 (m, 4H), 1.70-1.90 (m, 4H), 2.15-2.25 (m, 2H), 2.40-2.48 (m, 2H), 2.70-2.80 (m, 1 H), 3.05-3.25 (m, 6H), 3.47-3.62 (m, 2H), 6.38 (br s, 1 H), 7.20-7.40 (m, 9H), 7.80 (br m, 1 H).
MS m/z 509 [M+H]+
Example 1 may also be prepared according to the following method:
A mixture of 1-(4-chlorophenyl)cyclopentanecarboxylic acid (25.7 g, 114 mmol), 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium-3-oxid-hexafluoro phosphate (49.4 g, 130 mmol) and N,N-diisopropylethylamine (40 mL, 229 mmol) in DMF (475 mL) was stirred at room temperature for 15 minutes. To this mixture was added a solution of 1-(1-amino-3-phenylpropan-2-yl)-4-(trifluoromethyl)piperidin-4-ol (Preparation 2, 31.4 g, 104 mmol) in DMF (200 mL). The reaction was stirred at room temperature for 18 hours before partitioning between EtOAc (600 mL) and saturated aqueous sodium hydrogen carbonatesolution (600 mL). The aqueous layer was washed with EtOAc (2 x 600 mL). The combined organic layers were washed with water (600 mL), brine (600 mL), dried over sodium sulphate and concentrated in vacuo. The residue was purified using silica gel column chromatography eluting with 0: 1 to 1 : 1 EtOAc: heptanes to afford the title compound (44 g, 76%).
1H NMR (400MHz, CDCI3): δ ppm 1.35 (br s, 1 H), 1.49-1.85 (m, 6H), 1.90-1.99 (m, 2H), 2.25-2.55 (m, 7H), 2.56-2.70 (m, 1 H), 2.75-3.00 (m, 4H), 3.23-3.31 (m, 1 H), 5.87 (br s, 1 H), 7.07 (d, 2H), 7.16-7.30 (m, 7H).
MS m/z 509 [M+H]+
Examples 2 and 3
IS) and (R)-1-(4-Chlorophenyl)-N-f2-r4-hvdroxy-4-(trifluoromethyl)piperidin-1-vn-3-phenylpropyl)cyclopentanecarboxamide

Example 2
To a suspension of (S)-1-(1-amino-3-phenylpropan-2-yl)-4-(trifluoromethyl)piperidin-4-ol (Preparation 3, 70 mg, 0.232 mmol) and 1-(4-chlorophenyl)cyclopentanecarboxylic acid (57.3 mg, 0.255 mmol) in acetonitrile (0.8 mL) was added triethylamine (0.133 mL, 0.928 mmol) followed bypropylphosphonic anhydride (50% wt solution in EtOAc, 0.21 mL, 0.35 mmol). The reaction was stirred at room temperature for 1.5 hours after which the solution was purified directly by silica gel column chromatography eluting with 0-30% EtOAc in heptanes to afford the title compound (75 mg, 64%).
[a]D20 = +9.6 in DCM [20 mg/mL]
ee determination:
Column: ChiralTech AD-H, 250×4.6 mm, 5 micron.
Mobile phase A: CO2; Mobile phase B: MeOH with 0.2% ammonium hydroxide Gradient: 5% B at 0.00 mins, 60% B at 9.00 mins; hold to 9.5 mins and return to 5% B at 10 mins. Flow rate 3 mL/min.
Rt = 5.047 minutes, ee = 95%
Example 2 may also be prepared from rac-1-(4-chlorophenyl)-N-{2-[4-hydroxy-4- (trifluoromethyl)piperidin-1-yl]-3-phenylpropyl}cyclopentanecarboxamide(Example 1).
The racemate was separated into two enantiomers using preparative chiral chromatography as described below:
Chiralpak IA, 4.6x250mm, 5 micron.
Mobile phase: Hexane:DCM:EtOH:DEA 90:8:2:0.1
Flow rate: 1 mL/min
Rt = 8.351 minutes and Rt = 10.068 minutes
The first eluting isomer is Example 2: (S)-1-(4-chlorophenyl)-N-{2-[4-hydroxy-4-(trifluoromethyl)piperidin-1-yl]-3-phenylpropyl}cyclopentanecarboxamide. ee = 100% The second eluting isomer is Example 3: (R)-1-(4-chlorophenyl)-N-{2-[4-hydroxy-4-(trifluoromethyl)piperidin-1-yl]-3-phenylpropyl}cyclopentanecarboxamide. ee = 99.62% The compound of Example 2 prepared from the chiral separation method is identical by a-rotation and retention time to the compound of Example 2 prepared as the single enantiomer described above.
MS m/z 509 [M+H]+
1H NMR (400MHz, DMSO-d6): δ 1.30-1.80 (m, 10H), 2.20-2.30 (m, 1 H), 2.35-2.60 (m, 6H), 2.65-2.85 (m, 4H), 3.00-3.15 (m, 1 H), 5.50 (br s, 1 H), 6.95-7.00 (m, 1 H), 7.05-7.15 (m, 2H), 7.20-7.35 (m, 6H) ppm
PAPER
The discovery of a potent series of carboxamide TRPA1 antagonists
DOI: 10.1039/C6MD00387G, http://pubs.rsc.org/en/Content/ArticleLanding/2016/MD/C6MD00387G?utm_source=feedburner&utm_medium=feed&utm_campaign=Feed%3A+rss%2FMD+%28RSC+-+Med.+Chem.+Commun.+latest+articles%29#!divAbstract
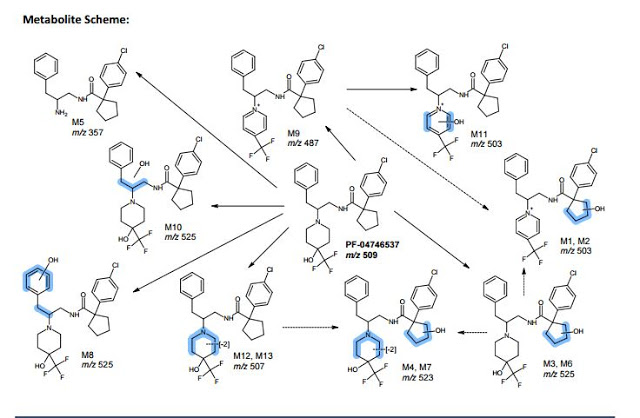
. Please note PF-6667294 is Compound 4 and PF-4746537 is Compound 8.
A series of potent and selective carboxamide TRPA1 antagonists were identified by a high throughput screen. Structure–activity relationship studies around this series are described, resulting in a highly potent example of the series. Pharmacokinetic and skin flux data are presented for this compound. Efficacy was observed in a topical cinnamaldehyde flare study, providing a topical proof of pharmacology for this mechanism. These data suggest TRPA1 antagonism could be a viable mechanism to treat topical conditions such as atopic dermatitis.



Discovery and development of TRPV1 antagonists
https://en.wikipedia.org/wiki/Discovery_and_development_of_TRPV1_antagonists
/////////////PF-04745637, PF 04745637, PF04745637, PFIZER, PRECLINICAL, TRPV1 antagonists, atopic dermatitis, 1917294-46-2
c1(ccccc1)CC(CNC(=O)C3(c2ccc(cc2)Cl)CCCC3)N4CCC(CC4)(O)C(F)(F)F
1, 2-Bis(4-(4-4-nitrophenyl)piperazin-1-yl)ethanone for androgen sensitive prostatic disorders
1, 2-Bis(4-(4-4-nitrophenyl)piperazin-1-yl)ethanone
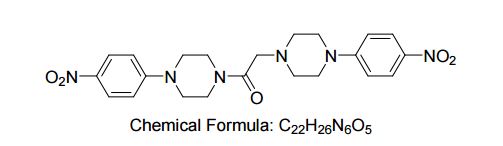
| Molecular Formula: | C22H26N6O5 |
|---|---|
| Molecular Weight: | 454.47904 g/mol |
![]()
CAS 330633-91-5
CDRI-?
For treatment of androgen sensitive prostatic disorders
![1,2-bis[4-(4-nitrophenyl)piperazin-1-yl]ethanone.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=2867018&t=l "A structure of 1,2-bis[4-(4-nitrophenyl)piperazin-1-yl]ethanone")
1, 2-Bis(4-(4-4-nitrophenyl)piperazin-1-yl)ethanone
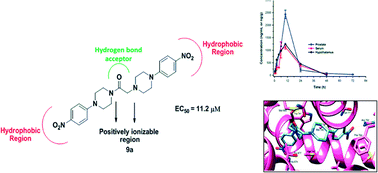
In the quest for novel scaffolds for the management of androgen sensitive prostatic disorders like prostate cancer and benign prostatic hyperplasia, a series of twenty-six aryl/heteroaryl piperazine derivatives have been described. Three compounds, 8a, 8c and 9a, exhibited good activity profiles against an androgen sensitive prostate cancer cell line (LNCaP) with EC50values of 9.8, 7.6 and 11.2 μM, respectively. These compounds caused a decrease in luciferase activity and a decline in PSA and Ca2+ levels, which are indicative of their anti-androgenic and α1A-adrenergic receptor blocking activities, respectively.
Compound 9a reduced the prostate weight of rats (47%) and in pharmacokinetic analysis at 10 mg kg−1 it demonstrated an MRT of ∼14 h post dose, exhibiting high levels in prostate. Compound 9a docked in a similar orientation to hydroxyflutamide on an androgen receptor and showed strong π–π interactions. These findings reveal that compound 9a is a promising candidate for management of prostatic disorders with anti-androgenic and α1A-blocking activities.
Design, synthesis and biological profiling of aryl piperazine based scaffolds for the management of androgen sensitive prostatic disorders
E-mail: vl_sharma@cdri.res.in, vlscdri@gmail.com
Fax: +91 522 2771941
Tel: +91 522 2772450 Ext. 4671
DOI: 10.1039/C6MD00426A, http://pubs.rsc.org/en/Content/ArticleLanding/2016/MD/C6MD00426A?utm_source=feedburner&utm_medium=feed&utm_campaign=Feed%3A+rss%2FMD+%28RSC+-+Med.+Chem.+Commun.+latest+articles%29#!divAbstract
1, 2-Bis(4-(4-4-nitrophenyl)piperazin-1-yl)ethanone (9a) To the mixture of 8a (0.3 g, 1.06 mmol) and Et3N (0.3 mL, 2.12 mmol) in CHCl3 (5 mL) was added 1-(4-nitrophenyl)piperazine (7a, 0.320 g, 1.59 mmol) in 5 mL CHCl3 dropwise within 1 h. After complete addition reaction mixture was further stirred in an oil bath at 80-85 °C for 15 h. The reaction mixture was cooled, washed with water (5 mL × 3) and the organic layer was separated. Combined organic layer was dried (anhyd. Na2SO4 and concentrated under reduced pressure in rotavapor. The solid obtained was purified by recrystallization using EtOAc/Hexane which furnished yellow crystals (yield 81%);
mp: 156-157 °C; IR (KBr) (cm-1): 3019, 2399, 1640, 1597, 1506, 1423, 1330;
1H NMR (400 MHz, CDCl3): 8.14-8.09 (4H, m), 6.84-6.81 (4H, m), 3.84-3.83 (4H, m), 3.49-3.44 (8H, m), 3.33 (2H, s), 2.72 (4H, t, J = 5.0 Hz);
13C NMR (75.4 MHz, CDCl3): 167.7, 154.7, 154.3, 138.8, 138.4, 125.9, 125.8, 112.9, 112.7, 60.8, 52.5, 46.9, 46.7, 44.6;
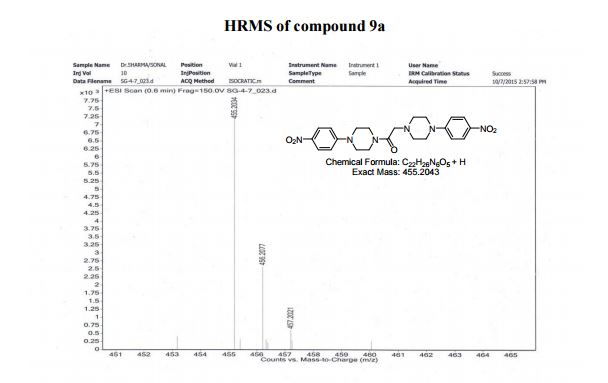
HRMS (ESI positive) m/z calcd. for C22H26N6O5 [M+H]+ : 455.2043, found: 455.2034;
Anal calcd. for C22H26N6O5: C, 58.14; H, 5.77; N, 18.49, found: C, 58.31; H, 5.92; N, 18.66.



SONAL GUPTA
Medicinal & Process Chemistry Division, CSIR-Central Drug Research Institute, Sector 10, Jankipuram ext., Lucknow-226031, India


Dr. VISHNU LAL SHARMA
http://www.cdriindia.org/VL_Sharma.htm
| Dr. VISHNU LAL SHARMA | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 |
Senior Principal Scientist (CSIR-CDRI ) / Professor (AcSIR) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| POST-DOCTORAL RESEARCH (ABROAD) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CURRENT AREAS OF INTEREST | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 From left to right upper row: Dr. S.T.V.S. Kiran Kumar, Dr. Lalit Kumar, Dr. V.L. Sharma, Dr. Nand Lal, Dr. Amit Sarswat Lower row: Dhanaraju Mandalapu, Sonal Gupta, Mrs. Tara Rawat (S.T.O.), Dr. Veenu bala, Dr. Santosh Jangir |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| THESIS SUPERVISED | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FORMER Ph.D. STUDENTS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FORMER PROJECT ASSISTANTS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PRESENT Ph.D. STUDENTS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FORMER POSTGRADUATE STUDENTS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MEMBERSHIP OF SOCIETES : | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PROJECTS: | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PUBLICATIONS & PATENTS- | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| SELECTED PUBLICATIONS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LIST OF PATENTS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
From left to right upper row: Dr. S.T.V.S. Kiran Kumar, Dr. Lalit Kumar, Dr. V.L. Sharma, Dr. Nand Lal, Dr. Amit Sarswat
Lower row: Dhanaraju Mandalapu, Sonal Gupta, Mrs. Tara Rawat (S.T.O.), Dr. Veenu bala, Dr. Santosh Jangir
///////////aryl piperazine, androgen sensitive prostatic disorders, 330633-91-5, CDRI-?
c1(ccc(cc1)[N+]([O-])=O)N2CCN(CC2)C(=O)CN3CCN(CC3)c4ccc(cc4)[N+]([O-])=O
ALMOREXANT REVISITED
| Almorexant; ACT-078573; (R)-2-((S)-6,7-Dimethoxy-1-(4-(trifluoromethyl)phenethyl)-3,4-dihydroisoquinolin-2(1H)-yl)-N-methyl-2-phenylacetamide; |
Almorexant (INN, codenamed ACT-078573) is an orexin antagonist, functioning as a competitive receptor antagonist of the OX1 and OX2 orexin receptors, which was being developed by the pharmaceutical companies Actelion and GSK for the treatment of insomnia. Development of the drug was abandoned in January 2011.[1]
Development
Originally developed by Actelion, from 2007 almorexant was being reported as a potential blockbuster drug, as its novel mechanism of action (orexin receptor antagonism) was thought to produce better quality sleep and fewer side effects than the traditionalbenzodiazepine and z drugs which dominated the multibillion-dollar insomnia medication market.[2][3]
In 2008, pharmaceutical giant GlaxoSmithKline bought the development and marketing rights for almorexant from Actelion for an initial payment of $147 million.[4] The deal was worth a potential $3.2billion if the drug were to successfully complete clinical development and obtain FDA approval.[5] GSK and Actelion continued to develop the drug together, and completed a Phase IIIclinical trial in November 2009.[6]
However, in January 2011 Actelion and GSK announced they were abandoning the development of almorexant because of its side effect profile.[1][7]
Mechanism of action
Almorexant is a competitive, dual OX1 and OX2 receptor antagonist and selectively inhibits the functional consequences of OX1 and OX2 receptor activation, such as intracellular Ca2+ mobilization.
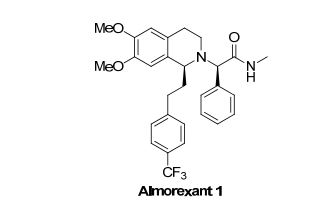

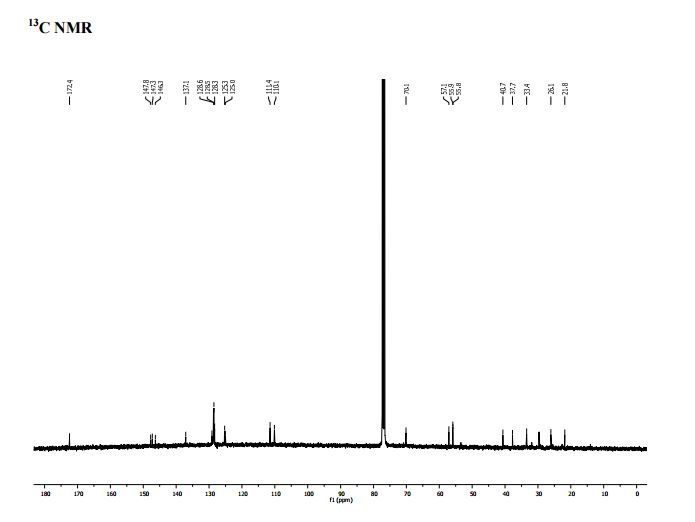
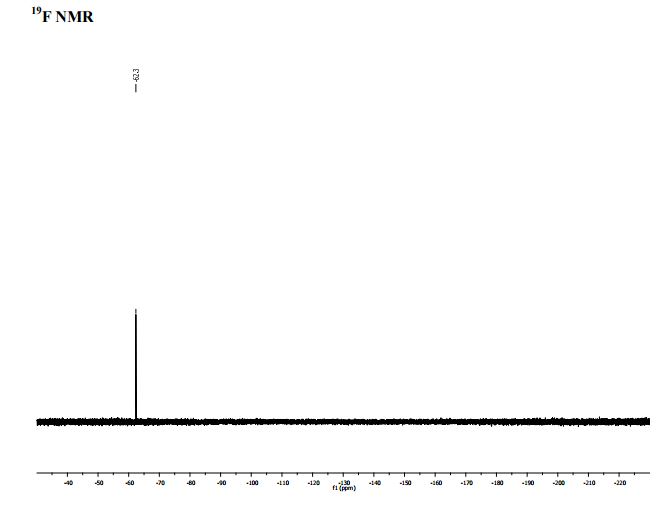




PAPER
http://pubs.rsc.org/en/content/articlelanding/2013/ob/c3ob40655e#!divAbstract
An enantioselective synthesis of almorexant, a potent antagonist of human orexin receptors, is presented. The chiral tetrahydroisoquinoline core structure was prepared via iridium-catalysed asymmetric intramolecular allylic amidation. Further key catalytic steps of the synthesis include an oxidative Heck reaction at room temperature and a hydrazine-mediated organocatalysedreduction.




https://www.google.com/patents/EP2227454A2?cl=en
Reaction scheme 5:

7*CH3COOH

Step 11 : synthesis of (2R)-2-{(-/S)-6,7-dimethoxy-1 -[2-(4-thfluoromethyl-phenyl)- ethyl]-3,4-dihydro-1 /-/-isoquinolin-2-yl}-Λ/-methyl-2-phenyl-acetamide (compound 8)

To the solution of the compound 7 in MIBK are added 1.2 equivalents of the compound 6, 1.1 equivalents caustic soda and 1.1 equivalents potassium carbonate and heated to 70-90 0C. After full conversion the solution is cooled to RT and water is added. Phase separation is followed by a second washing of the organic phase with water and again phase separation. Step 12: synthesis of (2R)-2-{(-/S)-6,7-dimethoxy-1 -[2-(4-trifluoromethyl-phenyl)- ethyl]-3,4-dihydro-1 /-/-isoquinolin-2-yl}-/\/-nnethyl-2-phenyl-acetannide hydrochloride acid (compound I)

To the organic phase of step 11 is added 1 equivalent aqueous hydrochloric acid and then the water removed by azeotropic distillation in vacuo. The precipitate is dissolved by addition of 2-propanol at 75 0C. Concentration of the solution leads to crystallisation and the suspension is then cooled to RT. To ensure complete crystallisation, the suspension is aged at RT, then filtered and washed with a MIBK-2-propanol mixture. The product is dried in vacuo at 50 0C.
PAPER

Several methods are presented for the enantioselective synthesis of the tetrahydroisoquinoline core of almorexant (ACT-078573A), a dual orexin receptor antagonist. Initial clinical supplies were secured by the Noyori Ru-catalyzed asymmetric transfer hydrogenation (Ru-Noyori ATH) of the dihydroisoquinoline precursor. Both the yield and enantioselectivity eroded upon scale-up. A broad screening exercise identified TaniaPhos as ligand for the iridium-catalyzed asymmetric hydrogenation with a dedicated catalyst pretreatment protocol, culminating in the manufacture of more than 6 t of the acetate salt of the tetrahydroisoquinoline. The major cost contributor was TaniaPhos. By switching the dihydroisoquinoline substrate of the Ru-Noyori ATH to its methanesulfonate salt, the ATH was later successfully reduced to practice, delivering several hundreds of kilograms of the tetrahydroisoquinoline, thereby reducing the catalyst cost contribution significantly. The two methods are compared with regard to green and efficiency metrics.
Catalytic Asymmetric Reduction of a 3,4-Dihydroisoquinoline for the Large-Scale Production of Almorexant: Hydrogenation or Transfer Hydrogenation?


References
- GSK and Actelion discontinue clinical development of almorexant – GSK press release, 28 Jan 2011
- Sleeping Beautifully – CBS Business Network 24 Sep 2007
- Is this FINALLY a cure for insomnia? The new pill which blocks the brain’s ‘stay awake’ messages – Daily Mail, 28 July 2008
- Actelion Sells Glaxo Almorexant Sleep Medicine Rights – Bloomberg, 14 July 2008
- Actelion’s top dollar deal leaves doubts, and little on the horizon – EP Vantage, 14 July 2008
- Almorexant in Adult Subjects With Chronic Primary Insomnia (RESTORA 1). ClinicalTrials.gov (February 3, 2010). Retrieved on May 6, 2010.
- Actelion and GSK Discontinue Clinical Development of Almorexant – Actelion press release, 28 Jan 2011
External links
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(2R)-2-[(1S)- 6,7-dimethoxy- 1-{2-[4-(trifluoromethyl)phenyl]ethyl}- 3,4-dihydroisoquinolin-2(1H)-yl]- N-methyl- 2-phenylacetamide
|
|
| Clinical data | |
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Metabolism | Hepatic |
| Identifiers | |
| CAS Number | 871224-64-5 |
| ATC code | none |
| PubChem | CID 23727689 |
| IUPHAR/BPS | 2886 |
| ChemSpider | 21377865 |
| UNII | 9KCW39P2EI |
| ChEMBL | CHEMBL455136 |
| Chemical data | |
| Formula | C29H31F3N2O3 |
| Molar mass | 512.6 g/mol (free base) |
///////Almorexant, ACT-078573
CNC(=O)C(C1=CC=CC=C1)N2CCC3=CC(=C(C=C3C2CCC4=CC=C(C=C4)C(F)(F)F)OC)OC
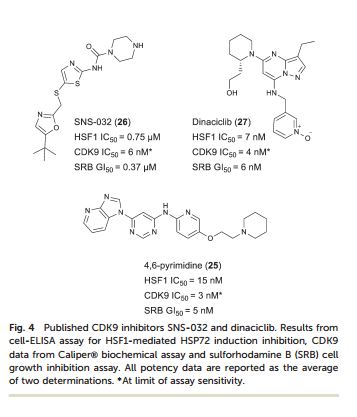
SNS-032, BMS-387032 A potent and selective Cdk inhibitor
SNS 032, BMS-387032
N-[5-[[[5-(1,1-Dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4-piperidinecarboxamide
Cas 345627-80-7, MP 165-167° C
M.Wt:380.53, Formula:C17H24N4O2S2
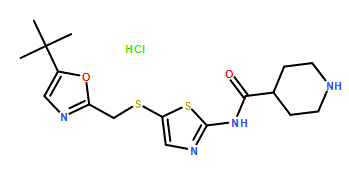
SNS 032, BMS-387032 HYDROCHLORIDE
| Formula | C17H24N4O2S2 . HCl |
|---|---|
| MW | 380.5 . 36.5 |
| CAS | 345627-90-9 |
A potent and selective Cdk inhibitor
Potent inhibitor of cyclin-dependent kinases (cdks) 9, 2 and 7 (IC50 values are 4, 38 and 62 nM respectively). Displays no activity against 190 additional kinases (IC50 >1000 nM). Arrests the cell cycle at G2/M; inhibits transcription, proliferation and colony formation, and induces apoptosis in RPMI-8226 multiple myeloma cells. Prevents tumor cell-induced VEGF secretion and in vitro angiogenesis. SNS-032 (BMS-387032) has firstly been described as a selective inhibitor of CDK2 with IC50 of 48 nM in cell-free assays and is 10- and 20-fold selective over CDK1/CDK4. It is also found to be sensitive to CDK7/9 with IC50 of 62 nM/4 nM, with little effect on CDK6. Phase 1.
Quality Control & MSDS
COA NMR HPLC Datasheet SDS/MSDS
- COA (Certificate Of Analysis)
- HPLC
- NMR (Nuclear Magnetic Resonance)
- MSDS (Material Safety Data Sheet)
SNS-032 (BMS-387032) is a potent and selective inhibitor of cyclin-dependent kinases (CDKs) 2, 7, and 9 [1], with IC50 values of 38 nM, 62 nM and 4 nM, respectively [2].
CDKs mean a family of serine/threonine kinases regulating cell cycle process. Some CDKs are related to transcription control and are often perturbed in cancer cells [3].
Decrease in the phosphorylation at Ser5 and Ser2 in the C-terminal domain (CTD) of RNA Pol II can indicate the inhibition to CDK9 and CDK7 [1]. Chronic lymphocytic leukemia (CLL) cells treated with SNS-032 for 6 or 24 hours showed a decrease in the phosphorylation of Ser2 and Ser5 of the CTD of RNA Pol II, this appeared to be both time- and concentration- dependent, and remarkably consistent among samples. For the phosphorylation of Ser2, the inhibition of SNS-032 was greater than that for the phosphorylation of Ser5, this was consistent with the fact that IC50 for the inhibition of CDK9 was lower compared with that for the inhibition of CDK7 (4 nM vs 62 nM). After 6 hours of SNS-032 exposure, protein levels of CDK7 and CDK9 were stable, but declined at 24 hours [4].
In patients with chronic lymphocytic leukemia (CLL), infusion of SNS-032 in a total dose of 75 mg/m2 resulted in a decrease in the phosphorylation at Ser5 and Ser2 in the C-terminal domain of RNA Pol II. This indicated the inhibition to Cdk9 and Cdk7 by SNS-032. This inhibition was first seen 2 hours after the beginning of the infusion with SNS-032, was pronounced after 6 hours and returned to baseline after 24 hours [1].

The cell cycle-regulated cyclin-dependent kinases (CDKs), CDK1, 2, and 4 have been extensively studied as potential therapeutic targets in cancer. Recent research has additionally underscored the potential role of several constitutively active CDKs including CDK7 and 9 as cancer targets. Phosphorylation of the c-terminal domain (CTD) of RNA Polymerase II by CDK7 and 9 are critical steps in transcriptional regulation. Inhibition of these kinases is predicted to have the greatest effect on the expression of proteins with short t½ and short-lived mRNA, including proteins involved in apoptotic regulation. CDK7 also activates cell-cycle CDKs 1, 2, 4 and 6. SNS-032 (formerly BMS-387032) has previously been described as a selective inhibitor of CDK2 with potent antitumor activity in animal models. Here we show that in addition to inhibition of CDK2, SNS-032 also inhibits CDK7/cyclinH and CDK9/cyclinT at low nanomolar concentrations in biochemical assays. The compound is highly selective for CDK inhibition; in a panel of 208 kinases, only four non-CDK proteins were inhibited by >50% at 1 μM SNS-032. The cellular pharmacology of SNS- 032 mirrors the biochemical data. Cells treated with SNS-032 show a rapid cell cycle arrest and onset of cell death that corresponds with inhibition of multiple substrates of CDK2, 7, and 9. For instance, inhibition of Rb phosphorylation, accumulation of cyclin E protein and cell-cycle arrest at GI and G2 are observed in multiple cell lines in a time and dose-dependent manner, consistent with inhibition of CDK2 and CDK7. Furthermore, SNS-032 inhibits CDK9-mediated phosphorylation of Ser2 in the CTD with an IC50 = 200 nM. Corresponding with inhibition of RNA polymerase II, the short half-life, anti-apoptotic protein Mcl-1 is rapidly depleted from cells, coincident with the phosphorylation of p53. Expression of Mcl-1 is a candidate predictor of aggressive disease and resistance to chemotherapy in CLL and is essential for survival of B-cell lymphoma and multiple myelomas, supporting the use of SNS-032 as a treatment for these diseases. SNS-032, a selective inhibitor of multiple CDKs involved in apoptosis and cell cycle regulation, has potential for antitumor activity in both solid and hematological cancers. SNS-032 is currently in phase 1 clinical studies.
SNS-032, was designed as a selective CDK2 inhibitor. Here, we show that in addition to CDK2, CDK 7 and 9 inhibitory activities also contribute to the biological activity of the molecule. The CDK2/cyclin E complex regulates entry of cells into S phase by phosphorylating Rb, a negative regulator of the transcription factor E2F. CDK2 phosphorylates a number of additional substrates, including cyclin E, signaling its degradation. Inhibiting CDK2 should therefore arrest cells in G1 and stabilize cyclin E. The cellcycle CDKs (CDK1, 2 4 and 6) are activated by phosphorylation by CDK7/cyclin H (also called CAK). Inhibition of CDK7 would therefore also result in cell-cycle arrest at multiple points in the cell cycle due to failure to activate the cell cycle CDKs. CDK 7 and 9 activate transcription by phosphorylating the CTD of RNA pol II. Inhibition of CTD phosphorylation has been shown to inhibit transcription and reduce expression of short lived proteins, including those involved in apoptosis regulation. Stalling of RNA polymerase has also been shown to activate p53, leading to apoptosis. Thus, the CDK7 and 9 inhibitory activities of SNS-032 are expected to cause cytotoxicity via induction of apoptosis.
SNS-032 is a selective CDK inhibitor, preferentially targeting CDK2, CDK7 and CDK9 in vitro. • In cell models, SNS-032 shows dual activity, targeting both cell cycle progression and apoptosis pathway proteins. • SNS-032 Inhibited CDK9 and 7-mediated phosphorylation of ser 2 and ser 5 of the CTD of RNA pol II and in turn downregulates the antiapoptotic protein Mcl-1. • SNS-032 induced a cell cycle arrest, and increased cyclin E levels are consistent with inhibition of cell cycle CDKs • Mcl-1 is a key survival factor in many B-cell malignancies. SNS-032 is being pursed as treatment for these diseases.
| Biological Activity | ||||||
|---|---|---|---|---|---|---|
| Description | SNS-032 is a novel, potent and selective CDK inhibitor of CDK2, CDK7 and CDK9 with IC50 of 38 nM, 62 nM and 4 nM, respectively. | |||||
| Targets | CDK2 | CDK7 | CDK9 | |||
| IC50 | 38 nM | 62 nM | 4 nM [1] | |||
| In Vitro | SNS-032 has low sensitivity to CDK1 and CDK4 with IC50 of 480 nM and 925 nM, respectively. SNS-032 effectively kills chronic lymphocytic leukemia cells in vitro regardless of prognostic indicators and treatment history. Compared with flavopiridol and roscovitine, SNS-032 is more potent, both in inhibition of RNA synthesis and at induction of apoptosis. SNS-032 activity is readily reversible; removal of SNS-032 reactivates RNA polymerase II, which led to resynthesis of Mcl-1 and cell survival. [1] SNS-032 inhibits three dimensional capillary network formations of endothelial cells. SNS-032 completely prevents U87MG cell–mediated capillary formation of HUVECs. In addition, SNS-032 significantly prevents the production of VEGF in both cell lines, SNS-032 prevents in vitro angiogenesis, and this action is attributable to blocking of VEGF. Preclinical studies have shown that SNS-032 induces cell cycle arrest and apoptosis across multiple cell lines. [2] SNS-032 blocks the cell cycle via inhibition of CDKs 2 and 7, and transcription via inhibition of CDKs 7 and 9. SNS-032 activity is unaffected by human serum. [3]SNS-032 induces a dose-dependent increase in annexin V staining and caspase-3 activation. At the molecular level, SNS-032 induces a marked dephosphorylation of serine 2 and 5 of RNA polymerase (RNA Pol) II and inhibits the expression of CDK2 and CDK9 and dephosphorylated CDK7. [4] | |||||
| In Vivo | SNS-032 prevents tumor cell-induced VEGF secretion in a tumor coculture model. [2] SNS-032, a new CDK inhibitor, is more selective and less cytotoxic and has been shown to prolong stable disease in solid tumors. [4] | |||||
| Clinical Trials | SNS-032 currently in phase I clinical trial for chronic lymphocytic leukemia (CLL) and multiple myeloma (MM). | |||||
| Description | SNS-032 is a selective inhibitor of CDK2 with IC50 of 48 nM. | |||||
| Targets | CDK2 | CDK7 | CDK9 | |||
| IC50 | 48 nM | 62 nM | 4 nM | |||
CLIP
http://www.mdpi.com/1420-3049/19/9/14366/htm#B39-molecules-19-14366
SNS032, previously called BMS-387032, has been developed by Sunesis. This compound, which contains a thiazole unit, selectively inhibits CDK2 (IC50: 38 nM), CDK7 (IC50: 62 nM) and CDK9 (IC50: 4 nM) [39]. Preclinical studies demonstrated that SNS032 was able to inhibit cell cycle activity along with transcription [20].
SNS032 is in phase I clinical trials for the treatment of chronic lymphoid leukemia along with multiple myeloma, and the mode of administration is intravenous [39]. The purpose is to evaluate the dose-escalation of SNS-032 along with its safety, pharmacokinetics, pharmacodynamic activity and clinical efficacy. Biomarker analyses demonstrated mechanism-based pharmacodynamic activity with inhibition of CDK7 and CDK9, although limited clinical activity in heavily pretreated patients was observed [39].
Tong, W.G.; Chen, R.; Plunkett, W.; Siegel, D.; Sinha, R.; Harvey, R.D.; Badros, A.Z.; Popplewell, L.; Coutre, S.; Fox, J.A.; et al. Phase I and pharmacologic study of SNS-032, a potent and selective CDK2, 7, and 9 inhibitor, in patients with advanced chronic lymphocytic leukemia and multiple myeloma. ASCO Annual Meeting. J. Clin. Oncol. 2010, 28, 3015–3022.

![Image result for N-(Cycloalkylamino)acyl-2-aminothiazole Inhibitors of Cyclin-Dependent Kinase 2. N-[5-[[[5-(1,1-Dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4- piperidinecarboxamide (BMS-387032), a Highly Efficacious and Selective Antitumor Agent,](https://journals.prous.com/journals/dof/20083311/html/df330932/images/sch01.gif)
SNS-032 (formerly BMS-387032) is a small-molecule cyclin-dependent kinase (CDK) inhibitor currently in phase I clinical trials for the treatment of B-cell malignancies and advanced solid tumors. Preclinical studies have shown that SNS-032 is a specific and potent inhibitor of CDK2, 7 and 9 which induces cell cycle arrest and apoptosis in tumor cell lines. It was shown to inhibit in vitro angiogenesis and prostaglandin E2 (PGE2) production, both strongly associated with tumorigenesis. Phase I clinical trials support the safety and tolerability of SNS-032 as evaluated in dose-escalation studies. The compound is currently administered by i.v. infusion but has shown promising potential for oral delivery.
![Image result for N-(Cycloalkylamino)acyl-2-aminothiazole Inhibitors of Cyclin-Dependent Kinase 2. N-[5-[[[5-(1,1-Dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4- piperidinecarboxamide (BMS-387032), a Highly Efficacious and Selective Antitumor Agent,](https://journals.prous.com/journals/dof/20083311/html/df330932/images/sch02.gif)
 NMR
NMR
CLIP
![Image result for N-(Cycloalkylamino)acyl-2-aminothiazole Inhibitors of Cyclin-Dependent Kinase 2. N-[5-[[[5-(1,1-Dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4- piperidinecarboxamide (BMS-387032), a Highly Efficacious and Selective Antitumor Agent,](https://i0.wp.com/www.rcsb.org/pdb/images/56H_600.gif)
![Image result for N-(Cycloalkylamino)acyl-2-aminothiazole Inhibitors of Cyclin-Dependent Kinase 2. N-[5-[[[5-(1,1-Dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4- piperidinecarboxamide (BMS-387032), a Highly Efficacious and Selective Antitumor Agent,](https://i0.wp.com/www.mdpi.com/ijms/ijms-14-21805/article_deploy/html/images/ijms-14-21805f2-1024.png)
The structures of representative protein kinases inhibitors based on the aminopyrazole scaffold.http://www.mdpi.com/1422-0067/14/11/21805/htm
CLIP
N-(Cycloalkylamino)acyl-2-aminothiazole Inhibitors of Cyclin-Dependent Kinase 2. N-[5-[[[5-(1,1-Dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4- piperidinecarboxamide (BMS-387032), a Highly Efficacious and Selective Antitumor Agent,

N-Acyl-2-aminothiazoles with nonaromatic acyl side chains containing a basic amine were found to be potent, selective inhibitors of CDK2/cycE which exhibit antitumor activity in mice. In particular, compound 21 {N-[5-[[[5-(1,1-dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4-piperidinecarboxamide, BMS-387032}, has been identified as an ATP-competitive and CDK2-selective inhibitor which has been selected to enter Phase 1 human clinical trials as an antitumor agent. In a cell-free enzyme assay, 21 showed a CDK2/cycE IC50 = 48 nM and was 10- and 20-fold selective over CDK1/cycB and CDK4/cycD, respectively. It was also highly selective over a panel of 12 unrelated kinases. Antiproliferative activity was established in an A2780 cellular cytotoxicity assay in which 21 showed an IC50 = 95 nM. Metabolism and pharmacokinetic studies showed that 21 exhibited a plasma half-life of 5−7 h in three species and moderately low protein binding in both mouse (69%) and human (63%) serum. Dosed orally to mouse, rat, and dog, 21showed 100%, 31%, and 28% bioavailability, respectively. As an antitumor agent in mice, 21administered at its maximum-tolerated dose exhibited a clearly superior efficacy profile when compared to flavopiridol in both an ip/ip P388 murine tumor model and in a sc/ip A2780 human ovarian carcinoma xenograft model.
CLIP

http://pubs.rsc.org/en/content/articlehtml/2016/md/c6md90040b
Heat shock factor 1 (HSF1) is a transcription factor that plays key roles in cancer, including providing a mechanism for cell survival under proteotoxic stress. Therefore, inhibition of the HSF1-stress pathway represents an exciting new opportunity in cancer treatment. We employed an unbiased phenotypic screen to discover inhibitors of the HSF1-stress pathway. Using this approach we identified an initial hit (1) based on a 4,6-pyrimidine scaffold (2.00 μM). Optimisation of cellular SAR led to an inhibitor with improved potency (25, 15 nM) in the HSF1 phenotypic assay. The 4,6-pyrimidine 25 was also shown to have high potency against the CDK9 enzyme (3 nM).

Discovery of 4,6-disubstituted pyrimidines as potent inhibitors of the heat shock factor 1 (HSF1) stress pathway and CDK9
E-mail: Paul.Workman@icr.ac.uk, Keith.Jones@icr.ac.uk
DOI: 10.1039/C6MD00159A
COMPD 25
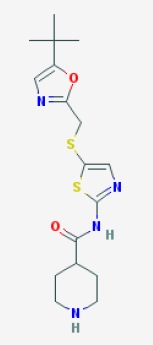
1H NMR (500 MHz, DMSO-d6) δ 10.38 (s, 1H), 9.21 (s, 1H), 8.74 (d, J = 0.9 Hz, 1H), 8.62 (dd, J = 8.2, 1.5 Hz, 1H), 8.56 (dd, J = 4.7, 1.5 Hz, 1H), 8.16-8.13 (m, 2H), 7.64 (br d, J = 8.6 Hz, 1H), 7.52-7.47 (m, 2H), 4.14 (t, J = 5.9 Hz, 2H), 2.66 (t, J = 5.9 Hz, 2H), 2.47-2.42 (m, 4H), 1.53-1.47 (m, 4H), 1.42 – 1.33 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 160.74, 158.32, 156.72, 154.88, 150.74, 146.47, 145.38, 143.74, 134.21, 125.02, 124.16, 122.29, 119.60, 114.32, 94.06, 66.49, 57.35, 54.35, 25.54, 23.88. HRMS (ESI+ ): calcd for C22H25N8O (M + H)+ , 417.2146; found 417.2163.
NOTE, THERE IS ERROR IN STRUCTURE ABOVE OF SNS 032
References
References:
[1]. Tong W.G., Chen R., Plunkett W., et al. Phase I and Pharmacologic Study of SNS-032, a Potent and Selective Cdk2, 7, and 9 Inhibitor, in Patients With Advanced Chronic Lymphocytic Leukemia and Multiple Myeloma. Journal of Clinical Oncology, 2010, 28(18):3015- 3022.
[2]. Chipumuro E., Marco E., Christensen C.L., et al. CDK7 Inhibition Suppresses Super-Enhancer-Linked Oncogenic Transcription in MYCN-Driven Cancer. Cell, 2014, 159:1-14.
[3]. Meng H., Jin Y.M., Liu H., et al. SNS-032 inhibits mTORC1/mTORC2 activity in acute myeloid leukemia cells and has synergistic activity with perifosine against Akt. Journal of Hematology & Oncology, 2013, 6:18.
[4]. Chen R., Wierda W.G., Chubb S., et al. Mechanism of action of SNS032, a novel cyclin-dependent kinase inhibitor, in chronic lymphocytic leukemia. Blood, 2009, 113(19):4637-4645.Chen et al (2010) Responses in mantle cell lymphoma cells to SNS-032 depend on the biological context of each cell line. Cancer Res. 70 6587. PMID: 20663900.
Conroy et al (2009) SNS-032 is a potent and selective CDK 2, 7 and 9 inhibitor that drives target modulation in patient samples. Cancer Chemother.Pharmacol. 64 723. PMID: 19169685.
Ali et al (2007) SNS-032 prevents tumor cell-induced angiogenesis by inhibiting vascular endothelial growth factor. Neoplasia 9 370. PMID: 17534442.
Misra et al (2004) N-(Cycloalkylamino)acyl-2-aminothiazole inhibitors of cyclin-dependent kinase 2. N-[5-[[[5-(1,1-Dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4- piperidinecarboxamide (BMS-387032), a highly efficacious and selective antitumor agent. J.Med.Chem. 47 1719. PMID: 15027863.
Abstract
SNS-032, a CDK inhibitor, exhibited modest to high anti-neuroblastoma activity against a panel of 109 neuroblastoma cell lines in the range of the therapeutic plasma levels reported for SNS-032 through a mechanism involving CDK7 and CDK9 inhibition-mediated down-regulation of XIAP, Mcl-1, BIRC2, cIAP-1 and surviving.
Abstract
The anti-AML mechanism of SNS-032, a cyclin-dependent kinase inhibitor, has been identified though characterizing in vitro effects of SNS-032 alone or in combination with perifosine.
Abstract
Although it induces apoptosis in cancer cells, SNS-032 has no significant effects on normal HSC and HPC in terms of self-renewal inhibition, differentiation suppression and apoptosis induction.
Abstract
The CDK7/9 inhibitor SNS-032-induced down-regulation of FIP1L1-PDGFRα and Bcr-Abl has the potential to be used to decrease the acquired resistant to imatinib.
Abstract
SNS-032, a CDK inhibitor, alone or in combination with Ara-C exhibited potent anti-AML activity, where down-regulation of antiapoptotic genes, cluding BCL2, XIAP amd MCL1, was associated with the synergistic anti-AML effect of the combination treatment.
CC(C)(C)C1=CN=C(O1)CSC2=CN=C(S2)NC(=O)C3CCNCC3
ミチグリニドカルシウム水和物 , Mitiglinide calcium hydrate, 快如妥/Glufast

- MF C19H25NO3
- MW 315.407 Da
Mitiglinide (INN, trade name Glufast) is a drug for the treatment of type 2 diabetes.[1]
Mitiglinide belongs to the meglitinide class of blood glucose-lowering drugs and is currently co-marketed in Japan by Kissei and Takeda. The North America rights to mitiglinide are held by Elixir Pharmaceuticals. Mitiglinide has not yet gained FDA approval.
Mitiglinide calcium hydrate was approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) on January 29, 2004. It was co-developed and co-marketed as Glufast® by Takeda and Kissei in Japan.
Mitiglinide is a rapid-acting insulin secretion-stimulating agent. It stimulates insulin secretion by closing the ATP-sensitive K+ (ATP) channels in pancreatic beta-cells. It is indicated for the treatment of type 2 diabetes mellitus.
Glufast® is available as tablet for oral use, containing 5 mg or 10 mg of Mitiglinide calcium hydrate. The recommended dose is 10 mg three times daily just before each meal (within 5 minutes).
China , Approved 2010-04-19, 快如妥/Glufast, Kissei
ミチグリニドカルシウム水和物

C38H48CaN2O6▪2H2O : 704.92
[207844-01-7]
Pharmacology
Mitiglinide is thought to stimulate insulin secretion by closing the ATP-sensitive K(+) K(ATP) channels in pancreatic beta-cells.

Dosage
Mitiglinide is delivered in tablet form.

| Molecular Weight | 333.42 |
| Formula | C19H27NO4 |
| CAS Number | 207844-01-7 |
Mitiglinide calcium hydrate




The condensation of dimethyl succinate (I) with benzaldehyde (II) by means of NaOMe in refluxing methanol followed by hydrolysis with NaOH in methanol/water gives 2-benzylidenesuccinic acid (III). Compound (III) is treated with refluxing Ac2O, yielding the corresponding anhydride (IV), which by reaction with cis-perhydroisoindole (V) in toluene affords the monoamide (VI). This amide is reduced with H2 over a chiral Rhodium catalyst and treated with (R)-1-phenylethylamine (VII) to provide the chiral salt (VIII) as a single diastereomer isolated by crystallization. Finally, this salt is treated first with aqueous NH4OH and then with aqueous CaCl2.

he optical resolution of racemic 2-benzylsuccinic acid (XV) using the chiral amines (R)-1-phenylethylamine (VII), (R)-1-(1-naphthyl)ethylamine (XIV) or (S)-1-phenyl-2-(4-tolyl)ethylamine (XVI) is carried out by fractional crystallization of the corresponding diastereomeric salts and treatment with 2N HCl, providing the desired enantiomer 2(S)-benzylsuccinic acid (XVII). Reaction of (XVII) with SOCl2 gives the corresponding acyl chloride (XVIII), which is treated with 4-nitrophenol (XIX) and TEA in dichloromethane to yield the activated diester (XX). The regioselective reaction of (XX) with cis-perhydroisoindole (V) in dichloromethane affords the monoamide (XXI), which by reaction with HCl and methanol provides the corresponding methyl ester (XXII). This ester is hydrolyzed with NaOH to the previously described chiral succinamic acid (XIII), which is finally converted into its calcium salt.

PATENT
https://www.google.com/patents/WO2009047797A2?cl=en
Perhydroisoindole derivative, (S)-mitiglinide of formula I is a potassium channel antagonist for the treatment of type 2 diabetes mellitus and is chemically known as (5)-2-benzyl-3-(cis-hexahydro-2- isoindolinylcarbonyl) propionic acid.
Formula I

It has potent oral hypoglycemic activity and is structurally different from the sulphonylureas, although it stimulates calcium influx by binding to the sulphonylurea receptor on pancreatic β-cells and closing K+ ATP channels. Perhydroisoindole derivatives including (S)-mitiglinide and salts thereof were first disclosed in US patent 5,202,335. This patent discloses preparation of (S)-mitiglinide by the reaction of (5)-3-benzyloxycarbonyl-4-phenylbutyric acid with cis-hexahydroisoindoline in the presence of N- methylmorpholine and isobutyl chloroformate followed by debenzylation with palladium on carbon in ethyl acetate to yield (5)-mitiglinide as viscous oil. (S)-Mitiglinide is isolated as its hemi calcium salt using calcium chloride in water which is further recrystallized with diisopropyl ether. Melting point of calcium salt of mitiglinide calcium dihydrate salt is herein reported as 179-185 0C. (S)-Mitiglinide prepared by the above process is obtained in low yields. Further, the synthetic method described in the patent does not enable the desired regioselectivity. Extensive purification steps are required to obtain the desired compound, which makes the process unattractive from industrial point of view. US patent 6,133,454 discloses a process for the preparation of (S)-mitiglinide by reacting dimethyl succinate with benzaldehyde in methanolic medium, to yield a diacid which is converted to corresponding anhydride and is further reacted with the perhydroisoindole to yield 2-[(cis- perhydroisomdol^-ytycarbonylmethyl^-phenylacrylic acid which is then subjected to catalytic hydrogenation using the complex rhodium/(2S,4S)-N-butoxycarbonyl-4-diphenylphosphino-2-diphenyl- phosphino-methylpyrrolidine (Rh/(S,S) BPPM) as asymmetric hydrogenation catalyst, followed by conversion to pharmaceutically acceptable salt of (S)-mitiglinide. The above patent utilizes ruthenium complex which is expensive, carcinogenic and toxicity, hence not recommended for industrial scale. European patent publication no. EP 0967204 discloses the preparation of mitiglinide by deprotecting benzyl-(S)-2-benzyl-3-(cis-hexahydro-2-isoindolinyl-carbonyl) propionate and converting the same to calcium dihydrate salt in crystalline form using calcium chloride, water and ethanol. The crystals of calcium salt are further recrystallized using ethanol and water. But the patent is silent about the crystalline form of mitiglinide calcium.
It will be appreciated by those skilled in the art that perhydroisoindole derivative, (S)-mitiglinide of formula I contains a chiral centre and therefore exists as enantiomers. Optically active compounds have increasingly gained importance since the technologies to develop optically active compounds in high purity have considerably improved. Obtaining asymmetric molecules has traditionally involved resolving the desired molecule from a racemic mixture using a chiral reagent, which is not profitable as it increases the cost and processing time. Alternatively, desired enantiomer can be obtained by selective recrystallization of one enantiomer. However such a process is considered inefficient, in that product recovery is often low, purity is uncertain and more than 50% of the material is lost. Enantiomers can also be resolved chromatographically, although the large amount of solvent required for conventional batch chromatography is cost prohibitive and results in the preparation of relatively dilute products. Limited throughput volumes also often make batch chromatography impractical for large-scale production. Even so, it is a common experience for those skilled in the art to find chiral separation of certain chiral mixtures to be inefficient or ineffective, thereby resulting in the efforts towards development of newer methodologies for asymmetric synthesis.
It would be of significant advantage to obtain (.S)-mitiglinide by development of reaction conditions necessary for productive manufacture of the required (5)-enantiomer, substantially free of the unwanted (R)-enantiomer, in large quantities that meet acceptable pharmaceutical standards. It is the property of the solid compounds to exist in different polymorphic form. By the term polymorphs mean to include different physical forms, crystal forms, crystalline/liquid crystalline/non-crystalline (amorphous) forms. This has especially become very interesting after observing that many antibiotics, antibacterials, tranquilizers etc, exhibit polymorphism and some/one of the polymorphic forms of a given drug exhibit superior bio-availability and consequently show much higher activity compared to other polymorphs. It has also been disclosed that the amorphous forms in a number of drugs exhibit different dissolution characteristics and in some cases different bioavailability patterns compared to the crystalline form [Konne T., Chem. Pharm. Bull. 38, 2003 (1990)]. The solubility of a material is also influenced by its solid-state properties, and it has been suggested that the solubility of an amorphous compound is 10 to 1600 times higher than that of its most stable crystalline structures (Bruno C. Hancock and Michael Parks, ‘What is the true solubility advantage for amorphous pharmaceuticals’, Pharmaceutical Research 2000, Apr; 17(4):397-404). Thus it can be concluded that amorphous products are in general more soluble and often show improved absorption in humans.
Thus, there is a widely recognized need for developing a stable polymorph, which would further offer advantages over crystalline forms in terms of better dissolution and the availability profiles. Also none of the prior art references disclose amorphous form of mitiglinide calcium. Thus present invention provides amorphous form of mitiglinide calcium.
It is also required that the final API like mitiglinide whether in the amorphous form or crystalline form must be free from the other impurities including the unwanted enantiomer, these can be side product and by product of the reaction, degradation products and starting materials. Impurities in final API are undesirable and in extreme cases, might even be harmful to a patient being treated with a dosage form containing the API. Therefore impurities introduced during commercial manufacturing processes must be limited to very small amounts and are preferably substantially absent. These limits are less than about 0.15 percent by weight of each identified impurity and 0.10 % by weight of unidentified and/or uncharacterized impurities. After the manufacture of APIs, the purity of the products, such as (S)- mitiglinide calcium dihydrate is required before commercialization, and in the manufacture of formulated pharmaceuticals. Therefore, pharmaceutical active compounds must be either free from these impurities or contain the impurities in acceptable limits. There is also a need for the isolation, characterization and identification of the impurities and their use as reference markers and reference standard. Thus, the present invention meets the need in the art for a novel, efficient and industrially advantageous process for providing optically pure perhydroisoindole derivatives, particularly (iS)-mitiglinide, which is unique with respect to its simplicity, scalability and involves controlling the steps of the reaction so that predominantly the desired (S)-enantiomer is produced in high yields and purity. The present invention also provides substantially pure (S)-mitiglinide and salts thereof having novel amide impurity in acceptable limit or free from this impurity.
Example 1: Preparation of (R) 4-benzyl-3-(3-phenylpropionv0-oxazolidin-2-one To a solution of (R)-4-benzyloxazolidin-2-one (50 g), 4-dimethylaminopyridine (4.85 g), 3-phenyl propionic acid (55.08 g) in dichloromethane (375 ml) under nitrogen atmosphere at 0-5 0C, dicyclohexylcarbodiimide (975.65 g) was added. The temperature was slowly raised to 25-30 0C and stirring was continued until no starting material was left as was confirmed by thin layer chromatography. Dicyclohexylurea formed during the reaction was filtered, washed with dichloromethane (200 ml) and the filtrate was washed with saturated solution of sodium bicarbonate (500 ml). The solution was dried over sodium sulphate and solvent was distilled off to obtained crude product which was purified from methanol (200 ml) at 10-15 °C and washed with methanol (50 ml) to obtain 81.0 g of the title compound. Example 2: Preparation of 3(5)-benzyl-4-(4-(J?)-benzyl-2-oxo-oxazolidin-3-yl)-4-oxo-butyrϊc acid tert-butyl ester
To a solution of (/?)-4-benzyl-3-(3-phenyl-propionyl)-oxazolidin-2-one (150 g) in anhydrous tetrahydrofuran (1.5 It) was added a solution of sodium hexamethyldisilazane (462 ml, 36-38% solution in tetrahydrofuran) with stirring at -85 to -95 0C for 60 minutes. Tert-butyl bromo acetate (137.5 g) in tetrahydrofuran (300 ml) was added to reaction mass and then stirred to 60 minutes at -85 to -95 0C. After completion of the reaction (monitored by TLC), the reaction mixture was poured into ammonium chloride solution (10%, 2.0 It) and extracted with ethyl acetate (2×750 ml). The combined organic layer was washed with demineralized water (1×750 ml) and dried over sodium sulphate. The solvent was evaporated under reduced pressure to obtain oily residue which was stirred with mixture of n-hexane (100 ml) and isopropyl alcohol (100 ml) at Oto -50C, filtered and dried under vacuum to obtain 153.12 g of title compound having chemical purity 99.41%, chiral purity 99.91% by HPLC, [α]D 20: (-)97.52° (c = 1, CHCl3) and M.P. : 117.1-118.20C.
Example 3: Preparation of 3(5)-benzyl-4-(4(i?)-benzyl-2-oxo-oxazolidin-3-yl)-4-oxobutyric acid Trifluoroacetic acid (100 g) was added to a solution of 3(5)-benzyl-4-(4-(/?)-benzyl-2-oxo-oxazolidin-3- yl)-4-oxobutyric acid tert-butyl ester (100 g) in dichloromethane (700 ml) at 25 0C and mixture was stirred further for about 12 hours ( when TLC indicated reaction to be complete). The reaction mixture was poured in to ammonium chloride solution (10%, 500 ml). The dichloromethane layer was separated and aqueous layer was extracted with dichloromethane (2 x 250 ml). The combined organic layer was dried over sodium sulphate and evaporated under reduced pressure to obtain title compound. The crude product was recrystallized from a mixture of ethyl acetate: n-hexane (1:4, 500 ml) to obtain 78.75g of the title compound having purity 99.56% by HPLC and M.P.: 145.9-146.40C.
Example 4: Preparation of (2S)-2-benzyl-l-((4R)-4-benzyl-2-oxo-oxazolidin-3-vI)-4-(hexahydro- isoindolin-2-yl)-butane-l,4-dione
To a solution of 3(5)-benzyl-4-(4-(/?)-benzyl-2-oxo-oxazolidin-3-yl)-4-oxo-butyric acid (50 g) in anhydrous dichloromethane (1.25 It) was added triethylamine (50 ml) with stirring at -20 to -30 0C and the stirred for 15 minutes. A solution of isobutylchloroformate (37.50g) in anhydrous dichloromethane (50 ml) was added at -20 to -30 0C and stirred for 60 minutes. Thereafter, a solution of cis- hexahydroisoindoline (32.50 g) in anhydrous dichloromethane (50 ml) was slowly added by maintaining temperature -20 to -300C. After the completion of the reaction (monitored by HPLC), the mixture was successively washed with 0.5N hydrochloric acid solution (500 ml), brine (300 ml) and dried over sodium sulphate. The solvent was evaporated under reduced pressure to obtain 102.0 g of the title compound having purity 94.39% by HPLC.
Example 5: Purification of r2S)-2-benzyl-l-((4R)-4-benzyl-2-oxo-oxazolidin-3-yl)-4-(hexahydro- isoindolin-2-vD-butane-l,4-dione
To the crude (2S)-2-benzyl-l-((4R)-4-benzyl-2-oxo-oxazolidin-3-yl)-4-(hexahydro-isoindolin-2-yl)- butane- 1,4-dione (51.0 g) was added methanol (150 ml) and the mixture was stirred for 5 hours at 0 to 5 0C. Solid that precipitated out was filtered, slurry washed with cold methanol (25 ml) and dried at 45 -50 0C under vacuum to obtain 28.80 g of pure title compound as a crystalline solid having purity of 99.71% by HPLC and M. P.: 104.1-105.70C.
Example 6: Preparation of calcium salt of (-SVmitiglinide. Step-1: Preparation of (-SVmitiglinide
(2S)-2-Benzyl- 1 -((4R)-4-benzyl-2-oxo-oxazolidin-3-yl)-4-(hexahydro-isoindolin-2-yl)-butane- 1 ,4-dione (28.0 g) was dissolved in tetrahydrofuran (196 ml) and a mixture of lithium hydroxide monohydrate (3.51 g) in demineralized water (56 ml) and hydrogen peroxide (40% solution, 5.5 ml) was added with stirring at 0 to 5 0C over a period of 30 minutes. The reaction mixture was further stirred at 0 to 5 0C till the completion of the reaction. After the completion of the reaction (monitored by TLC), the reaction was quenched with the addition of cooled sodium meta-bisulphate solution (25%, 168 ml) at 0 to 10 0C. The reaction mixture was extracted with ethyl acetate (2×112 ml), the layers were separated and the aqueous layer was discarded. The HPLC analysis of the aqueous layer shows 0.77% of amide impurity. The ethyl acetate layer was then extracted with aqueous ammonia solution (4%, 2×40 ml). The layers were separated and the aqueous layer was further extracted with ethyl acetate (2×280 ml). Combined ethyl acetate layer was discarded. This aqueous layer (280 ml) was used as such in the next stage. The aqueous layer display purity 96.19 % by HPLC and amide impurity 0.04% by HPLC. Step-2: Preparation of calcium salt of dSVmitiglinide
To the above stirred solution of (S)-mitiglinide in water and ammonia(280 ml), methanol (168 ml) was added, followed by calcium chloride (4.48 g) dissolved in demineralized water (56 ml) at ambient temperature and the mixture was stirred for 2 hours. The resulting precipitate was filtered, successively slurry washed with water (3 x 140 ml) and acetone (2 x 70 ml) and dried at 450C -500C under vacuum to obtain 16.1 g of title compound having purity 99.67% by HPLC and amide impurity 0.01% by HPLC. The title product was re-precipitated from a mixture of methanol and water and dried to obtain pure title compound.
Example 7: Preparation of (.SVmitiglinide
To a solution of (2S)-2-benzyl-l-((4R)-4-benzyl-2-oxo-oxazolidin-3-yl)-4-(hexahydro-isoindolin-2-yl)- butane- 1,4-dione (50 g) in tetrahydrofuran (350 ml) was added a solution of lithium hydroxide monohydrate (8.65 g) in demineralized water (100 ml) and hydrogen peroxide (30% w/w, 40 ml) with stirring at 5 to 10 0C over a period of 15 minutes. After the completion of reaction, sodium meta- bisulphate solution (40%, 500 ml) was added to the reaction mixture and the mixture was extracted with ethyl acetate (2 x 250 ml). The organic layer was dried over sodium sulphate and evaporated under vacuum to obtain 45.5 g of title compound having 35 % of R-benzyl oxozolidin-2-one as impurity. Example 8: Purification of (.S)-mitiglinide
Aqueous ammonia solution (4%, 300 ml) was added to the crude (5)-mitiglinide (30 g) and stirred. The reaction mixture was washed with ethyl acetate (3 x 300 ml). Thereafter the reaction mixture was acidified to pH 1 to 2 with IN hydrochloric acid solution (250 ml) and extracted with ethyl acetate (2 x 150 ml). The layers were separated and ethyl acetate layer was washed with demineralized water (2 x 150 ml), dried over sodium sulphate and then evaporated under reduced pressure to obtain 16.2 g of pure (5)-mitiglinide having purity 95.55% by HPLC Example 9: Preparation of calcium salt of (S)-mitiglinide
To a solution of (<S)-mitiglinide (15 g) in water (150 ml) and aqueous ammonia solution (25%, 15 ml) at 25 to 30 0C, a solution of calcium chloride (7.5 g) in demineralized water (37.5 ml) was added. The mixture was stirred for 1 hour to precipitate the calcium salt of (5)-mitiglinide dihydrate. The resulting precipitate was filtered, slurry washed with water (3 x 150ml) and dried at 45 to 50 0C to obtain 13.25 g of the title compound having purity of 98.84% by HPLC. Example 10: Purification of calcium salt of (5)-mitiglinide
(iS)-mitiglinide calcium (10 g) was dissolved in dimethylformamide (100 ml). This is followed by the addition of demineralized water (500 ml) at 25 to 30 0C. The mixture was stirred for 30 minutes. The precipitated solid was filtered, washed with water (10x 50ml) and dried at 45 to 50 0C under vacuum to obtain 8g of pure title compound as a crystalline solid having purity of 99.62% by HPLC. Example 11: Preparation of amorphous mitiglinide calcium
Crystalline mitiglinide calcium (2.0 g) was dissolved in tetrahydrofuran (20 ml) and filtered to remove undissolved and suspended particles. The solvent was then evaporated under vacuum to obtain a powder which was then dried under vacuum at 40-600C to obtain 1.70 g of the title compound. Example 12: Preparation of amorphous mitiglinide calcium
Crystalline mitiglinide calcium (2.0 g) was dissolved in dichloromethane (30 ml) and filtered to remove undissolved and suspended particles. The solvent was then evaporated under vacuum to obtain a powder which was then dried under vacuum at 40-600C to obtain 1.64 g of the title compound. Example 13: Preparation of amorphous mitiglinide calcium
Mitiglinide (2.0 g) was dissolved in methanol (20 ml) and methanolic ammonia (5.0 ml) solution was added to it. The solution was stirred at 25-30 0C and calcium chloride (1.5 g) dissolved in methanol was mixed with the solution of mitiglinide and ammonia in methanol and the solution was filtered to remove the suspended particles. The solvent was then evaporated under vacuum to obtain a powder which was then dried under vacuum at 40-600C to obtain 1.9 g of the title compound. Example 14: Preparation of amorphous mitiglinide calcium
Mitiglinide (2.0 g) was dissolved in dichloromethane (20 ml) and aqueous ammonia (3.6 ml, 25 % solution) was added to it. The solution was stirred at 25-300C and solid calcium chloride (1.5 g) was mixed with the solution of mitiglinide and ammonia in dichloromethane and the solution warmed at 30 – 35 0C. The solution was washed with water (2 xlO ml) and the clear solution was dried over sodium sulfate, filtered and evaporated under vacuum and finally dried at under vacuum at 40-60 0C to obtain 1.75 g of the title compound.
Example 15: Preparation of amorphous mitiglinide calcium
Crystalline mitiglinide calcium dihydrate (2.0 g) was dissolved in ethyl acetate (30 ml) and filtered to remove undissolved and suspended particles. Approimately. 60 % of the solvent was distilled off under vacuum to obtain a stirrable solution. The solution was then cooled to 15-2O0C, mixed with n-heptane (20 ml) and the mixture was stirred for 30 minutes. The resulting solid was filtered, washed with n-heptane and dried under vacuum at 45-600C to yield 1.72 g of the title compound. Example 16: Preparation of amorphous mitiglinide calcium
Crystalline mitiglinide calcium (2.Og) was dissolved in dichloromethane (30 ml) and filtered to remove undissolved and suspended particles. Approximately 60 % of the solvent was distilled off under vacuum to obtain a stirrable solution. The solution was then cooled to 15-200C and mixed with diisopropyl ether (20 ml). The mixture was stirred for 30 minutes and the resulting solid was filtered, washed with diisopropyl ether and dried under vacuum at 45-600C to obtain 1.70 g of the title compound. Example 17: Preparation of amorphous mitiglinide calcium
Mitiglinide (2.0 g) was dissolved in dichloromethane (20 ml) and aqueous ammonia (3.6 ml, 25 % solution) solution was added to it. The solution was stirred at 25-30 0C and mixed with solid calcium chloride (1.5 g) and the solution warmed at 30-35 0C and stirred for 30 minutes. The solution was washed with water (2 x 10 ml) and the clear solution was dried over sodium sulfate, and filtered. Approximately 60% of the solvent was distilled off under vacuum and the resulting viscous oil was cooled to 10-15 0C and mixed with diisopropyl ether (50 ml). The reaction mixture was stirred for 30-35 minutes and the resulting solid was filtered and dried at 40-600C to obtain 1.75 g of the title compound. Example 18: Conversion of amorphous mitiglinide calcium into crystalline mitiglinide calcium A suspension of amorphous mitiglinide calcium in diisopropyl ether (30 ml) was stirred for 2 hours at 25- 300C, filtered and dried under vacuum at 45-600C to obtain crystalline form of mitiglinide calcium. Example 19: Preparation of crystalline mitiglinide calcium
To a solution of mitiglinide (2.5 g) in water (2.5 ml), aqueous ammonia solution (approx 25%, 4.0 ml) and acetonitrile (2.5 ml) at 10-150C, calcium chloride (1.32 g) dissolved in demineralized water (15 ml) was added. The mixture was stirred for 2 hours. The resulting precipitate was filtered, slurry washed with water (3 x 25 ml) and acetone (2 x 5 ml) and dried at 45-500C under vacuum to obtain 2.12 g of title compound having purity: 99.72 % by HPLC.
Example 20: Preparation of crystalline mitiglinide calcium
To a solution of mitiglinide (2.5 g) in water (2.5 ml), aqueous ammonia solution (approx 25%, 4.0 ml) and tetrahydrofuran (2.5 ml) at 10-150C, calcium chloride (1.32 g) dissolved in demineralized water (15 ml) was added. The mixture was stirred for 2 hours. The resulting precipitate was filtered, slurry washed with water (3 x 25 ml) and acetone (2 x 5 ml) and dried at 45-500C under vacuum to obtain 1.95 g of title compound having purity: 99.52 % by HPLC.
Example 21; Preparation of crystalline mitiglinide calcium
To a solution of mitiglinide (30.0 g) in water (300 ml), aqueous ammonia solution (approx 25%, 48 ml) and acetone (300 ml) at 10-150C, calcium chloride (15.8 g) dissolved in demineralized water (180 ml) was added. The mixture was stirred for 2 hours. The resulting precipitate was filtered, slurry washed with water (3 x 300 ml) and acetone (2 x 60 ml) and dried at 45-500C under vacuum to obtain 24.32 g of title compound having purity: 99.42 % by HPLC.
Example 22: Preparation of crystalline mitiglinide calcium
To a solution of mitiglinide (3.0 g) in water (30 ml), aqueous ammonia solution (approx 25%, 4.8 ml) and isopropyl alcohol (300 ml) at 10-150C, calcium chloride (1.58 g) dissolved in demineralized water
(18 ml) was added. The mixture was stirred for 2 hours. The resulting precipitate was filtered, slurry washed with water (3 x 30 ml) and acetone (2 x 6 ml) and dried at 45-500C under vacuum to obtain 1.92 g of title compound having purity: 99.65 % by HPLC.
Example 23: Preparation of (2S)-2-benzyWV-((lR)-l-benzyl-2-hydroxy-ethyl)-4-(hexahvdro- isoindolin-2-yl)-4-oxo-buryramide
To a solution of (2S)-2-benzyl-l-((4R)-4-benzyl-2-oxo-oxazolidin-3-yl)-4-(hexahydro-isoindolin-2-yl)- butane-l,4-dione (20.0 g) in tetrahydrofuran (140 ml), a solution of lithium hydroxide monohydrate
(3.43 g,) in demineralized water (40 ml) was added and the reaction mixture was refluxed for 4 hours till the completion of the reactions (monitored by thin layer chromatography). After the completion of the reaction, the reaction mixture was poured into demineralized water (100 ml) and extracted with ethyl acetate (2 x 80 ml). The combined organic layer was washed with water (80 ml) and dried over sodium sulphate. The solvent was evaporated under reduced pressure to give residue which was stirred in isopropyl alcohol at 0-5 0C for 5 hours. The mixture was filtered and then dried at 40-45 0C under vacuum to obtain 12.48 g of title compound having purity 99.77 % by HPLC. Melting point = 77 – 800C.

PAPER
An Effective and Convenient Method for the Preparation of KAD-1229
Helvetica Chimica ActaVolume 87, Issue 8, Version of Record online: 27 AUG 2004
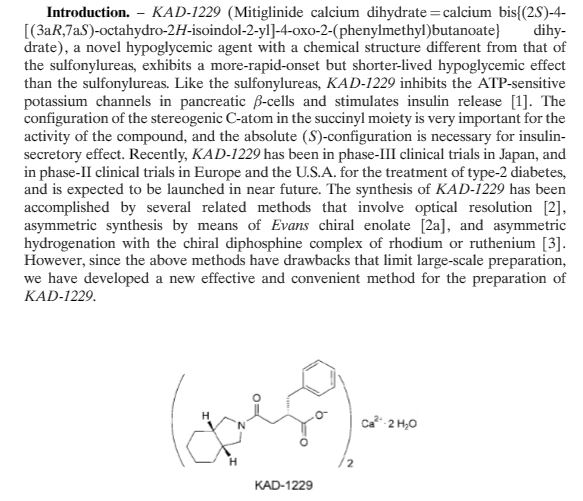

PAPER
asian journal of chemistry asian journal of chemistry
(S)-Mitiglinide calcium dihydrate is designated chemically … Identification, Synthesis and Characterization of Impurities of (S)-Mitiglinide Calcium Dihydrate………http://www.asianjournalofchemistry.co.in/(X(1))/User/ViewFreeArticle.aspx?ArticleID=26_9_51
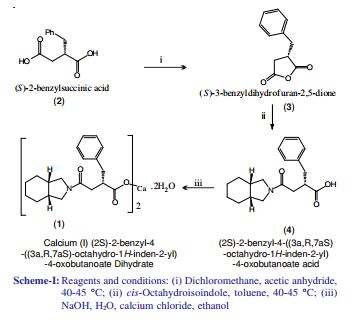
PATENT
CN 102382033

PATENT
https://www.google.com/patents/CN104311471A?cl=en
Mitiglinide calcium (mitiglinide calcium), the chemical name (2S) -2_ benzyl-3- (cis – hexahydro-2-isoindoline-carbonyl) propionic acid calcium salt dihydrate , for the treatment of type II diabetes. Kissei by Japanese pharmaceutical company research and development, and for the first time on sale in Japan in May 2004. Mitiglinide calcium is the second repaglinide, nateglinide after the first three columns MAG urea drugs, are ATP-dependent potassium channel blocker, is a derivative of phenylalanine, and its mechanism Similar sulfonylureas, but a faster onset of action and short half-life, is conducive to reducing postprandial blood glucose in diabetic patients, and avoid continuous glucose-induced low blood sugar, with the “in vitro pancreas” reputation.
郑德强 etc. on “Food and Drug” magazine was first disclosed the synthesis of calcium Mitiglinide, this method dimethyl succinate and benzaldehyde for raw materials, Stobble condensation, hydrolysis, dehydration anhydride, cis – perhydro isoindole reduced to give racemic acid after condensation, and then split, and salt get Mitiglinide calcium. Specific synthetic route the following equation. The method is relatively complex, in the preparation process to generate half of the unwanted enantiomer, which will waste a lot of cis – perhydro isoindole, and in the preparation of cis – to use science as a whole hydride hydrogen isoindole time reducing agent, the operation is more complicated, the cost is relatively high, and the chiral amine as a resolving agent split, the yield is low.

The patent discloses a CN201010573666 diethyl succinate and benzaldehyde, condensation occurs Stobble sodium ethoxide in ethanol and then hydrolyzed benzylidene succinic acid, succinic acid benzylidene get by catalytic hydrogenation DL-2-benzyl succinic acid, DL-2-benzyl succinic acid by (R) – a chiral amine resolving to give (S) -2- benzyl succinic acid, (S) -2- benzyl succinic acid anhydride to generate its role in the acetic anhydride, and the resulting acid anhydride and cis – hexahydro isoindole reaction of Mitiglinide acid, calcium chloride and ammonia most 后米格列奈 acid reacts with calcium Mitiglinide dihydrate. The synthesis route following formula. This method effectively avoids the expensive intermediate cis – perhydro isoindole waste, reduce costs, but still amounted to a six-step synthesis route much so that the reagent type, long cycle, low yield, and direct use in the synthesis process Sodium block protonated reagent preparation sodium methylate, generate a lot of flammable hydrogen gas, limiting the industrial application of the method.
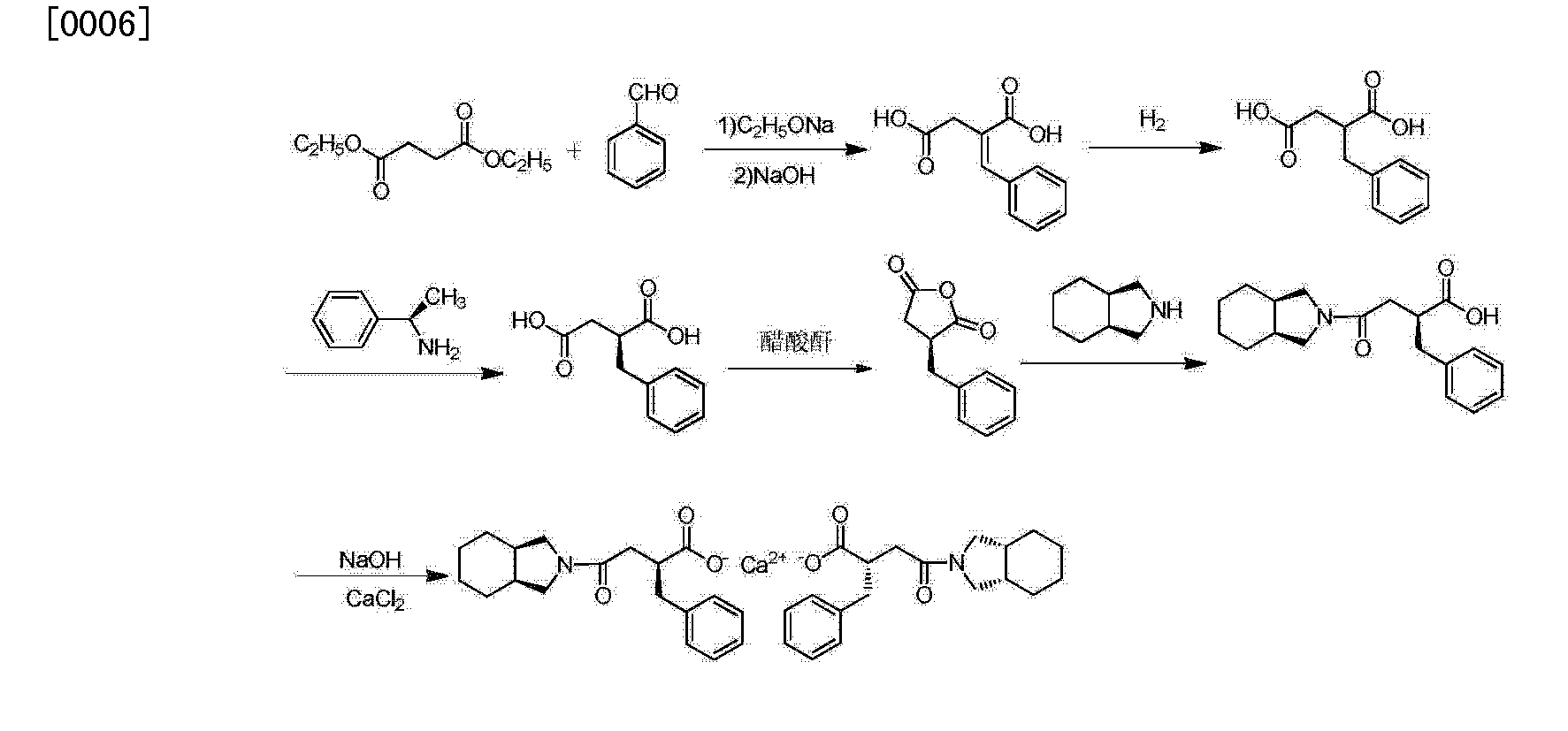
The present invention solves is to overcome the existing routes that exist in step lengthy reagent variety, low yield, long cycle, high cost, not suitable for industrial production shortcomings. The present invention provides the following formula preparation process route mitiglinide calcium, organic solvent for this preparation method uses less synthesis process is simple, high yield, good purity, suitable for industrial production.

An improved Mitiglinide calcium industrialized preparation method comprises the following steps: Step 1: Preparation of 2-benzylidene succinic acid; 2 steps: (S) prepared _2_ section succinic acid; Step 3: 2- (S) – section group _4_ oxo – (cis – perhydro isoindol-2-yl) butyric acid; Step 4: Preparation Mitiglinide calcium. Characterized in that: in step 1, using commercially available reagents protonated organic bases, protonation process using an organic alkali solution was slowly feeding methods. Step 2 chiral asymmetric reduction. Step 3 fails anhydride using direct selective amidation. Step 4 beating impurities using an aqueous solvent, prepared mitiglinide calcium dihydrate purification method.
The preparation step 1, using a commercially available organic bases as sodium methoxide or sodium ethoxide protonation agent. As optimization program, feeding method using sodium methoxide or sodium ethoxide solution formulated as the corresponding alcohol and the corresponding dialkyl succinate protonating a nucleophilic substitution reaction.
The preparation method described in Step 2, the use of Ru with BINAP homogeneous catalyst Ru (OAc) 2 [(S) -BINAP] as a chiral asymmetric synthesis of chiral reducing reagent.
The steps of the preparation method 3, using ethyl acetate as a reaction solvent, acid binding agent triethylamine do, imidazole and thionyl chloride selective amidation reagent, for cis – perhydro isoindole conduct Selective condensation title intermediate.
The step of preparing said 4, mitiglinide calcium crude product was slurried in 95% ethanol by suction, after simple preparation of high purity mitiglinide calcium dihydrate.
More specifically, the industrialized Mitiglinide calcium preparation, the following steps: Step 1: Preparation of succinate 2_ Benzylidene

Sodium methoxide (sodium ethoxide) was dissolved in methanol (ethanol), was added dropwise to dimethyl succinate (ethyl) ester, was heated at reflux for 30min, benzaldehyde was added dropwise under reflux, stirring at reflux completed the dropwise 3~5h, drops adding an aqueous solution of 4N NaOH dropwise Bi refluxed 4~6h, cooled to room temperature, adjusted with 6N HCl San PH 2, a solid precipitated, centrifuged, and dried to give the title intermediate 1. Step 2: Preparation of (S) -2- acid, benzyl butyl

Intermediate 1, methanol, and Ru (OAc) 2 [(S) -BINAP] into the reactor, the reactor with N2 the replacement air after heating to 50 ° C, a hydrogen pressure through 10h, cooled, filtered, The filtrate was concentrated to dryness to give the title intermediate 2. Step 3: 2- (S) – benzyl-4-oxo – (cis – perhydro isoindol-2-yl) butyric acid

Ethyl acetate was added to the reactor, triethylamine, imidazole and Intermediate 2, was stirred and cooled to -15~-5 ° C, was added dropwise thionyl chloride addition was complete, the -15 ° C~_5 ° C Under continued stirring 6h, a solution of cis – perhydro isoindole, drip completed, stirred at room temperature overnight, the reaction mixture was added IN hydrochloric acid, stirred Ih, separation, and the organic layer was washed with sodium hydroxide solution to extract IN The combined aqueous layer was washed with a small amount of ethyl acetate, the aqueous layer was adjusted with IN hydrochloric acid and the PH = 3, the aqueous layer was extracted with ethyl acetate, the organic layers combined, washed with water and saturated brine, and the organic layer was dried over anhydrous Na2SO4, filtered and the filtrate concentrated under reduced pressure to obtain the objective compound 3 billion Step 4: Preparation of calcium Mitiglinide

The 3 was dissolved in ethanol, was added 2N sodium hydroxide solution, after mixing the solution was added dropwise a 10% aqueous solution of calcium chloride, the reaction mixture was stirred vigorously 3~5h, ice-cooled, filtered, the filter cake with 95% ethanol beating crystallization, filtration, and dried in vacuo to give the title compound I.
Accordingly, the present invention is a method for preparing mitiglinide calcium has the following advantages:
1, Step 1, using commercially available sodium methylate (sodium ethanol) instead of sodium block as a proton agent, effectively avoid the risk of sodium block formed during the reaction a lot of flammable hydrogen gas, industrial production safer. Another use dropping protonated reagent feeding method can effectively avoid succinic acid alkyl ester of two methylene groups are protonated and reduce the incidence of side effects, so that the yield increased by nearly 20%.
2, Step 2, the selective reduction of chiral reagent (S) -BINAP instead of the original route after the first split reduction method, not only simplifies the reaction step, but low yield while avoiding split It leads to the risk of an increase in cost.
3, Step 3, the fixed selective amidation reaction conditions instead of the original first into anhydride after amidation reaction that simplifies the reaction steps to reduce the unit operations, shortening the production cycle, improve production efficiency.
4, Step 4, by using an aqueous solution of calcium Mitiglinide ethanol refining crude beating, then dried under reduced pressure to control the moisture content and reduce the difficulty of the operation, more conducive to industrial production.DETAILED DESCRIPTION The following examples further illustrate the invention, but the present invention is not limited thereto. Example One Step I: Preparation 2_ benzylidene succinic acid Sodium methoxide (9kg) and methanol (48L) into the 100L reactor, stirring to dissolve, into the high slot 50L. The dimethyl succinate (20kg) into the 200L reaction vessel, heated to reflux, methanol was added dropwise a solution of fast high tank of sodium methoxide, refluxed for reaction completion dropwise 30min, was added dropwise under reflux benzaldehyde (10. 9kg) dropwise with stirring at reflux completed 3~5h, HPLC detection benzaldehyde completion of the reaction, a solution of aqueous 4N NaOH (38L), Bi dropwise refluxed 4~6h, cooled to room temperature, 2, adjusted with 6N HCl and the precipitated solid was San PH, centrifugation, and dried in vacuo to give a pale yellow solid 19kg, i.e. an intermediate, yield 90%. Step 2: Preparation of (S) -2- butyric acid benzyl 200L detecting a high pressure hydrogenation reactor airtight, Intermediate I (19kg), methanol (95L) containing 5% Ru (0Ac) 2 [(S ) -BINAP] molecular sieve (SBA-15) supported catalyst (0. 95kg, homemade) into the reactor, purge the inside of the reactor with N2 atmosphere, followed by heating to 50 ° C, atmospheric pressure hydrogen-10h, cooled, filtered and the filtrate was concentrated to dryness under reduced pressure, the resulting solid was recrystallized from ethyl acetate and dried in vacuo to give an off-white solid 15. 5kg, i.e. intermediate 2, yield 81%, chiral purity 90. 5% θ. θ .. Step 3: 2- (S) – benzyl-4-oxo – (cis – perhydro isoindol-2-yl) butyric acid in 500L reaction vessel was charged with ethyl acetate (225L), triethylamine (1.8kg), imidazole (9. 8kg) and Intermediate 2 (15kg), stirred and cooled to -KTC, was added dropwise thionyl chloride (17. 2kg), the addition was complete, the -KTC~-5 ° C under Stirring was continued for 6h, a solution of cis – perhydro isoindole (9kg), drip completed, the reaction was stirred at room temperature for 18h, the reaction mixture was added IN HCl (150L) was stirred Ih, liquid separation, the organic layer was washed with IN sodium hydroxide solution (100LX3) extracted aqueous layers were combined, washed with ethyl acetate (50L) with, water layer was washed with IN of hydrochloric acid adjusted to PH = 3, the aqueous layer was extracted with ethyl acetate (IOOmLX 3), the combined organic layers , saturated brine (50LX 3) was washed, and the organic layer was dried over anhydrous Na2SO4, filtered, and the filtrate was concentrated under reduced pressure to give an oil 19. 8kg, i.e. Intermediate 3 Yield: 87%. Step 4: Preparation of mitiglinide calcium Intermediate 3 (. 19 8kg) and absolute ethanol (99L) into the 200L reactor, and stirred to dissolve, was added 2N sodium hydroxide solution (35L), minutes after mixing Batch into the high slot. The 500L reaction vessel was added 5% aqueous calcium chloride solution (155L), stirring was added dropwise a solution of the high slot, dropwise with vigorous stirring the reaction completion 3~5h, centrifuged, the cake was washed with 95% ethanol (99L) was recrystallized beating, centrifugation and dried in vacuo (50 ° C / 0. 09MPa), to give the title compound I 16. lkg, yield 73%.
PATENT
https://www.google.com/patents/CN102424664A?cl=en
Mitiglinide calcium Phenylalanine belong chiral compound synthesis routes according to different methods of constructing chiral center has the following three synthetic process:
① split method 😦 Document: CN 102101838A, CN 1844096, etc.)

In this method, diethyl succinate and benzaldehyde by Mobbe condensation, hydrolysis, dehydration anhydride, and after cis-hydrogenated isoindole condensation is reduced to give racemic acid, and then split, and salt to give Mitiglinide calcium. The first method step condensation reaction impurities, product separation and purification difficult, finally resolving the yield is low. This method is also a lack atom economy.
② asymmetric hydrogenation 😦 Document tetrahedron Letters, 1987,28 (17), 1905-1908; Tetrahedron Letters, 1989,30 (6), 735-738)

[0027] This method requires expensive rhodium complexes (Rh, (2S, 4Q-N_-butoxycarbonyl-4-diphenylphosphino _2_ diphenylphosphino-2-diphenylphosphino methylpyrrolidine alkyl), making the production cost is greatly improved, and the need for high-pressure hydrogenation reaction, is not conducive to industrial production.
③ chiral method 😦 Document: CN 1680321A)

The method uses phenylalanine as chiral starting materials, after diazotization, nucleophilic substitution, high temperature decarboxylation and condensation reaction product. Wherein the decarboxylation temperature is too low yield, making the overall process costs.
DISCLOSURE
The object of the present invention is to provide a simple, effective and easy-to-operate preparation Mitiglinide calcium.
The present invention provides a process for the preparation of calcium Mitiglinide, the synthesis route is as follows:

Step 1: D- phenylalanine in the acid hydrolysis of formula (¾ 2- hydroxy acid;
Step 2: formula (¾ 2- hydroxy acid under basic conditions to give protected hydroxyl sulfonate of formula (¾-hydroxyphenyl propionic acid ester;
Step 3: The formula (¾-hydroxyphenyl propionic acid ester in the acid-catalyzed carboxyl ester-protected formula (4) phenylalanine methyl sulfonate carboxylate;
Step 4: cis-hydrogen isoindole synthesis formula (6) perhydro isoindole halide;
Step 5: Under alkaline conditions, the formula ⑷ formula (6) nucleophilic substitution reaction formula (5) Mitiglinide acid
Step 6: Under alkaline conditions, the formula (¾ Mitiglinide ester hydrolysis to the calcium salt of formula (1) Mitiglinide calcium.
Preferably, the specific steps include:
Step 1: (D) – phenylalanine hydrolysis in a strong acid of formula (2) 2-hydroxyphenyl propionic acid
In (D) – phenylalanine as a starting material, in the presence of a strong acid such as sulfuric acid, _5 ° C _5 ° C hydrolysis, to give Formula (2) 2-hydroxyphenyl propionic acid White solid.
Step 2: The formula (¾ 2- hydroxy acid under basic conditions to protect the hydroxyl group sulfonic acid ester of formula (¾-hydroxyphenyl propionic acid ester
2-hydroxyphenyl propionic acid in an organic base such as triethylamine or pyridine, or an inorganic base such as sodium bicarbonate, sodium carbonate or potassium carbonate effect, p-hydroxybenzoic acid ester protecting performed, the protecting group used is an aliphatic or aromatic sulfonic acid group such as mesylate, tosylate or p-toluenesulfonic acid group, a sulfonic acid group is preferably methyl group or p-toluenesulfonic acid.
Step 3: Protect formula formula (¾-hydroxyphenyl propionic acid ester in the acid-catalyzed carboxyl ester group (4) benzenepropanoic
MitigIinide1 (I) carboxylic acid ester sulfonate
In the catalytic acid carboxyl benzenepropanoic acid ester group protection, the use of alcohol may be fatty alcohols or aromatic alcohols, preferably ethanol, t-butanol or benzyl alcohol.
Step 4: cis-hydrogen isoindole synthesis formula (6) perhydro isoindole halide
In the synthesis of perhydro isoindole halide in the haloacetyl halide can be used chloroacetyl chloride, bromoacetyl chloride or bromoacetyl bromide, chloroacetyl chloride is preferred.
Step 5: Under alkaline conditions, (4) and (6) a nucleophilic substitution reaction formula (¾ Mitiglinide acid
Under the conditions of a strong base, such as sodium alkoxide such as sodium ethoxide or sodium methylate, perhydro isoindole halide and phenylalanine sulfonate nucleophilic substitution reaction Mitiglinide ethyl reaction temperature of -10 ° C -25 ° c, preferably 0 ° C.
Step 6: Under alkaline conditions, the formula (¾ Mitiglinide ester hydrolysis to the calcium salt of formula (1) calcium Mitiglinide
Ethyl mitiglinide under basic conditions such as sodium hydroxide, potassium hydroxide, or an amine (ammonia) in the presence of an aqueous solution of calcium chloride, and hydrolyzed as calcium salt, in aqueous solution under conditions of heavy alcohol crystallization, high purity mitiglinide calcium.
The present invention and the prior art comparison, has the following advantages:
1, to find an innovative high-yield process for preparing calcium Mitiglinide route, a total yield of 47%;
2, with respect to the routing methods reported in the literature, the optical yield doubled, ee greater than 99%;
3. The process route of the raw materials are cheap, readily available, avoiding costly chiral resolving agents or the use of a catalyst;
4. The process route mild conditions, high temperature decarboxylation overcome the harsh reaction conditions.
In the present invention, (D) – phenylalanine as a starting material, after diazotization, a hydroxyl group and a carboxyl group protected, nucleophilic substitution, hydrolysis and other reactions prepared mitiglinide calcium, high yield. The present invention provides a process used by a wide range of raw materials, low prices, the total yield of 47%, optical purity greater than 99%, and mild reaction conditions, the reaction process is simple, avoid the literature, such as split, high-pressure hydrogenation method low yield, long reaction steps and other shortcomings, but also to overcome the harsh conditions of high temperature reaction deacidification, etc. for preparation and production of calcium Mitiglinide provides a new choice.
The process route mild conditions, high temperature decarboxylation overcome the harsh reaction conditions.
In the present invention, (D) – phenylalanine as a starting material, after diazotization, a hydroxyl group and a carboxyl group protected, nucleophilic substitution, hydrolysis and other reactions prepared mitiglinide calcium, high yield. The present invention provides a process used by a wide range of raw materials, low prices, the total yield of 47%, optical purity greater than 99%, and mild reaction conditions, the reaction process is simple, avoid the literature, such as split, high-pressure hydrogenation method low yield, long reaction steps and other shortcomings for Mitiglinide calcium preparation and production of a new choice.
Preferably, in the above embodiment, each step may be the following alternative, the embodiment can achieve the same advantageous effects to a third embodiment of embodiment:
Step 1: (D) – phenylalanine in the acid hydrolysis of formula (¾ 2- hydroxy acid
In (D) – phenylalanine as a starting material, in the presence of sulfuric acid, -50C _5 ° C hydrolysis, to give Formula O) 2-hydroxyphenyl propionic acid White solid.
Step 2: formula (¾ 2- hydroxy acid under basic conditions to give protected hydroxyl sulfonate of formula C3) hydroxyphenyl propionic acid ester
2-hydroxyphenyl propionic acid in an organic base such as triethylamine or pyridine, or an inorganic base such as sodium bicarbonate, sodium carbonate or potassium carbonate effect, p-hydroxybenzoic acid ester protecting performed, the protecting group used is an aliphatic or aromatic sulfonic acid group such as mesylate, tosylate or p-toluenesulfonic acid group, a sulfonic acid group is preferably methyl group or p-toluenesulfonic acid.
Step 3: Formula C3) hydroxyphenyl propionic acid ester in the acid-catalyzed carboxyl ester-protected formula (4) phenylalanine methyl sulfonate carboxylate [0118] In the acid-catalyzed, styrene-acrylic acid ester-protected carboxy, the use of alcohol may be fatty alcohols or aromatic alcohols, preferably ethanol, t-butanol or benzyl alcohol.
Step 4: cis-hydrogen isoindole synthesis formula (6) perhydro isoindole halide
In the synthesis of perhydro isoindole halide in the haloacetyl halide can be used chloroacetyl chloride, bromoacetyl chloride or bromoacetyl bromide, chloroacetyl chloride is preferred.
Step 5: Under alkaline conditions, the formula ⑷ formula (6) nucleophilic substitution reaction formula (5) Mitiglinide acid
Under the conditions of a strong base, such as sodium alkoxide such as sodium ethoxide or sodium methylate, perhydro isoindole halide and phenylalanine sulfonate nucleophilic substitution reaction Mitiglinide ethyl reaction temperature of -10 ° C -25 ° c, preferably 0 ° C.
Step 6: Under alkaline conditions, the formula (¾ Mitiglinide ester hydrolysis to the calcium salt of formula (1) calcium Mitiglinide
Ethyl mitiglinide under basic conditions such as sodium hydroxide, potassium hydroxide, or an amine (ammonia) in the presence of an aqueous solution of calcium chloride, and hydrolyzed as calcium salt, in aqueous solution under conditions of heavy alcohol crystallization, high purity mitiglinide calcium.

Patent
https://www.google.com/patents/CN103724253A?cl=en
bis [(2s) -2- benzyl-3- (cis – hexahydro isoindole-2-carbonyl) propionic acid] monocalcium dihydrate (mitiglinide calcium), the formula C38H48CaN206.2Η20 English called Mitiglinide Calcium Hydrate, structural formula (I) as

Mitiglinide Calcium is synthesized by Japan Orange Health Pharmaceutical Co., Ltd., in April 2004 in Japan, for through diet and exercise therapy can effectively control high blood sugar in type II diabetes patients.Mitiglinide calcium is the second repaglinide, nateglinide third after the United States and Glenn urea drugs belong phenylalanine derivatives. By closing ΑΤΡ Mitiglinide calcium-dependent pancreatic β cell membrane Κ channel, resulting in the Ca flow, increase intracellular Ca concentration of extracellular vesicles containing threshing leaving insulin, thereby stimulating the secretion of insulin.And only when the meal will be rapid and transient stimulates the pancreas to secrete insulin, sulphonylureas with the traditional Compared to the rapid onset and short duration of action, inhibition of postprandial hyperglycemia characteristic of type II diabetes, to avoid low blood sugar react, early first- and mild diabetes treatment, and well tolerated.
According to the literature and patent reports, prepared Mitiglinide calcium are the following methods.
Method I: 2_ (S) _ benzyl succinic acid as raw material, amides, reduction, calcium salt formation Mitiglinide this method, although fewer steps, but the chiral compound materials, expensive , the production cost is high, not suitable for industrial production. References: Sorbera LA, Leeson PA, Castaner RM, et al.Mitiglinidecalcium (KAD-1229) [J] .Drugs Future, 2000,25 (10):. 1034-1042 [0007] Method Two: succinate methyl ester with benzaldehyde for raw materials, Stobble condensation, hydrolysis, dehydration anhydride, cis – perhydro isoindole after condensation is reduced to give racemic acid, and then split into calcium salts and the like have Mitiglinide. This method is relatively complex and condensation reaction impurities, product separation and purification difficult, costly, and chiral separation time yield is low.[Reference: Zheng Dejiang, Liu Wentao, Wu Lihua synthetic calcium Mitiglinide [J] Food and Drug, 2007,9 (11): 13-15]
Method three: dimethyl succinate and benzaldehyde for raw materials, Stobbe condensation, reduction, split, with p-nitrophenol and dicyclohexyl carbodiimide activated calcium salt formation Mitiglinide This production cost is relatively high, and used column chromatography, suitable for industrial production. References: Synthesis Technology Zhang Hongmei Chen meritorious, Cao Xiaohui Mitiglinide of [J], modern chemicals, 2008,28 (8): 56-59.]
Example 1:
The cis – hexahydro-isoquinoline (250.4g, 2mol), anhydrous potassium carbonate (304.0g, 2.2mol), methylene burn (1000ml) was added to the reaction flask, keeping the temperature 0-5 ° C with vigorous stirring, dropwise acetyl chloride (271.0g, 2.4mol) in dichloromethane (500ml) solution, drip completed, room temperature 2.5h, point board monitoring, reaction complete, additional water 1000ml, organic layer was separated, water (1000ml), saturated brine (1000ml), dried over anhydrous sodium sulfate overnight, dichloromethane was distilled off under reduced pressure to give cis -N- chloroacetyl hexahydro isoindole (2) 357.4g oil close Rate: 88.6%.
The cis -N- chloroacetyl hexahydro isoindole (302.5g, 1.5mol), N_ within phenylpropionyl camphor sulfonamide (573.0g, 1.65mol), 70% sodium hydride (56.6g, 1.65 mol), Ν, Ν- dimethylformamide (900ml) was added to the reaction flask, at 50 ° C, the reaction was stirred vigorously 12h, to give the alkylated product, placed to room temperature before use.
100ml of water was slowly dropped to the above-mentioned system, drip complete, lithium hydroxide (39.5g, 1.65mol), tetrahydrofuran (600ml), at 0-5 ° C under a 30% solution of hydrogen peroxide solution 680ml, drop Albert, was transferred to the reaction was continued at room temperature for 18h, point board monitoring, reaction complete, additional water 1200ml, adjusting the pH to about 2_3, extracted with dichloromethane (900ml X 3), the combined organic phases with saturated brine (1500ml) wash, overnight over anhydrous sodium sulfate, the solvent was distilled off under reduced pressure to give a viscous liquid, to which was added ethyl acetate 250ml, stirred at room temperature, suction filtered, the filter cake with ethyl acetate (150ml) and dried to give (2s) – 2-benzyl-3- (cis – hexahydro isoindole-2-carbonyl) – propionic acid (6) as a white solid 231.8g, two steps yield: 49%. Compound 6 (230g, 0.73mol), water 1150ml, added to the reaction flask. After the whole solution, was added 2mol / L sodium hydroxide solution, 400ml, stirred at rt for 30min, was slowly added dropwise with vigorous stirring chloride (162.0g, 1.46mol) in water (320ml) solution dropwise was completed, the reaction was continued for 1.5h, filtration, water (200ml X 2) washing the filter cake to give a white solid, 60 ° C and dried under reduced pressure to 3h, the filter cake with 95% ethanol (2300ml) recrystallized Mitiglinide calcium (I) 430g, yield: 83.6%, mp: 178 ~ 183 ° C, FAB-MS: m / z316 [M + l] +; [α] D20 = + 5.45 ° (C = 1, methanol) [Document: m.ρ.: 179 ~ 185Ό, [α] d20 = + 5.64 ° (C = L 0, methanol)]; purity: 99.8% [HPLC normalization method : Column C18, mobile phase L OOmol / L potassium dihydrogen phosphate buffered saline – acetonitrile-water (20:35: 30) (adjusted pH = 2.10); detection wavelength 210nm]; iH-NMlUCDCldOOM), δ: 1.1 ~ 1.5 (16Η, m), 1.8 ~ 2.4 (6Η, m), 2.5 ~ 3.1 (14Η, m) 3.3 ~
3.8 (6H, m) 7.4 ~ 7.6 (10H, m); Elemental analysis (%):. C64.68, Η7.35, Ν3.94, Theory: C64.75, Η7.44, Ν3.97 yield : 36.05%, a purity of 99.8%.

PAPER
WEI HUANG,等: “Novel Convenient Synthesis of Mitiglinide“, 《SYNTHETIC COMMUNICATIONS》, vol. 37, no. 13, 3 July 2007 (2007-07-03), pages 2153 – 2157, XP055079498, DOI: doi:10.1080/00397910701392590
http://www.tandfonline.com/doi/abs/10.1080/00397910701392590
Abstract: A novel convenient synthesis of the hypoglycemic agent mitiglinide was developed. (2S)-4-[(3aR,7aS)-Octahydro-2H-isoindol-2-yl]-4-oxo-2-benzyl-butanoic acid (6) was prepared by selective hydrolysis of ethyl 4-[(3aR,7aS)-octahydro-2Hisoindol-2-yl]-4-oxo-2-benzyl-butanoate (5) using a-chymotrypsin; the latter was prepared by a novel facile route from (3aR,7aS)-octahydro-2H-isoindole. The overall yield was 25.6%.
Keywords: a-chymotrypsin, mitiglinide, synthesis
Mitiglinide (calcium bis[(2S)-4-[(3aR,7aS)-octahydro-2H–isoindol-2-yl]-4oxo-2-benzylbutanoate]dihydrate) is a novel oral hypoglycemic agent. It inhibits the adenosine triphosphate (ATP)-sensitive potassium channels in pancreatic b-cells and stimulates insulin release like sulfonylureas,[1] but has a rapid onset and short-lasting hypoglycemic effect as compared with the latter.
Mitiglinide has been synthesized by several related methods that involve optical resolution,[2] asymmetric synthesis,[2a,3] and diasteroselective alkylation using chiral auxiliary.[4]
In a previous article,[2] two optical resolution methods of the key compound racemic acid 4 were reported. One of them involves esterification with optically active alcohols, which are separated into the diastereomers by column chromatogeaphy and hydrolyzed. Only the diastereomeric (S)-Nbenzyl mandelamide ester could be separated; the overall yield was 28%,
The alternative method was optical resolution by optically active bases. The best result was 30.8% yield and 97% ee when using (R)-1-(1-naphthyl)-ethylamine as a base. In this article, we have developed a new optical resolution method of racemic ester 5 by a-chymotrypsin in 45.3% yield; the optical purity of (S)-acid (6) determined by chiral-phase high performance liquid chromatography (HPLC) on Sumichiral
OA3300 was 99.2% ee, and, the method can be used for scale-up preparation.
The synthesis of free acid 6 is shown in Scheme 1. (3aR,7aS)-Octahydro2H-isoindole was chloroacetylated in the presence of Et3N to afford (3aR, 7aS)-2-(chloro-acetyl)-octahydro-2H-isoindole (2), which was condensed with diethyl benzylmalonate followed by hydrolysis and decarbonylation to obtain 4-[(3aR,7aS)-octahydro-2H-isoindol-2-yl]-4-oxo-2-benzyl-butanoic acid (4). The overall yield of the three-step synthesis was 62.9%. The racemic acid (4) was esterified with SOCl2/EtOH to give the corresponding racemic ester (5). The (R)-ester was selectively hydrolyzed by a-chymotrypsin to separate out the (S)-ester, which was subjected to hydrolysis, giving 6.
The overall yield was 28.5% [based on (3aR,7aS)-octahydro-2H-isoindole].
Compound 6 was treated with calcium chloride and 25% ammonium hydroxide to give mitiglinide; after recrystallization from 95% EtOH, the pure product was obtained in 90% yield.
Patent
https://www.google.com/patents/WO2009047797A2?cl=en
EXAMPLES
Example 1: Preparation of (R) 4-benzyl-3-(3-phenylpropionv0-oxazolidin-2-one To a solution of (R)-4-benzyloxazolidin-2-one (50 g), 4-dimethylaminopyridine (4.85 g), 3-phenyl propionic acid (55.08 g) in dichloromethane (375 ml) under nitrogen atmosphere at 0-5 0C, dicyclohexylcarbodiimide (975.65 g) was added. The temperature was slowly raised to 25-30 0C and stirring was continued until no starting material was left as was confirmed by thin layer chromatography. Dicyclohexylurea formed during the reaction was filtered, washed with dichloromethane (200 ml) and the filtrate was washed with saturated solution of sodium bicarbonate (500 ml). The solution was dried over sodium sulphate and solvent was distilled off to obtained crude product which was purified from methanol (200 ml) at 10-15 °C and washed with methanol (50 ml) to obtain 81.0 g of the title compound. Example 2: Preparation of 3(5)-benzyl-4-(4-(J?)-benzyl-2-oxo-oxazolidin-3-yl)-4-oxo-butyrϊc acid tert-butyl ester
To a solution of (/?)-4-benzyl-3-(3-phenyl-propionyl)-oxazolidin-2-one (150 g) in anhydrous tetrahydrofuran (1.5 It) was added a solution of sodium hexamethyldisilazane (462 ml, 36-38% solution in tetrahydrofuran) with stirring at -85 to -95 0C for 60 minutes. Tert-butyl bromo acetate (137.5 g) in tetrahydrofuran (300 ml) was added to reaction mass and then stirred to 60 minutes at -85 to -95 0C. After completion of the reaction (monitored by TLC), the reaction mixture was poured into ammonium chloride solution (10%, 2.0 It) and extracted with ethyl acetate (2×750 ml). The combined organic layer was washed with demineralized water (1×750 ml) and dried over sodium sulphate. The solvent was evaporated under reduced pressure to obtain oily residue which was stirred with mixture of n-hexane (100 ml) and isopropyl alcohol (100 ml) at Oto -50C, filtered and dried under vacuum to obtain 153.12 g of title compound having chemical purity 99.41%, chiral purity 99.91% by HPLC, [α]D 20: (-)97.52° (c = 1, CHCl3) and M.P. : 117.1-118.20C.
Example 3: Preparation of 3(5)-benzyl-4-(4(i?)-benzyl-2-oxo-oxazolidin-3-yl)-4-oxobutyric acid Trifluoroacetic acid (100 g) was added to a solution of 3(5)-benzyl-4-(4-(/?)-benzyl-2-oxo-oxazolidin-3- yl)-4-oxobutyric acid tert-butyl ester (100 g) in dichloromethane (700 ml) at 25 0C and mixture was stirred further for about 12 hours ( when TLC indicated reaction to be complete). The reaction mixture was poured in to ammonium chloride solution (10%, 500 ml). The dichloromethane layer was separated and aqueous layer was extracted with dichloromethane (2 x 250 ml). The combined organic layer was dried over sodium sulphate and evaporated under reduced pressure to obtain title compound. The crude product was recrystallized from a mixture of ethyl acetate: n-hexane (1:4, 500 ml) to obtain 78.75g of the title compound having purity 99.56% by HPLC and M.P.: 145.9-146.40C.
Example 4: Preparation of (2S)-2-benzyl-l-((4R)-4-benzyl-2-oxo-oxazolidin-3-vI)-4-(hexahydro- isoindolin-2-yl)-butane-l,4-dione
To a solution of 3(5)-benzyl-4-(4-(/?)-benzyl-2-oxo-oxazolidin-3-yl)-4-oxo-butyric acid (50 g) in anhydrous dichloromethane (1.25 It) was added triethylamine (50 ml) with stirring at -20 to -30 0C and the stirred for 15 minutes. A solution of isobutylchloroformate (37.50g) in anhydrous dichloromethane (50 ml) was added at -20 to -30 0C and stirred for 60 minutes. Thereafter, a solution of cis- hexahydroisoindoline (32.50 g) in anhydrous dichloromethane (50 ml) was slowly added by maintaining temperature -20 to -300C. After the completion of the reaction (monitored by HPLC), the mixture was successively washed with 0.5N hydrochloric acid solution (500 ml), brine (300 ml) and dried over sodium sulphate. The solvent was evaporated under reduced pressure to obtain 102.0 g of the title compound having purity 94.39% by HPLC.
Example 5: Purification of r2S)-2-benzyl-l-((4R)-4-benzyl-2-oxo-oxazolidin-3-yl)-4-(hexahydro- isoindolin-2-vD-butane-l,4-dione
To the crude (2S)-2-benzyl-l-((4R)-4-benzyl-2-oxo-oxazolidin-3-yl)-4-(hexahydro-isoindolin-2-yl)- butane- 1,4-dione (51.0 g) was added methanol (150 ml) and the mixture was stirred for 5 hours at 0 to 5 0C. Solid that precipitated out was filtered, slurry washed with cold methanol (25 ml) and dried at 45 -50 0C under vacuum to obtain 28.80 g of pure title compound as a crystalline solid having purity of 99.71% by HPLC and M. P.: 104.1-105.70C.
Example 6: Preparation of calcium salt of (-SVmitiglinide. Step-1: Preparation of (-SVmitiglinide
(2S)-2-Benzyl- 1 -((4R)-4-benzyl-2-oxo-oxazolidin-3-yl)-4-(hexahydro-isoindolin-2-yl)-butane- 1 ,4-dione (28.0 g) was dissolved in tetrahydrofuran (196 ml) and a mixture of lithium hydroxide monohydrate (3.51 g) in demineralized water (56 ml) and hydrogen peroxide (40% solution, 5.5 ml) was added with stirring at 0 to 5 0C over a period of 30 minutes. The reaction mixture was further stirred at 0 to 5 0C till the completion of the reaction. After the completion of the reaction (monitored by TLC), the reaction was quenched with the addition of cooled sodium meta-bisulphate solution (25%, 168 ml) at 0 to 10 0C. The reaction mixture was extracted with ethyl acetate (2×112 ml), the layers were separated and the aqueous layer was discarded. The HPLC analysis of the aqueous layer shows 0.77% of amide impurity. The ethyl acetate layer was then extracted with aqueous ammonia solution (4%, 2×40 ml). The layers were separated and the aqueous layer was further extracted with ethyl acetate (2×280 ml). Combined ethyl acetate layer was discarded. This aqueous layer (280 ml) was used as such in the next stage. The aqueous layer display purity 96.19 % by HPLC and amide impurity 0.04% by HPLC. Step-2: Preparation of calcium salt of dSVmitiglinide
To the above stirred solution of (S)-mitiglinide in water and ammonia(280 ml), methanol (168 ml) was added, followed by calcium chloride (4.48 g) dissolved in demineralized water (56 ml) at ambient temperature and the mixture was stirred for 2 hours. The resulting precipitate was filtered, successively slurry washed with water (3 x 140 ml) and acetone (2 x 70 ml) and dried at 450C -500C under vacuum to obtain 16.1 g of title compound having purity 99.67% by HPLC and amide impurity 0.01% by HPLC. The title product was re-precipitated from a mixture of methanol and water and dried to obtain pure title compound.
Example 7: Preparation of (.SVmitiglinide
To a solution of (2S)-2-benzyl-l-((4R)-4-benzyl-2-oxo-oxazolidin-3-yl)-4-(hexahydro-isoindolin-2-yl)- butane- 1,4-dione (50 g) in tetrahydrofuran (350 ml) was added a solution of lithium hydroxide monohydrate (8.65 g) in demineralized water (100 ml) and hydrogen peroxide (30% w/w, 40 ml) with stirring at 5 to 10 0C over a period of 15 minutes. After the completion of reaction, sodium meta- bisulphate solution (40%, 500 ml) was added to the reaction mixture and the mixture was extracted with ethyl acetate (2 x 250 ml). The organic layer was dried over sodium sulphate and evaporated under vacuum to obtain 45.5 g of title compound having 35 % of R-benzyl oxozolidin-2-one as impurity. Example 8: Purification of (.S)-mitiglinide
Aqueous ammonia solution (4%, 300 ml) was added to the crude (5)-mitiglinide (30 g) and stirred. The reaction mixture was washed with ethyl acetate (3 x 300 ml). Thereafter the reaction mixture was acidified to pH 1 to 2 with IN hydrochloric acid solution (250 ml) and extracted with ethyl acetate (2 x 150 ml). The layers were separated and ethyl acetate layer was washed with demineralized water (2 x 150 ml), dried over sodium sulphate and then evaporated under reduced pressure to obtain 16.2 g of pure (5)-mitiglinide having purity 95.55% by HPLC Example 9: Preparation of calcium salt of (S)-mitiglinide
To a solution of (<S)-mitiglinide (15 g) in water (150 ml) and aqueous ammonia solution (25%, 15 ml) at 25 to 30 0C, a solution of calcium chloride (7.5 g) in demineralized water (37.5 ml) was added. The mixture was stirred for 1 hour to precipitate the calcium salt of (5)-mitiglinide dihydrate. The resulting precipitate was filtered, slurry washed with water (3 x 150ml) and dried at 45 to 50 0C to obtain 13.25 g of the title compound having purity of 98.84% by HPLC. Example 10: Purification of calcium salt of (5)-mitiglinide
(iS)-mitiglinide calcium (10 g) was dissolved in dimethylformamide (100 ml). This is followed by the addition of demineralized water (500 ml) at 25 to 30 0C. The mixture was stirred for 30 minutes. The precipitated solid was filtered, washed with water (10x 50ml) and dried at 45 to 50 0C under vacuum to obtain 8g of pure title compound as a crystalline solid having purity of 99.62% by HPLC. Example 11: Preparation of amorphous mitiglinide calcium
Crystalline mitiglinide calcium (2.0 g) was dissolved in tetrahydrofuran (20 ml) and filtered to remove undissolved and suspended particles. The solvent was then evaporated under vacuum to obtain a powder which was then dried under vacuum at 40-600C to obtain 1.70 g of the title compound. Example 12: Preparation of amorphous mitiglinide calcium
Crystalline mitiglinide calcium (2.0 g) was dissolved in dichloromethane (30 ml) and filtered to remove undissolved and suspended particles. The solvent was then evaporated under vacuum to obtain a powder which was then dried under vacuum at 40-600C to obtain 1.64 g of the title compound. Example 13: Preparation of amorphous mitiglinide calcium
Mitiglinide (2.0 g) was dissolved in methanol (20 ml) and methanolic ammonia (5.0 ml) solution was added to it. The solution was stirred at 25-30 0C and calcium chloride (1.5 g) dissolved in methanol was mixed with the solution of mitiglinide and ammonia in methanol and the solution was filtered to remove the suspended particles. The solvent was then evaporated under vacuum to obtain a powder which was then dried under vacuum at 40-600C to obtain 1.9 g of the title compound. Example 14: Preparation of amorphous mitiglinide calcium
Mitiglinide (2.0 g) was dissolved in dichloromethane (20 ml) and aqueous ammonia (3.6 ml, 25 % solution) was added to it. The solution was stirred at 25-300C and solid calcium chloride (1.5 g) was mixed with the solution of mitiglinide and ammonia in dichloromethane and the solution warmed at 30 – 35 0C. The solution was washed with water (2 xlO ml) and the clear solution was dried over sodium sulfate, filtered and evaporated under vacuum and finally dried at under vacuum at 40-60 0C to obtain 1.75 g of the title compound.
Example 15: Preparation of amorphous mitiglinide calcium
Crystalline mitiglinide calcium dihydrate (2.0 g) was dissolved in ethyl acetate (30 ml) and filtered to remove undissolved and suspended particles. Approimately. 60 % of the solvent was distilled off under vacuum to obtain a stirrable solution. The solution was then cooled to 15-2O0C, mixed with n-heptane (20 ml) and the mixture was stirred for 30 minutes. The resulting solid was filtered, washed with n-heptane and dried under vacuum at 45-600C to yield 1.72 g of the title compound. Example 16: Preparation of amorphous mitiglinide calcium
Crystalline mitiglinide calcium (2.Og) was dissolved in dichloromethane (30 ml) and filtered to remove undissolved and suspended particles. Approximately 60 % of the solvent was distilled off under vacuum to obtain a stirrable solution. The solution was then cooled to 15-200C and mixed with diisopropyl ether (20 ml). The mixture was stirred for 30 minutes and the resulting solid was filtered, washed with diisopropyl ether and dried under vacuum at 45-600C to obtain 1.70 g of the title compound. Example 17: Preparation of amorphous mitiglinide calcium
Mitiglinide (2.0 g) was dissolved in dichloromethane (20 ml) and aqueous ammonia (3.6 ml, 25 % solution) solution was added to it. The solution was stirred at 25-30 0C and mixed with solid calcium chloride (1.5 g) and the solution warmed at 30-35 0C and stirred for 30 minutes. The solution was washed with water (2 x 10 ml) and the clear solution was dried over sodium sulfate, and filtered. Approximately 60% of the solvent was distilled off under vacuum and the resulting viscous oil was cooled to 10-15 0C and mixed with diisopropyl ether (50 ml). The reaction mixture was stirred for 30-35 minutes and the resulting solid was filtered and dried at 40-600C to obtain 1.75 g of the title compound. Example 18: Conversion of amorphous mitiglinide calcium into crystalline mitiglinide calcium A suspension of amorphous mitiglinide calcium in diisopropyl ether (30 ml) was stirred for 2 hours at 25- 300C, filtered and dried under vacuum at 45-600C to obtain crystalline form of mitiglinide calcium. Example 19: Preparation of crystalline mitiglinide calcium
To a solution of mitiglinide (2.5 g) in water (2.5 ml), aqueous ammonia solution (approx 25%, 4.0 ml) and acetonitrile (2.5 ml) at 10-150C, calcium chloride (1.32 g) dissolved in demineralized water (15 ml) was added. The mixture was stirred for 2 hours. The resulting precipitate was filtered, slurry washed with water (3 x 25 ml) and acetone (2 x 5 ml) and dried at 45-500C under vacuum to obtain 2.12 g of title compound having purity: 99.72 % by HPLC.
Example 20: Preparation of crystalline mitiglinide calcium
To a solution of mitiglinide (2.5 g) in water (2.5 ml), aqueous ammonia solution (approx 25%, 4.0 ml) and tetrahydrofuran (2.5 ml) at 10-150C, calcium chloride (1.32 g) dissolved in demineralized water (15 ml) was added. The mixture was stirred for 2 hours. The resulting precipitate was filtered, slurry washed with water (3 x 25 ml) and acetone (2 x 5 ml) and dried at 45-500C under vacuum to obtain 1.95 g of title compound having purity: 99.52 % by HPLC.
Example 21; Preparation of crystalline mitiglinide calcium
To a solution of mitiglinide (30.0 g) in water (300 ml), aqueous ammonia solution (approx 25%, 48 ml) and acetone (300 ml) at 10-150C, calcium chloride (15.8 g) dissolved in demineralized water (180 ml) was added. The mixture was stirred for 2 hours. The resulting precipitate was filtered, slurry washed with water (3 x 300 ml) and acetone (2 x 60 ml) and dried at 45-500C under vacuum to obtain 24.32 g of title compound having purity: 99.42 % by HPLC.
Example 22: Preparation of crystalline mitiglinide calcium
To a solution of mitiglinide (3.0 g) in water (30 ml), aqueous ammonia solution (approx 25%, 4.8 ml) and isopropyl alcohol (300 ml) at 10-150C, calcium chloride (1.58 g) dissolved in demineralized water
(18 ml) was added. The mixture was stirred for 2 hours. The resulting precipitate was filtered, slurry washed with water (3 x 30 ml) and acetone (2 x 6 ml) and dried at 45-500C under vacuum to obtain 1.92 g of title compound having purity: 99.65 % by HPLC.
Example 23: Preparation of (2S)-2-benzyWV-((lR)-l-benzyl-2-hydroxy-ethyl)-4-(hexahvdro- isoindolin-2-yl)-4-oxo-buryramide
To a solution of (2S)-2-benzyl-l-((4R)-4-benzyl-2-oxo-oxazolidin-3-yl)-4-(hexahydro-isoindolin-2-yl)- butane-l,4-dione (20.0 g) in tetrahydrofuran (140 ml), a solution of lithium hydroxide monohydrate
(3.43 g,) in demineralized water (40 ml) was added and the reaction mixture was refluxed for 4 hours till the completion of the reactions (monitored by thin layer chromatography). After the completion of the reaction, the reaction mixture was poured into demineralized water (100 ml) and extracted with ethyl acetate (2 x 80 ml). The combined organic layer was washed with water (80 ml) and dried over sodium sulphate. The solvent was evaporated under reduced pressure to give residue which was stirred in isopropyl alcohol at 0-5 0C for 5 hours. The mixture was filtered and then dried at 40-45 0C under vacuum to obtain 12.48 g of title compound having purity 99.77 % by HPLC. Melting point = 77 – 800C.
PATENT
https://www.google.com/patents/CN102101838A?cl=en
Mitiglinide calcium (mitiglinide calcium), the chemical name (2S) _2_ benzyl _3_ (cis – hexahydro _2_ isoindolinyl-carbonyl) propionate dihydrate by Japanese pharmaceutical company developed Kissei ATP-dependent potassium channel blockers, 2004 for the first time in Japan for the treatment of type II diabetes.
Mitiglinide calcium is the second repaglinide, nateglinide after the first three columns MAG urea drugs, is a derivative of phenylalanine, which acts like mechanism sulfonylurea, but faster onset and the short half-life, is conducive to reducing postprandial blood glucose in diabetic patients, but also to avoid low blood sugar caused by continuous glucose, with “in vitro pancreas” reputation.
In recent years, synthetic methods as described in patent application number: Patent 200510200127 9, the synthesis process first synthesized racemic (±) 2_-benzyl-3- (cis – hexahydro iso-indole-2. carbonyl) propionic acid, and then split to give (2S) -2- benzyl-3- (cis – hexahydro isoindole-2-carbonyl) propionic acid, not a lot of waste material along _ hexahydro isoindole, and Chiral separation is not high.
DISCLOSURE
The technical problem to be solved by the present invention is to provide a material savings along _ hexahydro isoindole, and preparation of a high degree of chiral separation.
To solve the above technical problem, the technical solution of the present invention is employed as a method for preparing mitiglinide calcium, comprising the steps of:
Step 1 Synthesis, benzylidene succinic acid
With stirring, was added sodium metal in absolute ethanol, under an inert gas, the solution was heated to reflux with stirring, reflux for 45 fly 0 minutes, under reflux before the dropwise addition of benzaldehyde, and then added dropwise diethyl succinate esters, reaction stirring was continued for 2 to 3 hours, slowly reducing the LC-Ms detection, the ratio of formaldehyde starting material benzene, cooled to room temperature, after use 5 (T55wt% aqueous solution of NaOH to adjust the PH San 13.0, and then heated at reflux;. Γ4 hours, cooled to at room temperature, keeping the reaction solution temperature <25 ° C, pH adjusted with concentrated hydrochloric San 2.0, filtration, recrystallization cold tetrahydrofuran, wherein the molar ratio of sodium metal with benzaldehyde and diethyl succinate is: 0.3 … ~ 0 5: 1 2~1 5: 1; Step 2 synthesis, benzyl butyl acid

The benzylidene succinic acid into the reactor, 10% Pd / C and ethanol, evacuated, and then replaced with hydrogen three times, introducing hydrogen, atmospheric hydrogenation reaction 12~15 hours, the reaction solution suction After the filtrate was evaporated to dryness under reduced pressure, the resulting solid was recrystallized from ethyl acetate, wherein the mass ratio of benzylidene succinic acid with 10% Pd / C is 1: 0 0 15 ^ 20.
3 Synthesis [0006] step, (S) -2- acid, benzyl butyl

Benzyl succinic acid dissolved in methanol was added dropwise with stirring (R) – a chiral amine, stirred at room temperature 2 hours wide, and the precipitated solid was filtered and the solid dispersed in water, under stirring 6 mol / mL hydrochloric acid adjusted ρΗ = 1 (Γ2.0, stirred for 30 minutes, the solid by suction filtration, dried, and wherein the benzyl succinic acid (R) – chiral amine molar ratio of 1: 0~2 5 2;…
Said (R) – a chiral amine (R) -I- phenylethylamine, (R) -I- naphthylethylamine or (R) -I- phenyl-2-p-amine;
4 (S) synthesis step, -2-benzyl succinic anhydride

Reactor, has added (S) -2- benzyl succinic acid and acetic anhydride, at 7 5,0 ° C reaction 1 to 2 hours, isopropyl ether low temperature crystallization after cooling, heavy with ethyl acetate crystallization, wherein (S) -2- molar ratio of benzyl succinic acid and acetic anhydride: 1: 7 · 0 to 7 · 5;
Step 5, (2S) -2- benzyl-3- (cis – hexahydro isoindole-2-carbonyl) propionic acid Synthesis

Stirring, S- benzyl succinic anhydride is dissolved in dichloromethane, control the internal temperature <0 V, a solution of cis _ hexahydro isoindole, dropping it, keeping the internal temperature at <0! : Continue stirring for 2 to 3 hours, the reaction in 2 (T25 ° C 10~15 hours, concentrated to give a pale yellow viscous material, wherein the (S) -2- benzyl succinic anhydride and cis – hexahydro isoindole molar ratio of 1: 2 (Γ2 5; step 6, mitiglinide calcium synthesis.

To the reactor was added (2S) -2- benzyl-3- (cis – hexahydro isoindole-2-carbonyl) propionic acid, water and concentrated ammonia, stir until completely dissolved, a solution of anhydrous calcium chloride aqueous solution, gradually precipitated white solid was added dropwise and then stirred at room temperature 12~ after 15 hours, suction filtered, the filter cake washed with water, dried to give a white solid, i.e. Mitiglinide calcium crude, obtained crude product with methanol and water (volume Than 0.5 4~0 5: 1) and recrystallized as a white solid Mitiglinide calcium;
Beneficial effects: The invention provides a method for preparing calcium Mitiglinide not only saves raw material cis – hexahydro isoindole, and chiral separation is high.
Embodiment 1
Step 1 Synthesis, benzylidene succinic acid
Under stirring, sodium metal (1.7 g, 0. 072 mol) was added absolute ethanol (50 mL), and under argon, the solution was heated to reflux with stirring, reflux for 50 minutes under reflux before the dropwise addition of benzene Formaldehyde (23 mL, 0. 183 mol), and then added dropwise diethyl succinate (50 mL, 0. 275 mol), stirring was continued for 2.5 hours the reaction, reducing the slow LC-Ms detection, the ratio of formaldehyde starting material benzene , was cooled to room temperature, with 55wt.% aqueous NaOH solution adjusting pH ≥ 13.0, and then heated at reflux for 3 hours, cooled to room temperature, the reaction solution temperature maintained <25 ° C, with concentrated hydrochloric pH≤2.0, leaching, cryogenic tetrahydrofuran recrystallization, yield = 81.3%;
Step 2 synthesis, benzyl butyl acid
The benzylidene succinic acid (23. 7 g, 0. 114 mol) into the reactor, then add 10% Pd / C (4. 7g) and anhydrous ethanol (300 mL), evacuated, then Hydrogen replacement three times, introducing hydrogen, hydrogenated at atmospheric pressure for 14 hours, the reaction solution after filtration, evaporated to dryness under reduced pressure, the resulting solid was recrystallized from ethyl acetate, yield: 98% 9; step 3, (S). -2-butyric acid benzyl
Benzyl succinic acid (31. 2 g, 0. 156 mol) was dissolved in methanol (500 mL), and added dropwise with stirring (R) -I- phenylethylamine (41.2 g, 0. 343 mol), room temperature stirred for 1.5 hours, the precipitated solid was filtered, the solid dispersion to water (100 mL) and stirred at with 6 mol / mL hydrochloric acid adjusted ρΗ = 1. (Γ2. 0, stirred for 30 minutes, the solid was suction filtered, and dried Yield 87. 3%; 4 (S) synthesis step, -2-benzyl succinic anhydride
Reactor, has added (S) -2- benzylbutyl acid (27. 8 g, 0. 132 mol) and acetic anhydride (88 mL, 0. 964 mol), at 7 5,0 ° C for 1 hours, cooled and added to isopropyl ether (150 mL) low temperature crystallization, after recrystallization from ethyl acetate, yield: 73% 9;.
Under – (hexahydro-isoindole-2-carbonyl cis) acid synthesis stirring S- benzyl succinic anhydride (12. 7 g, 0 Step 5, (2S) -2- benzyl-3. 067 mol) was dissolved in dichloromethane (250 mL), to control the internal temperature <0 ° C, a solution of cis – hexahydro isoindole (18. 5 g, 0 154 mol), the addition was complete, maintaining the internal temperature in <0! : Continue stirring for 2.5 hours, the reaction in 2 (T25 ° C 12 hours, concentrated to give a pale yellow viscous material, yield: 83 1%; Step 6 Synthesis Mitiglinide calcium.
To the reactor was added (2S) -2- benzyl-3- (cis – hexahydro isoindole-2-carbonyl) propionic acid (. 28. 7 g, 0 091 mol), water (150 mL), and concentrated aqueous ammonia (12 mL), stirring until completely dissolved, a solution of anhydrous calcium chloride (12. 1 g, 0.109 mol) water (100 mL) solution was gradually precipitated white solid was added dropwise at room temperature and then stirred for 13 End hours, filtration, washing the filter cake, and drying to give a white solid, crude Mitiglinide calcium, derived from crude methanol and water (volume ratio 0.5 4~0 5: 1) and recrystallized as a white solid MIG Chennai column calcium, yield: 87.3%.
Second Embodiment
Example A similar experimental method steps 1 through 6 was carried out except in step 3, using (R) -1- naphthyl-amine (61. 4 g, 0. 359 mol) substituted (R) -I- phenylethylamine Other operating homogeneous reaction similar to this step of the synthesis yield: 87.3%.
Third Embodiment
Example A similar experimental method steps 1 through 6 was carried out except in step 3, using (R) -I- phenyl-2-p-tolyl-ethylamine (90. 2 g, 0. 374 mol) substituent (R ) -I- phenethylamine, other homogeneous reaction procedure similar to the synthesis yield of this step:. 83 4% ο
PATENT
WO 199832736

CLIP
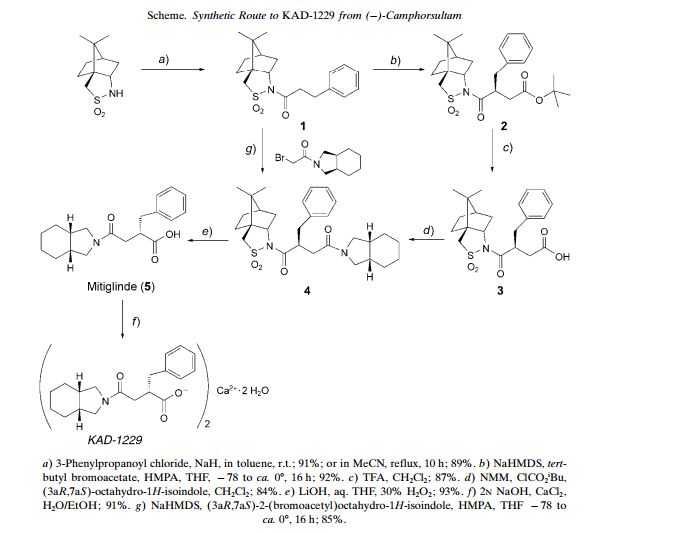
The process for the preparation of KAD-1229 starts from ()-camphorsultam ((3aS)-8,8-dimethylhexahydro-3a,6-methano-2,1-benzisothiazole 2,2-dioxide), readily available in 85% yield from ()- -camphor [4]. Treatment of ()-camphorsultam with excess 3-phenylpropionyl chloride in the presence of NaH in toluene at room temperature gave 1 in 91% yield (Scheme) [5]. An alternative procedure is to reflux camphorsultam with 1.1 to ca. 1.5 equiv. of 3-phenylpropionyl chloride in MeCN for 8 ± 10 h [6]. The crude product, acylsultam 1, purified by recrystallization from EtOH/H2O in 89% yield, was reacted with an equimolar amount of base to form the chiral enolate in dry ice/EtOH bath, followed by C()-re-alkylation [7] with tert-butyl bromoacetate to give 2. The choice of the organic base was very important: the reaction with BuLi, lithium diisopropylamide (LDA), or NaHMD (sodium hexamethyldisilazane) gave 2 in 30 ± 40%, 60%, or 90% yield, respectively, after recrystallization from MeOH. Alkylation promoted by these bases tends to give products with high diastereoselectivity, and the diastereoisomeric purity of crude product 2 was determined to be 93%. However, the reaction with NaHMDS as the base proceeded smoothly in high yield. The tert-butyl ester 2 was cleaved with TFA (CF3COOH) in CH2Cl2 to give the free acid 3 in 87% yield [8]. Acylation of (3aR,7aS)-octahydro-1H-isoindole with 3 by a mixed anhydride method afforded 4 in 84% yield [9]. Compound 4 can be also obtained in 85% yield via direct alkylation of 1 with (3aR,7aS)-2-(bromoacetyl)octahydro-1H-isoindole; however, the yield of the (2-bromoacetyl)octahydro-1H-isoindole prepared from 2-bromoacetyl bromide and cis-octahydro-1H-isoindole was only 40%. Nondestructive cleavage of 4 by hydroperoxide-assisted saponification (LiOH, aq. H2O2 , THF, r.t.) regenerated the camphorsultam (96% recovered yield) and gave mitiglinide (5) in 93% yield and high enantiomeric excess ( 99% by HPLC analysis of the corresponding methyl ester) [7]. Product 5 was treated with 2 NaOH, followed by treatment with CaCl2 . Recrystallization from aqueous EtOH gave KAD-1229 in 91% yield, with a melting point and specific rotation data identical to those in the literature [2b]. Co
(2S)-4-[(3aR,7aS)-Octahydro-2H-isoindol-2-yl]-4-oxo-2-(phenylmethyl)butanoic Acid ( Mitiglinide, 5). base
mitiglinide as a colorless viscous oil. The ee was determined to be 99.4% by HPLC analysis of the corresponding Me ester on a Chiralcel AS column (250 4.6 mm, flow rate 0.7 ml/min, UV 214 nm, n-hexane/i-PrOH 80 : 20 as the eluent).
20 Dalpha= -3.5 (c 1.0, MeOH).
1 H-NMR: 1.23 ± 1.63 (m, 8 H); 2.13 ± 2.22 (m, 2 H); 2.42 ± 2.52 (m, 2 H); 2.73 ± 3.32 (m, 7 H); 7.18 ± 7.32 (m, 5 H).
ESI-MS: 316.15 ( [M H]). Anal. calc. for C19H25NO3 (315.41): C 72.35, H 7.99, N 4.44; found: C 72.51, H 8.03, N 4.31.
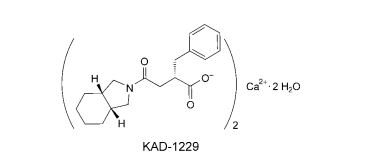
Calcium Bis{(2S)-4-[(3aR,7aS)-octahydro-2H-isoindol-2-yl]-4-oxo-2-(phenylmethyl)butanoate} Dihydrate (KAD-1229).
KAD-1229 as colorless crystals (0.82 g, 91%).
M.p. 179 ± 185 (lit. 179 ± 185 [2b]). 20 D 5.4 (c 0.6, MeOH) (lit. 20 D 5.7, c 1.0, MeOH [2b]).
1 H-NMR: 1.13 ± 1.39 (m, 16 H); 2.0 ± 2.3 (m, 6 H); 2.54 ± 2.83 (m, 14 H); 3.20 ± 3.22 (m, 6 H); 7.11 ± 7.28 (m, 10 H).
ESI-MS: 669.32 ( [M 2 H2O H]). Anal. calc. for C38H48CaN2O6 ¥2H2O (704.91): C 64.75, H 7.44, N 3.94; found: C 64.46, H 7.35, N 3.73.
REFERENCES for aboveclip
[1] H. Ohnota, T. Koizumi, N. Tsutsumi, M. Kobayashi, S. Inoue, S. J. Sato, Pharmacol. Exp. Ther. 1994, 269, 489; H. Ohnota, M. Kobayashi, T. Kiozumi, K. Katsuno, F. Sato, T. Azisawa, Biochem. Pharmacol. 1995, 49, 165; M. Kinukawa, H. Ohnota, T. Azisawa, Br. J. Pharmacol. 1996, 117, 17021.
[2] a) T. Yamaguchi, T. Yanagi, H. Hokari, Y. Mukaiyama, T. Kamijo, I. Yamamoto, Chem. Pharm. Bull. 1997, 45, 1518; b) T. Yamaguchi, T. Yanagi, H. Hokari, Y. Mukaiyama, T. Kamijo, I. Yamamoto, Chem. Pharm. Bull. 1998, 46, 337.
[3] J. P. Lecouve, C. Fugier, J. C. Souvie, Pat. WO9901430, 1999 (Chem. Abstr. 1999, 130, 110156r).
[4] M. Vandewalle, J. Van der Eycken, W. Oppolzer, C. Vullioud, Tetrahedron 1986, 42, 4035; F. A. Davis, J. C. Towson, M. C. Weismiller, S. Lal, P. J. Carroll, J. Am. Chem. Soc. 1988, 110, 8477.
[5] W. Oppolzer, O. Tamura, J. Deerberg, Helv. Chim. Acta 1992, 75, 1965.
[6] M. C. William, B. Corey, J. Org. Chem. 1998, 63, 6732.
[7] W. Oppolzer, R. Moretti, S. Thomi, Tetrahedron Lett. 1989, 30, 5603.
[8] H. Heitsch, R. Henning, H. W. Kleemann, W. Linz, W. U. Nicke, D. Ruppert, H. Urbach, A. Wagner, J. Med. Chem. 1993, 36, 2788.
[9] J. J. Plattner, P. A. Marcotte, H. D. Kleinert, H. H. Stein, J. Greer, G. Bolis, A. K. L. Fung, B. A. Bopp, J. R. Luly, J. Med. Chem. 1988, 31, 2277.
References
- Malaisse WJ (October 2008). “Mitiglinide: a rapid- and short-acting non-sulfonylurea insulinotropic agent for the treatment of type 2 diabetic patients”. Expert Opin Pharmacother. 9 (15): 2691–8. doi:10.1517/14656566.9.15.2691. PMID 18803455.
External links
- Elixir Pharmaceuticals – website of the U.S. rights holder for mitiglinide.
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| EP0507534A1 * | Mar 30, 1992 | Oct 7, 1992 | Kissei Pharmaceutical Co., Ltd. | Succinic acid compounds |
| EP0967204A1 * | Jan 22, 1998 | Dec 29, 1999 | Kissei Pharmaceutical Co Ltd | Process for producing benzylsuccinic acid derivatives |
| US6133454 * | Jul 1, 1998 | Oct 17, 2000 | Adir Et Compagnie | Method for preparing a substituted perhydroisoindole |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN102898348A * | Sep 8, 2012 | Jan 30, 2013 | 迪沙药业集团有限公司 | Preparation method for Mitiglinide calcium |
| CN102898348B * | Sep 8, 2012 | Sep 2, 2015 | 迪沙药业集团有限公司 | 一种米格列奈钙的制备方法 |
| CN103450069A * | Jun 24, 2013 | Dec 18, 2013 | 山西大同大学 | Preparation method of mitiglinide calcium |
| CN103724253A * | Dec 11, 2013 | Apr 16, 2014 | 苑振亭 | Preparation method for Mitiglinide calcium hydrate |
| CN103724253B * | Dec 11, 2013 | Jun 15, 2016 | 苑振亭 | 一种米格列奈钙的制备方法 |
| CN102659562A * | May 9, 2012 | Sep 12, 2012 | 山东铂源药业有限公司 | Synthesis method of mitiglinide calcium intermediate |
| CN102898348A * | Sep 8, 2012 | Jan 30, 2013 | 迪沙药业集团有限公司 | Preparation method for Mitiglinide calcium |
| CN102898348B * | Sep 8, 2012 | Sep 2, 2015 | 迪沙药业集团有限公司 | 一种米格列奈钙的制备方法 |
| CN103450069A * | Jun 24, 2013 | Dec 18, 2013 | 山西大同大学 | Preparation method of mitiglinide calcium |
| CN103709092A * | Nov 4, 2013 | Apr 9, 2014 | 河北科技大学 | High purity mitiglinide calcium preparation method |
| CN103709092B * | Nov 4, 2013 | Jul 6, 2016 | 河北科技大学 | 米格列奈钙的制备方法 |
| CN104311471A * | Sep 23, 2014 | Jan 28, 2015 | 山东省药学科学院 | Improved mitiglinide calcium industrialized preparation method |
| CN1616427A * | Nov 13, 2003 | May 18, 2005 | 中国科学院上海药物研究所 | New method for preparing medicine mitiglinide for treating diabetes |
| CN101270074A * | Mar 21, 2007 | Sep 24, 2008 | 北京德众万全药物技术开发有限公司 | Method for preparing high purity mitiglinide calcium |
| CN101492411A * | Jan 22, 2008 | Jul 29, 2009 | 北京华禧联合科技发展有限公司 | Improved method for preparation of mitiglinide |
| WO2009047797A2 * | Oct 7, 2008 | Apr 16, 2009 | Ind-Swift Laboratories Limited | Process for the preparation of perhydroisoindole derivative |
| Reference | ||
|---|---|---|
| 1 | * | 张永亮,等: “米格列奈合成方法研究“, 《化工中间体》, no. 1, 31 December 2009 (2009-12-31), pages 16 – 22 |
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN101492411A * | Jan 22, 2008 | Jul 29, 2009 | 北京华禧联合科技发展有限公司 | Improved method for preparation of mitiglinide |
| WO2005030719A1 * | Sep 24, 2004 | Apr 7, 2005 | Les Laboratoires Servier | Novel method for preparing cis-octahydro-isoindole |
| Reference | ||
|---|---|---|
| 1 | * | WEI HUANG,等: “Novel Convenient Synthesis of Mitiglinide“, 《SYNTHETIC COMMUNICATIONS》, vol. 37, no. 13, 3 July 2007 (2007-07-03), pages 2153 – 2157, XP055079498, DOI: doi:10.1080/00397910701392590 |
| 2 | * | 张永亮,等: “米格列奈合成方法研究“, 《化工中间体》, no. 1, 31 January 2009 (2009-01-31), pages 16 – 22 |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN103709092A * | Nov 4, 2013 | Apr 9, 2014 | 河北科技大学 | High purity mitiglinide calcium preparation method |
| CN103709092B * | Nov 4, 2013 | Jul 6, 2016 | 河北科技大学 | 米格列奈钙的制备方法 |
| EP0507534A1 * | Mar 30, 1992 | Oct 7, 1992 | Kissei Pharmaceutical Co., Ltd. | Succinic acid compounds |
| EP0967204A1 * | Jan 22, 1998 | Dec 29, 1999 | Kissei Pharmaceutical Co Ltd | Process for producing benzylsuccinic acid derivatives |
| US6133454 * | Jul 1, 1998 | Oct 17, 2000 | Adir Et Compagnie | Method for preparing a substituted perhydroisoindole |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN102898348A * | Sep 8, 2012 | Jan 30, 2013 | 迪沙药业集团有限公司 | Preparation method for Mitiglinide calcium |
| CN102898348B * | Sep 8, 2012 | Sep 2, 2015 | 迪沙药业集团有限公司 | 一种米格列奈钙的制备方法 |
| CN103450069A * | Jun 24, 2013 | Dec 18, 2013 | 山西大同大学 | Preparation method of mitiglinide calcium |
| CN103724253A * | Dec 11, 2013 | Apr 16, 2014 | 苑振亭 | Preparation method for Mitiglinide calcium hydrate |
| CN103724253B * | Dec 11, 2013 | Jun 15, 2016 | 苑振亭 | 一种米格列奈钙的制备方法 |
|
|
| Systematic (IUPAC) name | |
|---|---|
|
(2S)-2-benzyl-4-[(3aR,7aS)-octahydro-2H-isoindol- 2-yl]-4-oxobutanoic acid
|
|
| Clinical data | |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration |
oral |
| Identifiers | |
| CAS Number | 145375-43-5 |
| ATC code | A10BX08 (WHO) |
| PubChem | CID 121891 |
| DrugBank | DB01252 |
| ChemSpider | 108739 |
| UNII | D86I0XLB13 |
| KEGG | D01854 |
| ChEMBL | CHEMBL471498 |
| Chemical data | |
| Formula | C19H25NO3 |
| Molar mass | 315.41 g/mol |
/////////207844-01-7, 145525-41-3, KAD-1229, S-21403, MITIGLINIDE, Glufast, Kissei, 145375-43-5
Quality Control & MSDS
O=C(O)[C@@H](Cc1ccccc1)CC(=O)N3C[C@H]2CCCC[C@H]2C3
New aspects of developing a dry powder inhalation formulation applying the quality-by-design approach
DRUG REGULATORY AFFAIRS INTERNATIONAL

The current work outlines the application of an up-to-date and regulatory-based pharmaceutical quality management method, applied as a new development concept in the process of formulating dry powder inhalation systems (DPIs). According to the Quality by Design (QbD) methodology and Risk Assessment (RA) thinking, a mannitol based co-spray dried formula was produced as a model dosage form with meloxicam as the model active agent.
The concept and the elements of the QbD approach (regarding its systemic, scientific, risk-based, holistic, and proactive nature with defined steps for pharmaceutical development), as well as the experimental drug formulation (including the technological parameters assessed and the methods and processes applied) are described in the current paper.
Findings of the QbD based theoretical prediction and the results of the experimental development are compared and presented. Characteristics of the developed end-product were in correlation with the predictions, and all data were confirmed by the relevant results…
View original post 223 more words
ECA Task Force will publish Draft Data Integrity Guideline at Conference in October
DRUG REGULATORY AFFAIRS INTERNATIONAL

Data Integrity has become one of the most frequently observed GMP deviations at FDA and EU Inspections. For that reason the ECA Foundation decided to set up a Task Force on Data Integrity in December 2015 – with the goal to provide Guidance for the implementation in practice. Read more about the ECA Guidance on Data Integrity.
http://www.gmp-compliance.org/eca_mitt_05545_15488_n.html
Data Integrity has become one of the most frequently observed GMP deviations at FDA and EU Inspections. This is why the topic is currently in the centre of attention of both regulators and industry. And for that reason the ECA Foundation decided to set up a Task Force on Data Integrity in December 2015 – with the goal to provide Guidance for the implementation in practice.
The ECA Task Force will be comprised of members from both the IT Compliance Group and the Analytical QC Group. Current Members are:
– Dr. Wolfgang Schumacher…
View original post 177 more words
D2/5-HT2A receptor dual antagonist, (±)-SIPI 6360

(±)-SIPI 6360
D2/5-HT2A receptor dual antagonist
7-[3-[4-(6-fluoro-1,2-benzoxazol-3-yl)piperidin-1-yl]propoxy]-3-methyl-3,4-dihydro-1H-quinolin-2-one
2(1H)-Quinolinone, 7-[3-[4-(6-fluoro-1,2-benzisoxazol-3-yl)-1-piperidinyl]propoxy]-3,4-dihydro-3-methyl-
| Molecular Formula: | C25H28FN3O3 |
|---|---|
| Molecular Weight: | 437.506523 g/mol |
Schizophrenia is a common severe mental patients, mental illness is the most serious of all, the most dangerous kind, the worldwide incidence of about I%, with the accelerate pace of social life, The incidence was significantly increased. Most schizophrenic patients due to the long treatment period, high cost, side effects and give up the treatment, often lead to more serious social consequences.
Numerous studies show that the brain monoamine neurotransmitters, especially dopamine and 5-hydroxytryptamine system is closely related to the body’s normal mental activity, these two types of system disorder can lead to a variety of neuropsychiatric diseases such as schizophrenia , neuropathic pain, mania, anxiety disorders, all kinds of depression, Parkinson’s disease and the like.
The drugs currently used clinically primarily for conventional antipsychotics (such as dopamine D2 receptor antagonists) and atypical antipsychotics (such as D2 / 5-HT2a dual antagonist), where conventional antipsychotics because it is easy leads to extrapyramidal symptoms (EPS) and gradually phased out, atypical antipsychotics variety, but no one medication to improve the overall spectrum of schizophrenia has the absolute advantage, most of the positive or negative symptoms of a a symptom improvement, or reduced side effects. So look for low toxicity, rapid onset, treatment spectral width of new anti-schizophrenia drug has been a hot topic in the world pharmaceutical industry.
In recent years, scientists have found that the dopamine D2 partial agonist can over time reduce dopamine activity in the transfer of dopamine, but not all block; the other hand, when the low dopaminergic activity is caused by stimulating effect on both positive and negative symptoms of mental illness have a significant effect. 5-HT2a receptor antagonists can improve negative symptoms, while synergies D2 EPS side effects can be reduced to about 1% level (classical antipsychotic drugs EPS incidence is about 30%), part of the 5-HTla agonism and 5-HT2a and synergy can make in therapeutic doses under observation EPS decreased to undetectable levels, therefore, has D2 ,5-HT2a, 5HTla synergy targets three new anti-drugs are currently developed Jingshenfenlie focus and an important development direction.
The present invention relates to a quinoline derivative can stabilize the brain dopaminergic, serotonergic energy system, may for a variety of neurological and psychiatric diseases have improved and treatment can be used for neuropathic pain, mania, schizophrenia, anxiety disorders, all kinds of depression, Parkinson’s disease, especially in the treatment of schizophrenia.
DETAILS COMING……….
PATENT

PATENT

Example 1
1-1
7- (3- (4- (6-fluorophenyl and [d] different dumb-3-yl) piperidin-1-yl) propoxy) -3,4-dihydro-3-carboxylic acid -one – yl quinolin -2 (1H)
1) N- (3- methoxyphenyl) propionamide
3-methoxy-aniline (0.1mol), methylene chloride (30 mL), triethylamine (0.2mol), was added to the flask lOOmL three, propionyl chloride was added dropwise under ice (0.12mol) in methylene chloride 30 mL, temperature does not exceed 5 ° C, the addition was complete, the ice bath was removed and stirred at room temperature 0.5h, the system was washed with water, dilute hydrochloric acid, saturated brine, dried over anhydrous magnesium sulfate, and evaporated to dryness to give a white powdery solid 17.01g yield 95%.
2) 2-chloro-7-methoxy-3-methylquinoline
The DMF (20mL) was added to the three 250mL flask, was added dropwise under ice-salt bath of POCl 3 (100 mL), temperature does not exceed 0 ° C, the addition was completed stirring 0.5h, was added portionwise N- (3- methoxyphenoxy yl) propanamide powder (31.0g), was slowly warmed to 50 ° C, violent reaction, to be exothermic easing slowly warmed to reflux, the reaction was kept 2h, cooled to room temperature, the system was poured into 800 g of crushed ice to sodium carbonate to adjust the pH to 7 to precipitate a yellow solid with petroleum ether – ethyl acetate to give pure product 20.86g, yield 58%.
3) 3-methyl-7-methoxy-quinolin -2 (1H) – one
2-Chloro-7-methoxy-3-methyl-quinoline (20.76g), acetic acid (150 mL) placed in 250mL one-neck flask, heated at reflux for 24h, acetic acid recovery, and the residue was recrystallized from ethanol to 95%, white needle crystalline 16.08g, yield 85%.
4) 7-methoxy-3,4-dihydro-3-methyl-quinolin -2 (1H) – one
7-Methoxy-3-methyl-quinolin -2 (1H) – one (18.92g), acetic acid (150mL), 10% Pd / C (lg) was added to the three 250mL flask, the system was replaced with nitrogen air, and then the nitrogen was replaced with hydrogen, and then the reaction was heated to 80 ° C overnight, cooled to room temperature, filtered and the filtrate evaporated to dryness to give a white powder, washed with water once, 50 ° C and dried in vacuo 4h, as a white powdery solid 18.91g yield of 98.95%.
5) 7-hydroxy-3,4-dihydro-3-methyl-quinolin -2 (1H) – one
7-Methoxy-3,4-dihydro-3-methyl-quinolin -2 (1H) – one (19.12g), 40% hydrobromic acid (150 mL) placed in 250mL one-neck flask was heated at reflux for 12h cooled to room temperature, the precipitated solid was filtered, the filter cake successively with hydrobromic acid, washed with water, 50 ° C and dried in vacuo 4h, 14.60 g as a white powdery solid, yield 82.4%.
6) 3- (1- (3-chloropropyl) piperidin-4-yl) -6-fluorophenyl and [d] oxazole different dumb
6-fluoro-3- (piperidin-4-yl) benzo [d] isoxazol dummy oxazole (22.00g), 1- bromo-3-chloropropane (40mL), anhydrous potassium carbonate (40g), acetone ( 250mL) was added to a 500mL one-neck flask was refluxed overnight, cooled to room temperature, filtered, the filter cake was washed twice with hot acetone and the combined filtrate was added dropwise a solution of anhydrous hydrogen chloride in ethanol, the precipitated white solid was filtered, the filter cake washed with acetone after washing once, it was dissolved in 200mL of water, adjusted with sodium carbonate to pH 9, and filtered to obtain a white powdery solid 15.94 g, yield 48.0%
7) 7- (3- (4- (6-fluorobenzo [d] isoxazol-3-yl dummy) piperidin-1-yl) propoxy) -3,4-dihydro-3-methyl quinolin -2 (1H) – one
3- (1- (3-chloropropyl) piperidin-4-yl) -6-fluorophenyl and [d] oxazole different dumb (lmmol), 7- hydroxy-3,4-dihydro-3-carboxylic acid yl quinolin -2 (1H) – one (1.0 mmol), anhydrous potassium carbonate (3.0mmol) were added to the lOmLDMF, 60 ° C overnight the reaction, potassium carbonate was filtered off, the mother liquor evaporated to dryness to give a pale yellow solid, the filter cake recrystallized with 95% ethanol, 50 ° C and dried in vacuo 4h, as a white powdery solid 0.30g, 69% yield.
NMR IH (of DMSO-D . 6 ): L up to .27 (D, 3H, J = 9.2Hz), 2.06-2.32 (m, 9H), 2.67-2.69 (T, 2H), 2.95 (D * D, lH, J = 3.2Hz, 12.8Hz), 3.15-3.17 ( m, 2H), 4.05 (t, 2H, J = 6Hz), 6. 37 (d, lH, J = 2.4Hz), 6.56 (d * d, lH, J = 2.4Hz, 8.0Hz), 7.05-7.11 (m, 2H), 7.25-7.29 (m, lH), 7.73-7.76 (m, lH), 7.98 (s, lH), 11.43 (brs, lH)
ESI-MS: 438 (M + 1)
Example 2
Preparation 1-1 hydrochloride
7- (3- (4- (6-fluorophenyl and [d] different dumb-3-yl) piperidin-1-yl) propoxy) -3,4-dihydro-3-methyl-quinoline morpholine -2 (1H) – one (lmmol) was dissolved with ethyl acetate (50 mL) was slowly added dropwise a solution of anhydrous hydrogen chloride in ethyl acetate (lmol / L, 5mL), stirred for 2h, the precipitated solid was filtered, the filter cake washed with ethyl acetate, 50 ° C and dried in vacuo 4h, as a white powdery solid 0.436g, yield 92%.
ESI-MS: 438 (M + 1)
Elemental analysis results:
Calcd: C, 63.35%; H, 6.17%; Cl, 7.48%; F, 4.01%; N, 8.87%; O, 10.13%
Found: C, 63.29%; H, 6.24%; CI, 7.43%; F, 4.05%; N, 8.82%; O, 10.17%
Example 3
Preparation 1-1 methanesulfonate
The 1-1 (lmmol) was dissolved with ethyl acetate (50 mL) was slowly added dropwise a solution of methanesulfonic acid in ethyl acetate (lmol / L, 5mL), stirred for 2h, the precipitated solid was filtered, the filter cake with ethyl acetate wash, 50 ° C and dried in vacuo 4h, as a white powdery solid 0.487g, yield 91.3%.
ESI-MS: 438 (M + 1, positive mode), 95 (CH 3 the SO 3 -, negative mode) Elemental analysis:
Calcd: C, 58.52%; H, 6.04%; F, 3.56%; N, 7.87%; 0, 17.99%; S, 6.01%
Found: C, 58.49%; H, 6.09%; F, 3.50%; N, 7.81%; 0, 18.02%; S, 6.09%
PATENT
Paper
Development and Kilogram-Scale Synthesis of a D2/5-HT2A Receptor Dual Antagonist (±)-SIPI 6360

The kilogram-scale synthesis of a D2/5-HT2A receptor dual antagonist (±)-SIPI 6360 was developed as an alternative treatment for schizophrenia. Specifically, three conditions were modified and optimized, including the Vilsmeier conditions, to prepare quinoline 3. In addition, the palladium-catalyzed hydrogenation was modified to synthesize dihydroquinolin-2(1H)-one 5, and the reduction of β-chloroamide was altered to form 3-chloropropanamine 8. Ultimately these improvements led to the preparation of a 1.5 kg of (±)-SIPI 6360 batch in eight steps with an overall yield of 34% and purity of 99.8%.
//////// D2/5-HT2A receptor dual antagonist (±)-SIPI 6360, 1401333-14-9
c21CC(C(Nc1cc(cc2)OCCCN3CCC(CC3)c4c5ccc(cc5on4)F)=O)C
Fortune India presents award to Ajanta pharma

Fortune India has published list of 500 mid-size companies who ranked them on various parameters based on the results of 2013-2014. Ajanta pharma features very prominently in the lists. Ajanta’s ranking on various pararmeters is given below:
Ranked 3’d largest Wealth Creator on 5 year CAGR 93.14%
Ranked 10th on Capital Employed (ROCE)
Ranked 21st in Net profit .
Ranked 182nd in Sales
On 17’th August 2015, Fortune India organized an award function to present the awards to Top 10 largest weatth creator companies and Ajanta is one of those elite companies.
The awards were presented by Mr. Piyush Goyal, Minister of State-Power,Coal& New and Renewable Energy, Govt. of India to Mr. yogesh Agrawal, Managing Director and Mr.Rajesh Agrawal,Jt.Managing Director of the company.


Ajanta Pharma, “One of the Giants of Tomorrow” – Fortune India
We are pleased to share with you that Ajanta Pharma has been honoured as “ONE OF THE GIANTS OF TOMORROW” by prestigious Fortune India magazine on 19th August 2016 at New Delhi.
The honour was conferred to our Managing Director, Mr. Yogesh Agrawal and our Jt. Managing Director, Mr. Rajesh Agrawal at the hands of Hon. Mr. Nitin Gadkari, Union Minister for Road Transport and Highways and Shipping, Govt of India. This is the 2nd year in row where Ajanta has received the recognition from Fortune India.
Fortune India (June 2016 Issue) published the list of mid-size companies based on the financial year 2014-15 results and we are pleased to share with you that Ajanta has been ranked 3rd Top Wealth creator over last 5 years.


Ajanta Pharma Limited (APL) is a pharmaceutical company headquartered in Mumbai, India. It has strong presence in Branded Generic business in India & Emerging markets; and Generic business in USA. In India, company operates in selected therapeutic areas of Cardiology, Dermatology, Ophthalmology and Pain management. Its brands in each of sub-therapeutic areas or molecules hold leadership positions. In Emerging Markets, company has presences in Africa, Asia, Middle East, and CIS on broader therapeutic segments such as anti-malarial, gastro, antibiotics, cardiology, dermatology, pain management, etc. In USA, company has already no. of approved ANDA’s which are either commercialized or in process of being commercialized and large no. of ANDA’s are awaiting US FDA approval. We have state-of-the-art research facilities for formulation (finished product) and API development located at Mumbai, India. Our R&D capabilities are evident from number of products launched 1st to market by the company providing patients most needed compliance and convenience. A dedicated and focused team of more than 750 Ajantaites work for R&D, which is growing continuously. Ajanta has four formulations manufacturing facilities located in India and 1 in Mauritius. Besides that, we also have an API manufacturing facility located at Waluj, India. Ajanta’s flagship formulation facility at Paithan (Maharashtra, India) has approval of USFDA, WHO- Geneva (prequalification), UNICEF and many regulatory authorities from different parts of the world. We continuously invest in enhancing our existing manufacturing facilities to meet current cGMP requirements and also construct new facilities to meet the company’s growth requirements. We are in process of setting up 1 more formulations manufacturing facility for domestic and emerging markets at Guwahati, Assam. Please visit http://ajantapharma.com/ for more information. Contact: careers@ajantapharma.com
Specialties
Speciality Branded Generics, Generics, Complex Formulations
-
Website
-
Industry
Pharmaceuticals
-
Type
Public Company
-
Headquarters
98 Ajanta House Charkop, Kandivili West Mumbai,Maharashtra 400067 India
-
Company Size
5001-10,000 employees
-
Founded
1973
Rajesh Agrawal (left), Ajanta Pharma’s joint managing director, with brother Yogesh, who is also managing director of the company, at their Kandivli facility
Ajanta Pharma needed a shot of its own medicine, an energiser like 30-Plus. It found its antidote in the new generation of Agrawals: Mannalal’s sons, Yogesh and Rajesh.

“When I joined Ajanta (in 2000), and realised what was going on, I wanted to run away. I thought to myself, ‘Why did I return from the US? I could have had a job there,’” says Rajesh, 39, Ajanta’s joint managing director, who has a management degree from Bentley College, Massachusetts. “It was tough in the beginning, especially the situation with creditors and debtors.”
Together, Rajesh and his older brother Yogesh, 43, who is managing director, changed Ajanta’s trajectory by focusing on the ‘specialty’ generic drug market and putting an end to the company’s legacy businesses, which included OTC drug sales and supplying drugs to government health agencies in India and other countries.
This was a risky move, but it has paid off. Ajanta Pharma closed FY15 with a consolidated net sales of Rs 1,481 crore and a net profit of Rs 310 crore (this is a compound annual growth rate, or CAGR, of 57 percent for four years since 2011). In terms of net sales, it recorded a CAGR of 31 percent for the same period. This growth has come on a low base, but the signs are encouraging. Its market value currently stands at around Rs 13,500 crore; this is a 65-fold growth in 15 years.
References
https://www.linkedin.com/company/263285
/////////Ajanta Pharma, “One of the Giants of Tomorrow” , Fortune India, AWARD, Fortune India, RAJESH AGRAWAL
