WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ]
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
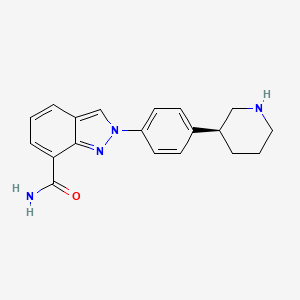
MK-4827(Niraparib) tosylate is a selective inhibitor of PARP1/PARP2 with IC50 of 3.8 nM/2.1 nM; with great activity in cancer cells with mutant BRCA-1 and BRCA-2; >330-fold selective against PARP3, V-PARP and Tank1. IC50 value: 3.8 nM/2.1 nM( PARP1/2) [1] Target: PARP1/2 in vitro: MK-4827 displays excellent PARP 1 and 2 inhibition with IC(50) = 3.8 and 2.1 nM, respectively, and in a whole cell assay, it inhibits PARP activity with EC(50) = 4 nM and inhibits proliferation of cancer cells with mutant BRCA-1 and BRCA-2 with CC(50) in the 10-100 nM range [1]. in vivo: MK-4827 is well tolerated in vivo and demonstrates efficacy as a single agent in a xenograft model of BRCA-1 deficient cancer [1]. In addition, MK-4827 strongly enhances the effect of radiation on a variety of human tumor xenografts, both p53 wild type and p53 mutant. The enhancement of radiation response is observed in clinically relevant radiation-dose fractionation schedules. The therapeutic window during which time MK-4827 interacts with radiation lasts for several hours. These biological attributes make translation of this therapeutic combination treatment feasible for translation to the treatment of a variety of human cancers [2].
An inhibitor of poly (ADP-ribose) polymerase (PARP) with potential antineoplastic activity. PARP Inhibitor MK4827 inhibits PARP activity, enhancing the accumulation of DNA strand breaks and promoting genomic instability and apoptosis. The PARP family of proteins detect and repair single strand DNA breaks by the base-excision repair (BER) pathway. The specific PARP family member target for PARP inhibitor MK4827 is unknown. (NCI Thesaurus)
Niraparib is due to be submitted for FDA approval (for maintenance therapy in ovarian cancer) later in 2016.[4]
Chemically, MK-4827 is C19H20N4O[5] (ignoring a possible tosylate group).[6]
A 2012 study found that PARP inhibitors exhibit cytotoxic effects not based solely on their enzymatic inhibition of PARP, but by their trapping of PARP on damaged DNA, and the strength of this trapping activity was ordered niraparib >> olaparib >> veliparib.[7]
MEDICINAL CHEMISTRY APPROACH
The Medicinal Chemistry approach to compound 1 is shown in Scheme ABOVE. The racemic piperidine 2 was accessed by reduction of the 3-aryl pyridine 3 and then resolved by salt formation with tartaric acid. Protection of the piperidine nitrogen in enantiomerically upgraded piperidine 2 and condensation with aldehyde 4 afforded imine 5 which, after displacement of the nitro group with sodium azide, underwent a thermally promoted cyclisation to afford the 2-aryl indazole 6. Conversion of the ester functionality to a primary amide and deprotection afforded the active pharmaceutical ingredient (API) as the hydrochloride salt. A final chiral HPLC purification was then required to upgrade the enantiomeric purity to >98% ee, followed by lyophilization to give the desired compound 1 as an amorphous HCl salt.
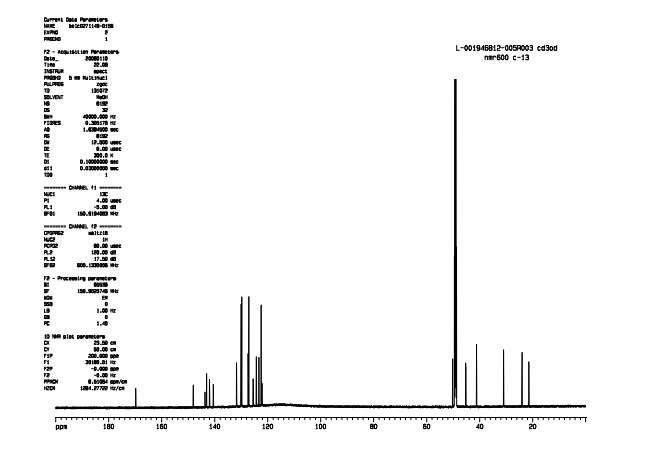
NMR CD3OD
Clinical trials
It has undergone a phase III trial for ovarian cancer.[8] It is reported that the primary endpoint (progression-free survival, PFS) was met.[4] Patients with and without a BRCA mutation both showed longer PFS.[4]
As of June 2016 seven clinical trials have been registered for MK-4827.[9]
Process development of the synthesis of the orally active poly(ADP-ribose)polymerase inhibitor niraparib is described. Two new asymmetric routes are reported, which converge on a high-yielding, regioselective, copper-catalyzed N-arylation of an indazole derivative as the late-stage fragment coupling step. Novel transaminase-mediated dynamic kinetic resolutions of racemic aldehyde surrogates provided enantioselective syntheses of the 3-aryl-piperidine coupling partner. Conversion of the C–N cross-coupling product to the final API was achieved by deprotection and salt metathesis to isolate the desired crystalline salt form.
Compound (1) a poly(ADP-ribose)polymerase (PARP) inhibitor has been made by a fit-for-purpose large-scale synthesis using either a classical resolution or chiral chromatographic separation. The development and relative merits of each route are discussed, along with operational improvements and extensive safety evaluations of potentially hazardous reactions.
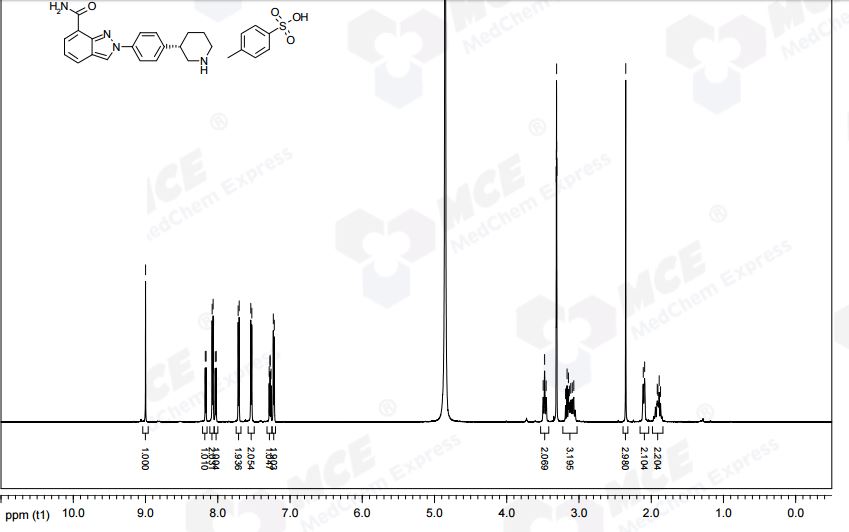
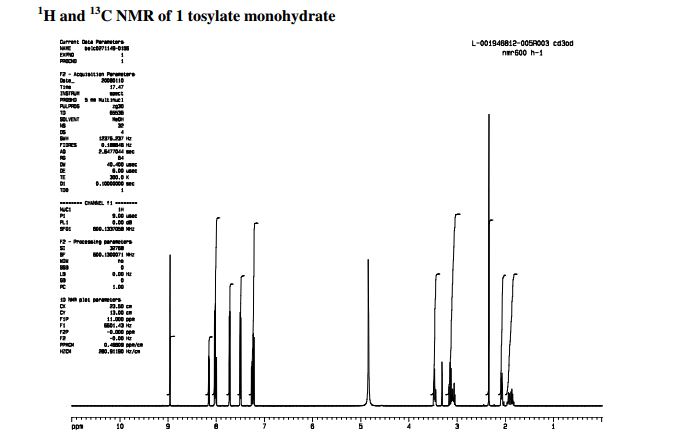
………………. The solid was collected and dried in vacuo at 40 °C to afford 1 as the tosylate monohydrate salt (797 g, 86%, >99 wt %, >99%ee) as a tan-coloured solid.
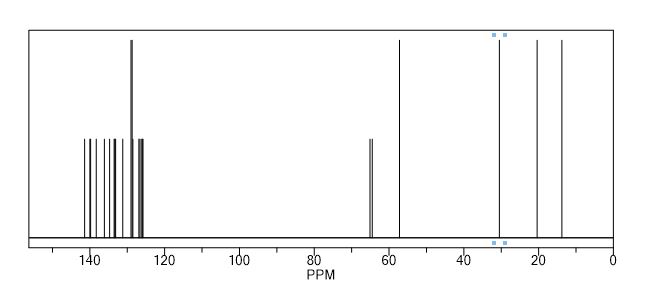
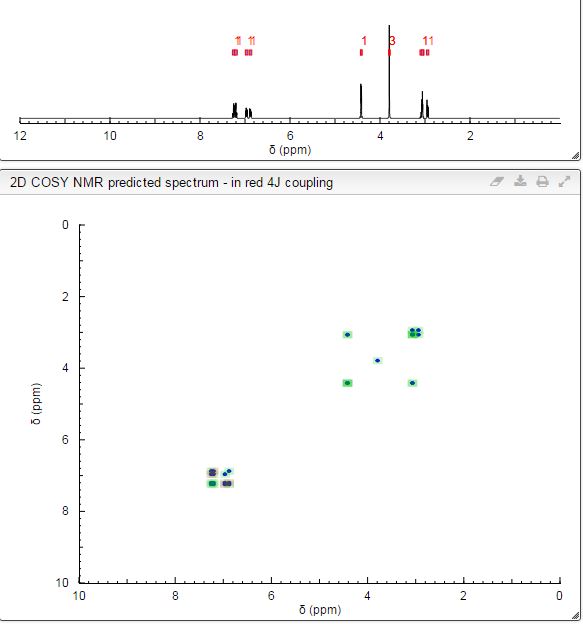
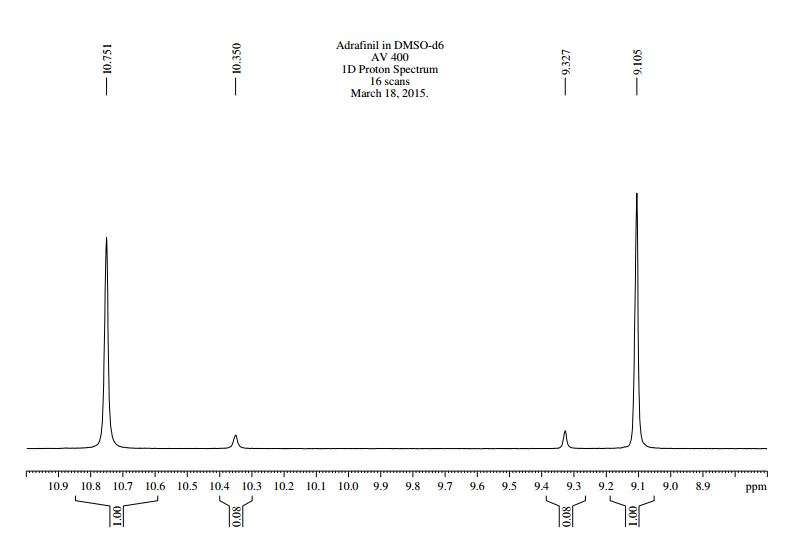
Mp = 144 °C. 1H NMR (600 MHz, CD3OD) δ 8.95 (1H, s), 8.15 (1H, dd, J = 7.1, 1.2 Hz), 8.02 (2H, m), 8.00 (1H, dd, J = 8.3, 1.2 Hz), 7.72 (2H, m), 7.49 (2H, m), 7.25 (1H, dd, J = 8.3, 7.1 Hz), 7.22 (2H, d, J = 8.0 Hz), 3.49–3.43 (2H, m), 3.16–3.04 (3H, m), 2.34 (3H, s), 2.09–2.05 (2H, m), 1.96–1.82 (2H, m).
Discovery of 2-{4-[(3S)-Piperidin-3-yl]phenyl}-2H-indazole-7-carboxamide (MK-4827): A Novel Oral Poly(ADP-ribose)polymerase (PARP) Inhibitor Efficacious in BRCA-1 and -2 Mutant Tumors
*To whom correspondence should be addressed. Current address: Department of Medicinal Chemistry, Merck Research Labs Boston, Avenue Louis Pasteur 33, Boston, MA 02115-5727. Phone: +1-617-992-2292. Fax: +1-617-992-2405. E-mail: philip_jones@merck.com.
Abstract
We disclose the development of a novel series of 2-phenyl-2H-indazole-7-carboxamides as poly(ADP-ribose)polymerase (PARP) 1 and 2 inhibitors. This series was optimized to improve enzyme and cellular activity, and the resulting PARP inhibitors display antiproliferation activities against BRCA-1 and BRCA-2 deficient cancer cells, with high selectivity over BRCA proficient cells. Extrahepatic oxidation by CYP450 1A1 and 1A2 was identified as a metabolic concern, and strategies to improve pharmacokinetic properties are reported. These efforts culminated in the identification of 2-{4-[(3S)-piperidin-3-yl]phenyl}-2H-indazole-7-carboxamide 56 (MK-4827), which displays good pharmacokinetic properties and is currently in phase I clinical trials. This compound displays excellent PARP 1 and 2 inhibition with IC50 = 3.8 and 2.1 nM, respectively, and in a whole cell assay, it inhibited PARP activity with EC50 = 4 nM and inhibited proliferation of cancer cells with mutant BRCA-1 and BRCA-2 with CC50 in the 10−100 nM range. Compound 56 was well tolerated in vivo and demonstrated efficacy as a single agent in a xenograft model of BRCA-1 deficient cancer.
A mixture of succinic anhydride 1 (110 g) and bromobenzene (695 mL) was cooled to below 5°C then added A1C13 (294 g). The slurry was allowed to warm to RT and then aged until the reaction was complete judged by HPLC. The reaction mixture was then transferred slowly into a cold HC1 solution resulting in the formation of a white precipitate. The white slurry was filtered through a fritted funnel rinsing with H20. To the off-white product was added MTBE and extracted with aq. NaOH. The aqueous layer was cooled in an ice bath. Concentrated HC1 was added drop wise to adjust the solution pH to 1 , resulting in the formation of a white slurry. The slurry was collected on a fritted funnel, rinsed with H20, and dried under vacuum with a N2 sweep at RT to give the target compound (265 g, 93% corrected yield) as a white powder.
1.2 Esterification
A mixture of the acid 2 (205 g), IPA (4 L) and cone. H2S04 (2.13 mL / 3.91 g) was heated to a gentle reflux until the reaction was complete judged by HPLC. The solution was then cooled to RT and concentrated to a volume of 350-400 mL. The residue was dissolved in
MTBE (1.2 L), washed with aq. Na2C03 followed by water. After dried over MgS04 , the filtrate was solvent-switched into heptane. The slurry was then filtered, and the cake was washed with cold heptane. After drying under vacuum, the target compound (223.5 g, 93% corrected yield) was obtained as a white powder.
1.3 Epoxidation
A mixture of Me3SOI (230 g) and DMSO (300 mL) was added KOt-Bu (113 g) followed by DMSO (300 mL). The mixture was aged for a further 1.5 hr. In a separate flask, ketone 3 (230 g) was dissolved in a mixture of THF (250 mL) and DMSO (150 mL), and the resulting solution was added drop wise to the ylide solution. The mixture was aged for 2 hr at RT, added hexanes (1 L), and then quenched by the addition of ice-water (600 mL). The layers were cut, and the organic layer was washed with water then with brine. The slightly cloudy yellow organic layer was dried over Na2S04 and filtered through a fritted funnel. Product solution assay was 176.1 g (76%> assay yield). This solution was carried forward into the rearrangement step. 1.4 Epoxide rearrangement and bisulfite formation
5 – not isolated
A solution of crude epoxide 4 (assay 59.5 g) in hexanes was solvent switched into PhMe, and added ZnBr2 (10.7 g). When the rearrangement was complete judged by HPLC, the slurry was filtered through a fritted funnel. The clear filtrate was washed with 10% aq. NaCl and then stirred with a solution of sodium bisulfite (NaHS03, 24.7 g) in H20 (140 mL) vigorously at RT for 3 hr. The cloudy aqueous layer was separated and washed with heptanes. By 1H-NMR assay, the aqueous solution contained 71.15 g bisulfite adduct 6 (30.4 wt % solution, 90%) yield from crude epoxide 4). This solution was used directly in the subsequent transaminase step.
1.5 Transaminase DKR
45 C, inert, 40-46 hrs 7
100 g/L as 17.16 wt % aq solution 99.3% ee
85-87% yield
To a cylindrical Labfors reactor was charged pyridoxal-5 -phosphate (1.4 g, 5.66 mmol), 452 ml 0.2 M borate buffer pH 10.5 containing 1M iPrNH2, 52 g transaminase (SEQ ID NO: 180), and 75 ml DMSO, and the resulting mixture was warmed to 45°C. The pH was controlled at pH 10.5 using 8 M aq iPrNH2. To this was added dropwise a mixture of 17.16 wt% aq solution of ester bi-sulfite 6 (147.2 g, 353 mmol) and 219 ml DMSO under N2 atmosphere. When the reaction was complete judged by HPLC, the reaction mixture was cooled and extracted with 1 volume of 3:2 IPA:IPAc. The aq/rag layer was extracted again with 1 volume of 3:7 IPATPAc. The organic layer was washed with brine at pH >9. Assay yield in solution was 78 g (87%); 99.3% ee. After dried over MgS04, and filtered through a fritted funnel, the crude solution was concentrated under vacuum flushing with IP Ac to remove IPA. The resulting slurry was concentrated to a final volume of -200 mL, cool to below 0°C, and filtered to collect the solid. The cake was washed with ice-cold IPAc and dried at RT under vacuum to give the desired product (84% corrected yield, 99.3 LCAP) as a white powder. 1.6. Reduction of amide
(S)-3-(4-bromophenyl)piperidine
The lactam 7 can be reduced to form the i eridine 8 as described below:
7 – crystalline
A mixture of lactam 7 (10.25 g at 97.5 wt %) in THF (100 mL) was cooled to < 10°C, and added NaBH4 (4.47 g). EtOH (6.89 mL) was then added slowly over 20 min. The slurry was aged for an additional 1 hr at 2°C after which BF3 THF (13.03 mL) was added over 1 hr. The slurry was slowly warmed to RT and aged until complete conversion judged by HPLC. The reaction was then cooled to < 5°C then slowly quenched with MeOH (7.96 mL), added HC1 (9.69 mL), then the reaction was heated to 45°C until decomplexation of product-borane complex was complete, as indicated by LC assay. The reaction was cooled, diluted with IPAc (75 mL) and water (80 mL), and then pH was adjusted with aqueous NH4OH to pH 8. The organic layer was separated, added 75 mL water, then pH adjusted to 10.5 with 50 wt % NaOH. The layers were separated and the organic layer was washed with brine. After solvent-switched to IPAc, LC Assay yield was 9.1g; 95.9%.
1.7 Tosylate salt formation The tosylate salt of the piperidine 8 can be formed as described below:
The crude piperidine 8 free base in IPA was heated to ~40°C. TsOH H20 solids was added portion-wise. The slurry was warmed to 50°C and held at that temperature for 2 h, and then slowly cooled to RT and aged overnight. Supernatant concentration was measured to be 2.5 g/ml (free base concentration). The solids were filtered and washed with IP Ac (3×15 mL) and dried at RT. Isolated solides: 14.85 g, 96% corrected isolated yield.
1.8 Boc protection
The piperi ine 8 tosylate salt can be protected as described below:
To a stirred slurry of the tosylate salt of piperidine 8 (25.03 g, 60.6 mmol) in MTBE (375 ml) was added NaOH (aq. 1.0 N, 72.7 ml, 72.7 mmol) at RT. To the mixture, (BOC)20 (13.36 ml, 57.6 mmol) was added slowly over 3 min. The resulting mixture was stirred for 4.5 hr at RT, and then the aqueous layer was separated. The MTBE layer was washed with water (100 ml X 2). The organic layer was filtered, and DMAC (100 ml) was added to the filtrate and
concentrated under vacuum. Product assay: 21.86 g, quantitative yield.
1.9 Terf-Butylamide Formation
N-(tert-butyl)- 1 H-indazole-7-carboxamide
10 11
Indazole-7-carboxylic acid 10 (50.3 g, 295 mmol) was dissolved in DMF, and added CDI (59.1 g, 354 mmol) at RT. After 1.5hr, tert-butylamine (62.5 ml, 589 mmol) was added to the reaction mixture. The resulting reaction mixture was warmed to 40 °C until complete
conversion, then cooled to RT. Water (600 ml) was added dropwise causing the mixture to form a thick slurry. Solid was collected by filtration and washed with 10% DMF in water (250 ml) followed by water. The solid was dried under vacuum. Beige solid: 55.31 g, 86%> isolated yield.
A mixture of the protected piperidine 9 (113 g, 18.23 wt%, 60.6 mmol) in DMAc (160 mL), compound 11 (13.82 g, 63.6 mmol), and K2CO3 (25.6 g, 182 mmol) was degassed by bubbling nitrogen. To the mixture was added CuBr (0.444 g, 3.03 mmol) and 8- hydroxyquinoline 12 (0.889 g, 6.06 mmol), and the resulting mixture was warmed to 110°C until complete conversion. The reaction mixture was then cooled, filtered through a pad of Celite, and rinsed with DMAc (100 ml). The filtrate was warmed to 35°C and added citric acid aqueous solution (10%) dropwise to form a light green slurry. After cooled to room temperature, the slurry was filtered, and the cake was washed with DMAc/Water (2/1, 150ml) followed by copious amount of water. The solid was dried under vacuum with nitrogen. Net weight: 27.24g. LC assay: 26.77g, 98.3 wt %. Assay yield: 93.6%.
1.11 Double deprotection
To compound 13 (20.0 g, 41.2 mmol) was added MSA (60 ml) and o-xylene (40 ml), and the the reaction mixture was warmed to 40°C until the complete conversion judged by HPLC. The reaction mixture was cooled to RT and added water (140 ml) slowly maintaining the temperature < 25°C. When the water addition was completed, the organic layer was removed, and the aq. layer was washed with toluene. The aqueous layer was filtered through a glass funnel, and the filtrate was added an aqueos solution of TsOH (11.77g in 23.5 ml) slowly at RT causing a thick slurry to form. Solid was collected by filtration, washed with water, and dried under vacuum. The titled compound was obtained as a white powder. Net weight: 20.6 g. LC assay: 20.0 g, 97.3 wt %. Assay yield: 95.2%.
EXAMPLE 2
The following Example 2 describes synthesis of the trifluoromethylacetate salt of compound 2-{4-[(3S)-Piperidin-3-yl]phenyl}-2H-indazole-7-carboxamide:
To the indazole-7-carboxylic acid 10 (400 mg, 2.47 mmol) in tetrahydrofuran (9.9 mL), was sequentially added HATU (1.13 g, 2.96 mmol), DIPEA (2.15 mL, 12.3 mmol), and cumylamine (500 mg, 3.70 mmol) at 50°C. The reaction was stirred overnight before being concentrated and loaded directly onto a silica column, eluting with 10-30% EtOAc/hexane. The product was collected and concentrated to afford the desired product as a colorless solid (557 mg, 81% yield).
A sealed vial containing the indazole-7-carboxamide 15 (50 mg, 0.18 mmol), copper(I) iodide (2.6 mg, 0.014 mmol), potassium phosphate tribasic (80 mg, 0.38 mmol), and aryl bromide 9 (73.1 mg, 0.215 mmol) was evacuated and backfilled with argon (x3). Trans-N,N’- dimethylcyclohexane-l,2-diamine (11.3 μΐ,, 0.072 mmol), and toluene (179 μΐ) were then added successively and the sealed vial was heated at 110 °C overnight. The vial was then cooled and toluene (0.30 mL) was added to the slurry. Crude LC/MS indicated >20: 1 selectivity for the desired indazole isomer. The crude product was purified by loading directly onto a Biotage Snap 10G silica column, eluting with 5-50% EtOAc/hexane. The product was collected and concentrated to afford the desired product as a colorless solid (78 mg, 81% yield).
2.3 Double deprotection
(5)-2-(4-(piperidin-3-yl)phenyl)-2H-indazole-7-carboxamide trifluoromethylacetate salt
16 17
To the piperidine-l-carboxylate 16 (45 mg, 0.084 mmol), was added triethylsilane (267 μί, 1.67 mmol) and TFA (0.965 mL, 12.5 mmol) at 25°C. The reaction was stirred for 4 hours and the reaction was concentrated in vacuo, and purified by mass triggered reverse phase HPLC (acetonitrile: water, with 0.1% TFA modifier). Lyphilization gave the desired product as the TFA salt and as a white solid (31 mg, 85% yield). HRMS (ESI) calc’d for Ci9H2iN40 [M+H]+: 321.1710, found 321.1710.
EXAMPLE 3
Following the conditions used in sections 2.1 and 2.2 of Example 2, this Example 3 shows regioselective N2 arylation of compound 9 using various amide protecting groups. The indazole-7-carboxylic acid 10 was reacted with various amines to generate a protected amide.
The amide protecting groups are indicated by the R group in Table 2. The amide coupling yield is provided in Table 2. The Cu-mediated carbon-nitrogen coupling of this indazole to compound 9 was then tested to determine if regioselective N2 arylation was possible. The arylation yield is also provided in Table 2. The data shows that various amide protecting groups on the indazole intermediate are suitable to generate efficient regioselective N2 arylation of compound 9.
The present invention relates to amide substituted indazoles which are inhibitors of the enzyme poly(ADP-ribose)polymerase (PARP), previously known as poly(ADP-ribose)synthase and poly(ADP-ribosyl)transferase. The compounds of the present invention are useful as monotherapies in tumors with specific defects in DNA-repair pathways and as enhancers of certain DNA-damaging agents such as anticancer agents and radiotherapy. Further, the compounds of the present invention are useful for reducing cell necrosis (in stroke and myocardial infarction), down regulating inflammation and tissue injury, treating retroviral infections and protecting against the toxicity of chemotherapy.
Poly(ADP-ribose) polymerase (PARP) constitute a super family of eighteen proteins containing PARP catalytic domains (Bioessays (2004) 26:1148). These proteins include PARP-1, PARP-2, PARP-3, tankyrase-1, tankyrase-2, vaultPARP and TiPARP. PARP-I, the founding member, consists of three main domains: an amino (N)-terminal DNA-binding domain (DBD) containing two zinc fingers, the automodification domain, and a carboxy (C)-terminal catalytic domain.
PARP are nuclear and cytoplasmic enzymes that cleave NAD+ to nicotinamide and ADP-ribose to form long and branched ADP-ribose polymers on target proteins, including
topoisomerases, histones and PARP itself (Biochem. Biophys. Res. Commun. (1998) 245:1-10).
Poly(ADP-ribosyl)ation has been implicated in several biological processes, including DNA repair, gene transcription, cell cycle progression, cell death, chromatin functions and genomic stability.
The catalytic activity of PARP-I and PARP-2 has been shown to be promptly stimulated by DNA strand breakages (see Pharmacological Research (2005) 52:25-33). In response to DNA damage, PARP-I binds to single and double DNA nicks. Under normal physiological conditions there is minimal PARP activity, however, upon DNA damage an immediate activation of PARP activity of up to 500-fold occurs. Both PARP-I and PARP-2 detect DNA strand interruptions acting as nick sensors, providing rapid signals to halt transcription and recruiting the enzymes required for DNA repair at the site of damage. Since radiotherapy and many chemotherapeutic approaches to cancer therapy act by inducing DNA damage, PARP inhibitors are useful as chemo- and radiosensitizers for cancer treatment. PARP inhibitors have been reported to be effective in radio sensitizing hypoxic tumor cells (US 5,032,617, US
5,215,738 and US 5,041,653).
Most of the biological effects of PARP relate to this poly (ADP-ribosyl)ation process which influences the properties and function of the target proteins; to the PAR oligomers that, when cleaved from poly(ADP-ribosyl)ated proteins, confer distinct cellular effects; the physical association of PARP with nuclear proteins to form functional complexes; and the lowering of the cellular level of its substrate NAD+ (Nature Review (2005) 4:421-440).
Besides being involved in DNA repair, PARP may also act as a mediator of cell death. Its excessive activation in pathological conditions such as ischemia and reperfusion injury can result in substantial depletion of the intercellular NAD+, which can lead to the impairment of several NAD+ dependent metabolic pathways and result in cell death (see Pharmacological Research (2005) 52:44-59). As a result of PARP activation, NAD+ levels significantly decline. Extensive PARP activation leads to severe depletion OfNAD+ in cells suffering from massive DNA damage. The short half-life of poly(ADP-ribose) results in a rapid turnover rate, as once poly(ADP-ribose) is formed, it is quickly degraded by the constitutively active poly(ADP-ribose) glycohydrolase (PARG). PARP and PARG form a cycle that converts a large amount OfNAD+ to ADP-ribose, causing a drop OfNAD+ and ATP to less than 20% of the normal level. Such a scenario is especially detrimental during ischemia when deprivation of oxygen has already drastically compromised cellular energy output. Subsequent free radical production during reperfusion is assumed to be a major cause of tissue damage. Part of the ATP drop, which is typical in many organs during ischemia and reperfusion, could be linked to NAD+ depletion due to poly(ADP-ribose) turnover. Thus, PARP inhibition is expected to preserve the cellular energy level thereby potentiating the survival of ischemic tissues after insult. Compounds which are inhibitors of PARP are therefore useful for treating conditions which result from PARP mediated cell death, including neurological conditions such as stroke, trauma and Parkinson’s disease.
PARP inhibitors have been demonstrated as being useful for the specific killing of BRCA-I and BRCA-2 deficient tumors {Nature (2005) 434:913-916 and 917-921; and Cancer Biology & Therapy (2005) 4:934-936).
PARP inhibitors have been shown to enhance the efficacy of anticancer drugs
{Pharmacological Research (2005) 52:25-33), including platinum compounds such as cisplatin and carboplatin {Cancer Chemother Pharmacol (1993) 33:157-162 and MoI Cancer Ther (2003) 2:371-382). PARP inhibitors have been shown to increase the antitumor activity of
topoisomerase I inhibitors such as Irinotecan and Topotecan (MoI Cancer Ther (2003) 2:371-382; and Clin Cancer Res (2000) 6:2860-2867) and this has been demonstrated in in vivo models (J Natl Cancer Inst (2004) 96:56-67).
PARP inhibitors have been shown to restore susceptibility to the cytotoxic and antiproliferative effects of temozolomide (TMZ) (see Curr Med Chem (2002) 9:1285-1301 and Med Chem Rev Online (2004) 1:144-150). This has been demonstrated in a number of in vitro models (Br J Cancer (1995) 72:849-856; Br J Cancer (1996) 74:1030-1036; MoI Pharmacol (1997) 52:249-258; Leukemia (1999) 13:901-909; GUa (2002) 40:44-54; and Clin Cancer Res (2000) 6:2860-2867 and (2004) 10:881-889) and in vivo models (Blood (2002) 99:2241-2244; Clin Cancer Res (2003) 9:5370-5379 and J Natl Cancer Inst (2004) 96:56-67). PAPR inhibitors have also been shown to prevent the appearance of necrosis induced by selective Λ3 -adenine methylating agents such as MeOSC>2(CH2)-lexitropsin (Me-Lex) {Pharmacological Research (2005) 52:25-33).
PARP inhibitors have been shown to act as radiation sensitizers. PARP inhibitors have been reported to be effective in radiosensitizing (hypoxic) tumor cells and effective in preventing tumor cells from recovering from potentially lethal {Br. J. Cancer (1984) 49(Suppl. VI):34-42; and Int. J. Radial Bioi. (1999) 75:91-100) and sub-lethal {Clin. Oncol. (2004) 16(l):29-39) damage of DNA after radiation therapy, presumably by their ability to prevent DNA strand break rejoining and by affecting several DNA damage signaling pathways.
PARP inhibitors have also been shown to be useful for treating acute and chronic myocardial diseases (see Pharmacological Research (2005) 52:34-43). For instance, it has been demonstrated that single injections of PARP inhibitors have reduced the infarct size caused by ischemia and reperfusion of the heart or skeletal muscle in rabbits. In these studies, a single injection of 3-amino-benzamide (10 mg/kg), either one minute before occlusion or one minute before reperfusion, caused similar reductions in infarct size in the heart (32-42%) while 1,5-dihydroxyisoquinoline (1 mg/kg), another PARP inhibitor, reduced infarct size by a comparable degree (38-48%). These results make it reasonable to assume that PARP inhibitors could salvage previously ischemic heart or reperfusion injury of skeletal muscle tissue {PNAS (1997) 94:679-683). Similar findings have also been reported in pigs {Eur. J. Pharmacol. (1998) 359:143-150 and Ann. Thorαc. Surg. (2002) 73:575-581) and in dogs (Shock. (2004) 21:426-32). PARP inhibitors have been demonstrated as being useful for treating certain vascular diseases, septic shock, ischemic injury and neurotoxicity {Biochim. Biophys. Actα (1989) 1014:1-7; J Clin. Invest. (1997) 100: 723-735). Oxygen radical DNA damage that leads to strand breaks in DNA, which are subsequently recognized by PARP, is a major contributing factor to such disease states as shown by PARP inhibitor studies (J Neurosci. Res. (1994) 39:38-46 and PNAS (1996) 93:4688-4692). PARP has also been demonstrated to play a role in the
pathogenesis of hemorrhagic shock {PNAS (2000) 97:10203-10208).
PARP inhibitors have been demonstrated as being useful for treatment of inflammation diseases (see Pharmacological Research (2005) 52:72-82 and 83-92).
It has also been demonstrated that efficient retroviral infection of mammalian cells is blocked by the inhibition of PARP activity. Such inhibition of recombinant retroviral vector infections has been shown to occur in various different cell types (J Virology, (1996)
70(6): 3992-4000). Inhibitors of PARP have thus been developed for use in anti- viral therapies and in cancer treatment (WO 91/18591).
In vitro and in vivo experiments have demonstrated that PARP inhibitors can be used for the treatment or prevention of autoimmune diseases such as Type I diabetes and diabetic complications {Pharmacological Research (2005) 52:60-71).
PARP inhibition has been speculated as delaying the onset of aging characteristics in human fibroblasts {Biochem. Biophys. Res. Comm. (1994) 201(2):665-672 and Pharmacological Research (2005) 52:93-99). This may be related to the role that PARP plays in controlling telomere function (Nature Gen., (1999) 23(l):76-80).
The vast majority of PARP inhibitors to date interact with the nicotinamide binding domain of the enzyme and behave as competitive inhibitors with respect to NAD+(Expert Opin. Ther. Patents (2004) 14:1531-1551). Structural analogues of nicotinamide, such as benzamide and derivatives were among the first compounds to be investigated as PARP inhibitors.
However, these molecules have a weak inhibitory activity and possess other effects unrelated to PARP inhibition. Thus, there is a need to provide potent inhibitors of the PARP enzyme.
Structurally related PARP inhibitors have previously been described. WO 1999/59973 discloses amide substituted benzene rings fused to 5 membered heteroaromatic rings;
WO2001/85687 discloses amide substituted indoles; WO 1997/04771, WO 2000/26192, WO 2000/32579, WO 2000/64878, WO 2000/68206, WO 2001/21615, WO 2002/068407, WO 2003/106430 and WO 2004/096793 disclose amide substituted benzo imidazoles; WO
2000/29384 discloses amide substituted benzoimidazoles and indoles; and EP 0879820 discloses amide substituted benzoxazoles.
It has now surprisingly been discovered that amide substituted indazoles of the present invention exhibit particularly high levels of inibition of the activity of poly(ADP-ribose)polymerase (PARP). Thus the compounds of the present invention are particularly useful as inhibitors of PARP-I and/or PARP-2. They also show particularly good levels of cellular activity, demonstrating good anti-proliferative effects in BRCAl and BRCA2 deficient cell lines.
The present invention provides compounds of formula I:
Scheme 1
A procedure to synthesize derivatives of those compounds of this invention is shown in scheme 1, whereby the substituted 2H-indazoles are prepared using a synthetic route similar to that described in WO 2005/066136. Following initial conversion of the 2-nitro-3-methyl-benzoic acid derivative into the corresponding ester, radical bromination of the methyl group using reagents like N-bromosuccinimide and benzoyl peroxide yields the key benzyl bromide derivative. Oxidation of this benzylic bromide to the corresponding benzaldehyde can be accomplished for instance using 7V-methylmorpholine-7V-oxide and molecular sieves. Following the condensation of the aldehyde with an amine, ring closure can be accomplished by treating the key intermediate with sodium azide at elevated temperature to introduce the final nitrogen atom and the resultant extrusion of nitrogen to furnish the indazole ring. A base such as lutidine can also be added to this reaction. Final conversion of the ester to the primary amide yields the desired derivatives. This can be accomplished either by heating the ester in an ammonia solution or by conversion to the corresponding carboxylic acid and then amide coupling.
Rx = C1-6alkyl
Oxidation
e.g. NMMO, mol sieves
NH3, THF or MeOH,
700C sealed tube, or
NaOH or KOH, NH3, HATU
or TBTU, DIPEA, DMF, RT
Scheme 1
Scheme 2
A variation of schemes 1 is shown below in scheme 2 and allows the introduction of substituents onto the indazole cores. When the required nitrobenzoic acid derivatives are not commercial available they can be prepared through nitration of the corresponding benzoic acid derivatives, for instance using potassium nitrate in concentrated sulphuric acid. Synthetic manipulations as decribed above allow the formation of the corresponding aniline which can either be cyclised to the indazole by firstly acetylation of the indazole and cyclisation with sodium nitrite in concentrated HCl acid at O0C. Alternatively, the aniline can be diazonitised with nitrosium tetrafluoroborate and the corresponding diazonium tetrafluoroborate salt decomposed at elevated temperatures to the corresponding dilfluorobenzene derivative by a Schiemann reaction
(Caution). Following the synthetic sequence as described in scheme 1 allows oxidation of the benzylic methyl group to the corresponding aldehyde and elaboration of the desired indazole derivatives by coupling with a (hetero)anilide and cyclisation with sodium azide.
Nitration Esterifi cation
KNO3, cone. e.g. AcCI, MeOH,
Reduction
H2, Pd/C
Scheme 2 Scheme 3
An alternative procedure involves functionalisation of the indazole at a late stage as shown in scheme 3. Here the indazole ester is first converted to the corresponding carboxamide and the subjected to nucleophilic aromatic substitution of the appropriate fluoro(hetero)aromatic bromide. This allows the preparation of a bromide derivative that can be cross coupled under Suzuki coupling conditions, for instance using tri(tert-butyl)phosphine and Pd2(dba)3 as catalysts in the presence of a base, such as sodium carbonate. Conversion to the desied piperidine moiety is then accomplished by a Fowler reaction using an acyl chloride, such as CBz-Cl and a reducing agent such as NaBH4. Final hydrogenation reaction can yield the corresponding piperidine derivatives.
A medicinal product authorization application requires comprehensive information on origin and quality of an active substance. What information is required was defined in two Guidelines so far: the Guideline “Chemistry of Active Substances” (3AQ5a) from 1987 and the “Guideline on the Chemistry of New Active Substances” from 2004. Because both Guidelines’ content do not take into account the ICH Guidelines Q8-11 issued in the meantime and do thus not meet the current state of the art in sciences and in regulatory practice, the EMA Quality Working Party (QWP) developed an updated document entitled “Guideline on the chemistry of active substances” (EMA/454576/2016), which was issued…
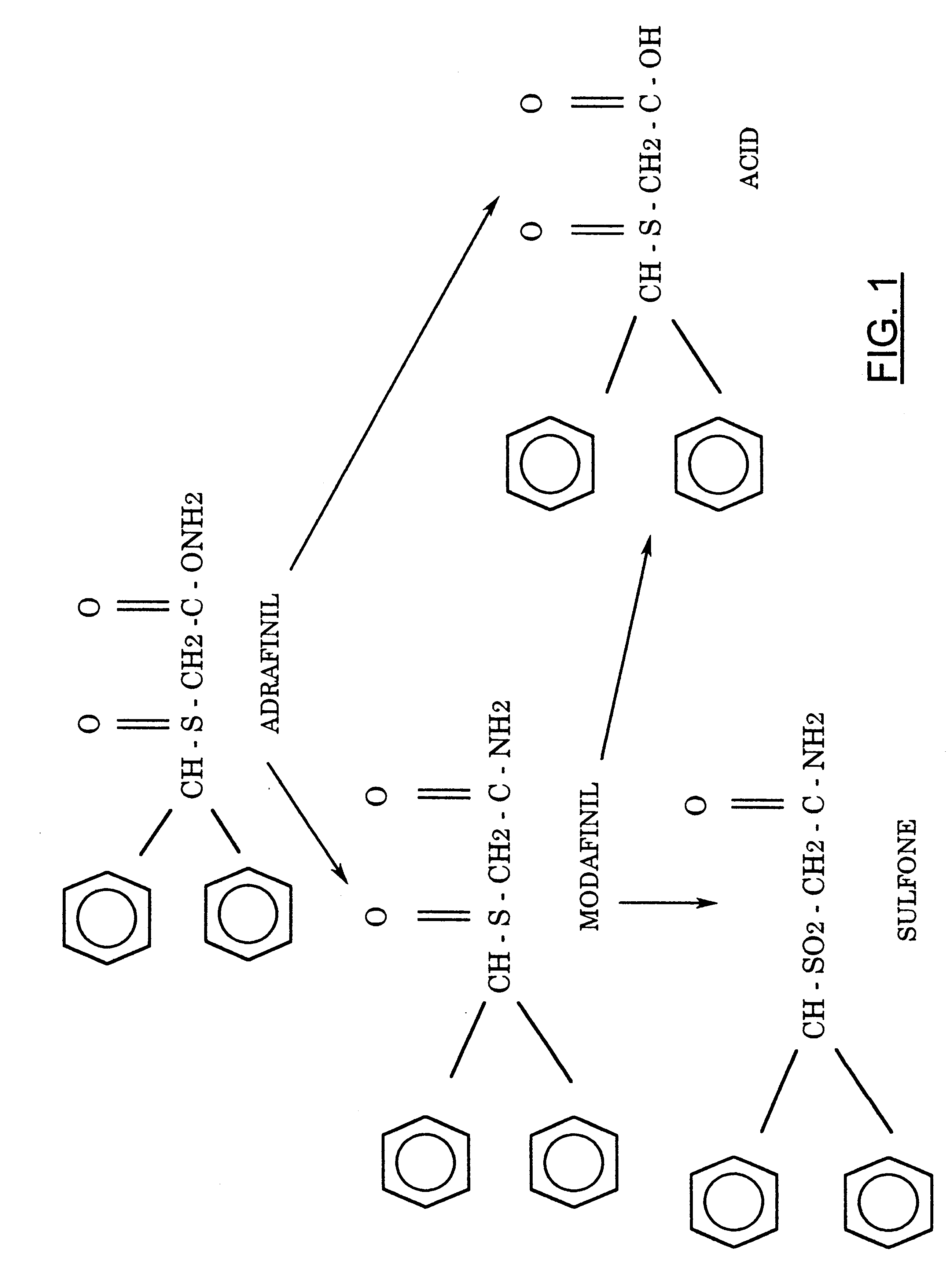
Although the mechanism of action of modafinil was initially unknown, it now appears that the drug acts as a selective, relatively weak, atypical dopamine reuptake inhibitor. However, it appears that other additional mechanisms may also be at play.
History
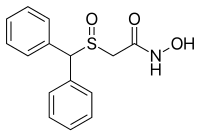
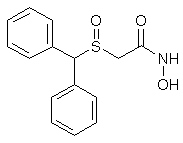
Modafinil was originally developed in France by neurophysiologist and emeritus experimental medicine professor Michel Jouvet and Lafon Laboratories. Modafinil originated with the late 1970s invention of a series of benzhydryl sulfinyl compounds, including adrafinil, which was first offered as an experimental treatment for narcolepsy in France in 1986. Modafinil is the primary metabolite of adrafinil, lacking the polar -OH group on its terminal amide,[77] and has similar activity to the parent drug but is much more widely used. It has been prescribed in France since 1994 under the name Modiodal, and in the US since 1998 as Provigil.
It was approved for use in the UK in December 2002. Modafinil is marketed in the US by Cephalon Inc., who originally leased the rights from Lafon, but eventually purchased the company in 2001.
Cephalon began to market the R-enantiomer armodafinil of modafinil in the U.S. in 2007. After protracted patent litigation and negotiations (see below), generic versions of modafinil became available in the U.S. in 2012.
That’s how it went…
2-benzhydryl-sulfanylacetamide.
Diphenylbromomethane (4,95g = 0.02 moles) and thiourea (1,52g=0.02moles) were refluxed in 20mls water for 30mins. As the synth from Rh’s says, a clear solution must have been formed in 5 mins, but in the end we still had a lot of oil at the bottom (the reasion to blame was old, semidecomposed diphenylbromomethane – when we opened the can, it emitted HBr). We were too lazy to separate the oil , so 2.5g (0.04moles) KOH in 15mls water was added straight and the reflux continued for 30 more mins. A disgusting stench filled the lab.
Thus obtained solution of potassium salt of diphenylmercaptane was cooled to 50-60 C and 1.9g (0.02moles) of chloroacetamide was added thereto. The mixtr was left to its own devices for 2hours – the precipitated oil crystallized. The xtals were filtered, washed thrice w/water, thrice w/ether (removing all benzhydrol). After drying there was obtained 1.9g (37%) of finely divided crystals with mp of 111 C.
With fresh diphenylbromomethane this will give not less than 80% – otherwise I’ll bee a reddish (this is an idiom which I am again unable to translate).
Modafinil
Into the solution of 3.6g benzhydrylsulfanylacetamide (0.014moles) in 15mls of GAA there was added 3mls (~0.03moles) 30% hydrogene peroxide. The mixture was left at RT (15 Ñ in our case, better not to heat above) for 20 hrs. Then into the solution there was added 30mls aqua, scratching the walls with a glass rod. After 1 hr the precipitate was filtered, washed w/water twice, then w/ether and dried. Yield – 2,3g (61%), mp – 158-159 C. After some time the mother liquor yielded some more product but we were too lazy to work it up.
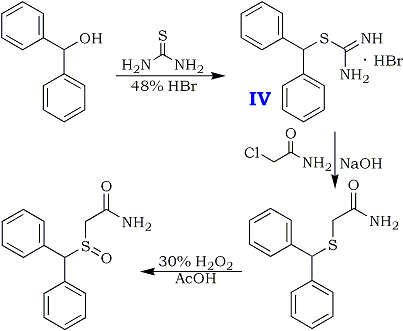
Diphenylmethanol (130 g, 0.7 mole) and thiourea (65 g, 0.85 mole) are added in 0.5 L reactor charging with water (325 ml). The mixture is heated to 95°C. (an emulsion is obtained) and 48% HBr (260 gr. 3.22 mole, 4.6 equivalents) is then added gradually during 0.5 hour. The mixture is heated under reflux {106-107°C) for 0.5 hour and cooled to 80-85°C. At this temperature, the mixture is seeded with several crystals of the product and the mixture is stirred at that temperature for 0.5 hour and then cooled to 25°C. The colorless crystals are collected by filtration, washed with water (200 ml) yielding about 240 gr. of wet crude isothiouronium salt.
Preparation of diphenylmethylthioacetamide.
A 2 L reactor was charged with diphenylmethylisothiouronium bromide crude wet obtained (240 gr.) and water {700 mL) under nitrogen. The suspension was heated to 60°C and 46% aqueous NaOH solution (98 ml, 1.68 mole, 2.4 eq.) was added. The reaction mixture was heated to 85°C and stirred until all the solid was dissolved. Then, it was cooled to 60°C and chloroacetamide (80 g, O.84 mole, 1.2 eq.) was added in five portions hour at 60-70°C during one hour. The suspension is stirred at 70°C for 4-5 hours. The mixture was filtered while warm and the cake was washed with hot water (250 ml). Diphenylmethylthioacetamide crude wet is obtained [220 gr., HPLC assay: 78%, HPLC purity: 95%, yield: 95% (from diphenylmethanol.)]
20 gr. of the product was recrystallized twice from ethyl acetate, dried in vacuo to give 15 gr. of pure title compound.
Preparation of Modafinil.
A 1.0 L reactor was charged with diphenylmethylthioacetamide crude wet (220 gr.) obtained above and glacial acetic acid (610 mL). The mixture was heated to 40°C and stirred until full dissolution is achieved. 5.8% H2O2 solution (500g, 1.2 eq) was added dropwise during 0.5 hours at 40-45°C. The reaction mixture was stirred at 40-45°C for 4 hours. Then sodium metabisulfite (18.3g) in 610 mL water was added in order to quench the unreacted H2O2 and the suspension was stirred for 0.5 hours. Then the reaction mixture was cooled to 15°C and filtered. The cake was washed with water (610 mL) and dried on air to obtain crude wet Modafinil (205 g). Reslurry in refluxing ethyl acetate, followed by recrystallization from methanol:water (4:1) solution afforded pure Modafinil [125 g, HPLC assay: 99.9%, HPLC purity: 99.9%, yield: 67% (from diphenylmethanol)].
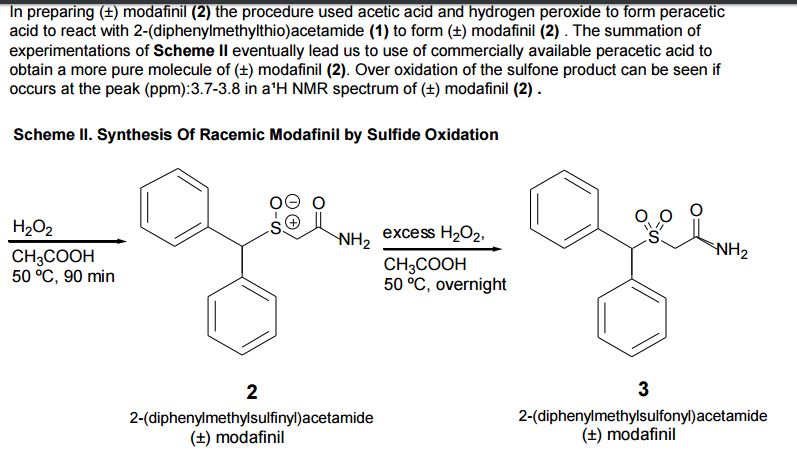
We report a facile procedure to synthesize racemic modafinil (diphenylmethylsulfinylacetamide), which is now being used in pharmacotherapy, and its achiral oxidized derivative (diphenylmethylsulfonyl acetamide). Modafinil is of interest more than for its potential anti-narcoleptic activity. It has also been reported to have neuroprotective properties and may potentially be effective in the enhancement of vigilance and cognitive performance. Finally, it may also protect from subclinical seizures that have been implicated as causative factors in autistic spectrum disorders and other neurodegenerative conditions. This agent can now be synthesized simply and in larger amounts than previously, making it more readily available for testing in various research modalities. The described procedure also lends itself to production of several other amides of potential interest. We are currently in the process of synthesizing and testing several new derivatives in this series. The anticonvulsant properties of modafinil and its sulfone derivative have not previously been extensively described in the literature. It may be of interest to note that the oxidized derivative of modafinil is also nontoxic and almost as effective as an anticonvulsant as the parent.
Experimental
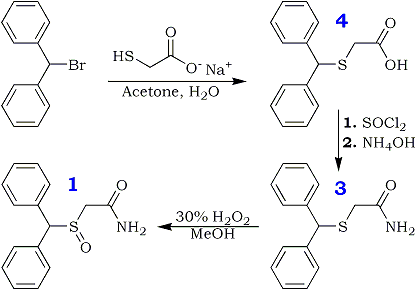
Diphenylmethylthioacetic Acid (3)
Benzhydryl bromide (14.78 gm, 0.059 mole) was dissolved in 75 ml of acetone in a 250-ml round-bottomed flask. To this solution was added dropwise sodium mercaptoacetate (6.59 g, 0.058 mole) in about 60 ml of H2O; the mixture was stirred under N2 for 2 h at room temperature and was thereafter warmed at about 60–70°C for 1 h. The reaction mixture was evaporated to dryness and taken up in CH2Cl2 and saturated aqueous NaHCO3. The organic extract was rejected, and the aqueous phase was treated with acid to pH 2 and chilled. Suction filtration gave the 6.9 g of the acid (3, 46%), mp 125°C. Rf 0.2. Recrystallization from MeOH/H2O gave mp 126–128°C.
Diphenylmethylthioacetamide (4)
Diphenylmethylthioacetic acid (19.5 g, 0.076 mole)
in 114 ml of dry benzene was taken in a 250-ml roundbottomed
flask attached to a reflux condenser, under N2 gas. To this was added thionyl chloride (19.5 ml, 0.097 mole) with a dropping funnel. The mixture was stirred at room temperature with a magnetic stirrer and refluxed for 1 h. Thereafter, the mixture was evaporated under low pressure to give a yellow oil that was taken up in about 100 ml of CH2Cl2 and filtered to yield a clear orange solution. This was chilled in ice water and added slowly to an ice-cold solution of concentrated NH4OH in H2O (40:40 ml). The ensuing mixture was stirred for 1 h and shaken well in a separatory funnel. The organic layer was dried (Na2SO4) and evaporated to dryness to give 14.39 g (54%) of the amide (4), mp 108–109°C (lit2 110°C). Rf 0.8. Recrystallization from CH3OH/H2O gave mp 109–110°C.
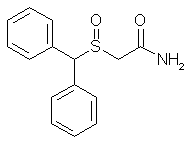
Diphenylmethylsulfinylacetamide (modafinil, 1)
Diphenylthioacetamide (3.46 g, 0.013 mole) was taken in glacial acetic acid (14 ml) with stirring; to this was added 1.34 ml of 30% H2O2 with chilling in ice water. The mixture was left in the refrigerator for 4 h and thereafter worked up by treating it with 70 ml of ice-cold water. The precipitated material was filtered under suction and washed with ice-cold water to give 1.5 g of white crystals (43%), mp 159–160°C. Rf 0.6. Recrystallization from hot MeOH gave mp 161–162°C
Diphenylmethylsufonylacetamide (2)
Diphenylmethylthioacetamide (2.5 g, 0.009 mole) (reg. No. 118779-53-6) was dissolved in about 12 ml of glacial acetic acid and 3 ml of 30% H2O2 and set aside overnight (16 h or more). The next day, the mixture was diluted with 100 ml of H2O and set aside to cool in the refrigerator. Upon filtration and drying, 2.1 g (80%) of 2 was obtained as a white powder. Rf 0.89. The melting point of sample after recrystallization from absolute EtOH was 195–197°C.
One aspect of our preparation of modafinil needs further mention. When diphenylmethylthioacetamide (4) is being oxidized by H2O2, care must be taken to keep the reaction mixture cool, and workup should be done in a timely manner. Allowing the reaction to go to 24 h or longer at room temperature results in the formation of the sulfone (2). The paper by Mu et al. (3) does not discuss this possibility. In our hands, the procedure stated therein led to the higher melting sulfone and not the modafinil. Our NMR data for the newly prepared modafinil preparation are in consonance with the data of the patented commercial product. It should be noted that the methylene protons in modafinil are geminally
coupled and appear as a pair of doublets. This is due to the fact that the adjacent sulfoxide moiety is chiral, and therefore the methylene protons adjacent to it wind up being diastereotopic with different chemical shifts and coupling. In the sulfone 2, the methylene protons appear as a singlet due to the fact that the adjacent sulfone moiety is achiral, thus making the two protons equivalent. Modafinil 1 is, however, an equal mixture of enantiomers, as in the reported patent and publication (2,3).
RESULTS
The chemical pathway leading to modafinil may be
represented in Scheme 1.
see pdf for further information and references,
CLIP
Synthesis and determination of the absolute configuration of the enantiomers of modafinil
Thomas Prisinzanoa, John Podobinskia, Kevin Tidgewella, Min Luoa and Dale Swensonb Tetrahedron: Asymmetry 15(6), 1053-1058 (2004) (../rhodium/pdf /modafinil.enantiomers.pdf)
DOI:10.1016/j.tetasy.2004.01.039
a Division of Medicinal & Natural Products Chemistry, College of Pharmacy, The University of Iowa, Iowa City, Iowa 52242-1112, USA b Department of Chemistry, The University of Iowa, Iowa City, Iowa 52242, USA
Abstract
The asymmetric synthesis of both enantiomers of modafinil, a unique CNS stimulant with a reduced abuse liability, is described. This approach effectively prepares modafinil on a multigram scale in several steps from benzhydrol. The described synthetic route has also been used to produce the more water soluble analogue, adrafinil. X-ray crystallographic analysis on (-)-(diphenylmethanesulfinyl)acetic acid has determined the absolute configuration to be R.
[alpha]D22 + 40.2 (c=1.11, MeOH)
Source of chirality: resolution via diastereomeric salt formation with (R)-(+)-alpha-methylbenzylamine
Absolute configuration: S CLIP
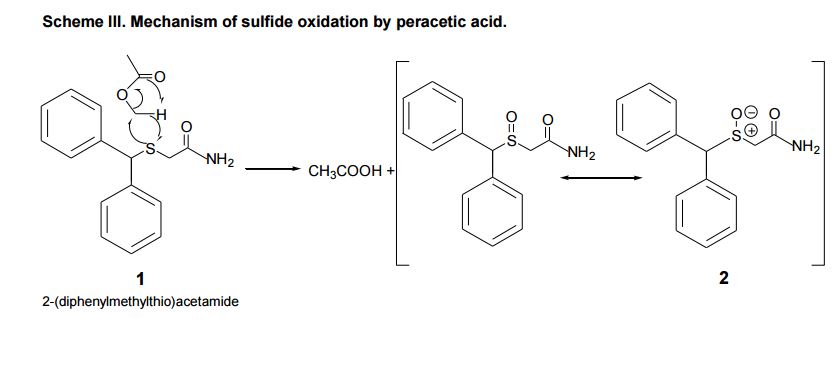
Narcolepsy is a debilitating neurological disorder which is characterized by chronic sleepiness and is marked to be disorganization of sleep and wake patterns. Every six out of ten thousand people in Western Europe and North America are affected by this disorder. Modafinil (Provigil®) is approved by the Food and Drug Administration for the treatment of narcolepsy. It is commonly used in opposition to Ritalin®, however Ritalin® has an associated dependency issue. Modafinil, a central nervous system stimulant, has reported to have little abuse potential. Modafinil has the ability to act like wake-promoting sympathomimetic agents which includes amphetamine. At relevant pharmacological concentrations modafinil lacks binding ability to receptors for sleep/wake regulation, which includes the ones used for norepinephrine and serotonin. The precise mechanism of action of modafinil is unknown and is presently being researched. Modafinil contains a chiral sulfoxide moiety but is prescribed as a racemate. In collaboration with faculty from the Psychology department at Western Michigan University we were to synthesize modafinil for behavioral studies with animals. Therefore a large scale of pure modafinil was synthesized.
The tetrahedral sulfur atom acts as a chiral center (being surrounded by two dissimilar carbon atoms, an oxygen atom and an electron lone pair (Figure 1). Unlike most analogous trisubstituted amines that undergo umbrella-like inversion at the nitrogen atom, sulfoxides are configurationally stable.
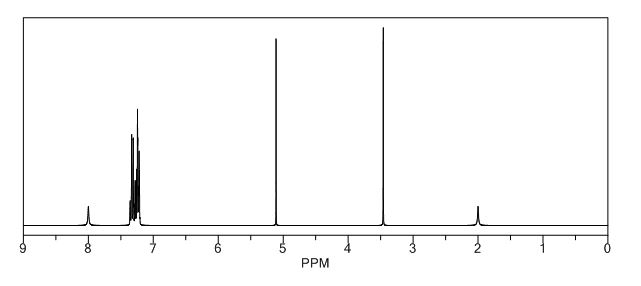
The initial target of this synthesis was to prepare the 2-(diphenylmethylthio)acetamide (1) (Scheme I). The reaction of benzyhydral chloride and thiourea are reacted with potassium iodide, water, heat, 30% sodium hydroxide, 2-chloroacetamide and triethylamine. The procedure required the 2-(diphenylmethylthio) acetamide (1) to be recrystallized to remove any impurities with methanol:water solution 60:40 . After recrystallization (Figure 2) the ¹H NMR spectrum of the synthesized 2-(diphenylmethylthio)acetamide (1) provides evidence that the recrystallization did not purify the compound. In addition recrystallization significantly reduced the percent yield from 78.3-79.2% to 56%. If the compound were pure it would only show peaks at the following locations (ppm): 3.05 (s, 2H), 5.18 (s, 1H), 6.53 (s, 1H), 7.21-7.44(m, 10H).
To produce pure 2-(diphenylmethylthio)acetamide (1) elimination of the recrystallization step and 2-(diphenylmethylthio)acetamide (1) was then purified via column chromatography using dichloromethane:ether 80:20 as an eluent with the stationary phase (silica gel). After testing several of the fractions from the column using thin layer chromatography the fractions where able to be identified that contained 2- (diphenylmethylthio)acetamide (1). Once 2-(diphenylmethylthio)acetamide (1) was isolated it was oxidized with peracetic acid. The oxidation process was extended to three hours due to lack of desired product (±) modafinil (Figure 1).
With the procedure we used and modified through experimentation a new procedure was developed that increased the percent yield from 56% to 78.3-79.2%. We encountered a few problems that lead to the removal of the recrystallization step and the use of column chromatography was performed to purify 2-(diphenylmethylthio)acetamide (1) . Over- oxidation could have occurred but would have showed up at 3.7-3.8 (ppm), this did not occur in our experiment. The peak at 1.5 (ppm) is a water peak that was not fully removed during the rotovep procedure. After a precise and confident procedure was perfected then we were able to upscale the reaction and sythesize12gs of pure (±) modafinil.
19.5g (0.076 mol) of benzhydrylthioaceticacid in 114 ml of benzene are placed in a three-necked flask provided with a condenser and a dropping funnel. The mixture is heated and 19 ml of thionyl chloride are added drop by drop. Once the addition is complete, the reflux is continued for about 1 hour, cooling and filtering are carried out and the benzene and the excess thionyl chloride and then evaporated. In this way, a clear orange oil is obtained.
Benzhydrylthioacetamide
35 ml of ammonia in 40 ml of water are introduced into a three-necked flask provided with a condenser and a dropping funnel and the benzhydrylthioacetyl chloride dissolved in about 100 ml of methylene chloride is added drop by drop. Once the addition is complete, the organic phase is washed with a dilute solution of soda and dried over Na2SO4, the solvent is evaporated and the residue is taken up in diisopropyl ether; in this way, the benzhydrylthioacetamide is crystallized. 16.8 g of product (yield 86%) are obtained. M.p. 110°C.
Modafinil (CRL 40,476)
14.39 g (0.056 mol) of benzhydrylthioacetamide are placed in a balloon flask and 60 ml of acetic acid and 5.6 ml of H2O2 (about 110 volumes, 33%) are added. The mixture is left in contact for one night at 40°C. and about 200 ml of water are then added; the CRL 40476 crystallizes. By recrystallization from methanol, 11.2 g of benzhydrylsulphinylacetamide are obtained. Yield: 73%. M.p. 164-66°C.
Novel Synthesis of Modafinil and its sulfone analog3
Our interest in synthesis of modified neuroactive compounds has led us to consider Modafinil (1), a stimulant and anti-narcoleptic agent that is finding increasing use in a number of neurological areas. The compound was originally prepared by a rather tedious route described in a procedure patented by L. Lafon2. More recently, its preparation has been reported by Mu et al.4 We believe that this compound has many interesting properties and possible alternative uses in addition to its recognized anti-narcoleptic actions.
Fig 1.
The chemical pathway leading to modafinil
Not having been able to obtain it from the patent holder, we proceeded to explore alternate synthetic pathways and settled on a convenient synthesis, which permitted us to produce this compound along with a primary derivative, the sulfone (2) in sufficient quantities for whole-animal studies. The current, more facile method starts with benzhydryl bromide and sodium thiolacetate in aqueous acetone, which reacts directly to form diphenylmethylthioacetic acid (3), possibly by an ionic mechanism. This resultant compound can be converted to its acid chloride that, in turn, may be used to acylate ammonia. The ensuing primary amide (4) may be gently oxidized by H2O2 to form the corresponding sulfoxide (Modafinil, 1) and, under more vigorous conditions, the modafinil sulfone (2), whose anticonvulsant and biological properties have not been described extensively in the literature. Additionally, this procedure is also uniquely suitable for large-scale preparation of Modafinil and its congeners.
One aspect of our preparation of modafinil needs further mention. When diphenylmethylthioacetamide (4) is being oxidized by H2O2, care must be taken to keep the reaction mixture cool, and workup should be done in a timely manner. Allowing the reaction to go to 24 h or longer at room temperature results in the formation of the sulfone (2). The paper by Mu et al.4 does not discuss this possibility. In our hands, the procedure stated therein led to the higher melting sulfone and not the modafinil. Our NMR data for the newly prepared modafinil preparation are in consonance with the data of the patented commercial product. It should be noted that the methylene protons in modafinil are geminally coupled and appear as a pair of doublets. This is due to the fact that the adjacent sulfoxide moiety is chiral, and therefore the methylene protons adjacent to it wind up being diastereotopic with different chemical shifts and coupling. In the sulfone 2, the methylene protons appear as a singlet due to the fact that the adjacent sulfone moiety is achiral, thus making the two protons equivalent. Modafinil 1 is, however, an equal mixture of enantiomers, as in the reported patent and publication2,4.
Experimental
The new compounds were prepared according to modified procedures published in the patent literature. Starting materials and solvents were obtained commercially from Fluka and/or Aldrich Chemical Corp. Thin layer chromatography (TLC) was performed on silica gel plates. Solvent system was EtOAc:MeOH:NH4OH, 100:10:3 by volume. Melting points are uncorrected.
Diphenylmethylthioacetic Acid (3)
Benzhydryl bromide (14.78 gm, 0.059 mole) was dissolved in 75 ml of acetone in a 250-ml round-bottomed flask. To this solution was added dropwise sodium mercaptoacetate (6.59 g, 0.058 mole) in about 60 ml of H2O; the mixture was stirred under N2 for 2 h at room temperature and was thereafter warmed at about 60–70°C for 1 h. The reaction mixture was evaporated to dryness and taken up in CH2Cl2 and saturated aqueous NaHCO3. The organic extract was rejected, and the aqueous phase was treated with acid to pH 2 and chilled. Suction filtration gave the 6.9 g of the acid (3, 46%), mp 125°C. Rf 0.2. Recrystallization from MeOH/H2O gave mp 126–128°C.
Diphenylmethylthioacetamide (4)
Diphenylmethylthioacetic acid 3 (19.5 g, 0.076 mole) in 114 ml of dry benzene was taken in a 250-ml roundbottomed flask attached to a reflux condenser, under N2 gas. To this was added thionyl chloride (19.5 ml, 0.097 mole) with a dropping funnel. The mixture was stirred at room temperature with a magnetic stirrer and refluxed for 1 h. Thereafter, the mixture was evaporated under low pressure to give a yellow oil that was taken up in about 100 ml of CH2Cl2 and filtered to yield a clear orange solution. This was chilled in ice water and added slowly to an ice-cold solution of concentrated NH4OH in H2O (40:40 ml). The ensuing mixture was stirred for 1 h and shaken well in a separatory funnel. The organic layer was dried (Na2SO4) and evaporated to dryness to give 14.39 g (54%) of the amide (4), mp 108–109°C (lit4 110°C). Rf 0.8. Recrystallization from CH3OH/H2O gave mp 109–110°C.
Diphenylmethylsulfinylacetamide (Modafinil, 1)
Diphenylmethylthioacetamide 4 (3.46 g, 0.013 mole) was taken in glacial acetic acid (14 ml) with stirring; to this was added 1.34 ml of 30% H2O2 with chilling in ice water. The mixture was left in the refrigerator for 4 h and thereafter worked up by treating it with 70 ml of ice-cold water. The precipitated material was filtered under suction and washed with ice-cold water to give 1.5 g of white crystals (43%), mp 159–160°C. Rf 0.6. Recrystallization from hot MeOH gave mp 161–162°C
Diphenylmethylsulfonylacetamide (2)
Diphenylmethylthioacetamide (2.5 g, 0.009 mole) (CAS No. 118779-53-6) was dissolved in about 12 ml of glacial acetic acid and 3 ml of 30% H2O2 and set aside overnight (16 h or more). The next day, the mixture was diluted with 100 ml of H2O and set aside to cool in the refrigerator. Upon filtration and drying, 2.1 g (80%) of 2 was obtained as a white powder. Rf 0.89. The melting point of sample after recrystallization from absolute EtOH was 195–197°C.
High-yield Synthesis of Modafinil from Benzhydrol5
A recent patent5 describes a very easy two-step route to the Modafinil precursor diphenylmethanethioacetamide from benzhydrol (diphenylmethanol) in 90% yield and with 95% purity. A 200g batch is made in a 2000 mL vessel using water as reaction medium and ethyl acetate for recrystallization of the product.
Diphenylmethylbromide is prepared in situ from benzhydrol and react it with thiourea in a one-pot reaction to form the corresponding isothiouronium salt. The crude salt is then reacted with chloroacetamide (by generating the thiolate cation in situ), and after filtration and washing, diphenylmethylthioacetamide is isolated in excellent yield and good purity. After oxidation of the thioacetamide with hydrogen peroxide, followed by recrystallization, the overall yield of Modafinil is 67% from the benzhydrol.
(Chimimanie’s Voice:) The synthesis works just as great without the nitrogen inert atmosphere (most patents do not use it at all), step two is only a hydrolysis of the thiouronium salt to the thiolate. You just have to put the salt, NaOH and heat till you got a homogenous solution, with no more solid material floating around. The following chloroacetamide SN2 reaction is a breeze too. Sometime a blue solution can bee obtained, it is nothing to worry about. In the final step, you just have to filter off the solid which did not dissolve when the crude thioacetamide is put in the GAA/H2O2, bee4 crashing the soluble one with water.
Do not forget to slurry the modafinil in EtOAc and then recrystallize it from aqueous MeOH, as the crystalline shape of modafinil is important for the kinetic and quality of effects, at least according to the patents EP0966962 and US2002043207.
Experimental
Preparation of isothiouronium Salt (IV)
Diphenylmethanol (130 g, 0.7 mole) and thiourea (65 g, 0.85 mole) are added in 0.5 L reactor charging with water (325 ml). The mixture is heated to 95°C. (an emulsion is obtained) and 48% HBr (260 gr. 3.22 mole, 4.6 equivalents) is then added gradually during 0.5 hour. The mixture is heated under reflux (106-107°C) for 0.5 hour and cooled to 80-85°C. At this temperature, the mixture is seeded with several crystals of the product and the mixture is stirred at that temperature for 0.5 hour and then cooled to 25°C. The colorless crystals are collected by filtration, washed with water (200 ml) yielding about 240 gr. of wet crude isothiouronium salt.
(Antoncho’s Voice:) Assholium successfully made Modafinil by this method, but there turned out to be a mistake in the original patent text – In the preparation of IV, the quantity of HBr stated here is excessive and leads to complete hydrolysis of the initially formed isothiouronium salt. The acid should bee added until the reaction mixture turns completely clear (about half as much as the patent says) – a sort of titration. Further addition will result in precipitation of heavy stinky oil, benzhydrylmethanethiol.
Preparation of diphenylmethylthioacetamide
A 2 L reactor was charged with diphenylmethylisothiouronium bromide crude wet obtained (240 gr.) and water (700 mL) under nitrogen. The suspension was heated to 60°C and 46% aqueous NaOH solution (98 ml, 1.68 mole, 2.4 eq.) was added. The reaction mixture was heated to 85°C and stirred until all the solid was dissolved. Then, it was cooled to 60°C and chloroacetamide (80 g, 0.84 mole, 1.2 eq.) was added in five portions hour at 60-70°C during one hour. The suspension is stirred at 70°C for 4-5 hours. The mixture was filtered while warm and the cake was washed with hot water (250 ml). Diphenylmethylthioacetamide crude wet is obtained [220 gr., HPLC assay: 78%, HPLC purity: 95%, yield: 95% from diphenylmethanol]. 20g of the product was recrystallized twice from ethyl acetate, dried in vacuo to give 15g of pure title compound.
Preparation of Modafinil
A 1.0 L reactor was charged with diphenylmethylthioacetamide crude wet (220 gr.) obtained above and glacial acetic acid (610 mL). The mixture was heated to 40°C and stirred until full dissolution is achieved. 5.8% H2O2 solution (500g, 1.2 eq) was added dropwise during 0.5 hours at 40-45°C. The reaction mixture was stirred at 40-45°C for 4 hours. Then sodium metabisulfite (18.3g) in 610 mL water was added in order to quench the unreacted H2O2 and the suspension was stirred for 0.5 hours. Then the reaction mixture was cooled to 15°C and filtered. The cake was washed with water (610 mL) and dried on air to obtain crude wet Modafinil (205 g). Reslurry in refluxing ethyl acetate, followed by recrystallization from methanol:water (4:1) solution afforded pure Modafinil [125 g, HPLC assay: 99.9%, HPLC purity: 99.9%, yield: 67% (from diphenylmethanol)].
Nithiananda Chatterjie, James P. Stables, Hsin Wang, and George J. Alexander, Anti-Narcoleptic Agent Modafinil and Its Sulfone: A Novel Facile Synthesis and Potential Anti-Epileptic Activity, Neurochemical Research, 29(8), 1481–1486 (2004)
Mu, B., Lei, G., He, X., and Du, X., Synthesis of central stimulant modafinil.Zhongguo Yaowu Huaxue Zazhi, 9(2), 132–134 (1999)
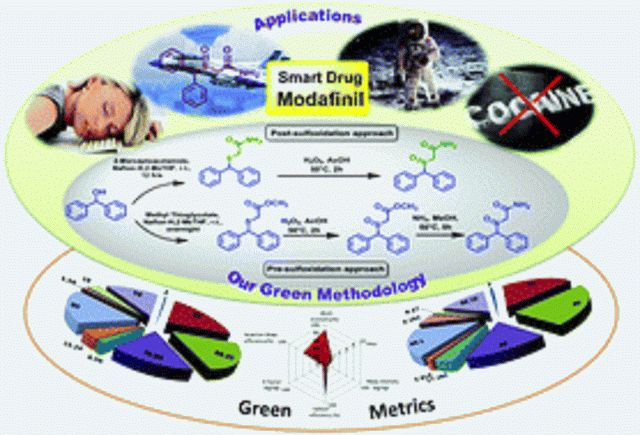
Green Chem., 2017, Advance Article DOI: 10.1039/C6GC02623K, Communication
Shivam Maurya, Dhiraj Yadav, Kemant Pratap, Atul Kumar
We developed a post-sulfoxidation protocol for the synthesis of Modafinil that exhibits improved sustainability credentials, utilizing the recyclable heterogeneous catalyst Nafion-H.
Efficient atom and step economic (EASE) synthesis of the “smart drug” Modafinil
aMedicinal & Process Chemistry Division, CSIR-Central Drug Research Institute, Sector 10, Jankipuram Extension, Sitapur Road, P.O. Box 173, Lucknow 226031, India E-mail: dratulsax@gmail.com, atul_kumar@cdri.res.in
bAcademy of Scientific and Innovative Research, New Delhi 110001, India
Modafinil (2-[(diphenylmethyl)sulfinyl]acetamide, MOD) is a key psychostimulant drug used for the treatment of narcolepsy and other sleep disorders that has a very low addiction liability. Recently, MOD has been clinically investigated for the treatment of cocaine addiction and used by astronauts in long-term space missions. We have developed a synthetic strategy for “smart drug” Modafinil. An efficient atom and step economic (EASE) synthesis has been carried out by the direct reaction of benzhydrol and 2-mercaptoacetamide using the recyclable heterogeneous catalyst Nafion-H along with post-sulfoxidation. This protocol exhibits improved sustainability credentials. We have also developed a superior pre-sulfoxidation approach for the synthesis of Modafinil.
Modafinil Physical State – White solid; M.p. 158-159ºC,
Green Chem., 2017, Advance Article DOI: 10.1039/C6GC02623K, Communication
Shivam Maurya, Dhiraj Yadav, Kemant Pratap, Atul Kumar
We developed a post-sulfoxidation protocol for the synthesis of Modafinil that exhibits improved sustainability credentials, utilizing the recyclable heterogeneous catalyst Nafion-H.
Efficient atom and step economic (EASE) synthesis of the “smart drug” Modafinil
aMedicinal & Process Chemistry Division, CSIR-Central Drug Research Institute, Sector 10, Jankipuram Extension, Sitapur Road, P.O. Box 173, Lucknow 226031, India E-mail: dratulsax@gmail.com, atul_kumar@cdri.res.in
bAcademy of Scientific and Innovative Research, New Delhi 110001, India
The ICH Q11 Guideline describing approaches to developing and understanding the manufacturing process of drug substances was finalised in May 2012. Since then the pharmaceutical industry and the drug substance manufacturers had time to get familiar with the principles outlined in this guideline. However, experience has shown that there is some need for clarification. Thus the Q11 Implementation Working Group recently issued a Questions and Answers Document.
The ICH Q11 Guideline describes approaches to developing and understanding the manufacturing process of drug substances. It was finalised in May 2012 and since then the pharmaceutical industry and the drug substance manufacturers had time to get familiar with the principles outlined in this guideline. However, experiences during implementation of these principles within this 4 years period have shown that there is need for clarification in particular with regard to the selection and justification of starting materials.
On 30 November 2016 the ICH published a Questions and Answers…
UNII F38R0JR742
CAS number 82186-77-4
Weight Average: 528.94
Monoisotopic: 527.154947772
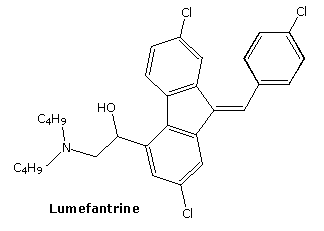
Chemical Formula C30H32Cl3NO
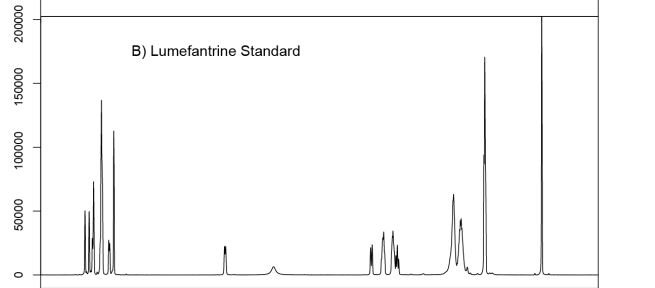
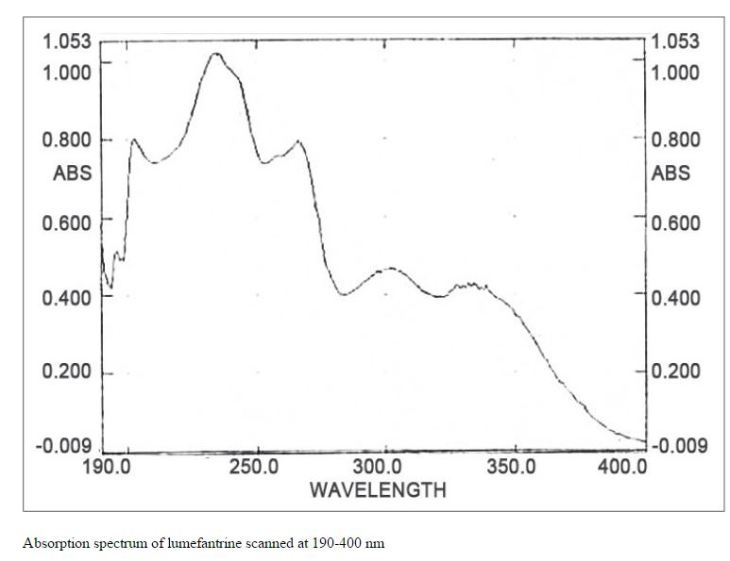
Lumefantrine (or benflumetol) is an antimalarial drug. It is only used in combination with artemether. The term “co-artemether” is sometimes used to describe this combination.[1] Lumefantrine has a much longer half-life compared to artemether and so is therefore thought to clear any residual parasites that remain after combination treatment.[2]
Lumefantrine, along with pyronaridine and naphtoquine, were synthesized in course of the Project 523 antimalaria drug research initiated in 1967; these compounds are all used in combination antimalaria therapies.[3][4][5]
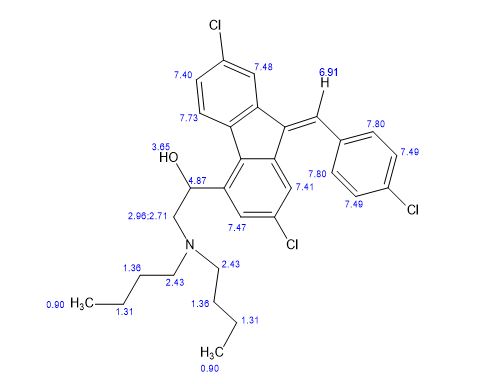
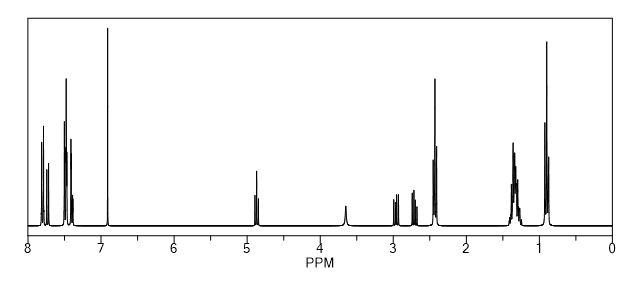
Lumefantrine is an antimalarial drug chemically known as 2-(dibutylamino)-1-[(9Z)-2, 7-dichloro-9-(4- chlorobenzylidene)-9H-floren-4-yl] ethanol, which is used in the prevention and treatment of Malaria in worm blooded animals. Lumefantrine is using the combination of β-Artemether in the treatment of Malaria
SYN
Synthetic Reference
Beutler, Ulrich; Fuenfschilling, Peter C.; Steinkemper, Andreas. An Improved Manufacturing Process for the Antimalaria Drug Coartem. Part II. Organic Process Research & Development. Volume 11. Issue 3. Pages 341-345. 2007.
SYN 2
Synthetic Reference
Rao, Dharmaraj Ramachandra; Kankan, Rajendra Narayanrao; Phull, Manjinder Singh. Process for preparation of lumefantrine as antimalarial agent with improved method. Assignee Cipla Co., Ltd., India. CN 1865227. (2006).
SYN 3
Synthetic Reference
Sethi, Madhuresh Kumar; Gonuguntla, Anantavena Rani; Arikatla, Siva Lakshmi Devi; Mulukutla, Suryanarayana; Yerramalla, Rajakrishna; Bontalakoti, Jaganmohanarao; Vemula, Lakshminarayana; Thirunavukarasu, Jayaprakash. Synthesis and characterization of novel related substances of Lumefantrine, an anti-malarial drug. Pharma Chemica. Volume 8. Issue 3. Pages 91-100. 2016.
SYN4
Synthetic Reference
Mathur, Prafull; Mathur, Suvigya; Vishwanath, Kannan; Mishra, Anand Kumar. Preparation of lumefantrine. Assignee Aanjaneya Lifecare Limited, India. IN 2013MU00611. (2015).
SYN 5
Synthetic Reference
Wu, Guang-liang; Dai, Ying-jie; Kang, Cong-min; Zi, Yan. A new synthetic technology of anti-malarial drug lumefantrine. Zhongguo Xinyao Zazhi. Volume 21. Issue 24. Pages 2944-2947. 2012.
SYN 6
Synthetic Reference
Krishna, Bettadapura Gundappa; Verma, Sudhakar; Krishna, Sujatha; Naik, Gajanan; Arulmoli, Thangavel. A process for preparation of lumefantrine. Assignee SeQuent Scientific Limited, India. IN 2012CH00470. (2012).
SYN 7
Synthetic Reference
Bansi, Lal; Genbhau, Gund Vitthal; Prabhakar, Bapat Chintamani; Popat, Bochiya Pravin; Banshi, Punde Dnyanadeo; Venkata, Reddy Prabhakar Gorla. Improved one pot process for the synthesis of lumefantrine. Assignee Calyx Chemicals and Pharmaceuticals Ltd., India. IN 2009MU01437. (2010).
SYN 8
Synthetic Reference
Shailesh, Singh; Dhaval, Vashi; Vinod, Gaikwad; Sanjay, Chowkekar; Sanjay, Bute. Process for the preparation of lumefantrine. Assignee Ajanta Pharma Ltd., India. IN 2008MU01677. (2010).
SYN 9
Synthetic Reference
Rawalnath, Sakhardande Rajiv; Kanji, Khatri Navin; Nilkanth, Firake Pandharinath; Vasant, Panchal Rajesh; Nagesh, Babrekar Chandan; Madhukar, Mohite Dhanaji. Preparation of lumefantrine. Assignee Saxena, Alok, India. IN 2006MU00260. (2007).
///////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
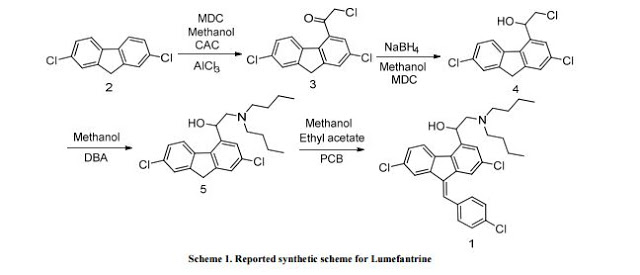
Embodiment 1:The synthesis of (the chloro- 2- of 2- (the chloro- 9H- fluorenes -4- bases of 2,7- bis-) ethyoxyl) trimethyl silane
2,7- dichloro fluorenes -4- oxirane (100g, 0.368mol), zinc oxide are sequentially added in 1000ml there-necked flasks (2.94g, 0.036mol) and 500ml dichloromethane, TMSCl (43g, 0.4mol) are added drop-wise in above-mentioned reaction solution, room temperature reaction 2h.Filtering, a small amount of dichloromethane washing of filter cake, filtrate washing, organic phase anhydrous sodium sulfate drying, filtering, filtrate are rotated to analysis After going out solid, stop revolving, stand and separate out a large amount of yellow solids, filtering, drain to obtain (the chloro- 2- of 2- (2,7- bis- chloro- 9H- fluorenes- 4- yls) ethyoxyl) trimethyl silane yellow solid 102g, yield 73%.δ(1HNMR,CDCl3):7.78-7.80(m,1H), 7.56(s,1H),7.48(s,1H),7.41(s,1H),7.32-7.35(m,1H),5.64-5.67(m,1H),4.03-4.11(m, 2H),3.84(m,2H),0.63(s,9H),ppm。
Embodiment 2:N- butyl-N- (1- (the chloro- 9H- fluorenes -4- bases of 2,7- bis-) -2- (trimethylsiloxy group) ethyl) butyl – The synthesis of 1- amine:
(the chloro- 2- of 2- (the chloro- 9H- fluorenes -4- bases of 2,7- bis-) ethyoxyl) trimethyl silicane is sequentially added in 1000ml there-necked flasks Alkane (100g, 0.26mol), di-n-butylamine (67g, 0.52mol), potassium carbonate (71.65g, 0.52mol) and 800ml acetonitriles.Put It is changed to nitrogen system, back flow reaction 16h.After being down to room temperature, filtering, a small amount of acetonitrile of filter cake washs, and filtrate is evaporated to obtain fourth containing N- The crude yellow oil of base-N- (1- (the chloro- 9H- fluorenes -4- bases of 2,7- bis-) -2- (trimethylsiloxy group) ethyl) butyl -1- amine. Crude product is directly used in be synthesized in next step, without purifying.MS:479(MH+)
Embodiment 3:The synthesis of 2- dibutyl aminos -2- [the chloro- 9H- fluorenes -4- bases of 2,7- bis-] ethanol
By-the N- of butyl containing N- obtained in the previous step (1- (the chloro- 9H- fluorenes -4- bases of 2,7- bis-) -2- (trimethylsiloxy group) second Base) crude yellow oil of butyl -1- amine is dissolved in 500ml tetrahydrofurans, concentrated hydrochloric acid (1ml), nitrogen protection, heating is added dropwise To 50 DEG C of reactions, TLC tracing detections to raw material have reacted completely, cool, and separate out solid, filtering, the washing of filter cake tetrahydrofuran, take out Dry, filtrate is abandoned, and filter cake is transferred in beaker, adds 200ml dichloromethane, and saturated aqueous sodium carbonate is adjusted pH=8, separated Machine phase, water layer are extracted once with 200ml dichloromethane again, merge organic phase, are washed, and are dried, are concentrated to give yellow oil 68g, two step yields 64%.δ(1HNMR,CDCl3):7.80(s,1H),7.56(s,1H),7.38(m,2H),7.25(s,1H), 4.7(brs,1H),4.03-4.11(m,2H),3.99(m,1H),3.56(m,2H),2.42-2.85(m,4H),1.44-1.48 (m,4H),1.23-1.28(m,4H),0.86-0.91(m,6H).ppm。
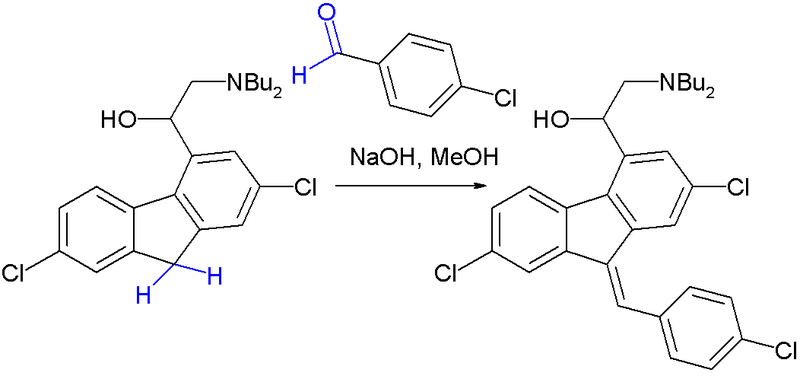
Embodiment 4:(RS, Z) -2- dibutyl aminos -2- [bis- chloro- 9- of 2,7- (4- chlorobenzenes methylene) -9H- fluorenes -4- bases] second The synthesis of alcohol
2- dibutyl aminos -2- [the chloro- 9H- fluorenes -4- bases of 2,7- bis-] ethanol (68g, 0.167mol) is dissolved in 200ml ethanol In, 4-chloro-benzaldehyde (28.2g, 0.2mol) and sodium hydroxide (2.68g, 0.067mol) are added, nitrogen protection, is heated to reflux anti- Answer 1h.Room temperature is down to, watery hydrochloric acid, which is added dropwise, makes product be filtered into salting out, and the washing of filter cake ethanol, drains to obtain yellow solid, shifts Into beaker, 200ml dichloromethane is added, adds saturated aqueous sodium carbonate to dissociate, be layered, aqueous phase uses 200ml dichloromethane again Extraction once, merges organic phase, washing, and anhydrous sodium sulfate drying 6h is filtered, and filtrate is concentrated to give (RS, Z) -2- dibutyl aminos -2- [2,7- bis- chloro- 9- (4- chlorobenzenes methylene) -9H- fluorenes -4- bases] ethanol be yellow foam shape solid 74g, yield 83%, 0-5 DEG C Preserve.δ(1HNMR,CDCl3):7.75(brs,1H),7.66(s,1H),7.55(s,1H),7.44-7.48(m,5H),7.28- 7.29(m,1H),7.30-7.31(m,1H),4.7(brs,2H),3.88-3.89(m,2H),2.71-2.72(m,2H),2.60- 2.63(m,2H),1.44-1.48(m,4H),1.23-1.31(m,4H),0.86-0.90(m,6H).ppm。
Process for high purity lumefantrine Dr KrishnaSarma Pathy,Ramesh.D,P atchutaramaiah,Ch Sivasubramanyam Abstract. The present invention relates to Process for the preparation of lumefantrine Lumefantrine is a dichlorobenzylidine derivative effective for the treatment of various types of malaria. Chemically lumefantrine is 2-Dibutylamino-1-[ 2, 7-dichloro-9-(4- chlorobenzylidene)-9H-fluoren-4-yl]-Ethanol (racemate) The antimalarial agent is active against multi-drug resistant strains of Plasmodium falciparum. In combination with artemether, the drug is also used for the treatment of uncomplicated falciparum malaria. It has primary action as blood schizontocidal and secondary action as inhibition of nucleic acid and protein synthesis within the malarial parasite thus having a longer duration of anti malarial action. Thus, today lumefantrine is a drug of choice in antimalarial treatment against P. faliciparum. Therefore, development of an appropriate analytical procedure for the quantitative analysis of lumefantrine is of considerable importance to pharmaceutical industry. The spectroscopic techniques were used to confirm the identity of lumefantrine. The IR spectra, showed strong absorption band at 3404.67 cm-1(OH), 2953.28 cm-1(aliphatic and aromatic CH), 1757.31 cm-1(-C=C-), 933 cm-1(alkanes) and 696.37-373.22 cm-1 (Cl). Thus, IR spectra confirmed the presence of these functional groups in the structure of lumefantrine. The mass spectrum showed a sharp molecular ion peak at 528.0 m/z in Q1 MS (m/z, parent ion) parameter at negative polarity confirming the molecular weight of lumefantrine. The NMR spectra observed triplet at 0.943-0.989 (methyl protons of alkyl chain); a multiplet at 1.372-1.498 (methylene protons of alkyl chains); a multiplet at 2.449-2.909 (methylene protons of alkyl chain); broad singlet at 4.573 (OH proton); and multiplet at 7.314-7.733 (aromatic proton), thus confirming identity of lumefantrine. Summary of the process Stage1 Cl Cl 2,7-dichloro-9H-fluorene ClCl O Cl 2-chloro-1-(2,7-dichloro-9H-fluoren-3-yl)ethanone AlCl3 Chloroacetyl chloride In a 1 liter 3-necked flask, equipped with stirrer, thermometer and reflux condenser450.0 ml MDC cool to 0C.Start addition of 59.4 gm chloro acetyl chloride at 0-5C and stir for 15 min.Start addition of aluminium chloride at 0-5C. Stir for 30.0 min at same temp.Start addition of 2,7-dichloro fluorene soln. preprepared in 300.0 ml MDC d 150.0 ml saturated brine for washing and stir for 15-20 min. at 50-55 C.Allow to settle the layers at 50- 55C for 30.0 min.Separate the ORGANIC LAYER. at 50-55C.Again repeat Abovestep. for one time.Collect ORGANIC LAYER. and distill out dibutyl amine under vac. At 60- 90C.Disconnect vac. And cool reaction mass to 60C add 400.0 ml methanol and reflux to 30.0 min. After maintaining the reaction mass cool reaction mass to 50C.Seed the reaction mass by adding 500 mg stage 2 material at . 50C.Cool reaction mass to 30- 35C.Stir reaction mass further at same temp. fot 2 hrs.Filter reaction mass at 30- 35C.Suck dry and wash with 50.0 ml methanol at 30-35C.Suck dry and unload. Dry at 50-55C. Wt. of wet cake : 139.0 gm Wt. of dry material : 123.0 gm . . . a b c . . . a b c M.P: 72-76C Stage3: ClCl N OH + Cl COH ClCl N OH Cl 2,7-dichloro-alpha-[(dibutyl amino) p-chloro benzaldehyde methyl]-9H-fluoren-4-yl]oxirane Mol. Wt. 406 Lumefantrine Mol. Wt. 140.5Mol. Wt. 528.5 Under Nitrogen In a 2 liter 3-necked flask, equipped with stirrer, thermometer and reflux.Charge 840.0 ml ethanol at 30-35C.Chrge 120.0gm stage2 material at 30-35C.Charge 27.96 gm sodium methoxide in 30 min. at 30C exothermobserved temp. rise to 45C.Charge 41.52 gm p- Chloro benzaldehyde .Maintain reaction mass temp. 30-35C for 24 hrs. Check TLC a : Stage 2 b : Co spot c : reaction mass Mobile phase Hexane : Ethyl acetate 9 : 1 If TLC complies 1) 2) 3) Filter reaction mass at 30-35C. Suck dry and wash with 100.0 ml methanol. Suck dry and unload wet cake. Dry material at 50-55 C . . . a bc Wt. of wet cake : 184.0 gm Wt. Of dry material : 141.0 gm M.P : 122-126C Purification : In a 1 liter 3-necked flask, equipped with stirrer, thermometer and reflux .Charge 700.0 ml ethyl acetate at 30-35C.Chrge crude 141.0 gm material at 30-35C.Heat reaction mass to refluxand main for 30.0 min. to dissolve the material.Filter reaction mass at 75-80C.Collect filtrate cool to reaction mass to 0- 5C.Maintain reaction mass at same temp. for 30.0 min.Filter the reaction mass at 0- 5C.Suck dry and wash with 100.0 ml ethyl acetate at 0-5C.Suck dry and unload. Dry material at 50-55 C Wt. of wet cake : 100.0 gm Wt. Of dry material : 70-80 gm (19) (PDF) Process for high purity lumefantrine. Available from: https://www.researchgate.net/publication/221933417_Process_for_high_purity_lumefantrine [accessed Feb 06 2022].
M.P: 128-132C RESULTS:= Lumefantrine 99.15% (by HPLC) 128 – 130 Purity Melting Pointo C Liquid chromatography HPLC-UV investigation of the impurity profiles was performed on a HPLC-PDA apparatus consisting of a Waters Alliance 2695 separation module and a Waters 2998 photodiode array detector with Empower 2 software for data acquisition . For PDA detection, the UV spectrum was recorded at 190-400 nm. Quantification was performed at 266 nm. The positive ion ESI and the collision-induced dissociation (CID) mass spectra were obtained from the LC-UV/MS apparatus consisting of a Spectra System SN4000 interface, a Spectra System SCM1000 degasser, a Spectra System P1000XR pump, a Spectra System AS3000 autosampler, and a Finnigan LCQ Classic ion trap mass spectrometer in positive ion mode mass to charge range m/z 100 to m/z 2000 at unit resolution and with a peak width of 0.25 daltons/z, equipped with a Waters 2487 dual wavelength UV detector and Xcalibur 2.0 software (Thermo) for data acquisition. ESI was conducted using a needle voltage of 4.5 kV. Nitrogen was used as sheath and auxiliary gas with the heated capillary set at 250C. UV-detection was used for quantification (at 266 nm), while ESI-ion trap MS detection was used for identification. LC determination of impurities in lumefantrine samples was performed using a Purospher STAR RP-18 endcapped (150 4.6 mm, 5 m particle size) column (Merck, Darmstadt, Germany) with guard column at 30C under isocratic conditions with a mobile phase consisting of ammonium acetate (pH 4.9; 0.1 M) and acetonitrile (10:90, v/v). The flow rate was set at 2.0 mL/min (minimal run time: 30 min.). The injection volume was 10 l. Under these conditions, lumefantrine elutes at approximately 22 min. System suitability tests (SSTs) were established as the plate number on lumefantrine (N 8.2 103) and the oxidative stress degradation product (N 2.4 103), the signal-to-noise ratio of 0.5% l.c. lumefantrine solution (S/N 30), the peak area ratio of the 0.5% l.c. versus 100% l.c. (between 0.4 and 0.6) and the relative position of the in-situ prepared N-oxide by H2O2 treatment (RRTbetween 0.12 and 0.22). The LC method was validated for the determination of lumefantrine and its related impurities. The selectivity of the developed chromatographic method was established by the separation of lumefantrine and its impurities. A correlation coefficient (r2) of 0.9998 for lumefantrine (0.0006 to 0.01 mg/ml) and 1.0 for impurity A and DBK (0.001 to 0.1 mg/ml) demonstrated that the HPLC method is linear in the lower range. LOD/LOQ values for lumefantrine, DBK and impurity A were calculated (S/N = 3 resp. 10): 0.004 mg/mland 0.026 mg/ml for lumefantrine (0.004% respectively 0.026% l.c.), 0.011 mg/ml and 0.040 mg/ml for DBK (0.012% respectively 0.042% l.c.) and 0.110 mg/ml and 0.393 mg/ml for impurity A (0.115% respectively 0.409% l.c.). The analytical stability of lumefantrine, impurity A and DBK was confirmed over a storage period of 24 hours at 5C, i.e. the sample compartment temperature. Accuracy and precision were evaluated by repeated analysis (n = 6), with 102.6% l.c. recovery and 2.1%, respectively 2.86%, for repeatability, respectively intermediate precision. Structural information for the observed and/or reported lumefantrine related impurities # Compound [formula, mono-isotopic mass]Structure Origin # Compound [formula, mono-isotopic mass]Structure Origin Alkaline stress Oxidative stress Oxidative stress Metabolite 1Desbenzylketo N-oxide [C23H27NO3Cl2, MW 435.14] 2 Lumefantrine (mono-)desbutyl derivative [C26H24NOCl3, MW 473.09] 3Lumefantrine N-oxide [C30H32NO2Cl3, MW 543.15] Oxidative stress Degradation 4 2,7-dichloro-4-[2-(di-n-butylamino)-1-hydroxyethyl]- 9H-fluoren-9-one; Desbenzylketo derivative (DBK) [C23H27NO2Cl2, MW 419.14] Oxidative stress Acidic stress Degradation 5 2-(di-n-butylamino)-1-[2,7-dichloro-9H-fluoren-4- yl]ethanol; Desbenzyl derivative [C23H29NOCl2,405.16] Synthesis 6 Synthesis impurity found in lumefantrine API; Lumefantrine oxide [C30H32NO2Cl3, MW 543.14] Synthesis 7 Synthesis impurity found in lumefantrine API; Lumefantrine oxide [C30H32NO2Cl3, MW 543.14] Synthesis 8 (RS,Z)-2-(Dibutylamino)-2-(2,7-dichloro-9-(4- chloro-benzylidene)-9H-fluoren-4-yl)ethanol (isomeric compound); Impurity A (Ph. Int./USP Salmous) [C30H32NOCl3, MW 527.15] Synthesis 9 Synthesis (19) (PDF) Process for high purity lumefantrine. Available from: https://www.researchgate.net/publication/221933417_Process_for_high_purity_lumefantrine [accessed Feb 06 2022].
mpurity found in lumefantrine API; Lumefantrine oxide [C30H32NO2Cl3, MW 543.14] Synthesis 10 (1S,3R,5R)-1,3-bis((EZ)-2,7-Dichloro-9-(4- chlorobenzyl-idene)-9H-fluoren-4-yl)-2,6- dioxabicyclo[3.1.0]hexane; Impurity BA(USP Salmous) [C44H24Cl6O2, 797.39] 2-((EZ)-2,6-Dichloro-9- (4-chlorobenzylidene)-9H- fluoren-4-yl)-3′-((EZ)-2,7-dichloro-9-(4- chlorobenzylidene)-9H-fluoren-4-yl)-2,2′-bioxirane; Impurity BB (USP Salmous) [C44H24Cl6O2, 797.39] Percentage maximum actual levels of lumefantrine related impuritiesobserved(1) Synthesis 11 Synthesis # CompoundAPIFPP Unstressed Stressed Release Accelerated Stressed 1.39 0.560.11 21.32 1 Desbenzylketo N-oxide 2 Monodesbutyl derivative 3 Lumefantrine N-oxide 4 Desbenzylketo derivative 5 Desbenzyl derivative 6Lumefantrine oxide (RRT ~ 0.49) 7Lumefantrine oxide (RRT ~ 0.52) 8 Impurity A 9Lumefantrine oxide (RRT ~ 0.59) (1) RT: reporting threshold = 0.10% 0.60 0.12 0.34 0.86 4.26 HPLC characteristics of lumefantrine related impurities # CompoundRT(1)RRT(2) Most abundant m/z observed 436.14 474.00 544.08 420.13 406.09 544.12 544.12 528.10 544.12 528.10 1 Desbenzylketo N-oxide 2 Monodesbutyl derivative 3.25 3 Lumefantrine N-oxide 4 Desbenzylketo derivative 7.41 5 Desbenzyl derivative 6 Lumefantrine oxide 7 Lumefantrine oxide 8 Impurity A 9 Lumefantrine oxide L Lumefantrine (1) Retention time (min.) (2) Relative retention time reference 1.790.08 0.15 0.17 0.33 0.34 0.49 0.52 0.58 0.59 1.00 3.96 7.69 10.96 11.45 12.70 12.97 22.28 1. De Spiegeleer B, Vergote V, Pezeshki A, Peremans K, Burvenich C. Impurity profiling quality control testing of synthetic peptides using liquid chromatography-photodiode array-fluorescence and liquid chromatography-electrospray ionization-mass spectrometry: The obestatin case. Anal Biochem. 2008;376:229234. doi: 10.1016/j.ab.2008.02.014.[ 2. Nicolas EC, Scholz TH. Active drug substance impurity profiling – Part I. LC/UV diode array spectral matching. J Pharm Biomed Anal. 1998;16:813824. doi: 10.1016/S0731-7085(97)00131- 3Roy J. Pharmaceutical impurities – A mini review. AAPS PharmSciTech. 2002;3:article 6. doi: 10.1208/pt030206. ICH guidelines – International Conference on Harmonization, Q3A(R2) Impurities in new drug substances CPMP/ICH/2737/99. (October 2006) http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC50000 2675.pdf [Accessed on 5 November 2010 at 11:06] 3. Authorized Lumefantrine USP Salmous Standard (Februari 2009) http://www.usp.org/pdf/EN/nonUSStandards/lumefantrine.pdf [Accessed on 24 October 2010 at 17:14] 4. Lumefantrine: Document QAS/06.186/FINAL (WHO Ph. Int. – July 2008) http://www.who.int/medicines/publications/pharmacopoeia/Lumef_monoFINALQAS06_186_July 08.pdf [Accessed on 24 October 2010 at 17:37] 5. Lee H. Pharmaceutical applications of liquid chromatography coupled with mass spectrometry (LC/MS) J Liq Chromatogr Relat Technol. 2005;28:11611202. doi: 10.1081/JLC-200053022.] 6. Cesar ID, Nogueira FHA, Pianetti GA. Simultaneous determination of artemether and lumefantrine in fixed dose combination tablets by HPLC with UV detection. J Pharm Biomed Anal. 2008;48:951954. doi: 10.1016/j.jpba.2008.05.022 7. Cesar ID, Nogueira FHA, Pianetti GA. Comparison of HPLC, UV spectrophotometry and potentiometric titration methods for the determination of lumefantrine in pharmaceutical products. J Pharm Biomed Anal. 2008;48:223226. doi: 10.1016/j.jpba.2008.05.006. 8. Patil KR, Rane VP, Sangshetti JN, Shinde DB. A Stability-Indicating LC Method for Lumefantrine. Chromatographia. 2009;69:375379. doi: 10.1365/s10337-008-0894-x. [Cross Ref] 9. Munjal V, Paliwal N, Chaursia BK, Varshney B, Ahmed T, Paliwal J. LC-tandem mass spectrometry method for quantification of lumefantrine in human plasma and its application to bioequivalence study. Chromatographia. 2010;71:505510. doi: 10.1365/s10337-009-1446-8. [Cross Ref] 10. ICH guidelines – International Conference on Harmonization, Q1A(R2) Stability testing of new drug substances and products CPMP/ICH/2736/99. (August 2003) http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC5 (19) (PDF) Process for high purity lumefantrine. Available from: https://www.researchgate.net/publication/221933417_Process_for_high_purity_lumefantrine [accessed Feb 06 2022].
[1] Ulrich Beutler, C Peter.; Fuenfschilling.; and Andreas, Steinkemper.; Novartis Pharma AG; Chemical and Analytical Development: CH-4002 Basel, Switzerland, Organic Process Research & Development 2007, 11, 341- 345.
[2] Boehm, M. Fuenfschilling.; Krieger, P. C.; Kuesters, E. M.; Struber, F.; Org. Process Res. DeV. 2007, 11, 336- 340.
[3] (a) Rao, D. R.; Kankan, R. N.; Phull, M. S.; Patent Application CN 1009-3724 20060424, 2005. (b) Deng, R.; Zhong, J.; Zhao, D.; Wang, J.; Yaoxue, X. 2000, 35 (1), 22. (c) Allmendinger, Th.; Wernsdorfer, W. H. PCT WO 99/67197.
[4] Perrumattam, J.; Shao, Ch.; Confer, W. L. Synthesis 1994, 1181.
[5] Fuenfschilling, P. C.; Hoehn P.; Mutz J.-P. Organic Process Res. Dev. 2007, 11, 13.
[6] Di Nunno, L.; Scilimati, A. Tetrahedron 1988, 44, 3639.
Preparation of 2-(dibutylamino)-1-[(9Z)-2, 7-dichloro-9-(4-chlorobenzylidene)-9H-floren-4-yl] ethanol (Lumefantrine) 1.
To a stirred solution of NaOH (1.97 g 0.0492 mol) in methanol (100 ml) there was added 1-(2, 7- dichloro-9 H-fluren-4-yl)-2-(dibutyl amino) ethanol (10 g, 0.0246 mol) and para chloro benzaldehyde (5.24 g 0.0372). The suspension obtained was stirred at reflux temperature till the absence of starting material by TLC. After confirming the product formation reaction mixture was cooled to room temperature and further stirred at same temperature for overnight. The precipitated solids were filtered and washed with methanol and dried under vacuum at 50°C to get desired compound. (Purity by HPLC: 99%).
[2–4] Thus, today lumefantrine is a drug of choice in antimalarial treatment against P. …. The NMRspectra observed triplet at 0.943-0.989 (methyl protons of alkyl …
The spectroscopic techniques were used to confirm the identity of lumefantrine. The IR spectra, showed strong absorption band at 3404.67 cm-1 (OH), 2953.28 cm-1 (aliphatic and aromatic CH), 1757.31 cm-1 (-C=C-), 933 cm-1 (alkanes) and 696.37-373.22 cm-1 (Cl). Thus, IR spectra confirmed the presence of these functional groups in the structure of lumefantrine.
The mass spectrum showed a sharp molecular ion peak at 528.0 m/z in Q1 MS (m/z, parent ion) parameter at negative polarity confirming the molecular weight of lumefantrine.
The NMR spectra observed triplet at 0.943-0.989 (methyl protons of alkyl chain); a multiplet at 1.372-1.498 (methylene protons of alkyl chains); a multiplet at 2.449-2.909 (methylene protons of alkyl chain); broad singlet at 4.573 (OH proton); and multiplet at 7.314-7.733 (aromatic proton), thus confirming identity of lumefantrine.
IH NMR PREDICT
13C NMR PREDICT
References
Jump up^Toovey S, Jamieson A, Nettleton G (2003). “Successful co-artemether (artemether-lumefantrine) clearance of falciparum malaria in a patient with severe cholera in Mozambique”. Travel medicine and infectious disease. 1 (3): 177–9. doi:10.1016/j.tmaid.2003.09.002. PMID17291911.
Jump up^White, Nicholas J.; van Vugt, Michele; Ezzet, Farkad (1999). “Clinical Pharmacokinetics and Pharmacodynamics of Artemether-Lumefantrine”. Clinical Pharmacokinetics. 37 (2): 105–125. doi:10.2165/00003088-199937020-00002. ISSN0312-5963.
Percent Composition: C 68.12%, H 6.10%, Cl 20.11%, N 2.65%, O 3.02%
Literature References: Racemic aryl alcohol originally synthesized in the 1970’s by the Academy of Military Medical Sciences in Beijing, China. Inhibits hemozoin formation. Prepn: R. Deng et al.,CN1042535 (1990 to Acad. Military Med. Sci., Microbiol. & Epidemic Dis. Instit.); C.A.114, 6046 (1991). LC determn in plasma: A. Annerberg et al.,J. Chromatogr. B822, 330 (2005). In vitro activity against Plasmodium falciparum: B. Pradines et al.,Antimicrob. Agents Chemother.43, 418 (1999).
Properties: Odorless, yellow powder. Poorly sol in water, oil, and most organic solvents. Sol in unsaturated fatty acids.
Derivative Type: Co-artemether
CAS Registry Number: 141204-94-6
Manufacturers’ Codes: CPG-56697
Trademarks: Coartem (Novartis); Riamet (Novartis)
Literature References: Fixed 6:1 mixture with artemether, q.v. Clinical pharmacokinetics and bioavailability: F. Ezzet et al.,Br. J. Clin. Pharmacol.46, 553 (1998). Clinical trial in children against P. falciparum malaria: C. Hatz et al.,Trop. Med. Int. Health3, 498 (1998); in adults: S. Looareesuwan et al.,Am. J. Trop. Med. Hyg.60, 238 (1999). Review of comparative clinical trials in malaria: A. A. Omari et al., Trop. Med. Int. Health9, 192-199 (2004).
By: Laura I. Mosquera-Giraldo and Lynne S. Taylor Imagine spending billions of dollars in the discovery of a new drug, and then realizing that it is impractical to administer it orally because it cannot reach the systemic circulation and achieve a therapeutic effect. This is the case for many emerging drugs that are insoluble in water, […]
Preparation of 1-(2-Methoxyphenoxy)-2,3-epoxypropane 4.
To a stirring solution of 2-methoxy phenol 2 (10 kg, 80.55 mol) and water (40 L) at about 30 °C was added sodium hydroxide (1.61 kg, 40.25 mol) and water (10 L). After stirring for 30−45 min, epichlorohydrin 3 (22.35 kg, 241.62 mol) was added and stirred for 10−12 h at 25−35 °C. Layers were separated, and water (40 L) was added to the organic layer (bottom layer) containing product. Sodium hydroxide solution (3.22 kg, 80.5 mol) and water (10 L) were added at 27 °C and stirred for 5−6 h at 27 °C.
The bottom product layer was separated and washed with sodium hydroxide solution (3.0 kg 75 mol) and water (30 L). Excess epichlorohydrin (3) was recovered by distillation of the product layer at below 90 °C under vacuum (650−700 mmHg) to give 13.65 kg (94%) of title compound with 98.3% purity by HPLC, 0.2% of 2- methoxy phenol 2, 0.1% of epichlorohydrin 3, 0.1% of chlorohydrin 11, 0.3% of dimer 12 and 0.3% of dihydroxy 13.
WATCH THIS POST AS DETAILS LIKE SYNTHESIS ARE UPDATED………….
Adrafinil is touted mainly for its stimulant properties and ability to provide alertness and wakefulness.
Stay up late/stay awake during normal sleeping hours: Adrafinil may be helpful for night workers who need a kick-start adapting their body’s natural circadian rhythm of wakefulness in the daytime and sleepiness in the evening to their job needs. This can also make it helpful for periodic late-night study sessions. Adrafinil is best taken in the afternoon or evening for nighttime wakefulness.
Boost energy, alertness, and focus during the day time: Adrafinil can also be used as an energy-boost during waking hours.
CONTACT SKYPE CATHERINESSPC WICKR
Adrafinil (INN) (brand name Olmifon)[2] is a discontinued wakefulness-promoting agent (or eugeroic) that was formerly used inFrance to promote vigilance (alertness), attention, wakefulness, mood, and other parameters, particularly in the elderly.[3][4] It was also used off-label by individuals who wished to avoid fatigue, such as night workers or others who needed to stay awake and alert for long periods of time. Additionally, “adrafinil is known to a larger nonscientific audience, where it is considered to be a nootropic agent.”[3] Adrafinil is a prodrug; it is primarily metabolizedin vivo to modafinil, resulting in very similar pharmacological effects.[3] Unlike modafinil, however, it takes time for the metabolite to accumulate to active levels in the bloodstream. Effects usually are apparent within 45–60 minutes when taken orally on an empty stomach. Adrafinil was marketed in France under the trade name Olmifon[2] until September 2011 when it was voluntarily discontinued.[4]
Pharmacology
Pharmacodynamics
Because α1-adrenergic receptorantagonists were found to block effects of adrafinil and modafinil in animals, “most investigators assume[d] that adrafinil and modafinil both serve as α1-adrenergic receptor agonists.”[3] However, adrafinil and modafinil have not been found to bind to the α1-adrenergic receptor and they lack peripheralsympathomimeticside effects associated with activation of this receptor;[5] hence, the evidence in support of this hypothesis is weak, and other mechanisms are probable.[3] Modafinil was subsequently screened at a variety of targets in 2009 and was found to act as a weak, atypical blocker of the dopamine transporter(and hence as a dopamine reuptake inhibitor), and this action may explain some or all of its pharmacological effects.[6][7][8] Relative to adrafinil, modafinil possesses greater specificity in its action, lacking or having a reduced incidence of many of the common side effects of the former (including stomach pain, skin irritation, anxiety, and elevated liverenzymes with prolonged use).[9][10][11] There is a case report of two patients that adrafinil may increase interest in sex.[3] A case report of adrafinil-induced orofacial dyskinesia exists.[12][13] Reports of this side effect also exist for modafinil.[12]
Pharmacokinetics
In addition to modafinil, adrafinil also produces modafinil acid (CRL-40467) and modafinil sulfone (CRL-41056) as metabolites, which form from metabolic modification of modafinil.
History
Adrafinil was discovered in 1974 by two chemists working for the French pharmaceutical company Laboratoires Lafon who were screening compounds in search of analgesics.[14] Pharmacological studies of adrafinil instead revealed psychostimulant-like effects such as hyperactivity and wakefulness in animals.[14] The substance was first tested in humans, specifically for the treatment of narcolepsy, in 1977–1978.[14] Introduced by Lafon (now Cephalon), it reached the market in France in 1984,[4] and for the treatment of narcolepsy in 1985.[14][15] In 1976, two years after the discovery of adrafinil, modafinil, its active metabolite, was discovered.[14] Modafinil appeared to be more potent than adrafinil in animal studies, and was selected for further clinical development, with both adrafinil and modafinil eventually reaching the market.[14] Modafinil was first approved in France in 1994, and then in the United States in 1998.[15] Lafon was acquired by Cephalon in 2001.[16] As of September 2011, Cephalon has discontinued Olmifon, its adrafinil product, while modafinil continues to be marketed.[4]
In 2005 a Medical Classification Committee in New Zealand recommended to MEDSAFE NZ that adrafinil be classified as a prescription medicine due to risks of it being used as a party drug. At that time adrafinil was not scheduled in New Zealand.[18]
Research
In a clinical trial with clomipramine and placebo as active comparators, adrafinil showed efficacy in the treatment of depression.[3] In contrast to clomipramine however, adrafinil was well-tolerated, and showed greater improvement in psychomotor retardation in comparison.[3] As such, “further investigations of the antidepressive effects of adrafinil are warranted.”[3]
/////////////
SYNTHESIS
Adrafinil (CAS NO.63547-13-7) was discovered in the late 1970s by scientists working with the French pharmaceutical company Group Lafon. First offered in France in 1986 as an experimental treatment for narcolepsy, Lafon later developed modafinil, the primary metabolite of adrafinil. Modafinil possesses greater selective alpha-1 adrenergic activity than adrafinil without the side effects of stomach pain, skin irritations, feelings of tension, and increases in liver enzyme levels.
It is important to monitor the liver of an individual using adrafinil. It can cause liver damage in some instances.
The Adrafinil with CAS registry number of 63547-13-7 is also known as 2-[(Diphenylmethyl)sulfinyl]-N-hydroxyacetamide. The IUPAC name is 2-Benzhydrylsulfinyl-N-hydroxyacetamide. It belongs to product categories of Aromatics Compounds; Aromatics; Intermediates & Fine Chemicals; Pharmaceuticals; Sulfur & Selenium Compounds. This chemical is a light pink solid and its EINECS registry number is 264-303-1. In addition, the formula is C15H15NO3S and the molecular weight is 289.35. This chemical is harmful if swallowed.
Preparation of Adrafinil: it is prepared by reaction of diphenylmethyl bromide with thiourea. This reaction needs reagent NaOH. After reacting with chloroacetic acid, hydrochloric acid amine and hydrogen peroxide, the product is obtained. The yield is about 73%.
Uses of Adrafinil: it is used as non-amphetamine-type psychostimulant and can wake up and raise awareness. For the elderly arousal disorder and depressive symptoms in symptomatic treatment.
15.2 g (0.2 mol) of thiourea and 150 ml of demineralized water are introduced into a 500 ml three-neck flask equipped with a central mechanical stirrer, and with a dropping funnel and a condenser on the (respective) side-necks.The temperature of the reaction mixture is brought to 50°and 49.4g (0.2 mol) of bromodiphenyl- methane are added all at once whilst continuing the heating. After refluxing for about 5 minutes, the solution, which has become limpid, is cooled to 20°C and 200 ml of 2.5 N NaOH are then added dropwise whilst maintaining the said temperature. The temperature is then again kept at the reflex for 30 minutes after which, when the mixture has returned to ordinary temperature (15-25°C), the aqueous solution is acidified with 45 ml of concentrated hydrochloric acid. The supernatant oil is extracted with 250 ml of diethyl ether and the organic phase is washed with 4×80 ml of water and then dried over magnesium sulphate. 39 g of crude diphenylmethane-thiol are thus obtained. Yield 97.5%.
Benzhydryl-thioacetic acid
10 g (0.05 mol) of diphenylmethane-thiol and 2g (0.05 mol) of NaOH dissolved in 60 ml of demineralised water are introduced successively into a 250 ml flask equipped with a magnetic stirrer and a reflux condenser. The reactants are left in contact for 10 minutes whilst stirring, and a solution consisting of 7g (0.075 mol) of chloroacetic acid, 3g (0.075 mol) of NaOH pellets and 60 ml of demineralized water is then added all at once. The aqueous solution is gently warmed to about 50°C for 15 minutes, washed with 50 ml of ether, decanted and acidified with concentrated hydrochloric acid. after filtration, 10.2g of benzhydryl-thioacetic acid are thus obtained. Melting point 129-130°C. Yield 79%.
Ethyl benzhydryl-thioacetate
The following reaction mixture is heated under reflux for 7 hours: 10.2 g (0.0395 mol) of benzhydryl-thioacetic acid, 100 ml of anhydrous ethanol and 2 ml of sulphuric acid. When heating has been completed, the ethanol isevaporated in vacuo; the oily residue is taken up in 100 ml of ethyl ether and the organic solution is then washed with water, with an aqueous sodium carbonate solution and then with water until the wash waters have a neutral pH. After drying over sodium sulphate, the solvent is evaporated. 10.5g of ethyl benzhydryl- thioacetate are thus obtained. Yield 93%.
Benzhydryl-thioacetohydroxamic acid
The following three solutions are prepared:
Ethyl Benzhydryl-thioacetate 10.8 g (0.0378 mol) in 40 ml methanol
Hydroxylamine hydrochloride 5.25 g (0.0756 mol) in 40 ml methanol
Potassium Hydroxide pellets 7.3 g (0.0134 mol) in 40 ml methanol
The solutions are heated, if necessary, until they become limpid, and when the temperatures have again fallen to below 40°C, the solution of potassium hydroxide in methanol is poured into the solution of hydroxylamine hydrochloride in alcohol. Finally, at a temperature of about 5° to 10°C, the solution of ethyl benzhydryl- thioacetate is added in its turn. After leaving the reactants in contact for 10 minutes, the sodium chloride is filtered off the limpid solution obtained is kept for about 15 hours at ordinary temperature. The methanol is then evaporated under reduced pressure, the residual oil is taken up in 100 ml of water and the aqueous solution is acidified with 3 N hydrochloric acid. The hydroxamic acid which has crystallized is filtered off, washed with water and then dried. 9.1 g of product are obtained. Yield = 87.5%. Melting point 118-120°C.
Adrafinil (CRL 40,028)
10.4g (0.038 mol) of benzhydryl-thioacetohydroxamic acid are oxidized at 40°C, over the course of 2 hours, by means of 3.8 ml (0.038 mol) of hydrogen peroxide of 110 volumes strength (33%), in 100 ml of acetic acid.
When the oxidation has ended, the acetic acid is evaporated under reduced pressure and the residual oil is taken up in 60 ml of ethyl acetate. The product which has crystallized is filtered off and then purified by recrystallisation from a 3:2 (by volume) mixture of ethyl acetate and isopropyl alcohol.
8g (73%) of Adrafinil, mp 159-160°C, are thus obtained. H2O Solubility
CLIP
Figure 2: GC/MS extracted ion chromatogram (a) and mass spectrum (b) of derivatized adrafinil in the electron ionization mode (monitoring the m/z 167, 165 and 152 ions; all the four peaks are derivatised adrafinil products).
Figure 4: LC/ESI-MS full scan chromatogram of adrafinil and its metabolites (a) (modafinil acid RT 3.8 min, adrafinil RT 4.0 min, modafinil RT 4.1 min), and LC/ESI-MS full scan mass spectra of modafinil acid (b), adrafinil (c), and (d) modafinil. (b, c and d showing the similar ions at m/z 167, 165, 152 together with the appropriate sodium and potassium adducts).
Jump up^Quisenberry AJ, Baker LE (Dec 2015). “Dopaminergic mediation of the discriminative stimulus functions of modafinil in rats”. Psychopharmacology. 232 (24): 4411–9.doi:10.1007/s00213-015-4065-0. PMID26374456.
Jump up^Ballas, Christos A; Deborah Kim; Claudia F Baldassano; Nicholas Hoeh (July 2002). “Modafinil: past, present and future”. Expert Review of Neurotherapeutics. 2 (4): 449–57.doi:10.1586/14737175.2.4.449. PMID19810941.
Percent Composition: C 62.26%, H 5.23%, N 4.84%, O 16.59%, S 11.08%
Literature References: a-Adrenergic agonist. Prepn, pharmacology: L. Lafon, BE846880; US4066686 (1977, 1978 both to Lafon). Mode of action: J. Duteil et al.,Eur. J. Pharmacol.59, 121 (1979); C. Rozé et al.,Arch. Int. Pharmacodyn. Ther.265, 119 (1983). Psychopharmacology in mice: F. A. Rambert et al.,J. Pharmacol.17, 37 (1986).
Properties: Crystals from ethyl acetate-isopropyl alcohol, mp 159-160°. Soly in water <1 g/l. LD50 in mice (mg/kg): <2048 i.p.; 1950 gastric admin (Lafon).
Generics: The US Food and Drug Administration (FDA) recently published a new Guidance regarding Prior Approval Supplements (PAS). Read more about FDA´s Guidance for Industry “ANDA Submissions – Prior Approval Supplements Under GDUFA“.
On October 14, 2016, the US Food and Drug Administration (FDA) published a new Guidance regarding Prior Approval Supplements (PAS).
FDA says that “this guidance is intended to assist applicants preparing to submit to FDA prior approval supplements (PASs) and amendments to PASs for abbreviated new drug applications (ANDAs)”.
Specifically, the guidance describes how the Generic Drug User Fee Amendments of 2012 (GDUFA) performance metric goals apply to:
A PAS subject to the refuse-to-receive (RTR) standards;
A PAS that requires an inspection;
A PAS for which an inspection is not required;
An amendment to a PAS;
Other PAS-related matters.
GDUFA is designed to speed the delivery of safe and effective generic drugs to the…

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....