
Home » Uncategorized (Page 144)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
CHINESE MEDICINE..Scutellaria baicalensis fights cancer

Scutellaria baicalensis (or Baikal Skullcap, as opposed to Scutellaria lateriflora, a Skullcap native to North America) is a species of flowering plant in the Lamiaceae family.
Traditional Chinese medicine
It is one of the 50 fundamental herbs used in traditional Chinese medicine, where it has the name huáng qín (Chinese: 黄芩).[2] As a Chinese traditional medicine, Huang Qin usually refers to the dried root of Scutellaria baicalensis Georgi, S. viscidula Bge., S. amoena C.H. Wright, and S. ikoninkovii Ju.
Chemistry
Several chemical compounds have been isolated from the root; among them, baicalein, baicalin, wogonin, norwogonin, oroxylin A[3] and β-sitosterol are the major ones.
Etymology confusion
It is important to note the Latin name of the Skullcap being used as there are over 200 varieties, some used for various ailments, each with varying degrees of effectiveness. Sometimes Scutellaria lateriflora (North American Skullcap) is mistaken for Scutellaria baicalensis (Baikal Skullcap). This confusion can result in the intake of the lateriflora variety which is often processed and contaminated with other plants with high enough levels of toxicity to be of concern.
Baikal skullcap (scientific name Scutellaria baicalensis) is a plant. The root is used to make medicine. Common substitutions for Baikal skullcap in Chinese medicine include related plants whose scientific names are Scutellaria viscidula, Scutellaria amonea, and Scutellaria ikoninikovii.
Baikal skullcap is used to treat respiratory infections, hay fever, and fever. It is also used for gastrointestinal (GI) infections, as well as liver problems including viralhepatitis and jaundice.
Some people use Baikal skullcap for HIV/AIDS, kidney infections, pelvic inflammation, and sores or swelling. It is also used for scarlet fever, headache, irritability, red eyes, flushed face, seizures, epilepsy, hysteria, nervous tension, and to relieve a bitter taste in the mouth.

The active ingredient in Baikal skullcap, baicalin, is used in combination with shung hua (ephedra) to treat upper respiratory tract infections. In combination with other herbs, Baikal skullcap is used to treat attention deficit-hyperactivity disorder (ADHD),prostate cancer, a lung condition called bronchiolitis, arthritis, and hemorrhoids.
Baikal skullcap is also sometimes applied to the skin for psoriasis.
How does it work?
It is thought that the active chemicals in Baikal skullcap might be able to decrease inflammation, stop tumor growth, and prevent tumor cell reproduction.

Scutellaria baicalensis , also called Chinese skullcap, is a member of the mint family and has long been used in traditional Chinese herbal medicine . Chinese skullcap has been incorporated in herbal formulas designed to treat such widely varying conditions as cancer, liver disease, allergies, skin conditions, and epilepsy. The root is the part used medicinally.
Note: Chinese skullcap is substantially different from American skullcap ( Scutellaria lateriflora ).
Skullcap (Scutellaria baicalensis) has been widely used as a dietary ingredient and traditional herbal medicine owing to its anti-inflammatory and anticancer properties. In this study, we investigated the anti-allergic effects of skullcap and its active compounds, focusing on T cell-mediated responses ex vivoand in vivo. Splenocytes from mice sensitized with ovalbumin (OVA) were isolated for analyses of cytokine production and cell viability. Mice sensitized with OVA were orally administered skullcap or wogonin for 16 days, and then immunoglobulin (Ig) and cytokine levels were measured by enzyme-linked immunosorbent assays. Treatment with skullcap significantly inhibited interleukin (IL)-4 production without reduction of cell viability. Moreover, wogonin, but not baicalin and baicalein, suppressed IL-4 and interferon-gamma production. In vivo, skullcap and wogonin downregulated OVA-induced Th2 immune responses, especially IgE and IL-5 prediction. Wogonin as an active component of skullcap may be applied as a therapeutic agent for IgE- and IL-5-mediated allergic disorders…….http://www.mdpi.com/1420-3049/19/2/2536

References
- “Scutellaria baicalensis information from NPGS/GRIN”. USDA. Retrieved 2008-02-19.
- Zhang XW, Li WF, Li WW, Ren KH, Fan CM, Chen YY, Shen YL (2011). “Protective effects of the aqueous extract of Scutellaria baicalensis against acrolein-induced oxidative stress in cultured human umbilical vein endothelial cells”. Pharm Biol 49 (3): 256–261. doi:10.3109/13880209.2010.501803. PMID 20979538.
- Isolation and purification of baicalein, wogonin and oroxylin A from the medicinal plant Scutellaria baicalensis by high-speed counter-current chromatography. Hua-Bin Li and Feng Chen, Journal of Chromatography A, 13 May 2005, Volume 1074, Issues 1–2, pages 107–110, doi:10.1016/j.chroma.2005.03.088
- Scutellaria baicalensis List of Chemicals (Dr. Duke’s Databases)
- Scutellaria baicalensis (Plants for a Future)
- Sung Mun Jung et al., “Reduction of urate crystal-induced inflammation by root extracts from traditional oriental medicinal plants: elevation of prostaglandin D2 levels”, Arthritis Research & Therapy 2007, 9:R64 doi:10.1186/ar2222. Considers anti-inflammatory properties of dried roots from the species Angelica sinensis (Dong Quai), Acanthopanax senticosus (now known as Eleutherococcus senticosus, or Siberian Ginseng), andScutellaria baicalensis (Baikal Skullcap).
GREEN CHEMISTRY…Reduction of amides without hydride reagents

Clostridium sporogenes
Essentially all medicines and current drug candidates contain at least one basic nitrogen atom. A common approach to the synthesis of amines is to reduce the corresponding amide with a hydride reagent such as LiAlH4, DIBAL, RedAl, B2H6, Et3SiH, or polymethylhydroxysilane (PMHS).
The reaction survey reported that reduction of amides to amines was used in only 0.6% of chemical transformations; this number would surely be higher if safer methods for use on scale were available. The survey indicated that the number of amide reductions was equally split between diborane and hydride reagents.
Lithium aluminium hydride,

having a molecular weight of 38 and four hydrides per molecule, has the highest hydride density and is frequently used, even though it co-generates an inorganic by-product (lithium aluminum hydroxide) which is difficult to separate from the product. The workup procedure recommended by one bulk supplier (Chemetall) is to precipitate and filter the aluminum hydroxide salts. However, slow filtrations and product loss through occlusion or adsorption are typical problems that can be encountered.
Options for disposal of the cake include dissolving in water and sending to a waste water treatment plant or drying the cake and sending to a chemical waste dump that accepts solids.1 Both options have an environmental impact. Therefore, a generally applicable, safe, environmentally benign and economically viable method for the reduction of amides to amines would have an appreciable benefit to numerous processes.
Hydrogen gas is the ideal reductant because the only by-product is water. Thus, much research has been directed towards discovery of a transition metal catalyst selective for hydrogenation of amides. However, even with the best catalysts, both high temperature (![[similar]](https://i0.wp.com/www.rsc.org/images/entities/char_223c.gif) 150 °C) and pressure (>100 bar) are required. These conditions involve expensive high pressure hydrogenation equipment not typically available in a common pharmaceutical manufacturing plant.
150 °C) and pressure (>100 bar) are required. These conditions involve expensive high pressure hydrogenation equipment not typically available in a common pharmaceutical manufacturing plant.
The harsh conditions also preclude the use of these catalysts with substrates that contain other reducible or thermally labile functional groups. Recent research has led to the discovery of catalysts that are effective at lower temperature and pressure, giving encouragement that the goal of finding a selective, low pressure/temperature catalyst is realistic.2
Another approach would be to use a biotransformation to reduce the amide. It is notable that a number of bacteria and fungi reduce carboxylic acids to aldehydes or ketones.3 The usual fate of amides in biological pathways is hydrolysis. However, an anaerobic bacteria, Clostridium sporogenes, has been reported to reduce benzamide to benzylamine. 4

A key challenge in this technology area is gaining a detailed understanding of these complex enzyme-catalysed processes that require ATP/NADPH co-factor recycling, and getting the enzymes cloned and produced on a large scale in suitable expression systems.
The acylation/reduction strategy for N-alkylation avoids the need to handle alkylating agents and would be more widely used if a safer, more atom economical or preferably catalytic method for amide reduction were developed. The solution to this problem could be either chemical or biochemical.
- Chemetall brochures, Lithium Aluminum Hydride… strong, concentrated and economical, Oct. 2000, pp. 18–19 Search PubMed .
- A. A. Smith, P. Dani, P. D. Higginson and A. J. Pettman, World Pat., WO2005/066112 A1, 2005 Search PubMed .
- (a) A. Hage, H. E. Schoemaker and J. A. Field, Appl. Microbiol. Biotechnol., 1999, 52, 834–838 CrossRef CAS Search PubMed ; (b) A. He, T. Li, L. Daniels, I. Fotheringham and J. P. N. Rosazza, Appl. Environ. Microbiol., 2004, 70, 1874–1881 CrossRef CAS Search PubMed .
- O. Dipeolu, J. Gardiner and G. Stephens, Biotechnol. Lett., 2005, 27, 1803–1807 CrossRef CAS Search PubMed .
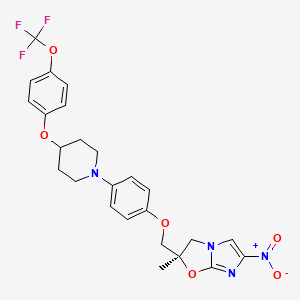
Delamanid……….an experimental drug for the treatment of multi-drug-resistant tuberculosis.

Delamanid
http://www.ama-assn.org/resources/doc/usan/delamanid.pdf
(2R)-2-Methyl-6-nitro-2-[(4-{4-[4-(trifluoromethoxy)phenoxy]-1-piperidinyl}phenoxy)methyl]-2,3-dihydroimidazo[2,1-b][1,3]oxazole
2(R)-Methyl-6-nitro-2-[4-[4-[4-(trifluoromethoxy)phenoxy]piperidin-1-yl]phenoxymethyl]-2,3-dihydroimidazo[2,1-b]oxazole
(R) -2-methyl-6-nitro-2- { 4- [4- (4- trifluoromethoxyphenoxy) piperidin-l-yl] phenoxymethyl } -2 , 3- dihydroimidazo [2 , 1-b] oxazole
Imidazo[2,1-b]oxazole, 2,3-dihydro-2-methyl-6-nitro-2-[[4-[4-[4-(trifluoromethoxy)phenoxy]-1-piperidinyl]phenoxy]methyl]-, (2R)-
(R)-2-methyl-6-nitro-2-{4-[4-(4-trifluoromethoxyphenoxy)piperidin-1-yl]phenoxymethyl}-2,3-dihydroimidazo[2,1-b]oxazole
681492-22-8 cas no
Delamanid, 681492-22-8, Delamanid (JAN/USAN), Delamanid [USAN:INN],UNII-8OOT6M1PC7,
- OPC 67683
- OPC-67683
- UNII-8OOT6M1PC7
Molecular Formula: C25H25F3N4O6
Molecular Weight: 534.48441
Clinical trials……….http://clinicaltrials.gov/search/intervention=OPC+67683+OR+Delamanid
CLINICAL TRIALS
Primary Sponsor: Otsuka Pharmaceutical Development & Commercialization, Inc.
Trial ID / Reg # / URL: http://clinicaltrials.gov/ct2/show/NCT00685360

Delamanid (USAN, codenamed OPC-67683) is an experimental drug for the treatment of multi-drug-resistant tuberculosis. It works by blocking the synthesis of mycolic acids in Mycobacterium tuberculosis, the organism which causes tuberculosis, thus destabilising its cell wall.[1][2][3]
In phase II clinical trials, the drug was used in combination with standard treatments, such as four or five of the drugs ethambutol, isoniazid,pyrazinamide, rifampicin, aminoglycoside antibiotics, and quinolones. Healing rates (measured as sputum culture conversion) were significantly better in patients who additionally took delamanid.[3][4]
The European Medicines Agency (EMA) recommended conditional marketing authorization for delamanid in adults with multidrug-resistant pulmonary tuberculosis without other treatment options because of resistance or tolerability. The EMA considered the data show that the benefits of delamanid outweigh the risks, but that additional studies were needed on the long-term effectiveness.[5]
Delamanid, an antibiotic active against Mycobacterium tuberculosis strains, has been filed for approval in the E.U. and by Otsuka for the treatment of multidrug-resistant tuberculosis. In 2013, a positive opinion was received in the E.U. for this indication. Phase III trials for treatment of multidrug-resistant tuberculosis are under way in the U.S. Phase II study for the pediatric use is undergone in the Europe.
The drug candidate’s antimycobacterial mechanism of action is via specific inhibition of the synthesis pathway of mycolic acid, which is a cell wall component unique to M. tuberculosis.
In 2008, orphan drug designation was received in Japan for the treatment of pulmonary tuberculosis.
Tuberculosis (TB), an airborne lung infection, still remains a major public health problem worldwide. It is estimated that about 32% of the world population is infected with TB bacillus, and of those, approximately 8.9 million people develop active TB and 1.7 million die as a result annually according to 2004 figures. Human immunodeficiency virus (HIV) infection has been a major contributing factor in the current resurgence of TB. HIV-associated TB is widespread, especially in sub-Saharan Africa, and such an infectious process may further accelerate the resurgence of TB.
Moreover, the recent emergence of multidrug-resistant (MDR) strains ofMycobacterium tuberculosis that are resistant to two major effective drugs, isonicotinic acid hydrazide (INH) and rifampicin (RFP), has further complicated the world situation.
The World Health Organization (WHO) has estimated that if the present conditions remain unchanged, more than 30 million lives will be claimed by TB between 2000 and 2020. As for subsequent drug development, not a single new effective compound has been launched as an antituberculosis agent since the introduction of RFP in 1965, despite the great advances that have been made in drug development technologies.
Although many effective vaccine candidates have been developed, more potent vaccines will not become immediately available. The current therapy consists of an intensive phase with four drugs, INH, RFP, pyrazinamide (PZA), and streptomycin (SM) or ethambutol (EB), administered for 2 months followed by a continuous phase with INH and RFP for 4 months. Thus, there exists an urgent need for the development of potent new antituberculosis agents with low-toxicity profiles that are effective against both drug-susceptible and drug-resistant strains of M. tuberculosis and that are capable of shortening the current duration of therapy.
………………………
(R)-2-bromo-4-nitro-1-(2-methyl-2-oxiranylmethyl)imidazole
4-[4-(4-Trifluoromethoxyphenoxy)piperidin-1-yl]phenol
ARE THE INTERMEDIATES
Example 1884
Production of (R)-2-methyl-6-nitro-2-{4-[4-(4-trifluoromethoxyphenoxy)piperidin-1-yl]phenoxymethyl}-2,3-dihydroimidazo[2,1-b]oxazole
4-[4-(4-Trifluoromethoxyphenoxy)piperidin-1-yl]phenol (693 mg, 1.96 mmol) was dissolved in N,N′-dimethylformamide (3 ml), and sodium hydride (86 mg, 2.16 mmol) was added while cooling on ice followed by stirring at 70-75° C. for 20 minutes. The mixture was cooled on ice. To the solution, a solution prepared by dissolving (R)-2-bromo-4-nitro-1-(2-methyl-2-oxiranylmethyl)imidazole (720 mg, 2.75 mmol) in N,N′-dimethylformamide (3 ml) was added followed by stirring at 70-75° C. for 20 minutes. The reaction mixture was allowed to return to room temperature, ice water (25 ml) was added, and the resultant solution was extracted with methylene chloride (50 ml) three times. The organic phases were combined, washed with water 3 times, and dried over magnesium sulfate. After filtration, the filtrate was concentrated, and the residue was purified by silica gel column chromatography (methylene chloride/ethyl acetate=3/1). Recrystallization from ethyl acetate/isopropyl ether gave (R)-2-methyl-6-nitro-2-{4-[4-(4-trifluoromethoxyphenoxy)piperidin-1-yl]phenoxymethyl}-2,3-dihydroimidazo[2,1-b]oxazole (343 mg, 33%) as a light yellow powder.
…………………………
WO 2010021409 AND http://worldwide.espacenet.com/publicationDetails/biblio?CC=IN&NR=203704A1&KC=A1&FT=D
FOR 2, 4 DINITROIMIDAZOLE
…………………………………………
These patent literatures disclose Reaction Schemes A and B below as the processes for producing the aforementioned 2, 3-dihydroimidazo [2, 1-b] oxazole compound.
Reaction Scheme A:

wherein R1 is a hydrogen atom or lower-alkyl group; R2 is a substituted pxperidyl group or a substituted piperazinyl group; and X1 is a halogen atom or a nitro group.
Reaction Scheme B:


wherein X2 is a halogen or a group causing a substitution reaction similar to that of a halogen; n is an integer from 1 to 6; and R1, R2 and X1 are the same as in Reaction Scheme A.
An oxazole com ound represented by Formula (la) :

, i.e., 2-methyl-6-nitro-2-{4- [4- (4- trifluoromethoxyphenoxy) piperidin-l-yl] phenoxymethyl }-2, 3- dihydroimidazo [2, 1-b] oxazole (hereunder, this compound may be simply referred to as “Compound la”) is produced, for example, by the method shown in the Reaction Scheme C below (Patent
Literature 3) . In this specification, the term “oxazole compound’ means an oxazole derivative that encompasses compounds that contain an oxazole ring or an oxazoline ring (dihydrooxazole ring) in the molecule.
Reaction Scheme C:


However, the aforementioned methods are unsatisfactory in terms of the yield of the objective compound. For example, the method of Reaction Scheme C allows the objective oxazole Compound (la) to be obtained from Compound (2a) at a yield as low as 35.9%. Therefore, alternative methods for producing the compound in an industrially advantageous manner are desired. Citation List
Patent Literature
PTL 1: WO2004/033463
PTL 2: WO2004/035547
PTL 3: WO2008/140090
Example 9
Production of (R) -2-methyl-6-nitro-2- { 4- [4- (4- trifluoromethoxyphenoxy) piperidin-l-yl] phenoxymethyl } -2 , 3- dihydroimidazo [2 , 1-b] oxazole
{R) -1- [ – {2 , 3-epoxy-2-methylpropoxy ) phenyl] -4- [4- ( trifluoromethoxy ) phenoxy ] piperidine (10.0 g, 23.6 mmol, optical purity of 94.3%ee), 2-chloro-4-nitroimidazole (4.0 g, 27.2 mmol), sodium acetate (0.4 g, 4.9 mmol), and t- butyl acetate (10 ml) were mixed and stirred at 100°C for 3.5 hours. Methanol (70 ml) was added to the reaction mixture, and then a 25% sodium hydroxide aqueous solution (6.3 g, 39.4 mmol) was added thereto dropwise while cooling with ice. The resulting mixture was stirred at 0°C for 1.5 hours, and further stirred at approximately room
temperature for 40 minutes. Water (15 ml) and ethyl acetate (5 ml) were added thereto, and the mixture was stirred at 45 to 55°C for 1 hour. The mixture was cooled to room temperature, and the precipitated crystals were collected by filtration. The precipitated crystals were subsequently washed with methanol (30 ml) and water (40 ml) . Methanol (100 ml) was added to the resulting
crystals, followed by stirring under reflux for 30 minutes. The mixture was cooled to room temperature. The crystals were then collected by filtration and washed with methanol (30 ml) . The resulting crystals were dried under reduced pressure, obtaining 9.3 g of the objective product (yield: 73%) .
Optical purity: 99.4%ee.
……………….
Synthesis and antituberculosis activity of a novel series of optically active 6-nitro-2,3-dihydroimidazo[2,1-b]oxazoles
J Med Chem 2006, 49(26): 7854
http://pubs.acs.org/doi/abs/10.1021/jm060957y
(R)-2-Methyl-6-nitro-2-{4-[4-(4-trifluoromethoxyphenoxy)piperidin-1-yl]phenoxymethyl}-2,3-dihydroimidazo[2,1-b]oxazole (19, DELAMANID).
To a mixture of 27 (127.56 g, 586.56 mmol) and 4-[4-(4-trifluoromethoxyphenoxy)piperidin-1-yl]phenol (28g) (165.70 g, 468.95 mmol) in N,N-dimethylformamide (1600 mL) was added 60% sodium hydride (22.51 g, 562.74 mmol) at 0 °C portionwise. After the mixture was stirred at 50 °C for 2 h under a nitrogen atmosphere, the reaction mixture was cooled in an ice bath and carefully quenched with ethyl acetate (230 mL) and ice water (50 mL). The thus-obtained mixture was poured into water (3000 mL) and stirred for 30 min. The resulting precipitates were collected by filtration, washed with water, and dried at 60 °C overnight. This crude product was purified by silica gel column chromatography using a dichloromethane and ethyl acetate mixture (5/1) as solvent. The appropriate fractions were combined and evaporated under reduced pressure. The residue was recrystallized from ethyl acetate (1300 mL)−isopropyl alcohol (150 mL) to afford 19 (119.11 g, 48%) as a pale yellow crystalline powder.
Mp 195−196 °C.
1H NMR (CDCl3) δ 1.77 (3H, s), 1.87−2.16 (4H, m), 2.95−3.05 (2H, m), 3.32−3.41 (2H, m), 4.02 (1H, d, J = 10.2 Hz), 4.04 (1H, d, J = 10.2 Hz), 4.18 (1H, J = 10.2 Hz), 4.36−4.45 (1H, m), 4.49 (1H, d, J = 10.2 Hz), 6.76 (2H, d, J = 6.7 Hz), 6.87−6.94 (4H, m), 7.14 (2H, d, J = 8.6 Hz), 7.55 (1H, s).
[α  −9.9° (c 1.01, CHCl3).
−9.9° (c 1.01, CHCl3).
MS (DI) m/z 535 (M+ + 1). Anal. (C25H25F3N4O6) C, H, N.
http://pubs.acs.org/doi/suppl/10.1021/jm060957y/suppl_file/jm060957ysi20061113_095044.pdf
References
- Matsumoto, M.; Hashizume, H.; Tomishige, T.; Kawasaki, M.; Tsubouchi, H.; Sasaki, H.; Shimokawa, Y.; Komatsu, M. (2006). “OPC-67683, a Nitro-Dihydro-Imidazooxazole Derivative with Promising Action against Tuberculosis in Vitro and in Mice”. PLoS Medicine 3 (11): e466.doi:10.1371/journal.pmed.0030466. PMC 1664607. PMID 17132069.
- Skripconoka, V.; Danilovits, M.; Pehme, L.; Tomson, T.; Skenders, G.; Kummik, T.; Cirule, A.; Leimane, V.; Kurve, A.; Levina, K.; Geiter, L. J.; Manissero, D.; Wells, C. D. (2012). “Delamanid Improves Outcomes and Reduces Mortality for Multidrug-Resistant Tuberculosis”. European Respiratory Journal41 (6): 1393–1400. doi:10.1183/09031936.00125812. PMC 3669462. PMID 23018916.
- H. Spreitzer (18 February 2013). “Neue Wirkstoffe – Bedaquilin und Delamanid”. Österreichische Apothekerzeitung (in German) (4/2013): 22.
- Gler, M. T.; Skripconoka, V.; Sanchez-Garavito, E.; Xiao, H.; Cabrera-Rivero, J. L.; Vargas-Vasquez, D. E.; Gao, M.; Awad, M.; Park, S. K.; Shim, T. S.; Suh, G. Y.; Danilovits, M.; Ogata, H.; Kurve, A.; Chang, J.; Suzuki, K.; Tupasi, T.; Koh, W. J.; Seaworth, B.; Geiter, L. J.; Wells, C. D. (2012). “Delamanid for Multidrug-Resistant Pulmonary Tuberculosis”. New England Journal of Medicine 366 (23): 2151–2160. doi:10.1056/NEJMoa1112433.PMID 22670901.
- Drug Discovery & Development. EMA Recommends Two New Tuberculosis Treatments. November 22, 2013.
- Synthesis and antituberculous activity of a novel series of optically active 6-nitro-2,3-dihydroimidazo[2,1-b]oxazoles
45th Intersci Conf Antimicrob Agents Chemother (ICAAC) (December 16-19, Washington DC) 2005, Abst F-1473
|
12-28-2006
|
Synthesis and antituberculosis activity of a novel series of optically active 6-nitro-2,3-dihydroimidazo[2,1-b]oxazoles.
|
Journal of medicinal chemistry
|
|
|
11-1-2006
|
OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice.
|
PLoS medicine
|
|
1-1-2008
|
New anti-tuberculosis drugs with novel mechanisms of action.
|
Current medicinal chemistry
|
|
11-11-2010
|
Synthesis and Structure-Activity Relationships of Aza- and Diazabiphenyl Analogues of the Antitubercular Drug (6S)-2-Nitro-6-{[4-(trifluoromethoxy)benzyl]oxy}-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (PA-824).
|
Journal of medicinal chemistry
|
|
5-1-2012
|
Tuberculosis: the drug development pipeline at a glance.
|
European journal of medicinal chemistry
|
|
|
1-12-2012
|
Structure-activity relationships for amide-, carbamate-, and urea-linked analogues of the tuberculosis drug (6S)-2-nitro-6-{[4-(trifluoromethoxy)benzyl]oxy}-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (PA-824).
|
Journal of medicinal chemistry
|
|
9-11-2009
|
Pharmaceutical Composition Achieving Excellent Absorbency of Pharmacologically Active Substance
|
|
|
1-16-2009
|
Sulfonamide Derivatives for the Treatment of Bacterial Infections
|
| WO2004033463A1 | Oct 10, 2003 | Apr 22, 2004 | Otsuka Pharma Co Ltd | 2,3-DIHYDRO-6-NITROIMIDAZO[2,1-b]OXAZOLES |
| WO2004035547A1 | Oct 14, 2003 | Apr 29, 2004 | Otsuka Pharma Co Ltd | 1-substituted 4-nitroimidazole compound and process for producing the same |
| WO2008140090A1 | May 7, 2008 | Nov 20, 2008 | Otsuka Pharma Co Ltd | Epoxy compound and method for manufacturing the same |
| JP2009269859A * | Title not available |
TB

It is estimated that a third of the world’s population is currently infected with tuberculosis, leading to 1.6 million deaths annually. The current drug regimen is 40 years old and takes 6-9 months to administer. In addition, the emergence of drug resistant strains and HIV co-infection mean that there is an urgent need for new anti-tuberculosis drugs. The twenty-first century has seen a revival in research and development activity in this area, with several new drug candidates entering clinical trials. This review considers new potential first-line anti-tuberculosis drug candidates, in particular those with novel mechanisms of action, as these are most likely to prove effective against resistant strains.
From among acid-fast bacteria, human Mycobacterium tuberculosis has been widely known. It is said that the one-third of the human population is infected with this bacterium. In addition to the human Mycobacterium tuberculosis, Mycobacterium africanum and Mycobacterium bovis have also been known to belong to the Mycobacterium tuberoculosis group. These bacteria are known as Mycobacteria having a strong pathogenicity to humans.
Against these tuberculoses, treatment is carried out using three agents, rifampicin, isoniazid, and ethambutol (or streptomycin) that are regarded as first-line agents, or using four agents such as the above three agents and pyrazinamide.
However, since the treatment of tuberculosis requires extremely long-term administration of agents, it might result in poor compliance, and the treatment often ends in failure.
Moreover, in respect of the above agents, it has been reported that: rifampicin causes hepatopathy, flu syndrome, drug allergy, and its concomitant administration with other drugs is contraindicated due to P450-associated enzyme induction; that isoniazid causes peripheral nervous system disorder and induces serious hepatopathy when used in combination with rifampicin; that ethambutol brings on failure of eyesight due to optic nerve disorder; that streptomycin brings on diminution of the hearing faculty due to the 8th cranial nerve disorder; and that pyrazinamide causes adverse reactions such a hepatopathy, gouty attack associated with increase of uric acid level, vomiting (A Clinician’s Guide To Tuberculosis, Michael D. Iseman 2000 by Lippincott Williams & Wilkins, printed in the USA, ISBN 0-7817-1749-3, Tuberculosis, 2nd edition, Fumiyuki Kuze and Takahide Izumi, Igaku-Shoin Ltd., 1992).
Actually, it has been reported that cases where the standard chemotherapy could not be carried out due to the adverse reactions to these agents made up 70% (approximately 23%, 52 cases) of the total cases where administration of the agents was discontinued (the total 228 hospitalized patients who were subject to the research) (Kekkaku, Vol. 74, 77-82, 1999).
In particular, hepatotoxicity, which is induced by rifampicin, isoniazid, and ethambutol out of the 5 agents used in combination for the aforementioned first-line treatment, is known as an adverse reaction that is developed most frequently. At the same time, Mycobacterium tuberculosis resistant to antitubercular agents, multi-drug-resistant Mycobacterium tuberculosis, and the like have been increasing, and the presence of these types of Mycobacterium tuberculosismakes the treatment more difficult.
According to the investigation made by WHO (1996 to 1999), the proportion ofMycobacterium tuberculosis that is resistant to any of the existing antitubercular agents to the total types of Mycobacterium tuberculosis that have been isolated over the world reaches 19%, and it has been published that the proportion of multi-drug-resistant Mycobacterium tuberculosis is 5.1%. The number of carriers infected with such multi-drug-resistant Mycobacterium tuberculosis is estimated to be 60,000,000, and concerns are still rising that multi-drug-resistantMycobacterium tuberculosis will increase in the future (April 2001 as a supplement to the journal Tuberculosis, the “Scientific Blueprint for TB Drug Development.”)
In addition, the major cause of death of AIDS patients is tuberculosis. It has been reported that the number of humans suffering from both tuberculosis and HIV reaches 10,700,000 at the time of year 1997 (Global Alliance for TB drug development). Moreover, it is considered that the mixed infection of tuberculosisand HIV has an at least 30 times higher risk of developing tuberculosis than the ordinary circumstances.
Taking into consideration the aforementioned current situation, the profiles of the desired antitubercular agent is as follows: (1) an agent, which is effective even for multi-drug-resistant Mycobacterium tuberculosis, (2) an agent enabling a short-term chemotherapy, (3) an agent with fewer adverse reactions, (4) an agent showing an efficacy to latent infecting Mycobacterium tuberculosis (i.e., latentMycobacterium tuberculosis), and (5) an orally administrable agent.
Examples of bacteria known to have a pathogenicity to humans include offending bacteria of recently increasing MAC infection (Mycobacterium avium—intracellulare complex infection) such as Mycobacterium avium andMycobacterium intracellulare, and atypical acid-fast bacteria such asMycobacterium kansasii, Mycobacterium marinum, Mycobacterium simiae, Mycobacterium scrofulaceum, Mycobacterium szulgai, Mycobacterium xenopi, Mycobacterium malmoense, Mycobacterium haemophilum, Mycobacterium ulcerans, Mycobacterium shimoidei, Mycobacterium fortuitum, Mycobacterium chelonae, Mycobacterium smegmatis, and Mycobacterium aurum.
Nowadays, there are few therapeutic agents effective for these atypical acid-fast bacterial infections. Under the presence circumstances, antitubercular agents such as rifampicin, isoniazid, ethambutol, streptomycin and kanamycin, a newquinolone agent that is a therapeutic agent for common bacterial infections, macrolide antibiotics, aminoglycoside antibiotics, and tetracycline antibiotics are used in combination.
However, when compared with the treatment of common bacterial infections, the treatment of atypical acid-fast bacterial infections requires a long-term administration-of agents, and there have been reported cases where the infection is changed to an intractable one, finally leading to death. To break the afore-mentioned current situation, the development of an agent having a stronger efficacy is desired.
For example, National Publication of International Patent Application No. 11-508270 (WO97/01562) discloses that a 6-nitro-1,2,3,4-tetrahydro[2,1-b]-imidazopyran compound has a bactericidal action in vitro to Mycobacterium tuberculosis (H37Rv strain) and multi-drug-resistant Mycobacterium tuberculosis, and that the above compound has a therapeutic effect to a tuberculosis-infected animal model when it is orally administered and thus useful as antitubercular agent.
Balsam for the bones: Chemists develop a nanopaste for the repair of bone defects
Following accidents or cancer surgery surgeons often have to transplant healthy bone tissue or synthetic material to repair the resulting bone defects. Unfortunately, these procedures do not always have the desired effect.
Now a professor for inorganic chemistry, Matthias Epple was attracted to the interface between biology and medical science. “We have been investigating the impact of mineral tissue such as teeth, bone and sea shells for many years and are now using the knowledge we have gained to produce new biomaterials.” To achieve this he has collaborated closely with medical scientists and his current project – carried out with three of his doctoral students – was no exception.
“The repair of bone defects presents a real challenge for surgeons,” he relates. “When possible they collect the patient’s own bone from various locations, such as the iliac crest, and implant it where needed to fill defects.” The researcher explained…
View original post 267 more words
Zabofloxacin
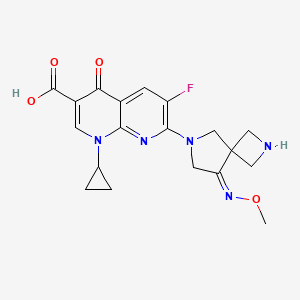
zabofloxacin, 219680-11-2
UNII-LV66BA6V2G, DW-224a
Molecular Formula: C19H20FN5O4
Molecular Weight: 401.391603
DONG WHA PHARMA SOUTH KOREA in phase 3
1-Cyclopropyl-6-fluoro-7-[8-(methoxyimino)-2,6-diazaspiro[3.4]oct-6-yl]-4-oxo-1,4-dihydro-1,8-naphthyridine-3-carboxylic acid
Zabofloxacin is being developed as a new fluoroquinolone antibiotic that is a potent and selective inhibitor of the essential bacterial type II topoisomerases and topoisomerase IV. Zabofloxacin is indicated for community-acquired respiratory infections due to Gram-positive bacteria. The aim of this study was to compare the pharmacokinetics (PK) of the zabofloxacin hydrochloride 400 mg capsule (DW224a, 366.7 mg aszabofloxacin) with the PK of the zabofloxacin aspartate 488 mg tablet (DW224aa, 366.5 mg as zabofloxacin) in healthy Korean male volunteers to assess the bioequivalence between the two drug formulations
Zabofloxacin hydrochloride is a fluoroquinolone antibiotic with enhanced in vitro activity against Streptococcus pneumoniae, including strains resistant to other antibiotics. The spectrum of activity of zabofloxacin includes bacterial strains that are responsible for most community-acquired respiratory infections. Phase III clinical studies are currently ongoing at Dong-Wha for the treatment of patients with acute bacterial exacerbation of chronic obstructive pulmonary disease. Phase II trials had been ongoing at IASO; however no recent developments have been reported.The product candidate was originated by Dong Wha. In 2007, Dong Wha granted PB BioSciences worldwide exclusive development and marketing rights, except in Japan, Korea, China, Taiwan, Singapore, Indonesia, India, Thailand, Malaysia, Vietnam, Hong Kong, Australia and New Zealand.
Zabofloxacin was separated using an isocratic elution on a Capcell Pak C18 column using an acetonitrile–methanol–phosphate buffer (1 g of KH2PO4 and 1 g of heptane sulfonic acid sodium salt in 720 mL of purified water) and a 1 M tetrabutylammonium dihydrogenphosphate solution (18.5:8.5:72:1, by volume) as a mobile phase at a flow rate of 0.25 mL/min with UV detection at 275 nm. The lower limit of quantification (LLOQ) and the upper limit of quantification (ULOQ) were 100 ng/mL and 20000 ng/mL, respectively, with acceptable linearity in the range from 100 to 20000 ng/mL (R > 0.999). The intra- and inter-day accuracy (RE) ranged from −8.2% to 1.8% and the intra- and inter-day precision (CV) ranged from 3.8% to 10.6% for zabofloxacin. In addition, stock solution stability, recovery, freeze–thaw effects, and short-term and long-term stability met the acceptance criteria.
…………………………
Example 1. l-Cyclopropyl-6-fluoro-7-[8-(methoxyimino)-2,6-diazaspiro[3,4]oct-6-yl]-4- oxo-l,4-dihydro[l,8]naphthyridine-3-carboxylic acid methanesulfonate
30 350mg of
7-[2-(t-buthoxycarbonyl)-8-(methoxyimino)-2,6-diazaspiro[3.4]oct-6-yl]-l- cyclopropyl-6-fluoro-4-oxo-l,4-dihydro[l,8]naphthyridine-3-carboxylic acid was dissolved in 5ml of dichloromethane and thereto 0.6ml of trifluoroacetic acid was dropped. The mixture was stirred for 5 hours at room temperature and thereto 10ml (if ethylether was added. It was stirred additionally for 1 hour and thus precipitated solid was filtered, dissolved in 5ml of diluted NaOH and neutralized with diluted hydrochloric acid. The precipitate thus obtained was filtered and dried. The resulting solid was added to 5ml of lN-methanesulfonic acid in ethanol and stirred for 1 hour. Thus obtained precipitate was filtered and dried to give 185g of the titled compound(yield : 47.8%). m.p. : 228- 229 °C
1H-NMR(DMSO-dG+CF3COOD, ppm): 0.97(s, 2H), 1.14(d, 2H), 2.48(s, 3H), 3.57(bs, IH), 3.88(s, 3H), 4.06-4.17(m, 411), 4.40(s, 2H), 4.49(s, 2H), 7.88(d, Hi, J=12.67Hz), 8.49(s, IH).
………………………………..
aspartate of 1-cyclopropyl-6-fluoro-7-(8-methoxyimino-2,6-diaza-spiro[3.4]oct-6-yl)-4-oxo-1,4-dihydro-[1,8]naphthyridine-3-carboxylic acid comprises a step of reacting 1-cyclopropyl-6-fluoro-7-(8-methoxyimino-2,6-diaza-spiro[3.4]oct-6-yl)-4-oxo-1,4-dihydro-[1,8]naphthyridine-3-carboxylic acid with aspartic acid in a solvent. The method can be represented by Scheme 1.

Example 1 Preparation of the D-Aspartic Acid Salt of 1-cyclopropyl-6-fluoro-7-(8-methoxyimino-2,6-diaza-spiro[3.4]oct-6-yl)-4-oxo-1,4-dihydro-[1,8]naphthyridine-3-carboxylic acid
1-Cyclopropyl-6-fluoro-7-(8-methoxyimino-2,6-diaza-spiro[3.4]oct-6-yl)-4-oxo-1,4-dihydro-[1,8]naphthyridine-3-carboxylic acid (5.0 g) was added to 50% ethanol (80 mL), and then the mixture was stirred at 50° C. for 10 minutes. D-Aspartic acid (2.0 g) was added and then the mixture was stirred at 50° C. for 1 hour. The mixture was cooled to room temperature, and then the resulting solid was collected by filtration. Ethanol (100 mL) was added to the filtrate, and then the mixture was stirred for 30 minutes. The resulting solid was collected by filtration to obtain a total of 5.55 g of the target compound (yield: 83%). Melting point: 200-201° C. 1H NMR (D2O): δ 0.97 (bs, 2H), 1.27 (d, 2H), 2.00 (dd, 1H, J=8.8, 17.6 Hz), 2.77 (dd, 1H, J=3.3, 17.0 Hz), 3.53 (bs, 1H), 3.84 (dd, 1H, J=3.3, 8.78 Hz), 4.01 (s, 3H), 4.31-4.45 (m, 8H), 7.46 (d, 1H, J=12.2 Hz), 8.42 (s, 1H).
Example 2 Preparation of L-Aspartic Acid Salt of 1-cyclopropyl-6-fluoro-7-(8-methoxyimino-2,6-diaza-spiro[3.4]oct-6-yl)-4-oxo-1,4-dihydro-[1,8]naphthyridine-3-carboxylic acid
1-Cyclopropyl-6-fluoro-7-(8-methoxyimino-2,6-diaza-spiro[3.4]oct-6-yl)-4-oxo-1,4-dihydro-[1,8]naphthyridine-3-carboxylic acid (500 mg) was added to 50% ethanol (20 mL), and then the mixture was stirred at 50° C. for 10 minutes. L-Aspartic acid (174 mg) was added and then the mixture was stirred at 50° C. for 1 hour. The mixture was cooled to room temperature. Ethanol (20 mL) was added to the reaction mixture, and then the mixture was stirred for 30 minutes. The resulting solid was collected by filtration to obtain 550 mg of the target compound (yield: 82%). Melting point: 205-206° C. 1H NMR (d6-DMSO): δ 0.93 (d, 2H, J=3.5 Hz), 1.20 (d, 2H, J=6.8 Hz), 2.42 (dd, 1H, J=9.2, 17.3 Hz), 2.59 (dd, 1H, J=3.3, 17.2 Hz), 3.50 (m, 1H), 3.59 (1H, dd, J=3.1, 9.1 Hz), 3.91 (s, 3H), 4.24 (m, 6H), 4.41 (br, 2H), 7.59 (d, 1H, J=12.4 Hz), 8.41 (s, 1H).
Example 3 Preparation of Hydrochloric Acid Salt, Phosphate Salt, and Formate Salt of 1-cyclopropyl-6-fluoro-7-(8-methoxyimino-2,6-diaza-spiro[3.4]oct-6-yl)-4-oxo-1,4-dihydro-[1,8]naphthyridine-3-carboxylic acid
3-1 Hydrochloric Acid Salt
Ethanol (3 mL) was cooled to 0° C. and acetyl chloride (1.13 mL) was added, and then the mixture was stirred for 30 minutes. 1-Cyclopropyl-6-fluoro-7-(8-methoxyimino-2,6-diaza-spiro[3.4]oct-6-yl)-4-oxo-1,4-dihydro-[1,8]naphthyridine-3-carboxylic acid (800 mg) was added to the reaction mixture, and then stirred at 0° C. for 30 minutes. Tetrahydrofuran (4 mL) was added, and then the mixture was stirred for 30 minutes. The resulting solid was collected by filtration and dried to obtain 776 mg of the target compound (yield: 89%). Melting point: 244-245° C. 1H NMR (d6-DMSO): δ 1.07 (d, 2H, J=4.7 Hz), 1.21 (d, 2H, J=6.8 Hz), 3.68 (m, 1H), 3.94 (s, 3H), 4.17 (m, 2H), 4.40 (s, 2H), 4.53 (s, 2H), 8.03 (d, 1H, J=12.5 Hz), 8.59 (s, 1H).
ref
Han J, Kim JC, Chung MK, Kim B, Choi DR.
Biol Pharm Bull. 2003 Jun;26(6):832-9
Jin HE, Kang IH, Shim CK.
J Pharm Pharm Sci. 2011;14(3):291-305.
Jin HE, Lee KR, Kang IH, Chung SJ, Shim CK.
J Pharm Biomed Anal. 2011 Mar 25;54(4):873-7. doi: 10.1016/j.jpba.2010.11.001. Epub 2010 Nov 9.
Kosowska-Shick, K.; Credito, K.; Pankuch, G.A.; Lin, G.; Bozdogan, B.; McGhee, P.; Dewasse, B.;
Choi, D.-R.; Ryu, J.M.; Appelbaum, P.C. Antipneumococcal activity of DW-224a, a new
quinolone, compared to those of eight other agents. Antimicrob. Agents Chemother. 2006, 50,
2064–2071.
Park, H.-S.; Kim, H.-J.; Seol, M.-J.; Choi, D.-R.; Choi, E.-C.; Kwak, J.-H. In vitro and in vivo
antibacterial activities of DW-224a, a new fluoronaphthyridone. Antimicrob. Agents Chemother.
2006, 50, 2261–2264.
Dong Wha Pharmaceutical Co. Ltd. A study to evaluate efficacy and safety profile of
Zabofloxacin tablet 400 mg and moxifloxacin tablet 400 mg. Available online:
http://www.clinicaltrials.gov/ct2/show/NCT01658020 (accessed on 15 July 2013).
Dong Wha Pharmaceutical Co. Ltd. A new quinolone antibiotic. Available online:
http://www.dong-wha.co.kr/english/rnd/rnd02_03.asp (accessed on 15 April 2013).
| US4957922 * | Mar 29, 1989 | Sep 18, 1990 | Bayer Aktiengesellschaft | Infusion solutions of 1-cyclopropyl-6-fluoro-1,4-di-hydro-4-oxo-7-(1-piperazinyl)-quinoline-3-carboxylic acid |
| US5563149 * | Aug 8, 1994 | Oct 8, 1996 | Cheil Foods & Chemicals, Inc. | Aqueous solutions of pyridone carboxylic acids |
| US6552196 * | Sep 6, 2001 | Apr 22, 2003 | Dong Wha Pharmaceutical Industrial Co., Ltd. | Quinolone carboxylic acid derivatives |
|
7-23-2010
|
ASPARTATE OF 1-CYCLOPROPYL-6-FLUORO-7-(8-METHOXYIMINO-2,6-DIAZA-SPIRO[3.4]OCT-6-YL)-4-OXO-1,4-DIHYDRO-[1,8]NAPHTHYRIDINE-3-CARBOXYLIC ACID, METHOD FOR PREPARING THE SAME, AND ANTIMICROBIAL PHARMACEUTICAL COMPOSITION COMPRISING THE SAME
|
Green…Asymmetric hydrogentation of unfunctionalised olefins/enamines/imines
Asymmetric hydrogentation of unfunctionalised olefins/enamines/imines
The reaction survey found that the predominant strategy for the introduction of chirality was through classical chemical resolutions as opposed to introductions through biotransformation or transition metal or organometallic catalytic means.
Asymmetric hydrogenation provides an elegant methodology for the introduction of chirality, meeting many of the goals of green chemistry and is finding increasing application in API synthesis.47
The efficiency of this approach is elegantly exemplified by the Merck second generation synthesis of sitagliptin 5 (Scheme ), where an unprecedented final stage asymmetric hydrogenation of the unprotected enamide 6 resulted in an increase in overall yield of almost 50% and produced 100 kg less waste per kg sitagliptin48 when compared with the first generation approach.49
 |
||
| Scheme The synthesis of sitagliptin. | ||
There are challenging areas remaining within the field, for example, the hydrogenation of enamides and related substrates in the synthesis of amino acids has numerous examples50 but few examples exist for unsubstitued enamines41 and imines. Some classes of alkene offer additional challenges.51 For the pharmaceutical industry, the limited time for synthetic route identification is an issue and access to catalyst and ligand diversity is required to ensure the application of this approach.52
Some pharmaceutical companies have synthesised their own ligands and have found very effective catalysts.53 The majority of academic asymmetric hydrogenation approaches are based on homogeneous catalysis to overcome issues of activation and mass transfer. For pharmaceutical use, efficient catalyst and ligand recovery, and eliminating heavy metal contamination of the API are significant requirements for the industry.
These controls are often easier to achieve with heterogeneous methodology where there are less examples.50 The demonstration of organocatalytic hydride transfer offers the possibility of future access to metal free asymmetric hydrogenations.54
- 47………V. Farina, J. T. Reeves, C. H. Senanayake and J. J. Song, Chem. Rev., 2006, 106, 2734–2793. See also Asymmetric Catalysis on Industrial Scale Challenges, Approaches and Solutions, ed. H.-U. Blaser and E. Schmidt, Wiley-VCH, Weinheim, 2004 Search PubMed .
- 48………..http://www.epa.gov/greenchemistry/pubs/pgcc/winners/gspa06.html .
- 49……K. B. Hansen, J. Balsells, S. Dreher, Y. Hsiao, M. Kubryk, M. Palucki, N. Rivera, D. Steinhuebel, J. D. Armstrong III, D. Askin and E. J. J. Grabowski, Org. Process Res. Dev., 2005, 9, 634–639 Search PubMed .
- 50………..M. Studer, H.-U. Blaser and C. Exner, Adv. Synth. Catal., 2003, 345, 45–65 CrossRef CAS Search PubMed .
- 51……..X. Cui and K. Burgess, Chem. Rev., 2005, 105, 3272–3296 CrossRef CAS Search PubMed
- 52……….I. C. Lennon and C. J. Pilkington, Synthesis, 2003, 1639–1642 CrossRef CAS Search PubMed .
- 53………G. Hoge, H.-P. Wu, W. S. Kissel, D. A. Plum, D. J. Greene and J. Bao, J. Am. Chem. Soc., 2004, 126, 5966–5967 CrossRef CAS Search PubMed .
- 54……..H. Adolfsson, Angew. Chem., Int. Ed., 2005, 44, 3340–3342 CrossRef CAS Search PubMed .
Garcinia Cambogia Kills 89% of Pancreatic Cancer Cells and Synergizes with Curcumin

Garcinia Cambogia Kills 89% of Pancreatic Cancer Cells & Synergizes with Curcumin: Garcinol, a compound found in the famous weight-loss fruit garcinia cambogia, was shown to reduce viability of human pancreatic cancer cells (Panc-1) by up to 89% in vitro.
This garcinia extract even beat out the super-herb curcumin, which killed only 63% of these cells. However, when garcinol and curcumin were combined, their effectiveness at killing the pancreatic cancer cells increased up to 10-fold at lower doses!
This means they may be effective at dose levels which are more easily achieved in cancer patients. Garcinol has also shown strong activity against breast cancer, lung cancer, head and neck cancer, colon cancer and leukemia. Garcinol, a powerful antioxidant, is also found in other fruit of the garcinia genus such as mangosteen.
Be aware, however, that many garcinia cambogia weight-loss supplements only concentrate hydroxycitric acid and leave out the garcinol, so as usual getting the whole food (or juice) brings the most benefit.
Remember that a healthy, balanced diet centred on organic vegetables, fruit, whole foods, spices and herbs is of central importance to good health. But unique fruits such as garcinia cambogia might make excellent additions to get that extra level of protection.http://www.ncbi.nlm.nih.gov/pubmed/22685460
Garcinia Cambogia Extract Explained
The latest in innovation in weight loss supplements is Garcinia Cambogia. It is unparalleled in its ability to help boost your body’s weight loss potential, and help you achieve your perfect weight.
There’s no wonder it’s quickly gained a huge following, with endorsements from celebrities to health experts, with scientifically proven ability to help you increase your fat burning power.
As with all supplements like this, there are questions as to how it works, and just how it can benefit you, with your health and in losing weight. This site’s goal is to hopefully answer some of these questions, and to show you just how you can benefit from this amazing supplement.

What is Garcinia Cambogia?
Garcinia Cambogia is a fruit, that is grown all over Asia, but originating in Indonesia and grows particularly well grows best with tropical conditions. It rose to prominence after appearance on the massively lauded American health show, Doctor Oz. It had recently been subject to a medical trial where the study scientifically proved it was highly effective in increasing burning up fat and aiding in overall weight loss.
Can Garcinia Cambogia Extract Help Me Lose Weight?
Well Garcinia Cambogia contains a useful compound called Hydroxycitric Acid, which I’ll refer to as HCA for ease of reference. Garcinia Cambogia contains one of the highest known concentrations of HCA, and this was why it was noticed as a potential weight loss supplement. HCA has two main mechanisms in which it works to boost your fat burning potential:

Firstly it will reduce the ability for the body to convert carbohydrates into fat cells, meaning that even without a calorific controlled diet; you will be able to aid your body’s ability to burn of existing fat, while not gaining additional fat.
Secondly it will also suppress your appetite, meaning that it will not only help reduce the weight you can put on by stopping putting on additional fat, it will also massively reduce the cravings and hunger that usually lead to breaking a diet and weight loss routine. This means that your body will just be burning off the existing fat, helping you to achieve that perfect weight!
What About Side Effects form Garcinia Cambogia?
The most amazing thing about Garcinia Cambogia is that the side effects of the product are almost non-existent in the all-natural extract. By this I mean an extract that contains purely Garcinia Cambogia extract without any additional additives that some unrepeatable sellers will try to pass off as the quality product. Those extracts that contain additives can cause side effect in users of Garcinia Cambogia, which are related to the different additives and binding agents added.
The cost of Garcinia Cambogia from a supplier, whom ensures a high quality and natural product, will range from $40-50 a bottle. There is however introductory offers from some suppliers, such as Miracle Garcinia Cambogia currently offering a free bottle of Garcinia Cambogia with every order.
This means the overall cost per bottle of this amazing product can drop as low as $28.99. Most of these offers unfortunately do have a limited stock and therefore won’t be around forever.
Garcinia gummi-gutta is a tropical[2] species of Garcinia native to Indonesia. Common names include garcinia cambogia (a former scientific name), as well as gambooge, brindleberry,[3] brindall berry, Malabar tamarind,[2] assam fruit, vadakkan puli (northern tamarind) and kudam puli (pot tamarind).[4] This fruit looks like a small pumpkin and is green to pale yellow in color. It has recently received considerable media attention because of its purported effects on weight loss, although there is no clinical evidence to support this claim.

Cultivation


Ripe fruit
Garcinia gummi-gutta is grown for its fruit in southeast Asia, coastal Karnataka/Kerala, India, and west and central Africa. It thrives in most moist forests.
Garcinia gummi-gutta is one of several closely related Garcinia species from the plant family Guttiferae.[5] With thin skin and deep vertical lobes, the fruit of G. gummi-gutta and related species range from about the size of an orange to that of a grapefruit; G. gummi-gutta looks more like a small yellowish, greenish or sometimes reddish pumpkin.[6] The color can vary considerably. When the rinds are dried and cured in preparation for storage and extraction, they are dark brown or black in color.
Along the west coast of South India, G. gummi-gutta is popularly termed “Malabar tamarind,” and shares culinary uses with the tamarind (Tamarindus indica). The latter is a small and the former a quite large evergreen tree. G. gummi-gutta is also called “goraka” or, in some areas, simply “kattcha puli” (souring fruit).
Uses
Cooking
Garcinia gummi-gutta is used in cooking, including in the preparation of curries. The fruit rind and extracts of Garcinia species are called for in many traditional recipes,[7] and various species of Garcinia are used similarly in food preparation in Assam (India), Thailand, Malaysia, Burma and other Southeast Asian countries. In the Indian Ayurvedic medicine, “sour” flavors are said to activate digestion. The extract and rind of Garcinia gummi-gutta is a curry condiment in India. It is an essential souring ingredient in the Southern Thai variant of kaeng som, a sour curry.
Garcinia gummi-gutta is employed commercially in fish curing, especially in Sri Lanka (Colombo curing) and South India, which makes use of the antibacterial qualities of the fruit.
The trees can be found in forested areas and also are protected in plantations otherwise given over to pepper, spice, and coffee production.
Traditional medicine
Aside from its use in food preparation and preservation, extracts of G. gummi-gutta are sometimes used in traditional medicine aspurgatives. The fruit rind is also used to make medicine.
Weight loss
In late 2012, a United States television personality, Dr. Oz, promoted Garcinia cambogia extract as a “magic” weight-loss aid. Dr. Oz’s previous endorsements have often led to a substantial increase in consumer interest in the promoted products. However, a dearth of scientific evidence and clinical trials do not support claims that Garcinia cambogia is an effective weight-loss aid.[8][9] A meta-analysis found a possible small, short-term weight loss effect (under 1 kilogram).[10] However, side effects—namely hepatotoxicity (chemical-driven liver damage)—led to one preparation being withdrawn from the market.[11][12]
A 1998 randomized controlled trial looked at the effects of hydroxycitric acid, the purported active component in Garcinia gummi-gutta, as a potential antiobesity agent in 135 people. The conclusion from this trial was that “Garcinia cambogia failed to produce significant weight loss and fat mass loss beyond that observed with placebo”.[13]

When the fruit is sun dried for several days, it becomes black with a shrivelled body
References
- “Garcinia gummi-gutta (L.) Roxb.”. The Plant List. Royal Botanic Gardens, Kew and Missouri Botanical Garden. Retrieved 1 June 2013.
- “USDA GRIN Taxonomy”.
- “Potential treatments for insulin resistance in the horse: A comparative multi-species review”. Science Direct. Retrieved 6 October 2013.
- “Meals that heal – Soul curry”. The Hindu. Retrieved 3 October 2013.
- Publications & Information Directorate, Council of Scientific & Industrial Research (1986). G. cambogia Desr. The Useful Plants of India. (New Delhi: Publications & Information Directorate, 1986) 229.
- “Fruit yellowish or reddish, size of an orange having six or eight deep longitudinal grooves in its fleshy pericarp. Pulp acid of a pleasant flavor. It is dried among the Singalese who use it in curries.” Uphof, J.C. Th. (1968).
- “The acid rinds of the ripe fruit are eaten, and in Ceylon are dried, and eaten as a condiment in curries.” Drury, Heber (1873). “Garcinia gambogia(Desrous) N. 0. Clusiaceae”. The Useful Plants of India, second edition. London: William H. Allen & Co. p. 220.
- Belluz, Julia; Hoffman, Steven J. (1 January 2013). “Dr. Oz’s Miraculous Medical Advice; Pay no attention to that man behind the curtain”. Slate. The Slate Group. Retrieved 31 May 2013.
- Márquez F1, Babio N, Bulló M, Salas-Salvadó J (2012). “Evaluation of the safety and efficacy of hydroxycitric acid or Garcinia cambogia extracts in humans”. Crit Rev Food Sci Nutr 52 (7): 585–94. doi:10.1080/10408398.2010.500551. PMID 22530711.
- Hepatotoxicity (from hepatic toxicity) implies driven liver damage.
- Lobb, A. (2009). “Hepatoxicity associated with weight-loss supplements: A case for better post-marketing surveillance”. World Journal of Gastroenterology 15 (14): 1786–1787. doi:10.3748/wjg.15.1786. PMC 2668789. PMID 19360927.
- Kim YJ1, Choi MS, Park YB, Kim SR, Lee MK, Jung UJ (2013). “Garcinia Cambogia attenuates diet-induced adiposity but exacerbates hepatic collagen accumulation and inflammation”. World J Gastroenterol 19 (29): 4689–701. doi:10.3748/wjg.v19.i29.4689. PMID 23922466.
- Heymsfield, S. B.; Allison, D. B.; Vasselli, J. R.; Pietrobelli, A.; Greenfield, D.; Nunez, C. (1998). “Garcinia cambogia (Hydroxycitric Acid) as a Potential Antiobesity Agent: A Randomized Controlled Trial”. JAMA: the Journal of the American Medical Association 280 (18): 1596–1600.doi:10.1001/jama.280.18.1596. PMID 9820262.
BERAPROST….Stable prostacyclin analog.

BERAPROST
https://www.ama-assn.org/resources/doc/usan/beraprost.pdf
2,3,3a,8b-tetrahydro-2-hydroxy-1-(3-hydroxy-4-methyl-1-octen-6-ynyl)-1H-cyclopenta(b)benzofuran-5-butanoic acid
(±)-(IR*,2R*,3aS*,8bS*)-2,3,3a,8b-tetrahydro-2-hydroxy-1-[(E)-(3S*)-3-hydroxy-4-methyl-1-octene-6-inyl]-1H-cyclopenta[b]benzofuran-5-butyric acid
rac-4-{(1R,2R,3aS,8bS)-2-hydroxy-1-[(1E,3S,4RS)-3-hydroxy-4-methyloct-1-en-6-ynyl]-2,3,3a,8b-tetrahydro-1H-cyclopenta[b][1]benzofuran-5-yl}butanoic acid
- Beraprost
- Beraprostum
- Beraprostum [INN-Latin]
- MDL 201229
- MDL-201229
- ML 1229
- ML-1229
- UNII-35E3NJJ4O6
Beraprostum, Beraprostum [INN-Latin], ML 1229, MDL 201229, 88430-50-6
Molecular Formula: C24H30O5
Molecular Weight: 398.492
CLINICAL TRIALS..http://clinicaltrials.gov/search/intervention=Beraprost
Beraprost is a synthetic analogue of prostacyclin, under clinical trials for the treatment of pulmonary hypertension. It is also being studied for use in avoiding reperfusion injury.
As an analogue of prostacyclin PGI2, beraprost effects vasodilation, which in turn lowers the blood pressure. Beraprost also inhibits plateletaggregation, though the role this phenomenon may play in relation to pulmonary hypertension has yet to be determined.
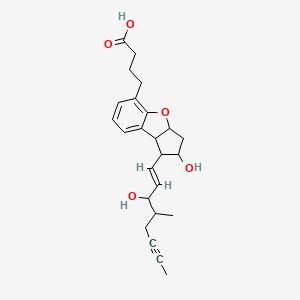
Beraprost …sodium salt
ML 1129; Procyclin; TRK 100 (CAS 88475-69-8)

Beraprost is an analog of prostacyclin in which the unstable enol-ether has been replaced by a benzofuran ether function. This modification increases the plasma half-life from 30 seconds to several hours, and permits the compound to be taken orally. Doses of 20-100 µg in humans, given 1 to 3 times per day, have been demonstrated to improve clinical end points in diseases responsive to prostacyclin. Oral beraprost therapy improved the survival and pulmonary hemodynamics of patients with primary pulmonary hypertension.1 Beraprost inhibits platelet aggregation in healthy subjects and in diabetic patients at similar doses.2,3
| Synonyms |
|
|---|---|
| Formal Name | 2,3,3a,8b-tetrahydro-2-hydroxy-1-(3-hydroxy-4-methyl-1-octen-6-ynyl)-1H-cyclopenta[b]benzofuran-5-butanoic acid, monosodium salt |
| CAS Number | 88475-69-8 |
| Molecular Formula | C24H29O5 · Na |
| Formula Weight | 420.5 |
- Beraprost sodium is a prostacyclin analog and an NOS3 expression enhancer that was first launched in 1992 in Japan pursuant to a collaboration between Astellas Pharma and Toray for the oral treatment of peripheral vascular disease (PVD), including Raynaud’s syndrome and Buerger’s disease. In 2000, the drug was commercialized for the treatment of pulmonary hypertension. Development for the oral treatment of intermittent claudication associated with arteriosclerosis obliterans (ASO) was discontinued at Kaken and United Therapeutics after the product failed to demonstrate statistically significant results in a phase III efficacy trial.
- In terms of clinical development, beraprost sodium is currently in phase II clinical trials at Kaken for the treatment of lumbar spinal canal stenosis and at Astellas Pharma for the oral treatment of primary chronic renal failure. The company is also conducting phase III trials for the treatment of nephrosclerosis. The drug has also been studied through phase II clinical trials at Kaken for the oral treatment of diabetic neuropathy, but recent progress reports for this indication have not been made available.
- Beraprost is an oral form of prostacyclin, a member of the family of lipid molecules known as eicosanoids. Prostacyclin is produced in the endothelial cells from prostaglandin H2 by the action of the enzyme prostacyclin synthase. It has been shown to keep blood vessels dilated and free of platelet aggregation.
- Beraprost sodium was originally developed at Toray in Japan, and rights to the drug were subsequently acquired by Astellas Pharma. A 1972 alliance between Toray and Kaken Pharmaceutical to develop and commercialize prostaglandin led to a later collaboration agreement for the development of beraprost. In 1990, Toray granted the right to market the drug to Sanofi (formerly known as sanofi-aventis), a licensing agreement that was later expanded to include Canada, the U.S., South America, Africa, Southeast Asia, South Asia, Korea and China. In September 1996, Bristol-Myers Squibb entered into separate agreements with Sanofi and Toray to acquire all development and marketing rights to beraprost in the U.S. and Canada. In January 1999, United Therapeutics and Toray agreed to cooperatively test the drug in North America, and in July 2000, a new agreement was signed pursuant to which United Therapeutics gained exclusive North American rights to develop and commercialize sustained-release formulations of beraprost for all vascular and cardiovascular diseases. In 1999, orphan drug designation was received in the U.S. for the treatment of pulmonary arterial hypertension associated with any New York Heart Association classification (Class I, II, III, or IV). In 2011, orphan drug designation was assigned in the U.S. for the treatment of pulmonary arterial hypertension.
-
The compound name of beraprost which is used as an antimetastasis agent of malignant tumors according to the present invention is (±)-(IR*,2R*,3aS*,8bS*)-2,3,3a,8b-tetrahydro-2-hydroxy-1-[(E)-(3S*)-3-hydroxy-4-methyl-1-octene-6-inyl]-1H-cyclopenta[b]benzofuran-5-butyric acid. This compound has the following structure.

Beraprost is described in Japanese Laid-open Patent Application (Kokai) Nos. 58-32277, 57-144276 and 58-124778 and the like as a PGI₂ derivative having a structure in which the exoenol moiety characteristic to beraprost is converted to inter-m-phenylene structure. However, it is not known that beraprost has an activity to inhibit metastasis of malignant tumors.
-
The beraprost which is an effective ingredient of the agent of the present invention includes not only racemic body, but also d-body and l-body. Beraprost can be produced by, for example, the method described in the above-mentioned Japanese Laid-open Patent Application (Kokai) No. 58-124778. The salts of beraprost include any pharmaceutically acceptable salts including alkaline metal salts such as sodium salt and potassium salt; alkaline earth metal salts such as magnesium salt and calcium salt; ammonium salt; primary, secondary and tertiary amine salts; and basic amino acid salts.

…………………..
EXAMPLE 6 Beraprost of the Formula (I)
0.246 g (0.6 mmol) of compound of the general formula (II) obtained in Example 5 is dissolved in 1 ml of methanol and 1 ml of 1 M aqueous sodium hydroxide solution is added dropwise slowly thereto. After stirring for an hour the methanol is distilled off from the reaction mixture in vacuum. The aqueous residue is diluted with 10 ml of water extracted with methyl-tert.butyl-ether and the combined organic phase is washed with saturated NaCl solution, dried on Na2SO4 and evaporated. The residue of evaporation is crystallized from ethylacetate-hexane mixture and the pure above mentioned title compound is obtained as colourless crystals.
Yield: 0.21 g (87%)
TLC-Rf (toluene-dioxan-acetic acid 20:10:1)=0.41
Melting point: 98–112° C.
1H NMR (400 MHz, CDCl3), δH (ppm): 1.00d, 1.03d [3H; J=6.8 Hz; 21-H3]; 1.79m [1H; 16-H]; 1.80t, 1.81t [3H, J=2.5,2.4 Hz; 20-H3]; 2.3–1.9m [5H, 3-H2, 10Hb, 17-H2]; 2.34t [1H; J=7.4 Hz; 2-H2]; 2.43m [1H; 12-H]; 2.64m [3H; 10-Ha, 4-H2]; 3.43t, 3.44t [1H, J=8.7,8.5 Hz; 8-H]; 3.92m [1H; 11-H]; 4.07t, 4.17t [1H, J=7.3,5.6 Hz; 15-H]; 4.3b [2H; OH]; 5.09m [1H, 9-H]; 5.58dd, 5.61dd [1H; J=15.3,6.5 Hz; 14-H]; 5.67dd, 5.68dd [1H; J=15.3,8.0 Hz; 13-H]; 6.77m [1H; 2′-H]; 6.95m [2H; 1′-H,3′-H]13C NMR (100 MHz, CDCl3), δC (ppm): 3.5, 3.6 [C-20]; 14.7, 15.8 [C-21]; 22.3, 22.6 [C-17]; 24.6 [C-2]; 29.1 [C-4]; 33.1 [C-3]; 38.2, 38.3 [C-16]; 41.2 [C-10]; 50.4 [C-8]; 58.8 [C-12]; 75.8, 76.3, 76.4 [C-11, C-15]; 77.2, 77.4 [C-18, C-19]; 84.5, 84.6 [C-9]; 120.6 [C-2′]; 121.9 [C-3′]; 123.2 [C-5]; 129.0 [C-1′]; 129.7 [C-7]; 132.3, 133.0, 133.8, 134.0 [C-13, C-14]; 157.2 [C-6]; 178.3 [C-1].
EXAMPLE 7 Beraprost Sodium Salt (The Sodium Salt of the Compound of Formula (I)
0.199 g of beraprost is dissolved in 2 ml of methanol, 0.5 ml of 1 M aqueous solution of sodium hydroxide is added thereto and after their mixing the solvent is evaporated in vacuum and thus the above title salt is obtained as colourless crystals.
Yield: 0.21 g (100%)
Melting point: >205° C.
1H NMR (400 MHz, DMSO-d6), δH (ppm): 0.90d, 0.92d [3H; J=6.7 Hz; 21-H3]; 1.75–1.55m [7H; 10Hb, 16-H, 3-H2, 20-H3]; 1.89t [2H, J=7.6 Hz; 2-H2]; 1.94m [1H; 17-Hb]; 2.16q [1H, J=8.5 Hz; 12-H]; 2.25m [1H; 17-Ha]; 2.44t [2H; J=7.5 Hz; 4-H2]; 2.50o [1H; 10-Ha]; 3.39t [1H, J=8.5 Hz; 8-H]; 3.72td [1H; J=8.5,6.1 Hz; 11-H]; 3.84t 3.96t [1H, J=6.5,6.0 Hz; 15-H]; 4.85b [2H, OH]; 5.01dt [1H, J=8.5,6.6 Hz; 9-H]; 5.46dd, 5.47dd [1H; J=15.4,6.5 Hz, J=15.4,6.0 Hz; 14-H]; 5.65dd, 5.66dd [1H; J=15.4,8.5 Hz; 13-H]; 6.71m [1H; 2′-H]; 6.92m [2H; 1′-H, 3′-H] During the above thin layer chromatography (TLC) procedures we used plates MERCK Kieselgel 60 F254, thickness of layer is 0.2 mm, length of plates is 5 cm.


…………….
-
Reaction Scheme A.




-
The starting material of bromocarboxylic acid, Compound 1, and the process for the preparation thereof are disclosed in Japanese Patent Application No. 29637/81.
-
Scheme B.
REACTION SCHEME B
-

- REACTION SCHEME C
-

 Org Lett 2012, 14(1): 299
Org Lett 2012, 14(1): 299
EP0024943A1 Sep 2, 1980 Mar 11, 1981 Toray Industries, Inc. 5,6,7-Trinor-4,8-inter-m-phenylene PGI2 derivatives and pharmaceutical compositions containing them EP0084856A1 Jan 19, 1983 Aug 3, 1983 Toray Industries, Inc. 5,6,7-Trinor-4, 8-inter-m-phenylene prostaglandin I2 derivatives JP3069909B Title not available
-
Sulfoaildenafil …. An analog of Sildenafil which has been used as an illegal adulterant in some dietary supplements

Sulfoaildenafil
An analog of Sildenafil which has been used as an illegal adulterant in some dietary supplements.
856190-47-1 cas no
5-(5-(((3R,5S)-3,5-Dimethylpiperazin-1-yl)sulfonyl)-2-ethoxyphenyl)-1-methyl-3-propyl-1H-pyrazolo[4,3-d]pyrimidine-7(4H)-thione
-
7H-Pyrazolo(4,3-d)pyrimidine-7-thione, 5-(5-(((3R,5S)-3,5-dimethyl-1-piperazinyl)sulfonyl)-2-ethoxyphenyl)-1,6-dihydro-1-methyl-3-propyl-, rel-
- Sildenafil thione
- Thioaildenafil
- UNII-33DX49E09G
-
-
C23-H32-N6-O3-S2
- 504.6768
-
Sulfoaildenafil (thioaildenafil) is a synthetic chemical compound that is a structural analog of sildenafil (Viagra).[1] It was first reported in 2005,[2] and it is not approved by any health regulation agency. Like sildenafil, sulfoaildenafil is a phosphodiesterase type 5 inhibitor.
Sulfoaildenafil has been found as an adulterant in a variety of supplements which are sold as “natural” or “herbal” sexual enhancement products.[3][4][5][6] A range of designer analogues of USA FDA-approved inhibitors of type-5 cGMP-specific phosphodiesterase (PDE5), such as sildenafil and vardenafil, have been detected in recent years as adulturants in over-the-counter herbal aphrodisiac products and dietary supplements,[7][8][9] in an apparent attempt to circumvent both the legal restrictions on sale of erectile dysfunction drugs, which are prescription-onlymedicines in most Western countries, and the patent protection which prevents sale of these drugs by competitors except under license to their inventors.

Figure 1. Biological pathway of penile erection
These compounds have been demonstrated to display PDE5 inhibitory activity in vitro and presumably have similar effects when consumed, but have undergone no formal testing in either humans or animals, and as such represent a significant health risk to consumers of these products due to their unknown safety profile.[10] Some attempts have been made to ban these drugs as unlicensed medicines, but progress has been slow so far, as even in those jurisdictions which have laws targeting designer drugs, the laws are drafted to ban analogues of illegal drugs of abuse, rather than analogues of prescription medicines. However at least one court case has resulted in a product being taken off the market.[11]

Figure 2. PDE5 domains

In December 2010, the United States Food and Drug Administration (FDA) issued a warning to consumers about such products stating, “The FDA has found many products marketed as dietary supplements for sexual enhancement during the past several years that can be harmful because they contain active ingredients in FDA-approved drugs or variations of these ingredients.”[12]

Figure 3. PDE5 Domains
An analog of aildenafil, which is a potent and highly selective inhibitor of phosphodiesterase 5, was found in a dietary supplement marketed for enhancement of sexual function. The compound was isolated by silica gel column chromatography, and its structure was identified by means of 13C-NMR spectrometry, 1H-NMR spectrometry, high-resolution MS, and X-ray structure determination. The compound was identified to be sulfoaildenafil(other names: thioaildenafil, dimethyl sildenafil thione, and thiomethisosildenafil). Sulfoaildenafil is very similar to the compound thiohomosildenafil. As it is difficult to distinguish between them by LC-photodiode array detector analysis, ultra-performance LC (UPLC)/MS, ion trap LC/MS/MS (LC/IT-MS/MS), and GC/MS were performed. The mass spectra of thiohomosildenafil by UPLC/MS and LC/IT-MS/MS showed mass fragments of m/z 58, 72, and 355, and the mass spectrum by GC/MS showed mass fragments of m/z 56, 72, and 420. Some of these fragments had low intensities, but they were useful for distinguishing between the two compounds. The relationship between aildenafil (other names: dimethylsildenafil and methisosildenafil) and homosildenafil is similar to that between sulfoaildenafil and thiohomosildenafil. Therefore, these compounds were also examined.http://www.ncbi.nlm.nih.gov/pubmed/22320083
……………………………………………………….
Volume 50, Issue 2, 8 September 2009, Pages 228–231
Phosphodiesterase type 5 (PDE-5) inhibitors represent a class of drugs used primarily in the treatment of erectile dysfunction. Currently, three PDE-5 inhibitors have been approved by the U.S. Food and Drug Administration (FDA) for use in the United States: sildenafil citrate, tadalafil, and vardenafil hydrochloride trihydrate. A bulk material, labeled as an ingredient for a dietary supplement, was analyzed for the presence of PDE-5 inhibitors. The compound that was detected displayed structural similarities to sildenafil, and was characterized further using LC–MSn, FTICRMS, X-ray crystallography and NMR. The compound was given the name sulfoaildenafil. When compared to sildenafil, sulfoaildenafil contains a sulfur atom substitution for the oxygen atom in the pyrazolopyrimidine portion of the molecule, and a 3,5-dimethyl substitution on the piperazine ring, rather than the 4-methyl moiety. The X-ray crystallographic data indicate that the material in this sample is comprised of two polymorphs, which may affect the chemical and/or biological properties of any product formulated with this compound.
……………..

http://www.theresonance.com/2012/categories/pharmaceutical/adulterated-natural-products
……………….
Herbal Supplement for Erectile Dysfunction Found to Contain Thio Structural Analog of Sildenafil (Viagra)
A herbal supplement marketed to alleviate erectile dysfunction was recently submitted for testing in our laboratory because it was surprisingly effective considering it should only contain the traditional herbals utilized for this problem such as Oyster, 2-Deoxy-D Glucose, Barberry, Snow Lotus, Bombyx Mori L., Ginger Root, Salfron Crocus.
http://process-nmr.com/WordPress/?cat=5
References
- Gratz, SR; Zeller, M; Mincey, DW; Flurer, CL (2009). “Structural characterization of sulfoaildenafil, an analog of sildenafil”. Journal of pharmaceutical and biomedical analysis 50 (2): 228–31. doi:10.1016/j.jpba.2009.04.003. PMID 19427155.
- Li, Shuxin; Ren, Jianping; Zhao, Yanjin; Lv, Qiujun; Guo, Jinhua. Pyrazolopyrimidinethione Derivatives, Salts and Solvates thereof, Preparation Methods and Use thereof. WO 2005058899
- Gryniewicz, CM; Reepmeyer, JC; Kauffman, JF; Buhse, LF (2009). “Detection of undeclared erectile dysfunction drugs and analogues in dietary supplements by ion mobility spectrometry”. Journal of pharmaceutical and biomedical analysis 49 (3): 601–6. doi:10.1016/j.jpba.2008.12.002. PMID 19150190.
- FDA warns consumers to avoid sexual enhancement pills, Sanjay Gupta, CNN, December 13th, 2010
- Reepmeyer JC, d’Avignon DA (January 2009). “Structure elucidation of thioketone analogues of sildenafil detected as adulterants in herbal aphrodisiacs”. Journal of Pharmaceutical and Biomedical Analysis 49 (1): 145–50. doi:10.1016/j.jpba.2008.10.007. PMID 19042103.
- Balayssac S, Trefi S, Gilard V, Malet-Martino M, Martino R, Delsuc MA (November 2008). “2D and 3D DOSY (1)H NMR, a useful tool for analysis of complex mixtures: Application to herbal drugs or dietary supplements for erectile dysfunction”. Journal of Pharmaceutical and Biomedical Analysis 50 (4): 602–12. doi:10.1016/j.jpba.2008.10.034. PMID 19108978.
- Zou P, Oh SS, Hou P, Low MY, Koh HL (February 2006). “Simultaneous determination of synthetic phosphodiesterase-5 inhibitors found in a dietary supplement and pre-mixed bulk powders for dietary supplements using high-performance liquid chromatography with diode array detection and liquid chromatography-electrospray ionization tandem mass spectrometry”. J Chromatogr A 1104 (1-2): 113–22. doi:10.1016/j.chroma.2005.11.103. PMID 16364350.
- Gratz SR, Gamble BM, Flurer RA (2006). “Accurate mass measurement using Fourier transform ion cyclotron resonance mass spectrometry for structure elucidation of designer drug analogs of tadalafil, vardenafil and sildenafil in herbal and pharmaceutical matrices”. Rapid Commun. Mass Spectrom. 20 (15): 2317–27. doi:10.1002/rcm.2594. PMID 16817245.
- Hou P, Zou P, Low MY, Chan E, Koh HL (September 2006). “Structural identification of a new acetildenafil analogue from pre-mixed bulk powder intended as a dietary supplement”. Food Addit Contam 23 (9): 870–5. doi:10.1080/02652030600803856. PMID 16901855.
- Oh, SS; Zou, P; Low, MY; Koh, HL (2006). “Detection of sildenafil analogues in herbal products for erectile dysfunction.”. Journal of toxicology and environmental health. Part A 69 (21): 1951–8.doi:10.1080/15287390600751355. PMID 16982533.
- Venhuis, BJ; Blok-Tip, L; De Kaste, D (2008). “Designer drugs in herbal aphrodisiacs.”. Forensic Science International 177 (2–3): e25–7. doi:10.1016/j.forsciint.2007.11.007. PMID 18178354.
- FDA warns consumers to avoid Man Up Now capsules, United States Food and Drug Administration, Dec. 15, 2010
