

| EudraCT | Title | Phase | Status | Date |
|---|---|---|---|---|
| 2019-003807-37 | A Double-Blind, Randomized, Placebo-Controlled Study to Evaluate the Efficacy and Safety of Odevixibat (A4250) in Children with Biliary Atresia Who Have Undergone a Kasai Hepatoportoenterostomy (BOLD) | Phase 3 | Ongoing | 2020-07-29 |
| 2015-001157-32 | An Exploratory Phase II Study to demonstrate the Safety and Efficacy of A4250 | Phase 2 | Completed | 2015-05-13 |
| 2014-004070-42 | An Exploratory, Phase IIa Cross-Over Study to Demonstrate the Efficacy | Phase 2 | Ongoing | 2014-12-09 |
| 2017-002325-38 | An Open-label Extension Study to Evaluate Long-term Efficacy and Safety of A4250 in Children with Progressive Familial Intrahepatic Cholestasis Types 1 and 2 (PEDFIC 2) | Phase 3 | Ongoing | |
| 2017-002338-21 | A Double-Blind, Randomized, Placebo-Controlled, Phase 3 Study to Demonstrate Efficacy and Safety of A4250 in Children with Progressive Familial Intrahepatic Cholestasis Types 1 and 2 (PEDFIC 1) | Phase 3 | Ongoing, Completed |
Home » PHASE 3 (Page 3)
Category Archives: PHASE 3
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
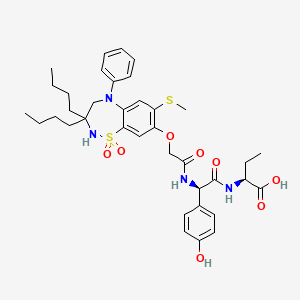
Odevixibat

Odevixibat
A-4250, AR-H 064974
CAS 501692-44-0
BUTANOIC ACID, 2-(((2R)-2-((2-((3,3-DIBUTYL-2,3,4,5-TETRAHYDRO-7-(METHYLTHIO)-1,1-DIOXIDO-5-PHENYL-1,2,5-BENZOTHIADIAZEPIN-8-YL)OXY)ACETYL)AMINO)-2-(4-HYDROXYPHENYL)ACETYL)AMINO)-, (2S)-
(2S)-2-[[(2R)-2-[[2-[(3,3-dibutyl-7-methylsulfanyl-1,1-dioxo-5-phenyl-2,4-dihydro-1λ6,2,5-benzothiadiazepin-8-yl)oxy]acetyl]amino]-2-(4-hydroxyphenyl)acetyl]amino]butanoic acid
| Molecular Formula | C37H48N4O8S2 |
| Molecular Weight | 740.929 |
-
-
-
- UPDATE 7/20/2021FDA APPROVED, To treat pruritus,
-
- New Drug Application (NDA): 215498
Company: ALBIREO PHARMA INC
-
- AZD8294WHO 10706AR-H064974HY-109120CS-0078340D11716US9694018, 5Originator Albireo AB
- Developer Albireo AB; Albireo Pharma
- ClassAcetamides; Butyric acids; Hepatoprotectants; Small molecules; Sulfones; Thiazepines
- Mechanism of Action Sodium-bile acid cotransporter inhibitors
- Orphan Drug Status Yes – Primary biliary cirrhosis; Biliary atresia; Intrahepatic cholestasis; Alagille syndrome
- New Molecular Entity Yes
- Phase III Biliary atresia; Intrahepatic cholestasis
- Phase II Alagille syndrome; Cholestasis; Primary biliary cirrhosis
- No development reported Non-alcoholic steatohepatitis
- 22 Jul 2020 Albireo initiates an expanded-access programme for Intrahepatic cholestasis in USA, Canada, Australia and Europe
- 14 Jul 2020 Phase-III clinical trials in Biliary atresia (In infants, In neonates) in Belgium (PO) after July 2020 (EudraCT2019-003807-37)
- 14 Jul 2020 Phase-III clinical trials in Biliary atresia (In infants, In neonates) in Germany, France, United Kingdom, Hungary (PO) (EudraCT2019-003807-37)
UPDATE Bylvay, FDA APPROVED2021/7/20 AND EMA 2021/7/16
Odevixibat, sold under the trade name Bylvay, is a medication for the treatment of progressive familial intrahepatic cholestasis (PFIC).[1]
The most common side effects include diarrhea, abdominal pain, hemorrhagic diarrhea, soft feces, and hepatomegaly (enlarged liver).[1]
Odevixibat is a reversible, potent, selective inhibitor of the ileal bile acid transporter (IBAT).[1][2]
In May 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) recommended granting a marketing authorization in the European Union for odevixibat for the treatment of PFIC in people aged six months or older.[1][3]
A-4250 (odevixibat) is a selective inhibitor of the ileal bile acid transporter (IBAT) that acts locally in the gut. Ileum absorbs glyco-and taurine-conjugated forms of the bile salts. IBAT is the first step in absorption at the brush-border membrane. A-4250 works by decreasing the re-absorption of bile acids from the small intestine to the liver, whichreduces the toxic levels of bile acids during the progression of the disease. It exhibits therapeutic intervention by checking the transport of bile acids. Studies show that A-4250 has the potential to decrease the damage in the liver cells and the development of fibrosis/cirrhosis of the liver known to occur in progressive familial intrahepatic cholestasis. A-4250 is a designated orphan drug in the USA for October 2012. A-4250 is a designated orphan drug in the EU for October 2016. A-4250 was awarded PRIME status for PFIC by EMA in October 2016. A-4250 is in phase II clinical trials by Albireo for the treatment of primary biliary cirrhosis (PBC) and cholestatic pruritus. In an open label Phase 2 study in children with cholestatic liver disease and pruritus, odevixibat showed reductions in serum bile acids and pruritus in most patients and exhibited a favorable overall tolerability profile.
![]()
Odevixibat is a highly potent, non-systemic ileal bile acid transport inhibitor (IBATi) that has has minimal systemic exposure and acts locally in the small intestine. Albireo is developing odevixibat to treat rare pediatric cholestatic liver diseases, including progressive familial intrahepatic cholestasis, biliary atresia and Alagille syndrome.
With normal function, approximately 95 percent of bile acids released from the liver into the bile ducts to aid in liver function are recirculated to the liver via the IBAT in a process called enterohepatic circulation. In people with cholestatic liver diseases, the bile flow is interrupted, resulting in elevated levels of toxic bile acids accumulating in the liver and serum. Accordingly, a product capable of inhibiting the IBAT could lead to a reduction in bile acids returning to the liver and may represent a promising approach for treating cholestatic liver diseases.
The randomized, double-blind, placebo-controlled, global multicenter PEDFIC 1 Phase 3 clinical trial of odevixibat in 62 patients, ages 6 months to 15.9 years, with PFIC type 1 or type 2 met its two primary endpoints demonstrating that odevixibat reduced serum bile acids (sBAs) (p=0.003) and improved pruritus (p=0.004), and was well tolerated with a low single digit diarrhea rate. These topline data substantiate the potential for odevixibat to be first drug for PFIC patients. The Company intends to complete regulatory filings in the EU and U.S. no later than early 2021, in anticipation of regulatory approval, issuance of a rare pediatric disease priority review voucher and launch in the second half of 2021.
Odevixibat is being evaluated in the ongoing PEDFIC 2 open-label trial (NCT03659916) designed to assess long-term safety and durability of response in a cohort of patients rolled over from PEDFIC 1 and a second cohort of PFIC patients who are not eligible for PEDFIC 1.
Odevixibat is also currently being evaluated in a second Phase 3 clinical trial, BOLD (NCT04336722), in patients with biliary atresia. BOLD, the largest prospective intervention trial ever conducted in biliary atresia, is a double-blind, randomized, placebo-controlled trial which will enroll approximately 200 patients at up to 75 sites globally to evaluate the efficacy and safety of odevixibat in children with biliary atresia who have undergone a Kasai procedure before age three months. The company also anticipates initiating a pivotal trial of odevixibat for Alagille syndrome by the end of 2020.
For more information about the PEDFIC 2 or BOLD studies, please visit ClinicalTrials.gov or contact medinfo@albireopharma.com.
The odevixibat PFIC program, or elements of it, have received fast track, rare pediatric disease and orphan drug designations in the United States. In addition, the FDA has granted orphan drug designation to odevixibat for the treatment of Alagille syndrome, biliary atresia and primary biliary cholangitis. The EMA has granted odevixibat orphan designation, as well as access to the PRIority MEdicines (PRIME) scheme for the treatment of PFIC. Its Paediatric Committee has agreed to Albireo’s odevixibat Pediatric Investigation Plan for PFIC. EMA has also granted orphan designation to odevixibat for the treatment of biliary atresia, Alagille syndrome and primary biliary cholangitis.

PATENT
https://patents.google.com/patent/US9694018B1/en
Example 5
1,1-Dioxo-3,3-dibutyl-5-phenyl-7-methylthio-8-(N—{(R)-α-[N—((S)-1-carboxypropyl) carbamoyl]-4-hydroxybenzyl}carbamoylmethoxy)-2,3,4,5-tetrahydro-1,2,5-benzothiadiazepine, Mw. 740.94.
This compound is prepared as described in Example 29 of WO3022286.
PATENT
https://patents.google.com/patent/WO2003022286A1/sv
Example 29
1,1-Dioxo-3,3-dibutyl-5-phenyl-7-methylthio-8-(N-((R)-α-[N-((S)- 1-carboxypropyl) carbamoyl]-4-hydroxybenzyl}carbamoylmethoxy)-2,3,4,5-tetrahydro-1,2,5-benzothiadiazepine
A solution of 1,1-dioxo-3,3-dibutyl-5-phenyl-7-methylthio-8-[N-((R)-α-carboxy-4-hydroxybenzyl)carbamoylmethoxy]-2,3,4,5-tetrahydro-1,2,5-benzothiadiazepine (Example 18; 0.075 g, 0.114 mmol), butanoic acid, 2-amino-, 1,1-dimethylethyl ester, hydrochloride, (2S)-(0.031 g, 0.160 mmol) and Ν-methylmorpholine (0.050 ml, 0.457 mmol) in DMF (4 ml) was stirred at RT for 10 min, after which TBTU (0.048 g, 0.149 mmol) was added. After 1h, the conversion to the ester was complete. M/z: 797.4. The solution was diluted with toluene and then concentrated. The residue was dissolved in a mixture of DCM (5 ml) and TFA (2 ml) and the mixture was stirred for 7h. The solvent was removed under reduced pressure. The residue was purified by preparative HPLC using a gradient of 20-60% MeCΝ in 0.1M ammonium acetate buffer as eluent. The title compound was obtained in 0.056 g (66 %) as a white solid. ΝMR (400 MHz, DMSO-d6): 0.70 (3H, t), 0.70-0.80 (6H, m), 0.85-1.75 (14H, m), 2.10 (3H, s), 3.80 (2H, brs), 4.00-4.15 (1H, m), 4.65 (1H, d(AB)), 4.70 (1H, d(AB)), 5.50 (1H, d), 6.60 (1H, s), 6.65-7.40 (11H, m), 8.35 (1H, d), 8.50 (1H, d) 9.40 (1H, brs).
PATENT
https://patents.google.com/patent/US20140323412A1/en
PATENT
https://patents.google.com/patent/WO2013063526A1/e
PATENT
https://patents.google.com/patent/WO2019245448A1/en
The compound l,l-dioxo-3,3-dibutyl-5-phenyl-7-methylthio-8-(A/-{(R)-a-[A/-((S)-l-carboxypropyl) carbamoyl]-4-hydroxybenzyl}carbamoylmethoxy)-2,3,4,5-tetrahydro-l,2,5-benzothiadiazepine (odevixibat; also known as A4250) is disclosed in WO 03/022286. The structure of odevixibat is shown below.

As an inhibitor of the ileal bile acid transporter (IBAT) mechanism, odevixibat inhibits the natural reabsorption of bile acids from the ileum into the hepatic portal circulation. Bile acids that are not reabsorbed from the ileum are instead excreted into the faeces. The overall removal of bile acids from the enterohepatic circulation leads to a decrease in the level of bile acids in serum and the liver. Odevixibat, or a pharmaceutically acceptable salt thereof, is therefore useful in the treatment or prevention of diseases such as dyslipidemia, constipation, diabetes and liver diseases, and especially liver diseases that are associated with elevated bile acid levels.
According to the experimental section of WO 03/022286, the last step in the preparation of odevixibat involves the hydrolysis of a tert-butyl ester under acidic conditions. The crude compound was obtained by evaporation of the solvent under reduced pressure followed by purification of the residue by preparative HPLC (Example 29). No crystalline material was identified.
Amorphous materials may contain high levels of residual solvents, which is highly undesirable for materials that should be used as pharmaceuticals. Also, because of their lower chemical and physical stability, as compared with crystalline material, amorphous materials may display faster
decomposition and may spontaneously form crystals with a variable degree of crystallinity. This may result in unreproducible solubility rates and difficulties in storing and handling the material. In pharmaceutical preparations, the active pharmaceutical ingredient (API) is for that reason preferably used in a highly crystalline state. Thus, there is a need for crystal modifications of odevixibat having improved properties with respect to stability, bulk handling and solubility. In particular, it is an object of the present invention to provide a stable crystal modification of odevixibat that does not contain high levels of residual solvents, that has improved chemical stability and can be obtained in high levels of crystallinity.
Example 1
Preparation of crystal modification 1
Absolute alcohol (100.42 kg) and crude odevixibat (18.16 kg) were charged to a 250-L GLR with stirring under nitrogen atmosphere. Purified water (12.71 kg) was added and the reaction mass was stirred under nitrogen atmosphere at 25 ± 5 °C for 15 minutes. Stirring was continued at 25 ± 5 °C for 3 to 60 minutes, until a clear solution had formed. The solution was filtered through a 5.0 m SS cartridge filter, followed by a 0.2 m PP cartridge filter and then transferred to a clean reactor.
Purified water (63.56 kg) was added slowly over a period of 2 to 3 hours at 25 ± 5 °C, and the solution was seeded with crystal modification 1 of odevixibat. The solution was stirred at 25 ± 5 °C for 12 hours. During this time, the solution turned turbid. The precipitated solids were filtered through centrifuge and the material was spin dried for 30 minutes. The material was thereafter vacuum dried in a Nutsche filter for 12 hours. The material was then dried in a vacuum tray drier at 25 ± 5 °C under vacuum (550 mm Hg) for 10 hours and then at 30 ± 5 °C under vacuum (550 mm Hg) for 16 hours. The material was isolated as an off-white crystalline solid. The isolated crystalline material was milled and stored in LDPE bags.
An overhydrated sample was analyzed with XRPD and the diffractogram is shown in Figure 2.
Another sample was dried at 50 °C in vacuum and thereafter analysed with XRPD. The diffractogram of the dried sample is shown in Figure 1.
The diffractograms for the drying of the sample are shown in Figures 3 and 4 for 2Q ranges 5 – 13 ° and 18 – 25 °, respectively (overhydrated sample at the bottom and dry sample at the top).
References
- ^ Jump up to:a b c d “First treatment for rare liver disease”. European Medicines Agency (EMA) (Press release). 21 May 2021. Retrieved 21 May 2021. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Odevixibat”. Albireo Pharma. Retrieved 21 May 2021.
- ^ “Bylvay: Pending EC decision”. European Medicines Agency (EMA). 19 May 2021. Retrieved 21 May 2021.
External links
- “Odevixibat”. Drug Information Portal. U.S. National Library of Medicine.
ClinicalTrials.gov
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| NCT04336722 | Efficacy and Safety of Odevixibat in Children With Biliary Atresia Who Have Undergone a Kasai HPE (BOLD) | Phase 3 | Recruiting | 2020-09-02 |
| NCT04483531 | Odevixibat for the Treatment of Progressive Familial Intrahepatic Cholestasis | Available | 2020-08-25 | |
| NCT03566238 | This Study Will Investigate the Efficacy and Safety of A4250 in Children With PFIC 1 or 2 | Phase 3 | Active, not recruiting | 2020-03-05 |
| NCT03659916 | Long Term Safety & Efficacy Study Evaluating The Effect of A4250 in Children With PFIC | Phase 3 | Recruiting | 2020-01-21 |
| NCT03608319 | Study of A4250 in Healthy Volunteers Under Fasting, Fed and Sprinkled Conditions | Phase 1 | Completed | 2018-09-19 |
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| NCT02630875 | A4250, an IBAT Inhibitor in Pediatric Cholestasis | Phase 2 | Completed | 2018-03-29 |
| NCT02360852 | IBAT Inhibitor A4250 for Cholestatic Pruritus | Phase 2 | Terminated | 2017-02-23 |
| NCT02963077 | A Safety and Pharmakokinetic Study of A4250 Alone or in Combination With A3384 | Phase 1 | Completed | 2016-11-16 |
EU Clinical Trials Register
.
 |
|
| Clinical data | |
|---|---|
| Trade names | Bylvay |
| Routes of administration |
By mouth |
| ATC code |
|
| Identifiers | |
| CAS Number |
|
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C37H48N4O8S2 |
| Molar mass | 740.93 g·mol−1 |
| 3D model (JSmol) | |
////////////odevixibat, Orphan Drug Status, phase 3, Albireo, A-4250, A 4250, AR-H 064974
CCCCC1(CN(C2=CC(=C(C=C2S(=O)(=O)N1)OCC(=O)NC(C3=CC=C(C=C3)O)C(=O)NC(CC)C(=O)O)SC)C4=CC=CC=C4)CCCC
| publicationnumber | |||||||
| US-2020046635-A1 | |||||||
| US-2020046636-A1 | US-2020046757-A1 | US-2020046758-A1 | US-2020002299-A1 | WO-2019245448-A1 | WO-2019245449-A1 | US-2019046451-A1 | US-2019070217-A1 |
| US-10441605-B2 | |||||||
| US-2017224720-A1 | |||||||
| US-2017224721-A1 | |||||||
| US-2018264029-A1 | |||||||
| US-2018360869-A1 | |||||||
| US-2018360870-A1 | |||||||
| US-2018360871-A1 | |||||||
| WO-2017133517-A1 | |||||||
| US-2017143738-A1 | |||||||
| US-2017143783-A1 | |||||||
| EP-2968230-A2 | |||||||
| EP-2968262-A1 | |||||||
| US-2014271734-A1 | |||||||
| US-2014275090-A1 | |||||||
| WO-2014144485-A1 | |||||||
| WO-2014144485-A9 | |||||||
| WO-2014144650-A2 | |||||||
| EP-2770990-A1 | |||||||
| EP-2771003-A1 | |||||||
| EP-2771003-B1 | |||||||
| EP-3266457-A1 | |||||||
| EP-3278796-A1 | |||||||
| US-10512657-B2 | |||||||
| US-2013108573-A1 | |||||||
| US-2013109671-A1 | |||||||
| US-2013338093-A1 | |||||||
| US-2014243281-A1 | |||||||
| US-2014323412-A1 | |||||||
| US-2016310518-A1 | |||||||
| US-2017368085-A1 | |||||||
| US-2019169217-A1 | |||||||
| US-2020069715-A1 | |||||||
| WO-2013063512-A1 | |||||||
| WO-2013063526-A1 | |||||||
| EP-2739286-A2 | |||||||
| WO-2013020108-A2 | |||||||
| EP-2637646-B1 | |||||||
| EP-2637668-B1 | |||||||
| EP-3023102-A1 | |||||||
| EP-3023102-B1 | |||||||
| EP-3400944-A1 | |||||||
| US-10000528-B2 | |||||||
| US-10011633-B2 | |||||||
| US-10093697-B2 | |||||||
| US-10221212-B2 | |||||||
| US-2012114588-A1 | |||||||
| US-2013225511-A1 | |||||||
| US-2013236541-A1 | |||||||
| US-2015031636-A1 | |||||||
| US-2015031637-A1 | |||||||
| US-2016193277-A1 | |||||||
| US-2016194353-A1 | |||||||
| US-2017182059-A1 | |||||||
| US-2017182115-A1 | |||||||
| US-2018022776-A1 | |||||||
| US-2018030088-A1 | |||||||
| US-2018030089-A1 | |||||||
| US-2018162904-A1 | |||||||
| US-2018362577-A1 | |||||||
| US-9688720-B2 | |||||||
| US-9694018-B1 | |||||||
| US-10555950-B2 | |||||||
| US-2016220577-A1 | |||||||
| US-9339480-B2 | |||||||
| WO-2008039829-A2 | |||||||
| US-2009069285-A1 | |||||||
| US-7842684-B2 | |||||||
| WO-2007051995-A2 | |||||||
| EP-1896408-A1 | |||||||
| EP-1896409-A1 | |||||||
| EP-1896457-A1 | |||||||
| US-2010048529-A1 | |||||||
| US-2010048530-A1 | |||||||
| US-2010137273-A1 | |||||||
| US-2010152156-A1 | |||||||
| US-2010168039-A1 | |||||||
| US-2010168075-A1 | |||||||
| US-7893048-B2 | |||||||
| US-7906502-B2 | |||||||
| WO-2006137792-A1 | |||||||
| WO-2006137794-A1 | |||||||
| WO-2006137795-A1 | |||||||
| US-2010216759-A1 | |||||||
| US-7863265-B2 | |||||||
| US-2008194494-A1 | |||||||
| US-2009186834-A1 | |||||||
| WO-2006102674-A2 | |||||||
| US-2009005321-A1 | |||||||
| EP-1831151-A1 | |||||||
| US-2008114064-A1 | |||||||
| WO-2006065214-A1 | |||||||
| EP-1699759-A1 | |||||||
| US-2007142304-A1 | |||||||
| US-2008064676-A1 | |||||||
| US-2010099657-A2 | |||||||
| US-7871998-B2 | |||||||
| WO-2005061452-A1 | |||||||
| EP-1638922-A1 | |||||||
| EP-1638926-A1 | |||||||
| EP-1638930-A1 | |||||||
| EP-1675820-A2 | |||||||
| EP-1676833-A1 | |||||||
| US-2005148656-A1 | |||||||
| US-2006142389-A1 | |||||||
| US-2006178432-A1 | |||||||
| US-2006194879-A1 | |||||||
| US-2006258866-A1 | |||||||
| US-2007099928-A1 | |||||||
| US-2007099997-A1 | |||||||
| US-2007244198-A1 | |||||||
| US-7309720-B2 | |||||||
| WO-2004110984-A1 | |||||||
| WO-2004113270-A2 | |||||||
| WO-2004113276-A1 | |||||||
| WO-2004113283-A1 | |||||||
| EP-1610770-A1 | |||||||
| EP-1610770-B1 | |||||||
| EP-1894564-A2 | |||||||
| US-2006199797-A1 | |||||||
| US-7514421-B2 | |||||||
| WO-2004089350-A1 | |||||||
| EP-1572626-A1 | |||||||
| US-2005131068-A1 | |||||||
| WO-2004056748-A1 | |||||||
| EP-1539120-A1 | |||||||
| US-2006083790-A1 | |||||||
| WO-2004006899-A1 | |||||||
| EP-1521742-A1 | |||||||
| US-2005239766-A1 | |||||||
| US-7470678-B2 | |||||||
| WO-2004005247-A1 | |||||||
| EP-1517679-A1 | |||||||
| EP-1517679-B1 | |||||||
| EP-1517883-A1 | |||||||
| EP-1517883-B1 | |||||||
| EP-1517883-B8 | |||||||
| US-2005222261-A1 | |||||||
| US-2005256198-A1 | |||||||
| US-2005267149-A1 | |||||||
| US-7351858-B2 | |||||||
| US-7355069-B2 | |||||||
| US-7521461-B2 | |||||||
| WO-2004000294-A1 | |||||||
| WO-2004000790-A1 | |||||||
| EP-1478368-A1 | |||||||
| US-2005124557-A1 | |||||||
| WO-03061663-A1 | |||||||
| EP-1458672-A1 | |||||||
| EP-1458672-B1 | |||||||
| EP-1458673-A1 | |||||||
| EP-1458673-B1 | |||||||
| EP-1458677-A1 | |||||||
| EP-1458677-B1 | |||||||
| US-2005113362-A1 | |||||||
| US-2005171204-A1 | |||||||
| US-2005215630-A1 | |||||||
| US-2005282822-A1 | |||||||
| US-7256307-B2 | |||||||
| US-7276539-B2 | |||||||
| US-7488844-B2 | |||||||
| US-7514471-B2 | |||||||
| WO-03051821-A1 | |||||||
| WO-03051822-A1 | |||||||
| WO-03051826-A1 | |||||||
| EP-1427423-B1 | |||||||
| EP-1427423-B9 | |||||||
| US-2005038009-A1 | |||||||
| US-7132416-B2 |
Desidustat


Ranjit Desai
Inventor of Oxemia (Desidustat), a breakthrough PHD inhibitor approved for Chronic Kidney Diseases (CKD) / Accomplished pharma executive / 4 INDs in 4 years, ZYDUS LIFESCIENCES
DESIDUSTAT
Formal Name
N-[[1-(cyclopropylmethoxy)-1,2-dihydro-4-hydroxy-2-oxo-3-quinolinyl]carbonyl]-glycine
CAS Number 1616690-16-4
Molecular Formula C16H16N2O6
Formula Weight 332.3
FormulationA crystalline solid
λmax233, 291, 335
2-(1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carboxamido)acetic acid
desidustat
Glycine, N-((1-(cyclopropylmethoxy)-1,2-dihydro-4-hydroxy-2-oxo-3-quinolinyl)carbonyl)-
N-(1-(Cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carbonyl)glycine
ZYAN1 compound
(1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carbonyl) glycine in 98% yield, as a solid. MS (ESI-MS): m/z 333.05 (M+H) +. 1H NMR (DMSO-d 6): 0.44-0.38 (m, 2H), 0.62-0.53 (m, 2H), 1.34-1.24 (m, 1H), 4.06-4.04 (d, 2H), 4.14-4.13 (d, 2H), 7.43-7.39 (t, 1H), 7.72-7.70 (d, 1H), 7.89-7.85 (m, 1H), 8.11-8.09 (dd, 1H), 10.27-10.24 (t, 1H), 12.97 (bs, 1H), 16.99 (s, 1H). HPLC Purity: 99.85%
Oxemia (Desidustat) has received approval from the Drug Controller General of India. This was an incredible team effort by Zydans across the organization and I am so proud of what we have accomplished. Oxemia is a breakthrough treatment for Anemia associated with Chronic Kidney Disease in Patients either on Dialysis or Not on Dialysis, and will help improve quality of life for CKD patients. Team #zydus , on to our next effort!

Desidustat (INN, also known as ZYAN1) is a drug for the treatment of anemia of chronic kidney disease. This drug with the brand name Oxemia is discovered and developed by Zydus Life Sciences.[1] The subject expert committee of CDSCO has recommended the grant of permission for manufacturing and marketing of Desidustat 25 mg and 50 mg tablets in India,based on some conditions related to package insert, phase 4 protocols, prescription details, and GCP.[2] Clinical trials on desidustat have been done in India and Australia.[3] In a Phase 2, randomized, double-blind, 6-week, placebo-controlled, dose-ranging, safety and efficacy study, a mean hemoglobin increase of 1.57, 2.22, and 2.92 g/dL in desidustat 100, 150, and 200 mg arms, respectively, was observed.[4] The Phase 3 clinical trials were conducted at additional lower doses as of 2019.[5] Desidustat is developed for the treatment of anemia as an oral tablet, where currently injections of erythropoietin and its analogues are drugs of choice. Desidustat is a HIF prolyl-hydroxylase inhibitor. In preclinical studies, effects of desidustat was assessed in normal and nephrectomized rats, and in chemotherapy-induced anemia. Desidustat demonstrated hematinic potential by combined effects on endogenous erythropoietin release and efficient iron utilization.[6][7] Desidustat can also be useful in treatment of anemia of inflammation since it causes efficient erythropoiesis and hepcidin downregulation.[8] In January 2020, Zydus entered into licensing agreement with China Medical System (CMS) Holdings for development and commercialization of desidustat in Greater China. Under the license agreement, CMS will pay Zydus an initial upfront payment, regulatory milestones, sales milestones and royalties on net sales of the product. CMS will be responsible for development, registration and commercialization of desidustat in Greater China.[9] It has been observed that desidustat protects against acute and chronic kidney injury by reducing inflammatory cytokines like IL-6 and oxidative stress [10] A clinical trial to evaluate the efficacy and safety of desidustat tablet for the management of Covid-19 patients is ongoing in Mexico, wherein desidustat has shown to prevent acute respiratory distress syndrome (ARDS) by inhibiting IL-6.[11] Zydus has also received approval from the US FDA to initiate clinical trials of desidustat in chemotherapy Induced anemia (CIA).[12]. Desidustat has met the primary endpoints in the phase 3 clinical trials and Zydus had filed the New Drug Application (NDA) to DCGI in November, 2021.[13]\
CLIP
Zydus receives DCGI approval for new drug Oxemia; what you need to know
The new drug is an oral, small molecule hypoxia-inducible factor-prolyl hydroxylase (HIF-PH) inhibitor, Zydus said in a statement.
Gujarat-based pharma company Zydus Lifesciences on Monday received the Drugs Controller General of India (DCGI) approval for its new drug application for a first-of-its-kind oral treatment for anemia associated with Chronic Kidney Disease (CKD) – Oxemia (Desidustat).
The new drug is an oral, small molecule hypoxia-inducible factor-prolyl hydroxylase (HIF-PH) inhibitor, the drug firm said in a statement.
Desidustat showed good safety profile, improved iron mobilization and LDL-C reduction in CKD patients in DREAM-D and DREAM-ND Phase III clinical trials, conducted in approximately 1,200 subjects. Desidustat provides CKD patients with an oral convenient therapeutic option for the treatment of anemia. The pharma major did not, however, declare the cost per dose if the drug is available in the market.
“After more than a decade of research and development into the science of HIF-PH inhibitors, results have demonstrated that Oxemia addresses this unmet need and additionally reduces hepcidin, inflammation and enables better iron mobilization. This advancement offers ease of convenience for the patient and will also reduce the disease burden by providing treatment at an affordable cost, thereby improving the quality of life for patients suffering from Chronic Kidney Disease,” Chairman of Zydus Lifesciences Pankaj Patel said.
Chronic Kidney Disease (CKD) is a progressive medical condition characterised by a gradual loss of kidney function and is accompanied by comorbidities like anemia, cardiovascular diseases (hypertension, heart failure and stroke), diabetes mellitus, eventually leading to kidney failure.
PATENT
|
Scheme 3:
|
Step 1′a Process for Preparation of ethyl 2-iodobenzoate (XI-a)
Step-2 Process for the Preparation of ethyl 2-((tert-butoxycarbonyl)(cyclopropylmethoxy)aminolbenzoate (XII-a)
Step 3 Process for the Preparation of ethyl 2-((cyclopropylmethoxy)amino)benzoate (XIII-a)
Step 4 Process for the Preparation of ethyl 24N-(cyclopropylinethoxy)-3-ethoxy-3-oxopropanamido)benzoate (XIV-a)
Step 5: Process for the Preparation of ethyl 1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2 dihydroquinolline-3-carboxylate (XY-a)
Purification
Step 6 Process for the Preparation of ethyl (1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carbonyl)glycinate (XVI-a)
Purification
Step 7: Process for the Preparation of (1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carbonyl)glycine (I-a)
Polymorphic Data (XRPD):
References[edit]
- ^ “Zydus receives DCGI approval for new drug Oxemia; what you need to know”.
- ^ CDSCO, SEC Committee. “SEC meeting to examine IND proposals, dated 29.12.2021”. CDSCO website Govt of India. CDSCO. Retrieved 19 January 2022.
- ^ Kansagra KA, Parmar D, Jani RH, Srinivas NR, Lickliter J, Patel HV, et al. (January 2018). “Phase I Clinical Study of ZYAN1, A Novel Prolyl-Hydroxylase (PHD) Inhibitor to Evaluate the Safety, Tolerability, and Pharmacokinetics Following Oral Administration in Healthy Volunteers”. Clinical Pharmacokinetics. 57 (1): 87–102. doi:10.1007/s40262-017-0551-3. PMC 5766731. PMID 28508936.
- ^ Parmar DV, Kansagra KA, Patel JC, Joshi SN, Sharma NS, Shelat AD, Patel NB, Nakrani VB, Shaikh FA, Patel HV; on behalf of the ZYAN1 Trial Investigators. Outcomes of Desidustat Treatment in People with Anemia and Chronic Kidney Disease: A Phase 2 Study. Am J Nephrol. 2019 May 21;49(6):470-478. doi: 10.1159/000500232.
- ^ “Zydus Cadila announces phase III clinical trials of Desidustat”. 17 April 2019. Retrieved 20 April 2019 – via The Hindu BusinessLine.
- ^ Jain MR, Joharapurkar AA, Pandya V, Patel V, Joshi J, Kshirsagar S, et al. (February 2016). “Pharmacological Characterization of ZYAN1, a Novel Prolyl Hydroxylase Inhibitor for the Treatment of Anemia”. Drug Research. 66 (2): 107–12. doi:10.1055/s-0035-1554630. PMID 26367279.
- ^ Joharapurkar AA, Pandya VB, Patel VJ, Desai RC, Jain MR (August 2018). “Prolyl Hydroxylase Inhibitors: A Breakthrough in the Therapy of Anemia Associated with Chronic Diseases”. Journal of Medicinal Chemistry. 61 (16): 6964–6982. doi:10.1021/acs.jmedchem.7b01686. PMID 29712435.
- ^ Jain M, Joharapurkar A, Patel V, Kshirsagar S, Sutariya B, Patel M, et al. (January 2019). “Pharmacological inhibition of prolyl hydroxylase protects against inflammation-induced anemia via efficient erythropoiesis and hepcidin downregulation”. European Journal of Pharmacology. 843: 113–120. doi:10.1016/j.ejphar.2018.11.023. PMID 30458168. S2CID 53943666.
- ^ Market, Capital (20 January 2020). “Zydus enters into licensing agreement with China Medical System Holdings”. Business Standard India. Retrieved 20 January 2020 – via Business Standard.
- ^ Joharapurkar, Amit; Patel, Vishal; Kshirsagar, Samadhan; Patel, Maulik; Savsani, Hardikkumar; Jain, Mukul (22 January 2021). “Prolyl hydroxylase inhibitor desidustat protects against acute and chronic kidney injury by reducing inflammatory cytokines and oxidative stress”. Drug Development Research. 82 (6): 852–860. doi:10.1002/ddr.21792. PMID 33480036. S2CID 231680317.
- ^ “Zydus’ trials of Desidustat shows positive results for Covid-19 management”. The Hindu Business Line. The Hindu. Retrieved 25 January 2021.
- ^ “Zydus receives approval from USFDA to initiate clinical trials of Desidustat in cancer patients receiving chemotherapy”. PipelineReview.com. La Merie Publishing. Retrieved 22 January 2021.
- ^ “Stock Share Price | Get Quote | BSE”.
|
WO – 30.04.2020
|
|
2.WO/2020/058882METHODS OF PRODUCING VENOUS ANGIOBLASTS AND SINUSOIDAL ENDOTHELIAL CELL-LIKE CELLS AND COMPOSITIONS THEREOF
WO – 26.03.2020
|
|
3.110876806APPLICATION OF HIF2ALPHA AGONIST AND ACER2 AGONIST IN PREPARATION OF MEDICINE FOR TREATING ATHEROSCLEROSIS
CN – 13.03.2020
|
|
US – 28.11.2019
|
|
WO – 06.09.2019
|
|
EA – 30.10.2015
Настоящее изобретение относится к новым соединениям общей формулы (I), фармацевтическим композициям, содержащим указанные соединения, применению этих соединений для лечения состояний, опосредованных пролилгидроксилазой HIF, и к способу лечения анемии, включающему введение заявленных соединений |
|
EP – 28.10.2015
|
|
US – 22.10.2015
The present invention relates to novel compounds of the general formula (I), their tautomeric forms, their stereoisomers, their pharmaceutically acceptable salts, pharmaceutical compositions containing them, methods for their preparation, use of these compounds in medicine and the intermediates involved in their preparation. |
|
WO – 03.07.2014
|
 |
|
| Clinical data | |
|---|---|
| Other names | ZYAN1 |
| Identifiers | |
| CAS Number | |
| UNII | |
| Chemical and physical data | |
| Formula | C16H16N2O6 |
| Molar mass | 332.312 g·mol−1 |
| 3D model (JSmol) | |
Date
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| NCT04215120 | Desidustat in the Treatment of Anemia in CKD on Dialysis Patients | Phase 3 | Recruiting | 2020-01-02 |
| NCT04012957 | Desidustat in the Treatment of Anemia in CKD | Phase 3 | Recruiting | 2019-12-24 |
////////// DESIDUSTAT, ZYDUS CADILA, COVID 19, CORONA VIRUS, PHASE 3, ZYAN 1, OXEMIA, APPROVALS 2022, INDIA 2022

Remdesivir, レムデシビル , ремдесивир , ريمديسيفير , 瑞德西韦 ,
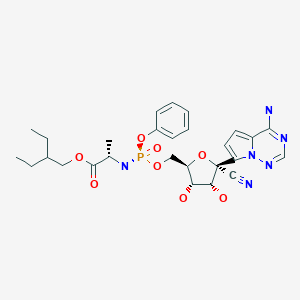
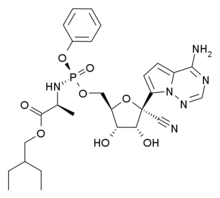
Remdesivir
| Formula |
C27H35N6O8P
|
|---|---|
| CAS |
1809249-37-3
|
| Mol weight |
602.576
|
レムデシビル
UNII:3QKI37EEHE
ремдесивир [Russian] [INN]
ريمديسيفير [Arabic] [INN]
瑞德西韦 [Chinese] [INN]
2-Ethylbutyl (2S)-2-{[(S)-{[(2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydro-2-furanyl]methoxy}(phenoxy)phosphoryl]amino}propanoate (non-preferred name)
L-Alanine, N-((S)-hydroxyphenoxyphosphinyl)-, 2-ethylbutyl ester, 6-ester with 2-C-(4-aminopyrrolo(2,1-f)(1,2,4)triazin-7-yl)-2,5-anhydro-D-altrononitrile
2-Ethylbutyl (2S)-2-(((S)-(((2R,3S,4R,5R)-5-(4-aminopyrrolo(2,1-f)(1,2,4)triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)propanoate
- 2-Ethylbutyl (2S)-2-[[(S)-[[(2R,3S,4R,5R)-5-(4-aminopyrrolo(2,1-f)(1,2,4)triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl]methoxy]phenoxyphosphoryl]amino]propanoate
- 2-Ethylbutyl (2S)-2-[[[[(2R,3S,4R,5R)-5-(4-aminopyrrolo(2,1-f)(1,2,4)triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl]methoxy]phenoxyphosphoryl]amino]propanoate
- 2-Ethylbutyl N-[(S)-[2-C-(4-aminopyrrolo(2,1-f)(1,2,4)triazin-7-yl)-2,5-anhydro-D-altrononitril-6-O-yl]phenoxyphosphoryl]-L-alaninate
- GS 5734
- L-Alanine, N-[(S)-hydroxyphenoxyphosphinyl)-, 2-ethylbutyl ester,6-ester with 2-C-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-2,5-anhydro-D-altrononitrile
GS-5734
Treatment of viral infections
Phase III, clinical trials for the treatment of hospitalized patients with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection (COVID-19). National Institute of Allergy and Infectious Diseases (NIAID) is evaluating remdesivir in phase II/III clinical trials for the treatment of Ebola virus infection.
The compound has been evaluated in preclinical studies for the potential treatment of Middle East respiratory syndrome coronavirus (MERS-CoV) and severe acute respiratory syndrome coronavirus (SARS-CoV) infections.
Remdesivir is a nucleoside analogue, with effective antiviral activity, with EC50s of 74 nM for ARS-CoV and MERS-CoV in HAE cells, and 30 nM for murine hepatitis virus in delayed brain tumor cells.
Remdesivir (development code GS-5734) is a novel antiviral drug in the class of nucleotide analogs. It was developed by Gilead Sciences as a treatment for Ebola virus disease and Marburg virus infections,[1] though it has subsequently also been found to show antiviral activity against other single stranded RNA viruses such as respiratory syncytial virus, Junin virus, Lassa fever virus, Nipah virus, Hendra virus, and the coronaviruses (including MERS and SARS viruses).[2][3] It is being studied for SARS-CoV-2 and Nipah and Hendra virus infections.[4][5][6] Based on success against other coronavirus infections, Gilead provided remdesivir to physicians who treated an American patient in Snohomish County, Washington in 2020, infected with SARS-CoV-2[7] and is providing the compound to China to conduct a pair of trials in infected individuals with and without severe symptoms.[8]
Research usage
Laboratory tests suggest remdesivir is effective against a wide range of viruses, including SARS-CoV and MERS-CoV. The medication was pushed to treat the West African Ebola virus epidemic of 2013–2016. Although the drug turned out to be safe, it was not particularly effective against filoviruses such as the Ebola virus.
Ebola virus
Remdesivir was rapidly pushed through clinical trials due to the West African Ebola virus epidemic of 2013–2016, eventually being used in at least one human patient despite its early development stage at the time. Preliminary results were promising and it was used in the emergency setting during the Kivu Ebola epidemic that started in 2018 along with further clinical trials, until August 2019, when Congolese health officials announced that it was significantly less effective than monoclonal antibody treatments such as mAb114 and REGN-EB3. The trials, however, established its safety profile.[9][10][11][12][13][14][15][16]
SARS-CoV-2
In response to the 2019–20 coronavirus outbreak induced by coronavirus SARS-CoV-2, Gilead provided remdesivir for a “small number of patients” in collaboration with Chinese medical authorities for studying its effects.[17]
Gilead also started laboratory testing of remdesivir against SARS-CoV-2. Gilead stated that remdesivir was “shown to be active” against SARS and MERS in animals.[3][18]
In late January 2020, remdesivir was administered to the first US patient to be confirmed to be infected by SARS-CoV-2, in Snohomish County, Washington, for “compassionate use” after he progressed to pneumonia. While no broad conclusions were made based on the single treatment, the patient’s condition improved dramatically the next day,[7] and he was eventually discharged.[19]
Also in late January 2020, Chinese medical researchers stated to the media that in exploratory research considering a selection of 30 drug candidates. Remdesivir and two other drugs, chloroquine and lopinavir/ritonavir, seemed to have “fairly good inhibitory effects” on SARS-CoV-2 at the cellular level. Requests to start clinical testing were submitted,[20][21]. On February 6, 2020, a clinical trial of remdesivir began in China.[22]
Other viruses
The active form of remdesivir, GS-441524, shows promise for treating feline coronavirus.[23]
Mechanism of action and resistance
Remdesivir is a prodrug that metabolizes into its active form GS-441524. GS-441524 is an adenosine nucleotide analog that confuses viral RNA polymerase and evades proofreading by viral exoribonuclease (ExoN), causing a decrease in viral RNA production. It was unknown whether it terminates RNA chains or causes mutations in them.[24]However, it has been learned that the RNA dependent RNA polymerase of ebolavirus is inhibited for the most part by delayed chain termination.[25]
Mutations in the mouse hepatitis virus RNA replicase that cause partial resistance were identified in 2018. These mutations make the viruses less effective in nature, and the researchers believe they will likely not persist where the drug is not being used.[24]
MORE SYNTHESIS COMING, WATCH THIS SPACE…………………..

SYNTHESIS
Remdesivir can be synthesized in multiple steps from ribose derivatives. The figure below is one of the synthesis route of remdesivir invented by Chun et al. from Gilead Sciences.[26]In this method, intermediate a is firstly prepared from L-alanine and phenyl phosphorodichloridate in presence of triethylamine and dichloromethane; triple benzyl-protected ribose is oxidized by dimethyl sulfoxide with acetic anhydride and give the lactone intermediate b; pyrrolo[2,1-f][1,2,4]triazin-4-amine is brominated, and the amine group is protected by excess trimethylsilyl chloride. n-Butyllithium undergoes a halogen-lithium exchange reaction with the bromide at -78 °C to yield the intermediate c. The intermediate b is then added to a solution containing intermediate c dropwise. After quenching the reaction in a weakly acidic aqueous solution, a mixture of 1: 1 anomers was obtained. It was then reacted with an excess of trimethylsilyl cyanide in dichloromethane at -78 °C for 10 minutes. Trimethylsilyl triflate was added and reacts for an additional 1 hour, and the mixture was quenched in an aqueous sodium hydrogen carbonate. A nitrile intermediate was obtained. The protective group, benzyl, was then removed with boron trichloride in dichloromethane at -20 °C. The excess of boron trichloride was quenched in a mixture of potassium carbonate and methanol. A benzyl-free intermediate was obtained. The isomers were then separated via reversed-phase HPLC. The optically pure compound and intermediate a are reacted with trimethyl phosphate and methylimidazole to obtain a diastereomer mixture of remdesivir. In the end, optically pure remdesivir can be obtained through methods such as chiral resolution.
The synthesis of Remdesivir was invented by Byoung Kwon Chun et al. from Gilead Sciences, Inc. and claimed in the patent, WO2016069826A1.
中文: 瑞德西韋的合成方法是由吉利德科學公司的 Byoung Kwon Chun等人所發明,並在WO2016069826A1中聲明專利。

PATENT
WO 2018204198
Prevention and treatment methods for some Arenaviridae , Coronaviridae , Filoviridae, Flaviviridae, and Paramyxoviridae viruses present challenges due to a lack of vaccine or post-exposure treatment modality for preventing or managing these infections. In some cases, patients only receive supportive and resource intensive therapy such as electrolyte and fluid balancing, oxygen, blood pressure maintenance, or treatment for secondary infections. Thus, there is a need for antiviral therapies having a potential for broad antiviral activity.
[0004] The compound (S)-2-ethylbutyl 2-(((S)-(((2R,3 S,4R,5R)-5-(4-aminopyrrolo[2, 1-f][l,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy) phosphoryl)amino)propanoate, referred herein as Compound 1 or Formula I, is known to exhibit antiviral properties against Arenaviridae, Coronaviridae, Filoviridae, and
Paramyxoviridae viruses as described in Warren, T. et al., Nature (2016) 531 :381-385 and antiviral activities against Flaviviridae viruses as described in co-pending United States provisional patent application no. 62/325,419 filed April 20, 2016.
[0005] (S)-2-Ethylbutyl 2-(((S)-(((2R,3S,4R,5R)-5-(4-aminopyrrolo[2, l-f][l,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)
propanoate or 2-ethylbutyl ((S)-(((2R,3 S,4R,5R)-5-(4-aminopyrrolo[2, l-f][l,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)-L-alaninate, (Formula I), has the following structure:
Formula I
PATENT
WO 2017184668
https://patents.google.com/patent/WO2017184668A1/en
A. Preparation of Compounds
Example 1. (2S)-ethyl 2-(chloro(phenoxy)phosphorylamino)pro anoate (Chloridate A)

[0246] Ethyl alanine ester hydrochloride salt (1.69 g, 11 mmol) was dissolved in anhydrous CH2CI2 (10 mL) and the mixture stirred with cooling to 0 °C under N2(g). Phenyl dichlorophosphate (1.49 mL, 10 mmol) was added followed by dropwise addition of Et3N over 10 min. The reaction mixture was then slowly warmed to RT and stirred for 12 h. Anhydrous Et20 (50 mL) was added and the mixture stirred for 30 min. The solid that formed was removed by filtration, and the filtrate concentrated under reduced pressure. The residue was subjected to silica gel chromatography eluting with 0-50% EtOAc in hexanes to provide intermediate A (1.13 g, 39%). H NMR (300 MHz, CDC13) δ 7.39-7.27 (m, 5H), 4.27 (m, 3H), 1.52 (m, 3H), 1.32 (m, 3H). 31P NMR (121.4 MHz, CDC13) δ 8.2, 7.8.
Example 2. (2S)-2-ethylbutyl 2-(chloro(phenoxy)phosphorylamino)propanoate
(Chloridate B

[0247] The 2-ethylbutyl alanine chlorophosphoramidate ester B was prepared using the same procedure as chloridate A except substituting 2-ethylbutyl alanine ester for ethyl alanine ester. The material is used crude in the next reaction. Treatment with methanol or ethanol forms the displaced product with the requisite LCMS signal.
Example 3. (2S)-isopropyl 2-(chloro(phenoxy)phosphorylamino)propanoate
(Chloridate C)

C
[0248] The isopropyl alanine chlorophosphoramidate ester C was prepared using the same procedure as chloridate A except substituting isopropyl alanine ester for the ethyl alanine ester. The material is used crude in the next reaction. Treatment with methanol or ethanol forms the displaced product with the requisite LCMS signal.
Example 4. (2S)-2-ethylbutyl 2-((((2R,3S,4R,5R)-5-(4-aminopyrrolo[l,2-firi,2,41triazin- 7-yl)-5-cvano-3,4-dihvdroxytetrahydrofuran-2- yl)methoxy)(phenoxy)phosphorylamino)propanoate (Compound 9)
[0249] Compound 9 can be prepared by several methods described below. Procedure 1

[0250] Prepared from Compound 1 and chloridate B according to the same method as for the preparation of compound 8 as described in PCT Publication no. WO 2012/012776. 1H NMR (300 MHz, CD3OD) δ 7.87 (m, 1H), 7.31-7.16 (m, 5H), 6.92-6.89 (m, 2H), 4.78 (m, 1H), 4.50-3.80 (m, 7H), 1.45-1.24 (m, 8H), 0.95-0.84 (m, 6H). 31P NMR (121.4 MHz, CD3OD) δ 3.7. LCMS m/z 603.1 [M+H], 601.0 [M-H].
Procedure 2

9
[0251] (2S)-2-ethylbutyl 2-(((((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,l-f][l,2,4]triazin-7- yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino) propanoate. (2S)-2-ethylbutyl 2-(((4-nitrophenoxy)(phenoxy)phosphoryl)amino)propanoate (1.08 g, 2.4 mmol) was dissolved in anhydrous DMF (9 mL) and stirred under a nitrogen atmosphere at RT. (2R,3R,4S,5R)-2-(4-aminopyrrolo[2,l-f][l,2,4]triazin-7-yl)-3,4- dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-carbonitrile (350 mg, 1.2 mmol) was added to the reaction mixture in one portion. A solution of i-butylmagnesium chloride in THF (1M, 1.8 mL, 1.8 mmol) was then added to the reaction drop wise over 10 minutes. The reaction was stirred for 2 h, at which point the reaction mixture was diluted with ethyl acetate (50 mL) and washed with saturated aqueous sodium bicarbonate solution (3 x 15 mL) followed by saturated aqueous sodium chloride solution (15 mL). The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The resulting oil was purified with silica gel column chromatography (0-10% MeOH in DCM) to afford (2S)-2- ethylbutyl 2-(((((2R,3S,4R,5R)-5-(4-aminopyrrolo[2, l-f][l,2,4]triazin-7-yl)-5-cyano-3,4- dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino) propanoate (311 mg, 43%, 1 :0.4 diastereomeric mixture at phosphorus) as a white solid. H NMR (400 MHz, CD3OD) δ 7.85 (m, 1H), 7.34 – 7.23 (m, 2H), 7.21 – 7.09 (m, 3H), 6.94 – 6.84 (m, 2H), 4.78 (d, / = 5.4 Hz, 1H), 4.46 – 4.33 (m, 2H), 4.33 – 4.24 (m, 1H), 4.18 (m, 1H), 4.05 – 3.80 (m, 3H), 1.52 – 1.39 (m, 1H), 1.38 – 1.20 (m, 7H), 0.85 (m, 6H). 31P NMR (162 MHz, CD3OD) δ 3.71, 3.65. LCMS m/z 603.1 [M+H], 600.9 [M-H]. HPLC (2-98% MeCN-H20 gradient with 0.1% TFA modifier over 8.5 min, 1.5mL/min, Column: Phenomenex Kinetex C18, 2.6 um 100 A, 4.6 x 100 mm ) tR = 5.544 min, 5.601 min
Separation of the (S) and (R) Diastereomers
[0252] (2S)-2-ethylbutyl 2-(((((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,l-f][l,2,4]triazin-7-yl)- 5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino) propanoate was dissolved in acetonitrile. The resulting solution was loaded onto Lux Cellulose-2 chiral column, equilibrated in acetonitrile, and eluted with isocratic
acetonitrile/methanol (95 :5 vol/vol). The first eluting diastereomer had a retention time of 17.4 min, and the second eluting diastereomer had a retention time of 25.0 min.
[0253] First Eluting Diastereomer is (S)-2-ethylbutyl 2-(((R)-(((2R,3S,4R,5R)-5-(4- aminopyrrolo[2, 1 -f] [ 1 ,2,4]triazin-7-yl)-5-cyano-3 ,4-dihydroxytetrahydrofuran-2- yl)methoxy)(phenoxy)phos horyl)amino)propanoate:

!HNMR (400 MHz, CD3OD) δ 8.05 (s, 1H), 7.36 (d, / = 4.8 Hz, 1H), 7.29 (br t, J = 7.8 Hz, 2H), 7.19 – 7.13 (m, 3H), 7.11 (d, / = 4.8 Hz, 1H), 4.73 (d, / = 5.2 Hz, 1H), 4.48 – 4.38 (m, 2H), 4.37 – 4.28 (m, 1H), 4.17 (t, / = 5.6 Hz, 1H), 4.08 – 3.94 (m, 2H), 3.94 – 3.80 (m, 1H), 1.48 (sep, / = 12.0, 6.1 Hz, 1H), 1.34 (p, / = 7.3 Hz, 4H), 1.29 (d, / = 7.2 Hz, 3H), 0.87 (t, / = 7.4 Hz, 6H). 31PNMR (162 MHz, CD3OD) δ 3.71 (s). HPLC (2-98% MeCN-H20 gradient with 0.1 % TFA modifier over 8.5 min, 1.5mL/min, Column: Phenomenex Kinetex C18, 2.6 um 100 A, 4.6 x 100 mm ) is = 5.585 min. [0254] Second Eluting Diastereomer is (S)-2-ethylbutyl 2-(((S)-(((2R,3S,4R,5R)-5-(4- aminopyrrolo[2, 1 -f] [ 1 ,2,4]triazin-7-yl)-5-cyano-3 ,4-dihydroxytetrahydrofuran-2- yl)methoxy)(phenoxy)phosphoryl)amino)propanoate:

HNMR (400 MHz, CD3OD) δ 8.08 (s, 1H), 7.36 – 7.28 (m, 3H), 7.23 – 7.14 (m, 3H), 7.08 (d, 7 = 4.8 Hz, 1H), 4.71 (d, 7 = 5.3 Hz, 1H), 4.45 – 4.34 (m, 2H), 4.32 – 4.24 (m, 1H), 4.14 (t, / = 5.8 Hz, 1H), 4.08 – 3.94 (m, 2H), 3.93 – 3.85 (m, 1H), 1.47 (sep, / = 6.2 Hz, 1H), 1.38 – 1.26 (m, 7H), 0.87 (t, / = 7.5 Hz, 6H). 31PNMR (162 MHz, CD3OD) δ 3.73 (s). HPLC (2- 98% MeCN-H20 gradient with 0.1% TFA modifier over 8.5 min, 1.5mL/min, Column: Phenomenex Kinetex C18, 2.6 urn 100 A, 4.6 x 100 mm ) tR = 5.629 min.
Example 5. (S)-2-ethylbutyl 2-(((S)-(((2R,3S,4R,5R)-5-(4-aminopyrrolor2J- f|[l,2,41triazin-7-yl)-5-cvano-3,4-dihvdroxytetrahvdrofuran-2- yl)methoxy)(phenoxy)phosphoryl)amino)propanoate (32)

[0255] The preparation of (S)-2-ethylbutyl 2-(((S)-(((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,l f][l,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2- yl)methoxy)(phenoxy)phosphoryl)amino)propanoate is described below.
Preparation of (3R,4R,5R)-3,4-bis(benzyloxy)-5-((benzyloxy)methyl)dihydrofuran-2(3H)- one.

[0256] (3R,4R,5R)-3,4-bis(benzyloxy)-5-((benzyloxy)methyl)tetrahydrofuran-2-ol (15.0g) was combined with MTBE (60.0 mL), KBr (424.5 mg), aqueous K2HP04solution (2.5M, 14.3 mL), and TEMPO (56 mg). This mixture was cooled to about 1 °C. Aqueous bleach solution (7.9%wt.) was slowly charged in portions until complete consumption of starting material as indicated through a starch/iodide test. The layers were separated, and the aqueous layer was extracted with MTBE. The combined organic phase was dried over MgS04 and concentrated under reduced pressure to yield the product as a solid.
Preparation (4-amino-7-iodopyrrolor2,l-fl ri,2,41triazine)

[0257] To a cold solution of 4-aminopyrrolo[2, l-f][l,2,4]-triazine (10.03 g; 74.8 mmol) in N,N-dimethylformamide (70.27 g), N-iodosuccinimide (17.01g; 75.6 mmol) was charged in portions, while keeping the contents at about 0 °C. Upon reaction completion (about 3 h at about 0 °C), the reaction mixture was transferred into a 1 M sodium hydroxide aqueous solution (11 g NaOH and 276 mL water) while keeping the contents at about 20-30 °C. The resulting slurry was agitated at about 22 °C for 1.5 h and then filtered. The solids are rinsed with water (50 mL) and dried at about 50 °C under vacuum to yield 4-amino-7- iodopyrrolo[2,l-f] [l,2,4]triazine as a solid. !H NMR (400 MHz, DMSO-d6) δ 7.90 (s, 1H), 7.78 (br s, 2H), 6.98 (d, J = 4.4 Hz, 1H), 6.82 (d, J = 4.4 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 155.7, 149.1, 118.8, 118.1, 104.4, 71.9. MS m/z = 260.97 [M+H].
Preparation (3R,4R,5R)-2-(4-aminopyrrolor2, l-firi,2,41triazin-7-yl)-3,4-bis(benzyloxy)-5- ((benzyloxy)methyl)tetrahvdrofuran-2-ol via (4-amino-7-iodopyrrolor2,l-fl ri,2,41triazine)

[0258] To a reactor under a nitrogen atmosphere was charged iodobase 2 (81 g) and THF (1.6 LV). The resulting solution was cooled to about 5 °C, and TMSC1 (68 g) was charged. PhMgCl (345mL, 1.8 M in THF) was then charged slowly while maintaining an internal temperature at about < 5°C. The reaction mixture was stirred at about 0°C for 30 min, and then cooled to about -15 °C. z‘PrMgCl-LiCl (311 mL, 1.1 M in THF) was charged slowly while maintaining an internal temperature below about -12 °C. After about 10 minutes of stirring at about -15 °C, the reaction mixture was cooled to about -20 °C, and a solution of lactone 1 (130 g) in THF (400 mL) was charged. The reaction mixture was then agitated at about -20 °C for about 1 h and quenched with AcOH (57 mL). The reaction mixture was warmed to about 0 °C and adjusted to pH 7-8 with aqueous NaHCC>3 (5 wt%, 1300 mL). The reaction mixture was then diluted with EtOAc (1300 mL), and the organic and aqueous layers were separated. The organic layer was washed with IN HC1 (1300 mL), aqueous NaHCC>3 (5 wt%, 1300 mL), and brine (1300 mL), and then dried over anhydrous Na2S04 and concentrated to dryness. Purification by silica gel column chromatography using a gradient consisting of a mixture of MeOH and EtOAc afforded the product.
Preparation ((2S)-2-ethylbutyl 2- (((perfluorophenoxy)(phenoxy)phosphoryl)amino)propanoate) (mixture of Sp and Rp):
1 ) phenyl dichlorophosphate
CH2CI2, -78 °C to ambient
2) pentafluorophenol
Et3N, 0 °C to ambient

[0259] L- Alanine 2-ethylbutyl ester hydrochloride (5.0 g, 23.84 mmol) was combined with methylene chloride (40 mL), cooled to about -78 °C, and phenyl dichlorophosphate (3.65 mL, 23.84 mmol) was added. Triethylamine (6.6 mL, 47.68 mmol) was added over about 60 min at about -78 °C and the resulting mixture was stirred at ambient temperature for 3h. The reaction mixture was cooled to about 0 °C and pentafluorophenol (4.4 g, 23.84 mmol) was added. Triethylamine (3.3 mL, 23.84 mmol) was added over about 60 min. The mixture was stirred for about 3h at ambient temperature and concentrated under reduced pressure. The residue was dissolved in EtOAc, washed with an aqueous sodium carbonate solution several times, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using a gradient of EtOAc and hexanes (0 to 30%). Product containing fractions were concentrated under reduced pressure to give (2S)-2-ethylbutyl 2-(((perfluorophenoxy)(phenoxy)phosphoryl)amino)propanoate as a solid. H NMR (400 MHz, Chloroform-d) δ 7.41 – 7.32 (m, 4H), 7.30 – 7.17 (m, 6H), 4.24 – 4.16 (m, 1H), 4.13 – 4.03 (m, 4H), 4.01 – 3.89 (m, 1H), 1.59 – 1.42 (m, 8H), 1.40 – 1.31 (m, 8H), 0.88 (t, J = 7.5 Hz, 12H). 31P NMR (162 MHz, Chloroform-d) δ – 1.52. 19F NMR (377 MHz, Chloroform-d) δ – 153.63, – 153.93 (m), – 160.05 (td, J = 21.9, 3.6 Hz), – 162.65 (qd, J = 22.4, 20.5, 4.5 Hz). MS m/z = 496 [M+H]. Preparation of Title Compound (mixture of Sp and Rp):
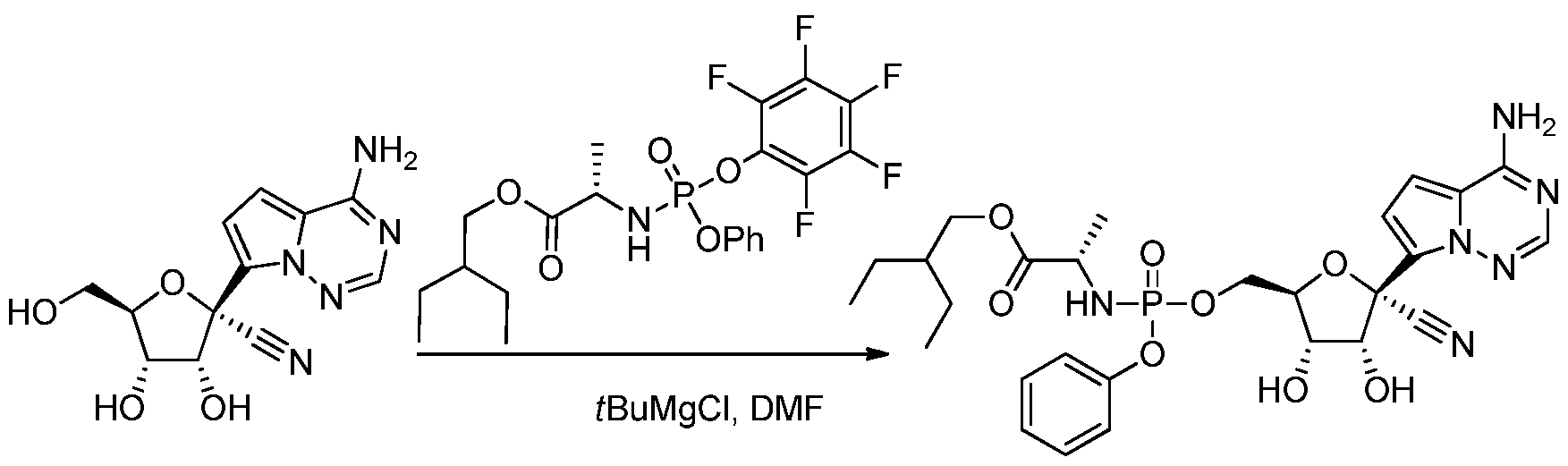
[0260] The nucleoside (29 mg, 0.1 mmol) and the phosphonamide (60 mg, 0.12 mmol) and N,N-dimethylformamide (2 mL) were combined at ambient temperature. 7¾ri-Butyl magnesiumchloride (1M in THF, 0.15 mL) was slowly added. After about lh, the reaction was diluted with ethyl acetate, washed with aqueous citric acid solution (5%wt.), aqueous saturated NaHC03 solution and saturated brine solution. The organic phase was dried over Na2S04 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using a gradient of methanol and CH2CI2 (0 to 5%). Product containing fractions were concentrated under reduced pressure to provide the product.
Preparation of (3aR,4R,6R,6aR)-4-(4-aminopyrrolor2, l-firi,2,41triazin-7-yl)-6- (hvdroxymethyl)-2,2-dimethyltetrahydrofuror3,4-diri,31dioxole-4-carbonitrile:

[0261] To a mixture of (2R,3R,4S,5R)-2-(4-aminopyrrolo[2, l-f] [l,2,4]triazin-7-yl)-3,4- dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-carbonitrile (5.8g, 0.02 mol), 2,2- dimethoxypropane (11.59 mL, 0.09 mol) and acetone (145 mL) at ambient temperature was added sulfuric acid (18M, 1.44 mL). The mixture was warmed to about 45 °C. After about 30 min, the mixture was cooled to ambient temperature and sodium bicarbonate (5.8 g) and water 5.8 mL) were added. After 15 min, the mixture was concentrated under reduced pressure. The residue was taken up in ethyl acetate (150 mL) and water (50 mL). The aqueous layer was extracted with ethyl acetate (2 x 50 mL). The combined organic phase was dried over sodium sulfate and concentrated under reduced pressure to give crude (2R,3R,4S,5R)-2-(4-aminopyrrolo[2, l-f] [l,2,4]triazin-7-yl)-3,4-dihydroxy-5- (hydroxymethyl)tetrahydrofuran-2-carbonitrile. !H NMR (400 MHz, CD3OD) δ 7.84 (s, 1H), 6.93 (d, / = 4.6 Hz, 1H), 6.89 (d, / = 4.6 Hz, 1H), 5.40 (d, / = 6.7 Hz, 1H), 5.00 (dd, / = 6.7, 3.3 Hz, 1H), 4.48 – 4.40 (m, 1H), 3.81 – 3.72 (m, 2H), 1.71 (s, 3H), 1.40 (s, 3H). MS m/z = 332.23 [M+l].
Preparation of (2S)-2-ethylbutyl 2-(((((2R,3S,4R,5R)-5-(4-aminopyrrolor2,l-firi,2,41triazin- 7-yl)-5-cvano-3,4-dihvdroxytetrahydrofuran-2- yl)methoxy)(phenoxy)phosphoryl)amino)propanoate:

[0262] Acetonitrile (100 mL) was combined with (2S)-2-ethylbutyl 2-(((4- nitrophenoxy)(phenoxy)phosphoryl)-amino)propanoate (9.6 g, 21.31 mmol), the substrate alcohol (6.6 g, 0.02 mol), magnesium chloride (1.9 g, 19.91 mmol) at ambient temperature. The mixture was agitated for about 15 min and N,N-diisopropylethylamine (8.67 mL, 49.78 mmol) was added. After about 4h, the reaction was diluted with ethyl acetate (100 mL), cooled to about 0 °C and combined with aqueous citric acid solution (5%wt., 100 mL). The organic phase was washed with aqueous citric acid solution (5%wt., 100 mL) and aqueous saturated ammonium chloride solution (40 mL), aqueous potassium carbonate solution
(10%wt., 2 x 100 mL), and aqueous saturated brine solution (100 mL). The organic phase was dried with sodium sulfate and concentrated under reduced pressure to provide crude product. !H NMR (400 MHz, CD3OD) δ 7.86 (s, 1H), 7.31 – 7.22 (m, 2H), 7.17 – 7.09 (m, 3H), 6.93 – 6.84 (m, 2H), 5.34 (d, / = 6.7 Hz, 1H), 4.98 (dd, / = 6.6, 3.5 Hz, 1H), 4.59 – 4.50 (m, 1H), 4.36 – 4.22 (m, 2H), 4.02 (dd, / = 10.9, 5.7 Hz, 1H), 3.91 (dd, / = 10.9, 5.7 Hz, 1H), 3.83 (dq, / = 9.7, 7.1 Hz, 1H), 1.70 (s, 3H), 1.50 – 1.41 (m, 1H), 1.39 (s, 3H), 1.36 – 1.21 (m, 7H), 0.86 (t, / = 7.4 Hz, 6H). MS m/z = 643.21 [M+l]. Preparation of (S)-2-ethylbutyl 2-(((S)-(((2R.3S.4R.5R)-5-(4-aminopyrrolor2.1- firi,2,41triazin-7-yl)-5-cvano-3,4-ditivdroxytetratiydrofuran-2- yl)methoxy)( henoxy)phosphoryl)amino)propanoate (Compound 32)

Compound 32
[0263] The crude acetonide (12.85 g) was combined with tetrahydrofuran (50 mL) and concentrated under reduced pressure. The residue was taken up in tetrahydrofuran (100 mL), cooled to about 0 °C and concentrated HC1 (20 mL) was slowly added. The mixture was allowed to warm to ambient temperature. After consumption of the starting acetonide as indicated by HPLC analysis, water (100 mL) was added followed by aqueous saturated sodium bicarbonate solution (200 mL). The mixture was extracted with ethyl acetate (100 mL), the organic phase washed with aqueous saturated brine solution (50 mL), dried over sodium sulfated and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using a gradient of methanol and ethyl acetate (0 to 20%).
Product containing fractions were concentrated under reduced pressure to provide the product.
PATENT
US 20170071964
US 20160122374
PAPER
Journal of Medicinal Chemistry (2017), 60(5), 1648-1661.
https://pubs.acs.org/doi/full/10.1021/acs.jmedchem.6b01594

The recent Ebola virus (EBOV) outbreak in West Africa was the largest recorded in history with over 28,000 cases, resulting in >11,000 deaths including >500 healthcare workers. A focused screening and lead optimization effort identified 4b (GS-5734) with anti-EBOV EC50 = 86 nM in macrophages as the clinical candidate. Structure activity relationships established that the 1′-CN group and C-linked nucleobase were critical for optimal anti-EBOV potency and selectivity against host polymerases. A robust diastereoselective synthesis provided sufficient quantities of 4b to enable preclinical efficacy in a non-human-primate EBOV challenge model. Once-daily 10 mg/kg iv treatment on days 3–14 postinfection had a significant effect on viremia and mortality, resulting in 100% survival of infected treated animals [ Nature 2016, 531, 381−385]. A phase 2 study (PREVAIL IV) is currently enrolling and will evaluate the effect of 4b on viral shedding from sanctuary sites in EBOV survivors.
(S)-2-Ethylbutyl 2-(((S)-(((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)propanoate (4b)
Compound 4b was prepared from 4 and 22b as described previously.(17)1H NMR (400 MHz, methanol-d4): δ 7.86 (s, 1H), 7.33–7.26 (m, 2H), 7.21–7.12 (m, 3H), 6.91 (d, J = 4.6 Hz, 1H), 6.87 (d, J = 4.6 Hz, 1H), 4.79 (d, J = 5.4 Hz, 1H), 4.43–4.34 (m, 2H), 4.28 (ddd, J = 10.3, 5.9, 4.2 Hz, 1H), 4.17 (t, J = 5.6 Hz, 1H), 4.02 (dd, J = 10.9, 5.8 Hz, 1H), 3.96–3.85 (m, 2H), 1.49–1.41 (m, 1H), 1.35–1.27 (m, 8H), 0.85 (t, J = 7.4 Hz, 6H).
13C NMR (100 MHz, methanol-d4): δ 174.98, 174.92, 157.18, 152.14, 152.07, 148.27, 130.68, 126.04, 125.51, 121.33, 121.28, 117.90, 117.58, 112.29, 102.60, 84.31, 84.22, 81.26, 75.63, 71.63, 68.10, 67.17, 67.12, 51.46, 41.65, 24.19, 20.56, 20.50, 11.33, 11.28.
31P NMR (162 MHz, methanol-d4): δ 3.66 (s).
HRMS (m/z): [M]+ calcd for C27H35N6O8P, 602.2254; found, 602.2274.
[α]21D – 21 (c 1.0, MeOH).
PAPER
Nature (London, United Kingdom) (2016), 531(7594), 381-385.
https://www.nature.com/articles/nature17180
 |
|
| Clinical data | |
|---|---|
| Other names | GS-5734 |
| Routes of administration |
By mouth, insufflation |
| ATC code |
|
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C27H35N6O8P |
| Molar mass | 602.585 g·mol−1 |
| 3D model (JSmol) | |
|
|
| Clinical data | |
|---|---|
| Other names | GS-5734 |
| Routes of administration |
By mouth, insufflation |
| ATC code |
|
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C27H35N6O8P |
| Molar mass | 602.585 g·mol−1 |
| 3D model (JSmol) | |
//////////////Remdesivir, レムデシビル , UNII:3QKI37EEHE, ремдесивир , ريمديسيفير , 瑞德西韦 , GS-5734 , GS 5734, PHASE 3 , CORONOVIRUS, COVID-19
CCC(CC)COC(=O)[C@H](C)N[P@](=O)(OC[C@H]1O[C@](C#N)([C@H](O)[C@@H]1O)c2ccc3c(N)ncnn23)Oc4ccccc4
NEW DRUG APPROVALS
ONE TIME
$10.00
Coblopasvir

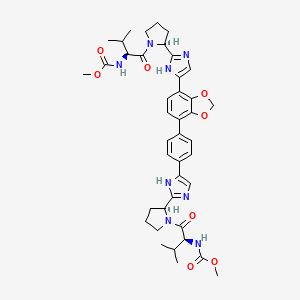
Coblopasvir
CAS: 1312608-46-0
Chemical Formula: C41H50N8O8
Molecular Weight: 782.89
methyl {(2S)-1-[(2S)-2-(4-{4-[7-(2-[(2S)-1-{(2S)-2- [(methoxycarbonyl)amino]-3-methylbutanoyl}pyrrolidin-2-yl]-1H-imidazol-4-yl)-2H-1,3-benzodioxol-4-yl]phenyl}-1Himidazol-2-yl)pyrrolidin-1-yl]-3-methyl-1-oxobutan-2-yl}carbamate
hepatitis C virus infection
KW-136
Coblopasvir is an antiviral drug candidate.
Coblopasvir dihydrochloride

CAS 1966138-53-3
| C41 H50 N8 O8 . 2 Cl H | |
| Molecular Weight | 855.806 |
| PHASE 3 | Beijing Kawin Technology Share-Holding |
Hepatitis C virus (HCV), or hepatitis C virus infection, is a chronic blood-borne infection. Studies have shown that 40% of chronic liver diseases are associated with HCV infection, and an estimated 8,000-10,000 people die each year. HCV-related end-stage liver disease is the most common indication for liver transplantation in adults.
In the past ten years, antiviral therapy for chronic liver disease has developed rapidly, and significant improvement has been seen in the treatment effect. However, even with the combination therapy with pegylated IFN-α plus ribavirin, 40% to 50% of patients fail to treat, that is, they are non-responders or relapsers. These patients do not currently have an effective treatment alternative. Because the risk of HCV-related chronic liver disease is related to the duration of HCV infection, and the risk of cirrhosis increases in patients who have been infected for more than 20 years, chronic liver disease often progresses to advanced stages with cirrhosis, ascites, jaundice, and rupture of varicose veins. , Brain disease, and progressive liver failure, and the risk of liver cancer is also significantly increased.
HCV is a enveloped positive-strand RNA virus of the Flaviviridae family. The single-stranded HCV RNA genome is approximately 9500 nucleotides in length and has a single open reading frame (ORF) that encodes a single open reading frame (ORF) of approximately 3,000 amino acids. Mostly polyprotein. In infected cells, cellular and viral proteases cleave this polyprotein at multiple sites to produce viral structural and non-structural (NS) proteins. There are two viral proteases that affect the production of mature non-structural proteins (NS2, NS3, NS4, NS4A, NS4B, NS5A, and NS5B). The first viral protease is cleaved at the NS2-NS3 junction of the polyprotein; the second viral protease is A “NS3 protease” that mediates all subsequent cleavage events at a site downstream of the NS3 position relative to the polyprotein (ie, the site between the C-terminus of NS3 and the C-terminus of the polyprotein). The NS3 protease exhibits cis-activity at the NS3-NS4 cleavage site and, conversely, exhibits trans-activity at the remaining NS4A-NS4B, NS4B-NS5A, and NS5A-NS5B sites. The NS4A protein is thought to provide multiple functions, such as acting as a cofactor for the NS3 protease, and may promote membrane localization of NS3 and other viral replicase components. The formation of a complex between NS3 and NS4A may be necessary for NS3-mediated processing events and improves the proteolytic efficiency at all sites recognized by NS3. NS3 protease may also exhibit nucleotide triphosphatase and RNA helicase activity. NS5B is an RNA-dependent RNA polymerase involved in HCV RNA replication. In addition, compounds that inhibit the effects of NS5A in viral replication may be useful for treating HCV.
Beijing Kawin Technology Share-Holding, in collaboration with Beijing Fu Rui Tiancheng Biotechnology and Ginkgo Pharma , is developing coblopasvir as an oral capsule formulation of dihydrochloride salt (KW-136), for treating hepatitis C virus infection. In June 2018, an NDA was filed in China by Beijing Kawin Technology and Sichuan Qingmu Pharmaceutical . In August 2018, the application was granted Priority Review in China . Also, Beijing Kawin is investigating a tablet formulation of coblopasvir dihydrochloride.
PATENT
WO2011075607 , claiming substituted heterocyclic derivatives as HCV replication inhibitors useful for treating HCV infection and liver fibrosis, assigned to Beijing Kawin Technology Share-Holding Co Ltd and InterMune Inc ,
PATENT
CN 108675998
PATENT
WO-2020001089
Novel crystalline and amorphous forms of methyl carbamate compound, particularly coblopasvir dihydrochloride , (designated as Forms H) processes for their preparation, compositions and combinations comprising them are claimed. Also claim is an article or kit comprising a container and a package insert, wherein the container contains coblopasvir dihydrochloride.

Step 7
To a solution of compound 1-IXf (250 mg, 0.31 mmol) in toluene (10.0 mL) was added NH4OAc (4.0 g, 50 mmol) and the mixture was refluxed for 16 hours. The reaction mixture was diluted with ethyl acetate and washed with water and brine. The solvent was removed and the residue was purified by preparative HPLC to give Compound I (43.5 mg, yield 20%) as a white solid. MS (ESI) m / z (M + H) + 783.4.
Example 2 Preparation of a compound of formula II
Compound of formula (I) N-[(2S) -1-[(2S) -2- {4- [7- (4- {2-[(2S) -1-[(2S) -2-[(A Oxycarbonyl) amino] -3-methylbutanoyl] pyrrolidin-2-yl] -1H-imidazol-4-yl} phenyl) -2H-1,3-benzodioxo-4-yl] Preparation of -1H-imidazol-2-yl} pyrrolidin-1-yl] -3-methyl-1-oxobutane-2-yl] carbamate dihydrochloride
At room temperature, a solution of the pure product of structural formula I (800 g, 1.0 eq) and ethyl acetate (8 L) were sequentially added to a 20 L bottle and stirred. A 11.2% HCl / ethyl acetate solution (839 g) was added dropwise to the system, the temperature of the system was controlled at 15 ° C to 25 ° C, and the mixture was stirred for more than 3 hours to stop the reaction. The filter cake was filtered with ethyl acetate (2L). Wash the cake, bake the cake at a controlled temperature of 40-60 ° C, sample and test until the ethyl acetate residue is <0.5%, (about 73 hours of baking), to obtain the compound of formula II, off-white solid powder or granules, 774 g, HPLC Purity: 98.65%, yield: 88.5%, tested XRPD as amorphous.
///////////////Coblopasvir , KW-136, hepatitis C virus infection, CHINA, Beijing Kawin Technology, NDA, Phase III
O=C(OC)N[C@@H](C(C)C)C(N1[C@H](C2=NC(C3=CC=C(C4=C5OCOC5=C(C6=CNC([C@H]7N(C([C@@H](NC(OC)=O)C(C)C)=O)CCC7)=N6)C=C4)C=C3)=CN2)CCC1)=O
Benvitimod, Tapinarof, тапинароф , تابيناروف , 他匹那罗 ,
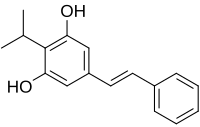
Benvitimod, Tapinarof
- Molecular FormulaC17H18O2
- Average mass254.324 Da
3,5-dihydroxy-4-isopropyl-trans-stilbene
Launched – 2019 CHINA, Psoriasis, Tianji Pharma
тапинароф [Russian] [INN]WBI-1001
تابيناروف [Arabic] [INN]
他匹那罗 [Chinese] [INN]
(E)-2-(1-Methylethyl)-5-(2-phenylethenyl)-1,3-benzenediol
1,3-Benzenediol, 2-(1-methylethyl)-5-(2-phenylethenyl)-, (E)-
1,3-Benzenediol, 2-(1-methylethyl)-5-[(E)-2-phenylethenyl]-
10253
2-Isopropyl-5-[(E)-2-phenylvinyl]-1,3-benzenediol
3,5-Dihydroxy-4-isopropyl-trans-stilbene
5-[(E)-2-phenylethenyl]-2-(propan-2-yl)benzene-1,3-diol
79338-84-4 [RN]
84HW7D0V04
Research Code:WB-1001; WBI-1001
Trade Name:MOA:NSAID
Indication:Atopic dermatitis; PsoriasisStatus:
Phase III (Active)
Company:GlaxoSmithKline (Originator), Welichem Biotech (Originator), 天济药业 (Originator)
2894512
DMVT-505
GSK-2894512
RVT-505
WB-1001
WBI-1001
84HW7D0V04 (UNII code)
DMVT-505
GSK-2894512
RVT-505
WB-1001
WBI-1001
84HW7D0V04 (UNII code)
In May 2019, the drug was appoved in China for the treatment of moderate stable psoriasis vulgaris in adults and, in July 2019, Tianji Pharma (subsidiary of Guanhao Biotech) launched the product in China for the treatment of moderate stable psoriasis vulgaris in adults.
Benvitimod is in phase III clinical trials, Dermavant Sciences for the treatment of atopic dermatitis and psoriasis.
The compound was co-developed by Welichem Biotech and Stiefel Laboratories (subsidiary of GSK). However, Shenzhen Celestial Pharmaceuticals acquired the developement rights in China, Taiwan, Macao and Hong Kong.
Benvitimod (also known as Tapinarof or 3,5-dihydroxy-4-isopropyl-trans-stilbene) is a bacterial stilbenoid produced in Photorhabdus bacterial symbionts of Heterorhabditis nematodes.It is a product of an alternative ketosynthase-directed stilbenoids biosynthesis pathway. It is derived from the condensation of two β-ketoacyl thioesters. It is produced by the Photorhabdus luminescens bacterial symbiont species of the entomopathogenic nematode, Heterorhabditis megidis.
Benvitimod (also known as tapinarof or 3,5-dihydroxy-4-isopropyl-trans-stilbene) is a bacterial stilbenoid produced in Photorhabdus bacterial symbionts of Heterorhabditis nematodes. It is a product of an alternative ketosynthase-directed stilbenoids biosynthesis pathway. It is derived from the condensation of two β-ketoacyl thioesters .[1] It is produced by the Photorhabdus luminescens bacterial symbiont species of the entomopathogenic nematode, Heterorhabditis megidis. Experiments with infected larvae of Galleria mellonella, the wax moth, support the hypothesis that the compound has antibiotic properties that help minimize competition from other microorganisms and prevents the putrefaction of the nematode-infected insect cadaver.[2]
Tapinarof is a non-steroidal anti-inflammatory drug originated by Welichem Biotech. Dermavant Sciences is developing the product outside China in phase III clinical trials for the treatment of plaque psoriasis. The company is also conducting phase II clinical trials for the treatment of atopic dermatitis. Phase II studies had also been conducted by Welichem Biotech and Stiefel (subsidiary of GlaxoSmithKline) for these indications.
Tapinarof was originated at Welichem Biotech, from which Tianji Pharma and Shenzen Celestial Pharmaceuticals obtained rights to the product in the Greater China region in 2005. In 2012, Welichem licensed development and commercialization rights in all other regions to Stiefel. In 2013, Welichem entered into an asset purchase agreement to regain Greater China rights to the product from Tianji Pharma and Celestial; however, this agreement was terminated in 2014. In 2018, Stiefel transferred its product license to Dermavant Sciences.
_Poinar,_1975.jpg)
Entomopathogenic nematodesemerging from a wax moth cadaver
//////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Medical research
Benvitimod is being studied in clinical trials for the treatment of plaque psoriasis.[3]
PATENTS
Route 1


1. US2003171429A1.
2. US2005059733A1.
Route 2


Reference:1. CN103265412A.


Patent
https://patents.google.com/patent/CN103992212A/en
phenalkenyl Maude (Benvitimod) is a new generation of anti-inflammatory drugs, are useful for treating a variety of major autoimmune diseases, such as psoriasis, eczema, hair and more concentrated colitis allergic diseases.Phenalkenyl Maud stilbene compound, comprising cis and trans isomers, the trans alkenyl benzene Maude has a strong physiological activity, stability and physical and chemical properties, and cis alkenyl benzene Modesto predominantly trans phenalkenyl Maud byproducts during synthesis, conventional methods such as benzene alkenyl Maude Wittig reaction of cis-isomer impurity is inevitable.

[0004] benzyl trans-alkenyl Maude as main impurities in the synthesis, whether a drug is detected, or monitored during the reaction, the synthesis and analysis methods established cis alkenyl benzene Maude has very important significance.Phenalkenyl Maud conventional synthetic methods the impurity content is very low, and the properties of the cis compound is extremely unstable, easily converted to trans-structure, the synthetic method according to the preceding, the cis compound difficult to separate. The synthesis method has not been reported before in the literature. Thus, to find a synthesis route of cis-alkenyl benzene Maude critical.
[0005] The synthesis of compounds of cis-stilbene, in the prior art, there have been many reports, however, the prior art method of synthesizing a reaction product of the cis starting materials and reagents difficult source, the catalyst used is expensive higher costs, operational difficulties, is not conducive to large-scale production, such as:
① Gaukroger K, John A.Hadfield.Novel syntheses of cis and trans isomers ofcombretastatin A-4 [J] .J.0rg.Chemj 2001, (66): 8135-8138, instead of styrene and substituted phenyl bromide boric acid as the raw material, the Suzuki coupling reaction is a palladium catalyst, to give the cis compound, the reaction follows the formula:

Yield and selectivity of the process the structure is good, but the reaction is difficult source of raw materials, catalyst more expensive, limiting the use of this method.
[0006] ② Felix N, Ngassaj Erick A, Lindsey, Brandon Ej Haines.The first Cu- and
amine-free Sonogashira-type cross-coupling in the C_6 -alkynylation of protected
2, -deoxyadenosine [J] .Tetrahedron Letters, 2009, (65): 4085-4091, with a substituted phenethyl m
Alkynyl easily catalyst Pd / CaC03, Fe2 (CO) 9, Pd (OAc) 2 and the like produce cis compound to catalytic reduction. The reaction follows the formula:

Advantage of this method is stereospecific reduction of alkynes in the catalyst, to overcome the phenomenon of cis-trans isomerization of the Wittig reaction, but the reaction requires at _78 ° C, is not conducive to the operation, and the reagent sources difficult, expensive than high cost increase is not conducive to mass production.
[0007] ③ Belluci G, Chiappe C, Moro G L0.Crown ether catalyzed stereospecificsynthesis of Z_and E-stilbenes by Wittig reaction in a solid-liquid two-phasessystem [J] .Tetrahedron Letters, 1996, (37): 4225-4228 using Pd (PPh3) 4 as catalyst, an organic zinc reagent with a halide compound of cis-coupling reaction formula as follows:

The advantage of this method is that selective, high yield to give cis; deficiency is difficult to handle, the catalyst is expensive.
[0008] ④ new Wang, Zhangxue Jing, Zhou Yue, Zouyong Shun, trans-3,4 ‘, 5-trihydroxy-stilbene China Pharmaceutical Synthesis, 2005, 14 (4);. 204-208, reported that the trans compound of formula was dissolved in DMSO solution at a concentration dubbed, ultraviolet irradiation was reacted at 365nm, converted into cis compounds, see the following reaction formula:

However, the concentration of the solution preparation method, the reaction time is more stringent requirements.

The synthesis of cis-alkenyl benzene Maude application embodiments Example 1 A synthesis of cis-alkenyl Maude benzene and benzene-cis-ene prepared Maude, the reaction was carried out according to the following scheme:

Specific preparation process steps performed in the following order:
(O methylation reaction
The 195.12g (Imol) of 3, 5-hydroxy-4-isopropyl benzoic acid, 414.57g (3mol) in DMF was added 5000ml anhydrous potassium carbonate, mixing, stirred at room temperature, then cooled in an ice-salt bath next, slowly added dropwise 425.85g (3mol) of iodomethane, warmed to room temperature after the addition was complete, the reaction 2h, after completion of the reaction was stirred with water, extracted with ethyl acetate, and concentrated to give 3,5-dimethoxy-4- isopropyl benzoate; yield 93%, purity of 99%.
[0033] (2) a reduction reaction
3000ml tetrahydrofuran and 240g (Imol) 3,5-dimethoxy-4-isopropyl benzoate, 151.40g (4mol) mixing at room temperature sodium borohydride was stirred and heated to reflux was slowly added dropwise 400ml methanol, reaction 4h, was added 3L of water was stirred, extracted with ethyl acetate, washed with water, the solvent was removed by rotary evaporation to give a white solid, to give 3,5-dimethoxy-4-isopropylbenzene methanol; 96% yield purity was 99%.
[0034] (3) the oxidation reaction
The 212g (ImoI) of 3,5-dimethoxy-4-isopropylbenzene methanol, DMSO 800ml and 500ml of acetic anhydride were mixed and stirred at rt After 2h, stirred with water, extracted with ethyl acetate, washed with water, dried , and concentrated to give 3,5-dimethoxy-4-isopropyl-benzaldehyde; 94% yield, 99% purity.
[0035] (4) a condensation reaction
The mixture was 209.18g (lmol) of 3,5-dimethoxy-4-isopropyl-benzoic awake and 136.15g (Imol) phenylacetic acid was added 5000ml of acetic anhydride, stirred to dissolve, sodium acetate was added 246.09g , heating to 135 ° C, the reaction after 6h, cooled to room temperature after adjusting the dilute acid 2 was added, extracted with ethyl acetate, the pH was concentrated, added saturated sodium bicarbonate solution adjusted to pH 7, stirred 2h, and extracted with dichloromethane , adding dilute aqueous hydrochloric acid pH 2, the yellow solid was filtered, to obtain 3,5-dimethoxy-4-isopropyl-stilbene acid; 96% yield, 80% purity.
[0036] (5) decarboxylation reaction
The 327g (Imol) of 3,5-dimethoxy-4-isopropyl-stilbene acid and 384g (6mol) of copper powder were added to 5000ml of quinoline, 180 ° C reaction 3h, cooled to room temperature ethyl acetate was added with stirring, filtered, and the filtrate was washed with dilute hydrochloric acid to the aqueous layer was colorless and the aqueous phase was extracted with ethyl acetate inverted, the organic layers were combined, washed with water and saturated brine until neutral, i.e., spin-dried to give 3,5 – dimethoxy-4-isopropyl-stilbene; 92% yield, 77% purity.
[0037] (6) Demethylation
The 282.32g (Imol) of 3,5-dimethoxy-4-isopropyl-stilbene 4000ml toluene was placed in an ice bath and stirring, was cooled to 0 ° C, and dissolved slowly added 605.9g (5mol after) in N, N- dimethylaniline, was added 666.7g (5mol) of anhydrous aluminum chloride. after stirring for 0.5h, warmed to room temperature, the reaction was heated to 100 ° C 2h, cooled to 60 ° C , hot toluene layer was separated, diluted hydrochloric acid was added to the aqueous phase with stirring to adjust the PH value of 2, extracted with ethyl acetate, washed with water, and concentrated to give the cis-alkenyl benzene Modesto; crude yield 95%, purity 74 %.After separation by column chromatography using 300-400 mesh silica gel, benzene-cis-ene was isolated Maude pure, 68% yield, 98.5% purity. The resulting cis-alkenyl benzene Maud NMR shown in Figure 1, NMR data are as follows:
1HNMR (CDCl3, 500 Hz, δ: ppm), 7.255 (m, 5H), 6.558 (d, 1H), 6.402 (d, 1H), 6.218 (s, 2H), 4.872 (s, 2H), 3.423 (m , 1H), 1.359 (q, 6H). Coupling constants / = 12.
[0038] trans-alkenyl benzene Maud NMR shown in Figure 2, the following NMR data:
1HNMR (CDCl3, 500 Hz, δ: ppm), 7.477 (d, 2H), 7.360 (t, 2H), 6.969 (q, 2H), 6.501 (s, 1H), 4.722 (s, 2H), 3.486 (m , 1H), 1.380 (t, 6H). Coupling constants / = 16.
[0039] HPLC conditions a cis alkenyl benzene Maude pure product: column was Nucleosil 5 C18; column temperature was 20 ° C; detection wavelength 318nm; mobile phase consisting of 50:50 by volume of acetonitrile and water; flow rate It was 0.6mL / min, injection volume of 5 μ L; cis phenalkenyl Maude 18.423min retention time of a peak in an amount of 96.39%, see Figure 3. Trans phenalkenyl Maude 17.630min retention time of a peak, the content was 99.8%, see Figure 4.After mixing the two, trans-alkenyl benzene Maude 17.664min retention time of the peak, cis-alkenyl benzene Maude 18.458min retention time of the peak, see Figure 5.
PATENT
https://patents.google.com/patent/CN103172497A/en

phenalkenyl Maude is a natural product, a metabolite as to be symbionts.Phenalkenyl Maud Escherichia coli, Staphylococcus aureus has a very significant inhibitory effect, in addition, there is a styrenic Maude suppression of inflammation and its reactive derivative with immunomodulating activity. Alkenyl benzene Modesto topical ointment as an active ingredient, as a class of drugs has been completed two clinical treatment of psoriasis and eczema, the results of ongoing clinical phase III clinical studies, it has been shown to be completed in both psoriasis and eczema clearly effect, together with a styrenic Maude is a non-hormonal natural small molecule compounds, can be prepared synthetically prepared, therefore, it exhibits good market prospect.
[0004] a styrenic Maude initial synthesis route is as follows:
[0005]

[0006] The reaction conditions for each step: 1) isopropanol, 80% sulfuric acid, 60 ° C, 65% .2) sodium borohydride, boron trifluoride, tetrahydrofuran, 0 ° C, 90% .3). of thionyl chloride, heated under reflux, 85% .4). triethyl phosphate, 120 ° C, 80% .5). benzaldehyde, sodium hydride, 85% .6) pyridine hydrochloride, 190 ° C, 60 %.
[0007] The chemical synthesis route, although ultimately obtained a styrenic Maude, but the overall yield is low, part of the reaction step is not suitable for industrial production, due to process conditions result in the synthesis of certain byproducts produced is difficult to remove impurities, difficult to achieve the quality standard APIs.
Preparation of 4-isopropyl-dimethoxy-benzoic acid [0011] 1,3,5_
[0012] 1000 l reactor 200 liters of 80% sulfuric acid formulation (V / V), the temperature was lowered to room temperature, put 80 kg 3,5_-dimethoxybenzoate ,, stirring gradually warmed to 60 ° C, in was added dropwise within 25 kg of isopropanol I hour, the reaction was complete after 5 hours, 500 liters of hot water, filtered, the filter cake was washed with a small amount of hot water I th, crushed cake was removed and dried. The dried powder was recrystallized from toluene, the product was filtered to give 78 kg `, yield 86%. Preparation 2,3,5_ dimethoxy-4-isopropylbenzene methanol
[0013] 1000 l reactor was added 50 kg 3,5_ _4_ isopropyl dimethoxy benzoic acid, 24 kg of potassium borohydride, 400 l of THF, at room temperature was slowly added dropwise 65 kg BF3.Et2O was stirred 12 hours, the reaction was complete, pure water was added dropwise to destroy excess BF3, filtered, concentrated to dryness, methanol – water to give an off-white recrystallized 40.3 kg, yield 90.1%.
[0014] Preparation of 3,3,5-_ ■ methoxy _4- isopropyl group gas section
[0015] 1000 l autoclave, 100 kg of 3,5-dimethoxy-4-isopropylbenzene methanol, 220 l of DMF, 0 ° C and added dropwise with stirring and 50 l of thionyl chloride, 24 hours after the reaction was complete, 300 liters of water and 300 liters of ethyl acetate, the aqueous phase was stirred layered discharged, and then washed with 200 liters of water was added 3 times, until complete removal of DMF, was added concentrated crystallized from petroleum ether to give 98 kg of white solid was filtered and dried a yield of 91%.
Preparation of methyl-dimethoxy-4-isopropylbenzene of diethyl [0016] 4,3,5_
[0017] 500 l autoclave, 98 kg 3,5_ _4_ isopropyl dimethoxy benzyl chloride and 120 l of triethyl phosphite, the reaction at 120 ° C 5h, fear distilled off under reduced pressure, the collection 145-155 ° C / 4mmHg fear minutes, cured at room temperature to give a colorless light solid was 118 kg, yield 81.6%.
, 3- [0018] 5, E-1 _ ■ methoxy-2-isopropyl-5- (2-phenylethyl lean-yl) – benzene
[0019] 500 l autoclave, 33 kg 3,5_-dimethoxy-4-isopropylbenzene acid diethyl ester, 10.8 kg of benzaldehyde, and 120 l of tetrahydrofuran, at 40 ° C, and nitrogen with stirring, was added dropwise a solution of 11.8 kg potassium tert-butoxide in 50 liters of tetrahydrofuran, the temperature dropping control not to exceed 50 ° C. after the dropwise addition stirring was continued for I h, the reaction was complete, 150 liters of ethyl acetate and extracted , washed twice with 150 liters of water, 100 l I washed with brine, and the organic phase was dried and concentrated, methanol – water (I: D as a white crystalline solid 25.3 kg, yield 91%.
[0020] 6> 1, 3 ~ _ ■ Light-2-isopropyl-5- (2-phenylethyl lean-yl) – benzene (I), (De Dae dilute benzene)
[0021] 100 l autoclave, 10 kg 1,3_-dimethoxy-2-isopropyl-5- (2-styryl) benzene _ pyridine hydrochloride and 25 kg nitrogen atmosphere was heated to 180 -190 ° C, stirred for 3 hours after the reaction was completed, 20 l HCl (2N) cooling to 100 ° C, and 20 liters of ethyl acetate the product was extracted, dried and concentrated to give the product 7.3 kg, 83% yield.
[0022] The method for purifying:
[0023] 100 l added to the reaction vessel 15.5 kg of crude product and 39 liters of toluene, heated to the solid all dissolved completely, filtered hot and left to crystallize, after crystallization, filtration, the crystals with cold toluene 10 washed liter at 60 ° C, protected from light vacuo dried for 24 hours, to obtain 14 kg of white needle crystals, yield 90%.
CLIP
https://www.eosmedchem.com/article/237.html
Design new synthesis of Route of Benvitimod
Nov 26, 2018
1.Benvitimod and intermediates
Benvitimod 79338-84-4 intermediate: 1999-10-5
Benvitimod 79338-84-4 intermediate: 2150-37-0
Benvitimod 79338-84-4 intermediate: 344396-17-4
Benvitimod 79338-84-4 intermediate: 344396-18-5
Benvitimod 79338-84-4 intermediate: 344396-19-6
Benvitimod 79338-84-4 intermediate: 1080-32-6
Benvitimod 79338-84-4 intermediate: 678986-73-7
Benvitimod 79338-84-4 intermediate: 55703-81-6
Benvitimod 79338-84-4 intermediate: 1190122-19-0
Benvitimod 79338-84-4 intermediate: 443982-76-1
Benvitimod 79338-84-4 intermediate: 100-52-72.ROS-Benvitimod
(1)

(2)
 3.
3.
Name: Benvitimod
CAS#: 79338-84-4
Chemical Formula: C17H18O2
Exact Mass: 254.1307
Molecular Weight: 254.329
Elemental Analysis: C, 80.28; H, 7.13; O, 12.58
Benvitimod 79338-84-4 intermediate: 1999-10-5
Benvitimod 79338-84-4 intermediate: 2150-37-0
Benvitimod 79338-84-4 intermediate: 344396-17-4
Benvitimod 79338-84-4 intermediate: 344396-18-5
Benvitimod 79338-84-4 intermediate: 344396-19-6
Benvitimod 79338-84-4 intermediate: 1080-32-6
Benvitimod 79338-84-4 intermediate: 678986-73-7
Benvitimod 79338-84-4 intermediate: 55703-81-6
Benvitimod 79338-84-4 intermediate: 1190122-19-0
Benvitimod 79338-84-4 intermediate: 443982-76-1
Benvitimod 79338-84-4 intermediate: 100-52-72.ROS-Benvitimod
(1)
(2)
Name: Benvitimod
CAS#: 79338-84-4
Chemical Formula: C17H18O2
Exact Mass: 254.1307
Molecular Weight: 254.329
Elemental Analysis: C, 80.28; H, 7.13; O, 12.58
References
- ^ Joyce SA; Brachmann AO; Glazer I; Lango L; Schwär G; Clarke DJ; Bode HB (2008). “Bacterial biosynthesis of a multipotent stilbene”. Angew Chem Int Ed Engl. 47 (10): 1942–5. doi:10.1002/anie.200705148. PMID 18236486.
- ^ Hu, K; Webster, JM (2000). “Antibiotic production in relation to bacterial growth and nematode development in Photorhabdus–Heterorhabditis infected Galleria mellonella larvae”. FEMS Microbiology Letters. 189 (2): 219–23. doi:10.1111/j.1574-6968.2000.tb09234.x. PMID 10930742.
- ^ “New Topical for Mild to Moderate Psoriasis in the Works”. Medscape. March 5, 2017.
- https://onlinelibrary.wiley.com/action/downloadSupplement?doi=10.1002%2Fanie.201814016&file=anie201814016-sup-0001-misc_information.pdf
///Benvitimod, Tapinarof, WBI-1001, тапинароф , تابيناروف , 他匹那罗 , Welichem Biotech, Stiefel Laboratories, Shenzhen Celestial Pharmaceuticals,CHINA 2019 , Psoriasis, Tianji Pharma, Dermavant Sciences, PHASE 3, fda 2022, approvals 2022, vtama, tapinarof
update….
5/23/2022 fda approved, To treat plaque psoriasis, vtama, tapinarof
 |
|
| Names | |
|---|---|
| Preferred IUPAC name
5-[(E)-2-Phenylethen-1-yl]-2-(propan-2-yl)benzene-1,3-diol
|
|
| Other names
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChemSpider | |
|
PubChem CID
|
|
| UNII | |
| Properties | |
| C17H18O2 | |
| Molar mass | 254.329 g·mol−1 |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
Benvitimod (also known as tapinarof or 3,5-dihydroxy-4-isopropyl-trans-stilbene) is a bacterial stilbenoid produced in Photorhabdus bacterial symbionts of Heterorhabditis nematodes. It is a product of an alternative ketosynthase-directed stilbenoid biosynthesis pathway. It is derived from the condensation of two β-ketoacyl thioesters.[1] It is produced by the Photorhabdus luminescens bacterial symbiont species of the entomopathogenic nematode, Heterorhabditis megidis. Experiments with infected larvae of Galleria mellonella, the wax moth, support the hypothesis that the compound has antibiotic properties that help minimize competition from other microorganisms and prevents the putrefaction of the nematode-infected insect cadaver.[2]
Medical research
Benvitimod is being studied in clinical trials for the treatment of plaque psoriasis.[3]
See also
- Pinosylvin, a molecule produced in pines that does not bear the isopropyl alkylation.
References
- ^ Joyce SA; Brachmann AO; Glazer I; Lango L; Schwär G; Clarke DJ; Bode HB (2008). “Bacterial biosynthesis of a multipotent stilbene”. Angew Chem Int Ed Engl. 47 (10): 1942–5. CiteSeerX 10.1.1.603.247. doi:10.1002/anie.200705148. PMID 18236486.
- ^ Hu, K; Webster, JM (2000). “Antibiotic production in relation to bacterial growth and nematode development in Photorhabdus–Heterorhabditis infected Galleria mellonella larvae”. FEMS Microbiology Letters. 189 (2): 219–23. doi:10.1111/j.1574-6968.2000.tb09234.x. PMID 10930742.
- ^ “New Topical for Mild to Moderate Psoriasis in the Works”. Medscape. March 5, 2017.
