
Home » EU PIPELINE (Page 2)
Category Archives: EU PIPELINE
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Teva Pharmaceutical has been given a green light by the European Commission (EC) for Lonquex, a rival to Amgen’s blockbuster Neulasta.

lipegfilgrastim
C 864 H 1369 N 225 O 258 S 9 [C 2 H 4 O] N
pegylated granulocyte colony stimulating factor; O3.133-[N5-(N-{[ω-methoxypoly (oxyethylene)] carbonyl} glycyl)-α-neuraminyl-(2 → 6)-α-D-galactopyranosyl]-L-methionyl -des-1-L-alanine-des-37-L-valine-des-38-L-serine-des-39-L-glutamic acid-human granulocyte colony-stimulating factor (G-CSF, pluripoietin)
Lonquex (lipegfilgrastim) has been approved to reduce the duration of neutropaenia (low white blood cell counts) and febrile neutropaenia in patients undergoing cytotoxic chemotherapy for cancer, and is given as a single subcutaneous dose per cycle of chemotherapy.
Like Neulasta (pegfilgrastim), Lonquex is a long-acting recombinant granulocyte colony-stimulating factor (G-CSF) and is dosed at the same frequency as Amgen’s drug.
http://www.pmlive.com/pharma_news/neulasta_rival_from_teva_cleared_in_eu_495953
Biogen’s multiple sclerosis drug Tecfidera obtains EU approval
US-based Biogen Idec has received approval from the European Commission (EC) for its Tecfidera (dimethyl fumarate) as a first-line oral treatment for people with relapsing-remitting multiple sclerosis (RRMS), the most common form of multiple sclerosis (MS).
Biogen’s multiple sclerosis drug Tecfidera obtains EU approval click here
GSK’s diabetes drug Eperzan moves towards approval in Europe
GlaxoSmithKline has received a positive opinion from the European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) for albiglutide, under the brand name Eperzan, for treatment of type 2 diabetes.
CLICK ON TITLE
GSK’s diabetes drug Eperzan moves towards approval in Europe
Dolutegravir approved by the EU Commission (synthesis included in this post)

Dolutegravir
2H-Pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxamide, N-[(2,4-difluorophenyl)methyl]-3,4,6,8,12,12a-hexahydro-7-hydroxy-4-methyl-6,8-dioxo-, (4R,12aS)
(3R,11aS)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide
(4R,12aS)-N-(2,4-difluorobenzyl)-7-hydroxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxamide
Trade Name:Tivicay
Synonym:GSK1349572, S-349572, GSK572
Date of Approval: August 12, 2013 (US)
Indication:HIV infection
Drug class: Integrase strand transfer inhibitor
Company: ViiV Healthcare,GlaxoSmithKline
INNOVATOR …ViiV Healthcare
CAS number: 1051375-16-6
MF:C20H19F2N3O5
MW:419.4
Chemical Name: (4R,12aS)-N-[(2,4-difluorophenyl)methyl]-7-hydroxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a- hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxamide
Patent: US8129385
Patent expiration date: Oct 5, 2027
PCT patent application: W02006116764
Dolutegravir (DTG, GSK1349572) is an integrase inhibitor being developed for the treatment of human immunodeficiency virus (HIV)-1 infection by GlaxoSmithKline (GSK) on behalf of Shionogi-ViiV Healthcare LLC. DTG is metabolized primarily by uridine diphosphate glucuronyltransferase (UGT)1A1, with a minor role of cytochrome P450 (CYP)3A, and with renal elimination of unchanged drug being extremely low (< 1% of the dose).
The European Commission has on 21 January 2014 Dolutegravir (Tivicay, ViiV) permit as part of combination therapy for the treatment of HIV-infected persons over the age of 12 years.Dolutegravir (Tivicay, ViiV) is an integrase inhibitor, in combination with other antiretroviral drugs in adults and adolescents can be used from 12 years for the treatment of HIV infection.
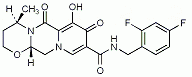
Source: Communication from the European Commission
Dolutegravir[1] is a FDA-approved drug[2] for the treatment of HIV infection. Dolutegravir is an integrase inhibitor. Known as S/GSK1349572 or just “572” the drug is marketed as Tivicay[3] by GlaxoSmithKline (GSK). In February, 2013 the Food and Drug Administration announced that it would fast track dolutegravir’s approval process.[4] On August 13, 2013, dolutegravir was approved by the FDA. On November 4, 2013, dolutegravir was approved by Health Canada.[5]
The oral HIV integrase inhibitor S-349572 was originated by Shionogi-GlaxoSmithKline and Shionogi-ViiV Healthcare. In 2013, the product was approved and launched in the U.S. for the treatment of HIV-1 in adults and children aged 12 years and older, in combination with other antiretroviral agents. A positive opinion was received in the E.U for this indication and, in 2014, approval was attained in Europe for this indication. Registration is pending in Japan.
In 2013, orphan drug designation in Japan was assigned to the compound.
Dolutegravir is approved for use in a broad population of HIV-infected patients. It can be used to treat HIV-infected adults who have never taken HIV therapy (treatment-naïve) and HIV-infected adults who have previously taken HIV therapy (treatment-experienced), including those who have been treated with other integrase strand transfer inhibitors. Tivicay is also approved for children ages 12 years and older weighing at least 40 kilograms (kg) who are treatment-naïve or treatment-experienced but have not previously taken other integrase strand transfer inhibitors.[6]
Dolutegravir has also been compared head-to-head with a preferred regimen from the DHHS guidelines in each of the three classes (i.e. 1.) nuc + non-nuc, 2.) nuc + boosted PI, and 3.) nuc + integrase inhibitor).
SPRING-2 compared dolutegravir to another integrase inhibitor, raltegravir, with both coformulated with a choice of TDF/FTC orABC/3TC. After 48 weeks of treatment 88% of those on dolutegravir had less than 50 copies of HIV per mL compared to 85% in the raltegravir group, thus demonstrating non-inferiority.[9]
The FLAMINGO study has been presented at scientific meetings but as of early 2014 has not yet been published. It is an open-label trial of dolutegravir versus darunavir boosted with ritonavir. In this trial 90% of those on dolutegravir based regimens had viral loads < 50 at 48 weeks compared to 83% in the darunavir/r.[10] This 7% difference was statistically significant for superiority of the dolutegravir based regimens.
Another trial comparing dolutegravir to efavirenz, SINGLE, was the first trial to show statistical superiority to an efavirenz/FTC/TDF coformulated regimen for treatment naive patients.[11] After 48 weeks of treatment, 88% of the dolutegravir group had HIV RNA levels < 50 copies / mL versus 81% of the efavirenz group. This has led one commentator to predict that it may replace efavirenz as the first line choice for initial therapy as it can also be formulated in one pill, once-a-day regimens.[12]
Doultegravir has also been studied in patients who have been on previous antiretroviral medications. The VIKING trial looked at patients who had known resistance to the first generation integrase inhibitor raltegravir. After 24 weeks 41% of patients on 50mg dolutegravir once daily and 75% of patients on 50mg twice daily (both along with an optimized background regimen) achieved an HIV RNA viral load of < 50 copies per mL. This demonstrated that there was little clinical cross-resistance between the two integrase inhibitors. [13]
Dolutegravir (also known as S/GSK1349572), a second-generation integrase inhibitor under development by GlaxoSmithKline and its Japanese partner Shionogi for the treatment of HIV infection, was given priority review status from the US Food and Drug Administration (FDA) in February, 2013.
GlaxoSmithKline marketed the first HIV drug Retrovir in 1987 before losing out to Gilead Sciences Inc. (GILD) as the world’s biggest maker of AIDS medicines. The virus became resistant to Retrovir when given on its own, leading to the development of therapeutic cocktails.
The new once-daily drug Dolutegravir, which belongs to a novel class known as integrase inhibitors that block the virus causing AIDS from entering cells, is owned by ViiV Healthcare, a joint venture focused on HIV in which GSK is the largest shareholder.
Raltegravir (brand name Isentress) received approval by the U.S. Food and Drug Administration (FDA) on 12 October 2007, the first of a new class of HIV drugs, the integrase inhibitors, to receive such approval. it is a potent and well tolerated antiviral agent. However, it has the limitations of twice-daily dosing and a relatively modest genetic barrier to the development of resistance, prompting the search for agents with once-daily dosing.
Elvitegravir, approved by the FDA on August 27, 2012 as part of theelvitegravir/cobicistat/tenofovir disoproxil fumarate/emtricitabine fixed-dose combination pill (Quad pill, brand name Stribild) has the benefit of being part of a one-pill, once-daily regimen, but suffers from extensive cross-resistance with raltegravir.
Gilead’s Atripla (Emtricitabine/Tenofovir/efavirenz), approved in 2006 with loss of patent protection in 20121, is the top-selling HIV treatment. The $3.2 billion medicine combines three drugs in one pill, two compounds that make up Gilead’s Truvada (Emtricitabine/Tenofovir) and Bristol- Myers Squibb Co.’s Sustiva (Efavirenz).
A three-drug combination containing dolutegravir and ViiV’s older two-in-one treatment Epzicom(Abacavir/Lamivudine, marketed outside US as Kivexa) proved better than Gilead’s market-leading Atripla in a clinical trial released in July, 2012 (See the Full Conference Report Here), suggesting it may supplant the world’s best-selling AIDS medicine as the preferred front-line therapy. In the latest Phase III study, after 48 weeks of treatment, 88% of patients taking the dolutegravir-based regimen had reduced viral levels to the goal compared with 81% of patients taking Atripla. More patients taking Atripla dropped out of the study because of adverse events compared with those taking dolutegravir — 10% versus just 2% — which was the main driver of the difference in efficacy. The result was the second positive final-stage clinical read-out for dolutegravir, following encouraging results against U.S. company Merck & Co’s rival Isentress in April, 2012 (See the Conference Abstract Here)..
Dolutegravir is viewed by analysts as a potential multibillion-dollar-a-year seller, as its once-daily dosing is likely to be attractive to patients. The FDA is scheduled to issue a decision on the drug’s approval by August 17。
TIVICAY contains dolutegravir, as dolutegravir sodium, an HIV INSTI. The chemical name of dolutegravir sodium is sodium (4R,12aS)-9-{[(2,4-difluorophenyl)methyl]carbamoyl}-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazin-7-olate. The empirical formula is C20H18F2N3NaO5 and the molecular weight is 441.36 g/mol. It has the following structural formula:
 |
Dolutegravir sodium is a white to light yellow powder and is slightly soluble in water.
Each film-coated tablet of TIVICAY for oral administration contains 52.6 mg of dolutegravir sodium, which is equivalent to 50 mg dolutegravir free acid, and the following inactive ingredients: D-mannitol, microcrystalline cellulose, povidone K29/32, sodium starch glycolate, and sodium stearyl fumarate. The tablet film-coating contains the inactive ingredients iron oxide yellow, macrogol/PEG, polyvinyl alcohol-part hydrolyzed, talc, and titanium dioxide.
……………………………………
INTRODUCTION
Among viruses, human immunodeficiency virus (HIV), a kind of retrovirus, is known to cause acquired immunodeficiency syndrome (AIDS). The therapeutic agent for AIDS is mainly selected from a group of reverse transcriptase inhibitors (e.g., AZT, 3TC) and protease inhibitors (e.g., Indinavir), but they are proved to be accompanied by side effects such as nephropathy and the emergence of resistant viruses. Thus, the development of anti-HIV agents having the other mechanism of action has been desired.
On the other hand, a combination therapy is reported to be efficient in treatment for AIDS because of the frequent emergence of the resistant mutant. Reverse transcriptase inhibitors and protease inhibitors are clinically used as an anti-HIV agent, however agents having the same mechanism of action often exhibit cross-resistance or only an additional activity. Therefore, anti-HIV agents having the other mechanism of action are desired.
Under the circumstances above, an HIV integrase inhibitor has been focused on as an anti-HIV agent having a novel mechanism of action (Ref: Patent Documents 1 and 2). As an anti-HIV agent having such a mechanism of action, known are carbamoyl-substituted hydroxypyrimidinone derivative (Ref: Patent Documents 3 and 4) and carbamoyl-substituted hydroxypyrrolidione derivative (Ref: Patent Document 5). Further, a patent application concerning carbamoyl-substituted hydroxypyridone derivative has been filed (Ref: Patent Document 6, Example 8).
Other known carbamoylpyridone derivatives include 5-alkoxypyridine-3-carboxamide derivatives and γ-pyrone-3-carboxamide derivatives, which are a plant growth inhibitor or herbicide (Ref: Patent Documents 7-9).
Other HIV integrase inhibitors include N-containing condensed cyclic compounds (Ref: Patent Document 10).
- [Patent Document 1] WO03/0166275
- [Patent Document 2] WO2004/024693
- [Patent Document 3] WO03/035076
- [Patent Document 4] WO03/035076
- [Patent Document 5] WO2004/004657
- [Patent Document 6] JP Patent Application 2003-32772
- [Patent Document 7] JP Patent Publication 1990-108668
- [Patent Document 8] JP Patent Publication 1990-108683
- [Patent Document 9] JP Patent Publication 1990-96506
- [Patent Document 10] WO2005/016927
-
Patent Document 1 describes compounds (I) and (II), which are useful as anti-HIV drugs and shown by formulae:

-
This document describes the following reaction formula as a method of producing compound (I).


-
Furthermore, Patent Documents 2 to 6 describe the following reaction formula as an improved method of producing compound (I).


-
- [Patent Document 1] International publication No.2006/116764 pamphlet
- [Patent Document 2] International publication No.2010/011812 pamphlet
- [Patent Document 3] International publication No.2010/011819 pamphlet
- [Patent Document 4] International publication No.2010/068262 pamphlet
- [Patent Document 5] International publication No.2010/067176 pamphlet
- [Patent Document 6] International publication No.2010/068253 pamphlet
- [Patent Document 7] US Patent 4769380A
- [Patent Document 8] International applicationPCT/JP2010/055316
- [PATENT DOCUMENTS]
[NON-PATENT DOCUMENTS]
-
- [Non-Patent Document 1] Journal of Organic Chemistry, 1991, 56(16), 4963-4967
- [Non-Patent Document 2] Science of Synthesis, 2005, 15, 285-387
- [Non-Patent Document 3] Journal of Chemical Society Parkin Transaction. 1, 1997, Issue. 2, 163-169
-



…………………………………………
Dolutegravir synthesis (EP2602260, 2013). LiHMDS as the non-nucleophilic strong base pulling compound 1 carbonyl group proton alpha position with an acid chloride after 2 and ring closure reaction to obtain 3 , 3 via primary amine 4 ring opening ring closure to obtain 5 , NBS the bromine under acidic conditions to obtain aldehyde acetal becomes 6 , 6 of the aldehyde and amino alcohols 7 and turn off the condensation reaction obtained by the ring 8 , alkaline hydrolysis 8 of bromine into a hydroxyl group and hydrolyzable ester obtained 9 after the 10 occurred acid condensation Dolutegravir.
………………………………………………………
Synthesis of Dolutegravir (S/GSK1349572, GSK1349572)

………………………
SYNTHESIS

2H-Pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxamide, N-[(2,4-difluorophenyl)methyl]-3,4,6,8,12,12a-hexahydro-7-hydroxy-4-methyl-6,8-dioxo-, (4R,12aS) ………..dolutegravir
PATENT US8129385

Desired isomer
Example Z-1
(3R,11aS)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide sodium salt

a)
(3R,11aS)—N-[(2,4-Difluorophenyl)methyl]-3-methyl-5,7-dioxo-6-[(phenylmethyl)oxy]-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide. To a solution of 16a (409 mg, 0.87 mmol) in dichloroethane (20 mL) was added (2R)-2-amino-1-propanol (0.14 mL, 1.74 mmol) and 10 drops of glacial acetic acid. The resultant solution was heated at reflux for 2 h. Upon cooling, Celite was added to the mixture and the solvents removed in vacuo and the material was purified via silica gel chromatography (2% CH3OH/CH2Cl2 gradient elution) to give (3R,11aS)—N-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6-[(phenylmethyl)oxy]-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (396 mg, 92%) as a glass. 1H NMR (CDCl3) δ 10.38 (m, 1H), 8.42 (s, 1H), 7.54-7.53 (m, 2H), 7.37-7.24 (m, 4H), 6.83-6.76 (m, 2H), 5.40 (d, J=10.0 Hz, 1H), 5.22 (d, J=10.0 Hz, 1H), 5.16 (dd, J=9.6, 6.0 Hz, 1H), 4.62 (m, 2H), 4.41 (m, 1H), 4.33-4.30 (m, 2H), 3.84 (dd, J=12.0, 10.0 Hz, 1H), 3.63 (dd, J=8.4, 7.2 Hz, 1H), 1.37 (d, J=6.0 Hz, 3H); ES+MS: 496 (M+1).
b)
(3R,11aS)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide sodium salt. To a solution of (3R,11aS)—N-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6-[(phenylmethyl)oxy]-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (396 mg, 0.80 mmol) in methanol (30 mL) was added 10% Pd/C (25 mg). Hydrogen was bubbled through the reaction mixture via a balloon for 2 h. The resultant mixture was filtered through Celite with methanol and dichloromethane.
The filtrate was concentrated in vacuo to give (3R,11aS)—N-[(2,4-difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide , DOLUTEGRAVIR as a pink tinted white solid (278 mg, 86%).
1H NMR (CDCl3) δ 11.47 (m, 1H), 10.29 (m, 1H), 8.32 (s, 1H), 7.36 (m, 1H), 6.82 (m, 2H), 5.31 (dd, J=9.6, 3.6 Hz, 1H), 4.65 (m, 2H), 4.47-4.38 (m, 3H), 3.93 (dd, J=12.0, 10.0 Hz, 1H), 3.75 (m, 1H), 1.49 (d, J=5.6 Hz, 3H); ES+ MS: 406 (M+1).
DOLUTEGRAVIR NA SALT
The above material (278 mg, 0.66 mmol) was taken up in ethanol (10 mL) and treated with 1 N sodium hydroxide (aq) (0.66 ml, 0.66 mmol). The resulting suspension was stirred at room temperature for 30 min. Ether was added and the liquids were collected to provide the sodium salt of the title compound as a white powder (291 mg, 99%). 1H NMR (DMSO-d6) δ 10.68 (m, 1H), 7.90 (s, 1H), 7.35 (m, 1H), 7.20 (m, 1H), 7.01 (m, 1H), 5.20 (m, 1H), 4.58 (m, 1H), 4.49 (m, 2H), 4.22 (m, 2H), 3.74 (dd, J=11.2, 10.4 Hz, 1H), 3.58 (m, 1H), 1.25 (d, J=4.4 Hz, 3H).
UNDESIRED ISOMER
Example Z-9
(3S,11aR)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide sodium salt

The title compound was made in two steps using a similar process to that described in example Z-1. 16a (510 mg, 1.08 mmol) and (25)-2-amino-1-propanol (0.17 mL, 2.17 mmol) were reacted in 1,2-dichloroethane (20 mL) with acetic acid to give (3S,11aR)—N-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6-[(phenylmethyl)oxy]-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (500 mg, 93%). This material was hydrogenated in a second step as described in example Z-1 to give (3S,11aR)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (386 mg, 94%) as a tinted white solid. 1H NMR (CDCl3) δ 11.46 (m, 1H), 10.28 (m, 1H), 8.32 (s, 1H), 7.35 (m, 1H), 6.80 (m, 2H), 5.30 (dd, J=10.0, 4.0 Hz, 1H), 4.63 (m, 2H), 4.48-4.37 (m, 3H), 3.91 (dd, J=12.0, 10.0 Hz, 1H), 3.73 (m, 1H), 1.48 (d, J=6.0 Hz, 3H); ES+ MS: 406 (M+1). This material (385 mg, 0.95 mmol) was treated with sodium hydroxide (0.95 mL, 1.0 M, 0.95 mmol) in ethanol (15 mL) as described in example Z-1 to provide its corresponding sodium salt (381 mg, 94%) as a white solid. 1H NMR (DMSO-d6) δ 10.66 (m, 1H), 7.93 (s, 1H), 7.33 (m, 1H), 7.20 (m, 1H), 7.01 (m, 1H), 5.19 (m, 1H), 4.59 (m, 1H), 4.48 (m, 2H), 4.22 (m, 2H), 3.75 (m, 1 H), 3.57 (m, 1H), 1.24 (d, J=5.6 Hz, 3H).
SYNTHESIS OF INTERMEDIATES

IN ABOVE SCHEME SYNTHESIS UPTO COMPD 9 MAY BE USEFUL IN SYNTHESIS BUT READERS DISCRETION IS SOUGHT IN THIS ?????????????????
1) Maltol 1 (189 g, 1.5 mol) was dissolved in dimethylformamide (1890 ml), and benzyl bromide (184 ml, 1.5 mol) was added. After the solution was stirred at 80° C. for 15 minutes, potassium carbonate (228 g, 1.65 mol) was added, and the mixture was stirred for 1 hour. After the reaction solution was cooled to room temperature, an inorganic salt was filtered, and the filtrate was distilled off under reduced pressure. To the again precipitated inorganic salt was added tetrahydrofuran (1000 ml), this was filtered, and the filtrate was distilled off under reduced pressure to obtain the crude product (329 g, >100%) of 3-benzyloxy-2-methyl-pyran-4-one 2 as a brown oil.
NMR (CDCl3) δ: 2.09 (3H, s), 5.15 (2H, s), 6.36 (1H, d, J=5.6 Hz), 7.29-7.41 (5H, m), 7.60 (1H, d, J=5.6 Hz).
2) The compound 2 (162.2 g, 750 mmol) was dissolved in ethanol (487 ml), and aqueous ammonia (28%, 974 ml) and a 6N aqueous sodium hydroxide solution (150 ml, 900 mmol) were added. After the reaction solution was stirred at 90° C. for 1 hour, this was cooled to under ice-cooling, and ammonium chloride (58 g, 1080 mmol) was added. To the reaction solution was added chloroform, this was extracted, and the organic layer was washed with an aqueous saturated sodium bicarbonate solution, and dried with anhydrous sodium sulfate. The solvent was distilled off under reduced pressure, isopropyl alcohol and diethyl ether were added to the residue, and precipitated crystals were filtered to obtain 3-benzyloxy-2-methyl-1H-pyridine-4-one 3 (69.1 g, 43%) as a pale yellow crystal.
NMR (DMSO-d6) δ: 2.05 (3H, s), 5.04 (2H, s), 6.14 (1H, d, J=7.0 Hz), 7.31-7.42 (5H, m), 7.46 (1H, d, J=7.2 Hz), 11.29 (1H, brs).
3) The above compound 3 (129 g, 699 mmol) was suspended in acetonitrile (1300 ml), and N-bromosuccinic acid imide (117 g, 659 mmol) was added, followed by stirring at room temperature for 90 minutes. Precipitated crystals were filtered, and washed with acetonitrile and diethyl ether to obtain 3-benzyloxy-5-bromo-2-methyl-pyridine-4-ol 4 (154 g, 88%) as a colorless crystal.
NMR (DMSO-d6) δ: 2.06 (3H, s), 5.04 (2H, s), 7.32-7.42 (5H, m), 8.03 (1H, d, J=5.5 Hz), 11.82 (1H, brs).
4) To a solution of the compound 4 (88 g, 300 mmol), palladium acetate (13.4 g, 60 mmol) and 1,3-bis(diphenylphosphino)propane (30.8 g, 516 mmol) in dimethylformamide (660 ml) were added methanol (264 ml) and triethylamine (210 ml, 1.5 mol) at room temperature. The interior of a reaction vessel was replaced with carbon monoxide, and the material was stirred at room temperature for 30 minutes, and stirred at 80 degree for 18 hours. A vessel to which ethyl acetate (1500 ml), an aqueous saturated ammonium chloride solution (1500 ml) and water (1500 ml) had been added was stirred under ice-cooling, and the reaction solution was added thereto. Precipitates were filtered, and washed with water (300 ml), ethyl acetate (300 ml) and diethyl ether (300 ml) to obtain 5-benzyloxy-4-hydroxy-6-methyl-nicotinic acid methyl ester 5 (44.9 g, 55%) as a colorless crystal.
NMR (DMSO-d6) δ: 2.06 (3H, s), 3.72 (3H, s), 5.02 (2H, s), 7.33-7.42 (5H, m), 8.07 (1H, s).
5) After a solution of the compound 5 (19.1 g, 70 mmol) in acetic anhydride (134 ml) was stirred at 130° C. for 40 minutes, the solvent was distilled off under reduced pressure to obtain 4-acetoxy-5-benzyloxy-6-methyl-nicotinic acid methyl ester 6 (19.9 g, 90%) as a flesh colored crystal.
NMR (CDCl3) δ: 2.29 (3H, s), 2.52 (3H, s), 3.89 (3H, s), 4.98 (2H, s), 7.36-7.41 (5H, m), 8.85 (1H, s).
6) To a solution of the compound 6 (46.2 g, 147 mmol) in chloroform (370 ml) was added metachloroperbenzoic acid (65%) (42.8 g, 161 mmol) in portions under ice-cooling, and this was stirred at room temperature for 90 minutes. To the reaction solution was added a 10% aqueous potassium carbonate solution, and this was stirred for 10 minutes, followed by extraction with chloroform. The organic layer was washed with successively with a 10% aqueous potassium carbonate solution, an aqueous saturated ammonium chloride solution, and an aqueous saturated sodium chloride solution, and dried with anhydrous sodium sulfate. The solvent was distilled off under induced pressure, and the residue was washed with diisopropyl ether to obtain 4-acetoxy-5-benzyloxy-6-methyl-1-oxy-nicotinic acid methyl ester 7 (42.6 g, 87%) as a colorless crystal.
NMR (CDCl3) δ: 2.30 (3H, s), 2.41 (3H, s), 3.90 (3H, s), 5.02 (2H, s), 7.37-7.39 (5H, m), 8.70 (1H, s).
7) To acetic anhydride (500 ml) which had been heated to stir at 130° C. was added the compound 7 (42.6 g, 129 mmol) over 2 minutes, and this was stirred for 20 minutes. The solvent was distilled off under reduced pressure to obtain 4-acetoxy-6-acetoxymethyl-5-benzyloxy-nicotinic acid methyl ester 8 (49.6 g, >100%) as a black oil.
NMR (CDCl3) δ: 2.10 (3H, s), 2.28 (3H, s), 3.91 (3H, s), 5.07 (2H, s), 5.20 (2H, s), 7.35-7.41 (5H, m), 8.94 (1H, s).
8) To a solution of the compound 8 (46.8 g, 125 mmol) in methanol (140 ml) was added a 2N aqueous sodium hydroxide solution (376 ml) under ice-cooling, and this was stirred at 50° C. for 40 minutes. To the reaction solution were added diethyl ether and 2N hydrochloric acid under ice-cooling, and precipitated crystals were filtered. Resulting crystals were washed with water and diethyl ether to obtain 5-benzyloxy-4-hydroxy-6-hydroxymethyl-nicotinic acid 9 (23.3 g, 68%) as a colorless crystal.
NMR (DMSO-d6) δ: 4.49 (2H, s), 5.19 (2H, s), 5.85 (1H, brs), 7.14-7.20 (2H, m), 7.33-7.43 (7H, m), 8.30 (1H, s), 10.73 (1H, t, J=5.8 Hz), 11.96 (1H, brs).
9) To a solution of the compound 9 (131 g, 475 mmol), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (219 g, 1140 mmol) and 1-hydroxybenzotriazole (128 g, 950 mmol) in dimethylformamide (1300 ml) was added 4-fluorobenzylamine (109 ml, 950 mmol), and this was stirred at 80° C. for 1.5 hours. After the reaction solution was cooled to room temperature, hydrochloric acid was added, followed by extraction with ethyl acetate. The extract was washed with a 5% aqueous potassium carbonate solution, an aqueous saturated ammonium chloride solution, and an aqueous saturated sodium chloride solution, and dried with anhydrous sodium sulfate. The solvent was distilled off under reduced pressure to obtain a mixture (175 g) of 10 and 11. the resulting mixture was dissolved in acetic acid (1050 ml) and water (1050 ml), and zinc (31.1 g, 475 mmol) was added, followed by heating to reflux for 1 hour. After the reaction solution was cooled to room temperature, a 10% aqueous potassium carbonate solution was added, followed by extraction with ethyl acetate. The extract was washed with an aqueous saturated ammonium chloride solution, and an aqueous saturated sodium chloride solution, and dried with anhydrous sodium sulfate. After the solvent was distilled off under reduced pressure, this was washed with diethyl ether to obtain 5-benzyloxy-N-(4-fluoro-benzyl)-4-hydroxy-6-hydroxymethyl-nicotinic acid amide 10 (107 g, 59%) as a colorless crystal.
NMR (DMSO-d6) δ: 4.45 (2H, d, J=4.3 Hz), 4.52 (2H, d, J=5.8 Hz), 5.09 (2H, s), 6.01 (1H, brs), 7.36-7.43 (5H, m), 8.31 (1H, s), 12.63 (1H, brs).
………………..
SYNTHESIS
- Example 3

3H IS DOLUTEGRAVIR
Step 1
-
N,N-dimethylformamide dimethyl acetal (4.9 ml, 36.5 mmol) was added dropwise to compound 3A (5.0 g, 30.4 mmol) under cooling at 0°C. After stirring at 0°C for 1 hour, 100 ml of ethyl acetate was added to the reaction solution, and the organic layer was washed with a 0.5 N aqueous hydrochloric acid solution (50 ml). The aqueous layer was separated, followed by extraction with ethyl acetate (50 ml). The organic layers were combined, washed with a saturated aqueous solution of sodium bicarbonate and saturated saline in this order, and then dried over anhydrous sodium sulfate. The solvent was distilled off, and the obtained residue was purified by silica gel column chromatography (n-hexane-ethyl acetate: 1:1 (v/v) → ethyl acetate) to obtain 4.49 g (yield: 67%) of compound 3B as an oil.
1H-NMR (CDCl3)δ:1.32 (3H, t, J = 7.1 Hz), 2.90 (3H, br s), 3.29 (3H, br s), 4.23 (2H, q, J = 7.1 Hz), 4.54 (2H, s), 7.81 (1H, s).
Step 2
-
Lithium hexamethyldisilazide (1.0 M solution in toluene, 49 ml, 49.0 mmol) was diluted with tetrahydrofuran (44 ml). A tetrahydrofuran (10 ml) solution of compound 3B (4.49 g, 20.4 mmol) was added dropwise thereto under cooling at -78°C, and a tetrahydrofuran (10 ml) solution of ethyl oxalyl chloride (3.35 g, 24.5 mmol) was then added dropwise to the mixture. The mixture was stirred at -78°C for 2 hours and then heated to 0°C. 2 N hydrochloric acid was added to the reaction solution, and the mixture was stirred for 20 minutes, followed by extraction with ethyl acetate (200 ml x 2). The organic layer was washed with a saturated aqueous solution of sodium bicarbonate and saturated saline and then dried over anhydrous sodium sulfate. The solvent was distilled off, and the obtained residue was purified by silica gel column chromatography (n-hexane-ethyl acetate: 7:3 → 5:5 → 0:10 (v/v)) to obtain 1.77 g (yield: 31%) of compound 3C as a white solid.
1H-NMR (CDCl3)δ:1.36-1.46 (6H, m), 4.35-4.52 (8H, m), 8.53 (1H, s).
Step 3
-
Aminoacetaldehyde dimethyl acetal (0.13 ml, 1.20 mmol) was added to an ethanol (6 ml) solution of compound 3C (300 mg, 1.09 mmol) at 0°C, and the mixture was stirred at 0°C for 1.5 hours, then at room temperature for 18 hours, and at 60°C for 4 hours. The solvent in the reaction solution was distilled off under reduced pressure, and the obtained residue was then purified by silica gel column chromatography (n-hexane-ethyl acetate: 5:5 → 0:10 (v/v)) to obtain 252 mg (yield: 64%) of compound 3D as an oil.
1H-NMR (CDCl3)δ:1.36-1.47 (6H, m), 3.42 (6H, s), 3.90 (2H, d, J = 5.2 Hz), 4.37 (3H, q, J = 7.2 Hz), 4.50 (2H, q, J = 7.2 Hz), 8.16 (1H, s).
Step 4
-
62% H2SO4 (892 mg, 5.64 mmol) was added to a formic acid (10 ml) solution of compound 3D (1.02 g, 2.82 mmol), and the mixture was stirred at room temperature for 16 hours. The formic acid was distilled off under reduced pressure. To the residue, methylene chloride was added, and the mixture was pH-adjusted to 6.6 by the addition of a saturated aqueous solution of sodium bicarbonate. The methylene chloride layer was separated, while the aqueous layer was subjected to extraction with methylene chloride. The methylene chloride layers were combined and dried over anhydrous sodium sulfate. The solvent was distilled off to obtain 531.8 mg of compound 3E as a yellow oil.
1H-NMR (CDCl3) δ: 1.28-1.49 (6H, m), 4.27-4.56 (4H, m), 4.84 (2H, s), 8.10 (1H, s), 9.72 (1H, s).
Step 5
-
Methanol (0.20 ml, 5.0 mmol), (R)-3-amino-butan-1-ol (179 mg, 2.0 mmol), and acetic acid (0.096 ml, 1.70 mmol) were added to a toluene (5 ml) solution of compound 3E (531 mg, 1.68 mmol), and the mixture was heated to reflux for 4 hours. The reaction solution was cooled to room temperature, then diluted with chloroform, and then washed with a saturated aqueous solution of sodium bicarbonate. The aqueous layer was subjected to extraction with chloroform. The chloroform layers were combined, washed with saturated saline, and then dried over anhydrous sodium sulfate. The solvent was distilled off, and the obtained residue was purified by silica gel column chromatography (chloroform-methanol: 100:0 → 90:10) to obtain 309.4 mg of compound 3F as a brown oil.
1H-NMR (CDCl3) δ: 1.40 (3H, t, J = 7.1 Hz), 1.40 (3H, d, J = 7.1 Hz), 1.55-1.61 (1H, m), 2.19-2.27 (1H, m), 4.00 (1H, d, J = 1.5 Hz), 4.03 (1H, d, J = 2.5 Hz), 4.10 (1H, dd, J = 13.2, 6.3 Hz), 4.26 (1H, dd, J = 13.2, 3.8 Hz), 4.38 (2H, q, J = 7.1 Hz), 5.00-5.05 (1H, m), 5.31 (1H, dd, J = 6.4, 3.9 Hz), 8.10 (1H, s).
Step 6
-
Potassium trimethylsilanolate (333 mg, 2.34 mmol) was added to a 1,2-dimethoxyethane (2 ml) solution of compound 3F (159 mg, 0.47 mmol), and the mixture was stirred at room temperature for 7 hours. 1 N hydrochloric acid and saturated saline were added to the reaction solution, followed by extraction with chloroform. The chloroform layers were combined and dried over anhydrous sodium sulfate. The solvent was distilled off to obtain 34.4 mg (yield: 25%) of compound 3G as an orange powder.
1H-NMR (CDCl3) δ: 1.46 (3H, d, J = 3.5 Hz), 1.58-1.65 (1H, m), 2.26-2.30 (1H,m), 4.06-4.10 (2H, m), 4.31 (1H, dd, J = 13.8, 5.6 Hz), 4.48 (1H, dd, J = 13.6, 3.9 Hz), 5.03 (1H, t, J = 6.4 Hz), 5.36 (1H, dd, J = 5.5, 4.0 Hz), 8.44 (1H, s), 12.80 (1H, s), 14.90 (1H, s).
Step 7
-
Compound 3G (16 mg, 0.054 mmol) and 2,4-difluorobenzylamine (17 mg, 0.12 mmol) were dissolved in N,N-dimethylformamide (1 ml). To the solution, N,N,N’,N’-tetramethyl-O-(7-aza-benzotriazol-1-yl)uronium hexafluorophosphate (HATU) (53 mg, 0.14 mmol) and N-methylmorpholine (0.031 ml, 0.28 mmol) were added, and the mixture was stirred at room temperature for 16 hours. 2,4-difluorobenzylamine (17 mg, 0.12 mmol), HATU (64 mg, 0.17 mmol), and N-methylmorpholine (0.037 ml, 0.34 mmol) were further added thereto, and the mixture was stirred at room temperature for additional 16 hours. 0.5 N hydrochloric acid was added to the reaction solution, followed by extraction with ethyl acetate. The ethyl acetate layers were combined, washed with 0.5 N hydrochloric acid and then with saturated saline, and then dried over anhydrous sodium sulfate. The solvent was distilled off, and the obtained residue was purified by preparative high-performance liquid chromatography to obtain 12.5 mg (yield: 55%) of compound 3H as an orange solid.
- DOLUTEGRAVIR
-
1H-NMR (DMSO-d6) δ: 1.36 (3H, d, J = 6.9 Hz), 1.55-1.60 (1H, m), 2.01-2.05 (1H, m), 3.92-3.94 (1H, m), 4.04 (1H, t, J = 12.6 Hz), 4.38-4.41 (1H, m), 4.57-4.60 (1H, m), 4.81-4.83 (1H, m), 5.46-5.49 (1H, m), 7.08-7.11 (1H, m), 7.25-7.30 (1H, m), 7.41 (1H, dd, J = 15.3, 8.7 Hz), 8.53 (1H, s), 10.38 (1H, s), 12.53 (1H, s).
ISOMERS OF DOLUTEGRAVIR
- Reference Example 1

Step 1
-
Acetic acid (180 mg, 3.00 mmol) was added to a toluene (90 ml) solution of compound A-1 (4.39 g, 9.33 mmol) and (R)-3-aminobutan-1-ol (998 mg, 11.2 mmol), and the mixture was stirred at 50°C for 90 minutes. The reaction solution was allowed to cool to room temperature and then poured to a saturated aqueous solution of sodium bicarbonate. The organic layer was separated, while the aqueous layer was subjected to extraction three times with ethyl acetate. The combined extracts were washed with saturated saline and then dried over sodium sulfate. The solvent was distilled off to obtain 4.29 g of crude product A-2.
Step 2
-
The crude product A-2 obtained in the preceding step was dissolved in ethanol (40 ml). To the solution, a 2 N aqueous sodium hydroxide solution (20 ml) was added at room temperature, and the mixture was stirred at the same temperature for 2 hours. The reaction solution was neutralized to pH 7 using a 2 N aqueous hydrochloric acid solution. The solvent was directly distilled off. The obtained crude product A-3 was subjected to azeotropy with toluene (100 ml) and used in the next step without being purified.
Step 3
-
HOBt (1.65 g, 12.2 mmol) and WSC HCl (2.34 g, 12.2 mmol) were added at room temperature to a DMF (100 ml) solution of the crude product A-3 obtained in the preceding step, and the mixture was stirred at the same temperature for 15 hours. Water was added to the reaction solution, followed by extraction three times with ethyl acetate. The combined extracts were washed with water three times and then dried over sodium sulfate. The solvent was distilled off, and the obtained oil was subjected to silica gel column chromatography for purification. Elution was performed first with n-hexane-ethyl acetate (3:7, v/v) and then with only ethyl acetate. The fraction of interest was concentrated, and the obtained oil was then dissolved in ethyl acetate. The solution was crystallized with diisopropyl ether as a poor solvent. The obtained crystals were collected by filtration and dissolved again in ethyl acetate. The solution was recrystallized to obtain 1.84 g of compound A-4.
1HNMR (CDCl3) δ: 1.49 (3H, d, J = 6.6 Hz), 1.88-1.96 (1H, m), 2.13-2.26 (1H, m), 3.90-4.17 (4H, m), 4.42-4.47 (1H, m), 4.63 (2H, d, J = 6.0 Hz), 5.12-5.17 (1H, m), 5.17 (1H, d, J = 9.9 Hz), 5.33 (1H, d, J = 9.9 Hz), 6.77-6.87 (2H, m), 7.27-7.42 (4H, m), 7.59-7.62 (2H, m), 8.35 (1H, s), 10.41 (1H, t, J = 5.7 Hz).
Step 4
-
The compound A-4 was subjected to the hydroxy deprotection reaction described in Step F of the paragraph [0088] to obtain compound A-5.
1HNMR (DMSO-d6) δ:1.41 (3H, d, J = 6.3 Hz), 1.85-1.92 (1H, m), 1.50-1.75 (1H, m), 4.02-4.09 (3H, m), 4.28-4.34 (1H, m), 4.53 (2H, d, J = 5.7 Hz), 4.64 (1H, dd, J = 3.9 Hz, 12.6 Hz), 5.45 (1H, dd, J = 3.6 Hz, 9.3 Hz), 7.06 (1H, ddd, J = 2.7 Hz, 8.4 Hz, 8.4 Hz), 7.20-7.28 (1H, m), 7.35-7.42 (1H, m), 8.43 (1H, s),10.37 (1H, t, J = 6.0 Hz),12.37 (1H, brs).
- Reference Example 2
-

-
Compound A-1 was reacted with (S)-3-aminobutan-1-ol in Step 1. Compound B-5 was obtained in the same way as in Reference Example 1.
1HNMR (DMSO-d6) δ:1.41 (3H, d, J = 6.3 Hz), 1.85-1.92 (1H, m), 1.50-1.75 (1H, m), 4.02-4.09 (3H, m), 4.28-4.34 (1H, m), 4.53 (2H, d, J = 5.7 Hz), 4.64 (1H, dd, J = 3.9 Hz, 12.6 Hz), 5.45 (1H, dd, J = 3.6 Hz, 9.3 Hz), 7.06 (1H, ddd, J = 2.7 Hz, 8.4 Hz, 8.4 Hz), 7.20-7.28 (1H, m), 7.35-7.42 (1H, m), 8.43 (1H, s),10.37 (1H, t, J = 6.0 Hz),12.37 (1H, brs).

……………..

ENTRY 68
………………………….
WO 2010068262
…………………………
WO 2010068253
…………………………………
WO 2011119566
…………………………..
Synthesis
Example 3

I was under cooling added dropwise at 0 ℃ (4.9 ml, 36.5 mmol) and N, N-dimethylformamide dimethyl acetal (5.0 g, 30.4 mmol) in the first step compound 3A. After stirring for 1 hour at 0 ℃, ethyl acetate was added to 100ml, the reaction mixture was washed with 0.5N aqueous hydrochloric acid (50 ml). Was extracted with ethyl acetate (50ml) and solution was separated and the aqueous layer. The organic layers were combined, washed successively with saturated aqueous sodium bicarbonate solution and saturated brine, and then dried over anhydrous sodium sulfate. After the solvent was distilled off, silica gel column chromatography and the residue obtained was – and purified by (n-hexane (v / v) → ethyl acetate 1:1) to an oil (67% yield) of Compound 3B 4.49 g I got a thing.
1 H-NMR (CDCl 3) δ: 1.32 (3H, t, J = 7.1 Hz), 2.90 (3H, br s), 3.29 (3H, br s), 4.23 (2H, q, J = 7.1 Hz), 4.54 (2H, s), 7.81 (1H, s).
Diluted with tetrahydrofuran (44 ml) (1.0M toluene solution, 49 ml, 49.0 mmol) the second step lithium hexamethyldisilazide, under cooling at -78 ℃, compound 3B (4.49 g, 20.4 mmol) in this After dropwise tetrahydrofuran (10 ml) was added dropwise tetrahydrofuran (3.35 g, 24.5 mmol) of ethyl oxalyl chloride and (10 ml) solution. After stirring for 2 hours at -78 ℃, I was warmed to 0 ℃. After washing (200 ml x 2), saturated aqueous sodium bicarbonate solution and the organic layer with saturated brine After stirring for 20 minutes, extracted with ethyl acetate by adding 2N hydrochloric acid, the reaction solution was dried over anhydrous sodium sulfate. After removal of the solvent, silica gel column chromatography and the residue obtained – was purified (n-hexane (v / v) ethyl acetate 7:3 → 5:5 → 0:10), compound 3C 1.77 g (yield I as a white solid 31%).
1 H-NMR (CDCl 3) δ :1.36-1 .46 (6H, m), 4.35-4.52 (8H, m), 8.53 (1H, s).
Was added at 0 ℃ (0.13 ml, 1.20 mmol) the aminoacetaldehyde dimethyl acetal ethanol (300 mg, 1.09 mmol) of the third step compound 3C to (6 ml) solution, 1 hour and 30 minutes at 0 ℃, 18 hours at room temperature , then I was stirred for 4 hours at 60 ℃. After the solvent was evaporated under reduced pressure and the reaction mixture by silica gel column chromatography and the residue obtained was – and purified by (n-hexane (v / v) ethyl acetate 5:5 → 0:10), compound 3D 252 mg (yield: I got as an oil 64%) rate.
1 H-NMR (CDCl 3) δ :1.36-1 .47 (6H, m), 3.42 (6H, s), 3.90 (2H, d, J = 5.2 Hz), 4.37 (3H, q, J = 7.2 Hz), 4.50 (2H, q, J = 7.2 Hz), 8.16 (1H, s).
Was added (892 mg, 5.64 mmol) and 2 SO 4 62-H% formic acid (1.02 g, 2.82 mmol) in a fourth step the compound for 3D (10 ml) solution was stirred at room temperature for 16 hours. Methylene chloride was added to the residue Shi distilled off under reduced pressure and formic acid was adjusted to pH = 6.6 by addition of saturated aqueous sodium bicarbonate. The solution was separated methylene chloride layer was extracted with methylene chloride and the aqueous layer. I was dried over anhydrous sodium sulfate combined methylene chloride layers. The solvent was then distilled off and was obtained as a yellow oil 531.8 mg compound 3E.
1H-NMR (CDCl3) δ: 1.28-1.49 (6H, m), 4.27-4.56 (4H, m), 4.84 (2H, s), 8.10 (1H, s), 9.72 (1H, s).
Amino – – butane – 1 – ol (179 mg, 2.0 mmol), methanol (0.20 ml, 5.0 mmol), (R) -3 toluene (531 mg, 1.68 mmol) in the fifth step to compound 3E (5 ml) solution was added (0.096 ml, 1.70 mmol) acetic acid was heated under reflux for 4 hours. After dilution with chloroform, cooled to room temperature, the reaction mixture was washed with a saturated aqueous sodium bicarbonate solution, and the aqueous layer was extracted with chloroform. After washing with saturated brine combined chloroform layer was dried over anhydrous sodium sulfate. The solvent was then distilled off, silica gel column chromatography and the residue obtained – and (chloroform methanol 100:0 → 90:10), was obtained as a brown oil 309.4 mg compound 3F.
1H-NMR (CDCl3) δ: 1.40 (3H, t, J = 7.1 Hz), 1.40 (3H, d, J = 7.1 Hz), 1.55-1.61 (1H, m), 2.19-2.27 (1H, m), 4.00 (1H, d, J = 1.5 Hz), 4.03 (1H, d, J = 2.5 Hz), 4.10 (1H, dd, J = 13.2, 6.3 Hz), 4.26 (1H, dd, J = 13.2, 3.8 Hz ), 4.38 (2H, q, J = 7.1 Hz), 5.00-5.05 (1H, m), 5.31 (1H, dd, J = 6.4, 3.9 Hz), 8.10 (1H, s).
1,2 (159 mg, 0.47 mmol) in the sixth step compound 3F – was added (333 mg, 2.34 mmol) and potassium trimethylsilanolate dimethoxyethane (2 ml) solution was stirred for 7 hours at room temperature. Brine was added to the 1N-hydrochloric acid to the reaction mixture, followed by extraction with chloroform. The combined chloroform layer was dried over anhydrous sodium sulfate. The solvent was removed by distillation, and I as an orange powder (25% yield) of compound 3G 34.4 mg.
1H-NMR (CDCl3) δ: 1.46 (3H, d, J = 3.5 Hz), 1.58-1.65 (1H, m), 2.26-2.30 (1H, m), 4.06-4.10 (2H, m), 4.31 (1H , dd, J = 13.8, 5.6 Hz), 4.48 (1H, dd, J = 13.6, 3.9 Hz), 5.03 (1H, t, J = 6.4 Hz), 5.36 (1H, dd, J = 5.5, 4.0 Hz) , 8.44 (1H, s), 12.80 (1H, s), 14.90 (1H, s).
2,4 (16 mg, 0.054 mmol) and the seventh step compound 3G – was dissolved in N, N-dimethylformamide (1 ml) (17 mg, 0.12 mmol) difluorobenzyl amine, N, N, N ‘, N was added (0.031 ml, 0.28 mmol) and N-methylmorpholine uronium hexafluorophosphate (HATU) (53 mg, 0.14 mmol), and ‘- tetramethyl-O-(yl 7 – aza – – benzo triazolopyrimidine -1) I was stirred at room temperature for 16 h. 2,4 – was added (0.037 ml, 0.34 mmol) and N-methylmorpholine (64 mg, 0.17 mmol) and (17 mg, 0.12 mmol), HATU difluorobenzylamine, and the mixture was stirred for 16 hours at room temperature. I was extracted with ethyl acetate addition of 0.5N-hydrochloric acid to the reaction mixture. 0.5N-hydrochloric acid and then was washed with saturated brine, and dried over anhydrous sodium sulfate and combined ethyl acetate layer. The solvent was then distilled off, and purified by preparative high performance liquid chromatography residue was obtained as an orange solid (55% yield) of compound 3H 12.5 mg.
1H-NMR (DMSO-d6) δ: 1.36 (3H, d, J = 6.9 Hz), 1.55-1.60 (1H, m), 2.01-2.05 (1H, m), 3.92-3.94 (1H, m), 4.04 (1H, t, J = 12.6 Hz), 4.38-4.41 (1H, m), 4.57-4.60 (1H, m), 4.81-4.83 (1H, m), 5.46-5.49 (1H, m), 7.08-7.11 (1H, m), 7.25-7.30 (1H, m), 7.41 (1H, dd, J = 15.3, 8.7 Hz), 8.53 (1H, s), 10.38 (1H, s), 12.53 (1H, s)
References
- [1] American Medical Association (AMA), STATEMENT ON A NONPROPRIETARY NAME ADOPTED BY THE USAN COUNCIL (Dolutegravir) Accessed 3 December 2011.
- FDA approves new drug to treat HIV infection http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm364744.htm Aug. 12, 2013
- “U.S. FDA approves GlaxoSmithKline’s HIV drug Tivicay”. Reuters. 12 August 2013. Retrieved 13 February 2013.
- “GSK wins priority status for new HIV drug in U.S”. Reuters. 16 February 2013. Retrieved 18 February 2013.
- “ViiV Healthcare receives approval for Tivicay™ (dolutegravir) in Canada for the treatment of HIV”. Retrieved 11 November 2013.
- FDA approves new drug to treat HIV infection http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm364744.htm Aug. 12, 2013
- U.S. FDA approves GlaxoSmithKline’s HIV drug Tivicay http://www.reuters.com/article/2013/08/12/us-glaxosmithkline-hivdrug-idUSBRE97B0WU20130812 Mon Aug 12, 2013 6:40pm EDT
- “Dolutegravir Prescribing Information”. Retrieved 3 January 2014.
- Raffi, F; Jaeger, H; Quiros-Roldan, E; Albrecht, H; Belonosova, E; Gatell, JM; Baril, JG; Domingo, P; Brennan, C; Almond, S; Min, S; extended SPRING-2 Study, Group (Nov 2013). “Once-daily dolutegravir versus twice-daily raltegravir in antiretroviral-naive adults with HIV-1 infection (SPRING-2 study): 96 week results from a randomised, double-blind, non-inferiority trial.”. The Lancet infectious diseases 13 (11): 927–35. PMID 24074642.
- http://www.natap.org/2013/ICAAC/ICAAC_24.htm
- Walmsley, Sharon L.; Antela, Antonio; Clumeck, Nathan; Duiculescu, Dan; Eberhard, Andrea; Gutiérrez, Felix; Hocqueloux, Laurent; Maggiolo, Franco; Sandkovsky, Uriel; Granier, Catherine; Pappa, Keith; Wynne, Brian; Min, Sherene; Nichols, Garrett (7 November 2013). “Dolutegravir plus Abacavir–Lamivudine for the Treatment of HIV-1 Infection”. New England Journal of Medicine 369 (19): 1807–1818. doi:10.1056/NEJMoa1215541.
- Sax, Paul. “SINGLE Study Underscores Waning of the Efavirenz Era — But Probably Just in the USA – See more at:http://blogs.jwatch.org/hiv-id-observations/index.php/single-study-underscores-waning-of-the-efavirenz-era-but-probably-just-in-the-usa/2013/11/06/#sthash.A39SderN.dpuf”. Retrieved 19 December 2013.
- Eron, JJ; Clotet, B; Durant, J; Katlama, C; Kumar, P; Lazzarin, A; Poizot-Martin, I; Richmond, G; Soriano, V; Ait-Khaled, M; Fujiwara, T; Huang, J; Min, S; Vavro, C; Yeo, J; VIKING Study, Group (2013 Mar 1). “Safety and efficacy of dolutegravir in treatment-experienced subjects with raltegravir-resistant HIV type 1 infection: 24-week results of the VIKING Study.”. The Journal of infectious diseases 207 (5): 740–8. PMID 23225901.
-
WO2010011812A1 * Jul 23, 2009 Jan 28, 2010 Smithkline Beecham Corporation Chemical compounds WO2010011819A1 * Jul 23, 2009 Jan 28, 2010 Smithkline Beecham Corporation Chemical compounds -
-
- [Patent Document 1] International publication No.2006/116764 pamphlet
- [Patent Document 2] International publication No.2010/011812 pamphlet
- [Patent Document 3] International publication No.2010/011819 pamphlet
- [Patent Document 4] International publication No.2010/068262 pamphlet
- [Patent Document 5] International publication No.2010/067176 pamphlet
- [Patent Document 6] International publication No.2010/068253 pamphlet
- [Patent Document 7] US Patent 4769380A
- [Patent Document 8] International applicationPCT/JP2010/055316
[NON-PATENT DOCUMENTS]
-
- [Non-Patent Document 1] Journal of Organic Chemistry, 1991, 56(16), 4963-4967
- [Non-Patent Document 2] Science of Synthesis, 2005, 15, 285-387
- [Non-Patent Document 3] Journal of Chemical Society Parkin Transaction. 1, 1997, Issue. 2, 163-169
-
…………………
Sources:
Johns, Brian Alvin; Kawasuji, Takashi; Taishi, Teruhiko; Taoda, Yoshiyuki ; Polycyclic carbamoylpyridone derivative having HIV integrase inhibitory activity and their preparation; PCT Int. Appl., WO2006116764, 02 Nov 2006
Johns, Brian Alvin; Weatherhead, Jason Gordon;Tricyclic heterocyclic compounds as antiviral agents and their preparation and use in the treatment of HIV infection; PCT Int. Appl., WO2010011812, 28 Jan 2010
Johns, Brian Alvin; Weatherhead, Jason Gordon; Tricyclic heterocyclic compounds as antiviral agents and their preparation and use in the treatment of HIV infection;PCT Int. Appl., WO2010011819, 28 Jan 2010
Yoshida, Hiroshi; Taoda, Yoshiyuki; Johns, Brian Alvin; Synthesis of fused tricyclic carbamoylpyridone HIV integrase inhibitors and intermediates;PCT Int. Appl.,WO2010068253, 17 Jun 2010
Johns, Brian Alvin; Duan, Maosheng; Hakogi, Toshikazu;Processes and intermediates for fused tricyclic carbamoylpyridone HIV integrase inhibitors;PCT Int. Appl., WO2010068262, 17 Jun 2010
Sumino, Yukihito; Okamoto, Kazuya; Masui, Moriyasu; Yamada, Daisuke; Ikarashi, Fumiya;Preparation of compounds having HIV integrase inhibitory activity; PCT Int. Appl.,WO2012018065, 09 Feb 2012
Kawasuji, Takashi; Johns, Brian A.;Discovery of dolutegravir and S/GSK1265744: Carbamoyl pyridone HIV-1 integrase inhibitors;Abstracts, 64th Southeast Regional Meeting of the American Chemical Society, Raleigh, NC, United States, November 14-17 (2012), SERM-176.
Kawasuji, Takashi; Johns, Brian A.; Yoshida, Hiroshi; Weatherhead, Jason G.; Akiyama, Toshiyuki; Taishi, Teruhiko; Taoda, Yoshiyuki; Mikamiyama-Iwata, Minako; Murai, Hitoshi; Kiyama, Ryuichi; Fuji, Masahiro; Tanimoto, Norihiko; Yoshinaga, Tomokazu; Seki, Takahiro; Kobayashi, Masanori; Sato, Akihiko; Garvey, Edward P.; Fujiwara, Tamio; Carbamoyl Pyridone HIV-1 Integrase Inhibitors. 2. Bi- and Tricyclic Derivatives Result in Superior Antiviral and Pharmacokinetic Profiles;Journal of Medicinal Chemistry (2013), 56(3), 1124-1135
Walmsley S et al. Dolutegravir (DTG; S/GSK1349572) + abacavir/lamivudine once daily statistically superior to tenofovir/emtricitabine/efavirenz: 48-week results – SINGLE (ING114467). 52nd ICAAC, 9-12 September 2012, San Francisco. Abstract H-556b.
http://www.abstractsonline.com/Plan/ViewAbstract.aspx?sKey=e1c18d5b-830f-4b4e-8671-35bcfb20eed5&cKey=af219b7d-2171-46b2-91ef-b8049552c9e5&mKey=%7b6B114A1D-85A4-4054-A83B-04D8B9B8749F%7d
http://www.natap.org/2012/ICAAC/ICAAC_06.htm
http://i-base.info/htb/20381
Raffi F et al. Once-daily dolutegravir (DTG; S/GSK1349572) is non-inferior to raltegravir (RAL) in antiretroviral-naive adults: 48 week results from SPRING-2 (ING113086). 19th International AIDS Conference. 22-27 July 2012, Washington. Late breaker oral presentation THLBB04.
http://pag.aids2012.org/abstracts.aspx?aid=20990
National Institutes of Health (U.S.). A trial comparing GSK1349572 50 mg plus abacavir/lamivudine once daily to Atripla (also called the SINGLE trial). Available from:http://clinicaltrials.gov/ct2/show/NCT01263015.
Stellbrink HJ, Reynes J, Lazzarin A, et al. Dolutegravir in combination therapy exhibits rapid and sustained antiviral response in ARV-naïve adults: 96-week results from SPRING-1 (ING112276) (Abstract 102LB). Paper presented at: 19th Conference on Retroviruses and Opportunistic Infections; 2012 March 5–8; Seattle, WA. Available from:http://www.retroconference.org/2012b/Abstracts/45432.html
Gilead’s HCV drug Sovaldi gets Europe OK
Gilead Sciences’ closely-watched hepatitis C drug Sovaldi has been given the green light in Europe.
The European Commission has granted marketing authorisation for Sovaldi (sofosbuvir) 400mg tablets
which, as part of HCV combination therapy with peg-interferon and ribavirin, offers cure rates of around 90% in previously-untreated adults. However, most significant is that the once-daily nucleotide analogue polymerase inhibitor is the first all-oral treatment option for up to 24 weeks for patients unsuitable for interferon.
Read more at: http://www.pharmatimes.com/Article/14-01-20/Gilead_s_HCV_drug_Sovaldi_gets_Europe_OK.aspx#ixzz2qwHI3iJi
SYNTHESIS
-
sofosbuvir » All About Drugs
http://www.allfordrugs.com/tag/sofosbuvir/ALL ABOUT DRUGS BY DR ANTHONY MELVIN CRASTO, WORLD DRUG TRACKER HELPING … US Approves Breakthrough Hepatitis C Drug,Sofosbuvir.
DAPAGLIFLOZIN SEES LIGHT

DAPAGLIFLOZIN, BMS-512148
(2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol,
cas 461432-26-8
| Molecular Formula: C21H25ClO6 |
| Molecular Weight: 408.87 |
Bristol-Myers Squibb (Originator)
AstraZeneca
TYPE 2 DIABETES,SGLT-2 Inhibitors
launched 2012, as forxiga in EU

Dapagliflozin propanediol is a solvate containing 1:1:1 ratio of the dapagliflozin, (S)-(+)-1,2-propanediol, and water.
US——-In 2011, the product was not recommended for approval by the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee. In 2011, the FDA assigned a complete response letter to the application. A new application was resubmitted in 2013 by Bristol-Myers Squibb and AstraZeneca in the U.S
WILMINGTON, Del. & PRINCETON, N.J.--(BUSINESS WIRE)--December 12, 2013--
AstraZeneca (NYSE:AZN) and Bristol-Myers Squibb Company (NYSE:BMY) today announced the U.S. Food and Drug Administration’s (FDA) Endocrinologic and Metabolic Drugs Advisory Committee (EMDAC) voted 13-1 that the benefits of dapagliflozin use outweigh identified risks and support marketing of dapagliflozin as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus. The Advisory Committee also voted 10-4 that the data provided sufficient evidence that dapagliflozin, relative to comparators, has an acceptable cardiovascular risk profile.
The FDA is not bound by the Advisory Committee’s recommendation but takes its advice into consideration when reviewing the application for an investigational agent. The Prescription Drug User Fee Act (PDUFA) goal date for dapagliflozin is Jan. 11, 2014.

Dapagliflozin is being reviewed by the FDA for use as monotherapy, and in combination with other antidiabetic agents, as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes. It is a selective and reversible inhibitor of sodium-glucose cotransporter 2 (SGLT2) that works independently of insulin to help remove excess glucose from the body. Dapagliflozin, an investigational compound in the U.S., was the first SGLT2 inhibitor to be approved anywhere in the world. Dapagliflozin is currently approved under the trade name [Forxiga](TM) for the treatment of adults with type 2 diabetes, along with diet and exercise, in 38 countries, including the European Union and Australia.
http://online.wsj.com/article/PR-CO-20131212-910828.html?dsk=y
………………………………………………………………..
PATENTS
WO 2010138535
WO 2011060256
WO 2012041898
WO 2012163990
WO 2013068850
WO 2012163546
WO 2013068850
WO 2013079501

Dapagliflozin (INN/USAN,[1] trade name Forxiga) is a drug used to treat type 2 diabetes. It was developed by Bristol-Myers Squibb in partnership with AstraZeneca. Although dapagliflozin’s method of action would operate on both types of diabetes[1] and other conditions resulting inhyperglycemia, the current clinical trials specifically exclude participants with type 1 diabetes.[2][3]
In July 2011 an US Food and Drug Administration (FDA) committee recommended against approval until more data was available.[4] The Prescription Drug User Fee Act (PDUFA) date for dapagliflozin for the treatment of Type 2 diabetes was extended three months by the FDA to January 28, 2012.
In April 2012, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency issued a positive opinion on the drug. It is now marketed in a number of European countries including the UK and Germany.
Dapagliflozin inhibits subtype 2 of the sodium-glucose transport proteins (SGLT2), which is responsible for at least 90% of the glucose reabsorption in the kidney. Blocking this transporter causes blood glucose to be eliminated through the urine.[5] The efficacy of the this medication class has yet to be determined, but in initial clinical trials, dapagliflozin lowers HbA1c by 0.90 percentage points when added to metformin.[6]
Type II diabetes is the most common form of diabetes accounting for 90% of diabetes cases. Over 100 million people worldwide have type-2 diabetes (nearly 17 million in the U.S.) and the prevalence is increasing dramatically in both the developed and developing worlds. Type-II diabetes is a lifelong illness, which generally starts in middle age or later part of life, but can start at any age. Patients with type-2 diabetes do not respond properly to insulin, the hormone that normally allows the body to convert blood glucose into energy or store it in cells to be used later. The problem in type-2 diabetes is a condition called insulin resistance where the body produces insulin, in normal or even high amounts, but certain mechanisms prevent insulin from moving glucose into cells. Because the body does not use insulin properly, glucose rises to unsafe levels in the blood, the condition known as hyperglycemia.
Hyperglycemia, that is, elevated plasma glucose, is a hallmark of diabetes. Plasma glucose is normally filtered in the kidney in the glomerulus but is actively reabsorbed in the proximal tubule (kidney). Sodium-dependent glucose co-transporter SGLT2 appears to be the major transporter responsible for the reuptake of glucose at this site. The SGLT inhibitor phlorizin, and closely related analogs, inhibit this reuptake process in diabetic rodents and dogs, resulting in normalization of plasma glucose levels by promoting glucose excretion without hypoglycemic side effects. Long term (6 month) treatment of Zucker diabetic rats with an SGLT2 inhibitor has been reported to improve insulin response to glycemia, improve insulin sensitivity, and delay the onset of nephropathy and neuropathy in these animals, with no detectable pathology in the kidney and no electrolyte imbalance in plasma. Selective inhibition of SGLT2 in diabetic patients would be expected to normalize plasma glucose by enhancing the excretion of glucose in the urine, thereby improving insulin sensitivity and delaying the development of diabetic complications.
The treatment of diabetes is an important health concern and despite a wide range of available therapies, the epidemic continues. Type 2 diabetes (T2DM) is a progressive disease caused by insulin resistance and decreased pancreatic β-cell function. Insulin is produced by the pancreatic β-cell and mediates cellular glucose uptake and clearance. Insulin resistance is characterized by the lack of response to the actions of this hormone which results in decreased cellular clearance of glucose from the circulation and overproduction of glucose by the liver.
The currently available therapies to treat type 2 diabetes augment the action or delivery of insulin to lower blood glucose. However, despite therapy, many patients do not achieve control of their type 2 diabetes. According to the National Health and Nutrition Examination Survey (NHANES) III, only 36% of type 2 diabetics achieve glycemic control defined as a A1C<7.0% with current therapies. In an effort to treat type 2 diabetes, aggressive therapy with multiple pharmacologic agents may be prescribed. The use of insulin plus oral agents has increased from approximately 3 to 11% from NHANES II to III.
Thus, treatment of hyperglycemia in type 2 diabetes (T2DM) remains a major challenge, particularly in patients who require insulin as the disease progresses. Various combinations of insulin with oral anti-diabetic agents (OADs) have been investigated in recent years, and an increasing number of patients have been placed on these regimens. Poulsen, M. K. et al., “The combined effect of triple therapy with rosiglitazone, metformin, and insulin in type 2 diabetic patients”,Diabetes Care, 26 (12):3273-3279 (2003); Buse, J., “Combining insulin and oral agents”, Am. J. Med., 108 (Supp. 6a):23S-32S (2000). Often, these combination therapies become less effective in controlling hyperglycemia over time, particularly as weight gain and worsening insulin resistance impair insulin response pathways.
Hypoglycemia, weight gain, and subsequent increased insulin resistance are significant factors that limit optimal titration and effectiveness of insulin. (Holman, R. R. et al., “Addition of biphasic, prandial, or basal insulin to oral therapy in type 2 diabetes”, N. Engl. J. Med., 357 (17):1716-1730 (2007)). Weight gain with insulin therapy is predominantly a consequence of the reduction of glucosuria, and is thought to be proportional to the correction of glycemia. (Makimattila, S. et al., “Causes of weight gain during insulin therapy with and without metformin in patients with Type II diabetes mellitus”, Diabetologia, 42 (4):406-412 (1999)). Insulin drives weight gain when used alone or with OADs. (Buse, J., supra). In some cases, intensive insulin therapy may worsen lipid overload and complicate progression of the disease through a spiral of caloric surplus, hyperinsulinemia, increased lipogenesis, increased adipocity, increased insulin resistance, beta-cell toxicity, and hyperglycemia. (Unger, R. H., “Reinventing type 2 diabetes: pathogenesis, treatment, and prevention”, JAMA, 299 (10):1185-1187 (2008)). Among commonly used OADs, thiazolidinediones (TZDs) and sulfonylureas intrinsically contribute to weight gain as glucosuria dissipates with improved glycemic control. Weight gain is less prominent with metformin, acting through suppression of hepatic glucose output, or with incretin-based DPP-4 inhibitors. Overall, there is a pressing need for novel agents that can be safely added to insulin-dependent therapies to help achieve glycemic targets without increasing the risks of weight gain or hypoglycemia.
A novel approach to treating hyperglycemia involves targeting transporters for glucose reabsorption in the kidney. (Kanai, Y. et al., “The human kidney low affinity Na+/glucose cotransporter SGLT2. Delineation of the major renal reabsorptive mechanism for D-glucose”, J. Clin. Invest., 93 (1):397-404 (1994)). Agents that selectively block the sodium-glucose cotransporter 2 (SGLT2) located in the proximal tubule of the kidney can inhibit reabsorption of glucose and induce its elimination through urinary excretion. (Brown, G. K., “Glucose transporters: structure, function and consequences of deficiency”, J. Inherit. Metab. Dis., 23 (3):237-246 (2000)). SGLT2 inhibition has been shown in pre-clinical models to lower blood glucose independently of insulin. (Han, S. et al., “Dapagliflozin, a selective SGLT2 inhibitor, improves glucose homeostasis in normal and diabetic rats”, Diabetes, 57 (6):1723-1729 (2008); Katsuno, K. et al., “Sergliflozin, a novel selective inhibitor of low-affinity sodium glucose cotransporter (SGLT2), validates the critical role of SGLT2 in renal glucose reabsorption and modulates plasma glucose level”, J. Pharmacol. Exp. Ther., 320 (1):323-330 (2007)).
Dapagliflozin(BMS-512148) is a potent sodium-glucose transport proteins inhibitor with IC50 of 1.1 nM and 1.4uM for SGLT2 and SGLT1, respectively. Dapagliflozin (BMS-512148) inhibits subtype 2 of the sodium-glucose transport proteins (SGLT2), which is responsible for at least 90% of the glucose reabsorption in the kidney. Blocking this transporter causes blood glucose to be eliminated through the urine. Symptoms of hypoglycaemia occurred in similar proportions of patients in the dapagliflozin (2~4%) and placebo groups (3%). Signs, symptoms, and other reports suggestive of genital infections were more frequent in the dapagliflozin groups (2•5 mg, [8%]; 5 mg, [13%]; 10 mg, [9%]) than in the placebo group ( [5%]).
Dapagliflozin (which is disclosed in U.S. Pat. No. 6,515,117) is an inhibitor of sodium-glucose reabsorption by the kidney, by inhibiting SGLT2, which results in an increased excretion of glucose in the urine. This effect lowers plasma glucose in an insulin-independent manner.
Dapagliflozin is currently undergoing clinical development for treatment of type 2 diabetes. (Han, S. et al., supra; Meng, W. et al., “Discovery of dapagliflozin: a potent, selective renal sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes”, J. Med. Chem., 51 (5):1145-1149 (2008)). Phase 2a and 2b studies with dapagliflozin have demonstrated efficacy in reducing hyperglycemia either alone or in combination with metformin in patients with T2DM. (Komoroski, B. et al., “Dapagliflozin, a novel, selective SGLT2 inhibitor, improved glycemic control over 2 weeks in patients with type 2 diabetes mellitus”, Clin. Pharmacol. Ther., 85 (5):513-519 (2009); List, J. F. et al., “Dapagliflozin-induced glucosuria is accompanied by weight loss in type 2 diabetic patients”, 68th Scientific Sessions of the American Diabetes Association, San Francisco, Calif., Jun. 6-10, 2008, Presentation No. 0461P).
It has been found that dapagliflozin does not act through insulin signaling pathways and is effective in controlling blood sugar in patients whose insulin signaling pathways do not work well. This applies to extremes of insulin resistance, in type 2 diabetes as well as in insulin resistance syndromes, caused by, for example, mutations in the insulin receptor.
Since dapagliflozin leads to heavy glycosuria (sometimes up to about 70 grams per day) it can lead to rapid weight loss and tiredness. The glucose acts as an osmotic diuretic (this effect is the cause of polyuria in diabetes) which can lead to dehydration. The increased amount of glucose in the urine can also worsen the infections already associated with diabetes, particularly urinary tract infections and thrush (candidiasis). Dapagliflozin is also associated with hypotensive reactions.

The IC50 for SGLT2 is less than one thousandth of the IC50 for SGLT1 (1.1 versus 1390 nmol/l), so that the drug does not interfere with the intestinal glucose absorption.[7]
- Statement on a nonproprietory name adopted by the USAN council
- Efficacy and Safety of Dapagliflozin, Added to Therapy of Patients With Type 2 Diabetes With Inadequate Glycemic Control on Insulin, ClinicalTrials.gov, April 2009
- Trial Details for Trial MB102-020, Bristol-Myers Squibb, May 2009
- “FDA panel advises against approval of dapagliflozin”. 19 July 2011.
- Prous Science: Molecule of the Month November 2007
- UEndocrine: Internet Endocrinology Community
- Schubert-Zsilavecz, M, Wurglics, M, Neue Arzneimittel 2008/2009
- more1) Pal, Manojit et al; Improved Process for the preparation of SGLT2 inhibitor dapagliflozin via glycosylation of 5-bromo-2-Chloro-4′-ethoxydiphenylmethane with Gluconolactone ;. Indian Pat Appl,. 2010CH03942 , 19 Oct 20122) Lemaire, Sebastien et al; Stereoselective C-Glycosylation Reactions with Arylzinc Reagents ;Organic Letters , 2012, 14 (6), 1480-1483;3) Zhuo, Biqin and Xing, Xijuan; Process for preparation of Dapagliflozin amino acid cocrystals ;Faming Zhuanli Shenqing , 102 167 715, 31 Aug 20114) Shao, Hua et al; Total synthesis of SGLT2 inhibitor Dapagliflozin ; Hecheng Huaxue , 18 (3), 389-392; 2010
5) Liou, Jason et al; Processes for the preparation of C-Aryl glycoside amino acid complexes as potential SGLT2 Inhibitors ;. PCT Int Appl,. WO2010022313
6) Seed, Brian et al; Preparation of Deuterated benzyl-benzene glycosides having an inhibitory Effect on sodium-dependent glucose co-transporter; . PCT Int Appl,. WO2010009243
7) Song, Yanli et al; Preparation of benzylbenzene glycoside Derivatives as antidiabetic Agents ;. PCT Int Appl,. WO2009026537
8) Meng, Wei et al; D iscovery of Dapagliflozin: A Potent, Selective Renal Sodium-Dependent Glucose cotransporter 2 (SGLT2) Inhibitor for the Treatment of Type 2 Diabetes ; Journal of Medicinal chemistr y, 2008, 51 (5), 1145 -1149;
9) Gougoutas, Jack Z. et al; Solvates Crystalline complexes of amino acid with (1S)-1 ,5-anhydro-LC (3 – ((phenyl) methyl) phenyl)-D-glucitol were prepared as for SGLT2 Inhibitors the treatment of Diabetes ;. PCT Int Appl,. WO2008002824
10) Deshpande, Prashant P. et al; Methods of producing C-Aryl glucoside SGLT2 Inhibitors ;.. U.S. Pat Appl Publ,. 20,040,138,439

dapagliflozin being an inhibitor of sodiumdependent glucose transporters found in the intestine and kidney (SGLT2) and to a method for treating diabetes, especially type II diabetes, as well as hyperglycemia, hyperinsulinemia, obesity, hypertriglyceridemia, Syndrome X, diabetic
complications, atherosclerosis and related diseases, employing such C-aryl glucosides alone or in combination with one, two or more other type antidiabetic agent and/or one, two or more other type therapeutic agents such as hypolipidemic agents.
Approximately 100 million people worldwide suffer from type II diabetes (NIDDM – non-insulin-dependent diabetes mellitus), which is characterized by hyperglycemia due to excessive hepatic glucose production and peripheral insulin resistance, the root causes for which are as yet unknown. Hyperglycemia is considered to be the major risk factor for the development of diabetic complications, and is likely to contribute directly to the impairment of insulin secretion seen in advanced NIDDM. Normalization of plasma glucose in NIDDM patients would be predicted to improve insulin action, and to offset the development of diabetic complications. An inhibitor of the sodium-dependent glucose transporter SGLT2 in the kidney would be expected to aid in the normalization of plasma glucose levels, and perhaps body weight, by enhancing glucose excretion.
Dapagliflozin can be prepared using similar procedures as described in U.S. Pat. No. 6,515,117 or international published applications no. WO 03/099836 and WO 2008/116179
WO 03/099836 A1 refers to dapagliflozin having the structure according to formula 1 .

formula 1
WO 03/099836 A1 discloses a route of synthesis on pages 8-10, whereby one major step is the purification of a compound of formula 2

formula 2
The compound of formula 2 provides a means of purification for providing a compound of formula 1 since it crystallizes. Subsequently the crystalline form of the compound of formula 2 can be deprotected and converted to dapagliflozin. Using this process, dapagliflozin is obtained as an amorphous glassy off-white solid containing 0.1 1 mol% of EtOAc. Crystallization of a pharmaceutical drug is usually advantageous as it provides means for purification also suitable for industrial scale preparation. However, for providing an active pharmaceutical drug a very high purity is required. In particular, organic impurities such as EtOAc either need to be avoided or further purification steps are needed to provide the drug in a
pharmaceutically acceptable form, i.e. substantially free of organic solvents. Thus, there is the need in the art to obtain pure and crystalline dapagliflozinwhich is substantially free of organic solvents.
WO 2008/002824 A1 discloses several alternative solid forms of dapagliflozin, such as e.g. solvates containing organic alcohols or co-crystals with amino acids such as proline and phenylalanine. For instance, the document discloses crystalline
dapagliflozin solvates which additionally contain water molecules (see e.g.
Examples 3-6), but is silent about solid forms of dapagliflozin which do not contain impurities such as organic alcohols. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps. In contrast, the document relates to dapagliflozin solvates where an alcohol and water are both incorporated into the crystal lattice. Hence, there is the need in the art to obtain pure and crystalline dapagliflozin suitable for pharmaceutical production.
WO 2008/1 16179 A1 refers to an immediate release pharmaceutical composition comprising dapagliflozin and propylene glycol. Propylene glycol is a chiral
substance and (S)-propylene glycol used is very expensive. Consequently, also the immediate release pharmaceutical composition is more expensive.
Crystalline forms (in comparision to the amorphous form) often show desired different physical and/or biological characteristics which may assist in the manufacture or formulation of the active compound, to the purity levels and uniformity required for regulatory approval. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps.
…..
WO 2008/ 1 16179 Al seems to disclose an immediate release formulation comprising dapagliflozin and propylene glycol hydrate. WO 2008/ 116195 A2 refers to the use of an SLGT2 inhibitor in the treatment of obesity
http://www.google.com/patents/US20120282336
http://www.tga.gov.au/pdf/auspar/auspar-dapagliflozin-propanediol-monohydrate-130114.pdf
Example 2 Dapagliflozin (S) PGS—(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (S)-propane-1,2-diol hydrate (1:1:1)
Dapagliflozin (S) propylene glycol hydrate (1:1:1) can be prepared using similar procedures as described in published applications WO 08/002824 and WO 2008/116179, the disclosures of which are herein incorporated by reference in their entirety for any purpose. SGLT2 EC50=1.1 nM.
Example 3 Dapagliflozin (R) PGS—(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (R)-propane-1,2-diol hydrate (1:1:1)
Dapagliflozin (R) propylene glycol hydrate (1:1:1) can be prepared using similar procedures as described in WO 08/002824 and WO 2008/116179, the disclosures of which are herein incorporated by reference in their entirety for any purpose. SGLT2 EC50=1.1 nM.
WO 2008/002824 A1 discloses several alternative solid forms of dapagliflozin, such as e.g. solvates containing organic alcohols or co-crystals with amino acids such as proline and phenylalanine. For instance, the document discloses crystalline
dapagliflozin solvates which additionally contain water molecules (see e.g.
Examples 3-6), but is silent about solid forms of dapagliflozin which do not contain impurities such as organic alcohols. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps. In contrast, the document relates to dapagliflozin solvates where an alcohol and water are both incorporated into the crystal lattice. Hence, there is the need in the art to obtain pure and crystalline dapagliflozin suitable for pharmaceutical production.
WO 2008/1 16179 A1 refers to an immediate release pharmaceutical composition comprising dapagliflozin and propylene glycol. Propylene glycol is a chiral
substance and (S)-propylene glycol used is very expensive. Consequently, also the immediate release pharmaceutical composition is more expensive.
Surprisingly, amorphous dapagliflozin can be purified with the process of the present invention. For instance amorphous dapagliflozin having a purity of 99,0% can be converted to crystalline dapagliflozin hydrate having a purity of 100% (see examples of the present application). Moreover, said crystalline dapagliflozin hydrate does not contain any additional solvent which is desirable. Thus, the process of purifying dapagliflozin according to the present invention is superior compared with the process of WO 03/099836 A1 .
Additionally, the dapagliflozin hydrate obtained is crystalline which is advantageous with respect to the formulation of a pharmaceutical composition. The use of expensive diols such as (S)-propanediol for obtaining an immediate release pharmaceutical composition as disclosed in WO 2008/1 16179 A1 can be avoided
………………………………
In Vitro Characterization and Pharmacokinetics of Dapagliflozin …
dmd.aspetjournals.org/content/…/DMD29165_supplemental_data_.doc

Dapagliflozin (BMS-512148), (2S,3R,4R,5S,6R)-2-(3-(4-Ethoxybenzyl)-4-chlorophenyl)
-6-hydroxymethyl-tetrahydro-2H-pyran-3,4,5-triol. 1H NMR (500 MHz, CD3OD) δ 7.33
(d, J = 6.0, 1H), 7.31 (d, J = 2.2, 1H), 7.31 (dd, J = 2.2, 6.0, 1H), 7.07 (d, J = 8.8, 2H),
6.78 (d, J = 8.8, 2H), 4.07-3.90 (m, 7H), 3.85 (d, J = 10.6, 1H), 3.69 (dd, J = 5.3, 10.6,
1H), 3.42-3.25 (m, 4H), 1.34 (t, J = 7.0, 3H). 13C NMR (125 MHz, CD3OD) δ 158.8,
140.0, 139.9, 134.4, 132.9, 131.9, 130.8, 130.1, 128.2, 115.5, 82.9, 82.2, 79.7, 76.4, 71.9,
64.5, 63.1, 39.2, 15.2.
HRMS calculated for C21H25ClNaO6 (M+Na)+
For C21H25ClO6: C, 61.68; H, 6.16. Found: C, 61.16; H, 6.58.
: 431.1237; found 431.1234. Anal. Calcd
SECOND SET
1H NMR (500 MHz, CD3OD) δ 7.33 (d, J = 6.0, 1H), 7.31 (d, J = 2.2, 1H), 7.31 (dd, J = 2.2, 6.0, 1H), 7.07 (d, J = 8.8, 2H), 6.78 (d, J = 8.8, 2H), 4.07–3.90 (m, 7H), 3.85 (d, J = 10.6, 1H), 3.69 (dd, J = 5.3, 10.6, 1H), 3.42–3.25 (m, 4H), 1.34 (t, J = 7.0, 3H);
13C NMR (125 MHz, CD3OD) δ 158.8, 140.0, 139.9, 134.4, 132.9, 131.9, 130.8, 130.1, 128.2, 115.5, 82.9, 82.2, 79.7, 76.4, 71.9, 64.5, 63.1, 39.2, 15.2;
HRMS calcd for C21H25ClNaO6 (M + Na)+ 431.1237, found 431.1234. Anal. Calcd for C21H25ClO6: C, 61.68; H, 6.16. Found: C, 61.16; H, 6.58.
………………………
HPLC
-
HPLC measurements were performed with an Agilent 1100 series instrument equipped with a UV-vis detector set to 240 nm according to the following method:
Column: Ascentis Express RP-Amide 4.6 x 150 mm, 2.7 mm;
Column temperature: 25 °C
– Eluent A: 0.1 % formic acid in water
– Eluent B: 0.1 % formic acid in acetonitrile
– Injection volume: 3 mL
– Flow: 0.7 mL/min
– Gradient:Time [min] [%] B 0.0 25 25.0 65 26.0 70 29.0 70 29.5 25 35.0 25 ……………………..

……..
http://www.google.com/patents/WO2013068850A2?cl=en
EXAMPLE 24 – Synthesis of 2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(4-chloro-3-(4- ethoxybenzyl)phenyl)- -D-glucopyranoside 2,4-di-6>-TBDPS-dapagliflozin; (IVj”))

[0229] l-(5-Bromo-2-chlorobenzyl)-4-ethoxybenzene (1.5 g, 4.6 mmol) and magnesium powder (0.54 g, 22.2 mmol) were placed in a suitable reactor, followed by THF (12 mL) and 1,2- dibromoethane (0.16 mL). The mixture was heated to reflux. After the reaction had initiated, a solution of l-(5-bromo-2-chlorobenzyl)-4-ethoxybenzene (4.5 g, 13.8 mmol) in THF (28 mL) was added dropwise. The mixture was allowed to stir for another hour under reflux, and was then cooled to ambient temperature, and then titrated to determine the concentration. The above prepared 4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl magnesium bromide (31 mL, 10 mmol, 0.32 M in THF) and A1C13 (0.5 M in THF, 8.0 mL, 4.0 mmol) were mixed at ambient temperature to give a black solution, which was stirred at ambient temperature for 1 hour. To a solution of
I, 6-anhydro-2,4-di-6>-ieri-butyldiphenylsilyl- -D-glucopyranose (0.64 g, 1.0 mmol) in PhOMe (3.0 mL) at ambient temperature was added phenylmagnesium bromide (0.38 mL, 1.0 mmol, 2.6 M solution in Et20). After stirring for about 5 min the solution was then added into the above prepared aluminum mixture via syringe, followed by additional PhOMe (1.0 mL) to rinse the flask. The mixture was concentrated under reduced pressure (50 torr) at 60 °C (external bath temperature) to remove low-boiling point ethereal solvents and then PhOMe (6mL) was added. The reaction mixture was heated at 130 °C (external bath temperature) for 8 hours at which time HPLC assay analysis indicated a 51% yield of 2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(4-chloro-3- (4-ethoxybenzyl)phenyl)- -D-glucopyranoside. After cooling to ambient temperature, the reaction was treated with 10% aqueous NaOH (1 mL), THF (10 mL) and diatomaceous earth at ambient temperature, then the mixture was filtered and the filter cake was washed with THF. The combined filtrates were concentrated and the crude product was purified by silica gel column chromatography (eluting with 1:30 EtOAc/77-heptane) affording the product 2,4-di-6>- ieri-butyldiphenylsilyl- 1 – -(4-chloro-3 -(4-ethoxybenzyl)phenyl)- β-D-glucopyranoside (0.30 g, 34%) as a white powder.
1H NMR (400 MHz, CDC13) δ 7.56-7.54 (m, 2H), 7.43-7.31 (m, 13H), 7.29-7.22 (m, 6H), 7.07- 7.04 (m, 2H), 7.00 (d, J= 2.0 Hz, IH), 6.87 (dd, J= 8.4, 2.0 Hz, IH), 6.83-6.81 (m, 2H), 4.18 (d, J= 9.6 Hz, IH), 4.02 (q, J= 6.9 Hz, 2H), 3.96 (d, J= 10.8 Hz, 2H), 3.86 (ddd, J= 11.3, 7.7, 1.1 Hz, IH), 3.76 (ddd, J= 8.4, 8.4, 4.8 Hz, IH), 3.56 (ddd, J= 9.0, 6.4, 2.4 Hz, IH), 3.50 (dd, J=
I I.4, 5.4 Hz, IH), 3.44 (dd, J= 9.4, 8.6 Hz, IH), 3.38 (dd, J= 8.8, 8.8 Hz, IH), 1.70 (dd, J= 7.8, 5.4 Hz, IH, OH), 1.42 (t, J= 6.8 Hz, 3H), 1.21 (d, J= 5.2 Hz, IH, OH), 1.00 (s, 9H), 0.64 (s, 9H); 13C NMR (100 MHz, CDC13) δ 157.4 (C), 138.8 (C), 137.4 (C), 136.3 (CH x2), 136.1 (CH x2), 135.2 (CH x2), 135.0 (C), 134.9 (CH x2), 134.8 (C), 134.2 (C), 132.8 (C), 132.0 (C), 131.6 (CH), 131.1 (C), 129.9 (CH x2), 129.7 (CH), 129.6 (CH), 129.5 (CH), 129.4 (CH), 129.2 (CH), 127.58 (CH x2), 127.57 (CH x2), 127.54 (CH x2), 127.31 (CH), 127.28 (CH x2), 114.4 (CH x2), 82.2 (CH), 80.5 (CH), 79.3 (CH), 76.3 (CH), 72.7 (CH), 63.4 (CH2), 62.7 (CH2), 38.2 (CH2), 27.2 (CH3 x3), 26.6 (CH3 x3), 19.6 (C), 19.2 (C), 14.9 (CH3). EXAMPLE 25 -Synthesis of dapagliflozin ((25,3R,4R,55,6/?)-2-[4-chloro-3-(4- ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol; (Ij))

IVj’ U
[0230] A solution of the 2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(4-chloro-3-(4- ethoxybenzyl)phenyl)- -D-glucopyranoside (60 mg, 0.068 mmol) in THF (3.0 mL) and TBAF (3.0 mL, 3.0 mmol, 1.0 M in THF) was stirred at ambient temperature for 15 hours. CaC03 (0.62 g), Dowex^ 50WX8-400 ion exchange resin (1.86 g) and MeOH (5mL) were added to the product mixture and the suspension was stirred at ambient temperature for 1 hour and then the mixture was filtrated through a pad of diatomaceous earth. The filter cake was rinsed with MeOH and the combined filtrates was evaporated under vacuum and the resulting residue was purified by column chromatography (eluting with 1 : 10 MeOH/DCM) affording dapagliflozin (30 mg).
1H NMR (400 MHz, CD3OD) δ 7.37-7.34 (m, 2H), 7.29 (dd, J= 8.2, 2.2 Hz, 1H), 7.12-7.10 (m, 2H), 6.82-6.80 (m, 2H), 4.10 (d, J= 9.6 Hz, 2H), 4.04 (d, J= 9.2 Hz, 2H), 4.00 (q, J= 7.1 Hz, 2H), 3.91-3.87 (m, 1H), 3.73-3.67(m, 1H), 3.47-3.40 (m, 3H), 3.31-3.23 (m, 2H), 1.37 (t, J= 7.0 Hz, 3H);
13C NMR (100 MHz, CD3OD) δ 157.4 (C), 138.6 (C), 138.5 (C), 133.1 (C), 131.5 (C), 130.5 (CH), 129.4 (CH x2), 128.7 (CH), 126.8 (CH), 114.0 (CH x2), 80.5 (CH), 80.8 (CH), 78.3 (CH), 75.0 (CH), 70.4 (CH), 63.0 (CH2), 61.7 (CH2), 37.8 (CH2), 13.8 (CH3);
LCMS (ESI) m/z 426 (100, [M+NH4]+), 428 (36, [M+NH4+2]+), 447 (33, [M+K]+).
Example 1 – Synthesis of l,6-anhydro-2,4-di-6>-ieri-butyldiphenylsilyl- -D-glucopyranose (II”)

III II”
[0206] To a suspension solution of l,6-anhydro- -D-glucopyranose (1.83 g, 11.3 mmol) and imidazole (3.07 g, 45.2 mmol) in THF (10 mL) at 0 °C was added dropwise a solution of TBDPSC1 (11.6 mL, 45.2 mmol) in THF (10 mL). After the l,6-anhydro-P-D-gJucopyranose was consumed, water (10 mL) was added and the mixture was extracted twice with EtOAc (20 mL each), washed with brine (10 mL), dried (Na2S04) and concentrated. Column
chromatography (eluting with 1 :20 EtOAc/rc-heptane) afforded 2,4-di-6>-ieri-butyldiphenylsilyl- l,6-anhydro- “D-glucopyranose (5.89 g, 81%).
1H NMR (400 MHz, CDC13) δ 7.82-7.70 (m, 8H), 7.49-7.36 (m, 12H), 5.17 (s, IH), 4.22 (d, J= 4.8 Hz, IH), 3.88-3.85 (m, IH), 3.583-3.579 (m, IH), 3.492-3.486 (m, IH), 3.47-3.45 (m, IH), 3.30 (dd, J= 7.4, 5.4 Hz, IH), 1.71 (d, J= 6.0 Hz, IH), 1.142 (s, 9H), 1.139 (s, 9H); 13C NMR (100 MHz, CDCI3) δ 135.89 (CH x2), 135.87 (CH x2), 135.85 (CH x2), 135.83 (CH x2), 133.8 (C), 133.5 (C), 133.3 (C), 133.2 (C), 129.94 (CH), 129.92 (CH), 129.90 (CH), 129.88 (CH), 127.84 (CH2 x2), 127.82 (CH2 x2), 127.77 (CH2 x4), 102.4 (CH), 76.9 (CH), 75.3 (CH), 73.9 (CH), 73.5 (CH), 65.4 (CH2), 27.0 (CH3 x6), 19.3 (C x2).
Otsuka and Lundbeck’s Once-Monthly Abilify Maintena(R) (Aripiprazole) Now Approved in Europe
Otsuka and Lundbeck’s Once-Monthly Abilify Maintena(R) (Aripiprazole) Now Approved in Europe for Maintenance Treatment of Schizophrenia in Adult Patients Stabilized with Oral Aripiprazole
do not forget to see my old blog post, contains synthesis
Preventing relapse is critical in the treatment of schizophrenia. Pivotal
studies, that supported the EU submission, demonstrate that Abilify
Maintena can reduce the risk of relapse relative to placebo and is
non-inferior to oral aripiprazole in patients with schizophrenia1,2
-- The approval of Abilify Maintena now provides patients with schizophrenia
access to a once-monthly formulation with established efficacy together
with a tolerability profile that is comparable to that of oral ABILIFY(R)
(aripiprazole)2
-- At the end of the randomized, double-blind treatment phase in one of the
pivotal trials, 93 percent of patients reported being extremely, very or
somewhat satisfied with Abilify Maintena treatment3
-- Abilify Maintena is the only dopamine D2 partial agonist in a
once-monthly, injectable form to receive marketing authorization in
Europe for maintenance treatment of schizophrenia
-- Abilify Maintena will be the first commercialized product in Europe from
the global alliance between Otsuka and Lundbeck which is focused on
developing Central Nervous System (CNS) therapies worldwide
TOKYO & COPENHAGEN, Denmark--(BUSINESS WIRE)--November 21, 2013--
Otsuka Pharmaceutical Co., Ltd. (Otsuka) and H. Lundbeck A/S (Lundbeck) today announced marketing authorization approval from the European Commission for Abilify Maintena (aripiprazole), an intramuscular (IM) once-monthly injectable formulation for maintenance treatment of schizophrenia in adult patients stabilized with oral aripiprazole.
Abilify Maintena reduces the risk of relapse relative to placebo over the long-term and provides effective treatment of schizophrenia.(1,2) It has a tolerability profile similar to oral aripiprazole(1) , and demonstrated statistically significant benefits on patients’ personal and social functioning as compared to placebo.(1,4) 93 percent of patients treated with Abilify Maintena were extremely, very or somewhat satisfied with their treatment at the end of the double-blind treatment phase.(4)
“We strongly believe the schizophrenia community will welcome the availability of Abilify Maintena to help improve outcomes for patients living with schizophrenia. As a company, our focus is to develop treatments to help protect against relapse and preserve brain function,” said Ole Vahlgren, CEO & President, Otsuka Europe. “We must partner with health care professionals and caregivers to help patients get the best treatments that focus on reducing the risk of relapse.”
“Studies have shown that the early use of long-acting injectables can prevent a person with schizophrenia from experiencing a relapse,”(5) said Ole Chrintz, SVP International Markets & Europe, Lundbeck. “Efficacy is important, but a treatment for a chronic condition such as schizophrenia also needs to be well tolerated so patients will stay on it over the long-term. We believe Abilify Maintena meets this need.”(1,2,3)
About the Studies
The efficacy of Abilify Maintena was demonstrated in two double-blind, Phase III, randomized trials. The safety profile for Abilify Maintena was demonstrated to be similar to that of oral ABILIFY. The most frequently observed adverse drug reactions (ADRs) reported in >=5 percent of patients in two double-blind controlled clinical trials of Abilify Maintena were weight increases (9.0 percent), akathisia (7.9 percent), insomnia (5.8 percent), and injection site pain (5.1 percent).(1,2)
About Abilify Maintena
Abilify Maintena is the only dopamine D2 partial agonist in once-monthly, injectable form to receive marketing authorization for maintenance treatment in schizophrenia. Physicians now have an alternative treatment option, with a tolerability profile comparable to the well-established oral ABILIFY, to address the on-going need to reduce the risk of relapse in patients with schizophrenia.
The European label states that Abilify Maintena is a powder and solvent for prolonged-release suspension for intra-muscular (IM) injection. It is a once-monthly formulation of aripiprazole in a sterile lyophilized powder that is reconstituted with sterile water. Abilify Maintena is indicated for maintenance treatment of schizophrenia in adult patients stabilized with oral aripiprazole.
After the first injection, treatment with 10 mg to 20 mg oral aripiprazole should be continued for 14 consecutive days to maintain therapeutic aripiprazole concentrations during initiation of therapy.
About Schizophrenia and Disease Relapse
Schizophrenia is a disease characterized by a distortion in the process of thinking and of emotional responsiveness. It most commonly manifests as hallucinations, paranoid or bizarre delusions, or disorganized speech and thinking, and is accompanied by significant social or occupational dysfunction. Onset of symptoms typically occurs in young adulthood, and the condition is chronic, often requiring lifelong treatment to mitigate symptoms.
Relapse of schizophrenia refers to an exacerbation or acute psychotic break that is characterised primarily by the emergence of positive symptoms such as hallucinations, delusions and disordered thinking.(6)
Relapse can occur when a patient no longer responds to antipsychotic medication, does not take the medication as prescribed, or stops taking their medication altogether. There are many reasons patients stop taking their medication, including poor insight about their illness, side effects from current treatments, complicated medication regimen or lack of support from family.(7) Abilify Maintena is able to significantly reduce the risk of relapse in patients with schizophrenia.(1)
It has been estimated that schizophrenia affects approximately one percent of the adult population in the U.S. and Europe, and approximately 24 million people worldwide.(8,9) In Europe there are approximately 4.4 million adults with schizophrenia,(10) prevalent equally in both genders.(11,12) While there is no cure for the disease, symptoms and risk of relapse can be managed in most patients with appropriate antipsychotic treatment. However, when the disease is not managed, patients are at increased risk of disease relapse, which can cause the re-emergence or worsening of psychotic symptoms.(13)
Schizophrenia places a significant burden on society. It is regarded among the most financially costly illnesses and is according to the World Health Organization (WHO), the 8(th) leading cause of DALYs (lost healthy years) worldwide among patients between the age of 15-44.(11) With 50 percent of patients not receiving appropriate care and 80 percent of patients relapsing within the first 5 years,(14) there is a significant unmet need to be addressed in schizophrenia.
About the Lundbeck and Otsuka Global Alliance
Lundbeck and Otsuka established a global alliance in November 2011 to bring to bear their considerable experience and resources in the CNS area to introduce next-generation treatments for conditions such as schizophrenia, depression, Alzheimer’s disease and alcohol dependency.
About Otsuka Pharmaceutical Co., Ltd.
Otsuka Pharmaceutical Co., Ltd. is a global healthcare company with the corporate philosophy: ‘Otsuka-people creating new products for better health worldwide.’ Otsuka researches, develops, manufactures and markets innovative and original products, with a focus on pharmaceutical products for the treatment of diseases and nutraceutical products for the maintenance of everyday health.
In pharmaceuticals, Otsuka is a leading firm in the challenging area of mental health and also has research programs for several under-addressed diseases including tuberculosis, a significant global public health issue. These commitments illustrate more powerfully than words how Otsuka is a “big venture” company at heart, applying a youthful spirit of creativity in everything it does.
Otsuka is a wholly owned subsidiary of Otsuka Holdings Co., Ltd., the holding company for the Otsuka Group. The chairman Akihiko Otsuka is the third generation of Otsuka family members to lead the business, whose origins date from 1921. The Otsuka Group employs approximately 42,000 people globally and its products are available in more than 80 countries worldwide. Consolidated sales were approximately EUR10 billion or USD 13 billion for fiscal year 2012 (4/1/2012-3/31/2013). Otsuka Pharmaceutical welcomes you to visit its global website at https://www.otsuka.co.jp/en/
About H. Lundbeck A/S
Lundbeck is a global pharmaceutical company highly committed to improving the quality of life of people living with brain diseases. For this purpose, Lundbeck is engaged in the entire value chain throughout research, development, production, marketing and sales of pharmaceuticals across the world. The company’s products are targeted at disorders such as depression and anxiety, psychotic disorders, epilepsy, Huntington’s, Alzheimer’s and Parkinson’s diseases. Lundbeck’s pipeline consists of several mid- to late-stage development programmes.
Lundbeck employs more than 5,800 people worldwide, 2,000 of whom are based in Denmark. We have research in 57 countries and our products are registered in more than 100 countries. We have research centers in Denmark, China and the United States and production facilities in Italy, France, Mexico, China and Denmark. Lundbeck generated revenue of approximately DKK 15 billion in 2012. For additional information, we encourage you to visit our corporate site http://www.lundbeck.com.
REFERENCES:
1. Kane, JM et al. Aripiprazole intramuscular depot as maintenance treatment in
patients with schizophrenia: a 52-week, multicenter, randomized,
double-blind, placebo-controlled study. J Clin Psychiatry 2012;73(5):617-62
2. Fleischhacker WW, Sanchez R, Perry PP, et al. Aripiprazole once-monthly for
the treatment of schizophrenia: a double-blind, randomized, non-inferiority
study vs. oral aripiprazole. Annual Meeting of the American Psychiatric
Association, 18--22 May, 2013 (poster).
3. Sanchez R, et al. Patient-reported Outcomes with
Aripiprazole-Intramuscular-Depot for Long-term Maintenance Treatment in
Schiziphrenia. NR6-42 2012 (poster)
4. Carson WH, Perry P, Sanchez R, et al. Effects of a Once-Monthly Formulation
of Aripiprazole on Secondary Efficacy Outcomes in Maintenance Treatment of
Schizophrenia. Institute on Psychiatric Services meeting, October 4--7, 2012
(Poster)
5. Long-acting injectable antipsychotics in the treatment of schizophrenia:
their role in relapse prevention. Agid O, et al. Expert Opin. Pharmacother.
(2010) 11(14):2301-2317
6. Ayuso-Gutierrez JL, del Rio Vega JM. Factors influencing relapse in the
long-term course of schizophrenia. Schizophr Res. 1997; 28(2-3): 199-206
7. Baloush-Kleinman V, et al. Adherence to antipsychotic drug treatment in
early-episode schizophrenia: a six-month naturalistic follow-up study.
Schizophr Res. 2011;130(1-3):176-81.
8. National Institute of Mental Health (NIMH). Health Topics: Statistics.
Available at http://www.nimh.nih.gov/statistics/1SCHIZ.shtml. Accessed
October 22, 2013
9. World Health Organization (WHO). Schizophrenia Fact Sheet. 2010. Available
at: http://www.who.int/mental_health/management/schizophrenia/en/. Accessed
October 22, 2013.
10. World Health Organization (WHO) The global burden of disease:2004 update
(2008)
11. National Institute of Mental Health (NIMH). The Numbers Count: Mental
Disorders in America. 2010. Available at
http://www.nimh.nih.gov/health/publications/the-numbers-count-mental-disorde
rs-in-America/index.shtml#Schizophrenia. Accessed October 22, 2013
12. Regier DA, et al. The de facto US mental and addictive disorders service
system. Epidemiologic catchment area prospective 1-year prevalence rates of
disorders and services. Arch Gen Psychiatry. 1993;50(2):85-94.
13. American Psychiatric Association. Practice guideline for the treatment of
patients with schizophrenia. Second edition. 2004. Available at
http://psychiatryonline.org/content.aspx?bookid=28§ionid=1665359.
Accessed October 28, 2013
14. Robinson D, et al. Predictors of relapse following response from a first
episode of schizophrenia or schizoaffective disorder. Arch Gen Psychiatry.
1999;56(3):241-247
read my earlier blog post
https://newdrugapprovals.wordpress.com/2013/02/07/the-european-medicines-agency-ema-approves-otsukas-aripiprazole-abilify-for-the-treatment-of-moderate-to-severe-manic-episodes-in-bipolar-i-disorder-in-adolescents/
European Commission Approves Gilead’s VitektaTM, an Integrase Inhibitor for the Treatment of HIV-1 Infection

Elvitegravir
697761-98-1 CAS
FOSTER CITY, Calif.–(BUSINESS WIRE)–Nov. 18, 2013– Gilead Sciences, Inc. (Nasdaq: GILD) today announced that the European Commission has granted marketing authorization for VitektaTM (elvitegravir 85 mg and 150 mg) tablets, an integrase inhibitor for the treatment of HIV-1 infection in adults without known mutations associated with resistance to elvitegravir. Vitekta is indicated for use as part of HIV treatment regimens that include a ritonavir-boosted protease inhibitor.http://www.pharmalive.com/eu-oks-gileads-vitekta Vitekta interferes with HIV replication by blocking the virus from integrating into the genetic material of human cells. In clinical trials, Vitekta was effective in suppressing HIV among patients with drug-resistant strains of HIV.http://www.pharmalive.com/eu-oks-gileads-vitekta

Elvitegravir (EVG, formerly GS-9137) is a drug used for the treatment of HIV infection. It acts as an integrase inhibitor. It was developed[1] by the pharmaceutical company Gilead Sciences, which licensed EVG from Japan Tobacco in March 2008.[2][3][4] The drug gained approval by U.S. Food and Drug Administration on August 27, 2012 for use in adult patients starting HIV treatment for the first time as part of the fixed dose combination known as Stribild.[5]
According to the results of the phase II clinical trial, patients taking once-daily elvitegravir boosted by ritonavir had greater reductions in viral load after 24 weeks compared to individuals randomized to receive a ritonavir-boosted protease inhibitor.[6]
Human immunodeficiency virus type 1 (HIV-1) is the causative agent of acquired immunodeficiency disease syndrome (AIDS). After over 26 years of efforts, there is still not a therapeutic cure or an effective vaccine against HIV/AIDS. The clinical management of HIV-1 infected people largely relies on antiretroviral therapy (ART). Although highly active antiretroviral therapy (HAART) has provided an effective way to treat AIDS patients, the huge burden of ART in developing countries, together with the increasing incidence of drug resistant viruses among treated people, calls for continuous efforts for the development of anti-HIV-1 drugs. Currently, four classes of over 30 licensed antiretrovirals (ARVs) and combination regimens of these ARVs are in use clinically including: reverse transcriptase inhibitors (RTIs) (e.g. nucleoside reverse transcriptase inhibitors, NRTIs; and non-nucleoside reverse transcriptase inhibitors, NNRTIs), protease inhibitors (PIs), integrase inhibitors and entry inhibitors (e.g. fusion inhibitors and CCR5 antagonists).

- Gilead Press Release Phase III Clinical Trial of Elvitegravir July 22, 2008
- Gilead Press Release Gilead and Japan Tobacco Sign Licensing Agreement for Novel HIV Integrase Inhibitor March 22, 2008
- Shimura K, Kodama E, Sakagami Y, et al. (2007). “Broad Anti-Retroviral Activity and Resistance Profile of a Novel Human Immunodeficiency Virus Integrase Inhibitor, Elvitegravir (JTK-303/GS-9137)”. J Virol 82 (2): 764. doi:10.1128/JVI.01534-07. PMC 2224569. PMID 17977962.
- Stellbrink HJ (2007). “Antiviral drugs in the treatment of AIDS: what is in the pipeline ?”. Eur. J. Med. Res. 12 (9): 483–95. PMID 17933730.
- Sax, P. E.; Dejesus, E.; Mills, A.; Zolopa, A.; Cohen, C.; Wohl, D.; Gallant, J. E.; Liu, H. C.; Zhong, L.; Yale, K.; White, K.; Kearney, B. P.; Szwarcberg, J.; Quirk, E.; Cheng, A. K.; Gs-Us-236-0102 Study, T. (2012). “Co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir versus co-formulated efavirenz, emtricitabine, and tenofovir for initial treatment of HIV-1 infection: A randomised, double-blind, phase 3 trial, analysis of results after 48 weeks”.The Lancet 379 (9835): 2439–2448. doi:10.1016/S0140-6736(12)60917-9. PMID 22748591. edit
- Thaczuk, Derek and Carter, Michael. ICAAC: Best response to elvitegravir seen when used with T-20 and other active agents Aidsmap.com. 19 Sept. 2007.

The life cycle of HIV-1. 1. HIV-1 gp120 binds to CD4 and co-receptor CCR5/CXCR4 on target cell; 2. HIV-1 gp41 mediates fusion with target cell; 3. Nucleocapsid containing viral genome and enzymes enters cells; 4. Viral genome and enzymes are released; 5. Viral reverse transcriptase catalyzes reverse transcription of ssRNA, forming RNA-DNA hybrids; 6. RNA template is degraded by ribonuclease H followed by the synthesis of HIV dsDNA; 7. Viral dsDNA is transported into the nucleus and integrated into the host chromosomal DNA by the viral integrase enzyme; 8. Transcription of proviral DNA into genomic ssRNA and mRNAs formation after processing; 9. Viral RNA is exported to cytoplasm; 10. Synthesis of viral precursor proteins under the catalysis of host-cell ribosomes; 11. Viral protease cleaves the precursors into viral proteins; 12. HIV ssRNA and proteins assemble under host cell membrane, into which gp120 and gp41 are inserted; 13. Membrane of host-cell buds out, forming the viral envelope; 14. Matured viral particle is released
Elvitegravir, also known as GS 9137 or JTK 303, is an investigational new drug and a novel oral integrase inhibitor that is being evaluated for the treatment of HIV-1 infection. After HIVs genetic material is deposited inside a cell, its RNA must be converted (reverse transcribed) into DNA. A viral enzyme called integrase then helps to hide HIVs DNA inside the cell’s DNA. Once this happens, the cell can begin producing genetic material for new viruses. Integrase inhibitors, such as elvitegravir, are designed to block the activity of the integrase enzyme and to prevent HIV DNA from entering healthy cell DNA. Elvitegravir has the chemical name: 6-(3-chloro-2-fluorobenzyl)-1-[(S)-1 -hydroxy -methyl-2- methylpropyl]-7-methoxy-4-oxo-1, 4-dihydroquinoline-3-carboxylic acid and has the following structural formula:

WO 2000040561 , WO 2000040563 and WO 2001098275 disclose 4-oxo-1 , 4-dihydro-3- quinoline which is useful as antiviral agents. WO2004046115 provides certain 4- oxoquinoline compounds that are useful as HIV Integrase inhibitors.
US 7176220 patent discloses elvitegravir, solvate, stereoisomer, tautomer, pharmaceutically acceptable salt thereof or pharmaceutical composition containing them and their method of treatment. The chemistry involved in the above said patent is depicted below in the Scheme A. Scheme-A
Toluene, DIPEA
SOCl2 ,COCl (S)-(+)-Valinol
Toluene
,4-Difluoro-5-iodo- benzoic acid


THF
dichlorobis(triphenylphosphine)
palladium argon stream,

Elvitegravir Form ] Elvitegravir (residue) US 7635704 patent discloses certain specific crystalline forms of elvitegravir. The specific crystalline forms are reported to have superior physical and chemical stability compared to other physical forms of the compound. Further, process for the preparation of elvitegravir also disclosed and is depicted below in the Scheme B. The given processes involve the isolation of the intermediates at almost all the stages.
Scheme B
2,
–

Zn THF,
CK Br THF CU “ZnBr dιchlorobis(trιphenylphos
phine)palladium

Elvitegravir WO 2007102499 discloses a compound which is useful as an intermediate for the synthesis of an anti-HIV agent having an integrase-inhibiting activity; a process for production of the compound; and a process for production of an anti-HIV agent using the intermediate.
WO 2009036161 also discloses synthetic processes and synthetic intermediates that can be used to prepare 4-oxoquinolone compounds having useful integrase inhibiting properties.
The said processes are tedious in making and the purity of the final compound is affected because of the number of steps, their isolation, purification etc., thus, there is a need for new synthetic methods for producing elvitegravir which process is cost effective, easy to practice, increase the yield and purity of the final compound, or that eliminate the use of toxic or costly reagents.
US Patent No 7176220 discloses Elvitegravir, solvate, stereoisomer, tautomer, pharmaceutically acceptable salt thereof or pharmaceutical composition containing them and ■ their method of treatment. US Patent No 7635704 discloses Elvitegravir Form II, Form III and processes for their preparation. The process for the preparation of Form Il disclosed in the said patent is mainly by three methods – a) dissolution of Elvitegravir followed by seeding with Form II, b) recrystallisation of Elvitegravir, and c) anti-solvent method.
The process for the preparation of Form III in the said patent is mainly by three methods – a) dissolution of Form Il in isobutyl acetate by heating followed by cooling the reaction mass, b) dissolution of Form Il in isobutyl acetate by heating followed by seeding with Form III, and c) dissolving Form Il in 2-propanol followed by seeding with Form III.
Amorphous materials are becoming more prevalent in the pharmaceutical industry. In order to overcome the solubility and potential bioavailability issues, amorphous solid forms are becoming front-runners. Of special importance is the distinction between amorphous and crystalline forms, as they have differing implications on drug substance stability, as well as drug product stability and efficacy.
An estimated 50% of all drug molecules used in medicinal therapy are administered as salts. A drug substance often has certain suboptimal physicochemical or biopharmaceutical properties that can be overcome by pairing a basic or acidic drug molecule with a counter- ion to create a salt version of the drug. The process is a simple way to modify the properties of a drug with ionizable functional groups to overcome undesirable features of the parent drug. Salt forms of drugs have a large effect on the drugs’ quality, safety, and performance. The properties of salt-forming species significantly affect the pharmaceutical properties of a drug and can greatly benefit chemists and formulators in various facets of drug discovery and development.



chemical synthesis from a carboxylic acid 1 starts after conversion to the acid chloride iodide NIS 2 , and with three condensation 4 . 4 and the amino alcohol 5 addition-elimination reaction occurs 6 , 6 off under alkaline conditions with TBS protected hydroxy get the ring 7 , 7 and zinc reagent 8 Negishi coupling occurs to get 9 , the last 9 hydrolysis and methoxylated
Elvitegravir dimer impurity, WO2011004389A2
Isolation of 1-[(2S)-1-({3-carboxy-6-(3-chloro-2-fluorobenzyl)-1 -[(2S)-I- hydroxy-3-methylbutan-2-yl]-4-oxo-1 , 4-dihydroquinolin-7-yl}oxy)-3- methylbutan-2-yl 6-(3-chloro-2-fluorobenzyl)-7-methoxy-4-oxo-1 , 4-dihydroquinoline-3-carboxylic acid (elvitegravir dimer impurity, 13)
After isolation of the elvitegravir from the mixture of ethyl acetate-hexane, solvent from the filtrate was removed under reduced pressure. The resultant residue purified by column chromatography using a mixture of ethyl acetate-hexane (gradient, 20-80% EtOAc in hexane) as an eluent. Upon concentration of the required fractions, a thick solid was obtained which was further purified on slurry washing with ethyl acetate to get pure elvitegravir dimer impurity (13). The 1H-NMR, 13C-NMR and mass spectral data complies with proposed structure.

1H-NMR (DMSO-Cf6, 300 MHz, ppm) – δ 0.79 (m, d=6.3 Hz, 6H, 20 & 2O’)\ 1.18 & 1.20 (d, J=6.3 Hz & J=6.2 Hz, 6H, 21 & 21′)1, 2.42-2.49 (m, 2H, 19 & 19′), 3.81-3.89 (m, 3H, T & 17’Ha), 3.94-4.01 (m, 1 H, 17’Hb), 4.01 (s, 3H, 23), 4.11 (s, 2H, 7), 4.83-4.85 (m, 3H, 17 & 18′), 5.22 (t, J=4.7 Hz, 1H, OH), 5.41-5.44 (m, 1 H, 18), 6.73-6.78 (t, J=7.1 Hz, 1 H, 11)1‘ 2, 6.92-6.98 (t, J=8.0 Hz, 1H, 3′) 1‘2, 7.12-7.22 (m, 2H, 1 & 3), 7.34-7.39 (m, 1H, 2′),
7.45-7.48 (m, 1 H, 2), 7.49, 7.56 (s, 2H, 15 & 15′), 7.99, 8.02 (s, 2H, 9 & 9′), 8.89, 9.01 (s, 2H, 13 & 13′), 15.30, 15.33 (s, 2H, COOH’ & COOH”).
13C-NMR (DMSO-Cf6, 75 MHz, ppm)- δ 18.87, 19.03 (2OC, 20’C), 19.11 , 19.24 (21 C, 21 ‘C), 27.94 (7’C), 28.40 (7C), 28.91 , 30.08 (19C, 19’C), 56.80(23C), 60.11 (171C), 63.59 (18C), 66.52 (18’C), 68.53 (17C), 97.86, 98.97 (15, 15′), 107.43, 108.16 (12C, 12’C),
118.77, 119.38 (1OC, 10’C), 119.57 (d, J=17.6 Hz, 41C), 119.61 (d, J=17.9 Hz, 4C),
124.88 (d, J=4.3 Hz, 31C), 125.18 (d, J=4.2 Hz, 3C), 126.59, 126.96 (9C1 9’C), 127.14 (8’C), 127.62 (d, J=15.9 Hz, 61C), 127.73 (8C), 127.99 (d, J=15.2 Hz, 6C), 128.66 (2’C),
128.84 (11C), 128.84 (2C), 130.03 (d, J=3.4 Hz, 1C), 142.14, 142.44 (14C, 14’C), 144.37, 145.56 (13C, 131C), 155.24 (d, J=245.1 Hz, 5’C)1 155.61 (d, J=245.1 Hz, 5C),
160.17 (16’C), 162.04 (16C), 166.00, 166.14 (22C, 22’C), 176.17, 176.22 (11C, 111C).
DIP MS: m/z (%)- 863 [M+H]+, 885 [M+Na]+.
Merck’s New Drug Application for an Investigational Intravenous (IV) Formulation of NOXAFIL® (posaconazole) Receives FDA Priority Review

Posaconazole, SCH 56592, Noxafil (Schering-Plough)
Posaconazole is a triazole antifungal drug that is used to treat invasive infections by Candida species and Aspergillus species in severely immunocompromised patients.
For prophylaxis of invasive Aspergillus and Candida infections in patients, 13 years of age and older, who are at high risk of developing these infections due to being severely immunocompromised as a result of procedures such as hematopoietic stem cell transplant (HSCT) recipients with graft-versus-host disease (GVHD), or due to hematologic malignancies with prolonged neutropenia from chemotherapy. Also for the treatment of oropharyngeal candidiasis, including oropharyngeal candidiasis refractory to itraconazole and/or fluconazole. Posaconazole is used as an alternative treatment for invasive aspergillosis, Fusarium infections, and zygomycosis in patients who are intolerant of, or whose disease is refractory to, other antifungals
Posaconazole is designated chemically as 4-[4-[4-[4-[[ (3R,5R)-5- (2,4-difluorophenyl)tetrahydro-5-(1H-1,2,4-triazol-1 -ylmethyl)-3-furanyl]methoxy]phenyl]-1 -piperazinyl]phenyl]-2-[ (1S,2S)-1 -ethyl-2- hydroxypropyl]-2,4-dihydro-3H-1,2,4-triazol-3-one with an empirical formula of C37H42F2N8O4 and a molecular weight of 700.8.
Posaconazole is used, for example, to prevent and/or treat invasive fungal infections caused by Candida species, Mucor species, Aspergillus species,Fusarium species, or Coccidioides species in immunocompromised patients and/or in patients where the disease is refractory to other antifungal agents such as amphothericin B, fluconazole, or itraconazole, and/or in patients who do not tolerate these antifungal agents.
CAS No. 171228-49-2
Posaconazole compounds have been described inU.S. Pat. Appl. No. 2003/0055067 for “Antifungal Composition with Enhanced Bioavailability,” U.S. Pat. Appl. No. 2004/0058974 for “Treating Fungal Infections,” and European Patent Publication1372394 (A1 ) for “Liquid Suspensions of Posaconazole (SCH 56592) with Enhanced Bioavailability for Treating Fungal Infections.”
| Synonyms: | Pcz;Pos;Noxafil;Sch 56592;Aids058495;Aids-058495;Posconazole;Posaconazole;Posaconazole for research;HYDROXYPROPYL]-2,4-DIHYDRO-3H-1,2,4-TRIAZOL-3-ONE |
| Molecular Formula: | C37H42F2N8O4 |
| Formula Weight: | 700.78 |
Merck’s New Drug Application for an Investigational Intravenous (IV) Formulation of NOXAFIL® (posaconazole) Receives FDA Priority Review
Marketing Authorization Application also Filed with the European Medicines Agency
WHITEHOUSE STATION, N.J., Nov. 18, 2013–(BUSINESS WIRE)–Merck (NYSE:MRK), known as MSD outside the United States and Canada, today announced that its New Drug Application for an investigational intravenous (IV) solution formulation of the company’s antifungal agent, NOXAFIL® (posaconazole), has been accepted for priority review by the U.S. Food and Drug Administration (FDA).http://www.pharmalive.com/mercks-noxafil-nda-gets-fda-priority-review
Posaconazole (CAS Registry Number 171228-49-2; CAS Name: 2,5-anhydro-1 ,3,4-trideoxy-2- C-(2,4-difluorophenyl)-4-[[4-[4-[4-[1-[(1S,2S)-1-ethyl-2-hydroxypropyl]-1 ,5-dihydro-5-oxo-4H- 1 ,2,4-triazol-4-yl]phenyl]-1-piperazinyl]phenoxy]methyl]-1-(1 H-1 ,2,4-triazol-1-yl)-D-threo-pentitol) which is represented by the following general formula (I)

(I)
is known as an antifungal agent. It is available as an oral suspension (40 mg/ml) under the trademark NOXAFIL® from Schering Corporation, Kenilworth, NJ. WO95/17407 and WO 96/38443 disclose the compound having the general formula (I) and its use in treating fungal infections. Various pharmaceutical compositions comprising posaconazole and being adapted for oral, topical or parenteral use are described e.g. in WO 02/80678, U.S. Patent No. 5,972,381 , U.S. Patent No. 5,834,472, U.S. Patent No. 4,957,730 and WO 2005/117831. As was mentioned above, WO 95/17407 and WO 96/38443 disclose the compound having the general formula (I). However, during prosecution of the subsequently filed European patent application no. 98951994.7, now European patent EP 1 021 439 B1 , the applicant declared that the methods disclosed in these publications only lead to the compound of formula (I) as an amorphous solid.
Polymorphism is a phenomenon relating to the occurrence of different crystal forms for one molecule. There may be several different crystalline forms for the same molecule with distinct crystal structures and distinct and varying physical properties like melting point, XRPD pattern, IR-spectrum and solubility profile. These polymorphs are thus distinct solid forms which share the molecular formula of the compound from which the crystals are made up, however, they may have distinct advantageous physical properties which can have a direct effect on the ability to process and/or manufacture the drug product, like flowability, as well as physical properties such as solubility, stability and dissolution properties which can have a direct effect on drug product stability, solubility, dissolution, and bioavailability.
Three polymorphic forms of posaconazole designated as forms I, Il and III are described and characterized in WO 99/18097 (US-B-6,713,481 , US-B-6,958,337). Crystalline forms Il and III were found to be unstable under the conditions investigated, so that crystalline form I was considered to be useful in the development of a pharmaceutical product.
A. K. Saksena et al., WO 9517407; eidem, US 5661151 (1995, 1997 both to Schering);
eidem, Tetrahedron Lett. 37, 5657 (1996).
SCH-56592, a novel orally active broad spectrum antifungal agent35th Intersci Conf Antimicrob Agents Chemother (Sept 17-20, San Francisco) 1995,Abst F61
seeSaksena, A.K.; Girijavallabhan, V.M.; Lovey, R.G.; Pike, R.E.; Wang, H.; Liu, Y.-T.; Ganguly, A.K.; Bennett, F. (Schering Corp.) EP 0736030; JP 1997500658; US 5661151; US 5703079; WO 9517407
Process for the preparation of triazolonesWO 9633178
Mono N-arylation of piperazine(III): Metal-catalyzed N-arylation and its application to the novel preparations of the antifungal posaconazole and its advanced intermediateTetrahedron Lett 2002,43(18),3359
Comparative antifungal spectrum: A. Cacciapuoti et al., Antimicrob. Agents Chemother. 44, 2017 (2000).
Pharmacokinetics, safety and tolerability: R. Courtney et al., ibid. 47, 2788 (2003).
HPLC determn in serum: H. Kim et al., J. Chromatogr. B 738, 93 (2000).
Review of development: A. K. Saksena et al. inAnti-Infectives: Recent Advances in Chemistry and Structure Activity Relationships (Royal Soc. Chem., Cambridge, 1997) pp 180-199; and clinical efficacy in fungal infections: R. Herbrecht, Int. J. Clin. Pract. 58, 612-624 (2004).
synthesis 1

……………..

Synthesis of intermediate (XX): The reaction of 2-chloro-2′,4′-difluoroacetophenone (I) with sodium acetate and NaI in DMF gives 2-acetoxy-2′,4′-difluoroacetophenone (II), which by methylenation with methyltriphenylphosphonium bromide and sodium bis(trimethylsilyl)amide in THF yields 2-(2,4-difluorophenyl)-2-propen-1-ol acetate ester (III). The hydrolysis of (III) with KOH in dioxane/water affords the corresponding alcohol (IV), which is regioselectively epoxidized with titanium tetraisopropoxide and L-(+)-diethyl tartrate in dichloromethane to (S)-(-)-2-(2,4-difluorophenyl)oxirane-2-methanol (V). The reaction of (V) with 1,2,4-triazole (VI) in DMF affords (R)-2-(2,4-difluorophenyl)-3-(1,2,4-triazol-1-yl)propane-1,2-diol (VII), which is selectively mesylated with methanesulfonyl chloride and triethylamine to the monomesylate (VIII). The cyclization of (VIII) with NaH in DMF gives the oxirane (IX), which is condensed with diethyl malonate (X) by means of NaH in DMSO to yield a mixture of (5R-cis)- and (5R-trans)-5-(2,4-difluorophenyl)-2-oxo-5-(1,2,4-triazol-1-ylmethyl) tetrahydrofuran-3-carboxylic acid ethyl ester (XI). The reduction of (XI) with NaBH4 and LiCl in ethanol affords (R)-4-(2,4-difluorophenyl)-2-(hydroxymethyl)-5-(1,2,4-triazol-1-yl) pentane-1,4-diol (XII), which is selectively tosylated with tosyl chloride and triethylamine in THF to the bistosylate (XIII). The cyclization of (XIII) by means of NaH in refluxing toluene gives (5R-cis)-5-(2,4-difluorophenyl)-5-(1,2,4-triazol-1-ylmethyl) tetrahydrofuran-3-methanol tosylate ester (XIV). The reaction of (XIV) with 1-(4-hydroxyphenyl)-4-(4-nitrophenyl)piperazine (XV) to obtain compound (XVI), and the following reaction sequence (XVI) to (XVII) to (XVIII) to (XIX) to (5R-cis)-4-[4-[4-[4-[5-(2,4-difluorophenyl)-5-(1,2,4-triazol-1-ylmethyl)tetrahydrofuran-3-ylmethoxy]phenyl]piperazin-1-yl]phenyl-3,4-dihydro-2H-1,2,4-triazol-3-one (XX) has been performed according to J Med Chem 1984, 27: 894-900.
………………….pat approved expiry
| United States | 5661151 | 1999-07-19 | 2019-07-19 |
| Canada | 2305803 | 2009-12-22 | 2018-10-05 |
| Canada | 2179396 | 2001-04-17 | 2014-12-20 |
| United States | 5703079 | 1994-08-26 | 2014-08-26 |
MORE INFO
| US Patent No | Patent expiry | |
|---|---|---|
| 5661151 | Jul 19, 2019 | |
| 5703079 | Aug 26, 2014 | |
| 6958337 | Oct 5, 2018 | |
| 8263600 | Apr 1, 2022 |
- Cornely OA, Maertens J, Winston DJ, Perfect J, Ullmann AJ, Walsh TJ, Helfgott D, Holowiecki J, Stockelberg D, Goh YT, Petrini M, Hardalo C, Suresh R, Angulo-Gonzalez D: Posaconazole vs. fluconazole or itraconazole prophylaxis in patients with neutropenia. N Engl J Med. 2007 Jan 25;356(4):348-59. Pubmed
- Ullmann AJ, Lipton JH, Vesole DH, Chandrasekar P, Langston A, Tarantolo SR, Greinix H, Morais de Azevedo W, Reddy V, Boparai N, Pedicone L, Patino H, Durrant S: Posaconazole or fluconazole for prophylaxis in severe graft-versus-host disease. N Engl J Med. 2007 Jan 25;356(4):335-47. Pubmed
- Bhattacharya M, Rajeshwari K, Dhingra B: Posaconazole. J Postgrad Med. 2010 Apr-Jun;56(2):163-7. Pubmed
- Frampton JE, Scott LJ: Posaconazole : a review of its use in the prophylaxis of invasive fungal infections. Drugs. 2008;68(7):993-1016.Pubmed
- Schiller DS, Fung HB: Posaconazole: an extended-spectrum triazole antifungal agent. Clin Ther. 2007 Sep;29(9):1862-86. Pubmed
- Kwon DS, Mylonakis E: Posaconazole: a new broad-spectrum antifungal agent. Expert Opin Pharmacother. 2007 Jun;8(8):1167-78.Pubmed
- Groll AH, Walsh TJ: Posaconazole: clinical pharmacology and potential for management of fungal infections. Expert Rev Anti Infect Ther. 2005 Aug;3(4):467-87. Pubmed
- Rachwalski EJ, Wieczorkiewicz JT, Scheetz MH: Posaconazole: an oral triazole with an extended spectrum of activity. Ann Pharmacother. 2008 Oct;42(10):1429-38. Epub 2008 Aug 19. Pubmed
- Li Y, Theuretzbacher U, Clancy CJ, Nguyen MH, Derendorf H: Pharmacokinetic/pharmacodynamic profile of posaconazole. Clin Pharmacokinet. 2010 Jun;49(6):379-96. doi: 10.2165/11319340-000000000-00000. Pubmed
YERVOY®(ipilimumab) Receives Marketing Authorisation for First-Line Treatment of Adult Patients with Advanced Melanoma in Europe
PARIS, France, November 8, 2013 /PRNewswire/ —
Yervoy, an innovative immuno-oncology therapy that has demonstrated durable long-term survival in some patients, [1] , [2] is now approved for use in previously-untreated patients
Bristol-Myers Squibb today announced that the European Commission (EC) has approved YERVOY® (ipilimumab) for the first-line treatment of adult patients with advanced (unresectable or metastatic) melanoma.[3] When initially approved in Europe in July 2011 for the treatment of adult patients with previously-treated advanced melanoma, ipilimumab represented the first major treatment advance in this disease in more than 30 years, providing the first overall survival benefit ever seen in the treatment of metastatic melanoma in a phase III study.[ 1 ]
http://www.pharmalive.com/ec-approves-yervoy
![]()
Ipilimumab
by Todd Campbell, The Motley Fool Sep 28th 2013 1:00PM
Updated Sep 28th 2013 1:02PM
In early 2011, the Food and Drug Administration approved Bristol-Myers Squibb‘s drug Yervoy as a treatment for skin cancer melanoma. The drug marked the first approved treatment proven to extend the life of a person diagnosed with the disease. It marked a big leap forward in medicine as an early leader in immunotherapy, or the unleashing of the body’s immune system on cancer.
read all at
http://www.dailyfinance.com/2013/09/28/yervoy-battles-melanoma-but-can-it-become-a-blockb/

Ipilimumab’s molecular target is CTLA-4 (Uniprot: P16410; canSAR ; PFAM: P16410), a negative regulator of T-cell activation. Ipilimumab augments T-cell activation and proliferation by binding to CTLA-4 and preventing its interaction with its ligands (CD80 and CD86). CTLA-4 is a membrane-bound, 223 amino acid long, T-cell protein. It contains an immunoglobulin V-type domain (PFAM:PF07686). The structure of CTLA-4 is determined (see e.g. PDBe:3osk)
Ipilimumab (i pi lim′ ue mab; also known as MDX-010 and MDX-101), marketed asYervoy, is a drug used for the treatment of melanoma, a type of skin cancer. It is a U.S. Food and Drug Administration (FDA) approved human monoclonal antibody developed byBristol-Myers Squibb, and works by activating the immune system by targeting CTLA-4.
Cytotoxic T lymphocytes (CTLs) can recognize and destroy cancer cells. However, there is also an inhibitory mechanism that interrupts this destruction. Ipilimumab turns off this inhibitory mechanism and allows CTLs to continue to destroy cancer cells.
In addition to melanoma, ipilimumab is undergoing clinical trials for the treatment of non-small cell lung carcinoma (NSCLC), small cell lung cancer (SCLC) and metastatic hormone-refractory prostate cancer.
Yervoy is a monoclonal antibody drug indicated for treating metastatic melanoma. The drug was developed by Bristol-Myers Squibb.
In March 2011, The US Food and Drug Administration (FDA) approved Yervoy to treat patients with newly diagnosed or previously-treated unresectable or metastatic melanoma. Yervoy is the first drug approved vor the treatment of metastatic melanoma in the US.
Bristol-Myers Squibb submitted a marketing authorisation application to the European Medicines Agency in May 2010. The drug received approval from the European Commission in July 2011.
Approval from Australia’s Therapeutic Goods Association was received in July 2011. The drug is currently being reviewed by Health Canada.
Metastatic melanoma
Melanoma responsible for majority of skin cancer deaths in the US. In metastatic melanoma the cancer spreads to other parts of the body from its starting point. It becomes difficult to treat the disease once it spreads beyond the skin to other parts of the body. The disease is also known as stage IV melanoma.
If the melanoma spreads to the lungs then the patient faces breathing problems. The patients with metastatic melanoma may feel symptoms of fatigue, loss of weight, and appetite and bowel problems.
The incidence of the disease has increased steadily in the US after 1970s. The American Cancer Society (ACS) estimated that more than 68,000 new cases of melanoma were registered in the US in 2009. The ACS estimated that the number of deaths occurred due to melanoma in 2010 was more than 8,700.
Yervoy mechanism
Yervoy treats metastatic melanoma by activating the immune system. The drug works by binding or inhibiting cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), a molecule that plays vital role in relating natural immune responses. The presence or absence of CTLA-4 can curb or increase the immune system’s T-cell response in fighting disease.
The drug also works by blocking a complex set of interactions in the immune system. It is designed to inhibit the activity of CTLA-4, thereby sustaining an active immune response in its attack on cancer cells.
Approvals and indications
Ipilimumab was approved by the FDA in March 2011 to treat patients with late-stage melanoma that has spread or cannot be removed by surgery. On February 1, 2012, Health Canada approved ipilimumab for “treatment of unresectable or metastatic melanoma in patients who have failed or do not tolerate other systemic therapy for advanced disease.” Additionally Ipilimumab was approved in the European Union (EU), for second line treatment of metastatic melanoma, November 2012

