
Home » 0rphan drug status (Page 19)
Category Archives: 0rphan drug status
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
FDA Approves Ryanodex for the Treatment of Malignant Hyperthermia

Dantrolene sodium
1-[[[5-(4-nitrophenyl)-2-furanyl]methylene]amino]-2,4-imidazolidinedione
VIEW THIS POST AT BELOW LINK UNTIL FORMATTING IS FIXED
http://www.allfordrugs.com/2014/07/24/fda-approves-ryanodex-for
-the-treatment-of-malignant-hyperthermia/
FDA Approves Ryanodex for the Treatment of Malignant Hyperthermia
WOODCLIFF LAKE, N.J.(BUSINESS WIRE) July 23, 2014 —
Eagle Pharmaceuticals, Inc. (“Eagle” or “the Company”)
(Nasdaq:EGRX) today announced that the U. S. Food and Drug Administration (FDA)
has approved Ryanodex (dantrolene sodium) for injectable
suspension indicated for
the treatment of malignant hyperthermia (MH), along
with the appropriate supportive measures.
MH is an inherited and potentially fatal disorder triggered
by certain anesthesia agents
in genetically susceptible individuals. FDA had designated
Ryanodex as an Orphan Drug in
August 2013. Eagle has been informed by the FDA that it will learn over the next four to
six weeks if it has been granted the seven year Orphan Drug market exclusivity.
read at
http://www.drugs.com/newdrugs/fda-approves-ryanodex-malignant-
hyperthermia-4058.html?utm_source=ddc&utm_medium=email&utm_
news+summary+-+July+23%2C+2014
READ MORE AT
PATENTS, CAS NO ETC
http://www.allfordrugs.com/2014/07/24/fda-approves-ryanodex-
Beloranib, 성분명 벨로라닙 ZGN-433….Zafgen’s Prader-Willi syndrome therapy receives orphan drug designation in Europe


Beloranib
CAS 251111-30-5 (beloranib),529511-79-3 (beloranib hemioxalate)
(E)-(3R,4S,5S,6R)-5-methoxy-4-((2R,3R)-2-methyl-3-(3-methylbut-2-en-1-yl)oxiran-2-yl)-1-oxaspiro[2.5]octan-6-yl 3-(4-(2-(dimethylamino)ethoxy)phenyl)acrylate
6-O-(4-dimethylaminoethoxy)cinnamoyl fumagillol
Mechanism of Action:methionine aminopeptidase 2 (MetAP2) inhibitor
Indication:Obesity US Patent : US6063812 Patent Exp Date: May 13, 2019
Originator: Chong Kun Dang (CKD) Pharma (종근당) Chong Kun Dang Pharm Corp
Developer: Zafgen Inc. (자프젠)Zafgen Corporation
Zafgen’s Prader-Willi syndrome therapy receives orphan drug designation in Europe The European Commission (EC) has granted orphan drug designation to US-based Zafgen for its beloranib for treating Prader-Willi syndrome. Beloranib is a potent inhibitor of Methionine aminopeptidase-2 that reduces hunger while stimulating the use of stored fat as an energy source (MetAP2). MetAP2 is an enzyme that modulates the activity of key cellular processes that control metabolism. http://www.pharmaceutical-technology.com/news/newszafgens-prader-willi-syndrome-therapy-receives-orphan-drug-designation-in-europe-4316842?WT.mc_id=DN_News
INTRODUCTION Beloranib is an experimental drug candidate for the treatment of obesity. It was discovered by CKD Pharmaceuticals and is currently being developed by Zafgen. Beloranib, an analog of the natural chemical compound fumagillin, is an inhibitor of the enzyme METAP2. It was originally designed as angiogenesis inhibitor for the treatment of cancer. However, once the potential anti-obesity effects of METAP2 inhibition became apparent, the clinical development began to focus on these effects and beloranib has shown positive results in preliminary clinical trials for this indication. At such low doses, says Thomas E. Hughes, president and chief executive officer of Zafgen, toxicity concerns tend to evaporate, in part because so little opportunity exists to inhibit off-target proteins.
Zafgen, a small pharmaceutical company in Cambridge, Mass., sees high selectivity and low toxicity with its covalent molecule for treating obesity, beloranib hemioxalate, also known as ZGN-433. “You’re passing a wave of the molecule through the body,” he says. “It hits the different tissues, silences the target enzyme where it finds it, and then it goes away.” Zafgen’s drug candidate inhibits an enzyme called methionine aminopeptidase 2 (MetAP2), which had been of interest in oncology circles until it turned out to be a poor target for treating cancer in mice. However, animals treated with a MetAP2 inhibitor lost weight. Zafgen pursued the enzyme as a target for obesity. Its drug candidate contains a spiroepoxide that bonds with a histidine in the protein’s active site.
ZGN-433 has undergone a Phase I clinical trial, in which obese volunteers lost up to 2 lb per week. It will enter Phase II trials within a year, Hughes says, funded by $33 million the company raised from investors. With dosing of up to 2 mg twice per week, ZGN-433 reaches a maximum concentration in the body of just a few nanomolar for several hours before the body quickly eliminates it, Hughes says. During that time, the drug is much more likely to interact with MetAP2 than with anything else. “You’re flying under the radar of a lot of concerns,” he says. “Drug-drug interactions are not an issue. There’s just not enough inhibitor to go around.
The same is true for off-target inhibition: The chance of off-target toxicity is largely gone.” Proponents of covalent inhibitors are quick to point out that dozens of such drugs are already on the market. They include aspirin, the world’s most widely used medicine; penicillin and related antibiotics; and recently developed blockbusters such as Plavix, Prevacid, and Nexium. The drugs treat a broad range of conditions, and many have minimal side effects, even when taken for years. By one count, of the marketed drugs that inhibit enzymes, more than one-third work by covalent modification (Biochemistry, DOI: 10.1021/bi050247e).
![]()
6-O-(4-dimethylaminoethoxy) cinnamoyl fumagillol hemioxalate
| Beloranib, ZGN-433, CKD-732 | |
|---|---|
 |
|
| Identifiers | |
| CAS number | 251111-30-5 |
| PubChem | 6918502 |
| ChemSpider | 26286923 |
| UNII | FI471K8BU6 |
| Jmol-3D images | Image 1 |
| Properties | |
| Molecular formula | C29H41NO6 |
| Molar mass | 499.64 g mol−1 |
| Except where noted otherwise, data are given for materials in their standard state (at 25 °C (77 °F), 100 kPa) | |
Beloranib (previously known as CKD-732; ZGN-433), a methionine aminopeptidase 2 (MetAP2) inhibitor originally designed as an anticancer agent, is being developed by Zafgen as a first-in-class obesity therapy. Beloranib, a twice-daily injection, is discovered by korean company Chong Kun Dang (CKD) Pharmaceuticals and was licensed to Cambridge, MA-based startup Zafgen, Inc. Zafgen holds exclusive worldwide rights (exclusive of Korea) for development and commercialization of beloranib. Beloranib, an analog of the antimicrobial agent fumagillin, is an inhibitor of the enzyme METAP2 involved in fatty acid production. It was originally designed as angiogenesis inhibitor for the treatment of cancer. However, once the potential anti-obesity effects of METAP2 inhibition became apparent, the clinical development began to focus on these effects.
Zafgen has chosen to develop beloranib not for the folks that need to shed a few pounds, but for severely obese people, and smaller groups of patients with rare and dangerous conditions. In January 2013, beloranib was granted orphan drug designation by the U.S. Food and Drug Administration to treat a rare genetic condition known as Prader-Willi Syndrome (PWS) that causes obesity through compulsive eating. Zafgen plans to seek the same designation for beloranib in craniopharyngioma (a rare benign brain tumor) related obesity as well. By going after these orphan indications, Zafgen can get onto the market quicker and cheaper than if it went straight for the larger obesity market. Zafgen recently completed two Phase 2a clinical trials evaluating beloranib’s ability to reduce body weight and to improve hyperphagia, one in PWS patients and one in severely obese patients. In its Phase 2a clinical trials, Zafgen observed reductions in body weight, body mass and body fat content in both patient populations and reductions in hyperphagia-related behaviors in PWS patients.
On June 19, 2014, Zafgen Inc. raised $96 million in its initial public offering (IPO) on the Nasdaq under the symbol “ZFGN” amid strong demand from investors. With its IPO cash, Zafgen plans to initiate its Phase 3 clinical program, consisting of two Phase 3 clinical trials, of beloranib in PWS patients, with the first Phase 3 trial to start in the second half of 2014, after finalizing the program design based on ongoing conversations with the FDA and certain European regulatory authorities. Zafgen is also planning a phase 2a trial in craniopharyngioma, and a Phase 2b trila in patients with severe obesity, all this year. The composition of matter patent (US6063812) on beloranib will each expire in May 2019. Zafgen owns two issued U.S. patents relating to beloranib polymorph compositions of matter that will expire in 2031 and two issued U.S. patents to methods of treating obesity that will expire in 2029.  Beloranib is an experimental drug candidate for the treatment of obesity. It was discovered by CKD Pharmaceuticals and is currently being developed by Zafgen.[1] Beloranib, an analog of the natural chemical compound fumagillin, is an inhibitor of the enzyme METAP2.[2] It was originally designed as angiogenesis inhibitor for the treatment of cancer.[3] However, once the potential anti-obesity effects of METAP2 inhibition became apparent, the clinical development began to focus on these effects and beloranib has shown positive results in preliminary clinical trials for this indication.[4][5]
Beloranib is an experimental drug candidate for the treatment of obesity. It was discovered by CKD Pharmaceuticals and is currently being developed by Zafgen.[1] Beloranib, an analog of the natural chemical compound fumagillin, is an inhibitor of the enzyme METAP2.[2] It was originally designed as angiogenesis inhibitor for the treatment of cancer.[3] However, once the potential anti-obesity effects of METAP2 inhibition became apparent, the clinical development began to focus on these effects and beloranib has shown positive results in preliminary clinical trials for this indication.[4][5]
………………………………..
http://www.google.com/patents/WO2005082349A1?cl=en
compound O-(4- dimethylaminoethoxycinnamoyl)fumagillol can be used in the form of a salt, e.g., acetate, lactate, benzoate, salicylate, mandelate, oxalate, methanesulfonate, or p- toluenesulfonate. Korean Patent No. 0357542 and its corresponding patents (U.S. Patent No. 6,063,812, Japanese Patent No. 3370985, and European Patent No. 1077964), filed by the present applicant, disclose fumagiUol derivatives, including the compounds used in the present invention. The composition of the present invention can be prepared in combination with pharmaceutically acceptable carriers commonly used in pharmaceutical formulations.
………………………..
http://www.google.com/patents/WO2012064838A1?cl=en
MetAP2 encodes a protein that functions at least in part by enzymatically removing the amino terminal methionine residue from certain newly translated proteins, such as, glyceraldehyde-3- phosphate dehydrogenase (Warder et al. (2008) J Proteome Res 7:4807). Increased expression of the MetAP2 gene has been historically associated with various forms of cancer. Molecules inhibiting the enzymatic activity of MetAP2 have been identified and have been explored for their utility in the treatment of various tumor types (Wang et al. (2003) Cancer Res 63:7861) and infectious diseases, such as, microsporidiosis, leishmaniasis, and malaria (Zhang et al. (2002) J. Biomed Sci. 9:34). Notably, inhibition of MetAP2 activity in obese and obese-diabetic animals leads to a reduction in body weight in part by increasing the oxidation of fat and in part by reducing the consumption of food (Rupnick et al. (2002) Proc Natl Acad Sci USA 99: 10730). [0003] 6-O-(4-Dimethylaminoethoxy)cinnamoyl fumagillol is a METAP2 inhibitor and is useful in the treatment of, e.g., obesity. 6-O-(4-Dimethylaminoethoxy)cinnamoyl fumagillol is characterized by formula I:

Example 1 [0060] Crystalline, Form A material of 6-O-(4-dimethylaminoethoxy)cinnamoyl fumagillol was prepared as follows: [0061] Approximately 423 mg of amorphous gum/oil-like 6-O-(4- dimethylaminoethoxy)cinnamoyl fumagillol free base compound was dissolved in ca. 6 mL of diisopropylether (IPE). The solution was allowed to stir for ca. 24 hours at ambient temperature (18-22°C) during which time solid precipitated. The resulting solid was isolated by filtration and dried under vacuum at ambient for ca. 4 hours (yield 35.8 %).
…………………..

………………….
http://www.google.com/patents/WO1999059986A1?cl=en
Example 14 : 0-(4-dimethylaminocinnamoyl)fumagillol 1) To a solution of 4-dimethylaminocinnamic acid (950 mg) in toluene (20 ml), dipyridyl disulfide (1.64 g) and triphenyl phosphine (1.97 g) were added, and the mixture was stirred for 12 hours. 2) The resultant solution of 1) was added to fumagillol (500 mg) at room temperature. Sodium hydride (142 mg) was added thereto, and the reaction mixture was stirred for 30 minutes. After adding saturated ammonium chloride solution (20 ml), the reaction mixture was extracted with ethyl acetate (100 ml). The organic layer was washed with brine and dried over anhydrous magnesium sulfate. After filtering, the solvent was distilled off under reduced pressure, and the residue was purified by column chromatography (eluent: ethyl acetate/ n-hexane = 1/2) to obtain yellow solid (470 mg). ‘H-NMR (CDCI3) δ : 7.60 (d, IH, J=15.8Hz), 7.41 (d, 2H, J=8.9Hz), 6.67 (d, 2H, J=8.9Hz), 6.27 (d, IH, J=15.8Hz), 5.71 (m, IH), 5.22 (bit, IH), 3.70 (dd, IH, J=2.8, 11.0Hz), 3.45 (s, 3H), 3.02 (s, 6H), 3.01 (d, IH, J=4.3Hz), 2.63 (t, IH, J=6.3Hz), 2.56 (d, IH, J=4.3Hz), 2.41 – 1.81 (m, 6H), 1.75 (s, 3H), 1.67 (s, 3H), 1.22 (s, 3H), 1.15 – 1.06 (m, IH)
………..
Organic Letters, 16(3), 792-795; 2014

An efficient, two-step construction of highly complex alkaloid-like compounds from the natural product fumagillol is described. This approach, which mimics a biosynthetic cyclase/oxidase sequence, allows for rapid and efficient structure elaboration of the basic fumagillol scaffold with a variety of readily available coupling partners. Mechanistic experiments leading to the discovery of an oxygen-directed oxidative Mannich reaction are also described.
References
- “News Release: Zafgen Secures $33 Million Series C Financing”. Zafgen, Inc. July 7, 2011.
- Chun, E; Han, CK; Yoon, JH; Sim, TB; Kim, YK; Lee, KY (2005). “Novel inhibitors targeted to methionine aminopeptidase 2 (MetAP2) strongly inhibit the growth of cancers in xenografted nude model”. International Journal of Cancer. Journal International Du Cancer 114 (1): 124–30. doi:10.1002/ijc.20687. PMID 15523682.
- Kim, EJ; Shin, WH (2005). “General pharmacology of CKD-732, a new anticancer agent: effects on central nervous, cardiovascular, and respiratory system”. Biological & Pharmaceutical Bulletin 28 (2): 217–23. doi:10.1248/bpb.28.217. PMID 15684472.
- “Zafgen Announces Positive Topline Phase 1b Data for ZGN-433 in Obesity”. MedNews. Drugs.com. 5 January 2011.
- “Fat-busting pill helps obese to shed two pounds a week – without changing their diets”. UK Daily Mail. 11 January 2011.
MORE REF Grenning, Alexander J. et al.Remodeling of Fumagillol: Discovery of an Oxygen-Directed Oxidative Mannich Reaction.Organic Letters, 16(3), 792-795; 2014
Hughes, T. E.; Kim, D. D.; Marjason, J.; Proietto, J.; Whitehead, J. P.; Vath, J. E. Ascending dose-controlled trial of beloranib, a novel obesity treatment for safety, tolerability, and weight loss in obese women. Obesity (2013), 21(9), 1782-1788.
Chung Il Hong, Jung Woo Kim, Sang Joon Lee, Soon Kil Ahn, Nam Song Choi, Ryung Kee Hong, Hyoung Sik Chun, Seung Kee Moon, Cheol Kyu Han. Angiogenesis inhibitors, antiarthritic agents and anticarcinogenic agents plus synthesis. US patent Number US6063812 A, Also published as CA2331873A1, CA2331873C, CN1301260A, CN100352810C, DE69903279D1, DE69903279T2, EP1077964A1,EP1077964B1,WO1999059986A1, Filing date: May 13, 1999.Original Assignee:Chong Kun Dang Corporation Crawford, Thomas; Reece, Hayley A.Preparation of crystalline forms of 6-O-(4-dimethylaminoethoxy)cinnamoylfumagillol.PCT Int. Appl. (2012), WO2012064838 A1, 20120518
Egorov, Maxim et al. Preparation of fumagillol derivatives useful for the treatment or prevention of bone tumors. PCT Int. Appl., WO2012130906, 04 Oct 2012
Stevenson, Cheri A.; Akullian, Laura C.; Petter, Russell C.; Kane, John J.; Hammond, Charles E.; Yin, Mao; Yurkovetskiy, Aleksandr.Preparation of biocompatible biodegradable fumagillin analog conjugates for the treatment of cancer. PCT Int. Appl. (2009), WO2009073445 A2, 20090611
Lee, Hong Woo et al.Design, synthesis, and antiangiogenic effects of a series of potent novel fumagillin analogues.Chemical & Pharmaceutical Bulletin, 55(7), 1024-1029; 2007
Lee, Hong Woo et al.Selective N-demethylation of tertiary aminofumagillols with selenium dioxide via a non-classical Polonovski type reaction.Heterocycles, 68(5), 915-932; 2006
|
References OTHERS |
1: Yin SQ, Wang JJ, Zhang CM, Liu ZP. The development of MetAP-2 inhibitors in cancer treatment. Curr Med Chem. 2012;19(7):1021-35. Review. PubMed PMID: 22229417.
2: Shin SJ, Ahn JB, Park KS, Lee YJ, Hong YS, Kim TW, Kim HR, Rha SY, Roh JK, Kim DH, Kim C, Chung HC. A Phase Ib pharmacokinetic study of the anti-angiogenic agent CKD-732 used in combination with capecitabine and oxaliplatin (XELOX) in metastatic colorectal cancer patients who progressed on irinotecan-based chemotherapy. Invest New Drugs. 2012 Apr;30(2):672-80. doi: 10.1007/s10637-010-9625-x. Epub 2010 Dec 29. PubMed PMID: 21188464.
3: Shin SJ, Jeung HC, Ahn JB, Rha SY, Roh JK, Park KS, Kim DH, Kim C, Chung HC. A phase I pharmacokinetic and pharmacodynamic study of CKD-732, an antiangiogenic agent, in patients with refractory solid cancer. Invest New Drugs. 2010 Oct;28(5):650-8. doi: 10.1007/s10637-009-9287-8. Epub 2009 Jul 8. PubMed PMID: 19585083.
4: Rhee Y, Park SY, Kim YM, Lee S, Lim SK. Angiogenesis inhibitor attenuates parathyroid hormone-induced anabolic effect. Biomed Pharmacother. 2009 Jan;63(1):63-8. doi: 10.1016/j.biopha.2007.10.013. Epub 2007 Nov 20. PubMed PMID: 18457934.
5: Kim YM, An JJ, Jin YJ, Rhee Y, Cha BS, Lee HC, Lim SK. Assessment of the anti-obesity effects of the TNP-470 analog, CKD-732. J Mol Endocrinol. 2007 Apr;38(4):455-65. PubMed PMID: 17446235.
6: Kim EJ, Shin WH. General pharmacology of CKD-732, a new anticancer agent: effects on central nervous, cardiovascular, and respiratory system. Biol Pharm Bull. 2005 Feb;28(2):217-23. PubMed PMID: 15684472.
7: Chun E, Han CK, Yoon JH, Sim TB, Kim YK, Lee KY. Novel inhibitors targeted to methionine aminopeptidase 2 (MetAP2) strongly inhibit the growth of cancers in xenografted nude model. Int J Cancer. 2005 Mar 10;114(1):124-30. PubMed PMID: 15523682.
8: Lee HS, Choi WK, Son HJ, Lee SS, Kim JK, Ahn SK, Hong CI, Min HK, Kim M, Myung SW. Absorption, distribution, metabolism, and excretion of CKD-732, a novel antiangiogenic fumagillin derivative, in rats, mice, and dogs. Arch Pharm Res. 2004 Feb;27(2):265-72. PubMed PMID: 15029870.
9: Kim JH, Lee SK, Ki MH, Choi WK, Ahn SK, Shin HJ, Hong CI. Development of parenteral formulation for a novel angiogenesis inhibitor, CKD-732 through complexation with hydroxypropyl-beta-cyclodextrin. Int J Pharm. 2004 Mar 19;272(1-2):79-89. PubMed PMID: 15019071.
10: Myung SW, Kim HY, Min HK, Kim DH, Kim M, Cho HW, Lee HS, Kim JK, Hong CI. The identification of in vitro metabolites of CKD-732 by liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom. 2002;16(21):2048-53. PubMed PMID: 12391579.
| WO2007072083A1 | Dec 22, 2006 | Jun 28, 2007 | Prosidion Ltd | Treatment of type 2 diabetes with a combination of dpiv inhibitor and metformin or thiazolidinedione |
| WO2011085201A1 * | Jan 7, 2011 | Jul 14, 2011 | Zafgen Corporation | Fumagillol type compounds and methods of making and using same |
| WO2011088055A2 * | Jan 11, 2011 | Jul 21, 2011 | Zafgen Corporation | Methods and compositions for treating cardiovascular disorders |
| WO2012064838A1 | Nov 9, 2011 | May 18, 2012 | Zafgen Corporation | Crystalline solids of a metap-2 inhibitor and methods of making and using same |
| WO2013169727A1 * | May 7, 2013 | Nov 14, 2013 | Zafgen, Inc. | Polymorphic salt of the oxalate salt of 6 – o – ( 4 – dimethylaminoethoxy) cinnarnoyl fumagillol and methods of making and using same |
| WO2013169857A1 * | May 8, 2013 | Nov 14, 2013 | Zafgen, Inc. | Treating hypothalamic obesity with metap2 inhibitors |
| EP2317845A1 * | Jul 17, 2009 | May 11, 2011 | Zafgen, Inc. | Methods of treating an overweight or obese subject |
| US8349891 | Aug 7, 2012 | Jan 8, 2013 | Zafgen, Inc. | Crystalline solids of a MetAP-2 inhibitor and methods of making and using same |
| US8367721 | Aug 7, 2012 | Feb 5, 2013 | Zafgen, Inc. | Methods of treating an overweight or obese subject |
| US8642650 | Dec 4, 2009 | Feb 4, 2014 | Zafgen, Inc. | Methods of treating an overweight or obese subject |
| US8735447 | Nov 16, 2012 | May 27, 2014 | Zafgen, Inc. | Crystalline solids of a MetAP-2 inhibitor and methods of making and using same |
| US20130018095 * | Jan 7, 2011 | Jan 17, 2013 | Vath James E | Fumigillol Type Compounds and Methods of Making and Using Same |
| WO2003027104A1 * | Jun 11, 2002 | Apr 3, 2003 | Byung-Ha Chang | Fumagillol derivatives and preparing method thereof |
| EP0682020A1 * | Aug 31, 1989 | Nov 15, 1995 | Takeda Chemical Industries, Ltd. | Fumagillol derivatives useful as angiogenesis inhibitors |
| US6040337 * | May 13, 1999 | Mar 21, 2000 | Chong Kun Dang Corporation | 5-demethoxyfumagillol derivatives and processes for preparing the same |
| US6063812 * | May 13, 1999 | May 16, 2000 | Chong Kun Dang Corporation | Angiogenesis inhibitors, antiarthritic agents and anticarcinogenic agents plus synthesis |
| WO1999059986A1 * | May 11, 1999 | Nov 25, 1999 | Soon Kil Ahn | Fumagillol derivatives and processes for preparing the same |
| WO2005082349A1 | Feb 25, 2005 | Sep 9, 2005 | Chong Kun Dang Pharm Corp | Composition for the treatment of obesity comprising fumagillol derivative |
| WO2010065883A2 | Dec 4, 2009 | Jun 10, 2010 | Zafgen Corporation | Method of treating an overweight or obese subject |
| KIM ET AL. JOURNAL OF MOLECULAR ENDOCRINOLOGY vol. 38, 2007, pages 455 – 465 | ||
| 2 | RUPNICK ET AL. PROC NATL ACAD SCI USA vol. 99, 2002, page 10730 | |
| 3 | WANG ET AL. CANCER RES vol. 63, 2003, page 7861 | |
| 4 | WARDER ET AL. J PROTEOME RES vol. 7, 2008, page 4807 | |
| 5 | * | YOO MEE KIM ET AL: “Assessment of the anti-obesity effects of the TNP-470 analog, CKD-732“, JOURNAL OF MOLECULAR ENDOCRINOLOGY, SOCIETY FOR ENDOCRINOLOGY, GB, vol. 38, no. 4, 1 April 2007 (2007-04-01), pages 455-465, XP002632891, ISSN: 0952-5041, DOI: 10.1677/JME.1.02165 |
| 6 | ZHANG ET AL. J. BIOMED SCI. vol. 9, 2002, page 34 |
………

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
MOBILE-+91 9323115463
GLENMARK SCIENTIST , INDIA
web link
web link
ABOUTME, BRAND ANTHONYCRASTO, COLLECTION OF SITE LINKS, BRAVESITES, GRAVATAR, JIMDO, SKILLPAGES, MIXXT, APNACIRCLE, ZIC ZAC, ZING ME, SCOOP IT, scribd, Stumbleupon, Delicious, pinnterest, tumblr, Newsvine, SLIDESHARE, ACADEMIA.EDU, GOOGLE PLUS, FACEBOOK, TWITTER, ISSUU, DIIGO, YOLASITE, Authorstream, Symbaloo, ISSUU, BLOGLOVIN, FRIENDFEED, NEWSVINE
Congratulations! Your presentation titled “Anthony Cra sto Glenmark scientist, helping millions with websites” has just crossed MILLION views.
アンソニー 安东尼 Энтони 안토니 أنتوني
blogs are

New Drug Approvals, ALL ABOUT DRUGS, WORLD DRUG TRACKER
MEDICINAL CHEM INTERNATIONAL, DRUG SYN INTERNATIONAL
SCALEUP OF DRUGS, ALL FOR DRUGS ON WEB,
MY CHINA, VIETNAM AND JAPAN BLOGS
ICELAND, RUSSIA, ARAB
BOBRDOBR, BLAND ICELAND, 100zakladok, adfty
GROUPS
you can post articles and will be administered by me on the google group which is very popular across the world
OPD GROUPSPACES, SCOOP OCI, organic-process-development GOOGLE, TVINX, MENDELEY WDT, SCIPEOPLE OPD, EPERNICUS OPD, SYNTHETIC ORGANIC CHEMISTRYLinkedIn group, DIIGO OPD, LINKEDIN OPD, WDT LINKEDIN, WDTI ZING


Trifarotene
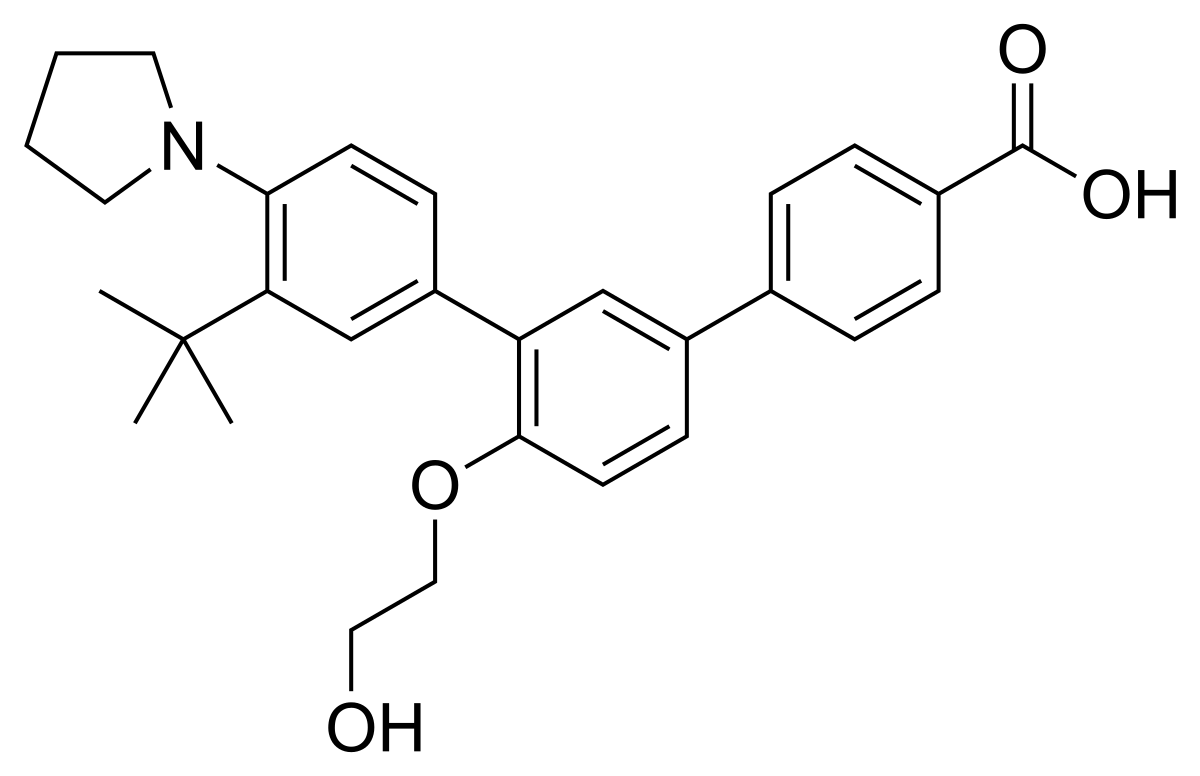
Trifarotene
CAS 895542-09-3
3”-Tert-butyl-4′-(2-hydroxyethoxy)-4”-(pyrrolidin-1-yl)(1,1′:3′,1”)terphenyl-4-carboxylic acid
3′-[3-tert-butyl-4-(pyrrolidin-1-yl)phenyl]-4′-(2-hydroxyethoxy)-[1,1′-biphenyl]-4-carboxylic acid
[1,1′:3′,1”-Terphenyl]-4-carboxylic acid, 3”-(1,1-dimethylethyl)-4′-(2-hydroxyethoxy)-4”-(1-pyrrolidinyl)-
0J8RN2W0HK
4′-(2-Hydroxyethoxy)-3”-(2-methyl-2-propanyl)-4”-(1-pyrrolidinyl)-1,1′:3′,1”-terphenyl-4-carboxylic acid
UNII-0J8RN2W0HK,
Galderma Research & Development
459.5766
C29 H33 N O4
- CD-5789
- CD5789
трифаротен [Russian] [INN]
تريفاروتين [Arabic] [INN]
曲法罗汀 [Chinese] [INN]
Trifarotene, sold under the brand name Aklief, is a medication for the topical treatment of acne vulgaris in those nine years of age and older.[1] It is a retinoid;[2] more specifically, it is a fourth generation selective retinoic acid receptor (RAR)-γ agonist.[3]
It was approved for medical use in the United States in 2019,[1][4][5] but is not approved in the European Union as of January 2021.[6] Trifarotene was granted orphan drug designation for the treatment of congenital ichthyosis by both the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA).[7][8]
State Solid
Experimental Properties
| PROPERTY | VALUE | SOURCE |
|---|---|---|
| melting point (°C) | 245C | FDA Label |
| pKa | 5.69 (pKa1) | FDA Label |
USFDA
The drug substance, trifarotene, a terphenyl acid derivative, is a retinoic
acid receptor (RAR) aQonist and is classified as a rotenoid. Trifarotene
intended as a drug for the treatment of acne vulgaris. Since trifarotene
has not been previously approved as an active ingredient in any drug
product in the United States, it is classified as a new molecular entity
(NME).
Trifarotene is produced as a white to off-white to slightly yellow crystalline
powder. It is slightly soluble in acetone, ethanol, and toluene, very slight
soluble in isopropanol, and practically insoluble in water (tiJT4
1
Cb><“JTrifarotene is nonhygroscopic and has pKa1 of 5.69 and pKa2 of 4.55. The chemical name
for trifarotene is 4-{3-[3-tert-butyl-4-(pyrrolidin-1-yl) phenyl]-4-(2-
hydroxyethoxy) phenyl} benzoic acid. It has the chemical formula of
C29H33NQ4, the molecular weiQht of 459.59, …………https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/211527Orig1s000ChemR.pdf
Prescription Products
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Aklief | Cream | 50 mcg | Topical | Galderma | 2019-11-28 | Not applicable |  |
|
| Aklief | Cream | 50 ug/1g | Topical | Galderma Laboratories, L.P. | 2019-10-04 | Not applicable |  |
Showing 1 to 2 of 2 entries
For treatment of congenital ichthyosis, PRECLINICAL, Galderma Res & Dev,
Galderma announced that the U.S. Food and Drug Administration (FDA) granted Orphan Drug Designation status for the company’s trifarotene molecule for the treatment of congenital ichthyosis. Based on this decision, Galderma plans to implement a clinical development plan, reinforcing its commitment to exploring new treatment options for rare diseases, as well as meeting the needs of all patients with skin diseases over the course of their lives.
Galderma治療先天性魚鱗癬的Trifarotene分子取得FDA的孤兒藥資格認定
http://news.msn.com.tw/market3773054.aspx

The company’s molecule trifarotene is a selective agonist of the gamma retinoic acid receptor (RAR-gamma), which is currently in clinical development for use in other more common dermatological conditions. It is the drug’s retinoid functionality and potent keratolytic properties that make it a potentially viable treatment of the lamellar ichthyosis pathology. Galderma has already initiated the program for investigating the treatment of lamellar ichthyosis with trifarotene and is currently working in collaboration with regulatory authorities to implement an innovative and expedient clinical development plan.
Ichthyoses comprise a large group of skin scaling disorders with diverse etiologies. The stereotypic pathophysiology is epidermal hyperplasia and abnormal desquamation, leading to visible accumulation of squames (scales) on the skin’s surface. Congenital ichthyosis is a term used to refer to a specific group of rare inherited forms of ichthyoses that are generally more severe than non-inherited forms of the disease. Lamellar ichthyosis is one such disorder that falls within the congenital ichthyosis category. Lamellar ichthyosis is recognized as a severe disease which persists throughout life. After birth, during the first post-natal weeks, the hyperkeratotic (colloidion) membrane patients are typically born with, is gradually shed and is replaced by scaling and lichenification that involves the entire body, including face, scalp, palms and soles. While usually not life threatening, lamellar ichthyosis can result in disability, partial deafness, poor adaptation to environmental conditions (due to hypohydrosis), severe discomfort (pruritus, fissuring of the skin), and significant psycho-social impact. The estimated prevalence of LI in the US is in the range of 1 per 100,000 to 1 per 200,000 persons.
Synthesis Reference
Thoreau, E. et. al. Structure-based design of Trifarotene (CD5789), a potent and selective RARγ agonist for the treatment of acne. Bioorganic & Medicinal Chemistry Letters, Volume 28, Issue 10. 2018. Pages 1736-1741
https://www.sciencedirect.com/science/article/abs/pii/S0960894X18303482
Trifarotene – Synthetic Route 1

Synthetic Description
Reference: Biadatti, Thibaud; Dumais, Laurence; Soulet, Catherine; Talano, Sandrine; Daver, Sebastien. Preparation of [1,1′:3′,1”]terphenyl-4-carboxylic acid and esters a novel ligands modulating retinoic acid receptors (RAR), and use thereof in human medicine and in cosmetics. Assignee Galderma Research & Development, S.N.C., Fr. WO 2006066978. (2016).
Showing 1 to 4 of 4 entries
PATENT
WO 2006066978
http://www.google.com/patents/WO2006066978A1?cl=en
Example 25 – 3″-ter.-Butyl-4′-(2-hvdroxyethoxy)-4″-pyrrolidin-1-ylM,1′:3′,1″1- terphenyl-4-carboxylic acid

In a manner similar to that of Example 6b, by reacting 500 mg (0.9 mmol) of ethyl 4′-(2- acetoxyethoxy)-3″-terf-butyl-4″-pyrrolidin-1 -yl[1 , 1 ‘;3’, 1 “]terphenyl-4-carboxylate with
300 mg (8 mmol) of sodium hydroxide, 242 mg of 3″-tert-butyl-4′-(2-hydroxyethoxy)-4″- pyrrolidin-1-yl[1l1′;3′,1″]terphenyl-4-carboxylic acid are obtained (yield = 55 %) in the form of a white solid (m.p. = 2230C).
1H NMR (DMSO. 400 MHz): 1.43 (s, 9H); 1.90 (m, 4H); 3.0 (m, 4H); 3.73 (d, J=4.7Hz, 2H); 4.1 (m, 2H); 4.7 (s, 1H); 7.2 (d, 1H, J=8.6Hz); 7.48 (m, 2H); 7.59 (d, J=1.6Hz, 1H); 7.64 (d, J=UHz, 1H); 7.68 (dd, J=2Hz, 7.8Hz, 1H); 7.82 (d, J=8.3Hz, 2H); 7.99 (d, J=8.4Hz, 2H).
PATENT
WO 2013178759
http://www.google.com/patents/WO2013178759A1?cl=en
PATENT
WO 2013178758
http://www.google.com/patents/WO2013178758A1?cl=en
PATENT
WO 2013178760
http://www.google.com/patents/WO2013178760A1?cl=en
The details of skin application are given in the table below.

SYN
New Drug Approvals for 2019: Synthesis and Clinical Applications
New Drug Approvals for 2019: Synthesis and Clinical Applications
Shuo Yuan, Bin Yu, Hong-Min Liu
PII: S0223-5234(20)30639-5
DOI: https://doi.org/10.1016/j.ejmech.2020.112667
Reference: EJMECH 112667
To appear in: European Journal of Medicinal Chemistry
Trifarotene (Aklief). In October 2019, trifarotene, a topical retinoid that
selectively targets retinoic acid receptor gamma (RAR-γ), was approved by the FDA
for the treatment of acne vulgaris [142]. The drug was developed and marketed by
Galderma Pharmaceutical in Switzerland. Trifarotene is considered as the first of the
‘fourth-generation’ retinoids due to its uniquely selective agonism at RAR-γ. The
selective agonism leads to downstream alterations, confering improved efficacy and
reduced side effects [143]. In two phase 3 clinical trials of 2420 patients with
moderate acne on the face and trunk, trifarotene was well tolerated and significantly
reduced inflammatory lesions as early as two weeks on the face and four weeks on the
back, shoulders and chest compared to vehicle (p<0.05) [144].
The synthetic approach of this drug was disclosed by Galderma Research &
Development (Scheme 25) [145]. Bromination of commercially available
2-(tert-butyl)aniline 171 gave 4-bromo-2-(tert-butyl)aniline 172 in quantitative yield,
which then reacted with 1-dibromobutane 173 to give phenylpyrrolidine 174 in 52%
yield. Miyaura reaction of 174 was realized by employing n-BuLi and triisopropyl
borate (TIPB) followed by washed with aqueous HCl, resulting in arylboronic acid
adduct 175 in 66% yield. Treatment of 175 with aromatic bromide 176 in the presence
of Pd(PPh3)4 gave the coupling product 177 in 47% yield, which then underwent
hydrolysis delivering trifarotene (XIX) in 55% yield.
The preparation of coupling partner 176 is depicted in Scheme 26. Esterification of
4-hydroxy-4-biphenylcarboxylic acid 178 gave ethyl benzoate derivative 179 upon
treatment with catalytic H2SO4 in the refluxing EtOH [145]. The resulting ester was
subjected to treatment with tetrabutylammonium bromide (TBAB) in THF, resulting
in bromide 180 in good yields, further NaH-mediated Williamson ether synthesis with
2-bromoethyl acetate 181 gave 176 in 95% yield.

[142] L.J. Scott, Trifarotene: first approval, Drugs 79 (2019) 1905-1909.
[143] E. Thoreau, J.M. Arlabosse, C. Bouix-Peter, S. Chambon, L. Chantalat, S.
Daver, L. Dumais, G. Duvert, A. Feret, G. Ouvry, J. Pascau, C. Raffin, N.
Rodeville, C. Soulet, S. Tabet, S. Talano, T. Portal, Structure-based design of
trifarotene (CD5789), a potent and selective RARγ agonist for the treatment of
acne, Bioorg. Med. Chem. Lett. 28 (2018) 1736-1741.
[144] J. Tan, D. Thiboutot, G. Popp, M. Gooderham, C. Lynde, J.D. Rosso, J. Weiss,
U. Blume-Peytavi, J. Weglovska, S. Johnson, L. Parish, D. Witkowska, N.S.
Colon, A.A. Saenz, F. Ahmad, M. Graeber, L.S. Gold, Randomized phase 3
evaluation of trifarotene 50 µg/g cream treatment of moderate facial and truncal
acne, J. Am. Acad. Dermatol. 80 (2019) 1691-1699.
[145] T. Biadatti, L. Dumais, C. Soulet, S. Talano, S. Daver, Novel ligands that
modulate rar receptors, and use thereof in human medicine and in cosmetics,
2006. WO2006066978.
| WO2006066978A1 * | Dec 21, 2005 | Jun 29, 2006 | Galderma Res & Dev | Novel ligands that modulate rar receptors, and use thereof in human medicine and in cosmetics |
| EP0826366A2 | Aug 1, 1997 | Mar 4, 1998 | Unilever N.V. | Cosmetic compositions containing hydroxy acid or retinoid |
| EP0989846A2 | Sep 22, 1998 | Apr 5, 2000 | E-L Management Corp. | Non-irritating cosmetic and pharmaceutical compositions |
| EP1831149A1 | Dec 21, 2005 | Sep 12, 2007 | Galderma Research & Development | Novel ligands that modulate rar receptors and use thereof in human medicine and in cosmetics |
| FR2915682A1 * | Title not available | |||
| US5851538 | Dec 29, 1995 | Dec 22, 1998 | Advanced Polymer Systems, Inc. | Retinoid formulations in porous microspheres for reduced irritation and enhanced stability |
| WO1999010308A1 * | Aug 21, 1998 | Mar 4, 1999 | Bernardon Jean Michel | Biphenyl derivatives substituted by an aromatic or heteroaromatic radical and pharmaceutical and cosmetic compositions containing same |
| US6150413 * | May 26, 1998 | Nov 21, 2000 | Centre International De Recherches Dermatologiques | Treatment of dermatological, rheumatic, respiratory, cardiovascular, bone and ophthalmological disorders, as well as mammalian skin and hair conditions; 4-(4-(biphenyl-2-yl)but-3-en-1-ynyl)benzoic acid, for example |
 |
|
| Clinical data | |
|---|---|
| Trade names | Aklief |
| Other names | CD5789 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a620004 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
Topical |
| Drug class | Skin and mucous membrane agents |
| ATC code | |
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.278.901 |
| Chemical and physical data | |
| Formula | C29H33NO4 |
| Molar mass | 459.586 g·mol−1 |
| 3D model (JSmol) | |
References
- ^ Jump up to:a b “Drug Trials Snapshots: Aklief”. U.S. Food and Drug Administration (FDA). 11 October 2019. Archived from the original on 19 November 2019. Retrieved 18 November 2019.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Trifarotene Monograph
- ^ Scott LJ (November 2019). “Trifarotene: First Approval”. Drugs. 79 (17): 1905–1909. doi:10.1007/s40265-019-01218-6. PMID 31713811.
- ^ “Aklief (trifarotene) FDA Approval History”. Drugs.com. 7 October 2019. Retrieved 19 November 2019.
- ^ “Drug Approval Package: Aklief”. U.S. Food and Drug Administration (FDA). 21 October 2019. Archived from the original on 19 November 2019. Retrieved 18 November 2019.
- ^ “Trifarotene”. European Medicines Agency. Retrieved 17 June 2020.
- ^ “Trifarotene Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 24 December 1999. Retrieved 19 August 2020.
- ^ “EU/3/20/2264”. European Medicines Agency (EMA). 12 August 2020. Retrieved 19 August 2020.
External links
- “Trifarotene”. Drug Information Portal. U.S. National Library of Medicine (NLM).
- Aubert J, Piwnica D, Bertino B, Blanchet-Rethore S, Carlavan I, Deret S, Dreno B, Gamboa B, Jomard A, Luzy AP, Mauvais P, Mounier C, Pascau J, Pelisson I, Portal T, Rivier M, Rossio P, Thoreau E, Vial E, Voegel JJ: Nonclinical and human pharmacology of the potent and selective topical retinoic acid receptor-gamma agonist trifarotene. Br J Dermatol. 2018 Aug;179(2):442-456. doi: 10.1111/bjd.16719. Epub 2018 Jul 4. [PubMed:29974453]
- Balak DMW: Topical trifarotene: a new retinoid. Br J Dermatol. 2018 Aug;179(2):231-232. doi: 10.1111/bjd.16733. [PubMed:30141539]
- Blume-Peytavi U, Fowler J, Kemeny L, Draelos Z, Cook-Bolden F, Dirschka T, Eichenfield L, Graeber M, Ahmad F, Alio Saenz A, Rich P, Tanghetti E: Long-term safety and efficacy of trifarotene 50 mug/g cream, a first-in-class RAR-gamma selective topical retinoid, in patients with moderate facial and truncal acne. J Eur Acad Dermatol Venereol. 2019 Jul 15. doi: 10.1111/jdv.15794. [PubMed:31306527]
- Tan J, Thiboutot D, Popp G, Gooderham M, Lynde C, Del Rosso J, Weiss J, Blume-Peytavi U, Weglovska J, Johnson S, Parish L, Witkowska D, Sanchez Colon N, Alio Saenz A, Ahmad F, Graeber M, Stein Gold L: Randomized phase 3 evaluation of trifarotene 50 mug/g cream treatment of moderate facial and truncal acne. J Am Acad Dermatol. 2019 Jun;80(6):1691-1699. doi: 10.1016/j.jaad.2019.02.044. Epub 2019 Feb 22. [PubMed:30802558]
- Chien A: Retinoids in Acne Management: Review of Current Understanding, Future Considerations, and Focus on Topical Treatments J Drugs Dermatol. 2018 Dec 1;17(12):s51-55. [PubMed:30586483]
- FDA Approved Drugs: Aklief® [Link]
трифаротен ,
تريفاروتين ,
曲法罗汀 ,
DOSAGE
| FORM | ROUTE | STRENGTH |
|---|---|---|
| Cream | Topical | 50 mcg |
| Cream | Topical | 50 ug/1g |
| Cream | Topical | 50 MICROGRAMMI/G |
Showing 1 to 3 of 3 entries
CLINICAL
| PHASE | STATUS | PURPOSE | CONDITIONS | COUNT |
|---|---|---|---|---|
| 4 | Enrolling by Invitation | Treatment | Acne Vulgaris | 1 |
| 3 | Completed | Treatment | Acne Vulgaris | 4 |
| 2 | Completed | Treatment | Acne Vulgaris | 1 |
| 2 | Recruiting | Treatment | Lamellar Ichthyosis | 1 |
| 1 | Completed | Treatment | Malignant Lymphomas | 1 |
Showing 1 to 5 of 5 entries
FDA Approves Beleodaq (belinostat) for Peripheral T-Cell Lymphoma

Belinostat (PXD101)
FAST TRACK FDA , ORPHAN STATUS
July 3, 2014 — The U.S. Food and Drug Administration today approved Beleodaq (belinostat) for the treatment of patients with peripheral T-cell lymphoma (PTCL), a rare and fast-growing type of non-Hodgkin lymphoma (NHL). The action was taken under the agency’s accelerated approval program.
- PDX101
- PX 105684
- PXD-101
- PXD101
- UNII-F4H96P17NZ
Belinostat (PXD101) is a novel HDAC inhibitor with IC50 of 27 nM, with activity demonstrated in cisplatin-resistant tumors.
CLINICAL TRIALS…http://clinicaltrials.gov/search/intervention=Belinostat+OR+PXD101

| Identifiers | |
|---|---|
| CAS | 414864-00-9 |
| PubChem | 6918638 |
| ChemSpider | 5293831 |
| UNII | F4H96P17NZ |
| ChEBI | CHEBI:61076 |
| ChEMBL | CHEMBL408513 |
| Jmol-3D images | Image 1 |
| Properties | |
| Molecular formula | C15H14N2O4S |
| Molar mass | 318.35 g mol−1 |
Belinostat inhibits the growth of tumor cells (A2780, HCT116, HT29, WIL, CALU-3, MCF7, PC3 and HS852) with IC50 from 0.2-0.66 μM. PD101 shows low activity in A2780/cp70 and 2780AD cells. Belinostat inhibits bladder cancer cell growth, especially in 5637 cells, which shows accumulation of G0-G1 phase, decrease in S phase, and increase in G2-M phase. Belinostat also shows enhanced tubulin acetylation in ovarian cancer cell lines. A recent study shows that Belinostat activates protein kinase A in a TGF-β signaling-dependent mechanism and decreases survivin mRNA.
PTCL comprises a diverse group of rare diseases in which lymph nodes become cancerous. In 2014, the National Cancer Institute estimates that 70,800 Americans will be diagnosed with NHL and 18,990 will die. PTCL represents about 10 to 15 percent of NHLs in North America.
Beleodaq works by stopping enzymes that contribute to T-cells, a type of immune cell, becoming cancerous. It is intended for patients whose disease returned after treatment (relapsed) or did not respond to previous treatment (refractory).
“This is the third drug that has been approved since 2009 for the treatment of peripheral T-cell lymphoma,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Today’s approval expands the number of treatment options available to patients with serious and life-threatening diseases.”
The FDA granted accelerated approval to Folotyn (pralatrexate) in 2009 for use in patients with relapsed or refractory PTCL and Istodax (romidepsin) in 2011 for the treatment of PTCL in patients who received at least one prior therapy.
The safety and effectiveness of Beleodaq was evaluated in a clinical study involving 129 participants with relapsed or refractory PTCL. All participants were treated with Beleodaq until their disease progressed or side effects became unacceptable. Results showed 25.8 percent of participants had their cancer disappear (complete response) or shrink (partial response) after treatment.
The most common side effects seen in Beleodaq-treated participants were nausea, fatigue, fever (pyrexia), low red blood cells (anemia), and vomiting.
The FDA’s accelerated approval program allows for approval of a drug based on surrogate or intermediate endpoints reasonably likely to predict clinical benefit for patients with serious conditions with unmet medical needs. Drugs receiving accelerated approval are subject to confirmatory trials verifying clinical benefit. Beleodaq also received orphan product designation by the FDA because it is intended to treat a rare disease or condition.
Beleodaq and Folotyn are marketed by Spectrum Pharmaceuticals, Inc., based in Henderson, Nevada. Istodax is marketed by Celgene Corporation based in Summit, New Jersey.
| MW 318.07 | |
| MF | C15H14N2O4S |
414864-00-9 cas no
866323-14-0
(2E)-N-hydroxy-3-[3-(phenylsulfamoyl)phenyl]acrylamide
A novel HDAC inhibitor
…………………………
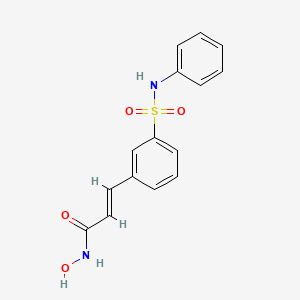
Belinostat (PXD101) is experimental drug candidate under development byTopoTarget for the treatment of hematological malignancies and solid tumors. It is a histone deacetylase inhibitor.[1]
A hydroxamate-type inhibitor of histone deacetylase.
NCI: A novel hydroxamic acid-type histone deacetylase (HDAC) inhibitor with antineoplastic activity. Belinostat targets HDAC enzymes, thereby inhibiting tumor cell proliferation, inducing apoptosis, promoting cellular differentiation, and inhibiting angiogenesis. This agent may sensitize drug-resistant tumor cells to other antineoplastic agents, possibly through a mechanism involving the down-regulation of thymidylate synthase
In 2007 preliminary results were released from the Phase II clinical trial of intravenous belinostat in combination with carboplatin and paclitaxel for relapsedovarian cancer.[2] Final results in late 2009 of a phase II trial for T cell lymphomawere encouraging.[3] Belinostat has been granted orphan drug and fast trackdesignation by the FDA.[4]

The study of inhibitors of histone deacetylases indicates that these enzymes play an important role in cell proliferation and differentiation. The inhibitor Trichostatin A (TSA) (Yoshida et al., 1990a) causes cell cycle arrest at both G1 and G2 phases (Yoshida and Beppu, 1988), reverts the transformed phenotype of different cell lines, and induces differentiation of Friend leukaemia cells and others (Yoshida et al., 1990b). TSA (and SAHA) have been reported to inhibit cell growth, induce terminal differentiation, and prevent the formation of tumours in mice (Finnin et al., 1999).
Trichostatin A (TSA)

Suberoylanilide Hydroxamic Acid (SAHA)

Cell cycle arrest by TSA correlates with an increased expression of gelsolin (Hoshikawa et al., 1994), an actin regulatory protein that is down regulated in malignant breast cancer (Mielnicki et al., 1999). Similar effects on cell cycle and differentiation have been observed with a number of deacetylase inhibitors (Kim et al., 1999). Trichostatin A has also been reported to be useful in the treatment of fibrosis, e.g., liver fibrosis and liver cirrhosis. See, e.g., Geerts et al., 1998.
Recently, certain compounds that induce differentiation have been reported to inhibit histone deacetylases. Several experimental antitumour compounds, such as trichostatin A (TSA), trapoxin, suberoylanilide hydroxamic acid (SAHA), and phenylbutyrate have been reported to act, at least in part, by inhibiting histone deacetylase (see, e.g., Yoshida et al., 1990; Richon et al., 1998; Kijima et al., 1993). Additionally, diallyl sulfide and related molecules (see, e.g., Lea et al., 1999), oxamflatin (see, e.g., Kim et al., 1999), MS-27-275, a synthetic benzamide derivative (see, e.g., Saito et al., 1999; Suzuki et al., 1999; note that MS-27-275 was later re-named as MS-275), butyrate derivatives (see, e.g., Lea and Tulsyan, 1995), FR901228 (see, e.g., Nokajima et al., 1998), depudecin (see, e.g., Kwon et al., 1998), and m-carboxycinnamic acid bishydroxamide (see, e.g., Richon et al., 1998) have been reported to inhibit histone deacetylases. In vitro, some of these compounds are reported to inhibit the growth of fibroblast cells by causing cell cycle arrest in the G1 and G2 phases, and can lead to the terminal differentiation and loss of transforming potential of a variety of transformed cell lines (see, e.g., Richon et al, 1996; Kim et al., 1999; Yoshida et al., 1995; Yoshida & Beppu, 1988). In vivo, phenybutyrate is reported to be effective in the treatment of acute promyelocytic leukemia in conjunction with retinoic acid (see, e.g., Warrell et al., 1998). SAHA is reported to be effective in preventing the formation of mammary tumours in rats, and lung tumours in mice (see, e.g., Desai et al., 1999).
The clear involvement of HDACs in the control of cell proliferation and differentiation suggest that aberrant HDAC activity may play a role in cancer. The most direct demonstration that deacetylases contribute to cancer development comes from the analysis of different acute promyelocytic leukaemias (APL). In most APL patients, a translocation of chromosomes 15 and 17 (t(15;17)) results in the expression of a fusion protein containing the N-terminal portion of PML gene product linked to most of RARσ (retinoic acid receptor). In some cases, a different translocation (t(11 ;17)) causes the fusion between the zinc finger protein PLZF and RARα. In the absence of ligand, the wild type RARα represses target genes by tethering HDAC repressor complexes to the promoter DNA. During normal hematopoiesis, retinoic acid (RA) binds RARα and displaces the repressor complex, allowing expression of genes implicated in myeloid differentiation. The RARα fusion proteins occurring in APL patients are no longer responsive to physiological levels of RA and they interfere with the expression of the RA- inducible genes that promote myeloid differentiation. This results in a clonal expansion of promyelocytic cells and development of leukaemia. In vitro experiments have shown that TSA is capable of restoring RA-responsiveness to the fusion RARα proteins and of allowing myeloid differentiation. These results establish a link between HDACs and oncogenesis and suggest that HDACs are potential targets for pharmaceutical intervention in APL patients. (See, for example, Kitamura et al., 2000; David et al., 1998; Lin et al., 1998).
BELINOSTAT
Furthermore, different lines of evidence suggest that HDACs may be important therapeutic targets in other types of cancer. Cell lines derived from many different cancers (prostate, coloreetal, breast, neuronal, hepatic) are induced to differentiate by HDAC inhibitors (Yoshida and Horinouchi, 1999). A number of HDAC inhibitors have been studied in animal models of cancer. They reduce tumour growth and prolong the lifespan of mice bearing different types of transplanted tumours, including melanoma, leukaemia, colon, lung and gastric carcinomas, etc. (Ueda et al., 1994; Kim et al., 1999).
Psoriasis is a common chronic disfiguring skin disease which is characterised by well-demarcated, red, hardened scaly plaques: these may be limited or widespread. The prevalence rate of psoriasis is approximately 2%, i.e., 12.5 million sufferers in the triad countries (US/Europe/Japan). While the disease is rarely fatal, it clearly has serious detrimental effects upon the quality of life of the patient: this is further compounded by the lack of effective therapies. Present treatments are either ineffective, cosmetically unacceptable, or possess undesired side effects. There is therefore a large unmet clinical need for effective and safe drugs for this condition. Psoriasis is a disease of complex etiology. Whilst there is clearly a genetic component, with a number of gene loci being involved, there are also undefined environmental triggers. Whatever the ultimate cause of psoriasis, at the cellular level, it is characterised by local T-cell mediated inflammation, by keratinocyte hyperproliferation, and by localised angiogenesis. These are all processes in which histone deacetylases have been implicated (see, e.g., Saunders et al., 1999; Bernhard et al, 1999; Takahashi et al, 1996; Kim et al , 2001 ). Therefore HDAC inhibitors may be of use in therapy for psoriasis. Candidate drugs may be screened, for example, using proliferation assays with T-cells and/or keratinocytes.
………………………………………………………………………..
PXD101/Belinostat®
(E)-N-hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide, also known as PXD101 and Belinostat®, shown below, is a well known histone deacetylate (HDAC) inhibitor. It is being developed for treatment of a range of disorders mediated by HDAC, including proliferative conditions (such as cancer and psoriasis), malaria, etc.

PXD101 was first described in WO 02/30879 A2. That document describes a multi-step method of synthesis which may conveniently be illustrated by the following scheme.


…………………………………..
GENERAL SYNTHESIS
IGNORE 10

ENTRY 45 IS BELINOSTAT
Scheme 1

By using amines instead of aniline, the corresponding products may be obtained. The use of aniline, 4-methoxyaniline, 4-methylaniline, 4-bromoaniline, 4-chloroaniline, 4-benzylamine, and 4-phenethyamine, among others, is described in the Examples below.
In another method, a suitable amino acid (e.g., ω-amino acid) having a protected carboxylic acid (e.g., as an ester) and an unprotected amino group is reacted with a sulfonyl chloride compound (e.g., RSO2CI) to give the corresponding sulfonamide having a protected carboxylic acid. The protected carboxylic acid is then deprotected using base to give the free carboxylic acid, which is then reacted with, for example, hydroxylamine 2-chlorotrityl resin followed by acid (e.g., trifluoroacetic acid), to give the desired carbamic acid.
One example of this approach is illustrated below, in Scheme 2, wherein the reaction conditions are as follows: (i) RSO2CI, pyridine, DCM, room temperature, 12 hours; (ii) 1 M LiOH or 1 M NaOH, dioxane, room temperature, 3-48 hours; (iii) hydroxylamine 2-chlorotrityl resin, HOAt, HATU, DIPEA, DCM, room temperature, 16 hours; and (iv) TFA/DCM (5:95, v/v), room temperature, 1.5 hours.
Scheme 2

Additional methods for the synthesis of compounds of the present invention are illustrated below and are exemplified in the examples below.
Scheme 3A

Scheme 3B

Scheme 4


Scheme 8

Scheme 9

……………………………………………………………………..
SYNTHESIS
Example 1
3-Formylbenzenesulfonic acid, sodium salt (1)

Oleum (5 ml) was placed in a reaction vessel and benzaldehyde (2.00 g, 18.84 mmol) was slowly added not exceeding the temperature of the reaction mixture more than 30°C. The obtained solution was stirred at 40°C for ten hours and at ambient temperature overnight. The reaction mixture was poured into ice and extracted with ethyl acetate. The aqueous phase was treated with CaC03 until the evolution of C02 ceased (pH~6-7), then the precipitated CaSO4was filtered off and washed with water. The filtrate was treated with Na2CO3 until the pH of the reaction medium increased to pH 8, obtained CaCO3 was filtered off and water solution was evaporated in vacuum. The residue was washed with methanol, the washings were evaporated and the residue was dried in desiccator over P2Oβ affording the title compound (2.00 g, 51%). 1H NMR (D20), δ: 7.56-8.40 (4H, m); 10.04 ppm (1 H, s).
Example 2 3-(3-Sulfophenyl)acrylic acid methyl ester, sodium salt (2)

Sodium salt of 3-formylbenzenesulfonic acid (1) (1.00 g, 4.80 mmol), potassium carbonate (1.32 g, 9.56 mmol), trimethyl phosphonoacetate (1.05 g, 5.77 mmol) and water (2 ml) were stirred at ambient temperature for 30 min., precipitated solid was filtered and washed with methanol. The filtrate was evaporated and the title compound (2) was obtained as a white solid (0.70 g, 55%). 1H NMR (DMSO- dβl HMDSO), δ: 3.68 (3H, s); 6.51 (1 H, d, J=16.0 Hz); 7.30-7.88 (5H, m).
Example 3 3-(3-Chlorosulfonylphenyl)acrylic acid methyl ester (3)

To the sodium salt of 3-(3-sulfophenyl)acrylic acid methyl ester (2) (0.670 g, 2.53 mmol) benzene (2 ml), thionyl chloride (1.508 g, 0.9 ml, 12.67 mmol) and 3 drops of dimethylformamide were added and the resultant suspension was stirred at reflux for one hour. The reaction mixture was evaporated, the residue was dissolved in benzene (3 ml), filtered and the filtrate was evaporated to give the title compound (0.6’40 g, 97%).
Example 4 3-(3-Phenylsulfamoylphenyl)acrylic acid methyl ester (4a)

A solution of 3-(3-chlorosulfonylphenyl)acrylic acid methyl ester (3) (0.640 g, 2.45 mmol) in dichloromethane (2 ml) was added to a mixture of aniline (0.465 g, 4.99 mmol) and pyridine (1 ml), and the resultant solution was stirred at 50°C for one hour. The reaction mixture was evaporated and the residue was partitioned between ethyl acetate and 10% HCI. The organic layer was washed successively with water, saturated NaCl, and dried (Na2S0 ). The solvent was removed and the residue was chromatographed on silica gel with chloroform-ethyl acetate (7:1 , v/v) as eluent. The obtained product was washed with diethyl ether to give the title compound (0.226 g, 29%). 1H NMR (CDCI3, HMDSO), δ: 3.72 (3H, s); 6.34 (1H, d, J=16.0 Hz); 6.68 (1 H, br s); 6.92-7.89 (10H, m).
Example 5 3-(3-Phenylsulfamoylphenyl)acrylic acid (5a)

3-(3-Phenylsulfamoylphenyl)acrylic acid methyl ester (4a) (0.220 g, 0.69 mmol) was dissolved in methanol (3 ml), 1N NaOH (2.08 ml, 2.08 mmol) was added and the resultant solution was stirred at ambient temperature overnight. The reaction mixture was partitioned between ethyl acetate and water. The aqueous layer was acidified with 10% HCI and stirred for 30 min. The precipitated solid was filtered, washed with water and dried in desiccator over P2Os to give the title compound as a white solid (0.173 g, 82%). Example 6 3-(3-Phenylsulfamoylphenyl)acryloyl chloride (6a)

To a suspension of 3-(3-phenylsulfamoylphenyl)acrylic acid (5a) (0.173 g, 0.57 mmol) in dichloromethane (2.3 ml) oxalyl chloride (0.17 ml, 1.95 mmol) and one drop of dimethylformamide were added. The reaction mixture was stirred at 40°C for one hour and concentrated under reduced pressure to give crude title compound (0.185 g).
Example 7
N-Hydroxy-3-(3-phenylsulfamoylphenyl)acrylamide (7a) (PX105684) BELINOSTAT

To a suspension of hydroxylamine hydrochloride (0.200 g, 2.87 mmol) in tetrahydrofuran (3.5 ml) a saturated NaHCOβ solution (2.5 ml) was added and the resultant mixture was stirred at ambient temperature for 10 min. To the reaction mixture a 3-(3-phenylsulfamoylphenyl)acryloyl chloride (6a) (0.185 g) solution in tetrahydrofuran (2.3 ml) was added and stirred at ambient temperature for one hour. The reaction mixture was partitioned between ethyl acetate and 2N HCI. The organic layer was washed successively with water and saturated NaCl, the solvent was removed and the residue was washed with acetonitrile and diethyl ether.
The title compound was obtained as a white solid (0.066 g, 36%), m.p. 172°C. BELINOSTAT

1H NMR (DMSO-d6, HMDSO), δ: 6.49 (1 H, d, J=16.0 Hz); 7.18-8.05 (10H, m); 9.16 (1 H, br s); 10.34 (1 H, s); 10.85 ppm (1 H, br s).
HPLC analysis on Symmetry C18column: impurities 4% (column size 3.9×150 mm; mobile phase acetonitrile – 0.1 M phosphate buffer (pH 2.5), 40:60; sample concentration 1 mg/ml; flow rate 0.8 ml/ min; detector UV 220 nm).
Anal. Calcd for C15Hι4N204S, %: C 56.59, H 4.43, N 8.80. Found, %: C 56.28, H 4.44, N 8.56.
……………………………………………………………………….
SYNTHESIS
US20100286279

…………………………………………………….
SYNTHESIS AND SPECTRAL DATA
Journal of Medicinal Chemistry, 2011 , vol. 54, 13 pg. 4694 – 4720
(E)-N-Hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide (28, belinostat, PXD101).
http://pubs.acs.org/doi/full/10.1021/jm2003552
http://pubs.acs.org/doi/suppl/10.1021/jm2003552/suppl_file/jm2003552_si_001.pdf
The methyl ester (27) (8.0 g) was prepared according to reported synthetic route,
(Watkins, C. J.; Romero-Martin, M.-R.; Moore, K. G.; Ritchie, J.; Finn, P. W.; Kalvinsh, I.;
Loza, E.; Dikvoska, K.; Gailite, V.; Vorona, M.; Piskunova, I.; Starchenkov, I.; Harris, C. J.;
Duffy, J. E. S. Carbamic acid compounds comprising a sulfonamide linkage as HDAC
inhibitors. PCT Int. Appl. WO200230879A2, April 18, 2002.)
but using procedure D (Experimental Section) or method described for 26 to convert the methyl ester to crude
hydroxamic acid which was further purified by chromatography (silica, MeOH/DCM = 1:10) to
afford 28 (PXD101) as off-white or pale yellow powder (2.5 g, 31%).
LC–MS m/z 319.0 ([M +H]+).
1H NMR (DMSO-d6) 12–9 (very broad, 2H), 7.90 (s, 1H), 7.76 (d, J = 7.7 Hz, 1H), 7.70 (d, J
= 7.8 Hz, 1H), 7.56 (t, J = 7.8 Hz, 1H), 7.44 (d, J = 15.8 Hz, 1H), 7.22 (t, J = 7.8 Hz, 2H), 7.08 (d,
J = 7.8 Hz, 2H), 7.01 (t, J = 7.3 Hz, 1H), 6.50 (d, J = 15.8 Hz, 1H);
13C NMR (DMSO-d6) 162.1,
140.6, 138.0, 136.5, 135.9, 131.8, 130.0, 129.2, 127.1, 124.8, 124.1, 121.3, 120.4.
Anal.
(C15H14N2O4S) C, H, N
………………………………………………..
SYNTHESIS
PXDIOI / Belinostat®
(E)-N-hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide, also known as PXD101 and Belinostat®, shown below, is a well known histone deacetylate (HDAC) inhibitor. It is being developed for treatment of a range of disorders mediated by HDAC, including proliferative conditions (such as cancer and psoriasis), malaria, etc.

PXD101 was first described in WO 02/30879 A2. That document describes a multi-step method of synthesis which may conveniently be illustrated by the following scheme.
Scheme 1
Not isolated

ed on (A)
on (D)

d on (H)

There is a need for alternative methods for the synthesis of PXD101 and related compounds for example, methods which are simpler and/or employ fewer steps and/or permit higher yields and/or higher purity product.
Scheme 5

DMAP, toluene



Synthesis 1 3-Bromo-N-phenyl-benzenesulfonamide (3)

To a 30 gallon (-136 L) reactor was charged aniline (2) (4.01 kg; 93.13 g/mol; 43 mol), toluene (25 L), and 4-(dimethylamino)pyridine (DMAP) (12 g), and the mixture was heated to 50-600C. 3-Bromobenzenesulfonyl chloride (1) (5 kg; 255.52 g/mol; 19.6 mol) was charged into the reactor over 30 minutes at 50-600C and progress of the reaction was monitored by HPLC. After 19 hours, toluene (5 L) was added due to losses overnight through the vent line and the reaction was deemed to be complete with no compound (1) being detected by HPLC. The reaction mixture was diluted with toluene (10 L) and then quenched with 2 M aqueous hydrochloric acid (20 L). The organic and aqueous layers were separated, the aqueous layer was discarded, and the organic layer was washed with water (20 L), and then 5% (w/w) sodium bicarbonate solution (20 L), while maintaining the batch temperature at 45-55°C. The batch was then used in the next synthesis.
Synthesis 2 (E)-3-(3-Phenylsulfamoyl-phenyl)-acrylic acid ethyl ester (5)

To the batch containing 3-bromo-N-phenyl-benzenesulfonamide (3) (the treated organic layer obtained in the previous synthesis) was added triethylamine (2.97 kg; 101.19 g/mol; 29.4 mol), tri(o-tolyl)phosphine (119 g; 304.37 g/mol; 0.4 mol), and palladium (II) acetate (44 g; 224.51 g/mol; 0.2 mol), and the resulting mixture was degassed four times with a vacuum/nitrogen purge at 45-55°C. Catalytic palladium (0) was formed in situ. The batch was then heated to 80-900C and ethyl acrylate (4) (2.16 kg; 100.12 g/mol; 21.6 mol) was slowly added over 2.75 hours. The batch was sampled after a further 2 hours and was deemed to be complete with no compound (3) being detected by HPLC. The batch was cooled to 45-55°C and for convenience was left at this temperature overnight.
The batch was then reduced in volume under vacuum to 20-25 L, at a batch temperature of 45-55°C, and ethyl acetate (20 L) was added. The batch was filtered and the residue washed with ethyl acetate (3.5 L). The residue was discarded and the filtrates were sent to a 100 gallon (-454 L) reactor, which had been pre-heated to 600C. The 30 gallon (-136 L) reactor was then cleaned to remove any residual Pd, while the batch in the 100 gallon (-454 L) reactor was washed with 2 M aqueous hydrochloric acid and water at 45-55°C. Once the washes were complete and the 30 gallon (-136 L) reactor was clean, the batch was transferred from the 100 gallon (-454 L) reactor back to the 30 gallon (-136 L) reactor and the solvent was swapped under vacuum from ethyl acetate/toluene to toluene while maintaining a batch temperature of 45-55°C (the volume was reduced to 20-25 L). At this point, the batch had precipitated and heptanes (10 L) were added to re-dissolve it. The batch was then cooled to 0-100C and held at this temperature over the weekend in order to precipitate the product. The batch was filtered and the residue was washed with heptanes (5 L). A sample of the wet-cake was taken for Pd analysis. The Pd content of the crude product (5) was determined to be 12.9 ppm.
The wet-cake was then charged back into the 30 gallon (-136 L) reactor along with ethyl acetate (50 L) and heated to 40-500C in order to obtain a solution. A sparkler filter loaded with 12 impregnated Darco G60® carbon pads was then connected to the reactor and the solution was pumped around in a loop through the sparkler filter. After 1 hour, a sample was taken and evaporated to dryness and analysed for Pd content. The amount of Pd was found to be 1.4 ppm. A second sample was taken after 2 hours and evaporated to dryness and analysed for Pd content. The amount of Pd had been reduced to 0.6 ppm. The batch was blown back into the reactor and held at 40-500C overnight before the solvent was swapped under vacuum from ethyl acetate to toluene while maintaining a batch temperature of 45-55°C (the volume was reduced to 20-25 L). At this point, the batch had precipitated and heptanes (10 L) were added to re-dissolve it and the batch was cooled to 0-100C and held at this temperature overnight in order to precipitate the product. The batch was filtered and the residue was washed with heptanes (5 L). The filtrate was discarded and the residue was dried at 45-55°C under vacuum for 25 hours. A first lot of the title compound (5) was obtained as an off-white solid (4.48 kg, 69% overall yield from 3-bromobenzenesulfonyl chloride (1)) with a Pd content of 0.4 ppm and a purity of 99.22% (AUC) by HPLC.
Synthesis 3 (E)-3-(3-Phenylsulfamoyl-phenyl)-acrvlic acid (6)

To the 30 gallon (-136 L) reactor was charged the (E)-3-(3-phenylsulfamoyl-phenyl)- acrylic acid ethyl ester (5) (4.48 kg; 331.39 g/mol; 13.5 mol) along with 2 M aqueous sodium hydroxide (17.76 L; -35 mol). The mixture was heated to 40-50°C and held at this temperature for 2 hours before sampling, at which point the reaction was deemed to be complete with no compound (5) being detected by HPLC. The batch was adjusted to pH 2.2 using 1 M aqueous hydrochloric acid while maintaining the batch temperature between 40-500C. The product had precipitated and the batch was cooled to 20-300C and held at this temperature for 1 hour before filtering and washing the cake with water (8.9 L). The filtrate was discarded. The batch was allowed to condition on the filter overnight before being charged back into the reactor and slurried in water (44.4 L) at 40-500C for 2 hours. The batch was cooled to 15-20°C, held for 1 hour, and then filtered and the residue washed with water (8.9 L). The filtrate was discarded. The crude title compound (6) was transferred to an oven for drying at 45-55°C under vacuum with a slight nitrogen bleed for 5 days (this was done for convenience) to give a white solid (3.93 kg, 97% yield). The moisture content of the crude material was measured using Karl Fischer (KF) titration and found to be <0.1% (w/w). To the 30 gallon (-136 L) reactor was charged the crude compound (6) along with acetonitrile (47.2 L). The batch was heated to reflux (about 80°C) and held at reflux for 2 hours before cooling to 0-10°C and holding at this temperature overnight in order to precipitate the product. The batch was filtered and the residue was washed with cold acetonitrile (7.9 L). The filtrate was discarded and the residue was dried under vacuum at 45-55°C for 21.5 hours. The title compound (6) was obtained as a fluffy white solid (3.37 kg, 84% yield with respect to compound (5)) with a purity of 99.89% (AUC) by HPLC.
Synthesis 4 (E)-N-Hvdroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide (PXD101) BELINOSTAT

To the 30 gallon (-136 L) reactor was charged (E)-3-(3-phenylsulfamoyl-phenyl)-acrylic acid (6) (3.37 kg; 303.34 g/mol; 11.1 mol) and a pre-mixed solution of 1 ,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in isopropyl acetate (IPAc) (27 g in 30 L; 152.24 g/mol; 0.18 mol). The slurry was stirred and thionyl chloride (SOCI2) (960 mL; density ~1.631 g/mL; 118.97 g/mol; -13 mol) was added to the reaction mixture and the batch was stirred at 20-300C overnight. After 18.5 hours, the batch was sampled and deemed to be complete with no compound (6) being detected by HPLC. The resulting solution was transferred to a 100 L Schott reactor for temporary storage while the
30 gallon (-136 L) reactor was rinsed with isopropyl acetate (IPAc) and water. Deionized water (28.9 L) was then added to the 30 gallon (-136 L) reactor followed by 50% (w/w) hydroxylamine (6.57 L; -1.078 g/mL; 33.03 g/mol; -214 mol) and another charge of deionized water (1.66 L) to rinse the lines free of hydroxylamine to make a 10% (w/w) hydroxylamine solution. Tetrahydrofuran (THF) (6.64 L) was then charged to the
30 gallon (-136 L) reactor and the mixture was stirred and cooled to 0-100C. The acid chloride solution (from the 100 L Schott reactor) was then slowly charged into the hydroxylamine solution over 1 hour maintaining a batch temperature of 0-10°C during the addition. The batch was then allowed to warm to 20-300C. The aqueous layer was separated and discarded. The organic layer was then reduced in volume under vacuum while maintaining a batch temperature of less than 300C. The intention was to distill out 10-13 L of solvent, but this level was overshot. A larger volume of isopropyl acetate (IPAc) (16.6 L) was added and about 6 L of solvent was distilled out. The batch had precipitated and heptanes (24.9 L) were added and the batch was held at 20-30°C overnight. The batch was filtered and the residue was washed with heptanes (6.64 L). The filtrate was discarded and the residue was dried at 45-55°C under vacuum with a slight nitrogen bleed over the weekend. The title compound (PXD101) was obtained as a light orange solid (3.11 kg, 89% yield with respect to compound (6)) with a purity of 99.25% (AUC) by HPLC.
The title compound (PXD101) (1.2 kg, 3.77 mol) was dissolved in 8 volumes of 1:1 (EtOH/water) at 600C. Sodium bicarbonate (15.8 g, 5 mol%) was added to the solution. Water (HPLC grade) was then added at a rate of 65 mL/min while keeping the internal temperature >57°C. After water (6.6 L) had been added, crystals started to form and the water addition was stopped. The reaction mixture was then cooled at a rate of 10°C/90 min to a temperature of 0-10cC and then stirred at ambient temperature overnight. The crystals were then filtered and collected. The filter cake was washed by slurrying in water (2 x 1.2 L) and then dried in an oven at 45°C for 60 hours with a slight nitrogen bleed. 1.048 kg (87% recovery) of a light orange solid was recovered. Microscopy and XRPD data showed a conglomerate of irregularly shaped birefringant crystalline particles. The compound was found to contain 0.02% water.
As discussed above: the yield of compound (5) with respect to compound (1) was 69%. the yield of compound (6) with respect to compound (5) was 84%. the yield of PXD101 with respect to compound (6) was 89%.
……………….
FORMULATION
Formulation Studies
These studies demonstrate a substantial enhancement of HDACi solubility (on the order of a 500-fold increase for PXD-101) using one or more of: cyclodextrin, arginine, and meglumine. The resulting compositions are stable and can be diluted to the desired target concentration without the risk of precipitation. Furthermore, the compositions have a pH that, while higher than ideal, is acceptable for use.

UV Absorbance
The ultraviolet (UV absorbance E\ value for PXD-101 was determined by plotting a calibration curve of PXD-101 concentration in 50:50 methanol/water at the λmax for the material, 269 nm. Using this method, the E1i value was determined as 715.7.
Methanol/water was selected as the subsequent diluting medium for solubility studies rather than neat methanol (or other organic solvent) to reduce the risk of precipitation of the cyclodextrin.
Solubility in Demineralised Water
The solubility of PXD-101 was determined to be 0.14 mg/mL for demineralised water. Solubility Enhancement with Cvclodextrins
Saturated samples of PXD-101 were prepared in aqueous solutions of two natural cyclodextrins (α-CD and γ-CD) and hydroxypropyl derivatives of the α, β and Y cyclodextrins (HP-α-CD, HP-β-CD and HP-γ-CD). All experiments were completed with cyclodextrin concentrations of 250 mg/mL, except for α-CD, where the solubility of the cyclodextrin was not sufficient to achieve this concentration. The data are summarised in the following table. HP-β-CD offers the best solubility enhancement for PXD-101.

Phase Solubility Determination of HP-β-CD
The phase solubility diagram for HP-β-CD was prepared for concentrations of cyclodextrin between 50 and 500 mg/mL (5-50% w/v). The calculated saturated solubilities of the complexed HDACi were plotted against the concentration of cyclodextrin. See Figure 1.
………………………..

- Plumb, Jane A.; Finn, Paul W.; Williams, Robert J.; Bandara, Morwenna J.; Romero, M. Rosario; Watkins, Claire J.; La Thangue, Nicholas B.; Brown, Robert (2003). “Pharmacodynamic Response and Inhibition of Growth of Human Tumor Xenografts by the Novel Histone Deacetylase Inhibitor PXD101”. Molecular Cancer Therapeutics 2 (8): 721–728. PMID 12939461.
- “CuraGen Corporation (CRGN) and TopoTarget A/S Announce Presentation of Belinostat Clinical Trial Results at AACR-NCI-EORTC International Conference”. October 2007.
- Final Results of a Phase II Trial of Belinostat (PXD101) in Patients with Recurrent or Refractory Peripheral or Cutaneous T-Cell Lymphoma, December 2009
- “Spectrum adds to cancer pipeline with $350M deal.”. February 2010.
- Helvetica Chimica Acta, 2005 , vol. 88, 7 PG. 1630 – 1657, MP 172
- WO2009/40517 A2, ….
- WO2006/120456 A1, …..
- Synthetic Communications, 2010 , vol. 40, 17 PG. 2520 – 2524, MP 172
- Journal of Medicinal Chemistry, 2011 , vol. 54, 13 PG. 4694 – 4720, NMR IN SUP INFO
| US2008274120 | 11-7-2008 | Histone Deacetylase (Hdac) Inhibitors (Pxd101) for the Treatment of Cancer Alone or in Combination With Chemotherapeutic Agent |
| US2008227845 | 9-19-2008 | CYCLOOXYGENASE-2 INHIBITOR/HISTONE DEACETYLASE INHIBITOR COMBINATION |
| US2008213399 | 9-5-2008 | Combination Therapies Using Hdac Inhibitors |
| US2008194690 | 8-15-2008 | Pharmaceutical Formulations Of Hdac Inhibitors |
| US7407988 | 8-6-2008 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US7402603 | 7-23-2008 | Cyclooxygenase-2 inhibitor/histone deacetylase inhibitor combination |
| US7183298 | 2-28-2007 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US2005107445 | 5-20-2005 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US6888027 | 5-4-2005 | Carbamic acid compounds comprising a sulfonamide linkage as hdac inhibitors |
| WO2002030879A2 | Sep 27, 2001 | Apr 18, 2002 | Prolifix Ltd | Carbamic acid compounds comprising asulfonamide linkage as hdac inhibitors |
| US7973181 | 7-6-2011 | HYDROXAMIC ACID DERIVATIVES AS INHIBITORS OF HDAC ENZYMATIC ACTIVITY |
| US7928081 | 4-20-2011 | Combined Use of Prame Inhibitors and Hdac Inhibitors |
| US2011077305 | 3-32-2011 | 5-LIPOXYGENASE INHIBITORS |
| US2011003777 | 1-7-2011 | Methods of Treatment Employing Prolonged Continuous Infusion of Belinostat |
| US2010286279 | 11-12-2010 | Methods of Synthesis of Certain Hydroxamic Acid Compounds |
| US2010190694 | 7-30-2010 | Methods for identifying patients who will respond well to cancer treatment |
| US2010010010 | 1-15-2010 | HDAC INHIBITORS |
| US2009312311 | 12-18-2009 | COMBINATION OF ORGANIC COMPOUNDS |
| US2009192211 | 7-31-2009 | CYCLOOXYGENASE-2 INHIBITOR/HISTONE DEACETYLASE INHIBITOR COMBINATION |
| US7557140 | 7-8-2009 | CARBAMIC ACID COMPOUNDS COMPRISING A SULFONAMIDE LINKAGE AS HDAC INHIBITORS |
| WO1998038859A1 * | Mar 4, 1998 | Sep 11, 1998 | Thomas E Barta | Sulfonyl divalent aryl or heteroaryl hydroxamic acid compounds |
| WO1999024399A1 * | Nov 12, 1998 | May 20, 1999 | Darwin Discovery Ltd | Hydroxamic and carboxylic acid derivatives having mmp and tnf inhibitory activity |
| WO2000056704A1 * | Mar 22, 2000 | Sep 28, 2000 | Duncan Batty | Hydroxamic and carboxylic acid derivatives |
| WO2000069819A1 * | May 12, 2000 | Nov 23, 2000 | Thomas E Barta | Hydroxamic acid derivatives as matrix metalloprotease inhibitors |
| WO2001038322A1 * | Nov 22, 2000 | May 31, 2001 | Methylgene Inc | Inhibitors of histone deacetylase |
| EP0570594A1 * | Dec 7, 1992 | Nov 24, 1993 | SHIONOGI & CO., LTD. | Hydroxamic acid derivative based on aromatic sulfonamide |
| EP0931788A2 * | Dec 16, 1998 | Jul 28, 1999 | Pfizer Inc. | Metalloprotease inhibitors |
| GB2312674A * | Title not available |
| WO2002030879A2 | Sep 27, 2001 | Apr 18, 2002 | Prolifix Ltd | Carbamic acid compounds comprising a sulfonamide linkage as hdac inhibitors |
| WO2005063806A1 | Dec 30, 2003 | Jul 14, 2005 | Council Scient Ind Res | Arginine hydrochloride enhances chaperone-like activity of alpha crystallin |
| US4642316 | May 20, 1985 | Feb 10, 1987 | Warner-Lambert Company | Parenteral phenytoin preparations |
| WO2008090585A2 * | Jan 25, 2008 | Jul 31, 2008 | Univ Roma | Soluble forms of inclusion complexes of histone deacetylase inhibitors and cyclodextrins, their preparation processes and uses in the pharmaceutical field |
| WO2009109861A1 * | Mar 6, 2009 | Sep 11, 2009 | Topotarget A/S | Methods of treatment employing prolonged continuous infusion of belinostat |
| WO2010048332A2 * | Oct 21, 2009 | Apr 29, 2010 | Acucela, Inc. | Compounds for treating ophthalmic diseases and disorders |
| WO2011064663A1 | Nov 24, 2010 | Jun 3, 2011 | Festuccia, Claudio | Combination treatment employing belinostat and bicalutamide |
| US20110003777 * | Mar 6, 2009 | Jan 6, 2011 | Topotarget A/S | Methods of Treatment Employing Prolonged Continuous Infusion of Belinostat |
………………………..
SPECTRUM
Tiny Biotech With Three Cancer Drugs Is More Alluring Takeover Bet Now
Forbes
The drug is one of Spectrum’s two drugs undergoing phase 3 clinical trials. Allergan paid Spectrum $41.5 million and will make additional payments of up to $304 million based on achieving certain milestones. So far, Raj Shrotriya, Spectrum’s chairman, …
……………………………..
Copenhagen, December 10, 2013
Topotarget announces the submission of a New Drug Application (NDA) for belinostat for the treatment of relapsed or refractory (R/R) peripheral T-cell lymphoma (PTCL) to the US Food and Drug Administration (FDA). The NDA has been filed for Accelerated Approval with a request for Priority Review. Response from the FDA regarding acceptance to file is expected within 60 days from the FDA receipt date.
read all this here
…………………….


SEE COMPILATION ON SIMILAR COMPOUNDS AT …………..http://drugsynthesisint.blogspot.in/p/nostat-series.html
PTC Therapeutics Initiates Confirmatory Phase 3 Clinical Trial of Translarna™ (ataluren) in Patients with Nonsense Mutation Cystic Fibrosis (nmCF)

ATALUREN
PTC 124
3-[5-(2-Fluorophenyl)-1,2,4-oxadiazol-3-yl]benzoic acid
| MF C15H9FN2O3 | ||
| Molecular Weight | 284.24 | |
| CAS Registry Number | 775304-57-9 |
PTC Therapeutics Initiates Confirmatory Phase 3 Clinical Trial of Translarna™ (ataluren) in Patients with Nonsense Mutation Cystic Fibrosis (nmCF) – MarketWatch

Ataluren, formerly known as PTC124, is a small-molecular agent designed by PTC Therapeutics and sold under the trade nameTranslarna. It makes ribosomes less sensitive to premature stop codons (referred to as “read-through”). This may be beneficial in diseases such as Duchenne muscular dystrophy where the mRNA contains a mutation causing premature stop codons or nonsense codons. There is ongoing debate over whether Ataluren is truly a functional drug (inducing codon read-through), or if it is nonfunctional, and the result was a false-positive hit from a biochemical screen based on luciferase.[1]
Ataluren has been tested on healthy humans and humans carrying genetic disorders caused by nonsense mutations,[2][3] such as some people with cystic fibrosis and Duchenne muscular dystrophy. In 2010, PTC Therapeutics released preliminary results of its phase 2b clinical trial for Duchenne muscular dystrophy, with participants not showing a significant improvement in the six minute walk distance after the 48 weeks of the trial.[4] This failure resulted in the termination of a $100 million deal with Genzyme to pursue the drug. However, other phase 2 clinical trials were successful for cystic fibrosis in Israel, France and Belgium.[5] Multicountry phase 3 clinical trials are currently in progress for cystic fibrosis in Europe and the USA.[6]
In cystic fibrosis, early studies of ataluren show that it improves nasal potential difference.[7]
Ataluren appears to be most effective for the stop codon ‘UGA’.[2]
On 23 May 2014 ataluren received a positive opinion from the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA).[8]

It is not that ataluren is a complex molecule. To judge from one of the patents, synthesis is straightforward starting from 2-cyanobenoic acid and 2-fluorobenzoyl chloride, both commercially available. The synthetic steps are methylation of 2-cyanobenoic acid (iodomethane), nitrile hydrolysis with hydroxylamine, esterification with the fluoro acid chloride using DIPEA, high-temperature dehydration to the oxadiazole and finally ester hydrolysis (NaOH).


References
- Derek (2013-09-18). “The Arguing Over PTC124 and Duchenne Muscular Dystrophy. In the Pipeline:”. Pipeline.corante.com. Retrieved 2013-11-28.
- Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, Paushkin S, Patel M, Trotta CR, Hwang S, Wilde RG, Karp G, Takasugi J, Chen G, Jones S, Ren H, Moon YC, Corson D, Turpoff AA, Campbell JA, Conn MM, Khan A, Almstead NG, Hedrick J, Mollin A, Risher N, Weetall M, Yeh S, Branstrom AA, Colacino JM, Babiak J, Ju WD, Hirawat S, Northcutt VJ, Miller LL, Spatrick P, He F, Kawana M, Feng H, Jacobson A, Peltz SW, Sweeney HL (May 2007). “PTC124 targets genetic disorders caused by nonsense mutations”. Nature 447 (7140): 87–91.doi:10.1038/nature05756. PMID 17450125.
- Hirawat S, Welch EM, Elfring GL, Northcutt VJ, Paushkin S, Hwang S, Leonard EM, Almstead NG, Ju W, Peltz SW, Miller LL (Apr 2007). “Safety, tolerability, and pharmacokinetics of PTC124, a nonaminoglycoside nonsense mutation suppressor, following single- and multiple-dose administration to healthy male and female adult volunteers”. Journal of clinical pharmacology 47 (4): 430–444. doi:10.1177/0091270006297140. PMID 17389552.
- “PTC THERAPEUTICS AND GENZYME CORPORATION ANNOUNCE PRELIMINARY RESULTS FROM THE PHASE 2B CLINICAL TRIAL OF ATALUREN FOR NONSENSE MUTATION DUCHENNE/BECKER MUSCULAR DYSTROPHY (NASDAQ:PTCT)”. Ptct.client.shareholder.com. Retrieved 2013-11-28.
- Wilschanski, M.; Miller, L. L.; Shoseyov, D.; Blau, H.; Rivlin, J.; Aviram, M.; Cohen, M.; Armoni, S.; Yaakov, Y.; Pugatsch, T.; Cohen-Cymberknoh, M.; Miller, N. L.; Reha, A.; Northcutt, V. J.; Hirawat, S.; Donnelly, K.; Elfring, G. L.; Ajayi, T.; Kerem, E. (2011). “Chronic ataluren (PTC124) treatment of nonsense mutation cystic fibrosis”. European Respiratory Journal 38 (1): 59–69. doi:10.1183/09031936.00120910. PMID 21233271. Sermet-Gaudelus, I.; Boeck, K. D.; Casimir, G. J.; Vermeulen, F.; Leal, T.; Mogenet, A.; Roussel, D.; Fritsch, J.; Hanssens, L.; Hirawat, S.; Miller, N. L.; Constantine, S.; Reha, A.; Ajayi, T.; Elfring, G. L.; Miller, L. L. (November 2010). “Ataluren (PTC124) induces cystic fibrosis transmembrane conductance regulator protein expression and activity in children with nonsense mutation cystic fibrosis”. American Journal of Respiratory and Critical Care Medicine 182 (10): 1262–1272.doi:10.1164/rccm.201001-0137OC. PMID 20622033.
- “PTC Therapeutics Completes Enrollment of Phase 3 Trial of Ataluren in Patients with Cystic Fibrosis (NASDAQ:PTCT)”. Ptct.client.shareholder.com. 2010-12-21. Retrieved 2013-11-28.
- Wilschanski, M. (2013). “Novel therapeutic approaches for cystic fibrosis”. Discovery medicine 15 (81): 127–133. PMID 23449115.
- http://www.marketwatch.com/story/ptc-therapeutics-receives-positive-opinion-from-chmp-for-translarna-ataluren-2014-05-23
External links
other sources
rINN: Ataluren
Other Names
PTC124®, 3-[5-(2-fluorophenyl)-1,2,4-oxadiazol-3-yl]benzoic acid
Pharmacological Information
Pharmacology Images

Ataluren Molecule

Web information on Ataluren
Relevant Clinical Literature
UK Guidance
Regulatory Literature
Other Literature
Orphan drug under investigation for treatment of genetic conditions where nonsense mutations result in premature termination of polypeptides. This drug, which is convenient to deliver orally, appears to allow ribosomal transcription ofRNA to continue past premature termination codon mutations with correct reading of the full normal transcript which then terminates at the proper stop codon. Problematically it has been postulated that assay artifact may have complicated evaluation of its efficacy which appears to be less than gentamicin.[1] Faults of this class in the transcription process are involved in several inherited diseases.
Some forms of cystic fibrosis and Duchenne muscular dystrophy are being targeted in the development stage of the drug.[2] Phase I and II trials are promising for cystic fibrosis.[3][4] In a mouse model of Duchenne muscular dystrophy, restoration of muscle function occurred.[5]
A potential issue is that there may be parts of the human genome whose optimal gene function through evolution has resulted from relatively recent in evolutionary terms insertion of a premature termination codon and so functional suboptimal transcripts of other proteins or functional RNAs might result.
References
- ↑ Roberts RG. A read-through drug put through its paces. PLoS biology. 2013; 11(6):e1001458.(Link to article – subscription may be required.)
- ↑ Hirawat S, Welch EM, Elfring GL, Northcutt VJ, Paushkin S, Hwang S, Leonard EM, Almstead NG, Ju W, Peltz SW, Miller LL. Safety, tolerability, and pharmacokinetics of PTC124, a nonaminoglycoside nonsense mutation suppressor, following single- and multiple-dose administration to healthy male and female adult volunteers. Journal of clinical pharmacology. 2007 Apr; 47(4):430-44.(Link to article– subscription may be required.)
- ↑ Kerem E, Hirawat S, Armoni S, Yaakov Y, Shoseyov D, Cohen M, Nissim-Rafinia M, Blau H, Rivlin J, Aviram M, Elfring GL, Northcutt VJ, Miller LL, Kerem B, Wilschanski M. Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: a prospective phase II trial. Lancet. 2008 Aug 30; 372(9640):719-27.(Link to article – subscription may be required.)
- ↑ Sermet-Gaudelus I, Boeck KD, Casimir GJ, Vermeulen F, Leal T, Mogenet A, Roussel D, Fritsch J, Hanssens L, Hirawat S, Miller NL, Constantine S, Reha A, Ajayi T, Elfring GL, Miller LL. Ataluren (PTC124) Induces Cystic Fibrosis Transmembrane Conductance Regulator Protein Expression and Activity in Children with Nonsense Mutation Cystic Fibrosis. American journal of respiratory and critical care medicine. 2010 Nov 15; 182(10):1262-72.(Link to article – subscription may be required.)
- ↑ Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, Paushkin S, Patel M, Trotta CR, Hwang S, Wilde RG, Karp G, Takasugi J, Chen G, Jones S, Ren H, Moon YC, Corson D, Turpoff AA, Campbell JA, Conn MM, Khan A, Almstead NG, Hedrick J, Mollin A, Risher N, Weetall M, Yeh S, Branstrom AA, Colacino JM, Babiak J, Ju WD, Hirawat S, Northcutt VJ, Miller LL, Spatrick P, He F, Kawana M, Feng H, Jacobson A, Peltz SW, Sweeney HL. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007 May 3; 447(7140):87-91.(Link to article – subscription may be required.)
old cut paste
A large-scale, multinational, phase 3 trial of the experimental drug ataluren has opened its first trial site, in Cincinnati, Ohio.
The trial is recruiting boys with Duchenne muscular dystrophy (DMD) or Becker muscular dystrophy (BMD) caused by anonsense mutation — also known as a premature stop codon — in the dystrophin gene. This type of mutation causes cells to stop synthesizing a protein before the process is complete, resulting in a short, nonfunctional protein. Nonsense mutations are believed to cause DMD or BMD in approximately 10 to 15 percent of boys with these disorders.
Ataluren — sometimes referred to as a stop codon read-through drug — has the potential to overcome the effects of a nonsense mutation and allow functional dystrophin — the muscle protein that’s missing in Duchenne MD and deficient in Becker MD — to be produced.
The orally delivered drug is being developed by PTC Therapeutics, a South Plainfield, N.J., biotechnology company, to whichMDA gave a $1.5 million grant in 2005.
PTC124 has been developed by PTC Therapeutics.
US Orphan Drug Market Outlook 2018 ……….download available

US Orphan Drug Market Outlook 2018
Academia.edu
US Orphan Drug Pipeline Insight by Phase & Indication 5.1 Research 5.2 Preclinical 5.3 Phase I 5.4 Phase I/II 5.5 Phase II 5.6 Phase II/III 5.7 Phase III …

http://www.academia.edu/7453102/US_Orphan_Drug_Market_Outlook_2018 …………… download at this site
![]()
Market Overview
In the largest market for orphan drugs, USA, there was a shortage of adequate therapies for treating many rare diseases. These therapies were not developed as companies did not expect these drugs to be highly profitable. Hence there was a lack of interest and thus investment on the part of pharma companies in the USA. Therefore, the FDA introduced incentives for developing such drugs. This step taken by the FDA was successful in creating a thriving market for orphan drugs. It was in the USA first that a special law exclusively for governing orphan drugs was framed in the form of the Orphan Drug Act of 1983. This led to an increase in the popularity of orphan drugs. The FDA also has been continuously increasing its efforts to support this market by providing significant financial and non-financial incentives to the pharmaceutical companies to attract them. This has been one of the major drivers of growth for the US orphan drugs market.
Figure 3-1: US Orphan Drug Market (US$ Billion), 2012-2018
2012201320142015201620172018
Source: KuicK Research
see my profile
http://ictmumbai.academia.edu/AnthonyMelvinCrastoPhD

DARA BioSciences receives FDA orphan drug designation for KRN5500 (SPK 241) …..Antitumor agent

KRN5500
Antitumor agent
151276-95-8 cas
IUPAC/Chemical name:
(2E,4E)-N-(2-(((2R,3R,4R,5R,6S)-6-((7H-purin-6-yl)amino)-2-((S)-1,2-dihydroxyethyl)-4,5-dihydroxytetrahydro-2H-pyran-3-yl)amino)-2-oxoethyl)tetradeca-2,4-dienamide
C28H43N7O7
Exact Mass: 589.32240
L-glycero-beta-L-manno-Heptopyranosylamine, 4-deoxy-4-((((1-oxo-2,4-tetradecadienyl)amino)acetyl)amino)-N-1H-purin-6-yl-, (E,E)-
L-glycero-beta-L-manno-Heptopyranosylamine, 4-deoxy-4-(((((2E,4E)-1-oxo-2,4-tetradecadienyl)amino)acetyl)amino)-N-1H-purin-6-yl-
(6-[4-Deoxy-4-[(2E,4E)-tetradecadienoylglycyl]amino-L-glycero-ß-L-manno-heptopyranosyl]amino-9H-purine)
NSC-650426, SPK-241, KRN-5500
N6-[4-Deoxy-4-[N2-[2(E),4(E)-tetradecadienoyl]glycylamino]-L-glycero-beta-L-manno-heptopyranosyl]adenine; N6-[4-Deoxy-4-[2-[tetradeca-2(E),4(E)-dienamido]acetamido]-L-glycero-beta-L-manno-heptopyranosyl]adenine
Kirin Brewery (Originator), National Cancer Institute (Codevelopment)
Antibiotics and Alkaloids, Antineoplastic Antibiotics, Colorectal Cancer Therapy, ONCOLYTIC DRUGS
-
- (1) Melting point: 182-183 °C,
- (2) Specific rotation [a]0 2S = 0 (c = 0.1, in methanol),
- (3) Elementary analysis:

- (4) FD mass spectrum (m/z): 590 (M + H) , C28 H4 3 N707
- (5) Infrared spectrum (KBr disc): 3250 cm-1, 1650 cm-1, 1620 cm-1,
- (6) Proton nuclear magnetic resonance spectrum (500 MHz, in CD30D) δH: 0.89 (3H, t, J = 7.3 Hz), 1.20-1.50 (14H, m), 2.18 (2H, dt, J = 7.3, 7.3 Hz), 3.6-3.8 (5H, m), 3.95 (1 H, d, J = 16.3 Hz), 3.98 (1H, d, J = 16.3 Hz), 4.00 (1H, dd, J = <1, 2.9 Hz), 4.15 (1H, dd, J = 10.8, 10.8 Hz), 5.66 (1 H, brs), 5.98 (1 H, d, J = 15.7 Hz), 6.12 (1 H, dt, J = 7.3, 15.7 Hz), 6.22 (1 H, dd, J = 10.0, 15.7 Hz), 7.17 (1 H, dd, J = 10.0, 15.7 Hz), 8.15 (1 H, s), 8.30 (1 H, s).
- EP 0525479; JP 1993186494; US 5461036; US 5631238

DARA BioSciences receives FDA orphan drug designation for KRN5500
DARA BioSciences has received orphan drug designation from the US Food and Drug Administration’s (FDA) Office of Orphan Products Development for KRN5500, for treating multiple myeloma
Multiple myeloma is a hematologic cancer or cancer of the blood.
KRN5500 is a non-opioid, non-narcotic compound that is currently being tested in Phase I clinical trial.
Earlier this year, KRN5500 received orphan status to be developed for the parenteral treatment of painful, chronic, chemotherapy-induced peripheral neuropathy (CCIPN) that is refractory to conventional analgesics in patients with cancer.
“We believe this myeloma-specific orphan designation enhances both the viability and the future market opportunity for this valuable pipeline product.”
DARA BioSciences MD, CEO and chief medical officer David J Drutz said: “It is noteworthy in this regard that up to 20% of myeloma patients have intrinsic peripheral neuropathy, an incidence that increases to the range of 75% in patients treated with neurotoxic drugs such as thalidomide or bortezomib.
KRN5500 is a semisynthetic derivative of the nucleoside-like antineoplastic antibiotic spicamycin, originally isolated from the bacterium Streptomyces alanosinicus. KRN 5500 inhibits protein synthesis by interfering with endoplasmic reticulum and Golgi apparatus functions. This agent also induces cell differentiation and caspase-dependent apoptosis.
KRN5500 is available as a solution for intravenous (IV) administration. KRN5500 was discovered in an effort to identify new agents that induced differentiation of myeloid leukemia cells.
Safety and efficacy data from Phase I trials have been leveraged to support DARA Therapeutics’ active IND and ongoing Phase 2a clinical trial. The objective of this Phase 2a feasibility study is to determine the potential of KRN5500 (a spicamycin analogue) to be a breakthrough medicine for the treatment of neuropathic pain in cancer patients.
Four clinical trials have been conducted in cancer patients, including one in Japan and 3 in the United States. Three of these studies are complete; the fourth was closed to patient accrual and treatment in December 2004.
A total of 91 patients with solid tumors have been treated with single IV KRN5500 doses of up to 21 mg/m2 and weekly doses of up to 42 mg/m2. While KRN5500 has not shown anti-cancer efficacy in any trial, its use in pain elimination is encouraging. (source: http://www.darabiosciences.com/krn5500.htm).

Chemical structures of KRN5500 and its known metabolites.
………………..
http://www.google.com/patents/EP0525479A1?cl=en
spk 241
- 6-[4′-N-(N’-trans,trans-2,4-tridecadienylglycyl)spicamynyl-amino]purine,
- (20) SPK241:


Example 52: Preparation of SPK241
-
[0214]To trans-2-dodecenal (4.5 g) dissolved in methylene chloride (80 ml) was added (carbomethoxymethylene)triphenylphosphorane (8.3 g), and the mixture was stirred for 2 hours. The reaction mixture was subjected to chromatography on a silica gel column with eluent systems of n-hexane- ethyl acetate (from 100:1 to 20:1) to give the methyl ester of trans,trans-2,4-tetradecadienoic acid (5.4 g). Potassium hydroxide (6.5 g) was dissolved in a mixed solvent of ethanol-water (1:1) (100 ml). The methyl ester of trans,trans-2,4-tetradecadienoic acid (5.4 g) was added to the mixture, and the resulting mixture was stirred at 60 °C for 40 minutes. After the reaction mixture was cooled, it was adjusted to the weak acidic range of pH with citric acid and extracted with ethyl acetate. The ethyl acetate layer was dried over anhydrous sodium sulfate and concentrated to give trans,trans-2,4-tetradecadienoic acid (4.4 g). Hereafter, the title compound can be synthesized by the two methods described below.
-
[0215]In the first method, trans,trans-2,4-tetradecadienoic acid (4.3 g) is first dissolved in N,N-dimethylformamide (DMF, 50 ml). Para-nitrophenol (2.67 g) and N,N’-dicyclohexylcarbodiimide (3.9 g) were added to trans,trans-2,4-tetradecadienoic acid solution, and the mixture was stirred for 12 hours. After precipitates produced were removed by filtration and the solvent (DMF) was removed by distillation, the residue was subjected to chromatography on a silica gel column with eluent systems of n-hexane-ethyl acetate (from 200:1 to 50:1) to give the active ester of trans,trans-2,4-tetradecadienoic acid (5.1 g). To the active ester (500 mg) dissolved in DMF (30 ml) were added 6-(4′-N-glycyl-spicamynyl-amino)purine hydrochloride (556 mg) and triethylamine (1.2 ml). The mixture was stirred for 12 hours. After the solvent was removed by distillation, the residue was subjected to chromatography on a silica gel column with eluent systems of chloroform-methanol (from 7:1 to 5:1) to give SPK241 in the yield of 398 mg.
-
[0216]In the second method, trans,trans-2,4-tetradecadienoic acid (99.6 g) was dissolved in thionyl chloride (87 ml), and the mixture was stirred at room temperature. The excessive thionyl chloride was removed by distillation to give trans,trans-2,4-tetradecadienoic acid chloride (102.0 g). To glycine (66.8 g) dissolved in an aqueous 2N sodium hydroxide solution (540 ml) were added at the same time trans,trans-2,4-tetradecadienoic acid chloride (102.0 g) and 2N sodium hydroxide (270 ml) with 1/10 portions at a 3 minute interval. After the addition was completed, the mixture was warmed to room temperature, stirred for 15 minutes and acidified with concentrated hydrochloric acid (140 ml) under ice-cooling. Precipitates thus produced were collected by filtration and desiccated to give trans,trans-2,4-tetradecadienoyl glycine (75.0 g). To the solution of trans,trans-2,4-tetradecadienoyl glycine (4.7 g) and 6-(4′-N-glycyl-spicamynyl-amino)-purine (5.1 g) in N,N-dimethylformamide (DMF, 60 ml) was added N-hydroxysuccinimide (2.1 g), and the mixture was ice-cooled. 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (3.4 g) dissolved in DMF (100 ml) was added dropwise to the mixture. After the addition was completed, the mixture was heated to room temperature and stirred for 12 hours. Water (500 ml) was added to the reaction mixture, and precipitates produced were collected by filtration and desiccated. Sodium methoxide (3.1 g) was added to a suspension of the precipitates in methanol (100 ml), and the mixture was stirred at room temperature, then ice-cooled and acidified by adding dropwise thereto a 10% methanolic hydrochloric acid solution. Precipitates produced were filtered, dried and subjected to chromatography on a silica gel column with eluent systems of chloroform-methanol (from 7:1 to 5:1) to give SPK241 in the yield of 5.00 g.
Physicochemical properties of SPK241
-
[0217]
- (1) Melting point: 182-183 °C,
- (2) Specific rotation [a]0 2S = 0 (c = 0.1, in methanol),
- (3) Elementary analysis:
- (4) FD mass spectrum (m/z): 590 (M + H) , C28 H4 3 N707
- (5) Infrared spectrum (KBr disc): 3250 cm-1, 1650 cm-1, 1620 cm-1,
- (6) Proton nuclear magnetic resonance spectrum (500 MHz, in CD30D) δH: 0.89 (3H, t, J = 7.3 Hz), 1.20-1.50 (14H, m), 2.18 (2H, dt, J = 7.3, 7.3 Hz), 3.6-3.8 (5H, m), 3.95 (1 H, d, J = 16.3 Hz), 3.98 (1H, d, J = 16.3 Hz), 4.00 (1H, dd, J = <1, 2.9 Hz), 4.15 (1H, dd, J = 10.8, 10.8 Hz), 5.66 (1 H, brs), 5.98 (1 H, d, J = 15.7 Hz), 6.12 (1 H, dt, J = 7.3, 15.7 Hz), 6.22 (1 H, dd, J = 10.0, 15.7 Hz), 7.17 (1 H, dd, J = 10.0, 15.7 Hz), 8.15 (1 H, s), 8.30 (1 H, s).
……………………………….
EP 0525479; JP 1993186494; US 5461036; US 5631238

Spicamycin derivs. and the use thereof
The hydrolysis of the spicamycin mixture (I) with R = alkyl by means of HCl in alcohol or water gives 6-(spicaminylamino)purine (II). (The hydrolysis can also be performed with other inorganic acids such as H2SO4 or organic ones such as acetic acid or formic acid.) The condensation of (II) with N-(tert-butoxycarbonyl)glycine (III) by the active ester method yields the protected glycyl derivative (IV), which is deprotected with TFA (or methanolic HCl) to afford the glycyl derivative (V). Finally, this compound is condensed with tetradeca-2(E),4(E)-dienoic acid (VI) by the active ester method to provide the target carboxamide derivative.
Otake, N.; Kawai, H.; Kawasaki, T.; Odagawa, A.; Kamishohara, M.; Sakai, T. (Kirin Brewery Co., Ltd.)
EP 0525479; JP 1993186494; US 5461036; US 5631238
…………….
| DE3407979A1 * | Mar 3, 1984 | Sep 6, 1984 | Kirin Brewery | Spicamycin sowie verfahren zu seiner herstellung |
| JPS59161396A | Title not available | |||
| US3155647 | Jul 24, 1963 | Nov 3, 1964 | Olin Mathieson | Septaciding and derivatives thereof |
| WO1990015811A1 | Jun 14, 1990 | Dec 27, 1990 | Kirin Brewery | Spicamycin x and its use |
| EP1328236A2 * | Sep 20, 2001 | Jul 23, 2003 | The General Hospital Corporation | Methods of decreasing or preventing pain using spicamycin derivatives |
| EP2305264A1 * | Sep 20, 2001 | Apr 6, 2011 | The General Hospital Corporation | Spicamycin derivatives for use in decreasing or preventing pain |
| EP2349285A2 * | Oct 9, 2009 | Aug 3, 2011 | Dara Biosciences, Inc. | Methods for treating or preventing pain using spicamycin derivatives |
| EP2597082A1 | Nov 24, 2011 | May 29, 2013 | Symrise AG | Compounds for masking an unpleasant taste |
| US5905069 * | Jan 26, 1998 | May 18, 1999 | The General Hospital Corporation | Methods of decreasing or preventing pain using spicamycin or derivatives thereof |
| US7196071 | Sep 20, 2001 | Mar 27, 2007 | The General Hospital Corporation | Methods of decreasing or preventing pain using spicamycin derivatives |
| US7375094 | Mar 15, 2007 | May 20, 2008 | The General Hospital Corporation | Produced via Streptomyces; antitumor agents; time-release agents; for opiod-resistant pain; drug screening |
| US7632825 | Apr 30, 2008 | Dec 15, 2009 | Bayer Pharmaceuticals Corporation | Methods of decreasing or preventing pain using spicamycin derivatives |
|
References 1: Mizumura Y. [Spicamycin derivative]. Nippon Rinsho. 2006 Feb;64(2):322-8. Review. Japanese. PubMed PMID: 16454188. 2: Bayés M, Rabasseda X, Prous JR. Gateways to clinical trials. Methods Find Exp Clin Pharmacol. 2004 Apr;26(3):211-44. PubMed PMID: 15148527. 3: Borsook D, Edwards AD. Antineuropathic effects of the antibiotic derivative spicamycin KRN5500. Pain Med. 2004 Mar;5(1):104-8. PubMed PMID: 14996243. 4: Bayés M, Rabasseda X, Prous JR. Gateways to clinical trials. Methods Find Exp Clin Pharmacol. 2003 Dec;25(10):831-55. PubMed PMID: 14735233. 5: Bayes M, Rabasseda X, Prous JR. Gateways to clinical trials. Methods Find Exp Clin Pharmacol. 2003 Nov;25(9):747-71. PubMed PMID: 14685303. 6: Supko JG, Eder JP Jr, Ryan DP, Seiden MV, Lynch TJ, Amrein PC, Kufe DW, Clark JW. Phase I clinical trial and pharmacokinetic study of the spicamycin analog KRN5500 administered as a 1-hour intravenous infusion for five consecutive days to patients with refractory solid tumors. Clin Cancer Res. 2003 Nov 1;9(14):5178-86. PubMed PMID: 14613997. 7: Yamamoto N, Tamura T, Kamiya Y, Ono H, Kondoh H, Shirao K, Matsumura Y, Tanigawara Y, Shimada Y. Phase I and pharmacokinetic study of KRN5500, a spicamycin derivative, for patients with advanced solid tumors. Jpn J Clin Oncol. 2003 Jun;33(6):302-8. PubMed PMID: 12913085. 8: Kobierski LA, Abdi S, DiLorenzo L, Feroz N, Borsook D. A single intravenous injection of KRN5500 (antibiotic spicamycin) produces long-term decreases in multiple sensory hypersensitivities in neuropathic pain. Anesth Analg. 2003 Jul;97(1):174-82, table of contents. PubMed PMID: 12818962. 9: Gadgeel SM, Boinpally RR, Heilbrun LK, Wozniak A, Jain V, Redman B, Zalupski M, Wiegand R, Parchment R, LoRusso PM. A phase I clinical trial of spicamycin derivative KRN5500 (NSC 650426) using a phase I accelerated titration “2B” design. Invest New Drugs. 2003 Feb;21(1):63-74. PubMed PMID: 12795531. 10: Byrd JC, Lucas DM, Mone AP, Kitner JB, Drabick JJ, Grever MR. KRN5500: a novel therapeutic agent with in vitro activity against human B-cell chronic lymphocytic leukemia cells mediates cytotoxicity via the intrinsic pathway of apoptosis. Blood. 2003 Jun 1;101(11):4547-50. Epub 2003 Feb 20. PubMed PMID: 12595316. |
Critical Outcome Technologies receives orphan drug designation for COTI-2

N’-(5,6,7,8-Tetrahydroquinolin-8-ylidene)-4-(2-pyridyl)piperazine-1-carbothiohydrazide
http://criticaloutcome.com/110819_COTI-2%20Fact%20Sheet.pdf
http://www.slideshare.net/trevorheisler/about-coti2
MW 366.483, C19 H22 N6 S
| PATENTS | WO 2008083491, WO 2010006438 |
Caspase 9 Activators
PKB beta/Akt2 Inhibitors
Critical Outcome Technologies,
Critical Outcome Technologies (COTI) (Originator) preclinical for ovary cancer
Critical Outcome Technologies has announced that the US Food and Drug Administration (FDA) has granted COTI-2 an Orphan Drug Designation for the treatment of ovarian cancer.
Critical Outcome Technologies president and CEO Dr Wayne Danter said that receiving the Orphan Drug Designation for COTI-2 speaks to the need for new treatment options for patients with ovarian cancer.
- COTI-2 | A Potential Breakthrough Therapy for Many Cancers June 11, 2013
- About COTI-2 Late preclinical drug candidate discovered using CHEMSAS® – the company’s proprietary, artificial intelligence-based drug discovery technology 2
- COTI-2 highlights 1 Potential breakthrough therapy for many cancers 2 Active against many cancers with mutations of the p53 gene 3 > 50% of all human cancers have a p53 mutation 3
- Why p53 is important? p53 is a tumour suppressing gene If mutated, cancers can develop & grow without control A mutation of the p53 gene is the most common mutation found in human cancer cells 4
- The future of cancer treatments COTI-2 targets and primarily destroys tumor cells Traditional chemotherapy kills growing & dividing cells, cancer or healthy COTI-2 would treat genetic mutations common in many types of cancer Most current treatments are organ specific (i.e. treatment for lung cancer, colon cancer, etc.) 5
- COTI-2 development progress Easily synthesized oral formulation with no stability issues Effective alone or in combination with approved cancer drugs In final two-species toxicity studies prior to FDA filing enabling human trials 6
http://www.google.com/patents/WO2008083491A1?cl=en
EXAMPLES
Synthesis of COTI-2 The synthesis of COTI-2, as depicted above, was conducted according to the following synthetic methodology:

DCM R T

H2N-NH2

lmidazol-1 -yl-(4-pyridin-2-yl-piperazin-1 -yl)-methanethione (or intermediate 3 above) was formed as follows. Λ/-(2-pyridyl) piperazine (MW 163.22, 0.91 ml, 6.0 mmoles, 1 eq) 2 was added to a solution of 1 ,1 ‘- thiocarbonyldiimidazole (MW 178.22, 1.069 g, 6.0 mmoles, 1 eq) 1 in 50 ml of dichloromethane at room temperature. The reaction mixture was stirred overnight at room temperature. The mixture was washed with water, dried \ over sodium sulfate, filtered and concentrated to provide imidazol-1-yl-(4- pyridin-2-yl-piperazin-1-yl)-methanethione (MW 273.36, 1.354 g, 4.95 mmol, 83% yield) 3, which was used without further purification. TLC (CH2CI2/MeOH: 95/5): Rf = 0.60, Product UV and Ninhydrin stain active. 1H-NMR (400 MHz, CDCI3), δ ppm: 3.72 (s, 4H), 4.02 (s, 4H), 6.67 (d, 1 H, J = 7 Hz), 6.72 (dd, 1 H, J = 7 and 5 Hz), 7.11 (s, 1 H), 7.24 (s, 1 H), 7.54 (t, 1 H, J = 7 Hz), 7.91 (s, 1 H), 8.20 (d, 1 H, J = 5 Hz).
Hydrazine hydrate (MW 50.06, 0.26 ml, 5.44 mmoles, 1.1 eq) was added to a solution of imidazol-1-yl-(4-pyridin-2-yl-piperazin-1-yl)- methanethione 3 (MW 210.30, 1.040 g, 4.95 mmol, 1 eq) in 30 ml of ethanol at room temperature. The reaction mixture was stirred under reflux for 2 hours. A white precipitate formed. This white solid was filtered off and rinsed with diethyl ether to yield 1-[Λ/-(2-pyridyl)-piperazine)-carbothioic acid hydrazide (MW 237.33, 0.86 g, 3.62 mmol, 73% yield) 4 as a white solid, and used without further purification. TLC (CH2CI2/MeOH: 95/5): Rf = 0.20, Product UV and Ninhydrin stain active. 1H-NMR (400 MHz, DMSO-d6), δ ppm: 3.53 (s, 4H), 3.85 (s, 4H), 6.66 (dd, 1 H, J = 8 and 5 Hz), 6.82 (d, 1 H, J = 8 Hz), 7.55 (t, 1 H, J = 8 Hz), 8.12 (d, 1 H, J = 5 Hz).


COTI-2
Finally, COTI-2 was formed as follows. 1-[Λ/-(2-pyridyl)-piperazine)- carbothioic acid hydrazide (MW 237.33, 0.475 g, 2.0 mmol, 1 eq) 4 and 6,7- dihydro-5H-quinolin-8-one (MW 147.18, 0.306 g, 2.0 mmol, 1 eq) 5 was dissolved in 15 ml of ethanol at room temperature. The mixture was then stirred under reflux for 20 hours. A yellow solid precipitated out of the solution. This solid was filtered off then rinsed with methanol and diethyl ether to yield COTI-2 (MW 366.48, 0.60 g, 1.64 mmol, 82% yield) as a yellow solid. TLC (CH2CI2/MeOH: 95/5): Rf = 0.75, Product UV and Ninhydrine stain active. HPLC analysis showed a mixture of isomers (approximately in 80/20 ratio), and >98% purity. During the HPLC Method Development, as expected, this product tends to be hydrolyzed in presence of TFA in mobile phase solution. MS (ESI+, 0.025% TFA in 50/50 MeOH/H2O): [M+H]+ = 367.1 , [M+Na]+ = 389.1 ; 1H-NMR (400 MHz, CDCI3), δ ppm (Major isomer): 2.09 (m, 2H), 2.92 (m, 4H), 3.67 (m, 4H), 4.27 (m, 4H), 6.69 (dd, 1 H, J = 8 and 5 Hz)1 7.25 (dd,
1 H, J = 8 and 5 Hz), 7.55 (d, 2H, J = 8 Hz), 8.23 (d, 1 H, J = 5 Hz), 8.63 (d, 1 H, \ J = 5 Hz), 14.76 (s, 1 H). δ ppm (Minor isomer): 2.09 (m, 2H), 3.14 (t, 4H, J = 6 Hz), 3.80 (m, 4H), 4.27 (m, 4H), 6.66 (m, 1 H), 7.31 (dd, 1 H, J = 8 and 5 Hz), 7.52 (m, 1 H), 7.70 (d, 1 H, J = 8 Hz), 8.23 (d, 1 H, J = 5 Hz), 8.53 (d, 1 H, J = 5 Hz), 15.65 (s, 1 H).
…………………………..
| WO2010006438A1 * | Jul 17, 2009 | Jan 21, 2010 | Critical Outcome Technologies Inc. | Thiosemicarbazone inhibitor compounds and cancer treatment methods |
| See also references of EP2121681A1 | ||
| 2 | * | SHRIDHAR ET AL.: ‘Synthesis & antiparasite activity of some new 1-(6/7-nitrobenzoxazin-3-yl)-4-substituted- 3-thiosemicarbazides & 4-disubstituted 3-(6-acetylbenzoxazin-3-one)thiosemicarbazo nes‘ INDIAN J. OF CHEM. vol. 26B, June 1987, pages 596 – 598, XP008109697 |
| 3 | * | WINKELMANN ET AL.: ‘Antimalarial and anticoccidial activity of 3-aryl-7-chloro-3,4-dihydriacridine-1,9-(2h ,10H)-diones‘ ARZHEIM.-FORSCH./DRUG RES. vol. 37, no. 6, 1987, pages 647 – 661, XP008109793 |
Selexipag Meets Primary Endpoint in Pivitol Phase III Griphon Outcome Study in Patients with Pulmonary Arterial Hypertension
June 16, 2014
Actelion Ltd today announced the top-line results of the pivotal Phase III GRIPHON study in 1,156 patients with pulmonary arterial hypertension (PAH) with selexipag, the first selective oral prostacyclin IP receptor agonist. Initial analysis shows that the event-driven outcome study has met its primary efficacy endpoint with high statistical significance.
June 16, 2014
Actelion Ltd today announced the top-line results of the pivotal Phase III GRIPHON study in 1,156 patients with pulmonary arterial hypertension (PAH) with selexipag, the first selective oral prostacyclin IP receptor agonist. Initial analysis shows that the event-driven outcome study has met its primary efficacy endpoint with high statistical significance.

Selexipag
N-[2-[4-[N-(5,6-Diphenylpyrazin-2-yl)-N-isopropylamino]butoxy]acetyl]methanesulfonamide
2-[4-[N-(5,6-Diphenylpyrazin-2-yl)-N-isopropylamino]butoxy]-N-(methylsulfonyl)acetamide
phase 3 pulmonary hypertention
Selexipag (ACT-293987, NS-304) is a drug currently in development by Actelion as a treatment of pulmonary arterial hypertension. Selexipag and its active metabolite, ACT-333679, are agonists at the PGI2 prostaglandin receptor, which leads to vasodilation in the pulmonary circulation
Selexipag, originally discovered and synthesized by Nippon Shinyaku, is a potent, orally available, selective prostacyclin IP receptor agonist.
Selexipag selectively targets the prostacyclin receptor (also called IP-receptor). The IP receptor is one of
5 types of prostanoid receptor. Prostacyclin activates the IP receptor inducing vasodilation and inhibiting proliferation of vascular smooth muscle cells. Selexipag, unlike prostacyclin analogs, is selective for the IP receptor over other prostanoid receptors.
In April 2008, Actelion and Nippon Shinyaku signed a licensing agreement, under which Actelion will be responsible for the global development and commercialization of selexipag outside Japan, and the two companies will co-develop and co-commercialize the drug in Japan.
http://www1.actelion.com/sites/en/scientists/development-pipeline/phase-3/selexipag.page
ABOUT THE ACTELION / NIPPON SHINYAKU ALLIANCE
Actelion and Nippon Shinyaku entered into an exclusive worldwide alliance in April 2008 to collaborate on selexipag, a first-in-class orally-available, selective IP receptor agonist for patients suffering from pulmonary arterial hypertension (PAH). This compound was originally discovered and synthesized by Nippon Shinyaku. Phase II evaluation has been completed, and a Phase III program in PAH patients has been initiated. Actelion is responsible for global development and commercialization of selexipag outside Japan, while the two companies will co-develop and co-commercialize in Japan. Nippon Shinyaku will receive milestone payments based on development stage and sales milestones as well as royalties on any sales of selexipag.
| Selexipag | |
|---|---|
 |
|
| Identifiers | |
| CAS number | 475086-01-2 |
| PubChem | 9913767 |
| ChemSpider | 8089417 |
| UNII | 5EXC0E384L |
| KEGG | D09994 |
| Jmol-3D images | Image 1 |
| Properties | |
| Molecular formula | C26H32N4O4S |
| Molar mass | 496.6 g·mol−1 |
NS-304 (ACT-293987), an orally available long acting non-prostanoid prostaglandin I2 (PGI-2) receptor agonist, is in phase III clinical trials at Actelion for the oral treatment of pulmonary hypertension. Nippon Shinyaku is conducting phase III clinical trials with NS-304 for this indication in Europe. In Japan, phase II clinical trials are ongoing for the treatment of pulmonary hypertension and chronic thromboembolic pulmonary hypertension.
Originally discovered and synthesized by Nippon Shinyaku, NS-304 stimulates PGI-2 receptors in blood vessels and exerts vasodilating effects.
In 2008, the compound was licensed to Actelion by Nippon Shinyaku on a worldwide basis with the exception of Japan for the oral treatment of pulmonary arterial hypertension (PAH). According to the final licensing agreement, Actelion will be responsible for global development and commercialization of NS-304 outside Japan, while the two companies will codevelop and co-commercialize the product candidate in Japan. In 2005, orphan drug designation was assigned in the E.U. by Nippon Shinyaku for the treatment of pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension.
…………………….
US2012/101276
http://www.google.st/patents/US20120101276?hl=pt-PT&cl=en
The present invention relates to a crystal of 2-{4-[N-(5,6-diphenylpyrazin-2-yl)-N-isopropylamino]butyloxy}-N-(methylsulfonyl)acetamide (hereinafter referred to as “compound A”).

BACKGROUND OF THE INVENTION
Compound A has an excellent PGI2 agonistic effect and shows a platelet aggregation inhibitory effect, a vasodilative effect, a bronchodilative effect, a lipid deposition inhibitory effect, a leukocyte activation inhibitory effect, etc. (see, for example, in WO 2002/088084 (“WO ‘084”)).
Specifically, compound A is useful as preventive or therapeutic agents for transient ischemic attack (TIA), diabetic neuropathy, diabetic gangrene, peripheral circulatory disturbance (e.g., chronic arterial occlusion, intermittent claudication, peripheral embolism, vibration syndrome, Raynaud’s disease), connective tissue disease (e.g., systemic lupus erythematosus, scleroderma, mixed connective tissue disease, vasculitic syndrome), reocclusion/restenosis after percutaneous transluminal coronary angioplasty (PTCA), arteriosclerosis, thrombosis (e.g., acute-phase cerebral thrombosis, pulmonary embolism), hypertension, pulmonary hypertension, ischemic disorder (e.g., cerebral infarction, myocardial infarction), angina (e.g., stable angina, unstable angina), glomerulonephritis, diabetic nephropathy, chronic renal failure, allergy, bronchial asthma, ulcer, pressure ulcer (bedsore), restenosis after coronary intervention such as atherectomy and stent implantation, thrombocytopenia by dialysis, the diseases in which fibrosis of organs or tissues is involved [e.g., Renal diseases (e.g., tuburointerstitial nephritis), respiratory diseases (e.g., interstitial pneumonia (pulmonary fibrosis), chronic obstructive pulmonary disease), digestive diseases (e.g., hepatocirrhosis, viral hepatitis, chronic pancreatitis and scirrhous stomachic cancer), cardiovascular diseases (e.g, myocardial fibrosis), bone and articular diseases (e.g, bone marrow fibrosis and rheumatoid arthritis), skin diseases (e.g, cicatrix after operation, scalded cicatrix, keloid, and hypertrophic cicatrix), obstetric diseases (e.g., hysteromyoma), urinary diseases (e.g., prostatic hypertrophy), other diseases (e.g., Alzheimer’s disease, sclerosing peritonitis; type I diabetes and organ adhesion after operation)], erectile dysfunction (e.g., diabetic erectile dysfunction, psychogenic erectile dysfunction, psychotic erectile dysfunction, erectile dysfunction associated with chronic renal failure, erectile dysfunction after intrapelvic operation for removing prostata, and vascular erectile dysfunction associated with aging and arteriosclerosis), inflammatory bowel disease (e.g., ulcerative colitis, Crohn’s disease, intestinal tuberculosis, ischemic colitis and intestinal ulcer associated with Behcet disease), gastritis, gastric ulcer, ischemic ophthalmopathy (e.g., retinal artery occlusion, retinal vein occlusion, ischemic optic neuropathy), sudden hearing loss, avascular necrosis of bone, intestinal damage caused by administration of a non-steroidal anti-inflammatory agent (e.g., diclofenac, meloxicam, oxaprozin, nabumetone, indomethacin, ibuprofen, ketoprofen, naproxen, celecoxib) (there is no particular limitation for the intestinal damage so far as it is damage appearing in duodenum, small intestine and large intestine and examples thereof include mucosal damage such as erosion and ulcer generated in duodenum, small intestine and large intestine), and symptoms associated with lumbar spinal canal stenosis (e.g., paralysis, dullness in sensory perception, pain, numbness, lowering in walking ability, etc. associated with cervical spinal canal stenosis, thoracic spinal canal stenosis, lumbar spinal canal stenosis, diffuse spinal canal stenosis or sacral stenosis) etc. (see, for example, in WO ‘084, WO 2009/157396, WO 2009/107736, WO 2009/154246, WO 2009/157397, and WO 2009/157398).
In addition, compound A is useful as an accelerating agent for angiogenic therapy such as gene therapy or autologous bone marrow transplantation, an accelerating agent for angiogenesis in restoration of peripheral artery or angiogenic therapy, etc. (see, for example, in WO ‘084).
Production of Compound A
Compound A can be produced, for example, according to the method described in WO ‘084, and, it can also be produced according to the production method mentioned below.

Step 1:
6-Iodo-2,3-diphenylpyrazine can be produced from 6-chloro-2,3-diphenylpyrazine by reacting it with sodium iodide. The reaction is carried out in the presence of an acid in an organic solvent (e.g., ethyl acetate, acetonitrile, acetone, methyl ethyl ketone, or their mixed solvent). The acid to be used is, for example, acetic acid, sulfuric acid, or their mixed acid. The amount of sodium iodide to be used is generally within a range of from 1 to 10 molar ratio relative to 6-chloro-2,3-diphenylpyrazine, preferably within a range of from 2 to 3 molar ratio. The reaction temperature varies depending on the kinds of the solvent and the acid to be used, but may be generally within a range of from 60° C. to 90° C. The reaction time varies depending on the kinds of the solvent and the acid to be used and on the reaction temperature, but may be generally within a range of from 9 hours to 15 hours.
Step 2:
5,6-Diphenyl-2-[(4-hydroxybutyl(isopropyl)amino]pyrazine can be produced from 6-iodo-2,3-diphenylpyrazine by reacting it with 4-hydroxybutyl(isopropyl)amine. The reaction is carried out in the presence of a base in an organic solvent (e.g., sulfolane, N-methylpyrrolidone, N,N-dimethylimidazolidinone, dimethyl sulfoxide or their mixed solvent). The base to be used is, for example, sodium hydrogencarbonate, potassium hydrogencarbonate, potassium carbonate, sodium carbonate or their mixed base. The amount of 4-hydroxybutyl(isopropyl)amine to be used may be generally within a range of from 1.5 to 5.0 molar ratio relative to 6-iodo-2,3-diphenylpyrazine, preferably within a range of from 2 to 3 molar ratio. The reaction temperature varies depending on the kinds of the solvent and the base to be used, but may be generally within a range of from 170° C. to 200° C. The reaction time varies depending on the kinds of the solvent and the base to be used and on the reaction temperature, but may be generally within a range of from 5 hours to 9 hours.
Step 3:
Compound A can be produced from 5,6-diphenyl-2-[4-hydroxybutyl(isopropyl)amino]pyrazine by reacting it with N-(2-chloroacetyl)methanesulfonamide. The reaction is carried out in the presence of a base in a solvent (N-methylpyrrolidone, 2-methyl-2-propanol or their mixed solvent). The base to be used is, for example, potassium t-butoxide, sodium t-butoxide or their mixed base. The amount of N-(2-chloroacetyl)methanesulfonamide to be used may be generally within a range of from 2 to 4 molar ratio relative to 5,6-diphenyl-2-[4-hydroxybutyl(isopropyl)amino]pyrazine, preferably within a range of from 2 to 3 molar ratio. The reaction temperature varies depending on the kinds of the solvent and the base to be used, but may be generally within a range of from −20° C. to 20° C. The reaction time varies depending on the kinds of the solvent and the base to be used and on the reaction temperature, but may be generally within a range of from 0.5 hours to 2 hours.
The compounds to be used as the starting materials in the above-mentioned production method for compound A are known compounds, or can be produced by known methods.
…………………………………
WO 2002088084
and
http://www.google.fm/patents/WO2009157398A1?cl=en
………………………
Bioorganic and Medicinal Chemistry, 2007 , vol. 15, 21 p. 6692 – 6704
compd 31
……………………
Bioorganic and Medicinal Chemistry, 2007 , vol. 15, 24 p. 7720 – 7725
 2a isthe drug
2a isthe drug
N-Acylsulfonamide and N-acylsulfonylurea derivatives of the carboxylic acid prostacyclin receptor agonist 1 were synthesized and their potential as prodrug forms of the carboxylic acid was evaluated in vitro and in vivo. These compounds were converted to the active compound 1 by hepatic microsomes from rats, dogs, monkeys, and humans, and some of the compounds were shown to yield sustained plasma concentrations of 1 when they were orally administered to monkeys. These types of analogues, including NS-304 (2a), are potentially useful prodrugs of 1.
http://www.sciencedirect.com/science/article/pii/S0968089607007614
References
- Kuwano et al. NS-304, an orally available and long-acting prostacyclin receptor agonist prodrug. J Pharmacol Exp Ther 2007;322:1181-1188.
- Kuwano et al. A long-acting and highly selective prostacyclin receptor agonist prodrug, NS-304, ameliorates rat pulmonary hypertension with unique relaxant responses of its active form MRE-269 on rat pulmonary artery. J Pharmacol Exp Ther 2008;326:691-699.
- Simonneau G, Lang I, Torbicki A, Hoeper MM, Delcroix M, Karlocai K, Galie N. Selexipag, an oral, selective IP receptor agonist for the treatment of pulmonary arterial hypertension Eur Respir J 2012; 40: 874-880
- Mubarak KK. A review of prostaglandin analogs in the management of patients with pulmonary arterial hypertension. Respir Med 2010;104:9-21.
- Sitbon, O.; Morrell, N. (2012). “Pathways in pulmonary arterial hypertension: The future is here”. European Respiratory Review 21 (126): 321–327. doi:10.1183/09059180.00004812. PMID 23204120.
ABOUT SELEXIPAG
Selexipag, originally discovered and synthesized by Nippon Shinyaku, is a potent, orally available, selective prostacyclin IP receptor agonist.
Selexipag selectively targets the prostacyclin receptor (also called IP-receptor). The IP receptor is one of 5 types of prostanoid receptor. Prostacyclin activates the IP receptor inducing vasodilation and inhibiting proliferation of vascular smooth muscle cells. Selexipag, unlike prostacyclin analogs, is selective for the IP receptor over other prostanoid receptors. In preclinical models selective IP receptor agonism has shown to maintain efficacy and reduce the risk of side effects mediated by activation of other prostanoid receptors, such as EP1 and EP3 receptors. [2,4,5]
Selexipag was previously evaluated in a Phase II, 43-patient, placebo-controlled, double-blind study, where patients were randomized in a 3:1 ratio receiving selexipag or placebo on top of PDE-5 inhibitor and/or ERA [6]
SELEXIPAG
Selexipag, originally discovered and synthesized by Nippon Shinyaku, is a potent, orally available, selective prostacyclin IP receptor agonist.
Selexipag selectively targets the prostacyclin receptor (also called IP-receptor). The IP receptor is one of 5 types of prostanoid receptor. Prostacyclin activates the IP receptor inducing vasodilation and inhibiting proliferation of vascular smooth muscle cells. Selexipag, unlike prostacyclin analogs, is selective for the IP receptor over other prostanoid receptors. In preclinical models selective IP receptor agonism has shown to maintain efficacy and reduce the risk of side effects mediated by activation of other prostanoid receptors, such as EP1 and EP3 receptors. [2,4,5]
Selexipag was previously evaluated in a Phase II, 43-patient, placebo-controlled, double-blind study, where patients were randomized in a 3:1 ratio receiving selexipag or placebo on top of PDE-5 inhibitor and/or ERA [6]
SELEXIPAG
Selexipag, originally discovered and synthesized by Nippon Shinyaku, is a potent, orally available, selective prostacyclin IP receptor agonist.
Selexipag selectively targets the prostacyclin receptor (also called IP-receptor). The IP receptor is one of 5 types of prostanoid receptor. Prostacyclin activates the IP receptor inducing vasodilation and inhibiting proliferation of vascular smooth muscle cells. Selexipag, unlike prostacyclin analogs, is selective for the IP receptor over other prostanoid receptors. In preclinical models selective IP receptor agonism has shown to maintain efficacy and reduce the risk of side effects mediated by activation of other prostanoid receptors, such as EP1 and EP3 receptors. [2,4,5]
Selexipag was previously evaluated in a Phase II, 43-patient, placebo-controlled, double-blind study, where patients were randomized in a 3:1 ratio receiving selexipag or placebo on top of PDE-5 inhibitor and/or ERA [6]
SELEXIPAG
Selexipag, originally discovered and synthesized by Nippon Shinyaku, is a potent, orally available, selective prostacyclin IP receptor agonist.
Selexipag selectively targets the prostacyclin receptor (also called IP-receptor). The IP receptor is one of 5 types of prostanoid receptor. Prostacyclin activates the IP receptor inducing vasodilation and inhibiting proliferation of vascular smooth muscle cells. Selexipag, unlike prostacyclin analogs, is selective for the IP receptor over other prostanoid receptors. In preclinical models selective IP receptor agonism has shown to maintain efficacy and reduce the risk of side effects mediated by activation of other prostanoid receptors, such as EP1 and EP3 receptors. [2,4,5]
Selexipag was previously evaluated in a Phase II, 43-patient, placebo-controlled, double-blind study, where patients were randomized in a 3:1 ratio receiving selexipag or placebo on top of PDE-5 inhibitor and/or ERA [6]
Provectus Phase III Melanoma Trial Results Earlier Than Planned?


Rose Bengal disodium
4 ,5,6,7-Tetrachloro-2′,4′,5′,7′-tetraiodo-3-oxo-3H-spiro[2-benzofuran-1,9′-xanthene]-3′,6′-diolate disodium salt
| cas | 632-69-9 |
C20 H2 Cl4 I4 O5 . 2 Na, mw 1017.36, PH 10
| innovator | Provectus |
![]()
The FDA has granted PV-10 Orphan Drug Status for the treatment of highly lethal metastatic melanoma and metastatic liver cancer. It has a successful and expanding Compassionate Use Program in operation and successfully completed trials on metastatic cancer of the breast, liver and melanoma, with positive results in all three. Positive effects in this context is that, if you inject PV-10 into a solid tumor, it kills cancer cells, usually within a week and doesn’t harm normal tissue. Many injected tumors actually disappear while others shrink and stop growing. The dual action of the drug is that the destruction of the cancer by direct injection of PV-10 serves to sensitize the patient’s immune system to seek out and kill similar cancer throughout the body. There is convincing evidence that untreated cancer distant from the treated cancer is attacked by the patient’s immune system after treatment.
PROVECTUS COMPANY OVERVIEW
Provectus (PVCT) is a clinical stage bio-pharmaceutical drug development company. There are 3 key scientific managers running the business along with the CFO, who is also the Chief Operating Officer. They preside over a stable of expert and specialized consultants. The company has two lead drug candidates: PH-10 for significant, often severe, and common skin disorders and PV-10, a dual action, local ablation and immunological anti-cancer drug. PH-10 is currently the subject of post-Phase II trial research into mode of action. PV-10 has successfully completed Phase II trials for malignant melanoma, is currently the subject of independent research on mode of actioRose Bengal disodium is in early clinical trials at Provectus for the topical treatment of psoriasis and atopic dermatitis. An intralesional injectable formulation is also in early clinical development as breast cancer, liver cancer and melanoma therapy. Development for the treatment of actinic keratosis had been ongoing; however, no recent development for this indication has been reported. The company is seeking approval in the U.S. to begin clinical evaluation of this formulation for the treatment of liver and prostate cancer. A compassionate use program is under way for Rose Bengal disodium for the treatment of non-visceral cancers.
The drug’s mechanism of action is believed to be characterized by the creation of free radicals upon activation, which eliminate diseased cells. The compound concentrates in tumors at cytotoxic levels while quickly dissipating from healthy tissue. Simultaneously, the drug triggers an immune response that can eliminate metastatic tumor tissue.
In 2007, orphan drug designation was assigned to Rose Bengal disodium by the FDA for the treatment of metastatic melanoma. This designation was also assigned to the compound in the U.S. in 2011 for the treatment of hepatocellular carcinoma.n and efficacy in conjunction with radiation, and it will have a Phase III pivotal trial starting shortly.
SUMMARY
1. If PV-10 and the Chemotherapies act as the prior data indicate, an NDA for melanoma may be submitted by Provectus in the first half of 2015.
2. If this occurs, the FDA denial of the Breakthrough Therapy designation will not have slowed PV-10’s progress to commercialization.
3. Given the relative safety and efficacy of the different drugs, if the trial is not stopped very early for humanitarian reasons, the planned Interim Analysis is likely to result in the cancellation of the trial, prior to the end of 2015.
4. Given PV-10’s superior safety and lack of significant side effects, if it is only as good as Chemotherapy, it will deserve FDA approval.
The Phase III pivotal trial will demonstrate the safety and efficacy of PV-10 to the market and to prospective acquirers a lot earlier than many have presumed.
Rose bengal (4,5,6,7-tetrachloro-2′,4′,5′,7′-tetraiodofluorescein) is a stain. Its sodium salt is commonly used in eye drops to stain damaged conjunctival and corneal cells and thereby identify damage to the eye. The stain is also used in the preparation of Foraminifera for microscopic analysis, allowing the distinction between forms that were alive or dead at the time of collection.
A form of Rose Bengal is also being studied as a treatment for certain cancers and skin conditions. The cancer formulation of the drug, known as PV-10, is currently undergoing clinical trials for melanoma and breast cancer. The company also has formulated a drug based on Rose Bengal for the treatment of eczema and psoriasis; this drug, PH-10, is currently in clinical trials as well.
| Rose bengal | |
|---|---|
 |
|
| Identifiers | |
| CAS number | 11121-48-5 |
| ATC code | S01 |
| Jmol-3D images | Image 1 |
| Properties | |
| Molecular formula | C20H4Cl4I4O5 |
| Molar mass | 973.67 g mol−1 |
Chemical applications

Light microscopy image of the undescribed species of Spinoloricus from Loricifera stained with Rose Bengal.
Rose Bengal is also used in synthetic chemistry to generate singlet oxygen from triplet oxygen. The singlet oxygen can then undergo a variety of useful reactions, particularly [2 + 2] cycloadditions with alkenes and similar systems.
Rose Bengal can be used to form many derivatives that have important medical functions. One such derivative was created so to be sonosensative but photoinsensative, so that with a high intensity focused ultrasound, it could be used in the treatment of cancer. The derivative was formed by amidation of Rose Bengal, which turned off the fluorescent and photosensitive properties of Rose Bengal, leading to a usable compound, named in the study as RB2.[1]
Salts of Rose Bengal can also be formed, with the molecular formula C20 H4 Cl4 I4 O5 . 2 Na, molecular weight of 1017.64 g/mol and CAS # 632-69-9. Known as Rose Bengal Sodium Salt, this compound has its own unique uses and properties, but also functions as a dye.[2]
Biological applications
PV-10 was found to cause an observable response in 60 percent of tumors treated, according to researchers in a phase II melanoma study. Locoregional disease control was observed in 75 percent of patients. Also confirmed was a “bystander effect”, previously observed in the phase I trial, whereby untreated lesions responded to treatment as well, potentially due to immune system response. These data were based on the interim results of the first 40 patients treated in an 80 patient study.[3] Rose Bengal has been shown to not just prevent the growth and spread of ovarian cancer, but also to cause apoptotic cell death of the cancer cells. This has been proven in vitro, in order to prove that Rose Bengal is still a possible option in the treatment of cancer, and further research should be done.[4]
Rose Bengal is also used in animal models of ischemic stroke (photothrombotic stroke models) in biomedical research. A bolus of the compound is injected into the venous system. Then the region of interest (e.g., the cerebral cortex) is exposed and illuminated by LASER light of 561 nm. A thrombus is formed in the illuminated blood vessels, causing a stroke in the dependent brain tissue.[5][6]
Rose bengal has been used for 50 years to diagnose liver and eye cancer. It has also been used as an insecticide.[7][8]
Rose Bengal is able to stain cells whenever the surface epithelium is not being properly protected by the preocular tear film, because Rose Bengal has been proven to not be able to stain cells because of the protective functioning of these preocular tear films.[9] This is why Rose Bengal is often useful as a stain in diagnosing certain medical issues, such as conjunctival and lid disorders.[10]
Rose Bengal has been used for ocular surface staining to study the efficacy of punctal plugs in the treatment of keratoconjunctivitis sicca. [11]
Rose Bengal is being researched as an agent in creating nano sutures.[12] Wounds are painted on both sides with it and then illuminated with an intense light. This links the tiny collagen fibers together sealing the wound.[13][14][15] Healing is faster and the seal reduces chances of infection.[16][17]
Rose Bengal is used in several microbiological media, including Cooke’s Rose Bengal agar, to suppress bacterial growth.
Rose Bengal has been used as a protoplasm stain to discriminate between living and dead micro-organisms, particularly Foraminifera, since the 1950s when Bill Walton developed the technique.[18]
Electronic applications
Rose Bengal demonstrates at least six distinct electronic properties[19] which are otherwise hidden in the molecule. Rose Bengal is a double planar molecule and relative rotation of the planes generate unique electronics. Therefore, Rose Bengal is a suitable candidate for molecular electronics.
History
Rose Bengal was originally prepared in 1884 by Gnehm, as an analogue of fluorescein.[20] The name is due to its similarity to alta, a dye that women in Bengal have used for centuries to colour their feet red during weddings and festivals.
References
- Kim, Y; Valentina Rubio, Jianjun Qi, Rongmin Xia, Zheng-Zheng Shi, Leif Peterson, Ching-Hsuan Tung, and Brian E. O’Neill (2012). “Cancer treatment using an optically inert Rose Bengal derivative combined with pulsed focused ultrasound”. AIP Conference Proceedings 1481: 175.
- “Rose Bengal Sodium Salt”. Sigma-Aldrich. Sigma Aldrich Co. Retrieved 12 November 2013.
- Metastatic Melanoma PV-10 Trial Results Encouraging Says Drug Company, Medical News Today, 09 Jun 2009
- Koevary, S (2012). “Selective toxicity of rose bengal to ovarian cancer cells in vitro”. International Journal of Physiology, Pathophysiology and Pharmacology 4: 99–107.
- Salber D, et al. (2006). “Differential uptake of [18F]FET and [3H]l-methionine in focal cortical ischemia”. Nuclear Medicine and Biology 33 (8): 1029–1035. doi:10.1016/j.nucmedbio.2006.09.004. PMID 17127177.
- Watson BD, Dietrich WD, Busto R, Wachtel MS, Ginsberg MD (1985). “Induction of reproducible brain infarction by photochemically initiated thrombosis”. Ann Neurol 17 (5): 497–504. doi:10.1002/ana.410170513. PMID 4004172.
- Capinera, John L.; Squitier, Jason M. (2000). “Insecticidal Activity of Photoactive Dyes to American and Migratory Grasshoppers (Orthoptera: Acrididae)”. Journal of Economic Entomology 93 (3): 662–666. doi:10.1603/0022-0493-93.3.662. PMID 10902313.
- Martin, Phyllis; Mischke, Sue; Schroder, Robert (1998). “Compatibility of Photoactive Dyes with Insect Biocontrol Agents”. Biocontrol Science and Technology 8 (4): 501–508. doi:10.1080/09583159830018.
- Feenstra, R; Tseng, S (July 1992). “What is actually stained by rose bengal?”. Arch Ophthalmol 110: 984–993. doi:10.1001/archopht.1992.01080190090035.
- Yokoi, Norihiko (2012). “Vital staining for disorders of conjunctiva and lids”. Atarashii Ganka 29: 1599–1605.
- Ervin AM, Wojciechowski R, Schein O (2010). “Punctal occlusion for dry eye syndrome”. Cochrane Database Syst Rev 9: CD006775. doi:10.1002/14651858.CD006775.pub2. PMID 20824852.
- Chan, B; Chan, O; So, K (2008). “Effects of photochemical crosslinking on the microstructure of collagen and a feasibility study on controlled protein release”. Acta Biomaterialia 4 (6): 1627–1636. doi:10.1016/j.actbio.2008.06.007. PMID 18640085.
- O’Neill A.C., Winograd J.M, Zeballos J.M., Johnson T.S., Randolph M.A., Bujold K.E., Kochevar I.E., Redmond R.W. (2007). “Microvascular anastomosis using a photochemical tissue bonding technique”. Lasers in Surgery and Medicine 39 (9): 716–722. doi:10.1002/lsm.20548. PMID 17960755.
- Mulroy L., Kim J., Wu I., Scharper P., Melki S.A., Azar D.A., Redmond R.W., Kochevar I.E. (2000). “Photochemical keratodesmos for repair of lamellar corneal incisions”. Invest Ophthalmol Vis Sci 41 (11): 3335–3340. PMID 11006222.
- Proano C.E., Mulroy L., Erika Jones E., Azar D.A., Redmond R.W., Kochevar I.E. (2004). Invest Ophthalmol Vis Sci: 2177–2181.
- Laser Show in the Surgical Suite, Technology Review, March/April 2009
- Laser Show in the Surgical Suite, Technology Review, 02.11.2009
- Walton, W. (1952), Techniques for recognition of living foraminifera, Contrib. Cushman Found. Foraminiferal Res., 3, 56 – 60
- A new approach to extract multiple distinct conformers and co-existing distinct electronic properties of a single molecule by point-contact method Anirban Bandyopadhyay, Satyajit Sahu, Daisuke Fujita and Yutaka Wakayama, Phys. Chem. Chem. Phys., 2010 view highlights in Royal Society of Chemistry,
- Alexander, Walter (2010). “American Society of Clinical Oncology, 2010 Annual Meeting and Rose Bengal: From a Wool Dye to a Cancer Therapy”. Pharmacy and Therapeutics 35 (8): 469–474. PMC 2935646. Retrieved 5 November 2013.
- US 2010021566
- WO 2011050164
- US 2011250296
External links
- Rose Bengal at the US National Library of Medicine Medical Subject Headings (MeSH)
- Absorption and extinction data
