
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 88)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
E 2212
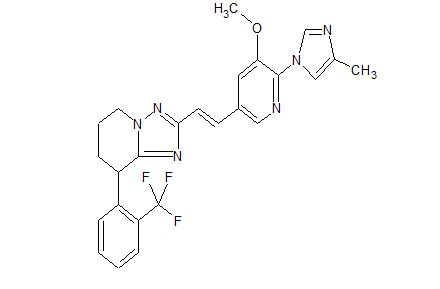
C25 H23 F3 N6 O, 480.48
CAS 1123197-68-1
(+) -2-{(E)-2-[5-methoxy-6-(4-methyl-1H-imidazol-1-yl)pyridin-3-yl]vinyl}-8-(2-trifluoromethylphenyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[1,5-a]pyridine
- (+)-5,6,7,8-Tetrahydro-2-[(1E)-2-[5-methoxy-6-(4-methyl-1H-imidazol-1-yl)-3-pyridinyl]ethenyl]-8-[2-(trifluoromethyl)phenyl][1,2,4]triazolo[1,5-a]pyridine
- (+)-2-[(E)-2-[5-Methoxy-6-(4-methyl-1H-imidazol-1-yl)pyridin-3-yl]ethenyl]-8-(2-trifluoromethylphenyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[1,5-a]pyridine

E2212
CAS 1123197-82-9
- C25 H23 F3 N6 O . 3/2 C4 H6 O6
- [1,2,4]Triazolo[1,5-a]pyridine, 5,6,7,8-tetrahydro-2-[(1E)-2-[6-methoxy-5-(4-methyl-1H-imidazol-1-yl)-2-pyridinyl]ethenyl]-8-[2-(trifluoromethyl)phenyl]-, (8S)-, (2S,3S)-2,3-dihydroxybutanedioate (2:3)
PATENT
https://patents.google.com/patent/US9453000B2/en
Examples 394 and 395 Synthesis of (+) and (−)-2-{(E)-2-[5-methoxy-6-(4-methyl-1H-imidazol-1-yl)pyridin-3-yl]vinyl}-8-(2-trifluoromethylphenyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[1,5-a]pyridine

230 mg of the racemic title compound was obtained from 1-amino-3-(2-trifluoromethylphenyl)piperidin-2-one (343 mg) and (E)-3-[5-methoxy-6-(4-methyl-1H-imidazol-1-yl)pyridin-3-yl]acrylic acid (500 mg) by the same method as in Examples 194 and 195. The racemic title compound (220 mg) was separated by CHIRALPAK™ IC manufactured by Daicel Chemical Industries, Ltd. (2 cm×25 cm; mobile phase: methanol) to obtain the title optically active compound with positive optical rotation and a retention time of 16 minutes (92 mg) and the title optically active compound with negative optical rotation and a retention time of 19 minutes (79 mg).
The property value of the title optically active compound with a retention time of 16 minutes is as follows.
ESI-MS; m/z 481 [M++H].
The property values of the title optically active compound with a retention time of 19 minutes are as follows.
ESI-MS; m/z 481 [M++H]. 1H-NMR (CDCl3) δ (ppm): 1.90-2.01 (m, 1H), 2.10-2.35 (m, 2H), 2.29 (s, 3H), 2.43-2.52 (m, 1H), 3.95 (s, 3H), 4.27-4.41 (m, 2H), 4.69 (dd, J=6.0, 8.4 Hz, 1H), 7.02 (d, J=8.0 Hz, 1H), 7.08 (d, J=16.4 Hz, 1H), 7.40 (dd, J=7.6, 7.6 Hz, 1H), 7.44-7.53 (m, 4H), 7.73 (d, J=8.0 Hz, 1H), 8.13 (d, J=1.6 Hz, 1H), 8.34 (s, 1H).
PATENT
WO2009028588
https://patentscope.wipo.int/search/en/detail.jsf;jsessionid=D6AD22B6CC7302560AE1ADCED305CDCE.wapp2nC?docId=WO2009028588&tab=FULLTEXT&queryString=%28PA%2Feisai%29%2520&recNum=93&maxRec=725
(+)および(-)-2-{(E)-2-[5-メトキシ-6-(4-メチル-1H-イミダゾール-1-イル)ピリジン-3-イル]ビニル}-8-(2-トリフルオロメチルフェニル)-5,6,7,8-テトラヒドロ-[1,2,4]トリアゾロ[1,5-a]ピリジンの合成
[化221]
実施例194および実施例195と同様の方法により、1-アミノ-3-(2-トリフルオロメチルフェニル)ピペリジン-2-オン(343mg)および(E)-3-[5-メトキシ-6-(4-メチル-1H-イミダゾール-1-イル)ピリジン-3-イル]アクリル酸(500mg)から、ラセミ体の表題化合物を230mg得た。ラセミ体の表題化合物(220mg)をダイセル製CHIRALPAK TM IC(2cm×25cm:移動相;メタノール)にて分取し、(+)の旋光性を有する保持時間16分の表題光学活性化合物(92mg)および(-)の旋光性を有する保持時間19分の表題光学活性化合物(79mg)を得た。
保持時間16分の表題光学活性体の物性値は以下の通りである。
ESI-MS;m/z 481[M ++H].
保持時間19分の表題光学活性体の物性値は以下の通りである。
ESI-MS;m/z 481[M ++H]. 1H-NMR(CDCl 3)δ(ppm):1.90-2.01(m,1H),2.10-2.35(m,2H),2.29(s,3H),2.43-2.52(m,1H),3.95(s,3H),4.27-4.41(m,2H),4.69(dd,J=6.0,8.4Hz,1H),7.02(d,J=8.0Hz,1H),7.08(d,J=16.4Hz,1H),7.40(dd,J=7.6,7.6Hz,1H),7.44-7.53(m,4H),7.73(d,J=8.0Hz,1H),8.13(d,J=1.6Hz,1H),8.34(s,1H).
Example 394 and Example 395
(+) and (−)-2-{(E) -2- [5-methoxy-6- (4-methyl-1H-imidazol-1-yl) pyridin-3-yl] Synthesis of vinyl} -8- (2-trifluoromethylphenyl) -5,6,7,8-tetrahydro- [1,2,4] triazolo [1,5-a] pyridine [Formula
221]
Example 194 and By a method similar to Example 195, 1-amino-3- (2-trifluoromethylphenyl) piperidin-2-one (343 mg) and (E) -3- [5-methoxy-6- (4-methyl-) 1 H-Imidazol-1-yl) pyridin-3-yl] acrylic acid (500 mg) gave 230 mg of the racemic title compound. Racemic title compound (220 mg) a Daicel CHIRALPAK TM IC (2 cm × 25 cm: mobile phase; methanol) was collected by min (+) title optically active compound of the retention time of 16 minutes with a optical rotation of (92 mg) The title optically active compound (79 mg) having a polarizability of (−) and a retention time of 19 minutes was obtained.
The physical property values of the title optically active substance with a retention time of 16 minutes are as follows.
ESI-MS; m / z 481 [M + + H].
The physical property values of the title optically active substance with a retention time of 19 minutes are as follows.
ESI-MS; m / z 481 [M + + H]. 1 H-NMR (CDCl 3)) Δ (ppm): 1.90 to 2.01 (m, 1 H), 2.10 to 2.35 (m, 2 H), 2.29 (s, 3 H), 2.43 to 2.52 (m) , 1 H), 3.95 (s, 3 H), 4.27-4. 41 (m, 2 H), 4.69 (dd, J = 6.0, 8.4 Hz, 1 H), 7.02 (d , J = 8.0 Hz, 1 H), 7.08 (d, J = 16.4 Hz, 1 H), 7.40 (dd, J = 7.6, 7.6 Hz, 1 H), 7.44-7. 53 (m, 4H), 7.73 (d, J = 8.0 Hz, 1 H), 8.13 (d, J = 1.6 Hz, 1 H), 8.34 (s, 1 H).
PATENT
https://patents.google.com/patent/WO2010098490A1/it


As a novel compound that has an effect of reducing the production of Aβ40 and
42 and is expected as a therapeutic or prophylactic agent for Alzheimer’s disease or the like, the present inventors have found a compound represented by the following formula (1) (compound
(D): [Formula 1]
and filed a patent application for the invention (PCT/JP08/065365).
Generally, properties of salts of compounds and those crystals that are useful as pharmaceuticals are highly important for the development of pharmaceuticals, because the properties greatly affect bioavailability of drugs, purity of drug substances, formulation of preparations, and the like. Therefore, it is necessary to research which salts and crystal forms of the compound of the formula (1) are most excellent as pharmaceuticals. Specifically, since their properties depend on the character of the individual compounds, it is generally difficult to estimate salts and crystal forms for drug substances having excellent properties and it is demanded to actually make various studies for each compound.
EXAMPLES [0023] The present invention will be described in detail below with reference to reference examples and examples; however, the present invention is not limited to these reference examples and examples. [0024]
The following abbreviations are used in the following reference examples and examples.
DMF: N,N’-dimethylformamide
THF: Tetrahydrofuran
EDC: lrEmyl-S-β-dimemylammopropytycarbodiimide hydrochloride HOBT: 1-Hydroxybenzotriazole IPEA: Diisopropylethylamine [0025]
In powder X-ray diffractometry of the crystals produced in the following examples, the resulting crystals were placed on a sample stage of a powder X-ray diffractometer and analyzed under the following conditions. [0026] Measurement conditions
Sample holder: Aluminum Target: Copper
Detector: Scintillation counter Tube voltage: 50 kV Tube current: 300 mA
Slit: DS 0.5 mm (Height limiting slit 2 mm), SS Open, RS Open Scanning rate : 5 °/min
Sampling interval: 0.02° Scan range: 5 to 35° Goniometer: Horizontal goniometer [0027] Reference Example 1
Svnmesis ofr8SV2-(fE)-246-memoxy-5-(4-memyl-lH-imidazol-l-vnpyridin-2-yllvmvU-8-(2-trifluoromethylphenyl‘)-5,6J,8-tetrahvdro-[1.2,41triazolo[l.,5-a]pyridine
[Formula 2]
Synthesis of l-amino-3-(2-trifluoromemylphenyl)piperidin-2-one Thionyl chloride (2.72 mL) was added to a solution of 2-trifluoromethylphenylacetic acid (1.9 g) in methanol (38 mL), followed by stirring at room temperature for three hours. The reaction solution was concentrated under reduced pressure. The resulting residue was diluted with DMF. Sodium hydride (containing 40% mineral oil, 410 mg) was added under ice-cooling, followed by stirring for 10 minutes. The reaction solution was further stirred for 30 minutes and then ice-cooled again. l-Chloro-3-iodopropane (1.02 mL) was added to the reaction mixture, and the reaction solution was stirred at room temperature overnight. Water and ethyl acetate were added to the reaction mixture, and the organic layer was separated. The resulting organic layer was washed with saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The resulting residue was diluted with ethanol (26.6 mL). Hydrazine monohydrate (7.6 mL) was added, and the reaction solution was stirred at room temperature for two hours and then at 60°C for further three hours. The reaction mixture was concentrated under reduced pressure. Saturated aqueous sodium bicarbonate and ethyl acetate and were added to the residue, and the organic layer was separated. The resulting organic layer was washed with saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography (carrier: Chromatorex NH; elution solvent: heptane-ethyl acetate system) to obtain 1.68 g of the title compound. The property values of the compound are as follows.
ESI-MS; m/z 259 [M+H-H]. 1H-NMR (CDCl3) δ (ppm): 1.82-2.10 (m, 3H), 2.18-2.26 (m, IH), 3.58-3.76 (m, 2H), 4.07 (dd, J = 10.0, 5.6 Hz, IH), 4.60 (s, 2H), 7.24 (d, J = 7.6 Hz, IH), 7.35 (t, J = 7.6 Hz, IH), 7.51 (t, J = 7.6 Hz, IH)5 7.66 (d, J = 7.6 Hz, IH). [0028] Synthesis of (EV3-[6-methoxy-5-(4-methyl- 1 H-imidazol- 1 -yl)pyridin-2-yl]-N-f2-oxo-3 -(2-trifluoromethylphenyl)piperidin- 1 -yl]acrylamide
EDC (834 mg), HOBT (588 mg) and IPEA (2.03 mL) were added to a suspension of (E)-3-[6-methoxy-5-(4-methyl-lH-imidazol-l-yl)pyridm-2-yl]acrylic acid trifluoroacetate (1.42 g) and l-amήio-3-(2-trifluoromethylphenyl)piperidin-2-one (750 mg) in DMF (30 mL). After stirring at room temperature for 14 hours, a saturated sodium bicarbonate solution and ethyl acetate were added to the reaction solution, and the organic layer was separated. The resulting organic layer was dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography (carrier: Chromatorex NH; elution solvent: ethyl acetate-methanol system) to obtain 1.23 g of the title compound. The property values of the compound are as follows. ESI-MS; m/z 500 [M1H-HJ. [0029]
Synthesis of r8S>-2-(fEV2-r6-methoxy-5-r4-methyl-lH-imidazol-l-vnpyridm’2-vnvinvU-8-(2-trifluoromethvlphenvD-5.6.7.8-tetrahvdro-ri.2.41triazoloπ.5-a1pvridine Phosphorus oxychloride (24.2 mL) was added to (E)-3~[6-methoxy-5-(4-methyl-lH-imidazol-l-yl)pyridin-2-yl]-N-[2-oxo-3-(2-trifluoromethylphenyl)piperidin-l-yl]acrylamide (1.2 g). The reaction solution was stirred at 1000C for one hour and then concentrated under reduced pressure. Subsequently, the residue was diluted with acetic acid (24.2 mL) and then ammonium acetate (1.9 g) was added, followed by stirring at 1500C for two hours. The reaction solution was left to cool to room temperature and then concentrated under reduced pressure. A saturated sodium bicarbonate solution and ethyl acetate were added to the resulting residue, and the organic layer was separated. The resulting organic layer was dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography (carrier: Chromatorex NH; elution solvent: heptane-ethyl acetate system) to obtain a racemate of the title compound (750 mg). The resulting racemate (410 mg) was separated by CHIRALP AK™ IA manufactured by Daicel Chemical Industries, Ltd. (2 cm x 25 cm, mobile phase: hexane:ethanol = 8:2, flow rate: 10 mL/min) to obtain the title compound with a retention time of 33 minutes and negative optical rotation (170 mg) as crystals. The property values of the title compound are as follows.
1H-NMR (CDCl3) δ (ppm): 1.90-2.01 (m, IH), 2.10-2.35 (m, 2H), 2.29 (d, J = 1.2 Hz, 3H), 2.42-2.51 (m, IH), 4.03 (s, 3H), 4.28-4.41 (m, 2H), 4.70 (dd, J = 8.4, 6.0 Hz, IH), 6.92 (d, J = 8.0 Hz, IH), 6.95 (t, J = 1.2 Hz, IH), 7.01 (d, J = 7.6 Hz, IH), 7.39 (t, J = 7.6 Hz5 IH), 7.44 (d, J = 16.0 Hz, IH), 7.45 (d, J = 8.0 Hz, IH), 7.49 (t, J = 7.6 Hz, IH), 7.63 (d, J = 16.0 Hz5 IH), 7.72 (d, J = 7.6 Hz, IH), 7.76 (d, J = 1.2 Hz, IH). [0030]
(8S)-2-{(E)-2-[6-Methoxy-5-(4-methyl-lH-imidazol-l-yl)ρyridin-2-yl]vinyl}-8-(2-trifluoromethylphenyl)-5,6,7,8-tetrahydro-[l,2,4]triazolo[l,5-a]pyridine synthesized according to the above reference example was used for the following synthesis of salts. [0031] Example 1
Synthesis of r8SV2-{rEV2-[6-methoxy-5-(4-methyl-lH-imidazol-l-vπpyridin-2-vnvinvU-8-f2-trifluoromethylphenyl)-5.6.7.8-tetrahvdro-fl,2,4]triazolo[l.,5-a]pyridine 1.5 D-tartrate
(8S)-2-{(E)-2-[6-methoxy-5-(4-methyl-lH-imidazol-l-yl)pyridin-2-yl]vinyl}-8-(2-trifluoromethylphenyl)-5,6,7,8-tetrahydro-[l,2,4]triazolo[l,5-a]pyridine (33.70 mg) was dissolved in 285 μL of a D-tartaric acid-ethanol solution (110.92 mg/3 mL) with stirring at room temperature. The oil was precipitated when 1 mL of heptane was added. Accordingly, the oily substance was dissolved by adding 1 mL of ethanol. Further, 0.5 mL of heptane was added, and the mixture was transferred to a low temperature laboratory at about 50C (under shading) and continuously stirred for 24 hours. Thus, partial gelation occurred. Thereafter, the mixture was brought back to room temperature and continuously stirred, resulting in precipitation of a solid. The solid was collected by filtration through a glass filter and dried under reduced pressure at room temperature to obtain 21.25 mg of the title compound as white solid crystals. 1H-NMR (600 MHz, DMSOd6) δ (ppm): 1.96 (m, IH), 2.14 (s, 3H), 2.16 (m, 2H), 2.29 (m, IH), 3.98 (s, 3H), 4.28 (m, 2H), 4.29 (s, 3H), 4.51 (dd, J = 9, 6 Hz, IH), 7.22 (s, IH), 7.25 (brd, J = 8 Hz, IH), 7.27 (d, J = 8 Hz, IH), 7.32 (d, J = 16 Hz, IH)5 7.46 (d, J = 16 Hz, IH), 7.49 (brdd, J = 8 Hz, IH), 7.61 (brdd, J = 8 Hz5 IH), 7.77 (brd, J = 8 Hz, IH), 7.78 (d, J = 8 Hz, IH), 7.91 (s, IH). [0032] Example 2
Synthesis of (‘8SV2-l(Ε)-2-f6-methoxy-5-(4-methyl-lH-imidazol-l-vnpyridm-2-yllvinyl>-8-f2-trifluoromethylphenylV5,6J,8-tetrahvdro-[l ,2,4]triazolo[l ,5-a]pyridine di-D-tartrate
(8S)-2-{(E)-2-[6-methoxy-5-(4-methyl-lH-imidazol-l-yl)pyridin-2-yl]vinyl}-8-(2-trifluoromethylphenyl)-5,657,8-tetrahydro-[l ,2,4]triazolo[l ,5-a]ρyridine (810.18 mg) was dissolved in 8 mL of a D-tartaric acid-ethanol solution (751.13 mg/10 mL) with stirring at room temperature. The oil was precipitated when 2 mL of heptane was added. Accordingly, the oily substance was dissolved by ultrasonic treatment to prepare a clear solution. Several mg of crystals of the 1.5 D-tartrate prepared according to Example 1 were added, followed by stirring at room temperature. Stirring for about one hour resulted in gelation and subsequent precipitation of a solid. Further, stirring was continued while gradually adding 14 mL of heptane. A part of the suspension (2 mL) was separated and the solid was collected by filtration through a glass filter. The solid was dried under reduced pressure at room temperature to obtain 71.14 mg of the title compound as white solid crystals. 1H-NMR (400 MHz, DMSOd6) δ (ppm): 1.97 (m, IH), 2.15 (s, 3H), 2.16 (m, 2H), 2.30 (m, IH), 3.98 (s, 3H), 4.28 (m, 2H), 4.29 (s, 4H), 4.51 (dd, J = 9, 6 Hz, IH), 7.22 (brs, IH), 7.25 (brd, J = 8 Hz, IH), 7.27 (d, J = 8 Hz, IH), 7.32 (d, J = 16 Hz, IH), 7.46 (d, J = 16 Hz, IH), 7.49 (brdd, J – 8 Hz, IH), 7.61 (brdd, J = 8 Hz, IH), 7.77 (brd, J = 8 Hz, IH), 7.78 (d, J = 8 Hz, IH), 7.91 (brs, IH). [0033] Example 3
Synthesis of r8SV2-(rE)-2-r6-methoxy-5-r4-methyl-lH-imidazol-l-vnpyridin-2-yl1vinvU-8-α-trifluoromethylphenyl)-5,6J,8-tetrahydro-[1.2,4]triazolo[l,5-a]pyridine disulfate
Concentrated sulfuric acid (11.5 μL) was added to a solution of (8S)-2-{(E)-2-[6- methoxy-5-(4-methyl-lH-imidazol-l-yl)pyridin-2-yl]vinyl}-8-(2-txifluoromethylphenyl)-5,6,7,8-tetrahydro-[l52,4]triazolo[l55-a]pyridine (98.09 mg) in ethanol (1 mL), and 1 mL of ethyl acetate was added with stirring at room temperature. Since the oily portion was confirmed on the bottom of the recovery flask, the oily substance was dissolved by ultrasonic treatment. Stirring at room temperature under shading for about 30 minutes resulted in precipitation of a solid. The solid was collected by filtration through a glass filter and dried under reduced pressure at room temperature to obtain 127.94 mg of the title compound as white solid crystals. 1H-NMR (400 MHz, DMSOd6) δ (ppm): 1.97 (m, IH), 2.17 (m, 2H), 2.30 (m, IH), 2.34 (brd, J = 1 Hz, 3H), 4.01 (s, 3H), 4.29 (m, 2H), 4.52 (dd, J = 9, 6 Hz, IH)5 7.25 (brd, J = 8 Hz, IH), 7.37 (d, J = 16 Hz, IH), 7.40 (d, J = 8 Hz, IH), 7.50 (brdd, J = 8 Hz, IH), 7.55 (d, J = 16 Hz, IH), 7.61 (brdd, J = 8 Hz, IH), 7.77 (m, IH), 7.78 (m, IH), 8.00 (d, J = 8 Hz, IH), 9.36 (d, J = 2 Hz, IH). [0034] Example 4 Synthesis of (8SV2-((E)-2-[“6-methoxy-5-(4-methyl-lH-imidazol-l-ylN)ρyridin-2-yllvinvU-8-(‘2-trifluoromethylphenyl)-5,6,7,8-tetrahvdiO-[1.2,41triazolo[l,5-a]pyridine dihydrobromide
Concentrated hydrobromic acid (24.8 μL) was added to a solution of (8S)-2-{(E)-2-[6-methoxy-5-(4-methyl-lH-imidazol-l-yl)pyridin-2-yl]vinyl}-8-(2-trifluoromethylphenyl)-5,6,7,84etrahydro-[l,254]triazolo[l55-a]pyridine (51.42 mg) m ethanol (1 mL), and 1 mL of heptane was added with stirring at room temperature. After several minutes, 1 mL of heptane was further added to the solution and stirring was continued. The solution was stirred at room temperature for one hour and then further stirred at about 50C for 20 minutes. The precipitated solid was collected by filtration through a glass filter and dried under reduced pressure at room temperature to obtain 49.24 mg of the title compound as white solid crystals. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 1.99 (m, IH), 2.17 (m, 2H), 2.30 (m, IH), 2.34 (brd, J = 1 Hz5 3H), 4.01 (s, 3H), 4.30 (m, 2H), 4.52 (dd, J = 9, 6 Hz5 IH), 7.25 (brd, J = 8 Hz5 IH), 7.37 (d, J = 16 Hz, IH), 7.40 (d, J = 7 Hz, IH)57.50 (brdd, J = 8 Hz, IH), 7.55 (d, J = 16 Hz, IH), 7.61 (brdd, J = 8 Hz5 IH), 7.77 (m, IH)5 7.78 (m, IH), 8.00 (d, J = 7 Hz, IH), 9.37 (d, J = 2 Hz, IH). [0035] Example 5
Synthesis of r8SV2-((Ε)-2-r6-methoxy-5-r4-methyl-lH-imidazol-l-yl)ρyridin-2-vnvinyl}-8-r2-trifluoromethylphenyl)-5,6J,8-tetrahvdro-[1.2,41triazolo[1.5-alpyridine hydrochloride
Concentrated hydrochloric acid (3.6 μL) was added to a solution of (8S)-2-{(E)- 2-[6-methoxy-5-(4-metiiyl-lH-imidazol-l-yl)pyridin-2-yl]vinyl}-8-(2-trifluoromethylplienyl)-5,6,7,8-te1xahydro-[l,2,4]triazolo[l,5-a]pyridme (19.80 mg) in 2-propanol (1 mL), and a total of 4 mL of heptane was added in 1 mL portions with stirring at room temperature. The solution was stirred at room temperature under shading for five days. The precipitated solid was collected by filtration through a glass filter and dried under reduced pressure at room temperature to obtain 7.45 mg of the title compound as white solid crystals. 1H-NMR (400 MHz, DMSOd6) δ (ppm): 1.97 (m, IH), 2.17 (m, 2H)5 2.30 (m, IH), 2.30 (s, 3H), 4.00 (s, 3H), 4.30 (m, 2H)5 4.52 (dd, J = 9, 6 Hz5 IH), 7.25 (brd, J – 8 Hz5 IH), 7.36 (d, J = 16 Hz5 IH), 7.37 (d5 J = 8 Hz, IH), 7.50 (brt, J = 8 Hz5 IH)5 7.53 (d, J = 16 Hz5 IH)5 7.61 (brt, J = 8 Hz5 IH)5 7.66 (brs, IH), 7.77 (brd, J = 8 Hz, IH)5 7.96 (d, J = 8 Hz5 IH), 9.06 (brs, IH). [0036] Example 6
Synthesis of (8S)-2-((ΕV2-r6-methoxy-5-(‘4-methyl-lH-imidazol-l-yl)pyridin-2-yl1vinvU-8-(2-trifluoromethylphenyl)-5.6,7,8-tetrahvdro-[l,2,4]triazolo[L5-a1pyridine hydrochloride Concentrated hydrochloric acid (14.3 μL) and heptane (7 mL) were added to a solution of (8S)-2-{(E)-2-[6-methoxy-5-(4-methyl-lH-imidazol-l-yl)pyridin-2-yl]vinyl}-8-(2-trifluoromethylphenyl)-5,657,8-tetrahydro-[l,2,4]triazolo[l,5-a]pyridine (79.77 mg) in 2-propanol (3 mL). A small amount of the crystals obtained in Example 5 were added as seed crystals with stirring at room temperature. The mixture was transferred to a low temperature laboratory at about 50C and stirred for one hour. Thereafter, 1 mL of heptane was further added, followed by stirring for several minutes. When the precipitated solid was collected by filtration through a glass filter, the solid was precipitated in the filtrate. The precipitated solid was collected by filtration through a glass filter and dried under reduced pressure at room temperature to obtain 38.02 mg of the title compound as white solid crystals. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 1.97 (m, IH), 2.17 (m5 2H), 2.29 (m, IH), 2.32 (brd, J = 1 Hz, 3H), 4.00 (s, 3H), 4.30 (m, 2H), 4.52 (dd, J = 9, 6 Hz, IH), 7.25 (brd, J = 8 Hz, IH), 7.37 (d, J = 16 Hz5 IH), 7.38 (d, J = 8 Hz, IH), 7.50 (brdd, J = 8 Hz, IH)5 7.54 (d, J = 16 Hz, IH), 7.61 (brdd, J = 8 Hz, IH), 7.72 (brs, IH), 7.77 (brd, J = 8 Hz, IH), 7.98 (d, J = 8 Hz5 IH)5 9.24 (brs, IH). [0037] Example 7
SvnJhesis off8SV2-f(E>2-r6-memoxy-5-(4-mefovπ trifluoromethylt>henylV5,6,7,8-tetrahvdro-[l,2,4]triazolo[l,5-a]pyridine mesylate
Mesylic acid (0.8 μL) was added to a mixed solution of (8S)-2-{(E)-2-[6- methoxy-5-(4-methyl-lH-imidazol-l-yl)pyridin-2-yl]vinyl}-8-(2-trifluoromethylphen^ tetrahydro-[l52,4]triazolo[l,5-a]pyridine (50 mg) in t-butyl methyl ether (0.8 mL)-ethaαol (0.1 mL). The mixture was solidified as a result of stirring at room temperature for two hours. The solid was collected by filtration through a glass filter. The solid was washed with t-butyl methyl ether-ethanol (8:1) and then dried under reduced pressure at room temperature to obtain 51.9 mg of the title compound as pale yellow solid crystals.
1H-NMR (DMSO-d6) δ (ppm): 1.90-2.05 (m, IH)3 2.10-2.22 (m, 2H), 2.28-2.40 (m, IH), 2.31 (s, 3H), 2.35 (s, 3H)5 4.02 (s, 3H)5 4.25-4.39 (m, 2H), 4.50-4.55 (m, IH), 7.27 (d5 J = 8.0 Hz5 IH)5 7.38 (d, J = 16.0 Hz5 IH)5 7.41 (d, J = 8.0 Hz, IH)5 7.51 (t5 J = 8.0 Hz5 IH)5 7.55 (d, J = 16.0 Hz5 IH), 7.63 (t, J = 8.0 Hz5 IH)5 7.78 (d, J = 8.0 Hz5 IH)5 7.79 (s, IH), 8.01 (d, J = 8.0 Hz5 IH), 9.37 (s, IH). [0038] Example 8 Synthesis of (8S)-2-((ΕV2-r6-methoxy-5-(4-methyl-lH-imidazol-l-vnpyridin-2-vnvinvn-8-r2-trifluoromethylphenyl)-5.6,7,8-tetrahydro-[l.,2,4‘|triazolo[l,5-a]pyridine diphosphate
A solution of phosphoric acid (52.8 mg) in acetonitrile (0.2 mL) was added to a solution of (8S)-2-{(E)-2-[6-methoxy-5-(4-methyl-lH-mτidazol-l-yl)ρyridin-2-yl]vinyl}-8-(2-trifluoromethylphenyl)-5565758-tetrahydro-[l5254]triazolo[l,5-a]pyridine (100 mg) in acetonitrile (0.8 mL) at room temperature. The precipitated oil was solidified as a result of stirring with spatula. The solid was collected by filtration through a glass filter. The solid was washed with ice-cold acetonitrile, air-dried at room temperature for 10 minutes and then dried under reduced pressure at room temperature to obtain 120 mg of the title compound as white solid crystals. 1H-NMR (DMSO-d6) δ (ppm): 1.90-2.05 (m, IH), 2.11-2.20 (m, 2H), 2.15 (s, 3H), 2.25-2.35 (m, IH), 3.99 (s, 3H)5 4.24-4.39 (m, 2H), 4.50-4.55 (m, IH)5 7.23 (s, IH), 7.26 (d, J = 7.0 Hz, IH), 7.28 (d, J = 8.0 Hz, IH), 7.33 (d, J = 16.0 Hz5 IH), 7.47 (d, J = 16.0 Hz5 IH), 7.51 (t, J = 7.0 Hz, IH), 7.63 (t, J = 7.0 Hz, IH), 7.78 (d, J = 7.0 Hz, IH), 7.79 (d, J = 8.0 Hz, IH), 7.90 (s, IH). [0039] Example 9 Svnmesis of(8SV2-{(E)-2-[6-memoxy-5-(4-methyl-lH-irnidazol-l-yl)pyridin-2-yl1vinvU-8-(2-trifluoromethylphenyl)-5,6,7,8-tetrahvdro-[l .2.41triazolo[l .5-a]pyridine diphosphate
A solution of phosphoric acid (13.2 mg) in ethanol (0.05 mL) was added to a mixed solution of (8S)-2-{(E)-2-[6-methoxy-5-(4-methyl-lH-imidazol-l-yl)pyridin-2-yl]vinyl}-8-(2-trifluoromethylphenyl)-5,6,7,8-tetrahydro-[l,2!,4]triazolo[l,5-a]pyridme (50 mg) in heptane (0.6 mL)-ethanol (0.15 mL) at room temperature. The reaction solution was stirred at room temperature, and the precipitated solid was collected by filtration through a glass filter. The solid was washed with heptane-ethanol (3:1) and then dried under reduced pressure at room temperature to obtain 37.6 mg of the title compound as white solid crystals. 1H-NMR (DMSOd6) δ (ppm): 1.90-2.05 (m, IH), 2.11-2.20 (m, 2H), 2.15 (s, 3H), 2.25-2.35 (m, IH), 3.99 (s, 3H), 4.24-4.39 (m, 2H), 4.50-4.55 (m, IH), 7.23 (s, IH), 7.26 (d, J = 7.0 Hz, IH), 7.28 (d, J = 8.0 Hz, IH), 7.33 (d, J = 16.0 Hz, IH), 7.47 (d, J = 16.0 Hz, IH), 7.51 (t, J = 7.0 Hz, IH), 7.63 (t, J = 7.0 Hz, IH), 7.78 (d, J = 7.0 Hz, IH), 7.79 (d, J = 8.0 Hz, IH), 7.90 (s, IH).
CLIP
Development of an Efficient Manufacturing Process for E2212 toward Rapid Clinical Introduction
 , Taiju Nakamura‡, Atsushi Kamada†, Takeo Sasaki§, Toshiyuki Uemura§, Yorihisa Hoshino†, Masaaki Matsuda†, Yongbo Hu∥, Daiju Hasegawa§, Kazato Inanaga‡, Nobuaki Sato§, Kazuhiro Yoshizawa‡, George A. Moniz∥, Gordon D. Wilkie∥, Francis G. Fang∥, Yoshihiro Nishikawa‡, and Katsuya Tagami*‡
, Taiju Nakamura‡, Atsushi Kamada†, Takeo Sasaki§, Toshiyuki Uemura§, Yorihisa Hoshino†, Masaaki Matsuda†, Yongbo Hu∥, Daiju Hasegawa§, Kazato Inanaga‡, Nobuaki Sato§, Kazuhiro Yoshizawa‡, George A. Moniz∥, Gordon D. Wilkie∥, Francis G. Fang∥, Yoshihiro Nishikawa‡, and Katsuya Tagami*‡
Process studies of E2212 (1) toward rapid clinical introduction are described. Through comprehensive route-finding studies and optimization of key condensation and cyclization steps, a racemate-based manufacturing route was established and successfully scaled-up to the hundred kilogram scale. For the rapid delivery of a drug substance containing the Z isomer for preclinical safety studies, the successful scale-up of the photoisomerization of an olefin in a flow system is also presented.
https://pubs.acs.org/doi/10.1021/acs.oprd.8b00444
E2212 (1) (18.0 kg, 92.5% yield) as a white solid. Mother liquor 3 were recycled according to the procedure described below. FTIR (cm–1, KBr) 3461, 3173, 2956, 1734, 1584, 1536, 1476, 1309, 1130, 835, 765, 752; 1H NMR (600 MHz, DMSO-d6) δ 7.91 (s, 1H), 7.78 (d, J = 8.4 Hz, 1H), 7.77 (br d, J = 8.4 Hz, 1H), 7.61 (br dd, J = 7.8, 7.8 Hz, 1H), 7.49 (br dd, J = 7.8, 7.8 Hz, 1H), 7.46 (d, J= 15.6 Hz, 1H), 7.32 (d, J = 15.6 Hz, 1H), 7.27 (d, J = 7.8 Hz, 1H), 7.25 (br d, J = 7.8 Hz, 1H), 7.22 (s, 1H), 4.51 (dd, J = 9.0, 6.0 Hz, 1H), 4.29 (s, 3H), 4.28 (m, 2H), 3.98 (s, 3H), 2.29 (m, 1H), 2.14 (s, 3H), 2.16 (m, 2H), 1.96 (m, 1H); 13C NMR (150 MHz, DMSO-d6) δ 173.3, 159.3, 155.4, 155.0, 150.1, 141.1, 137.1, 136.9, 133.6, 132.9, 131.0, 130.5, 127.6, 127.1 (q, JC–F = 30 Hz), 125.8 (q, JC–F = 5.6 Hz), 124.7 (q, JC–F = 270 Hz), 122.2, 120.7, 117.2, 116.5, 72.3, 53.7, 47.0, 37.6, 30.7, 21.3, 13.6; HRMS (ESI+) calcd for C25H23F3N6O ([M + H]+) 481.1958, found 481.1953.




///////////E2212, E 2212
Certolizumab pegol, セルトリズマブペゴル (遺伝子組換え)

>Amino acid sequence of the light chain DIQMTQSPSSLSASVGDRVTITCKASQNVGTNVAWYQQKPGKAPKALIYSASFLYSGVPY RFSGSGSGTDFTLTISSLQPEDFATYYCQQYNIYPLTFGQGTKVEIKRTVAAPSVFIFPP SDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLT LSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC
>Amino acid sequence of the heavy chain EVQLVESGGGLVQPGGSLRLSCAASGYVFTDYGMNWVRQAPGKGLEWMGWINTYIGEPIY ADSVKGRFTFSLDTSKSTAYLQMNSLRAEDTAVYYCARGYRSYAMDYWGQGTLVTVSSAS TKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGL YSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHTCAA
Certolizumab pegol
CAS: 428863-50-7
セルトリズマブペゴル (遺伝子組換え)
CDP 870 / CDP-870 / CDP870 / PHA-738144
| Formula |
C2115H3252N556O673S16
|
|---|---|
| Cas |
428863-50-7
|
| Mol weight |
47748.8128
|
Reducing signs and symptoms of Crohn’s disease and treatment of moderately to severely active rheumatoid arthritis (RA).
Certolizumab pegol is a recombinant Fab’ antibody fragment against tumor necrosis factor alpha which is conjugated to an approximately 40kDa polyethylene glycol (PEG2MAL40K). Polyethylene glycol helps to delay the metabolism and elimination of the drugs. Chemically, the light chain is made up of 214 amino acid residues while the heavy chain is composed of 229 amino acid residues. The molecular mass of the Fab’ antibody fragment itself is 47.8 kDa. It is used for the treatment of rheumatoid arthritis and Crohn’s disease. FDA approved on April 22, 2008
Certolizumab pegol (CDP870, tradename Cimzia) is a biologic medication for the treatment of Crohn’s disease,[1][2] rheumatoid arthritis, psoriatic arthritis and ankylosing spondylitis. It is a fragment of a monoclonal antibody specific to tumor necrosis factor alpha(TNF-α) and is manufactured by UCB.[3][4][5]
Medical uses
- Crohn’s Disease
- On April 22, 2008, the U.S. FDA approved Cimzia for the treatment of Crohn’s disease in people who did not respond sufficiently or adequately to standard therapy.[4][6][7]
- Rheumatoid arthritis
- On June 26, 2009, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) issued a positive opinion recommending that the European Commission grant a marketing authorisation for Cimzia for the treatment of rheumatoid arthritis only – the CHMP refused approval for the treatment of Crohn’s disease. The marketing authorisation was granted to UCB Pharma SA on October 1, 2009.[8]
- Psoriatic arthritis
- On September 27, 2013, the U.S. FDA approved Cimzia for the treatment of adult patients with active psoriatic arthritis.[9]
Method of action
Certolizumab pegol is a monoclonal antibody directed against tumor necrosis factor alpha. More precisely, it is a PEGylated Fabfragment of a humanized TNF inhibitor monoclonal antibody.[10]
Clinical trials
- Crohn’s disease
- Positive results have been demonstrated in two phase III trials (PRECiSE 1 and 2) of certolizumab pegol versus placebo in moderate to severe active Crohn’s disease.[1][10][11][12]
- Axial spondyloarthritis
- In 2013, a phase 3 double blind randomized placebo-controlled study found significantly positive results in patient self-reported questionnaires, with rapid improvement of function and pain reduction, in patients with axial spondyloarthritis.[13]
- Rheumatoid arthritis
- Certolizumab appears beneficial in those with rheumatoid arthritis.[14]
Side effects
Significant side effects occur in 2% of people who take the medication.[14]
References
- ^ Jump up to:a b Sandborn WJ, Feagan BG, Stoinov S, et al. (July 2007). “Certolizumab pegol for the treatment of Crohn’s disease”. N. Engl. J. Med. 357 (3): 228–38. doi:10.1056/NEJMoa067594. PMC 3187683. PMID 17634458.
- ^ Goel, Niti; Sue Stephens (2010). “Certolizumab pegol”. mAbs. 2 (2): 137–147. doi:10.4161/mabs.2.2.11271. PMC 2840232. PMID 20190560.
- ^ Kaushik VV, Moots RJ (April 2005). “CDP-870 (certolizumab) in rheumatoid arthritis”. Expert Opinion on Biological Therapy. 5 (4): 601–6. doi:10.1517/14712598.5.4.601. PMID 15934837.
- ^ Jump up to:a b index.cfm?fuseaction=Search.Label_ApprovalHistory “Cimzia Label and Approval History” Check
|url=value (help). Drugs@FDA. U.S. Food and Drug Administration(FDA). Retrieved 2009-11-15. - ^ “Cimzia Prescribing Information” (PDF). US Food and Drug Administration (FDA). April 2016. Retrieved 2016-08-21.
- ^ UCB press release – Cimzia Approved in the US for the Treatment of Moderate to Severe Crohn’s Disease. Retrieved April 22, 2008.
- ^ Waknine, Yael (May 1, 2008). “FDA Approvals: Patanase, Actonel, Cimzia”. Medscape. Retrieved 2008-05-01.
- ^ “Cimzia European Public Assessment Report”. European Medicines Agency. Retrieved November 15, 2009.
- ^ “Cimzia (certolizumab pegol) approved by the U.S. FDA for treatment of adult patients with active psoriatic arthritis”. Archived from the original on October 1, 2013. Retrieved October 1, 2013.
- ^ Jump up to:a b Schreiber S. et al., Certolizumab pegol, a humanised anti-TNF pegylated FAb’ fragment, is safe and effective in the maintenance of response and remission following induction in active Crohn’s disease: a phase 3 study (precise), Gut, 2005, 54, suppl7, A82
- ^ Sandborn et al., Certolizumab pegol administered subcutaneously is effective and well tolerated in patients with active Crohn’s disease: results from a 26-week, placebo-controlled Phase 3 study (PRECiSE 1), Gastroenterology, 2006, 130, A107
- ^ “New Analysis Shows Cimzia (Certolizumab Pegol) Maintained Remission and Response in Recent Onset Crohn’s Disease” (Press release). UCB. October 23, 2006. Retrieved 2009-11-15.
- ^ Sieper J, Tubergen A, Coteur G, Woltering F, Landewe R (May 2013). “PMS50 – Rapid Improvements In Patient-Reported Outcomes With Certolizumab Pegol In Patients With Axial Spondyloarthritis, Including Ankylosing Spondylitis And Non-Radiographic Axial Spondyloarthritis: 24-Week Results Of A Phase 3 Double Blind Randomized Placebo-Controlled Study”. Value in Health. 16 (3): A227. doi:10.1016/j.jval.2013.03.1150.
- ^ Jump up to:a b Ruiz Garcia, V; Jobanputra, P; Burls, A; Vela Casasempere, P; Bort-Marti, S; Bernal, JA (Sep 8, 2017). “Certolizumab pegol (CDP870) for rheumatoid arthritis in adults”(PDF). The Cochrane Database of Systematic Reviews. 9: CD007649. doi:10.1002/14651858.CD007649.pub4. PMID 28884785.
External links
- certolizumab+pegol at the US National Library of Medicine Medical Subject Headings (MeSH)
- Cimzia Website
FDA approves treatment Cimzia (certolizumab pegol) for patients with a type of inflammatory arthritis
March 28, 2019
Release
The U.S. Food and Drug Administration today approved Cimzia (certolizumab pegol) injection for treatment of adults with a certain type of inflammatory arthritis called non-radiographic axial spondyloarthritis (nr-axSpA), with objective signs of inflammation. This is the first time that the FDA has approved a treatment for nr-axSpA.
“Today’s approval of Cimzia fulfills an unmet need for patients suffering from non-radiographic axial spondyloarthritis as there has been no FDA-approved treatments until now,” said Nikolay Nikolov, M.D., associate director for rheumatology of the Division of Pulmonary, Allergy, and Rheumatology Products in the FDA’s Center for Drug Evaluation and Research.
Nr-axSpA is a type of inflammatory arthritis that causes inflammation in the spine and other symptoms. There is no visible damage seen on x-rays, so it is referred to as non-radiographic.
The efficacy of Cimzia for the treatment of nr-axSpA was studied in a randomized clinical trial in 317 adult patients with nr-axSpA with objective signs of inflammation, indicated by elevated C-reactive protein (CRP) levels and/or sacroiliitis (inflammation of the sacroiliac joints) on MRI. The trial measured the improvement response on the Ankylosing Spondylitis Disease Activity Score, a composite scoring system that assesses disease activity including patient-reported outcomes and CRP levels. Responses were greater for patients treated with Cimzia compared to patients treated with placebo. The overall safety profile observed in the Cimzia treatment group was consistent with the known safety profile of Cimzia.
The prescribing information for Cimzia includes a Boxed Warning to advise health care professionals and patients about the increased risk of serious infections leading to hospitalization or death including tuberculosis (TB), bacterial sepsis (infection in the blood steam), invasive fungal infections (such as histoplasmosis, an infection that affects the lungs), and other infections. Cimzia should be discontinued if a patient develops a serious infection or sepsis. Health care providers are advised to perform testing for latent TB and, if positive, to start treatment for TB prior to starting Cimzia. All patients should be monitored for active TB during treatment, even if the initial latent TB test is negative. The Boxed Warning also advises that lymphoma (cancer in blood cells) and other malignancies, some fatal, have been reported in children and adolescent patients treated with tumor necrosis factor (TNF) blockers, of which Cimzia is a member. Cimzia is not indicated for use in pediatric patients. Cimzia must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.
Cimzia was originally approved in 2008 and is also indicated for adult patients with Crohn’s disease, moderate-to-severe rheumatoid arthritis, active ankylosing spondylitis (AS) and moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy.
The FDA granted the approval of Cimzia to UCB.

Syringe with 200mg Certolizumab pegol
|
|
| Monoclonal antibody | |
|---|---|
| Type | Fab’ fragment |
| Source | Humanized (from mouse) |
| Target | TNF alpha |
| Clinical data | |
| Trade names | Cimzia |
| AHFS/Drugs.com | Consumer Drug Information |
| MedlinePlus | a608041 |
| License data | |
| Pregnancy category |
|
| Routes of administration |
Subcutaneous |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Elimination half-life | about 11 days |
| Excretion | Renal (PEG only) |
| Identifiers | |
| CAS Number | |
| ChemSpider |
|
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C2115H3252N556O673S16 |
| Molar mass | 47,750 g/mol g·mol−1 |
///////////////FDA 2019, Cimzia, certolizumab pegol, inflammatory arthritis, UCB
FDA approves treatment Cimzia (certolizumab pegol) for patients with a type of inflammatory arthritis
FDA approves treatment Cimzia (certolizumab pegol) for patients with a type of inflammatory arthritis
March 28, 2019
Release
The U.S. Food and Drug Administration today approved Cimzia (certolizumab pegol) injection for treatment of adults with a certain type of inflammatory arthritis called non-radiographic axial spondyloarthritis (nr-axSpA), with objective signs of inflammation. This is the first time that the FDA has approved a treatment for nr-axSpA.
“Today’s approval of Cimzia fulfills an unmet need for patients suffering from non-radiographic axial spondyloarthritis as there has been no FDA-approved treatments until now,” said Nikolay Nikolov, M.D., associate director for rheumatology of the Division of Pulmonary, Allergy, and Rheumatology Products in the FDA’s Center for Drug Evaluation and Research.
Nr-axSpA is a type of inflammatory arthritis that causes inflammation in the spine and other symptoms. There is no visible damage seen on x-rays, so it is referred to as non-radiographic.
The efficacy of Cimzia for the treatment of nr-axSpA was studied in a randomized clinical trial in 317 adult patients with nr-axSpA with objective signs of inflammation, indicated by elevated C-reactive protein (CRP) levels and/or sacroiliitis (inflammation of the sacroiliac joints) on MRI. The trial measured the improvement response on the Ankylosing Spondylitis Disease Activity Score, a composite scoring system that assesses disease activity including patient-reported outcomes and CRP levels. Responses were greater for patients treated with Cimzia compared to patients treated with placebo. The overall safety profile observed in the Cimzia treatment group was consistent with the known safety profile of Cimzia.
The prescribing information for Cimzia includes a Boxed Warning to advise health care professionals and patients about the increased risk of serious infections leading to hospitalization or death including tuberculosis (TB), bacterial sepsis (infection in the blood steam), invasive fungal infections (such as histoplasmosis, an infection that affects the lungs), and other infections. Cimzia should be discontinued if a patient develops a serious infection or sepsis. Health care providers are advised to perform testing for latent TB and, if positive, to start treatment for TB prior to starting Cimzia. All patients should be monitored for active TB during treatment, even if the initial latent TB test is negative. The Boxed Warning also advises that lymphoma (cancer in blood cells) and other malignancies, some fatal, have been reported in children and adolescent patients treated with tumor necrosis factor (TNF) blockers, of which Cimzia is a member. Cimzia is not indicated for use in pediatric patients. Cimzia must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.
Cimzia was originally approved in 2008 and is also indicated for adult patients with Crohn’s disease, moderate-to-severe rheumatoid arthritis, active ankylosing spondylitis (AS) and moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy.
The FDA granted the approval of Cimzia to UCB.
///////////////FDA 2019, Cimzia, certolizumab pegol, inflammatory arthritis, UCB
FDA approves new oral treatment for multiple sclerosis, Mavenclad (cladribine)
March 29, 2019
Release
The U.S. Food and Drug Administration today approved Mavenclad (cladribine) tablets to treat relapsing forms of multiple sclerosis (MS) in adults, to include relapsing-remitting disease and active secondary progressive disease. Mavenclad is not recommended for MS patients with clinically isolated syndrome. Because of its safety profile, the use of Mavenclad is generally recommended for patients who have had an inadequate response to, or are unable to tolerate, an alternate drug indicated for the treatment of MS.
“We are committed to supporting the development of safe and effective treatments for patients with multiple sclerosis,” said Billy Dunn, M.D., director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research. “The approval of Mavenclad represents an additional option for patients who have tried another treatment without success.”
MS is a chronic, inflammatory, autoimmune disease of the central nervous system that disrupts communications between the brain and other parts of the body. Most people experience their first symptoms of MS between the ages of 20 and 40. MS is among the most common causes of neurological disability in young adults and occurs more frequently in women than in men.
For most people, MS starts with a relapsing-remitting course, in which episodes of worsening function (relapses) are followed by recovery periods (remissions). These remissions may not be complete and may leave patients with some degree of residual disability. Many, but not all, patients with MS experience some degree of persistent disability that gradually worsens over time. In some patients, disability may progress independent of relapses, a process termed secondary progressive multiple sclerosis (SPMS). In the first few years of this process, many patients continue to experience relapses, a phase of the disease described as active SPMS. Active SPMS is one of the relapsing forms of MS, and drugs approved for the treatment of relapsing forms of MS can be used to treat active SPMS.
The efficacy of Mavenclad was shown in a clinical trial in 1,326 patients with relapsing forms of MS who had least one relapse in the previous 12 months. Mavenclad significantly decreased the number of relapses experienced by these patients compared to placebo. Mavenclad also reduced the progression of disability compared to placebo.
Mavenclad must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks. Mavenclad has a Boxed Warning for an increased risk of malignancy and fetal harm. Mavenclad is not to be used in patients with current malignancy. In patients with prior malignancy or with increased risk of malignancy, health care professionals should evaluate the benefits and risks of the use of Mavenclad on an individual patient basis. Health care professionals should follow standard cancer screening guidelines in patients treated with Mavenclad. The drug should not be used in pregnant women and in women and men of reproductive potential who do not plan to use effective contraception during treatment and for six months after the course of therapy because of the potential for fetal harm. Mavenclad should be stopped if the patient becomes pregnant.
Other warnings include the risk of decreased lymphocyte (white blood cell) counts; lymphocyte counts should be monitored before, during and after treatment. Mavenclad may increase the risk of infections; health care professionals should screen patients for infections and treatment with Mavenclad should be delayed if necessary. Mavenclad may cause hematologic toxicity and bone marrow suppression so health care professionals should measure a patient’s complete blood counts before, during and after therapy. The drug has been associated with graft-versus-host-disease following blood transfusions with non-irradiated blood. Mavenclad may cause liver injury and treatment should be interrupted or discontinued, as appropriate, if clinically significant liver injury is suspected.
The most common adverse reactions reported by patients receiving Mavenclad in the clinical trials include upper respiratory tract infections, headache and decreased lymphocyte counts.
The FDA granted approval of Mavenclad to EMD Serono, Inc.
Cevimeline, セビメリン
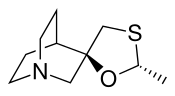
Cevimeline
セビメリン
- Molecular FormulaC10H17NOS
- Average mass199.313 Da
Cevimeline hydrochloride
-
- Synonyms:AF-102B, SNI-2011, SNK-508, Evoxac
- ATC:N07
- Use:cognition disorder, treatment of Sjogren’s syndrome, muscarinic M3-receptor agonist
- Chemical name:(2′R,3R)-rel-2′-methylspiro[1-azabicyclo[2.2.2]octane-3,5′-[1,3]oxathiolane] hydrochloride hydrate (2:2:1)
- Formula:C10H17NOS • HCl • 1/2H2O
- MW:489.57 g/mol
- CAS-RN:153504-70-2
- InChI Key:SURWTGAXEIEOGY-GHXDPTCOSA-N
- InChI:InChI=1S/C10H17NOS.ClH/c1-8-12-10(7-13-8)6-11-4-2-9(10)3-5-11;/h8-9H,2-7H2,1H3;1H/t8-,10-;/m1./s1
Derivatives
base
- Formula:C10H17NOS
- MW:199.32 g/mol
- CAS-RN:107233-08-9
anhydrous hydrochloride
- Formula:C10H17NOS • HCl
- MW:235.78 g/mol
- CAS-RN:107220-28-0
Cevimeline is cis-2′-methylspiro {1-azabicyclo [2.2.2] octane-3, 5′ -[1,3] oxathiolane} hydro-chloride, hydrate (2:1). Its empirical formula is C10H17NOS•HCl•½ H2O, and its structural formula is:
 Cevimeline has a molecular weight of 244.79. It is a white to off white crystalline powder with a melting point range of 201 to 203°C. It is freely soluble in alcohol and chloroform, very soluble in water, and virtually insoluble in ether. The pH of a 1% solution ranges from 4.6 to 5.6. Inactive ingredients include lactose monohydrate, hydroxypropyl cellulose, and magnesium stearate.
Cevimeline has a molecular weight of 244.79. It is a white to off white crystalline powder with a melting point range of 201 to 203°C. It is freely soluble in alcohol and chloroform, very soluble in water, and virtually insoluble in ether. The pH of a 1% solution ranges from 4.6 to 5.6. Inactive ingredients include lactose monohydrate, hydroxypropyl cellulose, and magnesium stearate.
Cevimeline hydrochloride [USAN]
RN: 153504-70-2
(+-)-cis-2-Methylspiro(1,3-oxathiolane-5,3′-quinuclidine) hydrochloride, hemihydrate
Cevimeline (trade name Evoxac) is a parasympathomimetic and muscarinic agonist,[1] with particular effect on M1 and M3 receptors. It is used in the treatment of dry mouth and especially associated with Sjögren’s syndrome.
Mechanism of action
By activating the M3 receptors of the parasympathetic nervous system, cevimeline stimulates secretion by the salivary glands, thereby alleviating dry mouth.
Side effects
Known side effects include nausea, vomiting, diarrhea, excessive sweating, rash, headache, runny nose, cough, drowsiness, hot flashes, blurred vision, and difficulty sleeping.[2]
Contraindications include asthma and angle closure glaucoma.
Clip
https://www.sciencedirect.com/science/article/abs/pii/S0731708515302260



CLIP
https://www.sciencedirect.com/science/article/pii/S0040403913005042



CLIP
CLIP

- Reaction of quinuclidin-3-one (I) with trimethylsulfoxonium iodide and NaH in DMSO gives epoxide (II), which is opened with SH2 in NaOH/water, yielding 3-hydroxy-3-(sulfanylmethyl)quinuclidine (III). The cyclization of compound (III) with acetaldehyde (IV) catalyzed by boron trifluoride ethearate or by SnCl4, POCl3, H3PO4 or p-toluenesulfonic acid affords a mixture of two diastereomeric spiroracemates, the (?-trans (V) and (?-cis (cevimeline). This mixture is separated by fractional recrystallization in acetone or by TLC chromatography, and treated with hydrochloric acid. The (?-trans-compound (V) can be isomerized to cevimeline by treatment with an acidic catalyst such as an organic sulfonic acid (trifluoromethanesulfonic acid, p-toluenesulfonic acid or methanesulfonic acid), a Lewis acid (SnCl4, FeCl3, BF3 or AlCl3) or sulfuric acid in refluxing toluene, hexane or CHCl3. Cevimeline hydrochloride hemihydrate is obtained from the above mentioned hydrochloride by a complex work-up using water, isopropanol and n-hexane.
- Synthesis of Cevimeline Hydrochloride (EN:134916): Reaction of quinuclidin-3-one (I) with trimethylsulfoxonium iodide and NaH in DMSO gives epoxide (II), which is opened with SH2 in NaOH/water, yielding 3-hydroxy-3-(sulfanylmethyl)quinuclidine (III) (1,2). The cyclization of compound (III) with acetaldehyde (IV) catalyzed by boron trifluoride ethearate (1) or by SnCl4, POCl3, H3PO4 or p-toluenesulfonic acid (2) affords a mixture of two diastereomeric spiro-racemates, the (?-trans (V) and (?-cis (cevimeline). This mixture is separated by fractional recrystallization in acetone or by TLC chromatography, and treated with hydrochloric acid (1,2). The (?-trans-compound (V) can be isomerized to cevimeline by treatment with an acidic catalyst such as an organic sulfonic acid (trifluoromethanesulfonic acid, p-toluenesulfonic acid or methanesulfonic acid), a Lewis acid (SnCl4, FeCl3, BF3 or AlCl3) or sulfuric acid in refluxing toluene, hexane or CHCl3 (2,3). Cevimeline hydrochloride hemihydrate is obtained from the above mentioned hydrochloride by a complex work-up using water, isopropanol and n-hexane (4).(Scheme 13491601a) Description M.p. 203 C (4). Sources Discovered by Israel Institute for Biological Research, Ness-Ziona (IL) and licensed to Snow Brand Milk Products Co. Ltd. (JP). In the U.S., comarketed by Snow Brand Milk Products and Daiichi Pharmaceutical Co., Ltd. In Japan, codeveloped with Nippon Kayaku Co. Ltd. Ishihara Sangyo Co., Ltd. (JP) is the bulk supplier. References 1. Fisher, A., Heldman, E., Grunfeld, Y., Karton, I., Levy, A. (Israel Institute for Biological Research); Derivs. of quinuclidine; EP 0205247, JP 1986280497, US 4855290. 2. Hayashi, K., Tokumoto, S., Yoshizawa, H., Isogai, T. (Ishihara Sangyo Kaisha, Ltd.); Method for producing 2-methylspiro(1,3-oxathiolan-5,3′)quinuclidine; EP 0683168, US 5571918. 3. Haga, T., Koyanagi, T., Hara, K., Maeda, M., Shigehara, I. (Ishihara Sangyo Kaisha, Ltd.); Method for isomerization of trans-form 2-methylspiro(1,3-oxathiolane-5,3′)quinuclidine or acid addition salts thereof; EP 0298491, US 4861886. 4. Saito, K., Ono, T., Honda, N. (Snow Brand Milk Products Co., Ltd.); Preparation method of cis-2-methylspiro(1,3-oxathiolane-5,3′)quinuclidine hydrochloride.1/2 hydrate capable of disgregating easily; JP 1992108792.
PATENT
https://patents.google.com/patent/US8080663B2/en
The present invention refers to a novel, industrially advantageous process for the preparation of an intermediate useful for the preparation of Cevimeline hydrochloride (1, cis-2-methylspiro(1,3-oxathiolane-5,3′)quiniclidine, Scheme 1). This pharmaceutical is useful for the treatment of diseases of the central nervous system due to disturbances of central cholinergic function and autoimmune system (Sjörgen’s syndrome) and is marketed as Evoxac®.
U.S. Pat. No. 4,855,290 describes a process for preparation of 2-methylspiro(1,3-oxathiolane-5,3′)quiniclidine (1). The process comprises the preparation of the epoxide of 3-methylenequiniclidine, which is subsequently reacted with hydrogen sulfide to produce 3-hydroxy-3-mercaptomethylquiniclidine and condensed with acetaldehyde in the presence of a Lewis acid (boron trifluoride etherate) to provide 2-methylspiro(1,3-oxathiolane-5,3′)quiniclidine. This process is depicted in Scheme I.

This process suffers from major disadvantages when transiting to industrial scale. These include the use of the highly hazardous and difficult to handle hydrogen sulfide gas. Also, boron trifluoride etherate is employed during the condensation step with acetaldehyde. The boron trifluoride etherate reagent is an air and moisture sensitive Lewis acid which has to be used under anhydrous conditions, thus creating a serious disadvantage in industrial settings. Another drawback of this process is the use of sodium hydride. U.S. Pat. Nos. 5,571,918 and 4,861,886 relate to the isomerization of the trans- to cis-form of 2-methylspiro(1,3-oxathiolane-5,3′)quiniclidine but do not describe methods for its preparation. Thus, an industrially acceptable and cost-effective method for the preparation of Cevimeline hydrochloride which overcomes the deficiencies of the prior art is required.
Further and other objects of the invention will be realized by those skilled in the art from the following Summary of the Invention and Detailed Description of Preferred Embodiments of the Invention thereof.
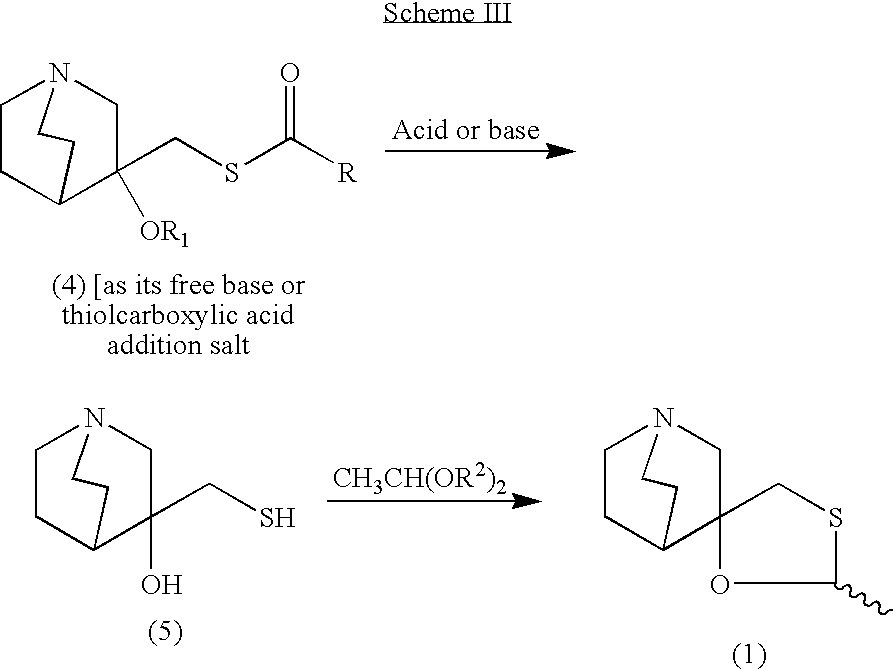
According to one aspect of the invention, a novel process is provided for the preparation of 2-methylspiro(1,3-oxathiolane-5,3′)quiniclidine (1). The process is industrially practical, efficient, safe and economical, as well as being environmentally friendly. The general method is shown in the Scheme II.

wherein R is selected from C1 to C6 alkyl and aryl groups, most preferably a methyl, ethyl or propyl group; R1 is hydrogen or a C2 to C7 alkyl or aryl carbonyl group; R2 is a C1 to C6 alkyl group, preferably methyl, ethyl, propyl, or butyl group.
EXAMPLE I Preparation of the Epoxide of 3-methylenequiniclidine (3)
A mixture of the hydrochloric salt of 3-quiniclidinone (2, 120 g, 795.7 mmol) and trimethylsulfoxonium iodide (219 g, 993.3 mmol) in dimethylsulfoxide (91.0 g, 0.63 mol) was cooled to 0-5° C. in an ice/water bath under nitrogen atmosphere. A solution of potassium tert-butoxide (201 g, 1789.1 mmol) in dimethylsulfoxide (500 mL) was added dropwise over 45 minutes. The mixture was warmed gradually to room temperature and stirred for an additional 16 hours at room temperature. After cooling to 0-5° C. (ice/water bath) the mixture was poured into an ice/water mixture (500 g) and then sodium chloride (300 g) was added. The mixture was stirred for 30 minutes and extracted with toluene (3×400 mL). The toluene phase was dried over sodium sulfate, filtered and evaporated to furnish the epoxide of 3-methylenequiniclidine (60 g, 431.7 mmol, 54% yield) as a yellow oil. The product could be used in the next step neat or as toluene solution after the extraction without further purification.
1H NMR (400 MHz, CDCl3): δ=3.10 (d, 1H, J=14.6 Hz); 2.98-2.77 (m, 5H); 2.74 (d, 1H, J=4.8 Hz); 2.70 (d, 1H, J=4.8 Hz); 1.96-1.89 (m, 1H); 1.79-1.62 (m, 2H); 1.60-1.54 (m, 1H); 1.38-1.36 (m,1H).
LRMS (ES+): 140.0 (100, M+H+).
EXAMPLE II Preparation of the Thiolacetic Acid Salt of 3-hydroxy-3-acetoxymercaptomethylquiniclidine (4)
A solution of the epoxide of 3-methylenequiniclidine (3, 54 g, 388.5 mmol) in toluene (200 mL) was cooled to 0-5° C. (ice/water bath). Thiolacetic acid was added dropwise over 10-15 minutes. The mixture was stirred at 0-5° C. for 30 minutes and then allowed to come to room temperature. After stirring at room temperature for 2 hours the formed precipitate was filtered and washed with toluene (2×100 mL) to give the 3-hydroxy-3-acetoxymercaptomethylquiniclidine thiolacetic acid salt (4 wherein R1 is H and R is methyl, 77 g, 264.6 mmol, 68%) as a light yellow solid. The product was used in the next step without any further purification.
1H NMR (400 MHz CD3OD): δ=3.47 (d, 1H, J=14.1 Hz); 3.37-3.18 (m, 7H); 2.40 (s, 3H); 2.38 (s, 3H); 2.36-2.27 (m, 1H), 2.14-2.05 (m, 2H); 2.03-1.93 (m, 1H); 1.81-1.78 (m, 1H).
LRMS (ES+): 216.1 (100, M−[SCOCH3]−+H+).
EXAMPLE III Preparation of 2-methylspiro(1,3-oxathiolane-5,3′)quiniclidine using p-toluenesulfonic acid (1)
To a solution of 3-hydroxy-3-acetoxymercaptomethylquiniclidine thiolacetic acid salt (4 wherein R1 is H and R is methyl, 3 g, 10.3 mmol) in iso-propanol (50 mL) was added p-toluenesulfonic acid monohydrate (5.9 g, 30.9 mmol) and the mixture was heated to reflux for 3.5 hours. The mixture was cooled to room temperature and acetaldehyde diethyl acetal (6.1 g, 51.5 mmol) was added. The mixture was heated to reflux and stirred for an additional 3 hours. The solvent was evaporated and the residue was dissolved in dichloromethane (50 mL). The mixture was cooled to 0-5° C. and a 25% aqueous solution of sodium hydroxide (80 mL) was added. The mixture was stirred for 10-15 minutes and the phases were separated. The aqueous phase was extracted with dichloromethane (3×50 mL). The organic phases were combined and extracted with 5% aqueous solution of sulfuric acid (3×50 mL). The acidic aqueous phases were combined and the pH was adjusted to 12 with a 25% aqueous solution of sodium hydroxide. The aqueous phase was extracted with heptane (3×50 mL) and the organic phases were combined, dried over sodium sulfate and the solvent was evaporated to give 2-methylspiro(1,3-oxathiolane-5,3′)quiniclidine (1.8 g, 9.2 mmol, 89% yield) as a 3:1 cis/trans ratio mixture of diastereomers (determined by 1H NMR).
LRMS (ES+): 200.1 (100, M+H+).
EXAMPLE IV Preparation of 2-methylspiro(1,3-oxathiolane-5,3′)quiniclidine (1) using racemic camphorsulfonic acid
In a similar experiment as Example III, racemic camphorsulfonic acid (7.2 g, 30.9 mmol) was added to a solution of 3-hydroxy-3-acetoxymercaptomethylquiniclidine thiolacetic acid salt (4 wherein R1 is H and R is methyl, 3 g, 10.3 mmol) in iso-propanol (50 mL). The mixture was refluxed for 5 h, cooled to room temperature and acetaldehyde diethyl acetal (6.1 g, 51.5 mmol) was added. The mixture was refluxed for an additional an 8 hours and processed according to Example III to give 2-methylspiro(1,3-oxathiolane-5,3′)quiniclidine (1.32 g, 6.63 mmol, 64% yield) in a 3.5:1 cis/trans ratio mixture of diastereomers (determined by 1H NMR).
EXAMPLE V Preparation of 2-methylspiro(1,3-oxathiolane-5,3′)quiniclidine (1) using phenyl sulfonic acid
In a similar experiment as Example III, to a solution of 3-hydroxy-3-acetoxymercaptomethylquiniclidine thiolacetic acid salt (4 wherein R1 is H and R is methyl, 3 g, 10.3 mmol) in iso-propanol (50 mL) was added phenyl sulfonic acid (4.9 g, 30.9 mmol) and the mixture was refluxed 5 h, cooled to room temperature and acetaldehyde diethyl acetal (6.1 g, 51.5 mmol) was added. The mixture was refluxed for an additional 8 hours and worked up in a manner similar to Example III to furnish 2-methylspiro(1,3-oxathiolane-5,3′)quiniclidine (1.6 g, 8.2 mmol, 80% yield) as a 2.5:1 cis/trans ratio mixture of diastereomers (determined by 1H NMR).
EXAMPLE VI Preparation of 2-methylspiro(1,3-oxathiolane-5,3′)quiniclidine (1) using p-toluenesulfonic acid in butanol
To a solution of 3-hydroxy-3-acetoxymercaptomethylquiniclidine thiolacetic acid salt (4 wherein R1 is H and R is methyl, 3 g, 10.3 mmol) in butanol (100 mL) was added of p-toluenesulfonic acid monohydrate (5.9 g, 30.9 mmol) and the mixture was refluxed for 3 hours with a Dean-Stark apparatus attached to the flask. The reaction mixture was cooled to room temperature and acetaldehyde diethyl acetal (6.1 g, 51.5 mmol) was added. The mixture was heated to 80° C. for an additional 8 h and worked up according to Example III to afford 2-methylspiro(1,3-oxathiolane-5,3′)quiniclidine (1.8 g, 9.2 mmol, 89% yield) as a 3:1 cis/trans ratio mixture of diastereomers (determined by 1H NMR).
References
- ^ Ono M, Takamura E, Shinozaki K, et al. (July 2004). “Therapeutic effect of cevimeline on dry eye in patients with Sjögren’s syndrome: a randomized, double-blind clinical study”. Am. J. Ophthalmol. 138 (1): 6–17. doi:10.1016/j.ajo.2004.02.010. PMID 15234277.
- ^ [1] MedicineNet: Cevimeline. Accessed 10/12/2007
-
- US 4 855 290 (Israel Institute for Biological Research; 8.8.1989; IL-prior. 10.5.1985).
- US 4 876 260 (Israel Institute for Biological Research; 24.10.1989; USA-prior. 28.10.1987).
- EP 683 168 (Ishihara Sangyo Kaisha; appl. 19.5.1995; J-prior. 19.5.1994).
-
Method for isomerization of trans-isomer:
- US 4 861 886 (Ishihara Sangyo Kaisha; 29.8.1989; J-prior. 10.7.1987).
-
Method of separation:
- IL 81 652 (Israel Institute for Biological Research; 12.5.1991; appl. 23.2.1987).
- JP 01 290 680 (Ishihara Sangyo Kaisha; 22.11.1989; J-prior. 18.5.1988).
-
Synthesis of enantiomerically pure (S)-3-hydroxy-3-mercaptomethylquinuclidine (S)-II:
- Bos, M.; Canesso, R.: Heterocycles (HTCYAM) 38 (8), 1889 (1994).
-
Synthesis of 3-quinuclidone:
- Sternbach, L.H.; Kaiser, S.: J. Am. Chem. Soc. (JACSAT) 74, 2215 (1952).
-
External links
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Evoxac |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a608025 |
| Pregnancy category |
|
| Routes of administration |
By mouth (capsules) |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Protein binding | <20% |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C10H17NOS |
| Molar mass | 199.31308 g/mol g·mol−1 |
| 3D model (JSmol) | |
/////////// Cevimeline, AF-102B, SNI-2011, SNK-508, Evoxac, セビメリン
FDA approves new oral testosterone capsule for treatment of men with certain forms of hypogonadism
FDA approves new oral testosterone capsule (testosterone undecanoate) for treatment of men with certain forms of hypogonadism
March 27, 2019
Release
The U.S. Food and Drug Administration today approved Jatenzo (testosterone undecanoate), an oral testosterone capsule to treat men with certain forms of hypogonadism. These men have low testosterone levels due to specific medical conditions, such as genetic disorders like Klinefelter syndrome or tumors that have damaged the pituitary gland. Jatenzo should not be used to treat men with “age-related hypogonadism,” in which testosterone levels decline due to aging, even if these men have symptoms that appear to be related to low testosterone. Jatenzo’s benefits do not outweigh its risks for that use.
“Jatenzo’s oral route of administration provides an important addition to current treatment options available for men with certain hypogonadal conditions who up until now have most commonly been treated with testosterone products that are applied to the skin or injected,” said Hylton V. Joffe, M.D, M.M.Sc., director of the Division of Bone, Reproductive and Urologic Products in the FDA’s Center for Drug Evaluation and Research. “But it’s important to emphasize that this drug should not, like other testosterone treatments, be used to treat older men with ‘age-related hypogonadism.’ The benefits of testosterone therapy, including Jatenzo, have not been established for this use, and Jatenzo’s effects on raising blood pressure can increase the risks of heart attack, stroke and cardiovascular death in this population.”
The efficacy of Jatenzo was demonstrated in a four-month clinical trial involving 166 men with hypogonadism. Study participants initially were given Jatenzo at a dose of 237 mg twice per day, and the dose was adjusted downward or upward to a maximum of 396 mg twice per day on the basis of testosterone levels. Eighty-seven percent of Jatenzo-treated men achieved an average testosterone level within the normal range, which was the primary study endpoint.
Jatenzo contains a boxed warning on its labeling stating that the drug can cause blood pressure to rise, increasing the risk of heart attack, stroke and cardiovascular death. Health care providers should consider a patient’s individual heart disease risks and ensure that blood pressure is adequately controlled before prescribing Jatenzo; they should also periodically monitor patient blood pressure during treatment. Jatenzo is currently one of two testosterone products that have this boxed warning. The FDA is requiring all testosterone product manufacturers to conduct blood pressure postmarketing trials to more clearly address whether these products increase blood pressure.
Common side effects, occurring in more than 2 percent of patients in the Jatenzo clinical trial, included headache, an increase in hematocrit (red blood cell count), a decrease in high-density lipoprotein cholesterol (“good” cholesterol), high blood pressure and nausea. An increase in prostate specific antigen (PSA) was also observed. Patients should have their hematocrit, cholesterol and PSA monitored regularly to check for changes. Those with benign prostate hyperplasia should be monitored for worsening of symptoms.
The FDA granted the approval of Jatenzo to Clarus Therapeutics.
//////////FDA 2019, Jatenzo, Clarus Therapeutics, (testosterone undecanoate,
Batimastat, バチマスタット

| Formula |
C23H31N3O4S2
|
|---|---|
| cas |
130370-60-4
|
| Mol weight |
477.6399
|
Batimastat (INN/USAN, codenamed BB-94) is an anticancer drug that belongs to the family of drugs called angiogenesis inhibitors. It acts as a matrix metalloproteinase inhibitor (MMPI) by mimicking natural MMPI peptides.
Batimastat was the first MMPI that went into clinical trials. First results of a Phase I trial appeared in 1994. The drug reached Phase III but was never marketed; mainly because it couldn’t be administered orally (as opposed to the newer and chemically similar MMPI marimastat), and injection into the peritoneum caused peritonitis.[1]
SYN
U.S. Patent 5,453,438
U.S. Patent 5,240,958
U.S. Patent 5,530,161

SYN
US 5240958; US 5310763; WO 9005719
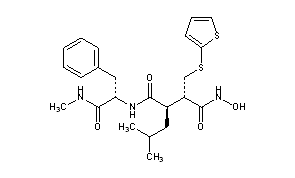
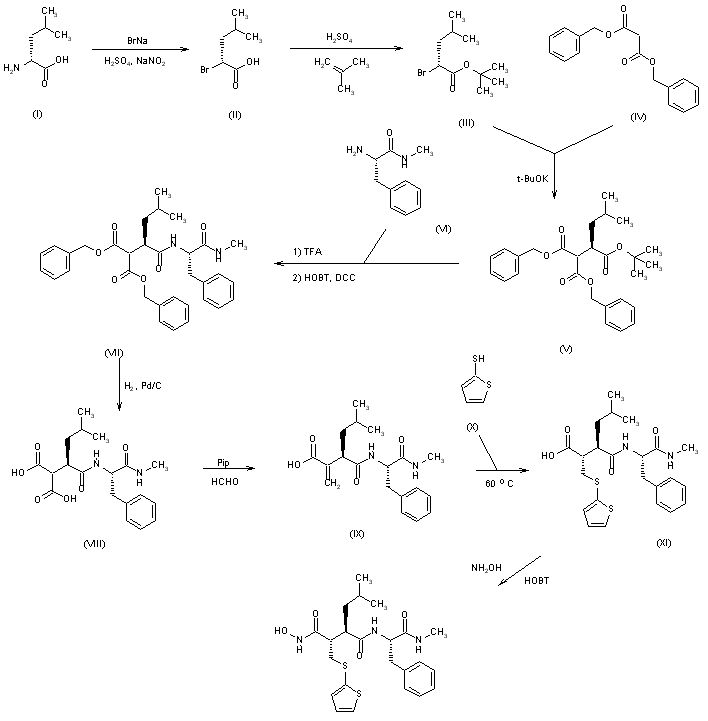
The treatment of D-leucine (I) with NaNO2, H2SO4 and NaBr gives 2(R)-bromo-5-methylpentanoic acid (II), which is esterified with isobutene and H2SO4 to the corresponding tert-butyl ester (III). The condensation of (III) with dibenzyl malonate (IV) by means of potassium tert-butoxide in DMF yields the malonyl derivative (V), which is treated with trifluoroacetic acid to hydrolyze the tert-butyl ester, and without isolation is condensed with L-phenylalanine methyl amide (VI) by means of hydroxybenzotriazole (HOBT) and dicyclohexylcarbodiimide (DCC), affording 4-benzyloxy-3-(benzyloxycarbonyl)-2(R)-isobutylsuccinyl-L-phenylalanine methylamide (VII). The elimination of the benzyl groups of (VII) by hydrogenolysis over Pd/C in ethanol gives the dicarboxylic acid (VIII), which by partial decarboxylation and reaction with aqueous formaldehyde and piperidine yields 4-hydroxy-2(R)-isobutyl-3-methylenesuccinyl-L-phenylalanine methylamide (IX). The addition of thiophene-2-thiol (X) to the double bond of (IX) affords 4-hydroxy-2(R)-isobutyl-3(S)-(2-thienylsulfanylmethyl)succinyl-L-phenylalanine methylamide (XI), which is finally treated with hydroxylamine and hydroxybenzotriazole in dichloromethane/DMF.
SPEC

HPLC

References
- ^ Rothenberg, M. L.; Nelson, A. R.; Hande, K. R. (1999). “New Drugs on the Horizon: Matrix Metalloproteinase Inhibitors”. Stem Cells. 17 (4): 237–240. doi:10.1002/stem.170237. PMID 10437989.
 |
|
| Clinical data | |
|---|---|
| Pregnancy category |
|
| Routes of administration |
Injection into pleural space or abdomen |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| ECHA InfoCard | 100.222.897 |
| Chemical and physical data | |
| Formula | C23H31N3O4S2 |
| Molar mass | 477.64 g/mol g·mol−1 |
| 3D model (JSmol) | |
//////////Batimastat, BB-94, バチマスタット ,
[H][C@@](CC1=CC=CC=C1)(NC(=O)[C@]([H])(CC(C)C)[C@]([H])(CSC1=CC=CS1)C(=O)NO)C(=O)NC
Abikoviromycin
- Molecular FormulaC10H11NO
- Average mass161.200 Da
Journal of Antibiotics (2003), 56, (9), 801-804.
“ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This is a compilation for educational purposes only. P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
READ
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on Facebook 

 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
Industry-Oriented Route Evaluation and Process Optimization for the Preparation of Brexpiprazole

Efforts toward route evaluation and process optimization for the preparation of brexpiprazole (1) are described. Starting from commercially available dihydroquinolinone 11, a three-step synthesis route composed of O-alkylation, oxidation, and N-alkylation was selected for industry-oriented process development aiming to reduce side reactions and achieve better impurity profiles. The reaction conditions of the three steps were investigated, and the control strategy for the process-related impurities was established. The optimized process was validated on the kilogram scale and now is viable for commercialization, with the results of not less than 99.90% purity of 1 (by HPLC) and not more than 0.05% of persistent impurities 15 and 16
Industry-Oriented Route Evaluation and Process Optimization for the Preparation of Brexpiprazole
, Yifeng Nian∥, Haji A. Aisa*† , and Jingshan Shen*§ 

ブレキサノロン , Brexanolone, Allopregnanolone

Brexanolone
318.501 g/mol, C21H34O2
CAS: 516-54-1
ブレキサノロン
The U.S. Food and Drug Administration today approved Zulresso (brexanolone) injection for intravenous (IV) use for the treatment of postpartum depression (PPD) in adult women. This is the first drug approved by the FDA specifically for PPD.
March 19, 2019
Release
The U.S. Food and Drug Administration today approved Zulresso (brexanolone) injection for intravenous (IV) use for the treatment of postpartum depression (PPD) in adult women. This is the first drug approved by the FDA specifically for PPD.
“Postpartum depression is a serious condition that, when severe, can be life-threatening. Women may experience thoughts about harming themselves or harming their child. Postpartum depression can also interfere with the maternal-infant bond. This approval marks the first time a drug has been specifically approved to treat postpartum depression, providing an important new treatment option,” said Tiffany Farchione, M.D., acting director of the Division of Psychiatry Products in the FDA’s Center for Drug Evaluation and Research. “Because of concerns about serious risks, including excessive sedation or sudden loss of consciousness during administration, Zulresso has been approved with a Risk Evaluation and Mitigation Strategy (REMS) and is only available to patients through a restricted distribution program at certified health care facilities where the health care provider can carefully monitor the patient.”
PPD is a major depressive episode that occurs following childbirth, although symptoms can start during pregnancy. As with other forms of depression, it is characterized by sadness and/or loss of interest in activities that one used to enjoy and a decreased ability to feel pleasure (anhedonia) and may present with symptoms such as cognitive impairment, feelings of worthlessness or guilt, or suicidal ideation.
Zulresso will be available only through a restricted program called the Zulresso REMS Program that requires the drug be administered by a health care provider in a certified health care facility. The REMS requires that patients be enrolled in the program prior to administration of the drug. Zulresso is administered as a continuous IV infusion over a total of 60 hours (2.5 days). Because of the risk of serious harm due to the sudden loss of consciousness, patients must be monitored for excessive sedation and sudden loss of consciousness and have continuous pulse oximetry monitoring (monitors oxygen levels in the blood). While receiving the infusion, patients must be accompanied during interactions with their child(ren). The need for these steps is addressed in a Boxed Warning in the drug’s prescribing information. Patients will be counseled on the risks of Zulresso treatment and instructed that they must be monitored for these effects at a health care facility for the entire 60 hours of infusion. Patients should not drive, operate machinery, or do other dangerous activities until feelings of sleepiness from the treatment have completely gone away.
The efficacy of Zulresso was shown in two clinical studies in participants who received a 60-hour continuous intravenous infusion of Zulresso or placebo and were then followed for four weeks. One study included patients with severe PPD and the other included patients with moderate PPD. The primary measure in the study was the mean change from baseline in depressive symptoms as measured by a depression rating scale. In both placebo controlled studies, Zulresso demonstrated superiority to placebo in improvement of depressive symptoms at the end of the first infusion. The improvement in depression was also observed at the end of the 30-day follow-up period.
The most common adverse reactions reported by patients treated with Zulresso in clinical trials include sleepiness, dry mouth, loss of consciousness and flushing. Health care providers should consider changing the therapeutic regimen, including discontinuing Zulresso in patients whose PPD becomes worse or who experience emergent suicidal thoughts and behaviors.
The FDA granted this application Priority Review and Breakthrough Therapydesignation.
Approval of Zulresso was granted to Sage Therapeutics, Inc.
Allopregnanolone, also known as 5α-pregnan-3α-ol-20-one or 3α,5α-tetrahydroprogesterone (3α,5α-THP), as well as brexanolone (USAN),[1] is an endogenous inhibitory pregnane neurosteroid[2] which has been approved by the FDA as a treatment for post-partum depression. It is synthesized from progesterone, and is a potent positive allosteric modulator of the action of γ-aminobutyric acid (GABA) at GABAA receptor.[2] Allopregnanolone has effects similar to those of other positive allosteric modulators of the GABA action at GABAA receptor such as the benzodiazepines, including anxiolytic, sedative, and anticonvulsant activity.[2][3][4] Endogenously produced allopregnanolone exerts a pivotal neurophysiological role by fine-tuning of GABAA receptor and modulating the action of several positive allosteric modulators and agonists at GABAA receptor.[5] The 21-hydroxylated derivative of this compound, tetrahydrodeoxycorticosterone (THDOC), is an endogenous inhibitory neurosteroid with similar properties to those of allopregnanolone, and the 3β-methyl analogue of allopregnanolone, ganaxolone, is under development to treat epilepsy and other conditions, including post-traumatic stress disorder (PTSD).[2]
Biochemistry
Biosynthesis
The biosynthesis of allopregnanolone in the brain starts with the conversion of progesterone into 5α-dihydroprogesterone by 5α-reductase type I. After that, 3α-hydroxysteroid dehydrogenase converts this intermediate into allopregnanolone.[2] Allopregnanolone in the brain is produced by cortical and hippocampus pyramidal neurons and pyramidal-like neurons of the basolateral amygdala.[6]
Biological activity
Allopregnanolone acts as a highly potent positive allosteric modulator of the GABAA receptor.[2] While allopregnanolone, like other inhibitory neurosteroids such as THDOC, positively modulates all GABAA receptor isoforms, those isoforms containing δ subunitsexhibit the greatest potentiation.[7] Allopregnanolone has also been found to act as a positive allosteric modulator of the GABAA-ρ receptor, though the implications of this action are unclear.[8][9] In addition to its actions on GABA receptors, allopregnanolone, like progesterone, is known to be a negative allosteric modulator of nACh receptors,[10] and also appears to act as a negative allosteric modulator of the 5-HT3 receptor.[11] Along with the other inhibitory neurosteroids, allopregnanolone appears to have little or no action at other ligand-gated ion channels, including the NMDA, AMPA, kainate, and glycine receptors.[12]
Unlike progesterone, allopregnanolone is inactive at the nuclear progesterone receptor (nPR).[12] However, allopregnanolone can be intracellularly oxidized into 5α-dihydroprogesterone, which is an agonist of the nPR, and thus/in accordance, allopregnanolone does appear to have indirect nPR-mediated progestogenic effects.[13] In addition, allopregnanolone has recently been found to be an agonist of the newly discovered membrane progesterone receptors (mPR), including mPRδ, mPRα, and mPRβ, with its activity at these receptors about a magnitude more potent than at the GABAA receptor.[14][15] The action of allopregnanolone at these receptors may be related, in part, to its neuroprotective and antigonadotropic properties.[14][16] Also like progesterone, recent evidence has shown that allopregnanolone is an activator of the pregnane X receptor.[12][17]
Similarly to many other GABAA receptor positive allosteric modulators, allopregnanolone has been found to act as an inhibitor of L-type voltage-gated calcium channels (L-VGCCs),[18] including α1 subtypes Cav1.2 and Cav1.3.[19] However, the threshold concentration of allopregnanolone to inhibit L-VGCCs was determined to be 3 μM (3,000 nM), which is far greater than the concentration of 5 nM that has been estimated to be naturally produced in the human brain.[19] Thus, inhibition of L-VGCCs is unlikely of any actual significance in the effects of endogenous allopregnanolone.[19] Also, allopregnanolone, along with several other neurosteroids, has been found to activate the G protein-coupled bile acid receptor (GPBAR1, or TGR5).[20] However, it is only able to do so at micromolar concentrations, which, similarly to the case of the L-VGCCs, are far greater than the low nanomolar concentrations of allopregnanolone estimated to be present in the brain.[20]
Biological function
Allopregnanolone possesses a wide variety of effects, including, in no particular order, antidepressant, anxiolytic, stress-reducing, rewarding,[21] prosocial,[22] antiaggressive,[23]prosexual,[22] sedative, pro-sleep,[24] cognitive, memory-impairment, analgesic,[25] anesthetic, anticonvulsant, neuroprotective, and neurogenic effects.[2] Fluctuations in the levels of allopregnanolone and the other neurosteroids seem to play an important role in the pathophysiology of mood, anxiety, premenstrual syndrome, catamenial epilepsy, and various other neuropsychiatric conditions.[26][27][28]
Increased levels of allopregnanolone can produce paradoxical effects, including negative mood, anxiety, irritability, and aggression.[29][30][31] This appears to be because allopregnanolone possesses biphasic, U-shaped actions at the GABAA receptor – moderate level increases (in the range of 1.5–2 nM/L total allopregnanolone, which are approximately equivalent to luteal phase levels) inhibit the activity of the receptor, while lower and higher concentration increases stimulate it.[29][30] This seems to be a common effect of many GABAA receptor positive allosteric modulators.[26][31] In accordance, acute administration of low doses of micronized progesterone (which reliably elevates allopregnanolone levels) has been found to have negative effects on mood, while higher doses have a neutral effect.[32]
During pregnancy, allopregnanolone and pregnanolone are involved in sedation and anesthesia of the fetus.[33][34]
Chemistry
Allopregnanolone is a pregnane (C21) steroid and is also known as 5α-pregnan-3α-ol-20-one, 3α-hydroxy-5α-pregnan-20-one, or 3α,5α-tetrahydroprogesterone (3α,5α-THP). It is very closely related structurally to 5-pregnenolone (pregn-5-en-3β-ol-20-dione), progesterone (pregn-4-ene-3,20-dione), the isomers of pregnanedione (5-dihydroprogesterone; 5-pregnane-3,20-dione), the isomers of 4-pregnenolone (3-dihydroprogesterone; pregn-4-en-3-ol-20-one), and the isomers of pregnanediol (5-pregnane-3,20-diol). In addition, allopregnanolone is one of four isomers of pregnanolone (3,5-tetrahydroprogesterone), with the other three isomers being pregnanolone (5β-pregnan-3α-ol-20-one), isopregnanolone(5α-pregnan-3β-ol-20-one), and epipregnanolone (5β-pregnan-3β-ol-20-one).
Derivatives
A variety of synthetic derivatives and analogues of allopregnanolone with similar activity and effects exist, including alfadolone (3α,21-dihydroxy-5α-pregnane-11,20-dione), alfaxolone (3α-hydroxy-5α-pregnane-11,20-dione), ganaxolone (3α-hydroxy-3β-methyl-5α-pregnan-20-one), hydroxydione (21-hydroxy-5β-pregnane-3,20-dione), minaxolone (11α-(dimethylamino)-2β-ethoxy-3α-hydroxy-5α-pregnan-20-one), Org 20599 (21-chloro-3α-hydroxy-2β-morpholin-4-yl-5β-pregnan-20-one), Org 21465 (2β-(2,2-dimethyl-4-morpholinyl)-3α-hydroxy-11,20-dioxo-5α-pregnan-21-yl methanesulfonate), and renanolone (3α-hydroxy-5β-pregnan-11,20-dione).
Research
Allopregnanolone and the other endogenous inhibitory neurosteroids have short terminal half-lives and poor oral bioavailability, and for these reason, have not been pursued for clinical use as oral therapies, although development as a parenteral therapy for multiple indications has been carried out. However, synthetic analogs with improved pharmacokineticprofiles have been synthesized and are being investigated as potential oral therapeutic agents.
In other studies of compounds related to allopregnanolone, exogenous progesterone, such as oral micronized progesterone (OMP), elevates allopregnanolone levels in the body with good dose-to-serum level correlations.[35] Due to this, it has been suggested that OMP could be described as a prodrug of sorts for allopregnanolone.[35] As a result, there has been some interest in using OMP to treat catamenial epilepsy,[36] as well as other menstrual cycle-related and neurosteroid-associated conditions. In addition to OMP, oral pregnenolonehas also been found to act as a prodrug of allopregnanolone,[37][38][39] though also of pregnenolone sulfate.[40]
Allopregnanolone has been under development by Sage Therapeutics as an intravenously administered drug for the treatment of super-refractory status epilepticus, postpartum depression, and essential tremor.[41] As of 19 March 2019 the FDA has approved allopregnanolone for postpartum depression.
References
- ^ “ChemIDplus – 516-54-1 – AURFZBICLPNKBZ-SYBPFIFISA-N – Brexanolone [USAN] – Similar structures search, synonyms, formulas, resource links, and other chemical information”. NIH Toxnet. Retrieved 26 December 2017.
- ^ Jump up to:a b c d e f g Reddy DS (2010). Neurosteroids: endogenous role in the human brain and therapeutic potentials. Prog. Brain Res. Progress in Brain Research. 186. pp. 113–37. doi:10.1016/B978-0-444-53630-3.00008-7. ISBN 9780444536303. PMC 3139029. PMID 21094889.
- ^ Reddy DS, Rogawski MA (2012). “Neurosteroids — Endogenous Regulators of Seizure Susceptibility and Role in the Treatment of Epilepsy”. Jasper’s Basic Mechanisms of the Epilepsies, 4th Edition: 984–1002. doi:10.1093/med/9780199746545.003.0077. ISBN 9780199746545.
- ^ T. G. Kokate, B. E. Svensson & M. A. Rogawski (September 1994). “Anticonvulsant activity of neurosteroids: correlation with γ-aminobutyric acid-evoked chloride current potentiation”. The Journal of Pharmacology and Experimental Therapeutics. 270 (3): 1223–1229. PMID 7932175.
- ^ Pinna, G; Uzunova, V; Matsumoto, K; Puia, G; Mienville, J. -M; Costa, E; Guidotti, A (2000-03-01). “Brain allopregnanolone regulates the potency of the GABAA receptor agonist muscimol”. Neuropharmacology. 39 (3): 440–448. doi:10.1016/S0028-3908(99)00149-5. PMID 10698010.
- ^ Agís-Balboa, Roberto C.; Pinna, Graziano; Zhubi, Adrian; Maloku, Ekrem; Veldic, Marin; Costa, Erminio; Guidotti, Alessandro (2006-09-26). “Characterization of brain neurons that express enzymes mediating neurosteroid biosynthesis”. Proceedings of the National Academy of Sciences. 103 (39): 14602–14607. doi:10.1073/pnas.0606544103. ISSN 0027-8424. PMC 1600006. PMID 16984997.
- ^ Mousavi Nik A, Pressly B, Singh V, Antrobus S, Hulsizer S, Rogawski MA, Wulff H, Pessah IN (2017). “Rapid Throughput Analysis of GABAA Receptor Subtype Modulators and Blockers Using DiSBAC1(3) Membrane Potential Red Dye”. Mol. Pharmacol. 92 (1): 88–99. doi:10.1124/mol.117.108563. PMC 5452057. PMID 28428226.
- ^ Morris KD, Moorefield CN, Amin J (October 1999). “Differential modulation of the gamma-aminobutyric acid type C receptor by neuroactive steroids”. Mol. Pharmacol. 56 (4): 752–9. PMID 10496958.
- ^ Li W, Jin X, Covey DF, Steinbach JH (October 2007). “Neuroactive steroids and human recombinant rho1 GABAC receptors”. J. Pharmacol. Exp. Ther. 323 (1): 236–47. doi:10.1124/jpet.107.127365. PMC 3905684. PMID 17636008.
- ^ Bullock AE, Clark AL, Grady SR, et al. (June 1997). “Neurosteroids modulate nicotinic receptor function in mouse striatal and thalamic synaptosomes”. J. Neurochem. 68 (6): 2412–23. doi:10.1046/j.1471-4159.1997.68062412.x. PMID 9166735.
- ^ Wetzel CH, Hermann B, Behl C, et al. (September 1998). “Functional antagonism of gonadal steroids at the 5-hydroxytryptamine type 3 receptor”. Mol. Endocrinol. 12 (9): 1441–51. doi:10.1210/mend.12.9.0163. PMID 9731711.
- ^ Jump up to:a b c Mellon SH (October 2007). “Neurosteroid regulation of central nervous system development”. Pharmacol. Ther. 116 (1): 107–24. doi:10.1016/j.pharmthera.2007.04.011. PMC 2386997. PMID 17651807.
- ^ Rupprecht R, Reul JM, Trapp T, et al. (September 1993). “Progesterone receptor-mediated effects of neuroactive steroids”. Neuron. 11 (3): 523–30. doi:10.1016/0896-6273(93)90156-l. PMID 8398145.
- ^ Jump up to:a b Thomas P, Pang Y (2012). “Membrane progesterone receptors: evidence for neuroprotective, neurosteroid signaling and neuroendocrine functions in neuronal cells”. Neuroendocrinology. 96 (2): 162–71. doi:10.1159/000339822. PMC 3489003. PMID 22687885.
- ^ Pang Y, Dong J, Thomas P (January 2013). “Characterization, neurosteroid binding and brain distribution of human membrane progesterone receptors δ and {epsilon} (mPRδ and mPR{epsilon}) and mPRδ involvement in neurosteroid inhibition of apoptosis”. Endocrinology. 154 (1): 283–95. doi:10.1210/en.2012-1772. PMC 3529379. PMID 23161870.
- ^ Sleiter N, Pang Y, Park C, et al. (August 2009). “Progesterone receptor A (PRA) and PRB-independent effects of progesterone on gonadotropin-releasing hormone release”. Endocrinology. 150 (8): 3833–44. doi:10.1210/en.2008-0774. PMC 2717864. PMID 19423765.
- ^ Lamba V, Yasuda K, Lamba JK, et al. (September 2004). “PXR (NR1I2): splice variants in human tissues, including brain, and identification of neurosteroids and nicotine as PXR activators”. Toxicol. Appl. Pharmacol. 199 (3): 251–65. doi:10.1016/j.taap.2003.12.027. PMID 15364541.
- ^ Hu AQ, Wang ZM, Lan DM, et al. (July 2007). “Inhibition of evoked glutamate release by neurosteroid allopregnanolone via inhibition of L-type calcium channels in rat medial prefrontal cortex”. Neuropsychopharmacology. 32 (7): 1477–89. doi:10.1038/sj.npp.1301261. PMID 17151597.
- ^ Jump up to:a b c Earl DE, Tietz EI (April 2011). “Inhibition of recombinant L-type voltage-gated calcium channels by positive allosteric modulators of GABAA receptors”. J. Pharmacol. Exp. Ther. 337 (1): 301–11. doi:10.1124/jpet.110.178244. PMC 3063747. PMID 21262851.
- ^ Jump up to:a b Keitel V, Görg B, Bidmon HJ, et al. (November 2010). “The bile acid receptor TGR5 (Gpbar-1) acts as a neurosteroid receptor in brain”. Glia. 58 (15): 1794–805. doi:10.1002/glia.21049. PMID 20665558.
- ^ Rougé-Pont F, Mayo W, Marinelli M, Gingras M, Le Moal M, Piazza PV (July 2002). “The neurosteroid allopregnanolone increases dopamine release and dopaminergic response to morphine in the rat nucleus accumbens”. Eur. J. Neurosci. 16 (1): 169–73. doi:10.1046/j.1460-9568.2002.02084.x. PMID 12153544.
- ^ Jump up to:a b Frye CA (December 2009). “Neurosteroids’ effects and mechanisms for social, cognitive, emotional, and physical functions”. Psychoneuroendocrinology. 34 Suppl 1: S143–61. doi:10.1016/j.psyneuen.2009.07.005. PMC 2898141. PMID 19656632.
- ^ Pinna G, Costa E, Guidotti A (February 2005). “Changes in brain testosterone and allopregnanolone biosynthesis elicit aggressive behavior”. Proc. Natl. Acad. Sci. U.S.A. 102 (6): 2135–40. doi:10.1073/pnas.0409643102. PMC 548579. PMID 15677716.
- ^ Terán-Pérez G, Arana-Lechuga Y, Esqueda-León E, Santana-Miranda R, Rojas-Zamorano JÁ, Velázquez Moctezuma J (October 2012). “Steroid hormones and sleep regulation”. Mini Rev Med Chem. 12 (11): 1040–8. doi:10.2174/138955712802762167. PMID 23092405.
- ^ Patte-Mensah C, Meyer L, Taleb O, Mensah-Nyagan AG (February 2014). “Potential role of allopregnanolone for a safe and effective therapy of neuropathic pain”. Prog. Neurobiol. 113: 70–8. doi:10.1016/j.pneurobio.2013.07.004. PMID 23948490.
- ^ Jump up to:a b Bäckström T, Andersson A, Andreé L, et al. (December 2003). “Pathogenesis in menstrual cycle-linked CNS disorders”. Ann. N. Y. Acad. Sci. 1007: 42–53. doi:10.1196/annals.1286.005. PMID 14993039.
- ^ Guille C, Spencer S, Cavus I, Epperson CN (July 2008). “The role of sex steroids in catamenial epilepsy and premenstrual dysphoric disorder: implications for diagnosis and treatment”. Epilepsy Behav. 13 (1): 12–24. doi:10.1016/j.yebeh.2008.02.004. PMC 4112568. PMID 18346939.
- ^ Finocchi C, Ferrari M (May 2011). “Female reproductive steroids and neuronal excitability”. Neurol. Sci. 32 Suppl 1: S31–5. doi:10.1007/s10072-011-0532-5. PMID 21533709.
- ^ Jump up to:a b Bäckström T, Haage D, Löfgren M, et al. (September 2011). “Paradoxical effects of GABA-A modulators may explain sex steroid induced negative mood symptoms in some persons”. Neuroscience. 191: 46–54. doi:10.1016/j.neuroscience.2011.03.061. PMID 21600269.
- ^ Jump up to:a b Andréen L, Nyberg S, Turkmen S, van Wingen G, Fernández G, Bäckström T (September 2009). “Sex steroid induced negative mood may be explained by the paradoxical effect mediated by GABAA modulators”. Psychoneuroendocrinology. 34 (8): 1121–32. doi:10.1016/j.psyneuen.2009.02.003. PMID 19272715.
- ^ Jump up to:a b Bäckström T, Bixo M, Johansson M, et al. (February 2014). “Allopregnanolone and mood disorders”. Prog. Neurobiol. 113: 88–94. doi:10.1016/j.pneurobio.2013.07.005. PMID 23978486.
- ^ Andréen L, Sundström-Poromaa I, Bixo M, Nyberg S, Bäckström T (August 2006). “Allopregnanolone concentration and mood–a bimodal association in postmenopausal women treated with oral progesterone”. Psychopharmacology. 187 (2): 209–21. doi:10.1007/s00213-006-0417-0. PMID 16724185.
- ^ Mellor DJ, Diesch TJ, Gunn AJ, Bennet L (2005). “The importance of ‘awareness’ for understanding fetal pain”. Brain Res. Brain Res. Rev. 49 (3): 455–71. doi:10.1016/j.brainresrev.2005.01.006. PMID 16269314.
- ^ Lagercrantz H, Changeux JP (2009). “The emergence of human consciousness: from fetal to neonatal life”. Pediatr. Res. 65 (3): 255–60. doi:10.1203/PDR.0b013e3181973b0d. PMID 19092726.
[…] the fetus is sedated by the low oxygen tension of the fetal blood and the neurosteroid anesthetics pregnanolone and the sleep-inducing prostaglandin D2 provided by the placenta (36).
- ^ Jump up to:a b Andréen L, Spigset O, Andersson A, Nyberg S, Bäckström T (June 2006). “Pharmacokinetics of progesterone and its metabolites allopregnanolone and pregnanolone after oral administration of low-dose progesterone”. Maturitas. 54 (3): 238–44. doi:10.1016/j.maturitas.2005.11.005. PMID 16406399.
- ^ Orrin Devinsky; Steven Schachter; Steven Pacia (1 January 2005). Complementary and Alternative Therapies for Epilepsy. Demos Medical Publishing. pp. 378–. ISBN 978-1-934559-08-6.
- ^ Saudan C, Desmarchelier A, Sottas PE, Mangin P, Saugy M (2005). “Urinary marker of oral pregnenolone administration”. Steroids. 70 (3): 179–83. doi:10.1016/j.steroids.2004.12.007. PMID 15763596.
- ^ Piper T, Schlug C, Mareck U, Schänzer W (2011). “Investigations on changes in ¹³C/¹²C ratios of endogenous urinary steroids after pregnenolone administration”. Drug Test Anal. 3(5): 283–90. doi:10.1002/dta.281. PMID 21538944.
- ^ Sripada RK, Marx CE, King AP, Rampton JC, Ho SS, Liberzon I (2013). “Allopregnanolone elevations following pregnenolone administration are associated with enhanced activation of emotion regulation neurocircuits”. Biol. Psychiatry. 73 (11): 1045–53. doi:10.1016/j.biopsych.2012.12.008. PMC 3648625. PMID 23348009.
- ^ Ducharme N, Banks WA, Morley JE, Robinson SM, Niehoff ML, Mattern C, Farr SA (2010). “Brain distribution and behavioral effects of progesterone and pregnenolone after intranasal or intravenous administration”. Eur. J. Pharmacol. 641 (2–3): 128–34. doi:10.1016/j.ejphar.2010.05.033. PMC 3008321. PMID 20570588.
- ^ “Brexanolone – Sage Therapeutics”. AdisInsight.
Further reading
- Herd, MB; Belelli, D; Lambert, JJ (2007). “Neurosteroid modulation of synaptic and extrasynaptic GABA(A) receptors”. Pharmacology & Therapeutics. 116 (1): 20–34. doi:10.1016/j.pharmthera.2007.03.007. PMID 17531325.
 |
|
 |
|
| Names | |
|---|---|
| IUPAC name
1-(3-Hydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl)ethanone
|
|
| Other names
ALLO; Allo; ALLOP; AlloP; Brexanolone; 5α-Pregnan-3α-ol-20-one; 3α-Hydroxy-5α-pregnan-20-one; 3α,5α-Tetrahydroprogesterone; 3α,5α-THP; Zulresso
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChEMBL | |
| ChemSpider | |
|
PubChemCID
|
|
| UNII | |
| Properties | |
| C21H34O2 | |
| Molar mass | 318.501 g·mol−1 |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
//////////Brexanolone, Priority Review, Breakthrough Therapy designation, Zulresso, Sage Therapeutics Inc, FDA 2019, ブレキサノロン , Brexanolone, Allopregnanolone
CC(=O)C1CCC2C1(CCC3C2CCC4C3(CCC(C4)O)C)C

{kind=link}