

The VNRX-7145 combination is now in Phase I studies to treat resistant urinary tract infections.
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 87)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
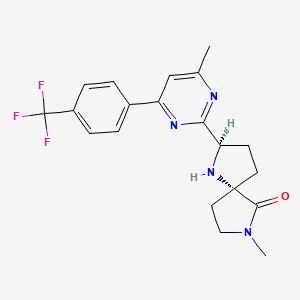
BIIB-095

BIIB-095
ROTATION (+)
1493790-64-9 CAS free form,
1493772-48-7 cas Hcl salt
cas 1493790-65-0, 1496563-32-6 ,SULPHATE ???
cas 1496563-31-5 SULFATE 1;1
cas 1496563-32-6 SULFATE HYDRATE 1;1;1
(2R,5S)-7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1 ,7-diazaspiro[4.4]nonan-6-one
1,7-Diazaspiro[4.4]nonan-6-one, 7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)phenyl]-2-pyrimidinyl]-, (2R,5S)-
C20 H21 F3 N4 O, 390.40
- Originator Biogen
- Class Analgesics
- Mechanism of Action Nav1.7 voltage-gated sodium channel inhibitors
- Phase I Neuropathic pain
- 29 Mar 2018 Phase-I clinical trials in Neuropathic pain (In volunteers) in United Kingdom (PO) (NCT03454126)
- 05 Mar 2018 Biogen plans a phase I trial for Pain, including Neuropathic pain (In volunteers) in USA (PO) (NCT03454126)
- 05 Mar 2018 Preclinical trials in Neuropathic Pain in USA (PO), before March 2018
In March 2018, a randomized, double blind, placebo controlled, single and multiple-ascending dose, dose-escalation phase I study ( NCT03454126; 255HV101; 2017-003982-90) was initiated in the UK in healthy subjects (expected n = 80) to evaluate the safety, tolerability and pharmacokinetics of BIIB-095. At that time, the trial was expected to complete in December 2018
Biogen is developing BIIB-095, a voltage-gated sodium channel 1.7 inhibitor, for the potential oral treatment of neuropathic pain [2027279], [2027426]. In March 2018, a phase I trial was initiated in healthy subjects
Biogen is developing oral agent BIIB-095 for the treatment of chronic pain, including neuropathic pain. A phase I clinical trial is under way in healthy volunteers.
The compound was first claimed in WO2013175205 , for treating schizophrenia, assigned to subsidiary Convergence Pharmaceuticals Limited , naming some of the inventors. This might present the structure of BIIB-095 , a voltage-gated sodium channel 1.7 inhibitor, being developed by Biogen for the oral treatment of neuropathic pain; in March 2018, a phase I trial was initiated in healthy subjects.
PATENT
WO2013175205

CONTD………………

INTERMEDIATE

WO 2013175206
US 20150119404
https://patents.google.com/patent/US20150119404
Patent
WO-2019067961
Novel salts (citrate, mesylate, hydrosulfate, saccharinate and oxalate) forms of 7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1,7-diazaspiro[4.4]nonan-6-one, processes for their preparation and compositions comprising them are claimed. Also claimed are their use for treating diseases and conditions mediated by modulation of voltage-gated sodium channels.
Voltage-gated sodium channels are responsible for the initial phase of the action potential, which is a wave of electrical depolarisation usually initiated at the soma of the neuron and propagated along the axon to the terminals. At the terminals, the action potential triggers the influx of calcium and the release of neurotransmitter. Drugs, such as lidocaine, that block voltage-gated sodium channels are used as local anaesthetics. Other sodium channel blockers, such as lamotrigine and carbamazepine are used to treat epilepsy. In the latter case, partial inhibition of voltage-gated sodium channels reduces neuronal excitability and reduces seizure propagation. In the case of local anaesthetics, regional block of sodium channels on sensory neurons prevents the conduction of painful stimuli. A key feature of these drugs is their state-dependent mechanism of action. The drugs are thought to stabilise an inactivated conformation of the channel that is adopted rapidly after the channel opens. This inactivated state provides a refractory period before the channel returns to its resting (closed) state ready to be reactivated. As a result, state-dependent sodium channel blockers inhibit the firing of neurons at high frequency, for example in response to painful stimuli, and will help to prevent repetitive firing during periods of prolonged neuronal depolarisation that might occur, for example, during a seizure. Action potentials triggered at lower frequencies, for example in the heart, will not be significantly affected by these drugs, although the safety margin differs in each case, since at high enough concentrations each of these drugs is capable of blocking the resting or open states of the channels.
The voltage-gated sodium channel family is made up of 9 subtypes, four of which are found in the brain, NaV1.1 , 1.2, 1.3 and 1.6. Of the other subtypes, NaV1.4 is found only in skeletal muscle, NaV1.5 is specific to cardiac muscle, and NaV1.7, 1.8, and 1.9 are found
predominantly in sensory neurons. The hypothesised binding site for state-dependent sodium channel blockers is the local anaesthetic (LA) binding site in the inner vestibule of the pore on transmembrane S6 of domain IV. Critical residues are located in a highly conserved region among the different subtypes, thus presenting a challenge for the design of new subtype selective drugs. Drugs such as lidocaine, lamotrigine and carbamazepine do not distinguish between the subtypes. However, selectivity can be achieved, and can be further enhanced functionally, as a result of the different frequencies at which the channels operate.
Drugs that block voltage-gated sodium channels in a state-dependent manner are also used in the treatment of bipolar disorder, either to reduce symptoms of mania or depression, or as mood stabilisers to prevent the emergence of mood episodes. Clinical and preclinical evidence also suggests that state-dependent sodium channel blockers may help to reduce the symptoms of schizophrenia. For example, lamotrigine has been shown to reduce symptoms of psychosis induced by ketamine in healthy human volunteers, and furthermore, studies in patients suggest that the drug can augment the antipsychotic efficacy of some atypical antipsychotic drugs, such as clozapine or olanzapine. It is hypothesised that efficacy in these psychiatric disorders may result in part from a reduction of excessive glutamate release. The reduction in glutamate release is thought to be a consequence of sodium channel inhibition in key brain areas, such as the frontal cortex. However, interaction with voltage-gated calcium channels may also contribute to the efficacy of these drugs.
WO 2013/175205 (Convergence Pharmaceuticals Limited) describes (2R,5S)-7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1 ,7-diazaspiro[4.4]nonan-6-one hydrochloride, sulfuric acid salt and sulfuric acid salt hydrate which are claimed to be modulators of voltage-gated sodium channels. The object of the invention is to identify alternative salts of said compound which have advantageous properties.
Example 1
(2R,5S)-7-Methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1,7-diazaspiro[4.4]nonan-6-
To a solution of (2R,5S)-7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1 ,7-diazaspiro[4.4]nonan-6-one (which may be prepared in accordance with the procedure described in Example 1 of WO 2013/175205) (4.45g, 0.0114 mol) dissolved in absolute ethanol (66.82 ml, 15 vol) at 45 °C was added a solution of citric acid in ethanol (1 M, 1.05 equiv. 12 ml) over a period of 2-3 minutes. The solution was aged at 45 °C for a period of 1 hour. After 30 minutes a seed of citrate salt (0.1 wt%) was added and the mixture allowed to cool over approximately 2 hours and mature for 18 hours at ambient temperature (approximately 10-15 °C). Following maturation the salt was noted to be a very thick suspension (white) that required mobilisation with 20 ml additional ethanol and a further maturation period of 2 hours at ambient temperature. Filtration was carried out under vacuum and the vessel and cake rinsed with 15 ml ethanol. The de-liquored cake was dried further in a vacuum oven at 50 °C to provide 6.0 g of crystalline white solid (91 % yield).
H NMR (400MHz, DMSO-D6): δΗ 1.90-2.05 (2H, m), 2.10-2.20 (2H, m,), 2.20-2.30 (1 H, m), -2.50 (1 H, m, partially masked by solvent)), 2.55-2.68 (4H, m), 2.56 (3H, s), 2.79 (3H, s),
3.28-3.40 (2H, m), 4.79 (1 H, t, J= 8.0 Hz), 7.92 (2H, d, J = 8.4 Hz), 8.03 (1 H, s), 8.45 (2H, d, J= 8.8Hz) ppm, (exchangeables not reported)
Characterisation of Example 1
The XRPD of Example 1 is presented in FIG. 1 and the DSC/TGA of Example 1 is presented in FIG. 2. The citrate salt of Example 1 displayed the following characteristics:
1 endotherm onset: 171.82°C
peak maximum: 174.55°C
There was an endotherm post the main endotherm.
There was no weight reduction until ca 168°C had been reached. The weight reduction commenced with the start of the main endotherm and coincided with the endotherm post the main endotherm which indicated that this thermal event was the onset of compound decomposition and loss of citric acid. Thermal events >220°C were due to compound decomposition.
The XPRD data in FIG. 1 demonstrated that under different extremes of humidity indicate a stable crystalline form of the citrate salt of Example 1 with no tendency to form hydrates. This is supported by DSC/TGA data in FIG. 2 which show clear transitions and no evidence of solvates.
Aqueous solubility of the citrate salt (Example 1) = 22mg/ml (25°C).
Example 2
(2R,5S)-7-Methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1,7-diazaspiro[4.4]nonan-6-one ) salt (E2)
To a solution of (2R,5S)-7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1 ,7-diazaspiro[4.4]nonan-6-one (which may be prepared in accordance with the procedure described in Example 1 of WO 2013/175205) (4.45g, 0.0114 mol) dissolved in absolute ethanol (66.82 ml, 15 vol) at 45 °C was added a solution of methanesulfonic acid in ethanol (1 M, 1.05 equiv. 12 ml) over a period of 2-3 minutes. The solution was aged at 45 °C for a period of 1 hour. After 10 minutes nucleation and gradual crystallisation was noted to afford a thick mixture. Additional ethanol was added (10 ml) to mobilise the suspension that was then allowed to cool over approximately 2 hours and mature for 18 hours at ambient temperature (approximately 10-15 °C). Following maturation the salt was noted to be a thin, mobile suspension (white) that was filtered under vacuum and the vessel and cake rinsed with 15 ml ethanol. The de-liquored cake was dried further in a vacuum oven at 50 °C to provide 4.0 g of crystalline white solid (72% yield).
H NMR (400MHz, DMSO-D6): δΗ 2.1-2.45 (4H, m), 2.27 (3H, s), 2.50-2.75 (2H, m), 2.61 (3H, s), 2.86 (3H, s), 3.35-3.50 (2H, m), 5.20 (1 H, t, J = 8 Hz), 7.96 (2H, d, J = 8.8 Hz), 8.17 (1 H, s), 8.51 (2H, d, J = 8.4Hz), 9.45 (1 H, br), 10.16 (1 H, br) ppm.
Characterisation of Example 2
The XRPD of Example 2 is presented in FIG. 3 and the DSC/TGA of Example 2 is presented in FIG. 4. The DSC thermograph of the methanesulfonate (mesylate) (Example 2) displayed the following characteristics:
One distinct endotherm onset: 247.34°C
peak maximum: 250.34°C
The TGA thermograph showed no weight reduction until ca 250°C had been reached. The weight reduction commenced with the start of the main endotherm and indicated that this thermal event was the onset of compound decomposition. There is no evidence of entrapped solvents or water.
The XPRD data in FIG. 3 demonstrated that under different extremes of humidity indicate a stable crystalline form of the mesylate salt of Example 2 with no tendency to form hydrates. This is supported by DSC/TGA data in FIG. 4 which show clear transitions and no evidence of solvates.
Aqueous solubility of the mesylate salt (Example 2) = 65mg/ml (25°C).
Example 3
Preparation of (2R,5S)-7-methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1,7-diazaspiro[4.4]nonan-6-one hydrosulfate single crystals: 25.0 mg of (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluorome
one hydrosulfate was added to 4 mL vial. 1.000 mL of anhydrous EtOH was added, and the sample was filtered. Anhydrous hexanes were added dropwise until the solution neared the precipitation point. The vial was sealed and left undisturbed for 24 hr, after which time a crop of single crystals was evident. The sample was sent for single crystal analysis and confirmed as the anhydrous (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one hydrosulfate form (FIGs. 5A-5B).
Example 4
Preparation of (2R,5S)-7-methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1,7-diazaspiro[4.4]nonan-6-one freebase: 8.00 g of (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one hydrosulfate (JM Lot R-2017-4323 D 301) was added to a 1 L Erlenmeyer flask and suspended and stirred vigorously in 400 mL of THF. 20% K2C03 (250 mL) was added and dissolved. The mixture was transferred to 1 L sep. funnel. 100 mL EtOAc was added and the aqueous and organic layers were separated. The aqueous layer was re-extracted with 50 mL of EtOAc and the combined organics were back-extracted with brine (100 mL) and water (100 mL). Due to fairly poor separation, a significant quantity of MgSCU was required to dry the solution. The solution was reduced via Rotavap (45 °C) to -50 mL, transferred to a 100 mL RB flask, reduced down to -10 mL, transferred to 20 mL scintillation vial and continued to be reduced to a thick oil. The oil was left on the Rotavap for another hour and a “wet” solid was obtained. Loosened solids on the bottom of the vial were left on the Rotavap for 1 hr with no heat applied to obtain a chunky solid. The contents was transferred to a mortar and pestle, ground to powder and fine granules, placed back in a 20 mL scintillation vial and left on a Rotavap overnight to obtain a dry solid (5.1 g). The XRPD pattern of (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one freebase is shown in FIG. 6.
Example 5
Preparation of (2R,5S)-7-methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1,7-diazaspiro[4.4]nonan-6-one saccharinate: 199.7 mg of (2R,5S)-7-Methyl-2-(4-
methyl-6-(4-(trifluoromethyl)phenyl)pyrirnidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one free base (0.5115 mmol) was dissolved in 4.2 mL of 2-Me-THF. 98.1 mg of saccharin (0.5106 mmol) was dissolved in 4.2 mL of 2-Me-THF. Saccharin was added to the freebase, and after 15 seconds the mixture began to precipitate and solidify. 10 mL of 2-Me-THF was added and stirred at max rpm as to provide a thick white suspension in 10 min. The suspension was filtered, air dried under vacuum for 10 min on frit, then dried under a stream of nitrogen for 30 min resulting in 215 mg of white solid product. The XRPD pattern for (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one saccharinate is shown in FIG. 7.
Example 6
Preparation of (2R,5S)-7-methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1,7-diazaspiro[4.4]nonan-6-one oxalate: 403 mg of (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one freebase was dissolved in 4.03 mL EtOH. 1.000 mL of this solution was added to a 4 mL vial. 23.8 mg of oxalic acid was dissolved in 1.000 mL of EtOH and added dropwise to the stirring (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one freebase solution. After 5 min, a white precipitate was evident and 2.000 mL of EtOH was added to the slurry to aid stirring. The resulting suspension was stirred overnight. The following day the suspension was filtered and dried on a frit under vacuum for 10 min yielding 106 mg of white solid. The XRPD pattern for (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one oxalate is shown in FIG. 8.
Example 7
The single crystal structural information and refinement parameters for (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one hydrosulfate are shown in Table 1.
Table 1.
Largest peak, hole / e A-3 0.363, -0.264
The most prominent XRPD diffraction peaks were (2Θ): 7.8±0.2°, 8.1±0.2°, 12.6±0.2°, 14.3±0.2°, 16.5±0.2°, 18.5±0.2°, 19.6±0.2°, 24.8±0.2° and 25.3±0.2°.
PATENTS
US2018360833NOVEL PYRIMIDINYL-DIAZOSPIRO COMPOUNDS2018-06-27
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2017304303 | Novel Pyrimidinyl-DiazoSpiro Compounds | 2017-07-11 | |
| US9737536 | Novel Pyrimidinyl-DiazoSpiro Compounds | 2016-05-25 | 2016-09-15 |
| US2016184306 | Novel Pyrimidinyl-DiazoSpiro Compounds | 2016-02-15 | 2016-06-30 |
| US9309254 | NOVEL COMPOUNDS | 2013-05-22 | 2015-04-30 |
| US9376445 | NOVEL COMPOUNDS | 2013-05-22 | 2015-06-18 |
////////////////BIIB-095, BIIB095, BIIB 095, PHASE 1
CC1=NC(=NC(=C1)C2=CC=C(C=C2)C(F)(F)F)C3CCC4(N3)CCN(C4=O)C
VNRX-7145
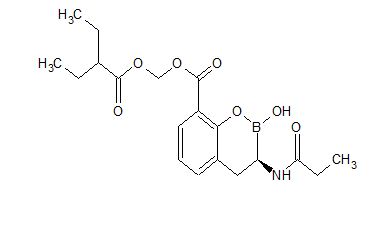

CAS 1842399-68-1
MF C19 H26 B N O7
MW 391.22
2H-1,2-Benzoxaborin-8-carboxylic acid, 3,4-dihydro-2-hydroxy-3-[(1-oxopropyl)amino]-, (2-ethyl-1-oxobutoxy)methyl ester, (3R)-
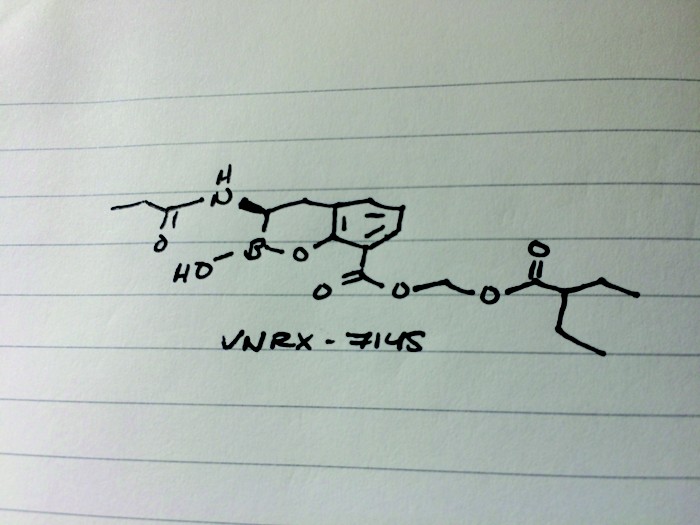
VNRX-7145
PATENT
WO 2015191907
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015191907
ntibiotics are the most effective drugs for curing bacteria-infectious diseases clinically. They have a wide market due to their advantages of good antibacterial effect with limited side effects. Among them, the beta-lactam class of antibiotics (for example, penicillins, cephalosporins, and carbapenems) is widely used because they have a strong bactericidal effect and low toxicity.
[0005] To counter the efficacy of the various beta-lactams, bacteria have evolved to produce variants of beta-lactam deactivating enzymes called beta-lactamases, and in the ability to share this tool inter- and intra-species. These beta-lactamases are categorized as“serine” or“metallo” based, respectively, on presence of a key serine or zinc in the enzyme active site. The rapid spread of this mechanism of bacterial resistance can severely limit beta-lactam treatment options in the hospital and in the community.
SCHEME 1
SCHEME 2
SCHEME 3
[00390] Alternatively, (II) can be obtained by treatment of (I) with hydrochloric acid (around 3-5 Molar in dioxane) in an alcohol solvent such as methanol, ethanol, or n-butanol at a temperature between room temperature and 120 ºC (SCHEME 4).
SCHEME 4
SCHEME 5
EXAMPLE 62: (R)-2-Hydroxy-3-propionylamino-3,4-dihydro-2H-benzo[e][1,2]oxaborinine-8-carboxylic acid
Step 1. Synthesis of 2-Methoxy-3-[2-propionylamino-2-(2,9,9-trimethyl-3,5-dioxa-4-bora-tricyclo[6.1.1.02,6]dec-4-yl)-ethyl]-benzoic acid tert-butyl ester.
[00540] Prepared from [(1S)-2-(3-tert-butoxycarbonyl-2-methoxy-phenyl)-1-chloro-ethyl]boronic acid (+) pinanediol ester and propionic acid following the procedure in Step 2 of Example 1. The crude product was purified by flash chromatography on silica gel (25-100% EtOAc/Hexane). ESI-MS m/z 486 (MH)+.
Step 2. Synthesis of (R)-2-Hydroxy-3-propionylamino-3,4-dihydro-2H-benzo[e][1,2]oxaborinine-8-carboxylic acid.
[00541] Prepared from 2-Methoxy-3-[2-propionylamino-2-(2,9,9-trimethyl-3,5-dioxa-4-bora-tricyclo[6.1.1.02,6]dec-4-yl)-ethyl]-benzoic acid tert-butyl ester following the procedure described in Step 3 of Example 1. The crude product was purified by reverse phase preparative HPLC and dried using lyophilization. ESI-MS m/z 264 (MH)+
CLIP
Candidate: VNRX-7145

Credit: Tien Nguyen/C&EN
Presenter: Christopher John Burns, president and chief executive officer of VenatoRx Pharmaceuticals
Target: β-lactamases
Disease: Resistant urinary tract infections
Reporter’s notes: Having unveiled an antibacterial candidate at last spring’s first time disclosures session, Burns was back with another, this time the molecule can be taken orally. Both VenatoRx (pronounced Ven-a-tor-ix) compounds resuscitate the activity of β-lactam drugs, which make up more than 60% of all antibiotics prescribed. Unfortunately, many bacteria have grown resistant to these antibiotics. The new compounds rescue the old antibacterials by inhibiting β-lactamases, enzymes that chew up the antibiotics. To test the activity of new β-lactamase-targeting compounds, the researchers settled on several “sentinel” bacteria strains. Then to find a candidate with oral bioavailability, the team focused on molecules with low polarity and low molecular weight. They found VNRX-7145, developed as a prodrug in which esterases in the liver clip off the tips of the molecule to reveal the active drug. VNRX-5133, disclosed at last year’s meeting, had to be delivered intravenously along with another IV-antibiotic Cefepime, and targeted serine and metallo β-lactamases. The new oral candidate VNRX-7145 inhibits serine β-lactamases with Ceftibuten as its partner. The VNRX-7145 combination is now in Phase I studies to treat resistant urinary tract infections.
////////////VNRX-7145, VNRX7145, VNRX 7145, Phase I, VenatoRx
CCC(CC)C(=O)OCOC(=O)c1cccc2C[C@H](NC(=O)CC)B(O)Oc12
CCC(CC)C(=O)OCOC(=O)c1cccc2C[C@H](NC(=O)CC)B(O)Oc12
Acefylline
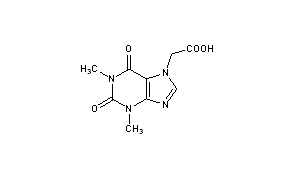
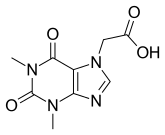
Acefylline
- Molecular FormulaC9H10N4O4
- Average mass238.200 Da
(1,3-Dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)acetic acid
1,2,3,6-Tetrahydro-1,3-dimethyl-2,6-dioxo-7H-purine-7-acetic Acid
1,3-Dimethylxanthine-7-acetic acid
211-490-2 [EINECS]
652-37-9 [RN]
7-(Carboxymethyl)theophylline
7H-Purine-7-acetic acid, 1,2,3,6-tetrahydro-1,3-dimethyl-2,6-dioxo-
CAS Registry Number: 652-37-9
CAS Name: 1,2,3,6-Tetrahydro-1,3-dimethyl-2,6-dioxopurine-7-acetic acid
Additional Names: carboxymethyltheophylline; 7-theophyllineacetic acid
Molecular Formula: C9H10N4O4
Molecular Weight: 238.20
Percent Composition: C 45.38%, H 4.23%, N 23.52%, O 26.87%
Literature References: Prepn: DE 352980 (1922 to E. Merck); Frdl. 14, 1320; S. M. Ride et al., Pharmazie 32, 672 (1977). Prepn of salts: J. Baisse, Bull. Soc. Chim. Fr. 1949, 769; M. Milletti, F. Virgili, Chimica 6, 394 (1951), C.A. 46, 8615h (1952). GC determn in urine: J. Zuidema, H. Hilbers, J. Chromatogr. 182, 445 (1980). HPLC determn in serum and pharmacokinetics: S. Sved et al.,Biopharm. Drug Dispos. 2, 177 (1981).
Properties: Crystals from water, mp 271°.
Melting point: mp 271°
Derivative Type: Sodium salt
CAS Registry Number: 837-27-4
Molecular Formula: C9H9N4NaO4
Molecular Weight: 260.18
Percent Composition: C 41.55%, H 3.49%, N 21.53%, Na 8.84%, O 24.60%
Properties: Silky needles, mp >300°.
Melting point: mp >300°
Derivative Type: Compd with piperazine
Additional Names: Acefylline piperazine; acepifylline
Trademarks: Dynaphylline (Welcker-Lyster); Etaphylline (Delalande); Etafillina (Delalande)
Properties: Undefined mixture of the 1:1 and 2:1 salts; contains 75-78% theophylline acetic acid and 22-25% anhydrous piperazine.
Therap-Cat: Bronchodilator.
Keywords: Bronchodilator; Xanthine Derivatives.
Acefylline (INN),[1] also known as acetyloxytheophylline, is a stimulant drug of the xanthine chemical class. It acts as an adenosine receptor antagonist. It is combined with diphenhydramine in the pharmaceutical preparation etanautine to help offset diphenhydramine induced drowsiness.[2]
Synthesis
DE 352980 (1922 to E. Merck); Frdl. 14, 1320; S. M. Ride et al., Pharmazie 32, 672 (1977).
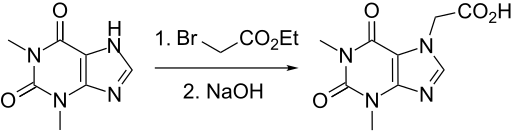
Acefylline
- Use:cardiotonic, diuretic, antispasmodic, bronchodilator
- Chemical name:1,2,3,6-tetrahydro-1,3-dimethyl-2,6-dioxo-7H-purine-7-acetic acid
- Formula:C9H10N4O4
- MW:238.20 g/mol
- CAS-RN:652-37-9
- EINECS:211-490-2
- LD50:1180 mg/kg (M, i.p.); 2733 mg/kg (M, p.o.)
Acepifylline
- Use:
- Chemical name:1,2,3,6-tetrahydro-1,3-dimethyl-2,6-dioxo-7H-purine-7-acetic acid compd. with piperazine
- Formula:C9H10N4O4 • xC4H10N2
- MW:unspecified
- CAS-RN:18833-13-1
- EINECS:242-614-3
Acefylline heptaminol
- Use:
- Chemical name:1,2,3,6-tetrahydro-1,3-dimethyl-2,6-dioxo-7H-purine-7-acetic acid compd. with 6-amino-2-methyl-2-heptaminol (1:1)
- Formula:C9H10N4O3 • C8H19NO
- MW:367.45 g/mol
- CAS-RN:59989-20-7
- EINECS:262-012-4
References
- ^ “International Nonproprietary Names for Pharmaceutical Substances (INN). Recommended International Nonproprietary Names (Rec. INN): List 21” (PDF). World Health Organization. Retrieved 29 December 2016.
- ^ Zuidema, Jan. (1978). “Biofarmaceutische en farmacokinetische aspecten van theofylline en acefylline”. Thesis (doctoral)–Universiteit van Amsterdam. References
Baisse, J.: Bull. Soc. Chim. Fr. (BSCFAS) 1949, 769.
DE 352 980 (E. Merck; 1922).
 |
|
 |
|
| Clinical data | |
|---|---|
| ATC code | |
| Identifiers | |
| CAS Number | |
| PubChemCID | |
| ChemSpider | |
| UNII | |
| ChEMBL | |
| ECHA InfoCard | 100.010.447 |
| Chemical and physical data | |
| Formula | C9H10N4O4 |
| Molar mass | 238.20 g/mol g·mol−1 |
| 3D model (JSmol) | |
////////Acefylline
LHC 165
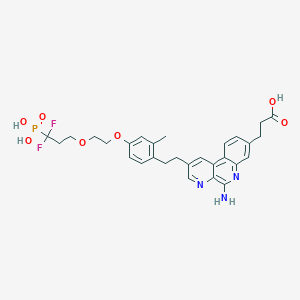

LHC165
3-[5-amino-2-[2-[4-[2-(3,3-difluoro-3-phosphonopropoxy)ethoxy]-2-methylphenyl]ethyl]benzo[f][1,7]naphthyridin-8-yl]propanoic acid
C29H32F2N3O7P, 603.56 g/mol
CAS 1258595-14-0
5-Amino-2-[2-[4-[2-(3,3-difluoro-3-phosphonopropoxy)ethoxy]-2-methylphenyl]ethyl]benzo[f][1,7]naphthyridine-8-propanoic acid
Benzo[f][1,7]naphthyridine-8-propanoic acid, 5-amino-2-[2-[4-[2-(3,3-difluoro-3-phosphonopropoxy)ethoxy]-2-methylphenyl]ethyl]-
- Originator Novartis
- Class Antineoplastics
- Mechanism of Action
- Undefined mechanism
- Phase I Solid tumours
- 31 Jan 2018 Phase-I clinical trials in Solid tumours (Combination therapy, Inoperable/Unresectable, Late-stage disease, Metastatic disease, Second-line therapy or greater) in USA, Belgium, Italy, Japan (Intratumoural) (NCT03301896)
- 31 Jan 2018 Phase-I clinical trials in Solid tumours (Inoperable/Unresectable, Late-stage disease, Metastatic disease, Monotherapy, Second-line therapy or greater) in USA, Japan, Italy, Belgium (Intratumoural) (NCT03301896)
- 10 Oct 2017 Novartis plans a phase I trial for Solid tumours (Monotherapy, Combination therapy, Inoperable/Unresectable, Late-stage disease, Metastatic disease, Second-line therapy or greater) in USA, Belgium, Canada, France, Germany, Italy, South Korea and Spain in November 2017 (Intratumoural) (NCT03301896)
PATENT
PATENT
US 20110053893
PATENT
WO 2011130379
PATENT
Scheme (III)
Scheme (IV)
Scheme (V)
Example 19 (Table 1: Compound 19): Synthesis of ‘3-(5-amino-2-(4-(2-(3,3-difluoro-3-phosphonopropoxy)ethoxy)-2-methylphenethyl)benzo[f][ 1, 7]naphthyridin-8-yl)propanoic acid (19)
Scheme 6
Step 1: (E)-ethyl 3-(3-(tert-butoxycarbonylamino)-4-chlorophenyl)acrylate (6-3)
[517] To a solution of tert-butyl 5-bromo-2-chlorophenylcarbamate (6-1) (1.0 equiv.) in acetonitrile (0.3 M) and EtOH (0.5 M) was added K2C03 (2.0 equiv.). The reaction was degassed and flushed with N , then added (E)-ethyl 3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)acrylate (6-2) (1.2 equiv.) and Pd(PPh3)4 (0.1 equiv.). The reaction was flushed again with N2 and stirred at 100 °C overnight. After cooling to room temperature, hexane was added, and the mixture was filtered through a pad of silica, eluting with EA/Hex (1 : 1) until the product was completely eluted. The filtrate was concentrated and purified on Combiflash, eluting with 0-15% EA in Hex to give (E)-ethyl 3-(3-(tert-butoxycarbonylamino)-4-chlorophenyl)acrylate (6-3) as a white solid.
Step 2: ethyl 3-(3-(tert-butoxycarbonylamino)-4-chlorophenyl)propanoate (6-4)
[518] To a solution of (E)-ethyl 3-(3-(tert-butoxycarbonylamino)-4-chlorophenyl)acrylate (6-3) (1.0 equiv.) in ethyl acetate/ethanol (1 : 1 , 0.3 M) was added Wilkinson’s catalyst (0.10 equiv.).
Hydrogen gas was introduced via a ballon, and the reaction was stirred at room temperature for 24 hours. The mixture was filtered through a pad of celite, washing with dichloromethane. The filtrate was concentrated in vacuo and purified by Combiflash using 0-10% ethyl acetate in hexane to give ethyl 3-(3-(tert-butoxycarbonylamino)-4-chlorophenyl)propanoate (6-4) as a solid.
Step 3: ethyl 3-(3-(tert-butoxycarbonylamino)-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)propanoate (6-5)
[519] A solution of ethyl 3-(3-(tert-butoxycarbonylamino)-4-chlorophenyl)propanoate (6-4) (1 .0 equiv.), 4,4,4,,4′,5,5,5′,5′-octamethyl-2,2′-bi(l ,3,2-dioxaborolane) (2.0 equiv.), tris(dibenzylideneacetone)dipalladium(0) (0.05 equiv.), 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl (0.20 equiv.), and potassium acetate (2.0 equiv.) in 1 ,4-dioxane (0.2 M) was degassed and stirred at 100 °C overnight. After cooling to ambient temperature, the reaction content was concentrated in vacuo. The crude material was purified by Combiflash using 0-50% ethyl acetate in hexane to afford ethyl 3-(3-(tert-butoxycarbonylamino)-4-(4,4,5,5-tetramethyl- 1 ,3,2-dioxaborolan-2-yl)phenyl)propanoate (6-5) as a brown oil. The product was stored at -20°C and used within a month of synthesis.
Step 4: l-bromo-4-(methoxymethoxy)-2-methylbenzene (6-7)
[520] To a solution of 4-bromo-3-methylphenol (6-6) (1.0 equiv.) in DMF (0.5 M) at 0 °C was added portionwise 60% wt NaH (1.5 equiv.). The addition was controlled such that internal reaction temperature never went above 10 °C. The reaction was stirred at room temperature for 45 minutes, then a solution of chloro(methoxy)methane (1.2 equiv.) in DMF (3 M) was added dropwise via additional funnel. The reaction was stirred at room temperature for 3.5 hours, and then quenched by pouring into ice. The resulting mixture was stirred at room temperature for 1 hour. Ether was added, and the two layers were separated. The aqueous layer was extracted (lx) with ether. The combined organic layers were washed with water (2x), brine, dried over MgS04, and concentrated to give 1 -bromo-4-(methoxymethoxy)-2-methylbenzene (6-7) as a colorless oil. The crude material was used in the next step without further purification.
Step 5: triethylf (4-(methoxymethoxy)-2-methylphenyl)ethynyl)silane
[521] A solution of l -bromo-4-(methoxymethoxy)-2-methylbenzene (1.0 equiv.), triethylamine (5.0 equiv.) in DMF (0.5 M) was degassed and flushed with nitrogen. To the reaction was added TES-acetylene (1.05 equiv.), Cul (0.098 equiv.), and Pd(PPh3)2Cl2 (0.098 equiv.). The reaction was heated to 60 °C and stirred overnight. After cooling to room temperature, water and ether were added. The layers were separated, and the organic layer was washed with water (2x). The organic layer was separated and passed through a pad of silica (packed with hexane). The silica was eluted with 10% EA in Hex. The fractions were combined and concentrated to give triethyl((4-(methoxymethoxy)-2-methylphenyl)ethynyl)silane as a black oil. The crude material was used in the next step without further purification.
Step 6: l-ethynyl-4-(methoxymethoxy)-2-methylbenzene (6-8)
[522] To a solution of triethyl((4-(methoxymethoxy)-2-methylphenyl)ethynyl)silane (1.0 equiv.) at
0 °C was slowly added tetrabutylammonium fluoride (1M solution in THF, 0.20 equiv.). At this
point, the ice-bath was removed and the reaction mixture was allowed to stir at room temperature for 45 minutes. The reaction mixture was then passed through a pad of silica (packed with hexane) and eluted with 20% EtOAc in Hexanes to remove insoluble salts. The crude product was then purified by Combiflash using 0-10% EtOAc in Hexanes to give 1 -ethynyl-4-(methoxymethoxy)-2-methylbenzene (6-8) as a slightly brown liquid.
Step 7: 3-chloro-5-((4-(methoxymethoxy)-2-methylphenyl)ethynyl)picolinonitrile (6-10)
[523] A solution of l -ethynyl-4-(methoxymethoxy)-2-methylbenzene (6-8) (1 .0 equiv.), 3,5-dichloropicolinonitrile (6-9) (0.90 equiv.), Cul (0.10 equiv.), and Pd(PPh3)2CI2 (0.10 equiv.), and triethylamine (5.0 equiv.) in DMF (0.25 M) was degassed and flushed with nitrogen. The reaction mixture was then heated to 60 °C and stirred overnight. After cooling to room temperature, water was added. The mixture was extracted with EA (2x). The combined organic layers were washed with 10% aq NH4OH (2x), brine, and concentrated. The crude material was filtered through a pad of silica (wetted with hexane). The silica was eluted with 10% EA in Hex. The fractions were combined and concentrated. The resulting solids were washed in hot ether and filtered to give a yellow solid, which was used in the next step without further purification. The filtrate was concentrated and purified by Combiflash using 0- 10% EtOAc in Hexanes to give 3-chloro-5-((4-(methoxymethoxy)-2-methylphenyl)ethynyl)picolinonitrile (6-10) as a yellow solid.
Step 8: ethyl 3-(5-amino-2-((4-(methoxymethoxy)-2-methylphenyl)ethynyl)-ben∑o fJfl, 7J
naphthyridin-8-yl)propanoate (6-11)
[524] A solution of 3-chloro-5-((4-(methoxymethoxy)-2-methylphenyl)ethynyl)picolinonitrile (6-10) (1 .0 equiv.), ethyl 3-(3-(tert-butoxycarbonylamino)-4-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)phenyl)propanoate (6-5) (1.25 equiv.), tris(dibenzylideneacetone)dipalladium(0) (0.10 equiv.), dicyclohexyl(2′,6′-dimethoxybiphenyl-2-yl)phosphine (0.20 equiv.), and sodium bicarbonate (3.0 equiv.) in «-butanol /H20 (5: 1 , 0.2 M) was degassed and stirred at 100 °C overnight. After cooling to ambient temperature, the reaction content was diluted with ethyl acetate and water. The two phases were separated, and the aqueous layer was extracted twice with ethyl acetate. The combined organic layers were washed with brine, dried over anhydrous MgS04, and concentrated in vacuo. The crude material was purified by flash chromatography on a COMBIFLASH® system (1SCO) using 0-40% ethyl acetate in DCM first to remove the impurity, then 0-4% MeOH in DCM to give ethyl 3-(5-amino-2-((4-(methoxymethoxy)-2-methylphenyl)ethynyl)-benzo[f][l ,7]naphthyridin-8-yl) propanoate (6-11). Further purification was accomplished by precipitating and washing in hot ether.
Step 9: ethyl 3-(5-amino-2-(4-(methoxymethoxy)-2-methylphenethyl)benzo[fl[l ]naphthyridin-8-yl)propanoate (6-12)
[525] A solution of ethyl 3-(5-amino-2-((4-(methoxymethoxy)-2-methylphenyl)ethynyl)-benzo[f][l ,7]naphthyridin-8-yl)propanoate (6-11) (1.0 equiv.) in EtOH/THF (3: 1 , 0.16 M) was flushed with nitrogen. Then, 10% wt Pd/C (0.20 equiv. by weight) was added. The reaction was flushed with hydrogen (2x) and stirred under a hydrogen balloon. After 24 hours, the reaction was filtered through a pad of celite, washing with 5%MeOH in DCM. The filtrate was checked for the presence of starting material using LCMS. The hydrogenation reaction was repeated until no more
of the alkyne starting material or alkene intermediate was detected. The crude product was purified by Combiflash using 0-4% eOH in DCM to give ethyl 3-(5-amino-2-(4-(methoxymethoxy)-2-methylphenethyl)benzo[f][l ,7]naphthyridin-8-yl)propanoate (6-12) as a white solid.
Step 10: ethyl 3-(5-amino-2-(4-hydroxy-2-methylphenethyl)benzo[fl[l ]naphthyridin-8-yl)propanoate (6-13)
[526] Ethyl 3-(5-amino-2-(4-(methoxymethoxy)-2-methylphenethyl)benzo[fJ[l ,7]naphthyridin-8-yl)propanoate (6-12) (1 .0 equiv.) was dissolved in EtOH (0.2 M), then added a solution of 4M HC1 in dioxane (0.2 M). The product precipitated out as a yellow salt. After stirring for 3 hours, the reaction was poured into a stirring solution of ether. The mixture was stirred for 10 minutes, then filtered and washed with ether. Ethyl 3-(5-amino-2-(4-hydroxy-2-methylphenethyl)benzo[fJ[l ,7]naphthyridin-8-yl)propanoate (6-13) was obtained as a yellow solid which was dried on vacuum overnight (bis-HCl salt). Alternatively, the crude product was purified by Combiflash using 0-5% MeOH in DCM to give the free base.
Step 11: ethyl 3-(5-amino-2-(4-(2-(3-(diethoxyphosphoryl)-3,3-difluoropropoxy)ethoxy)-2-methylphenethyl)benzo[f] [1 , 7]naphthyridin-8-yl)propanoate ( 6-15)
[527] To a solution of ethyl 3-(5-amino-2-(4-hydroxy-2-methylphenethyl)benzo[fJ [ l ,7]naphthyridin-8-yl)propanoate (6-13) (1.0 equiv.) dissolved in DMF (0.14 M) was added a solution of diethyl 3-(2-bromoethoxy)-l ,l -difluoropropylphosphonate (6-14: described in Example 7 – Step 1) (1 .3 equiv.) in DMF (0.7 M) and cesium carbonate (4 equiv.). The reaction was stirred at 60 °C. After 1.5 hours (or until reaction is complete by LCMS), DCM (2 volume equivalent) was added to the reaction. The solids (inorganic) were filtered, and the filtrate was concentration. The crude product was purified by Combiflash using 0-5%MeOH in DCM to give ethyl 3-(5-amino-2-(4-(2-(3-(diethoxyphosphoryl)-3,3-difluoropropoxy)ethoxy)-2-methylphenethyl)benzo[fJ
[1 ,7]naphthyridin-8-yl)propanoate (6-15) as an oil which upon standing became a white solid.
Step 12: 3-(5-amino-2-(4-(2-(3,3-difluoro-3-phosphompropoxy)ethoxy)-2-methylphenethyl)be o[f]
[1, 7]naphthyridin-8-yl)propanoic acid (19)
[528] To a solution of ethyl 3-(5-amino-2-(4-(2-(3-(diethoxyphosphoryl)-3,3-difluoropropoxy)ethoxy)-2-methylphenethyl)benzo[f][l ,7]naphthyridin-8-yl)propanoate (6-15) (1.0 equiv.) in DCM (0.16 M) at 0 °C was added slowly TMSBr (10 equiv.). The reaction was stirred at room temperature overnight. Additional TMSBr (5.0 equiv.) was added at 0 °C, and the reaction was again stirred at room temperature overnight. The solvent was removed by evaporation and the crude orange solids dried on hi-vac briefly. The solids were suspended in EtOH (0.5 M) and added 2.5 N
NaOH (10.0 equiv.). The reaction was stirred at 80 °C for 3 hours. After cooling to room temperature, the mixture was adjusted to pH 9 to 10 and directly purified on RP-HPLC using a CI 8 column, eluting with 10-40% 95:5 (MeCN/5mM NH4OAc) in l OmM NH4OAc (pH 9) gradient. The fractions containing the product were combined and concentrated in vacuo. The resulting white gel was dissolved in refluxing 1 :1 EtOH/water (0.04 M) with the addition of a few drops of ammonium hydroxide. While hot, the mixture was slowly poured into a stirring hot solution of acetone (0.009
M) preheated at 50 °C. The acetone suspension was slowly cooled to room temperature for 15 minutes with continued stirring, and then sat in an ice bath for 10 minutes. The solids were filtered and washed successively with acetone (2x) and ether (2x). The solids were dried on hi-vac overnight to give the 3-(5-amino-2-(4-(2-(3,3-difluoro-3-phosphonopropoxy)ethoxy)-2-methylphenethyl)benzo [fj[l ,7]naphthyridin-8-yl)propanoic acid (19) as a solid. Ή NMR (Dimethylsulfoxide-d6): δ 9.02 (s, 1 H), 8.82 (s, 1H), 8.55 (d, 1H, J = 8.4 Hz), 7.58 (s, 1H), 7.48 (d, 1 H, J = 8.4 Hz), 7.07 (d, 1H, J = 8.4 Hz), 6.75 (s, 1 H), 6.68 (d, 1H, J = 8.4 Hz), 4.03-4.00 (m, 2H), 3.72-3.68 (m, 4H), 3.16-3.12 (m, 2H), 3.03-2.96 (m, 4H), 2.67-2.64 (m, 2H), 2.33-2.32 (m, 2H), 2.26 (s, 3H). LRMS [M+H] = 604.2
PATENT
US 20120237546
PATENT
WO 2012031140
PATENT
Toll-like receptors (TLRs) are pattern recognition receptors which play an essential role in the innate immunity, by recognizing invasion of microbial pathogens and initiating intracellular signal transduction pathways to trigger expression of genes, the products of which can control innate immune responses. Specifically, Toll like receptor (TLR) agonists activate innate immune cells through the TLR-MyD88-NFk and IRF3/7 pathways. TLR7, TLR8, and TLR9 belong to a subfamily of TLRs based on their genomic structure, sequence similarities, and homology. TLR7, TLR8, and TLR9 are located in intracellular endolysosomal compartments and show a unique pattern of cell type-specific expression that is thought to be responsible for different pathogen response profiles.
Small molecule agonists of TLR7 and/or TLR8 have been reported and shown to activate innate immune responses by inducing selected cytokine biosynthesis, the induction of co-stimulatory molecules, and by increased antigen-presenting capacity. Such compounds include imidazoquinoline amine derivatives (U.S. Patent No. 4689338), imidazopyridine amine derivative (U.S. Patent No. 5446153), imidazonaphthyridine derivative (U.S. Patent No.
6194425), oxazoloquinoline amine derivatives (U.S. Patent No. 61 10929); thiazoloquinoline amine derivatives (U.S. Patent No. 61 10929), selenazoloquinoline amine derivatives (U.S. Patent No. 61 10929), pyrazolopyridine derivatives (U.S. Patent No. 9145410), and
benzonaphthyridine amine derivatives (U.S. Patent Nos. 8466167 and 9045470).
The synthetic TLR7 agonist, Imiquimod (1 -(2-methylpropyl)-1 H-imidazo[ 4,5-c]quinolin-4-amine) is FDA-approved in a cream formulation for the topical treatment of cutaneous basal cell carcinoma, actinic keratosis and genital warts, and has limited activity against cutaneous melanoma and breast tumors (J. Immunol. 2014, 193(9) : 4722^1-731 ). Systemic administration of Imiquimod, and structurally similar Resiquimod, is limited by cytokine- mediated adverse effects including severe flu-like symptoms (Expert Opin. Emerging Drugs (2010), 15:544-555). Consequently, Imiquimod is used exclusively in topical applications and is not used to treat deep, non-cutaneous tumors such as melanoma or solid tumors.
An injectable lipid modified imidazoquinoline (TLR7/8 dual agonist) that forms a tissue depot with gradual, sustained release which allows for local TLR triggering activity without systemic cytokine release has been reported (J. Immunol. 2014, 193(9): 4722^731 ). However, this compound was shown to be ineffective for large tumors and in addition the serum concentration of this compound 24 hours post subcutaneous administration decreased by approximately 50% (Journal for ImmunoTherapy of Cancer, 2014, 2:12). Therefore, there remains a need for intratumor administration of a TLR7 agonist with prolonged sustained release, which may benefit the treatment of large tumors.
clip
Candidate: LHC165

Credit: Tien Nguyen/C&EN
Presenter: Alex Cortez, senior Investigator I at the Genomics Institute of the Novartis Research Foundation
Target: Toll-like receptor 7 (TLR7)
Disease: Solid tumors
Reporter’s notes: Cortez shared another story in the realm of immuno-oncology, although the program that yielded this compound actually started in the world of vaccines. Cortez’s team had been focusing on vaccine adjuvants, small molecules that turn on the immune system to enhance a vaccine’s effect. They developed one such class of compound that activates toll-like receptor 7 (TLR7), a protein in the immune system that recognizes dangerous-looking molecules and can trigger the release of infection-clearing proteins. After observing TLR7 agonists’ ability to induce an immune response with vaccines, the researchers wondered whether the molecules could also be effective in immuno-oncology.
They found that LHC165 adsorbed to aluminum hydroxide reduced tumor growth in mice and, intriguingly, showed signs of an abscopal effect, in which untreated tumors shrink concurrently with treated tumors. The implication is that if the immune system recognizes one tumor site, it can recognize others. As with several of the candidates presented throughout the day, LHC165 bears a phosphate group and is injected into the tumor. It’s currently in Phase I trials in patients with advanced malignancies, which means they’ve already tried second and third line therapies, as a single agent and in combination with the checkpoint inhibitor PDR001.
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US9597326 | BENZONAPTHYRIDINE COMPOSITIONS AND USES THEREOF | 2011-04-13 | 2013-05-16 |
| US9950062 | COMPOUNDS AND COMPOSITIONS AS TLR ACTIVITY MODULATORS | 2010-09-01 | 2012-09-20 |
| US9517263 | BENZONAPHTHYRIDINE-CONTAINING VACCINES | 2010-06-10 | 2012-10-18 |
| US2015225432 | COMPOUNDS AND COMPOSITIONS AS TLR ACTIVITY MODULATORS | 2015-04-24 | 2015-08-13 |
| US9315530 | ADSORPTION OF IMMUNOPOTENTIATORS TO INSOLUBLE METAL SALTS | 2011-09-01 |
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2016213776 | ADSORPTION OF IMMUNOPOTENTIATORS TO INSOLUBLE METAL SALTS | 2016-04-07 | 2016-07-28 |
| US2012177681 | Formulation of immunopotentiators | 2011-09-01 | 2012-07-12 |
| US9045470 | COMPOUNDS AND COMPOSITIONS AS TLR ACTIVITY MODULATORS | 2011-03-03 | |
| US2018169204 | COMBINATION VACCINES WITH LOWER DOSES OF ANTIGEN AND/OR ADJUVANT | 2018-02-02 | |
| US9375471 | ADJUVANTED FORMULATIONS OF BOOSTER VACCINES | 2013-03-08 | 2013-09-12 |
//////LHC165, LHC 165, LHC -165, Phase I, Solid tumours, novartis
O=P(O)(O)C(F)(F)CCOCCOc4ccc(CCc1cc2c3ccc(CCC(=O)O)cc3nc(N)c2nc1)c(C)c4
CC1=C(C=CC(=C1)OCCOCCC(F)(F)P(=O)(O)O)CCC2=CN=C3C(=C2)C4=C(C=C(C=C4)CCC(=O)O)N=C3N
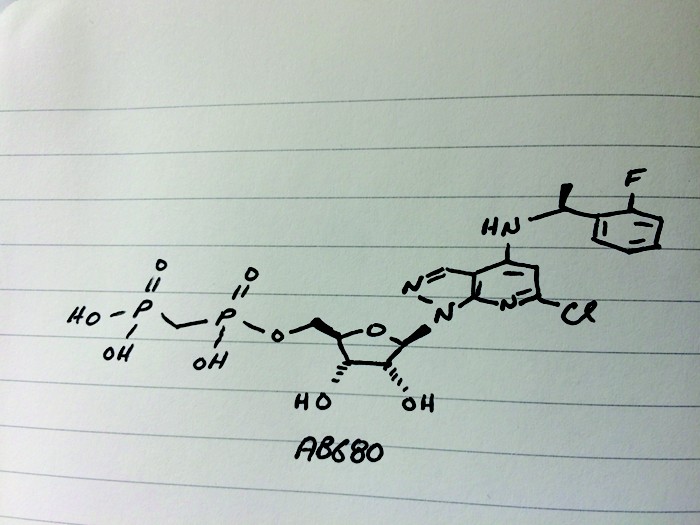
AB 680
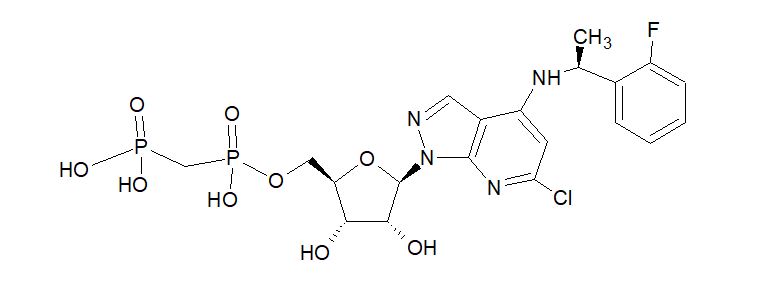
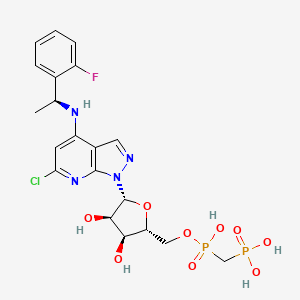
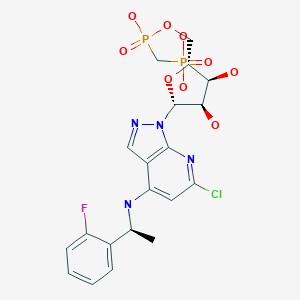

AB 680
C20H24ClFN4O9P2, 580.827 g/mol
Cas 2105904-82-1
1H-Pyrazolo[3,4-b]pyridin-4-amine, 6-chloro-N-[(1S)-1-(2-fluorophenyl)ethyl]-1-[5-O-[hydroxy(phosphonomethyl)phosphinyl]-β-D-ribofuranosyl]-
[[(2R,3S,4R,5R)-5-[6-chloro-4-[[(1S)-1-(2-fluorophenyl)ethyl]amino]pyrazolo[3,4-b]pyridin-1-yl]-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]methylphosphonic acid
[({[(2R,3S,4R,5R)-5-(6-chloro-4-{[(1S)-1-(2-fluorophenyl)ethyl]amino}-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-3,4-dihydroxyoxolan-2-yl]methoxy}(hydroxy)phosphoryl)methyl]phosphonic Acid
- Originator C
- Class Antineoplastics; Small molecules
- Mechanism of Action 5-nucleotidase inhibitors; Adenosine A2 receptor antagonists
- Phase I Cancer
- 19 Nov 2018 Arcus Biosciences plans to initiate a clinical trial in Cancer in first half of 2019
- 16 Oct 2018 Phase-I clinical trials in Cancer (In volunteers) in Australia (IV) (NCT03677973)
- 30 Sep 2018 Preclinical pharmacodynamics data in Cancer presented at 4th CRI-CIMT-EATI-AACR International Cancer Immunotherapy Conference (CRI-CIMT-EATI-AACR – 2018)
Clip
Credit: Tien Nguyen/C&EN
Presenter: Kenneth V. Lawson, senior scientist at Arcus Biosciences
Target: Ecto-5’-nucleotidase (CD73)
Disease: Cancer
Reporter’s notes: In the first talk of the day, Lawson introduced the idea of cancer drugs that target the host’s immune system. “Checkpoint inhibitors changed the way we think of treating cancer,” he said. These drugs successfully disrupt the binding interaction between a protein and a checkpoint protein that stops immune T cells from killing cancer cells. As a result, these drugs turn immune cells loose to attack tumor cells. But the drugs work only in about 30-40% of patients—an issue pharmaceutical companies like Arcus hope to address with new immunotherapies that can be taken in combination with checkpoint inhibitors.
Lawson’s team set out to inhibit an enzyme commonly found in tumors called CD73, the second of two enzymes which break down extracellular adenosine trisphosphate (ATP) to adenosine. Adenosine then binds to immunosuppressive receptors on immune cells and shuts them down. Yet developing a small molecule inhibitor of CD73 proved challenging, Lawson said. After striking out with high-throughput screening, the team turned to CD73’s natural substrate for inspiration. However, the molecule possessed more than one phosphate group, which is notoriously a liability for drug molecules because small molecules with such negative changes struggle to cross cell membranes. The team’s goal was to remove the phosphate groups, Lawson says, but things didn’t exactly go according to plan. After showing the audience a series of compounds from structure-activity relationship (SAR) studies—slides no medicinal chemistry talk would be complete without—Lawson revealed the structure of their final clinical compound AB680 as the sound of people flipping notebook sheets rippled across the room. Synthesized in 34% overall yield, the candidate ultimately included two phosphate groups—a feature that surprised audience members.
Tests revealed that AB680 can be given intravenously but the compound also showed moderate oral bioavailability. Lawson suggested a possible route for how the molecule might pass from the digestive tract to the bloodstream, a paracellular mechanism by which molecules cross the epithelium by passing through the space between cells. AB680 showed “extraordinary potency,” inhibiting CD73 in human T-cells at a concentration of 0.008 nM. The compound has a 4 day half-life, which means it could be dosed every two weeks, coinciding with the dosing schedule for patients who receive a checkpoint inhibitor. AB680 is currently in Phase 1 clinical trials with healthy patients.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US204141996&tab=PCTDESCRIPTION&maxRec=1000
|
Purinergic signaling, a type of extracellular signaling mediated by purine nucleotides and nucleosides such as ATP and adenosine, involves the activation of purinergic receptors in the cell and/or in nearby cells, resulting in the regulation of cellular functions. Most cells have the ability to release nucleotides, which generally occurs via regulated exocytosis (see Praetorius, H. A.; Leipziger, J. (1 Mar. 2010) Ann Rev Physiology 72(1): 377-393). The released nucleotides can then be hydrolyzed extracellularly by a variety of cellular membrane-bound enzymes referred to as ectonucleotidases.
|
Example 92
Synthesis of [({[(2R,3S,4R,5R)-5-(6-chloro-4-{[(1S)-1-(2-fluorophenyl)ethyl]amino}-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-3,4-dihydroxyoxolan-2-yl]methoxy}(hydroxy)phosphoryl)methyl]phosphonic Acid
PATENT

////////////////ARCUS, AB 680, AB680, AB-680, PHASE 1
https://www.arcusbio.com/wp-content/uploads/2018/04/AACR_AB680_1756_final_90x42-abstract-4886.pdf
Fc1ccccc1[C@H](C)Nc4cc(Cl)nc3c4cnn3[C@@H]2O[C@H](COP(=O)(O)CP(=O)(O)O)[C@@H](O)[C@H]2O
CC(C1=CC=CC=C1F)NC2=CC(=NC3=C2C=NN3C4C(C(C(O4)COP(=O)(CP(=O)(O)O)O)O)O)Cl
Solriamfetol hydrochloride, ソルリアムフェトル塩酸塩 , солриамфетол , سولريامفيتول , 索安非托 ,
Solriamfetol hydrochloride
FDA APPROVED 2019/3/20, Sunosi
ソルリアムフェトル塩酸塩; R228060, R 228060
| Formula |
C10H14N2O2. HCl
|
|---|---|
| CAS |
178429-65-7 HCL
|
| Mol weight |
230.6913
|
(2R)-2-Amino-3-phenylpropyl carbamate
(2R)-2-Amino-3-phenylpropylcarbamat
10117
178429-62-4 [RN] FREE FORM
Benzenepropanol, β-amino-, carbamate (ester), (βR)- [
солриамфетол [Russian] [INN]
سولريامفيتول [Arabic] [INN]
索安非托 [Chinese] [INN]
JZP-110
Originator SK Holdings
- Developer Jazz Pharmaceuticals plc; SK biopharmaceuticals
- Class Carbamates; Sleep disorder therapies; Small molecules
- Mechanism of Action Adrenergic uptake inhibitors; Dopamine uptake inhibitors
- Orphan Drug Status Yes – Narcolepsy
- Registered Hypersomnia
- Discontinued Depressive disorders
- 26 Mar 2019 Discontinued – Phase-I for Depressive disorders (Adjunctive treatment) in USA (PO) (Jazz Pharmaceuticals pipeline, March 2019)
- 20 Mar 2019 Registered for Hypersomnia (excessive daytime sleepiness) in patients with obstructive sleep apnoea and narcolepsy in USA (PO) – First global approval
- 20 Mar 2019 US FDA approves solriamfetol to improve wakefulness in adult patients with excessive daytime sleepiness associated with narcolepsy or obstructive sleep apnoea(OSA)
- New Drug Application (NDA): 211230
Company: JAZZ PHARMA IRELAND LTD
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Solriamfetol hydrochloride | K7RO88SP7A | 178429-65-7 | KAOVAAHCFNYXNJ-SBSPUUFOSA-N |
Solriamfetol, sold under the brand name Sunosi, is a medication used for the treatment of excessive sleepiness associated with narcolepsy and sleep apnea.[1]
Common side effects include headache, nausea, anxiety, and trouble sleeping.[1] It is a norepinephrine–dopamine reuptake inhibitor(NDRI). It is derived from phenylalanine and its chemical name is (R)-2-amino-3-phenylpropylcarbamate hydrochloride.[2]
The drug was discovered by a subsidiary of SK Group, which licensed rights outside of 11 countries in Asia to Aerial Pharma in 2011.[3]
History
The drug was discovered by a subsidiary of SK Group, which licensed rights outside of 11 countries in Asia to Aerial Pharma in 2011.[3]Aerial ran two Phase II trials of the drug in narcolepsy[4] before selling the license to solriamfetol to Jazz in 2014; Jazz Pharmaceuticalspaid Aerial $125 million up front and will pay Aerial and SK up to $272 million in milestone payments, and will pay double digit royalties to SK.[3][5]
In March 2019 the FDA accepted SK’s and Jazz’ NDA for use of solriamfetol to treat excessive sleepiness in people with narcolepsy or obstructuve sleep apnea; the drug has an orphan designation for narcolepsy.[3][6]
Names
During development it has been called SKL-N05, ADX-N05, ARL-N05, and JZP-110.[6]
Research
Solriamfetol had also been tested in animal models of depression, but as of 2017 that work had not been advanced to clinical trials.[7]
PATENT
WO 9607637
https://patents.google.com/patent/WO1996007637A1/e
Organic alkyl carbamates have been effectively used for controlling various central nervous system (CNS) disorders. For example, U.S. Pat. Nos . 2,884,444, 2,937,119 and 3,313,697 disclose function of carbamate in CNS disorders, especially as antiepileptic and centrally acting muscle relaxant.
Phenylethylamine derivatives, one important class of therapeutical medicines useful for managing CNS diseases, have been used mainly to treat obesity, narcolepsy, minimal brain dysfunction and mild depression.
Recent design of pharmacologically useful compounds has been based on amino acids or the derivatives thereof, which is mainly attributable to the fact that many of the compounds found in biological systems come from amino acids or the derivatives thereof. In addition, in most cases, the function of a pharmaceutically useful compound is effected after it binds to an enzyme or receptor, which may trigger the regulatory mechanisms of the enzyme or receptor.
REACTION SCHEME I
REACTION SCHEME II
REACTION SCHEME III
EXAMPLE I
Preparation of N-Benzyloxycarbonyl-D-phenylalaninol
In a 500 mL RB flask equipped with a mechanical stirrer and a dropping funnel, D-phenylalaninol (45.4 g, 300 mmol) was dissolved in 220 mL of distilled water, and cooled in an ice-bath. The pH of the solution was adjusted with 50 % sodium hydroxide to 14. Benzyl chloroformate (49.3 mL, 345 mmol) was charged into the dropping funnel and added slowly to the well stirred solution over 0.5 hr. After the completion of the addition, the reaction mixture was stirred for 1 hr. at 0 *C. The product precipitated from the reaction mixture as a white solid. It was collected by filtration and washed completely with distilled water. After being dried in vacuo, the solid thus obtained weighed 104 grams without any further purification: 99.8% Yield.
Melting point = 90 – 92 *C
[α]D20 = + 43.4 (c = 1.0, EtOH)
Analysis calc: C, 71.56; H, 6.71; N,4.91
Found: C, 71.35; H, 6.71; N,4.91
EXAMPLE II
Preparation of N-Benzyloxycarbonyl-D-phenylalaninol
carbamate
In a 500 mL RB flask, N-benzyloxycarbonyl-D- phenylalaninol (13.56 g, 50 mmol) was charged with antipyrine (11.29 g, 60 mmol) in 250 mL of dry THF under a nitrogen atmosphere. The reaction mixture was cooled in an ice-bath and phosgene (30.3 mL of 1.93 M solution in toluene, 58.5 mmol) was added quickly while vigorously stirring. After stirring for 1 hr. , the formation of a corresponding chloroformate from the starting material was monitored by TLC. The chloroformate solution thus prepared, was slowly added to a well stirred and ice-chilled aqueous ammonium hydroxide solution (75 mL, 28-30 %, 1,190 mmol) via cannula over 0.5 hr. The resulting reaction mixture was stirred for an extra 0.5 hr. The organic phase separated was collected. The aqueous phase was extracted twice with methylene chloride (100 mL). The combined organic phase was washed with brine (50 mL), dried over sodium sulfate, and concentrated to yield 17.8 g (113%) of foamy solid. It was purified a flash column chromatography to give 14.8 g of the title compound, white solid: 94% Yield.
Melting point = 121 – 125 *C
[α]D20 = + 28.6 (c = 2.0, EtOH)
Analysis calc. : C, 65.84; H, 6.14; N, 8.53
Found: C, 66.68; H, 6.21; N, 7.80
EXAMPLE III
Preparation of D-Phenylalaninol carbamate hydrochloric
acid salt In a 160 mL Parr reactor, N-benzyloxycarbonyl-D-phenylalaninol carbamate (9.43 g) was added with 75 mL of anhydrous methanol and 10 % palladium on charcoal (0.32 g). Then, the reactor was closed and purged with hydrogen for 1 in. The reaction was completed in 2 hrs . under 40 psi pressure of hydrogen at 45 #C. The catalyst was filtered off. Thereafter, the organic layer was concentrated into 5.97 g (102 %) of pale yellow thick liquid. The liquid was poured in 50 mL of anhydrous THF and cooled to 0 “C. Anhydrous hydrogen chloride gas was then purged through the solution with slowly stirring for
0.5 hr. 50 mL of anhydrous ether was added, to give a precipitate. Filtration with THF-ether (1:1) mixture provided 6.1 g of the title compound as a white solid: 88 % Yield.
Melting point = 172 – 174 “C
[α]D20 = – 12.9 (c = 2.0, H20)
Analysis calc. : C, 52.60; H, 6.55; N, 12.14; Cl, 15.37
Found: C, 51.90; H, 6.60; N, 12.15; Cl ,
15.52
EXAMPLE IV
Preparation of N-benzyloxγcarbonyl-L-Phenγlalaninol
The title compound was prepared in the same manner as that of Example I, except that (L)-phenylalaninol was used as the starting material.
Melting point = 90 – 92 *C
[α]D20 = – 42.0 (c = 1.0, EtOH)
Analysis calc. : C, 71.56; H, 6.71; N,4.91
Found: C, 70.98; H, 6.67; N,4.95
EXAMPLE V
Preparation of -N-benzyloxycarbonyl-L-Phenylalaninol
carbamate
The title compound was prepared in the same manner as that of Example II, except that N-benzyloxycarbonyl-L-phenylalaninol was used as the starting material.
Melting point = 121 – 128 ‘C
[α]D20 = – 28.9 (c = 2.0, EtOH)
Analysis calc: C, 65.84; H, 6.14; N, 8.53
Found: C, 65.45; H, 6.15; N, 8.32
EXAMPLE VI
Preparation of L-Phenylalaninol carbamate hydrochloric
acid salt
The title compound was prepared in the same manner as that of Example III, except that N-benzyloxycarbonyl-L-phenylalaninol carbamate was used as the starting material.
Melting point = 175 – 177 *C [α]D20 = + 13.1 (c = 1.0, H20)
Analysis calc : C, 52.60; H, 6.55; N, 12.14; Cl, 15.37
Found: C, 51.95; H, 6.58; N, 12.09; Cl , 15.37
EXAMPLE VII
Preparation of N-benzyloxycarbonyl-D,L-Phenylalaninol
The title compound was prepared in the same manner as that of Example I, except that (D,L)-phenylalaninol was used as the starting material.
Melting point = 72 – 75 #C
Analysis calc: C, 71.56; H, 6.71; N,4.91
Found: C, 71.37; H, 6.74; N,4.84
EXAMPLE VIII
Preparation of N-benzyloxycarbonyl-D,L-Phenylalaninol
carbamate
The title compound was prepared in the same manner as that of Example II, except that N-benzyloxycarbonyl-D,L-phenylalaninol was used as the starting material.
Melting point = 130 – 133 *C
Analysis calc: C, 65.84; H, 6.14; N, 8.53
Found: C, 65.85; H, 6.14; N, 8.49 EXAMPLE IX
Preparation of D,L-Phenylalaninol carbamate hydrochloric
acid salt
The title compound was prepared in the same manner as that of Example III, except that N-benzyloxycarbonyl-D,L-phenylalaninol carbamate was used as the starting material.
Melting point = 163 – 165 *C
Analysis calc: C, 52.60; H, 6.55; N, 12.14; Cl, 15.37
Found: C, 51.92; H, 6.56; N, 11.95; Cl , 15.82
PATENT
US 20050080268
PATENT
WO 2018133703
https://patents.google.com/patent/WO2018133703A1/en
Excessive daytime sleepiness (Excessive Daytime Sleepiness, EDS) or pathological somnolence refers to excessive daytime sleep and wakefulness associated with various sleep disorders. These disorders can be the basis for a sleep disorder or sleep have side effects caused by some other medical conditions. Excessive daytime sleep, also known as narcolepsy, sleep clinics is seen mainly in patients with disease that affects 12% of the general population. EDS patients may be manifested as mental distress, poor work or school performance, increasing the risk of accidents, the impact of EDS can debilitating, even life-threatening.
R228060, also known JZP-110, is a selective dopamine and norepinephrine reuptake inhibitor, originally developed by R & D, SK biopharmaceutical, 2014 Sir ownership of the pharmaceutical compound. R228060 has the potential to treat narcolepsy and sleep apnea syndrome, in three multi-center study in two global reached the primary endpoint, and achieved positive results, significantly improved adult obstructive sleep apnea patients excessive sleepiness in patients with narcolepsy and excessive sleep problems.
R228060 chemical name is O- carbamoyl – (D) – phenylalaninol, as shown in the structural formula of formula (I):

Solid Form different chemicals, can cause varying their solubility and stability, and thus affects the absorption and bioavailability of the drug, and can lead to differences in clinical efficacy. Improve the candidate compound has a solubility by salt way become an important means of drug development. Compared to the free form of the drug, suitable pharmaceutically acceptable salts can improve the solubility of the drug type, increased physical and chemical stability, and also to improve the drug-salt having a melting point, hygroscopicity, crystal type and other physical properties, further development of the pharmaceutical dosage form It plays an important role. Patent Document WO1996007637A1 discloses R228060 hydrochloride and its preparation method, and other characteristics of the obtained having a melting point of 172-174 deg.] C as a white solid, the solid was not given in the text data. Further, the present inventors found no other relevant R228060 hydrochloride polymorph or patent literature. Accordingly, the present need in the art to develop a comprehensive system R228060 hydrochloride polymorph, found to be suitable to the development of crystalline form. The present inventors after many experiments, found that polymorph CS1 R228060 hydrochloride CS2 and a melting point polymorph, Form CS1 and CS2 is Form 183 ℃, much higher than the melting point disclosed in prior art solid. It provides a better alternative preparation of pharmaceutical preparations containing R228060 is, has very important implications for drug development.
PATENT
WO 2019027941
(i?)-2-amino-3-phenylpropyl carbamate (APC) is a phenylalanine analog that has been demonstrated to be useful in the treatment of a variety of disorders, including excessive daytime sleepiness, cataplexy, narcolepsy, fatigue, depression, bipolar disorder, fibromyalgia, and others. See, for example, US Patent Nos. 8,232,315; 8,440,715; 8,552,060; 8,623,913; 8,729,120; 8,741,950; 8,895,609; 8,927,602; 9,226,910; and 9,359,290; and U.S. Publication Nos. 2012/0004300 and 2015/0018414. Methods for producing APC (which also has other names) and related compounds can be found in US Patent Nos. 5,955,499; 5,705,640; 6,140,532 and 5,756,817. All of the above patents and applications are hereby incorporated by reference in their entireties for all purposes.
EXAMPLE 1
Synthesis of Compounds
Compound 8 (110CR002)
1 B 110CR002
[0083] tert- utyl (if)-(l-(Carbamothioyloxy)-3-phenylpropan-2-yl)carbamate (IB): A
60% dispersion of sodium hydride (0.36 g, 4.78 mmol, 1.2 equiv) in mineral oil was added in portions to compound 1A (1.0 g. 3.98 mmol, 1 equiv) in THF (20 mL) at 0 °C. After stirring for 1 hour, carbon disulfide (0.191 g, 4.78 mmol, 1.2 equiv) was added at 0 °C. After an additional hour of stirring, methyl iodide (0.3 mL, 4.78 mmol, 1.2 equiv) was added and the reaction was warmed to room temperature. After stirring two additional hours, concentrated ammonium hydroxide (1.6 mL, 7.98 mmol, 2 equiv) was added and the reaction was stirred overnight at room temperature. The reaction was diluted with water (50 mL) and extracted with dichloromethane (3 x 50 mL). The combined organic layers were dried over sodium sulfate and concentrated under reduced pressure to give crude compound IB. The solid was triturated in diethyl ether (20 mL) to give compound IB (0.17 g, 14% yield) as a light yellow solid.
[0084] (R)-0-(2-Amino-3-phenylpropyl) carbamothioate dihydrochloride (110CR002):
4M HCI in dioxane (0.68 mL, 2.74 mmol, 5 equiv) was added to neat compound IB (0.17 g, 0.548 mmol, 1 equiv) and the reaction was stirred overnight. The solution was diluted with diethyl ether (20 mL) and the resulting suspension was filtered. The solid was triturated in diethyl ether (20 mL) and the filtered solid was dried under vacuum at room temperature for two hours to give compound 110CR003 (140 mg, 93% yield, 96.9% purity) as a white solid.
Compound 9 (110CR003)
Scheme 2
2A 2B 110CR003
[0085] (R)-2-((ter^Butoxycarbonyl)amino)-3-phenylpropyl sulfamate (2B): A solution of sulfamoyl chloride (1.15 g, 9.95 mmol, 2.5 equiv) in acetonitrile (2 mL) was added dropwise to a solution of compound 2 A (1.0 g, 3.98 mmol, 1 equiv) and triethylamine (2.1 mL, 14.95 mmol, 3.75 equiv) in N,N-dimethylacetamide (20 mL) at 0 °C. After stirring at room temperature for 4 hours, additional triethylamine (2.1 mL, 14.95 mmol, 3.75 equiv) and sulfamoyl chloride (1.15 g, 9.95 mmol, 2.5 equiv) in acetonitrile (2 mL) was added at 0 °C. The reaction was stirred at room temperature overnight, at which point LCMS indicated a 3 :2 mixture of product to starting material. Additional triethylamine (2.1 mL, 14.95 mmol, 3.75 equiv) and sulfamoyl chloride (1.15 g, 9.95 mmol, 2.5 equiv) in acetonitrile (2 mL) was added at 0 °C and the reaction was stirred at room temperature for an additional 6 hours. LCMS indicated a 4: 1 mixture of product to starting material. The reaction was quenched with saturated sodium bicarbonate (5 mL) and stirred for an additional hour at room temperature. The reaction was diluted with saturated sodium bicarbonate (25 mL) and extracted with ethyl acetate (3 x 50 mL). The combined organic layers were dried over sodium sulfate and concentrated under reduced pressure. The product still contained unreacted starting material which could not be easily separated. Sulfamoyl chloride (1.15 g, 9.95 mmol, 2.5 equiv) in acetonitrile (2 mL) was added dropwise to a solution of crude compound 2B (0.9 g) and triethylamine (2.1 mL, 14.95 mmol, 3.75 equiv) in N,N-dimethylacetamide (20 mL) at 0 °C. After stirring at room temperature for two hours, the reaction was quenched with saturated sodium bicarbonate (5 mL) and the reaction was stirred for an additional hour at room temperature. The reaction was diluted with saturated sodium bicarbonate (25 mL) and extracted with ethyl acetate (3 x 50 mL). The combined organic layers were dried over sodium sulfate and concentrated under reduced pressure. The residue was purified on an AnaLogix automated system (Redisep 24 g silica gel column), eluting with a gradient of 25 to 50% ethyl acetate in heptanes, to give compound 2B (0.37 g, 28% yield) as a white solid.
[0086] (R)-2-Amino-3-phenylpropyl sulfamate hydrochloride (110CR003): 4M HC1 in dioxane (1.4 mL, 5.6 mmol, 5 equiv) was added to neat compound 2B (0.37 g, 1.12 mmol, 1 equiv) and the reaction was stirred overnight. The solution was diluted with diethyl ether (20 mL) and the resulting suspension was filtered. The solid was triturated in diethyl ether (20 mL) and the filtered solid was dried under a vacuum at room temperature for two hours to give compound 110CR003 (250 mg, 84% yield, 97.8% purity) as a white solid.
Com ound 3 (110CR007)
[0087] (Benzyl (R)-(l-phenyl-3-ureidopropan-2-yl)carbamate) (3B): Concentrated hydrochloric acid (0.06 mL, 0.68 mmol, 0.12 equiv) was added to a solution of benzyl (ft)-(l -amino-3-phenylpropan-2-yl)carbamate ( 1.5 g, 5.28 mmol, 1 equiv) and urea (1.26 g, 21.21 mmol, 4 equiv) in toluene (150 mL) under nitrogen. After refluxing overnight, LCMS indicated the reaction was complete. The reaction was concentrated under reduced pressure, diluted with water (150 mL) and stirred for 30 minutes. The resulting solid was filtered and washed with water (25 mL) to give crude compound 3B (1.4 g, 4.27 mmol, 80% yield) as a white solid, which was used sequentially.
[0088] ((R)-l-(2-mino-3-phenylpropyl)urea) (3C): Compound 3B (0.5 g, 1.5 mmol, 1 equiv) and 10% palladium on carbon (0.09 g) in methanol (60 mL) was hydrogenated at 30 psi for 1 hour at which time LC-MS determined that the reaction was incomplete. The solution was filtered and fresh catalyst (0.09 g) was added. The solution was hydrogenated at 30 psi for an additional 45 minutes resulting in complete conversion. Two identical scale reactions were run for 105 minutes each, both resulting in complete conversion. The three runs were combined and filtered through celite, which was washed with methanol (50 mL). The filtrate was concentrated under reduced pressure to give crude compound 3C (0.9 g), which was used sequentially.
[0089] (R)-l-(2-Amino-3-phenylpropyI)urea hydrochloride (110CR007): Compound 3C (0.88 g, 4.58 mmol, 1 equiv) was dissolved diethyl ether (10 mL) and 4 N HCl in dioxane (2.31 mL, 9.27 mmol, 2 equiv) was added. The reaction was stirred overnight and then concentrated under reduced pressure to give crude 110CR007 as a white solid. The material was twice recrystallized from 10% methanol in ethanol (30 mL) to give 110CR007 (0.163 g, 16 % yield, 93.7 % purity) as a white solid.
Compound 4 (110CR009)
Scheme 4
[0090] Ethyl (R^)-4-((tert-butoxycarbonyI)amino)-5-phenylpent-2-enoate (4B): A solution of compound 4A (4.0 g, 16.1 mmol, 1 equiv) and ethyl (triphenylphos-phoranylidene)acetate (5.6 g, 16.1 mmol, 1 equiv) in dichloromethane (40 mL) was stirred at room temperature overnight. The reaction was concentrated under reduce pressure to remove the organic solvent and the resulting residue was purified on an AnaLogix automated system (40 g Sorbtech silica gel column), eluting with gradient of 50 to 100% ethyl acetate in heptanes, to give compound 4B (4.8 g, 94% yield) as a white solid.
[0091] (R^E)-4-((te *i-ButoxycarbonyI)amino)-5-phenylpent-2-enoic acid (4C): Lithium hydroxide (1.4 g, 60 mmol, 4 equiv) in water (15 mL) was added to compound 4B (4.8 g, 15 mmol, 1 equiv) in THF (60 mL) at room temperature and the reaction was stirred overnight. After 16 hours, the reaction was adjusted to pH 4 with IN hydrochloric acid. The organic layer was removed and the aqueous layer was extracted with ethyl acetate (2 x 50 mL). The combined organic layers was washed with saturated brine (50 mL), dried over sodium sulfate and concentrated under reduced pressure to give compound 4C (4.2 g, 97% yield) as a light cream solid, which was used subsequently.
[0092] Methyl (R E)-4-((½ -i-butoxycarbonyl)amino)-5-phenylpent-2-enoate (4D1):
Isobutyl chloro formate (1.3 mL, 10 mmol, 1 equiv) in THF (4 mL) was added dropwise to a solution of compound 4C (3.0 g, 10 mmol, 1 equiv) and N-methyl-morpholine (1.1 mL, 10 mmol, 1 equiv) in THF (12 mL) at -15 °C. After 30 minutes of stirring, LCMS indicated complete conversion to the anhydride intermediate. 2M Ammonia in methanol (5 mL, 10 mmol, 1 equiv) was added dropwise over 20 minutes, keeping the internal temperature between -25 to -15 °C. After 30 minutes of stirring, the reaction was warmed to room
temperature and stirred overnight. The reaction mixture was concentrated at reduced pressure to remove the organic solvent. The resulting residue was dissolved in ethyl acetate (50 mL) and washed with water (100 mL). The aqueous layer was extracted with ethyl acetate (2 x 50 mL). The combined organic layers were washed with saturated brine (50 mL), dried over sodium sulfate and concentrated under reduced pressure. The residue was purified on an AnaLogix automated system (80 g Sorbtech silica gel column), eluting with a gradient of 25 to 50% ethyl acetate in heptanes, to give compound 4D1 (1.1 g, 35 % yield) as a white solid.
[0093] Methyl (S)-4-((te^-butoxycarbonyl)amino)-5-phenylpentanoate (4D2): A mixture of compound 4D1 (1.1 g, 3.6 mmol, 1 equiv) and 10% palladium on carbon (0.33 g, 50% wet) in methanol (40 mL) was hydrogenated at 40 psi at room temperature for 4 hours. The mixture was filtered through celite, which was washed with methanol (100 mL). The filtrate was concentrated under reduced pressure to give compound 4D2 (1.1 g, 99% yield) as a white solid.
[0094] (S)-4-((ii? i-Butoxycarbonyl)amino)-5-phenylpentanoic acid (4D3): Lithium hydroxide (73 mg, 3 mmol, 1.5 equiv) in water (1 mL) was added to compound 4B (0.6 g, 2 mmol, 1 equiv) in THF (9 mL) at room temperature. After stirring overnight, the reaction was adjusted to pH 4 with IN hydrochloric acid. The organic layer was removed and the aqueous layer was extracted with ethyl acetate (3 x 25 mL). The combined organic layers was washed with saturated brine (25 mL), dried over sodium sulfate and concentrated under reduced pressure to give compound 4D3 (0.56 g, 98% yield) as a white solid, which was used subsequently.
[0095] tert-Butyl (S)-(5-amino-5-oxo-l-phenylpentan-2-yl)carbamate (4E): Isobutyl chloroformate (0.23 mL, 1.8 mmol, 1 equiv) in THF (0.5 mL) was added drop-wise to a solution of compound 4C (0.54 g, 1.8 mmol, 1 equiv) and N-methylmorpholine (0.2 mL, 1.8 mmol, 1 equiv) in THF (1 mL) at -15 °C. After 20 minutes of stirring, LCMS indicated complete conversion to the anhydride intermediate. 0.4M Ammonia in THF (9 mL, 3.6 mmol, 2 equiv) was added drop-wise over 20 minutes, keeping the internal temperature between -25 to -15 °C. After 30 minutes of stirring the reaction was warmed to room temperature and stirred overnight. The reaction mixture was concentrated under reduced pressure to remove the organic solvent. The resulting residue was dissolved in ethyl acetate (25 mL) and washed with water (25 mL). The organic layer was separated and the aqueous layer was extracted with ethyl acetate (2 x 25 mL). The combined organic layers were washed with saturated brine (50 mL), dried over sodium sulfate and concentrated under
reduced pressure to give compound 4E (0.5 g, 93% yield) as a white solid, which was used subsequently.
[0096] (S)-4-Amino-5-phenylpentanamide hydrochloride (110CR009): 4M HC1 in dioxane (6 mL, 25 mmol, 10 equiv) was added to compound 4E (0.73 g, 1.12 mmol, 1 equiv) After stirring overnight at room temperature, the reaction was diluted with diethyl ether (20 mL) and stirred for 6 hours. The resulting suspension was filtered and the solid was washed with diethyl ether (20 mL). The filtered solid was dried under vacuum at room temperature for two hours to give compound 110CR009 (340 mg, 60% yield, 97.9 % purity) as a white solid.
Compound 10 (110CR012)
[0097] tert-Butyl (R)-(l-(carbamoylthio)-3-phenyIpropan-2-yI)carbamate (5B):
Compound 5 A (0.15 g, 0.56 mmol, 1 equiv) was dissolved in THF (8 mL) and sparged with nitrogen for 15 minutes. Trichloroacetyl isocyanate (0.1 mL, 0.84 mmol, 1.5 equiv) was added and the solution stirred for 3 hours, at which point TLC (30% ethyl acetate in heptane) indicated absence of starting material. The reaction was cooled to 0°C and concentrated ammonium hydroxide (0.15 mL) was added. After stirring overnight at room temperature, TLC indicated that the reaction was complete. The reaction was washed with a 10% ammonium hydroxide (10 mL). The organic layer was concentrated under reduced pressure. The residue was purified on an AnaLogix automated system (12 g silica gel column), eluting with a gradient of 0 to 30% ethyl acetate in heptane, to give compound 5B. This reaction was repeated an additional two times 0.15 g and 0.18 g). The products were to give compound 5B (0.35 g, 1.12 mmol, 62.2% yield) as a white solid.
[0098] (R)-S-(2-Amino-3-phenylpropyl) carbamothioate hydrochloride (110CR012):
Compound 5B (0.35 g, 1.12 mmol, 1 equiv) was dissolved in 4N HCI in dioxane (2 mL). The reaction was stirred for two hours and then concentrated under reduced pressure to give crude 110CR012 as a white solid. The material was triturated in diethyl ether (15 mL) to give 110CR012 (0.215 g, 78 % yield, 98.0 % purity) as a white solid.
References
- ^ Jump up to:a b “SUNOSI™ (solriamfetol) Tablets, for Oral Use. Full Prescribing Information” (PDF). Jazz Pharmaceuticals. 2019. Retrieved 21 March2019.
- ^ Abad, VC; Guilleminault, C (2017). “New developments in the management of narcolepsy”. Nature and Science of Sleep. 9: 39–57. doi:10.2147/NSS.S103467. PMC 5344488. PMID 28424564.
- ^ Jump up to:a b c d Ji-young, Sohn (5 March 2018). “SK Biopharmaceuticals’ narcolepsy drug on track to hitting US market”. The Korea Herald.
- ^ Sullivan, SS; Guilleminault, C (2015). “Emerging drugs for common conditions of sleepiness: obstructive sleep apnea and narcolepsy”. Expert Opinion on Emerging Drugs. 20 (4): 571–82. doi:10.1517/14728214.2015.1115480. PMID 26558298.
- ^ Garde, Damian (January 14, 2014). “Jazz bets up to $397M on Aerial’s narcolepsy drug”. FierceBiotech.
- ^ Jump up to:a b “Solriamfetol – Jazz Pharmaceuticals/SK Biopharmaceuticals”. AdisInsight. Retrieved 15 April 2018.
- ^ de Biase, S; Nilo, A; Gigli, GL; Valente, M (August 2017). “Investigational therapies for the treatment of narcolepsy”. Expert Opinion on Investigational Drugs. 26 (8): 953–963. doi:10.1080/13543784.2017.1356819. PMID 28726523.
 |
|
| Clinical data | |
|---|---|
| Trade names | Sunosi |
| Synonyms | SKL-N05, ADX-N05, ARL-N05, and JZP-110; (R)-2-amino-3-phenylpropylcarbamate hydrochloride |
| Routes of administration |
By mouth |
| ATC code | |
| Pharmacokinetic data | |
| Bioavailability | ~95% |
| Protein binding | 13.3–19.4% |
| Metabolism | negligible |
| Elimination half-life | ~7.1 h |
| Excretion | urine (95% unchanged) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C10H14N2O2 |
| Molar mass | 194.234 g/mol g·mol−1 |
| 3D model (JSmol) | |
///////////Solriamfetol hydrochloride, Solriamfetol, ソルリアムフェトル塩酸塩; солриамфетол , سولريامفيتول , 索安非托 , JZP-110, Orphan Drug, fda 2019, R228060, R 228060
UPDATE MAR 2022
Solriamfetol, sold under the brand name Sunosi, is a medication used for the treatment of excessive sleepiness associated with narcolepsy and sleep apnea.[1] It is derived from d-phenylalanine and its chemical name is (R)-2-amino-3-phenylpropylcarbamate hydrochloride.[3] It is a norepinephrine–dopamine reuptake inhibitor (NDRI). Common side effects include headache, nausea, anxiety, and trouble sleeping.[1]
The drug was discovered by a subsidiary of SK Group, which licensed rights outside of eleven countries in Asia to Aerial Pharma in 2011.[4]
Synthetic Description
Reference: Choi, Yong-Moon; Kim, Min Woo. Process for preparing O-carbamoyl amino alcohols by treatment of amino alcohols with cyanates in the presence of acid. US 20050080268. (2005).
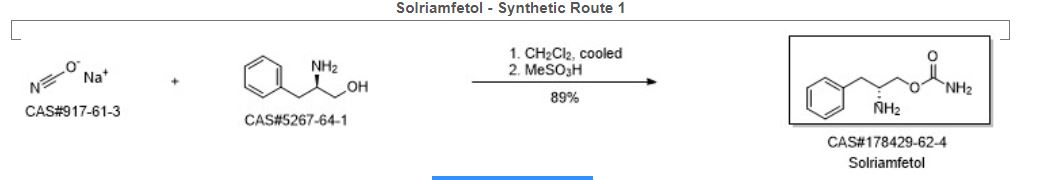
SYN
https://www.researchgate.net/figure/Synthesis-of-solriamfetol-173_fig37_344079894

SYN
Cite this article
Yin, Z., Hu, W., Zhang, W. et al. Tailor-made amino acid-derived pharmaceuticals approved by the FDA in 2019. Amino Acids 52, 1227–1261 (2020). https://doi.org/10.1007/s00726-020-02887-4
Solriamfetol (Sunosi™) Solriamfetol (Sunosi™) (173), formerly known as JZP-110, is a selective dopamine and norepinephrine reuptake inhibitor (DNRI) (Fig. 23). It was discovered by SK Biopharmaceuticals and developed by Jazz Pharmaceuticals (Markham 2019c). The afnity of solriamfetol for these monoamine transporters dopamine transporter (DAT, Ki=14.2 μM), norepinephrine transporter (NET, Ki=3.7 μM), and serotonin transporter (SERT, Ki=81.5 μM) was lower than that of cocaine in transfected cells and inhibits dopamine and norepinephrine reuptake with low potency (IC50=2.9 and 4.4 μM, respectively) (Baladi et al. 2018). In 2019, US FDA approved solriamfetol for using as an oral drug to improve wakefulness in adult patients with excessive daytime sleepiness associated with narcolepsy or obstructive sleep apnoea (OSA). It was granted as an orphan drug (Schweitzer et al. 2019). The systematic name of solriamfetol is (R)-2-amino3-phenylpropylcarbamate hydrochloride, which contains a phenylalanine (171)-derived (R)-2-amino-3-phenylpropan1-ol (172) moiety (Fig. 23). Some alkyl carbamates have been introduced for controlling various central nervous system (CNS) disorders. Phenylethylamine derivatives are one of the important class of therapeutical medicines, useful for managing CNS diseases. After an intensive research, these two skeletons were combined to produce solriamfetol (173) as a drug for the treatment of CNS disorder, especially for depression. The compound 174 with a (S) carbon center showed almost no activity at all, which the racemic compound 175 displayed a half potency of the activity (Fig. 24) (Yang and Gao 2019; Choi and Byun 1996). Solriamfetol (173) was discovered and patented by SK Biopharmaceuticals in 1996 (Choi and Byun 1996). The synthesis of solriamfetol using (D)-phenylalaninol (176) as a starting material is highlighted in Scheme 21. (D)-Phenylalaninol (176) was frst converted to Cbz-protected D-phenylalaninol 177 by reacting with benzyl chloroformate. Carbamoylation of 177 with phosgene followed by ammonolysis with excess of concentrated ammonium hydroxide aqueous solation aforded (D)-O-carbamoyl-N-benzyloxycarbonylphenylalaninol 178. Hydrogenolysis removal of the Cbz protection group gave solriamfetol 173 which was treated

with HCl (gas) to provide (D)-O-carbamoylphenylalaninol hydrochloride salt. In 2020, the Zhang lab reported a method of Ni-catalyzehd asymmetric hydrogenation of 2-amidoacrylates for making solriamfetol (173) (Hu et al. 2020). In this method, o-methoxybenzoyl chloride reacted with glycine methyl ester hydrochloride 179 under a base condition and then hydrolysised in the presence of NaOH to aford desired o-methoxyhippuric acid 180. The one-step construction of oxazolone 181 was accomplished by cyclization and condensation of 180 with benzaldehyde in acetic anhydride and PPh3. Oxazolone 181 was then treated with MeOH and NaOMe to aford 2-amidoacrylate 182. Hydrogenation of 182 using Ni salt and ligand (S)-DM-MeO-BIPHEP gave product 183 in 92% ee. The reduction of 183 with LiBH4 followed by hydrolysised in the presence of NaOH provided intermediate (D)-phenylalaninol 184. Then, (D)-phenylalaninol 184 was reacted with NaOCN yielded solriamfetol (173) in 91% ee (Scheme 22). As a general comment related to this and other chiral compounds discussed here, we would like to emphasize the growing awareness about the Self-Disproportionation of Enantiomers (SDE) phenomenon and the problems related to accurate determination of the stereochemical outcome of enantioselective catalytic reactions (Han et al. 2018, 2019b, 2011a; Soloshonok et al. 2017; Sorochinsky et al. 2013c,
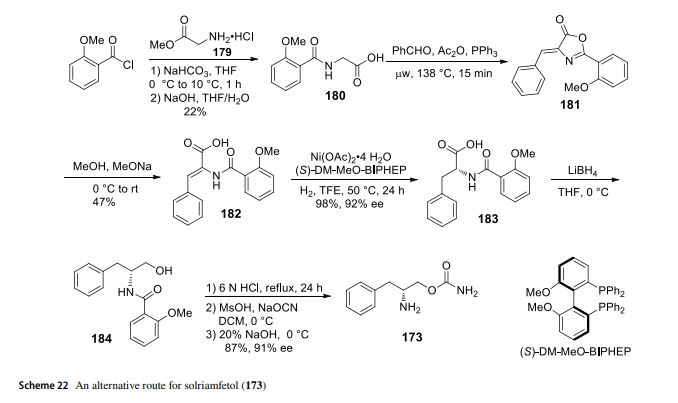
2013d). It was demonstrated that the SDE phenomenon is ubiquitous, being manifested virtually by all types of chiral compounds subjected to physicochemical phase transfer under totally achiral conditions (Han et al. 2019b; Sorochinsky et al. 2013c, d). One of the most frequent cases is a separation of more and less enantiomerically enriched fractions as compared with the original enantiomeric purity of a chiral compound. Consequently, to ensure the accuracy in the %ee determination, it was suggested to perform SDE tests, in particular, under the conditions of achiral column chromatography (Soroshinsky et al. 2013c) and sublimation (Han et al. 2011a).
(Soroshinsky et al. 2013c) Sorochinsky AE, Katagiri T, Ono T, Wzorek A, Aceña JL, Soloshonok VA (2013c) Optical purifcations via self-disproportionation of enantiomers by achiral chromatography; case study of a series of α-CF3-containing secondary alcohols. Chirality 25:365–368
SYN
Substances Referenced in Synthesis Path
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 63-91-2 | C9H11NO2 | D-Phenylalanine | |
| 5267-64-1 | C9H13NO | D-Phenylalaninol | |
| 75-44-5 | CCl2O | phosgene |
SYN
European Journal of Medicinal Chemistry
Volume 205, 1 November 2020, 112667
Solriamfetol (Sunosi). Solriamfetol is a norepinephrine-dopamine reuptake inhibitor (NDRI) that was developed by SK Biopharmaceuticals [124]. This drug was approved by the FDA for the treatment of EDS associated with obstructive sleep apnea (OSA) and narcolepsy [125]. In 12-week Phase III trials in this patient population, solriamfetol (75 mg or 150 mg once daily) could provide significantly great improvement [126]. Furthermore, solriamfetol was evaluated in more than 900 adults with EDS, and was maintained its effect relative to placebo after six months of use. The specific mechanism is still unknown, possibly through inhibiting dopamine and norepinephrine reuptakes [127,128].
A single-step kilogram-scale synthetic approach to solriamfetol is depicted in Scheme 21 [129]. D-Phenylalaninol 136 reacted with sodium cyanate in the presence of acid to give solriamfetol (XVI) in 89% yield.
[124] P.J. Strollo, J. Hedner, N. Collop, D.G. Lorch, D. Chen, L.P. Carter, Y. Lu, L. Lee, J. Black, J.L. Pepin, S. Redline, Solriamfetol for the treatment of excessive sleepiness in OSA: a placebo-controlled randomized withdrawal study, Chest 155 (2019) 364-374.
[125] A. Markham, Solriamfetol: first global approval, Drugs 79 (2019) 785-790.
[126] A. Malhotra, C. Shapiro, J.L. Pepin, J. Hedner, M. Ahmed, N. Foldvary-Schaefer, P.J. Strollo, G. Mayer, K. Sarmiento, M. Baladi, P. Chandler, L. Lee, R. Schwab, Long-term study of the safety and maintenance of efficacy of solriamfetol (JZP-110) in the treatment of excessive sleepiness in participants with narcolepsy or obstructive sleep apnea, Sleep 43 (2020) 220-230.
[127] M.J. Thorpy, C. Shapiro, G. Mayer, B.C. Corser, H. Emsellem, G. Plazzi, D. Chen, L.P. Carter, H. Wang, Y. Lu, J. Black, Y. Dauvilliers, A randomized study of solriamfetol for excessive sleepiness in narcolepsy, Ann. Neurol. 85 (2019) 359-370.
[128] P.K. Schweitzer, R. Rosenberg, G.K. Zammit, M. Gotfried, D. Chen, L.P. Carter, H. Wang, Y. Lu, J. Black, A. Malhotra, K.P. Strohl, Solriamfetol for excessive sleepiness in obstructive sleep apnea (TONES 3). A randomized controlled trial, Am. J. Respir. Crit. Care Med. 199 (2019) 1421-1431.
[129] Y.M. Choi, M.W. Kim, Process of preparing o-carbamoyl compounds in the presence of active amine group, 2005. WO2005033064.

Pharmacology
Pharmacodynamics
Solriamfetol is a norepinephrine–dopamine reuptake inhibitor (NDRI).[1] It binds to the dopamine transporter and the norepinephrine transporter with affinities (Ki) of 14.2 μM and 3.7 μM, respectively).[1] It inhibits the reuptake of dopamine and norepinephrine with IC50 values of 2.9 μM and 4.4 μM, respectively.[1] It has weak affinity for the serotonin transporter (Ki = 81.5 μM) and does not appreciably inhibit serotonin reuptake (IC50 > 100 μM).[1] Solriamfetol has no appreciable affinity for a variety of other targets, including the dopamine, serotonin, adrenergic, GABA, adenosine, histamine, orexin, benzodiazepine, and acetylcholine receptors.[1]
Pharmacokinetics
The elimination half-life of solriamfetol is about 7.1 hours.[1]
History
The drug was discovered by a subsidiary of SK Group, which licensed rights outside of eleven countries in Asia to Aerial Pharma in 2011.[4] Aerial ran two Phase II trials of the drug in narcolepsy[5] before selling the license to solriamfetol to Jazz in 2014; Jazz Pharmaceuticals paid Aerial $125 million up front and will pay Aerial and SK up to $272 million in milestone payments, and will pay double-digit royalties to SK.[4][6]
In 2019, solriamfetol was approved in the United States to improve wakefulness in adults with narcolepsy or obstructive sleep apnea (OSA).[7][8] It was granted orphan drug designation.[9]
The U.S. Food and Drug Administration (FDA) approved solriamfetol based primarily on evidence from five clinical trials (Trial 1/NCT02348593, Trial 2/NCT02348606, Trial 3/NCT02348619, Trial 4/NCT02348632, Trial 5 NCT01681121) of 622 patients with narcolepsy or obstructive sleep apnea (OSA).[7] The trials were conducted in Canada, Europe, and the United States.[7]
Solriamfetol was approved for medical use in the European Union in January 2020.[2]
Society and culture
Names
During development it has been called SKL-N05, ADX-N05, ARL-N05, and JZP-110.[10]
Legal status
In the United States, solriamfetol is a Schedule IV controlled substance,[1] meaning that it has an accepted medical use and a low potential for abuse, but that abuse may lead to physical or psychological dependence.[11] A prescription is required, and can only be refilled up to five times in a six-month period.[12] In countries of the European Union, a prescription is required.[2]
Research
A case report of solriamfetol for the treatment of attention deficit hyperactivity disorder (ADHD) exists.[13]
References
- ^ Jump up to:abcdefghijklmn “Sunosi – solriamfetol tablet, film coated”. DailyMed. 16 October 2019. Retrieved 24 November 2019.
- ^ Jump up to:abc “Sunosi EPAR”. European Medicines Agency (EMA). 12 November 2019. Retrieved 26 September 2020.
- ^ Abad VC, Guilleminault C (2017). “New developments in the management of narcolepsy”. Nature and Science of Sleep. 9: 39–57. doi:10.2147/NSS.S103467. PMC5344488. PMID28424564.
- ^ Jump up to:abc Ji-young S (5 March 2018). “SK Biopharmaceuticals’ narcolepsy drug on track to hitting US market”. The Korea Herald.
- ^ Sullivan SS, Guilleminault C (2015). “Emerging drugs for common conditions of sleepiness: obstructive sleep apnea and narcolepsy”. Expert Opinion on Emerging Drugs. 20 (4): 571–82. doi:10.1517/14728214.2015.1115480. PMID26558298. S2CID7951307.
- ^ Garde D (14 January 2014). “Jazz bets up to $397M on Aerial’s narcolepsy drug”. FierceBiotech.
- ^ Jump up to:abc “Drug Trials Snapshots: Sunosi”. U.S. Food and Drug Administration (FDA). 16 April 2019. Archived from the original on 28 September 2019. Retrieved 24 November 2019.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Drug Approval Package: Sunosi”. U.S. Food and Drug Administration (FDA). 29 April 2019. Retrieved 24 November 2019.This article incorporates text from this source, which is in the public domain.
- ^ “Solriamfetol Orphan Drug Approval”. U.S. Food and Drug Administration (FDA). Retrieved 24 November 2019.This article incorporates text from this source, which is in the public domain.
- ^ “Solriamfetol – Jazz Pharmaceuticals/SK Biopharmaceuticals”. AdisInsight. Retrieved 15 April 2018.
- ^ 21 U.S.C.§ 812 – Schedules of controlled substances
- ^ “Manuals – Practitioner’s Manual – Section V”. Retrieved 2014-01-07
- ^ Naguy A, El-Sheshaie A, Elsori DH, Alamiri B (April 2021). “Solriamfetol for attention deficit hyperactivity disorder”. CNS Spectr: 1–2. doi:10.1017/S1092852921000328. PMID33870884.
External links
- “Solriamfetol”. Drug Information Portal. U.S. National Library of Medicine.
- * “Solriamfetol hydrochloride”. Drug Information Portal. U.S. National Library of Medicine.
//////////////////
N[C@@H](COC(N)=O)CC1=CC=CC=C1
DESLORATADINE, デスロラタジン
Desloratadine
- Molecular FormulaC19H19ClN2
- Average mass310.821 Da
100643-71-8 [RN]
5H-Benzo[5,6]cyclohepta[1,2-b]pyridine, 8-chloro-6,11-dihydro-11-(4-piperidinylidene)-
7817
Desloratadine, Descarboethoxyloratadine, Sch-34117, DCL, Denosin, Clarinex RediTabs, Allex, Desalex, Opulis, Clarinex, Neoclarityn, Aerius, MK-4117
Desloratadine (trade name Clarinex and Aerius) is a tricyclic H1-antihistamine that is used to treat allergies. It is an active metaboliteof loratadine.
It was patented in 1984 and came into medical use in 2001.[1]
Medical uses
Desloratadine is used to treat allergic rhinitis, nasal congestion and chronic idiopathic urticaria (hives).[2] It is the major metabolite of loratadine and the two drugs are similar in safety and effectiveness.[2] Desloratadine is available in many dosage forms and under many trade names worldwide.[3]
An emerging indication for desloratadine is in the treatment of acne, as an inexpensive adjuvant to isotretinoin and possibly as maintenance therapy or monotherapy.[4][5]
Side effects
The most common side-effects are fatigue, dry mouth, and headache.[2]
Interactions
A number of drugs and other substances that are prone to interactions, such as ketoconazole, erythromycin and grapefruit juice, have shown no influence on desloratadine concentrations in the body. Desloratadine is judged to have a low potential for interactions.[6]
Pharmacology
Pharmacodynamics
Desloratadine is a selective H1–antihistamine which functions as an inverse agonist at the histamine H1 receptor.[7]
At very high doses, is also an antagonist at various subtypes of the muscarinic acetylcholine receptors. This effect is not relevant for the drug’s action at therapeutic doses.[8]
Pharmacokinetics
Desloratadine is well absorbed from the gut and reaches highest blood plasma concentrations after about three hours. In the bloodstream, 83 to 87% of the substance are bound to plasma proteins.[6]
Desloratadine is metabolized to 3-hydroxydesloratadine in a three-step sequence in normal metabolizers. First, n-glucuronidation of desloratadine by UGT2B10; then, 3-hydroxylation of desloratadine N-glucuronide by CYP2C8; and finally, a non-enzymatic deconjugation of 3-hydroxydesloratadine N-glucuronide.[9] Both desloratadine and 3-hydroxydesloratadine are eliminated via urine and feces with a half-life of 27 hours in normal metabolizers.[6][10]

3-Hydroxydesloratadine, the main metabolite
It exhibits only peripheral activity since it does not readily cross the blood-brain barrier; hence, it does not normally cause drowsiness because it does not readily enter the central nervous system.[11]
Desloratadine does not have a strong effect on a number of tested enzymes in the cytochrome P450 system. It was found to weakly inhibit CYP2B6, CYP2D6, and CYP3A4/CYP3A5, and not to inhibit CYP1A2, CYP2C8, CYP2C9, or CYP2C19. Desloratadine was found to be a potent and relatively selective inhibitor of UGT2B10, a weak to moderate inhibitor of UGT2B17, UGT1A10, and UGT2B4, and not to inhibit UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A9, UGT2B7, UGT2B15, UGT1A7, and UGT1A8.[9]
Pharmacogenomics
2% of Caucasian people and 18% of people from African descent are desloratadine poor metabolizers. In these people, the drug reaches threefold highest plasma concentrations six to seven hours after intake, and has a half-life of about 89 hours. However, the safety profile for these subjects is not worse than for extensive (normal) metabolizers.[6][10]
Clip
https://www.beilstein-journals.org/bjoc/articles/9/265
The value of substituted 3-picoline precursors is illustrated in the synthesis of clarinex (1.22, Desloratadine, Scheme 5), a dual antagonist of platelet activating factor (PAF) and of histamine used in the treatment of allergies. This compound consists of a highly functional tricyclic core with an unsaturated linkage to a pendant piperidine ring. The picoline derivative 1.23 is first treated with two equivalents of n-butyllithium (n-BuLi) followed by alkylation with benzyl chloride to give the chain elongated adduct [27]. The tert-butylamide 1.24 is then dehydrated with phosphorous oxychloride at elevated temperatures to yield the nitrile derivative 1.25. Introduction of the piperidine ring is achieved by utilisation of the appropriately substituted Grignard reagent 1.26. A Friedel–Crafts type acylation promoted by either triflic acid or polyphosphoric acid (PPA) furnishes the tricyclic structure 1.28 which upon N-demethylation affords clarinex (1.22).

CLIP
FTIR

SYN
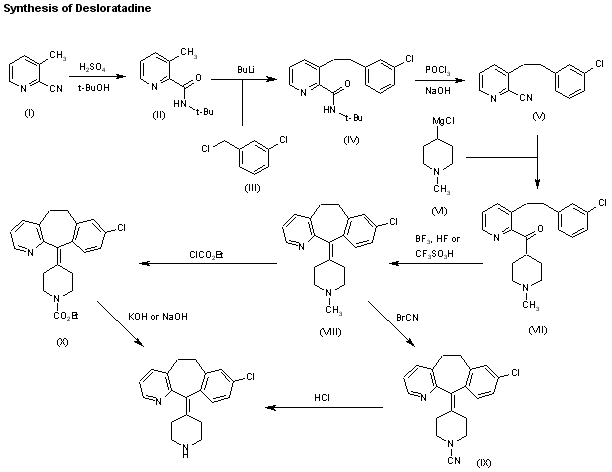
Alcoholysis of 3-methylpyridine-2-carbonitrile (I) with hot tert-butanol and H2SO4 gives the N-tert-butylcarboxamide (II), which is alkylated with 3-chlorobenzyl chloride (III) and BuLi in THF, yielding N-tert-butyl-3-[2-(3-chlorophenyl)ethyl]pyridine-2-carboxamide (IV). The reaction of (IV) with refluxing POCl3 and then with NaOH affords the corresponding nitrile (V), which is condensed with 1-methylpiperidin-4-ylmagnesium chloride (VI) in THF to give the ketone (VII). Cyclization of (VII) by means of either BF3 in HF or trifluoromethanesulfonic acid yields 8-chloro-11-(1-methylpiperidin-4-ylidene)-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridine (VIII), which is reacted with cyanogen bromide in benzene to give the N-cyano compound (IX). Finally, this compound is treated with HCl in refluxing acetic acid/water. Alternatively, 8-chloro-11-(1-methylpiperidin-4-ylidene)-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridine (VIII) is treated with ethyl chloroformate in hot toluene, affording the carbamate (X) (2), which is finally decarboxylated with KOH or NaOH in refluxing ethanol/water.
SYN
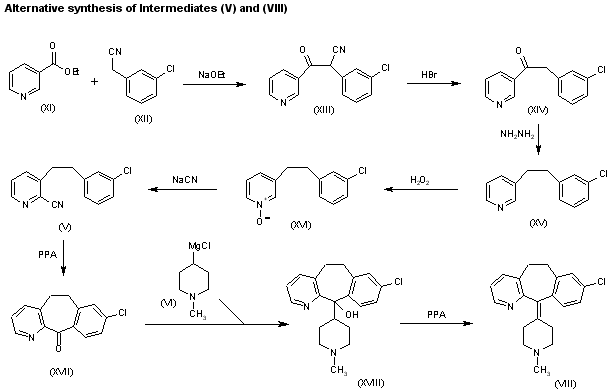
Condensation of ethyl nicotinate (XI) with 3-chlorophenylacetonitrile (XII) by means of sodium ethoxide in ethanol gives 2-(3-chlorophenyl)-3-oxo-3-(3-pyridyl)propionitrile (XIII), which by refluxing with concentrated HBr yields 2-(3-chlorophenyl)-1-(3-pyridyl)ethanone (XIV). The reduction of (XIV) with hydrazine hydrate and NaOH in diethylene glycol at 235-40 C affords 3-(2-phenylethyl) pyridine (XV), which is oxidized with H2O2 in hot acetic acid to provide the corresponding N-oxide (XVI). Reaction of (XVI) with NaCN and dimethyl sulfate in water affords the previously described 3-(2-phenylethyl)pyridine-2-carbonitrile (V), which can be worked up as previously described or cyclized with polyphosphoric acid (PPA) at 180 C to give 8-chloro-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin-11-one (XVII). The condensation of (XVII) with 1-methylpiperidin-4-ylmagnesium chloride (VI) in THF yields the corresponding carbinol (XVIII), which is dehydrated with PPA at 170 C to afford the previously reported 8-chloro-11-(1-methylpiperidin-4-ylidene)-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridine (VIII).
SYN
|
|
| Condensation of ethyl nicotinate (XI) with 3-chlorophenylacetonitrile (XII) by means of sodium ethoxide in ethanol gives 2-(3-chlorophenyl)-3-oxo-3-(3-pyridyl)propionitrile (XIII), which by refluxing with concentrated HBr yields 2-(3-chlorophenyl)-1-(3-pyridyl)ethanone (XIV). The reduction of (XIV) with hydrazine hydrate and NaOH in diethylene glycol at 235-40 C affords 3-(2-phenylethyl) pyridine (XV), which is oxidized with H2O2 in hot acetic acid to provide the corresponding N-oxide (XVI). Reaction of (XVI) with NaCN and dimethyl sulfate in water affords the previously described 3-(2-phenylethyl)pyridine-2-carbonitrile (V), which can be worked up as previously described or cyclized with polyphosphoric acid (PPA) at 180 C to give 8-chloro-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin-11-one (XVII). The condensation of (XVII) with 1-methylpiperidin-4-ylmagnesium chloride (VI) in THF yields the corresponding carbinol (XVIII), which is dehydrated with PPA at 170 C to afford the previously reported 8-chloro-11-(1-methylpiperidin-4-ylidene)-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridine (VIII). |
Syn
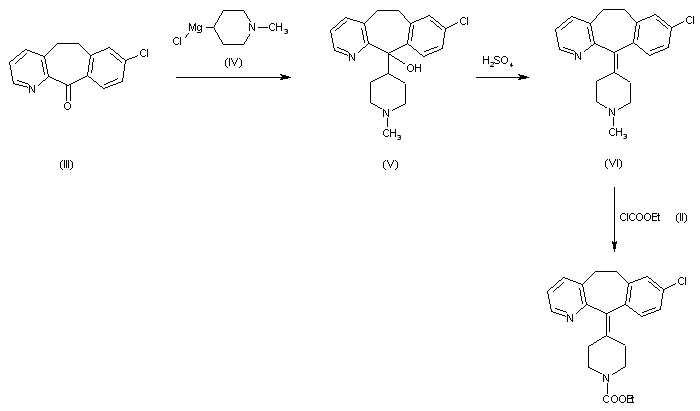
2) By reaction of 8-chloro-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin-11-one (III) with the Grignard reagent (IV) to give the tertiary carbinol (V), which is dehydrated with 85% H2SO4 affording 8-chloro-11-piperidinylidene derivative (VI). Finally, cornpound (VI) is treated with ethyl chloroformate (II) in toluene.
SYN
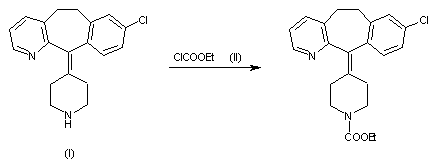
1) By carboxylation of 8-chloro-6,11-dihydro-11-(4-piperidylidene)-5H-benzo[5,6]cyctohepta[1,2-b]pyridine (I) with ethyl chloroformate (II) in refluxing benzene.
SYN
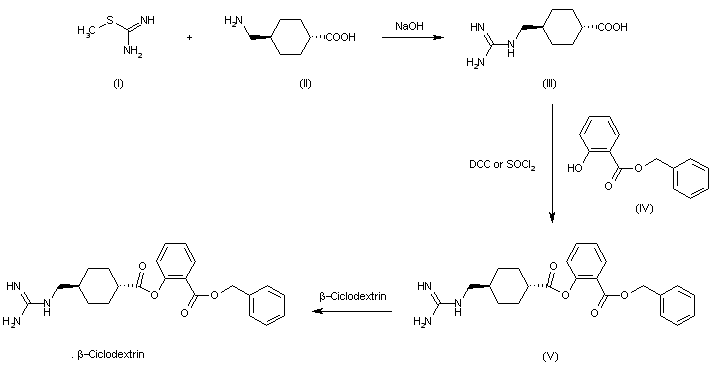
The condensation of S-methylisothiourea (I) with trans-4-(aminomethyl)cyclohexanecarboxylic acid (II) by means of NaOH in water gives trans-4-(guanidinomethyl)cyclohexanecarboxylic acid (III) (I), which is esterified with benzyl salicylate (IV) by means of dicyclohexylcarbodiimide (DCC) or SOCl2 yielding 2-benzyloxycarbonylphenyl trans-4-(guanidinomethyl)cyclohexanecarboxylate (V). Finally, this compound is treated with cyclodextrin in aqueous solution to afford the corresponding complex.
SPECTROSCOPY
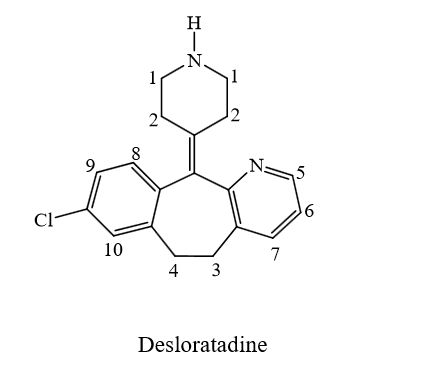
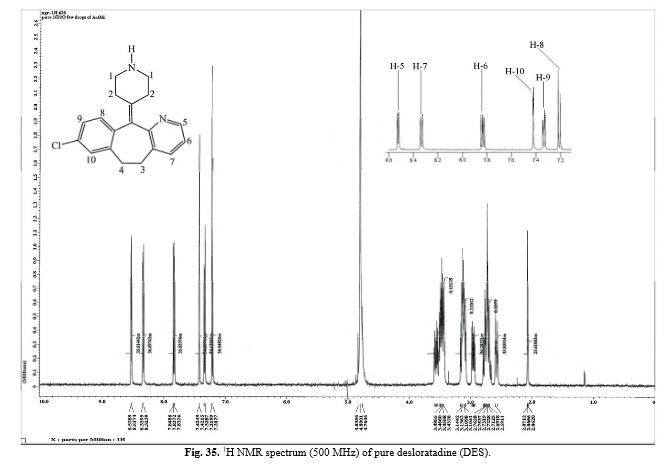
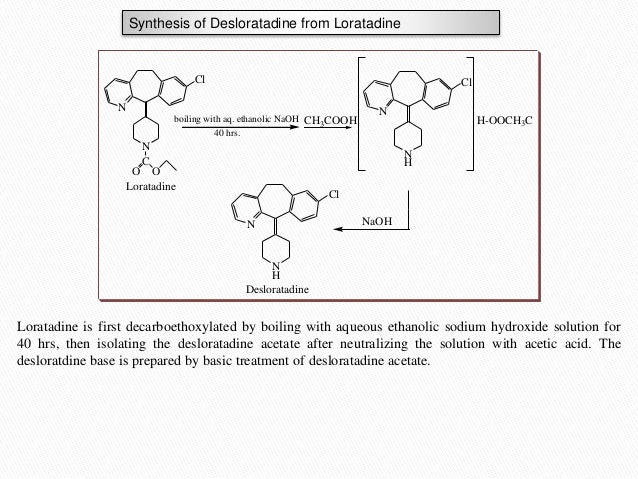
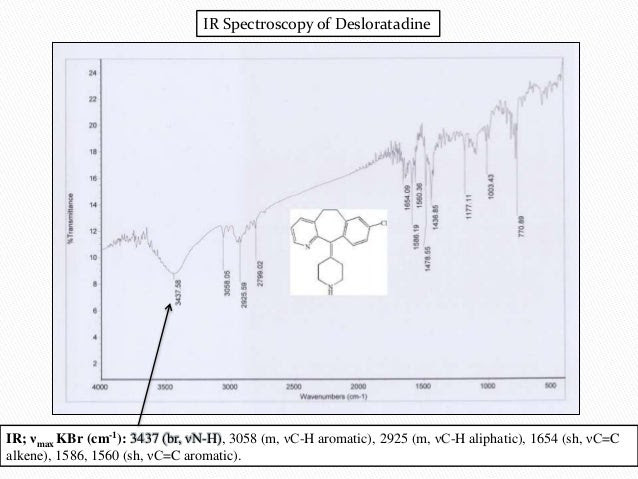


[0052] Table 1, desloratadine sample IH-NMR data of the DMS0_d6

[0055] The desloratadine 1H spectra of the samples were assigned:
[0056] (I) 1H spectra show that there are 10 groups of hydrogen from low field to high field integral hydrogen ratio was 1: 1: 1: 1: 1: 1: 2: 4:
2: 4, and desloratadine structure match.
[0057] (2) δ 8.334 处 hydrogen as a set of double doublet, number of protons is I, attributed to two hydrogen;
[0058] (3) δ 7.560 处 hydrogen as a set of double doublet, number of protons is I, attributed to four hydrogen;
[0059] (4) δ 7.282 处 doublet hydrogen as a group, the number of protons is I, 12 attributed to hydrogen.
[0060] (5) δ 7.198 处 hydrogen as a set of double doublet, number of protons is I, 14 attributed to hydrogen;
[0061] (6) δ 7.174 处 hydrogen as a set of double doublet, number of protons is I, attributed to three hydrogen;
[0062] (7) δ 7.064 处 doublet hydrogen as a group, the number of protons is I, 15 attributed to hydrogen;
[0063] (8) δ 3.314 处 hydrogen as a group multiplet, 2 protons attributable to 10 hydrogen;
[0064] (9) δ 2.831,2.554 hydrogen groups at multiplet, protons of 4, 18, 20, the home position is hydrogen;
[0065] (10) δ 2.819 处 hydrogen as a group multiplet, 2 protons attributable to 11 hydrogen;
[0066] (11) δ 2.108 处 hydrogen as a single peak, the number of protons is I, home to 19 active hydrogen;
[0067] (12) δ 2.205, 2.002 处 two hydrogen multiplet, protons of 4, 17, 21 bits attributed to hydrogen; [0068] From the foregoing, 1H-NMR spectrum data and the resulting product in this embodiment is of he will be loratadine same structure as the target product.
http://www.google.com/patents/CN103755682A?cl=en
References
- ^ Fischer, Jnos; Ganellin, C. Robin (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 549. ISBN 9783527607495.
- ^ Jump up to:a b c See S (2003). “Desloratadine for allergic rhinitis”. Am Fam Physician. 68 (10): 2015–6. PMID 14655812.
- ^ Drugs.com Desloratadine entry at drugs.com international Page accessed May 4, 2015
- ^ Lee HE, Chang IK, Lee Y, Kim CD, Seo YJ, Lee JH, Im M (2014). “Effect of antihistamine as an adjuvant treatment of isotretinoin in acne: a randomized, controlled comparative study”. J Eur Acad Dermatol Venereol. 28 (12): 1654–60. doi:10.1111/jdv.12403. PMID 25081735.
- ^ Layton AM (2016). “Top Ten List of Clinical Pearls in the Treatment of Acne Vulgaris”. Dermatol Clin. 34 (2): 147–57. doi:10.1016/j.det.2015.11.008. PMID 27015774.
- ^ Jump up to:a b c d “Aerius: EPAR – Product Information” (PDF). European Medicines Agency. 2017-06-07.
- ^ Canonica GW, Blaiss M (2011). “Antihistaminic, anti-inflammatory, and antiallergic properties of the nonsedating second-generation antihistamine desloratadine: a review of the evidence”. World Allergy Organ J. 4 (2): 47–53. doi:10.1097/WOX.0b013e3182093e19. PMC 3500039. PMID 23268457.
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Clarinex (US), Aerius, Dasselta, Deslordis (EU), others |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a602002 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
Oral (tablets, solution) |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | Rapidly absorbed |
| Protein binding | 83 to 87% |
| Metabolism | UGT2B10, CYP2C8 |
| Metabolites | 3-Hydroxydesloratadine |
| Onset of action | within 1 hour |
| Elimination half-life | 27 hours |
| Duration of action | up to 24 hours |
| Excretion | 40% as conjugated metabolites into urine Similar amount into the feces |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| ECHA InfoCard | 100.166.554 |
| Chemical and physical data | |
| Formula | C19H19ClN2 |
| Molar mass | 310.82 g/mol g·mol−1 |
| 3D model (JSmol) | |
//////////Desloratadine, Descarboethoxyloratadine, Sch-34117, DCL, Denosin, Clarinex RediTabs, Allex, Desalex, Opulis, Clarinex, Neoclarityn, Aerius, MK-4117
CMX-8521, CMX-521
CMX-8521, CMX-521
MF C13 H17 N5 O5, MW 323.30
CAS Number 2077178-99-3
7H-Pyrrolo[2,3-d]pyrimidine-5-carboxamide, 4-amino-2-methyl-7-β-D-ribofuranosyl-
Nucleoside analogs (oral, norovirus infection), Chimerix
4-amino-7-((2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-2-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carboxamide
4-amino-7-[(2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-2-methylpyrrolo[2,3-d]pyrimidine-5-carboxamide
CMX8521 is a nucleoside analog that inhibits the norovirus RNA polymerase. CMX8521 has in vitro activity against mouse and human norovirus.Where possible, Chimerix uses its lipid conjugate technology to build nucleoside-analog antivirals that are orally absorbed and have favorable tissue penetration.
CMX-8521 (presumed to be CMX-521) being developed by Chimerix for treating norovirus infection. In June 2018, a phase II efficacy trial was planned in 2019.
In January 2016, preclinical data were presented at the 34th Annual JP Morgan Healthcare Conference in San Francisco, CA. CMX-8521 had in vitro activity against mouse and human norovirus (EC50 = 2.1; CC50 = 114 microM). A 7-day non GLP toxicology/toxicokinetic study was completed in-life with no clinical or gross post mortem signs of toxicity. No off-target pharmacology was observed in vitro when screened against a panel of 87 receptors, transporters and enzymes associated with adverse pharmacology
PATENT
WO2017024310
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017024310
Scheme 1: General Synthesis of Compounds of the Invention

Scheme 2: General Synthesis of Compounds of the Invention

Example 7– Synthesis of Compound 1

[00315] Step 1 (Protocol #1): To a 100-L jacketed reactor were charged 4-amino-6- bromo-2-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (3.00 kg), (3R,4R,5R)-2-acetoxy-5- ((benzoyloxy)methyl)tetrahydrofuran-3,4-diyl dibenzoate (6.60 kg) and DCE (18.89 kg). Stirring was started and DBU (3.61) kg was added. Over a period of 03 h and 14 min, TMSOTf (8.01 kg) was added between 30.6 °C and 37.3 °C. IPC after 01 h and 30 min at approx.32 °C showed 4% of 4-amino-6-bromo-2-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (3.00 kg),
(3R,4R,5R)-2-acetoxy-5-((benzoyloxy)methyl)tetrahydrofuran-3,4-diyl dibenzoate remaining. IPC after 03h and 16 min at approx.32 °C showed 2% 4-amino-6-bromo-2-methyl-7H- pyrrolo[2,3-d]pyrimidine-5-carbonitrile (3.00 kg), (3R,4R,5R)-2-acetoxy-5- ((benzoyloxy)methyl)tetrahydrofuran-3,4-diyl dibenzoate remaining (spec:≤3%). The reaction mixture was diluted with DCM (39.81 kg) and quenched with potable water (15.02 kg) over an 11 min period between 9.5 °C and 15.6 °C. The extractive work-up (at approx.22 °C) was completed by a back extraction of the aqueous phase with DCM (19.90 kg), a wash with sat NaHCO3 (1.3 kg NaHCO3 in 14.9 kg potable water), a back extraction of the bicarbonate phase with DCM (19.71 kg) and a wash with brine (4.5 kg NaCl in 14.9 kg potable water). Note: the reactor was cleaned with potable water, acetone and DCM after each wash/back extraction.
[00316] The drummed organic phase containing the product was charged to the 100-L jacketed reactor through an in-line filter followed by a DCM rinse of the drum and filter with DCM (2.48 kg). The contents of the reactor were distilled to 31 L with the aid of vacuum over a period of 06 h and 04 min with a maximum temperature of 50.1 °C. At this point a thick suspension had formed. Next, over a period of 39 min, IPAc (41.88 kg) was added between 44.5 °C and 49.5 °C and the contents of the reactor were heated to 76.9 °C over a period of 01 h and 25 min. Next, the contents of the reactor were cooled to 9.9 °C over a period of 04 h and 21 min and stirred for 12 h and 26 min with a minimum temperature of 1.6 °C.
[00317] Step 1 (Protocol # 2): To a 100-L jacketed reactor were charged 4-amino-6- bromo-2-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (3.00 kg), (3R,4R,5R)-2-acetoxy-5- ((benzoyloxy)methyl)tetrahydrofuran-3,4-diyl dibenzoate (6.60 kg) and DCE (18.80 kg). Stirring was started and DBU (3.59) kg was added. Over a period of 01 h and 46 min, TMSOTf (7.90 kg) was added between 30.4 °C and 34.2 °C. IPC after 02 h and 49 min at approx.34 °C showed 1% of 4-amino-6-bromo-2-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile remaining (spec: ≤3%). The reaction mixture was diluted with DCM (40/70 kg) and quenched with potable water (14.97 kg) over an 04 min period between 9.9 °C and 18.0 °C. The extractive work-up (at approx.22 °C) was completed by a back extraction of the aqueous phase with DCM (20.34 kg), a wash with sat NaHCO3 (1.30 kg NaHCO3 in 14.90 kg potable water), a back extraction of the bicarbonate phase with DCM (20.65 kg) and a wash with brine (4.50 kg NaCl in 14.96 kg potable water). Note: the reactor was cleaned with potable water, acetone and DCM after each wash/back extraction.
[00318] The drummed organic phase containing the product was charged to the 100-L jacketed reactor through an in-line filter followed by a DCM rinse of the drum and filter with DCM (1.49 kg). The contents of the reactor were distilled to with the aid of vacuum over a period of 04 h and 49 min with a maximum temperature of 45.6 °C. At this point a thick suspension had formed. Next, over a period of 27 min, IPAc (41.70 kg) was added between 45.6 °C and 48.2 °C and the contents of the reactor were heated to 75.7 °C over a period of 01 h and 20 min. Next, the contents of the reactor were cooled to 9.4 °C over a period of 04 h and 15 min and stirred overnight with a minimum temperature of 2.3 °C.
[00319] Step 2: To the reactor were charged (2R,3R,4R,5R)-2-(4-amino-6-bromo-5- cyano-2-methyl-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-5-((benzoyloxy)methyl)tetrahydrofuran-3,4- diyl dibenzoate (10.0 kg), 10% Pd on C (Degussa, Type E101NE/W), trimethylamine (7.3 kg) and THF (44.5 kg). Hydrogen was submitted to the reactor and the mixture was stirred for 03 h and 54 min between 24.7 °C and 19.6 °C at approx.30.8 psig. IPC (HPLC) showed that
(2R,3R,4R,5R)-2-(4-amino-6-bromo-5-cyano-2-methyl-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-5- ((benzoyloxy)methyl)tetrahydrofuran-3,4-diyl dibenzoate could no longer be detected.
[00320] The reaction mixture was filtered over Celite (7.2 kg) and a polish filter and the filter residue was washed with THF (5.2 kg). The combined filtrate and wash was transferred to a 100-L jacketed reactor with the aid of a THF wash (2.12 kg). The contents of the reactor were vacuum distilled with a maximum batch temperature of 30.0 °C over a period of 05 h and 38 min to a final volume of 27 L. IPA (31.48 kg) was charged over a 40 min period to the reactor between 39.7 °C and 53.2 °C. The contents of the reactor were vacuum distilled with a maximum batch temperature of 53.2 °C over a period of 03 h and 02 min to a final volume of 33 L. IPA (48.99 kg) was charged over a 43 min period to the reactor between 53.1 °C and 57.1 °C. The contents of the reactor were heated to 60.2 °C, agitated for 12 min and cooled over a period of 04 and 28 min to 5.4 °C. Cold stirring was continued for a period of 08 h and 55 min with a minimum temperature of 1.1 °C. The slurry was filtered and washed with IPA (9.41 kg, at approx.4.5 °C). The residue was dried under vacuum with a nitrogen bleed for a period of 11 h and 44 min at a maximum temperature of 44.0 °C to provide an LOD of 0.36%. Yield: 6.58 kg (73.9 %).1H NMR confirms structure. Purity: 97.78 % (HPLC, AUC).
[00321] Step 3:

1100 g NaOH dissolved in potable water to a total volume of 1 L; 2 Diluted 500 mL conc. HCl in 2 L total with potable water [00322] A solution of (2R,3R,4R,5R)-2-(4-amino-5-cyano-2-methyl-7H-pyrrolo[2,3- d]pyrimidin-7-yl)-5-((benzoyloxy)methyl)tetrahydrofuran-3,4-diyl dibenzoate and THF was heated to 54 °C and the addition of 2.5 M NaOH was started. The initial addition gave a biphasic mixture and endothermic response (the temperature dropped to 50 °C) but as the addition continued a single phased, clear solution formed which was accompanied by a fast exotherm to 61 °C; the reaction temperature was maintained at 60 °C to 61 °C during the rest of the addition and for an additional 2 ½ h. IPC showed that no (2R,3R,4R,5R)-2-(4-amino-5-cyano-2-methyl- 7H-pyrrolo[2,3-d]pyrimidin-7-yl)-5-((benzoyloxy)methyl)tetrahydrofuran-3,4-diyl dibenzoate was left.
[00323] The reaction mixture was cooled to 21 °C and neutralized with 3 N HCl with external cooling to pH = 7.06 (Denver Instrument UB-10 pH meter equipped with a Sartorius P- P11 pH electrode, the electrode was checked with buffer solutions of pH = 4.00 and pH = 7.00); the mixture continued to cool to 8°C. The resulting neutralized mixture was distilled under vacuum with a pot temperature of 45 °C to 50 °C until the emergence of solids were observed in the pot. The suspension was cooled and stirred for 2 h at 2 °C. The beige suspension was filtered to afford a dark filtrate; the off-white residue was washed once with cold water (500 mL, 5 °C). A first LOD after 16 h gave a value of 18.73 %. HPLC) of the drying material showed the presence of 1.6% benzoate.
[00324] A brief rework study for compound 1, (containing 1.6% benzoic acid per AUC, HPLC) was executed in 10 vol of water (1 g in 10 mL):
● 3 h slurry at ambient
● 3h slurry at 50 °C
● 24 h slurry at ambient
[00325] All three experiments gave compound 1 with less than 0.1 % benzoic acid (UAC, HPLC). The slurries were fluid, were easily stirred and filtration was fast. Short term drying on the filter gave a powder-like solid indicating that a displacement wash with an organic solvent is not needed. Without wishing to be bound by theory, a loss of NMT than 1% is expected
(solubility 1 mg/mL).HPLC data for compound 1 were obtained with a method suitable for polar compounds using a Zorbax Eclipse Plus C18 column (water / ACN / TFA, 97.5 / 2.5 / 0.05). This is the same column used for steps 1 and 2.
[00326] The cold product suspension was filtered and the reactor and residue were washed with cold IPAc (approx.7.5 °C, 13.16 kg and 13.62 kg) until a colorless filtrate had been obtained. The residue was dried under vacuum and a nitrogen bleed≤ 45 °C for a period of 65 h and 19 min to an LOD of 0 %. Yield: 5.87 kg (70.7 %), 1H NMR confirmed identity; HPLC purity 98.84% (AUC). EQUIVALENTS
[0001] The disclosure can be embodied in other specific forms without departing from the spirit or essential characteristics thereof. The foregoing embodiments are therefore to be considered in all respects illustrative rather than limiting on the disclosure described herein. Scope of the disclosure is thus indicated by the appended claims rather than by the foregoing description, and all changes that come within the meaning and range of equivalency of the claims are intended to be embraced therein.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019060692&tab=PCTDESCRIPTION&maxRec=1000
Novel crystalline forms of 4-amino-7-((2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl) tetrahydrofuran-2-yl)-2-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carboxamide and their stable hemihydrate crystalline forms (designated as Form A-G), processes for their preparation and compositions comprising them are claimed. Also claimed is their use for treating viral infection.
Viral infections can have serious adverse effects on individuals and society as a whole. In addition to fatal viral infections such as Ebola, even non-fatal infections can have serious societal and economic consequences. For example, human noroviruses (NV) are the most common cause of epidemic acute gastroenteritis worldwide with an estimated 19-21 million cases each year in the United States including 56,000-71,000 hospitalizations and 570-800 deaths (Hall et al., Emerg.Infect.Dis. 2013 Aug; 19(8): 1198-205).
[0004] 4-amino-7-((2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl) tetrahydrofuran-2-yl)-2-methyl-7H-pyrrolo [2,3-d]pyrimidine-5-carboxamide (Compound 1) is an antiviral drug.
Formula 1
[0065] As used herein, “Formula I” is understood to encompass all diastereomers of 4-amino-7-(3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-2-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carboxamide, and pharmaceutically acceptable salts and solvates thereof. The structure of Formula I is shown below:
(Formula I).
[0066] In some embodiments, a compound of Formula I can be 4-amino-7-((2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-2-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carboxamide (“Compound 1”), or a pharmaceutically acceptable salt solvate, or isomers (e.g., enantiomers and diastereomers) thereof. The structure of Compound 1 is shown below:
| atent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US9701706 | Pyrrolopyrimidine nucleosides and analogs thereof | 2016-11-22 | 2017-07-11 |
| US9708359 | PYRROLOPYRIMIDINE NUCLEOSIDES AND ANALOGS THEREOF | 2016-08-08 | |
| US2017253628 | PYRROLOPYRIMIDINE NUCLEOSIDES AND ANALOGS THEREOF | 2017-05-18 |
///////////CMX-8521, CMX 8521, CMX-521, PHASE 1
NC(=O)c2cn([C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O)c3nc(C)nc(N)c23
RISDIPLAM , リスジプラム
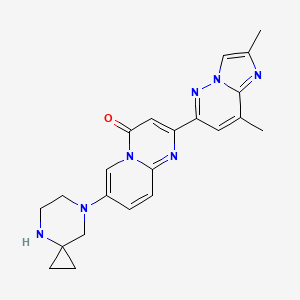
RISDIPLAM
RG-7916, RO-7034067, リスジプラム
| Formula |
C22H23N7O
|
|---|---|
| Cas |
1825352-65-5
|
| Mol weight |
401.4643
|
| US9969754 |
7-(4,7-diazaspiro[2.5]octan-7-yl)-2-(2,8-dimethylimidazo[1,2-b]pyridazin-6-yl)pyrido[1,2-a]pyrimidin-4-one
The compound was originally claimed in WO2015173181 , for treating spinal muscular atrophy (SMA). Roche , under license from PTC Therapeutics , and Chugai , are developing risdiplam (RO-7034067; RG-7916), a small-molecule survival motor neuron (SMN)2 gene splicing modulator and a lead from an SMN2 gene modulator program initiated by PTC Therapeutics in collaboration with the SMA Foundation , for the oral treatment of spinal muscular atrophy
The product was granted orphan drug designation in the U.S., E.U. and in Japan for the treatment of spinal muscular atrophy. In 2018, it also received PRIME designation in the E.U. for the same indication.
Risdiplam (RG7916, RO7034067) is a highly potent, selective and orally active small molecule experimental drug being developed by F. Hoffmann-La Roche, PTC Therapeutics and SMA Foundation to treat spinal muscular atrophy (SMA). It is a pyridazine derivative that works by increasing the amount of functional survival of motor neuron protein produced by the SMN2 gene through modifying its splicing pattern.[1][2]
As of September 2018, risdiplam is undergoing late-stage clinical trials across the spectrum of spinal muscular atrophy[3][4][5] where it has shown promising preliminary results.[6][7]
PATENT
WO2015173181
Example 20
7-(4,7-diazaspiro[2.5]octan-7-yl)-2-(2,8-dimethylimidazo[l,2-b]pyridazin-6- yl)pyrido[l,2-a]pyrimidin-4-one
In a sealed tube, 2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)-7-fluoro-pyrido[l,2-a]pyrimidin-4-one (Intermediate 2; 50 mg, 0.162 mmol), DIPEA (0.22 mL, 1.29 mmol, 4 eq.) and 4,7-diazaspiro[2.5]octane dihydrochloride (32 mg, 0.320 mmol, 3.0 eq.) were stirred in
DMSO (2 mL) at 130°C for 48 hours. The solvent was removed under high vacuum. The residue was taken up in CH2CI2 and washed with an aqueous saturated solution of NaHC03. The organic layer was separated and dried over Na2S04 and concentrated in vacuo. The crude was purified by column chromatography (Si02, CH2Cl2/MeOH=98/2 to 95/5) to afford the title product (12 mg, 18%) as a light yellow solid. MS m/z 402.3 [M+H+].

PATENT
WO-2019057740
Process for the preparation of risdiplam and its derivatives.
Scheme 1:
Scheme 3:
Scheme 4:
xample 1: tert-Butyl 7-(6-chloro-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate
5-Bromo-2-chloropyridine (85.0 g, 442 mmol), tert-butyl 4,7-diazaspiro[2.5]octane-4-carboxylate (102 g, 442 mmol) and Me-THF (722 g) were charged into a reaction vessel. After 10 minutes stirring, most of the solids were dissolved and [Pd(Xantphos)Cl2] (3.34 g) was added followed after 5 minutes by a solution of sodium tert-butanolate (56.3 g, 574 mmol) in Me-THF (173 g). The reaction mixture was stirred at 70 °C for 1.25 hours, cooled to room temperature and water (595 g) and 1-propylacetate (378 g) were added. After vigorous stirring, the phases were separated, the organic phase was washed with a second portion of water (425 g) and with a mixture of water (425 g) and brine (25 mL). The organic phase was treated with active charcoal (6.8 g), filtered and concentrated under reduced pressure to afford a brown oil, which was dissolved in tert-amyl-methyl-ether (347 g) at reflux. The solution was cooled slowly to room temperature. After stirring 18 hours at room temperature, n-heptane (205 g) was added and the suspension was further cooled to -10 °C. The precipitate was filtered off and dried under high vacuum to afford tert-butyl 7-(6-chloro-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate (110.9 g, 77.5%) as a beige solid.
Ή-ΝΜΡν (CDC13, 600 MHz): 7.95 (d, 1H); 7.18 – 7.14 (m, 1H); 7.13 – 7.09 (m, 1H); 3.79 – 3.63 (m, 2H); 3.24 – 3.12 (m, 2H); 2.96 (s, 2H); 1.47 (s, 9H); 1.11 – 1.04 (m, 2H); 0.90 -0.79 (m, 2H); LCMS: 324.15, 326.15 (M+H+)
Example 2: tert-butyl 7-(6-amino-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate
An autoclave equipped with an ascending pipe was filled with ammonia (78.7 g, 15 eq; 10 eq are sufficient) at -70 °C. Another autoclave was charged with tert-butyl 7-(6-chloro-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate (100 g, 309 mmol), sodium tert-butanolate (32.6 g, 340 mmol) and dioxane (800 mL). After 10 minutes stirring at room temperature under Ar, a solution of Pd2(dba)3 (1.41 g, 1.54 mmol) and tBuBrettPhos (1.50 g, 3.09 mmol) in dioxane (180 mL) was added. Thereafter, the connected ammonia vessel was warmed with a warm water bath and the connecting valve was opened. The autoclave was warmed to 30 °C and the reaction mixture stirred 5 hours at this temperature. The ammonia vessel was closed and disconnected. The excess ammonia was washed out of the autoclave with Argon. The reaction solution was poured into a separating funnel, the autoclave washed with ethyl acetate (300 mL) and water (100 mL) and these two solvent portions were added to the separating funnel. The biphasic mixture was further diluted with ethyl acetate (900 mL) and water (1000 mL). After vigorous stirring, the phases were separated. The organic phase was washed with a mixture of water (500 mL) and brine (10 mL). The combined aqueous phases were extracted twice with ethyl acetate (500 mL). The combined organic phases were treated with active charcoal (3.70 g, 309 mmol), filtered and the filtrate was concentrated under reduced pressure to afford a thick brown oil. This oil was dissolved in 1 -propyl acetate (160 mL) at 45-50°C and n-heptane (940 mL) was added drop wise within 1.5 hours. The suspension was cooled slowly to -5°C, stirred 4 hours at -5 °C and filtered. The precipitate was washed with cold n-heptane and dried under high vacuum at 50°C to afford tert-butyl 7-(6-amino-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate (81.4 g, 86.5%) as a beige solid.
Ή-ΝΜΡν (CDCb, 600 MHz): 7.71 (d, 1H); 7.12 (dd, 1H); 6.47 (d, 1H); 4.18 (br s, 2H); 3.74 – 3.58 (m, 2H); 3.09 – 2.94 (m, 2H); 2.81 (s, 2H); 1.52 – 1.39 (m, 9H); 1.17 – 0.98 (m, 2H); 0.92 – 0.75 (m, 2H); LCMS: 305.20 (M+H+)
Example 3: tert-butyl 7-(6-amino-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate
An autoclave was charged with tert-butyl 7-(6-chloro-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate (339 mg, 1 mmol), sodium tert-butanolate (109 mg, 1.1 mmol) and dioxane (5 mL). After 5 minutes stirring at room temperature under Argon [Pd(allyl)(tBuBrettPhos)]OTf (4 mg, 5 μιηοΐ) was added. Thereafter, the autoclave was closed and connected to an ammonia tank, the valve was open and ammonia (230 mg, 13.5 mmol) was introduced into the autoclave. The valve was closed and the autoclave disconnected. The autoclave was warmed to 30 °C and the reaction mixture stirred 4 hours at this temperature. Then the autoclave was opened and the excess ammonia was washed out of the autoclave with Argon. The reaction solution was poured into a flask and taken to dryness under reduced pressure. The residue was purified by chromatography over silica gel (eluent: dichloromethane/ethyl acetate to dichloromethane/methanol). After evaporation of the solvents tert-butyl 7-(6-amino-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate (283 mg, 93%) was isolated as a brown oil containing 4% dichloromethane and 3% ethyl acetate.
Example 4: tert-butyl 7-(6-nitro-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate
tert-Butyl 4,7-diazaspiro[2.5]octane-4-carboxylate oxalate salt (2.46 kg, 8.13 mol), 5-bromo-2-nitro-pyridine (1.50 kg, 7.39 mol) and dimethyl sulfoxide (7.80 L) were char; into a reaction vessel pre-heated to 35 °C. With stirring, and keeping the temperature below 40°C, lithium chloride (1.25 kg, 25.6 mol) was added portion- wise followed by tetramethylguanidine (2.98 kg, 25.9 mol). Dimethyl sulfoxide (450 mL) was used to rinse the feed line. The reaction mixture was stirred at 79 °C for 8 hours, cooled to 70°C and water (2.48 L) was added within 2 hours. After stirring at 70 °C for an additional 1 hour, the precipitate was filtered off and washed with water (4.5 L) three times. The precipitate was dissolved in ethyl acetate (15 L) and water (7.5 L) at reflux temperature. The phases were separated at 60°C and n-heptane (7.5 L) was added to the organic layer at 60°C within 30 minutes. The solution was cooled to 0°C in 2 hours and further stirred at 0°C for 1 hour. The precipitate was filtered off, washed with a mixture of ethyl acetate (750 mL)/n-heptane (375 mL) twice and dried under reduced pressure to afford 1.89 kg (76.4%) of tert-butyl 7-(6-nitro-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate as a yellow to light brown solid.
!H-NMR (CDCls, 600 MHz): 8.16 (d, 1H); 8.07 (d, 1H); 7.15 (dd, 1H); 3.80 – 3.72 (m, 2H); 3.49 – 3.41 (m, 2H); 3.23 (s, 2H); 1.48 (s, 9H); 1.16 – 1.08 (m, 2H); 0.92 – 0.85 (m, 2H); LCMS: 335.17 (M+H+)
Example 5: tert-butyl 7-(2-hydroxy-4-oxo-pyrido[l,2-a]pyrimidin-7-yl)-4,7-diazaspiro[2.5]octane-4-carboxylate
tert-Butyl 7-(6-amino-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate (80.0 g, 263 mmol) was dissolved in anisole (800 mL) and di-tert-butyl malonate (71.1 g, 315 mmol) was added. The solution was stirred 3.5 hours at 145 °C then cooled to room temperature. The precipitate was filtered off, washed with toluene (in portions, 320 mL in total) and dried under high vacuum at 50°C to afford tert-butyl 7-(2-hydroxy-4-oxo-pyrido[l,2-a]pyrimidin-7-yl)-4,7-diazaspiro[2.5]octane-4-carboxylate (65.6 g, 67%) as a light pink powder.
Ή-ΝΜΡν (CDCI3, 600 MHz): 8.46 (d, 1H); 7.74 (dd, 1H); 7.52 (d, 1H); 5.37 (s, 2H); 3.83 – 3.69 (m, 2H); 3.23 (t, 2H); 3.01 (s, 2H); 1.48 (s, 9H); 1.17 – 1.03 (m, 2H); 0.95 – 0.75 (m, 2H); LCMS: 373.19 (M+H+)
Example 6: tert-butyl 7-(2-hydroxy-4-oxo-pyrido[l,2-a]pyrimidin-7-yl)-4,7-diazaspiro[2.5]octane-4-carboxylate
tert-Butyl 7-(6-nitro-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate (950 g, 2.84 mol), Pt 1%, V 2% on active charcoal (95.1 g, 2 mmol) and ethyl acetate (9.5 L) were charged into an autoclave that was pressurized with hydrogen gas to 3 bar. The reaction mixture was stirred at room temperature for 6 hours. The excess hydrogen was vented. The reaction mixture was filtered, the catalyst was washed with ethyl acetate (0.95 L) three times. The filtrate was concentrated under reduced pressure and the solvent exchanged to anisole (add two portions of 2.85 L and 5.18 L) by distillation. Di tert-butyl malonate (921.7 g, 4.26 mol) was added and the charging line was rinsed with anisole (618 mL) and the reaction mixture was stirred at 125-135 °C for 8 hours. It may be necessary to distill off the by-product tert-butanol to reach this temperature. The progress of the reaction was followed eg.by HPLC. If the reaction stalls, the temperature is increased to 135-145°C and checked for progress after 1 hour. When the reaction was complete, the batch was cooled to room temperature and stirred at room temperature for 4 hours. The precipitate was filtered off, washed with toluene (3.55 L) and dried under vacuum at 60°C to afford tert-butyl 7-(2-hydroxy-4-oxo-pyrido[l,2-a]pyrimidin-7-yl)-4,7-diazaspiro[2.5]octane-4-carboxylate (861.0 g, 81.4%) as a yellow to light brown solid.
Example 7: tert-butyl 7-[4-oxo-2-(p-tolylsulfonyloxy)pyrido[l,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5]octane-4-carboxylate
A reactor was charged with tert-butyl 7-(2-hydroxy-4-oxo-pyrido[l,2-a]pyrimidin-7-yl)-4,7-diazaspiro[2.5]octane-4-carboxylate (920 g, 2.47 mol) and then triethylamine (325 g, 3.21 mol), followed by tosyl chloride (527.1 g, 2.77 mol) and dichloromethane (4.6 L). The reaction mixture was stirred at 20-25 °C for at least three hours. Upon complete reaction, the organic solution was washed with a prepared solution of HC1 (32%, 247.8 mL) and water (4.6 L), followed by a prepared solution of sodium hydroxide (432.3 mL of a 30% stock solution) and water (3.9 L) in that order. The organic phase was finally washed with water (4.8 L) and then dichloromethane was nearly completely distilled off under reduced pressure at 50-55°C. Ethyl acetate (920 mL) was added and distilled twice at this temperature under reduced pressure, and then ethyl acetate (4.8 L) was added and the suspension cooled to 20-25 °C over two hours. n-Heptane (944.4 mL) was added and the mixture was cooled to 0-5 °C and then stirred for an additional 3 hours. The precipitate was filtered off, washed with a prepared solution of ethyl acetate (772.8 mL) and n-heptane (147.2 mL), and then twice with n-heptane (2.6 L). The solid was dried under vacuum at 45-50°C to afford 1122.6 g (86.3%) tert-butyl 7-[4-oxo-2-(p-tolylsulfonyloxy)pyrido[l,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5]octane-4-carboxylate as yellow crystals.
!H-NMR (CDCls, 600 MHz): 8.32 (d, 1H); 8.00 – 7.89 (m, 2H); 7.66 (dd, 1H); 7.50 (d, 1H); 7.36 (d, 2H); 6.04 (s, 1H); 3.80 – 3.68 (m, 2H); 3.23 (t, 2H); 3.01 (s, 2H); 1.48 (s, 9H); 1.15 – 1.04 (m, 2H); 0.92 – 0.82 (m, 2H); LCMS: 527.20 (M+H+)
Example 8: 2,8-dimethyl-6-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)imidazo[l,2-b]pyridazine
6-Chloro-2,8-dimethylimidazo[l,2-b]pyridazine (40.0 g, 220 mmol), bis pinacol diborane (69.9 g, 275 mmol) and potassium acetate (43.2 g, 440 mmol) were suspended in acetonitrile (440 mL). The suspension was heated to reflux and stirred 30 minutes at reflux, then a suspension of PdCl2(dppf) (4.03 g, 5.51 mmol) and dppf (610 mg, 1.1 mmol) in acetonitrile (40 mL) was added. The vessel was rinsed with acetonitrile (20 mL), which were also poured into the reaction mixture. The orange suspension was further stirred at reflux, whereby acetonitrile (50 mL) were distilled off. After 4 hours, the reaction mixture was filtered off, the filter was washed with several portions of acetonitrile (in total 150 mL). The filtrate was diluted to obtain a volume of 700 mL. The 314 mmolar solution of 2,8-dimethyl-6-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)imidazo[l,2-b]pyridazine in acetonitrile was used as such in the next step.
Example 9: 2,8-dimethyl-6-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)imidazo[l,2-b]pyridazine
6-chloro-2,8-dimethylimidazo[l,2-b]pyridazine (29.0 g, 22.8 mmol), bis pinacol diborane (44.6, 25.1 mmol) and potassium acetate (31.3 g, 45.6 mmol) were suspended in 1-propyl acetate (365 mL). The suspension was heated to 80°C and a solution of
tricyclohexylphosphine (448 mg, 0.23 mmol) and Pd(OAc)2 (179 mg, 0.11 mmol) in 1-propyl acetate (37 mL) was added within 20 minutes. After 2.5 hours further stirring at 80°C, the suspension was cooled to 40°C and filtered at this temperature. The precipitate was washed with 1-propyl acetate (200 mL). The filtrate corresponds to 516.4 g of a 8.5% solution of 2,8-dimethyl-6-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)imidazo[l,2-b]pyridazine in 1 -propyl acetate.
Example 10: Isolation of 2,8-dimethyl-6-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)imidazo[ 1 ,2-b]pyridazine
In another experiment, the above solution obtained was cooled to 0-5 °C within 3 hours. The precipitate was filtered off, washed with cold 1 -propyl acetate and dried under high vacuum at 60°C to afford 2,8-dimethyl-6-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)imidazo[l,2-b]pyridazine (24. Og, 55%) as a colourless solid.
lH NMR (CDCls, 600 MHz, ) δ ppm 7.86 (d, J=0.7 Hz, 1 H), 7.20 (d, J=1.0 Hz, 1 H), 2.63 (d, J=1.0 Hz, 3 H), 2.51 (d, J=0.7 Hz, 3 H), 1.33 – 1.49 (m, 12 H)
Example 11: (step 6) tert-butyl 7-[2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)-4-oxo-pyrido[l,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5]octane-4-carboxylate
tert-Butyl 7-[4-oxo-2-(p-tolylsulfonyloxy)pyrido[l,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5] octane-4-carboxylate (25 g, 47.5 mmol), 2,8-dimethyl-6-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)imidazo[l,2-b]pyridazine (314 mM in acetonitrile, 191 mL, 59.8 mmol), PdCi2(dppf) (868 mg, 1.19 mmol) and aqueous potassium carbonate 4.07 M (17.1 mL, 69.8 mmol) were charged into a reaction vessel. The reaction mixture was stirred at reflux for 3 hours, cooled overnight to room temperature and filtered. The precipitate was washed with several portions of acetonitrile (146 mL in total), then suspended in methyl-THF (750 mL) and methanol (75 mL). Aqueous sodium hydrogen carbonate 5% (250 mL) was added, the mixture was vigorously stirred at 35°C. The phases were separated, the organic phase was washed again with aqueous sodium hydrogen carbonate 5% (250 mL). The organic phase was treated with active charcoal for 1 hour at room temperature, filtered and the filtrate was concentrated under reduced pressure at 60 °C to a volume of 225 mL, heated to reflux then cooled to room temperature, stirred at room temperature for 16 hours, then cooled to 0°C and stirred at 0°C for 3 hours. The precipitate was filtered off, washed with n-heptane (60 mL) and dried under high vacuum at 55°C to afford tert-butyl 7-[2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)-4-oxo-pyrido[l,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5]octane-4-carboxylate (20.13 g, 84.5%) as a yellow solid.
This solid could be recrystallized in the following manner: 15 g of the above solid was dissolved at reflux in toluene (135 mL) and ethanol (15 mL). The solution was slowly cooled to room temperature, stirred 16 hours at room temperature, then cooled to 0°C and stirred at 0°C for 4 hours. The precipitate was filtered off, washed with cold toluene and dried under high vacuum at 55°C to afford tert-butyl 7-[2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)-4-oxo-pyrido[l,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5]octane-4-carboxylate (11.92 g, 79.5%) as a yellow-green solid.
!H-NMR (CDCls, 600 MHz): 8.44 (d, 1H); 7.93 (d, 1H); 7.96 – 7.89 (m, 1H); 7.80 (d, 1H); 7.76 – 7.72 (m, 1H); 7.70 – 7.63 (m, 1H); 7.38 (s, 1H); 3.85 – 3.69 (m, 2H); 3.28 (t, 2H); 3.07 (s, 2H); 2.74 (d, 3H); 2.55 (s, 3H); 1.49 (s, 9H); 1.16 – 1.09 (m, 2H); 0.93 – 0.86 (m, 2H); LCMS: 502.26 (M+H+)
Example 12: tert-butyl 7-[2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)-4-oxo-pyrido[l,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5]octane-4-carboxylate
6-chloro-2,8-dimethylimidazo[l,2-b]pyridazine (4.14 g, 22.8 mmol), bis pinacol diborane (6.37g, 25.1 mmol) and potassium acetate (4.47 g, 45.6 mmol) were suspended in 1-propyl acetate (59 mL). The suspension was heated to 80°C and a solution of
tricyclohexylphosphine (63.9 mg, 0.23 mmol) and Pd(OAc)2 (25.6 mg, 0.11 mmol) in 1-propyl acetate (6 mL) was added within 20 minutes. After 2.5 hours further stirring at 80°C, the suspension was cooled to 40°C and filtered at this temperature. The precipitate was washed with 1-propyl acetate (32 mL). The filtrate corresponds to 74.6 g of a 8.5% solution of 2,8-dimethyl-6-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)imidazo[l,2-b]pyridazine in 1-propyl acetate.
A reaction vessel was charged with tert-butyl 7-[4-oxo-2-(p-tolylsulfonyloxy)pyrido[l,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5]octane-4-carboxylate (10.0 g, 19.0 mmol), tricyclohexylphosphine (58.6 mg, 0.21 mmol) and Pd(OAc)2 (21.3 mg, 0.10 mmol) and 1-propyl acetate (42 mL) and a solution of potassium carbonate (5.25 g, 38.0 mmol) in water (19.0 mL) was added. The suspension was heated to 70°C and the solution of 2,8-dimethyl-6-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)imidazo[l,2-b]pyridazine in 1-propyl acetate was added within 30 minutes. The mixture was stirred for 2 hours at 70-75°C. The suspension was cooled to 40°C, water (10 mL) was added. The suspension was aged for 30 minutes. The crude product was filtered off and rinsed with 1-propyl acetate (41 mL). The crude product was taken up in toluene (100 mL), 5% aqueous NaHC03-solution (30 mL) and 1-propanol (20.0 mL). The mixture was heated to 60-65 °C, the phases were separated and the organic phase was washed with 2 more portions of water (30.0 mL). The organic phase was filtered on active charcoal, the filter washed with toluene (60.0 mL). The filtrate was concentrated under reduced pressure to a volume of ca. 120 mL, heated to reflux and 1-propanol (0.8 mL) was added to obtain a solution. The solution was cooled to 0-5°C within 4-6 hours, stirred at 0-5°C for 1 hour. The precipitate was filtered off, washed with toluene (30 mL) and dried under reduced pressure at 70-80°C to afford tert-butyl 7-[2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)-4-oxo-pyrido[l,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5]octane-4-carboxylate (7.7 g, 80.8%) as a yellowish solid.
Example 13: 7-(4,7-diazaspiro[2.5]octan-7-yl)-2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)pyrido[l,2-a]pyrimidin-4-one di-hydrochloride salt
To prepare a solution of HC1 in in 1-propyl acetate/ 1-propanol, acetyl chloride (15.8 g, 199 mmol) was slowly added to a mixture of 1-propyl acetate (60 mL) and 1-propanol (30 mL) at 0°C, and stirring was pursued for an additional 2 hours at room temperature.
tert-Butyl 7-[2-(2,8-dimethylimidazo[ 1 ,2-b]pyridazin-6-yl)-4-oxo-pyrido[ 1 ,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5]octane-4-carboxylate (20 g, 39.9 mmol) was suspended in 1-propyl acetate (60 mL) and 1-propanol (30 mL) at room temperature and the HC1 solution in 1-propyl acetate and 1-propanol was added. The reaction mixture was heated within 3 hours to 70°C and stirred 16 hours at this temperature, then cooled to 20°C. The precipitate was filtered off, washed with 1-propyl acetate (50 mL) in several portions and dried under vacuum at 55 °C to afford 7-(4,7-diazaspiro[2.5]octan-7-yl)-2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)pyrido[l,2-a]pyrimidin-4-one hydrochloride salt (18.8 g, 99%) as yellow crystals.
^-NMR (CDCls, 600 MHz): 8.34 (s, 1H); 8.22(s, 1H); 8.05 (s, 1H); 8.01 (dd, 1H); 7.80 (d, 1H); 7.16 (s, 1H); 3.71 – 3.67 (m, 2H); 3.64 – 3.59 (m, 2H); 3.52 (s, 2H); 2.69 (s, 3H); 2.54 (s, 3H); 1.23- 1.20 (m, 2H); 1.14 – 1.08 (m, 2H); LCMS: 402.20 (M+H+)
Example 14: 7-(4,7-diazaspiro[2.5]octan-7-yl)-2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)pyrido[ 1 ,2-a]pyrimidin-4-one
To a suspension of tert-butyl 7-[2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)-4-oxo-pyrido[l,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5]octane-4-carboxylate (25 g, 50 mmol) in 1-propyl acetate (375 mL) was added a solution of HC1 in 1-propanol (prepared by adding slowly at 5°C acetyl chloride (18.0 mL) to 1-propanol (37.6 mL) and stirring 1 hour at room temperature). The stirred suspension was heated to 75°C within 10 hours and stirred a further 5 hours at 75 °C. Water (160.0 mL) was added and the phases were separated at 75°C. Aqueous sodium hydroxide 32% (27.8 mL) was added to the aqueous phase. The suspension obtained was cooled to room temperature within 5 hours and stirred one hour at room temperature. The precipitate was filtered off, washed with water (100.0 mL) and dried under reduced pressure at 50°C for 18 hours to afford 7-(4,7-diazaspiro[2.5]octan-7-yl)-2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)pyrido[l,2-a]pyrimidin-4-one (19.7 g, 98.3%) as yellow crystals.
!H-NMR (CDCb, 600 MHz): 8. 45 (d, 1H); 7.92 (d, 1H); 7.80 (s, 1H); 7.75 – 7.71 (m, 1H); 7.71 – 7.67 (m, 1H); 7.37 (s, 1H); 3.31 – 3.24 (m, 2H); 3.22 – 3.16 (m, 2H); 3.09 (s, 2H); 2.73 (s, 3H); 2.55 (s, 3H); 0.82- 0.76 (m, 2H); 0.71 – 0.63 (m, 2H); LCMS: 402.20
(M+H+)
Example 15: 7-(4,7-diazaspiro[2.5]octan-7-yl)-2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)pyrido[ 1 ,2-a]pyrimidin-4-one
A suspension of tert-butyl 7-[2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)-4-oxo-pyrido[l,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5]octane-4-carboxylate (13.5 g, 26.9
in toluene (237.0 g) was stirred at 75°C and a 21.9% solution of HCl in 1-propanol (21.4 g, 134.5 mmol) was added within 2.5 hours. The reaction mixture was stirred further at 75 °C until complete conversion. The reaction mixture was cooled to 20-25°C. Water (70 g) was added. The biphasic mixture was stirred another 10 minutes at 20-25 °C and the phases were separated. The organic phase was extracted with water (17 g) twice and the combined aqueous phases were added into mixture of aqueous sodium hydroxide 28% (15.0 g) and water (45.0 g). The suspension obtained was cooled to 20°C. The precipitate was filtered off , washed with water (25 g) three times and dried under reduced pressure at 60°C to afford 7-(4,7-diazaspiro[2.5]octan-7-yl)-2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)pyrido[l,2-a]pyrimidin-4-one (9.5 g, 95.1%) as yellow crystals.
Example 16: 4-bromo-6-chloro-pyridazin-3-amine
3-amino-6-chloropyridazine (20 g, 154 mmol), sodium bicarbonate (25.9 g, 309 mmol) and methanol (158 g) were charged in a reaction vessel and cooled to 0-10°C. Bromine (34.5 g, 216 mmol) was added drop wise and the reaction mixture was stirred 3 days at room temperature. 10% Aqueous sodium sulfate was added. The suspension was filtered off. The filtrate was washed with ethyl acetate (300 mL) twice. The combined organic layers were dried and evaporated. A suspension of the residue in methanol (50 mL) was heated to reflux, water (120 mL) was added and the suspension was stirred 16 hours at room temperature. The precipitate was filtered off and dried. The residue was suspended in n-heptane (50 mL), stirred 2 hours at room temperature, filtered off and dried to afford 4-bromo-6-chloro-pyridazin-3-amine (14.5 g, 46.2%) as a light brown solid.
!H-NMR (CDCls, 600 MHz): 7.55 (s, 1H); 5.83-4.89 (m, 2H); LCMS: 209.93 (M+H+)
Example 17: 4-bromo-6-chloro-pyridazin-3-amine
3-amino-6-chloropyridazine (50 g, 360 mmol), acetic acid (5.8 g, 96.5 mmol), sodium acetate (28.7 g, 289.5 mmol) and methanol (395 g) were charged in a reaction vessel and heated to 25-35°C. Dibromodimethylhydatoin (66.0 g, 231.6 mmol) was added in several portions and the reaction mixture was stirred 3 hours at 30°C. Completion is checked by IPC and if the conversion is incomplete, dibromodimethylhydantoin is added (5.5g). At reaction completion, 38% aqueous sodium sulfate (77.2 mmol NaHS03) was added slowly. The suspension was concentrated under reduced pressure and water (500 g) was added slowly at 45°C, then 30% aqueous sodium hydroxide (31.5 g, 231.6 mmol NaOH) was added at 20°C to adjust pH to 7-8. The precipitate was filtered off, washed with water and dried under reduced pressure to afford 4-bromo-6-chloro-pyridazin-3-amine (50.2 g, 62.5%) as a grey solid.
Example 18: 6-chloro-4-methyl-pyridazin-3-amine
4-bromo-6-chloro-pyridazin-3-amine (3.0 g, 14.4 mmol) and
tetrakis(triphenylphosphine)palladium (1666 mg, 144 μιηοΐ) were suspended in THF (13.2 g) and a solution of zinc chloride in Me-THF (2.0 M, 9 mL, 18 mmol) was added. The reaction mixture was cooled to -5°C and methyllithium in diethoxymethane (3.1 M, 11.6 mL, 36 mmol) was added. The reaction mixture was stirred at 45°C for 4 hours. Sodium sulfate decahydrate (11.7 g, 36 mmol) was added at room temperature, the mixture was stirred 1.5 hours at 60°C, diluted with water (100 mL) and after 30 minutes the precipitate was filtered off. The precipitate was dissolved in aqueous HC1 2M (100 mL) and ethyl acetate (140 mL). The biphasic system was filtered, the phases were separated and the pH of the water layer adjusted to 7 with aqueous NaOH 32% (18 mL). The precipitate was filtered and dried. The solid obtained was digested twice in methanol (20 mL) at room temperature. The two filtrates were combined, evaporated and dried under high vacuum to afford 6-chloro-4-methyl-pyridazin-3-amine (1.2 g, 58.1%) as a red solid.
Ή-ΝΜΡν (CDCb, 600 MHz): 7.09 (d, 1H); 4.90 (br s, 2H), 2.17 (d, 3H)
Example 19: 6-chloro-4-methyl-pyridazin-3-amine
4-bromo-6-chloro-pyridazin-3-amine (30.02 g, 143 mmol) and THF (180 mL) were charged into a reaction vessel. Methylmagnesium chloride (22% in THF, 50.0 mL, 1.03 eq.) was added at 20°C over 60 minutes, followed by zinc chloride in Me-THF (25%, 37 mL, 0.50 eq.) and palladium tetrakis(triphenyphosphine) (1.66 g, lmol%). The reaction mixture was heated to 50°C and methylmagnesium chloride (22% in THF, 81 mL, 1.7 eq.) was added slowly. The reaction mixture was stirred at 50°C until complete conversion, then at 10°C for 14.5 hours and poured into a mixture of water (90 g), aqueous HCl 33% (52.5 g) and toluene (150 mL) maintained at 20-30°C. The aqueous phase was separated and the organic phase was extracted with a solution of aqueous HCl 33% (2.0 g) and water (45 g). The aqueous layers were combined and washed with toluene (30 mL) twice and the pH was adjusted by addition of 25% aqueous ammonia solution. When a pH of 2.4 was reached, seeding crystals were added, the mixture was stirred further for 15 minutes and thereafter the pH was brought to 4.0. The suspension was stirred at 20°C for 2 hours, the precipitate was filtered off, washed with water (20 mL) three times to afford crude 6-chloro-4-methyl-pyridazin-3-amine (29 g) as a brown solid.
29 g crude product was transferred to a reaction vessel and methanol (20 mL) was added. The mixture was refluxed for 30 minutes and 12 g water was added. The solution was cooled to 0°C and stirred for 2 hours at this temperature. The precipitate was filtered off, washed with water three times and dried under reduced pressure at 40°C to afford purified 6-chloro-4-methyl-pyridazin-3-amine (13.8 g, 66%) as a light brown solid.
Alternative purification:
50 g crude 6-chloro-4-methyl-pyridazin-3-amine were dissolved in methanol (250 mL) and active charcoal (4.0 g) and diatomaceous earth (2.5 g) were added. The suspension was stirred at 45°C for 1 hour, cooled to 30°C and potassium hydrogenophosphate (2.1 g) was added. The suspension was stirred at 30°C for another 90 minutes, filtered and the precipitate washed with methanol (100 mL). The filtrate was concentrated to a residual volume of 175 mL and water (120 mL) was added. The resulting suspension was heated
to reflux affording a solution which was cooled to 20°C resulting in a suspension. The precipitate was filtered off, washed with water (90 mL) and dried under reduced pressure to afford pure 6-chloro-4-methyl-pyridazin-3-amine (38 g, 76%) as a light yellow solid.
Example 20: 6-chloro-2,8-dimethyl-imidazo[l,2-b]pyridazine
6-chloro-4-methyl-pyridazin-3-amine (70.95 kg, 494.2 mol), sodium bromide (35 kg, 345.9 mol), isopropyl acetate (611 kg), isopropanol (28 kg and water (35 kg) were charged into a reaction vessel. The reaction mixture was stirred at 80-85 °C for 8 hours. Isopropyl acetate (310 kg) and water (420 kg) were added. 30% Aqueous NaOH was added at 45-55 °C and the system was stirred for 2 hours. The phases were separated at 25-35 °C. The organic layer was washed with water (370 kg), filtered on diatomite (7 kg) and the filter washed with isopropyl acetate (35 kg). The organic phase was extracted with two portions of 5.4% aqueous sulfuric acid (910 kg followed by 579 kg). The combined aqueous phases were basified with 30% aqueous NaOH (158 kg). The suspension was stirred 2 hours at 15-25 °C. The precipitate was isolated by centrifugation in three portions, each washed with water (31 kg). The wet solid was dissolved in isopropyl acetate (980 kg) at 25-35 °C, the solution washed with water (210 kg), three times. The organic phase was treated with active charcoal for 12 hours at 45-50 °C, concentrated to ca. 300 kg and heated to 70-80 °C to obtain a clear solution. This solution was cooled to 50-60 °C, stirred at this temperature for 1 hour, n-heptane (378 kg) was added and stirring was pursued for 1 hour. The mixture was cooled to -10- -5°C and stirred for another 3 hours. The precipitate was isolated by centrifuging, washed with n-heptane (33 kg) and dried under reduced pressure at 30-50 °C for 15 hours to afford 67.4 kg (76%) 6-chloro-2,8-dimethyl-imidazo[l,2-b]pyridazine as an off-white solid.
XH-NMR (CDCls, 600 MHz): 7.67 (s, 1H); 6.86 (s, 1H); 2.65 (s, 3H), 2.50 (s, 3H)
Paper
https://pubs.acs.org/doi/pdf/10.1021/acs.jmedchem.8b00741

SMA is an inherited disease that leads to loss of motor function and ambulation and a reduced life expectancy. We have been working to develop orally administrated, systemically distributed small molecules to increase levels of functional SMN protein. Compound 2 was the first SMN2 splicing modifier tested in clinical trials in healthy volunteers and SMA patients. It was safe and well tolerated and increased SMN protein levels up to 2-fold in patients. Nevertheless, its development was stopped as a precautionary measure because retinal toxicity was observed in cynomolgus monkeys after chronic daily oral dosing (39 weeks) at exposures in excess of those investigated in patients. Herein, we describe the discovery of 1 (risdiplam, RG7916, RO7034067) that focused on thorough pharmacology, DMPK and safety characterization and optimization. This compound is undergoing pivotal clinical trials and is a promising medicine for the treatment of patients in all ages and stages with SMA.
7-(4,7-diazaspiro[2.5]octan-7-yl)-2-(2,8-dimethylimidazo[1,2-b]pyridazin-6-yl)pyrido[1,2-a]pyrimidin-4-one 1 (12 mg, 18%) as a pale yellow solid. 1H NMR (600 MHz,CDCl3) δ ppm 8.45 (d, J = 2.4 Hz, 1H), 7.92 (d, J = 1.0 Hz, 1H), 7.73 (d, J = 9.6 Hz, 1H) 7.80 (s, 1H), 7.70 (dd, J = 9.7, 2.5 Hz, 1H), 7.38 (s, 1H), 3.31–3.22 (m, 2H), 3.20–3.16 (m, 2H), 3.08 (s, 2H), 2.74 (d, J = 0.9 Hz, 3H) 2.55 (s, 3H), 1.68 (br s, 1H), 0.77–0.75 (m, 2H), 0.67–0.64 (m, 2 H);
13C NMR (151 MHz,CDCl3) δ ppm 158.2, 156.3, 148.5, 147.2, 144.1, 142.2, 140.0, 135.6, 131.2, 126.7, 114.9, 114.7, 110.1, 99.3, 56.7, 49.9, 44.5, 36.5, 16.9, 15.0, 13.0. LC–HRMS: m/z = 402.2051 [(M + H)+ calcd for C22H24N7O, 402.2042; Diff 0.9 mDa].


References
- ^ Maria Joao Almeida (2016-09-08). “RG7916”. BioNews Services. Retrieved 2017-10-08.
- ^ Zhao, Xin; Feng, Zhihua; Ling, Karen K. Y; Mollin, Anna; Sheedy, Josephine; Yeh, Shirley; Petruska, Janet; Narasimhan, Jana; Dakka, Amal; Welch, Ellen M; Karp, Gary; Chen, Karen S; Metzger, Friedrich; Ratni, Hasane; Lotti, Francesco; Tisdale, Sarah; Naryshkin, Nikolai A; Pellizzoni, Livio; Paushkin, Sergey; Ko, Chien-Ping; Weetall, Marla (2016). “Pharmacokinetics, pharmacodynamics, and efficacy of a small-molecule SMN2 splicing modifier in mouse models of spinal muscular atrophy”. Human Molecular Genetics. 25 (10): 1885. doi:10.1093/hmg/ddw062. PMC 5062580. PMID 26931466.
- ^ “Genentech/Roche Releases Clinical Trial Update for RG7916”. CureSMA. 2017-09-15. Retrieved 2017-10-08.
- ^ “A Study to Investigate the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics and Efficacy of RO7034067 in Infants With Type1 Spinal Muscular Atrophy (Firefish)”.
- ^ “A Study to Investigate the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics and Efficacy of RO7034067 in Type 2 and 3 Spinal Muscular Atrophy Participants (Sunfish)”.
- ^ “Updated Preliminary Data from SMA FIREFISH Program in Type 1 Babies Presented at the CureSMA Conference”. http://www.prnewswire.com. Retrieved 2018-09-11.
 |
|
| Clinical data | |
|---|---|
| Synonyms | RG7916; RO7034067 |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C22H23N7O |
| Molar mass | 401.474 g/mol g·mol−1 |
| 3D model (JSmol) | |
///////////RISDIPLAM, RG-7916, RO-7034067, リスジプラム , PHASE 3, PRIME designation, ORPHAN DRUG
76RS4S2ET1 (UNII code)
CC1=CC(=NN2C1=NC(=C2)C)C3=CC(=O)N4C=C(C=CC4=N3)N5CCNC6(C5)CC6
Cladribine, クラドリビン
Cladribine
クラドリビン
Leustatin
クラドリビン
RWJ 26251 / RWJ-26251
- Molecular FormulaC10H12ClN5O3
- Average mass285.687 Da
2-chloro-6-amino-9-(2-deoxy-β-D-erythro-pentofuranosyl)purine
2-Chlorodeoxyadenosine
4291-63-8 [RN]
6997
adenosine, 2-chloro-2′-deoxy- [ACD/Index Name]
AU7357560
CDA
(2R,3S,5R)-5-(6-Amino-2-chlor-9H-purin-9-yl)-2-(hydroxymethyl)tetrahydrofuran-3-ol
Leustatin (Trade name)
Litak (Trade name)
MLS000759397
Movectro (Trade name)
Mylinax
QA-1968
LAUNCHED, 1993, USA Ortho Biotech, Janssen Biotech
Cladribine, sold under the brand name Leustatin and Mavenclad among others, is a medication used to treat hairy cell leukemia(HCL, leukemic reticuloendotheliosis), B-cell chronic lymphocytic leukemia and relapsing-remitting multiple sclerosis.[4][5] Its chemical name is 2-chloro-2′-deoxyadenosine (2CdA).
Cladribine, a deoxyadenosine derivative developed by Ortho Biotech (currently Janssen), was first launched in the U.S. in 1993 as an intravenous treatment for hairy cell leukemia
Cladribine has been granted orphan drug designation in the U.S. in 1990 for the treatment of acute myeloid leukemia (AML) and hairy cell leukemia
As a purine analog, it is a synthetic chemotherapy agent that targets lymphocytes and selectively suppresses the immune system. Chemically, it mimics the nucleoside adenosine. However, unlike adenosine it is relatively resistant to breakdown by the enzyme adenosine deaminase, which causes it to accumulate in cells and interfere with the cell’s ability to process DNA. Cladribine is taken up cells via a transporter. Once inside a cell cladribine is activated mostly in lymphocytes, when it is triphosphorylated by the enzyme deoxyadenosine kinase (dCK). Various phosphatases dephosphorylate cladribine. Activated, triphosphorylated, cladribine is incorporated into mitochondrial and nuclear DNA, which triggers apoptosis. Non-activated cladribine is removed quickly from all other cells. This means that there is very little non-target cell loss.[4][6]
Medical uses
Cladribine is used for as a first and second-line treatment for symptomatic hairy cell leukemia and for B-cell chronic lymphocytic leukemia and is administered by intravenous or subcutaneous infusion.[5][7]
Since 2017, cladribine is approved as an oral formulation (10 mg tablet) for the treatment of RRMS in Europe, UAE, Argentina, Chile, Canada and Australia. Marketing authorization in the US was obtained in March 2019[8].
Some investigators have used the parenteral formulation orally to treat patients with HCL. It is important to note that approximately 40% of oral cladribine in bioavailable orally. It used, often in combination with other cytotoxic agents, to treat various kinds of histiocytosis, including Erdheim–Chester disease[9] and Langerhans cell histiocytosis,[10]
Cladribine can cause fetal harm when administered to a pregnant woman and is listed by the FDA as Pregnancy Category D; safety and efficacy in children has not been established.[7]
Adverse effects
Injectable cladribine suppresses the body’s ability to make new lymphocytes, natural killer cells and neutrophils (called myelosuppression); data from HCL studies showed that about 70% of people taking the drug had fewer white blood cells and about 30% developed infections and some of those progressed to septic shock; about 40% of people taking the drug had fewer red blood cells and became severely anemic; and about 10% of people had too few platelets.[7]
At the dosage used to treat HCL in two clinical trials, 16% of people had rashes and 22% had nausea, the nausea generally did not lead to vomiting.[7]
In comparison, in MS, cladribine is associated with a 6% rate of severe lymphocyte suppression (lymphopenia) (levels lower than 50% of normal). Other common side effects include headache (75%), sore throat (56%), common cold-like illness (42%) and nausea (39%)[11]
Mechanism of Action
As a purine analogue, it is taken up into rapidly proliferating cells like lymphocytes to be incorporated into DNA synthesis. Unlike adenosine, cladribine has a chlorine molecule at position 2, which renders it partially resistant to breakdown by adenosine deaminase (ADA). In cells it is phosphorylated into its toxic form, deoxyadenosine triphosphate, by the enzyme deoxycytidine kinase (DCK). This molecule is then incorporated into the DNA synthesis pathway, where it causes strand breakage. This is followed by the activation of transcription factor p53, the release of cytochrome c from mitochondria and eventual programmed cell death (apoptosis).[12] This process occurs over approximately 2 months, with a peak level of cell depletion 4–8 weeks after treatment[13]
Within the lymphocyte pool, cladribine targets B cells more than T cells. Both HCL and B-cell chronic lymphocytic leukaemia are types of B cell blood cancers. In MS, its effectiveness may be due to its ability to effectively deplete B cells, in particular memory B cells[14] In the pivotal phase 3 clinical trial of oral cladribine in MS, CLARITY, cladribine selectively depleted 80% of peripheral B cells, compared to only 40-50% of total T cells.[15] More recently, cladribine has been shown to induce long term, selective suppression of certain subtypes of B cells, especially memory B cells.[16]
Another family of enzymes, the 5´nucleotidase (5NCT) family, is also capable of dephosphorylating cladribine, making it inactive. The most important subtype of this group appears to be 5NCT1A, which is cytosolically active and specific for purine analogues. When DCK gene expression is expressed as a ratio with 5NCT1A, the cells with the highest ratios are B cells, especially germinal centre and naive B cells.[16] This again helps to explain which B cells are more vulnerable to cladribine-mediated apoptosis.
Although cladribine is selective for B cells, the long term suppression of memory B cells, which may contribute to its effect in MS, is not explained by gene or protein expression. Instead, cladribine appears to deplete the entire B cell department. However, while naive B cells rapidly move from lymphoid organs, the memory B cell pool repopulates very slowly from the bone marrow.
History
Ernest Beutler and Dennis A. Carson had studied adenosine deaminase deficiency and recognized that because the lack of adenosine deaminase led to the destruction of B cell lymphocytes, a drug designed to inhibit adenosine deaminase might be useful in lymphomas. Carson then synthesized cladribine, and through clinical research at Scripps starting in the 1980s, Beutler tested it as intravenous infusion and found it was especially useful to treat hairy cell leukemia (HCL). No pharmaceutical companies were interested in selling the drug because HCL was an orphan disease, so Beutler’s lab synthesized and packaged it and supplied it to the hospital pharmacy; the lab also developed a test to monitor blood levels. This was the first treatment that led to prolonged remission of HCL, which was previously untreatable.[17]:14–15
In February 1991 Scripps began a collaboration with Johnson & Johnson to bring intravenous cladribine to market and by December of that year J&J had filed an NDA; cladrabine was approved by the FDA in 1993 for HCL as an orphan drug,[18] and was approved in Europe later that year.[19]:2
The subcutaneous formulation was developed in Switzerland in the early 1990s and it was commercialized by Lipomed GmbH in the 2000s.[19]:2[20]
Multiple sclerosis
In the mid-1990s Beutler, in collaboration with Jack Sipe, a neurologist at Scripps, ran several clinical trials exploring the utility of cladribine in multiple sclerosis, based on the drug’s immunosuppressive effects. Sipe’s insight into MS, and Beutler’s interest in MS due to his sister’s having had it, led a very productive collaboration.[17]:17[21] Ortho-Clinical, a subsidiary of J&J, filed an NDA for cladribine for MS in 1997 but withdrew it in the late 1990s after discussion with the FDA proved that more clinical data would be needed.[22][23]
Ivax acquired the rights for oral administration of cladribine to treat MS from Scripps in 2000,[24] and partnered with Serono in 2002.[23] Ivax was acquired by Teva in 2006,[25][26] and Merck KGaA acquired control of Serono’s drug business in 2006.[27]
An oral formulation of the drug with cyclodextrin was developed[28]:16 and Ivax and Serono, and then Merck KGaA conducted several clinical studies. Merck KGaA submitted an application to the European Medicines Agency in 2009, which was rejected in 2010, and an appeal was denied in 2011.[28]:4–5 Likewise Merck KGaA’s NDA with the FDA rejected in 2011.[29] The concerns were that several cases of cancer had arisen, and the ratio of benefit to harm was not clear to regulators.[28]:54–55 The failures with the FDA and the EMA were a blow to Merck KGaA and were one of a series of events that led to a reorganization, layoffs, and closing the Swiss facility where Serono had arisen.[30][31] However, several MS clinical trials were still ongoing at the time of the rejections, and Merck KGaA committed to completing them.[29] A meta-analysis of data from clinical trials showed that cladiribine did not increase the risk of cancer at the doses used in the clinical trials.[32]
In 2015 Merck KGaA announced it would again seek regulatory approval with data from the completed clinical trials in hand,[30] and in 2016 the EMA accepted its application for review.[33] On June 22, 2017, the EMA’s Committee for Medicinal Products for Human Use (CHMP) adopted a positive opinion, recommending the granting of a marketing authorisation for the treatment of relapsing forms of multiple sclerosis.[34]
Finally, after all these problems it was approved in Europe on August 2017 for highly active RRMS.[35]
Efficacy
Cladribine is an effective treatment for relapsing remitting MS, with a reduction in the annual rate of relapses of 54.5%.[11] These effects may be sustained up to 4 years after initial treatment, even if no further doses are given.[36] Thus, cladribine is considered to be a highly effective immune reconstitution therapy in MS. Similar to alemtuzumab, cladribine is given as two courses approximately one year apart. Each course consists of 4-5 tablets given over a week in the first month, followed by a second dosing of another 4-5 tablets the following month[37] During this time and after the final dose patients are monitored for adverse effects and signs of relapse.
https://www.merckneurology.co.uk/wp-content/uploads/2017/08/mavenclad-table-1.jpg
Safety
Compared to alemtuzumab, cladribine is associated with a lower rate of severe lymphopenia. It also appears to have a lower rate of common adverse events, especially mild to moderate infections[11][36] As cladribine is not a recombinant biological therapy, it is not associated with the development of antibodies against the drug, which might reduce the effectiveness of future doses. Also, unlike alemtuzumab, cladribine is not associated with secondary autoimmunity.[38]
This is probably due to the fact cladribine more selectively targets B cells. Unlike alemtuzumab, cladribine is not associated with a rapid repopulation of the peripheral blood B cell pool, which then ´overshoots´ the original number by up to 30%.[39] Instead, B cells repopulate more slowly, reaching near normal total B cells numbers at 1 year. This phenomenon and the relative sparing of T cells, some of which might be important in regulating the system against other autoimmune reactions, is thought to explain the lack of secondary autoimmunity.
Use in clinical practice
The decision to start cladribine in MS depends on the degree of disease activity (as measured by number of relapses in the past year and T1 gadolinium-enhancing lesions on MRI), the failure of previous disease-modifying therapies, the potential risks and benefits and patient choice.
In the UK, the National Institute for Clinical Excellence (NICE) recommends cladribine for treating highly active RRMS in adults if the persons has:
rapidly evolving severe relapsing–remitting multiple sclerosis, that is, at least 2 relapses in the previous year and at least 1 T1 gadolinium-enhancing lesion at baseline MRI or
relapsing–remitting multiple sclerosis that has responded inadequately to treatment with disease-modifying therapy, defined as 1 relapse in the previous year and MRI evidence of disease activity.[40]
People with MS require counselling on the intended benefits of cladribine in reducing the risk of relapse and disease progression, versus the risk of adverse effects such as headaches, nausea and mild to moderate infections. Women of childbearing age also require counselling that they should not conceive while taking cladribine, due to the risk of harm to the fetus.
Cladribine, as the 10 mg oral preparation Mavenclad, is administered as two courses of tablets approximately one year apart. Each course consists of four to five treatment days in the first month, followed by an additional four to five treatment days in the second month. The recommended dose of Mavenclad is 3.5 mg/kg over 2 years, given in two treatment courses of 1.75 mg/kg/year. Therefore, the number of tablets administered on each treatment day depends on the person’s weight. A full guide to the dosing strategy can be found below:
https://www.merckneurology.co.uk/mavenclad/mavenclad-efficacy/
After treatment, people with MS are monitored with regular blood tests, looking specifically at the white cell count and liver function. Patients should be followed up regularly by their treating neurologist to assess efficacy, and should be able to contact their MS service in the case of adverse effects or relapse. After the first two years of active treatment no further therapy may need to be given, as cladribine has been shown to be efficacious for up to last least four years after treatment. However, if patients fail to respond, options include switching to other highly effective disease-modifying therapies such as alemtuzumab, fingolimod or natalizumab.
Research directions
Cladribine has been studied as part of a multi-drug chemotherapy regimen for drug-resistant T-cell prolymphocytic leukemia.[41]
REF
A universal biocatalyst for the preparation of base- and sugar-modified nucleosides via an enzymatic transglycosylation
Helv Chim Acta 2002, 85(7): 1901
Synthesis of 2-chloro-2′-deoxyadenosine by microbiological transglycosylation
Nucleosides Nucleotides 1993, 12(3-4): 417
Synthesis of 2-chloro-2′-deoxyadenosine by washed cells of E. coli
Biotechnol Lett 1992, 14(8): 669
Efficient syntheses of 2-chloro-2′-deoxyadenosine (cladribine) from 2′-deoxyguanosine
J Org Chem 2003, 68(3): 989
WO 2004028462
Synthesis of 2′-deoxytubercidin, 2′-deoxyadenosine, and related 2′-deoxynucleosides via a novel direct stereospecific sodium salt glycosylation procedure
J Am Chem Soc 1984, 106(21): 6379
WO 2011113476
A stereoselective process for the manufacture of a 2′-deoxy-beta-D-ribonucleoside using the vorbruggen glycosylation
Org Process Res Dev 2013, 17(11): 1419
A new synthesis of 2-chloro-2′-deoxyadenosine (Cladribine), CdA)
Nucleosides Nucleotides Nucleic Acids 2011, 30(5): 353
A dramatic concentration effect on the stereoselectivity of N-glycosylation for the synthesis of 2′-deoxy-beta-ribonucleosides
Chem Commun (London) 2012, 48(56): 7097
CN 105367616
PATENT
https://patents.google.com/patent/EP2891660A1/en
Previously Robins and Robins (Robins, M. J. and Robins, R. K., J. Am. Chem. Soc. 1965, 87, 4934-4940) reported that acid-catalyzed fusion of 1,3,5-tri-O-acety-2-deoxy-D-ribofuranose and 2,6-dichloropurine gave a 65% yield of an anomeric mixture 2,6-dichloro-9-(3′,5′-di-O-acetyl-2′-deoxy-α-,β-D-ribofuranosyl)-purines from which the α-anomer was obtained as a pure crystalline product by fractional crystallization from ethanol in 32% yield and the equivalent β-anomer remained in the mother liquor (see Scheme 1). The β-anomer, which could have been used to synthesize cladribine, wasn’t isolated further. The α-anomer was treated with methanolic ammonia which resulted in simultaneous deacetylation and amination to give 6-amino-2-chloro-9-(2′-deoxy-α-D-ribofuranosyl)-purine, which is a diastereomer of cladribine.

[0004]
Broom et al. (Christensen, L. F., Broom, A. D., Robins, M. J., and Bloch, A., J. Med. Chem. 1972, 15, 735-739) adapted Robins et al.’s method by treating the acetylated mixture (viz., 2,6-dichloro-9-(3′,5′-di-O-acety-2′-deoxy-α,β-D-ribofuranosyl)-purine) with liquid ammonia and reacylating the resulting 2′-deoxy-α-and –β-adenosines with p-toluoyl chloride (see Scheme 2). The desired 2-chloro-9-(3′,5′-di-O–p-toluoyl-2′-deoxy-β-D-ribofuranosyl)-adenine was then separated by chromatography and removal of the p-toluoyl group resulted in cladribine in 9% overall yield based on the fusion of 1,3,5-tri-O-acety-2-deoxy-D-ribofuranose and 2,6-dichloropurine.

[0005]
To increase the stereoselectivity in favour of the β-anomer, Robins et al.(Robins, R. L. et al., J. Am. Chem. Soc. 1984, 106, 6379-6382, US4760137 , EP0173059 ) provided an improved method in which the sodium salt of 2,6-dichloropurine was coupled with 1-chloro-2-deoxy-3,5-di-O–p-toluoyl-α-D-ribofuranose in acetonitrile (MeCN) to give the protected β-nucleoside in 59% isolated yield, following chromatography and crystallisation, in addition to 13% of the undesired N-7 regioisomer (see Scheme 3). The apparently higher selectivity in this coupling reaction is attributed to it being a direct SN2 displacement of the chloride ion by the purine sodium salt. The protected N-9 2′-deoxy-β-nucleoside was treated with methanolic ammonia at 100°C to give cladribine in an overall 42% yield. The drawback of this process is that the nucleophilic 7- position nitrogen competes in the SN2 reaction against the nucleophilic 9- position, leading to a mixture of the N-7 and N-9 glycosyl isomers as well as the need for chromatography and crystallisation to obtain the pure desired isomer.

[0006]
Gerszberg and Alonso (Gerszberg S. and Alonso, D. WO0064918 , and US20020052491 ) also utilised an SN2 approach with 1-chloro-2-deoxy-3,5-di-O–p-toluoyl-α-D-ribofuranose but instead coupled it with the sodium salt of 2-chloroadenine in acetone giving the desired β-anomer of the protected cladribine in 60% yield following crystallisation from ethanol (see Scheme 4). After the deprotection step using ammonia in methanol (MeOH), the β-anomer of cladribine was isolated in an overall 42% yield based on the 1-chlorosugar, and 30% if calculated based on the sodium salt since this was used in a 2.3 molar excess.

[0007]
To increase the regioselectivity towards glycosylation of the N-9 position, Gupta and Munk recently ( Gupta, P. K. and Munk, S. A., US20040039190 , WO2004018490 and CA2493724 ) conducted an SN2 reaction using the anomerically pure α-anomer, 1-chloro-2-deoxy-3,5-di-O–p-toluoyl-α-D-ribofuranose but coupling it with the potassium salt of a 6-heptanoylamido modified purine (see Scheme 5). The bulky alkyl group probably imparted steric hindrance around the N-7 position, resulting in the reported improved regioselectivity. Despite this, following deprotection, the overall yield of cladribine based on the 1-chlorosugar was 43%, showing no large improvement in overall yield on related methods. Moreover 2-chloroadenine required prior acylation with heptanoic anhydride at high temperature (130°C) in 72% yield, and the coupling required cryogenic cooling (-30°C) and the use of the strong base potassium hexamethyldisilazide and was followed by column chromatography to purify the product protected cladribine.

[0008]
More recently Robins et al. (Robins, M. J. et al., J. Org. Chem. 2006, 71, 7773-7779, US20080207891 ) published a procedure for synthesis of cladribine that purports to achieve almost quantitative yields in the N-9-regioselective glycosylation of 6-(substituted-imidazol-1-yl)-purine sodium salts with 1-chloro-2-deoxy-3,5-di-O–p-toluoyl-α-D-ribofuranose in MeCN/dichloromethane (DCM) mixtures to give small or no detectable amounts of the undesired α-anomer (see Scheme 6). In actuality this was only demonstrated on the multi-milligram to several grams scale, and whilst the actual coupling yield following chromatography of the desired N-9-β-anomer was high (83% to quantitative), the protected 6-(substituted-imidazol-1-yl)-products were obtained in 55% to 76% yield after recrystallisation. Following this, toxic benzyl iodide was used to activate the 6-(imidazole-1-yl) groups which were then subsequently displaced by ammonia at 60-80°C in methanolic ammonia to give cladribine in 59-70% yield following ion exchange chromatography and multiple crystallisations, or following extraction with DCM and crystallisation. Although high anomeric and regioselective glycosylation was demonstrated the procedure is longer than the prior arts, atom uneconomic and not readily applicable to industrial synthesis of cladribine such as due to the reliance on chromatography and the requirement for a pressure vessel in the substitution of the 6-(substituted-imidazole-1-yl) groups.

[0009]
Therefore, there is a need for a more direct, less laborious process, which will produce cladribine in good yield and high purity that is applicable to industrial scales.
EXAMPLE 1 Preparation of 2-chloro-6-trimethylsilylamino-9-[3,5-di-O-(4-chlorobenzoyl)-2-deoxy-β-D-ribofuranosyl]-purine
- [0052]
2-Chloroadenine (75 g, 0.44 mol, 1.0 eq.), MeCN (900 mL, 12 P), and BSTFA (343.5 g, 1.33 mol, 3.0 eq.) were stirred and heated under reflux until the mixture was almost turned clear. The mixture was cooled to 60°C and TfOH (7.9 mL, 0.089 mol, 0.2 eq.) and then 1-O-acetyl-3,5-di-O-(4-chlorobenzoyl)-2-deoxy-D-ribofuranose (III; 200.6 g, 1.0 eq.) were added into the mixture, and then the mixture was stirred at 60°C. After 1 hour, some solid precipitated from the solution and the mixture was heated for at least a further 10 hours. The mixture was cooled to r.t. and stirred for 2 hours. The solid was filtered and dried in vacuo at 60°C to give 180.6 g in 64% yield of a mixture of 2-chloro-6-trimethylsilylamino-9-[3,5-di-O-(4-chlorobenzoyl)-2-deoxy-β-D-ribofurano syl]-purine (IVa) with 95.4% HPLC purity and its non-silylated derivative 2-chloro-6-amino-9-[3,5-di-O-(4-chlorobenzoyl)-2′-deoxy-β-D-ribofuranosyl]-purine (IVb) with 1.1 % HPLC purity.
EXAMPLE 2 Preparation of 2-chloro-6-trimethylsilylamino-9-[3,5-di-O-(4-chlorobenzoyl)-2-deoxy-β-D-ribofuranosyl]-purine by isomerisation of a mixture of 2-chloro-6-amino-9-[3,5-di-O-(4-chlorobenzoyl)-2-deoxy-α,β-D-ribofuranosyl]-purine mixture
- [0053]
50.0 g of 2-chloro-6-amino-9-[3,5-di-O-(4-chlorobenzoyl)-2-deoxy-α,β-D-ribofuranosyl]-purine as a 0.6:1.0 mixture of the β-anomer IVb and α-anomer Vb(83.16 mmol, assay of α-anomer was 58.6% (52.06 mmol) and β-anomer was 34.3% (31.10 mmol, 17.15 g)), 68.6 g BSTFA (266.5 mmol) and 180 mL of MeCN (3.6 P) were charged into a dried 4-necked flask. The mixture was heated to 60°C under N2 for about 3 h and then 2.67 g of TfOH (17.8 mmol) was added. The mixture was stirred at 60°C for 15 h and was then cooled to about 25°C and stirred for a further 2 h, and then filtered. The filter cake was washed twice with MeCN (20 mL each) and dried at 60°C in vacuo for 6 h to give 24 g of off-white solid (the assay of 2-chloro-6-amino-9-[3,5-di-O-(4-chlorobenzoyl)-2-deoxy-α-D-ribofuranosyl]-purine was 1.4% (0.60 mmol, 0.34 g),
2-chloro-6-amino-9-[3,5-di-O-(4-chlorobenzoyl)-2-deoxy-β-D-ribofuranosyl]-purine was 8.4% (3.18 mmol, 2.02 g) and
2-chloro-6-trimethylsilylamino-9-[3,5-di-O-(4-chlorobenzoyl)-2-deoxy-β-D-ribofuranosyl]-purine was 86.6% (32.73 mmol, 20.78 g)).
Analysis of the 274.8 g of the mother liquor by assay showed that it in addition to the α-anomer it contained 0.5% (1.37 g, 2.43 mmol) of
2-chloro-6-amino-9-[3,5-di-O-(4-chlorobenzoyl)-2-deoxy-β-D-ribofuranosyl]-purine and 0.01% (0.027 g, 0.05 mmol) of
2-chloro-6-trimethylsilylamino-9-[3,5-di-O-(4-chlorobenzoyl)-2-deoxy-β-D-ribofuranosyl]-purine.
EXAMPLE 3 Preparation of 2-chloro-2′-deoxy-adenosine (cladribine)
- [0054]
To the above prepared mixture of 2-chloro-6-trimethylsilylamino-9-[3,5-di-O-(4-chlorobenzoyl)-2-deoxy-β-D-ribofurano syl]- purine (IVa) and 2-chloro-6-amino-9-[3,5-di-O-(4-chlorobenzoyl)-2′-deoxy-β-D-ribofuranosyl]-purine (IVb) (179 g, >95.4% HPLC purity) in MeOH (895 mL, 5 P) was added 29% MeONa/MeOH solution (5.25 g, 0.1 eq.) at 20-30°C. The mixture was stirred at 20-30°C for 6 hours, the solid was filtered, washed with MeOH (60 mL, 0.34 P) and then dried in vacuo at 50°C for 6 hour to give 72 g white to off-white crude cladribine with 98.9% HPLC purity in ca. 93% yield.
EXAMPLE 4 Recrystallisation
- [0055]
Crude cladribine (70 g), H2O (350 mL, 5 P), MeOH (350 mL, 5 P) and 29% MeONa/MeOH solution (0.17 g) were stirred and heated under reflux until the mixture turned clear. The mixture was stirred for 3 hour and was then filtered to remove the precipitates at 74-78°C. The mixture was stirred and heated under reflux until the mixture turned clear and was then cooled. Crystals started to form at ca. 45°C. The slurry was stirred for 2 hour at the cloudy point. The slurry was cooled slowly at a rate of 5°C/0.5 hour. The slurry was stirred at 10-20°C for 4-8 hours and then filtered. The filter cake was washed three times with MeOH (50 mL each) and dried at 50°C in vacuo for 6 hours to give 62.7 g of 99.9% HPLC pure cladribine in ca. 90% yield.
EXAMPLE 5 Preparation of 2-chloro-6-trimethylsilylamino-9-[3,5-di-O-(4-chlorobenzoyl)-2-deoxy-β-D-ribofuranosyl]-purine
- [0056]
2-Chloroadenine (2.2 Kg, 13.0 mol, 1.0 eq.), MeCN (20.7 Kg, 12 P), and BSTFA (10.0 Kg, 38.9 mol, 3.0 eq.) were stirred and heated under reflux for 3 hours and then filtered through celite and was cooled to about 60°C. TfOH (0.40 Kg, 2.6 mol, 0.2 eq.) and 1-O-acetyl-3,5-di-O-(4-chlorobenzoyl)-2-deoxy-D-ribofuranose (III; 5.87 Kg, 13.0 mol, 1.0 eq.) were added into the filtrate and the mixture was stirred at about 60°C for 29.5 hours. The slurry was cooled to about 20°C and stirred for 2 hours. The solids were filtered and washed with MeCN (2.8 Kg) twice and dried in vacuo at 60°C to give 5.17 Kg with a 96.5% HPLC purity in 62% yield of a mixture of 2-chloro-6-trimethylsilylamino-9-[3,5-di-O-(4-chlorobenzoyl)-2-deoxy-β-D-ribofurano syl]-purine (IVa), and non-silylated derivative 2-chloro-6-amino-9-[3,5-di-O-(4-chlorobenzoyl)-2′-deoxy-β-D-ribofuranosyl]-purine (IVb).
EXAMPLE 6 Preparation of 2-chloro-2′-deoxy-adenosine (cladribine)
- [0057]
To a mixture of 25% sodium methoxide in MeOH (0.11 Kg, 0.5 mol, 0.1 eq.) and MeOH (14.8 Kg, 5 P) at about at 25°C was added 2-chloro-6-trimethylsilylamino-9-[3,5-di-O-(4-chlorobenzoyl)-2-deoxy-β-D-ribofurano syl]-purine (IVa) and non-silylated derivative 2-chloro-6-amino-9-[3,5-di-O-(4-chlorobenzoyl)-2′-deoxy-β-D-ribofuranosyl]-purine (IVb) (3.70 Kg, combined HPLC purity of >96.3%) and the mixture was agitated at about 25°C for 2 hours. The solids were filtered, washed with MeOH (1.11 Kg, 0.4 P) and then dried in vacuo at 60°C for 4 hours to give 1.43 Kg of a crude cladribine with 97.8% HPLC purity in ca. 87% yield.
EXAMPLE 7 Recrystallisation of crude cladribine
- [0058]
A mixture of crude cladribine (1.94 Kg, >96.0% HPLC purity), MeOH (7.77 Kg, 5 P), process purified water (9.67 Kg, 5 P) and 25% sodium methoxide in MeOH (32 g, 0.15 mol) were stirred and heated under reflux until the solids dissolved. The solution was cooled to about 70°C and treated with activated carbon (0.16 Kg) and celite for 1 hour at about 70°C, rinsed with a mixture of preheated MeOH and process purified water (W/W = 1:1.25, 1.75 Kg). The filtrate was cooled to about 45°C and maintained at this temperature for 1 hours, and then cooled to about 15°C and agitated at this temperature for 2 hours. The solids were filtered and washed with MeOH (1.0 Kg, 0.7 P) three times and were then dried in vacuo at 60°C for 4 hours giving API grade cladribine (1.5 Kg, 5.2 mol) in 80% yield with 99.84% HPLC purity.
EXAMPLE 8 Recrystallisation of crude cladribine
- [0059]
A mixture of crude cladribine (1.92 Kg, >95.7% HPLC purity), MeOH (7.76 Kg, 5 P), process purified water (9.67 Kg, 5 P) and 25% sodium methoxide in MeOH (36 g, 0.17 mol) were stirred and heated under reflux until the solids dissolved. The solution was cooled to about 70°C and treated with activated carbon (0.15 Kg) and celite for 1 hour at about 70°C, rinsed with a mixture of preheated MeOH and process purified water (1:1.25, 1.74 Kg). The filtrate was cooled to about 45°C and maintained at this temperature for 1 hour, and then cooled to about 15°C and agitated at this temperature for 2 hours. The solids were filtered and washed with MeOH (1.0 Kg, 0.7 P) three times and were giving damp cladribine (1.83 Kg). A mixture of this cladribine (1.83 Kg), MeOH (7.33 Kg, 5 P) and process purified water (9.11 Kg, 5 P) were stirred and heated under reflux until the solids dissolved and was then cooled to about 45°C and maintained at this temperature for 1 hours. The slurry was further cooled to about 15°C and agitated at this temperature for 2 hours. The solids were filtered and washed with MeOH (0.9 Kg, 0.7 P) three times and were then dried in vacuo at 60°C for 4 hours giving API grade cladribine (1.38 Kg, 4.8 mol) in 75% yield with 99.86% HPLC purity.
SYN

Cladribine can be got from 2-Deoxy-D-ribose. The detail is as follows:

SYN
https://www.tandfonline.com/doi/abs/10.1080/15257770.2015.1071848?journalCode=lncn20
clip
FDA approves new oral treatment for multiple sclerosis, Mavenclad (cladribine)
The U.S. Food and Drug Administration today approved Mavenclad (cladribine) tablets to treat
relapsing forms of multiple sclerosis (MS) in adults, to include relapsing-remitting disease and active secondary progressive disease. Mavenclad is not recommended for MS patients with clinically isolated syndrome. Because of its safety profile, the use of Mavenclad is generally recommended for patients who have had an inadequate response to…
March 29, 2019
Release
The U.S. Food and Drug Administration today approved Mavenclad (cladribine) tablets to treat relapsing forms of multiple sclerosis (MS) in adults, to include relapsing-remitting disease and active secondary progressive disease. Mavenclad is not recommended for MS patients with clinically isolated syndrome. Because of its safety profile, the use of Mavenclad is generally recommended for patients who have had an inadequate response to, or are unable to tolerate, an alternate drug indicated for the treatment of MS.
“We are committed to supporting the development of safe and effective treatments for patients with multiple sclerosis,” said Billy Dunn, M.D., director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research. “The approval of Mavenclad represents an additional option for patients who have tried another treatment without success.”
MS is a chronic, inflammatory, autoimmune disease of the central nervous system that disrupts communications between the brain and other parts of the body. Most people experience their first symptoms of MS between the ages of 20 and 40. MS is among the most common causes of neurological disability in young adults and occurs more frequently in women than in men.
For most people, MS starts with a relapsing-remitting course, in which episodes of worsening function (relapses) are followed by recovery periods (remissions). These remissions may not be complete and may leave patients with some degree of residual disability. Many, but not all, patients with MS experience some degree of persistent disability that gradually worsens over time. In some patients, disability may progress independent of relapses, a process termed secondary progressive multiple sclerosis (SPMS). In the first few years of this process, many patients continue to experience relapses, a phase of the disease described as active SPMS. Active SPMS is one of the relapsing forms of MS, and drugs approved for the treatment of relapsing forms of MS can be used to treat active SPMS.
The efficacy of Mavenclad was shown in a clinical trial in 1,326 patients with relapsing forms of MS who had least one relapse in the previous 12 months. Mavenclad significantly decreased the number of relapses experienced by these patients compared to placebo. Mavenclad also reduced the progression of disability compared to placebo.
Mavenclad must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks. Mavenclad has a Boxed Warning for an increased risk of malignancy and fetal harm. Mavenclad is not to be used in patients with current malignancy. In patients with prior malignancy or with increased risk of malignancy, health care professionals should evaluate the benefits and risks of the use of Mavenclad on an individual patient basis. Health care professionals should follow standard cancer screening guidelines in patients treated with Mavenclad. The drug should not be used in pregnant women and in women and men of reproductive potential who do not plan to use effective contraception during treatment and for six months after the course of therapy because of the potential for fetal harm. Mavenclad should be stopped if the patient becomes pregnant.
Other warnings include the risk of decreased lymphocyte (white blood cell) counts; lymphocyte counts should be monitored before, during and after treatment. Mavenclad may increase the risk of infections; health care professionals should screen patients for infections and treatment with Mavenclad should be delayed if necessary. Mavenclad may cause hematologic toxicity and bone marrow suppression so health care professionals should measure a patient’s complete blood counts before, during and after therapy. The drug has been associated with graft-versus-host-disease following blood transfusions with non-irradiated blood. Mavenclad may cause liver injury and treatment should be interrupted or discontinued, as appropriate, if clinically significant liver injury is suspected.
The most common adverse reactions reported by patients receiving Mavenclad in the clinical trials include upper respiratory tract infections, headache and decreased lymphocyte counts.
The FDA granted approval of Mavenclad to EMD Serono, Inc.
References
- ^ Drugs.com International trade names for Cladribine Page accessed Jan 14, 2015
- ^ Jump up to:a b c d “PRODUCT INFORMATION LITAK© 2 mg/mL solution for injection” (PDF). TGA eBusiness Services. St Leonards, Australia: Orphan Australia Pty. Ltd. 10 May 2010. Retrieved 27 November 2014.
- ^ Liliemark, Jan (1997). “The Clinical Pharmacokinetics of Cladribine”. Clinical Pharmacokinetics. 32 (2): 120–131. doi:10.2165/00003088-199732020-00003. PMID 9068927.
- ^ Jump up to:a b “European Medicines Agency – – Litak”. http://www.ema.europa.eu.
- ^ Jump up to:a b “Leustat Injection. – Summary of Product Characteristics (SPC) – (eMC)”. http://www.medicines.org.uk.
- ^ Leist, TP; Weissert, R (2010). “Cladribine: mode of action and implications for treatment of multiple sclerosis”. Clinical Neuropharmacology. 34 (1): 28–35. doi:10.1097/wnf.0b013e318204cd90. PMID 21242742.
- ^ Jump up to:a b c d Cladribine label, last updated July 2012. Page accessed January 14, 2015
- ^ https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm634837.htm
- ^ Histiocytosis Association Erdheim-Chester Disease Page accessed Aug 20, 2016
- ^ Aricò M (2016). “Langerhans cell histiocytosis in children: from the bench to bedside for an updated therapy”. Br J Haematol. 173 (5): 663–70. doi:10.1111/bjh.13955. PMID 26913480.
The combination of cytarabine and cladribine is the current standard for second-line therapy of refractory cases with vital organ dysfunction.
- ^ Jump up to:a b c Giovannoni, G; Comi, G; Cook, S; Rammohan, K; Rieckmann, P; Soelberg Sørensen, P; Vermersch, P; Chang, P; Hamlett, A; Musch, B; Greenberg, SJ; CLARITY Study, Group. (4 February 2010). “A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis”. The New England Journal of Medicine. 362 (5): 416–26. doi:10.1056/NEJMoa0902533. PMID 20089960.
- ^ Johnston, JB (June 2011). “Mechanism of action of pentostatin and cladribine in hairy cell leukemia”. Leukemia & Lymphoma. 52 Suppl 2: 43–5. doi:10.3109/10428194.2011.570394. PMID 21463108.
- ^ Beutler, E; Piro, LD; Saven, A; Kay, AC; McMillan, R; Longmire, R; Carrera, CJ; Morin, P; Carson, DA (1991). “2-Chlorodeoxyadenosine (2-CdA): A Potent Chemotherapeutic and Immunosuppressive Nucleoside”. Leukemia & Lymphoma. 5 (1): 1–8. doi:10.3109/10428199109068099. PMID 27463204.
- ^ Baker, D; Marta, M; Pryce, G; Giovannoni, G; Schmierer, K (February 2017). “Memory B Cells are Major Targets for Effective Immunotherapy in Relapsing Multiple Sclerosis”. EBioMedicine. 16: 41–50. doi:10.1016/j.ebiom.2017.01.042. PMC 5474520. PMID 28161400.
- ^ Baker, D; Herrod, SS; Alvarez-Gonzalez, C; Zalewski, L; Albor, C; Schmierer, K (July 2017). “Both cladribine and alemtuzumab may effect MS via B-cell depletion”. Neurology: Neuroimmunology & Neuroinflammation. 4 (4): e360. doi:10.1212/NXI.0000000000000360. PMC 5459792. PMID 28626781.
- ^ Jump up to:a b Ceronie, B; Jacobs, BM; Baker, D; Dubuisson, N; Mao, Z; Ammoscato, F; Lock, H; Longhurst, HJ; Giovannoni, G; Schmierer, K (May 2018). “Cladribine treatment of multiple sclerosis is associated with depletion of memory B cells”. Journal of Neurology. 265 (5): 1199–1209. doi:10.1007/s00415-018-8830-y. PMC 5937883. PMID 29550884.
- ^ Jump up to:a b Marshall A. Lichtman Biographical Memoir: Ernest Beutler 1928–2008 National Academy of Sciences, 2012
- ^ Staff, The Pink Sheet Mar 8, 1993 Ortho Biotech’s Leustatin For Hairy Cell Leukemia
- ^ Jump up to:a b EMA 2004 Litak EMA package: Scientific Discussion
- ^ EMA 2004 Litak: Background Information one the Procedure
- ^ Eric Sauter and Mika Ono for Scripps News and Views. Vol 9. Issue 18. June 1, 2009 A Potential New MS Treatment’s Long and Winding Road
- ^ Tortorella C, Rovaris M, Filippi M (2001). “Cladribine. Ortho Biotech Inc”. Curr Opin Investig Drugs. 2 (12): 1751–6. PMID 11892941.
- ^ Jump up to:a b Carey Sargent for Dow Jones Newswires in the Wall Street Journal. Oct. 31, 2002 Serono Purchases Rights To Experimental MS Drug
- ^ Reuters. Dec 4, 2000. Ivax to Develop Cladribine for Multiple Sclerosis
- ^ Jennifer Bayot for the New York Times. July 26, 2005 Teva to Acquire Ivax, Another Maker of Generic Drugs
- ^ Teva Press Release, 2006. Teva Completes Acquisition of Ivax
- ^ Staff, First Word Pharma. Sept 21, 2006 Merck KGaA to acquire Serono
- ^ Jump up to:a b c EMA. 2011 Withdrawal Assessment Report for Movectro Procedure No. EMEA/H/C/001197
- ^ Jump up to:a b John Gever for MedPage Today June 22, 2011 06.22.2011 0 Merck KGaA Throws in Towel on Cladribine for MS
- ^ Jump up to:a b John Carroll for FierceBiotech Sep 11, 2015 Four years after a transatlantic slapdown, Merck KGaA will once again seek cladribine OK
- ^ Connolly, Allison (24 April 2012). “Merck KGaA to Close Merck Serono Site in Geneva, Cut Jobs”. Bloomberg.
- ^ Pakpoor, J; et al. (December 2015). “No evidence for higher risk of cancer in patients with multiple sclerosis taking cladribine”. Neurology: Neuroimmunology & Neuroinflammation. 2 (6): e158. doi:10.1212/nxi.0000000000000158. PMC 4592538. PMID 26468472.
- ^ Press release
- ^ Merck. “Cladribine Tablets Receives Positive CHMP Opinion for Treatment of Relapsing Forms of Multiple Sclerosis”. http://www.prnewswire.co.uk. Retrieved 2017-08-22.
- ^ Cladribine approved in Europe, Press Release
- ^ Jump up to:a b Giovannoni, G; Soelberg Sorensen, P; Cook, S; Rammohan, K; Rieckmann, P; Comi, G; Dangond, F; Adeniji, AK; Vermersch, P (1 August 2017). “Safety and efficacy of cladribine tablets in patients with relapsing-remitting multiple sclerosis: Results from the randomized extension trial of the CLARITY study”. Multiple Sclerosis (Houndmills, Basingstoke, England): 1352458517727603. doi:10.1177/1352458517727603. PMID 28870107.
- ^ “Sustained Efficacy – Merck Neurology”. Merck Neurology. Retrieved 28 September2018.
- ^ Guarnera, C; Bramanti, P; Mazzon, E (2017). “Alemtuzumab: a review of efficacy and risks in the treatment of relapsing remitting multiple sclerosis”. Therapeutics and Clinical Risk Management. 13: 871–879. doi:10.2147/TCRM.S134398. PMC 5522829. PMID 28761351.
- ^ Baker, D; Herrod, SS; Alvarez-Gonzalez, C; Giovannoni, G; Schmierer, K (1 August 2017). “Interpreting Lymphocyte Reconstitution Data From the Pivotal Phase 3 Trials of Alemtuzumab”. JAMA Neurology. 74 (8): 961–969. doi:10.1001/jamaneurol.2017.0676. PMC 5710323. PMID 28604916.
- ^ “Cladribine tablets for treating relapsing–remitting multiple sclerosis”. National Institute for Clinical Excellence. Retrieved 23 September 2018.
- ^ Hasanali, Zainul S.; Saroya, Bikramajit Singh; Stuart, August; Shimko, Sara; Evans, Juanita; Shah, Mithun Vinod; Sharma, Kamal; Leshchenko, Violetta V.; Parekh, Samir (24 June 2015). “Epigenetic therapy overcomes treatment resistance in T cell prolymphocytic leukemia”. Science Translational Medicine. 7 (293): 293ra102. doi:10.1126/scitranslmed.aaa5079. ISSN 1946-6234. PMC 4807901. PMID 26109102.
 |
|
| Clinical data | |
|---|---|
| Trade names | Leustatin, others[1] |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a693015 |
| License data | |
| Pregnancy category |
|
| Routes of administration |
Intravenous, subcutaneous(liquid) |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | 100% (i.v.); 37 to 51% (orally)[3] |
| Protein binding | 25% (range 5-50%)[2] |
| Metabolism | Mostly via intracellularkinases; 15-18% is excreted unchanged[2] |
| Elimination half-life | Terminal elimination half-life: Approximately 10 hours after both intravenous infusion an subcutaneous bolus injection[2] |
| Excretion | Urinary[2] |
| Identifiers | |
| CAS Number | |
| PubChemCID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| ECHA InfoCard | 100.164.726 |
| Chemical and physical data | |
| Formula | C10H12ClN5O3 |
| Molar mass | 285.687 g/mol g·mol−1 |
| 3D model (JSmol) | |
Cladribine
CAS Registry Number: 4291-63-8
CAS Name: 2-Chloro-2¢-deoxyadenosine
Additional Names: 2-chloro-6-amino-9-(2-deoxy-b-D-erythro-pentofuranosyl)purine; 2-chlorodeoxyadenosine; 2-CdA; CldAdo
Manufacturers’ Codes: NSC-105014-F
Trademarks: Leustatin (Ortho Biotech)
Molecular Formula: C10H12ClN5O3
Molecular Weight: 285.69
Percent Composition: C 42.04%, H 4.23%, Cl 12.41%, N 24.51%, O 16.80%
Literature References: Substituted purine nucleoside with antileukemic activity. Prepn as intermediate in synthesis of 2-deoxynucleosides: H. Venner, Ber. 93, 140 (1960); M. Ikehara, H. Tada, J. Am. Chem. Soc. 85, 2344 (1963); eidem, ibid. 87, 606 (1965). Synthesis and biological activity: L. F. Christensen et al., J. Med. Chem. 15, 735 (1972). Stereospecific synthesis: Z. Kazimierczuk et al., J. Am. Chem. Soc. 106, 6379 (1984); R. K. Robins, G. R. Revankar, EP 173059; eidem, US 4760137 (1986, 1988 both to Brigham Young Univ.). Specific toxicity to lymphocytes: D. A. Carson et al., Proc. Natl. Acad. Sci. USA 77, 6865 (1980); eidem, Blood 62, 737 (1983). Mechanism of action: S. Seto et al., J. Clin. Invest. 75, 377 (1985). Clinical evaluation in chronic lymphocytic leukemia: L. D. Piro et al., Blood 72, 1069 (1988); in hairy cell leukemia: eidem, N. Engl. J. Med. 322, 1117 (1990).
Properties: Crystals from water, softens at 210-215°, solidifies and turns brown (Christensen). Also reported as crystals from ethanol, mp 220° (softens), resolidifies, turns brown and does not melt below 300° (Kazimierczuk). [a]D25 -18.8° (c = 1 in DMF). uv max in 0.1N NaOH: 265 nm; in 0.1N HCl: 265 nm.
Melting point: mp 220° (softens), resolidifies, turns brown and does not melt below 300°
Optical Rotation: [a]D25 -18.8° (c = 1 in DMF)
Absorption maximum: uv max in 0.1N NaOH: 265 nm; in 0.1N HCl: 265 nm
Therap-Cat: Antineoplastic.
Keywords: Antineoplastic; Antimetabolites; Purine Analogs.
////////////fda 2019, Mavenclad, cladribine, multiple sclerosis, EMD Serono, クラドリビン , Leustatin, クラドリビン , orphan drug designation
NC1=C2N=CN([C@H]3C[C@H](O)[C@@H](CO)O3)C2=NC(Cl)=N1

{kind=link}